User login
Widespread Hyperkeratotic Papules in a Transplant Recipient
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
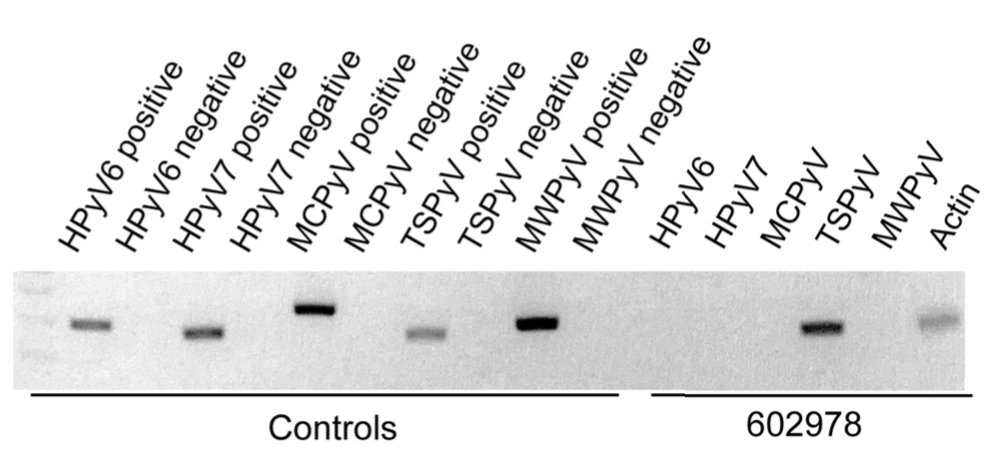
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
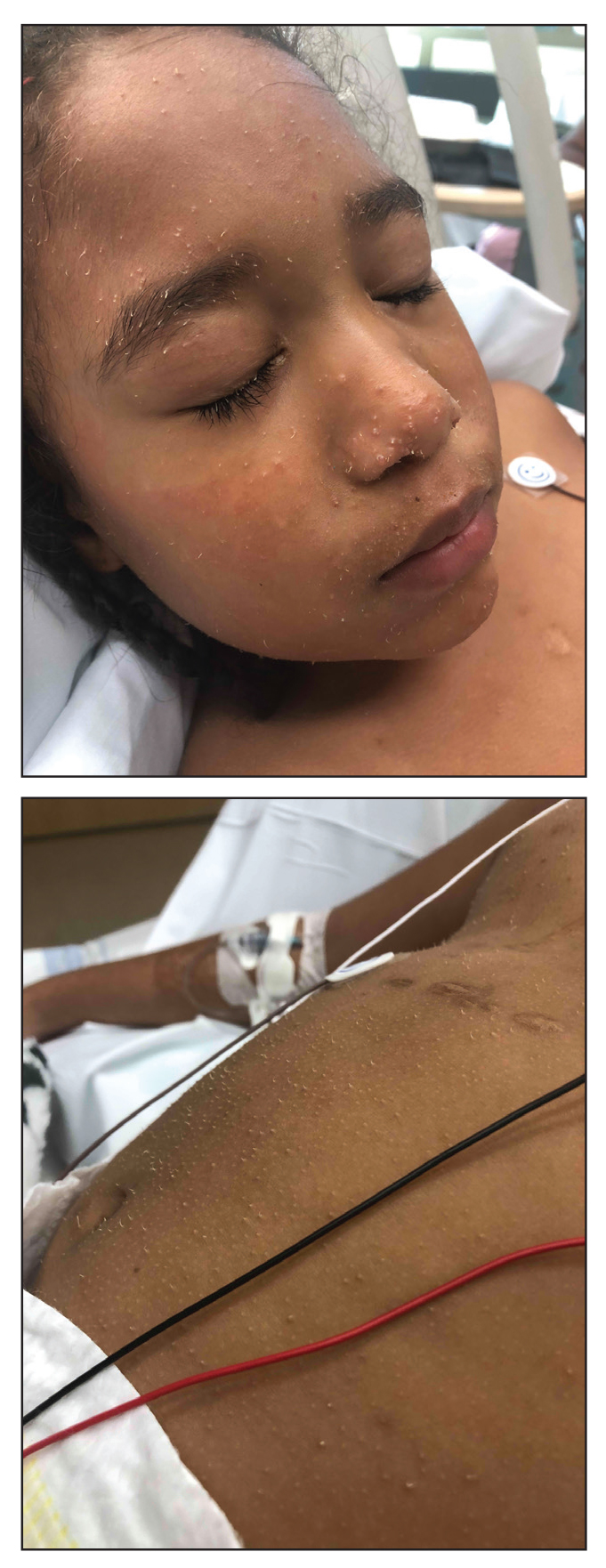
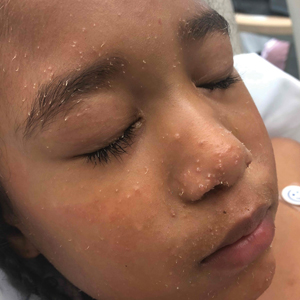
A 4-year-old girl with a history of cardiac transplantation 1 year prior for dilated cardiomyopathy presented to the dermatology consultation service with widespread hyperkeratotic papules of 2 months’ duration. The eruption initially had appeared on the face with subsequent involvement of the trunk and extremities. Her immunosuppressive medications included oral tacrolimus and mycophenolate mofetil. No over-the-counter or prescription treatments had been used for the eruption; the patient’s mother had been manually extracting the spicules from the nose, cheeks, and forehead with tweezers. The lesions were asymptomatic with only mild follicular erythema. Physical examination revealed multiple folliculocentric keratinous spicules on the nose, cheeks, forehead (top), trunk (bottom), arms, and legs.
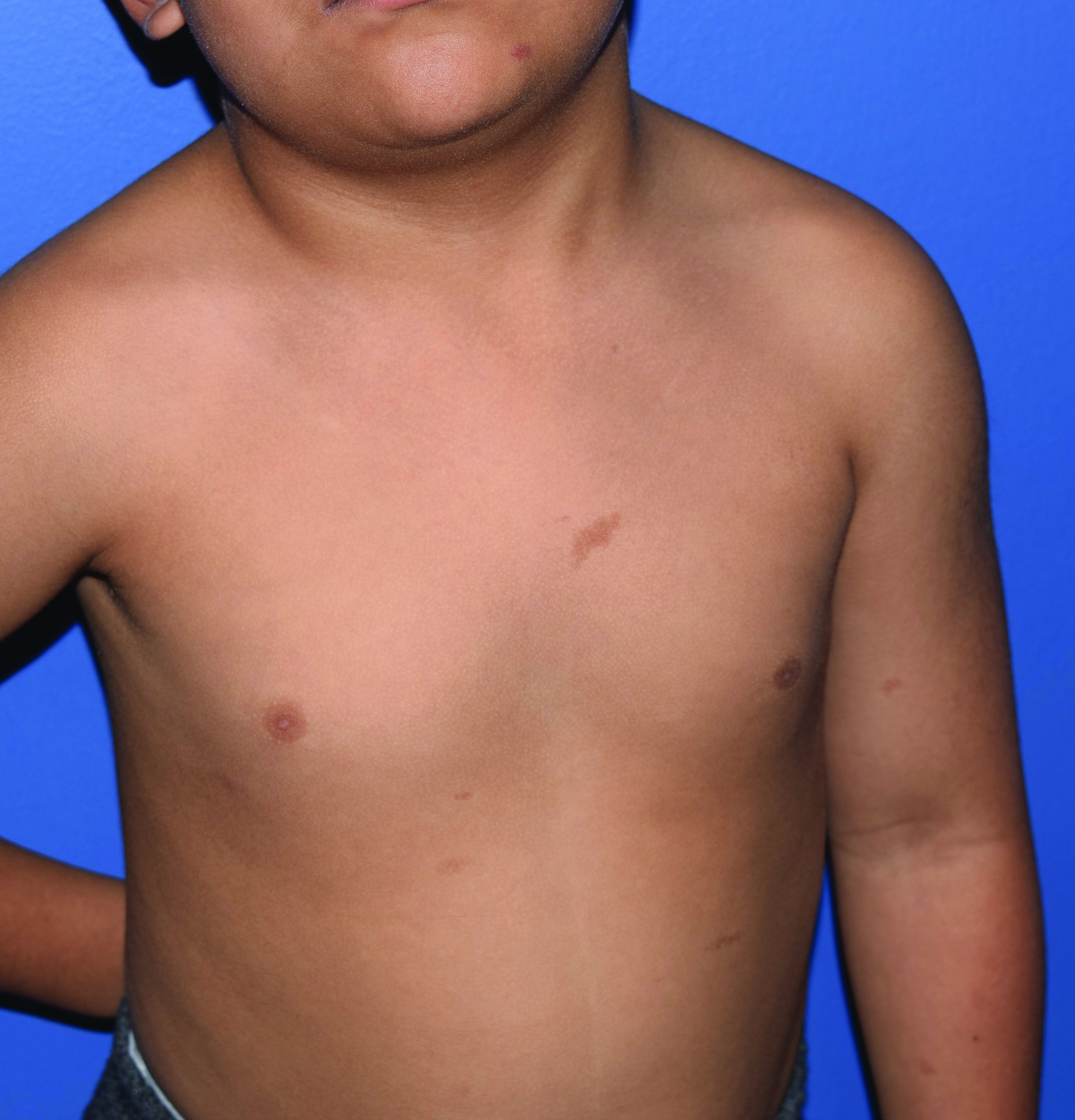
A 4-year-old presented to our pediatric dermatology clinic for evaluation of asymptomatic "brown spots."
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1

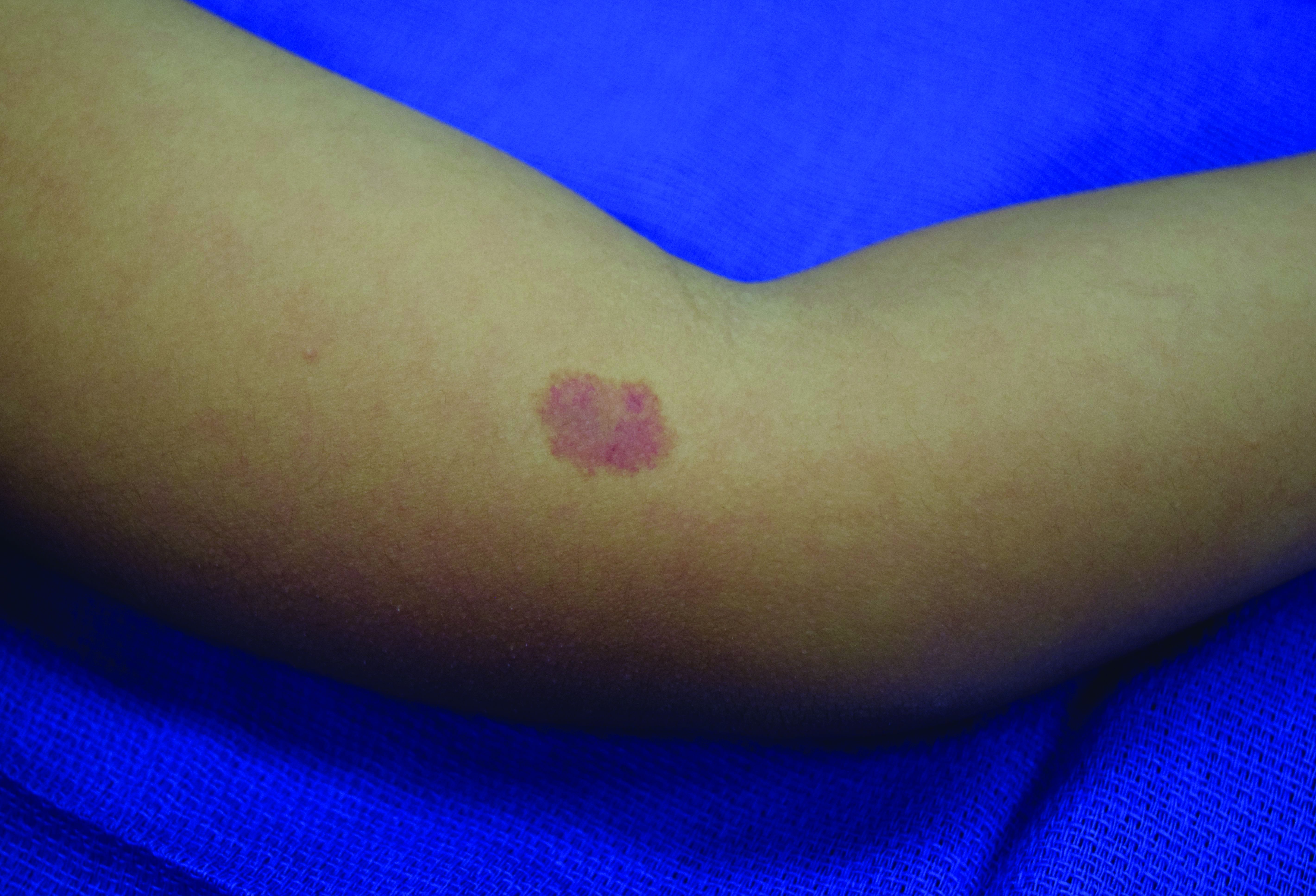
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4

It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.

Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4
It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.
Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4
It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.
Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.