User login
FDA panel not ready to recommend quizartinib approval for FLT3-ITD+ AML
SILVER SPRING, MD. – Daiichi Sankyo failed to make the case for approval of its investigational tyrosine kinase inhibitor quizartinib for patients with acute myeloid leukemia bearing the FLT3 internal tandem duplication (ITD) mutation.

Members of the Oncologic Drugs Advisory Committee (ODAC) of the Food and Drug Administration voted 8-3 not to recommend approval of the drug at this time, despite the prevailing sentiment among oncologists on the panel that, as one stated, “I need this drug. I want this drug.”
The prevailing majority of committee members agreed that the drug may have a place in the treatment of patients with FLT3-mutated AML, but that more robust data were needed to prove it.
Currently, only one agent, gilteritinib (Xospata) is approved by the FDA for the treatment of patients with relapsed or refractory FLT3-mutated AML.
QuANTUM-R
Daiichi Sankyo sought approval for quizartinib based on results of the phase 3 randomized QuANTUM-R trial. In this trial, single-agent therapy with quizartinib slightly but significantly prolonged survival – compared with salvage chemotherapy – of patients with relapsed/refractory FLT3-ITD positive AML.
Median overall survival (OS), the trial’s primary endpoint, was 6.2 months for 245 patients randomized to quizartinib, compared with 4.7 months for 122 patients assigned to salvage chemotherapy, a difference that translated into a hazard ratio (HR) for death of 0.76 (P = .0177).
The patients were randomly assigned on a 2:1 basis to receive either quizartinib or salvage chemotherapy. Quizartinib was dosed 30 mg per day for 15 days, which could be titrated upward to 60 mg daily if the corrected QT interval by Fredericia (QTcF) was 450 ms or less on day 16.
Chemotherapy was the investigator’s choice of one of three specified regimens: either low-dose cytarabine (LoDAC); mitoxantrone, etoposide, and intermediate-dose cytarabine (MEC); or fludarabine, cytarabine, and granulocyte-colony stimulating factor (G-CSF) with idarubicin (FLAG-IDA). Up to 2 cycles of MEC or FLAG-IDA were permitted; quizartinib and LoDAC were given until lack of benefit, unacceptable toxicity, or until the patient went on to hematopoietic stem cell transplant (HSCT).
Principal investigator Jorge Cortes, MD, from the University of Texas MD Anderson Cancer Center in Houston, speaking in support of the application, said that combined with the phase 2 study results, “these data support a clear and clinically meaningful benefit of quizartinib in this patient population.”
Mark Levis, MD. PhD, from the Johns Hopkins Sidney Kimmel Cancer Center in Baltimore, also spoke in support of the FLT3 inhibitor.
“I have studied both in the lab and in the clinic most FLT3 inhibitors that have been developed, including lestaurtinib, midostaurin, sorafenib and gilteritinib. Quizartinib is the most highly potent and selective FLT3 inhibitor I have ever worked with,” Dr. Levis said.
FDA: Data not up to snuff
But as FDA staff member Kunthel By, PhD, a statistical reviewer in the Office of Biostatistics, pointed out, the upper limit of the hazard ratio favoring quizartinib over chemotherapy was 0.99, and the difference in median overall survival was just 6.5 weeks.
Additionally, the trial data lacked internal consistency, showing no benefits for the drug in either event-free survival (EFS) or in complete response rates.
There were also imbalances in the number of patients with subsequent HSCT between the arms, with more patients on quizartinib undergoing HSCT despite not having a complete remission, than in the chemotherapy group. Also, there were differences in the number of patients who were randomized but not treated and in those censored early. And statistical stress tests indicated “a lack of robustness in the estimated treatment effect,” he said.
Safety issues raised in QuANTUM-R included slow potassium channel (IKs) blockade and related cardiac toxicitites, as well as the differentiation syndrome, acute febrile neutrophilic dermatosis, and cytopenias, said Aviva Krauss, MD, a clinical reviewer in the FDA’s Office of Hematology and Oncology Products.
“Quizartinib therapy is associated with significant and unique safety concerns in the [proposed population], including the risk of fatal cardiac events that cannot be predicted with certainty using routine QTc measurements,” she said.
She noted that the events occurred in QuANTUM-R despite dose modifications and concomitant medications guidelines in the study protocol.
Reviewers recommended that should the drug receive approval, the package labeling should include contraindication for use with other QT-prolonging agents, and a recommendation for prophylactic beta blockage, although the panelists in general felt that the latter recommendation was not necessary.
‘I believe in this drug’
The ODAC meeting was convened to answer questions about whether the overall survival results were credible based on a single clinical trial and outweighed the risks of treatment with quizartinib, and to assess risk strategies for reducing risks of potentially fatal cardiac toxicities, primarily prolongation of the QT interval.
A. Michael Lincoff, MD, a cardiologist at Case Western Reserve University and the Cleveland Clinic, both in Cleveland, Ohio, voted in favor of approval.
“I’m less concerned about the risk and I do think on the balance there is benefit,” he said.
But most committee members echoed the comments of Anthony D. Sung, MD, from the division of hematologic malignancies and cellular therapy at Duke University in Durham, N.C.
“My vote is based purely on the data I’m shown, and my vote is no,” he said. “But I want the FDA to know that I believe in this drug, and I think it should get approved, and I want to use it.”
The trial was sponsored by Daiichi Sankyo. Dr. Cortes reported research funding from Daiichi Sankyo, Pfizer, Arog, Astellas Pharma and Novartis, and consulting activities for all of the same companies except Arog. Dr. Levis is a paid consultant for Daiichi Sankyo. He and Dr. Cortes stated that they had no financial interests in the outcome of the ODAC meeting.
SILVER SPRING, MD. – Daiichi Sankyo failed to make the case for approval of its investigational tyrosine kinase inhibitor quizartinib for patients with acute myeloid leukemia bearing the FLT3 internal tandem duplication (ITD) mutation.
Members of the Oncologic Drugs Advisory Committee (ODAC) of the Food and Drug Administration voted 8-3 not to recommend approval of the drug at this time, despite the prevailing sentiment among oncologists on the panel that, as one stated, “I need this drug. I want this drug.”
The prevailing majority of committee members agreed that the drug may have a place in the treatment of patients with FLT3-mutated AML, but that more robust data were needed to prove it.
Currently, only one agent, gilteritinib (Xospata) is approved by the FDA for the treatment of patients with relapsed or refractory FLT3-mutated AML.
QuANTUM-R
Daiichi Sankyo sought approval for quizartinib based on results of the phase 3 randomized QuANTUM-R trial. In this trial, single-agent therapy with quizartinib slightly but significantly prolonged survival – compared with salvage chemotherapy – of patients with relapsed/refractory FLT3-ITD positive AML.
Median overall survival (OS), the trial’s primary endpoint, was 6.2 months for 245 patients randomized to quizartinib, compared with 4.7 months for 122 patients assigned to salvage chemotherapy, a difference that translated into a hazard ratio (HR) for death of 0.76 (P = .0177).
The patients were randomly assigned on a 2:1 basis to receive either quizartinib or salvage chemotherapy. Quizartinib was dosed 30 mg per day for 15 days, which could be titrated upward to 60 mg daily if the corrected QT interval by Fredericia (QTcF) was 450 ms or less on day 16.
Chemotherapy was the investigator’s choice of one of three specified regimens: either low-dose cytarabine (LoDAC); mitoxantrone, etoposide, and intermediate-dose cytarabine (MEC); or fludarabine, cytarabine, and granulocyte-colony stimulating factor (G-CSF) with idarubicin (FLAG-IDA). Up to 2 cycles of MEC or FLAG-IDA were permitted; quizartinib and LoDAC were given until lack of benefit, unacceptable toxicity, or until the patient went on to hematopoietic stem cell transplant (HSCT).
Principal investigator Jorge Cortes, MD, from the University of Texas MD Anderson Cancer Center in Houston, speaking in support of the application, said that combined with the phase 2 study results, “these data support a clear and clinically meaningful benefit of quizartinib in this patient population.”
Mark Levis, MD. PhD, from the Johns Hopkins Sidney Kimmel Cancer Center in Baltimore, also spoke in support of the FLT3 inhibitor.
“I have studied both in the lab and in the clinic most FLT3 inhibitors that have been developed, including lestaurtinib, midostaurin, sorafenib and gilteritinib. Quizartinib is the most highly potent and selective FLT3 inhibitor I have ever worked with,” Dr. Levis said.
FDA: Data not up to snuff
But as FDA staff member Kunthel By, PhD, a statistical reviewer in the Office of Biostatistics, pointed out, the upper limit of the hazard ratio favoring quizartinib over chemotherapy was 0.99, and the difference in median overall survival was just 6.5 weeks.
Additionally, the trial data lacked internal consistency, showing no benefits for the drug in either event-free survival (EFS) or in complete response rates.
There were also imbalances in the number of patients with subsequent HSCT between the arms, with more patients on quizartinib undergoing HSCT despite not having a complete remission, than in the chemotherapy group. Also, there were differences in the number of patients who were randomized but not treated and in those censored early. And statistical stress tests indicated “a lack of robustness in the estimated treatment effect,” he said.
Safety issues raised in QuANTUM-R included slow potassium channel (IKs) blockade and related cardiac toxicitites, as well as the differentiation syndrome, acute febrile neutrophilic dermatosis, and cytopenias, said Aviva Krauss, MD, a clinical reviewer in the FDA’s Office of Hematology and Oncology Products.
“Quizartinib therapy is associated with significant and unique safety concerns in the [proposed population], including the risk of fatal cardiac events that cannot be predicted with certainty using routine QTc measurements,” she said.
She noted that the events occurred in QuANTUM-R despite dose modifications and concomitant medications guidelines in the study protocol.
Reviewers recommended that should the drug receive approval, the package labeling should include contraindication for use with other QT-prolonging agents, and a recommendation for prophylactic beta blockage, although the panelists in general felt that the latter recommendation was not necessary.
‘I believe in this drug’
The ODAC meeting was convened to answer questions about whether the overall survival results were credible based on a single clinical trial and outweighed the risks of treatment with quizartinib, and to assess risk strategies for reducing risks of potentially fatal cardiac toxicities, primarily prolongation of the QT interval.
A. Michael Lincoff, MD, a cardiologist at Case Western Reserve University and the Cleveland Clinic, both in Cleveland, Ohio, voted in favor of approval.
“I’m less concerned about the risk and I do think on the balance there is benefit,” he said.
But most committee members echoed the comments of Anthony D. Sung, MD, from the division of hematologic malignancies and cellular therapy at Duke University in Durham, N.C.
“My vote is based purely on the data I’m shown, and my vote is no,” he said. “But I want the FDA to know that I believe in this drug, and I think it should get approved, and I want to use it.”
The trial was sponsored by Daiichi Sankyo. Dr. Cortes reported research funding from Daiichi Sankyo, Pfizer, Arog, Astellas Pharma and Novartis, and consulting activities for all of the same companies except Arog. Dr. Levis is a paid consultant for Daiichi Sankyo. He and Dr. Cortes stated that they had no financial interests in the outcome of the ODAC meeting.
SILVER SPRING, MD. – Daiichi Sankyo failed to make the case for approval of its investigational tyrosine kinase inhibitor quizartinib for patients with acute myeloid leukemia bearing the FLT3 internal tandem duplication (ITD) mutation.
Members of the Oncologic Drugs Advisory Committee (ODAC) of the Food and Drug Administration voted 8-3 not to recommend approval of the drug at this time, despite the prevailing sentiment among oncologists on the panel that, as one stated, “I need this drug. I want this drug.”
The prevailing majority of committee members agreed that the drug may have a place in the treatment of patients with FLT3-mutated AML, but that more robust data were needed to prove it.
Currently, only one agent, gilteritinib (Xospata) is approved by the FDA for the treatment of patients with relapsed or refractory FLT3-mutated AML.
QuANTUM-R
Daiichi Sankyo sought approval for quizartinib based on results of the phase 3 randomized QuANTUM-R trial. In this trial, single-agent therapy with quizartinib slightly but significantly prolonged survival – compared with salvage chemotherapy – of patients with relapsed/refractory FLT3-ITD positive AML.
Median overall survival (OS), the trial’s primary endpoint, was 6.2 months for 245 patients randomized to quizartinib, compared with 4.7 months for 122 patients assigned to salvage chemotherapy, a difference that translated into a hazard ratio (HR) for death of 0.76 (P = .0177).
The patients were randomly assigned on a 2:1 basis to receive either quizartinib or salvage chemotherapy. Quizartinib was dosed 30 mg per day for 15 days, which could be titrated upward to 60 mg daily if the corrected QT interval by Fredericia (QTcF) was 450 ms or less on day 16.
Chemotherapy was the investigator’s choice of one of three specified regimens: either low-dose cytarabine (LoDAC); mitoxantrone, etoposide, and intermediate-dose cytarabine (MEC); or fludarabine, cytarabine, and granulocyte-colony stimulating factor (G-CSF) with idarubicin (FLAG-IDA). Up to 2 cycles of MEC or FLAG-IDA were permitted; quizartinib and LoDAC were given until lack of benefit, unacceptable toxicity, or until the patient went on to hematopoietic stem cell transplant (HSCT).
Principal investigator Jorge Cortes, MD, from the University of Texas MD Anderson Cancer Center in Houston, speaking in support of the application, said that combined with the phase 2 study results, “these data support a clear and clinically meaningful benefit of quizartinib in this patient population.”
Mark Levis, MD. PhD, from the Johns Hopkins Sidney Kimmel Cancer Center in Baltimore, also spoke in support of the FLT3 inhibitor.
“I have studied both in the lab and in the clinic most FLT3 inhibitors that have been developed, including lestaurtinib, midostaurin, sorafenib and gilteritinib. Quizartinib is the most highly potent and selective FLT3 inhibitor I have ever worked with,” Dr. Levis said.
FDA: Data not up to snuff
But as FDA staff member Kunthel By, PhD, a statistical reviewer in the Office of Biostatistics, pointed out, the upper limit of the hazard ratio favoring quizartinib over chemotherapy was 0.99, and the difference in median overall survival was just 6.5 weeks.
Additionally, the trial data lacked internal consistency, showing no benefits for the drug in either event-free survival (EFS) or in complete response rates.
There were also imbalances in the number of patients with subsequent HSCT between the arms, with more patients on quizartinib undergoing HSCT despite not having a complete remission, than in the chemotherapy group. Also, there were differences in the number of patients who were randomized but not treated and in those censored early. And statistical stress tests indicated “a lack of robustness in the estimated treatment effect,” he said.
Safety issues raised in QuANTUM-R included slow potassium channel (IKs) blockade and related cardiac toxicitites, as well as the differentiation syndrome, acute febrile neutrophilic dermatosis, and cytopenias, said Aviva Krauss, MD, a clinical reviewer in the FDA’s Office of Hematology and Oncology Products.
“Quizartinib therapy is associated with significant and unique safety concerns in the [proposed population], including the risk of fatal cardiac events that cannot be predicted with certainty using routine QTc measurements,” she said.
She noted that the events occurred in QuANTUM-R despite dose modifications and concomitant medications guidelines in the study protocol.
Reviewers recommended that should the drug receive approval, the package labeling should include contraindication for use with other QT-prolonging agents, and a recommendation for prophylactic beta blockage, although the panelists in general felt that the latter recommendation was not necessary.
‘I believe in this drug’
The ODAC meeting was convened to answer questions about whether the overall survival results were credible based on a single clinical trial and outweighed the risks of treatment with quizartinib, and to assess risk strategies for reducing risks of potentially fatal cardiac toxicities, primarily prolongation of the QT interval.
A. Michael Lincoff, MD, a cardiologist at Case Western Reserve University and the Cleveland Clinic, both in Cleveland, Ohio, voted in favor of approval.
“I’m less concerned about the risk and I do think on the balance there is benefit,” he said.
But most committee members echoed the comments of Anthony D. Sung, MD, from the division of hematologic malignancies and cellular therapy at Duke University in Durham, N.C.
“My vote is based purely on the data I’m shown, and my vote is no,” he said. “But I want the FDA to know that I believe in this drug, and I think it should get approved, and I want to use it.”
The trial was sponsored by Daiichi Sankyo. Dr. Cortes reported research funding from Daiichi Sankyo, Pfizer, Arog, Astellas Pharma and Novartis, and consulting activities for all of the same companies except Arog. Dr. Levis is a paid consultant for Daiichi Sankyo. He and Dr. Cortes stated that they had no financial interests in the outcome of the ODAC meeting.
Master trial seeks to aid drug development for pediatric AML
NEW ORLEANS – Researchers are organizing a master trial in an attempt to improve the treatment of pediatric acute myeloid leukemia (AML).
The Pediatric Acute Leukemia (PedAL) trial is an effort to collect data on all pediatric AML patients. The plan is to use these data to match patients to clinical trials, better understand pediatric AML, and bring new treatments to this population.
E. Anders Kolb, MD, of Nemours Center for Cancer and Blood Disorders in Wilmington, Del., described the initiative at the annual meeting of the American Society of Pediatric Hematology/Oncology.
Dr. Kolb noted that several drugs have been approved to treat adult AML in the last 2 years, but most of them are not approved for use in children.
“What we see in childhood AML is a lot different than what we see in adult AML, and this challenges the paradigm that we have traditionally followed where we use the adult as the 'preclinical model' for pediatric AML,” he said. “I think we are learning more and more that children have a unique disease, unique targets, and need unique therapies.”
The PedAL initiative is an attempt to address these unique needs. PedAL is part of the Leukemia & Lymphoma Society’s Children’s Initiative, and it involves researchers from academic centers and the Children’s Oncology Group.
The PedAL initiative includes preclinical, biomarker, and informatics research, as well as the master clinical trial. The main goal of the master trial is to collect genomic, proteomic, metabolomic, flow cytometry, and clinical data from all children with AML and use these data to match patients to clinical trials.
The PedAL trial will leverage Project:EveryChild, an effort by the Children’s Oncology Group to study every child with cancer. Each child enrolled in this program has an identification number that follows the child through all clinical interventions.
The goal is that Project:EveryChild will capture all pediatric AML patients at the time of diagnosis, although patients can join the project at any time. Then, sequencing, clinical, and other data will be collected from these patients and stored in a data commons.
If patients relapse after standard or other therapies, the GEARBOX algorithm (genomic eligibility algorithm at relapse for better outcomes) can be used to match the patient’s information to clinical trial eligibility criteria and provide a list of appropriate trials.
Dr. Kolb said this process should reduce logistical barriers and get relapsed patients to trials more quickly. Additionally, the data collected through PedAL should help researchers design better trials for pediatric patients with relapsed AML.
“Ultimately, we’ll create the largest data set that will give us a better understanding of all the risks and benefits associated with postrelapse AML,” Dr. Kolb said. “No matter what happens to the patient, no matter where that patient enrolls, we’re going to have the capacity to collect data and present that data to the community for analysis for improved understanding of outcomes.”
Dr. Kolb and his colleagues are already working with researchers in Europe and Japan to make this a global effort and create an international data commons. In addition, the researchers are planning to collaborate with the pharmaceutical industry to unite efforts in pediatric AML drug development.
“We can’t just test drugs in kids because they worked in adults,” Dr. Kolb said. “We really need to maintain the integrity of the science and ask relevant questions in children but do so with the intent to make sure these drugs are licensed for use in kids.”
Dr. Kolb reported having no conflicts of interest. The PedAL trial is sponsored by the Leukemia & Lymphoma Society.
NEW ORLEANS – Researchers are organizing a master trial in an attempt to improve the treatment of pediatric acute myeloid leukemia (AML).
The Pediatric Acute Leukemia (PedAL) trial is an effort to collect data on all pediatric AML patients. The plan is to use these data to match patients to clinical trials, better understand pediatric AML, and bring new treatments to this population.
E. Anders Kolb, MD, of Nemours Center for Cancer and Blood Disorders in Wilmington, Del., described the initiative at the annual meeting of the American Society of Pediatric Hematology/Oncology.
Dr. Kolb noted that several drugs have been approved to treat adult AML in the last 2 years, but most of them are not approved for use in children.
“What we see in childhood AML is a lot different than what we see in adult AML, and this challenges the paradigm that we have traditionally followed where we use the adult as the 'preclinical model' for pediatric AML,” he said. “I think we are learning more and more that children have a unique disease, unique targets, and need unique therapies.”
The PedAL initiative is an attempt to address these unique needs. PedAL is part of the Leukemia & Lymphoma Society’s Children’s Initiative, and it involves researchers from academic centers and the Children’s Oncology Group.
The PedAL initiative includes preclinical, biomarker, and informatics research, as well as the master clinical trial. The main goal of the master trial is to collect genomic, proteomic, metabolomic, flow cytometry, and clinical data from all children with AML and use these data to match patients to clinical trials.
The PedAL trial will leverage Project:EveryChild, an effort by the Children’s Oncology Group to study every child with cancer. Each child enrolled in this program has an identification number that follows the child through all clinical interventions.
The goal is that Project:EveryChild will capture all pediatric AML patients at the time of diagnosis, although patients can join the project at any time. Then, sequencing, clinical, and other data will be collected from these patients and stored in a data commons.
If patients relapse after standard or other therapies, the GEARBOX algorithm (genomic eligibility algorithm at relapse for better outcomes) can be used to match the patient’s information to clinical trial eligibility criteria and provide a list of appropriate trials.
Dr. Kolb said this process should reduce logistical barriers and get relapsed patients to trials more quickly. Additionally, the data collected through PedAL should help researchers design better trials for pediatric patients with relapsed AML.
“Ultimately, we’ll create the largest data set that will give us a better understanding of all the risks and benefits associated with postrelapse AML,” Dr. Kolb said. “No matter what happens to the patient, no matter where that patient enrolls, we’re going to have the capacity to collect data and present that data to the community for analysis for improved understanding of outcomes.”
Dr. Kolb and his colleagues are already working with researchers in Europe and Japan to make this a global effort and create an international data commons. In addition, the researchers are planning to collaborate with the pharmaceutical industry to unite efforts in pediatric AML drug development.
“We can’t just test drugs in kids because they worked in adults,” Dr. Kolb said. “We really need to maintain the integrity of the science and ask relevant questions in children but do so with the intent to make sure these drugs are licensed for use in kids.”
Dr. Kolb reported having no conflicts of interest. The PedAL trial is sponsored by the Leukemia & Lymphoma Society.
NEW ORLEANS – Researchers are organizing a master trial in an attempt to improve the treatment of pediatric acute myeloid leukemia (AML).
The Pediatric Acute Leukemia (PedAL) trial is an effort to collect data on all pediatric AML patients. The plan is to use these data to match patients to clinical trials, better understand pediatric AML, and bring new treatments to this population.
E. Anders Kolb, MD, of Nemours Center for Cancer and Blood Disorders in Wilmington, Del., described the initiative at the annual meeting of the American Society of Pediatric Hematology/Oncology.
Dr. Kolb noted that several drugs have been approved to treat adult AML in the last 2 years, but most of them are not approved for use in children.
“What we see in childhood AML is a lot different than what we see in adult AML, and this challenges the paradigm that we have traditionally followed where we use the adult as the 'preclinical model' for pediatric AML,” he said. “I think we are learning more and more that children have a unique disease, unique targets, and need unique therapies.”
The PedAL initiative is an attempt to address these unique needs. PedAL is part of the Leukemia & Lymphoma Society’s Children’s Initiative, and it involves researchers from academic centers and the Children’s Oncology Group.
The PedAL initiative includes preclinical, biomarker, and informatics research, as well as the master clinical trial. The main goal of the master trial is to collect genomic, proteomic, metabolomic, flow cytometry, and clinical data from all children with AML and use these data to match patients to clinical trials.
The PedAL trial will leverage Project:EveryChild, an effort by the Children’s Oncology Group to study every child with cancer. Each child enrolled in this program has an identification number that follows the child through all clinical interventions.
The goal is that Project:EveryChild will capture all pediatric AML patients at the time of diagnosis, although patients can join the project at any time. Then, sequencing, clinical, and other data will be collected from these patients and stored in a data commons.
If patients relapse after standard or other therapies, the GEARBOX algorithm (genomic eligibility algorithm at relapse for better outcomes) can be used to match the patient’s information to clinical trial eligibility criteria and provide a list of appropriate trials.
Dr. Kolb said this process should reduce logistical barriers and get relapsed patients to trials more quickly. Additionally, the data collected through PedAL should help researchers design better trials for pediatric patients with relapsed AML.
“Ultimately, we’ll create the largest data set that will give us a better understanding of all the risks and benefits associated with postrelapse AML,” Dr. Kolb said. “No matter what happens to the patient, no matter where that patient enrolls, we’re going to have the capacity to collect data and present that data to the community for analysis for improved understanding of outcomes.”
Dr. Kolb and his colleagues are already working with researchers in Europe and Japan to make this a global effort and create an international data commons. In addition, the researchers are planning to collaborate with the pharmaceutical industry to unite efforts in pediatric AML drug development.
“We can’t just test drugs in kids because they worked in adults,” Dr. Kolb said. “We really need to maintain the integrity of the science and ask relevant questions in children but do so with the intent to make sure these drugs are licensed for use in kids.”
Dr. Kolb reported having no conflicts of interest. The PedAL trial is sponsored by the Leukemia & Lymphoma Society.
REPORTING FROM 2019 ASPHO CONFERENCE

Researchers propose new risk groups for NK-AML
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
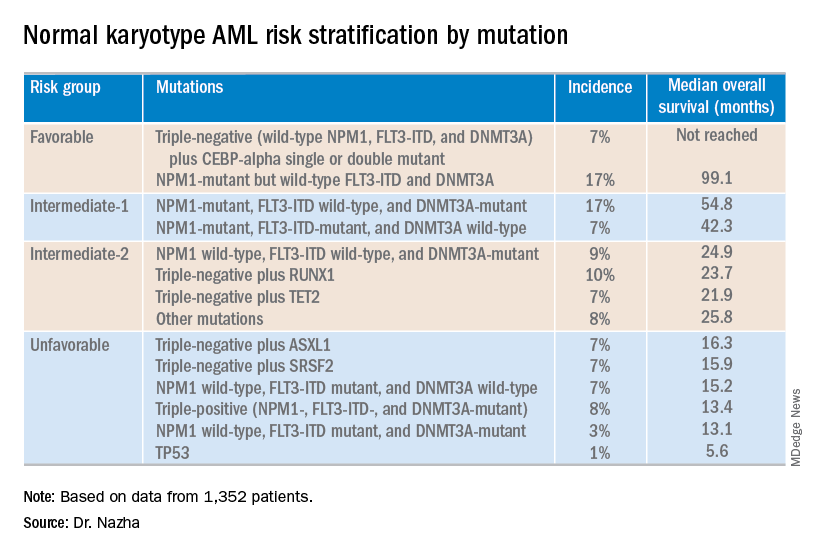
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
REPORTING FROM ALF 2019
Combo proves most effective in HMA-naive, higher-risk MDS
NEWPORT BEACH, CALIF. – The combination of oral rigosertib and azacitidine is proceeding to a phase 3 trial in patients with myelodysplastic syndromes (MDS), but it isn’t clear if the combination will continue to be developed for acute myeloid leukemia (AML).
In a phase 1/2 trial, oral rigosertib plus azacitidine produced a 90% response rate in higher-risk MDS patients who were naive to hypomethylating agents (HMAs), a 54% response rate in higher-risk MDS patients who had failed HMA therapy, and a 50% response rate in patients with AML.
Genitourinary toxicities were initially a concern in this trial, but researchers found ways to mitigate the risk of these toxicities, according to Richard Woodman, MD, chief medical officer and senior vice president of research and development at Onconova Therapeutics, the company developing rigosertib.
Dr. Woodman and his colleagues presented results from the phase 1/2 trial in two posters at the Acute Leukemia Forum of Hemedicus.
Results in AML
The researchers reported phase 1 results in 17 patients with AML. Eleven patients had AML, according to investigator assessment, and six patients had refractory anemia with excess blasts in transformation, according to French American British criteria, as well as least 20% excess blasts at baseline.
The median age of the patients was 73 years, and 53% were men. Two patients had received no prior therapies, six patients had relapsed disease, and nine were refractory to their last therapy.
Patients received oral rigosertib at escalating doses twice daily on days 1-21 of a 28-day cycle. The recommended phase 2 dose was 840 mg daily (560 mg in the morning and 280 mg in the afternoon), but there were two expansion cohorts in which patients received 1,120 mg daily (560 mg twice a day or 840 mg in the morning and 280 mg in the afternoon). The patients also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
Patients received a median of three treatment cycles. Fifteen of the 17 patients (88%) discontinued treatment, most because of progressive disease (n = 5), toxicity (n = 4), or death (n = 3).
Twelve patients were evaluable for response, and six (50%) responded. One patient achieved a morphologic complete remission (CR), three achieved a morphologic leukemia-free state, and two had a partial response.
The most common treatment-emergent adverse events (TEAEs) were fatigue (53%), diarrhea (53%), nausea (53%), constipation (47%), back pain (41%), pyrexia (41%), and pneumonia (35%). Grade 3 or higher TEAEs included pneumonia (35%) and anemia (24%).
These results haven’t provided a clear way forward for oral rigosertib and azacitidine in AML. Dr. Woodman said the researchers will have to review past studies and evaluate how AML patients (with at least 20% blasts) have responded to intravenous rigosertib, consult experts in the field, and then decide how they will move forward with oral rigosertib and azacitidine in AML.
Results in MDS
Dr. Woodman and his colleagues presented data on 74 patients with higher-risk MDS. The median age was 69 years, and 59% were men. Most patients were high risk (n = 23) or very high risk (n = 33), according to the Revised International Prognostic Scoring System.
The patients received oral rigosertib at a dose of 840 mg/day or higher on days 1-21 of a 28-day cycle. They also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
The median duration of treatment was 7.8 months in patients who were HMA naive and 4.9 months in patients who failed HMA therapy. The most common reasons for treatment discontinuation in the HMA-naive patients were toxicity (n = 8), progression (n = 7), and patient request (n = 7). The most common reasons for discontinuation in patients who had failed HMA therapy were progression (n = 12), toxicity (n = 5), and investigator decision (n = 4).
In total, 55 patients were evaluable for response, 26 who had failed HMA therapy and 29 who were HMA naive.
“The best responses, not surprisingly, were in patients that were HMA naive,” Dr. Woodman said.
In the HMA-naive patients, the overall response rate was 90%. Ten patients had a CR, five had a marrow CR with hematologic improvement, three had hematologic improvement alone, eight had a marrow CR alone, and three patients had stable disease. None of the patients progressed.
In the patients who had failed HMA therapy, the overall response rate was 54%. One patient achieved a CR, one had a partial response, five had a marrow CR with hematologic improvement, two had hematologic improvement alone, five had a marrow CR alone, seven had stable disease, and five progressed.
The median duration of response was 10.8 months in patients who failed HMA therapy and 12.2 months in the HMA-naive patients.
The most common TEAEs in the entire MDS cohort were hematuria (45%), constipation (43%), diarrhea (42%), fatigue (42%), dysuria (38%), pyrexia (36%), nausea (35%), neutropenia (31%), and thrombocytopenia (30%).
Grade 3 or higher TEAEs were neutropenia (27%), thrombocytopenia (26%), hematuria (9%), dysuria (9%), diarrhea (5%), fatigue (4%), and pyrexia (1%).
Dr. Woodman said patients who were most likely to be at risk for genitourinary toxicities (hematuria and dysuria) were those who weren’t well hydrated, took rigosertib at night, and didn’t void their bladders before bedtime. He said the researchers’ hypothesis is that there is some local bladder irritation in that setting.
However, the researchers found ways to mitigate the risk of genitourinary toxicities, including:
- Requiring the second dose of rigosertib to be taken in the afternoon rather than evening (about 3 p.m.).
- Asking patients to consume at least 2 liters of fluid per day.
- Having patients empty their bladders before bedtime.
- Assessing urine pH roughly 2 hours after the morning dose of rigosertib and prescribing sodium bicarbonate if the pH is less than 7.5.
Dr. Woodman said the phase 2 results in MDS patients have prompted the development of a phase 3 trial in which researchers will compare oral rigosertib plus azacitidine to azacitidine plus placebo.
Dr. Woodman is employed by Onconova Therapeutics, which sponsored the phase 1/2 trial. The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – The combination of oral rigosertib and azacitidine is proceeding to a phase 3 trial in patients with myelodysplastic syndromes (MDS), but it isn’t clear if the combination will continue to be developed for acute myeloid leukemia (AML).
In a phase 1/2 trial, oral rigosertib plus azacitidine produced a 90% response rate in higher-risk MDS patients who were naive to hypomethylating agents (HMAs), a 54% response rate in higher-risk MDS patients who had failed HMA therapy, and a 50% response rate in patients with AML.
Genitourinary toxicities were initially a concern in this trial, but researchers found ways to mitigate the risk of these toxicities, according to Richard Woodman, MD, chief medical officer and senior vice president of research and development at Onconova Therapeutics, the company developing rigosertib.
Dr. Woodman and his colleagues presented results from the phase 1/2 trial in two posters at the Acute Leukemia Forum of Hemedicus.
Results in AML
The researchers reported phase 1 results in 17 patients with AML. Eleven patients had AML, according to investigator assessment, and six patients had refractory anemia with excess blasts in transformation, according to French American British criteria, as well as least 20% excess blasts at baseline.
The median age of the patients was 73 years, and 53% were men. Two patients had received no prior therapies, six patients had relapsed disease, and nine were refractory to their last therapy.
Patients received oral rigosertib at escalating doses twice daily on days 1-21 of a 28-day cycle. The recommended phase 2 dose was 840 mg daily (560 mg in the morning and 280 mg in the afternoon), but there were two expansion cohorts in which patients received 1,120 mg daily (560 mg twice a day or 840 mg in the morning and 280 mg in the afternoon). The patients also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
Patients received a median of three treatment cycles. Fifteen of the 17 patients (88%) discontinued treatment, most because of progressive disease (n = 5), toxicity (n = 4), or death (n = 3).
Twelve patients were evaluable for response, and six (50%) responded. One patient achieved a morphologic complete remission (CR), three achieved a morphologic leukemia-free state, and two had a partial response.
The most common treatment-emergent adverse events (TEAEs) were fatigue (53%), diarrhea (53%), nausea (53%), constipation (47%), back pain (41%), pyrexia (41%), and pneumonia (35%). Grade 3 or higher TEAEs included pneumonia (35%) and anemia (24%).
These results haven’t provided a clear way forward for oral rigosertib and azacitidine in AML. Dr. Woodman said the researchers will have to review past studies and evaluate how AML patients (with at least 20% blasts) have responded to intravenous rigosertib, consult experts in the field, and then decide how they will move forward with oral rigosertib and azacitidine in AML.
Results in MDS
Dr. Woodman and his colleagues presented data on 74 patients with higher-risk MDS. The median age was 69 years, and 59% were men. Most patients were high risk (n = 23) or very high risk (n = 33), according to the Revised International Prognostic Scoring System.
The patients received oral rigosertib at a dose of 840 mg/day or higher on days 1-21 of a 28-day cycle. They also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
The median duration of treatment was 7.8 months in patients who were HMA naive and 4.9 months in patients who failed HMA therapy. The most common reasons for treatment discontinuation in the HMA-naive patients were toxicity (n = 8), progression (n = 7), and patient request (n = 7). The most common reasons for discontinuation in patients who had failed HMA therapy were progression (n = 12), toxicity (n = 5), and investigator decision (n = 4).
In total, 55 patients were evaluable for response, 26 who had failed HMA therapy and 29 who were HMA naive.
“The best responses, not surprisingly, were in patients that were HMA naive,” Dr. Woodman said.
In the HMA-naive patients, the overall response rate was 90%. Ten patients had a CR, five had a marrow CR with hematologic improvement, three had hematologic improvement alone, eight had a marrow CR alone, and three patients had stable disease. None of the patients progressed.
In the patients who had failed HMA therapy, the overall response rate was 54%. One patient achieved a CR, one had a partial response, five had a marrow CR with hematologic improvement, two had hematologic improvement alone, five had a marrow CR alone, seven had stable disease, and five progressed.
The median duration of response was 10.8 months in patients who failed HMA therapy and 12.2 months in the HMA-naive patients.
The most common TEAEs in the entire MDS cohort were hematuria (45%), constipation (43%), diarrhea (42%), fatigue (42%), dysuria (38%), pyrexia (36%), nausea (35%), neutropenia (31%), and thrombocytopenia (30%).
Grade 3 or higher TEAEs were neutropenia (27%), thrombocytopenia (26%), hematuria (9%), dysuria (9%), diarrhea (5%), fatigue (4%), and pyrexia (1%).
Dr. Woodman said patients who were most likely to be at risk for genitourinary toxicities (hematuria and dysuria) were those who weren’t well hydrated, took rigosertib at night, and didn’t void their bladders before bedtime. He said the researchers’ hypothesis is that there is some local bladder irritation in that setting.
However, the researchers found ways to mitigate the risk of genitourinary toxicities, including:
- Requiring the second dose of rigosertib to be taken in the afternoon rather than evening (about 3 p.m.).
- Asking patients to consume at least 2 liters of fluid per day.
- Having patients empty their bladders before bedtime.
- Assessing urine pH roughly 2 hours after the morning dose of rigosertib and prescribing sodium bicarbonate if the pH is less than 7.5.
Dr. Woodman said the phase 2 results in MDS patients have prompted the development of a phase 3 trial in which researchers will compare oral rigosertib plus azacitidine to azacitidine plus placebo.
Dr. Woodman is employed by Onconova Therapeutics, which sponsored the phase 1/2 trial. The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – The combination of oral rigosertib and azacitidine is proceeding to a phase 3 trial in patients with myelodysplastic syndromes (MDS), but it isn’t clear if the combination will continue to be developed for acute myeloid leukemia (AML).
In a phase 1/2 trial, oral rigosertib plus azacitidine produced a 90% response rate in higher-risk MDS patients who were naive to hypomethylating agents (HMAs), a 54% response rate in higher-risk MDS patients who had failed HMA therapy, and a 50% response rate in patients with AML.
Genitourinary toxicities were initially a concern in this trial, but researchers found ways to mitigate the risk of these toxicities, according to Richard Woodman, MD, chief medical officer and senior vice president of research and development at Onconova Therapeutics, the company developing rigosertib.
Dr. Woodman and his colleagues presented results from the phase 1/2 trial in two posters at the Acute Leukemia Forum of Hemedicus.
Results in AML
The researchers reported phase 1 results in 17 patients with AML. Eleven patients had AML, according to investigator assessment, and six patients had refractory anemia with excess blasts in transformation, according to French American British criteria, as well as least 20% excess blasts at baseline.
The median age of the patients was 73 years, and 53% were men. Two patients had received no prior therapies, six patients had relapsed disease, and nine were refractory to their last therapy.
Patients received oral rigosertib at escalating doses twice daily on days 1-21 of a 28-day cycle. The recommended phase 2 dose was 840 mg daily (560 mg in the morning and 280 mg in the afternoon), but there were two expansion cohorts in which patients received 1,120 mg daily (560 mg twice a day or 840 mg in the morning and 280 mg in the afternoon). The patients also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
Patients received a median of three treatment cycles. Fifteen of the 17 patients (88%) discontinued treatment, most because of progressive disease (n = 5), toxicity (n = 4), or death (n = 3).
Twelve patients were evaluable for response, and six (50%) responded. One patient achieved a morphologic complete remission (CR), three achieved a morphologic leukemia-free state, and two had a partial response.
The most common treatment-emergent adverse events (TEAEs) were fatigue (53%), diarrhea (53%), nausea (53%), constipation (47%), back pain (41%), pyrexia (41%), and pneumonia (35%). Grade 3 or higher TEAEs included pneumonia (35%) and anemia (24%).
These results haven’t provided a clear way forward for oral rigosertib and azacitidine in AML. Dr. Woodman said the researchers will have to review past studies and evaluate how AML patients (with at least 20% blasts) have responded to intravenous rigosertib, consult experts in the field, and then decide how they will move forward with oral rigosertib and azacitidine in AML.
Results in MDS
Dr. Woodman and his colleagues presented data on 74 patients with higher-risk MDS. The median age was 69 years, and 59% were men. Most patients were high risk (n = 23) or very high risk (n = 33), according to the Revised International Prognostic Scoring System.
The patients received oral rigosertib at a dose of 840 mg/day or higher on days 1-21 of a 28-day cycle. They also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
The median duration of treatment was 7.8 months in patients who were HMA naive and 4.9 months in patients who failed HMA therapy. The most common reasons for treatment discontinuation in the HMA-naive patients were toxicity (n = 8), progression (n = 7), and patient request (n = 7). The most common reasons for discontinuation in patients who had failed HMA therapy were progression (n = 12), toxicity (n = 5), and investigator decision (n = 4).
In total, 55 patients were evaluable for response, 26 who had failed HMA therapy and 29 who were HMA naive.
“The best responses, not surprisingly, were in patients that were HMA naive,” Dr. Woodman said.
In the HMA-naive patients, the overall response rate was 90%. Ten patients had a CR, five had a marrow CR with hematologic improvement, three had hematologic improvement alone, eight had a marrow CR alone, and three patients had stable disease. None of the patients progressed.
In the patients who had failed HMA therapy, the overall response rate was 54%. One patient achieved a CR, one had a partial response, five had a marrow CR with hematologic improvement, two had hematologic improvement alone, five had a marrow CR alone, seven had stable disease, and five progressed.
The median duration of response was 10.8 months in patients who failed HMA therapy and 12.2 months in the HMA-naive patients.
The most common TEAEs in the entire MDS cohort were hematuria (45%), constipation (43%), diarrhea (42%), fatigue (42%), dysuria (38%), pyrexia (36%), nausea (35%), neutropenia (31%), and thrombocytopenia (30%).
Grade 3 or higher TEAEs were neutropenia (27%), thrombocytopenia (26%), hematuria (9%), dysuria (9%), diarrhea (5%), fatigue (4%), and pyrexia (1%).
Dr. Woodman said patients who were most likely to be at risk for genitourinary toxicities (hematuria and dysuria) were those who weren’t well hydrated, took rigosertib at night, and didn’t void their bladders before bedtime. He said the researchers’ hypothesis is that there is some local bladder irritation in that setting.
However, the researchers found ways to mitigate the risk of genitourinary toxicities, including:
- Requiring the second dose of rigosertib to be taken in the afternoon rather than evening (about 3 p.m.).
- Asking patients to consume at least 2 liters of fluid per day.
- Having patients empty their bladders before bedtime.
- Assessing urine pH roughly 2 hours after the morning dose of rigosertib and prescribing sodium bicarbonate if the pH is less than 7.5.
Dr. Woodman said the phase 2 results in MDS patients have prompted the development of a phase 3 trial in which researchers will compare oral rigosertib plus azacitidine to azacitidine plus placebo.
Dr. Woodman is employed by Onconova Therapeutics, which sponsored the phase 1/2 trial. The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
REPORTING FROM ALF 2019

More abnormal cells linked to poorer ASCT outcomes in MDS
NEWPORT BEACH, CALIF. – Researchers say they’ve found an association between the percentage of cytogenetically abnormal cells at allogeneic stem cell transplant (ASCT) and posttransplant outcomes in patients with myelodysplastic syndromes (MDS).
Patients who had more than 60% cytogenetically abnormal cells at ASCT had significantly inferior overall survival (OS) and relapse-free survival (RFS), compared to patients with fewer abnormal cells.
Dipenkumar Modi, MD, of Barbara Ann Karmanos Cancer Institute at Wayne State University in Detroit, and his colleagues conducted this research and presented the results at the Acute Leukemia Forum of Hemedicus.
The researchers studied 109 adult MDS patients who underwent ASCT from January 2000 through December 2016. The patients were divided into three groups based on the percentage of cytogenetically abnormal cells at ASCT:
- Group 1 had less than 30% (n = 22)
- Group 2 had 30%-60% (n = 23)
- Group 3 had greater than 60% (n = 64).
Baseline characteristics were largely similar between the groups. However, patients in group 3 were significantly more likely than those in groups 1 and 2 to have del(5q) and monosomy 5+7 (P = .048).
Patients in group 1 had a significantly higher percentage of bone marrow transplants (as opposed to peripheral blood stem cell transplants) than patients in groups 2 and 3 (P = .039). And patients in group 1 had significantly fewer blasts at ASCT than patients in groups 2 and 3 (P = .011).
The researchers found no significant between-group differences in relapse and nonrelapse mortality, but there were significant differences in OS and RFS.
Patients in group 3 had inferior RFS compared to patients in group 1, which was the reference group. The hazard ratio (HR) was 2.503 (P = .013) in a univariable analysis and 2.196 (P = .049) in a multivariable analysis.
Group 3 also had inferior OS compared to group 1. The hazard ratio was 2.589 (P = .021) in a univariable analysis and 2.478 (P = .040) in a multivariable analysis.
There was no significant difference in RFS or OS between groups 1 and 2. The HR for RFS in group 2 was 1.879 (P = .148) in a univariable analysis and 1.365 (P = .506) in a multivariable analysis. The HR for OS was 1.997 (P = .155) and 1.413 (P = .511), respectively.
Dr. Modi said these results suggest patients with greater than 60% cytogenetically abnormal cells at ASCT should be monitored more closely after transplant, and their immunosuppressive medication should be tapered as soon as possible.
Dr. Modi and his colleagues reported having no conflicts of interest relevant to this research.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – Researchers say they’ve found an association between the percentage of cytogenetically abnormal cells at allogeneic stem cell transplant (ASCT) and posttransplant outcomes in patients with myelodysplastic syndromes (MDS).
Patients who had more than 60% cytogenetically abnormal cells at ASCT had significantly inferior overall survival (OS) and relapse-free survival (RFS), compared to patients with fewer abnormal cells.
Dipenkumar Modi, MD, of Barbara Ann Karmanos Cancer Institute at Wayne State University in Detroit, and his colleagues conducted this research and presented the results at the Acute Leukemia Forum of Hemedicus.
The researchers studied 109 adult MDS patients who underwent ASCT from January 2000 through December 2016. The patients were divided into three groups based on the percentage of cytogenetically abnormal cells at ASCT:
- Group 1 had less than 30% (n = 22)
- Group 2 had 30%-60% (n = 23)
- Group 3 had greater than 60% (n = 64).
Baseline characteristics were largely similar between the groups. However, patients in group 3 were significantly more likely than those in groups 1 and 2 to have del(5q) and monosomy 5+7 (P = .048).
Patients in group 1 had a significantly higher percentage of bone marrow transplants (as opposed to peripheral blood stem cell transplants) than patients in groups 2 and 3 (P = .039). And patients in group 1 had significantly fewer blasts at ASCT than patients in groups 2 and 3 (P = .011).
The researchers found no significant between-group differences in relapse and nonrelapse mortality, but there were significant differences in OS and RFS.
Patients in group 3 had inferior RFS compared to patients in group 1, which was the reference group. The hazard ratio (HR) was 2.503 (P = .013) in a univariable analysis and 2.196 (P = .049) in a multivariable analysis.
Group 3 also had inferior OS compared to group 1. The hazard ratio was 2.589 (P = .021) in a univariable analysis and 2.478 (P = .040) in a multivariable analysis.
There was no significant difference in RFS or OS between groups 1 and 2. The HR for RFS in group 2 was 1.879 (P = .148) in a univariable analysis and 1.365 (P = .506) in a multivariable analysis. The HR for OS was 1.997 (P = .155) and 1.413 (P = .511), respectively.
Dr. Modi said these results suggest patients with greater than 60% cytogenetically abnormal cells at ASCT should be monitored more closely after transplant, and their immunosuppressive medication should be tapered as soon as possible.
Dr. Modi and his colleagues reported having no conflicts of interest relevant to this research.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – Researchers say they’ve found an association between the percentage of cytogenetically abnormal cells at allogeneic stem cell transplant (ASCT) and posttransplant outcomes in patients with myelodysplastic syndromes (MDS).
Patients who had more than 60% cytogenetically abnormal cells at ASCT had significantly inferior overall survival (OS) and relapse-free survival (RFS), compared to patients with fewer abnormal cells.
Dipenkumar Modi, MD, of Barbara Ann Karmanos Cancer Institute at Wayne State University in Detroit, and his colleagues conducted this research and presented the results at the Acute Leukemia Forum of Hemedicus.
The researchers studied 109 adult MDS patients who underwent ASCT from January 2000 through December 2016. The patients were divided into three groups based on the percentage of cytogenetically abnormal cells at ASCT:
- Group 1 had less than 30% (n = 22)
- Group 2 had 30%-60% (n = 23)
- Group 3 had greater than 60% (n = 64).
Baseline characteristics were largely similar between the groups. However, patients in group 3 were significantly more likely than those in groups 1 and 2 to have del(5q) and monosomy 5+7 (P = .048).
Patients in group 1 had a significantly higher percentage of bone marrow transplants (as opposed to peripheral blood stem cell transplants) than patients in groups 2 and 3 (P = .039). And patients in group 1 had significantly fewer blasts at ASCT than patients in groups 2 and 3 (P = .011).
The researchers found no significant between-group differences in relapse and nonrelapse mortality, but there were significant differences in OS and RFS.
Patients in group 3 had inferior RFS compared to patients in group 1, which was the reference group. The hazard ratio (HR) was 2.503 (P = .013) in a univariable analysis and 2.196 (P = .049) in a multivariable analysis.
Group 3 also had inferior OS compared to group 1. The hazard ratio was 2.589 (P = .021) in a univariable analysis and 2.478 (P = .040) in a multivariable analysis.
There was no significant difference in RFS or OS between groups 1 and 2. The HR for RFS in group 2 was 1.879 (P = .148) in a univariable analysis and 1.365 (P = .506) in a multivariable analysis. The HR for OS was 1.997 (P = .155) and 1.413 (P = .511), respectively.
Dr. Modi said these results suggest patients with greater than 60% cytogenetically abnormal cells at ASCT should be monitored more closely after transplant, and their immunosuppressive medication should be tapered as soon as possible.
Dr. Modi and his colleagues reported having no conflicts of interest relevant to this research.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
REPORTING FROM ALF 2019

FDA approves ivosidenib frontline for certain AML patients
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.

In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
Biomarker testing may transform treatment of acute GVHD
NEWPORT BEACH, CALIF. – Researchers say they have identified biomarkers that may help guide early treatment decisions in patients with acute graft-versus-host disease (GVHD).
The biomarkers, ST2 and REG3-alpha, were measured during the first month of GVHD treatment and proved more accurate than clinical response for predicting 6-month nonrelapse mortality (NRM). In fact, biomarker assessment revealed patients who responded to treatment but had a high risk of NRM and nonresponders who had a low risk of NRM.
The researchers also found that biomarkers changed over the first month of treatment but remained significant predictors of NRM. This suggests that modifying treatment according to biomarker findings at various time points could result in better outcomes for patients.
“We think this is going to transform the way we treat graft-versus-host disease,” said James L.M. Ferrara, MD, DSc, of the Icahn School of Medicine at Mount Sinai, New York.
Dr. Ferrara and Hrishikesh Srinagesh, along with their colleagues at Mount Sinai, have conducted extensive research with these biomarkers and presented some of their findings at the Acute Leukemia Forum of Hemedicus.
Comparing biomarkers and response
In one study, the researchers evaluated 355 patients who had undergone allogeneic hematopoietic stem cell transplant at 1 of 20 Mount Sinai Acute GVHD International Consortium (MAGIC) centers between January 2016 and February 2018. All patients developed acute GVHD and received systemic steroids as treatment.
Patients provided blood samples weekly for the first month of treatment, and concentrations of ST2 and REG3-alpha were measured in each sample. Both biomarker concentrations were used to calculate the biomarker probability of NRM.
“The concentration of those two biomarkers are put into a computer, and we get … a single number, and that gives us the probability of mortality,” Dr. Ferrara said. “[W]e call this the MAGIC algorithm probability, or MAP. And when a MAP is low, the patient has a very low chance of dying from graft-versus-host disease, when it’s intermediate, they have an intermediate risk, and when it’s high, they have a high risk.”
The researchers then compared the MAP and clinical response for their ability to predict 6-month NRM throughout the first month of therapy for acute GVHD.
MAP bests response
After 1 month of therapy, the MAP was more accurate than clinical response for predicting 6-month NRM. The area under the curve was 0.84 and 0.65, respectively (P less than .001).
Likewise, the MAP after 1 week of therapy was more accurate than clinical response at 1 month for predicting 6-month NRM. The area under the curve was 0.80 and 0.65, respectively (P less than .001).
“[T]he clinical responses were good, but not great, at predicting long-term outcome, where the biomarker, the MAP, was significantly better,” Dr. Ferrara said. “[A]t every time point we tested, the biomarkers were better than the clinical responses.”
The researchers also identified subgroups of clinical responders and nonresponders for whom MAP more accurately predicted 6-month NRM.
The team found that 61% of clinical nonresponders were actually low risk according to MAP. And the incidence of 6-month NRM was significantly lower in the MAP-designated low-risk patients than in MAP-designated high-risk patients – 22% and 56%, respectively (P less than .001).
On the other hand, 10% of clinical responders were high risk according to MAP. The incidence of 6-month NRM was significantly higher in the high-risk patients than in the low-risk patients – 40% and 13%, respectively (P less than .001).
Assessing changes over time
The researchers found that patients who were initially high risk by MAP but had not experienced NRM by 6 months had significant decreases in their MAP after 4 weeks of treatment (P = .003). Patients who did experience NRM had a significant increase in their MAP whether their initial MAP was low (P = .007) or high (P = .024).
“What we found was that patients who lived tended to either have low biomarkers at the start of treatment and stay low or start out with high biomarkers and have reductions over the first month of therapy,” Mr. Srinagesh said. “Conversely, patients who tended to do worse were those who had either increases in their biomarkers or stayed high at all time points.”
The researchers identified a threshold – 0.290 – for separating patients by mortality risk.
“Patients who started out above the threshold and then went below it had a 5-fold reduction in mortality, whereas patients who started out below the threshold and rose above it had a 5-fold increase in mortality,” Mr. Srinagesh said.
MAP in clinical trials and practice
Based on these findings and results from related studies, the researchers theorize that MAP would be a better endpoint for clinical trials than clinical response.
At present, there are three trials in which researchers are using MAP as an endpoint to assess the efficacy of treatment for GVHD (NCT02133924, NCT03459040, and NCT03846479). Dr. Ferrara said a fourth trial is set to begin this summer.
Additionally, MAP is being used in clinical practice. A company called Viracor Eurofins Clinical Diagnostics licensed the MAGIC algorithm and provides three related tests for consumer use.
Viracor’s aGVHD Pre-Symptomatic Algorithm assigns patients to high- and low-risk groups based on results from samples collected 7 days after transplant. The aGVHD Symptomatic Onset Algorithm assigns patients to high-, intermediate-, and low-risk groups. The aGVHD Post-Treatment Algorithm, which can be used 7 days or more after GVHD treatment initiation, stratifies steroid-resistant patients into high- or low-risk groups for both NRM and overall survival.
“We are still in early days of figuring out how to use [the biomarker tests], but … what I’ve heard is that people are finding them to be useful in their clinical practice,” Dr. Ferrara said.
Dr. Ferrara has an ownership interest in and receives royalties from Viracor. Mr. Srinagesh reported having no relevant conflicts of interest. The research was supported by grants from the National Cancer Institute and the American Cancer Society.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – Researchers say they have identified biomarkers that may help guide early treatment decisions in patients with acute graft-versus-host disease (GVHD).
The biomarkers, ST2 and REG3-alpha, were measured during the first month of GVHD treatment and proved more accurate than clinical response for predicting 6-month nonrelapse mortality (NRM). In fact, biomarker assessment revealed patients who responded to treatment but had a high risk of NRM and nonresponders who had a low risk of NRM.
The researchers also found that biomarkers changed over the first month of treatment but remained significant predictors of NRM. This suggests that modifying treatment according to biomarker findings at various time points could result in better outcomes for patients.
“We think this is going to transform the way we treat graft-versus-host disease,” said James L.M. Ferrara, MD, DSc, of the Icahn School of Medicine at Mount Sinai, New York.
Dr. Ferrara and Hrishikesh Srinagesh, along with their colleagues at Mount Sinai, have conducted extensive research with these biomarkers and presented some of their findings at the Acute Leukemia Forum of Hemedicus.
Comparing biomarkers and response
In one study, the researchers evaluated 355 patients who had undergone allogeneic hematopoietic stem cell transplant at 1 of 20 Mount Sinai Acute GVHD International Consortium (MAGIC) centers between January 2016 and February 2018. All patients developed acute GVHD and received systemic steroids as treatment.
Patients provided blood samples weekly for the first month of treatment, and concentrations of ST2 and REG3-alpha were measured in each sample. Both biomarker concentrations were used to calculate the biomarker probability of NRM.
“The concentration of those two biomarkers are put into a computer, and we get … a single number, and that gives us the probability of mortality,” Dr. Ferrara said. “[W]e call this the MAGIC algorithm probability, or MAP. And when a MAP is low, the patient has a very low chance of dying from graft-versus-host disease, when it’s intermediate, they have an intermediate risk, and when it’s high, they have a high risk.”
The researchers then compared the MAP and clinical response for their ability to predict 6-month NRM throughout the first month of therapy for acute GVHD.
MAP bests response
After 1 month of therapy, the MAP was more accurate than clinical response for predicting 6-month NRM. The area under the curve was 0.84 and 0.65, respectively (P less than .001).
Likewise, the MAP after 1 week of therapy was more accurate than clinical response at 1 month for predicting 6-month NRM. The area under the curve was 0.80 and 0.65, respectively (P less than .001).
“[T]he clinical responses were good, but not great, at predicting long-term outcome, where the biomarker, the MAP, was significantly better,” Dr. Ferrara said. “[A]t every time point we tested, the biomarkers were better than the clinical responses.”
The researchers also identified subgroups of clinical responders and nonresponders for whom MAP more accurately predicted 6-month NRM.
The team found that 61% of clinical nonresponders were actually low risk according to MAP. And the incidence of 6-month NRM was significantly lower in the MAP-designated low-risk patients than in MAP-designated high-risk patients – 22% and 56%, respectively (P less than .001).
On the other hand, 10% of clinical responders were high risk according to MAP. The incidence of 6-month NRM was significantly higher in the high-risk patients than in the low-risk patients – 40% and 13%, respectively (P less than .001).
Assessing changes over time
The researchers found that patients who were initially high risk by MAP but had not experienced NRM by 6 months had significant decreases in their MAP after 4 weeks of treatment (P = .003). Patients who did experience NRM had a significant increase in their MAP whether their initial MAP was low (P = .007) or high (P = .024).
“What we found was that patients who lived tended to either have low biomarkers at the start of treatment and stay low or start out with high biomarkers and have reductions over the first month of therapy,” Mr. Srinagesh said. “Conversely, patients who tended to do worse were those who had either increases in their biomarkers or stayed high at all time points.”
The researchers identified a threshold – 0.290 – for separating patients by mortality risk.
“Patients who started out above the threshold and then went below it had a 5-fold reduction in mortality, whereas patients who started out below the threshold and rose above it had a 5-fold increase in mortality,” Mr. Srinagesh said.
MAP in clinical trials and practice
Based on these findings and results from related studies, the researchers theorize that MAP would be a better endpoint for clinical trials than clinical response.
At present, there are three trials in which researchers are using MAP as an endpoint to assess the efficacy of treatment for GVHD (NCT02133924, NCT03459040, and NCT03846479). Dr. Ferrara said a fourth trial is set to begin this summer.
Additionally, MAP is being used in clinical practice. A company called Viracor Eurofins Clinical Diagnostics licensed the MAGIC algorithm and provides three related tests for consumer use.
Viracor’s aGVHD Pre-Symptomatic Algorithm assigns patients to high- and low-risk groups based on results from samples collected 7 days after transplant. The aGVHD Symptomatic Onset Algorithm assigns patients to high-, intermediate-, and low-risk groups. The aGVHD Post-Treatment Algorithm, which can be used 7 days or more after GVHD treatment initiation, stratifies steroid-resistant patients into high- or low-risk groups for both NRM and overall survival.
“We are still in early days of figuring out how to use [the biomarker tests], but … what I’ve heard is that people are finding them to be useful in their clinical practice,” Dr. Ferrara said.
Dr. Ferrara has an ownership interest in and receives royalties from Viracor. Mr. Srinagesh reported having no relevant conflicts of interest. The research was supported by grants from the National Cancer Institute and the American Cancer Society.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – Researchers say they have identified biomarkers that may help guide early treatment decisions in patients with acute graft-versus-host disease (GVHD).
The biomarkers, ST2 and REG3-alpha, were measured during the first month of GVHD treatment and proved more accurate than clinical response for predicting 6-month nonrelapse mortality (NRM). In fact, biomarker assessment revealed patients who responded to treatment but had a high risk of NRM and nonresponders who had a low risk of NRM.
The researchers also found that biomarkers changed over the first month of treatment but remained significant predictors of NRM. This suggests that modifying treatment according to biomarker findings at various time points could result in better outcomes for patients.
“We think this is going to transform the way we treat graft-versus-host disease,” said James L.M. Ferrara, MD, DSc, of the Icahn School of Medicine at Mount Sinai, New York.
Dr. Ferrara and Hrishikesh Srinagesh, along with their colleagues at Mount Sinai, have conducted extensive research with these biomarkers and presented some of their findings at the Acute Leukemia Forum of Hemedicus.
Comparing biomarkers and response
In one study, the researchers evaluated 355 patients who had undergone allogeneic hematopoietic stem cell transplant at 1 of 20 Mount Sinai Acute GVHD International Consortium (MAGIC) centers between January 2016 and February 2018. All patients developed acute GVHD and received systemic steroids as treatment.
Patients provided blood samples weekly for the first month of treatment, and concentrations of ST2 and REG3-alpha were measured in each sample. Both biomarker concentrations were used to calculate the biomarker probability of NRM.
“The concentration of those two biomarkers are put into a computer, and we get … a single number, and that gives us the probability of mortality,” Dr. Ferrara said. “[W]e call this the MAGIC algorithm probability, or MAP. And when a MAP is low, the patient has a very low chance of dying from graft-versus-host disease, when it’s intermediate, they have an intermediate risk, and when it’s high, they have a high risk.”
The researchers then compared the MAP and clinical response for their ability to predict 6-month NRM throughout the first month of therapy for acute GVHD.
MAP bests response
After 1 month of therapy, the MAP was more accurate than clinical response for predicting 6-month NRM. The area under the curve was 0.84 and 0.65, respectively (P less than .001).
Likewise, the MAP after 1 week of therapy was more accurate than clinical response at 1 month for predicting 6-month NRM. The area under the curve was 0.80 and 0.65, respectively (P less than .001).
“[T]he clinical responses were good, but not great, at predicting long-term outcome, where the biomarker, the MAP, was significantly better,” Dr. Ferrara said. “[A]t every time point we tested, the biomarkers were better than the clinical responses.”
The researchers also identified subgroups of clinical responders and nonresponders for whom MAP more accurately predicted 6-month NRM.
The team found that 61% of clinical nonresponders were actually low risk according to MAP. And the incidence of 6-month NRM was significantly lower in the MAP-designated low-risk patients than in MAP-designated high-risk patients – 22% and 56%, respectively (P less than .001).
On the other hand, 10% of clinical responders were high risk according to MAP. The incidence of 6-month NRM was significantly higher in the high-risk patients than in the low-risk patients – 40% and 13%, respectively (P less than .001).
Assessing changes over time
The researchers found that patients who were initially high risk by MAP but had not experienced NRM by 6 months had significant decreases in their MAP after 4 weeks of treatment (P = .003). Patients who did experience NRM had a significant increase in their MAP whether their initial MAP was low (P = .007) or high (P = .024).
“What we found was that patients who lived tended to either have low biomarkers at the start of treatment and stay low or start out with high biomarkers and have reductions over the first month of therapy,” Mr. Srinagesh said. “Conversely, patients who tended to do worse were those who had either increases in their biomarkers or stayed high at all time points.”
The researchers identified a threshold – 0.290 – for separating patients by mortality risk.
“Patients who started out above the threshold and then went below it had a 5-fold reduction in mortality, whereas patients who started out below the threshold and rose above it had a 5-fold increase in mortality,” Mr. Srinagesh said.
MAP in clinical trials and practice
Based on these findings and results from related studies, the researchers theorize that MAP would be a better endpoint for clinical trials than clinical response.
At present, there are three trials in which researchers are using MAP as an endpoint to assess the efficacy of treatment for GVHD (NCT02133924, NCT03459040, and NCT03846479). Dr. Ferrara said a fourth trial is set to begin this summer.
Additionally, MAP is being used in clinical practice. A company called Viracor Eurofins Clinical Diagnostics licensed the MAGIC algorithm and provides three related tests for consumer use.
Viracor’s aGVHD Pre-Symptomatic Algorithm assigns patients to high- and low-risk groups based on results from samples collected 7 days after transplant. The aGVHD Symptomatic Onset Algorithm assigns patients to high-, intermediate-, and low-risk groups. The aGVHD Post-Treatment Algorithm, which can be used 7 days or more after GVHD treatment initiation, stratifies steroid-resistant patients into high- or low-risk groups for both NRM and overall survival.
“We are still in early days of figuring out how to use [the biomarker tests], but … what I’ve heard is that people are finding them to be useful in their clinical practice,” Dr. Ferrara said.
Dr. Ferrara has an ownership interest in and receives royalties from Viracor. Mr. Srinagesh reported having no relevant conflicts of interest. The research was supported by grants from the National Cancer Institute and the American Cancer Society.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
REPORTING FROM ALF 2019

Idelalisib shows long-term safety, efficacy for relapsed CLL
For patients with relapsed/refractory chronic lymphocytic leukemia (CLL), long-term treatment with the phosphoinositol 3-kinase inhibitor idelalisib appears safe and effective, according to investigators.
Final results from a phase 3 trial confirmed survival advantages when idelalisib is used in combination with rituximab, reported lead author Jeff P. Sharman, MD, of Willamette Valley Cancer Institute and Research Center in Springfield, Ore., and colleagues.
During follow-up, which exceeded 5 years in some patients, no new idelalisib-related adverse events were encountered, supporting the safety of long-term use, the investigators noted. The report is in the Journal of Clinical Oncology.
This study was “pivotal” for treating elderly patients with relapsed CLL, the investigators wrote, as these patients previously had few treatment options beyond supportive or palliative care.
Earlier results from the study showed that adding idelalisib to rituximab raised overall response rates from about 15.5% to 83.6% and median progression-free survival from 6.5 months to 19.4 months, resulting in “significantly better clinical outcomes compared with those seen with rituximab alone,” leading to approval by the Food and Drug Administration.
During the primary study, 110 patients received a combination of idelalisib and rituximab, while 108 patients received rituximab and placebo. The median patient age was 71 years, with a median of three lines of prior therapy. The present analysis focused on the 110 patients in the combination group who received at least one dose of idelalisib, whether or not they elected to participate in the extension phase.
After a median follow-up of 18 months, ranging from 0.3 months to 67.6 months, the overall response rate was 85.5% and the median progression-free survival was 20.3 months, both of which are similar to earlier findings. Median overall survival was 40.6 months.
With a median duration of exposure of 16.2 months, the safety analysis revealed no new idelalisib-related adverse events.
However, the investigators pointed out that prolonged therapy often led to diarrhea, which ultimately occurred in about half of patients (46.4%). Roughly equal amounts of patients experienced grade 2 (17.3%) or grade 3 or greater diarrhea (16.4%). In cases of grade 3 or greater diarrhea, steroid therapy was recommended, typically resulting in symptom resolution within 2 weeks; however, “there were insufficient numbers of patients to determine if steroid therapy affected the duration of symptoms,” the investigators wrote.
“The longer-term data presented here confirm the previously reported efficacy of targeting PI3K with idelalisib in patients with relapsed/refractory CLL and support the use of [idelalisib and rituximab] in this patient population with careful management of potential [adverse events],” they wrote.
Gilead Sciences funded the study. Dr. Sharman reported financial relationships with Gilead and other companies.
SOURCE: Sharman JP et al. J Clin Oncol. 2019 Apr 17. doi: 10.1200/JCO.18.01460.
For patients with relapsed/refractory chronic lymphocytic leukemia (CLL), long-term treatment with the phosphoinositol 3-kinase inhibitor idelalisib appears safe and effective, according to investigators.
Final results from a phase 3 trial confirmed survival advantages when idelalisib is used in combination with rituximab, reported lead author Jeff P. Sharman, MD, of Willamette Valley Cancer Institute and Research Center in Springfield, Ore., and colleagues.
During follow-up, which exceeded 5 years in some patients, no new idelalisib-related adverse events were encountered, supporting the safety of long-term use, the investigators noted. The report is in the Journal of Clinical Oncology.
This study was “pivotal” for treating elderly patients with relapsed CLL, the investigators wrote, as these patients previously had few treatment options beyond supportive or palliative care.
Earlier results from the study showed that adding idelalisib to rituximab raised overall response rates from about 15.5% to 83.6% and median progression-free survival from 6.5 months to 19.4 months, resulting in “significantly better clinical outcomes compared with those seen with rituximab alone,” leading to approval by the Food and Drug Administration.
During the primary study, 110 patients received a combination of idelalisib and rituximab, while 108 patients received rituximab and placebo. The median patient age was 71 years, with a median of three lines of prior therapy. The present analysis focused on the 110 patients in the combination group who received at least one dose of idelalisib, whether or not they elected to participate in the extension phase.
After a median follow-up of 18 months, ranging from 0.3 months to 67.6 months, the overall response rate was 85.5% and the median progression-free survival was 20.3 months, both of which are similar to earlier findings. Median overall survival was 40.6 months.
With a median duration of exposure of 16.2 months, the safety analysis revealed no new idelalisib-related adverse events.
However, the investigators pointed out that prolonged therapy often led to diarrhea, which ultimately occurred in about half of patients (46.4%). Roughly equal amounts of patients experienced grade 2 (17.3%) or grade 3 or greater diarrhea (16.4%). In cases of grade 3 or greater diarrhea, steroid therapy was recommended, typically resulting in symptom resolution within 2 weeks; however, “there were insufficient numbers of patients to determine if steroid therapy affected the duration of symptoms,” the investigators wrote.
“The longer-term data presented here confirm the previously reported efficacy of targeting PI3K with idelalisib in patients with relapsed/refractory CLL and support the use of [idelalisib and rituximab] in this patient population with careful management of potential [adverse events],” they wrote.
Gilead Sciences funded the study. Dr. Sharman reported financial relationships with Gilead and other companies.
SOURCE: Sharman JP et al. J Clin Oncol. 2019 Apr 17. doi: 10.1200/JCO.18.01460.
For patients with relapsed/refractory chronic lymphocytic leukemia (CLL), long-term treatment with the phosphoinositol 3-kinase inhibitor idelalisib appears safe and effective, according to investigators.
Final results from a phase 3 trial confirmed survival advantages when idelalisib is used in combination with rituximab, reported lead author Jeff P. Sharman, MD, of Willamette Valley Cancer Institute and Research Center in Springfield, Ore., and colleagues.
During follow-up, which exceeded 5 years in some patients, no new idelalisib-related adverse events were encountered, supporting the safety of long-term use, the investigators noted. The report is in the Journal of Clinical Oncology.
This study was “pivotal” for treating elderly patients with relapsed CLL, the investigators wrote, as these patients previously had few treatment options beyond supportive or palliative care.
Earlier results from the study showed that adding idelalisib to rituximab raised overall response rates from about 15.5% to 83.6% and median progression-free survival from 6.5 months to 19.4 months, resulting in “significantly better clinical outcomes compared with those seen with rituximab alone,” leading to approval by the Food and Drug Administration.
During the primary study, 110 patients received a combination of idelalisib and rituximab, while 108 patients received rituximab and placebo. The median patient age was 71 years, with a median of three lines of prior therapy. The present analysis focused on the 110 patients in the combination group who received at least one dose of idelalisib, whether or not they elected to participate in the extension phase.
After a median follow-up of 18 months, ranging from 0.3 months to 67.6 months, the overall response rate was 85.5% and the median progression-free survival was 20.3 months, both of which are similar to earlier findings. Median overall survival was 40.6 months.
With a median duration of exposure of 16.2 months, the safety analysis revealed no new idelalisib-related adverse events.
However, the investigators pointed out that prolonged therapy often led to diarrhea, which ultimately occurred in about half of patients (46.4%). Roughly equal amounts of patients experienced grade 2 (17.3%) or grade 3 or greater diarrhea (16.4%). In cases of grade 3 or greater diarrhea, steroid therapy was recommended, typically resulting in symptom resolution within 2 weeks; however, “there were insufficient numbers of patients to determine if steroid therapy affected the duration of symptoms,” the investigators wrote.
“The longer-term data presented here confirm the previously reported efficacy of targeting PI3K with idelalisib in patients with relapsed/refractory CLL and support the use of [idelalisib and rituximab] in this patient population with careful management of potential [adverse events],” they wrote.
Gilead Sciences funded the study. Dr. Sharman reported financial relationships with Gilead and other companies.
SOURCE: Sharman JP et al. J Clin Oncol. 2019 Apr 17. doi: 10.1200/JCO.18.01460.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Tagraxofusp produces high response rate in BPDCN
Tagraxofusp demonstrated efficacy in a phase 2 trial of patients with previously treated or untreated blastic plasmacytoid dendritic cell neoplasm (BPDCN).
The overall response rate was 90% in previously untreated patients who received the highest dose of tagraxofusp and 67% in patients with relapsed/refractory BPDCN.
The researchers wrote that capillary leak syndrome (CLS) was an important adverse event in this trial, as it caused two deaths. However, the researchers developed strategies that appear to mitigate the risk of CLS in patients taking tagraxofusp.
Naveen Pemmaraju, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues conducted the trial and reported the results in the New England Journal of Medicine.
The trial included 47 patients – 32 with previously untreated BPDCN and 15 with relapsed/refractory BPDCN. The patients’ median age at baseline was 70 years and 83% were men.
Three patients (all previously untreated) received tagraxofusp at 7 mcg/kg per day, and 44 patients received a 12 mcg/kg per day dose. All patients were treated on days 1-5 of a 21-day cycle.
Response and survival
In the 29 previously untreated patients who received the 12 mcg/kg dose of tagraxofusp, the overall response rate was 90%. The rate of complete response plus clinical complete response in these patients was 72%.
In the 15 patients with relapsed/refractory BPDCN, the overall response rate was 67%, and the rate of complete response plus clinical complete response was 33%.
A total of 14 patients, 13 of whom had previously untreated BPDCN, went on to transplant.
In the 29 previously untreated patients, the median overall survival was not reached at a median follow-up of 25 months. The overall survival rate was 62% at 12 months, 59% at 18 months, and 52% at 24 months.
In the 15 previously treated patients, the median overall survival was 8.5 months.
Safety
Common adverse events in this trial were ALT increase (64%), AST increase (60%), hypoalbuminemia (55%), peripheral edema (51%), thrombocytopenia (49%), nausea (45%), pyrexia (45%), and fatigue (45%).
Among the 44 patients who received the 12 mcg/kg dose of tagraxofusp, 8 (18%) developed CLS. Six patients had grade 2 CLS, one had grade 4, and one had grade 5. There was an additional CLS-related death in a patient who received tagraxofusp at 7 mcg/kg.
After the first death, the trial protocol was amended to reduce CLS risk. Inclusion criteria were changed so that patients must have normal cardiac function, adequate kidney function, and serum albumin of at least 3.2 g/dL. Additionally, the researchers began monitoring patients’ weight, albumin levels, and kidney function. The team withheld tagraxofusp if patients experienced rapid weight gain or if their serum albumin or systolic blood pressure fell too low.
The trial was sponsored by Stemline Therapeutics. The researchers reported relationships with Stemline and other companies.
Tagraxofusp demonstrated efficacy in a phase 2 trial of patients with previously treated or untreated blastic plasmacytoid dendritic cell neoplasm (BPDCN).
The overall response rate was 90% in previously untreated patients who received the highest dose of tagraxofusp and 67% in patients with relapsed/refractory BPDCN.
The researchers wrote that capillary leak syndrome (CLS) was an important adverse event in this trial, as it caused two deaths. However, the researchers developed strategies that appear to mitigate the risk of CLS in patients taking tagraxofusp.
Naveen Pemmaraju, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues conducted the trial and reported the results in the New England Journal of Medicine.
The trial included 47 patients – 32 with previously untreated BPDCN and 15 with relapsed/refractory BPDCN. The patients’ median age at baseline was 70 years and 83% were men.
Three patients (all previously untreated) received tagraxofusp at 7 mcg/kg per day, and 44 patients received a 12 mcg/kg per day dose. All patients were treated on days 1-5 of a 21-day cycle.
Response and survival
In the 29 previously untreated patients who received the 12 mcg/kg dose of tagraxofusp, the overall response rate was 90%. The rate of complete response plus clinical complete response in these patients was 72%.
In the 15 patients with relapsed/refractory BPDCN, the overall response rate was 67%, and the rate of complete response plus clinical complete response was 33%.
A total of 14 patients, 13 of whom had previously untreated BPDCN, went on to transplant.
In the 29 previously untreated patients, the median overall survival was not reached at a median follow-up of 25 months. The overall survival rate was 62% at 12 months, 59% at 18 months, and 52% at 24 months.
In the 15 previously treated patients, the median overall survival was 8.5 months.
Safety
Common adverse events in this trial were ALT increase (64%), AST increase (60%), hypoalbuminemia (55%), peripheral edema (51%), thrombocytopenia (49%), nausea (45%), pyrexia (45%), and fatigue (45%).
Among the 44 patients who received the 12 mcg/kg dose of tagraxofusp, 8 (18%) developed CLS. Six patients had grade 2 CLS, one had grade 4, and one had grade 5. There was an additional CLS-related death in a patient who received tagraxofusp at 7 mcg/kg.
After the first death, the trial protocol was amended to reduce CLS risk. Inclusion criteria were changed so that patients must have normal cardiac function, adequate kidney function, and serum albumin of at least 3.2 g/dL. Additionally, the researchers began monitoring patients’ weight, albumin levels, and kidney function. The team withheld tagraxofusp if patients experienced rapid weight gain or if their serum albumin or systolic blood pressure fell too low.
The trial was sponsored by Stemline Therapeutics. The researchers reported relationships with Stemline and other companies.
Tagraxofusp demonstrated efficacy in a phase 2 trial of patients with previously treated or untreated blastic plasmacytoid dendritic cell neoplasm (BPDCN).
The overall response rate was 90% in previously untreated patients who received the highest dose of tagraxofusp and 67% in patients with relapsed/refractory BPDCN.
The researchers wrote that capillary leak syndrome (CLS) was an important adverse event in this trial, as it caused two deaths. However, the researchers developed strategies that appear to mitigate the risk of CLS in patients taking tagraxofusp.
Naveen Pemmaraju, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues conducted the trial and reported the results in the New England Journal of Medicine.
The trial included 47 patients – 32 with previously untreated BPDCN and 15 with relapsed/refractory BPDCN. The patients’ median age at baseline was 70 years and 83% were men.
Three patients (all previously untreated) received tagraxofusp at 7 mcg/kg per day, and 44 patients received a 12 mcg/kg per day dose. All patients were treated on days 1-5 of a 21-day cycle.
Response and survival
In the 29 previously untreated patients who received the 12 mcg/kg dose of tagraxofusp, the overall response rate was 90%. The rate of complete response plus clinical complete response in these patients was 72%.
In the 15 patients with relapsed/refractory BPDCN, the overall response rate was 67%, and the rate of complete response plus clinical complete response was 33%.
A total of 14 patients, 13 of whom had previously untreated BPDCN, went on to transplant.
In the 29 previously untreated patients, the median overall survival was not reached at a median follow-up of 25 months. The overall survival rate was 62% at 12 months, 59% at 18 months, and 52% at 24 months.
In the 15 previously treated patients, the median overall survival was 8.5 months.
Safety
Common adverse events in this trial were ALT increase (64%), AST increase (60%), hypoalbuminemia (55%), peripheral edema (51%), thrombocytopenia (49%), nausea (45%), pyrexia (45%), and fatigue (45%).
Among the 44 patients who received the 12 mcg/kg dose of tagraxofusp, 8 (18%) developed CLS. Six patients had grade 2 CLS, one had grade 4, and one had grade 5. There was an additional CLS-related death in a patient who received tagraxofusp at 7 mcg/kg.
After the first death, the trial protocol was amended to reduce CLS risk. Inclusion criteria were changed so that patients must have normal cardiac function, adequate kidney function, and serum albumin of at least 3.2 g/dL. Additionally, the researchers began monitoring patients’ weight, albumin levels, and kidney function. The team withheld tagraxofusp if patients experienced rapid weight gain or if their serum albumin or systolic blood pressure fell too low.
The trial was sponsored by Stemline Therapeutics. The researchers reported relationships with Stemline and other companies.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Tagraxofusp produced responses in patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN).
Major finding: The overall response rate was 90% in previously untreated patients who received the highest dose of tagraxofusp and 67% in patients with relapsed/refractory BPDCN.
Study details: A phase 2 trial of 47 patients, 32 with previously untreated BPDCN and 15 with relapsed/refractory BPDCN.
Disclosures: The trial was sponsored by Stemline Therapeutics. The researchers reported relationships with Stemline and other companies.
Source: Pemmaraju N et al. N Engl J Med. 2019;380:1628-37.
Complementary medicine use common among patients on TKIs
GLASGOW – Many patients receiving tyrosine kinase inhibitors (TKIs) are taking complementary therapies or eating foods that interfere with TKI metabolism, based on results of a British survey of patients with chronic myeloid leukemia.

About one out of three patients with chronic myeloid leukemia (CML) reported taking complementary medicines, according to lead author David Sparksman, MD, of Norfolk and Norwich (England) University Hospital, and his colleagues.
Only a minority of patients were aware of the potential for dietary interactions with TKIs. However, even knowing the potential risk, about a quarter of patients still didn’t exclude these foods from their diets.
“These worrying results are unlikely to be confined to patients with CML,” the investigators wrote in an abstract presented at the annual meeting of the British Society for Haematology. “TKIs are used in the treatment of many other haematological malignancies.”
Because TKIs are metabolized by cytochrome P450 enzymes, inhibition of these enzymes by complementary therapies and foods may alter metabolism, and therefore, safety and efficacy of TKIs, according to the investigators.
“Use of complementary medicines and belief in their effectiveness is common,” the investigators wrote. “In a recent YouGov poll, 51% of those asked believed herbal medicine to be an effective treatment for illness.”
To investigate the prevalence of these beliefs and practices in a subset of cancer patients, the investigators identified 78 patients with CML undergoing follow-up at Norfolk and Norwich University Hospital. The median age of patients was 60 years. Eleven patients were excluded because they were not receiving a TKI and 6 patients declined to participate, leaving 61 patients in the final survey group.
Of these respondents, 41% had considered taking a complementary therapy and 34% were actively doing so. Further questioning revealed that about half of the patients taking a complementary medicine (52%) were taking a drug with known potential to interact with their TKI. Of these 11 patients, 5 were taking a complementary drug that would reduce serum concentrations of their TKI, potentially making it less effective. Conversely, six patients were taking a complementary drug that would increase serum concentrations, potentially increasing the risk of TKI side effects.
About 39% of respondents were aware of possible dietary interactions with TKIs, such as grapefruit. “Surprisingly,” the investigators said, 25% of patients with this knowledge still included such foods in their diet.
Dietary questioning revealed that among the patients who were unaware of food interactions, 67% were consuming foods that interact with TKIs.
Considering these results, the investigators offered some advice on patient communication and management.
“The use of complementary medicine should be discussed with all patients when starting TKIs and written information given to patients should highlight the potential dangers posed by substances which many patients currently regard as harmless,” thy wrote. “Since most patients will remain on treatment for many years, re-discussion about food and drug interactions should take place periodically to remind them of the potential risks.”
The investigators reported having no conflicts of interest.
GLASGOW – Many patients receiving tyrosine kinase inhibitors (TKIs) are taking complementary therapies or eating foods that interfere with TKI metabolism, based on results of a British survey of patients with chronic myeloid leukemia.
About one out of three patients with chronic myeloid leukemia (CML) reported taking complementary medicines, according to lead author David Sparksman, MD, of Norfolk and Norwich (England) University Hospital, and his colleagues.
Only a minority of patients were aware of the potential for dietary interactions with TKIs. However, even knowing the potential risk, about a quarter of patients still didn’t exclude these foods from their diets.
“These worrying results are unlikely to be confined to patients with CML,” the investigators wrote in an abstract presented at the annual meeting of the British Society for Haematology. “TKIs are used in the treatment of many other haematological malignancies.”
Because TKIs are metabolized by cytochrome P450 enzymes, inhibition of these enzymes by complementary therapies and foods may alter metabolism, and therefore, safety and efficacy of TKIs, according to the investigators.
“Use of complementary medicines and belief in their effectiveness is common,” the investigators wrote. “In a recent YouGov poll, 51% of those asked believed herbal medicine to be an effective treatment for illness.”
To investigate the prevalence of these beliefs and practices in a subset of cancer patients, the investigators identified 78 patients with CML undergoing follow-up at Norfolk and Norwich University Hospital. The median age of patients was 60 years. Eleven patients were excluded because they were not receiving a TKI and 6 patients declined to participate, leaving 61 patients in the final survey group.
Of these respondents, 41% had considered taking a complementary therapy and 34% were actively doing so. Further questioning revealed that about half of the patients taking a complementary medicine (52%) were taking a drug with known potential to interact with their TKI. Of these 11 patients, 5 were taking a complementary drug that would reduce serum concentrations of their TKI, potentially making it less effective. Conversely, six patients were taking a complementary drug that would increase serum concentrations, potentially increasing the risk of TKI side effects.
About 39% of respondents were aware of possible dietary interactions with TKIs, such as grapefruit. “Surprisingly,” the investigators said, 25% of patients with this knowledge still included such foods in their diet.
Dietary questioning revealed that among the patients who were unaware of food interactions, 67% were consuming foods that interact with TKIs.
Considering these results, the investigators offered some advice on patient communication and management.
“The use of complementary medicine should be discussed with all patients when starting TKIs and written information given to patients should highlight the potential dangers posed by substances which many patients currently regard as harmless,” thy wrote. “Since most patients will remain on treatment for many years, re-discussion about food and drug interactions should take place periodically to remind them of the potential risks.”
The investigators reported having no conflicts of interest.
GLASGOW – Many patients receiving tyrosine kinase inhibitors (TKIs) are taking complementary therapies or eating foods that interfere with TKI metabolism, based on results of a British survey of patients with chronic myeloid leukemia.
About one out of three patients with chronic myeloid leukemia (CML) reported taking complementary medicines, according to lead author David Sparksman, MD, of Norfolk and Norwich (England) University Hospital, and his colleagues.
Only a minority of patients were aware of the potential for dietary interactions with TKIs. However, even knowing the potential risk, about a quarter of patients still didn’t exclude these foods from their diets.
“These worrying results are unlikely to be confined to patients with CML,” the investigators wrote in an abstract presented at the annual meeting of the British Society for Haematology. “TKIs are used in the treatment of many other haematological malignancies.”
Because TKIs are metabolized by cytochrome P450 enzymes, inhibition of these enzymes by complementary therapies and foods may alter metabolism, and therefore, safety and efficacy of TKIs, according to the investigators.
“Use of complementary medicines and belief in their effectiveness is common,” the investigators wrote. “In a recent YouGov poll, 51% of those asked believed herbal medicine to be an effective treatment for illness.”
To investigate the prevalence of these beliefs and practices in a subset of cancer patients, the investigators identified 78 patients with CML undergoing follow-up at Norfolk and Norwich University Hospital. The median age of patients was 60 years. Eleven patients were excluded because they were not receiving a TKI and 6 patients declined to participate, leaving 61 patients in the final survey group.
Of these respondents, 41% had considered taking a complementary therapy and 34% were actively doing so. Further questioning revealed that about half of the patients taking a complementary medicine (52%) were taking a drug with known potential to interact with their TKI. Of these 11 patients, 5 were taking a complementary drug that would reduce serum concentrations of their TKI, potentially making it less effective. Conversely, six patients were taking a complementary drug that would increase serum concentrations, potentially increasing the risk of TKI side effects.
About 39% of respondents were aware of possible dietary interactions with TKIs, such as grapefruit. “Surprisingly,” the investigators said, 25% of patients with this knowledge still included such foods in their diet.
Dietary questioning revealed that among the patients who were unaware of food interactions, 67% were consuming foods that interact with TKIs.
Considering these results, the investigators offered some advice on patient communication and management.
“The use of complementary medicine should be discussed with all patients when starting TKIs and written information given to patients should highlight the potential dangers posed by substances which many patients currently regard as harmless,” thy wrote. “Since most patients will remain on treatment for many years, re-discussion about food and drug interactions should take place periodically to remind them of the potential risks.”
The investigators reported having no conflicts of interest.
REPORTING FROM BSH 2019