User login
Courts temporarily block Title X changes

U.S. District Judge Stanley Bastian for the District of Eastern Washington on April 25 approved a temporary nationwide ban against the program changes in response to legal a challenge by Washington state. The same day, U.S. District Judge for the District of Oregon Michael J. McShane also preliminarily barred the restrictions from taking effect in response to a legal challenge by the American Medical Association and the Planned Parenthood Federation of America.
Judge McShane called the program restrictions “arbitrary and capricious,” and wrote that the rules ignore comprehensive, ethical, and evidence-based health care, and impermissibly interfere with the patient-doctor relationship. Judge Bastian agreed, writing in his order that the plaintiffs have demonstrated that the restrictions violate the central purpose of Title X, which is to equalize access to comprehensive, evidence-based, and voluntary family planning.
“Plaintiffs have demonstrated they are likely to suffer irreparable harm in the absence of a preliminary injunction by presenting facts and argument that the final rule may or likely will: seriously disrupt or destroy the existing network of Title X providers in both the State of Washington and throughout the entire nation,” Judge Bastian wrote in his order.
Changes to the Title X program – scheduled to take effect May 3 – would have made health clinics ineligible for Title X funding if they offer, promote, or support abortion as a method of family planning. Title X grants generally go to health centers that provide reproductive health care – such as STD-testing, cancer screenings, and contraception – to low-income families. Under the rule, the government would withdraw financial assistance to clinics if they allow counseling or referrals associated with abortion, regardless of whether the money is used for other health care services.
HHS officials said that the final rule will provide for clear financial and physical separation between Title X and non–Title X activities, reduce confusion on the part of Title X clinics and the public about permissible Title X activities, and improve program transparency by requiring more complete reporting by grantees about their partnerships with referral agencies.
Washington state and the National Family Planning & Reproductive Health Association sued the U.S. Department of Health & Human Services in early March to block the agency from enforcing the modifications. A separate lawsuit was filed by the American Medical Association and the Planned Parenthood Federation of America to stop the funding changes, and 22 states issued a third legal challenge. The Title X changes impose a “government gag rule” on what information physicians can provide to their patients, according to the plaintiffs.
The American College of Physicians (ACP) and other groups, including the American Academy of Family Physicians, the American College of Obstetricians and Gynecologists, and the American Academy of Pediatrics have voiced their opposition to the Title X restrictions. In a joint court brief, the medical societies wrote that the Trump administration’s limitations to the Title X program will create cultural, geographic, and financial barriers to care; erode the physician-patient relationship; and cause extreme, immediate, and irreparable harm to millions of patients.
Washington Attorney General Bob Ferguson said the nationwide ban ensures that clinics across the nation can remain open and continue to provide quality, unbiased health care to women
“Trump’s ‘gag rule’ would have jeopardized health care access to women across the country,” he said in a statement. “Title X clinics, such as Planned Parenthood, provide essential services – now they can keep serving women while we continue to fight to keep the federal government out of the exam room.”
AMA President Barbara L. McAneny, MD, praised Judge McShane’s order. “The new rule would have placed obstacles to health care for low-income patients,” Dr. McAneny said in a statement. “We are pleased the judge shared the AMA’s concern about the physician-patient relationship that the rule would have jeopardized.”
The Trump administration had not said at press time whether it would appeal the order.
Antiabortion organizations, such as the Susan B. Anthony List, have expressed strong support of the Title X funding restrictions.
“The rule advances President Trump’s promise to stop taxpayer funding of abortion businesses like Planned Parenthood,” SBA List President Marjorie Dannenfelser said in a statement. “The Protect Life Rule does not cut family planning funding by a single dime, and instead directs tax dollars to entities that provide health care to women but do not perform abortions.”
agallegos@mdedge.com
U.S. District Judge Stanley Bastian for the District of Eastern Washington on April 25 approved a temporary nationwide ban against the program changes in response to legal a challenge by Washington state. The same day, U.S. District Judge for the District of Oregon Michael J. McShane also preliminarily barred the restrictions from taking effect in response to a legal challenge by the American Medical Association and the Planned Parenthood Federation of America.
Judge McShane called the program restrictions “arbitrary and capricious,” and wrote that the rules ignore comprehensive, ethical, and evidence-based health care, and impermissibly interfere with the patient-doctor relationship. Judge Bastian agreed, writing in his order that the plaintiffs have demonstrated that the restrictions violate the central purpose of Title X, which is to equalize access to comprehensive, evidence-based, and voluntary family planning.
“Plaintiffs have demonstrated they are likely to suffer irreparable harm in the absence of a preliminary injunction by presenting facts and argument that the final rule may or likely will: seriously disrupt or destroy the existing network of Title X providers in both the State of Washington and throughout the entire nation,” Judge Bastian wrote in his order.
Changes to the Title X program – scheduled to take effect May 3 – would have made health clinics ineligible for Title X funding if they offer, promote, or support abortion as a method of family planning. Title X grants generally go to health centers that provide reproductive health care – such as STD-testing, cancer screenings, and contraception – to low-income families. Under the rule, the government would withdraw financial assistance to clinics if they allow counseling or referrals associated with abortion, regardless of whether the money is used for other health care services.
HHS officials said that the final rule will provide for clear financial and physical separation between Title X and non–Title X activities, reduce confusion on the part of Title X clinics and the public about permissible Title X activities, and improve program transparency by requiring more complete reporting by grantees about their partnerships with referral agencies.
Washington state and the National Family Planning & Reproductive Health Association sued the U.S. Department of Health & Human Services in early March to block the agency from enforcing the modifications. A separate lawsuit was filed by the American Medical Association and the Planned Parenthood Federation of America to stop the funding changes, and 22 states issued a third legal challenge. The Title X changes impose a “government gag rule” on what information physicians can provide to their patients, according to the plaintiffs.
The American College of Physicians (ACP) and other groups, including the American Academy of Family Physicians, the American College of Obstetricians and Gynecologists, and the American Academy of Pediatrics have voiced their opposition to the Title X restrictions. In a joint court brief, the medical societies wrote that the Trump administration’s limitations to the Title X program will create cultural, geographic, and financial barriers to care; erode the physician-patient relationship; and cause extreme, immediate, and irreparable harm to millions of patients.
Washington Attorney General Bob Ferguson said the nationwide ban ensures that clinics across the nation can remain open and continue to provide quality, unbiased health care to women
“Trump’s ‘gag rule’ would have jeopardized health care access to women across the country,” he said in a statement. “Title X clinics, such as Planned Parenthood, provide essential services – now they can keep serving women while we continue to fight to keep the federal government out of the exam room.”
AMA President Barbara L. McAneny, MD, praised Judge McShane’s order. “The new rule would have placed obstacles to health care for low-income patients,” Dr. McAneny said in a statement. “We are pleased the judge shared the AMA’s concern about the physician-patient relationship that the rule would have jeopardized.”
The Trump administration had not said at press time whether it would appeal the order.
Antiabortion organizations, such as the Susan B. Anthony List, have expressed strong support of the Title X funding restrictions.
“The rule advances President Trump’s promise to stop taxpayer funding of abortion businesses like Planned Parenthood,” SBA List President Marjorie Dannenfelser said in a statement. “The Protect Life Rule does not cut family planning funding by a single dime, and instead directs tax dollars to entities that provide health care to women but do not perform abortions.”
agallegos@mdedge.com
U.S. District Judge Stanley Bastian for the District of Eastern Washington on April 25 approved a temporary nationwide ban against the program changes in response to legal a challenge by Washington state. The same day, U.S. District Judge for the District of Oregon Michael J. McShane also preliminarily barred the restrictions from taking effect in response to a legal challenge by the American Medical Association and the Planned Parenthood Federation of America.
Judge McShane called the program restrictions “arbitrary and capricious,” and wrote that the rules ignore comprehensive, ethical, and evidence-based health care, and impermissibly interfere with the patient-doctor relationship. Judge Bastian agreed, writing in his order that the plaintiffs have demonstrated that the restrictions violate the central purpose of Title X, which is to equalize access to comprehensive, evidence-based, and voluntary family planning.
“Plaintiffs have demonstrated they are likely to suffer irreparable harm in the absence of a preliminary injunction by presenting facts and argument that the final rule may or likely will: seriously disrupt or destroy the existing network of Title X providers in both the State of Washington and throughout the entire nation,” Judge Bastian wrote in his order.
Changes to the Title X program – scheduled to take effect May 3 – would have made health clinics ineligible for Title X funding if they offer, promote, or support abortion as a method of family planning. Title X grants generally go to health centers that provide reproductive health care – such as STD-testing, cancer screenings, and contraception – to low-income families. Under the rule, the government would withdraw financial assistance to clinics if they allow counseling or referrals associated with abortion, regardless of whether the money is used for other health care services.
HHS officials said that the final rule will provide for clear financial and physical separation between Title X and non–Title X activities, reduce confusion on the part of Title X clinics and the public about permissible Title X activities, and improve program transparency by requiring more complete reporting by grantees about their partnerships with referral agencies.
Washington state and the National Family Planning & Reproductive Health Association sued the U.S. Department of Health & Human Services in early March to block the agency from enforcing the modifications. A separate lawsuit was filed by the American Medical Association and the Planned Parenthood Federation of America to stop the funding changes, and 22 states issued a third legal challenge. The Title X changes impose a “government gag rule” on what information physicians can provide to their patients, according to the plaintiffs.
The American College of Physicians (ACP) and other groups, including the American Academy of Family Physicians, the American College of Obstetricians and Gynecologists, and the American Academy of Pediatrics have voiced their opposition to the Title X restrictions. In a joint court brief, the medical societies wrote that the Trump administration’s limitations to the Title X program will create cultural, geographic, and financial barriers to care; erode the physician-patient relationship; and cause extreme, immediate, and irreparable harm to millions of patients.
Washington Attorney General Bob Ferguson said the nationwide ban ensures that clinics across the nation can remain open and continue to provide quality, unbiased health care to women
“Trump’s ‘gag rule’ would have jeopardized health care access to women across the country,” he said in a statement. “Title X clinics, such as Planned Parenthood, provide essential services – now they can keep serving women while we continue to fight to keep the federal government out of the exam room.”
AMA President Barbara L. McAneny, MD, praised Judge McShane’s order. “The new rule would have placed obstacles to health care for low-income patients,” Dr. McAneny said in a statement. “We are pleased the judge shared the AMA’s concern about the physician-patient relationship that the rule would have jeopardized.”
The Trump administration had not said at press time whether it would appeal the order.
Antiabortion organizations, such as the Susan B. Anthony List, have expressed strong support of the Title X funding restrictions.
“The rule advances President Trump’s promise to stop taxpayer funding of abortion businesses like Planned Parenthood,” SBA List President Marjorie Dannenfelser said in a statement. “The Protect Life Rule does not cut family planning funding by a single dime, and instead directs tax dollars to entities that provide health care to women but do not perform abortions.”
agallegos@mdedge.com

The genesis of vaginal anomalies
According to our guest author Marc R. Laufer, MD, the “development of the female genital tract is a complex process that is dependent upon a series of events involving cellular differentiation, migration, fusion, and canalization. Failure of any one of these processes results in a congenital anomaly.”1

In 1933, A.K. Koff coined the terms sinovaginal bulb and vaginal plate. He proposed that the upper 80% of the vagina is derived from Müllerian epithelium and the lower 20% derived from urogenital sinus epithelium.2 In 1957, D. Bulmer proposed that vaginal epithelium derives solely from urogenital sinus epithelium.3 And in 2017, Robboy et al. supported Bulmer’s proposal that human vaginal epithelium derives solely from urogenital sinus epithelium and differs from mouse vaginal development.4
Beginning at 3 weeks of embryogenesis and continuing into the second trimester of pregnancy, development of the female genital tract takes place. The sinovaginal bulbs originate in the urogenital sinus at the distal aspect of the Müllerian tubercle. At approximately 13 weeks, these two solid evaginations grow out of the pelvic part of the urogenital sinus and proliferate into the caudal end of the uterovaginal canal to become a solid vaginal plate. Degeneration of the central cells of this vaginal plate, which occur in a cephalad direction, enables creation of the lower vagina. Canalization is generally completed by 20 weeks’ gestation.
Agenesis or absence of the lower vagina is usually associated with normal development of the upper vagina, cervix, uterus, and ovaries. It is the result of abnormal development of the sinovaginal bulbs and vaginal plate.
The hymenal membrane separates the vaginal lumen from the urogenital sinus. Secondary to degeneration of the central epithelial cells, the hymen typically ruptures, leaving a thin fold of mucous membrane around the vaginal introitus. Hymenal anatomic variants include microperforate, septate, or cribriform. They occur secondary to incomplete degeneration of the central portion of the hymen.
Dr. Laufer is chief of the division of gynecology, codirector of the Center for Young Women’s Health, and director of the Boston Center for Endometriosis, all at Boston Children’s Hospital. He also is professor of obstetrics, gynecology, and reproductive biology at Harvard Medical School, Boston. Dr. Laufer is an acclaimed physician, surgeon, clinical researcher, author, and teacher, and it is truly my pleasure to welcome him to this edition of the Master Class in Gynecologic Surgery.
Dr. Miller is a clinical associate professor at the University of Illinois at Chicago and past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in metropolitan Chicago and the director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. He reported no disclosures relevant to this Master Class. Email him at pdnews@mdedge.com.
References
1. Laufer M. Congenital anomalies of the hymen and vagina. Uptodate (accessed April 2019).
2. Contrib Embryol. 1933 Sep;24(140):59-91.
3. J Anat. 1957 Oct;91(4):490-509.
4. Differentiation. 2017 Sep-Oct;97:9-22.
According to our guest author Marc R. Laufer, MD, the “development of the female genital tract is a complex process that is dependent upon a series of events involving cellular differentiation, migration, fusion, and canalization. Failure of any one of these processes results in a congenital anomaly.”1
In 1933, A.K. Koff coined the terms sinovaginal bulb and vaginal plate. He proposed that the upper 80% of the vagina is derived from Müllerian epithelium and the lower 20% derived from urogenital sinus epithelium.2 In 1957, D. Bulmer proposed that vaginal epithelium derives solely from urogenital sinus epithelium.3 And in 2017, Robboy et al. supported Bulmer’s proposal that human vaginal epithelium derives solely from urogenital sinus epithelium and differs from mouse vaginal development.4
Beginning at 3 weeks of embryogenesis and continuing into the second trimester of pregnancy, development of the female genital tract takes place. The sinovaginal bulbs originate in the urogenital sinus at the distal aspect of the Müllerian tubercle. At approximately 13 weeks, these two solid evaginations grow out of the pelvic part of the urogenital sinus and proliferate into the caudal end of the uterovaginal canal to become a solid vaginal plate. Degeneration of the central cells of this vaginal plate, which occur in a cephalad direction, enables creation of the lower vagina. Canalization is generally completed by 20 weeks’ gestation.
Agenesis or absence of the lower vagina is usually associated with normal development of the upper vagina, cervix, uterus, and ovaries. It is the result of abnormal development of the sinovaginal bulbs and vaginal plate.
The hymenal membrane separates the vaginal lumen from the urogenital sinus. Secondary to degeneration of the central epithelial cells, the hymen typically ruptures, leaving a thin fold of mucous membrane around the vaginal introitus. Hymenal anatomic variants include microperforate, septate, or cribriform. They occur secondary to incomplete degeneration of the central portion of the hymen.
Dr. Laufer is chief of the division of gynecology, codirector of the Center for Young Women’s Health, and director of the Boston Center for Endometriosis, all at Boston Children’s Hospital. He also is professor of obstetrics, gynecology, and reproductive biology at Harvard Medical School, Boston. Dr. Laufer is an acclaimed physician, surgeon, clinical researcher, author, and teacher, and it is truly my pleasure to welcome him to this edition of the Master Class in Gynecologic Surgery.
Dr. Miller is a clinical associate professor at the University of Illinois at Chicago and past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in metropolitan Chicago and the director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. He reported no disclosures relevant to this Master Class. Email him at pdnews@mdedge.com.
References
1. Laufer M. Congenital anomalies of the hymen and vagina. Uptodate (accessed April 2019).
2. Contrib Embryol. 1933 Sep;24(140):59-91.
3. J Anat. 1957 Oct;91(4):490-509.
4. Differentiation. 2017 Sep-Oct;97:9-22.
According to our guest author Marc R. Laufer, MD, the “development of the female genital tract is a complex process that is dependent upon a series of events involving cellular differentiation, migration, fusion, and canalization. Failure of any one of these processes results in a congenital anomaly.”1
In 1933, A.K. Koff coined the terms sinovaginal bulb and vaginal plate. He proposed that the upper 80% of the vagina is derived from Müllerian epithelium and the lower 20% derived from urogenital sinus epithelium.2 In 1957, D. Bulmer proposed that vaginal epithelium derives solely from urogenital sinus epithelium.3 And in 2017, Robboy et al. supported Bulmer’s proposal that human vaginal epithelium derives solely from urogenital sinus epithelium and differs from mouse vaginal development.4
Beginning at 3 weeks of embryogenesis and continuing into the second trimester of pregnancy, development of the female genital tract takes place. The sinovaginal bulbs originate in the urogenital sinus at the distal aspect of the Müllerian tubercle. At approximately 13 weeks, these two solid evaginations grow out of the pelvic part of the urogenital sinus and proliferate into the caudal end of the uterovaginal canal to become a solid vaginal plate. Degeneration of the central cells of this vaginal plate, which occur in a cephalad direction, enables creation of the lower vagina. Canalization is generally completed by 20 weeks’ gestation.
Agenesis or absence of the lower vagina is usually associated with normal development of the upper vagina, cervix, uterus, and ovaries. It is the result of abnormal development of the sinovaginal bulbs and vaginal plate.
The hymenal membrane separates the vaginal lumen from the urogenital sinus. Secondary to degeneration of the central epithelial cells, the hymen typically ruptures, leaving a thin fold of mucous membrane around the vaginal introitus. Hymenal anatomic variants include microperforate, septate, or cribriform. They occur secondary to incomplete degeneration of the central portion of the hymen.
Dr. Laufer is chief of the division of gynecology, codirector of the Center for Young Women’s Health, and director of the Boston Center for Endometriosis, all at Boston Children’s Hospital. He also is professor of obstetrics, gynecology, and reproductive biology at Harvard Medical School, Boston. Dr. Laufer is an acclaimed physician, surgeon, clinical researcher, author, and teacher, and it is truly my pleasure to welcome him to this edition of the Master Class in Gynecologic Surgery.
Dr. Miller is a clinical associate professor at the University of Illinois at Chicago and past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in metropolitan Chicago and the director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. He reported no disclosures relevant to this Master Class. Email him at pdnews@mdedge.com.
References
1. Laufer M. Congenital anomalies of the hymen and vagina. Uptodate (accessed April 2019).
2. Contrib Embryol. 1933 Sep;24(140):59-91.
3. J Anat. 1957 Oct;91(4):490-509.
4. Differentiation. 2017 Sep-Oct;97:9-22.

Vaginal anomalies and their surgical correction
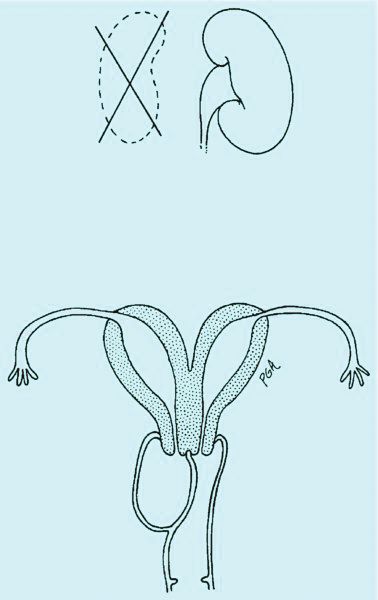
Congenital obstructive anomalies of the vagina are unusual and can be challenging to diagnose and manage. Two of the most challenging are obstructive hemivagina with ipsilateral renal agenesis (Figure 1a) and agenesis of the lower vagina (Figure 1b), the latter of which must be differentiated most commonly from imperforate hymen (Figure 1c). Evaluation and treatment of these anomalies is dependent upon the age of the patient, as well as the symptoms, and the timing of treatment should be individualized.
Agenesis of the lower vagina
Agenesis of the lower vagina and imperforate hymen may present either in the newborn period as a bulging introitus caused by mucocolpos from vaginal secretions stimulated by maternal estradiol or during adolescence at the time of menarche. In neonates, it often is best not to intervene when obstructive anomalies are suspected as long as there is no fever; pain; or compromise of respiration, urinary and bowel function, and other functionality. It will be easier to differentiate agenesis of the lower vagina and imperforate hymen – the latter of which is one of the most common obstructive lesions of the female genital tract – later on. And if the hymen remains imperforate, the mucus will be reabsorbed and the patient usually will remain asymptomatic until menarche.
In the adolescent time period, both anomalies often are identified when the patient presents with pelvic pain – usually cyclic pelvic pain with primary amenorrhea. Because the onset of menses typically occurs 2-3 years after the onset of estrogenization and breast development, evaluating breast development can help us to determine the timing of expected menarche. An obstructive anomaly should be suspected in an adolescent who presents with pain during this time period, after evaluation for an acute abdomen (Figure 2a).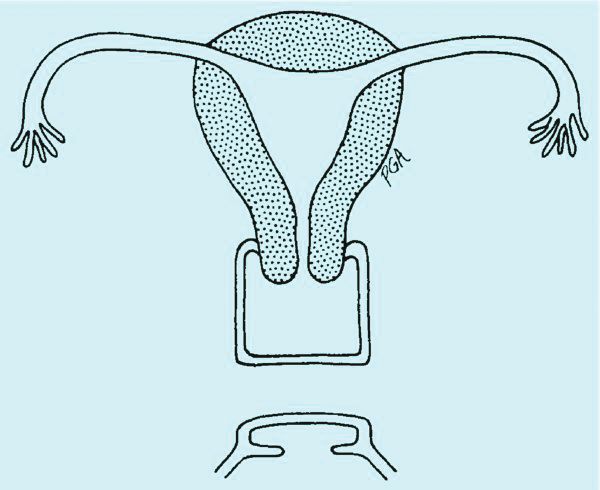
When a vaginal orifice is visualized upon evaluation of the external genitalia and separation of the labia, a higher anomaly such as a transverse vaginal septum should be suspected. When an introitus cannot be visualized, evaluation for an imperforate hymen or agenesis of the lower vagina is necessary (Figure 1b and 1c).
The simplest way to differentiate imperforate hymen from agenesis of the lower vagina is with visualization of the obstructing tissue on exam and usage of transperitoneal ultrasound. With the transducer placed on the vulva, we can evaluate the distance from the normal location of an introitus to the level of the obstruction. If the distance is in millimeters, then typically there is an imperforate hymen. If the distance is larger – more than several millimeters – then the differential diagnosis typically is agenesis of the lower vagina, an anomaly that results from abnormal development of the sinovaginal bulbs and vaginal plate.
The distance as measured by transperitoneal ultrasound also will indicate whether or not pull-through vaginoplasty (Figure 2b) – our standard treatment for lower vaginal agenesis – is possible using native vaginal mucosa from the upper vagina. Most commonly, the distance is less than 5 cm and we are able to make a transverse incision where the hymenal ring should be located, carry the dissection to the upper vagina, drain the obstruction, and mobilize the upper vaginal mucosa, suturing it to the newly created introitus to formulate a patent vaginal tract.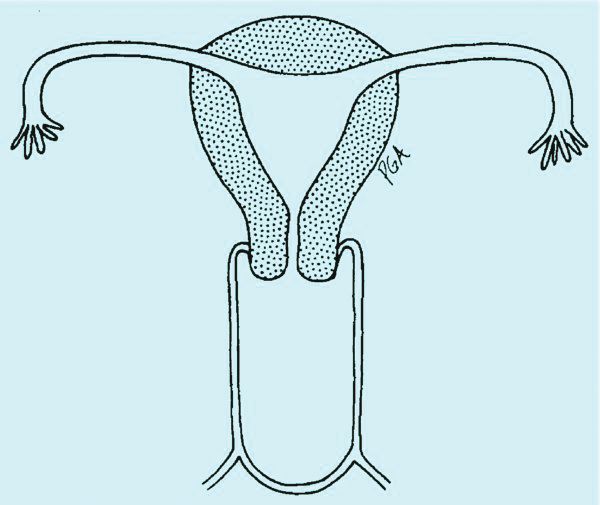
A rectoabdominal examination similarly can be helpful in making the diagnosis of lower vaginal agenesis and in determining whether there is enough tissue available for a pull-through procedure (Figures 2a and 2b). Because patients with this anomaly generally have normal cyclic pituitary-ovarian-endometrial function at menarche, the upper vagina will distend with blood products and secretions that can be palpated on the rectoabdominal exam. If the obstructed vaginal tissue is not felt with the rectal finger at midline, the obstructed agenesis of the vagina probably is too high for a straightforward pull-through procedure. Alternatively, the patient may have a unicornuate system with agenesis of the lower vagina; in this case, the obstructed upper vaginal tissue will not be in the midline but off to one side. MRI also may be helpful for defining the pelvic anatomy.
The optimal timing for a pull-through vaginoplasty (Figure 2b) is when a large hematocolpos (Figure 2a) is present, as the blood acts as a natural expander of the native vaginal tissue, increasing the amount of tissue available for a primary reanastomosis. This emphasizes the importance of an accurate initial diagnosis. Too often, obstructions that are actually lower vaginal agenesis are presumed to be imperforate hymen, and the hematocolpos is subsequently evacuated after a transverse incision and dissection of excess tissue, causing the upper vagina to retract and shrink. This mistake can result in the formation of a fistulous tract from the previously obstructed upper vagina to the level of the introitus.
The vaginoplasty is carried out with the patient in the dorsal lithotomy position. A Foley catheter is placed into the bladder to avoid an inadvertent anterior entry into the posterior wall of the bladder, and the labia are grasped and pulled down and out.
The hymenal tissue should be visible. A transverse incision is made, with electrocautery, where the introitus should be located, and a dissection is carried out to reach the obstructed upper vaginal tissue. Care is needed to keep the dissection in the midline and avoid the bladder above and the rectum below. In cases in which it is difficult to identify the area of obstruction, intraoperative ultrasound can be helpful. A spinal needle with a 10-cc syringe also can be used to identify a track through which to access the fluid.
The linear incision then is made with electrocautery and the obstructed hemivagina is entered. Allis clamps are used to grasp the vaginal mucosa from the previously obstructed upper vagina to help identify the tissue that needs to be mobilized. The tissue then is further dissected to free the upper vagina, and the edges are pulled down to the level of the introitus with Allis clamps. “Relaxing” incisions are made at 1, 5,7, and 11 o’clock to avoid a circumferential scar. The upper vaginal mucosa is sewn to the newly created introitus with a 2-0 vicryl suture on a UR6 (a smaller curved urology needle).
When the distance from normal introitus location to obstruction is greater than 5 cm, we sometimes use vaginal dilators to lessen the distance and reach the obstruction for a pull-through procedure. Alternatively, the upper vagina may be mobilized from above either robotically or laparoscopically so that the upper vaginal mucosa may be pulled down without entering the bladder. Occasionally, with greater distances over 5 cm, the vaginoplasty may require utilization of a buccal mucosal graft or a bowel segment.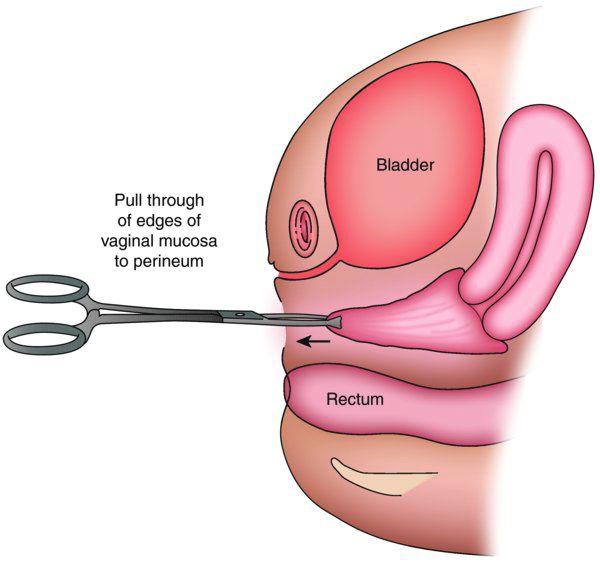
Intraoperative ultrasound can be especially helpful for locating the obstructed vagina in women with a unicornuate system because the upper vagina will not be in the midline and ultrasound can help determine the appropriate angle for dissection.
Prophylactic antibiotics initiated postoperatively are important with pull-through vaginoplasty, because the uterus and fallopian tubes may contain blood (an excellent growth media) and because there is a risk of bacteria ascending into what becomes an open system.
Postoperatively, we guide patients on the use of flexible Milex dilators (CooperSurgical) to ensure that the vagina heals without restenosis. The length of postoperative dilation therapy can vary from 2-12 months, depending on healing. The dilator is worn 24 hours a day, 7 days a week, and is removed only for urination, defecation, and cleaning. With adequate postoperative dilation, patients will have normal sexual and reproductive function, and vaginal delivery should be possible.
Obstructed hemivagina
An obstructed hemivagina, an uncommon Müllerian duct anomaly, occurs most often with ipsilateral renal agenesis and is commonly referred to as OHVIRA. Because the formation of the reproductive system is closely associated with the development of the urinary system, it is not unusual for renal anomalies to occur alongside Müllerian anomalies and vaginal anomalies. There should be a high index of suspicion for a reproductive tract anomaly in any patient known to have a horseshoe kidney, duplex collecting system, unilateral renal agenesis, or other renal anomaly.
Patients with obstructed hemivagina typically present in adolescence with pelvic pain or dysmenorrhea, and commonly are misdiagnosed as having endometriomas or vaginal cysts. On vaginal examination, the obstructed hemivagina may be visualized as a bulge coming from the lateral vaginal sidewall. While only one cervix is appreciated on a vaginal exam, an ultrasound examination often will show two uteri and two cervices. MRI also is helpful for diagnosis.
Obstructed hemivagina requires surgical correction to open the obstruction, excise the excess vaginal tissue, and create one vagina with access to the second cervix. Great care must be taken to avoid not only the bladder and rectum but the cervices. It is not unusual for the two cervices to be at different levels, with one cervix sharing medial aspects of the vaginal wall of the second vagina (Figure 1a). The tissue between the two cervices should be left in place to avoid compromising their blood supply.
We manage this anomaly primarily through a single-stage vaginoplasty. For the nonobstructed side to be visualized, a longitudinal incision into the obstructed hemivagina should be made at the point at which it is most easily palpated. As with agenesis of the lower vagina, the fluid to be drained tends to be old menstrual blood that is thick and dark brown. It is useful to set up two suction units at the time of surgery because tubing can become clogged.
The use of vaginal side wall retractors helps with visualization. Alternatively, I tend to use malleable abdominal wall retractors vaginally, as they can be bent to conform to the angle needed and come in different widths. When it is difficult to identify the area of obstruction, a spinal needle with a 10-cc syringe again can be used to identify a track for accessing the fluid. The linear incision then is made with electrocautery, and the obstructed hemivagina is entered.
Allis clamps are used to grasp the vaginal mucosa from the previously obstructed hemivagina to help identify the tissue that needs to be excised. Once the fluid is evacuated, a finger also can be placed into the obstructed vagina is help identify excess tissue. This three-dimensional elliptical area is similar to a septum but becomes the obstructed hemivagina as it attaches to the vaginal wall (Figure 1a).
Retrograde menses and endometriosis occur commonly with obstructive anomalies like obstructed hemivagina and agenesis of the lower vagina, but laparoscopy with the goal of treating endometriosis is not indicated. We discourage its use at the time of repair because there is evidence that almost all endometriosis will completely resorb on its own once the anomalies are corrected.1,2
As with repair of lower vagina agenesis, antibiotics to prevent an ascending infection should be taken after surgical correction of obstructed hemivagina. Patients with obstructed hemivagina can have a vaginal delivery if there are no other contraindications. Women with obstructed hemivagina and ipsilateral renal anomaly have essentially two unicornuate systems and thus are at risk of breech presentation and preterm delivery.
Dr. Laufer is chief of the division of gynecology, codirector of the Center for Young Women’s Health, and director of the Boston Center for Endometriosis, all at Boston Children’s Hospital. He also is professor of obstetrics, gynecology, and reproductive biology at Harvard Medical School, Boston.
References
1. Am J Obstet Gynecol. 1986;154:39.
2. J Pediatr Adolesc Gynecol. 2010;23(2):e89.
Congenital obstructive anomalies of the vagina are unusual and can be challenging to diagnose and manage. Two of the most challenging are obstructive hemivagina with ipsilateral renal agenesis (Figure 1a) and agenesis of the lower vagina (Figure 1b), the latter of which must be differentiated most commonly from imperforate hymen (Figure 1c). Evaluation and treatment of these anomalies is dependent upon the age of the patient, as well as the symptoms, and the timing of treatment should be individualized.
Agenesis of the lower vagina
Agenesis of the lower vagina and imperforate hymen may present either in the newborn period as a bulging introitus caused by mucocolpos from vaginal secretions stimulated by maternal estradiol or during adolescence at the time of menarche. In neonates, it often is best not to intervene when obstructive anomalies are suspected as long as there is no fever; pain; or compromise of respiration, urinary and bowel function, and other functionality. It will be easier to differentiate agenesis of the lower vagina and imperforate hymen – the latter of which is one of the most common obstructive lesions of the female genital tract – later on. And if the hymen remains imperforate, the mucus will be reabsorbed and the patient usually will remain asymptomatic until menarche.
In the adolescent time period, both anomalies often are identified when the patient presents with pelvic pain – usually cyclic pelvic pain with primary amenorrhea. Because the onset of menses typically occurs 2-3 years after the onset of estrogenization and breast development, evaluating breast development can help us to determine the timing of expected menarche. An obstructive anomaly should be suspected in an adolescent who presents with pain during this time period, after evaluation for an acute abdomen (Figure 2a).
When a vaginal orifice is visualized upon evaluation of the external genitalia and separation of the labia, a higher anomaly such as a transverse vaginal septum should be suspected. When an introitus cannot be visualized, evaluation for an imperforate hymen or agenesis of the lower vagina is necessary (Figure 1b and 1c).
The simplest way to differentiate imperforate hymen from agenesis of the lower vagina is with visualization of the obstructing tissue on exam and usage of transperitoneal ultrasound. With the transducer placed on the vulva, we can evaluate the distance from the normal location of an introitus to the level of the obstruction. If the distance is in millimeters, then typically there is an imperforate hymen. If the distance is larger – more than several millimeters – then the differential diagnosis typically is agenesis of the lower vagina, an anomaly that results from abnormal development of the sinovaginal bulbs and vaginal plate.
The distance as measured by transperitoneal ultrasound also will indicate whether or not pull-through vaginoplasty (Figure 2b) – our standard treatment for lower vaginal agenesis – is possible using native vaginal mucosa from the upper vagina. Most commonly, the distance is less than 5 cm and we are able to make a transverse incision where the hymenal ring should be located, carry the dissection to the upper vagina, drain the obstruction, and mobilize the upper vaginal mucosa, suturing it to the newly created introitus to formulate a patent vaginal tract.
A rectoabdominal examination similarly can be helpful in making the diagnosis of lower vaginal agenesis and in determining whether there is enough tissue available for a pull-through procedure (Figures 2a and 2b). Because patients with this anomaly generally have normal cyclic pituitary-ovarian-endometrial function at menarche, the upper vagina will distend with blood products and secretions that can be palpated on the rectoabdominal exam. If the obstructed vaginal tissue is not felt with the rectal finger at midline, the obstructed agenesis of the vagina probably is too high for a straightforward pull-through procedure. Alternatively, the patient may have a unicornuate system with agenesis of the lower vagina; in this case, the obstructed upper vaginal tissue will not be in the midline but off to one side. MRI also may be helpful for defining the pelvic anatomy.
The optimal timing for a pull-through vaginoplasty (Figure 2b) is when a large hematocolpos (Figure 2a) is present, as the blood acts as a natural expander of the native vaginal tissue, increasing the amount of tissue available for a primary reanastomosis. This emphasizes the importance of an accurate initial diagnosis. Too often, obstructions that are actually lower vaginal agenesis are presumed to be imperforate hymen, and the hematocolpos is subsequently evacuated after a transverse incision and dissection of excess tissue, causing the upper vagina to retract and shrink. This mistake can result in the formation of a fistulous tract from the previously obstructed upper vagina to the level of the introitus.
The vaginoplasty is carried out with the patient in the dorsal lithotomy position. A Foley catheter is placed into the bladder to avoid an inadvertent anterior entry into the posterior wall of the bladder, and the labia are grasped and pulled down and out.
The hymenal tissue should be visible. A transverse incision is made, with electrocautery, where the introitus should be located, and a dissection is carried out to reach the obstructed upper vaginal tissue. Care is needed to keep the dissection in the midline and avoid the bladder above and the rectum below. In cases in which it is difficult to identify the area of obstruction, intraoperative ultrasound can be helpful. A spinal needle with a 10-cc syringe also can be used to identify a track through which to access the fluid.
The linear incision then is made with electrocautery and the obstructed hemivagina is entered. Allis clamps are used to grasp the vaginal mucosa from the previously obstructed upper vagina to help identify the tissue that needs to be mobilized. The tissue then is further dissected to free the upper vagina, and the edges are pulled down to the level of the introitus with Allis clamps. “Relaxing” incisions are made at 1, 5,7, and 11 o’clock to avoid a circumferential scar. The upper vaginal mucosa is sewn to the newly created introitus with a 2-0 vicryl suture on a UR6 (a smaller curved urology needle).
When the distance from normal introitus location to obstruction is greater than 5 cm, we sometimes use vaginal dilators to lessen the distance and reach the obstruction for a pull-through procedure. Alternatively, the upper vagina may be mobilized from above either robotically or laparoscopically so that the upper vaginal mucosa may be pulled down without entering the bladder. Occasionally, with greater distances over 5 cm, the vaginoplasty may require utilization of a buccal mucosal graft or a bowel segment.
Intraoperative ultrasound can be especially helpful for locating the obstructed vagina in women with a unicornuate system because the upper vagina will not be in the midline and ultrasound can help determine the appropriate angle for dissection.
Prophylactic antibiotics initiated postoperatively are important with pull-through vaginoplasty, because the uterus and fallopian tubes may contain blood (an excellent growth media) and because there is a risk of bacteria ascending into what becomes an open system.
Postoperatively, we guide patients on the use of flexible Milex dilators (CooperSurgical) to ensure that the vagina heals without restenosis. The length of postoperative dilation therapy can vary from 2-12 months, depending on healing. The dilator is worn 24 hours a day, 7 days a week, and is removed only for urination, defecation, and cleaning. With adequate postoperative dilation, patients will have normal sexual and reproductive function, and vaginal delivery should be possible.
Obstructed hemivagina
An obstructed hemivagina, an uncommon Müllerian duct anomaly, occurs most often with ipsilateral renal agenesis and is commonly referred to as OHVIRA. Because the formation of the reproductive system is closely associated with the development of the urinary system, it is not unusual for renal anomalies to occur alongside Müllerian anomalies and vaginal anomalies. There should be a high index of suspicion for a reproductive tract anomaly in any patient known to have a horseshoe kidney, duplex collecting system, unilateral renal agenesis, or other renal anomaly.
Patients with obstructed hemivagina typically present in adolescence with pelvic pain or dysmenorrhea, and commonly are misdiagnosed as having endometriomas or vaginal cysts. On vaginal examination, the obstructed hemivagina may be visualized as a bulge coming from the lateral vaginal sidewall. While only one cervix is appreciated on a vaginal exam, an ultrasound examination often will show two uteri and two cervices. MRI also is helpful for diagnosis.
Obstructed hemivagina requires surgical correction to open the obstruction, excise the excess vaginal tissue, and create one vagina with access to the second cervix. Great care must be taken to avoid not only the bladder and rectum but the cervices. It is not unusual for the two cervices to be at different levels, with one cervix sharing medial aspects of the vaginal wall of the second vagina (Figure 1a). The tissue between the two cervices should be left in place to avoid compromising their blood supply.
We manage this anomaly primarily through a single-stage vaginoplasty. For the nonobstructed side to be visualized, a longitudinal incision into the obstructed hemivagina should be made at the point at which it is most easily palpated. As with agenesis of the lower vagina, the fluid to be drained tends to be old menstrual blood that is thick and dark brown. It is useful to set up two suction units at the time of surgery because tubing can become clogged.
The use of vaginal side wall retractors helps with visualization. Alternatively, I tend to use malleable abdominal wall retractors vaginally, as they can be bent to conform to the angle needed and come in different widths. When it is difficult to identify the area of obstruction, a spinal needle with a 10-cc syringe again can be used to identify a track for accessing the fluid. The linear incision then is made with electrocautery, and the obstructed hemivagina is entered.
Allis clamps are used to grasp the vaginal mucosa from the previously obstructed hemivagina to help identify the tissue that needs to be excised. Once the fluid is evacuated, a finger also can be placed into the obstructed vagina is help identify excess tissue. This three-dimensional elliptical area is similar to a septum but becomes the obstructed hemivagina as it attaches to the vaginal wall (Figure 1a).
Retrograde menses and endometriosis occur commonly with obstructive anomalies like obstructed hemivagina and agenesis of the lower vagina, but laparoscopy with the goal of treating endometriosis is not indicated. We discourage its use at the time of repair because there is evidence that almost all endometriosis will completely resorb on its own once the anomalies are corrected.1,2
As with repair of lower vagina agenesis, antibiotics to prevent an ascending infection should be taken after surgical correction of obstructed hemivagina. Patients with obstructed hemivagina can have a vaginal delivery if there are no other contraindications. Women with obstructed hemivagina and ipsilateral renal anomaly have essentially two unicornuate systems and thus are at risk of breech presentation and preterm delivery.
Dr. Laufer is chief of the division of gynecology, codirector of the Center for Young Women’s Health, and director of the Boston Center for Endometriosis, all at Boston Children’s Hospital. He also is professor of obstetrics, gynecology, and reproductive biology at Harvard Medical School, Boston.
References
1. Am J Obstet Gynecol. 1986;154:39.
2. J Pediatr Adolesc Gynecol. 2010;23(2):e89.
Congenital obstructive anomalies of the vagina are unusual and can be challenging to diagnose and manage. Two of the most challenging are obstructive hemivagina with ipsilateral renal agenesis (Figure 1a) and agenesis of the lower vagina (Figure 1b), the latter of which must be differentiated most commonly from imperforate hymen (Figure 1c). Evaluation and treatment of these anomalies is dependent upon the age of the patient, as well as the symptoms, and the timing of treatment should be individualized.
Agenesis of the lower vagina
Agenesis of the lower vagina and imperforate hymen may present either in the newborn period as a bulging introitus caused by mucocolpos from vaginal secretions stimulated by maternal estradiol or during adolescence at the time of menarche. In neonates, it often is best not to intervene when obstructive anomalies are suspected as long as there is no fever; pain; or compromise of respiration, urinary and bowel function, and other functionality. It will be easier to differentiate agenesis of the lower vagina and imperforate hymen – the latter of which is one of the most common obstructive lesions of the female genital tract – later on. And if the hymen remains imperforate, the mucus will be reabsorbed and the patient usually will remain asymptomatic until menarche.
In the adolescent time period, both anomalies often are identified when the patient presents with pelvic pain – usually cyclic pelvic pain with primary amenorrhea. Because the onset of menses typically occurs 2-3 years after the onset of estrogenization and breast development, evaluating breast development can help us to determine the timing of expected menarche. An obstructive anomaly should be suspected in an adolescent who presents with pain during this time period, after evaluation for an acute abdomen (Figure 2a).
When a vaginal orifice is visualized upon evaluation of the external genitalia and separation of the labia, a higher anomaly such as a transverse vaginal septum should be suspected. When an introitus cannot be visualized, evaluation for an imperforate hymen or agenesis of the lower vagina is necessary (Figure 1b and 1c).
The simplest way to differentiate imperforate hymen from agenesis of the lower vagina is with visualization of the obstructing tissue on exam and usage of transperitoneal ultrasound. With the transducer placed on the vulva, we can evaluate the distance from the normal location of an introitus to the level of the obstruction. If the distance is in millimeters, then typically there is an imperforate hymen. If the distance is larger – more than several millimeters – then the differential diagnosis typically is agenesis of the lower vagina, an anomaly that results from abnormal development of the sinovaginal bulbs and vaginal plate.
The distance as measured by transperitoneal ultrasound also will indicate whether or not pull-through vaginoplasty (Figure 2b) – our standard treatment for lower vaginal agenesis – is possible using native vaginal mucosa from the upper vagina. Most commonly, the distance is less than 5 cm and we are able to make a transverse incision where the hymenal ring should be located, carry the dissection to the upper vagina, drain the obstruction, and mobilize the upper vaginal mucosa, suturing it to the newly created introitus to formulate a patent vaginal tract.
A rectoabdominal examination similarly can be helpful in making the diagnosis of lower vaginal agenesis and in determining whether there is enough tissue available for a pull-through procedure (Figures 2a and 2b). Because patients with this anomaly generally have normal cyclic pituitary-ovarian-endometrial function at menarche, the upper vagina will distend with blood products and secretions that can be palpated on the rectoabdominal exam. If the obstructed vaginal tissue is not felt with the rectal finger at midline, the obstructed agenesis of the vagina probably is too high for a straightforward pull-through procedure. Alternatively, the patient may have a unicornuate system with agenesis of the lower vagina; in this case, the obstructed upper vaginal tissue will not be in the midline but off to one side. MRI also may be helpful for defining the pelvic anatomy.
The optimal timing for a pull-through vaginoplasty (Figure 2b) is when a large hematocolpos (Figure 2a) is present, as the blood acts as a natural expander of the native vaginal tissue, increasing the amount of tissue available for a primary reanastomosis. This emphasizes the importance of an accurate initial diagnosis. Too often, obstructions that are actually lower vaginal agenesis are presumed to be imperforate hymen, and the hematocolpos is subsequently evacuated after a transverse incision and dissection of excess tissue, causing the upper vagina to retract and shrink. This mistake can result in the formation of a fistulous tract from the previously obstructed upper vagina to the level of the introitus.
The vaginoplasty is carried out with the patient in the dorsal lithotomy position. A Foley catheter is placed into the bladder to avoid an inadvertent anterior entry into the posterior wall of the bladder, and the labia are grasped and pulled down and out.
The hymenal tissue should be visible. A transverse incision is made, with electrocautery, where the introitus should be located, and a dissection is carried out to reach the obstructed upper vaginal tissue. Care is needed to keep the dissection in the midline and avoid the bladder above and the rectum below. In cases in which it is difficult to identify the area of obstruction, intraoperative ultrasound can be helpful. A spinal needle with a 10-cc syringe also can be used to identify a track through which to access the fluid.
The linear incision then is made with electrocautery and the obstructed hemivagina is entered. Allis clamps are used to grasp the vaginal mucosa from the previously obstructed upper vagina to help identify the tissue that needs to be mobilized. The tissue then is further dissected to free the upper vagina, and the edges are pulled down to the level of the introitus with Allis clamps. “Relaxing” incisions are made at 1, 5,7, and 11 o’clock to avoid a circumferential scar. The upper vaginal mucosa is sewn to the newly created introitus with a 2-0 vicryl suture on a UR6 (a smaller curved urology needle).
When the distance from normal introitus location to obstruction is greater than 5 cm, we sometimes use vaginal dilators to lessen the distance and reach the obstruction for a pull-through procedure. Alternatively, the upper vagina may be mobilized from above either robotically or laparoscopically so that the upper vaginal mucosa may be pulled down without entering the bladder. Occasionally, with greater distances over 5 cm, the vaginoplasty may require utilization of a buccal mucosal graft or a bowel segment.
Intraoperative ultrasound can be especially helpful for locating the obstructed vagina in women with a unicornuate system because the upper vagina will not be in the midline and ultrasound can help determine the appropriate angle for dissection.
Prophylactic antibiotics initiated postoperatively are important with pull-through vaginoplasty, because the uterus and fallopian tubes may contain blood (an excellent growth media) and because there is a risk of bacteria ascending into what becomes an open system.
Postoperatively, we guide patients on the use of flexible Milex dilators (CooperSurgical) to ensure that the vagina heals without restenosis. The length of postoperative dilation therapy can vary from 2-12 months, depending on healing. The dilator is worn 24 hours a day, 7 days a week, and is removed only for urination, defecation, and cleaning. With adequate postoperative dilation, patients will have normal sexual and reproductive function, and vaginal delivery should be possible.
Obstructed hemivagina
An obstructed hemivagina, an uncommon Müllerian duct anomaly, occurs most often with ipsilateral renal agenesis and is commonly referred to as OHVIRA. Because the formation of the reproductive system is closely associated with the development of the urinary system, it is not unusual for renal anomalies to occur alongside Müllerian anomalies and vaginal anomalies. There should be a high index of suspicion for a reproductive tract anomaly in any patient known to have a horseshoe kidney, duplex collecting system, unilateral renal agenesis, or other renal anomaly.
Patients with obstructed hemivagina typically present in adolescence with pelvic pain or dysmenorrhea, and commonly are misdiagnosed as having endometriomas or vaginal cysts. On vaginal examination, the obstructed hemivagina may be visualized as a bulge coming from the lateral vaginal sidewall. While only one cervix is appreciated on a vaginal exam, an ultrasound examination often will show two uteri and two cervices. MRI also is helpful for diagnosis.
Obstructed hemivagina requires surgical correction to open the obstruction, excise the excess vaginal tissue, and create one vagina with access to the second cervix. Great care must be taken to avoid not only the bladder and rectum but the cervices. It is not unusual for the two cervices to be at different levels, with one cervix sharing medial aspects of the vaginal wall of the second vagina (Figure 1a). The tissue between the two cervices should be left in place to avoid compromising their blood supply.
We manage this anomaly primarily through a single-stage vaginoplasty. For the nonobstructed side to be visualized, a longitudinal incision into the obstructed hemivagina should be made at the point at which it is most easily palpated. As with agenesis of the lower vagina, the fluid to be drained tends to be old menstrual blood that is thick and dark brown. It is useful to set up two suction units at the time of surgery because tubing can become clogged.
The use of vaginal side wall retractors helps with visualization. Alternatively, I tend to use malleable abdominal wall retractors vaginally, as they can be bent to conform to the angle needed and come in different widths. When it is difficult to identify the area of obstruction, a spinal needle with a 10-cc syringe again can be used to identify a track for accessing the fluid. The linear incision then is made with electrocautery, and the obstructed hemivagina is entered.
Allis clamps are used to grasp the vaginal mucosa from the previously obstructed hemivagina to help identify the tissue that needs to be excised. Once the fluid is evacuated, a finger also can be placed into the obstructed vagina is help identify excess tissue. This three-dimensional elliptical area is similar to a septum but becomes the obstructed hemivagina as it attaches to the vaginal wall (Figure 1a).
Retrograde menses and endometriosis occur commonly with obstructive anomalies like obstructed hemivagina and agenesis of the lower vagina, but laparoscopy with the goal of treating endometriosis is not indicated. We discourage its use at the time of repair because there is evidence that almost all endometriosis will completely resorb on its own once the anomalies are corrected.1,2
As with repair of lower vagina agenesis, antibiotics to prevent an ascending infection should be taken after surgical correction of obstructed hemivagina. Patients with obstructed hemivagina can have a vaginal delivery if there are no other contraindications. Women with obstructed hemivagina and ipsilateral renal anomaly have essentially two unicornuate systems and thus are at risk of breech presentation and preterm delivery.
Dr. Laufer is chief of the division of gynecology, codirector of the Center for Young Women’s Health, and director of the Boston Center for Endometriosis, all at Boston Children’s Hospital. He also is professor of obstetrics, gynecology, and reproductive biology at Harvard Medical School, Boston.
References
1. Am J Obstet Gynecol. 1986;154:39.
2. J Pediatr Adolesc Gynecol. 2010;23(2):e89.
FDA orders companies to cease all sales of transvaginal mesh for POP repair

The mandate came after Boston Scientific and Coloplast failed to provide adequate safety and efficacy information to the federal regulatory body in the wake of a 2016 reclassification to Class III (high-risk) devices, according to an FDA press statement. Both companies were required to submit a premarket approval application to continue marketing the mesh in the United States. Boston Scientific did file two PMAs, one for each of its transvaginal mesh products, but the FDA said the applications did not contain the required efficacy and safety data.
Both companies will have 10 days to submit their plan to withdraw these products from the market.
“In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” said Jeffrey Shuren, MD, director of FDA’s Center for Devices and Radiological Health. “Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions. The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
The deadline for submitting premarket approval applications for POP repair with transvaginal mesh was July 5, 2018. Manufacturers that did not file PMAs were required to pull their devices from the market. Those that did could keep selling the mesh while FDA reviewed their PMAs.
Boston Scientific submitted PMAs for its two devices, the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair System. Coloplast filed a PMA for its device, Restorelle DirectFix Anterior. But in February, the FDA convened an advisory panel to discuss just how to evaluate the safety and efficacy of the products.
To prove efficacy, the panel concluded, transvaginal POP repair with mesh should be better than repair with native tissue at 36 months, and the safety should be superior to repair with native tissue repair. The FDA agreed. However, the submitted premarket approval application did not include these kinds of data. Therefore, the agency declined to approve the devices.
In addition to stopping U.S. sales, FDA has required Boston Scientific and Coloplast to continue safety and efficacy follow-up of all women included in their 522 studies.
Coloplast did not have a press or public statement on its website as of April 16. Boston Scientific did have one.
“Up to 50% of women in the U.S. will suffer from POP during their lives, and we believe these women should have access to safe and effective treatment options,” according to the statement. “As a global leader in the pelvic floor space, we remain steadfast in our commitment to helping women live better and healthier lives. We also remain confident in the benefits and safety of our treatments for POP, and we look forward to continuing to work with the FDA on our PMAs for the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair Matrix, which are currently under review.”
The FDA statement also included advice to women who have had the mesh procedure for POP, and for their physicians
“Women who have had transvaginal mesh placed for the surgical repair of POP should continue with their annual and other routine check-ups and follow-up care. There is no need to take additional action if they are satisfied with their surgery and are not having complications or symptoms. Patients should notify their health care professionals if they have complications or symptoms, including persistent vaginal bleeding or discharge, pelvic or groin pain, or pain with sex. They should also let their health care professional know if they have surgical mesh, especially if they plan to have another surgery or other medical procedures. Women who were planning to have mesh placed transvaginally for the repair of POP should discuss other treatment options with their doctors.”
The Food and Drug Administration’s decision ordering manufacturers to remove mesh for transvaginal repair of prolapse from the market was based on the products’ effectiveness and safety profile, compared with vaginal native tissue repairs. Previous studies have shown that polypropylene mesh for anterior repair had similar or slightly higher success, compared with native tissue repairs. This was not a sufficient benefit considering the potential adverse events that include mesh exposure, and the pelvic pain and dyspareunia associated with using these products. There is no additional benefit of using polypropylene mesh in the posterior compartment.
It would be interesting to review the information provided by manufacturers as part of the premarket approval. What were the primary endpoints for efficacy that were used? What were the rates of complications for mesh exposure, pelvic pain, and dyspareunia? How did the rates of pelvic pain and dyspareunia compare with native tissue repair.
Gynecologic surgeons still have a number of options for treating vaginal prolapse, which include vaginal native tissue repairs, and laparoscopic and abdominal surgeries that involve native tissue or polypropylene mesh. It will be interesting to see how the FDA’s Medical Device Safety action plan will affect future innovations for treating vaginal prolapse, while at the same time providing women and their physicians with products that are safe and effective.
Jose S. Maceda, MD, is a urogynecologist at Axia Women’s Health in King of Prussia, Penn. Dr. Maceda, who was asked to comment on the FDA decision, has no relevant financial disclosures.
The Food and Drug Administration’s decision ordering manufacturers to remove mesh for transvaginal repair of prolapse from the market was based on the products’ effectiveness and safety profile, compared with vaginal native tissue repairs. Previous studies have shown that polypropylene mesh for anterior repair had similar or slightly higher success, compared with native tissue repairs. This was not a sufficient benefit considering the potential adverse events that include mesh exposure, and the pelvic pain and dyspareunia associated with using these products. There is no additional benefit of using polypropylene mesh in the posterior compartment.
It would be interesting to review the information provided by manufacturers as part of the premarket approval. What were the primary endpoints for efficacy that were used? What were the rates of complications for mesh exposure, pelvic pain, and dyspareunia? How did the rates of pelvic pain and dyspareunia compare with native tissue repair.
Gynecologic surgeons still have a number of options for treating vaginal prolapse, which include vaginal native tissue repairs, and laparoscopic and abdominal surgeries that involve native tissue or polypropylene mesh. It will be interesting to see how the FDA’s Medical Device Safety action plan will affect future innovations for treating vaginal prolapse, while at the same time providing women and their physicians with products that are safe and effective.
Jose S. Maceda, MD, is a urogynecologist at Axia Women’s Health in King of Prussia, Penn. Dr. Maceda, who was asked to comment on the FDA decision, has no relevant financial disclosures.
The Food and Drug Administration’s decision ordering manufacturers to remove mesh for transvaginal repair of prolapse from the market was based on the products’ effectiveness and safety profile, compared with vaginal native tissue repairs. Previous studies have shown that polypropylene mesh for anterior repair had similar or slightly higher success, compared with native tissue repairs. This was not a sufficient benefit considering the potential adverse events that include mesh exposure, and the pelvic pain and dyspareunia associated with using these products. There is no additional benefit of using polypropylene mesh in the posterior compartment.
It would be interesting to review the information provided by manufacturers as part of the premarket approval. What were the primary endpoints for efficacy that were used? What were the rates of complications for mesh exposure, pelvic pain, and dyspareunia? How did the rates of pelvic pain and dyspareunia compare with native tissue repair.
Gynecologic surgeons still have a number of options for treating vaginal prolapse, which include vaginal native tissue repairs, and laparoscopic and abdominal surgeries that involve native tissue or polypropylene mesh. It will be interesting to see how the FDA’s Medical Device Safety action plan will affect future innovations for treating vaginal prolapse, while at the same time providing women and their physicians with products that are safe and effective.
Jose S. Maceda, MD, is a urogynecologist at Axia Women’s Health in King of Prussia, Penn. Dr. Maceda, who was asked to comment on the FDA decision, has no relevant financial disclosures.
The mandate came after Boston Scientific and Coloplast failed to provide adequate safety and efficacy information to the federal regulatory body in the wake of a 2016 reclassification to Class III (high-risk) devices, according to an FDA press statement. Both companies were required to submit a premarket approval application to continue marketing the mesh in the United States. Boston Scientific did file two PMAs, one for each of its transvaginal mesh products, but the FDA said the applications did not contain the required efficacy and safety data.
Both companies will have 10 days to submit their plan to withdraw these products from the market.
“In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” said Jeffrey Shuren, MD, director of FDA’s Center for Devices and Radiological Health. “Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions. The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
The deadline for submitting premarket approval applications for POP repair with transvaginal mesh was July 5, 2018. Manufacturers that did not file PMAs were required to pull their devices from the market. Those that did could keep selling the mesh while FDA reviewed their PMAs.
Boston Scientific submitted PMAs for its two devices, the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair System. Coloplast filed a PMA for its device, Restorelle DirectFix Anterior. But in February, the FDA convened an advisory panel to discuss just how to evaluate the safety and efficacy of the products.
To prove efficacy, the panel concluded, transvaginal POP repair with mesh should be better than repair with native tissue at 36 months, and the safety should be superior to repair with native tissue repair. The FDA agreed. However, the submitted premarket approval application did not include these kinds of data. Therefore, the agency declined to approve the devices.
In addition to stopping U.S. sales, FDA has required Boston Scientific and Coloplast to continue safety and efficacy follow-up of all women included in their 522 studies.
Coloplast did not have a press or public statement on its website as of April 16. Boston Scientific did have one.
“Up to 50% of women in the U.S. will suffer from POP during their lives, and we believe these women should have access to safe and effective treatment options,” according to the statement. “As a global leader in the pelvic floor space, we remain steadfast in our commitment to helping women live better and healthier lives. We also remain confident in the benefits and safety of our treatments for POP, and we look forward to continuing to work with the FDA on our PMAs for the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair Matrix, which are currently under review.”
The FDA statement also included advice to women who have had the mesh procedure for POP, and for their physicians
“Women who have had transvaginal mesh placed for the surgical repair of POP should continue with their annual and other routine check-ups and follow-up care. There is no need to take additional action if they are satisfied with their surgery and are not having complications or symptoms. Patients should notify their health care professionals if they have complications or symptoms, including persistent vaginal bleeding or discharge, pelvic or groin pain, or pain with sex. They should also let their health care professional know if they have surgical mesh, especially if they plan to have another surgery or other medical procedures. Women who were planning to have mesh placed transvaginally for the repair of POP should discuss other treatment options with their doctors.”
The mandate came after Boston Scientific and Coloplast failed to provide adequate safety and efficacy information to the federal regulatory body in the wake of a 2016 reclassification to Class III (high-risk) devices, according to an FDA press statement. Both companies were required to submit a premarket approval application to continue marketing the mesh in the United States. Boston Scientific did file two PMAs, one for each of its transvaginal mesh products, but the FDA said the applications did not contain the required efficacy and safety data.
Both companies will have 10 days to submit their plan to withdraw these products from the market.
“In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term,” said Jeffrey Shuren, MD, director of FDA’s Center for Devices and Radiological Health. “Patient safety is our highest priority, and women must have access to safe medical devices that provide relief from symptoms and better management of their medical conditions. The FDA has committed to taking forceful new actions to enhance device safety and encourage innovations that lead to safer medical devices, so that patients have access to safe and effective medical devices and the information they need to make informed decisions about their care.”
The deadline for submitting premarket approval applications for POP repair with transvaginal mesh was July 5, 2018. Manufacturers that did not file PMAs were required to pull their devices from the market. Those that did could keep selling the mesh while FDA reviewed their PMAs.
Boston Scientific submitted PMAs for its two devices, the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair System. Coloplast filed a PMA for its device, Restorelle DirectFix Anterior. But in February, the FDA convened an advisory panel to discuss just how to evaluate the safety and efficacy of the products.
To prove efficacy, the panel concluded, transvaginal POP repair with mesh should be better than repair with native tissue at 36 months, and the safety should be superior to repair with native tissue repair. The FDA agreed. However, the submitted premarket approval application did not include these kinds of data. Therefore, the agency declined to approve the devices.
In addition to stopping U.S. sales, FDA has required Boston Scientific and Coloplast to continue safety and efficacy follow-up of all women included in their 522 studies.
Coloplast did not have a press or public statement on its website as of April 16. Boston Scientific did have one.
“Up to 50% of women in the U.S. will suffer from POP during their lives, and we believe these women should have access to safe and effective treatment options,” according to the statement. “As a global leader in the pelvic floor space, we remain steadfast in our commitment to helping women live better and healthier lives. We also remain confident in the benefits and safety of our treatments for POP, and we look forward to continuing to work with the FDA on our PMAs for the Uphold LITE Vaginal Support System and the Xenform Soft Tissue Repair Matrix, which are currently under review.”
The FDA statement also included advice to women who have had the mesh procedure for POP, and for their physicians
“Women who have had transvaginal mesh placed for the surgical repair of POP should continue with their annual and other routine check-ups and follow-up care. There is no need to take additional action if they are satisfied with their surgery and are not having complications or symptoms. Patients should notify their health care professionals if they have complications or symptoms, including persistent vaginal bleeding or discharge, pelvic or groin pain, or pain with sex. They should also let their health care professional know if they have surgical mesh, especially if they plan to have another surgery or other medical procedures. Women who were planning to have mesh placed transvaginally for the repair of POP should discuss other treatment options with their doctors.”
Energy-based devices for vaginal rejuvenation described in FDA adverse event reports
The use of was implicated in nearly four dozen adverse event reports found in the agency’s medical device adverse event reporting database, researchers report.
The 45 unique event reports, submitted to the FDA during October 2015–January 2019, described 46 patients in total, of whom 33 reported long-term effects including pain, numbness, and burning, said the researchers, led by Jusleen Ahluwalia, MD, of the department of dermatology at the University of California, San Diego, and her coauthors. They included 31 that were reported by the patients, 8 reported by the manufacturer; 4 reported by the distributor, and 2 not specified.
These findings emphasize the need for clinical trials to evaluate the safety and efficacy of the lasers and radiofrequency devices that have been marketed and used for so-called vaginal rejuvenation procedures, they wrote in Lasers in Surgery and Medicine. The coauthors are Arisa Ortiz, MD, also with the University of California, San Diego, and Mathew M. Avram, MD, director of the Massachusetts General Hospital Dermatology Laser & Cosmetic Center, Boston. “Randomized studies are necessary to compare these therapies with standard modalities and to establish the safety of these devices,” they wrote.
In July 2018, the FDA issued a safety communication alerting patients and health care providers that the safety and effectiveness of energy-based devices has not been established for procedures described as “vaginal rejuvenation.” Scott Gottlieb, MD, FDA commissioner at the time, issued a statement decrying “deceptive health claims and significant risks” related to devices marketed for those medical procedures. In a November 2018 update, the FDA said they contacted some device manufacturers to express concerns that the devices were being marketed inappropriately and that manufacturers they had contacted so far “responded with adequate corrections.”
In their report, Dr. Ahluwalia and her associates noted that “vaginal rejuvenation” is an ill-defined term that may encompass a variety of procedures related to tightening; dyspareunia; dysuria; urinary incontinence; vulvar issues including irritation, dryness, and atrophy; and orgasmic dysfunction.
They found a total of 58 records in their review of the Manufacturer and User Facility Device Experience database, of which 25 were reported prior to the FDA’s July 2018 statement. Of 45 unique event descriptions found in those records, 39 were categorized as patient-related injuries, while 2 were operator-related injuries, 2 were device malfunctions, and 2 were not specified.
Pain was the most commonly adverse event, accounting for 19 reports in their analysis, while 11 patients reported numbness or burning.
Among the laser- and energy-based devices specifically described in the 39 patient-report injuries, the MonaLisa Touch had the highest number of adverse event reports (16), the data show. “However, this may be reflective of length of time bias as it is one of the first devices utilized to promote vaginal rejuvenation,” the authors pointed out.
In light of these findings, the authors advised clinicians to ask patients about their reasons for seeking vaginal rejuvenation procedures. “Normal variety of female genital appearances should also be reviewed when patients express cosmetic concerns,” they added. Concerns about related to genitourinary syndrome of menopause “or optimizing sexual function may be alleviated by exploring nonprocedural, conservative approaches, such as hormonal creams, if not contraindicated, and/or counseling,” they noted.
The authors provided conflict of interest disclosures related to Zalea, Inmode, Cytrellis, Zeltiq Aesthetics, Soliton, Sciton, Allergan, and Sienna Biopharmaceuticals, among others.
Adverse events related to devices and drugs can be reported to the FDA’s Medwatch program.
SOURCE: Ahluwalia J et al. Lasers Surg Med. 2019 Mar 29. doi: 10.1002/lsm.23084.
The use of was implicated in nearly four dozen adverse event reports found in the agency’s medical device adverse event reporting database, researchers report.
The 45 unique event reports, submitted to the FDA during October 2015–January 2019, described 46 patients in total, of whom 33 reported long-term effects including pain, numbness, and burning, said the researchers, led by Jusleen Ahluwalia, MD, of the department of dermatology at the University of California, San Diego, and her coauthors. They included 31 that were reported by the patients, 8 reported by the manufacturer; 4 reported by the distributor, and 2 not specified.
These findings emphasize the need for clinical trials to evaluate the safety and efficacy of the lasers and radiofrequency devices that have been marketed and used for so-called vaginal rejuvenation procedures, they wrote in Lasers in Surgery and Medicine. The coauthors are Arisa Ortiz, MD, also with the University of California, San Diego, and Mathew M. Avram, MD, director of the Massachusetts General Hospital Dermatology Laser & Cosmetic Center, Boston. “Randomized studies are necessary to compare these therapies with standard modalities and to establish the safety of these devices,” they wrote.
In July 2018, the FDA issued a safety communication alerting patients and health care providers that the safety and effectiveness of energy-based devices has not been established for procedures described as “vaginal rejuvenation.” Scott Gottlieb, MD, FDA commissioner at the time, issued a statement decrying “deceptive health claims and significant risks” related to devices marketed for those medical procedures. In a November 2018 update, the FDA said they contacted some device manufacturers to express concerns that the devices were being marketed inappropriately and that manufacturers they had contacted so far “responded with adequate corrections.”
In their report, Dr. Ahluwalia and her associates noted that “vaginal rejuvenation” is an ill-defined term that may encompass a variety of procedures related to tightening; dyspareunia; dysuria; urinary incontinence; vulvar issues including irritation, dryness, and atrophy; and orgasmic dysfunction.
They found a total of 58 records in their review of the Manufacturer and User Facility Device Experience database, of which 25 were reported prior to the FDA’s July 2018 statement. Of 45 unique event descriptions found in those records, 39 were categorized as patient-related injuries, while 2 were operator-related injuries, 2 were device malfunctions, and 2 were not specified.
Pain was the most commonly adverse event, accounting for 19 reports in their analysis, while 11 patients reported numbness or burning.
Among the laser- and energy-based devices specifically described in the 39 patient-report injuries, the MonaLisa Touch had the highest number of adverse event reports (16), the data show. “However, this may be reflective of length of time bias as it is one of the first devices utilized to promote vaginal rejuvenation,” the authors pointed out.
In light of these findings, the authors advised clinicians to ask patients about their reasons for seeking vaginal rejuvenation procedures. “Normal variety of female genital appearances should also be reviewed when patients express cosmetic concerns,” they added. Concerns about related to genitourinary syndrome of menopause “or optimizing sexual function may be alleviated by exploring nonprocedural, conservative approaches, such as hormonal creams, if not contraindicated, and/or counseling,” they noted.
The authors provided conflict of interest disclosures related to Zalea, Inmode, Cytrellis, Zeltiq Aesthetics, Soliton, Sciton, Allergan, and Sienna Biopharmaceuticals, among others.
Adverse events related to devices and drugs can be reported to the FDA’s Medwatch program.
SOURCE: Ahluwalia J et al. Lasers Surg Med. 2019 Mar 29. doi: 10.1002/lsm.23084.
The use of was implicated in nearly four dozen adverse event reports found in the agency’s medical device adverse event reporting database, researchers report.
The 45 unique event reports, submitted to the FDA during October 2015–January 2019, described 46 patients in total, of whom 33 reported long-term effects including pain, numbness, and burning, said the researchers, led by Jusleen Ahluwalia, MD, of the department of dermatology at the University of California, San Diego, and her coauthors. They included 31 that were reported by the patients, 8 reported by the manufacturer; 4 reported by the distributor, and 2 not specified.
These findings emphasize the need for clinical trials to evaluate the safety and efficacy of the lasers and radiofrequency devices that have been marketed and used for so-called vaginal rejuvenation procedures, they wrote in Lasers in Surgery and Medicine. The coauthors are Arisa Ortiz, MD, also with the University of California, San Diego, and Mathew M. Avram, MD, director of the Massachusetts General Hospital Dermatology Laser & Cosmetic Center, Boston. “Randomized studies are necessary to compare these therapies with standard modalities and to establish the safety of these devices,” they wrote.
In July 2018, the FDA issued a safety communication alerting patients and health care providers that the safety and effectiveness of energy-based devices has not been established for procedures described as “vaginal rejuvenation.” Scott Gottlieb, MD, FDA commissioner at the time, issued a statement decrying “deceptive health claims and significant risks” related to devices marketed for those medical procedures. In a November 2018 update, the FDA said they contacted some device manufacturers to express concerns that the devices were being marketed inappropriately and that manufacturers they had contacted so far “responded with adequate corrections.”
In their report, Dr. Ahluwalia and her associates noted that “vaginal rejuvenation” is an ill-defined term that may encompass a variety of procedures related to tightening; dyspareunia; dysuria; urinary incontinence; vulvar issues including irritation, dryness, and atrophy; and orgasmic dysfunction.
They found a total of 58 records in their review of the Manufacturer and User Facility Device Experience database, of which 25 were reported prior to the FDA’s July 2018 statement. Of 45 unique event descriptions found in those records, 39 were categorized as patient-related injuries, while 2 were operator-related injuries, 2 were device malfunctions, and 2 were not specified.
Pain was the most commonly adverse event, accounting for 19 reports in their analysis, while 11 patients reported numbness or burning.
Among the laser- and energy-based devices specifically described in the 39 patient-report injuries, the MonaLisa Touch had the highest number of adverse event reports (16), the data show. “However, this may be reflective of length of time bias as it is one of the first devices utilized to promote vaginal rejuvenation,” the authors pointed out.
In light of these findings, the authors advised clinicians to ask patients about their reasons for seeking vaginal rejuvenation procedures. “Normal variety of female genital appearances should also be reviewed when patients express cosmetic concerns,” they added. Concerns about related to genitourinary syndrome of menopause “or optimizing sexual function may be alleviated by exploring nonprocedural, conservative approaches, such as hormonal creams, if not contraindicated, and/or counseling,” they noted.
The authors provided conflict of interest disclosures related to Zalea, Inmode, Cytrellis, Zeltiq Aesthetics, Soliton, Sciton, Allergan, and Sienna Biopharmaceuticals, among others.
Adverse events related to devices and drugs can be reported to the FDA’s Medwatch program.
SOURCE: Ahluwalia J et al. Lasers Surg Med. 2019 Mar 29. doi: 10.1002/lsm.23084.
FROM LASERS IN SURGERY AND MEDICINE
Key clinical point: Nearly four dozen distinct adverse event reports related to energy-based devices used for vaginal rejuvenation were found in an analysis of an FDA database.
Major finding: The 45 unique event reports, disclosed to FDA during October 2015–January 2019, described 46 patients in total, of whom 33 reported long-term effects including pain, numbness, and burning.
Study details: Cross-sectional analysis of records in the Manufacturer and User Facility Device Experience database entered during October 2015–January 2019.
Disclosures: Authors provided conflict of interest disclosures related to ZALEA, InMode, Cytrellis, Zeltiq Aesthetics, Soliton, Sciton, Allergan, and Sienna Biopharmaceuticals, among others.
Source: Ahluwalia J et al. Lasers Surg Med. 2019 Mar 29. doi: 10.1002/lsm.23084.
Furosemide speeds ureteral patency confirmation, but is time savings worth the risk?
TUCSON, ARIZ. – according to results from a new randomized, controlled trial.

“It does make a difference, but is that really a [meaningful] difference? Every medication has adverse effects, so is it worth that extra time [savings] to take on that potential for side effects? It highlights the importance of statistical significance versus clinical significance, and I think [the clinical significance] can just be answered by each individual physician,” Simon Patton, MD, said in an interview.
Dr. Patton is a urogynecologist at Ascension Via Christi Medical Group in Wichita, Kan. He presented the study, which was conducted during his time as a fellow at the University of South Florida, Tampa, at the annual scientific meeting of the Society of Gynecologic Surgeons. Dr. Patton isn’t sure just how often physicians use furosemide during routine cystoscopy. “It would be great to do a survey to find out how many people use it in their practice,” he said.
Cystoscopy is used to during a surgery to ensure that no injury has been done to the bladder or the urethra, and the American Urogynecological Society recommends that it be performed during any pelvic reconstructive surgery. A key element of the test is confirming that the ureters are open. By increasing urine flow, furosemide can reduce the time to confirmation. But after conferring with a colleague who used the procedure, Dr. Patton looked for some data to support the practice and couldn’t find any.
Although the cystoscopy itself generally is safe, furosemide can cause hypotension, change in renal function, and even dehydration at higher doses. During the question-and-answer period, one attendee noted these issues and pointed out that furosemide can potentiate renal failure, especially among patients taking cephalosporins. “If you’re going to do this sort of trial, you have to consider potential adverse events. A single dose is probably not going to [cause an issue], but in the context of a study you want to monitor the adverse events that have been reported,” this attendee said.
The researchers did not observe any of these adverse events during the study, but Dr. Patton noted that the study was not powered to detect them. “We felt that with the low-dose, single-time [exposure], it was appropriate to not worry too much about those side effects,” he replied.
Another potential concern is that the increased urine flow could mask a kink in the ureter by forcing it open.
In the study, his team randomized 145 patients with a planned cystoscopy as part of a procedure to receive 10-mg furosemide (1 cc) or saline (1 cc) during a cystoscopy performed by an attending or a fellow. The median time to confirmation of ureteral patency was 86.5 seconds in the furosemide group, compared with 165.0 seconds in the saline group (difference, 78.5 seconds; P less than .001). The time to the first ureteral jet was 59 seconds versus 74 seconds, respectively (P less than .006). A Kaplan-Meier survival curve analysis also showed a significant improvement in time to ureteral patency confirmation (log-rank P less than .001).
The study was not funded. Dr. Patton has no relevant financial disclosures.
SOURCE: Patton S et al. SGS 2019, Abstract 10.
TUCSON, ARIZ. – according to results from a new randomized, controlled trial.
“It does make a difference, but is that really a [meaningful] difference? Every medication has adverse effects, so is it worth that extra time [savings] to take on that potential for side effects? It highlights the importance of statistical significance versus clinical significance, and I think [the clinical significance] can just be answered by each individual physician,” Simon Patton, MD, said in an interview.
Dr. Patton is a urogynecologist at Ascension Via Christi Medical Group in Wichita, Kan. He presented the study, which was conducted during his time as a fellow at the University of South Florida, Tampa, at the annual scientific meeting of the Society of Gynecologic Surgeons. Dr. Patton isn’t sure just how often physicians use furosemide during routine cystoscopy. “It would be great to do a survey to find out how many people use it in their practice,” he said.
Cystoscopy is used to during a surgery to ensure that no injury has been done to the bladder or the urethra, and the American Urogynecological Society recommends that it be performed during any pelvic reconstructive surgery. A key element of the test is confirming that the ureters are open. By increasing urine flow, furosemide can reduce the time to confirmation. But after conferring with a colleague who used the procedure, Dr. Patton looked for some data to support the practice and couldn’t find any.
Although the cystoscopy itself generally is safe, furosemide can cause hypotension, change in renal function, and even dehydration at higher doses. During the question-and-answer period, one attendee noted these issues and pointed out that furosemide can potentiate renal failure, especially among patients taking cephalosporins. “If you’re going to do this sort of trial, you have to consider potential adverse events. A single dose is probably not going to [cause an issue], but in the context of a study you want to monitor the adverse events that have been reported,” this attendee said.
The researchers did not observe any of these adverse events during the study, but Dr. Patton noted that the study was not powered to detect them. “We felt that with the low-dose, single-time [exposure], it was appropriate to not worry too much about those side effects,” he replied.
Another potential concern is that the increased urine flow could mask a kink in the ureter by forcing it open.
In the study, his team randomized 145 patients with a planned cystoscopy as part of a procedure to receive 10-mg furosemide (1 cc) or saline (1 cc) during a cystoscopy performed by an attending or a fellow. The median time to confirmation of ureteral patency was 86.5 seconds in the furosemide group, compared with 165.0 seconds in the saline group (difference, 78.5 seconds; P less than .001). The time to the first ureteral jet was 59 seconds versus 74 seconds, respectively (P less than .006). A Kaplan-Meier survival curve analysis also showed a significant improvement in time to ureteral patency confirmation (log-rank P less than .001).
The study was not funded. Dr. Patton has no relevant financial disclosures.
SOURCE: Patton S et al. SGS 2019, Abstract 10.
TUCSON, ARIZ. – according to results from a new randomized, controlled trial.
“It does make a difference, but is that really a [meaningful] difference? Every medication has adverse effects, so is it worth that extra time [savings] to take on that potential for side effects? It highlights the importance of statistical significance versus clinical significance, and I think [the clinical significance] can just be answered by each individual physician,” Simon Patton, MD, said in an interview.
Dr. Patton is a urogynecologist at Ascension Via Christi Medical Group in Wichita, Kan. He presented the study, which was conducted during his time as a fellow at the University of South Florida, Tampa, at the annual scientific meeting of the Society of Gynecologic Surgeons. Dr. Patton isn’t sure just how often physicians use furosemide during routine cystoscopy. “It would be great to do a survey to find out how many people use it in their practice,” he said.
Cystoscopy is used to during a surgery to ensure that no injury has been done to the bladder or the urethra, and the American Urogynecological Society recommends that it be performed during any pelvic reconstructive surgery. A key element of the test is confirming that the ureters are open. By increasing urine flow, furosemide can reduce the time to confirmation. But after conferring with a colleague who used the procedure, Dr. Patton looked for some data to support the practice and couldn’t find any.
Although the cystoscopy itself generally is safe, furosemide can cause hypotension, change in renal function, and even dehydration at higher doses. During the question-and-answer period, one attendee noted these issues and pointed out that furosemide can potentiate renal failure, especially among patients taking cephalosporins. “If you’re going to do this sort of trial, you have to consider potential adverse events. A single dose is probably not going to [cause an issue], but in the context of a study you want to monitor the adverse events that have been reported,” this attendee said.
The researchers did not observe any of these adverse events during the study, but Dr. Patton noted that the study was not powered to detect them. “We felt that with the low-dose, single-time [exposure], it was appropriate to not worry too much about those side effects,” he replied.
Another potential concern is that the increased urine flow could mask a kink in the ureter by forcing it open.
In the study, his team randomized 145 patients with a planned cystoscopy as part of a procedure to receive 10-mg furosemide (1 cc) or saline (1 cc) during a cystoscopy performed by an attending or a fellow. The median time to confirmation of ureteral patency was 86.5 seconds in the furosemide group, compared with 165.0 seconds in the saline group (difference, 78.5 seconds; P less than .001). The time to the first ureteral jet was 59 seconds versus 74 seconds, respectively (P less than .006). A Kaplan-Meier survival curve analysis also showed a significant improvement in time to ureteral patency confirmation (log-rank P less than .001).
The study was not funded. Dr. Patton has no relevant financial disclosures.
SOURCE: Patton S et al. SGS 2019, Abstract 10.
REPORTING FROM SGS 2019
Women with FXI deficiency face hemorrhage risk with gyn surgery
Women with factor XI (FXI) deficiency, particularly those with a history of bleeding and low plasmatic FXI levels, are at risk of experiencing obstetric and gynecologic hemorrhage, according to a retrospective analysis.
“[W]hile spontaneous bleeding episodes are unusual, FXI-deficient patients tend to bleed excessively after trauma or surgery, especially when highly fibrinolytic tissues are involved, such as rhino/oropharyngeal and genitourinary mucosa,” wrote Carlos Bravo-Perez, MD, of Universidad de Murcia in Spain, along with his colleagues. The report is in Medicina Clínica. “Menstruation, pregnancy and birth labour constitute intrinsic haemostatic challenges for FXI deficient women,” they noted.
The researchers conducted a retrospective analysis of 95 women with FXI deficiency. Cohort data was collected over a period of 20 years (1994 to 2014) from the city of Yecla, Spain.
Clinical information obtained included blood group, age, sex, aPTT ratio, and FXI coagulant levels, among others. The team completed a comprehensive molecular and biochemical analysis on all study patients.
“Surgical interventions and postoperative bleeding were recorded, as well as the use of antihemorrhagic prophylaxis or treatment,” the researchers wrote.
Bleeding occurred in 27.4% of women with FXI deficiency, with the majority of events being a single episode. The team reported that 52.5% of hemorrhagic events were provoked by surgical, obstetric, or dental procedures, while 47.5% were spontaneous bleeding events.
Among women who experienced bleeding events, 73.1% were gynecologic or obstetric hemorrhagic episodes. These included abnormal uterine bleeding, postpartum hemorrhage, excessive bleeding after a miscarriage, and bleeding after gynecologic surgery.
Overall, the proportion of abnormal uterine bleeding was 12.6%, the proportion of postpartum hemorrhage was 6.6%, and the proportion of gynecologic postoperative hemorrhage was 16%.
While the proportion of these bleeding events is higher than what has been observed in the general population, it is lower than what has been reported in other reviews for women with FXI deficiency, the researchers noted.
They also found that a positive history of bleeding and low plasmatic FXI levels were associated with a higher prevalence of gynecologic or obstetric bleeding.
The study was funded by Fundación Española de Trombosis y Hemostasia and Sociedad Española de Hematología y Hemoterapia in Spain. The authors reported having no conflicts of interest.
SOURCE: Bravo-Perez C et al. Med Clin (Barc). 2019 Mar 26. doi: 10.1016/j.medcli.2019.01.029.
Women with factor XI (FXI) deficiency, particularly those with a history of bleeding and low plasmatic FXI levels, are at risk of experiencing obstetric and gynecologic hemorrhage, according to a retrospective analysis.
“[W]hile spontaneous bleeding episodes are unusual, FXI-deficient patients tend to bleed excessively after trauma or surgery, especially when highly fibrinolytic tissues are involved, such as rhino/oropharyngeal and genitourinary mucosa,” wrote Carlos Bravo-Perez, MD, of Universidad de Murcia in Spain, along with his colleagues. The report is in Medicina Clínica. “Menstruation, pregnancy and birth labour constitute intrinsic haemostatic challenges for FXI deficient women,” they noted.
The researchers conducted a retrospective analysis of 95 women with FXI deficiency. Cohort data was collected over a period of 20 years (1994 to 2014) from the city of Yecla, Spain.
Clinical information obtained included blood group, age, sex, aPTT ratio, and FXI coagulant levels, among others. The team completed a comprehensive molecular and biochemical analysis on all study patients.
“Surgical interventions and postoperative bleeding were recorded, as well as the use of antihemorrhagic prophylaxis or treatment,” the researchers wrote.
Bleeding occurred in 27.4% of women with FXI deficiency, with the majority of events being a single episode. The team reported that 52.5% of hemorrhagic events were provoked by surgical, obstetric, or dental procedures, while 47.5% were spontaneous bleeding events.
Among women who experienced bleeding events, 73.1% were gynecologic or obstetric hemorrhagic episodes. These included abnormal uterine bleeding, postpartum hemorrhage, excessive bleeding after a miscarriage, and bleeding after gynecologic surgery.
Overall, the proportion of abnormal uterine bleeding was 12.6%, the proportion of postpartum hemorrhage was 6.6%, and the proportion of gynecologic postoperative hemorrhage was 16%.
While the proportion of these bleeding events is higher than what has been observed in the general population, it is lower than what has been reported in other reviews for women with FXI deficiency, the researchers noted.
They also found that a positive history of bleeding and low plasmatic FXI levels were associated with a higher prevalence of gynecologic or obstetric bleeding.
The study was funded by Fundación Española de Trombosis y Hemostasia and Sociedad Española de Hematología y Hemoterapia in Spain. The authors reported having no conflicts of interest.
SOURCE: Bravo-Perez C et al. Med Clin (Barc). 2019 Mar 26. doi: 10.1016/j.medcli.2019.01.029.
Women with factor XI (FXI) deficiency, particularly those with a history of bleeding and low plasmatic FXI levels, are at risk of experiencing obstetric and gynecologic hemorrhage, according to a retrospective analysis.
“[W]hile spontaneous bleeding episodes are unusual, FXI-deficient patients tend to bleed excessively after trauma or surgery, especially when highly fibrinolytic tissues are involved, such as rhino/oropharyngeal and genitourinary mucosa,” wrote Carlos Bravo-Perez, MD, of Universidad de Murcia in Spain, along with his colleagues. The report is in Medicina Clínica. “Menstruation, pregnancy and birth labour constitute intrinsic haemostatic challenges for FXI deficient women,” they noted.
The researchers conducted a retrospective analysis of 95 women with FXI deficiency. Cohort data was collected over a period of 20 years (1994 to 2014) from the city of Yecla, Spain.
Clinical information obtained included blood group, age, sex, aPTT ratio, and FXI coagulant levels, among others. The team completed a comprehensive molecular and biochemical analysis on all study patients.
“Surgical interventions and postoperative bleeding were recorded, as well as the use of antihemorrhagic prophylaxis or treatment,” the researchers wrote.
Bleeding occurred in 27.4% of women with FXI deficiency, with the majority of events being a single episode. The team reported that 52.5% of hemorrhagic events were provoked by surgical, obstetric, or dental procedures, while 47.5% were spontaneous bleeding events.
Among women who experienced bleeding events, 73.1% were gynecologic or obstetric hemorrhagic episodes. These included abnormal uterine bleeding, postpartum hemorrhage, excessive bleeding after a miscarriage, and bleeding after gynecologic surgery.
Overall, the proportion of abnormal uterine bleeding was 12.6%, the proportion of postpartum hemorrhage was 6.6%, and the proportion of gynecologic postoperative hemorrhage was 16%.
While the proportion of these bleeding events is higher than what has been observed in the general population, it is lower than what has been reported in other reviews for women with FXI deficiency, the researchers noted.
They also found that a positive history of bleeding and low plasmatic FXI levels were associated with a higher prevalence of gynecologic or obstetric bleeding.
The study was funded by Fundación Española de Trombosis y Hemostasia and Sociedad Española de Hematología y Hemoterapia in Spain. The authors reported having no conflicts of interest.
SOURCE: Bravo-Perez C et al. Med Clin (Barc). 2019 Mar 26. doi: 10.1016/j.medcli.2019.01.029.
FROM MEDICINA CLÍNICA
Stress incontinence surgery improves sexual dysfunction
TUCSON, ARIZ. –

The finding comes from a secondary analysis of two randomized, controlled trials comparing Burch colposuspension, autologous fascial slings, retropubic midurethral polypropylene slings, and transobturator midurethral polypropylene slings. The analysis looked at outcomes at 24 months after surgery. Stephanie Glass Clark, MD, a resident at Virginia Commonwealth University, Richmond, presented the results at the annual scientific meeting of the Society of Gynecologic Surgeons.
In the secondary analysis of the Stress Incontinence Surgical Treatment Efficacy Trial (SISTEr) and the Trial of Midurethral Slings (TOMUS) trials, Dr. Clark and her fellow researchers looked at the effect of surgical failure on sexual dysfunction outcomes. Subjective failure was defined as self-reported SUI symptoms or self-reported leakage by 3-day voiding diary beyond 3 months after the surgery. Objective failure was defined as any treatment for SUI after the surgery or a positive stress test or pad test beyond 3 months after the surgery.
Participants were excluded from the two studies if they were sexually inactive in the previous 6 months at baseline, at 12 months post baseline, or at 24 months. The studies employed the short form of the Pelvic Organ Prolapse/Urinary Incontinence Sexual Questionnaire (PISQ-12), which had 12 questions with scores ranging from 0 to 4. The secondary analysis sample included 488 women from SISTEr and 436 women from TOMUS.
There were some baseline differences among groups between the two trials, including vaginal deliveries, race/ethnicity, stage of prolapse, and concomitant surgeries performed at time of the anti-incontinence procedure.
All four surgeries were associated with improvements in sexual function, with no statistically significant between-group differences. Mean PISQ-12 scores improved from a range of 31-33 to a range of 36-38 at 24 months. Although there is no published minimum important difference for PISQ-12 scores, an improvement of at least one-half of a standard deviation is generally accepted as clinically meaningful. “In this case, the standard deviation at baseline was just under 3 and so the improvement of each treatment group by more than 1.5 is a clinically meaningful improvement in their sexual function,” Dr. Clark said.
“Sexual dysfunction is a much more common problem than we previously thought, so we’ve been trying to figure out if patients with pelvic floor disorders like stress incontinence are going to have any improvement in sexual dysfunction by surgically treating their stress incontinence. Previously published data had been pretty conflicting,” Dr. Clark added in an interview.
That previous research was mostly retrospective and could have been impacted by patient selection bias. By analyzing clinical trials, the researchers hoped to test their idea that the pelvic floor symptoms themselves may be key to sexual dysfunction and that treating it surgically would improve matters.
The positive result is encouraging, but it still leaves unanswered questions about the mechanism behind the relationship. Dr. Clark wondered whether leaking urine leakage during sex might be the culprit, or whether it is fear or shame associated with the condition.
The answer may come from further analysis of women who were sexually inactive at baseline, but became sexually active over the course of the studies. “I think looking at that patient population in particular is going to be an interesting area of research. Is it that it was completely related to their pelvic floor disorder, and then we fixed it [so] they could have a more fulfilling sexual life?” speculated Dr. Clark.
The study received some funding from the National Institutes of Health. Dr. Clark reported no relevant financial disclosures.
SOURCE: Clark SG et al. SGS 2019, Oral Presentation 11.
TUCSON, ARIZ. –
The finding comes from a secondary analysis of two randomized, controlled trials comparing Burch colposuspension, autologous fascial slings, retropubic midurethral polypropylene slings, and transobturator midurethral polypropylene slings. The analysis looked at outcomes at 24 months after surgery. Stephanie Glass Clark, MD, a resident at Virginia Commonwealth University, Richmond, presented the results at the annual scientific meeting of the Society of Gynecologic Surgeons.
In the secondary analysis of the Stress Incontinence Surgical Treatment Efficacy Trial (SISTEr) and the Trial of Midurethral Slings (TOMUS) trials, Dr. Clark and her fellow researchers looked at the effect of surgical failure on sexual dysfunction outcomes. Subjective failure was defined as self-reported SUI symptoms or self-reported leakage by 3-day voiding diary beyond 3 months after the surgery. Objective failure was defined as any treatment for SUI after the surgery or a positive stress test or pad test beyond 3 months after the surgery.
Participants were excluded from the two studies if they were sexually inactive in the previous 6 months at baseline, at 12 months post baseline, or at 24 months. The studies employed the short form of the Pelvic Organ Prolapse/Urinary Incontinence Sexual Questionnaire (PISQ-12), which had 12 questions with scores ranging from 0 to 4. The secondary analysis sample included 488 women from SISTEr and 436 women from TOMUS.
There were some baseline differences among groups between the two trials, including vaginal deliveries, race/ethnicity, stage of prolapse, and concomitant surgeries performed at time of the anti-incontinence procedure.
All four surgeries were associated with improvements in sexual function, with no statistically significant between-group differences. Mean PISQ-12 scores improved from a range of 31-33 to a range of 36-38 at 24 months. Although there is no published minimum important difference for PISQ-12 scores, an improvement of at least one-half of a standard deviation is generally accepted as clinically meaningful. “In this case, the standard deviation at baseline was just under 3 and so the improvement of each treatment group by more than 1.5 is a clinically meaningful improvement in their sexual function,” Dr. Clark said.
“Sexual dysfunction is a much more common problem than we previously thought, so we’ve been trying to figure out if patients with pelvic floor disorders like stress incontinence are going to have any improvement in sexual dysfunction by surgically treating their stress incontinence. Previously published data had been pretty conflicting,” Dr. Clark added in an interview.
That previous research was mostly retrospective and could have been impacted by patient selection bias. By analyzing clinical trials, the researchers hoped to test their idea that the pelvic floor symptoms themselves may be key to sexual dysfunction and that treating it surgically would improve matters.
The positive result is encouraging, but it still leaves unanswered questions about the mechanism behind the relationship. Dr. Clark wondered whether leaking urine leakage during sex might be the culprit, or whether it is fear or shame associated with the condition.
The answer may come from further analysis of women who were sexually inactive at baseline, but became sexually active over the course of the studies. “I think looking at that patient population in particular is going to be an interesting area of research. Is it that it was completely related to their pelvic floor disorder, and then we fixed it [so] they could have a more fulfilling sexual life?” speculated Dr. Clark.
The study received some funding from the National Institutes of Health. Dr. Clark reported no relevant financial disclosures.
SOURCE: Clark SG et al. SGS 2019, Oral Presentation 11.
TUCSON, ARIZ. –
The finding comes from a secondary analysis of two randomized, controlled trials comparing Burch colposuspension, autologous fascial slings, retropubic midurethral polypropylene slings, and transobturator midurethral polypropylene slings. The analysis looked at outcomes at 24 months after surgery. Stephanie Glass Clark, MD, a resident at Virginia Commonwealth University, Richmond, presented the results at the annual scientific meeting of the Society of Gynecologic Surgeons.
In the secondary analysis of the Stress Incontinence Surgical Treatment Efficacy Trial (SISTEr) and the Trial of Midurethral Slings (TOMUS) trials, Dr. Clark and her fellow researchers looked at the effect of surgical failure on sexual dysfunction outcomes. Subjective failure was defined as self-reported SUI symptoms or self-reported leakage by 3-day voiding diary beyond 3 months after the surgery. Objective failure was defined as any treatment for SUI after the surgery or a positive stress test or pad test beyond 3 months after the surgery.
Participants were excluded from the two studies if they were sexually inactive in the previous 6 months at baseline, at 12 months post baseline, or at 24 months. The studies employed the short form of the Pelvic Organ Prolapse/Urinary Incontinence Sexual Questionnaire (PISQ-12), which had 12 questions with scores ranging from 0 to 4. The secondary analysis sample included 488 women from SISTEr and 436 women from TOMUS.
There were some baseline differences among groups between the two trials, including vaginal deliveries, race/ethnicity, stage of prolapse, and concomitant surgeries performed at time of the anti-incontinence procedure.
All four surgeries were associated with improvements in sexual function, with no statistically significant between-group differences. Mean PISQ-12 scores improved from a range of 31-33 to a range of 36-38 at 24 months. Although there is no published minimum important difference for PISQ-12 scores, an improvement of at least one-half of a standard deviation is generally accepted as clinically meaningful. “In this case, the standard deviation at baseline was just under 3 and so the improvement of each treatment group by more than 1.5 is a clinically meaningful improvement in their sexual function,” Dr. Clark said.
“Sexual dysfunction is a much more common problem than we previously thought, so we’ve been trying to figure out if patients with pelvic floor disorders like stress incontinence are going to have any improvement in sexual dysfunction by surgically treating their stress incontinence. Previously published data had been pretty conflicting,” Dr. Clark added in an interview.
That previous research was mostly retrospective and could have been impacted by patient selection bias. By analyzing clinical trials, the researchers hoped to test their idea that the pelvic floor symptoms themselves may be key to sexual dysfunction and that treating it surgically would improve matters.
The positive result is encouraging, but it still leaves unanswered questions about the mechanism behind the relationship. Dr. Clark wondered whether leaking urine leakage during sex might be the culprit, or whether it is fear or shame associated with the condition.
The answer may come from further analysis of women who were sexually inactive at baseline, but became sexually active over the course of the studies. “I think looking at that patient population in particular is going to be an interesting area of research. Is it that it was completely related to their pelvic floor disorder, and then we fixed it [so] they could have a more fulfilling sexual life?” speculated Dr. Clark.
The study received some funding from the National Institutes of Health. Dr. Clark reported no relevant financial disclosures.
SOURCE: Clark SG et al. SGS 2019, Oral Presentation 11.
REPORTING FROM SGS 2019
Older women have good functional recovery after POP surgery
TUCSON, ARIZ. – according to a new study.

Functional status was defined as the ability to perform activities essential to self-care, independent living, and recreation.
Patients with the highest level of function at baseline actually had a slight decrease in functionality scores, although this difference was not clinically meaningful.
“We can tell patients: You’re high functioning, your prolapse is bothering you in other ways, but we’re going to keep you as high functioning as you are now. I think that’s really important for older women who are retired and want to stay active. They want to make sure they don’t have a surgery that’s going to make them less so,” Daniel Lee, MD, of the University of Pennsylvania, Philadelphia, said in an interview. Dr. Lee presented the study at the annual scientific meeting of the Society of Gynecologic Surgeons.
The study used data from multiple centers across the United States and a range of patient ethnicities. “That helped strengthen the conclusions,” Dr. Lee said. Most previous studies used generalized questionnaires rather than functional outcome questionnaires to determine patient outcomes.
One confounder is the potential presence of cognitive dysfunction, which can occur sometimes in older women following surgery. The study relied on surveys and excluded patients who weren’t able to understand them. Two patients were much worse after the surgery, and Dr. Lee speculated that cognitive dysfunction could have been the cause, although the team could not confirm that. “There’s a possibility that preexisting dementia or cognitive dysfunction could be unmasked by the surgery,” he said. The team is working to incorporate accelerometers to more objectively measure outcomes and will soon publish a feasibility study of their use in older women with cognitive dysfunction.
One limitation of the study was that it excluded women who were considered poor candidates for surgery. But that also suggests that patient selection is working as intended. “I think it just shows that surgeons do a very good job of figuring out who is a good surgical candidate, that they turn out to be better off [functionally] or that their prolapse gets improved,” Dr. Lee said.
The researchers analyzed questionnaire data from 176 women. The mean age was 72 years, and mean body mass index was 27 kg/m2; 87% of the women were Caucasian and 10% were black. Using the Activities Assessment Scale (AAS), which covers sedentary, ambulatory, and work/exercise domains, as well as the Patient Health Questionnaire to measure depression, the researchers found that, by 3 months, 59% of patients had an improved functional status, 35% had returned to within one standard deviation of baseline, and 6% had worsened, compared with their baseline scores.
Patients in the improved group started at a mean baseline total AAS score of 85 and improved to 100 at 3 months. Those who returned to baseline started at 100 on average and returned to 100 at 3 months, while those in the worsened group had a mean baseline score of 100 and a mean score of 93 at 3 months (P less than .001 for all comparisons).
The study was funded by the Fellows Pelvic Research Network. The investigators reported no relevant financial disclosures.
SOURCE: Lee D et al. SGS 2019, Abstract 09.
TUCSON, ARIZ. – according to a new study.
Functional status was defined as the ability to perform activities essential to self-care, independent living, and recreation.
Patients with the highest level of function at baseline actually had a slight decrease in functionality scores, although this difference was not clinically meaningful.
“We can tell patients: You’re high functioning, your prolapse is bothering you in other ways, but we’re going to keep you as high functioning as you are now. I think that’s really important for older women who are retired and want to stay active. They want to make sure they don’t have a surgery that’s going to make them less so,” Daniel Lee, MD, of the University of Pennsylvania, Philadelphia, said in an interview. Dr. Lee presented the study at the annual scientific meeting of the Society of Gynecologic Surgeons.
The study used data from multiple centers across the United States and a range of patient ethnicities. “That helped strengthen the conclusions,” Dr. Lee said. Most previous studies used generalized questionnaires rather than functional outcome questionnaires to determine patient outcomes.
One confounder is the potential presence of cognitive dysfunction, which can occur sometimes in older women following surgery. The study relied on surveys and excluded patients who weren’t able to understand them. Two patients were much worse after the surgery, and Dr. Lee speculated that cognitive dysfunction could have been the cause, although the team could not confirm that. “There’s a possibility that preexisting dementia or cognitive dysfunction could be unmasked by the surgery,” he said. The team is working to incorporate accelerometers to more objectively measure outcomes and will soon publish a feasibility study of their use in older women with cognitive dysfunction.
One limitation of the study was that it excluded women who were considered poor candidates for surgery. But that also suggests that patient selection is working as intended. “I think it just shows that surgeons do a very good job of figuring out who is a good surgical candidate, that they turn out to be better off [functionally] or that their prolapse gets improved,” Dr. Lee said.
The researchers analyzed questionnaire data from 176 women. The mean age was 72 years, and mean body mass index was 27 kg/m2; 87% of the women were Caucasian and 10% were black. Using the Activities Assessment Scale (AAS), which covers sedentary, ambulatory, and work/exercise domains, as well as the Patient Health Questionnaire to measure depression, the researchers found that, by 3 months, 59% of patients had an improved functional status, 35% had returned to within one standard deviation of baseline, and 6% had worsened, compared with their baseline scores.
Patients in the improved group started at a mean baseline total AAS score of 85 and improved to 100 at 3 months. Those who returned to baseline started at 100 on average and returned to 100 at 3 months, while those in the worsened group had a mean baseline score of 100 and a mean score of 93 at 3 months (P less than .001 for all comparisons).
The study was funded by the Fellows Pelvic Research Network. The investigators reported no relevant financial disclosures.
SOURCE: Lee D et al. SGS 2019, Abstract 09.
TUCSON, ARIZ. – according to a new study.
Functional status was defined as the ability to perform activities essential to self-care, independent living, and recreation.
Patients with the highest level of function at baseline actually had a slight decrease in functionality scores, although this difference was not clinically meaningful.
“We can tell patients: You’re high functioning, your prolapse is bothering you in other ways, but we’re going to keep you as high functioning as you are now. I think that’s really important for older women who are retired and want to stay active. They want to make sure they don’t have a surgery that’s going to make them less so,” Daniel Lee, MD, of the University of Pennsylvania, Philadelphia, said in an interview. Dr. Lee presented the study at the annual scientific meeting of the Society of Gynecologic Surgeons.
The study used data from multiple centers across the United States and a range of patient ethnicities. “That helped strengthen the conclusions,” Dr. Lee said. Most previous studies used generalized questionnaires rather than functional outcome questionnaires to determine patient outcomes.
One confounder is the potential presence of cognitive dysfunction, which can occur sometimes in older women following surgery. The study relied on surveys and excluded patients who weren’t able to understand them. Two patients were much worse after the surgery, and Dr. Lee speculated that cognitive dysfunction could have been the cause, although the team could not confirm that. “There’s a possibility that preexisting dementia or cognitive dysfunction could be unmasked by the surgery,” he said. The team is working to incorporate accelerometers to more objectively measure outcomes and will soon publish a feasibility study of their use in older women with cognitive dysfunction.
One limitation of the study was that it excluded women who were considered poor candidates for surgery. But that also suggests that patient selection is working as intended. “I think it just shows that surgeons do a very good job of figuring out who is a good surgical candidate, that they turn out to be better off [functionally] or that their prolapse gets improved,” Dr. Lee said.
The researchers analyzed questionnaire data from 176 women. The mean age was 72 years, and mean body mass index was 27 kg/m2; 87% of the women were Caucasian and 10% were black. Using the Activities Assessment Scale (AAS), which covers sedentary, ambulatory, and work/exercise domains, as well as the Patient Health Questionnaire to measure depression, the researchers found that, by 3 months, 59% of patients had an improved functional status, 35% had returned to within one standard deviation of baseline, and 6% had worsened, compared with their baseline scores.
Patients in the improved group started at a mean baseline total AAS score of 85 and improved to 100 at 3 months. Those who returned to baseline started at 100 on average and returned to 100 at 3 months, while those in the worsened group had a mean baseline score of 100 and a mean score of 93 at 3 months (P less than .001 for all comparisons).
The study was funded by the Fellows Pelvic Research Network. The investigators reported no relevant financial disclosures.
SOURCE: Lee D et al. SGS 2019, Abstract 09.
REPORTING FROM SGS 2019
Brexanolone approval ‘marks an important milestone’
In March 2019, the Food and Drug Administration approved a novel medication, Zulresso (brexanolone), for the treatment of postpartum depression. Brexanolone is the first FDA-approved medication for the treatment of postpartum depression, a serious illness that affects nearly one in nine women soon after giving birth.1

Mothers with postpartum depression experience feelings of sadness, irritability, and anxiety, as well as isolation from their loved ones (including their new baby) and exhaustion. The feelings of sadness and anxiety can be extreme, and can interfere with a woman’s ability to care for herself or her family. In some cases, these symptoms can be life threatening. Indeed, the most common cause of maternal death after childbirth in the developed world is suicide.2 Because of the severity of the symptoms and their impact on the family, postpartum depression usually requires treatment.
Until now, there have been no drugs specifically approved to treat postpartum depression. Commonly, postpartum depression is treated with medications that previously were approved for the treatment of major depressive disorder, despite limited evidence documenting their efficacy for postpartum depression. Other putative treatment alternatives include psychotherapy, estrogen therapy, and neuromodulation, such as electroconvulsive therapy and repetitive transcranial magnetic stimulation. Each of these treatments can take weeks or longer to take effect, time that is of elevated importance given the rapidly developing mother-infant relationship in the early postpartum period. Brexanolone addresses both the issue of efficacy and speed of onset, representing a major step forward in the care of women suffering from postpartum depression.
Importantly, the approval of brexanolone marks an important milestone for the psychiatric research community in general and the National Institute of Mental Health in particular, as it represents a compelling example of successful bench-to-bedside translation of basic neuroscience findings to benefit patients. As we have noted elsewhere,3 the research underlying the discovery of endogenous neurosteroids and their role in modulating GABA receptors laid the foundation for the development of brexanolone, an intravenous formulation of the neurosteroid allopregnanolone. The recognition that allopregnanolone was a protective factor induced by stress, that it derived from progesterone, and that its peripheral blood levels were dramatically reduced in the early postpartum period led to the hypothesis that it might be useful as a treatment for postpartum depression.
Sage Therapeutics took on the task of testing this hypothesis, designing a program, in consultation with the FDA, to test the efficacy of allopregnanolone in women with postpartum depression in a series of randomized, placebo-controlled studies assessing brexanolone. 4,5 It is a significant accomplishment of Sage Therapeutics to not only successfully complete the therapeutic program of studies (given past experience with difficulties recruiting these women for placebo-controlled treatment trials) but as well to demonstrate a robust therapeutic effect.
Although the FDA’s approval of a new and novel treatment is exciting for many women, there are still limitations to the broader use of brexanolone. It is delivered intravenously, requires an overnight stay in a certified medical center, and is likely to be considerably expensive, according to early reports – potentially limiting the access to the treatment. There also are potentially serious side effects, such as sedation, dizziness, or sudden loss of consciousness. Nonetheless, this is a promising first step and hopefully will spur further efforts to identify and optimize additional strategies to treat postpartum depression. In fact, other formulations of allopregnanolone and novel analogs to treat postpartum depression already are under study, including some that are orally bioavailable.6,7,8
Several important questions remain to be answered about both brexanolone and postpartum depression: What is the underlying mechanism through which allopregnanolone acts in the brain and reduces depressive symptoms? Is the mechanism unique to postpartum women, or might brexanolone also be effective in nonreproductive depressions in women and men? What causes postpartum depression, and what are the risk factors involved for women who develop this serious condition? Future work will focus on these and other important questions to the benefit of women who have suffered with this condition.
The FDA approval of brexanolone represents the second approval in a month of a new antidepressant treatment targeting different molecules in the brain. In early March 2019, the agency approved Spravato (esketamine) nasal spray as a therapy for treatment-resistant depression. Like brexanolone, esketamine is a fast-acting antidepressant that works through a novel mechanism, completely different from other antidepressants. These new treatment approvals are encouraging, as there has been a paucity for many years in approving new effective treatments for mood disorders.
However, treatment development in psychiatry still has a long way to go and the full underlying neurobiology of mood disorders, including postpartum depression, remains poorly understood. Many challenges are ahead of us in our efforts to develop new treatments and increase our understanding of mental illnesses. Nevertheless, the approval of brexanolone is an important milestone, giving hope to the many women who suffer from postpartum depression, and paving the way for the development of additional novel and effective medications to treat this serious and sometimes life-threatening condition.
Dr. Gordon is the director of the National Institute of Mental Health (NIMH), the lead federal agency for research on mental disorders. He oversees an extensive research portfolio of basic and clinical research that seeks to transform the understanding and treatment of mental illnesses, paving the way for prevention, recovery, and cure. Dr. Hillefors works at the NIMH and oversees the Translational Therapeutics Program in the division of translational research, focusing on the development of novel treatments and biomarkers and early phase clinical trials. She received her MD and PhD in neuroscience at the Karolinska Institute, Sweden. Dr. Schmidt joined the NIMH in 1986 after completing his psychiatric residency at the University of Toronto. He is the chief of the Section on Behavioral Endocrinology, within the Intramural Research Program at the NIMH, where his laboratory studies the relationship between hormones, stress, and mood – particularly in the areas of postpartum depression, severe premenstrual dysphoria, and perimenopausal depression.
References
1. J Psychiatric Res. 2018 Sep;104:235-48.
2. Br J Psychiatry. 2003 Oct;183:279-81.
3. NIMH Director’s Messages. 2019 Mar 20.
4. Lancet. 2017 Jul 29;390(10093):480-9.
5. Lancet. 2018 Sep 22; 392(10152):1058-70.
6. Sage Therapeutics News. 2019 Jan 7.
7. Marinus Pharmaceuticals. 2017 Jun 27.
8. ClinicalTrials.gov Identifier: NCT03460756. 2019 Mar.
In March 2019, the Food and Drug Administration approved a novel medication, Zulresso (brexanolone), for the treatment of postpartum depression. Brexanolone is the first FDA-approved medication for the treatment of postpartum depression, a serious illness that affects nearly one in nine women soon after giving birth.1
Mothers with postpartum depression experience feelings of sadness, irritability, and anxiety, as well as isolation from their loved ones (including their new baby) and exhaustion. The feelings of sadness and anxiety can be extreme, and can interfere with a woman’s ability to care for herself or her family. In some cases, these symptoms can be life threatening. Indeed, the most common cause of maternal death after childbirth in the developed world is suicide.2 Because of the severity of the symptoms and their impact on the family, postpartum depression usually requires treatment.
Until now, there have been no drugs specifically approved to treat postpartum depression. Commonly, postpartum depression is treated with medications that previously were approved for the treatment of major depressive disorder, despite limited evidence documenting their efficacy for postpartum depression. Other putative treatment alternatives include psychotherapy, estrogen therapy, and neuromodulation, such as electroconvulsive therapy and repetitive transcranial magnetic stimulation. Each of these treatments can take weeks or longer to take effect, time that is of elevated importance given the rapidly developing mother-infant relationship in the early postpartum period. Brexanolone addresses both the issue of efficacy and speed of onset, representing a major step forward in the care of women suffering from postpartum depression.
Importantly, the approval of brexanolone marks an important milestone for the psychiatric research community in general and the National Institute of Mental Health in particular, as it represents a compelling example of successful bench-to-bedside translation of basic neuroscience findings to benefit patients. As we have noted elsewhere,3 the research underlying the discovery of endogenous neurosteroids and their role in modulating GABA receptors laid the foundation for the development of brexanolone, an intravenous formulation of the neurosteroid allopregnanolone. The recognition that allopregnanolone was a protective factor induced by stress, that it derived from progesterone, and that its peripheral blood levels were dramatically reduced in the early postpartum period led to the hypothesis that it might be useful as a treatment for postpartum depression.
Sage Therapeutics took on the task of testing this hypothesis, designing a program, in consultation with the FDA, to test the efficacy of allopregnanolone in women with postpartum depression in a series of randomized, placebo-controlled studies assessing brexanolone. 4,5 It is a significant accomplishment of Sage Therapeutics to not only successfully complete the therapeutic program of studies (given past experience with difficulties recruiting these women for placebo-controlled treatment trials) but as well to demonstrate a robust therapeutic effect.
Although the FDA’s approval of a new and novel treatment is exciting for many women, there are still limitations to the broader use of brexanolone. It is delivered intravenously, requires an overnight stay in a certified medical center, and is likely to be considerably expensive, according to early reports – potentially limiting the access to the treatment. There also are potentially serious side effects, such as sedation, dizziness, or sudden loss of consciousness. Nonetheless, this is a promising first step and hopefully will spur further efforts to identify and optimize additional strategies to treat postpartum depression. In fact, other formulations of allopregnanolone and novel analogs to treat postpartum depression already are under study, including some that are orally bioavailable.6,7,8
Several important questions remain to be answered about both brexanolone and postpartum depression: What is the underlying mechanism through which allopregnanolone acts in the brain and reduces depressive symptoms? Is the mechanism unique to postpartum women, or might brexanolone also be effective in nonreproductive depressions in women and men? What causes postpartum depression, and what are the risk factors involved for women who develop this serious condition? Future work will focus on these and other important questions to the benefit of women who have suffered with this condition.
The FDA approval of brexanolone represents the second approval in a month of a new antidepressant treatment targeting different molecules in the brain. In early March 2019, the agency approved Spravato (esketamine) nasal spray as a therapy for treatment-resistant depression. Like brexanolone, esketamine is a fast-acting antidepressant that works through a novel mechanism, completely different from other antidepressants. These new treatment approvals are encouraging, as there has been a paucity for many years in approving new effective treatments for mood disorders.
However, treatment development in psychiatry still has a long way to go and the full underlying neurobiology of mood disorders, including postpartum depression, remains poorly understood. Many challenges are ahead of us in our efforts to develop new treatments and increase our understanding of mental illnesses. Nevertheless, the approval of brexanolone is an important milestone, giving hope to the many women who suffer from postpartum depression, and paving the way for the development of additional novel and effective medications to treat this serious and sometimes life-threatening condition.
Dr. Gordon is the director of the National Institute of Mental Health (NIMH), the lead federal agency for research on mental disorders. He oversees an extensive research portfolio of basic and clinical research that seeks to transform the understanding and treatment of mental illnesses, paving the way for prevention, recovery, and cure. Dr. Hillefors works at the NIMH and oversees the Translational Therapeutics Program in the division of translational research, focusing on the development of novel treatments and biomarkers and early phase clinical trials. She received her MD and PhD in neuroscience at the Karolinska Institute, Sweden. Dr. Schmidt joined the NIMH in 1986 after completing his psychiatric residency at the University of Toronto. He is the chief of the Section on Behavioral Endocrinology, within the Intramural Research Program at the NIMH, where his laboratory studies the relationship between hormones, stress, and mood – particularly in the areas of postpartum depression, severe premenstrual dysphoria, and perimenopausal depression.
References
1. J Psychiatric Res. 2018 Sep;104:235-48.
2. Br J Psychiatry. 2003 Oct;183:279-81.
3. NIMH Director’s Messages. 2019 Mar 20.
4. Lancet. 2017 Jul 29;390(10093):480-9.
5. Lancet. 2018 Sep 22; 392(10152):1058-70.
6. Sage Therapeutics News. 2019 Jan 7.
7. Marinus Pharmaceuticals. 2017 Jun 27.
8. ClinicalTrials.gov Identifier: NCT03460756. 2019 Mar.
In March 2019, the Food and Drug Administration approved a novel medication, Zulresso (brexanolone), for the treatment of postpartum depression. Brexanolone is the first FDA-approved medication for the treatment of postpartum depression, a serious illness that affects nearly one in nine women soon after giving birth.1
Mothers with postpartum depression experience feelings of sadness, irritability, and anxiety, as well as isolation from their loved ones (including their new baby) and exhaustion. The feelings of sadness and anxiety can be extreme, and can interfere with a woman’s ability to care for herself or her family. In some cases, these symptoms can be life threatening. Indeed, the most common cause of maternal death after childbirth in the developed world is suicide.2 Because of the severity of the symptoms and their impact on the family, postpartum depression usually requires treatment.
Until now, there have been no drugs specifically approved to treat postpartum depression. Commonly, postpartum depression is treated with medications that previously were approved for the treatment of major depressive disorder, despite limited evidence documenting their efficacy for postpartum depression. Other putative treatment alternatives include psychotherapy, estrogen therapy, and neuromodulation, such as electroconvulsive therapy and repetitive transcranial magnetic stimulation. Each of these treatments can take weeks or longer to take effect, time that is of elevated importance given the rapidly developing mother-infant relationship in the early postpartum period. Brexanolone addresses both the issue of efficacy and speed of onset, representing a major step forward in the care of women suffering from postpartum depression.
Importantly, the approval of brexanolone marks an important milestone for the psychiatric research community in general and the National Institute of Mental Health in particular, as it represents a compelling example of successful bench-to-bedside translation of basic neuroscience findings to benefit patients. As we have noted elsewhere,3 the research underlying the discovery of endogenous neurosteroids and their role in modulating GABA receptors laid the foundation for the development of brexanolone, an intravenous formulation of the neurosteroid allopregnanolone. The recognition that allopregnanolone was a protective factor induced by stress, that it derived from progesterone, and that its peripheral blood levels were dramatically reduced in the early postpartum period led to the hypothesis that it might be useful as a treatment for postpartum depression.
Sage Therapeutics took on the task of testing this hypothesis, designing a program, in consultation with the FDA, to test the efficacy of allopregnanolone in women with postpartum depression in a series of randomized, placebo-controlled studies assessing brexanolone. 4,5 It is a significant accomplishment of Sage Therapeutics to not only successfully complete the therapeutic program of studies (given past experience with difficulties recruiting these women for placebo-controlled treatment trials) but as well to demonstrate a robust therapeutic effect.
Although the FDA’s approval of a new and novel treatment is exciting for many women, there are still limitations to the broader use of brexanolone. It is delivered intravenously, requires an overnight stay in a certified medical center, and is likely to be considerably expensive, according to early reports – potentially limiting the access to the treatment. There also are potentially serious side effects, such as sedation, dizziness, or sudden loss of consciousness. Nonetheless, this is a promising first step and hopefully will spur further efforts to identify and optimize additional strategies to treat postpartum depression. In fact, other formulations of allopregnanolone and novel analogs to treat postpartum depression already are under study, including some that are orally bioavailable.6,7,8
Several important questions remain to be answered about both brexanolone and postpartum depression: What is the underlying mechanism through which allopregnanolone acts in the brain and reduces depressive symptoms? Is the mechanism unique to postpartum women, or might brexanolone also be effective in nonreproductive depressions in women and men? What causes postpartum depression, and what are the risk factors involved for women who develop this serious condition? Future work will focus on these and other important questions to the benefit of women who have suffered with this condition.
The FDA approval of brexanolone represents the second approval in a month of a new antidepressant treatment targeting different molecules in the brain. In early March 2019, the agency approved Spravato (esketamine) nasal spray as a therapy for treatment-resistant depression. Like brexanolone, esketamine is a fast-acting antidepressant that works through a novel mechanism, completely different from other antidepressants. These new treatment approvals are encouraging, as there has been a paucity for many years in approving new effective treatments for mood disorders.
However, treatment development in psychiatry still has a long way to go and the full underlying neurobiology of mood disorders, including postpartum depression, remains poorly understood. Many challenges are ahead of us in our efforts to develop new treatments and increase our understanding of mental illnesses. Nevertheless, the approval of brexanolone is an important milestone, giving hope to the many women who suffer from postpartum depression, and paving the way for the development of additional novel and effective medications to treat this serious and sometimes life-threatening condition.
Dr. Gordon is the director of the National Institute of Mental Health (NIMH), the lead federal agency for research on mental disorders. He oversees an extensive research portfolio of basic and clinical research that seeks to transform the understanding and treatment of mental illnesses, paving the way for prevention, recovery, and cure. Dr. Hillefors works at the NIMH and oversees the Translational Therapeutics Program in the division of translational research, focusing on the development of novel treatments and biomarkers and early phase clinical trials. She received her MD and PhD in neuroscience at the Karolinska Institute, Sweden. Dr. Schmidt joined the NIMH in 1986 after completing his psychiatric residency at the University of Toronto. He is the chief of the Section on Behavioral Endocrinology, within the Intramural Research Program at the NIMH, where his laboratory studies the relationship between hormones, stress, and mood – particularly in the areas of postpartum depression, severe premenstrual dysphoria, and perimenopausal depression.
References
1. J Psychiatric Res. 2018 Sep;104:235-48.
2. Br J Psychiatry. 2003 Oct;183:279-81.
3. NIMH Director’s Messages. 2019 Mar 20.
4. Lancet. 2017 Jul 29;390(10093):480-9.
5. Lancet. 2018 Sep 22; 392(10152):1058-70.
6. Sage Therapeutics News. 2019 Jan 7.
7. Marinus Pharmaceuticals. 2017 Jun 27.
8. ClinicalTrials.gov Identifier: NCT03460756. 2019 Mar.