User login
Preventing Wrong-Patient Electronic Orders in the Emergency Department
From SBH Health System, Bronx, NY.
Abstract
- Objective: To decrease the number of near-miss wrong-patient orders in a computerized physician order entry (CPOE) system.
- Methods: A CPOE alert was built that prompted the ordering clinician to reaffirm the identity of the patient by entering the patient’s initials and year of birth prior to placing an order. We used a retract and reorder tool to measure the frequency of near-miss wrong-patient order errors before and after implementation of the alert.
- Results: The ID reentry function decreased near-miss wrong-patient orders in the ED by 35% during the 8-week pilot period. The system was also successful in helping to decrease the percentage of all CPOE near-miss events by 49%.
- Conclusion: An alert that requires the prescriber to enter the patient’s initials and birth year is effective in decreasing wrong-patient orders in the CPOE system.
Key words: CPOE, near miss, patient safety, medical errors, wrong-patient errors.
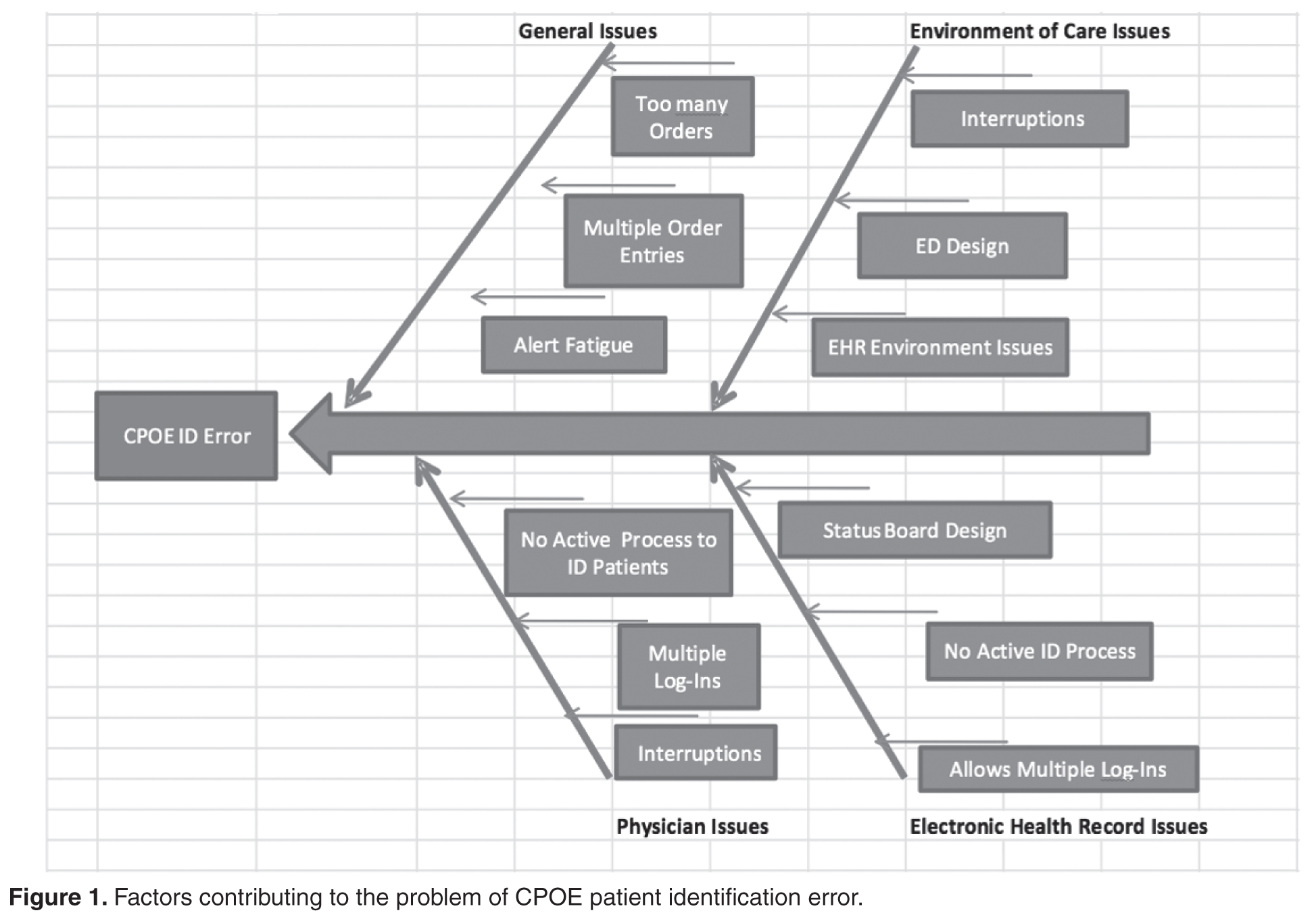
Computerized provider order entry (CPOE) systems are commonly used to place orders. CPOE has been shown to reduce errors [1–4]. However, medication errors also can be caused or exacerbated by the CPOE system [5–7]. One type of error that can occur is placing orders on the wrong patient [8]. Wrong-patient CPOE errors can lead to significant morbidity and mortality [8–11]. To reduce wrong-patient CPOE errors in our health system, we developed an alert that required the ordering clinician to verify the identity of the patient. In this paper, we describe our project and outcomes attained.
Methods
Setting
SBH Health System is a not for profit health system located in Bronx, New York. The SBH Health System also has academic affiliations, and AOA and ACGME residency and fellowship programs. St. Barnabas Hospital, SBH Health System’s acute care facility, is a safety net hospital, Level 1 trauma center, primary stroke center, and STEMI receiving center. St. Barnabas Hospital has 422 licensed beds and had a total of 91,476 emergency department visits in 2015. The electronic health record in use at the time of the project was Allscripts 6.1. The Allscripts product, including its CPOE functions, has been in use in the SBH emergency department (ED) since 2011.
Review of Current Process
A team of multidisciplinary stakeholders was assembled comprised of hospital senior leadership, ED leadership, and front-line staff. Representatives from all disciplines involved in the CPOE process were invited, including nursing, pharmacy, radiology, clinical laboratory, and information technology.
Next, we assessed our current error rate using a “retract and reorder” tool, which flags orders that have been placed for one patient, then erased and added to another patient’s file by the same clinician within a 10-minute time frame [8]. This tool, developed by Adelman et al, picks up near-miss errors, self-caught by the provider before causing harm [8]. Safety research has demonstrated that near-miss errors share the same causal pathway; therefore, measuring and preventing near-miss wrong-patient errors should reduce related errors that reach the patient.
For the period October–December 2014, we tabulated 231 near-miss wrong-patient orders that occured throughout the health system, of which 37% occurred in the ED. This translated to about 1 near-miss event per day in the ED. Given this data, the ED was the location for our quality improvement project.
Intervention

Outcomes
After a beta testing period of 1 week, the system was implemented on 3 November 2015. To assess the effectiveness of the alert system to prevent ordering errors, we used the retract and reorder tool to measure the rate of wrong-patient order entries for the 8-week period November–December 2015 and compared this with the preimplementation rate. Prior to the intervention, the average number of wrong patient order entries in the ED was 6.125 events per week. After implementation, the average number decreased to 4 events per week, a 35% decrease, and the proportion of near-miss ID errors in the ED relative to all such errors within the health system decreased from 37% to 19%.
Discussion
The original aim of the project was to decrease “wrong patient, right order” near-miss events by 30% in 3 months in the ED using an order-based patient ID reentry function. The goal was rapid improvement using a hard-wired EHR process, which is why a 3-month time frame was chosen. During our 8-week project, we surpassed this goal, documenting a 35% decrease in near-miss wrong-patient orders in the ED. This rate was similar to that achieved by Adelman et al [8] and Green et al [10]. Adelman et al found a 41% error reduction, while Green et al found a short-term 30% reduction in CPOE wrong-patient orders utilizing a 2.5-second mandatory delay before continuing the order entry for the purposes of patient verification.
Resident and attending staff conveyed to us anecdotally during both beta testing and implementation that the ID reentry function made them aware of incorrect patient selection even before entering the required initials and birth year. They then cancelled the order session on the wrong patient and chose the correct patient. This is consistent with the findings of Green’s study, which noted that ED practitioners backed out of appropriately 1 in 200 order entry sessions due to wrong patient selection [10].
We also assessed the additional time added to each order entry session. Initially, using observational data, the CPOE ID reentry function added 6.2 seconds to each order entry session. However, providers that were more familiar with the system took an average of 4.0 seconds. While this added time per order entry session does not seem like much of an issue or delay, in a busy 12-hour shift in the ED it could be seen as significant. Adelman reported 6.6 seconds additional time required in for the ID reentry function used in his study [8], while Green’s study was designed using a 2.5-second mandatory delay before users could close the verification dialogue box [10].
The biggest challenges in implementing our project were unforeseen IT issues. The “go-live” date for ICD-10 was the same as the date we were to start the ID reentry requirement. IT personnel were needed to help in the EHR ICD-10 development and support, which delayed our start date. Additionally, other IT issues were identified. For example, the initial implementation of this project was to begin in the ED involving active ED patients only. At the project’s onset, the ID reentry function erroneously became active in all hospital locations. To fix this error, the entire double ID system alert, including the ED location, had to be removed and adjusted.
In addition to the above challenges, the team discovered errors that needed to be addressed during beta testing. For example, some clinicians would enter an order but no alert asking for the identifying data appeared. The order was entered and completed without the use of the double ID. Once discovered, IT was able to identify and correct the error. Beta testing also revealed an error in the system where providers who incorrectly identified a patient were “locking-out” of the CPOE system for that particular patient during the patient’s entire encounter. This issue was also quickly identified and resolved.
Despite the effectiveness of this system in reducing the rate of near-miss wrong-patient orders in the ED, errors still occur. It is possible that providers are entering the patient’s initials and year of birth without carefully verifying the patient’s identity [9].The CPOE double ID system alert is about three-quarters the size of the monitor screen. Thus, the clinician is able to verify the patient’s initials and year of birth using the patient’s header on the screen behind the patient identification alert. If the provider simply types the initials and year of birth on the patient’s header, then an identification error can occur.
More work is needed to decrease CPOE-related patient identification errors. Possible improvements may include single sign-ons and a no-interruption policy when writing orders. During our investigation, it was found that some clinicians would have multiple EHR sign-on sessions open at one computer terminal. These multiple EHR sign-on sessions were sometimes the root cause of a wrong patient error. With multiple sign-on sessions open, clinicians could toggle back and forth between patients on the same computer terminal and mistakenly complete an order on the wrong patient.
No-interruption zones and policies have been proven to be an effective way of decreasing interruptions and enhancing safety during medication preparation [13,14]. Utilization of no-interruption zones for CPOE may also be effective. Potentially, the EHR background color could change when a clinician selects the “enter order” tab within the EHR. The new background color would signify to those around the clinician that he/she is not to be interrupted during that time.
After the success of this initial quality improvement project in the ED, the intensive care unit has been added as a location for the CPOE double identification system. The data and results for this phase of the project are being tabulated and seem promising. In addition, SBH Health System is exploring single sign-on software to both help clinicians provide service and enhance patient safety.
Corresponding author: Daniel Lombardi, DO, 4422 Third Ave., Bronx, NY 10457.
Financial disclosures: None.
1. Bates, DW, Leape L, Cullen DJ, et al. Effect of computerized physician order entry and a team intervention on prevention of serious medication errors. JAMA 1998;280:1311–16.
2. Bates, DW, Teich JM, Lee J, et al. The impact of computerized physician order entry on medication error prevention. J Am Med Inform Assoc 1999;6:313–21.
3. Kaushal R, Shojania KG, Bates DW. Effects of computerized physician order entry and clinical decision support systems on medication safety: a systematic review. Arch Intern Med 2003;163:1409–16.
4. Reckmann, MH, Westbrook JI, Koh Y, et al. Does computeized provider order entry reduce prescribing errors for hospital inpatients? A systematic review. J Am Med Inform Assoc 2009;16:613–23.
5. Koppel R, Metlay JP, Cohen A, et al. Role of computerized physician order entry systems in facilitating medication errors. JAMA 2005;293:1197–203.
6. Broder C. Study: CPOE can increase risk of medication errors. Health IT News. March 9, 2005.
7. Schiff GD, Amato MG, Eguale T, et al. Computerised physician order entry-related medication errors: analysis of reported errors and vulnerability testing of current systems. BMJ Qual Saf 2015;24:264–71.
8. Adelman, JS, Kalkut GE, Schechter CB, et al. Understanding and preventing wrong-patient electronic orders: a randomized controlled trial. JAM Med Inform Assoc 2013;20:305–10.
9. Yang A, Grissinger M. Pennsylvania Patient Safety Authority. Wrong-patient medication errors: an analysis of event reports in Pennsylvania and strategies for prevention. PA Patient Saf Advis 2013 June;10:41–9.
10. Green RA, Hripcsak G, Salmasian H, et al. Intercepting wrong-patient orders in a computerized provider order entry system. Ann Emerg Med 2015;65:679–86.
11. Institute for Safe Medication Practices. Oops, sorry, wrong patient! A patient verification process is needed everywhere, not just at the bedside. 10 Mar 2011. Accessed at www.ismp.org/Newsletters/acutecare/articles/20110310.asp.
12. Anthony K, Wiencek C, Bauer C, et al. No interruptions please: impact of a No Interruption Zone on medication safety in intensive care units. Crit Care Nurse 2010;30:21–9.
13. Institute for Safe Medication Practices. Side tracks on the safety express. interruptions lead to errors and unfinished…wait, what was i doing? 29 Nov 2012. Accessed at www.ismp.org/Newsletters/acutecare/showarticle.aspx?id=37.
From SBH Health System, Bronx, NY.
Abstract
- Objective: To decrease the number of near-miss wrong-patient orders in a computerized physician order entry (CPOE) system.
- Methods: A CPOE alert was built that prompted the ordering clinician to reaffirm the identity of the patient by entering the patient’s initials and year of birth prior to placing an order. We used a retract and reorder tool to measure the frequency of near-miss wrong-patient order errors before and after implementation of the alert.
- Results: The ID reentry function decreased near-miss wrong-patient orders in the ED by 35% during the 8-week pilot period. The system was also successful in helping to decrease the percentage of all CPOE near-miss events by 49%.
- Conclusion: An alert that requires the prescriber to enter the patient’s initials and birth year is effective in decreasing wrong-patient orders in the CPOE system.
Key words: CPOE, near miss, patient safety, medical errors, wrong-patient errors.
Computerized provider order entry (CPOE) systems are commonly used to place orders. CPOE has been shown to reduce errors [1–4]. However, medication errors also can be caused or exacerbated by the CPOE system [5–7]. One type of error that can occur is placing orders on the wrong patient [8]. Wrong-patient CPOE errors can lead to significant morbidity and mortality [8–11]. To reduce wrong-patient CPOE errors in our health system, we developed an alert that required the ordering clinician to verify the identity of the patient. In this paper, we describe our project and outcomes attained.
Methods
Setting
SBH Health System is a not for profit health system located in Bronx, New York. The SBH Health System also has academic affiliations, and AOA and ACGME residency and fellowship programs. St. Barnabas Hospital, SBH Health System’s acute care facility, is a safety net hospital, Level 1 trauma center, primary stroke center, and STEMI receiving center. St. Barnabas Hospital has 422 licensed beds and had a total of 91,476 emergency department visits in 2015. The electronic health record in use at the time of the project was Allscripts 6.1. The Allscripts product, including its CPOE functions, has been in use in the SBH emergency department (ED) since 2011.
Review of Current Process
A team of multidisciplinary stakeholders was assembled comprised of hospital senior leadership, ED leadership, and front-line staff. Representatives from all disciplines involved in the CPOE process were invited, including nursing, pharmacy, radiology, clinical laboratory, and information technology.
Next, we assessed our current error rate using a “retract and reorder” tool, which flags orders that have been placed for one patient, then erased and added to another patient’s file by the same clinician within a 10-minute time frame [8]. This tool, developed by Adelman et al, picks up near-miss errors, self-caught by the provider before causing harm [8]. Safety research has demonstrated that near-miss errors share the same causal pathway; therefore, measuring and preventing near-miss wrong-patient errors should reduce related errors that reach the patient.
For the period October–December 2014, we tabulated 231 near-miss wrong-patient orders that occured throughout the health system, of which 37% occurred in the ED. This translated to about 1 near-miss event per day in the ED. Given this data, the ED was the location for our quality improvement project.
Intervention
Outcomes
After a beta testing period of 1 week, the system was implemented on 3 November 2015. To assess the effectiveness of the alert system to prevent ordering errors, we used the retract and reorder tool to measure the rate of wrong-patient order entries for the 8-week period November–December 2015 and compared this with the preimplementation rate. Prior to the intervention, the average number of wrong patient order entries in the ED was 6.125 events per week. After implementation, the average number decreased to 4 events per week, a 35% decrease, and the proportion of near-miss ID errors in the ED relative to all such errors within the health system decreased from 37% to 19%.
Discussion
The original aim of the project was to decrease “wrong patient, right order” near-miss events by 30% in 3 months in the ED using an order-based patient ID reentry function. The goal was rapid improvement using a hard-wired EHR process, which is why a 3-month time frame was chosen. During our 8-week project, we surpassed this goal, documenting a 35% decrease in near-miss wrong-patient orders in the ED. This rate was similar to that achieved by Adelman et al [8] and Green et al [10]. Adelman et al found a 41% error reduction, while Green et al found a short-term 30% reduction in CPOE wrong-patient orders utilizing a 2.5-second mandatory delay before continuing the order entry for the purposes of patient verification.
Resident and attending staff conveyed to us anecdotally during both beta testing and implementation that the ID reentry function made them aware of incorrect patient selection even before entering the required initials and birth year. They then cancelled the order session on the wrong patient and chose the correct patient. This is consistent with the findings of Green’s study, which noted that ED practitioners backed out of appropriately 1 in 200 order entry sessions due to wrong patient selection [10].
We also assessed the additional time added to each order entry session. Initially, using observational data, the CPOE ID reentry function added 6.2 seconds to each order entry session. However, providers that were more familiar with the system took an average of 4.0 seconds. While this added time per order entry session does not seem like much of an issue or delay, in a busy 12-hour shift in the ED it could be seen as significant. Adelman reported 6.6 seconds additional time required in for the ID reentry function used in his study [8], while Green’s study was designed using a 2.5-second mandatory delay before users could close the verification dialogue box [10].
The biggest challenges in implementing our project were unforeseen IT issues. The “go-live” date for ICD-10 was the same as the date we were to start the ID reentry requirement. IT personnel were needed to help in the EHR ICD-10 development and support, which delayed our start date. Additionally, other IT issues were identified. For example, the initial implementation of this project was to begin in the ED involving active ED patients only. At the project’s onset, the ID reentry function erroneously became active in all hospital locations. To fix this error, the entire double ID system alert, including the ED location, had to be removed and adjusted.
In addition to the above challenges, the team discovered errors that needed to be addressed during beta testing. For example, some clinicians would enter an order but no alert asking for the identifying data appeared. The order was entered and completed without the use of the double ID. Once discovered, IT was able to identify and correct the error. Beta testing also revealed an error in the system where providers who incorrectly identified a patient were “locking-out” of the CPOE system for that particular patient during the patient’s entire encounter. This issue was also quickly identified and resolved.
Despite the effectiveness of this system in reducing the rate of near-miss wrong-patient orders in the ED, errors still occur. It is possible that providers are entering the patient’s initials and year of birth without carefully verifying the patient’s identity [9].The CPOE double ID system alert is about three-quarters the size of the monitor screen. Thus, the clinician is able to verify the patient’s initials and year of birth using the patient’s header on the screen behind the patient identification alert. If the provider simply types the initials and year of birth on the patient’s header, then an identification error can occur.
More work is needed to decrease CPOE-related patient identification errors. Possible improvements may include single sign-ons and a no-interruption policy when writing orders. During our investigation, it was found that some clinicians would have multiple EHR sign-on sessions open at one computer terminal. These multiple EHR sign-on sessions were sometimes the root cause of a wrong patient error. With multiple sign-on sessions open, clinicians could toggle back and forth between patients on the same computer terminal and mistakenly complete an order on the wrong patient.
No-interruption zones and policies have been proven to be an effective way of decreasing interruptions and enhancing safety during medication preparation [13,14]. Utilization of no-interruption zones for CPOE may also be effective. Potentially, the EHR background color could change when a clinician selects the “enter order” tab within the EHR. The new background color would signify to those around the clinician that he/she is not to be interrupted during that time.
After the success of this initial quality improvement project in the ED, the intensive care unit has been added as a location for the CPOE double identification system. The data and results for this phase of the project are being tabulated and seem promising. In addition, SBH Health System is exploring single sign-on software to both help clinicians provide service and enhance patient safety.
Corresponding author: Daniel Lombardi, DO, 4422 Third Ave., Bronx, NY 10457.
Financial disclosures: None.
From SBH Health System, Bronx, NY.
Abstract
- Objective: To decrease the number of near-miss wrong-patient orders in a computerized physician order entry (CPOE) system.
- Methods: A CPOE alert was built that prompted the ordering clinician to reaffirm the identity of the patient by entering the patient’s initials and year of birth prior to placing an order. We used a retract and reorder tool to measure the frequency of near-miss wrong-patient order errors before and after implementation of the alert.
- Results: The ID reentry function decreased near-miss wrong-patient orders in the ED by 35% during the 8-week pilot period. The system was also successful in helping to decrease the percentage of all CPOE near-miss events by 49%.
- Conclusion: An alert that requires the prescriber to enter the patient’s initials and birth year is effective in decreasing wrong-patient orders in the CPOE system.
Key words: CPOE, near miss, patient safety, medical errors, wrong-patient errors.
Computerized provider order entry (CPOE) systems are commonly used to place orders. CPOE has been shown to reduce errors [1–4]. However, medication errors also can be caused or exacerbated by the CPOE system [5–7]. One type of error that can occur is placing orders on the wrong patient [8]. Wrong-patient CPOE errors can lead to significant morbidity and mortality [8–11]. To reduce wrong-patient CPOE errors in our health system, we developed an alert that required the ordering clinician to verify the identity of the patient. In this paper, we describe our project and outcomes attained.
Methods
Setting
SBH Health System is a not for profit health system located in Bronx, New York. The SBH Health System also has academic affiliations, and AOA and ACGME residency and fellowship programs. St. Barnabas Hospital, SBH Health System’s acute care facility, is a safety net hospital, Level 1 trauma center, primary stroke center, and STEMI receiving center. St. Barnabas Hospital has 422 licensed beds and had a total of 91,476 emergency department visits in 2015. The electronic health record in use at the time of the project was Allscripts 6.1. The Allscripts product, including its CPOE functions, has been in use in the SBH emergency department (ED) since 2011.
Review of Current Process
A team of multidisciplinary stakeholders was assembled comprised of hospital senior leadership, ED leadership, and front-line staff. Representatives from all disciplines involved in the CPOE process were invited, including nursing, pharmacy, radiology, clinical laboratory, and information technology.
Next, we assessed our current error rate using a “retract and reorder” tool, which flags orders that have been placed for one patient, then erased and added to another patient’s file by the same clinician within a 10-minute time frame [8]. This tool, developed by Adelman et al, picks up near-miss errors, self-caught by the provider before causing harm [8]. Safety research has demonstrated that near-miss errors share the same causal pathway; therefore, measuring and preventing near-miss wrong-patient errors should reduce related errors that reach the patient.
For the period October–December 2014, we tabulated 231 near-miss wrong-patient orders that occured throughout the health system, of which 37% occurred in the ED. This translated to about 1 near-miss event per day in the ED. Given this data, the ED was the location for our quality improvement project.
Intervention
Outcomes
After a beta testing period of 1 week, the system was implemented on 3 November 2015. To assess the effectiveness of the alert system to prevent ordering errors, we used the retract and reorder tool to measure the rate of wrong-patient order entries for the 8-week period November–December 2015 and compared this with the preimplementation rate. Prior to the intervention, the average number of wrong patient order entries in the ED was 6.125 events per week. After implementation, the average number decreased to 4 events per week, a 35% decrease, and the proportion of near-miss ID errors in the ED relative to all such errors within the health system decreased from 37% to 19%.
Discussion
The original aim of the project was to decrease “wrong patient, right order” near-miss events by 30% in 3 months in the ED using an order-based patient ID reentry function. The goal was rapid improvement using a hard-wired EHR process, which is why a 3-month time frame was chosen. During our 8-week project, we surpassed this goal, documenting a 35% decrease in near-miss wrong-patient orders in the ED. This rate was similar to that achieved by Adelman et al [8] and Green et al [10]. Adelman et al found a 41% error reduction, while Green et al found a short-term 30% reduction in CPOE wrong-patient orders utilizing a 2.5-second mandatory delay before continuing the order entry for the purposes of patient verification.
Resident and attending staff conveyed to us anecdotally during both beta testing and implementation that the ID reentry function made them aware of incorrect patient selection even before entering the required initials and birth year. They then cancelled the order session on the wrong patient and chose the correct patient. This is consistent with the findings of Green’s study, which noted that ED practitioners backed out of appropriately 1 in 200 order entry sessions due to wrong patient selection [10].
We also assessed the additional time added to each order entry session. Initially, using observational data, the CPOE ID reentry function added 6.2 seconds to each order entry session. However, providers that were more familiar with the system took an average of 4.0 seconds. While this added time per order entry session does not seem like much of an issue or delay, in a busy 12-hour shift in the ED it could be seen as significant. Adelman reported 6.6 seconds additional time required in for the ID reentry function used in his study [8], while Green’s study was designed using a 2.5-second mandatory delay before users could close the verification dialogue box [10].
The biggest challenges in implementing our project were unforeseen IT issues. The “go-live” date for ICD-10 was the same as the date we were to start the ID reentry requirement. IT personnel were needed to help in the EHR ICD-10 development and support, which delayed our start date. Additionally, other IT issues were identified. For example, the initial implementation of this project was to begin in the ED involving active ED patients only. At the project’s onset, the ID reentry function erroneously became active in all hospital locations. To fix this error, the entire double ID system alert, including the ED location, had to be removed and adjusted.
In addition to the above challenges, the team discovered errors that needed to be addressed during beta testing. For example, some clinicians would enter an order but no alert asking for the identifying data appeared. The order was entered and completed without the use of the double ID. Once discovered, IT was able to identify and correct the error. Beta testing also revealed an error in the system where providers who incorrectly identified a patient were “locking-out” of the CPOE system for that particular patient during the patient’s entire encounter. This issue was also quickly identified and resolved.
Despite the effectiveness of this system in reducing the rate of near-miss wrong-patient orders in the ED, errors still occur. It is possible that providers are entering the patient’s initials and year of birth without carefully verifying the patient’s identity [9].The CPOE double ID system alert is about three-quarters the size of the monitor screen. Thus, the clinician is able to verify the patient’s initials and year of birth using the patient’s header on the screen behind the patient identification alert. If the provider simply types the initials and year of birth on the patient’s header, then an identification error can occur.
More work is needed to decrease CPOE-related patient identification errors. Possible improvements may include single sign-ons and a no-interruption policy when writing orders. During our investigation, it was found that some clinicians would have multiple EHR sign-on sessions open at one computer terminal. These multiple EHR sign-on sessions were sometimes the root cause of a wrong patient error. With multiple sign-on sessions open, clinicians could toggle back and forth between patients on the same computer terminal and mistakenly complete an order on the wrong patient.
No-interruption zones and policies have been proven to be an effective way of decreasing interruptions and enhancing safety during medication preparation [13,14]. Utilization of no-interruption zones for CPOE may also be effective. Potentially, the EHR background color could change when a clinician selects the “enter order” tab within the EHR. The new background color would signify to those around the clinician that he/she is not to be interrupted during that time.
After the success of this initial quality improvement project in the ED, the intensive care unit has been added as a location for the CPOE double identification system. The data and results for this phase of the project are being tabulated and seem promising. In addition, SBH Health System is exploring single sign-on software to both help clinicians provide service and enhance patient safety.
Corresponding author: Daniel Lombardi, DO, 4422 Third Ave., Bronx, NY 10457.
Financial disclosures: None.
1. Bates, DW, Leape L, Cullen DJ, et al. Effect of computerized physician order entry and a team intervention on prevention of serious medication errors. JAMA 1998;280:1311–16.
2. Bates, DW, Teich JM, Lee J, et al. The impact of computerized physician order entry on medication error prevention. J Am Med Inform Assoc 1999;6:313–21.
3. Kaushal R, Shojania KG, Bates DW. Effects of computerized physician order entry and clinical decision support systems on medication safety: a systematic review. Arch Intern Med 2003;163:1409–16.
4. Reckmann, MH, Westbrook JI, Koh Y, et al. Does computeized provider order entry reduce prescribing errors for hospital inpatients? A systematic review. J Am Med Inform Assoc 2009;16:613–23.
5. Koppel R, Metlay JP, Cohen A, et al. Role of computerized physician order entry systems in facilitating medication errors. JAMA 2005;293:1197–203.
6. Broder C. Study: CPOE can increase risk of medication errors. Health IT News. March 9, 2005.
7. Schiff GD, Amato MG, Eguale T, et al. Computerised physician order entry-related medication errors: analysis of reported errors and vulnerability testing of current systems. BMJ Qual Saf 2015;24:264–71.
8. Adelman, JS, Kalkut GE, Schechter CB, et al. Understanding and preventing wrong-patient electronic orders: a randomized controlled trial. JAM Med Inform Assoc 2013;20:305–10.
9. Yang A, Grissinger M. Pennsylvania Patient Safety Authority. Wrong-patient medication errors: an analysis of event reports in Pennsylvania and strategies for prevention. PA Patient Saf Advis 2013 June;10:41–9.
10. Green RA, Hripcsak G, Salmasian H, et al. Intercepting wrong-patient orders in a computerized provider order entry system. Ann Emerg Med 2015;65:679–86.
11. Institute for Safe Medication Practices. Oops, sorry, wrong patient! A patient verification process is needed everywhere, not just at the bedside. 10 Mar 2011. Accessed at www.ismp.org/Newsletters/acutecare/articles/20110310.asp.
12. Anthony K, Wiencek C, Bauer C, et al. No interruptions please: impact of a No Interruption Zone on medication safety in intensive care units. Crit Care Nurse 2010;30:21–9.
13. Institute for Safe Medication Practices. Side tracks on the safety express. interruptions lead to errors and unfinished…wait, what was i doing? 29 Nov 2012. Accessed at www.ismp.org/Newsletters/acutecare/showarticle.aspx?id=37.
1. Bates, DW, Leape L, Cullen DJ, et al. Effect of computerized physician order entry and a team intervention on prevention of serious medication errors. JAMA 1998;280:1311–16.
2. Bates, DW, Teich JM, Lee J, et al. The impact of computerized physician order entry on medication error prevention. J Am Med Inform Assoc 1999;6:313–21.
3. Kaushal R, Shojania KG, Bates DW. Effects of computerized physician order entry and clinical decision support systems on medication safety: a systematic review. Arch Intern Med 2003;163:1409–16.
4. Reckmann, MH, Westbrook JI, Koh Y, et al. Does computeized provider order entry reduce prescribing errors for hospital inpatients? A systematic review. J Am Med Inform Assoc 2009;16:613–23.
5. Koppel R, Metlay JP, Cohen A, et al. Role of computerized physician order entry systems in facilitating medication errors. JAMA 2005;293:1197–203.
6. Broder C. Study: CPOE can increase risk of medication errors. Health IT News. March 9, 2005.
7. Schiff GD, Amato MG, Eguale T, et al. Computerised physician order entry-related medication errors: analysis of reported errors and vulnerability testing of current systems. BMJ Qual Saf 2015;24:264–71.
8. Adelman, JS, Kalkut GE, Schechter CB, et al. Understanding and preventing wrong-patient electronic orders: a randomized controlled trial. JAM Med Inform Assoc 2013;20:305–10.
9. Yang A, Grissinger M. Pennsylvania Patient Safety Authority. Wrong-patient medication errors: an analysis of event reports in Pennsylvania and strategies for prevention. PA Patient Saf Advis 2013 June;10:41–9.
10. Green RA, Hripcsak G, Salmasian H, et al. Intercepting wrong-patient orders in a computerized provider order entry system. Ann Emerg Med 2015;65:679–86.
11. Institute for Safe Medication Practices. Oops, sorry, wrong patient! A patient verification process is needed everywhere, not just at the bedside. 10 Mar 2011. Accessed at www.ismp.org/Newsletters/acutecare/articles/20110310.asp.
12. Anthony K, Wiencek C, Bauer C, et al. No interruptions please: impact of a No Interruption Zone on medication safety in intensive care units. Crit Care Nurse 2010;30:21–9.
13. Institute for Safe Medication Practices. Side tracks on the safety express. interruptions lead to errors and unfinished…wait, what was i doing? 29 Nov 2012. Accessed at www.ismp.org/Newsletters/acutecare/showarticle.aspx?id=37.
Too Much of a Good Thing: Weakness, Dysphagia, and Stridor After Botulinum Toxin Injections
Case
A 68-year-old woman presented to the ED 5 days after receiving onabotulinumtoxinA cosmetic injections for wrinkles of the face and neck. She stated that she was unable to raise her head while in a supine position and that her head felt heavy when standing. She also experienced spasms and strain of the posterior cervical neck muscles. In addition, the patient described a constant need to swallow forcefully throughout the day, and felt an intermittent heavy sensation over her larynx that was associated with stridor. She noted these symptoms began 5 days after the onabotulinumtoxinA injections and had peaked 2 days prior to presentation. She also complained of dysphagia without odynophagia, but denied any changes in her voice.
The patient first began onabotulinumtoxinA injections 12 years earlier for aesthetic treatment of glabellar and peri-orbital wrinkles. She initially received the injections at a regular interval of 90 to 100 days. During the course of the first 2 years of treatment, the patient was under the care of a plastic surgeon; thereafter, she sought treatment at a physician-owned medical spa because it offered onabotulinumtoxinA at a lower price. The injections at the medical spa were administered by a physician assistant (PA). The patient stated that although the PA had steadily increased the dose of onabotulinumtoxinA to maintain the desired aesthetic effect, this was the first time she had experienced any side effects from the treatment.
The ED staff contacted the medical spa provider, who reviewed the patient’s medical record over the telephone. The PA stated that he had been the only practitioner at the facility to administer the onabotulinumtoxinA injections to the patient over her past 10 years there as a client. He further informed the emergency physician (EP) that 12 days prior to presentation, he had given the patient a total of 50 IU of onabotulinumtoxinA, in five separate injections, into the mid frontalis muscle; a total of 35 IU, in seven separate injections, into the glabellar region (procerus and corrugator muscles bilaterally); 20 IU into the lateral and inferior-lateral orbicularis oculi bilaterally, in four separate injections per side, (40 IU total); and a total of 100 IU in the anterior platysma, in 20 separate injections, for a total 1-day onabotulinumtoxinA dose of 225 IU.
The PA explained to the EP that he mixed the onabotulinumtoxinA in the patient’s room and had shown her the vials and dilution standard as recommended by the manufacturer because she had been requiring increased dosages and had previously questioned whether the onabotulinumtoxinA was diluted. The PA denied any other patients experiencing similar adverse events as those of the patient’s.
Over the last 10 years, the patient had received onabotulinumtoxinA in the nasolabial folds, upper and lower lip wrinkles, mentalis, depressor angular oris, buccal, nasalis, lateral brow, masseter, and calf muscles. The dosage of onabotulinumtoxinA at this most recent injection cycle was unchanged from her previous visit 3 months prior. According to the PA, the practice did not use abobotulinumtoxinA or incobotulinumtoxinA.
Regarding the patient’s medical history, she had no health issues suggestive of myasthenia gravis, multiple sclerosis, or Guillain-Barré syndrome. Examination of the face revealed decreased muscle excursion of the frontalis muscle from mid-brow to mid-brow, and stair-step wrinkle formation bilaterally. The procerus muscle was very weak, and the corrugator muscles were moderately diminished in strength. The lateral orbicularis oculi were very weak at each canthus. The extra-ocular muscles were intact. She had full mandibular excursion, and powerful movement of the tongue. The oropharynx and floor of the mouth were normal. She was noted to purposefully swallow and extend her neck every 90 to 120 seconds to “clear her throat,” though she did not drool and was able to handle her secretions and swallow fluids without aspiration. Her voice was normal and she was able to recite the letters “KKKKK,” “OOOOO,” and “EEEEE” in rapid fashion without breathiness or stridor. The rest of her facial muscles were normal.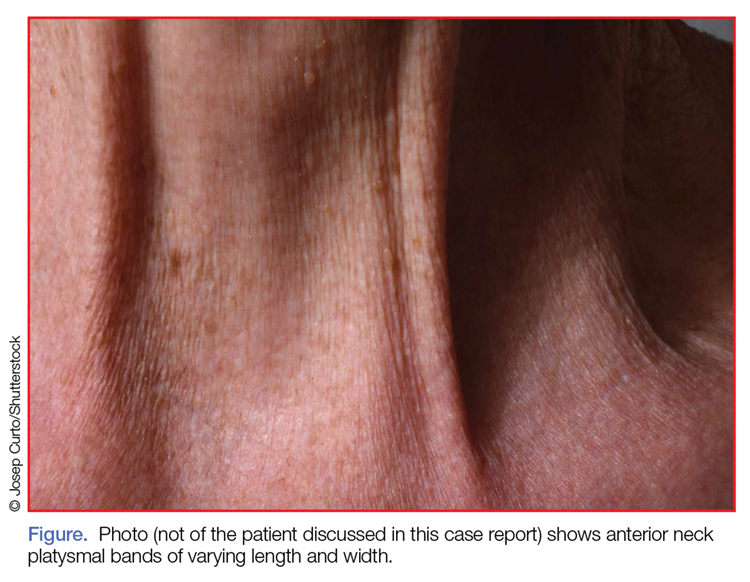
While examining the patient, the EP asked her to refrain from swallowing whenever she extended her neck. Upon complying with this request, her neck extension precipitated swallowing and, by not swallowing, she did not accumulate secretions. Once during the examination, the patient began swallowing and breathing rapidly with stridor. This less than 15-second episode was abated by full-neck extensions, which relieved the patient’s sensation of heaviness over the larynx. Her breathing and voice were normal immediately after this episode.
Examination of the anterior neck revealed four platysmal bands (Figure). One band measured 10 cm in length and extended from the mandible inferiorly; two bands measured 2 cm lateral to the midline bilaterally; and the fourth band extended 4 cm in length from the mandible immediately lateral to the longer platysmal band. The platysma and dermis were flaccid and redundant at rest and with exertion. The sternocleidomastoid muscles were weak with exertion. The larynx moved cephalad with swallowing. The posterior cervical neck and trapezius muscles were of normal tone and strength. No spasms or fasciculations were noted during the examination period.
While supine, the patient strained to lift her head and complained of a suffocating sensation over the larynx. She had no rashes or edema, and the remainder of the physical examination, vital signs, and pulse oximetry were normal. Laboratory evaluation, which included a complete blood count and serum electrolytes, was also normal.
An otolaryngologist consultation for laryngoscopy was obtained. After reviewing the patient’s case, the otolaryngologist concluded that given the patient’s history, intermittent stridor, and an absence of signs or symptoms suggestive of an impending upper airway obstruction (UAO), laryngoscopy was not warranted.
A plastic surgery consultation was then obtained. The patient’s examination was as noted above, and her vital signs and pulse oximetry remained normal throughout her ED stay. Although botulinum and botulinum antibody titers were ordered, the patient refused testing due to cost concerns. She was discharged home by plastic surgery services with a diagnosis of floppy neck and dysphagia secondary to aesthetic botulinum toxin paralysis of the bilateral sternocleidomastoid muscles and platysma. She was given a prescription for metoclopramide hydrochloride to stimulate motility of the upper gastrointestinal tract and to potentially improve swallowing.10
The patient was scheduled for a follow-up evaluation with the plastic surgeon 2 days after discharge. She was instructed to call 911 if she experienced stridor, shortness of breath, drooling, or if any airway issues arose. The patient did not return for her follow-up appointment with the plastic surgeon.
Discussion
Clostridium Botulinum Toxins
Clostridium botulinum is a gram-positive spore-forming anaerobic bacterium that produces extremely potent neuro-exotoxins. C botulinum is found in soil, contaminated foods, and in illicit injectable drugs (eg, heroin). Seven distinct antigenic botulinum toxins (A, B, C1, D, E, F, and G) are produced by several strains of C botulinum. Systemically, each neurotoxin is able to produce severe morbidity and mortality by causing generalized muscle paralysis and death by respiratory failure. The lethal dose of these agents is approximating 10(-9) g/kg body weight. Botulinum toxin type A is the most potent.1,2
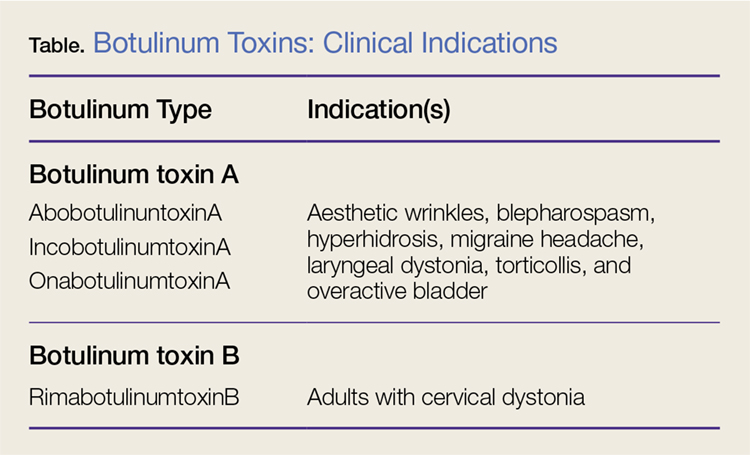
Nonetheless, botulinum toxin has been used clinically since the early 1970s. Currently, there are three FDA-approved botulinum toxin type A agents and one type B formulation (rimabotulinumtoxinB) (Table). Each formulation is unique, proprietary, and differs in molecular weight, toxin-complex size, protein content, and inactive ingredients. The effectiveness and adverse event profile for these four botulinum toxins is individually dependent upon the different dilutions and potency, onset of action, duration of effect, diffusion, and migration potential. Hence, the effective dose of one botulinum toxin does not equate to any other, resulting in a lack of interchangeability between botulinum toxins (eg, 5 IU of incobotulinumtoxinA does not equal 5 IU of onabotulinumtoxinA).
Aesthetic Indications
Historically, the use of botulinum toxin for aesthetic treatment of wrinkles and platysmal bands was first reported by Blitzer3 in 1993.Subsequently, the use of botulinum toxin for the aesthetic treatment of facial wrinkles, hypertrophic platysmal bands and horizontal neck lines gained popularity within the public and medical community.3-5
Anatomically, the platysma is a thin sheet-like muscle that originates in the superior fascia of the pectoralis and deltoid fascia, and extends over the full length of the neck up past the mandible and continuing into the submuscular aponeurotic system. The platysma is innervated by the seventh cranial nerve and functions to pull the jaw downward. The platysma muscle is attached directly to the skin. With normal aging, the anterior neck skin becomes flaccid, the central platysmal bands thicken and contract—forming bands, horizontal wrinkles, and loss of definition of the neck noticed at rest and with contraction of the platysma muscle. These vertical bands are known as platysmal bands. The platysmal bands are benign consequences of aging and as such are targets of correction through surgery or botulinum toxin injection.6,7
Mechanism of Action
Platysmal band and horizontal line injection techniques with botulinum toxin have been reported in the literature with dosages ranging from 15 IU to 200 IU used to block the Soluble N-ethylmaleimide-sensitive factor activating protein receptors. Typical onset of action begins at 3 days, with full paralytic effect at 7 days. Repeat injections every 3 to 4 months are required with prolonged effects seen with each subsequent injection due to chemodenervation-induced muscle atrophy.4,7,8
Adverse Effects
Commercial botulinum toxin type A has been associated with minor and transient side effects. Moderate complications seen in the neck region include transient soft-tissue edema, dermal ecchymoses, intramuscular hematoma, diffuse muscle soreness, neck flexor weakness, and headaches.4,8,9
The use of botulinum toxin for chemodenervation of the platysma can produce significant weakness of other neck muscles, including the sternocleidomastoid, cricothyroid, sternothyroid, and sternohyoid. Floppy neck and dysphagia may be due to diffusion of the toxin into the muscles of deglutition of the larynx; injection directly into the sternocleidomastoid muscle; or a result of the systemic effects of large dosages. Hoarseness, breathiness, and dysphagia may occur 3 to 4 days after injection, especially with doses over 75 IU.10
The recommended concentration of botulinum toxin type A causes a diffusion average of 1 cm in all directions from the injection sites. However, as the dilution increases, so does the zone of diffusion. Typical discharge instructions for platysma treatment include the overuse of the neck muscles for 2 to 4 hours after injection to encourage the botulinum toxin uptake for optimal result. Site manipulation (rubbing or massaging) also increases diffusion. For botulinum toxin type B, the zone of diffusion is greater because its molecular weight is less than the type A toxins, thus making it an undesirable agent for aesthetic facial chemodenervation.4,11
Toxin Resistance
Botulinum toxin resistance is a known complication that occurs normally as a result of the body recognizing the neurotoxin as a foreign substance and producing neutralizing antibodies (NAb). Primary botulinum toxin failure is known in patients who require high doses of the neurotoxin for treatment of neuromuscular disorders.12 Complete secondary therapy failure is known to occur in cosmetic patients after a single dose and those who have been receiving low-dose botulinum toxin regularly. The risk of NAb development increases with long-term treatment and high doses.12-18
Floppy Neck and Dysphagia
As previously noted, floppy neck and dysphagia are adverse clinical findings of botulinum toxin effect on the platysma, sternocleidomastoid, or the paralaryngeal muscles. In this case, the patient was fortunate to have only sustained weakness of the platysma and sternocleidomastoid muscles despite both a large neck and total body dose. Paralaryngeal muscle paralysis is not life-threatening, but the distress may precipitate paradoxical vocal cord motion and stridor.
Stridor
Stridor is typically a symptom of an upper airway obstruction (UAO) process. Typical UAO conditions encountered in the ED are infections (eg, epiglottitis, croup), foreign body, allergy, and laryngeal trauma. The age of the patient, onset of stridor, course of the stridor (ie, intermittent, continuous, worsening), associated symptoms (eg, fever, rash, swelling of oral soft tissues), and bruising must be ascertained.
In differentiating the etiology of stridor, the EP should observe the patient for any associated change in voice, inability to handle secretions, and position of comfort. Patients with stridor require admission and evaluation by an otolaryngologist as expeditiously as possible because impending UAO may quickly progress to complete UAO necessitating emergent intubation.
An atypical presentation of stridor to the ED is sporadic stridor. Sporadic attacks of stridor during activity have been associated with the entity of paradoxical vocal cord motion. Patients usually describe a choking sensation with inability to breathe resulting in an audible inspiratory and/or expiratory sound—ie, stridor. Wheezing may or may not be present. Patients may also describe tightness in the neck and sometimes in the chest. The attacks are usually seconds to minutes in duration. More often, there is a precipitating or an inducing factor such as hyperventilation, cough, panting, phonatory tasks, or the inhalation of irritants or perfume, or an oropharyngeal or laryngeal manipulation prior or postextubation. The feeling of stress alone is commonly reported prior to the attacks. When evaluating patients presenting with floppy neck, dysphagia, and stridor, it is imperative that the clinician conduct a thorough history and physical examination to determine if the symptoms are secondary to a systemic or local effect, and whether the patient will progress to an acute UAO (vocal cord paralysis) necessitating intubation in the ED and subsequent tracheostomy.19,20
Conclusion
The ready availability of botulinum toxins and their low-cost-benefit ratio continue to promote over-utilization for treatment of facial wrinkles, platysmal bands, and horizontal lines; migraine headache; and hyperhidrosis. Complications associated with overuse of botulinum toxins are due to either administration of a large single dose or from regional diffusion. With the increasing number of patients receiving botulinum injections, EPs should be aware of the four available toxin types onset of action, adverse events, and potential life-threatening complications of regional neck injections.
References
1. Huang W, Foster JA, Rogachefsky AS. Pharmacology of botulinum toxin. J Am Acad Dermatol. 2000;43(2 Pt 1):249-259. doi:10.1067/mjd.2000.105567.
2. Lamanna C. The most poisonous poison. Science. 1959;130(3378):763-772.
3. Blitzer A, Brin MF, Keen MS, Aviv JE. Botulinum toxin for the treatment of hyperfunctional lines of the face. Arch Otolaryngol Head Neck Surg. 1993;119(9):1018-1022.
4. Carruthers A, Carruthers J. Clinical indications and injection technique for the cosmetic use of botulinum A exotoxin. Dermatol Surg. 1998;24(11):1189-1194.
5. Carruthers J, Carruthers A. Botox use in the mid and lower face and neck. Semin Cutan Med Surg. 2001;20(2):85-92. doi:10.1053/sder.2001.25139
6. Hoefflin SM. Anatomy of the platysma and lip depressor muscles. A simplified mnemonic approach. Dermatol Surg. 1998;24(11):1225-1231.
7. Brandt FS, Bellman B. Cosmetic use of botulinum A exotoxin for the aging neck. Dermatol Surg. 1998;24(11):1232-1234.
8. Klein AW. Complications and adverse reactions with the use of botulinum toxin. Semin Cutan Med Surg. 2001;20(2):109-120. doi:10.1053/sder.2001.25964.
9. Carruthers A, Kiene K, Carruthers J. Botulinum A exotoxin use in clinical dermatology. J Am Acad Dermatol. 1996;34(5 Pt 1):788-797.
10. Howell K, Selber P, Graham HK, Reddihough D. Botulinum neurotoxin A: an unusual systemic effect. J Paediatr Child Health. 2007:43(6):499-501. doi:10.1111/j.1440-1754.2007.01122.x.
11. Carruthers A, Carruthers J. Toxins 99, new information about the botulinum neurotoxins. Dermatol Surg. 2000;26(3):174-176.
12. Dressler D, Adib Saberi F. New formulation of Botox: complete antibody-induced treatment failure in cervical dystonia. J Neurol Neurosurg Psychiatry. 2007;78(1):108-109. doi:10.1136/jnnp.2006.093419.
13. Borodic G. Immunologic resistance after repeated botulinum toxin type a injections for facial rhytides. Ophthal Plast Reconstr Surg. 2006;22:239-240. doi:10.1097/01.iop.0000217703.80859.a3.
14. Goschel H, Wohlfarth K, Frevert J, Dengler R, Bigalke H. Botulinum A toxin therapy: neutralizing and nonneutralizing antibodies—therapeutic consequences. Exp Neurol. 1997;147(1):96-102. doi:10.1006/exnr.1997.6580.
15. Hatheway CL. Toxigenic clostridia. Clin Microbiol Rev. 1990;3(1):66-98.
16. Smith LA. Development of recombinant vaccines for botulinum neurotoxin. Toxicon. 1998;36(11):1539-1548.
17. Houser MK, Sheean GL, Lees AJ. Further studies using higher doses of botulinum toxin type F for torticollis resistant to botulinum toxin type A. J Neurol Neurosurg Psychiatry. 1998;64(5):577-580.
18. Dressler D, Wohlfahrt K, Meyer-Rogge E, Wiest L, Bigalke H. Antibody-induced failure of botulinum toxin a therapy in cosmetic indications. Dermatol Surg. 2010;36 Suppl 4:2182-2187. doi:10.1111/j.1524-4725.2010.01710.x.
19. Maschka DA, Bauman NM, McCray PB Jr, Hoffman HT, Karnell MP, Smith RJ. A classification scheme for paradoxical vocal cord motion. Laryngoscope. 1997;107(11 Pt 1):1429-1435.
20. Altman KW, Simpson CB, Amin MR, Abaza M, Balkissoon R, Casiano RR. Cough and paradoxical vocal fold motion. Otolaryngol Head Neck Surg. 2002;127(6):501-511. doi:10.1067/mhn.2002.127589.
Case
A 68-year-old woman presented to the ED 5 days after receiving onabotulinumtoxinA cosmetic injections for wrinkles of the face and neck. She stated that she was unable to raise her head while in a supine position and that her head felt heavy when standing. She also experienced spasms and strain of the posterior cervical neck muscles. In addition, the patient described a constant need to swallow forcefully throughout the day, and felt an intermittent heavy sensation over her larynx that was associated with stridor. She noted these symptoms began 5 days after the onabotulinumtoxinA injections and had peaked 2 days prior to presentation. She also complained of dysphagia without odynophagia, but denied any changes in her voice.
The patient first began onabotulinumtoxinA injections 12 years earlier for aesthetic treatment of glabellar and peri-orbital wrinkles. She initially received the injections at a regular interval of 90 to 100 days. During the course of the first 2 years of treatment, the patient was under the care of a plastic surgeon; thereafter, she sought treatment at a physician-owned medical spa because it offered onabotulinumtoxinA at a lower price. The injections at the medical spa were administered by a physician assistant (PA). The patient stated that although the PA had steadily increased the dose of onabotulinumtoxinA to maintain the desired aesthetic effect, this was the first time she had experienced any side effects from the treatment.
The ED staff contacted the medical spa provider, who reviewed the patient’s medical record over the telephone. The PA stated that he had been the only practitioner at the facility to administer the onabotulinumtoxinA injections to the patient over her past 10 years there as a client. He further informed the emergency physician (EP) that 12 days prior to presentation, he had given the patient a total of 50 IU of onabotulinumtoxinA, in five separate injections, into the mid frontalis muscle; a total of 35 IU, in seven separate injections, into the glabellar region (procerus and corrugator muscles bilaterally); 20 IU into the lateral and inferior-lateral orbicularis oculi bilaterally, in four separate injections per side, (40 IU total); and a total of 100 IU in the anterior platysma, in 20 separate injections, for a total 1-day onabotulinumtoxinA dose of 225 IU.
The PA explained to the EP that he mixed the onabotulinumtoxinA in the patient’s room and had shown her the vials and dilution standard as recommended by the manufacturer because she had been requiring increased dosages and had previously questioned whether the onabotulinumtoxinA was diluted. The PA denied any other patients experiencing similar adverse events as those of the patient’s.
Over the last 10 years, the patient had received onabotulinumtoxinA in the nasolabial folds, upper and lower lip wrinkles, mentalis, depressor angular oris, buccal, nasalis, lateral brow, masseter, and calf muscles. The dosage of onabotulinumtoxinA at this most recent injection cycle was unchanged from her previous visit 3 months prior. According to the PA, the practice did not use abobotulinumtoxinA or incobotulinumtoxinA.
Regarding the patient’s medical history, she had no health issues suggestive of myasthenia gravis, multiple sclerosis, or Guillain-Barré syndrome. Examination of the face revealed decreased muscle excursion of the frontalis muscle from mid-brow to mid-brow, and stair-step wrinkle formation bilaterally. The procerus muscle was very weak, and the corrugator muscles were moderately diminished in strength. The lateral orbicularis oculi were very weak at each canthus. The extra-ocular muscles were intact. She had full mandibular excursion, and powerful movement of the tongue. The oropharynx and floor of the mouth were normal. She was noted to purposefully swallow and extend her neck every 90 to 120 seconds to “clear her throat,” though she did not drool and was able to handle her secretions and swallow fluids without aspiration. Her voice was normal and she was able to recite the letters “KKKKK,” “OOOOO,” and “EEEEE” in rapid fashion without breathiness or stridor. The rest of her facial muscles were normal.
While examining the patient, the EP asked her to refrain from swallowing whenever she extended her neck. Upon complying with this request, her neck extension precipitated swallowing and, by not swallowing, she did not accumulate secretions. Once during the examination, the patient began swallowing and breathing rapidly with stridor. This less than 15-second episode was abated by full-neck extensions, which relieved the patient’s sensation of heaviness over the larynx. Her breathing and voice were normal immediately after this episode.
Examination of the anterior neck revealed four platysmal bands (Figure). One band measured 10 cm in length and extended from the mandible inferiorly; two bands measured 2 cm lateral to the midline bilaterally; and the fourth band extended 4 cm in length from the mandible immediately lateral to the longer platysmal band. The platysma and dermis were flaccid and redundant at rest and with exertion. The sternocleidomastoid muscles were weak with exertion. The larynx moved cephalad with swallowing. The posterior cervical neck and trapezius muscles were of normal tone and strength. No spasms or fasciculations were noted during the examination period.
While supine, the patient strained to lift her head and complained of a suffocating sensation over the larynx. She had no rashes or edema, and the remainder of the physical examination, vital signs, and pulse oximetry were normal. Laboratory evaluation, which included a complete blood count and serum electrolytes, was also normal.
An otolaryngologist consultation for laryngoscopy was obtained. After reviewing the patient’s case, the otolaryngologist concluded that given the patient’s history, intermittent stridor, and an absence of signs or symptoms suggestive of an impending upper airway obstruction (UAO), laryngoscopy was not warranted.
A plastic surgery consultation was then obtained. The patient’s examination was as noted above, and her vital signs and pulse oximetry remained normal throughout her ED stay. Although botulinum and botulinum antibody titers were ordered, the patient refused testing due to cost concerns. She was discharged home by plastic surgery services with a diagnosis of floppy neck and dysphagia secondary to aesthetic botulinum toxin paralysis of the bilateral sternocleidomastoid muscles and platysma. She was given a prescription for metoclopramide hydrochloride to stimulate motility of the upper gastrointestinal tract and to potentially improve swallowing.10
The patient was scheduled for a follow-up evaluation with the plastic surgeon 2 days after discharge. She was instructed to call 911 if she experienced stridor, shortness of breath, drooling, or if any airway issues arose. The patient did not return for her follow-up appointment with the plastic surgeon.
Discussion
Clostridium Botulinum Toxins
Clostridium botulinum is a gram-positive spore-forming anaerobic bacterium that produces extremely potent neuro-exotoxins. C botulinum is found in soil, contaminated foods, and in illicit injectable drugs (eg, heroin). Seven distinct antigenic botulinum toxins (A, B, C1, D, E, F, and G) are produced by several strains of C botulinum. Systemically, each neurotoxin is able to produce severe morbidity and mortality by causing generalized muscle paralysis and death by respiratory failure. The lethal dose of these agents is approximating 10(-9) g/kg body weight. Botulinum toxin type A is the most potent.1,2
Nonetheless, botulinum toxin has been used clinically since the early 1970s. Currently, there are three FDA-approved botulinum toxin type A agents and one type B formulation (rimabotulinumtoxinB) (Table). Each formulation is unique, proprietary, and differs in molecular weight, toxin-complex size, protein content, and inactive ingredients. The effectiveness and adverse event profile for these four botulinum toxins is individually dependent upon the different dilutions and potency, onset of action, duration of effect, diffusion, and migration potential. Hence, the effective dose of one botulinum toxin does not equate to any other, resulting in a lack of interchangeability between botulinum toxins (eg, 5 IU of incobotulinumtoxinA does not equal 5 IU of onabotulinumtoxinA).
Aesthetic Indications
Historically, the use of botulinum toxin for aesthetic treatment of wrinkles and platysmal bands was first reported by Blitzer3 in 1993.Subsequently, the use of botulinum toxin for the aesthetic treatment of facial wrinkles, hypertrophic platysmal bands and horizontal neck lines gained popularity within the public and medical community.3-5
Anatomically, the platysma is a thin sheet-like muscle that originates in the superior fascia of the pectoralis and deltoid fascia, and extends over the full length of the neck up past the mandible and continuing into the submuscular aponeurotic system. The platysma is innervated by the seventh cranial nerve and functions to pull the jaw downward. The platysma muscle is attached directly to the skin. With normal aging, the anterior neck skin becomes flaccid, the central platysmal bands thicken and contract—forming bands, horizontal wrinkles, and loss of definition of the neck noticed at rest and with contraction of the platysma muscle. These vertical bands are known as platysmal bands. The platysmal bands are benign consequences of aging and as such are targets of correction through surgery or botulinum toxin injection.6,7
Mechanism of Action
Platysmal band and horizontal line injection techniques with botulinum toxin have been reported in the literature with dosages ranging from 15 IU to 200 IU used to block the Soluble N-ethylmaleimide-sensitive factor activating protein receptors. Typical onset of action begins at 3 days, with full paralytic effect at 7 days. Repeat injections every 3 to 4 months are required with prolonged effects seen with each subsequent injection due to chemodenervation-induced muscle atrophy.4,7,8
Adverse Effects
Commercial botulinum toxin type A has been associated with minor and transient side effects. Moderate complications seen in the neck region include transient soft-tissue edema, dermal ecchymoses, intramuscular hematoma, diffuse muscle soreness, neck flexor weakness, and headaches.4,8,9
The use of botulinum toxin for chemodenervation of the platysma can produce significant weakness of other neck muscles, including the sternocleidomastoid, cricothyroid, sternothyroid, and sternohyoid. Floppy neck and dysphagia may be due to diffusion of the toxin into the muscles of deglutition of the larynx; injection directly into the sternocleidomastoid muscle; or a result of the systemic effects of large dosages. Hoarseness, breathiness, and dysphagia may occur 3 to 4 days after injection, especially with doses over 75 IU.10
The recommended concentration of botulinum toxin type A causes a diffusion average of 1 cm in all directions from the injection sites. However, as the dilution increases, so does the zone of diffusion. Typical discharge instructions for platysma treatment include the overuse of the neck muscles for 2 to 4 hours after injection to encourage the botulinum toxin uptake for optimal result. Site manipulation (rubbing or massaging) also increases diffusion. For botulinum toxin type B, the zone of diffusion is greater because its molecular weight is less than the type A toxins, thus making it an undesirable agent for aesthetic facial chemodenervation.4,11
Toxin Resistance
Botulinum toxin resistance is a known complication that occurs normally as a result of the body recognizing the neurotoxin as a foreign substance and producing neutralizing antibodies (NAb). Primary botulinum toxin failure is known in patients who require high doses of the neurotoxin for treatment of neuromuscular disorders.12 Complete secondary therapy failure is known to occur in cosmetic patients after a single dose and those who have been receiving low-dose botulinum toxin regularly. The risk of NAb development increases with long-term treatment and high doses.12-18
Floppy Neck and Dysphagia
As previously noted, floppy neck and dysphagia are adverse clinical findings of botulinum toxin effect on the platysma, sternocleidomastoid, or the paralaryngeal muscles. In this case, the patient was fortunate to have only sustained weakness of the platysma and sternocleidomastoid muscles despite both a large neck and total body dose. Paralaryngeal muscle paralysis is not life-threatening, but the distress may precipitate paradoxical vocal cord motion and stridor.
Stridor
Stridor is typically a symptom of an upper airway obstruction (UAO) process. Typical UAO conditions encountered in the ED are infections (eg, epiglottitis, croup), foreign body, allergy, and laryngeal trauma. The age of the patient, onset of stridor, course of the stridor (ie, intermittent, continuous, worsening), associated symptoms (eg, fever, rash, swelling of oral soft tissues), and bruising must be ascertained.
In differentiating the etiology of stridor, the EP should observe the patient for any associated change in voice, inability to handle secretions, and position of comfort. Patients with stridor require admission and evaluation by an otolaryngologist as expeditiously as possible because impending UAO may quickly progress to complete UAO necessitating emergent intubation.
An atypical presentation of stridor to the ED is sporadic stridor. Sporadic attacks of stridor during activity have been associated with the entity of paradoxical vocal cord motion. Patients usually describe a choking sensation with inability to breathe resulting in an audible inspiratory and/or expiratory sound—ie, stridor. Wheezing may or may not be present. Patients may also describe tightness in the neck and sometimes in the chest. The attacks are usually seconds to minutes in duration. More often, there is a precipitating or an inducing factor such as hyperventilation, cough, panting, phonatory tasks, or the inhalation of irritants or perfume, or an oropharyngeal or laryngeal manipulation prior or postextubation. The feeling of stress alone is commonly reported prior to the attacks. When evaluating patients presenting with floppy neck, dysphagia, and stridor, it is imperative that the clinician conduct a thorough history and physical examination to determine if the symptoms are secondary to a systemic or local effect, and whether the patient will progress to an acute UAO (vocal cord paralysis) necessitating intubation in the ED and subsequent tracheostomy.19,20
Conclusion
The ready availability of botulinum toxins and their low-cost-benefit ratio continue to promote over-utilization for treatment of facial wrinkles, platysmal bands, and horizontal lines; migraine headache; and hyperhidrosis. Complications associated with overuse of botulinum toxins are due to either administration of a large single dose or from regional diffusion. With the increasing number of patients receiving botulinum injections, EPs should be aware of the four available toxin types onset of action, adverse events, and potential life-threatening complications of regional neck injections.
Case
A 68-year-old woman presented to the ED 5 days after receiving onabotulinumtoxinA cosmetic injections for wrinkles of the face and neck. She stated that she was unable to raise her head while in a supine position and that her head felt heavy when standing. She also experienced spasms and strain of the posterior cervical neck muscles. In addition, the patient described a constant need to swallow forcefully throughout the day, and felt an intermittent heavy sensation over her larynx that was associated with stridor. She noted these symptoms began 5 days after the onabotulinumtoxinA injections and had peaked 2 days prior to presentation. She also complained of dysphagia without odynophagia, but denied any changes in her voice.
The patient first began onabotulinumtoxinA injections 12 years earlier for aesthetic treatment of glabellar and peri-orbital wrinkles. She initially received the injections at a regular interval of 90 to 100 days. During the course of the first 2 years of treatment, the patient was under the care of a plastic surgeon; thereafter, she sought treatment at a physician-owned medical spa because it offered onabotulinumtoxinA at a lower price. The injections at the medical spa were administered by a physician assistant (PA). The patient stated that although the PA had steadily increased the dose of onabotulinumtoxinA to maintain the desired aesthetic effect, this was the first time she had experienced any side effects from the treatment.
The ED staff contacted the medical spa provider, who reviewed the patient’s medical record over the telephone. The PA stated that he had been the only practitioner at the facility to administer the onabotulinumtoxinA injections to the patient over her past 10 years there as a client. He further informed the emergency physician (EP) that 12 days prior to presentation, he had given the patient a total of 50 IU of onabotulinumtoxinA, in five separate injections, into the mid frontalis muscle; a total of 35 IU, in seven separate injections, into the glabellar region (procerus and corrugator muscles bilaterally); 20 IU into the lateral and inferior-lateral orbicularis oculi bilaterally, in four separate injections per side, (40 IU total); and a total of 100 IU in the anterior platysma, in 20 separate injections, for a total 1-day onabotulinumtoxinA dose of 225 IU.
The PA explained to the EP that he mixed the onabotulinumtoxinA in the patient’s room and had shown her the vials and dilution standard as recommended by the manufacturer because she had been requiring increased dosages and had previously questioned whether the onabotulinumtoxinA was diluted. The PA denied any other patients experiencing similar adverse events as those of the patient’s.
Over the last 10 years, the patient had received onabotulinumtoxinA in the nasolabial folds, upper and lower lip wrinkles, mentalis, depressor angular oris, buccal, nasalis, lateral brow, masseter, and calf muscles. The dosage of onabotulinumtoxinA at this most recent injection cycle was unchanged from her previous visit 3 months prior. According to the PA, the practice did not use abobotulinumtoxinA or incobotulinumtoxinA.
Regarding the patient’s medical history, she had no health issues suggestive of myasthenia gravis, multiple sclerosis, or Guillain-Barré syndrome. Examination of the face revealed decreased muscle excursion of the frontalis muscle from mid-brow to mid-brow, and stair-step wrinkle formation bilaterally. The procerus muscle was very weak, and the corrugator muscles were moderately diminished in strength. The lateral orbicularis oculi were very weak at each canthus. The extra-ocular muscles were intact. She had full mandibular excursion, and powerful movement of the tongue. The oropharynx and floor of the mouth were normal. She was noted to purposefully swallow and extend her neck every 90 to 120 seconds to “clear her throat,” though she did not drool and was able to handle her secretions and swallow fluids without aspiration. Her voice was normal and she was able to recite the letters “KKKKK,” “OOOOO,” and “EEEEE” in rapid fashion without breathiness or stridor. The rest of her facial muscles were normal.
While examining the patient, the EP asked her to refrain from swallowing whenever she extended her neck. Upon complying with this request, her neck extension precipitated swallowing and, by not swallowing, she did not accumulate secretions. Once during the examination, the patient began swallowing and breathing rapidly with stridor. This less than 15-second episode was abated by full-neck extensions, which relieved the patient’s sensation of heaviness over the larynx. Her breathing and voice were normal immediately after this episode.
Examination of the anterior neck revealed four platysmal bands (Figure). One band measured 10 cm in length and extended from the mandible inferiorly; two bands measured 2 cm lateral to the midline bilaterally; and the fourth band extended 4 cm in length from the mandible immediately lateral to the longer platysmal band. The platysma and dermis were flaccid and redundant at rest and with exertion. The sternocleidomastoid muscles were weak with exertion. The larynx moved cephalad with swallowing. The posterior cervical neck and trapezius muscles were of normal tone and strength. No spasms or fasciculations were noted during the examination period.
While supine, the patient strained to lift her head and complained of a suffocating sensation over the larynx. She had no rashes or edema, and the remainder of the physical examination, vital signs, and pulse oximetry were normal. Laboratory evaluation, which included a complete blood count and serum electrolytes, was also normal.
An otolaryngologist consultation for laryngoscopy was obtained. After reviewing the patient’s case, the otolaryngologist concluded that given the patient’s history, intermittent stridor, and an absence of signs or symptoms suggestive of an impending upper airway obstruction (UAO), laryngoscopy was not warranted.
A plastic surgery consultation was then obtained. The patient’s examination was as noted above, and her vital signs and pulse oximetry remained normal throughout her ED stay. Although botulinum and botulinum antibody titers were ordered, the patient refused testing due to cost concerns. She was discharged home by plastic surgery services with a diagnosis of floppy neck and dysphagia secondary to aesthetic botulinum toxin paralysis of the bilateral sternocleidomastoid muscles and platysma. She was given a prescription for metoclopramide hydrochloride to stimulate motility of the upper gastrointestinal tract and to potentially improve swallowing.10
The patient was scheduled for a follow-up evaluation with the plastic surgeon 2 days after discharge. She was instructed to call 911 if she experienced stridor, shortness of breath, drooling, or if any airway issues arose. The patient did not return for her follow-up appointment with the plastic surgeon.
Discussion
Clostridium Botulinum Toxins
Clostridium botulinum is a gram-positive spore-forming anaerobic bacterium that produces extremely potent neuro-exotoxins. C botulinum is found in soil, contaminated foods, and in illicit injectable drugs (eg, heroin). Seven distinct antigenic botulinum toxins (A, B, C1, D, E, F, and G) are produced by several strains of C botulinum. Systemically, each neurotoxin is able to produce severe morbidity and mortality by causing generalized muscle paralysis and death by respiratory failure. The lethal dose of these agents is approximating 10(-9) g/kg body weight. Botulinum toxin type A is the most potent.1,2
Nonetheless, botulinum toxin has been used clinically since the early 1970s. Currently, there are three FDA-approved botulinum toxin type A agents and one type B formulation (rimabotulinumtoxinB) (Table). Each formulation is unique, proprietary, and differs in molecular weight, toxin-complex size, protein content, and inactive ingredients. The effectiveness and adverse event profile for these four botulinum toxins is individually dependent upon the different dilutions and potency, onset of action, duration of effect, diffusion, and migration potential. Hence, the effective dose of one botulinum toxin does not equate to any other, resulting in a lack of interchangeability between botulinum toxins (eg, 5 IU of incobotulinumtoxinA does not equal 5 IU of onabotulinumtoxinA).
Aesthetic Indications
Historically, the use of botulinum toxin for aesthetic treatment of wrinkles and platysmal bands was first reported by Blitzer3 in 1993.Subsequently, the use of botulinum toxin for the aesthetic treatment of facial wrinkles, hypertrophic platysmal bands and horizontal neck lines gained popularity within the public and medical community.3-5
Anatomically, the platysma is a thin sheet-like muscle that originates in the superior fascia of the pectoralis and deltoid fascia, and extends over the full length of the neck up past the mandible and continuing into the submuscular aponeurotic system. The platysma is innervated by the seventh cranial nerve and functions to pull the jaw downward. The platysma muscle is attached directly to the skin. With normal aging, the anterior neck skin becomes flaccid, the central platysmal bands thicken and contract—forming bands, horizontal wrinkles, and loss of definition of the neck noticed at rest and with contraction of the platysma muscle. These vertical bands are known as platysmal bands. The platysmal bands are benign consequences of aging and as such are targets of correction through surgery or botulinum toxin injection.6,7
Mechanism of Action
Platysmal band and horizontal line injection techniques with botulinum toxin have been reported in the literature with dosages ranging from 15 IU to 200 IU used to block the Soluble N-ethylmaleimide-sensitive factor activating protein receptors. Typical onset of action begins at 3 days, with full paralytic effect at 7 days. Repeat injections every 3 to 4 months are required with prolonged effects seen with each subsequent injection due to chemodenervation-induced muscle atrophy.4,7,8
Adverse Effects
Commercial botulinum toxin type A has been associated with minor and transient side effects. Moderate complications seen in the neck region include transient soft-tissue edema, dermal ecchymoses, intramuscular hematoma, diffuse muscle soreness, neck flexor weakness, and headaches.4,8,9
The use of botulinum toxin for chemodenervation of the platysma can produce significant weakness of other neck muscles, including the sternocleidomastoid, cricothyroid, sternothyroid, and sternohyoid. Floppy neck and dysphagia may be due to diffusion of the toxin into the muscles of deglutition of the larynx; injection directly into the sternocleidomastoid muscle; or a result of the systemic effects of large dosages. Hoarseness, breathiness, and dysphagia may occur 3 to 4 days after injection, especially with doses over 75 IU.10
The recommended concentration of botulinum toxin type A causes a diffusion average of 1 cm in all directions from the injection sites. However, as the dilution increases, so does the zone of diffusion. Typical discharge instructions for platysma treatment include the overuse of the neck muscles for 2 to 4 hours after injection to encourage the botulinum toxin uptake for optimal result. Site manipulation (rubbing or massaging) also increases diffusion. For botulinum toxin type B, the zone of diffusion is greater because its molecular weight is less than the type A toxins, thus making it an undesirable agent for aesthetic facial chemodenervation.4,11
Toxin Resistance
Botulinum toxin resistance is a known complication that occurs normally as a result of the body recognizing the neurotoxin as a foreign substance and producing neutralizing antibodies (NAb). Primary botulinum toxin failure is known in patients who require high doses of the neurotoxin for treatment of neuromuscular disorders.12 Complete secondary therapy failure is known to occur in cosmetic patients after a single dose and those who have been receiving low-dose botulinum toxin regularly. The risk of NAb development increases with long-term treatment and high doses.12-18
Floppy Neck and Dysphagia
As previously noted, floppy neck and dysphagia are adverse clinical findings of botulinum toxin effect on the platysma, sternocleidomastoid, or the paralaryngeal muscles. In this case, the patient was fortunate to have only sustained weakness of the platysma and sternocleidomastoid muscles despite both a large neck and total body dose. Paralaryngeal muscle paralysis is not life-threatening, but the distress may precipitate paradoxical vocal cord motion and stridor.
Stridor
Stridor is typically a symptom of an upper airway obstruction (UAO) process. Typical UAO conditions encountered in the ED are infections (eg, epiglottitis, croup), foreign body, allergy, and laryngeal trauma. The age of the patient, onset of stridor, course of the stridor (ie, intermittent, continuous, worsening), associated symptoms (eg, fever, rash, swelling of oral soft tissues), and bruising must be ascertained.
In differentiating the etiology of stridor, the EP should observe the patient for any associated change in voice, inability to handle secretions, and position of comfort. Patients with stridor require admission and evaluation by an otolaryngologist as expeditiously as possible because impending UAO may quickly progress to complete UAO necessitating emergent intubation.
An atypical presentation of stridor to the ED is sporadic stridor. Sporadic attacks of stridor during activity have been associated with the entity of paradoxical vocal cord motion. Patients usually describe a choking sensation with inability to breathe resulting in an audible inspiratory and/or expiratory sound—ie, stridor. Wheezing may or may not be present. Patients may also describe tightness in the neck and sometimes in the chest. The attacks are usually seconds to minutes in duration. More often, there is a precipitating or an inducing factor such as hyperventilation, cough, panting, phonatory tasks, or the inhalation of irritants or perfume, or an oropharyngeal or laryngeal manipulation prior or postextubation. The feeling of stress alone is commonly reported prior to the attacks. When evaluating patients presenting with floppy neck, dysphagia, and stridor, it is imperative that the clinician conduct a thorough history and physical examination to determine if the symptoms are secondary to a systemic or local effect, and whether the patient will progress to an acute UAO (vocal cord paralysis) necessitating intubation in the ED and subsequent tracheostomy.19,20
Conclusion
The ready availability of botulinum toxins and their low-cost-benefit ratio continue to promote over-utilization for treatment of facial wrinkles, platysmal bands, and horizontal lines; migraine headache; and hyperhidrosis. Complications associated with overuse of botulinum toxins are due to either administration of a large single dose or from regional diffusion. With the increasing number of patients receiving botulinum injections, EPs should be aware of the four available toxin types onset of action, adverse events, and potential life-threatening complications of regional neck injections.
References
1. Huang W, Foster JA, Rogachefsky AS. Pharmacology of botulinum toxin. J Am Acad Dermatol. 2000;43(2 Pt 1):249-259. doi:10.1067/mjd.2000.105567.
2. Lamanna C. The most poisonous poison. Science. 1959;130(3378):763-772.
3. Blitzer A, Brin MF, Keen MS, Aviv JE. Botulinum toxin for the treatment of hyperfunctional lines of the face. Arch Otolaryngol Head Neck Surg. 1993;119(9):1018-1022.
4. Carruthers A, Carruthers J. Clinical indications and injection technique for the cosmetic use of botulinum A exotoxin. Dermatol Surg. 1998;24(11):1189-1194.
5. Carruthers J, Carruthers A. Botox use in the mid and lower face and neck. Semin Cutan Med Surg. 2001;20(2):85-92. doi:10.1053/sder.2001.25139
6. Hoefflin SM. Anatomy of the platysma and lip depressor muscles. A simplified mnemonic approach. Dermatol Surg. 1998;24(11):1225-1231.
7. Brandt FS, Bellman B. Cosmetic use of botulinum A exotoxin for the aging neck. Dermatol Surg. 1998;24(11):1232-1234.
8. Klein AW. Complications and adverse reactions with the use of botulinum toxin. Semin Cutan Med Surg. 2001;20(2):109-120. doi:10.1053/sder.2001.25964.
9. Carruthers A, Kiene K, Carruthers J. Botulinum A exotoxin use in clinical dermatology. J Am Acad Dermatol. 1996;34(5 Pt 1):788-797.
10. Howell K, Selber P, Graham HK, Reddihough D. Botulinum neurotoxin A: an unusual systemic effect. J Paediatr Child Health. 2007:43(6):499-501. doi:10.1111/j.1440-1754.2007.01122.x.
11. Carruthers A, Carruthers J. Toxins 99, new information about the botulinum neurotoxins. Dermatol Surg. 2000;26(3):174-176.
12. Dressler D, Adib Saberi F. New formulation of Botox: complete antibody-induced treatment failure in cervical dystonia. J Neurol Neurosurg Psychiatry. 2007;78(1):108-109. doi:10.1136/jnnp.2006.093419.
13. Borodic G. Immunologic resistance after repeated botulinum toxin type a injections for facial rhytides. Ophthal Plast Reconstr Surg. 2006;22:239-240. doi:10.1097/01.iop.0000217703.80859.a3.
14. Goschel H, Wohlfarth K, Frevert J, Dengler R, Bigalke H. Botulinum A toxin therapy: neutralizing and nonneutralizing antibodies—therapeutic consequences. Exp Neurol. 1997;147(1):96-102. doi:10.1006/exnr.1997.6580.
15. Hatheway CL. Toxigenic clostridia. Clin Microbiol Rev. 1990;3(1):66-98.
16. Smith LA. Development of recombinant vaccines for botulinum neurotoxin. Toxicon. 1998;36(11):1539-1548.
17. Houser MK, Sheean GL, Lees AJ. Further studies using higher doses of botulinum toxin type F for torticollis resistant to botulinum toxin type A. J Neurol Neurosurg Psychiatry. 1998;64(5):577-580.
18. Dressler D, Wohlfahrt K, Meyer-Rogge E, Wiest L, Bigalke H. Antibody-induced failure of botulinum toxin a therapy in cosmetic indications. Dermatol Surg. 2010;36 Suppl 4:2182-2187. doi:10.1111/j.1524-4725.2010.01710.x.
19. Maschka DA, Bauman NM, McCray PB Jr, Hoffman HT, Karnell MP, Smith RJ. A classification scheme for paradoxical vocal cord motion. Laryngoscope. 1997;107(11 Pt 1):1429-1435.
20. Altman KW, Simpson CB, Amin MR, Abaza M, Balkissoon R, Casiano RR. Cough and paradoxical vocal fold motion. Otolaryngol Head Neck Surg. 2002;127(6):501-511. doi:10.1067/mhn.2002.127589.
References
1. Huang W, Foster JA, Rogachefsky AS. Pharmacology of botulinum toxin. J Am Acad Dermatol. 2000;43(2 Pt 1):249-259. doi:10.1067/mjd.2000.105567.
2. Lamanna C. The most poisonous poison. Science. 1959;130(3378):763-772.
3. Blitzer A, Brin MF, Keen MS, Aviv JE. Botulinum toxin for the treatment of hyperfunctional lines of the face. Arch Otolaryngol Head Neck Surg. 1993;119(9):1018-1022.
4. Carruthers A, Carruthers J. Clinical indications and injection technique for the cosmetic use of botulinum A exotoxin. Dermatol Surg. 1998;24(11):1189-1194.
5. Carruthers J, Carruthers A. Botox use in the mid and lower face and neck. Semin Cutan Med Surg. 2001;20(2):85-92. doi:10.1053/sder.2001.25139
6. Hoefflin SM. Anatomy of the platysma and lip depressor muscles. A simplified mnemonic approach. Dermatol Surg. 1998;24(11):1225-1231.
7. Brandt FS, Bellman B. Cosmetic use of botulinum A exotoxin for the aging neck. Dermatol Surg. 1998;24(11):1232-1234.
8. Klein AW. Complications and adverse reactions with the use of botulinum toxin. Semin Cutan Med Surg. 2001;20(2):109-120. doi:10.1053/sder.2001.25964.
9. Carruthers A, Kiene K, Carruthers J. Botulinum A exotoxin use in clinical dermatology. J Am Acad Dermatol. 1996;34(5 Pt 1):788-797.
10. Howell K, Selber P, Graham HK, Reddihough D. Botulinum neurotoxin A: an unusual systemic effect. J Paediatr Child Health. 2007:43(6):499-501. doi:10.1111/j.1440-1754.2007.01122.x.
11. Carruthers A, Carruthers J. Toxins 99, new information about the botulinum neurotoxins. Dermatol Surg. 2000;26(3):174-176.
12. Dressler D, Adib Saberi F. New formulation of Botox: complete antibody-induced treatment failure in cervical dystonia. J Neurol Neurosurg Psychiatry. 2007;78(1):108-109. doi:10.1136/jnnp.2006.093419.
13. Borodic G. Immunologic resistance after repeated botulinum toxin type a injections for facial rhytides. Ophthal Plast Reconstr Surg. 2006;22:239-240. doi:10.1097/01.iop.0000217703.80859.a3.
14. Goschel H, Wohlfarth K, Frevert J, Dengler R, Bigalke H. Botulinum A toxin therapy: neutralizing and nonneutralizing antibodies—therapeutic consequences. Exp Neurol. 1997;147(1):96-102. doi:10.1006/exnr.1997.6580.
15. Hatheway CL. Toxigenic clostridia. Clin Microbiol Rev. 1990;3(1):66-98.
16. Smith LA. Development of recombinant vaccines for botulinum neurotoxin. Toxicon. 1998;36(11):1539-1548.
17. Houser MK, Sheean GL, Lees AJ. Further studies using higher doses of botulinum toxin type F for torticollis resistant to botulinum toxin type A. J Neurol Neurosurg Psychiatry. 1998;64(5):577-580.
18. Dressler D, Wohlfahrt K, Meyer-Rogge E, Wiest L, Bigalke H. Antibody-induced failure of botulinum toxin a therapy in cosmetic indications. Dermatol Surg. 2010;36 Suppl 4:2182-2187. doi:10.1111/j.1524-4725.2010.01710.x.
19. Maschka DA, Bauman NM, McCray PB Jr, Hoffman HT, Karnell MP, Smith RJ. A classification scheme for paradoxical vocal cord motion. Laryngoscope. 1997;107(11 Pt 1):1429-1435.
20. Altman KW, Simpson CB, Amin MR, Abaza M, Balkissoon R, Casiano RR. Cough and paradoxical vocal fold motion. Otolaryngol Head Neck Surg. 2002;127(6):501-511. doi:10.1067/mhn.2002.127589.
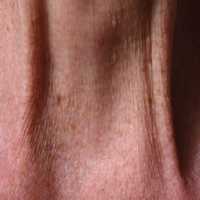
Do patients with submassive pulmonary embolism benefit from thrombolytic therapy?
For patients with submassive acute pulmonary embolism—the intermediate category between massive and low-risk—whether to give thrombolytic therapy is controversial. In general, patients with massive pulmonary embolism need this therapy, whereas those with low-risk pulmonary embolism do not—and neither do most of those with submassive embolism. But where should we draw the line?
More than 600,000 patients suffer pulmonary embolisms every year in the United States, and 50,000 to 200,000 people die of them.1–3 In various studies,4–6 within 1 year, 12.9% of patients had another pulmonary embolism, 7.3% developed chronic venous insufficiency, and 3.8% developed chronic thromboembolic pulmonary hypertension.
THREE CATEGORIES OF RISK
Episodes of acute pulmonary embolism are classified as low-risk (about 70% of cases), hemodynamically unstable or massive (5%), or submassive (25%).7,8
Low-risk acute pulmonary embolism is defined by the absence of right ventricular dysfunction and the absence of myocardial necrosis. The death rate in such cases is less than 1%.9 Its pharmacologic management includes parenteral anticoagulation and early initiation of long-term anticoagulation therapy, which the American College of Chest Physicians (ACCP) gives a grade IB recommendation (strong, based on moderate-quality evidence).10
Massive or hemodynamically unstable pulmonary embolism is characterized by any of the following, in the absence of other causes8:
- Sustained hypotension (systolic blood pressure < 90 mm Hg for ≥ 15 minutes)
- An absolute decrease in systolic blood pressure of 40 mm Hg or more
- Need for inotropic support
- Cardiac arrest
- Bradycardia (heart rate < 40 beats per minute).
The death rate is more than 30% in patients presenting with shock and approaches 70% in those presenting with cardiac arrest.11,12 Therefore, the consensus is that this category of pulmonary embolism merits aggressive treatment. Systemic thrombolytic therapy is recommended in those who are not at high risk of major bleeding, though the ACCP gives it only a grade 2C recommendation (weak, based on low-quality evidence).10
Submassive pulmonary embolism is defined by evidence of right ventricular dysfunction with normal blood pressure. According to the ACCP guidelines, thrombolytic therapy should be considered (grade 2C recommendation) for patients with acute pulmonary embolism without hypotension and with a low bleeding risk (with no renal failure and not on dual antiplatelet therapy), but at high risk of developing hypotension.10
DIAGNOSING SUBMASSIVE PULMONARY EMBOLISM, DELINEATING ITS SEVERITY
In managing acute pulmonary embolism, it is critical to recognize whether a patient is at high risk of clinical deterioration.
Blood pressure
The systolic blood pressure not only indicates whether the patient has hypotension (systolic blood pressure < 90 mm Hg) and therefore massive rather than submassive or low-risk pulmonary embolism; it is also important as a baseline value. A decrease in systolic blood pressure of 40 mm Hg or more is associated with worse outcomes.12
Right ventricular dysfunction
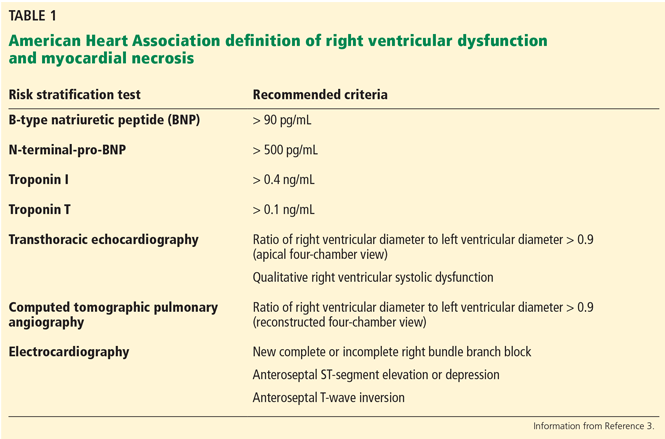
The physiologic response to occlusion of the pulmonary arteries can result in early myocardial injury and right ventricular dysfunction, which can be assessed by various methods (Table 1).
Electrocardiographic signs. Right heart strain may be recognized on electrocardiography as:
- Evidence of new complete or incomplete right bundle branch block
- T-wave inversion in the anterolateral leads V1 to V4
- S1Q3T3 (a large S wave in lead I, a Q wave in lead III, and an inverted T wave in lead III, the classic pattern of acute cor pulmonale).13
These findings add incremental prognostic value to echocardiographic findings in patients with submassive pulmonary embolism.14
Cardiac biomarkers such as B-type natriuretic peptide (BNP), N-terminal-pro-BNP (NT-pro-BNP), cardiac troponins, and heart-type fatty acid-binding protein (H-FABP) are also markers of right-sided myocardial damage and strain and can be used to identify patients with submassive pulmonary embolism.15 Abnormal levels of these substances are as follows:
- Troponin T greater than 0.1 ng/mL
- Troponin I greater than 0.4 ng/mL
- BNP greater than 90 pg/mL
- NT-pro-BNP greater than 500 pg/mL
- H-FABP less than 6 ng/mL.
These levels have prognostic value, identifying patients with submassive pulmonary embolism at risk of deterioration or death,14,16,17
Echocardiographic signs. Right ventricular dysfunction can be assessed quickly at the bedside with portable transthoracic echocardiography. A meta-analysis showed that close to 37% of hemodynamically stable patients with pulmonary embolism had echocardiographic evidence of right ventricular dysfunction on presentation and a higher short-term mortality rate.18 Evidence of right ventricular dysfunction includes the following:
- New-onset hypokinesis or akinesis
- Right ventricular dilation
- Right ventricular free-wall hypokinesis with apical sparing (the McConnell sign)
- Paradoxical movement of the interventricular septum
- Newly increased right ventricular systolic pressure
- Pulmonary hypertension, defined as tricuspid regurgitation jet velocity greater than 2.8 m/s.15,19
Computed tomographic (CT) angiography is widely available. Findings that have prognostic value in determining those at higher risk of death include the following20,21:
- A dilated right ventricle—ratio of right ventricle to left ventricle diameter (RV:LV ratio) greater than 0.9
- Interventricular septal bowing.
PESI and sPESI scores. The European Society of Cardiology 2014 guidelines stratify the risk in normotensive patients with pulmonary embolism according to their score on the Pulmonary Embolism Severity Index (PESI) or the simplified PESI (sPESI). There are five PESI classes. Those in PESI class III or IV or with an sPESI score of 1 or more (on a scale of 0 to 6) are considered at intermediate risk of clinical deterioration and are then further risk-stratified according to whether they have right ventricular dysfunction (based on echocardiography or computed tomography) and elevated cardiac biomarkers. These scoring systems are based on easily obtainable clinical information such as age, male sex, history of cancer, history of heart failure, history of chronic lung disease, heart rate, systolic blood pressure, respiratory rate, temperature, and altered mental status, and calculators are readily available.
Anticoagulation for all, plus thrombolysis for some
Patients with neither right ventricular dysfunction nor elevated cardiac biomarkers are at intermediate to low risk of clinical deterioration, and it is recommended that they be given anticoagulation therapy in an inpatient setting.
On the other hand, patients with both right ventricular dysfunction and elevated cardiac biomarkers are considered at intermediate to high risk of clinical deterioration; they should also be managed with anticoagulation and monitored closely for the need for rescue reperfusion therapy with thrombolytics.22
THROMBOLYTIC AGENTS
Thrombolytic agents are the cornerstone of management for patients presenting with pulmonary embolism who are at high risk. As noted above, these agents are recommended in massive pulmonary embolism, but their role in submassive pulmonary embolism remains controversial.
Thrombolytics work by activating endogenous plasminogen. The resulting plasmin promotes clot lysis, reducing the size of the thrombus, decreasing pulmonary vasculature resistance, and improving right ventricular function.23
To date, three thrombolytic agents have received US Food and Drug Administration approval for use in massive pulmonary embolism: alteplase, urokinase, and streptokinase. But only alteplase is still available in the United States. Alteplase is also the best tolerated, whereas streptokinase is highly antigenic and may cause further deterioration in an already unstable patient. Alteplase is also the most fibrin-specific and is considered the most potent of the three agents.24
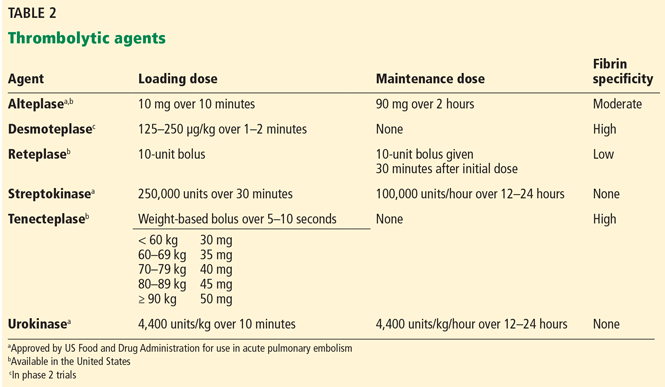
Additional thrombolytic agents under investigation for use in acute pulmonary embolism include reteplase, tenecteplase, and desmoteplase. These agents are more fibrin-specific than alteplase. Reteplase is a second-generation recombinant tissue-type plasminogen activator with a quicker onset of action and longer half-life than alteplase, allowing for bolus dosing. Tenecteplase, a variant of alteplase, is cleared more slowly and is 14 times more fibrin-specific than alteplase, also allowing for bolus dosing. Desmoteplase, a fibrin-specific agent currently in phase 2 trials, also has a longer half-life and appears to be more potent than alteplase. Table 2 lists the dosing and the degree of fibrin specificity of these agents.
Complications of thrombolytic therapy
Submassive pulmonary embolism has a low death rate, and the benefit of systemic thrombolytic therapy for this condition is controversial. Therefore, risk stratification is very important before pursuing this therapy.
A meta-analysis25 of 16 randomized controlled trials included 2,125 patients with pulmonary embolism:
- 210 (9.88%) in the low-risk category
- 1,499 (70.54%) in the submassive category
- 31 (1.46%) in the massive category
- 385 (18.11%) whose disease severity could not be determined.
Major bleeding occurred in:
- 98 (9.24%) of 1,061 patients receiving anticoagulation plus thrombolytics
- 36 (3.42%) of 1,054 patients receiving anticoagulation without thrombolytics (odds ratio [OR] 2.73, 95% confidence interval [CI] 1.91–3.91; number needed to harm [NNH] 18, 95% CI 13–27).
Intracranial hemorrhage occurred in:
- 15 (1.46%) of 2,014 patients on thrombolytic therapy
- 2 (0.19%) of 1,019 patients not on thrombolytic therapy (OR 4.63, 95% CI 1.78–12.04; NNH 78, 95% CI 48–206).
Of note, the incidence of major bleeding was not significantly increased in those age 65 or younger receiving thrombolytics (OR 1.25, 95% CI 0.5–3.14).

Comments. Definitions of major bleeding varied in the individual trials. Additionally, intracranial hemorrhage was included as a major bleeding end point in any trial in which it was not prespecified.
These findings emphasize the importance of risk stratification before pursuing thrombolytic therapy in submassive pulmonary embolism.
Table 3 lists absolute and relative contraindications to thrombolytic therapy.
MAJOR STUDIES IN SUBMASSIVE PULMONARY EMBOLISM
The MAPPET-3 trial
The Management Strategies and Prognosis of Pulmonary Embolism-3 (MAPPET-3) trial,26 in 2002, was the first major trial to study thrombolytic therapy in submassive pulmonary embolism.
In this prospective, randomized, double-blinded trial conducted in Germany, 118 patients received heparin with alteplase (100 mg over 2 hours) and 138 received heparin with placebo. The primary end point was in-hospital death or clinical deterioration requiring escalation of treatment. Secondary outcomes included recurrent pulmonary embolism, major bleeding, and stroke. Major bleeding was defined as fatal bleeding, hemorrhagic stroke, or drop in the hemoglobin concentration by more than 4 g/dL, with or without the need for red blood cell transfusion.
Right ventricular dysfunction was diagnosed by echocardiography in 30% of the participants, and the rest of the patients were classified as having submassive pulmonary embolism on the basis of electrocardiographic criteria alone. It is likely that the latter group had a less severe form of the disease and did not benefit from thrombolytic therapy as much as patients with echocardiographic findings of right ventricular dysfunction and elevated serum cardiac biomarkers.
Results. At 30 days, 11% of the alteplase-plus-heparin group had met the primary end point, compared with 24.6% of the placebo-plus-heparin group (P = .006). The difference was mostly driven by the need for secondary thrombolysis (7.6% vs 23.2%, P = .001), since 32 (23.2%) of the 138 patients in the control group required secondary thrombolysis, accounting for 94% of the 34 patients in this group who required escalation of treatment. Most cases of clinical deterioration in this group occurred within the first 5 days.
Mortality rates were 3.4% in the heparin-plus-alteplase group and 2.2% in the heparin-plus-placebo group, but the difference was not statistically significant (P = .71).
Major bleeding occurred in 1 patient in the heparin-plus-alteplase group and 5 patients in the heparin-plus-placebo group, but the trial’s definition of major bleeding may have resulted in underestimation of this event. The definition put forth by the International Society on Thrombosis and Haemostasis is less strict, defining bleeding in nonsurgical patients as major if it is fatal, symptomatic in a critical area or organ, or causing a fall in hemoglobin level of 2.0 g/dL or more, leading to transfusion of two or more units of whole blood or red cells.27
MOPETT trial
The Moderate Pulmonary Embolism Treated with Thrombolysis (MOPETT) trial28 was a single-center, randomized trial in 121 normotensive patients with “moderate” pulmonary embolism and right ventricular dysfunction. Moderate pulmonary embolism was defined as signs and symptoms of pulmonary embolism with evidence on computed tomographic angiography of greater than 70% involvement with thrombus in two or more lobes or left or right main pulmonary arteries, or by a high-probability ventilation-perfusion scan showing a mismatch in two or more lobes.
The authors defined right ventricular dysfunction by elevated cardiac markers or by findings on echocardiography. Only 20% of the participants were enrolled on the basis of right ventricular dysfunction on echocardiography, whereas almost 60% had elevated cardiac biomarkers.
The primary outcome was the development of pulmonary hypertension, based on echocardiography.
Patients were randomized to either anticoagulation alone (unfractionated heparin or low-molecular-weight heparin) or anticoagulation plus half-dose alteplase (0.5 mg/kg, to a maximum of 50 mg). Echocardiography was performed within 2 hours of study entry, at 48 hours, and every 6 months thereafter. The mean duration of follow-up was 28 months.
Results. Pulmonary hypertension developed in 16% of the anticoagulation-plus-alteplase group vs 57% of the anticoagulation-only group (P < .001). However, the clinical relevance of elevated right-sided pressures observed by echocardiography in asymptomatic patients is uncertain. Alteplase had no impact on the rates of death or recurrent pulmonary embolism.
PEITHO trial
The 2014 Pulmonary Embolism Thrombolysis (PEITHO) trial29 was a prospective, randomized, double-blinded, placebo-controlled trial conducted in 13 countries between 2007 and 2012. A total of 1,005 patients with submassive pulmonary embolism received unfractionated heparin and were randomized to also receive either tenecteplase or placebo.
The primary end point was death from any cause or hemodynamic compromise within 7 days of randomization. Secondary end points included death within 30 days, recurrence of pulmonary embolism, major bleeding, and stroke.
Echocardiography was strongly recommended for diagnosing right ventricular dysfunction in all patients. When this was unavailable, computed tomographic images were used to assess right ventricular dysfunction. Major bleeding was characterized as moderate or severe, and bleeding events were reported using the International Society on Thrombosis and Haemostasis criteria.
Results. The tenecteplase group had a lower rate of the primary end point at 7 days (2.6% vs 5.6%, P = .02), but no significant reduction in all-cause mortality at 30 days (2.4% vs 3.2%, P = .42). In addition, the tenecteplase group had higher rates of major extracranial bleeding (6.3% vs 1.2%, P < .001) and stroke (2.4% vs 0.2%, P = .004) at 7 days.
Although the PEITHO trial showed no reduction in mortality rates and showed a higher rate of major bleeding, this may have been related to using a higher dose of tenecteplase than needed in this population. Further studies should be conducted to confirm this theory.
TOPCOAT trial
The Tenecteplase or Placebo, Cardiopulmonary Outcomes at Three months (TOPCOAT) trial,30 published in 2014, was a multicenter, double-blind, intention-to-treat, randomized trial carried out in eight centers in the United States. The authors evaluated a composite outcome (death, circulatory shock, intubation, major bleeding, recurrent pulmonary embolism, and functional capacity) with the use of tenecteplase in submassive pulmonary embolism.
A total of 83 patients received low-molecular-weight heparin and were randomized to also receive either tenecteplase or placebo. Submassive pulmonary embolism was defined as evidence of right ventricular strain based on echocardiographic findings and elevated cardiac markers (troponin, BNP, or NT-pro-BNP).
Results. Adverse outcomes occurred in 37% of the patients in the placebo group compared with 15% of those in the tenecteplase group (P = .017). The study was terminated early because the lead author relocated to another institution.
Wang et al
In a prospective, randomized, open-label, multicenter study31 conducted in China between 2002 and 2006, 118 patients received low-molecular-weight heparin plus alteplase in a dose of either 100 mg or 50 mg over 2 hours.
Results. There were significantly fewer bleeding episodes in patients receiving half-dose alteplase in the subgroups that weighed less than 65 kg (14.8% vs 41.2%, P = .049) or who had a body mass index less than 24 kg/m2 (8.7% vs 42.9%, P = .014).
Meta-analysis
A subgroup analysis25 of patients with submassive pulmonary embolism from a 2014 meta-analysis of randomized controlled trials of thrombolytic therapy in pulmonary embolism found that thrombolysis was associated with a lower mortality rate (OR 0.48; 95% CI 0.25–0.92) but a higher rate of major bleeding (OR 3.19, 95% CI 2.07–4.92).
Is there a role for low-dose thrombolytic therapy?
The MOPETT study, discussed above, evaluated the effect of thrombolysis in a low (“safe”) dose in reducing pulmonary artery pressure in moderate pulmonary embolism.28 The primary end points were pulmonary hypertension and the composite end point of pulmonary hypertension and recurrent pulmonary embolism. In the thrombolysis group, the pulmonary arterial pressure fell immediately and was still lower at 28 months. As mentioned, although the incidence of pulmonary hypertension was lower with thrombolysis, no significant differences were noted in the rate of individual outcomes of death and recurrent pulmonary embolism when assessed independently. Furthermore, the definition of moderate pulmonary embolism used in this study is different from the submassive criteria.
Wang et al31 enrolled patients to receive low-molecular-weight heparin plus alteplase in a dose of either 50 or 100 mg. The rate of bleeding was lower with the 50-mg dose, but only in the subset of patients with lower weight and body mass index.
What is the role of catheter-guided therapy?
Catheter-directed therapy involves infusing thrombolytic agents directly into the pulmonary arteries where the clots are. The idea is to expose the patient to lower doses of systemic thrombolytics and thus decrease the risk of bleeding.
The ULTIMA study32 (Ultrasound-Assisted, Catheter-Directed Thrombolysis for Acute Intermediate-Risk Pulmonary Embolism) evaluated whether this treatment would reverse right ventricular dilation in intermediate-risk patients, compared with anticoagulation. Intermediate-risk pulmonary embolism was defined as an embolus located in at least one main or proximal lower lobe pulmonary artery and an RV:LV ratio of at least 1.0 obtained from the echocardiographic apical four-chamber view.
The study showed hemodynamic improvement as evidenced by a lower RV:LV ratio. However, at 90 days the mortality rate was not significantly lower in the treatment group than in the control group. Of note, no major bleeding events were reported in the treatment group.
The SEATTLE II trial,33 a nonrandomized study completed in April 2013, evaluated the efficacy and safety of ultrasonographically guided, catheter-based, low-dose fibrinolysis for patients with massive and submassive pulmonary embolism. Patients had CT evidence of proximal pulmonary embolism and a dilated right ventricle (RV:LV ratio ≥ 0.9). Patients received alteplase 24 mg, either as 1 mg/hour for 24 hours with a unilateral catheter or 1 mg/hour in each of two catheters for 12 hours.
At 48 hours after the procedure, the mean RV:LV ratio had decreased from 1.55 to 1.13, the mean pulmonary arterial systolic pressure had fallen, and the anatomical clot burden had decreased. A total of 15 patients (10%) experienced major bleeding but there were no reports of any fatal or intracranial bleeding. Patients with massive pulmonary embolism were more likely to experience major bleeding episodes than those with submassive pulmonary embolism (23% vs 7%, P = .02).
The weakness of this study is that it was a single-arm study and therefore limits comparisons with other therapies such as tissue plasminogen activator for massive pulmonary embolism or anticoagulation. Also, although there was an acute improvement in hemodynamics, it is unclear if that translates to improvement in mortality rate.
Based on the available literature,29,31,33 patients presenting with submassive pulmonary embolism who are of low body weight (body mass index < 24 kg/m2 or weight < 65 kg) or are over age 75 may benefit from low-dose catheter-guided thrombolysis therapy or low-dose systemic alteplase (50 mg). Further studies should be conducted comparing these two therapeutic strategies.
SURGICAL EMBOLECTOMY: STILL THE LAST RESORT
Surgery has been the last resort for patients with pulmonary embolism. Although recent reports show a decrease in mortality from advances in surgical embolectomy, the mortality rate is greater than 10%.34
- Indications for surgical embolectomy are35:
- Failure of or contraindications to thrombolytic therapy
- Continued hemodynamic instability despite maximal medical therapy
- Associated cardiac pathology such as patent foramen ovale, atrial septal defect, and free-floating right heart thrombi
- Inadequate time for systemic thrombolytics to take effect.
No large or randomized controlled trials of surgical embolectomy for submassive pulmonary embolism have been done. In one study, of 47 patients undergoing surgical embolectomy, 15 (32%) met the criteria for submassive pulmonary embolism based on right ventricular hemodynamic dysfunction. The report did not mention if biomarkers such as troponin and BNP were considered in the decision to operate.36
At this time, surgical embolectomy remains a last resort for patients with acute massive pulmonary embolism who have contraindications to thrombolysis or for whom it has failed. Given the risk of death associated with surgical embolectomy, large randomized controlled trials need to be done to see if there is any benefit in the submassive pulmonary embolism population.
ONE TREATMENT DOES NOT FIT ALL
Given the evidence to date, we do not recommend thrombolytic therapy for all patients with submassive pulmonary embolism. The risk of complications (hemorrhage) is significant, and the benefit is unclear. A one-treatment-for-all approach cannot be applied in this situation.

Furthermore, each of the trials performed so far defined submassive pulmonary embolism slightly differently (Table 4), and many were underpowered to detect a difference in mortality rates between the treatment groups. Further studies are needed to determine the exact subset of patients with submassive pulmonary embolism that may truly benefit from thrombolytic therapy.
As such, patients with submassive pulmonary embolism should be managed by a multidisciplinary team to determine the need for thrombolytic therapy, especially in low doses, on a case-by-case basis according to the patient’s risk of further clinical deterioration.
- Silverstein MD, Heit JA, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med 1998; 158:585–593.
- Stein PD, Matta F, Keyes DC, Willyerd GL. Impact of vena cava filters on in-hospital case fatality rate from pulmonary embolism. Am J Med 2012; 125:478–484.
- Wood KE. Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest 2002; 121:877–905.
- Heit JA, Mohr DN, Silverstein MD, Petterson TM, O’Fallon WM, Melton LJ 3rd. Predictors of recurrence after deep vein thrombosis and pulmonary embolism: a population-based cohort study. Arch Intern Med 2000; 160:761–768.
- Mohr DN, Silverstein MD, Heit JA, Petterson TM, O’Fallon WM, Melton LJ. The venous stasis syndrome after deep venous thrombosis or pulmonary embolism: a population-based study. Mayo Clin Proc 2000; 75:1249–1256.
- Pengo V, Lensing AW, Prins MH, et al; Thromboembolic Pulmonary Hypertension Study Group. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004; 350:2257–2264.
- Tapson VF. Acute pulmonary embolism. N Engl J Med 2008; 358:1037–1052.
- Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
- Kreit JW. The impact of right ventricular dysfunction on the prognosis and therapy of normotensive patients with pulmonary embolism. Chest 2004; 125:1539–1545.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Comess KA, DeRook FA, Russell ML, Tognazzi-Evans TA, Beach KW. The incidence of pulmonary embolism in unexplained sudden cardiac arrest with pulseless electrical activity. Am J Med 2000; 109:351–356.
- Kasper W, Konstantinides S, Geibel A, et al. Management strategies and determinants of outcome in acute major pulmonary embolism: results of a multicenter registry. J Am Coll Cardiol 1997; 30:1165–1171.
- Piazza G. Submassive pulmonary embolism. JAMA 2013; 309:171–180.
- Klok FA, Mos IC, Huisman MV. Brain-type natriuretic peptide levels in the prediction of adverse outcome in patients with pulmonary embolism: a systematic review and meta-analysis. Am J Respir Crit Care Med 2008; 178:425–430.
- Vanni S, Polidori G, Vergara R, et al. Prognostic value of ECG among patients with acute pulmonary embolism and normal blood pressure. Am J Med 2009; 122:257–264.
- Amorim S, Dias P, Rodrigues RA, et al. Troponin I as a marker of right ventricular dysfunction and severity of pulmonary embolism. Rev Port Cardiol 2006; 25:181–186.
- Dellas C, Puls M, Lankeit M, et al. Elevated heart-type fatty acid-binding protein levels on admission predict an adverse outcome in normotensive patients with acute pulmonary embolism. J Am Coll Cardiol 2010; 55:2150–2157.
- Cho JH, Kutti Sridharan G, Kim SH, et al. Right ventricular dysfunction as an echocardiographic prognostic factor in hemodynamically stable patients with acute pulmonary embolism: a meta-analysis. BMC Cardiovasc Disord 2014; 14:64.
- Nazeyrollas P, Metz D, Jolly D, et al. Use of transthoracic Doppler echocardiography combined with clinical and electrocardiographic data to predict acute pulmonary embolism. Eur Heart J 1996;17: 779–786.
- Wake N, Kumamaru KK, George E, et al. Computed tomography and echocardiography in patients with acute pulmonary embolism: part 1: correlation of findings of right ventricular enlargement. J Thorac Imaging 2014; 29:W1–W6.
- Becattini C, Agnelli G, Germini F, Vedovati MC. Computed tomography to assess risk of death in acute pulmonary embolism: a meta-analysis. Eur Respir J 2014; 43:1678–1690.
- Konstantinides SV, Torbicki A, Agnelli G, et al; Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). 2014 ESC guidelines on the diagnosis and management of acute pulmonary embolism. Eur Heart J 2014; 35:3033–3069, 69a–69k.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; American Heart Association Council on Peripheral Vascular Disease; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Daley MJ, Lat I. Clinical controversies in thrombolytic therapy for the management of acute pulmonary embolism. Pharmacotherapy 2012; 32:158–172.
- Chatterjee S, Chakraborty A, Weinberg I, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA 2014; 311:2414–2421.
- Konstantinides S, Geibel A, Heusel G, Heinrich F, Kasper W; Management Strategies and Prognosis of Pulmonary Embolism-3 Trial Investigators. Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism. N Engl J Med 2002; 347:1143–1150.
- Schulman S, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost 2005; 3:692–694.
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M; “MOPETT” Investigators. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 2013; 111:273–277.
- Meyer G, Vicaut E, Danays T, et al; PEITHO Investigators. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
- Kline JA, Nordenholz KE, Courtney DM, et al. Treatment of submassive pulmonary embolism with tenecteplase or placebo: cardiopulmonary outcomes at 3 months: multicenter double-blind, placebo-controlled randomized trial. J Thromb Haemost 2014; 12:459–468.
- Wang C, Zhai Z, Yang Y, et al; China Venous Thromboembolism (VTE) Study Group. Efficacy and safety of low dose recombinant tissue-type plasminogen activator for the treatment of acute pulmonary thromboembolism: a randomized, multicenter, controlled trial. Chest 2010; 137:254–262.
- Kucher N, Boekstegers P, Muller OJ, et al. Randomized, controlled trial of ultrasound-assisted catheter-directed thrombolysis for acute intermediate-risk pulmonary embolism. Circulation 2014; 129:479–486.
- Piazza G, Hohlfelder B, Jaff MR, et al; SEATTLE II Investigators. A prospective, single-arm, multicenter trial of ultrasound-facilitated, catheter-directed, low-dose fibrinolysis for acute massive and submassive pulmonary embolism (The SEATTLE II Study). JACC Cardiovasc Interv 2015; 8:1382–1392.
- Stein PD, Alnas M, Beemath A, Patel NR. Outcome of pulmonary embolectomy. Am J Cardiol 2007; 99:421–423.
- He C, Von Segesser LK, Kappetein PA, Mestres CA, Smith JA, Choong CK. Acute pulmonary embolectomy. Eur J Cardiothorac Surg 2013; 43:1087–1095.
- Leacche M, Unic D, Goldhaber SZ, et al. Modern surgical treatment of massive pulmonary embolism: results in 47 consecutive patients after rapid diagnosis and aggressive surgical approach. J Thorac Cardiovasc Surg 2005; 129:1018–1023.
For patients with submassive acute pulmonary embolism—the intermediate category between massive and low-risk—whether to give thrombolytic therapy is controversial. In general, patients with massive pulmonary embolism need this therapy, whereas those with low-risk pulmonary embolism do not—and neither do most of those with submassive embolism. But where should we draw the line?
More than 600,000 patients suffer pulmonary embolisms every year in the United States, and 50,000 to 200,000 people die of them.1–3 In various studies,4–6 within 1 year, 12.9% of patients had another pulmonary embolism, 7.3% developed chronic venous insufficiency, and 3.8% developed chronic thromboembolic pulmonary hypertension.
THREE CATEGORIES OF RISK
Episodes of acute pulmonary embolism are classified as low-risk (about 70% of cases), hemodynamically unstable or massive (5%), or submassive (25%).7,8
Low-risk acute pulmonary embolism is defined by the absence of right ventricular dysfunction and the absence of myocardial necrosis. The death rate in such cases is less than 1%.9 Its pharmacologic management includes parenteral anticoagulation and early initiation of long-term anticoagulation therapy, which the American College of Chest Physicians (ACCP) gives a grade IB recommendation (strong, based on moderate-quality evidence).10
Massive or hemodynamically unstable pulmonary embolism is characterized by any of the following, in the absence of other causes8:
- Sustained hypotension (systolic blood pressure < 90 mm Hg for ≥ 15 minutes)
- An absolute decrease in systolic blood pressure of 40 mm Hg or more
- Need for inotropic support
- Cardiac arrest
- Bradycardia (heart rate < 40 beats per minute).
The death rate is more than 30% in patients presenting with shock and approaches 70% in those presenting with cardiac arrest.11,12 Therefore, the consensus is that this category of pulmonary embolism merits aggressive treatment. Systemic thrombolytic therapy is recommended in those who are not at high risk of major bleeding, though the ACCP gives it only a grade 2C recommendation (weak, based on low-quality evidence).10
Submassive pulmonary embolism is defined by evidence of right ventricular dysfunction with normal blood pressure. According to the ACCP guidelines, thrombolytic therapy should be considered (grade 2C recommendation) for patients with acute pulmonary embolism without hypotension and with a low bleeding risk (with no renal failure and not on dual antiplatelet therapy), but at high risk of developing hypotension.10
DIAGNOSING SUBMASSIVE PULMONARY EMBOLISM, DELINEATING ITS SEVERITY
In managing acute pulmonary embolism, it is critical to recognize whether a patient is at high risk of clinical deterioration.
Blood pressure
The systolic blood pressure not only indicates whether the patient has hypotension (systolic blood pressure < 90 mm Hg) and therefore massive rather than submassive or low-risk pulmonary embolism; it is also important as a baseline value. A decrease in systolic blood pressure of 40 mm Hg or more is associated with worse outcomes.12
Right ventricular dysfunction
The physiologic response to occlusion of the pulmonary arteries can result in early myocardial injury and right ventricular dysfunction, which can be assessed by various methods (Table 1).
Electrocardiographic signs. Right heart strain may be recognized on electrocardiography as:
- Evidence of new complete or incomplete right bundle branch block
- T-wave inversion in the anterolateral leads V1 to V4
- S1Q3T3 (a large S wave in lead I, a Q wave in lead III, and an inverted T wave in lead III, the classic pattern of acute cor pulmonale).13
These findings add incremental prognostic value to echocardiographic findings in patients with submassive pulmonary embolism.14
Cardiac biomarkers such as B-type natriuretic peptide (BNP), N-terminal-pro-BNP (NT-pro-BNP), cardiac troponins, and heart-type fatty acid-binding protein (H-FABP) are also markers of right-sided myocardial damage and strain and can be used to identify patients with submassive pulmonary embolism.15 Abnormal levels of these substances are as follows:
- Troponin T greater than 0.1 ng/mL
- Troponin I greater than 0.4 ng/mL
- BNP greater than 90 pg/mL
- NT-pro-BNP greater than 500 pg/mL
- H-FABP less than 6 ng/mL.
These levels have prognostic value, identifying patients with submassive pulmonary embolism at risk of deterioration or death,14,16,17
Echocardiographic signs. Right ventricular dysfunction can be assessed quickly at the bedside with portable transthoracic echocardiography. A meta-analysis showed that close to 37% of hemodynamically stable patients with pulmonary embolism had echocardiographic evidence of right ventricular dysfunction on presentation and a higher short-term mortality rate.18 Evidence of right ventricular dysfunction includes the following:
- New-onset hypokinesis or akinesis
- Right ventricular dilation
- Right ventricular free-wall hypokinesis with apical sparing (the McConnell sign)
- Paradoxical movement of the interventricular septum
- Newly increased right ventricular systolic pressure
- Pulmonary hypertension, defined as tricuspid regurgitation jet velocity greater than 2.8 m/s.15,19
Computed tomographic (CT) angiography is widely available. Findings that have prognostic value in determining those at higher risk of death include the following20,21:
- A dilated right ventricle—ratio of right ventricle to left ventricle diameter (RV:LV ratio) greater than 0.9
- Interventricular septal bowing.
PESI and sPESI scores. The European Society of Cardiology 2014 guidelines stratify the risk in normotensive patients with pulmonary embolism according to their score on the Pulmonary Embolism Severity Index (PESI) or the simplified PESI (sPESI). There are five PESI classes. Those in PESI class III or IV or with an sPESI score of 1 or more (on a scale of 0 to 6) are considered at intermediate risk of clinical deterioration and are then further risk-stratified according to whether they have right ventricular dysfunction (based on echocardiography or computed tomography) and elevated cardiac biomarkers. These scoring systems are based on easily obtainable clinical information such as age, male sex, history of cancer, history of heart failure, history of chronic lung disease, heart rate, systolic blood pressure, respiratory rate, temperature, and altered mental status, and calculators are readily available.
Anticoagulation for all, plus thrombolysis for some
Patients with neither right ventricular dysfunction nor elevated cardiac biomarkers are at intermediate to low risk of clinical deterioration, and it is recommended that they be given anticoagulation therapy in an inpatient setting.
On the other hand, patients with both right ventricular dysfunction and elevated cardiac biomarkers are considered at intermediate to high risk of clinical deterioration; they should also be managed with anticoagulation and monitored closely for the need for rescue reperfusion therapy with thrombolytics.22
THROMBOLYTIC AGENTS
Thrombolytic agents are the cornerstone of management for patients presenting with pulmonary embolism who are at high risk. As noted above, these agents are recommended in massive pulmonary embolism, but their role in submassive pulmonary embolism remains controversial.
Thrombolytics work by activating endogenous plasminogen. The resulting plasmin promotes clot lysis, reducing the size of the thrombus, decreasing pulmonary vasculature resistance, and improving right ventricular function.23
To date, three thrombolytic agents have received US Food and Drug Administration approval for use in massive pulmonary embolism: alteplase, urokinase, and streptokinase. But only alteplase is still available in the United States. Alteplase is also the best tolerated, whereas streptokinase is highly antigenic and may cause further deterioration in an already unstable patient. Alteplase is also the most fibrin-specific and is considered the most potent of the three agents.24
Additional thrombolytic agents under investigation for use in acute pulmonary embolism include reteplase, tenecteplase, and desmoteplase. These agents are more fibrin-specific than alteplase. Reteplase is a second-generation recombinant tissue-type plasminogen activator with a quicker onset of action and longer half-life than alteplase, allowing for bolus dosing. Tenecteplase, a variant of alteplase, is cleared more slowly and is 14 times more fibrin-specific than alteplase, also allowing for bolus dosing. Desmoteplase, a fibrin-specific agent currently in phase 2 trials, also has a longer half-life and appears to be more potent than alteplase. Table 2 lists the dosing and the degree of fibrin specificity of these agents.
Complications of thrombolytic therapy
Submassive pulmonary embolism has a low death rate, and the benefit of systemic thrombolytic therapy for this condition is controversial. Therefore, risk stratification is very important before pursuing this therapy.
A meta-analysis25 of 16 randomized controlled trials included 2,125 patients with pulmonary embolism:
- 210 (9.88%) in the low-risk category
- 1,499 (70.54%) in the submassive category
- 31 (1.46%) in the massive category
- 385 (18.11%) whose disease severity could not be determined.
Major bleeding occurred in:
- 98 (9.24%) of 1,061 patients receiving anticoagulation plus thrombolytics
- 36 (3.42%) of 1,054 patients receiving anticoagulation without thrombolytics (odds ratio [OR] 2.73, 95% confidence interval [CI] 1.91–3.91; number needed to harm [NNH] 18, 95% CI 13–27).
Intracranial hemorrhage occurred in:
- 15 (1.46%) of 2,014 patients on thrombolytic therapy
- 2 (0.19%) of 1,019 patients not on thrombolytic therapy (OR 4.63, 95% CI 1.78–12.04; NNH 78, 95% CI 48–206).
Of note, the incidence of major bleeding was not significantly increased in those age 65 or younger receiving thrombolytics (OR 1.25, 95% CI 0.5–3.14).
Comments. Definitions of major bleeding varied in the individual trials. Additionally, intracranial hemorrhage was included as a major bleeding end point in any trial in which it was not prespecified.
These findings emphasize the importance of risk stratification before pursuing thrombolytic therapy in submassive pulmonary embolism.
Table 3 lists absolute and relative contraindications to thrombolytic therapy.
MAJOR STUDIES IN SUBMASSIVE PULMONARY EMBOLISM
The MAPPET-3 trial
The Management Strategies and Prognosis of Pulmonary Embolism-3 (MAPPET-3) trial,26 in 2002, was the first major trial to study thrombolytic therapy in submassive pulmonary embolism.
In this prospective, randomized, double-blinded trial conducted in Germany, 118 patients received heparin with alteplase (100 mg over 2 hours) and 138 received heparin with placebo. The primary end point was in-hospital death or clinical deterioration requiring escalation of treatment. Secondary outcomes included recurrent pulmonary embolism, major bleeding, and stroke. Major bleeding was defined as fatal bleeding, hemorrhagic stroke, or drop in the hemoglobin concentration by more than 4 g/dL, with or without the need for red blood cell transfusion.
Right ventricular dysfunction was diagnosed by echocardiography in 30% of the participants, and the rest of the patients were classified as having submassive pulmonary embolism on the basis of electrocardiographic criteria alone. It is likely that the latter group had a less severe form of the disease and did not benefit from thrombolytic therapy as much as patients with echocardiographic findings of right ventricular dysfunction and elevated serum cardiac biomarkers.
Results. At 30 days, 11% of the alteplase-plus-heparin group had met the primary end point, compared with 24.6% of the placebo-plus-heparin group (P = .006). The difference was mostly driven by the need for secondary thrombolysis (7.6% vs 23.2%, P = .001), since 32 (23.2%) of the 138 patients in the control group required secondary thrombolysis, accounting for 94% of the 34 patients in this group who required escalation of treatment. Most cases of clinical deterioration in this group occurred within the first 5 days.
Mortality rates were 3.4% in the heparin-plus-alteplase group and 2.2% in the heparin-plus-placebo group, but the difference was not statistically significant (P = .71).
Major bleeding occurred in 1 patient in the heparin-plus-alteplase group and 5 patients in the heparin-plus-placebo group, but the trial’s definition of major bleeding may have resulted in underestimation of this event. The definition put forth by the International Society on Thrombosis and Haemostasis is less strict, defining bleeding in nonsurgical patients as major if it is fatal, symptomatic in a critical area or organ, or causing a fall in hemoglobin level of 2.0 g/dL or more, leading to transfusion of two or more units of whole blood or red cells.27
MOPETT trial
The Moderate Pulmonary Embolism Treated with Thrombolysis (MOPETT) trial28 was a single-center, randomized trial in 121 normotensive patients with “moderate” pulmonary embolism and right ventricular dysfunction. Moderate pulmonary embolism was defined as signs and symptoms of pulmonary embolism with evidence on computed tomographic angiography of greater than 70% involvement with thrombus in two or more lobes or left or right main pulmonary arteries, or by a high-probability ventilation-perfusion scan showing a mismatch in two or more lobes.
The authors defined right ventricular dysfunction by elevated cardiac markers or by findings on echocardiography. Only 20% of the participants were enrolled on the basis of right ventricular dysfunction on echocardiography, whereas almost 60% had elevated cardiac biomarkers.
The primary outcome was the development of pulmonary hypertension, based on echocardiography.
Patients were randomized to either anticoagulation alone (unfractionated heparin or low-molecular-weight heparin) or anticoagulation plus half-dose alteplase (0.5 mg/kg, to a maximum of 50 mg). Echocardiography was performed within 2 hours of study entry, at 48 hours, and every 6 months thereafter. The mean duration of follow-up was 28 months.
Results. Pulmonary hypertension developed in 16% of the anticoagulation-plus-alteplase group vs 57% of the anticoagulation-only group (P < .001). However, the clinical relevance of elevated right-sided pressures observed by echocardiography in asymptomatic patients is uncertain. Alteplase had no impact on the rates of death or recurrent pulmonary embolism.
PEITHO trial
The 2014 Pulmonary Embolism Thrombolysis (PEITHO) trial29 was a prospective, randomized, double-blinded, placebo-controlled trial conducted in 13 countries between 2007 and 2012. A total of 1,005 patients with submassive pulmonary embolism received unfractionated heparin and were randomized to also receive either tenecteplase or placebo.
The primary end point was death from any cause or hemodynamic compromise within 7 days of randomization. Secondary end points included death within 30 days, recurrence of pulmonary embolism, major bleeding, and stroke.
Echocardiography was strongly recommended for diagnosing right ventricular dysfunction in all patients. When this was unavailable, computed tomographic images were used to assess right ventricular dysfunction. Major bleeding was characterized as moderate or severe, and bleeding events were reported using the International Society on Thrombosis and Haemostasis criteria.
Results. The tenecteplase group had a lower rate of the primary end point at 7 days (2.6% vs 5.6%, P = .02), but no significant reduction in all-cause mortality at 30 days (2.4% vs 3.2%, P = .42). In addition, the tenecteplase group had higher rates of major extracranial bleeding (6.3% vs 1.2%, P < .001) and stroke (2.4% vs 0.2%, P = .004) at 7 days.
Although the PEITHO trial showed no reduction in mortality rates and showed a higher rate of major bleeding, this may have been related to using a higher dose of tenecteplase than needed in this population. Further studies should be conducted to confirm this theory.
TOPCOAT trial
The Tenecteplase or Placebo, Cardiopulmonary Outcomes at Three months (TOPCOAT) trial,30 published in 2014, was a multicenter, double-blind, intention-to-treat, randomized trial carried out in eight centers in the United States. The authors evaluated a composite outcome (death, circulatory shock, intubation, major bleeding, recurrent pulmonary embolism, and functional capacity) with the use of tenecteplase in submassive pulmonary embolism.
A total of 83 patients received low-molecular-weight heparin and were randomized to also receive either tenecteplase or placebo. Submassive pulmonary embolism was defined as evidence of right ventricular strain based on echocardiographic findings and elevated cardiac markers (troponin, BNP, or NT-pro-BNP).
Results. Adverse outcomes occurred in 37% of the patients in the placebo group compared with 15% of those in the tenecteplase group (P = .017). The study was terminated early because the lead author relocated to another institution.
Wang et al
In a prospective, randomized, open-label, multicenter study31 conducted in China between 2002 and 2006, 118 patients received low-molecular-weight heparin plus alteplase in a dose of either 100 mg or 50 mg over 2 hours.
Results. There were significantly fewer bleeding episodes in patients receiving half-dose alteplase in the subgroups that weighed less than 65 kg (14.8% vs 41.2%, P = .049) or who had a body mass index less than 24 kg/m2 (8.7% vs 42.9%, P = .014).
Meta-analysis
A subgroup analysis25 of patients with submassive pulmonary embolism from a 2014 meta-analysis of randomized controlled trials of thrombolytic therapy in pulmonary embolism found that thrombolysis was associated with a lower mortality rate (OR 0.48; 95% CI 0.25–0.92) but a higher rate of major bleeding (OR 3.19, 95% CI 2.07–4.92).
Is there a role for low-dose thrombolytic therapy?
The MOPETT study, discussed above, evaluated the effect of thrombolysis in a low (“safe”) dose in reducing pulmonary artery pressure in moderate pulmonary embolism.28 The primary end points were pulmonary hypertension and the composite end point of pulmonary hypertension and recurrent pulmonary embolism. In the thrombolysis group, the pulmonary arterial pressure fell immediately and was still lower at 28 months. As mentioned, although the incidence of pulmonary hypertension was lower with thrombolysis, no significant differences were noted in the rate of individual outcomes of death and recurrent pulmonary embolism when assessed independently. Furthermore, the definition of moderate pulmonary embolism used in this study is different from the submassive criteria.
Wang et al31 enrolled patients to receive low-molecular-weight heparin plus alteplase in a dose of either 50 or 100 mg. The rate of bleeding was lower with the 50-mg dose, but only in the subset of patients with lower weight and body mass index.
What is the role of catheter-guided therapy?
Catheter-directed therapy involves infusing thrombolytic agents directly into the pulmonary arteries where the clots are. The idea is to expose the patient to lower doses of systemic thrombolytics and thus decrease the risk of bleeding.
The ULTIMA study32 (Ultrasound-Assisted, Catheter-Directed Thrombolysis for Acute Intermediate-Risk Pulmonary Embolism) evaluated whether this treatment would reverse right ventricular dilation in intermediate-risk patients, compared with anticoagulation. Intermediate-risk pulmonary embolism was defined as an embolus located in at least one main or proximal lower lobe pulmonary artery and an RV:LV ratio of at least 1.0 obtained from the echocardiographic apical four-chamber view.
The study showed hemodynamic improvement as evidenced by a lower RV:LV ratio. However, at 90 days the mortality rate was not significantly lower in the treatment group than in the control group. Of note, no major bleeding events were reported in the treatment group.
The SEATTLE II trial,33 a nonrandomized study completed in April 2013, evaluated the efficacy and safety of ultrasonographically guided, catheter-based, low-dose fibrinolysis for patients with massive and submassive pulmonary embolism. Patients had CT evidence of proximal pulmonary embolism and a dilated right ventricle (RV:LV ratio ≥ 0.9). Patients received alteplase 24 mg, either as 1 mg/hour for 24 hours with a unilateral catheter or 1 mg/hour in each of two catheters for 12 hours.
At 48 hours after the procedure, the mean RV:LV ratio had decreased from 1.55 to 1.13, the mean pulmonary arterial systolic pressure had fallen, and the anatomical clot burden had decreased. A total of 15 patients (10%) experienced major bleeding but there were no reports of any fatal or intracranial bleeding. Patients with massive pulmonary embolism were more likely to experience major bleeding episodes than those with submassive pulmonary embolism (23% vs 7%, P = .02).
The weakness of this study is that it was a single-arm study and therefore limits comparisons with other therapies such as tissue plasminogen activator for massive pulmonary embolism or anticoagulation. Also, although there was an acute improvement in hemodynamics, it is unclear if that translates to improvement in mortality rate.
Based on the available literature,29,31,33 patients presenting with submassive pulmonary embolism who are of low body weight (body mass index < 24 kg/m2 or weight < 65 kg) or are over age 75 may benefit from low-dose catheter-guided thrombolysis therapy or low-dose systemic alteplase (50 mg). Further studies should be conducted comparing these two therapeutic strategies.
SURGICAL EMBOLECTOMY: STILL THE LAST RESORT
Surgery has been the last resort for patients with pulmonary embolism. Although recent reports show a decrease in mortality from advances in surgical embolectomy, the mortality rate is greater than 10%.34
- Indications for surgical embolectomy are35:
- Failure of or contraindications to thrombolytic therapy
- Continued hemodynamic instability despite maximal medical therapy
- Associated cardiac pathology such as patent foramen ovale, atrial septal defect, and free-floating right heart thrombi
- Inadequate time for systemic thrombolytics to take effect.
No large or randomized controlled trials of surgical embolectomy for submassive pulmonary embolism have been done. In one study, of 47 patients undergoing surgical embolectomy, 15 (32%) met the criteria for submassive pulmonary embolism based on right ventricular hemodynamic dysfunction. The report did not mention if biomarkers such as troponin and BNP were considered in the decision to operate.36
At this time, surgical embolectomy remains a last resort for patients with acute massive pulmonary embolism who have contraindications to thrombolysis or for whom it has failed. Given the risk of death associated with surgical embolectomy, large randomized controlled trials need to be done to see if there is any benefit in the submassive pulmonary embolism population.
ONE TREATMENT DOES NOT FIT ALL
Given the evidence to date, we do not recommend thrombolytic therapy for all patients with submassive pulmonary embolism. The risk of complications (hemorrhage) is significant, and the benefit is unclear. A one-treatment-for-all approach cannot be applied in this situation.
Furthermore, each of the trials performed so far defined submassive pulmonary embolism slightly differently (Table 4), and many were underpowered to detect a difference in mortality rates between the treatment groups. Further studies are needed to determine the exact subset of patients with submassive pulmonary embolism that may truly benefit from thrombolytic therapy.
As such, patients with submassive pulmonary embolism should be managed by a multidisciplinary team to determine the need for thrombolytic therapy, especially in low doses, on a case-by-case basis according to the patient’s risk of further clinical deterioration.
For patients with submassive acute pulmonary embolism—the intermediate category between massive and low-risk—whether to give thrombolytic therapy is controversial. In general, patients with massive pulmonary embolism need this therapy, whereas those with low-risk pulmonary embolism do not—and neither do most of those with submassive embolism. But where should we draw the line?
More than 600,000 patients suffer pulmonary embolisms every year in the United States, and 50,000 to 200,000 people die of them.1–3 In various studies,4–6 within 1 year, 12.9% of patients had another pulmonary embolism, 7.3% developed chronic venous insufficiency, and 3.8% developed chronic thromboembolic pulmonary hypertension.
THREE CATEGORIES OF RISK
Episodes of acute pulmonary embolism are classified as low-risk (about 70% of cases), hemodynamically unstable or massive (5%), or submassive (25%).7,8
Low-risk acute pulmonary embolism is defined by the absence of right ventricular dysfunction and the absence of myocardial necrosis. The death rate in such cases is less than 1%.9 Its pharmacologic management includes parenteral anticoagulation and early initiation of long-term anticoagulation therapy, which the American College of Chest Physicians (ACCP) gives a grade IB recommendation (strong, based on moderate-quality evidence).10
Massive or hemodynamically unstable pulmonary embolism is characterized by any of the following, in the absence of other causes8:
- Sustained hypotension (systolic blood pressure < 90 mm Hg for ≥ 15 minutes)
- An absolute decrease in systolic blood pressure of 40 mm Hg or more
- Need for inotropic support
- Cardiac arrest
- Bradycardia (heart rate < 40 beats per minute).
The death rate is more than 30% in patients presenting with shock and approaches 70% in those presenting with cardiac arrest.11,12 Therefore, the consensus is that this category of pulmonary embolism merits aggressive treatment. Systemic thrombolytic therapy is recommended in those who are not at high risk of major bleeding, though the ACCP gives it only a grade 2C recommendation (weak, based on low-quality evidence).10
Submassive pulmonary embolism is defined by evidence of right ventricular dysfunction with normal blood pressure. According to the ACCP guidelines, thrombolytic therapy should be considered (grade 2C recommendation) for patients with acute pulmonary embolism without hypotension and with a low bleeding risk (with no renal failure and not on dual antiplatelet therapy), but at high risk of developing hypotension.10
DIAGNOSING SUBMASSIVE PULMONARY EMBOLISM, DELINEATING ITS SEVERITY
In managing acute pulmonary embolism, it is critical to recognize whether a patient is at high risk of clinical deterioration.
Blood pressure
The systolic blood pressure not only indicates whether the patient has hypotension (systolic blood pressure < 90 mm Hg) and therefore massive rather than submassive or low-risk pulmonary embolism; it is also important as a baseline value. A decrease in systolic blood pressure of 40 mm Hg or more is associated with worse outcomes.12
Right ventricular dysfunction
The physiologic response to occlusion of the pulmonary arteries can result in early myocardial injury and right ventricular dysfunction, which can be assessed by various methods (Table 1).
Electrocardiographic signs. Right heart strain may be recognized on electrocardiography as:
- Evidence of new complete or incomplete right bundle branch block
- T-wave inversion in the anterolateral leads V1 to V4
- S1Q3T3 (a large S wave in lead I, a Q wave in lead III, and an inverted T wave in lead III, the classic pattern of acute cor pulmonale).13
These findings add incremental prognostic value to echocardiographic findings in patients with submassive pulmonary embolism.14
Cardiac biomarkers such as B-type natriuretic peptide (BNP), N-terminal-pro-BNP (NT-pro-BNP), cardiac troponins, and heart-type fatty acid-binding protein (H-FABP) are also markers of right-sided myocardial damage and strain and can be used to identify patients with submassive pulmonary embolism.15 Abnormal levels of these substances are as follows:
- Troponin T greater than 0.1 ng/mL
- Troponin I greater than 0.4 ng/mL
- BNP greater than 90 pg/mL
- NT-pro-BNP greater than 500 pg/mL
- H-FABP less than 6 ng/mL.
These levels have prognostic value, identifying patients with submassive pulmonary embolism at risk of deterioration or death,14,16,17
Echocardiographic signs. Right ventricular dysfunction can be assessed quickly at the bedside with portable transthoracic echocardiography. A meta-analysis showed that close to 37% of hemodynamically stable patients with pulmonary embolism had echocardiographic evidence of right ventricular dysfunction on presentation and a higher short-term mortality rate.18 Evidence of right ventricular dysfunction includes the following:
- New-onset hypokinesis or akinesis
- Right ventricular dilation
- Right ventricular free-wall hypokinesis with apical sparing (the McConnell sign)
- Paradoxical movement of the interventricular septum
- Newly increased right ventricular systolic pressure
- Pulmonary hypertension, defined as tricuspid regurgitation jet velocity greater than 2.8 m/s.15,19
Computed tomographic (CT) angiography is widely available. Findings that have prognostic value in determining those at higher risk of death include the following20,21:
- A dilated right ventricle—ratio of right ventricle to left ventricle diameter (RV:LV ratio) greater than 0.9
- Interventricular septal bowing.
PESI and sPESI scores. The European Society of Cardiology 2014 guidelines stratify the risk in normotensive patients with pulmonary embolism according to their score on the Pulmonary Embolism Severity Index (PESI) or the simplified PESI (sPESI). There are five PESI classes. Those in PESI class III or IV or with an sPESI score of 1 or more (on a scale of 0 to 6) are considered at intermediate risk of clinical deterioration and are then further risk-stratified according to whether they have right ventricular dysfunction (based on echocardiography or computed tomography) and elevated cardiac biomarkers. These scoring systems are based on easily obtainable clinical information such as age, male sex, history of cancer, history of heart failure, history of chronic lung disease, heart rate, systolic blood pressure, respiratory rate, temperature, and altered mental status, and calculators are readily available.
Anticoagulation for all, plus thrombolysis for some
Patients with neither right ventricular dysfunction nor elevated cardiac biomarkers are at intermediate to low risk of clinical deterioration, and it is recommended that they be given anticoagulation therapy in an inpatient setting.
On the other hand, patients with both right ventricular dysfunction and elevated cardiac biomarkers are considered at intermediate to high risk of clinical deterioration; they should also be managed with anticoagulation and monitored closely for the need for rescue reperfusion therapy with thrombolytics.22
THROMBOLYTIC AGENTS
Thrombolytic agents are the cornerstone of management for patients presenting with pulmonary embolism who are at high risk. As noted above, these agents are recommended in massive pulmonary embolism, but their role in submassive pulmonary embolism remains controversial.
Thrombolytics work by activating endogenous plasminogen. The resulting plasmin promotes clot lysis, reducing the size of the thrombus, decreasing pulmonary vasculature resistance, and improving right ventricular function.23
To date, three thrombolytic agents have received US Food and Drug Administration approval for use in massive pulmonary embolism: alteplase, urokinase, and streptokinase. But only alteplase is still available in the United States. Alteplase is also the best tolerated, whereas streptokinase is highly antigenic and may cause further deterioration in an already unstable patient. Alteplase is also the most fibrin-specific and is considered the most potent of the three agents.24
Additional thrombolytic agents under investigation for use in acute pulmonary embolism include reteplase, tenecteplase, and desmoteplase. These agents are more fibrin-specific than alteplase. Reteplase is a second-generation recombinant tissue-type plasminogen activator with a quicker onset of action and longer half-life than alteplase, allowing for bolus dosing. Tenecteplase, a variant of alteplase, is cleared more slowly and is 14 times more fibrin-specific than alteplase, also allowing for bolus dosing. Desmoteplase, a fibrin-specific agent currently in phase 2 trials, also has a longer half-life and appears to be more potent than alteplase. Table 2 lists the dosing and the degree of fibrin specificity of these agents.
Complications of thrombolytic therapy
Submassive pulmonary embolism has a low death rate, and the benefit of systemic thrombolytic therapy for this condition is controversial. Therefore, risk stratification is very important before pursuing this therapy.
A meta-analysis25 of 16 randomized controlled trials included 2,125 patients with pulmonary embolism:
- 210 (9.88%) in the low-risk category
- 1,499 (70.54%) in the submassive category
- 31 (1.46%) in the massive category
- 385 (18.11%) whose disease severity could not be determined.
Major bleeding occurred in:
- 98 (9.24%) of 1,061 patients receiving anticoagulation plus thrombolytics
- 36 (3.42%) of 1,054 patients receiving anticoagulation without thrombolytics (odds ratio [OR] 2.73, 95% confidence interval [CI] 1.91–3.91; number needed to harm [NNH] 18, 95% CI 13–27).
Intracranial hemorrhage occurred in:
- 15 (1.46%) of 2,014 patients on thrombolytic therapy
- 2 (0.19%) of 1,019 patients not on thrombolytic therapy (OR 4.63, 95% CI 1.78–12.04; NNH 78, 95% CI 48–206).
Of note, the incidence of major bleeding was not significantly increased in those age 65 or younger receiving thrombolytics (OR 1.25, 95% CI 0.5–3.14).
Comments. Definitions of major bleeding varied in the individual trials. Additionally, intracranial hemorrhage was included as a major bleeding end point in any trial in which it was not prespecified.
These findings emphasize the importance of risk stratification before pursuing thrombolytic therapy in submassive pulmonary embolism.
Table 3 lists absolute and relative contraindications to thrombolytic therapy.
MAJOR STUDIES IN SUBMASSIVE PULMONARY EMBOLISM
The MAPPET-3 trial
The Management Strategies and Prognosis of Pulmonary Embolism-3 (MAPPET-3) trial,26 in 2002, was the first major trial to study thrombolytic therapy in submassive pulmonary embolism.
In this prospective, randomized, double-blinded trial conducted in Germany, 118 patients received heparin with alteplase (100 mg over 2 hours) and 138 received heparin with placebo. The primary end point was in-hospital death or clinical deterioration requiring escalation of treatment. Secondary outcomes included recurrent pulmonary embolism, major bleeding, and stroke. Major bleeding was defined as fatal bleeding, hemorrhagic stroke, or drop in the hemoglobin concentration by more than 4 g/dL, with or without the need for red blood cell transfusion.
Right ventricular dysfunction was diagnosed by echocardiography in 30% of the participants, and the rest of the patients were classified as having submassive pulmonary embolism on the basis of electrocardiographic criteria alone. It is likely that the latter group had a less severe form of the disease and did not benefit from thrombolytic therapy as much as patients with echocardiographic findings of right ventricular dysfunction and elevated serum cardiac biomarkers.
Results. At 30 days, 11% of the alteplase-plus-heparin group had met the primary end point, compared with 24.6% of the placebo-plus-heparin group (P = .006). The difference was mostly driven by the need for secondary thrombolysis (7.6% vs 23.2%, P = .001), since 32 (23.2%) of the 138 patients in the control group required secondary thrombolysis, accounting for 94% of the 34 patients in this group who required escalation of treatment. Most cases of clinical deterioration in this group occurred within the first 5 days.
Mortality rates were 3.4% in the heparin-plus-alteplase group and 2.2% in the heparin-plus-placebo group, but the difference was not statistically significant (P = .71).
Major bleeding occurred in 1 patient in the heparin-plus-alteplase group and 5 patients in the heparin-plus-placebo group, but the trial’s definition of major bleeding may have resulted in underestimation of this event. The definition put forth by the International Society on Thrombosis and Haemostasis is less strict, defining bleeding in nonsurgical patients as major if it is fatal, symptomatic in a critical area or organ, or causing a fall in hemoglobin level of 2.0 g/dL or more, leading to transfusion of two or more units of whole blood or red cells.27
MOPETT trial
The Moderate Pulmonary Embolism Treated with Thrombolysis (MOPETT) trial28 was a single-center, randomized trial in 121 normotensive patients with “moderate” pulmonary embolism and right ventricular dysfunction. Moderate pulmonary embolism was defined as signs and symptoms of pulmonary embolism with evidence on computed tomographic angiography of greater than 70% involvement with thrombus in two or more lobes or left or right main pulmonary arteries, or by a high-probability ventilation-perfusion scan showing a mismatch in two or more lobes.
The authors defined right ventricular dysfunction by elevated cardiac markers or by findings on echocardiography. Only 20% of the participants were enrolled on the basis of right ventricular dysfunction on echocardiography, whereas almost 60% had elevated cardiac biomarkers.
The primary outcome was the development of pulmonary hypertension, based on echocardiography.
Patients were randomized to either anticoagulation alone (unfractionated heparin or low-molecular-weight heparin) or anticoagulation plus half-dose alteplase (0.5 mg/kg, to a maximum of 50 mg). Echocardiography was performed within 2 hours of study entry, at 48 hours, and every 6 months thereafter. The mean duration of follow-up was 28 months.
Results. Pulmonary hypertension developed in 16% of the anticoagulation-plus-alteplase group vs 57% of the anticoagulation-only group (P < .001). However, the clinical relevance of elevated right-sided pressures observed by echocardiography in asymptomatic patients is uncertain. Alteplase had no impact on the rates of death or recurrent pulmonary embolism.
PEITHO trial
The 2014 Pulmonary Embolism Thrombolysis (PEITHO) trial29 was a prospective, randomized, double-blinded, placebo-controlled trial conducted in 13 countries between 2007 and 2012. A total of 1,005 patients with submassive pulmonary embolism received unfractionated heparin and were randomized to also receive either tenecteplase or placebo.
The primary end point was death from any cause or hemodynamic compromise within 7 days of randomization. Secondary end points included death within 30 days, recurrence of pulmonary embolism, major bleeding, and stroke.
Echocardiography was strongly recommended for diagnosing right ventricular dysfunction in all patients. When this was unavailable, computed tomographic images were used to assess right ventricular dysfunction. Major bleeding was characterized as moderate or severe, and bleeding events were reported using the International Society on Thrombosis and Haemostasis criteria.
Results. The tenecteplase group had a lower rate of the primary end point at 7 days (2.6% vs 5.6%, P = .02), but no significant reduction in all-cause mortality at 30 days (2.4% vs 3.2%, P = .42). In addition, the tenecteplase group had higher rates of major extracranial bleeding (6.3% vs 1.2%, P < .001) and stroke (2.4% vs 0.2%, P = .004) at 7 days.
Although the PEITHO trial showed no reduction in mortality rates and showed a higher rate of major bleeding, this may have been related to using a higher dose of tenecteplase than needed in this population. Further studies should be conducted to confirm this theory.
TOPCOAT trial
The Tenecteplase or Placebo, Cardiopulmonary Outcomes at Three months (TOPCOAT) trial,30 published in 2014, was a multicenter, double-blind, intention-to-treat, randomized trial carried out in eight centers in the United States. The authors evaluated a composite outcome (death, circulatory shock, intubation, major bleeding, recurrent pulmonary embolism, and functional capacity) with the use of tenecteplase in submassive pulmonary embolism.
A total of 83 patients received low-molecular-weight heparin and were randomized to also receive either tenecteplase or placebo. Submassive pulmonary embolism was defined as evidence of right ventricular strain based on echocardiographic findings and elevated cardiac markers (troponin, BNP, or NT-pro-BNP).
Results. Adverse outcomes occurred in 37% of the patients in the placebo group compared with 15% of those in the tenecteplase group (P = .017). The study was terminated early because the lead author relocated to another institution.
Wang et al
In a prospective, randomized, open-label, multicenter study31 conducted in China between 2002 and 2006, 118 patients received low-molecular-weight heparin plus alteplase in a dose of either 100 mg or 50 mg over 2 hours.
Results. There were significantly fewer bleeding episodes in patients receiving half-dose alteplase in the subgroups that weighed less than 65 kg (14.8% vs 41.2%, P = .049) or who had a body mass index less than 24 kg/m2 (8.7% vs 42.9%, P = .014).
Meta-analysis
A subgroup analysis25 of patients with submassive pulmonary embolism from a 2014 meta-analysis of randomized controlled trials of thrombolytic therapy in pulmonary embolism found that thrombolysis was associated with a lower mortality rate (OR 0.48; 95% CI 0.25–0.92) but a higher rate of major bleeding (OR 3.19, 95% CI 2.07–4.92).
Is there a role for low-dose thrombolytic therapy?
The MOPETT study, discussed above, evaluated the effect of thrombolysis in a low (“safe”) dose in reducing pulmonary artery pressure in moderate pulmonary embolism.28 The primary end points were pulmonary hypertension and the composite end point of pulmonary hypertension and recurrent pulmonary embolism. In the thrombolysis group, the pulmonary arterial pressure fell immediately and was still lower at 28 months. As mentioned, although the incidence of pulmonary hypertension was lower with thrombolysis, no significant differences were noted in the rate of individual outcomes of death and recurrent pulmonary embolism when assessed independently. Furthermore, the definition of moderate pulmonary embolism used in this study is different from the submassive criteria.
Wang et al31 enrolled patients to receive low-molecular-weight heparin plus alteplase in a dose of either 50 or 100 mg. The rate of bleeding was lower with the 50-mg dose, but only in the subset of patients with lower weight and body mass index.
What is the role of catheter-guided therapy?
Catheter-directed therapy involves infusing thrombolytic agents directly into the pulmonary arteries where the clots are. The idea is to expose the patient to lower doses of systemic thrombolytics and thus decrease the risk of bleeding.
The ULTIMA study32 (Ultrasound-Assisted, Catheter-Directed Thrombolysis for Acute Intermediate-Risk Pulmonary Embolism) evaluated whether this treatment would reverse right ventricular dilation in intermediate-risk patients, compared with anticoagulation. Intermediate-risk pulmonary embolism was defined as an embolus located in at least one main or proximal lower lobe pulmonary artery and an RV:LV ratio of at least 1.0 obtained from the echocardiographic apical four-chamber view.
The study showed hemodynamic improvement as evidenced by a lower RV:LV ratio. However, at 90 days the mortality rate was not significantly lower in the treatment group than in the control group. Of note, no major bleeding events were reported in the treatment group.
The SEATTLE II trial,33 a nonrandomized study completed in April 2013, evaluated the efficacy and safety of ultrasonographically guided, catheter-based, low-dose fibrinolysis for patients with massive and submassive pulmonary embolism. Patients had CT evidence of proximal pulmonary embolism and a dilated right ventricle (RV:LV ratio ≥ 0.9). Patients received alteplase 24 mg, either as 1 mg/hour for 24 hours with a unilateral catheter or 1 mg/hour in each of two catheters for 12 hours.
At 48 hours after the procedure, the mean RV:LV ratio had decreased from 1.55 to 1.13, the mean pulmonary arterial systolic pressure had fallen, and the anatomical clot burden had decreased. A total of 15 patients (10%) experienced major bleeding but there were no reports of any fatal or intracranial bleeding. Patients with massive pulmonary embolism were more likely to experience major bleeding episodes than those with submassive pulmonary embolism (23% vs 7%, P = .02).
The weakness of this study is that it was a single-arm study and therefore limits comparisons with other therapies such as tissue plasminogen activator for massive pulmonary embolism or anticoagulation. Also, although there was an acute improvement in hemodynamics, it is unclear if that translates to improvement in mortality rate.
Based on the available literature,29,31,33 patients presenting with submassive pulmonary embolism who are of low body weight (body mass index < 24 kg/m2 or weight < 65 kg) or are over age 75 may benefit from low-dose catheter-guided thrombolysis therapy or low-dose systemic alteplase (50 mg). Further studies should be conducted comparing these two therapeutic strategies.
SURGICAL EMBOLECTOMY: STILL THE LAST RESORT
Surgery has been the last resort for patients with pulmonary embolism. Although recent reports show a decrease in mortality from advances in surgical embolectomy, the mortality rate is greater than 10%.34
- Indications for surgical embolectomy are35:
- Failure of or contraindications to thrombolytic therapy
- Continued hemodynamic instability despite maximal medical therapy
- Associated cardiac pathology such as patent foramen ovale, atrial septal defect, and free-floating right heart thrombi
- Inadequate time for systemic thrombolytics to take effect.
No large or randomized controlled trials of surgical embolectomy for submassive pulmonary embolism have been done. In one study, of 47 patients undergoing surgical embolectomy, 15 (32%) met the criteria for submassive pulmonary embolism based on right ventricular hemodynamic dysfunction. The report did not mention if biomarkers such as troponin and BNP were considered in the decision to operate.36
At this time, surgical embolectomy remains a last resort for patients with acute massive pulmonary embolism who have contraindications to thrombolysis or for whom it has failed. Given the risk of death associated with surgical embolectomy, large randomized controlled trials need to be done to see if there is any benefit in the submassive pulmonary embolism population.
ONE TREATMENT DOES NOT FIT ALL
Given the evidence to date, we do not recommend thrombolytic therapy for all patients with submassive pulmonary embolism. The risk of complications (hemorrhage) is significant, and the benefit is unclear. A one-treatment-for-all approach cannot be applied in this situation.
Furthermore, each of the trials performed so far defined submassive pulmonary embolism slightly differently (Table 4), and many were underpowered to detect a difference in mortality rates between the treatment groups. Further studies are needed to determine the exact subset of patients with submassive pulmonary embolism that may truly benefit from thrombolytic therapy.
As such, patients with submassive pulmonary embolism should be managed by a multidisciplinary team to determine the need for thrombolytic therapy, especially in low doses, on a case-by-case basis according to the patient’s risk of further clinical deterioration.
- Silverstein MD, Heit JA, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med 1998; 158:585–593.
- Stein PD, Matta F, Keyes DC, Willyerd GL. Impact of vena cava filters on in-hospital case fatality rate from pulmonary embolism. Am J Med 2012; 125:478–484.
- Wood KE. Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest 2002; 121:877–905.
- Heit JA, Mohr DN, Silverstein MD, Petterson TM, O’Fallon WM, Melton LJ 3rd. Predictors of recurrence after deep vein thrombosis and pulmonary embolism: a population-based cohort study. Arch Intern Med 2000; 160:761–768.
- Mohr DN, Silverstein MD, Heit JA, Petterson TM, O’Fallon WM, Melton LJ. The venous stasis syndrome after deep venous thrombosis or pulmonary embolism: a population-based study. Mayo Clin Proc 2000; 75:1249–1256.
- Pengo V, Lensing AW, Prins MH, et al; Thromboembolic Pulmonary Hypertension Study Group. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004; 350:2257–2264.
- Tapson VF. Acute pulmonary embolism. N Engl J Med 2008; 358:1037–1052.
- Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
- Kreit JW. The impact of right ventricular dysfunction on the prognosis and therapy of normotensive patients with pulmonary embolism. Chest 2004; 125:1539–1545.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Comess KA, DeRook FA, Russell ML, Tognazzi-Evans TA, Beach KW. The incidence of pulmonary embolism in unexplained sudden cardiac arrest with pulseless electrical activity. Am J Med 2000; 109:351–356.
- Kasper W, Konstantinides S, Geibel A, et al. Management strategies and determinants of outcome in acute major pulmonary embolism: results of a multicenter registry. J Am Coll Cardiol 1997; 30:1165–1171.
- Piazza G. Submassive pulmonary embolism. JAMA 2013; 309:171–180.
- Klok FA, Mos IC, Huisman MV. Brain-type natriuretic peptide levels in the prediction of adverse outcome in patients with pulmonary embolism: a systematic review and meta-analysis. Am J Respir Crit Care Med 2008; 178:425–430.
- Vanni S, Polidori G, Vergara R, et al. Prognostic value of ECG among patients with acute pulmonary embolism and normal blood pressure. Am J Med 2009; 122:257–264.
- Amorim S, Dias P, Rodrigues RA, et al. Troponin I as a marker of right ventricular dysfunction and severity of pulmonary embolism. Rev Port Cardiol 2006; 25:181–186.
- Dellas C, Puls M, Lankeit M, et al. Elevated heart-type fatty acid-binding protein levels on admission predict an adverse outcome in normotensive patients with acute pulmonary embolism. J Am Coll Cardiol 2010; 55:2150–2157.
- Cho JH, Kutti Sridharan G, Kim SH, et al. Right ventricular dysfunction as an echocardiographic prognostic factor in hemodynamically stable patients with acute pulmonary embolism: a meta-analysis. BMC Cardiovasc Disord 2014; 14:64.
- Nazeyrollas P, Metz D, Jolly D, et al. Use of transthoracic Doppler echocardiography combined with clinical and electrocardiographic data to predict acute pulmonary embolism. Eur Heart J 1996;17: 779–786.
- Wake N, Kumamaru KK, George E, et al. Computed tomography and echocardiography in patients with acute pulmonary embolism: part 1: correlation of findings of right ventricular enlargement. J Thorac Imaging 2014; 29:W1–W6.
- Becattini C, Agnelli G, Germini F, Vedovati MC. Computed tomography to assess risk of death in acute pulmonary embolism: a meta-analysis. Eur Respir J 2014; 43:1678–1690.
- Konstantinides SV, Torbicki A, Agnelli G, et al; Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). 2014 ESC guidelines on the diagnosis and management of acute pulmonary embolism. Eur Heart J 2014; 35:3033–3069, 69a–69k.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; American Heart Association Council on Peripheral Vascular Disease; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Daley MJ, Lat I. Clinical controversies in thrombolytic therapy for the management of acute pulmonary embolism. Pharmacotherapy 2012; 32:158–172.
- Chatterjee S, Chakraborty A, Weinberg I, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA 2014; 311:2414–2421.
- Konstantinides S, Geibel A, Heusel G, Heinrich F, Kasper W; Management Strategies and Prognosis of Pulmonary Embolism-3 Trial Investigators. Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism. N Engl J Med 2002; 347:1143–1150.
- Schulman S, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost 2005; 3:692–694.
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M; “MOPETT” Investigators. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 2013; 111:273–277.
- Meyer G, Vicaut E, Danays T, et al; PEITHO Investigators. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
- Kline JA, Nordenholz KE, Courtney DM, et al. Treatment of submassive pulmonary embolism with tenecteplase or placebo: cardiopulmonary outcomes at 3 months: multicenter double-blind, placebo-controlled randomized trial. J Thromb Haemost 2014; 12:459–468.
- Wang C, Zhai Z, Yang Y, et al; China Venous Thromboembolism (VTE) Study Group. Efficacy and safety of low dose recombinant tissue-type plasminogen activator for the treatment of acute pulmonary thromboembolism: a randomized, multicenter, controlled trial. Chest 2010; 137:254–262.
- Kucher N, Boekstegers P, Muller OJ, et al. Randomized, controlled trial of ultrasound-assisted catheter-directed thrombolysis for acute intermediate-risk pulmonary embolism. Circulation 2014; 129:479–486.
- Piazza G, Hohlfelder B, Jaff MR, et al; SEATTLE II Investigators. A prospective, single-arm, multicenter trial of ultrasound-facilitated, catheter-directed, low-dose fibrinolysis for acute massive and submassive pulmonary embolism (The SEATTLE II Study). JACC Cardiovasc Interv 2015; 8:1382–1392.
- Stein PD, Alnas M, Beemath A, Patel NR. Outcome of pulmonary embolectomy. Am J Cardiol 2007; 99:421–423.
- He C, Von Segesser LK, Kappetein PA, Mestres CA, Smith JA, Choong CK. Acute pulmonary embolectomy. Eur J Cardiothorac Surg 2013; 43:1087–1095.
- Leacche M, Unic D, Goldhaber SZ, et al. Modern surgical treatment of massive pulmonary embolism: results in 47 consecutive patients after rapid diagnosis and aggressive surgical approach. J Thorac Cardiovasc Surg 2005; 129:1018–1023.
- Silverstein MD, Heit JA, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med 1998; 158:585–593.
- Stein PD, Matta F, Keyes DC, Willyerd GL. Impact of vena cava filters on in-hospital case fatality rate from pulmonary embolism. Am J Med 2012; 125:478–484.
- Wood KE. Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest 2002; 121:877–905.
- Heit JA, Mohr DN, Silverstein MD, Petterson TM, O’Fallon WM, Melton LJ 3rd. Predictors of recurrence after deep vein thrombosis and pulmonary embolism: a population-based cohort study. Arch Intern Med 2000; 160:761–768.
- Mohr DN, Silverstein MD, Heit JA, Petterson TM, O’Fallon WM, Melton LJ. The venous stasis syndrome after deep venous thrombosis or pulmonary embolism: a population-based study. Mayo Clin Proc 2000; 75:1249–1256.
- Pengo V, Lensing AW, Prins MH, et al; Thromboembolic Pulmonary Hypertension Study Group. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004; 350:2257–2264.
- Tapson VF. Acute pulmonary embolism. N Engl J Med 2008; 358:1037–1052.
- Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
- Kreit JW. The impact of right ventricular dysfunction on the prognosis and therapy of normotensive patients with pulmonary embolism. Chest 2004; 125:1539–1545.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Comess KA, DeRook FA, Russell ML, Tognazzi-Evans TA, Beach KW. The incidence of pulmonary embolism in unexplained sudden cardiac arrest with pulseless electrical activity. Am J Med 2000; 109:351–356.
- Kasper W, Konstantinides S, Geibel A, et al. Management strategies and determinants of outcome in acute major pulmonary embolism: results of a multicenter registry. J Am Coll Cardiol 1997; 30:1165–1171.
- Piazza G. Submassive pulmonary embolism. JAMA 2013; 309:171–180.
- Klok FA, Mos IC, Huisman MV. Brain-type natriuretic peptide levels in the prediction of adverse outcome in patients with pulmonary embolism: a systematic review and meta-analysis. Am J Respir Crit Care Med 2008; 178:425–430.
- Vanni S, Polidori G, Vergara R, et al. Prognostic value of ECG among patients with acute pulmonary embolism and normal blood pressure. Am J Med 2009; 122:257–264.
- Amorim S, Dias P, Rodrigues RA, et al. Troponin I as a marker of right ventricular dysfunction and severity of pulmonary embolism. Rev Port Cardiol 2006; 25:181–186.
- Dellas C, Puls M, Lankeit M, et al. Elevated heart-type fatty acid-binding protein levels on admission predict an adverse outcome in normotensive patients with acute pulmonary embolism. J Am Coll Cardiol 2010; 55:2150–2157.
- Cho JH, Kutti Sridharan G, Kim SH, et al. Right ventricular dysfunction as an echocardiographic prognostic factor in hemodynamically stable patients with acute pulmonary embolism: a meta-analysis. BMC Cardiovasc Disord 2014; 14:64.
- Nazeyrollas P, Metz D, Jolly D, et al. Use of transthoracic Doppler echocardiography combined with clinical and electrocardiographic data to predict acute pulmonary embolism. Eur Heart J 1996;17: 779–786.
- Wake N, Kumamaru KK, George E, et al. Computed tomography and echocardiography in patients with acute pulmonary embolism: part 1: correlation of findings of right ventricular enlargement. J Thorac Imaging 2014; 29:W1–W6.
- Becattini C, Agnelli G, Germini F, Vedovati MC. Computed tomography to assess risk of death in acute pulmonary embolism: a meta-analysis. Eur Respir J 2014; 43:1678–1690.
- Konstantinides SV, Torbicki A, Agnelli G, et al; Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). 2014 ESC guidelines on the diagnosis and management of acute pulmonary embolism. Eur Heart J 2014; 35:3033–3069, 69a–69k.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; American Heart Association Council on Peripheral Vascular Disease; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Daley MJ, Lat I. Clinical controversies in thrombolytic therapy for the management of acute pulmonary embolism. Pharmacotherapy 2012; 32:158–172.
- Chatterjee S, Chakraborty A, Weinberg I, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA 2014; 311:2414–2421.
- Konstantinides S, Geibel A, Heusel G, Heinrich F, Kasper W; Management Strategies and Prognosis of Pulmonary Embolism-3 Trial Investigators. Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism. N Engl J Med 2002; 347:1143–1150.
- Schulman S, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost 2005; 3:692–694.
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M; “MOPETT” Investigators. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 2013; 111:273–277.
- Meyer G, Vicaut E, Danays T, et al; PEITHO Investigators. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
- Kline JA, Nordenholz KE, Courtney DM, et al. Treatment of submassive pulmonary embolism with tenecteplase or placebo: cardiopulmonary outcomes at 3 months: multicenter double-blind, placebo-controlled randomized trial. J Thromb Haemost 2014; 12:459–468.
- Wang C, Zhai Z, Yang Y, et al; China Venous Thromboembolism (VTE) Study Group. Efficacy and safety of low dose recombinant tissue-type plasminogen activator for the treatment of acute pulmonary thromboembolism: a randomized, multicenter, controlled trial. Chest 2010; 137:254–262.
- Kucher N, Boekstegers P, Muller OJ, et al. Randomized, controlled trial of ultrasound-assisted catheter-directed thrombolysis for acute intermediate-risk pulmonary embolism. Circulation 2014; 129:479–486.
- Piazza G, Hohlfelder B, Jaff MR, et al; SEATTLE II Investigators. A prospective, single-arm, multicenter trial of ultrasound-facilitated, catheter-directed, low-dose fibrinolysis for acute massive and submassive pulmonary embolism (The SEATTLE II Study). JACC Cardiovasc Interv 2015; 8:1382–1392.
- Stein PD, Alnas M, Beemath A, Patel NR. Outcome of pulmonary embolectomy. Am J Cardiol 2007; 99:421–423.
- He C, Von Segesser LK, Kappetein PA, Mestres CA, Smith JA, Choong CK. Acute pulmonary embolectomy. Eur J Cardiothorac Surg 2013; 43:1087–1095.
- Leacche M, Unic D, Goldhaber SZ, et al. Modern surgical treatment of massive pulmonary embolism: results in 47 consecutive patients after rapid diagnosis and aggressive surgical approach. J Thorac Cardiovasc Surg 2005; 129:1018–1023.
KEY POINTS
- Most patients with submassive pulmonary embolism do not need thrombolytic therapy.
- Identifying patients with submassive pulmonary embolism at highest risk of clinical deterioration can guide physicians to consider thrombolytic therapy.
- In clinical trials, thrombolytic therapy reduced the rates of secondary outcomes but did not reduce the rate of death in this patient population.
Phlegmasia cerulea dolens from radiation-induced venous stenosis
A 77-year-old man presented with a 5-day history of painful swelling of his right leg. He reported no trauma, no recent surgery, no history of thrombophilic disorder, and no prolonged immobilization. However, he had a history of prostate cancer, treated 10 years earlier with pelvic radiation.
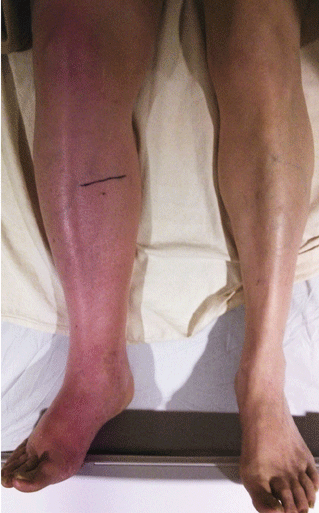
Examination revealed massive right leg swelling extending from the thigh to the ankle, along with bluish-red skin discoloration (Figure 1). Doppler ultrasonography demonstrated acute thrombosis involving the right iliofemoral veins. These findings were consistent with phlegmasia cerulea dolens.
Urgent percutaneous catheter-directed thrombolysis was performed. Venography revealed extensive thrombosis of the femoral vein (Figure 2A) extending into the right external iliac vein. This was treated with catheter-directed pharmacomechanical thrombectomy.
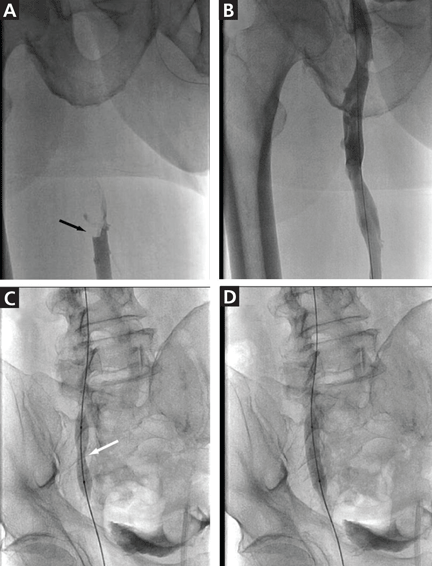
Venography after this procedure showed significant improvement in venous blood flow (Figure 2B). However, stenosis of the right external iliac vein was also noted (Figure 2C) and was treated with balloon angioplasty (Figure 2D) followed by placement of a stent (14 × 40 mm).
In the immediate postprocedural period, there was marked reduction in swelling and normalization of skin color (Figure 3). The patient did not experience significant bleeding during or after the procedure. Treatment with intravenous unfractionated heparin was continued during the hospital stay, and he was discharged on warfarin with a therapeutic international normalized ratio. At a follow-up visit 3 months later, he was asymptomatic.
A RARE BUT SEVERE TYPE OF ACUTE DEEP VEIN THROMBOSIS
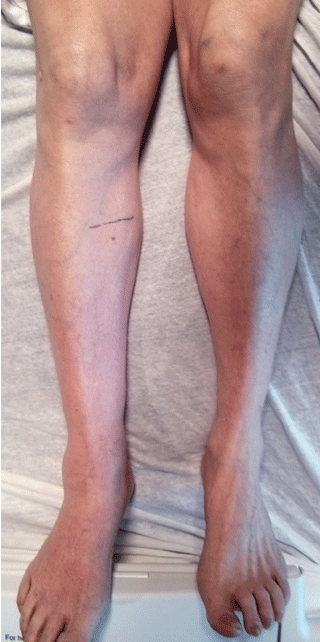
Phlegmasia cerulea dolens (painful cyanotic swollen leg) is a rare and severe form of acute deep vein thrombosis (DVT) characterized by marked limb pain, swelling, and blue discoloration.1 DVT is the most common cause of acute-onset unilateral leg pain, swelling, and skin discoloration.2
The differential diagnosis
The differential diagnosis includes infection (cellulitis, necrotizing fasciitis), compartment syndrome from limb injury, musculoskeletal conditions such as ruptured Baker cyst, venous stasis due to external compression (May-Thurner syndrome, iliac vein compression syndrome, pelvic tumor), acute limb ischemia from arterial obstruction, and complex regional pain syndrome (reflex sympathetic dystrophy).
Management recommendations
As in most cases of DVT, initial treatment of phlegmasia cerulea dolens involves systemic anticoagulation with heparin, elevation of the affected extremity, and fluid resuscitation if the patient is hypotensive. However, phlegmasia cerulea dolens is a major indication for catheter-directed thrombolysis,3,4 so an urgent vascular surgery or interventional cardiology consultation is also required. The American College of Chest Physicians recommends catheter-directed thrombolysis for acute DVT of the iliofemoral veins in patients with symptoms for less than 14 days, good functional capacity, and a life expectancy beyond 1 year.5 This intervention results in reduced incidence of postthrombotic syndrome and improved quality of life5,6 compared with anticoagulation therapy alone.
Who is at risk?
Risk factors for phlegmasia cerulea dolens include a history of malignancy, inherited or acquired thrombophilia, surgery, radiation therapy, trauma, placement of an inferior vena cava filter, and pregnancy. In our patient, the iliac vein stenosis most likely was the result of the radiation therapy he had undergone for prostate cancer.
Arterial stenosis is a well-known complication of radiation therapy and is associated with an increased risk of cardiovascular events.7,8 Radiation induces endothelial damage followed by proliferation of smooth muscle cells, resulting in luminal stenosis and thrombosis. At the cellular level, radiation leads to an acute increase in pro-inflammatory cytokines and endothelial adhesion molecules, causing the recruitment of inflammatory cells to radiation-exposed vessels and chronic activation of transcription factor NF-kappa B, leading to long-term inflammation and angiogenesis.9
Carotid, coronary, and iliac artery stenosis are known to occur around 10 years after radiation therapy to the head, neck, breast, and pelvis. Radiation-induced iliac vein stenosis is rare and can manifest as acute proximal DVT.
- Mumoli N, Invernizzi C, Luschi R, Carmignani G, Camaiti A, Cei M. Phlegmasia cerulea dolens. Circulation 2012; 125:1056–1057.
- Ely JW, Osheroff JA, Chambliss ML, Ebell MH. Approach to leg edema of unclear etiology. J Am Board Fam Med 2006; 19:148–160.
- Casey ET, Murad MH, Zumaeta-Garcia M, et al. Treatment of acute iliofemoral deep vein thrombosis. J Vasc Surg. 2012; 55:1463–1473.
- Chinsakchai K, Ten Duis K, Moll FL, de Borst GJ. Trends in management of phlegmasia cerulea dolens. Vasc Endovascular Surg 2011; 45:5–14.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):454S–545S.
- Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
- Hooning MJ, Botma A, Aleman BM, et al. Long-term risk of cardiovascular disease in 10-year survivors of breast cancer. J Natl Cancer Inst 2007; 99:365–375.
- Weintraub NL, Jones WK, Manka D. Understanding radiation-induced vascular disease. J Am Coll Cardiol 2010; 55:1237–1239.
- Halle M, Gabrielsen A, Paulsson-Berne G, et al. Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J Am Coll Cardiol 2010; 55:1227–1236.
A 77-year-old man presented with a 5-day history of painful swelling of his right leg. He reported no trauma, no recent surgery, no history of thrombophilic disorder, and no prolonged immobilization. However, he had a history of prostate cancer, treated 10 years earlier with pelvic radiation.
Examination revealed massive right leg swelling extending from the thigh to the ankle, along with bluish-red skin discoloration (Figure 1). Doppler ultrasonography demonstrated acute thrombosis involving the right iliofemoral veins. These findings were consistent with phlegmasia cerulea dolens.
Urgent percutaneous catheter-directed thrombolysis was performed. Venography revealed extensive thrombosis of the femoral vein (Figure 2A) extending into the right external iliac vein. This was treated with catheter-directed pharmacomechanical thrombectomy.
Venography after this procedure showed significant improvement in venous blood flow (Figure 2B). However, stenosis of the right external iliac vein was also noted (Figure 2C) and was treated with balloon angioplasty (Figure 2D) followed by placement of a stent (14 × 40 mm).
In the immediate postprocedural period, there was marked reduction in swelling and normalization of skin color (Figure 3). The patient did not experience significant bleeding during or after the procedure. Treatment with intravenous unfractionated heparin was continued during the hospital stay, and he was discharged on warfarin with a therapeutic international normalized ratio. At a follow-up visit 3 months later, he was asymptomatic.
A RARE BUT SEVERE TYPE OF ACUTE DEEP VEIN THROMBOSIS
Phlegmasia cerulea dolens (painful cyanotic swollen leg) is a rare and severe form of acute deep vein thrombosis (DVT) characterized by marked limb pain, swelling, and blue discoloration.1 DVT is the most common cause of acute-onset unilateral leg pain, swelling, and skin discoloration.2
The differential diagnosis
The differential diagnosis includes infection (cellulitis, necrotizing fasciitis), compartment syndrome from limb injury, musculoskeletal conditions such as ruptured Baker cyst, venous stasis due to external compression (May-Thurner syndrome, iliac vein compression syndrome, pelvic tumor), acute limb ischemia from arterial obstruction, and complex regional pain syndrome (reflex sympathetic dystrophy).
Management recommendations
As in most cases of DVT, initial treatment of phlegmasia cerulea dolens involves systemic anticoagulation with heparin, elevation of the affected extremity, and fluid resuscitation if the patient is hypotensive. However, phlegmasia cerulea dolens is a major indication for catheter-directed thrombolysis,3,4 so an urgent vascular surgery or interventional cardiology consultation is also required. The American College of Chest Physicians recommends catheter-directed thrombolysis for acute DVT of the iliofemoral veins in patients with symptoms for less than 14 days, good functional capacity, and a life expectancy beyond 1 year.5 This intervention results in reduced incidence of postthrombotic syndrome and improved quality of life5,6 compared with anticoagulation therapy alone.
Who is at risk?
Risk factors for phlegmasia cerulea dolens include a history of malignancy, inherited or acquired thrombophilia, surgery, radiation therapy, trauma, placement of an inferior vena cava filter, and pregnancy. In our patient, the iliac vein stenosis most likely was the result of the radiation therapy he had undergone for prostate cancer.
Arterial stenosis is a well-known complication of radiation therapy and is associated with an increased risk of cardiovascular events.7,8 Radiation induces endothelial damage followed by proliferation of smooth muscle cells, resulting in luminal stenosis and thrombosis. At the cellular level, radiation leads to an acute increase in pro-inflammatory cytokines and endothelial adhesion molecules, causing the recruitment of inflammatory cells to radiation-exposed vessels and chronic activation of transcription factor NF-kappa B, leading to long-term inflammation and angiogenesis.9
Carotid, coronary, and iliac artery stenosis are known to occur around 10 years after radiation therapy to the head, neck, breast, and pelvis. Radiation-induced iliac vein stenosis is rare and can manifest as acute proximal DVT.
A 77-year-old man presented with a 5-day history of painful swelling of his right leg. He reported no trauma, no recent surgery, no history of thrombophilic disorder, and no prolonged immobilization. However, he had a history of prostate cancer, treated 10 years earlier with pelvic radiation.
Examination revealed massive right leg swelling extending from the thigh to the ankle, along with bluish-red skin discoloration (Figure 1). Doppler ultrasonography demonstrated acute thrombosis involving the right iliofemoral veins. These findings were consistent with phlegmasia cerulea dolens.
Urgent percutaneous catheter-directed thrombolysis was performed. Venography revealed extensive thrombosis of the femoral vein (Figure 2A) extending into the right external iliac vein. This was treated with catheter-directed pharmacomechanical thrombectomy.
Venography after this procedure showed significant improvement in venous blood flow (Figure 2B). However, stenosis of the right external iliac vein was also noted (Figure 2C) and was treated with balloon angioplasty (Figure 2D) followed by placement of a stent (14 × 40 mm).
In the immediate postprocedural period, there was marked reduction in swelling and normalization of skin color (Figure 3). The patient did not experience significant bleeding during or after the procedure. Treatment with intravenous unfractionated heparin was continued during the hospital stay, and he was discharged on warfarin with a therapeutic international normalized ratio. At a follow-up visit 3 months later, he was asymptomatic.
A RARE BUT SEVERE TYPE OF ACUTE DEEP VEIN THROMBOSIS
Phlegmasia cerulea dolens (painful cyanotic swollen leg) is a rare and severe form of acute deep vein thrombosis (DVT) characterized by marked limb pain, swelling, and blue discoloration.1 DVT is the most common cause of acute-onset unilateral leg pain, swelling, and skin discoloration.2
The differential diagnosis
The differential diagnosis includes infection (cellulitis, necrotizing fasciitis), compartment syndrome from limb injury, musculoskeletal conditions such as ruptured Baker cyst, venous stasis due to external compression (May-Thurner syndrome, iliac vein compression syndrome, pelvic tumor), acute limb ischemia from arterial obstruction, and complex regional pain syndrome (reflex sympathetic dystrophy).
Management recommendations
As in most cases of DVT, initial treatment of phlegmasia cerulea dolens involves systemic anticoagulation with heparin, elevation of the affected extremity, and fluid resuscitation if the patient is hypotensive. However, phlegmasia cerulea dolens is a major indication for catheter-directed thrombolysis,3,4 so an urgent vascular surgery or interventional cardiology consultation is also required. The American College of Chest Physicians recommends catheter-directed thrombolysis for acute DVT of the iliofemoral veins in patients with symptoms for less than 14 days, good functional capacity, and a life expectancy beyond 1 year.5 This intervention results in reduced incidence of postthrombotic syndrome and improved quality of life5,6 compared with anticoagulation therapy alone.
Who is at risk?
Risk factors for phlegmasia cerulea dolens include a history of malignancy, inherited or acquired thrombophilia, surgery, radiation therapy, trauma, placement of an inferior vena cava filter, and pregnancy. In our patient, the iliac vein stenosis most likely was the result of the radiation therapy he had undergone for prostate cancer.
Arterial stenosis is a well-known complication of radiation therapy and is associated with an increased risk of cardiovascular events.7,8 Radiation induces endothelial damage followed by proliferation of smooth muscle cells, resulting in luminal stenosis and thrombosis. At the cellular level, radiation leads to an acute increase in pro-inflammatory cytokines and endothelial adhesion molecules, causing the recruitment of inflammatory cells to radiation-exposed vessels and chronic activation of transcription factor NF-kappa B, leading to long-term inflammation and angiogenesis.9
Carotid, coronary, and iliac artery stenosis are known to occur around 10 years after radiation therapy to the head, neck, breast, and pelvis. Radiation-induced iliac vein stenosis is rare and can manifest as acute proximal DVT.
- Mumoli N, Invernizzi C, Luschi R, Carmignani G, Camaiti A, Cei M. Phlegmasia cerulea dolens. Circulation 2012; 125:1056–1057.
- Ely JW, Osheroff JA, Chambliss ML, Ebell MH. Approach to leg edema of unclear etiology. J Am Board Fam Med 2006; 19:148–160.
- Casey ET, Murad MH, Zumaeta-Garcia M, et al. Treatment of acute iliofemoral deep vein thrombosis. J Vasc Surg. 2012; 55:1463–1473.
- Chinsakchai K, Ten Duis K, Moll FL, de Borst GJ. Trends in management of phlegmasia cerulea dolens. Vasc Endovascular Surg 2011; 45:5–14.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):454S–545S.
- Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
- Hooning MJ, Botma A, Aleman BM, et al. Long-term risk of cardiovascular disease in 10-year survivors of breast cancer. J Natl Cancer Inst 2007; 99:365–375.
- Weintraub NL, Jones WK, Manka D. Understanding radiation-induced vascular disease. J Am Coll Cardiol 2010; 55:1237–1239.
- Halle M, Gabrielsen A, Paulsson-Berne G, et al. Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J Am Coll Cardiol 2010; 55:1227–1236.
- Mumoli N, Invernizzi C, Luschi R, Carmignani G, Camaiti A, Cei M. Phlegmasia cerulea dolens. Circulation 2012; 125:1056–1057.
- Ely JW, Osheroff JA, Chambliss ML, Ebell MH. Approach to leg edema of unclear etiology. J Am Board Fam Med 2006; 19:148–160.
- Casey ET, Murad MH, Zumaeta-Garcia M, et al. Treatment of acute iliofemoral deep vein thrombosis. J Vasc Surg. 2012; 55:1463–1473.
- Chinsakchai K, Ten Duis K, Moll FL, de Borst GJ. Trends in management of phlegmasia cerulea dolens. Vasc Endovascular Surg 2011; 45:5–14.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):454S–545S.
- Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
- Hooning MJ, Botma A, Aleman BM, et al. Long-term risk of cardiovascular disease in 10-year survivors of breast cancer. J Natl Cancer Inst 2007; 99:365–375.
- Weintraub NL, Jones WK, Manka D. Understanding radiation-induced vascular disease. J Am Coll Cardiol 2010; 55:1237–1239.
- Halle M, Gabrielsen A, Paulsson-Berne G, et al. Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J Am Coll Cardiol 2010; 55:1227–1236.
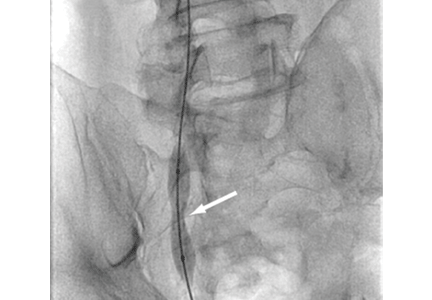
Thrombolysis in submassive pulmonary embolism: Finding the balance
In this issue of the Journal, Ataya et al1 provide a comprehensive review of thrombolysis in submassive pulmonary embolism, a subject of much debate. In massive pulmonary embolism, thrombolytic therapy is usually indicated2; in submassive pulmonary embolism, the decision is not so clear. Which patients with submassive embolism would benefit from thrombolysis, and which patients require only anticoagulant therapy? The answer lies in finding the balance between the potential benefit of thrombolytic therapy—preventing death or hemodynamic collapse—and the numerically low but potentially catastrophic risk of intracranial bleeding.
In general, submassive pulmonary embolism refers to an acute pulmonary embolus serious enough to cause evidence of right ventricular dysfunction or necrosis but not hemodynamic instability (ie, with systolic blood pressure > 90 mm Hg) on presentation.3 It accounts for about 25% of cases of pulmonary embolism,4,5 and perhaps 0.5 to 1% of patients admitted to intensive care units across the country.6 The 30-day mortality rate can be as high as 30%, making it a condition that requires prompt identification and appropriate management.
But clinical trials have failed to demonstrate a substantial improvement in mortality rates with thrombolytic therapy in patients with submassive pulmonary embolism, and have shown improvement only in other clinical end points.7 Part of the problem is that this is a heterogeneous condition, posing a challenge for the optimal design and interpretation of studies.
WHO IS AT RISK OF DEATH OR DETERIORATION?
If clinicians could ascertain in each patient whether the risk-benefit ratio is favorable for thrombolytic therapy, it would be easier to provide optimal care. This is not a straightforward task, and it requires integration of clinical judgment, high index of suspicion for deterioration, and clinical tools.
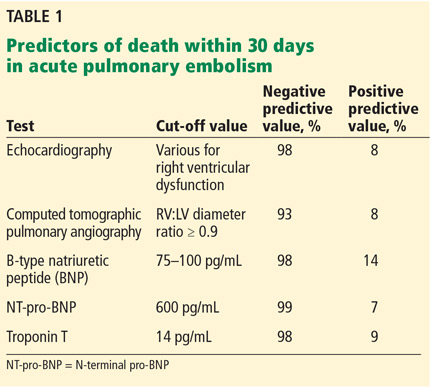
One of the challenges is that it is difficult to identify normotensive patients at the highest risk of poor outcomes. Several factors are associated with a higher risk of death within 30 days (Table 1). While each of these has a negative predictive value of about 95% or even higher (meaning that it is very good at predicting who will not die), they all have very low positive predictive values (meaning that none of them, by itself, is very good at predicting who will die).
For this reason, a multimodal approach to risk stratification has emerged. For example, Jiménez et al8 showed that normotensive patients with acute pulmonary embolism and a combination of abnormal Simplified Pulmonary Embolism Severity Index, elevated B-type natriuretic peptide level, elevated troponin level, and lower-extremity deep vein thrombosis had a 26% rate of complications (death, hemodynamic collapse, or recurrent pulmonary embolism) within 30 days.
Bova et al9 showed that the combination of borderline low systolic blood pressure (90–100 mm Hg), tachycardia (heart rate ≥ 110 beats per minute), elevated troponin, and right ventricular dysfunction by echocardiography or computed tomography allowed for the separation of three groups with significantly different rates of poor outcomes.
WHO IS AT RISK OF BLEEDING?
Estimation of the risk of bleeding is currently less sophisticated, and we need a bleeding score to use in the setting of acute pulmonary embolism. A few studies have shed some light on this issue beyond the known absolute and relative contraindications to thrombolysis.
Ataya et al1 note a meta-analysis10 showing that systemic thrombolytic therapy was not associated with an increased risk of major bleeding in patients age 65 or younger. Similarly, a large observational study showed a strong association between the risk of intracerebral hemorrhage and increasing age11 and also identified comorbidities such as kidney disease as risk factors. While the frequently cited Pulmonary Embolism Thrombolysis trial12 showed a significantly higher risk of stroke with tenecteplase, careful review of its data reveals that all 10 of the 506 patients in the tenecteplase group who sustained a hemorrhagic stroke were age 65 or older.12
A TEAM APPROACH
Thus, in patients with acute pulmonary embolism, clinicians face the difficult task of assessing the patient’s risk of death and clinical worsening and balancing that risk against the risk of bleeding, to identify those who may benefit from early reperfusion therapies, including systemic thrombolysis, catheter-directed thrombolysis, mechanical treatment, and surgical embolectomy.
Given the absence of high-quality evidence to guide these decisions, several institutions have developed multidisciplinary pulmonary embolism response teams to provide rapid evaluation and risk stratification and to recommend and implement advanced therapies, as appropriate. This is a novel concept that is still evolving but holds promise, as it integrates the experience and expertise of physicians in multiple specialties, such as pulmonary and critical care medicine, vascular medicine, interventional radiology, interventional cardiology, emergency medicine, and cardiothoracic surgery, who can then fill the currently existing knowledge gaps for clinical care and, possibly, research.13
Early published experience has documented the feasibility of this multidisciplinary approach.14 The first 95 patients treated at Cleveland Clinic had a 30-day mortality rate of 3.2%, which was lower than the expected 9% rate predicted by the Pulmonary Embolism Severity Index score (unpublished observation).
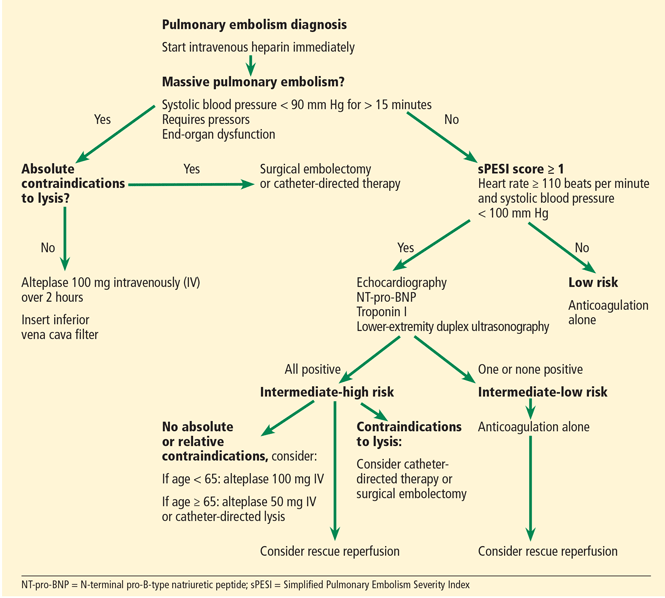
Figure 1 shows the algorithm currently used by Cleveland Clinic’s pulmonary embolism response team, with the caveat that no algorithm can fully capture the extent of the complexities and discussions that each case triggers within the team.
TOWARD BETTER UNDERSTANDING
As Ataya et al point out,1 the current state of the evidence does not allow a clear, simplistic, one-size-fits-all approach. A question that arises from this controversial topic is whether we should look for markers of right ventricular dysfunction in every patient admitted with a diagnosis of pulmonary embolism, or only in those with a significant anatomic burden of clot on imaging. Would testing everyone be an appropriate way to identify patients at risk of further deterioration early and therefore prevent adverse outcomes in a timely manner? Or would it not be cost-effective and translate into ordering more diagnostic testing, as well as an increase in downstream workup with higher healthcare costs?
Once we better understand this condition and the factors that predict a higher risk of deterioration, we should be able to design prospective studies that can help elucidate the most appropriate diagnostic and therapeutic approach for such challenging cases. In the meantime, it is important to appraise the evidence in a critical way, as Ataya et al have done in their review.
- Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. The role of thrombolytic therapy in patients with submassive pulmonary embolism. Cleve Clin J Med 2016; 83:923–932.
- Kucher N, Goldhaber SZ. Management of massive pulmonary embolism. Circulation 2005; 112:e28–e32.
- Busse LW, Vourlekis JS. Submassive pulmonary embolism. Crit Care Clin 2014; 30:447–473.
- Tapson VF. Acute pulmonary embolism. N Engl J Med 2008; 358:1037–1052.
- Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
- Bahloul M, Chaari A, Kallel H, et al. Pulmonary embolism in intensive care unit: predictive factors, clinical manifestations and outcome. Ann Thorac Med 2010; 5:97–103.
- Piazza G, Goldhaber SZ. Fibrinolysis for acute pulmonary embolism. Vasc Med 2010; 15:419–428.
- Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med 2014; 189:718–726.
- Bova C, Sanchez O, Prandoni P, et al. Identification of intermediate-risk patients with acute symptomatic pulmonary embolism. Eur Respir J 2014; 44:694–703.
- Chatterjee S, Chakraborty A, Weinberg I, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA 2014; 311:2414–2421.
- Stein PD, Matta F, Steinberger DS, Keyes DC. Intracerebral hemorrhage with thrombolytic therapy for acute pulmonary embolism. Am J Med 2012; 125:50–56.
- Meyer G, Vicaut E, Danays T, et al. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
- Dudzinski DM, Piazza G. Multidisciplinary pulmonary embolism response teams. Circulation 2016; 133:98–103.
- Kabrhel C, Rosovsky R, Channick R, et al. A multidisciplinary pulmonary embolism response team: initial 30-month experience with a novel approach to delivery of care to patients with submassive and massive pulmonary embolism. Chest 2016; 150:384–393.
In this issue of the Journal, Ataya et al1 provide a comprehensive review of thrombolysis in submassive pulmonary embolism, a subject of much debate. In massive pulmonary embolism, thrombolytic therapy is usually indicated2; in submassive pulmonary embolism, the decision is not so clear. Which patients with submassive embolism would benefit from thrombolysis, and which patients require only anticoagulant therapy? The answer lies in finding the balance between the potential benefit of thrombolytic therapy—preventing death or hemodynamic collapse—and the numerically low but potentially catastrophic risk of intracranial bleeding.
In general, submassive pulmonary embolism refers to an acute pulmonary embolus serious enough to cause evidence of right ventricular dysfunction or necrosis but not hemodynamic instability (ie, with systolic blood pressure > 90 mm Hg) on presentation.3 It accounts for about 25% of cases of pulmonary embolism,4,5 and perhaps 0.5 to 1% of patients admitted to intensive care units across the country.6 The 30-day mortality rate can be as high as 30%, making it a condition that requires prompt identification and appropriate management.
But clinical trials have failed to demonstrate a substantial improvement in mortality rates with thrombolytic therapy in patients with submassive pulmonary embolism, and have shown improvement only in other clinical end points.7 Part of the problem is that this is a heterogeneous condition, posing a challenge for the optimal design and interpretation of studies.
WHO IS AT RISK OF DEATH OR DETERIORATION?
If clinicians could ascertain in each patient whether the risk-benefit ratio is favorable for thrombolytic therapy, it would be easier to provide optimal care. This is not a straightforward task, and it requires integration of clinical judgment, high index of suspicion for deterioration, and clinical tools.
One of the challenges is that it is difficult to identify normotensive patients at the highest risk of poor outcomes. Several factors are associated with a higher risk of death within 30 days (Table 1). While each of these has a negative predictive value of about 95% or even higher (meaning that it is very good at predicting who will not die), they all have very low positive predictive values (meaning that none of them, by itself, is very good at predicting who will die).
For this reason, a multimodal approach to risk stratification has emerged. For example, Jiménez et al8 showed that normotensive patients with acute pulmonary embolism and a combination of abnormal Simplified Pulmonary Embolism Severity Index, elevated B-type natriuretic peptide level, elevated troponin level, and lower-extremity deep vein thrombosis had a 26% rate of complications (death, hemodynamic collapse, or recurrent pulmonary embolism) within 30 days.
Bova et al9 showed that the combination of borderline low systolic blood pressure (90–100 mm Hg), tachycardia (heart rate ≥ 110 beats per minute), elevated troponin, and right ventricular dysfunction by echocardiography or computed tomography allowed for the separation of three groups with significantly different rates of poor outcomes.
WHO IS AT RISK OF BLEEDING?
Estimation of the risk of bleeding is currently less sophisticated, and we need a bleeding score to use in the setting of acute pulmonary embolism. A few studies have shed some light on this issue beyond the known absolute and relative contraindications to thrombolysis.
Ataya et al1 note a meta-analysis10 showing that systemic thrombolytic therapy was not associated with an increased risk of major bleeding in patients age 65 or younger. Similarly, a large observational study showed a strong association between the risk of intracerebral hemorrhage and increasing age11 and also identified comorbidities such as kidney disease as risk factors. While the frequently cited Pulmonary Embolism Thrombolysis trial12 showed a significantly higher risk of stroke with tenecteplase, careful review of its data reveals that all 10 of the 506 patients in the tenecteplase group who sustained a hemorrhagic stroke were age 65 or older.12
A TEAM APPROACH
Thus, in patients with acute pulmonary embolism, clinicians face the difficult task of assessing the patient’s risk of death and clinical worsening and balancing that risk against the risk of bleeding, to identify those who may benefit from early reperfusion therapies, including systemic thrombolysis, catheter-directed thrombolysis, mechanical treatment, and surgical embolectomy.
Given the absence of high-quality evidence to guide these decisions, several institutions have developed multidisciplinary pulmonary embolism response teams to provide rapid evaluation and risk stratification and to recommend and implement advanced therapies, as appropriate. This is a novel concept that is still evolving but holds promise, as it integrates the experience and expertise of physicians in multiple specialties, such as pulmonary and critical care medicine, vascular medicine, interventional radiology, interventional cardiology, emergency medicine, and cardiothoracic surgery, who can then fill the currently existing knowledge gaps for clinical care and, possibly, research.13
Early published experience has documented the feasibility of this multidisciplinary approach.14 The first 95 patients treated at Cleveland Clinic had a 30-day mortality rate of 3.2%, which was lower than the expected 9% rate predicted by the Pulmonary Embolism Severity Index score (unpublished observation).
Figure 1 shows the algorithm currently used by Cleveland Clinic’s pulmonary embolism response team, with the caveat that no algorithm can fully capture the extent of the complexities and discussions that each case triggers within the team.
TOWARD BETTER UNDERSTANDING
As Ataya et al point out,1 the current state of the evidence does not allow a clear, simplistic, one-size-fits-all approach. A question that arises from this controversial topic is whether we should look for markers of right ventricular dysfunction in every patient admitted with a diagnosis of pulmonary embolism, or only in those with a significant anatomic burden of clot on imaging. Would testing everyone be an appropriate way to identify patients at risk of further deterioration early and therefore prevent adverse outcomes in a timely manner? Or would it not be cost-effective and translate into ordering more diagnostic testing, as well as an increase in downstream workup with higher healthcare costs?
Once we better understand this condition and the factors that predict a higher risk of deterioration, we should be able to design prospective studies that can help elucidate the most appropriate diagnostic and therapeutic approach for such challenging cases. In the meantime, it is important to appraise the evidence in a critical way, as Ataya et al have done in their review.
In this issue of the Journal, Ataya et al1 provide a comprehensive review of thrombolysis in submassive pulmonary embolism, a subject of much debate. In massive pulmonary embolism, thrombolytic therapy is usually indicated2; in submassive pulmonary embolism, the decision is not so clear. Which patients with submassive embolism would benefit from thrombolysis, and which patients require only anticoagulant therapy? The answer lies in finding the balance between the potential benefit of thrombolytic therapy—preventing death or hemodynamic collapse—and the numerically low but potentially catastrophic risk of intracranial bleeding.
In general, submassive pulmonary embolism refers to an acute pulmonary embolus serious enough to cause evidence of right ventricular dysfunction or necrosis but not hemodynamic instability (ie, with systolic blood pressure > 90 mm Hg) on presentation.3 It accounts for about 25% of cases of pulmonary embolism,4,5 and perhaps 0.5 to 1% of patients admitted to intensive care units across the country.6 The 30-day mortality rate can be as high as 30%, making it a condition that requires prompt identification and appropriate management.
But clinical trials have failed to demonstrate a substantial improvement in mortality rates with thrombolytic therapy in patients with submassive pulmonary embolism, and have shown improvement only in other clinical end points.7 Part of the problem is that this is a heterogeneous condition, posing a challenge for the optimal design and interpretation of studies.
WHO IS AT RISK OF DEATH OR DETERIORATION?
If clinicians could ascertain in each patient whether the risk-benefit ratio is favorable for thrombolytic therapy, it would be easier to provide optimal care. This is not a straightforward task, and it requires integration of clinical judgment, high index of suspicion for deterioration, and clinical tools.
One of the challenges is that it is difficult to identify normotensive patients at the highest risk of poor outcomes. Several factors are associated with a higher risk of death within 30 days (Table 1). While each of these has a negative predictive value of about 95% or even higher (meaning that it is very good at predicting who will not die), they all have very low positive predictive values (meaning that none of them, by itself, is very good at predicting who will die).
For this reason, a multimodal approach to risk stratification has emerged. For example, Jiménez et al8 showed that normotensive patients with acute pulmonary embolism and a combination of abnormal Simplified Pulmonary Embolism Severity Index, elevated B-type natriuretic peptide level, elevated troponin level, and lower-extremity deep vein thrombosis had a 26% rate of complications (death, hemodynamic collapse, or recurrent pulmonary embolism) within 30 days.
Bova et al9 showed that the combination of borderline low systolic blood pressure (90–100 mm Hg), tachycardia (heart rate ≥ 110 beats per minute), elevated troponin, and right ventricular dysfunction by echocardiography or computed tomography allowed for the separation of three groups with significantly different rates of poor outcomes.
WHO IS AT RISK OF BLEEDING?
Estimation of the risk of bleeding is currently less sophisticated, and we need a bleeding score to use in the setting of acute pulmonary embolism. A few studies have shed some light on this issue beyond the known absolute and relative contraindications to thrombolysis.
Ataya et al1 note a meta-analysis10 showing that systemic thrombolytic therapy was not associated with an increased risk of major bleeding in patients age 65 or younger. Similarly, a large observational study showed a strong association between the risk of intracerebral hemorrhage and increasing age11 and also identified comorbidities such as kidney disease as risk factors. While the frequently cited Pulmonary Embolism Thrombolysis trial12 showed a significantly higher risk of stroke with tenecteplase, careful review of its data reveals that all 10 of the 506 patients in the tenecteplase group who sustained a hemorrhagic stroke were age 65 or older.12
A TEAM APPROACH
Thus, in patients with acute pulmonary embolism, clinicians face the difficult task of assessing the patient’s risk of death and clinical worsening and balancing that risk against the risk of bleeding, to identify those who may benefit from early reperfusion therapies, including systemic thrombolysis, catheter-directed thrombolysis, mechanical treatment, and surgical embolectomy.
Given the absence of high-quality evidence to guide these decisions, several institutions have developed multidisciplinary pulmonary embolism response teams to provide rapid evaluation and risk stratification and to recommend and implement advanced therapies, as appropriate. This is a novel concept that is still evolving but holds promise, as it integrates the experience and expertise of physicians in multiple specialties, such as pulmonary and critical care medicine, vascular medicine, interventional radiology, interventional cardiology, emergency medicine, and cardiothoracic surgery, who can then fill the currently existing knowledge gaps for clinical care and, possibly, research.13
Early published experience has documented the feasibility of this multidisciplinary approach.14 The first 95 patients treated at Cleveland Clinic had a 30-day mortality rate of 3.2%, which was lower than the expected 9% rate predicted by the Pulmonary Embolism Severity Index score (unpublished observation).
Figure 1 shows the algorithm currently used by Cleveland Clinic’s pulmonary embolism response team, with the caveat that no algorithm can fully capture the extent of the complexities and discussions that each case triggers within the team.
TOWARD BETTER UNDERSTANDING
As Ataya et al point out,1 the current state of the evidence does not allow a clear, simplistic, one-size-fits-all approach. A question that arises from this controversial topic is whether we should look for markers of right ventricular dysfunction in every patient admitted with a diagnosis of pulmonary embolism, or only in those with a significant anatomic burden of clot on imaging. Would testing everyone be an appropriate way to identify patients at risk of further deterioration early and therefore prevent adverse outcomes in a timely manner? Or would it not be cost-effective and translate into ordering more diagnostic testing, as well as an increase in downstream workup with higher healthcare costs?
Once we better understand this condition and the factors that predict a higher risk of deterioration, we should be able to design prospective studies that can help elucidate the most appropriate diagnostic and therapeutic approach for such challenging cases. In the meantime, it is important to appraise the evidence in a critical way, as Ataya et al have done in their review.
- Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. The role of thrombolytic therapy in patients with submassive pulmonary embolism. Cleve Clin J Med 2016; 83:923–932.
- Kucher N, Goldhaber SZ. Management of massive pulmonary embolism. Circulation 2005; 112:e28–e32.
- Busse LW, Vourlekis JS. Submassive pulmonary embolism. Crit Care Clin 2014; 30:447–473.
- Tapson VF. Acute pulmonary embolism. N Engl J Med 2008; 358:1037–1052.
- Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
- Bahloul M, Chaari A, Kallel H, et al. Pulmonary embolism in intensive care unit: predictive factors, clinical manifestations and outcome. Ann Thorac Med 2010; 5:97–103.
- Piazza G, Goldhaber SZ. Fibrinolysis for acute pulmonary embolism. Vasc Med 2010; 15:419–428.
- Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med 2014; 189:718–726.
- Bova C, Sanchez O, Prandoni P, et al. Identification of intermediate-risk patients with acute symptomatic pulmonary embolism. Eur Respir J 2014; 44:694–703.
- Chatterjee S, Chakraborty A, Weinberg I, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA 2014; 311:2414–2421.
- Stein PD, Matta F, Steinberger DS, Keyes DC. Intracerebral hemorrhage with thrombolytic therapy for acute pulmonary embolism. Am J Med 2012; 125:50–56.
- Meyer G, Vicaut E, Danays T, et al. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
- Dudzinski DM, Piazza G. Multidisciplinary pulmonary embolism response teams. Circulation 2016; 133:98–103.
- Kabrhel C, Rosovsky R, Channick R, et al. A multidisciplinary pulmonary embolism response team: initial 30-month experience with a novel approach to delivery of care to patients with submassive and massive pulmonary embolism. Chest 2016; 150:384–393.
- Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. The role of thrombolytic therapy in patients with submassive pulmonary embolism. Cleve Clin J Med 2016; 83:923–932.
- Kucher N, Goldhaber SZ. Management of massive pulmonary embolism. Circulation 2005; 112:e28–e32.
- Busse LW, Vourlekis JS. Submassive pulmonary embolism. Crit Care Clin 2014; 30:447–473.
- Tapson VF. Acute pulmonary embolism. N Engl J Med 2008; 358:1037–1052.
- Kucher N, Rossi E, De Rosa M, Goldhaber SZ. Massive pulmonary embolism. Circulation 2006; 113:577–582.
- Bahloul M, Chaari A, Kallel H, et al. Pulmonary embolism in intensive care unit: predictive factors, clinical manifestations and outcome. Ann Thorac Med 2010; 5:97–103.
- Piazza G, Goldhaber SZ. Fibrinolysis for acute pulmonary embolism. Vasc Med 2010; 15:419–428.
- Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med 2014; 189:718–726.
- Bova C, Sanchez O, Prandoni P, et al. Identification of intermediate-risk patients with acute symptomatic pulmonary embolism. Eur Respir J 2014; 44:694–703.
- Chatterjee S, Chakraborty A, Weinberg I, et al. Thrombolysis for pulmonary embolism and risk of all-cause mortality, major bleeding, and intracranial hemorrhage: a meta-analysis. JAMA 2014; 311:2414–2421.
- Stein PD, Matta F, Steinberger DS, Keyes DC. Intracerebral hemorrhage with thrombolytic therapy for acute pulmonary embolism. Am J Med 2012; 125:50–56.
- Meyer G, Vicaut E, Danays T, et al. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
- Dudzinski DM, Piazza G. Multidisciplinary pulmonary embolism response teams. Circulation 2016; 133:98–103.
- Kabrhel C, Rosovsky R, Channick R, et al. A multidisciplinary pulmonary embolism response team: initial 30-month experience with a novel approach to delivery of care to patients with submassive and massive pulmonary embolism. Chest 2016; 150:384–393.
Case Studies in Toxicology: Somehow…It’s Always Lupus
Case
A 14-year-old girl with no known medical history presented to the ED via emergency medical services (EMS) approximately 1.5 hours after intentionally ingesting what she described as “a handful or two” of her mother’s lupus prescription medication in a suicide attempt. Initial vital signs and physical examination were normal, and her only complaint was nausea.
Thirty minutes after presentation, the patient suffered acute cardiovascular (CV) collapse: blood pressure, 57/39 mm Hg; heart rate, 90 beats/min. An initial electrocardiogram (ECG) revealed QRS duration of 123 milliseconds and QTc duration of 510 milliseconds, along with nonspecific T-wave abnormalities. A 150-mEq intravenous (IV) bolus of sodium bicarbonate and a 40-mEq potassium chloride IV infusion were administered, and both epinephrine and norepinephrine IV infusions were also initiated. A basic metabolic panel obtained prior to medication administration showed a potassium concentration of 1.9 mmol/L.
What is the differential diagnosis of toxicological hypokalemia?
Hypokalemia may be reflective of diminished whole body potassium stores or a transient alteration of intravascular potassium concentrations. In acute ingestions and overdose, the etiology of the hypokalemia is often electrolyte redistribution through either blockade of constitutive outward potassium leakage (eg, barium, insulin, quinine) or through increased activity of the Na+/K+-ATPase pump (eg, catecholamines, insulin, methylxanthines). This activity has little effect on whole body potassium stores, but can result in a profound fall in the serum potassium. While mild hypokalemia is generally well tolerated, more severe potassium abnormalities can cause muscular weakness, areflexic paralysis, respiratory failure, and life-threatening dysrhythmias. Common ECG findings include decreased T-wave amplitudes, ST-segment depression, and the presence or amplification of U waves.
Case Continuation
While the emergency physicians were stabilizing the patient, her mother provided additional information. Approximately 30 minutes after the exposure, the patient had become nauseated and told her mother what she had done. Her mother called EMS, and the patient was transported to the hospital, where she rapidly became symptomatic. Despite CV decompensation, she remained neurologically intact. On further questioning, the patient admitted to ingesting 6 g of her mother’s prescription of hydroxychloroquine (HCQ) in a suicide attempt but denied taking any other medications. She was stabilized on vasopressors and admitted to the intensive care unit.
What characterizes hydroxychloroquine toxicity?
Hydroxychloroquine is an aminoquinoline antibiotic that is classically used as an antimalarial to treat infection with Plasmodium vivax, P ovale, P malariae, and susceptible strains of P falciparum. In the United States, it is more commonly used to manage both rheumatoid arthritis and systemic lupus erythematosus (SLE), debilitating diseases which are estimated to affect anywhere from 161,000 to 322,000 Americans.1 Hydroxychloroquine is considered first-line therapy for SLE, but its mechanism of action in treating SLE-associated arthralgias is unclear.
Hydroxychloroquine is structurally similar to quinine and chloroquine (CQ), and not surprisingly exerts quinidine-like effects on the CV system with resultant negative inotropy and vasodilation. Its toxicity is characterized by rapid onset of clinical effects including central nervous system depression, seizures, apnea, hypotension, and arrhythmia. After large overdoses, cardiac arrest and death can occur within hours.
Hypokalemia is another hallmark of HCQ toxicity. It is thought to develop secondary to potassium channel blockade, which slows the constitutive release of potassium from the myocytes.2 As noted, the hypokalemia is transient and does not reflect whole-body depletion. With CQ, which is considered more toxic, there appears to be a correlation between the quantity of CQ ingested and both the degree of hypokalemia and the severity of the outcome. It is reasonable to assume the same for HCQ. There are little data to support that hypokalemia itself causes cardiotoxicity in patients with CQ or HCQ overdose.
Although lethal doses are not well established, animal studies suggest that HCQ is much less toxic than CQ, for which the clinical toxicity is better understood due to its more widespread use in overdose abroad.3 In children, the reported therapeutic dose is 10 mg/kg, but the minimum reported lethal dose was a single 300-mg tablet (30 mg/kg in a toddler). In adults, the toxic dose is reported as 20 mg/kg with lethal doses suggested to be as low as 30 mg/kg.
What are the treatment modalities for patients with hydroxychloroquine toxicity?
By analogy with the treatment of CQ poisoning, the mainstay of HCQ therapy is supportive care, including early intubation and ventilation to minimize metabolic demand. Direct-acting inotropes and vasopressors should be administered after saline to treat hypotension. Because of its large volume of distribution, extracorporeal removal has not proved to be of clinical value.4,5 Though data are sparse to determine its efficacy, there may be a role for giving activated charcoal, particularly following large overdoses—if it is given early after exposure and the patient has normal consciousness. If the patient is intubated and aspiration risk is minimized, gastric lavage may also be beneficial—especially when performed within an hour of the overdose. Syrup of ipecac should not be used.
High-dose diazepam is typically recommended, again by analogy with CQ, although there is no clear mechanism of action and its use remains controversial. Its protective effect in patients with CQ overdose is based on swine and rat models that demonstrated dose dependent relationships between diazepam and survival.6,7 A prospective study of CQ toxicity in humans reported improved survival rates when high-dose diazepam was given in combination with epinephrine.8 However, this study is limited by its comparison of prospectively studied patients with a retrospective control. A subsequent prospective study of moderately CQ-intoxicated patients did not find a benefit from treatment with diazepam.9 Furthermore, it remains unclear if the proposed benefit from high-dose diazepam in CQ toxicity may be extrapolated to HCQ, and cases of even massive HCQ ingestions report good outcomes without the use of high-dose diazepam.10
How aggressively should hypokalemia in hydroxychloroquine toxicity be treated?
As noted earlier, hypokalemia resulting from HCQ toxicity is transient, and aggressive repletion may result in rebound hyperkalemia once toxicity resolves. However, these dangers should be balanced with risks of hypokalemia-induced ventricular arrhythmias. Additionally, hypokalemia may be worsened by sodium bicarbonate that is administered to correct QRS prolongations, increasing the risk of dysrhythmia. Correction of hypokalemia in these cases is necessary but should be done with care and monitoring of serum potassium concentrations to minimize risk of hyperkalemia-induced ventricular arrhythmia.11
Case Conclusion
Throughout treatment, the patient remained neurologically intact. She did not receive benzodiazepines. The epinephrine and norepinephrine infusions were weaned, and she was discharged on hospital day 3 with no neurological or cardiac sequelae. She received an inpatient psychiatric evaluation and was referred to outpatient services for ongoing care.
1. Helmick CG, Felson DT, Lawrence RC, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part I. Arthritis Rheum. 2008;58(1):15-25. doi:10.1002/art.23177.
2. Clemessy JL, Favier C, Borron SW, Hantson PE, Vicaut E, Baud FJ. Hypokalaemia related to acute chloroquine ingestion. Lancet. 1995;3469(8979):877-880.
3. McChesney EW. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am J Med. 1983;75(suppl 1A):11-18.
4. Carmichael SJ, Charles B, Tett SE. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther Drug Monit. 2003;25(6):671-681.
5. Marquardt K, Albertson TE. Treatment of hydroxychloroquine overdose. Am J Emerg Med. 2001;19(5):420-424.
6. Crouzette J, Vicaut E, Palombo S, Girre C, Fournier PE. Experimental assessment of the protective activity of diazepam on the acute toxicity of chloroquine. J Toxicol Clin Toxicol. 1983;20(3):271-279.
7. Riou B, Lecarpentier Y, Barriot P, Viars P. Diazepam does not improve the mechanical performance of rat cardiac papillary muscle exposed to chloroquine in vitro. Intensive Care Med. 1989;15:390-3955.
8. Riou B, Barriot P, Rimailho A, Baud FJ. Treatment of severe chloroquine poisoning. N Engl J Med. 1988;318(1):1-6.
9. Clemessy JL, Angel G, Borron SW, et al. Therapeutic trial of diazepam versus placebo in acute chloroquine intoxications of moderate gravity. Intensive Care Med. 1996;22:1400-1405.
10. Yanturali S. Diazepam for treatment of massive chloroquine intoxication. Resuscitation. 2004;63(3):347-348.
11. Ling Ngan Wong A, Tsz Fung Cheung I, Graham CA. Hydroxychloroquine overdose: case report and recommendations for management. Eur J Emerg Med. 2008;15(1):16-8. doi:10.1097/MEJ.0b013e3280adcb56.
Case
A 14-year-old girl with no known medical history presented to the ED via emergency medical services (EMS) approximately 1.5 hours after intentionally ingesting what she described as “a handful or two” of her mother’s lupus prescription medication in a suicide attempt. Initial vital signs and physical examination were normal, and her only complaint was nausea.
Thirty minutes after presentation, the patient suffered acute cardiovascular (CV) collapse: blood pressure, 57/39 mm Hg; heart rate, 90 beats/min. An initial electrocardiogram (ECG) revealed QRS duration of 123 milliseconds and QTc duration of 510 milliseconds, along with nonspecific T-wave abnormalities. A 150-mEq intravenous (IV) bolus of sodium bicarbonate and a 40-mEq potassium chloride IV infusion were administered, and both epinephrine and norepinephrine IV infusions were also initiated. A basic metabolic panel obtained prior to medication administration showed a potassium concentration of 1.9 mmol/L.
What is the differential diagnosis of toxicological hypokalemia?
Hypokalemia may be reflective of diminished whole body potassium stores or a transient alteration of intravascular potassium concentrations. In acute ingestions and overdose, the etiology of the hypokalemia is often electrolyte redistribution through either blockade of constitutive outward potassium leakage (eg, barium, insulin, quinine) or through increased activity of the Na+/K+-ATPase pump (eg, catecholamines, insulin, methylxanthines). This activity has little effect on whole body potassium stores, but can result in a profound fall in the serum potassium. While mild hypokalemia is generally well tolerated, more severe potassium abnormalities can cause muscular weakness, areflexic paralysis, respiratory failure, and life-threatening dysrhythmias. Common ECG findings include decreased T-wave amplitudes, ST-segment depression, and the presence or amplification of U waves.
Case Continuation
While the emergency physicians were stabilizing the patient, her mother provided additional information. Approximately 30 minutes after the exposure, the patient had become nauseated and told her mother what she had done. Her mother called EMS, and the patient was transported to the hospital, where she rapidly became symptomatic. Despite CV decompensation, she remained neurologically intact. On further questioning, the patient admitted to ingesting 6 g of her mother’s prescription of hydroxychloroquine (HCQ) in a suicide attempt but denied taking any other medications. She was stabilized on vasopressors and admitted to the intensive care unit.
What characterizes hydroxychloroquine toxicity?
Hydroxychloroquine is an aminoquinoline antibiotic that is classically used as an antimalarial to treat infection with Plasmodium vivax, P ovale, P malariae, and susceptible strains of P falciparum. In the United States, it is more commonly used to manage both rheumatoid arthritis and systemic lupus erythematosus (SLE), debilitating diseases which are estimated to affect anywhere from 161,000 to 322,000 Americans.1 Hydroxychloroquine is considered first-line therapy for SLE, but its mechanism of action in treating SLE-associated arthralgias is unclear.
Hydroxychloroquine is structurally similar to quinine and chloroquine (CQ), and not surprisingly exerts quinidine-like effects on the CV system with resultant negative inotropy and vasodilation. Its toxicity is characterized by rapid onset of clinical effects including central nervous system depression, seizures, apnea, hypotension, and arrhythmia. After large overdoses, cardiac arrest and death can occur within hours.
Hypokalemia is another hallmark of HCQ toxicity. It is thought to develop secondary to potassium channel blockade, which slows the constitutive release of potassium from the myocytes.2 As noted, the hypokalemia is transient and does not reflect whole-body depletion. With CQ, which is considered more toxic, there appears to be a correlation between the quantity of CQ ingested and both the degree of hypokalemia and the severity of the outcome. It is reasonable to assume the same for HCQ. There are little data to support that hypokalemia itself causes cardiotoxicity in patients with CQ or HCQ overdose.
Although lethal doses are not well established, animal studies suggest that HCQ is much less toxic than CQ, for which the clinical toxicity is better understood due to its more widespread use in overdose abroad.3 In children, the reported therapeutic dose is 10 mg/kg, but the minimum reported lethal dose was a single 300-mg tablet (30 mg/kg in a toddler). In adults, the toxic dose is reported as 20 mg/kg with lethal doses suggested to be as low as 30 mg/kg.
What are the treatment modalities for patients with hydroxychloroquine toxicity?
By analogy with the treatment of CQ poisoning, the mainstay of HCQ therapy is supportive care, including early intubation and ventilation to minimize metabolic demand. Direct-acting inotropes and vasopressors should be administered after saline to treat hypotension. Because of its large volume of distribution, extracorporeal removal has not proved to be of clinical value.4,5 Though data are sparse to determine its efficacy, there may be a role for giving activated charcoal, particularly following large overdoses—if it is given early after exposure and the patient has normal consciousness. If the patient is intubated and aspiration risk is minimized, gastric lavage may also be beneficial—especially when performed within an hour of the overdose. Syrup of ipecac should not be used.
High-dose diazepam is typically recommended, again by analogy with CQ, although there is no clear mechanism of action and its use remains controversial. Its protective effect in patients with CQ overdose is based on swine and rat models that demonstrated dose dependent relationships between diazepam and survival.6,7 A prospective study of CQ toxicity in humans reported improved survival rates when high-dose diazepam was given in combination with epinephrine.8 However, this study is limited by its comparison of prospectively studied patients with a retrospective control. A subsequent prospective study of moderately CQ-intoxicated patients did not find a benefit from treatment with diazepam.9 Furthermore, it remains unclear if the proposed benefit from high-dose diazepam in CQ toxicity may be extrapolated to HCQ, and cases of even massive HCQ ingestions report good outcomes without the use of high-dose diazepam.10
How aggressively should hypokalemia in hydroxychloroquine toxicity be treated?
As noted earlier, hypokalemia resulting from HCQ toxicity is transient, and aggressive repletion may result in rebound hyperkalemia once toxicity resolves. However, these dangers should be balanced with risks of hypokalemia-induced ventricular arrhythmias. Additionally, hypokalemia may be worsened by sodium bicarbonate that is administered to correct QRS prolongations, increasing the risk of dysrhythmia. Correction of hypokalemia in these cases is necessary but should be done with care and monitoring of serum potassium concentrations to minimize risk of hyperkalemia-induced ventricular arrhythmia.11
Case Conclusion
Throughout treatment, the patient remained neurologically intact. She did not receive benzodiazepines. The epinephrine and norepinephrine infusions were weaned, and she was discharged on hospital day 3 with no neurological or cardiac sequelae. She received an inpatient psychiatric evaluation and was referred to outpatient services for ongoing care.
Case
A 14-year-old girl with no known medical history presented to the ED via emergency medical services (EMS) approximately 1.5 hours after intentionally ingesting what she described as “a handful or two” of her mother’s lupus prescription medication in a suicide attempt. Initial vital signs and physical examination were normal, and her only complaint was nausea.
Thirty minutes after presentation, the patient suffered acute cardiovascular (CV) collapse: blood pressure, 57/39 mm Hg; heart rate, 90 beats/min. An initial electrocardiogram (ECG) revealed QRS duration of 123 milliseconds and QTc duration of 510 milliseconds, along with nonspecific T-wave abnormalities. A 150-mEq intravenous (IV) bolus of sodium bicarbonate and a 40-mEq potassium chloride IV infusion were administered, and both epinephrine and norepinephrine IV infusions were also initiated. A basic metabolic panel obtained prior to medication administration showed a potassium concentration of 1.9 mmol/L.
What is the differential diagnosis of toxicological hypokalemia?
Hypokalemia may be reflective of diminished whole body potassium stores or a transient alteration of intravascular potassium concentrations. In acute ingestions and overdose, the etiology of the hypokalemia is often electrolyte redistribution through either blockade of constitutive outward potassium leakage (eg, barium, insulin, quinine) or through increased activity of the Na+/K+-ATPase pump (eg, catecholamines, insulin, methylxanthines). This activity has little effect on whole body potassium stores, but can result in a profound fall in the serum potassium. While mild hypokalemia is generally well tolerated, more severe potassium abnormalities can cause muscular weakness, areflexic paralysis, respiratory failure, and life-threatening dysrhythmias. Common ECG findings include decreased T-wave amplitudes, ST-segment depression, and the presence or amplification of U waves.
Case Continuation
While the emergency physicians were stabilizing the patient, her mother provided additional information. Approximately 30 minutes after the exposure, the patient had become nauseated and told her mother what she had done. Her mother called EMS, and the patient was transported to the hospital, where she rapidly became symptomatic. Despite CV decompensation, she remained neurologically intact. On further questioning, the patient admitted to ingesting 6 g of her mother’s prescription of hydroxychloroquine (HCQ) in a suicide attempt but denied taking any other medications. She was stabilized on vasopressors and admitted to the intensive care unit.
What characterizes hydroxychloroquine toxicity?
Hydroxychloroquine is an aminoquinoline antibiotic that is classically used as an antimalarial to treat infection with Plasmodium vivax, P ovale, P malariae, and susceptible strains of P falciparum. In the United States, it is more commonly used to manage both rheumatoid arthritis and systemic lupus erythematosus (SLE), debilitating diseases which are estimated to affect anywhere from 161,000 to 322,000 Americans.1 Hydroxychloroquine is considered first-line therapy for SLE, but its mechanism of action in treating SLE-associated arthralgias is unclear.
Hydroxychloroquine is structurally similar to quinine and chloroquine (CQ), and not surprisingly exerts quinidine-like effects on the CV system with resultant negative inotropy and vasodilation. Its toxicity is characterized by rapid onset of clinical effects including central nervous system depression, seizures, apnea, hypotension, and arrhythmia. After large overdoses, cardiac arrest and death can occur within hours.
Hypokalemia is another hallmark of HCQ toxicity. It is thought to develop secondary to potassium channel blockade, which slows the constitutive release of potassium from the myocytes.2 As noted, the hypokalemia is transient and does not reflect whole-body depletion. With CQ, which is considered more toxic, there appears to be a correlation between the quantity of CQ ingested and both the degree of hypokalemia and the severity of the outcome. It is reasonable to assume the same for HCQ. There are little data to support that hypokalemia itself causes cardiotoxicity in patients with CQ or HCQ overdose.
Although lethal doses are not well established, animal studies suggest that HCQ is much less toxic than CQ, for which the clinical toxicity is better understood due to its more widespread use in overdose abroad.3 In children, the reported therapeutic dose is 10 mg/kg, but the minimum reported lethal dose was a single 300-mg tablet (30 mg/kg in a toddler). In adults, the toxic dose is reported as 20 mg/kg with lethal doses suggested to be as low as 30 mg/kg.
What are the treatment modalities for patients with hydroxychloroquine toxicity?
By analogy with the treatment of CQ poisoning, the mainstay of HCQ therapy is supportive care, including early intubation and ventilation to minimize metabolic demand. Direct-acting inotropes and vasopressors should be administered after saline to treat hypotension. Because of its large volume of distribution, extracorporeal removal has not proved to be of clinical value.4,5 Though data are sparse to determine its efficacy, there may be a role for giving activated charcoal, particularly following large overdoses—if it is given early after exposure and the patient has normal consciousness. If the patient is intubated and aspiration risk is minimized, gastric lavage may also be beneficial—especially when performed within an hour of the overdose. Syrup of ipecac should not be used.
High-dose diazepam is typically recommended, again by analogy with CQ, although there is no clear mechanism of action and its use remains controversial. Its protective effect in patients with CQ overdose is based on swine and rat models that demonstrated dose dependent relationships between diazepam and survival.6,7 A prospective study of CQ toxicity in humans reported improved survival rates when high-dose diazepam was given in combination with epinephrine.8 However, this study is limited by its comparison of prospectively studied patients with a retrospective control. A subsequent prospective study of moderately CQ-intoxicated patients did not find a benefit from treatment with diazepam.9 Furthermore, it remains unclear if the proposed benefit from high-dose diazepam in CQ toxicity may be extrapolated to HCQ, and cases of even massive HCQ ingestions report good outcomes without the use of high-dose diazepam.10
How aggressively should hypokalemia in hydroxychloroquine toxicity be treated?
As noted earlier, hypokalemia resulting from HCQ toxicity is transient, and aggressive repletion may result in rebound hyperkalemia once toxicity resolves. However, these dangers should be balanced with risks of hypokalemia-induced ventricular arrhythmias. Additionally, hypokalemia may be worsened by sodium bicarbonate that is administered to correct QRS prolongations, increasing the risk of dysrhythmia. Correction of hypokalemia in these cases is necessary but should be done with care and monitoring of serum potassium concentrations to minimize risk of hyperkalemia-induced ventricular arrhythmia.11
Case Conclusion
Throughout treatment, the patient remained neurologically intact. She did not receive benzodiazepines. The epinephrine and norepinephrine infusions were weaned, and she was discharged on hospital day 3 with no neurological or cardiac sequelae. She received an inpatient psychiatric evaluation and was referred to outpatient services for ongoing care.
1. Helmick CG, Felson DT, Lawrence RC, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part I. Arthritis Rheum. 2008;58(1):15-25. doi:10.1002/art.23177.
2. Clemessy JL, Favier C, Borron SW, Hantson PE, Vicaut E, Baud FJ. Hypokalaemia related to acute chloroquine ingestion. Lancet. 1995;3469(8979):877-880.
3. McChesney EW. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am J Med. 1983;75(suppl 1A):11-18.
4. Carmichael SJ, Charles B, Tett SE. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther Drug Monit. 2003;25(6):671-681.
5. Marquardt K, Albertson TE. Treatment of hydroxychloroquine overdose. Am J Emerg Med. 2001;19(5):420-424.
6. Crouzette J, Vicaut E, Palombo S, Girre C, Fournier PE. Experimental assessment of the protective activity of diazepam on the acute toxicity of chloroquine. J Toxicol Clin Toxicol. 1983;20(3):271-279.
7. Riou B, Lecarpentier Y, Barriot P, Viars P. Diazepam does not improve the mechanical performance of rat cardiac papillary muscle exposed to chloroquine in vitro. Intensive Care Med. 1989;15:390-3955.
8. Riou B, Barriot P, Rimailho A, Baud FJ. Treatment of severe chloroquine poisoning. N Engl J Med. 1988;318(1):1-6.
9. Clemessy JL, Angel G, Borron SW, et al. Therapeutic trial of diazepam versus placebo in acute chloroquine intoxications of moderate gravity. Intensive Care Med. 1996;22:1400-1405.
10. Yanturali S. Diazepam for treatment of massive chloroquine intoxication. Resuscitation. 2004;63(3):347-348.
11. Ling Ngan Wong A, Tsz Fung Cheung I, Graham CA. Hydroxychloroquine overdose: case report and recommendations for management. Eur J Emerg Med. 2008;15(1):16-8. doi:10.1097/MEJ.0b013e3280adcb56.
1. Helmick CG, Felson DT, Lawrence RC, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part I. Arthritis Rheum. 2008;58(1):15-25. doi:10.1002/art.23177.
2. Clemessy JL, Favier C, Borron SW, Hantson PE, Vicaut E, Baud FJ. Hypokalaemia related to acute chloroquine ingestion. Lancet. 1995;3469(8979):877-880.
3. McChesney EW. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am J Med. 1983;75(suppl 1A):11-18.
4. Carmichael SJ, Charles B, Tett SE. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther Drug Monit. 2003;25(6):671-681.
5. Marquardt K, Albertson TE. Treatment of hydroxychloroquine overdose. Am J Emerg Med. 2001;19(5):420-424.
6. Crouzette J, Vicaut E, Palombo S, Girre C, Fournier PE. Experimental assessment of the protective activity of diazepam on the acute toxicity of chloroquine. J Toxicol Clin Toxicol. 1983;20(3):271-279.
7. Riou B, Lecarpentier Y, Barriot P, Viars P. Diazepam does not improve the mechanical performance of rat cardiac papillary muscle exposed to chloroquine in vitro. Intensive Care Med. 1989;15:390-3955.
8. Riou B, Barriot P, Rimailho A, Baud FJ. Treatment of severe chloroquine poisoning. N Engl J Med. 1988;318(1):1-6.
9. Clemessy JL, Angel G, Borron SW, et al. Therapeutic trial of diazepam versus placebo in acute chloroquine intoxications of moderate gravity. Intensive Care Med. 1996;22:1400-1405.
10. Yanturali S. Diazepam for treatment of massive chloroquine intoxication. Resuscitation. 2004;63(3):347-348.
11. Ling Ngan Wong A, Tsz Fung Cheung I, Graham CA. Hydroxychloroquine overdose: case report and recommendations for management. Eur J Emerg Med. 2008;15(1):16-8. doi:10.1097/MEJ.0b013e3280adcb56.
Man Thrown From Balky Bike

ANSWER
The radiograph does not demonstrate any evidence of acute chest or intrathoracic injury. Of note, it appears that the left humerus is dislocated anteriorly and inferiorly.
Dedicated shoulder radiographs were obtained, which confirmed the dislocation with no evidence of fracture. Orthopedics was consulted for evaluation and subsequent reduction.
ANSWER
The radiograph does not demonstrate any evidence of acute chest or intrathoracic injury. Of note, it appears that the left humerus is dislocated anteriorly and inferiorly.
Dedicated shoulder radiographs were obtained, which confirmed the dislocation with no evidence of fracture. Orthopedics was consulted for evaluation and subsequent reduction.
ANSWER
The radiograph does not demonstrate any evidence of acute chest or intrathoracic injury. Of note, it appears that the left humerus is dislocated anteriorly and inferiorly.
Dedicated shoulder radiographs were obtained, which confirmed the dislocation with no evidence of fracture. Orthopedics was consulted for evaluation and subsequent reduction.
A 55-year-old man is brought to your facility following a motorcycle accident. He was a helmeted rider who was inadvertently thrown from the motorcycle when the accelerator got stuck. Bystanders reported he had brief loss of consciousness, but upon arrival to your facility, he is awake.
History is limited as the patient is confused and repetitive in his speech, indicating he is post concussive. He is complaining of head, face, and chest wall pain. His vital signs are all within normal limits; he is hemodynamically stable. His O2 saturation is 98% on room air. Breath sounds are clear.
As you are completing your primary survey, portable chest and pelvic radiographs are obtained. The chest radiograph is shown. What is your impression?
A Computer-Assisted Process to Reduce Discharge of Emergency Department Patients with Abnormal Vital Signs
From Case Western Reserve University, MetroHealth Medical Center, Cleveland, OH.
Abstract
- Objective: To describe a computer-assisted process for reducing the number of patients discharged from the emergency department with abnormal vital signs.
- Methods: We devised a best practice alert in the Epic electronic medical record that triggers when the clinician attempts to print an after visit summary (discharge paperwork) at the time of discharge from the emergency department.
- Results: We saw no change in the percentage of patients discharged with elevated blood pressures, consistent with national recommendations. Removing that category of patients, we saw a decrease in the percentage of patients discharged with abnormal vital signs, primarily driven by a decrease in the percentage of patients discharged with tachycardia.
- Conclusion: A computer-assisted process can reduce the percentage of patients discharged with abnormal vital signs. Since based on national recommendations ED physicians do not address most elevated blood pressures in the ED, hypertension should not trigger an alert.
Abnormal vital signs in the emergency department (ED) have been associated with adverse outcomes [1,2]. While most patients discharged from the ED do well, some studies have found that the death rate within days to weeks post–ED discharge may be as high as 200 per 100,000 visits, although other studies have found a much lower rate [1]. A study by Sklar et al, although not specifically focused on vital signs at discharge, found that unexpected death within 7 days of ED discharge occurred at a rate of 30 per 100,000 patients. Abnormal vital signs, most commonly tachycardia, were present in 83% of cases [2].
In busy EDs, the combination of patient volume, frequent interruptions, and the intensity of tasks can result in deficiencies in vital sign monitoring [3–5] as well as abnormal vital signs not being recognized by the clinician at the time of patient discharge [6]. The importance of addressing this quality problem has been recognized. Prior efforts to address the problem have included nurses using manual methods to alert the physician to the presence of abnormal vital signs at the time of discharge [7]. Recommendations have been made to use electronic medical record (EMR) functions for prospectively addressing the problem of ED discharge with abnormal vital signs [8]. The utility of the EMR to identify potentially septic patients earlier and reduce mortality from sepsis via an algorithm that incorporated vital signs and other clinical crieria has been demonstrated [9,10]. In addition, automated vital signs advisories have been associated with increased survival on general hospital wards [11].
An adverse event that occurred at our institution prompted us to review this issue for our ED. We designed an EMR-assisted intervention to reduce the rate of patients discharged from the ED with abnormal vital signs.
Methods
Setting
Our ED is a busy, urban, Level 1 trauma center within a teaching facility. It sees over 100,000 patients per year and is segmented into resuscitation, high acuity, moderate acuity, and fast tract areas, in addition to the observation unit. Our organization uses the Epic (Madison, Wisconsin) electronic health record, which we have been using for over a decade.
Discharge Instructions—Old Process
In our ED, providers, attending physicians, residents and advance practice nurses enter and print their own discharge instructions, which are given to nursing staff to review with patients. Prior to the project, nurses were expected to notify a physician if they thought a vital sign was abnormal. Each nurse made independent decisions on what constituted a vital sign abnormality based on the patient’s condition and could communicate that to the provider at their discretion prior to discharge. This process created inconsistencies in care.

Development of Alert

We created an alert that appears within the provider workflow at the time the provider attempts to print discharge instructions. Based on literature review and operational leadership consensus, we set the parameters for abnormal vital signs (Table 1). We chose parameters to identify abnormalities important to 
The alert displays when a user attempts to print discharge instructions on a patient whose last recorded vital signs are not all normal. The display informs that there are abnormal vital signs (Figure). Upon display of the alert, the user can click on the message, which would take them to the vital sign entry activity in the EMR, or they can proceed with printing by clicking the print button (not visible in the Figure). The alert is not a forcing function; the user can proceed with printing the discharge instructions without addressing the abnormality that triggered the alert.
Pre-Post Evaluation
We would have liked to have determined how often the abnormal vital signs alert triggered, how it was responded to, and whether the patient was subsequently discharged with normal vital signs; however, our system does not record these events. Instead, we used the system to compare the percentage of adult patients who were discharged with abnormal vital signs for 2 time periods: the period prior to our December 2014 implementation (1 Oct to 1 Dec 2014) and the post implementation period (15 Dec 2014 to 15 Feb 2015). Our presumption was that the use of the alert system would reduce the percentage of patients discharged with abnormal vital signs, including an abnormal pulse oximetry.
To conduct our analysis, we identified adult patients seen during the 2 time periods. We eliminated those patients who died, left without being seen, eloped, were admitted or were transferred to other institutions. This resulted in 3664 patients, with 2179 in the pre-implementation group and 1485 in the post-implementation group. The higher volume in the pre group reflects the early occurrence of influenza season in our area during the study period, along with our generally busier time in late fall compared to winter.
The analysis was performed as a likelihood ratio chi-square analysis using SAS (Cary, NC) software.
Results
The analysis demonstrated that physicians were, by and large, following recommendations consistent with policies of the American College of Emergency Physicians regarding the management of elevated blood pressures, which do not mandate that patients with asymptomatic elevations of blood pressure receive medical intervention in the ED [12]. In our analysis, the percentage of patients discharged with elevated blood pressures actually increased from 7.5% to 9.9% following the intervention. Importantly, however, the percentage of patients discharged with low blood pressures decreased from 6.9% to 5.0% (P < 0.01).Tthe percent of patients discharged with an elevated heart rate, decreased from 58% in the pre alert group to 42% in the post alert group (P < 0.02). 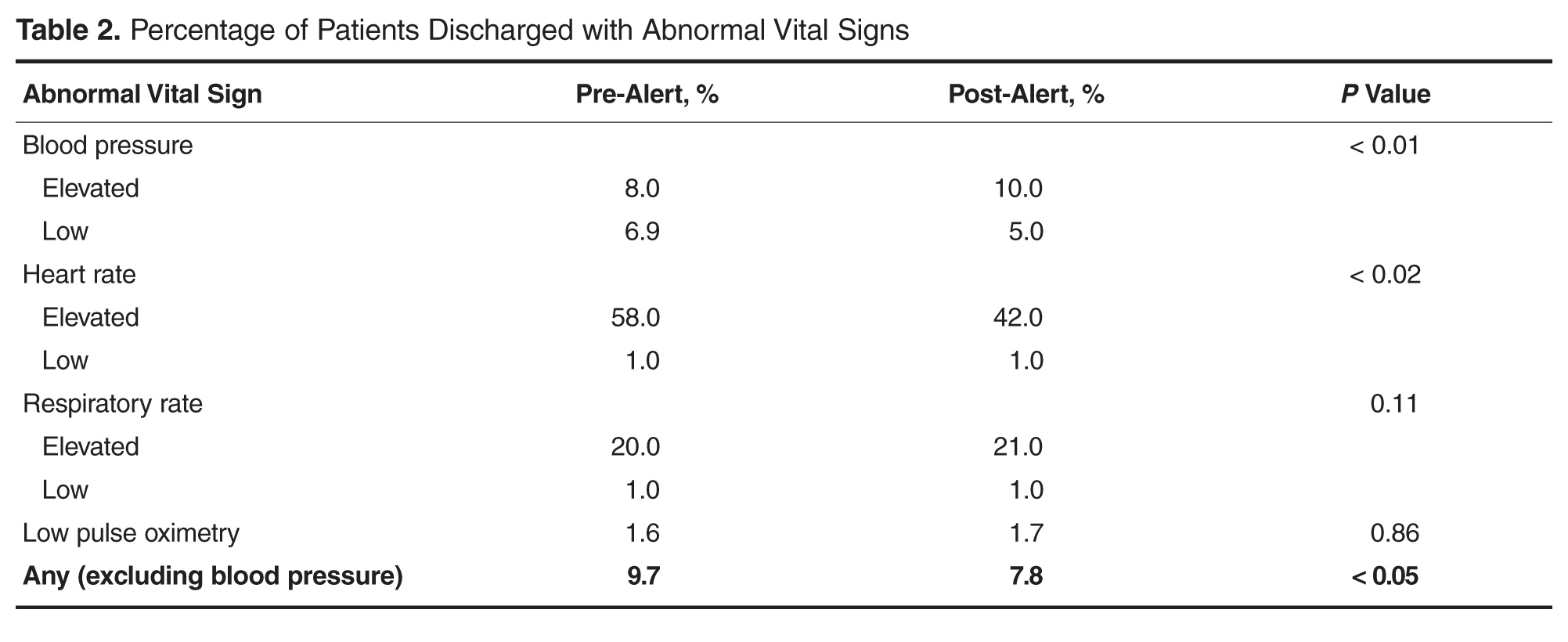
Discussion
In our study, we used features of the EMR prospectively to affect discharge and then used the database functions of the EMR to assess the effectiveness of those efforts. Previous studies that have looked at the incidence of abnormal vital signs at discharge have been manual, retrospective reviews of records. We are not aware of any studies reporting the results of introducing an EMR alert to prospectively identify patients with abnormal vital signs prior to discharge
While we found that this intervention was successful in reducing clinically relevant abnormal vital signs at discharge, we have realized that the elevated blood pressure alert was unnecessary and we have eliminated it from the programming. We will revisit our strategy to determine if further reducing the high blood pressure alerts can lead to greater improvements in reducing the percentage of patients discharged with abnormal vital signs.
Future plans include a review of the re-visit or hospitalization rate for patients discharged with abnormal vital signs. A companion study evaluating a similar approach to the care of children is under consideration. We are also considering including a field in the EMR for the clinician to document why they discharged a patient with abnormal vital signs.
Corresponding author: Jonathan E. Siff, MD 2500 MetroHealth Drive BG3-65 Cleveland, Ohio 44109 jsiff@metrohealth.org.
Financial disclosures: None.
1. Gunnarsdottir OS, Rafnsson V. Death within 8 days after discharge to home from the emergency department. Eur J of Public Health 2008;18:522–6.
2. Sklar DP, Crandall CS, Loeliger E, et al. Unanticipated death after discharge home from the emergency department. Ann Emerg Med 2007;49:735–45.
3. Johnson KD, Winkelman C, Burant CJ, et al. The factors that affect the frequency of vital sign monitoring in the emergency department. J Emerg Nurs 2014;40:27–35.
4. Gravel J, Opatrny L, Gouin S. High rate of missing vital signs data at triage in a paediatric emergency department. Paediatr Child Health 2006;11:211–5.
5. Depinet HE, Iyer SB, Hornung R, et al. The effect of emergency department crowding on reassessment of children with critically abnormal vital signs. Acad Emerg Med 2014;21:1116–20.
6. Hafner JW, Parrish SE, Hubler JR, et al. Repeat assessment of abnormal vital signs and patient re-examination in us emergency department patients. Ann Emerg Med 2006;48(4 Suppl):S66.
7. Domagala SE. Discharge vital signs: an enhancement to ED quality and patient outcomes. J Emerg Nursing 2009;35:138–40.
8. Welch S. Red flags: abnormal vital signs at discharge.Emerg Med News 2011:33;7–8.
9. Nguyen SQ, Mwakalindile E, Booth JS, et al. Automated electronic medical record sepsis detection in the emergency department. PeerJ 2014;2:e343.
10. Narayanan N, Gross AK, Pintens M, et al. Effect of an electronic medical record alert for severe sepsis among ED patients. Am J Emerg Med 2016;34:185–8.
11. Bellomo R, Ackerman M, Bailey M, et al. A controlled trial of electronic automated advisory vital signs monitoring in general hospital wards. Crit Care Med 2012;40:2349–61.
12. Wolf SJ, Lo B, Smith MD, et al. Clinical policy: critical issues in the evaluation and management of adult patients in the emergency department with asymptomatic elevated blood pressure. Ann Emerg Med 2013:62;59–68.
From Case Western Reserve University, MetroHealth Medical Center, Cleveland, OH.
Abstract
- Objective: To describe a computer-assisted process for reducing the number of patients discharged from the emergency department with abnormal vital signs.
- Methods: We devised a best practice alert in the Epic electronic medical record that triggers when the clinician attempts to print an after visit summary (discharge paperwork) at the time of discharge from the emergency department.
- Results: We saw no change in the percentage of patients discharged with elevated blood pressures, consistent with national recommendations. Removing that category of patients, we saw a decrease in the percentage of patients discharged with abnormal vital signs, primarily driven by a decrease in the percentage of patients discharged with tachycardia.
- Conclusion: A computer-assisted process can reduce the percentage of patients discharged with abnormal vital signs. Since based on national recommendations ED physicians do not address most elevated blood pressures in the ED, hypertension should not trigger an alert.
Abnormal vital signs in the emergency department (ED) have been associated with adverse outcomes [1,2]. While most patients discharged from the ED do well, some studies have found that the death rate within days to weeks post–ED discharge may be as high as 200 per 100,000 visits, although other studies have found a much lower rate [1]. A study by Sklar et al, although not specifically focused on vital signs at discharge, found that unexpected death within 7 days of ED discharge occurred at a rate of 30 per 100,000 patients. Abnormal vital signs, most commonly tachycardia, were present in 83% of cases [2].
In busy EDs, the combination of patient volume, frequent interruptions, and the intensity of tasks can result in deficiencies in vital sign monitoring [3–5] as well as abnormal vital signs not being recognized by the clinician at the time of patient discharge [6]. The importance of addressing this quality problem has been recognized. Prior efforts to address the problem have included nurses using manual methods to alert the physician to the presence of abnormal vital signs at the time of discharge [7]. Recommendations have been made to use electronic medical record (EMR) functions for prospectively addressing the problem of ED discharge with abnormal vital signs [8]. The utility of the EMR to identify potentially septic patients earlier and reduce mortality from sepsis via an algorithm that incorporated vital signs and other clinical crieria has been demonstrated [9,10]. In addition, automated vital signs advisories have been associated with increased survival on general hospital wards [11].
An adverse event that occurred at our institution prompted us to review this issue for our ED. We designed an EMR-assisted intervention to reduce the rate of patients discharged from the ED with abnormal vital signs.
Methods
Setting
Our ED is a busy, urban, Level 1 trauma center within a teaching facility. It sees over 100,000 patients per year and is segmented into resuscitation, high acuity, moderate acuity, and fast tract areas, in addition to the observation unit. Our organization uses the Epic (Madison, Wisconsin) electronic health record, which we have been using for over a decade.
Discharge Instructions—Old Process
In our ED, providers, attending physicians, residents and advance practice nurses enter and print their own discharge instructions, which are given to nursing staff to review with patients. Prior to the project, nurses were expected to notify a physician if they thought a vital sign was abnormal. Each nurse made independent decisions on what constituted a vital sign abnormality based on the patient’s condition and could communicate that to the provider at their discretion prior to discharge. This process created inconsistencies in care.
Development of Alert
We created an alert that appears within the provider workflow at the time the provider attempts to print discharge instructions. Based on literature review and operational leadership consensus, we set the parameters for abnormal vital signs (Table 1). We chose parameters to identify abnormalities important to
The alert displays when a user attempts to print discharge instructions on a patient whose last recorded vital signs are not all normal. The display informs that there are abnormal vital signs (Figure). Upon display of the alert, the user can click on the message, which would take them to the vital sign entry activity in the EMR, or they can proceed with printing by clicking the print button (not visible in the Figure). The alert is not a forcing function; the user can proceed with printing the discharge instructions without addressing the abnormality that triggered the alert.
Pre-Post Evaluation
We would have liked to have determined how often the abnormal vital signs alert triggered, how it was responded to, and whether the patient was subsequently discharged with normal vital signs; however, our system does not record these events. Instead, we used the system to compare the percentage of adult patients who were discharged with abnormal vital signs for 2 time periods: the period prior to our December 2014 implementation (1 Oct to 1 Dec 2014) and the post implementation period (15 Dec 2014 to 15 Feb 2015). Our presumption was that the use of the alert system would reduce the percentage of patients discharged with abnormal vital signs, including an abnormal pulse oximetry.
To conduct our analysis, we identified adult patients seen during the 2 time periods. We eliminated those patients who died, left without being seen, eloped, were admitted or were transferred to other institutions. This resulted in 3664 patients, with 2179 in the pre-implementation group and 1485 in the post-implementation group. The higher volume in the pre group reflects the early occurrence of influenza season in our area during the study period, along with our generally busier time in late fall compared to winter.
The analysis was performed as a likelihood ratio chi-square analysis using SAS (Cary, NC) software.
Results
The analysis demonstrated that physicians were, by and large, following recommendations consistent with policies of the American College of Emergency Physicians regarding the management of elevated blood pressures, which do not mandate that patients with asymptomatic elevations of blood pressure receive medical intervention in the ED [12]. In our analysis, the percentage of patients discharged with elevated blood pressures actually increased from 7.5% to 9.9% following the intervention. Importantly, however, the percentage of patients discharged with low blood pressures decreased from 6.9% to 5.0% (P < 0.01).Tthe percent of patients discharged with an elevated heart rate, decreased from 58% in the pre alert group to 42% in the post alert group (P < 0.02).
Discussion
In our study, we used features of the EMR prospectively to affect discharge and then used the database functions of the EMR to assess the effectiveness of those efforts. Previous studies that have looked at the incidence of abnormal vital signs at discharge have been manual, retrospective reviews of records. We are not aware of any studies reporting the results of introducing an EMR alert to prospectively identify patients with abnormal vital signs prior to discharge
While we found that this intervention was successful in reducing clinically relevant abnormal vital signs at discharge, we have realized that the elevated blood pressure alert was unnecessary and we have eliminated it from the programming. We will revisit our strategy to determine if further reducing the high blood pressure alerts can lead to greater improvements in reducing the percentage of patients discharged with abnormal vital signs.
Future plans include a review of the re-visit or hospitalization rate for patients discharged with abnormal vital signs. A companion study evaluating a similar approach to the care of children is under consideration. We are also considering including a field in the EMR for the clinician to document why they discharged a patient with abnormal vital signs.
Corresponding author: Jonathan E. Siff, MD 2500 MetroHealth Drive BG3-65 Cleveland, Ohio 44109 jsiff@metrohealth.org.
Financial disclosures: None.
From Case Western Reserve University, MetroHealth Medical Center, Cleveland, OH.
Abstract
- Objective: To describe a computer-assisted process for reducing the number of patients discharged from the emergency department with abnormal vital signs.
- Methods: We devised a best practice alert in the Epic electronic medical record that triggers when the clinician attempts to print an after visit summary (discharge paperwork) at the time of discharge from the emergency department.
- Results: We saw no change in the percentage of patients discharged with elevated blood pressures, consistent with national recommendations. Removing that category of patients, we saw a decrease in the percentage of patients discharged with abnormal vital signs, primarily driven by a decrease in the percentage of patients discharged with tachycardia.
- Conclusion: A computer-assisted process can reduce the percentage of patients discharged with abnormal vital signs. Since based on national recommendations ED physicians do not address most elevated blood pressures in the ED, hypertension should not trigger an alert.
Abnormal vital signs in the emergency department (ED) have been associated with adverse outcomes [1,2]. While most patients discharged from the ED do well, some studies have found that the death rate within days to weeks post–ED discharge may be as high as 200 per 100,000 visits, although other studies have found a much lower rate [1]. A study by Sklar et al, although not specifically focused on vital signs at discharge, found that unexpected death within 7 days of ED discharge occurred at a rate of 30 per 100,000 patients. Abnormal vital signs, most commonly tachycardia, were present in 83% of cases [2].
In busy EDs, the combination of patient volume, frequent interruptions, and the intensity of tasks can result in deficiencies in vital sign monitoring [3–5] as well as abnormal vital signs not being recognized by the clinician at the time of patient discharge [6]. The importance of addressing this quality problem has been recognized. Prior efforts to address the problem have included nurses using manual methods to alert the physician to the presence of abnormal vital signs at the time of discharge [7]. Recommendations have been made to use electronic medical record (EMR) functions for prospectively addressing the problem of ED discharge with abnormal vital signs [8]. The utility of the EMR to identify potentially septic patients earlier and reduce mortality from sepsis via an algorithm that incorporated vital signs and other clinical crieria has been demonstrated [9,10]. In addition, automated vital signs advisories have been associated with increased survival on general hospital wards [11].
An adverse event that occurred at our institution prompted us to review this issue for our ED. We designed an EMR-assisted intervention to reduce the rate of patients discharged from the ED with abnormal vital signs.
Methods
Setting
Our ED is a busy, urban, Level 1 trauma center within a teaching facility. It sees over 100,000 patients per year and is segmented into resuscitation, high acuity, moderate acuity, and fast tract areas, in addition to the observation unit. Our organization uses the Epic (Madison, Wisconsin) electronic health record, which we have been using for over a decade.
Discharge Instructions—Old Process
In our ED, providers, attending physicians, residents and advance practice nurses enter and print their own discharge instructions, which are given to nursing staff to review with patients. Prior to the project, nurses were expected to notify a physician if they thought a vital sign was abnormal. Each nurse made independent decisions on what constituted a vital sign abnormality based on the patient’s condition and could communicate that to the provider at their discretion prior to discharge. This process created inconsistencies in care.
Development of Alert
We created an alert that appears within the provider workflow at the time the provider attempts to print discharge instructions. Based on literature review and operational leadership consensus, we set the parameters for abnormal vital signs (Table 1). We chose parameters to identify abnormalities important to
The alert displays when a user attempts to print discharge instructions on a patient whose last recorded vital signs are not all normal. The display informs that there are abnormal vital signs (Figure). Upon display of the alert, the user can click on the message, which would take them to the vital sign entry activity in the EMR, or they can proceed with printing by clicking the print button (not visible in the Figure). The alert is not a forcing function; the user can proceed with printing the discharge instructions without addressing the abnormality that triggered the alert.
Pre-Post Evaluation
We would have liked to have determined how often the abnormal vital signs alert triggered, how it was responded to, and whether the patient was subsequently discharged with normal vital signs; however, our system does not record these events. Instead, we used the system to compare the percentage of adult patients who were discharged with abnormal vital signs for 2 time periods: the period prior to our December 2014 implementation (1 Oct to 1 Dec 2014) and the post implementation period (15 Dec 2014 to 15 Feb 2015). Our presumption was that the use of the alert system would reduce the percentage of patients discharged with abnormal vital signs, including an abnormal pulse oximetry.
To conduct our analysis, we identified adult patients seen during the 2 time periods. We eliminated those patients who died, left without being seen, eloped, were admitted or were transferred to other institutions. This resulted in 3664 patients, with 2179 in the pre-implementation group and 1485 in the post-implementation group. The higher volume in the pre group reflects the early occurrence of influenza season in our area during the study period, along with our generally busier time in late fall compared to winter.
The analysis was performed as a likelihood ratio chi-square analysis using SAS (Cary, NC) software.
Results
The analysis demonstrated that physicians were, by and large, following recommendations consistent with policies of the American College of Emergency Physicians regarding the management of elevated blood pressures, which do not mandate that patients with asymptomatic elevations of blood pressure receive medical intervention in the ED [12]. In our analysis, the percentage of patients discharged with elevated blood pressures actually increased from 7.5% to 9.9% following the intervention. Importantly, however, the percentage of patients discharged with low blood pressures decreased from 6.9% to 5.0% (P < 0.01).Tthe percent of patients discharged with an elevated heart rate, decreased from 58% in the pre alert group to 42% in the post alert group (P < 0.02).
Discussion
In our study, we used features of the EMR prospectively to affect discharge and then used the database functions of the EMR to assess the effectiveness of those efforts. Previous studies that have looked at the incidence of abnormal vital signs at discharge have been manual, retrospective reviews of records. We are not aware of any studies reporting the results of introducing an EMR alert to prospectively identify patients with abnormal vital signs prior to discharge
While we found that this intervention was successful in reducing clinically relevant abnormal vital signs at discharge, we have realized that the elevated blood pressure alert was unnecessary and we have eliminated it from the programming. We will revisit our strategy to determine if further reducing the high blood pressure alerts can lead to greater improvements in reducing the percentage of patients discharged with abnormal vital signs.
Future plans include a review of the re-visit or hospitalization rate for patients discharged with abnormal vital signs. A companion study evaluating a similar approach to the care of children is under consideration. We are also considering including a field in the EMR for the clinician to document why they discharged a patient with abnormal vital signs.
Corresponding author: Jonathan E. Siff, MD 2500 MetroHealth Drive BG3-65 Cleveland, Ohio 44109 jsiff@metrohealth.org.
Financial disclosures: None.
1. Gunnarsdottir OS, Rafnsson V. Death within 8 days after discharge to home from the emergency department. Eur J of Public Health 2008;18:522–6.
2. Sklar DP, Crandall CS, Loeliger E, et al. Unanticipated death after discharge home from the emergency department. Ann Emerg Med 2007;49:735–45.
3. Johnson KD, Winkelman C, Burant CJ, et al. The factors that affect the frequency of vital sign monitoring in the emergency department. J Emerg Nurs 2014;40:27–35.
4. Gravel J, Opatrny L, Gouin S. High rate of missing vital signs data at triage in a paediatric emergency department. Paediatr Child Health 2006;11:211–5.
5. Depinet HE, Iyer SB, Hornung R, et al. The effect of emergency department crowding on reassessment of children with critically abnormal vital signs. Acad Emerg Med 2014;21:1116–20.
6. Hafner JW, Parrish SE, Hubler JR, et al. Repeat assessment of abnormal vital signs and patient re-examination in us emergency department patients. Ann Emerg Med 2006;48(4 Suppl):S66.
7. Domagala SE. Discharge vital signs: an enhancement to ED quality and patient outcomes. J Emerg Nursing 2009;35:138–40.
8. Welch S. Red flags: abnormal vital signs at discharge.Emerg Med News 2011:33;7–8.
9. Nguyen SQ, Mwakalindile E, Booth JS, et al. Automated electronic medical record sepsis detection in the emergency department. PeerJ 2014;2:e343.
10. Narayanan N, Gross AK, Pintens M, et al. Effect of an electronic medical record alert for severe sepsis among ED patients. Am J Emerg Med 2016;34:185–8.
11. Bellomo R, Ackerman M, Bailey M, et al. A controlled trial of electronic automated advisory vital signs monitoring in general hospital wards. Crit Care Med 2012;40:2349–61.
12. Wolf SJ, Lo B, Smith MD, et al. Clinical policy: critical issues in the evaluation and management of adult patients in the emergency department with asymptomatic elevated blood pressure. Ann Emerg Med 2013:62;59–68.
1. Gunnarsdottir OS, Rafnsson V. Death within 8 days after discharge to home from the emergency department. Eur J of Public Health 2008;18:522–6.
2. Sklar DP, Crandall CS, Loeliger E, et al. Unanticipated death after discharge home from the emergency department. Ann Emerg Med 2007;49:735–45.
3. Johnson KD, Winkelman C, Burant CJ, et al. The factors that affect the frequency of vital sign monitoring in the emergency department. J Emerg Nurs 2014;40:27–35.
4. Gravel J, Opatrny L, Gouin S. High rate of missing vital signs data at triage in a paediatric emergency department. Paediatr Child Health 2006;11:211–5.
5. Depinet HE, Iyer SB, Hornung R, et al. The effect of emergency department crowding on reassessment of children with critically abnormal vital signs. Acad Emerg Med 2014;21:1116–20.
6. Hafner JW, Parrish SE, Hubler JR, et al. Repeat assessment of abnormal vital signs and patient re-examination in us emergency department patients. Ann Emerg Med 2006;48(4 Suppl):S66.
7. Domagala SE. Discharge vital signs: an enhancement to ED quality and patient outcomes. J Emerg Nursing 2009;35:138–40.
8. Welch S. Red flags: abnormal vital signs at discharge.Emerg Med News 2011:33;7–8.
9. Nguyen SQ, Mwakalindile E, Booth JS, et al. Automated electronic medical record sepsis detection in the emergency department. PeerJ 2014;2:e343.
10. Narayanan N, Gross AK, Pintens M, et al. Effect of an electronic medical record alert for severe sepsis among ED patients. Am J Emerg Med 2016;34:185–8.
11. Bellomo R, Ackerman M, Bailey M, et al. A controlled trial of electronic automated advisory vital signs monitoring in general hospital wards. Crit Care Med 2012;40:2349–61.
12. Wolf SJ, Lo B, Smith MD, et al. Clinical policy: critical issues in the evaluation and management of adult patients in the emergency department with asymptomatic elevated blood pressure. Ann Emerg Med 2013:62;59–68.
Serotonin syndrome: Preventing, recognizing, and treating it
With a substantial increase in antidepressant use in the United States over the last 2 decades, serotonin syndrome has become an increasingly common and significant clinical concern. In 1999, 6.5% of adults age 18 and older were taking antidepressants; by 2010, the percentage had increased to 10.4%.1 Though the true incidence of serotonin syndrome is difficult to determine, the number of ingestions of selective serotonin reuptake inhibitors (SSRIs) associated with moderate to major effects reported to US poison control centers increased from 7,349 in 20022 to 8,585 in 2005.3
Though the clinical manifestations are often mild to moderate, patients with serotonin syndrome can deteriorate rapidly and require intensive care. Unlike neuroleptic malignant syndrome, serotonin syndrome should not be considered an extremely rare idiosyncratic reaction to medication, but rather a progression of serotonergic toxicity based on increasing concentration levels that can occur in any patient regardless of age.4
Because it has a nonspecific prodrome and protean manifestations, serotonin syndrome can easily be overlooked, misdiagnosed, or exacerbated if not carefully assessed. Diagnosis requires a low threshold for suspicion and a meticulous history and physical examination. In the syndrome’s mildest stage, symptoms are often misattributed to other causes, and in its most severe form, it can easily be mistaken for neuroleptic malignant syndrome.
WHAT IS SEROTONIN SYNDROME?
Serotonin syndrome classically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. These symptoms are a result of increased serotonin levels affecting the central and peripheral nervous systems. Serotonin affects a family of receptors that has seven members, of which 5-HT1A and 5-HT2A are most often responsible for serotonin syndrome.5
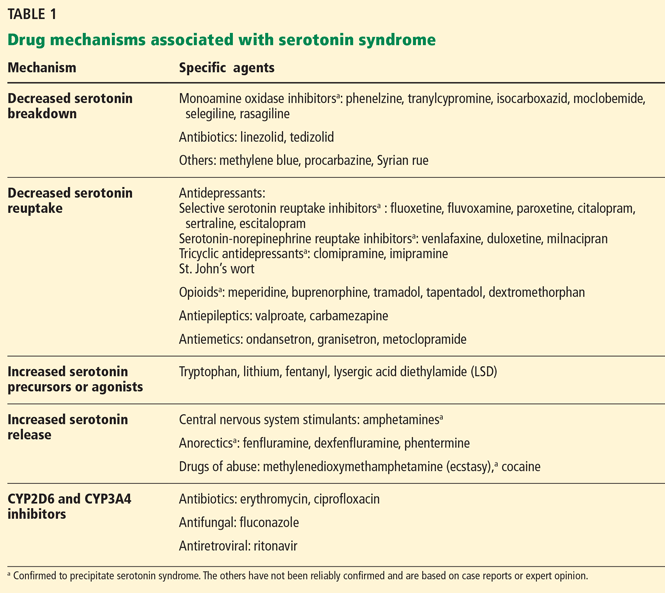
Conditions that can alter the regulation of serotonin include therapeutic doses, drug interactions, intentional or unintentional overdoses, and overlapping transitions between medications. As a result, drugs that have been associated with serotonin syndrome can be classified into the following five categories as shown below and in Table 1:
Drugs that decrease serotonin breakdown include monoamine oxidase inhibitors (MAOIs), linezolid,6 methylene blue, procarbazine, and Syrian rue.
Drugs that decrease serotonin reuptake include SSRIs, serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants, opioids (meperidine, buprenorphine, tramadol, tapentadol, dextromethorphan), antiepileptics (carbamazepine, valproate), and antiemetics (ondansetron, granisetron, metoclopramide), and the herbal preparation St. John’s wort.
Drugs that increase serotonin precursors or agonists include tryptophan, lithium, fentanyl, and lysergic acid diethylamide (LSD).
Drugs that increase serotonin release include fenfluramine, amphetamines, and methylenedioxymethamphetamine (ecstasy).
Drugs that prevent breakdown of the agents listed above are CYP2D6 and CYP3A4 inhibitors, eg, erythromycin,7 ciprofloxacin, fluconazole, ritonavir, and grapefruit juice.
However, the only drugs that have been reliably confirmed to precipitate serotonin syndrome are MAOIs, SSRIs, SNRIs, and serotonin releasers. Other listed drug interactions are based on case reports and have not been thoroughly evaluated.6–9
Currently, SSRIs are the most commonly prescribed antidepressant medications and, consequently, they are the ones most often implicated in serotonergic toxicity.1,10 An estimated 15% of SSRI overdoses lead to mild or moderate serotonin toxicity.11 Serotonergic agents used in conjunction can increase the risk for severe serotonin syndrome; an SSRI and an MAOI in combination poses the greatest risk.5
Ultimately, the incidence of serotonin syndrome is difficult to assess, but it is believed to be underreported because it is easy to misdiagnose and mild symptoms may be dismissed.
WHO IS AT RISK OF SEROTONIN SYNDROME?
Long-term antidepressant use has disproportionately increased in middle-aged and older adults and non-Hispanic whites.1,12,13 Intuitively, as the risk for depression increases dramatically in patients with chronic medical conditions, serotonin syndrome should be more prevalent among the elderly. In addition, patients with multiple comorbidities take more medications, increasing the risk of polypharmacy and adverse drug reactions.14
Although the epidemiology of serotonin syndrome has yet to be extensively studied, the combination of age and comorbidities may increase the risk for this condition.
HOW DOES IT PRESENT?
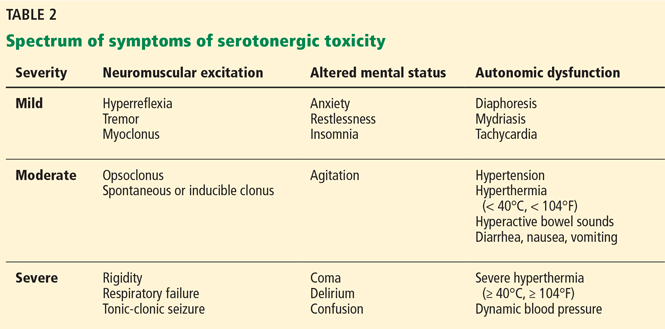
Serotonin syndrome characteristically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. However, these symptoms may not occur simultaneously: autonomic dysfunction is present in 40% of patients, neuromuscular excitation in 50%, and altered mental status in 40%.15 The symptoms can range from mild to life-threatening (Table 2).16
Autonomic dysfunction. Diaphoresis is present in 48.8% of cases, tachycardia in 44%, nausea and vomiting in 26.8%, and mydriasis in 19.5%. Other signs are hyperactive bowel sounds, diarrhea, and flushing.16
Neuromuscular excitation. Myoclonus is present in 48.8%, hyperreflexia in 41%, hyperthermia in 26.8%, and hypertonicity and rigidity in 19.5%. Other signs are spontaneous or inducible clonus, ocular clonus (continuous rhythmic oscillations of gaze), and tremor.
Altered mental status. Confusion is present in 41.2% and agitation in 36.5%. Other signs are anxiety, lethargy, and coma.
Symptoms of serotonin toxicity arise within an hour of a precipitating event (eg, ingestion) in approximately 28% of patients, and within 6 hours in 61%.16 Highly diagnostic features include hyperreflexia and induced or spontaneous clonus that are generally more pronounced in the lower limbs.11 Clonus can be elicited with ankle dorsiflexion.
In mild toxicity, patients may present with tremor or twitching and anxiety, as well as with hyperreflexia, tachycardia, diaphoresis, and mydriasis. Further investigation may uncover a recently initiated antidepressant or a cold-and-cough medication that contains dextromethorphan.15,17
In moderate toxicity, patients present in significant distress, with agitation and restlessness. Features may include hyperreflexia and clonus of the lower extremities, opsoclonus, hyperactive bowel sounds, diarrhea, nausea, vomiting, tachycardia, hypertension, diaphoresis, mydriasis, and hyperthermia (< 40°C, 104°F). The patient’s history may reveal use of ecstasy or combined treatment with serotonin-potentiating agents such as an antidepressant with a proserotonergic opioid, antiepileptic, or CYP2D6 or CYP3A4 inhibitor.15
Severe serotonin toxicity is a life-threatening condition that can lead to multiorgan failure within hours. It can be characterized by muscle rigidity, which can cause the body temperature to elevate rapidly to over 40°C. This hypertonicity can mask the classic and diagnostic signs of hyperreflexia and clonus. Patients may have unstable and dynamic vital signs with confusion or delirium and can experience tonic-clonic seizures.
If the muscle rigidity and resulting hyperthermia are not managed properly, patients can develop cellular damage and enzyme dysfunction leading to rhabdomyolysis, myoglobinuria, renal failure, metabolic acidosis, acute respiratory distress syndrome, and disseminated intravascular coagulation.16,18
Serotonin crisis is usually caused by the co-ingestion of multiple serotonergic agents, such as an antidepressant with an aforementioned opioid and antiemetic19; combining an SSRI and an MAOI poses the greatest risk. Alternatively, patients may have recently switched antidepressants without observing a safe washout period, leading to an overlap of serotonin levels.16
HOW DO WE DIAGNOSE SEROTONIN SYNDROME?
Serotonin syndrome is a clinical diagnosis and therefore requires a thorough review of medications and physical examination. Serum serotonin levels are an unreliable indicator of toxicity and do not correlate well with the clinical presentation.16

(based on information in reference 9).
Currently, there are two clinical tools for diagnosing serotonin syndrome: the Hunter serotonin toxicity criteria (Figure 1) and the Sternbach criteria.
The Hunter criteria are based more heavily on physical findings. The patient must have taken a serotonergic agent and have one of the following:
- Spontaneous clonus
- Inducible clonus plus agitation or diaphoresis
- Ocular clonus plus agitation or diaphoresis
- Inducible clonus or ocular clonus, plus hypertonia and hyperthermia
- Tremor plus hyperreflexia.
The Sternbach criteria. The patient must be using a serotonergic agent, must have no other causes of symptoms, must not have recently used a neuroleptic agent, and must have three of the following:
- Mental status changes
- Agitation
- Hyperreflexia
- Myoclonus
- Diaphoresis
- Shivering
- Tremor
- Diarrhea
- Incoordination
- Fever
The Hunter criteria are recommended and are more specific (97% vs 96%) and more sensitive (84% vs 75%) than the Sternbach criteria when compared with the gold standard of diagnosis by a clinical toxicologist.1 The Hunter criteria are also less likely to yield false-positive results.11
Differential diagnosis
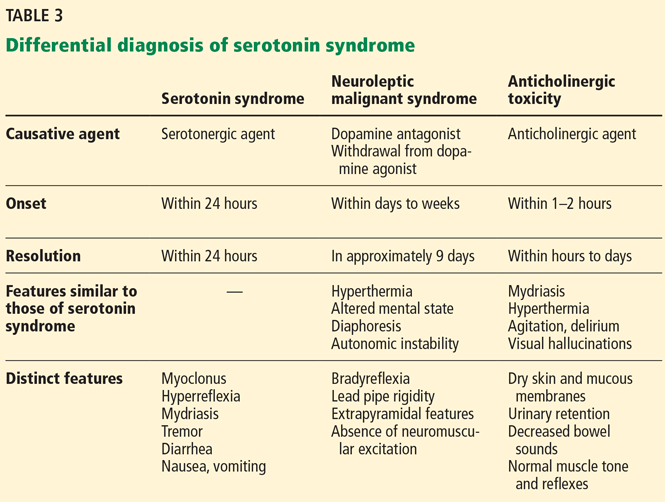
The differential diagnosis for serotonin syndrome includes neuroleptic malignant syndrome, anticholinergic poisoning (Table 3), metastatic carcinoma, central nervous system infection, gastroenteritis, and sepsis.
Neuroleptic malignant syndrome, the disorder most often misdiagnosed as serotonin syndrome, is an idiosyncratic reaction to a dopamine antagonist (eg, haloperidol, fluphenazine) that develops over days to weeks.20 In 70% of patients, agitated delirium with confusion appears first, followed by lead pipe rigidity and cogwheel tremor, then hyperthermia with body temperature greater than 40°C, and finally, profuse diaphoresis, tachycardia, hypertension, and tachypnea.21
Key elements that distinguish neuroleptic malignant syndrome are the timeline of the clinical course, bradyreflexia, and the absence of clonus. Prodromal symptoms of nausea, vomiting, and diarrhea are also rare in neuroleptic malignant syndrome. Neuroleptic malignant syndrome typically requires an average of 9 days to resolve.
Anticholinergic poisoning usually develops within 1 to 2 hours of oral ingestion. Symptoms include flushing, anhidrosis, anhidrotic hyperthermia, mydriasis, urinary retention, decreased bowel sounds, agitated delirium, and visual hallucinations. In contrast to serotonin syndrome, reflexes and muscle tone are normal with anticholinergic poisoning.
HOW CAN WE TREAT SEROTONIN SYNDROME?
The two mainstays of serotonin syndrome management are to discontinue the serotonergic agent and to give supportive care. Most patients improve within 24 hours of stopping the precipitating drug and starting therapy.16
For mild serotonin syndrome, treatment involves discontinuing the offending agent and supportive therapy with intravenous fluids, correction of vital signs, and symptomatic treatment with a benzodiazepine. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For moderate serotonin syndrome, treatment also involves stopping the serotonergic agent and giving supportive care. Symptomatic treatment with a benzodiazepine and nonserotonergic antiemetics is recommended, and standard cooling measures should be implemented for hyperthermia. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For severe serotonin toxicity, treatment should focus on management of airway, breathing, and circulation—ie, the “ABCs.” The two primary life-threatening concerns are hyperthermia (temperature > 40°C or 104°F) and rigidity, which can lead to hypoventilation.1,22 Controlling hyperthermia and rigidity can prevent other grave complications. Patients with severe serotonin toxicity should be sedated, paralyzed, and intubated.21 This will reverse ventilatory hypertonia and allow for mechanical ventilation. Paralysis will also prevent the exacerbation of hyperthermia, which is caused by muscle rigidity. Antipyretics have no role in the treatment of serotonin syndrome since the hyperthermia is not caused by a change in the hypothalamic temperature set point.21 Standard cooling measures should be used to manage hyperthermia.
Serotonin antagonists
Serotonin antagonists have had some success in case reports, but further studies are needed to confirm this.4,23,24
Cyproheptadine is a potent 5-HT2A antagonist; patients usually respond within 1 to 2 hours of administration. Signs and symptoms have resolved completely within times ranging from 20 minutes to 48 hours, depending on the severity of toxicity.
The recommended initial dose of cyproheptadine is 12 mg, followed by 2 mg every 2 hours if symptoms continue.16 Maintenance dosing with 8 mg every 6 hours should be prescribed once stabilization is achieved. The total daily dose for adults should not exceed 0.5 mg/kg/day. Cyproheptadine is available only in oral form but can be crushed and administered via a nasogastric tube.21
Chlorpromazine is a 5-HT1A and 5-HT2A antagonist and can be given intramuscularly. Despite case reports citing its effectiveness, the risk of hypotension, dystonic reactions, and neuroleptic malignant syndrome may make it a less desirable option.4,25
Cyproheptadine, chlorpromazine, and other serotonin receptor antagonists require further investigation beyond individual case reports to determine their effectiveness and reliability in treating serotonin syndrome.
Other agents
Benzodiazepines are considered a mainstay for symptomatic relief because of their anxiolytic and muscle relaxant effects.26 However, animal studies showed that treatment with benzodiazepines attenuated hyperthermia but had no effect on time to recovery or outcome.27
Neuromuscular blocking agents. The suggested neuromuscular blocking agent for severe toxicity is a nondepolarizing agent such as vecuronium. Succinylcholine should be avoided, as it can exacerbate rhabdomyolysis and hyperkalemia.21
Dantrolene has also been suggested for its muscle-relaxing effects and use in malignant hyperthermia. However, this treatment has not been successful in isolated case reports and has been ineffective in animal models.4,28
Physical restraints are ill-advised, since isometric muscle contractions can exacerbate hyperthermia and lactic acidosis in agitated patients.21 If physical restraints are necessary to deliver medications, they should be removed as soon as possible.
HOW CAN WE PREVENT SEROTONIN SYNDROME?
Prevention of serotonin syndrome begins with improving education and awareness in patients and healthcare providers. Patients should be primarily concerned with taking their medications carefully as prescribed and recognizing early signs and symptoms of serotonin toxicity.
As use of antidepressants among an aging population continues to increase, and as physicians in multiple disciplines prescribe them for evolving indications (eg, duloxetine to treat osteoarthritis, diabetic neuropathy, fibromyalgia, and chemotherapy-induced peripheral neuropathy), healthcare providers need to be prepared to see more cases of serotonin syndrome and its deleterious effects.29–31 Physicians should be vigilant in minimizing unnecessary use of serotonergic agents and reviewing drug regimens regularly to limit polypharmacy.
Electronic ordering systems should be designed to detect and alert the prescriber to possible interactions that can potentiate serotonin syndrome, and to not place the order until the prescriber overrides the alert. Combinations of SSRIs and MAOIs have the highest risk for inducing severe serotonin syndrome and should always be avoided.
If a patient is transitioning between serotonergic agents, physicians should observe a safe washout period to prevent overlap.16,32 Washout periods may differ among medications depending on their half-lives. For example, sertraline has a washout period of 2 weeks, while fluoxetine requires a washout period of 5 to 6 weeks.33 Consulting a pharmacist may be helpful when considering half-lives and washout periods.
We believe that educating both patients and physicians regarding prevention will help minimize the risk for serotonergic syndrome and will improve efficiency in assessment and management should toxicity develop.
- Mojtabai R, Olfson M. National trends in long-term use of antidepressant medications: results from the US National Health and Nutrition Examination Survey. J Clin Psychiatry 2014; 75:169–177.
- Watson WA, Litovitz TL, Rodgers GC Jr, et al. 2002 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2003;21:353–421.
- Lai MW, Klein-Schwartz W, Rodgers GC, et al. 2005 Annual Report of the American Association of Poison Control Centers’ national poisoning and exopsure database. Clin Toxicol (Phila) 2006; 44:803–932.
- Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol 1999; 13:100–109.
- Isbister GK, Buckley NA. The pathophysiology of serotonin toxicity in animals and humans: implications for diagnosis and treatment. Clin Neuropharmacol 2005; 28:205–214.
- Woytowish MR, Maynor LM. Clinical relevance of linezolid-associated serotonin toxicity. Ann Pharmacother 2013; 47:388–397.
- Lee DO, Lee CD. Serotonin syndrome in a child associated with erythromycin and sertraline. Pharmacotherapy 1999; 19:894–896.
- Gillman PK. Triptans, serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache 2010; 50:264–272.
- Isbister GK, Buckley NA, Whyte IM. Serotonin toxicity: a practical approach to diagnosis and treatment. Med J Aust 2007; 187:361–365.
- Mowry JB, Spyker DA, Cantilena LR Jr, McMillan N, Ford M. 2013 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 31st Annual Report. Clin Toxicol (Phila) 2014; 52:1032–1283.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Karkare SU, Bhattacharjee S, Kamble P, Aparasu R. Prevalence and predictors of antidepressant prescribing in nursing home residents in the United States. Am J Geriatr Pharmacother 2011; 9:109–119.
- Weissman J, Meyers BS, Ghosh S, Bruce ML. Demographic, clinical, and functional factors associated with antidepressant use in the home healthcare elderly. Am J Geriatr Psychiatry 2011; 19:1042–1045.
- Caughey GE, Roughead EE, Shakib S, Vitry AI, Gilbert AL. Co-morbidity and potential treatment conflicts in elderly heart failure patients: a retrospective, cross-sectional study of administrative claims data. Drugs Aging 2011; 28:575–581.
- Iqbal MM, Basil MJ, Kaplan J, Iqbal MT. Overview of serotonin syndrome. Ann Clin Psychiatry 2012; 24:310–318.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- Prakash S, Patel V, Kakked S, Patel I, Yadav R. Mild serotonin syndrome: a report of 12 cases. Ann Indian Acad Neurol 2015; 18:226–230.
- Davies O, Batajoo-Shrestha B, Sosa-Popoteur J, Olibrice M. Full recovery after severe serotonin syndrome, severe rhabdomyolysis, multi-organ failure and disseminated intravascular coagulopathy from MDMA. Heart Lung 2014; 43:117–119.
- Pedavally S, Fugate JE, Rabinstein AA. Serotonin syndrome in the intensive care unit: clinical presentations and precipitating medications. Neurocrit Care 2014; 21:108–113.
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Velamoor VR, Norman RM, Caroff SN, Mann SC, Sullivan KA, Antelo RE. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994; 182:168–173.
- Isbister GK, Hackett LP, Dawson AH, Whyte IM, Smith AJ. Moclobemide poisoning: toxicokinetics and occurrence of serotonin toxicity. Br J Clin Pharmacol 2003; 56:441–450.
- Graudins A, Stearman A, Chan B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med 1998; 16:615–619.
- Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med 1994; 331:1021–1022.
- Gillman PK. Successful treatment of serotonin syndrome with chlorpromazine. Med J Aust 1996; 165:345–346.
- Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ 2014; 348:g1626.
- Nisijima K, Shioda K, Yoshino T, Takano K, Kato S. Diazepam and chlormethiazole attenuate the development of hyperthermia in an animal model of the serotonin syndrome. Neurochem Int 2003; 43:155–164.
- Nisijima K, Yoshino T, Yui K, Katoh S. Potent serotonin (5-HT)(2A) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res 2001; 890:23–31.
- Micca JL, Ruff D, Ahl J, Wohlreich MM. Safety and efficacy of duloxetine treatment in older and younger patients with osteoarthritis knee pain: a post hoc, subgroup analysis of two randomized, placebo-controlled trials. BMC Musculoskelet Disord 2013; 14:137.
- Smith EM, Pang H, Cirrincione C, et al; Alliance for Clinical Trials in Oncology. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA 2013; 309:1359–1367.
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev 2014; 1:CD007115.
- Caughey GE, Roughead EE, Shakib S, McDermott RA, Vitry AI, Gilbert AL. Comorbidity of chronic disease and potential treatment conflicts in older people dispensed antidepressants. Age Ageing 2010; 39:488–494.
- Gury C, Cousin F. Pharmacokinetics of SSRI antidepressants: half-life and clinical applicability. Encephale 1999; 25:470–476. French.
With a substantial increase in antidepressant use in the United States over the last 2 decades, serotonin syndrome has become an increasingly common and significant clinical concern. In 1999, 6.5% of adults age 18 and older were taking antidepressants; by 2010, the percentage had increased to 10.4%.1 Though the true incidence of serotonin syndrome is difficult to determine, the number of ingestions of selective serotonin reuptake inhibitors (SSRIs) associated with moderate to major effects reported to US poison control centers increased from 7,349 in 20022 to 8,585 in 2005.3
Though the clinical manifestations are often mild to moderate, patients with serotonin syndrome can deteriorate rapidly and require intensive care. Unlike neuroleptic malignant syndrome, serotonin syndrome should not be considered an extremely rare idiosyncratic reaction to medication, but rather a progression of serotonergic toxicity based on increasing concentration levels that can occur in any patient regardless of age.4
Because it has a nonspecific prodrome and protean manifestations, serotonin syndrome can easily be overlooked, misdiagnosed, or exacerbated if not carefully assessed. Diagnosis requires a low threshold for suspicion and a meticulous history and physical examination. In the syndrome’s mildest stage, symptoms are often misattributed to other causes, and in its most severe form, it can easily be mistaken for neuroleptic malignant syndrome.
WHAT IS SEROTONIN SYNDROME?
Serotonin syndrome classically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. These symptoms are a result of increased serotonin levels affecting the central and peripheral nervous systems. Serotonin affects a family of receptors that has seven members, of which 5-HT1A and 5-HT2A are most often responsible for serotonin syndrome.5
Conditions that can alter the regulation of serotonin include therapeutic doses, drug interactions, intentional or unintentional overdoses, and overlapping transitions between medications. As a result, drugs that have been associated with serotonin syndrome can be classified into the following five categories as shown below and in Table 1:
Drugs that decrease serotonin breakdown include monoamine oxidase inhibitors (MAOIs), linezolid,6 methylene blue, procarbazine, and Syrian rue.
Drugs that decrease serotonin reuptake include SSRIs, serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants, opioids (meperidine, buprenorphine, tramadol, tapentadol, dextromethorphan), antiepileptics (carbamazepine, valproate), and antiemetics (ondansetron, granisetron, metoclopramide), and the herbal preparation St. John’s wort.
Drugs that increase serotonin precursors or agonists include tryptophan, lithium, fentanyl, and lysergic acid diethylamide (LSD).
Drugs that increase serotonin release include fenfluramine, amphetamines, and methylenedioxymethamphetamine (ecstasy).
Drugs that prevent breakdown of the agents listed above are CYP2D6 and CYP3A4 inhibitors, eg, erythromycin,7 ciprofloxacin, fluconazole, ritonavir, and grapefruit juice.
However, the only drugs that have been reliably confirmed to precipitate serotonin syndrome are MAOIs, SSRIs, SNRIs, and serotonin releasers. Other listed drug interactions are based on case reports and have not been thoroughly evaluated.6–9
Currently, SSRIs are the most commonly prescribed antidepressant medications and, consequently, they are the ones most often implicated in serotonergic toxicity.1,10 An estimated 15% of SSRI overdoses lead to mild or moderate serotonin toxicity.11 Serotonergic agents used in conjunction can increase the risk for severe serotonin syndrome; an SSRI and an MAOI in combination poses the greatest risk.5
Ultimately, the incidence of serotonin syndrome is difficult to assess, but it is believed to be underreported because it is easy to misdiagnose and mild symptoms may be dismissed.
WHO IS AT RISK OF SEROTONIN SYNDROME?
Long-term antidepressant use has disproportionately increased in middle-aged and older adults and non-Hispanic whites.1,12,13 Intuitively, as the risk for depression increases dramatically in patients with chronic medical conditions, serotonin syndrome should be more prevalent among the elderly. In addition, patients with multiple comorbidities take more medications, increasing the risk of polypharmacy and adverse drug reactions.14
Although the epidemiology of serotonin syndrome has yet to be extensively studied, the combination of age and comorbidities may increase the risk for this condition.
HOW DOES IT PRESENT?
Serotonin syndrome characteristically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. However, these symptoms may not occur simultaneously: autonomic dysfunction is present in 40% of patients, neuromuscular excitation in 50%, and altered mental status in 40%.15 The symptoms can range from mild to life-threatening (Table 2).16
Autonomic dysfunction. Diaphoresis is present in 48.8% of cases, tachycardia in 44%, nausea and vomiting in 26.8%, and mydriasis in 19.5%. Other signs are hyperactive bowel sounds, diarrhea, and flushing.16
Neuromuscular excitation. Myoclonus is present in 48.8%, hyperreflexia in 41%, hyperthermia in 26.8%, and hypertonicity and rigidity in 19.5%. Other signs are spontaneous or inducible clonus, ocular clonus (continuous rhythmic oscillations of gaze), and tremor.
Altered mental status. Confusion is present in 41.2% and agitation in 36.5%. Other signs are anxiety, lethargy, and coma.
Symptoms of serotonin toxicity arise within an hour of a precipitating event (eg, ingestion) in approximately 28% of patients, and within 6 hours in 61%.16 Highly diagnostic features include hyperreflexia and induced or spontaneous clonus that are generally more pronounced in the lower limbs.11 Clonus can be elicited with ankle dorsiflexion.
In mild toxicity, patients may present with tremor or twitching and anxiety, as well as with hyperreflexia, tachycardia, diaphoresis, and mydriasis. Further investigation may uncover a recently initiated antidepressant or a cold-and-cough medication that contains dextromethorphan.15,17
In moderate toxicity, patients present in significant distress, with agitation and restlessness. Features may include hyperreflexia and clonus of the lower extremities, opsoclonus, hyperactive bowel sounds, diarrhea, nausea, vomiting, tachycardia, hypertension, diaphoresis, mydriasis, and hyperthermia (< 40°C, 104°F). The patient’s history may reveal use of ecstasy or combined treatment with serotonin-potentiating agents such as an antidepressant with a proserotonergic opioid, antiepileptic, or CYP2D6 or CYP3A4 inhibitor.15
Severe serotonin toxicity is a life-threatening condition that can lead to multiorgan failure within hours. It can be characterized by muscle rigidity, which can cause the body temperature to elevate rapidly to over 40°C. This hypertonicity can mask the classic and diagnostic signs of hyperreflexia and clonus. Patients may have unstable and dynamic vital signs with confusion or delirium and can experience tonic-clonic seizures.
If the muscle rigidity and resulting hyperthermia are not managed properly, patients can develop cellular damage and enzyme dysfunction leading to rhabdomyolysis, myoglobinuria, renal failure, metabolic acidosis, acute respiratory distress syndrome, and disseminated intravascular coagulation.16,18
Serotonin crisis is usually caused by the co-ingestion of multiple serotonergic agents, such as an antidepressant with an aforementioned opioid and antiemetic19; combining an SSRI and an MAOI poses the greatest risk. Alternatively, patients may have recently switched antidepressants without observing a safe washout period, leading to an overlap of serotonin levels.16
HOW DO WE DIAGNOSE SEROTONIN SYNDROME?
Serotonin syndrome is a clinical diagnosis and therefore requires a thorough review of medications and physical examination. Serum serotonin levels are an unreliable indicator of toxicity and do not correlate well with the clinical presentation.16
(based on information in reference 9).
Currently, there are two clinical tools for diagnosing serotonin syndrome: the Hunter serotonin toxicity criteria (Figure 1) and the Sternbach criteria.
The Hunter criteria are based more heavily on physical findings. The patient must have taken a serotonergic agent and have one of the following:
- Spontaneous clonus
- Inducible clonus plus agitation or diaphoresis
- Ocular clonus plus agitation or diaphoresis
- Inducible clonus or ocular clonus, plus hypertonia and hyperthermia
- Tremor plus hyperreflexia.
The Sternbach criteria. The patient must be using a serotonergic agent, must have no other causes of symptoms, must not have recently used a neuroleptic agent, and must have three of the following:
- Mental status changes
- Agitation
- Hyperreflexia
- Myoclonus
- Diaphoresis
- Shivering
- Tremor
- Diarrhea
- Incoordination
- Fever
The Hunter criteria are recommended and are more specific (97% vs 96%) and more sensitive (84% vs 75%) than the Sternbach criteria when compared with the gold standard of diagnosis by a clinical toxicologist.1 The Hunter criteria are also less likely to yield false-positive results.11
Differential diagnosis
The differential diagnosis for serotonin syndrome includes neuroleptic malignant syndrome, anticholinergic poisoning (Table 3), metastatic carcinoma, central nervous system infection, gastroenteritis, and sepsis.
Neuroleptic malignant syndrome, the disorder most often misdiagnosed as serotonin syndrome, is an idiosyncratic reaction to a dopamine antagonist (eg, haloperidol, fluphenazine) that develops over days to weeks.20 In 70% of patients, agitated delirium with confusion appears first, followed by lead pipe rigidity and cogwheel tremor, then hyperthermia with body temperature greater than 40°C, and finally, profuse diaphoresis, tachycardia, hypertension, and tachypnea.21
Key elements that distinguish neuroleptic malignant syndrome are the timeline of the clinical course, bradyreflexia, and the absence of clonus. Prodromal symptoms of nausea, vomiting, and diarrhea are also rare in neuroleptic malignant syndrome. Neuroleptic malignant syndrome typically requires an average of 9 days to resolve.
Anticholinergic poisoning usually develops within 1 to 2 hours of oral ingestion. Symptoms include flushing, anhidrosis, anhidrotic hyperthermia, mydriasis, urinary retention, decreased bowel sounds, agitated delirium, and visual hallucinations. In contrast to serotonin syndrome, reflexes and muscle tone are normal with anticholinergic poisoning.
HOW CAN WE TREAT SEROTONIN SYNDROME?
The two mainstays of serotonin syndrome management are to discontinue the serotonergic agent and to give supportive care. Most patients improve within 24 hours of stopping the precipitating drug and starting therapy.16
For mild serotonin syndrome, treatment involves discontinuing the offending agent and supportive therapy with intravenous fluids, correction of vital signs, and symptomatic treatment with a benzodiazepine. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For moderate serotonin syndrome, treatment also involves stopping the serotonergic agent and giving supportive care. Symptomatic treatment with a benzodiazepine and nonserotonergic antiemetics is recommended, and standard cooling measures should be implemented for hyperthermia. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For severe serotonin toxicity, treatment should focus on management of airway, breathing, and circulation—ie, the “ABCs.” The two primary life-threatening concerns are hyperthermia (temperature > 40°C or 104°F) and rigidity, which can lead to hypoventilation.1,22 Controlling hyperthermia and rigidity can prevent other grave complications. Patients with severe serotonin toxicity should be sedated, paralyzed, and intubated.21 This will reverse ventilatory hypertonia and allow for mechanical ventilation. Paralysis will also prevent the exacerbation of hyperthermia, which is caused by muscle rigidity. Antipyretics have no role in the treatment of serotonin syndrome since the hyperthermia is not caused by a change in the hypothalamic temperature set point.21 Standard cooling measures should be used to manage hyperthermia.
Serotonin antagonists
Serotonin antagonists have had some success in case reports, but further studies are needed to confirm this.4,23,24
Cyproheptadine is a potent 5-HT2A antagonist; patients usually respond within 1 to 2 hours of administration. Signs and symptoms have resolved completely within times ranging from 20 minutes to 48 hours, depending on the severity of toxicity.
The recommended initial dose of cyproheptadine is 12 mg, followed by 2 mg every 2 hours if symptoms continue.16 Maintenance dosing with 8 mg every 6 hours should be prescribed once stabilization is achieved. The total daily dose for adults should not exceed 0.5 mg/kg/day. Cyproheptadine is available only in oral form but can be crushed and administered via a nasogastric tube.21
Chlorpromazine is a 5-HT1A and 5-HT2A antagonist and can be given intramuscularly. Despite case reports citing its effectiveness, the risk of hypotension, dystonic reactions, and neuroleptic malignant syndrome may make it a less desirable option.4,25
Cyproheptadine, chlorpromazine, and other serotonin receptor antagonists require further investigation beyond individual case reports to determine their effectiveness and reliability in treating serotonin syndrome.
Other agents
Benzodiazepines are considered a mainstay for symptomatic relief because of their anxiolytic and muscle relaxant effects.26 However, animal studies showed that treatment with benzodiazepines attenuated hyperthermia but had no effect on time to recovery or outcome.27
Neuromuscular blocking agents. The suggested neuromuscular blocking agent for severe toxicity is a nondepolarizing agent such as vecuronium. Succinylcholine should be avoided, as it can exacerbate rhabdomyolysis and hyperkalemia.21
Dantrolene has also been suggested for its muscle-relaxing effects and use in malignant hyperthermia. However, this treatment has not been successful in isolated case reports and has been ineffective in animal models.4,28
Physical restraints are ill-advised, since isometric muscle contractions can exacerbate hyperthermia and lactic acidosis in agitated patients.21 If physical restraints are necessary to deliver medications, they should be removed as soon as possible.
HOW CAN WE PREVENT SEROTONIN SYNDROME?
Prevention of serotonin syndrome begins with improving education and awareness in patients and healthcare providers. Patients should be primarily concerned with taking their medications carefully as prescribed and recognizing early signs and symptoms of serotonin toxicity.
As use of antidepressants among an aging population continues to increase, and as physicians in multiple disciplines prescribe them for evolving indications (eg, duloxetine to treat osteoarthritis, diabetic neuropathy, fibromyalgia, and chemotherapy-induced peripheral neuropathy), healthcare providers need to be prepared to see more cases of serotonin syndrome and its deleterious effects.29–31 Physicians should be vigilant in minimizing unnecessary use of serotonergic agents and reviewing drug regimens regularly to limit polypharmacy.
Electronic ordering systems should be designed to detect and alert the prescriber to possible interactions that can potentiate serotonin syndrome, and to not place the order until the prescriber overrides the alert. Combinations of SSRIs and MAOIs have the highest risk for inducing severe serotonin syndrome and should always be avoided.
If a patient is transitioning between serotonergic agents, physicians should observe a safe washout period to prevent overlap.16,32 Washout periods may differ among medications depending on their half-lives. For example, sertraline has a washout period of 2 weeks, while fluoxetine requires a washout period of 5 to 6 weeks.33 Consulting a pharmacist may be helpful when considering half-lives and washout periods.
We believe that educating both patients and physicians regarding prevention will help minimize the risk for serotonergic syndrome and will improve efficiency in assessment and management should toxicity develop.
With a substantial increase in antidepressant use in the United States over the last 2 decades, serotonin syndrome has become an increasingly common and significant clinical concern. In 1999, 6.5% of adults age 18 and older were taking antidepressants; by 2010, the percentage had increased to 10.4%.1 Though the true incidence of serotonin syndrome is difficult to determine, the number of ingestions of selective serotonin reuptake inhibitors (SSRIs) associated with moderate to major effects reported to US poison control centers increased from 7,349 in 20022 to 8,585 in 2005.3
Though the clinical manifestations are often mild to moderate, patients with serotonin syndrome can deteriorate rapidly and require intensive care. Unlike neuroleptic malignant syndrome, serotonin syndrome should not be considered an extremely rare idiosyncratic reaction to medication, but rather a progression of serotonergic toxicity based on increasing concentration levels that can occur in any patient regardless of age.4
Because it has a nonspecific prodrome and protean manifestations, serotonin syndrome can easily be overlooked, misdiagnosed, or exacerbated if not carefully assessed. Diagnosis requires a low threshold for suspicion and a meticulous history and physical examination. In the syndrome’s mildest stage, symptoms are often misattributed to other causes, and in its most severe form, it can easily be mistaken for neuroleptic malignant syndrome.
WHAT IS SEROTONIN SYNDROME?
Serotonin syndrome classically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. These symptoms are a result of increased serotonin levels affecting the central and peripheral nervous systems. Serotonin affects a family of receptors that has seven members, of which 5-HT1A and 5-HT2A are most often responsible for serotonin syndrome.5
Conditions that can alter the regulation of serotonin include therapeutic doses, drug interactions, intentional or unintentional overdoses, and overlapping transitions between medications. As a result, drugs that have been associated with serotonin syndrome can be classified into the following five categories as shown below and in Table 1:
Drugs that decrease serotonin breakdown include monoamine oxidase inhibitors (MAOIs), linezolid,6 methylene blue, procarbazine, and Syrian rue.
Drugs that decrease serotonin reuptake include SSRIs, serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants, opioids (meperidine, buprenorphine, tramadol, tapentadol, dextromethorphan), antiepileptics (carbamazepine, valproate), and antiemetics (ondansetron, granisetron, metoclopramide), and the herbal preparation St. John’s wort.
Drugs that increase serotonin precursors or agonists include tryptophan, lithium, fentanyl, and lysergic acid diethylamide (LSD).
Drugs that increase serotonin release include fenfluramine, amphetamines, and methylenedioxymethamphetamine (ecstasy).
Drugs that prevent breakdown of the agents listed above are CYP2D6 and CYP3A4 inhibitors, eg, erythromycin,7 ciprofloxacin, fluconazole, ritonavir, and grapefruit juice.
However, the only drugs that have been reliably confirmed to precipitate serotonin syndrome are MAOIs, SSRIs, SNRIs, and serotonin releasers. Other listed drug interactions are based on case reports and have not been thoroughly evaluated.6–9
Currently, SSRIs are the most commonly prescribed antidepressant medications and, consequently, they are the ones most often implicated in serotonergic toxicity.1,10 An estimated 15% of SSRI overdoses lead to mild or moderate serotonin toxicity.11 Serotonergic agents used in conjunction can increase the risk for severe serotonin syndrome; an SSRI and an MAOI in combination poses the greatest risk.5
Ultimately, the incidence of serotonin syndrome is difficult to assess, but it is believed to be underreported because it is easy to misdiagnose and mild symptoms may be dismissed.
WHO IS AT RISK OF SEROTONIN SYNDROME?
Long-term antidepressant use has disproportionately increased in middle-aged and older adults and non-Hispanic whites.1,12,13 Intuitively, as the risk for depression increases dramatically in patients with chronic medical conditions, serotonin syndrome should be more prevalent among the elderly. In addition, patients with multiple comorbidities take more medications, increasing the risk of polypharmacy and adverse drug reactions.14
Although the epidemiology of serotonin syndrome has yet to be extensively studied, the combination of age and comorbidities may increase the risk for this condition.
HOW DOES IT PRESENT?
Serotonin syndrome characteristically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. However, these symptoms may not occur simultaneously: autonomic dysfunction is present in 40% of patients, neuromuscular excitation in 50%, and altered mental status in 40%.15 The symptoms can range from mild to life-threatening (Table 2).16
Autonomic dysfunction. Diaphoresis is present in 48.8% of cases, tachycardia in 44%, nausea and vomiting in 26.8%, and mydriasis in 19.5%. Other signs are hyperactive bowel sounds, diarrhea, and flushing.16
Neuromuscular excitation. Myoclonus is present in 48.8%, hyperreflexia in 41%, hyperthermia in 26.8%, and hypertonicity and rigidity in 19.5%. Other signs are spontaneous or inducible clonus, ocular clonus (continuous rhythmic oscillations of gaze), and tremor.
Altered mental status. Confusion is present in 41.2% and agitation in 36.5%. Other signs are anxiety, lethargy, and coma.
Symptoms of serotonin toxicity arise within an hour of a precipitating event (eg, ingestion) in approximately 28% of patients, and within 6 hours in 61%.16 Highly diagnostic features include hyperreflexia and induced or spontaneous clonus that are generally more pronounced in the lower limbs.11 Clonus can be elicited with ankle dorsiflexion.
In mild toxicity, patients may present with tremor or twitching and anxiety, as well as with hyperreflexia, tachycardia, diaphoresis, and mydriasis. Further investigation may uncover a recently initiated antidepressant or a cold-and-cough medication that contains dextromethorphan.15,17
In moderate toxicity, patients present in significant distress, with agitation and restlessness. Features may include hyperreflexia and clonus of the lower extremities, opsoclonus, hyperactive bowel sounds, diarrhea, nausea, vomiting, tachycardia, hypertension, diaphoresis, mydriasis, and hyperthermia (< 40°C, 104°F). The patient’s history may reveal use of ecstasy or combined treatment with serotonin-potentiating agents such as an antidepressant with a proserotonergic opioid, antiepileptic, or CYP2D6 or CYP3A4 inhibitor.15
Severe serotonin toxicity is a life-threatening condition that can lead to multiorgan failure within hours. It can be characterized by muscle rigidity, which can cause the body temperature to elevate rapidly to over 40°C. This hypertonicity can mask the classic and diagnostic signs of hyperreflexia and clonus. Patients may have unstable and dynamic vital signs with confusion or delirium and can experience tonic-clonic seizures.
If the muscle rigidity and resulting hyperthermia are not managed properly, patients can develop cellular damage and enzyme dysfunction leading to rhabdomyolysis, myoglobinuria, renal failure, metabolic acidosis, acute respiratory distress syndrome, and disseminated intravascular coagulation.16,18
Serotonin crisis is usually caused by the co-ingestion of multiple serotonergic agents, such as an antidepressant with an aforementioned opioid and antiemetic19; combining an SSRI and an MAOI poses the greatest risk. Alternatively, patients may have recently switched antidepressants without observing a safe washout period, leading to an overlap of serotonin levels.16
HOW DO WE DIAGNOSE SEROTONIN SYNDROME?
Serotonin syndrome is a clinical diagnosis and therefore requires a thorough review of medications and physical examination. Serum serotonin levels are an unreliable indicator of toxicity and do not correlate well with the clinical presentation.16
(based on information in reference 9).
Currently, there are two clinical tools for diagnosing serotonin syndrome: the Hunter serotonin toxicity criteria (Figure 1) and the Sternbach criteria.
The Hunter criteria are based more heavily on physical findings. The patient must have taken a serotonergic agent and have one of the following:
- Spontaneous clonus
- Inducible clonus plus agitation or diaphoresis
- Ocular clonus plus agitation or diaphoresis
- Inducible clonus or ocular clonus, plus hypertonia and hyperthermia
- Tremor plus hyperreflexia.
The Sternbach criteria. The patient must be using a serotonergic agent, must have no other causes of symptoms, must not have recently used a neuroleptic agent, and must have three of the following:
- Mental status changes
- Agitation
- Hyperreflexia
- Myoclonus
- Diaphoresis
- Shivering
- Tremor
- Diarrhea
- Incoordination
- Fever
The Hunter criteria are recommended and are more specific (97% vs 96%) and more sensitive (84% vs 75%) than the Sternbach criteria when compared with the gold standard of diagnosis by a clinical toxicologist.1 The Hunter criteria are also less likely to yield false-positive results.11
Differential diagnosis
The differential diagnosis for serotonin syndrome includes neuroleptic malignant syndrome, anticholinergic poisoning (Table 3), metastatic carcinoma, central nervous system infection, gastroenteritis, and sepsis.
Neuroleptic malignant syndrome, the disorder most often misdiagnosed as serotonin syndrome, is an idiosyncratic reaction to a dopamine antagonist (eg, haloperidol, fluphenazine) that develops over days to weeks.20 In 70% of patients, agitated delirium with confusion appears first, followed by lead pipe rigidity and cogwheel tremor, then hyperthermia with body temperature greater than 40°C, and finally, profuse diaphoresis, tachycardia, hypertension, and tachypnea.21
Key elements that distinguish neuroleptic malignant syndrome are the timeline of the clinical course, bradyreflexia, and the absence of clonus. Prodromal symptoms of nausea, vomiting, and diarrhea are also rare in neuroleptic malignant syndrome. Neuroleptic malignant syndrome typically requires an average of 9 days to resolve.
Anticholinergic poisoning usually develops within 1 to 2 hours of oral ingestion. Symptoms include flushing, anhidrosis, anhidrotic hyperthermia, mydriasis, urinary retention, decreased bowel sounds, agitated delirium, and visual hallucinations. In contrast to serotonin syndrome, reflexes and muscle tone are normal with anticholinergic poisoning.
HOW CAN WE TREAT SEROTONIN SYNDROME?
The two mainstays of serotonin syndrome management are to discontinue the serotonergic agent and to give supportive care. Most patients improve within 24 hours of stopping the precipitating drug and starting therapy.16
For mild serotonin syndrome, treatment involves discontinuing the offending agent and supportive therapy with intravenous fluids, correction of vital signs, and symptomatic treatment with a benzodiazepine. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For moderate serotonin syndrome, treatment also involves stopping the serotonergic agent and giving supportive care. Symptomatic treatment with a benzodiazepine and nonserotonergic antiemetics is recommended, and standard cooling measures should be implemented for hyperthermia. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For severe serotonin toxicity, treatment should focus on management of airway, breathing, and circulation—ie, the “ABCs.” The two primary life-threatening concerns are hyperthermia (temperature > 40°C or 104°F) and rigidity, which can lead to hypoventilation.1,22 Controlling hyperthermia and rigidity can prevent other grave complications. Patients with severe serotonin toxicity should be sedated, paralyzed, and intubated.21 This will reverse ventilatory hypertonia and allow for mechanical ventilation. Paralysis will also prevent the exacerbation of hyperthermia, which is caused by muscle rigidity. Antipyretics have no role in the treatment of serotonin syndrome since the hyperthermia is not caused by a change in the hypothalamic temperature set point.21 Standard cooling measures should be used to manage hyperthermia.
Serotonin antagonists
Serotonin antagonists have had some success in case reports, but further studies are needed to confirm this.4,23,24
Cyproheptadine is a potent 5-HT2A antagonist; patients usually respond within 1 to 2 hours of administration. Signs and symptoms have resolved completely within times ranging from 20 minutes to 48 hours, depending on the severity of toxicity.
The recommended initial dose of cyproheptadine is 12 mg, followed by 2 mg every 2 hours if symptoms continue.16 Maintenance dosing with 8 mg every 6 hours should be prescribed once stabilization is achieved. The total daily dose for adults should not exceed 0.5 mg/kg/day. Cyproheptadine is available only in oral form but can be crushed and administered via a nasogastric tube.21
Chlorpromazine is a 5-HT1A and 5-HT2A antagonist and can be given intramuscularly. Despite case reports citing its effectiveness, the risk of hypotension, dystonic reactions, and neuroleptic malignant syndrome may make it a less desirable option.4,25
Cyproheptadine, chlorpromazine, and other serotonin receptor antagonists require further investigation beyond individual case reports to determine their effectiveness and reliability in treating serotonin syndrome.
Other agents
Benzodiazepines are considered a mainstay for symptomatic relief because of their anxiolytic and muscle relaxant effects.26 However, animal studies showed that treatment with benzodiazepines attenuated hyperthermia but had no effect on time to recovery or outcome.27
Neuromuscular blocking agents. The suggested neuromuscular blocking agent for severe toxicity is a nondepolarizing agent such as vecuronium. Succinylcholine should be avoided, as it can exacerbate rhabdomyolysis and hyperkalemia.21
Dantrolene has also been suggested for its muscle-relaxing effects and use in malignant hyperthermia. However, this treatment has not been successful in isolated case reports and has been ineffective in animal models.4,28
Physical restraints are ill-advised, since isometric muscle contractions can exacerbate hyperthermia and lactic acidosis in agitated patients.21 If physical restraints are necessary to deliver medications, they should be removed as soon as possible.
HOW CAN WE PREVENT SEROTONIN SYNDROME?
Prevention of serotonin syndrome begins with improving education and awareness in patients and healthcare providers. Patients should be primarily concerned with taking their medications carefully as prescribed and recognizing early signs and symptoms of serotonin toxicity.
As use of antidepressants among an aging population continues to increase, and as physicians in multiple disciplines prescribe them for evolving indications (eg, duloxetine to treat osteoarthritis, diabetic neuropathy, fibromyalgia, and chemotherapy-induced peripheral neuropathy), healthcare providers need to be prepared to see more cases of serotonin syndrome and its deleterious effects.29–31 Physicians should be vigilant in minimizing unnecessary use of serotonergic agents and reviewing drug regimens regularly to limit polypharmacy.
Electronic ordering systems should be designed to detect and alert the prescriber to possible interactions that can potentiate serotonin syndrome, and to not place the order until the prescriber overrides the alert. Combinations of SSRIs and MAOIs have the highest risk for inducing severe serotonin syndrome and should always be avoided.
If a patient is transitioning between serotonergic agents, physicians should observe a safe washout period to prevent overlap.16,32 Washout periods may differ among medications depending on their half-lives. For example, sertraline has a washout period of 2 weeks, while fluoxetine requires a washout period of 5 to 6 weeks.33 Consulting a pharmacist may be helpful when considering half-lives and washout periods.
We believe that educating both patients and physicians regarding prevention will help minimize the risk for serotonergic syndrome and will improve efficiency in assessment and management should toxicity develop.
- Mojtabai R, Olfson M. National trends in long-term use of antidepressant medications: results from the US National Health and Nutrition Examination Survey. J Clin Psychiatry 2014; 75:169–177.
- Watson WA, Litovitz TL, Rodgers GC Jr, et al. 2002 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2003;21:353–421.
- Lai MW, Klein-Schwartz W, Rodgers GC, et al. 2005 Annual Report of the American Association of Poison Control Centers’ national poisoning and exopsure database. Clin Toxicol (Phila) 2006; 44:803–932.
- Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol 1999; 13:100–109.
- Isbister GK, Buckley NA. The pathophysiology of serotonin toxicity in animals and humans: implications for diagnosis and treatment. Clin Neuropharmacol 2005; 28:205–214.
- Woytowish MR, Maynor LM. Clinical relevance of linezolid-associated serotonin toxicity. Ann Pharmacother 2013; 47:388–397.
- Lee DO, Lee CD. Serotonin syndrome in a child associated with erythromycin and sertraline. Pharmacotherapy 1999; 19:894–896.
- Gillman PK. Triptans, serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache 2010; 50:264–272.
- Isbister GK, Buckley NA, Whyte IM. Serotonin toxicity: a practical approach to diagnosis and treatment. Med J Aust 2007; 187:361–365.
- Mowry JB, Spyker DA, Cantilena LR Jr, McMillan N, Ford M. 2013 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 31st Annual Report. Clin Toxicol (Phila) 2014; 52:1032–1283.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Karkare SU, Bhattacharjee S, Kamble P, Aparasu R. Prevalence and predictors of antidepressant prescribing in nursing home residents in the United States. Am J Geriatr Pharmacother 2011; 9:109–119.
- Weissman J, Meyers BS, Ghosh S, Bruce ML. Demographic, clinical, and functional factors associated with antidepressant use in the home healthcare elderly. Am J Geriatr Psychiatry 2011; 19:1042–1045.
- Caughey GE, Roughead EE, Shakib S, Vitry AI, Gilbert AL. Co-morbidity and potential treatment conflicts in elderly heart failure patients: a retrospective, cross-sectional study of administrative claims data. Drugs Aging 2011; 28:575–581.
- Iqbal MM, Basil MJ, Kaplan J, Iqbal MT. Overview of serotonin syndrome. Ann Clin Psychiatry 2012; 24:310–318.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- Prakash S, Patel V, Kakked S, Patel I, Yadav R. Mild serotonin syndrome: a report of 12 cases. Ann Indian Acad Neurol 2015; 18:226–230.
- Davies O, Batajoo-Shrestha B, Sosa-Popoteur J, Olibrice M. Full recovery after severe serotonin syndrome, severe rhabdomyolysis, multi-organ failure and disseminated intravascular coagulopathy from MDMA. Heart Lung 2014; 43:117–119.
- Pedavally S, Fugate JE, Rabinstein AA. Serotonin syndrome in the intensive care unit: clinical presentations and precipitating medications. Neurocrit Care 2014; 21:108–113.
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Velamoor VR, Norman RM, Caroff SN, Mann SC, Sullivan KA, Antelo RE. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994; 182:168–173.
- Isbister GK, Hackett LP, Dawson AH, Whyte IM, Smith AJ. Moclobemide poisoning: toxicokinetics and occurrence of serotonin toxicity. Br J Clin Pharmacol 2003; 56:441–450.
- Graudins A, Stearman A, Chan B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med 1998; 16:615–619.
- Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med 1994; 331:1021–1022.
- Gillman PK. Successful treatment of serotonin syndrome with chlorpromazine. Med J Aust 1996; 165:345–346.
- Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ 2014; 348:g1626.
- Nisijima K, Shioda K, Yoshino T, Takano K, Kato S. Diazepam and chlormethiazole attenuate the development of hyperthermia in an animal model of the serotonin syndrome. Neurochem Int 2003; 43:155–164.
- Nisijima K, Yoshino T, Yui K, Katoh S. Potent serotonin (5-HT)(2A) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res 2001; 890:23–31.
- Micca JL, Ruff D, Ahl J, Wohlreich MM. Safety and efficacy of duloxetine treatment in older and younger patients with osteoarthritis knee pain: a post hoc, subgroup analysis of two randomized, placebo-controlled trials. BMC Musculoskelet Disord 2013; 14:137.
- Smith EM, Pang H, Cirrincione C, et al; Alliance for Clinical Trials in Oncology. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA 2013; 309:1359–1367.
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev 2014; 1:CD007115.
- Caughey GE, Roughead EE, Shakib S, McDermott RA, Vitry AI, Gilbert AL. Comorbidity of chronic disease and potential treatment conflicts in older people dispensed antidepressants. Age Ageing 2010; 39:488–494.
- Gury C, Cousin F. Pharmacokinetics of SSRI antidepressants: half-life and clinical applicability. Encephale 1999; 25:470–476. French.
- Mojtabai R, Olfson M. National trends in long-term use of antidepressant medications: results from the US National Health and Nutrition Examination Survey. J Clin Psychiatry 2014; 75:169–177.
- Watson WA, Litovitz TL, Rodgers GC Jr, et al. 2002 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2003;21:353–421.
- Lai MW, Klein-Schwartz W, Rodgers GC, et al. 2005 Annual Report of the American Association of Poison Control Centers’ national poisoning and exopsure database. Clin Toxicol (Phila) 2006; 44:803–932.
- Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol 1999; 13:100–109.
- Isbister GK, Buckley NA. The pathophysiology of serotonin toxicity in animals and humans: implications for diagnosis and treatment. Clin Neuropharmacol 2005; 28:205–214.
- Woytowish MR, Maynor LM. Clinical relevance of linezolid-associated serotonin toxicity. Ann Pharmacother 2013; 47:388–397.
- Lee DO, Lee CD. Serotonin syndrome in a child associated with erythromycin and sertraline. Pharmacotherapy 1999; 19:894–896.
- Gillman PK. Triptans, serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache 2010; 50:264–272.
- Isbister GK, Buckley NA, Whyte IM. Serotonin toxicity: a practical approach to diagnosis and treatment. Med J Aust 2007; 187:361–365.
- Mowry JB, Spyker DA, Cantilena LR Jr, McMillan N, Ford M. 2013 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 31st Annual Report. Clin Toxicol (Phila) 2014; 52:1032–1283.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Karkare SU, Bhattacharjee S, Kamble P, Aparasu R. Prevalence and predictors of antidepressant prescribing in nursing home residents in the United States. Am J Geriatr Pharmacother 2011; 9:109–119.
- Weissman J, Meyers BS, Ghosh S, Bruce ML. Demographic, clinical, and functional factors associated with antidepressant use in the home healthcare elderly. Am J Geriatr Psychiatry 2011; 19:1042–1045.
- Caughey GE, Roughead EE, Shakib S, Vitry AI, Gilbert AL. Co-morbidity and potential treatment conflicts in elderly heart failure patients: a retrospective, cross-sectional study of administrative claims data. Drugs Aging 2011; 28:575–581.
- Iqbal MM, Basil MJ, Kaplan J, Iqbal MT. Overview of serotonin syndrome. Ann Clin Psychiatry 2012; 24:310–318.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- Prakash S, Patel V, Kakked S, Patel I, Yadav R. Mild serotonin syndrome: a report of 12 cases. Ann Indian Acad Neurol 2015; 18:226–230.
- Davies O, Batajoo-Shrestha B, Sosa-Popoteur J, Olibrice M. Full recovery after severe serotonin syndrome, severe rhabdomyolysis, multi-organ failure and disseminated intravascular coagulopathy from MDMA. Heart Lung 2014; 43:117–119.
- Pedavally S, Fugate JE, Rabinstein AA. Serotonin syndrome in the intensive care unit: clinical presentations and precipitating medications. Neurocrit Care 2014; 21:108–113.
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Velamoor VR, Norman RM, Caroff SN, Mann SC, Sullivan KA, Antelo RE. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994; 182:168–173.
- Isbister GK, Hackett LP, Dawson AH, Whyte IM, Smith AJ. Moclobemide poisoning: toxicokinetics and occurrence of serotonin toxicity. Br J Clin Pharmacol 2003; 56:441–450.
- Graudins A, Stearman A, Chan B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med 1998; 16:615–619.
- Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med 1994; 331:1021–1022.
- Gillman PK. Successful treatment of serotonin syndrome with chlorpromazine. Med J Aust 1996; 165:345–346.
- Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ 2014; 348:g1626.
- Nisijima K, Shioda K, Yoshino T, Takano K, Kato S. Diazepam and chlormethiazole attenuate the development of hyperthermia in an animal model of the serotonin syndrome. Neurochem Int 2003; 43:155–164.
- Nisijima K, Yoshino T, Yui K, Katoh S. Potent serotonin (5-HT)(2A) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res 2001; 890:23–31.
- Micca JL, Ruff D, Ahl J, Wohlreich MM. Safety and efficacy of duloxetine treatment in older and younger patients with osteoarthritis knee pain: a post hoc, subgroup analysis of two randomized, placebo-controlled trials. BMC Musculoskelet Disord 2013; 14:137.
- Smith EM, Pang H, Cirrincione C, et al; Alliance for Clinical Trials in Oncology. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA 2013; 309:1359–1367.
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev 2014; 1:CD007115.
- Caughey GE, Roughead EE, Shakib S, McDermott RA, Vitry AI, Gilbert AL. Comorbidity of chronic disease and potential treatment conflicts in older people dispensed antidepressants. Age Ageing 2010; 39:488–494.
- Gury C, Cousin F. Pharmacokinetics of SSRI antidepressants: half-life and clinical applicability. Encephale 1999; 25:470–476. French.
KEY POINTS
- Serotonin syndrome is caused by elevated serotonin levels in the central and peripheral nervous systems.
- The classic presentation is the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. These symptoms vary based on the severity of serotonergic toxicity and often do not present concomitantly.
- Early recognition is critical to ensure appropriate resuscitative measures and to limit further use of drugs that can exacerbate symptoms.

Bilateral earlobe creases and coronary artery disease
A 70-year-old man with hypertension and hypercholesterolemia presented to the emergency department after the acute onset of substernal, pressure-like chest pain while climbing a flight of stairs. His physical examination was normal, but he was noted to have bilateral diagonal earlobe creases (the Frank sign) (Figure 1), considered by some to indicate risk of coronary artery disease.1–5
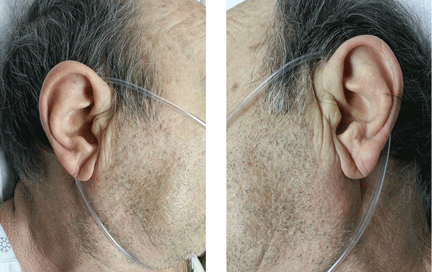
Electrocardiography showed atrial fibrillation with a ventricular rate of 149 beats per minute, ST-segment elevation in leads V1 and aVR, and ST-segment depression in leads V3 to V6, II, III, and aVF.
Urgent coronary arteriography showed severe coronary artery disease (Figure 2). Left ventriculography showed an ejection fraction of 50% with mild anterior wall hypokinesis. A drug-eluting stent was placed in the mid-left anterior descending artery. The patient tolerated the procedure well, and his chest pain resolved afterward.
A STILL-UNCLEAR ASSOCIATION

Sanders T. Frank, in 1973, first described a diagonal wrinkle-like line on the earlobe as a sign of coronary artery disease.1 Subsequently, autopsy studies suggested that deep bilateral earlobe creases could be an important sign of coronary atherosclerosis.2 Diagonal earlobe creases have been shown to be independently associated with increased prevalence, extent, and severity of coronary artery disease.3,4 They are also associated with major adverse cardiovascular events4 and ischemic stroke.5 The mechanism linking diagonal earlobe creases and atherosclerotic disease is not yet clear.
This patient’s presentation and evaluation remind us that bilateral earlobe creases may be useful to include in the clinical examination of patients with suspected coronary artery disease and may facilitate early recognition of disease in a patient at high risk.
- Frank ST. Aural sign of coronary-artery disease. N Engl J Med 1973; 289:327–328.
- Patel V, Champ C, Andrews PS, Gostelow BE, Gunasekara NP, Davidson AR. Diagonal earlobe creases and atheromatous disease: a postmortem study. J R Coll Phys Lond 1992; 26:274–277.
- Kaukola S, Manninen V, Valle M, Halonen PI. Ear-lobe crease and coronary atherosclerosis. Lancet 1979; 2:1377.
- Shmilovich H, Cheng VY, Rajani R, et al. Relation of diagonal ear lobe crease to the presence, extent, and severity of coronary artery disease determined by coronary computed tomography angiography. Am J Cardiol 2012; 109:1283–1287.
- Zapata-Wainberg G, Vivancos J. Images in clinical medicine: bilateral earlobe creases. N Engl J Med 2013; 368:e32.
A 70-year-old man with hypertension and hypercholesterolemia presented to the emergency department after the acute onset of substernal, pressure-like chest pain while climbing a flight of stairs. His physical examination was normal, but he was noted to have bilateral diagonal earlobe creases (the Frank sign) (Figure 1), considered by some to indicate risk of coronary artery disease.1–5
Electrocardiography showed atrial fibrillation with a ventricular rate of 149 beats per minute, ST-segment elevation in leads V1 and aVR, and ST-segment depression in leads V3 to V6, II, III, and aVF.
Urgent coronary arteriography showed severe coronary artery disease (Figure 2). Left ventriculography showed an ejection fraction of 50% with mild anterior wall hypokinesis. A drug-eluting stent was placed in the mid-left anterior descending artery. The patient tolerated the procedure well, and his chest pain resolved afterward.
A STILL-UNCLEAR ASSOCIATION
Sanders T. Frank, in 1973, first described a diagonal wrinkle-like line on the earlobe as a sign of coronary artery disease.1 Subsequently, autopsy studies suggested that deep bilateral earlobe creases could be an important sign of coronary atherosclerosis.2 Diagonal earlobe creases have been shown to be independently associated with increased prevalence, extent, and severity of coronary artery disease.3,4 They are also associated with major adverse cardiovascular events4 and ischemic stroke.5 The mechanism linking diagonal earlobe creases and atherosclerotic disease is not yet clear.
This patient’s presentation and evaluation remind us that bilateral earlobe creases may be useful to include in the clinical examination of patients with suspected coronary artery disease and may facilitate early recognition of disease in a patient at high risk.
A 70-year-old man with hypertension and hypercholesterolemia presented to the emergency department after the acute onset of substernal, pressure-like chest pain while climbing a flight of stairs. His physical examination was normal, but he was noted to have bilateral diagonal earlobe creases (the Frank sign) (Figure 1), considered by some to indicate risk of coronary artery disease.1–5
Electrocardiography showed atrial fibrillation with a ventricular rate of 149 beats per minute, ST-segment elevation in leads V1 and aVR, and ST-segment depression in leads V3 to V6, II, III, and aVF.
Urgent coronary arteriography showed severe coronary artery disease (Figure 2). Left ventriculography showed an ejection fraction of 50% with mild anterior wall hypokinesis. A drug-eluting stent was placed in the mid-left anterior descending artery. The patient tolerated the procedure well, and his chest pain resolved afterward.
A STILL-UNCLEAR ASSOCIATION
Sanders T. Frank, in 1973, first described a diagonal wrinkle-like line on the earlobe as a sign of coronary artery disease.1 Subsequently, autopsy studies suggested that deep bilateral earlobe creases could be an important sign of coronary atherosclerosis.2 Diagonal earlobe creases have been shown to be independently associated with increased prevalence, extent, and severity of coronary artery disease.3,4 They are also associated with major adverse cardiovascular events4 and ischemic stroke.5 The mechanism linking diagonal earlobe creases and atherosclerotic disease is not yet clear.
This patient’s presentation and evaluation remind us that bilateral earlobe creases may be useful to include in the clinical examination of patients with suspected coronary artery disease and may facilitate early recognition of disease in a patient at high risk.
- Frank ST. Aural sign of coronary-artery disease. N Engl J Med 1973; 289:327–328.
- Patel V, Champ C, Andrews PS, Gostelow BE, Gunasekara NP, Davidson AR. Diagonal earlobe creases and atheromatous disease: a postmortem study. J R Coll Phys Lond 1992; 26:274–277.
- Kaukola S, Manninen V, Valle M, Halonen PI. Ear-lobe crease and coronary atherosclerosis. Lancet 1979; 2:1377.
- Shmilovich H, Cheng VY, Rajani R, et al. Relation of diagonal ear lobe crease to the presence, extent, and severity of coronary artery disease determined by coronary computed tomography angiography. Am J Cardiol 2012; 109:1283–1287.
- Zapata-Wainberg G, Vivancos J. Images in clinical medicine: bilateral earlobe creases. N Engl J Med 2013; 368:e32.
- Frank ST. Aural sign of coronary-artery disease. N Engl J Med 1973; 289:327–328.
- Patel V, Champ C, Andrews PS, Gostelow BE, Gunasekara NP, Davidson AR. Diagonal earlobe creases and atheromatous disease: a postmortem study. J R Coll Phys Lond 1992; 26:274–277.
- Kaukola S, Manninen V, Valle M, Halonen PI. Ear-lobe crease and coronary atherosclerosis. Lancet 1979; 2:1377.
- Shmilovich H, Cheng VY, Rajani R, et al. Relation of diagonal ear lobe crease to the presence, extent, and severity of coronary artery disease determined by coronary computed tomography angiography. Am J Cardiol 2012; 109:1283–1287.
- Zapata-Wainberg G, Vivancos J. Images in clinical medicine: bilateral earlobe creases. N Engl J Med 2013; 368:e32.