User login
Immunotherapy’s cardiac effects require early monitoring, management
WASHINGTON – Unquestionably, immunotherapy is revolutionizing the care of patients with various solid tumors and hematologic malignancies.

But it’s equally true that there’s no such thing as either a free lunch or a cancer therapy free of side effects, whether it’s increased risk for heart failure associated with anthracycline-based chemotherapy, or inflammatory conditions, arrhythmias, and thromboembolic events associated with immune checkpoint inhibitors, said R. Frank Cornell, MD, of Vanderbilt University Medical Center in Nashville, Tenn.
“Early awareness and intervention is critical for improved outcomes, and a multidisciplinary approach between oncology, cardiology, the clinic nurse, and other health care providers is critical in managing these patients with these complicated therapies,” he said at the American College of Cardiology’s Advancing the Cardiovascular Care of the Oncology Patient meeting.
Checkpoint inhibitors and the heart
Toxicities associated with immune checkpoint inhibitors such as the programmed death 1/ligand 1 (PD-1/PD-L1) inhibitors nivolumab (Opdivo) and pembrolizumab (Keytruda) and the cytotoxic T-lymphocyte antigen 4 antibody ipilimumab (Yervoy) tend to mimic autoimmune conditions, Dr. Cornell said.
Cardiovascular events associated with these agents, while uncommon, include myocarditis, pericarditis, arrhythmias, impaired ventricular function with heart failure, vasculitis, and venous thromboembolism, he said, citing an American Society of Clinical Oncology (ASCO) clinical practice guideline (J Clin Oncol 2018;36[17]:1714-68).
Dr. Cornell described the case of a 63-year-old woman with disseminated metastatic melanoma who presented to the emergency department 10 days after starting on combination therapy with ipilimumab and nivolumab. She had developed shortness of breath, pleuritic chest pain, and a mild cough for 1 or 2 days.
Her cardiac laboratory markers had been normal at baseline, but were markedly elevated on presentation, and electrocardiograms showed complete heart block and subsequent ventricular tachycardia.
The patient was started on high-dose prednisone, but she died in hospital, and an autopsy showed that the cause of death was infiltration into the myocardium of CD3-positive and CD8-positive T lymphocytes.
“So how do we manage this? This is a good opportunity, I think, for further cardiology and oncology collaboration to develop more robust guidelines for what we can do to best prevent this,” Dr. Cornell said.
Patients started on the ipilimumab/nivolumab combination should be tested weekly for cardiac troponin, creatine kinase (CK) and CK-muscle/brain (CK-MB) weekly for the first 3-4 weeks of therapy. Therapy should be stopped if troponin levels continue to rise, and the patient should be started on high-dose steroids, he said.
The role of other anti-inflammatory agents such as infliximab (Remicade and biosimilars) is unclear and needs further study, he added.
Dr. Cornell cited a 2018 letter to The Lancet by Javid J. Moslehi, MD, and colleagues from Vanderbilt describing an increase in reports of fatal myocarditis among patients treated with checkpoint inhibitors.
“We highlight the high mortality rate with severe immune checkpoint inhibitor–related myocarditis, which is more frequent with combination PD-1 and CTLA-4 blockade, but can also occur with monotherapy. Myocarditis was observed across immune checkpoint inhibitor regimens, although it remains too early to determine whether the incidence differs between use of anti-PD1 and anti-PD-L1 drugs. Furthermore, this condition occurs early on during therapy and across cancer types,” they wrote.
Most of the patients had no preexisting cardiovascular disease, and most were not taking medications for hypertension, cardiovascular disease, or diabetes.
CAR-T cells and cardiac disease
The primary cardiac complications associated with CAR-T cell therapy are related to the cytokine release syndrome (CRS), a condition marked by progressive elevation in inflammatory cytokines that in turn leads to marked elevations in C-reactive protein (CRP), interferon gamma, tumor necrosis factor al, and release of pro-inflammatory cytokines including interleukin (IL) 6, IL-10, IL-12, and IL-1 beta.
In rare instances, CRS can lead to disseminated intravascular coagulation (DIC), capillary leak syndrome, and a hemophagocytic lymphohistiocytosis-like (HLH) syndrome, Dr. Cornell said.
Package inserts for the two Food and Drug Administration–approved CAR-T cell products, axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) show that each was associated in clinical trials with a high incidence of CRS.
Among patients treated with axicabtagene ciloleucel, 94% developed CRS, which was grade 3 or greater in severity in 13%. The median time to onset was 2 days, and the median duration was 7 days. Cardiovascular adverse events included grade 3 or greater tachycardia in 2%, arrhythmias in 7%, edema in 1%, dyspnea in 3%, pleural effusion in 2%, hypotension in 15%, hypertension in 6%, and thrombosis in 1%.
Among patients treated with tisagenlecleucel, 79% treated for B-cell acute lymphoblastic leukemia (B-ALL) and 74% treated for diffuse large B cell lymphoma (DLBCL) developed CRS, which was grade 3 or greater in 49% and 23% of patients, respectively. The median time to onset was 3 days, and the median duration of CRS was 8 days.
Cardiovascular adverse events of grade 3 or greater among these patients included tachycardia in 4%, fluid overload in 7%, edema in 1%, dyspnea in 12%, pulmonary edema in 4%, hypotension in 22%, and hypertension in 6%.
Risk factors for CRS include high pre-infusion tumor burden, active infections, and concurrent inflammatory processes, Dr. Cornell said.
Prevention of cardiovascular complications of CAR-T cell therapy requires management of CRS. Patients with grade 2 or greater CRS should receive the anti-IL-6 agent tocilizumab (Actemra) 8 mg/kg intravenously over 1 hour to a maximum dose of 800 mg. Tocilizumab infusions can be repeated every 8 hours as needed if the patient is not responsive to intravenous fluids or increasing supplement oxygen, but should be limited to a maximum of three doses over 24 hours, and a maximum total of four doses.
Patients with grade 3 CRS should also receive intravenous methylprednisolone 1 mg/kg twice daily or the equivalent amount of dexamethasone, with corticosteroids continued until the severity of CRS is grade 1 or less, then tapered over 3 days,
Patients with grade 4 CRS should also receive IV methylprednisolone 1,000 mg per day for 3 days, and if symptoms improve, continue management as per grade 3, Dr. Cornell said.
Dr. Cornell reported having nothing to disclose.
WASHINGTON – Unquestionably, immunotherapy is revolutionizing the care of patients with various solid tumors and hematologic malignancies.
But it’s equally true that there’s no such thing as either a free lunch or a cancer therapy free of side effects, whether it’s increased risk for heart failure associated with anthracycline-based chemotherapy, or inflammatory conditions, arrhythmias, and thromboembolic events associated with immune checkpoint inhibitors, said R. Frank Cornell, MD, of Vanderbilt University Medical Center in Nashville, Tenn.
“Early awareness and intervention is critical for improved outcomes, and a multidisciplinary approach between oncology, cardiology, the clinic nurse, and other health care providers is critical in managing these patients with these complicated therapies,” he said at the American College of Cardiology’s Advancing the Cardiovascular Care of the Oncology Patient meeting.
Checkpoint inhibitors and the heart
Toxicities associated with immune checkpoint inhibitors such as the programmed death 1/ligand 1 (PD-1/PD-L1) inhibitors nivolumab (Opdivo) and pembrolizumab (Keytruda) and the cytotoxic T-lymphocyte antigen 4 antibody ipilimumab (Yervoy) tend to mimic autoimmune conditions, Dr. Cornell said.
Cardiovascular events associated with these agents, while uncommon, include myocarditis, pericarditis, arrhythmias, impaired ventricular function with heart failure, vasculitis, and venous thromboembolism, he said, citing an American Society of Clinical Oncology (ASCO) clinical practice guideline (J Clin Oncol 2018;36[17]:1714-68).
Dr. Cornell described the case of a 63-year-old woman with disseminated metastatic melanoma who presented to the emergency department 10 days after starting on combination therapy with ipilimumab and nivolumab. She had developed shortness of breath, pleuritic chest pain, and a mild cough for 1 or 2 days.
Her cardiac laboratory markers had been normal at baseline, but were markedly elevated on presentation, and electrocardiograms showed complete heart block and subsequent ventricular tachycardia.
The patient was started on high-dose prednisone, but she died in hospital, and an autopsy showed that the cause of death was infiltration into the myocardium of CD3-positive and CD8-positive T lymphocytes.
“So how do we manage this? This is a good opportunity, I think, for further cardiology and oncology collaboration to develop more robust guidelines for what we can do to best prevent this,” Dr. Cornell said.
Patients started on the ipilimumab/nivolumab combination should be tested weekly for cardiac troponin, creatine kinase (CK) and CK-muscle/brain (CK-MB) weekly for the first 3-4 weeks of therapy. Therapy should be stopped if troponin levels continue to rise, and the patient should be started on high-dose steroids, he said.
The role of other anti-inflammatory agents such as infliximab (Remicade and biosimilars) is unclear and needs further study, he added.
Dr. Cornell cited a 2018 letter to The Lancet by Javid J. Moslehi, MD, and colleagues from Vanderbilt describing an increase in reports of fatal myocarditis among patients treated with checkpoint inhibitors.
“We highlight the high mortality rate with severe immune checkpoint inhibitor–related myocarditis, which is more frequent with combination PD-1 and CTLA-4 blockade, but can also occur with monotherapy. Myocarditis was observed across immune checkpoint inhibitor regimens, although it remains too early to determine whether the incidence differs between use of anti-PD1 and anti-PD-L1 drugs. Furthermore, this condition occurs early on during therapy and across cancer types,” they wrote.
Most of the patients had no preexisting cardiovascular disease, and most were not taking medications for hypertension, cardiovascular disease, or diabetes.
CAR-T cells and cardiac disease
The primary cardiac complications associated with CAR-T cell therapy are related to the cytokine release syndrome (CRS), a condition marked by progressive elevation in inflammatory cytokines that in turn leads to marked elevations in C-reactive protein (CRP), interferon gamma, tumor necrosis factor al, and release of pro-inflammatory cytokines including interleukin (IL) 6, IL-10, IL-12, and IL-1 beta.
In rare instances, CRS can lead to disseminated intravascular coagulation (DIC), capillary leak syndrome, and a hemophagocytic lymphohistiocytosis-like (HLH) syndrome, Dr. Cornell said.
Package inserts for the two Food and Drug Administration–approved CAR-T cell products, axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) show that each was associated in clinical trials with a high incidence of CRS.
Among patients treated with axicabtagene ciloleucel, 94% developed CRS, which was grade 3 or greater in severity in 13%. The median time to onset was 2 days, and the median duration was 7 days. Cardiovascular adverse events included grade 3 or greater tachycardia in 2%, arrhythmias in 7%, edema in 1%, dyspnea in 3%, pleural effusion in 2%, hypotension in 15%, hypertension in 6%, and thrombosis in 1%.
Among patients treated with tisagenlecleucel, 79% treated for B-cell acute lymphoblastic leukemia (B-ALL) and 74% treated for diffuse large B cell lymphoma (DLBCL) developed CRS, which was grade 3 or greater in 49% and 23% of patients, respectively. The median time to onset was 3 days, and the median duration of CRS was 8 days.
Cardiovascular adverse events of grade 3 or greater among these patients included tachycardia in 4%, fluid overload in 7%, edema in 1%, dyspnea in 12%, pulmonary edema in 4%, hypotension in 22%, and hypertension in 6%.
Risk factors for CRS include high pre-infusion tumor burden, active infections, and concurrent inflammatory processes, Dr. Cornell said.
Prevention of cardiovascular complications of CAR-T cell therapy requires management of CRS. Patients with grade 2 or greater CRS should receive the anti-IL-6 agent tocilizumab (Actemra) 8 mg/kg intravenously over 1 hour to a maximum dose of 800 mg. Tocilizumab infusions can be repeated every 8 hours as needed if the patient is not responsive to intravenous fluids or increasing supplement oxygen, but should be limited to a maximum of three doses over 24 hours, and a maximum total of four doses.
Patients with grade 3 CRS should also receive intravenous methylprednisolone 1 mg/kg twice daily or the equivalent amount of dexamethasone, with corticosteroids continued until the severity of CRS is grade 1 or less, then tapered over 3 days,
Patients with grade 4 CRS should also receive IV methylprednisolone 1,000 mg per day for 3 days, and if symptoms improve, continue management as per grade 3, Dr. Cornell said.
Dr. Cornell reported having nothing to disclose.
WASHINGTON – Unquestionably, immunotherapy is revolutionizing the care of patients with various solid tumors and hematologic malignancies.
But it’s equally true that there’s no such thing as either a free lunch or a cancer therapy free of side effects, whether it’s increased risk for heart failure associated with anthracycline-based chemotherapy, or inflammatory conditions, arrhythmias, and thromboembolic events associated with immune checkpoint inhibitors, said R. Frank Cornell, MD, of Vanderbilt University Medical Center in Nashville, Tenn.
“Early awareness and intervention is critical for improved outcomes, and a multidisciplinary approach between oncology, cardiology, the clinic nurse, and other health care providers is critical in managing these patients with these complicated therapies,” he said at the American College of Cardiology’s Advancing the Cardiovascular Care of the Oncology Patient meeting.
Checkpoint inhibitors and the heart
Toxicities associated with immune checkpoint inhibitors such as the programmed death 1/ligand 1 (PD-1/PD-L1) inhibitors nivolumab (Opdivo) and pembrolizumab (Keytruda) and the cytotoxic T-lymphocyte antigen 4 antibody ipilimumab (Yervoy) tend to mimic autoimmune conditions, Dr. Cornell said.
Cardiovascular events associated with these agents, while uncommon, include myocarditis, pericarditis, arrhythmias, impaired ventricular function with heart failure, vasculitis, and venous thromboembolism, he said, citing an American Society of Clinical Oncology (ASCO) clinical practice guideline (J Clin Oncol 2018;36[17]:1714-68).
Dr. Cornell described the case of a 63-year-old woman with disseminated metastatic melanoma who presented to the emergency department 10 days after starting on combination therapy with ipilimumab and nivolumab. She had developed shortness of breath, pleuritic chest pain, and a mild cough for 1 or 2 days.
Her cardiac laboratory markers had been normal at baseline, but were markedly elevated on presentation, and electrocardiograms showed complete heart block and subsequent ventricular tachycardia.
The patient was started on high-dose prednisone, but she died in hospital, and an autopsy showed that the cause of death was infiltration into the myocardium of CD3-positive and CD8-positive T lymphocytes.
“So how do we manage this? This is a good opportunity, I think, for further cardiology and oncology collaboration to develop more robust guidelines for what we can do to best prevent this,” Dr. Cornell said.
Patients started on the ipilimumab/nivolumab combination should be tested weekly for cardiac troponin, creatine kinase (CK) and CK-muscle/brain (CK-MB) weekly for the first 3-4 weeks of therapy. Therapy should be stopped if troponin levels continue to rise, and the patient should be started on high-dose steroids, he said.
The role of other anti-inflammatory agents such as infliximab (Remicade and biosimilars) is unclear and needs further study, he added.
Dr. Cornell cited a 2018 letter to The Lancet by Javid J. Moslehi, MD, and colleagues from Vanderbilt describing an increase in reports of fatal myocarditis among patients treated with checkpoint inhibitors.
“We highlight the high mortality rate with severe immune checkpoint inhibitor–related myocarditis, which is more frequent with combination PD-1 and CTLA-4 blockade, but can also occur with monotherapy. Myocarditis was observed across immune checkpoint inhibitor regimens, although it remains too early to determine whether the incidence differs between use of anti-PD1 and anti-PD-L1 drugs. Furthermore, this condition occurs early on during therapy and across cancer types,” they wrote.
Most of the patients had no preexisting cardiovascular disease, and most were not taking medications for hypertension, cardiovascular disease, or diabetes.
CAR-T cells and cardiac disease
The primary cardiac complications associated with CAR-T cell therapy are related to the cytokine release syndrome (CRS), a condition marked by progressive elevation in inflammatory cytokines that in turn leads to marked elevations in C-reactive protein (CRP), interferon gamma, tumor necrosis factor al, and release of pro-inflammatory cytokines including interleukin (IL) 6, IL-10, IL-12, and IL-1 beta.
In rare instances, CRS can lead to disseminated intravascular coagulation (DIC), capillary leak syndrome, and a hemophagocytic lymphohistiocytosis-like (HLH) syndrome, Dr. Cornell said.
Package inserts for the two Food and Drug Administration–approved CAR-T cell products, axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) show that each was associated in clinical trials with a high incidence of CRS.
Among patients treated with axicabtagene ciloleucel, 94% developed CRS, which was grade 3 or greater in severity in 13%. The median time to onset was 2 days, and the median duration was 7 days. Cardiovascular adverse events included grade 3 or greater tachycardia in 2%, arrhythmias in 7%, edema in 1%, dyspnea in 3%, pleural effusion in 2%, hypotension in 15%, hypertension in 6%, and thrombosis in 1%.
Among patients treated with tisagenlecleucel, 79% treated for B-cell acute lymphoblastic leukemia (B-ALL) and 74% treated for diffuse large B cell lymphoma (DLBCL) developed CRS, which was grade 3 or greater in 49% and 23% of patients, respectively. The median time to onset was 3 days, and the median duration of CRS was 8 days.
Cardiovascular adverse events of grade 3 or greater among these patients included tachycardia in 4%, fluid overload in 7%, edema in 1%, dyspnea in 12%, pulmonary edema in 4%, hypotension in 22%, and hypertension in 6%.
Risk factors for CRS include high pre-infusion tumor burden, active infections, and concurrent inflammatory processes, Dr. Cornell said.
Prevention of cardiovascular complications of CAR-T cell therapy requires management of CRS. Patients with grade 2 or greater CRS should receive the anti-IL-6 agent tocilizumab (Actemra) 8 mg/kg intravenously over 1 hour to a maximum dose of 800 mg. Tocilizumab infusions can be repeated every 8 hours as needed if the patient is not responsive to intravenous fluids or increasing supplement oxygen, but should be limited to a maximum of three doses over 24 hours, and a maximum total of four doses.
Patients with grade 3 CRS should also receive intravenous methylprednisolone 1 mg/kg twice daily or the equivalent amount of dexamethasone, with corticosteroids continued until the severity of CRS is grade 1 or less, then tapered over 3 days,
Patients with grade 4 CRS should also receive IV methylprednisolone 1,000 mg per day for 3 days, and if symptoms improve, continue management as per grade 3, Dr. Cornell said.
Dr. Cornell reported having nothing to disclose.
REPORTING FROM ACC CARDIO-ONCOLOGY
Key clinical point: Monitor for cardiac symptoms and treat or interrupt immunotherapy as needed.
Major finding: Immune checkpoint inhibitors and CAR T-cell therapies are associated with distinct cardiovascular adverse events.
Study details: Review of strategies for managing the cardiovascular consequences of cancer immunotherapies.
Disclosures: Dr. Cornell reported having nothing to disclose.
Novel bispecific CAR shows promise in B-cell malignancies
SAN DIEGO – A chimeric antigen receptor (CAR) targeting both CD19 and CD22 shows promising safety and efficacy for the treatment of relapsed or refractory B-cell malignancies in adults, according to early findings from a phase 1 trial of the novel bispecific CAR.
Of six patients with diffuse large B-cell lymphoma (DLBCL) and two patients with B-cell acute lymphoblastic leukemia (B-ALL) enrolled in the single-institution dose escalation study and available for safety analysis after the bispecific CAR T-cell infusion, five developed reversible grade 1 cytokine release syndrome (CRS) and one developed grade 2 CRS requiring treatment with tocilizumab, Nasheed Hossain, MD, reported at the annual meeting of the American Society of Hematology.
Additionally, two patients developed grade 1 neurotoxicity, and one developed grade 2 neurotoxicity requiring treatment with dexamethasone.
“But no dose-limiting toxicities have been encountered thus far,” said Dr. Hossain of Loyola University Medical Center, Chicago. “With regard to efficacy, the DLBCL overall response rate is 60%, with 1 [complete response] and 2 [partial responses] at day 28 and day 90, and the ALL overall response rate is 100%, with 1 CR and 1 PR at day 28.
“With longer follow-up, five patients have relapsed and biopsies at the time of progression all showed ongoing CD19 expression,” he said, adding that all enrolled patients are alive, except for one patient who died from disease progression.
Study participants were adults aged 35-75 years with DLBCL or B-ALL that was refractory to standard therapies.
“Our primary objectives are twofold: One is to determine the feasibility of making our CAR ... and [the other] is to assess the safety using an escalating CAR dose following standard cyclophosphamide/fludarabine conditioning,” Dr. Hossain said.
The dose assessed in the current analysis was 1 x 106 CAR T cells/kg; other planned doses include 3 x 106 CAR T cells/kg and 1 x 107 CAR T cells/kg, he said.
All patients underwent lymphodepletion with cyclophosphamide (500 mg/m2 daily x 3 doses) and fludarabine (30 mg/m2 daily x 3 doses) followed by CAR T-cell infusion 2 days later.
The findings of this ongoing study – the first in-human study of a bispecific loop CAR in the United States – suggest that the novel CAR has low toxicity and promising efficacy, Dr. Hossain said.
Currently approved therapies target CD19 alone, he said, noting that they all use the same anti-CD19 domain, but different costimulatory domains, and have good clinical efficacy of greater than 70% CRs in ALL and up to 52% CRs in DLBCL.
“But questions remain about determining the durability of response and the causes of therapy failure,” he said.
One common cause of treatment failure is CD19 antigen loss, and efforts to reduce such antigen loss using bispecific loop CARs targeting both CD19 and CD22 have shown promise. The CAR construct evaluated in this study was developed to target CD19 and CD22 with intracellular signaling domains incorporating 4-1BB and CD3-zeta to overcome CD19 immune escape.
“We have now escalated the dose to 3 x 106 CAR T cells/kg ... and an expansion study of 60 patients will follow,” Dr. Hossain said.
A companion phase 1 pediatric trial using the same CAR construct is also underway, with preliminary data presented at the ASH meeting demonstrating safety and tolerability in children with relapsed or refractory B-cell ALL.
Dr. Hossain reported having no financial disclosures.
SOURCE: Hossain N et al. ASH 2018, Abstract 490.
SAN DIEGO – A chimeric antigen receptor (CAR) targeting both CD19 and CD22 shows promising safety and efficacy for the treatment of relapsed or refractory B-cell malignancies in adults, according to early findings from a phase 1 trial of the novel bispecific CAR.
Of six patients with diffuse large B-cell lymphoma (DLBCL) and two patients with B-cell acute lymphoblastic leukemia (B-ALL) enrolled in the single-institution dose escalation study and available for safety analysis after the bispecific CAR T-cell infusion, five developed reversible grade 1 cytokine release syndrome (CRS) and one developed grade 2 CRS requiring treatment with tocilizumab, Nasheed Hossain, MD, reported at the annual meeting of the American Society of Hematology.
Additionally, two patients developed grade 1 neurotoxicity, and one developed grade 2 neurotoxicity requiring treatment with dexamethasone.
“But no dose-limiting toxicities have been encountered thus far,” said Dr. Hossain of Loyola University Medical Center, Chicago. “With regard to efficacy, the DLBCL overall response rate is 60%, with 1 [complete response] and 2 [partial responses] at day 28 and day 90, and the ALL overall response rate is 100%, with 1 CR and 1 PR at day 28.
“With longer follow-up, five patients have relapsed and biopsies at the time of progression all showed ongoing CD19 expression,” he said, adding that all enrolled patients are alive, except for one patient who died from disease progression.
Study participants were adults aged 35-75 years with DLBCL or B-ALL that was refractory to standard therapies.
“Our primary objectives are twofold: One is to determine the feasibility of making our CAR ... and [the other] is to assess the safety using an escalating CAR dose following standard cyclophosphamide/fludarabine conditioning,” Dr. Hossain said.
The dose assessed in the current analysis was 1 x 106 CAR T cells/kg; other planned doses include 3 x 106 CAR T cells/kg and 1 x 107 CAR T cells/kg, he said.
All patients underwent lymphodepletion with cyclophosphamide (500 mg/m2 daily x 3 doses) and fludarabine (30 mg/m2 daily x 3 doses) followed by CAR T-cell infusion 2 days later.
The findings of this ongoing study – the first in-human study of a bispecific loop CAR in the United States – suggest that the novel CAR has low toxicity and promising efficacy, Dr. Hossain said.
Currently approved therapies target CD19 alone, he said, noting that they all use the same anti-CD19 domain, but different costimulatory domains, and have good clinical efficacy of greater than 70% CRs in ALL and up to 52% CRs in DLBCL.
“But questions remain about determining the durability of response and the causes of therapy failure,” he said.
One common cause of treatment failure is CD19 antigen loss, and efforts to reduce such antigen loss using bispecific loop CARs targeting both CD19 and CD22 have shown promise. The CAR construct evaluated in this study was developed to target CD19 and CD22 with intracellular signaling domains incorporating 4-1BB and CD3-zeta to overcome CD19 immune escape.
“We have now escalated the dose to 3 x 106 CAR T cells/kg ... and an expansion study of 60 patients will follow,” Dr. Hossain said.
A companion phase 1 pediatric trial using the same CAR construct is also underway, with preliminary data presented at the ASH meeting demonstrating safety and tolerability in children with relapsed or refractory B-cell ALL.
Dr. Hossain reported having no financial disclosures.
SOURCE: Hossain N et al. ASH 2018, Abstract 490.
SAN DIEGO – A chimeric antigen receptor (CAR) targeting both CD19 and CD22 shows promising safety and efficacy for the treatment of relapsed or refractory B-cell malignancies in adults, according to early findings from a phase 1 trial of the novel bispecific CAR.
Of six patients with diffuse large B-cell lymphoma (DLBCL) and two patients with B-cell acute lymphoblastic leukemia (B-ALL) enrolled in the single-institution dose escalation study and available for safety analysis after the bispecific CAR T-cell infusion, five developed reversible grade 1 cytokine release syndrome (CRS) and one developed grade 2 CRS requiring treatment with tocilizumab, Nasheed Hossain, MD, reported at the annual meeting of the American Society of Hematology.
Additionally, two patients developed grade 1 neurotoxicity, and one developed grade 2 neurotoxicity requiring treatment with dexamethasone.
“But no dose-limiting toxicities have been encountered thus far,” said Dr. Hossain of Loyola University Medical Center, Chicago. “With regard to efficacy, the DLBCL overall response rate is 60%, with 1 [complete response] and 2 [partial responses] at day 28 and day 90, and the ALL overall response rate is 100%, with 1 CR and 1 PR at day 28.
“With longer follow-up, five patients have relapsed and biopsies at the time of progression all showed ongoing CD19 expression,” he said, adding that all enrolled patients are alive, except for one patient who died from disease progression.
Study participants were adults aged 35-75 years with DLBCL or B-ALL that was refractory to standard therapies.
“Our primary objectives are twofold: One is to determine the feasibility of making our CAR ... and [the other] is to assess the safety using an escalating CAR dose following standard cyclophosphamide/fludarabine conditioning,” Dr. Hossain said.
The dose assessed in the current analysis was 1 x 106 CAR T cells/kg; other planned doses include 3 x 106 CAR T cells/kg and 1 x 107 CAR T cells/kg, he said.
All patients underwent lymphodepletion with cyclophosphamide (500 mg/m2 daily x 3 doses) and fludarabine (30 mg/m2 daily x 3 doses) followed by CAR T-cell infusion 2 days later.
The findings of this ongoing study – the first in-human study of a bispecific loop CAR in the United States – suggest that the novel CAR has low toxicity and promising efficacy, Dr. Hossain said.
Currently approved therapies target CD19 alone, he said, noting that they all use the same anti-CD19 domain, but different costimulatory domains, and have good clinical efficacy of greater than 70% CRs in ALL and up to 52% CRs in DLBCL.
“But questions remain about determining the durability of response and the causes of therapy failure,” he said.
One common cause of treatment failure is CD19 antigen loss, and efforts to reduce such antigen loss using bispecific loop CARs targeting both CD19 and CD22 have shown promise. The CAR construct evaluated in this study was developed to target CD19 and CD22 with intracellular signaling domains incorporating 4-1BB and CD3-zeta to overcome CD19 immune escape.
“We have now escalated the dose to 3 x 106 CAR T cells/kg ... and an expansion study of 60 patients will follow,” Dr. Hossain said.
A companion phase 1 pediatric trial using the same CAR construct is also underway, with preliminary data presented at the ASH meeting demonstrating safety and tolerability in children with relapsed or refractory B-cell ALL.
Dr. Hossain reported having no financial disclosures.
SOURCE: Hossain N et al. ASH 2018, Abstract 490.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: Grade 1 cytokine release syndrome occurred in five patients, and grade 2 CRS occurred in one patient; there were no dose-limiting toxicities.
Study details: A phase 1 dose escalation study of nine patients.
Disclosures: Dr. Hossain reported having no financial disclosures.
Source: Hossain N et al. ASH 2018, Abstract 490.
Uninterrupted ibrutinib with CAR T could improve CLL outcomes
SAN DIEGO – Ibrutinib treatment continued before, during, and after infusion of the CD19-specific chimeric antigen receptor (CAR) T-cell therapy JCAR014 in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) appears to improve patient responses and decrease the risk of severe cytokine release syndrome.

The findings come from a comparison of sequential cohorts from a phase 1/2 study.
At 4 weeks after infusion, the approach was highly efficacious; overall response rates by 2008 International Workshop on CLL (IWCLL) criteria were 83% in 24 patients who received the uninterrupted ibrutinib regimen along with the JCAR014 therapy – a combination of CD4 and CD8 T cells – and 65% in 19 patients from a prior cohort who did not receive continuous ibrutinib, Jordan Gauthier, MD, reported at the annual meeting of the American Society of Hematology.
Concurrent ibrutinib was generally well tolerated, with 13 of 19 patients in the ibrutinib cohort receiving treatment as planned without discontinuation. The rates of grade 1 or higher cytokine release syndrome (CRS) were statistically similar in the ibrutinib and no-ibrutinib cohorts (74% and 92%, respectively). However, the rates of severe CRS (grade 3 or higher) were, strikingly, 0% and 25%, respectively, said Dr. Gauthier, a senior fellow in the Turtle Lab at Fred Hutchinson Cancer Center, Seattle.
Neurotoxicity occurred in 32% and 42% of patients in the groups; severe neurotoxicity occurred in 26% and 29%, respectively.
In the ibrutinib cohort, one patient with grade 2 CRS developed fatal presumed cardiac arrhythmia; in the no-ibrutinib cohort, one patient died from a CAR T cell–related toxicity.
Notably, a trend toward better expansion of CD8 CAR T cells and a significantly greater expansion of CD4 CAR T cells was observed in the ibrutinib cohort, he said.
The study was designed to assess JCAR014, and based on the initial cohort findings published in 2017, established a regimen of cyclophosphamide and fludarabine (Cy/Flu) lymphodepletion followed by JCAR014 infusion at 2 x 106 CAR T cells/kg. The study was not a randomized, head-to-head comparison but the groups were similar with respect to both patient and disease characteristics, Dr. Gauthier noted.
The outcomes in the first cohort were then compared retrospectively with those from the subsequent cohort of patients who received Cy/Flu with 2 x 106 CAR T cells/kg with concurrent ibrutinib administered at 420 mg per day from at least 2 weeks prior to leukapheresis until at least 3 months after JCAR014 infusion.
The rationale for uninterrupted ibrutinib in relapsed/refractory CLL patients receiving JCAR014 included potential prevention of tumor flare, mobilization of CLL cells into the blood from the lymph nodes, improvement of CAR T-cell function, and a decrease in CAR T-cell related toxicity, he said.
The concurrent administration of ibrutinib and JCAR014 was feasible for most patients. “[It] induced high response rates and deep responses early on at 4 weeks, and it was associated with higher in vivo expansion of CD4 CAR T cells and with lower rates of severe toxicity,” Dr. Gauthier said. “The next step is to hopefully validate these findings in a prospective phase 1/2 study.”
Dr. Gauthier reported having no financial disclosures.
SOURCE: Gauthier J et al. ASH 18, Abstract 299.
SAN DIEGO – Ibrutinib treatment continued before, during, and after infusion of the CD19-specific chimeric antigen receptor (CAR) T-cell therapy JCAR014 in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) appears to improve patient responses and decrease the risk of severe cytokine release syndrome.
The findings come from a comparison of sequential cohorts from a phase 1/2 study.
At 4 weeks after infusion, the approach was highly efficacious; overall response rates by 2008 International Workshop on CLL (IWCLL) criteria were 83% in 24 patients who received the uninterrupted ibrutinib regimen along with the JCAR014 therapy – a combination of CD4 and CD8 T cells – and 65% in 19 patients from a prior cohort who did not receive continuous ibrutinib, Jordan Gauthier, MD, reported at the annual meeting of the American Society of Hematology.
Concurrent ibrutinib was generally well tolerated, with 13 of 19 patients in the ibrutinib cohort receiving treatment as planned without discontinuation. The rates of grade 1 or higher cytokine release syndrome (CRS) were statistically similar in the ibrutinib and no-ibrutinib cohorts (74% and 92%, respectively). However, the rates of severe CRS (grade 3 or higher) were, strikingly, 0% and 25%, respectively, said Dr. Gauthier, a senior fellow in the Turtle Lab at Fred Hutchinson Cancer Center, Seattle.
Neurotoxicity occurred in 32% and 42% of patients in the groups; severe neurotoxicity occurred in 26% and 29%, respectively.
In the ibrutinib cohort, one patient with grade 2 CRS developed fatal presumed cardiac arrhythmia; in the no-ibrutinib cohort, one patient died from a CAR T cell–related toxicity.
Notably, a trend toward better expansion of CD8 CAR T cells and a significantly greater expansion of CD4 CAR T cells was observed in the ibrutinib cohort, he said.
The study was designed to assess JCAR014, and based on the initial cohort findings published in 2017, established a regimen of cyclophosphamide and fludarabine (Cy/Flu) lymphodepletion followed by JCAR014 infusion at 2 x 106 CAR T cells/kg. The study was not a randomized, head-to-head comparison but the groups were similar with respect to both patient and disease characteristics, Dr. Gauthier noted.
The outcomes in the first cohort were then compared retrospectively with those from the subsequent cohort of patients who received Cy/Flu with 2 x 106 CAR T cells/kg with concurrent ibrutinib administered at 420 mg per day from at least 2 weeks prior to leukapheresis until at least 3 months after JCAR014 infusion.
The rationale for uninterrupted ibrutinib in relapsed/refractory CLL patients receiving JCAR014 included potential prevention of tumor flare, mobilization of CLL cells into the blood from the lymph nodes, improvement of CAR T-cell function, and a decrease in CAR T-cell related toxicity, he said.
The concurrent administration of ibrutinib and JCAR014 was feasible for most patients. “[It] induced high response rates and deep responses early on at 4 weeks, and it was associated with higher in vivo expansion of CD4 CAR T cells and with lower rates of severe toxicity,” Dr. Gauthier said. “The next step is to hopefully validate these findings in a prospective phase 1/2 study.”
Dr. Gauthier reported having no financial disclosures.
SOURCE: Gauthier J et al. ASH 18, Abstract 299.
SAN DIEGO – Ibrutinib treatment continued before, during, and after infusion of the CD19-specific chimeric antigen receptor (CAR) T-cell therapy JCAR014 in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) appears to improve patient responses and decrease the risk of severe cytokine release syndrome.
The findings come from a comparison of sequential cohorts from a phase 1/2 study.
At 4 weeks after infusion, the approach was highly efficacious; overall response rates by 2008 International Workshop on CLL (IWCLL) criteria were 83% in 24 patients who received the uninterrupted ibrutinib regimen along with the JCAR014 therapy – a combination of CD4 and CD8 T cells – and 65% in 19 patients from a prior cohort who did not receive continuous ibrutinib, Jordan Gauthier, MD, reported at the annual meeting of the American Society of Hematology.
Concurrent ibrutinib was generally well tolerated, with 13 of 19 patients in the ibrutinib cohort receiving treatment as planned without discontinuation. The rates of grade 1 or higher cytokine release syndrome (CRS) were statistically similar in the ibrutinib and no-ibrutinib cohorts (74% and 92%, respectively). However, the rates of severe CRS (grade 3 or higher) were, strikingly, 0% and 25%, respectively, said Dr. Gauthier, a senior fellow in the Turtle Lab at Fred Hutchinson Cancer Center, Seattle.
Neurotoxicity occurred in 32% and 42% of patients in the groups; severe neurotoxicity occurred in 26% and 29%, respectively.
In the ibrutinib cohort, one patient with grade 2 CRS developed fatal presumed cardiac arrhythmia; in the no-ibrutinib cohort, one patient died from a CAR T cell–related toxicity.
Notably, a trend toward better expansion of CD8 CAR T cells and a significantly greater expansion of CD4 CAR T cells was observed in the ibrutinib cohort, he said.
The study was designed to assess JCAR014, and based on the initial cohort findings published in 2017, established a regimen of cyclophosphamide and fludarabine (Cy/Flu) lymphodepletion followed by JCAR014 infusion at 2 x 106 CAR T cells/kg. The study was not a randomized, head-to-head comparison but the groups were similar with respect to both patient and disease characteristics, Dr. Gauthier noted.
The outcomes in the first cohort were then compared retrospectively with those from the subsequent cohort of patients who received Cy/Flu with 2 x 106 CAR T cells/kg with concurrent ibrutinib administered at 420 mg per day from at least 2 weeks prior to leukapheresis until at least 3 months after JCAR014 infusion.
The rationale for uninterrupted ibrutinib in relapsed/refractory CLL patients receiving JCAR014 included potential prevention of tumor flare, mobilization of CLL cells into the blood from the lymph nodes, improvement of CAR T-cell function, and a decrease in CAR T-cell related toxicity, he said.
The concurrent administration of ibrutinib and JCAR014 was feasible for most patients. “[It] induced high response rates and deep responses early on at 4 weeks, and it was associated with higher in vivo expansion of CD4 CAR T cells and with lower rates of severe toxicity,” Dr. Gauthier said. “The next step is to hopefully validate these findings in a prospective phase 1/2 study.”
Dr. Gauthier reported having no financial disclosures.
SOURCE: Gauthier J et al. ASH 18, Abstract 299.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: Severe cytokine release syndrome occurred in 0% versus 25% of patients in the ibrutinib and no-ibrutinib cohorts, respectively.
Study details: A retrospective comparison of 43 patients in two cohorts from a phase 1/2 study.
Disclosures: Dr. Gauthier reported having no financial disclosures.
Source: Gauthier J et al. ASH 2018, Abstract 299.
Armored CAR protects T cells, induces remissions
SAN DIEGO – A second-generation CD19-specific “armored” chimeric antigen receptor (CAR) T-cell construct was associated with high complete remission rates in diffuse large B-cell lymphoma (DLBCL) and indolent non-Hodgkin lymphoma (NHL) in a phase 1 trial.

The CAR T construct – labeled 1928z-41BBL – also induced “encouraging” complete remission rates in patients with chronic lymphocytic leukemia (CLL) with Richter’s transformation, reported Jae H. Park, MD, of Memorial Sloan Kettering Cancer Center (MSKCC), New York, and his colleagues.
“Interestingly and encouragingly, severe [cytokine release syndrome] was not seen and grade 3 neurotoxicity was observed in less than 10%, with no grade 4 neurotoxicity, so there appears to be a favorable side effect profile,” Dr. Park said at the annual meeting of the American Society of Hematology.
Just as armored cars are designed to protect their valuable contents from people with bad intent, armored CAR T cells are engineered to protect the modified T-cells from a hostile tumor microenvironment and simultaneously recruit non-modified T cells to the target to produce a more robust immune response against malignant cells.
MSKCC investigators had previously shown that in contrast to other CAR T-cell constructs, the 1928z-41BBL configuration, which consists of two signaling domains (CD28 and CD3zeta) and the 4-1BB ligand, hit the sweet spot between tumor-killing function and T-cell persistence (Cancer Cell. 2015 Oct 12;28[4]:415-28).
In the current study, they enrolled 35 adults with relapsed or refractory CD19-positive hematologic malignancies, 29 of whom eventually underwent CAR T-cell infusions. The treated population comprised 14 patients with CLL (4 of whom had Richter’s transformation), 9 with DLBCL, 5 with indolent NHL, and 1 with acute lymphoblastic leukemia.
The patients with CLL had received a median of 5.5 prior lines of therapy, including ibrutinib (Imbruvica) and venetoclax (Venclexta).
There were 15 complete remissions (CR), with CR rates of 78% in DLBCL, 20% in CLL, 67% in CLL with Richter’s transformation, 60% in patients with indolent NHL, as well as CR in the single patient with ALL.
There were eight partial remissions. One patient with CLL had stable disease, and four patients had disease progression (one patient each with DLBCL, CLL, CLL with Richter’s, and indolent NHL).
Dr. Park noted that T cells are being detected in peripheral blood more than 6 months after T-cell infusion.
There were no cases of severe cytokine release syndrome, defined as requiring vasopressors and/or mechanical ventilation for hypoxia, and just three cases of grade 3 neurotoxicity. There were no cases of grade 4 neurotoxicity, no deaths related to neurotoxicity, and no cases of cerebral edema – a serious complication that has been seen in earlier CAR T-cell studies.
Split or multiple infusions of CAR T cells or incorporation of the technique into earlier lines of therapy might generate higher response rates, Dr. Park said.
The study was supported by Juno Therapeutics. Dr. Park reported consulting for and research funding from Juno, and financial relationships with other companies.
SOURCE: Park JH et al. ASH 2018, Abstract 224.
SAN DIEGO – A second-generation CD19-specific “armored” chimeric antigen receptor (CAR) T-cell construct was associated with high complete remission rates in diffuse large B-cell lymphoma (DLBCL) and indolent non-Hodgkin lymphoma (NHL) in a phase 1 trial.
The CAR T construct – labeled 1928z-41BBL – also induced “encouraging” complete remission rates in patients with chronic lymphocytic leukemia (CLL) with Richter’s transformation, reported Jae H. Park, MD, of Memorial Sloan Kettering Cancer Center (MSKCC), New York, and his colleagues.
“Interestingly and encouragingly, severe [cytokine release syndrome] was not seen and grade 3 neurotoxicity was observed in less than 10%, with no grade 4 neurotoxicity, so there appears to be a favorable side effect profile,” Dr. Park said at the annual meeting of the American Society of Hematology.
Just as armored cars are designed to protect their valuable contents from people with bad intent, armored CAR T cells are engineered to protect the modified T-cells from a hostile tumor microenvironment and simultaneously recruit non-modified T cells to the target to produce a more robust immune response against malignant cells.
MSKCC investigators had previously shown that in contrast to other CAR T-cell constructs, the 1928z-41BBL configuration, which consists of two signaling domains (CD28 and CD3zeta) and the 4-1BB ligand, hit the sweet spot between tumor-killing function and T-cell persistence (Cancer Cell. 2015 Oct 12;28[4]:415-28).
In the current study, they enrolled 35 adults with relapsed or refractory CD19-positive hematologic malignancies, 29 of whom eventually underwent CAR T-cell infusions. The treated population comprised 14 patients with CLL (4 of whom had Richter’s transformation), 9 with DLBCL, 5 with indolent NHL, and 1 with acute lymphoblastic leukemia.
The patients with CLL had received a median of 5.5 prior lines of therapy, including ibrutinib (Imbruvica) and venetoclax (Venclexta).
There were 15 complete remissions (CR), with CR rates of 78% in DLBCL, 20% in CLL, 67% in CLL with Richter’s transformation, 60% in patients with indolent NHL, as well as CR in the single patient with ALL.
There were eight partial remissions. One patient with CLL had stable disease, and four patients had disease progression (one patient each with DLBCL, CLL, CLL with Richter’s, and indolent NHL).
Dr. Park noted that T cells are being detected in peripheral blood more than 6 months after T-cell infusion.
There were no cases of severe cytokine release syndrome, defined as requiring vasopressors and/or mechanical ventilation for hypoxia, and just three cases of grade 3 neurotoxicity. There were no cases of grade 4 neurotoxicity, no deaths related to neurotoxicity, and no cases of cerebral edema – a serious complication that has been seen in earlier CAR T-cell studies.
Split or multiple infusions of CAR T cells or incorporation of the technique into earlier lines of therapy might generate higher response rates, Dr. Park said.
The study was supported by Juno Therapeutics. Dr. Park reported consulting for and research funding from Juno, and financial relationships with other companies.
SOURCE: Park JH et al. ASH 2018, Abstract 224.
SAN DIEGO – A second-generation CD19-specific “armored” chimeric antigen receptor (CAR) T-cell construct was associated with high complete remission rates in diffuse large B-cell lymphoma (DLBCL) and indolent non-Hodgkin lymphoma (NHL) in a phase 1 trial.
The CAR T construct – labeled 1928z-41BBL – also induced “encouraging” complete remission rates in patients with chronic lymphocytic leukemia (CLL) with Richter’s transformation, reported Jae H. Park, MD, of Memorial Sloan Kettering Cancer Center (MSKCC), New York, and his colleagues.
“Interestingly and encouragingly, severe [cytokine release syndrome] was not seen and grade 3 neurotoxicity was observed in less than 10%, with no grade 4 neurotoxicity, so there appears to be a favorable side effect profile,” Dr. Park said at the annual meeting of the American Society of Hematology.
Just as armored cars are designed to protect their valuable contents from people with bad intent, armored CAR T cells are engineered to protect the modified T-cells from a hostile tumor microenvironment and simultaneously recruit non-modified T cells to the target to produce a more robust immune response against malignant cells.
MSKCC investigators had previously shown that in contrast to other CAR T-cell constructs, the 1928z-41BBL configuration, which consists of two signaling domains (CD28 and CD3zeta) and the 4-1BB ligand, hit the sweet spot between tumor-killing function and T-cell persistence (Cancer Cell. 2015 Oct 12;28[4]:415-28).
In the current study, they enrolled 35 adults with relapsed or refractory CD19-positive hematologic malignancies, 29 of whom eventually underwent CAR T-cell infusions. The treated population comprised 14 patients with CLL (4 of whom had Richter’s transformation), 9 with DLBCL, 5 with indolent NHL, and 1 with acute lymphoblastic leukemia.
The patients with CLL had received a median of 5.5 prior lines of therapy, including ibrutinib (Imbruvica) and venetoclax (Venclexta).
There were 15 complete remissions (CR), with CR rates of 78% in DLBCL, 20% in CLL, 67% in CLL with Richter’s transformation, 60% in patients with indolent NHL, as well as CR in the single patient with ALL.
There were eight partial remissions. One patient with CLL had stable disease, and four patients had disease progression (one patient each with DLBCL, CLL, CLL with Richter’s, and indolent NHL).
Dr. Park noted that T cells are being detected in peripheral blood more than 6 months after T-cell infusion.
There were no cases of severe cytokine release syndrome, defined as requiring vasopressors and/or mechanical ventilation for hypoxia, and just three cases of grade 3 neurotoxicity. There were no cases of grade 4 neurotoxicity, no deaths related to neurotoxicity, and no cases of cerebral edema – a serious complication that has been seen in earlier CAR T-cell studies.
Split or multiple infusions of CAR T cells or incorporation of the technique into earlier lines of therapy might generate higher response rates, Dr. Park said.
The study was supported by Juno Therapeutics. Dr. Park reported consulting for and research funding from Juno, and financial relationships with other companies.
SOURCE: Park JH et al. ASH 2018, Abstract 224.
REPORTING FROM ASH 2018
Key clinical point: The 1928z-41BBL CAR T-cell construct induced high rates of complete remissions.
Major finding: The CAR T product was associated with a 78% complete remission rate in patients with heavily pretreated diffuse large B-cell lymphoma.
Study details: A phase 1 trial in 29 patients with CD19-positive hematologic malignancies.
Disclosures: Juno Therapeutics supported the study. Dr. Park reported consulting for and research funding from Juno, and financial relationships with other companies.
Source: Park JH et al. ASH 2018, Abstract 224.
Long-term side effects of CAR T cells mostly mild
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.

Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.
Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.
Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The most frequent adverse event was hypogammaglobulinemia in 60% of evaluable patients.
Study details: Prospective observational study of 60 patients with relapsed/refractory CLL or NHL.
Disclosures: Dr. Cordeiro reported having no relevant conflicts of interest.
Source: Cordeiro A et al. ASH 2018, Abstract 223.
Age-related gene expression may affect responses to RCC therapy
Older patients with ccRCC may respond better than younger patients to phosphoinositide 3-kinase (PI3K) or checkpoint inhibition because of age-related changes in gene expression, according to investigators.
This possibility was raised by in silico results from a broader study of gene expression patterns in clear cell renal carcinoma (ccRCC) and normal kidney tissues, reported lead author, Lara Feulner, MD, of the department of human genetics at McGill University and Genome Quebec Innovation Centre in Montreal.
“Several factors could contribute to the interindividual diversity among cancer patients,” the investigators wrote in a report published in Urologic Oncology.
“Their disease course could be affected not only by cell-intrinsic factors, but also by age-related changes impacting the vasculature, immune system and stroma. Little is known in this regard about ccRCC, a disease which affects adults across a wide age spectrum. Whether and how aging and comorbidities such as atherosclerosis may affect the biology and therapy of ccRCC has scarcely been considered,” they wrote.
The investigators explored this territory by analyzing datasets from The Cancer Genome Atlas (TCGA) and the Cancer Genomics of the Kidney (CAGEKID) program of the International Cancer Genome Consortium. Using regression, pathway enrichment, and connectivity mapping analyses, they were able to determine associations between age and gene expression, cellular processes, and drug treatment responses, respectively.
The investigators reported that age-related gene expression patterns occurred commonly in both normal and tumor tissues. Associations were reproducible between TCGA and CAGEKID datasets for both classes of tissue (tumor samples, R equal to 0.416, P less than 2.2 x 10-16; normal samples, R equal to 0.403, P less than 2.2 x 10-16). Out of the top 1,000 age-associated genes in tumor samples from each dataset, 383 were commonly downregulated with age and 294 were commonly upregulated with age in both datasets (P less than 2.2 x 10-16).
Among cellular pathways, the investigators found opposite age-relationship patterns. For example, normal tissues upregulated extracellular matrix and cell adhesion pathways with age, whereas tumor tissues downregulated the same pathways. Similar patterns of opposition were found in metabolism and oxidation pathways. Other age-related patterns were noted in some immune pathways, such as upregulation of toll-like receptor and tumor necrosis factor 2 noncanonical NF-kappa-B signaling in tumors, which became more common with age. A closer look showed that upregulation of tumor necrosis factor signaling was more common in female patients, who also downregulated Notch pathways more often than men.
Analysis of treatment responses showed possible relationships with age-dependent gene expression and immunotherapy. Specifically, of 532 genes tied to programmed cell death protein 1 (PD-1) resistance, 69 were among the 383 genes downregulated in older patients with ccRCC (P less than 2.2 x 10-16; 4.05 fold-enrichment), suggesting that older patients may respond better to anti-PD-1 therapy than younger patients. Similarly, connectivity map analysis showed that age-dependent gene expression may improve candidacy of older ccRCC patients for PI3K inhibition.
“We now have evidence that there are notable differences in tumor-associated pathway regulation between younger and older ccRCC patients, which may be therapeutically actionable,” the authors concluded.
The study was funded by the Cancer Research Society operation grant, a Canadian Cancer Society Research Institute Innovation-to-Impact grant, and a Canadian Institutes of Health Research Foundation grant. The authors reported no conflicts of interest.
SOURCE: Feulner et al. Urol Onc. 2018 Nov 23. doi: 10.1016/j.urolonc.2018.11.006.
Older patients with ccRCC may respond better than younger patients to phosphoinositide 3-kinase (PI3K) or checkpoint inhibition because of age-related changes in gene expression, according to investigators.
This possibility was raised by in silico results from a broader study of gene expression patterns in clear cell renal carcinoma (ccRCC) and normal kidney tissues, reported lead author, Lara Feulner, MD, of the department of human genetics at McGill University and Genome Quebec Innovation Centre in Montreal.
“Several factors could contribute to the interindividual diversity among cancer patients,” the investigators wrote in a report published in Urologic Oncology.
“Their disease course could be affected not only by cell-intrinsic factors, but also by age-related changes impacting the vasculature, immune system and stroma. Little is known in this regard about ccRCC, a disease which affects adults across a wide age spectrum. Whether and how aging and comorbidities such as atherosclerosis may affect the biology and therapy of ccRCC has scarcely been considered,” they wrote.
The investigators explored this territory by analyzing datasets from The Cancer Genome Atlas (TCGA) and the Cancer Genomics of the Kidney (CAGEKID) program of the International Cancer Genome Consortium. Using regression, pathway enrichment, and connectivity mapping analyses, they were able to determine associations between age and gene expression, cellular processes, and drug treatment responses, respectively.
The investigators reported that age-related gene expression patterns occurred commonly in both normal and tumor tissues. Associations were reproducible between TCGA and CAGEKID datasets for both classes of tissue (tumor samples, R equal to 0.416, P less than 2.2 x 10-16; normal samples, R equal to 0.403, P less than 2.2 x 10-16). Out of the top 1,000 age-associated genes in tumor samples from each dataset, 383 were commonly downregulated with age and 294 were commonly upregulated with age in both datasets (P less than 2.2 x 10-16).
Among cellular pathways, the investigators found opposite age-relationship patterns. For example, normal tissues upregulated extracellular matrix and cell adhesion pathways with age, whereas tumor tissues downregulated the same pathways. Similar patterns of opposition were found in metabolism and oxidation pathways. Other age-related patterns were noted in some immune pathways, such as upregulation of toll-like receptor and tumor necrosis factor 2 noncanonical NF-kappa-B signaling in tumors, which became more common with age. A closer look showed that upregulation of tumor necrosis factor signaling was more common in female patients, who also downregulated Notch pathways more often than men.
Analysis of treatment responses showed possible relationships with age-dependent gene expression and immunotherapy. Specifically, of 532 genes tied to programmed cell death protein 1 (PD-1) resistance, 69 were among the 383 genes downregulated in older patients with ccRCC (P less than 2.2 x 10-16; 4.05 fold-enrichment), suggesting that older patients may respond better to anti-PD-1 therapy than younger patients. Similarly, connectivity map analysis showed that age-dependent gene expression may improve candidacy of older ccRCC patients for PI3K inhibition.
“We now have evidence that there are notable differences in tumor-associated pathway regulation between younger and older ccRCC patients, which may be therapeutically actionable,” the authors concluded.
The study was funded by the Cancer Research Society operation grant, a Canadian Cancer Society Research Institute Innovation-to-Impact grant, and a Canadian Institutes of Health Research Foundation grant. The authors reported no conflicts of interest.
SOURCE: Feulner et al. Urol Onc. 2018 Nov 23. doi: 10.1016/j.urolonc.2018.11.006.
Older patients with ccRCC may respond better than younger patients to phosphoinositide 3-kinase (PI3K) or checkpoint inhibition because of age-related changes in gene expression, according to investigators.
This possibility was raised by in silico results from a broader study of gene expression patterns in clear cell renal carcinoma (ccRCC) and normal kidney tissues, reported lead author, Lara Feulner, MD, of the department of human genetics at McGill University and Genome Quebec Innovation Centre in Montreal.
“Several factors could contribute to the interindividual diversity among cancer patients,” the investigators wrote in a report published in Urologic Oncology.
“Their disease course could be affected not only by cell-intrinsic factors, but also by age-related changes impacting the vasculature, immune system and stroma. Little is known in this regard about ccRCC, a disease which affects adults across a wide age spectrum. Whether and how aging and comorbidities such as atherosclerosis may affect the biology and therapy of ccRCC has scarcely been considered,” they wrote.
The investigators explored this territory by analyzing datasets from The Cancer Genome Atlas (TCGA) and the Cancer Genomics of the Kidney (CAGEKID) program of the International Cancer Genome Consortium. Using regression, pathway enrichment, and connectivity mapping analyses, they were able to determine associations between age and gene expression, cellular processes, and drug treatment responses, respectively.
The investigators reported that age-related gene expression patterns occurred commonly in both normal and tumor tissues. Associations were reproducible between TCGA and CAGEKID datasets for both classes of tissue (tumor samples, R equal to 0.416, P less than 2.2 x 10-16; normal samples, R equal to 0.403, P less than 2.2 x 10-16). Out of the top 1,000 age-associated genes in tumor samples from each dataset, 383 were commonly downregulated with age and 294 were commonly upregulated with age in both datasets (P less than 2.2 x 10-16).
Among cellular pathways, the investigators found opposite age-relationship patterns. For example, normal tissues upregulated extracellular matrix and cell adhesion pathways with age, whereas tumor tissues downregulated the same pathways. Similar patterns of opposition were found in metabolism and oxidation pathways. Other age-related patterns were noted in some immune pathways, such as upregulation of toll-like receptor and tumor necrosis factor 2 noncanonical NF-kappa-B signaling in tumors, which became more common with age. A closer look showed that upregulation of tumor necrosis factor signaling was more common in female patients, who also downregulated Notch pathways more often than men.
Analysis of treatment responses showed possible relationships with age-dependent gene expression and immunotherapy. Specifically, of 532 genes tied to programmed cell death protein 1 (PD-1) resistance, 69 were among the 383 genes downregulated in older patients with ccRCC (P less than 2.2 x 10-16; 4.05 fold-enrichment), suggesting that older patients may respond better to anti-PD-1 therapy than younger patients. Similarly, connectivity map analysis showed that age-dependent gene expression may improve candidacy of older ccRCC patients for PI3K inhibition.
“We now have evidence that there are notable differences in tumor-associated pathway regulation between younger and older ccRCC patients, which may be therapeutically actionable,” the authors concluded.
The study was funded by the Cancer Research Society operation grant, a Canadian Cancer Society Research Institute Innovation-to-Impact grant, and a Canadian Institutes of Health Research Foundation grant. The authors reported no conflicts of interest.
SOURCE: Feulner et al. Urol Onc. 2018 Nov 23. doi: 10.1016/j.urolonc.2018.11.006.
FROM UROLOGIC ONCOLOGY
Key clinical point: Older patients with ccRCC may respond better than younger patients to phosphoinositide 3-kinase or checkpoint inhibition due to age-related changes in gene expression.
Major finding: Out of the top 1,000 age-associated genes in tumor samples, 383 were commonly downregulated with age and 294 were commonly upregulated with age in two large data sets (P less than 2.2 x 10-16).
Study details: An analysis of data from The Cancer Genome Atlas (TCGA) and the Cancer Genomics of the Kidney (CAGEKID) program of the International Cancer Genome Consortium.
Disclosures: The study was funded by the Cancer Research Society operation grant, a Canadian Cancer Society Research Institute Innovation-to-Impact grant, and a Canadian Institutes of Health Research Foundation grant.
Source: Feulner et al. Urol Oncol. 2018 Nov 23. doi: 10.1016/j.urolonc.2018.11.006.
Immunotherapy may hold the key to defeating virally associated cancers
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.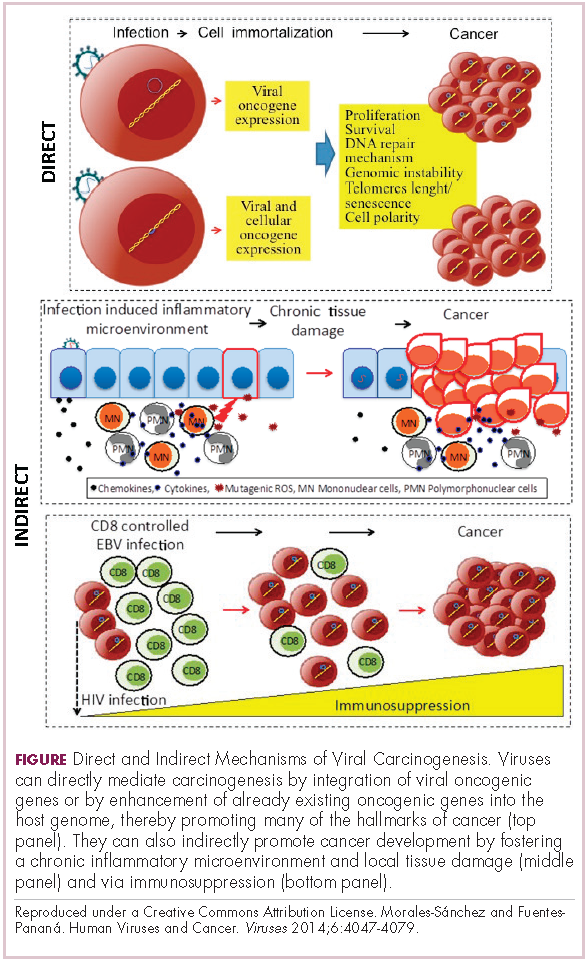
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).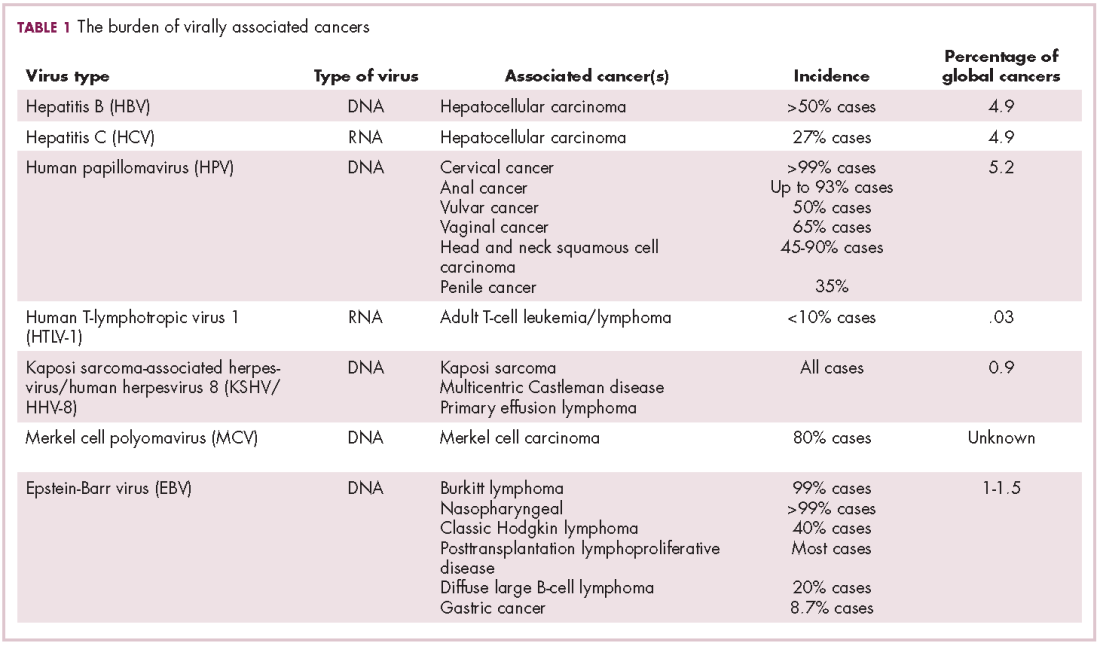
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9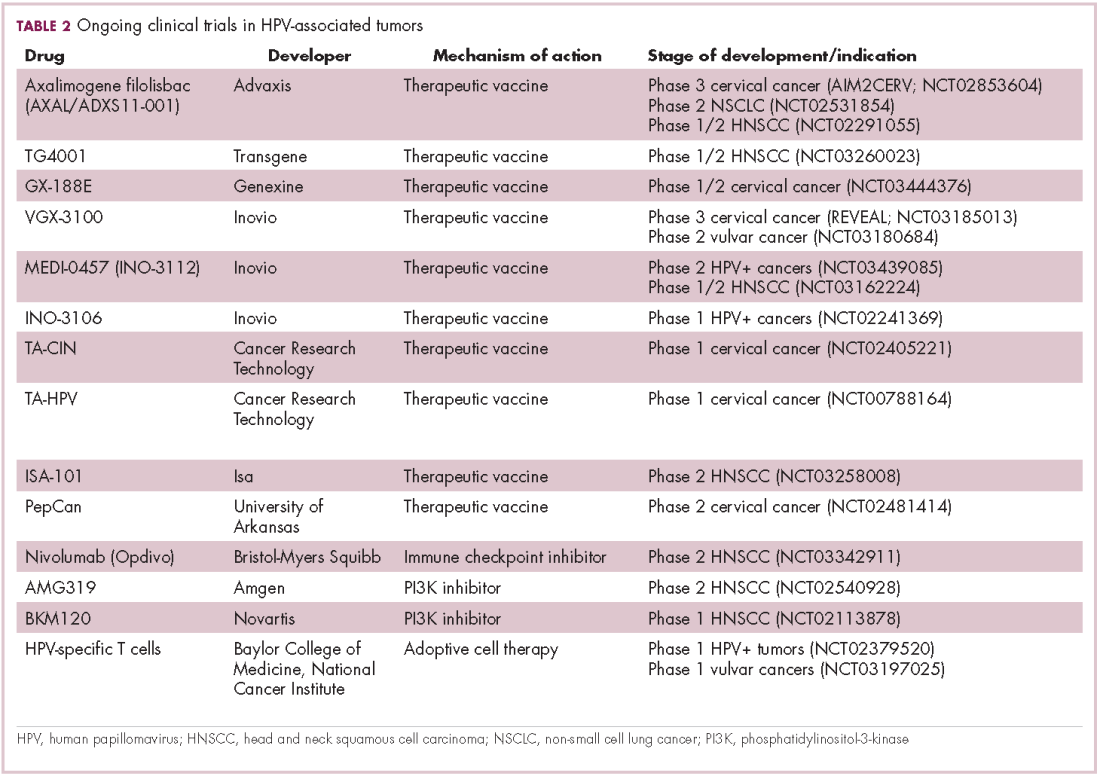
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).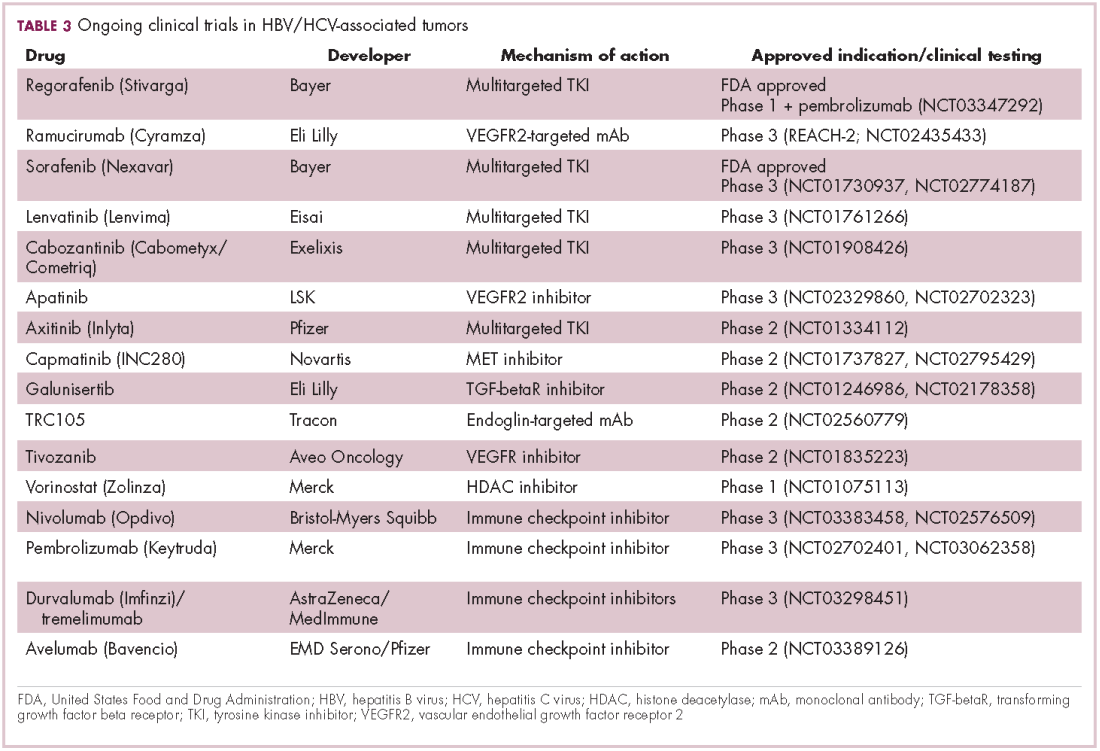
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).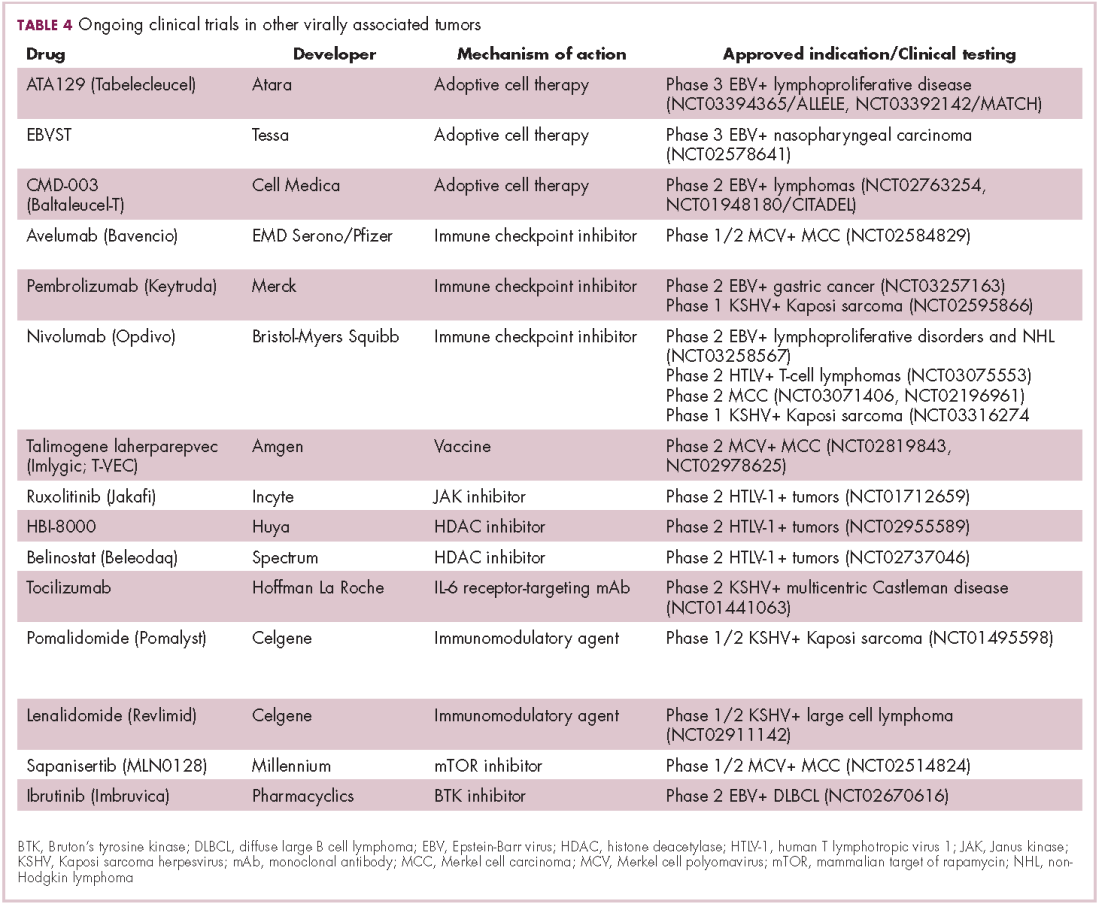
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
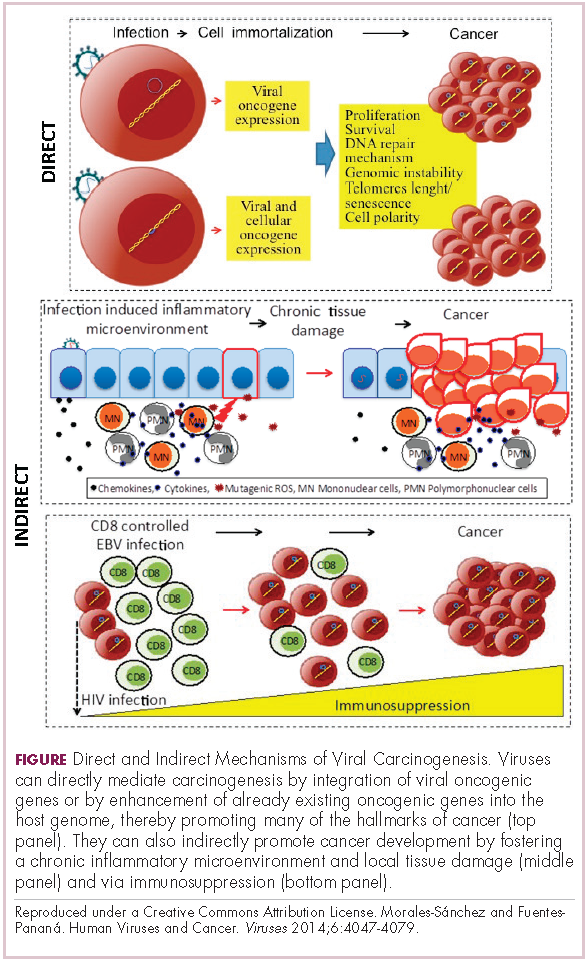
ASH preview: Studies target CAR T-cell improvements
, according to investigators in two separate studies.

Meanwhile, responses to tisagenlecleucel appear to be even more durable with longer follow-up, according to preliminary results from two more CAR T-cell therapy studies slated for presentation at the annual meeting of the American Society of Hematology.
A fifth study will show that bone marrow transplant may effectively consolidate remission after CAR T-cell therapy, according to Robert A. Brodsky, MD, ASH secretary, who highlighted the studies during a media briefing.
The ibrutinib study (abstract 299) shows that administering this BTK inhibitor starting 2 weeks prior to leukapheresis and continuing until 3 months after JCAR014 could improve responses and may decrease the incidence of severe cytokine release syndrome in patients with relapsed or refractory chronic lymphocytic leukemia (CLL).
Of 16 patients in an ibrutinib cohort and 18 patients in a no-ibrutinib cohort, the proportion of responders was 88% and 56%, respectively, according to preliminary data reported in the abstract. Grade 3-5 cytokine release syndrome occurred in 5 of 19 patients in the no-ibrutinib cohort, and 0 of 17 patients in the ibrutinib cohort.
Those findings are “early and preliminary, but very exciting” for ibrutinib in combination with this CD-19 specific CAR T-cell therapy said Dr. Brodsky, director of the division of hematology at Johns Hopkins University in Baltimore.
Early results of the checkpoint inhibitor study (abstract 556) suggest that pembrolizumab or nivolumab may augment CD19-directed CAR T-cell therapy in patients with relapsed B-cell acute lymphoblastic leukemia (ALL).
In data to date, 14 patients with early CAR T-cell loss, partial response, or no response to CAR T-cell therapy received a PD-1 inhibitor.
“The idea was if you can give pembrolizumab, you can take the brakes off, and maybe you can reinitiate the immune attack,” Dr. Brodsky said. “Sure enough, they were able to see that in roughly half of the patients. So again, very small, preliminary data, but very exciting that it is safe to give checkpoint inhibitors with CAR T-cells and it may be efficacious at getting the immune response back.”
One of the two tisagenlecleucel updates (abstract 895) showed that in the ELIANA trial, which included pediatric and young adults patients with relapsed/refractory ALL, the probability of relapse-free survival at 18 months was 66%.
“These are some very fast-growing tumors and these are refractory resistant patients, so as we get further and further out, it’s more encouraging to see that there are durable responses,” Dr. Brodsky said.
In the other tisagenlecleucel update (abstract 1684), investigators showed sustained disease control in Juliet, the global trial including adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), with 40% of patients still in remission at 18 months, according to Dr. Brodsky.
One more report Dr. Brodsky highlighted (abstract 967) will look at long-term follow-up after administration of SCRI-CAR19v1, a CD19-specific CAR T-cell product. Preliminary data suggest a survival advantage when hematopoietic stem cell transplantation is done after CAR T-cell induced remission.
“This study is very small and it’s retrospective, but it suggests that bone marrow transplant is a good way to consolidate the remission after CAR T-cell therapy,” Dr. Brodsky said.
, according to investigators in two separate studies.
Meanwhile, responses to tisagenlecleucel appear to be even more durable with longer follow-up, according to preliminary results from two more CAR T-cell therapy studies slated for presentation at the annual meeting of the American Society of Hematology.
A fifth study will show that bone marrow transplant may effectively consolidate remission after CAR T-cell therapy, according to Robert A. Brodsky, MD, ASH secretary, who highlighted the studies during a media briefing.
The ibrutinib study (abstract 299) shows that administering this BTK inhibitor starting 2 weeks prior to leukapheresis and continuing until 3 months after JCAR014 could improve responses and may decrease the incidence of severe cytokine release syndrome in patients with relapsed or refractory chronic lymphocytic leukemia (CLL).
Of 16 patients in an ibrutinib cohort and 18 patients in a no-ibrutinib cohort, the proportion of responders was 88% and 56%, respectively, according to preliminary data reported in the abstract. Grade 3-5 cytokine release syndrome occurred in 5 of 19 patients in the no-ibrutinib cohort, and 0 of 17 patients in the ibrutinib cohort.
Those findings are “early and preliminary, but very exciting” for ibrutinib in combination with this CD-19 specific CAR T-cell therapy said Dr. Brodsky, director of the division of hematology at Johns Hopkins University in Baltimore.
Early results of the checkpoint inhibitor study (abstract 556) suggest that pembrolizumab or nivolumab may augment CD19-directed CAR T-cell therapy in patients with relapsed B-cell acute lymphoblastic leukemia (ALL).
In data to date, 14 patients with early CAR T-cell loss, partial response, or no response to CAR T-cell therapy received a PD-1 inhibitor.
“The idea was if you can give pembrolizumab, you can take the brakes off, and maybe you can reinitiate the immune attack,” Dr. Brodsky said. “Sure enough, they were able to see that in roughly half of the patients. So again, very small, preliminary data, but very exciting that it is safe to give checkpoint inhibitors with CAR T-cells and it may be efficacious at getting the immune response back.”
One of the two tisagenlecleucel updates (abstract 895) showed that in the ELIANA trial, which included pediatric and young adults patients with relapsed/refractory ALL, the probability of relapse-free survival at 18 months was 66%.
“These are some very fast-growing tumors and these are refractory resistant patients, so as we get further and further out, it’s more encouraging to see that there are durable responses,” Dr. Brodsky said.
In the other tisagenlecleucel update (abstract 1684), investigators showed sustained disease control in Juliet, the global trial including adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), with 40% of patients still in remission at 18 months, according to Dr. Brodsky.
One more report Dr. Brodsky highlighted (abstract 967) will look at long-term follow-up after administration of SCRI-CAR19v1, a CD19-specific CAR T-cell product. Preliminary data suggest a survival advantage when hematopoietic stem cell transplantation is done after CAR T-cell induced remission.
“This study is very small and it’s retrospective, but it suggests that bone marrow transplant is a good way to consolidate the remission after CAR T-cell therapy,” Dr. Brodsky said.
, according to investigators in two separate studies.
Meanwhile, responses to tisagenlecleucel appear to be even more durable with longer follow-up, according to preliminary results from two more CAR T-cell therapy studies slated for presentation at the annual meeting of the American Society of Hematology.
A fifth study will show that bone marrow transplant may effectively consolidate remission after CAR T-cell therapy, according to Robert A. Brodsky, MD, ASH secretary, who highlighted the studies during a media briefing.
The ibrutinib study (abstract 299) shows that administering this BTK inhibitor starting 2 weeks prior to leukapheresis and continuing until 3 months after JCAR014 could improve responses and may decrease the incidence of severe cytokine release syndrome in patients with relapsed or refractory chronic lymphocytic leukemia (CLL).
Of 16 patients in an ibrutinib cohort and 18 patients in a no-ibrutinib cohort, the proportion of responders was 88% and 56%, respectively, according to preliminary data reported in the abstract. Grade 3-5 cytokine release syndrome occurred in 5 of 19 patients in the no-ibrutinib cohort, and 0 of 17 patients in the ibrutinib cohort.
Those findings are “early and preliminary, but very exciting” for ibrutinib in combination with this CD-19 specific CAR T-cell therapy said Dr. Brodsky, director of the division of hematology at Johns Hopkins University in Baltimore.
Early results of the checkpoint inhibitor study (abstract 556) suggest that pembrolizumab or nivolumab may augment CD19-directed CAR T-cell therapy in patients with relapsed B-cell acute lymphoblastic leukemia (ALL).
In data to date, 14 patients with early CAR T-cell loss, partial response, or no response to CAR T-cell therapy received a PD-1 inhibitor.
“The idea was if you can give pembrolizumab, you can take the brakes off, and maybe you can reinitiate the immune attack,” Dr. Brodsky said. “Sure enough, they were able to see that in roughly half of the patients. So again, very small, preliminary data, but very exciting that it is safe to give checkpoint inhibitors with CAR T-cells and it may be efficacious at getting the immune response back.”
One of the two tisagenlecleucel updates (abstract 895) showed that in the ELIANA trial, which included pediatric and young adults patients with relapsed/refractory ALL, the probability of relapse-free survival at 18 months was 66%.
“These are some very fast-growing tumors and these are refractory resistant patients, so as we get further and further out, it’s more encouraging to see that there are durable responses,” Dr. Brodsky said.
In the other tisagenlecleucel update (abstract 1684), investigators showed sustained disease control in Juliet, the global trial including adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), with 40% of patients still in remission at 18 months, according to Dr. Brodsky.
One more report Dr. Brodsky highlighted (abstract 967) will look at long-term follow-up after administration of SCRI-CAR19v1, a CD19-specific CAR T-cell product. Preliminary data suggest a survival advantage when hematopoietic stem cell transplantation is done after CAR T-cell induced remission.
“This study is very small and it’s retrospective, but it suggests that bone marrow transplant is a good way to consolidate the remission after CAR T-cell therapy,” Dr. Brodsky said.
NICE looks likely to reject use of Kymriah for DLBCL
The National Institute for Health and Care Excellence (NICE) has issued draft guidance recommending against tisagenlecleucel (Kymriah) as a treatment for adults with diffuse large B-cell lymphoma (DLBCL).
Tisagenlecleucel is a chimeric antigen receptor (CAR) T-cell therapy that was recently approved by the European Commission to treat adults with relapsed or refractory DLBCL who have received two or more lines of systemic therapy.
Tisagenlecleucel is also European Commission–approved to treat patients up to age 25 years who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse posttransplant, or in second or later relapse.
In September 2018, the National Health Service (NHS) in England announced tisagenlecleucel will be made available for these ALL patients through the Cancer Drugs Fund.
However, in who have received two or more lines of systemic therapy. NICE noted that there is no standard treatment for this patient group, and that salvage chemotherapy is the most common treatment option.
Although the latest results from the JULIET trial suggest tisagenlecleucel can produce responses in patients with relapsed/refractory DLBCL, there are no data comparing tisagenlecleucel with salvage chemotherapy. Additionally, tisagenlecleucel cannot be considered a life-extending treatment at the end of life, according to NICE criteria.
All cost-effectiveness estimates for tisagenlecleucel are above the range NICE normally considers acceptable, and tisagenlecleucel does not meet criteria for inclusion in the Cancer Drugs Fund.
The list price for tisagenlecleucel is 282,000 pounds. However, Novartis, the company developing tisagenlecleucel, has a confidential commercial arrangement with the NHS that lowers the price of tisagenlecleucel for the ALL indication. This arrangement would apply if tisagenlecleucel were recommended for the DLBCL indication.
In August, NICE issued a similar draft guidance document recommending against use of another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta). Axicabtagene ciloleucel is approved in Europe for the treatment of patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy.
The National Institute for Health and Care Excellence (NICE) has issued draft guidance recommending against tisagenlecleucel (Kymriah) as a treatment for adults with diffuse large B-cell lymphoma (DLBCL).
Tisagenlecleucel is a chimeric antigen receptor (CAR) T-cell therapy that was recently approved by the European Commission to treat adults with relapsed or refractory DLBCL who have received two or more lines of systemic therapy.
Tisagenlecleucel is also European Commission–approved to treat patients up to age 25 years who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse posttransplant, or in second or later relapse.
In September 2018, the National Health Service (NHS) in England announced tisagenlecleucel will be made available for these ALL patients through the Cancer Drugs Fund.
However, in who have received two or more lines of systemic therapy. NICE noted that there is no standard treatment for this patient group, and that salvage chemotherapy is the most common treatment option.
Although the latest results from the JULIET trial suggest tisagenlecleucel can produce responses in patients with relapsed/refractory DLBCL, there are no data comparing tisagenlecleucel with salvage chemotherapy. Additionally, tisagenlecleucel cannot be considered a life-extending treatment at the end of life, according to NICE criteria.
All cost-effectiveness estimates for tisagenlecleucel are above the range NICE normally considers acceptable, and tisagenlecleucel does not meet criteria for inclusion in the Cancer Drugs Fund.
The list price for tisagenlecleucel is 282,000 pounds. However, Novartis, the company developing tisagenlecleucel, has a confidential commercial arrangement with the NHS that lowers the price of tisagenlecleucel for the ALL indication. This arrangement would apply if tisagenlecleucel were recommended for the DLBCL indication.
In August, NICE issued a similar draft guidance document recommending against use of another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta). Axicabtagene ciloleucel is approved in Europe for the treatment of patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy.
The National Institute for Health and Care Excellence (NICE) has issued draft guidance recommending against tisagenlecleucel (Kymriah) as a treatment for adults with diffuse large B-cell lymphoma (DLBCL).
Tisagenlecleucel is a chimeric antigen receptor (CAR) T-cell therapy that was recently approved by the European Commission to treat adults with relapsed or refractory DLBCL who have received two or more lines of systemic therapy.
Tisagenlecleucel is also European Commission–approved to treat patients up to age 25 years who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse posttransplant, or in second or later relapse.
In September 2018, the National Health Service (NHS) in England announced tisagenlecleucel will be made available for these ALL patients through the Cancer Drugs Fund.
However, in who have received two or more lines of systemic therapy. NICE noted that there is no standard treatment for this patient group, and that salvage chemotherapy is the most common treatment option.
Although the latest results from the JULIET trial suggest tisagenlecleucel can produce responses in patients with relapsed/refractory DLBCL, there are no data comparing tisagenlecleucel with salvage chemotherapy. Additionally, tisagenlecleucel cannot be considered a life-extending treatment at the end of life, according to NICE criteria.
All cost-effectiveness estimates for tisagenlecleucel are above the range NICE normally considers acceptable, and tisagenlecleucel does not meet criteria for inclusion in the Cancer Drugs Fund.
The list price for tisagenlecleucel is 282,000 pounds. However, Novartis, the company developing tisagenlecleucel, has a confidential commercial arrangement with the NHS that lowers the price of tisagenlecleucel for the ALL indication. This arrangement would apply if tisagenlecleucel were recommended for the DLBCL indication.
In August, NICE issued a similar draft guidance document recommending against use of another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta). Axicabtagene ciloleucel is approved in Europe for the treatment of patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy.
Kymriah cost effectiveness depends on long-term outcomes
The cost-effectiveness of tisagenlecleucel (Kymriah) depends on long-term clinical outcomes, which are presently unknown, according to investigators.

If the long-term outcomes are more modest than clinical trials suggest, then payers may be unwilling to cover the costly therapy, reported John K. Lin, MD, of the Center for Primary Care and Outcomes Research at Stanford (Calif.) University, and his colleagues. Lowering the price or setting up an outcomes-based pricing structure may be necessary to get insurers to cover the therapy.
Tisagenlecleucel is an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy that was approved by the Food and Drug Administration in August 2017 for relapsed or refractory pediatric B-cell acute lymphoblastic leukemia (ALL). In 2018, the FDA expanded the indication for tisagenlecleucel to include adults with relapsed or refractory large B-cell lymphoma, though outcomes from lymphoma trials are not analyzed in the current study.
At a wholesale acquisition cost of $475,000 per infusion, it is the most expensive existing oncology therapy to date, and can be accompanied by expensive, potentially fatal adverse effects. However, clinical trials suggest that tisagenlecleucel can offer years of relapse-free remission, thereby allowing patients to forgo other expensive therapies such as hematopoietic stem cell transplantation (HSCT).
“Although tisagenlecleucel-induced remission rates are promising, compared with those of established therapies (greater than 80% vs. less than 50%), only short-term follow-up data currently exist,” the investigators wrote in the Journal of Clinical Oncology. “Given the high cost and broad applicability in other malignancies of tisagenlecleucel, a pressing question for policy makers, payers, patients, and clinicians is whether the cost of therapy represents reasonable value.”
The study used a Markov model to assess various long-term clinical outcome rates and cost thresholds of tisagenlecleucel. The lifetime cost of therapy was assessed and compared with costs of existing therapies.
The results showed that a 5-year relapse free survival rate of 40% would make the present cost ($475,000) of tisagenlecleucel economically reasonable. In this scenario, the increased life expectancy would be 12.1 years and would result in an additional 5.07 quality-adjusted life years (QALY) gained at a cost of $61,000 per QALY, compared with blinatumomab.
But if long-term outcomes are less favorable, tisagenlecleucel becomes much less cost effective. A 5-year relapse-free survival rate of 20% would drop increased life expectancy to 3.8 years, resulting in 1.80 QALYs gained and raising the cost to $151,000 per QALY.
“Our results suggest that at tisagenlecleucel’s current price and payment structure, its economic value is uncertain,” the investigators wrote.
They suggested a price drop to $200,000 or $350,000, which would allow the drug to remain cost effective even in a worse-case scenario, in which patients relapse and tisagenlecleucel is a bridge to transplant. Another option is to move to outcomes-based pricing. Making payment conditional on 7 months of remission would make the treatment cost effective, according to the analysis.
“Price reductions of tisagenlecleucel or payment only for longer-term remissions would favorably influence cost-effectiveness, even if long-term clinical outcomes are modest,” the investigators wrote.
The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award. One of the study coauthors reported consulting and research funding from Novartis.
SOURCE: Lin et al. J Clin Oncol. 2018 Sep 13. doi: 10.1200/JCO.2018.79.0642.
The cost-effectiveness of tisagenlecleucel (Kymriah) depends on long-term clinical outcomes, which are presently unknown, according to investigators.
If the long-term outcomes are more modest than clinical trials suggest, then payers may be unwilling to cover the costly therapy, reported John K. Lin, MD, of the Center for Primary Care and Outcomes Research at Stanford (Calif.) University, and his colleagues. Lowering the price or setting up an outcomes-based pricing structure may be necessary to get insurers to cover the therapy.
Tisagenlecleucel is an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy that was approved by the Food and Drug Administration in August 2017 for relapsed or refractory pediatric B-cell acute lymphoblastic leukemia (ALL). In 2018, the FDA expanded the indication for tisagenlecleucel to include adults with relapsed or refractory large B-cell lymphoma, though outcomes from lymphoma trials are not analyzed in the current study.
At a wholesale acquisition cost of $475,000 per infusion, it is the most expensive existing oncology therapy to date, and can be accompanied by expensive, potentially fatal adverse effects. However, clinical trials suggest that tisagenlecleucel can offer years of relapse-free remission, thereby allowing patients to forgo other expensive therapies such as hematopoietic stem cell transplantation (HSCT).
“Although tisagenlecleucel-induced remission rates are promising, compared with those of established therapies (greater than 80% vs. less than 50%), only short-term follow-up data currently exist,” the investigators wrote in the Journal of Clinical Oncology. “Given the high cost and broad applicability in other malignancies of tisagenlecleucel, a pressing question for policy makers, payers, patients, and clinicians is whether the cost of therapy represents reasonable value.”
The study used a Markov model to assess various long-term clinical outcome rates and cost thresholds of tisagenlecleucel. The lifetime cost of therapy was assessed and compared with costs of existing therapies.
The results showed that a 5-year relapse free survival rate of 40% would make the present cost ($475,000) of tisagenlecleucel economically reasonable. In this scenario, the increased life expectancy would be 12.1 years and would result in an additional 5.07 quality-adjusted life years (QALY) gained at a cost of $61,000 per QALY, compared with blinatumomab.
But if long-term outcomes are less favorable, tisagenlecleucel becomes much less cost effective. A 5-year relapse-free survival rate of 20% would drop increased life expectancy to 3.8 years, resulting in 1.80 QALYs gained and raising the cost to $151,000 per QALY.
“Our results suggest that at tisagenlecleucel’s current price and payment structure, its economic value is uncertain,” the investigators wrote.
They suggested a price drop to $200,000 or $350,000, which would allow the drug to remain cost effective even in a worse-case scenario, in which patients relapse and tisagenlecleucel is a bridge to transplant. Another option is to move to outcomes-based pricing. Making payment conditional on 7 months of remission would make the treatment cost effective, according to the analysis.
“Price reductions of tisagenlecleucel or payment only for longer-term remissions would favorably influence cost-effectiveness, even if long-term clinical outcomes are modest,” the investigators wrote.
The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award. One of the study coauthors reported consulting and research funding from Novartis.
SOURCE: Lin et al. J Clin Oncol. 2018 Sep 13. doi: 10.1200/JCO.2018.79.0642.
The cost-effectiveness of tisagenlecleucel (Kymriah) depends on long-term clinical outcomes, which are presently unknown, according to investigators.
If the long-term outcomes are more modest than clinical trials suggest, then payers may be unwilling to cover the costly therapy, reported John K. Lin, MD, of the Center for Primary Care and Outcomes Research at Stanford (Calif.) University, and his colleagues. Lowering the price or setting up an outcomes-based pricing structure may be necessary to get insurers to cover the therapy.
Tisagenlecleucel is an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy that was approved by the Food and Drug Administration in August 2017 for relapsed or refractory pediatric B-cell acute lymphoblastic leukemia (ALL). In 2018, the FDA expanded the indication for tisagenlecleucel to include adults with relapsed or refractory large B-cell lymphoma, though outcomes from lymphoma trials are not analyzed in the current study.
At a wholesale acquisition cost of $475,000 per infusion, it is the most expensive existing oncology therapy to date, and can be accompanied by expensive, potentially fatal adverse effects. However, clinical trials suggest that tisagenlecleucel can offer years of relapse-free remission, thereby allowing patients to forgo other expensive therapies such as hematopoietic stem cell transplantation (HSCT).
“Although tisagenlecleucel-induced remission rates are promising, compared with those of established therapies (greater than 80% vs. less than 50%), only short-term follow-up data currently exist,” the investigators wrote in the Journal of Clinical Oncology. “Given the high cost and broad applicability in other malignancies of tisagenlecleucel, a pressing question for policy makers, payers, patients, and clinicians is whether the cost of therapy represents reasonable value.”
The study used a Markov model to assess various long-term clinical outcome rates and cost thresholds of tisagenlecleucel. The lifetime cost of therapy was assessed and compared with costs of existing therapies.
The results showed that a 5-year relapse free survival rate of 40% would make the present cost ($475,000) of tisagenlecleucel economically reasonable. In this scenario, the increased life expectancy would be 12.1 years and would result in an additional 5.07 quality-adjusted life years (QALY) gained at a cost of $61,000 per QALY, compared with blinatumomab.
But if long-term outcomes are less favorable, tisagenlecleucel becomes much less cost effective. A 5-year relapse-free survival rate of 20% would drop increased life expectancy to 3.8 years, resulting in 1.80 QALYs gained and raising the cost to $151,000 per QALY.
“Our results suggest that at tisagenlecleucel’s current price and payment structure, its economic value is uncertain,” the investigators wrote.
They suggested a price drop to $200,000 or $350,000, which would allow the drug to remain cost effective even in a worse-case scenario, in which patients relapse and tisagenlecleucel is a bridge to transplant. Another option is to move to outcomes-based pricing. Making payment conditional on 7 months of remission would make the treatment cost effective, according to the analysis.
“Price reductions of tisagenlecleucel or payment only for longer-term remissions would favorably influence cost-effectiveness, even if long-term clinical outcomes are modest,” the investigators wrote.
The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award. One of the study coauthors reported consulting and research funding from Novartis.
SOURCE: Lin et al. J Clin Oncol. 2018 Sep 13. doi: 10.1200/JCO.2018.79.0642.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point:
Major finding: If 40% of patients achieve 5-year remission without relapse, then tisagenlecleucel would cost $61,000 per quality-adjusted life year.
Study details: An economic analysis involving tisagenlecleucel costs and clinical trial outcomes.
Disclosures: The study was funded by a Veterans Affairs Office of Academic Affiliations advanced fellowship in health service and research development, and a National Center for Advancing Translational Science Clinical and Translational Science Award. One study coauthor reported consulting and research funding from Novartis.
Source: Lin JK et al. J Clin Oncol. 2018 Sep 13. doi: 10.1200/JCO.2018.79.0642.