User login
Bullous Amyloidosis Masquerading as Pseudoporphyria
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
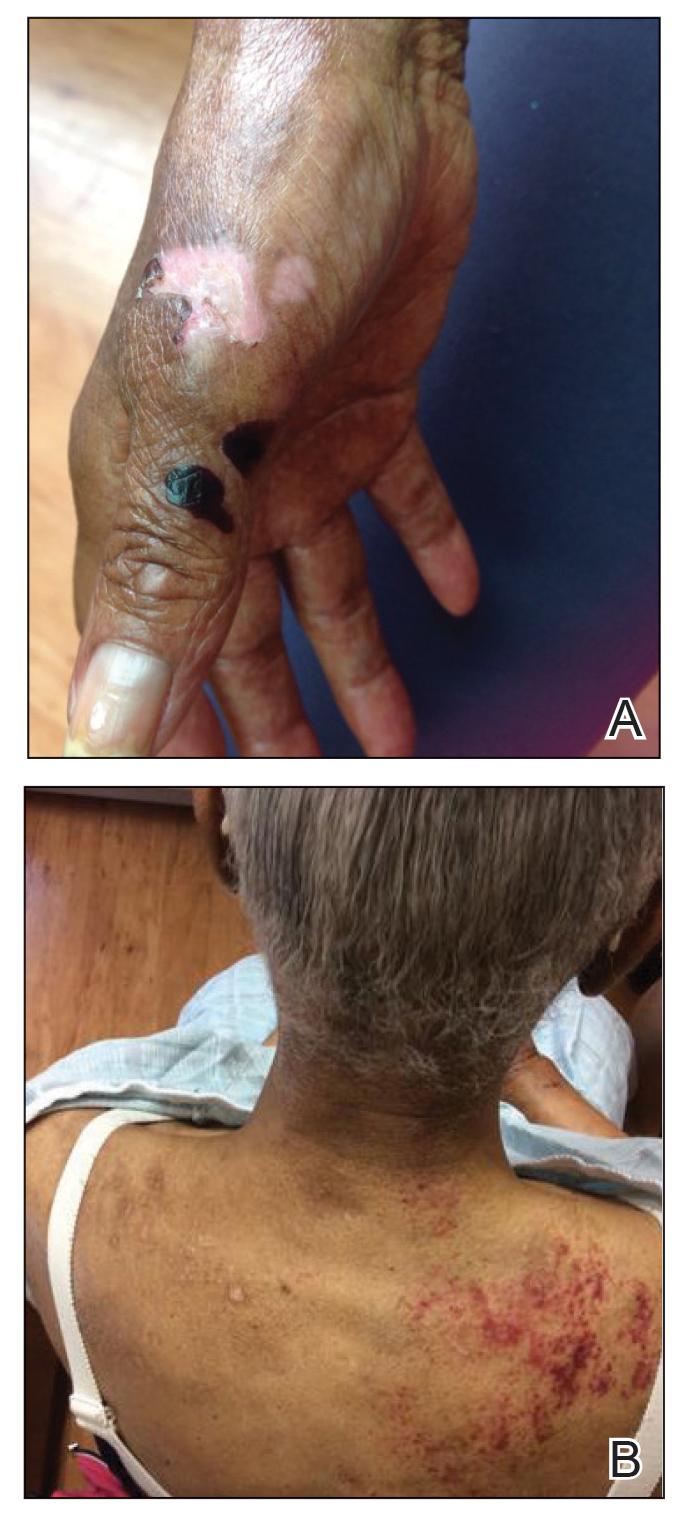
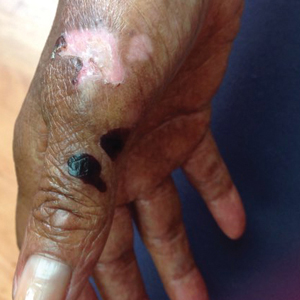
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.

Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
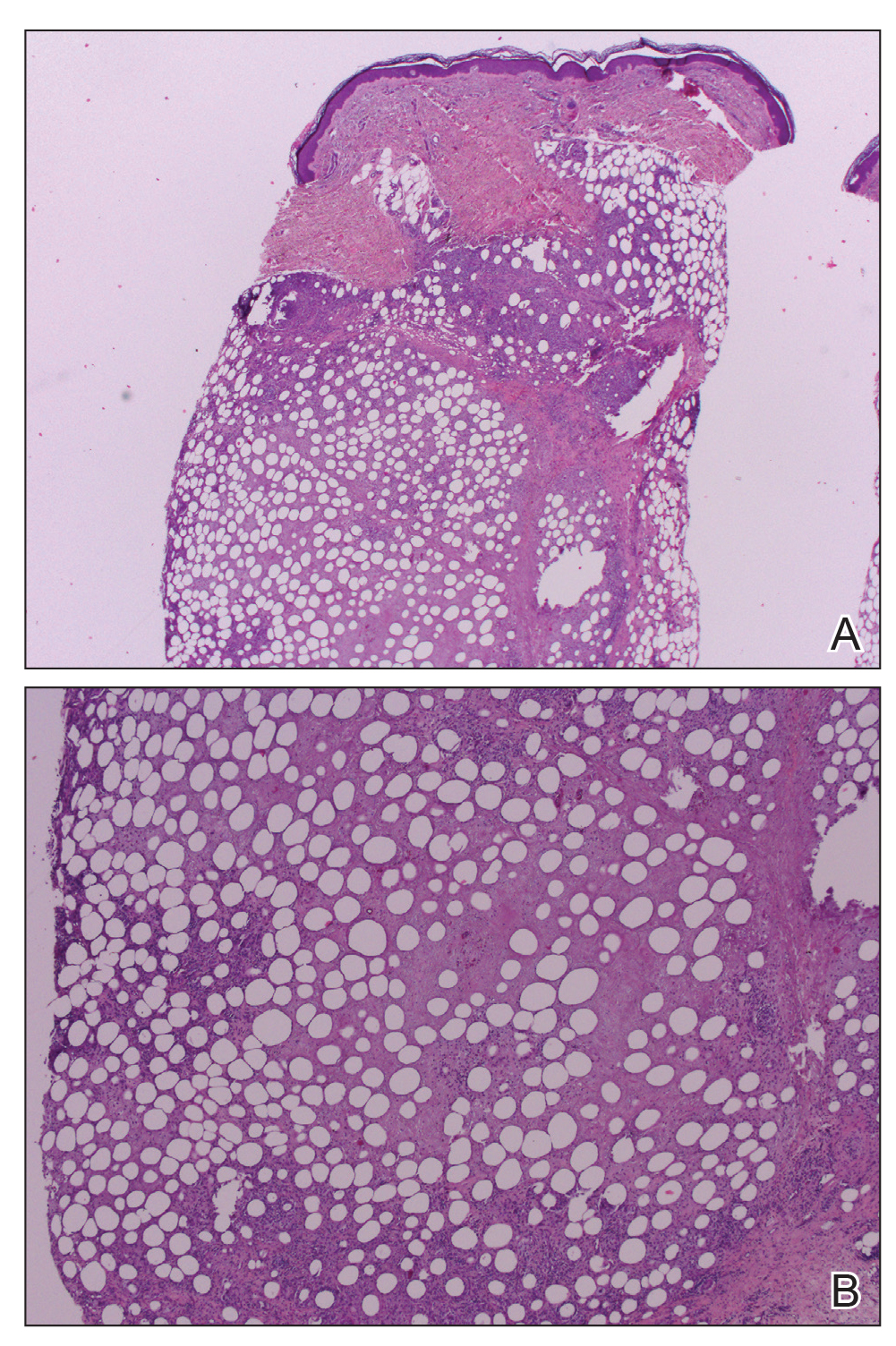
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Practice Points
- Primary systemic amyloidosis, including the rare cutaneous bullous amyloidosis, often is difficult to diagnose and has been associated with underlying plasma cell dyscrasia or multiple myeloma.
- When evaluating patients with initially convincing signs of pseudoporphyria, it is imperative to consider the diagnosis of bullous amyloidosis, which additionally can present with intraoral hemorrhagic vesicles and have confirmatory histopathologic features.
- Further investigation for multiple myeloma is warranted when patients with a chronic hemorrhagic bullous condition also present with symptoms of purpura, angina bullosa hemorrhagica, fatigue, weight loss, and/or macroglossia. Accurate diagnosis of bullous amyloidosis and timely treatment of its underlying cause will contribute to better, more proactive patient care.
Disseminated Erythema Induratum in a Patient With a History of Tuberculosis
To the Editor:
Erythema induratum, also known as nodular vasculitis, is a panniculitis that usually affects the lower extremities in middle-aged women. Classically, it has been described as a delayed-type hypersensitivity reaction to Mycobacterium tuberculosis, also known as a tuberculid.1,2 Other infections, however, also have been implicated as causes of erythema induratum, including bacillus Calmette-Guérin (BCG), the attenuated form of Mycobacterium bovis, which commonly is used for tuberculosis vaccination. Medications also may cause erythema induratum. The characteristic distribution of the nodules on the posterior calves helps to distinguish erythema induratum from other panniculitides. A PubMed search of articles indexed for MEDLINE using the term disseminated erythema induratum revealed few case reports documenting nodules on the arms, thighs, or chest, and only 1 case report of disseminated erythema induratum.3-8 We describe a rare combination of disseminated erythema induratum in a patient with remote exposure to tuberculosis and recent BCG exposure.
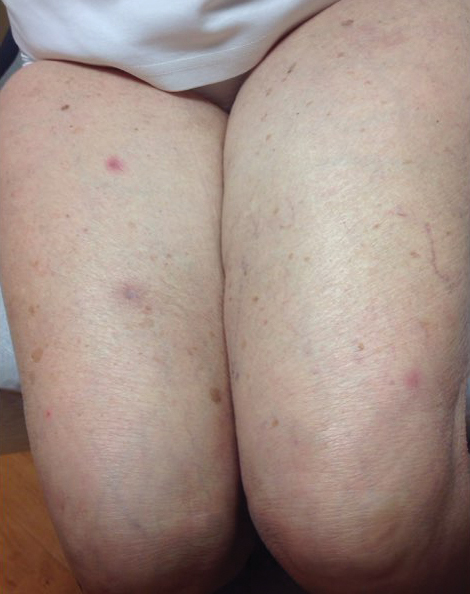
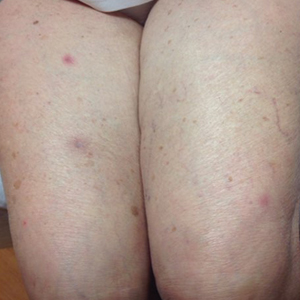
An 88-year-old woman presented for evaluation of violaceous, minimally tender, nonulcerated, subcutaneous nodules on the legs, arms, and trunk of several weeks’ duration (Figure 1). She had a remote history of tuberculosis as a child, prior to the advent of modern antituberculosis regimens. Her medical history also included hypertension, breast cancer treated with lymph node dissection, gastroesophageal reflux disease, and bladder cancer treated with intravesical BCG 10 years prior to the onset of the nodules. She reported minimal coughing and a 25-lb weight loss over the last year, but she denied night sweats, fever, or chills.
Workup included a biopsy, which showed a dense inflammatory infiltrate within the septae and lobules of the subcutaneous tissue (Figure 2A). Foci of necrosis were seen within the fat lobules (Figure 2B). The histologic diagnosis was erythema induratum. Tissue cultures for bacteria, fungi, and atypical mycobacteria were negative. Mycobacterium tuberculosis polymerase chain reaction (PCR) analysis also was negative. An IFN-γ release assay test was positive for infection with M tuberculosis, suggesting that the erythema induratum was due to tuberculosis rather than BCG exposure. A chest radiograph demonstrated a 22-mm nodule in the left lung (unchanged from a prior film) and a new 10-mm nodule in the left upper lobe.
The patient was referred to an infectious disease specialist who concurred that the erythema induratum and the new lung nodule likely represented a reactivation of tuberculosis. Sputum samples were found to be smear and culture negative for mycobacteria, but due to high clinical suspicion, she was started on a 4-drug tuberculosis regimen of isoniazid, rifampin, pyrazinamide, and ethambutol. Some lesions had started to improve prior to the institution of therapy; after initiation of treatment, all lesions resolved within 4 weeks of starting treatment without recurrence.
Erythema induratum was first described by Bazin9 in 1861. The disorder usually occurs in middle-aged women and is characterized by violaceous ulcerative plaques that classically present on the lower extremities, especially the calves. When the eruption occurs due to a nontuberculous etiology, the term nodular vasculitis is used.1,5 The distinction largely is historical, as most dermatologists today recognize erythema induratum and nodular vasculitis to be the same entity. Examples of nontuberculous causes include infections such as Nocardia, Pseudomonas, Fusarium, or other Mycobacterium species.10 Medications such as propylthiouracil also have been implicated.11 The classification of erythema induratum as a tuberculid suggests that the nodules are a reaction pattern rather than a primary infection, though the term tuberculid may be imprecise. The differential diagnosis of violaceous nodules on the lower extremities and trunk is broad and includes erythema nodosum, cutaneous polyarteritis nodosa, pancreatic panniculitis, subcutaneous T-cell lymphoma, and lupus profundus.1,11,12
Histologically, lesions classically demonstrate a mostly lobular panniculitis with varying degrees of septal fibrosis and focal necrosis. Neutrophils may predominate early, while adipocyte necrosis, epithelioid histiocytes, multinucleated giant cells, and lymphocytes may be found in older lesions. The presence of vasculitis as a requisite diagnostic criterion remains controversial.1,12
The incidence of erythema induratum has decreased since multidrug tuberculosis treatment has become more widespread.3 Our case displayed the disseminated variant of erythema induratum, an even rarer clinical entity.8 Interestingly, our patient had a history of tuberculosis and exposure to BCG prior to the development of lesions. Case reports have documented erythema induratum after BCG exposure but less frequently than in cases associated with tuberculosis.3,13
The use of BCG vaccines has necessitated the need for a more precise method of determining tuberculosis activity. The tuberculin skin test reacts positively with a history of BCG exposure, rendering it an inadequate test in a patient who is suspected of having an active or latent M tuberculosis infection.13,14 IFN-γ release assays are more specific in detecting latent or active tuberculosis than the tuberculin skin test. Such assays use early secretory antigenic target 6 and cultured filtrate protein 10 as antigens to determine sensitization to M tuberculosis.13,15 These antigens are not produced by BCG or Mycobacterium avium; however, other mycobacteria such as Mycobacterium marinum, Mycobacterium kansasii, and some strains of M bovis produce the aforementioned antigens, and exposure to these microbes may be confounding.13 Importantly, positive IFN-γ release assay results also have been documented after BCG exposure but occur at a much lower frequency than for tuberculosis.15 Thus, the combination of the positive IFN-γ release assay and new chest radiograph nodule in our patient provided strong evidence of reactivated tuberculosis as the precipitating cause of her skin disease.
Despite her negative PCR study, our patient’s presentation remains consistent with the diagnosis of disseminated erythema induratum.13,15 The value of PCR studies in establishing the diagnosis remains to be determined. Case reports have described positive PCR results detecting M tuberculosis in panniculitic nodules, suggesting that trace amounts of the organism are present in lesional tissue despite the negative culture result and immunostains.1 Tuberculid reactions, including lichen scrofulosorum, papulonecrotic tuberculid, and erythema induratum, historically are defined by the lack of positive cultures and immunostains, making positive PCR results difficult to reconcile pathophysiologically.1,13 Therefore, use of the term tuberculid altogether as a descriptor for pathogenesis of this disease may need to be avoided.16 Postulated explanations for the relationship of tuberculid diseases and negative cultures and immunostains include the presence of a small number of bacilli that escape routine laboratory detection, early destruction of organisms, or a reaction to circulating M tuberculosis fragments.2 Regardless, until the pathophysiology of erythema induratum has been fully elucidated, the value of PCR remains unclear.
Disseminated erythema induratum, an exceptionally rare variant of panniculitis, may be seen in patients with a remote history of M tuberculosis exposure and/or recent therapeutic BCG exposure. It is imperative to rule out active tuberculosis, especially in elderly patients whose disease predated the advent of modern antituberculosis therapy. Using an IFN-γ release assay in addition to chest radiographs and other clinical stigmata allows differentiation of the etiology of erythema induratum in those patients with tuberculosis who also were treated with BCG.
- Mascaro JM, Basalga E. Erythema induratum of Bazin. Dermatol Clin. 2008;28:439-445.
- Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
- Inoue T, Fukumoto T, Ansai S, et al. Erythema induratum of Bazin in an infant after bacilli Calmette-Guerin vaccination. J Dermatol. 2006;33:268-272.
- Degonda Halter M, Nebiker P, Hug B, et al. Atypical erythema induratum Bazin with tuberculous osteomyelitis. Internist. 2006;47:853-856.
- Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
- Sharma S, Sehgal VN, Bhattacharya SN, et al. Clinicopathologic spectrum of cutaneous tuberculosis: a retrospective analysis of 165 Indians. Am J Dermatopathol. 2015;37:444-450.
- Sethuraman G, Ramesh V. Cutaneous tuberculosis in children. Pediatr Dermatol. 2013;30:7-16.
- Teramura K, Fujimoto N, Nakanishi G, et al. Disseminated erythema induratum of Bazin. Eur J Dermatol. 2014;24:697-698.
- Bazin E. Extrait des Lecons Théoretiques et Cliniques sur le Scrofule. 2nd ed. Paris, France: Delhaye; 1861.
- Campbell SM, Winkelmann RR, Sammons DL. Erythema induratum caused by Mycobacterium chelonei in an immunocompetent patient. J Clin Aesthet Dermatol. 2013;6:38-40.
- Patterson JW. Panniculitis. In: Bolognia JL, Jorizzo J, Rapini RP, et al, eds. Dermatology. Barcelona, Spain: Mosby Elsevier; 2012:1641-1662.
- Segura S, Pujol R, Trinidade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
- Vera-Kellet C, Peters L, Elwood K, et al. Usefulness of interferon-γ release assays in the diagnosis of erythema induratum. Arch Dermatol. 2011;147:949-952.
- Prajapati V, Steed M, Grewal P, et al. Erythema induratum: case series illustrating the utility of the interferon-γ release assay in determining the association with tuberculosis. J Cutan Med Surg. 2013;17:S6-S11.
- Sim JH, Whang KU. Application of the QuantiFERON-Gold TB test in erythema induratum. J Dermatolog Treat. 2014;25:260-263.
- Wiebels D, Turnbull K, Steinkraus V, et al. Erythema induratum Bazin.”tuberculid” or tuberculosis? [in German]. Hautarzt. 2007;58:237-240.
To the Editor:
Erythema induratum, also known as nodular vasculitis, is a panniculitis that usually affects the lower extremities in middle-aged women. Classically, it has been described as a delayed-type hypersensitivity reaction to Mycobacterium tuberculosis, also known as a tuberculid.1,2 Other infections, however, also have been implicated as causes of erythema induratum, including bacillus Calmette-Guérin (BCG), the attenuated form of Mycobacterium bovis, which commonly is used for tuberculosis vaccination. Medications also may cause erythema induratum. The characteristic distribution of the nodules on the posterior calves helps to distinguish erythema induratum from other panniculitides. A PubMed search of articles indexed for MEDLINE using the term disseminated erythema induratum revealed few case reports documenting nodules on the arms, thighs, or chest, and only 1 case report of disseminated erythema induratum.3-8 We describe a rare combination of disseminated erythema induratum in a patient with remote exposure to tuberculosis and recent BCG exposure.
An 88-year-old woman presented for evaluation of violaceous, minimally tender, nonulcerated, subcutaneous nodules on the legs, arms, and trunk of several weeks’ duration (Figure 1). She had a remote history of tuberculosis as a child, prior to the advent of modern antituberculosis regimens. Her medical history also included hypertension, breast cancer treated with lymph node dissection, gastroesophageal reflux disease, and bladder cancer treated with intravesical BCG 10 years prior to the onset of the nodules. She reported minimal coughing and a 25-lb weight loss over the last year, but she denied night sweats, fever, or chills.
Workup included a biopsy, which showed a dense inflammatory infiltrate within the septae and lobules of the subcutaneous tissue (Figure 2A). Foci of necrosis were seen within the fat lobules (Figure 2B). The histologic diagnosis was erythema induratum. Tissue cultures for bacteria, fungi, and atypical mycobacteria were negative. Mycobacterium tuberculosis polymerase chain reaction (PCR) analysis also was negative. An IFN-γ release assay test was positive for infection with M tuberculosis, suggesting that the erythema induratum was due to tuberculosis rather than BCG exposure. A chest radiograph demonstrated a 22-mm nodule in the left lung (unchanged from a prior film) and a new 10-mm nodule in the left upper lobe.
The patient was referred to an infectious disease specialist who concurred that the erythema induratum and the new lung nodule likely represented a reactivation of tuberculosis. Sputum samples were found to be smear and culture negative for mycobacteria, but due to high clinical suspicion, she was started on a 4-drug tuberculosis regimen of isoniazid, rifampin, pyrazinamide, and ethambutol. Some lesions had started to improve prior to the institution of therapy; after initiation of treatment, all lesions resolved within 4 weeks of starting treatment without recurrence.
Erythema induratum was first described by Bazin9 in 1861. The disorder usually occurs in middle-aged women and is characterized by violaceous ulcerative plaques that classically present on the lower extremities, especially the calves. When the eruption occurs due to a nontuberculous etiology, the term nodular vasculitis is used.1,5 The distinction largely is historical, as most dermatologists today recognize erythema induratum and nodular vasculitis to be the same entity. Examples of nontuberculous causes include infections such as Nocardia, Pseudomonas, Fusarium, or other Mycobacterium species.10 Medications such as propylthiouracil also have been implicated.11 The classification of erythema induratum as a tuberculid suggests that the nodules are a reaction pattern rather than a primary infection, though the term tuberculid may be imprecise. The differential diagnosis of violaceous nodules on the lower extremities and trunk is broad and includes erythema nodosum, cutaneous polyarteritis nodosa, pancreatic panniculitis, subcutaneous T-cell lymphoma, and lupus profundus.1,11,12
Histologically, lesions classically demonstrate a mostly lobular panniculitis with varying degrees of septal fibrosis and focal necrosis. Neutrophils may predominate early, while adipocyte necrosis, epithelioid histiocytes, multinucleated giant cells, and lymphocytes may be found in older lesions. The presence of vasculitis as a requisite diagnostic criterion remains controversial.1,12
The incidence of erythema induratum has decreased since multidrug tuberculosis treatment has become more widespread.3 Our case displayed the disseminated variant of erythema induratum, an even rarer clinical entity.8 Interestingly, our patient had a history of tuberculosis and exposure to BCG prior to the development of lesions. Case reports have documented erythema induratum after BCG exposure but less frequently than in cases associated with tuberculosis.3,13
The use of BCG vaccines has necessitated the need for a more precise method of determining tuberculosis activity. The tuberculin skin test reacts positively with a history of BCG exposure, rendering it an inadequate test in a patient who is suspected of having an active or latent M tuberculosis infection.13,14 IFN-γ release assays are more specific in detecting latent or active tuberculosis than the tuberculin skin test. Such assays use early secretory antigenic target 6 and cultured filtrate protein 10 as antigens to determine sensitization to M tuberculosis.13,15 These antigens are not produced by BCG or Mycobacterium avium; however, other mycobacteria such as Mycobacterium marinum, Mycobacterium kansasii, and some strains of M bovis produce the aforementioned antigens, and exposure to these microbes may be confounding.13 Importantly, positive IFN-γ release assay results also have been documented after BCG exposure but occur at a much lower frequency than for tuberculosis.15 Thus, the combination of the positive IFN-γ release assay and new chest radiograph nodule in our patient provided strong evidence of reactivated tuberculosis as the precipitating cause of her skin disease.
Despite her negative PCR study, our patient’s presentation remains consistent with the diagnosis of disseminated erythema induratum.13,15 The value of PCR studies in establishing the diagnosis remains to be determined. Case reports have described positive PCR results detecting M tuberculosis in panniculitic nodules, suggesting that trace amounts of the organism are present in lesional tissue despite the negative culture result and immunostains.1 Tuberculid reactions, including lichen scrofulosorum, papulonecrotic tuberculid, and erythema induratum, historically are defined by the lack of positive cultures and immunostains, making positive PCR results difficult to reconcile pathophysiologically.1,13 Therefore, use of the term tuberculid altogether as a descriptor for pathogenesis of this disease may need to be avoided.16 Postulated explanations for the relationship of tuberculid diseases and negative cultures and immunostains include the presence of a small number of bacilli that escape routine laboratory detection, early destruction of organisms, or a reaction to circulating M tuberculosis fragments.2 Regardless, until the pathophysiology of erythema induratum has been fully elucidated, the value of PCR remains unclear.
Disseminated erythema induratum, an exceptionally rare variant of panniculitis, may be seen in patients with a remote history of M tuberculosis exposure and/or recent therapeutic BCG exposure. It is imperative to rule out active tuberculosis, especially in elderly patients whose disease predated the advent of modern antituberculosis therapy. Using an IFN-γ release assay in addition to chest radiographs and other clinical stigmata allows differentiation of the etiology of erythema induratum in those patients with tuberculosis who also were treated with BCG.
To the Editor:
Erythema induratum, also known as nodular vasculitis, is a panniculitis that usually affects the lower extremities in middle-aged women. Classically, it has been described as a delayed-type hypersensitivity reaction to Mycobacterium tuberculosis, also known as a tuberculid.1,2 Other infections, however, also have been implicated as causes of erythema induratum, including bacillus Calmette-Guérin (BCG), the attenuated form of Mycobacterium bovis, which commonly is used for tuberculosis vaccination. Medications also may cause erythema induratum. The characteristic distribution of the nodules on the posterior calves helps to distinguish erythema induratum from other panniculitides. A PubMed search of articles indexed for MEDLINE using the term disseminated erythema induratum revealed few case reports documenting nodules on the arms, thighs, or chest, and only 1 case report of disseminated erythema induratum.3-8 We describe a rare combination of disseminated erythema induratum in a patient with remote exposure to tuberculosis and recent BCG exposure.
An 88-year-old woman presented for evaluation of violaceous, minimally tender, nonulcerated, subcutaneous nodules on the legs, arms, and trunk of several weeks’ duration (Figure 1). She had a remote history of tuberculosis as a child, prior to the advent of modern antituberculosis regimens. Her medical history also included hypertension, breast cancer treated with lymph node dissection, gastroesophageal reflux disease, and bladder cancer treated with intravesical BCG 10 years prior to the onset of the nodules. She reported minimal coughing and a 25-lb weight loss over the last year, but she denied night sweats, fever, or chills.
Workup included a biopsy, which showed a dense inflammatory infiltrate within the septae and lobules of the subcutaneous tissue (Figure 2A). Foci of necrosis were seen within the fat lobules (Figure 2B). The histologic diagnosis was erythema induratum. Tissue cultures for bacteria, fungi, and atypical mycobacteria were negative. Mycobacterium tuberculosis polymerase chain reaction (PCR) analysis also was negative. An IFN-γ release assay test was positive for infection with M tuberculosis, suggesting that the erythema induratum was due to tuberculosis rather than BCG exposure. A chest radiograph demonstrated a 22-mm nodule in the left lung (unchanged from a prior film) and a new 10-mm nodule in the left upper lobe.
The patient was referred to an infectious disease specialist who concurred that the erythema induratum and the new lung nodule likely represented a reactivation of tuberculosis. Sputum samples were found to be smear and culture negative for mycobacteria, but due to high clinical suspicion, she was started on a 4-drug tuberculosis regimen of isoniazid, rifampin, pyrazinamide, and ethambutol. Some lesions had started to improve prior to the institution of therapy; after initiation of treatment, all lesions resolved within 4 weeks of starting treatment without recurrence.
Erythema induratum was first described by Bazin9 in 1861. The disorder usually occurs in middle-aged women and is characterized by violaceous ulcerative plaques that classically present on the lower extremities, especially the calves. When the eruption occurs due to a nontuberculous etiology, the term nodular vasculitis is used.1,5 The distinction largely is historical, as most dermatologists today recognize erythema induratum and nodular vasculitis to be the same entity. Examples of nontuberculous causes include infections such as Nocardia, Pseudomonas, Fusarium, or other Mycobacterium species.10 Medications such as propylthiouracil also have been implicated.11 The classification of erythema induratum as a tuberculid suggests that the nodules are a reaction pattern rather than a primary infection, though the term tuberculid may be imprecise. The differential diagnosis of violaceous nodules on the lower extremities and trunk is broad and includes erythema nodosum, cutaneous polyarteritis nodosa, pancreatic panniculitis, subcutaneous T-cell lymphoma, and lupus profundus.1,11,12
Histologically, lesions classically demonstrate a mostly lobular panniculitis with varying degrees of septal fibrosis and focal necrosis. Neutrophils may predominate early, while adipocyte necrosis, epithelioid histiocytes, multinucleated giant cells, and lymphocytes may be found in older lesions. The presence of vasculitis as a requisite diagnostic criterion remains controversial.1,12
The incidence of erythema induratum has decreased since multidrug tuberculosis treatment has become more widespread.3 Our case displayed the disseminated variant of erythema induratum, an even rarer clinical entity.8 Interestingly, our patient had a history of tuberculosis and exposure to BCG prior to the development of lesions. Case reports have documented erythema induratum after BCG exposure but less frequently than in cases associated with tuberculosis.3,13
The use of BCG vaccines has necessitated the need for a more precise method of determining tuberculosis activity. The tuberculin skin test reacts positively with a history of BCG exposure, rendering it an inadequate test in a patient who is suspected of having an active or latent M tuberculosis infection.13,14 IFN-γ release assays are more specific in detecting latent or active tuberculosis than the tuberculin skin test. Such assays use early secretory antigenic target 6 and cultured filtrate protein 10 as antigens to determine sensitization to M tuberculosis.13,15 These antigens are not produced by BCG or Mycobacterium avium; however, other mycobacteria such as Mycobacterium marinum, Mycobacterium kansasii, and some strains of M bovis produce the aforementioned antigens, and exposure to these microbes may be confounding.13 Importantly, positive IFN-γ release assay results also have been documented after BCG exposure but occur at a much lower frequency than for tuberculosis.15 Thus, the combination of the positive IFN-γ release assay and new chest radiograph nodule in our patient provided strong evidence of reactivated tuberculosis as the precipitating cause of her skin disease.
Despite her negative PCR study, our patient’s presentation remains consistent with the diagnosis of disseminated erythema induratum.13,15 The value of PCR studies in establishing the diagnosis remains to be determined. Case reports have described positive PCR results detecting M tuberculosis in panniculitic nodules, suggesting that trace amounts of the organism are present in lesional tissue despite the negative culture result and immunostains.1 Tuberculid reactions, including lichen scrofulosorum, papulonecrotic tuberculid, and erythema induratum, historically are defined by the lack of positive cultures and immunostains, making positive PCR results difficult to reconcile pathophysiologically.1,13 Therefore, use of the term tuberculid altogether as a descriptor for pathogenesis of this disease may need to be avoided.16 Postulated explanations for the relationship of tuberculid diseases and negative cultures and immunostains include the presence of a small number of bacilli that escape routine laboratory detection, early destruction of organisms, or a reaction to circulating M tuberculosis fragments.2 Regardless, until the pathophysiology of erythema induratum has been fully elucidated, the value of PCR remains unclear.
Disseminated erythema induratum, an exceptionally rare variant of panniculitis, may be seen in patients with a remote history of M tuberculosis exposure and/or recent therapeutic BCG exposure. It is imperative to rule out active tuberculosis, especially in elderly patients whose disease predated the advent of modern antituberculosis therapy. Using an IFN-γ release assay in addition to chest radiographs and other clinical stigmata allows differentiation of the etiology of erythema induratum in those patients with tuberculosis who also were treated with BCG.
- Mascaro JM, Basalga E. Erythema induratum of Bazin. Dermatol Clin. 2008;28:439-445.
- Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
- Inoue T, Fukumoto T, Ansai S, et al. Erythema induratum of Bazin in an infant after bacilli Calmette-Guerin vaccination. J Dermatol. 2006;33:268-272.
- Degonda Halter M, Nebiker P, Hug B, et al. Atypical erythema induratum Bazin with tuberculous osteomyelitis. Internist. 2006;47:853-856.
- Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
- Sharma S, Sehgal VN, Bhattacharya SN, et al. Clinicopathologic spectrum of cutaneous tuberculosis: a retrospective analysis of 165 Indians. Am J Dermatopathol. 2015;37:444-450.
- Sethuraman G, Ramesh V. Cutaneous tuberculosis in children. Pediatr Dermatol. 2013;30:7-16.
- Teramura K, Fujimoto N, Nakanishi G, et al. Disseminated erythema induratum of Bazin. Eur J Dermatol. 2014;24:697-698.
- Bazin E. Extrait des Lecons Théoretiques et Cliniques sur le Scrofule. 2nd ed. Paris, France: Delhaye; 1861.
- Campbell SM, Winkelmann RR, Sammons DL. Erythema induratum caused by Mycobacterium chelonei in an immunocompetent patient. J Clin Aesthet Dermatol. 2013;6:38-40.
- Patterson JW. Panniculitis. In: Bolognia JL, Jorizzo J, Rapini RP, et al, eds. Dermatology. Barcelona, Spain: Mosby Elsevier; 2012:1641-1662.
- Segura S, Pujol R, Trinidade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
- Vera-Kellet C, Peters L, Elwood K, et al. Usefulness of interferon-γ release assays in the diagnosis of erythema induratum. Arch Dermatol. 2011;147:949-952.
- Prajapati V, Steed M, Grewal P, et al. Erythema induratum: case series illustrating the utility of the interferon-γ release assay in determining the association with tuberculosis. J Cutan Med Surg. 2013;17:S6-S11.
- Sim JH, Whang KU. Application of the QuantiFERON-Gold TB test in erythema induratum. J Dermatolog Treat. 2014;25:260-263.
- Wiebels D, Turnbull K, Steinkraus V, et al. Erythema induratum Bazin.”tuberculid” or tuberculosis? [in German]. Hautarzt. 2007;58:237-240.
- Mascaro JM, Basalga E. Erythema induratum of Bazin. Dermatol Clin. 2008;28:439-445.
- Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
- Inoue T, Fukumoto T, Ansai S, et al. Erythema induratum of Bazin in an infant after bacilli Calmette-Guerin vaccination. J Dermatol. 2006;33:268-272.
- Degonda Halter M, Nebiker P, Hug B, et al. Atypical erythema induratum Bazin with tuberculous osteomyelitis. Internist. 2006;47:853-856.
- Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
- Sharma S, Sehgal VN, Bhattacharya SN, et al. Clinicopathologic spectrum of cutaneous tuberculosis: a retrospective analysis of 165 Indians. Am J Dermatopathol. 2015;37:444-450.
- Sethuraman G, Ramesh V. Cutaneous tuberculosis in children. Pediatr Dermatol. 2013;30:7-16.
- Teramura K, Fujimoto N, Nakanishi G, et al. Disseminated erythema induratum of Bazin. Eur J Dermatol. 2014;24:697-698.
- Bazin E. Extrait des Lecons Théoretiques et Cliniques sur le Scrofule. 2nd ed. Paris, France: Delhaye; 1861.
- Campbell SM, Winkelmann RR, Sammons DL. Erythema induratum caused by Mycobacterium chelonei in an immunocompetent patient. J Clin Aesthet Dermatol. 2013;6:38-40.
- Patterson JW. Panniculitis. In: Bolognia JL, Jorizzo J, Rapini RP, et al, eds. Dermatology. Barcelona, Spain: Mosby Elsevier; 2012:1641-1662.
- Segura S, Pujol R, Trinidade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
- Vera-Kellet C, Peters L, Elwood K, et al. Usefulness of interferon-γ release assays in the diagnosis of erythema induratum. Arch Dermatol. 2011;147:949-952.
- Prajapati V, Steed M, Grewal P, et al. Erythema induratum: case series illustrating the utility of the interferon-γ release assay in determining the association with tuberculosis. J Cutan Med Surg. 2013;17:S6-S11.
- Sim JH, Whang KU. Application of the QuantiFERON-Gold TB test in erythema induratum. J Dermatolog Treat. 2014;25:260-263.
- Wiebels D, Turnbull K, Steinkraus V, et al. Erythema induratum Bazin.”tuberculid” or tuberculosis? [in German]. Hautarzt. 2007;58:237-240.
Practice Points
- Erythema induratum is an uncommon panniculitis attributed to a delayed-type hypersensitivity reaction, classically to Mycobacterium tuberculosis.
- The workup for such patients with exposure to both M tuberculosis and bacillus Calmette-Guérin should include IFN-11γ release assays.
- Clinicians should be aware of the disseminated variant of erythema induratum and the laboratory testing needed to establish a cause and help direct treatment.
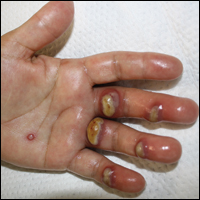
Tender Edematous Nodules on the Hand
The Diagnosis: Ecthyma Contagiosum (Orf)
Orf, or ecthyma contagiosum, is a zoonotic cutaneous infection caused by the orf DNA virus of the genus Parapoxvirus of the family Poxviridae. It is transmitted to humans through direct contact with infected animals, namely sheep and goats, and as such is most commonly seen in patients with occupational exposure to these animals such as butchers, farmers, veterinarians, and shepherds.1,2 Human-to-human transmission is exceedingly rare in immunocompetent patients.2,3 In affected animals, lesions usually are found around the mouth, muzzle, and eyes. In humans, hands are the most commonly affected site, and lesions occur 3 to 10 days after contact. Clinically, the lesions are nonspecific, and our patient presented with tender, erythematous, edematous nodules on the left hand. The differential diagnosis is broad and includes a milker's nodule, pyogenic granuloma, tularemia, anthrax, atypical mycobacterial infection, and sporotrichosis.1,4,5
The diagnosis usually is made with a thorough history and examination, but in cases of uncertainty, routine pathology with hematoxylin and eosin staining, electron microscopy, or real-time polymerase chain reaction may be used.2-4 Histopathologically, lesions demonstrate intraepidermal vesicles, vacuolization of keratinocytes of the upper epidermis with characteristic cytoplasmic inclusion bodies, rete ridge elongation, and dilated vessels in the intervening dermal papillae. Central necrosis may occur in well-developed lesions.2,6 Interestingly, our patient's biopsy exhibited all of these findings (Figure). Immunostains for cytomegalovirus and herpes simplex virus were negative, and Grocott-Gomori methenamine-silver and acid-fast bacillus stains also were negative.

Our patient also developed lymphangitic streaking suggestive of a bacterial superinfection and was treated with a course of intravenous antibiotics. She eventually was discharged with reassurance, wound care instructions, and outpatient antibiotics. She returned to an outside institution's emergency department for further evaluation, and she was admitted for workup. A lesional swab was sent for real-time polymerase chain reaction, which confirmed the diagnosis as orf. When the patient was contacted for follow-up 1 week after biopsy, the hand lesions had notably improved.
Orf is self-limited and typically resolves within 4 to 8 weeks after undergoing evolution through 5 described stages. The maculopapular stage is denoted by enlarging erythematous macule. The targetoid stage is described by a red center within a white halo surrounded by a broader red halo. The nodular stage is self-descriptive. The regenerative and regression stages describe the progressively improving, drier, and crusted nodules.3
Because orf is self-limited, no treatment is required, and patients should be counseled that their lesions should resolve within weeks. Complications include lymphangitis, secondary bacterial infection, and erythema multiforme.1,2,4,5 Immunocompromised patients may develop recalcitrant, giant, or multiple lesions that may be treated with topical imiquimod, topical cidofovir, intralesional interferon alfa, or surgical excision.1,2,4,7
We present a case of orf to remind practitioners of this rare entity. Although the disease is endemic worldwide, it likely is underreported due to its self-limited nature.2,4 A careful history may reveal the diagnosis, and overtreatment with antibiotics, many of which have their own significant side-effect profile, can then be avoided.
Acknowledgment
We thank Eric Behling, MD (Camden, New Jersey), for his contributions in obtaining the histologic images.
- Veraldi S, Nazzaro G, Vaira F, et al. Presentation of orf (ecthyma contagiosum) after sheep slaughtering for religious feasts. Infection. 2014;42:767-769.
- Al-Salam S, Nowotny N, Sohail MR, et al. Ecthyma contagiosum (orf)--report of a human case from the United Arab Emirates and review of the literature. J Cutan Pathol. 2008;35:603-607.
- Thurman RJ, Fitch RW. Images in clinical medicine. contagious ecthyma. N Engl J Med. 2015;372:E12.
- Meier R, Sommacal A, Stahel A, et al. Orf--an orphan disease? JRSM Open. 2015;6:2054270415593718.
- Joseph RH, Haddad FA, Matthews AL, et al. Erythema multiforme after orf virus infection: a report of two cases and literature review. Epidemiol Infect. 2015;143:385-390.
- Xu X, Yun SJ, Erikson L, et al. Diseases caused by viruses. In: Elder DE, Elenitsas R, Rosenbach M, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:781-815.
- Koufakis T, Katsaitis P, Gabranis I. Orf disease: a report of a case. Braz J Infect Dis. 2014;18:568-569.
The Diagnosis: Ecthyma Contagiosum (Orf)
Orf, or ecthyma contagiosum, is a zoonotic cutaneous infection caused by the orf DNA virus of the genus Parapoxvirus of the family Poxviridae. It is transmitted to humans through direct contact with infected animals, namely sheep and goats, and as such is most commonly seen in patients with occupational exposure to these animals such as butchers, farmers, veterinarians, and shepherds.1,2 Human-to-human transmission is exceedingly rare in immunocompetent patients.2,3 In affected animals, lesions usually are found around the mouth, muzzle, and eyes. In humans, hands are the most commonly affected site, and lesions occur 3 to 10 days after contact. Clinically, the lesions are nonspecific, and our patient presented with tender, erythematous, edematous nodules on the left hand. The differential diagnosis is broad and includes a milker's nodule, pyogenic granuloma, tularemia, anthrax, atypical mycobacterial infection, and sporotrichosis.1,4,5
The diagnosis usually is made with a thorough history and examination, but in cases of uncertainty, routine pathology with hematoxylin and eosin staining, electron microscopy, or real-time polymerase chain reaction may be used.2-4 Histopathologically, lesions demonstrate intraepidermal vesicles, vacuolization of keratinocytes of the upper epidermis with characteristic cytoplasmic inclusion bodies, rete ridge elongation, and dilated vessels in the intervening dermal papillae. Central necrosis may occur in well-developed lesions.2,6 Interestingly, our patient's biopsy exhibited all of these findings (Figure). Immunostains for cytomegalovirus and herpes simplex virus were negative, and Grocott-Gomori methenamine-silver and acid-fast bacillus stains also were negative.
Our patient also developed lymphangitic streaking suggestive of a bacterial superinfection and was treated with a course of intravenous antibiotics. She eventually was discharged with reassurance, wound care instructions, and outpatient antibiotics. She returned to an outside institution's emergency department for further evaluation, and she was admitted for workup. A lesional swab was sent for real-time polymerase chain reaction, which confirmed the diagnosis as orf. When the patient was contacted for follow-up 1 week after biopsy, the hand lesions had notably improved.
Orf is self-limited and typically resolves within 4 to 8 weeks after undergoing evolution through 5 described stages. The maculopapular stage is denoted by enlarging erythematous macule. The targetoid stage is described by a red center within a white halo surrounded by a broader red halo. The nodular stage is self-descriptive. The regenerative and regression stages describe the progressively improving, drier, and crusted nodules.3
Because orf is self-limited, no treatment is required, and patients should be counseled that their lesions should resolve within weeks. Complications include lymphangitis, secondary bacterial infection, and erythema multiforme.1,2,4,5 Immunocompromised patients may develop recalcitrant, giant, or multiple lesions that may be treated with topical imiquimod, topical cidofovir, intralesional interferon alfa, or surgical excision.1,2,4,7
We present a case of orf to remind practitioners of this rare entity. Although the disease is endemic worldwide, it likely is underreported due to its self-limited nature.2,4 A careful history may reveal the diagnosis, and overtreatment with antibiotics, many of which have their own significant side-effect profile, can then be avoided.
Acknowledgment
We thank Eric Behling, MD (Camden, New Jersey), for his contributions in obtaining the histologic images.
The Diagnosis: Ecthyma Contagiosum (Orf)
Orf, or ecthyma contagiosum, is a zoonotic cutaneous infection caused by the orf DNA virus of the genus Parapoxvirus of the family Poxviridae. It is transmitted to humans through direct contact with infected animals, namely sheep and goats, and as such is most commonly seen in patients with occupational exposure to these animals such as butchers, farmers, veterinarians, and shepherds.1,2 Human-to-human transmission is exceedingly rare in immunocompetent patients.2,3 In affected animals, lesions usually are found around the mouth, muzzle, and eyes. In humans, hands are the most commonly affected site, and lesions occur 3 to 10 days after contact. Clinically, the lesions are nonspecific, and our patient presented with tender, erythematous, edematous nodules on the left hand. The differential diagnosis is broad and includes a milker's nodule, pyogenic granuloma, tularemia, anthrax, atypical mycobacterial infection, and sporotrichosis.1,4,5
The diagnosis usually is made with a thorough history and examination, but in cases of uncertainty, routine pathology with hematoxylin and eosin staining, electron microscopy, or real-time polymerase chain reaction may be used.2-4 Histopathologically, lesions demonstrate intraepidermal vesicles, vacuolization of keratinocytes of the upper epidermis with characteristic cytoplasmic inclusion bodies, rete ridge elongation, and dilated vessels in the intervening dermal papillae. Central necrosis may occur in well-developed lesions.2,6 Interestingly, our patient's biopsy exhibited all of these findings (Figure). Immunostains for cytomegalovirus and herpes simplex virus were negative, and Grocott-Gomori methenamine-silver and acid-fast bacillus stains also were negative.
Our patient also developed lymphangitic streaking suggestive of a bacterial superinfection and was treated with a course of intravenous antibiotics. She eventually was discharged with reassurance, wound care instructions, and outpatient antibiotics. She returned to an outside institution's emergency department for further evaluation, and she was admitted for workup. A lesional swab was sent for real-time polymerase chain reaction, which confirmed the diagnosis as orf. When the patient was contacted for follow-up 1 week after biopsy, the hand lesions had notably improved.
Orf is self-limited and typically resolves within 4 to 8 weeks after undergoing evolution through 5 described stages. The maculopapular stage is denoted by enlarging erythematous macule. The targetoid stage is described by a red center within a white halo surrounded by a broader red halo. The nodular stage is self-descriptive. The regenerative and regression stages describe the progressively improving, drier, and crusted nodules.3
Because orf is self-limited, no treatment is required, and patients should be counseled that their lesions should resolve within weeks. Complications include lymphangitis, secondary bacterial infection, and erythema multiforme.1,2,4,5 Immunocompromised patients may develop recalcitrant, giant, or multiple lesions that may be treated with topical imiquimod, topical cidofovir, intralesional interferon alfa, or surgical excision.1,2,4,7
We present a case of orf to remind practitioners of this rare entity. Although the disease is endemic worldwide, it likely is underreported due to its self-limited nature.2,4 A careful history may reveal the diagnosis, and overtreatment with antibiotics, many of which have their own significant side-effect profile, can then be avoided.
Acknowledgment
We thank Eric Behling, MD (Camden, New Jersey), for his contributions in obtaining the histologic images.
- Veraldi S, Nazzaro G, Vaira F, et al. Presentation of orf (ecthyma contagiosum) after sheep slaughtering for religious feasts. Infection. 2014;42:767-769.
- Al-Salam S, Nowotny N, Sohail MR, et al. Ecthyma contagiosum (orf)--report of a human case from the United Arab Emirates and review of the literature. J Cutan Pathol. 2008;35:603-607.
- Thurman RJ, Fitch RW. Images in clinical medicine. contagious ecthyma. N Engl J Med. 2015;372:E12.
- Meier R, Sommacal A, Stahel A, et al. Orf--an orphan disease? JRSM Open. 2015;6:2054270415593718.
- Joseph RH, Haddad FA, Matthews AL, et al. Erythema multiforme after orf virus infection: a report of two cases and literature review. Epidemiol Infect. 2015;143:385-390.
- Xu X, Yun SJ, Erikson L, et al. Diseases caused by viruses. In: Elder DE, Elenitsas R, Rosenbach M, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:781-815.
- Koufakis T, Katsaitis P, Gabranis I. Orf disease: a report of a case. Braz J Infect Dis. 2014;18:568-569.
- Veraldi S, Nazzaro G, Vaira F, et al. Presentation of orf (ecthyma contagiosum) after sheep slaughtering for religious feasts. Infection. 2014;42:767-769.
- Al-Salam S, Nowotny N, Sohail MR, et al. Ecthyma contagiosum (orf)--report of a human case from the United Arab Emirates and review of the literature. J Cutan Pathol. 2008;35:603-607.
- Thurman RJ, Fitch RW. Images in clinical medicine. contagious ecthyma. N Engl J Med. 2015;372:E12.
- Meier R, Sommacal A, Stahel A, et al. Orf--an orphan disease? JRSM Open. 2015;6:2054270415593718.
- Joseph RH, Haddad FA, Matthews AL, et al. Erythema multiforme after orf virus infection: a report of two cases and literature review. Epidemiol Infect. 2015;143:385-390.
- Xu X, Yun SJ, Erikson L, et al. Diseases caused by viruses. In: Elder DE, Elenitsas R, Rosenbach M, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:781-815.
- Koufakis T, Katsaitis P, Gabranis I. Orf disease: a report of a case. Braz J Infect Dis. 2014;18:568-569.
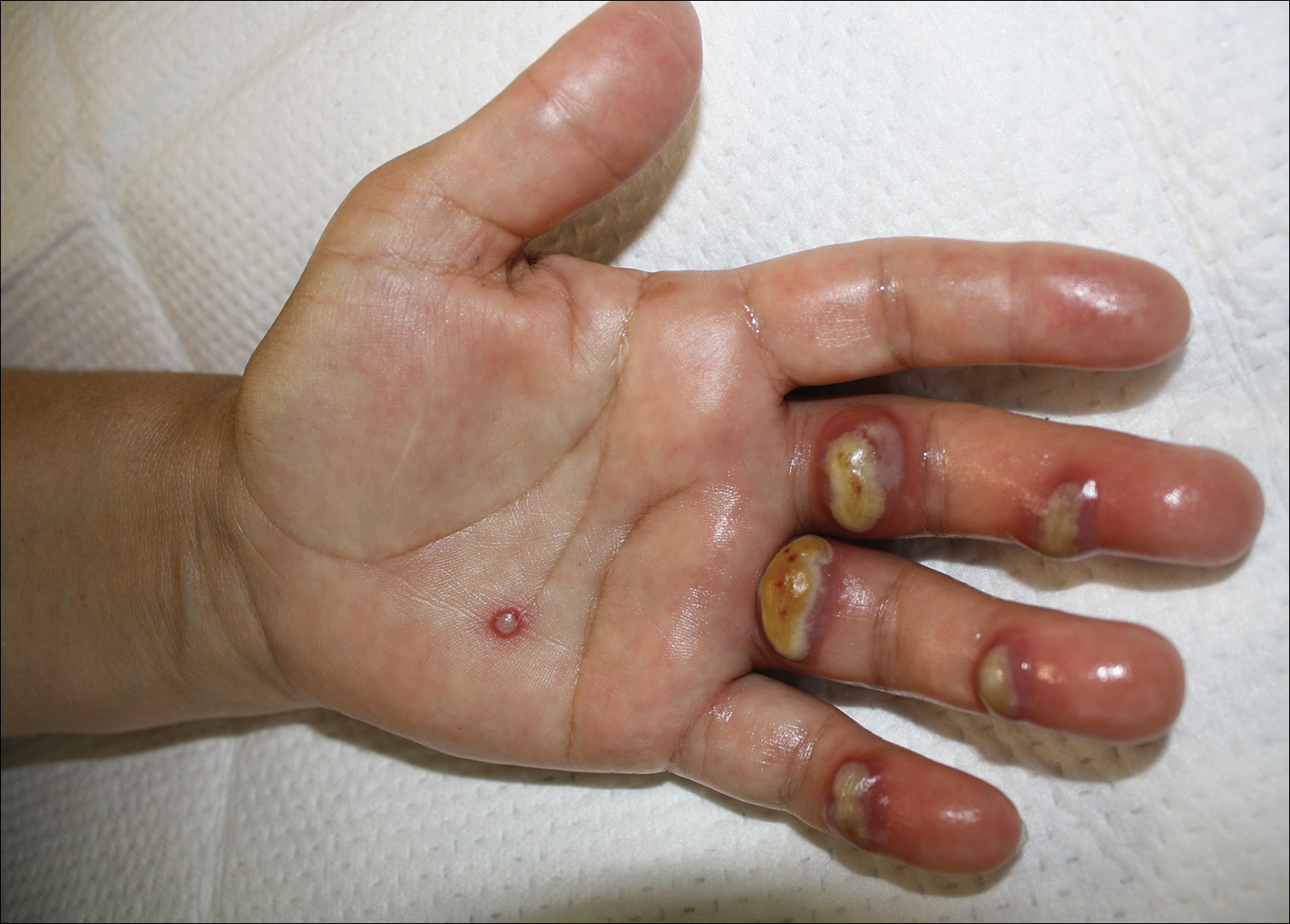
A 57-year-old woman presented to the emergency department (ED) for evaluation of a rash on the left hand of 2 weeks' duration. She described pinpoint red lesions on the left palm, as well as the third, fourth, and fifth fingers, which gradually enlarged and became painful. She denied any specific trauma but recalled cutting her hand on a piece of metal in the ground prior to the onset of the rash. She worked on a farm and bottle-fed sheep and chickens. Physical examination revealed tender edematous nodules with central gray pustules, and the left axillary lymph node was enlarged and tender. Ulceration was not appreciated. Various antibiotics including cephalexin, trimethoprim-sulfamethoxazole, and clindamycin were prescribed during prior ED visits, but she reported no improvement with these medications. She remained afebrile throughout the course of the hand rash, and laboratory workup was consistently unremarkable. Two sets of herpes simplex virus cultures from the ED visits showed no growth, and a hand radiograph also was normal. Medical history included coronary artery disease, myocardial infarction, mitral regurgitation, and hyperlipidemia.