User login
Early CAR T data on P-BCMA-101 in refractory myeloma
Early results from a phase 1 trial suggest the chimeric antigen receptor (CAR) T-cell therapy P-BCMA-101 can produce responses in patients with relapsed/refractory multiple myeloma.
All 11 patients treated have experienced some clinical response, with 8 patients achieving a partial response (PR) or better.
The most common adverse events were neutropenia and thrombocytopenia. One patient was suspected to have cytokine release syndrome (CRS), but the condition resolved without use of tocilizumab or steroids.
These results were presented at the 2018 CAR-TCR Summit by Eric Ostertag, MD, PhD, chief executive officer of Poseida Therapeutics Inc., the company developing P-BCMA-101.
Dr. Ostertag presented data on 11 patients with heavily pretreated multiple myeloma. They had a median of six prior therapies. The median age was 60 years, and most of the patients were considered high risk.
Prior to receiving P-BCMA-101, patients received conditioning with fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2) for 3 days.
Patients were then treated across three dose groups with average CAR T-cell doses of 51 x 106 (n = 3), 152 x 106 (n = 7), and 430 x 106 (n = 1).
As of Aug. 10, 2018, all 11 patients were still on study.
There were no dose-limiting toxicities. Eight patients developed neutropenia, and five had thrombocytopenia.
Researchers suspected CRS in one patient, but the condition resolved without tocilizumab or steroid treatment. There was no neurotoxicity reported, and none of the patients required admission to an intensive care unit.
All patients showed improvement in biomarkers following treatment.
Ten patients were evaluable for response by International Myeloma Working Group criteria. Seven of these patients achieved at least a PR, including very good partial responses (VGPRs) and stringent complete response (CR).
The eleventh patient also responded to treatment, but this patient has oligosecretory disease and was evaluable only by PET. The patient had a near-CR by PET.
Poseida Therapeutics would not disclose additional details regarding how many patients achieved a PR, VGPR, or CR, but the company plans to release more information on response at an upcoming meeting.
“The latest data results show that P-BCMA-101 induces deep responses in a heavily pretreated population with relapsed/refractory multiple myeloma, with some patients reaching VGPR and even stringent CR at early efficacy assessments,” Dr. Ostertag said.
This study (NCT03288493) is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.
Early results from a phase 1 trial suggest the chimeric antigen receptor (CAR) T-cell therapy P-BCMA-101 can produce responses in patients with relapsed/refractory multiple myeloma.
All 11 patients treated have experienced some clinical response, with 8 patients achieving a partial response (PR) or better.
The most common adverse events were neutropenia and thrombocytopenia. One patient was suspected to have cytokine release syndrome (CRS), but the condition resolved without use of tocilizumab or steroids.
These results were presented at the 2018 CAR-TCR Summit by Eric Ostertag, MD, PhD, chief executive officer of Poseida Therapeutics Inc., the company developing P-BCMA-101.
Dr. Ostertag presented data on 11 patients with heavily pretreated multiple myeloma. They had a median of six prior therapies. The median age was 60 years, and most of the patients were considered high risk.
Prior to receiving P-BCMA-101, patients received conditioning with fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2) for 3 days.
Patients were then treated across three dose groups with average CAR T-cell doses of 51 x 106 (n = 3), 152 x 106 (n = 7), and 430 x 106 (n = 1).
As of Aug. 10, 2018, all 11 patients were still on study.
There were no dose-limiting toxicities. Eight patients developed neutropenia, and five had thrombocytopenia.
Researchers suspected CRS in one patient, but the condition resolved without tocilizumab or steroid treatment. There was no neurotoxicity reported, and none of the patients required admission to an intensive care unit.
All patients showed improvement in biomarkers following treatment.
Ten patients were evaluable for response by International Myeloma Working Group criteria. Seven of these patients achieved at least a PR, including very good partial responses (VGPRs) and stringent complete response (CR).
The eleventh patient also responded to treatment, but this patient has oligosecretory disease and was evaluable only by PET. The patient had a near-CR by PET.
Poseida Therapeutics would not disclose additional details regarding how many patients achieved a PR, VGPR, or CR, but the company plans to release more information on response at an upcoming meeting.
“The latest data results show that P-BCMA-101 induces deep responses in a heavily pretreated population with relapsed/refractory multiple myeloma, with some patients reaching VGPR and even stringent CR at early efficacy assessments,” Dr. Ostertag said.
This study (NCT03288493) is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.
Early results from a phase 1 trial suggest the chimeric antigen receptor (CAR) T-cell therapy P-BCMA-101 can produce responses in patients with relapsed/refractory multiple myeloma.
All 11 patients treated have experienced some clinical response, with 8 patients achieving a partial response (PR) or better.
The most common adverse events were neutropenia and thrombocytopenia. One patient was suspected to have cytokine release syndrome (CRS), but the condition resolved without use of tocilizumab or steroids.
These results were presented at the 2018 CAR-TCR Summit by Eric Ostertag, MD, PhD, chief executive officer of Poseida Therapeutics Inc., the company developing P-BCMA-101.
Dr. Ostertag presented data on 11 patients with heavily pretreated multiple myeloma. They had a median of six prior therapies. The median age was 60 years, and most of the patients were considered high risk.
Prior to receiving P-BCMA-101, patients received conditioning with fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2) for 3 days.
Patients were then treated across three dose groups with average CAR T-cell doses of 51 x 106 (n = 3), 152 x 106 (n = 7), and 430 x 106 (n = 1).
As of Aug. 10, 2018, all 11 patients were still on study.
There were no dose-limiting toxicities. Eight patients developed neutropenia, and five had thrombocytopenia.
Researchers suspected CRS in one patient, but the condition resolved without tocilizumab or steroid treatment. There was no neurotoxicity reported, and none of the patients required admission to an intensive care unit.
All patients showed improvement in biomarkers following treatment.
Ten patients were evaluable for response by International Myeloma Working Group criteria. Seven of these patients achieved at least a PR, including very good partial responses (VGPRs) and stringent complete response (CR).
The eleventh patient also responded to treatment, but this patient has oligosecretory disease and was evaluable only by PET. The patient had a near-CR by PET.
Poseida Therapeutics would not disclose additional details regarding how many patients achieved a PR, VGPR, or CR, but the company plans to release more information on response at an upcoming meeting.
“The latest data results show that P-BCMA-101 induces deep responses in a heavily pretreated population with relapsed/refractory multiple myeloma, with some patients reaching VGPR and even stringent CR at early efficacy assessments,” Dr. Ostertag said.
This study (NCT03288493) is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.
FROM THE 2018 CAR-TCR SUMMIT
Key clinical point: The chimeric antigen receptor .
Major finding: All 11 patients have shown signs of response, and 8 patients achieved a partial response or better.
Study details: Eleven patients have been treated thus far in this phase 1 trial.
Disclosures: This trial is funded by the California Institute for Regenerative Medicine and Poseida Therapeutics.
Novartis nabs first CAR T approval in Canada
the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel is approved to treat patients aged 3-25 years who have B-cell acute lymphoblastic leukemia (ALL) and relapsed after allogenic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials – JULIET and ELIANA.
JULIET enrolled 165 adults with relapsed/refractory DLBCL, 111 of whom received a single infusion of tisagenlecleucel.
The overall response rate was 52% and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%; the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome.
These results were presented at the 2018 annual congress of the European Hematology Association in June.
The ELIANA trial included 75 children and young adults with relapsed/refractory ALL. All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The overall remission rate was 81%, with 60% of patients achieving a CR and 21% achieving CR with incomplete hematologic recovery. All patients whose best response was CR with incomplete hematologic recovery were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
About 95% of patients had adverse events thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 adverse eventss was 73% (N Engl J Med 2018; 378:439-48).
the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel is approved to treat patients aged 3-25 years who have B-cell acute lymphoblastic leukemia (ALL) and relapsed after allogenic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials – JULIET and ELIANA.
JULIET enrolled 165 adults with relapsed/refractory DLBCL, 111 of whom received a single infusion of tisagenlecleucel.
The overall response rate was 52% and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%; the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome.
These results were presented at the 2018 annual congress of the European Hematology Association in June.
The ELIANA trial included 75 children and young adults with relapsed/refractory ALL. All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The overall remission rate was 81%, with 60% of patients achieving a CR and 21% achieving CR with incomplete hematologic recovery. All patients whose best response was CR with incomplete hematologic recovery were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
About 95% of patients had adverse events thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 adverse eventss was 73% (N Engl J Med 2018; 378:439-48).
the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel is approved to treat patients aged 3-25 years who have B-cell acute lymphoblastic leukemia (ALL) and relapsed after allogenic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials – JULIET and ELIANA.
JULIET enrolled 165 adults with relapsed/refractory DLBCL, 111 of whom received a single infusion of tisagenlecleucel.
The overall response rate was 52% and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%; the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome.
These results were presented at the 2018 annual congress of the European Hematology Association in June.
The ELIANA trial included 75 children and young adults with relapsed/refractory ALL. All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The overall remission rate was 81%, with 60% of patients achieving a CR and 21% achieving CR with incomplete hematologic recovery. All patients whose best response was CR with incomplete hematologic recovery were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
About 95% of patients had adverse events thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 adverse eventss was 73% (N Engl J Med 2018; 378:439-48).
England green-lights coverage of one CAR T-cell therapy
The National Health Service (NHS) of England has announced that tisagenlecleucel (Kymriah), a chimeric antigen receptor (CAR) T-cell therapy, will soon be available for certain leukemia patients.
, and patients could potentially begin receiving the treatment within weeks.
NHS England struck a deal with Novartis to lower the price of tisagenlecleucel, which costs around £282,000 per patient at its full list price. The discount offered to the NHS is confidential.
Tisagenlecleucel was recently approved by the European Commission (EC) to treat patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
The EC also approved tisagenlecleucel to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
However, tisagenlecleucel will be available only for ALL patients in England, at least initially. A decision has not been made regarding funding for tisagenlecleucel in DLBCL, and Novartis previously decided to launch tisagenlecleucel in ALL first.
“It’s fantastic news for children and young people with this form of leukemia that CAR T-cell therapy will be made available on the NHS, making them the first in Europe to have routine access to this exciting new type of immunotherapy,” said Charles Swanton, Cancer Research UK’s chief clinician.
The first three NHS hospitals to go through the international accreditation process for the provision of tisagenlecleucel are in London, Manchester, and Newcastle. Subject to passing accreditation requirements, the first treatments could begin in a matter of weeks.
Another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta), has not fared as well as in England. The National Institute for Health and Care Excellence (NICE) recently issued draft guidance recommending against the use of axicabtagene ciloleucel in England.
Axicabtagene ciloleucel was approved by the EC to treat patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy. However, NICE said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy. NICE also said the price of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of NHS resources, and the therapy does not meet the criteria for inclusion in the Cancer Drugs Fund.
The National Health Service (NHS) of England has announced that tisagenlecleucel (Kymriah), a chimeric antigen receptor (CAR) T-cell therapy, will soon be available for certain leukemia patients.
, and patients could potentially begin receiving the treatment within weeks.
NHS England struck a deal with Novartis to lower the price of tisagenlecleucel, which costs around £282,000 per patient at its full list price. The discount offered to the NHS is confidential.
Tisagenlecleucel was recently approved by the European Commission (EC) to treat patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
The EC also approved tisagenlecleucel to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
However, tisagenlecleucel will be available only for ALL patients in England, at least initially. A decision has not been made regarding funding for tisagenlecleucel in DLBCL, and Novartis previously decided to launch tisagenlecleucel in ALL first.
“It’s fantastic news for children and young people with this form of leukemia that CAR T-cell therapy will be made available on the NHS, making them the first in Europe to have routine access to this exciting new type of immunotherapy,” said Charles Swanton, Cancer Research UK’s chief clinician.
The first three NHS hospitals to go through the international accreditation process for the provision of tisagenlecleucel are in London, Manchester, and Newcastle. Subject to passing accreditation requirements, the first treatments could begin in a matter of weeks.
Another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta), has not fared as well as in England. The National Institute for Health and Care Excellence (NICE) recently issued draft guidance recommending against the use of axicabtagene ciloleucel in England.
Axicabtagene ciloleucel was approved by the EC to treat patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy. However, NICE said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy. NICE also said the price of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of NHS resources, and the therapy does not meet the criteria for inclusion in the Cancer Drugs Fund.
The National Health Service (NHS) of England has announced that tisagenlecleucel (Kymriah), a chimeric antigen receptor (CAR) T-cell therapy, will soon be available for certain leukemia patients.
, and patients could potentially begin receiving the treatment within weeks.
NHS England struck a deal with Novartis to lower the price of tisagenlecleucel, which costs around £282,000 per patient at its full list price. The discount offered to the NHS is confidential.
Tisagenlecleucel was recently approved by the European Commission (EC) to treat patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
The EC also approved tisagenlecleucel to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
However, tisagenlecleucel will be available only for ALL patients in England, at least initially. A decision has not been made regarding funding for tisagenlecleucel in DLBCL, and Novartis previously decided to launch tisagenlecleucel in ALL first.
“It’s fantastic news for children and young people with this form of leukemia that CAR T-cell therapy will be made available on the NHS, making them the first in Europe to have routine access to this exciting new type of immunotherapy,” said Charles Swanton, Cancer Research UK’s chief clinician.
The first three NHS hospitals to go through the international accreditation process for the provision of tisagenlecleucel are in London, Manchester, and Newcastle. Subject to passing accreditation requirements, the first treatments could begin in a matter of weeks.
Another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta), has not fared as well as in England. The National Institute for Health and Care Excellence (NICE) recently issued draft guidance recommending against the use of axicabtagene ciloleucel in England.
Axicabtagene ciloleucel was approved by the EC to treat patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy. However, NICE said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy. NICE also said the price of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of NHS resources, and the therapy does not meet the criteria for inclusion in the Cancer Drugs Fund.
European Commission approves first CAR T-cell therapies
The , two chimeric antigen receptor (CAR) T-cell therapies.
Tisagenlecleucel is now approved for use in pediatric and young adult patients up to 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
Tisagenlecleucel is also approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
Axicabtagene ciloleucel is approved for adults with relapsed/refractory DLBCL and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy. The treatment is marketed by Kite, a Gilead company.
The axicabtagene ciloleucel approval is based on results from the single arm, ZUMA-1 trial. During the study of 101 patients who received a single infusion, 72% responded to therapy and 51% achieved a complete response. At 1 year, median overall survival had not been reached.
Novartis expects to launch tisagenlecleucel initially for pediatric ALL. The company said timing for tisagenlecleucel availability in each country will depend on multiple factors, including the onboarding of qualified treatment centers for the appropriate indications, as well as the completion of national reimbursement procedures.
The EC’s approval of tisagenlecleucel is based on results from the phase 2 JULIET and ELIANA trials.
Updated results from JULIET were presented at the annual congress of the European Hematology Association in June 2018. The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so because of disease progression or clinical deterioration.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. At the time of data cutoff, none of the responders had gone on to receive a stem cell transplant.
Updated results from ELIANA were published in New England Journal of Medicine (2018;378:439-48).
The trial included 75 children and young adults with relapsed/refractory ALL. The overall remission rate was 81% (61/75), with 60% of patients (n = 45) achieving a complete remission (CR) and 21% (n = 16) achieving a CR with incomplete hematologic recovery (CRi).
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
The , two chimeric antigen receptor (CAR) T-cell therapies.
Tisagenlecleucel is now approved for use in pediatric and young adult patients up to 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
Tisagenlecleucel is also approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
Axicabtagene ciloleucel is approved for adults with relapsed/refractory DLBCL and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy. The treatment is marketed by Kite, a Gilead company.
The axicabtagene ciloleucel approval is based on results from the single arm, ZUMA-1 trial. During the study of 101 patients who received a single infusion, 72% responded to therapy and 51% achieved a complete response. At 1 year, median overall survival had not been reached.
Novartis expects to launch tisagenlecleucel initially for pediatric ALL. The company said timing for tisagenlecleucel availability in each country will depend on multiple factors, including the onboarding of qualified treatment centers for the appropriate indications, as well as the completion of national reimbursement procedures.
The EC’s approval of tisagenlecleucel is based on results from the phase 2 JULIET and ELIANA trials.
Updated results from JULIET were presented at the annual congress of the European Hematology Association in June 2018. The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so because of disease progression or clinical deterioration.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. At the time of data cutoff, none of the responders had gone on to receive a stem cell transplant.
Updated results from ELIANA were published in New England Journal of Medicine (2018;378:439-48).
The trial included 75 children and young adults with relapsed/refractory ALL. The overall remission rate was 81% (61/75), with 60% of patients (n = 45) achieving a complete remission (CR) and 21% (n = 16) achieving a CR with incomplete hematologic recovery (CRi).
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
The , two chimeric antigen receptor (CAR) T-cell therapies.
Tisagenlecleucel is now approved for use in pediatric and young adult patients up to 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
Tisagenlecleucel is also approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
Axicabtagene ciloleucel is approved for adults with relapsed/refractory DLBCL and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy. The treatment is marketed by Kite, a Gilead company.
The axicabtagene ciloleucel approval is based on results from the single arm, ZUMA-1 trial. During the study of 101 patients who received a single infusion, 72% responded to therapy and 51% achieved a complete response. At 1 year, median overall survival had not been reached.
Novartis expects to launch tisagenlecleucel initially for pediatric ALL. The company said timing for tisagenlecleucel availability in each country will depend on multiple factors, including the onboarding of qualified treatment centers for the appropriate indications, as well as the completion of national reimbursement procedures.
The EC’s approval of tisagenlecleucel is based on results from the phase 2 JULIET and ELIANA trials.
Updated results from JULIET were presented at the annual congress of the European Hematology Association in June 2018. The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so because of disease progression or clinical deterioration.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. At the time of data cutoff, none of the responders had gone on to receive a stem cell transplant.
Updated results from ELIANA were published in New England Journal of Medicine (2018;378:439-48).
The trial included 75 children and young adults with relapsed/refractory ALL. The overall remission rate was 81% (61/75), with 60% of patients (n = 45) achieving a complete remission (CR) and 21% (n = 16) achieving a CR with incomplete hematologic recovery (CRi).
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Immunotherapy-related adverse effects: how to identify and treat them in the emergency department
DR HENRY I am pleased to be talking with Dr Maura Sammon, an emergency department (ED) physician, about identifying and treating immunotherapy-related side effects in the ED. This is a hot topic in oncology, and I was very interested in having an ED physician talk about what happens when treating oncologists send their patients to the ED, because a physician may think it is chemotherapy when it is immunotherapy. Let’s start with the example of an oncology patient going to the ED with some symptoms, and the ED physician asks the patient what they’re being treated with. The patient may or may not say the right thing – that is, inform you whether they are being treated with chemotherapy or immunotherapy. How do you morph over into knowing that they are not getting chemotherapy?
DR SAMMON Yes, that’s a big problem in the ED. Patients come to the ED and say they’re being treated for cancer. They say they’re on chemotherapy, when they’re actually on immunotherapy, and it can really send the treatment team down the wrong path. I have a metaphor to explain this. They say that Great Britain and the United States are two nations separated by a common language. For example, when a British person talks about football, they mean something very different than when an American talks about football. If someone in Great Britain asks you to come play football, you might show up with shoulder pads and a helmet rather than shin guards, and you’re left without having the right tools to participate in the game.
How this sometimes plays out with immunotherapy, unfortunately, is that a patient will present to the ED and say they’re having a cough and that they’re on chemotherapy for melanoma. Usually, this patient would be worked up for being in a potentially immunosuppressed state. You might get a white blood cell count. You might get a chest X-ray. You might see what looks to you like a new infiltrate on this chest X-ray and then start going down the path of treating someone whom you think is immunosuppressed with pneumonia and giving them antibiotics rather than what could be life-saving steroids, as would be the case if the patient were on immunotherapy.
It’s a real problem, because you have one word that patients may use meaning two very different things. It can get you into trouble if you are treating someone for potentially infectious causes rather than immunotherapy-related adverse reactions, which are much more similar to graft-versus-host disease than in the case of traditional chemotherapy.
DR HENRY That’s a very good point. I think, as we in oncology use these immunotherapies/checkpoint inhibitors more often, you will see them more often in the ED. Let’s get right into that. You’ve identified this patient as not getting a traditional chemotherapy – hopefully, all our records on these patients are available. You’ve decided to follow onto what might be a side effect of the immunotherapy, so I’m going to name the side effects that always occur to me: lung, gastrointestinal (GI) – which could be loose bowels or liver function – rash, endocrine problems. Let’s start with lung symptoms. You see the patient is short of breath and you identified immunotherapy. What’s your next step?
DR SAMMON That’s a great example, because the problem is that you see these patients with cough or shortness of breath and pulmonary complications, and pulmonary complications of immunotherapy, while rare, are potentially life threatening if they’re not identified quickly.
You can start with a chest X-ray on these patients knowing, however, that for a good percentage of them you won’t see findings on their chest X-ray (Figure 1, from Sammon M, Tobin T. Identification and management of immune-related adverse events in the emergency setting. Presented at: Advances in Cancer Immunotherapy – Society for Immunotherapy of Cancer (SITC); August 4, 2017; Philadelphia, PA). You need to proceed to computed tomography (CT), because the issue is that you can have protean findings on the CT related to immunotherapy treatment/adverse reactions. You need to have a very high index of suspicion regardless of what abnormal findings you’re seeing on this CT and erring toward withholding the drugs, starting treatment, and being more aggressive with this type of finding.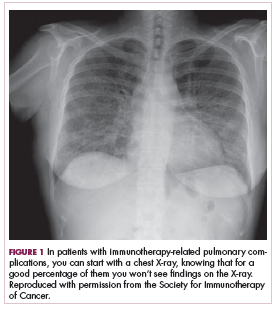
DR HENRY I’ve heard you talk at a conference about a patient with metastatic lung cancer, or some other tumor that may have existing disease in lung. The patient is aware of that, and the chart reflects that. Then you have this difficulty where the CT scan shows a pneumonitis, and it may not be tumor progression at all – it may be the drug. How do you work through that? Of course, your additional problem is you don’t have a whole lot of time. You must decide to whether you’re going to keep them in the ED, admit them, or send them home.
DR SAMMON Right. One of the first things is to get the oncologist involved at an early stage in treating this. We are all a team. We are all working together, and it’s very important to have that communication occur very early. I’m going to err on admitting those folks who have had symptoms and who have had findings on a chest CT, because they can progress. They can get much worse. I’m going to be getting the oncologist involved very early. We’re going to have the conversation about whether we should be starting steroids on them in the ED, getting them upstairs, and being aggressive in treating this.
DR HENRY So, time and therapy are very important.
DR SAMMON Yes.
DR HENRY You’ll get that steroid started as an antidote right away.
DR SAMMON Absolutely. For grade 2, you’re going to use methylprednisolone, 1 mg /kg daily. For anything higher than that – grade 3 or grade 4 – we’re going to start higher. We’re going to start at 2-4 mg/kg a day and get the patient upstairs and taper them slowly.
DR HENRY That’s worth empathizing. I, sadly, had a patient who ultimately did well, but had severe grade 4 pneumonitis. T
DR SAMMON Absolutely, but it’s very important to work together as a team to make these patients have good outcomes.
DR HENRY Agree. Let’s change over to the GI symptoms. The patient comes in and misidentifies him- or herself as having chemotherapy and diarrhea. We are used to causing nausea, vomiting, and diarrhea with some of our therapies. You realize, in talking to the oncologist, the patient is taking a checkpoint inhibitor. How would you approach the patient with diarrhea?
DR SAMMON First, I would talk to the patient. I would try to establish the baseline number of stools per day, because it’s not defined as a definite number of stools per day, it’s the number of stools above their baseline per day. If they’re having fewer than 4 stools above their baseline per day, I would send off some tests. We can send off a C diff (Clostridium difficile) test, we can send off a stool culture – all the parasites. Make sure the oncologist is going to be able to get these results and get them followed up, because these are results I’m not going to get back myself in the ED.
Then I’m going to talk to the patient about symptomatic treatment. I’m going to talk to them about oral hydration, a bland diet. Avoid using loperamide or any other antidiarrheal medicines, because that could decrease the frequency of stools but mask more severe symptoms that they may be having.
If I have a patient who is having more than 4 stools above their daily baseline and it’s been happening 4 to 6 stools a day for more than a week, I’m going to be sending those studies off, and I’m going to be having a conversation with the treating oncologist to find out if they want me to start the patient on steroids immediately, or if they want to wait for the test results to come back and have the steroids started as an outpatient.
These moderate patients can maybe wait a day until these test results come back. Those who are having more than 7 stools above baseline per day, peritoneal signs, ileus, or fever, are the patients you should worry about. You need to admit them for IV hydration. You need to do the stools, so you might need to keep them in the hospital until you find the results of the stool studies. You need to rule out perforation.
You may be starting steroids on these patients sooner rather than later. They’re going to be getting systemic corticosteroids at about 1-2 mg/kg of prednisone equivalent, assuming there is no perforation and their stool studies are negative. If they are unstable, though, they are really going to need high-dose corticosteroids. They are going to need methylprednisolone, 125 mg IV, to evaluate for their responsiveness. These folks really need to be treated as inpatients, and they need to have their oncologists involved early on with their treatment.
DR HENRY Yes. I couldn’t agree with you more. When I talk to the diarrhea side effect patients that we see, I tend to think it’s a curse. It’s volume. It’s calories. It’s electrolytes. The number of stools you’re mentioning, it is almost certainly going to need admission to rule out other causes. Then, if it’s the checkpoint inhibitor, the steroid antidotes.
Let’s move on to the rash. This is another organ system that can be affected by immunotherapy. What is your approach when you see a generalized body rash in a patient on one of these drugs who is sent to the ED?
DR SAMMON I am obviously going to be ruling out other causes first, but generally you’re going to see a maculopapular rash. It may be itchy. It may be burning. Patients will often describe it as just sort of having a tight sensation. I’m going to be looking at them a little bit like I look at a burn patient. What is their total body surface area that’s involved? If they’ve got less than 30% of their total body surface area involved, that’s considered a mild reaction.
For those folks, I’m not going to use systemic steroids, but I can give them some topical steroids, and I can give them some Benadryl, some diphenhydramine, and really treat them symptomatically as well as ensuring that they have early follow-up to make sure this isn’t progressing. Once we get between 30% and 50% body area, we’re talking about a moderate toxicity. If these patients are not improving rapidly with just withholding the drug, they need systemic corticosteroids.
We usually treat them at about 0.5-1 mg/kg body weight a day of prednisone equivalent. Just as with burn patients, these patients’ symptoms can become very severe. You can see signs of blistering, dermal ulceration, necrotic, bolus, hemorrhagic lesions (Figure 2, A-D).1 These folks can have very difficult-to-manage fluid balances, and they’re at very high risk for skin infections as well. They need to be treated as inpatients. If possible, you might want to consider sending these patients to a burn unit. They need systemic corticosteroids, 1-2 mg/kg per day, and they need careful monitoring for signs of dehydration, electrolytic abnormalities, and/or skin infections. They need excellent wound care.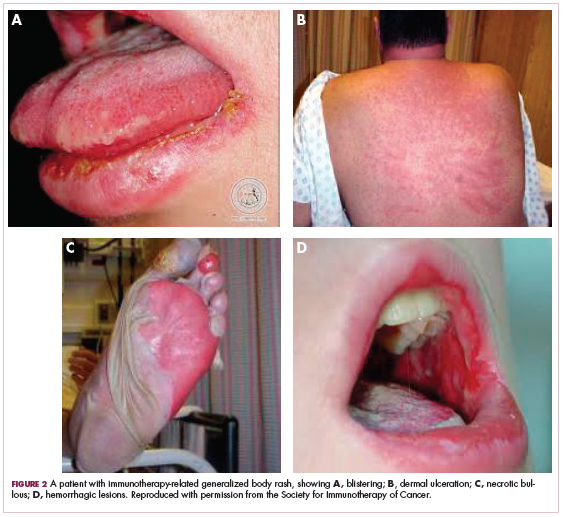
DR HENRY That’s very well put and always difficult, because there are so many causes of rash. That takes me to an area that has always been difficult for me, which is therapy-related endocrine problems. It’s interesting to note that these drugs can cause endocrine problems. I’ve heard you speak about the pituitary affecting vision, as well as thyroid or adrenal issues. Let’s start with how you’d approach vision difficulty in a patient on these drugs.
DR SAMMON The endocrinopathies that you can get with these checkpoint inhibitors really have a myriad of symptoms. Your patient may present saying that they’re feeling tired, that they’re feeling weak, or they may have a headache. If your patient is having actual pituitary enlargement, they can present with headaches, visual field defects, or cranial nerve defects. The reason for those symptoms is that the pituitary sits in the cavernous sinus, and you have various cranial nerves passing through that area as well as the optic chiasm just above the pituitary gland (Figure 3).1 Your patient may present with a bitemporal hemianopia. Or with diplopia. You are going to want to very quickly get either a CT scan or an MRI to find out if that is what’s going on (Figure 4).1 These folks need to be treated aggressively as well.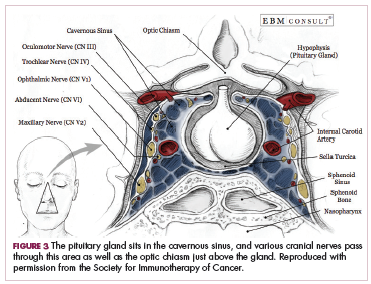
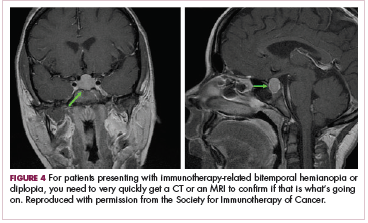
DR HENRY You’ll get your CT scan or your MRI and rule out an enlargement or a change to the visual field. I haven’t seen this yet, but certainly exciting when you see it to treat it. Would you get the radialis brevis involved, steroids involved? How would you manage that?
DR SAMMON It’s interesting, because you do want to use corticosteroids. One of the questions here is, which corticosteroid do you want to use? If you’re talking about someone who may have adrenal insufficiency, you may want to be able to do a stimulation test. In these patients, you may want to choose using dexamethasone, because you can still do the corticotropin stimulation test. However, if your patient is in frank shock because of what you think is an adrenal crisis, you’re going to want to use hydrocortisone. If a patient is truly hypotensive and unstable, the testing is at that point less important than the treatment.
DR HENRY Very interesting. We have covered what I would consider the major aspects of these fascinating drugs. We haven’t covered all of what they do when they work well, which hopefully we’re seeing more and more often, but we have covered very well what can happen when things go wrong in side effects. Anything else that you would like to add from the ED perspective or other side effects worth mentioning?
DR SAMMON The thing that I would most like to share with the oncology office is the importance of communicating with your patients that, when they’re on these drugs, they need to tell emergency physicians that they’re on immunotherapy, not chemotherapy. It might be helpful to give these patients a card stating that they’re on immunotherapy, not chemotherapy, and outlining some of the side effects that ED physicians should be looking out for in these patients.
DR HENRY That’s a great point. I’ve seen that some of the manufacturers have little cheat cards that the patient can carry naming the drug and the side effects, because not all ED doctors are aware of the side effects of these drugs.
DR SAMMON Absolutely. We love those cards.
DR HENRY Yes. I’ve also given some to the ED doctors at Pennsylvania Hospital, and they love it. I think we’ve covered everything in quite a bit of detail. Thank you, Dr Sammon, for sharing this information from the frontlines of the ED.
DR HENRY I am pleased to be talking with Dr Maura Sammon, an emergency department (ED) physician, about identifying and treating immunotherapy-related side effects in the ED. This is a hot topic in oncology, and I was very interested in having an ED physician talk about what happens when treating oncologists send their patients to the ED, because a physician may think it is chemotherapy when it is immunotherapy. Let’s start with the example of an oncology patient going to the ED with some symptoms, and the ED physician asks the patient what they’re being treated with. The patient may or may not say the right thing – that is, inform you whether they are being treated with chemotherapy or immunotherapy. How do you morph over into knowing that they are not getting chemotherapy?
DR SAMMON Yes, that’s a big problem in the ED. Patients come to the ED and say they’re being treated for cancer. They say they’re on chemotherapy, when they’re actually on immunotherapy, and it can really send the treatment team down the wrong path. I have a metaphor to explain this. They say that Great Britain and the United States are two nations separated by a common language. For example, when a British person talks about football, they mean something very different than when an American talks about football. If someone in Great Britain asks you to come play football, you might show up with shoulder pads and a helmet rather than shin guards, and you’re left without having the right tools to participate in the game.
How this sometimes plays out with immunotherapy, unfortunately, is that a patient will present to the ED and say they’re having a cough and that they’re on chemotherapy for melanoma. Usually, this patient would be worked up for being in a potentially immunosuppressed state. You might get a white blood cell count. You might get a chest X-ray. You might see what looks to you like a new infiltrate on this chest X-ray and then start going down the path of treating someone whom you think is immunosuppressed with pneumonia and giving them antibiotics rather than what could be life-saving steroids, as would be the case if the patient were on immunotherapy.
It’s a real problem, because you have one word that patients may use meaning two very different things. It can get you into trouble if you are treating someone for potentially infectious causes rather than immunotherapy-related adverse reactions, which are much more similar to graft-versus-host disease than in the case of traditional chemotherapy.
DR HENRY That’s a very good point. I think, as we in oncology use these immunotherapies/checkpoint inhibitors more often, you will see them more often in the ED. Let’s get right into that. You’ve identified this patient as not getting a traditional chemotherapy – hopefully, all our records on these patients are available. You’ve decided to follow onto what might be a side effect of the immunotherapy, so I’m going to name the side effects that always occur to me: lung, gastrointestinal (GI) – which could be loose bowels or liver function – rash, endocrine problems. Let’s start with lung symptoms. You see the patient is short of breath and you identified immunotherapy. What’s your next step?
DR SAMMON That’s a great example, because the problem is that you see these patients with cough or shortness of breath and pulmonary complications, and pulmonary complications of immunotherapy, while rare, are potentially life threatening if they’re not identified quickly.
You can start with a chest X-ray on these patients knowing, however, that for a good percentage of them you won’t see findings on their chest X-ray (Figure 1, from Sammon M, Tobin T. Identification and management of immune-related adverse events in the emergency setting. Presented at: Advances in Cancer Immunotherapy – Society for Immunotherapy of Cancer (SITC); August 4, 2017; Philadelphia, PA). You need to proceed to computed tomography (CT), because the issue is that you can have protean findings on the CT related to immunotherapy treatment/adverse reactions. You need to have a very high index of suspicion regardless of what abnormal findings you’re seeing on this CT and erring toward withholding the drugs, starting treatment, and being more aggressive with this type of finding.
DR HENRY I’ve heard you talk at a conference about a patient with metastatic lung cancer, or some other tumor that may have existing disease in lung. The patient is aware of that, and the chart reflects that. Then you have this difficulty where the CT scan shows a pneumonitis, and it may not be tumor progression at all – it may be the drug. How do you work through that? Of course, your additional problem is you don’t have a whole lot of time. You must decide to whether you’re going to keep them in the ED, admit them, or send them home.
DR SAMMON Right. One of the first things is to get the oncologist involved at an early stage in treating this. We are all a team. We are all working together, and it’s very important to have that communication occur very early. I’m going to err on admitting those folks who have had symptoms and who have had findings on a chest CT, because they can progress. They can get much worse. I’m going to be getting the oncologist involved very early. We’re going to have the conversation about whether we should be starting steroids on them in the ED, getting them upstairs, and being aggressive in treating this.
DR HENRY So, time and therapy are very important.
DR SAMMON Yes.
DR HENRY You’ll get that steroid started as an antidote right away.
DR SAMMON Absolutely. For grade 2, you’re going to use methylprednisolone, 1 mg /kg daily. For anything higher than that – grade 3 or grade 4 – we’re going to start higher. We’re going to start at 2-4 mg/kg a day and get the patient upstairs and taper them slowly.
DR HENRY That’s worth empathizing. I, sadly, had a patient who ultimately did well, but had severe grade 4 pneumonitis. T
DR SAMMON Absolutely, but it’s very important to work together as a team to make these patients have good outcomes.
DR HENRY Agree. Let’s change over to the GI symptoms. The patient comes in and misidentifies him- or herself as having chemotherapy and diarrhea. We are used to causing nausea, vomiting, and diarrhea with some of our therapies. You realize, in talking to the oncologist, the patient is taking a checkpoint inhibitor. How would you approach the patient with diarrhea?
DR SAMMON First, I would talk to the patient. I would try to establish the baseline number of stools per day, because it’s not defined as a definite number of stools per day, it’s the number of stools above their baseline per day. If they’re having fewer than 4 stools above their baseline per day, I would send off some tests. We can send off a C diff (Clostridium difficile) test, we can send off a stool culture – all the parasites. Make sure the oncologist is going to be able to get these results and get them followed up, because these are results I’m not going to get back myself in the ED.
Then I’m going to talk to the patient about symptomatic treatment. I’m going to talk to them about oral hydration, a bland diet. Avoid using loperamide or any other antidiarrheal medicines, because that could decrease the frequency of stools but mask more severe symptoms that they may be having.
If I have a patient who is having more than 4 stools above their daily baseline and it’s been happening 4 to 6 stools a day for more than a week, I’m going to be sending those studies off, and I’m going to be having a conversation with the treating oncologist to find out if they want me to start the patient on steroids immediately, or if they want to wait for the test results to come back and have the steroids started as an outpatient.
These moderate patients can maybe wait a day until these test results come back. Those who are having more than 7 stools above baseline per day, peritoneal signs, ileus, or fever, are the patients you should worry about. You need to admit them for IV hydration. You need to do the stools, so you might need to keep them in the hospital until you find the results of the stool studies. You need to rule out perforation.
You may be starting steroids on these patients sooner rather than later. They’re going to be getting systemic corticosteroids at about 1-2 mg/kg of prednisone equivalent, assuming there is no perforation and their stool studies are negative. If they are unstable, though, they are really going to need high-dose corticosteroids. They are going to need methylprednisolone, 125 mg IV, to evaluate for their responsiveness. These folks really need to be treated as inpatients, and they need to have their oncologists involved early on with their treatment.
DR HENRY Yes. I couldn’t agree with you more. When I talk to the diarrhea side effect patients that we see, I tend to think it’s a curse. It’s volume. It’s calories. It’s electrolytes. The number of stools you’re mentioning, it is almost certainly going to need admission to rule out other causes. Then, if it’s the checkpoint inhibitor, the steroid antidotes.
Let’s move on to the rash. This is another organ system that can be affected by immunotherapy. What is your approach when you see a generalized body rash in a patient on one of these drugs who is sent to the ED?
DR SAMMON I am obviously going to be ruling out other causes first, but generally you’re going to see a maculopapular rash. It may be itchy. It may be burning. Patients will often describe it as just sort of having a tight sensation. I’m going to be looking at them a little bit like I look at a burn patient. What is their total body surface area that’s involved? If they’ve got less than 30% of their total body surface area involved, that’s considered a mild reaction.
For those folks, I’m not going to use systemic steroids, but I can give them some topical steroids, and I can give them some Benadryl, some diphenhydramine, and really treat them symptomatically as well as ensuring that they have early follow-up to make sure this isn’t progressing. Once we get between 30% and 50% body area, we’re talking about a moderate toxicity. If these patients are not improving rapidly with just withholding the drug, they need systemic corticosteroids.
We usually treat them at about 0.5-1 mg/kg body weight a day of prednisone equivalent. Just as with burn patients, these patients’ symptoms can become very severe. You can see signs of blistering, dermal ulceration, necrotic, bolus, hemorrhagic lesions (Figure 2, A-D).1 These folks can have very difficult-to-manage fluid balances, and they’re at very high risk for skin infections as well. They need to be treated as inpatients. If possible, you might want to consider sending these patients to a burn unit. They need systemic corticosteroids, 1-2 mg/kg per day, and they need careful monitoring for signs of dehydration, electrolytic abnormalities, and/or skin infections. They need excellent wound care.
DR HENRY That’s very well put and always difficult, because there are so many causes of rash. That takes me to an area that has always been difficult for me, which is therapy-related endocrine problems. It’s interesting to note that these drugs can cause endocrine problems. I’ve heard you speak about the pituitary affecting vision, as well as thyroid or adrenal issues. Let’s start with how you’d approach vision difficulty in a patient on these drugs.
DR SAMMON The endocrinopathies that you can get with these checkpoint inhibitors really have a myriad of symptoms. Your patient may present saying that they’re feeling tired, that they’re feeling weak, or they may have a headache. If your patient is having actual pituitary enlargement, they can present with headaches, visual field defects, or cranial nerve defects. The reason for those symptoms is that the pituitary sits in the cavernous sinus, and you have various cranial nerves passing through that area as well as the optic chiasm just above the pituitary gland (Figure 3).1 Your patient may present with a bitemporal hemianopia. Or with diplopia. You are going to want to very quickly get either a CT scan or an MRI to find out if that is what’s going on (Figure 4).1 These folks need to be treated aggressively as well.
DR HENRY You’ll get your CT scan or your MRI and rule out an enlargement or a change to the visual field. I haven’t seen this yet, but certainly exciting when you see it to treat it. Would you get the radialis brevis involved, steroids involved? How would you manage that?
DR SAMMON It’s interesting, because you do want to use corticosteroids. One of the questions here is, which corticosteroid do you want to use? If you’re talking about someone who may have adrenal insufficiency, you may want to be able to do a stimulation test. In these patients, you may want to choose using dexamethasone, because you can still do the corticotropin stimulation test. However, if your patient is in frank shock because of what you think is an adrenal crisis, you’re going to want to use hydrocortisone. If a patient is truly hypotensive and unstable, the testing is at that point less important than the treatment.
DR HENRY Very interesting. We have covered what I would consider the major aspects of these fascinating drugs. We haven’t covered all of what they do when they work well, which hopefully we’re seeing more and more often, but we have covered very well what can happen when things go wrong in side effects. Anything else that you would like to add from the ED perspective or other side effects worth mentioning?
DR SAMMON The thing that I would most like to share with the oncology office is the importance of communicating with your patients that, when they’re on these drugs, they need to tell emergency physicians that they’re on immunotherapy, not chemotherapy. It might be helpful to give these patients a card stating that they’re on immunotherapy, not chemotherapy, and outlining some of the side effects that ED physicians should be looking out for in these patients.
DR HENRY That’s a great point. I’ve seen that some of the manufacturers have little cheat cards that the patient can carry naming the drug and the side effects, because not all ED doctors are aware of the side effects of these drugs.
DR SAMMON Absolutely. We love those cards.
DR HENRY Yes. I’ve also given some to the ED doctors at Pennsylvania Hospital, and they love it. I think we’ve covered everything in quite a bit of detail. Thank you, Dr Sammon, for sharing this information from the frontlines of the ED.
DR HENRY I am pleased to be talking with Dr Maura Sammon, an emergency department (ED) physician, about identifying and treating immunotherapy-related side effects in the ED. This is a hot topic in oncology, and I was very interested in having an ED physician talk about what happens when treating oncologists send their patients to the ED, because a physician may think it is chemotherapy when it is immunotherapy. Let’s start with the example of an oncology patient going to the ED with some symptoms, and the ED physician asks the patient what they’re being treated with. The patient may or may not say the right thing – that is, inform you whether they are being treated with chemotherapy or immunotherapy. How do you morph over into knowing that they are not getting chemotherapy?
DR SAMMON Yes, that’s a big problem in the ED. Patients come to the ED and say they’re being treated for cancer. They say they’re on chemotherapy, when they’re actually on immunotherapy, and it can really send the treatment team down the wrong path. I have a metaphor to explain this. They say that Great Britain and the United States are two nations separated by a common language. For example, when a British person talks about football, they mean something very different than when an American talks about football. If someone in Great Britain asks you to come play football, you might show up with shoulder pads and a helmet rather than shin guards, and you’re left without having the right tools to participate in the game.
How this sometimes plays out with immunotherapy, unfortunately, is that a patient will present to the ED and say they’re having a cough and that they’re on chemotherapy for melanoma. Usually, this patient would be worked up for being in a potentially immunosuppressed state. You might get a white blood cell count. You might get a chest X-ray. You might see what looks to you like a new infiltrate on this chest X-ray and then start going down the path of treating someone whom you think is immunosuppressed with pneumonia and giving them antibiotics rather than what could be life-saving steroids, as would be the case if the patient were on immunotherapy.
It’s a real problem, because you have one word that patients may use meaning two very different things. It can get you into trouble if you are treating someone for potentially infectious causes rather than immunotherapy-related adverse reactions, which are much more similar to graft-versus-host disease than in the case of traditional chemotherapy.
DR HENRY That’s a very good point. I think, as we in oncology use these immunotherapies/checkpoint inhibitors more often, you will see them more often in the ED. Let’s get right into that. You’ve identified this patient as not getting a traditional chemotherapy – hopefully, all our records on these patients are available. You’ve decided to follow onto what might be a side effect of the immunotherapy, so I’m going to name the side effects that always occur to me: lung, gastrointestinal (GI) – which could be loose bowels or liver function – rash, endocrine problems. Let’s start with lung symptoms. You see the patient is short of breath and you identified immunotherapy. What’s your next step?
DR SAMMON That’s a great example, because the problem is that you see these patients with cough or shortness of breath and pulmonary complications, and pulmonary complications of immunotherapy, while rare, are potentially life threatening if they’re not identified quickly.
You can start with a chest X-ray on these patients knowing, however, that for a good percentage of them you won’t see findings on their chest X-ray (Figure 1, from Sammon M, Tobin T. Identification and management of immune-related adverse events in the emergency setting. Presented at: Advances in Cancer Immunotherapy – Society for Immunotherapy of Cancer (SITC); August 4, 2017; Philadelphia, PA). You need to proceed to computed tomography (CT), because the issue is that you can have protean findings on the CT related to immunotherapy treatment/adverse reactions. You need to have a very high index of suspicion regardless of what abnormal findings you’re seeing on this CT and erring toward withholding the drugs, starting treatment, and being more aggressive with this type of finding.
DR HENRY I’ve heard you talk at a conference about a patient with metastatic lung cancer, or some other tumor that may have existing disease in lung. The patient is aware of that, and the chart reflects that. Then you have this difficulty where the CT scan shows a pneumonitis, and it may not be tumor progression at all – it may be the drug. How do you work through that? Of course, your additional problem is you don’t have a whole lot of time. You must decide to whether you’re going to keep them in the ED, admit them, or send them home.
DR SAMMON Right. One of the first things is to get the oncologist involved at an early stage in treating this. We are all a team. We are all working together, and it’s very important to have that communication occur very early. I’m going to err on admitting those folks who have had symptoms and who have had findings on a chest CT, because they can progress. They can get much worse. I’m going to be getting the oncologist involved very early. We’re going to have the conversation about whether we should be starting steroids on them in the ED, getting them upstairs, and being aggressive in treating this.
DR HENRY So, time and therapy are very important.
DR SAMMON Yes.
DR HENRY You’ll get that steroid started as an antidote right away.
DR SAMMON Absolutely. For grade 2, you’re going to use methylprednisolone, 1 mg /kg daily. For anything higher than that – grade 3 or grade 4 – we’re going to start higher. We’re going to start at 2-4 mg/kg a day and get the patient upstairs and taper them slowly.
DR HENRY That’s worth empathizing. I, sadly, had a patient who ultimately did well, but had severe grade 4 pneumonitis. T
DR SAMMON Absolutely, but it’s very important to work together as a team to make these patients have good outcomes.
DR HENRY Agree. Let’s change over to the GI symptoms. The patient comes in and misidentifies him- or herself as having chemotherapy and diarrhea. We are used to causing nausea, vomiting, and diarrhea with some of our therapies. You realize, in talking to the oncologist, the patient is taking a checkpoint inhibitor. How would you approach the patient with diarrhea?
DR SAMMON First, I would talk to the patient. I would try to establish the baseline number of stools per day, because it’s not defined as a definite number of stools per day, it’s the number of stools above their baseline per day. If they’re having fewer than 4 stools above their baseline per day, I would send off some tests. We can send off a C diff (Clostridium difficile) test, we can send off a stool culture – all the parasites. Make sure the oncologist is going to be able to get these results and get them followed up, because these are results I’m not going to get back myself in the ED.
Then I’m going to talk to the patient about symptomatic treatment. I’m going to talk to them about oral hydration, a bland diet. Avoid using loperamide or any other antidiarrheal medicines, because that could decrease the frequency of stools but mask more severe symptoms that they may be having.
If I have a patient who is having more than 4 stools above their daily baseline and it’s been happening 4 to 6 stools a day for more than a week, I’m going to be sending those studies off, and I’m going to be having a conversation with the treating oncologist to find out if they want me to start the patient on steroids immediately, or if they want to wait for the test results to come back and have the steroids started as an outpatient.
These moderate patients can maybe wait a day until these test results come back. Those who are having more than 7 stools above baseline per day, peritoneal signs, ileus, or fever, are the patients you should worry about. You need to admit them for IV hydration. You need to do the stools, so you might need to keep them in the hospital until you find the results of the stool studies. You need to rule out perforation.
You may be starting steroids on these patients sooner rather than later. They’re going to be getting systemic corticosteroids at about 1-2 mg/kg of prednisone equivalent, assuming there is no perforation and their stool studies are negative. If they are unstable, though, they are really going to need high-dose corticosteroids. They are going to need methylprednisolone, 125 mg IV, to evaluate for their responsiveness. These folks really need to be treated as inpatients, and they need to have their oncologists involved early on with their treatment.
DR HENRY Yes. I couldn’t agree with you more. When I talk to the diarrhea side effect patients that we see, I tend to think it’s a curse. It’s volume. It’s calories. It’s electrolytes. The number of stools you’re mentioning, it is almost certainly going to need admission to rule out other causes. Then, if it’s the checkpoint inhibitor, the steroid antidotes.
Let’s move on to the rash. This is another organ system that can be affected by immunotherapy. What is your approach when you see a generalized body rash in a patient on one of these drugs who is sent to the ED?
DR SAMMON I am obviously going to be ruling out other causes first, but generally you’re going to see a maculopapular rash. It may be itchy. It may be burning. Patients will often describe it as just sort of having a tight sensation. I’m going to be looking at them a little bit like I look at a burn patient. What is their total body surface area that’s involved? If they’ve got less than 30% of their total body surface area involved, that’s considered a mild reaction.
For those folks, I’m not going to use systemic steroids, but I can give them some topical steroids, and I can give them some Benadryl, some diphenhydramine, and really treat them symptomatically as well as ensuring that they have early follow-up to make sure this isn’t progressing. Once we get between 30% and 50% body area, we’re talking about a moderate toxicity. If these patients are not improving rapidly with just withholding the drug, they need systemic corticosteroids.
We usually treat them at about 0.5-1 mg/kg body weight a day of prednisone equivalent. Just as with burn patients, these patients’ symptoms can become very severe. You can see signs of blistering, dermal ulceration, necrotic, bolus, hemorrhagic lesions (Figure 2, A-D).1 These folks can have very difficult-to-manage fluid balances, and they’re at very high risk for skin infections as well. They need to be treated as inpatients. If possible, you might want to consider sending these patients to a burn unit. They need systemic corticosteroids, 1-2 mg/kg per day, and they need careful monitoring for signs of dehydration, electrolytic abnormalities, and/or skin infections. They need excellent wound care.
DR HENRY That’s very well put and always difficult, because there are so many causes of rash. That takes me to an area that has always been difficult for me, which is therapy-related endocrine problems. It’s interesting to note that these drugs can cause endocrine problems. I’ve heard you speak about the pituitary affecting vision, as well as thyroid or adrenal issues. Let’s start with how you’d approach vision difficulty in a patient on these drugs.
DR SAMMON The endocrinopathies that you can get with these checkpoint inhibitors really have a myriad of symptoms. Your patient may present saying that they’re feeling tired, that they’re feeling weak, or they may have a headache. If your patient is having actual pituitary enlargement, they can present with headaches, visual field defects, or cranial nerve defects. The reason for those symptoms is that the pituitary sits in the cavernous sinus, and you have various cranial nerves passing through that area as well as the optic chiasm just above the pituitary gland (Figure 3).1 Your patient may present with a bitemporal hemianopia. Or with diplopia. You are going to want to very quickly get either a CT scan or an MRI to find out if that is what’s going on (Figure 4).1 These folks need to be treated aggressively as well.
DR HENRY You’ll get your CT scan or your MRI and rule out an enlargement or a change to the visual field. I haven’t seen this yet, but certainly exciting when you see it to treat it. Would you get the radialis brevis involved, steroids involved? How would you manage that?
DR SAMMON It’s interesting, because you do want to use corticosteroids. One of the questions here is, which corticosteroid do you want to use? If you’re talking about someone who may have adrenal insufficiency, you may want to be able to do a stimulation test. In these patients, you may want to choose using dexamethasone, because you can still do the corticotropin stimulation test. However, if your patient is in frank shock because of what you think is an adrenal crisis, you’re going to want to use hydrocortisone. If a patient is truly hypotensive and unstable, the testing is at that point less important than the treatment.
DR HENRY Very interesting. We have covered what I would consider the major aspects of these fascinating drugs. We haven’t covered all of what they do when they work well, which hopefully we’re seeing more and more often, but we have covered very well what can happen when things go wrong in side effects. Anything else that you would like to add from the ED perspective or other side effects worth mentioning?
DR SAMMON The thing that I would most like to share with the oncology office is the importance of communicating with your patients that, when they’re on these drugs, they need to tell emergency physicians that they’re on immunotherapy, not chemotherapy. It might be helpful to give these patients a card stating that they’re on immunotherapy, not chemotherapy, and outlining some of the side effects that ED physicians should be looking out for in these patients.
DR HENRY That’s a great point. I’ve seen that some of the manufacturers have little cheat cards that the patient can carry naming the drug and the side effects, because not all ED doctors are aware of the side effects of these drugs.
DR SAMMON Absolutely. We love those cards.
DR HENRY Yes. I’ve also given some to the ED doctors at Pennsylvania Hospital, and they love it. I think we’ve covered everything in quite a bit of detail. Thank you, Dr Sammon, for sharing this information from the frontlines of the ED.
Meeting the potential of immunotherapy: new targets provide rational combinations
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.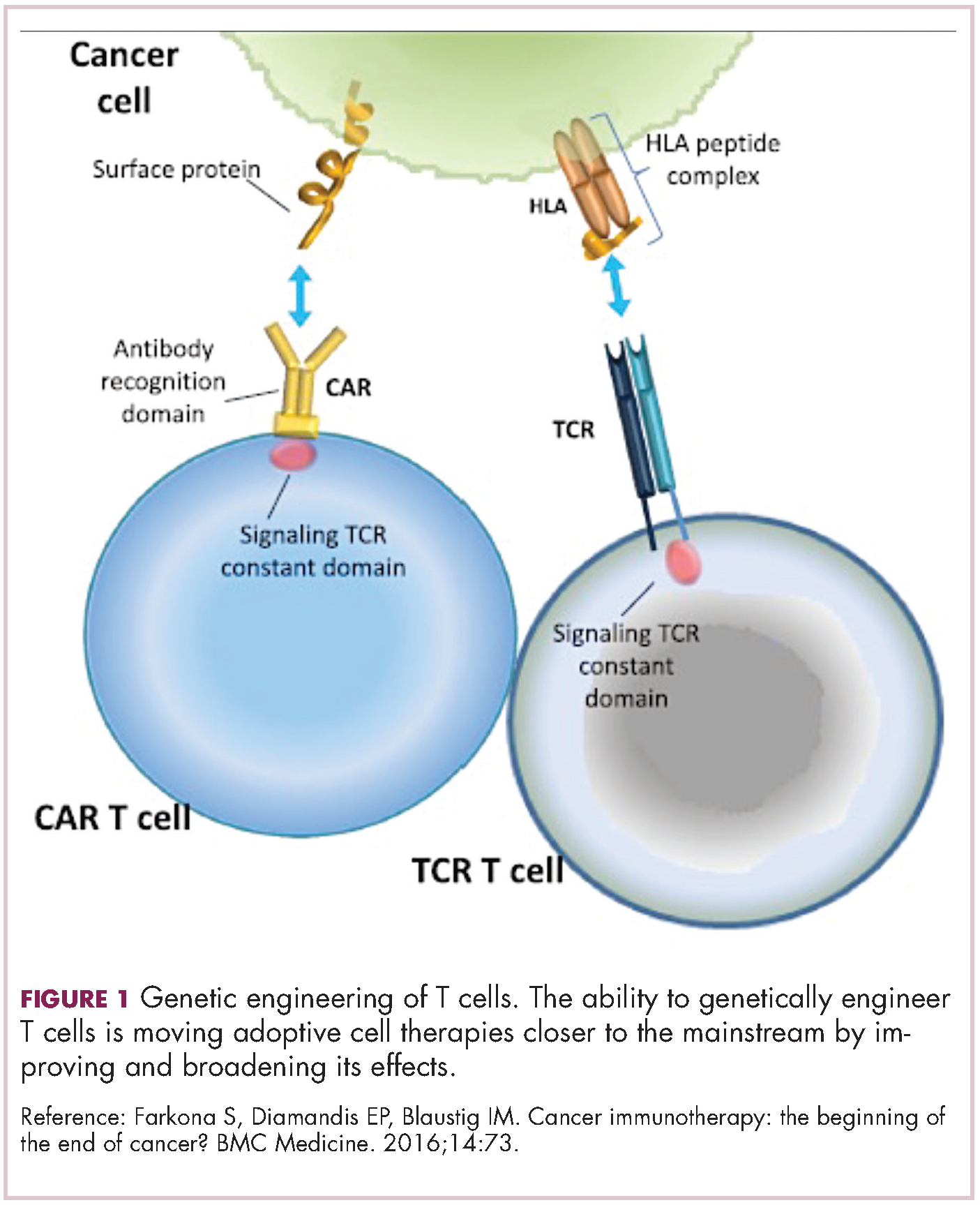
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17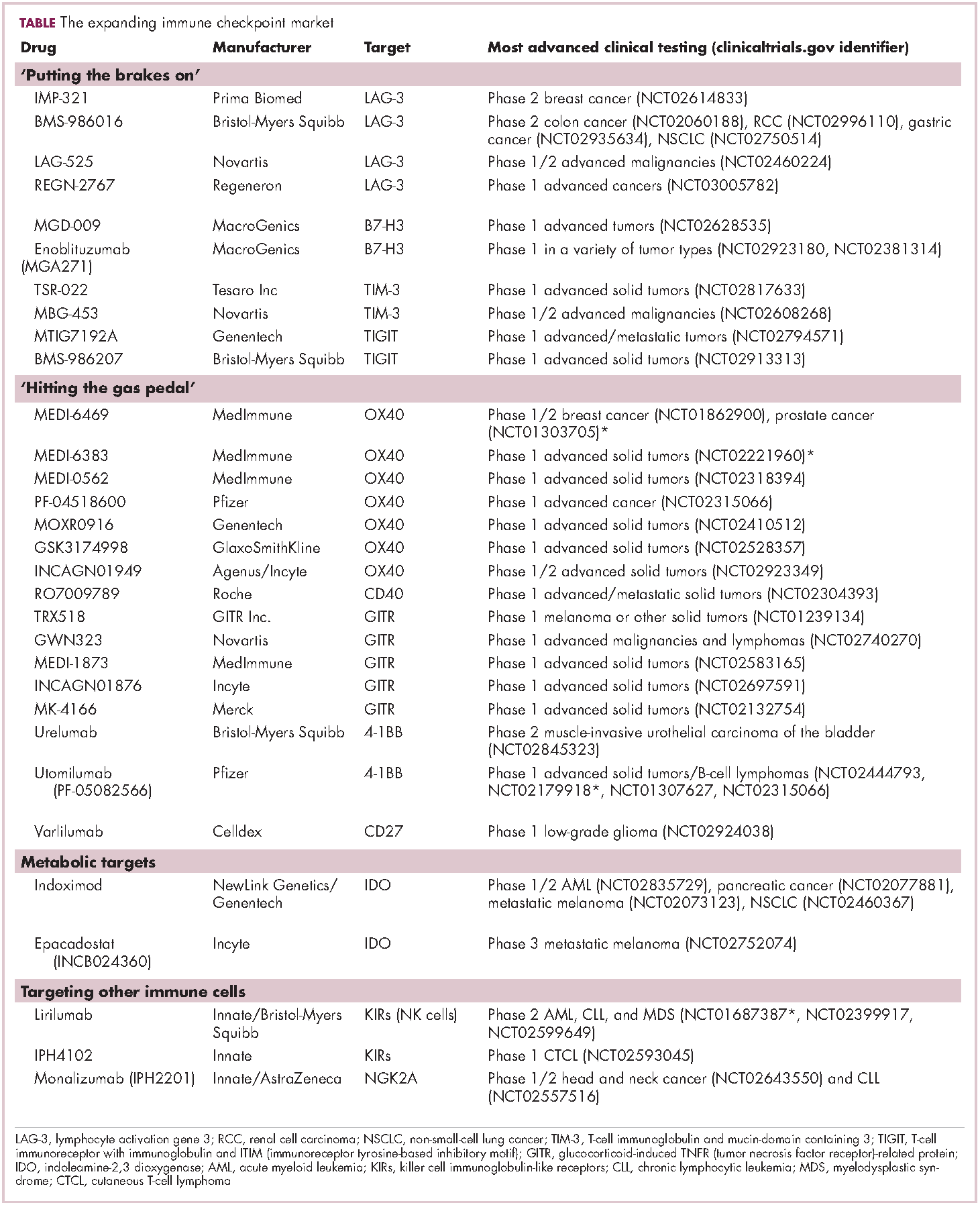
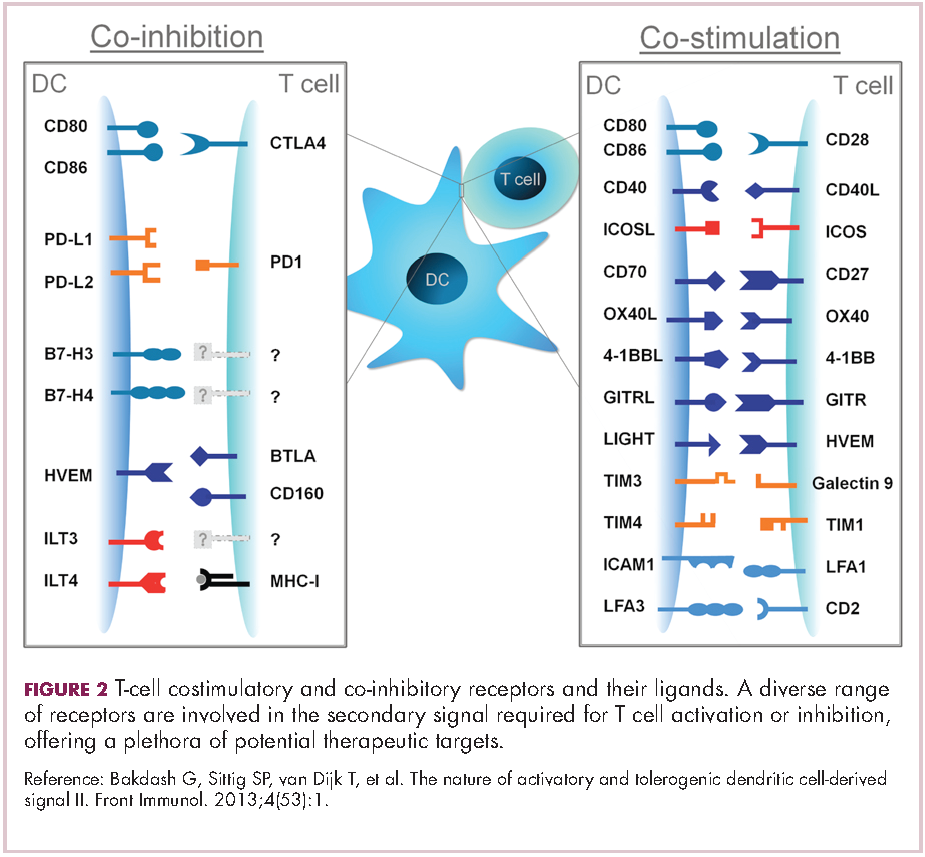
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).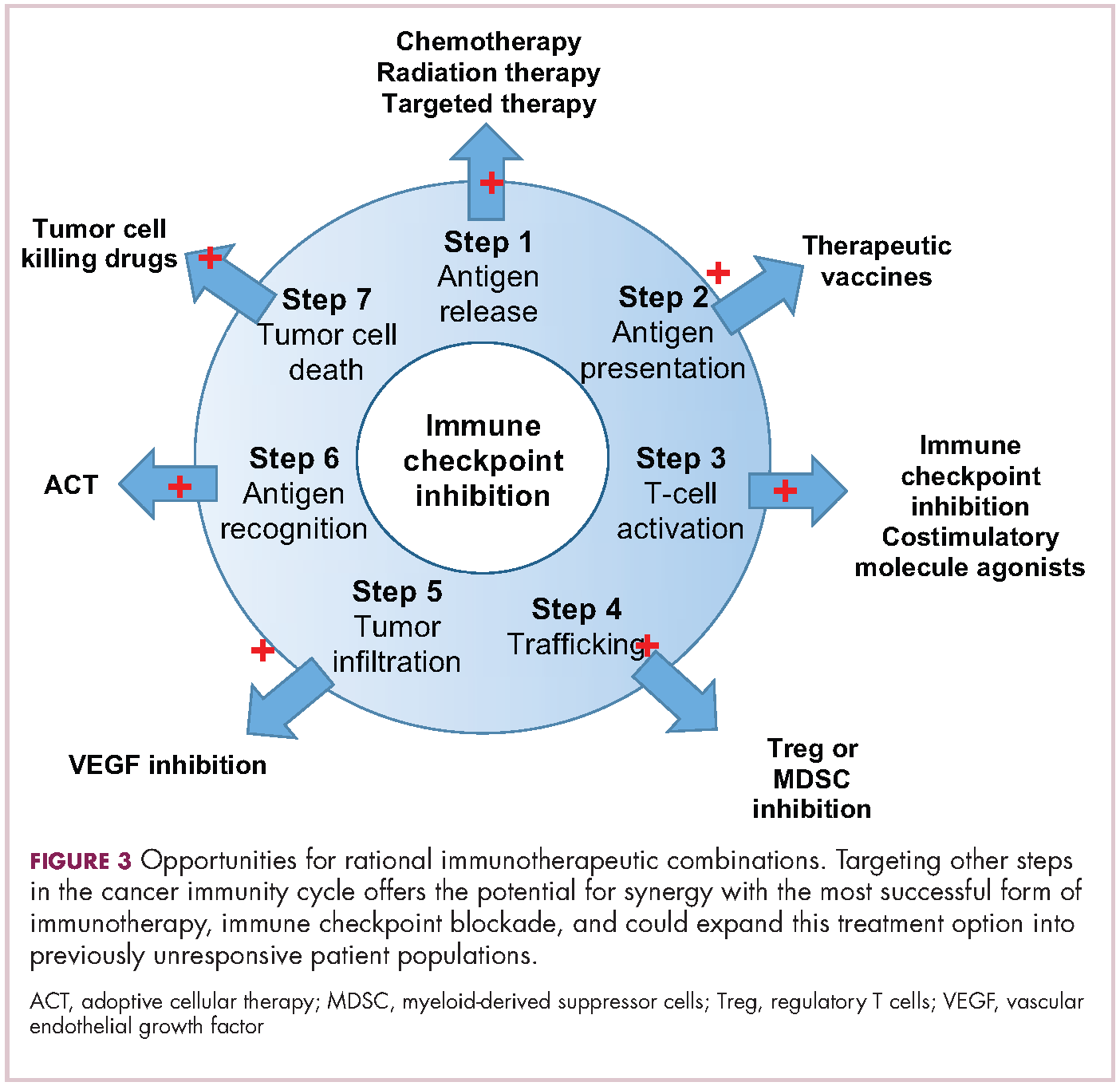
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
Immunotherapies shape the treatment landscape for hematologic malignancies
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.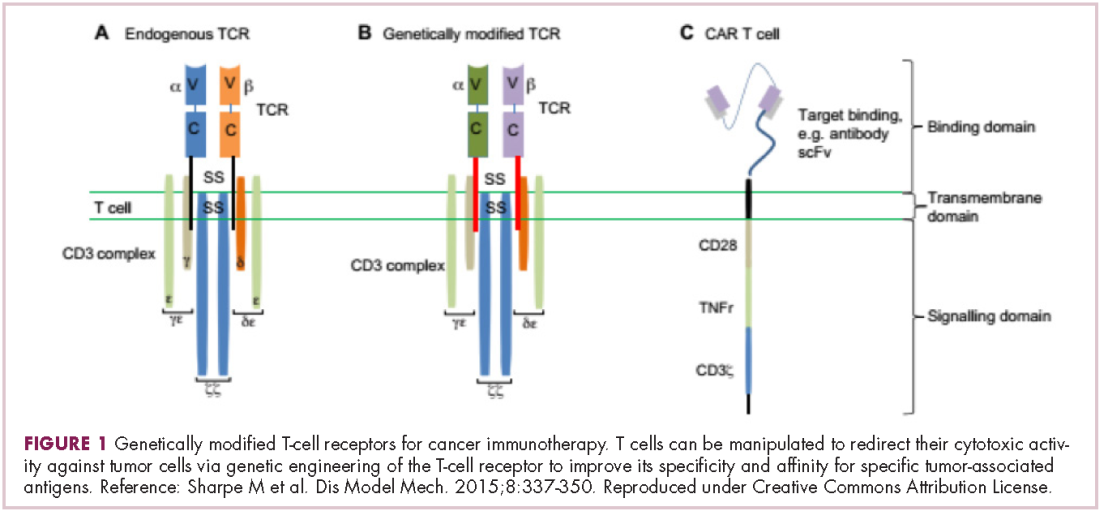
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5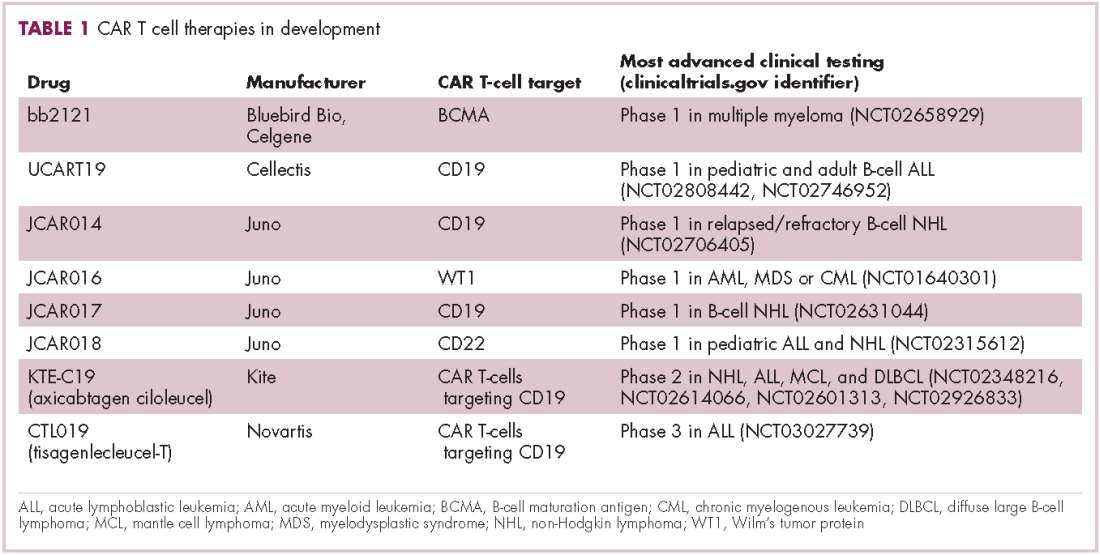
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12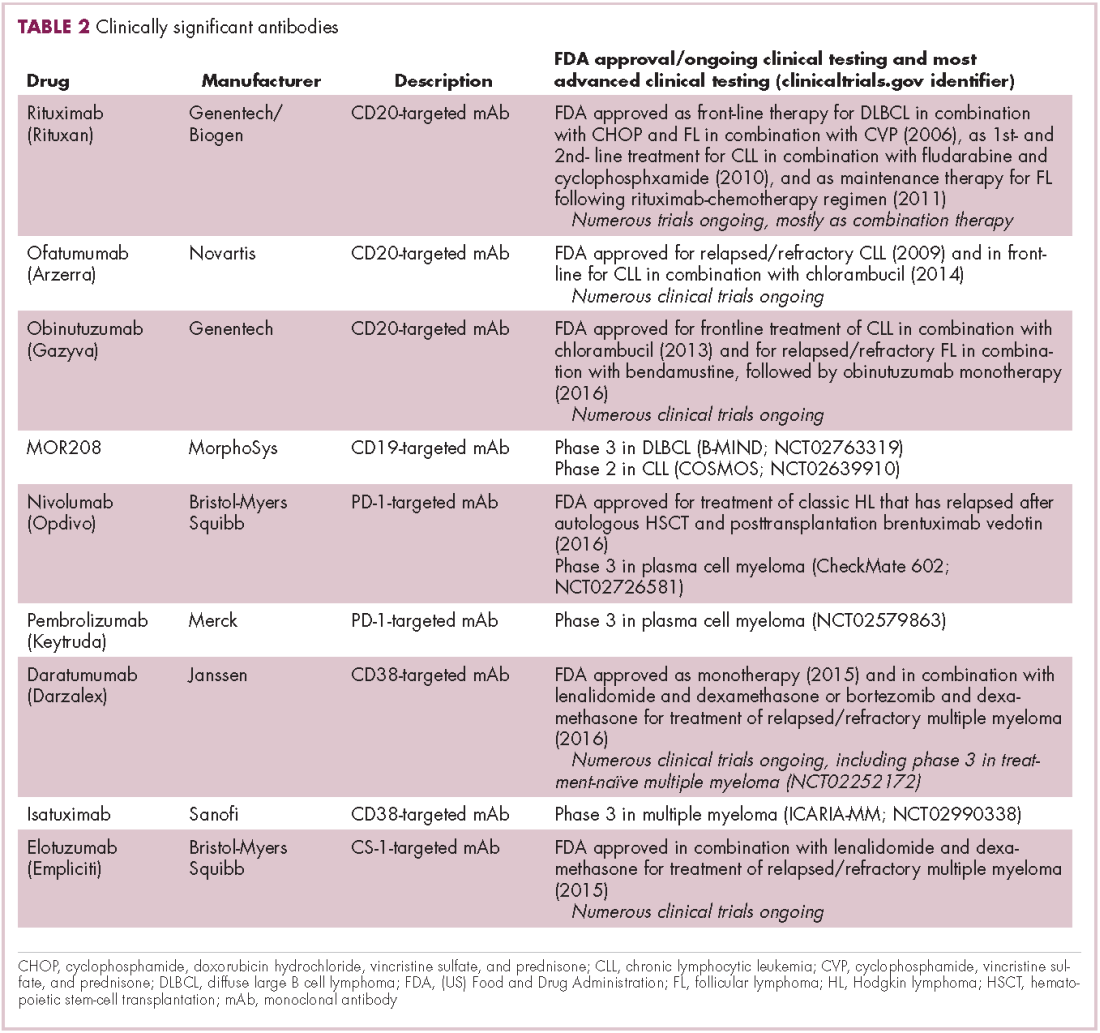
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20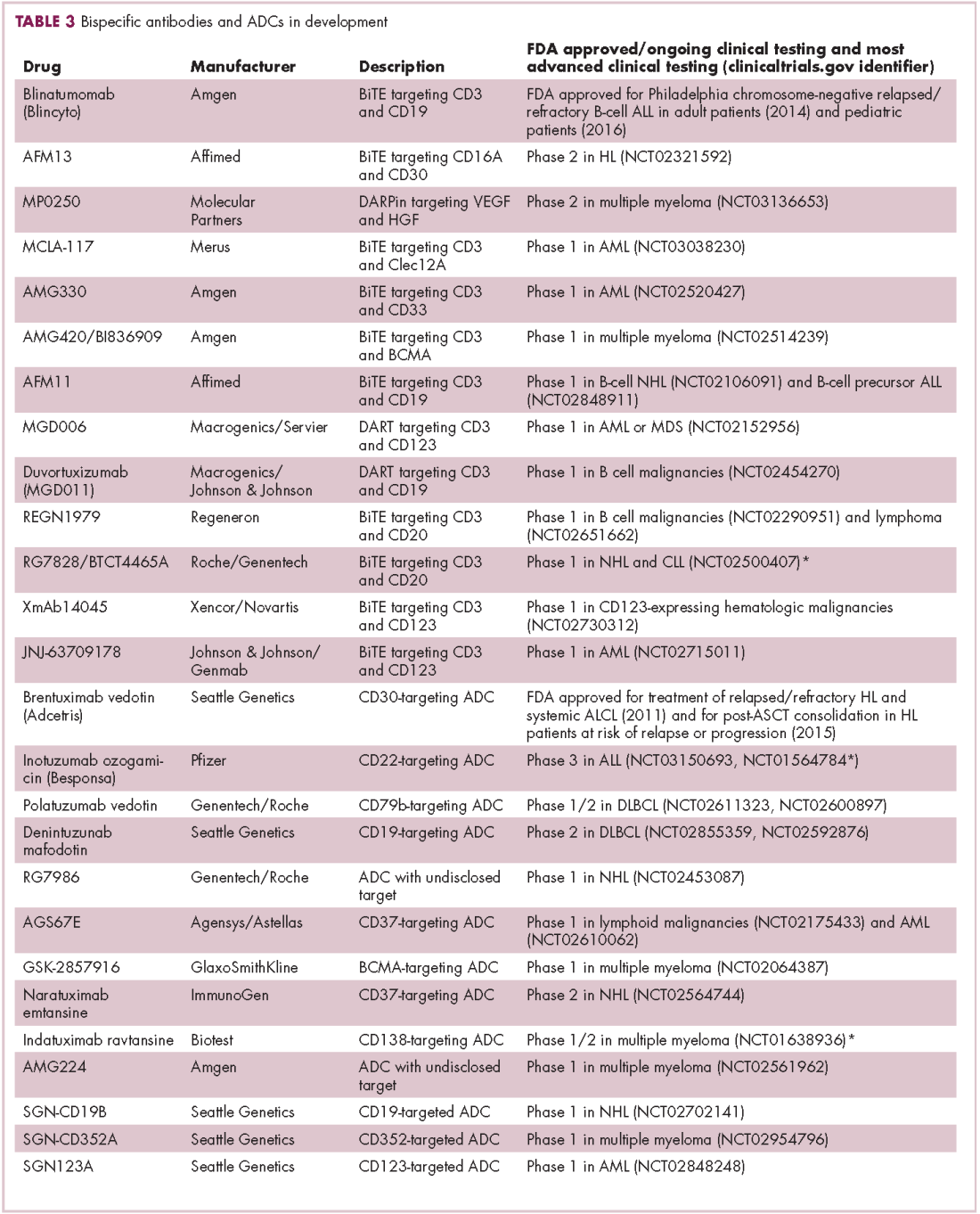
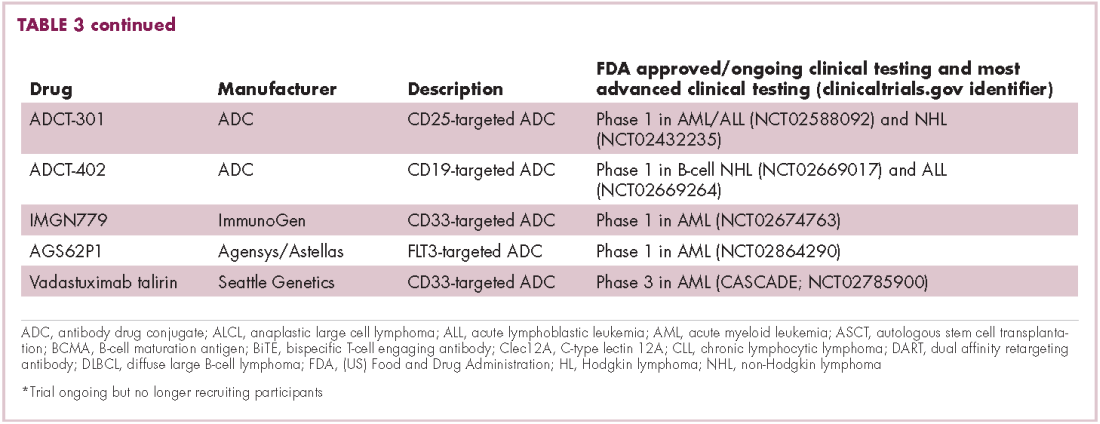
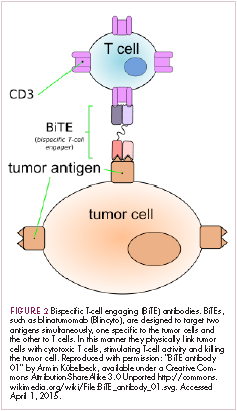
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.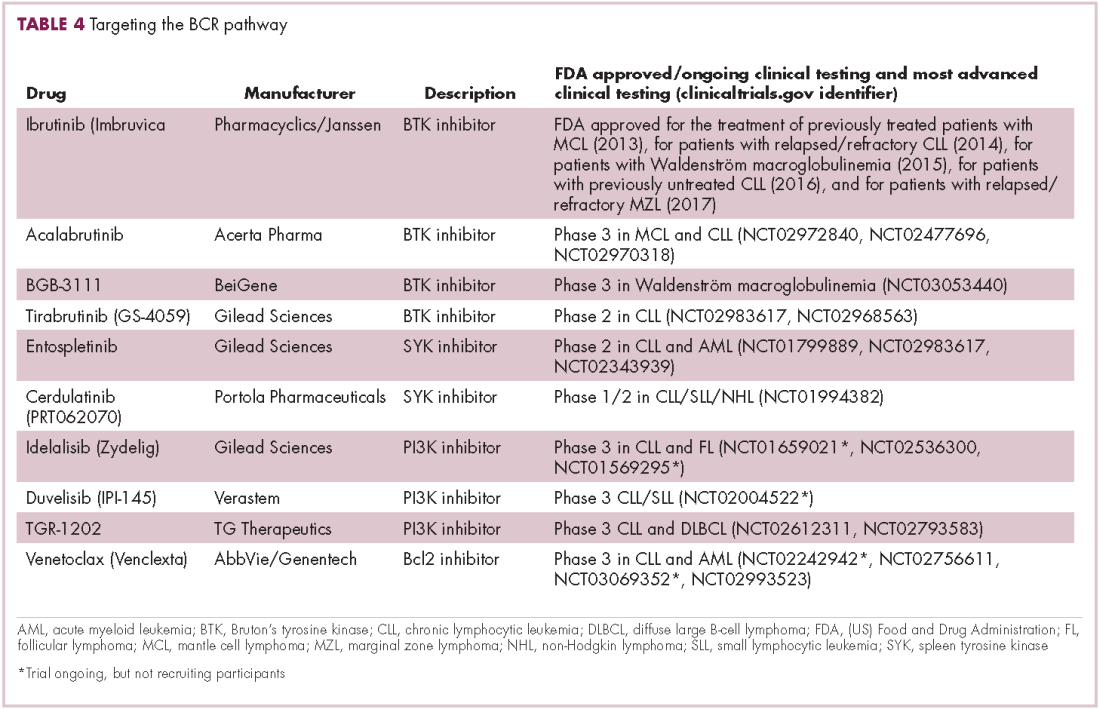
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
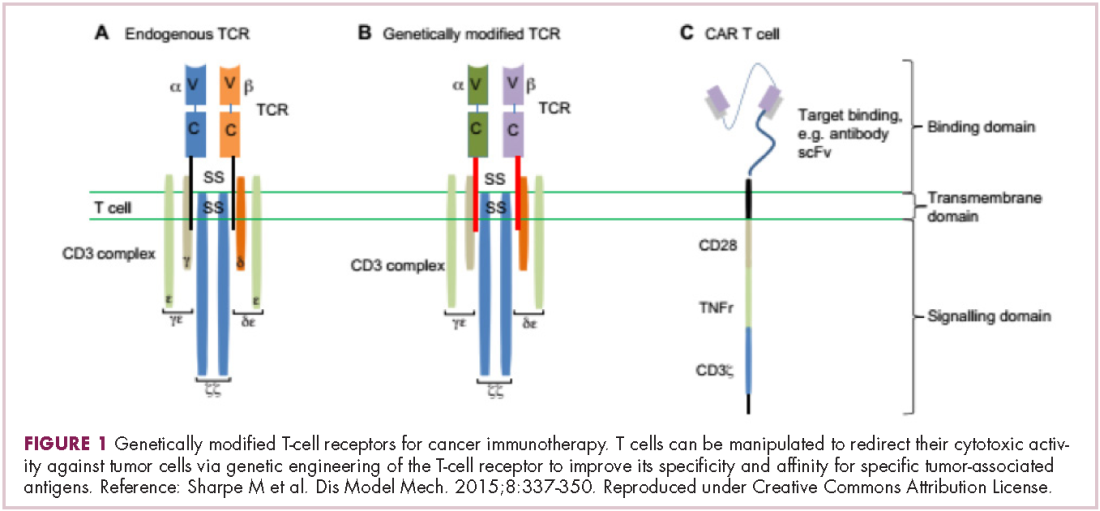
Groups release guidelines for CAR T treatment in children
emphasize the need for a flexible approach to detect early signs of serious complications for younger patients treated with this emerging class of medicines.

Researchers at the University of Texas MD Anderson Cancer Center, Houston, and the Pediatric Acute Lung Injury and Sepsis Investigators Network (PALISI) developed the guidelines, which were published in Nature Reviews Clinical Oncology. The recommendations build on the guidelines for more general use of these medicines from MD Anderson’s CARTOX Program, which Nature Reviews Clinical Oncology published in 2017.
Among the chief concerns with this new class of medicines are cytokine-release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES), according to Kris Michael Mahadeo, MD MPH, of the MD Anderson Cancer Center and his coauthors of the new paper.
Some of the tools used for older patients in screening for complications with CAR T drugs don’t work as well with younger ones, Dr. Mahadeo said in an interview. For instance, at MD Anderson, a handwriting sample is used to monitor patients for CAR T cell-related encephalopathy syndrome, which has symptoms of confusion and delirium. Patients provide a baseline handwriting sample of a single sentence that’s scanned into the medical record, and then they are asked to write this again during their time in the hospital, he said. But this tool may not work for children too young to write well.
The new guidelines suggest using the Cornell Assessment of Pediatric Delirium (CAPD) or to evaluate a child’s mental state, asking questions about eye contact, and level of awareness and mood, Dr. Mahadeo said. An alternative for patients aged 12 years and older with greater cognitive ability is the CARTOX-10 grading system.
“The nurses who spent most of the day with these patients will observe them over their shift and kind of get an idea of what was normal and answer a series of questions” through the CAPD tool, which is already used in ICUs, Dr. Mahadeo said. “It takes into consideration both the nurses’ perception and the parents, or whoever is at the bedside with the child. So that if they have a concern, it gives them a point that actually escalates things upward.”
The newly published recommendations also remind physicians and others caring for young patients to pay attention to these reports.
“Parent and/or caregiver concerns should be addressed because early signs or symptoms of CRS can be subtle and best recognized by those who know the child best,” Dr. Mahadeo and his colleagues wrote in a summary of key recommendations in the paper.
The recommendations also noted a need for close monitoring for complications such as hypotension, hypocalcemia, and catheter-related pain in young patients who require a leukapheresis catheter for cell collection. Infant and younger children “might not verbalize these symptoms,” according to the researchers.
Other recommendations include:
- Obtaining the child’s assent when appropriate, with psychological services often aiding in this goal. Dr. Mahadeo and his colleagues recommend considering “age-appropriate advance directives.”
- Maintaining high vigilance for sinus tachycardia as an early sign of CRS, using age-specific normal range or baseline values.
- Giving pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
- Considering participation with a prospective collaboration with intensive-care registries that could allow accurate data entry of cell-therapy variables into the Center for International Blood and Marrow Transplant Research registry by cell-therapy programs.
The Food and Drug Administration approved the first two CAR T-cell therapies in the United States in 2017: Novartis’ tisagenlecleucel (Kymriah) for children and young adults with B-cell precursor acute lymphoblastic leukemia and later for adults with large B-cell lymphoma; and axicabtagene ciloleucel (Yescarta), sold by Gilead, for adults with large B-cell lymphoma. The therapies involve reengineering a patient’s T cells such that they recognize the threat of cancer, and then introducing them back into the body. The European Medicines Agency’s Committee for Medicinal Products for Human Use in June recommended granting marketing authorization to these drugs.
In the new pediatric guidelines, Dr. Mahadeo and his colleagues noted the use of CAR T-cell therapies for treatment of solid tumors and other malignancies in children already “is being explored.” “Moreover, consideration of earlier or upfront use of CAR T-cell therapy might spare patients the acute and long-term toxicities associated with traditional chemotherapy and/or radiation regimens,” they wrote.

There’s been great interest in learning how to most safely use the CAR T cell therapies, said Helen Heslop, MD, of Baylor College of Medicine.
She pointed to a 2014 publication in the journal Blood from Daniel W. Lee and his colleagues as an earlier example of this research. By now, cancer centers will have worked out their own procedures for pediatric use of CAR T therapies, hewing to standards set by the Foundation for the Accreditation of Cellular Therapy (FACT), Dr. Heslop said.
Dr. Heslop also stressed the role of the FDA in requiring risk evaluation and management strategy programs for these drugs. All of this, including the new guidelines from Dr. Mahadeo and his colleagues, is part of a growing body of research into safe use of CAR T therapies, Dr. Heslop said.
“It’s an active area of research,” she said. “Most centers will look at all of it and then develop what works best in their own individual center for providing the best care for the patients.”
The newly published guidelines could prove an “important contribution” to managing the risk of CAR T therapies, Phyllis I. Warkentin, MD, chief medical officer for FACT, said in an interview, while stressing that they were not more or less important than other similar efforts. Physicians learning how to use the CAR T therapies may welcome new input, as most of what’s been published has been about adults, she said.
“You don’t have the luxury of a lot of time to be learning on the job, so to speak,” with CAR T therapies, she said. “Many of the toxicities are fairly severe and fairly sudden.”
Dr. Heslop has been on advisory board for Gilead and Novartis. Dr. Warkentin and Dr. Mahadeo each reported having no financial disclosures. Other authors of the guidelines paper reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
SOURCE: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
emphasize the need for a flexible approach to detect early signs of serious complications for younger patients treated with this emerging class of medicines.
Researchers at the University of Texas MD Anderson Cancer Center, Houston, and the Pediatric Acute Lung Injury and Sepsis Investigators Network (PALISI) developed the guidelines, which were published in Nature Reviews Clinical Oncology. The recommendations build on the guidelines for more general use of these medicines from MD Anderson’s CARTOX Program, which Nature Reviews Clinical Oncology published in 2017.
Among the chief concerns with this new class of medicines are cytokine-release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES), according to Kris Michael Mahadeo, MD MPH, of the MD Anderson Cancer Center and his coauthors of the new paper.
Some of the tools used for older patients in screening for complications with CAR T drugs don’t work as well with younger ones, Dr. Mahadeo said in an interview. For instance, at MD Anderson, a handwriting sample is used to monitor patients for CAR T cell-related encephalopathy syndrome, which has symptoms of confusion and delirium. Patients provide a baseline handwriting sample of a single sentence that’s scanned into the medical record, and then they are asked to write this again during their time in the hospital, he said. But this tool may not work for children too young to write well.
The new guidelines suggest using the Cornell Assessment of Pediatric Delirium (CAPD) or to evaluate a child’s mental state, asking questions about eye contact, and level of awareness and mood, Dr. Mahadeo said. An alternative for patients aged 12 years and older with greater cognitive ability is the CARTOX-10 grading system.
“The nurses who spent most of the day with these patients will observe them over their shift and kind of get an idea of what was normal and answer a series of questions” through the CAPD tool, which is already used in ICUs, Dr. Mahadeo said. “It takes into consideration both the nurses’ perception and the parents, or whoever is at the bedside with the child. So that if they have a concern, it gives them a point that actually escalates things upward.”
The newly published recommendations also remind physicians and others caring for young patients to pay attention to these reports.
“Parent and/or caregiver concerns should be addressed because early signs or symptoms of CRS can be subtle and best recognized by those who know the child best,” Dr. Mahadeo and his colleagues wrote in a summary of key recommendations in the paper.
The recommendations also noted a need for close monitoring for complications such as hypotension, hypocalcemia, and catheter-related pain in young patients who require a leukapheresis catheter for cell collection. Infant and younger children “might not verbalize these symptoms,” according to the researchers.
Other recommendations include:
- Obtaining the child’s assent when appropriate, with psychological services often aiding in this goal. Dr. Mahadeo and his colleagues recommend considering “age-appropriate advance directives.”
- Maintaining high vigilance for sinus tachycardia as an early sign of CRS, using age-specific normal range or baseline values.
- Giving pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
- Considering participation with a prospective collaboration with intensive-care registries that could allow accurate data entry of cell-therapy variables into the Center for International Blood and Marrow Transplant Research registry by cell-therapy programs.
The Food and Drug Administration approved the first two CAR T-cell therapies in the United States in 2017: Novartis’ tisagenlecleucel (Kymriah) for children and young adults with B-cell precursor acute lymphoblastic leukemia and later for adults with large B-cell lymphoma; and axicabtagene ciloleucel (Yescarta), sold by Gilead, for adults with large B-cell lymphoma. The therapies involve reengineering a patient’s T cells such that they recognize the threat of cancer, and then introducing them back into the body. The European Medicines Agency’s Committee for Medicinal Products for Human Use in June recommended granting marketing authorization to these drugs.
In the new pediatric guidelines, Dr. Mahadeo and his colleagues noted the use of CAR T-cell therapies for treatment of solid tumors and other malignancies in children already “is being explored.” “Moreover, consideration of earlier or upfront use of CAR T-cell therapy might spare patients the acute and long-term toxicities associated with traditional chemotherapy and/or radiation regimens,” they wrote.
There’s been great interest in learning how to most safely use the CAR T cell therapies, said Helen Heslop, MD, of Baylor College of Medicine.
She pointed to a 2014 publication in the journal Blood from Daniel W. Lee and his colleagues as an earlier example of this research. By now, cancer centers will have worked out their own procedures for pediatric use of CAR T therapies, hewing to standards set by the Foundation for the Accreditation of Cellular Therapy (FACT), Dr. Heslop said.
Dr. Heslop also stressed the role of the FDA in requiring risk evaluation and management strategy programs for these drugs. All of this, including the new guidelines from Dr. Mahadeo and his colleagues, is part of a growing body of research into safe use of CAR T therapies, Dr. Heslop said.
“It’s an active area of research,” she said. “Most centers will look at all of it and then develop what works best in their own individual center for providing the best care for the patients.”
The newly published guidelines could prove an “important contribution” to managing the risk of CAR T therapies, Phyllis I. Warkentin, MD, chief medical officer for FACT, said in an interview, while stressing that they were not more or less important than other similar efforts. Physicians learning how to use the CAR T therapies may welcome new input, as most of what’s been published has been about adults, she said.
“You don’t have the luxury of a lot of time to be learning on the job, so to speak,” with CAR T therapies, she said. “Many of the toxicities are fairly severe and fairly sudden.”
Dr. Heslop has been on advisory board for Gilead and Novartis. Dr. Warkentin and Dr. Mahadeo each reported having no financial disclosures. Other authors of the guidelines paper reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
SOURCE: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
emphasize the need for a flexible approach to detect early signs of serious complications for younger patients treated with this emerging class of medicines.
Researchers at the University of Texas MD Anderson Cancer Center, Houston, and the Pediatric Acute Lung Injury and Sepsis Investigators Network (PALISI) developed the guidelines, which were published in Nature Reviews Clinical Oncology. The recommendations build on the guidelines for more general use of these medicines from MD Anderson’s CARTOX Program, which Nature Reviews Clinical Oncology published in 2017.
Among the chief concerns with this new class of medicines are cytokine-release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES), according to Kris Michael Mahadeo, MD MPH, of the MD Anderson Cancer Center and his coauthors of the new paper.
Some of the tools used for older patients in screening for complications with CAR T drugs don’t work as well with younger ones, Dr. Mahadeo said in an interview. For instance, at MD Anderson, a handwriting sample is used to monitor patients for CAR T cell-related encephalopathy syndrome, which has symptoms of confusion and delirium. Patients provide a baseline handwriting sample of a single sentence that’s scanned into the medical record, and then they are asked to write this again during their time in the hospital, he said. But this tool may not work for children too young to write well.
The new guidelines suggest using the Cornell Assessment of Pediatric Delirium (CAPD) or to evaluate a child’s mental state, asking questions about eye contact, and level of awareness and mood, Dr. Mahadeo said. An alternative for patients aged 12 years and older with greater cognitive ability is the CARTOX-10 grading system.
“The nurses who spent most of the day with these patients will observe them over their shift and kind of get an idea of what was normal and answer a series of questions” through the CAPD tool, which is already used in ICUs, Dr. Mahadeo said. “It takes into consideration both the nurses’ perception and the parents, or whoever is at the bedside with the child. So that if they have a concern, it gives them a point that actually escalates things upward.”
The newly published recommendations also remind physicians and others caring for young patients to pay attention to these reports.
“Parent and/or caregiver concerns should be addressed because early signs or symptoms of CRS can be subtle and best recognized by those who know the child best,” Dr. Mahadeo and his colleagues wrote in a summary of key recommendations in the paper.
The recommendations also noted a need for close monitoring for complications such as hypotension, hypocalcemia, and catheter-related pain in young patients who require a leukapheresis catheter for cell collection. Infant and younger children “might not verbalize these symptoms,” according to the researchers.
Other recommendations include:
- Obtaining the child’s assent when appropriate, with psychological services often aiding in this goal. Dr. Mahadeo and his colleagues recommend considering “age-appropriate advance directives.”
- Maintaining high vigilance for sinus tachycardia as an early sign of CRS, using age-specific normal range or baseline values.
- Giving pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
- Considering participation with a prospective collaboration with intensive-care registries that could allow accurate data entry of cell-therapy variables into the Center for International Blood and Marrow Transplant Research registry by cell-therapy programs.
The Food and Drug Administration approved the first two CAR T-cell therapies in the United States in 2017: Novartis’ tisagenlecleucel (Kymriah) for children and young adults with B-cell precursor acute lymphoblastic leukemia and later for adults with large B-cell lymphoma; and axicabtagene ciloleucel (Yescarta), sold by Gilead, for adults with large B-cell lymphoma. The therapies involve reengineering a patient’s T cells such that they recognize the threat of cancer, and then introducing them back into the body. The European Medicines Agency’s Committee for Medicinal Products for Human Use in June recommended granting marketing authorization to these drugs.
In the new pediatric guidelines, Dr. Mahadeo and his colleagues noted the use of CAR T-cell therapies for treatment of solid tumors and other malignancies in children already “is being explored.” “Moreover, consideration of earlier or upfront use of CAR T-cell therapy might spare patients the acute and long-term toxicities associated with traditional chemotherapy and/or radiation regimens,” they wrote.
There’s been great interest in learning how to most safely use the CAR T cell therapies, said Helen Heslop, MD, of Baylor College of Medicine.
She pointed to a 2014 publication in the journal Blood from Daniel W. Lee and his colleagues as an earlier example of this research. By now, cancer centers will have worked out their own procedures for pediatric use of CAR T therapies, hewing to standards set by the Foundation for the Accreditation of Cellular Therapy (FACT), Dr. Heslop said.
Dr. Heslop also stressed the role of the FDA in requiring risk evaluation and management strategy programs for these drugs. All of this, including the new guidelines from Dr. Mahadeo and his colleagues, is part of a growing body of research into safe use of CAR T therapies, Dr. Heslop said.
“It’s an active area of research,” she said. “Most centers will look at all of it and then develop what works best in their own individual center for providing the best care for the patients.”
The newly published guidelines could prove an “important contribution” to managing the risk of CAR T therapies, Phyllis I. Warkentin, MD, chief medical officer for FACT, said in an interview, while stressing that they were not more or less important than other similar efforts. Physicians learning how to use the CAR T therapies may welcome new input, as most of what’s been published has been about adults, she said.
“You don’t have the luxury of a lot of time to be learning on the job, so to speak,” with CAR T therapies, she said. “Many of the toxicities are fairly severe and fairly sudden.”
Dr. Heslop has been on advisory board for Gilead and Novartis. Dr. Warkentin and Dr. Mahadeo each reported having no financial disclosures. Other authors of the guidelines paper reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
SOURCE: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
FROM NATURE REVIEWS CLINICAL ONCOLOGY
Key clinical point: Multidisciplinary approach aids in managing CAR T-cell therapy’s severe potential toxicities in children.
Major finding: The guideline calls for pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
Study details: Consensus guidelines on the care of children receiving CAR T-cell therapy from the Pediatric Acute Lung Injury and Sepsis Investigators and the MD Anderson Cancer Center CARTOX program.
Disclosures: Dr. Mahadeo reported having no financial disclosures. Other coauthors reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
Source: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
Melanoma survival shorter in those given high dose glucocorticoids for ipilimumab-induced hypophysitis
than did patients taking low-dose steroids for the adverse event, according to a new retrospective analysis in Cancer.
“Treatment with high-dose glucocorticoids does not appear to confer any obvious advantage to patients with IH (ipilimumab-induced hypophysitis) and may negatively affect tumor response to CPI (checkpoint-inhibitor therapy),” wrote Alexander Faje, MD, a neuroendocrinologist at Massachusetts General Hospital, Boston. “We recommend against the routine use of higher doses in these patients and that such treatment should be reserved for clinical indications like visual compromise or perhaps for intractable headache.”
Hypophysitis after treatment with a CTLA-4 inhibitor, such as ipilimumab, can approach 12%, though it is much less common with the checkpoint inhibitors that target PD-1 and PD-L1. Past studies examining the effects of glucocorticoids for immune-related adverse events have compared patients with severe events to those with minimal or no events. Since emergence of hypophysitis correlates with better overall survival, this is a flawed approach, the researchers said.
For their study, the researchers compared groups of patients with the same immune-related adverse events who received treatment with varying amounts of glucocorticoids.
They reviewed outcomes for 64 melanoma patients who had received ipilimumab monotherapy and were diagnosed with ipilimumab-induced hypophysitis treated in the Partners Healthcare system. Fourteen patients had received low-dose glucocorticoids, defined as a maximum average daily dose of 7.5 mg of prednisone or the equivalent. Fifty patients received high-dose glucocorticoids, defined as anything above that amount.
Overall survival and time to treatment failure were significantly higher in the low-dose group than the high-dose group (P = .002 for OS and P = .001 for TTF). Median overall survival was 23.3 months and time to treatment failure was 11.4 months in those given high-dose steroids. Median overall survival had not been reached in those given low-dose steroids.
While the findings are preliminary, the authors noted they may have implications for managing other immune-related adverse events. “Although the use of lower doses of immunosuppressive medications may be less of an option in many circumstances for other (immune-related adverse events), therapeutic parsimony would seem desirable with more tailored regimens as the biologic mechanisms underpinning these processes are further elucidated.”
SOURCE: Faje AT et al. Cancer. 2018 Jul 5.
The study results provide further evidence that hypophysitis appears to be an adverse effect of ipilimumab therapy that is linked with improved outcomes in melanoma. As hypophysitis tends to be self-limited, it can be treated safely with replacement therapy rather than high-dose steroid therapy, which was associated with reduced overall survival.
But the low number of patients on low-dose glucocorticoids – just 14 – is a limitation of the study and the group’s favorable outcomes could have been due to chance alone. Further, the 7.5-mg cut-off for high- vs. low-dose steroid therapy is somewhat arbitrary.
In support of the study’s overall conclusions, however, exploratory analyses have produced similar findings at somewhat higher cut-offs.
As the mechanism of hypophysitis is somewhat distinct compared with other immune-checkpoint toxicities, it might not be appropriate to generalize these findings to other toxic responses to these drugs.
The mechanisms of toxicities related to immune-checkpoint inhibitors are still not well defined. Unraveling those mechanisms may identify patients at high risk and hold the potential for aiding in the design of novel therapeutics that unleash antitumor immunity. Glucocorticoids are a fairly effective treatment for immune-related toxicities but remain a blunt, nonspecific way to suppress aberrant immunity. Designing inhibitors of culprit cellular populations or cytokines may combat toxicity without compromising the efficacy of immune therapy or promoting systemic immunosuppression.
Douglas B. Johnson, MD, is with Vanderbilt University Medical Center and Vanderbilt Ingram Cancer Center, Nashville, Tenn. He made his remarks in an editorial (Cancer 2018 Jul 5. doi: 10.1002/cncr.31627.
The study results provide further evidence that hypophysitis appears to be an adverse effect of ipilimumab therapy that is linked with improved outcomes in melanoma. As hypophysitis tends to be self-limited, it can be treated safely with replacement therapy rather than high-dose steroid therapy, which was associated with reduced overall survival.
But the low number of patients on low-dose glucocorticoids – just 14 – is a limitation of the study and the group’s favorable outcomes could have been due to chance alone. Further, the 7.5-mg cut-off for high- vs. low-dose steroid therapy is somewhat arbitrary.
In support of the study’s overall conclusions, however, exploratory analyses have produced similar findings at somewhat higher cut-offs.
As the mechanism of hypophysitis is somewhat distinct compared with other immune-checkpoint toxicities, it might not be appropriate to generalize these findings to other toxic responses to these drugs.
The mechanisms of toxicities related to immune-checkpoint inhibitors are still not well defined. Unraveling those mechanisms may identify patients at high risk and hold the potential for aiding in the design of novel therapeutics that unleash antitumor immunity. Glucocorticoids are a fairly effective treatment for immune-related toxicities but remain a blunt, nonspecific way to suppress aberrant immunity. Designing inhibitors of culprit cellular populations or cytokines may combat toxicity without compromising the efficacy of immune therapy or promoting systemic immunosuppression.
Douglas B. Johnson, MD, is with Vanderbilt University Medical Center and Vanderbilt Ingram Cancer Center, Nashville, Tenn. He made his remarks in an editorial (Cancer 2018 Jul 5. doi: 10.1002/cncr.31627.
The study results provide further evidence that hypophysitis appears to be an adverse effect of ipilimumab therapy that is linked with improved outcomes in melanoma. As hypophysitis tends to be self-limited, it can be treated safely with replacement therapy rather than high-dose steroid therapy, which was associated with reduced overall survival.
But the low number of patients on low-dose glucocorticoids – just 14 – is a limitation of the study and the group’s favorable outcomes could have been due to chance alone. Further, the 7.5-mg cut-off for high- vs. low-dose steroid therapy is somewhat arbitrary.
In support of the study’s overall conclusions, however, exploratory analyses have produced similar findings at somewhat higher cut-offs.
As the mechanism of hypophysitis is somewhat distinct compared with other immune-checkpoint toxicities, it might not be appropriate to generalize these findings to other toxic responses to these drugs.
The mechanisms of toxicities related to immune-checkpoint inhibitors are still not well defined. Unraveling those mechanisms may identify patients at high risk and hold the potential for aiding in the design of novel therapeutics that unleash antitumor immunity. Glucocorticoids are a fairly effective treatment for immune-related toxicities but remain a blunt, nonspecific way to suppress aberrant immunity. Designing inhibitors of culprit cellular populations or cytokines may combat toxicity without compromising the efficacy of immune therapy or promoting systemic immunosuppression.
Douglas B. Johnson, MD, is with Vanderbilt University Medical Center and Vanderbilt Ingram Cancer Center, Nashville, Tenn. He made his remarks in an editorial (Cancer 2018 Jul 5. doi: 10.1002/cncr.31627.
than did patients taking low-dose steroids for the adverse event, according to a new retrospective analysis in Cancer.
“Treatment with high-dose glucocorticoids does not appear to confer any obvious advantage to patients with IH (ipilimumab-induced hypophysitis) and may negatively affect tumor response to CPI (checkpoint-inhibitor therapy),” wrote Alexander Faje, MD, a neuroendocrinologist at Massachusetts General Hospital, Boston. “We recommend against the routine use of higher doses in these patients and that such treatment should be reserved for clinical indications like visual compromise or perhaps for intractable headache.”
Hypophysitis after treatment with a CTLA-4 inhibitor, such as ipilimumab, can approach 12%, though it is much less common with the checkpoint inhibitors that target PD-1 and PD-L1. Past studies examining the effects of glucocorticoids for immune-related adverse events have compared patients with severe events to those with minimal or no events. Since emergence of hypophysitis correlates with better overall survival, this is a flawed approach, the researchers said.
For their study, the researchers compared groups of patients with the same immune-related adverse events who received treatment with varying amounts of glucocorticoids.
They reviewed outcomes for 64 melanoma patients who had received ipilimumab monotherapy and were diagnosed with ipilimumab-induced hypophysitis treated in the Partners Healthcare system. Fourteen patients had received low-dose glucocorticoids, defined as a maximum average daily dose of 7.5 mg of prednisone or the equivalent. Fifty patients received high-dose glucocorticoids, defined as anything above that amount.
Overall survival and time to treatment failure were significantly higher in the low-dose group than the high-dose group (P = .002 for OS and P = .001 for TTF). Median overall survival was 23.3 months and time to treatment failure was 11.4 months in those given high-dose steroids. Median overall survival had not been reached in those given low-dose steroids.
While the findings are preliminary, the authors noted they may have implications for managing other immune-related adverse events. “Although the use of lower doses of immunosuppressive medications may be less of an option in many circumstances for other (immune-related adverse events), therapeutic parsimony would seem desirable with more tailored regimens as the biologic mechanisms underpinning these processes are further elucidated.”
SOURCE: Faje AT et al. Cancer. 2018 Jul 5.
than did patients taking low-dose steroids for the adverse event, according to a new retrospective analysis in Cancer.
“Treatment with high-dose glucocorticoids does not appear to confer any obvious advantage to patients with IH (ipilimumab-induced hypophysitis) and may negatively affect tumor response to CPI (checkpoint-inhibitor therapy),” wrote Alexander Faje, MD, a neuroendocrinologist at Massachusetts General Hospital, Boston. “We recommend against the routine use of higher doses in these patients and that such treatment should be reserved for clinical indications like visual compromise or perhaps for intractable headache.”
Hypophysitis after treatment with a CTLA-4 inhibitor, such as ipilimumab, can approach 12%, though it is much less common with the checkpoint inhibitors that target PD-1 and PD-L1. Past studies examining the effects of glucocorticoids for immune-related adverse events have compared patients with severe events to those with minimal or no events. Since emergence of hypophysitis correlates with better overall survival, this is a flawed approach, the researchers said.
For their study, the researchers compared groups of patients with the same immune-related adverse events who received treatment with varying amounts of glucocorticoids.
They reviewed outcomes for 64 melanoma patients who had received ipilimumab monotherapy and were diagnosed with ipilimumab-induced hypophysitis treated in the Partners Healthcare system. Fourteen patients had received low-dose glucocorticoids, defined as a maximum average daily dose of 7.5 mg of prednisone or the equivalent. Fifty patients received high-dose glucocorticoids, defined as anything above that amount.
Overall survival and time to treatment failure were significantly higher in the low-dose group than the high-dose group (P = .002 for OS and P = .001 for TTF). Median overall survival was 23.3 months and time to treatment failure was 11.4 months in those given high-dose steroids. Median overall survival had not been reached in those given low-dose steroids.
While the findings are preliminary, the authors noted they may have implications for managing other immune-related adverse events. “Although the use of lower doses of immunosuppressive medications may be less of an option in many circumstances for other (immune-related adverse events), therapeutic parsimony would seem desirable with more tailored regimens as the biologic mechanisms underpinning these processes are further elucidated.”
SOURCE: Faje AT et al. Cancer. 2018 Jul 5.
FROM CANCER
Key clinical point: Significantly lower overall survival and shorter time to treatment failure was seen in melanoma patients taking higher doses of glucocorticoids for ipilimumab-induced hypophysitis than in those taking lower doses.
Major finding: Median overall survival was 23.3 months and time to treatment failure was 11.4 months in those given high-dose steroids. Median overall survival had not been reached in those given low-dose steroids.
Study details: A retrospective review of 64 melanoma patients on single-agent ipilimumab therapy who were given glucocorticoids for ipilimumab-induced hypophysitis.
Disclosures: No funding source disclosed. The authors made no disclosures related to the submitted work.
Source: Faje AT et al. Cancer 2018 Jul 5.
Checkpoint inhibitors in autoimmune disease: More flares, better cancer outcomes
AMSTERDAM – In patients with autoimmune diseases, cancer treatment with checkpoint inhibitor immunotherapy increases the risk of flares, but these flares are associated with improved cancer outcomes, according to a multicenter, retrospective study presented at the European Congress of Rheumatology.
“Survival was longer in patients who experienced a flare of their preexisting autoimmune disease or any other immune-related adverse event, but this gain was lost if an immunosuppressive therapy was used,” reported Alice Tison, a resident in rheumatology at the Centre Hospitalier Universitaire, Brest, France.

These were some of the mixed messages from this evaluation, which involved 112 patients with preexisting autoimmune disease (PAD) whose data were collected from 11 tertiary care centers in France. Of the cases of PAD represented, the majority involved joint diseases, including psoriatic arthritis (28%), rheumatoid arthritis (18%), and spondyloarthritis (4.5%). However, other types of PAD, including inflammatory bowel disease (13%), were included in the series.
Only 33% of the patients had active disease at the time that checkpoint inhibitor therapy was initiated, and only 21% were taking an immunosuppressive therapy for their disease. Of those on therapy, the majority were taking steroids, but about a third of those on therapy were taking a disease-modifying antirheumatic drug, such as methotrexate.
With the initiation of checkpoint inhibitors, which were offered primarily for the treatment of melanoma (59%) and non–small cell lung cancer (36%), 42% of patients with PAD developed a disease flare. Of these, 30% were considered severe. Other immune-related events not considered related to the underlying disease, such as colitis, were also observed but at rates not clearly different than those observed in patients without PAD.
The activity of checkpoint inhibitors did not appear to be different than that observed in non-PAD patients. For example, the overall response rate was 48% in those with melanoma and 54% in those with non–small cell lung cancer. After a median of 8 months of follow-up, the median progression-free survival was 12.4 months and 9.7 months for the two diseases, respectively. Median overall survival had not been reached in either disease.
However, those with a flare or another immune-related adverse event had significantly better progression-free survival (P = .016) and overall survival (P = .004) when compared with those who did not flare or have an immune-related adverse event. According to Ms. Tison, this has been reported before, but a more surprising finding was that the gain in progression-free survival and overall survival was lost in those treated with an immunosuppressive drug.
Even though non-PAD patients commonly receive steroids for immune-related adverse events such as colitis, the loss of benefit in PAD patients who received immunosuppressive therapies may be caused by, at least in part, cross-reactivity between tumor antigens and autoantigens, Ms. Tison speculated.
Ms. Tison was cautious in drawing conclusions about specific strategies to optimize benefits from checkpoint inhibitors in PAD based on this limited series of patients. However, she did suggest that discontinuation of immunosuppressive therapies prior to initiating checkpoint inhibitors may be prudent in PAD patients, particularly those with inactive disease.
Overall, she emphasized that checkpoint inhibitors “have revolutionized the management of several cancers” and should not be denied to PAD patients who are otherwise appropriate candidates. Although flares are common, more than half of PAD patients in this series did not flare and flares were mild to moderate in most of those who did.
“The response to checkpoint inhibitors in PAD patients is good,” Ms. Tison advised. For those who do flare, “we need prospective studies to understand which strategies provide a good balance of benefit to risk” for cancer immunotherapy and for the options to manage immune-related adverse events.
The study was not industry funded. Ms. Tison reported no potential conflicts of interest.
SOURCE: Tison A et al. Ann Rheum Dis. 2018;77(Suppl 2):147. EULAR Congress 2018, Abstract OP0196.
AMSTERDAM – In patients with autoimmune diseases, cancer treatment with checkpoint inhibitor immunotherapy increases the risk of flares, but these flares are associated with improved cancer outcomes, according to a multicenter, retrospective study presented at the European Congress of Rheumatology.
“Survival was longer in patients who experienced a flare of their preexisting autoimmune disease or any other immune-related adverse event, but this gain was lost if an immunosuppressive therapy was used,” reported Alice Tison, a resident in rheumatology at the Centre Hospitalier Universitaire, Brest, France.
These were some of the mixed messages from this evaluation, which involved 112 patients with preexisting autoimmune disease (PAD) whose data were collected from 11 tertiary care centers in France. Of the cases of PAD represented, the majority involved joint diseases, including psoriatic arthritis (28%), rheumatoid arthritis (18%), and spondyloarthritis (4.5%). However, other types of PAD, including inflammatory bowel disease (13%), were included in the series.
Only 33% of the patients had active disease at the time that checkpoint inhibitor therapy was initiated, and only 21% were taking an immunosuppressive therapy for their disease. Of those on therapy, the majority were taking steroids, but about a third of those on therapy were taking a disease-modifying antirheumatic drug, such as methotrexate.
With the initiation of checkpoint inhibitors, which were offered primarily for the treatment of melanoma (59%) and non–small cell lung cancer (36%), 42% of patients with PAD developed a disease flare. Of these, 30% were considered severe. Other immune-related events not considered related to the underlying disease, such as colitis, were also observed but at rates not clearly different than those observed in patients without PAD.
The activity of checkpoint inhibitors did not appear to be different than that observed in non-PAD patients. For example, the overall response rate was 48% in those with melanoma and 54% in those with non–small cell lung cancer. After a median of 8 months of follow-up, the median progression-free survival was 12.4 months and 9.7 months for the two diseases, respectively. Median overall survival had not been reached in either disease.
However, those with a flare or another immune-related adverse event had significantly better progression-free survival (P = .016) and overall survival (P = .004) when compared with those who did not flare or have an immune-related adverse event. According to Ms. Tison, this has been reported before, but a more surprising finding was that the gain in progression-free survival and overall survival was lost in those treated with an immunosuppressive drug.
Even though non-PAD patients commonly receive steroids for immune-related adverse events such as colitis, the loss of benefit in PAD patients who received immunosuppressive therapies may be caused by, at least in part, cross-reactivity between tumor antigens and autoantigens, Ms. Tison speculated.
Ms. Tison was cautious in drawing conclusions about specific strategies to optimize benefits from checkpoint inhibitors in PAD based on this limited series of patients. However, she did suggest that discontinuation of immunosuppressive therapies prior to initiating checkpoint inhibitors may be prudent in PAD patients, particularly those with inactive disease.
Overall, she emphasized that checkpoint inhibitors “have revolutionized the management of several cancers” and should not be denied to PAD patients who are otherwise appropriate candidates. Although flares are common, more than half of PAD patients in this series did not flare and flares were mild to moderate in most of those who did.
“The response to checkpoint inhibitors in PAD patients is good,” Ms. Tison advised. For those who do flare, “we need prospective studies to understand which strategies provide a good balance of benefit to risk” for cancer immunotherapy and for the options to manage immune-related adverse events.
The study was not industry funded. Ms. Tison reported no potential conflicts of interest.
SOURCE: Tison A et al. Ann Rheum Dis. 2018;77(Suppl 2):147. EULAR Congress 2018, Abstract OP0196.
AMSTERDAM – In patients with autoimmune diseases, cancer treatment with checkpoint inhibitor immunotherapy increases the risk of flares, but these flares are associated with improved cancer outcomes, according to a multicenter, retrospective study presented at the European Congress of Rheumatology.
“Survival was longer in patients who experienced a flare of their preexisting autoimmune disease or any other immune-related adverse event, but this gain was lost if an immunosuppressive therapy was used,” reported Alice Tison, a resident in rheumatology at the Centre Hospitalier Universitaire, Brest, France.
These were some of the mixed messages from this evaluation, which involved 112 patients with preexisting autoimmune disease (PAD) whose data were collected from 11 tertiary care centers in France. Of the cases of PAD represented, the majority involved joint diseases, including psoriatic arthritis (28%), rheumatoid arthritis (18%), and spondyloarthritis (4.5%). However, other types of PAD, including inflammatory bowel disease (13%), were included in the series.
Only 33% of the patients had active disease at the time that checkpoint inhibitor therapy was initiated, and only 21% were taking an immunosuppressive therapy for their disease. Of those on therapy, the majority were taking steroids, but about a third of those on therapy were taking a disease-modifying antirheumatic drug, such as methotrexate.
With the initiation of checkpoint inhibitors, which were offered primarily for the treatment of melanoma (59%) and non–small cell lung cancer (36%), 42% of patients with PAD developed a disease flare. Of these, 30% were considered severe. Other immune-related events not considered related to the underlying disease, such as colitis, were also observed but at rates not clearly different than those observed in patients without PAD.
The activity of checkpoint inhibitors did not appear to be different than that observed in non-PAD patients. For example, the overall response rate was 48% in those with melanoma and 54% in those with non–small cell lung cancer. After a median of 8 months of follow-up, the median progression-free survival was 12.4 months and 9.7 months for the two diseases, respectively. Median overall survival had not been reached in either disease.
However, those with a flare or another immune-related adverse event had significantly better progression-free survival (P = .016) and overall survival (P = .004) when compared with those who did not flare or have an immune-related adverse event. According to Ms. Tison, this has been reported before, but a more surprising finding was that the gain in progression-free survival and overall survival was lost in those treated with an immunosuppressive drug.
Even though non-PAD patients commonly receive steroids for immune-related adverse events such as colitis, the loss of benefit in PAD patients who received immunosuppressive therapies may be caused by, at least in part, cross-reactivity between tumor antigens and autoantigens, Ms. Tison speculated.
Ms. Tison was cautious in drawing conclusions about specific strategies to optimize benefits from checkpoint inhibitors in PAD based on this limited series of patients. However, she did suggest that discontinuation of immunosuppressive therapies prior to initiating checkpoint inhibitors may be prudent in PAD patients, particularly those with inactive disease.
Overall, she emphasized that checkpoint inhibitors “have revolutionized the management of several cancers” and should not be denied to PAD patients who are otherwise appropriate candidates. Although flares are common, more than half of PAD patients in this series did not flare and flares were mild to moderate in most of those who did.
“The response to checkpoint inhibitors in PAD patients is good,” Ms. Tison advised. For those who do flare, “we need prospective studies to understand which strategies provide a good balance of benefit to risk” for cancer immunotherapy and for the options to manage immune-related adverse events.
The study was not industry funded. Ms. Tison reported no potential conflicts of interest.
SOURCE: Tison A et al. Ann Rheum Dis. 2018;77(Suppl 2):147. EULAR Congress 2018, Abstract OP0196.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point: Cancer patients who take a checkpoint inhibitor and have a preexisting autoimmune disease were significantly more likely to have a disease flare but also a better cancer outcome than were those without preexisting disease.
Major finding: In those with a disease flare, progression-free and overall survival were significantly improved (P = .016 and P = .004, respectively).
Study details: Retrospective multicenter study.
Disclosures: The study was not industry funded. Ms. Tison reported no potential conflicts of interest.
Source: Tison A et al. Ann Rheum Dis. 2018;77(Suppl 2):147. EULAR Congress 2018, Abstract OP0196.