User login
Opioid therapy and sleep apnea
To the Editor: I enjoyed Dr. Galicia-Castillo’s article about long-term opioid therapy in older adults,1 which reaffirmed the imperative to “start low and go slow” to minimize the risk of addiction. However, the article missed an opportunity to raise awareness regarding another extremely important side effect of chronic prescription opioid consumption, that of ingestion prior to sleep, with consequent cessation of breathing leading to death.
According to the Drug Enforcement Administration,2 most narcotic deaths are a result of respiratory depression. And the American Pain Society has stated, “No patient has succumbed to [opioid] respiratory depression while awake.”3
Dr. Galicia-Castillo noted that the prevalence of central sleep apnea in chronic opioid users is 24%, based on a review by Correa et al.4 As alarming as this number is, other investigators have estimated it to be even higher—as high as 50% to 90%.5
Walker et al,6 in a study of 60 patients, found that the higher the opioid dose the patients were on, the more episodes of obstructive sleep apnea and central sleep apnea per hour they had. Yet prescribing a low dose does not adequately protect the chronic opioid user. Farney et al7 reported that oxygen saturation dropped precipitously—from 98% to 70%—15 minutes after a patient took just 7.5 mg of hydrocodone in the middle of the night. Mogri et al8 reported that a patient had 91 apnea events within 1 hour of taking 15 mg of oxycodone at 2 am.
Opioids, benzodiazepines, barbiturates, and ethanol individually and additively suppress medullary reflex ventilatory drive during sleep, especially during non–rapid-eye-movement (non-REM) sleep.6 During waking hours, in contrast, there is redundant backup of cerebral cortical drive, ensuring that we keep breathing. Therefore, people are most vulnerable to dying of opioid ingestion during sleep.
Moreover, oxygen desaturation during episodes of sleep apnea may precipitate seizures (which may be lethal) or coronary vasospasm with consequent malignant arrhythmias and myocardial ischemia.
Continuous positive airway pressure protects against obstructive sleep apnea, but not against central sleep apnea.9
Patients need to be aware of the danger, and physicians need to consider the pharmacokinetic profiles of the opioid preparations they prescribe. If patients are taking an opioid that has a short half-life, such as immediate-release oxycodone, they should not take it within 5 hours of sleep. Longer-lasting preparations need a longer interval, and some, such as extended-release tramadol, may need to be taken only on awakening.
Safe sleep can be facilitated by medications that are sedating but do not compromise ventilation. Optimal agents also enhance restorative REM and stage III and IV deep-sleep duration, and some may have the additional benefit of reducing the risk of cancer.10,11 Such agents may include baclofen, cyproheptadine, gabapentin, mirtazepine, and melatonin. Nonpharmacologic measures include sleep hygiene, aerobic exercise, and cognitive behavioral therapy.
A retrospective study12 found that 301 (60.4%) of 498 patients who died while on opioid therapy and whose death was judged to be related to the opioid were also taking benzodiazepines. Patients who take opioids should avoid taking benzodiazepines, barbiturates, or alcohol before going to sleep, and physicians should be extremely cautious about prescribing benzodiazepines and barbiturates to patients who are on opioids.
- Galicia-Castillo M. Opioids for persistent pain in older adults. Cleve Clin J Med 2016; 83:443–451.
- Drug Enforcement Administration. Drugs of Abuse. 2005 Edition. Washington, DC: US Government Printing Office, 2005:19.
- American Pain Society, Principles of Analgesic Use in the Treatment of Acute Pain and Cancer Pain, 4th ed. Glenview, IL: American Pain Society, 1999:30.
- Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg 2015; 120:1273–1285.
- Panagiotou I, Mystakidou K. Non-analgesic effects of opioids: opioids’ effects on sleep (including sleep apnea). Curr Pharm Des 2012; 18:6025–6033.
- Walker JM, Farney RJ, Rhondeau SM, et al. Chronic opioid use is a risk factor for the development of central sleep apnea and ataxic breathing. J Clin Sleep Med 2007; 3:455–461. Erratum in J Clin Sleep Med 2007; 3:table of contents.
- Farney RJ, Walker JM, Cloward TV, Rhondeau S. Sleep-disordered breathing associated with long-term opioid therapy. Chest 2003; 123:632–639.
- Mogri M, Khan MI, Grant BJ, Mador MJ. Central sleep apnea induced by acute ingestion of opioids. Chest 2008; 133:1484–1488.
- Guilleminault C, Cao M, Yue HJ, Chawla P. Obstructive sleep apnea and chronic opioid use. Lung 2010; 188:459–468.
- Kao CH, Sun LM, Liang JA, Chang SN, Sung FC, Muo CH. Relationship of zolpidem and cancer risk: a Taiwanese population-based cohort study. Mayo Clin Proc 2012; 87:430–436.
- Kripke DF. Hypnotic drug risks of mortality, infection, depression, and cancer: but lack of benefit. F1000Res 2016; 5:918.
- Gomes T, Mamdani MM, Dhalla IA, Paterson JM, Juurlink DN. Opioid dose and drug-related mortality in patients with nonmalignant pain. Arch Intern Med 2011; 171:686–691.
To the Editor: I enjoyed Dr. Galicia-Castillo’s article about long-term opioid therapy in older adults,1 which reaffirmed the imperative to “start low and go slow” to minimize the risk of addiction. However, the article missed an opportunity to raise awareness regarding another extremely important side effect of chronic prescription opioid consumption, that of ingestion prior to sleep, with consequent cessation of breathing leading to death.
According to the Drug Enforcement Administration,2 most narcotic deaths are a result of respiratory depression. And the American Pain Society has stated, “No patient has succumbed to [opioid] respiratory depression while awake.”3
Dr. Galicia-Castillo noted that the prevalence of central sleep apnea in chronic opioid users is 24%, based on a review by Correa et al.4 As alarming as this number is, other investigators have estimated it to be even higher—as high as 50% to 90%.5
Walker et al,6 in a study of 60 patients, found that the higher the opioid dose the patients were on, the more episodes of obstructive sleep apnea and central sleep apnea per hour they had. Yet prescribing a low dose does not adequately protect the chronic opioid user. Farney et al7 reported that oxygen saturation dropped precipitously—from 98% to 70%—15 minutes after a patient took just 7.5 mg of hydrocodone in the middle of the night. Mogri et al8 reported that a patient had 91 apnea events within 1 hour of taking 15 mg of oxycodone at 2 am.
Opioids, benzodiazepines, barbiturates, and ethanol individually and additively suppress medullary reflex ventilatory drive during sleep, especially during non–rapid-eye-movement (non-REM) sleep.6 During waking hours, in contrast, there is redundant backup of cerebral cortical drive, ensuring that we keep breathing. Therefore, people are most vulnerable to dying of opioid ingestion during sleep.
Moreover, oxygen desaturation during episodes of sleep apnea may precipitate seizures (which may be lethal) or coronary vasospasm with consequent malignant arrhythmias and myocardial ischemia.
Continuous positive airway pressure protects against obstructive sleep apnea, but not against central sleep apnea.9
Patients need to be aware of the danger, and physicians need to consider the pharmacokinetic profiles of the opioid preparations they prescribe. If patients are taking an opioid that has a short half-life, such as immediate-release oxycodone, they should not take it within 5 hours of sleep. Longer-lasting preparations need a longer interval, and some, such as extended-release tramadol, may need to be taken only on awakening.
Safe sleep can be facilitated by medications that are sedating but do not compromise ventilation. Optimal agents also enhance restorative REM and stage III and IV deep-sleep duration, and some may have the additional benefit of reducing the risk of cancer.10,11 Such agents may include baclofen, cyproheptadine, gabapentin, mirtazepine, and melatonin. Nonpharmacologic measures include sleep hygiene, aerobic exercise, and cognitive behavioral therapy.
A retrospective study12 found that 301 (60.4%) of 498 patients who died while on opioid therapy and whose death was judged to be related to the opioid were also taking benzodiazepines. Patients who take opioids should avoid taking benzodiazepines, barbiturates, or alcohol before going to sleep, and physicians should be extremely cautious about prescribing benzodiazepines and barbiturates to patients who are on opioids.
To the Editor: I enjoyed Dr. Galicia-Castillo’s article about long-term opioid therapy in older adults,1 which reaffirmed the imperative to “start low and go slow” to minimize the risk of addiction. However, the article missed an opportunity to raise awareness regarding another extremely important side effect of chronic prescription opioid consumption, that of ingestion prior to sleep, with consequent cessation of breathing leading to death.
According to the Drug Enforcement Administration,2 most narcotic deaths are a result of respiratory depression. And the American Pain Society has stated, “No patient has succumbed to [opioid] respiratory depression while awake.”3
Dr. Galicia-Castillo noted that the prevalence of central sleep apnea in chronic opioid users is 24%, based on a review by Correa et al.4 As alarming as this number is, other investigators have estimated it to be even higher—as high as 50% to 90%.5
Walker et al,6 in a study of 60 patients, found that the higher the opioid dose the patients were on, the more episodes of obstructive sleep apnea and central sleep apnea per hour they had. Yet prescribing a low dose does not adequately protect the chronic opioid user. Farney et al7 reported that oxygen saturation dropped precipitously—from 98% to 70%—15 minutes after a patient took just 7.5 mg of hydrocodone in the middle of the night. Mogri et al8 reported that a patient had 91 apnea events within 1 hour of taking 15 mg of oxycodone at 2 am.
Opioids, benzodiazepines, barbiturates, and ethanol individually and additively suppress medullary reflex ventilatory drive during sleep, especially during non–rapid-eye-movement (non-REM) sleep.6 During waking hours, in contrast, there is redundant backup of cerebral cortical drive, ensuring that we keep breathing. Therefore, people are most vulnerable to dying of opioid ingestion during sleep.
Moreover, oxygen desaturation during episodes of sleep apnea may precipitate seizures (which may be lethal) or coronary vasospasm with consequent malignant arrhythmias and myocardial ischemia.
Continuous positive airway pressure protects against obstructive sleep apnea, but not against central sleep apnea.9
Patients need to be aware of the danger, and physicians need to consider the pharmacokinetic profiles of the opioid preparations they prescribe. If patients are taking an opioid that has a short half-life, such as immediate-release oxycodone, they should not take it within 5 hours of sleep. Longer-lasting preparations need a longer interval, and some, such as extended-release tramadol, may need to be taken only on awakening.
Safe sleep can be facilitated by medications that are sedating but do not compromise ventilation. Optimal agents also enhance restorative REM and stage III and IV deep-sleep duration, and some may have the additional benefit of reducing the risk of cancer.10,11 Such agents may include baclofen, cyproheptadine, gabapentin, mirtazepine, and melatonin. Nonpharmacologic measures include sleep hygiene, aerobic exercise, and cognitive behavioral therapy.
A retrospective study12 found that 301 (60.4%) of 498 patients who died while on opioid therapy and whose death was judged to be related to the opioid were also taking benzodiazepines. Patients who take opioids should avoid taking benzodiazepines, barbiturates, or alcohol before going to sleep, and physicians should be extremely cautious about prescribing benzodiazepines and barbiturates to patients who are on opioids.
- Galicia-Castillo M. Opioids for persistent pain in older adults. Cleve Clin J Med 2016; 83:443–451.
- Drug Enforcement Administration. Drugs of Abuse. 2005 Edition. Washington, DC: US Government Printing Office, 2005:19.
- American Pain Society, Principles of Analgesic Use in the Treatment of Acute Pain and Cancer Pain, 4th ed. Glenview, IL: American Pain Society, 1999:30.
- Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg 2015; 120:1273–1285.
- Panagiotou I, Mystakidou K. Non-analgesic effects of opioids: opioids’ effects on sleep (including sleep apnea). Curr Pharm Des 2012; 18:6025–6033.
- Walker JM, Farney RJ, Rhondeau SM, et al. Chronic opioid use is a risk factor for the development of central sleep apnea and ataxic breathing. J Clin Sleep Med 2007; 3:455–461. Erratum in J Clin Sleep Med 2007; 3:table of contents.
- Farney RJ, Walker JM, Cloward TV, Rhondeau S. Sleep-disordered breathing associated with long-term opioid therapy. Chest 2003; 123:632–639.
- Mogri M, Khan MI, Grant BJ, Mador MJ. Central sleep apnea induced by acute ingestion of opioids. Chest 2008; 133:1484–1488.
- Guilleminault C, Cao M, Yue HJ, Chawla P. Obstructive sleep apnea and chronic opioid use. Lung 2010; 188:459–468.
- Kao CH, Sun LM, Liang JA, Chang SN, Sung FC, Muo CH. Relationship of zolpidem and cancer risk: a Taiwanese population-based cohort study. Mayo Clin Proc 2012; 87:430–436.
- Kripke DF. Hypnotic drug risks of mortality, infection, depression, and cancer: but lack of benefit. F1000Res 2016; 5:918.
- Gomes T, Mamdani MM, Dhalla IA, Paterson JM, Juurlink DN. Opioid dose and drug-related mortality in patients with nonmalignant pain. Arch Intern Med 2011; 171:686–691.
- Galicia-Castillo M. Opioids for persistent pain in older adults. Cleve Clin J Med 2016; 83:443–451.
- Drug Enforcement Administration. Drugs of Abuse. 2005 Edition. Washington, DC: US Government Printing Office, 2005:19.
- American Pain Society, Principles of Analgesic Use in the Treatment of Acute Pain and Cancer Pain, 4th ed. Glenview, IL: American Pain Society, 1999:30.
- Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg 2015; 120:1273–1285.
- Panagiotou I, Mystakidou K. Non-analgesic effects of opioids: opioids’ effects on sleep (including sleep apnea). Curr Pharm Des 2012; 18:6025–6033.
- Walker JM, Farney RJ, Rhondeau SM, et al. Chronic opioid use is a risk factor for the development of central sleep apnea and ataxic breathing. J Clin Sleep Med 2007; 3:455–461. Erratum in J Clin Sleep Med 2007; 3:table of contents.
- Farney RJ, Walker JM, Cloward TV, Rhondeau S. Sleep-disordered breathing associated with long-term opioid therapy. Chest 2003; 123:632–639.
- Mogri M, Khan MI, Grant BJ, Mador MJ. Central sleep apnea induced by acute ingestion of opioids. Chest 2008; 133:1484–1488.
- Guilleminault C, Cao M, Yue HJ, Chawla P. Obstructive sleep apnea and chronic opioid use. Lung 2010; 188:459–468.
- Kao CH, Sun LM, Liang JA, Chang SN, Sung FC, Muo CH. Relationship of zolpidem and cancer risk: a Taiwanese population-based cohort study. Mayo Clin Proc 2012; 87:430–436.
- Kripke DF. Hypnotic drug risks of mortality, infection, depression, and cancer: but lack of benefit. F1000Res 2016; 5:918.
- Gomes T, Mamdani MM, Dhalla IA, Paterson JM, Juurlink DN. Opioid dose and drug-related mortality in patients with nonmalignant pain. Arch Intern Med 2011; 171:686–691.

In reply: Opioid therapy and sleep apnea
In Reply: Dr. Geller makes some excellent points about sleep and opioid use.
Opioids pose risks,1 just like any other type of medication. In particular, opioids have been linked to sleep-disordered breathing, which affects 70% to 85% of patients taking opioids.2–4
Other options can be used in some older adults, but they are not always successful. Ideally, nonpharmacologic strategies and nonopioid medications such as acetaminophen, nonsteroidal anti-inflammatory agents, antidepressants, and anticonvulsants should be used, although these medications have their own side effects. Optimum pain control may offer the potential for significant improvement in function, and opioids are but one tool in the clinician’s kit.
Ongoing discussions of the risks and benefits are necessary, along with continuous re-evaluation of the need for and effect of opioids.
- Davis MP, Mehta Z. Opioids and chronic pain: where is the balance? Curr Oncol Rep 2016; 18:71.
- Jungquist CR, Flannery M, Perlis ML, Grace JT. Relationship of chronic pain and opioid use with respiratory disturbance during sleep. Pain Manag Nurs 2012; 13:70–79.
- Webster LR, Choi Y, Desai H, Webster L, Grant BJ. Sleep-disordered breathing and chronic opioid therapy. Pain Med 2008; 9:425–432.
- Mogri M, Khan MI, Grant BJ, Mador MJ. Central sleep apnea induced by acute ingestion of opioid. Chest 2008; 133:1484–1488.
In Reply: Dr. Geller makes some excellent points about sleep and opioid use.
Opioids pose risks,1 just like any other type of medication. In particular, opioids have been linked to sleep-disordered breathing, which affects 70% to 85% of patients taking opioids.2–4
Other options can be used in some older adults, but they are not always successful. Ideally, nonpharmacologic strategies and nonopioid medications such as acetaminophen, nonsteroidal anti-inflammatory agents, antidepressants, and anticonvulsants should be used, although these medications have their own side effects. Optimum pain control may offer the potential for significant improvement in function, and opioids are but one tool in the clinician’s kit.
Ongoing discussions of the risks and benefits are necessary, along with continuous re-evaluation of the need for and effect of opioids.
In Reply: Dr. Geller makes some excellent points about sleep and opioid use.
Opioids pose risks,1 just like any other type of medication. In particular, opioids have been linked to sleep-disordered breathing, which affects 70% to 85% of patients taking opioids.2–4
Other options can be used in some older adults, but they are not always successful. Ideally, nonpharmacologic strategies and nonopioid medications such as acetaminophen, nonsteroidal anti-inflammatory agents, antidepressants, and anticonvulsants should be used, although these medications have their own side effects. Optimum pain control may offer the potential for significant improvement in function, and opioids are but one tool in the clinician’s kit.
Ongoing discussions of the risks and benefits are necessary, along with continuous re-evaluation of the need for and effect of opioids.
- Davis MP, Mehta Z. Opioids and chronic pain: where is the balance? Curr Oncol Rep 2016; 18:71.
- Jungquist CR, Flannery M, Perlis ML, Grace JT. Relationship of chronic pain and opioid use with respiratory disturbance during sleep. Pain Manag Nurs 2012; 13:70–79.
- Webster LR, Choi Y, Desai H, Webster L, Grant BJ. Sleep-disordered breathing and chronic opioid therapy. Pain Med 2008; 9:425–432.
- Mogri M, Khan MI, Grant BJ, Mador MJ. Central sleep apnea induced by acute ingestion of opioid. Chest 2008; 133:1484–1488.
- Davis MP, Mehta Z. Opioids and chronic pain: where is the balance? Curr Oncol Rep 2016; 18:71.
- Jungquist CR, Flannery M, Perlis ML, Grace JT. Relationship of chronic pain and opioid use with respiratory disturbance during sleep. Pain Manag Nurs 2012; 13:70–79.
- Webster LR, Choi Y, Desai H, Webster L, Grant BJ. Sleep-disordered breathing and chronic opioid therapy. Pain Med 2008; 9:425–432.
- Mogri M, Khan MI, Grant BJ, Mador MJ. Central sleep apnea induced by acute ingestion of opioid. Chest 2008; 133:1484–1488.

Submassive pulmonary embolism
To the Editor: I read with interest the review on submassive pulmonary embolism by Ataya et al1 in the December 2016 issue. I had 3 questions or observations for the authors
First, systemic thrombolytic therapy for massive or hemodynamically unstable pulmonary embolism is given a grade 2C recommendation, similar to the level for select patients with submassive pulmonary embolism with low bleeding risk but at high risk of developing hypotension. The reference for this is the 2012 American College of Chest Physicians guidelines.2 I would like to point out that these guidelines were updated and published in February 2016,3 and systemic thrombolytic therapy for massive pulmonary embolism now carries a grade 2B recommendation. Thrombolytic therapy still has a grade 2C recommendation for select patients with submassive pulmonary embolism.
Second, the Moderate Pulmonary Embolism Treated With Thrombolysis (MOPETT) trial is described as a randomized trial in patients with moderate pulmonary hypertension and right ventricular dysfunction. I would like to point out that right ventricular dysfunction was not a criterion for enrollment in the trial.4
Finally, catheter-directed thrombolytic therapy is mentioned as an option for select patients with submassive and massive pulmonary embolism. The advantage is believed to be due to local action of the drug with fewer systemic effects. Since the protocol involves alteplase for 12 or 24 hours with a maximum dose of 24 mg, and since in most cases pulmonary embolism originates in the lower extremity, are we not exposing these patients to further clot propagation for 12 or 24 hours without the benefit of concomitant systemic anticoagulation or an inferior vena cava filter?
- Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. Do patients with submassive pulmonary embolism benefit from thrombolytic therapy? Cleve Clin J Med 2016; 83:923–932.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149:315–352.
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M; “MOPETT” Investigators. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 2013; 111:273–277.
To the Editor: I read with interest the review on submassive pulmonary embolism by Ataya et al1 in the December 2016 issue. I had 3 questions or observations for the authors
First, systemic thrombolytic therapy for massive or hemodynamically unstable pulmonary embolism is given a grade 2C recommendation, similar to the level for select patients with submassive pulmonary embolism with low bleeding risk but at high risk of developing hypotension. The reference for this is the 2012 American College of Chest Physicians guidelines.2 I would like to point out that these guidelines were updated and published in February 2016,3 and systemic thrombolytic therapy for massive pulmonary embolism now carries a grade 2B recommendation. Thrombolytic therapy still has a grade 2C recommendation for select patients with submassive pulmonary embolism.
Second, the Moderate Pulmonary Embolism Treated With Thrombolysis (MOPETT) trial is described as a randomized trial in patients with moderate pulmonary hypertension and right ventricular dysfunction. I would like to point out that right ventricular dysfunction was not a criterion for enrollment in the trial.4
Finally, catheter-directed thrombolytic therapy is mentioned as an option for select patients with submassive and massive pulmonary embolism. The advantage is believed to be due to local action of the drug with fewer systemic effects. Since the protocol involves alteplase for 12 or 24 hours with a maximum dose of 24 mg, and since in most cases pulmonary embolism originates in the lower extremity, are we not exposing these patients to further clot propagation for 12 or 24 hours without the benefit of concomitant systemic anticoagulation or an inferior vena cava filter?
To the Editor: I read with interest the review on submassive pulmonary embolism by Ataya et al1 in the December 2016 issue. I had 3 questions or observations for the authors
First, systemic thrombolytic therapy for massive or hemodynamically unstable pulmonary embolism is given a grade 2C recommendation, similar to the level for select patients with submassive pulmonary embolism with low bleeding risk but at high risk of developing hypotension. The reference for this is the 2012 American College of Chest Physicians guidelines.2 I would like to point out that these guidelines were updated and published in February 2016,3 and systemic thrombolytic therapy for massive pulmonary embolism now carries a grade 2B recommendation. Thrombolytic therapy still has a grade 2C recommendation for select patients with submassive pulmonary embolism.
Second, the Moderate Pulmonary Embolism Treated With Thrombolysis (MOPETT) trial is described as a randomized trial in patients with moderate pulmonary hypertension and right ventricular dysfunction. I would like to point out that right ventricular dysfunction was not a criterion for enrollment in the trial.4
Finally, catheter-directed thrombolytic therapy is mentioned as an option for select patients with submassive and massive pulmonary embolism. The advantage is believed to be due to local action of the drug with fewer systemic effects. Since the protocol involves alteplase for 12 or 24 hours with a maximum dose of 24 mg, and since in most cases pulmonary embolism originates in the lower extremity, are we not exposing these patients to further clot propagation for 12 or 24 hours without the benefit of concomitant systemic anticoagulation or an inferior vena cava filter?
- Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. Do patients with submassive pulmonary embolism benefit from thrombolytic therapy? Cleve Clin J Med 2016; 83:923–932.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149:315–352.
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M; “MOPETT” Investigators. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 2013; 111:273–277.
- Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. Do patients with submassive pulmonary embolism benefit from thrombolytic therapy? Cleve Clin J Med 2016; 83:923–932.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149:315–352.
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M; “MOPETT” Investigators. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 2013; 111:273–277.
In reply: Submassive pulmonary embolism
In Reply: We thank Dr. Katyal for his thoughtful comments.
Dr. Katyal points out that the grade of recommendation for thrombolysis in patients with massive pulmonary embolism was upgraded from 2C to 2B in the 2016 American College of Chest Physicians (ACCP) guidelines1 compared with the 2012 guidelines2 that we cited. The upgrade in this recommendation was owing to 2 small trials and 1 large randomized controlled trial that included patients with submassive pulmonary embolism.3–5 Interestingly, these 3 studies led to an upgrade in the level of recommendation for thrombolysis in the treatment of massive pulmonary embolism, perhaps more from a safety aspect (in view of the incidence of major bleeding vs mortality). Regardless, Dr. Katyal is correct in highlighting that the new 2016 ACCP guidelines now give a grade of 2B for thrombolytic therapy in the treatment of massive pulmonary embolism. These guidelines had not been published at the time of submission of our manuscript.
Dr. Katyal is also correct that patients were not required to have right ventricular dysfunction to be enrolled in the MOPETT trial.3 As we pointed out, “Only 20% of the participants were enrolled on the basis of right ventricular dysfunction on echocardiography, whereas almost 60% had elevated cardiac biomarkers.”6
Regarding catheter-directed therapy, patients who received low-dose catheter-directed alteplase were also concurrently anticoagulated with systemic unfractionated heparin in the Ultrasound-Assisted, Catheter-Directed Thrombolysis for Acute Intermediate-Risk Pulmonary Embolism (ULTIMA) trial.7 The ULTIMA trial authors commented that unfractionated heparin was started with an 80-U/kg bolus followed by an 18-U/kg/hour infusion to target an anti-factor Xa level of 0.3 to 0.7 μg/mL, which is considered therapeutic anticoagulation. The investigators in the SEATTLE II trial8 continued systemic unfractionated heparin but targeted a lower “intermediate” anticoagulation target (an augmented partial thromboplastin time of 40–60 seconds), so these patients weren’t completely without systemic anticoagulation either. At our institution, the current practice is to target an anti-Xa level of 0.3 to 0.7 μg/mL in patients receiving catheter-directed therapy for large-volume pulmonary embolism.
- Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149:315–352.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M; “MOPETT” Investigators. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 2013; 111:273–277.
- Meyer G, Vicaut E, Danays T, et al; PEITHO Investigators. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
- Kline JA, Nordenholz KE, Courtney DM, et al. Treatment of submassive pulmonary embolism with tenecteplase or placebo: cardiopulmonary outcomes at 3 months: multicenter double-blind, placebo-controlled randomized trial. J Thromb Haemost 2014; 12:459–468.
- Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. Do patients with submassive pulmonary embolism benefit from thrombolytic therapy? Cleve Clin J Med 2016; 83:923–932.
- Kucher N, Boekstegers P, Muller OJ, et al. Randomized, controlled trial of ultrasound-assisted catheter-directed thrombolysis for acute intermediate-risk pulmonary embolism. Circulation 2014; 129:479–486.
- Piazza G, Hohlfelder B, Jaff MR, et al; SEATTLE II Investigators. A prospective, single-arm, multicenter trial of ultrasound-facilitated, catheter-directed, low-dose fibrinolysis for acute massive and submassive pulmonary embolism (The SEATTLE II Study). JACC Cardiovasc Interv 2015; 8:1382–1392.
In Reply: We thank Dr. Katyal for his thoughtful comments.
Dr. Katyal points out that the grade of recommendation for thrombolysis in patients with massive pulmonary embolism was upgraded from 2C to 2B in the 2016 American College of Chest Physicians (ACCP) guidelines1 compared with the 2012 guidelines2 that we cited. The upgrade in this recommendation was owing to 2 small trials and 1 large randomized controlled trial that included patients with submassive pulmonary embolism.3–5 Interestingly, these 3 studies led to an upgrade in the level of recommendation for thrombolysis in the treatment of massive pulmonary embolism, perhaps more from a safety aspect (in view of the incidence of major bleeding vs mortality). Regardless, Dr. Katyal is correct in highlighting that the new 2016 ACCP guidelines now give a grade of 2B for thrombolytic therapy in the treatment of massive pulmonary embolism. These guidelines had not been published at the time of submission of our manuscript.
Dr. Katyal is also correct that patients were not required to have right ventricular dysfunction to be enrolled in the MOPETT trial.3 As we pointed out, “Only 20% of the participants were enrolled on the basis of right ventricular dysfunction on echocardiography, whereas almost 60% had elevated cardiac biomarkers.”6
Regarding catheter-directed therapy, patients who received low-dose catheter-directed alteplase were also concurrently anticoagulated with systemic unfractionated heparin in the Ultrasound-Assisted, Catheter-Directed Thrombolysis for Acute Intermediate-Risk Pulmonary Embolism (ULTIMA) trial.7 The ULTIMA trial authors commented that unfractionated heparin was started with an 80-U/kg bolus followed by an 18-U/kg/hour infusion to target an anti-factor Xa level of 0.3 to 0.7 μg/mL, which is considered therapeutic anticoagulation. The investigators in the SEATTLE II trial8 continued systemic unfractionated heparin but targeted a lower “intermediate” anticoagulation target (an augmented partial thromboplastin time of 40–60 seconds), so these patients weren’t completely without systemic anticoagulation either. At our institution, the current practice is to target an anti-Xa level of 0.3 to 0.7 μg/mL in patients receiving catheter-directed therapy for large-volume pulmonary embolism.
In Reply: We thank Dr. Katyal for his thoughtful comments.
Dr. Katyal points out that the grade of recommendation for thrombolysis in patients with massive pulmonary embolism was upgraded from 2C to 2B in the 2016 American College of Chest Physicians (ACCP) guidelines1 compared with the 2012 guidelines2 that we cited. The upgrade in this recommendation was owing to 2 small trials and 1 large randomized controlled trial that included patients with submassive pulmonary embolism.3–5 Interestingly, these 3 studies led to an upgrade in the level of recommendation for thrombolysis in the treatment of massive pulmonary embolism, perhaps more from a safety aspect (in view of the incidence of major bleeding vs mortality). Regardless, Dr. Katyal is correct in highlighting that the new 2016 ACCP guidelines now give a grade of 2B for thrombolytic therapy in the treatment of massive pulmonary embolism. These guidelines had not been published at the time of submission of our manuscript.
Dr. Katyal is also correct that patients were not required to have right ventricular dysfunction to be enrolled in the MOPETT trial.3 As we pointed out, “Only 20% of the participants were enrolled on the basis of right ventricular dysfunction on echocardiography, whereas almost 60% had elevated cardiac biomarkers.”6
Regarding catheter-directed therapy, patients who received low-dose catheter-directed alteplase were also concurrently anticoagulated with systemic unfractionated heparin in the Ultrasound-Assisted, Catheter-Directed Thrombolysis for Acute Intermediate-Risk Pulmonary Embolism (ULTIMA) trial.7 The ULTIMA trial authors commented that unfractionated heparin was started with an 80-U/kg bolus followed by an 18-U/kg/hour infusion to target an anti-factor Xa level of 0.3 to 0.7 μg/mL, which is considered therapeutic anticoagulation. The investigators in the SEATTLE II trial8 continued systemic unfractionated heparin but targeted a lower “intermediate” anticoagulation target (an augmented partial thromboplastin time of 40–60 seconds), so these patients weren’t completely without systemic anticoagulation either. At our institution, the current practice is to target an anti-Xa level of 0.3 to 0.7 μg/mL in patients receiving catheter-directed therapy for large-volume pulmonary embolism.
- Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149:315–352.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M; “MOPETT” Investigators. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 2013; 111:273–277.
- Meyer G, Vicaut E, Danays T, et al; PEITHO Investigators. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
- Kline JA, Nordenholz KE, Courtney DM, et al. Treatment of submassive pulmonary embolism with tenecteplase or placebo: cardiopulmonary outcomes at 3 months: multicenter double-blind, placebo-controlled randomized trial. J Thromb Haemost 2014; 12:459–468.
- Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. Do patients with submassive pulmonary embolism benefit from thrombolytic therapy? Cleve Clin J Med 2016; 83:923–932.
- Kucher N, Boekstegers P, Muller OJ, et al. Randomized, controlled trial of ultrasound-assisted catheter-directed thrombolysis for acute intermediate-risk pulmonary embolism. Circulation 2014; 129:479–486.
- Piazza G, Hohlfelder B, Jaff MR, et al; SEATTLE II Investigators. A prospective, single-arm, multicenter trial of ultrasound-facilitated, catheter-directed, low-dose fibrinolysis for acute massive and submassive pulmonary embolism (The SEATTLE II Study). JACC Cardiovasc Interv 2015; 8:1382–1392.
- Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149:315–352.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e419S–e494S.
- Sharifi M, Bay C, Skrocki L, Rahimi F, Mehdipour M; “MOPETT” Investigators. Moderate pulmonary embolism treated with thrombolysis (from the “MOPETT” Trial). Am J Cardiol 2013; 111:273–277.
- Meyer G, Vicaut E, Danays T, et al; PEITHO Investigators. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 2014; 370:1402–1411.
- Kline JA, Nordenholz KE, Courtney DM, et al. Treatment of submassive pulmonary embolism with tenecteplase or placebo: cardiopulmonary outcomes at 3 months: multicenter double-blind, placebo-controlled randomized trial. J Thromb Haemost 2014; 12:459–468.
- Ataya A, Cope J, Shahmohammadi A, Alnuaimat H. Do patients with submassive pulmonary embolism benefit from thrombolytic therapy? Cleve Clin J Med 2016; 83:923–932.
- Kucher N, Boekstegers P, Muller OJ, et al. Randomized, controlled trial of ultrasound-assisted catheter-directed thrombolysis for acute intermediate-risk pulmonary embolism. Circulation 2014; 129:479–486.
- Piazza G, Hohlfelder B, Jaff MR, et al; SEATTLE II Investigators. A prospective, single-arm, multicenter trial of ultrasound-facilitated, catheter-directed, low-dose fibrinolysis for acute massive and submassive pulmonary embolism (The SEATTLE II Study). JACC Cardiovasc Interv 2015; 8:1382–1392.
Case Studies in Toxicology: The Perils of Playing Catch-up
Case
A 16-year-old girl, who recently emigrated from Haiti, was brought to the pediatric ED by her mother for evaluation of a 2-hour history of gastric discomfort. Upon arrival at the ED waiting area, the patient experienced a sudden onset of generalized tonic-clonic movement with altered sensorium, though she did not fall to the ground and was not injured. Vital signs from triage were: blood pressure, 110/76 mm Hg; heart rate, 112 beats/min; respiratory rate, 22 breaths/min; and temperature, 97°F. Oxygen saturation was 98% on room air.
The patient was immediately attached to a cardiac monitor, given oxygen via a face mask, and received airway suctioning. Despite receiving a total of 4 mg of lorazepam, the seizure continued. Physical examination revealed no signs of external injury, but the ongoing generalized status epilepticus made the examination difficult.
What are the causes of refractory seizures in an adolescent patient?
The differential diagnosis for pediatric patients presenting with refractory seizure is the same as that for adult patients and should include treatment noncompliance, infection, vascular event (eg, stroke, hemorrhage), trauma (eg, cerebral contusions), metabolic and electrolyte disturbances, anticonvulsant toxicity, and exposure to a convulsant toxin.
While certain drugs (eg, cocaine) may cause status epilepticus through a secondary effect such as ischemia or a bleed, some drugs can directly cause refractory seizures. A few drugs and toxins are responsible for the majority of such seizures: bupropion; carbon monoxide; diphenhydramine; ethanol (withdrawal); hypoglycemics; lead; theophylline; tramadol; and certain antibiotics, including cephalosporins, penicillins, quinolones, and, in particular, isoniazid (INH).1
Case Continuation
Upon further history-taking, the patient’s mother informed the ED staff that during a recent visit to a local clinic, her daughter tested positive on routine screening for tuberculosis and was given “some medications.” The patient’s mother further noted that her daughter was scheduled for a follow-up appointment at the same clinic later this morning. She believed the patient had taken “a few” of the prescribed pills at once to “catch-up” on missed doses prior to that appointment, and provided the ED staff with an empty bottle of INH that she had found in her daughter’s purse.
What are the signs and symptoms of acute isoniazid toxicity?
Isoniazid toxicity should be suspected in any patient who has access to INH—even if the drug was prescribed for someone other than the patient. Acute toxicity develops rapidly after the ingestion of supratherapeutic doses of INH and includes nausea, abdominal discomfort, vomiting, dizziness, and excessive fatigue or lethargy. Patients can present with tachycardia, stupor, agitation, mydriasis, increased anion gap metabolic acidosis, and encephalopathy.
Seizures occur due to an INH-induced functional pyridoxine deficiency. Isoniazid inhibits pyridoxine phosphokinase, the enzyme that converts pyridoxine (vitamin B6) to its physiologically active form, pyridoxal 5’-phosphate (PLP). Because the conversion of glutamate (an excitatory neurotransmitter) to gamma-aminobutyric acid (GABA; the body’s main inhibitory neurotransmitter) is dependent on PLP, an excess of glutamate and a deficiency of GABA occurs following INH overdose. The result is neuroexcitation, which manifests as generalized seizures in affected patients.
The most consequential effect of INH overdose, however, is the development of seizure refractory to conventional therapy, such as benzodiazepines. This occurs because benzodiazepines are indirect-acting GABA agonists, and require the presence of GABA to elicit their effect. Therefore, due to the impairment of GABA synthesis, benzodiazepines are limited or ineffective as anticonvulsants. Although INH doses in excess of 20 mg/kg may result in neuroexcitation, refractory seizures are uncommon with doses <70 mg/kg.
Complications of chronic INH use include hepatotoxicity, and patients will present with jaundice, hepatomegaly, and right upper quadrant pain and tenderness. Isoniazid must be discontinued rapidly in
How is acute isoniazid-induced seizure managed?
Management of patients with refractory seizure should initially include an assessment and management of the patient’s airway, breathing, and circulation. Although seizures induced by INH toxicity are often resistant to benzodiazepines, these agents remain the first-line therapy. For patients who fail to respond to a reasonable trial of benzodiazepines (eg, lorazepam 6 mg intravenously [IV]), pyridoxine should be administered.3 The recommended dose is 1 g pyridoxine per every 1 g of INH ingested—if the initial dose ingested is known—with a maximum dose of 5 g pyridoxine. If the initial dose of INH is not known, 70 mg/kg of pyridoxine, up to 5 g, is recommended. Repeated doses of pyridoxine can be administered if the seizure continues, up to a total dose of 10 g in an adult. At extremely high doses, pyridoxine itself can be neurotoxic, limiting the maximal antidotal dose.
Rapid initiation of pyridoxine is a challenge since typical stocks in most EDs are not in an adequate supply required for treatment. Additionally, a typical vial of pyridoxine contains 100 mg, highlighting the rare need to open dozens of vials for a single patient. Drawing up adequate doses of the IV formulation can be a challenge and time-consuming.
Regardless, the most reliable and rapid route of administration for pyridoxine is IV, at a rate of 0.5 to 1 g/min. Even if the seizure resolves prior to completion of the initial dose, the remaining doses should still be administered over a 4- to 6-hour period. Oral or (more likely) nasogastric administration of pyridoxine can be administered if the IV formulation is not available, but neither are optimal routes of delivery. Every effort should be made to stock pyridoxine in the antidote supply in the ED to avoid time delays involving finding, preparing, and administering the drug in these scenarios. Previous studies have found that most EDs are not prepared to handle pyridoxine replacement.4,5
Since benzodiazepines and barbiturates are GABA agonists with complementary mechanisms of actions to pyridoxine, they should be administered to potentiate the antiseizure effect of pyridoxine. If the seizure does not terminate, the use of propofol or general anesthesia may be required. Once the seizure is terminated, oral activated charcoal can be administered if the ingestion occurred within several hours of presentation. Given the rapid onset of effect of a large dose of INH, most patients will develop seizure shortly after exposure, limiting the benefits of both aggressive gastrointestinal decontamination and delayed activated charcoal. Charcoal also can be used for patients who overdose on INH but do not develop seizures.
Although the utility of a head computed tomography (CT) scan or laboratory studies is limited given the context of the exposure, these are generally obtained for patients with new-onset seizure. Since many patients with INH toxicity do not seize, such a patient may have a lower seizure threshold due to the existence of a subclinical cerebral lesion or metabolic abnormality.
Case Conclusion
The patient’s INH-induced refractory seizure was treated with pyridoxine. Her history suggested that she had ingested an unknown number of INH tablets within an hour. On this initial basis, an IV dose of 5,000 mg of pyridoxine was administered. The patient’s seizures terminated within 2 minutes of the infusion, and no additional doses of pyridoxine were required. Given the lack of concern for self-harm, an acetaminophen concentration was not obtained. A urine toxicology screen was negative for cocaine and amphetamines, and a CT scan of the head was negative for any abnormality. The patient was admitted to the pediatric intensive care unit for status epileptics and was discharged home on hospital day 2 after an uneventful stay.
1. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82. doi:10.1016/j.yebeh.2015.04.034.
2. Latent tuberculosis infection: a guide for primary health care providers. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/tb/publications/LTBI/treatment.htm. Updated August 5, 2016. Accessed December 13, 2016.
3. Howland MA. Antidotes in depth: pyridoxine. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:797-799.
4. Shah BR, Santucci K, Sinert R, Steiner P. Acute isoniazid neurotoxicity in an urban hospital. Pediatrics. 1995;95(5):700-704.
5. Santucci KA, Shah BR, Linakis JG. Acute isoniazid exposures and antidote availability. Pediatr Emerg Care. 1999;15(2):99-101.
Case
A 16-year-old girl, who recently emigrated from Haiti, was brought to the pediatric ED by her mother for evaluation of a 2-hour history of gastric discomfort. Upon arrival at the ED waiting area, the patient experienced a sudden onset of generalized tonic-clonic movement with altered sensorium, though she did not fall to the ground and was not injured. Vital signs from triage were: blood pressure, 110/76 mm Hg; heart rate, 112 beats/min; respiratory rate, 22 breaths/min; and temperature, 97°F. Oxygen saturation was 98% on room air.
The patient was immediately attached to a cardiac monitor, given oxygen via a face mask, and received airway suctioning. Despite receiving a total of 4 mg of lorazepam, the seizure continued. Physical examination revealed no signs of external injury, but the ongoing generalized status epilepticus made the examination difficult.
What are the causes of refractory seizures in an adolescent patient?
The differential diagnosis for pediatric patients presenting with refractory seizure is the same as that for adult patients and should include treatment noncompliance, infection, vascular event (eg, stroke, hemorrhage), trauma (eg, cerebral contusions), metabolic and electrolyte disturbances, anticonvulsant toxicity, and exposure to a convulsant toxin.
While certain drugs (eg, cocaine) may cause status epilepticus through a secondary effect such as ischemia or a bleed, some drugs can directly cause refractory seizures. A few drugs and toxins are responsible for the majority of such seizures: bupropion; carbon monoxide; diphenhydramine; ethanol (withdrawal); hypoglycemics; lead; theophylline; tramadol; and certain antibiotics, including cephalosporins, penicillins, quinolones, and, in particular, isoniazid (INH).1
Case Continuation
Upon further history-taking, the patient’s mother informed the ED staff that during a recent visit to a local clinic, her daughter tested positive on routine screening for tuberculosis and was given “some medications.” The patient’s mother further noted that her daughter was scheduled for a follow-up appointment at the same clinic later this morning. She believed the patient had taken “a few” of the prescribed pills at once to “catch-up” on missed doses prior to that appointment, and provided the ED staff with an empty bottle of INH that she had found in her daughter’s purse.
What are the signs and symptoms of acute isoniazid toxicity?
Isoniazid toxicity should be suspected in any patient who has access to INH—even if the drug was prescribed for someone other than the patient. Acute toxicity develops rapidly after the ingestion of supratherapeutic doses of INH and includes nausea, abdominal discomfort, vomiting, dizziness, and excessive fatigue or lethargy. Patients can present with tachycardia, stupor, agitation, mydriasis, increased anion gap metabolic acidosis, and encephalopathy.
Seizures occur due to an INH-induced functional pyridoxine deficiency. Isoniazid inhibits pyridoxine phosphokinase, the enzyme that converts pyridoxine (vitamin B6) to its physiologically active form, pyridoxal 5’-phosphate (PLP). Because the conversion of glutamate (an excitatory neurotransmitter) to gamma-aminobutyric acid (GABA; the body’s main inhibitory neurotransmitter) is dependent on PLP, an excess of glutamate and a deficiency of GABA occurs following INH overdose. The result is neuroexcitation, which manifests as generalized seizures in affected patients.
The most consequential effect of INH overdose, however, is the development of seizure refractory to conventional therapy, such as benzodiazepines. This occurs because benzodiazepines are indirect-acting GABA agonists, and require the presence of GABA to elicit their effect. Therefore, due to the impairment of GABA synthesis, benzodiazepines are limited or ineffective as anticonvulsants. Although INH doses in excess of 20 mg/kg may result in neuroexcitation, refractory seizures are uncommon with doses <70 mg/kg.
Complications of chronic INH use include hepatotoxicity, and patients will present with jaundice, hepatomegaly, and right upper quadrant pain and tenderness. Isoniazid must be discontinued rapidly in
How is acute isoniazid-induced seizure managed?
Management of patients with refractory seizure should initially include an assessment and management of the patient’s airway, breathing, and circulation. Although seizures induced by INH toxicity are often resistant to benzodiazepines, these agents remain the first-line therapy. For patients who fail to respond to a reasonable trial of benzodiazepines (eg, lorazepam 6 mg intravenously [IV]), pyridoxine should be administered.3 The recommended dose is 1 g pyridoxine per every 1 g of INH ingested—if the initial dose ingested is known—with a maximum dose of 5 g pyridoxine. If the initial dose of INH is not known, 70 mg/kg of pyridoxine, up to 5 g, is recommended. Repeated doses of pyridoxine can be administered if the seizure continues, up to a total dose of 10 g in an adult. At extremely high doses, pyridoxine itself can be neurotoxic, limiting the maximal antidotal dose.
Rapid initiation of pyridoxine is a challenge since typical stocks in most EDs are not in an adequate supply required for treatment. Additionally, a typical vial of pyridoxine contains 100 mg, highlighting the rare need to open dozens of vials for a single patient. Drawing up adequate doses of the IV formulation can be a challenge and time-consuming.
Regardless, the most reliable and rapid route of administration for pyridoxine is IV, at a rate of 0.5 to 1 g/min. Even if the seizure resolves prior to completion of the initial dose, the remaining doses should still be administered over a 4- to 6-hour period. Oral or (more likely) nasogastric administration of pyridoxine can be administered if the IV formulation is not available, but neither are optimal routes of delivery. Every effort should be made to stock pyridoxine in the antidote supply in the ED to avoid time delays involving finding, preparing, and administering the drug in these scenarios. Previous studies have found that most EDs are not prepared to handle pyridoxine replacement.4,5
Since benzodiazepines and barbiturates are GABA agonists with complementary mechanisms of actions to pyridoxine, they should be administered to potentiate the antiseizure effect of pyridoxine. If the seizure does not terminate, the use of propofol or general anesthesia may be required. Once the seizure is terminated, oral activated charcoal can be administered if the ingestion occurred within several hours of presentation. Given the rapid onset of effect of a large dose of INH, most patients will develop seizure shortly after exposure, limiting the benefits of both aggressive gastrointestinal decontamination and delayed activated charcoal. Charcoal also can be used for patients who overdose on INH but do not develop seizures.
Although the utility of a head computed tomography (CT) scan or laboratory studies is limited given the context of the exposure, these are generally obtained for patients with new-onset seizure. Since many patients with INH toxicity do not seize, such a patient may have a lower seizure threshold due to the existence of a subclinical cerebral lesion or metabolic abnormality.
Case Conclusion
The patient’s INH-induced refractory seizure was treated with pyridoxine. Her history suggested that she had ingested an unknown number of INH tablets within an hour. On this initial basis, an IV dose of 5,000 mg of pyridoxine was administered. The patient’s seizures terminated within 2 minutes of the infusion, and no additional doses of pyridoxine were required. Given the lack of concern for self-harm, an acetaminophen concentration was not obtained. A urine toxicology screen was negative for cocaine and amphetamines, and a CT scan of the head was negative for any abnormality. The patient was admitted to the pediatric intensive care unit for status epileptics and was discharged home on hospital day 2 after an uneventful stay.
Case
A 16-year-old girl, who recently emigrated from Haiti, was brought to the pediatric ED by her mother for evaluation of a 2-hour history of gastric discomfort. Upon arrival at the ED waiting area, the patient experienced a sudden onset of generalized tonic-clonic movement with altered sensorium, though she did not fall to the ground and was not injured. Vital signs from triage were: blood pressure, 110/76 mm Hg; heart rate, 112 beats/min; respiratory rate, 22 breaths/min; and temperature, 97°F. Oxygen saturation was 98% on room air.
The patient was immediately attached to a cardiac monitor, given oxygen via a face mask, and received airway suctioning. Despite receiving a total of 4 mg of lorazepam, the seizure continued. Physical examination revealed no signs of external injury, but the ongoing generalized status epilepticus made the examination difficult.
What are the causes of refractory seizures in an adolescent patient?
The differential diagnosis for pediatric patients presenting with refractory seizure is the same as that for adult patients and should include treatment noncompliance, infection, vascular event (eg, stroke, hemorrhage), trauma (eg, cerebral contusions), metabolic and electrolyte disturbances, anticonvulsant toxicity, and exposure to a convulsant toxin.
While certain drugs (eg, cocaine) may cause status epilepticus through a secondary effect such as ischemia or a bleed, some drugs can directly cause refractory seizures. A few drugs and toxins are responsible for the majority of such seizures: bupropion; carbon monoxide; diphenhydramine; ethanol (withdrawal); hypoglycemics; lead; theophylline; tramadol; and certain antibiotics, including cephalosporins, penicillins, quinolones, and, in particular, isoniazid (INH).1
Case Continuation
Upon further history-taking, the patient’s mother informed the ED staff that during a recent visit to a local clinic, her daughter tested positive on routine screening for tuberculosis and was given “some medications.” The patient’s mother further noted that her daughter was scheduled for a follow-up appointment at the same clinic later this morning. She believed the patient had taken “a few” of the prescribed pills at once to “catch-up” on missed doses prior to that appointment, and provided the ED staff with an empty bottle of INH that she had found in her daughter’s purse.
What are the signs and symptoms of acute isoniazid toxicity?
Isoniazid toxicity should be suspected in any patient who has access to INH—even if the drug was prescribed for someone other than the patient. Acute toxicity develops rapidly after the ingestion of supratherapeutic doses of INH and includes nausea, abdominal discomfort, vomiting, dizziness, and excessive fatigue or lethargy. Patients can present with tachycardia, stupor, agitation, mydriasis, increased anion gap metabolic acidosis, and encephalopathy.
Seizures occur due to an INH-induced functional pyridoxine deficiency. Isoniazid inhibits pyridoxine phosphokinase, the enzyme that converts pyridoxine (vitamin B6) to its physiologically active form, pyridoxal 5’-phosphate (PLP). Because the conversion of glutamate (an excitatory neurotransmitter) to gamma-aminobutyric acid (GABA; the body’s main inhibitory neurotransmitter) is dependent on PLP, an excess of glutamate and a deficiency of GABA occurs following INH overdose. The result is neuroexcitation, which manifests as generalized seizures in affected patients.
The most consequential effect of INH overdose, however, is the development of seizure refractory to conventional therapy, such as benzodiazepines. This occurs because benzodiazepines are indirect-acting GABA agonists, and require the presence of GABA to elicit their effect. Therefore, due to the impairment of GABA synthesis, benzodiazepines are limited or ineffective as anticonvulsants. Although INH doses in excess of 20 mg/kg may result in neuroexcitation, refractory seizures are uncommon with doses <70 mg/kg.
Complications of chronic INH use include hepatotoxicity, and patients will present with jaundice, hepatomegaly, and right upper quadrant pain and tenderness. Isoniazid must be discontinued rapidly in
How is acute isoniazid-induced seizure managed?
Management of patients with refractory seizure should initially include an assessment and management of the patient’s airway, breathing, and circulation. Although seizures induced by INH toxicity are often resistant to benzodiazepines, these agents remain the first-line therapy. For patients who fail to respond to a reasonable trial of benzodiazepines (eg, lorazepam 6 mg intravenously [IV]), pyridoxine should be administered.3 The recommended dose is 1 g pyridoxine per every 1 g of INH ingested—if the initial dose ingested is known—with a maximum dose of 5 g pyridoxine. If the initial dose of INH is not known, 70 mg/kg of pyridoxine, up to 5 g, is recommended. Repeated doses of pyridoxine can be administered if the seizure continues, up to a total dose of 10 g in an adult. At extremely high doses, pyridoxine itself can be neurotoxic, limiting the maximal antidotal dose.
Rapid initiation of pyridoxine is a challenge since typical stocks in most EDs are not in an adequate supply required for treatment. Additionally, a typical vial of pyridoxine contains 100 mg, highlighting the rare need to open dozens of vials for a single patient. Drawing up adequate doses of the IV formulation can be a challenge and time-consuming.
Regardless, the most reliable and rapid route of administration for pyridoxine is IV, at a rate of 0.5 to 1 g/min. Even if the seizure resolves prior to completion of the initial dose, the remaining doses should still be administered over a 4- to 6-hour period. Oral or (more likely) nasogastric administration of pyridoxine can be administered if the IV formulation is not available, but neither are optimal routes of delivery. Every effort should be made to stock pyridoxine in the antidote supply in the ED to avoid time delays involving finding, preparing, and administering the drug in these scenarios. Previous studies have found that most EDs are not prepared to handle pyridoxine replacement.4,5
Since benzodiazepines and barbiturates are GABA agonists with complementary mechanisms of actions to pyridoxine, they should be administered to potentiate the antiseizure effect of pyridoxine. If the seizure does not terminate, the use of propofol or general anesthesia may be required. Once the seizure is terminated, oral activated charcoal can be administered if the ingestion occurred within several hours of presentation. Given the rapid onset of effect of a large dose of INH, most patients will develop seizure shortly after exposure, limiting the benefits of both aggressive gastrointestinal decontamination and delayed activated charcoal. Charcoal also can be used for patients who overdose on INH but do not develop seizures.
Although the utility of a head computed tomography (CT) scan or laboratory studies is limited given the context of the exposure, these are generally obtained for patients with new-onset seizure. Since many patients with INH toxicity do not seize, such a patient may have a lower seizure threshold due to the existence of a subclinical cerebral lesion or metabolic abnormality.
Case Conclusion
The patient’s INH-induced refractory seizure was treated with pyridoxine. Her history suggested that she had ingested an unknown number of INH tablets within an hour. On this initial basis, an IV dose of 5,000 mg of pyridoxine was administered. The patient’s seizures terminated within 2 minutes of the infusion, and no additional doses of pyridoxine were required. Given the lack of concern for self-harm, an acetaminophen concentration was not obtained. A urine toxicology screen was negative for cocaine and amphetamines, and a CT scan of the head was negative for any abnormality. The patient was admitted to the pediatric intensive care unit for status epileptics and was discharged home on hospital day 2 after an uneventful stay.
1. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82. doi:10.1016/j.yebeh.2015.04.034.
2. Latent tuberculosis infection: a guide for primary health care providers. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/tb/publications/LTBI/treatment.htm. Updated August 5, 2016. Accessed December 13, 2016.
3. Howland MA. Antidotes in depth: pyridoxine. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:797-799.
4. Shah BR, Santucci K, Sinert R, Steiner P. Acute isoniazid neurotoxicity in an urban hospital. Pediatrics. 1995;95(5):700-704.
5. Santucci KA, Shah BR, Linakis JG. Acute isoniazid exposures and antidote availability. Pediatr Emerg Care. 1999;15(2):99-101.
1. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82. doi:10.1016/j.yebeh.2015.04.034.
2. Latent tuberculosis infection: a guide for primary health care providers. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/tb/publications/LTBI/treatment.htm. Updated August 5, 2016. Accessed December 13, 2016.
3. Howland MA. Antidotes in depth: pyridoxine. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:797-799.
4. Shah BR, Santucci K, Sinert R, Steiner P. Acute isoniazid neurotoxicity in an urban hospital. Pediatrics. 1995;95(5):700-704.
5. Santucci KA, Shah BR, Linakis JG. Acute isoniazid exposures and antidote availability. Pediatr Emerg Care. 1999;15(2):99-101.

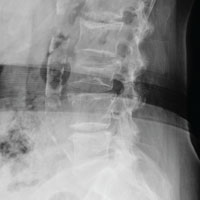
From Hydroplane to Ankle Pain
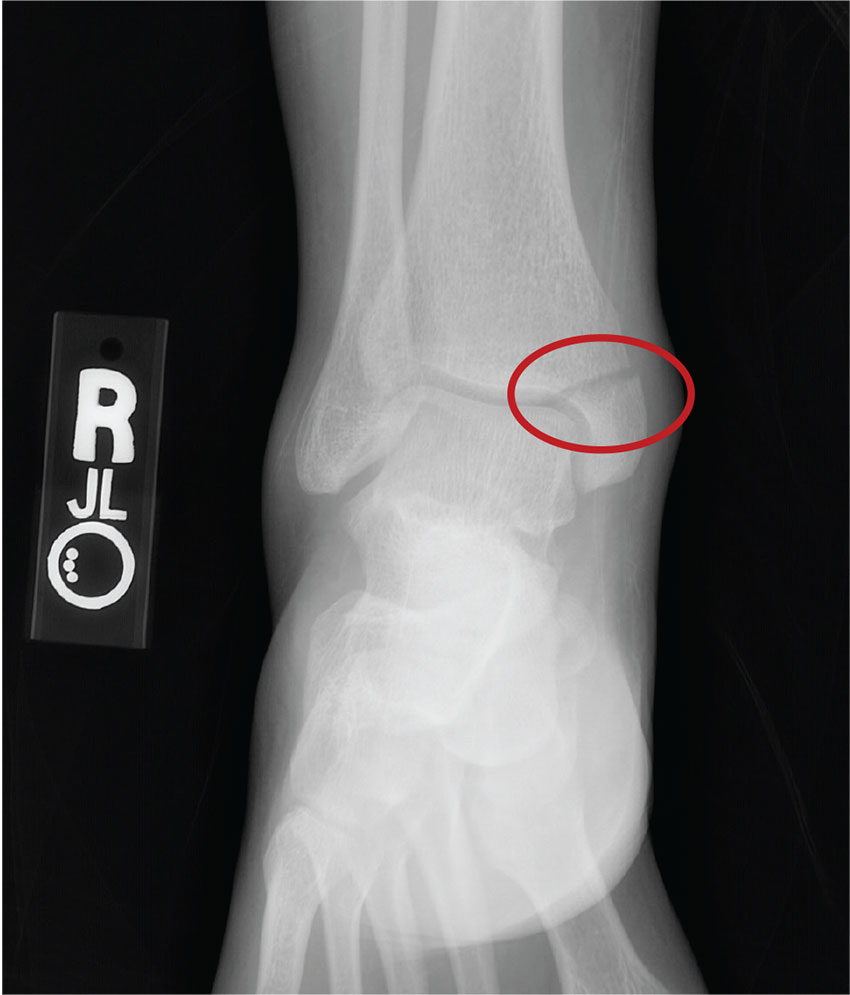
ANSWER
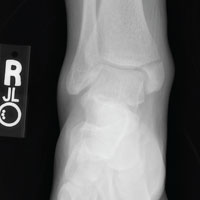
The radiograph shows an acute fracture of the medial malleolus. It is minimally displaced. The mortise joint appears intact. The patient was placed in a short leg splint for immobilization, and prompt orthopedic follow-up was arranged.
ANSWER
The radiograph shows an acute fracture of the medial malleolus. It is minimally displaced. The mortise joint appears intact. The patient was placed in a short leg splint for immobilization, and prompt orthopedic follow-up was arranged.
ANSWER
The radiograph shows an acute fracture of the medial malleolus. It is minimally displaced. The mortise joint appears intact. The patient was placed in a short leg splint for immobilization, and prompt orthopedic follow-up was arranged.

A 40-year-old woman presents to urgent care for evaluation of ankle pain following a car accident. She was a restrained driver who lost control of her vehicle while driving on wet roads. Her vehicle hit a telephone pole head on. There was no air bag deployment. Initially, she thought she was fine and declined EMS transport to a local hospital. But when she experienced severe pain bearing weight on her right foot, she opted to have it evaluated.
She denies any other complaints. Her medical history is otherwise unremarkable, and vital signs are normal. Physical examination of her right ankle demonstrates general soft-tissue swelling but no obvious deformity. She has moderate tenderness on both the medial and lateral aspects of her ankle. She has limited dorsiflexion and plantar flexion secondary to pain. Good distal pulses are palpable, and good capillary refill is noted in all of the toes.
A radiograph of the ankle is shown. What is your impression?
Parsimonious blood use and lower transfusion triggers: What is the evidence?
For decades, physicians believed in the benefit of prompt transfusion of blood to keep the hemoglobin level at arbitrary, optimum levels, ie, close to normal values, especially in the critically ill, the elderly, and those with coronary syndromes, stroke, or renal failure.
However, the evidence supporting arbitrary hemoglobin values as an indication for transfusion was weak or nonexistent. Also, blood transfusion can have complications and adverse effects, and blood is costly and scarce. These considerations prompted research into when blood transfusion should be considered, and recommendations that it should be used more sparingly than in the past.
This review offers a perspective on the evidence supporting restrictive blood use. First, we focus on hemodilution studies that demonstrated that humans can tolerate anemia. Then, we look at studies that compared a restrictive transfusion strategy with a liberal one in patients with critical illness and active bleeding. We conclude with current recommendations for blood transfusion.
EVIDENCE FROM HEMODILUTION STUDIES
Hemoglobin is essential for tissue oxygenation, but the serum hemoglobin concentration is just one of several factors involved.1–5 In anemia, the body can adapt not only by increasing production of red blood cells, but also by:
- Increasing cardiac output
- Increasing synthesis of 2,3-diphosphoglycerate (2,3-DPG), with a consequent shift in the oxyhemoglobin dissociation curve to the right, allowing enhanced release of oxygen at the tissue level
- Moving more carbon dioxide into the blood (the Bohr effect), which decreases pH and also shifts the dissociation curve to the right.
Just 20 years ago, physicians were using arbitrary cutoffs such as hemoglobin 10 g/dL or hematocrit 30% as indications for blood transfusion, without reasonable evidence to support these values. Not until acute normovolemic hemodilution studies were performed were we able to progressively appraise how well patients could tolerate lower levels of hemoglobin without significant adverse outcomes.
Acute normovolemic hemodilution involves withdrawing blood and replacing it with crystalloid or colloid solution to maintain the volume.6
Initial studies were done in animals and focused on the safety of acute anemia regarding splanchnic perfusion. Subsequently, studies proved that healthy, elderly, and stable cardiac patients can tolerate acute anemia with normal cardiovascular response. The targets in these studies were modest at first, but researchers aimed progressively for more aggressive hemodilution with lower hemoglobin targets and demonstrated that the body can tolerate and adapt to more severe anemia.6–8
Studies in healthy patients
Weiskopf et al9 assessed the effect of severe anemia in 32 conscious healthy patients (11 presurgical patients and 21 volunteers not undergoing surgery) by performing acute normovolemic hemodilution with 5% human albumin, autologous plasma, or both, with a target hemoglobin level of 5 g/dL. The process was done gradually, obtaining aliquots of blood of 500 to 900 mL. Cardiac index increased, along with a mild increase in oxygen consumption with no increase in plasma lactate levels, suggesting that in conscious healthy patients, tissue oxygenation remains adequate even in severe anemia.
Leung et al10 addressed the electrocardiographic changes that occur with severe anemia (hemoglobin 5 g/dL) in 55 healthy volunteers. Three developed transient, reversible ST-segment depression, which was associated with a higher heart rate than in the volunteers with no electrocardiographic changes; however, the changes were reversible and asymptomatic, and thus were considered physiologic and benign.
Hemodilution in healthy elderly patients
Spahn et al11 performed 6 and 12 mL/kg isovolemic exchange of blood for 6% hydroxyethyl starch in 20 patients older than 65 years (mean age 76, range 65–88) without underlying coronary disease.
The patients’ mean hemoglobin level decreased from 11.6 g/dL to 8.8 g/dL. Their cardiac index and oxygen extraction values increased adequately, with stable oxygen consumption during hemodilution. There were no electrocardiographic signs of ischemia.
Hemodilution in coronary artery disease
Spahn et al12 performed hemodilution studies in 60 patients (ages 35–81) with coronary artery disease managed chronically with beta-blockers who were scheduled for coronary artery bypass graft surgery. Hemodilution was performed with 6- and 12-mL/kg isovolemic exchange of blood for 6% hydroxyethyl starch maintaining normovolemia and stable filling pressures. Hemoglobin levels decreased from 12.6 g/dL to 9.9 g/dL. The hemodilution process was done before the revascularization. The authors monitored hemodynamic variables, ST-segment deviation, and oxygen consumption before and after each hemodilution.
There was a compensatory increase in cardiac index and oxygen extraction with consequent stable oxygen consumption. These changes were independent of patient age or left ventricular function. In addition, there were no electrocardiographic signs of ischemia.
Licker et al13 studied the hemodynamic effect of preoperative hemodilution in 50 patients with coronary artery disease undergoing coronary artery bypass graft surgery, performing transesophageal echocardiography before and after hemodilution. The patients underwent isovolemic exchange with iso-oncotic starch to target a hematocrit of 28%.
Acute normovolemic hemodilution triggered an increase in cardiac stroke volume, which had a direct correlation with an increase in the central venous pressure and the left ventricular end-diastolic area. No signs of ischemia were seen in these patients on electrocardiography or echocardiography (eg, left ventricular wall-motion abnormalities).
Hemodilution in mitral regurgitation
Spahn et al14 performed acute isovolemic hemodilution with 6% hydroxyethyl starch in 20 patients with mitral regurgitation. The cardiac filling pressures were stable before and after hemodilution; the mean hemoglobin value decreased from 13 to 10.3 g/dL. The cardiac index and oxygen extraction increased proportionally, with stable oxygen consumption; these findings were the same regardless of whether the patient was in normal sinus rhythm or atrial fibrillation.
Effect of hemodilution on cognition
Weiskopf et al15 assessed the effect of anemia on executive and memory function by inducing progressive acute isovolemic anemia in 90 healthy volunteers (age 29 ± 5), reducing their hemoglobin values to 7, 6, and 5 g/dL and performing repetitive neuropsychological and memory testing before and after the hemodilution, as well as after autologous blood transfusion to return their hemoglobin level to 7 g/dL.
There were no changes in reaction time or error rate at a hemoglobin concentration of 7 g/dL compared with the performance at a baseline hemoglobin concentration of 14 g/dL. The volunteers got slower on a mathematics test at hemoglobin levels of 6 g/dL and 5 g/dL, but their error rate did not increase. Immediate and delayed memory were significantly impaired at hemoglobin of 5 g/dL but not at 6 g/dL. All tests normalized with blood transfusion once the hemoglobin level reached 7 g/dL.15
Weiskopf et al16 subsequently investigated whether giving supplemental oxygen to raise the arterial partial pressure of oxygen (Pao2) to 350 mm Hg or greater would overcome the neurocognitive effects of severe acute anemia. They followed a protocol similar to the one in the earlier study15 and induced anemia in 31 healthy volunteers, age 28 ± 4 years, with a mean baseline hemoglobin concentration of 12.7 g/dL.
When the volunteers reached a hemoglobin concentration of 5.7 ± 0.3 g/dL, they were significantly slower on the mathematics test, and their delayed memory was significantly impaired. Then, in a double-blind fashion, they were given either room air or oxygen. Oxygen increased the Pao2 to 406 mm Hg and normalized neurocognitive performance.
Hemodilution studies in surgical patients
A 2015 meta-analysis17 of 63 studies involving 3,819 surgical patients compared the risk of perioperative allogeneic blood transfusion as well as the overall volume of transfused blood in patients undergoing preoperative acute normovolemic hemodilution vs a control group. Though the overall data showed that the patients who underwent acute normovolemic hemodilution needed fewer transfusions and less blood (relative risk [RR] 0.74, 95% confidence interval [CI] 0.63–0.88, P = .0006), the authors noted significant heterogeneity and publication bias.
However, the hemodilution studies paved the way for justifying a more conservative and restrictive transfusion strategy, with a hemoglobin cutoff value of 7 g/dL, and in acute anemia, using oxygen to overcome acute neurocognitive effects while searching for and correcting the cause of the anemia.
STUDIES OF RESTRICTIVE VS LIBERAL TRANSFUSION STRATEGIES
Studies in critical care and high-risk patients
Hébert et al18 randomized 418 critical care patients to a restrictive transfusion approach (in which they were given red blood cells if their hemoglobin concentration dropped below 7.0 g/dL) and 420 patients to a liberal strategy (given red blood cells if their hemoglobin concentration dropped below 10.0 g/dL). Mortality rates (restrictive vs liberal strategy) were as follows:
- Overall at 30 days 18.7% vs 23.3%, P = .11
- In the subgroup with less-severe disease (Acute Physiology and Chronic Health Evaluation II [APACHE II] score < 20), 8.7% vs 16.1%, P = .03
- In the subgroup under age 55, 5.7% vs 13%, P = .02
- In the subgroup with clinically significant cardiac disease, 20.5% vs 22.9%, P = .69
- In the hospital, 22.2% vs 28.1%; P = .05.
This study demonstrated that parsimonious blood use did not worsen clinical outcomes in critical care patients.
Carson et al19 evaluated 2,016 patients age 50 and older who had a history of or risk factors for cardiovascular disease and a baseline hemoglobin level below 10 g/dL who underwent surgery for hip fracture. Patients were randomized to two transfusion strategies based on threshold hemoglobin level: restrictive (< 8 g/dL) or liberal (< 10 g/dL). The primary outcome was death or inability to walk without assistance at 60-day follow-up. The median number of units of blood used was 2 in the liberal group and 0 in the restrictive group.
There was no significant difference in the rates of the primary outcome (odds ratio [OR] 1.01, 95% CI 0.84–1.22), infection, venous thromboembolism, or reoperation. This study demonstrated that a liberal transfusion strategy offered no benefit over a restrictive one.
Rao et al20 analyzed the impact of blood transfusion in 24,112 patients with acute coronary syndromes enrolled in three large trials. Ten percent of the patients received at least 1 blood transfusion during their hospitalization, and they were older and had more complex comorbidity.
At 30 days, the group that had received blood had higher rates of death (adjusted hazard ratio [HR] 3.94, 95% CI 3.26–4.75) and the combined outcome of death or myocardial infarction (HR 2.92, 95% CI 2.55–3.35). Transfusion in patients whose nadir hematocrit was higher than 25% was associated with worse outcomes.
This study suggests being cautious about routinely transfusing blood in stable patients with ischemic heart disease solely on the basis of arbitrary hematocrit levels.
Carson et al,21 however, in a later trial, found a trend toward worse outcomes with a restrictive strategy than with a liberal one. Here, 110 patients with acute coronary syndrome or stable angina undergoing cardiac catheterization were randomized to a target hemoglobin level of either at least 8 mg/dL or at least 10 g/dL. The primary outcome (a composite of death, myocardial infarction, or unscheduled revascularization 30 days after randomization) occurred in 14 patients (25.5%) in the restrictive group and 6 patients (10.9%) in the liberal group (P = .054), and 7 (13.0%) vs 1 (1.8%) of the patients died (P = .032).
Murphy et al22 similarly found trends toward worse outcomes with a restrictive strategy in cardiac patients. The investigators randomized 2,007 elective cardiac surgery patients with a postoperative hemoglobin level lower than 9 g/dL to a hemoglobin transfusion threshold of either 7.5 or 9 g/dL. Outcomes (restrictive vs liberal strategies):
- Transfusion rates 53.4% vs 92.2%
- Rates of the primary outcome (a serious infection [sepsis or wound infection] or ischemic event [stroke, myocardial infarction, mesenteric ischemia, or acute kidney injury] within 3 months):
35.1% vs 33.0%, OR 1.11, 95% CI 0.91–1.34, P = .30) - Mortality rates 4.2% vs 2.6%, HR 1.64, 95% CI 1.00–2.67, P = .045
- Total costs did not differ significantly between the groups.
These studies21,22 suggest the need for more definitive trials in patients with active coronary disease and in cardiac surgery patients.
Holst et al23 randomized 998 intensive care patients in septic shock to hemoglobin thresholds for transfusion of 7 vs 9 g/dL. Mortality rates at 90 days (the primary outcome) were 43.0% vs 45.0%, RR 0.94, 95% CI 0.78–1.09, P = .44.
This study suggests that even in septic shock, a liberal transfusion strategy has no advantage over a parsimonious one.
Active bleeding, especially active gastrointestinal bleeding, poses a significant stress that may trigger empirical transfusion even without evidence of the real hemoglobin level.
Villanueva et al24 randomized 921 patients with severe acute upper-gastrointestinal bleeding to two groups, with hemoglobin transfusion triggers of 7 vs 9 g/dL. The findings were impressive:
- Freedom from transfusion 51% vs 14% (P < .001)
- Survival rates at 6 weeks 95% vs 91% (HR 0.55, 95% CI 0.33–0.92, P = .02)
- Rebleeding 10% vs 16% (P = .01).
Patients with peptic ulcer disease as well as those with cirrhosis stage Child-Pugh class A or B had higher survival rates with a restrictive transfusion strategy.
The RELIEVE trial25 compared the effect of a restrictive transfusion strategy in elderly patients on mechanical ventilation in 6 intensive care units in the United Kingdom. Transfusion triggers were hemoglobin 7 vs 9 g/dL, and the mortality rate at 180 days was 55% vs 37%, RR 0.68, 95% CI 0.44–1.05, P = .073.
Meta-analyses and observational studies
Rohde et al26 performed a systematic review and meta-analysis of 17 trials with 7,456 patients, which revealed that a restrictive strategy is associated with a lower risk of nosocomial infection, including pneumonia, wound infection, and sepsis.
The pooled risk of all serious infections was 10.6% in the restrictive group and 12.7% in the liberal group. Even after adjusting for the use of leukocyte reduction, the risk of infection was lower in the restrictive strategy group (RR 0.83, 95% CI 0.69–0.99). With a hemoglobin threshold of less than 7.0 g/dL, the risk of serious infection was 14% lower. Although this was not statistically significant overall (RR 0.86, 95% CI 0.72–1.02), the difference was statistically significant in the subgroup undergoing orthopedic surgery (RR 0.72, 95% CI 0.53–0.97) and the subgroup presenting with sepsis (RR 0.51, 95% CI 0.28–0.95).
Salpeter et al27 performed a meta-analysis and systematic review of three randomized trials (N = 2,364) comparing a restrictive hemoglobin transfusion trigger (hemoglobin < 7 g/dL) vs a more liberal trigger. The groups with restrictive transfusion triggers had lower rates of:
- In-hospital mortality (RR 0.74, 95% CI 0.60–0.92)
- Total mortality (RR 0.80, 95% CI 0.65–0.98)
- Rebleeding (RR 0.64, 95% CI 0.45–0.90)
- Acute coronary syndrome (RR 0.44, 95% CI 0.22–0.89)
- Pulmonary edema (RR 0.48, 95% CI 0.33–0.72)
- Bacterial infections (RR 0.86, 95% CI 0.73–1.00).
Wang et al28 performed a meta-analysis of 4 randomized controlled trials in patients with upper-gastrointestinal bleeding comparing restrictive (hemoglobin < 7 g/dL) vs liberal transfusion strategies. The primary outcomes were death and rebleeding. The restrictive strategy was associated with:
- A lower mortality rate (OR 0.52, 95% CI 0.31–0.87, P = .01)
- A lower rebleeding rate (OR 0.26, 95% CI 0.03–2.10, P = .21)
- Shorter hospitalizations (P = .009)
- Less blood transfused (P = .0005).
Vincent et al,29 in a prospective observational study of 3,534 patients in intensive care units in 146 facilities in Western Europe, found a correlation between transfusion and mortality. Transfusion was done most often in elderly patients and those with a longer stay in the intensive care unit. The 28-day mortality rate was 22.7% in patients who received a transfusion and 17.1% in those who did not (P = .02). The more units of blood the patients received, the more likely they were to die, and receiving more than 4 units was associated with worse outcomes (P = .01).
Dunne et al30 performed a study of 6,301 noncardiac surgical patients in the Veterans Affairs Maryland Healthcare System from the National Veterans Administration Surgical Quality Improvement Program from 1995 to 2000. Multiple logistic regression analysis revealed that the composite of low hematocrit before and after surgery and high transfusion rates (> 4 units per hospitalization) were associated with higher rates of death (P < .01) and postoperative pneumonia (P ≤ .05) and longer hospitalizations (P < .05). The risk of pneumonia increased proportionally with the decrease in hematocrit.
These findings support pharmacologic optimization of anemia with hematinic supplementation before surgery to decrease the risk of needing a transfusion, often with parenteral iron. The fact that the patient’s hemoglobin can be optimized preoperatively by nontransfusional means may decrease the likelihood of blood transfusion, as the hemoglobin will potentially remain above the transfusion threshold. For example, if a patient has a preoperative hemoglobin level of 10 g/dL, and it is optimized up to 12, then if postoperatively the hemoglobin level drops 3 g/dL instead of reaching the threshold of 7 g/dL, the nadir will be just 9 g/dL, far above that transfusion threshold.
Brunskill et al,31 in a Cochrane review of 6 trials with 2,722 patients undergoing surgery for hip fracture, found no difference in rates of mortality, functional recovery or postoperative morbidity with a restrictive transfusion strategy (hemoglobin target > 8 g/dL vs a liberal one (> 10 g/dL). However, the quality of evidence was rated as low. The authors concluded that there is no justification for liberal red blood cell transfusion thresholds (10 g/dL), and a more restrictive transfusion threshold is preferable.
Weinberg et al32 found that, in trauma patients, receiving more than 6 units of blood was associated with poor prognosis, and outcomes were worse when the blood was older than 2 weeks. However, the effect of blood age is not significant when using smaller transfusion volumes (1 to 2 units of red blood cells).
Studies in sickle cell disease
Sickle cell disease patients have high levels of hemoglobin S, which causes erythrocyte sickling and increases blood viscosity. Transfusion with normal erythrocytes increases the amount of hemoglobin A (the normal variant).33,34
In trials in surgical patients,35,36 conservative strategies for preoperative blood transfusion aiming at a hemoglobin level of 10 g/dL were as effective in preventing postoperative complications as decreasing the hemoglobin S levels to 30% by aggressive exchange transfusion.35
In nonsurgical patients, blood transfusion should be based on formal risk-benefit assessments. Therefore, the expert panel report on sickle cell management advises against blood transfusion in sickle cell patients with uncomplicated vaso-occlusive crises, priapism, asymptomatic anemia, or acute kidney injury in the absence of multisystem organ failure.34
Is hemoglobin the most relevant marker?
Most studies that compared restrictive and liberal transfusion strategies focused on using a lower hemoglobin threshold as the transfusion trigger, not on using fewer units of blood. Is the amount of blood transfused more important than the hemoglobin threshold? Perhaps a study focused both on a restrictive vs liberal strategy and also on the minimum amount of blood that each patient may benefit from would help to answer this question.
We should beware of routinely using the hemoglobin concentration as a threshold for transfusion and a surrogate marker of transfusion benefit because changes in hemoglobin concentration may not reflect changes in absolute red cell mass.37 Changes in plasma volume (an increase or decrease) affect the hematocrit concentration without necessarily affecting the total red cell mass. Unfortunately, red cell mass is very difficult to measure; hence, the hemoglobin and hematocrit values are used instead. Studies addressing changes in red cell mass may be needed, perhaps even to validate using the hemoglobin concentration as the sole indicator for transfusion.
Is fresh blood better than old blood?
Using blood that is more than 14 days old may be associated with poor outcomes, for several possible reasons. Red blood cells age rapidly in refrigeration, and usually just 75% may remain viable 24 hours after phlebotomy. Adenosine triphosphate and 2,3-DPG levels steadily decrease, with a consequent decrease in capacity for appropriate tissue oxygen delivery. In addition, loss of membrane phospholipids causes progressive rigidity of the red cell membrane with consequent formation of echynocytes after 14 to 21 days.38,39
The use of blood more than 14 days old in cardiac surgery patients has been associated with worse outcomes, including higher rates of death, prolonged intubation, acute renal failure, and sepsis.40 Similar poor outcomes have been seen in trauma patients.32
Lacroix et al,41 in a multicenter, randomized trial in critically ill adults, compared the outcomes of transfusion of fresh packed red cells (stored < 8 days) or old blood (stored for a mean of 22 days). The primary outcome was the mortality rate at 90 days: 37.0% in the fresh-blood group vs 35.3% in the old-blood group (HR 1.1, 95% CI 0.9–1.2, P = .38).
The authors concluded that using fresh blood compared with old blood was not associated with a lower 90-day mortality rate in critically ill adults.
RISKS ASSOCIATED WITH TRANSFUSION
Infections
The risk of infection from blood transfusion is small. Human immunodeficiency virus (HIV) is transmitted in 1 in 1.5 million transfused blood components, and hepatitis C virus in 1 in 1.1 million; these odds are similar to those of having a fatal airplane accident (1 in 1.7 million per flight). Hepatitis B virus infection is more common, the reported incidence being 1 in 357,000.42
Noninfectious complications
Transfusion-associated circulatory overload occurs in 4% to 6% of patients who receive a transfusion. Therefore, circulatory overload is a greater danger from transfusion than infection is.42
Febrile nonhemolytic transfusion reactions occur in 1.1% of patients with prestorage leukoreduction.
Transfusion-associated acute lung injury occurs in 0.8 per 10,000 blood components transfused.
Errors associated with blood transfusion include, in decreasing order of frequency, transfusion of the wrong blood component, handling and storage errors, inappropriate administration of anti-D immunoglobulin, and avoidable, delayed, or insufficient transfusions.43
Surgery and condition-specific complications of red blood cell transfusion
Cardiovascular surgery. Transfusion is associated with a higher risk of postoperative stroke, respiratory failure, acute respiratory distress syndrome, prolonged intubation time, reintubation, in-hospital death, sepsis, and longer postoperative length of stay.44
Malignancy. The use of blood in this setting has been found to be an independent predictor of recurrence, decreased survival, and increased risk of lymphoplasmacytic and marginal-zone lymphomas.44–47
Vascular, orthopedic, and other surgeries. Transfusion is associated with a higher risk of death, thromboembolic events, acute kidney injury, death, composite morbidity, reoperation, sepsis, and pulmonary complications.44
ST-segment elevation myocardial infarction, sepsis, and intensive care unit admissions. Transfusion is associated with an increased risk of rebleeding, death, and secondary infections.44
COST OF RED BLOOD CELL TRANSFUSION
Up to 85 million units of red blood cells are transfused per year worldwide, 15 million of them in the United States.42 At our hospital in 2013, 1 unit of leukocyte-reduced red blood cells cost $957.27, which included the costs of acquisition, processing, banking, patient testing, administration, and monitoring.
The Premier Healthcare Alliance48 analyzed data from 7.4 million discharges from 464 hospitals between April 2011 and March 2012. Blood use varied significantly among hospitals, and the hospitals in the lowest quartile of blood use had better patient outcomes. If all the hospitals used as little blood as those in the lowest quartile and had outcomes as good, blood product use would be reduced by 802,716 units, with savings of up to $165 million annually.
In addition to the economic cost of blood transfusion, the clinician must be aware of the cost in terms of comorbidities caused by unnecessary blood transfusion.49,50
RECOMMENDATIONS FROM THE AABB
In view of all the current compelling evidence, a restrictive approach to transfusion is the single best strategy to minimize adverse outcomes.51 Below, we outline the current recommendations from the AABB (formerly the American Association of Blood Banks),42 which are similar to the national clinical guideline on blood transfusion in the United Kingdom,52 and have recently been updated, confirming the initial recommendations.53
In critical care patients, transfusion should be considered if the hemoglobin concentration is 7 g/dL or less.
In postoperative patients and hospitalized patients with preexisting cardiovascular disease, transfusion should be considered if the hemoglobin concentration is 8 g/dL or less or if the patient has signs or symptoms of anemia such as chest pain, orthostatic hypotension, or tachycardia unresponsive to fluid resuscitation, or heart failure.
In hemodynamically stable patients with acute coronary syndrome, there is not enough evidence to allow a formal recommendation for or against a liberal or restrictive transfusion threshold.
Consider both the hemoglobin concentration and the symptoms when deciding whether to give a transfusion. This recommendation is shared by a National Institutes of Health consensus conference,54 which indicates that multiple factors related to the patient’s clinical status and oxygen delivery should be considered before deciding to transfuse red blood cells.
The Society of Hospital Medicine55 and the American Society of Hematology56 concur with a parsimonious approach to blood use in their Choosing Wisely campaigns. The American Society of Hematology recommends that if transfusion of red blood cells is necessary, the minimum number of units should be given that relieve the symptoms of anemia or achieve a safe hemoglobin range (7–8 g/dL in stable noncardiac inpatients).57
New electronic tools can monitor the ordering and use of blood products in real time and can identify the hemoglobin level used as the trigger for transfusion. They also provide data on blood use by physician, hospital, and department. These tools can reveal current practice at a glance and allow sharing of best practices among peers and institutions.52
CONSIDER TRANSFUSION FOR HEMOGLOBIN BELOW 7 G/DL
The routine use of blood has come under scrutiny, given its association with increased healthcare costs and morbidity. The accepted practice in stable medical patients is a restrictive threshold approach for blood transfusion, which is to consider (not necessarily give) a single unit of packed red blood cells for a hemoglobin less than 7 g/dL.
However, studies in acute coronary syndrome patients and postoperative cardiac surgery patients have not shown the restrictive threshold to be superior to a liberal threshold in terms of outcomes and costs. This variability suggests the need for further studies to determine the best course of action in different patient subpopulations (eg, surgical, oncologic, trauma, critical illness).
Also, a limitation of most of the clinical studies was that only the hemoglobin concentration was used as a marker of anemia, with no strict assessment of changes in red cell mass with transfusion.
Despite the variability in certain populations, the overall weight of current evidence favors a restrictive approach to blood transfusion (hemoglobin < 7 g/dL), although perhaps in patients who have active coronary disease or are undergoing cardiac surgery, a more lenient threshold (< 8 g/dL) for transfusion should be considered.
- Shander A, Gross I, Hill S, Javidroozi M, Sledge S; College of American Pathologists; American Society of Anesthesiologists; Society of Thoracic Surgeons and Society of Cardiovascular Anesthesiologists; Society of Critical Care Medicine; Italian Society of Transfusion Medicine and Immunohaematology; American Association of Blood Banks. A new perspective on best transfusion practices. Blood Transfus 2013; 11:193–202.
- Madjdpour C, Spahn DR. Allogeneic red blood cell transfusion: physiology of oxygen transport. Best Pract Res Clin Anaesthesiol 2007; 21:163–171.
- Tánczos K, Molnár Z. The oxygen supply-demand balance: a monitoring challenge. Best Pract Res Clin Anaesthesiol 2013; 27:201–207.
- Hebert PC, Van der Linden P, Biro G, Hu LQ. Physiologic aspects of anemia. Crit Care Clin 2004; 20:187–212.
- Spinelli E, Bartlett RH. Anemia and transfusion in critical care: physiology and management. J Intensive Care Med 2016; 31:295–306.
- Jamnicki M, Kocian R, Van Der Linden P, Zaugg M, Spahn DR. Acute normovolemic hemodilution: physiology, limitations, and clinical use. J Cardiothorac Vasc Anesth 2003; 17:747–754.
- Monk TG. Acute normovolemic hemodilution. Anesthesiol Clin North America 2005; 23:271–281.
- Shander A, Rijhwani TS. Acute normovolemic hemodilution. Transfusion 2004; 44(suppl 2):26S–34S.
- Weiskopf RB, Viele MK, Feiner J, et al. Human cardiovascular and metabolic response to acute, severe isovolemic anemia. JAMA 1998; 279:217–221.
- Leung JM, Weiskopf RB, Feiner J, et al. Electrocardiographic ST-segment changes during acute, severe isovolemic hemodilution in humans. Anesthesiology 2000; 93:1004–1010.
- Spahn DR, Zollinger A, Schlumpf RB, et al. Hemodilution tolerance in elderly patients without known cardiac disease. Anesth Analg 1996; 82:681–686.
- Spahn DR, Schmid ER, Seifert B, Pasch T. Hemodilution tolerance in patients with coronary artery disease who are receiving chronic beta-adrenergic blocker therapy. Anesth Analg 1996; 82:687–694.
- Licker M, Ellenberger C, Sierra J, Christenson J, Diaper J, Morel D. Cardiovascular response to acute normovolemic hemodilution in patients with coronary artery diseases: assessment with transesophageal echocardiography. Crit Care Med 2005; 33:591–597.
- Spahn DR, Seifert B, Pasch T, Schmid ER. Haemodilution tolerance in patients with mitral regurgitation. Anaesthesia 1998; 53:20–24.
- Weiskopf RB, Kramer JH, Viele M, et al. Acute severe isovolemic anemia impairs cognitive function and memory in humans. Anesthesiology 2000; 92:1646–1652.
- Weiskopf RB, Feiner J, Hopf HW, et al. Oxygen reverses deficits of cognitive function and memory and increased heart rate induced by acute severe isovolemic anemia. Anesthesiology 2002; 96:871–877.
- Zhou X, Zhang C, Wang Y, Yu L, Yan M. Preoperative acute normovolemic hemodilution for minimizing allogeneic blood transfusion: a meta-analysis. Anesth Analg 2015; 121:1443–1455.
- Hébert P, Wells G, Blajchman M, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999: 340:409–417.
- Carson JL, Terrin ML, Noveck H, et al; FOCUS Investigators. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med 2011; 365:2453–2462.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
- Carson JL, Brooks MM, Abbott JD, et al. Liberal versus restrictive transfusion thresholds for patients with symptomatic coronary artery disease. Am Heart J 2013; 165:964.e1–971.e1.
- Murphy GJ, Pike K, Rogers CA, et al; TITRe2 Investigators. Liberal or restrictive transfusion after cardiac surgery. N Engl J Med 2015; 372:997–1008.
- Holst LB, Haase N, Wetterslev J, et al; TRISS Trial Group; Scandinavian Critical Care Trials Group. Lower versus higher hemoglobin threshold for transfusion in septic shock. N Engl J Med 2014; 371:1381–1391.
- Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013; 368:11–21.
- Walsh TS, Boyd JA, Watson D, et al; RELIEVE Investigators. Restrictive versus liberal transfusion strategies for older mechanically ventilated critically ill patients: a randomized pilot trial. Crit Care Med 2013; 41:2354–2363.
- Rohde JM, Dimcheff DE, Blumberg N, et al. Health care–associated infection after red blood cell transfusion. JAMA 2014; 311:1317–1326.
- Salpeter SR, Buckley JS, Chatterjee S. Impact of more restrictive blood transfusion strategies on clinical outcomes: a meta-analysis and systematic review. Am J Med 2014; 127:124.e3–131.e3.
- Wang J, Bao YX, Bai M, Zhang YG, Xu WD, Qi XS. Restrictive vs liberal transfusion for upper gastrointestinal bleeding: a meta-analysis of randomized controlled trials. World J Gastroenterol 2013; 19:6919–6927.
- Vincent JL, Baron JF, Reinhart K, et al; ABC (Anemia and Blood Transfusion in Critical Care) Investigators. Anemia and blood transfusion in critically ill patients. JAMA 2002; 288:1499–1507.
- Dunne JR, Malone D, Tracy JK, Gannon C, Napolitano LM. Perioperative anemia: an independent risk factor for infection, mortality, and resource utilization in surgery. J Surg Res 2002; 102:237–244.
- Brunskill SJ, Millette SL, Shokoohi A, et al. Red blood cell transfusion for people undergoing hip fracture surgery. Cochrane Database Syst Rev 2015; 4:CD009699.
- Weinberg JA, McGwin G Jr, Griffin RL, et al. Age of transfused blood: an independent predictor of mortality despite universal leukoreduction. J Trauma 2008; 65:279–284.
- Steinberg M. Management of sickle cell disease. N Engl J Med 1999; 340:1021–1030.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease. JAMA 2014; 312:1033–1048.
- Vichinsky EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med 1995; 333:206–213.
- Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet 2013; 381:930–938.
- Goodnough LT, Levy JH, Murphy MF. Concepts of blood transfusion in adults. Lancet 2013; 381:1845–1854.
- Holme S. Current issues related to the quality of stored RBCs. Transfus Apher Sci 2005; 33:55–61.
- Hovav T, Yedgar S, Manny N, Barshtein G. Alteration of red cell aggregability and shape during blood storage. Transfusion 1999; 39:277–281.
- Koch CG, Li L, Sessler DI, et al. Duration of red-cell storage and complications after cardiac surgery. N Engl J Med 2008; 358:1229–1239.
- Lacroix J, Hebert PC, Fergusson DA, et al. Age of transfused blood in critically ill adults. N Engl J Med 2015; 372:1410–1418.
- Carson JL, Grossman BJ, Kleinman S, et al; Clinical Transfusion Medicine Committee of the AABB. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Intern Med 2012; 157:49–58.
- Bolton-Maggs P, Watt A, Poles D, et al, on behalf of the Serious Hazards of Transfusion (SHOT) Steering Group. The 2015 Annual SHOT Report. www.shotuk.org/wp-content/uploads/SHOT-2015-Annual-Report-Web-Edition-Final-bookmarked.pdf. Accessed November 30, 2016.
- Shander A, Javidroozi M, Ozawa S, Hare GMT. What is really dangerous: anaemia or transfusion? Br J Anaesth 2011; 107(suppl 1):i41–i59.
- Reeh M, Ghadban T, Dedow J, et al. Allogenic blood transfusion is associated with poor perioperative and long-term outcome in esophageal cancer. World J Surg 2016 Oct 11. [Epub ahead of print]
- Elmi M, Mahar A, Kagedan D, et al. The impact of blood transfusion on perioperative outcomes following gastric cancer resection: an analysis of the American College of Surgeons National Surgical Quality Improvement Program database. Can J Surg 2016; 59:322–329.
- Aquina CT, Blumberg N, Becerra AZ, et al. Association among blood transfusion, sepsis, and decreased long-term survival after colon cancer resection. Ann Surg 2016; Sep 14. [Epub ahead of print] PubMed PMID: 27631770.
- Premiere Analysis. Standardization of blood utilization practices could provide opportunity for improved outcomes, reduced costs. A Premiere Healthcare Alliance Analysis. 2012.
- Simeone F, Franchi F, Cevenini G, et al. A simple clinical model for planning transfusion quantities in heart surgery. BMC Med Inform Decis Mak 2011; 11:44.
- Spahn DR, Goodnough LT. Alternatives to blood transfusion. Lancet 2013; 381:1855–1865.
- Holst LB, Petersen MW, Haase N, Perner A, Wetterslev J. Restrictive versus liberal transfusion strategy for red blood cell transfusion: systematic review of randomised trials with meta-analysis and trial sequential analysis. BMJ 2015; 350:h1354.
- National Institute for Health and Care Excellence: Clinical Guidelines. London: National Institute for Health and Care Excellence (UK). www.ncbi.nlm.nih.gov/books/NBK11822/.
- Carson JL, Guyatt G, Heddle NM, et al. Clinical practice guidelines from the AABB: red blood cell transfusion thresholds and storage. JAMA 2016 Oct 12. doi: 10.1001/jama.2016.9185. [Epub ahead of print]
- Consensus conference. Perioperative red blood cell transfusion. JAMA 1988; 260:2700–2703.
- Bulger J, Nickel W, Messler J, et al. Choosing wisely in adult hospital medicine: five opportunities for improved healthcare value. J Hosp Med 2013; 8:486–492.
- Hicks LK, Bering H, Carson KR, et al. The ASH Choosing Wisely® campaign: five hematologic tests and treatments to question. Blood 2013; 122:3879–3883.
- Haemonetics IMPACT Online. The Blood Management Company. www.haemonetics.com/Products/Services/Consulting Services/IMPACT Online.aspx. Accessed November 30, 2016.
For decades, physicians believed in the benefit of prompt transfusion of blood to keep the hemoglobin level at arbitrary, optimum levels, ie, close to normal values, especially in the critically ill, the elderly, and those with coronary syndromes, stroke, or renal failure.
However, the evidence supporting arbitrary hemoglobin values as an indication for transfusion was weak or nonexistent. Also, blood transfusion can have complications and adverse effects, and blood is costly and scarce. These considerations prompted research into when blood transfusion should be considered, and recommendations that it should be used more sparingly than in the past.
This review offers a perspective on the evidence supporting restrictive blood use. First, we focus on hemodilution studies that demonstrated that humans can tolerate anemia. Then, we look at studies that compared a restrictive transfusion strategy with a liberal one in patients with critical illness and active bleeding. We conclude with current recommendations for blood transfusion.
EVIDENCE FROM HEMODILUTION STUDIES
Hemoglobin is essential for tissue oxygenation, but the serum hemoglobin concentration is just one of several factors involved.1–5 In anemia, the body can adapt not only by increasing production of red blood cells, but also by:
- Increasing cardiac output
- Increasing synthesis of 2,3-diphosphoglycerate (2,3-DPG), with a consequent shift in the oxyhemoglobin dissociation curve to the right, allowing enhanced release of oxygen at the tissue level
- Moving more carbon dioxide into the blood (the Bohr effect), which decreases pH and also shifts the dissociation curve to the right.
Just 20 years ago, physicians were using arbitrary cutoffs such as hemoglobin 10 g/dL or hematocrit 30% as indications for blood transfusion, without reasonable evidence to support these values. Not until acute normovolemic hemodilution studies were performed were we able to progressively appraise how well patients could tolerate lower levels of hemoglobin without significant adverse outcomes.
Acute normovolemic hemodilution involves withdrawing blood and replacing it with crystalloid or colloid solution to maintain the volume.6
Initial studies were done in animals and focused on the safety of acute anemia regarding splanchnic perfusion. Subsequently, studies proved that healthy, elderly, and stable cardiac patients can tolerate acute anemia with normal cardiovascular response. The targets in these studies were modest at first, but researchers aimed progressively for more aggressive hemodilution with lower hemoglobin targets and demonstrated that the body can tolerate and adapt to more severe anemia.6–8
Studies in healthy patients
Weiskopf et al9 assessed the effect of severe anemia in 32 conscious healthy patients (11 presurgical patients and 21 volunteers not undergoing surgery) by performing acute normovolemic hemodilution with 5% human albumin, autologous plasma, or both, with a target hemoglobin level of 5 g/dL. The process was done gradually, obtaining aliquots of blood of 500 to 900 mL. Cardiac index increased, along with a mild increase in oxygen consumption with no increase in plasma lactate levels, suggesting that in conscious healthy patients, tissue oxygenation remains adequate even in severe anemia.
Leung et al10 addressed the electrocardiographic changes that occur with severe anemia (hemoglobin 5 g/dL) in 55 healthy volunteers. Three developed transient, reversible ST-segment depression, which was associated with a higher heart rate than in the volunteers with no electrocardiographic changes; however, the changes were reversible and asymptomatic, and thus were considered physiologic and benign.
Hemodilution in healthy elderly patients
Spahn et al11 performed 6 and 12 mL/kg isovolemic exchange of blood for 6% hydroxyethyl starch in 20 patients older than 65 years (mean age 76, range 65–88) without underlying coronary disease.
The patients’ mean hemoglobin level decreased from 11.6 g/dL to 8.8 g/dL. Their cardiac index and oxygen extraction values increased adequately, with stable oxygen consumption during hemodilution. There were no electrocardiographic signs of ischemia.
Hemodilution in coronary artery disease
Spahn et al12 performed hemodilution studies in 60 patients (ages 35–81) with coronary artery disease managed chronically with beta-blockers who were scheduled for coronary artery bypass graft surgery. Hemodilution was performed with 6- and 12-mL/kg isovolemic exchange of blood for 6% hydroxyethyl starch maintaining normovolemia and stable filling pressures. Hemoglobin levels decreased from 12.6 g/dL to 9.9 g/dL. The hemodilution process was done before the revascularization. The authors monitored hemodynamic variables, ST-segment deviation, and oxygen consumption before and after each hemodilution.
There was a compensatory increase in cardiac index and oxygen extraction with consequent stable oxygen consumption. These changes were independent of patient age or left ventricular function. In addition, there were no electrocardiographic signs of ischemia.
Licker et al13 studied the hemodynamic effect of preoperative hemodilution in 50 patients with coronary artery disease undergoing coronary artery bypass graft surgery, performing transesophageal echocardiography before and after hemodilution. The patients underwent isovolemic exchange with iso-oncotic starch to target a hematocrit of 28%.
Acute normovolemic hemodilution triggered an increase in cardiac stroke volume, which had a direct correlation with an increase in the central venous pressure and the left ventricular end-diastolic area. No signs of ischemia were seen in these patients on electrocardiography or echocardiography (eg, left ventricular wall-motion abnormalities).
Hemodilution in mitral regurgitation
Spahn et al14 performed acute isovolemic hemodilution with 6% hydroxyethyl starch in 20 patients with mitral regurgitation. The cardiac filling pressures were stable before and after hemodilution; the mean hemoglobin value decreased from 13 to 10.3 g/dL. The cardiac index and oxygen extraction increased proportionally, with stable oxygen consumption; these findings were the same regardless of whether the patient was in normal sinus rhythm or atrial fibrillation.
Effect of hemodilution on cognition
Weiskopf et al15 assessed the effect of anemia on executive and memory function by inducing progressive acute isovolemic anemia in 90 healthy volunteers (age 29 ± 5), reducing their hemoglobin values to 7, 6, and 5 g/dL and performing repetitive neuropsychological and memory testing before and after the hemodilution, as well as after autologous blood transfusion to return their hemoglobin level to 7 g/dL.
There were no changes in reaction time or error rate at a hemoglobin concentration of 7 g/dL compared with the performance at a baseline hemoglobin concentration of 14 g/dL. The volunteers got slower on a mathematics test at hemoglobin levels of 6 g/dL and 5 g/dL, but their error rate did not increase. Immediate and delayed memory were significantly impaired at hemoglobin of 5 g/dL but not at 6 g/dL. All tests normalized with blood transfusion once the hemoglobin level reached 7 g/dL.15
Weiskopf et al16 subsequently investigated whether giving supplemental oxygen to raise the arterial partial pressure of oxygen (Pao2) to 350 mm Hg or greater would overcome the neurocognitive effects of severe acute anemia. They followed a protocol similar to the one in the earlier study15 and induced anemia in 31 healthy volunteers, age 28 ± 4 years, with a mean baseline hemoglobin concentration of 12.7 g/dL.
When the volunteers reached a hemoglobin concentration of 5.7 ± 0.3 g/dL, they were significantly slower on the mathematics test, and their delayed memory was significantly impaired. Then, in a double-blind fashion, they were given either room air or oxygen. Oxygen increased the Pao2 to 406 mm Hg and normalized neurocognitive performance.
Hemodilution studies in surgical patients
A 2015 meta-analysis17 of 63 studies involving 3,819 surgical patients compared the risk of perioperative allogeneic blood transfusion as well as the overall volume of transfused blood in patients undergoing preoperative acute normovolemic hemodilution vs a control group. Though the overall data showed that the patients who underwent acute normovolemic hemodilution needed fewer transfusions and less blood (relative risk [RR] 0.74, 95% confidence interval [CI] 0.63–0.88, P = .0006), the authors noted significant heterogeneity and publication bias.
However, the hemodilution studies paved the way for justifying a more conservative and restrictive transfusion strategy, with a hemoglobin cutoff value of 7 g/dL, and in acute anemia, using oxygen to overcome acute neurocognitive effects while searching for and correcting the cause of the anemia.
STUDIES OF RESTRICTIVE VS LIBERAL TRANSFUSION STRATEGIES
Studies in critical care and high-risk patients
Hébert et al18 randomized 418 critical care patients to a restrictive transfusion approach (in which they were given red blood cells if their hemoglobin concentration dropped below 7.0 g/dL) and 420 patients to a liberal strategy (given red blood cells if their hemoglobin concentration dropped below 10.0 g/dL). Mortality rates (restrictive vs liberal strategy) were as follows:
- Overall at 30 days 18.7% vs 23.3%, P = .11
- In the subgroup with less-severe disease (Acute Physiology and Chronic Health Evaluation II [APACHE II] score < 20), 8.7% vs 16.1%, P = .03
- In the subgroup under age 55, 5.7% vs 13%, P = .02
- In the subgroup with clinically significant cardiac disease, 20.5% vs 22.9%, P = .69
- In the hospital, 22.2% vs 28.1%; P = .05.
This study demonstrated that parsimonious blood use did not worsen clinical outcomes in critical care patients.
Carson et al19 evaluated 2,016 patients age 50 and older who had a history of or risk factors for cardiovascular disease and a baseline hemoglobin level below 10 g/dL who underwent surgery for hip fracture. Patients were randomized to two transfusion strategies based on threshold hemoglobin level: restrictive (< 8 g/dL) or liberal (< 10 g/dL). The primary outcome was death or inability to walk without assistance at 60-day follow-up. The median number of units of blood used was 2 in the liberal group and 0 in the restrictive group.
There was no significant difference in the rates of the primary outcome (odds ratio [OR] 1.01, 95% CI 0.84–1.22), infection, venous thromboembolism, or reoperation. This study demonstrated that a liberal transfusion strategy offered no benefit over a restrictive one.
Rao et al20 analyzed the impact of blood transfusion in 24,112 patients with acute coronary syndromes enrolled in three large trials. Ten percent of the patients received at least 1 blood transfusion during their hospitalization, and they were older and had more complex comorbidity.
At 30 days, the group that had received blood had higher rates of death (adjusted hazard ratio [HR] 3.94, 95% CI 3.26–4.75) and the combined outcome of death or myocardial infarction (HR 2.92, 95% CI 2.55–3.35). Transfusion in patients whose nadir hematocrit was higher than 25% was associated with worse outcomes.
This study suggests being cautious about routinely transfusing blood in stable patients with ischemic heart disease solely on the basis of arbitrary hematocrit levels.
Carson et al,21 however, in a later trial, found a trend toward worse outcomes with a restrictive strategy than with a liberal one. Here, 110 patients with acute coronary syndrome or stable angina undergoing cardiac catheterization were randomized to a target hemoglobin level of either at least 8 mg/dL or at least 10 g/dL. The primary outcome (a composite of death, myocardial infarction, or unscheduled revascularization 30 days after randomization) occurred in 14 patients (25.5%) in the restrictive group and 6 patients (10.9%) in the liberal group (P = .054), and 7 (13.0%) vs 1 (1.8%) of the patients died (P = .032).
Murphy et al22 similarly found trends toward worse outcomes with a restrictive strategy in cardiac patients. The investigators randomized 2,007 elective cardiac surgery patients with a postoperative hemoglobin level lower than 9 g/dL to a hemoglobin transfusion threshold of either 7.5 or 9 g/dL. Outcomes (restrictive vs liberal strategies):
- Transfusion rates 53.4% vs 92.2%
- Rates of the primary outcome (a serious infection [sepsis or wound infection] or ischemic event [stroke, myocardial infarction, mesenteric ischemia, or acute kidney injury] within 3 months):
35.1% vs 33.0%, OR 1.11, 95% CI 0.91–1.34, P = .30) - Mortality rates 4.2% vs 2.6%, HR 1.64, 95% CI 1.00–2.67, P = .045
- Total costs did not differ significantly between the groups.
These studies21,22 suggest the need for more definitive trials in patients with active coronary disease and in cardiac surgery patients.
Holst et al23 randomized 998 intensive care patients in septic shock to hemoglobin thresholds for transfusion of 7 vs 9 g/dL. Mortality rates at 90 days (the primary outcome) were 43.0% vs 45.0%, RR 0.94, 95% CI 0.78–1.09, P = .44.
This study suggests that even in septic shock, a liberal transfusion strategy has no advantage over a parsimonious one.
Active bleeding, especially active gastrointestinal bleeding, poses a significant stress that may trigger empirical transfusion even without evidence of the real hemoglobin level.
Villanueva et al24 randomized 921 patients with severe acute upper-gastrointestinal bleeding to two groups, with hemoglobin transfusion triggers of 7 vs 9 g/dL. The findings were impressive:
- Freedom from transfusion 51% vs 14% (P < .001)
- Survival rates at 6 weeks 95% vs 91% (HR 0.55, 95% CI 0.33–0.92, P = .02)
- Rebleeding 10% vs 16% (P = .01).
Patients with peptic ulcer disease as well as those with cirrhosis stage Child-Pugh class A or B had higher survival rates with a restrictive transfusion strategy.
The RELIEVE trial25 compared the effect of a restrictive transfusion strategy in elderly patients on mechanical ventilation in 6 intensive care units in the United Kingdom. Transfusion triggers were hemoglobin 7 vs 9 g/dL, and the mortality rate at 180 days was 55% vs 37%, RR 0.68, 95% CI 0.44–1.05, P = .073.
Meta-analyses and observational studies
Rohde et al26 performed a systematic review and meta-analysis of 17 trials with 7,456 patients, which revealed that a restrictive strategy is associated with a lower risk of nosocomial infection, including pneumonia, wound infection, and sepsis.
The pooled risk of all serious infections was 10.6% in the restrictive group and 12.7% in the liberal group. Even after adjusting for the use of leukocyte reduction, the risk of infection was lower in the restrictive strategy group (RR 0.83, 95% CI 0.69–0.99). With a hemoglobin threshold of less than 7.0 g/dL, the risk of serious infection was 14% lower. Although this was not statistically significant overall (RR 0.86, 95% CI 0.72–1.02), the difference was statistically significant in the subgroup undergoing orthopedic surgery (RR 0.72, 95% CI 0.53–0.97) and the subgroup presenting with sepsis (RR 0.51, 95% CI 0.28–0.95).
Salpeter et al27 performed a meta-analysis and systematic review of three randomized trials (N = 2,364) comparing a restrictive hemoglobin transfusion trigger (hemoglobin < 7 g/dL) vs a more liberal trigger. The groups with restrictive transfusion triggers had lower rates of:
- In-hospital mortality (RR 0.74, 95% CI 0.60–0.92)
- Total mortality (RR 0.80, 95% CI 0.65–0.98)
- Rebleeding (RR 0.64, 95% CI 0.45–0.90)
- Acute coronary syndrome (RR 0.44, 95% CI 0.22–0.89)
- Pulmonary edema (RR 0.48, 95% CI 0.33–0.72)
- Bacterial infections (RR 0.86, 95% CI 0.73–1.00).
Wang et al28 performed a meta-analysis of 4 randomized controlled trials in patients with upper-gastrointestinal bleeding comparing restrictive (hemoglobin < 7 g/dL) vs liberal transfusion strategies. The primary outcomes were death and rebleeding. The restrictive strategy was associated with:
- A lower mortality rate (OR 0.52, 95% CI 0.31–0.87, P = .01)
- A lower rebleeding rate (OR 0.26, 95% CI 0.03–2.10, P = .21)
- Shorter hospitalizations (P = .009)
- Less blood transfused (P = .0005).
Vincent et al,29 in a prospective observational study of 3,534 patients in intensive care units in 146 facilities in Western Europe, found a correlation between transfusion and mortality. Transfusion was done most often in elderly patients and those with a longer stay in the intensive care unit. The 28-day mortality rate was 22.7% in patients who received a transfusion and 17.1% in those who did not (P = .02). The more units of blood the patients received, the more likely they were to die, and receiving more than 4 units was associated with worse outcomes (P = .01).
Dunne et al30 performed a study of 6,301 noncardiac surgical patients in the Veterans Affairs Maryland Healthcare System from the National Veterans Administration Surgical Quality Improvement Program from 1995 to 2000. Multiple logistic regression analysis revealed that the composite of low hematocrit before and after surgery and high transfusion rates (> 4 units per hospitalization) were associated with higher rates of death (P < .01) and postoperative pneumonia (P ≤ .05) and longer hospitalizations (P < .05). The risk of pneumonia increased proportionally with the decrease in hematocrit.
These findings support pharmacologic optimization of anemia with hematinic supplementation before surgery to decrease the risk of needing a transfusion, often with parenteral iron. The fact that the patient’s hemoglobin can be optimized preoperatively by nontransfusional means may decrease the likelihood of blood transfusion, as the hemoglobin will potentially remain above the transfusion threshold. For example, if a patient has a preoperative hemoglobin level of 10 g/dL, and it is optimized up to 12, then if postoperatively the hemoglobin level drops 3 g/dL instead of reaching the threshold of 7 g/dL, the nadir will be just 9 g/dL, far above that transfusion threshold.
Brunskill et al,31 in a Cochrane review of 6 trials with 2,722 patients undergoing surgery for hip fracture, found no difference in rates of mortality, functional recovery or postoperative morbidity with a restrictive transfusion strategy (hemoglobin target > 8 g/dL vs a liberal one (> 10 g/dL). However, the quality of evidence was rated as low. The authors concluded that there is no justification for liberal red blood cell transfusion thresholds (10 g/dL), and a more restrictive transfusion threshold is preferable.
Weinberg et al32 found that, in trauma patients, receiving more than 6 units of blood was associated with poor prognosis, and outcomes were worse when the blood was older than 2 weeks. However, the effect of blood age is not significant when using smaller transfusion volumes (1 to 2 units of red blood cells).
Studies in sickle cell disease
Sickle cell disease patients have high levels of hemoglobin S, which causes erythrocyte sickling and increases blood viscosity. Transfusion with normal erythrocytes increases the amount of hemoglobin A (the normal variant).33,34
In trials in surgical patients,35,36 conservative strategies for preoperative blood transfusion aiming at a hemoglobin level of 10 g/dL were as effective in preventing postoperative complications as decreasing the hemoglobin S levels to 30% by aggressive exchange transfusion.35
In nonsurgical patients, blood transfusion should be based on formal risk-benefit assessments. Therefore, the expert panel report on sickle cell management advises against blood transfusion in sickle cell patients with uncomplicated vaso-occlusive crises, priapism, asymptomatic anemia, or acute kidney injury in the absence of multisystem organ failure.34
Is hemoglobin the most relevant marker?
Most studies that compared restrictive and liberal transfusion strategies focused on using a lower hemoglobin threshold as the transfusion trigger, not on using fewer units of blood. Is the amount of blood transfused more important than the hemoglobin threshold? Perhaps a study focused both on a restrictive vs liberal strategy and also on the minimum amount of blood that each patient may benefit from would help to answer this question.
We should beware of routinely using the hemoglobin concentration as a threshold for transfusion and a surrogate marker of transfusion benefit because changes in hemoglobin concentration may not reflect changes in absolute red cell mass.37 Changes in plasma volume (an increase or decrease) affect the hematocrit concentration without necessarily affecting the total red cell mass. Unfortunately, red cell mass is very difficult to measure; hence, the hemoglobin and hematocrit values are used instead. Studies addressing changes in red cell mass may be needed, perhaps even to validate using the hemoglobin concentration as the sole indicator for transfusion.
Is fresh blood better than old blood?
Using blood that is more than 14 days old may be associated with poor outcomes, for several possible reasons. Red blood cells age rapidly in refrigeration, and usually just 75% may remain viable 24 hours after phlebotomy. Adenosine triphosphate and 2,3-DPG levels steadily decrease, with a consequent decrease in capacity for appropriate tissue oxygen delivery. In addition, loss of membrane phospholipids causes progressive rigidity of the red cell membrane with consequent formation of echynocytes after 14 to 21 days.38,39
The use of blood more than 14 days old in cardiac surgery patients has been associated with worse outcomes, including higher rates of death, prolonged intubation, acute renal failure, and sepsis.40 Similar poor outcomes have been seen in trauma patients.32
Lacroix et al,41 in a multicenter, randomized trial in critically ill adults, compared the outcomes of transfusion of fresh packed red cells (stored < 8 days) or old blood (stored for a mean of 22 days). The primary outcome was the mortality rate at 90 days: 37.0% in the fresh-blood group vs 35.3% in the old-blood group (HR 1.1, 95% CI 0.9–1.2, P = .38).
The authors concluded that using fresh blood compared with old blood was not associated with a lower 90-day mortality rate in critically ill adults.
RISKS ASSOCIATED WITH TRANSFUSION
Infections
The risk of infection from blood transfusion is small. Human immunodeficiency virus (HIV) is transmitted in 1 in 1.5 million transfused blood components, and hepatitis C virus in 1 in 1.1 million; these odds are similar to those of having a fatal airplane accident (1 in 1.7 million per flight). Hepatitis B virus infection is more common, the reported incidence being 1 in 357,000.42
Noninfectious complications
Transfusion-associated circulatory overload occurs in 4% to 6% of patients who receive a transfusion. Therefore, circulatory overload is a greater danger from transfusion than infection is.42
Febrile nonhemolytic transfusion reactions occur in 1.1% of patients with prestorage leukoreduction.
Transfusion-associated acute lung injury occurs in 0.8 per 10,000 blood components transfused.
Errors associated with blood transfusion include, in decreasing order of frequency, transfusion of the wrong blood component, handling and storage errors, inappropriate administration of anti-D immunoglobulin, and avoidable, delayed, or insufficient transfusions.43
Surgery and condition-specific complications of red blood cell transfusion
Cardiovascular surgery. Transfusion is associated with a higher risk of postoperative stroke, respiratory failure, acute respiratory distress syndrome, prolonged intubation time, reintubation, in-hospital death, sepsis, and longer postoperative length of stay.44
Malignancy. The use of blood in this setting has been found to be an independent predictor of recurrence, decreased survival, and increased risk of lymphoplasmacytic and marginal-zone lymphomas.44–47
Vascular, orthopedic, and other surgeries. Transfusion is associated with a higher risk of death, thromboembolic events, acute kidney injury, death, composite morbidity, reoperation, sepsis, and pulmonary complications.44
ST-segment elevation myocardial infarction, sepsis, and intensive care unit admissions. Transfusion is associated with an increased risk of rebleeding, death, and secondary infections.44
COST OF RED BLOOD CELL TRANSFUSION
Up to 85 million units of red blood cells are transfused per year worldwide, 15 million of them in the United States.42 At our hospital in 2013, 1 unit of leukocyte-reduced red blood cells cost $957.27, which included the costs of acquisition, processing, banking, patient testing, administration, and monitoring.
The Premier Healthcare Alliance48 analyzed data from 7.4 million discharges from 464 hospitals between April 2011 and March 2012. Blood use varied significantly among hospitals, and the hospitals in the lowest quartile of blood use had better patient outcomes. If all the hospitals used as little blood as those in the lowest quartile and had outcomes as good, blood product use would be reduced by 802,716 units, with savings of up to $165 million annually.
In addition to the economic cost of blood transfusion, the clinician must be aware of the cost in terms of comorbidities caused by unnecessary blood transfusion.49,50
RECOMMENDATIONS FROM THE AABB
In view of all the current compelling evidence, a restrictive approach to transfusion is the single best strategy to minimize adverse outcomes.51 Below, we outline the current recommendations from the AABB (formerly the American Association of Blood Banks),42 which are similar to the national clinical guideline on blood transfusion in the United Kingdom,52 and have recently been updated, confirming the initial recommendations.53
In critical care patients, transfusion should be considered if the hemoglobin concentration is 7 g/dL or less.
In postoperative patients and hospitalized patients with preexisting cardiovascular disease, transfusion should be considered if the hemoglobin concentration is 8 g/dL or less or if the patient has signs or symptoms of anemia such as chest pain, orthostatic hypotension, or tachycardia unresponsive to fluid resuscitation, or heart failure.
In hemodynamically stable patients with acute coronary syndrome, there is not enough evidence to allow a formal recommendation for or against a liberal or restrictive transfusion threshold.
Consider both the hemoglobin concentration and the symptoms when deciding whether to give a transfusion. This recommendation is shared by a National Institutes of Health consensus conference,54 which indicates that multiple factors related to the patient’s clinical status and oxygen delivery should be considered before deciding to transfuse red blood cells.
The Society of Hospital Medicine55 and the American Society of Hematology56 concur with a parsimonious approach to blood use in their Choosing Wisely campaigns. The American Society of Hematology recommends that if transfusion of red blood cells is necessary, the minimum number of units should be given that relieve the symptoms of anemia or achieve a safe hemoglobin range (7–8 g/dL in stable noncardiac inpatients).57
New electronic tools can monitor the ordering and use of blood products in real time and can identify the hemoglobin level used as the trigger for transfusion. They also provide data on blood use by physician, hospital, and department. These tools can reveal current practice at a glance and allow sharing of best practices among peers and institutions.52
CONSIDER TRANSFUSION FOR HEMOGLOBIN BELOW 7 G/DL
The routine use of blood has come under scrutiny, given its association with increased healthcare costs and morbidity. The accepted practice in stable medical patients is a restrictive threshold approach for blood transfusion, which is to consider (not necessarily give) a single unit of packed red blood cells for a hemoglobin less than 7 g/dL.
However, studies in acute coronary syndrome patients and postoperative cardiac surgery patients have not shown the restrictive threshold to be superior to a liberal threshold in terms of outcomes and costs. This variability suggests the need for further studies to determine the best course of action in different patient subpopulations (eg, surgical, oncologic, trauma, critical illness).
Also, a limitation of most of the clinical studies was that only the hemoglobin concentration was used as a marker of anemia, with no strict assessment of changes in red cell mass with transfusion.
Despite the variability in certain populations, the overall weight of current evidence favors a restrictive approach to blood transfusion (hemoglobin < 7 g/dL), although perhaps in patients who have active coronary disease or are undergoing cardiac surgery, a more lenient threshold (< 8 g/dL) for transfusion should be considered.
For decades, physicians believed in the benefit of prompt transfusion of blood to keep the hemoglobin level at arbitrary, optimum levels, ie, close to normal values, especially in the critically ill, the elderly, and those with coronary syndromes, stroke, or renal failure.
However, the evidence supporting arbitrary hemoglobin values as an indication for transfusion was weak or nonexistent. Also, blood transfusion can have complications and adverse effects, and blood is costly and scarce. These considerations prompted research into when blood transfusion should be considered, and recommendations that it should be used more sparingly than in the past.
This review offers a perspective on the evidence supporting restrictive blood use. First, we focus on hemodilution studies that demonstrated that humans can tolerate anemia. Then, we look at studies that compared a restrictive transfusion strategy with a liberal one in patients with critical illness and active bleeding. We conclude with current recommendations for blood transfusion.
EVIDENCE FROM HEMODILUTION STUDIES
Hemoglobin is essential for tissue oxygenation, but the serum hemoglobin concentration is just one of several factors involved.1–5 In anemia, the body can adapt not only by increasing production of red blood cells, but also by:
- Increasing cardiac output
- Increasing synthesis of 2,3-diphosphoglycerate (2,3-DPG), with a consequent shift in the oxyhemoglobin dissociation curve to the right, allowing enhanced release of oxygen at the tissue level
- Moving more carbon dioxide into the blood (the Bohr effect), which decreases pH and also shifts the dissociation curve to the right.
Just 20 years ago, physicians were using arbitrary cutoffs such as hemoglobin 10 g/dL or hematocrit 30% as indications for blood transfusion, without reasonable evidence to support these values. Not until acute normovolemic hemodilution studies were performed were we able to progressively appraise how well patients could tolerate lower levels of hemoglobin without significant adverse outcomes.
Acute normovolemic hemodilution involves withdrawing blood and replacing it with crystalloid or colloid solution to maintain the volume.6
Initial studies were done in animals and focused on the safety of acute anemia regarding splanchnic perfusion. Subsequently, studies proved that healthy, elderly, and stable cardiac patients can tolerate acute anemia with normal cardiovascular response. The targets in these studies were modest at first, but researchers aimed progressively for more aggressive hemodilution with lower hemoglobin targets and demonstrated that the body can tolerate and adapt to more severe anemia.6–8
Studies in healthy patients
Weiskopf et al9 assessed the effect of severe anemia in 32 conscious healthy patients (11 presurgical patients and 21 volunteers not undergoing surgery) by performing acute normovolemic hemodilution with 5% human albumin, autologous plasma, or both, with a target hemoglobin level of 5 g/dL. The process was done gradually, obtaining aliquots of blood of 500 to 900 mL. Cardiac index increased, along with a mild increase in oxygen consumption with no increase in plasma lactate levels, suggesting that in conscious healthy patients, tissue oxygenation remains adequate even in severe anemia.
Leung et al10 addressed the electrocardiographic changes that occur with severe anemia (hemoglobin 5 g/dL) in 55 healthy volunteers. Three developed transient, reversible ST-segment depression, which was associated with a higher heart rate than in the volunteers with no electrocardiographic changes; however, the changes were reversible and asymptomatic, and thus were considered physiologic and benign.
Hemodilution in healthy elderly patients
Spahn et al11 performed 6 and 12 mL/kg isovolemic exchange of blood for 6% hydroxyethyl starch in 20 patients older than 65 years (mean age 76, range 65–88) without underlying coronary disease.
The patients’ mean hemoglobin level decreased from 11.6 g/dL to 8.8 g/dL. Their cardiac index and oxygen extraction values increased adequately, with stable oxygen consumption during hemodilution. There were no electrocardiographic signs of ischemia.
Hemodilution in coronary artery disease
Spahn et al12 performed hemodilution studies in 60 patients (ages 35–81) with coronary artery disease managed chronically with beta-blockers who were scheduled for coronary artery bypass graft surgery. Hemodilution was performed with 6- and 12-mL/kg isovolemic exchange of blood for 6% hydroxyethyl starch maintaining normovolemia and stable filling pressures. Hemoglobin levels decreased from 12.6 g/dL to 9.9 g/dL. The hemodilution process was done before the revascularization. The authors monitored hemodynamic variables, ST-segment deviation, and oxygen consumption before and after each hemodilution.
There was a compensatory increase in cardiac index and oxygen extraction with consequent stable oxygen consumption. These changes were independent of patient age or left ventricular function. In addition, there were no electrocardiographic signs of ischemia.
Licker et al13 studied the hemodynamic effect of preoperative hemodilution in 50 patients with coronary artery disease undergoing coronary artery bypass graft surgery, performing transesophageal echocardiography before and after hemodilution. The patients underwent isovolemic exchange with iso-oncotic starch to target a hematocrit of 28%.
Acute normovolemic hemodilution triggered an increase in cardiac stroke volume, which had a direct correlation with an increase in the central venous pressure and the left ventricular end-diastolic area. No signs of ischemia were seen in these patients on electrocardiography or echocardiography (eg, left ventricular wall-motion abnormalities).
Hemodilution in mitral regurgitation
Spahn et al14 performed acute isovolemic hemodilution with 6% hydroxyethyl starch in 20 patients with mitral regurgitation. The cardiac filling pressures were stable before and after hemodilution; the mean hemoglobin value decreased from 13 to 10.3 g/dL. The cardiac index and oxygen extraction increased proportionally, with stable oxygen consumption; these findings were the same regardless of whether the patient was in normal sinus rhythm or atrial fibrillation.
Effect of hemodilution on cognition
Weiskopf et al15 assessed the effect of anemia on executive and memory function by inducing progressive acute isovolemic anemia in 90 healthy volunteers (age 29 ± 5), reducing their hemoglobin values to 7, 6, and 5 g/dL and performing repetitive neuropsychological and memory testing before and after the hemodilution, as well as after autologous blood transfusion to return their hemoglobin level to 7 g/dL.
There were no changes in reaction time or error rate at a hemoglobin concentration of 7 g/dL compared with the performance at a baseline hemoglobin concentration of 14 g/dL. The volunteers got slower on a mathematics test at hemoglobin levels of 6 g/dL and 5 g/dL, but their error rate did not increase. Immediate and delayed memory were significantly impaired at hemoglobin of 5 g/dL but not at 6 g/dL. All tests normalized with blood transfusion once the hemoglobin level reached 7 g/dL.15
Weiskopf et al16 subsequently investigated whether giving supplemental oxygen to raise the arterial partial pressure of oxygen (Pao2) to 350 mm Hg or greater would overcome the neurocognitive effects of severe acute anemia. They followed a protocol similar to the one in the earlier study15 and induced anemia in 31 healthy volunteers, age 28 ± 4 years, with a mean baseline hemoglobin concentration of 12.7 g/dL.
When the volunteers reached a hemoglobin concentration of 5.7 ± 0.3 g/dL, they were significantly slower on the mathematics test, and their delayed memory was significantly impaired. Then, in a double-blind fashion, they were given either room air or oxygen. Oxygen increased the Pao2 to 406 mm Hg and normalized neurocognitive performance.
Hemodilution studies in surgical patients
A 2015 meta-analysis17 of 63 studies involving 3,819 surgical patients compared the risk of perioperative allogeneic blood transfusion as well as the overall volume of transfused blood in patients undergoing preoperative acute normovolemic hemodilution vs a control group. Though the overall data showed that the patients who underwent acute normovolemic hemodilution needed fewer transfusions and less blood (relative risk [RR] 0.74, 95% confidence interval [CI] 0.63–0.88, P = .0006), the authors noted significant heterogeneity and publication bias.
However, the hemodilution studies paved the way for justifying a more conservative and restrictive transfusion strategy, with a hemoglobin cutoff value of 7 g/dL, and in acute anemia, using oxygen to overcome acute neurocognitive effects while searching for and correcting the cause of the anemia.
STUDIES OF RESTRICTIVE VS LIBERAL TRANSFUSION STRATEGIES
Studies in critical care and high-risk patients
Hébert et al18 randomized 418 critical care patients to a restrictive transfusion approach (in which they were given red blood cells if their hemoglobin concentration dropped below 7.0 g/dL) and 420 patients to a liberal strategy (given red blood cells if their hemoglobin concentration dropped below 10.0 g/dL). Mortality rates (restrictive vs liberal strategy) were as follows:
- Overall at 30 days 18.7% vs 23.3%, P = .11
- In the subgroup with less-severe disease (Acute Physiology and Chronic Health Evaluation II [APACHE II] score < 20), 8.7% vs 16.1%, P = .03
- In the subgroup under age 55, 5.7% vs 13%, P = .02
- In the subgroup with clinically significant cardiac disease, 20.5% vs 22.9%, P = .69
- In the hospital, 22.2% vs 28.1%; P = .05.
This study demonstrated that parsimonious blood use did not worsen clinical outcomes in critical care patients.
Carson et al19 evaluated 2,016 patients age 50 and older who had a history of or risk factors for cardiovascular disease and a baseline hemoglobin level below 10 g/dL who underwent surgery for hip fracture. Patients were randomized to two transfusion strategies based on threshold hemoglobin level: restrictive (< 8 g/dL) or liberal (< 10 g/dL). The primary outcome was death or inability to walk without assistance at 60-day follow-up. The median number of units of blood used was 2 in the liberal group and 0 in the restrictive group.
There was no significant difference in the rates of the primary outcome (odds ratio [OR] 1.01, 95% CI 0.84–1.22), infection, venous thromboembolism, or reoperation. This study demonstrated that a liberal transfusion strategy offered no benefit over a restrictive one.
Rao et al20 analyzed the impact of blood transfusion in 24,112 patients with acute coronary syndromes enrolled in three large trials. Ten percent of the patients received at least 1 blood transfusion during their hospitalization, and they were older and had more complex comorbidity.
At 30 days, the group that had received blood had higher rates of death (adjusted hazard ratio [HR] 3.94, 95% CI 3.26–4.75) and the combined outcome of death or myocardial infarction (HR 2.92, 95% CI 2.55–3.35). Transfusion in patients whose nadir hematocrit was higher than 25% was associated with worse outcomes.
This study suggests being cautious about routinely transfusing blood in stable patients with ischemic heart disease solely on the basis of arbitrary hematocrit levels.
Carson et al,21 however, in a later trial, found a trend toward worse outcomes with a restrictive strategy than with a liberal one. Here, 110 patients with acute coronary syndrome or stable angina undergoing cardiac catheterization were randomized to a target hemoglobin level of either at least 8 mg/dL or at least 10 g/dL. The primary outcome (a composite of death, myocardial infarction, or unscheduled revascularization 30 days after randomization) occurred in 14 patients (25.5%) in the restrictive group and 6 patients (10.9%) in the liberal group (P = .054), and 7 (13.0%) vs 1 (1.8%) of the patients died (P = .032).
Murphy et al22 similarly found trends toward worse outcomes with a restrictive strategy in cardiac patients. The investigators randomized 2,007 elective cardiac surgery patients with a postoperative hemoglobin level lower than 9 g/dL to a hemoglobin transfusion threshold of either 7.5 or 9 g/dL. Outcomes (restrictive vs liberal strategies):
- Transfusion rates 53.4% vs 92.2%
- Rates of the primary outcome (a serious infection [sepsis or wound infection] or ischemic event [stroke, myocardial infarction, mesenteric ischemia, or acute kidney injury] within 3 months):
35.1% vs 33.0%, OR 1.11, 95% CI 0.91–1.34, P = .30) - Mortality rates 4.2% vs 2.6%, HR 1.64, 95% CI 1.00–2.67, P = .045
- Total costs did not differ significantly between the groups.
These studies21,22 suggest the need for more definitive trials in patients with active coronary disease and in cardiac surgery patients.
Holst et al23 randomized 998 intensive care patients in septic shock to hemoglobin thresholds for transfusion of 7 vs 9 g/dL. Mortality rates at 90 days (the primary outcome) were 43.0% vs 45.0%, RR 0.94, 95% CI 0.78–1.09, P = .44.
This study suggests that even in septic shock, a liberal transfusion strategy has no advantage over a parsimonious one.
Active bleeding, especially active gastrointestinal bleeding, poses a significant stress that may trigger empirical transfusion even without evidence of the real hemoglobin level.
Villanueva et al24 randomized 921 patients with severe acute upper-gastrointestinal bleeding to two groups, with hemoglobin transfusion triggers of 7 vs 9 g/dL. The findings were impressive:
- Freedom from transfusion 51% vs 14% (P < .001)
- Survival rates at 6 weeks 95% vs 91% (HR 0.55, 95% CI 0.33–0.92, P = .02)
- Rebleeding 10% vs 16% (P = .01).
Patients with peptic ulcer disease as well as those with cirrhosis stage Child-Pugh class A or B had higher survival rates with a restrictive transfusion strategy.
The RELIEVE trial25 compared the effect of a restrictive transfusion strategy in elderly patients on mechanical ventilation in 6 intensive care units in the United Kingdom. Transfusion triggers were hemoglobin 7 vs 9 g/dL, and the mortality rate at 180 days was 55% vs 37%, RR 0.68, 95% CI 0.44–1.05, P = .073.
Meta-analyses and observational studies
Rohde et al26 performed a systematic review and meta-analysis of 17 trials with 7,456 patients, which revealed that a restrictive strategy is associated with a lower risk of nosocomial infection, including pneumonia, wound infection, and sepsis.
The pooled risk of all serious infections was 10.6% in the restrictive group and 12.7% in the liberal group. Even after adjusting for the use of leukocyte reduction, the risk of infection was lower in the restrictive strategy group (RR 0.83, 95% CI 0.69–0.99). With a hemoglobin threshold of less than 7.0 g/dL, the risk of serious infection was 14% lower. Although this was not statistically significant overall (RR 0.86, 95% CI 0.72–1.02), the difference was statistically significant in the subgroup undergoing orthopedic surgery (RR 0.72, 95% CI 0.53–0.97) and the subgroup presenting with sepsis (RR 0.51, 95% CI 0.28–0.95).
Salpeter et al27 performed a meta-analysis and systematic review of three randomized trials (N = 2,364) comparing a restrictive hemoglobin transfusion trigger (hemoglobin < 7 g/dL) vs a more liberal trigger. The groups with restrictive transfusion triggers had lower rates of:
- In-hospital mortality (RR 0.74, 95% CI 0.60–0.92)
- Total mortality (RR 0.80, 95% CI 0.65–0.98)
- Rebleeding (RR 0.64, 95% CI 0.45–0.90)
- Acute coronary syndrome (RR 0.44, 95% CI 0.22–0.89)
- Pulmonary edema (RR 0.48, 95% CI 0.33–0.72)
- Bacterial infections (RR 0.86, 95% CI 0.73–1.00).
Wang et al28 performed a meta-analysis of 4 randomized controlled trials in patients with upper-gastrointestinal bleeding comparing restrictive (hemoglobin < 7 g/dL) vs liberal transfusion strategies. The primary outcomes were death and rebleeding. The restrictive strategy was associated with:
- A lower mortality rate (OR 0.52, 95% CI 0.31–0.87, P = .01)
- A lower rebleeding rate (OR 0.26, 95% CI 0.03–2.10, P = .21)
- Shorter hospitalizations (P = .009)
- Less blood transfused (P = .0005).
Vincent et al,29 in a prospective observational study of 3,534 patients in intensive care units in 146 facilities in Western Europe, found a correlation between transfusion and mortality. Transfusion was done most often in elderly patients and those with a longer stay in the intensive care unit. The 28-day mortality rate was 22.7% in patients who received a transfusion and 17.1% in those who did not (P = .02). The more units of blood the patients received, the more likely they were to die, and receiving more than 4 units was associated with worse outcomes (P = .01).
Dunne et al30 performed a study of 6,301 noncardiac surgical patients in the Veterans Affairs Maryland Healthcare System from the National Veterans Administration Surgical Quality Improvement Program from 1995 to 2000. Multiple logistic regression analysis revealed that the composite of low hematocrit before and after surgery and high transfusion rates (> 4 units per hospitalization) were associated with higher rates of death (P < .01) and postoperative pneumonia (P ≤ .05) and longer hospitalizations (P < .05). The risk of pneumonia increased proportionally with the decrease in hematocrit.
These findings support pharmacologic optimization of anemia with hematinic supplementation before surgery to decrease the risk of needing a transfusion, often with parenteral iron. The fact that the patient’s hemoglobin can be optimized preoperatively by nontransfusional means may decrease the likelihood of blood transfusion, as the hemoglobin will potentially remain above the transfusion threshold. For example, if a patient has a preoperative hemoglobin level of 10 g/dL, and it is optimized up to 12, then if postoperatively the hemoglobin level drops 3 g/dL instead of reaching the threshold of 7 g/dL, the nadir will be just 9 g/dL, far above that transfusion threshold.
Brunskill et al,31 in a Cochrane review of 6 trials with 2,722 patients undergoing surgery for hip fracture, found no difference in rates of mortality, functional recovery or postoperative morbidity with a restrictive transfusion strategy (hemoglobin target > 8 g/dL vs a liberal one (> 10 g/dL). However, the quality of evidence was rated as low. The authors concluded that there is no justification for liberal red blood cell transfusion thresholds (10 g/dL), and a more restrictive transfusion threshold is preferable.
Weinberg et al32 found that, in trauma patients, receiving more than 6 units of blood was associated with poor prognosis, and outcomes were worse when the blood was older than 2 weeks. However, the effect of blood age is not significant when using smaller transfusion volumes (1 to 2 units of red blood cells).
Studies in sickle cell disease
Sickle cell disease patients have high levels of hemoglobin S, which causes erythrocyte sickling and increases blood viscosity. Transfusion with normal erythrocytes increases the amount of hemoglobin A (the normal variant).33,34
In trials in surgical patients,35,36 conservative strategies for preoperative blood transfusion aiming at a hemoglobin level of 10 g/dL were as effective in preventing postoperative complications as decreasing the hemoglobin S levels to 30% by aggressive exchange transfusion.35
In nonsurgical patients, blood transfusion should be based on formal risk-benefit assessments. Therefore, the expert panel report on sickle cell management advises against blood transfusion in sickle cell patients with uncomplicated vaso-occlusive crises, priapism, asymptomatic anemia, or acute kidney injury in the absence of multisystem organ failure.34
Is hemoglobin the most relevant marker?
Most studies that compared restrictive and liberal transfusion strategies focused on using a lower hemoglobin threshold as the transfusion trigger, not on using fewer units of blood. Is the amount of blood transfused more important than the hemoglobin threshold? Perhaps a study focused both on a restrictive vs liberal strategy and also on the minimum amount of blood that each patient may benefit from would help to answer this question.
We should beware of routinely using the hemoglobin concentration as a threshold for transfusion and a surrogate marker of transfusion benefit because changes in hemoglobin concentration may not reflect changes in absolute red cell mass.37 Changes in plasma volume (an increase or decrease) affect the hematocrit concentration without necessarily affecting the total red cell mass. Unfortunately, red cell mass is very difficult to measure; hence, the hemoglobin and hematocrit values are used instead. Studies addressing changes in red cell mass may be needed, perhaps even to validate using the hemoglobin concentration as the sole indicator for transfusion.
Is fresh blood better than old blood?
Using blood that is more than 14 days old may be associated with poor outcomes, for several possible reasons. Red blood cells age rapidly in refrigeration, and usually just 75% may remain viable 24 hours after phlebotomy. Adenosine triphosphate and 2,3-DPG levels steadily decrease, with a consequent decrease in capacity for appropriate tissue oxygen delivery. In addition, loss of membrane phospholipids causes progressive rigidity of the red cell membrane with consequent formation of echynocytes after 14 to 21 days.38,39
The use of blood more than 14 days old in cardiac surgery patients has been associated with worse outcomes, including higher rates of death, prolonged intubation, acute renal failure, and sepsis.40 Similar poor outcomes have been seen in trauma patients.32
Lacroix et al,41 in a multicenter, randomized trial in critically ill adults, compared the outcomes of transfusion of fresh packed red cells (stored < 8 days) or old blood (stored for a mean of 22 days). The primary outcome was the mortality rate at 90 days: 37.0% in the fresh-blood group vs 35.3% in the old-blood group (HR 1.1, 95% CI 0.9–1.2, P = .38).
The authors concluded that using fresh blood compared with old blood was not associated with a lower 90-day mortality rate in critically ill adults.
RISKS ASSOCIATED WITH TRANSFUSION
Infections
The risk of infection from blood transfusion is small. Human immunodeficiency virus (HIV) is transmitted in 1 in 1.5 million transfused blood components, and hepatitis C virus in 1 in 1.1 million; these odds are similar to those of having a fatal airplane accident (1 in 1.7 million per flight). Hepatitis B virus infection is more common, the reported incidence being 1 in 357,000.42
Noninfectious complications
Transfusion-associated circulatory overload occurs in 4% to 6% of patients who receive a transfusion. Therefore, circulatory overload is a greater danger from transfusion than infection is.42
Febrile nonhemolytic transfusion reactions occur in 1.1% of patients with prestorage leukoreduction.
Transfusion-associated acute lung injury occurs in 0.8 per 10,000 blood components transfused.
Errors associated with blood transfusion include, in decreasing order of frequency, transfusion of the wrong blood component, handling and storage errors, inappropriate administration of anti-D immunoglobulin, and avoidable, delayed, or insufficient transfusions.43
Surgery and condition-specific complications of red blood cell transfusion
Cardiovascular surgery. Transfusion is associated with a higher risk of postoperative stroke, respiratory failure, acute respiratory distress syndrome, prolonged intubation time, reintubation, in-hospital death, sepsis, and longer postoperative length of stay.44
Malignancy. The use of blood in this setting has been found to be an independent predictor of recurrence, decreased survival, and increased risk of lymphoplasmacytic and marginal-zone lymphomas.44–47
Vascular, orthopedic, and other surgeries. Transfusion is associated with a higher risk of death, thromboembolic events, acute kidney injury, death, composite morbidity, reoperation, sepsis, and pulmonary complications.44
ST-segment elevation myocardial infarction, sepsis, and intensive care unit admissions. Transfusion is associated with an increased risk of rebleeding, death, and secondary infections.44
COST OF RED BLOOD CELL TRANSFUSION
Up to 85 million units of red blood cells are transfused per year worldwide, 15 million of them in the United States.42 At our hospital in 2013, 1 unit of leukocyte-reduced red blood cells cost $957.27, which included the costs of acquisition, processing, banking, patient testing, administration, and monitoring.
The Premier Healthcare Alliance48 analyzed data from 7.4 million discharges from 464 hospitals between April 2011 and March 2012. Blood use varied significantly among hospitals, and the hospitals in the lowest quartile of blood use had better patient outcomes. If all the hospitals used as little blood as those in the lowest quartile and had outcomes as good, blood product use would be reduced by 802,716 units, with savings of up to $165 million annually.
In addition to the economic cost of blood transfusion, the clinician must be aware of the cost in terms of comorbidities caused by unnecessary blood transfusion.49,50
RECOMMENDATIONS FROM THE AABB
In view of all the current compelling evidence, a restrictive approach to transfusion is the single best strategy to minimize adverse outcomes.51 Below, we outline the current recommendations from the AABB (formerly the American Association of Blood Banks),42 which are similar to the national clinical guideline on blood transfusion in the United Kingdom,52 and have recently been updated, confirming the initial recommendations.53
In critical care patients, transfusion should be considered if the hemoglobin concentration is 7 g/dL or less.
In postoperative patients and hospitalized patients with preexisting cardiovascular disease, transfusion should be considered if the hemoglobin concentration is 8 g/dL or less or if the patient has signs or symptoms of anemia such as chest pain, orthostatic hypotension, or tachycardia unresponsive to fluid resuscitation, or heart failure.
In hemodynamically stable patients with acute coronary syndrome, there is not enough evidence to allow a formal recommendation for or against a liberal or restrictive transfusion threshold.
Consider both the hemoglobin concentration and the symptoms when deciding whether to give a transfusion. This recommendation is shared by a National Institutes of Health consensus conference,54 which indicates that multiple factors related to the patient’s clinical status and oxygen delivery should be considered before deciding to transfuse red blood cells.
The Society of Hospital Medicine55 and the American Society of Hematology56 concur with a parsimonious approach to blood use in their Choosing Wisely campaigns. The American Society of Hematology recommends that if transfusion of red blood cells is necessary, the minimum number of units should be given that relieve the symptoms of anemia or achieve a safe hemoglobin range (7–8 g/dL in stable noncardiac inpatients).57
New electronic tools can monitor the ordering and use of blood products in real time and can identify the hemoglobin level used as the trigger for transfusion. They also provide data on blood use by physician, hospital, and department. These tools can reveal current practice at a glance and allow sharing of best practices among peers and institutions.52
CONSIDER TRANSFUSION FOR HEMOGLOBIN BELOW 7 G/DL
The routine use of blood has come under scrutiny, given its association with increased healthcare costs and morbidity. The accepted practice in stable medical patients is a restrictive threshold approach for blood transfusion, which is to consider (not necessarily give) a single unit of packed red blood cells for a hemoglobin less than 7 g/dL.
However, studies in acute coronary syndrome patients and postoperative cardiac surgery patients have not shown the restrictive threshold to be superior to a liberal threshold in terms of outcomes and costs. This variability suggests the need for further studies to determine the best course of action in different patient subpopulations (eg, surgical, oncologic, trauma, critical illness).
Also, a limitation of most of the clinical studies was that only the hemoglobin concentration was used as a marker of anemia, with no strict assessment of changes in red cell mass with transfusion.
Despite the variability in certain populations, the overall weight of current evidence favors a restrictive approach to blood transfusion (hemoglobin < 7 g/dL), although perhaps in patients who have active coronary disease or are undergoing cardiac surgery, a more lenient threshold (< 8 g/dL) for transfusion should be considered.
- Shander A, Gross I, Hill S, Javidroozi M, Sledge S; College of American Pathologists; American Society of Anesthesiologists; Society of Thoracic Surgeons and Society of Cardiovascular Anesthesiologists; Society of Critical Care Medicine; Italian Society of Transfusion Medicine and Immunohaematology; American Association of Blood Banks. A new perspective on best transfusion practices. Blood Transfus 2013; 11:193–202.
- Madjdpour C, Spahn DR. Allogeneic red blood cell transfusion: physiology of oxygen transport. Best Pract Res Clin Anaesthesiol 2007; 21:163–171.
- Tánczos K, Molnár Z. The oxygen supply-demand balance: a monitoring challenge. Best Pract Res Clin Anaesthesiol 2013; 27:201–207.
- Hebert PC, Van der Linden P, Biro G, Hu LQ. Physiologic aspects of anemia. Crit Care Clin 2004; 20:187–212.
- Spinelli E, Bartlett RH. Anemia and transfusion in critical care: physiology and management. J Intensive Care Med 2016; 31:295–306.
- Jamnicki M, Kocian R, Van Der Linden P, Zaugg M, Spahn DR. Acute normovolemic hemodilution: physiology, limitations, and clinical use. J Cardiothorac Vasc Anesth 2003; 17:747–754.
- Monk TG. Acute normovolemic hemodilution. Anesthesiol Clin North America 2005; 23:271–281.
- Shander A, Rijhwani TS. Acute normovolemic hemodilution. Transfusion 2004; 44(suppl 2):26S–34S.
- Weiskopf RB, Viele MK, Feiner J, et al. Human cardiovascular and metabolic response to acute, severe isovolemic anemia. JAMA 1998; 279:217–221.
- Leung JM, Weiskopf RB, Feiner J, et al. Electrocardiographic ST-segment changes during acute, severe isovolemic hemodilution in humans. Anesthesiology 2000; 93:1004–1010.
- Spahn DR, Zollinger A, Schlumpf RB, et al. Hemodilution tolerance in elderly patients without known cardiac disease. Anesth Analg 1996; 82:681–686.
- Spahn DR, Schmid ER, Seifert B, Pasch T. Hemodilution tolerance in patients with coronary artery disease who are receiving chronic beta-adrenergic blocker therapy. Anesth Analg 1996; 82:687–694.
- Licker M, Ellenberger C, Sierra J, Christenson J, Diaper J, Morel D. Cardiovascular response to acute normovolemic hemodilution in patients with coronary artery diseases: assessment with transesophageal echocardiography. Crit Care Med 2005; 33:591–597.
- Spahn DR, Seifert B, Pasch T, Schmid ER. Haemodilution tolerance in patients with mitral regurgitation. Anaesthesia 1998; 53:20–24.
- Weiskopf RB, Kramer JH, Viele M, et al. Acute severe isovolemic anemia impairs cognitive function and memory in humans. Anesthesiology 2000; 92:1646–1652.
- Weiskopf RB, Feiner J, Hopf HW, et al. Oxygen reverses deficits of cognitive function and memory and increased heart rate induced by acute severe isovolemic anemia. Anesthesiology 2002; 96:871–877.
- Zhou X, Zhang C, Wang Y, Yu L, Yan M. Preoperative acute normovolemic hemodilution for minimizing allogeneic blood transfusion: a meta-analysis. Anesth Analg 2015; 121:1443–1455.
- Hébert P, Wells G, Blajchman M, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999: 340:409–417.
- Carson JL, Terrin ML, Noveck H, et al; FOCUS Investigators. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med 2011; 365:2453–2462.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
- Carson JL, Brooks MM, Abbott JD, et al. Liberal versus restrictive transfusion thresholds for patients with symptomatic coronary artery disease. Am Heart J 2013; 165:964.e1–971.e1.
- Murphy GJ, Pike K, Rogers CA, et al; TITRe2 Investigators. Liberal or restrictive transfusion after cardiac surgery. N Engl J Med 2015; 372:997–1008.
- Holst LB, Haase N, Wetterslev J, et al; TRISS Trial Group; Scandinavian Critical Care Trials Group. Lower versus higher hemoglobin threshold for transfusion in septic shock. N Engl J Med 2014; 371:1381–1391.
- Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013; 368:11–21.
- Walsh TS, Boyd JA, Watson D, et al; RELIEVE Investigators. Restrictive versus liberal transfusion strategies for older mechanically ventilated critically ill patients: a randomized pilot trial. Crit Care Med 2013; 41:2354–2363.
- Rohde JM, Dimcheff DE, Blumberg N, et al. Health care–associated infection after red blood cell transfusion. JAMA 2014; 311:1317–1326.
- Salpeter SR, Buckley JS, Chatterjee S. Impact of more restrictive blood transfusion strategies on clinical outcomes: a meta-analysis and systematic review. Am J Med 2014; 127:124.e3–131.e3.
- Wang J, Bao YX, Bai M, Zhang YG, Xu WD, Qi XS. Restrictive vs liberal transfusion for upper gastrointestinal bleeding: a meta-analysis of randomized controlled trials. World J Gastroenterol 2013; 19:6919–6927.
- Vincent JL, Baron JF, Reinhart K, et al; ABC (Anemia and Blood Transfusion in Critical Care) Investigators. Anemia and blood transfusion in critically ill patients. JAMA 2002; 288:1499–1507.
- Dunne JR, Malone D, Tracy JK, Gannon C, Napolitano LM. Perioperative anemia: an independent risk factor for infection, mortality, and resource utilization in surgery. J Surg Res 2002; 102:237–244.
- Brunskill SJ, Millette SL, Shokoohi A, et al. Red blood cell transfusion for people undergoing hip fracture surgery. Cochrane Database Syst Rev 2015; 4:CD009699.
- Weinberg JA, McGwin G Jr, Griffin RL, et al. Age of transfused blood: an independent predictor of mortality despite universal leukoreduction. J Trauma 2008; 65:279–284.
- Steinberg M. Management of sickle cell disease. N Engl J Med 1999; 340:1021–1030.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease. JAMA 2014; 312:1033–1048.
- Vichinsky EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med 1995; 333:206–213.
- Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet 2013; 381:930–938.
- Goodnough LT, Levy JH, Murphy MF. Concepts of blood transfusion in adults. Lancet 2013; 381:1845–1854.
- Holme S. Current issues related to the quality of stored RBCs. Transfus Apher Sci 2005; 33:55–61.
- Hovav T, Yedgar S, Manny N, Barshtein G. Alteration of red cell aggregability and shape during blood storage. Transfusion 1999; 39:277–281.
- Koch CG, Li L, Sessler DI, et al. Duration of red-cell storage and complications after cardiac surgery. N Engl J Med 2008; 358:1229–1239.
- Lacroix J, Hebert PC, Fergusson DA, et al. Age of transfused blood in critically ill adults. N Engl J Med 2015; 372:1410–1418.
- Carson JL, Grossman BJ, Kleinman S, et al; Clinical Transfusion Medicine Committee of the AABB. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Intern Med 2012; 157:49–58.
- Bolton-Maggs P, Watt A, Poles D, et al, on behalf of the Serious Hazards of Transfusion (SHOT) Steering Group. The 2015 Annual SHOT Report. www.shotuk.org/wp-content/uploads/SHOT-2015-Annual-Report-Web-Edition-Final-bookmarked.pdf. Accessed November 30, 2016.
- Shander A, Javidroozi M, Ozawa S, Hare GMT. What is really dangerous: anaemia or transfusion? Br J Anaesth 2011; 107(suppl 1):i41–i59.
- Reeh M, Ghadban T, Dedow J, et al. Allogenic blood transfusion is associated with poor perioperative and long-term outcome in esophageal cancer. World J Surg 2016 Oct 11. [Epub ahead of print]
- Elmi M, Mahar A, Kagedan D, et al. The impact of blood transfusion on perioperative outcomes following gastric cancer resection: an analysis of the American College of Surgeons National Surgical Quality Improvement Program database. Can J Surg 2016; 59:322–329.
- Aquina CT, Blumberg N, Becerra AZ, et al. Association among blood transfusion, sepsis, and decreased long-term survival after colon cancer resection. Ann Surg 2016; Sep 14. [Epub ahead of print] PubMed PMID: 27631770.
- Premiere Analysis. Standardization of blood utilization practices could provide opportunity for improved outcomes, reduced costs. A Premiere Healthcare Alliance Analysis. 2012.
- Simeone F, Franchi F, Cevenini G, et al. A simple clinical model for planning transfusion quantities in heart surgery. BMC Med Inform Decis Mak 2011; 11:44.
- Spahn DR, Goodnough LT. Alternatives to blood transfusion. Lancet 2013; 381:1855–1865.
- Holst LB, Petersen MW, Haase N, Perner A, Wetterslev J. Restrictive versus liberal transfusion strategy for red blood cell transfusion: systematic review of randomised trials with meta-analysis and trial sequential analysis. BMJ 2015; 350:h1354.
- National Institute for Health and Care Excellence: Clinical Guidelines. London: National Institute for Health and Care Excellence (UK). www.ncbi.nlm.nih.gov/books/NBK11822/.
- Carson JL, Guyatt G, Heddle NM, et al. Clinical practice guidelines from the AABB: red blood cell transfusion thresholds and storage. JAMA 2016 Oct 12. doi: 10.1001/jama.2016.9185. [Epub ahead of print]
- Consensus conference. Perioperative red blood cell transfusion. JAMA 1988; 260:2700–2703.
- Bulger J, Nickel W, Messler J, et al. Choosing wisely in adult hospital medicine: five opportunities for improved healthcare value. J Hosp Med 2013; 8:486–492.
- Hicks LK, Bering H, Carson KR, et al. The ASH Choosing Wisely® campaign: five hematologic tests and treatments to question. Blood 2013; 122:3879–3883.
- Haemonetics IMPACT Online. The Blood Management Company. www.haemonetics.com/Products/Services/Consulting Services/IMPACT Online.aspx. Accessed November 30, 2016.
- Shander A, Gross I, Hill S, Javidroozi M, Sledge S; College of American Pathologists; American Society of Anesthesiologists; Society of Thoracic Surgeons and Society of Cardiovascular Anesthesiologists; Society of Critical Care Medicine; Italian Society of Transfusion Medicine and Immunohaematology; American Association of Blood Banks. A new perspective on best transfusion practices. Blood Transfus 2013; 11:193–202.
- Madjdpour C, Spahn DR. Allogeneic red blood cell transfusion: physiology of oxygen transport. Best Pract Res Clin Anaesthesiol 2007; 21:163–171.
- Tánczos K, Molnár Z. The oxygen supply-demand balance: a monitoring challenge. Best Pract Res Clin Anaesthesiol 2013; 27:201–207.
- Hebert PC, Van der Linden P, Biro G, Hu LQ. Physiologic aspects of anemia. Crit Care Clin 2004; 20:187–212.
- Spinelli E, Bartlett RH. Anemia and transfusion in critical care: physiology and management. J Intensive Care Med 2016; 31:295–306.
- Jamnicki M, Kocian R, Van Der Linden P, Zaugg M, Spahn DR. Acute normovolemic hemodilution: physiology, limitations, and clinical use. J Cardiothorac Vasc Anesth 2003; 17:747–754.
- Monk TG. Acute normovolemic hemodilution. Anesthesiol Clin North America 2005; 23:271–281.
- Shander A, Rijhwani TS. Acute normovolemic hemodilution. Transfusion 2004; 44(suppl 2):26S–34S.
- Weiskopf RB, Viele MK, Feiner J, et al. Human cardiovascular and metabolic response to acute, severe isovolemic anemia. JAMA 1998; 279:217–221.
- Leung JM, Weiskopf RB, Feiner J, et al. Electrocardiographic ST-segment changes during acute, severe isovolemic hemodilution in humans. Anesthesiology 2000; 93:1004–1010.
- Spahn DR, Zollinger A, Schlumpf RB, et al. Hemodilution tolerance in elderly patients without known cardiac disease. Anesth Analg 1996; 82:681–686.
- Spahn DR, Schmid ER, Seifert B, Pasch T. Hemodilution tolerance in patients with coronary artery disease who are receiving chronic beta-adrenergic blocker therapy. Anesth Analg 1996; 82:687–694.
- Licker M, Ellenberger C, Sierra J, Christenson J, Diaper J, Morel D. Cardiovascular response to acute normovolemic hemodilution in patients with coronary artery diseases: assessment with transesophageal echocardiography. Crit Care Med 2005; 33:591–597.
- Spahn DR, Seifert B, Pasch T, Schmid ER. Haemodilution tolerance in patients with mitral regurgitation. Anaesthesia 1998; 53:20–24.
- Weiskopf RB, Kramer JH, Viele M, et al. Acute severe isovolemic anemia impairs cognitive function and memory in humans. Anesthesiology 2000; 92:1646–1652.
- Weiskopf RB, Feiner J, Hopf HW, et al. Oxygen reverses deficits of cognitive function and memory and increased heart rate induced by acute severe isovolemic anemia. Anesthesiology 2002; 96:871–877.
- Zhou X, Zhang C, Wang Y, Yu L, Yan M. Preoperative acute normovolemic hemodilution for minimizing allogeneic blood transfusion: a meta-analysis. Anesth Analg 2015; 121:1443–1455.
- Hébert P, Wells G, Blajchman M, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999: 340:409–417.
- Carson JL, Terrin ML, Noveck H, et al; FOCUS Investigators. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med 2011; 365:2453–2462.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
- Carson JL, Brooks MM, Abbott JD, et al. Liberal versus restrictive transfusion thresholds for patients with symptomatic coronary artery disease. Am Heart J 2013; 165:964.e1–971.e1.
- Murphy GJ, Pike K, Rogers CA, et al; TITRe2 Investigators. Liberal or restrictive transfusion after cardiac surgery. N Engl J Med 2015; 372:997–1008.
- Holst LB, Haase N, Wetterslev J, et al; TRISS Trial Group; Scandinavian Critical Care Trials Group. Lower versus higher hemoglobin threshold for transfusion in septic shock. N Engl J Med 2014; 371:1381–1391.
- Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013; 368:11–21.
- Walsh TS, Boyd JA, Watson D, et al; RELIEVE Investigators. Restrictive versus liberal transfusion strategies for older mechanically ventilated critically ill patients: a randomized pilot trial. Crit Care Med 2013; 41:2354–2363.
- Rohde JM, Dimcheff DE, Blumberg N, et al. Health care–associated infection after red blood cell transfusion. JAMA 2014; 311:1317–1326.
- Salpeter SR, Buckley JS, Chatterjee S. Impact of more restrictive blood transfusion strategies on clinical outcomes: a meta-analysis and systematic review. Am J Med 2014; 127:124.e3–131.e3.
- Wang J, Bao YX, Bai M, Zhang YG, Xu WD, Qi XS. Restrictive vs liberal transfusion for upper gastrointestinal bleeding: a meta-analysis of randomized controlled trials. World J Gastroenterol 2013; 19:6919–6927.
- Vincent JL, Baron JF, Reinhart K, et al; ABC (Anemia and Blood Transfusion in Critical Care) Investigators. Anemia and blood transfusion in critically ill patients. JAMA 2002; 288:1499–1507.
- Dunne JR, Malone D, Tracy JK, Gannon C, Napolitano LM. Perioperative anemia: an independent risk factor for infection, mortality, and resource utilization in surgery. J Surg Res 2002; 102:237–244.
- Brunskill SJ, Millette SL, Shokoohi A, et al. Red blood cell transfusion for people undergoing hip fracture surgery. Cochrane Database Syst Rev 2015; 4:CD009699.
- Weinberg JA, McGwin G Jr, Griffin RL, et al. Age of transfused blood: an independent predictor of mortality despite universal leukoreduction. J Trauma 2008; 65:279–284.
- Steinberg M. Management of sickle cell disease. N Engl J Med 1999; 340:1021–1030.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease. JAMA 2014; 312:1033–1048.
- Vichinsky EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med 1995; 333:206–213.
- Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet 2013; 381:930–938.
- Goodnough LT, Levy JH, Murphy MF. Concepts of blood transfusion in adults. Lancet 2013; 381:1845–1854.
- Holme S. Current issues related to the quality of stored RBCs. Transfus Apher Sci 2005; 33:55–61.
- Hovav T, Yedgar S, Manny N, Barshtein G. Alteration of red cell aggregability and shape during blood storage. Transfusion 1999; 39:277–281.
- Koch CG, Li L, Sessler DI, et al. Duration of red-cell storage and complications after cardiac surgery. N Engl J Med 2008; 358:1229–1239.
- Lacroix J, Hebert PC, Fergusson DA, et al. Age of transfused blood in critically ill adults. N Engl J Med 2015; 372:1410–1418.
- Carson JL, Grossman BJ, Kleinman S, et al; Clinical Transfusion Medicine Committee of the AABB. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Intern Med 2012; 157:49–58.
- Bolton-Maggs P, Watt A, Poles D, et al, on behalf of the Serious Hazards of Transfusion (SHOT) Steering Group. The 2015 Annual SHOT Report. www.shotuk.org/wp-content/uploads/SHOT-2015-Annual-Report-Web-Edition-Final-bookmarked.pdf. Accessed November 30, 2016.
- Shander A, Javidroozi M, Ozawa S, Hare GMT. What is really dangerous: anaemia or transfusion? Br J Anaesth 2011; 107(suppl 1):i41–i59.
- Reeh M, Ghadban T, Dedow J, et al. Allogenic blood transfusion is associated with poor perioperative and long-term outcome in esophageal cancer. World J Surg 2016 Oct 11. [Epub ahead of print]
- Elmi M, Mahar A, Kagedan D, et al. The impact of blood transfusion on perioperative outcomes following gastric cancer resection: an analysis of the American College of Surgeons National Surgical Quality Improvement Program database. Can J Surg 2016; 59:322–329.
- Aquina CT, Blumberg N, Becerra AZ, et al. Association among blood transfusion, sepsis, and decreased long-term survival after colon cancer resection. Ann Surg 2016; Sep 14. [Epub ahead of print] PubMed PMID: 27631770.
- Premiere Analysis. Standardization of blood utilization practices could provide opportunity for improved outcomes, reduced costs. A Premiere Healthcare Alliance Analysis. 2012.
- Simeone F, Franchi F, Cevenini G, et al. A simple clinical model for planning transfusion quantities in heart surgery. BMC Med Inform Decis Mak 2011; 11:44.
- Spahn DR, Goodnough LT. Alternatives to blood transfusion. Lancet 2013; 381:1855–1865.
- Holst LB, Petersen MW, Haase N, Perner A, Wetterslev J. Restrictive versus liberal transfusion strategy for red blood cell transfusion: systematic review of randomised trials with meta-analysis and trial sequential analysis. BMJ 2015; 350:h1354.
- National Institute for Health and Care Excellence: Clinical Guidelines. London: National Institute for Health and Care Excellence (UK). www.ncbi.nlm.nih.gov/books/NBK11822/.
- Carson JL, Guyatt G, Heddle NM, et al. Clinical practice guidelines from the AABB: red blood cell transfusion thresholds and storage. JAMA 2016 Oct 12. doi: 10.1001/jama.2016.9185. [Epub ahead of print]
- Consensus conference. Perioperative red blood cell transfusion. JAMA 1988; 260:2700–2703.
- Bulger J, Nickel W, Messler J, et al. Choosing wisely in adult hospital medicine: five opportunities for improved healthcare value. J Hosp Med 2013; 8:486–492.
- Hicks LK, Bering H, Carson KR, et al. The ASH Choosing Wisely® campaign: five hematologic tests and treatments to question. Blood 2013; 122:3879–3883.
- Haemonetics IMPACT Online. The Blood Management Company. www.haemonetics.com/Products/Services/Consulting Services/IMPACT Online.aspx. Accessed November 30, 2016.
KEY POINTS
- In critical care patients, transfusion should be considered when the hemoglobin concentration reaches 7 g/dL or less.
- In postoperative patients and hospitalized patients with preexisting cardiovascular disease, transfusion should be considered at a hemoglobin concentration of 8 g/dL or less or for symptoms such as chest pain, orthostatic hypotension, or tachycardia unresponsive to fluid resuscitation, or heart failure.
- Consider both the hemoglobin concentration and the symptoms when deciding whether to give a patient a transfusion.

A patient with altered mental status and an acid-base disturbance
A 78-year-old black woman with a history of osteoarthrosis and chronic diffuse joint pain presents with altered mental status and tachypnea, which began 3 hours earlier. She lives alone, and her family suspects she abuses both alcohol and her pain medications. She has not been eating well and has lost approximately 10 pounds over the past 3 months. Her analgesic regimen includes acetaminophen and acetaminophen-oxycodone.
In the emergency department her temperature is 98.6°F (37.0°C), pulse 100 beats per minute and regular, respiratory rate 22 per minute, and blood pressure 136/98 mm Hg. She is obtunded but has no focal neurologic defects or meningismus. She has no signs of heart failure (jugular venous distention, cardiomegaly, or gallops), and examination of the lungs and abdomen is unremarkable.

Suspecting that the patient may have taken too much oxycodone, the physician gives her naloxone, but her mental status does not improve. Results of chest radiography and cranial computed tomography are unremarkable. The physician’s initial impression is that the patient has “metabolic encephalopathy of unknown etiology.”
The patient’s laboratory values are shown in Table 1.
WHICH ACID-BASE DISORDER DOES SHE HAVE?
1. Which acid-base disorder does this patient have?
- Metabolic acidosis and respiratory alkalosis
- Metabolic acidosis and respiratory acidosis
- Metabolic acidosis with an elevated anion gap
- A triple disturbance: metabolic acidosis, respiratory acidosis, and metabolic alkalosis
A 5-step approach
Acid-base disorders can be diagnosed and characterized using a systematic approach known as the “Rules of 5” (Table 2)1:
1. Determine the arterial pH status.
2. Determine whether the primary process is respiratory, metabolic, or both.
3. Calculate the anion gap.
4. Check the degree of compensation (respiratory or metabolic).
5. If the patient has metabolic acidosis with an elevated anion gap, check whether the bicarbonate level has decreased as much as the anion gap has increased (ie, whether there is a delta gap).
Let us apply this approach to the patient described above.
1. What is her pH status?
An arterial pH less than 7.40 is acidemic, whereas a pH higher than 7.44 is alkalemic. (Acidemia and alkalemia refer to the abnormal laboratory value, while acidosis and alkalosis refer to the process causing the abnormal value—a subtle distinction, but worth keeping in mind.)
Caveat. A patient may have a significant acid-base disorder even if the pH is normal. Therefore, even if the pH is normal, one should verify that the partial pressure of carbon dioxide (Pco2), bicarbonate level, and anion gap are normal. If they are not, the patient may have a mixed acid-base disorder such as respiratory acidosis superimposed on metabolic alkalosis.
Our patient’s pH is 7.25, which is in the acidemic range.
2. Is her acidosis respiratory, metabolic, or both?
Respiratory acidosis and alkalosis affect the Pco2. The Pco2 is high in respiratory acidosis (due to failure to get rid of excess carbon dioxide), whereas it is low in respiratory alkalosis (due to loss of too much carbon dioxide through hyperventilation).
Metabolic acidosis and alkalosis, on the other hand, affect the serum bicarbonate level. In metabolic acidosis the bicarbonate level is low, whereas in metabolic alkalosis the bicarbonate level is high.
Moreover, in mixed respiratory and metabolic acidosis, the bicarbonate level can be low and the Pco2 can be high. In mixed metabolic and respiratory alkalosis, the bicarbonate level can be high and the Pco2 can be low (Table 2).
Our patient’s serum bicarbonate level is low at 16.0 mmol/L, indicating that the process is metabolic. Her Pco2 is also low (28 mm Hg), which reflects an appropriate response to compensate for the acidosis.
3. What is her anion gap?
Always calculate the anion gap, ie, the serum sodium concentration minus the serum chloride and serum bicarbonate concentrations. If the patient’s serum albumin level is low, for every 1 gram it is below normal, an additional 2.5 mmol/L should be added to the calculated anion gap. We consider an anion gap of 10 mmol/L or less as normal.
Caveats. The blood sample used to calculate the anion gap should be drawn close in time to the arterial blood gas sample.
Although the anion gap is an effective tool in assessing acid-base disorders, further investigation is warranted if clinical judgment suggests that an anion gap calculation is inconsistent with the patient’s circumstances.2
Our patient’s anion gap is elevated (21 mmol/L). Her serum albumin level is in the normal range, so her anion gap does not need to be adjusted.
4. Is the degree of compensation appropriate for the primary acid-base disturbance?
The kidneys compensate for the lungs, and vice versa. That is, in respiratory acidosis or alkalosis, the kidneys adjust the bicarbonate levels, and in metabolic acidosis, the lungs adjust the Pco2 (although in metabolic alkalosis, it is hard for patients to breathe less, especially if they are already hypoxic).
In metabolic acidosis, people compensate by breathing harder to get rid of more carbon dioxide. For every 1-mmol/L decrease in the bicarbonate level, the Pco2 should decrease by 1.3 mm Hg.
Compensation does not return pH to normal; rather, it mitigates the impact of an acid or alkali excess or deficit. If the pH is normalized with an underlying acid-base disturbance, there may be mixed acid-base processes rather than compensation.
Our patient’s bicarbonate level is 16 mmol/L, which is 9 mmol/L lower than normal (for acid-base calculations, we use 25 mmol/L as the nominal normal level). If she is compensating appropriately, her Pco2 should decline from 40 mm Hg (the nominal normal level) by about 11.7 mm Hg (9 × 1.3), to approximately 28.3 mm Hg. Her Pco2 is, indeed, 28 mm Hg, indicating that she is compensating adequately for her metabolic acidosis.
If we use Winter’s formula instead (Pco2 = [1.5 × the bicarbonate level] + 8 ± 2),3 the lowest calculated Pco2 would be 30 mm Hg, which is within 2 mm Hg of the Rules of 5 calculation. Other formulas for calculating compensation are available.3
This information rules out the first two answers to question 1, ie, metabolic acidosis with respiratory alkalosis or acidosis.
5. Is there a delta gap?
Although we know the patient has metabolic acidosis with an elevated anion gap, we have not ruled out the possibility that she may have a triple disturbance. For this reason we need to check her delta gap.
In metabolic acidosis with an elevated anion gap, as the bicarbonate level decreases, the anion gap should increase by the same amount. If the bicarbonate level decreases more than the anion gap increases, the additional decline is the result of a second process—an additional normal-anion-gap acidosis. If the bicarbonate level does not decrease as much as the anion gap increases, there is an additional metabolic alkalosis.
Our patient’s bicarbonate level decreased 9 mmol/L (from the nominal normal level of 25 to 16), and therefore her anion gap should have increased approximately the same amount—and it did. (A normal anion gap for problem-solving is 10, and this patient’s anion gap has increased to 21. A difference of ± 2 is insignificant.) This conclusion verifies that a triple acid-base disturbance is not present, so the last answer is incorrect.
So, the correct answer to the question posed above is metabolic acidosis with an elevated anion gap (that is, metabolic acidosis with appropriate respiratory compensation).
‘MUD PILES’: FINDING THE CAUSE OF ANION GAP METABOLIC ACIDOSIS

The possible causes of metabolic acidosis with an elevated anion gap (as in our patient) can be summarized in the mnemonic MUD PILES (methanol, uremia, diabetes, paraldehyde, isoniazid, lactate, ethylene glycol, and salicylates), which has been used for many years. Parts of it are no longer useful, but rather than discard it, we propose to update it (Table 3).
Methanol and ethylene glycol
We will address toxic ingestion of methanol and ethylene glycol (the “M” and “E” of MUD PILES) at the same time.

In cases of suspected ingestion of toxic substances such as these, it is useful to examine the osmol gap, ie, the difference between the calculated and the measured serum osmolality. Serum osmolality (in mOsm/kg) is calculated as the sodium concentration in mmol/L times 2, plus the glucose concentration in mg/dL divided by 18, plus the blood urea nitrogen concentration in mg/dL divided by 2.8 (Table 4). If the measured osmolality is higher than this calculated value, the difference may be due to solutes in the blood that should not be there such as ethylene glycol, diethylene glycol, methanol, and their many metabolic products.
In our patient, ingestion of both methanol and ethylene glycol should be considered, since she lives alone and has been suspected of alcohol and opioid abuse. Her calculated osmol gap is 278 mOsm/kg. Her measured osmolality is 318 mOsm/kg (Table 1). The osmol gap is 40 mOsm/kg (normal is ≤ 10).4,5 Therefore, her osmol gap is elevated.
Identifying the specific substance the patient ingested that caused metabolic acidosis with anion gap may be difficult. Poisonings with these agents do not always increase the osmol gap.6 A high index of suspicion is essential. It is helpful to have the family search for any sources of ethylene glycol and methanol at home and initiate treatment early if an ingestion is suspected, using fomepizole (an alcohol dehydrogenase inhibitor) or parenteral ethanol and hemodialysis.7 Liquid chromatography identifies these two toxins, but results are not available emergently.
Diethylene glycol ingestion should also be considered.8 Since it is diagnosed and treated like ethylene glycol intoxication, it can be placed with the “E” of (di)ethylene glycol in the mnemonic.
Uremia
Renal failure can lead to metabolic acidosis.9 Our patient has no history of kidney disease, but her blood urea nitrogen and creatinine concentrations are above normal, and her estimated glomerular filtration rate by the Modification of Diet in Renal Disease formula is 48 mL/min/1.73 m2—low, but not uremic.
Rhabdomyolysis (suspected by elevated creatine kinase values) should be considered in any patient with mental status changes, suspected toxic ingestion, and metabolic acidosis (see the “I” in MUD PILES below). Compartment syndromes with muscle necrosis may present in a subtle fashion. Therefore, renal failure from rhabdomyolysis may complicate this patient’s course later, and should be kept in mind.
Diabetes
The patient has no history of diabetes and has a normal blood glucose level. Blood testing did not reveal ketones. She is not taking metformin (alleged to cause lactic acidosis) or a sodium-glucose cotransporter 2 inhibitor (which have been associated with ketoacidosis).10
There is another, less common cause of ketoacidosis: alcohol.11 Although alcoholism is common, alcoholic ketoacidosis is uncommon, even in heavy drinkers. Ethyl alcohol causing metabolic acidosis is similar to metabolic acidosis with (di)ethylene glycol and methanol, and if suspected it should be treated empirically (first with thiamine, then dextrose and saline, and correcting other electrolyte disturbances such as hypokalemia and hypomagnesemia) before specific identification is made. Ketones (predominantly beta-hydroxybutyrate) may persist up to 2 weeks after alcohol ingestion has stopped.11 Ketosis in the setting of alcoholic ketoacidosis is frequently accompanied by other markers of alcohol target organ injury: elevated bilirubin, aspartate aminotransferase, alanine aminotransferase, and gamma-glutamyl transferase levels. The term “ketohepatitis” has been suggested as an alternative to alcoholic ketosis.11
This patient did not have an elevated blood ethanol level, and her liver markers were otherwise normal.
THE NEW MUD PILES
2. Which of the following is (are) true? Regarding the remaining letters of the MUD PILES mnemonic:
- The “P” (paraldehyde) has been replaced by pyroglutamic acid (5-oxoproline) and propylene glycol.
- There are two isomers of lactate (dextro and levo), and consequently two clinical varieties of lactic acidosis.
- Isoniazid is no longer associated with metabolic acidosis with elevated anion gap.
- Salicylates can paradoxically be associated both with elevated and low anion gaps.
Isoniazid is still associated with metabolic acidosis with elevated anion gap, and so the third answer choice is false; the rest are true.
Paraldehyde, isoniazid, lactate
The “P,” “I,” and “L” (d-lactate) of the revamped MUD PILES acronym are less common than the others. They should be considered when the more typical causes of metabolic acidosis are not present, as in this patient.
UPDATING THE ‘P’ IN MUD PILES
Paraldehyde is rarely prescribed anymore. A PubMed search on December 21, 2015 applying the terms paraldehyde and metabolic acidosis yielded 17 results. Those specific to anion gap metabolic acidosis were from 1957 to 1986 (n = 9).12–20
Therefore, we can eliminate paraldehyde from the MUD PILES mnemonic and replace it with pyroglutamic acid and propylene glycol.
5-Oxoproline or pyroglutamic acid, a metabolite of acetaminophen
Acetaminophen depletes glutathione stores in acute overdoses, in patients with inborn errors of metabolism, and after chronic ingestion of excessive, frequent doses. Depletion of glutathione increases metabolic products, including pyroglutamic acid, which dissociates into hydrogen ions (leading to metabolic acidosis and an anion gap), and 5-oxoproline, (which can be detected in the urine).21,22
Risk factors for metabolic acidosis with acetaminophen ingestion include malnutrition, chronic alcoholism, liver disease, and female sex. In fact, most cases have been reported in females, and altered mental status has been common.
Metabolic acidosis with pyroglutamic acid can occur without elevated acetaminophen levels. Serum and urine levels of pyroglutamic acid may assist with diagnosis. Since identification of urine pyroglutamic acid usually requires outside laboratory assistance, a clinical diagnosis is often made initially and corroborated later by laboratory results. When the anion gap metabolic acidosis is multifactorial, as it was suspected to be in a case reported by Tan et al,23 the osmol gap may be elevated as a consequence of additional toxic ingestions, as it was in the reported patient.
No controlled studies of treatment have been done. n-Acetylcysteine may be of benefit. Occasional patients have been dialyzed for removal of excess pyroglutamic acid.
Propylene glycol, a component of parenteral lorazepam
Lorazepam is a hydrophobic drug, so when it is given parenterally, it must be mixed with a suitable solvent. A typical formulation adds propylene glycol. In patients receiving high doses of lorazepam as relaxation therapy for acute respiratory distress syndrome in the intensive care unit, or as treatment of alcohol withdrawal, the propylene glycol component can precipitate anion gap metabolic acidosis.24,25
Although nearly one-half of the administered propylene glycol is excreted by the kidneys, the remaining substrate is metabolized by alcohol dehydrogenase into d,l-lactaldehyde, then converted into d- or l-lactate. l-Lactate can be metabolized, but d-lactate cannot and leads to anion gap metabolic acidosis. This is another toxic metabolic acidosis associated with an elevated osmol gap. An increasing osmol gap in the intensive care unit can serve as a surrogate marker of excessive propylene glycol administration.23
Isoniazid
Although it is uncommon, there are reports of isoniazid-induced anion gap metabolic acidosis,26 either due to overdoses, or less commonly, with normal dosing. Isoniazid should therefore remain in the mnemonic MUD PILES and may be suspected when metabolic acidosis is accompanied by seizures unresponsive to usual therapy. The seizures respond to pyridoxine.
The “I” should also be augmented by newer causes of metabolic acidosis associated with “ingestions.” Ecstasy, or 3,4-methylenedioxymethamphetamine, can cause metabolic acidosis and seizures. Ecstasy has been associated with rhabdomyolysis and uremia, also leading to anion gap metabolic acidosis.27 A newer class of abused substances, synthetic cathinones (“bath salts”), are associated with metabolic acidosis, compartment syndrome, and renal failure.28
Lactic acidosis
Lactic acidosis and metabolic acidosis can result from hypoperfusion (type A) or other causes (type B). Not all lactic acidosis is contingent on l-lactate, which humans can metabolize. Metabolic acidosis may be a consequence of d-lactate (mammals have no d-lactate dehydrogenase). d-Lactic acidosis as a result of short bowel syndrome has been known for more than a generation.29 However, d-lactic acidosis occurs in another new setting. The new “P” in MUD PILES, propylene glycol, can generate substantial amounts of d-lactate.29
d-lactic metabolic acidosis is always accompanied by neurologic manifestations (slurred speech, confusion, somnolence, ataxia, abusive behavior, and others).30 With short bowel syndrome, the neurologic manifestations occur after eating and clear later.30
Although our patient’s anion gap is more than 20 mmol/L, her blood level of lactate is not elevated, and she had no history to suggest short-bowel syndrome.
Salicylates
Salicylate overdose can cause a mixed acid-base disorder: metabolic acidosis with elevated anion gap and respiratory alkalosis.
Although our patient does not have respiratory alkalosis, an aspirin overdose must be considered. A salicylate level was ordered; it was negative.
Despite the typical association of salicylates with an elevated anion gap, they may also cause a negative anion gap.31 Chloride-sensing ion-specific electrodes contain a membrane permeable to chloride. Salicylates can increase the chloride permeability of these membranes, generating pseudohyperchloremia, and consequently, a negative anion gap.
WHAT ELSE MUST BE CONSIDERED?
3. In view of her anion gap metabolic acidosis, elevated osmol gap, and absence of diabetes, renal failure, or lactate excess, what are the remaining diagnoses to consider in this patient? (Choose all that are potential sources of metabolic acidosis and an increased anion gap.)
- Methanol, ethylene, or diethylene glycol
- Excessive, chronic acetaminophen ingestion
- Salicylate toxicity
- Alcoholic ketoacidosis
All of the above can potentially contribute to metabolic acidosis.
A search of the patient’s home did not reveal a source of methanol or either ethylene or diethylene glycol. Similarly, no aspirin was found, and the patient’s salicylate levels were not elevated. The patient’s laboratory work did not reveal increased ketones.
Since none of the common causes of metabolic acidosis were discovered, and since the patient had been taking acetaminophen, the diagnosis of excessive chronic acetaminophen ingestion was suspected pending laboratory verification. Identification of 5-oxoproline in the urine may take a week or more since the sample is usually sent to special laboratories. Acetaminophen levels in this patient were significantly elevated, as were urinary oxyproline levels, which returned later.
The patient was diagnosed with pyroglutamic acid metabolic acidosis. She was treated supportively and with n-acetylcysteine intravenously, although there have been no controlled studies of the efficacy of this drug. Seventy-two hours after admission, she had improved. Her acid-base status returned to normal.
GOLD MARK: ANOTHER WAY TO REMEMBER
Another mnemonic device for remembering the causes of metabolic acidosis with elevated anion gap is “GOLD MARK”: glycols (ethylene and propylene), oxoproline (instead of pyroglutamic acid from acetaminophen), l-lactate, d-lactate, methanol, aspirin, renal failure, and ketoacidosis).32
ACID-BASE DISORDERS IN DIFFERENT DISEASES
Diverse diseases cause distinctive acid-base abnormalities. Matching the appropriate acid-base abnormality with its associated disease may lead to more timely diagnosis and treatment:
Type 2 diabetes mellitus, for example, can lead to lactic acidosis, ketoacidosis, or type 4 renal tubular acidosis.33
Heart failure, although not typically framed in the context of acid-base physiology, can lead to elevated lactate, which is associated with a worse prognosis.34
Acquired immunodeficiency syndrome. Abacavir can cause normal anion gap metabolic acidosis.35,36
Cancer37,38 can be associated with proximal tubular renal tubular acidosis and lactic acidosis.
An expanding array of toxic ingestions
Metabolic acidosis may be the most prominent and potentially lethal clinical acid-base disturbance. When metabolic acidosis occurs in certain disease states—lactic acidosis with hypoperfusion or methanol ingestion with metabolic acidosis, for example—there is increased morbidity and mortality.
As reflected in the revisions to MUD PILES and in the newer GOLD MARK acronym, the osmol gap has become more valuable in differential diagnosis of metabolic acidosis with an elevated anion gap consequent to an expanding array of toxic ingestions (methanol, propylene glycol, pyroglutamic acid-oxoproline, ethylene glycol, and diethylene glycol), which may accompany pyroglutamic acid-oxoproline.
- Whittier WL, Rutecki GW. Primer on clinical acid-base problem solving. Dis Mon 2004; 50:122–162.
- Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol 2007; 2:162–174.
- Adrogué HJ, Madias NE. Secondary responses to altered acid-base status: the rules of engagement. J Am Soc Nephrol 2010; 21:920–923.
- Krasowski MD, Wilcoxon RM, Miron J. A retrospective analysis of glycol and toxic alcohol ingestion: utility of anion and osmolal gaps. BMC Clin Pathol 2012;12:1.
- Latus J, Kimmel M, Alscher MD, Braun N. Ethylene glycol poisoning: a rare but life-threatening cause of metabolic acidosis—a single-centre experience. Clin Kidney J 2012; 5:120–123.
- Kraut JA. Diagnosis of toxic alcohols: limitations of present methods. Clin Toxicol (Phila) 2015; 53:589–595.
- Ghannoum M, Hoffman RS, Mowry JB, Lavergne V. Trends in toxic alcohol exposures in the United States from 2000 to 2013: a focus on the use of antidotes and extracorporeal treatments. Semin Dial 2014; 27:395–401.
- Schep LJ, Slaughter RJ, Temple WA, Beasley DM. Diethylene glycol poisoning. Clin Toxicol (Phila) 2009; 47:525–535.
- Kraut JA, Madias NE. Metabolic acidosis of CKD: an update. Am J Kidney Dis 2016; 67:307–317.
- Taylor SI, Blau JE, Rother KI. SGLT2 inhibitors may predispose to ketoacidosis. J Clin Endocrinol Metab 2015; 100:2849–2852.
- Yokoyama A, Yokoyama T, Mizukami T, et al. Alcoholic ketosis: prevalence, determinants, and ketohepatitis in Japanese alcoholic men. Alcohol Alcohol 2014; 49:618–625.
- Hayward JN, Boshell BR. Paraldehyde intoxication with metabolic acidosis; report of two cases, experimental data and a critical review of the literature. Am J Med 1957; 23:965–976.
- Elkinton JR, Huth EJ, Clark JK, Barker ES, Seligson D. Renal tubular acidosis with organic aciduria during paraldehyde ingestion; six year study of an unusual case. Am J Med 1957; 23:977–986.
- Waterhouse C, Stern EA. Metabolic acidosis occurring during administration of paraldehyde. Am J Med 1957; 23:987–989.
- Beier LS, Pitts WH, Gonick HC. Metabolic acidosis occurring during paraldehyde intoxication. Ann Intern Med 1963; 58:155–158.
- Hiemcke T. Metabolic acidosis due to paraldehyde. Ned Tijdschr Geneeskd 1964; 108:2165–2167. Dutch.
- Gailitis RJ. Paraldehyde acidosis syndrome. IMJ III Med J 1966; 129:258–262.
- Gutman RA, Burnell JM. Paraldehyde acidosis. Am J Med 1967; 42:435–440.
- Hadden JW, Metzner RJ. Pseudoketosis and hyperacetaldehydemia in paraldehyde acidosis. Am J Med 1969; 47:642–647.
- Linter CM, Linter SP. Severe lactic acidosis following paraldehyde administration. Br J Psychiatry 1986; 149:650–651.
- Zand L, Muriithi A, Nelsen E, et al. Severe anion gap metabolic acidosis from acetaminophen use secondary to 5-oxoproline (pyroglutamic acid) accumulation. Am J Med Sci 2012; 344:501–504.
- Abkur TM, Mohammed W, Ali M, Casserly L. Acetaminophen-induced anion gap metabolic acidosis secondary to 5-oxoproline: a case report. J Med Case Rep 2014; 8:409.
- Tan EM, Kalimullah E, Sohail MR, Ramar K. Diagnostic challenge in a patient with severe anion gap metabolic acidosis. Case Rep Crit Care 2015; 2015:272914.
- Jorens PG, Demey HE, Schepens PJ, et al. Unusual d-lactic acid acidosis from propylene glycol metabolism in overdose. J Toxicol Clin Toxicol 2004; 42:163–169.
- Barnes BJ, Gerst C, Smith JR, Terrell AR, Mullins ME. Osmol gap as a surrogate marker for serum propylene glycol concentrations in patients receiving lorazepam for sedation. Pharmacotherapy 2006; 26:23–33.
- Gokhale YA, Vaidya MS, Mehta AD, Rathod NN. Isoniazid toxicity presenting as status epilepticus and severe metabolic acidosis. J Assoc Physicians India 2009; 57:70–71.
- Ben-Abraham R, Szold O, Rudick V, Weinbroum AA. ‘Ecstasy’ intoxication: life-threatening manifestations and resuscitative measures in the intensive care setting. Eur J Emerg Med 2003; 10:309–313.
- German CL, Fleckenstein AE, Hanson GR. Bath salts and synthetic cathinones: an emerging designer drug phenomenon. Life Sci 2014; 97:2–8.
- Jorens PG, Demey HE, Schepens PJ, et al. Unusual d-lactic acidosis from propylene glycol metabolism in overdose. J Toxicol Clin Toxicol 2004; 42:163–169.
- Kang KP, Le S, Kang SK. d-Lactic acidosis in humans: review and update. Electrolyte Blood Press 2006; 4:53–56.
- Emmett M. Approach to the patient with a negative anion gap. Am J Kidney Dis 2016; 67:143–150.
- Mehta AN, Emmett JB, Emmett M. GOLD MARK: an anion gap mnemonic for the 21st Century. Lancet 2008; 372:892.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Park JJ, Choi DJ, Yoon CH, et al; KorHF Registry. The prognostic value of arterial blood gas analysis in high-risk acute heart failure patients: an analysis of the Korean Heart Failure (KorHF) registry. Eur J Heart Fail 2015; 17:601–611.
- Musso CG, Belloso WH, Glassock RJ. Water, electrolytes, and acid-base alterations in human immunodeficiency virus infected patients. World J Nephrol 2016; 5:33–42.
- Camara-Lemarroy CR, Flores-Cantu H, Calderon-Hernandez HJ, Diaz-Torres MA, Villareal-Velazquez HJ. Drug-induced haemolysis, renal failure, thrombocytopenia and lactic acidosis in patients with HIV and cryptococcal meningitis: a diagnostic challenge. Int J STD AIDS 2015; 26:1052–1054.
- Miltiadous G, Christidis D, Kalogirou M, Elisaf M. Causes and mechanisms of acid-base and electrolyte abnormalities in cancer. Eur J Intern Med 2008; 19:1–7.
- Vlachostergios PJ, Oikonomou KG, Gibilaro E, Apergis G. Elevated lactic acid is a negative prognostic factor in metastatic lung cancer. Cancer Biomark 2015; 15:725–734.
A 78-year-old black woman with a history of osteoarthrosis and chronic diffuse joint pain presents with altered mental status and tachypnea, which began 3 hours earlier. She lives alone, and her family suspects she abuses both alcohol and her pain medications. She has not been eating well and has lost approximately 10 pounds over the past 3 months. Her analgesic regimen includes acetaminophen and acetaminophen-oxycodone.
In the emergency department her temperature is 98.6°F (37.0°C), pulse 100 beats per minute and regular, respiratory rate 22 per minute, and blood pressure 136/98 mm Hg. She is obtunded but has no focal neurologic defects or meningismus. She has no signs of heart failure (jugular venous distention, cardiomegaly, or gallops), and examination of the lungs and abdomen is unremarkable.
Suspecting that the patient may have taken too much oxycodone, the physician gives her naloxone, but her mental status does not improve. Results of chest radiography and cranial computed tomography are unremarkable. The physician’s initial impression is that the patient has “metabolic encephalopathy of unknown etiology.”
The patient’s laboratory values are shown in Table 1.
WHICH ACID-BASE DISORDER DOES SHE HAVE?
1. Which acid-base disorder does this patient have?
- Metabolic acidosis and respiratory alkalosis
- Metabolic acidosis and respiratory acidosis
- Metabolic acidosis with an elevated anion gap
- A triple disturbance: metabolic acidosis, respiratory acidosis, and metabolic alkalosis
A 5-step approach
Acid-base disorders can be diagnosed and characterized using a systematic approach known as the “Rules of 5” (Table 2)1:
1. Determine the arterial pH status.
2. Determine whether the primary process is respiratory, metabolic, or both.
3. Calculate the anion gap.
4. Check the degree of compensation (respiratory or metabolic).
5. If the patient has metabolic acidosis with an elevated anion gap, check whether the bicarbonate level has decreased as much as the anion gap has increased (ie, whether there is a delta gap).
Let us apply this approach to the patient described above.
1. What is her pH status?
An arterial pH less than 7.40 is acidemic, whereas a pH higher than 7.44 is alkalemic. (Acidemia and alkalemia refer to the abnormal laboratory value, while acidosis and alkalosis refer to the process causing the abnormal value—a subtle distinction, but worth keeping in mind.)
Caveat. A patient may have a significant acid-base disorder even if the pH is normal. Therefore, even if the pH is normal, one should verify that the partial pressure of carbon dioxide (Pco2), bicarbonate level, and anion gap are normal. If they are not, the patient may have a mixed acid-base disorder such as respiratory acidosis superimposed on metabolic alkalosis.
Our patient’s pH is 7.25, which is in the acidemic range.
2. Is her acidosis respiratory, metabolic, or both?
Respiratory acidosis and alkalosis affect the Pco2. The Pco2 is high in respiratory acidosis (due to failure to get rid of excess carbon dioxide), whereas it is low in respiratory alkalosis (due to loss of too much carbon dioxide through hyperventilation).
Metabolic acidosis and alkalosis, on the other hand, affect the serum bicarbonate level. In metabolic acidosis the bicarbonate level is low, whereas in metabolic alkalosis the bicarbonate level is high.
Moreover, in mixed respiratory and metabolic acidosis, the bicarbonate level can be low and the Pco2 can be high. In mixed metabolic and respiratory alkalosis, the bicarbonate level can be high and the Pco2 can be low (Table 2).
Our patient’s serum bicarbonate level is low at 16.0 mmol/L, indicating that the process is metabolic. Her Pco2 is also low (28 mm Hg), which reflects an appropriate response to compensate for the acidosis.
3. What is her anion gap?
Always calculate the anion gap, ie, the serum sodium concentration minus the serum chloride and serum bicarbonate concentrations. If the patient’s serum albumin level is low, for every 1 gram it is below normal, an additional 2.5 mmol/L should be added to the calculated anion gap. We consider an anion gap of 10 mmol/L or less as normal.
Caveats. The blood sample used to calculate the anion gap should be drawn close in time to the arterial blood gas sample.
Although the anion gap is an effective tool in assessing acid-base disorders, further investigation is warranted if clinical judgment suggests that an anion gap calculation is inconsistent with the patient’s circumstances.2
Our patient’s anion gap is elevated (21 mmol/L). Her serum albumin level is in the normal range, so her anion gap does not need to be adjusted.
4. Is the degree of compensation appropriate for the primary acid-base disturbance?
The kidneys compensate for the lungs, and vice versa. That is, in respiratory acidosis or alkalosis, the kidneys adjust the bicarbonate levels, and in metabolic acidosis, the lungs adjust the Pco2 (although in metabolic alkalosis, it is hard for patients to breathe less, especially if they are already hypoxic).
In metabolic acidosis, people compensate by breathing harder to get rid of more carbon dioxide. For every 1-mmol/L decrease in the bicarbonate level, the Pco2 should decrease by 1.3 mm Hg.
Compensation does not return pH to normal; rather, it mitigates the impact of an acid or alkali excess or deficit. If the pH is normalized with an underlying acid-base disturbance, there may be mixed acid-base processes rather than compensation.
Our patient’s bicarbonate level is 16 mmol/L, which is 9 mmol/L lower than normal (for acid-base calculations, we use 25 mmol/L as the nominal normal level). If she is compensating appropriately, her Pco2 should decline from 40 mm Hg (the nominal normal level) by about 11.7 mm Hg (9 × 1.3), to approximately 28.3 mm Hg. Her Pco2 is, indeed, 28 mm Hg, indicating that she is compensating adequately for her metabolic acidosis.
If we use Winter’s formula instead (Pco2 = [1.5 × the bicarbonate level] + 8 ± 2),3 the lowest calculated Pco2 would be 30 mm Hg, which is within 2 mm Hg of the Rules of 5 calculation. Other formulas for calculating compensation are available.3
This information rules out the first two answers to question 1, ie, metabolic acidosis with respiratory alkalosis or acidosis.
5. Is there a delta gap?
Although we know the patient has metabolic acidosis with an elevated anion gap, we have not ruled out the possibility that she may have a triple disturbance. For this reason we need to check her delta gap.
In metabolic acidosis with an elevated anion gap, as the bicarbonate level decreases, the anion gap should increase by the same amount. If the bicarbonate level decreases more than the anion gap increases, the additional decline is the result of a second process—an additional normal-anion-gap acidosis. If the bicarbonate level does not decrease as much as the anion gap increases, there is an additional metabolic alkalosis.
Our patient’s bicarbonate level decreased 9 mmol/L (from the nominal normal level of 25 to 16), and therefore her anion gap should have increased approximately the same amount—and it did. (A normal anion gap for problem-solving is 10, and this patient’s anion gap has increased to 21. A difference of ± 2 is insignificant.) This conclusion verifies that a triple acid-base disturbance is not present, so the last answer is incorrect.
So, the correct answer to the question posed above is metabolic acidosis with an elevated anion gap (that is, metabolic acidosis with appropriate respiratory compensation).
‘MUD PILES’: FINDING THE CAUSE OF ANION GAP METABOLIC ACIDOSIS
The possible causes of metabolic acidosis with an elevated anion gap (as in our patient) can be summarized in the mnemonic MUD PILES (methanol, uremia, diabetes, paraldehyde, isoniazid, lactate, ethylene glycol, and salicylates), which has been used for many years. Parts of it are no longer useful, but rather than discard it, we propose to update it (Table 3).
Methanol and ethylene glycol
We will address toxic ingestion of methanol and ethylene glycol (the “M” and “E” of MUD PILES) at the same time.
In cases of suspected ingestion of toxic substances such as these, it is useful to examine the osmol gap, ie, the difference between the calculated and the measured serum osmolality. Serum osmolality (in mOsm/kg) is calculated as the sodium concentration in mmol/L times 2, plus the glucose concentration in mg/dL divided by 18, plus the blood urea nitrogen concentration in mg/dL divided by 2.8 (Table 4). If the measured osmolality is higher than this calculated value, the difference may be due to solutes in the blood that should not be there such as ethylene glycol, diethylene glycol, methanol, and their many metabolic products.
In our patient, ingestion of both methanol and ethylene glycol should be considered, since she lives alone and has been suspected of alcohol and opioid abuse. Her calculated osmol gap is 278 mOsm/kg. Her measured osmolality is 318 mOsm/kg (Table 1). The osmol gap is 40 mOsm/kg (normal is ≤ 10).4,5 Therefore, her osmol gap is elevated.
Identifying the specific substance the patient ingested that caused metabolic acidosis with anion gap may be difficult. Poisonings with these agents do not always increase the osmol gap.6 A high index of suspicion is essential. It is helpful to have the family search for any sources of ethylene glycol and methanol at home and initiate treatment early if an ingestion is suspected, using fomepizole (an alcohol dehydrogenase inhibitor) or parenteral ethanol and hemodialysis.7 Liquid chromatography identifies these two toxins, but results are not available emergently.
Diethylene glycol ingestion should also be considered.8 Since it is diagnosed and treated like ethylene glycol intoxication, it can be placed with the “E” of (di)ethylene glycol in the mnemonic.
Uremia
Renal failure can lead to metabolic acidosis.9 Our patient has no history of kidney disease, but her blood urea nitrogen and creatinine concentrations are above normal, and her estimated glomerular filtration rate by the Modification of Diet in Renal Disease formula is 48 mL/min/1.73 m2—low, but not uremic.
Rhabdomyolysis (suspected by elevated creatine kinase values) should be considered in any patient with mental status changes, suspected toxic ingestion, and metabolic acidosis (see the “I” in MUD PILES below). Compartment syndromes with muscle necrosis may present in a subtle fashion. Therefore, renal failure from rhabdomyolysis may complicate this patient’s course later, and should be kept in mind.
Diabetes
The patient has no history of diabetes and has a normal blood glucose level. Blood testing did not reveal ketones. She is not taking metformin (alleged to cause lactic acidosis) or a sodium-glucose cotransporter 2 inhibitor (which have been associated with ketoacidosis).10
There is another, less common cause of ketoacidosis: alcohol.11 Although alcoholism is common, alcoholic ketoacidosis is uncommon, even in heavy drinkers. Ethyl alcohol causing metabolic acidosis is similar to metabolic acidosis with (di)ethylene glycol and methanol, and if suspected it should be treated empirically (first with thiamine, then dextrose and saline, and correcting other electrolyte disturbances such as hypokalemia and hypomagnesemia) before specific identification is made. Ketones (predominantly beta-hydroxybutyrate) may persist up to 2 weeks after alcohol ingestion has stopped.11 Ketosis in the setting of alcoholic ketoacidosis is frequently accompanied by other markers of alcohol target organ injury: elevated bilirubin, aspartate aminotransferase, alanine aminotransferase, and gamma-glutamyl transferase levels. The term “ketohepatitis” has been suggested as an alternative to alcoholic ketosis.11
This patient did not have an elevated blood ethanol level, and her liver markers were otherwise normal.
THE NEW MUD PILES
2. Which of the following is (are) true? Regarding the remaining letters of the MUD PILES mnemonic:
- The “P” (paraldehyde) has been replaced by pyroglutamic acid (5-oxoproline) and propylene glycol.
- There are two isomers of lactate (dextro and levo), and consequently two clinical varieties of lactic acidosis.
- Isoniazid is no longer associated with metabolic acidosis with elevated anion gap.
- Salicylates can paradoxically be associated both with elevated and low anion gaps.
Isoniazid is still associated with metabolic acidosis with elevated anion gap, and so the third answer choice is false; the rest are true.
Paraldehyde, isoniazid, lactate
The “P,” “I,” and “L” (d-lactate) of the revamped MUD PILES acronym are less common than the others. They should be considered when the more typical causes of metabolic acidosis are not present, as in this patient.
UPDATING THE ‘P’ IN MUD PILES
Paraldehyde is rarely prescribed anymore. A PubMed search on December 21, 2015 applying the terms paraldehyde and metabolic acidosis yielded 17 results. Those specific to anion gap metabolic acidosis were from 1957 to 1986 (n = 9).12–20
Therefore, we can eliminate paraldehyde from the MUD PILES mnemonic and replace it with pyroglutamic acid and propylene glycol.
5-Oxoproline or pyroglutamic acid, a metabolite of acetaminophen
Acetaminophen depletes glutathione stores in acute overdoses, in patients with inborn errors of metabolism, and after chronic ingestion of excessive, frequent doses. Depletion of glutathione increases metabolic products, including pyroglutamic acid, which dissociates into hydrogen ions (leading to metabolic acidosis and an anion gap), and 5-oxoproline, (which can be detected in the urine).21,22
Risk factors for metabolic acidosis with acetaminophen ingestion include malnutrition, chronic alcoholism, liver disease, and female sex. In fact, most cases have been reported in females, and altered mental status has been common.
Metabolic acidosis with pyroglutamic acid can occur without elevated acetaminophen levels. Serum and urine levels of pyroglutamic acid may assist with diagnosis. Since identification of urine pyroglutamic acid usually requires outside laboratory assistance, a clinical diagnosis is often made initially and corroborated later by laboratory results. When the anion gap metabolic acidosis is multifactorial, as it was suspected to be in a case reported by Tan et al,23 the osmol gap may be elevated as a consequence of additional toxic ingestions, as it was in the reported patient.
No controlled studies of treatment have been done. n-Acetylcysteine may be of benefit. Occasional patients have been dialyzed for removal of excess pyroglutamic acid.
Propylene glycol, a component of parenteral lorazepam
Lorazepam is a hydrophobic drug, so when it is given parenterally, it must be mixed with a suitable solvent. A typical formulation adds propylene glycol. In patients receiving high doses of lorazepam as relaxation therapy for acute respiratory distress syndrome in the intensive care unit, or as treatment of alcohol withdrawal, the propylene glycol component can precipitate anion gap metabolic acidosis.24,25
Although nearly one-half of the administered propylene glycol is excreted by the kidneys, the remaining substrate is metabolized by alcohol dehydrogenase into d,l-lactaldehyde, then converted into d- or l-lactate. l-Lactate can be metabolized, but d-lactate cannot and leads to anion gap metabolic acidosis. This is another toxic metabolic acidosis associated with an elevated osmol gap. An increasing osmol gap in the intensive care unit can serve as a surrogate marker of excessive propylene glycol administration.23
Isoniazid
Although it is uncommon, there are reports of isoniazid-induced anion gap metabolic acidosis,26 either due to overdoses, or less commonly, with normal dosing. Isoniazid should therefore remain in the mnemonic MUD PILES and may be suspected when metabolic acidosis is accompanied by seizures unresponsive to usual therapy. The seizures respond to pyridoxine.
The “I” should also be augmented by newer causes of metabolic acidosis associated with “ingestions.” Ecstasy, or 3,4-methylenedioxymethamphetamine, can cause metabolic acidosis and seizures. Ecstasy has been associated with rhabdomyolysis and uremia, also leading to anion gap metabolic acidosis.27 A newer class of abused substances, synthetic cathinones (“bath salts”), are associated with metabolic acidosis, compartment syndrome, and renal failure.28
Lactic acidosis
Lactic acidosis and metabolic acidosis can result from hypoperfusion (type A) or other causes (type B). Not all lactic acidosis is contingent on l-lactate, which humans can metabolize. Metabolic acidosis may be a consequence of d-lactate (mammals have no d-lactate dehydrogenase). d-Lactic acidosis as a result of short bowel syndrome has been known for more than a generation.29 However, d-lactic acidosis occurs in another new setting. The new “P” in MUD PILES, propylene glycol, can generate substantial amounts of d-lactate.29
d-lactic metabolic acidosis is always accompanied by neurologic manifestations (slurred speech, confusion, somnolence, ataxia, abusive behavior, and others).30 With short bowel syndrome, the neurologic manifestations occur after eating and clear later.30
Although our patient’s anion gap is more than 20 mmol/L, her blood level of lactate is not elevated, and she had no history to suggest short-bowel syndrome.
Salicylates
Salicylate overdose can cause a mixed acid-base disorder: metabolic acidosis with elevated anion gap and respiratory alkalosis.
Although our patient does not have respiratory alkalosis, an aspirin overdose must be considered. A salicylate level was ordered; it was negative.
Despite the typical association of salicylates with an elevated anion gap, they may also cause a negative anion gap.31 Chloride-sensing ion-specific electrodes contain a membrane permeable to chloride. Salicylates can increase the chloride permeability of these membranes, generating pseudohyperchloremia, and consequently, a negative anion gap.
WHAT ELSE MUST BE CONSIDERED?
3. In view of her anion gap metabolic acidosis, elevated osmol gap, and absence of diabetes, renal failure, or lactate excess, what are the remaining diagnoses to consider in this patient? (Choose all that are potential sources of metabolic acidosis and an increased anion gap.)
- Methanol, ethylene, or diethylene glycol
- Excessive, chronic acetaminophen ingestion
- Salicylate toxicity
- Alcoholic ketoacidosis
All of the above can potentially contribute to metabolic acidosis.
A search of the patient’s home did not reveal a source of methanol or either ethylene or diethylene glycol. Similarly, no aspirin was found, and the patient’s salicylate levels were not elevated. The patient’s laboratory work did not reveal increased ketones.
Since none of the common causes of metabolic acidosis were discovered, and since the patient had been taking acetaminophen, the diagnosis of excessive chronic acetaminophen ingestion was suspected pending laboratory verification. Identification of 5-oxoproline in the urine may take a week or more since the sample is usually sent to special laboratories. Acetaminophen levels in this patient were significantly elevated, as were urinary oxyproline levels, which returned later.
The patient was diagnosed with pyroglutamic acid metabolic acidosis. She was treated supportively and with n-acetylcysteine intravenously, although there have been no controlled studies of the efficacy of this drug. Seventy-two hours after admission, she had improved. Her acid-base status returned to normal.
GOLD MARK: ANOTHER WAY TO REMEMBER
Another mnemonic device for remembering the causes of metabolic acidosis with elevated anion gap is “GOLD MARK”: glycols (ethylene and propylene), oxoproline (instead of pyroglutamic acid from acetaminophen), l-lactate, d-lactate, methanol, aspirin, renal failure, and ketoacidosis).32
ACID-BASE DISORDERS IN DIFFERENT DISEASES
Diverse diseases cause distinctive acid-base abnormalities. Matching the appropriate acid-base abnormality with its associated disease may lead to more timely diagnosis and treatment:
Type 2 diabetes mellitus, for example, can lead to lactic acidosis, ketoacidosis, or type 4 renal tubular acidosis.33
Heart failure, although not typically framed in the context of acid-base physiology, can lead to elevated lactate, which is associated with a worse prognosis.34
Acquired immunodeficiency syndrome. Abacavir can cause normal anion gap metabolic acidosis.35,36
Cancer37,38 can be associated with proximal tubular renal tubular acidosis and lactic acidosis.
An expanding array of toxic ingestions
Metabolic acidosis may be the most prominent and potentially lethal clinical acid-base disturbance. When metabolic acidosis occurs in certain disease states—lactic acidosis with hypoperfusion or methanol ingestion with metabolic acidosis, for example—there is increased morbidity and mortality.
As reflected in the revisions to MUD PILES and in the newer GOLD MARK acronym, the osmol gap has become more valuable in differential diagnosis of metabolic acidosis with an elevated anion gap consequent to an expanding array of toxic ingestions (methanol, propylene glycol, pyroglutamic acid-oxoproline, ethylene glycol, and diethylene glycol), which may accompany pyroglutamic acid-oxoproline.
A 78-year-old black woman with a history of osteoarthrosis and chronic diffuse joint pain presents with altered mental status and tachypnea, which began 3 hours earlier. She lives alone, and her family suspects she abuses both alcohol and her pain medications. She has not been eating well and has lost approximately 10 pounds over the past 3 months. Her analgesic regimen includes acetaminophen and acetaminophen-oxycodone.
In the emergency department her temperature is 98.6°F (37.0°C), pulse 100 beats per minute and regular, respiratory rate 22 per minute, and blood pressure 136/98 mm Hg. She is obtunded but has no focal neurologic defects or meningismus. She has no signs of heart failure (jugular venous distention, cardiomegaly, or gallops), and examination of the lungs and abdomen is unremarkable.
Suspecting that the patient may have taken too much oxycodone, the physician gives her naloxone, but her mental status does not improve. Results of chest radiography and cranial computed tomography are unremarkable. The physician’s initial impression is that the patient has “metabolic encephalopathy of unknown etiology.”
The patient’s laboratory values are shown in Table 1.
WHICH ACID-BASE DISORDER DOES SHE HAVE?
1. Which acid-base disorder does this patient have?
- Metabolic acidosis and respiratory alkalosis
- Metabolic acidosis and respiratory acidosis
- Metabolic acidosis with an elevated anion gap
- A triple disturbance: metabolic acidosis, respiratory acidosis, and metabolic alkalosis
A 5-step approach
Acid-base disorders can be diagnosed and characterized using a systematic approach known as the “Rules of 5” (Table 2)1:
1. Determine the arterial pH status.
2. Determine whether the primary process is respiratory, metabolic, or both.
3. Calculate the anion gap.
4. Check the degree of compensation (respiratory or metabolic).
5. If the patient has metabolic acidosis with an elevated anion gap, check whether the bicarbonate level has decreased as much as the anion gap has increased (ie, whether there is a delta gap).
Let us apply this approach to the patient described above.
1. What is her pH status?
An arterial pH less than 7.40 is acidemic, whereas a pH higher than 7.44 is alkalemic. (Acidemia and alkalemia refer to the abnormal laboratory value, while acidosis and alkalosis refer to the process causing the abnormal value—a subtle distinction, but worth keeping in mind.)
Caveat. A patient may have a significant acid-base disorder even if the pH is normal. Therefore, even if the pH is normal, one should verify that the partial pressure of carbon dioxide (Pco2), bicarbonate level, and anion gap are normal. If they are not, the patient may have a mixed acid-base disorder such as respiratory acidosis superimposed on metabolic alkalosis.
Our patient’s pH is 7.25, which is in the acidemic range.
2. Is her acidosis respiratory, metabolic, or both?
Respiratory acidosis and alkalosis affect the Pco2. The Pco2 is high in respiratory acidosis (due to failure to get rid of excess carbon dioxide), whereas it is low in respiratory alkalosis (due to loss of too much carbon dioxide through hyperventilation).
Metabolic acidosis and alkalosis, on the other hand, affect the serum bicarbonate level. In metabolic acidosis the bicarbonate level is low, whereas in metabolic alkalosis the bicarbonate level is high.
Moreover, in mixed respiratory and metabolic acidosis, the bicarbonate level can be low and the Pco2 can be high. In mixed metabolic and respiratory alkalosis, the bicarbonate level can be high and the Pco2 can be low (Table 2).
Our patient’s serum bicarbonate level is low at 16.0 mmol/L, indicating that the process is metabolic. Her Pco2 is also low (28 mm Hg), which reflects an appropriate response to compensate for the acidosis.
3. What is her anion gap?
Always calculate the anion gap, ie, the serum sodium concentration minus the serum chloride and serum bicarbonate concentrations. If the patient’s serum albumin level is low, for every 1 gram it is below normal, an additional 2.5 mmol/L should be added to the calculated anion gap. We consider an anion gap of 10 mmol/L or less as normal.
Caveats. The blood sample used to calculate the anion gap should be drawn close in time to the arterial blood gas sample.
Although the anion gap is an effective tool in assessing acid-base disorders, further investigation is warranted if clinical judgment suggests that an anion gap calculation is inconsistent with the patient’s circumstances.2
Our patient’s anion gap is elevated (21 mmol/L). Her serum albumin level is in the normal range, so her anion gap does not need to be adjusted.
4. Is the degree of compensation appropriate for the primary acid-base disturbance?
The kidneys compensate for the lungs, and vice versa. That is, in respiratory acidosis or alkalosis, the kidneys adjust the bicarbonate levels, and in metabolic acidosis, the lungs adjust the Pco2 (although in metabolic alkalosis, it is hard for patients to breathe less, especially if they are already hypoxic).
In metabolic acidosis, people compensate by breathing harder to get rid of more carbon dioxide. For every 1-mmol/L decrease in the bicarbonate level, the Pco2 should decrease by 1.3 mm Hg.
Compensation does not return pH to normal; rather, it mitigates the impact of an acid or alkali excess or deficit. If the pH is normalized with an underlying acid-base disturbance, there may be mixed acid-base processes rather than compensation.
Our patient’s bicarbonate level is 16 mmol/L, which is 9 mmol/L lower than normal (for acid-base calculations, we use 25 mmol/L as the nominal normal level). If she is compensating appropriately, her Pco2 should decline from 40 mm Hg (the nominal normal level) by about 11.7 mm Hg (9 × 1.3), to approximately 28.3 mm Hg. Her Pco2 is, indeed, 28 mm Hg, indicating that she is compensating adequately for her metabolic acidosis.
If we use Winter’s formula instead (Pco2 = [1.5 × the bicarbonate level] + 8 ± 2),3 the lowest calculated Pco2 would be 30 mm Hg, which is within 2 mm Hg of the Rules of 5 calculation. Other formulas for calculating compensation are available.3
This information rules out the first two answers to question 1, ie, metabolic acidosis with respiratory alkalosis or acidosis.
5. Is there a delta gap?
Although we know the patient has metabolic acidosis with an elevated anion gap, we have not ruled out the possibility that she may have a triple disturbance. For this reason we need to check her delta gap.
In metabolic acidosis with an elevated anion gap, as the bicarbonate level decreases, the anion gap should increase by the same amount. If the bicarbonate level decreases more than the anion gap increases, the additional decline is the result of a second process—an additional normal-anion-gap acidosis. If the bicarbonate level does not decrease as much as the anion gap increases, there is an additional metabolic alkalosis.
Our patient’s bicarbonate level decreased 9 mmol/L (from the nominal normal level of 25 to 16), and therefore her anion gap should have increased approximately the same amount—and it did. (A normal anion gap for problem-solving is 10, and this patient’s anion gap has increased to 21. A difference of ± 2 is insignificant.) This conclusion verifies that a triple acid-base disturbance is not present, so the last answer is incorrect.
So, the correct answer to the question posed above is metabolic acidosis with an elevated anion gap (that is, metabolic acidosis with appropriate respiratory compensation).
‘MUD PILES’: FINDING THE CAUSE OF ANION GAP METABOLIC ACIDOSIS
The possible causes of metabolic acidosis with an elevated anion gap (as in our patient) can be summarized in the mnemonic MUD PILES (methanol, uremia, diabetes, paraldehyde, isoniazid, lactate, ethylene glycol, and salicylates), which has been used for many years. Parts of it are no longer useful, but rather than discard it, we propose to update it (Table 3).
Methanol and ethylene glycol
We will address toxic ingestion of methanol and ethylene glycol (the “M” and “E” of MUD PILES) at the same time.
In cases of suspected ingestion of toxic substances such as these, it is useful to examine the osmol gap, ie, the difference between the calculated and the measured serum osmolality. Serum osmolality (in mOsm/kg) is calculated as the sodium concentration in mmol/L times 2, plus the glucose concentration in mg/dL divided by 18, plus the blood urea nitrogen concentration in mg/dL divided by 2.8 (Table 4). If the measured osmolality is higher than this calculated value, the difference may be due to solutes in the blood that should not be there such as ethylene glycol, diethylene glycol, methanol, and their many metabolic products.
In our patient, ingestion of both methanol and ethylene glycol should be considered, since she lives alone and has been suspected of alcohol and opioid abuse. Her calculated osmol gap is 278 mOsm/kg. Her measured osmolality is 318 mOsm/kg (Table 1). The osmol gap is 40 mOsm/kg (normal is ≤ 10).4,5 Therefore, her osmol gap is elevated.
Identifying the specific substance the patient ingested that caused metabolic acidosis with anion gap may be difficult. Poisonings with these agents do not always increase the osmol gap.6 A high index of suspicion is essential. It is helpful to have the family search for any sources of ethylene glycol and methanol at home and initiate treatment early if an ingestion is suspected, using fomepizole (an alcohol dehydrogenase inhibitor) or parenteral ethanol and hemodialysis.7 Liquid chromatography identifies these two toxins, but results are not available emergently.
Diethylene glycol ingestion should also be considered.8 Since it is diagnosed and treated like ethylene glycol intoxication, it can be placed with the “E” of (di)ethylene glycol in the mnemonic.
Uremia
Renal failure can lead to metabolic acidosis.9 Our patient has no history of kidney disease, but her blood urea nitrogen and creatinine concentrations are above normal, and her estimated glomerular filtration rate by the Modification of Diet in Renal Disease formula is 48 mL/min/1.73 m2—low, but not uremic.
Rhabdomyolysis (suspected by elevated creatine kinase values) should be considered in any patient with mental status changes, suspected toxic ingestion, and metabolic acidosis (see the “I” in MUD PILES below). Compartment syndromes with muscle necrosis may present in a subtle fashion. Therefore, renal failure from rhabdomyolysis may complicate this patient’s course later, and should be kept in mind.
Diabetes
The patient has no history of diabetes and has a normal blood glucose level. Blood testing did not reveal ketones. She is not taking metformin (alleged to cause lactic acidosis) or a sodium-glucose cotransporter 2 inhibitor (which have been associated with ketoacidosis).10
There is another, less common cause of ketoacidosis: alcohol.11 Although alcoholism is common, alcoholic ketoacidosis is uncommon, even in heavy drinkers. Ethyl alcohol causing metabolic acidosis is similar to metabolic acidosis with (di)ethylene glycol and methanol, and if suspected it should be treated empirically (first with thiamine, then dextrose and saline, and correcting other electrolyte disturbances such as hypokalemia and hypomagnesemia) before specific identification is made. Ketones (predominantly beta-hydroxybutyrate) may persist up to 2 weeks after alcohol ingestion has stopped.11 Ketosis in the setting of alcoholic ketoacidosis is frequently accompanied by other markers of alcohol target organ injury: elevated bilirubin, aspartate aminotransferase, alanine aminotransferase, and gamma-glutamyl transferase levels. The term “ketohepatitis” has been suggested as an alternative to alcoholic ketosis.11
This patient did not have an elevated blood ethanol level, and her liver markers were otherwise normal.
THE NEW MUD PILES
2. Which of the following is (are) true? Regarding the remaining letters of the MUD PILES mnemonic:
- The “P” (paraldehyde) has been replaced by pyroglutamic acid (5-oxoproline) and propylene glycol.
- There are two isomers of lactate (dextro and levo), and consequently two clinical varieties of lactic acidosis.
- Isoniazid is no longer associated with metabolic acidosis with elevated anion gap.
- Salicylates can paradoxically be associated both with elevated and low anion gaps.
Isoniazid is still associated with metabolic acidosis with elevated anion gap, and so the third answer choice is false; the rest are true.
Paraldehyde, isoniazid, lactate
The “P,” “I,” and “L” (d-lactate) of the revamped MUD PILES acronym are less common than the others. They should be considered when the more typical causes of metabolic acidosis are not present, as in this patient.
UPDATING THE ‘P’ IN MUD PILES
Paraldehyde is rarely prescribed anymore. A PubMed search on December 21, 2015 applying the terms paraldehyde and metabolic acidosis yielded 17 results. Those specific to anion gap metabolic acidosis were from 1957 to 1986 (n = 9).12–20
Therefore, we can eliminate paraldehyde from the MUD PILES mnemonic and replace it with pyroglutamic acid and propylene glycol.
5-Oxoproline or pyroglutamic acid, a metabolite of acetaminophen
Acetaminophen depletes glutathione stores in acute overdoses, in patients with inborn errors of metabolism, and after chronic ingestion of excessive, frequent doses. Depletion of glutathione increases metabolic products, including pyroglutamic acid, which dissociates into hydrogen ions (leading to metabolic acidosis and an anion gap), and 5-oxoproline, (which can be detected in the urine).21,22
Risk factors for metabolic acidosis with acetaminophen ingestion include malnutrition, chronic alcoholism, liver disease, and female sex. In fact, most cases have been reported in females, and altered mental status has been common.
Metabolic acidosis with pyroglutamic acid can occur without elevated acetaminophen levels. Serum and urine levels of pyroglutamic acid may assist with diagnosis. Since identification of urine pyroglutamic acid usually requires outside laboratory assistance, a clinical diagnosis is often made initially and corroborated later by laboratory results. When the anion gap metabolic acidosis is multifactorial, as it was suspected to be in a case reported by Tan et al,23 the osmol gap may be elevated as a consequence of additional toxic ingestions, as it was in the reported patient.
No controlled studies of treatment have been done. n-Acetylcysteine may be of benefit. Occasional patients have been dialyzed for removal of excess pyroglutamic acid.
Propylene glycol, a component of parenteral lorazepam
Lorazepam is a hydrophobic drug, so when it is given parenterally, it must be mixed with a suitable solvent. A typical formulation adds propylene glycol. In patients receiving high doses of lorazepam as relaxation therapy for acute respiratory distress syndrome in the intensive care unit, or as treatment of alcohol withdrawal, the propylene glycol component can precipitate anion gap metabolic acidosis.24,25
Although nearly one-half of the administered propylene glycol is excreted by the kidneys, the remaining substrate is metabolized by alcohol dehydrogenase into d,l-lactaldehyde, then converted into d- or l-lactate. l-Lactate can be metabolized, but d-lactate cannot and leads to anion gap metabolic acidosis. This is another toxic metabolic acidosis associated with an elevated osmol gap. An increasing osmol gap in the intensive care unit can serve as a surrogate marker of excessive propylene glycol administration.23
Isoniazid
Although it is uncommon, there are reports of isoniazid-induced anion gap metabolic acidosis,26 either due to overdoses, or less commonly, with normal dosing. Isoniazid should therefore remain in the mnemonic MUD PILES and may be suspected when metabolic acidosis is accompanied by seizures unresponsive to usual therapy. The seizures respond to pyridoxine.
The “I” should also be augmented by newer causes of metabolic acidosis associated with “ingestions.” Ecstasy, or 3,4-methylenedioxymethamphetamine, can cause metabolic acidosis and seizures. Ecstasy has been associated with rhabdomyolysis and uremia, also leading to anion gap metabolic acidosis.27 A newer class of abused substances, synthetic cathinones (“bath salts”), are associated with metabolic acidosis, compartment syndrome, and renal failure.28
Lactic acidosis
Lactic acidosis and metabolic acidosis can result from hypoperfusion (type A) or other causes (type B). Not all lactic acidosis is contingent on l-lactate, which humans can metabolize. Metabolic acidosis may be a consequence of d-lactate (mammals have no d-lactate dehydrogenase). d-Lactic acidosis as a result of short bowel syndrome has been known for more than a generation.29 However, d-lactic acidosis occurs in another new setting. The new “P” in MUD PILES, propylene glycol, can generate substantial amounts of d-lactate.29
d-lactic metabolic acidosis is always accompanied by neurologic manifestations (slurred speech, confusion, somnolence, ataxia, abusive behavior, and others).30 With short bowel syndrome, the neurologic manifestations occur after eating and clear later.30
Although our patient’s anion gap is more than 20 mmol/L, her blood level of lactate is not elevated, and she had no history to suggest short-bowel syndrome.
Salicylates
Salicylate overdose can cause a mixed acid-base disorder: metabolic acidosis with elevated anion gap and respiratory alkalosis.
Although our patient does not have respiratory alkalosis, an aspirin overdose must be considered. A salicylate level was ordered; it was negative.
Despite the typical association of salicylates with an elevated anion gap, they may also cause a negative anion gap.31 Chloride-sensing ion-specific electrodes contain a membrane permeable to chloride. Salicylates can increase the chloride permeability of these membranes, generating pseudohyperchloremia, and consequently, a negative anion gap.
WHAT ELSE MUST BE CONSIDERED?
3. In view of her anion gap metabolic acidosis, elevated osmol gap, and absence of diabetes, renal failure, or lactate excess, what are the remaining diagnoses to consider in this patient? (Choose all that are potential sources of metabolic acidosis and an increased anion gap.)
- Methanol, ethylene, or diethylene glycol
- Excessive, chronic acetaminophen ingestion
- Salicylate toxicity
- Alcoholic ketoacidosis
All of the above can potentially contribute to metabolic acidosis.
A search of the patient’s home did not reveal a source of methanol or either ethylene or diethylene glycol. Similarly, no aspirin was found, and the patient’s salicylate levels were not elevated. The patient’s laboratory work did not reveal increased ketones.
Since none of the common causes of metabolic acidosis were discovered, and since the patient had been taking acetaminophen, the diagnosis of excessive chronic acetaminophen ingestion was suspected pending laboratory verification. Identification of 5-oxoproline in the urine may take a week or more since the sample is usually sent to special laboratories. Acetaminophen levels in this patient were significantly elevated, as were urinary oxyproline levels, which returned later.
The patient was diagnosed with pyroglutamic acid metabolic acidosis. She was treated supportively and with n-acetylcysteine intravenously, although there have been no controlled studies of the efficacy of this drug. Seventy-two hours after admission, she had improved. Her acid-base status returned to normal.
GOLD MARK: ANOTHER WAY TO REMEMBER
Another mnemonic device for remembering the causes of metabolic acidosis with elevated anion gap is “GOLD MARK”: glycols (ethylene and propylene), oxoproline (instead of pyroglutamic acid from acetaminophen), l-lactate, d-lactate, methanol, aspirin, renal failure, and ketoacidosis).32
ACID-BASE DISORDERS IN DIFFERENT DISEASES
Diverse diseases cause distinctive acid-base abnormalities. Matching the appropriate acid-base abnormality with its associated disease may lead to more timely diagnosis and treatment:
Type 2 diabetes mellitus, for example, can lead to lactic acidosis, ketoacidosis, or type 4 renal tubular acidosis.33
Heart failure, although not typically framed in the context of acid-base physiology, can lead to elevated lactate, which is associated with a worse prognosis.34
Acquired immunodeficiency syndrome. Abacavir can cause normal anion gap metabolic acidosis.35,36
Cancer37,38 can be associated with proximal tubular renal tubular acidosis and lactic acidosis.
An expanding array of toxic ingestions
Metabolic acidosis may be the most prominent and potentially lethal clinical acid-base disturbance. When metabolic acidosis occurs in certain disease states—lactic acidosis with hypoperfusion or methanol ingestion with metabolic acidosis, for example—there is increased morbidity and mortality.
As reflected in the revisions to MUD PILES and in the newer GOLD MARK acronym, the osmol gap has become more valuable in differential diagnosis of metabolic acidosis with an elevated anion gap consequent to an expanding array of toxic ingestions (methanol, propylene glycol, pyroglutamic acid-oxoproline, ethylene glycol, and diethylene glycol), which may accompany pyroglutamic acid-oxoproline.
- Whittier WL, Rutecki GW. Primer on clinical acid-base problem solving. Dis Mon 2004; 50:122–162.
- Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol 2007; 2:162–174.
- Adrogué HJ, Madias NE. Secondary responses to altered acid-base status: the rules of engagement. J Am Soc Nephrol 2010; 21:920–923.
- Krasowski MD, Wilcoxon RM, Miron J. A retrospective analysis of glycol and toxic alcohol ingestion: utility of anion and osmolal gaps. BMC Clin Pathol 2012;12:1.
- Latus J, Kimmel M, Alscher MD, Braun N. Ethylene glycol poisoning: a rare but life-threatening cause of metabolic acidosis—a single-centre experience. Clin Kidney J 2012; 5:120–123.
- Kraut JA. Diagnosis of toxic alcohols: limitations of present methods. Clin Toxicol (Phila) 2015; 53:589–595.
- Ghannoum M, Hoffman RS, Mowry JB, Lavergne V. Trends in toxic alcohol exposures in the United States from 2000 to 2013: a focus on the use of antidotes and extracorporeal treatments. Semin Dial 2014; 27:395–401.
- Schep LJ, Slaughter RJ, Temple WA, Beasley DM. Diethylene glycol poisoning. Clin Toxicol (Phila) 2009; 47:525–535.
- Kraut JA, Madias NE. Metabolic acidosis of CKD: an update. Am J Kidney Dis 2016; 67:307–317.
- Taylor SI, Blau JE, Rother KI. SGLT2 inhibitors may predispose to ketoacidosis. J Clin Endocrinol Metab 2015; 100:2849–2852.
- Yokoyama A, Yokoyama T, Mizukami T, et al. Alcoholic ketosis: prevalence, determinants, and ketohepatitis in Japanese alcoholic men. Alcohol Alcohol 2014; 49:618–625.
- Hayward JN, Boshell BR. Paraldehyde intoxication with metabolic acidosis; report of two cases, experimental data and a critical review of the literature. Am J Med 1957; 23:965–976.
- Elkinton JR, Huth EJ, Clark JK, Barker ES, Seligson D. Renal tubular acidosis with organic aciduria during paraldehyde ingestion; six year study of an unusual case. Am J Med 1957; 23:977–986.
- Waterhouse C, Stern EA. Metabolic acidosis occurring during administration of paraldehyde. Am J Med 1957; 23:987–989.
- Beier LS, Pitts WH, Gonick HC. Metabolic acidosis occurring during paraldehyde intoxication. Ann Intern Med 1963; 58:155–158.
- Hiemcke T. Metabolic acidosis due to paraldehyde. Ned Tijdschr Geneeskd 1964; 108:2165–2167. Dutch.
- Gailitis RJ. Paraldehyde acidosis syndrome. IMJ III Med J 1966; 129:258–262.
- Gutman RA, Burnell JM. Paraldehyde acidosis. Am J Med 1967; 42:435–440.
- Hadden JW, Metzner RJ. Pseudoketosis and hyperacetaldehydemia in paraldehyde acidosis. Am J Med 1969; 47:642–647.
- Linter CM, Linter SP. Severe lactic acidosis following paraldehyde administration. Br J Psychiatry 1986; 149:650–651.
- Zand L, Muriithi A, Nelsen E, et al. Severe anion gap metabolic acidosis from acetaminophen use secondary to 5-oxoproline (pyroglutamic acid) accumulation. Am J Med Sci 2012; 344:501–504.
- Abkur TM, Mohammed W, Ali M, Casserly L. Acetaminophen-induced anion gap metabolic acidosis secondary to 5-oxoproline: a case report. J Med Case Rep 2014; 8:409.
- Tan EM, Kalimullah E, Sohail MR, Ramar K. Diagnostic challenge in a patient with severe anion gap metabolic acidosis. Case Rep Crit Care 2015; 2015:272914.
- Jorens PG, Demey HE, Schepens PJ, et al. Unusual d-lactic acid acidosis from propylene glycol metabolism in overdose. J Toxicol Clin Toxicol 2004; 42:163–169.
- Barnes BJ, Gerst C, Smith JR, Terrell AR, Mullins ME. Osmol gap as a surrogate marker for serum propylene glycol concentrations in patients receiving lorazepam for sedation. Pharmacotherapy 2006; 26:23–33.
- Gokhale YA, Vaidya MS, Mehta AD, Rathod NN. Isoniazid toxicity presenting as status epilepticus and severe metabolic acidosis. J Assoc Physicians India 2009; 57:70–71.
- Ben-Abraham R, Szold O, Rudick V, Weinbroum AA. ‘Ecstasy’ intoxication: life-threatening manifestations and resuscitative measures in the intensive care setting. Eur J Emerg Med 2003; 10:309–313.
- German CL, Fleckenstein AE, Hanson GR. Bath salts and synthetic cathinones: an emerging designer drug phenomenon. Life Sci 2014; 97:2–8.
- Jorens PG, Demey HE, Schepens PJ, et al. Unusual d-lactic acidosis from propylene glycol metabolism in overdose. J Toxicol Clin Toxicol 2004; 42:163–169.
- Kang KP, Le S, Kang SK. d-Lactic acidosis in humans: review and update. Electrolyte Blood Press 2006; 4:53–56.
- Emmett M. Approach to the patient with a negative anion gap. Am J Kidney Dis 2016; 67:143–150.
- Mehta AN, Emmett JB, Emmett M. GOLD MARK: an anion gap mnemonic for the 21st Century. Lancet 2008; 372:892.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Park JJ, Choi DJ, Yoon CH, et al; KorHF Registry. The prognostic value of arterial blood gas analysis in high-risk acute heart failure patients: an analysis of the Korean Heart Failure (KorHF) registry. Eur J Heart Fail 2015; 17:601–611.
- Musso CG, Belloso WH, Glassock RJ. Water, electrolytes, and acid-base alterations in human immunodeficiency virus infected patients. World J Nephrol 2016; 5:33–42.
- Camara-Lemarroy CR, Flores-Cantu H, Calderon-Hernandez HJ, Diaz-Torres MA, Villareal-Velazquez HJ. Drug-induced haemolysis, renal failure, thrombocytopenia and lactic acidosis in patients with HIV and cryptococcal meningitis: a diagnostic challenge. Int J STD AIDS 2015; 26:1052–1054.
- Miltiadous G, Christidis D, Kalogirou M, Elisaf M. Causes and mechanisms of acid-base and electrolyte abnormalities in cancer. Eur J Intern Med 2008; 19:1–7.
- Vlachostergios PJ, Oikonomou KG, Gibilaro E, Apergis G. Elevated lactic acid is a negative prognostic factor in metastatic lung cancer. Cancer Biomark 2015; 15:725–734.
- Whittier WL, Rutecki GW. Primer on clinical acid-base problem solving. Dis Mon 2004; 50:122–162.
- Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol 2007; 2:162–174.
- Adrogué HJ, Madias NE. Secondary responses to altered acid-base status: the rules of engagement. J Am Soc Nephrol 2010; 21:920–923.
- Krasowski MD, Wilcoxon RM, Miron J. A retrospective analysis of glycol and toxic alcohol ingestion: utility of anion and osmolal gaps. BMC Clin Pathol 2012;12:1.
- Latus J, Kimmel M, Alscher MD, Braun N. Ethylene glycol poisoning: a rare but life-threatening cause of metabolic acidosis—a single-centre experience. Clin Kidney J 2012; 5:120–123.
- Kraut JA. Diagnosis of toxic alcohols: limitations of present methods. Clin Toxicol (Phila) 2015; 53:589–595.
- Ghannoum M, Hoffman RS, Mowry JB, Lavergne V. Trends in toxic alcohol exposures in the United States from 2000 to 2013: a focus on the use of antidotes and extracorporeal treatments. Semin Dial 2014; 27:395–401.
- Schep LJ, Slaughter RJ, Temple WA, Beasley DM. Diethylene glycol poisoning. Clin Toxicol (Phila) 2009; 47:525–535.
- Kraut JA, Madias NE. Metabolic acidosis of CKD: an update. Am J Kidney Dis 2016; 67:307–317.
- Taylor SI, Blau JE, Rother KI. SGLT2 inhibitors may predispose to ketoacidosis. J Clin Endocrinol Metab 2015; 100:2849–2852.
- Yokoyama A, Yokoyama T, Mizukami T, et al. Alcoholic ketosis: prevalence, determinants, and ketohepatitis in Japanese alcoholic men. Alcohol Alcohol 2014; 49:618–625.
- Hayward JN, Boshell BR. Paraldehyde intoxication with metabolic acidosis; report of two cases, experimental data and a critical review of the literature. Am J Med 1957; 23:965–976.
- Elkinton JR, Huth EJ, Clark JK, Barker ES, Seligson D. Renal tubular acidosis with organic aciduria during paraldehyde ingestion; six year study of an unusual case. Am J Med 1957; 23:977–986.
- Waterhouse C, Stern EA. Metabolic acidosis occurring during administration of paraldehyde. Am J Med 1957; 23:987–989.
- Beier LS, Pitts WH, Gonick HC. Metabolic acidosis occurring during paraldehyde intoxication. Ann Intern Med 1963; 58:155–158.
- Hiemcke T. Metabolic acidosis due to paraldehyde. Ned Tijdschr Geneeskd 1964; 108:2165–2167. Dutch.
- Gailitis RJ. Paraldehyde acidosis syndrome. IMJ III Med J 1966; 129:258–262.
- Gutman RA, Burnell JM. Paraldehyde acidosis. Am J Med 1967; 42:435–440.
- Hadden JW, Metzner RJ. Pseudoketosis and hyperacetaldehydemia in paraldehyde acidosis. Am J Med 1969; 47:642–647.
- Linter CM, Linter SP. Severe lactic acidosis following paraldehyde administration. Br J Psychiatry 1986; 149:650–651.
- Zand L, Muriithi A, Nelsen E, et al. Severe anion gap metabolic acidosis from acetaminophen use secondary to 5-oxoproline (pyroglutamic acid) accumulation. Am J Med Sci 2012; 344:501–504.
- Abkur TM, Mohammed W, Ali M, Casserly L. Acetaminophen-induced anion gap metabolic acidosis secondary to 5-oxoproline: a case report. J Med Case Rep 2014; 8:409.
- Tan EM, Kalimullah E, Sohail MR, Ramar K. Diagnostic challenge in a patient with severe anion gap metabolic acidosis. Case Rep Crit Care 2015; 2015:272914.
- Jorens PG, Demey HE, Schepens PJ, et al. Unusual d-lactic acid acidosis from propylene glycol metabolism in overdose. J Toxicol Clin Toxicol 2004; 42:163–169.
- Barnes BJ, Gerst C, Smith JR, Terrell AR, Mullins ME. Osmol gap as a surrogate marker for serum propylene glycol concentrations in patients receiving lorazepam for sedation. Pharmacotherapy 2006; 26:23–33.
- Gokhale YA, Vaidya MS, Mehta AD, Rathod NN. Isoniazid toxicity presenting as status epilepticus and severe metabolic acidosis. J Assoc Physicians India 2009; 57:70–71.
- Ben-Abraham R, Szold O, Rudick V, Weinbroum AA. ‘Ecstasy’ intoxication: life-threatening manifestations and resuscitative measures in the intensive care setting. Eur J Emerg Med 2003; 10:309–313.
- German CL, Fleckenstein AE, Hanson GR. Bath salts and synthetic cathinones: an emerging designer drug phenomenon. Life Sci 2014; 97:2–8.
- Jorens PG, Demey HE, Schepens PJ, et al. Unusual d-lactic acidosis from propylene glycol metabolism in overdose. J Toxicol Clin Toxicol 2004; 42:163–169.
- Kang KP, Le S, Kang SK. d-Lactic acidosis in humans: review and update. Electrolyte Blood Press 2006; 4:53–56.
- Emmett M. Approach to the patient with a negative anion gap. Am J Kidney Dis 2016; 67:143–150.
- Mehta AN, Emmett JB, Emmett M. GOLD MARK: an anion gap mnemonic for the 21st Century. Lancet 2008; 372:892.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Park JJ, Choi DJ, Yoon CH, et al; KorHF Registry. The prognostic value of arterial blood gas analysis in high-risk acute heart failure patients: an analysis of the Korean Heart Failure (KorHF) registry. Eur J Heart Fail 2015; 17:601–611.
- Musso CG, Belloso WH, Glassock RJ. Water, electrolytes, and acid-base alterations in human immunodeficiency virus infected patients. World J Nephrol 2016; 5:33–42.
- Camara-Lemarroy CR, Flores-Cantu H, Calderon-Hernandez HJ, Diaz-Torres MA, Villareal-Velazquez HJ. Drug-induced haemolysis, renal failure, thrombocytopenia and lactic acidosis in patients with HIV and cryptococcal meningitis: a diagnostic challenge. Int J STD AIDS 2015; 26:1052–1054.
- Miltiadous G, Christidis D, Kalogirou M, Elisaf M. Causes and mechanisms of acid-base and electrolyte abnormalities in cancer. Eur J Intern Med 2008; 19:1–7.
- Vlachostergios PJ, Oikonomou KG, Gibilaro E, Apergis G. Elevated lactic acid is a negative prognostic factor in metastatic lung cancer. Cancer Biomark 2015; 15:725–734.
Imaging suggestive, but symptoms atypical
A 66-year-old man with chronic obstructive pulmonary disease (COPD) was brought to the emergency department with a 2-week history of progressive dyspnea followed by altered mental status. He had no history of diabetes mellitus, hypertension, or drug abuse.
On physical examination, he was stuporous. He had no fever or hypotension, but his pulse and breathing were rapid, and he had central cyanosis, bilateral conjunctival congestion, a puffy face, generalized wheezing, basilar crackles in both lungs, and leg edema.
Laboratory testing showed hypoxia and severe hypercarbia. His hematocrit was 65% (reference range 39–51) and his hemoglobin level was 21.5 g/dL (13–17).
The patient was diagnosed with an exacerbation of COPD. He was intubated, placed on mechanical ventilation, and admitted to the intensive care unit.
Computed tomography (CT) performed because of his decreased level of consciousness (Figure 1) showed increased attenuation in the ambient cistern and the lateral aspect of the lateral cerebral fissure, suggesting subarachnoid hemorrhage. The attenuation value in these areas was 89 Hounsfield units (typical values for brain tissue are in the 20s to 30s, and for blood in the 30s to 40s). To further evaluate for subarachnoid hemorrhage, lumbar puncture was performed, but analysis of the fluid sample showed normal protein and glucose levels and no cells.
Based on the results of cerebrospinal fluid evaluation and on the CT attenuation value, a diagnosis of pseudosubarachnoid hemorrhage due to polycythemia was made.
SUBARACHNOID VS PSEUDOSUBARACHNOID HEMORRHAGE
Subarachnoid hemorrhage typically begins with a “thunder-clap” headache (beginning suddenly and described by patients as “the worst headache ever.”) While not all patients have this presentation, if imaging suggests subarachnoid hemorrhage but the patient has atypical signs and symptoms (eg, other than headache), then pseudosubarachnoid hemorrhage should be considered.
Brain CT is one of the most reliable tools for diagnosing subarachnoid hemorrhage in the emergency department. Done within 6 hours of symptom onset, it has a sensitivity of 98.7% and a specificity of 99.9%.1 Magnetic resonance imaging can also visualize subarachnoid hemorrhage within the first 12 hours, typically as a hyperintensity in the subarachnoid space on fluid-attenuated inversion-recovery sequences2 and on susceptibility-weighted sequences.
Lumbar puncture is also an important diagnostic tool but carries a risk of brain herniation in patients with brain edema.
Pseudosubarachnoid hemorrhage is an artifact of CT imaging. It is rare, and its prevalence is unknown.3 However, it may be seen in up to 20% of patients after resuscitation, as a result of diffuse cerebral edema that lowers the attenuation of brain tissue on CT, making the vessels relatively conspicuous. It can also be seen in purulent meningitis (due to proteinaceous influx after blood-brain barrier disruption),4 in meningeal leukemia (due to increased cellularity in the leptomeninges), and in severe polycythemia (from a higher concentration of blood and hemoglobin in the vessels).3,5–7
Although the level of attenuation on CT may help distinguish subarachnoid from pseudosubarachnoid hemorrhage, its accuracy has not been defined. Inspecting the CT images may clarify whether areas with high attenuation look like blood vessels vs subarachnoid hemorrhage.
Our patient recovered and had an uneventful follow-up. The cause of his elevated hematocrit was likely chronic hypoxia from COPD.
Acknowledgment: We thank Dr. Saeide Khanbagi and Dr. Azade Nasr-lari for their cooperation.
- Dubosh NM, Bellolio MF, Rabinstein AA, Edlow JA. Sensitivity of early brain computed tomography to exclude aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. Stroke 2016; 47:750–755.
- Sohn CH, Baik SK, Lee HJ, et al. MR imaging of hyperacute subarachnoid and intraventricular hemorrhage at 3T: a preliminary report of gradient echo T2*-weighted sequences. AJNR Am J Neuroradiol 2005; 26:662–665.
- Yuzawa H, Higano S, Mugikura S, et al. Pseudo-subarachnoid hemorrhage found in patients with postresuscitation encephalopathy: characteristics of CT findings and clinical importance. AJNR Am J Neuroradiol 2008; 29:1544–1549.
- Given CA 2nd, Burdette JH, Elster AD, Williams DW 3rd. Pseudo-subarachnoid hemorrhage: a potential imaging pitfall associated with diffuse cerebral edema. AJNR Am J Neuroradiol 2003; 24:254–256.
- Avrahami E, Katz R, Rabin A, Friedman V. CT diagnosis of non-traumatic subarachnoid haemorrhage in patients with brain edema. Eur J Radiol 1998; 28:222–225.
- Ben Salem D, Osseby GV, Rezaizadeh-Bourdariat K, et al. Spontaneous hyperdense intracranial vessels seen on CT scan in polycythemia cases. J Radiol 2003; 84:605–608. French.
- Hsieh SW, Khor GT, Chen CN, Huang P. Pseudo subarachnoid hemorrhage in meningeal leukemia. J Emerg Med 2012; 42:e109–e111.
A 66-year-old man with chronic obstructive pulmonary disease (COPD) was brought to the emergency department with a 2-week history of progressive dyspnea followed by altered mental status. He had no history of diabetes mellitus, hypertension, or drug abuse.
On physical examination, he was stuporous. He had no fever or hypotension, but his pulse and breathing were rapid, and he had central cyanosis, bilateral conjunctival congestion, a puffy face, generalized wheezing, basilar crackles in both lungs, and leg edema.
Laboratory testing showed hypoxia and severe hypercarbia. His hematocrit was 65% (reference range 39–51) and his hemoglobin level was 21.5 g/dL (13–17).
The patient was diagnosed with an exacerbation of COPD. He was intubated, placed on mechanical ventilation, and admitted to the intensive care unit.
Computed tomography (CT) performed because of his decreased level of consciousness (Figure 1) showed increased attenuation in the ambient cistern and the lateral aspect of the lateral cerebral fissure, suggesting subarachnoid hemorrhage. The attenuation value in these areas was 89 Hounsfield units (typical values for brain tissue are in the 20s to 30s, and for blood in the 30s to 40s). To further evaluate for subarachnoid hemorrhage, lumbar puncture was performed, but analysis of the fluid sample showed normal protein and glucose levels and no cells.
Based on the results of cerebrospinal fluid evaluation and on the CT attenuation value, a diagnosis of pseudosubarachnoid hemorrhage due to polycythemia was made.
SUBARACHNOID VS PSEUDOSUBARACHNOID HEMORRHAGE
Subarachnoid hemorrhage typically begins with a “thunder-clap” headache (beginning suddenly and described by patients as “the worst headache ever.”) While not all patients have this presentation, if imaging suggests subarachnoid hemorrhage but the patient has atypical signs and symptoms (eg, other than headache), then pseudosubarachnoid hemorrhage should be considered.
Brain CT is one of the most reliable tools for diagnosing subarachnoid hemorrhage in the emergency department. Done within 6 hours of symptom onset, it has a sensitivity of 98.7% and a specificity of 99.9%.1 Magnetic resonance imaging can also visualize subarachnoid hemorrhage within the first 12 hours, typically as a hyperintensity in the subarachnoid space on fluid-attenuated inversion-recovery sequences2 and on susceptibility-weighted sequences.
Lumbar puncture is also an important diagnostic tool but carries a risk of brain herniation in patients with brain edema.
Pseudosubarachnoid hemorrhage is an artifact of CT imaging. It is rare, and its prevalence is unknown.3 However, it may be seen in up to 20% of patients after resuscitation, as a result of diffuse cerebral edema that lowers the attenuation of brain tissue on CT, making the vessels relatively conspicuous. It can also be seen in purulent meningitis (due to proteinaceous influx after blood-brain barrier disruption),4 in meningeal leukemia (due to increased cellularity in the leptomeninges), and in severe polycythemia (from a higher concentration of blood and hemoglobin in the vessels).3,5–7
Although the level of attenuation on CT may help distinguish subarachnoid from pseudosubarachnoid hemorrhage, its accuracy has not been defined. Inspecting the CT images may clarify whether areas with high attenuation look like blood vessels vs subarachnoid hemorrhage.
Our patient recovered and had an uneventful follow-up. The cause of his elevated hematocrit was likely chronic hypoxia from COPD.
Acknowledgment: We thank Dr. Saeide Khanbagi and Dr. Azade Nasr-lari for their cooperation.
A 66-year-old man with chronic obstructive pulmonary disease (COPD) was brought to the emergency department with a 2-week history of progressive dyspnea followed by altered mental status. He had no history of diabetes mellitus, hypertension, or drug abuse.
On physical examination, he was stuporous. He had no fever or hypotension, but his pulse and breathing were rapid, and he had central cyanosis, bilateral conjunctival congestion, a puffy face, generalized wheezing, basilar crackles in both lungs, and leg edema.
Laboratory testing showed hypoxia and severe hypercarbia. His hematocrit was 65% (reference range 39–51) and his hemoglobin level was 21.5 g/dL (13–17).
The patient was diagnosed with an exacerbation of COPD. He was intubated, placed on mechanical ventilation, and admitted to the intensive care unit.
Computed tomography (CT) performed because of his decreased level of consciousness (Figure 1) showed increased attenuation in the ambient cistern and the lateral aspect of the lateral cerebral fissure, suggesting subarachnoid hemorrhage. The attenuation value in these areas was 89 Hounsfield units (typical values for brain tissue are in the 20s to 30s, and for blood in the 30s to 40s). To further evaluate for subarachnoid hemorrhage, lumbar puncture was performed, but analysis of the fluid sample showed normal protein and glucose levels and no cells.
Based on the results of cerebrospinal fluid evaluation and on the CT attenuation value, a diagnosis of pseudosubarachnoid hemorrhage due to polycythemia was made.
SUBARACHNOID VS PSEUDOSUBARACHNOID HEMORRHAGE
Subarachnoid hemorrhage typically begins with a “thunder-clap” headache (beginning suddenly and described by patients as “the worst headache ever.”) While not all patients have this presentation, if imaging suggests subarachnoid hemorrhage but the patient has atypical signs and symptoms (eg, other than headache), then pseudosubarachnoid hemorrhage should be considered.
Brain CT is one of the most reliable tools for diagnosing subarachnoid hemorrhage in the emergency department. Done within 6 hours of symptom onset, it has a sensitivity of 98.7% and a specificity of 99.9%.1 Magnetic resonance imaging can also visualize subarachnoid hemorrhage within the first 12 hours, typically as a hyperintensity in the subarachnoid space on fluid-attenuated inversion-recovery sequences2 and on susceptibility-weighted sequences.
Lumbar puncture is also an important diagnostic tool but carries a risk of brain herniation in patients with brain edema.
Pseudosubarachnoid hemorrhage is an artifact of CT imaging. It is rare, and its prevalence is unknown.3 However, it may be seen in up to 20% of patients after resuscitation, as a result of diffuse cerebral edema that lowers the attenuation of brain tissue on CT, making the vessels relatively conspicuous. It can also be seen in purulent meningitis (due to proteinaceous influx after blood-brain barrier disruption),4 in meningeal leukemia (due to increased cellularity in the leptomeninges), and in severe polycythemia (from a higher concentration of blood and hemoglobin in the vessels).3,5–7
Although the level of attenuation on CT may help distinguish subarachnoid from pseudosubarachnoid hemorrhage, its accuracy has not been defined. Inspecting the CT images may clarify whether areas with high attenuation look like blood vessels vs subarachnoid hemorrhage.
Our patient recovered and had an uneventful follow-up. The cause of his elevated hematocrit was likely chronic hypoxia from COPD.
Acknowledgment: We thank Dr. Saeide Khanbagi and Dr. Azade Nasr-lari for their cooperation.
- Dubosh NM, Bellolio MF, Rabinstein AA, Edlow JA. Sensitivity of early brain computed tomography to exclude aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. Stroke 2016; 47:750–755.
- Sohn CH, Baik SK, Lee HJ, et al. MR imaging of hyperacute subarachnoid and intraventricular hemorrhage at 3T: a preliminary report of gradient echo T2*-weighted sequences. AJNR Am J Neuroradiol 2005; 26:662–665.
- Yuzawa H, Higano S, Mugikura S, et al. Pseudo-subarachnoid hemorrhage found in patients with postresuscitation encephalopathy: characteristics of CT findings and clinical importance. AJNR Am J Neuroradiol 2008; 29:1544–1549.
- Given CA 2nd, Burdette JH, Elster AD, Williams DW 3rd. Pseudo-subarachnoid hemorrhage: a potential imaging pitfall associated with diffuse cerebral edema. AJNR Am J Neuroradiol 2003; 24:254–256.
- Avrahami E, Katz R, Rabin A, Friedman V. CT diagnosis of non-traumatic subarachnoid haemorrhage in patients with brain edema. Eur J Radiol 1998; 28:222–225.
- Ben Salem D, Osseby GV, Rezaizadeh-Bourdariat K, et al. Spontaneous hyperdense intracranial vessels seen on CT scan in polycythemia cases. J Radiol 2003; 84:605–608. French.
- Hsieh SW, Khor GT, Chen CN, Huang P. Pseudo subarachnoid hemorrhage in meningeal leukemia. J Emerg Med 2012; 42:e109–e111.
- Dubosh NM, Bellolio MF, Rabinstein AA, Edlow JA. Sensitivity of early brain computed tomography to exclude aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. Stroke 2016; 47:750–755.
- Sohn CH, Baik SK, Lee HJ, et al. MR imaging of hyperacute subarachnoid and intraventricular hemorrhage at 3T: a preliminary report of gradient echo T2*-weighted sequences. AJNR Am J Neuroradiol 2005; 26:662–665.
- Yuzawa H, Higano S, Mugikura S, et al. Pseudo-subarachnoid hemorrhage found in patients with postresuscitation encephalopathy: characteristics of CT findings and clinical importance. AJNR Am J Neuroradiol 2008; 29:1544–1549.
- Given CA 2nd, Burdette JH, Elster AD, Williams DW 3rd. Pseudo-subarachnoid hemorrhage: a potential imaging pitfall associated with diffuse cerebral edema. AJNR Am J Neuroradiol 2003; 24:254–256.
- Avrahami E, Katz R, Rabin A, Friedman V. CT diagnosis of non-traumatic subarachnoid haemorrhage in patients with brain edema. Eur J Radiol 1998; 28:222–225.
- Ben Salem D, Osseby GV, Rezaizadeh-Bourdariat K, et al. Spontaneous hyperdense intracranial vessels seen on CT scan in polycythemia cases. J Radiol 2003; 84:605–608. French.
- Hsieh SW, Khor GT, Chen CN, Huang P. Pseudo subarachnoid hemorrhage in meningeal leukemia. J Emerg Med 2012; 42:e109–e111.
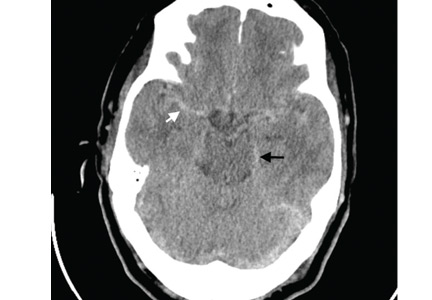
Does Chronic Complaining Mask Acute Problem?
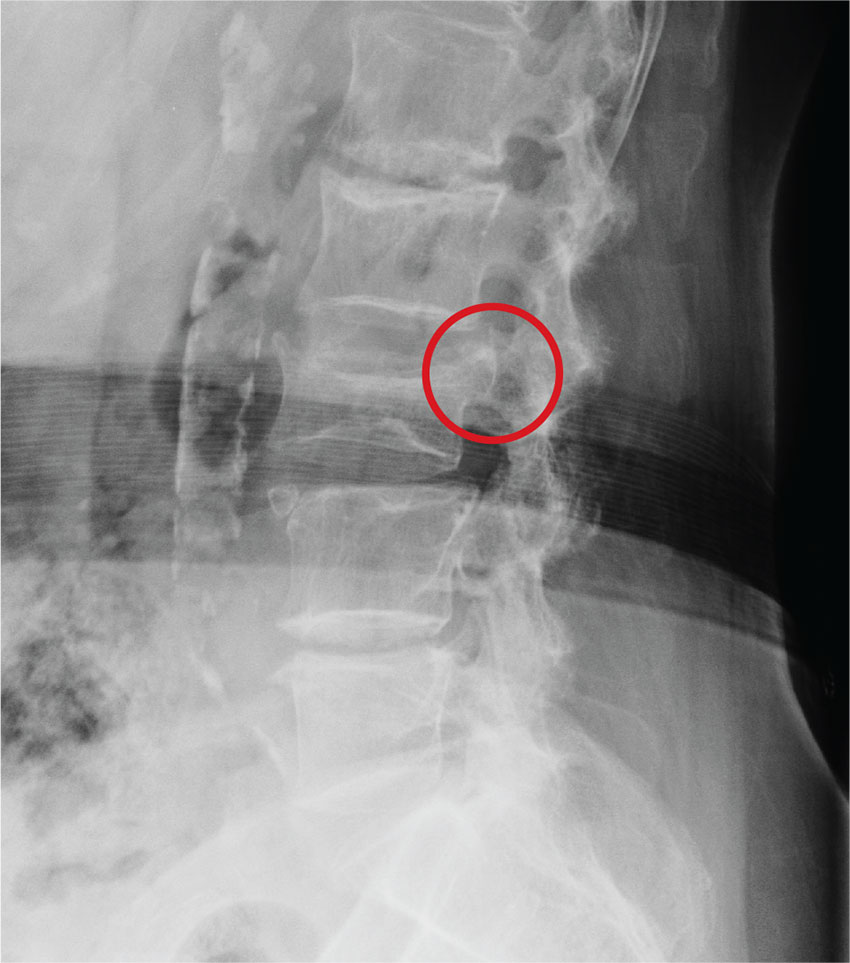
ANSWER
The radiograph demonstrates age-related degenerative changes. Of note is a moderate compression fracture of L3 with close to 50% loss of height. It is difficult to say for sure the fracture is acute; by plain film alone, it would be deemed age indeterminate.
Of concern, though, is the posterior portion of the superior endplate of L3, which appears to be posteriorly displaced into the spinal canal. This finding suggests possible retropulsion and, if this fracture was in fact acute, would suggest a possible unstable fracture. Further imaging is warranted.
The patient underwent noncontrast CT of the lumbar spine, which demonstrated that the fracture was acute. It also indicated that there was dorsal retropulsion causing close to 50% canal compromise. The patient was subsequently admitted for further workup and evaluation.
ANSWER
The radiograph demonstrates age-related degenerative changes. Of note is a moderate compression fracture of L3 with close to 50% loss of height. It is difficult to say for sure the fracture is acute; by plain film alone, it would be deemed age indeterminate.
Of concern, though, is the posterior portion of the superior endplate of L3, which appears to be posteriorly displaced into the spinal canal. This finding suggests possible retropulsion and, if this fracture was in fact acute, would suggest a possible unstable fracture. Further imaging is warranted.
The patient underwent noncontrast CT of the lumbar spine, which demonstrated that the fracture was acute. It also indicated that there was dorsal retropulsion causing close to 50% canal compromise. The patient was subsequently admitted for further workup and evaluation.
ANSWER
The radiograph demonstrates age-related degenerative changes. Of note is a moderate compression fracture of L3 with close to 50% loss of height. It is difficult to say for sure the fracture is acute; by plain film alone, it would be deemed age indeterminate.
Of concern, though, is the posterior portion of the superior endplate of L3, which appears to be posteriorly displaced into the spinal canal. This finding suggests possible retropulsion and, if this fracture was in fact acute, would suggest a possible unstable fracture. Further imaging is warranted.
The patient underwent noncontrast CT of the lumbar spine, which demonstrated that the fracture was acute. It also indicated that there was dorsal retropulsion causing close to 50% canal compromise. The patient was subsequently admitted for further workup and evaluation.
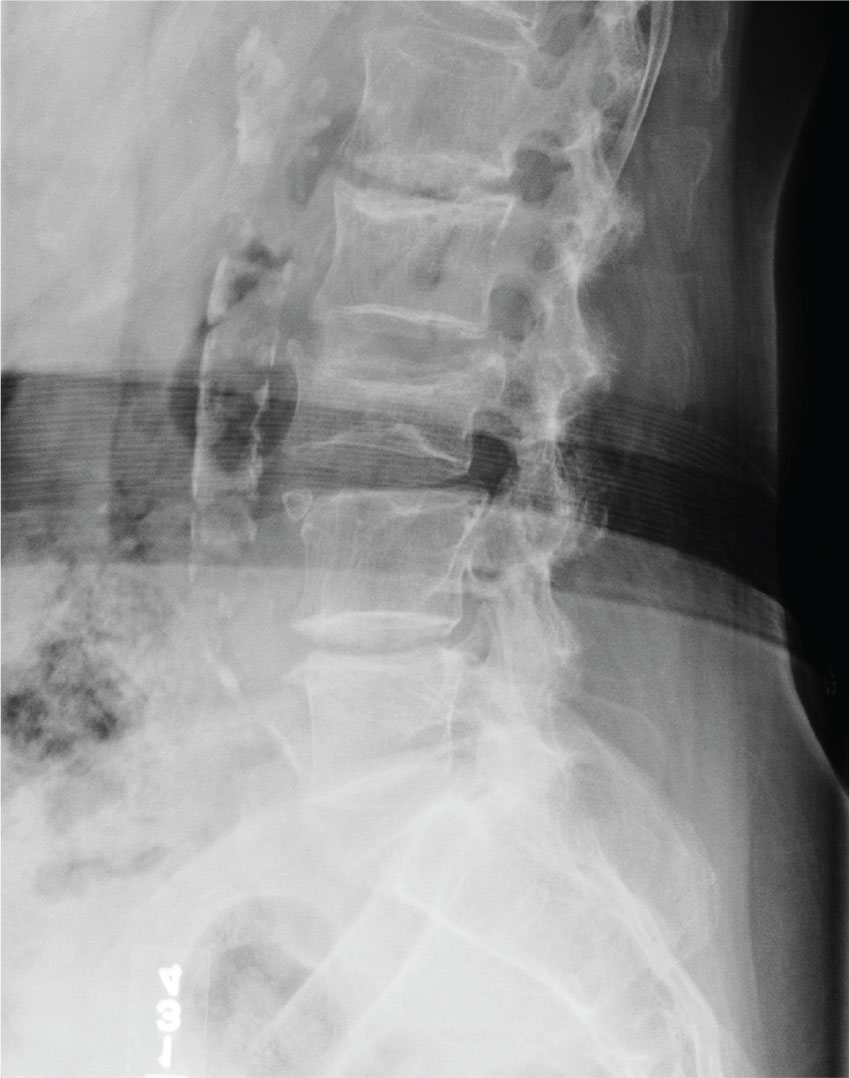
An 80-year-old woman who resides in a nursing home is sent to urgent care for evaluation of back pain. There have been no witnessed injuries or falls. The notes from the nursing home staff indicate that the patient, who has severe dementia, tends to chronically complain about one ailment or another. However, the patient’s family seems to feel she is complaining more than usual.
Her only other medical history is controlled hypertension. Her vital signs are stable. On physical exam, you note an elderly female who is pleasant but very confused. She is able to follow simple commands and appears to be able to move all her extremities well, with no obvious neurologic compromise. Inspection of her back does not demonstrate any obvious wounds, bruising, or step-offs. She does have generalized tenderness as you palpate her lumbar region.
The triage nurse already ordered lumbar spine radiographs, which have been completed. The lateral view is shown. What is your impression?