User login
Brentuximab vedotin sBLA receives priority review

The US Food and Drug Administration (FDA) has accepted for priority review a supplemental biologics license application (sBLA) for brentuximab vedotin (ADCETRIS).
With this sBLA, Seattle Genetics, Inc., is seeking approval for brentuximab vedotin in combination with chemotherapy for frontline treatment of patients with advanced classical Hodgkin lymphoma (HL).
The FDA expects to make a decision on the sBLA by May 1, 2018.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The priority review for this sBLA is based on positive results from the phase 3 ECHELON-1 trial.
The FDA previously granted brentuximab vedotin breakthrough therapy designation based on ECHELON-1 results.
Breakthrough therapy designation is intended to expedite the development and review of promising drug candidates for serious or life-threatening conditions. It is based upon clinical evidence of substantial improvement over existing therapies in one or more clinically significant endpoints.
ECHELON-1
Result from ECHELON-1 were presented at the 2017 ASH Annual Meeting and simultaneously published in NEJM.
In this trial, researchers compared brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine (A+AVD) to doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) as frontline treatment for 1334 patients with advanced HL.
The study’s primary endpoint was modified progression-free survival (PFS), which was defined as time to progression, death, or evidence of non-complete response after completion of frontline therapy followed by subsequent anticancer therapy.
According to an independent review facility, A+AVD provided a significant improvement in modified PFS compared to ABVD. The hazard ratio was 0.77 (P=0.035), which corresponds to a 23% reduction in the risk of progression, death, or the need for additional anticancer therapy.
The 2-year modified PFS rate was 82.1% in the A+AVD arm and 77.2% in the ABVD arm.
There was no significant difference between the treatment arms when it came to response rates or overall survival.
The objective response rate was 86% in the A+AVD arm and 83% in the ABVD arm (P=0.12). The complete response rate was 73% and 70%, respectively (P=0.22).
The interim 2-year overall survival rate was 97% in the A+AVD arm and 95% in the ABVD arm (hazard ratio=0.72; P=0.19).
The overall incidence of adverse events (AEs) was 99% in the A+AVD arm and 98% in the ABVD arm. The incidence of grade 3 or higher AEs was 83% and 66%, respectively, and the incidence of serious AEs was 43% and 27%, respectively.
Neutropenia, febrile neutropenia, and peripheral neuropathy were more common with A+AVD, while pulmonary toxicity was more common with ABVD.
About brentuximab vedotin
Brentuximab vedotin is already FDA-approved to treat adults with:
- Classical HL who have failed autologous hematopoietic stem cell transplant (auto-HSCT) or, in those who are not auto-HSCT candidates, have failed at least 2 prior multi-agent chemotherapy regimens.
- Classical HL at high risk of relapse or progression as post-auto-HSCT consolidation.
- Primary cutaneous anaplastic large-cell lymphoma (ALCL) or CD30-expressing mycosis fungoides who have received prior systemic therapy.
- Systemic ALCL who have failed at least 1 prior multi-agent chemotherapy regimen. (The drug has accelerated approval for this indication, based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.)

The US Food and Drug Administration (FDA) has accepted for priority review a supplemental biologics license application (sBLA) for brentuximab vedotin (ADCETRIS).
With this sBLA, Seattle Genetics, Inc., is seeking approval for brentuximab vedotin in combination with chemotherapy for frontline treatment of patients with advanced classical Hodgkin lymphoma (HL).
The FDA expects to make a decision on the sBLA by May 1, 2018.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The priority review for this sBLA is based on positive results from the phase 3 ECHELON-1 trial.
The FDA previously granted brentuximab vedotin breakthrough therapy designation based on ECHELON-1 results.
Breakthrough therapy designation is intended to expedite the development and review of promising drug candidates for serious or life-threatening conditions. It is based upon clinical evidence of substantial improvement over existing therapies in one or more clinically significant endpoints.
ECHELON-1
Result from ECHELON-1 were presented at the 2017 ASH Annual Meeting and simultaneously published in NEJM.
In this trial, researchers compared brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine (A+AVD) to doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) as frontline treatment for 1334 patients with advanced HL.
The study’s primary endpoint was modified progression-free survival (PFS), which was defined as time to progression, death, or evidence of non-complete response after completion of frontline therapy followed by subsequent anticancer therapy.
According to an independent review facility, A+AVD provided a significant improvement in modified PFS compared to ABVD. The hazard ratio was 0.77 (P=0.035), which corresponds to a 23% reduction in the risk of progression, death, or the need for additional anticancer therapy.
The 2-year modified PFS rate was 82.1% in the A+AVD arm and 77.2% in the ABVD arm.
There was no significant difference between the treatment arms when it came to response rates or overall survival.
The objective response rate was 86% in the A+AVD arm and 83% in the ABVD arm (P=0.12). The complete response rate was 73% and 70%, respectively (P=0.22).
The interim 2-year overall survival rate was 97% in the A+AVD arm and 95% in the ABVD arm (hazard ratio=0.72; P=0.19).
The overall incidence of adverse events (AEs) was 99% in the A+AVD arm and 98% in the ABVD arm. The incidence of grade 3 or higher AEs was 83% and 66%, respectively, and the incidence of serious AEs was 43% and 27%, respectively.
Neutropenia, febrile neutropenia, and peripheral neuropathy were more common with A+AVD, while pulmonary toxicity was more common with ABVD.
About brentuximab vedotin
Brentuximab vedotin is already FDA-approved to treat adults with:
- Classical HL who have failed autologous hematopoietic stem cell transplant (auto-HSCT) or, in those who are not auto-HSCT candidates, have failed at least 2 prior multi-agent chemotherapy regimens.
- Classical HL at high risk of relapse or progression as post-auto-HSCT consolidation.
- Primary cutaneous anaplastic large-cell lymphoma (ALCL) or CD30-expressing mycosis fungoides who have received prior systemic therapy.
- Systemic ALCL who have failed at least 1 prior multi-agent chemotherapy regimen. (The drug has accelerated approval for this indication, based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.)
The US Food and Drug Administration (FDA) has accepted for priority review a supplemental biologics license application (sBLA) for brentuximab vedotin (ADCETRIS).
With this sBLA, Seattle Genetics, Inc., is seeking approval for brentuximab vedotin in combination with chemotherapy for frontline treatment of patients with advanced classical Hodgkin lymphoma (HL).
The FDA expects to make a decision on the sBLA by May 1, 2018.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The priority review for this sBLA is based on positive results from the phase 3 ECHELON-1 trial.
The FDA previously granted brentuximab vedotin breakthrough therapy designation based on ECHELON-1 results.
Breakthrough therapy designation is intended to expedite the development and review of promising drug candidates for serious or life-threatening conditions. It is based upon clinical evidence of substantial improvement over existing therapies in one or more clinically significant endpoints.
ECHELON-1
Result from ECHELON-1 were presented at the 2017 ASH Annual Meeting and simultaneously published in NEJM.
In this trial, researchers compared brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine (A+AVD) to doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) as frontline treatment for 1334 patients with advanced HL.
The study’s primary endpoint was modified progression-free survival (PFS), which was defined as time to progression, death, or evidence of non-complete response after completion of frontline therapy followed by subsequent anticancer therapy.
According to an independent review facility, A+AVD provided a significant improvement in modified PFS compared to ABVD. The hazard ratio was 0.77 (P=0.035), which corresponds to a 23% reduction in the risk of progression, death, or the need for additional anticancer therapy.
The 2-year modified PFS rate was 82.1% in the A+AVD arm and 77.2% in the ABVD arm.
There was no significant difference between the treatment arms when it came to response rates or overall survival.
The objective response rate was 86% in the A+AVD arm and 83% in the ABVD arm (P=0.12). The complete response rate was 73% and 70%, respectively (P=0.22).
The interim 2-year overall survival rate was 97% in the A+AVD arm and 95% in the ABVD arm (hazard ratio=0.72; P=0.19).
The overall incidence of adverse events (AEs) was 99% in the A+AVD arm and 98% in the ABVD arm. The incidence of grade 3 or higher AEs was 83% and 66%, respectively, and the incidence of serious AEs was 43% and 27%, respectively.
Neutropenia, febrile neutropenia, and peripheral neuropathy were more common with A+AVD, while pulmonary toxicity was more common with ABVD.
About brentuximab vedotin
Brentuximab vedotin is already FDA-approved to treat adults with:
- Classical HL who have failed autologous hematopoietic stem cell transplant (auto-HSCT) or, in those who are not auto-HSCT candidates, have failed at least 2 prior multi-agent chemotherapy regimens.
- Classical HL at high risk of relapse or progression as post-auto-HSCT consolidation.
- Primary cutaneous anaplastic large-cell lymphoma (ALCL) or CD30-expressing mycosis fungoides who have received prior systemic therapy.
- Systemic ALCL who have failed at least 1 prior multi-agent chemotherapy regimen. (The drug has accelerated approval for this indication, based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.)
BTK inhibitor zanubrutinib active in non-Hodgkin lymphomas
ATLANTA – , according to data presented at the annual meeting of the American Society of Hematology.
Response rates ranged from 31% to 88% depending on the lymphoma subtype. Overall, approximately 10% of patients discontinued the drug because of adverse events, reported Constantine S. Tam, MBBS, MD, of Peter MacCallum Cancer Centre & St. Vincent’s Hospital, Melbourne.
“There was encouraging activity against all the spectrum of indolent and aggressive NHL subtypes … and durable responses were observed across a variety of histologies,” Dr. Tam said.
Zanubrutinib is a second-generation BTK inhibitor that, based on biochemical assays, has higher selectivity against BTK than ibrutinib, Dr. Tam said.
He presented results of an open-label, multicenter, phase 1b study of daily or twice-daily zanubrutinib in patients with B-cell malignancies, most of them relapsed or refractory to prior therapies. The lymphoma subtypes he presented included diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), mantle cell lymphoma (MCL), and marginal zone lymphoma (MZL).
For 34 patients with indolent lymphomas (FL and MZL), the most frequent adverse events were petechiae/purpura/contusion and upper respiratory tract infection. Eleven grade 3-5 adverse events were reported, including neutropenia, infection, nausea, urinary tract infection, and abdominal pain.
Atrial fibrillation was observed in two patients in the aggressive NHL cohort, for an overall AF rate of approximately 2%, Dr. Tam said.
For 65 patients with aggressive lymphomas (DLBCL and MCL), the most frequent adverse events were petechiae/purpura/contusion and diarrhea; 27 grade 3-5 adverse events were reported, including neutropenia, pneumonia, and anemia.
The highest overall response rate reported was for MCL, at 88% (28 of 32 patients) followed by MZL at 78% (7 of 9 patients), FL at 41% (7 of 17 patients), and DLBCL 31% (8 of 26 patients).
The recommended phase 2 dose for zanubrutinib is either 320 mg/day once daily or a split dose of 160 mg twice daily, Dr. Tam said.
Based on this experience, investigators started a registration trial of zanubrutinib in combination with obinutuzumab for FL, and additional trials are planned, according to Dr. Tam.
There are also registration trials in Waldenstrom macroglobulinemia and chronic lymphocytic leukemia based on other data suggesting activity of zanubrutinib in those disease types, he added.
Zanubrutinib is a product of BeiGene. Dr. Tam reported disclosures related to Roche, Janssen Cilag, Abbvie, Celgene, Pharmacyclics, Onyx, and Amgen.
SOURCE: Tam C et al, ASH 2017, Abstract 152
ATLANTA – , according to data presented at the annual meeting of the American Society of Hematology.
Response rates ranged from 31% to 88% depending on the lymphoma subtype. Overall, approximately 10% of patients discontinued the drug because of adverse events, reported Constantine S. Tam, MBBS, MD, of Peter MacCallum Cancer Centre & St. Vincent’s Hospital, Melbourne.
“There was encouraging activity against all the spectrum of indolent and aggressive NHL subtypes … and durable responses were observed across a variety of histologies,” Dr. Tam said.
Zanubrutinib is a second-generation BTK inhibitor that, based on biochemical assays, has higher selectivity against BTK than ibrutinib, Dr. Tam said.
He presented results of an open-label, multicenter, phase 1b study of daily or twice-daily zanubrutinib in patients with B-cell malignancies, most of them relapsed or refractory to prior therapies. The lymphoma subtypes he presented included diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), mantle cell lymphoma (MCL), and marginal zone lymphoma (MZL).
For 34 patients with indolent lymphomas (FL and MZL), the most frequent adverse events were petechiae/purpura/contusion and upper respiratory tract infection. Eleven grade 3-5 adverse events were reported, including neutropenia, infection, nausea, urinary tract infection, and abdominal pain.
Atrial fibrillation was observed in two patients in the aggressive NHL cohort, for an overall AF rate of approximately 2%, Dr. Tam said.
For 65 patients with aggressive lymphomas (DLBCL and MCL), the most frequent adverse events were petechiae/purpura/contusion and diarrhea; 27 grade 3-5 adverse events were reported, including neutropenia, pneumonia, and anemia.
The highest overall response rate reported was for MCL, at 88% (28 of 32 patients) followed by MZL at 78% (7 of 9 patients), FL at 41% (7 of 17 patients), and DLBCL 31% (8 of 26 patients).
The recommended phase 2 dose for zanubrutinib is either 320 mg/day once daily or a split dose of 160 mg twice daily, Dr. Tam said.
Based on this experience, investigators started a registration trial of zanubrutinib in combination with obinutuzumab for FL, and additional trials are planned, according to Dr. Tam.
There are also registration trials in Waldenstrom macroglobulinemia and chronic lymphocytic leukemia based on other data suggesting activity of zanubrutinib in those disease types, he added.
Zanubrutinib is a product of BeiGene. Dr. Tam reported disclosures related to Roche, Janssen Cilag, Abbvie, Celgene, Pharmacyclics, Onyx, and Amgen.
SOURCE: Tam C et al, ASH 2017, Abstract 152
ATLANTA – , according to data presented at the annual meeting of the American Society of Hematology.
Response rates ranged from 31% to 88% depending on the lymphoma subtype. Overall, approximately 10% of patients discontinued the drug because of adverse events, reported Constantine S. Tam, MBBS, MD, of Peter MacCallum Cancer Centre & St. Vincent’s Hospital, Melbourne.
“There was encouraging activity against all the spectrum of indolent and aggressive NHL subtypes … and durable responses were observed across a variety of histologies,” Dr. Tam said.
Zanubrutinib is a second-generation BTK inhibitor that, based on biochemical assays, has higher selectivity against BTK than ibrutinib, Dr. Tam said.
He presented results of an open-label, multicenter, phase 1b study of daily or twice-daily zanubrutinib in patients with B-cell malignancies, most of them relapsed or refractory to prior therapies. The lymphoma subtypes he presented included diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), mantle cell lymphoma (MCL), and marginal zone lymphoma (MZL).
For 34 patients with indolent lymphomas (FL and MZL), the most frequent adverse events were petechiae/purpura/contusion and upper respiratory tract infection. Eleven grade 3-5 adverse events were reported, including neutropenia, infection, nausea, urinary tract infection, and abdominal pain.
Atrial fibrillation was observed in two patients in the aggressive NHL cohort, for an overall AF rate of approximately 2%, Dr. Tam said.
For 65 patients with aggressive lymphomas (DLBCL and MCL), the most frequent adverse events were petechiae/purpura/contusion and diarrhea; 27 grade 3-5 adverse events were reported, including neutropenia, pneumonia, and anemia.
The highest overall response rate reported was for MCL, at 88% (28 of 32 patients) followed by MZL at 78% (7 of 9 patients), FL at 41% (7 of 17 patients), and DLBCL 31% (8 of 26 patients).
The recommended phase 2 dose for zanubrutinib is either 320 mg/day once daily or a split dose of 160 mg twice daily, Dr. Tam said.
Based on this experience, investigators started a registration trial of zanubrutinib in combination with obinutuzumab for FL, and additional trials are planned, according to Dr. Tam.
There are also registration trials in Waldenstrom macroglobulinemia and chronic lymphocytic leukemia based on other data suggesting activity of zanubrutinib in those disease types, he added.
Zanubrutinib is a product of BeiGene. Dr. Tam reported disclosures related to Roche, Janssen Cilag, Abbvie, Celgene, Pharmacyclics, Onyx, and Amgen.
SOURCE: Tam C et al, ASH 2017, Abstract 152
REPORTING FROM ASH 2017
Key clinical point: Monotherapy with the BTK inhibitor zanubrutinib (BGB-3111) was active and well tolerated in patients with a variety of non-Hodgkin lymphoma (NHL) subtypes.
Major finding: Response rates ranged from 31% to 88% depending on the lymphoma subtype.
Data source: Preliminary results of an open-label, multicenter, phase 1b study including 99 patients with relapsed or refractory diffuse large B-cell lymphoma, follicular lymphoma, mantle cell lymphoma, or marginal zone lymphoma.
Disclosures: Zanubrutinib is a product of BeiGene. Constantine S. Tam, MBBS, MD, reported disclosures related to Roche, Janssen Cilag, Abbvie, Celgene, Pharmacyclics, Onyx, and Amgen.
Source: Tam C et al. ASH 2017, Abstract 152.
Daratumumab looks good in light chain amyloidosis
ATLANTA – In patients with previously treated immunoglobulin light chain (AL) amyloidosis, daratumumab monotherapy produced deep, rapid hematologic responses, based on initial results from a phase 2 trial.
So far, the response rate is about twice the rate seen with daratumumab in relapsed/refractory multiple myeloma, Murielle Roussel, MD, of IUCT-Oncopole, Toulouse, France, said at the annual meeting of the American Society of Hematology. “We observed deep and rapid clonal responses, even after the first infusion.”
“Daratumumab also had a good safety profile characterized by nonsevere adverse events after initial infusion. There was only one drug-related serious adverse event, grade 3 lymphopenia,” she said.
In a second study, the risk for daratumumab infusion reactions was low when patients received a prophylactic regimen initiated about an hour before daratumumab infusion.
Daratumumab, a novel, fully humanized IgG1-kappa monoclonal antibody with high affinity for CD38, is approved for treating relapsed/refractory multiple myeloma. In AL amyloidosis, as in myeloma, monoclonal light chains nearly always originate from plasma cells that consistently express CD38.
Data from small studies indicate that daratumumab effectively treats AL amyloidosis. To further evaluate safety and efficacy, 36 adults with previously treated disease received 28-day cycles of daratumumab (16 mg/kg IV) weekly for two cycles and then every other week for four cycles. Most patients had received three prior lines of therapy, about two-thirds had cardiac involvement (median baseline NT-proBNP 1,118 ng/L; range, 60-6,825), and about 60% had renal involvement.
At data cutoff in mid-November 2017, fifteen patients had completed all six treatment cycles. Three stopped treatment because of progression. Two died, one of progressive cardiac amyloidosis and one of unrelated lung cancer.
Eleven patients had grade 1-2 infusion reactions at first injection. Among 17 grade 3 or higher adverse events, only lymphopenia was deemed treatment related.
At 6 months, 15 of 32 evaluable patients (44%) had a very good partial response (VGPR; at least a 40% drop in baseline difference in involved and uninvolved free light chains (dFLC). Another 16% had a partial response, and 41% did not respond.
Patients with durable responses tended to have about a 70% drop in dFLC after the first daratumumab dose. Baseline variables did not seem to predict durability of response, Dr. Roussel said. “Further studies in amyloidosis are warranted in relapsed or refractory patients and also in the frontline setting.”
The second trial focused on preventing infusion reactions to daratumumab. In early trials of daratumumab for relapsed/recalcitrant multiple myeloma, patients developed moderate to severe bronchospasm, laryngeal or pulmonary edema, hypoxia, and hypertension, noted Vaishali Sanchorawala, MD, of Boston Medical Center. Since those trials, prophylactic therapies have been used to reduce the risk of infusion reactions.
Dr. Sanchorawala’s study enrolled 12 patients with previously treated AL amyloidosis and cardiac biomarker stage II or stage III disease. About 60% of patients were refractory to their last treatment. Median NT-proBNP level was 1,357 pg/mL (range, 469-3,962), median urine protein excretion was 0.44 g (0-10.1), and median dFLC was 105 mg/dL (3.8-854).
Patients received 16 mg/kg daratumumab IV weekly for 8 weeks, then every 2 weeks for 16 weeks, and then every 4 weeks for up to 24 months. About an hour before infusion, they received acetaminophen, diphenhydramine, loratadine famotidine, montelukast, and methylprednisolone (100 mg for two infusions; 60 mg thereafter). Ondansetron also was added to control mild nausea and vomiting. Two hours into the infusion, patients received diphenhydramine, famotidine, and methylprednisolone (40 mg). They received methylprednisolone (20 mg) and montelukast 1-2 days after the first two infusions, after which montelukast was optional. All received prophylactic acyclovir.
At the Nov. 15, 2017 data cutoff, 11 patients remained on study and one left after disease progressed. This patient’s disease was refractory to many prior therapies and had a complete response to autologous stem cell transplant, said Dr. Sanchorawala.
There were no grade 3-4 infusion reactions. Nine evaluable patients at 3 months had two complete hematologic responses, six VGPRs (at least a 65% drop in dFLC), and one partial response. One-third had at least a 30% improvement in NT-proBNP at 3 months, as did three of four evaluable patients at 6 months. About half had least a 30% drop in urine protein excretion at 6 months.
First infusions lasted a median of 7 hours, making them doable during a clinic day if bloods are drawn beforehand, Dr. Sanchorawala said. Second and subsequent infusions took about 4 hours.
“Preliminary data suggest a rapid hematologic response after one dose of daratumumab and high rates of response at 3 and 6 months, ” she concluded. “Since the plasma cell clone is so low in amyloidosis, single-agent daratumumab has a very positive, strong effect. We may not need to combine other agents with this therapy.”
Both presentations sparked substantial interest during the discussion period after the presentations, especially because daratumumab was given as monotherapy. “This would be a new indication for daratumumab,” said session moderator Dan Vogl, MD, director of the Abramson Cancer Center Clinical Research Unit, University of Pennsylvania, Philadelphia.
Janssen makes daratumumab and provided partial funding for both studies. Dr. Sanchorawala had no conflicts of interest. Dr. Roussel disclosed honoraria and research funding from Janssen.
SOURCES: Sanchorawala V et al. ASH 2017 Abstract 507; Roussel M et al. ASH 2017 Abstract 508.
ATLANTA – In patients with previously treated immunoglobulin light chain (AL) amyloidosis, daratumumab monotherapy produced deep, rapid hematologic responses, based on initial results from a phase 2 trial.
So far, the response rate is about twice the rate seen with daratumumab in relapsed/refractory multiple myeloma, Murielle Roussel, MD, of IUCT-Oncopole, Toulouse, France, said at the annual meeting of the American Society of Hematology. “We observed deep and rapid clonal responses, even after the first infusion.”
“Daratumumab also had a good safety profile characterized by nonsevere adverse events after initial infusion. There was only one drug-related serious adverse event, grade 3 lymphopenia,” she said.
In a second study, the risk for daratumumab infusion reactions was low when patients received a prophylactic regimen initiated about an hour before daratumumab infusion.
Daratumumab, a novel, fully humanized IgG1-kappa monoclonal antibody with high affinity for CD38, is approved for treating relapsed/refractory multiple myeloma. In AL amyloidosis, as in myeloma, monoclonal light chains nearly always originate from plasma cells that consistently express CD38.
Data from small studies indicate that daratumumab effectively treats AL amyloidosis. To further evaluate safety and efficacy, 36 adults with previously treated disease received 28-day cycles of daratumumab (16 mg/kg IV) weekly for two cycles and then every other week for four cycles. Most patients had received three prior lines of therapy, about two-thirds had cardiac involvement (median baseline NT-proBNP 1,118 ng/L; range, 60-6,825), and about 60% had renal involvement.
At data cutoff in mid-November 2017, fifteen patients had completed all six treatment cycles. Three stopped treatment because of progression. Two died, one of progressive cardiac amyloidosis and one of unrelated lung cancer.
Eleven patients had grade 1-2 infusion reactions at first injection. Among 17 grade 3 or higher adverse events, only lymphopenia was deemed treatment related.
At 6 months, 15 of 32 evaluable patients (44%) had a very good partial response (VGPR; at least a 40% drop in baseline difference in involved and uninvolved free light chains (dFLC). Another 16% had a partial response, and 41% did not respond.
Patients with durable responses tended to have about a 70% drop in dFLC after the first daratumumab dose. Baseline variables did not seem to predict durability of response, Dr. Roussel said. “Further studies in amyloidosis are warranted in relapsed or refractory patients and also in the frontline setting.”
The second trial focused on preventing infusion reactions to daratumumab. In early trials of daratumumab for relapsed/recalcitrant multiple myeloma, patients developed moderate to severe bronchospasm, laryngeal or pulmonary edema, hypoxia, and hypertension, noted Vaishali Sanchorawala, MD, of Boston Medical Center. Since those trials, prophylactic therapies have been used to reduce the risk of infusion reactions.
Dr. Sanchorawala’s study enrolled 12 patients with previously treated AL amyloidosis and cardiac biomarker stage II or stage III disease. About 60% of patients were refractory to their last treatment. Median NT-proBNP level was 1,357 pg/mL (range, 469-3,962), median urine protein excretion was 0.44 g (0-10.1), and median dFLC was 105 mg/dL (3.8-854).
Patients received 16 mg/kg daratumumab IV weekly for 8 weeks, then every 2 weeks for 16 weeks, and then every 4 weeks for up to 24 months. About an hour before infusion, they received acetaminophen, diphenhydramine, loratadine famotidine, montelukast, and methylprednisolone (100 mg for two infusions; 60 mg thereafter). Ondansetron also was added to control mild nausea and vomiting. Two hours into the infusion, patients received diphenhydramine, famotidine, and methylprednisolone (40 mg). They received methylprednisolone (20 mg) and montelukast 1-2 days after the first two infusions, after which montelukast was optional. All received prophylactic acyclovir.
At the Nov. 15, 2017 data cutoff, 11 patients remained on study and one left after disease progressed. This patient’s disease was refractory to many prior therapies and had a complete response to autologous stem cell transplant, said Dr. Sanchorawala.
There were no grade 3-4 infusion reactions. Nine evaluable patients at 3 months had two complete hematologic responses, six VGPRs (at least a 65% drop in dFLC), and one partial response. One-third had at least a 30% improvement in NT-proBNP at 3 months, as did three of four evaluable patients at 6 months. About half had least a 30% drop in urine protein excretion at 6 months.
First infusions lasted a median of 7 hours, making them doable during a clinic day if bloods are drawn beforehand, Dr. Sanchorawala said. Second and subsequent infusions took about 4 hours.
“Preliminary data suggest a rapid hematologic response after one dose of daratumumab and high rates of response at 3 and 6 months, ” she concluded. “Since the plasma cell clone is so low in amyloidosis, single-agent daratumumab has a very positive, strong effect. We may not need to combine other agents with this therapy.”
Both presentations sparked substantial interest during the discussion period after the presentations, especially because daratumumab was given as monotherapy. “This would be a new indication for daratumumab,” said session moderator Dan Vogl, MD, director of the Abramson Cancer Center Clinical Research Unit, University of Pennsylvania, Philadelphia.
Janssen makes daratumumab and provided partial funding for both studies. Dr. Sanchorawala had no conflicts of interest. Dr. Roussel disclosed honoraria and research funding from Janssen.
SOURCES: Sanchorawala V et al. ASH 2017 Abstract 507; Roussel M et al. ASH 2017 Abstract 508.
ATLANTA – In patients with previously treated immunoglobulin light chain (AL) amyloidosis, daratumumab monotherapy produced deep, rapid hematologic responses, based on initial results from a phase 2 trial.
So far, the response rate is about twice the rate seen with daratumumab in relapsed/refractory multiple myeloma, Murielle Roussel, MD, of IUCT-Oncopole, Toulouse, France, said at the annual meeting of the American Society of Hematology. “We observed deep and rapid clonal responses, even after the first infusion.”
“Daratumumab also had a good safety profile characterized by nonsevere adverse events after initial infusion. There was only one drug-related serious adverse event, grade 3 lymphopenia,” she said.
In a second study, the risk for daratumumab infusion reactions was low when patients received a prophylactic regimen initiated about an hour before daratumumab infusion.
Daratumumab, a novel, fully humanized IgG1-kappa monoclonal antibody with high affinity for CD38, is approved for treating relapsed/refractory multiple myeloma. In AL amyloidosis, as in myeloma, monoclonal light chains nearly always originate from plasma cells that consistently express CD38.
Data from small studies indicate that daratumumab effectively treats AL amyloidosis. To further evaluate safety and efficacy, 36 adults with previously treated disease received 28-day cycles of daratumumab (16 mg/kg IV) weekly for two cycles and then every other week for four cycles. Most patients had received three prior lines of therapy, about two-thirds had cardiac involvement (median baseline NT-proBNP 1,118 ng/L; range, 60-6,825), and about 60% had renal involvement.
At data cutoff in mid-November 2017, fifteen patients had completed all six treatment cycles. Three stopped treatment because of progression. Two died, one of progressive cardiac amyloidosis and one of unrelated lung cancer.
Eleven patients had grade 1-2 infusion reactions at first injection. Among 17 grade 3 or higher adverse events, only lymphopenia was deemed treatment related.
At 6 months, 15 of 32 evaluable patients (44%) had a very good partial response (VGPR; at least a 40% drop in baseline difference in involved and uninvolved free light chains (dFLC). Another 16% had a partial response, and 41% did not respond.
Patients with durable responses tended to have about a 70% drop in dFLC after the first daratumumab dose. Baseline variables did not seem to predict durability of response, Dr. Roussel said. “Further studies in amyloidosis are warranted in relapsed or refractory patients and also in the frontline setting.”
The second trial focused on preventing infusion reactions to daratumumab. In early trials of daratumumab for relapsed/recalcitrant multiple myeloma, patients developed moderate to severe bronchospasm, laryngeal or pulmonary edema, hypoxia, and hypertension, noted Vaishali Sanchorawala, MD, of Boston Medical Center. Since those trials, prophylactic therapies have been used to reduce the risk of infusion reactions.
Dr. Sanchorawala’s study enrolled 12 patients with previously treated AL amyloidosis and cardiac biomarker stage II or stage III disease. About 60% of patients were refractory to their last treatment. Median NT-proBNP level was 1,357 pg/mL (range, 469-3,962), median urine protein excretion was 0.44 g (0-10.1), and median dFLC was 105 mg/dL (3.8-854).
Patients received 16 mg/kg daratumumab IV weekly for 8 weeks, then every 2 weeks for 16 weeks, and then every 4 weeks for up to 24 months. About an hour before infusion, they received acetaminophen, diphenhydramine, loratadine famotidine, montelukast, and methylprednisolone (100 mg for two infusions; 60 mg thereafter). Ondansetron also was added to control mild nausea and vomiting. Two hours into the infusion, patients received diphenhydramine, famotidine, and methylprednisolone (40 mg). They received methylprednisolone (20 mg) and montelukast 1-2 days after the first two infusions, after which montelukast was optional. All received prophylactic acyclovir.
At the Nov. 15, 2017 data cutoff, 11 patients remained on study and one left after disease progressed. This patient’s disease was refractory to many prior therapies and had a complete response to autologous stem cell transplant, said Dr. Sanchorawala.
There were no grade 3-4 infusion reactions. Nine evaluable patients at 3 months had two complete hematologic responses, six VGPRs (at least a 65% drop in dFLC), and one partial response. One-third had at least a 30% improvement in NT-proBNP at 3 months, as did three of four evaluable patients at 6 months. About half had least a 30% drop in urine protein excretion at 6 months.
First infusions lasted a median of 7 hours, making them doable during a clinic day if bloods are drawn beforehand, Dr. Sanchorawala said. Second and subsequent infusions took about 4 hours.
“Preliminary data suggest a rapid hematologic response after one dose of daratumumab and high rates of response at 3 and 6 months, ” she concluded. “Since the plasma cell clone is so low in amyloidosis, single-agent daratumumab has a very positive, strong effect. We may not need to combine other agents with this therapy.”
Both presentations sparked substantial interest during the discussion period after the presentations, especially because daratumumab was given as monotherapy. “This would be a new indication for daratumumab,” said session moderator Dan Vogl, MD, director of the Abramson Cancer Center Clinical Research Unit, University of Pennsylvania, Philadelphia.
Janssen makes daratumumab and provided partial funding for both studies. Dr. Sanchorawala had no conflicts of interest. Dr. Roussel disclosed honoraria and research funding from Janssen.
SOURCES: Sanchorawala V et al. ASH 2017 Abstract 507; Roussel M et al. ASH 2017 Abstract 508.
REPORTING FROM ASH 2017
Key clinical point: Major finding: Rates of very good partial response or complete response were 44% and 33%, respectively, at 6 months.
Data source: Two phase 2 trials of daratumumab monotherapy in patients with previously treated light chain amyloidosis (NCT02816476 [36 patients] and NCT02841033 [12 patients]).
Disclosures: Janssen makes daratumumab and provided partial funding for both studies. Dr. Roussel disclosed honoraria and research funding from Janssen. Dr. Sanchorawala had no conflicts of interest.
Sources: Sanchorawala V et al. ASH 2017 Abstract 507; Roussel M et al. ASH 2017 Abstract 508.
Update reveals ongoing responses in ZUMA-1

ATLANTA—The chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (axi-cel; KTE-C19) is showing consistent, ongoing responses more than a year after infusion.
An updated analysis of the phase 1/2 ZUMA-1 trial showed that 42% of patients who received axi-cel maintained an objective response at a median follow-up of 15.4 months.
Forty percent of patients have maintained a complete response (CR).
This compares with a 44% objective response rate and a 39% CR rate in the primary analysis of phase 2 ZUMA-1 data, when the median follow-up was 8.7 months.
Sattva S. Neelapu, MD, of MD Anderson Cancer Center in Houston, Texas, reported the long-term results from ZUMA-1 at the 2017 ASH Annual Meeting (abstract 578). The findings were published simultaneously in NEJM.
The primary phase 2 analysis was previously presented at the AACR Annual Meeting 2017.
At ASH 2017, Dr Neelapu disclosed that he has received research funding and served as a consultant for Kite Pharma, the developer of axi-cel. Kite Pharma and the Leukemia & Lymphoma Society Therapy Acceleration Program supported ZUMA-1.
Study schema and patient characteristics
Phase 1 of ZUMA-1 enrolled 7 patients with diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), or transformed follicular lymphoma (TFL).
In phase 2, 101 patients were grouped into 2 cohorts—77 with refractory DLBCL and 24 with refractory PMBCL/TFL.
A total of 108 patients were treated in phases 1 and 2 and were included in the long-term pooled analysis.
Patients received a conditioning regimen of cyclophosphamide and fludarabine and, 2 days later, a fixed dose of axi-cel at 2 x 106 CAR T cells/kg.
“Importantly, the product could be manufactured for 99% of enrolled patients,” Dr Neelapu said. “Moreover, 91% of the enrolled patients were dosed with axi-cel, and there were no patients lost to follow-up.”
Patients in the pooled analysis were a median age of 58 (range, 23–76), and 27 (25%) were 65 or older.
Seventy-three patients (68%) were male, 62 (57%) had an ECOG status of 1, 90 (83%) had stage III or IV disease, and 48 (44%) had an IPI score of 3 to 4.
Seventy-six patients (70%) had received 3 or more prior therapies.
Eighty patients (74%) were refractory to their second or later line of therapy, and 70 (65%) had progressive disease as their best response to their last prior therapy. Twenty-five patients (23%) had relapsed after autologous stem cell transplant.
Response
The data cutoff for the long-term analysis was August 11, 2017.
In addition to the ongoing responses mentioned above, the best objective response was 82% in both the phase 2 primary analysis and the long-term analysis for phases 1 and 2.
CR as the best objective response increased from 54% in the primary analysis to 58% at the longer follow-up.
“We did observe deepening of the responses over time,” Dr Neelapu said. “At the time of the first tumor assessment, 60 patients had either partial remission or stable disease. But 23 of those 60 eventually achieved a complete remission up to 15 months post-infusion without any additional therapy.”
The median time to conversion from partial response to CR was 64 days (range, 49–242).
“The durability of these responses was observed consistently across key covariates,” Dr Neelapu added, “including the refractory subgroups, the disease stage groups, IPI risk groups. The CD19 status at baseline did not matter, nor did the cell of origin, or the CD4/CD8 ratio of the product.”
Furthermore, the investigators observed no differences in patients who received tocilizumab or corticosteroids.
The median duration of response for all patients was 11.1 months. For those who achieved CR, the median duration of response has not yet been reached.
Three of the 7 patients (43%) in the phase 1 part of the trial had an ongoing CR at 24 months.
At the median follow-up of 15.4 months, 42% of patients were progression-free, and 56% were alive.
The median overall survival has not been reached. Investigators estimated the 18-month overall survival to be 52%.
Safety
Adverse events (AEs) of grade 3 or higher occurred in 97% of patients, and serious AEs of grade 3 or higher occurred in 46% of patients in the updated analysis.
No new axi-cel-related AEs of cytokine release syndrome, neurologic events, or grade 5 AEs have arisen since the primary analysis.
There were four grade 5 events, 2 of which were related to axi-cel.
“All these four grade 5 events were previously reported—three in the phase 2 and one in the phase 1 trial,” Dr Neelapu said.
Most patients experienced hypogammaglobulinemia and B-cell aplasia. Eight percent of patients had IVIG support during the study.
Infections, such as pneumonia, influenza, and viral infection, were the most common new-onset treatment-emergent serious AEs occurring after 6 months in 10 patients. All were manageable and resolved prior to the data cut-off.
Persistence and resistance
“We observed long-term persistence of the CAR T cells,” Dr Neelapu said.
CAR T cells persisted in 71% of patients still responding at 1 year. And durable responses were observed in patients with and without detectable CAR T cells.
A central review committee analyzed biopsies of 21 evaluable patients at progression to try to determine the mechanism of resistance.
Fourteen of 21 (67%) biopsies were CD19-positive. Of these, 9 were PD-L1-positive, 4 were PD-L1-negative, and 1 was not evaluable.
Seven patients (33%) were CD19-negative compared to baseline. Of these, 4 were PD-L1-positive, 2 were PD-L1-negative, and 1 was not evaluable.
“This PD-L1 expression was observed in both CD19-positive relapses and CD19-negative relapses,” Dr Neelapu emphasized.
Of the 21 patients, 62% were PD-L1-positive.
Investigators hypothesize that 2 potential mechanisms could contribute to relapse: loss of CD19 and expression of PD-L1.
Axi-cel (Yescarta™) was approved by the US Food and Drug Administration in October for the treatment of adults with relapsed or refractory large B-cell lymphoma. ![]()
ATLANTA—The chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (axi-cel; KTE-C19) is showing consistent, ongoing responses more than a year after infusion.
An updated analysis of the phase 1/2 ZUMA-1 trial showed that 42% of patients who received axi-cel maintained an objective response at a median follow-up of 15.4 months.
Forty percent of patients have maintained a complete response (CR).
This compares with a 44% objective response rate and a 39% CR rate in the primary analysis of phase 2 ZUMA-1 data, when the median follow-up was 8.7 months.
Sattva S. Neelapu, MD, of MD Anderson Cancer Center in Houston, Texas, reported the long-term results from ZUMA-1 at the 2017 ASH Annual Meeting (abstract 578). The findings were published simultaneously in NEJM.
The primary phase 2 analysis was previously presented at the AACR Annual Meeting 2017.
At ASH 2017, Dr Neelapu disclosed that he has received research funding and served as a consultant for Kite Pharma, the developer of axi-cel. Kite Pharma and the Leukemia & Lymphoma Society Therapy Acceleration Program supported ZUMA-1.
Study schema and patient characteristics
Phase 1 of ZUMA-1 enrolled 7 patients with diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), or transformed follicular lymphoma (TFL).
In phase 2, 101 patients were grouped into 2 cohorts—77 with refractory DLBCL and 24 with refractory PMBCL/TFL.
A total of 108 patients were treated in phases 1 and 2 and were included in the long-term pooled analysis.
Patients received a conditioning regimen of cyclophosphamide and fludarabine and, 2 days later, a fixed dose of axi-cel at 2 x 106 CAR T cells/kg.
“Importantly, the product could be manufactured for 99% of enrolled patients,” Dr Neelapu said. “Moreover, 91% of the enrolled patients were dosed with axi-cel, and there were no patients lost to follow-up.”
Patients in the pooled analysis were a median age of 58 (range, 23–76), and 27 (25%) were 65 or older.
Seventy-three patients (68%) were male, 62 (57%) had an ECOG status of 1, 90 (83%) had stage III or IV disease, and 48 (44%) had an IPI score of 3 to 4.
Seventy-six patients (70%) had received 3 or more prior therapies.
Eighty patients (74%) were refractory to their second or later line of therapy, and 70 (65%) had progressive disease as their best response to their last prior therapy. Twenty-five patients (23%) had relapsed after autologous stem cell transplant.
Response
The data cutoff for the long-term analysis was August 11, 2017.
In addition to the ongoing responses mentioned above, the best objective response was 82% in both the phase 2 primary analysis and the long-term analysis for phases 1 and 2.
CR as the best objective response increased from 54% in the primary analysis to 58% at the longer follow-up.
“We did observe deepening of the responses over time,” Dr Neelapu said. “At the time of the first tumor assessment, 60 patients had either partial remission or stable disease. But 23 of those 60 eventually achieved a complete remission up to 15 months post-infusion without any additional therapy.”
The median time to conversion from partial response to CR was 64 days (range, 49–242).
“The durability of these responses was observed consistently across key covariates,” Dr Neelapu added, “including the refractory subgroups, the disease stage groups, IPI risk groups. The CD19 status at baseline did not matter, nor did the cell of origin, or the CD4/CD8 ratio of the product.”
Furthermore, the investigators observed no differences in patients who received tocilizumab or corticosteroids.
The median duration of response for all patients was 11.1 months. For those who achieved CR, the median duration of response has not yet been reached.
Three of the 7 patients (43%) in the phase 1 part of the trial had an ongoing CR at 24 months.
At the median follow-up of 15.4 months, 42% of patients were progression-free, and 56% were alive.
The median overall survival has not been reached. Investigators estimated the 18-month overall survival to be 52%.
Safety
Adverse events (AEs) of grade 3 or higher occurred in 97% of patients, and serious AEs of grade 3 or higher occurred in 46% of patients in the updated analysis.
No new axi-cel-related AEs of cytokine release syndrome, neurologic events, or grade 5 AEs have arisen since the primary analysis.
There were four grade 5 events, 2 of which were related to axi-cel.
“All these four grade 5 events were previously reported—three in the phase 2 and one in the phase 1 trial,” Dr Neelapu said.
Most patients experienced hypogammaglobulinemia and B-cell aplasia. Eight percent of patients had IVIG support during the study.
Infections, such as pneumonia, influenza, and viral infection, were the most common new-onset treatment-emergent serious AEs occurring after 6 months in 10 patients. All were manageable and resolved prior to the data cut-off.
Persistence and resistance
“We observed long-term persistence of the CAR T cells,” Dr Neelapu said.
CAR T cells persisted in 71% of patients still responding at 1 year. And durable responses were observed in patients with and without detectable CAR T cells.
A central review committee analyzed biopsies of 21 evaluable patients at progression to try to determine the mechanism of resistance.
Fourteen of 21 (67%) biopsies were CD19-positive. Of these, 9 were PD-L1-positive, 4 were PD-L1-negative, and 1 was not evaluable.
Seven patients (33%) were CD19-negative compared to baseline. Of these, 4 were PD-L1-positive, 2 were PD-L1-negative, and 1 was not evaluable.
“This PD-L1 expression was observed in both CD19-positive relapses and CD19-negative relapses,” Dr Neelapu emphasized.
Of the 21 patients, 62% were PD-L1-positive.
Investigators hypothesize that 2 potential mechanisms could contribute to relapse: loss of CD19 and expression of PD-L1.
Axi-cel (Yescarta™) was approved by the US Food and Drug Administration in October for the treatment of adults with relapsed or refractory large B-cell lymphoma. ![]()
ATLANTA—The chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (axi-cel; KTE-C19) is showing consistent, ongoing responses more than a year after infusion.
An updated analysis of the phase 1/2 ZUMA-1 trial showed that 42% of patients who received axi-cel maintained an objective response at a median follow-up of 15.4 months.
Forty percent of patients have maintained a complete response (CR).
This compares with a 44% objective response rate and a 39% CR rate in the primary analysis of phase 2 ZUMA-1 data, when the median follow-up was 8.7 months.
Sattva S. Neelapu, MD, of MD Anderson Cancer Center in Houston, Texas, reported the long-term results from ZUMA-1 at the 2017 ASH Annual Meeting (abstract 578). The findings were published simultaneously in NEJM.
The primary phase 2 analysis was previously presented at the AACR Annual Meeting 2017.
At ASH 2017, Dr Neelapu disclosed that he has received research funding and served as a consultant for Kite Pharma, the developer of axi-cel. Kite Pharma and the Leukemia & Lymphoma Society Therapy Acceleration Program supported ZUMA-1.
Study schema and patient characteristics
Phase 1 of ZUMA-1 enrolled 7 patients with diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), or transformed follicular lymphoma (TFL).
In phase 2, 101 patients were grouped into 2 cohorts—77 with refractory DLBCL and 24 with refractory PMBCL/TFL.
A total of 108 patients were treated in phases 1 and 2 and were included in the long-term pooled analysis.
Patients received a conditioning regimen of cyclophosphamide and fludarabine and, 2 days later, a fixed dose of axi-cel at 2 x 106 CAR T cells/kg.
“Importantly, the product could be manufactured for 99% of enrolled patients,” Dr Neelapu said. “Moreover, 91% of the enrolled patients were dosed with axi-cel, and there were no patients lost to follow-up.”
Patients in the pooled analysis were a median age of 58 (range, 23–76), and 27 (25%) were 65 or older.
Seventy-three patients (68%) were male, 62 (57%) had an ECOG status of 1, 90 (83%) had stage III or IV disease, and 48 (44%) had an IPI score of 3 to 4.
Seventy-six patients (70%) had received 3 or more prior therapies.
Eighty patients (74%) were refractory to their second or later line of therapy, and 70 (65%) had progressive disease as their best response to their last prior therapy. Twenty-five patients (23%) had relapsed after autologous stem cell transplant.
Response
The data cutoff for the long-term analysis was August 11, 2017.
In addition to the ongoing responses mentioned above, the best objective response was 82% in both the phase 2 primary analysis and the long-term analysis for phases 1 and 2.
CR as the best objective response increased from 54% in the primary analysis to 58% at the longer follow-up.
“We did observe deepening of the responses over time,” Dr Neelapu said. “At the time of the first tumor assessment, 60 patients had either partial remission or stable disease. But 23 of those 60 eventually achieved a complete remission up to 15 months post-infusion without any additional therapy.”
The median time to conversion from partial response to CR was 64 days (range, 49–242).
“The durability of these responses was observed consistently across key covariates,” Dr Neelapu added, “including the refractory subgroups, the disease stage groups, IPI risk groups. The CD19 status at baseline did not matter, nor did the cell of origin, or the CD4/CD8 ratio of the product.”
Furthermore, the investigators observed no differences in patients who received tocilizumab or corticosteroids.
The median duration of response for all patients was 11.1 months. For those who achieved CR, the median duration of response has not yet been reached.
Three of the 7 patients (43%) in the phase 1 part of the trial had an ongoing CR at 24 months.
At the median follow-up of 15.4 months, 42% of patients were progression-free, and 56% were alive.
The median overall survival has not been reached. Investigators estimated the 18-month overall survival to be 52%.
Safety
Adverse events (AEs) of grade 3 or higher occurred in 97% of patients, and serious AEs of grade 3 or higher occurred in 46% of patients in the updated analysis.
No new axi-cel-related AEs of cytokine release syndrome, neurologic events, or grade 5 AEs have arisen since the primary analysis.
There were four grade 5 events, 2 of which were related to axi-cel.
“All these four grade 5 events were previously reported—three in the phase 2 and one in the phase 1 trial,” Dr Neelapu said.
Most patients experienced hypogammaglobulinemia and B-cell aplasia. Eight percent of patients had IVIG support during the study.
Infections, such as pneumonia, influenza, and viral infection, were the most common new-onset treatment-emergent serious AEs occurring after 6 months in 10 patients. All were manageable and resolved prior to the data cut-off.
Persistence and resistance
“We observed long-term persistence of the CAR T cells,” Dr Neelapu said.
CAR T cells persisted in 71% of patients still responding at 1 year. And durable responses were observed in patients with and without detectable CAR T cells.
A central review committee analyzed biopsies of 21 evaluable patients at progression to try to determine the mechanism of resistance.
Fourteen of 21 (67%) biopsies were CD19-positive. Of these, 9 were PD-L1-positive, 4 were PD-L1-negative, and 1 was not evaluable.
Seven patients (33%) were CD19-negative compared to baseline. Of these, 4 were PD-L1-positive, 2 were PD-L1-negative, and 1 was not evaluable.
“This PD-L1 expression was observed in both CD19-positive relapses and CD19-negative relapses,” Dr Neelapu emphasized.
Of the 21 patients, 62% were PD-L1-positive.
Investigators hypothesize that 2 potential mechanisms could contribute to relapse: loss of CD19 and expression of PD-L1.
Axi-cel (Yescarta™) was approved by the US Food and Drug Administration in October for the treatment of adults with relapsed or refractory large B-cell lymphoma. ![]()
Pemphigus associated with higher risk of hematologic malignancies
, based on the findings of a retrospective study conducted at the Rambam Health Care Campus, Haifa, Israel.
Although the findings are preliminary, the possible associations should be considered when treating pemphigus patients, the investigators reported in the Journal of the American Academy of Dermatology.
Khalaf Kridin, MD, of the Rambam Health Care Campus department of dermatology and his fellow investigators conducted a cross-sectional, retrospective, controlled study of 11,859 patients gathered from the Clait Health Services computerized database. A total of 1,985 pemphigus patients and 9,874 control patients were included. Patients were 72 years old on average, and most were female (60%) and Jewish (90%).
Dr. Kridin and his colleagues measured the prevalence of acute and chronic leukemia, Hodgkin and non-Hodgkin lymphoma, multiple myeloma, and polycythemia vera.
The pemphigus patients, compared with the control group, had a significantly higher prevalence of chronic leukemia (0.9% vs 0.4% [P = .007]), multiple myeloma (0.8% vs 0.4% [P = .009]), and non-Hodgkin lymphoma (1.8% vs 1.2% [P = .040]).
In a sensitivity analysis, patients with pemphigus were twice as likely to have chronic leukemia (odds ratio = 2.1; 95% confidence interval, 1.2-3.6) and multiple myeloma (OR = 2.2; 95% CI, 1.2-3.9) and were one and a half times as likely to have non-Hodgkin lymphoma (OR = 1.5; 95% CI, 1.0-2.2).
Dr. Kridin and his fellow investigators hypothesized that the risks may be related to some pemphigus treatments.
“Certain immunosuppressive treatments for pemphigus, such as azathioprine, could increase the risk of developing hematologic malignancies,” they wrote. “Controlling for immunosuppressive agents attenuated the association of pemphigus with non-Hodgkin lymphoma and multiple myeloma, hinting that they play a role in the higher prevalence.”
Chronic immune stimulation also may be influencing a higher prevalence of hematologic cancers in pemphigus patients “by randomly introducing pro-oncogenic mutations in rapidly dividing cells,” they said.
Investigators were limited by a lack of data on patients’ immunopathological subtype, clinical features, severity of pemphigus, and precise histological type of leukemia and lymphoma.
Dr. Kridin and his fellow investigators reported no relevant financial disclosures.
ezimmerman@frontlinemedcom.com
SOURCE: Kridin K et al. J Am Acad Dermatol. 2017 Dec 2. doi:10.1016/j.jaad.2017.11.039.
, based on the findings of a retrospective study conducted at the Rambam Health Care Campus, Haifa, Israel.
Although the findings are preliminary, the possible associations should be considered when treating pemphigus patients, the investigators reported in the Journal of the American Academy of Dermatology.
Khalaf Kridin, MD, of the Rambam Health Care Campus department of dermatology and his fellow investigators conducted a cross-sectional, retrospective, controlled study of 11,859 patients gathered from the Clait Health Services computerized database. A total of 1,985 pemphigus patients and 9,874 control patients were included. Patients were 72 years old on average, and most were female (60%) and Jewish (90%).
Dr. Kridin and his colleagues measured the prevalence of acute and chronic leukemia, Hodgkin and non-Hodgkin lymphoma, multiple myeloma, and polycythemia vera.
The pemphigus patients, compared with the control group, had a significantly higher prevalence of chronic leukemia (0.9% vs 0.4% [P = .007]), multiple myeloma (0.8% vs 0.4% [P = .009]), and non-Hodgkin lymphoma (1.8% vs 1.2% [P = .040]).
In a sensitivity analysis, patients with pemphigus were twice as likely to have chronic leukemia (odds ratio = 2.1; 95% confidence interval, 1.2-3.6) and multiple myeloma (OR = 2.2; 95% CI, 1.2-3.9) and were one and a half times as likely to have non-Hodgkin lymphoma (OR = 1.5; 95% CI, 1.0-2.2).
Dr. Kridin and his fellow investigators hypothesized that the risks may be related to some pemphigus treatments.
“Certain immunosuppressive treatments for pemphigus, such as azathioprine, could increase the risk of developing hematologic malignancies,” they wrote. “Controlling for immunosuppressive agents attenuated the association of pemphigus with non-Hodgkin lymphoma and multiple myeloma, hinting that they play a role in the higher prevalence.”
Chronic immune stimulation also may be influencing a higher prevalence of hematologic cancers in pemphigus patients “by randomly introducing pro-oncogenic mutations in rapidly dividing cells,” they said.
Investigators were limited by a lack of data on patients’ immunopathological subtype, clinical features, severity of pemphigus, and precise histological type of leukemia and lymphoma.
Dr. Kridin and his fellow investigators reported no relevant financial disclosures.
ezimmerman@frontlinemedcom.com
SOURCE: Kridin K et al. J Am Acad Dermatol. 2017 Dec 2. doi:10.1016/j.jaad.2017.11.039.
, based on the findings of a retrospective study conducted at the Rambam Health Care Campus, Haifa, Israel.
Although the findings are preliminary, the possible associations should be considered when treating pemphigus patients, the investigators reported in the Journal of the American Academy of Dermatology.
Khalaf Kridin, MD, of the Rambam Health Care Campus department of dermatology and his fellow investigators conducted a cross-sectional, retrospective, controlled study of 11,859 patients gathered from the Clait Health Services computerized database. A total of 1,985 pemphigus patients and 9,874 control patients were included. Patients were 72 years old on average, and most were female (60%) and Jewish (90%).
Dr. Kridin and his colleagues measured the prevalence of acute and chronic leukemia, Hodgkin and non-Hodgkin lymphoma, multiple myeloma, and polycythemia vera.
The pemphigus patients, compared with the control group, had a significantly higher prevalence of chronic leukemia (0.9% vs 0.4% [P = .007]), multiple myeloma (0.8% vs 0.4% [P = .009]), and non-Hodgkin lymphoma (1.8% vs 1.2% [P = .040]).
In a sensitivity analysis, patients with pemphigus were twice as likely to have chronic leukemia (odds ratio = 2.1; 95% confidence interval, 1.2-3.6) and multiple myeloma (OR = 2.2; 95% CI, 1.2-3.9) and were one and a half times as likely to have non-Hodgkin lymphoma (OR = 1.5; 95% CI, 1.0-2.2).
Dr. Kridin and his fellow investigators hypothesized that the risks may be related to some pemphigus treatments.
“Certain immunosuppressive treatments for pemphigus, such as azathioprine, could increase the risk of developing hematologic malignancies,” they wrote. “Controlling for immunosuppressive agents attenuated the association of pemphigus with non-Hodgkin lymphoma and multiple myeloma, hinting that they play a role in the higher prevalence.”
Chronic immune stimulation also may be influencing a higher prevalence of hematologic cancers in pemphigus patients “by randomly introducing pro-oncogenic mutations in rapidly dividing cells,” they said.
Investigators were limited by a lack of data on patients’ immunopathological subtype, clinical features, severity of pemphigus, and precise histological type of leukemia and lymphoma.
Dr. Kridin and his fellow investigators reported no relevant financial disclosures.
ezimmerman@frontlinemedcom.com
SOURCE: Kridin K et al. J Am Acad Dermatol. 2017 Dec 2. doi:10.1016/j.jaad.2017.11.039.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: There may be an association between pemphigus and developing certain cancers.
Major finding: Prevalence of chronic leukemia, multiple myeloma, and non-Hodgkin lymphoma was 0.9%, 0.8%, and 1.8%, respectively. The prevalence in controls was 0.4%, 0.4%, and 1.2%, respectively.
Study details: Cross-sectional study of 1,985 pemphigus patients and 9,874 control subjects gathered from the Clait Health Services computerized database.
Disclosures: The investigators reported no relevant disclosures.
Source: Kridin K et al. J Am Acad Dermatol. 2017 Dec 2. doi: 10.1016/j.jaad.2017.11.039.
Research explains why cisplatin causes hearing loss
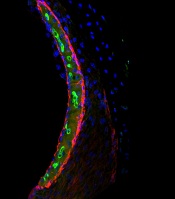
Researchers have gained new insight into hearing loss caused by cisplatin.
By measuring and mapping cisplatin retention in mouse and human inner ear tissues, the researchers found that cisplatin builds up in the inner ear and can remain there for years.
The team also found that a region in the inner ear called the stria vascularis could be targeted to prevent hearing loss resulting from cisplatin.
Lisa L. Cunningham, PhD, of the National Institute on Deafness and other Communications Disorders (NIDCD) in Bethesda, Maryland, and her colleagues reported these findings in Nature Communications.
The researchers noted that cisplatin can cause permanent hearing loss in 40% to 80% of treated patients. The team’s new findings help explain why.
The researchers found that, in most areas of the body, cisplatin is eliminated within days or weeks of treatment, but, in the inner ear, the drug remains much longer.
The team developed a mouse model that represents cisplatin-induced hearing loss seen in human patients.
By looking at inner ear tissue of mice after the first, second, and third cisplatin treatment, the researchers saw that cisplatin remained in the mouse inner ear much longer than in most other body tissues, and the drug builds up with each successive treatment.
The team also studied inner ear tissue donated by deceased adults who had been treated with cisplatin and found the drug is retained in the inner ear months or years after treatment.
When the researchers examined inner ear tissue from a child, they found cisplatin buildup that was even higher than that seen in adults.
Taken together, these results suggest the inner ear readily takes up cisplatin but has limited ability to remove the drug.
In mice and human tissues, the researchers saw the highest buildup of cisplatin in a part of the inner ear called the stria vascularis, which helps maintain the positive electrical charge in inner ear fluid that certain cells need to detect sound.
The team found the accumulation of cisplatin in the stria vascularis contributed to cisplatin-related hearing loss.
“Our findings suggest that if we can prevent cisplatin from entering the stria vascularis in the inner ear during treatment, we may be able to protect cancer patients from developing cisplatin-induced hearing loss,” Dr Cunningham said. ![]()
Researchers have gained new insight into hearing loss caused by cisplatin.
By measuring and mapping cisplatin retention in mouse and human inner ear tissues, the researchers found that cisplatin builds up in the inner ear and can remain there for years.
The team also found that a region in the inner ear called the stria vascularis could be targeted to prevent hearing loss resulting from cisplatin.
Lisa L. Cunningham, PhD, of the National Institute on Deafness and other Communications Disorders (NIDCD) in Bethesda, Maryland, and her colleagues reported these findings in Nature Communications.
The researchers noted that cisplatin can cause permanent hearing loss in 40% to 80% of treated patients. The team’s new findings help explain why.
The researchers found that, in most areas of the body, cisplatin is eliminated within days or weeks of treatment, but, in the inner ear, the drug remains much longer.
The team developed a mouse model that represents cisplatin-induced hearing loss seen in human patients.
By looking at inner ear tissue of mice after the first, second, and third cisplatin treatment, the researchers saw that cisplatin remained in the mouse inner ear much longer than in most other body tissues, and the drug builds up with each successive treatment.
The team also studied inner ear tissue donated by deceased adults who had been treated with cisplatin and found the drug is retained in the inner ear months or years after treatment.
When the researchers examined inner ear tissue from a child, they found cisplatin buildup that was even higher than that seen in adults.
Taken together, these results suggest the inner ear readily takes up cisplatin but has limited ability to remove the drug.
In mice and human tissues, the researchers saw the highest buildup of cisplatin in a part of the inner ear called the stria vascularis, which helps maintain the positive electrical charge in inner ear fluid that certain cells need to detect sound.
The team found the accumulation of cisplatin in the stria vascularis contributed to cisplatin-related hearing loss.
“Our findings suggest that if we can prevent cisplatin from entering the stria vascularis in the inner ear during treatment, we may be able to protect cancer patients from developing cisplatin-induced hearing loss,” Dr Cunningham said. ![]()
Researchers have gained new insight into hearing loss caused by cisplatin.
By measuring and mapping cisplatin retention in mouse and human inner ear tissues, the researchers found that cisplatin builds up in the inner ear and can remain there for years.
The team also found that a region in the inner ear called the stria vascularis could be targeted to prevent hearing loss resulting from cisplatin.
Lisa L. Cunningham, PhD, of the National Institute on Deafness and other Communications Disorders (NIDCD) in Bethesda, Maryland, and her colleagues reported these findings in Nature Communications.
The researchers noted that cisplatin can cause permanent hearing loss in 40% to 80% of treated patients. The team’s new findings help explain why.
The researchers found that, in most areas of the body, cisplatin is eliminated within days or weeks of treatment, but, in the inner ear, the drug remains much longer.
The team developed a mouse model that represents cisplatin-induced hearing loss seen in human patients.
By looking at inner ear tissue of mice after the first, second, and third cisplatin treatment, the researchers saw that cisplatin remained in the mouse inner ear much longer than in most other body tissues, and the drug builds up with each successive treatment.
The team also studied inner ear tissue donated by deceased adults who had been treated with cisplatin and found the drug is retained in the inner ear months or years after treatment.
When the researchers examined inner ear tissue from a child, they found cisplatin buildup that was even higher than that seen in adults.
Taken together, these results suggest the inner ear readily takes up cisplatin but has limited ability to remove the drug.
In mice and human tissues, the researchers saw the highest buildup of cisplatin in a part of the inner ear called the stria vascularis, which helps maintain the positive electrical charge in inner ear fluid that certain cells need to detect sound.
The team found the accumulation of cisplatin in the stria vascularis contributed to cisplatin-related hearing loss.
“Our findings suggest that if we can prevent cisplatin from entering the stria vascularis in the inner ear during treatment, we may be able to protect cancer patients from developing cisplatin-induced hearing loss,” Dr Cunningham said. ![]()
Survival improvements lag for young Hispanic patients with myeloma
ATLANTA –
Among U.S. adults diagnosed with multiple myeloma by age 40 years, 5-year and 10-year survival improved significantly (P less than .0001) for non-Hispanic blacks and whites, but not for Hispanics (5-year survival, P = .08; 10-year survival, P = .13), Abdel-Ghani Azzouqa, MD, and colleagues reported in a poster at the annual meeting of the American Society of Hematology.
Other population-based studies have uncovered racial and ethnic disparities in myeloma outcomes but had not honed in on the experience of young adult patients, who make up a growing proportion of diagnosed patients, said Dr. Azzouqa.
He and his associates analyzed Surveillance Epidemiology and End Results (SEER) data on patients diagnosed between ages 18 and 40 years with histologically confirmed multiple myeloma. The dataset spanned 1973-2014 and included 1,460 patients, of whom about 60% were male. Median age at diagnosis was 37 years; 47% of patients were non-Hispanic white, 28% were non-Hispanic black, 18% were Hispanic, 5.5% were Asian, and about 1% were of other ethnicities.
For young Hispanic patients with myeloma, 5-year survival improved from 39% before 1996, when stem cell transplants and novel therapies became available, to 56% from 2002 onward. This change was not statistically significant (P = .08), and 10-year survival rates also did not change significantly (from 21% to 33%; P = .13).
Five-year and 10-year survival did improve significantly for both genders (P = .0001) and among non-Hispanic blacks (P = .0001) and non-Hispanic whites (P = .0001).
Racial/ethnic subgroups did not differ significantly by median age at diagnosis, gender distribution, or listed cause of death, Dr. Azzouqa noted. Thus, reasons for the difference in survival for Hispanic patients remain unclear. Perhaps they reflect differences in disease biology, treatment response, or access or use of effective novel therapies, he said.
The researchers had no external funding sources. Dr. Azzouqa had no conflicts of interest. Lead author Dr. Sikander Ailawadhi disclosed ties to funding Pharmacyclics, Amgen, Novartis, and Takeda.
SOURCE: Ailawadhi S et al. ASH Abstract 2149
ATLANTA –
Among U.S. adults diagnosed with multiple myeloma by age 40 years, 5-year and 10-year survival improved significantly (P less than .0001) for non-Hispanic blacks and whites, but not for Hispanics (5-year survival, P = .08; 10-year survival, P = .13), Abdel-Ghani Azzouqa, MD, and colleagues reported in a poster at the annual meeting of the American Society of Hematology.
Other population-based studies have uncovered racial and ethnic disparities in myeloma outcomes but had not honed in on the experience of young adult patients, who make up a growing proportion of diagnosed patients, said Dr. Azzouqa.
He and his associates analyzed Surveillance Epidemiology and End Results (SEER) data on patients diagnosed between ages 18 and 40 years with histologically confirmed multiple myeloma. The dataset spanned 1973-2014 and included 1,460 patients, of whom about 60% were male. Median age at diagnosis was 37 years; 47% of patients were non-Hispanic white, 28% were non-Hispanic black, 18% were Hispanic, 5.5% were Asian, and about 1% were of other ethnicities.
For young Hispanic patients with myeloma, 5-year survival improved from 39% before 1996, when stem cell transplants and novel therapies became available, to 56% from 2002 onward. This change was not statistically significant (P = .08), and 10-year survival rates also did not change significantly (from 21% to 33%; P = .13).
Five-year and 10-year survival did improve significantly for both genders (P = .0001) and among non-Hispanic blacks (P = .0001) and non-Hispanic whites (P = .0001).
Racial/ethnic subgroups did not differ significantly by median age at diagnosis, gender distribution, or listed cause of death, Dr. Azzouqa noted. Thus, reasons for the difference in survival for Hispanic patients remain unclear. Perhaps they reflect differences in disease biology, treatment response, or access or use of effective novel therapies, he said.
The researchers had no external funding sources. Dr. Azzouqa had no conflicts of interest. Lead author Dr. Sikander Ailawadhi disclosed ties to funding Pharmacyclics, Amgen, Novartis, and Takeda.
SOURCE: Ailawadhi S et al. ASH Abstract 2149
ATLANTA –
Among U.S. adults diagnosed with multiple myeloma by age 40 years, 5-year and 10-year survival improved significantly (P less than .0001) for non-Hispanic blacks and whites, but not for Hispanics (5-year survival, P = .08; 10-year survival, P = .13), Abdel-Ghani Azzouqa, MD, and colleagues reported in a poster at the annual meeting of the American Society of Hematology.
Other population-based studies have uncovered racial and ethnic disparities in myeloma outcomes but had not honed in on the experience of young adult patients, who make up a growing proportion of diagnosed patients, said Dr. Azzouqa.
He and his associates analyzed Surveillance Epidemiology and End Results (SEER) data on patients diagnosed between ages 18 and 40 years with histologically confirmed multiple myeloma. The dataset spanned 1973-2014 and included 1,460 patients, of whom about 60% were male. Median age at diagnosis was 37 years; 47% of patients were non-Hispanic white, 28% were non-Hispanic black, 18% were Hispanic, 5.5% were Asian, and about 1% were of other ethnicities.
For young Hispanic patients with myeloma, 5-year survival improved from 39% before 1996, when stem cell transplants and novel therapies became available, to 56% from 2002 onward. This change was not statistically significant (P = .08), and 10-year survival rates also did not change significantly (from 21% to 33%; P = .13).
Five-year and 10-year survival did improve significantly for both genders (P = .0001) and among non-Hispanic blacks (P = .0001) and non-Hispanic whites (P = .0001).
Racial/ethnic subgroups did not differ significantly by median age at diagnosis, gender distribution, or listed cause of death, Dr. Azzouqa noted. Thus, reasons for the difference in survival for Hispanic patients remain unclear. Perhaps they reflect differences in disease biology, treatment response, or access or use of effective novel therapies, he said.
The researchers had no external funding sources. Dr. Azzouqa had no conflicts of interest. Lead author Dr. Sikander Ailawadhi disclosed ties to funding Pharmacyclics, Amgen, Novartis, and Takeda.
SOURCE: Ailawadhi S et al. ASH Abstract 2149
REPORTING FROM ASH 2017
Key clinical point: Recent improvements in multiple myeloma survival have left young Hispanics behind.
Major finding: Five-year and 10-year survival have improved significantly among young blacks and non-Hispanic whites with multiple myeloma (P less than .0001 for all comparisons) but not Hispanics (5-year survival P = .08; 10-year survival P = .13).
Data source: Surveillance Epidemiology and End Results (SEER) data for 1,460 adults up to 40 years old when diagnosed with multiple myeloma.
Disclosures: The researchers had no external funding sources. Dr. Azzouqa had no conflicts of interest. Lead author Dr. Sikander Ailawadhi disclosed funding from Pharmacyclics, Amgen, Novartis, and Takeda.
Source: Ailawadhi S et al. ASH Abstract 2149.
Drug receives fast track, orphan designations for PTCL

The US Food and Drug Administration (FDA) has granted orphan drug and fast track designations to tenalisib (RP6530) for the treatment of peripheral T-cell lymphoma (PTCL).
Tenalisib is a dual PI3K delta/gamma inhibitor being developed by Rhizen Pharmaceuticals.
Research has shown that tenalisib inhibits the growth of immortalized cancerous cell lines and primary leukemia/lymphoma cells.
In preclinical studies, tenalisib reprogrammed macrophages from an immunosuppressive M2-like phenotype (pro-tumor) to an inflammatory M1-like state (anti-tumor).
Researchers are currently conducting a phase 1 study of tenalisib in patients with relapsed/refractory PTCL. Results from this study were presented at the 2017 ASH Annual Meeting (abstract 2791*).
The presentation included data on 50 patients—24 with PTCL and 26 with cutaneous T-cell lymphoma (CTCL).
For the PTCL patients, the median age was 63 (range, 40-89), and 67% were male. The median number of prior therapies was 3 (range, 1-7). All patients had an ECOG status of 0 (n=14) or 1 (n=10). More patients had relapsed disease (n=17, 58%) than refractory disease (n=10, 42%).
For the CTCL patients, the median age was 67 (range, 37-84), and 46% were male. The median number of prior therapies was 5.5 (range, 2-15). All patients had an ECOG status of 0 (n=23) or 1 (n=3). More patients had refractory disease (n=15, 58%) than relapsed disease (n=11, 42%).
In the dose-escalation portion of the study, patients received tenalisib at 200 mg twice daily (BID), 400 mg BID, 800 mg BID fasting, or 800 mg BID fed. The maximum tolerated dose was 800 mg BID fasting, so this dose is being used in the expansion cohort.
Twelve PTCL patients were evaluable for efficacy. The overall response rate in these patients was 58% (7/12), with a 25% complete response rate (3/12).
Sixteen CTCL patients were evaluable for efficacy. The overall response rate was 56% (9/16). All responders had partial responses.
In both PTCL and CTCL patients, treatment-related grade 3 or higher adverse events (AEs) included transaminitis (22%), rash (6%), neutropenia (6%), hypophosphatemia (2%), increased international normalized ratio (2%), diplopia secondary to neuropathy (2%), and sepsis (2%).
Treatment-related serious AEs included sepsis, increased international normalized ratio, diplopia secondary to neuropathy, and pyrexia. Five patients discontinued treatment due to AEs.
About orphan and fast track designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of new drug applications for medicines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss all aspects of development to support a drug’s approval, and also provides the opportunity to submit sections of a new drug application on a rolling basis as data become available. ![]()
*Data in the abstract differ from the presentation.
The US Food and Drug Administration (FDA) has granted orphan drug and fast track designations to tenalisib (RP6530) for the treatment of peripheral T-cell lymphoma (PTCL).
Tenalisib is a dual PI3K delta/gamma inhibitor being developed by Rhizen Pharmaceuticals.
Research has shown that tenalisib inhibits the growth of immortalized cancerous cell lines and primary leukemia/lymphoma cells.
In preclinical studies, tenalisib reprogrammed macrophages from an immunosuppressive M2-like phenotype (pro-tumor) to an inflammatory M1-like state (anti-tumor).
Researchers are currently conducting a phase 1 study of tenalisib in patients with relapsed/refractory PTCL. Results from this study were presented at the 2017 ASH Annual Meeting (abstract 2791*).
The presentation included data on 50 patients—24 with PTCL and 26 with cutaneous T-cell lymphoma (CTCL).
For the PTCL patients, the median age was 63 (range, 40-89), and 67% were male. The median number of prior therapies was 3 (range, 1-7). All patients had an ECOG status of 0 (n=14) or 1 (n=10). More patients had relapsed disease (n=17, 58%) than refractory disease (n=10, 42%).
For the CTCL patients, the median age was 67 (range, 37-84), and 46% were male. The median number of prior therapies was 5.5 (range, 2-15). All patients had an ECOG status of 0 (n=23) or 1 (n=3). More patients had refractory disease (n=15, 58%) than relapsed disease (n=11, 42%).
In the dose-escalation portion of the study, patients received tenalisib at 200 mg twice daily (BID), 400 mg BID, 800 mg BID fasting, or 800 mg BID fed. The maximum tolerated dose was 800 mg BID fasting, so this dose is being used in the expansion cohort.
Twelve PTCL patients were evaluable for efficacy. The overall response rate in these patients was 58% (7/12), with a 25% complete response rate (3/12).
Sixteen CTCL patients were evaluable for efficacy. The overall response rate was 56% (9/16). All responders had partial responses.
In both PTCL and CTCL patients, treatment-related grade 3 or higher adverse events (AEs) included transaminitis (22%), rash (6%), neutropenia (6%), hypophosphatemia (2%), increased international normalized ratio (2%), diplopia secondary to neuropathy (2%), and sepsis (2%).
Treatment-related serious AEs included sepsis, increased international normalized ratio, diplopia secondary to neuropathy, and pyrexia. Five patients discontinued treatment due to AEs.
About orphan and fast track designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of new drug applications for medicines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss all aspects of development to support a drug’s approval, and also provides the opportunity to submit sections of a new drug application on a rolling basis as data become available. ![]()
*Data in the abstract differ from the presentation.
The US Food and Drug Administration (FDA) has granted orphan drug and fast track designations to tenalisib (RP6530) for the treatment of peripheral T-cell lymphoma (PTCL).
Tenalisib is a dual PI3K delta/gamma inhibitor being developed by Rhizen Pharmaceuticals.
Research has shown that tenalisib inhibits the growth of immortalized cancerous cell lines and primary leukemia/lymphoma cells.
In preclinical studies, tenalisib reprogrammed macrophages from an immunosuppressive M2-like phenotype (pro-tumor) to an inflammatory M1-like state (anti-tumor).
Researchers are currently conducting a phase 1 study of tenalisib in patients with relapsed/refractory PTCL. Results from this study were presented at the 2017 ASH Annual Meeting (abstract 2791*).
The presentation included data on 50 patients—24 with PTCL and 26 with cutaneous T-cell lymphoma (CTCL).
For the PTCL patients, the median age was 63 (range, 40-89), and 67% were male. The median number of prior therapies was 3 (range, 1-7). All patients had an ECOG status of 0 (n=14) or 1 (n=10). More patients had relapsed disease (n=17, 58%) than refractory disease (n=10, 42%).
For the CTCL patients, the median age was 67 (range, 37-84), and 46% were male. The median number of prior therapies was 5.5 (range, 2-15). All patients had an ECOG status of 0 (n=23) or 1 (n=3). More patients had refractory disease (n=15, 58%) than relapsed disease (n=11, 42%).
In the dose-escalation portion of the study, patients received tenalisib at 200 mg twice daily (BID), 400 mg BID, 800 mg BID fasting, or 800 mg BID fed. The maximum tolerated dose was 800 mg BID fasting, so this dose is being used in the expansion cohort.
Twelve PTCL patients were evaluable for efficacy. The overall response rate in these patients was 58% (7/12), with a 25% complete response rate (3/12).
Sixteen CTCL patients were evaluable for efficacy. The overall response rate was 56% (9/16). All responders had partial responses.
In both PTCL and CTCL patients, treatment-related grade 3 or higher adverse events (AEs) included transaminitis (22%), rash (6%), neutropenia (6%), hypophosphatemia (2%), increased international normalized ratio (2%), diplopia secondary to neuropathy (2%), and sepsis (2%).
Treatment-related serious AEs included sepsis, increased international normalized ratio, diplopia secondary to neuropathy, and pyrexia. Five patients discontinued treatment due to AEs.
About orphan and fast track designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of new drug applications for medicines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss all aspects of development to support a drug’s approval, and also provides the opportunity to submit sections of a new drug application on a rolling basis as data become available. ![]()
*Data in the abstract differ from the presentation.
Single-agent daratumumab active in smoldering multiple myeloma
ATLANTA – Daratumumab monotherapy led to durable partial responses among intermediate to high-risk patients with smoldering multiple myeloma, according to results from the phase II CENTAURUS trial.
Although less than 5% of patients had complete responses, 27% had at least a very good partial response to long-term therapy (up to 20 treatment cycles lasting 8 weeks each), Craig C. Hofmeister, MD, of the Ohio State University Comprehensive Cancer Center, Columbus, said at the annual meeting of the American Society of Hematology. The coprimary endpoint, median progression-free survival, exceeded 24 months in all dose cohorts, and was the longest when patients were treated longest.
Current guidelines recommend monitoring smoldering multiple myeloma every 3-6 months and treating only after patients progress. However, some experts pursue earlier treatment in the premalignant setting.
In CENTAURUS, 123 adults with smoldering multiple myeloma were randomly assigned to receive daratumumab (16 mg/kg IV) in 8-week cycles according to a long, intermediate, or short/intense schedule. The long schedule consisted of treatment weekly for cycle 1, every other week for cycles 2-3, monthly for cycles 4-7, and once every 8 weeks for up to 13 more cycles. The intermediate schedule consisted of treatment weekly in cycle 1 and every 8 weeks for up to 20 cycles. The short, intense schedule consisted of weekly treatment for 8 weeks (one cycle). Patients were followed for up to 4 years or until they progressed to multiple myeloma based on International Myeloma Working Group guidelines.
Over a median follow-up period of 15.8 months (range, 0 to 24 months), rates of complete response were 2% in the long treatment arm, 5% in the intermediate treatment arm, and 0% in the short treatment arm. Rates of at least very good partial response were 29%, 24%, and 15%, respectively. Overall response rates were 56%, 54%, and 38%, respectively. Median PFS was not reached in any arm, exceeding 24 months.
Treatment was generally well tolerated, said Dr. Hofmeister. The most common treatment-related adverse effects were fatigue, cough, upper respiratory tract infection, headache, and insomnia. Hypertension and hyperglycemia were the most common grade 3-4 treatment-emergent adverse events, affecting up to 5% of patients per arm. Fewer than 10% of patients in any arm developed treatment-emergent hematologic adverse events, and fewer than 5% developed grade 3-4 pneumonia or sepsis. There were three cases of a second primary malignancy, including one case of breast cancer and two cases of melanoma.
Rates of infusion-related reactions did not correlate with treatment duration. Grade 3-4 infusion-related reactions affected 0% to 3% of patients per arm. The sole death in this trial resulted from disease progression in a patient from the short treatment arm. “Taken together, efficacy and safety data support long dosing compared to intermediate and short dosing,” Dr. Hofmeister said.
The three arms were demographically similar. Patients tended to be white, in their late 50s to 60s, and to have ECOG scores of 0 with at least two risk factors for progression. About 70% had IgG disease and nearly half had less than 20% plasma cells in bone marrow.
Janssen, the maker of daratumumab, sponsored the trial. Dr. Hofmeister disclosed research funding from Janssen and research support, honoraria, and advisory relationships with Adaptive Biotechnologies, Thrasos, Celgene, Karyopharm, Takeda, and other pharmaceutical companies.
SOURCE: Hofmeister C et al, ASH 2017, Abstract 510.
ATLANTA – Daratumumab monotherapy led to durable partial responses among intermediate to high-risk patients with smoldering multiple myeloma, according to results from the phase II CENTAURUS trial.
Although less than 5% of patients had complete responses, 27% had at least a very good partial response to long-term therapy (up to 20 treatment cycles lasting 8 weeks each), Craig C. Hofmeister, MD, of the Ohio State University Comprehensive Cancer Center, Columbus, said at the annual meeting of the American Society of Hematology. The coprimary endpoint, median progression-free survival, exceeded 24 months in all dose cohorts, and was the longest when patients were treated longest.
Current guidelines recommend monitoring smoldering multiple myeloma every 3-6 months and treating only after patients progress. However, some experts pursue earlier treatment in the premalignant setting.
In CENTAURUS, 123 adults with smoldering multiple myeloma were randomly assigned to receive daratumumab (16 mg/kg IV) in 8-week cycles according to a long, intermediate, or short/intense schedule. The long schedule consisted of treatment weekly for cycle 1, every other week for cycles 2-3, monthly for cycles 4-7, and once every 8 weeks for up to 13 more cycles. The intermediate schedule consisted of treatment weekly in cycle 1 and every 8 weeks for up to 20 cycles. The short, intense schedule consisted of weekly treatment for 8 weeks (one cycle). Patients were followed for up to 4 years or until they progressed to multiple myeloma based on International Myeloma Working Group guidelines.
Over a median follow-up period of 15.8 months (range, 0 to 24 months), rates of complete response were 2% in the long treatment arm, 5% in the intermediate treatment arm, and 0% in the short treatment arm. Rates of at least very good partial response were 29%, 24%, and 15%, respectively. Overall response rates were 56%, 54%, and 38%, respectively. Median PFS was not reached in any arm, exceeding 24 months.
Treatment was generally well tolerated, said Dr. Hofmeister. The most common treatment-related adverse effects were fatigue, cough, upper respiratory tract infection, headache, and insomnia. Hypertension and hyperglycemia were the most common grade 3-4 treatment-emergent adverse events, affecting up to 5% of patients per arm. Fewer than 10% of patients in any arm developed treatment-emergent hematologic adverse events, and fewer than 5% developed grade 3-4 pneumonia or sepsis. There were three cases of a second primary malignancy, including one case of breast cancer and two cases of melanoma.
Rates of infusion-related reactions did not correlate with treatment duration. Grade 3-4 infusion-related reactions affected 0% to 3% of patients per arm. The sole death in this trial resulted from disease progression in a patient from the short treatment arm. “Taken together, efficacy and safety data support long dosing compared to intermediate and short dosing,” Dr. Hofmeister said.
The three arms were demographically similar. Patients tended to be white, in their late 50s to 60s, and to have ECOG scores of 0 with at least two risk factors for progression. About 70% had IgG disease and nearly half had less than 20% plasma cells in bone marrow.
Janssen, the maker of daratumumab, sponsored the trial. Dr. Hofmeister disclosed research funding from Janssen and research support, honoraria, and advisory relationships with Adaptive Biotechnologies, Thrasos, Celgene, Karyopharm, Takeda, and other pharmaceutical companies.
SOURCE: Hofmeister C et al, ASH 2017, Abstract 510.
ATLANTA – Daratumumab monotherapy led to durable partial responses among intermediate to high-risk patients with smoldering multiple myeloma, according to results from the phase II CENTAURUS trial.
Although less than 5% of patients had complete responses, 27% had at least a very good partial response to long-term therapy (up to 20 treatment cycles lasting 8 weeks each), Craig C. Hofmeister, MD, of the Ohio State University Comprehensive Cancer Center, Columbus, said at the annual meeting of the American Society of Hematology. The coprimary endpoint, median progression-free survival, exceeded 24 months in all dose cohorts, and was the longest when patients were treated longest.
Current guidelines recommend monitoring smoldering multiple myeloma every 3-6 months and treating only after patients progress. However, some experts pursue earlier treatment in the premalignant setting.
In CENTAURUS, 123 adults with smoldering multiple myeloma were randomly assigned to receive daratumumab (16 mg/kg IV) in 8-week cycles according to a long, intermediate, or short/intense schedule. The long schedule consisted of treatment weekly for cycle 1, every other week for cycles 2-3, monthly for cycles 4-7, and once every 8 weeks for up to 13 more cycles. The intermediate schedule consisted of treatment weekly in cycle 1 and every 8 weeks for up to 20 cycles. The short, intense schedule consisted of weekly treatment for 8 weeks (one cycle). Patients were followed for up to 4 years or until they progressed to multiple myeloma based on International Myeloma Working Group guidelines.
Over a median follow-up period of 15.8 months (range, 0 to 24 months), rates of complete response were 2% in the long treatment arm, 5% in the intermediate treatment arm, and 0% in the short treatment arm. Rates of at least very good partial response were 29%, 24%, and 15%, respectively. Overall response rates were 56%, 54%, and 38%, respectively. Median PFS was not reached in any arm, exceeding 24 months.
Treatment was generally well tolerated, said Dr. Hofmeister. The most common treatment-related adverse effects were fatigue, cough, upper respiratory tract infection, headache, and insomnia. Hypertension and hyperglycemia were the most common grade 3-4 treatment-emergent adverse events, affecting up to 5% of patients per arm. Fewer than 10% of patients in any arm developed treatment-emergent hematologic adverse events, and fewer than 5% developed grade 3-4 pneumonia or sepsis. There were three cases of a second primary malignancy, including one case of breast cancer and two cases of melanoma.
Rates of infusion-related reactions did not correlate with treatment duration. Grade 3-4 infusion-related reactions affected 0% to 3% of patients per arm. The sole death in this trial resulted from disease progression in a patient from the short treatment arm. “Taken together, efficacy and safety data support long dosing compared to intermediate and short dosing,” Dr. Hofmeister said.
The three arms were demographically similar. Patients tended to be white, in their late 50s to 60s, and to have ECOG scores of 0 with at least two risk factors for progression. About 70% had IgG disease and nearly half had less than 20% plasma cells in bone marrow.
Janssen, the maker of daratumumab, sponsored the trial. Dr. Hofmeister disclosed research funding from Janssen and research support, honoraria, and advisory relationships with Adaptive Biotechnologies, Thrasos, Celgene, Karyopharm, Takeda, and other pharmaceutical companies.
SOURCE: Hofmeister C et al, ASH 2017, Abstract 510.
REPORTING FROM ASH 2017
Key clinical point: Single-agent daratumumab therapy was active and its safety profile was acceptable in patients with smoldering multiple myeloma.
Major finding: Rates of at least very good partial response were 29%, 24%, and 15% among patients who received long, intermediate, and short/intense treatment schedules, respectively. Median progression-free survival exceeded 24 months in all three arms.
Data source: CENTAURUS, a phase II trial of 123 patients with smoldering multiple myeloma.
Disclosures: Janssen sponsored the trial. Dr. Hofmeister disclosed research funding from Janssen and research support, honoraria, and advisory relationships with Adaptive Biotechnologies, Thrasos, Celgene, Karyopharm, Takeda, and other pharmaceutical companies.
Source: Hofmeister C et al, ASH 2017, Abstract 510.
Study identifies predictors of acquired von Willebrand disease
ATLANTA – Waldenström macroglobulinemia can present as acquired von Willebrand disease (VWD), and when it does, the finding strongly correlates with high serum IgM levels and the presence of CXCR4 mutations, according to the results of a large, single-center retrospective study.
Further, successfully treating Waldenström macroglobulinemia often resolves acquired VWD, and the depth of treatment response predicts the degree of improvement, Jorge J. Castillo, MD, and his associates wrote in a poster presented at the annual meeting of the American Society of Hematology.
Acquired VWD is an uncommon, poorly understood presentation of Waldenström macroglobulinemia. Because affected patients require treatment, better characterizing this subgroup is important, the investigators noted.
At the Bing Center for Waldenström Macroglobulinemia at Dana-Farber Cancer Institute in Boston, the researchers retrospectively studied 320 individuals with newly diagnosed Waldenström macroglobulinemia and used logistic regression analysis to seek predictors of acquired VWD, which they evaluated by measuring levels of VW factor antigen, VW factor activity, and factor VIII. Levels under 30% were considered VWD and levels between 30% and 50% were considered low-level VWD.
In all, 49 individuals had acquired VWD while 271 patients did not. These two groups were similar in terms of age, sex, hemoglobin level, platelet count, and bone marrow involvement. However, 45% of patients with acquired VWD had serum IgM levels above 6,000 mg/dL versus 6% of patients without acquired VWD (P less than .001), and 47% of patients with acquired VWD had serum IgM levels between 3,000 and 5,999 versus 31% of patients without acquired VWD (P less than .001). Also, 77% of patients with acquired VWD tested positive for CXCR4 mutation versus 37% of patients without acquired VWD (P less than .001).
A significantly higher proportion of patients without acquired VWD had white blood cell concentrations above 6,000/mcL (29% vs. 50%; P = .006). This finding lost statistical significance in the logistic regression model, but all the other variables remained significantly associated. Serum IgM levels above 6,000 mg/dL conferred a 55-fold increase in the odds of having acquired VWD (95% confidence interval, 17-177; P less than .001), and serum IgM levels between 3,000 and 5,999 mg/dL led to an 11-fold increase in these odds (95% CI, 4-34). The presence of CXCR4 mutations was associated with a sixfold increased odds of acquired VWD (95% CI, 2-15). The P value for each of these three associations was at or below .001.
Therapy for Waldenström macroglobulinemia led to statistically significant increases in levels of factor VIII, VW factor antigen, and VW factor activity (P less than .001) and the median of each level improved by at least 35% after treatment. After treatment, 78% of patients with acquired VWD had levels of all three measures above 50% (versus 0% before treatment; P less than .001). Patients with acquired VWD with the best responses to treatment had about a 90% decrease in IgM levels, while those with a partial response had about a two-thirds decrease and patients with stable disease had about a 20% decrease. A linear regression model confirmed that depth of treatment response, based on change in IgM level, correlated with degree of improvement in VWD – that is, the extent of improvement in levels of VW factor antigen, VW factor activity, and factor VIII.
No external funding sources were reported. Dr. Castillo disclosed consulting ties and research funding from Pharmacyclics and Janssen, and research funding from Millenium and Abbvie.
SOURCE: Castillo J, et al. ASH 2017 Abstract 1088.
ATLANTA – Waldenström macroglobulinemia can present as acquired von Willebrand disease (VWD), and when it does, the finding strongly correlates with high serum IgM levels and the presence of CXCR4 mutations, according to the results of a large, single-center retrospective study.
Further, successfully treating Waldenström macroglobulinemia often resolves acquired VWD, and the depth of treatment response predicts the degree of improvement, Jorge J. Castillo, MD, and his associates wrote in a poster presented at the annual meeting of the American Society of Hematology.
Acquired VWD is an uncommon, poorly understood presentation of Waldenström macroglobulinemia. Because affected patients require treatment, better characterizing this subgroup is important, the investigators noted.
At the Bing Center for Waldenström Macroglobulinemia at Dana-Farber Cancer Institute in Boston, the researchers retrospectively studied 320 individuals with newly diagnosed Waldenström macroglobulinemia and used logistic regression analysis to seek predictors of acquired VWD, which they evaluated by measuring levels of VW factor antigen, VW factor activity, and factor VIII. Levels under 30% were considered VWD and levels between 30% and 50% were considered low-level VWD.
In all, 49 individuals had acquired VWD while 271 patients did not. These two groups were similar in terms of age, sex, hemoglobin level, platelet count, and bone marrow involvement. However, 45% of patients with acquired VWD had serum IgM levels above 6,000 mg/dL versus 6% of patients without acquired VWD (P less than .001), and 47% of patients with acquired VWD had serum IgM levels between 3,000 and 5,999 versus 31% of patients without acquired VWD (P less than .001). Also, 77% of patients with acquired VWD tested positive for CXCR4 mutation versus 37% of patients without acquired VWD (P less than .001).
A significantly higher proportion of patients without acquired VWD had white blood cell concentrations above 6,000/mcL (29% vs. 50%; P = .006). This finding lost statistical significance in the logistic regression model, but all the other variables remained significantly associated. Serum IgM levels above 6,000 mg/dL conferred a 55-fold increase in the odds of having acquired VWD (95% confidence interval, 17-177; P less than .001), and serum IgM levels between 3,000 and 5,999 mg/dL led to an 11-fold increase in these odds (95% CI, 4-34). The presence of CXCR4 mutations was associated with a sixfold increased odds of acquired VWD (95% CI, 2-15). The P value for each of these three associations was at or below .001.
Therapy for Waldenström macroglobulinemia led to statistically significant increases in levels of factor VIII, VW factor antigen, and VW factor activity (P less than .001) and the median of each level improved by at least 35% after treatment. After treatment, 78% of patients with acquired VWD had levels of all three measures above 50% (versus 0% before treatment; P less than .001). Patients with acquired VWD with the best responses to treatment had about a 90% decrease in IgM levels, while those with a partial response had about a two-thirds decrease and patients with stable disease had about a 20% decrease. A linear regression model confirmed that depth of treatment response, based on change in IgM level, correlated with degree of improvement in VWD – that is, the extent of improvement in levels of VW factor antigen, VW factor activity, and factor VIII.
No external funding sources were reported. Dr. Castillo disclosed consulting ties and research funding from Pharmacyclics and Janssen, and research funding from Millenium and Abbvie.
SOURCE: Castillo J, et al. ASH 2017 Abstract 1088.
ATLANTA – Waldenström macroglobulinemia can present as acquired von Willebrand disease (VWD), and when it does, the finding strongly correlates with high serum IgM levels and the presence of CXCR4 mutations, according to the results of a large, single-center retrospective study.
Further, successfully treating Waldenström macroglobulinemia often resolves acquired VWD, and the depth of treatment response predicts the degree of improvement, Jorge J. Castillo, MD, and his associates wrote in a poster presented at the annual meeting of the American Society of Hematology.
Acquired VWD is an uncommon, poorly understood presentation of Waldenström macroglobulinemia. Because affected patients require treatment, better characterizing this subgroup is important, the investigators noted.
At the Bing Center for Waldenström Macroglobulinemia at Dana-Farber Cancer Institute in Boston, the researchers retrospectively studied 320 individuals with newly diagnosed Waldenström macroglobulinemia and used logistic regression analysis to seek predictors of acquired VWD, which they evaluated by measuring levels of VW factor antigen, VW factor activity, and factor VIII. Levels under 30% were considered VWD and levels between 30% and 50% were considered low-level VWD.
In all, 49 individuals had acquired VWD while 271 patients did not. These two groups were similar in terms of age, sex, hemoglobin level, platelet count, and bone marrow involvement. However, 45% of patients with acquired VWD had serum IgM levels above 6,000 mg/dL versus 6% of patients without acquired VWD (P less than .001), and 47% of patients with acquired VWD had serum IgM levels between 3,000 and 5,999 versus 31% of patients without acquired VWD (P less than .001). Also, 77% of patients with acquired VWD tested positive for CXCR4 mutation versus 37% of patients without acquired VWD (P less than .001).
A significantly higher proportion of patients without acquired VWD had white blood cell concentrations above 6,000/mcL (29% vs. 50%; P = .006). This finding lost statistical significance in the logistic regression model, but all the other variables remained significantly associated. Serum IgM levels above 6,000 mg/dL conferred a 55-fold increase in the odds of having acquired VWD (95% confidence interval, 17-177; P less than .001), and serum IgM levels between 3,000 and 5,999 mg/dL led to an 11-fold increase in these odds (95% CI, 4-34). The presence of CXCR4 mutations was associated with a sixfold increased odds of acquired VWD (95% CI, 2-15). The P value for each of these three associations was at or below .001.
Therapy for Waldenström macroglobulinemia led to statistically significant increases in levels of factor VIII, VW factor antigen, and VW factor activity (P less than .001) and the median of each level improved by at least 35% after treatment. After treatment, 78% of patients with acquired VWD had levels of all three measures above 50% (versus 0% before treatment; P less than .001). Patients with acquired VWD with the best responses to treatment had about a 90% decrease in IgM levels, while those with a partial response had about a two-thirds decrease and patients with stable disease had about a 20% decrease. A linear regression model confirmed that depth of treatment response, based on change in IgM level, correlated with degree of improvement in VWD – that is, the extent of improvement in levels of VW factor antigen, VW factor activity, and factor VIII.
No external funding sources were reported. Dr. Castillo disclosed consulting ties and research funding from Pharmacyclics and Janssen, and research funding from Millenium and Abbvie.
SOURCE: Castillo J, et al. ASH 2017 Abstract 1088.
AT ASH 2017
Key clinical point: Successfully treating Waldenström macroglobulinemia often resolves acquired von Willebrand disease.
Major finding: Therapy for Waldenström macroglobulinemia led to statistically significant increases in levels of factor VIII, VW factor antigen, and VW factor activity (P less than .001) and the median of each level improved by at least 35% after treatment.
Data source: A single-center retrospective study of 320 patients with newly diagnosed Waldenström macroglobulinemia.
Disclosures: No external funding sources were reported. Dr. Castillo disclosed consultancy and research funding from Pharmacyclics and Janssen. He also disclosed research funding from Millenium and Abbvie.
Source: Castillo J, et al. ASH 2017 Abstract 1088.