User login
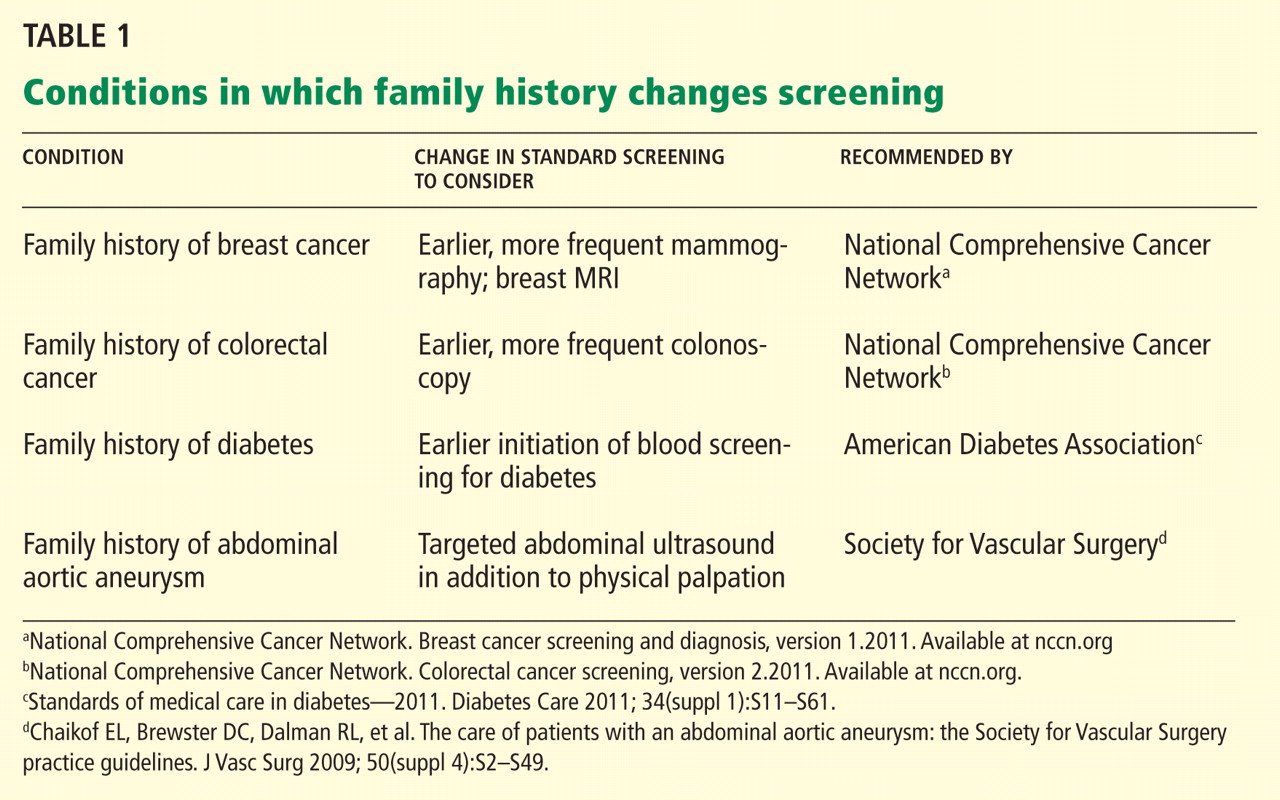
Family history: Still relevant in the genomics era
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
KEY POINTS
- The family history is an underused tool for predicting the risk of disease and for personalizing preventive care.
- Barriers to the appropriate collection and use of the family history include concerns over the reliability of patient reporting, a lack of time and reimbursement, and provider knowledge gaps.
- Use of family history to inform genetic testing for hereditary cancer syndromes has been shown to improve outcomes and may reduce overall health care costs.
- Future solutions need to focus on creating time-effective ways to collect and analyze the family history, and on developing innovative methods of educating medical providers at all levels of training as to how to apply the family history in clinical practice.
Personalizing patient care
The concept and promise of personalized health care have been anticipated for decades. Yet in its breadth and in the way we would like it practiced, it is in its infancy.
Personalized health care aims to individualize care by integrating a person’s unique clinical, molecular (ie, genetic, genomic), and environmental information. Applied not only when the patient is sick but also when he or she is well, it builds on and enhances our current standards of care.
As Sir William Osler recognized more than a century ago, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”1
PREDICTING THE RISK OF DISEASE
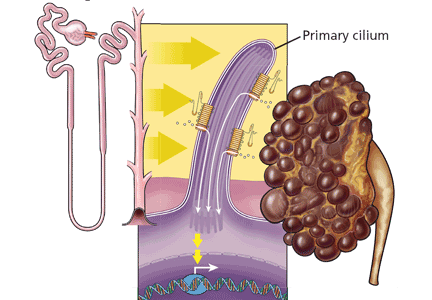
For years, we have attempted to predict and stratify the risk of disease, and the Human Genome Project has given us a new set of tools to help understand the complexity of disease and its variability.
We know and have known for decades—and in some cultures, for centuries—that family history is the most clinically validated tool for predicting the risk of disease.
Nevertheless, evidence suggests that we physicians are not collecting adequate family histories, and because of this, we are missing opportunities to intervene and prevent diseases predicted by the family history. The current standard of care is to use family medical histories to hone genetic differential diagnoses, and based on the differential diagnoses, to target specific genes to test in the setting of genetic counseling. Current genetic testing is used for molecular diagnoses and predictive testing so that gene-specific clinical management can be subsequently tailored.
PREDICTING RESPONSE TO TREATMENT
The use of personalized health care to predict response to treatment is a novel and constantly evolving practice.
ABO blood typing is a form of genetics-based personalization of safe transfusion that dates back to World War II.
A prominent, recent success story is in cancer treatment. For example, the American Society of Clinical Oncology now recommends that tumors from patients with node-negative, estrogen-receptor-positive breast cancer be evaluated with the Oncotype DX assay.2 This test measures the expression of 21 genes, and the score obtained identifies patients most likely to benefit from adjuvant chemotherapy. A similar 12-gene expression signature has been developed for colon cancer, and others have been developed for hematologic cancers.2,3 As with other new but apparently valid tests, the risk scores derived are sensitive at the extremes but ambiguous in the mid-ranges. We anticipate many more developments in this field.
In the field of pharmacogenomics, there is evidence to suggest that prior knowledge of CYP2C9 and VKORC1 genotypes enhances outcomes for patients starting treatment with warfarin. The US Food and Drug Administration revised the label on warfarin in February 2010, suggesting that genotypes be taken into consideration when the drug is prescribed.4
However, clinicians have been slow to adopt genotype testing when prescribing warfarin. Some cite the paucity of large, randomized, controlled trials demonstrating clinical utility of genotype-informed prescribing. Others cite concern that warfarin will soon become obsolete with the arrival of newer anticoagulants (such as factor X inhibitors) that do not carry warfarin’s adverse effects, and these genotypes will therefore become moot. Perhaps, as we move forward and new drugs are developed, companion genotype tests could be developed at the same time to be used with them.5
IMPROVING CARE, SAVING MONEY, AND EMPOWERING PATIENTS
The goal of personalized health care, by customizing treatments (medication types and dosages) and preventive strategies, is to optimize medical care and improve outcomes for each patient. It could improve the quality of care by targeting interventions and reducing adverse events, topics that are important to all of us in the current environment of health care reform.
A personalized approach might also, in the long run, decrease the cost of health care by driving appropriate utilization of resources.
Lastly, the true value of personalized health care may be in its potential to improve patient satisfaction and to empower our patients to work with us towards better health.
WE LAUNCH A NEW SERIES
To keep physicians up-to-date on progress in personalized health care, the Cleveland Clinic Journal of Medicine will present a series of articles on the topic. The series, to run once a quarter, begins in this issue, on page 331, with an article on the importance of the family history as a piece of genetic information that can help to predict the risk of disease and inform preventive care plans. Future topics will include the role of genetics and genomics in personalized care of patients with breast and colorectal cancers; the genetic counselor as a part of the health care team; pharmacogenomics; and ethical, legal, and societal considerations.
Our goal in this series is to provide practical information to help our readers incorporate personalized approaches into daily practice. In addition, as patients become more interested in and informed about personalized health care, we hope this information will help clinicians to effectively coach them about its potential benefits and risks. We also hope this information will enable our readers to ask the right questions so that patient and health care provider can work together to help the patient grow old gracefully.
As the series unfolds, we ask you to send us feedback and to suggest other topics in personalized health care you would like us to cover in this series.
- Osler W. Aequanimitas, With Other Addresses to Medical Students, Nurses and Practitioners of Medicine. 2nd edition. Philadelphia, PA: P. Blakiston’s Sone & Co, 1906:348.
- McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med 2011; 364:340–350.
- Eng C. Microenvironmental protection in diffuse large-B-cell lymphoma. N Engl J Med 2008; 359:2379–2381.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med 2010; 363:301–304.
The concept and promise of personalized health care have been anticipated for decades. Yet in its breadth and in the way we would like it practiced, it is in its infancy.
Personalized health care aims to individualize care by integrating a person’s unique clinical, molecular (ie, genetic, genomic), and environmental information. Applied not only when the patient is sick but also when he or she is well, it builds on and enhances our current standards of care.
As Sir William Osler recognized more than a century ago, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”1
PREDICTING THE RISK OF DISEASE
For years, we have attempted to predict and stratify the risk of disease, and the Human Genome Project has given us a new set of tools to help understand the complexity of disease and its variability.
We know and have known for decades—and in some cultures, for centuries—that family history is the most clinically validated tool for predicting the risk of disease.
Nevertheless, evidence suggests that we physicians are not collecting adequate family histories, and because of this, we are missing opportunities to intervene and prevent diseases predicted by the family history. The current standard of care is to use family medical histories to hone genetic differential diagnoses, and based on the differential diagnoses, to target specific genes to test in the setting of genetic counseling. Current genetic testing is used for molecular diagnoses and predictive testing so that gene-specific clinical management can be subsequently tailored.
PREDICTING RESPONSE TO TREATMENT
The use of personalized health care to predict response to treatment is a novel and constantly evolving practice.
ABO blood typing is a form of genetics-based personalization of safe transfusion that dates back to World War II.
A prominent, recent success story is in cancer treatment. For example, the American Society of Clinical Oncology now recommends that tumors from patients with node-negative, estrogen-receptor-positive breast cancer be evaluated with the Oncotype DX assay.2 This test measures the expression of 21 genes, and the score obtained identifies patients most likely to benefit from adjuvant chemotherapy. A similar 12-gene expression signature has been developed for colon cancer, and others have been developed for hematologic cancers.2,3 As with other new but apparently valid tests, the risk scores derived are sensitive at the extremes but ambiguous in the mid-ranges. We anticipate many more developments in this field.
In the field of pharmacogenomics, there is evidence to suggest that prior knowledge of CYP2C9 and VKORC1 genotypes enhances outcomes for patients starting treatment with warfarin. The US Food and Drug Administration revised the label on warfarin in February 2010, suggesting that genotypes be taken into consideration when the drug is prescribed.4
However, clinicians have been slow to adopt genotype testing when prescribing warfarin. Some cite the paucity of large, randomized, controlled trials demonstrating clinical utility of genotype-informed prescribing. Others cite concern that warfarin will soon become obsolete with the arrival of newer anticoagulants (such as factor X inhibitors) that do not carry warfarin’s adverse effects, and these genotypes will therefore become moot. Perhaps, as we move forward and new drugs are developed, companion genotype tests could be developed at the same time to be used with them.5
IMPROVING CARE, SAVING MONEY, AND EMPOWERING PATIENTS
The goal of personalized health care, by customizing treatments (medication types and dosages) and preventive strategies, is to optimize medical care and improve outcomes for each patient. It could improve the quality of care by targeting interventions and reducing adverse events, topics that are important to all of us in the current environment of health care reform.
A personalized approach might also, in the long run, decrease the cost of health care by driving appropriate utilization of resources.
Lastly, the true value of personalized health care may be in its potential to improve patient satisfaction and to empower our patients to work with us towards better health.
WE LAUNCH A NEW SERIES
To keep physicians up-to-date on progress in personalized health care, the Cleveland Clinic Journal of Medicine will present a series of articles on the topic. The series, to run once a quarter, begins in this issue, on page 331, with an article on the importance of the family history as a piece of genetic information that can help to predict the risk of disease and inform preventive care plans. Future topics will include the role of genetics and genomics in personalized care of patients with breast and colorectal cancers; the genetic counselor as a part of the health care team; pharmacogenomics; and ethical, legal, and societal considerations.
Our goal in this series is to provide practical information to help our readers incorporate personalized approaches into daily practice. In addition, as patients become more interested in and informed about personalized health care, we hope this information will help clinicians to effectively coach them about its potential benefits and risks. We also hope this information will enable our readers to ask the right questions so that patient and health care provider can work together to help the patient grow old gracefully.
As the series unfolds, we ask you to send us feedback and to suggest other topics in personalized health care you would like us to cover in this series.
The concept and promise of personalized health care have been anticipated for decades. Yet in its breadth and in the way we would like it practiced, it is in its infancy.
Personalized health care aims to individualize care by integrating a person’s unique clinical, molecular (ie, genetic, genomic), and environmental information. Applied not only when the patient is sick but also when he or she is well, it builds on and enhances our current standards of care.
As Sir William Osler recognized more than a century ago, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”1
PREDICTING THE RISK OF DISEASE
For years, we have attempted to predict and stratify the risk of disease, and the Human Genome Project has given us a new set of tools to help understand the complexity of disease and its variability.
We know and have known for decades—and in some cultures, for centuries—that family history is the most clinically validated tool for predicting the risk of disease.
Nevertheless, evidence suggests that we physicians are not collecting adequate family histories, and because of this, we are missing opportunities to intervene and prevent diseases predicted by the family history. The current standard of care is to use family medical histories to hone genetic differential diagnoses, and based on the differential diagnoses, to target specific genes to test in the setting of genetic counseling. Current genetic testing is used for molecular diagnoses and predictive testing so that gene-specific clinical management can be subsequently tailored.
PREDICTING RESPONSE TO TREATMENT
The use of personalized health care to predict response to treatment is a novel and constantly evolving practice.
ABO blood typing is a form of genetics-based personalization of safe transfusion that dates back to World War II.
A prominent, recent success story is in cancer treatment. For example, the American Society of Clinical Oncology now recommends that tumors from patients with node-negative, estrogen-receptor-positive breast cancer be evaluated with the Oncotype DX assay.2 This test measures the expression of 21 genes, and the score obtained identifies patients most likely to benefit from adjuvant chemotherapy. A similar 12-gene expression signature has been developed for colon cancer, and others have been developed for hematologic cancers.2,3 As with other new but apparently valid tests, the risk scores derived are sensitive at the extremes but ambiguous in the mid-ranges. We anticipate many more developments in this field.
In the field of pharmacogenomics, there is evidence to suggest that prior knowledge of CYP2C9 and VKORC1 genotypes enhances outcomes for patients starting treatment with warfarin. The US Food and Drug Administration revised the label on warfarin in February 2010, suggesting that genotypes be taken into consideration when the drug is prescribed.4
However, clinicians have been slow to adopt genotype testing when prescribing warfarin. Some cite the paucity of large, randomized, controlled trials demonstrating clinical utility of genotype-informed prescribing. Others cite concern that warfarin will soon become obsolete with the arrival of newer anticoagulants (such as factor X inhibitors) that do not carry warfarin’s adverse effects, and these genotypes will therefore become moot. Perhaps, as we move forward and new drugs are developed, companion genotype tests could be developed at the same time to be used with them.5
IMPROVING CARE, SAVING MONEY, AND EMPOWERING PATIENTS
The goal of personalized health care, by customizing treatments (medication types and dosages) and preventive strategies, is to optimize medical care and improve outcomes for each patient. It could improve the quality of care by targeting interventions and reducing adverse events, topics that are important to all of us in the current environment of health care reform.
A personalized approach might also, in the long run, decrease the cost of health care by driving appropriate utilization of resources.
Lastly, the true value of personalized health care may be in its potential to improve patient satisfaction and to empower our patients to work with us towards better health.
WE LAUNCH A NEW SERIES
To keep physicians up-to-date on progress in personalized health care, the Cleveland Clinic Journal of Medicine will present a series of articles on the topic. The series, to run once a quarter, begins in this issue, on page 331, with an article on the importance of the family history as a piece of genetic information that can help to predict the risk of disease and inform preventive care plans. Future topics will include the role of genetics and genomics in personalized care of patients with breast and colorectal cancers; the genetic counselor as a part of the health care team; pharmacogenomics; and ethical, legal, and societal considerations.
Our goal in this series is to provide practical information to help our readers incorporate personalized approaches into daily practice. In addition, as patients become more interested in and informed about personalized health care, we hope this information will help clinicians to effectively coach them about its potential benefits and risks. We also hope this information will enable our readers to ask the right questions so that patient and health care provider can work together to help the patient grow old gracefully.
As the series unfolds, we ask you to send us feedback and to suggest other topics in personalized health care you would like us to cover in this series.
- Osler W. Aequanimitas, With Other Addresses to Medical Students, Nurses and Practitioners of Medicine. 2nd edition. Philadelphia, PA: P. Blakiston’s Sone & Co, 1906:348.
- McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med 2011; 364:340–350.
- Eng C. Microenvironmental protection in diffuse large-B-cell lymphoma. N Engl J Med 2008; 359:2379–2381.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med 2010; 363:301–304.
- Osler W. Aequanimitas, With Other Addresses to Medical Students, Nurses and Practitioners of Medicine. 2nd edition. Philadelphia, PA: P. Blakiston’s Sone & Co, 1906:348.
- McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med 2011; 364:340–350.
- Eng C. Microenvironmental protection in diffuse large-B-cell lymphoma. N Engl J Med 2008; 359:2379–2381.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med 2010; 363:301–304.
Exploring the human genome, and relearning genetics by necessity

The ability to scan the entire human genome and to recognize variations in specific nucleotides within recognized genes is more than a technologic feat. It is now possible to assess the risk of some genetic diseases before they are phenotypically expressed. We are increasingly able to predict whether specific drugs will be effective or pose higher risks of adverse effects in individual patients, a field called pharmacogenomics. How much pharmacogenomics can and should be incorporated into our practice as part of personalized medicine remains to be determined,
Genome-wide association studies can answer certain research questions, but also raise additional ones. In some ways, these studies are like molecular epidemiology—they can demonstrate a statistical association between a risk factor and a clinical event such as a heart attack, but just as in traditional epidemiologic studies, association does not always equate with causation.
As discussed by Drs. Manace and Babyatsky in this issue of the Journal, additional techniques can be used to try to sort out the issue of association vs causation—in this case, whether C-reactive protein (CRP) is merely associated with cardiovascular events or is a cause of them. Using the tools of traditional clinical research, it would be ideal to demonstrate that the use of a highly specific inhibitor of the risk factor (CRP) prevents the disease. CRP levels can be lowered with statins, but these drugs also reduce levels of low-density lipoprotein cholesterol, which will lower the risk of cardiac events. Thus, statins do not have the specificity to prove that CRP causes myocardial infarction.
This paper is one of the first in the Journal to discuss advances in genomics that may affect our practice. Beginning in May, the Journal will begin a new series on personalized medicine to highlight the role that genetics and molecular medicine can play in our clinical practice and in our understanding of pathophysiology.
The ability to scan the entire human genome and to recognize variations in specific nucleotides within recognized genes is more than a technologic feat. It is now possible to assess the risk of some genetic diseases before they are phenotypically expressed. We are increasingly able to predict whether specific drugs will be effective or pose higher risks of adverse effects in individual patients, a field called pharmacogenomics. How much pharmacogenomics can and should be incorporated into our practice as part of personalized medicine remains to be determined,
Genome-wide association studies can answer certain research questions, but also raise additional ones. In some ways, these studies are like molecular epidemiology—they can demonstrate a statistical association between a risk factor and a clinical event such as a heart attack, but just as in traditional epidemiologic studies, association does not always equate with causation.
As discussed by Drs. Manace and Babyatsky in this issue of the Journal, additional techniques can be used to try to sort out the issue of association vs causation—in this case, whether C-reactive protein (CRP) is merely associated with cardiovascular events or is a cause of them. Using the tools of traditional clinical research, it would be ideal to demonstrate that the use of a highly specific inhibitor of the risk factor (CRP) prevents the disease. CRP levels can be lowered with statins, but these drugs also reduce levels of low-density lipoprotein cholesterol, which will lower the risk of cardiac events. Thus, statins do not have the specificity to prove that CRP causes myocardial infarction.
This paper is one of the first in the Journal to discuss advances in genomics that may affect our practice. Beginning in May, the Journal will begin a new series on personalized medicine to highlight the role that genetics and molecular medicine can play in our clinical practice and in our understanding of pathophysiology.
The ability to scan the entire human genome and to recognize variations in specific nucleotides within recognized genes is more than a technologic feat. It is now possible to assess the risk of some genetic diseases before they are phenotypically expressed. We are increasingly able to predict whether specific drugs will be effective or pose higher risks of adverse effects in individual patients, a field called pharmacogenomics. How much pharmacogenomics can and should be incorporated into our practice as part of personalized medicine remains to be determined,
Genome-wide association studies can answer certain research questions, but also raise additional ones. In some ways, these studies are like molecular epidemiology—they can demonstrate a statistical association between a risk factor and a clinical event such as a heart attack, but just as in traditional epidemiologic studies, association does not always equate with causation.
As discussed by Drs. Manace and Babyatsky in this issue of the Journal, additional techniques can be used to try to sort out the issue of association vs causation—in this case, whether C-reactive protein (CRP) is merely associated with cardiovascular events or is a cause of them. Using the tools of traditional clinical research, it would be ideal to demonstrate that the use of a highly specific inhibitor of the risk factor (CRP) prevents the disease. CRP levels can be lowered with statins, but these drugs also reduce levels of low-density lipoprotein cholesterol, which will lower the risk of cardiac events. Thus, statins do not have the specificity to prove that CRP causes myocardial infarction.
This paper is one of the first in the Journal to discuss advances in genomics that may affect our practice. Beginning in May, the Journal will begin a new series on personalized medicine to highlight the role that genetics and molecular medicine can play in our clinical practice and in our understanding of pathophysiology.
Gene-based, rational drug-dosing: An evolving, complex opportunity

For some drugs there is a key step in metabolism, often in a rate-limiting pathway, with an enzyme that has known and detectable polymorphisms that differ dramatically in their ability to affect the drug’s degradation. In theory, by determining the patient’s specific genotype ahead of time, the initial dose of the drug can be determined more rationally. In this issue of the Journal, Kitzmiller et al describe several drugs for which this may be true.
However, for this approach to be practical and cost-effective, several conditions should be met. The drug must be one that needs to be dosed to its therapeutic level rapidly: if there is time to titrate slowly, then there is little need for the extra expense associated with genotyping in order to titrate it more rapidly. Also, it should be proven that dosing based on advance knowledge of the genotype of the target actually results in safer or more efficacious dosing.
For carbamazepine (Tegretol, Equetro) and allopurinol (Zyloprim), specific human leukocyte antigen haplotypes are associated with a strikingly increased frequency of serious hypersensitivity reactions. In some patients, these should be checked before giving the drug.
But the concept of pharmacogenomics is broad, and it may yet explain many vagaries of drug-responsiveness in individual patients. Polymorphisms in renal anion transporters may dictate the level of anionic drugs. Drug-receptor polymorphisms may determine the affinity of a drug for its target and, hence, its efficacy. Cell-membrane transporters, which may have functionally different stable alleles or polymorphisms, may regulate intracellular drug levels by pumping the drug into or out of cells with different efficiencies.
As the entire human genome is dissected and analyzed, and as more and more genes (with their polymorphisms) are linked to specific functions readily detectable in specific patients, we will have more opportunities to match the right drug and dose to the right patient. We are not there yet, but that day is coming.
For some drugs there is a key step in metabolism, often in a rate-limiting pathway, with an enzyme that has known and detectable polymorphisms that differ dramatically in their ability to affect the drug’s degradation. In theory, by determining the patient’s specific genotype ahead of time, the initial dose of the drug can be determined more rationally. In this issue of the Journal, Kitzmiller et al describe several drugs for which this may be true.
However, for this approach to be practical and cost-effective, several conditions should be met. The drug must be one that needs to be dosed to its therapeutic level rapidly: if there is time to titrate slowly, then there is little need for the extra expense associated with genotyping in order to titrate it more rapidly. Also, it should be proven that dosing based on advance knowledge of the genotype of the target actually results in safer or more efficacious dosing.
For carbamazepine (Tegretol, Equetro) and allopurinol (Zyloprim), specific human leukocyte antigen haplotypes are associated with a strikingly increased frequency of serious hypersensitivity reactions. In some patients, these should be checked before giving the drug.
But the concept of pharmacogenomics is broad, and it may yet explain many vagaries of drug-responsiveness in individual patients. Polymorphisms in renal anion transporters may dictate the level of anionic drugs. Drug-receptor polymorphisms may determine the affinity of a drug for its target and, hence, its efficacy. Cell-membrane transporters, which may have functionally different stable alleles or polymorphisms, may regulate intracellular drug levels by pumping the drug into or out of cells with different efficiencies.
As the entire human genome is dissected and analyzed, and as more and more genes (with their polymorphisms) are linked to specific functions readily detectable in specific patients, we will have more opportunities to match the right drug and dose to the right patient. We are not there yet, but that day is coming.
For some drugs there is a key step in metabolism, often in a rate-limiting pathway, with an enzyme that has known and detectable polymorphisms that differ dramatically in their ability to affect the drug’s degradation. In theory, by determining the patient’s specific genotype ahead of time, the initial dose of the drug can be determined more rationally. In this issue of the Journal, Kitzmiller et al describe several drugs for which this may be true.
However, for this approach to be practical and cost-effective, several conditions should be met. The drug must be one that needs to be dosed to its therapeutic level rapidly: if there is time to titrate slowly, then there is little need for the extra expense associated with genotyping in order to titrate it more rapidly. Also, it should be proven that dosing based on advance knowledge of the genotype of the target actually results in safer or more efficacious dosing.
For carbamazepine (Tegretol, Equetro) and allopurinol (Zyloprim), specific human leukocyte antigen haplotypes are associated with a strikingly increased frequency of serious hypersensitivity reactions. In some patients, these should be checked before giving the drug.
But the concept of pharmacogenomics is broad, and it may yet explain many vagaries of drug-responsiveness in individual patients. Polymorphisms in renal anion transporters may dictate the level of anionic drugs. Drug-receptor polymorphisms may determine the affinity of a drug for its target and, hence, its efficacy. Cell-membrane transporters, which may have functionally different stable alleles or polymorphisms, may regulate intracellular drug levels by pumping the drug into or out of cells with different efficiencies.
As the entire human genome is dissected and analyzed, and as more and more genes (with their polymorphisms) are linked to specific functions readily detectable in specific patients, we will have more opportunities to match the right drug and dose to the right patient. We are not there yet, but that day is coming.
Pharmacogenomics for the primary care provider: Why should we care?
Since the human genome was sequenced in 2000, the American public has continued to hold hope that our growing understanding of genetics will revolutionize the practice of medicine.
One way genetics promises to improve the quality and value of health care is in personalized medicine, by helping us tailor treatment to a person’s individual genetic makeup. One such approach is called pharmacogenomics.
Pharmacogenomics uses knowledge of a person’s genetics to understand how a particular drug will work, or not work, in his or her body. For instance, some people might carry genes that make them more sensitive than average to a drug, and therefore they would require a lower dose. Others might have genes that make them resistant to the drug, meaning the drug is ineffective unless they receive a higher dose.
Adverse drug reactions are a leading cause of death in hospitalized patients in the United States and are responsible for billions of dollars in health care costs.1,2 Our current practice of prescribing for adult patients is largely trial-and-error, with the same dose given to all patients, in many cases with little regard even to sex, height, or weight.
Pharmacogenomics promises to change this way of prescribing to a customized approach that uses genetic information to predict an individual’s response to medications. It is one piece of an overall initiative to personalize patient care based on the patient’s individual characteristics and preferences.
OVERCOMING BARRIERS TO USING PHARMACOGENOMICS IN PRACTICE
If personalized medicine has promised to improve the quality and value of health care for our patients, why have we been so slow to adopt this information in clinical practice?
The usual barriers to clinical adoption certainly exist. We need further studies to determine whether genetic-based prescribing is truly valid, and for which patient populations. We need to determine whether this approach is cost-effective and better than the current standard of care. We need to work on payment options.
However, one of the largest barriers for busy primary care physicians is the lack of time to keep up with new information. Many practicing physicians were taught little about formal genetics in medical school. The body of scientific literature on pharmacogenomics is fragmented, and it crosses disease states and specialties, making it difficult to unite. Given the breadth of diseases treated and drugs prescribed by primary care physicians, it is unrealistic for most to keep track of the vast body of literature of pharmacogenomic testing and to decipher how to apply this to clinical practice.
In this issue of the Journal, Kitzmiller et al3 provide one solution to this problem, giving an overview of pharmacogenomic applications that might be pertinent to practicing physicians. However, as we try to make pharmacogenomics accessible to busy physicians, we need other solutions to integrate pharmacogenomic information efficiently into the clinical work flow. One approach might be to build pharmacogenomics into the electronic medical record. We can also store the integrated information in research databases and provide clinical recommendations on Internet sites such as www.pharmgkb.org, and we can develop applications to run on cell phones and iPads.
QUESTIONS REMAIN
Kitzmiller et al discuss an important step in this process, highlighting several key questions:
Should we seek genetics-based information to personalize drug selection? Based on the information presented in the literature and in the Kitzmiller paper, there may be circumstances when it is appropriate to consider doing so. While the evidence is not yet compelling to order these tests on a regular basis in clinical practice, this information might be helpful in some situations, such as for patients who have had adverse effects from minimal doses of antidepressants.
For now, clinicians should not abandon their current practice of personalizing patient care on the basis of personal, cultural, and economic preferences. Rather, they should consider pharmacogenomic information an additional piece of information when selecting drug therapy. We should also encourage health care systems and interested providers to be early adopters and to study how their outcomes compare with the standard of care.
Once we have this information, what is our obligation to use it? An increasing number of patients already have genetic information in their health record, either ordered by or provided to their physicians. However, there is little in the scientific literature to guide us in this arena.
Yet most of us would agree that if we have information (genetic or otherwise) that can help to select a drug type or dose or reduce adverse events or costs, we should consider this information in our decision-making. Several circumstances are documented in this paper and in the literature in which prior knowledge about drug metabolism can help in selecting a dose of medication. One example would be the 50% recommended reduction in tricyclic antidepressant dose if the patient is a CYP2D6 poor metabolizer.4
MOVING FORWARD AS A TEAM
In summary, Kitzmiller et al bring to light the promise and the uncertainties that currently exist in the field of pharmacogenomics. While it is unclear if we should incorporate pharmacogenomic tests into standard medical practice at this time, it is clear that this information is becoming more readily available, whether or not we have requested it. Some would argue that, once we have the information, we have an obligation to use it, just as we use other information in our clinical decision-making. This means we need to develop tools and resources to help practitioners evaluate pharmacogenomic data and incorporate it into clinical care in an efficient manner.
The authors also highlight the need for more education about drug metabolism in general, and they cite several instances in which knowledge of drug interactions and metabolism can clearly influence decision-making. An example is paroxetine (Paxil) inhibition of tamoxifen (Nolvadex).5
Lastly, regardless of our personal feelings about the clinical usefulness of genetic testing in large populations, we need to work together to determine clinical utility and validity and to develop efficient ways to put into practice findings that could affect patient care. As we move forward, we need to work as a team, utilizing our clinical partners—pharmacists, pharmacologists, metabolism and health information technology experts, and medical geneticists. Working as a team, pooling our resources and tools, we move closer to providing world-class personalized health care.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events among older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice. Why drugs work in some patients but not others. Cleve Clin J Med 2011; 78:243–257.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Schwarz EB, McNamara M, Miller RG, Walsh JM. Update in women’s health for the general internist. J Gen Intern Med201; 26:207–213.
Since the human genome was sequenced in 2000, the American public has continued to hold hope that our growing understanding of genetics will revolutionize the practice of medicine.
One way genetics promises to improve the quality and value of health care is in personalized medicine, by helping us tailor treatment to a person’s individual genetic makeup. One such approach is called pharmacogenomics.
Pharmacogenomics uses knowledge of a person’s genetics to understand how a particular drug will work, or not work, in his or her body. For instance, some people might carry genes that make them more sensitive than average to a drug, and therefore they would require a lower dose. Others might have genes that make them resistant to the drug, meaning the drug is ineffective unless they receive a higher dose.
Adverse drug reactions are a leading cause of death in hospitalized patients in the United States and are responsible for billions of dollars in health care costs.1,2 Our current practice of prescribing for adult patients is largely trial-and-error, with the same dose given to all patients, in many cases with little regard even to sex, height, or weight.
Pharmacogenomics promises to change this way of prescribing to a customized approach that uses genetic information to predict an individual’s response to medications. It is one piece of an overall initiative to personalize patient care based on the patient’s individual characteristics and preferences.
OVERCOMING BARRIERS TO USING PHARMACOGENOMICS IN PRACTICE
If personalized medicine has promised to improve the quality and value of health care for our patients, why have we been so slow to adopt this information in clinical practice?
The usual barriers to clinical adoption certainly exist. We need further studies to determine whether genetic-based prescribing is truly valid, and for which patient populations. We need to determine whether this approach is cost-effective and better than the current standard of care. We need to work on payment options.
However, one of the largest barriers for busy primary care physicians is the lack of time to keep up with new information. Many practicing physicians were taught little about formal genetics in medical school. The body of scientific literature on pharmacogenomics is fragmented, and it crosses disease states and specialties, making it difficult to unite. Given the breadth of diseases treated and drugs prescribed by primary care physicians, it is unrealistic for most to keep track of the vast body of literature of pharmacogenomic testing and to decipher how to apply this to clinical practice.
In this issue of the Journal, Kitzmiller et al3 provide one solution to this problem, giving an overview of pharmacogenomic applications that might be pertinent to practicing physicians. However, as we try to make pharmacogenomics accessible to busy physicians, we need other solutions to integrate pharmacogenomic information efficiently into the clinical work flow. One approach might be to build pharmacogenomics into the electronic medical record. We can also store the integrated information in research databases and provide clinical recommendations on Internet sites such as www.pharmgkb.org, and we can develop applications to run on cell phones and iPads.
QUESTIONS REMAIN
Kitzmiller et al discuss an important step in this process, highlighting several key questions:
Should we seek genetics-based information to personalize drug selection? Based on the information presented in the literature and in the Kitzmiller paper, there may be circumstances when it is appropriate to consider doing so. While the evidence is not yet compelling to order these tests on a regular basis in clinical practice, this information might be helpful in some situations, such as for patients who have had adverse effects from minimal doses of antidepressants.
For now, clinicians should not abandon their current practice of personalizing patient care on the basis of personal, cultural, and economic preferences. Rather, they should consider pharmacogenomic information an additional piece of information when selecting drug therapy. We should also encourage health care systems and interested providers to be early adopters and to study how their outcomes compare with the standard of care.
Once we have this information, what is our obligation to use it? An increasing number of patients already have genetic information in their health record, either ordered by or provided to their physicians. However, there is little in the scientific literature to guide us in this arena.
Yet most of us would agree that if we have information (genetic or otherwise) that can help to select a drug type or dose or reduce adverse events or costs, we should consider this information in our decision-making. Several circumstances are documented in this paper and in the literature in which prior knowledge about drug metabolism can help in selecting a dose of medication. One example would be the 50% recommended reduction in tricyclic antidepressant dose if the patient is a CYP2D6 poor metabolizer.4
MOVING FORWARD AS A TEAM
In summary, Kitzmiller et al bring to light the promise and the uncertainties that currently exist in the field of pharmacogenomics. While it is unclear if we should incorporate pharmacogenomic tests into standard medical practice at this time, it is clear that this information is becoming more readily available, whether or not we have requested it. Some would argue that, once we have the information, we have an obligation to use it, just as we use other information in our clinical decision-making. This means we need to develop tools and resources to help practitioners evaluate pharmacogenomic data and incorporate it into clinical care in an efficient manner.
The authors also highlight the need for more education about drug metabolism in general, and they cite several instances in which knowledge of drug interactions and metabolism can clearly influence decision-making. An example is paroxetine (Paxil) inhibition of tamoxifen (Nolvadex).5
Lastly, regardless of our personal feelings about the clinical usefulness of genetic testing in large populations, we need to work together to determine clinical utility and validity and to develop efficient ways to put into practice findings that could affect patient care. As we move forward, we need to work as a team, utilizing our clinical partners—pharmacists, pharmacologists, metabolism and health information technology experts, and medical geneticists. Working as a team, pooling our resources and tools, we move closer to providing world-class personalized health care.
Since the human genome was sequenced in 2000, the American public has continued to hold hope that our growing understanding of genetics will revolutionize the practice of medicine.
One way genetics promises to improve the quality and value of health care is in personalized medicine, by helping us tailor treatment to a person’s individual genetic makeup. One such approach is called pharmacogenomics.
Pharmacogenomics uses knowledge of a person’s genetics to understand how a particular drug will work, or not work, in his or her body. For instance, some people might carry genes that make them more sensitive than average to a drug, and therefore they would require a lower dose. Others might have genes that make them resistant to the drug, meaning the drug is ineffective unless they receive a higher dose.
Adverse drug reactions are a leading cause of death in hospitalized patients in the United States and are responsible for billions of dollars in health care costs.1,2 Our current practice of prescribing for adult patients is largely trial-and-error, with the same dose given to all patients, in many cases with little regard even to sex, height, or weight.
Pharmacogenomics promises to change this way of prescribing to a customized approach that uses genetic information to predict an individual’s response to medications. It is one piece of an overall initiative to personalize patient care based on the patient’s individual characteristics and preferences.
OVERCOMING BARRIERS TO USING PHARMACOGENOMICS IN PRACTICE
If personalized medicine has promised to improve the quality and value of health care for our patients, why have we been so slow to adopt this information in clinical practice?
The usual barriers to clinical adoption certainly exist. We need further studies to determine whether genetic-based prescribing is truly valid, and for which patient populations. We need to determine whether this approach is cost-effective and better than the current standard of care. We need to work on payment options.
However, one of the largest barriers for busy primary care physicians is the lack of time to keep up with new information. Many practicing physicians were taught little about formal genetics in medical school. The body of scientific literature on pharmacogenomics is fragmented, and it crosses disease states and specialties, making it difficult to unite. Given the breadth of diseases treated and drugs prescribed by primary care physicians, it is unrealistic for most to keep track of the vast body of literature of pharmacogenomic testing and to decipher how to apply this to clinical practice.
In this issue of the Journal, Kitzmiller et al3 provide one solution to this problem, giving an overview of pharmacogenomic applications that might be pertinent to practicing physicians. However, as we try to make pharmacogenomics accessible to busy physicians, we need other solutions to integrate pharmacogenomic information efficiently into the clinical work flow. One approach might be to build pharmacogenomics into the electronic medical record. We can also store the integrated information in research databases and provide clinical recommendations on Internet sites such as www.pharmgkb.org, and we can develop applications to run on cell phones and iPads.
QUESTIONS REMAIN
Kitzmiller et al discuss an important step in this process, highlighting several key questions:
Should we seek genetics-based information to personalize drug selection? Based on the information presented in the literature and in the Kitzmiller paper, there may be circumstances when it is appropriate to consider doing so. While the evidence is not yet compelling to order these tests on a regular basis in clinical practice, this information might be helpful in some situations, such as for patients who have had adverse effects from minimal doses of antidepressants.
For now, clinicians should not abandon their current practice of personalizing patient care on the basis of personal, cultural, and economic preferences. Rather, they should consider pharmacogenomic information an additional piece of information when selecting drug therapy. We should also encourage health care systems and interested providers to be early adopters and to study how their outcomes compare with the standard of care.
Once we have this information, what is our obligation to use it? An increasing number of patients already have genetic information in their health record, either ordered by or provided to their physicians. However, there is little in the scientific literature to guide us in this arena.
Yet most of us would agree that if we have information (genetic or otherwise) that can help to select a drug type or dose or reduce adverse events or costs, we should consider this information in our decision-making. Several circumstances are documented in this paper and in the literature in which prior knowledge about drug metabolism can help in selecting a dose of medication. One example would be the 50% recommended reduction in tricyclic antidepressant dose if the patient is a CYP2D6 poor metabolizer.4
MOVING FORWARD AS A TEAM
In summary, Kitzmiller et al bring to light the promise and the uncertainties that currently exist in the field of pharmacogenomics. While it is unclear if we should incorporate pharmacogenomic tests into standard medical practice at this time, it is clear that this information is becoming more readily available, whether or not we have requested it. Some would argue that, once we have the information, we have an obligation to use it, just as we use other information in our clinical decision-making. This means we need to develop tools and resources to help practitioners evaluate pharmacogenomic data and incorporate it into clinical care in an efficient manner.
The authors also highlight the need for more education about drug metabolism in general, and they cite several instances in which knowledge of drug interactions and metabolism can clearly influence decision-making. An example is paroxetine (Paxil) inhibition of tamoxifen (Nolvadex).5
Lastly, regardless of our personal feelings about the clinical usefulness of genetic testing in large populations, we need to work together to determine clinical utility and validity and to develop efficient ways to put into practice findings that could affect patient care. As we move forward, we need to work as a team, utilizing our clinical partners—pharmacists, pharmacologists, metabolism and health information technology experts, and medical geneticists. Working as a team, pooling our resources and tools, we move closer to providing world-class personalized health care.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events among older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice. Why drugs work in some patients but not others. Cleve Clin J Med 2011; 78:243–257.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Schwarz EB, McNamara M, Miller RG, Walsh JM. Update in women’s health for the general internist. J Gen Intern Med201; 26:207–213.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events among older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice. Why drugs work in some patients but not others. Cleve Clin J Med 2011; 78:243–257.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Schwarz EB, McNamara M, Miller RG, Walsh JM. Update in women’s health for the general internist. J Gen Intern Med201; 26:207–213.
Pharmacogenomic testing: Relevance in medical practice
In many patients, certain drugs do not work as well as expected, whereas in other patients they cause toxic effects, even at lower doses. For some patients, the reason may be genetic.
Sizeable minorities of the population carry genetic variants—polymorphisms— that affect their response to various drugs. Thanks to genetic research, our understanding of the variability of drug response has advanced markedly in the last decade. Many relevant polymorphisms have been identified, and tests for some of them are available.
Armed with the knowledge of their patients’ genetic status, physicians could predict their response to certain drugs, leading to better efficacy, fewer adverse drug reactions, and a better cost-benefit ratio.
The possible impact is substantial, since more than half of the drugs most commonly involved in adverse drug reactions are metabolized by polymorphic enzymes.1 Adverse drug reactions remain a significant detriment to public health, having a substantial impact on rates of morbidity and death and on healthcare costs.2–8 In the United States, adverse drug reactions are a leading cause of death in hospitalized patients4 and are annually responsible for hundreds of thousands of deaths and hundreds of billions of dollars in added costs.2,4,6–8

In the meantime, physicians can educate their patients and promote efforts to incorporate genomic information into standard clinical decision-making.
This article offers an overview of pharmacogenomic testing, discussing implications and limitations of a few validated tests. Specifically, we will discuss testing that is relevant when using warfarin (Coumadin), clopidogrel (Plavix), statins, tamoxifen (Nolvadex), codeine, and psychotropic medications, as well as the future role of pharmacogenomic testing in medicine.
WHAT IS PHARMACOGENOMICS?
Pharmacogenomics is the study of how genetic factors relate to interindividual variability of drug response.
Many clinicians may not be familiar with the background and terminology used in the pharmacogenomic literature. Below, a brief review of the terminology is followed by a schematic describing the various stages of research involved in pharmacogenomics and the advancement of a test into standard practice.
The review and schematic may be helpful for evaluating the clinical significance of pharmacogenomics-related articles.
From genotype to phenotype
Genotype refers to the coding sequence of DNA base pairs for a particular gene, and phenotype (eg, disease or drug response) refers to a trait resulting from the protein product encoded by the gene. The name of a gene often refers to its protein product and is italicized (eg, the CYP3A4 gene encodes for the CYP3A4 enzyme).
Two alleles per autosomal gene (one paternal and one maternal) form the genotype. Heterozygotes possess two different alleles, and homozygotes possess two of the same alleles. The most common allele in a population is referred to as the wild type, and allele frequencies can vary greatly in different populations.9
Most sequence variations are single nucleotide polymorphisms (SNPs, pronounced “snips”), a single DNA base pair substitution that may result in a different gene product. SNPs can be classified as structural RNA polymorphisms (srSNPs), regulatory polymorphisms (rSNPs), or polymorphisms in coding regions (cSNPs)10: srSNPs alter mRNA processing and translation, rSNPs alter transcription, and cSNPs alter protein sequence and function.
Recently, genetic associations with a phenotype have been done on a large scale, with millions of SNPs measured in each of many subjects. This approach, called a genomewide association study or GWAS, has revealed countless candidate genes for clinical traits, but only a few have resulted in a practical clinical application. SNPs may by themselves exert a pharmacokinetic effect (ie, how the body processes the drug), a pharmacodynamic effect (ie, how the drug affects the body), or both, or they may act in concert with other genetic factors. Pharmacodynamic effects can result from a pharmacokinetic effect or can result from variations in a pharmacologic target.
Establishing a genotype-phenotype association can involve clinical studies, animal transgenic studies, or molecular and cellular functional assays.
Clinical applications are emerging

WARFARIN: IMPORTANCE OF CYP2C9, VKORC1
Warfarin is used for the long-term treatment and prevention of thromboembolic events.
This drug has a narrow therapeutic window and shows substantial interpatient dose variability. The start of warfarin therapy is associated with one of the highest rates of adverse events and emergency room visits of any single drug.12 More than 2 million patients start warfarin each year in the United States alone,13 and about 20% of them are hospitalized within the first 6 months because of bleeding due to overanticoagulation.14
The findings from a recent study suggest that pharmacogenomic testing may eventually allow more patients to safely benefit from warfarin therapy. In this large, nationwide, prospective study, hospitalization rates were 30% lower when pharmacogenomic testing was used.14 However, no reduction was seen in the time needed to reach the target international normalized ratio (INR) or in the need for INR checks at 6 months. Furthermore, this study used historical control data, and some or all of the reduction in hospitalization rates may be attributed to more frequent INR checks in the patients who underwent testing than in the historical control group.
A relationship between warfarin dose requirements and the genetic status of CYP2C9, which encodes a major drug-metabolizing enzyme, has been demonstrated in retrospective and prospective studies.15–17
S-warfarin is metabolized by CYP2C9, which is polymorphic
Warfarin contains equal amounts of two isomers, designated S and R. S-warfarin, which is more potent, is metabolized principally by CYP2C9, while R-warfarin is metabolized by CYP1A2, CYP2C19, and CYP3A4.
People who possess two copies of the wild type CYP2C9 gene CYP2C9*1 metabolize warfarin very well and so are called “extensive warfarin metabolizers.” Carriers of the allelic variants CYP2C9*2 and CYP2C9*3 (which have point mutations in exons 3 and 7 of CYP2C9, respectively), have less capacity. Compared with those who are homozygous for the wild-type gene, homozygous carriers of CYP2C9* 3 clear S-warfarin at a rate that is 90% lower, and those with the CYP2C9*1/*3, CYP2C9* 1/*2, CYP2C9*2/*2, or CYP2C9*2/*3 genotypes clear it at a rate 50% to 75% lower. A meta-analysis of 12 studies found that the CYP2C9 genotype accounted for 12% of the interindividual variability of warfarin dose requirements.18
About 8% of whites carry at least one copy of CYP2C9*2, as do 1% of African Americans; the allele is rare in Asian populations. The frequency of CYP2C9*3 is 6% in whites, 1% in African Americans, and 3% in Asians.19,20 People with CYP2C9*4 or CYP2C9*5 have a diminished capacity to clear warfarin; however, these variants occur so infrequently that their clinical relevance may be minimal.
Warfarin’s target, VKOR, is also polymorphic
Genetic variation in warfarin’s pharmacologic target, vitamin K 2,3-epoxide reductase (VKOR), also influences dose requirements. Warfarin decreases the synthesis of vitamin-K-dependent clotting factors by inhibiting VKOR. This inhibition depends on the patient’s C1 subunit gene, VKORC1. Patients with a guanine-to-adenine SNP 1,639 bases upstream of VKORC1 (−1639G>A) need lower warfarin doses—an average of 25% lower in those with the GA genotype (ie, one allele has guanine in the −1639 position and the other allele has adenine in that position) and 50% lower in those with the AA genotype compared with the wild-type genotype GG.21 This promoter SNP, positioned upstream (ie, before the gene-coding region), greatly influences VKORC1 expression.
A meta-analysis of 10 studies found that the VKORC1 polymorphism accounts for 25% of the interindividual variation in warfarin dose.18 In one study, the frequency of the AA genotype in a white population was 14%, AG 47%, and GG 39%; in a Chinese population the frequency of AA was 82%, AG 18%, and GG 0.35%.22
CYP4F2 and GGCX also affect warfarin’s dose requirements
Genetic variations in the enzymes CYP4F2 and gamma-glutamyl carboxylase (GGCX) also influence warfarin dose requirements. Although the data are limited and the effects are smaller than those of CYP2C9 and VKORC1, people with a SNP in CYP4F2 need 8% higher doses of warfarin, while those with a SNP in GGCX need 6% lower doses.23
CYP2C9 and VKORC1 testing is available
Currently, the clinical pharmacogenetic tests relevant for warfarin use are for CYP2C9 and VKORC1.10
The FDA has approved four warfarin pharmacogenetic test kits, but most third-party payers are reluctant to reimburse for testing because it is not currently considered a standard of care. Testing typically costs a few hundred dollars, but it should become less expensive as it becomes more commonplace. The current FDA-approved product label for warfarin does not recommend routine pharmacogenomic testing for determining initial or maintenance doses, but it does acknowledge that dose requirements are influenced by CYP2C9 and VKORC1 and states that genotype information, when available, can assist in selecting the starting dose.24
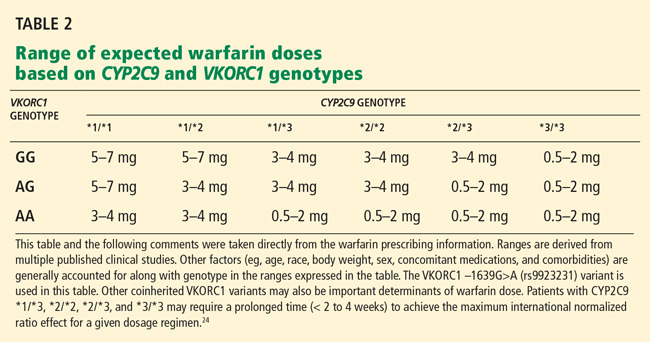
Clinical trials of warfarin pharmacogenomic testing are under way
Although genetic status can greatly influence an individual patient’s warfarin dosing requirement, routine prospective pharmacogenomic testing is not endorsed by the FDA or by other expert panels26 because there is currently insufficient evidence to recommend for or against it.
Several large prospective trials are under way. For example, the National Heart, Lung, and Blood Institute began a prospective trial in about 1,200 patients to evaluate the use of clinical plus genetic information to guide the initiation of warfarin therapy and to improve anticoagulation control for patients.27 The results, expected in September 2011, and those of other large prospective trials should provide adequate evidence for making recommendations about the clinical utility of routine pharmacogenetic testing for guiding warfarin therapy.
Several recent cost-utility and cost-effectiveness studies have attempted to quantify the value of pharmacogenomic testing for warfarin therapy,28–30 but their analyses are largely limited because the benefit (clinical utility) is yet to be sufficiently characterized.
The relevance of such analyses may soon be drastically diminished, as several non-vitamin-K-dependent blood thinners such as rivaroxaban (Xarelto), dabigatran (Pradaxa), and apixaban are poised to enter clinical practice.31
CLOPIDOGREL IS ACTIVATED BY CYP2C19
Clopidogrel, taken by about 40 million patients worldwide, is used to prevent atherothrombotic events and cardiac stent thrombosis when given along with aspirin.
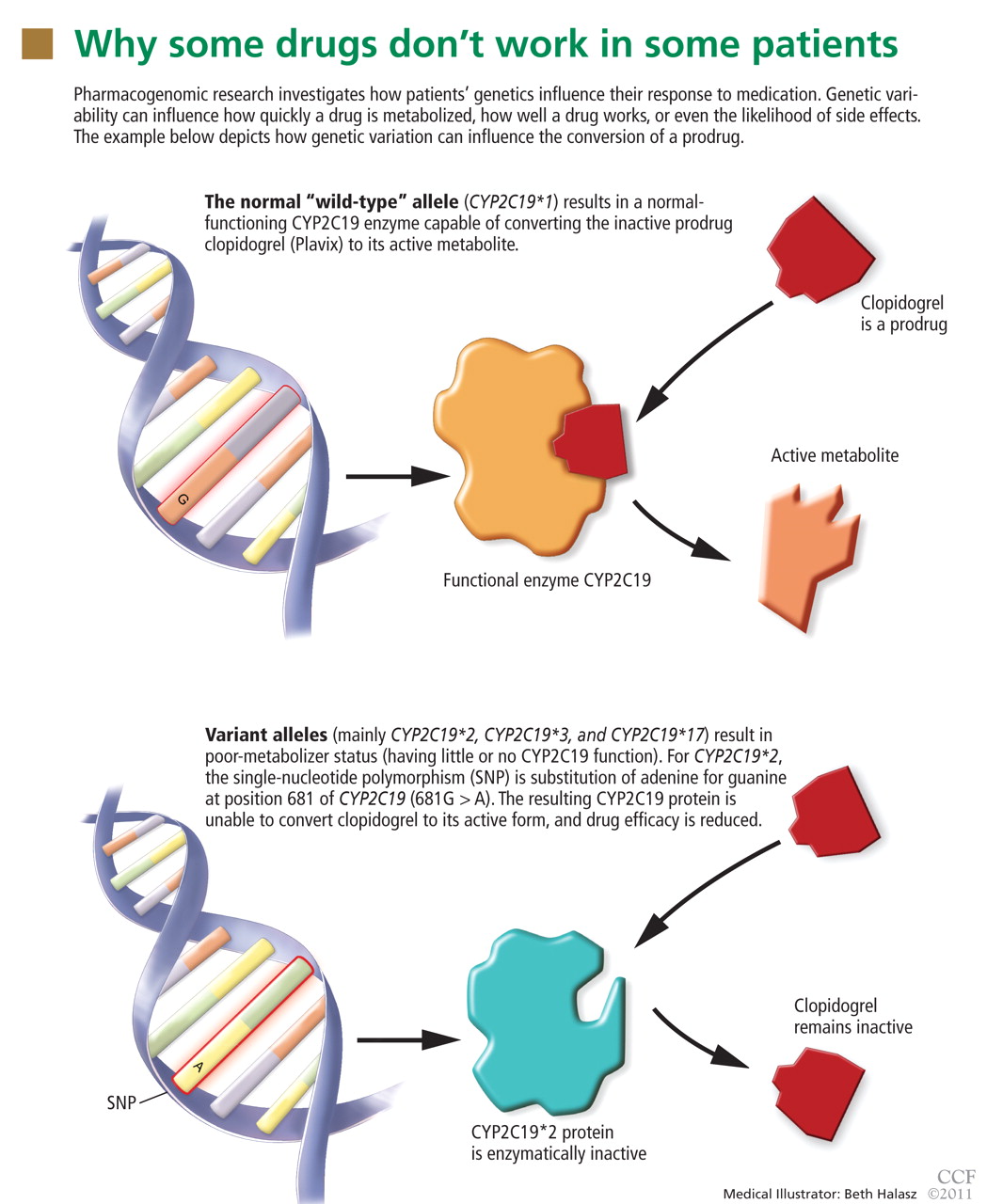
Studies of clopidogrel pharmacogenomics
A recent genome-wide association study conducted in a cohort of 429 healthy Amish persons revealed a SNP in CYP2C19 to be associated with a diminished response to clopidogrel and to account for 12% of the variation in drug response.33 Traditional factors (the patient’s age, body-mass index, and cholesterol level) combined accounted for less than 10% of the variation.
Findings were similar in a subsequent investigation in 227 cardiac patients receiving clopidogrel: 21% of those with the variant had a cardiovascular ischemic event or died during a 1-year follow-up period compared with 10% of those without the variant (hazard ratio 2.42, P = .02).33
A 12-year prospective study investigating clopidogrel efficacy in 300 cardiac patients under the age of 45 used cardiovascular death, nonfatal myocardial infarction, and urgent coronary revascularization as end points. It concluded that the only independent predictor of these events was the patient’s CYP2C19 status.34
A study in 2,200 patients with recent myocardial infarction examined whether any of the known allelic variations that modulate clopidogrel’s absorption (ABCB1), metabolic activation (CYP3A4/5 and CYP2C19), or biologic activity (P2RY12 and ITGB3) was associated with a higher rate of the combined end point of all-cause mortality, nonfatal myocardial infarction, or stroke. None of the SNPs in CYP3A4/5, P2RY12, or ITGB3 that were evaluated was associated with a higher risk at 1 year. However, the allelic variations modulating clopidogrel’s absorption (ABCB1) and metabolism (CYP2C19) were associated with higher event rates. Patients with two variant ABCB1 alleles had a higher adjusted hazard ratio (95% confidence interval [CI] 1.2–2.47) than those with the wild-type allele. Patients who had one or two CYP2C19 loss-of-function alleles had a higher event rate than those with two wild-type alleles (95% CI 1.10–3.58 and 1.71–7.51, respectively).35
Conversely, researchers from the Population Health Research Institute found no association between poor-metabolizer status and treatment outcomes when CYP2C19 analysis was retrospectively added to the findings of two large clinical trials (combined N > 5,000). However, patients with acute coronary syndrome benefited more from clopidogrel treatment if they were ultra-rapid metabolizers (possessing the gain-of-function allele CYP2C19*17).36
Current status of clopidogrel testing: Uncertain
A current FDA boxed warning states that poor CYP2C19 metabolizers may not benefit from clopidogrel and recommends that prescribers consider alternative treatment for patients in this category.37 However, routine CYP2C19 testing is not recommended, and no firm recommendations have been established regarding dose adjustments for CYP2C19 status.

In 2010, the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association collectively pronounced the current evidence base insufficient for recommending routine pharmacogenomic testing.39
Needed are large-scale studies examining the cost-effectiveness and clinical utility of genotype-guided clopidogrel therapy compared with other therapy options such as prasugrel (Effient), an analogue not metabolized by CYP2C19. One such study, sponsored by Medco Health Solutions, plans to enroll 14,600 cardiac patients and has an estimated completion date in June 2011.40 The expectation that clopidogrel will be available in generic form in 2012 adds to the uncertainty regarding the cost-effectiveness of CYP2C19 testing for clopidogrel therapy.
STATINS: SLC01B1*5 INCREASES MYOPATHY RISK
Statins lower the concentration of low-density lipoprotein cholesterol (LDL-C), resulting in a relative-risk reduction of about 20% for each 1 mmol/L (39 mg/dL) decrement in LDL-C.41 They are one of the most commonly prescribed classes of drugs, but their side effects can limit their appeal: statin-induced myopathy occurs in about 1:1,000 to 1:10,000 patients and is difficult to predict.
SLC01B1. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH), a genome-wide association study, recently found a SNP (SLCO1B1* 5) in the SLC01B1 gene to be associated with a higher risk of statin-induced myopathy in cardiac patients receiving simvastatin (Zocor) 40 or 80 mg daily.42 The SLC01B1 gene, located on chromosome 12, influences the extent of the drug’s hepatic uptake and its serum concentration. Only the SLC01B1*5 SNP emerged as a predictor of statin-induced myopathy across the entire genome.42
The authors believe the findings are likely to apply to other statins. The mechanisms leading to statin-induced myopathy and the impact of statin pharmacogenomics are still unclear.43
CYP3A4. Other genetic variants may play a vital role in determining response to statin therapy. Carriers of a newly identified CYP3A4 polymorphism (intron 6 SNP, rs35599367, C>T) required significantly lower statin doses (0.2–0.6 times less) for optimal lipid control. The analyses included atorvastatin (Lipitor), simvastatin, and lovastatin (Mevacor), and the association was robust (P = .019).44
Statin pharmacogenomic testing is not routinely recommended
Routine pharmacogenomic testing for statin therapy is not recommended. Additional studies are needed to determine the clinical utility and cost-effectiveness of pharmacogenomic testing (involving a combination of several polymorphisms) in various patient populations delineated by type of statin, dose, and concomitant use of other drugs.
TAMOXIFEN IS ACTIVATED BY CYP2D6
Tamoxifen is prescribed to prevent the recurrence of estrogen-receptor-positive breast cancer, to treat metastatic breast cancer, to prevent cancer in high-risk populations, and to treat ductal carcinoma in situ.
Tamoxifen is metabolized to form endoxifen, which has much higher potency and higher systemic levels than tamoxifen.45 Both CYP2D6 and CYP3A4/5 are required to produce endoxifen via two intermediates, but CYP2D6 catalyzes the critical step leading to metabolic activation.
The CYP2D6 gene is highly polymorphic, with more than 75 allelic variants identified. Extensive literature is available describing the influence of CYP2D6 polymorphisms on tamoxifen metabolism and therapy outcomes.46–52 Several CYP2D6 variants result in reduced or no enzyme activity, and people who have more than two normally functioning alleles have exaggerated enzyme activity (gene amplification).
Classification of CYP2D6 status
Several systems have been developed to categorize the phenotypic activity of CYP2D6 based on genotype.
A genetic basis for the observed diversity in the metabolism of cytochrome P450 substrates was recognized more than 30 years ago. People were categorized as either extensive or poor metabolizers, reflecting normal vs impaired ability to metabolize the CYP2D6 substrates sparteine and debrisoquine. Later work expanded this system to include categories for intermediate (between poor and extensive) and ultra-rapid (better than extensive) metabolizers.
The genetic basis for these categories includes homozygosity for dysfunctional variants (the poor-metabolizer group) or extra copies of normal functioning variants (the ultra-rapid-metabolizer group).
Newer systems have been described for characterizing the CYP2D6 activity phenotype whereby CYP2D6 variants are assigned activity scores.53–56 The various scoring systems have been reviewed by Kirchheiner.57

A recent version of the activity scoring system also takes into consideration the many drugs that inhibit CYP2D6, such as amiodarone (Cordarone) and fluoxetine (Prozac) that can reduce the action of tamoxifen if given with it (Table 4).58 For example, the tamoxifen exposure (as predicted by the CYP2D6-activity score) experienced by a CYP2D6 extensive metabolizer taking a CYP2D6-inhibiting drug may be similar to the exposure experienced by a CYP2D6 poor metabolizer receiving the same tamoxifen dose but not taking a CYP2D6-inhibiting drug.
Likewise, the activity score of a CYP2D6 intermediate metabolizer taking a CYP2D6-inducing drug may be similar to that of a CYP2D6 ultra-rapid metabolizer not taking a CYP2D6-inducing drug. Examples of CYP2D6 inducers are dexamethasone, rifampin, and hyperforin (St. John’s wort).
While the newer systems are reported to provide better correlations between genotype and phenotype scores, the older scoring systems and the categorical names are still widely used (eg, in the FDA-approved AmpliChip CYP450 test from Roche,59 which includes genotype data for CYP2D6 and CYP2C19).
No firm recommendations for CYP2D6 testing in tamoxifen users
The different genotypes and phenotypes vary in prevalence in different ethnic groups, and significantly different activity levels for endoxifen formation are observed. Tamoxifen lacks efficacy in those who are poor CYP2D6 metabolizers—ie, about 7% of the white population.
However, the FDA has not made firm recommendations about CYP2D6 testing for prescribing tamoxifen because the evidence of benefit, although suggestive, has been considered insufficient.
Clinicians should be aware that tamoxifen’s efficacy is greatly reduced by concomitant therapy with CYP2D6-inhibiting drugs (Table 4).
Other genes affecting tamoxifen: CYP3A4/5, SULT1A1, and UGT2B15
Some investigators propose that polymorphisms in additional genes encoding enzymes in the tamoxifen metabolic and elimination pathways (eg, CYP3A4/5, SULT1A1, and UGT2B15) also need to be considered to account adequately for interindividual variation in drug response.
For example, CYP3A4 and CYP3A5 are also polymorphic, and large interindividual variation exists in their enzyme activities. These enzymes have overlapping substrate specificities, represent the most abundant drug-metabolizing enzymes in the human liver, and are involved in the biotransformation of a broad range of endogenous substrates and most drugs.60
Clinical studies evaluating the impact of CYP3A4/5 polymorphisms have been inconsistent in their conclusions, which is generally attributed to the relatively low functional impact or the low prevalence of the SNPs evaluated. Many of the nearly 100 CYP3A4/5 polymorphisms identified have not yet been characterized regarding their functional impact on enzyme expression or activity. CYP-3A4/5 enzyme activity is highly variable between individuals and warrants further study of its role in outcomes of tamoxifen therapy. Ongoing and future prospective clinical trials evaluating CYP2D6, CYP3A4/5, and other relevant polymorphisms are necessary to define their clinical relevance before routine genetic testing for tamoxifen can be justified.
CODEINE IS ALSO ACTIVATED BY CYP2D6
Codeine also depends on the CYP2D6 gene, as it must be activated to its more potent opioid metabolites, including morphine. Poor CYP2D6 metabolizers do not benefit from codeine therapy.
The pharmacogenomics of codeine has become a hot topic, especially regarding breast-feeding mothers. The debate was ignited with the publication in 2006 of a case report of an infant’s death, apparently the result of metabolic polymorphisms.61 The evolution of this debate and the outcome of the case may be noteworthy to clinicians, as they illustrate the gravity of public and patient interest in pharmacogenomic testing. In this case, the breast-feeding mother had taken codeine regularly for about 14 days when her 13-day-old infant died from toxic levels of morphine. Unknown to her and the prescriber, both the mother and infant were ultra-rapid CYP2D6 metabolizers, resulting in a more rapid and extensive conversion of codeine to morphine.
A logical strategy for preventing similar deaths would be routine CYP2D6 genotyping when prescribing codeine to breast-feeding mothers. However, after several investigations examined the metabolic and excretion pathways of codeine in their entirety, the FDA did not recommend routine CYP2D6 testing when prescribing codeine to breastfeeding mothers because several other factors, including rare genetic variations of other enzymes, proved necessary for reaching the opioid toxicity leading to the infant’s death.62
PHARMACOGENOMICS OF PSYCHOTROPIC DRUGS
Pharmacogenomic testing has clinical utility for some psychotropic drugs.
HLA-B and carbamazepine
Considered a standard of care, HLA-B genotyping is appropriate before prescribing carbamazepine (Tegretol, Equetro) to patients in populations in which HLAB*1502 is likely to be present, such as Asians. Carriers of HLAB* 1502 are at higher risk of life-threatening skin reactions such as Stevens-Johnson syndrome.11
Several other pharmacogenomic applications for psychotropic medications have been suggested, but routine testing has not been recommended by the FDA or endorsed by any expert panel because sufficient clinical utility and cost-effectiveness have not been demonstrated. A brief summary of study findings and a few practical suggestions follow.
Polymorphisms in metabolizing enzymes have been investigated in patients receiving psychotropic drugs.
CYP2D6 and antidepressants
Many antidepressants show significant differences in plasma drug levels with CYP2D6 polymorphisms (in descending order of influence)55:
- Imipramine (Tofranil)
- Doxepin (Adapin, Silenor, Sinequan)
- Maprotiline (Deprilept, Ludiomil, Psymion)
- Trimipramine (Surmontil)
- Desipramine (Noraprim)
- Nortriptyline (Aventyl, Pamelor)
- Clomipramine (Anafranil)
- Paroxetine (Paxil)
- Venlafaxine (Effexor)
- Amitriptyline (Elavil)
- Mianserin
- Trazadone (Desyrel)
- Bupropion (Wellbutrin)
- Nefazodone (Serzone)
- Citalopram (Celexa)
- Sertraline (Zoloft).
CYP2D6 and antipsychotics
Several antipsychotics are also influenced by CYP2D6 polymorphisms (also in descending order of influence)55:
- Perphenazine (Trilafon)
- Thioridazine (Mellaril)
- Olanzapine (Zyprexa)
- Zuclopenthixol (Cisordinol, Clopixol, Acuphase)
- Aripiprazole (Abilify)
- Flupentixol (Depixol, Fluanxol)
- Haloperidol (Haldol)
- Perazine (Taxilan)
- Risperidone (Risperdal)
- Pimozide (Orap).
CYP2C19 and antidepressants
CYP2C19 polymorphisms are likewise associated with differences in drug metabolism for many antidepressants, such as (in descending order of CYP2C19-mediated influence)55:
- Trimipramine
- Doxepin
- Amitriptyline
- Imipramine
- Citalopram (Celexa)
- Clomipramine
- Moclobemide (Aurorix, Manerix)
- Sertraline
- Fluvoxamine (Luvox).
Clinical relevance of CYP2D6 and CYP2C19
Several studies have demonstrated that poor and intermediate CYP2D6 metabolizers have a higher incidence of adverse effects when taking CYP2D6-dependent antidepressants63–68; however, an almost equal number of studies did not find statistically significant associations.69–72 Likewise, several studies have found an association between ultra-rapid CYP2D6 metabolizer status and diminished response to antidepressants,65,73,74 but no association was found in a larger retrospective study.75
Routine CYP2D6 and CYP2C19 screening is not recommended when prescribing psychotropic drugs. However, reviews of the pharmacokinetic data have suggested a few practical applications when genetic status is already known. In general, clinicians can consider reducing the dose of tricyclic antidepressants by about 50% when prescribing to CYP2D6-poor-metabolizers.55,76–78
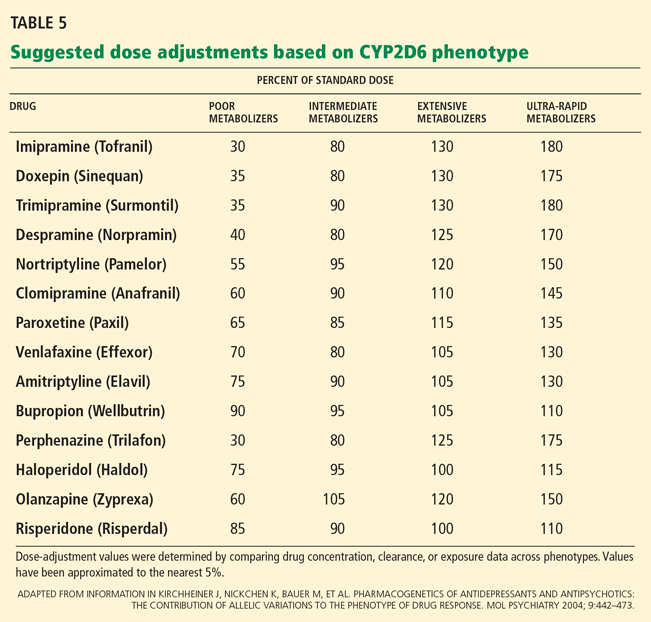
Genes that affect serotonin metabolism
Several genes in the serotonin pathway have been investigated to determine whether they influence patients’ susceptibility to depression and adverse effects and response to psychotropic medications.
SLC6A4. Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 appear to influence the treatment response and side-effect profiles of selective serotonin reuptake inhibitors (SSRIs). Carriers of the SLC6A4 5-HTTLPR L alleles have fewer side effects79 and better response to SSRI treatment, and carriers of the S allele have a higher incidence of antidepressant-induced mania80 and poorer response to SSRI treatment.81
5-HT. Polymorphisms in serotonin receptors (2A and 2C subtypes) appear to influence SSRI response and side effects. Carriers of 5-HT 2A C alleles had more severe adverse effects from paroxetine,71 but another 5-HT 2A polymorphism common to Asians is associated with better response to antidepressant therapy.82 A 5-HT 2C polymorphism was associated with a lower incidence of antipsychotic-induced weight gain.83
Although the understanding of these relationships is incomplete and routine pharmacogenomic testing is not currently recommended, reviews of the pharmacodynamic data have suggested a few practical applications when a patient’s genetic status is already known. One should consider:
- Selecting treatments other than SSRIs for depressed patients known to possess the SLC6A4 variant
- Selecting citalopram for depressed patients known to carry the 5-HT 2A polymorphism
- Avoiding treatment with antipsychotic drugs for patients known to possess the 5-HT 2C polymorphism.
THE FUTURE OF PHARMACOGENOMIC TESTING
The examples discussed in this article provide some insight about how pharmacogenomic testing is maturing and slowly being integrated into the practice of medicine. They also illustrate the complexity of the multiple stages of research that pharmacogenomic applications must go through in order to be adopted as standard practice.
In the future, pharmacogenomic data will continue to accumulate, and the clinical utility of many other pharmacogenomic tests may be uncovered. The FDA provides information on emerging pharmacogenomic tests at its Web site, www.fda.gov.11 Its up-to-date “Table of Valid Genomic Biomarkers in the Context of Approved Drug Labels” includes boxed warnings, recommendations, research outcomes, and relevant population genetics.
If the FDA continues its current policy, prospective randomized trials that show improvement in patient outcomes will remain the gold standard for determining the clinical significance of a pharmacogenomic test. Furthermore, cost-benefit analyses are likely to continue dictating policy regarding pharmacogenomic testing, and cost-benefit profiles should improve as technology advances and as information gathered from a single test becomes applicable to multiple medications and clinical scenarios.
In the meantime, physicians should become familiar with the terms used in medical genetics and pharmacogenomics and begin to understand genetic contributions to the outcomes of drug therapy. For example, understanding the consequences of metabolizer status and the frequency of variants in a given population can be tremendously helpful when advising our patients about anticipating potential problems when taking specific medications and about making informed decisions about pharmacogenomic testing.
This exchange of information alone may go a long way in improving therapy outcomes even when prospective pharmacogenomic testing is not routinely performed. Furthermore, an increasing number of patients will already have genotyping information available when they come to us, and clinicians need to be aware of the many pharmacogenomic applications recommended by the FDA when genetic status is known.10
- King HC, Sinha AA. Gene expression profile analysis by DNA micro-arrays: promise and pitfalls. JAMA 2001; 286:2280–2288.
- Nuckols TK, Paddock SM, Bower AG, et al. Costs of intravenous adverse drug events in academic and nonacademic intensive care units. Med Care 2008; 46:17–24.
- Gurwitz JH, Field TS, Judge J, et al. The incidence of adverse events in two long-term care facilities. Am J Med 2005; 118:251–258.
- Vargas E, Terleira A, Hernando F, et al. Effects of adverse drug reactions on length of stay in surgical intensive care units. Crit Care Med 2003; 31:694–698.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events in older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997; 277:301–306.
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 2001; 41:192–199.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002; 3:229–243.
- Sadee W. Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med 2009; 1:116.
- US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm. Accessed 1/18/2011.
- Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med 2007; 147:755–765.
- Elias DJ, Topol EJ. Warfarin pharmacogenomics: a big step forward for individualized medicine: enlightened dosing of warfarin. Eur J Hum Genet 2008; 16:532–534.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev 2008; 40:355–375.
- García-Martín E, Martínez C, Ladero JM, Agúndez JA. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10:29–40.
- Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6:341–349.
- Wen MS, Lee M, Chen JJ, et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther 2008; 84:83–89.
- Larramendy-Gozalo C, Yang JQ, Verstuyft C, et al. Genetic polymorphism of vitamin K epoxide reductase (VKORC1) 1173C>T in a Chinese and a Caucasian population. Basic Clin Pharmacol Toxicol 2006; 98:611–613.
- Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111:4106–4112.
- Bristol-Myers Squibb. Coumadin (warfarin sodium) Prescribing Information. January 2010.
- Barnes-Jewish Hospital at Washington University Medical Center. Warfarin dosing. http://warfarindosing.orgAccessed 1/20/2011.
- Flockhart DA, O’Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008; 10:139–150.
- National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed January 20, 2011.
- Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 2009; 150:73–83.
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks, and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 2010; 28:61–74.
- Patrick AR, Avron J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes 2009; 2:429–436.
- Haas S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 2008; 25:52–60.
- Bhatt DL. Tailoring antiplatelet therapy based on pharmacogenomics: how well do the data fit? JAMA 2009; 302:896–897.
- Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849–857.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363:1704–1714.
- Bristol-Myers Squibb/Sanofi Pharmaceutical Partnership. Plavix (clopidogrel bisulfate) prescribing information. August 2010.
- P450 Drug Interaction Table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed 1/21/2011.
- Society for Cardiovascular Angiography and Interventions; Holmes DR, Dehmer GJ, Kaul S, Leifer D, O’Gara PT, Stein CM. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010; 122:537–557.
- National Institutes of Health. Genotype Guided Comparison of Clopidogrel and Prasugrel Outcomes Study. http://clinicaltrialsfeeds.org/clinical-trials/show/NCT00995514. Accessed 1/20/2011.
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8:453–463.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 2010; 10:1–11.
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 2010; Apr 13 [Epub ahead of print].
- Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005; 23:9312–9318.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J Pharm Sci 2007; 96:2224–2231.
- Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol 2008; 6:493–494.
- Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin Pharmacol Ther 2008; 83:160–166.
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med 2008; 10:e34.
- Dezentjé VO, Guchelaar HJ, Nortier JW, van del Velde CJ, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009; 15:15–21.
- Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw 2009; 7:203–213.
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009; 9:576–586.
- Steimer W, Zöpf K, von Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clinical Cancer 2004; 50:1623–1633.
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharm Ther 2008; 83:234–242.
- Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004; 9:442–473.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J 2007; 7:257–265.
- Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008; 83:225–227.
- Borges S, Desta Z, Jin Y, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol 2010; 50:450–458.
- Hoffmann-La Roche Ltd. AmpliChip CYP450 Test. http://www.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. Accessed 1/21/2011.
- Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001; 58:737–747.
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368:704.
- Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharm Ther 2009; 86:634–643.
- Grzesiak M, Beszłej A, Lebioda A, Jonkisz A, Dobosz T, Kienja A. [Retrospective assessment of the antidepressants tolerance in the group of patients with diagnosis of depression and different CYP2D6 genotype.] [In Polish] Psychiatr Pol 2003; 37:433–444.
- Laika B, Leucht S, Heres S, Steimer W. Intermediate metabolizer: increased side effects in psychoactive drug therapy. The key to cost-effectiveness of pretreatment CYP2D6 screening? Pharmacogenomics J 2009; 9:395–403.
- Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: Impact on adverse effects and nonresponse during treatment with antidepressants—a pilot study. Clin Pharm Ther 2004; 75:386–393.
- McAlpine DE, O’Kane DJ, Black JL, Mrazek DA. Cytochrome P450 2D6 genotype variation and venlafaxine dosage. Mayo Clin Proc 2007; 82:1065–1068.
- Chen S, Chou WH, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther 1996; 60:522–534.
- Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006; 31:493–502.
- Whyte EM, Romkes M, Mulsant BH, et al. CYP2D6 genotype and venlafaxine-XR concentrations in depressed elderly. Int J Geriatr Psychiatry 2006; 21:542–549.
- Roberts RL, Mulder RT, Joyce PR, Luty SE, Kennedy MA. No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2004; 19:17–23.
- Murphy GM, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 2003; 160:1830–1835.
- Gillman PK. Re: no evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2005; 20:61–62.
- Gex-Fabry M, Eap CB, Oneda B, et al. CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther Drug Monit 2008; 30:474–482.
- Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol 2004; 59:803–807.
- Serretti A, Calati R, Massat I, et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 2009; 24:250–256.
- de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47:75–85.
- de Leon J, Susce MT, Johnson M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr 2009; 14:19–34.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther 2009; 124:57–73.
- Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009; 112:267–272.
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry 2007; 12:247–257.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15:473–500.
- Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003; 160:677–679.
In many patients, certain drugs do not work as well as expected, whereas in other patients they cause toxic effects, even at lower doses. For some patients, the reason may be genetic.
Sizeable minorities of the population carry genetic variants—polymorphisms— that affect their response to various drugs. Thanks to genetic research, our understanding of the variability of drug response has advanced markedly in the last decade. Many relevant polymorphisms have been identified, and tests for some of them are available.
Armed with the knowledge of their patients’ genetic status, physicians could predict their response to certain drugs, leading to better efficacy, fewer adverse drug reactions, and a better cost-benefit ratio.
The possible impact is substantial, since more than half of the drugs most commonly involved in adverse drug reactions are metabolized by polymorphic enzymes.1 Adverse drug reactions remain a significant detriment to public health, having a substantial impact on rates of morbidity and death and on healthcare costs.2–8 In the United States, adverse drug reactions are a leading cause of death in hospitalized patients4 and are annually responsible for hundreds of thousands of deaths and hundreds of billions of dollars in added costs.2,4,6–8
In the meantime, physicians can educate their patients and promote efforts to incorporate genomic information into standard clinical decision-making.
This article offers an overview of pharmacogenomic testing, discussing implications and limitations of a few validated tests. Specifically, we will discuss testing that is relevant when using warfarin (Coumadin), clopidogrel (Plavix), statins, tamoxifen (Nolvadex), codeine, and psychotropic medications, as well as the future role of pharmacogenomic testing in medicine.
WHAT IS PHARMACOGENOMICS?
Pharmacogenomics is the study of how genetic factors relate to interindividual variability of drug response.
Many clinicians may not be familiar with the background and terminology used in the pharmacogenomic literature. Below, a brief review of the terminology is followed by a schematic describing the various stages of research involved in pharmacogenomics and the advancement of a test into standard practice.
The review and schematic may be helpful for evaluating the clinical significance of pharmacogenomics-related articles.
From genotype to phenotype
Genotype refers to the coding sequence of DNA base pairs for a particular gene, and phenotype (eg, disease or drug response) refers to a trait resulting from the protein product encoded by the gene. The name of a gene often refers to its protein product and is italicized (eg, the CYP3A4 gene encodes for the CYP3A4 enzyme).
Two alleles per autosomal gene (one paternal and one maternal) form the genotype. Heterozygotes possess two different alleles, and homozygotes possess two of the same alleles. The most common allele in a population is referred to as the wild type, and allele frequencies can vary greatly in different populations.9
Most sequence variations are single nucleotide polymorphisms (SNPs, pronounced “snips”), a single DNA base pair substitution that may result in a different gene product. SNPs can be classified as structural RNA polymorphisms (srSNPs), regulatory polymorphisms (rSNPs), or polymorphisms in coding regions (cSNPs)10: srSNPs alter mRNA processing and translation, rSNPs alter transcription, and cSNPs alter protein sequence and function.
Recently, genetic associations with a phenotype have been done on a large scale, with millions of SNPs measured in each of many subjects. This approach, called a genomewide association study or GWAS, has revealed countless candidate genes for clinical traits, but only a few have resulted in a practical clinical application. SNPs may by themselves exert a pharmacokinetic effect (ie, how the body processes the drug), a pharmacodynamic effect (ie, how the drug affects the body), or both, or they may act in concert with other genetic factors. Pharmacodynamic effects can result from a pharmacokinetic effect or can result from variations in a pharmacologic target.
Establishing a genotype-phenotype association can involve clinical studies, animal transgenic studies, or molecular and cellular functional assays.
Clinical applications are emerging
WARFARIN: IMPORTANCE OF CYP2C9, VKORC1
Warfarin is used for the long-term treatment and prevention of thromboembolic events.
This drug has a narrow therapeutic window and shows substantial interpatient dose variability. The start of warfarin therapy is associated with one of the highest rates of adverse events and emergency room visits of any single drug.12 More than 2 million patients start warfarin each year in the United States alone,13 and about 20% of them are hospitalized within the first 6 months because of bleeding due to overanticoagulation.14
The findings from a recent study suggest that pharmacogenomic testing may eventually allow more patients to safely benefit from warfarin therapy. In this large, nationwide, prospective study, hospitalization rates were 30% lower when pharmacogenomic testing was used.14 However, no reduction was seen in the time needed to reach the target international normalized ratio (INR) or in the need for INR checks at 6 months. Furthermore, this study used historical control data, and some or all of the reduction in hospitalization rates may be attributed to more frequent INR checks in the patients who underwent testing than in the historical control group.
A relationship between warfarin dose requirements and the genetic status of CYP2C9, which encodes a major drug-metabolizing enzyme, has been demonstrated in retrospective and prospective studies.15–17
S-warfarin is metabolized by CYP2C9, which is polymorphic
Warfarin contains equal amounts of two isomers, designated S and R. S-warfarin, which is more potent, is metabolized principally by CYP2C9, while R-warfarin is metabolized by CYP1A2, CYP2C19, and CYP3A4.
People who possess two copies of the wild type CYP2C9 gene CYP2C9*1 metabolize warfarin very well and so are called “extensive warfarin metabolizers.” Carriers of the allelic variants CYP2C9*2 and CYP2C9*3 (which have point mutations in exons 3 and 7 of CYP2C9, respectively), have less capacity. Compared with those who are homozygous for the wild-type gene, homozygous carriers of CYP2C9* 3 clear S-warfarin at a rate that is 90% lower, and those with the CYP2C9*1/*3, CYP2C9* 1/*2, CYP2C9*2/*2, or CYP2C9*2/*3 genotypes clear it at a rate 50% to 75% lower. A meta-analysis of 12 studies found that the CYP2C9 genotype accounted for 12% of the interindividual variability of warfarin dose requirements.18
About 8% of whites carry at least one copy of CYP2C9*2, as do 1% of African Americans; the allele is rare in Asian populations. The frequency of CYP2C9*3 is 6% in whites, 1% in African Americans, and 3% in Asians.19,20 People with CYP2C9*4 or CYP2C9*5 have a diminished capacity to clear warfarin; however, these variants occur so infrequently that their clinical relevance may be minimal.
Warfarin’s target, VKOR, is also polymorphic
Genetic variation in warfarin’s pharmacologic target, vitamin K 2,3-epoxide reductase (VKOR), also influences dose requirements. Warfarin decreases the synthesis of vitamin-K-dependent clotting factors by inhibiting VKOR. This inhibition depends on the patient’s C1 subunit gene, VKORC1. Patients with a guanine-to-adenine SNP 1,639 bases upstream of VKORC1 (−1639G>A) need lower warfarin doses—an average of 25% lower in those with the GA genotype (ie, one allele has guanine in the −1639 position and the other allele has adenine in that position) and 50% lower in those with the AA genotype compared with the wild-type genotype GG.21 This promoter SNP, positioned upstream (ie, before the gene-coding region), greatly influences VKORC1 expression.
A meta-analysis of 10 studies found that the VKORC1 polymorphism accounts for 25% of the interindividual variation in warfarin dose.18 In one study, the frequency of the AA genotype in a white population was 14%, AG 47%, and GG 39%; in a Chinese population the frequency of AA was 82%, AG 18%, and GG 0.35%.22
CYP4F2 and GGCX also affect warfarin’s dose requirements
Genetic variations in the enzymes CYP4F2 and gamma-glutamyl carboxylase (GGCX) also influence warfarin dose requirements. Although the data are limited and the effects are smaller than those of CYP2C9 and VKORC1, people with a SNP in CYP4F2 need 8% higher doses of warfarin, while those with a SNP in GGCX need 6% lower doses.23
CYP2C9 and VKORC1 testing is available
Currently, the clinical pharmacogenetic tests relevant for warfarin use are for CYP2C9 and VKORC1.10
The FDA has approved four warfarin pharmacogenetic test kits, but most third-party payers are reluctant to reimburse for testing because it is not currently considered a standard of care. Testing typically costs a few hundred dollars, but it should become less expensive as it becomes more commonplace. The current FDA-approved product label for warfarin does not recommend routine pharmacogenomic testing for determining initial or maintenance doses, but it does acknowledge that dose requirements are influenced by CYP2C9 and VKORC1 and states that genotype information, when available, can assist in selecting the starting dose.24
Clinical trials of warfarin pharmacogenomic testing are under way
Although genetic status can greatly influence an individual patient’s warfarin dosing requirement, routine prospective pharmacogenomic testing is not endorsed by the FDA or by other expert panels26 because there is currently insufficient evidence to recommend for or against it.
Several large prospective trials are under way. For example, the National Heart, Lung, and Blood Institute began a prospective trial in about 1,200 patients to evaluate the use of clinical plus genetic information to guide the initiation of warfarin therapy and to improve anticoagulation control for patients.27 The results, expected in September 2011, and those of other large prospective trials should provide adequate evidence for making recommendations about the clinical utility of routine pharmacogenetic testing for guiding warfarin therapy.
Several recent cost-utility and cost-effectiveness studies have attempted to quantify the value of pharmacogenomic testing for warfarin therapy,28–30 but their analyses are largely limited because the benefit (clinical utility) is yet to be sufficiently characterized.
The relevance of such analyses may soon be drastically diminished, as several non-vitamin-K-dependent blood thinners such as rivaroxaban (Xarelto), dabigatran (Pradaxa), and apixaban are poised to enter clinical practice.31
CLOPIDOGREL IS ACTIVATED BY CYP2C19
Clopidogrel, taken by about 40 million patients worldwide, is used to prevent atherothrombotic events and cardiac stent thrombosis when given along with aspirin.
Studies of clopidogrel pharmacogenomics
A recent genome-wide association study conducted in a cohort of 429 healthy Amish persons revealed a SNP in CYP2C19 to be associated with a diminished response to clopidogrel and to account for 12% of the variation in drug response.33 Traditional factors (the patient’s age, body-mass index, and cholesterol level) combined accounted for less than 10% of the variation.
Findings were similar in a subsequent investigation in 227 cardiac patients receiving clopidogrel: 21% of those with the variant had a cardiovascular ischemic event or died during a 1-year follow-up period compared with 10% of those without the variant (hazard ratio 2.42, P = .02).33
A 12-year prospective study investigating clopidogrel efficacy in 300 cardiac patients under the age of 45 used cardiovascular death, nonfatal myocardial infarction, and urgent coronary revascularization as end points. It concluded that the only independent predictor of these events was the patient’s CYP2C19 status.34
A study in 2,200 patients with recent myocardial infarction examined whether any of the known allelic variations that modulate clopidogrel’s absorption (ABCB1), metabolic activation (CYP3A4/5 and CYP2C19), or biologic activity (P2RY12 and ITGB3) was associated with a higher rate of the combined end point of all-cause mortality, nonfatal myocardial infarction, or stroke. None of the SNPs in CYP3A4/5, P2RY12, or ITGB3 that were evaluated was associated with a higher risk at 1 year. However, the allelic variations modulating clopidogrel’s absorption (ABCB1) and metabolism (CYP2C19) were associated with higher event rates. Patients with two variant ABCB1 alleles had a higher adjusted hazard ratio (95% confidence interval [CI] 1.2–2.47) than those with the wild-type allele. Patients who had one or two CYP2C19 loss-of-function alleles had a higher event rate than those with two wild-type alleles (95% CI 1.10–3.58 and 1.71–7.51, respectively).35
Conversely, researchers from the Population Health Research Institute found no association between poor-metabolizer status and treatment outcomes when CYP2C19 analysis was retrospectively added to the findings of two large clinical trials (combined N > 5,000). However, patients with acute coronary syndrome benefited more from clopidogrel treatment if they were ultra-rapid metabolizers (possessing the gain-of-function allele CYP2C19*17).36
Current status of clopidogrel testing: Uncertain
A current FDA boxed warning states that poor CYP2C19 metabolizers may not benefit from clopidogrel and recommends that prescribers consider alternative treatment for patients in this category.37 However, routine CYP2C19 testing is not recommended, and no firm recommendations have been established regarding dose adjustments for CYP2C19 status.
In 2010, the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association collectively pronounced the current evidence base insufficient for recommending routine pharmacogenomic testing.39
Needed are large-scale studies examining the cost-effectiveness and clinical utility of genotype-guided clopidogrel therapy compared with other therapy options such as prasugrel (Effient), an analogue not metabolized by CYP2C19. One such study, sponsored by Medco Health Solutions, plans to enroll 14,600 cardiac patients and has an estimated completion date in June 2011.40 The expectation that clopidogrel will be available in generic form in 2012 adds to the uncertainty regarding the cost-effectiveness of CYP2C19 testing for clopidogrel therapy.
STATINS: SLC01B1*5 INCREASES MYOPATHY RISK
Statins lower the concentration of low-density lipoprotein cholesterol (LDL-C), resulting in a relative-risk reduction of about 20% for each 1 mmol/L (39 mg/dL) decrement in LDL-C.41 They are one of the most commonly prescribed classes of drugs, but their side effects can limit their appeal: statin-induced myopathy occurs in about 1:1,000 to 1:10,000 patients and is difficult to predict.
SLC01B1. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH), a genome-wide association study, recently found a SNP (SLCO1B1* 5) in the SLC01B1 gene to be associated with a higher risk of statin-induced myopathy in cardiac patients receiving simvastatin (Zocor) 40 or 80 mg daily.42 The SLC01B1 gene, located on chromosome 12, influences the extent of the drug’s hepatic uptake and its serum concentration. Only the SLC01B1*5 SNP emerged as a predictor of statin-induced myopathy across the entire genome.42
The authors believe the findings are likely to apply to other statins. The mechanisms leading to statin-induced myopathy and the impact of statin pharmacogenomics are still unclear.43
CYP3A4. Other genetic variants may play a vital role in determining response to statin therapy. Carriers of a newly identified CYP3A4 polymorphism (intron 6 SNP, rs35599367, C>T) required significantly lower statin doses (0.2–0.6 times less) for optimal lipid control. The analyses included atorvastatin (Lipitor), simvastatin, and lovastatin (Mevacor), and the association was robust (P = .019).44
Statin pharmacogenomic testing is not routinely recommended
Routine pharmacogenomic testing for statin therapy is not recommended. Additional studies are needed to determine the clinical utility and cost-effectiveness of pharmacogenomic testing (involving a combination of several polymorphisms) in various patient populations delineated by type of statin, dose, and concomitant use of other drugs.
TAMOXIFEN IS ACTIVATED BY CYP2D6
Tamoxifen is prescribed to prevent the recurrence of estrogen-receptor-positive breast cancer, to treat metastatic breast cancer, to prevent cancer in high-risk populations, and to treat ductal carcinoma in situ.
Tamoxifen is metabolized to form endoxifen, which has much higher potency and higher systemic levels than tamoxifen.45 Both CYP2D6 and CYP3A4/5 are required to produce endoxifen via two intermediates, but CYP2D6 catalyzes the critical step leading to metabolic activation.
The CYP2D6 gene is highly polymorphic, with more than 75 allelic variants identified. Extensive literature is available describing the influence of CYP2D6 polymorphisms on tamoxifen metabolism and therapy outcomes.46–52 Several CYP2D6 variants result in reduced or no enzyme activity, and people who have more than two normally functioning alleles have exaggerated enzyme activity (gene amplification).
Classification of CYP2D6 status
Several systems have been developed to categorize the phenotypic activity of CYP2D6 based on genotype.
A genetic basis for the observed diversity in the metabolism of cytochrome P450 substrates was recognized more than 30 years ago. People were categorized as either extensive or poor metabolizers, reflecting normal vs impaired ability to metabolize the CYP2D6 substrates sparteine and debrisoquine. Later work expanded this system to include categories for intermediate (between poor and extensive) and ultra-rapid (better than extensive) metabolizers.
The genetic basis for these categories includes homozygosity for dysfunctional variants (the poor-metabolizer group) or extra copies of normal functioning variants (the ultra-rapid-metabolizer group).
Newer systems have been described for characterizing the CYP2D6 activity phenotype whereby CYP2D6 variants are assigned activity scores.53–56 The various scoring systems have been reviewed by Kirchheiner.57
A recent version of the activity scoring system also takes into consideration the many drugs that inhibit CYP2D6, such as amiodarone (Cordarone) and fluoxetine (Prozac) that can reduce the action of tamoxifen if given with it (Table 4).58 For example, the tamoxifen exposure (as predicted by the CYP2D6-activity score) experienced by a CYP2D6 extensive metabolizer taking a CYP2D6-inhibiting drug may be similar to the exposure experienced by a CYP2D6 poor metabolizer receiving the same tamoxifen dose but not taking a CYP2D6-inhibiting drug.
Likewise, the activity score of a CYP2D6 intermediate metabolizer taking a CYP2D6-inducing drug may be similar to that of a CYP2D6 ultra-rapid metabolizer not taking a CYP2D6-inducing drug. Examples of CYP2D6 inducers are dexamethasone, rifampin, and hyperforin (St. John’s wort).
While the newer systems are reported to provide better correlations between genotype and phenotype scores, the older scoring systems and the categorical names are still widely used (eg, in the FDA-approved AmpliChip CYP450 test from Roche,59 which includes genotype data for CYP2D6 and CYP2C19).
No firm recommendations for CYP2D6 testing in tamoxifen users
The different genotypes and phenotypes vary in prevalence in different ethnic groups, and significantly different activity levels for endoxifen formation are observed. Tamoxifen lacks efficacy in those who are poor CYP2D6 metabolizers—ie, about 7% of the white population.
However, the FDA has not made firm recommendations about CYP2D6 testing for prescribing tamoxifen because the evidence of benefit, although suggestive, has been considered insufficient.
Clinicians should be aware that tamoxifen’s efficacy is greatly reduced by concomitant therapy with CYP2D6-inhibiting drugs (Table 4).
Other genes affecting tamoxifen: CYP3A4/5, SULT1A1, and UGT2B15
Some investigators propose that polymorphisms in additional genes encoding enzymes in the tamoxifen metabolic and elimination pathways (eg, CYP3A4/5, SULT1A1, and UGT2B15) also need to be considered to account adequately for interindividual variation in drug response.
For example, CYP3A4 and CYP3A5 are also polymorphic, and large interindividual variation exists in their enzyme activities. These enzymes have overlapping substrate specificities, represent the most abundant drug-metabolizing enzymes in the human liver, and are involved in the biotransformation of a broad range of endogenous substrates and most drugs.60
Clinical studies evaluating the impact of CYP3A4/5 polymorphisms have been inconsistent in their conclusions, which is generally attributed to the relatively low functional impact or the low prevalence of the SNPs evaluated. Many of the nearly 100 CYP3A4/5 polymorphisms identified have not yet been characterized regarding their functional impact on enzyme expression or activity. CYP-3A4/5 enzyme activity is highly variable between individuals and warrants further study of its role in outcomes of tamoxifen therapy. Ongoing and future prospective clinical trials evaluating CYP2D6, CYP3A4/5, and other relevant polymorphisms are necessary to define their clinical relevance before routine genetic testing for tamoxifen can be justified.
CODEINE IS ALSO ACTIVATED BY CYP2D6
Codeine also depends on the CYP2D6 gene, as it must be activated to its more potent opioid metabolites, including morphine. Poor CYP2D6 metabolizers do not benefit from codeine therapy.
The pharmacogenomics of codeine has become a hot topic, especially regarding breast-feeding mothers. The debate was ignited with the publication in 2006 of a case report of an infant’s death, apparently the result of metabolic polymorphisms.61 The evolution of this debate and the outcome of the case may be noteworthy to clinicians, as they illustrate the gravity of public and patient interest in pharmacogenomic testing. In this case, the breast-feeding mother had taken codeine regularly for about 14 days when her 13-day-old infant died from toxic levels of morphine. Unknown to her and the prescriber, both the mother and infant were ultra-rapid CYP2D6 metabolizers, resulting in a more rapid and extensive conversion of codeine to morphine.
A logical strategy for preventing similar deaths would be routine CYP2D6 genotyping when prescribing codeine to breast-feeding mothers. However, after several investigations examined the metabolic and excretion pathways of codeine in their entirety, the FDA did not recommend routine CYP2D6 testing when prescribing codeine to breastfeeding mothers because several other factors, including rare genetic variations of other enzymes, proved necessary for reaching the opioid toxicity leading to the infant’s death.62
PHARMACOGENOMICS OF PSYCHOTROPIC DRUGS
Pharmacogenomic testing has clinical utility for some psychotropic drugs.
HLA-B and carbamazepine
Considered a standard of care, HLA-B genotyping is appropriate before prescribing carbamazepine (Tegretol, Equetro) to patients in populations in which HLAB*1502 is likely to be present, such as Asians. Carriers of HLAB* 1502 are at higher risk of life-threatening skin reactions such as Stevens-Johnson syndrome.11
Several other pharmacogenomic applications for psychotropic medications have been suggested, but routine testing has not been recommended by the FDA or endorsed by any expert panel because sufficient clinical utility and cost-effectiveness have not been demonstrated. A brief summary of study findings and a few practical suggestions follow.
Polymorphisms in metabolizing enzymes have been investigated in patients receiving psychotropic drugs.
CYP2D6 and antidepressants
Many antidepressants show significant differences in plasma drug levels with CYP2D6 polymorphisms (in descending order of influence)55:
- Imipramine (Tofranil)
- Doxepin (Adapin, Silenor, Sinequan)
- Maprotiline (Deprilept, Ludiomil, Psymion)
- Trimipramine (Surmontil)
- Desipramine (Noraprim)
- Nortriptyline (Aventyl, Pamelor)
- Clomipramine (Anafranil)
- Paroxetine (Paxil)
- Venlafaxine (Effexor)
- Amitriptyline (Elavil)
- Mianserin
- Trazadone (Desyrel)
- Bupropion (Wellbutrin)
- Nefazodone (Serzone)
- Citalopram (Celexa)
- Sertraline (Zoloft).
CYP2D6 and antipsychotics
Several antipsychotics are also influenced by CYP2D6 polymorphisms (also in descending order of influence)55:
- Perphenazine (Trilafon)
- Thioridazine (Mellaril)
- Olanzapine (Zyprexa)
- Zuclopenthixol (Cisordinol, Clopixol, Acuphase)
- Aripiprazole (Abilify)
- Flupentixol (Depixol, Fluanxol)
- Haloperidol (Haldol)
- Perazine (Taxilan)
- Risperidone (Risperdal)
- Pimozide (Orap).
CYP2C19 and antidepressants
CYP2C19 polymorphisms are likewise associated with differences in drug metabolism for many antidepressants, such as (in descending order of CYP2C19-mediated influence)55:
- Trimipramine
- Doxepin
- Amitriptyline
- Imipramine
- Citalopram (Celexa)
- Clomipramine
- Moclobemide (Aurorix, Manerix)
- Sertraline
- Fluvoxamine (Luvox).
Clinical relevance of CYP2D6 and CYP2C19
Several studies have demonstrated that poor and intermediate CYP2D6 metabolizers have a higher incidence of adverse effects when taking CYP2D6-dependent antidepressants63–68; however, an almost equal number of studies did not find statistically significant associations.69–72 Likewise, several studies have found an association between ultra-rapid CYP2D6 metabolizer status and diminished response to antidepressants,65,73,74 but no association was found in a larger retrospective study.75
Routine CYP2D6 and CYP2C19 screening is not recommended when prescribing psychotropic drugs. However, reviews of the pharmacokinetic data have suggested a few practical applications when genetic status is already known. In general, clinicians can consider reducing the dose of tricyclic antidepressants by about 50% when prescribing to CYP2D6-poor-metabolizers.55,76–78
Genes that affect serotonin metabolism
Several genes in the serotonin pathway have been investigated to determine whether they influence patients’ susceptibility to depression and adverse effects and response to psychotropic medications.
SLC6A4. Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 appear to influence the treatment response and side-effect profiles of selective serotonin reuptake inhibitors (SSRIs). Carriers of the SLC6A4 5-HTTLPR L alleles have fewer side effects79 and better response to SSRI treatment, and carriers of the S allele have a higher incidence of antidepressant-induced mania80 and poorer response to SSRI treatment.81
5-HT. Polymorphisms in serotonin receptors (2A and 2C subtypes) appear to influence SSRI response and side effects. Carriers of 5-HT 2A C alleles had more severe adverse effects from paroxetine,71 but another 5-HT 2A polymorphism common to Asians is associated with better response to antidepressant therapy.82 A 5-HT 2C polymorphism was associated with a lower incidence of antipsychotic-induced weight gain.83
Although the understanding of these relationships is incomplete and routine pharmacogenomic testing is not currently recommended, reviews of the pharmacodynamic data have suggested a few practical applications when a patient’s genetic status is already known. One should consider:
- Selecting treatments other than SSRIs for depressed patients known to possess the SLC6A4 variant
- Selecting citalopram for depressed patients known to carry the 5-HT 2A polymorphism
- Avoiding treatment with antipsychotic drugs for patients known to possess the 5-HT 2C polymorphism.
THE FUTURE OF PHARMACOGENOMIC TESTING
The examples discussed in this article provide some insight about how pharmacogenomic testing is maturing and slowly being integrated into the practice of medicine. They also illustrate the complexity of the multiple stages of research that pharmacogenomic applications must go through in order to be adopted as standard practice.
In the future, pharmacogenomic data will continue to accumulate, and the clinical utility of many other pharmacogenomic tests may be uncovered. The FDA provides information on emerging pharmacogenomic tests at its Web site, www.fda.gov.11 Its up-to-date “Table of Valid Genomic Biomarkers in the Context of Approved Drug Labels” includes boxed warnings, recommendations, research outcomes, and relevant population genetics.
If the FDA continues its current policy, prospective randomized trials that show improvement in patient outcomes will remain the gold standard for determining the clinical significance of a pharmacogenomic test. Furthermore, cost-benefit analyses are likely to continue dictating policy regarding pharmacogenomic testing, and cost-benefit profiles should improve as technology advances and as information gathered from a single test becomes applicable to multiple medications and clinical scenarios.
In the meantime, physicians should become familiar with the terms used in medical genetics and pharmacogenomics and begin to understand genetic contributions to the outcomes of drug therapy. For example, understanding the consequences of metabolizer status and the frequency of variants in a given population can be tremendously helpful when advising our patients about anticipating potential problems when taking specific medications and about making informed decisions about pharmacogenomic testing.
This exchange of information alone may go a long way in improving therapy outcomes even when prospective pharmacogenomic testing is not routinely performed. Furthermore, an increasing number of patients will already have genotyping information available when they come to us, and clinicians need to be aware of the many pharmacogenomic applications recommended by the FDA when genetic status is known.10
In many patients, certain drugs do not work as well as expected, whereas in other patients they cause toxic effects, even at lower doses. For some patients, the reason may be genetic.
Sizeable minorities of the population carry genetic variants—polymorphisms— that affect their response to various drugs. Thanks to genetic research, our understanding of the variability of drug response has advanced markedly in the last decade. Many relevant polymorphisms have been identified, and tests for some of them are available.
Armed with the knowledge of their patients’ genetic status, physicians could predict their response to certain drugs, leading to better efficacy, fewer adverse drug reactions, and a better cost-benefit ratio.
The possible impact is substantial, since more than half of the drugs most commonly involved in adverse drug reactions are metabolized by polymorphic enzymes.1 Adverse drug reactions remain a significant detriment to public health, having a substantial impact on rates of morbidity and death and on healthcare costs.2–8 In the United States, adverse drug reactions are a leading cause of death in hospitalized patients4 and are annually responsible for hundreds of thousands of deaths and hundreds of billions of dollars in added costs.2,4,6–8
In the meantime, physicians can educate their patients and promote efforts to incorporate genomic information into standard clinical decision-making.
This article offers an overview of pharmacogenomic testing, discussing implications and limitations of a few validated tests. Specifically, we will discuss testing that is relevant when using warfarin (Coumadin), clopidogrel (Plavix), statins, tamoxifen (Nolvadex), codeine, and psychotropic medications, as well as the future role of pharmacogenomic testing in medicine.
WHAT IS PHARMACOGENOMICS?
Pharmacogenomics is the study of how genetic factors relate to interindividual variability of drug response.
Many clinicians may not be familiar with the background and terminology used in the pharmacogenomic literature. Below, a brief review of the terminology is followed by a schematic describing the various stages of research involved in pharmacogenomics and the advancement of a test into standard practice.
The review and schematic may be helpful for evaluating the clinical significance of pharmacogenomics-related articles.
From genotype to phenotype
Genotype refers to the coding sequence of DNA base pairs for a particular gene, and phenotype (eg, disease or drug response) refers to a trait resulting from the protein product encoded by the gene. The name of a gene often refers to its protein product and is italicized (eg, the CYP3A4 gene encodes for the CYP3A4 enzyme).
Two alleles per autosomal gene (one paternal and one maternal) form the genotype. Heterozygotes possess two different alleles, and homozygotes possess two of the same alleles. The most common allele in a population is referred to as the wild type, and allele frequencies can vary greatly in different populations.9
Most sequence variations are single nucleotide polymorphisms (SNPs, pronounced “snips”), a single DNA base pair substitution that may result in a different gene product. SNPs can be classified as structural RNA polymorphisms (srSNPs), regulatory polymorphisms (rSNPs), or polymorphisms in coding regions (cSNPs)10: srSNPs alter mRNA processing and translation, rSNPs alter transcription, and cSNPs alter protein sequence and function.
Recently, genetic associations with a phenotype have been done on a large scale, with millions of SNPs measured in each of many subjects. This approach, called a genomewide association study or GWAS, has revealed countless candidate genes for clinical traits, but only a few have resulted in a practical clinical application. SNPs may by themselves exert a pharmacokinetic effect (ie, how the body processes the drug), a pharmacodynamic effect (ie, how the drug affects the body), or both, or they may act in concert with other genetic factors. Pharmacodynamic effects can result from a pharmacokinetic effect or can result from variations in a pharmacologic target.
Establishing a genotype-phenotype association can involve clinical studies, animal transgenic studies, or molecular and cellular functional assays.
Clinical applications are emerging
WARFARIN: IMPORTANCE OF CYP2C9, VKORC1
Warfarin is used for the long-term treatment and prevention of thromboembolic events.
This drug has a narrow therapeutic window and shows substantial interpatient dose variability. The start of warfarin therapy is associated with one of the highest rates of adverse events and emergency room visits of any single drug.12 More than 2 million patients start warfarin each year in the United States alone,13 and about 20% of them are hospitalized within the first 6 months because of bleeding due to overanticoagulation.14
The findings from a recent study suggest that pharmacogenomic testing may eventually allow more patients to safely benefit from warfarin therapy. In this large, nationwide, prospective study, hospitalization rates were 30% lower when pharmacogenomic testing was used.14 However, no reduction was seen in the time needed to reach the target international normalized ratio (INR) or in the need for INR checks at 6 months. Furthermore, this study used historical control data, and some or all of the reduction in hospitalization rates may be attributed to more frequent INR checks in the patients who underwent testing than in the historical control group.
A relationship between warfarin dose requirements and the genetic status of CYP2C9, which encodes a major drug-metabolizing enzyme, has been demonstrated in retrospective and prospective studies.15–17
S-warfarin is metabolized by CYP2C9, which is polymorphic
Warfarin contains equal amounts of two isomers, designated S and R. S-warfarin, which is more potent, is metabolized principally by CYP2C9, while R-warfarin is metabolized by CYP1A2, CYP2C19, and CYP3A4.
People who possess two copies of the wild type CYP2C9 gene CYP2C9*1 metabolize warfarin very well and so are called “extensive warfarin metabolizers.” Carriers of the allelic variants CYP2C9*2 and CYP2C9*3 (which have point mutations in exons 3 and 7 of CYP2C9, respectively), have less capacity. Compared with those who are homozygous for the wild-type gene, homozygous carriers of CYP2C9* 3 clear S-warfarin at a rate that is 90% lower, and those with the CYP2C9*1/*3, CYP2C9* 1/*2, CYP2C9*2/*2, or CYP2C9*2/*3 genotypes clear it at a rate 50% to 75% lower. A meta-analysis of 12 studies found that the CYP2C9 genotype accounted for 12% of the interindividual variability of warfarin dose requirements.18
About 8% of whites carry at least one copy of CYP2C9*2, as do 1% of African Americans; the allele is rare in Asian populations. The frequency of CYP2C9*3 is 6% in whites, 1% in African Americans, and 3% in Asians.19,20 People with CYP2C9*4 or CYP2C9*5 have a diminished capacity to clear warfarin; however, these variants occur so infrequently that their clinical relevance may be minimal.
Warfarin’s target, VKOR, is also polymorphic
Genetic variation in warfarin’s pharmacologic target, vitamin K 2,3-epoxide reductase (VKOR), also influences dose requirements. Warfarin decreases the synthesis of vitamin-K-dependent clotting factors by inhibiting VKOR. This inhibition depends on the patient’s C1 subunit gene, VKORC1. Patients with a guanine-to-adenine SNP 1,639 bases upstream of VKORC1 (−1639G>A) need lower warfarin doses—an average of 25% lower in those with the GA genotype (ie, one allele has guanine in the −1639 position and the other allele has adenine in that position) and 50% lower in those with the AA genotype compared with the wild-type genotype GG.21 This promoter SNP, positioned upstream (ie, before the gene-coding region), greatly influences VKORC1 expression.
A meta-analysis of 10 studies found that the VKORC1 polymorphism accounts for 25% of the interindividual variation in warfarin dose.18 In one study, the frequency of the AA genotype in a white population was 14%, AG 47%, and GG 39%; in a Chinese population the frequency of AA was 82%, AG 18%, and GG 0.35%.22
CYP4F2 and GGCX also affect warfarin’s dose requirements
Genetic variations in the enzymes CYP4F2 and gamma-glutamyl carboxylase (GGCX) also influence warfarin dose requirements. Although the data are limited and the effects are smaller than those of CYP2C9 and VKORC1, people with a SNP in CYP4F2 need 8% higher doses of warfarin, while those with a SNP in GGCX need 6% lower doses.23
CYP2C9 and VKORC1 testing is available
Currently, the clinical pharmacogenetic tests relevant for warfarin use are for CYP2C9 and VKORC1.10
The FDA has approved four warfarin pharmacogenetic test kits, but most third-party payers are reluctant to reimburse for testing because it is not currently considered a standard of care. Testing typically costs a few hundred dollars, but it should become less expensive as it becomes more commonplace. The current FDA-approved product label for warfarin does not recommend routine pharmacogenomic testing for determining initial or maintenance doses, but it does acknowledge that dose requirements are influenced by CYP2C9 and VKORC1 and states that genotype information, when available, can assist in selecting the starting dose.24
Clinical trials of warfarin pharmacogenomic testing are under way
Although genetic status can greatly influence an individual patient’s warfarin dosing requirement, routine prospective pharmacogenomic testing is not endorsed by the FDA or by other expert panels26 because there is currently insufficient evidence to recommend for or against it.
Several large prospective trials are under way. For example, the National Heart, Lung, and Blood Institute began a prospective trial in about 1,200 patients to evaluate the use of clinical plus genetic information to guide the initiation of warfarin therapy and to improve anticoagulation control for patients.27 The results, expected in September 2011, and those of other large prospective trials should provide adequate evidence for making recommendations about the clinical utility of routine pharmacogenetic testing for guiding warfarin therapy.
Several recent cost-utility and cost-effectiveness studies have attempted to quantify the value of pharmacogenomic testing for warfarin therapy,28–30 but their analyses are largely limited because the benefit (clinical utility) is yet to be sufficiently characterized.
The relevance of such analyses may soon be drastically diminished, as several non-vitamin-K-dependent blood thinners such as rivaroxaban (Xarelto), dabigatran (Pradaxa), and apixaban are poised to enter clinical practice.31
CLOPIDOGREL IS ACTIVATED BY CYP2C19
Clopidogrel, taken by about 40 million patients worldwide, is used to prevent atherothrombotic events and cardiac stent thrombosis when given along with aspirin.
Studies of clopidogrel pharmacogenomics
A recent genome-wide association study conducted in a cohort of 429 healthy Amish persons revealed a SNP in CYP2C19 to be associated with a diminished response to clopidogrel and to account for 12% of the variation in drug response.33 Traditional factors (the patient’s age, body-mass index, and cholesterol level) combined accounted for less than 10% of the variation.
Findings were similar in a subsequent investigation in 227 cardiac patients receiving clopidogrel: 21% of those with the variant had a cardiovascular ischemic event or died during a 1-year follow-up period compared with 10% of those without the variant (hazard ratio 2.42, P = .02).33
A 12-year prospective study investigating clopidogrel efficacy in 300 cardiac patients under the age of 45 used cardiovascular death, nonfatal myocardial infarction, and urgent coronary revascularization as end points. It concluded that the only independent predictor of these events was the patient’s CYP2C19 status.34
A study in 2,200 patients with recent myocardial infarction examined whether any of the known allelic variations that modulate clopidogrel’s absorption (ABCB1), metabolic activation (CYP3A4/5 and CYP2C19), or biologic activity (P2RY12 and ITGB3) was associated with a higher rate of the combined end point of all-cause mortality, nonfatal myocardial infarction, or stroke. None of the SNPs in CYP3A4/5, P2RY12, or ITGB3 that were evaluated was associated with a higher risk at 1 year. However, the allelic variations modulating clopidogrel’s absorption (ABCB1) and metabolism (CYP2C19) were associated with higher event rates. Patients with two variant ABCB1 alleles had a higher adjusted hazard ratio (95% confidence interval [CI] 1.2–2.47) than those with the wild-type allele. Patients who had one or two CYP2C19 loss-of-function alleles had a higher event rate than those with two wild-type alleles (95% CI 1.10–3.58 and 1.71–7.51, respectively).35
Conversely, researchers from the Population Health Research Institute found no association between poor-metabolizer status and treatment outcomes when CYP2C19 analysis was retrospectively added to the findings of two large clinical trials (combined N > 5,000). However, patients with acute coronary syndrome benefited more from clopidogrel treatment if they were ultra-rapid metabolizers (possessing the gain-of-function allele CYP2C19*17).36
Current status of clopidogrel testing: Uncertain
A current FDA boxed warning states that poor CYP2C19 metabolizers may not benefit from clopidogrel and recommends that prescribers consider alternative treatment for patients in this category.37 However, routine CYP2C19 testing is not recommended, and no firm recommendations have been established regarding dose adjustments for CYP2C19 status.
In 2010, the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association collectively pronounced the current evidence base insufficient for recommending routine pharmacogenomic testing.39
Needed are large-scale studies examining the cost-effectiveness and clinical utility of genotype-guided clopidogrel therapy compared with other therapy options such as prasugrel (Effient), an analogue not metabolized by CYP2C19. One such study, sponsored by Medco Health Solutions, plans to enroll 14,600 cardiac patients and has an estimated completion date in June 2011.40 The expectation that clopidogrel will be available in generic form in 2012 adds to the uncertainty regarding the cost-effectiveness of CYP2C19 testing for clopidogrel therapy.
STATINS: SLC01B1*5 INCREASES MYOPATHY RISK
Statins lower the concentration of low-density lipoprotein cholesterol (LDL-C), resulting in a relative-risk reduction of about 20% for each 1 mmol/L (39 mg/dL) decrement in LDL-C.41 They are one of the most commonly prescribed classes of drugs, but their side effects can limit their appeal: statin-induced myopathy occurs in about 1:1,000 to 1:10,000 patients and is difficult to predict.
SLC01B1. The Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH), a genome-wide association study, recently found a SNP (SLCO1B1* 5) in the SLC01B1 gene to be associated with a higher risk of statin-induced myopathy in cardiac patients receiving simvastatin (Zocor) 40 or 80 mg daily.42 The SLC01B1 gene, located on chromosome 12, influences the extent of the drug’s hepatic uptake and its serum concentration. Only the SLC01B1*5 SNP emerged as a predictor of statin-induced myopathy across the entire genome.42
The authors believe the findings are likely to apply to other statins. The mechanisms leading to statin-induced myopathy and the impact of statin pharmacogenomics are still unclear.43
CYP3A4. Other genetic variants may play a vital role in determining response to statin therapy. Carriers of a newly identified CYP3A4 polymorphism (intron 6 SNP, rs35599367, C>T) required significantly lower statin doses (0.2–0.6 times less) for optimal lipid control. The analyses included atorvastatin (Lipitor), simvastatin, and lovastatin (Mevacor), and the association was robust (P = .019).44
Statin pharmacogenomic testing is not routinely recommended
Routine pharmacogenomic testing for statin therapy is not recommended. Additional studies are needed to determine the clinical utility and cost-effectiveness of pharmacogenomic testing (involving a combination of several polymorphisms) in various patient populations delineated by type of statin, dose, and concomitant use of other drugs.
TAMOXIFEN IS ACTIVATED BY CYP2D6
Tamoxifen is prescribed to prevent the recurrence of estrogen-receptor-positive breast cancer, to treat metastatic breast cancer, to prevent cancer in high-risk populations, and to treat ductal carcinoma in situ.
Tamoxifen is metabolized to form endoxifen, which has much higher potency and higher systemic levels than tamoxifen.45 Both CYP2D6 and CYP3A4/5 are required to produce endoxifen via two intermediates, but CYP2D6 catalyzes the critical step leading to metabolic activation.
The CYP2D6 gene is highly polymorphic, with more than 75 allelic variants identified. Extensive literature is available describing the influence of CYP2D6 polymorphisms on tamoxifen metabolism and therapy outcomes.46–52 Several CYP2D6 variants result in reduced or no enzyme activity, and people who have more than two normally functioning alleles have exaggerated enzyme activity (gene amplification).
Classification of CYP2D6 status
Several systems have been developed to categorize the phenotypic activity of CYP2D6 based on genotype.
A genetic basis for the observed diversity in the metabolism of cytochrome P450 substrates was recognized more than 30 years ago. People were categorized as either extensive or poor metabolizers, reflecting normal vs impaired ability to metabolize the CYP2D6 substrates sparteine and debrisoquine. Later work expanded this system to include categories for intermediate (between poor and extensive) and ultra-rapid (better than extensive) metabolizers.
The genetic basis for these categories includes homozygosity for dysfunctional variants (the poor-metabolizer group) or extra copies of normal functioning variants (the ultra-rapid-metabolizer group).
Newer systems have been described for characterizing the CYP2D6 activity phenotype whereby CYP2D6 variants are assigned activity scores.53–56 The various scoring systems have been reviewed by Kirchheiner.57
A recent version of the activity scoring system also takes into consideration the many drugs that inhibit CYP2D6, such as amiodarone (Cordarone) and fluoxetine (Prozac) that can reduce the action of tamoxifen if given with it (Table 4).58 For example, the tamoxifen exposure (as predicted by the CYP2D6-activity score) experienced by a CYP2D6 extensive metabolizer taking a CYP2D6-inhibiting drug may be similar to the exposure experienced by a CYP2D6 poor metabolizer receiving the same tamoxifen dose but not taking a CYP2D6-inhibiting drug.
Likewise, the activity score of a CYP2D6 intermediate metabolizer taking a CYP2D6-inducing drug may be similar to that of a CYP2D6 ultra-rapid metabolizer not taking a CYP2D6-inducing drug. Examples of CYP2D6 inducers are dexamethasone, rifampin, and hyperforin (St. John’s wort).
While the newer systems are reported to provide better correlations between genotype and phenotype scores, the older scoring systems and the categorical names are still widely used (eg, in the FDA-approved AmpliChip CYP450 test from Roche,59 which includes genotype data for CYP2D6 and CYP2C19).
No firm recommendations for CYP2D6 testing in tamoxifen users
The different genotypes and phenotypes vary in prevalence in different ethnic groups, and significantly different activity levels for endoxifen formation are observed. Tamoxifen lacks efficacy in those who are poor CYP2D6 metabolizers—ie, about 7% of the white population.
However, the FDA has not made firm recommendations about CYP2D6 testing for prescribing tamoxifen because the evidence of benefit, although suggestive, has been considered insufficient.
Clinicians should be aware that tamoxifen’s efficacy is greatly reduced by concomitant therapy with CYP2D6-inhibiting drugs (Table 4).
Other genes affecting tamoxifen: CYP3A4/5, SULT1A1, and UGT2B15
Some investigators propose that polymorphisms in additional genes encoding enzymes in the tamoxifen metabolic and elimination pathways (eg, CYP3A4/5, SULT1A1, and UGT2B15) also need to be considered to account adequately for interindividual variation in drug response.
For example, CYP3A4 and CYP3A5 are also polymorphic, and large interindividual variation exists in their enzyme activities. These enzymes have overlapping substrate specificities, represent the most abundant drug-metabolizing enzymes in the human liver, and are involved in the biotransformation of a broad range of endogenous substrates and most drugs.60
Clinical studies evaluating the impact of CYP3A4/5 polymorphisms have been inconsistent in their conclusions, which is generally attributed to the relatively low functional impact or the low prevalence of the SNPs evaluated. Many of the nearly 100 CYP3A4/5 polymorphisms identified have not yet been characterized regarding their functional impact on enzyme expression or activity. CYP-3A4/5 enzyme activity is highly variable between individuals and warrants further study of its role in outcomes of tamoxifen therapy. Ongoing and future prospective clinical trials evaluating CYP2D6, CYP3A4/5, and other relevant polymorphisms are necessary to define their clinical relevance before routine genetic testing for tamoxifen can be justified.
CODEINE IS ALSO ACTIVATED BY CYP2D6
Codeine also depends on the CYP2D6 gene, as it must be activated to its more potent opioid metabolites, including morphine. Poor CYP2D6 metabolizers do not benefit from codeine therapy.
The pharmacogenomics of codeine has become a hot topic, especially regarding breast-feeding mothers. The debate was ignited with the publication in 2006 of a case report of an infant’s death, apparently the result of metabolic polymorphisms.61 The evolution of this debate and the outcome of the case may be noteworthy to clinicians, as they illustrate the gravity of public and patient interest in pharmacogenomic testing. In this case, the breast-feeding mother had taken codeine regularly for about 14 days when her 13-day-old infant died from toxic levels of morphine. Unknown to her and the prescriber, both the mother and infant were ultra-rapid CYP2D6 metabolizers, resulting in a more rapid and extensive conversion of codeine to morphine.
A logical strategy for preventing similar deaths would be routine CYP2D6 genotyping when prescribing codeine to breast-feeding mothers. However, after several investigations examined the metabolic and excretion pathways of codeine in their entirety, the FDA did not recommend routine CYP2D6 testing when prescribing codeine to breastfeeding mothers because several other factors, including rare genetic variations of other enzymes, proved necessary for reaching the opioid toxicity leading to the infant’s death.62
PHARMACOGENOMICS OF PSYCHOTROPIC DRUGS
Pharmacogenomic testing has clinical utility for some psychotropic drugs.
HLA-B and carbamazepine
Considered a standard of care, HLA-B genotyping is appropriate before prescribing carbamazepine (Tegretol, Equetro) to patients in populations in which HLAB*1502 is likely to be present, such as Asians. Carriers of HLAB* 1502 are at higher risk of life-threatening skin reactions such as Stevens-Johnson syndrome.11
Several other pharmacogenomic applications for psychotropic medications have been suggested, but routine testing has not been recommended by the FDA or endorsed by any expert panel because sufficient clinical utility and cost-effectiveness have not been demonstrated. A brief summary of study findings and a few practical suggestions follow.
Polymorphisms in metabolizing enzymes have been investigated in patients receiving psychotropic drugs.
CYP2D6 and antidepressants
Many antidepressants show significant differences in plasma drug levels with CYP2D6 polymorphisms (in descending order of influence)55:
- Imipramine (Tofranil)
- Doxepin (Adapin, Silenor, Sinequan)
- Maprotiline (Deprilept, Ludiomil, Psymion)
- Trimipramine (Surmontil)
- Desipramine (Noraprim)
- Nortriptyline (Aventyl, Pamelor)
- Clomipramine (Anafranil)
- Paroxetine (Paxil)
- Venlafaxine (Effexor)
- Amitriptyline (Elavil)
- Mianserin
- Trazadone (Desyrel)
- Bupropion (Wellbutrin)
- Nefazodone (Serzone)
- Citalopram (Celexa)
- Sertraline (Zoloft).
CYP2D6 and antipsychotics
Several antipsychotics are also influenced by CYP2D6 polymorphisms (also in descending order of influence)55:
- Perphenazine (Trilafon)
- Thioridazine (Mellaril)
- Olanzapine (Zyprexa)
- Zuclopenthixol (Cisordinol, Clopixol, Acuphase)
- Aripiprazole (Abilify)
- Flupentixol (Depixol, Fluanxol)
- Haloperidol (Haldol)
- Perazine (Taxilan)
- Risperidone (Risperdal)
- Pimozide (Orap).
CYP2C19 and antidepressants
CYP2C19 polymorphisms are likewise associated with differences in drug metabolism for many antidepressants, such as (in descending order of CYP2C19-mediated influence)55:
- Trimipramine
- Doxepin
- Amitriptyline
- Imipramine
- Citalopram (Celexa)
- Clomipramine
- Moclobemide (Aurorix, Manerix)
- Sertraline
- Fluvoxamine (Luvox).
Clinical relevance of CYP2D6 and CYP2C19
Several studies have demonstrated that poor and intermediate CYP2D6 metabolizers have a higher incidence of adverse effects when taking CYP2D6-dependent antidepressants63–68; however, an almost equal number of studies did not find statistically significant associations.69–72 Likewise, several studies have found an association between ultra-rapid CYP2D6 metabolizer status and diminished response to antidepressants,65,73,74 but no association was found in a larger retrospective study.75
Routine CYP2D6 and CYP2C19 screening is not recommended when prescribing psychotropic drugs. However, reviews of the pharmacokinetic data have suggested a few practical applications when genetic status is already known. In general, clinicians can consider reducing the dose of tricyclic antidepressants by about 50% when prescribing to CYP2D6-poor-metabolizers.55,76–78
Genes that affect serotonin metabolism
Several genes in the serotonin pathway have been investigated to determine whether they influence patients’ susceptibility to depression and adverse effects and response to psychotropic medications.
SLC6A4. Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 appear to influence the treatment response and side-effect profiles of selective serotonin reuptake inhibitors (SSRIs). Carriers of the SLC6A4 5-HTTLPR L alleles have fewer side effects79 and better response to SSRI treatment, and carriers of the S allele have a higher incidence of antidepressant-induced mania80 and poorer response to SSRI treatment.81
5-HT. Polymorphisms in serotonin receptors (2A and 2C subtypes) appear to influence SSRI response and side effects. Carriers of 5-HT 2A C alleles had more severe adverse effects from paroxetine,71 but another 5-HT 2A polymorphism common to Asians is associated with better response to antidepressant therapy.82 A 5-HT 2C polymorphism was associated with a lower incidence of antipsychotic-induced weight gain.83
Although the understanding of these relationships is incomplete and routine pharmacogenomic testing is not currently recommended, reviews of the pharmacodynamic data have suggested a few practical applications when a patient’s genetic status is already known. One should consider:
- Selecting treatments other than SSRIs for depressed patients known to possess the SLC6A4 variant
- Selecting citalopram for depressed patients known to carry the 5-HT 2A polymorphism
- Avoiding treatment with antipsychotic drugs for patients known to possess the 5-HT 2C polymorphism.
THE FUTURE OF PHARMACOGENOMIC TESTING
The examples discussed in this article provide some insight about how pharmacogenomic testing is maturing and slowly being integrated into the practice of medicine. They also illustrate the complexity of the multiple stages of research that pharmacogenomic applications must go through in order to be adopted as standard practice.
In the future, pharmacogenomic data will continue to accumulate, and the clinical utility of many other pharmacogenomic tests may be uncovered. The FDA provides information on emerging pharmacogenomic tests at its Web site, www.fda.gov.11 Its up-to-date “Table of Valid Genomic Biomarkers in the Context of Approved Drug Labels” includes boxed warnings, recommendations, research outcomes, and relevant population genetics.
If the FDA continues its current policy, prospective randomized trials that show improvement in patient outcomes will remain the gold standard for determining the clinical significance of a pharmacogenomic test. Furthermore, cost-benefit analyses are likely to continue dictating policy regarding pharmacogenomic testing, and cost-benefit profiles should improve as technology advances and as information gathered from a single test becomes applicable to multiple medications and clinical scenarios.
In the meantime, physicians should become familiar with the terms used in medical genetics and pharmacogenomics and begin to understand genetic contributions to the outcomes of drug therapy. For example, understanding the consequences of metabolizer status and the frequency of variants in a given population can be tremendously helpful when advising our patients about anticipating potential problems when taking specific medications and about making informed decisions about pharmacogenomic testing.
This exchange of information alone may go a long way in improving therapy outcomes even when prospective pharmacogenomic testing is not routinely performed. Furthermore, an increasing number of patients will already have genotyping information available when they come to us, and clinicians need to be aware of the many pharmacogenomic applications recommended by the FDA when genetic status is known.10
- King HC, Sinha AA. Gene expression profile analysis by DNA micro-arrays: promise and pitfalls. JAMA 2001; 286:2280–2288.
- Nuckols TK, Paddock SM, Bower AG, et al. Costs of intravenous adverse drug events in academic and nonacademic intensive care units. Med Care 2008; 46:17–24.
- Gurwitz JH, Field TS, Judge J, et al. The incidence of adverse events in two long-term care facilities. Am J Med 2005; 118:251–258.
- Vargas E, Terleira A, Hernando F, et al. Effects of adverse drug reactions on length of stay in surgical intensive care units. Crit Care Med 2003; 31:694–698.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events in older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997; 277:301–306.
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 2001; 41:192–199.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002; 3:229–243.
- Sadee W. Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med 2009; 1:116.
- US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm. Accessed 1/18/2011.
- Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med 2007; 147:755–765.
- Elias DJ, Topol EJ. Warfarin pharmacogenomics: a big step forward for individualized medicine: enlightened dosing of warfarin. Eur J Hum Genet 2008; 16:532–534.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev 2008; 40:355–375.
- García-Martín E, Martínez C, Ladero JM, Agúndez JA. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10:29–40.
- Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6:341–349.
- Wen MS, Lee M, Chen JJ, et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther 2008; 84:83–89.
- Larramendy-Gozalo C, Yang JQ, Verstuyft C, et al. Genetic polymorphism of vitamin K epoxide reductase (VKORC1) 1173C>T in a Chinese and a Caucasian population. Basic Clin Pharmacol Toxicol 2006; 98:611–613.
- Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111:4106–4112.
- Bristol-Myers Squibb. Coumadin (warfarin sodium) Prescribing Information. January 2010.
- Barnes-Jewish Hospital at Washington University Medical Center. Warfarin dosing. http://warfarindosing.orgAccessed 1/20/2011.
- Flockhart DA, O’Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008; 10:139–150.
- National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed January 20, 2011.
- Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 2009; 150:73–83.
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks, and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 2010; 28:61–74.
- Patrick AR, Avron J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes 2009; 2:429–436.
- Haas S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 2008; 25:52–60.
- Bhatt DL. Tailoring antiplatelet therapy based on pharmacogenomics: how well do the data fit? JAMA 2009; 302:896–897.
- Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849–857.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363:1704–1714.
- Bristol-Myers Squibb/Sanofi Pharmaceutical Partnership. Plavix (clopidogrel bisulfate) prescribing information. August 2010.
- P450 Drug Interaction Table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed 1/21/2011.
- Society for Cardiovascular Angiography and Interventions; Holmes DR, Dehmer GJ, Kaul S, Leifer D, O’Gara PT, Stein CM. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010; 122:537–557.
- National Institutes of Health. Genotype Guided Comparison of Clopidogrel and Prasugrel Outcomes Study. http://clinicaltrialsfeeds.org/clinical-trials/show/NCT00995514. Accessed 1/20/2011.
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8:453–463.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 2010; 10:1–11.
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 2010; Apr 13 [Epub ahead of print].
- Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005; 23:9312–9318.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J Pharm Sci 2007; 96:2224–2231.
- Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol 2008; 6:493–494.
- Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin Pharmacol Ther 2008; 83:160–166.
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med 2008; 10:e34.
- Dezentjé VO, Guchelaar HJ, Nortier JW, van del Velde CJ, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009; 15:15–21.
- Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw 2009; 7:203–213.
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009; 9:576–586.
- Steimer W, Zöpf K, von Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clinical Cancer 2004; 50:1623–1633.
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharm Ther 2008; 83:234–242.
- Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004; 9:442–473.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J 2007; 7:257–265.
- Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008; 83:225–227.
- Borges S, Desta Z, Jin Y, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol 2010; 50:450–458.
- Hoffmann-La Roche Ltd. AmpliChip CYP450 Test. http://www.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. Accessed 1/21/2011.
- Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001; 58:737–747.
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368:704.
- Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharm Ther 2009; 86:634–643.
- Grzesiak M, Beszłej A, Lebioda A, Jonkisz A, Dobosz T, Kienja A. [Retrospective assessment of the antidepressants tolerance in the group of patients with diagnosis of depression and different CYP2D6 genotype.] [In Polish] Psychiatr Pol 2003; 37:433–444.
- Laika B, Leucht S, Heres S, Steimer W. Intermediate metabolizer: increased side effects in psychoactive drug therapy. The key to cost-effectiveness of pretreatment CYP2D6 screening? Pharmacogenomics J 2009; 9:395–403.
- Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: Impact on adverse effects and nonresponse during treatment with antidepressants—a pilot study. Clin Pharm Ther 2004; 75:386–393.
- McAlpine DE, O’Kane DJ, Black JL, Mrazek DA. Cytochrome P450 2D6 genotype variation and venlafaxine dosage. Mayo Clin Proc 2007; 82:1065–1068.
- Chen S, Chou WH, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther 1996; 60:522–534.
- Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006; 31:493–502.
- Whyte EM, Romkes M, Mulsant BH, et al. CYP2D6 genotype and venlafaxine-XR concentrations in depressed elderly. Int J Geriatr Psychiatry 2006; 21:542–549.
- Roberts RL, Mulder RT, Joyce PR, Luty SE, Kennedy MA. No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2004; 19:17–23.
- Murphy GM, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 2003; 160:1830–1835.
- Gillman PK. Re: no evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2005; 20:61–62.
- Gex-Fabry M, Eap CB, Oneda B, et al. CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther Drug Monit 2008; 30:474–482.
- Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol 2004; 59:803–807.
- Serretti A, Calati R, Massat I, et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 2009; 24:250–256.
- de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47:75–85.
- de Leon J, Susce MT, Johnson M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr 2009; 14:19–34.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther 2009; 124:57–73.
- Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009; 112:267–272.
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry 2007; 12:247–257.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15:473–500.
- Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003; 160:677–679.
- King HC, Sinha AA. Gene expression profile analysis by DNA micro-arrays: promise and pitfalls. JAMA 2001; 286:2280–2288.
- Nuckols TK, Paddock SM, Bower AG, et al. Costs of intravenous adverse drug events in academic and nonacademic intensive care units. Med Care 2008; 46:17–24.
- Gurwitz JH, Field TS, Judge J, et al. The incidence of adverse events in two long-term care facilities. Am J Med 2005; 118:251–258.
- Vargas E, Terleira A, Hernando F, et al. Effects of adverse drug reactions on length of stay in surgical intensive care units. Crit Care Med 2003; 31:694–698.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events in older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 1997; 277:301–306.
- Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc (Wash) 2001; 41:192–199.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002; 3:229–243.
- Sadee W. Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med 2009; 1:116.
- US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm. Accessed 1/18/2011.
- Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med 2007; 147:755–765.
- Elias DJ, Topol EJ. Warfarin pharmacogenomics: a big step forward for individualized medicine: enlightened dosing of warfarin. Eur J Hum Genet 2008; 16:532–534.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Au N, Rettie AE. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab Rev 2008; 40:355–375.
- García-Martín E, Martínez C, Ladero JM, Agúndez JA. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol Diagn Ther 2006; 10:29–40.
- Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6:341–349.
- Wen MS, Lee M, Chen JJ, et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther 2008; 84:83–89.
- Larramendy-Gozalo C, Yang JQ, Verstuyft C, et al. Genetic polymorphism of vitamin K epoxide reductase (VKORC1) 1173C>T in a Chinese and a Caucasian population. Basic Clin Pharmacol Toxicol 2006; 98:611–613.
- Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008; 111:4106–4112.
- Bristol-Myers Squibb. Coumadin (warfarin sodium) Prescribing Information. January 2010.
- Barnes-Jewish Hospital at Washington University Medical Center. Warfarin dosing. http://warfarindosing.orgAccessed 1/20/2011.
- Flockhart DA, O’Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008; 10:139–150.
- National Institutes of Health. ClinicalTrials.gov. http://clinicaltrials.gov. Accessed January 20, 2011.
- Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med 2009; 150:73–83.
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks, and costs of warfarin pharmacogenomic testing. Pharmacoeconomics 2010; 28:61–74.
- Patrick AR, Avron J, Choudhry NK. Cost-effectiveness of genotype-guided warfarin dosing for patients with atrial fibrillation. Circ Cardiovasc Qual Outcomes 2009; 2:429–436.
- Haas S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 2008; 25:52–60.
- Bhatt DL. Tailoring antiplatelet therapy based on pharmacogenomics: how well do the data fit? JAMA 2009; 302:896–897.
- Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302:849–857.
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Paré G, Mehta SR, Yusuf S, et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363:1704–1714.
- Bristol-Myers Squibb/Sanofi Pharmaceutical Partnership. Plavix (clopidogrel bisulfate) prescribing information. August 2010.
- P450 Drug Interaction Table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/table.asp. Accessed 1/21/2011.
- Society for Cardiovascular Angiography and Interventions; Holmes DR, Dehmer GJ, Kaul S, Leifer D, O’Gara PT, Stein CM. ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the American Heart Association. Circulation 2010; 122:537–557.
- National Institutes of Health. Genotype Guided Comparison of Clopidogrel and Prasugrel Outcomes Study. http://clinicaltrialsfeeds.org/clinical-trials/show/NCT00995514. Accessed 1/20/2011.
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8:453–463.
- SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 2010; 10:1–11.
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J 2010; Apr 13 [Epub ahead of print].
- Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005; 23:9312–9318.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J Pharm Sci 2007; 96:2224–2231.
- Flockhart D. CYP2D6 genotyping and the pharmacogenetics of tamoxifen. Clin Adv Hematol Oncol 2008; 6:493–494.
- Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clin Pharmacol Ther 2008; 83:160–166.
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med 2008; 10:e34.
- Dezentjé VO, Guchelaar HJ, Nortier JW, van del Velde CJ, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009; 15:15–21.
- Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw 2009; 7:203–213.
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 2009; 9:576–586.
- Steimer W, Zöpf K, von Amelunxen S, et al. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clinical Cancer 2004; 50:1623–1633.
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharm Ther 2008; 83:234–242.
- Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004; 9:442–473.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J 2007; 7:257–265.
- Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008; 83:225–227.
- Borges S, Desta Z, Jin Y, et al. Composite functional genetic and comedication CYP2D6 activity score in predicting tamoxifen drug exposure among breast cancer patients. J Clin Pharmacol 2010; 50:450–458.
- Hoffmann-La Roche Ltd. AmpliChip CYP450 Test. http://www.roche.com/assays/Pages/AmpliChipCYP450Test.aspx. Accessed 1/21/2011.
- Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 2001; 58:737–747.
- Koren G, Cairns J, Chitayat D, Gaedigk A, Leeder SJ. Pharmacogenetics of morphine poisoning in a breastfed neonate of a codeine-prescribed mother. Lancet 2006; 368:704.
- Willmann S, Edginton AN, Coboeken K, Ahr G, Lippert J. Risk to the breast-fed neonate from codeine treatment to the mother: a quantitative mechanistic modeling study. Clin Pharm Ther 2009; 86:634–643.
- Grzesiak M, Beszłej A, Lebioda A, Jonkisz A, Dobosz T, Kienja A. [Retrospective assessment of the antidepressants tolerance in the group of patients with diagnosis of depression and different CYP2D6 genotype.] [In Polish] Psychiatr Pol 2003; 37:433–444.
- Laika B, Leucht S, Heres S, Steimer W. Intermediate metabolizer: increased side effects in psychoactive drug therapy. The key to cost-effectiveness of pretreatment CYP2D6 screening? Pharmacogenomics J 2009; 9:395–403.
- Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: Impact on adverse effects and nonresponse during treatment with antidepressants—a pilot study. Clin Pharm Ther 2004; 75:386–393.
- McAlpine DE, O’Kane DJ, Black JL, Mrazek DA. Cytochrome P450 2D6 genotype variation and venlafaxine dosage. Mayo Clin Proc 2007; 82:1065–1068.
- Chen S, Chou WH, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther 1996; 60:522–534.
- Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006; 31:493–502.
- Whyte EM, Romkes M, Mulsant BH, et al. CYP2D6 genotype and venlafaxine-XR concentrations in depressed elderly. Int J Geriatr Psychiatry 2006; 21:542–549.
- Roberts RL, Mulder RT, Joyce PR, Luty SE, Kennedy MA. No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2004; 19:17–23.
- Murphy GM, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 2003; 160:1830–1835.
- Gillman PK. Re: no evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum Psychopharmacol 2005; 20:61–62.
- Gex-Fabry M, Eap CB, Oneda B, et al. CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther Drug Monit 2008; 30:474–482.
- Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol 2004; 59:803–807.
- Serretti A, Calati R, Massat I, et al. Cytochrome P450 CYP1A2, CYP2C9, CYP2C19 and CYP2D6 genes are not associated with response and remission in a sample of depressive patients. Int Clin Psychopharmacol 2009; 24:250–256.
- de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006; 47:75–85.
- de Leon J, Susce MT, Johnson M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr 2009; 14:19–34.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Horstmann S, Binder EB. Pharmacogenomics of antidepressant drugs. Pharmacol Ther 2009; 124:57–73.
- Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord 2009; 112:267–272.
- Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry 2007; 12:247–257.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15:473–500.
- Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT(2C) receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003; 160:677–679.
KEY POINTS
- Polymorphisms that affect the pharmacokinetics and pharmacodynamics of specific drugs are common.
- Testing for certain polymorphisms before prescribing certain drugs could help avoid adverse drug effects and improve efficacy.
- Pharmacogenomic testing has only recently begun to enter clinical practice, and routine testing is currently limited to a few clinical scenarios. However, additional applications may be just around the corner.
- Many pharmacogenomic tests are available, but testing has not yet been recommended for most drugs. Needed are large-scale trials to show that routine testing improves patient outcomes.
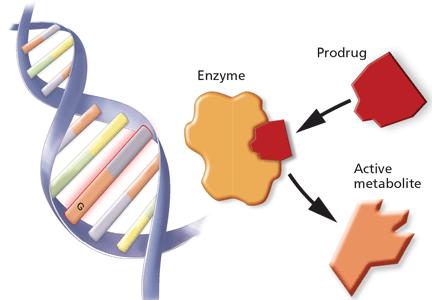
Bony bridge of a bifid rib
A 21-year-old man has had cough and hemoptysis for 3 days. For the past 3 years he has smoked one pack of cigarettes a day. His medical history is unremarkable, and he has had no chest trauma or thoracic surgery.
The patient says he was born full-term, and he has never been aware of any congenital anomalies.
Q: Which is the most likely diagnosis?
- Fractured rib
- Poland syndrome
- Paget disease
- Bifid rib
A: Bifid rib, a congenital anomaly, is the correct answer (see below).
Fractured rib. The patient has no history of chest trauma or thoracic surgery, nor any evidence on chest x-ray to suggest a fractured rib. Also, he has no evidence of osteoporosis to suspect a spontaneous rib fracture. His hemoptysis is most likely due to acute bronchitis.
Poland syndrome is a unilateral deficiency of the pectoralis muscle, variably associated with ipsilateral thoracic and upper limb anomalies. Bilateral hypoplasia or aplasia of the pectoralis muscle and upper-limb defects in association with variable thoracic muscles, chest wall deformities, and lower-limb defects has been infrequently reported in the literature. The diagnosis is usually based on the physical examination (asymmetric chest) or on chest x-ray (unilateral hyperlucent lung).1 This is not the case in our patient.
Paget disease is a chronic, abnormal bone-remodeling process that leads to enlarged, less-dense, brittle bones. The spine, femur, pelvis, skull, clavicle, and humerus are most commonly affected. In the United States, the prevalence is 3% to 4% in people over age 40. Black Americans have a higher prevalence rate than black Africans, and the disease is rare in Asians.
Pain is the most common symptom, but Paget disease is usually asymptomatic. Paget disease can lead to insufficiency fractures, pathologic fractures, secondary arthritis, and nerve impingement in the spine or the base of the skull. Sarcomatous degeneration of the affected bone has been reported, but is rare.
Radiographic findings are often diagnostic. The skull and long bones typically show evidence of osteolysis from the epiphysis and advancing along the diaphysis. Radiographic findings in the sclerotic phase typically involve the axial skeleton and include trabecular coarsening and distortion and cortical thickening.
Rib abnormalities may be observed; these may either be isolated or may be a sign of multi-system malformations. However, in our patient, the radiographic finding of a bony bridge does not fit the description of Paget disease.2
BIFID RIB
The overall prevalence of bifid rib is estimated at 0.15% to 3.4% (mean 2%), and it accounts for up to 20% of all congenital rib anomalies.3 It is usually unilateral. Wattanasirichaigoon et al4 described patterns of rib defects in 47 cases, with bifid rib accounting for 28% of cases.
As with Paget disease, rib anomalies may occur in isolation or in association with multi-system malformations. Since the ribs originate from the mesoderm, it is not surprising that the costal defects are associated with malformations in other organs of the same origin, such as the heart and the kidneys.3 Bifid ribs are also seen in several genetic disorders such as Gorlin-Goltz (ie, basal cell nevus) syndrome, which affects multiple organs including bones, skin, eye, and neural system.5 Occasionally, it is encountered as a part of Jobs syndrome (ie, high levels of immunoglobulin E and recurrent infections),6 and Kindler syndrome, a rare genodermatosis.7
The literature contains little information about the clinical significance of bifid rib. Patients should undergo a thorough physical examination, including oral and cutaneous evaluation, to rule out a genetic syndrome. Physical findings such as palmar pits, subcutaneous calcifications, or odontogenic cyst warrant a more intensive radiologic and genetic investigation.5 If the physical examination is normal and if the patient is asymptomatic, additional clinical or radiologic investigation is of low yield. And as in our patient, the anomaly may go unnoticed on computed tomography of the chest.
- Allam SR, Yadav R, Meziane M, Mehta AC. A middle-aged man with asymptomatic chest wall asymmetry. Cleve Clin J Med 2006; 73:754–756.
- Hung HC, Ou HY, Huang JS, Chuang MC, Wu TJ. Tumor-associated hypercalcemia in a patient with Paget’s disease. Kaohsiung J Med Sci 2008; 24:152–156.
- Charles I, Scott J. Pectoral girdle, spine, ribs, and pelvic girdle. In:Stevenson RE, Hall JG, Goodmann RM, eds. Human Malformations and Related Anomalies, vol 2. Oxford University Press: New York, 1993:655–697.
- Wattanasirichaigoon D, Prasad C, Schneider G, Evans JA, Korf BR. Rib defects in patterns of multiple malformations: a retrospective review and phenotypic analysis of 47 cases. Am J Med Genet A 2003; 122A:63–69.
- Rai S, Gauba K. Jaw cyst-basal cell nevus-bifid rib syndrome: a case report. J Indian Soc Pedod Prev Dent 2007; 25:137–139.
- Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am 2008; 28:277–291.
- Sharma RC, Mahajan V, Sharma NL, Sharma AK. Kindler syndrome. Int J Dermatol 2003; 42:727–732.
A 21-year-old man has had cough and hemoptysis for 3 days. For the past 3 years he has smoked one pack of cigarettes a day. His medical history is unremarkable, and he has had no chest trauma or thoracic surgery.
The patient says he was born full-term, and he has never been aware of any congenital anomalies.
Q: Which is the most likely diagnosis?
- Fractured rib
- Poland syndrome
- Paget disease
- Bifid rib
A: Bifid rib, a congenital anomaly, is the correct answer (see below).
Fractured rib. The patient has no history of chest trauma or thoracic surgery, nor any evidence on chest x-ray to suggest a fractured rib. Also, he has no evidence of osteoporosis to suspect a spontaneous rib fracture. His hemoptysis is most likely due to acute bronchitis.
Poland syndrome is a unilateral deficiency of the pectoralis muscle, variably associated with ipsilateral thoracic and upper limb anomalies. Bilateral hypoplasia or aplasia of the pectoralis muscle and upper-limb defects in association with variable thoracic muscles, chest wall deformities, and lower-limb defects has been infrequently reported in the literature. The diagnosis is usually based on the physical examination (asymmetric chest) or on chest x-ray (unilateral hyperlucent lung).1 This is not the case in our patient.
Paget disease is a chronic, abnormal bone-remodeling process that leads to enlarged, less-dense, brittle bones. The spine, femur, pelvis, skull, clavicle, and humerus are most commonly affected. In the United States, the prevalence is 3% to 4% in people over age 40. Black Americans have a higher prevalence rate than black Africans, and the disease is rare in Asians.
Pain is the most common symptom, but Paget disease is usually asymptomatic. Paget disease can lead to insufficiency fractures, pathologic fractures, secondary arthritis, and nerve impingement in the spine or the base of the skull. Sarcomatous degeneration of the affected bone has been reported, but is rare.
Radiographic findings are often diagnostic. The skull and long bones typically show evidence of osteolysis from the epiphysis and advancing along the diaphysis. Radiographic findings in the sclerotic phase typically involve the axial skeleton and include trabecular coarsening and distortion and cortical thickening.
Rib abnormalities may be observed; these may either be isolated or may be a sign of multi-system malformations. However, in our patient, the radiographic finding of a bony bridge does not fit the description of Paget disease.2
BIFID RIB
The overall prevalence of bifid rib is estimated at 0.15% to 3.4% (mean 2%), and it accounts for up to 20% of all congenital rib anomalies.3 It is usually unilateral. Wattanasirichaigoon et al4 described patterns of rib defects in 47 cases, with bifid rib accounting for 28% of cases.
As with Paget disease, rib anomalies may occur in isolation or in association with multi-system malformations. Since the ribs originate from the mesoderm, it is not surprising that the costal defects are associated with malformations in other organs of the same origin, such as the heart and the kidneys.3 Bifid ribs are also seen in several genetic disorders such as Gorlin-Goltz (ie, basal cell nevus) syndrome, which affects multiple organs including bones, skin, eye, and neural system.5 Occasionally, it is encountered as a part of Jobs syndrome (ie, high levels of immunoglobulin E and recurrent infections),6 and Kindler syndrome, a rare genodermatosis.7
The literature contains little information about the clinical significance of bifid rib. Patients should undergo a thorough physical examination, including oral and cutaneous evaluation, to rule out a genetic syndrome. Physical findings such as palmar pits, subcutaneous calcifications, or odontogenic cyst warrant a more intensive radiologic and genetic investigation.5 If the physical examination is normal and if the patient is asymptomatic, additional clinical or radiologic investigation is of low yield. And as in our patient, the anomaly may go unnoticed on computed tomography of the chest.
A 21-year-old man has had cough and hemoptysis for 3 days. For the past 3 years he has smoked one pack of cigarettes a day. His medical history is unremarkable, and he has had no chest trauma or thoracic surgery.
The patient says he was born full-term, and he has never been aware of any congenital anomalies.
Q: Which is the most likely diagnosis?
- Fractured rib
- Poland syndrome
- Paget disease
- Bifid rib
A: Bifid rib, a congenital anomaly, is the correct answer (see below).
Fractured rib. The patient has no history of chest trauma or thoracic surgery, nor any evidence on chest x-ray to suggest a fractured rib. Also, he has no evidence of osteoporosis to suspect a spontaneous rib fracture. His hemoptysis is most likely due to acute bronchitis.
Poland syndrome is a unilateral deficiency of the pectoralis muscle, variably associated with ipsilateral thoracic and upper limb anomalies. Bilateral hypoplasia or aplasia of the pectoralis muscle and upper-limb defects in association with variable thoracic muscles, chest wall deformities, and lower-limb defects has been infrequently reported in the literature. The diagnosis is usually based on the physical examination (asymmetric chest) or on chest x-ray (unilateral hyperlucent lung).1 This is not the case in our patient.
Paget disease is a chronic, abnormal bone-remodeling process that leads to enlarged, less-dense, brittle bones. The spine, femur, pelvis, skull, clavicle, and humerus are most commonly affected. In the United States, the prevalence is 3% to 4% in people over age 40. Black Americans have a higher prevalence rate than black Africans, and the disease is rare in Asians.
Pain is the most common symptom, but Paget disease is usually asymptomatic. Paget disease can lead to insufficiency fractures, pathologic fractures, secondary arthritis, and nerve impingement in the spine or the base of the skull. Sarcomatous degeneration of the affected bone has been reported, but is rare.
Radiographic findings are often diagnostic. The skull and long bones typically show evidence of osteolysis from the epiphysis and advancing along the diaphysis. Radiographic findings in the sclerotic phase typically involve the axial skeleton and include trabecular coarsening and distortion and cortical thickening.
Rib abnormalities may be observed; these may either be isolated or may be a sign of multi-system malformations. However, in our patient, the radiographic finding of a bony bridge does not fit the description of Paget disease.2
BIFID RIB
The overall prevalence of bifid rib is estimated at 0.15% to 3.4% (mean 2%), and it accounts for up to 20% of all congenital rib anomalies.3 It is usually unilateral. Wattanasirichaigoon et al4 described patterns of rib defects in 47 cases, with bifid rib accounting for 28% of cases.
As with Paget disease, rib anomalies may occur in isolation or in association with multi-system malformations. Since the ribs originate from the mesoderm, it is not surprising that the costal defects are associated with malformations in other organs of the same origin, such as the heart and the kidneys.3 Bifid ribs are also seen in several genetic disorders such as Gorlin-Goltz (ie, basal cell nevus) syndrome, which affects multiple organs including bones, skin, eye, and neural system.5 Occasionally, it is encountered as a part of Jobs syndrome (ie, high levels of immunoglobulin E and recurrent infections),6 and Kindler syndrome, a rare genodermatosis.7
The literature contains little information about the clinical significance of bifid rib. Patients should undergo a thorough physical examination, including oral and cutaneous evaluation, to rule out a genetic syndrome. Physical findings such as palmar pits, subcutaneous calcifications, or odontogenic cyst warrant a more intensive radiologic and genetic investigation.5 If the physical examination is normal and if the patient is asymptomatic, additional clinical or radiologic investigation is of low yield. And as in our patient, the anomaly may go unnoticed on computed tomography of the chest.
- Allam SR, Yadav R, Meziane M, Mehta AC. A middle-aged man with asymptomatic chest wall asymmetry. Cleve Clin J Med 2006; 73:754–756.
- Hung HC, Ou HY, Huang JS, Chuang MC, Wu TJ. Tumor-associated hypercalcemia in a patient with Paget’s disease. Kaohsiung J Med Sci 2008; 24:152–156.
- Charles I, Scott J. Pectoral girdle, spine, ribs, and pelvic girdle. In:Stevenson RE, Hall JG, Goodmann RM, eds. Human Malformations and Related Anomalies, vol 2. Oxford University Press: New York, 1993:655–697.
- Wattanasirichaigoon D, Prasad C, Schneider G, Evans JA, Korf BR. Rib defects in patterns of multiple malformations: a retrospective review and phenotypic analysis of 47 cases. Am J Med Genet A 2003; 122A:63–69.
- Rai S, Gauba K. Jaw cyst-basal cell nevus-bifid rib syndrome: a case report. J Indian Soc Pedod Prev Dent 2007; 25:137–139.
- Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am 2008; 28:277–291.
- Sharma RC, Mahajan V, Sharma NL, Sharma AK. Kindler syndrome. Int J Dermatol 2003; 42:727–732.
- Allam SR, Yadav R, Meziane M, Mehta AC. A middle-aged man with asymptomatic chest wall asymmetry. Cleve Clin J Med 2006; 73:754–756.
- Hung HC, Ou HY, Huang JS, Chuang MC, Wu TJ. Tumor-associated hypercalcemia in a patient with Paget’s disease. Kaohsiung J Med Sci 2008; 24:152–156.
- Charles I, Scott J. Pectoral girdle, spine, ribs, and pelvic girdle. In:Stevenson RE, Hall JG, Goodmann RM, eds. Human Malformations and Related Anomalies, vol 2. Oxford University Press: New York, 1993:655–697.
- Wattanasirichaigoon D, Prasad C, Schneider G, Evans JA, Korf BR. Rib defects in patterns of multiple malformations: a retrospective review and phenotypic analysis of 47 cases. Am J Med Genet A 2003; 122A:63–69.
- Rai S, Gauba K. Jaw cyst-basal cell nevus-bifid rib syndrome: a case report. J Indian Soc Pedod Prev Dent 2007; 25:137–139.
- Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am 2008; 28:277–291.
- Sharma RC, Mahajan V, Sharma NL, Sharma AK. Kindler syndrome. Int J Dermatol 2003; 42:727–732.
A 40-year-old man with spells of generalized weakness and paresthesias
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
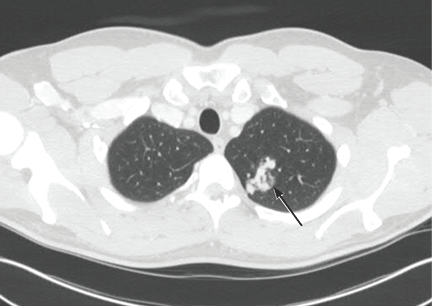
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
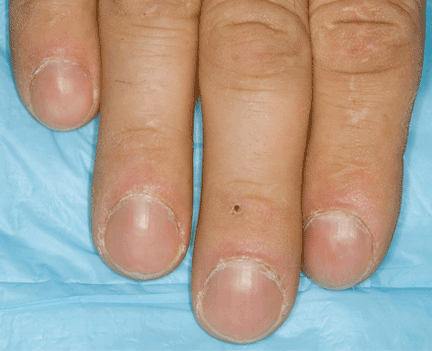
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
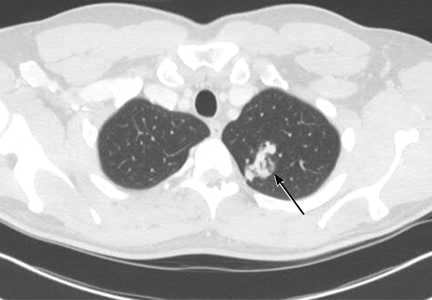
Autosomal dominant polycystic kidney disease: Emerging concepts of pathogenesis and new treatments
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
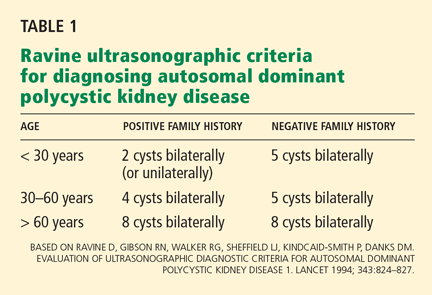
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
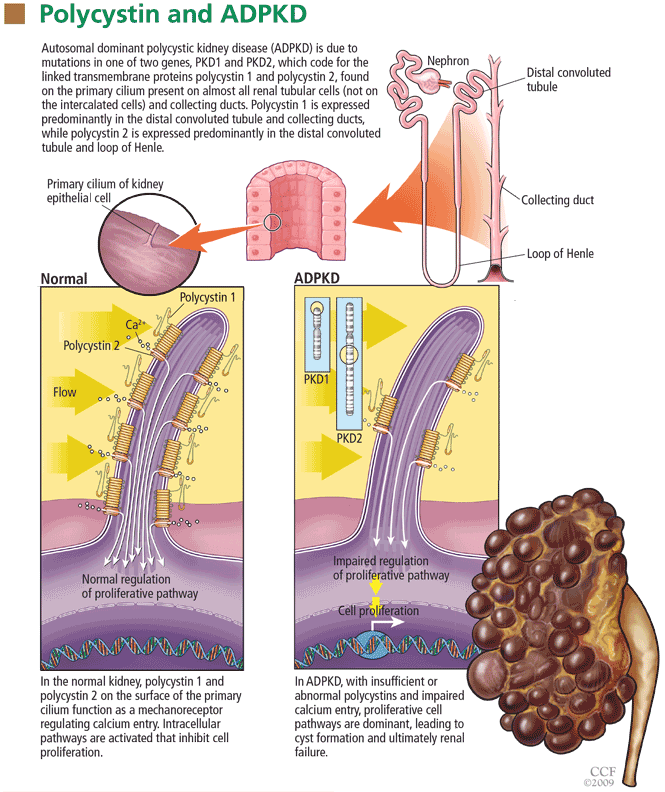
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
KEY POINTS
- In ADPKD the expanding cysts destroy normally functioning kidney tissue, causing hypertension, pain, and other complications, but renal function remains relatively stable until kidney volumes reach a critical size.
- Testing for genetic defects that cause ADPKD is available. The specific mutation involved (PKD1 or PKD2) affects the age of onset and therefore the rate of disease progression as well as the likelihood of cardiovascular complications. Other factors include somatic mutations (“second hits”) of the normal paired chromosome.
- Intracranial aneurysms are a key noncystic feature and may present with a very severe (“sentinel” or “thunderclap”) headache requiring immediate medical attention. Their occurrence is strongly influenced by family history.
- Basic research indicates that patients may be advised to increase their water intake, limit their sodium intake, and avoid caffeine and methylxanthine derivatives.