User login
Vaccines in the Pipeline For Diseases Affecting Children Worldwide
VAIL, COLO. – Effective vaccines for two of the top causes of mortality and morbidity in children worldwide – malaria and dengue fever – are now in pivotal phase III clinical trials and could reach the market within 3 years.
In addition, promising vaccines for tuberculosis and group B streptococcus are in earlier-stage clinical trials, Dr. Edwin J. Asturias noted at a conference on pediatric infectious diseases, which was sponsored by the Children’s Hospital Colorado in Aurora.
Malaria
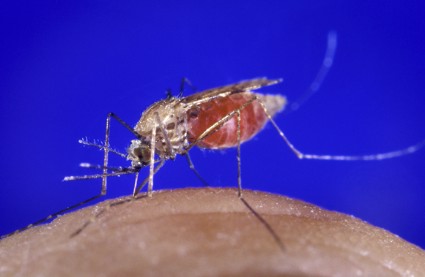
"The malaria vaccine is coming. It is close. It will be the most important vaccine for killer diseases in children. There is a lot of hope for that vaccine," according to Dr. Asturias of the center for global health at the University of Colorado, Denver.

Malaria is endemic in 99 countries. An estimated 216 million new cases and 655,000 deaths resulting from the infection occurred in 2010. More than 90% of the world’s malaria deaths are in Africa, with most of them in children younger than age 5 years.
The malaria vaccine – now in the pivotal phase III MAL059 trial that’s expected to be completed by 2014 – has been in development by GlaxoSmithKline for 2 decades. The vaccine is known as RTS,S. It is designed for children living in endemic areas. The vaccine focuses on the pre-erythrocytic stage of infection, aiming to prevent the malaria parasite from multiplying in the liver and reentering the bloodstream.
In an interim report from the phase III trial, the three-dose series of RTS,S achieved a 56% reduction in clinical malaria among 5- to 17-month-olds (N. Engl. J. Med. 2008;359:2521-32).
"The question everybody has is, is 56% enough, or should we aim for higher efficacy in malaria?" the pediatric infectious disease specialist said.
For him, the answer is clear: "If we can reduce mortality from malaria by 50%, that would be a great success in terms of preventing this important killer around the world," Dr. Asturias said.
But the benefits will extend well beyond mortality reduction to include nationwide favorable impacts upon lost days of productivity and strain on health care systems. "In countries where malaria is endemic, every case of fever gets treated as malaria, and that’s a huge burden in terms of health resource utilization," he added.
It’s also hypothetically possible that herd immunity could amplify the clinical efficacy of RTS,S beyond that 56% figure. It all depends upon how good the vaccine is at quelling parasitemia, an issue that hasn’t been well studied as yet.
Frequent boosting may be necessary for several years after the three-dose series is given. In studies in African infants and children, the protective effect has been sustained for only about 18 months.
Much of the credit for the recent major advances in developing new vaccines for important childhood diseases worldwide belongs to an ongoing UNICEF/WHO initiative and to the Bill and Melinda Gates Foundation, which Dr. Asturias said "has been a game changer." During this decade, the foundation’s declared aim is to utilize existing vaccines and to support development of new ones in order to prevent the deaths of 7.6 million children younger than age 5 years.
Dengue Virus
Sanofi Pasteur’s chimeric flavivirus vaccine is in phase III studies totaling more than 30,000 participants in Latin America and Asia. These pivotal trials will be completed in 2014. The company is sufficiently confident that it has a winner that it has built a $440 million factory to produce the vaccine.
In a phase IIB clinical trial conducted in Thai children, the vaccine – which has received fast-track development status from the Food and Drug Administration – protected against three of the four dengue virus serotypes, which means it should decrease the incidence of dengue hemorrhagic fever, the most feared manifestation of the viral infection.
Other dengue vaccines using alternative immunization strategies are in earlier stages of development.
In countries where the mosquito-borne dengue virus is endemic and reporting is adequate, the incidence of infection is about 5% per year (Vaccine 2002;20:3043-6). Three-quarters of infections are asymptomatic or entail only a few days of mild flulike illness. Among the 25% of infections that are symptomatic, 98%-99% result in dengue fever, also known as "breakbone fever," whereas 1%-2% manifest as dengue hemorrhagic fever, which has a mortality rate of up to 5% depending upon the availability of supportive care. The infection is more severe in school-age youths and in individuals infected for the second time.
Tuberculosis
Twelve new candidate TB vaccines have entered clinical trials. Seven are subunit vaccines, two utilize immunotherapy as an adjunct to chemotherapy, and three are recombinant bacille Calmette-Guérin vaccines designed to replace the current BCG vaccine, which doesn’t protect adults from reactivation or reinfection decades after their first exposure to the pathogen. The two most advanced candidate vaccines are now in phase IIB trials; they are designed to enhance immunity in individuals who received BCG vaccine early in life. Phase II studies are likely to continue to 2020.
"We’re still far away from a good tuberculosis vaccine at this point," according to Dr. Asturias.
The most pressing need now is to define markers for TB infection and reactivation so that researchers can demonstrate vaccine efficacy without having to wait 30 years for pulmonary TB or other clinical disease to develop, he added.
Group B Streptococcus
The push to develop a group B strep vaccine stems from the fact that in developing countries, this infection is a leading cause of sepsis and meningitis in the first 3 months of life. And worldwide, the proportion of deaths in children younger than age 5 years that occur during the neonatal period hasn’t changed in 2 decades. A UNICEF/WHO estimate concluded that of 8.8 million deaths in children younger than age 5 years in 2008, 42% occurred in neonates. A maternal group B strep vaccine is key to reducing that disturbing figure, in Dr. Asturias’s view.
There are at least 10 group B strep serotypes, but most disease is caused by three of them. A Novartis trivalent vaccine showed "very promising" results in a phase I study; a phase III clinical trial for prevention of disease in pregnant women is scheduled to get underway late next year, he said.
Dr. Asturias reported having conducted vaccine research studies sponsored by Sanofi Pasteur and Crucell, serving on data safety monitoring boards for Inviragen, Sanofi Pasteur, and PATH, and acting as a consultant to the Bill and Melinda Gates Foundation.
VAIL, COLO. – Effective vaccines for two of the top causes of mortality and morbidity in children worldwide – malaria and dengue fever – are now in pivotal phase III clinical trials and could reach the market within 3 years.
In addition, promising vaccines for tuberculosis and group B streptococcus are in earlier-stage clinical trials, Dr. Edwin J. Asturias noted at a conference on pediatric infectious diseases, which was sponsored by the Children’s Hospital Colorado in Aurora.
Malaria
"The malaria vaccine is coming. It is close. It will be the most important vaccine for killer diseases in children. There is a lot of hope for that vaccine," according to Dr. Asturias of the center for global health at the University of Colorado, Denver.
Malaria is endemic in 99 countries. An estimated 216 million new cases and 655,000 deaths resulting from the infection occurred in 2010. More than 90% of the world’s malaria deaths are in Africa, with most of them in children younger than age 5 years.
The malaria vaccine – now in the pivotal phase III MAL059 trial that’s expected to be completed by 2014 – has been in development by GlaxoSmithKline for 2 decades. The vaccine is known as RTS,S. It is designed for children living in endemic areas. The vaccine focuses on the pre-erythrocytic stage of infection, aiming to prevent the malaria parasite from multiplying in the liver and reentering the bloodstream.
In an interim report from the phase III trial, the three-dose series of RTS,S achieved a 56% reduction in clinical malaria among 5- to 17-month-olds (N. Engl. J. Med. 2008;359:2521-32).
"The question everybody has is, is 56% enough, or should we aim for higher efficacy in malaria?" the pediatric infectious disease specialist said.
For him, the answer is clear: "If we can reduce mortality from malaria by 50%, that would be a great success in terms of preventing this important killer around the world," Dr. Asturias said.
But the benefits will extend well beyond mortality reduction to include nationwide favorable impacts upon lost days of productivity and strain on health care systems. "In countries where malaria is endemic, every case of fever gets treated as malaria, and that’s a huge burden in terms of health resource utilization," he added.
It’s also hypothetically possible that herd immunity could amplify the clinical efficacy of RTS,S beyond that 56% figure. It all depends upon how good the vaccine is at quelling parasitemia, an issue that hasn’t been well studied as yet.
Frequent boosting may be necessary for several years after the three-dose series is given. In studies in African infants and children, the protective effect has been sustained for only about 18 months.
Much of the credit for the recent major advances in developing new vaccines for important childhood diseases worldwide belongs to an ongoing UNICEF/WHO initiative and to the Bill and Melinda Gates Foundation, which Dr. Asturias said "has been a game changer." During this decade, the foundation’s declared aim is to utilize existing vaccines and to support development of new ones in order to prevent the deaths of 7.6 million children younger than age 5 years.
Dengue Virus
Sanofi Pasteur’s chimeric flavivirus vaccine is in phase III studies totaling more than 30,000 participants in Latin America and Asia. These pivotal trials will be completed in 2014. The company is sufficiently confident that it has a winner that it has built a $440 million factory to produce the vaccine.
In a phase IIB clinical trial conducted in Thai children, the vaccine – which has received fast-track development status from the Food and Drug Administration – protected against three of the four dengue virus serotypes, which means it should decrease the incidence of dengue hemorrhagic fever, the most feared manifestation of the viral infection.
Other dengue vaccines using alternative immunization strategies are in earlier stages of development.
In countries where the mosquito-borne dengue virus is endemic and reporting is adequate, the incidence of infection is about 5% per year (Vaccine 2002;20:3043-6). Three-quarters of infections are asymptomatic or entail only a few days of mild flulike illness. Among the 25% of infections that are symptomatic, 98%-99% result in dengue fever, also known as "breakbone fever," whereas 1%-2% manifest as dengue hemorrhagic fever, which has a mortality rate of up to 5% depending upon the availability of supportive care. The infection is more severe in school-age youths and in individuals infected for the second time.
Tuberculosis
Twelve new candidate TB vaccines have entered clinical trials. Seven are subunit vaccines, two utilize immunotherapy as an adjunct to chemotherapy, and three are recombinant bacille Calmette-Guérin vaccines designed to replace the current BCG vaccine, which doesn’t protect adults from reactivation or reinfection decades after their first exposure to the pathogen. The two most advanced candidate vaccines are now in phase IIB trials; they are designed to enhance immunity in individuals who received BCG vaccine early in life. Phase II studies are likely to continue to 2020.
"We’re still far away from a good tuberculosis vaccine at this point," according to Dr. Asturias.
The most pressing need now is to define markers for TB infection and reactivation so that researchers can demonstrate vaccine efficacy without having to wait 30 years for pulmonary TB or other clinical disease to develop, he added.
Group B Streptococcus
The push to develop a group B strep vaccine stems from the fact that in developing countries, this infection is a leading cause of sepsis and meningitis in the first 3 months of life. And worldwide, the proportion of deaths in children younger than age 5 years that occur during the neonatal period hasn’t changed in 2 decades. A UNICEF/WHO estimate concluded that of 8.8 million deaths in children younger than age 5 years in 2008, 42% occurred in neonates. A maternal group B strep vaccine is key to reducing that disturbing figure, in Dr. Asturias’s view.
There are at least 10 group B strep serotypes, but most disease is caused by three of them. A Novartis trivalent vaccine showed "very promising" results in a phase I study; a phase III clinical trial for prevention of disease in pregnant women is scheduled to get underway late next year, he said.
Dr. Asturias reported having conducted vaccine research studies sponsored by Sanofi Pasteur and Crucell, serving on data safety monitoring boards for Inviragen, Sanofi Pasteur, and PATH, and acting as a consultant to the Bill and Melinda Gates Foundation.
VAIL, COLO. – Effective vaccines for two of the top causes of mortality and morbidity in children worldwide – malaria and dengue fever – are now in pivotal phase III clinical trials and could reach the market within 3 years.
In addition, promising vaccines for tuberculosis and group B streptococcus are in earlier-stage clinical trials, Dr. Edwin J. Asturias noted at a conference on pediatric infectious diseases, which was sponsored by the Children’s Hospital Colorado in Aurora.
Malaria
"The malaria vaccine is coming. It is close. It will be the most important vaccine for killer diseases in children. There is a lot of hope for that vaccine," according to Dr. Asturias of the center for global health at the University of Colorado, Denver.
Malaria is endemic in 99 countries. An estimated 216 million new cases and 655,000 deaths resulting from the infection occurred in 2010. More than 90% of the world’s malaria deaths are in Africa, with most of them in children younger than age 5 years.
The malaria vaccine – now in the pivotal phase III MAL059 trial that’s expected to be completed by 2014 – has been in development by GlaxoSmithKline for 2 decades. The vaccine is known as RTS,S. It is designed for children living in endemic areas. The vaccine focuses on the pre-erythrocytic stage of infection, aiming to prevent the malaria parasite from multiplying in the liver and reentering the bloodstream.
In an interim report from the phase III trial, the three-dose series of RTS,S achieved a 56% reduction in clinical malaria among 5- to 17-month-olds (N. Engl. J. Med. 2008;359:2521-32).
"The question everybody has is, is 56% enough, or should we aim for higher efficacy in malaria?" the pediatric infectious disease specialist said.
For him, the answer is clear: "If we can reduce mortality from malaria by 50%, that would be a great success in terms of preventing this important killer around the world," Dr. Asturias said.
But the benefits will extend well beyond mortality reduction to include nationwide favorable impacts upon lost days of productivity and strain on health care systems. "In countries where malaria is endemic, every case of fever gets treated as malaria, and that’s a huge burden in terms of health resource utilization," he added.
It’s also hypothetically possible that herd immunity could amplify the clinical efficacy of RTS,S beyond that 56% figure. It all depends upon how good the vaccine is at quelling parasitemia, an issue that hasn’t been well studied as yet.
Frequent boosting may be necessary for several years after the three-dose series is given. In studies in African infants and children, the protective effect has been sustained for only about 18 months.
Much of the credit for the recent major advances in developing new vaccines for important childhood diseases worldwide belongs to an ongoing UNICEF/WHO initiative and to the Bill and Melinda Gates Foundation, which Dr. Asturias said "has been a game changer." During this decade, the foundation’s declared aim is to utilize existing vaccines and to support development of new ones in order to prevent the deaths of 7.6 million children younger than age 5 years.
Dengue Virus
Sanofi Pasteur’s chimeric flavivirus vaccine is in phase III studies totaling more than 30,000 participants in Latin America and Asia. These pivotal trials will be completed in 2014. The company is sufficiently confident that it has a winner that it has built a $440 million factory to produce the vaccine.
In a phase IIB clinical trial conducted in Thai children, the vaccine – which has received fast-track development status from the Food and Drug Administration – protected against three of the four dengue virus serotypes, which means it should decrease the incidence of dengue hemorrhagic fever, the most feared manifestation of the viral infection.
Other dengue vaccines using alternative immunization strategies are in earlier stages of development.
In countries where the mosquito-borne dengue virus is endemic and reporting is adequate, the incidence of infection is about 5% per year (Vaccine 2002;20:3043-6). Three-quarters of infections are asymptomatic or entail only a few days of mild flulike illness. Among the 25% of infections that are symptomatic, 98%-99% result in dengue fever, also known as "breakbone fever," whereas 1%-2% manifest as dengue hemorrhagic fever, which has a mortality rate of up to 5% depending upon the availability of supportive care. The infection is more severe in school-age youths and in individuals infected for the second time.
Tuberculosis
Twelve new candidate TB vaccines have entered clinical trials. Seven are subunit vaccines, two utilize immunotherapy as an adjunct to chemotherapy, and three are recombinant bacille Calmette-Guérin vaccines designed to replace the current BCG vaccine, which doesn’t protect adults from reactivation or reinfection decades after their first exposure to the pathogen. The two most advanced candidate vaccines are now in phase IIB trials; they are designed to enhance immunity in individuals who received BCG vaccine early in life. Phase II studies are likely to continue to 2020.
"We’re still far away from a good tuberculosis vaccine at this point," according to Dr. Asturias.
The most pressing need now is to define markers for TB infection and reactivation so that researchers can demonstrate vaccine efficacy without having to wait 30 years for pulmonary TB or other clinical disease to develop, he added.
Group B Streptococcus
The push to develop a group B strep vaccine stems from the fact that in developing countries, this infection is a leading cause of sepsis and meningitis in the first 3 months of life. And worldwide, the proportion of deaths in children younger than age 5 years that occur during the neonatal period hasn’t changed in 2 decades. A UNICEF/WHO estimate concluded that of 8.8 million deaths in children younger than age 5 years in 2008, 42% occurred in neonates. A maternal group B strep vaccine is key to reducing that disturbing figure, in Dr. Asturias’s view.
There are at least 10 group B strep serotypes, but most disease is caused by three of them. A Novartis trivalent vaccine showed "very promising" results in a phase I study; a phase III clinical trial for prevention of disease in pregnant women is scheduled to get underway late next year, he said.
Dr. Asturias reported having conducted vaccine research studies sponsored by Sanofi Pasteur and Crucell, serving on data safety monitoring boards for Inviragen, Sanofi Pasteur, and PATH, and acting as a consultant to the Bill and Melinda Gates Foundation.
EXPERT ANALYSIS FROM A CONFERENCE ON PEDIATRIC INFECTIOUS DISEASES
Breast Cancer Chemoprevention: Hit It Harder
ESTES PARK, COLO. – Primary care physicians appear to have dropped the ball when it comes to primary chemoprevention of breast cancer, studies suggest.
Chemoprevention is enormously underutilized. An estimated 2 million American premenopausal women are candidates for primary chemoprevention of breast cancer with tamoxifen, an agent shown to reduce their risk by 50%. Yet only 4% of them are on this safe and generally well-tolerated estrogen receptor antagonist, Dr. Jennifer R. Diamond said at an update in internal medicine sponsored by the University of Colorado at Denver.

Moreover, when she informally polled her audience at the outset of her talk, 93% indicated via electronic clicker that they were not comfortable with chemoprevention for breast cancer and didn’t commonly use it in appropriately selected patients in their practice.
"The most common side effect I see with tamoxifen for primary chemoprevention is hot flashes. But many young women will do fine. They don’t have any problems. And the opportunity to reduce the risk of breast cancer by 50% in these young women makes this an option I think you really need to be offering to young women" who are at increased risk for breast cancer, said Dr. Diamond, a medical oncologist at the university.
Nearly 230,000 American women will receive a new diagnosis of breast cancer in 2012. More than 39,000 women will die this year from the malignancy.
Secondary chemoprevention is most effective for women who have been diagnosed with lobular carcinoma in situ. The recommendation to embark on secondary chemoprevention usually comes from the patient’s medical oncologist.
In contrast, most candidates for primary chemoprevention have never seen a medical oncologist. Thus, primary chemoprevention falls within the bailiwick of primary care physicians. The latest guidelines from the National Comprehensive Cancer Network identify appropriate candidates for primary chemoprevention as women who have a first-degree relative with breast or ovarian cancer; a history of thoracic irradiation; mutations predisposing to breast cancer; or a 5-year risk of breast cancer of at least 1.7% according to the breast cancer risk assessment tool (the Gail model).
A 5-year risk of 1.7% or greater is not a high bar. It equates on the Gail model to being 65 years old without additional risk factors. "You’ll find that many of your patients are candidates," Dr. Diamond predicted.
Two options are available for primary chemoprevention; the determining factor as to which is best in a given high-risk individual is menopausal status. Premenopausal women can benefit from 5 years of oral tamoxifen at 20 mg/day. In postmenopausal women, however, exemestane (Aromasin), an aromatase inhibitor, is the safer, better choice, even though the drug’s use in chemoprevention is, for now, off label, she continued.
The oft-cited figure of a 50% reduction in breast cancer risk conferred by tamoxifen comes from the landmark randomized, placebo-controlled NSABP (National Surgical Adjuvant Breast and Bowel Project) P-1 trial (J. Natl. Cancer Inst. 1998;90:1371-88). The protective effect continued after treatment ended. The number needed to treat at 20 mg/day for 5 years in order to prevent one additional case of breast cancer was calculated at 95 in 5 years and 56 in 10 years. Women younger than age 50 years experienced no excess risk of serious adverse events.
"The risks of venous thromboembolism and endometrial cancer are what we all worry about with tamoxifen, but they’re actually incredibly uncommon" in women younger than age 50, she said. "I don’t recommend that you use tamoxifen in your postmenopausal women; that’s where you get into trouble with endometrial cancer and thromboembolic events. I believe that you can safely use tamoxifen in premenopausal women without a significantly increased risk of thromboembolic events. I equate the risk to that of birth control pills."
Raloxifene joins tamoxifen as the only other proved drug with Food and Drug Administration approval for primary breast cancer chemoprevention. The NSABP STAR (Study of Tamoxifen and Raloxifene) trial showed that the two medications are equivalent for prevention of invasive breast cancer, but that raloxifene is less effective in preventing ductal carcinoma in situ (JAMA 2006;295:2727-41). For this reason, Dr. Diamond and her colleagues don’t use it.
For postmenopausal women who are candidates for primary chemoprevention, Dr. Diamond said that she turns to exemestane on the basis of the National Cancer Institute of Canada CTG MAP-3 trial, which showed a 65% reduction in the annual incidence of breast cancer with 5 years of the aromatase inhibitor, compared with placebo. (N. Engl. J. Med. 2011;364:2381-91).
The number needed to treat with exemestane at 25 mg/day for 5 years in order to prevent one invasive breast cancer was 94 in 3 years and 26 in 5 years.
"I would argue that this is a really low NNT, and a lot of patients in your practice probably would fall into the category of qualifying for the MAP-3 study," Dr. Diamond said.
There were no serious risks associated with exemestane in MAP-3. Hot flashes and other menopausal symptoms were more common than with placebo, yet reassuringly there was no difference between the exemestane and control groups on quality of life measures.
Bone mineral density decreased over time in the exemestane group; however, patients on the aromatase inhibitor had no increased risk of fractures. Dr. Diamond advised monitoring bone density in exemestane-treated women, and if it drops significantly, consider prescribing denosumab as a subcutaneous injection once every 6 months. Denosumab is already FDA approved to increase bone mass in patients on adjuvant aromatase inhibitor therapy for breast cancer.
Dr. Diamond reported having no financial conflicts.
ESTES PARK, COLO. – Primary care physicians appear to have dropped the ball when it comes to primary chemoprevention of breast cancer, studies suggest.
Chemoprevention is enormously underutilized. An estimated 2 million American premenopausal women are candidates for primary chemoprevention of breast cancer with tamoxifen, an agent shown to reduce their risk by 50%. Yet only 4% of them are on this safe and generally well-tolerated estrogen receptor antagonist, Dr. Jennifer R. Diamond said at an update in internal medicine sponsored by the University of Colorado at Denver.
Moreover, when she informally polled her audience at the outset of her talk, 93% indicated via electronic clicker that they were not comfortable with chemoprevention for breast cancer and didn’t commonly use it in appropriately selected patients in their practice.
"The most common side effect I see with tamoxifen for primary chemoprevention is hot flashes. But many young women will do fine. They don’t have any problems. And the opportunity to reduce the risk of breast cancer by 50% in these young women makes this an option I think you really need to be offering to young women" who are at increased risk for breast cancer, said Dr. Diamond, a medical oncologist at the university.
Nearly 230,000 American women will receive a new diagnosis of breast cancer in 2012. More than 39,000 women will die this year from the malignancy.
Secondary chemoprevention is most effective for women who have been diagnosed with lobular carcinoma in situ. The recommendation to embark on secondary chemoprevention usually comes from the patient’s medical oncologist.
In contrast, most candidates for primary chemoprevention have never seen a medical oncologist. Thus, primary chemoprevention falls within the bailiwick of primary care physicians. The latest guidelines from the National Comprehensive Cancer Network identify appropriate candidates for primary chemoprevention as women who have a first-degree relative with breast or ovarian cancer; a history of thoracic irradiation; mutations predisposing to breast cancer; or a 5-year risk of breast cancer of at least 1.7% according to the breast cancer risk assessment tool (the Gail model).
A 5-year risk of 1.7% or greater is not a high bar. It equates on the Gail model to being 65 years old without additional risk factors. "You’ll find that many of your patients are candidates," Dr. Diamond predicted.
Two options are available for primary chemoprevention; the determining factor as to which is best in a given high-risk individual is menopausal status. Premenopausal women can benefit from 5 years of oral tamoxifen at 20 mg/day. In postmenopausal women, however, exemestane (Aromasin), an aromatase inhibitor, is the safer, better choice, even though the drug’s use in chemoprevention is, for now, off label, she continued.
The oft-cited figure of a 50% reduction in breast cancer risk conferred by tamoxifen comes from the landmark randomized, placebo-controlled NSABP (National Surgical Adjuvant Breast and Bowel Project) P-1 trial (J. Natl. Cancer Inst. 1998;90:1371-88). The protective effect continued after treatment ended. The number needed to treat at 20 mg/day for 5 years in order to prevent one additional case of breast cancer was calculated at 95 in 5 years and 56 in 10 years. Women younger than age 50 years experienced no excess risk of serious adverse events.
"The risks of venous thromboembolism and endometrial cancer are what we all worry about with tamoxifen, but they’re actually incredibly uncommon" in women younger than age 50, she said. "I don’t recommend that you use tamoxifen in your postmenopausal women; that’s where you get into trouble with endometrial cancer and thromboembolic events. I believe that you can safely use tamoxifen in premenopausal women without a significantly increased risk of thromboembolic events. I equate the risk to that of birth control pills."
Raloxifene joins tamoxifen as the only other proved drug with Food and Drug Administration approval for primary breast cancer chemoprevention. The NSABP STAR (Study of Tamoxifen and Raloxifene) trial showed that the two medications are equivalent for prevention of invasive breast cancer, but that raloxifene is less effective in preventing ductal carcinoma in situ (JAMA 2006;295:2727-41). For this reason, Dr. Diamond and her colleagues don’t use it.
For postmenopausal women who are candidates for primary chemoprevention, Dr. Diamond said that she turns to exemestane on the basis of the National Cancer Institute of Canada CTG MAP-3 trial, which showed a 65% reduction in the annual incidence of breast cancer with 5 years of the aromatase inhibitor, compared with placebo. (N. Engl. J. Med. 2011;364:2381-91).
The number needed to treat with exemestane at 25 mg/day for 5 years in order to prevent one invasive breast cancer was 94 in 3 years and 26 in 5 years.
"I would argue that this is a really low NNT, and a lot of patients in your practice probably would fall into the category of qualifying for the MAP-3 study," Dr. Diamond said.
There were no serious risks associated with exemestane in MAP-3. Hot flashes and other menopausal symptoms were more common than with placebo, yet reassuringly there was no difference between the exemestane and control groups on quality of life measures.
Bone mineral density decreased over time in the exemestane group; however, patients on the aromatase inhibitor had no increased risk of fractures. Dr. Diamond advised monitoring bone density in exemestane-treated women, and if it drops significantly, consider prescribing denosumab as a subcutaneous injection once every 6 months. Denosumab is already FDA approved to increase bone mass in patients on adjuvant aromatase inhibitor therapy for breast cancer.
Dr. Diamond reported having no financial conflicts.
ESTES PARK, COLO. – Primary care physicians appear to have dropped the ball when it comes to primary chemoprevention of breast cancer, studies suggest.
Chemoprevention is enormously underutilized. An estimated 2 million American premenopausal women are candidates for primary chemoprevention of breast cancer with tamoxifen, an agent shown to reduce their risk by 50%. Yet only 4% of them are on this safe and generally well-tolerated estrogen receptor antagonist, Dr. Jennifer R. Diamond said at an update in internal medicine sponsored by the University of Colorado at Denver.
Moreover, when she informally polled her audience at the outset of her talk, 93% indicated via electronic clicker that they were not comfortable with chemoprevention for breast cancer and didn’t commonly use it in appropriately selected patients in their practice.
"The most common side effect I see with tamoxifen for primary chemoprevention is hot flashes. But many young women will do fine. They don’t have any problems. And the opportunity to reduce the risk of breast cancer by 50% in these young women makes this an option I think you really need to be offering to young women" who are at increased risk for breast cancer, said Dr. Diamond, a medical oncologist at the university.
Nearly 230,000 American women will receive a new diagnosis of breast cancer in 2012. More than 39,000 women will die this year from the malignancy.
Secondary chemoprevention is most effective for women who have been diagnosed with lobular carcinoma in situ. The recommendation to embark on secondary chemoprevention usually comes from the patient’s medical oncologist.
In contrast, most candidates for primary chemoprevention have never seen a medical oncologist. Thus, primary chemoprevention falls within the bailiwick of primary care physicians. The latest guidelines from the National Comprehensive Cancer Network identify appropriate candidates for primary chemoprevention as women who have a first-degree relative with breast or ovarian cancer; a history of thoracic irradiation; mutations predisposing to breast cancer; or a 5-year risk of breast cancer of at least 1.7% according to the breast cancer risk assessment tool (the Gail model).
A 5-year risk of 1.7% or greater is not a high bar. It equates on the Gail model to being 65 years old without additional risk factors. "You’ll find that many of your patients are candidates," Dr. Diamond predicted.
Two options are available for primary chemoprevention; the determining factor as to which is best in a given high-risk individual is menopausal status. Premenopausal women can benefit from 5 years of oral tamoxifen at 20 mg/day. In postmenopausal women, however, exemestane (Aromasin), an aromatase inhibitor, is the safer, better choice, even though the drug’s use in chemoprevention is, for now, off label, she continued.
The oft-cited figure of a 50% reduction in breast cancer risk conferred by tamoxifen comes from the landmark randomized, placebo-controlled NSABP (National Surgical Adjuvant Breast and Bowel Project) P-1 trial (J. Natl. Cancer Inst. 1998;90:1371-88). The protective effect continued after treatment ended. The number needed to treat at 20 mg/day for 5 years in order to prevent one additional case of breast cancer was calculated at 95 in 5 years and 56 in 10 years. Women younger than age 50 years experienced no excess risk of serious adverse events.
"The risks of venous thromboembolism and endometrial cancer are what we all worry about with tamoxifen, but they’re actually incredibly uncommon" in women younger than age 50, she said. "I don’t recommend that you use tamoxifen in your postmenopausal women; that’s where you get into trouble with endometrial cancer and thromboembolic events. I believe that you can safely use tamoxifen in premenopausal women without a significantly increased risk of thromboembolic events. I equate the risk to that of birth control pills."
Raloxifene joins tamoxifen as the only other proved drug with Food and Drug Administration approval for primary breast cancer chemoprevention. The NSABP STAR (Study of Tamoxifen and Raloxifene) trial showed that the two medications are equivalent for prevention of invasive breast cancer, but that raloxifene is less effective in preventing ductal carcinoma in situ (JAMA 2006;295:2727-41). For this reason, Dr. Diamond and her colleagues don’t use it.
For postmenopausal women who are candidates for primary chemoprevention, Dr. Diamond said that she turns to exemestane on the basis of the National Cancer Institute of Canada CTG MAP-3 trial, which showed a 65% reduction in the annual incidence of breast cancer with 5 years of the aromatase inhibitor, compared with placebo. (N. Engl. J. Med. 2011;364:2381-91).
The number needed to treat with exemestane at 25 mg/day for 5 years in order to prevent one invasive breast cancer was 94 in 3 years and 26 in 5 years.
"I would argue that this is a really low NNT, and a lot of patients in your practice probably would fall into the category of qualifying for the MAP-3 study," Dr. Diamond said.
There were no serious risks associated with exemestane in MAP-3. Hot flashes and other menopausal symptoms were more common than with placebo, yet reassuringly there was no difference between the exemestane and control groups on quality of life measures.
Bone mineral density decreased over time in the exemestane group; however, patients on the aromatase inhibitor had no increased risk of fractures. Dr. Diamond advised monitoring bone density in exemestane-treated women, and if it drops significantly, consider prescribing denosumab as a subcutaneous injection once every 6 months. Denosumab is already FDA approved to increase bone mass in patients on adjuvant aromatase inhibitor therapy for breast cancer.
Dr. Diamond reported having no financial conflicts.
EXPERT ANALYSIS FROM AN UPDATE IN INTERNAL MEDICINE SPONSORED BY THE UNIVERSITY OF COLORADO

Prior Thoracic Radiotherapy Warrants Annual Breast MRI
ESTES PARK, COLO. – Primary care physicians are uniquely positioned to help their younger adult female patients who have a history of thoracic radiation therapy for lymphoma by arranging for them to begin undergoing annual screening breast MRI, Dr. Jennifer R. Diamond said.
"That mantle cell radiation includes the upper inner quadrants of both breasts; those patients are at extremely high risk for developing breast cancer," explained Dr. Diamond, a medical oncologist at the University of Colorado, Aurora. "Many times, they’re no longer being followed by their oncologist, and it’s up to their primary care physician to pull the trigger and order the test."

Many patients likely wouldn’t know that they should do breast cancer screening at such an early age, she added, because breast cancer wasn’t an anticipated outcome back when they had their radiotherapy.
National Comprehensive Cancer Network guidelines call for annual breast MRI and mammography along with clinical breast exams every 6-12 months, beginning 8-10 years after thoracic radiotherapy or at age 25 years, whichever occurs later. Dr. Diamond typically has patients stagger the two imaging modalities 6 months apart.
Both annual breast MRI and mammography are necessary. Screening MRI is far more sensitive than mammography alone in high-risk women, and it leads to cancer diagnosis at an earlier stage. However, MRI can miss ductal carcinoma in situ that would be evident as abnormal calcifications on screening mammography, she continued.
Annual breast MRI, in addition to mammography, is also indicated for women at high breast cancer risk as defined by a lifetime estimated risk in excess of 20%, or because they possess a BRCA 1 or 2 mutation, according to the NCCN. Dr. Diamond recommended BRCAPRO as a very useful tool for estimating the lifetime risk of breast cancer in women with no previous breast cancer.
In response to an audience question, she said she’s had no problem in getting insurers to pay for annual breast MRI to supplement annual mammography, so long as she clearly documents in her note that the patient has a greater than 20% lifetime estimated risk of breast cancer.
"They won’t cover it even a week earlier than annually, though," Dr. Diamond added.
She is a big fan of breast tomosynthesis, also known as three-dimensional mammography. The Food and Drug Administration–licensed technology produces a 3-D view of the breast tissue in addition to the standard two-dimensional images, all obtained in one compression with only about 2 seconds of additional compression time. Studies have shown that this tool substantially reduces patient recall rates for nondefinitive mammograms, Dr. Diamond said.
"It allows the mammographer to scroll through the breast, looking up and down sort of like with a CT scan, but without the same radiation exposure," she explained. "It’s a really great new technology, and I think most mammography centers will increasingly turn to it."
Dr. Diamond reported having no financial conflicts.
ESTES PARK, COLO. – Primary care physicians are uniquely positioned to help their younger adult female patients who have a history of thoracic radiation therapy for lymphoma by arranging for them to begin undergoing annual screening breast MRI, Dr. Jennifer R. Diamond said.
"That mantle cell radiation includes the upper inner quadrants of both breasts; those patients are at extremely high risk for developing breast cancer," explained Dr. Diamond, a medical oncologist at the University of Colorado, Aurora. "Many times, they’re no longer being followed by their oncologist, and it’s up to their primary care physician to pull the trigger and order the test."
Many patients likely wouldn’t know that they should do breast cancer screening at such an early age, she added, because breast cancer wasn’t an anticipated outcome back when they had their radiotherapy.
National Comprehensive Cancer Network guidelines call for annual breast MRI and mammography along with clinical breast exams every 6-12 months, beginning 8-10 years after thoracic radiotherapy or at age 25 years, whichever occurs later. Dr. Diamond typically has patients stagger the two imaging modalities 6 months apart.
Both annual breast MRI and mammography are necessary. Screening MRI is far more sensitive than mammography alone in high-risk women, and it leads to cancer diagnosis at an earlier stage. However, MRI can miss ductal carcinoma in situ that would be evident as abnormal calcifications on screening mammography, she continued.
Annual breast MRI, in addition to mammography, is also indicated for women at high breast cancer risk as defined by a lifetime estimated risk in excess of 20%, or because they possess a BRCA 1 or 2 mutation, according to the NCCN. Dr. Diamond recommended BRCAPRO as a very useful tool for estimating the lifetime risk of breast cancer in women with no previous breast cancer.
In response to an audience question, she said she’s had no problem in getting insurers to pay for annual breast MRI to supplement annual mammography, so long as she clearly documents in her note that the patient has a greater than 20% lifetime estimated risk of breast cancer.
"They won’t cover it even a week earlier than annually, though," Dr. Diamond added.
She is a big fan of breast tomosynthesis, also known as three-dimensional mammography. The Food and Drug Administration–licensed technology produces a 3-D view of the breast tissue in addition to the standard two-dimensional images, all obtained in one compression with only about 2 seconds of additional compression time. Studies have shown that this tool substantially reduces patient recall rates for nondefinitive mammograms, Dr. Diamond said.
"It allows the mammographer to scroll through the breast, looking up and down sort of like with a CT scan, but without the same radiation exposure," she explained. "It’s a really great new technology, and I think most mammography centers will increasingly turn to it."
Dr. Diamond reported having no financial conflicts.
ESTES PARK, COLO. – Primary care physicians are uniquely positioned to help their younger adult female patients who have a history of thoracic radiation therapy for lymphoma by arranging for them to begin undergoing annual screening breast MRI, Dr. Jennifer R. Diamond said.
"That mantle cell radiation includes the upper inner quadrants of both breasts; those patients are at extremely high risk for developing breast cancer," explained Dr. Diamond, a medical oncologist at the University of Colorado, Aurora. "Many times, they’re no longer being followed by their oncologist, and it’s up to their primary care physician to pull the trigger and order the test."
Many patients likely wouldn’t know that they should do breast cancer screening at such an early age, she added, because breast cancer wasn’t an anticipated outcome back when they had their radiotherapy.
National Comprehensive Cancer Network guidelines call for annual breast MRI and mammography along with clinical breast exams every 6-12 months, beginning 8-10 years after thoracic radiotherapy or at age 25 years, whichever occurs later. Dr. Diamond typically has patients stagger the two imaging modalities 6 months apart.
Both annual breast MRI and mammography are necessary. Screening MRI is far more sensitive than mammography alone in high-risk women, and it leads to cancer diagnosis at an earlier stage. However, MRI can miss ductal carcinoma in situ that would be evident as abnormal calcifications on screening mammography, she continued.
Annual breast MRI, in addition to mammography, is also indicated for women at high breast cancer risk as defined by a lifetime estimated risk in excess of 20%, or because they possess a BRCA 1 or 2 mutation, according to the NCCN. Dr. Diamond recommended BRCAPRO as a very useful tool for estimating the lifetime risk of breast cancer in women with no previous breast cancer.
In response to an audience question, she said she’s had no problem in getting insurers to pay for annual breast MRI to supplement annual mammography, so long as she clearly documents in her note that the patient has a greater than 20% lifetime estimated risk of breast cancer.
"They won’t cover it even a week earlier than annually, though," Dr. Diamond added.
She is a big fan of breast tomosynthesis, also known as three-dimensional mammography. The Food and Drug Administration–licensed technology produces a 3-D view of the breast tissue in addition to the standard two-dimensional images, all obtained in one compression with only about 2 seconds of additional compression time. Studies have shown that this tool substantially reduces patient recall rates for nondefinitive mammograms, Dr. Diamond said.
"It allows the mammographer to scroll through the breast, looking up and down sort of like with a CT scan, but without the same radiation exposure," she explained. "It’s a really great new technology, and I think most mammography centers will increasingly turn to it."
Dr. Diamond reported having no financial conflicts.
EXPERT ANALYSIS FROM AN UPDATE IN INTERNAL MEDICINE SPONSORED BY THE UNIVERSITY OF COLORADO

Novel Estrogen Combination Shows Favorable Coagulation Profile
HOUSTON – The novel investigational tissue selective estrogen complex comprising bazedoxifene plus conjugated estrogens displayed an overall favorable coagulation profile in a pooled analysis of three randomized, placebo-controlled, double-blind, phase III clinical trials.
Moreover, the risk of venous thromboembolism or cerebrovascular events was low, at 0.2% or less during up to 2 years of treatment, a rate not significantly different from that of control patients, Dr. Sven O. Skouby of Herlev (Denmark) Hospital said at the annual meeting of the Endocrine Society.
He presented an analysis of serial coagulation parameters obtained in 1,978 postmenopausal women with intact uteri who were randomized to bazedoxifene at 20 mg per day plus either 0.45 or 0.625 mg of conjugated estrogens or to placebo in the first, fourth, and fifth SMART (Selective Estrogens, Menopause, and Response to Therapy) trials. SMART-1 assessed changes in coagulation variables at 12 and 24 months, while SMART-4 and -5 did so at 12 months.
Changes from baseline in protein C activity, d-dimer level, and prothrombin and partial thromboplastin times were similar in the two bazedoxifene/conjugated estrogens study arms and in placebo-treated controls.
The two active treatment arms showed modest but not clinically meaningful decreases in plasminogen activator-1, antithrombin III, and fibrinogen, compared with placebo. Similarly, plasminogen activity in the two active treatment groups increased by a mean of 0.1 L/L over the course of 24 months, a statistically greater increase than the 0.06 L/L seen in the control group, but not a clinically significant difference, Dr. Skouby observed.
SMART-1, -4, and -5 included 3,549 randomized patients with follow-up data on hard clinical thromboembolic events. Three patients on bazedoxifene/conjugated estrogens experienced deep vein thrombosis during 2 years of therapy, as did one on placebo. There were no cases of pulmonary embolism. Two of the treated patients and no controls had a cerebrovascular event.
In a separate presentation, Dr. Sebastian Mirkin reported on a pooled analysis of lipid changes in 4,409 postmenopausal women in SMART-1 through -5.
At 24 months, LDL cholesterol fell by a mean 7.4% from baseline in the bazedoxifene 20 mg/conjugated estrogens 0.45 mg group, and by 8.4% in those on bazedoxifene 20 mg/conjugated estrogens 0.625 mg, compared with a 2% increase in controls.
HDL levels rose by 7.3% in the lower-dose and 7.9% in the higher-dose groups, a significantly greater benefit than the 2.8% increase with placebo. Apolipoprotein B declined by a mean of 0.4% in the lower- and 0.5% in the higher-dose bazedoxifene/conjugated estrogens group, compared with a 4.3% increase in controls.
The one negative was that triglyceride levels increased by 20.3% in the lower- and 18.7% in the higher-dose groups, significantly greater than the 7.1% rise with placebo, reported Dr. Mirkin.
Bazedoxifene is not approved in the United States. Pfizer submitted a new drug application to the Food and Drug Administration in 2006, and in 2007 got an "approvable letter" that asked for more information. There is no public record of Pfizer’s response. Pfizer has announced that it will submit an application to the FDA for the bazedoxifene/conjugated estrogen combination this year.
The SMART trials were sponsored by Pfizer. Dr. Skouby serves as a consultant to the company. Dr. Mirkin is a Pfizer employee.
HOUSTON – The novel investigational tissue selective estrogen complex comprising bazedoxifene plus conjugated estrogens displayed an overall favorable coagulation profile in a pooled analysis of three randomized, placebo-controlled, double-blind, phase III clinical trials.
Moreover, the risk of venous thromboembolism or cerebrovascular events was low, at 0.2% or less during up to 2 years of treatment, a rate not significantly different from that of control patients, Dr. Sven O. Skouby of Herlev (Denmark) Hospital said at the annual meeting of the Endocrine Society.
He presented an analysis of serial coagulation parameters obtained in 1,978 postmenopausal women with intact uteri who were randomized to bazedoxifene at 20 mg per day plus either 0.45 or 0.625 mg of conjugated estrogens or to placebo in the first, fourth, and fifth SMART (Selective Estrogens, Menopause, and Response to Therapy) trials. SMART-1 assessed changes in coagulation variables at 12 and 24 months, while SMART-4 and -5 did so at 12 months.
Changes from baseline in protein C activity, d-dimer level, and prothrombin and partial thromboplastin times were similar in the two bazedoxifene/conjugated estrogens study arms and in placebo-treated controls.
The two active treatment arms showed modest but not clinically meaningful decreases in plasminogen activator-1, antithrombin III, and fibrinogen, compared with placebo. Similarly, plasminogen activity in the two active treatment groups increased by a mean of 0.1 L/L over the course of 24 months, a statistically greater increase than the 0.06 L/L seen in the control group, but not a clinically significant difference, Dr. Skouby observed.
SMART-1, -4, and -5 included 3,549 randomized patients with follow-up data on hard clinical thromboembolic events. Three patients on bazedoxifene/conjugated estrogens experienced deep vein thrombosis during 2 years of therapy, as did one on placebo. There were no cases of pulmonary embolism. Two of the treated patients and no controls had a cerebrovascular event.
In a separate presentation, Dr. Sebastian Mirkin reported on a pooled analysis of lipid changes in 4,409 postmenopausal women in SMART-1 through -5.
At 24 months, LDL cholesterol fell by a mean 7.4% from baseline in the bazedoxifene 20 mg/conjugated estrogens 0.45 mg group, and by 8.4% in those on bazedoxifene 20 mg/conjugated estrogens 0.625 mg, compared with a 2% increase in controls.
HDL levels rose by 7.3% in the lower-dose and 7.9% in the higher-dose groups, a significantly greater benefit than the 2.8% increase with placebo. Apolipoprotein B declined by a mean of 0.4% in the lower- and 0.5% in the higher-dose bazedoxifene/conjugated estrogens group, compared with a 4.3% increase in controls.
The one negative was that triglyceride levels increased by 20.3% in the lower- and 18.7% in the higher-dose groups, significantly greater than the 7.1% rise with placebo, reported Dr. Mirkin.
Bazedoxifene is not approved in the United States. Pfizer submitted a new drug application to the Food and Drug Administration in 2006, and in 2007 got an "approvable letter" that asked for more information. There is no public record of Pfizer’s response. Pfizer has announced that it will submit an application to the FDA for the bazedoxifene/conjugated estrogen combination this year.
The SMART trials were sponsored by Pfizer. Dr. Skouby serves as a consultant to the company. Dr. Mirkin is a Pfizer employee.
HOUSTON – The novel investigational tissue selective estrogen complex comprising bazedoxifene plus conjugated estrogens displayed an overall favorable coagulation profile in a pooled analysis of three randomized, placebo-controlled, double-blind, phase III clinical trials.
Moreover, the risk of venous thromboembolism or cerebrovascular events was low, at 0.2% or less during up to 2 years of treatment, a rate not significantly different from that of control patients, Dr. Sven O. Skouby of Herlev (Denmark) Hospital said at the annual meeting of the Endocrine Society.
He presented an analysis of serial coagulation parameters obtained in 1,978 postmenopausal women with intact uteri who were randomized to bazedoxifene at 20 mg per day plus either 0.45 or 0.625 mg of conjugated estrogens or to placebo in the first, fourth, and fifth SMART (Selective Estrogens, Menopause, and Response to Therapy) trials. SMART-1 assessed changes in coagulation variables at 12 and 24 months, while SMART-4 and -5 did so at 12 months.
Changes from baseline in protein C activity, d-dimer level, and prothrombin and partial thromboplastin times were similar in the two bazedoxifene/conjugated estrogens study arms and in placebo-treated controls.
The two active treatment arms showed modest but not clinically meaningful decreases in plasminogen activator-1, antithrombin III, and fibrinogen, compared with placebo. Similarly, plasminogen activity in the two active treatment groups increased by a mean of 0.1 L/L over the course of 24 months, a statistically greater increase than the 0.06 L/L seen in the control group, but not a clinically significant difference, Dr. Skouby observed.
SMART-1, -4, and -5 included 3,549 randomized patients with follow-up data on hard clinical thromboembolic events. Three patients on bazedoxifene/conjugated estrogens experienced deep vein thrombosis during 2 years of therapy, as did one on placebo. There were no cases of pulmonary embolism. Two of the treated patients and no controls had a cerebrovascular event.
In a separate presentation, Dr. Sebastian Mirkin reported on a pooled analysis of lipid changes in 4,409 postmenopausal women in SMART-1 through -5.
At 24 months, LDL cholesterol fell by a mean 7.4% from baseline in the bazedoxifene 20 mg/conjugated estrogens 0.45 mg group, and by 8.4% in those on bazedoxifene 20 mg/conjugated estrogens 0.625 mg, compared with a 2% increase in controls.
HDL levels rose by 7.3% in the lower-dose and 7.9% in the higher-dose groups, a significantly greater benefit than the 2.8% increase with placebo. Apolipoprotein B declined by a mean of 0.4% in the lower- and 0.5% in the higher-dose bazedoxifene/conjugated estrogens group, compared with a 4.3% increase in controls.
The one negative was that triglyceride levels increased by 20.3% in the lower- and 18.7% in the higher-dose groups, significantly greater than the 7.1% rise with placebo, reported Dr. Mirkin.
Bazedoxifene is not approved in the United States. Pfizer submitted a new drug application to the Food and Drug Administration in 2006, and in 2007 got an "approvable letter" that asked for more information. There is no public record of Pfizer’s response. Pfizer has announced that it will submit an application to the FDA for the bazedoxifene/conjugated estrogen combination this year.
The SMART trials were sponsored by Pfizer. Dr. Skouby serves as a consultant to the company. Dr. Mirkin is a Pfizer employee.
AT THE ANNUAL MEETING OF THE ENDOCRINE SOCIETY
Major Finding: An overall favorable coagulation profile was documented in women on an investigational tissue-selective estrogen complex for up to 2 years.
Data Source: This was a pooled analysis of several thousand postmenopausal women with intact uteri who were randomized to bazedoxifene 20 mg/conjugated estrogens 0.45 mg, bazedoxifene 20 mg/conjugated estrogens 0.625 mg, or placebo for up to 2 years in three phase III clinical trials.
Disclosures: The SMART trials were sponsored by Pfizer. Dr. Skouby serves as a consultant to the company. Dr. Mirkin is a Pfizer employee.
Routine Infant Meningococcal Immunization - It's Complicated!
VAIL, COLO. – When the Advisory Committee on Immunization Practices meets in late October to review the recently approved meningococcal groups C and Y and Haemophilus influenzae type b tetanus toxoid conjugate for infant immunization, it will not recommend the vaccine’s routine use in all infants, Dr. Marsha S. Anderson predicted.
The Hib-MenCY-TT vaccine is marketed as MenHibrix.

For the past couple of years, the ACIP Working Group for Meningococcal Disease has been discussing whether infants should be routinely vaccinated against meningococcal disease. The group indicated at its June 2012 meeting that the time isn’t right for such a practice recommendation, in spite of the Food and Drug Administration’s approval the very same month of MenHibrix as the first-ever meningococcal vaccine licensed for infant immunization starting as early as age 6 weeks.
Routine infant meningococcal vaccination is a complicated issue, with arguments for and against, and there is no right answer, Dr. Anderson said at a conference on pediatric infectious diseases, which was sponsored by Children’s Hospital Colorado in Aurora. But when she ran down the pros and cons, she caused a seismic shift in audience opinion.
At the outset of her talk, when she polled her audience to see who favored routine meningococcal vaccination in infancy given the current licensed vaccines, two-thirds of the audience signaled via electronic clicker that they were for it. Afterward, however, 82% were opposed.
Meningococcal disease is the top cause of bacterial meningitis in children and young adults in the United States. Outbreaks cause widespread public fear and panic. The mortality rate of meningococcal meningitis is 10%, with death typically coming within 24-48 hours after symptom onset. Up to 20% of survivors of bacterial meningitis have learning disabilities, permanent hearing loss, or other serious sequelae, noted Dr. Anderson of the University of Colorado at Denver, Aurora.
Arguments supporting routine infant immunization include the fact that the meningococcal disease incidence rate is much higher in infants than in other age groups. The current ACIP recommendation for meningococcal conjugate vaccination, which calls for routine vaccination at age 11-12 years with a booster dose at age 16, doesn’t address that epidemiologic reality. Moreover, the MenHibrix vaccine was shown to be safe and effective in prelicensure studies.
"And as physicians and nurses, we all want to protect every single child against this devastating disease," she observed.
On the con side, 50%-60% of all infant disease in the United States involves serogroup B meningococcus – and protection against serogroup B isn’t included in any licensed U.S. vaccine. This is not for lack of trying. The problem is that the polysaccharide capsule of serogroup B meningococcus is poorly immunogenic. Vaccines that were developed in New Zealand, Cuba, and Australia to protect against serogroup B in those isolated nations turned out to be strain specific and don’t protect against U.S. strains.
On a more optimistic note, promising studies are underway in other countries using vaccines made from multiple strains and with several antigenic targets. It’s quite likely that an effective serogroup B–protective vaccine will eventually get here, according to Dr. Anderson.
Another argument against routine immunization in infancy is that the annual incidence rate of meningococcal disease in the United States is at an all-time low, with an incidence rate only one-fifth that in the early 1980s. A Centers for Disease Control and Prevention modeling analysis estimated that routine administration of a four-dose infant series of meningococcal conjugate vaccine in the current low-incidence era might prevent 44 cases of meningococcal disease annually. The number of infants who would need to be vaccinated in order to prevent one case was estimated at 76,000. The number needed to be vaccinated in order to prevent one death was put at 642,000. That’s not very cost effective.
There also are practical stumbling blocks standing in the way of routine infant immunization. MenHibrix is given in a four-dose series at 2, 4, 6, and 12 months.
"I’m guessing everybody is shuddering at trying to figure out how to fit that into the current vaccine schedule," Dr. Anderson guessed.
Actually, though, it would not necessarily require any additional shots, she noted. It could be administered in a three-shot visit together with DTaP/IPV/Hep B (Pediatrix) and PCV13 (Prevnar13), although that necessitates switching from other combination Hib products.
Another option is the vaccination of toddlers with MenACWY-D (Menactra), a two-dose series given at 9 and 12 months. The difficulty here is that at present there is no routine office visit at 9 months of age. Plus, most kids won’t be protected against meningococcus until the second dose, so they will remain vulnerable to the infection through most of infancy.
"Complete control of meningococcal disease in the United States is not likely to be achieved until an effective conjugate vaccine with broad-based coverage to include serogroup B is introduced," Dr. Anderson concluded.
She reported having no financial conflicts.
VAIL, COLO. – When the Advisory Committee on Immunization Practices meets in late October to review the recently approved meningococcal groups C and Y and Haemophilus influenzae type b tetanus toxoid conjugate for infant immunization, it will not recommend the vaccine’s routine use in all infants, Dr. Marsha S. Anderson predicted.
The Hib-MenCY-TT vaccine is marketed as MenHibrix.
For the past couple of years, the ACIP Working Group for Meningococcal Disease has been discussing whether infants should be routinely vaccinated against meningococcal disease. The group indicated at its June 2012 meeting that the time isn’t right for such a practice recommendation, in spite of the Food and Drug Administration’s approval the very same month of MenHibrix as the first-ever meningococcal vaccine licensed for infant immunization starting as early as age 6 weeks.
Routine infant meningococcal vaccination is a complicated issue, with arguments for and against, and there is no right answer, Dr. Anderson said at a conference on pediatric infectious diseases, which was sponsored by Children’s Hospital Colorado in Aurora. But when she ran down the pros and cons, she caused a seismic shift in audience opinion.
At the outset of her talk, when she polled her audience to see who favored routine meningococcal vaccination in infancy given the current licensed vaccines, two-thirds of the audience signaled via electronic clicker that they were for it. Afterward, however, 82% were opposed.
Meningococcal disease is the top cause of bacterial meningitis in children and young adults in the United States. Outbreaks cause widespread public fear and panic. The mortality rate of meningococcal meningitis is 10%, with death typically coming within 24-48 hours after symptom onset. Up to 20% of survivors of bacterial meningitis have learning disabilities, permanent hearing loss, or other serious sequelae, noted Dr. Anderson of the University of Colorado at Denver, Aurora.
Arguments supporting routine infant immunization include the fact that the meningococcal disease incidence rate is much higher in infants than in other age groups. The current ACIP recommendation for meningococcal conjugate vaccination, which calls for routine vaccination at age 11-12 years with a booster dose at age 16, doesn’t address that epidemiologic reality. Moreover, the MenHibrix vaccine was shown to be safe and effective in prelicensure studies.
"And as physicians and nurses, we all want to protect every single child against this devastating disease," she observed.
On the con side, 50%-60% of all infant disease in the United States involves serogroup B meningococcus – and protection against serogroup B isn’t included in any licensed U.S. vaccine. This is not for lack of trying. The problem is that the polysaccharide capsule of serogroup B meningococcus is poorly immunogenic. Vaccines that were developed in New Zealand, Cuba, and Australia to protect against serogroup B in those isolated nations turned out to be strain specific and don’t protect against U.S. strains.
On a more optimistic note, promising studies are underway in other countries using vaccines made from multiple strains and with several antigenic targets. It’s quite likely that an effective serogroup B–protective vaccine will eventually get here, according to Dr. Anderson.
Another argument against routine immunization in infancy is that the annual incidence rate of meningococcal disease in the United States is at an all-time low, with an incidence rate only one-fifth that in the early 1980s. A Centers for Disease Control and Prevention modeling analysis estimated that routine administration of a four-dose infant series of meningococcal conjugate vaccine in the current low-incidence era might prevent 44 cases of meningococcal disease annually. The number of infants who would need to be vaccinated in order to prevent one case was estimated at 76,000. The number needed to be vaccinated in order to prevent one death was put at 642,000. That’s not very cost effective.
There also are practical stumbling blocks standing in the way of routine infant immunization. MenHibrix is given in a four-dose series at 2, 4, 6, and 12 months.
"I’m guessing everybody is shuddering at trying to figure out how to fit that into the current vaccine schedule," Dr. Anderson guessed.
Actually, though, it would not necessarily require any additional shots, she noted. It could be administered in a three-shot visit together with DTaP/IPV/Hep B (Pediatrix) and PCV13 (Prevnar13), although that necessitates switching from other combination Hib products.
Another option is the vaccination of toddlers with MenACWY-D (Menactra), a two-dose series given at 9 and 12 months. The difficulty here is that at present there is no routine office visit at 9 months of age. Plus, most kids won’t be protected against meningococcus until the second dose, so they will remain vulnerable to the infection through most of infancy.
"Complete control of meningococcal disease in the United States is not likely to be achieved until an effective conjugate vaccine with broad-based coverage to include serogroup B is introduced," Dr. Anderson concluded.
She reported having no financial conflicts.
VAIL, COLO. – When the Advisory Committee on Immunization Practices meets in late October to review the recently approved meningococcal groups C and Y and Haemophilus influenzae type b tetanus toxoid conjugate for infant immunization, it will not recommend the vaccine’s routine use in all infants, Dr. Marsha S. Anderson predicted.
The Hib-MenCY-TT vaccine is marketed as MenHibrix.
For the past couple of years, the ACIP Working Group for Meningococcal Disease has been discussing whether infants should be routinely vaccinated against meningococcal disease. The group indicated at its June 2012 meeting that the time isn’t right for such a practice recommendation, in spite of the Food and Drug Administration’s approval the very same month of MenHibrix as the first-ever meningococcal vaccine licensed for infant immunization starting as early as age 6 weeks.
Routine infant meningococcal vaccination is a complicated issue, with arguments for and against, and there is no right answer, Dr. Anderson said at a conference on pediatric infectious diseases, which was sponsored by Children’s Hospital Colorado in Aurora. But when she ran down the pros and cons, she caused a seismic shift in audience opinion.
At the outset of her talk, when she polled her audience to see who favored routine meningococcal vaccination in infancy given the current licensed vaccines, two-thirds of the audience signaled via electronic clicker that they were for it. Afterward, however, 82% were opposed.
Meningococcal disease is the top cause of bacterial meningitis in children and young adults in the United States. Outbreaks cause widespread public fear and panic. The mortality rate of meningococcal meningitis is 10%, with death typically coming within 24-48 hours after symptom onset. Up to 20% of survivors of bacterial meningitis have learning disabilities, permanent hearing loss, or other serious sequelae, noted Dr. Anderson of the University of Colorado at Denver, Aurora.
Arguments supporting routine infant immunization include the fact that the meningococcal disease incidence rate is much higher in infants than in other age groups. The current ACIP recommendation for meningococcal conjugate vaccination, which calls for routine vaccination at age 11-12 years with a booster dose at age 16, doesn’t address that epidemiologic reality. Moreover, the MenHibrix vaccine was shown to be safe and effective in prelicensure studies.
"And as physicians and nurses, we all want to protect every single child against this devastating disease," she observed.
On the con side, 50%-60% of all infant disease in the United States involves serogroup B meningococcus – and protection against serogroup B isn’t included in any licensed U.S. vaccine. This is not for lack of trying. The problem is that the polysaccharide capsule of serogroup B meningococcus is poorly immunogenic. Vaccines that were developed in New Zealand, Cuba, and Australia to protect against serogroup B in those isolated nations turned out to be strain specific and don’t protect against U.S. strains.
On a more optimistic note, promising studies are underway in other countries using vaccines made from multiple strains and with several antigenic targets. It’s quite likely that an effective serogroup B–protective vaccine will eventually get here, according to Dr. Anderson.
Another argument against routine immunization in infancy is that the annual incidence rate of meningococcal disease in the United States is at an all-time low, with an incidence rate only one-fifth that in the early 1980s. A Centers for Disease Control and Prevention modeling analysis estimated that routine administration of a four-dose infant series of meningococcal conjugate vaccine in the current low-incidence era might prevent 44 cases of meningococcal disease annually. The number of infants who would need to be vaccinated in order to prevent one case was estimated at 76,000. The number needed to be vaccinated in order to prevent one death was put at 642,000. That’s not very cost effective.
There also are practical stumbling blocks standing in the way of routine infant immunization. MenHibrix is given in a four-dose series at 2, 4, 6, and 12 months.
"I’m guessing everybody is shuddering at trying to figure out how to fit that into the current vaccine schedule," Dr. Anderson guessed.
Actually, though, it would not necessarily require any additional shots, she noted. It could be administered in a three-shot visit together with DTaP/IPV/Hep B (Pediatrix) and PCV13 (Prevnar13), although that necessitates switching from other combination Hib products.
Another option is the vaccination of toddlers with MenACWY-D (Menactra), a two-dose series given at 9 and 12 months. The difficulty here is that at present there is no routine office visit at 9 months of age. Plus, most kids won’t be protected against meningococcus until the second dose, so they will remain vulnerable to the infection through most of infancy.
"Complete control of meningococcal disease in the United States is not likely to be achieved until an effective conjugate vaccine with broad-based coverage to include serogroup B is introduced," Dr. Anderson concluded.
She reported having no financial conflicts.
EXPERT ANALYSIS FROM A CONFERENCE ON PEDIATRIC INFECTIOUS DISEASES SPONSORED BY CHILDREN'S HOSPITAL COLORADO

Finally! The Dessert-With-Breakfast Diet
HOUSTON – A high-protein, high-carbohydrate breakfast that included a daily dessert such as cake or a cookie resulted in significantly greater weight loss through 32 weeks than an identically low-calorie weight loss diet featuring a low-carbohydrate breakfast.
That’s what Dr. Daniela Jakubowicz found in a randomized trial presented at the annual meeting of the Endocrine Society.

"To achieve long-term weight loss, meal timing and composition must counteract weight loss compensatory mechanisms that encourage weight regain after weight loss. A high-carbohydrate and protein breakfast may prevent weight regain by reducing diet-induced compensatory changes in hunger, craving, and ghrelin suppression," said Dr. Jakubowicz of the Wolfson Medical Center at Tel Aviv University.
She reported on 193 obese, sedentary, nondiabetic subjects with an average age of 47 years and a body mass index of 32 kg/m2 who were randomized to one of two weight-loss diets. Both incorporated 1,600 calories daily for men and 1,400 for women. The dessert-for-breakfast diet for women allotted 600 calories at breakfast, 500 at lunch, and 300 at dinner, while the low-carbohydrate breakfast comprised 300 calories in the morning, 500 at lunch, and 600 at dinner. The big-breakfast group consumed 60 g of carbohydrate at breakfast, compared with 15 g in the comparison group.
The two diets proved similarly effective through the first 16 weeks, with average weight losses of 15.1 kg in the low-carbohydrate breakfast group and 13.5 kg in the dessert-with-breakfast eaters. At that point, however, the trajectories diverged. Over the next 16 weeks, the low-calorie breakfast group regained an average of 11.6 kg, while the big-breakfast group lost a further 6.9 kg.

Thus, the net weight loss at 32 weeks was 20.4 kg (45 pounds) in the dessert-with-breakfast group, compared with a scant 3.5 kg (7.7 lb) in the low-carbohydrate group.
What’s the explanation?
"More important than the speed of weight loss is that patients feel satiety – less hunger – and that they prevent craving," the endocrinologist said. "Several studies have now shown that diets that avoid chocolates and cookies and carbohydrates increase the addiction to carbohydrates – they increase the craving. So instead, if you eat a snack in the morning every day, after a while you become less addicted to these foods. Also, protein in the morning increases satiety. We know that if you have enough protein in the morning you’re not hungry all day."
Indeed, these points were born out in patient self-assessments conducted using visual analog scales at weeks 16 and 32. Scores for hunger were significantly lower and satiety scores significantly higher in the dessert-for-breakfast group at both time points. In addition, this group had significantly lower craving scores for fatty foods, sweets, and fast food. Dietary compliance was significantly better in the big-breakfast group than in controls from week 8 on. At week 32, for example, the big-breakfast group averaged 5 episodes of noncompliance per week, compared with 15 in controls.
Moreover, levels of ghrelin – the so-called hunger hormone – were suppressed by 45% in the dessert-with-breakfast group 2 hours after breakfast at week 32, compared with a 30% reduction in controls. The insulin response to a standardized test meal was significantly lower in the big-breakfast group as well.
In a separate randomized trial Dr. Jakubowicz conducted in 93 obese, sedentary, nondiabetic women, placing the big meal of the day at breakfast resulted in significantly greater weight loss, waist circumference shrinkage, and salutary lipid changes than when dinner was the big meal.
Both groups were placed on a 1,400-calorie-per-day diet. One diet entailed 700 calories at breakfast and 200 at dinner, while the other featured 200 calories at breakfast and 700 at dinner. After 12 weeks, the big-breakfast group lost an average of 8.7 kg, while the big-dinner group lost 3.3 kg. Waist circumference decreased by 7 cm from a baseline of 110 cm in the big-breakfast group, compared with a 2-cm reduction in controls.
Triglyceride levels improved from 180 mg/dL at baseline to 129 mg/dL at 12 weeks in the big-breakfast group, while they worsened from a baseline of 178 mg/dL to 195 mg/dL in the big-dinner group.
"It’s important to note that triglycerides increased on the big-dinner diet even though the people lost weight. The management of lipids after eating carbohydrates at night is worse than with the same carbohydrates in the morning," she concluded.
The average LDL cholesterol level in the big-breakfast group dropped from 160 mg/dL at baseline to 151 mg/dL, a significantly greater effect than seen in the big-dinner group, whose LDL went from 164 mg/dL at baseline to 162 mg/dL at 12 weeks.
Dr. Jakubowicz reported having no financial conflicts.
HOUSTON – A high-protein, high-carbohydrate breakfast that included a daily dessert such as cake or a cookie resulted in significantly greater weight loss through 32 weeks than an identically low-calorie weight loss diet featuring a low-carbohydrate breakfast.
That’s what Dr. Daniela Jakubowicz found in a randomized trial presented at the annual meeting of the Endocrine Society.
"To achieve long-term weight loss, meal timing and composition must counteract weight loss compensatory mechanisms that encourage weight regain after weight loss. A high-carbohydrate and protein breakfast may prevent weight regain by reducing diet-induced compensatory changes in hunger, craving, and ghrelin suppression," said Dr. Jakubowicz of the Wolfson Medical Center at Tel Aviv University.
She reported on 193 obese, sedentary, nondiabetic subjects with an average age of 47 years and a body mass index of 32 kg/m2 who were randomized to one of two weight-loss diets. Both incorporated 1,600 calories daily for men and 1,400 for women. The dessert-for-breakfast diet for women allotted 600 calories at breakfast, 500 at lunch, and 300 at dinner, while the low-carbohydrate breakfast comprised 300 calories in the morning, 500 at lunch, and 600 at dinner. The big-breakfast group consumed 60 g of carbohydrate at breakfast, compared with 15 g in the comparison group.
The two diets proved similarly effective through the first 16 weeks, with average weight losses of 15.1 kg in the low-carbohydrate breakfast group and 13.5 kg in the dessert-with-breakfast eaters. At that point, however, the trajectories diverged. Over the next 16 weeks, the low-calorie breakfast group regained an average of 11.6 kg, while the big-breakfast group lost a further 6.9 kg.
Thus, the net weight loss at 32 weeks was 20.4 kg (45 pounds) in the dessert-with-breakfast group, compared with a scant 3.5 kg (7.7 lb) in the low-carbohydrate group.
What’s the explanation?
"More important than the speed of weight loss is that patients feel satiety – less hunger – and that they prevent craving," the endocrinologist said. "Several studies have now shown that diets that avoid chocolates and cookies and carbohydrates increase the addiction to carbohydrates – they increase the craving. So instead, if you eat a snack in the morning every day, after a while you become less addicted to these foods. Also, protein in the morning increases satiety. We know that if you have enough protein in the morning you’re not hungry all day."
Indeed, these points were born out in patient self-assessments conducted using visual analog scales at weeks 16 and 32. Scores for hunger were significantly lower and satiety scores significantly higher in the dessert-for-breakfast group at both time points. In addition, this group had significantly lower craving scores for fatty foods, sweets, and fast food. Dietary compliance was significantly better in the big-breakfast group than in controls from week 8 on. At week 32, for example, the big-breakfast group averaged 5 episodes of noncompliance per week, compared with 15 in controls.
Moreover, levels of ghrelin – the so-called hunger hormone – were suppressed by 45% in the dessert-with-breakfast group 2 hours after breakfast at week 32, compared with a 30% reduction in controls. The insulin response to a standardized test meal was significantly lower in the big-breakfast group as well.
In a separate randomized trial Dr. Jakubowicz conducted in 93 obese, sedentary, nondiabetic women, placing the big meal of the day at breakfast resulted in significantly greater weight loss, waist circumference shrinkage, and salutary lipid changes than when dinner was the big meal.
Both groups were placed on a 1,400-calorie-per-day diet. One diet entailed 700 calories at breakfast and 200 at dinner, while the other featured 200 calories at breakfast and 700 at dinner. After 12 weeks, the big-breakfast group lost an average of 8.7 kg, while the big-dinner group lost 3.3 kg. Waist circumference decreased by 7 cm from a baseline of 110 cm in the big-breakfast group, compared with a 2-cm reduction in controls.
Triglyceride levels improved from 180 mg/dL at baseline to 129 mg/dL at 12 weeks in the big-breakfast group, while they worsened from a baseline of 178 mg/dL to 195 mg/dL in the big-dinner group.
"It’s important to note that triglycerides increased on the big-dinner diet even though the people lost weight. The management of lipids after eating carbohydrates at night is worse than with the same carbohydrates in the morning," she concluded.
The average LDL cholesterol level in the big-breakfast group dropped from 160 mg/dL at baseline to 151 mg/dL, a significantly greater effect than seen in the big-dinner group, whose LDL went from 164 mg/dL at baseline to 162 mg/dL at 12 weeks.
Dr. Jakubowicz reported having no financial conflicts.
HOUSTON – A high-protein, high-carbohydrate breakfast that included a daily dessert such as cake or a cookie resulted in significantly greater weight loss through 32 weeks than an identically low-calorie weight loss diet featuring a low-carbohydrate breakfast.
That’s what Dr. Daniela Jakubowicz found in a randomized trial presented at the annual meeting of the Endocrine Society.
"To achieve long-term weight loss, meal timing and composition must counteract weight loss compensatory mechanisms that encourage weight regain after weight loss. A high-carbohydrate and protein breakfast may prevent weight regain by reducing diet-induced compensatory changes in hunger, craving, and ghrelin suppression," said Dr. Jakubowicz of the Wolfson Medical Center at Tel Aviv University.
She reported on 193 obese, sedentary, nondiabetic subjects with an average age of 47 years and a body mass index of 32 kg/m2 who were randomized to one of two weight-loss diets. Both incorporated 1,600 calories daily for men and 1,400 for women. The dessert-for-breakfast diet for women allotted 600 calories at breakfast, 500 at lunch, and 300 at dinner, while the low-carbohydrate breakfast comprised 300 calories in the morning, 500 at lunch, and 600 at dinner. The big-breakfast group consumed 60 g of carbohydrate at breakfast, compared with 15 g in the comparison group.
The two diets proved similarly effective through the first 16 weeks, with average weight losses of 15.1 kg in the low-carbohydrate breakfast group and 13.5 kg in the dessert-with-breakfast eaters. At that point, however, the trajectories diverged. Over the next 16 weeks, the low-calorie breakfast group regained an average of 11.6 kg, while the big-breakfast group lost a further 6.9 kg.
Thus, the net weight loss at 32 weeks was 20.4 kg (45 pounds) in the dessert-with-breakfast group, compared with a scant 3.5 kg (7.7 lb) in the low-carbohydrate group.
What’s the explanation?
"More important than the speed of weight loss is that patients feel satiety – less hunger – and that they prevent craving," the endocrinologist said. "Several studies have now shown that diets that avoid chocolates and cookies and carbohydrates increase the addiction to carbohydrates – they increase the craving. So instead, if you eat a snack in the morning every day, after a while you become less addicted to these foods. Also, protein in the morning increases satiety. We know that if you have enough protein in the morning you’re not hungry all day."
Indeed, these points were born out in patient self-assessments conducted using visual analog scales at weeks 16 and 32. Scores for hunger were significantly lower and satiety scores significantly higher in the dessert-for-breakfast group at both time points. In addition, this group had significantly lower craving scores for fatty foods, sweets, and fast food. Dietary compliance was significantly better in the big-breakfast group than in controls from week 8 on. At week 32, for example, the big-breakfast group averaged 5 episodes of noncompliance per week, compared with 15 in controls.
Moreover, levels of ghrelin – the so-called hunger hormone – were suppressed by 45% in the dessert-with-breakfast group 2 hours after breakfast at week 32, compared with a 30% reduction in controls. The insulin response to a standardized test meal was significantly lower in the big-breakfast group as well.
In a separate randomized trial Dr. Jakubowicz conducted in 93 obese, sedentary, nondiabetic women, placing the big meal of the day at breakfast resulted in significantly greater weight loss, waist circumference shrinkage, and salutary lipid changes than when dinner was the big meal.
Both groups were placed on a 1,400-calorie-per-day diet. One diet entailed 700 calories at breakfast and 200 at dinner, while the other featured 200 calories at breakfast and 700 at dinner. After 12 weeks, the big-breakfast group lost an average of 8.7 kg, while the big-dinner group lost 3.3 kg. Waist circumference decreased by 7 cm from a baseline of 110 cm in the big-breakfast group, compared with a 2-cm reduction in controls.
Triglyceride levels improved from 180 mg/dL at baseline to 129 mg/dL at 12 weeks in the big-breakfast group, while they worsened from a baseline of 178 mg/dL to 195 mg/dL in the big-dinner group.
"It’s important to note that triglycerides increased on the big-dinner diet even though the people lost weight. The management of lipids after eating carbohydrates at night is worse than with the same carbohydrates in the morning," she concluded.
The average LDL cholesterol level in the big-breakfast group dropped from 160 mg/dL at baseline to 151 mg/dL, a significantly greater effect than seen in the big-dinner group, whose LDL went from 164 mg/dL at baseline to 162 mg/dL at 12 weeks.
Dr. Jakubowicz reported having no financial conflicts.
AT THE ANNUAL MEETING OF THE ENDOCRINE SOCIETY
Major Finding: Obese, nondiabetic patients assigned to a weight-loss diet emphasizing a high-protein, high-carbohydrate breakfast including a dessert lost an average of 45 pounds through 32 weeks of follow-up, compared with a 7.7-pound weight loss in patients randomized to an isocaloric low-carb diet.
Data Source: The 32-week randomized trial included 193 obese, sedentary, nondiabetic men and women.
Disclosures: Dr. Jakubowicz reported having no financial conflicts.

Earlier Kawasaki Diagnosis and Treatment Needed
VAIL, COLO. – The majority of coronary artery abnormalities occurring in a large series of children with Kawasaki disease were detected at the time of hospital admission, prior to treatment.
What this means is that, contrary to the conventional wisdom, there is no such thing as a safe window for diagnosis and treatment of Kawasaki disease, Dr. Samuel R. Dominguez stressed at a conference on pediatric infectious diseases sponsored by the Children’s Hospital Colorado.

The "safe window" concept dates back to a classic 26-year-old study that concluded that treating patients with Kawasaki disease by day 10 of their illness greatly reduced the incidence of coronary artery aneurysms, the most serious disease complication (N. Engl. J. Med. 1986;315:341-7).
Anecdotal experience to the contrary convinced Dr. Dominguez and coworkers at Children’s Hospital Colorado in Aurora that it was time to take a formal look at their institutional experience.
"It was our gestalt that some kids had coronary artery abnormalities much earlier in the course of their illness than what we’d thought from the literature. We had this growing sense that the development of coronary artery lesions was less common after discharge than we’d initially thought," the pediatric infectious disease specialist explained.
That proved to be the case.
Among all 210 patients who were admitted for Kawasaki disease over a 4-year period (all of whom were appropriately treated with intravenous immunoglobulin and aspirin), 27% had coronary artery abnormalities detected during their acute illness or subsequent outpatient follow-up. In 81% of affected kids, the coronary artery abnormalities were identified on the basis of a z score of 2.5 or above on the initial echocardiogram that was obtained at the time of admission. The coronary lesions were identified in 21% of affected children on or before day 5 of their illness, in 60% on or before day 7, and in 80% on or before day 10 of their illness.
The Colorado findings are supported by other fairly recent studies, according to Dr. Dominguez. An analysis of the Pediatric Health Information System database that included nearly 5,200 admissions for Kawasaki disease at 27 U.S. pediatric hospitals during 2001-2006 found that 3.3% of patients developed coronary artery aneurysms, 81% of which were detected during their initial hospitalization (Pediatrics 2009;124:1-8). And a Pediatric Heart Network study concluded that Kawasaki disease patients with a normal echocardiogram on admission had only a 6% incidence of developing coronary lesions at a later time, meaning that most coronary abnormalities were present at admission (Circulation 2007;116:174-9).
Intriguingly, fully 46% of Kawasaki disease patients in the Colorado study who had coronary lesions on admission had incomplete Kawasaki disease.
"That’s a much higher rate than we think of," Dr. Dominguez said. "It raises the concern that many cases of Kawasaki disease may currently be undiagnosed and not treated."
Incomplete Kawasaki disease is a diagnostic category that was created in the revised American Heart Association guidelines in an effort to identify subsets of Kawasaki disease patients earlier so treatment can be started expeditiously (Circulation 2004;110:2747-71). The revision was made in response to recognition that infants often fail to meet the classic diagnostic criteria for Kawasaki disease, yet they have a high incidence of coronary artery aneurysms.
The revised guidelines basically state that infants aged 6 months or younger on day 7 of fever without other explanation should undergo laboratory testing, even if they don’t have a generalized rash, bilateral nonexudative conjunctivitis, or any of the other clinical criteria for classic Kawasaki disease. If lab results yield evidence of systemic inflammation, then echocardiography is warranted (Pediatrics 2004;114:1708-33).
Dr. Dominguez said the clear implication of the Colorado study is that earlier diagnosis and treatment are needed in order to reduce the incidence of coronary artery abnormalities in children with Kawasaki disease. Increased clinical suspicion, greater use of the published algorithm for incomplete Kawasaki disease, and earlier resort to echocardiography in the initial work-up may result in more rapid therapy.
Although there is no guarantee that earlier diagnosis and treatment will prevent coronary lesions, that is the hope, he added.
The Children’s Hospital Colorado study was recently published (Pediatr. Infect. Dis. J. 2012 July 3 [doi:10.1097/INF.0b013e318266bcf9]).
Dr. Dominguez reported having no financial conflicts.
VAIL, COLO. – The majority of coronary artery abnormalities occurring in a large series of children with Kawasaki disease were detected at the time of hospital admission, prior to treatment.
What this means is that, contrary to the conventional wisdom, there is no such thing as a safe window for diagnosis and treatment of Kawasaki disease, Dr. Samuel R. Dominguez stressed at a conference on pediatric infectious diseases sponsored by the Children’s Hospital Colorado.
The "safe window" concept dates back to a classic 26-year-old study that concluded that treating patients with Kawasaki disease by day 10 of their illness greatly reduced the incidence of coronary artery aneurysms, the most serious disease complication (N. Engl. J. Med. 1986;315:341-7).
Anecdotal experience to the contrary convinced Dr. Dominguez and coworkers at Children’s Hospital Colorado in Aurora that it was time to take a formal look at their institutional experience.
"It was our gestalt that some kids had coronary artery abnormalities much earlier in the course of their illness than what we’d thought from the literature. We had this growing sense that the development of coronary artery lesions was less common after discharge than we’d initially thought," the pediatric infectious disease specialist explained.
That proved to be the case.
Among all 210 patients who were admitted for Kawasaki disease over a 4-year period (all of whom were appropriately treated with intravenous immunoglobulin and aspirin), 27% had coronary artery abnormalities detected during their acute illness or subsequent outpatient follow-up. In 81% of affected kids, the coronary artery abnormalities were identified on the basis of a z score of 2.5 or above on the initial echocardiogram that was obtained at the time of admission. The coronary lesions were identified in 21% of affected children on or before day 5 of their illness, in 60% on or before day 7, and in 80% on or before day 10 of their illness.
The Colorado findings are supported by other fairly recent studies, according to Dr. Dominguez. An analysis of the Pediatric Health Information System database that included nearly 5,200 admissions for Kawasaki disease at 27 U.S. pediatric hospitals during 2001-2006 found that 3.3% of patients developed coronary artery aneurysms, 81% of which were detected during their initial hospitalization (Pediatrics 2009;124:1-8). And a Pediatric Heart Network study concluded that Kawasaki disease patients with a normal echocardiogram on admission had only a 6% incidence of developing coronary lesions at a later time, meaning that most coronary abnormalities were present at admission (Circulation 2007;116:174-9).
Intriguingly, fully 46% of Kawasaki disease patients in the Colorado study who had coronary lesions on admission had incomplete Kawasaki disease.
"That’s a much higher rate than we think of," Dr. Dominguez said. "It raises the concern that many cases of Kawasaki disease may currently be undiagnosed and not treated."
Incomplete Kawasaki disease is a diagnostic category that was created in the revised American Heart Association guidelines in an effort to identify subsets of Kawasaki disease patients earlier so treatment can be started expeditiously (Circulation 2004;110:2747-71). The revision was made in response to recognition that infants often fail to meet the classic diagnostic criteria for Kawasaki disease, yet they have a high incidence of coronary artery aneurysms.
The revised guidelines basically state that infants aged 6 months or younger on day 7 of fever without other explanation should undergo laboratory testing, even if they don’t have a generalized rash, bilateral nonexudative conjunctivitis, or any of the other clinical criteria for classic Kawasaki disease. If lab results yield evidence of systemic inflammation, then echocardiography is warranted (Pediatrics 2004;114:1708-33).
Dr. Dominguez said the clear implication of the Colorado study is that earlier diagnosis and treatment are needed in order to reduce the incidence of coronary artery abnormalities in children with Kawasaki disease. Increased clinical suspicion, greater use of the published algorithm for incomplete Kawasaki disease, and earlier resort to echocardiography in the initial work-up may result in more rapid therapy.
Although there is no guarantee that earlier diagnosis and treatment will prevent coronary lesions, that is the hope, he added.
The Children’s Hospital Colorado study was recently published (Pediatr. Infect. Dis. J. 2012 July 3 [doi:10.1097/INF.0b013e318266bcf9]).
Dr. Dominguez reported having no financial conflicts.
VAIL, COLO. – The majority of coronary artery abnormalities occurring in a large series of children with Kawasaki disease were detected at the time of hospital admission, prior to treatment.
What this means is that, contrary to the conventional wisdom, there is no such thing as a safe window for diagnosis and treatment of Kawasaki disease, Dr. Samuel R. Dominguez stressed at a conference on pediatric infectious diseases sponsored by the Children’s Hospital Colorado.
The "safe window" concept dates back to a classic 26-year-old study that concluded that treating patients with Kawasaki disease by day 10 of their illness greatly reduced the incidence of coronary artery aneurysms, the most serious disease complication (N. Engl. J. Med. 1986;315:341-7).
Anecdotal experience to the contrary convinced Dr. Dominguez and coworkers at Children’s Hospital Colorado in Aurora that it was time to take a formal look at their institutional experience.
"It was our gestalt that some kids had coronary artery abnormalities much earlier in the course of their illness than what we’d thought from the literature. We had this growing sense that the development of coronary artery lesions was less common after discharge than we’d initially thought," the pediatric infectious disease specialist explained.
That proved to be the case.
Among all 210 patients who were admitted for Kawasaki disease over a 4-year period (all of whom were appropriately treated with intravenous immunoglobulin and aspirin), 27% had coronary artery abnormalities detected during their acute illness or subsequent outpatient follow-up. In 81% of affected kids, the coronary artery abnormalities were identified on the basis of a z score of 2.5 or above on the initial echocardiogram that was obtained at the time of admission. The coronary lesions were identified in 21% of affected children on or before day 5 of their illness, in 60% on or before day 7, and in 80% on or before day 10 of their illness.
The Colorado findings are supported by other fairly recent studies, according to Dr. Dominguez. An analysis of the Pediatric Health Information System database that included nearly 5,200 admissions for Kawasaki disease at 27 U.S. pediatric hospitals during 2001-2006 found that 3.3% of patients developed coronary artery aneurysms, 81% of which were detected during their initial hospitalization (Pediatrics 2009;124:1-8). And a Pediatric Heart Network study concluded that Kawasaki disease patients with a normal echocardiogram on admission had only a 6% incidence of developing coronary lesions at a later time, meaning that most coronary abnormalities were present at admission (Circulation 2007;116:174-9).
Intriguingly, fully 46% of Kawasaki disease patients in the Colorado study who had coronary lesions on admission had incomplete Kawasaki disease.
"That’s a much higher rate than we think of," Dr. Dominguez said. "It raises the concern that many cases of Kawasaki disease may currently be undiagnosed and not treated."
Incomplete Kawasaki disease is a diagnostic category that was created in the revised American Heart Association guidelines in an effort to identify subsets of Kawasaki disease patients earlier so treatment can be started expeditiously (Circulation 2004;110:2747-71). The revision was made in response to recognition that infants often fail to meet the classic diagnostic criteria for Kawasaki disease, yet they have a high incidence of coronary artery aneurysms.
The revised guidelines basically state that infants aged 6 months or younger on day 7 of fever without other explanation should undergo laboratory testing, even if they don’t have a generalized rash, bilateral nonexudative conjunctivitis, or any of the other clinical criteria for classic Kawasaki disease. If lab results yield evidence of systemic inflammation, then echocardiography is warranted (Pediatrics 2004;114:1708-33).
Dr. Dominguez said the clear implication of the Colorado study is that earlier diagnosis and treatment are needed in order to reduce the incidence of coronary artery abnormalities in children with Kawasaki disease. Increased clinical suspicion, greater use of the published algorithm for incomplete Kawasaki disease, and earlier resort to echocardiography in the initial work-up may result in more rapid therapy.
Although there is no guarantee that earlier diagnosis and treatment will prevent coronary lesions, that is the hope, he added.
The Children’s Hospital Colorado study was recently published (Pediatr. Infect. Dis. J. 2012 July 3 [doi:10.1097/INF.0b013e318266bcf9]).
Dr. Dominguez reported having no financial conflicts.
AT A CONFERENCE ON PEDIATRIC INFECTIOUS DISEASES SPONSORED BY THE CHILDREN'S HOSPITAL COLORADO
Major Finding: Among a large group of children with Kawasaki disease who developed coronary artery abnormalities during their acute illness or convalescence, the coronary abnormalities were noted on the initial echocardiogram obtained at the time of admission in 81% of cases.
Data Source: This was a retrospective study involving all 210 patients with Kawasaki disease at a single pediatric hospital during a 4-year period.
Disclosures: The presenter reported having no financial conflicts.

Enthesis-Related Arthritis Has Two Faces
BERLIN – Recent evidence strongly suggests that enthesis-related arthritis in adolescents has two distinct clinical phenotypes, according to Dr. John Ioannou of University College London.
"The traditional view of enthesis-related arthritis has been that peripheral joint involvement and entheseal disease are common at presentation and then sacroiliitis occurs after many years as a late feature. I think that’s a paradigm that has been borne out not to be true. We can confidently shred that up and throw it away," he declared at the annual European Congress of Rheumatology.

The increasing use of MRI in the evaluation of patients who meet revised International League of Associations for Rheumatology diagnostic criteria for enthesis-related arthritis (ERA) has demonstrated that sacroiliitis appears early in the course of the disease in many of them. This early-onset sacroiliitis doesn’t show up on standard radiography, he explained.
ERA is an understudied subtype of juvenile idiopathic arthritis. The true incidence and prevalence of ERA aren’t known. Dr. Ioannou and his coworkers are conducting a long-term observational study at University College London. Analysis of the first 68 patients in the ERA cohort who have undergone MRI scanning indicated that 36, or 62%, had axial disease while the remainder did not.
The axial phenotype of ERA is characterized by a strong association with HLA-B27-positivity; more than 80% of patients with axial ERA were HLA-B27 positive, as was true for only a minority of those with the peripheral ERA phenotype. Extra-articular manifestations of ERA – inflammatory bowel disease and acute anterior uveitis – occurred only in HLA-B27-positive patients with axial disease.
Patients who were unaffected by axial disease had a strong proclivity for ankle arthritis, which was present in 80% of that group, compared with one-fourth of patients with axial ERA.
Fifty-eight of the 68 patients in the ERA cohort are male. Their median age at disease onset was 11 years and 1 month. The median time from disease onset to the appearance of inflammatory spinal symptoms was 2 years and 8 months.
The London experience matches that described by Italian investigators at the University of Florence, who are following a cohort of 59 patients with ERA. Twenty-one of the 59 reported symptoms of inflammatory back pain beginning a median of 1 year and 3 months after onset of ERA. MRI revealed the presence of sacroiliitis in 17 of the 21, all of whom had negative X-rays of the sacroiliac joints. As in the London series, the Italian patients with axial involvement were far more likely to be HLA-B27-positive, while those without axial disease had a greater frequency of ankle arthritis (J. Rheumatol. 2010;37:2395-401).
Twenty-nine of the 68 patients in the London cohort are on anti–tumor necrosis factor (anti-TNF) therapy plus methotrexate or another nonbiologic disease-modifying antirheumatic drug.
"Our anecdotal experience is that anti-TNF therapy tends to work pretty well in patients with ERA, but the remission rate is low. We’ve not been able to discontinue anti-TNF therapy in our patients," Dr. Ioannou noted.
This is consistent with the experience of a Dutch group. They reported that two-thirds of 22 ERA patients treated with an anti-TNF agent had inactive arthritis and enthesis after 15 months; however, none was able to discontinue biologic therapy without flaring of their disease (J. Rheumatol. 2011;38:2258-63).
The pathogenic mechanisms driving the two clinical patterns of ERA aren’t understood yet. Defining those mechanisms will provide a rationale for deciding on the best treatments for patients with one clinical phenotype or the other. At present, treatment is largely based upon extrapolation from data gathered in treating other forms of juvenile idiopathic arthritis or adult ankylosing spondylitis, the rheumatologist said.
Dr. Ioannou reported having no financial conflicts.
BERLIN – Recent evidence strongly suggests that enthesis-related arthritis in adolescents has two distinct clinical phenotypes, according to Dr. John Ioannou of University College London.
"The traditional view of enthesis-related arthritis has been that peripheral joint involvement and entheseal disease are common at presentation and then sacroiliitis occurs after many years as a late feature. I think that’s a paradigm that has been borne out not to be true. We can confidently shred that up and throw it away," he declared at the annual European Congress of Rheumatology.
The increasing use of MRI in the evaluation of patients who meet revised International League of Associations for Rheumatology diagnostic criteria for enthesis-related arthritis (ERA) has demonstrated that sacroiliitis appears early in the course of the disease in many of them. This early-onset sacroiliitis doesn’t show up on standard radiography, he explained.
ERA is an understudied subtype of juvenile idiopathic arthritis. The true incidence and prevalence of ERA aren’t known. Dr. Ioannou and his coworkers are conducting a long-term observational study at University College London. Analysis of the first 68 patients in the ERA cohort who have undergone MRI scanning indicated that 36, or 62%, had axial disease while the remainder did not.
The axial phenotype of ERA is characterized by a strong association with HLA-B27-positivity; more than 80% of patients with axial ERA were HLA-B27 positive, as was true for only a minority of those with the peripheral ERA phenotype. Extra-articular manifestations of ERA – inflammatory bowel disease and acute anterior uveitis – occurred only in HLA-B27-positive patients with axial disease.
Patients who were unaffected by axial disease had a strong proclivity for ankle arthritis, which was present in 80% of that group, compared with one-fourth of patients with axial ERA.
Fifty-eight of the 68 patients in the ERA cohort are male. Their median age at disease onset was 11 years and 1 month. The median time from disease onset to the appearance of inflammatory spinal symptoms was 2 years and 8 months.
The London experience matches that described by Italian investigators at the University of Florence, who are following a cohort of 59 patients with ERA. Twenty-one of the 59 reported symptoms of inflammatory back pain beginning a median of 1 year and 3 months after onset of ERA. MRI revealed the presence of sacroiliitis in 17 of the 21, all of whom had negative X-rays of the sacroiliac joints. As in the London series, the Italian patients with axial involvement were far more likely to be HLA-B27-positive, while those without axial disease had a greater frequency of ankle arthritis (J. Rheumatol. 2010;37:2395-401).
Twenty-nine of the 68 patients in the London cohort are on anti–tumor necrosis factor (anti-TNF) therapy plus methotrexate or another nonbiologic disease-modifying antirheumatic drug.
"Our anecdotal experience is that anti-TNF therapy tends to work pretty well in patients with ERA, but the remission rate is low. We’ve not been able to discontinue anti-TNF therapy in our patients," Dr. Ioannou noted.
This is consistent with the experience of a Dutch group. They reported that two-thirds of 22 ERA patients treated with an anti-TNF agent had inactive arthritis and enthesis after 15 months; however, none was able to discontinue biologic therapy without flaring of their disease (J. Rheumatol. 2011;38:2258-63).
The pathogenic mechanisms driving the two clinical patterns of ERA aren’t understood yet. Defining those mechanisms will provide a rationale for deciding on the best treatments for patients with one clinical phenotype or the other. At present, treatment is largely based upon extrapolation from data gathered in treating other forms of juvenile idiopathic arthritis or adult ankylosing spondylitis, the rheumatologist said.
Dr. Ioannou reported having no financial conflicts.
BERLIN – Recent evidence strongly suggests that enthesis-related arthritis in adolescents has two distinct clinical phenotypes, according to Dr. John Ioannou of University College London.
"The traditional view of enthesis-related arthritis has been that peripheral joint involvement and entheseal disease are common at presentation and then sacroiliitis occurs after many years as a late feature. I think that’s a paradigm that has been borne out not to be true. We can confidently shred that up and throw it away," he declared at the annual European Congress of Rheumatology.
The increasing use of MRI in the evaluation of patients who meet revised International League of Associations for Rheumatology diagnostic criteria for enthesis-related arthritis (ERA) has demonstrated that sacroiliitis appears early in the course of the disease in many of them. This early-onset sacroiliitis doesn’t show up on standard radiography, he explained.
ERA is an understudied subtype of juvenile idiopathic arthritis. The true incidence and prevalence of ERA aren’t known. Dr. Ioannou and his coworkers are conducting a long-term observational study at University College London. Analysis of the first 68 patients in the ERA cohort who have undergone MRI scanning indicated that 36, or 62%, had axial disease while the remainder did not.
The axial phenotype of ERA is characterized by a strong association with HLA-B27-positivity; more than 80% of patients with axial ERA were HLA-B27 positive, as was true for only a minority of those with the peripheral ERA phenotype. Extra-articular manifestations of ERA – inflammatory bowel disease and acute anterior uveitis – occurred only in HLA-B27-positive patients with axial disease.
Patients who were unaffected by axial disease had a strong proclivity for ankle arthritis, which was present in 80% of that group, compared with one-fourth of patients with axial ERA.
Fifty-eight of the 68 patients in the ERA cohort are male. Their median age at disease onset was 11 years and 1 month. The median time from disease onset to the appearance of inflammatory spinal symptoms was 2 years and 8 months.
The London experience matches that described by Italian investigators at the University of Florence, who are following a cohort of 59 patients with ERA. Twenty-one of the 59 reported symptoms of inflammatory back pain beginning a median of 1 year and 3 months after onset of ERA. MRI revealed the presence of sacroiliitis in 17 of the 21, all of whom had negative X-rays of the sacroiliac joints. As in the London series, the Italian patients with axial involvement were far more likely to be HLA-B27-positive, while those without axial disease had a greater frequency of ankle arthritis (J. Rheumatol. 2010;37:2395-401).
Twenty-nine of the 68 patients in the London cohort are on anti–tumor necrosis factor (anti-TNF) therapy plus methotrexate or another nonbiologic disease-modifying antirheumatic drug.
"Our anecdotal experience is that anti-TNF therapy tends to work pretty well in patients with ERA, but the remission rate is low. We’ve not been able to discontinue anti-TNF therapy in our patients," Dr. Ioannou noted.
This is consistent with the experience of a Dutch group. They reported that two-thirds of 22 ERA patients treated with an anti-TNF agent had inactive arthritis and enthesis after 15 months; however, none was able to discontinue biologic therapy without flaring of their disease (J. Rheumatol. 2011;38:2258-63).
The pathogenic mechanisms driving the two clinical patterns of ERA aren’t understood yet. Defining those mechanisms will provide a rationale for deciding on the best treatments for patients with one clinical phenotype or the other. At present, treatment is largely based upon extrapolation from data gathered in treating other forms of juvenile idiopathic arthritis or adult ankylosing spondylitis, the rheumatologist said.
Dr. Ioannou reported having no financial conflicts.
EXPERT ANALYSIS FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY

GnRH Antagonist Improves Rheumatoid Arthritis
HOUSTON – Use of a gonadotropin-releasing hormone antagonist showed early promise as a novel therapy for rheumatoid arthritis in a brief, proof-of-concept study that was presented at the annual meeting of the Endocrine Society.
Five days of subcutaneous injections of cetrorelix (Cetrotide) that were aimed at rapidly reducing levels of luteinizing hormone and follicle-stimulating hormone safely improved signs and symptoms of RA and reduced tumor necrosis factor–alpha levels in a 99-patient, placebo-controlled, randomized trial. Larger and longer-term clinical trials of GnRH antagonists as a potential treatment for RA are planned, according to Dr. Anita Kass of Betanien Hospital in Skien, Norway.
All study participants had active RA and were on stable doses of disease-modifying antirheumatic drugs. They were randomized to placebo or subcutaneous cetrorelix dosed at 5 mg on days 1-3 and 3 mg on days 4-5.
Outcomes were assessed on day 5, when LH and FSH levels in the cetrorelix arm were at their nadir. At that time, an ACR 20 (American College of Rheumatology scale, based on a 20% improvement in specified criteria) response was documented in 40% of patients on cetrorelix, compared with 18% of controls. Remission according to the DAS28 (Disease Activity Score, including a 28-joint count) was achieved in 13% of patients on the GnRH antagonist, compared with none on placebo. The erythrocyte sedimentation rate had declined by a mean of 1.06 mm/hour, compared with baseline, in the active treatment group, compared with a 5.04-mm/hour increase in controls.
In addition, the cetrorelix group had a mean 0.58 log pg/mL decrease in TNF-alpha compared with baseline, whereas controls experienced a 0.02 log pg/mL reduction. All of these differences between the cetrorelix and control groups were statistically significant.
The reduction in DAS28 score by day 5 was 0.82 in the cetrorelix group and 0.57 in controls, a trend that didn’t reach significance.
Adverse events in the two study arms did not differ.
Dr. Kass credited the impetus for this line of clinical investigation to earlier work by other investigators at the Weizmann Institute of Science in Rehovot, Israel. They showed that GnRH and GnRH-II are produced by T cells, and that the hormones have roles that extend beyond the reproductive system to include immune system regulation. The hormones trigger T-cell chemotaxis and homing to specific organs (Nat. Med. 2002;8:1421-6).
This proof-of-concept study was supported by the Norwegian Health and Rehabilitation Organization. The presenter reported having no financial conflicts.
HOUSTON – Use of a gonadotropin-releasing hormone antagonist showed early promise as a novel therapy for rheumatoid arthritis in a brief, proof-of-concept study that was presented at the annual meeting of the Endocrine Society.
Five days of subcutaneous injections of cetrorelix (Cetrotide) that were aimed at rapidly reducing levels of luteinizing hormone and follicle-stimulating hormone safely improved signs and symptoms of RA and reduced tumor necrosis factor–alpha levels in a 99-patient, placebo-controlled, randomized trial. Larger and longer-term clinical trials of GnRH antagonists as a potential treatment for RA are planned, according to Dr. Anita Kass of Betanien Hospital in Skien, Norway.
All study participants had active RA and were on stable doses of disease-modifying antirheumatic drugs. They were randomized to placebo or subcutaneous cetrorelix dosed at 5 mg on days 1-3 and 3 mg on days 4-5.
Outcomes were assessed on day 5, when LH and FSH levels in the cetrorelix arm were at their nadir. At that time, an ACR 20 (American College of Rheumatology scale, based on a 20% improvement in specified criteria) response was documented in 40% of patients on cetrorelix, compared with 18% of controls. Remission according to the DAS28 (Disease Activity Score, including a 28-joint count) was achieved in 13% of patients on the GnRH antagonist, compared with none on placebo. The erythrocyte sedimentation rate had declined by a mean of 1.06 mm/hour, compared with baseline, in the active treatment group, compared with a 5.04-mm/hour increase in controls.
In addition, the cetrorelix group had a mean 0.58 log pg/mL decrease in TNF-alpha compared with baseline, whereas controls experienced a 0.02 log pg/mL reduction. All of these differences between the cetrorelix and control groups were statistically significant.
The reduction in DAS28 score by day 5 was 0.82 in the cetrorelix group and 0.57 in controls, a trend that didn’t reach significance.
Adverse events in the two study arms did not differ.
Dr. Kass credited the impetus for this line of clinical investigation to earlier work by other investigators at the Weizmann Institute of Science in Rehovot, Israel. They showed that GnRH and GnRH-II are produced by T cells, and that the hormones have roles that extend beyond the reproductive system to include immune system regulation. The hormones trigger T-cell chemotaxis and homing to specific organs (Nat. Med. 2002;8:1421-6).
This proof-of-concept study was supported by the Norwegian Health and Rehabilitation Organization. The presenter reported having no financial conflicts.
HOUSTON – Use of a gonadotropin-releasing hormone antagonist showed early promise as a novel therapy for rheumatoid arthritis in a brief, proof-of-concept study that was presented at the annual meeting of the Endocrine Society.
Five days of subcutaneous injections of cetrorelix (Cetrotide) that were aimed at rapidly reducing levels of luteinizing hormone and follicle-stimulating hormone safely improved signs and symptoms of RA and reduced tumor necrosis factor–alpha levels in a 99-patient, placebo-controlled, randomized trial. Larger and longer-term clinical trials of GnRH antagonists as a potential treatment for RA are planned, according to Dr. Anita Kass of Betanien Hospital in Skien, Norway.
All study participants had active RA and were on stable doses of disease-modifying antirheumatic drugs. They were randomized to placebo or subcutaneous cetrorelix dosed at 5 mg on days 1-3 and 3 mg on days 4-5.
Outcomes were assessed on day 5, when LH and FSH levels in the cetrorelix arm were at their nadir. At that time, an ACR 20 (American College of Rheumatology scale, based on a 20% improvement in specified criteria) response was documented in 40% of patients on cetrorelix, compared with 18% of controls. Remission according to the DAS28 (Disease Activity Score, including a 28-joint count) was achieved in 13% of patients on the GnRH antagonist, compared with none on placebo. The erythrocyte sedimentation rate had declined by a mean of 1.06 mm/hour, compared with baseline, in the active treatment group, compared with a 5.04-mm/hour increase in controls.
In addition, the cetrorelix group had a mean 0.58 log pg/mL decrease in TNF-alpha compared with baseline, whereas controls experienced a 0.02 log pg/mL reduction. All of these differences between the cetrorelix and control groups were statistically significant.
The reduction in DAS28 score by day 5 was 0.82 in the cetrorelix group and 0.57 in controls, a trend that didn’t reach significance.
Adverse events in the two study arms did not differ.
Dr. Kass credited the impetus for this line of clinical investigation to earlier work by other investigators at the Weizmann Institute of Science in Rehovot, Israel. They showed that GnRH and GnRH-II are produced by T cells, and that the hormones have roles that extend beyond the reproductive system to include immune system regulation. The hormones trigger T-cell chemotaxis and homing to specific organs (Nat. Med. 2002;8:1421-6).
This proof-of-concept study was supported by the Norwegian Health and Rehabilitation Organization. The presenter reported having no financial conflicts.
AT THE ANNUAL MEETING OF ENDOCRINE SOCIETY
Major Finding: Inhibition of GnRH improved the signs and symptoms of RA and reduced TNF-alpha levels.
Data Source: This was an exploratory pilot study involving 99 patients with active RA who were randomized to 5 days of the GnRH antagonist cetrorelix or placebo.
Disclosures: This proof-of-concept study was supported by the Norwegian Health and Rehabilitation Organization. The presenter reported having no financial conflicts.
Self-Testing Helps Stabilize Warfarin Therapeutic Range
CHICAGO ? The new oral anticoagulants for stroke prevention in atrial fibrillation may be garnering all the buzz, but don?t count out the old standby warfarin yet.
"It?s not just a knee-jerk reaction that all patients should be switched to the new agents. It?s dependent upon how well you as a physician are managing your patients on warfarin," Dr. Jack E. Ansell asserted at the annual meeting of the American College of Cardiology.

"Warfarin therapy is all about management. If it?s not managed well, you can compare it to anything, and anything is going to be better. And if it?s managed very well, then it?s very difficult to beat warfarin therapy," said Dr. Ansell, who is the chairman of the department of medicine at Lenox Hill Hospital in New York.
A growing body of evidence indicates that the new standard in high-quality management of warfarin therapy involves patient self-testing of International Normalized Ratios (INR) at home using a fingerstick blood sample and a portable point-of-care device.
As a case in point: Dr. Ansell presented highlights of the new STABLE study, in which he and his coinvestigators conducted a retrospective analysis of the real-world experience of more than 29,000 warfarin-treated patients enrolled in a national commercial comprehensive self-test support service. The study was reported in JACC 2012 March 27 [doi: 10.1016/S0735-1097(12)61865-8]).
Patients who performed frequent self-testing ? meaning more than 80% of their self-testing was done on a weekly basis ? had a mean time spent in the therapeutic INR range (TTR) of 74%. That?s unprecedented, he said.
By comparison, in the pivotal RE-LY randomized trial for dabigatran (Pradaxa), the control group on warfarin had a TTR of 64% (N. Engl. J. Med. 2009;361:1139-51). In the ROCKET-AF trial of rivaroxaban (Xarelto), warfarin controls had a TTR of 55% (N. Engl. J. Med. 2011;365:883-91). And in the ARISTOTLE study of apixaban (Eliquis), an agent expected to soon receive Food and Drug Administration marketing approval, the warfarin control group had a TTR of 62% (N. Engl. J. Med. 2011;365:981-92).
In all these major randomized trials involving the novel oral anticoagulants, patients assigned to warfarin were closely managed, but in traditional fashion ? home self-testing wasn?t involved.
In contrast, in the STABLE study, the overall TTR, including those patients who self-tested variably and inconsistently, was still 69.7%.
"This is important because the cost-effectiveness analyses done with dabigatran and the other new anticoagulants suggest that when you get up to a TTR above 70% with warfarin, the cost-effectiveness of the new agents diminishes and warfarin actually becomes more cost-effective," according to Dr. Ansell.
A particularly impressive finding in STABLE was that patients who did weekly self-testing had a 2.3% incidence of critical value INR results, defined as an INR below 1.5 or greater than 5.0. "This is really a phenomenally low result," he commented. It represented a 48% reduction from the 4.4% incidence in patients with variable self-testing frequency.
Participants in the STABLE study tested themselves at home, but their warfarin dosing was managed by their referring physicians or anticoagulation clinics. Thus, an individual?s TTR reflected the warfarin management expertise of the referral source.
There are several reasons why home monitoring achieves better TTRs and ? as shown in other studies ? lower major bleeding and thrombotic event rates than with usual care or anticoagulation clinics not utilizing patient self-monitoring, Dr. Ansell said.
Home testing is more frequent, timely, and consistent, and the immediate feedback regarding INR results is likely to promote adherence.
A variant of patient self-testing starting to catch on in the United States is patient self-management. This entails teaching patients how to manage their own warfarin dose on the basis of their home INR measurements.
The most recent American College of Chest Physicians clinical practice guidelines on antithrombotic therapy for atrial fibrillation give patient self-management of warfarin therapy a class 2B recommendation, stating, "For patients treated with vitamin K antagonists who are motivated and can demonstrate competency in self-management strategies, including the self-testing equipment, we suggest patient self-management rather than the usual outpatient INR monitoring" (CHEST 2012;141: 2 suppl. e531S-e575S [doi: 10.1378/chest.11-2304]).
Session cochair Dr. Samuel Z. Goldhaber agreed with Dr. Ansell that warfarin still has a place in anticoagulation therapy. The fact that it costs as little as $4 per month while dabigatran, for example, retails for 60 times that amount, is not to be shrugged off in an era of runaway health care spending, he said.
Plus, warfarin, for all its drawbacks, is a known quantity backed by more than a half century of clinical experience.
"Even though warfarin can cause horrible complications, there are no more surprises left about what warfarin can do," observed Dr. Goldhaber, who is a professor of medicine at Harvard Medical School and director of the venous thromboembolism research group at Brigham and Women?s Hospital, Boston.
A potential game changer for warfarin is the possibility that rapid pharmacogenetic testing will enable physicians to improve upon the current method of warfarin dosing.
One advantage warfarin has is that bleeding episodes can be reversed by administration of vitamin K. In contrast, there is as yet no reliable means of reversing major bleeding in patients on the novel anticoagulants. But Dr. Lars Wallentin said this limitation of the new agents is outweighed by the consistent finding that they have lower rates of intracranial hemorrhage than those of warfarin.
"There is no antidote to warfarin that has proven to have any effect in patients with ICH. And there is no evidence as far as I can see that however you control INR you can reach as low a level of ICH as with these new agents. I think this is a specific downside of warfarin that we can?t get away from," said Dr. Wallentin, who is a professor of cardiology at Uppsala (Sweden) University.
The STABLE study was funded by Alere Home Monitoring, Inc. Dr. Ansell is a consultant to the company.
Dr. Goldhaber has served as a consultant to numerous pharmaceutical companies developing cardiovascular medications.
Dr. Wallentin was principal investigator in the ARISTOTLE study of apixaban, funded by Pfizer and Bristol Myers Squibb, and has served as a consultant to those and other pharmaceutical companies.
CHICAGO ? The new oral anticoagulants for stroke prevention in atrial fibrillation may be garnering all the buzz, but don?t count out the old standby warfarin yet.
"It?s not just a knee-jerk reaction that all patients should be switched to the new agents. It?s dependent upon how well you as a physician are managing your patients on warfarin," Dr. Jack E. Ansell asserted at the annual meeting of the American College of Cardiology.
"Warfarin therapy is all about management. If it?s not managed well, you can compare it to anything, and anything is going to be better. And if it?s managed very well, then it?s very difficult to beat warfarin therapy," said Dr. Ansell, who is the chairman of the department of medicine at Lenox Hill Hospital in New York.
A growing body of evidence indicates that the new standard in high-quality management of warfarin therapy involves patient self-testing of International Normalized Ratios (INR) at home using a fingerstick blood sample and a portable point-of-care device.
As a case in point: Dr. Ansell presented highlights of the new STABLE study, in which he and his coinvestigators conducted a retrospective analysis of the real-world experience of more than 29,000 warfarin-treated patients enrolled in a national commercial comprehensive self-test support service. The study was reported in JACC 2012 March 27 [doi: 10.1016/S0735-1097(12)61865-8]).
Patients who performed frequent self-testing ? meaning more than 80% of their self-testing was done on a weekly basis ? had a mean time spent in the therapeutic INR range (TTR) of 74%. That?s unprecedented, he said.
By comparison, in the pivotal RE-LY randomized trial for dabigatran (Pradaxa), the control group on warfarin had a TTR of 64% (N. Engl. J. Med. 2009;361:1139-51). In the ROCKET-AF trial of rivaroxaban (Xarelto), warfarin controls had a TTR of 55% (N. Engl. J. Med. 2011;365:883-91). And in the ARISTOTLE study of apixaban (Eliquis), an agent expected to soon receive Food and Drug Administration marketing approval, the warfarin control group had a TTR of 62% (N. Engl. J. Med. 2011;365:981-92).
In all these major randomized trials involving the novel oral anticoagulants, patients assigned to warfarin were closely managed, but in traditional fashion ? home self-testing wasn?t involved.
In contrast, in the STABLE study, the overall TTR, including those patients who self-tested variably and inconsistently, was still 69.7%.
"This is important because the cost-effectiveness analyses done with dabigatran and the other new anticoagulants suggest that when you get up to a TTR above 70% with warfarin, the cost-effectiveness of the new agents diminishes and warfarin actually becomes more cost-effective," according to Dr. Ansell.
A particularly impressive finding in STABLE was that patients who did weekly self-testing had a 2.3% incidence of critical value INR results, defined as an INR below 1.5 or greater than 5.0. "This is really a phenomenally low result," he commented. It represented a 48% reduction from the 4.4% incidence in patients with variable self-testing frequency.
Participants in the STABLE study tested themselves at home, but their warfarin dosing was managed by their referring physicians or anticoagulation clinics. Thus, an individual?s TTR reflected the warfarin management expertise of the referral source.
There are several reasons why home monitoring achieves better TTRs and ? as shown in other studies ? lower major bleeding and thrombotic event rates than with usual care or anticoagulation clinics not utilizing patient self-monitoring, Dr. Ansell said.
Home testing is more frequent, timely, and consistent, and the immediate feedback regarding INR results is likely to promote adherence.
A variant of patient self-testing starting to catch on in the United States is patient self-management. This entails teaching patients how to manage their own warfarin dose on the basis of their home INR measurements.
The most recent American College of Chest Physicians clinical practice guidelines on antithrombotic therapy for atrial fibrillation give patient self-management of warfarin therapy a class 2B recommendation, stating, "For patients treated with vitamin K antagonists who are motivated and can demonstrate competency in self-management strategies, including the self-testing equipment, we suggest patient self-management rather than the usual outpatient INR monitoring" (CHEST 2012;141: 2 suppl. e531S-e575S [doi: 10.1378/chest.11-2304]).
Session cochair Dr. Samuel Z. Goldhaber agreed with Dr. Ansell that warfarin still has a place in anticoagulation therapy. The fact that it costs as little as $4 per month while dabigatran, for example, retails for 60 times that amount, is not to be shrugged off in an era of runaway health care spending, he said.
Plus, warfarin, for all its drawbacks, is a known quantity backed by more than a half century of clinical experience.
"Even though warfarin can cause horrible complications, there are no more surprises left about what warfarin can do," observed Dr. Goldhaber, who is a professor of medicine at Harvard Medical School and director of the venous thromboembolism research group at Brigham and Women?s Hospital, Boston.
A potential game changer for warfarin is the possibility that rapid pharmacogenetic testing will enable physicians to improve upon the current method of warfarin dosing.
One advantage warfarin has is that bleeding episodes can be reversed by administration of vitamin K. In contrast, there is as yet no reliable means of reversing major bleeding in patients on the novel anticoagulants. But Dr. Lars Wallentin said this limitation of the new agents is outweighed by the consistent finding that they have lower rates of intracranial hemorrhage than those of warfarin.
"There is no antidote to warfarin that has proven to have any effect in patients with ICH. And there is no evidence as far as I can see that however you control INR you can reach as low a level of ICH as with these new agents. I think this is a specific downside of warfarin that we can?t get away from," said Dr. Wallentin, who is a professor of cardiology at Uppsala (Sweden) University.
The STABLE study was funded by Alere Home Monitoring, Inc. Dr. Ansell is a consultant to the company.
Dr. Goldhaber has served as a consultant to numerous pharmaceutical companies developing cardiovascular medications.
Dr. Wallentin was principal investigator in the ARISTOTLE study of apixaban, funded by Pfizer and Bristol Myers Squibb, and has served as a consultant to those and other pharmaceutical companies.
CHICAGO ? The new oral anticoagulants for stroke prevention in atrial fibrillation may be garnering all the buzz, but don?t count out the old standby warfarin yet.
"It?s not just a knee-jerk reaction that all patients should be switched to the new agents. It?s dependent upon how well you as a physician are managing your patients on warfarin," Dr. Jack E. Ansell asserted at the annual meeting of the American College of Cardiology.
"Warfarin therapy is all about management. If it?s not managed well, you can compare it to anything, and anything is going to be better. And if it?s managed very well, then it?s very difficult to beat warfarin therapy," said Dr. Ansell, who is the chairman of the department of medicine at Lenox Hill Hospital in New York.
A growing body of evidence indicates that the new standard in high-quality management of warfarin therapy involves patient self-testing of International Normalized Ratios (INR) at home using a fingerstick blood sample and a portable point-of-care device.
As a case in point: Dr. Ansell presented highlights of the new STABLE study, in which he and his coinvestigators conducted a retrospective analysis of the real-world experience of more than 29,000 warfarin-treated patients enrolled in a national commercial comprehensive self-test support service. The study was reported in JACC 2012 March 27 [doi: 10.1016/S0735-1097(12)61865-8]).
Patients who performed frequent self-testing ? meaning more than 80% of their self-testing was done on a weekly basis ? had a mean time spent in the therapeutic INR range (TTR) of 74%. That?s unprecedented, he said.
By comparison, in the pivotal RE-LY randomized trial for dabigatran (Pradaxa), the control group on warfarin had a TTR of 64% (N. Engl. J. Med. 2009;361:1139-51). In the ROCKET-AF trial of rivaroxaban (Xarelto), warfarin controls had a TTR of 55% (N. Engl. J. Med. 2011;365:883-91). And in the ARISTOTLE study of apixaban (Eliquis), an agent expected to soon receive Food and Drug Administration marketing approval, the warfarin control group had a TTR of 62% (N. Engl. J. Med. 2011;365:981-92).
In all these major randomized trials involving the novel oral anticoagulants, patients assigned to warfarin were closely managed, but in traditional fashion ? home self-testing wasn?t involved.
In contrast, in the STABLE study, the overall TTR, including those patients who self-tested variably and inconsistently, was still 69.7%.
"This is important because the cost-effectiveness analyses done with dabigatran and the other new anticoagulants suggest that when you get up to a TTR above 70% with warfarin, the cost-effectiveness of the new agents diminishes and warfarin actually becomes more cost-effective," according to Dr. Ansell.
A particularly impressive finding in STABLE was that patients who did weekly self-testing had a 2.3% incidence of critical value INR results, defined as an INR below 1.5 or greater than 5.0. "This is really a phenomenally low result," he commented. It represented a 48% reduction from the 4.4% incidence in patients with variable self-testing frequency.
Participants in the STABLE study tested themselves at home, but their warfarin dosing was managed by their referring physicians or anticoagulation clinics. Thus, an individual?s TTR reflected the warfarin management expertise of the referral source.
There are several reasons why home monitoring achieves better TTRs and ? as shown in other studies ? lower major bleeding and thrombotic event rates than with usual care or anticoagulation clinics not utilizing patient self-monitoring, Dr. Ansell said.
Home testing is more frequent, timely, and consistent, and the immediate feedback regarding INR results is likely to promote adherence.
A variant of patient self-testing starting to catch on in the United States is patient self-management. This entails teaching patients how to manage their own warfarin dose on the basis of their home INR measurements.
The most recent American College of Chest Physicians clinical practice guidelines on antithrombotic therapy for atrial fibrillation give patient self-management of warfarin therapy a class 2B recommendation, stating, "For patients treated with vitamin K antagonists who are motivated and can demonstrate competency in self-management strategies, including the self-testing equipment, we suggest patient self-management rather than the usual outpatient INR monitoring" (CHEST 2012;141: 2 suppl. e531S-e575S [doi: 10.1378/chest.11-2304]).
Session cochair Dr. Samuel Z. Goldhaber agreed with Dr. Ansell that warfarin still has a place in anticoagulation therapy. The fact that it costs as little as $4 per month while dabigatran, for example, retails for 60 times that amount, is not to be shrugged off in an era of runaway health care spending, he said.
Plus, warfarin, for all its drawbacks, is a known quantity backed by more than a half century of clinical experience.
"Even though warfarin can cause horrible complications, there are no more surprises left about what warfarin can do," observed Dr. Goldhaber, who is a professor of medicine at Harvard Medical School and director of the venous thromboembolism research group at Brigham and Women?s Hospital, Boston.
A potential game changer for warfarin is the possibility that rapid pharmacogenetic testing will enable physicians to improve upon the current method of warfarin dosing.
One advantage warfarin has is that bleeding episodes can be reversed by administration of vitamin K. In contrast, there is as yet no reliable means of reversing major bleeding in patients on the novel anticoagulants. But Dr. Lars Wallentin said this limitation of the new agents is outweighed by the consistent finding that they have lower rates of intracranial hemorrhage than those of warfarin.
"There is no antidote to warfarin that has proven to have any effect in patients with ICH. And there is no evidence as far as I can see that however you control INR you can reach as low a level of ICH as with these new agents. I think this is a specific downside of warfarin that we can?t get away from," said Dr. Wallentin, who is a professor of cardiology at Uppsala (Sweden) University.
The STABLE study was funded by Alere Home Monitoring, Inc. Dr. Ansell is a consultant to the company.
Dr. Goldhaber has served as a consultant to numerous pharmaceutical companies developing cardiovascular medications.
Dr. Wallentin was principal investigator in the ARISTOTLE study of apixaban, funded by Pfizer and Bristol Myers Squibb, and has served as a consultant to those and other pharmaceutical companies.