User login
Hodgkin lymphoma survivors are at an increased risk of subsequent ER-negative breast cancer
Young women with primary Hodgkin lymphoma had an increased relative risk of estrogen receptor–positive breast cancer if they received radiotherapy and, irrespective of the type of treatment they got, an elevated risk of ER-negative breast cancer, based on results of a study based on patient records from 12 U.S. National Cancer Institute Surveillance, Epidemiology, and End Results registries.
Of 7,355 women diagnosed with primary Hodgkin lymphoma during 1973-2009 and aged 10-39 years, 377 patients subsequently were diagnosed with breast cancer at a mean age of 45 years; 57% of the cancers were ER positive, 34% were ER negative, and 9% had unknown/borderline ER status, Diana R. Withrow, PhD, and her colleagues from the radiation epidemiology branch, division of cancer epidemiology and genetics, National Cancer Institute reported in JAMA Oncology.
Survivors of Hodgkin lymphoma had a greater relative risk of ER-negative (standardized incidence ratio, 5.8; 95% confidence interval, 4.8-6.9) than ER-positive breast cancer (SIR, 3.1; 95% CI, 2.7-3.5; P less than .001 for the difference), the researchers wrote.
For ER-positive disease, the increased SIR was observed only among women who had received radiotherapy for their Hodgkin lymphoma (SIR, 3.9; 95% CI, 3.4-4.5). In this group, the SIR for ER-positive disease was lower in the chemotherapy than in the no/unknown chemotherapy group (P = .04), said the researchers.
The authors acknowledged that lack of information on patient risk factors such as family history, reproductive factors, and hormone therapy, as well as detailed treatment information such as radiotherapy dose, fields, specific chemotherapeutic agents, and subsequent therapy is a limitation of the current study. Further research, including comprehensive treatment records, will lead to a better understanding of the association between treatment and breast cancer subtype in these patients, the researchers concluded.
None of the study authors reported any conflicts of interest.
SOURCE: Withrow D et al. doi: 10.1001/jamaoncol.2017.4887.
Young women with primary Hodgkin lymphoma had an increased relative risk of estrogen receptor–positive breast cancer if they received radiotherapy and, irrespective of the type of treatment they got, an elevated risk of ER-negative breast cancer, based on results of a study based on patient records from 12 U.S. National Cancer Institute Surveillance, Epidemiology, and End Results registries.
Of 7,355 women diagnosed with primary Hodgkin lymphoma during 1973-2009 and aged 10-39 years, 377 patients subsequently were diagnosed with breast cancer at a mean age of 45 years; 57% of the cancers were ER positive, 34% were ER negative, and 9% had unknown/borderline ER status, Diana R. Withrow, PhD, and her colleagues from the radiation epidemiology branch, division of cancer epidemiology and genetics, National Cancer Institute reported in JAMA Oncology.
Survivors of Hodgkin lymphoma had a greater relative risk of ER-negative (standardized incidence ratio, 5.8; 95% confidence interval, 4.8-6.9) than ER-positive breast cancer (SIR, 3.1; 95% CI, 2.7-3.5; P less than .001 for the difference), the researchers wrote.
For ER-positive disease, the increased SIR was observed only among women who had received radiotherapy for their Hodgkin lymphoma (SIR, 3.9; 95% CI, 3.4-4.5). In this group, the SIR for ER-positive disease was lower in the chemotherapy than in the no/unknown chemotherapy group (P = .04), said the researchers.
The authors acknowledged that lack of information on patient risk factors such as family history, reproductive factors, and hormone therapy, as well as detailed treatment information such as radiotherapy dose, fields, specific chemotherapeutic agents, and subsequent therapy is a limitation of the current study. Further research, including comprehensive treatment records, will lead to a better understanding of the association between treatment and breast cancer subtype in these patients, the researchers concluded.
None of the study authors reported any conflicts of interest.
SOURCE: Withrow D et al. doi: 10.1001/jamaoncol.2017.4887.
Young women with primary Hodgkin lymphoma had an increased relative risk of estrogen receptor–positive breast cancer if they received radiotherapy and, irrespective of the type of treatment they got, an elevated risk of ER-negative breast cancer, based on results of a study based on patient records from 12 U.S. National Cancer Institute Surveillance, Epidemiology, and End Results registries.
Of 7,355 women diagnosed with primary Hodgkin lymphoma during 1973-2009 and aged 10-39 years, 377 patients subsequently were diagnosed with breast cancer at a mean age of 45 years; 57% of the cancers were ER positive, 34% were ER negative, and 9% had unknown/borderline ER status, Diana R. Withrow, PhD, and her colleagues from the radiation epidemiology branch, division of cancer epidemiology and genetics, National Cancer Institute reported in JAMA Oncology.
Survivors of Hodgkin lymphoma had a greater relative risk of ER-negative (standardized incidence ratio, 5.8; 95% confidence interval, 4.8-6.9) than ER-positive breast cancer (SIR, 3.1; 95% CI, 2.7-3.5; P less than .001 for the difference), the researchers wrote.
For ER-positive disease, the increased SIR was observed only among women who had received radiotherapy for their Hodgkin lymphoma (SIR, 3.9; 95% CI, 3.4-4.5). In this group, the SIR for ER-positive disease was lower in the chemotherapy than in the no/unknown chemotherapy group (P = .04), said the researchers.
The authors acknowledged that lack of information on patient risk factors such as family history, reproductive factors, and hormone therapy, as well as detailed treatment information such as radiotherapy dose, fields, specific chemotherapeutic agents, and subsequent therapy is a limitation of the current study. Further research, including comprehensive treatment records, will lead to a better understanding of the association between treatment and breast cancer subtype in these patients, the researchers concluded.
None of the study authors reported any conflicts of interest.
SOURCE: Withrow D et al. doi: 10.1001/jamaoncol.2017.4887.
FROM JAMA ONCOLOGY
Key clinical point: .
Major finding: Survivors of Hodgkin lymphoma had a greater relative risk of ER-negative (standardized incidence ratio, 5.8) than ER-positive breast cancer (SIR, 3.1).
Study details: 7,355 women diagnosed with first primary Hodgkin lymphoma during 1973-2009, who were aged 10-39 years, and reported to 12 U.S. National Cancer Institute SEER registries.
Disclosures: None reported.
Source: Withrow D et al. doi: 10.1001/jamaoncolo.2017.4887.
Frontline brentuximab vedotin improved Hodgkin lymphoma outcomes
Replacing bleomycin with brentuximab vedotin in the classic ABVD regimen improved a measure of progression-free survival and reduced pulmonary toxicity in patients with previously untreated Hodgkin lymphoma, findings from a randomized, phase 3 trial suggest.
Patients receiving brentuximab plus chemotherapy had a “statistically significant and clinically meaningful improvement” in the rate of modified progression-free survival, according to results published in the New England Journal of Medicine.
Pulmonary toxicity also occurred at a lower rate with the regimen containing brentuximab, an anti-CD30 antibody–drug conjugate, wrote Joseph M. Connors, MD, of the British Columbia Cancer Agency, Vancouver, and his coauthors.
Taken together, these findings suggest brentuximab vedotin and chemotherapy had “substantially less pulmonary toxicity and appears to be more effective for frontline treatment of advanced-stage classic Hodgkin lymphoma,” the researchers wrote.
Bleomycin is often omitted from later cycles of chemotherapy for patients with Hodgkin lymphoma due to pulmonary symptoms, and is sometimes associated with unpredictable or even fatal pulmonary toxicity, the researchers noted.
The brentuximab vedotin arm of the trial did have more neurotoxicity, which was largely reversible, and more myelotoxicity, though that “can be ameliorated with prophylactic granulocyte colony-stimulating factor (G-CSF),” the researchers wrote.
The study by Dr. Connors and colleagues, known as ECHELON-1, was an open-label, multicenter, randomized phase 3 trial including patients with previously untreated stage III or IV classic Hodgkin lymphoma. Among enrolled patients, 664 received brentuximab vedotin plus doxorubicin, vinblastine, and dacarbazine (AVD), and 670 received standard doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD).
The study used a primary endpoint of progression-free survival augmented to include not only time to disease progression or death, but also “modified progression,” which the researchers defined as evidence of noncomplete response at the end of frontline chemotherapy.
It is accepted practice to give more chemotherapy or radiotherapy in Hodgkin lymphoma patients who have a positive PET scan at the end of frontline therapy, since metabolically detectable residual disease reliably predicts imminent progression, Dr. Connors and coauthors wrote.
“In this context, the conventional endpoint of progression-free survival does not accurately assess the curative intent of frontline chemotherapy,” they wrote.
With a median 24.9-month follow-up, modified 2-year progression-free survival in the trial was 82.1% for patients receiving brentuximab plus AVD, versus 77.2% for ABVD (P = .03), a 23% risk reduction.
Pulmonary toxicity of grade 3 or higher occurred in less than 1% of patients in the brentuximab vedotin plus AVD arm of the trial and in 3% of the ABVD arm.
Neutropenia was reported in 58% and 45% in the brentuximab vedotin plus AVD and ABVD arms, respectively, while febrile neutropenia was reported in 19% and 8%, respectively.
In the brentuximab plus AVD group, the rate of febrile neutropenia was 11% for those patients who received primary prophylaxis with G-CSF, and 21% for patients who did not, the researchers noted.
Peripheral neuropathy was seen in 67% and 43% of the brentuximab vedotin plus AVD and ABVD arms, respectively, and about two-thirds of the patients in the brentuximab vedotin plus AVD arm had improvement or resolution at the final follow-up visit.
The study was supported by Millennium Pharmaceuticals and Seattle Genetics. Researchers reported ties to Millennium Pharmaceuticals, Takeda Pharmaceuticals, Seattle Genetics, and other companies.
hematologynews@frontlinemedcom.com
SOURCE: Connors JM et al. N Engl J Med. 2018;378:331-44.
The incorporation of the CD30 immunotoxin brentuximab vedotin into frontline therapy for Hodgkin lymphoma “has been eagerly anticipated, and the wait is over,” Dan L. Longo, MD, and Vincent T. DeVita Jr., MD, wrote in an editorial.
Results of the randomized phase 3 trial by Dr. Connors and colleagues suggest that, with a relatively short follow-up, adding brentuximab vedotin to AVD combination chemotherapy “merits consideration” as a first-line treatment, according to Dr. Longo and Dr. DeVita.
“Although it is too early to rule out unanticipated late side effects … brentuximab vedotin + AVD appears to be more effective than ABVD (and is unlikely to be less effective) and is associated with fewer, more treatable toxicities,” they wrote.
Adverse effects of ABVD are generally modest, but the bleomycin in the regimen can result in serious pulmonary toxicity. While the rate of serious pulmonary toxicity is low, “clinicians have the impression that it is unpredictable,” the authors noted.
Beyond a significant improvement in modified progression-free survival with a follow-up of 25 months, brentuximab vedotin plus AVD was associated with lower pulmonary toxicity, they noted.
While the brentuximab vedotin had more neutropenia and neuropathy, neutropenia could be addressed with G-CSF between doses, and neuropathy was mainly low grade and completely resolved most of the time, the authors said.
Brentuximab had promising single-agent activity in previous Hodgkin lymphoma studies, so substituting it for bleomycin “had the potential to improve on ABVD. And it did,” Dr. Longo and Dr. DeVita wrote.
Dr. Longo is with Dana-Farber Cancer Institute, Boston, and Dr. DeVita is with the Yale Cancer Center, New Haven, Conn. These comments are based on their editorial appearing in the New England Journal of Medicine (2018 Jan 24. doi: 10.1056/NEJMe1715141). Dr. DeVita reported no disclosures, and Dr. Longo reported employment as Deputy Editor with the New England Journal of Medicine.
The incorporation of the CD30 immunotoxin brentuximab vedotin into frontline therapy for Hodgkin lymphoma “has been eagerly anticipated, and the wait is over,” Dan L. Longo, MD, and Vincent T. DeVita Jr., MD, wrote in an editorial.
Results of the randomized phase 3 trial by Dr. Connors and colleagues suggest that, with a relatively short follow-up, adding brentuximab vedotin to AVD combination chemotherapy “merits consideration” as a first-line treatment, according to Dr. Longo and Dr. DeVita.
“Although it is too early to rule out unanticipated late side effects … brentuximab vedotin + AVD appears to be more effective than ABVD (and is unlikely to be less effective) and is associated with fewer, more treatable toxicities,” they wrote.
Adverse effects of ABVD are generally modest, but the bleomycin in the regimen can result in serious pulmonary toxicity. While the rate of serious pulmonary toxicity is low, “clinicians have the impression that it is unpredictable,” the authors noted.
Beyond a significant improvement in modified progression-free survival with a follow-up of 25 months, brentuximab vedotin plus AVD was associated with lower pulmonary toxicity, they noted.
While the brentuximab vedotin had more neutropenia and neuropathy, neutropenia could be addressed with G-CSF between doses, and neuropathy was mainly low grade and completely resolved most of the time, the authors said.
Brentuximab had promising single-agent activity in previous Hodgkin lymphoma studies, so substituting it for bleomycin “had the potential to improve on ABVD. And it did,” Dr. Longo and Dr. DeVita wrote.
Dr. Longo is with Dana-Farber Cancer Institute, Boston, and Dr. DeVita is with the Yale Cancer Center, New Haven, Conn. These comments are based on their editorial appearing in the New England Journal of Medicine (2018 Jan 24. doi: 10.1056/NEJMe1715141). Dr. DeVita reported no disclosures, and Dr. Longo reported employment as Deputy Editor with the New England Journal of Medicine.
The incorporation of the CD30 immunotoxin brentuximab vedotin into frontline therapy for Hodgkin lymphoma “has been eagerly anticipated, and the wait is over,” Dan L. Longo, MD, and Vincent T. DeVita Jr., MD, wrote in an editorial.
Results of the randomized phase 3 trial by Dr. Connors and colleagues suggest that, with a relatively short follow-up, adding brentuximab vedotin to AVD combination chemotherapy “merits consideration” as a first-line treatment, according to Dr. Longo and Dr. DeVita.
“Although it is too early to rule out unanticipated late side effects … brentuximab vedotin + AVD appears to be more effective than ABVD (and is unlikely to be less effective) and is associated with fewer, more treatable toxicities,” they wrote.
Adverse effects of ABVD are generally modest, but the bleomycin in the regimen can result in serious pulmonary toxicity. While the rate of serious pulmonary toxicity is low, “clinicians have the impression that it is unpredictable,” the authors noted.
Beyond a significant improvement in modified progression-free survival with a follow-up of 25 months, brentuximab vedotin plus AVD was associated with lower pulmonary toxicity, they noted.
While the brentuximab vedotin had more neutropenia and neuropathy, neutropenia could be addressed with G-CSF between doses, and neuropathy was mainly low grade and completely resolved most of the time, the authors said.
Brentuximab had promising single-agent activity in previous Hodgkin lymphoma studies, so substituting it for bleomycin “had the potential to improve on ABVD. And it did,” Dr. Longo and Dr. DeVita wrote.
Dr. Longo is with Dana-Farber Cancer Institute, Boston, and Dr. DeVita is with the Yale Cancer Center, New Haven, Conn. These comments are based on their editorial appearing in the New England Journal of Medicine (2018 Jan 24. doi: 10.1056/NEJMe1715141). Dr. DeVita reported no disclosures, and Dr. Longo reported employment as Deputy Editor with the New England Journal of Medicine.
Replacing bleomycin with brentuximab vedotin in the classic ABVD regimen improved a measure of progression-free survival and reduced pulmonary toxicity in patients with previously untreated Hodgkin lymphoma, findings from a randomized, phase 3 trial suggest.
Patients receiving brentuximab plus chemotherapy had a “statistically significant and clinically meaningful improvement” in the rate of modified progression-free survival, according to results published in the New England Journal of Medicine.
Pulmonary toxicity also occurred at a lower rate with the regimen containing brentuximab, an anti-CD30 antibody–drug conjugate, wrote Joseph M. Connors, MD, of the British Columbia Cancer Agency, Vancouver, and his coauthors.
Taken together, these findings suggest brentuximab vedotin and chemotherapy had “substantially less pulmonary toxicity and appears to be more effective for frontline treatment of advanced-stage classic Hodgkin lymphoma,” the researchers wrote.
Bleomycin is often omitted from later cycles of chemotherapy for patients with Hodgkin lymphoma due to pulmonary symptoms, and is sometimes associated with unpredictable or even fatal pulmonary toxicity, the researchers noted.
The brentuximab vedotin arm of the trial did have more neurotoxicity, which was largely reversible, and more myelotoxicity, though that “can be ameliorated with prophylactic granulocyte colony-stimulating factor (G-CSF),” the researchers wrote.
The study by Dr. Connors and colleagues, known as ECHELON-1, was an open-label, multicenter, randomized phase 3 trial including patients with previously untreated stage III or IV classic Hodgkin lymphoma. Among enrolled patients, 664 received brentuximab vedotin plus doxorubicin, vinblastine, and dacarbazine (AVD), and 670 received standard doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD).
The study used a primary endpoint of progression-free survival augmented to include not only time to disease progression or death, but also “modified progression,” which the researchers defined as evidence of noncomplete response at the end of frontline chemotherapy.
It is accepted practice to give more chemotherapy or radiotherapy in Hodgkin lymphoma patients who have a positive PET scan at the end of frontline therapy, since metabolically detectable residual disease reliably predicts imminent progression, Dr. Connors and coauthors wrote.
“In this context, the conventional endpoint of progression-free survival does not accurately assess the curative intent of frontline chemotherapy,” they wrote.
With a median 24.9-month follow-up, modified 2-year progression-free survival in the trial was 82.1% for patients receiving brentuximab plus AVD, versus 77.2% for ABVD (P = .03), a 23% risk reduction.
Pulmonary toxicity of grade 3 or higher occurred in less than 1% of patients in the brentuximab vedotin plus AVD arm of the trial and in 3% of the ABVD arm.
Neutropenia was reported in 58% and 45% in the brentuximab vedotin plus AVD and ABVD arms, respectively, while febrile neutropenia was reported in 19% and 8%, respectively.
In the brentuximab plus AVD group, the rate of febrile neutropenia was 11% for those patients who received primary prophylaxis with G-CSF, and 21% for patients who did not, the researchers noted.
Peripheral neuropathy was seen in 67% and 43% of the brentuximab vedotin plus AVD and ABVD arms, respectively, and about two-thirds of the patients in the brentuximab vedotin plus AVD arm had improvement or resolution at the final follow-up visit.
The study was supported by Millennium Pharmaceuticals and Seattle Genetics. Researchers reported ties to Millennium Pharmaceuticals, Takeda Pharmaceuticals, Seattle Genetics, and other companies.
hematologynews@frontlinemedcom.com
SOURCE: Connors JM et al. N Engl J Med. 2018;378:331-44.
Replacing bleomycin with brentuximab vedotin in the classic ABVD regimen improved a measure of progression-free survival and reduced pulmonary toxicity in patients with previously untreated Hodgkin lymphoma, findings from a randomized, phase 3 trial suggest.
Patients receiving brentuximab plus chemotherapy had a “statistically significant and clinically meaningful improvement” in the rate of modified progression-free survival, according to results published in the New England Journal of Medicine.
Pulmonary toxicity also occurred at a lower rate with the regimen containing brentuximab, an anti-CD30 antibody–drug conjugate, wrote Joseph M. Connors, MD, of the British Columbia Cancer Agency, Vancouver, and his coauthors.
Taken together, these findings suggest brentuximab vedotin and chemotherapy had “substantially less pulmonary toxicity and appears to be more effective for frontline treatment of advanced-stage classic Hodgkin lymphoma,” the researchers wrote.
Bleomycin is often omitted from later cycles of chemotherapy for patients with Hodgkin lymphoma due to pulmonary symptoms, and is sometimes associated with unpredictable or even fatal pulmonary toxicity, the researchers noted.
The brentuximab vedotin arm of the trial did have more neurotoxicity, which was largely reversible, and more myelotoxicity, though that “can be ameliorated with prophylactic granulocyte colony-stimulating factor (G-CSF),” the researchers wrote.
The study by Dr. Connors and colleagues, known as ECHELON-1, was an open-label, multicenter, randomized phase 3 trial including patients with previously untreated stage III or IV classic Hodgkin lymphoma. Among enrolled patients, 664 received brentuximab vedotin plus doxorubicin, vinblastine, and dacarbazine (AVD), and 670 received standard doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD).
The study used a primary endpoint of progression-free survival augmented to include not only time to disease progression or death, but also “modified progression,” which the researchers defined as evidence of noncomplete response at the end of frontline chemotherapy.
It is accepted practice to give more chemotherapy or radiotherapy in Hodgkin lymphoma patients who have a positive PET scan at the end of frontline therapy, since metabolically detectable residual disease reliably predicts imminent progression, Dr. Connors and coauthors wrote.
“In this context, the conventional endpoint of progression-free survival does not accurately assess the curative intent of frontline chemotherapy,” they wrote.
With a median 24.9-month follow-up, modified 2-year progression-free survival in the trial was 82.1% for patients receiving brentuximab plus AVD, versus 77.2% for ABVD (P = .03), a 23% risk reduction.
Pulmonary toxicity of grade 3 or higher occurred in less than 1% of patients in the brentuximab vedotin plus AVD arm of the trial and in 3% of the ABVD arm.
Neutropenia was reported in 58% and 45% in the brentuximab vedotin plus AVD and ABVD arms, respectively, while febrile neutropenia was reported in 19% and 8%, respectively.
In the brentuximab plus AVD group, the rate of febrile neutropenia was 11% for those patients who received primary prophylaxis with G-CSF, and 21% for patients who did not, the researchers noted.
Peripheral neuropathy was seen in 67% and 43% of the brentuximab vedotin plus AVD and ABVD arms, respectively, and about two-thirds of the patients in the brentuximab vedotin plus AVD arm had improvement or resolution at the final follow-up visit.
The study was supported by Millennium Pharmaceuticals and Seattle Genetics. Researchers reported ties to Millennium Pharmaceuticals, Takeda Pharmaceuticals, Seattle Genetics, and other companies.
hematologynews@frontlinemedcom.com
SOURCE: Connors JM et al. N Engl J Med. 2018;378:331-44.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Modified 2-year progression-free survival was 82.1% with the brentuximab vedotin–containing regimen versus 77.2% for ABVD (P = .03).
Study details: An open-label, multicenter, randomized phase 3 trial including 1,334 patients with previously untreated stage III or IV classic Hodgkin lymphoma.
Disclosures: The study was supported by Millennium Pharmaceuticals and Seattle Genetics. Researchers reported ties to Millennium Pharmaceuticals, Takeda Pharmaceuticals, Seattle Genetics, and other companies.
Source: Connors JM et al. N Engl J Med. 2018;378:331-44.
T-cell therapy produces durable responses in rel/ref HL

Engineered T cells can produce durable responses in patients with Epstein Barr virus–positive (EBV+), relapsed/refractory Hodgkin lymphoma (HL), according to research published in the Journal of Clinical Oncology.
These T cells, known as DNRII-LSTs, produced responses in 4 of the 8 patients studied.
This included 3 complete responses (CRs), the longest of which has exceeded 7 years.
What’s more, these responses were achieved without the use of lymphodepleting chemotherapy.
“While the study is small, its findings are incredibly encouraging for our [patients’] families and for the cancer field,” said study author Catherine M. Bollard, MD, MBChB, of Children’s National Health System in Washington, DC.
To engineer the DNRII-LSTs, Dr Bollard and her colleagues forced expression of a dominant-negative TGF-beta receptor type 2 (DNRII) on LMP-specific T cells (LSTs), which are T cells directed to the EBV latency-associated antigens LMP-1 and LMP-2.
The goal of forcing DNRII expression was to enable the LSTs to resist the hostile tumor environment so they could seek out and kill the tumor cells.
Dr Bollard and her colleagues administered DNRII-LSTs to 8 patients with EBV+ HL. The patients ranged in age from 27 to 47.
Seven of the 8 patients had active disease at the time of DNRII-LST infusion. Two patients had stage IVB HL, 1 had stage IIIB, and 2 had stage IIB. Four patients had nodular-sclerosing HL.
Six patients had relapsed twice. The remaining 2 patients had relapsed 3 and 4 times, respectively. All patients had previously received an autologous stem cell transplant and a range of multi-agent chemotherapy regimens (eg, ABVD, R-ICE, and MOPP).
For this study, the patients received 2 to 12 infusions of DNRII-LSTs, at doses ranging from 2 × 107 to 1.5 × 108 cells/m2.
Results
The researchers found that autologous DNRII-LSTs (given to 7 patients) did not cause autoimmunity, and donor-derived DNRII-LSTs (n=1) did not induce graft-vs-host disease.
The team also noted there were no toxicities resulting from cytokine release syndrome.
Four patients achieved a response to treatment—3 CRs and a partial response.
All complete responders are still in CR, but the partial responder progressed at 19 months and ultimately died of sepsis (2 years after the first dose of DNRII-LSTs).
The other 4 patients had stable disease (SD) for 4 months to 13 months after treatment with DNRII-LSTs.
One patient with SD died of disease progression 2 years after receiving DNRII-LSTs, and another died of transplant complications less than 2 years after the last dose of DNRII-LSTs.
One patient with SD went on to receive additional therapy and is still alive more than 6 years after receiving DNRII-LSTs (currently receiving nivolumab). Another SD patient went on to receive additional therapy, achieved a CR, and is still alive.
One of the patients who achieved a CR to DNRII-LSTs remains in CR more than 7 years after the last dose. Another patient’s CR has exceeded 2 years, and another’s has exceeded 5 years.
All 3 of these patients received doses of 2 × 107 cells/m2. The patients with the longest and shortest CRs each received 2 infusions of DNRII-LSTs. The patient with the CR exceeding 5 years received 12 infusions.
“These results come 18 years after this revolutionary approach was first conceptualized,” Dr Bollard said. “I started work in this area in 2000. At that time, the oncology community had little enthusiasm for the use of T-cell therapies to treat cancer.”
“Even then, when T-cell therapy was in its relative infancy, some research institutions began to see more than 90% complete responses and cure rates in some settings. This most recent study points to the potential of specialized T cells to fight even more types of immune-evading tumors.” ![]()
Engineered T cells can produce durable responses in patients with Epstein Barr virus–positive (EBV+), relapsed/refractory Hodgkin lymphoma (HL), according to research published in the Journal of Clinical Oncology.
These T cells, known as DNRII-LSTs, produced responses in 4 of the 8 patients studied.
This included 3 complete responses (CRs), the longest of which has exceeded 7 years.
What’s more, these responses were achieved without the use of lymphodepleting chemotherapy.
“While the study is small, its findings are incredibly encouraging for our [patients’] families and for the cancer field,” said study author Catherine M. Bollard, MD, MBChB, of Children’s National Health System in Washington, DC.
To engineer the DNRII-LSTs, Dr Bollard and her colleagues forced expression of a dominant-negative TGF-beta receptor type 2 (DNRII) on LMP-specific T cells (LSTs), which are T cells directed to the EBV latency-associated antigens LMP-1 and LMP-2.
The goal of forcing DNRII expression was to enable the LSTs to resist the hostile tumor environment so they could seek out and kill the tumor cells.
Dr Bollard and her colleagues administered DNRII-LSTs to 8 patients with EBV+ HL. The patients ranged in age from 27 to 47.
Seven of the 8 patients had active disease at the time of DNRII-LST infusion. Two patients had stage IVB HL, 1 had stage IIIB, and 2 had stage IIB. Four patients had nodular-sclerosing HL.
Six patients had relapsed twice. The remaining 2 patients had relapsed 3 and 4 times, respectively. All patients had previously received an autologous stem cell transplant and a range of multi-agent chemotherapy regimens (eg, ABVD, R-ICE, and MOPP).
For this study, the patients received 2 to 12 infusions of DNRII-LSTs, at doses ranging from 2 × 107 to 1.5 × 108 cells/m2.
Results
The researchers found that autologous DNRII-LSTs (given to 7 patients) did not cause autoimmunity, and donor-derived DNRII-LSTs (n=1) did not induce graft-vs-host disease.
The team also noted there were no toxicities resulting from cytokine release syndrome.
Four patients achieved a response to treatment—3 CRs and a partial response.
All complete responders are still in CR, but the partial responder progressed at 19 months and ultimately died of sepsis (2 years after the first dose of DNRII-LSTs).
The other 4 patients had stable disease (SD) for 4 months to 13 months after treatment with DNRII-LSTs.
One patient with SD died of disease progression 2 years after receiving DNRII-LSTs, and another died of transplant complications less than 2 years after the last dose of DNRII-LSTs.
One patient with SD went on to receive additional therapy and is still alive more than 6 years after receiving DNRII-LSTs (currently receiving nivolumab). Another SD patient went on to receive additional therapy, achieved a CR, and is still alive.
One of the patients who achieved a CR to DNRII-LSTs remains in CR more than 7 years after the last dose. Another patient’s CR has exceeded 2 years, and another’s has exceeded 5 years.
All 3 of these patients received doses of 2 × 107 cells/m2. The patients with the longest and shortest CRs each received 2 infusions of DNRII-LSTs. The patient with the CR exceeding 5 years received 12 infusions.
“These results come 18 years after this revolutionary approach was first conceptualized,” Dr Bollard said. “I started work in this area in 2000. At that time, the oncology community had little enthusiasm for the use of T-cell therapies to treat cancer.”
“Even then, when T-cell therapy was in its relative infancy, some research institutions began to see more than 90% complete responses and cure rates in some settings. This most recent study points to the potential of specialized T cells to fight even more types of immune-evading tumors.” ![]()
Engineered T cells can produce durable responses in patients with Epstein Barr virus–positive (EBV+), relapsed/refractory Hodgkin lymphoma (HL), according to research published in the Journal of Clinical Oncology.
These T cells, known as DNRII-LSTs, produced responses in 4 of the 8 patients studied.
This included 3 complete responses (CRs), the longest of which has exceeded 7 years.
What’s more, these responses were achieved without the use of lymphodepleting chemotherapy.
“While the study is small, its findings are incredibly encouraging for our [patients’] families and for the cancer field,” said study author Catherine M. Bollard, MD, MBChB, of Children’s National Health System in Washington, DC.
To engineer the DNRII-LSTs, Dr Bollard and her colleagues forced expression of a dominant-negative TGF-beta receptor type 2 (DNRII) on LMP-specific T cells (LSTs), which are T cells directed to the EBV latency-associated antigens LMP-1 and LMP-2.
The goal of forcing DNRII expression was to enable the LSTs to resist the hostile tumor environment so they could seek out and kill the tumor cells.
Dr Bollard and her colleagues administered DNRII-LSTs to 8 patients with EBV+ HL. The patients ranged in age from 27 to 47.
Seven of the 8 patients had active disease at the time of DNRII-LST infusion. Two patients had stage IVB HL, 1 had stage IIIB, and 2 had stage IIB. Four patients had nodular-sclerosing HL.
Six patients had relapsed twice. The remaining 2 patients had relapsed 3 and 4 times, respectively. All patients had previously received an autologous stem cell transplant and a range of multi-agent chemotherapy regimens (eg, ABVD, R-ICE, and MOPP).
For this study, the patients received 2 to 12 infusions of DNRII-LSTs, at doses ranging from 2 × 107 to 1.5 × 108 cells/m2.
Results
The researchers found that autologous DNRII-LSTs (given to 7 patients) did not cause autoimmunity, and donor-derived DNRII-LSTs (n=1) did not induce graft-vs-host disease.
The team also noted there were no toxicities resulting from cytokine release syndrome.
Four patients achieved a response to treatment—3 CRs and a partial response.
All complete responders are still in CR, but the partial responder progressed at 19 months and ultimately died of sepsis (2 years after the first dose of DNRII-LSTs).
The other 4 patients had stable disease (SD) for 4 months to 13 months after treatment with DNRII-LSTs.
One patient with SD died of disease progression 2 years after receiving DNRII-LSTs, and another died of transplant complications less than 2 years after the last dose of DNRII-LSTs.
One patient with SD went on to receive additional therapy and is still alive more than 6 years after receiving DNRII-LSTs (currently receiving nivolumab). Another SD patient went on to receive additional therapy, achieved a CR, and is still alive.
One of the patients who achieved a CR to DNRII-LSTs remains in CR more than 7 years after the last dose. Another patient’s CR has exceeded 2 years, and another’s has exceeded 5 years.
All 3 of these patients received doses of 2 × 107 cells/m2. The patients with the longest and shortest CRs each received 2 infusions of DNRII-LSTs. The patient with the CR exceeding 5 years received 12 infusions.
“These results come 18 years after this revolutionary approach was first conceptualized,” Dr Bollard said. “I started work in this area in 2000. At that time, the oncology community had little enthusiasm for the use of T-cell therapies to treat cancer.”
“Even then, when T-cell therapy was in its relative infancy, some research institutions began to see more than 90% complete responses and cure rates in some settings. This most recent study points to the potential of specialized T cells to fight even more types of immune-evading tumors.” ![]()
Treatment costs threaten cancer program growth

Treatment costs are the greatest threat to the growth of cancer programs, according to a survey of nearly 300 cancer program administrators and providers.
Sixty-eight percent of survey respondents said treatment costs were among the biggest threats to future cancer program growth at their organization.
Other top threats to growth included physician alignment around services and program goals—cited by 47% of respondents—and changes in healthcare coverage—cited by 46%.
This survey—the “2017 Trending Now in Cancer Care Survey”—was conducted by the Association of Community Cancer Centers (ACCC) and Advisory Board’s Oncology Roundtable. It was supported by Pfizer Oncology.
The survey was distributed via email on July 24, 2017. Respondents included 293 cancer program administrators and providers from 209 institutions. They submitted responses over 6 weeks.
Respondents identified the following “biggest threats” to cancer program growth:
- Cost of drugs and/or new treatment modalities—68%
- Physician alignment around services and program goals—47%
- Changes in healthcare coverage—46%
- Cuts to fee-for-service reimbursement—44%
- Shifting reimbursement to value-based care—43%
- Marketplace competition—35%
- Workforce planning (eg, managing staff shortages)—34%
- Network strategy and integration—33%
- Site of care policies issued by private payers—27%
- Access to capital—26%
- Quality reporting requirements—22%
- Health information technology—21%
- Other—6%.
When asked to identify their greatest opportunities for cost savings, respondents overwhelmingly pointed toward clinical standardization (63%) and drugs (62%).
Other opportunities included:
- Supplies—28%
- Capital expenses (eg, imaging technology)—24%
- Non-clinical staff (eg, billing and coding specialists)—22%
- Technology maintenance—20%
- Clinical staff—16%
- Retail pharmacy—14%
- Clinical research—10%
- Support services (eg, acupuncture)—9%
- Other—4%.
Respondents also said the investments most likely to yield a return for their cancer program were:
- Increasing the number of sub-specialists (eg, breast surgeons)—59%
- Marketing—41%
- Specialty pharmacy—36%
- Increasing the number of general oncology physicians—34%
- Screening services (eg, mobile screening unit)—29%
- Capital investments—24%
- Clinical research—16%
- Support services—15%
- Retail pharmacy—14%
- Building upgrades—14%.
More details on the “2017 Trending Now in Cancer Care Survey” can be found on the ACCC website. ![]()
Treatment costs are the greatest threat to the growth of cancer programs, according to a survey of nearly 300 cancer program administrators and providers.
Sixty-eight percent of survey respondents said treatment costs were among the biggest threats to future cancer program growth at their organization.
Other top threats to growth included physician alignment around services and program goals—cited by 47% of respondents—and changes in healthcare coverage—cited by 46%.
This survey—the “2017 Trending Now in Cancer Care Survey”—was conducted by the Association of Community Cancer Centers (ACCC) and Advisory Board’s Oncology Roundtable. It was supported by Pfizer Oncology.
The survey was distributed via email on July 24, 2017. Respondents included 293 cancer program administrators and providers from 209 institutions. They submitted responses over 6 weeks.
Respondents identified the following “biggest threats” to cancer program growth:
- Cost of drugs and/or new treatment modalities—68%
- Physician alignment around services and program goals—47%
- Changes in healthcare coverage—46%
- Cuts to fee-for-service reimbursement—44%
- Shifting reimbursement to value-based care—43%
- Marketplace competition—35%
- Workforce planning (eg, managing staff shortages)—34%
- Network strategy and integration—33%
- Site of care policies issued by private payers—27%
- Access to capital—26%
- Quality reporting requirements—22%
- Health information technology—21%
- Other—6%.
When asked to identify their greatest opportunities for cost savings, respondents overwhelmingly pointed toward clinical standardization (63%) and drugs (62%).
Other opportunities included:
- Supplies—28%
- Capital expenses (eg, imaging technology)—24%
- Non-clinical staff (eg, billing and coding specialists)—22%
- Technology maintenance—20%
- Clinical staff—16%
- Retail pharmacy—14%
- Clinical research—10%
- Support services (eg, acupuncture)—9%
- Other—4%.
Respondents also said the investments most likely to yield a return for their cancer program were:
- Increasing the number of sub-specialists (eg, breast surgeons)—59%
- Marketing—41%
- Specialty pharmacy—36%
- Increasing the number of general oncology physicians—34%
- Screening services (eg, mobile screening unit)—29%
- Capital investments—24%
- Clinical research—16%
- Support services—15%
- Retail pharmacy—14%
- Building upgrades—14%.
More details on the “2017 Trending Now in Cancer Care Survey” can be found on the ACCC website. ![]()
Treatment costs are the greatest threat to the growth of cancer programs, according to a survey of nearly 300 cancer program administrators and providers.
Sixty-eight percent of survey respondents said treatment costs were among the biggest threats to future cancer program growth at their organization.
Other top threats to growth included physician alignment around services and program goals—cited by 47% of respondents—and changes in healthcare coverage—cited by 46%.
This survey—the “2017 Trending Now in Cancer Care Survey”—was conducted by the Association of Community Cancer Centers (ACCC) and Advisory Board’s Oncology Roundtable. It was supported by Pfizer Oncology.
The survey was distributed via email on July 24, 2017. Respondents included 293 cancer program administrators and providers from 209 institutions. They submitted responses over 6 weeks.
Respondents identified the following “biggest threats” to cancer program growth:
- Cost of drugs and/or new treatment modalities—68%
- Physician alignment around services and program goals—47%
- Changes in healthcare coverage—46%
- Cuts to fee-for-service reimbursement—44%
- Shifting reimbursement to value-based care—43%
- Marketplace competition—35%
- Workforce planning (eg, managing staff shortages)—34%
- Network strategy and integration—33%
- Site of care policies issued by private payers—27%
- Access to capital—26%
- Quality reporting requirements—22%
- Health information technology—21%
- Other—6%.
When asked to identify their greatest opportunities for cost savings, respondents overwhelmingly pointed toward clinical standardization (63%) and drugs (62%).
Other opportunities included:
- Supplies—28%
- Capital expenses (eg, imaging technology)—24%
- Non-clinical staff (eg, billing and coding specialists)—22%
- Technology maintenance—20%
- Clinical staff—16%
- Retail pharmacy—14%
- Clinical research—10%
- Support services (eg, acupuncture)—9%
- Other—4%.
Respondents also said the investments most likely to yield a return for their cancer program were:
- Increasing the number of sub-specialists (eg, breast surgeons)—59%
- Marketing—41%
- Specialty pharmacy—36%
- Increasing the number of general oncology physicians—34%
- Screening services (eg, mobile screening unit)—29%
- Capital investments—24%
- Clinical research—16%
- Support services—15%
- Retail pharmacy—14%
- Building upgrades—14%.
More details on the “2017 Trending Now in Cancer Care Survey” can be found on the ACCC website. ![]()
Technique could aid treatment of CLL

Researchers say they have developed a new technique for assessing chromosomal abnormalities in chronic lymphocytic leukemia (CLL).
The team believes their method, called immuno-flowFISH, could be used at the time of CLL diagnosis for disease stratification and after treatment to assess residual disease.
Kathryn A. Fuller, PhD, of The University of Western Australia in Crawley, Australia, and her colleagues described immuno-flowFISH in the journal Methods.
The name “immuno-flowFISH” acknowledges what has been incorporated into this technology.
“Immuno” recognizes that immunology testing is used to identify the CLL cells. “Flow” is used because the machine is an imaging flow cytometer. And “FISH” is the test that identifies the chromosomes inside the cells.
The researchers said they found that immuno-flowFISH could detect trisomic chromosomal abnormalities in cells with the phenotype of CLL.
And immuno-flowFISH provided greater specificity and sensitivity than standard FISH.
In particular, the researchers were able to analyze 10,000 to 20,000 cells in each sample, which is 100 to 200 times greater than traditional FISH methods.
“The imaging cytometer can analyze samples at a rate of up to 2000 cells per second, which means we can investigate a large number of cells in a relatively short amount of time, giving us greater sensitivity,” Dr Fuller said.
“This immuno-flowFISH method is an exciting development in personalizing pathology testing for leukemia,” added study author Wendy N. Erber, MD, DPhil, PhD, of The University of Western Australia.
Dr Erber and her colleagues are now expanding immuno-flowFISH so it can be applied to other malignancies as well. ![]()
Researchers say they have developed a new technique for assessing chromosomal abnormalities in chronic lymphocytic leukemia (CLL).
The team believes their method, called immuno-flowFISH, could be used at the time of CLL diagnosis for disease stratification and after treatment to assess residual disease.
Kathryn A. Fuller, PhD, of The University of Western Australia in Crawley, Australia, and her colleagues described immuno-flowFISH in the journal Methods.
The name “immuno-flowFISH” acknowledges what has been incorporated into this technology.
“Immuno” recognizes that immunology testing is used to identify the CLL cells. “Flow” is used because the machine is an imaging flow cytometer. And “FISH” is the test that identifies the chromosomes inside the cells.
The researchers said they found that immuno-flowFISH could detect trisomic chromosomal abnormalities in cells with the phenotype of CLL.
And immuno-flowFISH provided greater specificity and sensitivity than standard FISH.
In particular, the researchers were able to analyze 10,000 to 20,000 cells in each sample, which is 100 to 200 times greater than traditional FISH methods.
“The imaging cytometer can analyze samples at a rate of up to 2000 cells per second, which means we can investigate a large number of cells in a relatively short amount of time, giving us greater sensitivity,” Dr Fuller said.
“This immuno-flowFISH method is an exciting development in personalizing pathology testing for leukemia,” added study author Wendy N. Erber, MD, DPhil, PhD, of The University of Western Australia.
Dr Erber and her colleagues are now expanding immuno-flowFISH so it can be applied to other malignancies as well. ![]()
Researchers say they have developed a new technique for assessing chromosomal abnormalities in chronic lymphocytic leukemia (CLL).
The team believes their method, called immuno-flowFISH, could be used at the time of CLL diagnosis for disease stratification and after treatment to assess residual disease.
Kathryn A. Fuller, PhD, of The University of Western Australia in Crawley, Australia, and her colleagues described immuno-flowFISH in the journal Methods.
The name “immuno-flowFISH” acknowledges what has been incorporated into this technology.
“Immuno” recognizes that immunology testing is used to identify the CLL cells. “Flow” is used because the machine is an imaging flow cytometer. And “FISH” is the test that identifies the chromosomes inside the cells.
The researchers said they found that immuno-flowFISH could detect trisomic chromosomal abnormalities in cells with the phenotype of CLL.
And immuno-flowFISH provided greater specificity and sensitivity than standard FISH.
In particular, the researchers were able to analyze 10,000 to 20,000 cells in each sample, which is 100 to 200 times greater than traditional FISH methods.
“The imaging cytometer can analyze samples at a rate of up to 2000 cells per second, which means we can investigate a large number of cells in a relatively short amount of time, giving us greater sensitivity,” Dr Fuller said.
“This immuno-flowFISH method is an exciting development in personalizing pathology testing for leukemia,” added study author Wendy N. Erber, MD, DPhil, PhD, of The University of Western Australia.
Dr Erber and her colleagues are now expanding immuno-flowFISH so it can be applied to other malignancies as well. ![]()
EC authorizes brentuximab vedotin for CTCL

The European Commission (EC) has extended the conditional marketing authorization for brentuximab vedotin (Adcetris®).
The drug is now approved for use in adults with CD30-positive cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
Brentuximab vedotin can be marketed for this indication in the member states of the European Union as well as in Norway, Liechtenstein, and Iceland.
Conditional marketing authorization from the EC is valid for 1 year and is reviewed annually.
With conditional authorization, drug developers are required to provide comprehensive data confirming a drug’s benefit-risk balance is positive. Once these data are available, a conditional marketing authorization may be converted to a standard marketing authorization.
Drugs are eligible for conditional marketing authorization if they are designated as orphan medicines, intended for use in emergency situations, or designed to treat, prevent, or diagnose seriously debilitating or life-threatening diseases.
The EC previously granted brentuximab vedotin conditional marketing authorization for the treatment of:
- Adults with CD30+ Hodgkin lymphoma who are at an increased risk of relapse or progression following autologous hematopoietic stem cell transplant (auto-HSCT)
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma after auto-HSCT or after at least 2 prior therapies when auto-HSCT or multi-agent chemotherapy is not a treatment option
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
Phase 3 data
The EC’s latest authorization for brentuximab vedotin is based on data from the phase 3 ALCANZA trial.
Updated results from ALCANZA were presented at the 2017 ASH Annual Meeting in December. Results were previously presented at the 9th Annual T-cell Lymphoma Forum in January 2017 and published in The Lancet in June 2017.
The trial included 128 evaluable patients with CD30-positive CTCL who had received at least 1 prior systemic therapy.
Sixty-four patients were assigned to receive brentuximab vedotin, and 64 were assigned to receive the investigator’s choice of methotrexate or bexarotene (control arm). Patients received treatment for up to 1 year.
For the update, the median follow-up was 33.9 months.
There was a significant improvement in the rate of objective response lasting at least 4 months (ORR4) in the brentuximab vedotin arm compared to the control arm. The ORR4 was 60.9% and 7.8%, respectively (P<0.001). The complete response rate was 18.8% and 0%, respectively (P<0.001).
The median progression-free survival was 15.8 months in the brentuximab vedotin arm and 3.6 months in the control arm (hazard ratio=0.373; 95% CI, 0.245-0.569; P<0.001).
At time of analysis, 73% of patients in the brentuximab vedotin arm and 75% in the control arm had received 1 or more subsequent skin-directed or systemic therapies. The median time to next treatment was 14.2 months in the brentuximab vedotin arm and 6.1 months in the control arm (P<0.001).
Peripheral neuropathy was the most commonly reported adverse event in patients who received brentuximab vedotin. The incidence was 67% in these patients and 6% in controls.
In the brentuximab arm, 86% of patients reported resolution or improvement in peripheral neuropathy. Eighteen patients had ongoing peripheral neuropathy events, including 15 patients with grade 1 and 3 patients with grade 2 events. ![]()
The European Commission (EC) has extended the conditional marketing authorization for brentuximab vedotin (Adcetris®).
The drug is now approved for use in adults with CD30-positive cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
Brentuximab vedotin can be marketed for this indication in the member states of the European Union as well as in Norway, Liechtenstein, and Iceland.
Conditional marketing authorization from the EC is valid for 1 year and is reviewed annually.
With conditional authorization, drug developers are required to provide comprehensive data confirming a drug’s benefit-risk balance is positive. Once these data are available, a conditional marketing authorization may be converted to a standard marketing authorization.
Drugs are eligible for conditional marketing authorization if they are designated as orphan medicines, intended for use in emergency situations, or designed to treat, prevent, or diagnose seriously debilitating or life-threatening diseases.
The EC previously granted brentuximab vedotin conditional marketing authorization for the treatment of:
- Adults with CD30+ Hodgkin lymphoma who are at an increased risk of relapse or progression following autologous hematopoietic stem cell transplant (auto-HSCT)
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma after auto-HSCT or after at least 2 prior therapies when auto-HSCT or multi-agent chemotherapy is not a treatment option
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
Phase 3 data
The EC’s latest authorization for brentuximab vedotin is based on data from the phase 3 ALCANZA trial.
Updated results from ALCANZA were presented at the 2017 ASH Annual Meeting in December. Results were previously presented at the 9th Annual T-cell Lymphoma Forum in January 2017 and published in The Lancet in June 2017.
The trial included 128 evaluable patients with CD30-positive CTCL who had received at least 1 prior systemic therapy.
Sixty-four patients were assigned to receive brentuximab vedotin, and 64 were assigned to receive the investigator’s choice of methotrexate or bexarotene (control arm). Patients received treatment for up to 1 year.
For the update, the median follow-up was 33.9 months.
There was a significant improvement in the rate of objective response lasting at least 4 months (ORR4) in the brentuximab vedotin arm compared to the control arm. The ORR4 was 60.9% and 7.8%, respectively (P<0.001). The complete response rate was 18.8% and 0%, respectively (P<0.001).
The median progression-free survival was 15.8 months in the brentuximab vedotin arm and 3.6 months in the control arm (hazard ratio=0.373; 95% CI, 0.245-0.569; P<0.001).
At time of analysis, 73% of patients in the brentuximab vedotin arm and 75% in the control arm had received 1 or more subsequent skin-directed or systemic therapies. The median time to next treatment was 14.2 months in the brentuximab vedotin arm and 6.1 months in the control arm (P<0.001).
Peripheral neuropathy was the most commonly reported adverse event in patients who received brentuximab vedotin. The incidence was 67% in these patients and 6% in controls.
In the brentuximab arm, 86% of patients reported resolution or improvement in peripheral neuropathy. Eighteen patients had ongoing peripheral neuropathy events, including 15 patients with grade 1 and 3 patients with grade 2 events. ![]()
The European Commission (EC) has extended the conditional marketing authorization for brentuximab vedotin (Adcetris®).
The drug is now approved for use in adults with CD30-positive cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
Brentuximab vedotin can be marketed for this indication in the member states of the European Union as well as in Norway, Liechtenstein, and Iceland.
Conditional marketing authorization from the EC is valid for 1 year and is reviewed annually.
With conditional authorization, drug developers are required to provide comprehensive data confirming a drug’s benefit-risk balance is positive. Once these data are available, a conditional marketing authorization may be converted to a standard marketing authorization.
Drugs are eligible for conditional marketing authorization if they are designated as orphan medicines, intended for use in emergency situations, or designed to treat, prevent, or diagnose seriously debilitating or life-threatening diseases.
The EC previously granted brentuximab vedotin conditional marketing authorization for the treatment of:
- Adults with CD30+ Hodgkin lymphoma who are at an increased risk of relapse or progression following autologous hematopoietic stem cell transplant (auto-HSCT)
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma after auto-HSCT or after at least 2 prior therapies when auto-HSCT or multi-agent chemotherapy is not a treatment option
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
Phase 3 data
The EC’s latest authorization for brentuximab vedotin is based on data from the phase 3 ALCANZA trial.
Updated results from ALCANZA were presented at the 2017 ASH Annual Meeting in December. Results were previously presented at the 9th Annual T-cell Lymphoma Forum in January 2017 and published in The Lancet in June 2017.
The trial included 128 evaluable patients with CD30-positive CTCL who had received at least 1 prior systemic therapy.
Sixty-four patients were assigned to receive brentuximab vedotin, and 64 were assigned to receive the investigator’s choice of methotrexate or bexarotene (control arm). Patients received treatment for up to 1 year.
For the update, the median follow-up was 33.9 months.
There was a significant improvement in the rate of objective response lasting at least 4 months (ORR4) in the brentuximab vedotin arm compared to the control arm. The ORR4 was 60.9% and 7.8%, respectively (P<0.001). The complete response rate was 18.8% and 0%, respectively (P<0.001).
The median progression-free survival was 15.8 months in the brentuximab vedotin arm and 3.6 months in the control arm (hazard ratio=0.373; 95% CI, 0.245-0.569; P<0.001).
At time of analysis, 73% of patients in the brentuximab vedotin arm and 75% in the control arm had received 1 or more subsequent skin-directed or systemic therapies. The median time to next treatment was 14.2 months in the brentuximab vedotin arm and 6.1 months in the control arm (P<0.001).
Peripheral neuropathy was the most commonly reported adverse event in patients who received brentuximab vedotin. The incidence was 67% in these patients and 6% in controls.
In the brentuximab arm, 86% of patients reported resolution or improvement in peripheral neuropathy. Eighteen patients had ongoing peripheral neuropathy events, including 15 patients with grade 1 and 3 patients with grade 2 events. ![]()
Ofatumumab to be pulled from non-US markets

Novartis is planning to stop marketing ofatumumab (Arzerra®) outside the US, but the drug will still be available for certain patients.
The company plans to work with regulatory authorities to establish compassionate use programs for chronic lymphocytic leukemia (CLL) patients outside the US who are currently receiving ofatumumab.
Patients accessing these programs will be offered continued treatment with ofatumumab for as long as they benefit from it, free of charge.
And Novartis will continue to market ofatumumab for CLL in the US.
“Novartis’s intention to transition Arzerra to compassionate use programs in the non-US markets reflects the fact that many more drugs have become available for CLL over the last 5 years and that there is a low number of patients using Arzerra outside of the US market,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
Ofatumumab is marketed under a collaboration agreement between Genmab and Novartis.
Because Novartis has decided to pull ofatumumab from non-US markets, the company will pay Genmab a lump sum of $50 million (USD) for lost potential milestones and royalties. Royalties will continue to be earned on net sales of the drug.
Novartis intends to start the transition to compassionate use programs as soon as the company and regulatory authorities have agreed to a plan.
The two phase 3 studies of ofatumumab in relapsing multiple sclerosis and the study in indolent non-Hodgkin lymphoma will not be affected by this change. All 3 trials will continue.
About ofatumumab
Ofatumumab is a monoclonal antibody designed to target CD20 on the surface of CLL cells and normal B lymphocytes.
In more than 60 countries worldwide, including the US and European Union member countries, ofatumumab is approved as monotherapy for CLL patients who are refractory to treatment with fludarabine and alemtuzumab.
Other approved indications for ofatumumab in the European Union include:
- In combination with chlorambucil or bendamustine to treat CLL patients who have not received prior therapy and are not eligible for fludarabine-based therapy
- In combination with fludarabine and cyclophosphamide to treat adults with relapsed CLL.
Other approved indications for ofatumumab in the US include:
- In combination with chlorambucil to treat previously untreated CLL patients for whom fludarabine-based therapy is considered inappropriate
- In combination with fludarabine and cyclophosphamide to treat patients with relapsed CLL
- For extended treatment of patients who are in complete or partial response after at least 2 lines of therapy for recurrent or progressive CLL.

Novartis is planning to stop marketing ofatumumab (Arzerra®) outside the US, but the drug will still be available for certain patients.
The company plans to work with regulatory authorities to establish compassionate use programs for chronic lymphocytic leukemia (CLL) patients outside the US who are currently receiving ofatumumab.
Patients accessing these programs will be offered continued treatment with ofatumumab for as long as they benefit from it, free of charge.
And Novartis will continue to market ofatumumab for CLL in the US.
“Novartis’s intention to transition Arzerra to compassionate use programs in the non-US markets reflects the fact that many more drugs have become available for CLL over the last 5 years and that there is a low number of patients using Arzerra outside of the US market,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
Ofatumumab is marketed under a collaboration agreement between Genmab and Novartis.
Because Novartis has decided to pull ofatumumab from non-US markets, the company will pay Genmab a lump sum of $50 million (USD) for lost potential milestones and royalties. Royalties will continue to be earned on net sales of the drug.
Novartis intends to start the transition to compassionate use programs as soon as the company and regulatory authorities have agreed to a plan.
The two phase 3 studies of ofatumumab in relapsing multiple sclerosis and the study in indolent non-Hodgkin lymphoma will not be affected by this change. All 3 trials will continue.
About ofatumumab
Ofatumumab is a monoclonal antibody designed to target CD20 on the surface of CLL cells and normal B lymphocytes.
In more than 60 countries worldwide, including the US and European Union member countries, ofatumumab is approved as monotherapy for CLL patients who are refractory to treatment with fludarabine and alemtuzumab.
Other approved indications for ofatumumab in the European Union include:
- In combination with chlorambucil or bendamustine to treat CLL patients who have not received prior therapy and are not eligible for fludarabine-based therapy
- In combination with fludarabine and cyclophosphamide to treat adults with relapsed CLL.
Other approved indications for ofatumumab in the US include:
- In combination with chlorambucil to treat previously untreated CLL patients for whom fludarabine-based therapy is considered inappropriate
- In combination with fludarabine and cyclophosphamide to treat patients with relapsed CLL
- For extended treatment of patients who are in complete or partial response after at least 2 lines of therapy for recurrent or progressive CLL.
Novartis is planning to stop marketing ofatumumab (Arzerra®) outside the US, but the drug will still be available for certain patients.
The company plans to work with regulatory authorities to establish compassionate use programs for chronic lymphocytic leukemia (CLL) patients outside the US who are currently receiving ofatumumab.
Patients accessing these programs will be offered continued treatment with ofatumumab for as long as they benefit from it, free of charge.
And Novartis will continue to market ofatumumab for CLL in the US.
“Novartis’s intention to transition Arzerra to compassionate use programs in the non-US markets reflects the fact that many more drugs have become available for CLL over the last 5 years and that there is a low number of patients using Arzerra outside of the US market,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
Ofatumumab is marketed under a collaboration agreement between Genmab and Novartis.
Because Novartis has decided to pull ofatumumab from non-US markets, the company will pay Genmab a lump sum of $50 million (USD) for lost potential milestones and royalties. Royalties will continue to be earned on net sales of the drug.
Novartis intends to start the transition to compassionate use programs as soon as the company and regulatory authorities have agreed to a plan.
The two phase 3 studies of ofatumumab in relapsing multiple sclerosis and the study in indolent non-Hodgkin lymphoma will not be affected by this change. All 3 trials will continue.
About ofatumumab
Ofatumumab is a monoclonal antibody designed to target CD20 on the surface of CLL cells and normal B lymphocytes.
In more than 60 countries worldwide, including the US and European Union member countries, ofatumumab is approved as monotherapy for CLL patients who are refractory to treatment with fludarabine and alemtuzumab.
Other approved indications for ofatumumab in the European Union include:
- In combination with chlorambucil or bendamustine to treat CLL patients who have not received prior therapy and are not eligible for fludarabine-based therapy
- In combination with fludarabine and cyclophosphamide to treat adults with relapsed CLL.
Other approved indications for ofatumumab in the US include:
- In combination with chlorambucil to treat previously untreated CLL patients for whom fludarabine-based therapy is considered inappropriate
- In combination with fludarabine and cyclophosphamide to treat patients with relapsed CLL
- For extended treatment of patients who are in complete or partial response after at least 2 lines of therapy for recurrent or progressive CLL.
Bright light therapy improves sleep in cancer survivors

Results of a pilot study suggest that systematic bright light exposure can improve sleep in fatigued cancer survivors.
Subjects who were exposed to bright light every morning for 4 weeks had a significantly greater improvement in sleep efficiency than those who were exposed to dim light over the same period.
In fact, subjects in the bright light group were able to achieve clinically normal levels of sleep efficiency, and subjects in the dim light group were not.
Sleep efficiency is the percentage of time in bed that subjects spent sleeping.
Lisa M. Wu, PhD, of Northwestern University in Chicago, Illinois, and her colleagues reported these results in the Journal of Clinical Sleep Medicine.
The team noted that cancer patients report sleep disturbances at a significantly higher rate than the general population. Between 23% and 44% of cancer patients experience insomnia symptoms even years after treatment.
With this in mind, the researchers studied 44 individuals who had completed cancer treatment and met criteria for clinically significant fatigue at screening.
The subjects had an average age of 53.6, and 75% percent were female. Roughly 55% (n=24) had been diagnosed with a hematologic malignancy.
The subjects were randomized to a bright white light intervention or a dim red light intervention. Subjects in both treatment arms were provided with a light box and instructed to use it every morning for 30 minutes for 4 weeks. Sleep was evaluated using wrist actigraphy and the Pittsburgh Sleep Quality Index.
At baseline, 52.6% of subjects in the dim light group and 60% in the bright light group exceeded the clinical cutoff for poor sleep efficiency (≤ 85%). The mean sleep efficiency was 81.8% and 82.8%, respectively.
During the study period, sleep efficiency improved significantly more among subjects exposed to the bright light than those exposed to the dim light (P=0.003).
The mean sleep efficiency was in the clinically normal range for subjects in the bright light group at the end of the intervention (86.06%) and 3 weeks after (85.77%).
However, the cutoff for poor sleep efficiency was not reached in the dim light group, either at the end of the intervention (mean=79.35%) or 3 weeks after (mean=80.88%).
Total sleep time tended to increase over the study period for subjects in the bright light group, but there was no significant difference in total sleep time between the bright light and dim light groups.
Likewise, there was no significant between-group difference in waking after sleep onset, although this outcome tended to decrease over the study period for subjects in the bright light group.
“Systematic light exposure using bright white light is a low-cost and easily disseminated intervention that offers a feasible and potentially effective alternative to improve sleep in cancer survivors,” Dr Wu said.
However, she and her colleagues noted that larger-scale studies are needed. ![]()
Results of a pilot study suggest that systematic bright light exposure can improve sleep in fatigued cancer survivors.
Subjects who were exposed to bright light every morning for 4 weeks had a significantly greater improvement in sleep efficiency than those who were exposed to dim light over the same period.
In fact, subjects in the bright light group were able to achieve clinically normal levels of sleep efficiency, and subjects in the dim light group were not.
Sleep efficiency is the percentage of time in bed that subjects spent sleeping.
Lisa M. Wu, PhD, of Northwestern University in Chicago, Illinois, and her colleagues reported these results in the Journal of Clinical Sleep Medicine.
The team noted that cancer patients report sleep disturbances at a significantly higher rate than the general population. Between 23% and 44% of cancer patients experience insomnia symptoms even years after treatment.
With this in mind, the researchers studied 44 individuals who had completed cancer treatment and met criteria for clinically significant fatigue at screening.
The subjects had an average age of 53.6, and 75% percent were female. Roughly 55% (n=24) had been diagnosed with a hematologic malignancy.
The subjects were randomized to a bright white light intervention or a dim red light intervention. Subjects in both treatment arms were provided with a light box and instructed to use it every morning for 30 minutes for 4 weeks. Sleep was evaluated using wrist actigraphy and the Pittsburgh Sleep Quality Index.
At baseline, 52.6% of subjects in the dim light group and 60% in the bright light group exceeded the clinical cutoff for poor sleep efficiency (≤ 85%). The mean sleep efficiency was 81.8% and 82.8%, respectively.
During the study period, sleep efficiency improved significantly more among subjects exposed to the bright light than those exposed to the dim light (P=0.003).
The mean sleep efficiency was in the clinically normal range for subjects in the bright light group at the end of the intervention (86.06%) and 3 weeks after (85.77%).
However, the cutoff for poor sleep efficiency was not reached in the dim light group, either at the end of the intervention (mean=79.35%) or 3 weeks after (mean=80.88%).
Total sleep time tended to increase over the study period for subjects in the bright light group, but there was no significant difference in total sleep time between the bright light and dim light groups.
Likewise, there was no significant between-group difference in waking after sleep onset, although this outcome tended to decrease over the study period for subjects in the bright light group.
“Systematic light exposure using bright white light is a low-cost and easily disseminated intervention that offers a feasible and potentially effective alternative to improve sleep in cancer survivors,” Dr Wu said.
However, she and her colleagues noted that larger-scale studies are needed. ![]()
Results of a pilot study suggest that systematic bright light exposure can improve sleep in fatigued cancer survivors.
Subjects who were exposed to bright light every morning for 4 weeks had a significantly greater improvement in sleep efficiency than those who were exposed to dim light over the same period.
In fact, subjects in the bright light group were able to achieve clinically normal levels of sleep efficiency, and subjects in the dim light group were not.
Sleep efficiency is the percentage of time in bed that subjects spent sleeping.
Lisa M. Wu, PhD, of Northwestern University in Chicago, Illinois, and her colleagues reported these results in the Journal of Clinical Sleep Medicine.
The team noted that cancer patients report sleep disturbances at a significantly higher rate than the general population. Between 23% and 44% of cancer patients experience insomnia symptoms even years after treatment.
With this in mind, the researchers studied 44 individuals who had completed cancer treatment and met criteria for clinically significant fatigue at screening.
The subjects had an average age of 53.6, and 75% percent were female. Roughly 55% (n=24) had been diagnosed with a hematologic malignancy.
The subjects were randomized to a bright white light intervention or a dim red light intervention. Subjects in both treatment arms were provided with a light box and instructed to use it every morning for 30 minutes for 4 weeks. Sleep was evaluated using wrist actigraphy and the Pittsburgh Sleep Quality Index.
At baseline, 52.6% of subjects in the dim light group and 60% in the bright light group exceeded the clinical cutoff for poor sleep efficiency (≤ 85%). The mean sleep efficiency was 81.8% and 82.8%, respectively.
During the study period, sleep efficiency improved significantly more among subjects exposed to the bright light than those exposed to the dim light (P=0.003).
The mean sleep efficiency was in the clinically normal range for subjects in the bright light group at the end of the intervention (86.06%) and 3 weeks after (85.77%).
However, the cutoff for poor sleep efficiency was not reached in the dim light group, either at the end of the intervention (mean=79.35%) or 3 weeks after (mean=80.88%).
Total sleep time tended to increase over the study period for subjects in the bright light group, but there was no significant difference in total sleep time between the bright light and dim light groups.
Likewise, there was no significant between-group difference in waking after sleep onset, although this outcome tended to decrease over the study period for subjects in the bright light group.
“Systematic light exposure using bright white light is a low-cost and easily disseminated intervention that offers a feasible and potentially effective alternative to improve sleep in cancer survivors,” Dr Wu said.
However, she and her colleagues noted that larger-scale studies are needed. ![]()
Risks of MGUS persist beyond 30 years
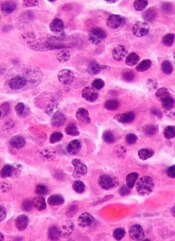
A long-term study showed that patients with monoclonal gammopathy of undetermined significance (MGUS) were still at risk of progressing to other plasma-cell or lymphoid disorders after more than 30 years of follow-up.
The risk of developing such disorders was nearly 7 times higher in MGUS patients than in matched control subjects.
Patients with MGUS also had a significantly shorter median survival than controls.
Researchers reported these findings in NEJM.
“Monoclonal gammopathy of undetermined significance is present in more than 3% of the general population age 50 and older,” said study author S. Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minnesota.
“In some cases, people with monoclonal gammopathy of undetermined significance go on to develop multiple myeloma.”
With this in mind, Dr Rajkumar and his colleagues studied 1384 patients—210 with IgM MGUS and 1129 with non-IgM MGUS. Patients were diagnosed with MGUS from 1960 through 1994, and their median age at diagnosis was 72.
The median follow-up was 34.1 years (range, 0.0 to 43.6), so there were 14,130 person-years of follow-up.
During that time, 147 patients progressed to another disorder, including:
- 97 to multiple myeloma
- 19 to non-Hodgkin lymphoma
- 14 to AL amyloidosis
- 13 to Waldenstrom’s macroglobulinemia
- 3 to chronic lymphocytic leukemia
- 1 to plasmacytoma.
The rate of progression in MGUS patients—11%—represented a risk of these disorders that was 6.5 times higher than the risk observed in an age- and sex-matched control population.
The risk of progression also increased over time for MGUS patients. Without accounting for death due to competing causes, the risk of progression was 10% at 10 years, 18% at 20 years, 28% at 30 years, and 36% at both 35 and 40 years.
“We also found that patients with monoclonal gammopathy of undetermined significance had shorter survival than comparable people without the condition, which raises the possibility there may be other disorders associated with monoclonal gammopathy of undetermined significance that still need further study,” Dr Rajkumar said.
The median survival was 8.1 years in MGUS patients and 12.4 years in controls (P<0.001).
Overall, 1300 MGUS patients (94%) had died at last follow-up. Of the 84 patients who were still alive, 5 had progressed. ![]()
A long-term study showed that patients with monoclonal gammopathy of undetermined significance (MGUS) were still at risk of progressing to other plasma-cell or lymphoid disorders after more than 30 years of follow-up.
The risk of developing such disorders was nearly 7 times higher in MGUS patients than in matched control subjects.
Patients with MGUS also had a significantly shorter median survival than controls.
Researchers reported these findings in NEJM.
“Monoclonal gammopathy of undetermined significance is present in more than 3% of the general population age 50 and older,” said study author S. Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minnesota.
“In some cases, people with monoclonal gammopathy of undetermined significance go on to develop multiple myeloma.”
With this in mind, Dr Rajkumar and his colleagues studied 1384 patients—210 with IgM MGUS and 1129 with non-IgM MGUS. Patients were diagnosed with MGUS from 1960 through 1994, and their median age at diagnosis was 72.
The median follow-up was 34.1 years (range, 0.0 to 43.6), so there were 14,130 person-years of follow-up.
During that time, 147 patients progressed to another disorder, including:
- 97 to multiple myeloma
- 19 to non-Hodgkin lymphoma
- 14 to AL amyloidosis
- 13 to Waldenstrom’s macroglobulinemia
- 3 to chronic lymphocytic leukemia
- 1 to plasmacytoma.
The rate of progression in MGUS patients—11%—represented a risk of these disorders that was 6.5 times higher than the risk observed in an age- and sex-matched control population.
The risk of progression also increased over time for MGUS patients. Without accounting for death due to competing causes, the risk of progression was 10% at 10 years, 18% at 20 years, 28% at 30 years, and 36% at both 35 and 40 years.
“We also found that patients with monoclonal gammopathy of undetermined significance had shorter survival than comparable people without the condition, which raises the possibility there may be other disorders associated with monoclonal gammopathy of undetermined significance that still need further study,” Dr Rajkumar said.
The median survival was 8.1 years in MGUS patients and 12.4 years in controls (P<0.001).
Overall, 1300 MGUS patients (94%) had died at last follow-up. Of the 84 patients who were still alive, 5 had progressed. ![]()
A long-term study showed that patients with monoclonal gammopathy of undetermined significance (MGUS) were still at risk of progressing to other plasma-cell or lymphoid disorders after more than 30 years of follow-up.
The risk of developing such disorders was nearly 7 times higher in MGUS patients than in matched control subjects.
Patients with MGUS also had a significantly shorter median survival than controls.
Researchers reported these findings in NEJM.
“Monoclonal gammopathy of undetermined significance is present in more than 3% of the general population age 50 and older,” said study author S. Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minnesota.
“In some cases, people with monoclonal gammopathy of undetermined significance go on to develop multiple myeloma.”
With this in mind, Dr Rajkumar and his colleagues studied 1384 patients—210 with IgM MGUS and 1129 with non-IgM MGUS. Patients were diagnosed with MGUS from 1960 through 1994, and their median age at diagnosis was 72.
The median follow-up was 34.1 years (range, 0.0 to 43.6), so there were 14,130 person-years of follow-up.
During that time, 147 patients progressed to another disorder, including:
- 97 to multiple myeloma
- 19 to non-Hodgkin lymphoma
- 14 to AL amyloidosis
- 13 to Waldenstrom’s macroglobulinemia
- 3 to chronic lymphocytic leukemia
- 1 to plasmacytoma.
The rate of progression in MGUS patients—11%—represented a risk of these disorders that was 6.5 times higher than the risk observed in an age- and sex-matched control population.
The risk of progression also increased over time for MGUS patients. Without accounting for death due to competing causes, the risk of progression was 10% at 10 years, 18% at 20 years, 28% at 30 years, and 36% at both 35 and 40 years.
“We also found that patients with monoclonal gammopathy of undetermined significance had shorter survival than comparable people without the condition, which raises the possibility there may be other disorders associated with monoclonal gammopathy of undetermined significance that still need further study,” Dr Rajkumar said.
The median survival was 8.1 years in MGUS patients and 12.4 years in controls (P<0.001).
Overall, 1300 MGUS patients (94%) had died at last follow-up. Of the 84 patients who were still alive, 5 had progressed.
CAR T-cell therapy on fast track in US, EU

The chimeric antigen receptor (CAR) T-cell therapy tisagenlecleucel (Kymriah, formerly CTL019) is getting fast-tracked in the United States (US) and European Union (EU).
The US Food and Drug Administration (FDA) has accepted for priority review the supplemental biologics license application (sBLA) for tisagenlecleucel for the treatment of adults with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) who are ineligible for, or relapse after, autologous hematopoietic stem cell transplant (auto-HSCT).
Meanwhile, the European Medicines Agency (EMA) has granted accelerated assessment to the marketing authorization application (MAA) for tisagenlecleucel for the treatment of children and young adults with R/R B-cell acute lymphoblastic leukemia (ALL) and for adults with R/R DLBCL who are ineligible for auto-HSCT.
If the sBLA and MAA are approved, tisagenlecleucel will be the first CAR T-cell therapy available for 2 distinct indications in non-Hodgkin lymphoma and B-cell ALL.
Tisagenlecleucel became the first CAR T-cell therapy to receive regulatory approval when it was approved by the FDA in August 2017 for use in patients up to 25 years of age who have B-cell precursor ALL that is refractory or in second or later relapse.
Supporting data
The regulatory applications for tisagenlecleucel in the US and EU are supported by data from the Novartis-sponsored global clinical trial program in children and young adults with R/R B-cell ALL and adults with R/R DLBCL.
Results from the phase 2 JULIET trial served as the basis of the sBLA and MAA for tisagenlecleucel in adults with R/R DLCBL. Data from this trial were presented at the 2017 ASH Annual Meeting in December.
Results from the phase 2 ELIANA study were submitted as part of the MAA for tisagenlecleucel in children and young adults with R/R B-cell ALL. Data from this trial were presented at the 2017 EHA Congress last June.
About priority review, accelerated assessment
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The FDA’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The EMA grants accelerated assessment when a product is expected to be of major public health interest, particularly from the point of view of therapeutic innovation.
Accelerated assessment shortens the review period from 210 days to 150 days.
The chimeric antigen receptor (CAR) T-cell therapy tisagenlecleucel (Kymriah, formerly CTL019) is getting fast-tracked in the United States (US) and European Union (EU).
The US Food and Drug Administration (FDA) has accepted for priority review the supplemental biologics license application (sBLA) for tisagenlecleucel for the treatment of adults with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) who are ineligible for, or relapse after, autologous hematopoietic stem cell transplant (auto-HSCT).
Meanwhile, the European Medicines Agency (EMA) has granted accelerated assessment to the marketing authorization application (MAA) for tisagenlecleucel for the treatment of children and young adults with R/R B-cell acute lymphoblastic leukemia (ALL) and for adults with R/R DLBCL who are ineligible for auto-HSCT.
If the sBLA and MAA are approved, tisagenlecleucel will be the first CAR T-cell therapy available for 2 distinct indications in non-Hodgkin lymphoma and B-cell ALL.
Tisagenlecleucel became the first CAR T-cell therapy to receive regulatory approval when it was approved by the FDA in August 2017 for use in patients up to 25 years of age who have B-cell precursor ALL that is refractory or in second or later relapse.
Supporting data
The regulatory applications for tisagenlecleucel in the US and EU are supported by data from the Novartis-sponsored global clinical trial program in children and young adults with R/R B-cell ALL and adults with R/R DLBCL.
Results from the phase 2 JULIET trial served as the basis of the sBLA and MAA for tisagenlecleucel in adults with R/R DLCBL. Data from this trial were presented at the 2017 ASH Annual Meeting in December.
Results from the phase 2 ELIANA study were submitted as part of the MAA for tisagenlecleucel in children and young adults with R/R B-cell ALL. Data from this trial were presented at the 2017 EHA Congress last June.
About priority review, accelerated assessment
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The FDA’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The EMA grants accelerated assessment when a product is expected to be of major public health interest, particularly from the point of view of therapeutic innovation.
Accelerated assessment shortens the review period from 210 days to 150 days.
The chimeric antigen receptor (CAR) T-cell therapy tisagenlecleucel (Kymriah, formerly CTL019) is getting fast-tracked in the United States (US) and European Union (EU).
The US Food and Drug Administration (FDA) has accepted for priority review the supplemental biologics license application (sBLA) for tisagenlecleucel for the treatment of adults with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) who are ineligible for, or relapse after, autologous hematopoietic stem cell transplant (auto-HSCT).
Meanwhile, the European Medicines Agency (EMA) has granted accelerated assessment to the marketing authorization application (MAA) for tisagenlecleucel for the treatment of children and young adults with R/R B-cell acute lymphoblastic leukemia (ALL) and for adults with R/R DLBCL who are ineligible for auto-HSCT.
If the sBLA and MAA are approved, tisagenlecleucel will be the first CAR T-cell therapy available for 2 distinct indications in non-Hodgkin lymphoma and B-cell ALL.
Tisagenlecleucel became the first CAR T-cell therapy to receive regulatory approval when it was approved by the FDA in August 2017 for use in patients up to 25 years of age who have B-cell precursor ALL that is refractory or in second or later relapse.
Supporting data
The regulatory applications for tisagenlecleucel in the US and EU are supported by data from the Novartis-sponsored global clinical trial program in children and young adults with R/R B-cell ALL and adults with R/R DLBCL.
Results from the phase 2 JULIET trial served as the basis of the sBLA and MAA for tisagenlecleucel in adults with R/R DLCBL. Data from this trial were presented at the 2017 ASH Annual Meeting in December.
Results from the phase 2 ELIANA study were submitted as part of the MAA for tisagenlecleucel in children and young adults with R/R B-cell ALL. Data from this trial were presented at the 2017 EHA Congress last June.
About priority review, accelerated assessment
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The FDA’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The EMA grants accelerated assessment when a product is expected to be of major public health interest, particularly from the point of view of therapeutic innovation.
Accelerated assessment shortens the review period from 210 days to 150 days.