User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Cannabis-using MS patients improve cognition with 28 days of abstinence
STOCKHOLM – The good news about cognitive impairment in patients with multiple sclerosis who’ve been using cannabis heavily for symptom relief – even for many years – is that their memory, executive function, and information processing speed will improve significantly once they’ve been off the drug for just 28 days, according to the results of a randomized trial presented at the annual congress of the European Committee for Treatment and Research in Multiple Sclerosis.

“It’s good for neurologists to know that, if they prescribe cannabis or their patient is self-medicating and chooses to stop, their cognition will improve considerably,” observed Cecilia Meza, a coinvestigator in the study led by Anthony Feinstein, MD, professor of psychiatry at the University of Toronto.
But there’s a surprise twist to this study, she explained in an interview: “We showed patients their results, and they also felt that their cognition was doing a lot better, but despite that, they would rather be using cannabis to feel better than to have their memory intact. The pain was that bad,” said Ms. Meza, a research coordinator at the university’s Sunnybrook Research Institute.
It’s known that cognitive impairment in healthy long-term cannabis users, provided they started as adults, is fully reversed after 28 days of abstinence. But disease-related cognitive dysfunction affects 40%-80% of patients with MS, and cannabis use may compound this impairment.
The study included 40 MS patients with global impairment of cognition, none of whom were cannabis users prior to their diagnosis. They typically started using it for MS symptom relief 2-3 years after receiving their diagnosis. By the time they were approached for study participation, they had been using cannabis four to five times per day or more for an average of 7 years for relief of symptoms, including incontinence, spasticity, poor sleep, headaches, and difficulties in eating.
All participants were willing to try 28 days of abstinence; half were randomized to do so, while the others stayed the course. Study endpoints included change from baseline to day 28 in the Brief Repeatable Neuropsychological Battery, functional MRI done while taking the Symbol Digit Modalities Test, and urine testing to assure compliance with abstinence.
By day 28, the abstinence group – and with one exception, urine testing confirmed they were bona fide cannabis quitters for the study duration – performed significantly better on the neuropsychological test battery than at baseline, with an associated significant increase in brain activation in the bilateral inferior frontal gyri, as well as the caudate and declive cerebellum while executing the Symbol Digit Modalities Test. The control group who kept on using cannabis showed no such improvements.
The full study details were published in conjunction with Ms. Meza’s presentation (Brain. 2019 Sep 1;142[9]:2800-12).
She reported having no financial conflicts regarding the study, funded by the Multiple Sclerosis Society of Canada.
SOURCE: Meza C. ECTRIMS 2019, Abstract P542.
STOCKHOLM – The good news about cognitive impairment in patients with multiple sclerosis who’ve been using cannabis heavily for symptom relief – even for many years – is that their memory, executive function, and information processing speed will improve significantly once they’ve been off the drug for just 28 days, according to the results of a randomized trial presented at the annual congress of the European Committee for Treatment and Research in Multiple Sclerosis.
“It’s good for neurologists to know that, if they prescribe cannabis or their patient is self-medicating and chooses to stop, their cognition will improve considerably,” observed Cecilia Meza, a coinvestigator in the study led by Anthony Feinstein, MD, professor of psychiatry at the University of Toronto.
But there’s a surprise twist to this study, she explained in an interview: “We showed patients their results, and they also felt that their cognition was doing a lot better, but despite that, they would rather be using cannabis to feel better than to have their memory intact. The pain was that bad,” said Ms. Meza, a research coordinator at the university’s Sunnybrook Research Institute.
It’s known that cognitive impairment in healthy long-term cannabis users, provided they started as adults, is fully reversed after 28 days of abstinence. But disease-related cognitive dysfunction affects 40%-80% of patients with MS, and cannabis use may compound this impairment.
The study included 40 MS patients with global impairment of cognition, none of whom were cannabis users prior to their diagnosis. They typically started using it for MS symptom relief 2-3 years after receiving their diagnosis. By the time they were approached for study participation, they had been using cannabis four to five times per day or more for an average of 7 years for relief of symptoms, including incontinence, spasticity, poor sleep, headaches, and difficulties in eating.
All participants were willing to try 28 days of abstinence; half were randomized to do so, while the others stayed the course. Study endpoints included change from baseline to day 28 in the Brief Repeatable Neuropsychological Battery, functional MRI done while taking the Symbol Digit Modalities Test, and urine testing to assure compliance with abstinence.
By day 28, the abstinence group – and with one exception, urine testing confirmed they were bona fide cannabis quitters for the study duration – performed significantly better on the neuropsychological test battery than at baseline, with an associated significant increase in brain activation in the bilateral inferior frontal gyri, as well as the caudate and declive cerebellum while executing the Symbol Digit Modalities Test. The control group who kept on using cannabis showed no such improvements.
The full study details were published in conjunction with Ms. Meza’s presentation (Brain. 2019 Sep 1;142[9]:2800-12).
She reported having no financial conflicts regarding the study, funded by the Multiple Sclerosis Society of Canada.
SOURCE: Meza C. ECTRIMS 2019, Abstract P542.
STOCKHOLM – The good news about cognitive impairment in patients with multiple sclerosis who’ve been using cannabis heavily for symptom relief – even for many years – is that their memory, executive function, and information processing speed will improve significantly once they’ve been off the drug for just 28 days, according to the results of a randomized trial presented at the annual congress of the European Committee for Treatment and Research in Multiple Sclerosis.
“It’s good for neurologists to know that, if they prescribe cannabis or their patient is self-medicating and chooses to stop, their cognition will improve considerably,” observed Cecilia Meza, a coinvestigator in the study led by Anthony Feinstein, MD, professor of psychiatry at the University of Toronto.
But there’s a surprise twist to this study, she explained in an interview: “We showed patients their results, and they also felt that their cognition was doing a lot better, but despite that, they would rather be using cannabis to feel better than to have their memory intact. The pain was that bad,” said Ms. Meza, a research coordinator at the university’s Sunnybrook Research Institute.
It’s known that cognitive impairment in healthy long-term cannabis users, provided they started as adults, is fully reversed after 28 days of abstinence. But disease-related cognitive dysfunction affects 40%-80% of patients with MS, and cannabis use may compound this impairment.
The study included 40 MS patients with global impairment of cognition, none of whom were cannabis users prior to their diagnosis. They typically started using it for MS symptom relief 2-3 years after receiving their diagnosis. By the time they were approached for study participation, they had been using cannabis four to five times per day or more for an average of 7 years for relief of symptoms, including incontinence, spasticity, poor sleep, headaches, and difficulties in eating.
All participants were willing to try 28 days of abstinence; half were randomized to do so, while the others stayed the course. Study endpoints included change from baseline to day 28 in the Brief Repeatable Neuropsychological Battery, functional MRI done while taking the Symbol Digit Modalities Test, and urine testing to assure compliance with abstinence.
By day 28, the abstinence group – and with one exception, urine testing confirmed they were bona fide cannabis quitters for the study duration – performed significantly better on the neuropsychological test battery than at baseline, with an associated significant increase in brain activation in the bilateral inferior frontal gyri, as well as the caudate and declive cerebellum while executing the Symbol Digit Modalities Test. The control group who kept on using cannabis showed no such improvements.
The full study details were published in conjunction with Ms. Meza’s presentation (Brain. 2019 Sep 1;142[9]:2800-12).
She reported having no financial conflicts regarding the study, funded by the Multiple Sclerosis Society of Canada.
SOURCE: Meza C. ECTRIMS 2019, Abstract P542.
REPORTING FROM ECTRIMS 2019
Labeling of medication warnings
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.

The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Four genetic variants link psychotic experiences to multiple mental disorders
Four genetic variations appear to link psychotic experiences with other psychiatric disorders, including schizophrenia, major depressive disorder, bipolar disorder, and neurodevelopmental disorders, a large genetic study has concluded.
, reported Sophie E. Legge, PhD, and colleagues. Their study was published in JAMA Psychiatry.
Although it is informative, the study is unlikely to expand the knowledge of schizophrenia-specific genetics.
“Consistent with other studies, the heritability estimate (1.71%) was low, and given that the variance explained in our polygenic risk analysis was also low, the finding suggests that understanding the genetics of psychotic experiences is unlikely to have an important effect on understanding the genetics of schizophrenia specifically,” wrote Dr. Legge, of the MRC Center for Neuropsychiatric Genetics and Genomics in the division of psychological medicine and clinical neurosciences at Cardiff (Wales) University, and colleagues.
The team conducted a genomewide association study (GWAS) using data from 127,966 individuals in the U.K. Biobank. Of these, 6,123 reported any psychotic experience, 2,143 reported distressing psychotic experiences, and 3,337 reported multiple experiences. The remainder served as controls. At the time of the biobank data collection, the subjects were a mean of 64 years of age; 56% were women.
First psychotic experience occurred at a mean of almost 32 years of age, but about a third reported that the first episode occurred before age 20, or that psychotic experiences had been happening ever since they could remember. Another third reported their first experience between ages 40 and 76 years.
The investigators conducted three GWAS studies: one for any psychotic experience, one for distressing experiences, and one for multiple experiences.
No significant genetic associations were found among those with multiple psychotic experiences, the authors said.
But they did find four variants significantly associated with the other experience categories.
Two variants were associated with any psychotic experience. Those with rs10994278, an intronic variant within Ankyrin-3 (ANK3), were 16% more likely to have a psychotic experience (odds ratio, 1.16). Those with intergenic variant rs549656827 were 39% less likely (OR, 0.61). “The ANK3 gene encodes ankyrin-G, a protein that has been shown to regulate the assembly of voltage-gated sodium channels and is essential for normal synaptic function,” the authors said. “ANK3 is one of strongest and most replicated genes for bipolar disorder, and variants within ANK3 have also been associated in the Psychiatric Genomics Consortium cross-disorder GWAS, and in a rare variant analysis of autism spectrum disorder.”
Two variants were linked to distressing psychotic experiences: rs75459873, intronic to cannabinoid receptor 2 (CNR2), decreased the risk by 34% (OR, 0.66). Intergenic variant rs3849810 increased the risk by 12% (OR, 1.12).
“CNR2 encodes for CB2, one of two well-characterized cannabinoid receptors. Several lines of evidence have implicated the endocannabinoid system in psychiatric disorders, including schizophrenia and depression. The main psychoactive agent of cannabis, tetrahydrocannabinol, can cause acute psychotic symptoms and cognitive impairment. Given that cannabis use is strongly associated with psychotic experiences, we tested, but found no evidence for, a mediating or moderating effect of cannabis use on the association of rs75459873 and distressing psychotic experiences. However, while no evidence was found in this study, a mediating effect of cannabis use cannot be ruled out given the relatively low power of such analyses and the potential measurement error.”
Also, significant genetic correlations were found between any psychotic experiences and major depressive disorder, autism spectrum disorder, ADHD, and schizophrenia. However, the polygenic risk scores for schizophrenia, major depressive disorder, bipolar disorder, ADHD, and autism spectrum disorder, were low.
“We also considered individual psychotic symptoms and found that polygenic risk scores for schizophrenia, bipolar disorder, depression, and ADHD were more strongly associated with delusions of persecution than with the other psychotic symptoms.”
Those with distressing psychotic experiences tended to have more copy number variations (CNVs) associated with schizophrenia (OR, 2.04) and neurodevelopmental disorders (OR, 1.75). The team also found significant associations between distressing experiences and major depressive disorder, ADHD, autism spectrum disorder, and schizophrenia.
“We found particular enrichment of these [polygenic risk scores] in distressing psychotic experiences and for delusions of persecution,” they noted. “ ... All schizophrenia-associated [copy number variations] are also associated with neurodevelopmental disorders such as intellectual disability and autism.”
The study’s strengths include its large sample size. Among its limitations, the researchers said, are the study’s retrospective measurement of psychotic experiences based on self-report from a questionnaire that was online. Gathering the data in that way raised the likelihood of possible error, they said.
Dr. Legge reported having no disclosures.
SOURCE: Legge SE et al. JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2508.
The genetic links uncovered in this study offer an intriguing, but incomplete look at the risks of psychotic experiences and their complicated intertwinings with other mental disorders, wrote Albert R. Powers III, MD, PhD.
“Penetrance of the genes in question likely depends at least in part on environmental influences, some of which have been studied extensively,” he wrote. “Recently, some have proposed risk stratification by exposome – a composite score of relevant exposures that may increase risk for psychosis and is analogous to the polygenic risk score used [here].
“The combination of environmental and genetic composite scores may lead to improved insight into individualized pathways toward psychotic experiences, highlighting genetic vulnerabilities to specific stressors likely to lead to phenotypic expression. Ultimately, this will require a more sophisticated mapping between phenomenology and biology than currently exists.”
One approach would be to combine deep phenotyping and behavioral analyses in a framework that could link all relevant levels from symptoms to neurophysiology.
“One such framework is predictive processing theory, which is linked closely with the free energy principle and the Bayesian brain hypothesis and attempts to explain perceptual and cognitive phenomena as manifestations of a drive to maintain as accurate an internal model of one’s surroundings as possible by minimizing prediction errors. This relatively simple scheme makes specific – and, importantly, falsifiable – assessments of the mathematical signatures of neurotypical processes and the ways they might break down to produce specific psychiatric symptoms.”
Dr. Powers is an assistant professor at the department of psychiatry at Yale University, New Haven, Conn., and serves as medical director of the PRIME Psychosis Research Clinic at Yale. His comments came in an accompanying editorial (JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2391 ).
The genetic links uncovered in this study offer an intriguing, but incomplete look at the risks of psychotic experiences and their complicated intertwinings with other mental disorders, wrote Albert R. Powers III, MD, PhD.
“Penetrance of the genes in question likely depends at least in part on environmental influences, some of which have been studied extensively,” he wrote. “Recently, some have proposed risk stratification by exposome – a composite score of relevant exposures that may increase risk for psychosis and is analogous to the polygenic risk score used [here].
“The combination of environmental and genetic composite scores may lead to improved insight into individualized pathways toward psychotic experiences, highlighting genetic vulnerabilities to specific stressors likely to lead to phenotypic expression. Ultimately, this will require a more sophisticated mapping between phenomenology and biology than currently exists.”
One approach would be to combine deep phenotyping and behavioral analyses in a framework that could link all relevant levels from symptoms to neurophysiology.
“One such framework is predictive processing theory, which is linked closely with the free energy principle and the Bayesian brain hypothesis and attempts to explain perceptual and cognitive phenomena as manifestations of a drive to maintain as accurate an internal model of one’s surroundings as possible by minimizing prediction errors. This relatively simple scheme makes specific – and, importantly, falsifiable – assessments of the mathematical signatures of neurotypical processes and the ways they might break down to produce specific psychiatric symptoms.”
Dr. Powers is an assistant professor at the department of psychiatry at Yale University, New Haven, Conn., and serves as medical director of the PRIME Psychosis Research Clinic at Yale. His comments came in an accompanying editorial (JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2391 ).
The genetic links uncovered in this study offer an intriguing, but incomplete look at the risks of psychotic experiences and their complicated intertwinings with other mental disorders, wrote Albert R. Powers III, MD, PhD.
“Penetrance of the genes in question likely depends at least in part on environmental influences, some of which have been studied extensively,” he wrote. “Recently, some have proposed risk stratification by exposome – a composite score of relevant exposures that may increase risk for psychosis and is analogous to the polygenic risk score used [here].
“The combination of environmental and genetic composite scores may lead to improved insight into individualized pathways toward psychotic experiences, highlighting genetic vulnerabilities to specific stressors likely to lead to phenotypic expression. Ultimately, this will require a more sophisticated mapping between phenomenology and biology than currently exists.”
One approach would be to combine deep phenotyping and behavioral analyses in a framework that could link all relevant levels from symptoms to neurophysiology.
“One such framework is predictive processing theory, which is linked closely with the free energy principle and the Bayesian brain hypothesis and attempts to explain perceptual and cognitive phenomena as manifestations of a drive to maintain as accurate an internal model of one’s surroundings as possible by minimizing prediction errors. This relatively simple scheme makes specific – and, importantly, falsifiable – assessments of the mathematical signatures of neurotypical processes and the ways they might break down to produce specific psychiatric symptoms.”
Dr. Powers is an assistant professor at the department of psychiatry at Yale University, New Haven, Conn., and serves as medical director of the PRIME Psychosis Research Clinic at Yale. His comments came in an accompanying editorial (JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2391 ).
Four genetic variations appear to link psychotic experiences with other psychiatric disorders, including schizophrenia, major depressive disorder, bipolar disorder, and neurodevelopmental disorders, a large genetic study has concluded.
, reported Sophie E. Legge, PhD, and colleagues. Their study was published in JAMA Psychiatry.
Although it is informative, the study is unlikely to expand the knowledge of schizophrenia-specific genetics.
“Consistent with other studies, the heritability estimate (1.71%) was low, and given that the variance explained in our polygenic risk analysis was also low, the finding suggests that understanding the genetics of psychotic experiences is unlikely to have an important effect on understanding the genetics of schizophrenia specifically,” wrote Dr. Legge, of the MRC Center for Neuropsychiatric Genetics and Genomics in the division of psychological medicine and clinical neurosciences at Cardiff (Wales) University, and colleagues.
The team conducted a genomewide association study (GWAS) using data from 127,966 individuals in the U.K. Biobank. Of these, 6,123 reported any psychotic experience, 2,143 reported distressing psychotic experiences, and 3,337 reported multiple experiences. The remainder served as controls. At the time of the biobank data collection, the subjects were a mean of 64 years of age; 56% were women.
First psychotic experience occurred at a mean of almost 32 years of age, but about a third reported that the first episode occurred before age 20, or that psychotic experiences had been happening ever since they could remember. Another third reported their first experience between ages 40 and 76 years.
The investigators conducted three GWAS studies: one for any psychotic experience, one for distressing experiences, and one for multiple experiences.
No significant genetic associations were found among those with multiple psychotic experiences, the authors said.
But they did find four variants significantly associated with the other experience categories.
Two variants were associated with any psychotic experience. Those with rs10994278, an intronic variant within Ankyrin-3 (ANK3), were 16% more likely to have a psychotic experience (odds ratio, 1.16). Those with intergenic variant rs549656827 were 39% less likely (OR, 0.61). “The ANK3 gene encodes ankyrin-G, a protein that has been shown to regulate the assembly of voltage-gated sodium channels and is essential for normal synaptic function,” the authors said. “ANK3 is one of strongest and most replicated genes for bipolar disorder, and variants within ANK3 have also been associated in the Psychiatric Genomics Consortium cross-disorder GWAS, and in a rare variant analysis of autism spectrum disorder.”
Two variants were linked to distressing psychotic experiences: rs75459873, intronic to cannabinoid receptor 2 (CNR2), decreased the risk by 34% (OR, 0.66). Intergenic variant rs3849810 increased the risk by 12% (OR, 1.12).
“CNR2 encodes for CB2, one of two well-characterized cannabinoid receptors. Several lines of evidence have implicated the endocannabinoid system in psychiatric disorders, including schizophrenia and depression. The main psychoactive agent of cannabis, tetrahydrocannabinol, can cause acute psychotic symptoms and cognitive impairment. Given that cannabis use is strongly associated with psychotic experiences, we tested, but found no evidence for, a mediating or moderating effect of cannabis use on the association of rs75459873 and distressing psychotic experiences. However, while no evidence was found in this study, a mediating effect of cannabis use cannot be ruled out given the relatively low power of such analyses and the potential measurement error.”
Also, significant genetic correlations were found between any psychotic experiences and major depressive disorder, autism spectrum disorder, ADHD, and schizophrenia. However, the polygenic risk scores for schizophrenia, major depressive disorder, bipolar disorder, ADHD, and autism spectrum disorder, were low.
“We also considered individual psychotic symptoms and found that polygenic risk scores for schizophrenia, bipolar disorder, depression, and ADHD were more strongly associated with delusions of persecution than with the other psychotic symptoms.”
Those with distressing psychotic experiences tended to have more copy number variations (CNVs) associated with schizophrenia (OR, 2.04) and neurodevelopmental disorders (OR, 1.75). The team also found significant associations between distressing experiences and major depressive disorder, ADHD, autism spectrum disorder, and schizophrenia.
“We found particular enrichment of these [polygenic risk scores] in distressing psychotic experiences and for delusions of persecution,” they noted. “ ... All schizophrenia-associated [copy number variations] are also associated with neurodevelopmental disorders such as intellectual disability and autism.”
The study’s strengths include its large sample size. Among its limitations, the researchers said, are the study’s retrospective measurement of psychotic experiences based on self-report from a questionnaire that was online. Gathering the data in that way raised the likelihood of possible error, they said.
Dr. Legge reported having no disclosures.
SOURCE: Legge SE et al. JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2508.
Four genetic variations appear to link psychotic experiences with other psychiatric disorders, including schizophrenia, major depressive disorder, bipolar disorder, and neurodevelopmental disorders, a large genetic study has concluded.
, reported Sophie E. Legge, PhD, and colleagues. Their study was published in JAMA Psychiatry.
Although it is informative, the study is unlikely to expand the knowledge of schizophrenia-specific genetics.
“Consistent with other studies, the heritability estimate (1.71%) was low, and given that the variance explained in our polygenic risk analysis was also low, the finding suggests that understanding the genetics of psychotic experiences is unlikely to have an important effect on understanding the genetics of schizophrenia specifically,” wrote Dr. Legge, of the MRC Center for Neuropsychiatric Genetics and Genomics in the division of psychological medicine and clinical neurosciences at Cardiff (Wales) University, and colleagues.
The team conducted a genomewide association study (GWAS) using data from 127,966 individuals in the U.K. Biobank. Of these, 6,123 reported any psychotic experience, 2,143 reported distressing psychotic experiences, and 3,337 reported multiple experiences. The remainder served as controls. At the time of the biobank data collection, the subjects were a mean of 64 years of age; 56% were women.
First psychotic experience occurred at a mean of almost 32 years of age, but about a third reported that the first episode occurred before age 20, or that psychotic experiences had been happening ever since they could remember. Another third reported their first experience between ages 40 and 76 years.
The investigators conducted three GWAS studies: one for any psychotic experience, one for distressing experiences, and one for multiple experiences.
No significant genetic associations were found among those with multiple psychotic experiences, the authors said.
But they did find four variants significantly associated with the other experience categories.
Two variants were associated with any psychotic experience. Those with rs10994278, an intronic variant within Ankyrin-3 (ANK3), were 16% more likely to have a psychotic experience (odds ratio, 1.16). Those with intergenic variant rs549656827 were 39% less likely (OR, 0.61). “The ANK3 gene encodes ankyrin-G, a protein that has been shown to regulate the assembly of voltage-gated sodium channels and is essential for normal synaptic function,” the authors said. “ANK3 is one of strongest and most replicated genes for bipolar disorder, and variants within ANK3 have also been associated in the Psychiatric Genomics Consortium cross-disorder GWAS, and in a rare variant analysis of autism spectrum disorder.”
Two variants were linked to distressing psychotic experiences: rs75459873, intronic to cannabinoid receptor 2 (CNR2), decreased the risk by 34% (OR, 0.66). Intergenic variant rs3849810 increased the risk by 12% (OR, 1.12).
“CNR2 encodes for CB2, one of two well-characterized cannabinoid receptors. Several lines of evidence have implicated the endocannabinoid system in psychiatric disorders, including schizophrenia and depression. The main psychoactive agent of cannabis, tetrahydrocannabinol, can cause acute psychotic symptoms and cognitive impairment. Given that cannabis use is strongly associated with psychotic experiences, we tested, but found no evidence for, a mediating or moderating effect of cannabis use on the association of rs75459873 and distressing psychotic experiences. However, while no evidence was found in this study, a mediating effect of cannabis use cannot be ruled out given the relatively low power of such analyses and the potential measurement error.”
Also, significant genetic correlations were found between any psychotic experiences and major depressive disorder, autism spectrum disorder, ADHD, and schizophrenia. However, the polygenic risk scores for schizophrenia, major depressive disorder, bipolar disorder, ADHD, and autism spectrum disorder, were low.
“We also considered individual psychotic symptoms and found that polygenic risk scores for schizophrenia, bipolar disorder, depression, and ADHD were more strongly associated with delusions of persecution than with the other psychotic symptoms.”
Those with distressing psychotic experiences tended to have more copy number variations (CNVs) associated with schizophrenia (OR, 2.04) and neurodevelopmental disorders (OR, 1.75). The team also found significant associations between distressing experiences and major depressive disorder, ADHD, autism spectrum disorder, and schizophrenia.
“We found particular enrichment of these [polygenic risk scores] in distressing psychotic experiences and for delusions of persecution,” they noted. “ ... All schizophrenia-associated [copy number variations] are also associated with neurodevelopmental disorders such as intellectual disability and autism.”
The study’s strengths include its large sample size. Among its limitations, the researchers said, are the study’s retrospective measurement of psychotic experiences based on self-report from a questionnaire that was online. Gathering the data in that way raised the likelihood of possible error, they said.
Dr. Legge reported having no disclosures.
SOURCE: Legge SE et al. JAMA Psychiatry. 2019 Sep 25. doi: 10.1001/jamapsychiatry.2019.2508.
FROM JAMA PSYCHIATRY
How does alcohol intake affect dementia risk in older adults?
Mild cognitive impairment (MCI) may influence the relationship between alcohol consumption and dementia risk, a study of more than 3,000 adults suggests. In addition, , according to the study, which was published in JAMA Network Open.
“The associations of self-reported alcohol consumption with dementia risk and cognitive decline were more consistently adverse among individuals with MCI than those with normal cognition,” reported Manja Koch, PhD, a researcher in the department of nutrition at Harvard T.H. Chan School of Public Health in Boston and colleagues. “This was particularly true for the subset of individuals [with MCI] who drank more than 14.0 servings per week, whose rate of cognitive decline and risk of dementia were the highest of any subgroup.”
Among older adults with normal cognition, the results generally were consistent with those of a recent meta-analysis that found a U-shaped relationship between drinking and dementia, the researchers said (Eur J Epidemiol. 2017 Jan;32[1]:31-42.).
“Our results did not show significant associations and clearly do not suffice to suggest a clinical benefit from even limited alcohol use,” said Dr. Koch and colleagues. “Nonetheless, our findings provide some reassurance that alcohol consumed within recommended limits was not associated with an increased risk of dementia among older adults with normal baseline cognition.”
GEMS data
To study whether alcohol consumption is associated with the risk of dementia and cognitive decline in older adults with and without MCI, the investigators analyzed data from the Ginkgo Evaluation of Memory Study (GEMS). GEMS was a randomized controlled trial conducted between 2000 and 2008 that found no overall association between ginkgo biloba and dementia prevention. During the trial, participants completed the Modified Mini-Mental State Examination, the Clinical Dementia Rating scale, and the cognitive portion of the Alzheimer’s Disease Assessment Scale.
In the present study, the investigators analyzed data from 3,021 participants aged 72 years and older who were free of dementia at baseline and had provided information about their alcohol intake. Their median age was 78 years, and 46.2% were female. Fifty-eight percent consumed alcohol, including 45% of the participants with MCI at baseline.
During follow-up, 512 cases of dementia occurred. Among the 473 participants with MCI at baseline, the adjusted hazard ratio (HR) for dementia was 1.72 for those who consumed more than 14 drinks per week, compared with light drinkers who consumed less than 1 drink per week. For participants who consumed between 7 and 14 drinks per week, the adjusted HR for dementia was 0.63 among those without MCI and 0.93 among those with MCI, relative to light drinkers who consumed less than 1 drink per week.
Among adults with normal cognition at baseline, daily low-quantity drinking was associated with lower dementia risk, compared with infrequent higher-quantity drinking (HR, 0.45).
Trial excluded adults with excessive alcohol use
Limitations of the study include a lack of data about any changes in alcohol consumption over time. In addition, the original trial excluded people with a known history of excessive alcohol use. Furthermore, it is possible that the “long preclinical phase of dementia” and other health issues affect drinking behavior, the authors said. “At present, our findings cannot be directly translated into clinical recommendations,” the authors said. Nevertheless, the results “suggest that, while caring for older adults, physicians should carefully assess the full dimensions of drinking behavior and cognition when providing guidance to patients about alcohol consumption,” they said.
The study was supported by grants from the National Center for Complementary and Alternative Medicine; the National Institute of Neurological Disorders and Stroke; the Office of Dietary Supplements of the National Institute on Aging; the National Heart, Lung, and Blood Institute; the University of Pittsburgh Alzheimer’s Disease Research Center; the Roena Kulynych Center for Memory and Cognition Research; and Wake Forest University School of Medicine. In addition, the researchers used plasma samples from the National Cell Repository for Alzheimer’s Disease, which receives support from the National Institute on Aging. Dr. Koch had no conflicts of interest. Coauthors disclosed university and government grants and personal fees from pharmaceutical companies outside the study. One author was an employee of Genentech at the time of publication, but Genentech did not contribute to the study.
SOURCE: Koch M et al. JAMA Network Open. 2019 Sep 27. doi: 10.1001/jamanetworkopen.2019.10319.
Mild cognitive impairment (MCI) may influence the relationship between alcohol consumption and dementia risk, a study of more than 3,000 adults suggests. In addition, , according to the study, which was published in JAMA Network Open.
“The associations of self-reported alcohol consumption with dementia risk and cognitive decline were more consistently adverse among individuals with MCI than those with normal cognition,” reported Manja Koch, PhD, a researcher in the department of nutrition at Harvard T.H. Chan School of Public Health in Boston and colleagues. “This was particularly true for the subset of individuals [with MCI] who drank more than 14.0 servings per week, whose rate of cognitive decline and risk of dementia were the highest of any subgroup.”
Among older adults with normal cognition, the results generally were consistent with those of a recent meta-analysis that found a U-shaped relationship between drinking and dementia, the researchers said (Eur J Epidemiol. 2017 Jan;32[1]:31-42.).
“Our results did not show significant associations and clearly do not suffice to suggest a clinical benefit from even limited alcohol use,” said Dr. Koch and colleagues. “Nonetheless, our findings provide some reassurance that alcohol consumed within recommended limits was not associated with an increased risk of dementia among older adults with normal baseline cognition.”
GEMS data
To study whether alcohol consumption is associated with the risk of dementia and cognitive decline in older adults with and without MCI, the investigators analyzed data from the Ginkgo Evaluation of Memory Study (GEMS). GEMS was a randomized controlled trial conducted between 2000 and 2008 that found no overall association between ginkgo biloba and dementia prevention. During the trial, participants completed the Modified Mini-Mental State Examination, the Clinical Dementia Rating scale, and the cognitive portion of the Alzheimer’s Disease Assessment Scale.
In the present study, the investigators analyzed data from 3,021 participants aged 72 years and older who were free of dementia at baseline and had provided information about their alcohol intake. Their median age was 78 years, and 46.2% were female. Fifty-eight percent consumed alcohol, including 45% of the participants with MCI at baseline.
During follow-up, 512 cases of dementia occurred. Among the 473 participants with MCI at baseline, the adjusted hazard ratio (HR) for dementia was 1.72 for those who consumed more than 14 drinks per week, compared with light drinkers who consumed less than 1 drink per week. For participants who consumed between 7 and 14 drinks per week, the adjusted HR for dementia was 0.63 among those without MCI and 0.93 among those with MCI, relative to light drinkers who consumed less than 1 drink per week.
Among adults with normal cognition at baseline, daily low-quantity drinking was associated with lower dementia risk, compared with infrequent higher-quantity drinking (HR, 0.45).
Trial excluded adults with excessive alcohol use
Limitations of the study include a lack of data about any changes in alcohol consumption over time. In addition, the original trial excluded people with a known history of excessive alcohol use. Furthermore, it is possible that the “long preclinical phase of dementia” and other health issues affect drinking behavior, the authors said. “At present, our findings cannot be directly translated into clinical recommendations,” the authors said. Nevertheless, the results “suggest that, while caring for older adults, physicians should carefully assess the full dimensions of drinking behavior and cognition when providing guidance to patients about alcohol consumption,” they said.
The study was supported by grants from the National Center for Complementary and Alternative Medicine; the National Institute of Neurological Disorders and Stroke; the Office of Dietary Supplements of the National Institute on Aging; the National Heart, Lung, and Blood Institute; the University of Pittsburgh Alzheimer’s Disease Research Center; the Roena Kulynych Center for Memory and Cognition Research; and Wake Forest University School of Medicine. In addition, the researchers used plasma samples from the National Cell Repository for Alzheimer’s Disease, which receives support from the National Institute on Aging. Dr. Koch had no conflicts of interest. Coauthors disclosed university and government grants and personal fees from pharmaceutical companies outside the study. One author was an employee of Genentech at the time of publication, but Genentech did not contribute to the study.
SOURCE: Koch M et al. JAMA Network Open. 2019 Sep 27. doi: 10.1001/jamanetworkopen.2019.10319.
Mild cognitive impairment (MCI) may influence the relationship between alcohol consumption and dementia risk, a study of more than 3,000 adults suggests. In addition, , according to the study, which was published in JAMA Network Open.
“The associations of self-reported alcohol consumption with dementia risk and cognitive decline were more consistently adverse among individuals with MCI than those with normal cognition,” reported Manja Koch, PhD, a researcher in the department of nutrition at Harvard T.H. Chan School of Public Health in Boston and colleagues. “This was particularly true for the subset of individuals [with MCI] who drank more than 14.0 servings per week, whose rate of cognitive decline and risk of dementia were the highest of any subgroup.”
Among older adults with normal cognition, the results generally were consistent with those of a recent meta-analysis that found a U-shaped relationship between drinking and dementia, the researchers said (Eur J Epidemiol. 2017 Jan;32[1]:31-42.).
“Our results did not show significant associations and clearly do not suffice to suggest a clinical benefit from even limited alcohol use,” said Dr. Koch and colleagues. “Nonetheless, our findings provide some reassurance that alcohol consumed within recommended limits was not associated with an increased risk of dementia among older adults with normal baseline cognition.”
GEMS data
To study whether alcohol consumption is associated with the risk of dementia and cognitive decline in older adults with and without MCI, the investigators analyzed data from the Ginkgo Evaluation of Memory Study (GEMS). GEMS was a randomized controlled trial conducted between 2000 and 2008 that found no overall association between ginkgo biloba and dementia prevention. During the trial, participants completed the Modified Mini-Mental State Examination, the Clinical Dementia Rating scale, and the cognitive portion of the Alzheimer’s Disease Assessment Scale.
In the present study, the investigators analyzed data from 3,021 participants aged 72 years and older who were free of dementia at baseline and had provided information about their alcohol intake. Their median age was 78 years, and 46.2% were female. Fifty-eight percent consumed alcohol, including 45% of the participants with MCI at baseline.
During follow-up, 512 cases of dementia occurred. Among the 473 participants with MCI at baseline, the adjusted hazard ratio (HR) for dementia was 1.72 for those who consumed more than 14 drinks per week, compared with light drinkers who consumed less than 1 drink per week. For participants who consumed between 7 and 14 drinks per week, the adjusted HR for dementia was 0.63 among those without MCI and 0.93 among those with MCI, relative to light drinkers who consumed less than 1 drink per week.
Among adults with normal cognition at baseline, daily low-quantity drinking was associated with lower dementia risk, compared with infrequent higher-quantity drinking (HR, 0.45).
Trial excluded adults with excessive alcohol use
Limitations of the study include a lack of data about any changes in alcohol consumption over time. In addition, the original trial excluded people with a known history of excessive alcohol use. Furthermore, it is possible that the “long preclinical phase of dementia” and other health issues affect drinking behavior, the authors said. “At present, our findings cannot be directly translated into clinical recommendations,” the authors said. Nevertheless, the results “suggest that, while caring for older adults, physicians should carefully assess the full dimensions of drinking behavior and cognition when providing guidance to patients about alcohol consumption,” they said.
The study was supported by grants from the National Center for Complementary and Alternative Medicine; the National Institute of Neurological Disorders and Stroke; the Office of Dietary Supplements of the National Institute on Aging; the National Heart, Lung, and Blood Institute; the University of Pittsburgh Alzheimer’s Disease Research Center; the Roena Kulynych Center for Memory and Cognition Research; and Wake Forest University School of Medicine. In addition, the researchers used plasma samples from the National Cell Repository for Alzheimer’s Disease, which receives support from the National Institute on Aging. Dr. Koch had no conflicts of interest. Coauthors disclosed university and government grants and personal fees from pharmaceutical companies outside the study. One author was an employee of Genentech at the time of publication, but Genentech did not contribute to the study.
SOURCE: Koch M et al. JAMA Network Open. 2019 Sep 27. doi: 10.1001/jamanetworkopen.2019.10319.
FROM JAMA NETWORK OPEN
FDA expands Dysport’s upper-limb spasticity indication to children
The Food and Drug Administration has expanded the indication of abobotulinumtoxinA (Dysport) for upper-limb spasticity to include patients aged 2 years and older, according to a release from Ipsen. This botulinum toxin product received approval for this indication in adults in 2015 and approval for lower-limb spasticity in patients aged 2 years and older in 2016. Notably, Orphan Drug Exclusivity prevents it from being indicated for patients with cerebral palsy because another botulinum toxin product, onabotulinumtoxinA (Botox), already was approved for the indication in June 2019.

Spasticity affects the muscles and joints of extremities, especially in growing children, and is usually caused by nerve damage, such as head trauma or spinal cord injury. The degree of spasticity can vary from mild muscle stiffness to severe, painful, and uncontrollable muscle spasms.
AbobotulinumtoxinA was evaluated for upper-limb spasticity in a phase 3, randomized, double-blind, low-dose controlled, multicenter study; the study enrolled 210 children aged 2-17 years with the condition and a Modified Ashworth Scale grade 2 or greater for elbow and wrist flexors. The children were randomized 1:1:1 to injections of either 8 units/kg, 16 units/kg, or 2 units/kg into the elbow flexors and wrist flexors. At 6 weeks, there were statistically significant improvements in Modified Ashworth Scale grade, the primary endpoint, with least-square mean changes from baseline of –2.0, –2.3, and –1.6, respectively.
AbobotulinumtoxinA and all other botulinum toxin products carry a boxed warning, the most serious warning the FDA issues. This warning refers to risk of botulism-like symptoms caused by the botulinum toxin spreading away from the injection area; these symptoms can included sometimes life-threatening difficulty swallowing or breathing. AbobotulinumtoxinA is contraindicated in patients with known hypersensitivity to any botulinum toxin or any of the components, those with presence of infection at proposed injection site(s), and those with known allergy to cow’s milk protein. It is also important to note that botulinum toxin preparations are not interchangeable; the potency units of one are not the same as those of another. Full prescribing information can be found on the Ipsen website.
The Food and Drug Administration has expanded the indication of abobotulinumtoxinA (Dysport) for upper-limb spasticity to include patients aged 2 years and older, according to a release from Ipsen. This botulinum toxin product received approval for this indication in adults in 2015 and approval for lower-limb spasticity in patients aged 2 years and older in 2016. Notably, Orphan Drug Exclusivity prevents it from being indicated for patients with cerebral palsy because another botulinum toxin product, onabotulinumtoxinA (Botox), already was approved for the indication in June 2019.
Spasticity affects the muscles and joints of extremities, especially in growing children, and is usually caused by nerve damage, such as head trauma or spinal cord injury. The degree of spasticity can vary from mild muscle stiffness to severe, painful, and uncontrollable muscle spasms.
AbobotulinumtoxinA was evaluated for upper-limb spasticity in a phase 3, randomized, double-blind, low-dose controlled, multicenter study; the study enrolled 210 children aged 2-17 years with the condition and a Modified Ashworth Scale grade 2 or greater for elbow and wrist flexors. The children were randomized 1:1:1 to injections of either 8 units/kg, 16 units/kg, or 2 units/kg into the elbow flexors and wrist flexors. At 6 weeks, there were statistically significant improvements in Modified Ashworth Scale grade, the primary endpoint, with least-square mean changes from baseline of –2.0, –2.3, and –1.6, respectively.
AbobotulinumtoxinA and all other botulinum toxin products carry a boxed warning, the most serious warning the FDA issues. This warning refers to risk of botulism-like symptoms caused by the botulinum toxin spreading away from the injection area; these symptoms can included sometimes life-threatening difficulty swallowing or breathing. AbobotulinumtoxinA is contraindicated in patients with known hypersensitivity to any botulinum toxin or any of the components, those with presence of infection at proposed injection site(s), and those with known allergy to cow’s milk protein. It is also important to note that botulinum toxin preparations are not interchangeable; the potency units of one are not the same as those of another. Full prescribing information can be found on the Ipsen website.
The Food and Drug Administration has expanded the indication of abobotulinumtoxinA (Dysport) for upper-limb spasticity to include patients aged 2 years and older, according to a release from Ipsen. This botulinum toxin product received approval for this indication in adults in 2015 and approval for lower-limb spasticity in patients aged 2 years and older in 2016. Notably, Orphan Drug Exclusivity prevents it from being indicated for patients with cerebral palsy because another botulinum toxin product, onabotulinumtoxinA (Botox), already was approved for the indication in June 2019.
Spasticity affects the muscles and joints of extremities, especially in growing children, and is usually caused by nerve damage, such as head trauma or spinal cord injury. The degree of spasticity can vary from mild muscle stiffness to severe, painful, and uncontrollable muscle spasms.
AbobotulinumtoxinA was evaluated for upper-limb spasticity in a phase 3, randomized, double-blind, low-dose controlled, multicenter study; the study enrolled 210 children aged 2-17 years with the condition and a Modified Ashworth Scale grade 2 or greater for elbow and wrist flexors. The children were randomized 1:1:1 to injections of either 8 units/kg, 16 units/kg, or 2 units/kg into the elbow flexors and wrist flexors. At 6 weeks, there were statistically significant improvements in Modified Ashworth Scale grade, the primary endpoint, with least-square mean changes from baseline of –2.0, –2.3, and –1.6, respectively.
AbobotulinumtoxinA and all other botulinum toxin products carry a boxed warning, the most serious warning the FDA issues. This warning refers to risk of botulism-like symptoms caused by the botulinum toxin spreading away from the injection area; these symptoms can included sometimes life-threatening difficulty swallowing or breathing. AbobotulinumtoxinA is contraindicated in patients with known hypersensitivity to any botulinum toxin or any of the components, those with presence of infection at proposed injection site(s), and those with known allergy to cow’s milk protein. It is also important to note that botulinum toxin preparations are not interchangeable; the potency units of one are not the same as those of another. Full prescribing information can be found on the Ipsen website.
Legislators disagree on new drug-pricing proposal
Legislators sparred about the best way to lower drug prices during a Sept. 25 hearing, bickering along partisan lines over new legislation by U.S. House Speaker Nancy Pelosi, (D-Calif.) that would require Medicare to negotiate drug prices with manufacturers.

The more than 4-hour hearing by the House Committee on Energy & Commerce Subcommittee on Health centered on HR 3, the “Lower Drug Costs Now Act of 2019,” introduced by Speaker Pelosi on Sept. 24, which would compel the Centers for Medicare & Medicaid Services to make deals with manufacturers on the maximum, reasonable price for the top 250 highest-cost drugs. If a drug maker refused to participate in the negotiation, the company would face a steep noncompliance fee, according to the bill, while manufacturers that overcharged Medicare or failed to offer the negotiated price would be subject to a civil penalty equal to 10 times the cost difference. The legislation includes a $2,000 out-of-pocket limit for Medicare patients.
If enacted, the legislation would transform the pricing landscape of medications and allow more families to access needed treatments, Speaker Pelosi said during the hearing.
“What it does, as you know; it ends the ban – imagine, there’s a ban on negotiating for lower drug prices,” she said. “So, it ends the ban for the [U.S. Department of Health and Human Services] Secretary now to have the opportunity to negotiate for lower prices. But, the even better news is that these drug prices will be lower not just for Medicare recipients, which was one of the original proposals, but for everyone. It will stop companies from ripping us off by charging five times, four times, three times what is charged in other countries.”
However, Republican legislators spent much of the hearing criticizing the bill, claiming the legislation was crafted behind closed doors by Democrats who made no efforts to gain Republican feedback.
“There is no debate that Republicans and Democrats want to work together to lower drug cost for consumers,” Ranking Member Greg Walden (R-Ore.) said during the hearing. “Madam chair, I have to strongly express my great frustration about the decision to sabotage both the tradition of this committee and the bipartisan work that you know was well underway to tackle high-cost drugs. ... We’ve worked in a bipartisan way up until now. I thought we were headed in a good faith down that same path until the speaker’s office dropped this partisan plan on our [progress]. This is partisan politics at its worst, and it’s an avoidable failure.”
Amid the partisan squabbling, legislators heard differing views from economists about the logistics of the legislation and whether the pricing negotiation model makes sense for the United States.
Gerard F. Anderson, PhD, a professor at Johns Hopkins University and director of the Johns Hopkins Center for Hospital Finance and Management in Baltimore told congressional leaders that negotiation between the government and drug makers is both possible and results in lower prices, even for drugs without therapeutic competition. He noted that the Medicaid program currently negotiates prices for supplemental rebates and that the Veterans Administration and the Department of Defense routinely negotiate discounts greater than the federal supply schedule. Dr. Anderson estimated that the Veterans Administration and the Department of Defense pay an average of 30%-40% less than Medicare prescription drug plans for the same medications.

“Allowing the federal government to negotiate prices for expensive drugs without competition in both the public and private sectors will be more effective in lowering drug prices for everyone,” Dr. Anderson said during testimony. “ Imposing financial penalties for drug companies will bring them to the table. International prices can be used to determine if the drug company is negotiating in good faith.”
But Benedic Ippolito, PhD, a research fellow for the American Enterprise Institute, Washington, raised concerns the legislation may cause reduced innovation. He said the United States accounts for about 60% of drug spending in the developed world, and that, because the market is so large, changes in spending will have first-order implications for the types of future drugs available, Dr. Ippolito said during testimony.
“Consider the incentives associated with the [bill’s] negotiation process,” he said. “Drugs that have no competitors would be subject to aggressive rate regulation by the [HHS] Secretary. The same is not true of drugs with at least one such competitor. It is entirely possible that being a second market entrant could prove substantially more profitable than bringing a novel therapeutic to market. Thus, the proposal could substantially depress incentives to pursue path-breaking drugs.”
Outside the hearing, Speaker Pelosi’s proposed bill is being praised by some physician groups, including the American College of Physicians. In a statement, ACP President Robert McLean, MD, said the college was pleased that the bill focuses on keeping drugs affordable for patients and includes provisions that would support research into new therapies and treatment options.
“ACP specifically supports the provisions that would allow Medicare to negotiate prices with manufacturers,” Dr. McLean said in the statement. “The College has longstanding policy supporting the ability of Medicare to leverage its purchasing power and directly negotiate with manufacturers for drug prices. Although ACP does not have policy on several of the specific provisions in the bill, we are supportive of its overall goals and direction, particularly the emphasis on negotiation and transparency in drug pricing.
Meanwhile, Senate Finance Chairman Charles E. Grassley, (R-Iowa) and Ranking Member Ron Wyden (D-Ore.) released the statutory text of their own drug-pricing legislation on Sept. 25, titled “the Prescription Drug Pricing Reduction Act of 2019 (S-2543).” The bill calls for a number of changes to the Medicare Part D program, such as reduced beneficiary cost sharing and linking drug price increases to the rate of inflation. The legislation would also make more information available regarding pharmacy benefit manager practices and change how Medicare calculates Part B prescription drug payment amounts to lower spending and out-of-pocket costs for patients.
“President Trump has called on Congress to work together on a bipartisan bill to lower prescription drug prices, [and] there’s only one bipartisan bill in Congress to lower prescription drug prices that’s passed the committee process,” Chairman Grassley said in a statement. “This is it. Making prescription drugs more affordable consistently ranks as a top issue for Americans from every corner of the country. I encourage my colleagues on both sides of the aisle to come to the table and work with us to get a bill passed and signed into law.”
Legislators sparred about the best way to lower drug prices during a Sept. 25 hearing, bickering along partisan lines over new legislation by U.S. House Speaker Nancy Pelosi, (D-Calif.) that would require Medicare to negotiate drug prices with manufacturers.
The more than 4-hour hearing by the House Committee on Energy & Commerce Subcommittee on Health centered on HR 3, the “Lower Drug Costs Now Act of 2019,” introduced by Speaker Pelosi on Sept. 24, which would compel the Centers for Medicare & Medicaid Services to make deals with manufacturers on the maximum, reasonable price for the top 250 highest-cost drugs. If a drug maker refused to participate in the negotiation, the company would face a steep noncompliance fee, according to the bill, while manufacturers that overcharged Medicare or failed to offer the negotiated price would be subject to a civil penalty equal to 10 times the cost difference. The legislation includes a $2,000 out-of-pocket limit for Medicare patients.
If enacted, the legislation would transform the pricing landscape of medications and allow more families to access needed treatments, Speaker Pelosi said during the hearing.
“What it does, as you know; it ends the ban – imagine, there’s a ban on negotiating for lower drug prices,” she said. “So, it ends the ban for the [U.S. Department of Health and Human Services] Secretary now to have the opportunity to negotiate for lower prices. But, the even better news is that these drug prices will be lower not just for Medicare recipients, which was one of the original proposals, but for everyone. It will stop companies from ripping us off by charging five times, four times, three times what is charged in other countries.”
However, Republican legislators spent much of the hearing criticizing the bill, claiming the legislation was crafted behind closed doors by Democrats who made no efforts to gain Republican feedback.
“There is no debate that Republicans and Democrats want to work together to lower drug cost for consumers,” Ranking Member Greg Walden (R-Ore.) said during the hearing. “Madam chair, I have to strongly express my great frustration about the decision to sabotage both the tradition of this committee and the bipartisan work that you know was well underway to tackle high-cost drugs. ... We’ve worked in a bipartisan way up until now. I thought we were headed in a good faith down that same path until the speaker’s office dropped this partisan plan on our [progress]. This is partisan politics at its worst, and it’s an avoidable failure.”
Amid the partisan squabbling, legislators heard differing views from economists about the logistics of the legislation and whether the pricing negotiation model makes sense for the United States.
Gerard F. Anderson, PhD, a professor at Johns Hopkins University and director of the Johns Hopkins Center for Hospital Finance and Management in Baltimore told congressional leaders that negotiation between the government and drug makers is both possible and results in lower prices, even for drugs without therapeutic competition. He noted that the Medicaid program currently negotiates prices for supplemental rebates and that the Veterans Administration and the Department of Defense routinely negotiate discounts greater than the federal supply schedule. Dr. Anderson estimated that the Veterans Administration and the Department of Defense pay an average of 30%-40% less than Medicare prescription drug plans for the same medications.
“Allowing the federal government to negotiate prices for expensive drugs without competition in both the public and private sectors will be more effective in lowering drug prices for everyone,” Dr. Anderson said during testimony. “ Imposing financial penalties for drug companies will bring them to the table. International prices can be used to determine if the drug company is negotiating in good faith.”
But Benedic Ippolito, PhD, a research fellow for the American Enterprise Institute, Washington, raised concerns the legislation may cause reduced innovation. He said the United States accounts for about 60% of drug spending in the developed world, and that, because the market is so large, changes in spending will have first-order implications for the types of future drugs available, Dr. Ippolito said during testimony.
“Consider the incentives associated with the [bill’s] negotiation process,” he said. “Drugs that have no competitors would be subject to aggressive rate regulation by the [HHS] Secretary. The same is not true of drugs with at least one such competitor. It is entirely possible that being a second market entrant could prove substantially more profitable than bringing a novel therapeutic to market. Thus, the proposal could substantially depress incentives to pursue path-breaking drugs.”
Outside the hearing, Speaker Pelosi’s proposed bill is being praised by some physician groups, including the American College of Physicians. In a statement, ACP President Robert McLean, MD, said the college was pleased that the bill focuses on keeping drugs affordable for patients and includes provisions that would support research into new therapies and treatment options.
“ACP specifically supports the provisions that would allow Medicare to negotiate prices with manufacturers,” Dr. McLean said in the statement. “The College has longstanding policy supporting the ability of Medicare to leverage its purchasing power and directly negotiate with manufacturers for drug prices. Although ACP does not have policy on several of the specific provisions in the bill, we are supportive of its overall goals and direction, particularly the emphasis on negotiation and transparency in drug pricing.
Meanwhile, Senate Finance Chairman Charles E. Grassley, (R-Iowa) and Ranking Member Ron Wyden (D-Ore.) released the statutory text of their own drug-pricing legislation on Sept. 25, titled “the Prescription Drug Pricing Reduction Act of 2019 (S-2543).” The bill calls for a number of changes to the Medicare Part D program, such as reduced beneficiary cost sharing and linking drug price increases to the rate of inflation. The legislation would also make more information available regarding pharmacy benefit manager practices and change how Medicare calculates Part B prescription drug payment amounts to lower spending and out-of-pocket costs for patients.
“President Trump has called on Congress to work together on a bipartisan bill to lower prescription drug prices, [and] there’s only one bipartisan bill in Congress to lower prescription drug prices that’s passed the committee process,” Chairman Grassley said in a statement. “This is it. Making prescription drugs more affordable consistently ranks as a top issue for Americans from every corner of the country. I encourage my colleagues on both sides of the aisle to come to the table and work with us to get a bill passed and signed into law.”
Legislators sparred about the best way to lower drug prices during a Sept. 25 hearing, bickering along partisan lines over new legislation by U.S. House Speaker Nancy Pelosi, (D-Calif.) that would require Medicare to negotiate drug prices with manufacturers.
The more than 4-hour hearing by the House Committee on Energy & Commerce Subcommittee on Health centered on HR 3, the “Lower Drug Costs Now Act of 2019,” introduced by Speaker Pelosi on Sept. 24, which would compel the Centers for Medicare & Medicaid Services to make deals with manufacturers on the maximum, reasonable price for the top 250 highest-cost drugs. If a drug maker refused to participate in the negotiation, the company would face a steep noncompliance fee, according to the bill, while manufacturers that overcharged Medicare or failed to offer the negotiated price would be subject to a civil penalty equal to 10 times the cost difference. The legislation includes a $2,000 out-of-pocket limit for Medicare patients.
If enacted, the legislation would transform the pricing landscape of medications and allow more families to access needed treatments, Speaker Pelosi said during the hearing.
“What it does, as you know; it ends the ban – imagine, there’s a ban on negotiating for lower drug prices,” she said. “So, it ends the ban for the [U.S. Department of Health and Human Services] Secretary now to have the opportunity to negotiate for lower prices. But, the even better news is that these drug prices will be lower not just for Medicare recipients, which was one of the original proposals, but for everyone. It will stop companies from ripping us off by charging five times, four times, three times what is charged in other countries.”
However, Republican legislators spent much of the hearing criticizing the bill, claiming the legislation was crafted behind closed doors by Democrats who made no efforts to gain Republican feedback.
“There is no debate that Republicans and Democrats want to work together to lower drug cost for consumers,” Ranking Member Greg Walden (R-Ore.) said during the hearing. “Madam chair, I have to strongly express my great frustration about the decision to sabotage both the tradition of this committee and the bipartisan work that you know was well underway to tackle high-cost drugs. ... We’ve worked in a bipartisan way up until now. I thought we were headed in a good faith down that same path until the speaker’s office dropped this partisan plan on our [progress]. This is partisan politics at its worst, and it’s an avoidable failure.”
Amid the partisan squabbling, legislators heard differing views from economists about the logistics of the legislation and whether the pricing negotiation model makes sense for the United States.
Gerard F. Anderson, PhD, a professor at Johns Hopkins University and director of the Johns Hopkins Center for Hospital Finance and Management in Baltimore told congressional leaders that negotiation between the government and drug makers is both possible and results in lower prices, even for drugs without therapeutic competition. He noted that the Medicaid program currently negotiates prices for supplemental rebates and that the Veterans Administration and the Department of Defense routinely negotiate discounts greater than the federal supply schedule. Dr. Anderson estimated that the Veterans Administration and the Department of Defense pay an average of 30%-40% less than Medicare prescription drug plans for the same medications.
“Allowing the federal government to negotiate prices for expensive drugs without competition in both the public and private sectors will be more effective in lowering drug prices for everyone,” Dr. Anderson said during testimony. “ Imposing financial penalties for drug companies will bring them to the table. International prices can be used to determine if the drug company is negotiating in good faith.”
But Benedic Ippolito, PhD, a research fellow for the American Enterprise Institute, Washington, raised concerns the legislation may cause reduced innovation. He said the United States accounts for about 60% of drug spending in the developed world, and that, because the market is so large, changes in spending will have first-order implications for the types of future drugs available, Dr. Ippolito said during testimony.
“Consider the incentives associated with the [bill’s] negotiation process,” he said. “Drugs that have no competitors would be subject to aggressive rate regulation by the [HHS] Secretary. The same is not true of drugs with at least one such competitor. It is entirely possible that being a second market entrant could prove substantially more profitable than bringing a novel therapeutic to market. Thus, the proposal could substantially depress incentives to pursue path-breaking drugs.”
Outside the hearing, Speaker Pelosi’s proposed bill is being praised by some physician groups, including the American College of Physicians. In a statement, ACP President Robert McLean, MD, said the college was pleased that the bill focuses on keeping drugs affordable for patients and includes provisions that would support research into new therapies and treatment options.
“ACP specifically supports the provisions that would allow Medicare to negotiate prices with manufacturers,” Dr. McLean said in the statement. “The College has longstanding policy supporting the ability of Medicare to leverage its purchasing power and directly negotiate with manufacturers for drug prices. Although ACP does not have policy on several of the specific provisions in the bill, we are supportive of its overall goals and direction, particularly the emphasis on negotiation and transparency in drug pricing.
Meanwhile, Senate Finance Chairman Charles E. Grassley, (R-Iowa) and Ranking Member Ron Wyden (D-Ore.) released the statutory text of their own drug-pricing legislation on Sept. 25, titled “the Prescription Drug Pricing Reduction Act of 2019 (S-2543).” The bill calls for a number of changes to the Medicare Part D program, such as reduced beneficiary cost sharing and linking drug price increases to the rate of inflation. The legislation would also make more information available regarding pharmacy benefit manager practices and change how Medicare calculates Part B prescription drug payment amounts to lower spending and out-of-pocket costs for patients.
“President Trump has called on Congress to work together on a bipartisan bill to lower prescription drug prices, [and] there’s only one bipartisan bill in Congress to lower prescription drug prices that’s passed the committee process,” Chairman Grassley said in a statement. “This is it. Making prescription drugs more affordable consistently ranks as a top issue for Americans from every corner of the country. I encourage my colleagues on both sides of the aisle to come to the table and work with us to get a bill passed and signed into law.”
Prevalence of developmental disabilities up significantly since 2009
The significant increase in developmental disability prevalence in U.S. children from 2009 to 2017 may have been driven by factors such as better identification and availability of services, according to analysis of National Health Interview Survey (NHIS) data.
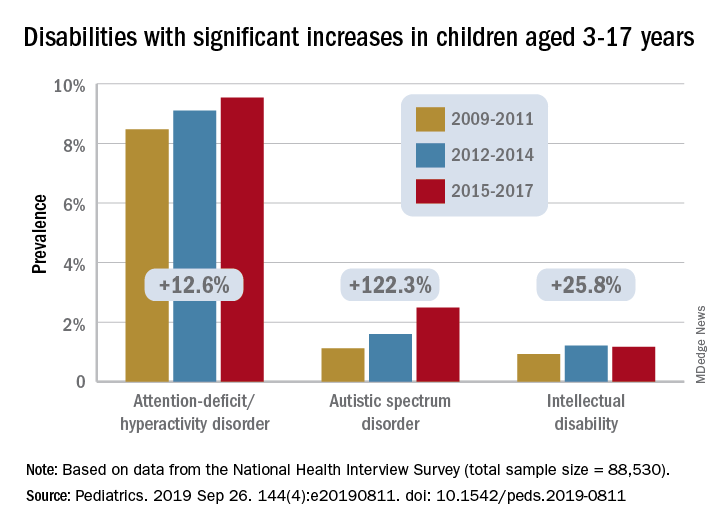
The prevalence of any developmental disability rose from 16% in 2009 to 18% in 2017 in children aged 3-17 years, for an increase of 9.5%, Benjamin Zablotsky, PhD, of the National Center for Health Statistics, Hyattsville, Md., and associates reported in Pediatrics.
Changes among the various conditions were not uniform, however, and most of the increase came from ADHD, autism spectrum disorder (ASD), and intellectual disability, which were up by 12.6%, 122.3%, and 25.8%, respectively, they said, based on data for 88,530 children from the NHIS.
Because other studies have shown that the “prevalence of ADHD symptoms and impairment has remained steady over time,” the increase according to NHIS data “could be driven by better identification of children who meet criteria for ADHD, as current estimates of diagnosed prevalence are in line with community-based studies,” Dr. Zablotsky and associates wrote.
Improved identification, “related to increasing parental awareness and changing provider practices” also may account for much of the rise in ASD prevalence, they said.
It also may be related to changes in the survey itself, as “an increase of about 80% was seen in the 2014 NHIS following changes to the wording and ordering of the question capturing ASD.”
The investigators offered a similar explanation for the increase in intellectual disability prevalence, which increased by 72% from 2011 to 2013 when the phrasing of the NHIS question was changed from “mental retardation” to “intellectual disability, also known as mental retardation.”
The other specific conditions – blindness, cerebral palsy, hearing loss, learning disability, seizures, and stuttering/stammering – all saw nonsignificant changes during the study period, with one exception. “Other developmental delay” dropped by a significant 13%. “It is possible that parents have become less likely to select this category because their children have increasingly been diagnosed with another specified condition on the survey,” Dr. Zablotsky and associates said.
“These findings have major implications for pediatric training and workforce needs and more broadly for public health policies and resources to meet the complex medical and educational needs of the rising number of children with disabilities and their families,” Maureen S. Durkin, PhD, DrPH, said in an accompanying editorial.
The trends reported by Dr. Zablotsky and associates, which have been seen in other countries, are the result of improved survival among children, so, “in this sense, a rise in the prevalence of developmental disabilities may be seen as a global indicator of progress in children’s health and pediatric care,” said Dr. Durkin, a epidemiologist in Madison, Wis.
Dr. Zablotsky and coauthors said that there was no external funding for the study and that they had no relevant financial relationships to disclose. Dr. Durkin said that she had no potential conflicts of interest.
SOURCES: Zablotsky B et al. Pediatrics. 2019 Sep 26. 144(4):e20190811. doi: 10.1542/peds.2019-0811; Durkin MS. Pediatrics. 2019 Sep 26. 144[4]:e20192005. doi: 10.1542/peds.2019-2005.
The significant increase in developmental disability prevalence in U.S. children from 2009 to 2017 may have been driven by factors such as better identification and availability of services, according to analysis of National Health Interview Survey (NHIS) data.
The prevalence of any developmental disability rose from 16% in 2009 to 18% in 2017 in children aged 3-17 years, for an increase of 9.5%, Benjamin Zablotsky, PhD, of the National Center for Health Statistics, Hyattsville, Md., and associates reported in Pediatrics.
Changes among the various conditions were not uniform, however, and most of the increase came from ADHD, autism spectrum disorder (ASD), and intellectual disability, which were up by 12.6%, 122.3%, and 25.8%, respectively, they said, based on data for 88,530 children from the NHIS.
Because other studies have shown that the “prevalence of ADHD symptoms and impairment has remained steady over time,” the increase according to NHIS data “could be driven by better identification of children who meet criteria for ADHD, as current estimates of diagnosed prevalence are in line with community-based studies,” Dr. Zablotsky and associates wrote.
Improved identification, “related to increasing parental awareness and changing provider practices” also may account for much of the rise in ASD prevalence, they said.
It also may be related to changes in the survey itself, as “an increase of about 80% was seen in the 2014 NHIS following changes to the wording and ordering of the question capturing ASD.”
The investigators offered a similar explanation for the increase in intellectual disability prevalence, which increased by 72% from 2011 to 2013 when the phrasing of the NHIS question was changed from “mental retardation” to “intellectual disability, also known as mental retardation.”
The other specific conditions – blindness, cerebral palsy, hearing loss, learning disability, seizures, and stuttering/stammering – all saw nonsignificant changes during the study period, with one exception. “Other developmental delay” dropped by a significant 13%. “It is possible that parents have become less likely to select this category because their children have increasingly been diagnosed with another specified condition on the survey,” Dr. Zablotsky and associates said.
“These findings have major implications for pediatric training and workforce needs and more broadly for public health policies and resources to meet the complex medical and educational needs of the rising number of children with disabilities and their families,” Maureen S. Durkin, PhD, DrPH, said in an accompanying editorial.
The trends reported by Dr. Zablotsky and associates, which have been seen in other countries, are the result of improved survival among children, so, “in this sense, a rise in the prevalence of developmental disabilities may be seen as a global indicator of progress in children’s health and pediatric care,” said Dr. Durkin, a epidemiologist in Madison, Wis.
Dr. Zablotsky and coauthors said that there was no external funding for the study and that they had no relevant financial relationships to disclose. Dr. Durkin said that she had no potential conflicts of interest.
SOURCES: Zablotsky B et al. Pediatrics. 2019 Sep 26. 144(4):e20190811. doi: 10.1542/peds.2019-0811; Durkin MS. Pediatrics. 2019 Sep 26. 144[4]:e20192005. doi: 10.1542/peds.2019-2005.
The significant increase in developmental disability prevalence in U.S. children from 2009 to 2017 may have been driven by factors such as better identification and availability of services, according to analysis of National Health Interview Survey (NHIS) data.
The prevalence of any developmental disability rose from 16% in 2009 to 18% in 2017 in children aged 3-17 years, for an increase of 9.5%, Benjamin Zablotsky, PhD, of the National Center for Health Statistics, Hyattsville, Md., and associates reported in Pediatrics.
Changes among the various conditions were not uniform, however, and most of the increase came from ADHD, autism spectrum disorder (ASD), and intellectual disability, which were up by 12.6%, 122.3%, and 25.8%, respectively, they said, based on data for 88,530 children from the NHIS.
Because other studies have shown that the “prevalence of ADHD symptoms and impairment has remained steady over time,” the increase according to NHIS data “could be driven by better identification of children who meet criteria for ADHD, as current estimates of diagnosed prevalence are in line with community-based studies,” Dr. Zablotsky and associates wrote.
Improved identification, “related to increasing parental awareness and changing provider practices” also may account for much of the rise in ASD prevalence, they said.
It also may be related to changes in the survey itself, as “an increase of about 80% was seen in the 2014 NHIS following changes to the wording and ordering of the question capturing ASD.”
The investigators offered a similar explanation for the increase in intellectual disability prevalence, which increased by 72% from 2011 to 2013 when the phrasing of the NHIS question was changed from “mental retardation” to “intellectual disability, also known as mental retardation.”
The other specific conditions – blindness, cerebral palsy, hearing loss, learning disability, seizures, and stuttering/stammering – all saw nonsignificant changes during the study period, with one exception. “Other developmental delay” dropped by a significant 13%. “It is possible that parents have become less likely to select this category because their children have increasingly been diagnosed with another specified condition on the survey,” Dr. Zablotsky and associates said.
“These findings have major implications for pediatric training and workforce needs and more broadly for public health policies and resources to meet the complex medical and educational needs of the rising number of children with disabilities and their families,” Maureen S. Durkin, PhD, DrPH, said in an accompanying editorial.
The trends reported by Dr. Zablotsky and associates, which have been seen in other countries, are the result of improved survival among children, so, “in this sense, a rise in the prevalence of developmental disabilities may be seen as a global indicator of progress in children’s health and pediatric care,” said Dr. Durkin, a epidemiologist in Madison, Wis.
Dr. Zablotsky and coauthors said that there was no external funding for the study and that they had no relevant financial relationships to disclose. Dr. Durkin said that she had no potential conflicts of interest.
SOURCES: Zablotsky B et al. Pediatrics. 2019 Sep 26. 144(4):e20190811. doi: 10.1542/peds.2019-0811; Durkin MS. Pediatrics. 2019 Sep 26. 144[4]:e20192005. doi: 10.1542/peds.2019-2005.
FROM PEDIATRICS
Key clinical point:
Major finding: Among U.S. children aged 3-17 years, prevalence of developmental disabilities increased by 9.5% from 2009 to 2017.
Study details: The sample from the National Health Interview Survey included 88,530 children.
Disclosures: The investigators said that there was no external funding for the study and that they had no relevant financial relationships to disclose.
Source: Zablotsky B et al. Pediatrics. 2019 Sep 26. 144(4):e20190811. doi: 10.1542/peds.2019-0811.
Neurostimulation device may treat vertigo in patients with vestibular migraine
according to a small, open-label study published online Sept. 25 in Neurology.
The retrospective, single-center study included data from 18 patients who had vestibular migraine and received acute treatment with a handheld stimulation device. The device used in the study, GammaCore, is approved for the treatment of migraine. There are no approved treatments for vestibular migraine.
“There’s a huge need for effective treatments for vestibular migraine attacks,” first author Shin C. Beh, MD, said in a news release. “People with vestibular migraine do not always have headaches, and when they do, they are often less severe than in typical migraine, so the pain-relieving drugs used for typical migraine often are not effective. People can take drugs that suppress the vertigo or the nausea, but those drugs cause drowsiness and make it hard for people to go about their usual activities.” Dr. Beh is an assistant professor of neurology and neurotherapeutics at University of Texas Southwestern Medical Center in Dallas.
The patients had an average age of about 46 years, and 16 were women. A total of 14 patients received treatment during an acute vestibular migraine attack, and 4 received treatment while they had bothersome interictal dizziness consistent with persistent perceptual postural dizziness.
After stimulation, vertigo improved in 13 of the 14 people with vestibular migraine attacks. In two of these patients, vertigo resolved completely, and five had at least a 50% improvement in vertigo symptoms. On a 0-10 scale, patients’ mean vertigo rating decreased from 5.2 before stimulation to 3.1 after stimulation.
Of five patients with vestibular migraine attacks who had headache, all experienced a reduction in headache pain. Mean headache severity decreased from 6 before stimulation to 2.4 after stimulation.
None of the four patients with interictal dizziness experienced a reduction in dizziness after stimulation.
Patients place the device against the neck to receive electrical stimulation, and patients received stimulation for 2 minutes on each side of the neck.
Participants reported a mild pulling sensation of the neck muscles during the stimulation but did not report pain or other side effects.
“Vestibular migraine is the most common neurologic cause of vertigo and can greatly interfere with a person’s daily life,” Dr. Beh said. “If these results can be confirmed with larger studies, not only could there finally be a treatment for vestibular migraine, such a treatment would also be easy to use.”
A randomized, double-blind, sham-controlled study is needed to further assess the use of noninvasive vagus nerve stimulation in treating vestibular migraine, the authors said.
The study had no targeted funding, and Dr. Beh had no relevant disclosures. Coauthor Deborah I. Friedman, MD, professor of neurology and neurotherapeutics and ophthalmology at UT Southwestern Medical Center, disclosed serving on advisory boards or speaking for pharmaceutical and medical device companies. Dr. Friedman is on the editorial advisory board of Neurology Reviews.
SOURCE: Beh SC et al. Neurology. 2019 Sep 25. doi: 10.1212/WNL.0000000000008388.
according to a small, open-label study published online Sept. 25 in Neurology.
The retrospective, single-center study included data from 18 patients who had vestibular migraine and received acute treatment with a handheld stimulation device. The device used in the study, GammaCore, is approved for the treatment of migraine. There are no approved treatments for vestibular migraine.
“There’s a huge need for effective treatments for vestibular migraine attacks,” first author Shin C. Beh, MD, said in a news release. “People with vestibular migraine do not always have headaches, and when they do, they are often less severe than in typical migraine, so the pain-relieving drugs used for typical migraine often are not effective. People can take drugs that suppress the vertigo or the nausea, but those drugs cause drowsiness and make it hard for people to go about their usual activities.” Dr. Beh is an assistant professor of neurology and neurotherapeutics at University of Texas Southwestern Medical Center in Dallas.
The patients had an average age of about 46 years, and 16 were women. A total of 14 patients received treatment during an acute vestibular migraine attack, and 4 received treatment while they had bothersome interictal dizziness consistent with persistent perceptual postural dizziness.
After stimulation, vertigo improved in 13 of the 14 people with vestibular migraine attacks. In two of these patients, vertigo resolved completely, and five had at least a 50% improvement in vertigo symptoms. On a 0-10 scale, patients’ mean vertigo rating decreased from 5.2 before stimulation to 3.1 after stimulation.
Of five patients with vestibular migraine attacks who had headache, all experienced a reduction in headache pain. Mean headache severity decreased from 6 before stimulation to 2.4 after stimulation.
None of the four patients with interictal dizziness experienced a reduction in dizziness after stimulation.
Patients place the device against the neck to receive electrical stimulation, and patients received stimulation for 2 minutes on each side of the neck.
Participants reported a mild pulling sensation of the neck muscles during the stimulation but did not report pain or other side effects.
“Vestibular migraine is the most common neurologic cause of vertigo and can greatly interfere with a person’s daily life,” Dr. Beh said. “If these results can be confirmed with larger studies, not only could there finally be a treatment for vestibular migraine, such a treatment would also be easy to use.”
A randomized, double-blind, sham-controlled study is needed to further assess the use of noninvasive vagus nerve stimulation in treating vestibular migraine, the authors said.
The study had no targeted funding, and Dr. Beh had no relevant disclosures. Coauthor Deborah I. Friedman, MD, professor of neurology and neurotherapeutics and ophthalmology at UT Southwestern Medical Center, disclosed serving on advisory boards or speaking for pharmaceutical and medical device companies. Dr. Friedman is on the editorial advisory board of Neurology Reviews.
SOURCE: Beh SC et al. Neurology. 2019 Sep 25. doi: 10.1212/WNL.0000000000008388.
according to a small, open-label study published online Sept. 25 in Neurology.
The retrospective, single-center study included data from 18 patients who had vestibular migraine and received acute treatment with a handheld stimulation device. The device used in the study, GammaCore, is approved for the treatment of migraine. There are no approved treatments for vestibular migraine.
“There’s a huge need for effective treatments for vestibular migraine attacks,” first author Shin C. Beh, MD, said in a news release. “People with vestibular migraine do not always have headaches, and when they do, they are often less severe than in typical migraine, so the pain-relieving drugs used for typical migraine often are not effective. People can take drugs that suppress the vertigo or the nausea, but those drugs cause drowsiness and make it hard for people to go about their usual activities.” Dr. Beh is an assistant professor of neurology and neurotherapeutics at University of Texas Southwestern Medical Center in Dallas.
The patients had an average age of about 46 years, and 16 were women. A total of 14 patients received treatment during an acute vestibular migraine attack, and 4 received treatment while they had bothersome interictal dizziness consistent with persistent perceptual postural dizziness.
After stimulation, vertigo improved in 13 of the 14 people with vestibular migraine attacks. In two of these patients, vertigo resolved completely, and five had at least a 50% improvement in vertigo symptoms. On a 0-10 scale, patients’ mean vertigo rating decreased from 5.2 before stimulation to 3.1 after stimulation.
Of five patients with vestibular migraine attacks who had headache, all experienced a reduction in headache pain. Mean headache severity decreased from 6 before stimulation to 2.4 after stimulation.
None of the four patients with interictal dizziness experienced a reduction in dizziness after stimulation.
Patients place the device against the neck to receive electrical stimulation, and patients received stimulation for 2 minutes on each side of the neck.
Participants reported a mild pulling sensation of the neck muscles during the stimulation but did not report pain or other side effects.
“Vestibular migraine is the most common neurologic cause of vertigo and can greatly interfere with a person’s daily life,” Dr. Beh said. “If these results can be confirmed with larger studies, not only could there finally be a treatment for vestibular migraine, such a treatment would also be easy to use.”
A randomized, double-blind, sham-controlled study is needed to further assess the use of noninvasive vagus nerve stimulation in treating vestibular migraine, the authors said.
The study had no targeted funding, and Dr. Beh had no relevant disclosures. Coauthor Deborah I. Friedman, MD, professor of neurology and neurotherapeutics and ophthalmology at UT Southwestern Medical Center, disclosed serving on advisory boards or speaking for pharmaceutical and medical device companies. Dr. Friedman is on the editorial advisory board of Neurology Reviews.
SOURCE: Beh SC et al. Neurology. 2019 Sep 25. doi: 10.1212/WNL.0000000000008388.
FROM NEUROLOGY
Key clinical point: Noninvasive vagus nerve stimulation may reduce the intensity of vertigo and headache in patients with acute vestibular migraine attacks.
Major finding: After stimulation, vertigo improved in 13 out of 14 people with vestibular migraine attacks. In 2 of these patients, vertigo resolved completely, and 5 had at least a 50% improvement in vertigo symptoms. On a 0-10 scale, patients’ mean vertigo rating decreased from 5.2 before stimulation to 3.1 after stimulation.
Study details: An open-label study of 18 patients with vestibular migraine.
Disclosures: The study had no targeted funding, and the first author had no relevant disclosures. A coauthor disclosed serving on advisory boards or speaking for pharmaceutical and medical device companies and is on the editorial advisory board of Neurology Reviews.
Source: Beh SC et al. Neurology. 2019 Sep 25. doi: 10.1212/WNL.0000000000008388.
Continuation of natalizumab treatment reduces risk of MS relapses during pregnancy
STOCKHOLM – according to research presented at the annual congress of the European Committee for Treatment and Research in Multiple Sclerosis. Continuation of treatment is not associated with major fetal risks.
Pregnancy influences the choice of treatment for patients with MS. Not all therapies are compatible with pregnancy; the newer, more effective treatments in particular may pose risks in this regard. “There is a need to accumulate new data on how to manage our highly active patients with MS – in particular, patients who are treated with natalizumab,” said Doriana Landi, MD, PhD, a research fellow at the University of Rome Tor Vergata.
Data suggest that suspension of natalizumab before pregnancy is associated with significant disease worsening. In 2018, Portaccio et al. found that suspending natalizumab treatment before conception is associated with a higher risk of relapses during pregnancy (Neurology. 2018 Mar 6;90[10]:e832-9). Suspending natalizumab treatment at conception, however, reduced the risk of relapse by a factor of three. Other data have suggested that continuing natalizumab treatment until the 30th week of gestation could protect mothers and guarantee fetal safety.
Comparing two cohorts of women with MS
To investigate this question further, Dr. Landi and colleagues investigated a cohort of 29 women with MS who were receiving natalizumab and became pregnant. They compared this cohort’s outcomes with those of the cohort that Portaccio et al. had studied. The investigators categorized participants in both cohorts into three groups according to the time of their last infusion of natalizumab in relation to their last menstrual period. Group 0 had their last infusion before their last menstrual period, group 1 had their last infusion during the first trimester of pregnancy, and group 2 continued treatment after the first trimester. All women restarted natalizumab post partum and had at least 12 months of follow-up after delivery.
Dr. Landi and colleagues calculated the annualized relapse rate during pregnancy and during 12 months post partum for all groups. They also compared the outcomes of newborns between groups.
Prematurity may have influenced rate of hematologic complications
In all, the researchers analyzed 90 completed pregnancies in 86 women with MS. There were no significant demographic differences between groups. The overall population’s mean age was approximately 31 years, and its median Expanded Disability Status Scale (EDSS) score was about 2. The women gave birth to 94 newborns. Mean gestational age was 38.42 weeks, mean birth weight was 2,878.08 mg, and mean length was 48.23 cm.
Group 0 included 31 patients, group 1 included 30 patients, and group 2 included 28 patients. The median interval between the last dose of natalizumab and last menstrual period was 70 days for group 0, 21 days for group 1, and 197 days for group 2. Group 2 received a median of five natalizumab infusions during pregnancy, with a median interval of 80 days between the last prepartum dose and delivery. The median interval between last prepartum dose and the first postpartum dose was 411 days in group 0, 288 in group 1, and 103 in group 2.
Most pregnancies in group 2 were exposed to natalizumab for at least 28 weeks of gestation. Women in group 2 restarted natalizumab treatment significantly earlier, compared with women in the other groups.
Group 0 had a significantly increased ARR during and after pregnancy, compared with the other groups, and group 2 had the lowest ARR during pregnancy. The mean ARR during pregnancy was 1.06 in group 0, 0.49 in group 1, and 0.09 in group 2. The ARR decreased to 0 in pregnancies exposed to natalizumab for more than 90 days. The mean ARR post partum was 0.39 in group 0, 0.23 in group 1, and 0.10 in group 2. Women in group 0 and group 1 had a higher EDSS during pregnancy than before conception.
Newborns’ mean gestational age, birth weight, and length did not differ significantly between groups. Five newborns in group 2, three of whom were premature, had anemia. This outcome is consistent with previous findings, and prematurity may be a confounding factor, said Dr. Landi. The frequency of birth defects was higher in group 2 than in the other groups, but most of them occurred in one twin, said Dr. Landi. The investigators observed malformations in one group 0 newborn (minor), four group 1 newborns (five minor and one major), and four (two minor and four major) group 2 newborns.
In pregnancy during which natalizumab treatment is prolonged until the third trimester, treatment should be restarted after delivery within 2 weeks of the last infusion to avoid potential doubled rebound, said Dr. Landi. Future studies should examine whether extended dosing is as effective as regular dosing. A larger sample size is needed to estimate risks correctly for newborns in exposed pregnancies and counsel patients appropriately.
Dr. Landi reported receiving travel funding from Biogen, Merck Serono, Sanofi-Genzyme, and Teva; honoraria for speaking from Sanofi-Genzyme and Teva; and consultation fees from Merck Serono and Teva. She is an investigator in clinical trials being conducted for Biogen, Merck Serono, Novartis, Roche, and Teva.
SOURCE: Landi D et al. ECTRIMS 2019, Abstract 338.
STOCKHOLM – according to research presented at the annual congress of the European Committee for Treatment and Research in Multiple Sclerosis. Continuation of treatment is not associated with major fetal risks.
Pregnancy influences the choice of treatment for patients with MS. Not all therapies are compatible with pregnancy; the newer, more effective treatments in particular may pose risks in this regard. “There is a need to accumulate new data on how to manage our highly active patients with MS – in particular, patients who are treated with natalizumab,” said Doriana Landi, MD, PhD, a research fellow at the University of Rome Tor Vergata.
Data suggest that suspension of natalizumab before pregnancy is associated with significant disease worsening. In 2018, Portaccio et al. found that suspending natalizumab treatment before conception is associated with a higher risk of relapses during pregnancy (Neurology. 2018 Mar 6;90[10]:e832-9). Suspending natalizumab treatment at conception, however, reduced the risk of relapse by a factor of three. Other data have suggested that continuing natalizumab treatment until the 30th week of gestation could protect mothers and guarantee fetal safety.
Comparing two cohorts of women with MS
To investigate this question further, Dr. Landi and colleagues investigated a cohort of 29 women with MS who were receiving natalizumab and became pregnant. They compared this cohort’s outcomes with those of the cohort that Portaccio et al. had studied. The investigators categorized participants in both cohorts into three groups according to the time of their last infusion of natalizumab in relation to their last menstrual period. Group 0 had their last infusion before their last menstrual period, group 1 had their last infusion during the first trimester of pregnancy, and group 2 continued treatment after the first trimester. All women restarted natalizumab post partum and had at least 12 months of follow-up after delivery.
Dr. Landi and colleagues calculated the annualized relapse rate during pregnancy and during 12 months post partum for all groups. They also compared the outcomes of newborns between groups.
Prematurity may have influenced rate of hematologic complications
In all, the researchers analyzed 90 completed pregnancies in 86 women with MS. There were no significant demographic differences between groups. The overall population’s mean age was approximately 31 years, and its median Expanded Disability Status Scale (EDSS) score was about 2. The women gave birth to 94 newborns. Mean gestational age was 38.42 weeks, mean birth weight was 2,878.08 mg, and mean length was 48.23 cm.
Group 0 included 31 patients, group 1 included 30 patients, and group 2 included 28 patients. The median interval between the last dose of natalizumab and last menstrual period was 70 days for group 0, 21 days for group 1, and 197 days for group 2. Group 2 received a median of five natalizumab infusions during pregnancy, with a median interval of 80 days between the last prepartum dose and delivery. The median interval between last prepartum dose and the first postpartum dose was 411 days in group 0, 288 in group 1, and 103 in group 2.
Most pregnancies in group 2 were exposed to natalizumab for at least 28 weeks of gestation. Women in group 2 restarted natalizumab treatment significantly earlier, compared with women in the other groups.
Group 0 had a significantly increased ARR during and after pregnancy, compared with the other groups, and group 2 had the lowest ARR during pregnancy. The mean ARR during pregnancy was 1.06 in group 0, 0.49 in group 1, and 0.09 in group 2. The ARR decreased to 0 in pregnancies exposed to natalizumab for more than 90 days. The mean ARR post partum was 0.39 in group 0, 0.23 in group 1, and 0.10 in group 2. Women in group 0 and group 1 had a higher EDSS during pregnancy than before conception.
Newborns’ mean gestational age, birth weight, and length did not differ significantly between groups. Five newborns in group 2, three of whom were premature, had anemia. This outcome is consistent with previous findings, and prematurity may be a confounding factor, said Dr. Landi. The frequency of birth defects was higher in group 2 than in the other groups, but most of them occurred in one twin, said Dr. Landi. The investigators observed malformations in one group 0 newborn (minor), four group 1 newborns (five minor and one major), and four (two minor and four major) group 2 newborns.
In pregnancy during which natalizumab treatment is prolonged until the third trimester, treatment should be restarted after delivery within 2 weeks of the last infusion to avoid potential doubled rebound, said Dr. Landi. Future studies should examine whether extended dosing is as effective as regular dosing. A larger sample size is needed to estimate risks correctly for newborns in exposed pregnancies and counsel patients appropriately.
Dr. Landi reported receiving travel funding from Biogen, Merck Serono, Sanofi-Genzyme, and Teva; honoraria for speaking from Sanofi-Genzyme and Teva; and consultation fees from Merck Serono and Teva. She is an investigator in clinical trials being conducted for Biogen, Merck Serono, Novartis, Roche, and Teva.
SOURCE: Landi D et al. ECTRIMS 2019, Abstract 338.
STOCKHOLM – according to research presented at the annual congress of the European Committee for Treatment and Research in Multiple Sclerosis. Continuation of treatment is not associated with major fetal risks.
Pregnancy influences the choice of treatment for patients with MS. Not all therapies are compatible with pregnancy; the newer, more effective treatments in particular may pose risks in this regard. “There is a need to accumulate new data on how to manage our highly active patients with MS – in particular, patients who are treated with natalizumab,” said Doriana Landi, MD, PhD, a research fellow at the University of Rome Tor Vergata.
Data suggest that suspension of natalizumab before pregnancy is associated with significant disease worsening. In 2018, Portaccio et al. found that suspending natalizumab treatment before conception is associated with a higher risk of relapses during pregnancy (Neurology. 2018 Mar 6;90[10]:e832-9). Suspending natalizumab treatment at conception, however, reduced the risk of relapse by a factor of three. Other data have suggested that continuing natalizumab treatment until the 30th week of gestation could protect mothers and guarantee fetal safety.
Comparing two cohorts of women with MS
To investigate this question further, Dr. Landi and colleagues investigated a cohort of 29 women with MS who were receiving natalizumab and became pregnant. They compared this cohort’s outcomes with those of the cohort that Portaccio et al. had studied. The investigators categorized participants in both cohorts into three groups according to the time of their last infusion of natalizumab in relation to their last menstrual period. Group 0 had their last infusion before their last menstrual period, group 1 had their last infusion during the first trimester of pregnancy, and group 2 continued treatment after the first trimester. All women restarted natalizumab post partum and had at least 12 months of follow-up after delivery.
Dr. Landi and colleagues calculated the annualized relapse rate during pregnancy and during 12 months post partum for all groups. They also compared the outcomes of newborns between groups.
Prematurity may have influenced rate of hematologic complications
In all, the researchers analyzed 90 completed pregnancies in 86 women with MS. There were no significant demographic differences between groups. The overall population’s mean age was approximately 31 years, and its median Expanded Disability Status Scale (EDSS) score was about 2. The women gave birth to 94 newborns. Mean gestational age was 38.42 weeks, mean birth weight was 2,878.08 mg, and mean length was 48.23 cm.
Group 0 included 31 patients, group 1 included 30 patients, and group 2 included 28 patients. The median interval between the last dose of natalizumab and last menstrual period was 70 days for group 0, 21 days for group 1, and 197 days for group 2. Group 2 received a median of five natalizumab infusions during pregnancy, with a median interval of 80 days between the last prepartum dose and delivery. The median interval between last prepartum dose and the first postpartum dose was 411 days in group 0, 288 in group 1, and 103 in group 2.
Most pregnancies in group 2 were exposed to natalizumab for at least 28 weeks of gestation. Women in group 2 restarted natalizumab treatment significantly earlier, compared with women in the other groups.
Group 0 had a significantly increased ARR during and after pregnancy, compared with the other groups, and group 2 had the lowest ARR during pregnancy. The mean ARR during pregnancy was 1.06 in group 0, 0.49 in group 1, and 0.09 in group 2. The ARR decreased to 0 in pregnancies exposed to natalizumab for more than 90 days. The mean ARR post partum was 0.39 in group 0, 0.23 in group 1, and 0.10 in group 2. Women in group 0 and group 1 had a higher EDSS during pregnancy than before conception.
Newborns’ mean gestational age, birth weight, and length did not differ significantly between groups. Five newborns in group 2, three of whom were premature, had anemia. This outcome is consistent with previous findings, and prematurity may be a confounding factor, said Dr. Landi. The frequency of birth defects was higher in group 2 than in the other groups, but most of them occurred in one twin, said Dr. Landi. The investigators observed malformations in one group 0 newborn (minor), four group 1 newborns (five minor and one major), and four (two minor and four major) group 2 newborns.
In pregnancy during which natalizumab treatment is prolonged until the third trimester, treatment should be restarted after delivery within 2 weeks of the last infusion to avoid potential doubled rebound, said Dr. Landi. Future studies should examine whether extended dosing is as effective as regular dosing. A larger sample size is needed to estimate risks correctly for newborns in exposed pregnancies and counsel patients appropriately.
Dr. Landi reported receiving travel funding from Biogen, Merck Serono, Sanofi-Genzyme, and Teva; honoraria for speaking from Sanofi-Genzyme and Teva; and consultation fees from Merck Serono and Teva. She is an investigator in clinical trials being conducted for Biogen, Merck Serono, Novartis, Roche, and Teva.
SOURCE: Landi D et al. ECTRIMS 2019, Abstract 338.
REPORTING FROM ECTRIMS 2019
Happiness in my solo practice
I don’t want to rule the world.

Some doctors do, albeit in a non–Attila-the-Hun sort of way. They want to have offices on every street corner, in every suburb of a given city, sometimes more than one city. Like the Starbucks of medicine.
That’s not me. I’m happy in my little one-office world.
Maybe I just don’t have the ambition, or the business mindset, or whatever it takes to want to do that. I understand it’s all part of wanting to be successful, and obviously those doctors are more driven in that direction than I am. The more offices, the more patients can be seen, and the more money you make.
It’s not quite that simple, though. No one can be in more than one place at the same time, so to see more patients at more places you need more doctors. To pay more doctors requires more money, which in turn requires more patients.
There’s nothing wrong with taking over the world (or at least a suburb) if you like that sort of thing. But to me, more money brings more headaches. More offices to rent, more staff to hire, more people to handle billing, IT, HR, payroll, accounting, contracts, and so on.
You can have it. I’ve taken over all the world I want, in my case a 1,200-square-foot suite on the second floor of a small-to-medium-size medical building. To some that may sound unambitious, but to me, it’s perfect.
I know where my Keurig, Sodastream, and office supplies are. Except for my secretary and her cheerfully rambunctious young daughter, I don’t have to worry about sharing stuff here, or if anyone wants a different carpet color, or what’s going on at a satellite office halfway across town.
If other doctors want to try and take over the world, more power to them, but I’m happy with this. Enough is as good as a feast.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I don’t want to rule the world.
Some doctors do, albeit in a non–Attila-the-Hun sort of way. They want to have offices on every street corner, in every suburb of a given city, sometimes more than one city. Like the Starbucks of medicine.
That’s not me. I’m happy in my little one-office world.
Maybe I just don’t have the ambition, or the business mindset, or whatever it takes to want to do that. I understand it’s all part of wanting to be successful, and obviously those doctors are more driven in that direction than I am. The more offices, the more patients can be seen, and the more money you make.
It’s not quite that simple, though. No one can be in more than one place at the same time, so to see more patients at more places you need more doctors. To pay more doctors requires more money, which in turn requires more patients.
There’s nothing wrong with taking over the world (or at least a suburb) if you like that sort of thing. But to me, more money brings more headaches. More offices to rent, more staff to hire, more people to handle billing, IT, HR, payroll, accounting, contracts, and so on.
You can have it. I’ve taken over all the world I want, in my case a 1,200-square-foot suite on the second floor of a small-to-medium-size medical building. To some that may sound unambitious, but to me, it’s perfect.
I know where my Keurig, Sodastream, and office supplies are. Except for my secretary and her cheerfully rambunctious young daughter, I don’t have to worry about sharing stuff here, or if anyone wants a different carpet color, or what’s going on at a satellite office halfway across town.
If other doctors want to try and take over the world, more power to them, but I’m happy with this. Enough is as good as a feast.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I don’t want to rule the world.
Some doctors do, albeit in a non–Attila-the-Hun sort of way. They want to have offices on every street corner, in every suburb of a given city, sometimes more than one city. Like the Starbucks of medicine.
That’s not me. I’m happy in my little one-office world.
Maybe I just don’t have the ambition, or the business mindset, or whatever it takes to want to do that. I understand it’s all part of wanting to be successful, and obviously those doctors are more driven in that direction than I am. The more offices, the more patients can be seen, and the more money you make.
It’s not quite that simple, though. No one can be in more than one place at the same time, so to see more patients at more places you need more doctors. To pay more doctors requires more money, which in turn requires more patients.
There’s nothing wrong with taking over the world (or at least a suburb) if you like that sort of thing. But to me, more money brings more headaches. More offices to rent, more staff to hire, more people to handle billing, IT, HR, payroll, accounting, contracts, and so on.
You can have it. I’ve taken over all the world I want, in my case a 1,200-square-foot suite on the second floor of a small-to-medium-size medical building. To some that may sound unambitious, but to me, it’s perfect.
I know where my Keurig, Sodastream, and office supplies are. Except for my secretary and her cheerfully rambunctious young daughter, I don’t have to worry about sharing stuff here, or if anyone wants a different carpet color, or what’s going on at a satellite office halfway across town.
If other doctors want to try and take over the world, more power to them, but I’m happy with this. Enough is as good as a feast.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.