User login
Aortic dissection: Prompt diagnosis and emergency treatment are critical
A 50-year-old man developed severe chest pain and collapsed to the floor. The pain was sudden in onset, was burning in quality, and was located in the center of his chest. Emergency medical services arrived a few minutes later and found the patient diaphoretic and cyanotic, with an initial blood pressure of 74/54 mm Hg and a heart rate of 125 beats per minute. He was rushed to the hospital.
His medical history was unremarkable. He smoked one pack of cigarettes per day for 20 years. His father died of a “heart attack” at age 52.
In the emergency department he underwent echocardiography with a portable handheld unit, which showed a pericardial effusion and cardiac tamponade. He was sent for emergency computed tomography of the chest, which revealed an aneurysm of the aortic root and acute type A (Stanford classification) aortic dissection with hemopericardium.
He underwent emergency cardiac surgery. At the time of surgery, he was in cardiogenic shock from aortic dissection complicated by severe aortic regurgitation and cardiac tamponade with hemopericardium. The aortic valve was trileaflet. A 27-mm St. Jude composite valve graft root replacement was performed.
The patient did well and was discharged home 7 days after surgery. Pathologic study of the aorta revealed cystic medial degeneration. He did not have any features of Marfan syndrome or Loeys-Dietz syndrome. His three children underwent evaluation, and each had a normal physical examination and echocardiographic test results.
A HIGH INDEX OF SUSPICION IS CRITICAL
Acute aortic dissection is the most common aortic catastrophe, with an incidence estimated at 5 to 30 per 1 million people per year, amounting to nearly 10,000 cases per year in the United States.1–4
The diagnosis of acute aortic dissection has many potential pitfalls.2,3 Aortic dissection may mimic other more common conditions, such as coronary ischemia, pleurisy, heart failure, stroke, and acute abdominal illness. Because acute aortic dissection may be rapidly fatal, one must maintain a high index of suspicion.2,3 Prompt diagnosis and emergency treatment are critical.
WHAT CAUSES AORTIC DISSECTION?
One hypothesis is that acute aortic dissection is caused by a primary tear in the aortic intima, with blood from the aortic lumen penetrating into the diseased media leading to dissection and creating a true and false lumen.2 Another is that rupture of the vasa vasorum leads to hemorrhage in the aortic wall with subsequent intimal disruption, creating the intimal tear and aortic dissection.
Once a dissection starts, pulsatile flow of blood within the aortic wall causes it to extend. The dissection flap may be localized, but it often spirals the entire length of the aorta. Distention of the false lumen with blood may cause the intimal flap to compress the true lumen and potentially lead to malperfusion syndromes.
CLASSIFIED ACCORDING TO LOCATION
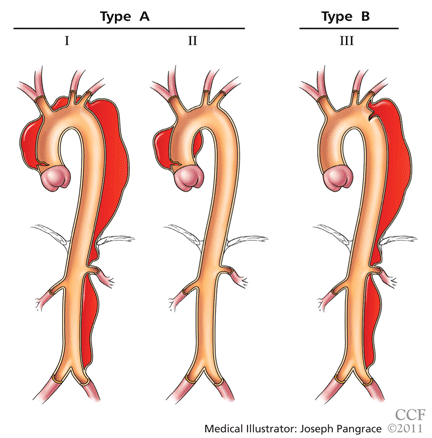
It is important to recognize the location of the dissection, as the prognosis and treatment depend on whether the ascending aorta is involved.2,3 For classification purposes, the ascending aorta is the portion proximal to the brachiocephalic artery, while the descending aorta is the portion distal to the left subclavian artery.3
The DeBakey classification defines a type I aortic dissection as one that begins in the ascending aorta and extends at least to the aortic arch or beyond. Type II dissections involve the ascending aorta only, while type III dissections begin in the descending aorta, most often just distal to the left subclavian artery.
The Stanford classification scheme divides dissections into type A and type B. Type A dissections involve the ascending aorta, while type B dissections do not involve the ascending aorta.
Which classification scheme is used is not important. However, identifying patients with dissection of the ascending aorta (DeBakey type I or type II or Stanford type A) is critical, as emergency cardiac surgery is recommended for this type of dissection.2,3 For the purposes of this paper, the Stanford classification scheme will be used.
Dissection that involves the ascending aorta most commonly occurs in people ages 50 to 60, whereas acute dissection of the descending aorta typically occurs in people 10 years older.1,2
An acute aortic dissection is one that has occurred within 2 weeks of symptom onset. A chronic dissection is one that occurred more than 2 weeks after symptoms began.
DISEASES AND CONDITIONS ASSOCIATED WITH AORTIC DISSECTION

Hypertension and disorders leading to disruption of the normal structure and function of the aortic wall. About 75% of patients with acute aortic dissection have underlying hypertension.1–3
Cystic medial degeneration is a common pathologic feature in many cases of aortic dissection.

Cocaine use and intense weight-lifting increase the shear stresses on the aorta.2,3
Inflammatory aortic diseases such as giant cell arteritis.
Pregnancy can be complicated by aortic dissection, usually in the setting of an underlying aortopathy.5
Iatrogenic aortic dissection accounts for about 4% of cases, as a result of cardiac surgery, catheterization, stenting, or use of an intra-aortic balloon pump.1
Aortic aneurysm. Patients with thoracic aortic aneurysm are at higher risk of aortic dissection, and the larger the aortic diameter, the higher the risk.2,3,6 In the International Registry of Acute Aortic Dissection (IRAD), the average size of the aorta was about 5.3 cm at the time of acute dissection. Importantly, about 40% of acute dissections of the ascending aorta occur in patients with ascending aortic diameters less than 5.0 cm.7,8
Thus, many factors are associated with acute dissection, and specific reasons leading to an individual’s susceptibility to sudden dissection are poorly understood.
CLINICAL FEATURES OF ACUTE AORTIC DISSECTION
Because the symptoms of acute dissection may mimic other, more common conditions, one of the most important factors in the diagnosis of aortic dissection is a high clinical suspicion.1–3
What is the pretest risk of disease?
Recently, the American College of Cardiology (ACC) and the American Heart Association (AHA) released joint guidelines on thoracic aortic disease.3 These guidelines provide an approach to patients who have complaints that may represent acute thoracic aortic dissection, the intent being to establish a pretest risk of disease to be used to guide decision-making.3
The focused evaluation includes specific questions about underlying conditions, symptoms, and findings on examination that may greatly increase the likelihood of acute dissection. These include:
- High-risk conditions and historical features associated with aortic dissection, such as Marfan syndrome and other genetic disorders (Table 2), bicuspid aortic valve, family history of thoracic aortic aneurysm or dissection, known thoracic aortic aneurysm, and recent aortic manipulation
- Pain in the chest, back, or abdomen with high-risk features (eg, abrupt onset, severe intensity, or a ripping or tearing quality)
- High-risk findings on examination (eg, pulse deficits, new aortic regurgitation, hypotension, shock, or systolic blood pressure differences).
Using this information, expedited aortic imaging and treatment algorithms have been devised to improve the diagnosis.3
Using the IRAD database of more than 2,500 acute dissections, the diagnostic algorithm proposed in the ACC/AHA guidelines was shown to be highly sensitive (about 95%) for detecting acute aortic dissection.4 In addition, using this score may expedite evaluation by classifying certain patients as being at high risk of acute dissection.3,4
Important to recognize is that almost two-thirds of patients who suffered dissection in this large database did not have one of the “high-risk conditions” associated with dissection.4 Additionally, the specificity of the ACC/AHA algorithm is unknown, and further testing is necessary.4
Acute onset of severe pain
More than 90% of acute dissections present with acute pain in the chest or the back, or both.1–3 The pain is usually severe, of sudden onset, and often described as sharp or, occasionally, tearing, ripping, or stabbing. The pain usually differs from that of coronary ischemia, being most severe at its onset as opposed to the less intense, crescendo-like pain of angina or myocardial infarction. The pain may migrate as the dissection progresses along the length of the aorta or to branch vessels. It may abate, leading to a false sense of security in the patient and the physician.3 “Painless” dissection occurs in a minority, usually in those with syncope, neurologic symptoms, or heart failure.1–3
The patient with acute dissection may be anxious and may feel a sense of doom.
Acute heart failure, related to severe aortic regurgitation, may be a predominant symptom in dissection of the ascending aorta.
Syncope may occur as a result of aortic rupture, hemopericardium with cardiac tamponade, or acute neurologic complications.
Vascular insufficiency may occur in any branch vessel, leading to clinical syndromes that include acute myocardial infarction, stroke, paraplegia, paraparesis, mesenteric ischemia, and limb ischemia.
PHYSICAL FINDINGS CAN VARY WIDELY
Findings on physical examination in acute aortic dissection vary widely depending on underlying conditions and on the specific complications of the dissection.
Although the classic presentation is acute, severe pain in the chest or back in a severely hypertensive patient with aortic regurgitation and pulse deficits, most patients do not have all these characteristics.4 Most patients with type B dissection are hypertensive on presentation, but many with type A dissection present with normal blood pressure or hypotension.1 Pulse deficits (unequal or absent pulses) are reported in 10% to 30% of acute dissections and may be intermittent as the dynamic movement of the dissection flap interferes with branch vessel perfusion.1–3
- Aortic leaflet prolapse or distortion of the leaflet alignment
- Malcoaptation of the aortic leaflets from dilation of the aortic root and annulus
- Prolapse of the intimal flap across the aortic valve, interfering with valve function
- Preexisting aortic regurgitation from underlying aortic root aneurysm or primary aortic valve disease (such as a bicuspid aortic valve).
Neurologic manifestations are most common in dissection of the ascending aorta and are particularly important to recognize, as they may dominate the clinical presentation and lead to delay in the diagnosis of dissection.2,3 Neurologic syndromes include:
- Persistent or transient ischemic stroke
- Spinal cord ischemia
- Ischemic neuropathy
- Hypoxic encephalopathy.
These manifestations are related to malperfusion to branches supplying the brain, spinal cord, or peripheral nerves.9
Syncope is relatively common in aortic dissection and may be related to acute hypotension caused by cardiac tamponade or aortic rupture, cerebral vessel obstruction, or activation of cerebral baroreceptors.2,9 It is important to consider aortic dissection in the differential diagnosis in cases of unexplained syncope.3
Aortic dissections may extend into the abdominal aorta, leading to vascular complications involving one or more branch vessels.10 The renal artery is involved in at least 5% to 10% of cases and may lead to renal ischemia, infarction, renal insufficiency, or refractory hypertension.2Mesenteric ischemia or infarction occurs in about 5% of dissections, may be difficult to diagnose, and is particularly dangerous.2,8 Aortic dissection may extend into the iliac arteries and may cause acute lower extremity ischemia.
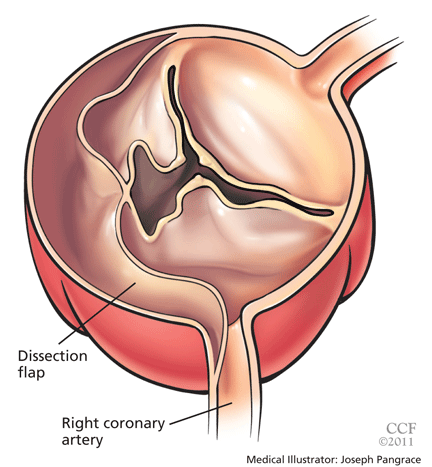
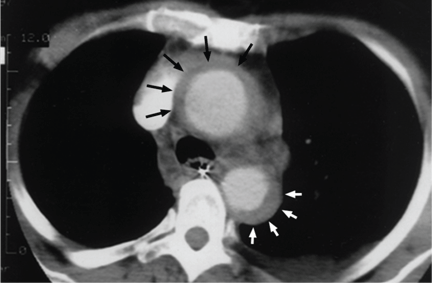
Acute myocardial infarction due to involvement of the dissection flap causing malperfusion of a coronary artery occurs in 1% to 7% of acute type A aortic dissections.1–3 The right coronary artery (Figure 2) is most commonly involved, leading to acute inferior myocardial infarction. Acute myocardial ischemia and infarction in the setting of dissection may lead to a delay in the diagnosis of dissection and to bleeding complications from antiplatelet and anticoagulant drugs given to treat the acute coronary syndrome.
Cardiac tamponade, occurring in about 10% of acute type A dissections, portends a higher risk of death.2,3
Additional clinical features of aortic dissection include a left-sided pleural effusion, usually related to an inflammatory response. An acute hemothorax may occur from rupture or leaking of a descending aortic dissection.
FINDINGS ON RADIOGRAPHY AND ELECTROCARDIOGRAPHY
Electrocardiography usually has normal or nonspecific findings, unless acute myocardial infarction complicates the dissection.
D-DIMER LEVELS
Biomarkers for the diagnosis of acute aortic dissection are of great interest.
D-dimer levels rise in acute aortic dissection as they do in pulmonary embolism.11 A D-dimer level greater than 1,600 ng/mL within the first 6 hours has a very high positive likelihood ratio for dissection, so this test may be useful in identifying patients with a high probability for dissection. In the first 24 hours after symptom onset, a D-dimer level of less than 500 ng/mL has a negative predictive value of 95%. Thus, elevations in D-dimer may help decide which imaging to perform in a patient presenting with chest pain or suspicion of dissection.11
However, D-dimer levels may not be elevated in dissection variants, such as aortic intramural hematoma or penetrating aortic ulcer. Additionally, once 24 hours have elapsed since the dissection started, D-dimer levels may no longer be elevated. The current ACC/AHA guidelines on thoracic aortic disease concluded that the D-dimer level cannot be used to rule out aortic dissection in high-risk individuals.3
Additional studies may clarify the appropriate role of the D-dimer assay in diagnosing aortic dissection.
DEFINITIVE IMAGING STUDIES: CT, MRI, TEE
Contrast-enhanced computed tomography (CT), magnetic resonance imaging (MRI), and transesophageal echocardiography (TEE) all have very high sensitivity and specificity for the diagnosis of aortic dissection.2,3 The choice of imaging study often depends on the availability of these studies, with CT and TEE being the most commonly performed initial studies.
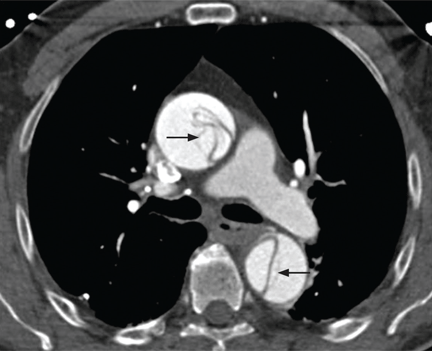
MRI is outstanding for detecting and following aortic dissection, but it is usually not the initial study performed because of the time required for image acquisition and because it is generally not available on an emergency basis.
While transthoracic echocardiography (TTE) can detect aortic dissection, its sensitivity is much lower than that of other imaging tests.2,3 Therefore, negative findings on TTE do not exclude aortic dissection.
MANAGEMENT OF AORTIC DISSECTION
When acute aortic dissection is diagnosed, multidisciplinary evaluation and treatment are necessary. Time is of the essence, as the death rate in acute dissection may be as high as 1% per hour during the first 24 hours.1–3 All patients with acute aortic dissection, whether type A or type B, should be transferred to a tertiary care center with a staff experienced in managing aortic dissection and its complications.3 Emergency surgery is recommended for type A aortic dissection, whereas type B dissection is generally treated medically unless complications occur.2,3
The cornerstone of drug therapy is the prompt reduction in blood pressure with a beta-blocker to reduce shear stresses on the aorta. Intravenous agents such as esmolol (Brevibloc) or labetalol (Normodyne) are usually chosen. Sodium nitroprusside may be added to beta-blocker therapy for rapid blood pressure control in appropriate patients. The patient may require multiple antihypertensive medications. If hypertension is refractory, one must consider renal artery hypertension due to the dissection causing renal malperfusion.2 Acute pain may also worsen hypertension, and appropriate analgesia should be used.
Definitive therapy in acute dissection
Patients with acute type A dissection require emergency surgery,2,3 as they are at risk for life-threatening complications including cardiac tamponade from hemopericardium, aortic rupture, stroke, visceral ischemia, and heart failure due to severe aortic regurgitation. When aortic regurgitation complicates acute type A dissection, some patients are adequately treated by resuspension of the aortic valve leaflets, while others require valve-sparing root replacement or prosthetic aortic valve replacement.
Surgical therapy is associated with a survival benefit compared with medical therapy in acute type A dissection.1 The 14-day mortality rate for acute type A dissection treated surgically is about 25%.1 Patients with high-risk features such as heart failure, shock, tamponade, and mesenteric ischemia have a worse prognosis compared with those without these features.2,12,13
Acute type B aortic dissection carries a lower rate of death than type A dissection.1–3 In the IRAD cohort, the early mortality rate in those with type B dissection treated medically was about 10%.1 However, when complications such as malperfusion, shock, or requirement for surgery occur in type B dissection, the mortality rate is much higher,2,14 with rates of 25% to 50% reported.2
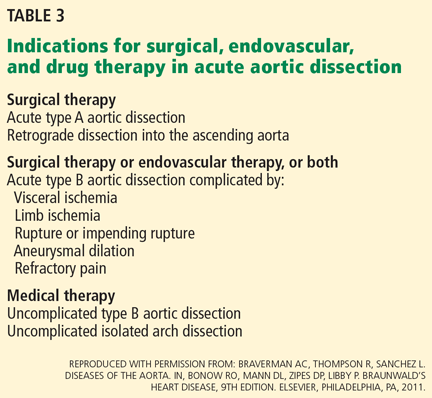
Thus, initial medical therapy is the preferred approach to acute type B dissection, and surgery or endovascular therapy is reserved for patients with acute complications.2,3 Typical indications for surgery or endovascular therapy in type B dissection include visceral or limb ischemia, aortic rupture, refractory pain, and aneurysmal dilation (Table 3).2
Endovascular therapy in aortic dissection
The high mortality rate with open surgery in acute type B dissection has spurred tremendous interest in endovascular treatments for complications involving the descending aorta and branch vessels.2
Fenestration of the aorta and stenting of branch vessels were the earliest techniques used in complicated type B dissection. By fenestrating (ie, opening) the intimal flap, blood can flow from the false lumen into the true lumen, decompressing the distended false lumen.
Endovascular stenting is used for acute aortic rupture, for malperfusion syndromes, and for rapidly enlarging false lumens. Endovascular grafts may cover the area of a primary intimal tear and thus eliminate the flow into the false channel and promote false-lumen thrombosis. Many patients with complicated type B dissection are treated with a hybrid approach, in which one segment of the aorta, such as the aortic arch, is treated surgically, while the descending aorta receives an endovascular graft.2
Patients with a type B dissection treated medically are at risk for late complications, including aneurysmal enlargement and subsequent aortic rupture. The Investigation of Stent Grafts in Aortic Dissection (INSTEAD) trial included 140 patients with uncomplicated type B dissection and compared drug therapy with endovascular stent grafting.15 After 2 years of follow-up, there was no difference in the rate of death between the two treatment groups. Patients receiving endovascular grafts had a higher rate of false-lumen thrombosis.
More studies are under way to examine the role of endovascular therapy in uncomplicated type B dissection.
AORTIC DISSECTION VARIANTS
Aortic intramural hematoma
Aortic intramural hematoma is a form of acute aortic syndrome in which a hematoma develops in the aortic media and no intimal flap is visualized either by imaging or at surgery.2,3,16 It is important to recognize this clinical entity in a patient presenting with acute chest or back pain, as sometimes it is mistaken for a “thrombus in a nonaneurysmal aorta.” Intramural hematoma accounts for 5% to 25% of acute aortic syndromes, depending on the study population (it is more common in Asian studies).2,3,17 It may present with symptoms similar to classic aortic dissection and is classified as type A or type B, depending on whether the ascending aorta is involved.
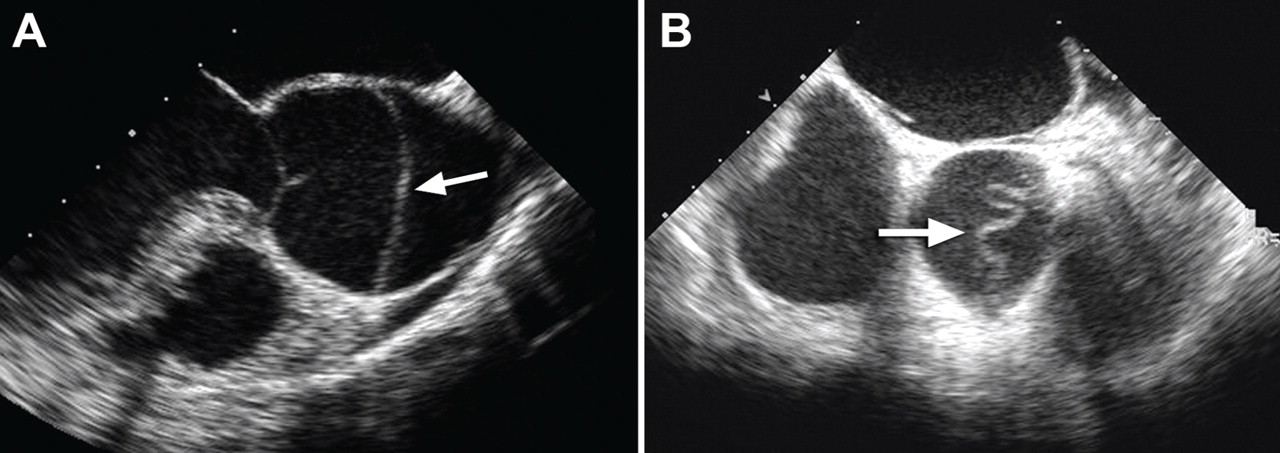
Patients with an intramural hematoma may progress to having complications such as hemopericardium, classic aortic dissection, aortic rupture, or aneurysmal dilation.2,3 However, many cases of type B aortic intramural hematoma result in complete resorption of the hematoma over time. In general, like classic aortic dissection, type A intramural hematoma is treated with emergency surgery and type B with initial medical therapy.2,3
There are reports from Southeast Asia of successful initial medical therapy for type A intramural hematoma, with surgery used for acute complications.18 In the Western literature, improved outcomes are reported with initial surgical therapy.17 Given the unpredictable nature of type A intramural hematoma, most experts recommend surgical therapy for appropriate candidates with acute type A intramural hematoma.2,3,19
Penetrating atherosclerotic ulcer of the aorta
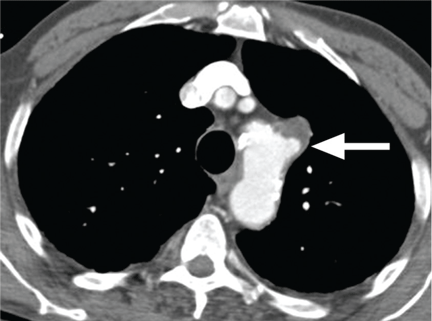
Penetrating atherosclerotic ulcer of the aorta, another acute aortic syndrome, results from acute penetration of an atherosclerotic aortic lesion through the internal elastic lamina into the media.2,3,20 It is often associated with bleeding into the media, or intramural hematoma. While the ulcer may be found incidentally on imaging studies, especially in patients with severe aortic atherosclerosis, the typical presentation is acute, severe chest or back pain. It occurs most often in the descending aorta and the abdominal aorta.
Penetrating atherosclerotic ulcer may lead to pseudoaneurysm formation, focal aortic dissection, aortic rupture, or late aortic aneurysm.2
LONG-TERM MANAGEMENT AFTER AORTIC DISSECTION
After hospital discharge, patients with aortic dissection require lifelong management. This includes blood pressure control, lifestyle modification, serial imaging of the aorta with CT or MRI, patient education about the condition, and, when appropriate, screening of family members for aortic disease.5,21
Reported survival rates after hospitalization for type A dissection are 52% to 94% at 1 year and 45% to 88% at 5 years.2,22 The 10-year actuarial survival rate for those with acute dissection who survive the acute hospitalization is reported as 30% to 60%. Long-term survival rates after acute type B dissection have been reported at 56% to 92% at 1 year and 48% to 82% at 5 years.23 Survival rates depend on many factors, including the underlying condition, the age of the patient, and comorbidities.
It is important to treat hypertension after aortic dissection, with a goal blood pressure of 120/80 mm Hg or less for most patients. Older studies found higher mortality rates with poorly controlled hypertension. Beta-blockers are the drugs of first choice. Even in the absence of hypertension, long-term beta-blocker therapy should be used to lessen the aortic stress and force of ventricular contraction.
Genetic evaluation
Genetically triggered causes of aortic dissection should be considered. In many circumstances, referral to a medical geneticist or other practitioner knowledgeable in these conditions is important when these disorders are being evaluated (Table 2).
Many of these disorders have an autosomal dominant inheritance, and the patient should be asked about a family history of aortic disease, aortic dissection, or unexplained sudden death. Features of Marfan syndrome, Loeys-Dietz syndrome, and familial thoracic aortic aneurysm syndromes should be sought. Through comprehensive family studies, it is now recognized that up to 20% of patients with thoracic aortic disease (such as aneurysm or dissection) have another first-degree relative with thoracic aortic disease.2,3,24 Thus, first-degree relatives of patients with aortic aneurysm or dissection should be screened for thoracic aortic aneurysm disease.
Research into molecular genetics is providing a better understanding of the genetics of aortic dissection.3 New mutations associated with aortic dissection are being discovered in signaling pathways as well as elements critical for the integrity of the vascular wall.2,3 However, at present, most patients with aortic dissection will not have a specific identifiable genetic defect.
Not only does genetic testing enable the accurate diagnosis of the affected individual, but also treatments are often based on this diagnosis.3 Importantly, the identification of a specific gene mutation (ie, in TGFBR1 or 2, FBN1, ACTA2, MYH11, and COL3A1) in an affected individual has the potential to identify other family members at risk.3
Follow-up imaging
It is important to continue to image the aorta after aortic dissection. Patients may develop progressive dilation or aneurysm formation of the dissected aorta, pseudoaneurysm formation after repair, or recurrent dissection. Many patients require additional surgery on the aorta because of late aneurysm formation.
CT or MRI is usually performed at least every 6 months in the first 2 years after dissection and at least annually thereafter. More centers are choosing MRI for long-term follow-up to avoid the repeated radiation exposure with serial CT.
Patient education
Besides receiving medical therapy and undergoing imaging, patients with aortic dissection should be educated about this condition.5,21 The patient should be aware of symptoms suggesting dissection and should be instructed to seek attention for any concerning symptoms.
Lifestyle modifications are also important. The patient should be educated about safe activity levels and to avoid heavy isometric exercise, such as weight-lifting. Some patients will have to cease their current occupation because of activity restrictions.
- Hagan PG, Nienaber CA, Isselbacher EM, et al. International Registry of Acute Aortic Dissection (IRAD): new insights from an old disease. JAMA 2000; 283:897–903.
- Braverman AC, Thompson R, Sanchez L. Diseases of the aorta. In:Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease, 9th Edition. Elsevier, Philadelphia, 2011.
- Hiratzka LF, Bakris GL, Beckman JA, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. Guidelines for the management of patients with thoracic aortic disease. Circulation 2010; 121:e266–e369.
- Rogers AM, Herman LK, Booher AM, et al. Sensitivity of the aortic dissection detection risk score, a novel guideline-based tool for identification of acute aortic dissection at initial presentation. Results from the International Registry of Acute Aortic Dissection. Circulation 2011; 123:2213–2228.
- Braverman AC. Acute aortic dissection: clinician update. Circulation 2010; 122:184–188.
- Davies RR, Gallo A, Coady MA, et al. Novel measurement of relative aortic size predicts rupture of thoracic aortic aneurysms. Ann Thorac Surg 2006; 81:169–177.
- Pape LA, Tsai TT, Isselbacher EM, et al. Aortic diameter >5.5 cm is not a good predictor of type A aortic dissection. Observations from the International Registry of Acute Aortic Dissection. Circulation 2007; 116:1120–1127.
- Parish LM, Gorman JH, Kahn S, et al. Aortic size in acute type A dissection: implications for preventative ascending aortic replacement. Eur J Cardiothorac Surg 2009; 35:941–945.
- Gaul C, Dietrich W, Erbguth FJ. Neurological symptoms in acute aortic dissection: a challenge for neurologists. Cerebrovasc Dis 2008; 26:1–8.
- Upchurch GR, Nienaber C, Fattori R, et al Acute aortic dissection presenting with primarily abdominal pain: a rare manifestation of a deadly disease. Ann Vasc Surg 2005; 19:367–373.
- Suzuki T, Distante A, Zizza A, et al. Diagnosis of acute aortic dissection by D-dimer: the International Registry of Acute Aortic Dissection substudy on biomarkers (IRAD-bio) experience. Circulation 2009; 119:2702–2707.
- Tsai TT, Trimarchi S, Neinaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg 2009; 37:149–159.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Contemporary results of surgery in acute type A aortic dissection: the International Registry of Acute Aortic Dissection experience. J Thorac Cardiovasc Surg 2005; 129:112–122.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Role and results of surgery in acute type B aortic dissection. Insights from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2006; 114(suppl 1):I-357–I-364.
- Nienaber CA, Rousseau H, Eggbrecht H, et al. Randomized comparison of strategies for type B aortic dissection. The Investigation of STEnt grafts in Aortic Dissection (INSTEAD) Trial. Circulation 2009; 120:2519–2528.
- Evangelista A, Mukherjee D, Mehta RH, et al. Acute intramural hematoma of the aorta. Circulation 2005; 111:1063–1070.
- Pelzel JM, Braverman AC, Hirsch AT, Harris KM. International heterogeneity in diagnostic frequency and clinical outcomes of ascending aortic intramural hematoma. J Am Soc Echo 2007; 20:1260–1268.
- Song JK, Yim JH, Ahn JM, et al. Outcomes of patients with acute type A aortic intramural hematoma. Circulation 2009; 120:2046–2052.
- Harris KM, Pelzel JM, Braverman AC. Letter regarding article, “Outcomes of patients with acute type A intramural hematoma.” Circulation 2010; 121:e456.
- Sundt TM. Intramural hematoma and penetrating atherosclerotic ulcer of the aorta. Ann Thorac Surg 2007; 83:S835–S841.
- Juang D, Braverman A, Eagle K. Aortic dissection. Circulation 2008; 118:e507–e510.
- Tsai TT, Evangelista A, Nienaber CA, et al. Long-term survival in patients presenting with type A acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114(suppl I):I-350–I-356.
- Tsai TT, Fattori R, Trimarchi S, et al. Long-term survival in patients presenting with type B acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114:2226–2231.
- Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections: incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg 2006; 82:1400–1405.
A 50-year-old man developed severe chest pain and collapsed to the floor. The pain was sudden in onset, was burning in quality, and was located in the center of his chest. Emergency medical services arrived a few minutes later and found the patient diaphoretic and cyanotic, with an initial blood pressure of 74/54 mm Hg and a heart rate of 125 beats per minute. He was rushed to the hospital.
His medical history was unremarkable. He smoked one pack of cigarettes per day for 20 years. His father died of a “heart attack” at age 52.
In the emergency department he underwent echocardiography with a portable handheld unit, which showed a pericardial effusion and cardiac tamponade. He was sent for emergency computed tomography of the chest, which revealed an aneurysm of the aortic root and acute type A (Stanford classification) aortic dissection with hemopericardium.
He underwent emergency cardiac surgery. At the time of surgery, he was in cardiogenic shock from aortic dissection complicated by severe aortic regurgitation and cardiac tamponade with hemopericardium. The aortic valve was trileaflet. A 27-mm St. Jude composite valve graft root replacement was performed.
The patient did well and was discharged home 7 days after surgery. Pathologic study of the aorta revealed cystic medial degeneration. He did not have any features of Marfan syndrome or Loeys-Dietz syndrome. His three children underwent evaluation, and each had a normal physical examination and echocardiographic test results.
A HIGH INDEX OF SUSPICION IS CRITICAL
Acute aortic dissection is the most common aortic catastrophe, with an incidence estimated at 5 to 30 per 1 million people per year, amounting to nearly 10,000 cases per year in the United States.1–4
The diagnosis of acute aortic dissection has many potential pitfalls.2,3 Aortic dissection may mimic other more common conditions, such as coronary ischemia, pleurisy, heart failure, stroke, and acute abdominal illness. Because acute aortic dissection may be rapidly fatal, one must maintain a high index of suspicion.2,3 Prompt diagnosis and emergency treatment are critical.
WHAT CAUSES AORTIC DISSECTION?
One hypothesis is that acute aortic dissection is caused by a primary tear in the aortic intima, with blood from the aortic lumen penetrating into the diseased media leading to dissection and creating a true and false lumen.2 Another is that rupture of the vasa vasorum leads to hemorrhage in the aortic wall with subsequent intimal disruption, creating the intimal tear and aortic dissection.
Once a dissection starts, pulsatile flow of blood within the aortic wall causes it to extend. The dissection flap may be localized, but it often spirals the entire length of the aorta. Distention of the false lumen with blood may cause the intimal flap to compress the true lumen and potentially lead to malperfusion syndromes.
CLASSIFIED ACCORDING TO LOCATION
It is important to recognize the location of the dissection, as the prognosis and treatment depend on whether the ascending aorta is involved.2,3 For classification purposes, the ascending aorta is the portion proximal to the brachiocephalic artery, while the descending aorta is the portion distal to the left subclavian artery.3
The DeBakey classification defines a type I aortic dissection as one that begins in the ascending aorta and extends at least to the aortic arch or beyond. Type II dissections involve the ascending aorta only, while type III dissections begin in the descending aorta, most often just distal to the left subclavian artery.
The Stanford classification scheme divides dissections into type A and type B. Type A dissections involve the ascending aorta, while type B dissections do not involve the ascending aorta.
Which classification scheme is used is not important. However, identifying patients with dissection of the ascending aorta (DeBakey type I or type II or Stanford type A) is critical, as emergency cardiac surgery is recommended for this type of dissection.2,3 For the purposes of this paper, the Stanford classification scheme will be used.
Dissection that involves the ascending aorta most commonly occurs in people ages 50 to 60, whereas acute dissection of the descending aorta typically occurs in people 10 years older.1,2
An acute aortic dissection is one that has occurred within 2 weeks of symptom onset. A chronic dissection is one that occurred more than 2 weeks after symptoms began.
DISEASES AND CONDITIONS ASSOCIATED WITH AORTIC DISSECTION
Hypertension and disorders leading to disruption of the normal structure and function of the aortic wall. About 75% of patients with acute aortic dissection have underlying hypertension.1–3
Cystic medial degeneration is a common pathologic feature in many cases of aortic dissection.
Cocaine use and intense weight-lifting increase the shear stresses on the aorta.2,3
Inflammatory aortic diseases such as giant cell arteritis.
Pregnancy can be complicated by aortic dissection, usually in the setting of an underlying aortopathy.5
Iatrogenic aortic dissection accounts for about 4% of cases, as a result of cardiac surgery, catheterization, stenting, or use of an intra-aortic balloon pump.1
Aortic aneurysm. Patients with thoracic aortic aneurysm are at higher risk of aortic dissection, and the larger the aortic diameter, the higher the risk.2,3,6 In the International Registry of Acute Aortic Dissection (IRAD), the average size of the aorta was about 5.3 cm at the time of acute dissection. Importantly, about 40% of acute dissections of the ascending aorta occur in patients with ascending aortic diameters less than 5.0 cm.7,8
Thus, many factors are associated with acute dissection, and specific reasons leading to an individual’s susceptibility to sudden dissection are poorly understood.
CLINICAL FEATURES OF ACUTE AORTIC DISSECTION
Because the symptoms of acute dissection may mimic other, more common conditions, one of the most important factors in the diagnosis of aortic dissection is a high clinical suspicion.1–3
What is the pretest risk of disease?
Recently, the American College of Cardiology (ACC) and the American Heart Association (AHA) released joint guidelines on thoracic aortic disease.3 These guidelines provide an approach to patients who have complaints that may represent acute thoracic aortic dissection, the intent being to establish a pretest risk of disease to be used to guide decision-making.3
The focused evaluation includes specific questions about underlying conditions, symptoms, and findings on examination that may greatly increase the likelihood of acute dissection. These include:
- High-risk conditions and historical features associated with aortic dissection, such as Marfan syndrome and other genetic disorders (Table 2), bicuspid aortic valve, family history of thoracic aortic aneurysm or dissection, known thoracic aortic aneurysm, and recent aortic manipulation
- Pain in the chest, back, or abdomen with high-risk features (eg, abrupt onset, severe intensity, or a ripping or tearing quality)
- High-risk findings on examination (eg, pulse deficits, new aortic regurgitation, hypotension, shock, or systolic blood pressure differences).
Using this information, expedited aortic imaging and treatment algorithms have been devised to improve the diagnosis.3
Using the IRAD database of more than 2,500 acute dissections, the diagnostic algorithm proposed in the ACC/AHA guidelines was shown to be highly sensitive (about 95%) for detecting acute aortic dissection.4 In addition, using this score may expedite evaluation by classifying certain patients as being at high risk of acute dissection.3,4
Important to recognize is that almost two-thirds of patients who suffered dissection in this large database did not have one of the “high-risk conditions” associated with dissection.4 Additionally, the specificity of the ACC/AHA algorithm is unknown, and further testing is necessary.4
Acute onset of severe pain
More than 90% of acute dissections present with acute pain in the chest or the back, or both.1–3 The pain is usually severe, of sudden onset, and often described as sharp or, occasionally, tearing, ripping, or stabbing. The pain usually differs from that of coronary ischemia, being most severe at its onset as opposed to the less intense, crescendo-like pain of angina or myocardial infarction. The pain may migrate as the dissection progresses along the length of the aorta or to branch vessels. It may abate, leading to a false sense of security in the patient and the physician.3 “Painless” dissection occurs in a minority, usually in those with syncope, neurologic symptoms, or heart failure.1–3
The patient with acute dissection may be anxious and may feel a sense of doom.
Acute heart failure, related to severe aortic regurgitation, may be a predominant symptom in dissection of the ascending aorta.
Syncope may occur as a result of aortic rupture, hemopericardium with cardiac tamponade, or acute neurologic complications.
Vascular insufficiency may occur in any branch vessel, leading to clinical syndromes that include acute myocardial infarction, stroke, paraplegia, paraparesis, mesenteric ischemia, and limb ischemia.
PHYSICAL FINDINGS CAN VARY WIDELY
Findings on physical examination in acute aortic dissection vary widely depending on underlying conditions and on the specific complications of the dissection.
Although the classic presentation is acute, severe pain in the chest or back in a severely hypertensive patient with aortic regurgitation and pulse deficits, most patients do not have all these characteristics.4 Most patients with type B dissection are hypertensive on presentation, but many with type A dissection present with normal blood pressure or hypotension.1 Pulse deficits (unequal or absent pulses) are reported in 10% to 30% of acute dissections and may be intermittent as the dynamic movement of the dissection flap interferes with branch vessel perfusion.1–3
- Aortic leaflet prolapse or distortion of the leaflet alignment
- Malcoaptation of the aortic leaflets from dilation of the aortic root and annulus
- Prolapse of the intimal flap across the aortic valve, interfering with valve function
- Preexisting aortic regurgitation from underlying aortic root aneurysm or primary aortic valve disease (such as a bicuspid aortic valve).
Neurologic manifestations are most common in dissection of the ascending aorta and are particularly important to recognize, as they may dominate the clinical presentation and lead to delay in the diagnosis of dissection.2,3 Neurologic syndromes include:
- Persistent or transient ischemic stroke
- Spinal cord ischemia
- Ischemic neuropathy
- Hypoxic encephalopathy.
These manifestations are related to malperfusion to branches supplying the brain, spinal cord, or peripheral nerves.9
Syncope is relatively common in aortic dissection and may be related to acute hypotension caused by cardiac tamponade or aortic rupture, cerebral vessel obstruction, or activation of cerebral baroreceptors.2,9 It is important to consider aortic dissection in the differential diagnosis in cases of unexplained syncope.3
Aortic dissections may extend into the abdominal aorta, leading to vascular complications involving one or more branch vessels.10 The renal artery is involved in at least 5% to 10% of cases and may lead to renal ischemia, infarction, renal insufficiency, or refractory hypertension.2Mesenteric ischemia or infarction occurs in about 5% of dissections, may be difficult to diagnose, and is particularly dangerous.2,8 Aortic dissection may extend into the iliac arteries and may cause acute lower extremity ischemia.
Acute myocardial infarction due to involvement of the dissection flap causing malperfusion of a coronary artery occurs in 1% to 7% of acute type A aortic dissections.1–3 The right coronary artery (Figure 2) is most commonly involved, leading to acute inferior myocardial infarction. Acute myocardial ischemia and infarction in the setting of dissection may lead to a delay in the diagnosis of dissection and to bleeding complications from antiplatelet and anticoagulant drugs given to treat the acute coronary syndrome.
Cardiac tamponade, occurring in about 10% of acute type A dissections, portends a higher risk of death.2,3
Additional clinical features of aortic dissection include a left-sided pleural effusion, usually related to an inflammatory response. An acute hemothorax may occur from rupture or leaking of a descending aortic dissection.
FINDINGS ON RADIOGRAPHY AND ELECTROCARDIOGRAPHY
Electrocardiography usually has normal or nonspecific findings, unless acute myocardial infarction complicates the dissection.
D-DIMER LEVELS
Biomarkers for the diagnosis of acute aortic dissection are of great interest.
D-dimer levels rise in acute aortic dissection as they do in pulmonary embolism.11 A D-dimer level greater than 1,600 ng/mL within the first 6 hours has a very high positive likelihood ratio for dissection, so this test may be useful in identifying patients with a high probability for dissection. In the first 24 hours after symptom onset, a D-dimer level of less than 500 ng/mL has a negative predictive value of 95%. Thus, elevations in D-dimer may help decide which imaging to perform in a patient presenting with chest pain or suspicion of dissection.11
However, D-dimer levels may not be elevated in dissection variants, such as aortic intramural hematoma or penetrating aortic ulcer. Additionally, once 24 hours have elapsed since the dissection started, D-dimer levels may no longer be elevated. The current ACC/AHA guidelines on thoracic aortic disease concluded that the D-dimer level cannot be used to rule out aortic dissection in high-risk individuals.3
Additional studies may clarify the appropriate role of the D-dimer assay in diagnosing aortic dissection.
DEFINITIVE IMAGING STUDIES: CT, MRI, TEE
Contrast-enhanced computed tomography (CT), magnetic resonance imaging (MRI), and transesophageal echocardiography (TEE) all have very high sensitivity and specificity for the diagnosis of aortic dissection.2,3 The choice of imaging study often depends on the availability of these studies, with CT and TEE being the most commonly performed initial studies.
MRI is outstanding for detecting and following aortic dissection, but it is usually not the initial study performed because of the time required for image acquisition and because it is generally not available on an emergency basis.
While transthoracic echocardiography (TTE) can detect aortic dissection, its sensitivity is much lower than that of other imaging tests.2,3 Therefore, negative findings on TTE do not exclude aortic dissection.
MANAGEMENT OF AORTIC DISSECTION
When acute aortic dissection is diagnosed, multidisciplinary evaluation and treatment are necessary. Time is of the essence, as the death rate in acute dissection may be as high as 1% per hour during the first 24 hours.1–3 All patients with acute aortic dissection, whether type A or type B, should be transferred to a tertiary care center with a staff experienced in managing aortic dissection and its complications.3 Emergency surgery is recommended for type A aortic dissection, whereas type B dissection is generally treated medically unless complications occur.2,3
The cornerstone of drug therapy is the prompt reduction in blood pressure with a beta-blocker to reduce shear stresses on the aorta. Intravenous agents such as esmolol (Brevibloc) or labetalol (Normodyne) are usually chosen. Sodium nitroprusside may be added to beta-blocker therapy for rapid blood pressure control in appropriate patients. The patient may require multiple antihypertensive medications. If hypertension is refractory, one must consider renal artery hypertension due to the dissection causing renal malperfusion.2 Acute pain may also worsen hypertension, and appropriate analgesia should be used.
Definitive therapy in acute dissection
Patients with acute type A dissection require emergency surgery,2,3 as they are at risk for life-threatening complications including cardiac tamponade from hemopericardium, aortic rupture, stroke, visceral ischemia, and heart failure due to severe aortic regurgitation. When aortic regurgitation complicates acute type A dissection, some patients are adequately treated by resuspension of the aortic valve leaflets, while others require valve-sparing root replacement or prosthetic aortic valve replacement.
Surgical therapy is associated with a survival benefit compared with medical therapy in acute type A dissection.1 The 14-day mortality rate for acute type A dissection treated surgically is about 25%.1 Patients with high-risk features such as heart failure, shock, tamponade, and mesenteric ischemia have a worse prognosis compared with those without these features.2,12,13
Acute type B aortic dissection carries a lower rate of death than type A dissection.1–3 In the IRAD cohort, the early mortality rate in those with type B dissection treated medically was about 10%.1 However, when complications such as malperfusion, shock, or requirement for surgery occur in type B dissection, the mortality rate is much higher,2,14 with rates of 25% to 50% reported.2
Thus, initial medical therapy is the preferred approach to acute type B dissection, and surgery or endovascular therapy is reserved for patients with acute complications.2,3 Typical indications for surgery or endovascular therapy in type B dissection include visceral or limb ischemia, aortic rupture, refractory pain, and aneurysmal dilation (Table 3).2
Endovascular therapy in aortic dissection
The high mortality rate with open surgery in acute type B dissection has spurred tremendous interest in endovascular treatments for complications involving the descending aorta and branch vessels.2
Fenestration of the aorta and stenting of branch vessels were the earliest techniques used in complicated type B dissection. By fenestrating (ie, opening) the intimal flap, blood can flow from the false lumen into the true lumen, decompressing the distended false lumen.
Endovascular stenting is used for acute aortic rupture, for malperfusion syndromes, and for rapidly enlarging false lumens. Endovascular grafts may cover the area of a primary intimal tear and thus eliminate the flow into the false channel and promote false-lumen thrombosis. Many patients with complicated type B dissection are treated with a hybrid approach, in which one segment of the aorta, such as the aortic arch, is treated surgically, while the descending aorta receives an endovascular graft.2
Patients with a type B dissection treated medically are at risk for late complications, including aneurysmal enlargement and subsequent aortic rupture. The Investigation of Stent Grafts in Aortic Dissection (INSTEAD) trial included 140 patients with uncomplicated type B dissection and compared drug therapy with endovascular stent grafting.15 After 2 years of follow-up, there was no difference in the rate of death between the two treatment groups. Patients receiving endovascular grafts had a higher rate of false-lumen thrombosis.
More studies are under way to examine the role of endovascular therapy in uncomplicated type B dissection.
AORTIC DISSECTION VARIANTS
Aortic intramural hematoma
Aortic intramural hematoma is a form of acute aortic syndrome in which a hematoma develops in the aortic media and no intimal flap is visualized either by imaging or at surgery.2,3,16 It is important to recognize this clinical entity in a patient presenting with acute chest or back pain, as sometimes it is mistaken for a “thrombus in a nonaneurysmal aorta.” Intramural hematoma accounts for 5% to 25% of acute aortic syndromes, depending on the study population (it is more common in Asian studies).2,3,17 It may present with symptoms similar to classic aortic dissection and is classified as type A or type B, depending on whether the ascending aorta is involved.
Patients with an intramural hematoma may progress to having complications such as hemopericardium, classic aortic dissection, aortic rupture, or aneurysmal dilation.2,3 However, many cases of type B aortic intramural hematoma result in complete resorption of the hematoma over time. In general, like classic aortic dissection, type A intramural hematoma is treated with emergency surgery and type B with initial medical therapy.2,3
There are reports from Southeast Asia of successful initial medical therapy for type A intramural hematoma, with surgery used for acute complications.18 In the Western literature, improved outcomes are reported with initial surgical therapy.17 Given the unpredictable nature of type A intramural hematoma, most experts recommend surgical therapy for appropriate candidates with acute type A intramural hematoma.2,3,19
Penetrating atherosclerotic ulcer of the aorta
Penetrating atherosclerotic ulcer of the aorta, another acute aortic syndrome, results from acute penetration of an atherosclerotic aortic lesion through the internal elastic lamina into the media.2,3,20 It is often associated with bleeding into the media, or intramural hematoma. While the ulcer may be found incidentally on imaging studies, especially in patients with severe aortic atherosclerosis, the typical presentation is acute, severe chest or back pain. It occurs most often in the descending aorta and the abdominal aorta.
Penetrating atherosclerotic ulcer may lead to pseudoaneurysm formation, focal aortic dissection, aortic rupture, or late aortic aneurysm.2
LONG-TERM MANAGEMENT AFTER AORTIC DISSECTION
After hospital discharge, patients with aortic dissection require lifelong management. This includes blood pressure control, lifestyle modification, serial imaging of the aorta with CT or MRI, patient education about the condition, and, when appropriate, screening of family members for aortic disease.5,21
Reported survival rates after hospitalization for type A dissection are 52% to 94% at 1 year and 45% to 88% at 5 years.2,22 The 10-year actuarial survival rate for those with acute dissection who survive the acute hospitalization is reported as 30% to 60%. Long-term survival rates after acute type B dissection have been reported at 56% to 92% at 1 year and 48% to 82% at 5 years.23 Survival rates depend on many factors, including the underlying condition, the age of the patient, and comorbidities.
It is important to treat hypertension after aortic dissection, with a goal blood pressure of 120/80 mm Hg or less for most patients. Older studies found higher mortality rates with poorly controlled hypertension. Beta-blockers are the drugs of first choice. Even in the absence of hypertension, long-term beta-blocker therapy should be used to lessen the aortic stress and force of ventricular contraction.
Genetic evaluation
Genetically triggered causes of aortic dissection should be considered. In many circumstances, referral to a medical geneticist or other practitioner knowledgeable in these conditions is important when these disorders are being evaluated (Table 2).
Many of these disorders have an autosomal dominant inheritance, and the patient should be asked about a family history of aortic disease, aortic dissection, or unexplained sudden death. Features of Marfan syndrome, Loeys-Dietz syndrome, and familial thoracic aortic aneurysm syndromes should be sought. Through comprehensive family studies, it is now recognized that up to 20% of patients with thoracic aortic disease (such as aneurysm or dissection) have another first-degree relative with thoracic aortic disease.2,3,24 Thus, first-degree relatives of patients with aortic aneurysm or dissection should be screened for thoracic aortic aneurysm disease.
Research into molecular genetics is providing a better understanding of the genetics of aortic dissection.3 New mutations associated with aortic dissection are being discovered in signaling pathways as well as elements critical for the integrity of the vascular wall.2,3 However, at present, most patients with aortic dissection will not have a specific identifiable genetic defect.
Not only does genetic testing enable the accurate diagnosis of the affected individual, but also treatments are often based on this diagnosis.3 Importantly, the identification of a specific gene mutation (ie, in TGFBR1 or 2, FBN1, ACTA2, MYH11, and COL3A1) in an affected individual has the potential to identify other family members at risk.3
Follow-up imaging
It is important to continue to image the aorta after aortic dissection. Patients may develop progressive dilation or aneurysm formation of the dissected aorta, pseudoaneurysm formation after repair, or recurrent dissection. Many patients require additional surgery on the aorta because of late aneurysm formation.
CT or MRI is usually performed at least every 6 months in the first 2 years after dissection and at least annually thereafter. More centers are choosing MRI for long-term follow-up to avoid the repeated radiation exposure with serial CT.
Patient education
Besides receiving medical therapy and undergoing imaging, patients with aortic dissection should be educated about this condition.5,21 The patient should be aware of symptoms suggesting dissection and should be instructed to seek attention for any concerning symptoms.
Lifestyle modifications are also important. The patient should be educated about safe activity levels and to avoid heavy isometric exercise, such as weight-lifting. Some patients will have to cease their current occupation because of activity restrictions.
A 50-year-old man developed severe chest pain and collapsed to the floor. The pain was sudden in onset, was burning in quality, and was located in the center of his chest. Emergency medical services arrived a few minutes later and found the patient diaphoretic and cyanotic, with an initial blood pressure of 74/54 mm Hg and a heart rate of 125 beats per minute. He was rushed to the hospital.
His medical history was unremarkable. He smoked one pack of cigarettes per day for 20 years. His father died of a “heart attack” at age 52.
In the emergency department he underwent echocardiography with a portable handheld unit, which showed a pericardial effusion and cardiac tamponade. He was sent for emergency computed tomography of the chest, which revealed an aneurysm of the aortic root and acute type A (Stanford classification) aortic dissection with hemopericardium.
He underwent emergency cardiac surgery. At the time of surgery, he was in cardiogenic shock from aortic dissection complicated by severe aortic regurgitation and cardiac tamponade with hemopericardium. The aortic valve was trileaflet. A 27-mm St. Jude composite valve graft root replacement was performed.
The patient did well and was discharged home 7 days after surgery. Pathologic study of the aorta revealed cystic medial degeneration. He did not have any features of Marfan syndrome or Loeys-Dietz syndrome. His three children underwent evaluation, and each had a normal physical examination and echocardiographic test results.
A HIGH INDEX OF SUSPICION IS CRITICAL
Acute aortic dissection is the most common aortic catastrophe, with an incidence estimated at 5 to 30 per 1 million people per year, amounting to nearly 10,000 cases per year in the United States.1–4
The diagnosis of acute aortic dissection has many potential pitfalls.2,3 Aortic dissection may mimic other more common conditions, such as coronary ischemia, pleurisy, heart failure, stroke, and acute abdominal illness. Because acute aortic dissection may be rapidly fatal, one must maintain a high index of suspicion.2,3 Prompt diagnosis and emergency treatment are critical.
WHAT CAUSES AORTIC DISSECTION?
One hypothesis is that acute aortic dissection is caused by a primary tear in the aortic intima, with blood from the aortic lumen penetrating into the diseased media leading to dissection and creating a true and false lumen.2 Another is that rupture of the vasa vasorum leads to hemorrhage in the aortic wall with subsequent intimal disruption, creating the intimal tear and aortic dissection.
Once a dissection starts, pulsatile flow of blood within the aortic wall causes it to extend. The dissection flap may be localized, but it often spirals the entire length of the aorta. Distention of the false lumen with blood may cause the intimal flap to compress the true lumen and potentially lead to malperfusion syndromes.
CLASSIFIED ACCORDING TO LOCATION
It is important to recognize the location of the dissection, as the prognosis and treatment depend on whether the ascending aorta is involved.2,3 For classification purposes, the ascending aorta is the portion proximal to the brachiocephalic artery, while the descending aorta is the portion distal to the left subclavian artery.3
The DeBakey classification defines a type I aortic dissection as one that begins in the ascending aorta and extends at least to the aortic arch or beyond. Type II dissections involve the ascending aorta only, while type III dissections begin in the descending aorta, most often just distal to the left subclavian artery.
The Stanford classification scheme divides dissections into type A and type B. Type A dissections involve the ascending aorta, while type B dissections do not involve the ascending aorta.
Which classification scheme is used is not important. However, identifying patients with dissection of the ascending aorta (DeBakey type I or type II or Stanford type A) is critical, as emergency cardiac surgery is recommended for this type of dissection.2,3 For the purposes of this paper, the Stanford classification scheme will be used.
Dissection that involves the ascending aorta most commonly occurs in people ages 50 to 60, whereas acute dissection of the descending aorta typically occurs in people 10 years older.1,2
An acute aortic dissection is one that has occurred within 2 weeks of symptom onset. A chronic dissection is one that occurred more than 2 weeks after symptoms began.
DISEASES AND CONDITIONS ASSOCIATED WITH AORTIC DISSECTION
Hypertension and disorders leading to disruption of the normal structure and function of the aortic wall. About 75% of patients with acute aortic dissection have underlying hypertension.1–3
Cystic medial degeneration is a common pathologic feature in many cases of aortic dissection.
Cocaine use and intense weight-lifting increase the shear stresses on the aorta.2,3
Inflammatory aortic diseases such as giant cell arteritis.
Pregnancy can be complicated by aortic dissection, usually in the setting of an underlying aortopathy.5
Iatrogenic aortic dissection accounts for about 4% of cases, as a result of cardiac surgery, catheterization, stenting, or use of an intra-aortic balloon pump.1
Aortic aneurysm. Patients with thoracic aortic aneurysm are at higher risk of aortic dissection, and the larger the aortic diameter, the higher the risk.2,3,6 In the International Registry of Acute Aortic Dissection (IRAD), the average size of the aorta was about 5.3 cm at the time of acute dissection. Importantly, about 40% of acute dissections of the ascending aorta occur in patients with ascending aortic diameters less than 5.0 cm.7,8
Thus, many factors are associated with acute dissection, and specific reasons leading to an individual’s susceptibility to sudden dissection are poorly understood.
CLINICAL FEATURES OF ACUTE AORTIC DISSECTION
Because the symptoms of acute dissection may mimic other, more common conditions, one of the most important factors in the diagnosis of aortic dissection is a high clinical suspicion.1–3
What is the pretest risk of disease?
Recently, the American College of Cardiology (ACC) and the American Heart Association (AHA) released joint guidelines on thoracic aortic disease.3 These guidelines provide an approach to patients who have complaints that may represent acute thoracic aortic dissection, the intent being to establish a pretest risk of disease to be used to guide decision-making.3
The focused evaluation includes specific questions about underlying conditions, symptoms, and findings on examination that may greatly increase the likelihood of acute dissection. These include:
- High-risk conditions and historical features associated with aortic dissection, such as Marfan syndrome and other genetic disorders (Table 2), bicuspid aortic valve, family history of thoracic aortic aneurysm or dissection, known thoracic aortic aneurysm, and recent aortic manipulation
- Pain in the chest, back, or abdomen with high-risk features (eg, abrupt onset, severe intensity, or a ripping or tearing quality)
- High-risk findings on examination (eg, pulse deficits, new aortic regurgitation, hypotension, shock, or systolic blood pressure differences).
Using this information, expedited aortic imaging and treatment algorithms have been devised to improve the diagnosis.3
Using the IRAD database of more than 2,500 acute dissections, the diagnostic algorithm proposed in the ACC/AHA guidelines was shown to be highly sensitive (about 95%) for detecting acute aortic dissection.4 In addition, using this score may expedite evaluation by classifying certain patients as being at high risk of acute dissection.3,4
Important to recognize is that almost two-thirds of patients who suffered dissection in this large database did not have one of the “high-risk conditions” associated with dissection.4 Additionally, the specificity of the ACC/AHA algorithm is unknown, and further testing is necessary.4
Acute onset of severe pain
More than 90% of acute dissections present with acute pain in the chest or the back, or both.1–3 The pain is usually severe, of sudden onset, and often described as sharp or, occasionally, tearing, ripping, or stabbing. The pain usually differs from that of coronary ischemia, being most severe at its onset as opposed to the less intense, crescendo-like pain of angina or myocardial infarction. The pain may migrate as the dissection progresses along the length of the aorta or to branch vessels. It may abate, leading to a false sense of security in the patient and the physician.3 “Painless” dissection occurs in a minority, usually in those with syncope, neurologic symptoms, or heart failure.1–3
The patient with acute dissection may be anxious and may feel a sense of doom.
Acute heart failure, related to severe aortic regurgitation, may be a predominant symptom in dissection of the ascending aorta.
Syncope may occur as a result of aortic rupture, hemopericardium with cardiac tamponade, or acute neurologic complications.
Vascular insufficiency may occur in any branch vessel, leading to clinical syndromes that include acute myocardial infarction, stroke, paraplegia, paraparesis, mesenteric ischemia, and limb ischemia.
PHYSICAL FINDINGS CAN VARY WIDELY
Findings on physical examination in acute aortic dissection vary widely depending on underlying conditions and on the specific complications of the dissection.
Although the classic presentation is acute, severe pain in the chest or back in a severely hypertensive patient with aortic regurgitation and pulse deficits, most patients do not have all these characteristics.4 Most patients with type B dissection are hypertensive on presentation, but many with type A dissection present with normal blood pressure or hypotension.1 Pulse deficits (unequal or absent pulses) are reported in 10% to 30% of acute dissections and may be intermittent as the dynamic movement of the dissection flap interferes with branch vessel perfusion.1–3
- Aortic leaflet prolapse or distortion of the leaflet alignment
- Malcoaptation of the aortic leaflets from dilation of the aortic root and annulus
- Prolapse of the intimal flap across the aortic valve, interfering with valve function
- Preexisting aortic regurgitation from underlying aortic root aneurysm or primary aortic valve disease (such as a bicuspid aortic valve).
Neurologic manifestations are most common in dissection of the ascending aorta and are particularly important to recognize, as they may dominate the clinical presentation and lead to delay in the diagnosis of dissection.2,3 Neurologic syndromes include:
- Persistent or transient ischemic stroke
- Spinal cord ischemia
- Ischemic neuropathy
- Hypoxic encephalopathy.
These manifestations are related to malperfusion to branches supplying the brain, spinal cord, or peripheral nerves.9
Syncope is relatively common in aortic dissection and may be related to acute hypotension caused by cardiac tamponade or aortic rupture, cerebral vessel obstruction, or activation of cerebral baroreceptors.2,9 It is important to consider aortic dissection in the differential diagnosis in cases of unexplained syncope.3
Aortic dissections may extend into the abdominal aorta, leading to vascular complications involving one or more branch vessels.10 The renal artery is involved in at least 5% to 10% of cases and may lead to renal ischemia, infarction, renal insufficiency, or refractory hypertension.2Mesenteric ischemia or infarction occurs in about 5% of dissections, may be difficult to diagnose, and is particularly dangerous.2,8 Aortic dissection may extend into the iliac arteries and may cause acute lower extremity ischemia.
Acute myocardial infarction due to involvement of the dissection flap causing malperfusion of a coronary artery occurs in 1% to 7% of acute type A aortic dissections.1–3 The right coronary artery (Figure 2) is most commonly involved, leading to acute inferior myocardial infarction. Acute myocardial ischemia and infarction in the setting of dissection may lead to a delay in the diagnosis of dissection and to bleeding complications from antiplatelet and anticoagulant drugs given to treat the acute coronary syndrome.
Cardiac tamponade, occurring in about 10% of acute type A dissections, portends a higher risk of death.2,3
Additional clinical features of aortic dissection include a left-sided pleural effusion, usually related to an inflammatory response. An acute hemothorax may occur from rupture or leaking of a descending aortic dissection.
FINDINGS ON RADIOGRAPHY AND ELECTROCARDIOGRAPHY
Electrocardiography usually has normal or nonspecific findings, unless acute myocardial infarction complicates the dissection.
D-DIMER LEVELS
Biomarkers for the diagnosis of acute aortic dissection are of great interest.
D-dimer levels rise in acute aortic dissection as they do in pulmonary embolism.11 A D-dimer level greater than 1,600 ng/mL within the first 6 hours has a very high positive likelihood ratio for dissection, so this test may be useful in identifying patients with a high probability for dissection. In the first 24 hours after symptom onset, a D-dimer level of less than 500 ng/mL has a negative predictive value of 95%. Thus, elevations in D-dimer may help decide which imaging to perform in a patient presenting with chest pain or suspicion of dissection.11
However, D-dimer levels may not be elevated in dissection variants, such as aortic intramural hematoma or penetrating aortic ulcer. Additionally, once 24 hours have elapsed since the dissection started, D-dimer levels may no longer be elevated. The current ACC/AHA guidelines on thoracic aortic disease concluded that the D-dimer level cannot be used to rule out aortic dissection in high-risk individuals.3
Additional studies may clarify the appropriate role of the D-dimer assay in diagnosing aortic dissection.
DEFINITIVE IMAGING STUDIES: CT, MRI, TEE
Contrast-enhanced computed tomography (CT), magnetic resonance imaging (MRI), and transesophageal echocardiography (TEE) all have very high sensitivity and specificity for the diagnosis of aortic dissection.2,3 The choice of imaging study often depends on the availability of these studies, with CT and TEE being the most commonly performed initial studies.
MRI is outstanding for detecting and following aortic dissection, but it is usually not the initial study performed because of the time required for image acquisition and because it is generally not available on an emergency basis.
While transthoracic echocardiography (TTE) can detect aortic dissection, its sensitivity is much lower than that of other imaging tests.2,3 Therefore, negative findings on TTE do not exclude aortic dissection.
MANAGEMENT OF AORTIC DISSECTION
When acute aortic dissection is diagnosed, multidisciplinary evaluation and treatment are necessary. Time is of the essence, as the death rate in acute dissection may be as high as 1% per hour during the first 24 hours.1–3 All patients with acute aortic dissection, whether type A or type B, should be transferred to a tertiary care center with a staff experienced in managing aortic dissection and its complications.3 Emergency surgery is recommended for type A aortic dissection, whereas type B dissection is generally treated medically unless complications occur.2,3
The cornerstone of drug therapy is the prompt reduction in blood pressure with a beta-blocker to reduce shear stresses on the aorta. Intravenous agents such as esmolol (Brevibloc) or labetalol (Normodyne) are usually chosen. Sodium nitroprusside may be added to beta-blocker therapy for rapid blood pressure control in appropriate patients. The patient may require multiple antihypertensive medications. If hypertension is refractory, one must consider renal artery hypertension due to the dissection causing renal malperfusion.2 Acute pain may also worsen hypertension, and appropriate analgesia should be used.
Definitive therapy in acute dissection
Patients with acute type A dissection require emergency surgery,2,3 as they are at risk for life-threatening complications including cardiac tamponade from hemopericardium, aortic rupture, stroke, visceral ischemia, and heart failure due to severe aortic regurgitation. When aortic regurgitation complicates acute type A dissection, some patients are adequately treated by resuspension of the aortic valve leaflets, while others require valve-sparing root replacement or prosthetic aortic valve replacement.
Surgical therapy is associated with a survival benefit compared with medical therapy in acute type A dissection.1 The 14-day mortality rate for acute type A dissection treated surgically is about 25%.1 Patients with high-risk features such as heart failure, shock, tamponade, and mesenteric ischemia have a worse prognosis compared with those without these features.2,12,13
Acute type B aortic dissection carries a lower rate of death than type A dissection.1–3 In the IRAD cohort, the early mortality rate in those with type B dissection treated medically was about 10%.1 However, when complications such as malperfusion, shock, or requirement for surgery occur in type B dissection, the mortality rate is much higher,2,14 with rates of 25% to 50% reported.2
Thus, initial medical therapy is the preferred approach to acute type B dissection, and surgery or endovascular therapy is reserved for patients with acute complications.2,3 Typical indications for surgery or endovascular therapy in type B dissection include visceral or limb ischemia, aortic rupture, refractory pain, and aneurysmal dilation (Table 3).2
Endovascular therapy in aortic dissection
The high mortality rate with open surgery in acute type B dissection has spurred tremendous interest in endovascular treatments for complications involving the descending aorta and branch vessels.2
Fenestration of the aorta and stenting of branch vessels were the earliest techniques used in complicated type B dissection. By fenestrating (ie, opening) the intimal flap, blood can flow from the false lumen into the true lumen, decompressing the distended false lumen.
Endovascular stenting is used for acute aortic rupture, for malperfusion syndromes, and for rapidly enlarging false lumens. Endovascular grafts may cover the area of a primary intimal tear and thus eliminate the flow into the false channel and promote false-lumen thrombosis. Many patients with complicated type B dissection are treated with a hybrid approach, in which one segment of the aorta, such as the aortic arch, is treated surgically, while the descending aorta receives an endovascular graft.2
Patients with a type B dissection treated medically are at risk for late complications, including aneurysmal enlargement and subsequent aortic rupture. The Investigation of Stent Grafts in Aortic Dissection (INSTEAD) trial included 140 patients with uncomplicated type B dissection and compared drug therapy with endovascular stent grafting.15 After 2 years of follow-up, there was no difference in the rate of death between the two treatment groups. Patients receiving endovascular grafts had a higher rate of false-lumen thrombosis.
More studies are under way to examine the role of endovascular therapy in uncomplicated type B dissection.
AORTIC DISSECTION VARIANTS
Aortic intramural hematoma
Aortic intramural hematoma is a form of acute aortic syndrome in which a hematoma develops in the aortic media and no intimal flap is visualized either by imaging or at surgery.2,3,16 It is important to recognize this clinical entity in a patient presenting with acute chest or back pain, as sometimes it is mistaken for a “thrombus in a nonaneurysmal aorta.” Intramural hematoma accounts for 5% to 25% of acute aortic syndromes, depending on the study population (it is more common in Asian studies).2,3,17 It may present with symptoms similar to classic aortic dissection and is classified as type A or type B, depending on whether the ascending aorta is involved.
Patients with an intramural hematoma may progress to having complications such as hemopericardium, classic aortic dissection, aortic rupture, or aneurysmal dilation.2,3 However, many cases of type B aortic intramural hematoma result in complete resorption of the hematoma over time. In general, like classic aortic dissection, type A intramural hematoma is treated with emergency surgery and type B with initial medical therapy.2,3
There are reports from Southeast Asia of successful initial medical therapy for type A intramural hematoma, with surgery used for acute complications.18 In the Western literature, improved outcomes are reported with initial surgical therapy.17 Given the unpredictable nature of type A intramural hematoma, most experts recommend surgical therapy for appropriate candidates with acute type A intramural hematoma.2,3,19
Penetrating atherosclerotic ulcer of the aorta
Penetrating atherosclerotic ulcer of the aorta, another acute aortic syndrome, results from acute penetration of an atherosclerotic aortic lesion through the internal elastic lamina into the media.2,3,20 It is often associated with bleeding into the media, or intramural hematoma. While the ulcer may be found incidentally on imaging studies, especially in patients with severe aortic atherosclerosis, the typical presentation is acute, severe chest or back pain. It occurs most often in the descending aorta and the abdominal aorta.
Penetrating atherosclerotic ulcer may lead to pseudoaneurysm formation, focal aortic dissection, aortic rupture, or late aortic aneurysm.2
LONG-TERM MANAGEMENT AFTER AORTIC DISSECTION
After hospital discharge, patients with aortic dissection require lifelong management. This includes blood pressure control, lifestyle modification, serial imaging of the aorta with CT or MRI, patient education about the condition, and, when appropriate, screening of family members for aortic disease.5,21
Reported survival rates after hospitalization for type A dissection are 52% to 94% at 1 year and 45% to 88% at 5 years.2,22 The 10-year actuarial survival rate for those with acute dissection who survive the acute hospitalization is reported as 30% to 60%. Long-term survival rates after acute type B dissection have been reported at 56% to 92% at 1 year and 48% to 82% at 5 years.23 Survival rates depend on many factors, including the underlying condition, the age of the patient, and comorbidities.
It is important to treat hypertension after aortic dissection, with a goal blood pressure of 120/80 mm Hg or less for most patients. Older studies found higher mortality rates with poorly controlled hypertension. Beta-blockers are the drugs of first choice. Even in the absence of hypertension, long-term beta-blocker therapy should be used to lessen the aortic stress and force of ventricular contraction.
Genetic evaluation
Genetically triggered causes of aortic dissection should be considered. In many circumstances, referral to a medical geneticist or other practitioner knowledgeable in these conditions is important when these disorders are being evaluated (Table 2).
Many of these disorders have an autosomal dominant inheritance, and the patient should be asked about a family history of aortic disease, aortic dissection, or unexplained sudden death. Features of Marfan syndrome, Loeys-Dietz syndrome, and familial thoracic aortic aneurysm syndromes should be sought. Through comprehensive family studies, it is now recognized that up to 20% of patients with thoracic aortic disease (such as aneurysm or dissection) have another first-degree relative with thoracic aortic disease.2,3,24 Thus, first-degree relatives of patients with aortic aneurysm or dissection should be screened for thoracic aortic aneurysm disease.
Research into molecular genetics is providing a better understanding of the genetics of aortic dissection.3 New mutations associated with aortic dissection are being discovered in signaling pathways as well as elements critical for the integrity of the vascular wall.2,3 However, at present, most patients with aortic dissection will not have a specific identifiable genetic defect.
Not only does genetic testing enable the accurate diagnosis of the affected individual, but also treatments are often based on this diagnosis.3 Importantly, the identification of a specific gene mutation (ie, in TGFBR1 or 2, FBN1, ACTA2, MYH11, and COL3A1) in an affected individual has the potential to identify other family members at risk.3
Follow-up imaging
It is important to continue to image the aorta after aortic dissection. Patients may develop progressive dilation or aneurysm formation of the dissected aorta, pseudoaneurysm formation after repair, or recurrent dissection. Many patients require additional surgery on the aorta because of late aneurysm formation.
CT or MRI is usually performed at least every 6 months in the first 2 years after dissection and at least annually thereafter. More centers are choosing MRI for long-term follow-up to avoid the repeated radiation exposure with serial CT.
Patient education
Besides receiving medical therapy and undergoing imaging, patients with aortic dissection should be educated about this condition.5,21 The patient should be aware of symptoms suggesting dissection and should be instructed to seek attention for any concerning symptoms.
Lifestyle modifications are also important. The patient should be educated about safe activity levels and to avoid heavy isometric exercise, such as weight-lifting. Some patients will have to cease their current occupation because of activity restrictions.
- Hagan PG, Nienaber CA, Isselbacher EM, et al. International Registry of Acute Aortic Dissection (IRAD): new insights from an old disease. JAMA 2000; 283:897–903.
- Braverman AC, Thompson R, Sanchez L. Diseases of the aorta. In:Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease, 9th Edition. Elsevier, Philadelphia, 2011.
- Hiratzka LF, Bakris GL, Beckman JA, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. Guidelines for the management of patients with thoracic aortic disease. Circulation 2010; 121:e266–e369.
- Rogers AM, Herman LK, Booher AM, et al. Sensitivity of the aortic dissection detection risk score, a novel guideline-based tool for identification of acute aortic dissection at initial presentation. Results from the International Registry of Acute Aortic Dissection. Circulation 2011; 123:2213–2228.
- Braverman AC. Acute aortic dissection: clinician update. Circulation 2010; 122:184–188.
- Davies RR, Gallo A, Coady MA, et al. Novel measurement of relative aortic size predicts rupture of thoracic aortic aneurysms. Ann Thorac Surg 2006; 81:169–177.
- Pape LA, Tsai TT, Isselbacher EM, et al. Aortic diameter >5.5 cm is not a good predictor of type A aortic dissection. Observations from the International Registry of Acute Aortic Dissection. Circulation 2007; 116:1120–1127.
- Parish LM, Gorman JH, Kahn S, et al. Aortic size in acute type A dissection: implications for preventative ascending aortic replacement. Eur J Cardiothorac Surg 2009; 35:941–945.
- Gaul C, Dietrich W, Erbguth FJ. Neurological symptoms in acute aortic dissection: a challenge for neurologists. Cerebrovasc Dis 2008; 26:1–8.
- Upchurch GR, Nienaber C, Fattori R, et al Acute aortic dissection presenting with primarily abdominal pain: a rare manifestation of a deadly disease. Ann Vasc Surg 2005; 19:367–373.
- Suzuki T, Distante A, Zizza A, et al. Diagnosis of acute aortic dissection by D-dimer: the International Registry of Acute Aortic Dissection substudy on biomarkers (IRAD-bio) experience. Circulation 2009; 119:2702–2707.
- Tsai TT, Trimarchi S, Neinaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg 2009; 37:149–159.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Contemporary results of surgery in acute type A aortic dissection: the International Registry of Acute Aortic Dissection experience. J Thorac Cardiovasc Surg 2005; 129:112–122.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Role and results of surgery in acute type B aortic dissection. Insights from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2006; 114(suppl 1):I-357–I-364.
- Nienaber CA, Rousseau H, Eggbrecht H, et al. Randomized comparison of strategies for type B aortic dissection. The Investigation of STEnt grafts in Aortic Dissection (INSTEAD) Trial. Circulation 2009; 120:2519–2528.
- Evangelista A, Mukherjee D, Mehta RH, et al. Acute intramural hematoma of the aorta. Circulation 2005; 111:1063–1070.
- Pelzel JM, Braverman AC, Hirsch AT, Harris KM. International heterogeneity in diagnostic frequency and clinical outcomes of ascending aortic intramural hematoma. J Am Soc Echo 2007; 20:1260–1268.
- Song JK, Yim JH, Ahn JM, et al. Outcomes of patients with acute type A aortic intramural hematoma. Circulation 2009; 120:2046–2052.
- Harris KM, Pelzel JM, Braverman AC. Letter regarding article, “Outcomes of patients with acute type A intramural hematoma.” Circulation 2010; 121:e456.
- Sundt TM. Intramural hematoma and penetrating atherosclerotic ulcer of the aorta. Ann Thorac Surg 2007; 83:S835–S841.
- Juang D, Braverman A, Eagle K. Aortic dissection. Circulation 2008; 118:e507–e510.
- Tsai TT, Evangelista A, Nienaber CA, et al. Long-term survival in patients presenting with type A acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114(suppl I):I-350–I-356.
- Tsai TT, Fattori R, Trimarchi S, et al. Long-term survival in patients presenting with type B acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114:2226–2231.
- Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections: incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg 2006; 82:1400–1405.
- Hagan PG, Nienaber CA, Isselbacher EM, et al. International Registry of Acute Aortic Dissection (IRAD): new insights from an old disease. JAMA 2000; 283:897–903.
- Braverman AC, Thompson R, Sanchez L. Diseases of the aorta. In:Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease, 9th Edition. Elsevier, Philadelphia, 2011.
- Hiratzka LF, Bakris GL, Beckman JA, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. Guidelines for the management of patients with thoracic aortic disease. Circulation 2010; 121:e266–e369.
- Rogers AM, Herman LK, Booher AM, et al. Sensitivity of the aortic dissection detection risk score, a novel guideline-based tool for identification of acute aortic dissection at initial presentation. Results from the International Registry of Acute Aortic Dissection. Circulation 2011; 123:2213–2228.
- Braverman AC. Acute aortic dissection: clinician update. Circulation 2010; 122:184–188.
- Davies RR, Gallo A, Coady MA, et al. Novel measurement of relative aortic size predicts rupture of thoracic aortic aneurysms. Ann Thorac Surg 2006; 81:169–177.
- Pape LA, Tsai TT, Isselbacher EM, et al. Aortic diameter >5.5 cm is not a good predictor of type A aortic dissection. Observations from the International Registry of Acute Aortic Dissection. Circulation 2007; 116:1120–1127.
- Parish LM, Gorman JH, Kahn S, et al. Aortic size in acute type A dissection: implications for preventative ascending aortic replacement. Eur J Cardiothorac Surg 2009; 35:941–945.
- Gaul C, Dietrich W, Erbguth FJ. Neurological symptoms in acute aortic dissection: a challenge for neurologists. Cerebrovasc Dis 2008; 26:1–8.
- Upchurch GR, Nienaber C, Fattori R, et al Acute aortic dissection presenting with primarily abdominal pain: a rare manifestation of a deadly disease. Ann Vasc Surg 2005; 19:367–373.
- Suzuki T, Distante A, Zizza A, et al. Diagnosis of acute aortic dissection by D-dimer: the International Registry of Acute Aortic Dissection substudy on biomarkers (IRAD-bio) experience. Circulation 2009; 119:2702–2707.
- Tsai TT, Trimarchi S, Neinaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg 2009; 37:149–159.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Contemporary results of surgery in acute type A aortic dissection: the International Registry of Acute Aortic Dissection experience. J Thorac Cardiovasc Surg 2005; 129:112–122.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Role and results of surgery in acute type B aortic dissection. Insights from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2006; 114(suppl 1):I-357–I-364.
- Nienaber CA, Rousseau H, Eggbrecht H, et al. Randomized comparison of strategies for type B aortic dissection. The Investigation of STEnt grafts in Aortic Dissection (INSTEAD) Trial. Circulation 2009; 120:2519–2528.
- Evangelista A, Mukherjee D, Mehta RH, et al. Acute intramural hematoma of the aorta. Circulation 2005; 111:1063–1070.
- Pelzel JM, Braverman AC, Hirsch AT, Harris KM. International heterogeneity in diagnostic frequency and clinical outcomes of ascending aortic intramural hematoma. J Am Soc Echo 2007; 20:1260–1268.
- Song JK, Yim JH, Ahn JM, et al. Outcomes of patients with acute type A aortic intramural hematoma. Circulation 2009; 120:2046–2052.
- Harris KM, Pelzel JM, Braverman AC. Letter regarding article, “Outcomes of patients with acute type A intramural hematoma.” Circulation 2010; 121:e456.
- Sundt TM. Intramural hematoma and penetrating atherosclerotic ulcer of the aorta. Ann Thorac Surg 2007; 83:S835–S841.
- Juang D, Braverman A, Eagle K. Aortic dissection. Circulation 2008; 118:e507–e510.
- Tsai TT, Evangelista A, Nienaber CA, et al. Long-term survival in patients presenting with type A acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114(suppl I):I-350–I-356.
- Tsai TT, Fattori R, Trimarchi S, et al. Long-term survival in patients presenting with type B acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114:2226–2231.
- Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections: incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg 2006; 82:1400–1405.
KEY POINTS
- Aortic surgery is the treatment of choice for dissection of the ascending aorta, whereas dissection of the descending aorta is initially managed medically.
- Look for an underlying genetic predisposition to aortic disease and, in many instances, screen first-degree relatives for aortic disease.
- Long-term management requires serial imaging of the aorta, blood pressure control, and, for many, future aortic procedures.
- Measuring the D-dimer levels may help in decision-making for appropriate imaging in patients presenting with chest pain, as an elevated level raises the suspicion of dissection. However, more study of this and other biomarkers is needed.
- Advances in molecular genetics and the biology of the aortic wall promise to improve the diagnosis and prognosis of aortic disease.
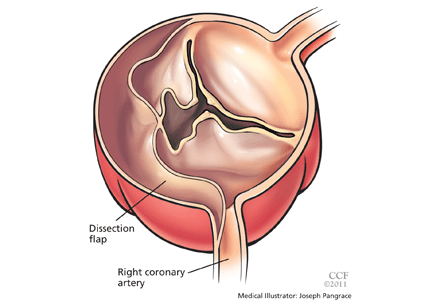
Post PICC Placement
ANSWER
The radiograph demonstrates a right PICC line terminating at the superior vena cava. There is no evidence of pneumothorax.
Of note, however, is a large oval density within the right upper lobe, measuring 4.5 x 7 cm. This lesion could represent a loculated mass such as an abscess or hematoma. Further workup with additional imaging is warranted.
The working theory on this patient was that the density likely represented abscess or infection. Contrast-enhanced CT of the chest suggested likely abscess. The patient then underwent successful CT-guided needle biopsy, which returned positive results for Cryptococcus.
ANSWER
The radiograph demonstrates a right PICC line terminating at the superior vena cava. There is no evidence of pneumothorax.
Of note, however, is a large oval density within the right upper lobe, measuring 4.5 x 7 cm. This lesion could represent a loculated mass such as an abscess or hematoma. Further workup with additional imaging is warranted.
The working theory on this patient was that the density likely represented abscess or infection. Contrast-enhanced CT of the chest suggested likely abscess. The patient then underwent successful CT-guided needle biopsy, which returned positive results for Cryptococcus.
ANSWER
The radiograph demonstrates a right PICC line terminating at the superior vena cava. There is no evidence of pneumothorax.
Of note, however, is a large oval density within the right upper lobe, measuring 4.5 x 7 cm. This lesion could represent a loculated mass such as an abscess or hematoma. Further workup with additional imaging is warranted.
The working theory on this patient was that the density likely represented abscess or infection. Contrast-enhanced CT of the chest suggested likely abscess. The patient then underwent successful CT-guided needle biopsy, which returned positive results for Cryptococcus.
A 31-year-old man is admitted to your facility with presumed cryptococcal meningitis. He has a one-month history of progressively worsening headaches, malaise, weight loss, and blurred vision. A lumbar puncture demonstrates an extremely elevated opening pressure and yields samples that are positive for Cryptococcus on microscopic examination. The patient denies any significant medical history. Specifically, there is no history of HIV, which was confirmed by recent serologic testing at another hospital. As a result of poor peripheral access and with anticipated need for lengthy IV antifungal therapy, a PICC (peripherally inserted central catheter) line is ordered. It is placed without incident at the bedside, and as per protocol, a post-placement portable chest radiograph is obtained. What is your impression?
Man Thrown From All-Terrain Vehicle
ANSWER
The image shows several things. First, there is a well-corticated lucency through the base of the odontoid process. This most likely represents what is referred to as an os odontoideum (congenital spinal variant). The other possibility is that it could be an old odontoid fracture with nonunion.
In addition, there are superior end-plate fractures noted in C6, C7, T1, and T2. However, no definite fracture lines are evident, suggesting these are subacute or old injuries. MRI can be performed to differentiate old versus new fractures; in this case, it was determined these were old.
ANSWER
The image shows several things. First, there is a well-corticated lucency through the base of the odontoid process. This most likely represents what is referred to as an os odontoideum (congenital spinal variant). The other possibility is that it could be an old odontoid fracture with nonunion.
In addition, there are superior end-plate fractures noted in C6, C7, T1, and T2. However, no definite fracture lines are evident, suggesting these are subacute or old injuries. MRI can be performed to differentiate old versus new fractures; in this case, it was determined these were old.
ANSWER
The image shows several things. First, there is a well-corticated lucency through the base of the odontoid process. This most likely represents what is referred to as an os odontoideum (congenital spinal variant). The other possibility is that it could be an old odontoid fracture with nonunion.
In addition, there are superior end-plate fractures noted in C6, C7, T1, and T2. However, no definite fracture lines are evident, suggesting these are subacute or old injuries. MRI can be performed to differentiate old versus new fractures; in this case, it was determined these were old.
A 57-year-old man is transferred to your facility after being thrown from an all-terrain vehicle. He was not wearing a helmet and was documented to have loss of consciousness. Upon arrival, his primary complaint is severe headache. He gives no significant medical history. Initial assessment shows a male on a backboard with full cervical spine immobilization. His Glasgow Coma Scale score is 14, with a blood pressure of 130/83 mm Hg and a heart rate of 87 beats/min. He has several abrasions on his face, but his pupils are equal and react well. His heart and lungs appear to be clear and the abdomen is benign. He is able to move all extremities well, with no obvious neurovascular compromise. After removal from the backboard, he is sent to the radiology department for multiple scans. A static sagittal image from CT of the cervical spine is shown. What is your impression?
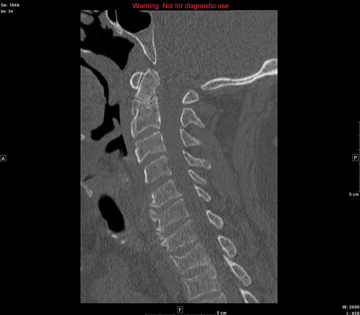
Grand Rounds: Man, 46, With a Curious Ear Pain
A 46-year-old man presented to a hospital emergency department (ED) with a four-day history of right ear pain. He described the pain as a constant, dull, burning pain radiating to the neck and face, associated with a feeling of congestion. The patient also stated that the right side of his face had felt numb for about one day.
Three days earlier, the man had been seen by his primary health care provider, who told him that his ear looked normal and free of infection. The day before his current presentation to the ED, however, he noticed what he described as an “acne-like” rash on his ear lobe. Shortly before coming to the ED, the patient also developed numbness over his right upper lip, which he likened to the effects of procaine during a dental visit. He reported drooling from the right side of his mouth while drinking water and difficulty blinking his right eye.
He denied any tinnitus, fever, headache, or change in hearing. A review of symptoms was positive only for mild dizziness during the previous two to three days.
The patient was a well-appearing white man. He was alert and oriented to identity, time, and place. His skin was warm, dry, and intact. The examiner noticed a small area of erythematous rash with vesicles on the man’s right ear lobe. The external auditory canals appeared within normal limits, with no erythema or edema, and were nontender bilaterally. The tympanic membranes were normal bilaterally, without bulging or discernible fluid levels.
The ocular exam was normal with no visual acuity changes and no fluorescein uptake; external ocular movements were intact. A slight droop was noted in the right eyelid, but there was no droop on the contralateral side of his face. When asked to puff up his cheeks, the patient found it difficult to do so on the right side of his mouth without releasing air from his lips.
The remainder of the cranial nerves were intact. Muscle strength was 5/5 in all extremities and equal bilaterally. The man’s gait was within normal limits, and the remaining findings in the physical exam were normal.
The initial diagnosis considered in the differential was otitis externa, because it is a common explanation for ear pain in patients who present to the ED.1,2 Also, in otitis, pain is characteristically present in the affected ear, and erythema is often found in the external auditory canal.3 However, this diagnosis was deemed unlikely because otitis externa would not explain the neurologic findings or the vesicular rash.1
Bell’s palsy was next in the differential, as it was considered consistent with the patient’s unilateral neurologic deficits.4 In addition to weakness or palsy of the facial nerve, many patients with Bell’s palsy complain of mastoid pain, which can be confused with a complaint of ear pain.5 However, patients with Bell’s palsy have no rash, and this diagnosis was considered unlikely.
The painful, burning rash on the patient’s face was characteristic of herpes zoster (shingles), which was next in the differential. Infrequently, shingles can also cause weakness in the nerve it affects. In the case patient, weakness that was evident in the affected nerve resembled that seen in Bell’s palsy. This combination of symptoms is referred to as Ramsay Hunt syndrome—which in this case was decided to be the correct diagnosis.
DISCUSSION
Ramsay Hunt syndrome (RHS, also known as geniculate herpes5,6) is caused by the varicella-zoster virus, most commonly known as the cause of chickenpox. In the United States, RHS is believed to affect only about one in 1,500 persons, although 20% to 30% of persons experience herpes zoster infection at some time.7
Soon after a chickenpox infection subsides, the virus spreads along the sensory nerve fibers of the peripheral and cranial nerves. The virus then becomes dormant in the dorsal root ganglion, where in some patients it later reactivates in the form of shingles.8
In RHS, the ganglia of cranial nerve VII (CN VII, the facial nerve, which innervates the facial muscles) are infected; for this reason, the condition is also referred to as zoster oticus.9 Because of the involvement and weakening of the facial nerve, the presentation of RHS often resembles that of Bell’s palsy or facial nerve palsy.
While most cases of Bell’s palsy are idiopathic,10,11 RHS can usually be attributed to viral infection—most commonly, infection with herpes simplex virus type 1 (HSV-1).12 RHS can be differentiated from Bell’s palsy by the presence of a rash on the ipsilateral side. The rash appears in the form of inflamed vesicles on an erythematous base and may be present around the ear (see figure), the eardrum, the hard and soft palate, or the tongue.6 When the rash is painful, it is often described as a burning pain. Loss of taste may occur in the anterior portion of the tongue.9,12
Unlike shingles, which usually manifests as a sensory neuropathy, RHS is distinguished by motor neuropathy.7 The patient usually reports weakness in the facial muscles on one side, leading to difficulty drinking water or puffing out the cheek and to drooling on one side of the face. A complaint of dryness in the ipsilateral eye may result from weakness or an inability to close the eyelid.
It is important to note that as in Bell’s palsy, RHS can be differentiated from stroke by the patient’s inability to wrinkle the forehead. The motor muscles of the forehead are innervated by both sides of the brain; in the case of stroke, only one side of the brain is affected, and movement of the forehead remains possible on the contralateral side. In facial nerve palsy, the nerve itself is affected; thus, no movement of the forehead is possible.13 Other common complaints in patients with facial nerve palsy include vertigo, hearing loss, and changes in facial sensation.
RHS was first described in 1907 as herpes zoster associated with Bell’s palsy by the neurologist J. Ramsay Hunt, for whom the condition is named.9,14 RHS is more common in men than women. It occurs most commonly in adults and is rare in children younger than 6.13,15
Diagnosis
In most cases, a diagnosis of RHS is made on a clinical basis.1 However, a polymerase chain reaction (PCR) assay can be performed on samples of tear fluid or submandibular saliva to detect the zoster virus.16,17 PCR can also be performed using exudates from the geniculate zone of the ear (a small area in the center of the auricle6,14), which is more sensitive than tears or blood.18,19 Findings from a complete blood count and the erythrocyte sedimentation rate can be used to differentiate between infectious and inflammatory causes.13
Head CT or MRI can be obtained to rule out any structural lesions. In one study, Kim et al20 examined MRI changes in patients with either Bell’s palsy or RHS. In both conditions, researchers were able to identify swelling of the labyrinthine segment of the facial nerve on temporal MRI scans.20 Although CT has not been shown to have any prognostic or diagnostic application, it can occasionally be used if decompression of the facial nerve is warranted.11
Treatment
Data used to support the use of corticosteroids for treatment of Bell’s palsy10,21,22 have been extrapolated to justify their use for treatment of RHS,23 and prednisolone is the most common choice.10 Steroids reduce the associated inflammation, resulting in decreased pain and neurologic symptoms. A daily dose for one to two weeks, followed by a slow taper, is the preferred prescribing method.10
The addition of acyclovir has been recommended to inhibit viral DNA replication9,23 (valacyclovir and famciclovir have also been mentioned12,18). If started within three days of symptom onset, acyclovir can help reduce pain and hasten resolution of symptoms.
In a large retrospective study, it was demonstrated that patients treated with prednisone at 1.0 mg/kg/d for five days, followed by a 10-day taper, combined with acyclovir, showed long-term improvement that was statistically significant.23 Complete facial recovery was reported in only 52% of patients, however. Risk factors for a poor prognosis include hypertension, diabetes mellitus, and advancing age.7
Artificial tears are also prescribed to keep the affected eye from becoming irritated and dry. The patient can be instructed to tape the eye closed at night.10
Early diagnosis and treatment (ie, within three days of symptom onset, and preferably with a combination of acyclovir and steroids23) is an important factor in a good prognosis.7,23 Because RHS-affected patients have only about a 50% chance of full recovery,23 proper follow-up care is extremely important. Follow-up visits are recommended at two weeks, six weeks, and three months.13 For optimal outcomes in patients with this neurologic diagnosis, referral to a neurologist is recommended for ongoing management. This practitioner is likely to detect subtle changes in patient presentation and can perform follow-up testing as needed.
THE CASE PATIENT
One week after the patient’s visit to the ED, he was contacted by hospital staff for a standard satisfaction and quality control survey. The patient (who had been treated with steroids and acyclovir, ibuprofen, and artificial tears) reported almost complete resolution of his pain; any mild pain, he said, was easily tolerated or could be resolved with OTC medication. He reported only minimal persistent facial weakness, stating that he was able to eat, drink, and speak normally.
The patient had not been seen by any health care provider for follow-up, but he agreed to make an appointment as soon as possible.
REFERENCES
1. Kim D, Bhimani M. Ramsay Hunt syndrome presenting as simple otitis externa. CJEM. 2008;10(3):247-250.
2. Agius AM, Pickles JM, Burch KL. A prospective study of otitis externa. Clin Otolaryngol. 1992;17(2):150-154.
3. Rosenfeld RM, Brown L, Cannon CR, et al; American Academy of Otolaryngology—Head and Neck Surgery Foundation. Clinical practice guideline: acute otitis externa. Otolaryngol Head Neck Surg. 2006;134(4 suppl):S4-S23.
4. Holland J, Bernstein J. Bell’s palsy. Clin Evid (Online). 2011 Mar 7;2011.pii:1204.
5. Jacewicz M. Bell’s palsy (2007). www.merckmanuals.com/professional/sec16/ch219/ch219i.html. Accessed May 26, 2011.
6. Harrison K. Discussion: the Ramsay Hunt Syndrome. Proc Royal Soc Med. 1953;47(371):11-24.
7. Bhupal HK. Ramsay Hunt syndrome presenting in primary care. Practitioner. 2010;254(1727):33-35.
8. Aizawa H, Ohtani F, Furuta Y, et al. Variable patterns of varicella-zoster virus reactivation in Ramsay Hunt syndrome. J Med Virol. 2004;74(2):355-360.
9. Gondivkar S, Parikh V, Parikh R. Herpes zoster oticus: a rare clinical entity. Contemp Clin Dent. 2010;1(2):127-129.
10. Sullivan FM, Swan IRC, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med. 2007;357(16):1598-1607.
11. Gilden DH. Bell’s palsy. N Engl J Med. 2004;351(13):1323-1331.
12. Diaz GA, Rakita RM, Koelle DM. A case of Ramsay Hunt–like syndrome caused by herpes simplex virus type 2. Clin Infect Dis. 2005;40(10):1545-1547.
13. Miravalle AA. Ramsay Hunt syndrome. http://emedicine.medscape.com/article/1166804-over iew. Accessed July 22, 2011.
14. Hunt JR. On herpetic inflammation of the geniculate ganglion: a new syndrome and its complications. J Nerv Ment Dis. 1907;34:73-96.
15. Sandoval CC, Núñez FA, Lizama CM, et al. Ramsay Hunt syndrome in children: four cases and review [in Spanish]. Rev Chilena Infectol. 2008; 25(6):458-464.
16. Murakami S, Nakashiro Y, Mizobuchi M, et al. Varicella-zoster virus distribution in Ramsay Hunt syndrome revealed by polymerase chain reaction. Acta Otolaryngol. 1998;118(2):145-149.
17. Hiroshige K, Ikeda M, Hondo R. Detection of varicella zoster virus DNA in tear fluid and saliva of patients with Ramsay Hunt syndrome. Otol Neurol. 2002;23(4):602-607.
18. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatr. 2001;71(2):148-154.
19. Murakami S, Honda N, Mizobuchi M, et al. Rapid diagnosis of varicella zoster virus infection in acute facial palsy. Neurology. 1998;51(4):1202-1205.
20. Kim IS, Shin SH, Kim J, et al. Correlation between MRI and operative findings in Bell’s palsy and Ramsay Hunt syndrome. Yonsei Med J. 2007;48(6):963-968.
21. Engström M, Berg T, Stjernquist-Desatnik A, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2008;7(11):993-1000.
22. Hato N, Yamada H, Kohno H, et al. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol. 2007;28(3):408-413.
23. Murakami S, Hato N, Horiuchi J, et al. Treatment of Ramsay Hunt syndrome with acyclovir-prednisone: significance of early diagnosis and treatment. Ann Neurol. 1997;41(3):353-357.
A 46-year-old man presented to a hospital emergency department (ED) with a four-day history of right ear pain. He described the pain as a constant, dull, burning pain radiating to the neck and face, associated with a feeling of congestion. The patient also stated that the right side of his face had felt numb for about one day.
Three days earlier, the man had been seen by his primary health care provider, who told him that his ear looked normal and free of infection. The day before his current presentation to the ED, however, he noticed what he described as an “acne-like” rash on his ear lobe. Shortly before coming to the ED, the patient also developed numbness over his right upper lip, which he likened to the effects of procaine during a dental visit. He reported drooling from the right side of his mouth while drinking water and difficulty blinking his right eye.
He denied any tinnitus, fever, headache, or change in hearing. A review of symptoms was positive only for mild dizziness during the previous two to three days.
The patient was a well-appearing white man. He was alert and oriented to identity, time, and place. His skin was warm, dry, and intact. The examiner noticed a small area of erythematous rash with vesicles on the man’s right ear lobe. The external auditory canals appeared within normal limits, with no erythema or edema, and were nontender bilaterally. The tympanic membranes were normal bilaterally, without bulging or discernible fluid levels.
The ocular exam was normal with no visual acuity changes and no fluorescein uptake; external ocular movements were intact. A slight droop was noted in the right eyelid, but there was no droop on the contralateral side of his face. When asked to puff up his cheeks, the patient found it difficult to do so on the right side of his mouth without releasing air from his lips.
The remainder of the cranial nerves were intact. Muscle strength was 5/5 in all extremities and equal bilaterally. The man’s gait was within normal limits, and the remaining findings in the physical exam were normal.
The initial diagnosis considered in the differential was otitis externa, because it is a common explanation for ear pain in patients who present to the ED.1,2 Also, in otitis, pain is characteristically present in the affected ear, and erythema is often found in the external auditory canal.3 However, this diagnosis was deemed unlikely because otitis externa would not explain the neurologic findings or the vesicular rash.1
Bell’s palsy was next in the differential, as it was considered consistent with the patient’s unilateral neurologic deficits.4 In addition to weakness or palsy of the facial nerve, many patients with Bell’s palsy complain of mastoid pain, which can be confused with a complaint of ear pain.5 However, patients with Bell’s palsy have no rash, and this diagnosis was considered unlikely.
The painful, burning rash on the patient’s face was characteristic of herpes zoster (shingles), which was next in the differential. Infrequently, shingles can also cause weakness in the nerve it affects. In the case patient, weakness that was evident in the affected nerve resembled that seen in Bell’s palsy. This combination of symptoms is referred to as Ramsay Hunt syndrome—which in this case was decided to be the correct diagnosis.
DISCUSSION
Ramsay Hunt syndrome (RHS, also known as geniculate herpes5,6) is caused by the varicella-zoster virus, most commonly known as the cause of chickenpox. In the United States, RHS is believed to affect only about one in 1,500 persons, although 20% to 30% of persons experience herpes zoster infection at some time.7
Soon after a chickenpox infection subsides, the virus spreads along the sensory nerve fibers of the peripheral and cranial nerves. The virus then becomes dormant in the dorsal root ganglion, where in some patients it later reactivates in the form of shingles.8
In RHS, the ganglia of cranial nerve VII (CN VII, the facial nerve, which innervates the facial muscles) are infected; for this reason, the condition is also referred to as zoster oticus.9 Because of the involvement and weakening of the facial nerve, the presentation of RHS often resembles that of Bell’s palsy or facial nerve palsy.
While most cases of Bell’s palsy are idiopathic,10,11 RHS can usually be attributed to viral infection—most commonly, infection with herpes simplex virus type 1 (HSV-1).12 RHS can be differentiated from Bell’s palsy by the presence of a rash on the ipsilateral side. The rash appears in the form of inflamed vesicles on an erythematous base and may be present around the ear (see figure), the eardrum, the hard and soft palate, or the tongue.6 When the rash is painful, it is often described as a burning pain. Loss of taste may occur in the anterior portion of the tongue.9,12
Unlike shingles, which usually manifests as a sensory neuropathy, RHS is distinguished by motor neuropathy.7 The patient usually reports weakness in the facial muscles on one side, leading to difficulty drinking water or puffing out the cheek and to drooling on one side of the face. A complaint of dryness in the ipsilateral eye may result from weakness or an inability to close the eyelid.
It is important to note that as in Bell’s palsy, RHS can be differentiated from stroke by the patient’s inability to wrinkle the forehead. The motor muscles of the forehead are innervated by both sides of the brain; in the case of stroke, only one side of the brain is affected, and movement of the forehead remains possible on the contralateral side. In facial nerve palsy, the nerve itself is affected; thus, no movement of the forehead is possible.13 Other common complaints in patients with facial nerve palsy include vertigo, hearing loss, and changes in facial sensation.
RHS was first described in 1907 as herpes zoster associated with Bell’s palsy by the neurologist J. Ramsay Hunt, for whom the condition is named.9,14 RHS is more common in men than women. It occurs most commonly in adults and is rare in children younger than 6.13,15
Diagnosis
In most cases, a diagnosis of RHS is made on a clinical basis.1 However, a polymerase chain reaction (PCR) assay can be performed on samples of tear fluid or submandibular saliva to detect the zoster virus.16,17 PCR can also be performed using exudates from the geniculate zone of the ear (a small area in the center of the auricle6,14), which is more sensitive than tears or blood.18,19 Findings from a complete blood count and the erythrocyte sedimentation rate can be used to differentiate between infectious and inflammatory causes.13
Head CT or MRI can be obtained to rule out any structural lesions. In one study, Kim et al20 examined MRI changes in patients with either Bell’s palsy or RHS. In both conditions, researchers were able to identify swelling of the labyrinthine segment of the facial nerve on temporal MRI scans.20 Although CT has not been shown to have any prognostic or diagnostic application, it can occasionally be used if decompression of the facial nerve is warranted.11
Treatment
Data used to support the use of corticosteroids for treatment of Bell’s palsy10,21,22 have been extrapolated to justify their use for treatment of RHS,23 and prednisolone is the most common choice.10 Steroids reduce the associated inflammation, resulting in decreased pain and neurologic symptoms. A daily dose for one to two weeks, followed by a slow taper, is the preferred prescribing method.10
The addition of acyclovir has been recommended to inhibit viral DNA replication9,23 (valacyclovir and famciclovir have also been mentioned12,18). If started within three days of symptom onset, acyclovir can help reduce pain and hasten resolution of symptoms.
In a large retrospective study, it was demonstrated that patients treated with prednisone at 1.0 mg/kg/d for five days, followed by a 10-day taper, combined with acyclovir, showed long-term improvement that was statistically significant.23 Complete facial recovery was reported in only 52% of patients, however. Risk factors for a poor prognosis include hypertension, diabetes mellitus, and advancing age.7
Artificial tears are also prescribed to keep the affected eye from becoming irritated and dry. The patient can be instructed to tape the eye closed at night.10
Early diagnosis and treatment (ie, within three days of symptom onset, and preferably with a combination of acyclovir and steroids23) is an important factor in a good prognosis.7,23 Because RHS-affected patients have only about a 50% chance of full recovery,23 proper follow-up care is extremely important. Follow-up visits are recommended at two weeks, six weeks, and three months.13 For optimal outcomes in patients with this neurologic diagnosis, referral to a neurologist is recommended for ongoing management. This practitioner is likely to detect subtle changes in patient presentation and can perform follow-up testing as needed.
THE CASE PATIENT
One week after the patient’s visit to the ED, he was contacted by hospital staff for a standard satisfaction and quality control survey. The patient (who had been treated with steroids and acyclovir, ibuprofen, and artificial tears) reported almost complete resolution of his pain; any mild pain, he said, was easily tolerated or could be resolved with OTC medication. He reported only minimal persistent facial weakness, stating that he was able to eat, drink, and speak normally.
The patient had not been seen by any health care provider for follow-up, but he agreed to make an appointment as soon as possible.
REFERENCES
1. Kim D, Bhimani M. Ramsay Hunt syndrome presenting as simple otitis externa. CJEM. 2008;10(3):247-250.
2. Agius AM, Pickles JM, Burch KL. A prospective study of otitis externa. Clin Otolaryngol. 1992;17(2):150-154.
3. Rosenfeld RM, Brown L, Cannon CR, et al; American Academy of Otolaryngology—Head and Neck Surgery Foundation. Clinical practice guideline: acute otitis externa. Otolaryngol Head Neck Surg. 2006;134(4 suppl):S4-S23.
4. Holland J, Bernstein J. Bell’s palsy. Clin Evid (Online). 2011 Mar 7;2011.pii:1204.
5. Jacewicz M. Bell’s palsy (2007). www.merckmanuals.com/professional/sec16/ch219/ch219i.html. Accessed May 26, 2011.
6. Harrison K. Discussion: the Ramsay Hunt Syndrome. Proc Royal Soc Med. 1953;47(371):11-24.
7. Bhupal HK. Ramsay Hunt syndrome presenting in primary care. Practitioner. 2010;254(1727):33-35.
8. Aizawa H, Ohtani F, Furuta Y, et al. Variable patterns of varicella-zoster virus reactivation in Ramsay Hunt syndrome. J Med Virol. 2004;74(2):355-360.
9. Gondivkar S, Parikh V, Parikh R. Herpes zoster oticus: a rare clinical entity. Contemp Clin Dent. 2010;1(2):127-129.
10. Sullivan FM, Swan IRC, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med. 2007;357(16):1598-1607.
11. Gilden DH. Bell’s palsy. N Engl J Med. 2004;351(13):1323-1331.
12. Diaz GA, Rakita RM, Koelle DM. A case of Ramsay Hunt–like syndrome caused by herpes simplex virus type 2. Clin Infect Dis. 2005;40(10):1545-1547.
13. Miravalle AA. Ramsay Hunt syndrome. http://emedicine.medscape.com/article/1166804-over iew. Accessed July 22, 2011.
14. Hunt JR. On herpetic inflammation of the geniculate ganglion: a new syndrome and its complications. J Nerv Ment Dis. 1907;34:73-96.
15. Sandoval CC, Núñez FA, Lizama CM, et al. Ramsay Hunt syndrome in children: four cases and review [in Spanish]. Rev Chilena Infectol. 2008; 25(6):458-464.
16. Murakami S, Nakashiro Y, Mizobuchi M, et al. Varicella-zoster virus distribution in Ramsay Hunt syndrome revealed by polymerase chain reaction. Acta Otolaryngol. 1998;118(2):145-149.
17. Hiroshige K, Ikeda M, Hondo R. Detection of varicella zoster virus DNA in tear fluid and saliva of patients with Ramsay Hunt syndrome. Otol Neurol. 2002;23(4):602-607.
18. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatr. 2001;71(2):148-154.
19. Murakami S, Honda N, Mizobuchi M, et al. Rapid diagnosis of varicella zoster virus infection in acute facial palsy. Neurology. 1998;51(4):1202-1205.
20. Kim IS, Shin SH, Kim J, et al. Correlation between MRI and operative findings in Bell’s palsy and Ramsay Hunt syndrome. Yonsei Med J. 2007;48(6):963-968.
21. Engström M, Berg T, Stjernquist-Desatnik A, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2008;7(11):993-1000.
22. Hato N, Yamada H, Kohno H, et al. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol. 2007;28(3):408-413.
23. Murakami S, Hato N, Horiuchi J, et al. Treatment of Ramsay Hunt syndrome with acyclovir-prednisone: significance of early diagnosis and treatment. Ann Neurol. 1997;41(3):353-357.
A 46-year-old man presented to a hospital emergency department (ED) with a four-day history of right ear pain. He described the pain as a constant, dull, burning pain radiating to the neck and face, associated with a feeling of congestion. The patient also stated that the right side of his face had felt numb for about one day.
Three days earlier, the man had been seen by his primary health care provider, who told him that his ear looked normal and free of infection. The day before his current presentation to the ED, however, he noticed what he described as an “acne-like” rash on his ear lobe. Shortly before coming to the ED, the patient also developed numbness over his right upper lip, which he likened to the effects of procaine during a dental visit. He reported drooling from the right side of his mouth while drinking water and difficulty blinking his right eye.
He denied any tinnitus, fever, headache, or change in hearing. A review of symptoms was positive only for mild dizziness during the previous two to three days.
The patient was a well-appearing white man. He was alert and oriented to identity, time, and place. His skin was warm, dry, and intact. The examiner noticed a small area of erythematous rash with vesicles on the man’s right ear lobe. The external auditory canals appeared within normal limits, with no erythema or edema, and were nontender bilaterally. The tympanic membranes were normal bilaterally, without bulging or discernible fluid levels.
The ocular exam was normal with no visual acuity changes and no fluorescein uptake; external ocular movements were intact. A slight droop was noted in the right eyelid, but there was no droop on the contralateral side of his face. When asked to puff up his cheeks, the patient found it difficult to do so on the right side of his mouth without releasing air from his lips.
The remainder of the cranial nerves were intact. Muscle strength was 5/5 in all extremities and equal bilaterally. The man’s gait was within normal limits, and the remaining findings in the physical exam were normal.
The initial diagnosis considered in the differential was otitis externa, because it is a common explanation for ear pain in patients who present to the ED.1,2 Also, in otitis, pain is characteristically present in the affected ear, and erythema is often found in the external auditory canal.3 However, this diagnosis was deemed unlikely because otitis externa would not explain the neurologic findings or the vesicular rash.1
Bell’s palsy was next in the differential, as it was considered consistent with the patient’s unilateral neurologic deficits.4 In addition to weakness or palsy of the facial nerve, many patients with Bell’s palsy complain of mastoid pain, which can be confused with a complaint of ear pain.5 However, patients with Bell’s palsy have no rash, and this diagnosis was considered unlikely.
The painful, burning rash on the patient’s face was characteristic of herpes zoster (shingles), which was next in the differential. Infrequently, shingles can also cause weakness in the nerve it affects. In the case patient, weakness that was evident in the affected nerve resembled that seen in Bell’s palsy. This combination of symptoms is referred to as Ramsay Hunt syndrome—which in this case was decided to be the correct diagnosis.
DISCUSSION
Ramsay Hunt syndrome (RHS, also known as geniculate herpes5,6) is caused by the varicella-zoster virus, most commonly known as the cause of chickenpox. In the United States, RHS is believed to affect only about one in 1,500 persons, although 20% to 30% of persons experience herpes zoster infection at some time.7
Soon after a chickenpox infection subsides, the virus spreads along the sensory nerve fibers of the peripheral and cranial nerves. The virus then becomes dormant in the dorsal root ganglion, where in some patients it later reactivates in the form of shingles.8
In RHS, the ganglia of cranial nerve VII (CN VII, the facial nerve, which innervates the facial muscles) are infected; for this reason, the condition is also referred to as zoster oticus.9 Because of the involvement and weakening of the facial nerve, the presentation of RHS often resembles that of Bell’s palsy or facial nerve palsy.
While most cases of Bell’s palsy are idiopathic,10,11 RHS can usually be attributed to viral infection—most commonly, infection with herpes simplex virus type 1 (HSV-1).12 RHS can be differentiated from Bell’s palsy by the presence of a rash on the ipsilateral side. The rash appears in the form of inflamed vesicles on an erythematous base and may be present around the ear (see figure), the eardrum, the hard and soft palate, or the tongue.6 When the rash is painful, it is often described as a burning pain. Loss of taste may occur in the anterior portion of the tongue.9,12
Unlike shingles, which usually manifests as a sensory neuropathy, RHS is distinguished by motor neuropathy.7 The patient usually reports weakness in the facial muscles on one side, leading to difficulty drinking water or puffing out the cheek and to drooling on one side of the face. A complaint of dryness in the ipsilateral eye may result from weakness or an inability to close the eyelid.
It is important to note that as in Bell’s palsy, RHS can be differentiated from stroke by the patient’s inability to wrinkle the forehead. The motor muscles of the forehead are innervated by both sides of the brain; in the case of stroke, only one side of the brain is affected, and movement of the forehead remains possible on the contralateral side. In facial nerve palsy, the nerve itself is affected; thus, no movement of the forehead is possible.13 Other common complaints in patients with facial nerve palsy include vertigo, hearing loss, and changes in facial sensation.
RHS was first described in 1907 as herpes zoster associated with Bell’s palsy by the neurologist J. Ramsay Hunt, for whom the condition is named.9,14 RHS is more common in men than women. It occurs most commonly in adults and is rare in children younger than 6.13,15
Diagnosis
In most cases, a diagnosis of RHS is made on a clinical basis.1 However, a polymerase chain reaction (PCR) assay can be performed on samples of tear fluid or submandibular saliva to detect the zoster virus.16,17 PCR can also be performed using exudates from the geniculate zone of the ear (a small area in the center of the auricle6,14), which is more sensitive than tears or blood.18,19 Findings from a complete blood count and the erythrocyte sedimentation rate can be used to differentiate between infectious and inflammatory causes.13
Head CT or MRI can be obtained to rule out any structural lesions. In one study, Kim et al20 examined MRI changes in patients with either Bell’s palsy or RHS. In both conditions, researchers were able to identify swelling of the labyrinthine segment of the facial nerve on temporal MRI scans.20 Although CT has not been shown to have any prognostic or diagnostic application, it can occasionally be used if decompression of the facial nerve is warranted.11
Treatment
Data used to support the use of corticosteroids for treatment of Bell’s palsy10,21,22 have been extrapolated to justify their use for treatment of RHS,23 and prednisolone is the most common choice.10 Steroids reduce the associated inflammation, resulting in decreased pain and neurologic symptoms. A daily dose for one to two weeks, followed by a slow taper, is the preferred prescribing method.10
The addition of acyclovir has been recommended to inhibit viral DNA replication9,23 (valacyclovir and famciclovir have also been mentioned12,18). If started within three days of symptom onset, acyclovir can help reduce pain and hasten resolution of symptoms.
In a large retrospective study, it was demonstrated that patients treated with prednisone at 1.0 mg/kg/d for five days, followed by a 10-day taper, combined with acyclovir, showed long-term improvement that was statistically significant.23 Complete facial recovery was reported in only 52% of patients, however. Risk factors for a poor prognosis include hypertension, diabetes mellitus, and advancing age.7
Artificial tears are also prescribed to keep the affected eye from becoming irritated and dry. The patient can be instructed to tape the eye closed at night.10
Early diagnosis and treatment (ie, within three days of symptom onset, and preferably with a combination of acyclovir and steroids23) is an important factor in a good prognosis.7,23 Because RHS-affected patients have only about a 50% chance of full recovery,23 proper follow-up care is extremely important. Follow-up visits are recommended at two weeks, six weeks, and three months.13 For optimal outcomes in patients with this neurologic diagnosis, referral to a neurologist is recommended for ongoing management. This practitioner is likely to detect subtle changes in patient presentation and can perform follow-up testing as needed.
THE CASE PATIENT
One week after the patient’s visit to the ED, he was contacted by hospital staff for a standard satisfaction and quality control survey. The patient (who had been treated with steroids and acyclovir, ibuprofen, and artificial tears) reported almost complete resolution of his pain; any mild pain, he said, was easily tolerated or could be resolved with OTC medication. He reported only minimal persistent facial weakness, stating that he was able to eat, drink, and speak normally.
The patient had not been seen by any health care provider for follow-up, but he agreed to make an appointment as soon as possible.
REFERENCES
1. Kim D, Bhimani M. Ramsay Hunt syndrome presenting as simple otitis externa. CJEM. 2008;10(3):247-250.
2. Agius AM, Pickles JM, Burch KL. A prospective study of otitis externa. Clin Otolaryngol. 1992;17(2):150-154.
3. Rosenfeld RM, Brown L, Cannon CR, et al; American Academy of Otolaryngology—Head and Neck Surgery Foundation. Clinical practice guideline: acute otitis externa. Otolaryngol Head Neck Surg. 2006;134(4 suppl):S4-S23.
4. Holland J, Bernstein J. Bell’s palsy. Clin Evid (Online). 2011 Mar 7;2011.pii:1204.
5. Jacewicz M. Bell’s palsy (2007). www.merckmanuals.com/professional/sec16/ch219/ch219i.html. Accessed May 26, 2011.
6. Harrison K. Discussion: the Ramsay Hunt Syndrome. Proc Royal Soc Med. 1953;47(371):11-24.
7. Bhupal HK. Ramsay Hunt syndrome presenting in primary care. Practitioner. 2010;254(1727):33-35.
8. Aizawa H, Ohtani F, Furuta Y, et al. Variable patterns of varicella-zoster virus reactivation in Ramsay Hunt syndrome. J Med Virol. 2004;74(2):355-360.
9. Gondivkar S, Parikh V, Parikh R. Herpes zoster oticus: a rare clinical entity. Contemp Clin Dent. 2010;1(2):127-129.
10. Sullivan FM, Swan IRC, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med. 2007;357(16):1598-1607.
11. Gilden DH. Bell’s palsy. N Engl J Med. 2004;351(13):1323-1331.
12. Diaz GA, Rakita RM, Koelle DM. A case of Ramsay Hunt–like syndrome caused by herpes simplex virus type 2. Clin Infect Dis. 2005;40(10):1545-1547.
13. Miravalle AA. Ramsay Hunt syndrome. http://emedicine.medscape.com/article/1166804-over iew. Accessed July 22, 2011.
14. Hunt JR. On herpetic inflammation of the geniculate ganglion: a new syndrome and its complications. J Nerv Ment Dis. 1907;34:73-96.
15. Sandoval CC, Núñez FA, Lizama CM, et al. Ramsay Hunt syndrome in children: four cases and review [in Spanish]. Rev Chilena Infectol. 2008; 25(6):458-464.
16. Murakami S, Nakashiro Y, Mizobuchi M, et al. Varicella-zoster virus distribution in Ramsay Hunt syndrome revealed by polymerase chain reaction. Acta Otolaryngol. 1998;118(2):145-149.
17. Hiroshige K, Ikeda M, Hondo R. Detection of varicella zoster virus DNA in tear fluid and saliva of patients with Ramsay Hunt syndrome. Otol Neurol. 2002;23(4):602-607.
18. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatr. 2001;71(2):148-154.
19. Murakami S, Honda N, Mizobuchi M, et al. Rapid diagnosis of varicella zoster virus infection in acute facial palsy. Neurology. 1998;51(4):1202-1205.
20. Kim IS, Shin SH, Kim J, et al. Correlation between MRI and operative findings in Bell’s palsy and Ramsay Hunt syndrome. Yonsei Med J. 2007;48(6):963-968.
21. Engström M, Berg T, Stjernquist-Desatnik A, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2008;7(11):993-1000.
22. Hato N, Yamada H, Kohno H, et al. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol. 2007;28(3):408-413.
23. Murakami S, Hato N, Horiuchi J, et al. Treatment of Ramsay Hunt syndrome with acyclovir-prednisone: significance of early diagnosis and treatment. Ann Neurol. 1997;41(3):353-357.
Man Presents with Altered Mental Status and Confusion
ANSWER
The chest radiograph demonstrates a fairly large right hilar mass with an associated right upper lobe infiltrate. This finding is very worrisome for a bronchogenic carcinoma.
Subsequent CT of the brain also demonstrated a right parietal mass. Thus, this lung lesion is most likely a primary neoplasm with metastatic involvement.
ANSWER
The chest radiograph demonstrates a fairly large right hilar mass with an associated right upper lobe infiltrate. This finding is very worrisome for a bronchogenic carcinoma.
Subsequent CT of the brain also demonstrated a right parietal mass. Thus, this lung lesion is most likely a primary neoplasm with metastatic involvement.
ANSWER
The chest radiograph demonstrates a fairly large right hilar mass with an associated right upper lobe infiltrate. This finding is very worrisome for a bronchogenic carcinoma.
Subsequent CT of the brain also demonstrated a right parietal mass. Thus, this lung lesion is most likely a primary neoplasm with metastatic involvement.
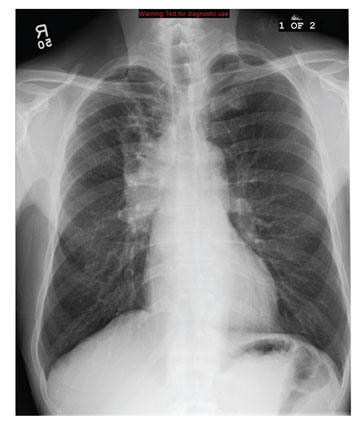
A 59-year-old man presents to your facility for evaluation of altered mental status and confusion that have progressively worsened in the past two months. He denies any injury or trauma. He states he has had associated headaches and dizziness. He denies any weight loss, shortness of breath, or malaise; however, he himself is able to notice that his mentation has “not been right.” His medical history is significant for mild hypertension and hyperlipidemia, both of which are well controlled. He discloses a 50–pack-year history of cigarette use. Physical exam reveals a male in no obvious distress but complaining of a moderate headache. His vital signs are stable. His oxygen saturation is 97% on room air. Breath sounds appear clear. The neurologic exam shows no focal deficits. You order noncontrast CT of the head, as well as a chest radiograph. The chest radiograph is shown. What is your impression?
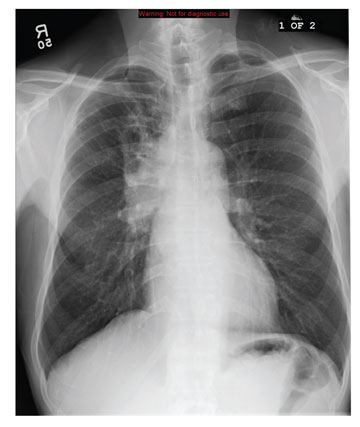
Hypothermia after cardiac arrest: Beneficial, but slow to be adopted
A 30-year-old man experienced an episode of syncope while at work. He fully recovered consciousness within 2 minutes, but the emergency services team was called. As he was being loaded into the ambulance he again lost consciousness, and he was noted to be in ventricular fibrillation. Advanced cardiac life support was immediately started and continued for 50 minutes before a hemodynamically stable spontaneous rhythm was obtained.
On arrival at the emergency department of the local hospital, he was intubated to protect his airway, as he was comatose. A 12-lead electrocardiogram showed ST-segment elevations in leads V1, V2, and V3 and a wide QRS complex with an rSR′ pattern, consistent with right bundle branch block.
Mild therapeutic hypothermia was initiated by infusing intravenous saline solution chilled to 4°C and by applying cooling blankets, and he was transferred to our hospital on an emergency basis for further management. Here, hypothermia was maintained using an intravenous cooling catheter.
HYPOTHERMIA: BENEFICIAL, BUT SLOW TO BE ADOPTED
Mild therapeutic hypothermia is a recommended therapeutic intervention for out-of-hospital cardiac arrest due to ventricular fibrillation. Nonetheless, first-responders, emergency-room staff, and intensive-care teams have been slow to adopt and integrate it into a comprehensive postresuscitation strategy. This article summarizes the evidence supporting this therapy and how it is performed.
PROPOSED MECHANISMS OF BENEFIT
Mild therapeutic hypothermia is thought to protect against anoxic brain injury in survivors of cardiac arrest via several mechanisms:
- Decreasing neuronal metabolism in the early stage of ischemic injury
- Decreasing glucose and oxygen consumption by the brain,1 which reduces supply-demand mismatch
- Decreasing the release of excitatory amino acids (eg, glutamate) that normally trigger cytotoxic cascades in the intermediate phase of injury2
- Reducing the production of harmful reactive oxygen species3
- Maintaining cellular pH4
- Reducing cell death5
- Slowing the breakdown of the blood-brain barrier that worsens cerebral edema.6
CLINICAL DATA SUPPORTING HYPOTHERMIA
There has been an interest in therapeutic hypothermia for several decades. In the 1950s, it was used in small numbers of cases in a variety of cardiac arrest situations.7,8 Interest was rekindled in the mid-1990s after a number of animal studies suggested it might be beneficial in prolonged cerebral ischemia and anoxia,9,10 and reports of case-series described its use in adults with out-of-hospital cardiac arrest.11,12
In October 2002, the International Liaison Committee on Resuscitation (ILCOR), made up of executive members of several organizations including the American Heart Association, recommended that “unconscious adult patients with spontaneous circulation after out-of-hospital cardiac arrest” should be cooled to 32°C to 34°C [89.6°F–93.2°F] for 12 to 24 hours “when the initial rhythm was ventricular fibrillation.”13
Two large randomized trials
This position statement was based largely on the results of two randomized clinical trials published simultaneously earlier in 2002.14,15 These two trials were important not only because they were the largest randomized trials of this therapy to that point, but also because they used meaningful, prospectively defined clinical end points: all-cause mortality and degree of cognitive preservation as assessed using the Glasgow-Pittsburgh Cerebral Performance Category (CPC) scale.

Bernard et al14 performed a randomized trial in four centers in Australia, assigning 77 patients either to a goal temperature of 32°C to 34°C or to normothermia for 12 hours, with all other resuscitative measures being the same in both groups. The primary outcome measured was survival to hospital discharge with sufficient neurologic function to be discharged to home or to a rehabilitation facility, ie, a CPC score of 1 or 2.
In the hypothermia group, 21 (49%) of the 43 patients survived and had an outcome that was considered “good” (ie, they were discharged home or to a rehabilitation facility), compared with 9 (26%) of the 34 patients in the normothermia group (unadjusted odds ratio 2.65, 95% confidence interval [CI] 1.02–6.88, P = .046). Proportionally fewer patients in the hypothermia group died—22 (51%) of 43 vs 23 (68%) of 34; however, the difference was not statistically significant (P = .145).
The Hypothermia After Cardiac Arrest Study Group15 screened 3,551 European patients who suffered out-of-hospital cardiac arrest15; 275 patients were randomized to mild therapeutic hypothermia or normothermia for 24 hours. The primary outcome was the percentage of patients who had a CPC score of 1 or 2 (vs 3 to 5) at 6 months, and the secondary outcome was the rate of death at 6 months.
At 6 months, 75 (55%) of the 136 patients in the hypothermia group had a CPC score of 1 or 2, compared with 54 (39%) of the 137 patients in the normothermia group (P = .009). The rate of death was also lower with hypothermia: 55% vs 41% (P = .02).
In both trials, patients were included only if their cardiac arrest was witnessed, if their initial cardiac rhythm was ventricular fibrillation or pulseless ventricular tachycardia, if circulation spontaneously returned within 60 minutes with standard basic and advanced cardiac life support protocols, and if they were still comatose on arrival at the hospital. They were excluded if they were over age 75, if they had suffered a cerebrovascular accident at the time of cardiac arrest, or if the arrest was caused by trauma or drug overdose. In addition, the European trial excluded patients who suffered another cardiac arrest after the initial return of spontaneous circulation but before cooling was started.
The standard of care
In view of the available clinical data, the 2002 ILCOR guidelines and a 2005 statement from the American Heart Association advocated mild therapeutic hypothermia for survivors of out-of-hospital ventricular tachycardia or fibrillation.16 Subsequently, this therapy has become more widely practiced and accepted as the standard of care among critical-care providers.
Of note, some public health officials and local governments are strongly promoting this treatment for survivors of cardiac arrest in the community.17 More and more of these groups are mandating that these patients be transported only to hospitals that have therapeutic hypothermia protocols in place, bypassing those not equipped to provide this treatment.18
INDICATIONS, CONTRAINDICATIONS, AND GRAY AREAS
What are the indications and contraindications to the use of hypothermia after out-of-hospital cardiac arrest? What are some of the “gray areas”?
Indications. This treatment is indicated for comatose adults who have had a witnessed cardiac arrest, whose initial cardiac rhythm was ventricular fibrillation or pulseless ventricular tachycardia, and whose circulation spontaneously returned in less than 60 minutes with basic and advanced cardiac life support. This carries a class I recommendation, level of evidence B, and was recently reinforced in the 2010 update to the American Heart Association guidelines for cardiopulmonary resuscitation.19

Absolute contraindications include hemorrhagic stroke (which must be proved by computed tomography) and cardiac arrest due to trauma (Table 2). Other major contraindications are a Glasgow Coma Scale score of 8 or higher before the initiation of mild therapeutic hypothermia, cardiac arrest due to drug overdose, and preexisting hypothermia (< 34°C) when first-responders arrive.
Relative contraindications include baseline coagulopathy and severe hypotension (mean atrial pressure < 60 mm Hg) that is not correctable by fluid infusion, vasopressors, or invasive hemodynamic support.
Gray areas. There are not enough data to make a firm recommendation about whether to apply mild therapeutic hypothermia if a witnessed cardiac arrest with ventricular fibrillation or ventricular tachycardia occurs in the hospital, but data from out-of-hospital cardiac arrest patients appear applicable for hospitalized patients.
The data are also quite limited and equivocal on its use for out-of-hospital cardiac arrest in patients whose initial cardiac rhythm is pulseless electrical activity or asystole,20,21 likely because of the competing risk of comorbidities and the resultant lower baseline survival rate in these patients.
Consequently, for in-hospital postarrest patients with any initial rhythm and for out-of-hospital cardiac arrest patients with rhythms other than ventricular tachycardia or ventricular fibrillation, the 2010 guideline recommendation on the use of mild therapeutic hypothermia is less enthusiastic (class IIb, level of evidence B).19
There are also few data on the use of mild therapeutic hypothermia in post-arrest patients in circulatory shock requiring vasopressors or intra-aortic balloon counterpulsation, largely limited to case series and comparisons with historical controls.22,23 Further investigation is clearly needed in these areas. Until then, it should be considered at the physician’s and the team’s discretion, on a case-by-case basis.
HYPOTHERMIA IN CASES OF VENTRICULAR FIBRILLATION AND ACUTE CORONARY SYNDROME
The value of coronary angiography after out-of-hospital cardiac arrest was first highlighted by Spaulding et al,24 who performed it urgently in 84 consecutive survivors of out-of-hospital cardiac arrest, 36 of whom had ST-segment elevation myocardial infarction (STEMI). Angiography uncovered an acute coronary occlusion in 40 (48%) of the 84 patients.
In this series, ST-segment elevation was a strong predictor of acute coronary occlusion (odds ratio 4.3; 95% CI 1.6–2.0; P = .004). However, 9 patients without chest pain or ST elevation were also found to have an occluded infarct-related artery. Successful angioplasty was an independent predictor of survival, highlighting the importance of an angiographic definition in this population.
These findings were recently confirmed in the larger Parisian Region Out of Hospital Cardiac Arrest (PROCAT) registry in 435 patients who had no obvious extracardiac cause of arrest, for whom successful culprit coronary angioplasty was associated with survival.25
Angioplasty comes first, but neither treatment need be delayed
Efforts to induce hypothermia must not be allowed to delay the door-to-balloon time of post-arrest patients in the setting of STEMI. The top priority is establishing patency of the infarct-related artery with a goal of salvaging ischemic myocardium and obtaining mechanical and electrical stabilization.
Fortunately, mild therapeutic hypothermia does not necessarily delay emergency revascularization if hypothermia protocols are well established. In fact, induction of mild therapeutic hypothermia prior to or on arrival at the catheterization laboratory has been shown to be feasible and safe.26,27
We believe that all centers performing primary percutaneous coronary intervention for STEMI should have immediate access to and expertise in mild therapeutic hypothermia. Regional planning and integration of STEMI and out-of-hospital cardiac arrest networks will ensure that most patients with STEMI have access to this treatment when it is indicated.
Does hypothermia help the heart? Does it increase bleeding?
Researchers have been interested in therapeutic hypothermia as a means of reducing myocardial infarct size,28,29 but clinical trials have not shown a clear-cut benefit in this regard. However, these investigations have also added to the evidence that antiplatelet and anticoagulation therapy in patients undergoing mild therapeutic hypothermia does not result in a statistically significant excess of major bleeding events, which is a potential concern.
Of note, these studies were neither powered nor specifically designed to evaluate for major bleeding as an end point. Therefore, these complications should still be carefully monitored for.
IS THERE AN OPTIMAL TIME TO BEGIN MILD THERAPEUTIC HYPOTHERMIA?
Experimental data suggest that mild therapeutic hypothermia should be started as soon as possible after a comprehensive clinical evaluation indicates the patient is eligible.30–33 However, clinical data are not robustly in favor of starting it before the patient reaches the hospital rather than on hospital arrival.
In a recent randomized trial in 2,334 survivors of out-of-hospital cardiac arrest, outcomes were no better if hypothermia was started by paramedics than if it was started on arrival at the hospital (47.5% vs 52.6% discharged to home or rehabilitation; 95% CI 0.70–1.17; P = .43).34
Earlier data from smaller studies had suggested that prehospital initiation of hypothermia (for example, using chilled intravenous saline infusions) in carefully selected patients with out-of-hospital cardiac arrest was safe and showed a nonsignificant trend toward better outcomes.20,35
The randomized controlled trials that showed hypothermia to be beneficial used very slow cooling methods; consequently, it is reasonable to allow up to 6 hours from initial presentation to first-responders to start it. There are, however, no conclusive data in humans for or against starting it later than 6 hours after presentation. Most experts believe that its potential neurologic and mortality benefits are largely lost if it is delayed more than 6 hours.
The overall message from these data seems to be that, in patients who survive cardiac arrest outside the hospital with ventricular tachycardia or fibrillation, mild therapeutic hypothermia is effective and safe and should be started as soon as possible after arrival at the hospital.
METHODS FOR INDUCING AND MAINTAINING HYPOTHERMIA
Cooling the patient
To cool the patient and keep him or her cold, caregivers have used ice packs placed around the head, groin, and axillae; intravenous infusion of saline maintained at 4°C (39°F); and cooling-air blankets. More recently, thermal wraps and intravascular cooling catheters have been used.36–38 The newer methods are more effective in rapidly bringing patients to the target temperature of 32 to 34°C (usually within 3 or 4 hours) and keeping them within this range, and they auto-adjust their output on the basis of measured core temperature.
The Pre ROSC Intranasal Cooling Effectiveness (PRINCE) trial demonstrated the safety and efficacy of nasopharyngeal cooling using a perfluorocarbon aerosol given via a nasopharyngeal cannula in patients with out-of-hospital cardiac arrest.39
Monitoring the core temperature
The patient’s core temperature is most commonly monitored with a probe in the esophagus, bladder, rectum, or pulmonary artery.40
Of these, the bladder and rectum are considered “intermediate” monitoring sites, as their temperatures tend to lag behind the core temperature. Furthermore, the bladder temperature can be significantly altered by the flow of urine, which can vary considerably during the cooling and rewarming process.
Esophageal temperature monitoring is relatively noninvasive and tends to reliably and accurately reflect core temperature as long as the probe is placed far enough down (about 45 cm from the nose in an average adult) that it is not affected by proximity to the trachea.
Pulmonary artery catheters are considered the gold standard for core temperature monitoring, but they pose risks such as bloodstream infection and large-vessel damage. In practice, many patients admitted to the coronary intensive care unit after out-of-hospital cardiac arrest require pulmonary artery catheterization anyway for other indications, and in these situations it is the preferred method of monitoring the core temperature.
However, no approach is ideal in terms of measuring the temperature in the critical end organs. Rather, core temperature monitoring serves as a guide to help ensure consistent clinical practice in attaining and maintaining mild therapeutic hypothermia.
Preventing shivering
To achieve and maintain the goal temperature, the body’s natural response to a decrease in core temperature—shivering—must be watched for and eliminated. A number of drugs may be used for this purpose.41
Paralytic drugs are used to reduce shivering; nursing staff must be trained to monitor for signs of occult shivering (eg, jaw vibration) and adjust the dose of paralytic drug accordingly. Since the patients are paralyzed, they must also receive continuous intravenous sedation.
Other commonly used drugs that decrease the hypothalamic drive to shiver include buspirone (BuSpar), a serotonin 5HT-1A partial agonist, and meperidine (Demerol), an opiate agonist of kappa and mu receptors.
Rewarming after 24 hours
Rewarming is conventionally started after 24 hours of mild therapeutic hypothermia, at a rate no greater than 0.5°C (1°F) per hour.
Because sedation is used during the hypothermia period of 24 hours, a washout period for these medications is necessary, and the neurologic prognosis of cardiac arrest patients who undergo mild therapeutic hypothermia cannot be adequately assessed until 72 hours after rewarming.
ADVERSE EFFECTS OF MILD THERAPEUTIC HYPOTHERMIA
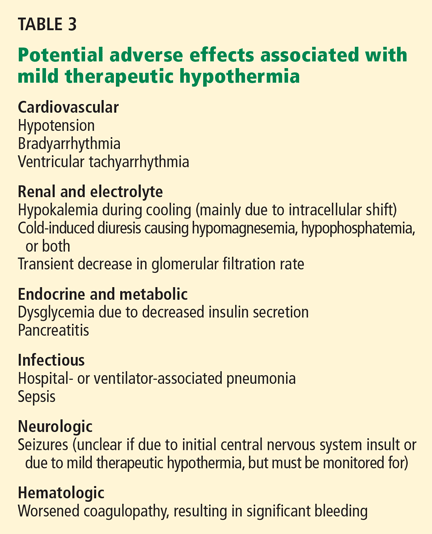
Some of these effects are predictable. Decreasing the body temperature causes potassium to shift into the cells, and this same potassium will leave the intracellular space during the rewarming phase. For this reason, aggressive potassium repletion for mild hypokalemia (potassium levels of 3.0–3.5 mmol/L) during mild therapeutic hypothermia can result in dangerous hyperkalemia during rewarming and should generally be avoided.
As another example, the enzymes involved in coagulation are less effective at lower temperatures. Thus, if it occurs, active bleeding requiring transfusion warrants consideration of stopping the hypothermia.
Adverse effects should be watched for (eg, by checking electrolyte levels frequently, monitoring blood glucose, continuous electroencephalographic monitoring during the cooling phase, and avoiding placement of intracardiac catheters once the goal temperature is reached) and addressed as they happen. However, in a recent review of this subject42 the balance of evidence continued to indicate that the benefit of this treatment exceeds its risks.
OUR PATIENT RECOVERS
After 24 hours of therapeutic hypothermia, our patient was gradually rewarmed to a normal temperature, and sedation and paralysis were discontinued.
Analysis of his prearrest and postarrest 12-lead electrocardiograms revealed a type I Brugada pattern (coved ST elevation and negative T waves in V1, V2, and V3, caused by abnormal repolarization due to inherited mutations in SCN5A). Cardiac catheterization revealed normal coronary arteries, and MRI revealed no evidence of arrhythmogenic right ventricular cardiomyopathy or other structural abnormalities.
In the next 72 hours the patient was successfully extubated, and he gradually returned to full neurologic function. Before he went home a few days later, a single-lead cardioverter-defibrillator was implanted to prevent sudden cardiac death. All of his first-degree relatives were encouraged to undergo genetic screening for SCN5A mutations. The patient is currently back to his previous high level of functioning as a marketing manager, husband, and father of two young children.
- Erecinska M, Thoresen M, Silver IA. Effects of hypothermia on energy metabolism in mammalian central nervous system. J Cereb Blood Flow Metab 2003; 23:513–530.
- Nakashima K, Todd MM. Effects of hypothermia on the rate of excitatory amino acid release after ischemic depolarization. Stroke 1996; 27:913–918.
- Thoresen M, Satas S, Puka-Sundvall M, et al. Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport 1997; 8:3359–3362.
- Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med 2009; 37(suppl 7):S186–S202.
- Yang D, Guo S, Zhang T, Li H. Hypothermia attenuates ischemia/reperfusion-induced endothelial cell apoptosis via alterations in apoptotic pathways and JNK signaling. FEBS Lett 2009; 583:2500–2506.
- Karibe H, Zarow GJ, Graham SH, Weinstein PR. Mild intraischemic hypothermia reduces postischemic hyperperfusion, delayed postischemic hypoperfusion, blood-brain barrier disruption, brain edema, and neuronal damage volume after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab 1994; 14:620–627.
- Benson DW, Williams GR, Spencer FC, Yates AJ. The use of hypothermia after cardiac arrest. Anesth Analg 1959; 38:423–428.
- Williams GR, Spencer FC. The clinical use of hypothermia following cardiac arrest. Ann Surg 1958; 148:462–468.
- Baker CJ, Onesti ST, Barth KN, Prestigiacomo CJ, Solomon RA. Hypothermic protection following middle cerebral artery occlusion in the rat. Surg Neurol 1991; 36:175–180.
- Ridenour TR, Warner DS, Todd MM, McAllister AC. Mild hypothermia reduces infarct size resulting from temporary but not permanent focal ischemia in rats. Stroke 1992; 23:733–738.
- Bernard SA, Jones BM, Horne MK. Clinical trial of induced hypothermia in comatose survivors of out-of-hospital cardiac arrest. Ann Emerg Med 1997; 30:146–153.
- Yanagawa Y, Ishihara S, Norio H, et al. Preliminary clinical outcome study of mild resuscitative hypothermia after out-of-hospital cardiopulmonary arrest. Resuscitation 1998; 39:61–66.
- Nolan JP, Morley PT, Vanden Hoek TL, et al; International Liaison Committee on Resuscitation. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation 2003; 108:118–121.
- Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 2002; 346:557–563.
- Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 2002; 346:549–556.
- ECC Committee, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2005; 112(suppl 24):IV1–IV203.
- Hartocollis A. “City Pushes Cooling Therapy for Cardiac Arrest”. New York Times, December4th, 2008,A1. http://www.nytimes.com/2008/12/04/nyregion/04cool.html. Accessed May 31, , 2011.
- Nichol G, Aufderheide TP, Eigel B, et al; American Heart Association Emergency Cardiovascular Care Committee. Regional systems of care for out-of-hospital cardiac arrest: A policy statement from the American Heart Association. Circulation 2010; 121:709–729.
- Field JM, Hazinski MF, Sayre MR, et al. Part 1: executive summary: 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation 2010; 122(suppl 3):S640–S56.
- Hachimi-Idrissi S, Corne L, Ebinger G, Michotte Y, Huyghens L. Mild hypothermia induced by a helmet device: a clinical feasibility study. Resuscitation 2001; 51:275–281.
- Kim F, Olsufka M, Longstreth WT, et al. Pilot randomized clinical trial of prehospital induction of mild hypothermia in out-of-hospital cardiac arrest patients with a rapid infusion of 4 degrees C normal saline. Circulation 2007; 115:3064–3070.
- Hovdenes J, Laake JH, Aaberge L, Haugaa H, Bugge JF. Therapeutic hypothermia after out-of-hospital cardiac arrest: experiences with patients treated with percutaneous coronary intervention and cardiogenic shock. Acta Anaesthesiol Scand 2007; 51:137–142.
- Skulec R, Kovarnik T, Dostalova G, Kolar J, Linhart A. Induction of mild hypothermia in cardiac arrest survivors presenting with cardiogenic shock syndrome. Acta Anaesthesiol Scand 2008; 52:188–194.
- Spaulding CM, Joly LM, Rosenberg A, et al. Immediate coronary angiography in survivors of out-of-hospital cardiac arrest. N Engl J Med 1997; 336:1629–1633.
- Dumas F, Cariou A, Manzo-Silberman S, et al. Immediate percutaneous coronary intervention is associated with better survival after out-of-hospital cardiac arrest: insights from the PROCAT (Parisian Region Out of hospital Cardiac ArresT) registry. Circ Cardiovasc Interv 2010; 3:200–207.
- Knafelj R, Radsel P, Ploj T, Noc M. Primary percutaneous coronary intervention and mild induced hypothermia in comatose survivors of ventricular fibrillation with ST-elevation acute myocardial infarction. Resuscitation 2007; 74:227–234.
- Wolfrum S, Pierau C, Radke PW, Schunkert H, Kurowski V. Mild therapeutic hypothermia in patients after out-of-hospital cardiac arrest due to acute ST-segment elevation myocardial infarction undergoing immediate percutaneous coronary intervention. Crit Care Med 2008; 36:1780–1786.
- O’Neill WW, on behalf of the COOL-MI Investigators. Cooling as an adjunct to primary PCI for myocardial infarction. Presented at Transcatheter Cardiovascular Therapeutics. Washington, DC; 2004.
- Grines CL, on behalf of the ICE-IT Investigators. Intravascular cooling adjunctive to percutaneous coronary intervention for acute myocardial infarction. Presented at Transcatheter Cardiovascular Therapeutics. Washington, DC; 2004.
- Weil MH, Gazmuri RJ. Hypothermia after cardiac arrest. Crit Care Med 1991; 19:315.
- Abella BS, Zhao D, Alvarado J, Hamann K, Vanden Hoek TL, Becker LB. Intra-arrest cooling improves outcomes in a murine cardiac arrest model. Circulation 2004; 109:2786–2791.
- Zhao D, Abella BS, Beiser DG, et al. Intra-arrest cooling with delayed reperfusion yields higher survival than earlier normothermic resuscitation in a mouse model of cardiac arrest. Resuscitation 2008; 77:242–249.
- Jia X, Koenig MA, Shin HC, et al. Improving neurological outcomes post-cardiac arrest in a rat model: immediate hypothermia and quantitative EEG monitoring. Resuscitation 2008; 76:431–442.
- Bernard SA, Smith K, Cameron P, et al; Rapid Infusion of Cold Hartmanns (RICH) Investigators. Induction of therapeutic hypothermia by paramedics after resuscitation from out-of-hospital ventricular fibrillation cardiac arrest: a randomized controlled trial. Circulation 2010; 122:737–742.
- Bruel C, Parienti JJ, Marie W, et al. Mild hypothermia during advanced life support: a preliminary study in out-of-hospital cardiac arrest. Crit Care 2008; 12:R31.
- Pichon N, Amiel JB, François B, Dugard A, Etchecopar C, Vignon P. Efficacy of and tolerance to mild induced hypothermia after out-of-hospital cardiac arrest using an endovascular cooling system. Crit Care 2007; 11:R71.
- Wolff B, Machill K, Schumacher D, Schulzki I, Werner D. Early achievement of mild therapeutic hypothermia and the neurologic outcome after cardiac arrest. Int J Cardiol 2009; 133:223–228.
- Heard KJ, Peberdy MA, Sayre MR, et al. A randomized controlled trial comparing the Arctic Sun to standard cooling for induction of hypothermia after cardiac arrest. Resuscitation 2010; 81:9–14.
- Castrén M, Nordberg P, Svensson L, et al. Intra-arrest transnasal evaporative cooling: a randomized, prehospital, multicenter study (PRINCE: Pre-ROSC IntraNasal Cooling Effectiveness). Circulation 2010; 122:729–736.
- Insler SR, Sessler DI. Perioperative thermoregulation and temperature monitoring. Anesthesiol Clin 2006; 24:823–837.
- Weant KA, Martin JE, Humphries RL, Cook AM. Pharmacologic options for reducing the shivering response to therapeutic hypothermia. Pharmacotherapy 2010; 30:830–841.
- Holzer M. Targeted temperature management for comatose survivors of cardiac arrest. N Engl J Med 2010; 363:1256–1264.
A 30-year-old man experienced an episode of syncope while at work. He fully recovered consciousness within 2 minutes, but the emergency services team was called. As he was being loaded into the ambulance he again lost consciousness, and he was noted to be in ventricular fibrillation. Advanced cardiac life support was immediately started and continued for 50 minutes before a hemodynamically stable spontaneous rhythm was obtained.
On arrival at the emergency department of the local hospital, he was intubated to protect his airway, as he was comatose. A 12-lead electrocardiogram showed ST-segment elevations in leads V1, V2, and V3 and a wide QRS complex with an rSR′ pattern, consistent with right bundle branch block.
Mild therapeutic hypothermia was initiated by infusing intravenous saline solution chilled to 4°C and by applying cooling blankets, and he was transferred to our hospital on an emergency basis for further management. Here, hypothermia was maintained using an intravenous cooling catheter.
HYPOTHERMIA: BENEFICIAL, BUT SLOW TO BE ADOPTED
Mild therapeutic hypothermia is a recommended therapeutic intervention for out-of-hospital cardiac arrest due to ventricular fibrillation. Nonetheless, first-responders, emergency-room staff, and intensive-care teams have been slow to adopt and integrate it into a comprehensive postresuscitation strategy. This article summarizes the evidence supporting this therapy and how it is performed.
PROPOSED MECHANISMS OF BENEFIT
Mild therapeutic hypothermia is thought to protect against anoxic brain injury in survivors of cardiac arrest via several mechanisms:
- Decreasing neuronal metabolism in the early stage of ischemic injury
- Decreasing glucose and oxygen consumption by the brain,1 which reduces supply-demand mismatch
- Decreasing the release of excitatory amino acids (eg, glutamate) that normally trigger cytotoxic cascades in the intermediate phase of injury2
- Reducing the production of harmful reactive oxygen species3
- Maintaining cellular pH4
- Reducing cell death5
- Slowing the breakdown of the blood-brain barrier that worsens cerebral edema.6
CLINICAL DATA SUPPORTING HYPOTHERMIA
There has been an interest in therapeutic hypothermia for several decades. In the 1950s, it was used in small numbers of cases in a variety of cardiac arrest situations.7,8 Interest was rekindled in the mid-1990s after a number of animal studies suggested it might be beneficial in prolonged cerebral ischemia and anoxia,9,10 and reports of case-series described its use in adults with out-of-hospital cardiac arrest.11,12
In October 2002, the International Liaison Committee on Resuscitation (ILCOR), made up of executive members of several organizations including the American Heart Association, recommended that “unconscious adult patients with spontaneous circulation after out-of-hospital cardiac arrest” should be cooled to 32°C to 34°C [89.6°F–93.2°F] for 12 to 24 hours “when the initial rhythm was ventricular fibrillation.”13
Two large randomized trials
This position statement was based largely on the results of two randomized clinical trials published simultaneously earlier in 2002.14,15 These two trials were important not only because they were the largest randomized trials of this therapy to that point, but also because they used meaningful, prospectively defined clinical end points: all-cause mortality and degree of cognitive preservation as assessed using the Glasgow-Pittsburgh Cerebral Performance Category (CPC) scale.
Bernard et al14 performed a randomized trial in four centers in Australia, assigning 77 patients either to a goal temperature of 32°C to 34°C or to normothermia for 12 hours, with all other resuscitative measures being the same in both groups. The primary outcome measured was survival to hospital discharge with sufficient neurologic function to be discharged to home or to a rehabilitation facility, ie, a CPC score of 1 or 2.
In the hypothermia group, 21 (49%) of the 43 patients survived and had an outcome that was considered “good” (ie, they were discharged home or to a rehabilitation facility), compared with 9 (26%) of the 34 patients in the normothermia group (unadjusted odds ratio 2.65, 95% confidence interval [CI] 1.02–6.88, P = .046). Proportionally fewer patients in the hypothermia group died—22 (51%) of 43 vs 23 (68%) of 34; however, the difference was not statistically significant (P = .145).
The Hypothermia After Cardiac Arrest Study Group15 screened 3,551 European patients who suffered out-of-hospital cardiac arrest15; 275 patients were randomized to mild therapeutic hypothermia or normothermia for 24 hours. The primary outcome was the percentage of patients who had a CPC score of 1 or 2 (vs 3 to 5) at 6 months, and the secondary outcome was the rate of death at 6 months.
At 6 months, 75 (55%) of the 136 patients in the hypothermia group had a CPC score of 1 or 2, compared with 54 (39%) of the 137 patients in the normothermia group (P = .009). The rate of death was also lower with hypothermia: 55% vs 41% (P = .02).
In both trials, patients were included only if their cardiac arrest was witnessed, if their initial cardiac rhythm was ventricular fibrillation or pulseless ventricular tachycardia, if circulation spontaneously returned within 60 minutes with standard basic and advanced cardiac life support protocols, and if they were still comatose on arrival at the hospital. They were excluded if they were over age 75, if they had suffered a cerebrovascular accident at the time of cardiac arrest, or if the arrest was caused by trauma or drug overdose. In addition, the European trial excluded patients who suffered another cardiac arrest after the initial return of spontaneous circulation but before cooling was started.
The standard of care
In view of the available clinical data, the 2002 ILCOR guidelines and a 2005 statement from the American Heart Association advocated mild therapeutic hypothermia for survivors of out-of-hospital ventricular tachycardia or fibrillation.16 Subsequently, this therapy has become more widely practiced and accepted as the standard of care among critical-care providers.
Of note, some public health officials and local governments are strongly promoting this treatment for survivors of cardiac arrest in the community.17 More and more of these groups are mandating that these patients be transported only to hospitals that have therapeutic hypothermia protocols in place, bypassing those not equipped to provide this treatment.18
INDICATIONS, CONTRAINDICATIONS, AND GRAY AREAS
What are the indications and contraindications to the use of hypothermia after out-of-hospital cardiac arrest? What are some of the “gray areas”?
Indications. This treatment is indicated for comatose adults who have had a witnessed cardiac arrest, whose initial cardiac rhythm was ventricular fibrillation or pulseless ventricular tachycardia, and whose circulation spontaneously returned in less than 60 minutes with basic and advanced cardiac life support. This carries a class I recommendation, level of evidence B, and was recently reinforced in the 2010 update to the American Heart Association guidelines for cardiopulmonary resuscitation.19
Absolute contraindications include hemorrhagic stroke (which must be proved by computed tomography) and cardiac arrest due to trauma (Table 2). Other major contraindications are a Glasgow Coma Scale score of 8 or higher before the initiation of mild therapeutic hypothermia, cardiac arrest due to drug overdose, and preexisting hypothermia (< 34°C) when first-responders arrive.
Relative contraindications include baseline coagulopathy and severe hypotension (mean atrial pressure < 60 mm Hg) that is not correctable by fluid infusion, vasopressors, or invasive hemodynamic support.
Gray areas. There are not enough data to make a firm recommendation about whether to apply mild therapeutic hypothermia if a witnessed cardiac arrest with ventricular fibrillation or ventricular tachycardia occurs in the hospital, but data from out-of-hospital cardiac arrest patients appear applicable for hospitalized patients.
The data are also quite limited and equivocal on its use for out-of-hospital cardiac arrest in patients whose initial cardiac rhythm is pulseless electrical activity or asystole,20,21 likely because of the competing risk of comorbidities and the resultant lower baseline survival rate in these patients.
Consequently, for in-hospital postarrest patients with any initial rhythm and for out-of-hospital cardiac arrest patients with rhythms other than ventricular tachycardia or ventricular fibrillation, the 2010 guideline recommendation on the use of mild therapeutic hypothermia is less enthusiastic (class IIb, level of evidence B).19
There are also few data on the use of mild therapeutic hypothermia in post-arrest patients in circulatory shock requiring vasopressors or intra-aortic balloon counterpulsation, largely limited to case series and comparisons with historical controls.22,23 Further investigation is clearly needed in these areas. Until then, it should be considered at the physician’s and the team’s discretion, on a case-by-case basis.
HYPOTHERMIA IN CASES OF VENTRICULAR FIBRILLATION AND ACUTE CORONARY SYNDROME
The value of coronary angiography after out-of-hospital cardiac arrest was first highlighted by Spaulding et al,24 who performed it urgently in 84 consecutive survivors of out-of-hospital cardiac arrest, 36 of whom had ST-segment elevation myocardial infarction (STEMI). Angiography uncovered an acute coronary occlusion in 40 (48%) of the 84 patients.
In this series, ST-segment elevation was a strong predictor of acute coronary occlusion (odds ratio 4.3; 95% CI 1.6–2.0; P = .004). However, 9 patients without chest pain or ST elevation were also found to have an occluded infarct-related artery. Successful angioplasty was an independent predictor of survival, highlighting the importance of an angiographic definition in this population.
These findings were recently confirmed in the larger Parisian Region Out of Hospital Cardiac Arrest (PROCAT) registry in 435 patients who had no obvious extracardiac cause of arrest, for whom successful culprit coronary angioplasty was associated with survival.25
Angioplasty comes first, but neither treatment need be delayed
Efforts to induce hypothermia must not be allowed to delay the door-to-balloon time of post-arrest patients in the setting of STEMI. The top priority is establishing patency of the infarct-related artery with a goal of salvaging ischemic myocardium and obtaining mechanical and electrical stabilization.
Fortunately, mild therapeutic hypothermia does not necessarily delay emergency revascularization if hypothermia protocols are well established. In fact, induction of mild therapeutic hypothermia prior to or on arrival at the catheterization laboratory has been shown to be feasible and safe.26,27
We believe that all centers performing primary percutaneous coronary intervention for STEMI should have immediate access to and expertise in mild therapeutic hypothermia. Regional planning and integration of STEMI and out-of-hospital cardiac arrest networks will ensure that most patients with STEMI have access to this treatment when it is indicated.
Does hypothermia help the heart? Does it increase bleeding?
Researchers have been interested in therapeutic hypothermia as a means of reducing myocardial infarct size,28,29 but clinical trials have not shown a clear-cut benefit in this regard. However, these investigations have also added to the evidence that antiplatelet and anticoagulation therapy in patients undergoing mild therapeutic hypothermia does not result in a statistically significant excess of major bleeding events, which is a potential concern.
Of note, these studies were neither powered nor specifically designed to evaluate for major bleeding as an end point. Therefore, these complications should still be carefully monitored for.
IS THERE AN OPTIMAL TIME TO BEGIN MILD THERAPEUTIC HYPOTHERMIA?
Experimental data suggest that mild therapeutic hypothermia should be started as soon as possible after a comprehensive clinical evaluation indicates the patient is eligible.30–33 However, clinical data are not robustly in favor of starting it before the patient reaches the hospital rather than on hospital arrival.
In a recent randomized trial in 2,334 survivors of out-of-hospital cardiac arrest, outcomes were no better if hypothermia was started by paramedics than if it was started on arrival at the hospital (47.5% vs 52.6% discharged to home or rehabilitation; 95% CI 0.70–1.17; P = .43).34
Earlier data from smaller studies had suggested that prehospital initiation of hypothermia (for example, using chilled intravenous saline infusions) in carefully selected patients with out-of-hospital cardiac arrest was safe and showed a nonsignificant trend toward better outcomes.20,35
The randomized controlled trials that showed hypothermia to be beneficial used very slow cooling methods; consequently, it is reasonable to allow up to 6 hours from initial presentation to first-responders to start it. There are, however, no conclusive data in humans for or against starting it later than 6 hours after presentation. Most experts believe that its potential neurologic and mortality benefits are largely lost if it is delayed more than 6 hours.
The overall message from these data seems to be that, in patients who survive cardiac arrest outside the hospital with ventricular tachycardia or fibrillation, mild therapeutic hypothermia is effective and safe and should be started as soon as possible after arrival at the hospital.
METHODS FOR INDUCING AND MAINTAINING HYPOTHERMIA
Cooling the patient
To cool the patient and keep him or her cold, caregivers have used ice packs placed around the head, groin, and axillae; intravenous infusion of saline maintained at 4°C (39°F); and cooling-air blankets. More recently, thermal wraps and intravascular cooling catheters have been used.36–38 The newer methods are more effective in rapidly bringing patients to the target temperature of 32 to 34°C (usually within 3 or 4 hours) and keeping them within this range, and they auto-adjust their output on the basis of measured core temperature.
The Pre ROSC Intranasal Cooling Effectiveness (PRINCE) trial demonstrated the safety and efficacy of nasopharyngeal cooling using a perfluorocarbon aerosol given via a nasopharyngeal cannula in patients with out-of-hospital cardiac arrest.39
Monitoring the core temperature
The patient’s core temperature is most commonly monitored with a probe in the esophagus, bladder, rectum, or pulmonary artery.40
Of these, the bladder and rectum are considered “intermediate” monitoring sites, as their temperatures tend to lag behind the core temperature. Furthermore, the bladder temperature can be significantly altered by the flow of urine, which can vary considerably during the cooling and rewarming process.
Esophageal temperature monitoring is relatively noninvasive and tends to reliably and accurately reflect core temperature as long as the probe is placed far enough down (about 45 cm from the nose in an average adult) that it is not affected by proximity to the trachea.
Pulmonary artery catheters are considered the gold standard for core temperature monitoring, but they pose risks such as bloodstream infection and large-vessel damage. In practice, many patients admitted to the coronary intensive care unit after out-of-hospital cardiac arrest require pulmonary artery catheterization anyway for other indications, and in these situations it is the preferred method of monitoring the core temperature.
However, no approach is ideal in terms of measuring the temperature in the critical end organs. Rather, core temperature monitoring serves as a guide to help ensure consistent clinical practice in attaining and maintaining mild therapeutic hypothermia.
Preventing shivering
To achieve and maintain the goal temperature, the body’s natural response to a decrease in core temperature—shivering—must be watched for and eliminated. A number of drugs may be used for this purpose.41
Paralytic drugs are used to reduce shivering; nursing staff must be trained to monitor for signs of occult shivering (eg, jaw vibration) and adjust the dose of paralytic drug accordingly. Since the patients are paralyzed, they must also receive continuous intravenous sedation.
Other commonly used drugs that decrease the hypothalamic drive to shiver include buspirone (BuSpar), a serotonin 5HT-1A partial agonist, and meperidine (Demerol), an opiate agonist of kappa and mu receptors.
Rewarming after 24 hours
Rewarming is conventionally started after 24 hours of mild therapeutic hypothermia, at a rate no greater than 0.5°C (1°F) per hour.
Because sedation is used during the hypothermia period of 24 hours, a washout period for these medications is necessary, and the neurologic prognosis of cardiac arrest patients who undergo mild therapeutic hypothermia cannot be adequately assessed until 72 hours after rewarming.
ADVERSE EFFECTS OF MILD THERAPEUTIC HYPOTHERMIA
Some of these effects are predictable. Decreasing the body temperature causes potassium to shift into the cells, and this same potassium will leave the intracellular space during the rewarming phase. For this reason, aggressive potassium repletion for mild hypokalemia (potassium levels of 3.0–3.5 mmol/L) during mild therapeutic hypothermia can result in dangerous hyperkalemia during rewarming and should generally be avoided.
As another example, the enzymes involved in coagulation are less effective at lower temperatures. Thus, if it occurs, active bleeding requiring transfusion warrants consideration of stopping the hypothermia.
Adverse effects should be watched for (eg, by checking electrolyte levels frequently, monitoring blood glucose, continuous electroencephalographic monitoring during the cooling phase, and avoiding placement of intracardiac catheters once the goal temperature is reached) and addressed as they happen. However, in a recent review of this subject42 the balance of evidence continued to indicate that the benefit of this treatment exceeds its risks.
OUR PATIENT RECOVERS
After 24 hours of therapeutic hypothermia, our patient was gradually rewarmed to a normal temperature, and sedation and paralysis were discontinued.
Analysis of his prearrest and postarrest 12-lead electrocardiograms revealed a type I Brugada pattern (coved ST elevation and negative T waves in V1, V2, and V3, caused by abnormal repolarization due to inherited mutations in SCN5A). Cardiac catheterization revealed normal coronary arteries, and MRI revealed no evidence of arrhythmogenic right ventricular cardiomyopathy or other structural abnormalities.
In the next 72 hours the patient was successfully extubated, and he gradually returned to full neurologic function. Before he went home a few days later, a single-lead cardioverter-defibrillator was implanted to prevent sudden cardiac death. All of his first-degree relatives were encouraged to undergo genetic screening for SCN5A mutations. The patient is currently back to his previous high level of functioning as a marketing manager, husband, and father of two young children.
A 30-year-old man experienced an episode of syncope while at work. He fully recovered consciousness within 2 minutes, but the emergency services team was called. As he was being loaded into the ambulance he again lost consciousness, and he was noted to be in ventricular fibrillation. Advanced cardiac life support was immediately started and continued for 50 minutes before a hemodynamically stable spontaneous rhythm was obtained.
On arrival at the emergency department of the local hospital, he was intubated to protect his airway, as he was comatose. A 12-lead electrocardiogram showed ST-segment elevations in leads V1, V2, and V3 and a wide QRS complex with an rSR′ pattern, consistent with right bundle branch block.
Mild therapeutic hypothermia was initiated by infusing intravenous saline solution chilled to 4°C and by applying cooling blankets, and he was transferred to our hospital on an emergency basis for further management. Here, hypothermia was maintained using an intravenous cooling catheter.
HYPOTHERMIA: BENEFICIAL, BUT SLOW TO BE ADOPTED
Mild therapeutic hypothermia is a recommended therapeutic intervention for out-of-hospital cardiac arrest due to ventricular fibrillation. Nonetheless, first-responders, emergency-room staff, and intensive-care teams have been slow to adopt and integrate it into a comprehensive postresuscitation strategy. This article summarizes the evidence supporting this therapy and how it is performed.
PROPOSED MECHANISMS OF BENEFIT
Mild therapeutic hypothermia is thought to protect against anoxic brain injury in survivors of cardiac arrest via several mechanisms:
- Decreasing neuronal metabolism in the early stage of ischemic injury
- Decreasing glucose and oxygen consumption by the brain,1 which reduces supply-demand mismatch
- Decreasing the release of excitatory amino acids (eg, glutamate) that normally trigger cytotoxic cascades in the intermediate phase of injury2
- Reducing the production of harmful reactive oxygen species3
- Maintaining cellular pH4
- Reducing cell death5
- Slowing the breakdown of the blood-brain barrier that worsens cerebral edema.6
CLINICAL DATA SUPPORTING HYPOTHERMIA
There has been an interest in therapeutic hypothermia for several decades. In the 1950s, it was used in small numbers of cases in a variety of cardiac arrest situations.7,8 Interest was rekindled in the mid-1990s after a number of animal studies suggested it might be beneficial in prolonged cerebral ischemia and anoxia,9,10 and reports of case-series described its use in adults with out-of-hospital cardiac arrest.11,12
In October 2002, the International Liaison Committee on Resuscitation (ILCOR), made up of executive members of several organizations including the American Heart Association, recommended that “unconscious adult patients with spontaneous circulation after out-of-hospital cardiac arrest” should be cooled to 32°C to 34°C [89.6°F–93.2°F] for 12 to 24 hours “when the initial rhythm was ventricular fibrillation.”13
Two large randomized trials
This position statement was based largely on the results of two randomized clinical trials published simultaneously earlier in 2002.14,15 These two trials were important not only because they were the largest randomized trials of this therapy to that point, but also because they used meaningful, prospectively defined clinical end points: all-cause mortality and degree of cognitive preservation as assessed using the Glasgow-Pittsburgh Cerebral Performance Category (CPC) scale.
Bernard et al14 performed a randomized trial in four centers in Australia, assigning 77 patients either to a goal temperature of 32°C to 34°C or to normothermia for 12 hours, with all other resuscitative measures being the same in both groups. The primary outcome measured was survival to hospital discharge with sufficient neurologic function to be discharged to home or to a rehabilitation facility, ie, a CPC score of 1 or 2.
In the hypothermia group, 21 (49%) of the 43 patients survived and had an outcome that was considered “good” (ie, they were discharged home or to a rehabilitation facility), compared with 9 (26%) of the 34 patients in the normothermia group (unadjusted odds ratio 2.65, 95% confidence interval [CI] 1.02–6.88, P = .046). Proportionally fewer patients in the hypothermia group died—22 (51%) of 43 vs 23 (68%) of 34; however, the difference was not statistically significant (P = .145).
The Hypothermia After Cardiac Arrest Study Group15 screened 3,551 European patients who suffered out-of-hospital cardiac arrest15; 275 patients were randomized to mild therapeutic hypothermia or normothermia for 24 hours. The primary outcome was the percentage of patients who had a CPC score of 1 or 2 (vs 3 to 5) at 6 months, and the secondary outcome was the rate of death at 6 months.
At 6 months, 75 (55%) of the 136 patients in the hypothermia group had a CPC score of 1 or 2, compared with 54 (39%) of the 137 patients in the normothermia group (P = .009). The rate of death was also lower with hypothermia: 55% vs 41% (P = .02).
In both trials, patients were included only if their cardiac arrest was witnessed, if their initial cardiac rhythm was ventricular fibrillation or pulseless ventricular tachycardia, if circulation spontaneously returned within 60 minutes with standard basic and advanced cardiac life support protocols, and if they were still comatose on arrival at the hospital. They were excluded if they were over age 75, if they had suffered a cerebrovascular accident at the time of cardiac arrest, or if the arrest was caused by trauma or drug overdose. In addition, the European trial excluded patients who suffered another cardiac arrest after the initial return of spontaneous circulation but before cooling was started.
The standard of care
In view of the available clinical data, the 2002 ILCOR guidelines and a 2005 statement from the American Heart Association advocated mild therapeutic hypothermia for survivors of out-of-hospital ventricular tachycardia or fibrillation.16 Subsequently, this therapy has become more widely practiced and accepted as the standard of care among critical-care providers.
Of note, some public health officials and local governments are strongly promoting this treatment for survivors of cardiac arrest in the community.17 More and more of these groups are mandating that these patients be transported only to hospitals that have therapeutic hypothermia protocols in place, bypassing those not equipped to provide this treatment.18
INDICATIONS, CONTRAINDICATIONS, AND GRAY AREAS
What are the indications and contraindications to the use of hypothermia after out-of-hospital cardiac arrest? What are some of the “gray areas”?
Indications. This treatment is indicated for comatose adults who have had a witnessed cardiac arrest, whose initial cardiac rhythm was ventricular fibrillation or pulseless ventricular tachycardia, and whose circulation spontaneously returned in less than 60 minutes with basic and advanced cardiac life support. This carries a class I recommendation, level of evidence B, and was recently reinforced in the 2010 update to the American Heart Association guidelines for cardiopulmonary resuscitation.19
Absolute contraindications include hemorrhagic stroke (which must be proved by computed tomography) and cardiac arrest due to trauma (Table 2). Other major contraindications are a Glasgow Coma Scale score of 8 or higher before the initiation of mild therapeutic hypothermia, cardiac arrest due to drug overdose, and preexisting hypothermia (< 34°C) when first-responders arrive.
Relative contraindications include baseline coagulopathy and severe hypotension (mean atrial pressure < 60 mm Hg) that is not correctable by fluid infusion, vasopressors, or invasive hemodynamic support.
Gray areas. There are not enough data to make a firm recommendation about whether to apply mild therapeutic hypothermia if a witnessed cardiac arrest with ventricular fibrillation or ventricular tachycardia occurs in the hospital, but data from out-of-hospital cardiac arrest patients appear applicable for hospitalized patients.
The data are also quite limited and equivocal on its use for out-of-hospital cardiac arrest in patients whose initial cardiac rhythm is pulseless electrical activity or asystole,20,21 likely because of the competing risk of comorbidities and the resultant lower baseline survival rate in these patients.
Consequently, for in-hospital postarrest patients with any initial rhythm and for out-of-hospital cardiac arrest patients with rhythms other than ventricular tachycardia or ventricular fibrillation, the 2010 guideline recommendation on the use of mild therapeutic hypothermia is less enthusiastic (class IIb, level of evidence B).19
There are also few data on the use of mild therapeutic hypothermia in post-arrest patients in circulatory shock requiring vasopressors or intra-aortic balloon counterpulsation, largely limited to case series and comparisons with historical controls.22,23 Further investigation is clearly needed in these areas. Until then, it should be considered at the physician’s and the team’s discretion, on a case-by-case basis.
HYPOTHERMIA IN CASES OF VENTRICULAR FIBRILLATION AND ACUTE CORONARY SYNDROME
The value of coronary angiography after out-of-hospital cardiac arrest was first highlighted by Spaulding et al,24 who performed it urgently in 84 consecutive survivors of out-of-hospital cardiac arrest, 36 of whom had ST-segment elevation myocardial infarction (STEMI). Angiography uncovered an acute coronary occlusion in 40 (48%) of the 84 patients.
In this series, ST-segment elevation was a strong predictor of acute coronary occlusion (odds ratio 4.3; 95% CI 1.6–2.0; P = .004). However, 9 patients without chest pain or ST elevation were also found to have an occluded infarct-related artery. Successful angioplasty was an independent predictor of survival, highlighting the importance of an angiographic definition in this population.
These findings were recently confirmed in the larger Parisian Region Out of Hospital Cardiac Arrest (PROCAT) registry in 435 patients who had no obvious extracardiac cause of arrest, for whom successful culprit coronary angioplasty was associated with survival.25
Angioplasty comes first, but neither treatment need be delayed
Efforts to induce hypothermia must not be allowed to delay the door-to-balloon time of post-arrest patients in the setting of STEMI. The top priority is establishing patency of the infarct-related artery with a goal of salvaging ischemic myocardium and obtaining mechanical and electrical stabilization.
Fortunately, mild therapeutic hypothermia does not necessarily delay emergency revascularization if hypothermia protocols are well established. In fact, induction of mild therapeutic hypothermia prior to or on arrival at the catheterization laboratory has been shown to be feasible and safe.26,27
We believe that all centers performing primary percutaneous coronary intervention for STEMI should have immediate access to and expertise in mild therapeutic hypothermia. Regional planning and integration of STEMI and out-of-hospital cardiac arrest networks will ensure that most patients with STEMI have access to this treatment when it is indicated.
Does hypothermia help the heart? Does it increase bleeding?
Researchers have been interested in therapeutic hypothermia as a means of reducing myocardial infarct size,28,29 but clinical trials have not shown a clear-cut benefit in this regard. However, these investigations have also added to the evidence that antiplatelet and anticoagulation therapy in patients undergoing mild therapeutic hypothermia does not result in a statistically significant excess of major bleeding events, which is a potential concern.
Of note, these studies were neither powered nor specifically designed to evaluate for major bleeding as an end point. Therefore, these complications should still be carefully monitored for.
IS THERE AN OPTIMAL TIME TO BEGIN MILD THERAPEUTIC HYPOTHERMIA?
Experimental data suggest that mild therapeutic hypothermia should be started as soon as possible after a comprehensive clinical evaluation indicates the patient is eligible.30–33 However, clinical data are not robustly in favor of starting it before the patient reaches the hospital rather than on hospital arrival.
In a recent randomized trial in 2,334 survivors of out-of-hospital cardiac arrest, outcomes were no better if hypothermia was started by paramedics than if it was started on arrival at the hospital (47.5% vs 52.6% discharged to home or rehabilitation; 95% CI 0.70–1.17; P = .43).34
Earlier data from smaller studies had suggested that prehospital initiation of hypothermia (for example, using chilled intravenous saline infusions) in carefully selected patients with out-of-hospital cardiac arrest was safe and showed a nonsignificant trend toward better outcomes.20,35
The randomized controlled trials that showed hypothermia to be beneficial used very slow cooling methods; consequently, it is reasonable to allow up to 6 hours from initial presentation to first-responders to start it. There are, however, no conclusive data in humans for or against starting it later than 6 hours after presentation. Most experts believe that its potential neurologic and mortality benefits are largely lost if it is delayed more than 6 hours.
The overall message from these data seems to be that, in patients who survive cardiac arrest outside the hospital with ventricular tachycardia or fibrillation, mild therapeutic hypothermia is effective and safe and should be started as soon as possible after arrival at the hospital.
METHODS FOR INDUCING AND MAINTAINING HYPOTHERMIA
Cooling the patient
To cool the patient and keep him or her cold, caregivers have used ice packs placed around the head, groin, and axillae; intravenous infusion of saline maintained at 4°C (39°F); and cooling-air blankets. More recently, thermal wraps and intravascular cooling catheters have been used.36–38 The newer methods are more effective in rapidly bringing patients to the target temperature of 32 to 34°C (usually within 3 or 4 hours) and keeping them within this range, and they auto-adjust their output on the basis of measured core temperature.
The Pre ROSC Intranasal Cooling Effectiveness (PRINCE) trial demonstrated the safety and efficacy of nasopharyngeal cooling using a perfluorocarbon aerosol given via a nasopharyngeal cannula in patients with out-of-hospital cardiac arrest.39
Monitoring the core temperature
The patient’s core temperature is most commonly monitored with a probe in the esophagus, bladder, rectum, or pulmonary artery.40
Of these, the bladder and rectum are considered “intermediate” monitoring sites, as their temperatures tend to lag behind the core temperature. Furthermore, the bladder temperature can be significantly altered by the flow of urine, which can vary considerably during the cooling and rewarming process.
Esophageal temperature monitoring is relatively noninvasive and tends to reliably and accurately reflect core temperature as long as the probe is placed far enough down (about 45 cm from the nose in an average adult) that it is not affected by proximity to the trachea.
Pulmonary artery catheters are considered the gold standard for core temperature monitoring, but they pose risks such as bloodstream infection and large-vessel damage. In practice, many patients admitted to the coronary intensive care unit after out-of-hospital cardiac arrest require pulmonary artery catheterization anyway for other indications, and in these situations it is the preferred method of monitoring the core temperature.
However, no approach is ideal in terms of measuring the temperature in the critical end organs. Rather, core temperature monitoring serves as a guide to help ensure consistent clinical practice in attaining and maintaining mild therapeutic hypothermia.
Preventing shivering
To achieve and maintain the goal temperature, the body’s natural response to a decrease in core temperature—shivering—must be watched for and eliminated. A number of drugs may be used for this purpose.41
Paralytic drugs are used to reduce shivering; nursing staff must be trained to monitor for signs of occult shivering (eg, jaw vibration) and adjust the dose of paralytic drug accordingly. Since the patients are paralyzed, they must also receive continuous intravenous sedation.
Other commonly used drugs that decrease the hypothalamic drive to shiver include buspirone (BuSpar), a serotonin 5HT-1A partial agonist, and meperidine (Demerol), an opiate agonist of kappa and mu receptors.
Rewarming after 24 hours
Rewarming is conventionally started after 24 hours of mild therapeutic hypothermia, at a rate no greater than 0.5°C (1°F) per hour.
Because sedation is used during the hypothermia period of 24 hours, a washout period for these medications is necessary, and the neurologic prognosis of cardiac arrest patients who undergo mild therapeutic hypothermia cannot be adequately assessed until 72 hours after rewarming.
ADVERSE EFFECTS OF MILD THERAPEUTIC HYPOTHERMIA
Some of these effects are predictable. Decreasing the body temperature causes potassium to shift into the cells, and this same potassium will leave the intracellular space during the rewarming phase. For this reason, aggressive potassium repletion for mild hypokalemia (potassium levels of 3.0–3.5 mmol/L) during mild therapeutic hypothermia can result in dangerous hyperkalemia during rewarming and should generally be avoided.
As another example, the enzymes involved in coagulation are less effective at lower temperatures. Thus, if it occurs, active bleeding requiring transfusion warrants consideration of stopping the hypothermia.
Adverse effects should be watched for (eg, by checking electrolyte levels frequently, monitoring blood glucose, continuous electroencephalographic monitoring during the cooling phase, and avoiding placement of intracardiac catheters once the goal temperature is reached) and addressed as they happen. However, in a recent review of this subject42 the balance of evidence continued to indicate that the benefit of this treatment exceeds its risks.
OUR PATIENT RECOVERS
After 24 hours of therapeutic hypothermia, our patient was gradually rewarmed to a normal temperature, and sedation and paralysis were discontinued.
Analysis of his prearrest and postarrest 12-lead electrocardiograms revealed a type I Brugada pattern (coved ST elevation and negative T waves in V1, V2, and V3, caused by abnormal repolarization due to inherited mutations in SCN5A). Cardiac catheterization revealed normal coronary arteries, and MRI revealed no evidence of arrhythmogenic right ventricular cardiomyopathy or other structural abnormalities.
In the next 72 hours the patient was successfully extubated, and he gradually returned to full neurologic function. Before he went home a few days later, a single-lead cardioverter-defibrillator was implanted to prevent sudden cardiac death. All of his first-degree relatives were encouraged to undergo genetic screening for SCN5A mutations. The patient is currently back to his previous high level of functioning as a marketing manager, husband, and father of two young children.
- Erecinska M, Thoresen M, Silver IA. Effects of hypothermia on energy metabolism in mammalian central nervous system. J Cereb Blood Flow Metab 2003; 23:513–530.
- Nakashima K, Todd MM. Effects of hypothermia on the rate of excitatory amino acid release after ischemic depolarization. Stroke 1996; 27:913–918.
- Thoresen M, Satas S, Puka-Sundvall M, et al. Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport 1997; 8:3359–3362.
- Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med 2009; 37(suppl 7):S186–S202.
- Yang D, Guo S, Zhang T, Li H. Hypothermia attenuates ischemia/reperfusion-induced endothelial cell apoptosis via alterations in apoptotic pathways and JNK signaling. FEBS Lett 2009; 583:2500–2506.
- Karibe H, Zarow GJ, Graham SH, Weinstein PR. Mild intraischemic hypothermia reduces postischemic hyperperfusion, delayed postischemic hypoperfusion, blood-brain barrier disruption, brain edema, and neuronal damage volume after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab 1994; 14:620–627.
- Benson DW, Williams GR, Spencer FC, Yates AJ. The use of hypothermia after cardiac arrest. Anesth Analg 1959; 38:423–428.
- Williams GR, Spencer FC. The clinical use of hypothermia following cardiac arrest. Ann Surg 1958; 148:462–468.
- Baker CJ, Onesti ST, Barth KN, Prestigiacomo CJ, Solomon RA. Hypothermic protection following middle cerebral artery occlusion in the rat. Surg Neurol 1991; 36:175–180.
- Ridenour TR, Warner DS, Todd MM, McAllister AC. Mild hypothermia reduces infarct size resulting from temporary but not permanent focal ischemia in rats. Stroke 1992; 23:733–738.
- Bernard SA, Jones BM, Horne MK. Clinical trial of induced hypothermia in comatose survivors of out-of-hospital cardiac arrest. Ann Emerg Med 1997; 30:146–153.
- Yanagawa Y, Ishihara S, Norio H, et al. Preliminary clinical outcome study of mild resuscitative hypothermia after out-of-hospital cardiopulmonary arrest. Resuscitation 1998; 39:61–66.
- Nolan JP, Morley PT, Vanden Hoek TL, et al; International Liaison Committee on Resuscitation. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation 2003; 108:118–121.
- Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 2002; 346:557–563.
- Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 2002; 346:549–556.
- ECC Committee, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2005; 112(suppl 24):IV1–IV203.
- Hartocollis A. “City Pushes Cooling Therapy for Cardiac Arrest”. New York Times, December4th, 2008,A1. http://www.nytimes.com/2008/12/04/nyregion/04cool.html. Accessed May 31, , 2011.
- Nichol G, Aufderheide TP, Eigel B, et al; American Heart Association Emergency Cardiovascular Care Committee. Regional systems of care for out-of-hospital cardiac arrest: A policy statement from the American Heart Association. Circulation 2010; 121:709–729.
- Field JM, Hazinski MF, Sayre MR, et al. Part 1: executive summary: 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation 2010; 122(suppl 3):S640–S56.
- Hachimi-Idrissi S, Corne L, Ebinger G, Michotte Y, Huyghens L. Mild hypothermia induced by a helmet device: a clinical feasibility study. Resuscitation 2001; 51:275–281.
- Kim F, Olsufka M, Longstreth WT, et al. Pilot randomized clinical trial of prehospital induction of mild hypothermia in out-of-hospital cardiac arrest patients with a rapid infusion of 4 degrees C normal saline. Circulation 2007; 115:3064–3070.
- Hovdenes J, Laake JH, Aaberge L, Haugaa H, Bugge JF. Therapeutic hypothermia after out-of-hospital cardiac arrest: experiences with patients treated with percutaneous coronary intervention and cardiogenic shock. Acta Anaesthesiol Scand 2007; 51:137–142.
- Skulec R, Kovarnik T, Dostalova G, Kolar J, Linhart A. Induction of mild hypothermia in cardiac arrest survivors presenting with cardiogenic shock syndrome. Acta Anaesthesiol Scand 2008; 52:188–194.
- Spaulding CM, Joly LM, Rosenberg A, et al. Immediate coronary angiography in survivors of out-of-hospital cardiac arrest. N Engl J Med 1997; 336:1629–1633.
- Dumas F, Cariou A, Manzo-Silberman S, et al. Immediate percutaneous coronary intervention is associated with better survival after out-of-hospital cardiac arrest: insights from the PROCAT (Parisian Region Out of hospital Cardiac ArresT) registry. Circ Cardiovasc Interv 2010; 3:200–207.
- Knafelj R, Radsel P, Ploj T, Noc M. Primary percutaneous coronary intervention and mild induced hypothermia in comatose survivors of ventricular fibrillation with ST-elevation acute myocardial infarction. Resuscitation 2007; 74:227–234.
- Wolfrum S, Pierau C, Radke PW, Schunkert H, Kurowski V. Mild therapeutic hypothermia in patients after out-of-hospital cardiac arrest due to acute ST-segment elevation myocardial infarction undergoing immediate percutaneous coronary intervention. Crit Care Med 2008; 36:1780–1786.
- O’Neill WW, on behalf of the COOL-MI Investigators. Cooling as an adjunct to primary PCI for myocardial infarction. Presented at Transcatheter Cardiovascular Therapeutics. Washington, DC; 2004.
- Grines CL, on behalf of the ICE-IT Investigators. Intravascular cooling adjunctive to percutaneous coronary intervention for acute myocardial infarction. Presented at Transcatheter Cardiovascular Therapeutics. Washington, DC; 2004.
- Weil MH, Gazmuri RJ. Hypothermia after cardiac arrest. Crit Care Med 1991; 19:315.
- Abella BS, Zhao D, Alvarado J, Hamann K, Vanden Hoek TL, Becker LB. Intra-arrest cooling improves outcomes in a murine cardiac arrest model. Circulation 2004; 109:2786–2791.
- Zhao D, Abella BS, Beiser DG, et al. Intra-arrest cooling with delayed reperfusion yields higher survival than earlier normothermic resuscitation in a mouse model of cardiac arrest. Resuscitation 2008; 77:242–249.
- Jia X, Koenig MA, Shin HC, et al. Improving neurological outcomes post-cardiac arrest in a rat model: immediate hypothermia and quantitative EEG monitoring. Resuscitation 2008; 76:431–442.
- Bernard SA, Smith K, Cameron P, et al; Rapid Infusion of Cold Hartmanns (RICH) Investigators. Induction of therapeutic hypothermia by paramedics after resuscitation from out-of-hospital ventricular fibrillation cardiac arrest: a randomized controlled trial. Circulation 2010; 122:737–742.
- Bruel C, Parienti JJ, Marie W, et al. Mild hypothermia during advanced life support: a preliminary study in out-of-hospital cardiac arrest. Crit Care 2008; 12:R31.
- Pichon N, Amiel JB, François B, Dugard A, Etchecopar C, Vignon P. Efficacy of and tolerance to mild induced hypothermia after out-of-hospital cardiac arrest using an endovascular cooling system. Crit Care 2007; 11:R71.
- Wolff B, Machill K, Schumacher D, Schulzki I, Werner D. Early achievement of mild therapeutic hypothermia and the neurologic outcome after cardiac arrest. Int J Cardiol 2009; 133:223–228.
- Heard KJ, Peberdy MA, Sayre MR, et al. A randomized controlled trial comparing the Arctic Sun to standard cooling for induction of hypothermia after cardiac arrest. Resuscitation 2010; 81:9–14.
- Castrén M, Nordberg P, Svensson L, et al. Intra-arrest transnasal evaporative cooling: a randomized, prehospital, multicenter study (PRINCE: Pre-ROSC IntraNasal Cooling Effectiveness). Circulation 2010; 122:729–736.
- Insler SR, Sessler DI. Perioperative thermoregulation and temperature monitoring. Anesthesiol Clin 2006; 24:823–837.
- Weant KA, Martin JE, Humphries RL, Cook AM. Pharmacologic options for reducing the shivering response to therapeutic hypothermia. Pharmacotherapy 2010; 30:830–841.
- Holzer M. Targeted temperature management for comatose survivors of cardiac arrest. N Engl J Med 2010; 363:1256–1264.
- Erecinska M, Thoresen M, Silver IA. Effects of hypothermia on energy metabolism in mammalian central nervous system. J Cereb Blood Flow Metab 2003; 23:513–530.
- Nakashima K, Todd MM. Effects of hypothermia on the rate of excitatory amino acid release after ischemic depolarization. Stroke 1996; 27:913–918.
- Thoresen M, Satas S, Puka-Sundvall M, et al. Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport 1997; 8:3359–3362.
- Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med 2009; 37(suppl 7):S186–S202.
- Yang D, Guo S, Zhang T, Li H. Hypothermia attenuates ischemia/reperfusion-induced endothelial cell apoptosis via alterations in apoptotic pathways and JNK signaling. FEBS Lett 2009; 583:2500–2506.
- Karibe H, Zarow GJ, Graham SH, Weinstein PR. Mild intraischemic hypothermia reduces postischemic hyperperfusion, delayed postischemic hypoperfusion, blood-brain barrier disruption, brain edema, and neuronal damage volume after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab 1994; 14:620–627.
- Benson DW, Williams GR, Spencer FC, Yates AJ. The use of hypothermia after cardiac arrest. Anesth Analg 1959; 38:423–428.
- Williams GR, Spencer FC. The clinical use of hypothermia following cardiac arrest. Ann Surg 1958; 148:462–468.
- Baker CJ, Onesti ST, Barth KN, Prestigiacomo CJ, Solomon RA. Hypothermic protection following middle cerebral artery occlusion in the rat. Surg Neurol 1991; 36:175–180.
- Ridenour TR, Warner DS, Todd MM, McAllister AC. Mild hypothermia reduces infarct size resulting from temporary but not permanent focal ischemia in rats. Stroke 1992; 23:733–738.
- Bernard SA, Jones BM, Horne MK. Clinical trial of induced hypothermia in comatose survivors of out-of-hospital cardiac arrest. Ann Emerg Med 1997; 30:146–153.
- Yanagawa Y, Ishihara S, Norio H, et al. Preliminary clinical outcome study of mild resuscitative hypothermia after out-of-hospital cardiopulmonary arrest. Resuscitation 1998; 39:61–66.
- Nolan JP, Morley PT, Vanden Hoek TL, et al; International Liaison Committee on Resuscitation. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation 2003; 108:118–121.
- Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 2002; 346:557–563.
- Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 2002; 346:549–556.
- ECC Committee, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2005; 112(suppl 24):IV1–IV203.
- Hartocollis A. “City Pushes Cooling Therapy for Cardiac Arrest”. New York Times, December4th, 2008,A1. http://www.nytimes.com/2008/12/04/nyregion/04cool.html. Accessed May 31, , 2011.
- Nichol G, Aufderheide TP, Eigel B, et al; American Heart Association Emergency Cardiovascular Care Committee. Regional systems of care for out-of-hospital cardiac arrest: A policy statement from the American Heart Association. Circulation 2010; 121:709–729.
- Field JM, Hazinski MF, Sayre MR, et al. Part 1: executive summary: 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation 2010; 122(suppl 3):S640–S56.
- Hachimi-Idrissi S, Corne L, Ebinger G, Michotte Y, Huyghens L. Mild hypothermia induced by a helmet device: a clinical feasibility study. Resuscitation 2001; 51:275–281.
- Kim F, Olsufka M, Longstreth WT, et al. Pilot randomized clinical trial of prehospital induction of mild hypothermia in out-of-hospital cardiac arrest patients with a rapid infusion of 4 degrees C normal saline. Circulation 2007; 115:3064–3070.
- Hovdenes J, Laake JH, Aaberge L, Haugaa H, Bugge JF. Therapeutic hypothermia after out-of-hospital cardiac arrest: experiences with patients treated with percutaneous coronary intervention and cardiogenic shock. Acta Anaesthesiol Scand 2007; 51:137–142.
- Skulec R, Kovarnik T, Dostalova G, Kolar J, Linhart A. Induction of mild hypothermia in cardiac arrest survivors presenting with cardiogenic shock syndrome. Acta Anaesthesiol Scand 2008; 52:188–194.
- Spaulding CM, Joly LM, Rosenberg A, et al. Immediate coronary angiography in survivors of out-of-hospital cardiac arrest. N Engl J Med 1997; 336:1629–1633.
- Dumas F, Cariou A, Manzo-Silberman S, et al. Immediate percutaneous coronary intervention is associated with better survival after out-of-hospital cardiac arrest: insights from the PROCAT (Parisian Region Out of hospital Cardiac ArresT) registry. Circ Cardiovasc Interv 2010; 3:200–207.
- Knafelj R, Radsel P, Ploj T, Noc M. Primary percutaneous coronary intervention and mild induced hypothermia in comatose survivors of ventricular fibrillation with ST-elevation acute myocardial infarction. Resuscitation 2007; 74:227–234.
- Wolfrum S, Pierau C, Radke PW, Schunkert H, Kurowski V. Mild therapeutic hypothermia in patients after out-of-hospital cardiac arrest due to acute ST-segment elevation myocardial infarction undergoing immediate percutaneous coronary intervention. Crit Care Med 2008; 36:1780–1786.
- O’Neill WW, on behalf of the COOL-MI Investigators. Cooling as an adjunct to primary PCI for myocardial infarction. Presented at Transcatheter Cardiovascular Therapeutics. Washington, DC; 2004.
- Grines CL, on behalf of the ICE-IT Investigators. Intravascular cooling adjunctive to percutaneous coronary intervention for acute myocardial infarction. Presented at Transcatheter Cardiovascular Therapeutics. Washington, DC; 2004.
- Weil MH, Gazmuri RJ. Hypothermia after cardiac arrest. Crit Care Med 1991; 19:315.
- Abella BS, Zhao D, Alvarado J, Hamann K, Vanden Hoek TL, Becker LB. Intra-arrest cooling improves outcomes in a murine cardiac arrest model. Circulation 2004; 109:2786–2791.
- Zhao D, Abella BS, Beiser DG, et al. Intra-arrest cooling with delayed reperfusion yields higher survival than earlier normothermic resuscitation in a mouse model of cardiac arrest. Resuscitation 2008; 77:242–249.
- Jia X, Koenig MA, Shin HC, et al. Improving neurological outcomes post-cardiac arrest in a rat model: immediate hypothermia and quantitative EEG monitoring. Resuscitation 2008; 76:431–442.
- Bernard SA, Smith K, Cameron P, et al; Rapid Infusion of Cold Hartmanns (RICH) Investigators. Induction of therapeutic hypothermia by paramedics after resuscitation from out-of-hospital ventricular fibrillation cardiac arrest: a randomized controlled trial. Circulation 2010; 122:737–742.
- Bruel C, Parienti JJ, Marie W, et al. Mild hypothermia during advanced life support: a preliminary study in out-of-hospital cardiac arrest. Crit Care 2008; 12:R31.
- Pichon N, Amiel JB, François B, Dugard A, Etchecopar C, Vignon P. Efficacy of and tolerance to mild induced hypothermia after out-of-hospital cardiac arrest using an endovascular cooling system. Crit Care 2007; 11:R71.
- Wolff B, Machill K, Schumacher D, Schulzki I, Werner D. Early achievement of mild therapeutic hypothermia and the neurologic outcome after cardiac arrest. Int J Cardiol 2009; 133:223–228.
- Heard KJ, Peberdy MA, Sayre MR, et al. A randomized controlled trial comparing the Arctic Sun to standard cooling for induction of hypothermia after cardiac arrest. Resuscitation 2010; 81:9–14.
- Castrén M, Nordberg P, Svensson L, et al. Intra-arrest transnasal evaporative cooling: a randomized, prehospital, multicenter study (PRINCE: Pre-ROSC IntraNasal Cooling Effectiveness). Circulation 2010; 122:729–736.
- Insler SR, Sessler DI. Perioperative thermoregulation and temperature monitoring. Anesthesiol Clin 2006; 24:823–837.
- Weant KA, Martin JE, Humphries RL, Cook AM. Pharmacologic options for reducing the shivering response to therapeutic hypothermia. Pharmacotherapy 2010; 30:830–841.
- Holzer M. Targeted temperature management for comatose survivors of cardiac arrest. N Engl J Med 2010; 363:1256–1264.
KEY POINTS
- This treatment is indicated for comatose adult patients who have had a witnessed cardiac arrest, whose initial cardiac rhythm is ventricular fibrillation or pulseless ventricular tachycardia, and who have return of spontaneous circulation with basic and advanced cardiac life support.
- Contraindications include hemorrhagic stroke, a Glasgow Coma Scale score of 8 or higher, cardiac arrest due to drug overdose, and preexisting hypothermia. Relative contraindications include baseline coagulopathy and severe hypotension (mean arterial pressure < 60 mm Hg) that is not correctable by fluid infusion, vasopressors, or invasive hemodynamic support.
- Adverse effects have included hypokalemia, bradyarrhythmia, ventricular tachycardia, hypotension, seizures, hyperglycemia, a transient decrease in the glomerular filtration rate, abnormal coagulation studies, and an increased incidence of pneumonia and sepsis.
Grand Rounds: Woman, 20, With Difficulty Walking
A 20-year-old woman presented to her primary care clinic with a chief complaint of lower leg weakness and difficulty walking. The weakness she described had been worsening over the previous four days, with progressively worsening tingling and numbness of her toes bilaterally.
The day before the patient presented, she noticed numbness and paresthesia in both calves. At the time of her presentation to the clinic, she complained of low back ache, paresthesia of both hands, numbness bilaterally to her groin, difficulty sitting upright, ataxia, and a numb, thick-feeling tongue. She denied fever, neck stiffness, shortness of breath, headache, or visual changes.
The patient stated that 10 days earlier, she had developed an upper respiratory infection for which she was seen at the clinic and treated with a seven-day course of amoxicillin/clavulanate 875/125 mg twice daily. She said that she had recovered completely.
A review of the patient’s systems revealed proximal muscle weakness bilaterally (2/5) and loss of touch-pressure in the lower extremities. She was experiencing paresthesia of the hands and mild weakness bilaterally (4/5). She also walked with an ataxic gait and had reduced deep tendon reflexes in the lower limbs. All cranial nerves were intact, and her vital signs were stable.
The woman’s medical history was positive only for asthma. Her family history included ischemic stroke in the maternal grandfather and brain tumor in the paternal grandfather. Social history was positive for alcohol intake (ranging from four to 12 beers per week). The patient said she had never smoked or used illicit drugs. She was an unmarried college student, living in a dorm on campus. She participated in track at school.
The patient was admitted to the hospital telemetry step-down unit, and a neurology consultation was requested. Tests were ordered, among them MRI of the head and spine and comprehensive blood work, to rule out neurologic, infectious, or metabolic causes of the patient’s weakness; urinalysis was also obtained. These tests all yielded negative results.
A lumbar puncture performed the following day revealed a cerebrospinal fluid (CSF) protein level of 570 mg/L (normal range, 150 to 450 mg/L). Leukocytes numbered 2 cells/mm3 (normal count, 0 to 10 cells/mm3).
Based on the patient’s presentation, history, and symptoms, a neurologist made a diagnosis of Guillain-Barré syndrome. It was decided that no electromyographic (EMG) study was required to rule out other disease processes (eg, spinal cord disease, multiple sclerosis, tumors).
The patient underwent a five-dose course of immunomodulatory therapy with IV immunoglobulin (IVIG). In the step-down unit, she experienced one incident of sinus bradycardia (ie, resting heart rate between 40 and 50 beats/min). Her blood pressure remained stable, as did her respiratory status, according to peak expiratory flow measured frequently at her bedside.
Physical therapy was initiated, consisting of passive and active range of motion, crossovers with the patient’s feet, and stair training. This was done in response to a complaint of ankle weakness, and it helped to strengthen weakened muscles and improve alignment while the patient was bedridden and in a weakened, fatigued state. Additionally, the patient was given enoxaparin, wore antiembolic hose, and used sequential compression devices while in bed. As a result of these measures, she never experienced a pulmonary embolus or deep vein thrombosis (DVT) as a result of being immobilized.
By the seventh day of hospitalization, the patient had stable vital signs and improved lower limb strength, and numbness was resolving in her hands and lower extremities. She was discharged to home, with physical therapy to resume on an outpatient basis.
Discussion
Guillain-Barré syndrome (GBS), an acute immune-mediated paralytic disorder,1 manifests in the form of weakness and diminished reflexes. Affecting the peripheral nerves, GBS is characterized by progressive symmetrical ascending weakness with varying degrees of sensory complaints.2,3
GBS occurs worldwide, and incidence is estimated between 1.1 and 1.8 cases per 100,000 persons.4 In the United States, GBS can be found in all age-groups, with peak incidence noted in elderly persons and young adults.5,6 Even with treatment, 3% to 10% of patients are reported to die of this illness, and 20% cannot walk six months after symptom onset.7 In one prospective population-based study of patients with confirmed GBS, 6% of patients died within 30 days of symptom onset, often as a result of respiratory complications.8
GBS is a postinfectious disorder, with cases developing several days or weeks after a viral or bacterial illness—most commonly, an upper respiratory infection or diarrhea (see Table 19-13). The most common trigger of GBS is infection with the bacterial microorganism Campylobacter jejuni (occurring in 15% to 40% of patients with GBS),9,14 a pathogen that can produce demyelination-causing antibodies. Other responsible pathogens include cytomegalovirus and Epstein-Barr virus.9 In a process called molecular mimicry, the immune system is unable to distinguish the amino acid of an infectious organism from the proteinaceous content of the peripheral nerve.15 Subsequently, the immune system attacks and destroys the myelin sheath.
An example of this is the apparent cross-reaction of the ganglioside GM1 with C jejuni lipopolysaccharide antigens.14,15 The resulting effect is immunologic damage to the peripheral nervous system. The flaccid paralysis that occurs in patients with GBS is thought to be caused by lymphocytic infiltration and complement activation of the spinal roots and peripheral nerves, where macrophages strip the myelin.5,15,16
Stages and Variants
Three stages characterize the course of GBS. The acute phase, which lasts one to four weeks, begins with onset of symptoms and persists until the associated neurologic deterioration has ceased. During the second phase, the plateau period, symptoms persist with no further deterioration; this stage can last several days to several weeks or months. The final phase, the recovery period, can last from four months to two years after symptom onset.15,17,18
The clinical course of GBS is highly variable and in many cases difficult to predict. Certain factors have been associated with a poor outcome: advancing age, previous presence of diarrhea, need for mechanical ventilation, an extended plateau phase, and a lower patient score on the Erasmus GBS Outcome Scale,19 when measured two weeks after GBS onset.8,20 This score can help predict the patient’s chance of independent walking after six months.15,19
Although the classic presenting symptom of GBS is symmetric ascending weakness, several disease variants have been identified, with differing symptoms and degrees of recovery. These variants also differ in terms of the muscle groups affected; in some, visual defects may be present at onset. GBS variants include21:
• Acute motor axonal neuropathy (AMAN)1,22
• Acute inflammatory demyelinating polyneuropathy (AIDP)1
• Pharyngeal-cervical-brachial variant23
• Purely sensory variant24
• Miller-Fisher syndrome, which manifests with ophthalmoplegia, in addition to ataxia and areflexia25
• Axonal form.5,21
AMAN and AIDP are the most common subtypes of GBS.1
Symptoms, Signs, and Disease Manifestations
Limb weakness, the classic presenting symptom of GBS, is both symmetrical and ascending. Weakness can develop acutely and progress over days to weeks.2,15 Hughes and Cornblath26 also note pain, numbness, and paresthesias among the initial symptoms of GBS. Others include sensory changes, cranial nerve involvement, various autonomic changes, and respiratory or oropharyngeal weakness. Reflexes, particularly the tendon reflexes, may be diminished or absent.15,18,21 In many cases, sensory changes (ie, pain) may precede the onset of weakness, often making diagnosis difficult.15
Cranial nerves most commonly affected are V, VI, VII, X, XI, and XII, with manifestations that include dysphagia, dysarthria, diplopia, limitation to eye movements, and facial droop and weakness. Usually facial and oropharyngeal weakness occur after the extremities and trunk are affected. Blindness may occur if demyelination of the optic nerve occurs; this is seen in Miller-Fisher syndrome.10,15,25,27
In GBS, many patients report pain, which can present as bilateral sciatica or as throbbing or aching in the large muscles of the upper legs, flanks, or back.28 This pain, which results from the demyelination of the sensory nerve fibers, can be severe.10
Patients with GBS may experience manifestations of autonomic nervous system dysfunction—for example, arrhythmias, hypotension or hypertension, urinary retention, cardiomyopathy, and paralytic ileus.10,20 Dysautonomia often impedes patients’ progress in inpatient rehabilitation. Patients may have persistent problems involving postural hypotension, hypertension, excessive sympathetic outflow, or bladder and bowel dysfunction.29
Blood pressure fluctuations, often attributed to changes in catecholamine levels and disturbances in the baroreceptor reflex pathway, are common and are considered characteristic of GBS. Transient or persistent hypotension is caused by the dysregulation of the parasympathetic and sympathetic systems, with subsequent alterations in venomotor tone.3 Additionally, an increased sensitivity to catecholamine can lead to cardiovascular disturbances, resulting in denervation hypersensitivity and impairment of the carotid sinus reflex.
Arrhythmias occur in perhaps half of patients with GBS. The most common is sustained sinus tachycardia, which usually requires no treatment. Bradycardia leading to atrioventricular blocks and asystole is believed to result from afferent baroreceptor reflex failure. Treatment may be required—either administration of atropine or insertion of a pacemaker, depending on the severity of the arrhythmia.3,10
Myocardial involvement can range from asymptomatic mycocarditis to neurogenic stunned myocardium and heart failure. Patients with ECG abnormalities should undergo two-dimensional echocardiographic studies and other testing to explore cardiac involvement. Acute coronary syndromes, including ST-segment elevation MI, have been reported, in some cases associated with IVIG treatment. In one patient, coronary spasm was reported, with clean coronary arteries found on cardiac catheterization.3
Patients with GBS are at risk for compromised neuromuscular respiratory function; demyelination of the nerves that innervate the intercostal muscles and the diaphragm can result in respiratory failure. Key clinical indicators of respiratory muscle fatigue include tachypnea, diaphoresis, and asynchronous movements of the abdomen and chest;10 other symptoms relevant to respiratory or oropharyngeal weakness include slurred speech, dyspnea (with or without exertion), difficulty swallowing, and inability to cough.2,10 Serial respiratory function testing is advisable to detect patients at risk for respiratory failure.30
Diagnosis
Guillain-Barré is a syndrome diagnosed by a collection of symptoms (see Table 22,21,31), including subacute developing paralysis, symmetrical bilateral weakness beginning at onset, and diminishing to absent reflexes.21,31 Other causes for rapidly developing weaknesses should be ruled out (see Table 310,21,26,31). Lumbar puncture typically shows increased protein levels with a normal white cell count; however, neither this test nor electrophysiologic evaluation offers significant value for diagnosis of GBS.21,26,31
During the acute phase of GBS (within three weeks of onset), there is found an elevation of CSF protein (> 550 mg/L) without an elevation in white blood cells. This phenomenon, called albuminocytologic dissociation, reflects inflammation of the nerve roots and is considered the hallmark of GBS.2
MRI can also facilitate the diagnosis of GBS; it demonstrates anterior and posterior intrathecal spinal nerve roots and cauda equina.32 In patients with GBS, evidence supporting breakdown of the blood–nerve barrier can be seen in abnormal gadolinium enhancement of the intrathecal nerve roots on MRI.33
When electrophysiologic studies are performed, they typically reveal slowing nerve conduction, prolonged distal latencies, and partial motor conduction block.34 The characteristic finding of early demyelination is conduction block, a reduction in the amplitude of the muscle action potential after stimulation of the distal, as opposed to the proximal, nerve.28 Nerve conduction studies may help in the diagnosis and classification of GBS—and, to a limited extent, formulation of a prognosis. Such alternative diagnoses as myositis and myasthenia gravis may be excluded by neurophysiology.26 Early in GBS, neurophysiologic abnormalities may be very mild or occasionally normal; test results may not correlate with clinical disability.35,36
The clinician cannot depend on clinical features alone to predict respiratory decline.31 Frequent evaluations of respiratory effort, by measurement of maximal inspiratory pressures and vital capacity, should be performed at the bedside to monitor diaphragmatic strength. Respiratory ventilation should be initiated if the patient becomes hypoxic or experiences a rapid decline in vital capacity (ie, below 60% of predicted value).10 Mechanical ventilation is more likely to be required in patients with a negative inspiratory force of less than 30 cm H2O.31
Treatment
Guillain-Barré syndrome has an acute onset and progression. Patients quickly become nonambulatory and may require total ventilation due to paralysis. Therapeutic options are IVIG or plasmapheresis (plasma exchange).37-40 Corticosteroids do not appear to benefit patients with GBS.41,42
Several mechanisms appear to contribute to the effectiveness of immunoglobulin.38,39 Infused IVIG interferes with antigen presentation, inhibits antibody production, neutralizes pathologic autoantibodies, and modulates other immunologic events involved in the pathogenesis of autoimmune neuromuscular diseases, including GBS.43 Adverse reactions, which are usually minor, include headache, fever, chills, myalgia, and malaise. In rare instances, anaphylaxis or renal failure may occur.15,44
In plasmapheresis, blood is removed from the body and dialyzed, with circulating antibodies and immunoglobulins removed from the plasma; fresh frozen plasma, albumin, or saline is administered. This treatment, performed via central venous catheter, should be initiated as soon as possible after onset of symptoms but can be implemented as late as 30 days after GBS onset. Plasmapheresis requires personnel trained in dialysis, which may not be performed in all hospitals. Possible adverse events include infection and hemorrhage. Laboratory values must be monitored for hypokalemia and hypocalcemia.45,46
Supportive Care
Patients with GBS require intensive care and very close monitoring for complications of respiratory difficulty and autonomic dysfunction. Individualized programs should be initiated for patients in the acute phase of GBS, aimed at the prevention of contractures and skin breakdown.10 Exercise programs, as conducted with the case patient, should also help relieve the fatigue syndromes that accompany GBS.
Immobilization associated with bed rest incurs a risk for pulmonary emboli and DVT; this has been found true during the first 12 weeks after symptom onset in patients with GBS who remain immobile.47 The use of antiembolic hose and sequential compression devices can help reduce the risk for thrombotic events.10 Use of enoxaparin or heparin is recommended for nonambulating patients until they are able to walk, with Gaber et al47 specifying the use of low-molecular-weight heparin to reduce, but not eliminate, the risk for DVT.
The pain associated with GBS can be severe. Narcotic analgesics may be administered with careful monitoring of autonomic denervation. Long-term management of neuropathic pain may require adjuvant therapy, such as tricyclic antidepressants, gabapentin, or tramadol hydrochloride.10 According to Pandey et al,48 gabapentin alone may suffice for pain control in GBS, with minimal adverse effects. In certain rehabilitation facilities, tricyclic antidepressants, capsaicin, and transcutaneous nerve stimulation have been reported effective; during the early stages of treatment, until these treatments reach their full effect, pain medications such as tramadol or narcotics can provide temporary relief.29
More than one-half of patients with GBS in the acute phase can develop ileus. Constipation can also occur as a result of pain medication use, prolonged bed rest, and poor intake. Auscultation of bowel sounds and abdominal assessment should be performed daily to monitor for ileus. Hughes et al10 do not recommend the use of promotility drugs in patients with dysautonomia.
After hospital discharge, easy fatigability can affect work and social activities. With continued physical therapy, occupational therapy, and monitoring, however, patients with GBS can expect to return to an optimal level of functioning. Speed of recovery varies with these patients from a few months to several years, depending on such factors as age and the extent to which axonal degeneration has occurred.6,49
The Case Patient
For several weeks after discharge, the case patient continued to experience fatigue, low back pain, and general muscle pain. With her family’s support, she continued to receive outpatient physical therapy, and within one month she had regained her ankle strength. She was soon able to resume her classes, despite some lingering fatigue.
Conclusion
Guillain-Barré syndrome is a potentially life-threatening disease whose symptoms health care providers need to recognize quickly to provide prompt treatment. Supportive care for both patient and family is of key importance for maximum rehabilitation and return to the previous lifestyle. The clinical course of GBS is highly variable and difficult to predict. The patient’s outcome depends on several factors, including age and severity of illness. GBS patients can experience long-term psychosocial effects.
References
1. Magira EE, Papaioakim M, Nachamkin I, et al. Differential distribution of HLA-DQ beta/DR beta epitopes in the two forms of Guillain-Barré syndrome, acute motor axonal neuropathy and acute inflammatory demyelinating polyneuropathy (AIDP): identification of DQ beta epitopes associated with susceptibility to and protection from AIDP. J Immunol. 2003;170(6):3074-3080.
2. Tremblay ME, Closon A, D’Anjou G, Bussières JF. Guillain-Barré syndrome following H1N1 immunization in a pediatric patient. Ann Pharmacother. 2010;44(7-8):1330-1333.
3. Mukerji S, Aloka F, Farooq MU, et al. Cardiovascular complications of the Guillain-Barré syndrome. Am J Cardiol. 2009;104(10):1452-1455.
4. McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barré syndrome worldwide: a systematic literature review. Neuroepidemiology. 2009;32(2):150-163.
5. Haber P, Sejvar J, Mikaeloff Y, DeStefano F. Vaccines and Guillain-Barré syndrome. Drug Saf. 2009; 32(4):309-323.
6. van Doorn PA. What’s new in Guillain-Barré syndrome in 2007-2008? J Periph Nerv Syst. 2009;14(2):72-74.
7. van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008;7(10):939-950.
8. Chiò A, Cocito D, Leone M, et al; Piemonte and alle d’Aosta Register for Guillain-Barré Syndrome. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003; 60(7):1146-1150.
9. Hadden RD, Karch H, Hartung HP, et al. Preceding infections, immune factors, and outcome in Guillain-Barré syndrome. Neurology. 2001;56(6):758-765.
10. Hughes RA, Wijdicks EF, Benson E, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol. 2005;62(8):1194-1198.
11. Aluka KJ, Turner PL, Fullum TM. Guillain-Barré syndrome and postbariatric surgery polyneuropathies. JSLS. 2009;13(2):250-253.
12. Brannagan TH 3rd, Zhou Y. HIV-associated Guillain-Barré syndrome. J Neurol Sci. 2003;208(1-2):39-42.
13. Lin WC, Lee PI, Lu CY, et al. Mycoplasma pneumoniae encephalitis in childhood. J Microbiol Immunol Infect. 2002;35(3):173-178.
14. Sivadon-Tardy V, Orlikowski D, Porcher R, et al. Detection of Campylobacter jejuni by culture and real-time PCR in a French cohort of patients with Guillain-Barre syndrome. J Clin Microbiol. 2010;48 (6):2278-2281.
15. van Doorn PA, Kuitwaard K, Walgaard C, et al. IVIG treatment and prognosis in Guillain-Barré syndrome. J Clin Immunol. 2010;30 suppl 1:S74-S78.
16. Kaida K, Kusunoki S. Guillan-Barré syndrome: update on immunobiology and treatment. Expert Rev Neurother. 2009;9(9):1307-1319.
17. Forsberg A, Press R, Einarsson U, et al. Disability and health-related quality of life in Guillain-Barré syndrome during the first two years after onset: a prospective study. Clin Rehabil. 2005;19(8):900-909.
18. Criteria for diagnosis of Guillain-Barré syndrome. Ann Neurol. 1978;3(6):565-566.
19. van Koningsveld R, Steyerberg EW, Hughes RA, et al. A clinical progostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007;6(7):589-594.
20. Koeppen S, Kraywinkel K, Wessendorf TE, et al. Long-term outcome of Guillain-Barré syndrome. Neurocrit Care. 2006;5(3)235-242.
21. Sheridan JM, Smith D. Atypical Guillain-Barré in the emergency department. West J Emerg Med. 2010;11(1):80-82.
22. Ogawara K, Kuwabara S, Koga M, et al. Anti-GM1b IgG antibody is associated with acute motor axonal neuropathy and Campylobacter jejuni infection. J Neurol Sci. 2003;210(1-2):41-45.
23. Nagashima T, Koga M, Odaka M, et al. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Arch Neurol. 2007;64(10):1519-1523.
24. Oh SJ, LaGanke C, Claussen GC. Sensory Guillain-Barré syndrome. Neurology. 2001;56(1):82-86.
25. Aráranyi Z, Kovács T, Sipos I, Bereczki D. Miller Fisher syndrome: brief overview and update with a focus on electrophysiological findings. Eur J Neurol. 2011 Jun 1. [Epub ahead of print]
26. Hughes RA, Cornblath, DR. Guillain-Barré syndrome. Lancet. 2005;366(9497):1653-1666.
27. Snyder LA, Rismondo V, Miller NR. The Fisher variant of Guillain-Barré syndrome (Fisher syndrome). J Neuroophthalmol. 2009;29(4):312-324.
28. Ropper AH. The Guillain-Barré syndrome. N Engl J Med.1992;326(17):1130-1136.
29. Meythaler JM. Rehabilitation of Guillain-Barré syndrome. Arch Phys Med Rehabil.1997;78(8):872-879.
30. Sharshar T, Chevret S, Bourdain F, et al; French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Early predictors of mechanical ventilation in Guillain-Barré syndrome. Crit Care Med. 2003; 31(1):278-283.
31. McGillicuddy DC, Walker O, Shapiro NI, et al. Guillain-Barré syndrome in the emergency department. Ann Emerg Med. 2006;47(4):390-393.
32. Yikilmaz A, Doganay S, Gumus H, et al. Magnetic resonance imaging of childhood Guillain-Barré syndrome. Childs Nerv Syst. 2010;26(8):1103-1108.
33. Gonzalez-Quevedo A, Carriera RF, O’Farrill ZL, et al. An appraisal of blood-cerebrospinal fluid barrier dysfunction during the course of Guillain-Barré syndrome. Neurol India. 2009;57(3):288-294.
34. Abai S, Kim SB, Kim JP, Lim YJ. Guillan-Barré syndrome combined with acute cervical myelopathy. J Korean Neurosurg Soc. 2010;48(3):298-300.
35. Uncini A, Yuki N. Electrophysiologic and immunopathologic correlates in Guillain-Barré syndrome subtypes. Expert Rev Neurother. 2009;9(6):869-884.
36. Hadden RD, Hughes RA. Management of inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2003;74 suppl 2:ii9-ii14.
37. Raphaël JC, Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2002;(2):CD001798.
38. Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Jun 16; (6):CD002063.
39. Human immunoglobulin and the Guillain-Barré syndrome: new indication. An alternative to plasmapheresis. Prescrire Int. 2000;9(49):142-143.
40. van der Meché FG, Schmitz PI; Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;327(17):1123-1129.
41. Hughes RA, Swan AV, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Feb 16;(2):CD001446.
42. Hahn AF. Guillain-Barré syndrome. Lancet. 1998; 352(9128):635-641.
43. Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;291(19):2367-2375.
44. Kuitwaard K, de Gelder J, Tio-Gillen AP, et al. Pharmacokenetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann Neurol. 2009;66(5):597-603.
45. Atkinson SB, Carr RL, Maybee P, Haynes D. The challenges of managing and treating Guillain-Barré syndrome during the acute phase. Dimens Crit Care Nurs. 2006;25(6):256-263.
46. van Doorn PA. Treatment of Guillain-Barré syndrome and CIDP. J Periph Nerv Syst. 2005;10(2):113-127.
47. Gaber TA, Kirker SGB, Jenner JR. Current practice of prophylactic anticoagulation in Guillain-Barré syndrome. Clin Rehabil. 2002;16(2):190-193.
48. Pandey CK, Bose N, Garg G, et al. Gabapentin for the treatment of pain in Guillain-Barré syndrome: a double-blinded, placebo-controlled, crossover study. Anesth Analg. 2002;95(6):1719-1723.
49. de Vries JM, Hagemans ML, Bussmann JB, et al. Fatigue in neuromuscular disorders: focus on Guillain-Barré syndrome and Pompe disease. Cell Mol Life Sci. 2010;67(5):701-713.
A 20-year-old woman presented to her primary care clinic with a chief complaint of lower leg weakness and difficulty walking. The weakness she described had been worsening over the previous four days, with progressively worsening tingling and numbness of her toes bilaterally.
The day before the patient presented, she noticed numbness and paresthesia in both calves. At the time of her presentation to the clinic, she complained of low back ache, paresthesia of both hands, numbness bilaterally to her groin, difficulty sitting upright, ataxia, and a numb, thick-feeling tongue. She denied fever, neck stiffness, shortness of breath, headache, or visual changes.
The patient stated that 10 days earlier, she had developed an upper respiratory infection for which she was seen at the clinic and treated with a seven-day course of amoxicillin/clavulanate 875/125 mg twice daily. She said that she had recovered completely.
A review of the patient’s systems revealed proximal muscle weakness bilaterally (2/5) and loss of touch-pressure in the lower extremities. She was experiencing paresthesia of the hands and mild weakness bilaterally (4/5). She also walked with an ataxic gait and had reduced deep tendon reflexes in the lower limbs. All cranial nerves were intact, and her vital signs were stable.
The woman’s medical history was positive only for asthma. Her family history included ischemic stroke in the maternal grandfather and brain tumor in the paternal grandfather. Social history was positive for alcohol intake (ranging from four to 12 beers per week). The patient said she had never smoked or used illicit drugs. She was an unmarried college student, living in a dorm on campus. She participated in track at school.
The patient was admitted to the hospital telemetry step-down unit, and a neurology consultation was requested. Tests were ordered, among them MRI of the head and spine and comprehensive blood work, to rule out neurologic, infectious, or metabolic causes of the patient’s weakness; urinalysis was also obtained. These tests all yielded negative results.
A lumbar puncture performed the following day revealed a cerebrospinal fluid (CSF) protein level of 570 mg/L (normal range, 150 to 450 mg/L). Leukocytes numbered 2 cells/mm3 (normal count, 0 to 10 cells/mm3).
Based on the patient’s presentation, history, and symptoms, a neurologist made a diagnosis of Guillain-Barré syndrome. It was decided that no electromyographic (EMG) study was required to rule out other disease processes (eg, spinal cord disease, multiple sclerosis, tumors).
The patient underwent a five-dose course of immunomodulatory therapy with IV immunoglobulin (IVIG). In the step-down unit, she experienced one incident of sinus bradycardia (ie, resting heart rate between 40 and 50 beats/min). Her blood pressure remained stable, as did her respiratory status, according to peak expiratory flow measured frequently at her bedside.
Physical therapy was initiated, consisting of passive and active range of motion, crossovers with the patient’s feet, and stair training. This was done in response to a complaint of ankle weakness, and it helped to strengthen weakened muscles and improve alignment while the patient was bedridden and in a weakened, fatigued state. Additionally, the patient was given enoxaparin, wore antiembolic hose, and used sequential compression devices while in bed. As a result of these measures, she never experienced a pulmonary embolus or deep vein thrombosis (DVT) as a result of being immobilized.
By the seventh day of hospitalization, the patient had stable vital signs and improved lower limb strength, and numbness was resolving in her hands and lower extremities. She was discharged to home, with physical therapy to resume on an outpatient basis.
Discussion
Guillain-Barré syndrome (GBS), an acute immune-mediated paralytic disorder,1 manifests in the form of weakness and diminished reflexes. Affecting the peripheral nerves, GBS is characterized by progressive symmetrical ascending weakness with varying degrees of sensory complaints.2,3
GBS occurs worldwide, and incidence is estimated between 1.1 and 1.8 cases per 100,000 persons.4 In the United States, GBS can be found in all age-groups, with peak incidence noted in elderly persons and young adults.5,6 Even with treatment, 3% to 10% of patients are reported to die of this illness, and 20% cannot walk six months after symptom onset.7 In one prospective population-based study of patients with confirmed GBS, 6% of patients died within 30 days of symptom onset, often as a result of respiratory complications.8
GBS is a postinfectious disorder, with cases developing several days or weeks after a viral or bacterial illness—most commonly, an upper respiratory infection or diarrhea (see Table 19-13). The most common trigger of GBS is infection with the bacterial microorganism Campylobacter jejuni (occurring in 15% to 40% of patients with GBS),9,14 a pathogen that can produce demyelination-causing antibodies. Other responsible pathogens include cytomegalovirus and Epstein-Barr virus.9 In a process called molecular mimicry, the immune system is unable to distinguish the amino acid of an infectious organism from the proteinaceous content of the peripheral nerve.15 Subsequently, the immune system attacks and destroys the myelin sheath.
An example of this is the apparent cross-reaction of the ganglioside GM1 with C jejuni lipopolysaccharide antigens.14,15 The resulting effect is immunologic damage to the peripheral nervous system. The flaccid paralysis that occurs in patients with GBS is thought to be caused by lymphocytic infiltration and complement activation of the spinal roots and peripheral nerves, where macrophages strip the myelin.5,15,16
Stages and Variants
Three stages characterize the course of GBS. The acute phase, which lasts one to four weeks, begins with onset of symptoms and persists until the associated neurologic deterioration has ceased. During the second phase, the plateau period, symptoms persist with no further deterioration; this stage can last several days to several weeks or months. The final phase, the recovery period, can last from four months to two years after symptom onset.15,17,18
The clinical course of GBS is highly variable and in many cases difficult to predict. Certain factors have been associated with a poor outcome: advancing age, previous presence of diarrhea, need for mechanical ventilation, an extended plateau phase, and a lower patient score on the Erasmus GBS Outcome Scale,19 when measured two weeks after GBS onset.8,20 This score can help predict the patient’s chance of independent walking after six months.15,19
Although the classic presenting symptom of GBS is symmetric ascending weakness, several disease variants have been identified, with differing symptoms and degrees of recovery. These variants also differ in terms of the muscle groups affected; in some, visual defects may be present at onset. GBS variants include21:
• Acute motor axonal neuropathy (AMAN)1,22
• Acute inflammatory demyelinating polyneuropathy (AIDP)1
• Pharyngeal-cervical-brachial variant23
• Purely sensory variant24
• Miller-Fisher syndrome, which manifests with ophthalmoplegia, in addition to ataxia and areflexia25
• Axonal form.5,21
AMAN and AIDP are the most common subtypes of GBS.1
Symptoms, Signs, and Disease Manifestations
Limb weakness, the classic presenting symptom of GBS, is both symmetrical and ascending. Weakness can develop acutely and progress over days to weeks.2,15 Hughes and Cornblath26 also note pain, numbness, and paresthesias among the initial symptoms of GBS. Others include sensory changes, cranial nerve involvement, various autonomic changes, and respiratory or oropharyngeal weakness. Reflexes, particularly the tendon reflexes, may be diminished or absent.15,18,21 In many cases, sensory changes (ie, pain) may precede the onset of weakness, often making diagnosis difficult.15
Cranial nerves most commonly affected are V, VI, VII, X, XI, and XII, with manifestations that include dysphagia, dysarthria, diplopia, limitation to eye movements, and facial droop and weakness. Usually facial and oropharyngeal weakness occur after the extremities and trunk are affected. Blindness may occur if demyelination of the optic nerve occurs; this is seen in Miller-Fisher syndrome.10,15,25,27
In GBS, many patients report pain, which can present as bilateral sciatica or as throbbing or aching in the large muscles of the upper legs, flanks, or back.28 This pain, which results from the demyelination of the sensory nerve fibers, can be severe.10
Patients with GBS may experience manifestations of autonomic nervous system dysfunction—for example, arrhythmias, hypotension or hypertension, urinary retention, cardiomyopathy, and paralytic ileus.10,20 Dysautonomia often impedes patients’ progress in inpatient rehabilitation. Patients may have persistent problems involving postural hypotension, hypertension, excessive sympathetic outflow, or bladder and bowel dysfunction.29
Blood pressure fluctuations, often attributed to changes in catecholamine levels and disturbances in the baroreceptor reflex pathway, are common and are considered characteristic of GBS. Transient or persistent hypotension is caused by the dysregulation of the parasympathetic and sympathetic systems, with subsequent alterations in venomotor tone.3 Additionally, an increased sensitivity to catecholamine can lead to cardiovascular disturbances, resulting in denervation hypersensitivity and impairment of the carotid sinus reflex.
Arrhythmias occur in perhaps half of patients with GBS. The most common is sustained sinus tachycardia, which usually requires no treatment. Bradycardia leading to atrioventricular blocks and asystole is believed to result from afferent baroreceptor reflex failure. Treatment may be required—either administration of atropine or insertion of a pacemaker, depending on the severity of the arrhythmia.3,10
Myocardial involvement can range from asymptomatic mycocarditis to neurogenic stunned myocardium and heart failure. Patients with ECG abnormalities should undergo two-dimensional echocardiographic studies and other testing to explore cardiac involvement. Acute coronary syndromes, including ST-segment elevation MI, have been reported, in some cases associated with IVIG treatment. In one patient, coronary spasm was reported, with clean coronary arteries found on cardiac catheterization.3
Patients with GBS are at risk for compromised neuromuscular respiratory function; demyelination of the nerves that innervate the intercostal muscles and the diaphragm can result in respiratory failure. Key clinical indicators of respiratory muscle fatigue include tachypnea, diaphoresis, and asynchronous movements of the abdomen and chest;10 other symptoms relevant to respiratory or oropharyngeal weakness include slurred speech, dyspnea (with or without exertion), difficulty swallowing, and inability to cough.2,10 Serial respiratory function testing is advisable to detect patients at risk for respiratory failure.30
Diagnosis
Guillain-Barré is a syndrome diagnosed by a collection of symptoms (see Table 22,21,31), including subacute developing paralysis, symmetrical bilateral weakness beginning at onset, and diminishing to absent reflexes.21,31 Other causes for rapidly developing weaknesses should be ruled out (see Table 310,21,26,31). Lumbar puncture typically shows increased protein levels with a normal white cell count; however, neither this test nor electrophysiologic evaluation offers significant value for diagnosis of GBS.21,26,31
During the acute phase of GBS (within three weeks of onset), there is found an elevation of CSF protein (> 550 mg/L) without an elevation in white blood cells. This phenomenon, called albuminocytologic dissociation, reflects inflammation of the nerve roots and is considered the hallmark of GBS.2
MRI can also facilitate the diagnosis of GBS; it demonstrates anterior and posterior intrathecal spinal nerve roots and cauda equina.32 In patients with GBS, evidence supporting breakdown of the blood–nerve barrier can be seen in abnormal gadolinium enhancement of the intrathecal nerve roots on MRI.33
When electrophysiologic studies are performed, they typically reveal slowing nerve conduction, prolonged distal latencies, and partial motor conduction block.34 The characteristic finding of early demyelination is conduction block, a reduction in the amplitude of the muscle action potential after stimulation of the distal, as opposed to the proximal, nerve.28 Nerve conduction studies may help in the diagnosis and classification of GBS—and, to a limited extent, formulation of a prognosis. Such alternative diagnoses as myositis and myasthenia gravis may be excluded by neurophysiology.26 Early in GBS, neurophysiologic abnormalities may be very mild or occasionally normal; test results may not correlate with clinical disability.35,36
The clinician cannot depend on clinical features alone to predict respiratory decline.31 Frequent evaluations of respiratory effort, by measurement of maximal inspiratory pressures and vital capacity, should be performed at the bedside to monitor diaphragmatic strength. Respiratory ventilation should be initiated if the patient becomes hypoxic or experiences a rapid decline in vital capacity (ie, below 60% of predicted value).10 Mechanical ventilation is more likely to be required in patients with a negative inspiratory force of less than 30 cm H2O.31
Treatment
Guillain-Barré syndrome has an acute onset and progression. Patients quickly become nonambulatory and may require total ventilation due to paralysis. Therapeutic options are IVIG or plasmapheresis (plasma exchange).37-40 Corticosteroids do not appear to benefit patients with GBS.41,42
Several mechanisms appear to contribute to the effectiveness of immunoglobulin.38,39 Infused IVIG interferes with antigen presentation, inhibits antibody production, neutralizes pathologic autoantibodies, and modulates other immunologic events involved in the pathogenesis of autoimmune neuromuscular diseases, including GBS.43 Adverse reactions, which are usually minor, include headache, fever, chills, myalgia, and malaise. In rare instances, anaphylaxis or renal failure may occur.15,44
In plasmapheresis, blood is removed from the body and dialyzed, with circulating antibodies and immunoglobulins removed from the plasma; fresh frozen plasma, albumin, or saline is administered. This treatment, performed via central venous catheter, should be initiated as soon as possible after onset of symptoms but can be implemented as late as 30 days after GBS onset. Plasmapheresis requires personnel trained in dialysis, which may not be performed in all hospitals. Possible adverse events include infection and hemorrhage. Laboratory values must be monitored for hypokalemia and hypocalcemia.45,46
Supportive Care
Patients with GBS require intensive care and very close monitoring for complications of respiratory difficulty and autonomic dysfunction. Individualized programs should be initiated for patients in the acute phase of GBS, aimed at the prevention of contractures and skin breakdown.10 Exercise programs, as conducted with the case patient, should also help relieve the fatigue syndromes that accompany GBS.
Immobilization associated with bed rest incurs a risk for pulmonary emboli and DVT; this has been found true during the first 12 weeks after symptom onset in patients with GBS who remain immobile.47 The use of antiembolic hose and sequential compression devices can help reduce the risk for thrombotic events.10 Use of enoxaparin or heparin is recommended for nonambulating patients until they are able to walk, with Gaber et al47 specifying the use of low-molecular-weight heparin to reduce, but not eliminate, the risk for DVT.
The pain associated with GBS can be severe. Narcotic analgesics may be administered with careful monitoring of autonomic denervation. Long-term management of neuropathic pain may require adjuvant therapy, such as tricyclic antidepressants, gabapentin, or tramadol hydrochloride.10 According to Pandey et al,48 gabapentin alone may suffice for pain control in GBS, with minimal adverse effects. In certain rehabilitation facilities, tricyclic antidepressants, capsaicin, and transcutaneous nerve stimulation have been reported effective; during the early stages of treatment, until these treatments reach their full effect, pain medications such as tramadol or narcotics can provide temporary relief.29
More than one-half of patients with GBS in the acute phase can develop ileus. Constipation can also occur as a result of pain medication use, prolonged bed rest, and poor intake. Auscultation of bowel sounds and abdominal assessment should be performed daily to monitor for ileus. Hughes et al10 do not recommend the use of promotility drugs in patients with dysautonomia.
After hospital discharge, easy fatigability can affect work and social activities. With continued physical therapy, occupational therapy, and monitoring, however, patients with GBS can expect to return to an optimal level of functioning. Speed of recovery varies with these patients from a few months to several years, depending on such factors as age and the extent to which axonal degeneration has occurred.6,49
The Case Patient
For several weeks after discharge, the case patient continued to experience fatigue, low back pain, and general muscle pain. With her family’s support, she continued to receive outpatient physical therapy, and within one month she had regained her ankle strength. She was soon able to resume her classes, despite some lingering fatigue.
Conclusion
Guillain-Barré syndrome is a potentially life-threatening disease whose symptoms health care providers need to recognize quickly to provide prompt treatment. Supportive care for both patient and family is of key importance for maximum rehabilitation and return to the previous lifestyle. The clinical course of GBS is highly variable and difficult to predict. The patient’s outcome depends on several factors, including age and severity of illness. GBS patients can experience long-term psychosocial effects.
References
1. Magira EE, Papaioakim M, Nachamkin I, et al. Differential distribution of HLA-DQ beta/DR beta epitopes in the two forms of Guillain-Barré syndrome, acute motor axonal neuropathy and acute inflammatory demyelinating polyneuropathy (AIDP): identification of DQ beta epitopes associated with susceptibility to and protection from AIDP. J Immunol. 2003;170(6):3074-3080.
2. Tremblay ME, Closon A, D’Anjou G, Bussières JF. Guillain-Barré syndrome following H1N1 immunization in a pediatric patient. Ann Pharmacother. 2010;44(7-8):1330-1333.
3. Mukerji S, Aloka F, Farooq MU, et al. Cardiovascular complications of the Guillain-Barré syndrome. Am J Cardiol. 2009;104(10):1452-1455.
4. McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barré syndrome worldwide: a systematic literature review. Neuroepidemiology. 2009;32(2):150-163.
5. Haber P, Sejvar J, Mikaeloff Y, DeStefano F. Vaccines and Guillain-Barré syndrome. Drug Saf. 2009; 32(4):309-323.
6. van Doorn PA. What’s new in Guillain-Barré syndrome in 2007-2008? J Periph Nerv Syst. 2009;14(2):72-74.
7. van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008;7(10):939-950.
8. Chiò A, Cocito D, Leone M, et al; Piemonte and alle d’Aosta Register for Guillain-Barré Syndrome. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003; 60(7):1146-1150.
9. Hadden RD, Karch H, Hartung HP, et al. Preceding infections, immune factors, and outcome in Guillain-Barré syndrome. Neurology. 2001;56(6):758-765.
10. Hughes RA, Wijdicks EF, Benson E, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol. 2005;62(8):1194-1198.
11. Aluka KJ, Turner PL, Fullum TM. Guillain-Barré syndrome and postbariatric surgery polyneuropathies. JSLS. 2009;13(2):250-253.
12. Brannagan TH 3rd, Zhou Y. HIV-associated Guillain-Barré syndrome. J Neurol Sci. 2003;208(1-2):39-42.
13. Lin WC, Lee PI, Lu CY, et al. Mycoplasma pneumoniae encephalitis in childhood. J Microbiol Immunol Infect. 2002;35(3):173-178.
14. Sivadon-Tardy V, Orlikowski D, Porcher R, et al. Detection of Campylobacter jejuni by culture and real-time PCR in a French cohort of patients with Guillain-Barre syndrome. J Clin Microbiol. 2010;48 (6):2278-2281.
15. van Doorn PA, Kuitwaard K, Walgaard C, et al. IVIG treatment and prognosis in Guillain-Barré syndrome. J Clin Immunol. 2010;30 suppl 1:S74-S78.
16. Kaida K, Kusunoki S. Guillan-Barré syndrome: update on immunobiology and treatment. Expert Rev Neurother. 2009;9(9):1307-1319.
17. Forsberg A, Press R, Einarsson U, et al. Disability and health-related quality of life in Guillain-Barré syndrome during the first two years after onset: a prospective study. Clin Rehabil. 2005;19(8):900-909.
18. Criteria for diagnosis of Guillain-Barré syndrome. Ann Neurol. 1978;3(6):565-566.
19. van Koningsveld R, Steyerberg EW, Hughes RA, et al. A clinical progostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007;6(7):589-594.
20. Koeppen S, Kraywinkel K, Wessendorf TE, et al. Long-term outcome of Guillain-Barré syndrome. Neurocrit Care. 2006;5(3)235-242.
21. Sheridan JM, Smith D. Atypical Guillain-Barré in the emergency department. West J Emerg Med. 2010;11(1):80-82.
22. Ogawara K, Kuwabara S, Koga M, et al. Anti-GM1b IgG antibody is associated with acute motor axonal neuropathy and Campylobacter jejuni infection. J Neurol Sci. 2003;210(1-2):41-45.
23. Nagashima T, Koga M, Odaka M, et al. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Arch Neurol. 2007;64(10):1519-1523.
24. Oh SJ, LaGanke C, Claussen GC. Sensory Guillain-Barré syndrome. Neurology. 2001;56(1):82-86.
25. Aráranyi Z, Kovács T, Sipos I, Bereczki D. Miller Fisher syndrome: brief overview and update with a focus on electrophysiological findings. Eur J Neurol. 2011 Jun 1. [Epub ahead of print]
26. Hughes RA, Cornblath, DR. Guillain-Barré syndrome. Lancet. 2005;366(9497):1653-1666.
27. Snyder LA, Rismondo V, Miller NR. The Fisher variant of Guillain-Barré syndrome (Fisher syndrome). J Neuroophthalmol. 2009;29(4):312-324.
28. Ropper AH. The Guillain-Barré syndrome. N Engl J Med.1992;326(17):1130-1136.
29. Meythaler JM. Rehabilitation of Guillain-Barré syndrome. Arch Phys Med Rehabil.1997;78(8):872-879.
30. Sharshar T, Chevret S, Bourdain F, et al; French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Early predictors of mechanical ventilation in Guillain-Barré syndrome. Crit Care Med. 2003; 31(1):278-283.
31. McGillicuddy DC, Walker O, Shapiro NI, et al. Guillain-Barré syndrome in the emergency department. Ann Emerg Med. 2006;47(4):390-393.
32. Yikilmaz A, Doganay S, Gumus H, et al. Magnetic resonance imaging of childhood Guillain-Barré syndrome. Childs Nerv Syst. 2010;26(8):1103-1108.
33. Gonzalez-Quevedo A, Carriera RF, O’Farrill ZL, et al. An appraisal of blood-cerebrospinal fluid barrier dysfunction during the course of Guillain-Barré syndrome. Neurol India. 2009;57(3):288-294.
34. Abai S, Kim SB, Kim JP, Lim YJ. Guillan-Barré syndrome combined with acute cervical myelopathy. J Korean Neurosurg Soc. 2010;48(3):298-300.
35. Uncini A, Yuki N. Electrophysiologic and immunopathologic correlates in Guillain-Barré syndrome subtypes. Expert Rev Neurother. 2009;9(6):869-884.
36. Hadden RD, Hughes RA. Management of inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2003;74 suppl 2:ii9-ii14.
37. Raphaël JC, Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2002;(2):CD001798.
38. Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Jun 16; (6):CD002063.
39. Human immunoglobulin and the Guillain-Barré syndrome: new indication. An alternative to plasmapheresis. Prescrire Int. 2000;9(49):142-143.
40. van der Meché FG, Schmitz PI; Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;327(17):1123-1129.
41. Hughes RA, Swan AV, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Feb 16;(2):CD001446.
42. Hahn AF. Guillain-Barré syndrome. Lancet. 1998; 352(9128):635-641.
43. Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;291(19):2367-2375.
44. Kuitwaard K, de Gelder J, Tio-Gillen AP, et al. Pharmacokenetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann Neurol. 2009;66(5):597-603.
45. Atkinson SB, Carr RL, Maybee P, Haynes D. The challenges of managing and treating Guillain-Barré syndrome during the acute phase. Dimens Crit Care Nurs. 2006;25(6):256-263.
46. van Doorn PA. Treatment of Guillain-Barré syndrome and CIDP. J Periph Nerv Syst. 2005;10(2):113-127.
47. Gaber TA, Kirker SGB, Jenner JR. Current practice of prophylactic anticoagulation in Guillain-Barré syndrome. Clin Rehabil. 2002;16(2):190-193.
48. Pandey CK, Bose N, Garg G, et al. Gabapentin for the treatment of pain in Guillain-Barré syndrome: a double-blinded, placebo-controlled, crossover study. Anesth Analg. 2002;95(6):1719-1723.
49. de Vries JM, Hagemans ML, Bussmann JB, et al. Fatigue in neuromuscular disorders: focus on Guillain-Barré syndrome and Pompe disease. Cell Mol Life Sci. 2010;67(5):701-713.
A 20-year-old woman presented to her primary care clinic with a chief complaint of lower leg weakness and difficulty walking. The weakness she described had been worsening over the previous four days, with progressively worsening tingling and numbness of her toes bilaterally.
The day before the patient presented, she noticed numbness and paresthesia in both calves. At the time of her presentation to the clinic, she complained of low back ache, paresthesia of both hands, numbness bilaterally to her groin, difficulty sitting upright, ataxia, and a numb, thick-feeling tongue. She denied fever, neck stiffness, shortness of breath, headache, or visual changes.
The patient stated that 10 days earlier, she had developed an upper respiratory infection for which she was seen at the clinic and treated with a seven-day course of amoxicillin/clavulanate 875/125 mg twice daily. She said that she had recovered completely.
A review of the patient’s systems revealed proximal muscle weakness bilaterally (2/5) and loss of touch-pressure in the lower extremities. She was experiencing paresthesia of the hands and mild weakness bilaterally (4/5). She also walked with an ataxic gait and had reduced deep tendon reflexes in the lower limbs. All cranial nerves were intact, and her vital signs were stable.
The woman’s medical history was positive only for asthma. Her family history included ischemic stroke in the maternal grandfather and brain tumor in the paternal grandfather. Social history was positive for alcohol intake (ranging from four to 12 beers per week). The patient said she had never smoked or used illicit drugs. She was an unmarried college student, living in a dorm on campus. She participated in track at school.
The patient was admitted to the hospital telemetry step-down unit, and a neurology consultation was requested. Tests were ordered, among them MRI of the head and spine and comprehensive blood work, to rule out neurologic, infectious, or metabolic causes of the patient’s weakness; urinalysis was also obtained. These tests all yielded negative results.
A lumbar puncture performed the following day revealed a cerebrospinal fluid (CSF) protein level of 570 mg/L (normal range, 150 to 450 mg/L). Leukocytes numbered 2 cells/mm3 (normal count, 0 to 10 cells/mm3).
Based on the patient’s presentation, history, and symptoms, a neurologist made a diagnosis of Guillain-Barré syndrome. It was decided that no electromyographic (EMG) study was required to rule out other disease processes (eg, spinal cord disease, multiple sclerosis, tumors).
The patient underwent a five-dose course of immunomodulatory therapy with IV immunoglobulin (IVIG). In the step-down unit, she experienced one incident of sinus bradycardia (ie, resting heart rate between 40 and 50 beats/min). Her blood pressure remained stable, as did her respiratory status, according to peak expiratory flow measured frequently at her bedside.
Physical therapy was initiated, consisting of passive and active range of motion, crossovers with the patient’s feet, and stair training. This was done in response to a complaint of ankle weakness, and it helped to strengthen weakened muscles and improve alignment while the patient was bedridden and in a weakened, fatigued state. Additionally, the patient was given enoxaparin, wore antiembolic hose, and used sequential compression devices while in bed. As a result of these measures, she never experienced a pulmonary embolus or deep vein thrombosis (DVT) as a result of being immobilized.
By the seventh day of hospitalization, the patient had stable vital signs and improved lower limb strength, and numbness was resolving in her hands and lower extremities. She was discharged to home, with physical therapy to resume on an outpatient basis.
Discussion
Guillain-Barré syndrome (GBS), an acute immune-mediated paralytic disorder,1 manifests in the form of weakness and diminished reflexes. Affecting the peripheral nerves, GBS is characterized by progressive symmetrical ascending weakness with varying degrees of sensory complaints.2,3
GBS occurs worldwide, and incidence is estimated between 1.1 and 1.8 cases per 100,000 persons.4 In the United States, GBS can be found in all age-groups, with peak incidence noted in elderly persons and young adults.5,6 Even with treatment, 3% to 10% of patients are reported to die of this illness, and 20% cannot walk six months after symptom onset.7 In one prospective population-based study of patients with confirmed GBS, 6% of patients died within 30 days of symptom onset, often as a result of respiratory complications.8
GBS is a postinfectious disorder, with cases developing several days or weeks after a viral or bacterial illness—most commonly, an upper respiratory infection or diarrhea (see Table 19-13). The most common trigger of GBS is infection with the bacterial microorganism Campylobacter jejuni (occurring in 15% to 40% of patients with GBS),9,14 a pathogen that can produce demyelination-causing antibodies. Other responsible pathogens include cytomegalovirus and Epstein-Barr virus.9 In a process called molecular mimicry, the immune system is unable to distinguish the amino acid of an infectious organism from the proteinaceous content of the peripheral nerve.15 Subsequently, the immune system attacks and destroys the myelin sheath.
An example of this is the apparent cross-reaction of the ganglioside GM1 with C jejuni lipopolysaccharide antigens.14,15 The resulting effect is immunologic damage to the peripheral nervous system. The flaccid paralysis that occurs in patients with GBS is thought to be caused by lymphocytic infiltration and complement activation of the spinal roots and peripheral nerves, where macrophages strip the myelin.5,15,16
Stages and Variants
Three stages characterize the course of GBS. The acute phase, which lasts one to four weeks, begins with onset of symptoms and persists until the associated neurologic deterioration has ceased. During the second phase, the plateau period, symptoms persist with no further deterioration; this stage can last several days to several weeks or months. The final phase, the recovery period, can last from four months to two years after symptom onset.15,17,18
The clinical course of GBS is highly variable and in many cases difficult to predict. Certain factors have been associated with a poor outcome: advancing age, previous presence of diarrhea, need for mechanical ventilation, an extended plateau phase, and a lower patient score on the Erasmus GBS Outcome Scale,19 when measured two weeks after GBS onset.8,20 This score can help predict the patient’s chance of independent walking after six months.15,19
Although the classic presenting symptom of GBS is symmetric ascending weakness, several disease variants have been identified, with differing symptoms and degrees of recovery. These variants also differ in terms of the muscle groups affected; in some, visual defects may be present at onset. GBS variants include21:
• Acute motor axonal neuropathy (AMAN)1,22
• Acute inflammatory demyelinating polyneuropathy (AIDP)1
• Pharyngeal-cervical-brachial variant23
• Purely sensory variant24
• Miller-Fisher syndrome, which manifests with ophthalmoplegia, in addition to ataxia and areflexia25
• Axonal form.5,21
AMAN and AIDP are the most common subtypes of GBS.1
Symptoms, Signs, and Disease Manifestations
Limb weakness, the classic presenting symptom of GBS, is both symmetrical and ascending. Weakness can develop acutely and progress over days to weeks.2,15 Hughes and Cornblath26 also note pain, numbness, and paresthesias among the initial symptoms of GBS. Others include sensory changes, cranial nerve involvement, various autonomic changes, and respiratory or oropharyngeal weakness. Reflexes, particularly the tendon reflexes, may be diminished or absent.15,18,21 In many cases, sensory changes (ie, pain) may precede the onset of weakness, often making diagnosis difficult.15
Cranial nerves most commonly affected are V, VI, VII, X, XI, and XII, with manifestations that include dysphagia, dysarthria, diplopia, limitation to eye movements, and facial droop and weakness. Usually facial and oropharyngeal weakness occur after the extremities and trunk are affected. Blindness may occur if demyelination of the optic nerve occurs; this is seen in Miller-Fisher syndrome.10,15,25,27
In GBS, many patients report pain, which can present as bilateral sciatica or as throbbing or aching in the large muscles of the upper legs, flanks, or back.28 This pain, which results from the demyelination of the sensory nerve fibers, can be severe.10
Patients with GBS may experience manifestations of autonomic nervous system dysfunction—for example, arrhythmias, hypotension or hypertension, urinary retention, cardiomyopathy, and paralytic ileus.10,20 Dysautonomia often impedes patients’ progress in inpatient rehabilitation. Patients may have persistent problems involving postural hypotension, hypertension, excessive sympathetic outflow, or bladder and bowel dysfunction.29
Blood pressure fluctuations, often attributed to changes in catecholamine levels and disturbances in the baroreceptor reflex pathway, are common and are considered characteristic of GBS. Transient or persistent hypotension is caused by the dysregulation of the parasympathetic and sympathetic systems, with subsequent alterations in venomotor tone.3 Additionally, an increased sensitivity to catecholamine can lead to cardiovascular disturbances, resulting in denervation hypersensitivity and impairment of the carotid sinus reflex.
Arrhythmias occur in perhaps half of patients with GBS. The most common is sustained sinus tachycardia, which usually requires no treatment. Bradycardia leading to atrioventricular blocks and asystole is believed to result from afferent baroreceptor reflex failure. Treatment may be required—either administration of atropine or insertion of a pacemaker, depending on the severity of the arrhythmia.3,10
Myocardial involvement can range from asymptomatic mycocarditis to neurogenic stunned myocardium and heart failure. Patients with ECG abnormalities should undergo two-dimensional echocardiographic studies and other testing to explore cardiac involvement. Acute coronary syndromes, including ST-segment elevation MI, have been reported, in some cases associated with IVIG treatment. In one patient, coronary spasm was reported, with clean coronary arteries found on cardiac catheterization.3
Patients with GBS are at risk for compromised neuromuscular respiratory function; demyelination of the nerves that innervate the intercostal muscles and the diaphragm can result in respiratory failure. Key clinical indicators of respiratory muscle fatigue include tachypnea, diaphoresis, and asynchronous movements of the abdomen and chest;10 other symptoms relevant to respiratory or oropharyngeal weakness include slurred speech, dyspnea (with or without exertion), difficulty swallowing, and inability to cough.2,10 Serial respiratory function testing is advisable to detect patients at risk for respiratory failure.30
Diagnosis
Guillain-Barré is a syndrome diagnosed by a collection of symptoms (see Table 22,21,31), including subacute developing paralysis, symmetrical bilateral weakness beginning at onset, and diminishing to absent reflexes.21,31 Other causes for rapidly developing weaknesses should be ruled out (see Table 310,21,26,31). Lumbar puncture typically shows increased protein levels with a normal white cell count; however, neither this test nor electrophysiologic evaluation offers significant value for diagnosis of GBS.21,26,31
During the acute phase of GBS (within three weeks of onset), there is found an elevation of CSF protein (> 550 mg/L) without an elevation in white blood cells. This phenomenon, called albuminocytologic dissociation, reflects inflammation of the nerve roots and is considered the hallmark of GBS.2
MRI can also facilitate the diagnosis of GBS; it demonstrates anterior and posterior intrathecal spinal nerve roots and cauda equina.32 In patients with GBS, evidence supporting breakdown of the blood–nerve barrier can be seen in abnormal gadolinium enhancement of the intrathecal nerve roots on MRI.33
When electrophysiologic studies are performed, they typically reveal slowing nerve conduction, prolonged distal latencies, and partial motor conduction block.34 The characteristic finding of early demyelination is conduction block, a reduction in the amplitude of the muscle action potential after stimulation of the distal, as opposed to the proximal, nerve.28 Nerve conduction studies may help in the diagnosis and classification of GBS—and, to a limited extent, formulation of a prognosis. Such alternative diagnoses as myositis and myasthenia gravis may be excluded by neurophysiology.26 Early in GBS, neurophysiologic abnormalities may be very mild or occasionally normal; test results may not correlate with clinical disability.35,36
The clinician cannot depend on clinical features alone to predict respiratory decline.31 Frequent evaluations of respiratory effort, by measurement of maximal inspiratory pressures and vital capacity, should be performed at the bedside to monitor diaphragmatic strength. Respiratory ventilation should be initiated if the patient becomes hypoxic or experiences a rapid decline in vital capacity (ie, below 60% of predicted value).10 Mechanical ventilation is more likely to be required in patients with a negative inspiratory force of less than 30 cm H2O.31
Treatment
Guillain-Barré syndrome has an acute onset and progression. Patients quickly become nonambulatory and may require total ventilation due to paralysis. Therapeutic options are IVIG or plasmapheresis (plasma exchange).37-40 Corticosteroids do not appear to benefit patients with GBS.41,42
Several mechanisms appear to contribute to the effectiveness of immunoglobulin.38,39 Infused IVIG interferes with antigen presentation, inhibits antibody production, neutralizes pathologic autoantibodies, and modulates other immunologic events involved in the pathogenesis of autoimmune neuromuscular diseases, including GBS.43 Adverse reactions, which are usually minor, include headache, fever, chills, myalgia, and malaise. In rare instances, anaphylaxis or renal failure may occur.15,44
In plasmapheresis, blood is removed from the body and dialyzed, with circulating antibodies and immunoglobulins removed from the plasma; fresh frozen plasma, albumin, or saline is administered. This treatment, performed via central venous catheter, should be initiated as soon as possible after onset of symptoms but can be implemented as late as 30 days after GBS onset. Plasmapheresis requires personnel trained in dialysis, which may not be performed in all hospitals. Possible adverse events include infection and hemorrhage. Laboratory values must be monitored for hypokalemia and hypocalcemia.45,46
Supportive Care
Patients with GBS require intensive care and very close monitoring for complications of respiratory difficulty and autonomic dysfunction. Individualized programs should be initiated for patients in the acute phase of GBS, aimed at the prevention of contractures and skin breakdown.10 Exercise programs, as conducted with the case patient, should also help relieve the fatigue syndromes that accompany GBS.
Immobilization associated with bed rest incurs a risk for pulmonary emboli and DVT; this has been found true during the first 12 weeks after symptom onset in patients with GBS who remain immobile.47 The use of antiembolic hose and sequential compression devices can help reduce the risk for thrombotic events.10 Use of enoxaparin or heparin is recommended for nonambulating patients until they are able to walk, with Gaber et al47 specifying the use of low-molecular-weight heparin to reduce, but not eliminate, the risk for DVT.
The pain associated with GBS can be severe. Narcotic analgesics may be administered with careful monitoring of autonomic denervation. Long-term management of neuropathic pain may require adjuvant therapy, such as tricyclic antidepressants, gabapentin, or tramadol hydrochloride.10 According to Pandey et al,48 gabapentin alone may suffice for pain control in GBS, with minimal adverse effects. In certain rehabilitation facilities, tricyclic antidepressants, capsaicin, and transcutaneous nerve stimulation have been reported effective; during the early stages of treatment, until these treatments reach their full effect, pain medications such as tramadol or narcotics can provide temporary relief.29
More than one-half of patients with GBS in the acute phase can develop ileus. Constipation can also occur as a result of pain medication use, prolonged bed rest, and poor intake. Auscultation of bowel sounds and abdominal assessment should be performed daily to monitor for ileus. Hughes et al10 do not recommend the use of promotility drugs in patients with dysautonomia.
After hospital discharge, easy fatigability can affect work and social activities. With continued physical therapy, occupational therapy, and monitoring, however, patients with GBS can expect to return to an optimal level of functioning. Speed of recovery varies with these patients from a few months to several years, depending on such factors as age and the extent to which axonal degeneration has occurred.6,49
The Case Patient
For several weeks after discharge, the case patient continued to experience fatigue, low back pain, and general muscle pain. With her family’s support, she continued to receive outpatient physical therapy, and within one month she had regained her ankle strength. She was soon able to resume her classes, despite some lingering fatigue.
Conclusion
Guillain-Barré syndrome is a potentially life-threatening disease whose symptoms health care providers need to recognize quickly to provide prompt treatment. Supportive care for both patient and family is of key importance for maximum rehabilitation and return to the previous lifestyle. The clinical course of GBS is highly variable and difficult to predict. The patient’s outcome depends on several factors, including age and severity of illness. GBS patients can experience long-term psychosocial effects.
References
1. Magira EE, Papaioakim M, Nachamkin I, et al. Differential distribution of HLA-DQ beta/DR beta epitopes in the two forms of Guillain-Barré syndrome, acute motor axonal neuropathy and acute inflammatory demyelinating polyneuropathy (AIDP): identification of DQ beta epitopes associated with susceptibility to and protection from AIDP. J Immunol. 2003;170(6):3074-3080.
2. Tremblay ME, Closon A, D’Anjou G, Bussières JF. Guillain-Barré syndrome following H1N1 immunization in a pediatric patient. Ann Pharmacother. 2010;44(7-8):1330-1333.
3. Mukerji S, Aloka F, Farooq MU, et al. Cardiovascular complications of the Guillain-Barré syndrome. Am J Cardiol. 2009;104(10):1452-1455.
4. McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barré syndrome worldwide: a systematic literature review. Neuroepidemiology. 2009;32(2):150-163.
5. Haber P, Sejvar J, Mikaeloff Y, DeStefano F. Vaccines and Guillain-Barré syndrome. Drug Saf. 2009; 32(4):309-323.
6. van Doorn PA. What’s new in Guillain-Barré syndrome in 2007-2008? J Periph Nerv Syst. 2009;14(2):72-74.
7. van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008;7(10):939-950.
8. Chiò A, Cocito D, Leone M, et al; Piemonte and alle d’Aosta Register for Guillain-Barré Syndrome. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003; 60(7):1146-1150.
9. Hadden RD, Karch H, Hartung HP, et al. Preceding infections, immune factors, and outcome in Guillain-Barré syndrome. Neurology. 2001;56(6):758-765.
10. Hughes RA, Wijdicks EF, Benson E, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol. 2005;62(8):1194-1198.
11. Aluka KJ, Turner PL, Fullum TM. Guillain-Barré syndrome and postbariatric surgery polyneuropathies. JSLS. 2009;13(2):250-253.
12. Brannagan TH 3rd, Zhou Y. HIV-associated Guillain-Barré syndrome. J Neurol Sci. 2003;208(1-2):39-42.
13. Lin WC, Lee PI, Lu CY, et al. Mycoplasma pneumoniae encephalitis in childhood. J Microbiol Immunol Infect. 2002;35(3):173-178.
14. Sivadon-Tardy V, Orlikowski D, Porcher R, et al. Detection of Campylobacter jejuni by culture and real-time PCR in a French cohort of patients with Guillain-Barre syndrome. J Clin Microbiol. 2010;48 (6):2278-2281.
15. van Doorn PA, Kuitwaard K, Walgaard C, et al. IVIG treatment and prognosis in Guillain-Barré syndrome. J Clin Immunol. 2010;30 suppl 1:S74-S78.
16. Kaida K, Kusunoki S. Guillan-Barré syndrome: update on immunobiology and treatment. Expert Rev Neurother. 2009;9(9):1307-1319.
17. Forsberg A, Press R, Einarsson U, et al. Disability and health-related quality of life in Guillain-Barré syndrome during the first two years after onset: a prospective study. Clin Rehabil. 2005;19(8):900-909.
18. Criteria for diagnosis of Guillain-Barré syndrome. Ann Neurol. 1978;3(6):565-566.
19. van Koningsveld R, Steyerberg EW, Hughes RA, et al. A clinical progostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007;6(7):589-594.
20. Koeppen S, Kraywinkel K, Wessendorf TE, et al. Long-term outcome of Guillain-Barré syndrome. Neurocrit Care. 2006;5(3)235-242.
21. Sheridan JM, Smith D. Atypical Guillain-Barré in the emergency department. West J Emerg Med. 2010;11(1):80-82.
22. Ogawara K, Kuwabara S, Koga M, et al. Anti-GM1b IgG antibody is associated with acute motor axonal neuropathy and Campylobacter jejuni infection. J Neurol Sci. 2003;210(1-2):41-45.
23. Nagashima T, Koga M, Odaka M, et al. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Arch Neurol. 2007;64(10):1519-1523.
24. Oh SJ, LaGanke C, Claussen GC. Sensory Guillain-Barré syndrome. Neurology. 2001;56(1):82-86.
25. Aráranyi Z, Kovács T, Sipos I, Bereczki D. Miller Fisher syndrome: brief overview and update with a focus on electrophysiological findings. Eur J Neurol. 2011 Jun 1. [Epub ahead of print]
26. Hughes RA, Cornblath, DR. Guillain-Barré syndrome. Lancet. 2005;366(9497):1653-1666.
27. Snyder LA, Rismondo V, Miller NR. The Fisher variant of Guillain-Barré syndrome (Fisher syndrome). J Neuroophthalmol. 2009;29(4):312-324.
28. Ropper AH. The Guillain-Barré syndrome. N Engl J Med.1992;326(17):1130-1136.
29. Meythaler JM. Rehabilitation of Guillain-Barré syndrome. Arch Phys Med Rehabil.1997;78(8):872-879.
30. Sharshar T, Chevret S, Bourdain F, et al; French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Early predictors of mechanical ventilation in Guillain-Barré syndrome. Crit Care Med. 2003; 31(1):278-283.
31. McGillicuddy DC, Walker O, Shapiro NI, et al. Guillain-Barré syndrome in the emergency department. Ann Emerg Med. 2006;47(4):390-393.
32. Yikilmaz A, Doganay S, Gumus H, et al. Magnetic resonance imaging of childhood Guillain-Barré syndrome. Childs Nerv Syst. 2010;26(8):1103-1108.
33. Gonzalez-Quevedo A, Carriera RF, O’Farrill ZL, et al. An appraisal of blood-cerebrospinal fluid barrier dysfunction during the course of Guillain-Barré syndrome. Neurol India. 2009;57(3):288-294.
34. Abai S, Kim SB, Kim JP, Lim YJ. Guillan-Barré syndrome combined with acute cervical myelopathy. J Korean Neurosurg Soc. 2010;48(3):298-300.
35. Uncini A, Yuki N. Electrophysiologic and immunopathologic correlates in Guillain-Barré syndrome subtypes. Expert Rev Neurother. 2009;9(6):869-884.
36. Hadden RD, Hughes RA. Management of inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2003;74 suppl 2:ii9-ii14.
37. Raphaël JC, Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2002;(2):CD001798.
38. Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Jun 16; (6):CD002063.
39. Human immunoglobulin and the Guillain-Barré syndrome: new indication. An alternative to plasmapheresis. Prescrire Int. 2000;9(49):142-143.
40. van der Meché FG, Schmitz PI; Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;327(17):1123-1129.
41. Hughes RA, Swan AV, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Feb 16;(2):CD001446.
42. Hahn AF. Guillain-Barré syndrome. Lancet. 1998; 352(9128):635-641.
43. Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;291(19):2367-2375.
44. Kuitwaard K, de Gelder J, Tio-Gillen AP, et al. Pharmacokenetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann Neurol. 2009;66(5):597-603.
45. Atkinson SB, Carr RL, Maybee P, Haynes D. The challenges of managing and treating Guillain-Barré syndrome during the acute phase. Dimens Crit Care Nurs. 2006;25(6):256-263.
46. van Doorn PA. Treatment of Guillain-Barré syndrome and CIDP. J Periph Nerv Syst. 2005;10(2):113-127.
47. Gaber TA, Kirker SGB, Jenner JR. Current practice of prophylactic anticoagulation in Guillain-Barré syndrome. Clin Rehabil. 2002;16(2):190-193.
48. Pandey CK, Bose N, Garg G, et al. Gabapentin for the treatment of pain in Guillain-Barré syndrome: a double-blinded, placebo-controlled, crossover study. Anesth Analg. 2002;95(6):1719-1723.
49. de Vries JM, Hagemans ML, Bussmann JB, et al. Fatigue in neuromuscular disorders: focus on Guillain-Barré syndrome and Pompe disease. Cell Mol Life Sci. 2010;67(5):701-713.
Elderly Man with Headaches and Weakness
ANSWER
The chest radiograph demonstrates evidence of previous sternotomy. No evidence of acute infiltrate is noted.
However, there is a prominence within the left hilar region. This finding is strongly suggestive of neoplasm until proven otherwise. The patient was promptly referred for CT of the chest, abdomen, and pelvis, which confirmed the lesion. Subsequent CT-guided biopsy was performed.
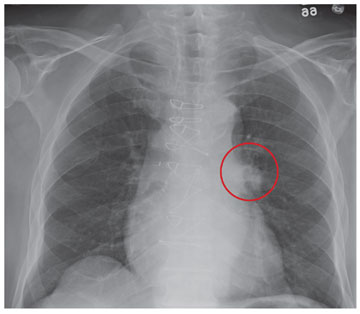
ANSWER
The chest radiograph demonstrates evidence of previous sternotomy. No evidence of acute infiltrate is noted.
However, there is a prominence within the left hilar region. This finding is strongly suggestive of neoplasm until proven otherwise. The patient was promptly referred for CT of the chest, abdomen, and pelvis, which confirmed the lesion. Subsequent CT-guided biopsy was performed.
ANSWER
The chest radiograph demonstrates evidence of previous sternotomy. No evidence of acute infiltrate is noted.
However, there is a prominence within the left hilar region. This finding is strongly suggestive of neoplasm until proven otherwise. The patient was promptly referred for CT of the chest, abdomen, and pelvis, which confirmed the lesion. Subsequent CT-guided biopsy was performed.
A 71-year-old man presents with complaints of headaches and weakness that have been ongoing for almost a month. He denies any fever, nausea, or vomiting. He has noticed an occasional cough and denies any weight loss. The patient has an extensive history of coronary artery disease, hypertension, and hyperlipidemia. History is also significant for coronary artery bypass grafting. He denies any history of smoking. The man is afebrile, and the rest of his vital signs, including pulse oximetry, are within normal limits. Physical exam shows an elderly, ill-appearing man in no obvious distress. Breath sounds bilaterally are clear. You order a chest radiograph along with some bloodwork. The chest radiograph is shown. What is your impression?
ST-segment depression and T-wave inversion: Classification, differential diagnosis, and caveats
Depression of the ST segment and inversion of the T wave are common electrocardiographic abnormalities. Knowing the various ischemic and nonischemic morphologic features is critical for a timely diagnosis of high-risk myocardial ischemia and electrolyte- or drug-related abnormalities. Moreover, it is important to recognize that true posterior infarction or subtle ST-segment elevation infarction may masquerade as ST-segment depression ischemia, and that pulmonary embolism may masquerade as anterior ischemia. These common electrocardiographic abnormalities are summarized in Table 1.
THE ST SEGMENT AND THE T WAVE: A PRIMER

The ST segment corresponds to the plateau phase of ventricular repolarization (phase 2 of the action potential), while the T wave corresponds to the phase of rapid ventricular repolarization (phase 3). ST-segment or T-wave changes may be secondary to abnormalities of depolarization, ie, pre-excitation or abnormalities of QRS voltage or duration.
On the other hand, ST-segment and T-wave abnormalities may be unrelated to any QRS abnormality, in which case they are called primary repolarization abnormalities. These are caused by ischemia, pericarditis, myocarditis, drugs (digoxin, antiarrhythmic drugs), and electrolyte abnormalities, particularly potassium abnormalities.
ST-segment deviation is usually measured at its junction with the end of the QRS complex, ie, the J point, and is referenced against the TP or PR segment.1 But some prefer to measure the magnitude of the ST-segment deviation 40 to 80 ms after the J point, when all myocardial fibers are expected to have reached the same level of membrane potential and to form an isoelectric ST segment; at the very onset of repolarization, small differences in membrane potential may normally be seen and may cause deviation of the J point and of the early portion of the ST segment.2
Although a diagnosis of ST-segment elevation myocardial infarction (STEMI) that mandates emergency reperfusion therapy requires ST-segment elevation greater than 1 mm in at least two contiguous leads,3 any ST-segment depression or elevation (≥ 0.5 mm, using the usual standard of 1.0 mV = 10 mm) may be abnormal, particularly when the clinical context or the shape of the ST segment suggests ischemia, or when other ischemic signs such as T-wave abnormalities, Q waves, or reciprocal ST-segment changes are concomitantly present. On the other hand, ST-segment depression of up to 0.5 mm in leads V2 and V3 and 1 mm in the other leads may be normal.1
In adults, the T wave normally is inverted in lead aVR; is upright or inverted in leads aVL, III, and V1; and is upright in leads I, II, aVF, and V2 through V6. The T wave is considered inverted when it is deeper than 1 mm; it is considered flat when its peak amplitude is between 1.0 mm and −1.0 mm.1
As we will discuss, certain features allow the various causes of ST-segment and T-wave abnormalities to be distinguished from one another.
SECONDARY ST-SEGMENT AND T-WAVE ABNORMALITIES
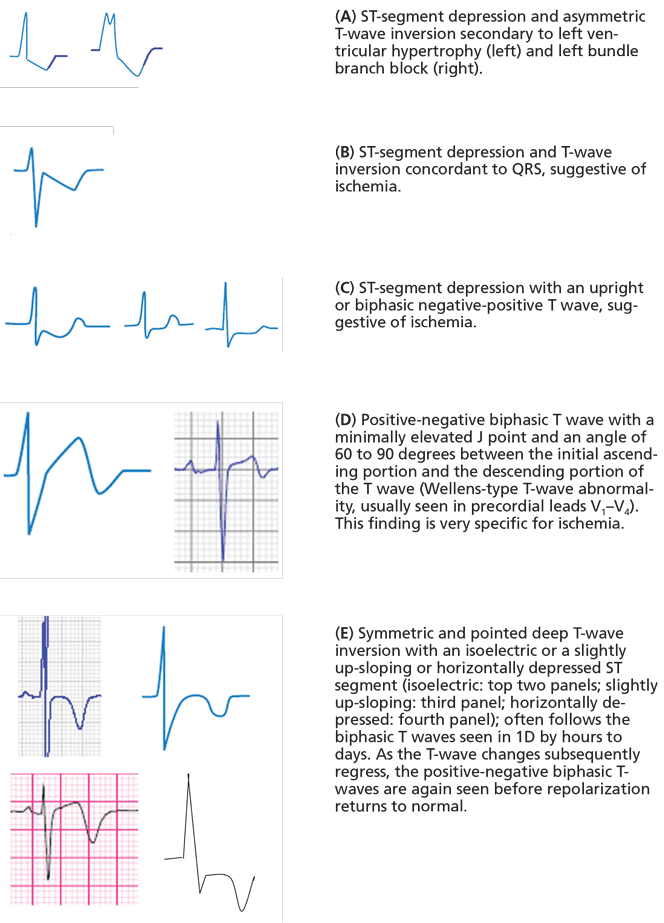
- The ST segment and T wave are directed opposite to the QRS: this is called discordance between the QRS complex and the ST-T abnormalities. In the case of right bundle branch block, the ST and T are directed opposite to the terminal portion of the QRS, ie, the part of the QRS deformed by the conduction abnormality.
- The ST segment and T wave are both abnormal and deviate in the same direction, ie, the ST segment is down-sloping and the T wave is inverted in leads with an upright QRS complex, which gives the ST-T complex a “reverse checkmark” asymmetric morphology.
- The ST and T abnormalities are not dynamic, ie, they do not change in the course of several hours to several days.
Thus, in cases of left ventricular hypertrophy or left bundle branch block, since the QRS complex is upright in the left lateral leads I, aVL, V5, and V6, the ST segment is characteristically depressed and the T wave is inverted in these leads (Figure 2). In cases of right ventricular hypertrophy or right bundle branch block, T waves are characteristically inverted in the right precordial leads V1, V2, and V3.
Left bundle branch block is always associated with secondary ST-T abnormalities, the absence of which suggests associated ischemia. Left and right ventricular hypertrophy, on the other hand, are not always associated with ST-T abnormalities, but when these are present, they correlate with more severe hypertrophy or ventricular systolic dysfunction,4 and have been called strain pattern. In addition, while these morphologic features are consistent with secondary abnormalities, they do not rule out ischemia in a patient with angina.
Some exceptions to these typical morphologic features:
- Right ventricular hypertrophy and right bundle branch block may be associated with isolated T-wave inversion without ST-segment depression in precordial leads V1, V2, and V3.
- Left ventricular hypertrophy may be associated with symmetric T-wave inversion without ST-segment depression or with a horizontally depressed ST segment. This may be the case in up to one-third of ST-T abnormalities secondary to left ventricular hypertrophy and is seen in hypertrophic cardiomyopathy, particularly the apical variant, in leads V3 through V6.5
ISCHEMIC ST-SEGMENT DEPRESSION, T-WAVE INVERSION, OR BOTH
ST-segment depression or T-wave inversion is consistent with ischemia if any of the following is true:
- The ST-segment depression or T-wave inversion is directed in the same direction as the QRS complex: this is called concordance between the QRS complex and the ST or T abnormality (Figure 1B).
- The ST segment is depressed but the T wave is upright (Figure 1C).
- The T wave has a positive-negative biphasic pattern (Figure 1D).
- The T wave is symmetrically inverted and has a pointed configuration, while the ST segment is not deviated or is upwardly bowed (coved) or horizontally depressed (Figure 1E).
- The magnitude of ST-segment depression progresses or regresses on serial tracings, or ST-segment depression progresses to T-wave abnormality during ischemia-free intervals (dynamic ST-segment depression).
Unlike ST-segment elevation, ST-segment depression does not localize ischemia.6 However, the extent and the magnitude of ST-segment depression correlate with the extent and the severity of ischemia. In fact, ST-segment depression in eight or more leads, combined with ST-segment elevation in leads aVR and V1 and occurring during ischemic pain, is associated with a 75% predictive accuracy for left main coronary artery or three-vessel disease (Figure 3).7,8 This finding may also be seen in cases of tight proximal stenosis of the left anterior descending coronary artery.9
Wellens syndrome
Wellens and his colleagues showed that 75% of patients who developed these T-wave abnormalities and who were treated medically without angiographic investigation went on to develop extensive anterior wall myocardial infarction within a mean of 8.5 days.10
In a later investigation of 1,260 patients presenting with unstable angina, 180 patients (14%) had this characteristic T-wave pattern.11 All of the latter patients had stenosis of 50% or more in the proximal left anterior descending artery, and 18% had total occlusion of the left anterior descending artery.
Thus, although medical management may provide symptomatic improvement at first, early coronary angiography and revascularization should be strongly considered in anyone with Wellens syndrome because it usually predicts impending anterior myocardial infarction.
Wellens syndrome is characterized by two patterns of T-wave changes. In 75% of cases, T waves are deeply (≥ 5 mm) and symmetrically inverted in leads V2 through V4 (Figures 1E, 4B). In 25% of cases, the T wave has a characteristic positive-negative biphasic morphology in leads V2 through V4 (Figures 1D, 4A).10 In both patterns, the ST segment is isoelectric or minimally elevated (< 1 mm) with a straight or convex morphology, the down-slope of the T wave is sharp, and the QT interval is often prolonged. These abnormalities are characteristically seen hours to days after the ischemic chest pain resolves. In fact, the ischemic episode is usually associated with transient ST-segment elevation or depression that progresses to the T-wave abnormality after the pain subsides.11
In Wellens’ original description, only 12% of patients had increases in their creatine kinase levels, and these were small. Therefore, the electrocardiogram may be the only indication of an impending large anterior infarction in a chest-pain-free patient.12
T waves that are symmetrically but less deeply inverted than Wellens-type T waves may still represent ischemia. However, this finding is less specific for ischemia and is associated with better outcomes than Wellens syndrome or ST-segment deviation, particularly when the T wave is less than 3 mm deep.14 In fact, one prospective cohort study found that isolated mild T-wave inversion in patients presenting with acute coronary syndrome is associated with a favorable long-term outcome, similar to that in patients with no electrocardiographic changes.15
FREQUENTLY MISSED DIAGNOSES MANIFESTING AS ST-SEGMENT DEPRESSION OR T-WAVE INVERSION
True posterior ST-segment elevation myocardial infarction
When accompanied by inferior STEMI, posterior infarction is easily recognized, but it can be difficult to diagnose when it occurs alone, the so-called true posterior STEMI.
In most cases of posterior infarction, the posterior chest leads V7, V8, and V9 reveal ST-segment elevation.19 One study found that ST-segment depression in the anterior precordial leads was as sensitive as ST-segment elevation in leads V7 through V9 in identifying posterior myocardial infarction (sensitivity 80%),20 while other studies found that ST-segment deviation on standard 12-lead electrocardiography has a lower sensitivity (about 60%) in identifying posterior infarction.18,21
Tall or wide (≥ 0.04-s) R waves in leads V1 or V2, particularly when associated with upright T waves, suggest posterior infarction and may further corroborate this diagnosis, but this finding may take up to 24 hours to manifest and is seen in only about 50% of patients with posterior infarction.21
Studies have shown that ST-segment elevation on standard 12-lead electrocardiography is found in fewer than 50% of patients with acute left circumflex occlusion and inferoposterior infarction,18 yet these are cases of “missed” STEMI that indeed benefit from emergency angiography and reperfusion. In addition, studies of non–ST-segment elevation acute coronary syndrome consistently identify patients who have epicardial vessel occlusion (about 15%–20% of cases),18 yet their initial angiography is usually delayed for hours or days after the initial presentation.
A subgroup analysis from TRITON–TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel Thrombolysis in Myocardial Infarction 38) evaluated patients with isolated anterior ST-segment depression. An occluded “culprit” artery was found 26% of the time, most often the left circumflex artery. Moreover, those patients had a significantly higher rate of death or myocardial infarction at 30-day follow-up than patients without a culprit artery, probably related to delayed revascularization.22
Recognizing that ST-segment depression that is greatest in leads V1, V2, or V3 represents posterior infarction helps identify a portion of the missed STEMIs in a timely fashion. In addition, in cases of anterior ST-segment depression and in cases of chest pain with nondiagnostic electrocardiography, the recording of ST elevation in leads V7, V8, and V9 is highly sensitive for detecting a true posterior injury.
Acute pulmonary embolism
An anterior ischemic pattern of symmetric T-wave inversion in the precordial leads V1 through V4 may also be a sign of acute or chronic right ventricular strain, particularly acute pulmonary embolism. Sinus tachycardia is usually present, but other signs of pulmonary embolism, such as right ventricular hypertrophy and right bundle branch block, may be absent. In fact, T-wave inversion in leads V1 through V4 is noted in 19% of patients with nonmassive pulmonary embolism and in 85% of patients with massive pulmonary embolism, and is the most sensitive and specific electrocardiographic finding in massive pulmonary embolism.23
In addition, acute pulmonary embolism may be associated with T-wave inversion in leads III and aVF,24 and changes of concomitant anterior and inferior ischemia should always raise the question of this diagnosis.
In one retrospective study of patients with acute pulmonary embolism, nonspecific ST-segment or T-wave changes were the most common finding on electrocardiography, noted in 49%.25 Rapid regression of these changes on serial tracings favors pulmonary embolism rather than myocardial infarction.
ST-segment depression reciprocal to a subtle ST-segment elevation
When ST-segment elevation occurs in two contiguous standard leads while ST-segment depression occurs in other leads, and when the ST-segment and T-wave abnormalities are ischemic rather than secondary to depolarization abnormalities, ST-segment elevation is considered the primary ischemic abnormality whereas ST-segment depression is often considered a reciprocal “mirror image” change. This “reciprocal” change may also represent remote ischemia in a distant territory in patients with multivessel coronary disease.26,27
Reciprocal ST-segment depression is present in all patients with inferior myocardial infarction and in 70% of patients with anterior myocardial infarction.28
Hypokalemia and digitalis effect
DIFFUSE (GLOBAL) T-WAVE INVERSION
Walder and Spodick36 have found this pattern to be caused most often by myocardial ischemia or neurologic events, particularly intracranial hemorrhage, and it seems more prevalent in women. Other causes include hypertrophic cardiomyopathy, stress-induced cardiomyopathy (takotsubo cardiomyopathy), cocaine abuse, pericarditis, pulmonary embolism, and advanced or complete atrioventricular block.36,37
The prognosis in patients with global T-wave inversion is determined by the underlying disease, and the striking T-wave changes per se do not imply a poor prognosis.38
OTHER CAUSES OF T-WAVE INVERSION OR ST-SEGMENT DEPRESSION
Various other entities may cause T-wave inversion, notably acute pericarditis or myocarditis, 41,42 memory T-wave phenomenon,43,44 and normal variants of repolarization (Table 1, Figure 9).45 Additionally, a nonpathologic junctional ST-segment depression may be seen in tachycardia (Figure 10).
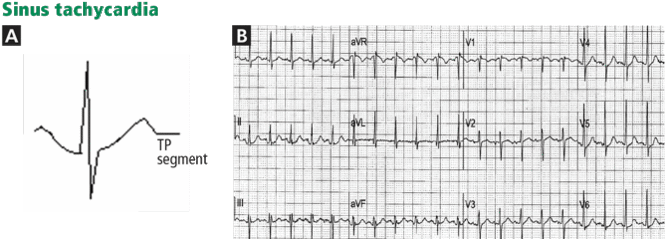
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Surawicz B, Knilans TK. Non-Q wave myocardial infarction, unstable angina pectoris, myocardial ischemia. In: Chou's Electrocardiography in Clinical Practice: Adult and Pediatric. 5th ed. Philadelphia: WB Saunders; 2001:194–207.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Okin PM, Devereux RB, Nieminen MS, et al; LIFE Study Investigators. Electrocardiographic strain pattern and prediction of new-onset congestive heart failure in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study. Circulation 2006; 113:67–73.
- Huwez FU, Pringle SD, Macfarlane PW. Variable patterns of ST-T abnormalities in patients with left ventricular hypertrophy and normal coronary arteries. Br Heart J 1992; 67:304–307.
- Li D, Li CY, Yong AC, Kilpatrick D. Source of electrocardiographic ST changes in subendocardial ischemia. Circ Res 1998; 82:957–970.
- Gorgels AP, Vos MA, Mulleneers R, de Zwaan C, Bär FW, Wellens HJ. Value of the electrocardiogram in diagnosing the number of severely narrowed coronary arteries in rest angina pectoris. Am J Cardiol 1993; 72:999–1003.
- Glancy DL. Electrocardiographic diagnosis of acute myocardial infarction. J La State Med Soc 2002; 154:66–75.
- Yamaji H, Iwasaki K, Kusachi S, et al. Prediction of acute left main coronary artery obstruction by 12-lead electrocardiography. ST segment elevation in lead aVR with less ST segment elevation in lead V(1). J Am Coll Cardiol 2001; 38:1348–1354.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- Lilaonitkul M, Robinson K, Roberts M. Wellens’ syndrome: significance of ECG pattern recognition in the emergency department. Emerg Med J 2009; 26:750–751.
- Glancy DL, Khuri B, Cospolich B. Heed the warning: Wellens’ type T-wave inversion is caused by proximal left anterior descending lesion. Proc (Bayl Univ Med Cent) 2000; 13:416–418.
- Savonitto S, Ardissino D, Granger CB, et al. Prognostic value of the admission electrocardiogram in acute coronary syndromes. JAMA 1999; 281:707–713.
- Mueller C, Neumann FJ, Perach W, Perruchoud AP, Buettner HJ. Prognostic value of the admission electrocardiogram in patients with unstable angina/non-ST-segment elevation myocardial infarction treated with very early revascularization. Am J Med 2004; 117:145–150.
- Boden WE, Spodick DH. Diagnostic significance of precordial ST-segment depression. Am J Cardiol 1989; 63:358–361.
- Shah A, Wagner GS, Green CL, et al. Electrocardiographic differentiation of the ST-segment depression of acute myocardial injury due to the left circumflex artery occlusion from that of myocardial ischemia of nonocclusive etiologies. Am J Cardiol 1997; 80:512–513.
- Krishnaswamy A, Lincoff AM, Menon V. Magnitude and consequences of missing the acute infarct-related circumflex artery. Am Heart J 2009; 158:706–712.
- Matetzky S, Freimark D, Feinberg MS, et al. Acute myocardial infarction with isolated ST-segment elevation in posterior chest leads V7-9: “hidden” ST-segment elevations revealing acute posterior infarction. J Am Coll Cardiol 1999; 34:748–753.
- Matetzky S, Freimark D, Chouraqui P, et al. Significance of ST segment elevations in posterior chest leads (V7 to V9) in patients with acute inferior myocardial infarction: application for thrombolytic therapy. J Am Coll Cardiol 1998; 31:506–511.
- Huey BL, Beller GA, Kaiser DL, Gibson RS. A comprehensive analysis of myocardial infarction due to left circumflex artery occlusion: comparison with infarction due to right coronary artery and left anterior descending artery occlusion. J Am Coll Cardiol 1988; 12:1156–1166.
- Gibson CM, Pride YB, Mohanavelu S, Wiviott SD, Antman EM, Braunwald E. Abstract 1999: Angiographic and clinical outcomes among patients with acute coronary syndrome presenting with isolated anterior ST-segment depressions. Circulation 2008; 118:S–654.
- Ferrari E, Imbert A, Chevalier T, Mihoubi A, Morand P, Baudouy M. The ECG in pulmonary embolism. Predictive value of negative T waves in precordial leads—80 case reports. Chest 1997; 111:537–543.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Norell MS, Lyons JP, Gardener JE, Layton CA, Balcon R. Significance of “reciprocal” ST segment depression: left ventriculographic observations during left anterior descending coronary angioplasty. J Am Coll Cardiol 1989; 13:1270–1274.
- Haraphongse M, Tanomsup S, Jugdutt BI. Inferior ST segment depression during acute anterior myocardial infarction: clinical and angiographic correlations. J Am Coll Cardiol 1984; 4:467–476.
- Surawicz B, Knilans TK. Acute ischemia: electrocardiographic patterns. In: Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric. 5th edition. Philadelphia: WB Saunders; 2001:122–153.
- Wagner GS, Macfarlane P, Wellens H, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part VI: acute ischemia/infarction: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:1003–1011.
- Brady WJ, Perron AD, Syverud SA, et al. Reciprocal ST segment depression: impact on the electrocardiographic diagnosis of ST segment elevation acute myocardial infarction. Am J Emerg Med 2002; 20:35–38.
- Surawicz B. Electrolytes and the electrocardiogram. Postgrad Med 1974; 55:123–129.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Glancy DL, Wang WL. ECG of the month. Abnormal electrocardiogram in a woman with a urinary tract infection. Sinus rhythm, rate 82/minute. Sagging ST segments, low T waves, and prominent U waves suggest hypokalemia. J La State Med Soc 2007; 159:5–7.
- Surawicz B, Braun HA, Crum WB, Kemp RL, Wagner S, Bellet S. Quantitative analysis of the electrocardiographic pattern of hypopotassemia. Circulation 1957; 16:750–763.
- Glancy DL, Rochon BJ, Ilie CC, Parker JM, Jones MB, Atluri P. Global T-wave inversion in a 77-year-old woman. Proc (Bayl Univ Med Cent) 2009; 22:81–82.
- Walder LA, Spodick DH. Global T wave inversion. J Am Coll Cardiol 1991; 17:1479–1485.
- Lui CY. Acute pulmonary embolism as the cause of global T wave inversion and QT prolongation. A case report. J Electrocardiol 1993; 26:91–95.
- Walder LA, Spodick DH. Global T wave inversion: long-term followup. J Am Coll Cardiol 1993; 21:1652–1656.
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Wittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352:539–548.
- Spodick DH. Electrocardiogram in acute pericarditis. Distributions of morphologic and axial changes by stages. Am J Cardiol 1974; 33:470–474.
- Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
- Rosenbaum MB, Blanco HH, Elizari MV, Lázzari JO, Davidenko JM. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol 1982; 50:213–222.
- Paparella N, Ouyang F, Fuca G, Kuck KH, Cappato R, Alboni P. Significance of newly acquired negative T waves after interruption of paroxysmal reentrant supraventricular tachycardia with narrow QRS complex. Am J Cardiol 2000; 85:261–263.
- Kaid KA, Maqsood A, Cohen M, Rothfeld E. Further characterization of the “persistent juvenile T-wave pattern” in adults. J Electrocardiol 2008; 41:644–645.
Depression of the ST segment and inversion of the T wave are common electrocardiographic abnormalities. Knowing the various ischemic and nonischemic morphologic features is critical for a timely diagnosis of high-risk myocardial ischemia and electrolyte- or drug-related abnormalities. Moreover, it is important to recognize that true posterior infarction or subtle ST-segment elevation infarction may masquerade as ST-segment depression ischemia, and that pulmonary embolism may masquerade as anterior ischemia. These common electrocardiographic abnormalities are summarized in Table 1.
THE ST SEGMENT AND THE T WAVE: A PRIMER
The ST segment corresponds to the plateau phase of ventricular repolarization (phase 2 of the action potential), while the T wave corresponds to the phase of rapid ventricular repolarization (phase 3). ST-segment or T-wave changes may be secondary to abnormalities of depolarization, ie, pre-excitation or abnormalities of QRS voltage or duration.
On the other hand, ST-segment and T-wave abnormalities may be unrelated to any QRS abnormality, in which case they are called primary repolarization abnormalities. These are caused by ischemia, pericarditis, myocarditis, drugs (digoxin, antiarrhythmic drugs), and electrolyte abnormalities, particularly potassium abnormalities.
ST-segment deviation is usually measured at its junction with the end of the QRS complex, ie, the J point, and is referenced against the TP or PR segment.1 But some prefer to measure the magnitude of the ST-segment deviation 40 to 80 ms after the J point, when all myocardial fibers are expected to have reached the same level of membrane potential and to form an isoelectric ST segment; at the very onset of repolarization, small differences in membrane potential may normally be seen and may cause deviation of the J point and of the early portion of the ST segment.2
Although a diagnosis of ST-segment elevation myocardial infarction (STEMI) that mandates emergency reperfusion therapy requires ST-segment elevation greater than 1 mm in at least two contiguous leads,3 any ST-segment depression or elevation (≥ 0.5 mm, using the usual standard of 1.0 mV = 10 mm) may be abnormal, particularly when the clinical context or the shape of the ST segment suggests ischemia, or when other ischemic signs such as T-wave abnormalities, Q waves, or reciprocal ST-segment changes are concomitantly present. On the other hand, ST-segment depression of up to 0.5 mm in leads V2 and V3 and 1 mm in the other leads may be normal.1
In adults, the T wave normally is inverted in lead aVR; is upright or inverted in leads aVL, III, and V1; and is upright in leads I, II, aVF, and V2 through V6. The T wave is considered inverted when it is deeper than 1 mm; it is considered flat when its peak amplitude is between 1.0 mm and −1.0 mm.1
As we will discuss, certain features allow the various causes of ST-segment and T-wave abnormalities to be distinguished from one another.
SECONDARY ST-SEGMENT AND T-WAVE ABNORMALITIES
- The ST segment and T wave are directed opposite to the QRS: this is called discordance between the QRS complex and the ST-T abnormalities. In the case of right bundle branch block, the ST and T are directed opposite to the terminal portion of the QRS, ie, the part of the QRS deformed by the conduction abnormality.
- The ST segment and T wave are both abnormal and deviate in the same direction, ie, the ST segment is down-sloping and the T wave is inverted in leads with an upright QRS complex, which gives the ST-T complex a “reverse checkmark” asymmetric morphology.
- The ST and T abnormalities are not dynamic, ie, they do not change in the course of several hours to several days.
Thus, in cases of left ventricular hypertrophy or left bundle branch block, since the QRS complex is upright in the left lateral leads I, aVL, V5, and V6, the ST segment is characteristically depressed and the T wave is inverted in these leads (Figure 2). In cases of right ventricular hypertrophy or right bundle branch block, T waves are characteristically inverted in the right precordial leads V1, V2, and V3.
Left bundle branch block is always associated with secondary ST-T abnormalities, the absence of which suggests associated ischemia. Left and right ventricular hypertrophy, on the other hand, are not always associated with ST-T abnormalities, but when these are present, they correlate with more severe hypertrophy or ventricular systolic dysfunction,4 and have been called strain pattern. In addition, while these morphologic features are consistent with secondary abnormalities, they do not rule out ischemia in a patient with angina.
Some exceptions to these typical morphologic features:
- Right ventricular hypertrophy and right bundle branch block may be associated with isolated T-wave inversion without ST-segment depression in precordial leads V1, V2, and V3.
- Left ventricular hypertrophy may be associated with symmetric T-wave inversion without ST-segment depression or with a horizontally depressed ST segment. This may be the case in up to one-third of ST-T abnormalities secondary to left ventricular hypertrophy and is seen in hypertrophic cardiomyopathy, particularly the apical variant, in leads V3 through V6.5
ISCHEMIC ST-SEGMENT DEPRESSION, T-WAVE INVERSION, OR BOTH
ST-segment depression or T-wave inversion is consistent with ischemia if any of the following is true:
- The ST-segment depression or T-wave inversion is directed in the same direction as the QRS complex: this is called concordance between the QRS complex and the ST or T abnormality (Figure 1B).
- The ST segment is depressed but the T wave is upright (Figure 1C).
- The T wave has a positive-negative biphasic pattern (Figure 1D).
- The T wave is symmetrically inverted and has a pointed configuration, while the ST segment is not deviated or is upwardly bowed (coved) or horizontally depressed (Figure 1E).
- The magnitude of ST-segment depression progresses or regresses on serial tracings, or ST-segment depression progresses to T-wave abnormality during ischemia-free intervals (dynamic ST-segment depression).
Unlike ST-segment elevation, ST-segment depression does not localize ischemia.6 However, the extent and the magnitude of ST-segment depression correlate with the extent and the severity of ischemia. In fact, ST-segment depression in eight or more leads, combined with ST-segment elevation in leads aVR and V1 and occurring during ischemic pain, is associated with a 75% predictive accuracy for left main coronary artery or three-vessel disease (Figure 3).7,8 This finding may also be seen in cases of tight proximal stenosis of the left anterior descending coronary artery.9
Wellens syndrome
Wellens and his colleagues showed that 75% of patients who developed these T-wave abnormalities and who were treated medically without angiographic investigation went on to develop extensive anterior wall myocardial infarction within a mean of 8.5 days.10
In a later investigation of 1,260 patients presenting with unstable angina, 180 patients (14%) had this characteristic T-wave pattern.11 All of the latter patients had stenosis of 50% or more in the proximal left anterior descending artery, and 18% had total occlusion of the left anterior descending artery.
Thus, although medical management may provide symptomatic improvement at first, early coronary angiography and revascularization should be strongly considered in anyone with Wellens syndrome because it usually predicts impending anterior myocardial infarction.
Wellens syndrome is characterized by two patterns of T-wave changes. In 75% of cases, T waves are deeply (≥ 5 mm) and symmetrically inverted in leads V2 through V4 (Figures 1E, 4B). In 25% of cases, the T wave has a characteristic positive-negative biphasic morphology in leads V2 through V4 (Figures 1D, 4A).10 In both patterns, the ST segment is isoelectric or minimally elevated (< 1 mm) with a straight or convex morphology, the down-slope of the T wave is sharp, and the QT interval is often prolonged. These abnormalities are characteristically seen hours to days after the ischemic chest pain resolves. In fact, the ischemic episode is usually associated with transient ST-segment elevation or depression that progresses to the T-wave abnormality after the pain subsides.11
In Wellens’ original description, only 12% of patients had increases in their creatine kinase levels, and these were small. Therefore, the electrocardiogram may be the only indication of an impending large anterior infarction in a chest-pain-free patient.12
T waves that are symmetrically but less deeply inverted than Wellens-type T waves may still represent ischemia. However, this finding is less specific for ischemia and is associated with better outcomes than Wellens syndrome or ST-segment deviation, particularly when the T wave is less than 3 mm deep.14 In fact, one prospective cohort study found that isolated mild T-wave inversion in patients presenting with acute coronary syndrome is associated with a favorable long-term outcome, similar to that in patients with no electrocardiographic changes.15
FREQUENTLY MISSED DIAGNOSES MANIFESTING AS ST-SEGMENT DEPRESSION OR T-WAVE INVERSION
True posterior ST-segment elevation myocardial infarction
When accompanied by inferior STEMI, posterior infarction is easily recognized, but it can be difficult to diagnose when it occurs alone, the so-called true posterior STEMI.
In most cases of posterior infarction, the posterior chest leads V7, V8, and V9 reveal ST-segment elevation.19 One study found that ST-segment depression in the anterior precordial leads was as sensitive as ST-segment elevation in leads V7 through V9 in identifying posterior myocardial infarction (sensitivity 80%),20 while other studies found that ST-segment deviation on standard 12-lead electrocardiography has a lower sensitivity (about 60%) in identifying posterior infarction.18,21
Tall or wide (≥ 0.04-s) R waves in leads V1 or V2, particularly when associated with upright T waves, suggest posterior infarction and may further corroborate this diagnosis, but this finding may take up to 24 hours to manifest and is seen in only about 50% of patients with posterior infarction.21
Studies have shown that ST-segment elevation on standard 12-lead electrocardiography is found in fewer than 50% of patients with acute left circumflex occlusion and inferoposterior infarction,18 yet these are cases of “missed” STEMI that indeed benefit from emergency angiography and reperfusion. In addition, studies of non–ST-segment elevation acute coronary syndrome consistently identify patients who have epicardial vessel occlusion (about 15%–20% of cases),18 yet their initial angiography is usually delayed for hours or days after the initial presentation.
A subgroup analysis from TRITON–TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel Thrombolysis in Myocardial Infarction 38) evaluated patients with isolated anterior ST-segment depression. An occluded “culprit” artery was found 26% of the time, most often the left circumflex artery. Moreover, those patients had a significantly higher rate of death or myocardial infarction at 30-day follow-up than patients without a culprit artery, probably related to delayed revascularization.22
Recognizing that ST-segment depression that is greatest in leads V1, V2, or V3 represents posterior infarction helps identify a portion of the missed STEMIs in a timely fashion. In addition, in cases of anterior ST-segment depression and in cases of chest pain with nondiagnostic electrocardiography, the recording of ST elevation in leads V7, V8, and V9 is highly sensitive for detecting a true posterior injury.
Acute pulmonary embolism
An anterior ischemic pattern of symmetric T-wave inversion in the precordial leads V1 through V4 may also be a sign of acute or chronic right ventricular strain, particularly acute pulmonary embolism. Sinus tachycardia is usually present, but other signs of pulmonary embolism, such as right ventricular hypertrophy and right bundle branch block, may be absent. In fact, T-wave inversion in leads V1 through V4 is noted in 19% of patients with nonmassive pulmonary embolism and in 85% of patients with massive pulmonary embolism, and is the most sensitive and specific electrocardiographic finding in massive pulmonary embolism.23
In addition, acute pulmonary embolism may be associated with T-wave inversion in leads III and aVF,24 and changes of concomitant anterior and inferior ischemia should always raise the question of this diagnosis.
In one retrospective study of patients with acute pulmonary embolism, nonspecific ST-segment or T-wave changes were the most common finding on electrocardiography, noted in 49%.25 Rapid regression of these changes on serial tracings favors pulmonary embolism rather than myocardial infarction.
ST-segment depression reciprocal to a subtle ST-segment elevation
When ST-segment elevation occurs in two contiguous standard leads while ST-segment depression occurs in other leads, and when the ST-segment and T-wave abnormalities are ischemic rather than secondary to depolarization abnormalities, ST-segment elevation is considered the primary ischemic abnormality whereas ST-segment depression is often considered a reciprocal “mirror image” change. This “reciprocal” change may also represent remote ischemia in a distant territory in patients with multivessel coronary disease.26,27
Reciprocal ST-segment depression is present in all patients with inferior myocardial infarction and in 70% of patients with anterior myocardial infarction.28
Hypokalemia and digitalis effect
DIFFUSE (GLOBAL) T-WAVE INVERSION
Walder and Spodick36 have found this pattern to be caused most often by myocardial ischemia or neurologic events, particularly intracranial hemorrhage, and it seems more prevalent in women. Other causes include hypertrophic cardiomyopathy, stress-induced cardiomyopathy (takotsubo cardiomyopathy), cocaine abuse, pericarditis, pulmonary embolism, and advanced or complete atrioventricular block.36,37
The prognosis in patients with global T-wave inversion is determined by the underlying disease, and the striking T-wave changes per se do not imply a poor prognosis.38
OTHER CAUSES OF T-WAVE INVERSION OR ST-SEGMENT DEPRESSION
Various other entities may cause T-wave inversion, notably acute pericarditis or myocarditis, 41,42 memory T-wave phenomenon,43,44 and normal variants of repolarization (Table 1, Figure 9).45 Additionally, a nonpathologic junctional ST-segment depression may be seen in tachycardia (Figure 10).
Depression of the ST segment and inversion of the T wave are common electrocardiographic abnormalities. Knowing the various ischemic and nonischemic morphologic features is critical for a timely diagnosis of high-risk myocardial ischemia and electrolyte- or drug-related abnormalities. Moreover, it is important to recognize that true posterior infarction or subtle ST-segment elevation infarction may masquerade as ST-segment depression ischemia, and that pulmonary embolism may masquerade as anterior ischemia. These common electrocardiographic abnormalities are summarized in Table 1.
THE ST SEGMENT AND THE T WAVE: A PRIMER
The ST segment corresponds to the plateau phase of ventricular repolarization (phase 2 of the action potential), while the T wave corresponds to the phase of rapid ventricular repolarization (phase 3). ST-segment or T-wave changes may be secondary to abnormalities of depolarization, ie, pre-excitation or abnormalities of QRS voltage or duration.
On the other hand, ST-segment and T-wave abnormalities may be unrelated to any QRS abnormality, in which case they are called primary repolarization abnormalities. These are caused by ischemia, pericarditis, myocarditis, drugs (digoxin, antiarrhythmic drugs), and electrolyte abnormalities, particularly potassium abnormalities.
ST-segment deviation is usually measured at its junction with the end of the QRS complex, ie, the J point, and is referenced against the TP or PR segment.1 But some prefer to measure the magnitude of the ST-segment deviation 40 to 80 ms after the J point, when all myocardial fibers are expected to have reached the same level of membrane potential and to form an isoelectric ST segment; at the very onset of repolarization, small differences in membrane potential may normally be seen and may cause deviation of the J point and of the early portion of the ST segment.2
Although a diagnosis of ST-segment elevation myocardial infarction (STEMI) that mandates emergency reperfusion therapy requires ST-segment elevation greater than 1 mm in at least two contiguous leads,3 any ST-segment depression or elevation (≥ 0.5 mm, using the usual standard of 1.0 mV = 10 mm) may be abnormal, particularly when the clinical context or the shape of the ST segment suggests ischemia, or when other ischemic signs such as T-wave abnormalities, Q waves, or reciprocal ST-segment changes are concomitantly present. On the other hand, ST-segment depression of up to 0.5 mm in leads V2 and V3 and 1 mm in the other leads may be normal.1
In adults, the T wave normally is inverted in lead aVR; is upright or inverted in leads aVL, III, and V1; and is upright in leads I, II, aVF, and V2 through V6. The T wave is considered inverted when it is deeper than 1 mm; it is considered flat when its peak amplitude is between 1.0 mm and −1.0 mm.1
As we will discuss, certain features allow the various causes of ST-segment and T-wave abnormalities to be distinguished from one another.
SECONDARY ST-SEGMENT AND T-WAVE ABNORMALITIES
- The ST segment and T wave are directed opposite to the QRS: this is called discordance between the QRS complex and the ST-T abnormalities. In the case of right bundle branch block, the ST and T are directed opposite to the terminal portion of the QRS, ie, the part of the QRS deformed by the conduction abnormality.
- The ST segment and T wave are both abnormal and deviate in the same direction, ie, the ST segment is down-sloping and the T wave is inverted in leads with an upright QRS complex, which gives the ST-T complex a “reverse checkmark” asymmetric morphology.
- The ST and T abnormalities are not dynamic, ie, they do not change in the course of several hours to several days.
Thus, in cases of left ventricular hypertrophy or left bundle branch block, since the QRS complex is upright in the left lateral leads I, aVL, V5, and V6, the ST segment is characteristically depressed and the T wave is inverted in these leads (Figure 2). In cases of right ventricular hypertrophy or right bundle branch block, T waves are characteristically inverted in the right precordial leads V1, V2, and V3.
Left bundle branch block is always associated with secondary ST-T abnormalities, the absence of which suggests associated ischemia. Left and right ventricular hypertrophy, on the other hand, are not always associated with ST-T abnormalities, but when these are present, they correlate with more severe hypertrophy or ventricular systolic dysfunction,4 and have been called strain pattern. In addition, while these morphologic features are consistent with secondary abnormalities, they do not rule out ischemia in a patient with angina.
Some exceptions to these typical morphologic features:
- Right ventricular hypertrophy and right bundle branch block may be associated with isolated T-wave inversion without ST-segment depression in precordial leads V1, V2, and V3.
- Left ventricular hypertrophy may be associated with symmetric T-wave inversion without ST-segment depression or with a horizontally depressed ST segment. This may be the case in up to one-third of ST-T abnormalities secondary to left ventricular hypertrophy and is seen in hypertrophic cardiomyopathy, particularly the apical variant, in leads V3 through V6.5
ISCHEMIC ST-SEGMENT DEPRESSION, T-WAVE INVERSION, OR BOTH
ST-segment depression or T-wave inversion is consistent with ischemia if any of the following is true:
- The ST-segment depression or T-wave inversion is directed in the same direction as the QRS complex: this is called concordance between the QRS complex and the ST or T abnormality (Figure 1B).
- The ST segment is depressed but the T wave is upright (Figure 1C).
- The T wave has a positive-negative biphasic pattern (Figure 1D).
- The T wave is symmetrically inverted and has a pointed configuration, while the ST segment is not deviated or is upwardly bowed (coved) or horizontally depressed (Figure 1E).
- The magnitude of ST-segment depression progresses or regresses on serial tracings, or ST-segment depression progresses to T-wave abnormality during ischemia-free intervals (dynamic ST-segment depression).
Unlike ST-segment elevation, ST-segment depression does not localize ischemia.6 However, the extent and the magnitude of ST-segment depression correlate with the extent and the severity of ischemia. In fact, ST-segment depression in eight or more leads, combined with ST-segment elevation in leads aVR and V1 and occurring during ischemic pain, is associated with a 75% predictive accuracy for left main coronary artery or three-vessel disease (Figure 3).7,8 This finding may also be seen in cases of tight proximal stenosis of the left anterior descending coronary artery.9
Wellens syndrome
Wellens and his colleagues showed that 75% of patients who developed these T-wave abnormalities and who were treated medically without angiographic investigation went on to develop extensive anterior wall myocardial infarction within a mean of 8.5 days.10
In a later investigation of 1,260 patients presenting with unstable angina, 180 patients (14%) had this characteristic T-wave pattern.11 All of the latter patients had stenosis of 50% or more in the proximal left anterior descending artery, and 18% had total occlusion of the left anterior descending artery.
Thus, although medical management may provide symptomatic improvement at first, early coronary angiography and revascularization should be strongly considered in anyone with Wellens syndrome because it usually predicts impending anterior myocardial infarction.
Wellens syndrome is characterized by two patterns of T-wave changes. In 75% of cases, T waves are deeply (≥ 5 mm) and symmetrically inverted in leads V2 through V4 (Figures 1E, 4B). In 25% of cases, the T wave has a characteristic positive-negative biphasic morphology in leads V2 through V4 (Figures 1D, 4A).10 In both patterns, the ST segment is isoelectric or minimally elevated (< 1 mm) with a straight or convex morphology, the down-slope of the T wave is sharp, and the QT interval is often prolonged. These abnormalities are characteristically seen hours to days after the ischemic chest pain resolves. In fact, the ischemic episode is usually associated with transient ST-segment elevation or depression that progresses to the T-wave abnormality after the pain subsides.11
In Wellens’ original description, only 12% of patients had increases in their creatine kinase levels, and these were small. Therefore, the electrocardiogram may be the only indication of an impending large anterior infarction in a chest-pain-free patient.12
T waves that are symmetrically but less deeply inverted than Wellens-type T waves may still represent ischemia. However, this finding is less specific for ischemia and is associated with better outcomes than Wellens syndrome or ST-segment deviation, particularly when the T wave is less than 3 mm deep.14 In fact, one prospective cohort study found that isolated mild T-wave inversion in patients presenting with acute coronary syndrome is associated with a favorable long-term outcome, similar to that in patients with no electrocardiographic changes.15
FREQUENTLY MISSED DIAGNOSES MANIFESTING AS ST-SEGMENT DEPRESSION OR T-WAVE INVERSION
True posterior ST-segment elevation myocardial infarction
When accompanied by inferior STEMI, posterior infarction is easily recognized, but it can be difficult to diagnose when it occurs alone, the so-called true posterior STEMI.
In most cases of posterior infarction, the posterior chest leads V7, V8, and V9 reveal ST-segment elevation.19 One study found that ST-segment depression in the anterior precordial leads was as sensitive as ST-segment elevation in leads V7 through V9 in identifying posterior myocardial infarction (sensitivity 80%),20 while other studies found that ST-segment deviation on standard 12-lead electrocardiography has a lower sensitivity (about 60%) in identifying posterior infarction.18,21
Tall or wide (≥ 0.04-s) R waves in leads V1 or V2, particularly when associated with upright T waves, suggest posterior infarction and may further corroborate this diagnosis, but this finding may take up to 24 hours to manifest and is seen in only about 50% of patients with posterior infarction.21
Studies have shown that ST-segment elevation on standard 12-lead electrocardiography is found in fewer than 50% of patients with acute left circumflex occlusion and inferoposterior infarction,18 yet these are cases of “missed” STEMI that indeed benefit from emergency angiography and reperfusion. In addition, studies of non–ST-segment elevation acute coronary syndrome consistently identify patients who have epicardial vessel occlusion (about 15%–20% of cases),18 yet their initial angiography is usually delayed for hours or days after the initial presentation.
A subgroup analysis from TRITON–TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel Thrombolysis in Myocardial Infarction 38) evaluated patients with isolated anterior ST-segment depression. An occluded “culprit” artery was found 26% of the time, most often the left circumflex artery. Moreover, those patients had a significantly higher rate of death or myocardial infarction at 30-day follow-up than patients without a culprit artery, probably related to delayed revascularization.22
Recognizing that ST-segment depression that is greatest in leads V1, V2, or V3 represents posterior infarction helps identify a portion of the missed STEMIs in a timely fashion. In addition, in cases of anterior ST-segment depression and in cases of chest pain with nondiagnostic electrocardiography, the recording of ST elevation in leads V7, V8, and V9 is highly sensitive for detecting a true posterior injury.
Acute pulmonary embolism
An anterior ischemic pattern of symmetric T-wave inversion in the precordial leads V1 through V4 may also be a sign of acute or chronic right ventricular strain, particularly acute pulmonary embolism. Sinus tachycardia is usually present, but other signs of pulmonary embolism, such as right ventricular hypertrophy and right bundle branch block, may be absent. In fact, T-wave inversion in leads V1 through V4 is noted in 19% of patients with nonmassive pulmonary embolism and in 85% of patients with massive pulmonary embolism, and is the most sensitive and specific electrocardiographic finding in massive pulmonary embolism.23
In addition, acute pulmonary embolism may be associated with T-wave inversion in leads III and aVF,24 and changes of concomitant anterior and inferior ischemia should always raise the question of this diagnosis.
In one retrospective study of patients with acute pulmonary embolism, nonspecific ST-segment or T-wave changes were the most common finding on electrocardiography, noted in 49%.25 Rapid regression of these changes on serial tracings favors pulmonary embolism rather than myocardial infarction.
ST-segment depression reciprocal to a subtle ST-segment elevation
When ST-segment elevation occurs in two contiguous standard leads while ST-segment depression occurs in other leads, and when the ST-segment and T-wave abnormalities are ischemic rather than secondary to depolarization abnormalities, ST-segment elevation is considered the primary ischemic abnormality whereas ST-segment depression is often considered a reciprocal “mirror image” change. This “reciprocal” change may also represent remote ischemia in a distant territory in patients with multivessel coronary disease.26,27
Reciprocal ST-segment depression is present in all patients with inferior myocardial infarction and in 70% of patients with anterior myocardial infarction.28
Hypokalemia and digitalis effect
DIFFUSE (GLOBAL) T-WAVE INVERSION
Walder and Spodick36 have found this pattern to be caused most often by myocardial ischemia or neurologic events, particularly intracranial hemorrhage, and it seems more prevalent in women. Other causes include hypertrophic cardiomyopathy, stress-induced cardiomyopathy (takotsubo cardiomyopathy), cocaine abuse, pericarditis, pulmonary embolism, and advanced or complete atrioventricular block.36,37
The prognosis in patients with global T-wave inversion is determined by the underlying disease, and the striking T-wave changes per se do not imply a poor prognosis.38
OTHER CAUSES OF T-WAVE INVERSION OR ST-SEGMENT DEPRESSION
Various other entities may cause T-wave inversion, notably acute pericarditis or myocarditis, 41,42 memory T-wave phenomenon,43,44 and normal variants of repolarization (Table 1, Figure 9).45 Additionally, a nonpathologic junctional ST-segment depression may be seen in tachycardia (Figure 10).
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Surawicz B, Knilans TK. Non-Q wave myocardial infarction, unstable angina pectoris, myocardial ischemia. In: Chou's Electrocardiography in Clinical Practice: Adult and Pediatric. 5th ed. Philadelphia: WB Saunders; 2001:194–207.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Okin PM, Devereux RB, Nieminen MS, et al; LIFE Study Investigators. Electrocardiographic strain pattern and prediction of new-onset congestive heart failure in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study. Circulation 2006; 113:67–73.
- Huwez FU, Pringle SD, Macfarlane PW. Variable patterns of ST-T abnormalities in patients with left ventricular hypertrophy and normal coronary arteries. Br Heart J 1992; 67:304–307.
- Li D, Li CY, Yong AC, Kilpatrick D. Source of electrocardiographic ST changes in subendocardial ischemia. Circ Res 1998; 82:957–970.
- Gorgels AP, Vos MA, Mulleneers R, de Zwaan C, Bär FW, Wellens HJ. Value of the electrocardiogram in diagnosing the number of severely narrowed coronary arteries in rest angina pectoris. Am J Cardiol 1993; 72:999–1003.
- Glancy DL. Electrocardiographic diagnosis of acute myocardial infarction. J La State Med Soc 2002; 154:66–75.
- Yamaji H, Iwasaki K, Kusachi S, et al. Prediction of acute left main coronary artery obstruction by 12-lead electrocardiography. ST segment elevation in lead aVR with less ST segment elevation in lead V(1). J Am Coll Cardiol 2001; 38:1348–1354.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- Lilaonitkul M, Robinson K, Roberts M. Wellens’ syndrome: significance of ECG pattern recognition in the emergency department. Emerg Med J 2009; 26:750–751.
- Glancy DL, Khuri B, Cospolich B. Heed the warning: Wellens’ type T-wave inversion is caused by proximal left anterior descending lesion. Proc (Bayl Univ Med Cent) 2000; 13:416–418.
- Savonitto S, Ardissino D, Granger CB, et al. Prognostic value of the admission electrocardiogram in acute coronary syndromes. JAMA 1999; 281:707–713.
- Mueller C, Neumann FJ, Perach W, Perruchoud AP, Buettner HJ. Prognostic value of the admission electrocardiogram in patients with unstable angina/non-ST-segment elevation myocardial infarction treated with very early revascularization. Am J Med 2004; 117:145–150.
- Boden WE, Spodick DH. Diagnostic significance of precordial ST-segment depression. Am J Cardiol 1989; 63:358–361.
- Shah A, Wagner GS, Green CL, et al. Electrocardiographic differentiation of the ST-segment depression of acute myocardial injury due to the left circumflex artery occlusion from that of myocardial ischemia of nonocclusive etiologies. Am J Cardiol 1997; 80:512–513.
- Krishnaswamy A, Lincoff AM, Menon V. Magnitude and consequences of missing the acute infarct-related circumflex artery. Am Heart J 2009; 158:706–712.
- Matetzky S, Freimark D, Feinberg MS, et al. Acute myocardial infarction with isolated ST-segment elevation in posterior chest leads V7-9: “hidden” ST-segment elevations revealing acute posterior infarction. J Am Coll Cardiol 1999; 34:748–753.
- Matetzky S, Freimark D, Chouraqui P, et al. Significance of ST segment elevations in posterior chest leads (V7 to V9) in patients with acute inferior myocardial infarction: application for thrombolytic therapy. J Am Coll Cardiol 1998; 31:506–511.
- Huey BL, Beller GA, Kaiser DL, Gibson RS. A comprehensive analysis of myocardial infarction due to left circumflex artery occlusion: comparison with infarction due to right coronary artery and left anterior descending artery occlusion. J Am Coll Cardiol 1988; 12:1156–1166.
- Gibson CM, Pride YB, Mohanavelu S, Wiviott SD, Antman EM, Braunwald E. Abstract 1999: Angiographic and clinical outcomes among patients with acute coronary syndrome presenting with isolated anterior ST-segment depressions. Circulation 2008; 118:S–654.
- Ferrari E, Imbert A, Chevalier T, Mihoubi A, Morand P, Baudouy M. The ECG in pulmonary embolism. Predictive value of negative T waves in precordial leads—80 case reports. Chest 1997; 111:537–543.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Norell MS, Lyons JP, Gardener JE, Layton CA, Balcon R. Significance of “reciprocal” ST segment depression: left ventriculographic observations during left anterior descending coronary angioplasty. J Am Coll Cardiol 1989; 13:1270–1274.
- Haraphongse M, Tanomsup S, Jugdutt BI. Inferior ST segment depression during acute anterior myocardial infarction: clinical and angiographic correlations. J Am Coll Cardiol 1984; 4:467–476.
- Surawicz B, Knilans TK. Acute ischemia: electrocardiographic patterns. In: Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric. 5th edition. Philadelphia: WB Saunders; 2001:122–153.
- Wagner GS, Macfarlane P, Wellens H, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part VI: acute ischemia/infarction: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:1003–1011.
- Brady WJ, Perron AD, Syverud SA, et al. Reciprocal ST segment depression: impact on the electrocardiographic diagnosis of ST segment elevation acute myocardial infarction. Am J Emerg Med 2002; 20:35–38.
- Surawicz B. Electrolytes and the electrocardiogram. Postgrad Med 1974; 55:123–129.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Glancy DL, Wang WL. ECG of the month. Abnormal electrocardiogram in a woman with a urinary tract infection. Sinus rhythm, rate 82/minute. Sagging ST segments, low T waves, and prominent U waves suggest hypokalemia. J La State Med Soc 2007; 159:5–7.
- Surawicz B, Braun HA, Crum WB, Kemp RL, Wagner S, Bellet S. Quantitative analysis of the electrocardiographic pattern of hypopotassemia. Circulation 1957; 16:750–763.
- Glancy DL, Rochon BJ, Ilie CC, Parker JM, Jones MB, Atluri P. Global T-wave inversion in a 77-year-old woman. Proc (Bayl Univ Med Cent) 2009; 22:81–82.
- Walder LA, Spodick DH. Global T wave inversion. J Am Coll Cardiol 1991; 17:1479–1485.
- Lui CY. Acute pulmonary embolism as the cause of global T wave inversion and QT prolongation. A case report. J Electrocardiol 1993; 26:91–95.
- Walder LA, Spodick DH. Global T wave inversion: long-term followup. J Am Coll Cardiol 1993; 21:1652–1656.
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Wittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352:539–548.
- Spodick DH. Electrocardiogram in acute pericarditis. Distributions of morphologic and axial changes by stages. Am J Cardiol 1974; 33:470–474.
- Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
- Rosenbaum MB, Blanco HH, Elizari MV, Lázzari JO, Davidenko JM. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol 1982; 50:213–222.
- Paparella N, Ouyang F, Fuca G, Kuck KH, Cappato R, Alboni P. Significance of newly acquired negative T waves after interruption of paroxysmal reentrant supraventricular tachycardia with narrow QRS complex. Am J Cardiol 2000; 85:261–263.
- Kaid KA, Maqsood A, Cohen M, Rothfeld E. Further characterization of the “persistent juvenile T-wave pattern” in adults. J Electrocardiol 2008; 41:644–645.
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Surawicz B, Knilans TK. Non-Q wave myocardial infarction, unstable angina pectoris, myocardial ischemia. In: Chou's Electrocardiography in Clinical Practice: Adult and Pediatric. 5th ed. Philadelphia: WB Saunders; 2001:194–207.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Okin PM, Devereux RB, Nieminen MS, et al; LIFE Study Investigators. Electrocardiographic strain pattern and prediction of new-onset congestive heart failure in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study. Circulation 2006; 113:67–73.
- Huwez FU, Pringle SD, Macfarlane PW. Variable patterns of ST-T abnormalities in patients with left ventricular hypertrophy and normal coronary arteries. Br Heart J 1992; 67:304–307.
- Li D, Li CY, Yong AC, Kilpatrick D. Source of electrocardiographic ST changes in subendocardial ischemia. Circ Res 1998; 82:957–970.
- Gorgels AP, Vos MA, Mulleneers R, de Zwaan C, Bär FW, Wellens HJ. Value of the electrocardiogram in diagnosing the number of severely narrowed coronary arteries in rest angina pectoris. Am J Cardiol 1993; 72:999–1003.
- Glancy DL. Electrocardiographic diagnosis of acute myocardial infarction. J La State Med Soc 2002; 154:66–75.
- Yamaji H, Iwasaki K, Kusachi S, et al. Prediction of acute left main coronary artery obstruction by 12-lead electrocardiography. ST segment elevation in lead aVR with less ST segment elevation in lead V(1). J Am Coll Cardiol 2001; 38:1348–1354.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- Lilaonitkul M, Robinson K, Roberts M. Wellens’ syndrome: significance of ECG pattern recognition in the emergency department. Emerg Med J 2009; 26:750–751.
- Glancy DL, Khuri B, Cospolich B. Heed the warning: Wellens’ type T-wave inversion is caused by proximal left anterior descending lesion. Proc (Bayl Univ Med Cent) 2000; 13:416–418.
- Savonitto S, Ardissino D, Granger CB, et al. Prognostic value of the admission electrocardiogram in acute coronary syndromes. JAMA 1999; 281:707–713.
- Mueller C, Neumann FJ, Perach W, Perruchoud AP, Buettner HJ. Prognostic value of the admission electrocardiogram in patients with unstable angina/non-ST-segment elevation myocardial infarction treated with very early revascularization. Am J Med 2004; 117:145–150.
- Boden WE, Spodick DH. Diagnostic significance of precordial ST-segment depression. Am J Cardiol 1989; 63:358–361.
- Shah A, Wagner GS, Green CL, et al. Electrocardiographic differentiation of the ST-segment depression of acute myocardial injury due to the left circumflex artery occlusion from that of myocardial ischemia of nonocclusive etiologies. Am J Cardiol 1997; 80:512–513.
- Krishnaswamy A, Lincoff AM, Menon V. Magnitude and consequences of missing the acute infarct-related circumflex artery. Am Heart J 2009; 158:706–712.
- Matetzky S, Freimark D, Feinberg MS, et al. Acute myocardial infarction with isolated ST-segment elevation in posterior chest leads V7-9: “hidden” ST-segment elevations revealing acute posterior infarction. J Am Coll Cardiol 1999; 34:748–753.
- Matetzky S, Freimark D, Chouraqui P, et al. Significance of ST segment elevations in posterior chest leads (V7 to V9) in patients with acute inferior myocardial infarction: application for thrombolytic therapy. J Am Coll Cardiol 1998; 31:506–511.
- Huey BL, Beller GA, Kaiser DL, Gibson RS. A comprehensive analysis of myocardial infarction due to left circumflex artery occlusion: comparison with infarction due to right coronary artery and left anterior descending artery occlusion. J Am Coll Cardiol 1988; 12:1156–1166.
- Gibson CM, Pride YB, Mohanavelu S, Wiviott SD, Antman EM, Braunwald E. Abstract 1999: Angiographic and clinical outcomes among patients with acute coronary syndrome presenting with isolated anterior ST-segment depressions. Circulation 2008; 118:S–654.
- Ferrari E, Imbert A, Chevalier T, Mihoubi A, Morand P, Baudouy M. The ECG in pulmonary embolism. Predictive value of negative T waves in precordial leads—80 case reports. Chest 1997; 111:537–543.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Norell MS, Lyons JP, Gardener JE, Layton CA, Balcon R. Significance of “reciprocal” ST segment depression: left ventriculographic observations during left anterior descending coronary angioplasty. J Am Coll Cardiol 1989; 13:1270–1274.
- Haraphongse M, Tanomsup S, Jugdutt BI. Inferior ST segment depression during acute anterior myocardial infarction: clinical and angiographic correlations. J Am Coll Cardiol 1984; 4:467–476.
- Surawicz B, Knilans TK. Acute ischemia: electrocardiographic patterns. In: Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric. 5th edition. Philadelphia: WB Saunders; 2001:122–153.
- Wagner GS, Macfarlane P, Wellens H, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part VI: acute ischemia/infarction: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:1003–1011.
- Brady WJ, Perron AD, Syverud SA, et al. Reciprocal ST segment depression: impact on the electrocardiographic diagnosis of ST segment elevation acute myocardial infarction. Am J Emerg Med 2002; 20:35–38.
- Surawicz B. Electrolytes and the electrocardiogram. Postgrad Med 1974; 55:123–129.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Glancy DL, Wang WL. ECG of the month. Abnormal electrocardiogram in a woman with a urinary tract infection. Sinus rhythm, rate 82/minute. Sagging ST segments, low T waves, and prominent U waves suggest hypokalemia. J La State Med Soc 2007; 159:5–7.
- Surawicz B, Braun HA, Crum WB, Kemp RL, Wagner S, Bellet S. Quantitative analysis of the electrocardiographic pattern of hypopotassemia. Circulation 1957; 16:750–763.
- Glancy DL, Rochon BJ, Ilie CC, Parker JM, Jones MB, Atluri P. Global T-wave inversion in a 77-year-old woman. Proc (Bayl Univ Med Cent) 2009; 22:81–82.
- Walder LA, Spodick DH. Global T wave inversion. J Am Coll Cardiol 1991; 17:1479–1485.
- Lui CY. Acute pulmonary embolism as the cause of global T wave inversion and QT prolongation. A case report. J Electrocardiol 1993; 26:91–95.
- Walder LA, Spodick DH. Global T wave inversion: long-term followup. J Am Coll Cardiol 1993; 21:1652–1656.
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Wittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352:539–548.
- Spodick DH. Electrocardiogram in acute pericarditis. Distributions of morphologic and axial changes by stages. Am J Cardiol 1974; 33:470–474.
- Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
- Rosenbaum MB, Blanco HH, Elizari MV, Lázzari JO, Davidenko JM. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol 1982; 50:213–222.
- Paparella N, Ouyang F, Fuca G, Kuck KH, Cappato R, Alboni P. Significance of newly acquired negative T waves after interruption of paroxysmal reentrant supraventricular tachycardia with narrow QRS complex. Am J Cardiol 2000; 85:261–263.
- Kaid KA, Maqsood A, Cohen M, Rothfeld E. Further characterization of the “persistent juvenile T-wave pattern” in adults. J Electrocardiol 2008; 41:644–645.
KEY POINTS
- ST-T abnormalities concordant to the QRS complex suggest ischemia.
- Deep T-wave inversion or positive-negative biphasic T waves in the anterior precordial leads reflect severe left anterior descending coronary artery stenosis.
- Two particular patterns of ST-segment depression reflect ST-segment elevation myocardial infarction rather than non–ST-segment elevation acute coronary syndrome: ST-segment depression that is reciprocal to a subtle and sometimes overlooked ST-segment elevation, and ST-segment depression that is maximal in leads V1–V3, suggesting true posterior infarction.
- T-wave inversion in the anterior precordial leads may be seen in cases of acute pulmonary embolism, while flattened T waves with prominent U waves and ST-segment depression may reflect hypokalemia or digitalis therapy.
Grand Rounds: Woman, 22, With Dizziness and Headache
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.