User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Dark Brown Hyperkeratotic Nodule on the Back
The Diagnosis: Seborrheic Keratosis-like Melanoma
Seborrheic keratosis (SK) is a benign neoplasm commonly encountered on the skin and frequently diagnosed by clinical examination alone. Seborrheic keratosis-like melanomas are melanomas that clinically or dermatoscopically resemble SKs and thus can be challenging to accurately diagnose. Melanomas can have a hyperkeratotic or verrucous appearance1-3 and can even exhibit dermatoscopic and microscopic features that are found in SKs such as comedolike openings and milialike cysts as well as acanthosis and pseudohorn cysts, respectively.2
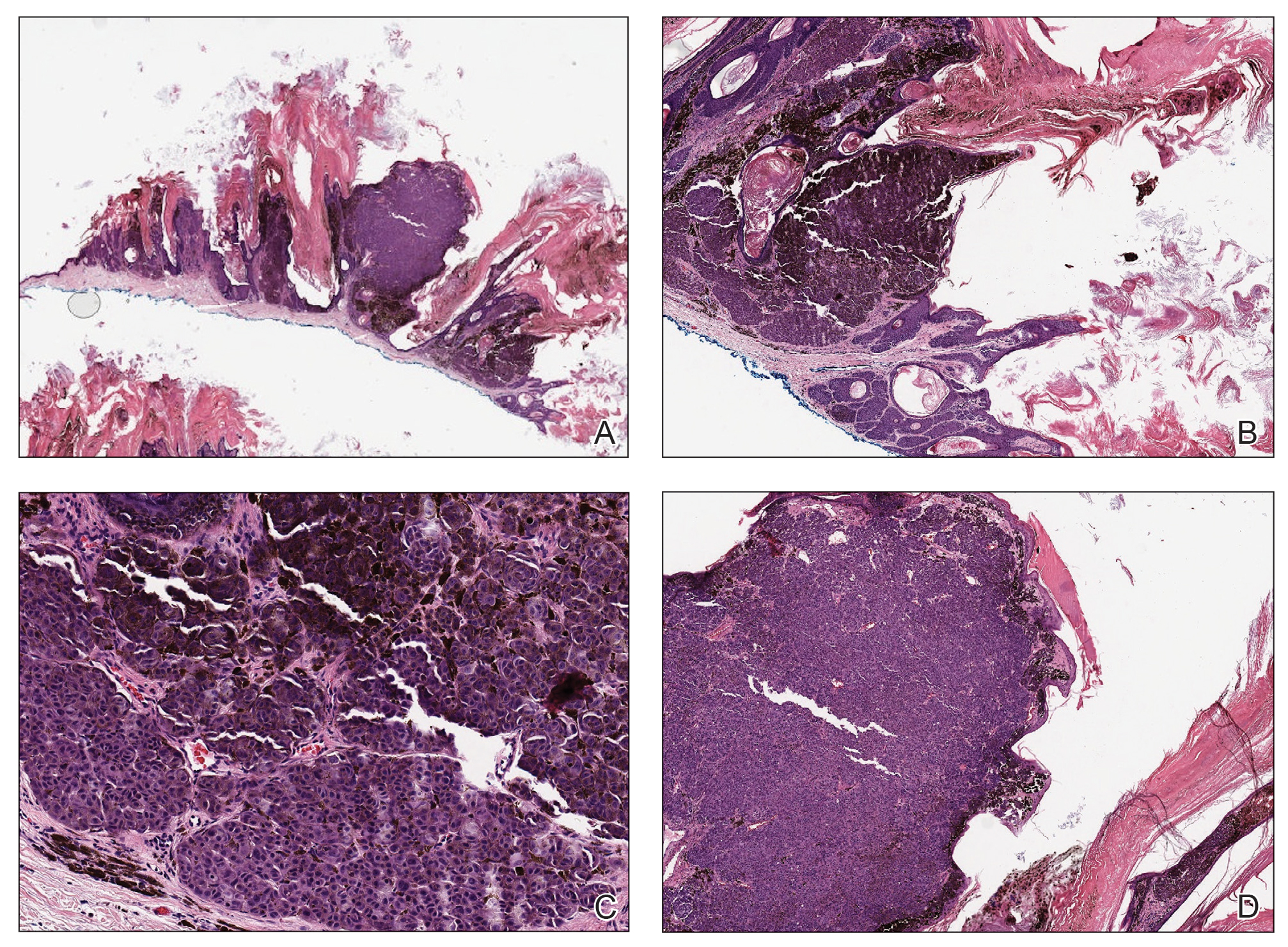
In our patient, histopathology revealed SK-like architecture with hyperorthokeratosis, papillomatosis, pseudohorn cyst formation, and basaloid acanthosis (Figure). However, within the lesion was an asymmetric proliferation of nested atypical melanocytes with melanin pigment production. The atypical melanocytes filled and expanded papillomatous projections without notable pagetoid growth and extended into the dermis. There was a background congenital nevus component. These findings were diagnostic of invasive malignant melanoma, extending to a Breslow depth of 5.5 mm. A follow-up sentinel lymph node biopsy was negative for metastatic melanoma. The clinical and histologic findings did not show melanoma in the surrounding skin to suggest colonization of an SK by an adjacent melanoma. The clinical history of a long-standing lesion in conjunction with a congenital nevus component on histology favored a diagnosis of melanoma arising in association with a congenital nevus with an SK-like architecture rather than arising in a preexisting SK or de novo melanoma.
Because our patient did not have multiple widespread SKs and reported rapid growth in the lesion in the last 6 months, there was concern for a malignant neoplasm. However, in patients with numerous SKs or areas of chronically sun-damaged skin, it can be difficult to identify suspicious lesions. It is important for clinicians to remain aware of SK-like melanomas and have a lower threshold for biopsy of any changing or symptomatic lesion that clinically resembles an SK. In our case, the history of change and the markedly different clinical appearance of the lesion in comparison to our patient's SKs prompted the biopsy. Criteria have been proposed to help differentiate these entities under dermoscopy, with melanoma showing the presence of the blue-black sign, pigment network, pseudopods or streaks, and/or the blue-white veil.4
Cutaneous metastases classically present as dermal nodules, plaques, or ulcers.5,6 A rare pigmented case of metastatic breast adenocarcinoma clinically mimicking melanoma has been reported.7 There is limited literature on the dermoscopic features of cutaneous metastases, but it appears that polymorphic vascular patterns are most common.5,8 The possibility of a metastatic melanoma involving an SK is a theoretical consideration, but there was no prior history of melanoma in our patient, and the histologic findings were consistent with primary melanoma. There was no histologic evidence of pigmented metastatic breast carcinoma or metastatic lung carcinoma.
Pigmented malignant hidroacanthoma simplex and pigmented porocarcinomas are rare malignant sweat gland tumors.9-11 Their benign counterparts are the more commonly encountered hidroacanthoma simplex (intraepidermal poroma) and poroma. Pigmented malignant hidroacanthoma simplex has been reported to clinically mimic an irritated SK.10 The histopathology of our case did not have features of malignant hidroacanthoma simplex or porocarcinoma. Pigmented squamous cell carcinoma is an uncommon variant of squamous cell carcinoma, and histopathology would reveal proliferation of atypical keratinocytes.12
- Saggini A, Cota C, Lora V, et al. Uncommon histopathological variants of malignant melanoma. part 2. Am J Dermatopathol. 2019;41:321-342.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019;80:178-188.
- Tran PT, Truong AK, Munday W, et al. Verrucous melanoma masquerading as a seborrheic keratosis. Dermatol Online J. 2019;25:13030/qt1m07k7fm.
- Carrera C, Segura S, Aguilera P. Dermoscopic clues for diagnosing melanomas that resemble seborrheic keratosis. JAMA Dermatol. 2017;153:544-551.
- Strickley JD, Jenson AB, Jung JY. Cutaneous metastasis. Hematol Oncol Clin North Am. 2019;33:173-197.
- Chernoff KA, Marghoob AA, Lacouture ME. Dermoscopic findings in cutaneous metastases. JAMA Dermatol. 2014;150:429-433.
- Marti N, Molina I, Monteagudo C, et al. Cutaneous metastasis of breast carcinoma mimicking malignant melanoma in scalp. Dermatol Online J. 2008;14:12.
- Kelati A, Gallouj S. Dermoscopy of skin metastases from breast cancer: two case reports. J Med Case Rep. 2018;12:273.
- Ishida M, Hotta M, Kushima R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38:227-231.
- Lee JY, Lin MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33:705-708.
- Ueo T, Kashima K, Daa T, et al. Porocarcinoma arising in pigmented hidroacanthoma simplex. Am J Dermatopathol. 2005;27:500-503.
- Motta de Morais P, Schettini A, Rocha J, et al. Pigmented squamous cell carcinoma: case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
The Diagnosis: Seborrheic Keratosis-like Melanoma
Seborrheic keratosis (SK) is a benign neoplasm commonly encountered on the skin and frequently diagnosed by clinical examination alone. Seborrheic keratosis-like melanomas are melanomas that clinically or dermatoscopically resemble SKs and thus can be challenging to accurately diagnose. Melanomas can have a hyperkeratotic or verrucous appearance1-3 and can even exhibit dermatoscopic and microscopic features that are found in SKs such as comedolike openings and milialike cysts as well as acanthosis and pseudohorn cysts, respectively.2
In our patient, histopathology revealed SK-like architecture with hyperorthokeratosis, papillomatosis, pseudohorn cyst formation, and basaloid acanthosis (Figure). However, within the lesion was an asymmetric proliferation of nested atypical melanocytes with melanin pigment production. The atypical melanocytes filled and expanded papillomatous projections without notable pagetoid growth and extended into the dermis. There was a background congenital nevus component. These findings were diagnostic of invasive malignant melanoma, extending to a Breslow depth of 5.5 mm. A follow-up sentinel lymph node biopsy was negative for metastatic melanoma. The clinical and histologic findings did not show melanoma in the surrounding skin to suggest colonization of an SK by an adjacent melanoma. The clinical history of a long-standing lesion in conjunction with a congenital nevus component on histology favored a diagnosis of melanoma arising in association with a congenital nevus with an SK-like architecture rather than arising in a preexisting SK or de novo melanoma.
Because our patient did not have multiple widespread SKs and reported rapid growth in the lesion in the last 6 months, there was concern for a malignant neoplasm. However, in patients with numerous SKs or areas of chronically sun-damaged skin, it can be difficult to identify suspicious lesions. It is important for clinicians to remain aware of SK-like melanomas and have a lower threshold for biopsy of any changing or symptomatic lesion that clinically resembles an SK. In our case, the history of change and the markedly different clinical appearance of the lesion in comparison to our patient's SKs prompted the biopsy. Criteria have been proposed to help differentiate these entities under dermoscopy, with melanoma showing the presence of the blue-black sign, pigment network, pseudopods or streaks, and/or the blue-white veil.4
Cutaneous metastases classically present as dermal nodules, plaques, or ulcers.5,6 A rare pigmented case of metastatic breast adenocarcinoma clinically mimicking melanoma has been reported.7 There is limited literature on the dermoscopic features of cutaneous metastases, but it appears that polymorphic vascular patterns are most common.5,8 The possibility of a metastatic melanoma involving an SK is a theoretical consideration, but there was no prior history of melanoma in our patient, and the histologic findings were consistent with primary melanoma. There was no histologic evidence of pigmented metastatic breast carcinoma or metastatic lung carcinoma.
Pigmented malignant hidroacanthoma simplex and pigmented porocarcinomas are rare malignant sweat gland tumors.9-11 Their benign counterparts are the more commonly encountered hidroacanthoma simplex (intraepidermal poroma) and poroma. Pigmented malignant hidroacanthoma simplex has been reported to clinically mimic an irritated SK.10 The histopathology of our case did not have features of malignant hidroacanthoma simplex or porocarcinoma. Pigmented squamous cell carcinoma is an uncommon variant of squamous cell carcinoma, and histopathology would reveal proliferation of atypical keratinocytes.12
The Diagnosis: Seborrheic Keratosis-like Melanoma
Seborrheic keratosis (SK) is a benign neoplasm commonly encountered on the skin and frequently diagnosed by clinical examination alone. Seborrheic keratosis-like melanomas are melanomas that clinically or dermatoscopically resemble SKs and thus can be challenging to accurately diagnose. Melanomas can have a hyperkeratotic or verrucous appearance1-3 and can even exhibit dermatoscopic and microscopic features that are found in SKs such as comedolike openings and milialike cysts as well as acanthosis and pseudohorn cysts, respectively.2
In our patient, histopathology revealed SK-like architecture with hyperorthokeratosis, papillomatosis, pseudohorn cyst formation, and basaloid acanthosis (Figure). However, within the lesion was an asymmetric proliferation of nested atypical melanocytes with melanin pigment production. The atypical melanocytes filled and expanded papillomatous projections without notable pagetoid growth and extended into the dermis. There was a background congenital nevus component. These findings were diagnostic of invasive malignant melanoma, extending to a Breslow depth of 5.5 mm. A follow-up sentinel lymph node biopsy was negative for metastatic melanoma. The clinical and histologic findings did not show melanoma in the surrounding skin to suggest colonization of an SK by an adjacent melanoma. The clinical history of a long-standing lesion in conjunction with a congenital nevus component on histology favored a diagnosis of melanoma arising in association with a congenital nevus with an SK-like architecture rather than arising in a preexisting SK or de novo melanoma.
Because our patient did not have multiple widespread SKs and reported rapid growth in the lesion in the last 6 months, there was concern for a malignant neoplasm. However, in patients with numerous SKs or areas of chronically sun-damaged skin, it can be difficult to identify suspicious lesions. It is important for clinicians to remain aware of SK-like melanomas and have a lower threshold for biopsy of any changing or symptomatic lesion that clinically resembles an SK. In our case, the history of change and the markedly different clinical appearance of the lesion in comparison to our patient's SKs prompted the biopsy. Criteria have been proposed to help differentiate these entities under dermoscopy, with melanoma showing the presence of the blue-black sign, pigment network, pseudopods or streaks, and/or the blue-white veil.4
Cutaneous metastases classically present as dermal nodules, plaques, or ulcers.5,6 A rare pigmented case of metastatic breast adenocarcinoma clinically mimicking melanoma has been reported.7 There is limited literature on the dermoscopic features of cutaneous metastases, but it appears that polymorphic vascular patterns are most common.5,8 The possibility of a metastatic melanoma involving an SK is a theoretical consideration, but there was no prior history of melanoma in our patient, and the histologic findings were consistent with primary melanoma. There was no histologic evidence of pigmented metastatic breast carcinoma or metastatic lung carcinoma.
Pigmented malignant hidroacanthoma simplex and pigmented porocarcinomas are rare malignant sweat gland tumors.9-11 Their benign counterparts are the more commonly encountered hidroacanthoma simplex (intraepidermal poroma) and poroma. Pigmented malignant hidroacanthoma simplex has been reported to clinically mimic an irritated SK.10 The histopathology of our case did not have features of malignant hidroacanthoma simplex or porocarcinoma. Pigmented squamous cell carcinoma is an uncommon variant of squamous cell carcinoma, and histopathology would reveal proliferation of atypical keratinocytes.12
- Saggini A, Cota C, Lora V, et al. Uncommon histopathological variants of malignant melanoma. part 2. Am J Dermatopathol. 2019;41:321-342.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019;80:178-188.
- Tran PT, Truong AK, Munday W, et al. Verrucous melanoma masquerading as a seborrheic keratosis. Dermatol Online J. 2019;25:13030/qt1m07k7fm.
- Carrera C, Segura S, Aguilera P. Dermoscopic clues for diagnosing melanomas that resemble seborrheic keratosis. JAMA Dermatol. 2017;153:544-551.
- Strickley JD, Jenson AB, Jung JY. Cutaneous metastasis. Hematol Oncol Clin North Am. 2019;33:173-197.
- Chernoff KA, Marghoob AA, Lacouture ME. Dermoscopic findings in cutaneous metastases. JAMA Dermatol. 2014;150:429-433.
- Marti N, Molina I, Monteagudo C, et al. Cutaneous metastasis of breast carcinoma mimicking malignant melanoma in scalp. Dermatol Online J. 2008;14:12.
- Kelati A, Gallouj S. Dermoscopy of skin metastases from breast cancer: two case reports. J Med Case Rep. 2018;12:273.
- Ishida M, Hotta M, Kushima R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38:227-231.
- Lee JY, Lin MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33:705-708.
- Ueo T, Kashima K, Daa T, et al. Porocarcinoma arising in pigmented hidroacanthoma simplex. Am J Dermatopathol. 2005;27:500-503.
- Motta de Morais P, Schettini A, Rocha J, et al. Pigmented squamous cell carcinoma: case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
- Saggini A, Cota C, Lora V, et al. Uncommon histopathological variants of malignant melanoma. part 2. Am J Dermatopathol. 2019;41:321-342.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019;80:178-188.
- Tran PT, Truong AK, Munday W, et al. Verrucous melanoma masquerading as a seborrheic keratosis. Dermatol Online J. 2019;25:13030/qt1m07k7fm.
- Carrera C, Segura S, Aguilera P. Dermoscopic clues for diagnosing melanomas that resemble seborrheic keratosis. JAMA Dermatol. 2017;153:544-551.
- Strickley JD, Jenson AB, Jung JY. Cutaneous metastasis. Hematol Oncol Clin North Am. 2019;33:173-197.
- Chernoff KA, Marghoob AA, Lacouture ME. Dermoscopic findings in cutaneous metastases. JAMA Dermatol. 2014;150:429-433.
- Marti N, Molina I, Monteagudo C, et al. Cutaneous metastasis of breast carcinoma mimicking malignant melanoma in scalp. Dermatol Online J. 2008;14:12.
- Kelati A, Gallouj S. Dermoscopy of skin metastases from breast cancer: two case reports. J Med Case Rep. 2018;12:273.
- Ishida M, Hotta M, Kushima R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38:227-231.
- Lee JY, Lin MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33:705-708.
- Ueo T, Kashima K, Daa T, et al. Porocarcinoma arising in pigmented hidroacanthoma simplex. Am J Dermatopathol. 2005;27:500-503.
- Motta de Morais P, Schettini A, Rocha J, et al. Pigmented squamous cell carcinoma: case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
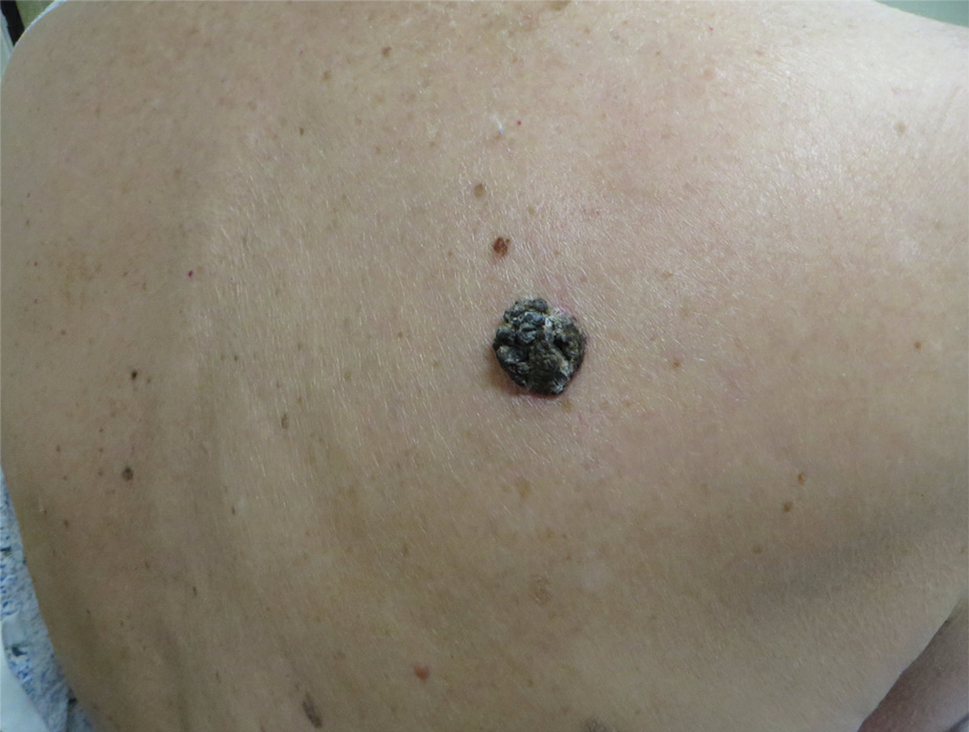
A 71-year-old woman presented with a persistent asymptomatic lesion on the right upper back that had recently increased in size and changed in color, shape, and texture. The lesion had been present for many years. Physical examination revealed a 1.5-cm, dark brown, hyperkeratotic nodule with no identifiable pigment network on dermatoscopy. The patient had no personal history of melanoma but did have a history of stage I non–small cell lung cancer. A review of systems was noncontributory. A shave biopsy of the lesion was performed.
Empowering Residents to Address Socioeconomic Disparities in Dermatology
Studding almost every inch of skin except the face are gray lichenified plaques coating a patient’s body like worn leather. Raking his nails across his arm, the patient reminds me how long he had waited to receive this referral and how early he had awoken for this appointment. He was well acquainted with the value of promptness; in his world, it might make the difference between sleeping on a cot and a night spent on concrete.
Over the last year, the patient had cycled through the few safety-net clinics scattered throughout the city. He had accumulated numerous different diagnoses from atopic dermatitis to disseminated tinea corporis. A few minutes, one #15 scalpel, and mineral oil were all it took for us to unravel the mystery. As the attending and I peered through the microscope at the scabies ovum, I couldn’t help but wonder about the alternative outcomes to his case. Left untreated, scabies compromises the skin barrier, paving the way for secondary infections such as cellulitis. Depending on the pathogen, this infection may in turn evolve into acute postinfectious glomerulonephritis.1-4 An elusive diagnosis can quietly escalate into considerable morbidity for patients. This case highlights the dire consequences of dermatologic health disparities and places medicine’s primordial function into sharp focus: the alleviation of suffering.
The Dermatologic Burden of Disease
As a major contributor to global disease burden, dermatologic disease is the fourth greatest cause of disability worldwide when mortality is factored out.5,6 Among global rural populations, dermatologic disease constitutes one of the leading causes of death and/or loss of professional capabilities.7 In the United States alone, nearly 27% of the population saw a physician for at least 1 dermatologic disease in 2013.5 The tremendous prevalence of skin disease magnifies discrepancies in access to dermatologic care, which has been observed to be influenced by age, socioeconomic background, rurality, and sex.8
There has been growing focus on the national shortage of dermatologists over the last 2 decades.9,10 With an aging population and rising incidence of skin cancer, this undersupply is projected to increase and disproportionately impact ethnic minorities as well as those from socioeconomically disadvantaged backgrounds.8,9,11-14 These trends are of particular importance to residents and medical trainees. Multiple studies have demonstrated that the patient demographic of hospital-based resident clinics includes primarily minority and disenfranchised populations with poorer overall health.15-17 In contrast to faculty clinics, residents treat patients who are more likely to be nonwhite and more likely to be reimbursed by Medicaid.18 The unique demographic makeup of hospital-based resident clinics raises questions about the preparedness and comfort of resident physicians in managing the nuances of health care delivery in these settings.10
Providing equitable care to marginalized populations within the constraints of 15- to 30-minute visits can be challenging to physicians and trainees. Even clinicians with the best of intentions may be impeded by a lack of familiarity with the daily realities of impoverished living conditions, implicit prejudice against people living in poverty, and adapting recommendations to varying levels of health literacy among patients.19,20 Contending with these daunting obstacles can be discouraging. Given how entrenched certain institutional barriers are, questioning them may seem an exercise in futility, yet history demonstrates that residents can and have been empowered to improve tangible outcomes for vulnerable populations. In reflecting on approaches of the general medical education system, The Josiah Macy Jr. Foundation President George E. Thibault, MD, observed that, “When appropriately trained, deployed and incented, [residents] can help achieve institutional goals to improve quality, safety and efficiency.”21
Start Small But Dream Big
Action begins with awareness. Medical school and teaching hospital curricula are increasingly integrating educational exercises regarding the social determinants of health and populations with unmet needs. Medical training presents an exclusive opportunity to gain exposure to and familiarity with patient populations that one might not otherwise encounter. Immersion programs provide invaluable experience in tailoring health care delivery to the needs of vulnerable communities. Although opportunities for international rotations abound, domestic rotations among underserved populations can be just as transformative, including correctional medicine, homeless clinics, the Indian Health Service, and rural communities.
Create Partnerships to Broaden Impact of Service
Affecting the largest and most visible organ, skin disease often presents a substantial concern for patients and can herald systemic disease. The nature of dermatologic disease engenders close collaboration between general practitioners and specialists. For example, while resident-run or safety-net clinics characteristically center on providing holistic care for patients through internal medicine or primary care, these overworked and understaffed clinics often are in need of evaluation by specialists for specific concerns. Some clinic models feature dermatology faculty who volunteer routinely (ie, every 2 weeks, every month) to examine all the clinic’s patients presenting with concerns pertinent to the specialty. Drawing on their respective areas of expertise, general practitioners and dermatologists therefore can collaborate to connect disadvantaged patients with the specialized care they need.
Challenges Present Opportunities for Innovation
Adhering to the social distancing requirements of the COVID-19 pandemic protocol has driven clinicians to utilize innovative approaches to patient care. The rural-urban misdistribution of the dermatologist workforce has long been established, with rural patients often experiencing lengthy wait times to see a specialist.9 Both synchronous and asynchronous teledermatology modalities provide an ideal platform for triaging patients with dermatologic concerns who otherwise have meager access to a dermatologist.
Final Thoughts
Residency training is a prime opportunity to gain exposure to the broad spectrum of disease within dermatology as well as the diverse range of affected patients. Drawing on the aforementioned strategies, residents can leverage this knowledge in the service of underserved patients.
- McCarthy JS, Kemp DJ, Walton SF, et al. Scabies: more than just an irritation. Postgrad Med J. 2004;80:382-387.
- Svartman M, Finklea JF, Earle DP, et al. Epidemic scabies and acute glomerulonephritis in Trinidad. Lancet. 1972;1:249-251.
- Hersch C. Acute glomerulonephritis due to skin disease, with special reference to scabies. S Afr Med J. 1967;41:29-34.
- Carapetis JR, Connors C, Yarmirr D, et al. Success of a scabies control program in an Australian aboriginal community. Pediatr Infect Dis J. 1997;16:494-499.
- Lim HW, Collins SAB, Resneck JS Jr, et al. The burden of skin disease in the United States [published online March 1, 2017]. J Am Acad Dermatol. 2017;76:958-972.e2.
- Karimkhani C, Dellavalle RP, Coffeng LE, et al. Global skin disease morbidity and mortality: an update from the Global Burden of Disease Study 2013. JAMA Dermatol. 2017;153:406-412.
- Morrone A. Poverty, dignity, and forgotten skin care: dermatology in the stream of human mobile population. Dermatol Clin. 2008;26:245-256, vi-vii.
- Tripathi R, Knusel KD, Ezaldein HH, et al. Association of demographic and socioeconomic characteristics with differences in use of outpatient dermatology services in the United States. JAMA Dermatol. 2018;154:1286-1291.
- Feng H, Berk-Krauss J, Feng PW, et al. Comparison of dermatologist density between urban and rural counties in the United States. JAMA Dermatol. 2018;154:1265-1271.
- Blanco G, Vasquez R, Nezafati K, et al. How residency programs can foster practice for the underserved. J Am Acad Dermatol. 2012;67:158-159.
- Kosmadaki MG, Gilchrest BA. The demographics of aging in the United States: implications for dermatology. Arch Dermatol. 2002;138:1427-1428.
- Donaldson MR, Coldiron BM. No end in sight: the skin cancer epidemic continues. Semin Cutan Med Surg. 2011;30:3-5.
- Dall TM, Gallo PD, Chakrabarti R, et al. An aging population and growing disease burden will require a large and specialized health care workforce by 2025. Health Aff (Millwood). 2013;32:2013-2020.
- Sauaia A, Dellavalle RP. Health care inequities: an introduction for dermatology providers. Dermatol Clin. 2009;27:103-107.
- Brook RH, Fink A, Kosecoff J, et al. Educating physicians and treating patients in the ambulatory setting. where are we going and how will we know when we arrive? Ann Intern Med. 1987;107:392-398.
- Yancy WS Jr, Macpherson DS, Hanusa BH, et al. Patient satisfaction in resident and attending ambulatory care clinics. J Gen Intern Med. 2001;16:755-762. 17. Fiebach NH,
- Wong JG. Taking care of patients in resident clinics: where do we stand? J Gen Intern Med. 2001;16:787-789.
- Loignon C, Boudreault-Fournier A, Truchon K, et al. Medical residents reflect on their prejudices toward poverty: a photovoice training project. BMC Med Educ. 2014;14:1050.
- Scheid D, Logue E, Gilchrist VJ, et al. Do we practice what we preach? comparing the patients of faculty and residents. Fam Med. 1995;27:519-524.
- Loignon C, Gottin T, Dupéré S, et al. General practitioners’ perspective on poverty: a qualitative study in Montreal, Canada. Fam Pract. 2018;35:105-110.
- Parks T. Empowered residents can help transform medical care. American Medical Association website. Published November 30, 2016. Accessed March 18, 2021. www.ama-assn.org/education/improve-gme/empowered-residents-can-help-transform-medical-care
Studding almost every inch of skin except the face are gray lichenified plaques coating a patient’s body like worn leather. Raking his nails across his arm, the patient reminds me how long he had waited to receive this referral and how early he had awoken for this appointment. He was well acquainted with the value of promptness; in his world, it might make the difference between sleeping on a cot and a night spent on concrete.
Over the last year, the patient had cycled through the few safety-net clinics scattered throughout the city. He had accumulated numerous different diagnoses from atopic dermatitis to disseminated tinea corporis. A few minutes, one #15 scalpel, and mineral oil were all it took for us to unravel the mystery. As the attending and I peered through the microscope at the scabies ovum, I couldn’t help but wonder about the alternative outcomes to his case. Left untreated, scabies compromises the skin barrier, paving the way for secondary infections such as cellulitis. Depending on the pathogen, this infection may in turn evolve into acute postinfectious glomerulonephritis.1-4 An elusive diagnosis can quietly escalate into considerable morbidity for patients. This case highlights the dire consequences of dermatologic health disparities and places medicine’s primordial function into sharp focus: the alleviation of suffering.
The Dermatologic Burden of Disease
As a major contributor to global disease burden, dermatologic disease is the fourth greatest cause of disability worldwide when mortality is factored out.5,6 Among global rural populations, dermatologic disease constitutes one of the leading causes of death and/or loss of professional capabilities.7 In the United States alone, nearly 27% of the population saw a physician for at least 1 dermatologic disease in 2013.5 The tremendous prevalence of skin disease magnifies discrepancies in access to dermatologic care, which has been observed to be influenced by age, socioeconomic background, rurality, and sex.8
There has been growing focus on the national shortage of dermatologists over the last 2 decades.9,10 With an aging population and rising incidence of skin cancer, this undersupply is projected to increase and disproportionately impact ethnic minorities as well as those from socioeconomically disadvantaged backgrounds.8,9,11-14 These trends are of particular importance to residents and medical trainees. Multiple studies have demonstrated that the patient demographic of hospital-based resident clinics includes primarily minority and disenfranchised populations with poorer overall health.15-17 In contrast to faculty clinics, residents treat patients who are more likely to be nonwhite and more likely to be reimbursed by Medicaid.18 The unique demographic makeup of hospital-based resident clinics raises questions about the preparedness and comfort of resident physicians in managing the nuances of health care delivery in these settings.10
Providing equitable care to marginalized populations within the constraints of 15- to 30-minute visits can be challenging to physicians and trainees. Even clinicians with the best of intentions may be impeded by a lack of familiarity with the daily realities of impoverished living conditions, implicit prejudice against people living in poverty, and adapting recommendations to varying levels of health literacy among patients.19,20 Contending with these daunting obstacles can be discouraging. Given how entrenched certain institutional barriers are, questioning them may seem an exercise in futility, yet history demonstrates that residents can and have been empowered to improve tangible outcomes for vulnerable populations. In reflecting on approaches of the general medical education system, The Josiah Macy Jr. Foundation President George E. Thibault, MD, observed that, “When appropriately trained, deployed and incented, [residents] can help achieve institutional goals to improve quality, safety and efficiency.”21
Start Small But Dream Big
Action begins with awareness. Medical school and teaching hospital curricula are increasingly integrating educational exercises regarding the social determinants of health and populations with unmet needs. Medical training presents an exclusive opportunity to gain exposure to and familiarity with patient populations that one might not otherwise encounter. Immersion programs provide invaluable experience in tailoring health care delivery to the needs of vulnerable communities. Although opportunities for international rotations abound, domestic rotations among underserved populations can be just as transformative, including correctional medicine, homeless clinics, the Indian Health Service, and rural communities.
Create Partnerships to Broaden Impact of Service
Affecting the largest and most visible organ, skin disease often presents a substantial concern for patients and can herald systemic disease. The nature of dermatologic disease engenders close collaboration between general practitioners and specialists. For example, while resident-run or safety-net clinics characteristically center on providing holistic care for patients through internal medicine or primary care, these overworked and understaffed clinics often are in need of evaluation by specialists for specific concerns. Some clinic models feature dermatology faculty who volunteer routinely (ie, every 2 weeks, every month) to examine all the clinic’s patients presenting with concerns pertinent to the specialty. Drawing on their respective areas of expertise, general practitioners and dermatologists therefore can collaborate to connect disadvantaged patients with the specialized care they need.
Challenges Present Opportunities for Innovation
Adhering to the social distancing requirements of the COVID-19 pandemic protocol has driven clinicians to utilize innovative approaches to patient care. The rural-urban misdistribution of the dermatologist workforce has long been established, with rural patients often experiencing lengthy wait times to see a specialist.9 Both synchronous and asynchronous teledermatology modalities provide an ideal platform for triaging patients with dermatologic concerns who otherwise have meager access to a dermatologist.
Final Thoughts
Residency training is a prime opportunity to gain exposure to the broad spectrum of disease within dermatology as well as the diverse range of affected patients. Drawing on the aforementioned strategies, residents can leverage this knowledge in the service of underserved patients.
Studding almost every inch of skin except the face are gray lichenified plaques coating a patient’s body like worn leather. Raking his nails across his arm, the patient reminds me how long he had waited to receive this referral and how early he had awoken for this appointment. He was well acquainted with the value of promptness; in his world, it might make the difference between sleeping on a cot and a night spent on concrete.
Over the last year, the patient had cycled through the few safety-net clinics scattered throughout the city. He had accumulated numerous different diagnoses from atopic dermatitis to disseminated tinea corporis. A few minutes, one #15 scalpel, and mineral oil were all it took for us to unravel the mystery. As the attending and I peered through the microscope at the scabies ovum, I couldn’t help but wonder about the alternative outcomes to his case. Left untreated, scabies compromises the skin barrier, paving the way for secondary infections such as cellulitis. Depending on the pathogen, this infection may in turn evolve into acute postinfectious glomerulonephritis.1-4 An elusive diagnosis can quietly escalate into considerable morbidity for patients. This case highlights the dire consequences of dermatologic health disparities and places medicine’s primordial function into sharp focus: the alleviation of suffering.
The Dermatologic Burden of Disease
As a major contributor to global disease burden, dermatologic disease is the fourth greatest cause of disability worldwide when mortality is factored out.5,6 Among global rural populations, dermatologic disease constitutes one of the leading causes of death and/or loss of professional capabilities.7 In the United States alone, nearly 27% of the population saw a physician for at least 1 dermatologic disease in 2013.5 The tremendous prevalence of skin disease magnifies discrepancies in access to dermatologic care, which has been observed to be influenced by age, socioeconomic background, rurality, and sex.8
There has been growing focus on the national shortage of dermatologists over the last 2 decades.9,10 With an aging population and rising incidence of skin cancer, this undersupply is projected to increase and disproportionately impact ethnic minorities as well as those from socioeconomically disadvantaged backgrounds.8,9,11-14 These trends are of particular importance to residents and medical trainees. Multiple studies have demonstrated that the patient demographic of hospital-based resident clinics includes primarily minority and disenfranchised populations with poorer overall health.15-17 In contrast to faculty clinics, residents treat patients who are more likely to be nonwhite and more likely to be reimbursed by Medicaid.18 The unique demographic makeup of hospital-based resident clinics raises questions about the preparedness and comfort of resident physicians in managing the nuances of health care delivery in these settings.10
Providing equitable care to marginalized populations within the constraints of 15- to 30-minute visits can be challenging to physicians and trainees. Even clinicians with the best of intentions may be impeded by a lack of familiarity with the daily realities of impoverished living conditions, implicit prejudice against people living in poverty, and adapting recommendations to varying levels of health literacy among patients.19,20 Contending with these daunting obstacles can be discouraging. Given how entrenched certain institutional barriers are, questioning them may seem an exercise in futility, yet history demonstrates that residents can and have been empowered to improve tangible outcomes for vulnerable populations. In reflecting on approaches of the general medical education system, The Josiah Macy Jr. Foundation President George E. Thibault, MD, observed that, “When appropriately trained, deployed and incented, [residents] can help achieve institutional goals to improve quality, safety and efficiency.”21
Start Small But Dream Big
Action begins with awareness. Medical school and teaching hospital curricula are increasingly integrating educational exercises regarding the social determinants of health and populations with unmet needs. Medical training presents an exclusive opportunity to gain exposure to and familiarity with patient populations that one might not otherwise encounter. Immersion programs provide invaluable experience in tailoring health care delivery to the needs of vulnerable communities. Although opportunities for international rotations abound, domestic rotations among underserved populations can be just as transformative, including correctional medicine, homeless clinics, the Indian Health Service, and rural communities.
Create Partnerships to Broaden Impact of Service
Affecting the largest and most visible organ, skin disease often presents a substantial concern for patients and can herald systemic disease. The nature of dermatologic disease engenders close collaboration between general practitioners and specialists. For example, while resident-run or safety-net clinics characteristically center on providing holistic care for patients through internal medicine or primary care, these overworked and understaffed clinics often are in need of evaluation by specialists for specific concerns. Some clinic models feature dermatology faculty who volunteer routinely (ie, every 2 weeks, every month) to examine all the clinic’s patients presenting with concerns pertinent to the specialty. Drawing on their respective areas of expertise, general practitioners and dermatologists therefore can collaborate to connect disadvantaged patients with the specialized care they need.
Challenges Present Opportunities for Innovation
Adhering to the social distancing requirements of the COVID-19 pandemic protocol has driven clinicians to utilize innovative approaches to patient care. The rural-urban misdistribution of the dermatologist workforce has long been established, with rural patients often experiencing lengthy wait times to see a specialist.9 Both synchronous and asynchronous teledermatology modalities provide an ideal platform for triaging patients with dermatologic concerns who otherwise have meager access to a dermatologist.
Final Thoughts
Residency training is a prime opportunity to gain exposure to the broad spectrum of disease within dermatology as well as the diverse range of affected patients. Drawing on the aforementioned strategies, residents can leverage this knowledge in the service of underserved patients.
- McCarthy JS, Kemp DJ, Walton SF, et al. Scabies: more than just an irritation. Postgrad Med J. 2004;80:382-387.
- Svartman M, Finklea JF, Earle DP, et al. Epidemic scabies and acute glomerulonephritis in Trinidad. Lancet. 1972;1:249-251.
- Hersch C. Acute glomerulonephritis due to skin disease, with special reference to scabies. S Afr Med J. 1967;41:29-34.
- Carapetis JR, Connors C, Yarmirr D, et al. Success of a scabies control program in an Australian aboriginal community. Pediatr Infect Dis J. 1997;16:494-499.
- Lim HW, Collins SAB, Resneck JS Jr, et al. The burden of skin disease in the United States [published online March 1, 2017]. J Am Acad Dermatol. 2017;76:958-972.e2.
- Karimkhani C, Dellavalle RP, Coffeng LE, et al. Global skin disease morbidity and mortality: an update from the Global Burden of Disease Study 2013. JAMA Dermatol. 2017;153:406-412.
- Morrone A. Poverty, dignity, and forgotten skin care: dermatology in the stream of human mobile population. Dermatol Clin. 2008;26:245-256, vi-vii.
- Tripathi R, Knusel KD, Ezaldein HH, et al. Association of demographic and socioeconomic characteristics with differences in use of outpatient dermatology services in the United States. JAMA Dermatol. 2018;154:1286-1291.
- Feng H, Berk-Krauss J, Feng PW, et al. Comparison of dermatologist density between urban and rural counties in the United States. JAMA Dermatol. 2018;154:1265-1271.
- Blanco G, Vasquez R, Nezafati K, et al. How residency programs can foster practice for the underserved. J Am Acad Dermatol. 2012;67:158-159.
- Kosmadaki MG, Gilchrest BA. The demographics of aging in the United States: implications for dermatology. Arch Dermatol. 2002;138:1427-1428.
- Donaldson MR, Coldiron BM. No end in sight: the skin cancer epidemic continues. Semin Cutan Med Surg. 2011;30:3-5.
- Dall TM, Gallo PD, Chakrabarti R, et al. An aging population and growing disease burden will require a large and specialized health care workforce by 2025. Health Aff (Millwood). 2013;32:2013-2020.
- Sauaia A, Dellavalle RP. Health care inequities: an introduction for dermatology providers. Dermatol Clin. 2009;27:103-107.
- Brook RH, Fink A, Kosecoff J, et al. Educating physicians and treating patients in the ambulatory setting. where are we going and how will we know when we arrive? Ann Intern Med. 1987;107:392-398.
- Yancy WS Jr, Macpherson DS, Hanusa BH, et al. Patient satisfaction in resident and attending ambulatory care clinics. J Gen Intern Med. 2001;16:755-762. 17. Fiebach NH,
- Wong JG. Taking care of patients in resident clinics: where do we stand? J Gen Intern Med. 2001;16:787-789.
- Loignon C, Boudreault-Fournier A, Truchon K, et al. Medical residents reflect on their prejudices toward poverty: a photovoice training project. BMC Med Educ. 2014;14:1050.
- Scheid D, Logue E, Gilchrist VJ, et al. Do we practice what we preach? comparing the patients of faculty and residents. Fam Med. 1995;27:519-524.
- Loignon C, Gottin T, Dupéré S, et al. General practitioners’ perspective on poverty: a qualitative study in Montreal, Canada. Fam Pract. 2018;35:105-110.
- Parks T. Empowered residents can help transform medical care. American Medical Association website. Published November 30, 2016. Accessed March 18, 2021. www.ama-assn.org/education/improve-gme/empowered-residents-can-help-transform-medical-care
- McCarthy JS, Kemp DJ, Walton SF, et al. Scabies: more than just an irritation. Postgrad Med J. 2004;80:382-387.
- Svartman M, Finklea JF, Earle DP, et al. Epidemic scabies and acute glomerulonephritis in Trinidad. Lancet. 1972;1:249-251.
- Hersch C. Acute glomerulonephritis due to skin disease, with special reference to scabies. S Afr Med J. 1967;41:29-34.
- Carapetis JR, Connors C, Yarmirr D, et al. Success of a scabies control program in an Australian aboriginal community. Pediatr Infect Dis J. 1997;16:494-499.
- Lim HW, Collins SAB, Resneck JS Jr, et al. The burden of skin disease in the United States [published online March 1, 2017]. J Am Acad Dermatol. 2017;76:958-972.e2.
- Karimkhani C, Dellavalle RP, Coffeng LE, et al. Global skin disease morbidity and mortality: an update from the Global Burden of Disease Study 2013. JAMA Dermatol. 2017;153:406-412.
- Morrone A. Poverty, dignity, and forgotten skin care: dermatology in the stream of human mobile population. Dermatol Clin. 2008;26:245-256, vi-vii.
- Tripathi R, Knusel KD, Ezaldein HH, et al. Association of demographic and socioeconomic characteristics with differences in use of outpatient dermatology services in the United States. JAMA Dermatol. 2018;154:1286-1291.
- Feng H, Berk-Krauss J, Feng PW, et al. Comparison of dermatologist density between urban and rural counties in the United States. JAMA Dermatol. 2018;154:1265-1271.
- Blanco G, Vasquez R, Nezafati K, et al. How residency programs can foster practice for the underserved. J Am Acad Dermatol. 2012;67:158-159.
- Kosmadaki MG, Gilchrest BA. The demographics of aging in the United States: implications for dermatology. Arch Dermatol. 2002;138:1427-1428.
- Donaldson MR, Coldiron BM. No end in sight: the skin cancer epidemic continues. Semin Cutan Med Surg. 2011;30:3-5.
- Dall TM, Gallo PD, Chakrabarti R, et al. An aging population and growing disease burden will require a large and specialized health care workforce by 2025. Health Aff (Millwood). 2013;32:2013-2020.
- Sauaia A, Dellavalle RP. Health care inequities: an introduction for dermatology providers. Dermatol Clin. 2009;27:103-107.
- Brook RH, Fink A, Kosecoff J, et al. Educating physicians and treating patients in the ambulatory setting. where are we going and how will we know when we arrive? Ann Intern Med. 1987;107:392-398.
- Yancy WS Jr, Macpherson DS, Hanusa BH, et al. Patient satisfaction in resident and attending ambulatory care clinics. J Gen Intern Med. 2001;16:755-762. 17. Fiebach NH,
- Wong JG. Taking care of patients in resident clinics: where do we stand? J Gen Intern Med. 2001;16:787-789.
- Loignon C, Boudreault-Fournier A, Truchon K, et al. Medical residents reflect on their prejudices toward poverty: a photovoice training project. BMC Med Educ. 2014;14:1050.
- Scheid D, Logue E, Gilchrist VJ, et al. Do we practice what we preach? comparing the patients of faculty and residents. Fam Med. 1995;27:519-524.
- Loignon C, Gottin T, Dupéré S, et al. General practitioners’ perspective on poverty: a qualitative study in Montreal, Canada. Fam Pract. 2018;35:105-110.
- Parks T. Empowered residents can help transform medical care. American Medical Association website. Published November 30, 2016. Accessed March 18, 2021. www.ama-assn.org/education/improve-gme/empowered-residents-can-help-transform-medical-care
Resident Pearl
- Even while in training, dermatology residents have the agency to impact their communities by connecting their expertise to the patients in greatest need.
Hyperpigmentation on the Head and Neck
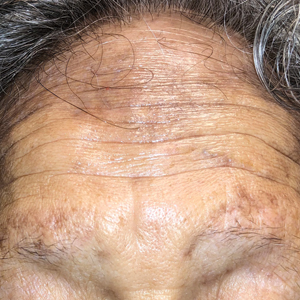
The Diagnosis: Frontal Fibrosing Alopecia Overlapping With Lichen Planus Pigmentosus
Microscopic examination revealed focal dermal pigmentation, papillary fibrosis, and epidermal atrophy. These clinical and histologic findings indicated a diagnosis of fully developed lichen planus pigmentosus (LPP) overlapping with frontal fibrosing alopecia (FFA). Other cases have demonstrated an association between LPP and FFA.1,2
Lichen planus pigmentosus is considered an uncommon variant of lichen planus, as it has similar histopathologic findings and occasional coexistence.3,4 It is characterized by hyperpigmented macules primarily located in sun-exposed and flexural areas of the skin. First described in India,5 this disease has a predilection for darker skin (Fitzpatrick skin types III-V),6,7 and it has been reported in other racial and ethnic groups including Latin Americans, Middle Eastern populations, Japanese, and Koreans.4,8 Typically, lesions initially appear as ill-defined, blue-grey, round to oval macules that coalesce into hyperpigmented patches. Involvement most commonly begins at the forehead and temples, which are affected in nearly all patients. Infrequently, LPP can be generalized or affect the oral mucosa; involvement of the palms, soles, and nails does not occur. Patients may be asymptomatic, but some experience mild pruritus and burning. The disease course is chronic and insidious, with new lesions appearing over time and old lesions progressively darkening and expanding.6,7,9
Although the pathogenesis of LPP is unknown, several exposures have been implicated, such as amla oil, mustard oil, henna, hair dye, and environmental pollutants.7 Because lesions characteristically occur in sun-exposed areas, UV light also may be involved. In addition, studies have suggested that LPP is associated with endocrinopathies such as diabetes mellitus and dyslipidemias, as in our patient, as well as autoimmune conditions such as vitiligo and systemic lupus erythematosus.10,11
Histopathologic findings are characterized by vacuolar degeneration of the basal layer in the epidermis as well as perivascular lymphohistiocytic infiltration and the presence of melanophages in the dermis.3,9 Lichen planus pigmentosus is difficult to treat, as no consistently effective modality has been established. Topical tacrolimus, topical corticosteroids, oral retinoids, lasers, and sun protection have been implemented with underwhelming results.12
Frontal fibrosing alopecia is a variant of lichen planopilaris that predominantly affects postmenopausal women and presents with frontotemporal hair loss in a bandlike distribution.5,13 Both terminal and vellus hairs are affected. Involvement of multiple hair-bearing sites of the skin have been reported, including the entire scalp, eyebrows, and eyelashes. Affected areas may display hypopigmentation and be accompanied by pruritus and trichodynia.14,15 The pathogenesis currently is under investigation, with studies demonstrating autoimmune, genetic, and possibly even endocrine predispositions.16-18 Biopsies of lesions are indistinguishable from lichen planopilaris, which shows follicular lymphocytic infiltration, perifollicular fibrosis, interface dermatitis of the follicular infundibulum and isthmus, and vertical fibrous tracks.5 Patients with FFA have demonstrated variable responses to treatments, with one study showing improvement with oral finasteride or dutasteride.14 Topical and intralesional corticosteroids have yielded suboptimal effects. Other modalities include hydroxychloroquine and mycophenolate mofetil.15,19
Co-occurrence of LPP and FFA primarily is seen in postmenopausal women with darker skin,14,15 as in our patient, though premenopausal cases have been reported. Lichen planus pigmentosus may serve as a harbinger in most patients.1,2 In a similar fashion, our patient presented with hyperpigmented macular lesions prior to the onset of frontotemporal hair loss.
Our patient was started on finasteride 2.5 mg daily, minoxidil foam 5%, clobetasol solution 0.05%, triamcinolone ointment 0.1%, and hydrocortisone ointment 2.5%. She was instructed to commence treatment and follow up in 6 months.
The differential diagnosis includes dermatologic conditions that mimic both LPP and FFA. Postinflammatory hyperpigmentation and fixed drug reaction were unlikely based on the patient's history. The lesions of ashy dermatosis are characteristically gray erythematous macules on the trunk and limbs. Riehl melanosis is a rare pigmented contact dermatitis that is associated with a history of repeated contact with sensitizing allergens. Although Hori nevus is characterized by small, blue-gray or brown macules on the face, lesions predominantly occur on the bony prominences of the cheeks. Melasma also presents with dark to gray macules that affect the face and less commonly the neck, as in our patient.2
Early discoid lupus erythematosus presents with round erythematous plaques with overlying scale extending into the hair follicles. In pseudopalade of Brocq, an idiopathic cicatricial alopecia, lesions typically are flesh colored. Biopsy also shows epidermal atrophy with additional dermal sclerosis and fibrosis. Folliculitis decalvans is a scarring form of alopecia associated with erythema and pustules, findings that were not present in our patient. Keratosis follicularis spinulosa decalvans is a rare, X-linked inherited ichthyosis manifesting as scarring alopecia with follicular depressions and papules on the scalp in younger males. Photophobia and other manifestations may be present. Alopecia mucinosa is a nonscarring alopecia with grouped follicular erythematous patches or plaques. Mucin sometimes can be squeezed from affected areas, and histopathologic examination shows mucin accumulation.4
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-442.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Rieder E, Kaplan J, Kamino H, et al. Lichen planus pigmentosus. Dermatol Online J. 2013;19:20713.
- Kashima A, Tajiri A, Yamashita A, et al. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol. 2007;46:740-742.
- Bhutani L, Bedi T, Pandhi R. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Kanwa AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514.
- Torres J, Guadalupe A, Reyes E, et al. Lichen planus pigmentosus in patients with endocrinopathies and hepatitis C. J Am Acad Dermatol. 2013;68:AB139.
- Kim JE, Won CH, Chang S, et al. Linear lichen planus pigmentosus of the forehead treated by neodymium:yttrium-aluminum-garnet laser and topical tacrolimus. J Dermatol. 2012;39:189-191.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-1257.
- Rodriguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
The Diagnosis: Frontal Fibrosing Alopecia Overlapping With Lichen Planus Pigmentosus
Microscopic examination revealed focal dermal pigmentation, papillary fibrosis, and epidermal atrophy. These clinical and histologic findings indicated a diagnosis of fully developed lichen planus pigmentosus (LPP) overlapping with frontal fibrosing alopecia (FFA). Other cases have demonstrated an association between LPP and FFA.1,2
Lichen planus pigmentosus is considered an uncommon variant of lichen planus, as it has similar histopathologic findings and occasional coexistence.3,4 It is characterized by hyperpigmented macules primarily located in sun-exposed and flexural areas of the skin. First described in India,5 this disease has a predilection for darker skin (Fitzpatrick skin types III-V),6,7 and it has been reported in other racial and ethnic groups including Latin Americans, Middle Eastern populations, Japanese, and Koreans.4,8 Typically, lesions initially appear as ill-defined, blue-grey, round to oval macules that coalesce into hyperpigmented patches. Involvement most commonly begins at the forehead and temples, which are affected in nearly all patients. Infrequently, LPP can be generalized or affect the oral mucosa; involvement of the palms, soles, and nails does not occur. Patients may be asymptomatic, but some experience mild pruritus and burning. The disease course is chronic and insidious, with new lesions appearing over time and old lesions progressively darkening and expanding.6,7,9
Although the pathogenesis of LPP is unknown, several exposures have been implicated, such as amla oil, mustard oil, henna, hair dye, and environmental pollutants.7 Because lesions characteristically occur in sun-exposed areas, UV light also may be involved. In addition, studies have suggested that LPP is associated with endocrinopathies such as diabetes mellitus and dyslipidemias, as in our patient, as well as autoimmune conditions such as vitiligo and systemic lupus erythematosus.10,11
Histopathologic findings are characterized by vacuolar degeneration of the basal layer in the epidermis as well as perivascular lymphohistiocytic infiltration and the presence of melanophages in the dermis.3,9 Lichen planus pigmentosus is difficult to treat, as no consistently effective modality has been established. Topical tacrolimus, topical corticosteroids, oral retinoids, lasers, and sun protection have been implemented with underwhelming results.12
Frontal fibrosing alopecia is a variant of lichen planopilaris that predominantly affects postmenopausal women and presents with frontotemporal hair loss in a bandlike distribution.5,13 Both terminal and vellus hairs are affected. Involvement of multiple hair-bearing sites of the skin have been reported, including the entire scalp, eyebrows, and eyelashes. Affected areas may display hypopigmentation and be accompanied by pruritus and trichodynia.14,15 The pathogenesis currently is under investigation, with studies demonstrating autoimmune, genetic, and possibly even endocrine predispositions.16-18 Biopsies of lesions are indistinguishable from lichen planopilaris, which shows follicular lymphocytic infiltration, perifollicular fibrosis, interface dermatitis of the follicular infundibulum and isthmus, and vertical fibrous tracks.5 Patients with FFA have demonstrated variable responses to treatments, with one study showing improvement with oral finasteride or dutasteride.14 Topical and intralesional corticosteroids have yielded suboptimal effects. Other modalities include hydroxychloroquine and mycophenolate mofetil.15,19
Co-occurrence of LPP and FFA primarily is seen in postmenopausal women with darker skin,14,15 as in our patient, though premenopausal cases have been reported. Lichen planus pigmentosus may serve as a harbinger in most patients.1,2 In a similar fashion, our patient presented with hyperpigmented macular lesions prior to the onset of frontotemporal hair loss.
Our patient was started on finasteride 2.5 mg daily, minoxidil foam 5%, clobetasol solution 0.05%, triamcinolone ointment 0.1%, and hydrocortisone ointment 2.5%. She was instructed to commence treatment and follow up in 6 months.
The differential diagnosis includes dermatologic conditions that mimic both LPP and FFA. Postinflammatory hyperpigmentation and fixed drug reaction were unlikely based on the patient's history. The lesions of ashy dermatosis are characteristically gray erythematous macules on the trunk and limbs. Riehl melanosis is a rare pigmented contact dermatitis that is associated with a history of repeated contact with sensitizing allergens. Although Hori nevus is characterized by small, blue-gray or brown macules on the face, lesions predominantly occur on the bony prominences of the cheeks. Melasma also presents with dark to gray macules that affect the face and less commonly the neck, as in our patient.2
Early discoid lupus erythematosus presents with round erythematous plaques with overlying scale extending into the hair follicles. In pseudopalade of Brocq, an idiopathic cicatricial alopecia, lesions typically are flesh colored. Biopsy also shows epidermal atrophy with additional dermal sclerosis and fibrosis. Folliculitis decalvans is a scarring form of alopecia associated with erythema and pustules, findings that were not present in our patient. Keratosis follicularis spinulosa decalvans is a rare, X-linked inherited ichthyosis manifesting as scarring alopecia with follicular depressions and papules on the scalp in younger males. Photophobia and other manifestations may be present. Alopecia mucinosa is a nonscarring alopecia with grouped follicular erythematous patches or plaques. Mucin sometimes can be squeezed from affected areas, and histopathologic examination shows mucin accumulation.4
The Diagnosis: Frontal Fibrosing Alopecia Overlapping With Lichen Planus Pigmentosus
Microscopic examination revealed focal dermal pigmentation, papillary fibrosis, and epidermal atrophy. These clinical and histologic findings indicated a diagnosis of fully developed lichen planus pigmentosus (LPP) overlapping with frontal fibrosing alopecia (FFA). Other cases have demonstrated an association between LPP and FFA.1,2
Lichen planus pigmentosus is considered an uncommon variant of lichen planus, as it has similar histopathologic findings and occasional coexistence.3,4 It is characterized by hyperpigmented macules primarily located in sun-exposed and flexural areas of the skin. First described in India,5 this disease has a predilection for darker skin (Fitzpatrick skin types III-V),6,7 and it has been reported in other racial and ethnic groups including Latin Americans, Middle Eastern populations, Japanese, and Koreans.4,8 Typically, lesions initially appear as ill-defined, blue-grey, round to oval macules that coalesce into hyperpigmented patches. Involvement most commonly begins at the forehead and temples, which are affected in nearly all patients. Infrequently, LPP can be generalized or affect the oral mucosa; involvement of the palms, soles, and nails does not occur. Patients may be asymptomatic, but some experience mild pruritus and burning. The disease course is chronic and insidious, with new lesions appearing over time and old lesions progressively darkening and expanding.6,7,9
Although the pathogenesis of LPP is unknown, several exposures have been implicated, such as amla oil, mustard oil, henna, hair dye, and environmental pollutants.7 Because lesions characteristically occur in sun-exposed areas, UV light also may be involved. In addition, studies have suggested that LPP is associated with endocrinopathies such as diabetes mellitus and dyslipidemias, as in our patient, as well as autoimmune conditions such as vitiligo and systemic lupus erythematosus.10,11
Histopathologic findings are characterized by vacuolar degeneration of the basal layer in the epidermis as well as perivascular lymphohistiocytic infiltration and the presence of melanophages in the dermis.3,9 Lichen planus pigmentosus is difficult to treat, as no consistently effective modality has been established. Topical tacrolimus, topical corticosteroids, oral retinoids, lasers, and sun protection have been implemented with underwhelming results.12
Frontal fibrosing alopecia is a variant of lichen planopilaris that predominantly affects postmenopausal women and presents with frontotemporal hair loss in a bandlike distribution.5,13 Both terminal and vellus hairs are affected. Involvement of multiple hair-bearing sites of the skin have been reported, including the entire scalp, eyebrows, and eyelashes. Affected areas may display hypopigmentation and be accompanied by pruritus and trichodynia.14,15 The pathogenesis currently is under investigation, with studies demonstrating autoimmune, genetic, and possibly even endocrine predispositions.16-18 Biopsies of lesions are indistinguishable from lichen planopilaris, which shows follicular lymphocytic infiltration, perifollicular fibrosis, interface dermatitis of the follicular infundibulum and isthmus, and vertical fibrous tracks.5 Patients with FFA have demonstrated variable responses to treatments, with one study showing improvement with oral finasteride or dutasteride.14 Topical and intralesional corticosteroids have yielded suboptimal effects. Other modalities include hydroxychloroquine and mycophenolate mofetil.15,19
Co-occurrence of LPP and FFA primarily is seen in postmenopausal women with darker skin,14,15 as in our patient, though premenopausal cases have been reported. Lichen planus pigmentosus may serve as a harbinger in most patients.1,2 In a similar fashion, our patient presented with hyperpigmented macular lesions prior to the onset of frontotemporal hair loss.
Our patient was started on finasteride 2.5 mg daily, minoxidil foam 5%, clobetasol solution 0.05%, triamcinolone ointment 0.1%, and hydrocortisone ointment 2.5%. She was instructed to commence treatment and follow up in 6 months.
The differential diagnosis includes dermatologic conditions that mimic both LPP and FFA. Postinflammatory hyperpigmentation and fixed drug reaction were unlikely based on the patient's history. The lesions of ashy dermatosis are characteristically gray erythematous macules on the trunk and limbs. Riehl melanosis is a rare pigmented contact dermatitis that is associated with a history of repeated contact with sensitizing allergens. Although Hori nevus is characterized by small, blue-gray or brown macules on the face, lesions predominantly occur on the bony prominences of the cheeks. Melasma also presents with dark to gray macules that affect the face and less commonly the neck, as in our patient.2
Early discoid lupus erythematosus presents with round erythematous plaques with overlying scale extending into the hair follicles. In pseudopalade of Brocq, an idiopathic cicatricial alopecia, lesions typically are flesh colored. Biopsy also shows epidermal atrophy with additional dermal sclerosis and fibrosis. Folliculitis decalvans is a scarring form of alopecia associated with erythema and pustules, findings that were not present in our patient. Keratosis follicularis spinulosa decalvans is a rare, X-linked inherited ichthyosis manifesting as scarring alopecia with follicular depressions and papules on the scalp in younger males. Photophobia and other manifestations may be present. Alopecia mucinosa is a nonscarring alopecia with grouped follicular erythematous patches or plaques. Mucin sometimes can be squeezed from affected areas, and histopathologic examination shows mucin accumulation.4
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-442.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Rieder E, Kaplan J, Kamino H, et al. Lichen planus pigmentosus. Dermatol Online J. 2013;19:20713.
- Kashima A, Tajiri A, Yamashita A, et al. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol. 2007;46:740-742.
- Bhutani L, Bedi T, Pandhi R. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Kanwa AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514.
- Torres J, Guadalupe A, Reyes E, et al. Lichen planus pigmentosus in patients with endocrinopathies and hepatitis C. J Am Acad Dermatol. 2013;68:AB139.
- Kim JE, Won CH, Chang S, et al. Linear lichen planus pigmentosus of the forehead treated by neodymium:yttrium-aluminum-garnet laser and topical tacrolimus. J Dermatol. 2012;39:189-191.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-1257.
- Rodriguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
- Dlova NC. Frontal fibrosing alopecia and lichen planus pigmentosus: is there a link? Br J Dermatol. 2013;168:439-442.
- Pirmez R, Duque-Estrada B, Donati A, et al. Clinical and dermoscopic features of lichen planus pigmentosus in 37 patients with frontal fibrosing alopecia. Br J Dermatol. 2016;175:1387-1390.
- Rieder E, Kaplan J, Kamino H, et al. Lichen planus pigmentosus. Dermatol Online J. 2013;19:20713.
- Kashima A, Tajiri A, Yamashita A, et al. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol. 2007;46:740-742.
- Bhutani L, Bedi T, Pandhi R. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Kanwa AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514.
- Torres J, Guadalupe A, Reyes E, et al. Lichen planus pigmentosus in patients with endocrinopathies and hepatitis C. J Am Acad Dermatol. 2013;68:AB139.
- Kim JE, Won CH, Chang S, et al. Linear lichen planus pigmentosus of the forehead treated by neodymium:yttrium-aluminum-garnet laser and topical tacrolimus. J Dermatol. 2012;39:189-191.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle's epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-1257.
- Rodriguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
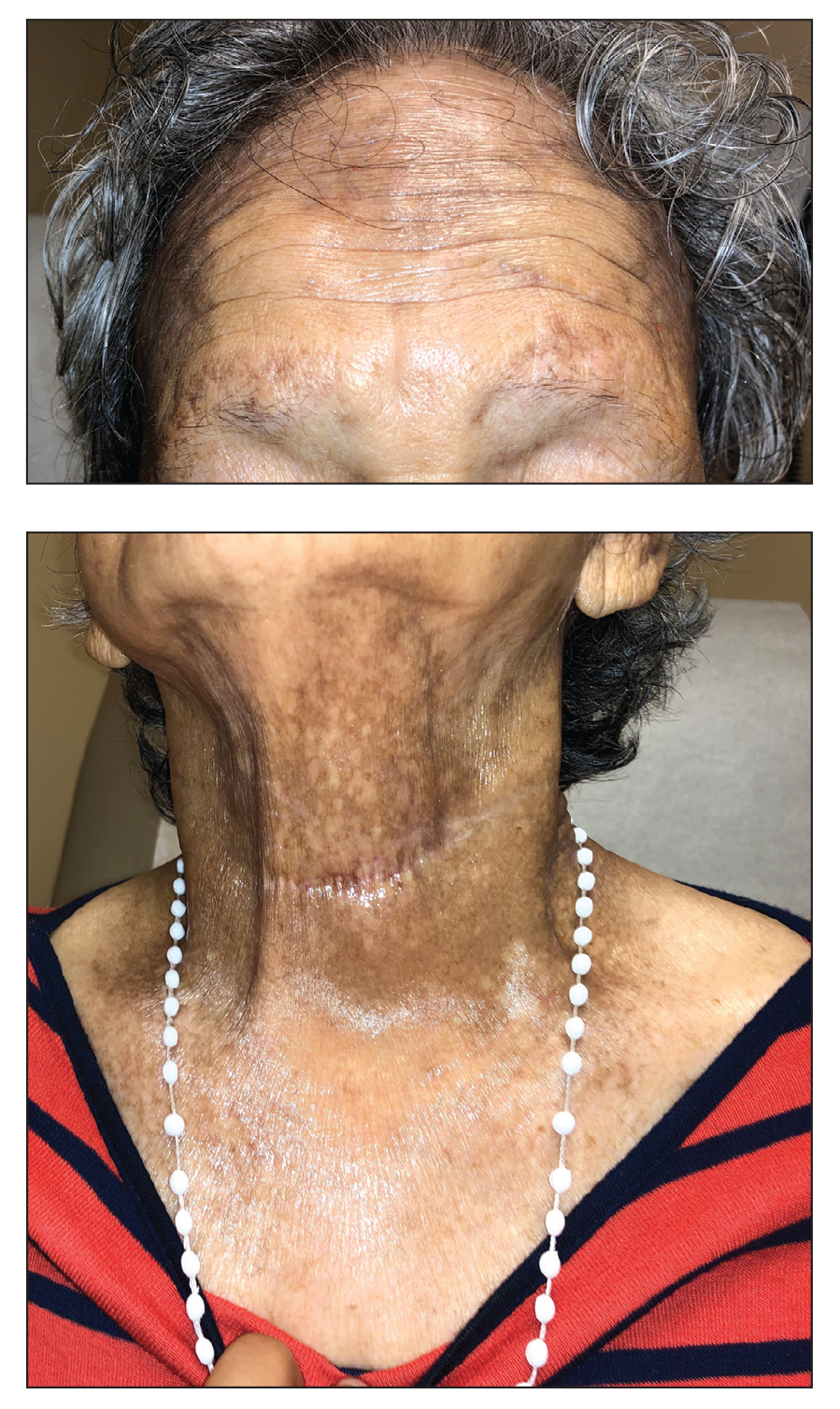
A 78-year-old Asian woman presented to the dermatology clinic with progressively worsening dark spots on the forehead and neck of 3 months’ duration. She noted mild pruritis and hair loss involving the eyebrows and anterior scalp. Her medical history was notable for type 2 diabetes mellitus. She denied any new medical conditions or medications and had no prior history of similar symptoms. Physical examination showed hyperpigmented brown macules and patches on the forehead (top) and anterior neck (bottom) with sparing of the posterior neck and lower face. Alopecia with areas of perifollicular erythema and hyperpigmentation with reduced follicular openings were present on the eyebrows and anterior forehead. Two punch biopsies of head and neck lesions were performed.
Tender, Diffuse, Edematous, and Erythematous Papules on the Face, Neck, Chest, and Extremities
The Diagnosis: Sweet Syndrome
Sweet syndrome, alternatively known as acute febrile neutrophilic dermatosis, typically presents with variably tender, erythematous papules, plaques, or nodules in middle-aged adults.1 Systemic symptoms such as fever, fatigue, and arthralgia often accompany these cutaneous findings.1,2 Although the pathophysiology has not been fully elucidated, this syndrome frequently is associated with infections, especially upper respiratory illnesses; medications; and malignancies. Among cases of malignancy-associated Sweet syndrome, hematologic malignancies, particularly acute myeloid leukemia and myelodysplastic syndrome, are more common than solid organ malignancies.1,2 Sweet syndrome may precede the associated malignancy by several months; thus, patients without an identifiable trigger for Sweet syndrome should be closely followed.2 Treatment with systemic steroids typically is effective.1,3 Typical histologic features include papillary dermal edema and a brisk neutrophilic infiltrate in the superficial to mid dermis (quiz image).4 Overlying epidermal spongiosis with or without vesiculation also can be seen.4 Leukocytoclasia and endothelial swelling without fibrinoid necrosis are typical, though full-blown leukocytoclastic vasculitis can be seen.3,4 A histiocytoid variant also has been described in which the dermal infiltrate is composed of mononuclear cells reminiscent of histiocytes that are thought to be immature cells of myeloid origin. This variant histologically can simulate leukemia cutis.5
Perniosis, also known as chilblains, typically presents with red to violaceous macules or papules on acral sites, particularly the distal fingers and toes.6,7 It tends to affect young women more frequently than other demographic groups. Although the pathophysiology is not fully understood, perniosis is thought to represent an abnormal inflammatory response to cold environmental conditions. It can occur as an idiopathic disorder or in association with various systemic illnesses including lupus erythematosus.6,7 The typical histologic findings include papillary dermal edema and a lymphocytic infiltrate in the superficial to deep dermis, often with perivascular and perieccrine accentuation (Figure 1).3,6 Other less common microscopic findings include sparse keratinocyte necrosis, basal layer vacuolar change, swelling of endothelial cells, and lymphocytic vasculitis.6 The lesions typically resolve spontaneously within a few weeks, but in some cases they may be chronic.3
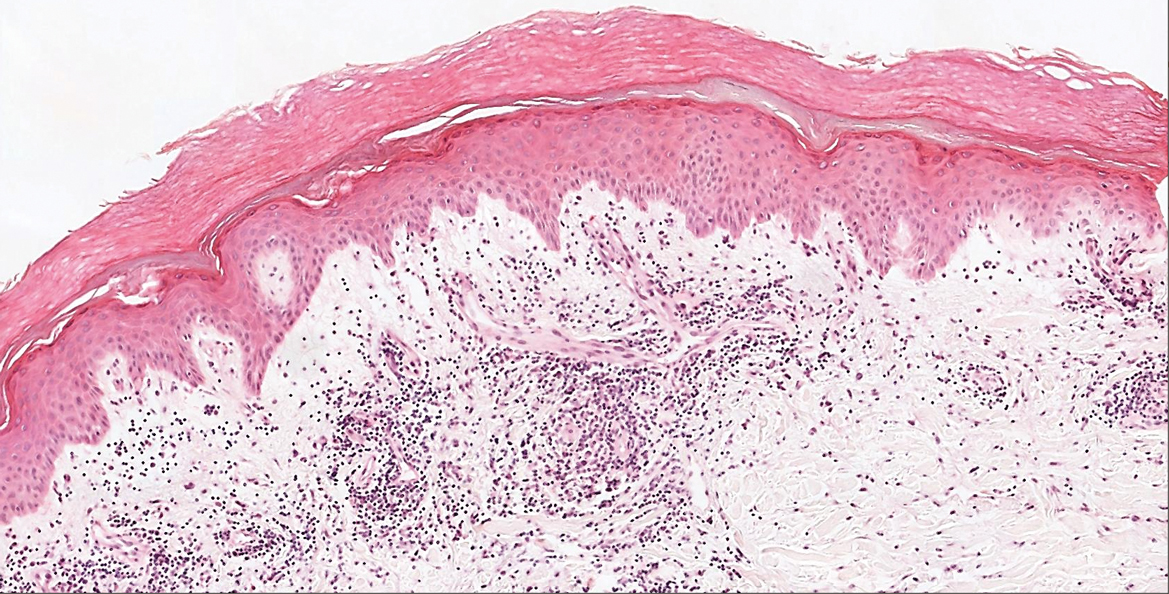
Polymorphous light eruption, a common photodermatosis induced by UV light exposure, typically presents in adolescence or early adulthood with a female predominance. Patients usually develop this pruritic rash on sun-exposed skin other than the face and dorsal aspects of the hands in the spring or early summer upon increased sun exposure after the winter season.3,8 Consistent sunlight exposure throughout the summer months results in decreased flares. Various cutaneous morphologies including papules, vesicles, and plaques can be seen.3,8 Histologic findings include papillary dermal edema and a perivascular lymphocytic infiltrate in the superficial to deep dermis (Figure 2).4
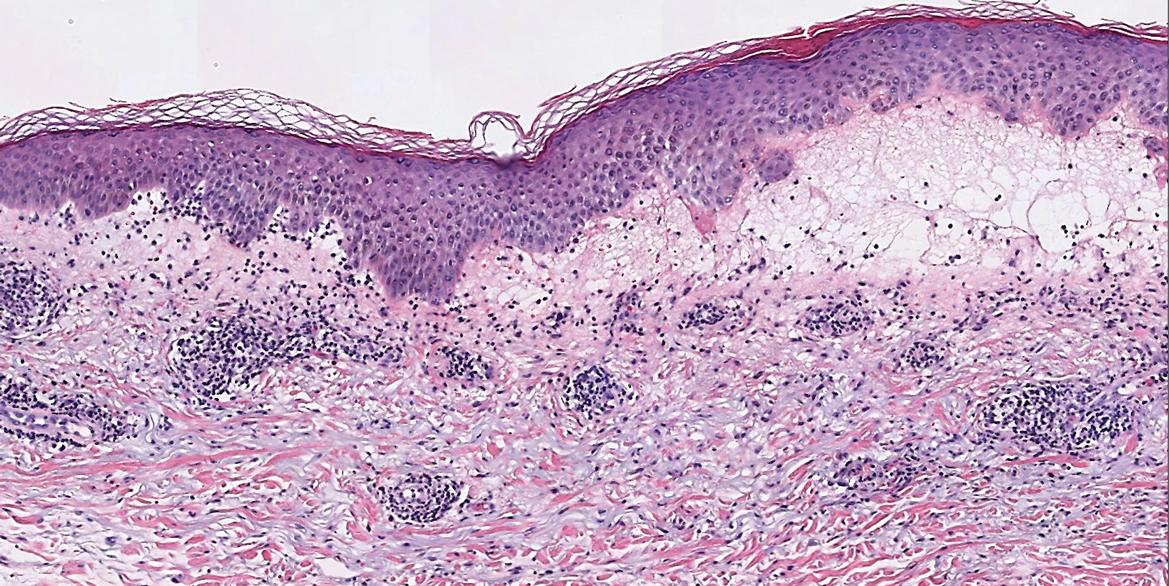
Tinea corporis, a superficial cutaneous dermatophyte infection, typically presents as annular scaly plaques with central clearing. Vesicles and pustules also can be seen.3 The diagnosis can be confirmed via fungal culture, identification of hyphae on microscopic examination of skin scrapings using potassium hydroxide, or cutaneous biopsy. Histologic clues to diagnosis include a "compact stratum corneum (either uniform or forming a layer beneath a basket weave stratum corneum), parakeratosis, mild spongiosis, and neutrophils in the stratum corneum" (Figure 3).9 Papillary dermal edema also may be present, though this finding less commonly is reported.9,10 Because fungal hyphae can be difficult to identify on hematoxylin and eosin-stained slides, special stains such as periodic acid-Schiff or Grocott-Gomori methenamine-silver may be helpful.9 These infections are managed with topical or oral antifungal medications.

Wells syndrome, also known as eosinophilic cellulitis, presents with an acute eruption that can clinically resemble bacterial cellulitis.3 It has been described in children and adults with various clinical morphologies including plaques, bullae, papulovesicles, and papulonodules. Peripheral eosinophilia may be present.11 The clinical lesions usually resolve spontaneously in a few weeks to months, but recurrences are typical.3,11 Histologic findings include papillary dermal edema with or without subepidermal bulla formation and epidermal spongiosis as well as a mixed inflammatory infiltrate with a predominance of eosinophils and flame figures (Figure 4).4 Flame figures are collagen fibers coated with major basic protein and other constituents of degranulated eosinophils.3 Although flame figures often are present in Wells syndrome, they are not specific to this condition.3,4 Some consider Wells syndrome an exaggerated reaction pattern rather than a specific entity.3
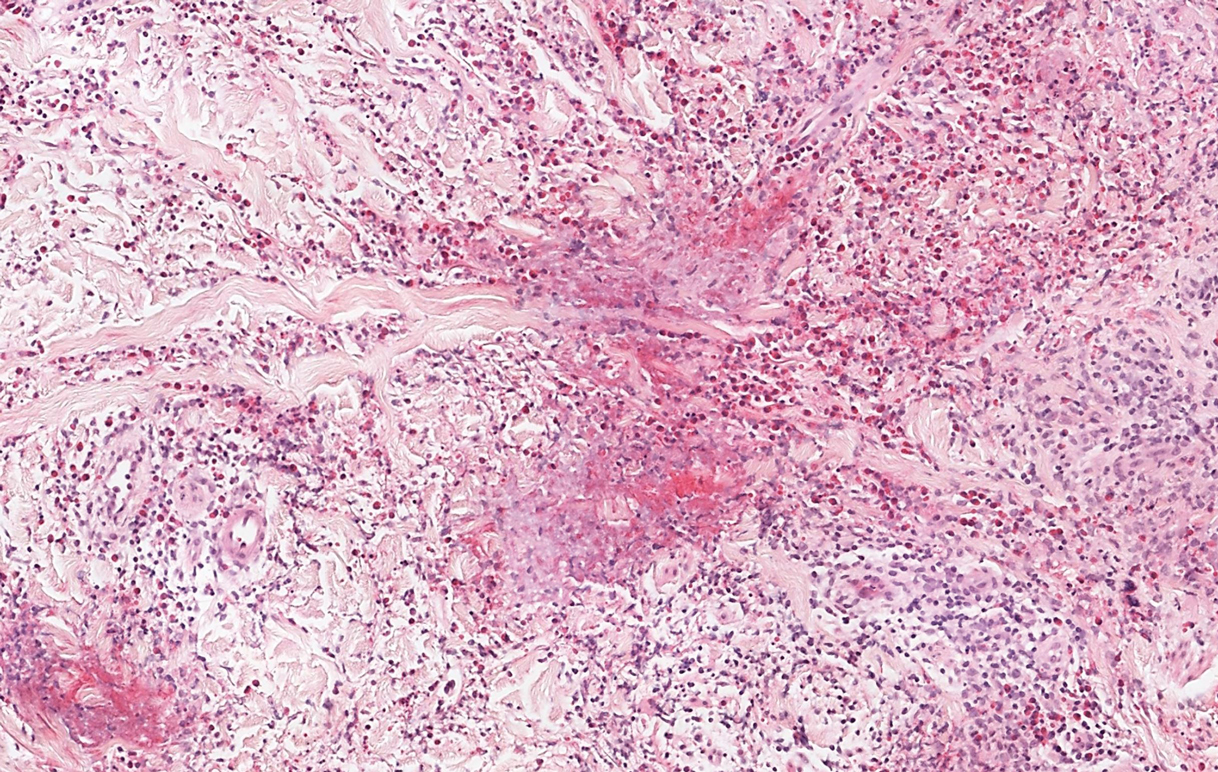
- Rochet N, Chavan R, Cappel M, et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557-564.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Elsevier; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. Elsevier Saunders; 2012.
- Alegría-Landa V, Rodríguez-Pinilla S, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Boada A, Bielsa I, Fernández-Figueras M, et al. Perniosis: clinical and histopathological analysis. Am J Dermatopathol. 2010;32:19-23.
- Takci Z, Vahaboglu G, Eksioglu H. Epidemiological patterns of perniosis, and its association with systemic disorder. Clin Exp Dermatol. 2012;37:844-849.
- Gruber-Wackernagel A, Byrne S, Wolf P. Polymorphous light eruption: clinic aspects and pathogenesis. Dermatol Clin. 2014;32:315-334.
- Elbendary A, Valdebran M, Gad A, et al. When to suspect tinea; a histopathologic study of 103 cases of PAS-positive tinea. J Cutan Pathol. 2016;46:852-857.
- Hoss D, Berke A, Kerr P, et al. Prominent papillary dermal edema in dermatophytosis (tinea corporis). J Cutan Pathol. 2010;37:237-242.
- Caputo R, Marzano A, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
The Diagnosis: Sweet Syndrome
Sweet syndrome, alternatively known as acute febrile neutrophilic dermatosis, typically presents with variably tender, erythematous papules, plaques, or nodules in middle-aged adults.1 Systemic symptoms such as fever, fatigue, and arthralgia often accompany these cutaneous findings.1,2 Although the pathophysiology has not been fully elucidated, this syndrome frequently is associated with infections, especially upper respiratory illnesses; medications; and malignancies. Among cases of malignancy-associated Sweet syndrome, hematologic malignancies, particularly acute myeloid leukemia and myelodysplastic syndrome, are more common than solid organ malignancies.1,2 Sweet syndrome may precede the associated malignancy by several months; thus, patients without an identifiable trigger for Sweet syndrome should be closely followed.2 Treatment with systemic steroids typically is effective.1,3 Typical histologic features include papillary dermal edema and a brisk neutrophilic infiltrate in the superficial to mid dermis (quiz image).4 Overlying epidermal spongiosis with or without vesiculation also can be seen.4 Leukocytoclasia and endothelial swelling without fibrinoid necrosis are typical, though full-blown leukocytoclastic vasculitis can be seen.3,4 A histiocytoid variant also has been described in which the dermal infiltrate is composed of mononuclear cells reminiscent of histiocytes that are thought to be immature cells of myeloid origin. This variant histologically can simulate leukemia cutis.5
Perniosis, also known as chilblains, typically presents with red to violaceous macules or papules on acral sites, particularly the distal fingers and toes.6,7 It tends to affect young women more frequently than other demographic groups. Although the pathophysiology is not fully understood, perniosis is thought to represent an abnormal inflammatory response to cold environmental conditions. It can occur as an idiopathic disorder or in association with various systemic illnesses including lupus erythematosus.6,7 The typical histologic findings include papillary dermal edema and a lymphocytic infiltrate in the superficial to deep dermis, often with perivascular and perieccrine accentuation (Figure 1).3,6 Other less common microscopic findings include sparse keratinocyte necrosis, basal layer vacuolar change, swelling of endothelial cells, and lymphocytic vasculitis.6 The lesions typically resolve spontaneously within a few weeks, but in some cases they may be chronic.3
Polymorphous light eruption, a common photodermatosis induced by UV light exposure, typically presents in adolescence or early adulthood with a female predominance. Patients usually develop this pruritic rash on sun-exposed skin other than the face and dorsal aspects of the hands in the spring or early summer upon increased sun exposure after the winter season.3,8 Consistent sunlight exposure throughout the summer months results in decreased flares. Various cutaneous morphologies including papules, vesicles, and plaques can be seen.3,8 Histologic findings include papillary dermal edema and a perivascular lymphocytic infiltrate in the superficial to deep dermis (Figure 2).4
Tinea corporis, a superficial cutaneous dermatophyte infection, typically presents as annular scaly plaques with central clearing. Vesicles and pustules also can be seen.3 The diagnosis can be confirmed via fungal culture, identification of hyphae on microscopic examination of skin scrapings using potassium hydroxide, or cutaneous biopsy. Histologic clues to diagnosis include a "compact stratum corneum (either uniform or forming a layer beneath a basket weave stratum corneum), parakeratosis, mild spongiosis, and neutrophils in the stratum corneum" (Figure 3).9 Papillary dermal edema also may be present, though this finding less commonly is reported.9,10 Because fungal hyphae can be difficult to identify on hematoxylin and eosin-stained slides, special stains such as periodic acid-Schiff or Grocott-Gomori methenamine-silver may be helpful.9 These infections are managed with topical or oral antifungal medications.
Wells syndrome, also known as eosinophilic cellulitis, presents with an acute eruption that can clinically resemble bacterial cellulitis.3 It has been described in children and adults with various clinical morphologies including plaques, bullae, papulovesicles, and papulonodules. Peripheral eosinophilia may be present.11 The clinical lesions usually resolve spontaneously in a few weeks to months, but recurrences are typical.3,11 Histologic findings include papillary dermal edema with or without subepidermal bulla formation and epidermal spongiosis as well as a mixed inflammatory infiltrate with a predominance of eosinophils and flame figures (Figure 4).4 Flame figures are collagen fibers coated with major basic protein and other constituents of degranulated eosinophils.3 Although flame figures often are present in Wells syndrome, they are not specific to this condition.3,4 Some consider Wells syndrome an exaggerated reaction pattern rather than a specific entity.3
The Diagnosis: Sweet Syndrome
Sweet syndrome, alternatively known as acute febrile neutrophilic dermatosis, typically presents with variably tender, erythematous papules, plaques, or nodules in middle-aged adults.1 Systemic symptoms such as fever, fatigue, and arthralgia often accompany these cutaneous findings.1,2 Although the pathophysiology has not been fully elucidated, this syndrome frequently is associated with infections, especially upper respiratory illnesses; medications; and malignancies. Among cases of malignancy-associated Sweet syndrome, hematologic malignancies, particularly acute myeloid leukemia and myelodysplastic syndrome, are more common than solid organ malignancies.1,2 Sweet syndrome may precede the associated malignancy by several months; thus, patients without an identifiable trigger for Sweet syndrome should be closely followed.2 Treatment with systemic steroids typically is effective.1,3 Typical histologic features include papillary dermal edema and a brisk neutrophilic infiltrate in the superficial to mid dermis (quiz image).4 Overlying epidermal spongiosis with or without vesiculation also can be seen.4 Leukocytoclasia and endothelial swelling without fibrinoid necrosis are typical, though full-blown leukocytoclastic vasculitis can be seen.3,4 A histiocytoid variant also has been described in which the dermal infiltrate is composed of mononuclear cells reminiscent of histiocytes that are thought to be immature cells of myeloid origin. This variant histologically can simulate leukemia cutis.5
Perniosis, also known as chilblains, typically presents with red to violaceous macules or papules on acral sites, particularly the distal fingers and toes.6,7 It tends to affect young women more frequently than other demographic groups. Although the pathophysiology is not fully understood, perniosis is thought to represent an abnormal inflammatory response to cold environmental conditions. It can occur as an idiopathic disorder or in association with various systemic illnesses including lupus erythematosus.6,7 The typical histologic findings include papillary dermal edema and a lymphocytic infiltrate in the superficial to deep dermis, often with perivascular and perieccrine accentuation (Figure 1).3,6 Other less common microscopic findings include sparse keratinocyte necrosis, basal layer vacuolar change, swelling of endothelial cells, and lymphocytic vasculitis.6 The lesions typically resolve spontaneously within a few weeks, but in some cases they may be chronic.3
Polymorphous light eruption, a common photodermatosis induced by UV light exposure, typically presents in adolescence or early adulthood with a female predominance. Patients usually develop this pruritic rash on sun-exposed skin other than the face and dorsal aspects of the hands in the spring or early summer upon increased sun exposure after the winter season.3,8 Consistent sunlight exposure throughout the summer months results in decreased flares. Various cutaneous morphologies including papules, vesicles, and plaques can be seen.3,8 Histologic findings include papillary dermal edema and a perivascular lymphocytic infiltrate in the superficial to deep dermis (Figure 2).4
Tinea corporis, a superficial cutaneous dermatophyte infection, typically presents as annular scaly plaques with central clearing. Vesicles and pustules also can be seen.3 The diagnosis can be confirmed via fungal culture, identification of hyphae on microscopic examination of skin scrapings using potassium hydroxide, or cutaneous biopsy. Histologic clues to diagnosis include a "compact stratum corneum (either uniform or forming a layer beneath a basket weave stratum corneum), parakeratosis, mild spongiosis, and neutrophils in the stratum corneum" (Figure 3).9 Papillary dermal edema also may be present, though this finding less commonly is reported.9,10 Because fungal hyphae can be difficult to identify on hematoxylin and eosin-stained slides, special stains such as periodic acid-Schiff or Grocott-Gomori methenamine-silver may be helpful.9 These infections are managed with topical or oral antifungal medications.
Wells syndrome, also known as eosinophilic cellulitis, presents with an acute eruption that can clinically resemble bacterial cellulitis.3 It has been described in children and adults with various clinical morphologies including plaques, bullae, papulovesicles, and papulonodules. Peripheral eosinophilia may be present.11 The clinical lesions usually resolve spontaneously in a few weeks to months, but recurrences are typical.3,11 Histologic findings include papillary dermal edema with or without subepidermal bulla formation and epidermal spongiosis as well as a mixed inflammatory infiltrate with a predominance of eosinophils and flame figures (Figure 4).4 Flame figures are collagen fibers coated with major basic protein and other constituents of degranulated eosinophils.3 Although flame figures often are present in Wells syndrome, they are not specific to this condition.3,4 Some consider Wells syndrome an exaggerated reaction pattern rather than a specific entity.3
- Rochet N, Chavan R, Cappel M, et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557-564.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Elsevier; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. Elsevier Saunders; 2012.
- Alegría-Landa V, Rodríguez-Pinilla S, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Boada A, Bielsa I, Fernández-Figueras M, et al. Perniosis: clinical and histopathological analysis. Am J Dermatopathol. 2010;32:19-23.
- Takci Z, Vahaboglu G, Eksioglu H. Epidemiological patterns of perniosis, and its association with systemic disorder. Clin Exp Dermatol. 2012;37:844-849.
- Gruber-Wackernagel A, Byrne S, Wolf P. Polymorphous light eruption: clinic aspects and pathogenesis. Dermatol Clin. 2014;32:315-334.
- Elbendary A, Valdebran M, Gad A, et al. When to suspect tinea; a histopathologic study of 103 cases of PAS-positive tinea. J Cutan Pathol. 2016;46:852-857.
- Hoss D, Berke A, Kerr P, et al. Prominent papillary dermal edema in dermatophytosis (tinea corporis). J Cutan Pathol. 2010;37:237-242.
- Caputo R, Marzano A, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Rochet N, Chavan R, Cappel M, et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557-564.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Elsevier; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. Elsevier Saunders; 2012.
- Alegría-Landa V, Rodríguez-Pinilla S, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Boada A, Bielsa I, Fernández-Figueras M, et al. Perniosis: clinical and histopathological analysis. Am J Dermatopathol. 2010;32:19-23.
- Takci Z, Vahaboglu G, Eksioglu H. Epidemiological patterns of perniosis, and its association with systemic disorder. Clin Exp Dermatol. 2012;37:844-849.
- Gruber-Wackernagel A, Byrne S, Wolf P. Polymorphous light eruption: clinic aspects and pathogenesis. Dermatol Clin. 2014;32:315-334.
- Elbendary A, Valdebran M, Gad A, et al. When to suspect tinea; a histopathologic study of 103 cases of PAS-positive tinea. J Cutan Pathol. 2016;46:852-857.
- Hoss D, Berke A, Kerr P, et al. Prominent papillary dermal edema in dermatophytosis (tinea corporis). J Cutan Pathol. 2010;37:237-242.
- Caputo R, Marzano A, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
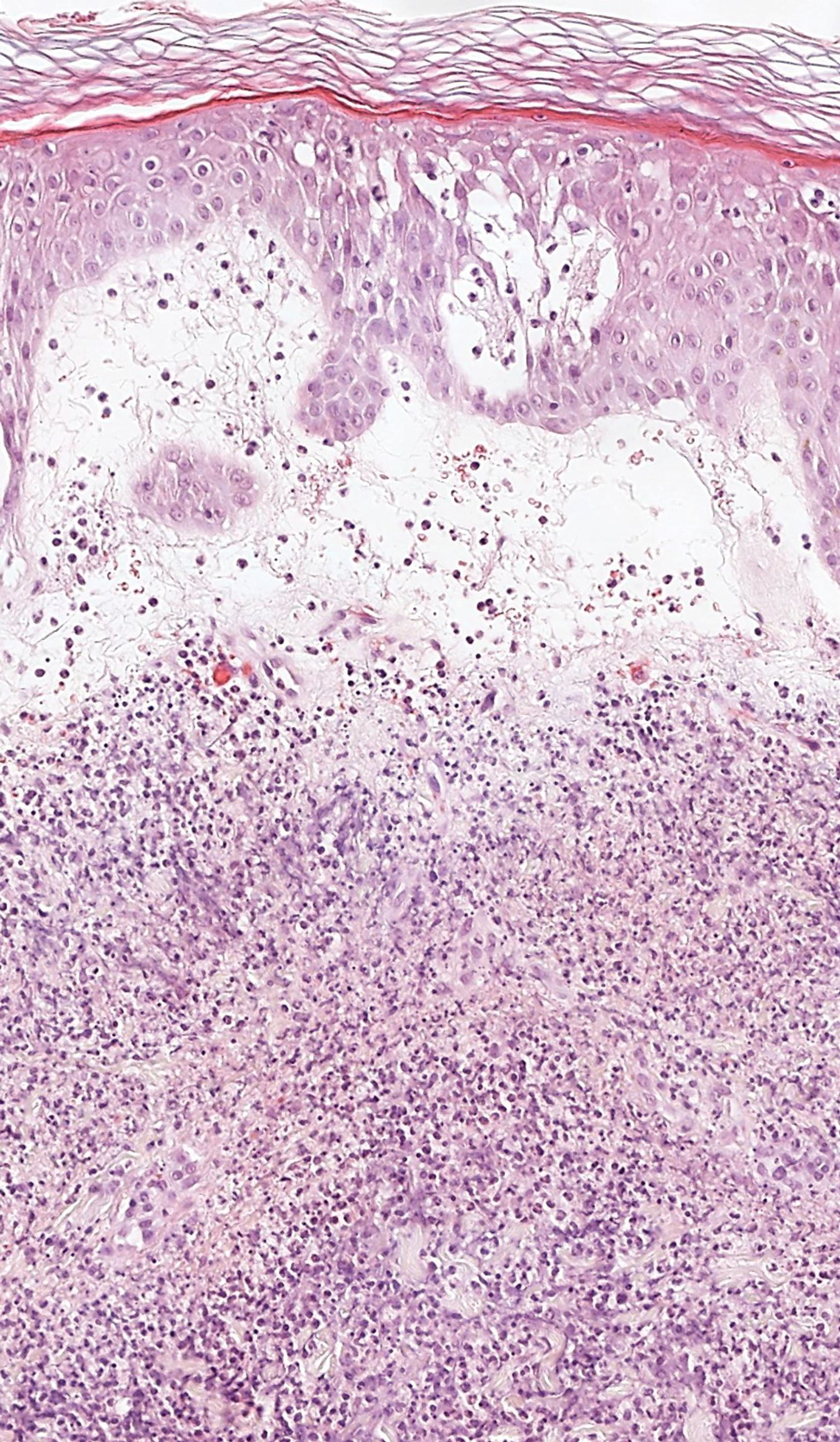
A 62-year-old woman presented with a tender diffuse eruption of erythematous and edematous papules and plaques on the face, neck, chest, and extremities, some appearing vesiculopustular.
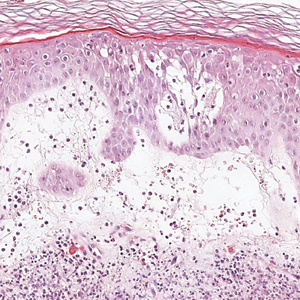
Asymptomatic Hemorrhagic Lesions in an Anemic Woman
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
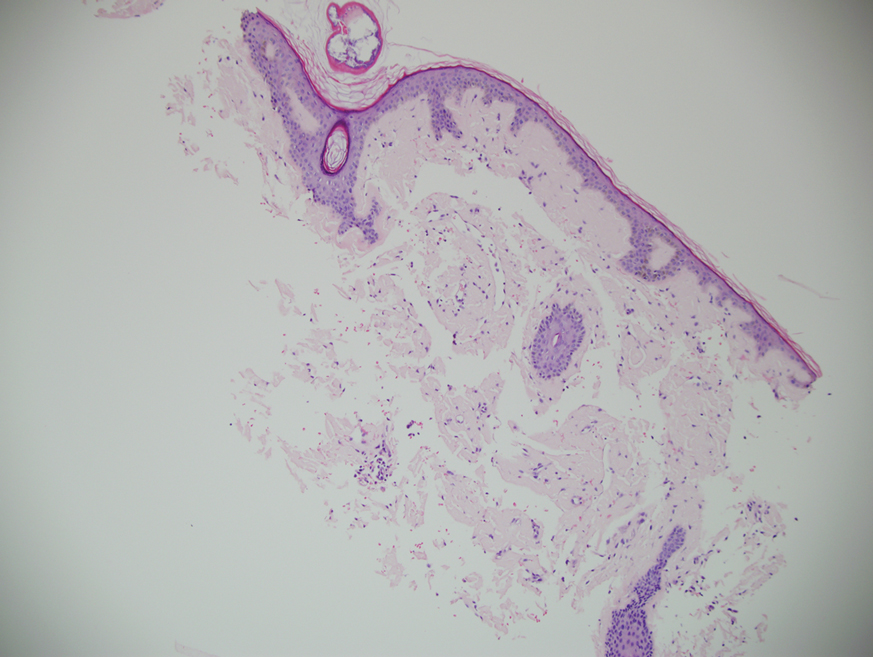
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
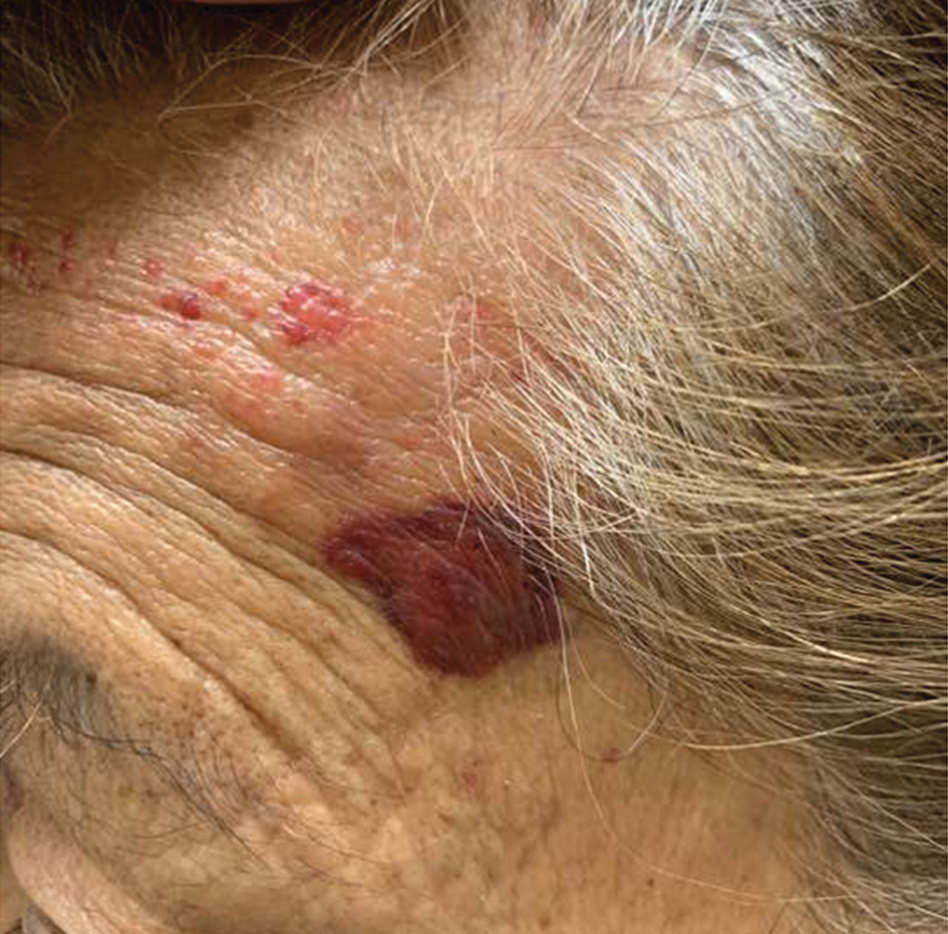
A 67-year-old woman with a medical history of type 2 diabetes mellitus, unspecified leukocytosis, and anemia presented to the dermatology clinic with asymptomatic hemorrhagic bullae on the face, chest, and tongue, as well as a large, tender, tense, hemorrhagic bulla on the groin of 3 to 4 months’ duration. A review of systems was negative for fever, chills, night sweats, malaise, shortness of breath, and dyspnea on exertion. A complete blood cell count showed mild leukocytosis, anemia, and thrombocytopenia. Her creatinine level was slightly elevated. Chest computed tomography showed early pulmonary fibrosis and coronary artery calcification. An echocardiogram showed diastolic dysfunction with moderate left ventricle thickening. A serum and urine electrophoresis demonstrated elevated free λ light chains with an M-spike. A punch biopsy was performed.
High-Potency Topical Steroid Treatment of Multiple Keratoacanthomas Associated With Prurigo Nodularis
Practice Gap
Multiple keratoacanthomas (KAs) of the legs often are a challenge to treat, especially when these lesions appear within a field of prurigo nodules. Multiple KAs associated with prurigo nodularis is a rarer finding; more often, the condition is reported on the lower limbs of elderly women with actinically damaged skin.1,2 At times, it can be difficult to distinguish between KA and prurigo nodularis in these patients, who often report notable pruritus and might have associated eczematous dermatitis.2
Keratoacanthomas often are treated with aggressive modalities, such as Mohs micrographic surgery, excision, and electrodesiccation and curettage. Some patients are hesitant to undergo surgical treatment, however, preferring a less invasive approach. Trauma from these aggressive modalities also can be associated with recurrence of existing lesions or development of new KAs, possibly related to stimulation of a local inflammatory response and upregulation of helper T cells.2-4
Acitretin and other systemic retinoids often are considered first-line therapy for multiple KAs. Cyclosporine has been added as adjunctive treatment in cases associated with prurigo nodularis or eczematous dermatitis1,2; however, these treatments have a high rate of discontinuation because of adverse effects, including transaminitis, xerostomia, alopecia (acitretin), and renal toxicity (cyclosporine).2
Another treatment option for patients with coexisting KA and prurigo nodularis is intralesional corticosteroids, often administered in combination with systemic retinoids.3 Topical 5-fluorouracil (5-FU) has been used successfully for KA, but topical treatment options are limited if 5-FU fails. Topical imiquimod and cryotherapy are thought to be of little benefit, and the appearance of new KA within imiquimod and cryotherapy treatment fields has been reported.1,2 Topical corticosteroids have been used as an adjuvant therapy for multiple KAs associated with prurigo nodularis; however, a PubMed search of articles indexed for MEDLINE using the terms keratoacanthoma and steroid and keratoacanthoma and prurigo nodularis yielded no published reports of successful use of topical corticosteroids as monotherapy.2
The Technique
For patients who want to continue topical treatment of coexisting KA and prurigo nodularis after topical 5-FU fails, we have found success applying a high-potency topical corticosteroid to affected areas under occlusion nightly for 6 to 8 weeks. This treatment not only leads to resolution of KA but also simultaneously treats prurigo nodules that might be clinically difficult to distinguish from KA in some presentations. This regimen has been implemented in our practice with remarkable reduction of KA burden and relief of pruritus.
In a 68-year-old woman who was treated with this technique, multiple biopsies had shown KA (or well-differentiated squamous cell carcinoma that appeared clinically as KA) on the shin (Figure, A) arising amid many lesions consistent with prurigo nodules. Topical 5-FU had failed, but the patient did not want to be treated with a more invasive modality, such as excision or injection.
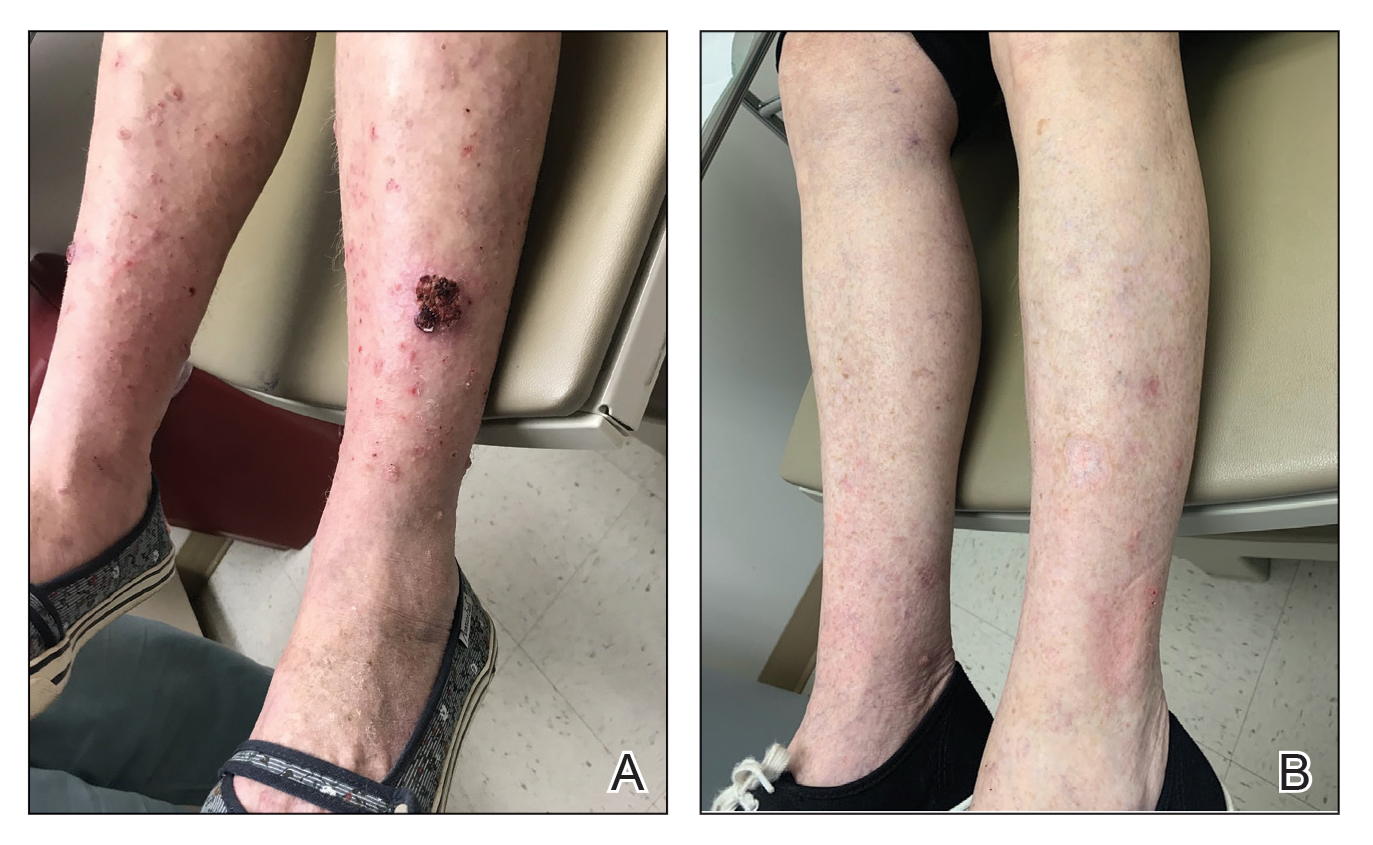
Instead, we treated the patient with clobetasol propionate ointment 0.05% under occlusion nightly for 6 weeks. This strategy produced resolution of both KA and prurigo nodules (Figure, B). When lesions recurred after a few months, they were successfully re-treated with topical clobetasol under occlusion in a second 6-week course.
Practical Implications
Treatment of multiple KAs associated with prurigo nodularis can present a distinct challenge. For the subset of patients who want to pursue topical treatment, options reported in the literature are limited. We have found success treating multiple KAs and associated prurigo nodules with a high-potency topical corticosteroid under occlusion, with minimal or no adverse effects. We believe that a topical corticosteroid can be implemented easily in clinical practice before a more invasive surgical or intralesional modality is considered.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233. doi:10.1016/j.jaad.2015.11.033
- Wu TP, Miller K, Cohen DE, et al. Keratoacanthomas arising in association with prurigo nodules in pruritic, actinically damaged skin. J Am Acad Dermatol. 2013;69:426-430. doi:10.1016/J.JAAD.2013.03.035
- Sanders S, Busam KJ, Halpern AC, et al. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958. doi:10.1046/j.1524-4725.2002.02069.x
- Lee S, Coutts I, Ryan A, et al. Keratoacanthoma formation after skin grafting: a brief report and pathophysiological hypothesis. Australas J Dermatol. 2017;58:E117-E119. doi:10.1111/ajd.12501
Practice Gap
Multiple keratoacanthomas (KAs) of the legs often are a challenge to treat, especially when these lesions appear within a field of prurigo nodules. Multiple KAs associated with prurigo nodularis is a rarer finding; more often, the condition is reported on the lower limbs of elderly women with actinically damaged skin.1,2 At times, it can be difficult to distinguish between KA and prurigo nodularis in these patients, who often report notable pruritus and might have associated eczematous dermatitis.2
Keratoacanthomas often are treated with aggressive modalities, such as Mohs micrographic surgery, excision, and electrodesiccation and curettage. Some patients are hesitant to undergo surgical treatment, however, preferring a less invasive approach. Trauma from these aggressive modalities also can be associated with recurrence of existing lesions or development of new KAs, possibly related to stimulation of a local inflammatory response and upregulation of helper T cells.2-4
Acitretin and other systemic retinoids often are considered first-line therapy for multiple KAs. Cyclosporine has been added as adjunctive treatment in cases associated with prurigo nodularis or eczematous dermatitis1,2; however, these treatments have a high rate of discontinuation because of adverse effects, including transaminitis, xerostomia, alopecia (acitretin), and renal toxicity (cyclosporine).2
Another treatment option for patients with coexisting KA and prurigo nodularis is intralesional corticosteroids, often administered in combination with systemic retinoids.3 Topical 5-fluorouracil (5-FU) has been used successfully for KA, but topical treatment options are limited if 5-FU fails. Topical imiquimod and cryotherapy are thought to be of little benefit, and the appearance of new KA within imiquimod and cryotherapy treatment fields has been reported.1,2 Topical corticosteroids have been used as an adjuvant therapy for multiple KAs associated with prurigo nodularis; however, a PubMed search of articles indexed for MEDLINE using the terms keratoacanthoma and steroid and keratoacanthoma and prurigo nodularis yielded no published reports of successful use of topical corticosteroids as monotherapy.2
The Technique
For patients who want to continue topical treatment of coexisting KA and prurigo nodularis after topical 5-FU fails, we have found success applying a high-potency topical corticosteroid to affected areas under occlusion nightly for 6 to 8 weeks. This treatment not only leads to resolution of KA but also simultaneously treats prurigo nodules that might be clinically difficult to distinguish from KA in some presentations. This regimen has been implemented in our practice with remarkable reduction of KA burden and relief of pruritus.
In a 68-year-old woman who was treated with this technique, multiple biopsies had shown KA (or well-differentiated squamous cell carcinoma that appeared clinically as KA) on the shin (Figure, A) arising amid many lesions consistent with prurigo nodules. Topical 5-FU had failed, but the patient did not want to be treated with a more invasive modality, such as excision or injection.
Instead, we treated the patient with clobetasol propionate ointment 0.05% under occlusion nightly for 6 weeks. This strategy produced resolution of both KA and prurigo nodules (Figure, B). When lesions recurred after a few months, they were successfully re-treated with topical clobetasol under occlusion in a second 6-week course.
Practical Implications
Treatment of multiple KAs associated with prurigo nodularis can present a distinct challenge. For the subset of patients who want to pursue topical treatment, options reported in the literature are limited. We have found success treating multiple KAs and associated prurigo nodules with a high-potency topical corticosteroid under occlusion, with minimal or no adverse effects. We believe that a topical corticosteroid can be implemented easily in clinical practice before a more invasive surgical or intralesional modality is considered.
Practice Gap
Multiple keratoacanthomas (KAs) of the legs often are a challenge to treat, especially when these lesions appear within a field of prurigo nodules. Multiple KAs associated with prurigo nodularis is a rarer finding; more often, the condition is reported on the lower limbs of elderly women with actinically damaged skin.1,2 At times, it can be difficult to distinguish between KA and prurigo nodularis in these patients, who often report notable pruritus and might have associated eczematous dermatitis.2
Keratoacanthomas often are treated with aggressive modalities, such as Mohs micrographic surgery, excision, and electrodesiccation and curettage. Some patients are hesitant to undergo surgical treatment, however, preferring a less invasive approach. Trauma from these aggressive modalities also can be associated with recurrence of existing lesions or development of new KAs, possibly related to stimulation of a local inflammatory response and upregulation of helper T cells.2-4
Acitretin and other systemic retinoids often are considered first-line therapy for multiple KAs. Cyclosporine has been added as adjunctive treatment in cases associated with prurigo nodularis or eczematous dermatitis1,2; however, these treatments have a high rate of discontinuation because of adverse effects, including transaminitis, xerostomia, alopecia (acitretin), and renal toxicity (cyclosporine).2
Another treatment option for patients with coexisting KA and prurigo nodularis is intralesional corticosteroids, often administered in combination with systemic retinoids.3 Topical 5-fluorouracil (5-FU) has been used successfully for KA, but topical treatment options are limited if 5-FU fails. Topical imiquimod and cryotherapy are thought to be of little benefit, and the appearance of new KA within imiquimod and cryotherapy treatment fields has been reported.1,2 Topical corticosteroids have been used as an adjuvant therapy for multiple KAs associated with prurigo nodularis; however, a PubMed search of articles indexed for MEDLINE using the terms keratoacanthoma and steroid and keratoacanthoma and prurigo nodularis yielded no published reports of successful use of topical corticosteroids as monotherapy.2
The Technique
For patients who want to continue topical treatment of coexisting KA and prurigo nodularis after topical 5-FU fails, we have found success applying a high-potency topical corticosteroid to affected areas under occlusion nightly for 6 to 8 weeks. This treatment not only leads to resolution of KA but also simultaneously treats prurigo nodules that might be clinically difficult to distinguish from KA in some presentations. This regimen has been implemented in our practice with remarkable reduction of KA burden and relief of pruritus.
In a 68-year-old woman who was treated with this technique, multiple biopsies had shown KA (or well-differentiated squamous cell carcinoma that appeared clinically as KA) on the shin (Figure, A) arising amid many lesions consistent with prurigo nodules. Topical 5-FU had failed, but the patient did not want to be treated with a more invasive modality, such as excision or injection.
Instead, we treated the patient with clobetasol propionate ointment 0.05% under occlusion nightly for 6 weeks. This strategy produced resolution of both KA and prurigo nodules (Figure, B). When lesions recurred after a few months, they were successfully re-treated with topical clobetasol under occlusion in a second 6-week course.
Practical Implications
Treatment of multiple KAs associated with prurigo nodularis can present a distinct challenge. For the subset of patients who want to pursue topical treatment, options reported in the literature are limited. We have found success treating multiple KAs and associated prurigo nodules with a high-potency topical corticosteroid under occlusion, with minimal or no adverse effects. We believe that a topical corticosteroid can be implemented easily in clinical practice before a more invasive surgical or intralesional modality is considered.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233. doi:10.1016/j.jaad.2015.11.033
- Wu TP, Miller K, Cohen DE, et al. Keratoacanthomas arising in association with prurigo nodules in pruritic, actinically damaged skin. J Am Acad Dermatol. 2013;69:426-430. doi:10.1016/J.JAAD.2013.03.035
- Sanders S, Busam KJ, Halpern AC, et al. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958. doi:10.1046/j.1524-4725.2002.02069.x
- Lee S, Coutts I, Ryan A, et al. Keratoacanthoma formation after skin grafting: a brief report and pathophysiological hypothesis. Australas J Dermatol. 2017;58:E117-E119. doi:10.1111/ajd.12501
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233. doi:10.1016/j.jaad.2015.11.033
- Wu TP, Miller K, Cohen DE, et al. Keratoacanthomas arising in association with prurigo nodules in pruritic, actinically damaged skin. J Am Acad Dermatol. 2013;69:426-430. doi:10.1016/J.JAAD.2013.03.035
- Sanders S, Busam KJ, Halpern AC, et al. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958. doi:10.1046/j.1524-4725.2002.02069.x
- Lee S, Coutts I, Ryan A, et al. Keratoacanthoma formation after skin grafting: a brief report and pathophysiological hypothesis. Australas J Dermatol. 2017;58:E117-E119. doi:10.1111/ajd.12501
Gynecologic and Obstetric Implications of Darier Disease: A Dermatologist’s Perspective
Darier disease (DD)(also known as dyskeratosis follicularis) is a rare, autosomal-dominant genodermatosis characterized by greasy, rough, keratotic papules; typical nail abnormalities; mucosal changes; and characteristic dyskeratotic acantholysis that is called corps ronds and grains on histopathologic analysis. Darier disease is caused by mutations of the ATP2A2 gene on chromosome 12q23-24.1,2
Because of the autosomal-dominant pattern of inheritance in DD, if either parent is affected by DD, approximately 50% of their offspring will have the disorder. Therefore, couples need to be offered genetic counseling at a preconception visit or early in pregnancy. Although penetrance of DD is complete, spontaneous mutations are frequent and expressivity is variable1; prenatal diagnosis, though available since the 1980s, is therefore unreliable in DD, given the considerable variation in phenotypic expressivity. Differing phenotypes underscore the importance of proper counseling by the treating dermatologist or other provider. Females with a mild or nearly undetectable phenotype can give birth to a child with severe disease.
Lack of clear understanding about the variable phenotypic expressivity of DD can cause considerable anger, anxiety, guilt, psychological trauma, and fear in parents, should their child later develop a severe phenotype. They may feel that they were not properly prepared for the outcome. The physician-parent or physician-patient relationship can be negatively impacted if ongoing counseling is inadequate.
Clinically, DD presents in early adolescence (age range, 6–20 years) in most patients, which means that the disease and female reproductive years are contemporaneous. However, gynecologic and obstetric issues and complications of DD rarely have been addressed.3 Oromucosal involvement in DD is reported in 13% to 50% of cases, yet vaginal and cervical mucosal involvement rarely has been described,4,5 likely due to underreporting. Therefore, in this rare disease, it is important to address these aspects so that the patients are provided with appropriate management options.
Implications for Cervical Screening and Papanicolaou Tests
Cytopathologic findings of a Papanicolaou test taken from a patient with DD can lead to erroneous diagnosis of a low-grade squamous intraepithelial lesion due to cervical involvement by the disease process; therefore, correct interpretation of a smear may be inappropriate and erroneous. The cytopathologist needs to be informed of the patient’s diagnosis of DD in advance for appropriate reporting.5,6
Obstetric Implications
Fertility is normal in DD patients, and pregnancy usually has a normal course; however, exacerbation and remission of disease have been reported. de la Rosa Carrillo7 reported a case of vegetating DD during pregnancy. He described it as an exacerbation with concurrent bacterial infection and bilateral external otitis.7 Spouge et al8 reported a case of a 58-year-old woman who was the mother of 4 DD patients. She experienced an exacerbation of DD during all 6 pregnancies but improved immediately postpartum.8 Espy et al9 evaluated 8 cases of women with DD and described spontaneous improvement of the disorder during pregnancy (1 case) or while taking an oral contraceptive (3 cases).
Prenatal Counseling
Women with DD should be encouraged to talk to their dermatologist, obstetrician, or other provider of prenatal care regarding plans for pregnancy, labor, and delivery, as these events might be affected by the disorder. During pregnancy, careful monitoring and self-care remain essential. Simple measures to reduce the impact of irritants on DD during pregnancy include keeping the skin cool, using a soothing moisturizer, applying photoprotection, and using sunscreen. Treatment with systemic retinoids must be avoided if pregnancy is planned.
Warty plaques and papules of DD can involve flexures (groin, vulva, and perineum), with resultant malodor and pruritus10 as well as the potential for (drug resistant) secondary infection (eg, Staphylococcus aureus, group B Streptococcus, viruses [eg, Kaposi varicelliform eruption]). Skin swabs should be taken for culture and susceptibility testing, and infection should be treated at the earliest sign.
Management Concerns During Pregnancy and Delivery
Because the benefits of treating DD might outweigh risk in certain cases, thorough discussion with the patient about options is recommended, including the following concerns:
• Because mucocutaneous elasticity of the birth canal, including the vulva, perineum, and groin, is essential for nontraumatic vaginal delivery, it might be necessary to schedule an elective cesarean delivery in DD patients in whom these regions are involved.11
• In females with lower abdominal lesions, using a Pfannenstiel-Kerr incision for cesarean delivery might be problematic.11
• A single case report has described successful anesthetic management of labor, delivery, and postpartum care in a DD patient.12 Involvement of the skin of the back might preclude safe administration of regional anesthesia; however, because DD lesions are considered noninfectious, the authors operatively administered a subarachnoid block at the L3-L4 interspace through a lesion-free area. Postpartum, the patient was observed in the intensive care unit. She and the baby remained stable; she did not develop infectious complications, including a central nervous system infection.12
•Mucosal involvement is relatively rare in DD and has not been reported to compromise airway management.8
Postnatal Considerations
Breastfeeding might have to be stopped early or withheld altogether if there is widespread involvement of the skin of the breast or the nipple.11 Darier disease has been associated with neuropsychiatric manifestations, including major depression (30%), suicide attempts (13%), suicidal thoughts (31%), cyclothymia, bipolar disorder (4%), and epilepsy (3%).13,14 Therefore, patients should be screened for postpartum psychiatric manifestations at an early follow-up visit.
Final Thoughts
Although the etiology of DD is well known, the gynelogic and obstretric implications of this genodermatosis have rarely been described. This brief commentary is an attempt to provide the important information to a practicing dermatologist for appropriate management of female DD patients.
- Bale SJ, Toro JR. Genetic basis of Darier-White disease: bad pumps cause bumps. J Cutan Med Surg. 2000;4:103-106. doi:10.1177/120347540000400212
- Kansal NK, Hazarika N, Rao S. Familial case of Darier disease with guttate leukoderma: a case series from India. Indian Dermatol Online J. 2018;9:62-63. doi:10.4103/idoj.IDOJ_52_17
- Lynch PJ. Vulvar dermatoses: the eczematous diseases. In: Black M, Ambros-Rudolph CM, Edwards L, Lynch P, eds. Obstetric and Gynecologic Dermatology. 3rd ed. Mosby-Elsevier; 2008:192-194.
- Adam AE. Ectopic Darier’s disease of the cervix: an extraordinary cause of an abnormal smear. Cytopathology. 1996;7:414-421. doi:10.1111/j.1365-2303.1996.tb00547.x
- Suárez-Peñaranda JM, Antúnez JR, Del Rio E, et al. Vaginal involvement in a woman with Darier’s disease: a case report. Acta Cytol. 2005;49:530-532. doi:10.1159/000326200
- Boon ME. Dr. Darier’s lesson: it can be advantageous to the patient to ignore evident cytonuclear changes. Acta Cytol. 2005;49:469-470. doi:10.1159/000326189
- de la Rosa Carrillo D. Vegetating Darier’s disease during pregnancy. Acta Derm Venereol. 2006;86:259-260. doi:10.2340/00015555-0066
- Spouge JD, Trott JR, Chesko G. Darier-White’s disease: a cause of white lesions of the mucosa. report of four cases. Oral Surg Oral Med Oral Pathol. 1966;21:441-457. doi:10.1016/0030-4220(66)90401-4
- Espy PD, Stone S, Jolly HW Jr. Hormonal dependency in Darier disease. Cutis. 1976;17:315-320.
- De D, Kanwar AJ, Saikia UN. Uncommon flexural presentation of Darier disease. J Cutan Med Surg. 2008;12:249-252. doi:10.2310/7750.2008.07035
- Quinlivan JA, O'Halloran LC. Darier’s disease and pregnancy. Dermatol Aspects. 2013;1:1-3. doi:10.7243/2053-5309-1-1
- Sharma R, Singh BP, Das SN. Anesthetic management of cesarean section in a parturient with Darier’s disease. Acta Anaesthesiol Taiwan. 2010;48:158-159. doi:10.1016/S1875-4597(10)60051-3
- Gordon-Smith K, Jones LA, Burge SM, et al. The neuropsychiatric phenotype in Darier disease. Br J Dermatol. 2010;163:515-522. doi:10.1111/j.1365-2133.2010.09834.x
- Dodiuk-Gad RP, Cohen-Barak E, Khayat M, et al. Darier disease in Israel: combined evaluation of genetic and neuropsychiatric aspects. Br J Dermatol. 2016;174:562-568. doi:10.1111/bjd.14220
Darier disease (DD)(also known as dyskeratosis follicularis) is a rare, autosomal-dominant genodermatosis characterized by greasy, rough, keratotic papules; typical nail abnormalities; mucosal changes; and characteristic dyskeratotic acantholysis that is called corps ronds and grains on histopathologic analysis. Darier disease is caused by mutations of the ATP2A2 gene on chromosome 12q23-24.1,2
Because of the autosomal-dominant pattern of inheritance in DD, if either parent is affected by DD, approximately 50% of their offspring will have the disorder. Therefore, couples need to be offered genetic counseling at a preconception visit or early in pregnancy. Although penetrance of DD is complete, spontaneous mutations are frequent and expressivity is variable1; prenatal diagnosis, though available since the 1980s, is therefore unreliable in DD, given the considerable variation in phenotypic expressivity. Differing phenotypes underscore the importance of proper counseling by the treating dermatologist or other provider. Females with a mild or nearly undetectable phenotype can give birth to a child with severe disease.
Lack of clear understanding about the variable phenotypic expressivity of DD can cause considerable anger, anxiety, guilt, psychological trauma, and fear in parents, should their child later develop a severe phenotype. They may feel that they were not properly prepared for the outcome. The physician-parent or physician-patient relationship can be negatively impacted if ongoing counseling is inadequate.
Clinically, DD presents in early adolescence (age range, 6–20 years) in most patients, which means that the disease and female reproductive years are contemporaneous. However, gynecologic and obstetric issues and complications of DD rarely have been addressed.3 Oromucosal involvement in DD is reported in 13% to 50% of cases, yet vaginal and cervical mucosal involvement rarely has been described,4,5 likely due to underreporting. Therefore, in this rare disease, it is important to address these aspects so that the patients are provided with appropriate management options.
Implications for Cervical Screening and Papanicolaou Tests
Cytopathologic findings of a Papanicolaou test taken from a patient with DD can lead to erroneous diagnosis of a low-grade squamous intraepithelial lesion due to cervical involvement by the disease process; therefore, correct interpretation of a smear may be inappropriate and erroneous. The cytopathologist needs to be informed of the patient’s diagnosis of DD in advance for appropriate reporting.5,6
Obstetric Implications
Fertility is normal in DD patients, and pregnancy usually has a normal course; however, exacerbation and remission of disease have been reported. de la Rosa Carrillo7 reported a case of vegetating DD during pregnancy. He described it as an exacerbation with concurrent bacterial infection and bilateral external otitis.7 Spouge et al8 reported a case of a 58-year-old woman who was the mother of 4 DD patients. She experienced an exacerbation of DD during all 6 pregnancies but improved immediately postpartum.8 Espy et al9 evaluated 8 cases of women with DD and described spontaneous improvement of the disorder during pregnancy (1 case) or while taking an oral contraceptive (3 cases).
Prenatal Counseling
Women with DD should be encouraged to talk to their dermatologist, obstetrician, or other provider of prenatal care regarding plans for pregnancy, labor, and delivery, as these events might be affected by the disorder. During pregnancy, careful monitoring and self-care remain essential. Simple measures to reduce the impact of irritants on DD during pregnancy include keeping the skin cool, using a soothing moisturizer, applying photoprotection, and using sunscreen. Treatment with systemic retinoids must be avoided if pregnancy is planned.
Warty plaques and papules of DD can involve flexures (groin, vulva, and perineum), with resultant malodor and pruritus10 as well as the potential for (drug resistant) secondary infection (eg, Staphylococcus aureus, group B Streptococcus, viruses [eg, Kaposi varicelliform eruption]). Skin swabs should be taken for culture and susceptibility testing, and infection should be treated at the earliest sign.
Management Concerns During Pregnancy and Delivery
Because the benefits of treating DD might outweigh risk in certain cases, thorough discussion with the patient about options is recommended, including the following concerns:
• Because mucocutaneous elasticity of the birth canal, including the vulva, perineum, and groin, is essential for nontraumatic vaginal delivery, it might be necessary to schedule an elective cesarean delivery in DD patients in whom these regions are involved.11
• In females with lower abdominal lesions, using a Pfannenstiel-Kerr incision for cesarean delivery might be problematic.11
• A single case report has described successful anesthetic management of labor, delivery, and postpartum care in a DD patient.12 Involvement of the skin of the back might preclude safe administration of regional anesthesia; however, because DD lesions are considered noninfectious, the authors operatively administered a subarachnoid block at the L3-L4 interspace through a lesion-free area. Postpartum, the patient was observed in the intensive care unit. She and the baby remained stable; she did not develop infectious complications, including a central nervous system infection.12
•Mucosal involvement is relatively rare in DD and has not been reported to compromise airway management.8
Postnatal Considerations
Breastfeeding might have to be stopped early or withheld altogether if there is widespread involvement of the skin of the breast or the nipple.11 Darier disease has been associated with neuropsychiatric manifestations, including major depression (30%), suicide attempts (13%), suicidal thoughts (31%), cyclothymia, bipolar disorder (4%), and epilepsy (3%).13,14 Therefore, patients should be screened for postpartum psychiatric manifestations at an early follow-up visit.
Final Thoughts
Although the etiology of DD is well known, the gynelogic and obstretric implications of this genodermatosis have rarely been described. This brief commentary is an attempt to provide the important information to a practicing dermatologist for appropriate management of female DD patients.
Darier disease (DD)(also known as dyskeratosis follicularis) is a rare, autosomal-dominant genodermatosis characterized by greasy, rough, keratotic papules; typical nail abnormalities; mucosal changes; and characteristic dyskeratotic acantholysis that is called corps ronds and grains on histopathologic analysis. Darier disease is caused by mutations of the ATP2A2 gene on chromosome 12q23-24.1,2
Because of the autosomal-dominant pattern of inheritance in DD, if either parent is affected by DD, approximately 50% of their offspring will have the disorder. Therefore, couples need to be offered genetic counseling at a preconception visit or early in pregnancy. Although penetrance of DD is complete, spontaneous mutations are frequent and expressivity is variable1; prenatal diagnosis, though available since the 1980s, is therefore unreliable in DD, given the considerable variation in phenotypic expressivity. Differing phenotypes underscore the importance of proper counseling by the treating dermatologist or other provider. Females with a mild or nearly undetectable phenotype can give birth to a child with severe disease.
Lack of clear understanding about the variable phenotypic expressivity of DD can cause considerable anger, anxiety, guilt, psychological trauma, and fear in parents, should their child later develop a severe phenotype. They may feel that they were not properly prepared for the outcome. The physician-parent or physician-patient relationship can be negatively impacted if ongoing counseling is inadequate.
Clinically, DD presents in early adolescence (age range, 6–20 years) in most patients, which means that the disease and female reproductive years are contemporaneous. However, gynecologic and obstetric issues and complications of DD rarely have been addressed.3 Oromucosal involvement in DD is reported in 13% to 50% of cases, yet vaginal and cervical mucosal involvement rarely has been described,4,5 likely due to underreporting. Therefore, in this rare disease, it is important to address these aspects so that the patients are provided with appropriate management options.
Implications for Cervical Screening and Papanicolaou Tests
Cytopathologic findings of a Papanicolaou test taken from a patient with DD can lead to erroneous diagnosis of a low-grade squamous intraepithelial lesion due to cervical involvement by the disease process; therefore, correct interpretation of a smear may be inappropriate and erroneous. The cytopathologist needs to be informed of the patient’s diagnosis of DD in advance for appropriate reporting.5,6
Obstetric Implications
Fertility is normal in DD patients, and pregnancy usually has a normal course; however, exacerbation and remission of disease have been reported. de la Rosa Carrillo7 reported a case of vegetating DD during pregnancy. He described it as an exacerbation with concurrent bacterial infection and bilateral external otitis.7 Spouge et al8 reported a case of a 58-year-old woman who was the mother of 4 DD patients. She experienced an exacerbation of DD during all 6 pregnancies but improved immediately postpartum.8 Espy et al9 evaluated 8 cases of women with DD and described spontaneous improvement of the disorder during pregnancy (1 case) or while taking an oral contraceptive (3 cases).
Prenatal Counseling
Women with DD should be encouraged to talk to their dermatologist, obstetrician, or other provider of prenatal care regarding plans for pregnancy, labor, and delivery, as these events might be affected by the disorder. During pregnancy, careful monitoring and self-care remain essential. Simple measures to reduce the impact of irritants on DD during pregnancy include keeping the skin cool, using a soothing moisturizer, applying photoprotection, and using sunscreen. Treatment with systemic retinoids must be avoided if pregnancy is planned.
Warty plaques and papules of DD can involve flexures (groin, vulva, and perineum), with resultant malodor and pruritus10 as well as the potential for (drug resistant) secondary infection (eg, Staphylococcus aureus, group B Streptococcus, viruses [eg, Kaposi varicelliform eruption]). Skin swabs should be taken for culture and susceptibility testing, and infection should be treated at the earliest sign.
Management Concerns During Pregnancy and Delivery
Because the benefits of treating DD might outweigh risk in certain cases, thorough discussion with the patient about options is recommended, including the following concerns:
• Because mucocutaneous elasticity of the birth canal, including the vulva, perineum, and groin, is essential for nontraumatic vaginal delivery, it might be necessary to schedule an elective cesarean delivery in DD patients in whom these regions are involved.11
• In females with lower abdominal lesions, using a Pfannenstiel-Kerr incision for cesarean delivery might be problematic.11
• A single case report has described successful anesthetic management of labor, delivery, and postpartum care in a DD patient.12 Involvement of the skin of the back might preclude safe administration of regional anesthesia; however, because DD lesions are considered noninfectious, the authors operatively administered a subarachnoid block at the L3-L4 interspace through a lesion-free area. Postpartum, the patient was observed in the intensive care unit. She and the baby remained stable; she did not develop infectious complications, including a central nervous system infection.12
•Mucosal involvement is relatively rare in DD and has not been reported to compromise airway management.8
Postnatal Considerations
Breastfeeding might have to be stopped early or withheld altogether if there is widespread involvement of the skin of the breast or the nipple.11 Darier disease has been associated with neuropsychiatric manifestations, including major depression (30%), suicide attempts (13%), suicidal thoughts (31%), cyclothymia, bipolar disorder (4%), and epilepsy (3%).13,14 Therefore, patients should be screened for postpartum psychiatric manifestations at an early follow-up visit.
Final Thoughts
Although the etiology of DD is well known, the gynelogic and obstretric implications of this genodermatosis have rarely been described. This brief commentary is an attempt to provide the important information to a practicing dermatologist for appropriate management of female DD patients.
- Bale SJ, Toro JR. Genetic basis of Darier-White disease: bad pumps cause bumps. J Cutan Med Surg. 2000;4:103-106. doi:10.1177/120347540000400212
- Kansal NK, Hazarika N, Rao S. Familial case of Darier disease with guttate leukoderma: a case series from India. Indian Dermatol Online J. 2018;9:62-63. doi:10.4103/idoj.IDOJ_52_17
- Lynch PJ. Vulvar dermatoses: the eczematous diseases. In: Black M, Ambros-Rudolph CM, Edwards L, Lynch P, eds. Obstetric and Gynecologic Dermatology. 3rd ed. Mosby-Elsevier; 2008:192-194.
- Adam AE. Ectopic Darier’s disease of the cervix: an extraordinary cause of an abnormal smear. Cytopathology. 1996;7:414-421. doi:10.1111/j.1365-2303.1996.tb00547.x
- Suárez-Peñaranda JM, Antúnez JR, Del Rio E, et al. Vaginal involvement in a woman with Darier’s disease: a case report. Acta Cytol. 2005;49:530-532. doi:10.1159/000326200
- Boon ME. Dr. Darier’s lesson: it can be advantageous to the patient to ignore evident cytonuclear changes. Acta Cytol. 2005;49:469-470. doi:10.1159/000326189
- de la Rosa Carrillo D. Vegetating Darier’s disease during pregnancy. Acta Derm Venereol. 2006;86:259-260. doi:10.2340/00015555-0066
- Spouge JD, Trott JR, Chesko G. Darier-White’s disease: a cause of white lesions of the mucosa. report of four cases. Oral Surg Oral Med Oral Pathol. 1966;21:441-457. doi:10.1016/0030-4220(66)90401-4
- Espy PD, Stone S, Jolly HW Jr. Hormonal dependency in Darier disease. Cutis. 1976;17:315-320.
- De D, Kanwar AJ, Saikia UN. Uncommon flexural presentation of Darier disease. J Cutan Med Surg. 2008;12:249-252. doi:10.2310/7750.2008.07035
- Quinlivan JA, O'Halloran LC. Darier’s disease and pregnancy. Dermatol Aspects. 2013;1:1-3. doi:10.7243/2053-5309-1-1
- Sharma R, Singh BP, Das SN. Anesthetic management of cesarean section in a parturient with Darier’s disease. Acta Anaesthesiol Taiwan. 2010;48:158-159. doi:10.1016/S1875-4597(10)60051-3
- Gordon-Smith K, Jones LA, Burge SM, et al. The neuropsychiatric phenotype in Darier disease. Br J Dermatol. 2010;163:515-522. doi:10.1111/j.1365-2133.2010.09834.x
- Dodiuk-Gad RP, Cohen-Barak E, Khayat M, et al. Darier disease in Israel: combined evaluation of genetic and neuropsychiatric aspects. Br J Dermatol. 2016;174:562-568. doi:10.1111/bjd.14220
- Bale SJ, Toro JR. Genetic basis of Darier-White disease: bad pumps cause bumps. J Cutan Med Surg. 2000;4:103-106. doi:10.1177/120347540000400212
- Kansal NK, Hazarika N, Rao S. Familial case of Darier disease with guttate leukoderma: a case series from India. Indian Dermatol Online J. 2018;9:62-63. doi:10.4103/idoj.IDOJ_52_17
- Lynch PJ. Vulvar dermatoses: the eczematous diseases. In: Black M, Ambros-Rudolph CM, Edwards L, Lynch P, eds. Obstetric and Gynecologic Dermatology. 3rd ed. Mosby-Elsevier; 2008:192-194.
- Adam AE. Ectopic Darier’s disease of the cervix: an extraordinary cause of an abnormal smear. Cytopathology. 1996;7:414-421. doi:10.1111/j.1365-2303.1996.tb00547.x
- Suárez-Peñaranda JM, Antúnez JR, Del Rio E, et al. Vaginal involvement in a woman with Darier’s disease: a case report. Acta Cytol. 2005;49:530-532. doi:10.1159/000326200
- Boon ME. Dr. Darier’s lesson: it can be advantageous to the patient to ignore evident cytonuclear changes. Acta Cytol. 2005;49:469-470. doi:10.1159/000326189
- de la Rosa Carrillo D. Vegetating Darier’s disease during pregnancy. Acta Derm Venereol. 2006;86:259-260. doi:10.2340/00015555-0066
- Spouge JD, Trott JR, Chesko G. Darier-White’s disease: a cause of white lesions of the mucosa. report of four cases. Oral Surg Oral Med Oral Pathol. 1966;21:441-457. doi:10.1016/0030-4220(66)90401-4
- Espy PD, Stone S, Jolly HW Jr. Hormonal dependency in Darier disease. Cutis. 1976;17:315-320.
- De D, Kanwar AJ, Saikia UN. Uncommon flexural presentation of Darier disease. J Cutan Med Surg. 2008;12:249-252. doi:10.2310/7750.2008.07035
- Quinlivan JA, O'Halloran LC. Darier’s disease and pregnancy. Dermatol Aspects. 2013;1:1-3. doi:10.7243/2053-5309-1-1
- Sharma R, Singh BP, Das SN. Anesthetic management of cesarean section in a parturient with Darier’s disease. Acta Anaesthesiol Taiwan. 2010;48:158-159. doi:10.1016/S1875-4597(10)60051-3
- Gordon-Smith K, Jones LA, Burge SM, et al. The neuropsychiatric phenotype in Darier disease. Br J Dermatol. 2010;163:515-522. doi:10.1111/j.1365-2133.2010.09834.x
- Dodiuk-Gad RP, Cohen-Barak E, Khayat M, et al. Darier disease in Israel: combined evaluation of genetic and neuropsychiatric aspects. Br J Dermatol. 2016;174:562-568. doi:10.1111/bjd.14220
Practice Points
- Because Darier disease (DD) manifests during reproductive years, systemic retinoids should be used carefully in female patients.
- For a Papanicolaou test to be properly interpreted in a patient with DD, the cytopathologist must be informed of the DD diagnosis.
- Darier disease may be exacerbated or relieved during pregnancy.
Botanical Briefs: Phytophotodermatitis Is an Occupational and Recreational Dermatosis in the Limelight
Phytophotodermatitis (PPD) is a nonallergic contact dermatitis and thus is independent of the immune system, so prior sensitization is not required.1-3 It sometimes is known by colorful names such as margarita photodermatitis, in which a slice of lime in a refreshing summer drink may be etiologic,4,5 or berloque dermatitis, caused by exposure to perfumes containing bergapten (5-methoxypsoralen).6,7 Phytophotodermatitis may develop when phototoxic agents such as furocoumarins, which protect plants from fungal pathogens, and psoralens are applied to the skin followed by exposure to UV light, more specifically in the UVA range of 320 to 400 nm. Thus, these chemicals produce a phototoxic rather than photoallergic reaction, leading to cellular damage. Furocoumarins and psoralens often are found in plants such as celery and figs as well as in citrus fruits such as limes, lemons, and grapefruits. Exposure may be cryptic, as the patient may not consider or mention the eruption as possibly caused by activities such as soaking one’s feet in a folk remedy containing fig leaves.7,8 Once these phototoxic agents come in contact with the skin, the symptoms of PPD may arise within 24 hours of exposure, beginning as an acute dermatitis with erythema, edema, vesicles, or bullae accompanied by pain and itching.
Etiology
Phytophotodermatitis is caused by exposure to several different types of plants, including Ficus carica (common fig), the genus Citrus (eg, lime, lemon), or Pastina sativa (wild parsnip). Each of these contain furocoumarins and psoralens—phototoxic agents that cause cellular damage with epidermal necrosis and resultant pain when the skin is exposed to UVA light.1-4 There are 2 types of photochemical reactions in PPD: type I reactions occur in the absence of oxygen, whereas oxygen is present in type II reactions. Both damage cell membranes and DNA, which then results in DNA interstrand cross-linking between the psoralen furan ring and the thymine or cytosine of DNA, activating arachidonic acid metabolic pathways to produce cell death.1
Epidemiology
The incidence of PPD is unknown due to the high variability of reactions in individuals spanning from children to the elderly. It can be caused by many different wild and domestic plants in many areas of the world and can affect any individual regardless of age, race, gender, or ethnicity. Some individuals may be affected by hyperpigmentation without prominent inflammation.8 Diagnosis of PPD can be challenging, and an occupation and recreational history of exposure or recent travel with possible contact with plants may be required.
Occupational Dermatitis
Recreational Dermatitis
Phytophotodermatitis may be caused by exposure to phototoxic agents during leisure activities. Recreational exposure can occur almost anywhere, including in the kitchen, backyard, park, or woods, as well as at the beach. One notable culprit in recreational PPD is cooking with limes, parsley, or parsnips—plants that often are employed as garnishes in dishes, allowing early exposure of juices on the hands. Individuals who garden recreationally should be aware of ornamental plants such as hogweed and figs, which are notorious for causing PPD.13 Children’s camp counselors should have knowledge of PPD, as children have considerable curiosity and may touch or play with attractive plants such as hogweed. Children enjoying sports in parks can accidentally fall onto or be exposed to wild parsnip or hogweed growing nearby and wake up the next day with erythema and burning.14 Photoprotection is important, but sunscreens containing carrot extract can produce PPD.15 Widespread PPD over 80% of the body surface area due to sunbathing after applying fig leaf tea as a tanning agent has been described.16 Eating figs does not cause photosensitization unless the juice is smeared onto the skin. Margarita dermatitis and “Mexican beer dermatitis” can occur due to limes and other citrus fruits being used as ingredients in summer drinks.5 Similarly, preparing sangria may produce PPD from lime and lemon juices.17 In one report, hiking in Corsica resulted in PPD following incidental contact with the endemic plant Peucedanum paniculatum.18
Perfume (Berloque) Dermatitis
Perfume dermatitis, or berloque dermatitis, is a type of PPD for which the name is derived from the German word berlock or the French word berloque meaning trinket or charm; it was first described in 1925 by Rosenthal7 with regard to pendantlike streaks of pigmentation on the neck, face, arms, or trunk. The dermatitis develops due to bergapten, a component of bergamot oil, which is derived from the rind of Citrus bergamia. Many perfumes contain bergamot oil, but the incidence of this condition has been diminished due to use of artificial bergamot oil.6
Clinical Manifestation
Phytophotodermatitis is first evident as erythematous patches that appear within 24 hours of initial exposure to a phototoxic agent and UVA light, sometimes with a burning sensation. Solar exposure within 48 hours of sufficient plant exposure is required. Perfuse sweating may enhance the reaction.19 Rarely, it first may be seen with the sudden appearance of
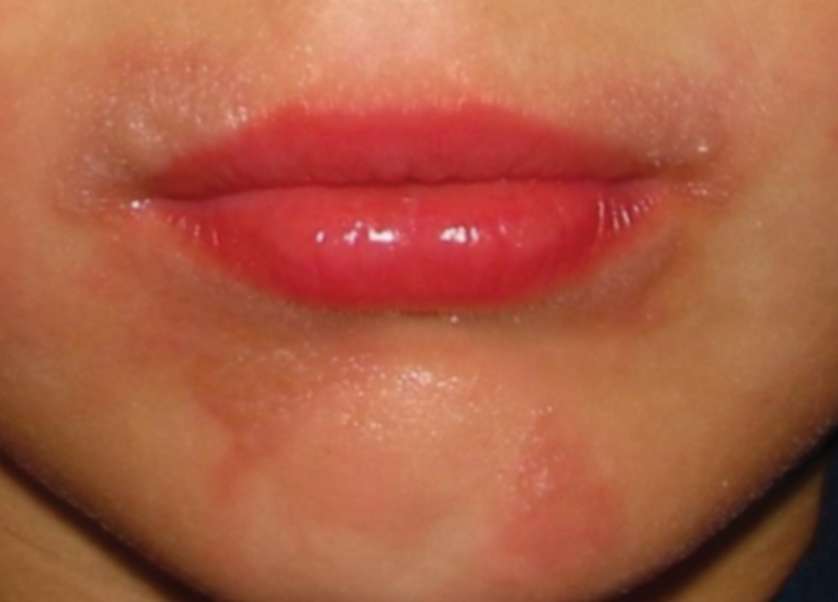
Differential Diagnosis
Phytophotodermatitis may resemble other types of dermatitis, particularly other forms of contact dermatitis such poison ivy, and occasionally other environmental simulants such as jellyfish stings.1-6,20,21 Photosensitizing disorders including porphyria cutanea tarda, pseudoporphyria, and lupus erythematosus must be distinguished from PPD.22-24 Photosensitizing medications such tetracyclines, thiazide diuretics, sulfonamides, griseofulvin, and sulfonylureas should be considered. Airborne contact dermatitis may resemble PPD, as when poison ivy is burned and is exposed to the skin in sites of airborne contact.20 Excessive solar exposure is popular, particularly among adolescents, so sunburn and sunburnlike reactions can be noteworthy.25,26
Treatment
Phytophotodermatitis can be treated with topical steroids, sometimes adding an oral antihistamine, and occasionally oral steroids.2-4 Localized pain or a burning sensation should respond to therapy. Alternatively, a cold compress applied to the skin can relieve the pain and pruritus, and the burn can be debrided and dressed daily with silver sulfadiazine plus an oral nonsteroidal anti-inflammatory drug. This eruption should be self-limited as long as it is recognized early and the cause avoided. Management of acute exposure includes prompt application of soap and water and avoidance of UV light exposure for 48 to 72 hours to prevent psoralen photoactivation.
Because PPD is essentially a chemical burn, a burn protocol and possible referral to a burn center may be needed, whether the reaction is acute or widespread.11,12,14,27,28 Surgical debridement and skin grafting rarely may be mandated.14 Postinflammatory hyperpigmentation may ensue as the dermatitis resolves but is not common.
The best approach for PPD is prevention (Figure 2). Individuals who are at risk should be aware of their surroundings and potential plants of concern and employ personal protective equipment to shield the skin from plant sap, which should be promptly removed if it comes in contact with the skin.

- Zhang R, Zhu W. Phytophotodermatitis due to Chinese herbal medicine decoction. Indian J Dermatol. 2011;56:329-331.
- Harshman J, Quan Y, Hsiang D. Phytophotodermatitis: rash with many faces. Can Fam Physician. 2017;63:938-940.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Hankinson A, Lloyd B, Alweis R. Lime-induced phytophotodermatitis [published online September 29, 2014]. J Community Hosp Intern Med Perspect. doi:10.3402/jchimp.v4.25090
- Abramowitz AI, Resnik KS, Cohen KR. Margarita photodermatitis. N Engl J Med. 2013;328:891.
- Quaak MS, Martens H, Hassing RJ, et al. The sunny side of lime. J Travel Med. 2012;19:327-328.
- Rosenthal O. Berloque dermatitis: Berliner Dermatologische Gesellschaft. Dermatol Zeitschrift. 1925;42:295.
- Choi JY, Hwang S, Lee SH, et al. Asymptomatic hyperpigmentation without preceding inflammation as a clinical feature of citrus fruits–induced phytophotodermatitis. Ann Dermatol. 2018;30:75-78.
- Wynn P, Bell S. Phytophotodermatitis in grounds operatives. Occup Med (Lond). 2005;55:393-395.
- Klimaszyk P, Klimaszyk D, Piotrowiak M, et al. Unusual complications after occupational exposure to giant hogweed (Heracleum mantegazzianum): a case report. Int J Occup Med Environ Health. 2014;27:141-144.
- Downs JW, Cumpston KL, Feldman MJ. Giant hogweed phytophotodermatitis. Clin Toxicol (Phila). 2019;57:822-823.
- Maso MJ, Ruszkowski AM, Bauerle J, et al. Celery phytophotodermatitis in a chef. Arch Dermatol. 1991;127:912-913.
- Derraik JG, Rademaker M. Phytophotodermatitis caused by contact with a fig tree (Ficus carica). New Zealand Med J. 2007;120:U2720.
- Chan JC, Sullivan PJ, O’Sullivan MJ, et al. Full thickness burn caused by exposure to giant hogweed: delayed presentation, histological features and surgical management. J Plast Reconstr Aesthet Surg. 2011;64:128-130.
- Bosanac SS, Clark AK, Sivamani RK. Phytophotodermatitis related to carrot extract–containing sunscreen. Dermatol Online J. 2018;24:1-3.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Mioduszewski M, Beecker J. Phytophotodermatitis from making sangria: a phototoxic reaction to lime and lemon juice. CMAJ. 2015;187:756.
- Torrents R, Schmitt C, Domangé B, et al. Phytophotodermatitis with Peucedanum paniculatum: an endemic species to Corsica. Clin Toxicol (Phila). 2019;57:68-69.
- Sarhane KA, Ibrahim A, Fagan SP, et al. Phytophotodermatitis. Eplasty. 2013;13:ic57.
- DeLeo VA, Suarez SM, Maso MJ. Photoallergic contact dermatitis. results of photopatch testing in New York, 1985 to 1990. Arch Dermatol. 1992;128:1513-1518.
- Kimyon RS, Warshaw EM. Airborne allergic contact dermatitis: management and responsible allergens on the American Contact Dermatitis Society Core Series. Dermatitis. 2019;30:106-115.
- Miteva L, Broshtilova V, Schwartz RA. Unusual clinical manifestations of chronic discoid lupus erythematosus. Serbian J Dermatol Venereol. 2014;6:69-72.
- Handler NS, Handler MZ, Stephany MP, et al. Porphyria cutanea tarda: an intriguing genetic disease and marker. Int J Dermatol. 2017;56:E106-E117.
- Papadopoulos AJ, Schwartz RA, Fekete Z, et al. Pseudoporphyria: an atypical variant resembling toxic epidermal necrolysis. J Cutan Med Surg. 2001;5:479-485.
- Jasterzbski TJ, Janniger EJ, Schwartz RA. Adolescent tanning practices: understanding the popularity of excessive ultraviolet light exposure. In: Or
anje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:177-185. - Lai YC, Janniger EJ, Schwartz RA. Solar protection policy in school children: proposals for progress. In: Oranje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:165-176.
- Lagey K, Duinslaeger L, Vanderkelen A. Burns induced by plants. Burns. 1995;21:542-543.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:e238745.
Phytophotodermatitis (PPD) is a nonallergic contact dermatitis and thus is independent of the immune system, so prior sensitization is not required.1-3 It sometimes is known by colorful names such as margarita photodermatitis, in which a slice of lime in a refreshing summer drink may be etiologic,4,5 or berloque dermatitis, caused by exposure to perfumes containing bergapten (5-methoxypsoralen).6,7 Phytophotodermatitis may develop when phototoxic agents such as furocoumarins, which protect plants from fungal pathogens, and psoralens are applied to the skin followed by exposure to UV light, more specifically in the UVA range of 320 to 400 nm. Thus, these chemicals produce a phototoxic rather than photoallergic reaction, leading to cellular damage. Furocoumarins and psoralens often are found in plants such as celery and figs as well as in citrus fruits such as limes, lemons, and grapefruits. Exposure may be cryptic, as the patient may not consider or mention the eruption as possibly caused by activities such as soaking one’s feet in a folk remedy containing fig leaves.7,8 Once these phototoxic agents come in contact with the skin, the symptoms of PPD may arise within 24 hours of exposure, beginning as an acute dermatitis with erythema, edema, vesicles, or bullae accompanied by pain and itching.
Etiology
Phytophotodermatitis is caused by exposure to several different types of plants, including Ficus carica (common fig), the genus Citrus (eg, lime, lemon), or Pastina sativa (wild parsnip). Each of these contain furocoumarins and psoralens—phototoxic agents that cause cellular damage with epidermal necrosis and resultant pain when the skin is exposed to UVA light.1-4 There are 2 types of photochemical reactions in PPD: type I reactions occur in the absence of oxygen, whereas oxygen is present in type II reactions. Both damage cell membranes and DNA, which then results in DNA interstrand cross-linking between the psoralen furan ring and the thymine or cytosine of DNA, activating arachidonic acid metabolic pathways to produce cell death.1
Epidemiology
The incidence of PPD is unknown due to the high variability of reactions in individuals spanning from children to the elderly. It can be caused by many different wild and domestic plants in many areas of the world and can affect any individual regardless of age, race, gender, or ethnicity. Some individuals may be affected by hyperpigmentation without prominent inflammation.8 Diagnosis of PPD can be challenging, and an occupation and recreational history of exposure or recent travel with possible contact with plants may be required.
Occupational Dermatitis
Recreational Dermatitis
Phytophotodermatitis may be caused by exposure to phototoxic agents during leisure activities. Recreational exposure can occur almost anywhere, including in the kitchen, backyard, park, or woods, as well as at the beach. One notable culprit in recreational PPD is cooking with limes, parsley, or parsnips—plants that often are employed as garnishes in dishes, allowing early exposure of juices on the hands. Individuals who garden recreationally should be aware of ornamental plants such as hogweed and figs, which are notorious for causing PPD.13 Children’s camp counselors should have knowledge of PPD, as children have considerable curiosity and may touch or play with attractive plants such as hogweed. Children enjoying sports in parks can accidentally fall onto or be exposed to wild parsnip or hogweed growing nearby and wake up the next day with erythema and burning.14 Photoprotection is important, but sunscreens containing carrot extract can produce PPD.15 Widespread PPD over 80% of the body surface area due to sunbathing after applying fig leaf tea as a tanning agent has been described.16 Eating figs does not cause photosensitization unless the juice is smeared onto the skin. Margarita dermatitis and “Mexican beer dermatitis” can occur due to limes and other citrus fruits being used as ingredients in summer drinks.5 Similarly, preparing sangria may produce PPD from lime and lemon juices.17 In one report, hiking in Corsica resulted in PPD following incidental contact with the endemic plant Peucedanum paniculatum.18
Perfume (Berloque) Dermatitis
Perfume dermatitis, or berloque dermatitis, is a type of PPD for which the name is derived from the German word berlock or the French word berloque meaning trinket or charm; it was first described in 1925 by Rosenthal7 with regard to pendantlike streaks of pigmentation on the neck, face, arms, or trunk. The dermatitis develops due to bergapten, a component of bergamot oil, which is derived from the rind of Citrus bergamia. Many perfumes contain bergamot oil, but the incidence of this condition has been diminished due to use of artificial bergamot oil.6
Clinical Manifestation
Phytophotodermatitis is first evident as erythematous patches that appear within 24 hours of initial exposure to a phototoxic agent and UVA light, sometimes with a burning sensation. Solar exposure within 48 hours of sufficient plant exposure is required. Perfuse sweating may enhance the reaction.19 Rarely, it first may be seen with the sudden appearance of
Differential Diagnosis
Phytophotodermatitis may resemble other types of dermatitis, particularly other forms of contact dermatitis such poison ivy, and occasionally other environmental simulants such as jellyfish stings.1-6,20,21 Photosensitizing disorders including porphyria cutanea tarda, pseudoporphyria, and lupus erythematosus must be distinguished from PPD.22-24 Photosensitizing medications such tetracyclines, thiazide diuretics, sulfonamides, griseofulvin, and sulfonylureas should be considered. Airborne contact dermatitis may resemble PPD, as when poison ivy is burned and is exposed to the skin in sites of airborne contact.20 Excessive solar exposure is popular, particularly among adolescents, so sunburn and sunburnlike reactions can be noteworthy.25,26
Treatment
Phytophotodermatitis can be treated with topical steroids, sometimes adding an oral antihistamine, and occasionally oral steroids.2-4 Localized pain or a burning sensation should respond to therapy. Alternatively, a cold compress applied to the skin can relieve the pain and pruritus, and the burn can be debrided and dressed daily with silver sulfadiazine plus an oral nonsteroidal anti-inflammatory drug. This eruption should be self-limited as long as it is recognized early and the cause avoided. Management of acute exposure includes prompt application of soap and water and avoidance of UV light exposure for 48 to 72 hours to prevent psoralen photoactivation.
Because PPD is essentially a chemical burn, a burn protocol and possible referral to a burn center may be needed, whether the reaction is acute or widespread.11,12,14,27,28 Surgical debridement and skin grafting rarely may be mandated.14 Postinflammatory hyperpigmentation may ensue as the dermatitis resolves but is not common.
The best approach for PPD is prevention (Figure 2). Individuals who are at risk should be aware of their surroundings and potential plants of concern and employ personal protective equipment to shield the skin from plant sap, which should be promptly removed if it comes in contact with the skin.
Phytophotodermatitis (PPD) is a nonallergic contact dermatitis and thus is independent of the immune system, so prior sensitization is not required.1-3 It sometimes is known by colorful names such as margarita photodermatitis, in which a slice of lime in a refreshing summer drink may be etiologic,4,5 or berloque dermatitis, caused by exposure to perfumes containing bergapten (5-methoxypsoralen).6,7 Phytophotodermatitis may develop when phototoxic agents such as furocoumarins, which protect plants from fungal pathogens, and psoralens are applied to the skin followed by exposure to UV light, more specifically in the UVA range of 320 to 400 nm. Thus, these chemicals produce a phototoxic rather than photoallergic reaction, leading to cellular damage. Furocoumarins and psoralens often are found in plants such as celery and figs as well as in citrus fruits such as limes, lemons, and grapefruits. Exposure may be cryptic, as the patient may not consider or mention the eruption as possibly caused by activities such as soaking one’s feet in a folk remedy containing fig leaves.7,8 Once these phototoxic agents come in contact with the skin, the symptoms of PPD may arise within 24 hours of exposure, beginning as an acute dermatitis with erythema, edema, vesicles, or bullae accompanied by pain and itching.
Etiology
Phytophotodermatitis is caused by exposure to several different types of plants, including Ficus carica (common fig), the genus Citrus (eg, lime, lemon), or Pastina sativa (wild parsnip). Each of these contain furocoumarins and psoralens—phototoxic agents that cause cellular damage with epidermal necrosis and resultant pain when the skin is exposed to UVA light.1-4 There are 2 types of photochemical reactions in PPD: type I reactions occur in the absence of oxygen, whereas oxygen is present in type II reactions. Both damage cell membranes and DNA, which then results in DNA interstrand cross-linking between the psoralen furan ring and the thymine or cytosine of DNA, activating arachidonic acid metabolic pathways to produce cell death.1
Epidemiology
The incidence of PPD is unknown due to the high variability of reactions in individuals spanning from children to the elderly. It can be caused by many different wild and domestic plants in many areas of the world and can affect any individual regardless of age, race, gender, or ethnicity. Some individuals may be affected by hyperpigmentation without prominent inflammation.8 Diagnosis of PPD can be challenging, and an occupation and recreational history of exposure or recent travel with possible contact with plants may be required.
Occupational Dermatitis
Recreational Dermatitis
Phytophotodermatitis may be caused by exposure to phototoxic agents during leisure activities. Recreational exposure can occur almost anywhere, including in the kitchen, backyard, park, or woods, as well as at the beach. One notable culprit in recreational PPD is cooking with limes, parsley, or parsnips—plants that often are employed as garnishes in dishes, allowing early exposure of juices on the hands. Individuals who garden recreationally should be aware of ornamental plants such as hogweed and figs, which are notorious for causing PPD.13 Children’s camp counselors should have knowledge of PPD, as children have considerable curiosity and may touch or play with attractive plants such as hogweed. Children enjoying sports in parks can accidentally fall onto or be exposed to wild parsnip or hogweed growing nearby and wake up the next day with erythema and burning.14 Photoprotection is important, but sunscreens containing carrot extract can produce PPD.15 Widespread PPD over 80% of the body surface area due to sunbathing after applying fig leaf tea as a tanning agent has been described.16 Eating figs does not cause photosensitization unless the juice is smeared onto the skin. Margarita dermatitis and “Mexican beer dermatitis” can occur due to limes and other citrus fruits being used as ingredients in summer drinks.5 Similarly, preparing sangria may produce PPD from lime and lemon juices.17 In one report, hiking in Corsica resulted in PPD following incidental contact with the endemic plant Peucedanum paniculatum.18
Perfume (Berloque) Dermatitis
Perfume dermatitis, or berloque dermatitis, is a type of PPD for which the name is derived from the German word berlock or the French word berloque meaning trinket or charm; it was first described in 1925 by Rosenthal7 with regard to pendantlike streaks of pigmentation on the neck, face, arms, or trunk. The dermatitis develops due to bergapten, a component of bergamot oil, which is derived from the rind of Citrus bergamia. Many perfumes contain bergamot oil, but the incidence of this condition has been diminished due to use of artificial bergamot oil.6
Clinical Manifestation
Phytophotodermatitis is first evident as erythematous patches that appear within 24 hours of initial exposure to a phototoxic agent and UVA light, sometimes with a burning sensation. Solar exposure within 48 hours of sufficient plant exposure is required. Perfuse sweating may enhance the reaction.19 Rarely, it first may be seen with the sudden appearance of
Differential Diagnosis
Phytophotodermatitis may resemble other types of dermatitis, particularly other forms of contact dermatitis such poison ivy, and occasionally other environmental simulants such as jellyfish stings.1-6,20,21 Photosensitizing disorders including porphyria cutanea tarda, pseudoporphyria, and lupus erythematosus must be distinguished from PPD.22-24 Photosensitizing medications such tetracyclines, thiazide diuretics, sulfonamides, griseofulvin, and sulfonylureas should be considered. Airborne contact dermatitis may resemble PPD, as when poison ivy is burned and is exposed to the skin in sites of airborne contact.20 Excessive solar exposure is popular, particularly among adolescents, so sunburn and sunburnlike reactions can be noteworthy.25,26
Treatment
Phytophotodermatitis can be treated with topical steroids, sometimes adding an oral antihistamine, and occasionally oral steroids.2-4 Localized pain or a burning sensation should respond to therapy. Alternatively, a cold compress applied to the skin can relieve the pain and pruritus, and the burn can be debrided and dressed daily with silver sulfadiazine plus an oral nonsteroidal anti-inflammatory drug. This eruption should be self-limited as long as it is recognized early and the cause avoided. Management of acute exposure includes prompt application of soap and water and avoidance of UV light exposure for 48 to 72 hours to prevent psoralen photoactivation.
Because PPD is essentially a chemical burn, a burn protocol and possible referral to a burn center may be needed, whether the reaction is acute or widespread.11,12,14,27,28 Surgical debridement and skin grafting rarely may be mandated.14 Postinflammatory hyperpigmentation may ensue as the dermatitis resolves but is not common.
The best approach for PPD is prevention (Figure 2). Individuals who are at risk should be aware of their surroundings and potential plants of concern and employ personal protective equipment to shield the skin from plant sap, which should be promptly removed if it comes in contact with the skin.
- Zhang R, Zhu W. Phytophotodermatitis due to Chinese herbal medicine decoction. Indian J Dermatol. 2011;56:329-331.
- Harshman J, Quan Y, Hsiang D. Phytophotodermatitis: rash with many faces. Can Fam Physician. 2017;63:938-940.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Hankinson A, Lloyd B, Alweis R. Lime-induced phytophotodermatitis [published online September 29, 2014]. J Community Hosp Intern Med Perspect. doi:10.3402/jchimp.v4.25090
- Abramowitz AI, Resnik KS, Cohen KR. Margarita photodermatitis. N Engl J Med. 2013;328:891.
- Quaak MS, Martens H, Hassing RJ, et al. The sunny side of lime. J Travel Med. 2012;19:327-328.
- Rosenthal O. Berloque dermatitis: Berliner Dermatologische Gesellschaft. Dermatol Zeitschrift. 1925;42:295.
- Choi JY, Hwang S, Lee SH, et al. Asymptomatic hyperpigmentation without preceding inflammation as a clinical feature of citrus fruits–induced phytophotodermatitis. Ann Dermatol. 2018;30:75-78.
- Wynn P, Bell S. Phytophotodermatitis in grounds operatives. Occup Med (Lond). 2005;55:393-395.
- Klimaszyk P, Klimaszyk D, Piotrowiak M, et al. Unusual complications after occupational exposure to giant hogweed (Heracleum mantegazzianum): a case report. Int J Occup Med Environ Health. 2014;27:141-144.
- Downs JW, Cumpston KL, Feldman MJ. Giant hogweed phytophotodermatitis. Clin Toxicol (Phila). 2019;57:822-823.
- Maso MJ, Ruszkowski AM, Bauerle J, et al. Celery phytophotodermatitis in a chef. Arch Dermatol. 1991;127:912-913.
- Derraik JG, Rademaker M. Phytophotodermatitis caused by contact with a fig tree (Ficus carica). New Zealand Med J. 2007;120:U2720.
- Chan JC, Sullivan PJ, O’Sullivan MJ, et al. Full thickness burn caused by exposure to giant hogweed: delayed presentation, histological features and surgical management. J Plast Reconstr Aesthet Surg. 2011;64:128-130.
- Bosanac SS, Clark AK, Sivamani RK. Phytophotodermatitis related to carrot extract–containing sunscreen. Dermatol Online J. 2018;24:1-3.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Mioduszewski M, Beecker J. Phytophotodermatitis from making sangria: a phototoxic reaction to lime and lemon juice. CMAJ. 2015;187:756.
- Torrents R, Schmitt C, Domangé B, et al. Phytophotodermatitis with Peucedanum paniculatum: an endemic species to Corsica. Clin Toxicol (Phila). 2019;57:68-69.
- Sarhane KA, Ibrahim A, Fagan SP, et al. Phytophotodermatitis. Eplasty. 2013;13:ic57.
- DeLeo VA, Suarez SM, Maso MJ. Photoallergic contact dermatitis. results of photopatch testing in New York, 1985 to 1990. Arch Dermatol. 1992;128:1513-1518.
- Kimyon RS, Warshaw EM. Airborne allergic contact dermatitis: management and responsible allergens on the American Contact Dermatitis Society Core Series. Dermatitis. 2019;30:106-115.
- Miteva L, Broshtilova V, Schwartz RA. Unusual clinical manifestations of chronic discoid lupus erythematosus. Serbian J Dermatol Venereol. 2014;6:69-72.
- Handler NS, Handler MZ, Stephany MP, et al. Porphyria cutanea tarda: an intriguing genetic disease and marker. Int J Dermatol. 2017;56:E106-E117.
- Papadopoulos AJ, Schwartz RA, Fekete Z, et al. Pseudoporphyria: an atypical variant resembling toxic epidermal necrolysis. J Cutan Med Surg. 2001;5:479-485.
- Jasterzbski TJ, Janniger EJ, Schwartz RA. Adolescent tanning practices: understanding the popularity of excessive ultraviolet light exposure. In: Or
anje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:177-185. - Lai YC, Janniger EJ, Schwartz RA. Solar protection policy in school children: proposals for progress. In: Oranje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:165-176.
- Lagey K, Duinslaeger L, Vanderkelen A. Burns induced by plants. Burns. 1995;21:542-543.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:e238745.
- Zhang R, Zhu W. Phytophotodermatitis due to Chinese herbal medicine decoction. Indian J Dermatol. 2011;56:329-331.
- Harshman J, Quan Y, Hsiang D. Phytophotodermatitis: rash with many faces. Can Fam Physician. 2017;63:938-940.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Hankinson A, Lloyd B, Alweis R. Lime-induced phytophotodermatitis [published online September 29, 2014]. J Community Hosp Intern Med Perspect. doi:10.3402/jchimp.v4.25090
- Abramowitz AI, Resnik KS, Cohen KR. Margarita photodermatitis. N Engl J Med. 2013;328:891.
- Quaak MS, Martens H, Hassing RJ, et al. The sunny side of lime. J Travel Med. 2012;19:327-328.
- Rosenthal O. Berloque dermatitis: Berliner Dermatologische Gesellschaft. Dermatol Zeitschrift. 1925;42:295.
- Choi JY, Hwang S, Lee SH, et al. Asymptomatic hyperpigmentation without preceding inflammation as a clinical feature of citrus fruits–induced phytophotodermatitis. Ann Dermatol. 2018;30:75-78.
- Wynn P, Bell S. Phytophotodermatitis in grounds operatives. Occup Med (Lond). 2005;55:393-395.
- Klimaszyk P, Klimaszyk D, Piotrowiak M, et al. Unusual complications after occupational exposure to giant hogweed (Heracleum mantegazzianum): a case report. Int J Occup Med Environ Health. 2014;27:141-144.
- Downs JW, Cumpston KL, Feldman MJ. Giant hogweed phytophotodermatitis. Clin Toxicol (Phila). 2019;57:822-823.
- Maso MJ, Ruszkowski AM, Bauerle J, et al. Celery phytophotodermatitis in a chef. Arch Dermatol. 1991;127:912-913.
- Derraik JG, Rademaker M. Phytophotodermatitis caused by contact with a fig tree (Ficus carica). New Zealand Med J. 2007;120:U2720.
- Chan JC, Sullivan PJ, O’Sullivan MJ, et al. Full thickness burn caused by exposure to giant hogweed: delayed presentation, histological features and surgical management. J Plast Reconstr Aesthet Surg. 2011;64:128-130.
- Bosanac SS, Clark AK, Sivamani RK. Phytophotodermatitis related to carrot extract–containing sunscreen. Dermatol Online J. 2018;24:1-3.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Mioduszewski M, Beecker J. Phytophotodermatitis from making sangria: a phototoxic reaction to lime and lemon juice. CMAJ. 2015;187:756.
- Torrents R, Schmitt C, Domangé B, et al. Phytophotodermatitis with Peucedanum paniculatum: an endemic species to Corsica. Clin Toxicol (Phila). 2019;57:68-69.
- Sarhane KA, Ibrahim A, Fagan SP, et al. Phytophotodermatitis. Eplasty. 2013;13:ic57.
- DeLeo VA, Suarez SM, Maso MJ. Photoallergic contact dermatitis. results of photopatch testing in New York, 1985 to 1990. Arch Dermatol. 1992;128:1513-1518.
- Kimyon RS, Warshaw EM. Airborne allergic contact dermatitis: management and responsible allergens on the American Contact Dermatitis Society Core Series. Dermatitis. 2019;30:106-115.
- Miteva L, Broshtilova V, Schwartz RA. Unusual clinical manifestations of chronic discoid lupus erythematosus. Serbian J Dermatol Venereol. 2014;6:69-72.
- Handler NS, Handler MZ, Stephany MP, et al. Porphyria cutanea tarda: an intriguing genetic disease and marker. Int J Dermatol. 2017;56:E106-E117.
- Papadopoulos AJ, Schwartz RA, Fekete Z, et al. Pseudoporphyria: an atypical variant resembling toxic epidermal necrolysis. J Cutan Med Surg. 2001;5:479-485.
- Jasterzbski TJ, Janniger EJ, Schwartz RA. Adolescent tanning practices: understanding the popularity of excessive ultraviolet light exposure. In: Or
anje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:177-185. - Lai YC, Janniger EJ, Schwartz RA. Solar protection policy in school children: proposals for progress. In: Oranje A, Al-Mutairi N, Shwayder T, eds. Practical Pediatric Dermatology. Controversies in Diagnosis and Treatment. Springer Verlag; 2016:165-176.
- Lagey K, Duinslaeger L, Vanderkelen A. Burns induced by plants. Burns. 1995;21:542-543.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:e238745.
Practice Points
- Phytophotodermatitis (PPD) can be both an occupational and recreational dermatosis.
- Phytophotodermatitis is a nonallergic contact dermatitis and thus is independent of the immune system, so prior sensitization is not required.
- Individuals who work with plants should be aware of PPD and methods of prevention.
- Phytophotodermatitis may be evident only as asymptomatic hyperpigmentation.
Squamous Cell Carcinoma in Hidradenitis Suppurativa Lesions Following Tumor Necrosis Factor α Inhibitors
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with high morbidity rates. Symptoms typically develop between puberty and the third decade of life, affecting twice as many females as males, with an overall disease prevalence of 1% to 4%.1 The pathogenesis is theorized to be related to an immune response to follicular occlusion and rupture in genetically susceptible individuals.
Among the complications associated with HS, the development of cutaneous squamous cell carcinoma (SCC) is 4.6-times more likely within HS lesions than in normal skin and typically is seen in the setting of long-standing disease, particularly in men with HS lesions located on the buttocks and genital region for more than 20 years.2 In 2015, the tumor necrosis factor (TNF) inhibitor adalimumab was approved by the US Food and Drug Administration for the treatment of HS. Tumor necrosis factor α inhibitors have been associated with an increased risk for skin cancer in other clinical settings.3,4 We present a case of locally advanced SCC that developed in a patient with HS who was treated with adalimumab and infliximab (both TNF-α inhibitors), ultimately leading to the patient’s death.
A 59-year-old man who smoked with a 40-year history of severe HS, who previously was lost to follow-up, presented to our dermatology clinic with lesions on the buttocks. Physical examination demonstrated confluent, indurated, boggy plaques; scattered sinus tracts with purulent drainage; scattered cystlike nodules; and tenderness to palpation consistent with Hurley stage III disease (Figure 1A). No involvement of the axillae or groin was noted. He was started on doxycycline and a prednisone taper with minimal improvement and subsequently was switched to adalimumab 3 months later. Adalimumab provided little relief and was discontinued; therapy was transitioned to infliximab 3 months later.
The patient returned to our clinic 3 months later with a severe flare and intractable pain after 4 infusions of infliximab. Physical examination showed a 7×5-cm deep malodorous ulcer with fibrinous exudate on the left buttock, several 2- to 3-cm shallow ulcers draining yellow exudate, and numerous fluctuant subcutaneous nodules on a background of scarring and sinus tracts. He was started again on doxycycline and a prednisone taper. At follow-up 2 weeks later, the largest ulcer had increased to 8 cm, and more indurated and tender subcutaneous nodules and scattered ulcerations developed (Figure 1B). Two punch biopsies of the left buttock revealed an invasive keratinizing carcinoma with no connection to the epidermis, consistent with SCC (Figure 2). Human papillomavirus (HPV) test results with probes for 37 HPV types—13 that were high risk (HPV-16, −18, −31, −33, −35, −39, −45, −51, −52, −56, −58, −59, −68)—were negative. Computerized tomography demonstrated diffuse thickening of the skin on the buttocks, inguinal adenopathy suspicious for nodal metastases, and no evidence of distant metastatic disease. Given the extent of the disease, surgical treatment was not an option, and he began receiving palliative radiotherapy. However, his health declined, and he developed aspiration pneumonia and hypotension requiring pressor support. He was transitioned to hospice care and died 3 months after presentation.
Tumor necrosis factor α antagonist treatment is being increasingly used to control HS but also may increase the risk for SCC development. We performed a search of PubMed articles indexed for MEDLINE as well as Web of Science using the terms hidradenitis suppurativa or acne inversa and one of the following—tumor necrosis factor inhibitor, infliximab, adalimumab, or etanercept—and squamous cell carcinoma or Marjolin ulcer. Seven cases of SCC arising in an HS patient treated with a TNF-α inhibitor have been reported (Table).5-10 Four cases were associated with infliximab use, 2 with adalimumab, and our case occurred after both adalimumab and infliximab treatment. All individuals were men with severe, long-standing disease of the anogenital region. In addition to smoking, HPV-16 positivity also has been reported as a risk factor for developing SCC in the setting of HS.11 In our patient, however, HPV testing did not cover all HPV strains, but several high-risk strains, including HPV-16, were negative.
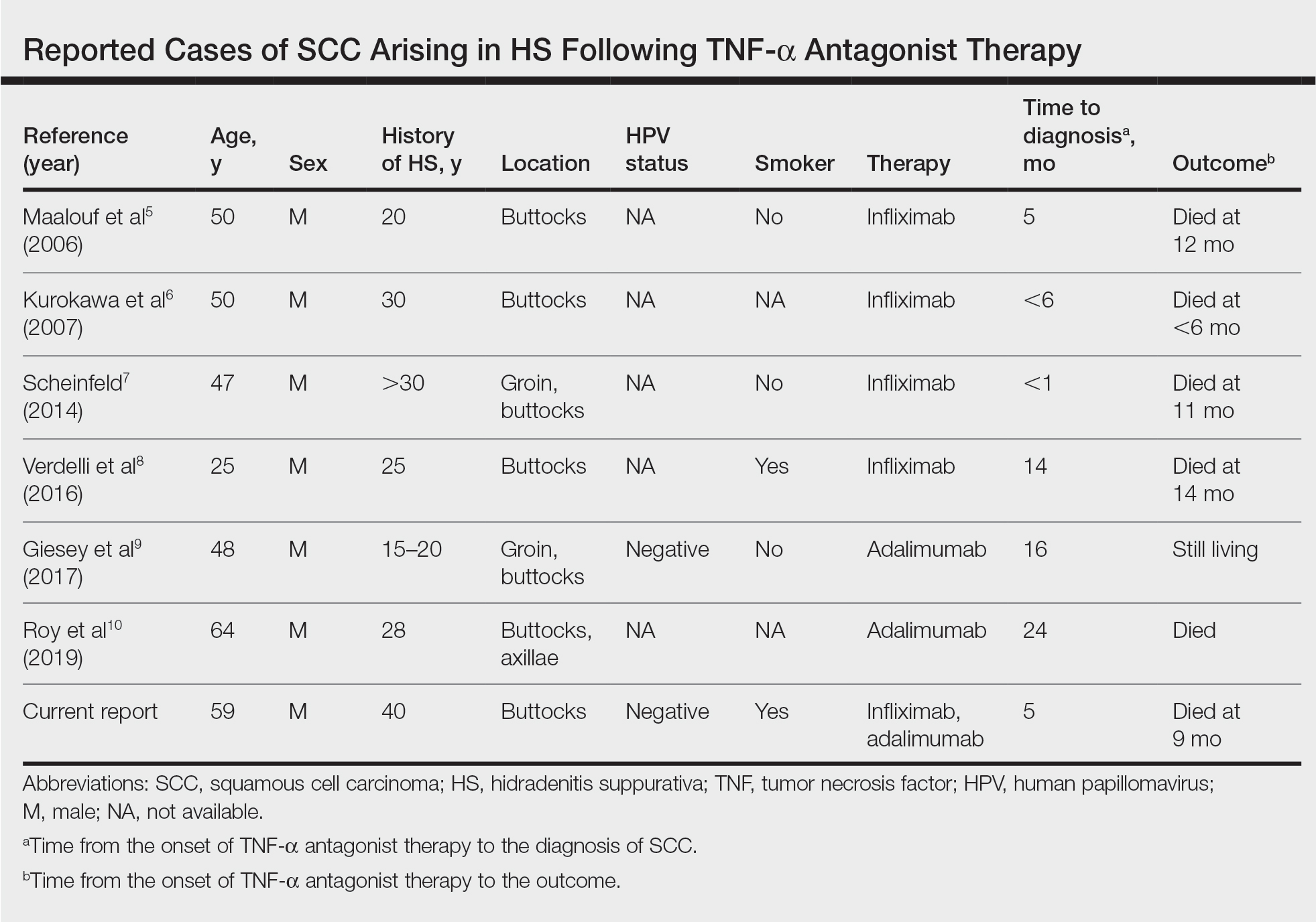
Hidradenitis suppurativa is caused by an immune response to ruptured follicles and TNF-α antagonists are useful in suppressing this response; however, immunosuppression can lead to an increased susceptibility to malignancy, especially in SCC. It is unclear whether the use of infliximab or adalimumab is causal, additive, or a confounder in the development of SCC in patients with severe HS. It is possible that these agents increase the rapidity of the development of SCC in already-susceptible patients. Although TNF-α antagonists can be an effective therapeutic option for patients with moderate to severe HS, the potential risk for contributing to skin cancer development should raise provider suspicion in high-risk patients. Given the findings in this report, it may be suitable for providers to consider a biopsy prior to initiating TNF-α therapy in men older than 20 years with moderate to severe HS of the groin or buttocks, in addition to more frequent monitoring and a lower threshold to biopsy lesions with rapid growth or ulceration.
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561; quiz 562-533.
- Lapins J, Ye W, Nyren O, et al. Incidence of cancer among patients with hidradenitis suppurativa. Arch Dermatol. 2001;137:730-734.
- Askling J, Fahrbach K, Nordstrom B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119-130.
- Mariette X, Matucci-Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta-analysis. Ann Rheum Dis. 2011;70:1895-1904.
- Maalouf E, Faye O, Poli F, et al. Fatal epidermoid carcinoma in hidradenitis suppurativa following treatment with infliximab. Ann Dermatol Venereol. 2006;133(5 pt 1):473-474.
- Kurokawa I, Nishimura K, Yamanaka K, et al. Cytokeratin expression in squamous cell carcinoma arising from hidradenitis suppurativa (acne inversa). J Cutan Pathol. 2007;34:675-678.
- Scheinfeld N. A case of a patient with stage III familial hidradenitis suppurativa treated with 3 courses of infliximab and died of metastatic squamous cell carcinoma. Dermatol Online J. 2014;20(3).
- Verdelli A, Antiga E, Bonciani D, et al. A fatal case of hidradenitis suppurativa associated with sepsis and squamous cell carcinoma. Int J Dermatol. 2016;55:E52-E53.
- Giesey R, Delost GR, Honaker J, et al. Metastatic squamous cell carcinoma in a patient treated with adalimumab for hidradenitis suppurativa. JAAD Case Rep. 2017;3:489-491.
- Roy C, Roy S, Ghazawi F, et al. Cutaneous squamous cell carcinoma arising in hidradenitis suppurativa: a case report. SAGE Open Med Case Rep. 2019;7:2050313X19847359.
- Lavogiez C, Delaporte E, Darras-Vercambre S, et al. Clinicopathological study of 13 cases of squamous cell carcinoma complicating hidradenitis suppurativa. Dermatology. 2010;220:147-153.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with high morbidity rates. Symptoms typically develop between puberty and the third decade of life, affecting twice as many females as males, with an overall disease prevalence of 1% to 4%.1 The pathogenesis is theorized to be related to an immune response to follicular occlusion and rupture in genetically susceptible individuals.
Among the complications associated with HS, the development of cutaneous squamous cell carcinoma (SCC) is 4.6-times more likely within HS lesions than in normal skin and typically is seen in the setting of long-standing disease, particularly in men with HS lesions located on the buttocks and genital region for more than 20 years.2 In 2015, the tumor necrosis factor (TNF) inhibitor adalimumab was approved by the US Food and Drug Administration for the treatment of HS. Tumor necrosis factor α inhibitors have been associated with an increased risk for skin cancer in other clinical settings.3,4 We present a case of locally advanced SCC that developed in a patient with HS who was treated with adalimumab and infliximab (both TNF-α inhibitors), ultimately leading to the patient’s death.
A 59-year-old man who smoked with a 40-year history of severe HS, who previously was lost to follow-up, presented to our dermatology clinic with lesions on the buttocks. Physical examination demonstrated confluent, indurated, boggy plaques; scattered sinus tracts with purulent drainage; scattered cystlike nodules; and tenderness to palpation consistent with Hurley stage III disease (Figure 1A). No involvement of the axillae or groin was noted. He was started on doxycycline and a prednisone taper with minimal improvement and subsequently was switched to adalimumab 3 months later. Adalimumab provided little relief and was discontinued; therapy was transitioned to infliximab 3 months later.
The patient returned to our clinic 3 months later with a severe flare and intractable pain after 4 infusions of infliximab. Physical examination showed a 7×5-cm deep malodorous ulcer with fibrinous exudate on the left buttock, several 2- to 3-cm shallow ulcers draining yellow exudate, and numerous fluctuant subcutaneous nodules on a background of scarring and sinus tracts. He was started again on doxycycline and a prednisone taper. At follow-up 2 weeks later, the largest ulcer had increased to 8 cm, and more indurated and tender subcutaneous nodules and scattered ulcerations developed (Figure 1B). Two punch biopsies of the left buttock revealed an invasive keratinizing carcinoma with no connection to the epidermis, consistent with SCC (Figure 2). Human papillomavirus (HPV) test results with probes for 37 HPV types—13 that were high risk (HPV-16, −18, −31, −33, −35, −39, −45, −51, −52, −56, −58, −59, −68)—were negative. Computerized tomography demonstrated diffuse thickening of the skin on the buttocks, inguinal adenopathy suspicious for nodal metastases, and no evidence of distant metastatic disease. Given the extent of the disease, surgical treatment was not an option, and he began receiving palliative radiotherapy. However, his health declined, and he developed aspiration pneumonia and hypotension requiring pressor support. He was transitioned to hospice care and died 3 months after presentation.
Tumor necrosis factor α antagonist treatment is being increasingly used to control HS but also may increase the risk for SCC development. We performed a search of PubMed articles indexed for MEDLINE as well as Web of Science using the terms hidradenitis suppurativa or acne inversa and one of the following—tumor necrosis factor inhibitor, infliximab, adalimumab, or etanercept—and squamous cell carcinoma or Marjolin ulcer. Seven cases of SCC arising in an HS patient treated with a TNF-α inhibitor have been reported (Table).5-10 Four cases were associated with infliximab use, 2 with adalimumab, and our case occurred after both adalimumab and infliximab treatment. All individuals were men with severe, long-standing disease of the anogenital region. In addition to smoking, HPV-16 positivity also has been reported as a risk factor for developing SCC in the setting of HS.11 In our patient, however, HPV testing did not cover all HPV strains, but several high-risk strains, including HPV-16, were negative.
Hidradenitis suppurativa is caused by an immune response to ruptured follicles and TNF-α antagonists are useful in suppressing this response; however, immunosuppression can lead to an increased susceptibility to malignancy, especially in SCC. It is unclear whether the use of infliximab or adalimumab is causal, additive, or a confounder in the development of SCC in patients with severe HS. It is possible that these agents increase the rapidity of the development of SCC in already-susceptible patients. Although TNF-α antagonists can be an effective therapeutic option for patients with moderate to severe HS, the potential risk for contributing to skin cancer development should raise provider suspicion in high-risk patients. Given the findings in this report, it may be suitable for providers to consider a biopsy prior to initiating TNF-α therapy in men older than 20 years with moderate to severe HS of the groin or buttocks, in addition to more frequent monitoring and a lower threshold to biopsy lesions with rapid growth or ulceration.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with high morbidity rates. Symptoms typically develop between puberty and the third decade of life, affecting twice as many females as males, with an overall disease prevalence of 1% to 4%.1 The pathogenesis is theorized to be related to an immune response to follicular occlusion and rupture in genetically susceptible individuals.
Among the complications associated with HS, the development of cutaneous squamous cell carcinoma (SCC) is 4.6-times more likely within HS lesions than in normal skin and typically is seen in the setting of long-standing disease, particularly in men with HS lesions located on the buttocks and genital region for more than 20 years.2 In 2015, the tumor necrosis factor (TNF) inhibitor adalimumab was approved by the US Food and Drug Administration for the treatment of HS. Tumor necrosis factor α inhibitors have been associated with an increased risk for skin cancer in other clinical settings.3,4 We present a case of locally advanced SCC that developed in a patient with HS who was treated with adalimumab and infliximab (both TNF-α inhibitors), ultimately leading to the patient’s death.
A 59-year-old man who smoked with a 40-year history of severe HS, who previously was lost to follow-up, presented to our dermatology clinic with lesions on the buttocks. Physical examination demonstrated confluent, indurated, boggy plaques; scattered sinus tracts with purulent drainage; scattered cystlike nodules; and tenderness to palpation consistent with Hurley stage III disease (Figure 1A). No involvement of the axillae or groin was noted. He was started on doxycycline and a prednisone taper with minimal improvement and subsequently was switched to adalimumab 3 months later. Adalimumab provided little relief and was discontinued; therapy was transitioned to infliximab 3 months later.
The patient returned to our clinic 3 months later with a severe flare and intractable pain after 4 infusions of infliximab. Physical examination showed a 7×5-cm deep malodorous ulcer with fibrinous exudate on the left buttock, several 2- to 3-cm shallow ulcers draining yellow exudate, and numerous fluctuant subcutaneous nodules on a background of scarring and sinus tracts. He was started again on doxycycline and a prednisone taper. At follow-up 2 weeks later, the largest ulcer had increased to 8 cm, and more indurated and tender subcutaneous nodules and scattered ulcerations developed (Figure 1B). Two punch biopsies of the left buttock revealed an invasive keratinizing carcinoma with no connection to the epidermis, consistent with SCC (Figure 2). Human papillomavirus (HPV) test results with probes for 37 HPV types—13 that were high risk (HPV-16, −18, −31, −33, −35, −39, −45, −51, −52, −56, −58, −59, −68)—were negative. Computerized tomography demonstrated diffuse thickening of the skin on the buttocks, inguinal adenopathy suspicious for nodal metastases, and no evidence of distant metastatic disease. Given the extent of the disease, surgical treatment was not an option, and he began receiving palliative radiotherapy. However, his health declined, and he developed aspiration pneumonia and hypotension requiring pressor support. He was transitioned to hospice care and died 3 months after presentation.
Tumor necrosis factor α antagonist treatment is being increasingly used to control HS but also may increase the risk for SCC development. We performed a search of PubMed articles indexed for MEDLINE as well as Web of Science using the terms hidradenitis suppurativa or acne inversa and one of the following—tumor necrosis factor inhibitor, infliximab, adalimumab, or etanercept—and squamous cell carcinoma or Marjolin ulcer. Seven cases of SCC arising in an HS patient treated with a TNF-α inhibitor have been reported (Table).5-10 Four cases were associated with infliximab use, 2 with adalimumab, and our case occurred after both adalimumab and infliximab treatment. All individuals were men with severe, long-standing disease of the anogenital region. In addition to smoking, HPV-16 positivity also has been reported as a risk factor for developing SCC in the setting of HS.11 In our patient, however, HPV testing did not cover all HPV strains, but several high-risk strains, including HPV-16, were negative.
Hidradenitis suppurativa is caused by an immune response to ruptured follicles and TNF-α antagonists are useful in suppressing this response; however, immunosuppression can lead to an increased susceptibility to malignancy, especially in SCC. It is unclear whether the use of infliximab or adalimumab is causal, additive, or a confounder in the development of SCC in patients with severe HS. It is possible that these agents increase the rapidity of the development of SCC in already-susceptible patients. Although TNF-α antagonists can be an effective therapeutic option for patients with moderate to severe HS, the potential risk for contributing to skin cancer development should raise provider suspicion in high-risk patients. Given the findings in this report, it may be suitable for providers to consider a biopsy prior to initiating TNF-α therapy in men older than 20 years with moderate to severe HS of the groin or buttocks, in addition to more frequent monitoring and a lower threshold to biopsy lesions with rapid growth or ulceration.
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561; quiz 562-533.
- Lapins J, Ye W, Nyren O, et al. Incidence of cancer among patients with hidradenitis suppurativa. Arch Dermatol. 2001;137:730-734.
- Askling J, Fahrbach K, Nordstrom B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119-130.
- Mariette X, Matucci-Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta-analysis. Ann Rheum Dis. 2011;70:1895-1904.
- Maalouf E, Faye O, Poli F, et al. Fatal epidermoid carcinoma in hidradenitis suppurativa following treatment with infliximab. Ann Dermatol Venereol. 2006;133(5 pt 1):473-474.
- Kurokawa I, Nishimura K, Yamanaka K, et al. Cytokeratin expression in squamous cell carcinoma arising from hidradenitis suppurativa (acne inversa). J Cutan Pathol. 2007;34:675-678.
- Scheinfeld N. A case of a patient with stage III familial hidradenitis suppurativa treated with 3 courses of infliximab and died of metastatic squamous cell carcinoma. Dermatol Online J. 2014;20(3).
- Verdelli A, Antiga E, Bonciani D, et al. A fatal case of hidradenitis suppurativa associated with sepsis and squamous cell carcinoma. Int J Dermatol. 2016;55:E52-E53.
- Giesey R, Delost GR, Honaker J, et al. Metastatic squamous cell carcinoma in a patient treated with adalimumab for hidradenitis suppurativa. JAAD Case Rep. 2017;3:489-491.
- Roy C, Roy S, Ghazawi F, et al. Cutaneous squamous cell carcinoma arising in hidradenitis suppurativa: a case report. SAGE Open Med Case Rep. 2019;7:2050313X19847359.
- Lavogiez C, Delaporte E, Darras-Vercambre S, et al. Clinicopathological study of 13 cases of squamous cell carcinoma complicating hidradenitis suppurativa. Dermatology. 2010;220:147-153.
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561; quiz 562-533.
- Lapins J, Ye W, Nyren O, et al. Incidence of cancer among patients with hidradenitis suppurativa. Arch Dermatol. 2001;137:730-734.
- Askling J, Fahrbach K, Nordstrom B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119-130.
- Mariette X, Matucci-Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta-analysis. Ann Rheum Dis. 2011;70:1895-1904.
- Maalouf E, Faye O, Poli F, et al. Fatal epidermoid carcinoma in hidradenitis suppurativa following treatment with infliximab. Ann Dermatol Venereol. 2006;133(5 pt 1):473-474.
- Kurokawa I, Nishimura K, Yamanaka K, et al. Cytokeratin expression in squamous cell carcinoma arising from hidradenitis suppurativa (acne inversa). J Cutan Pathol. 2007;34:675-678.
- Scheinfeld N. A case of a patient with stage III familial hidradenitis suppurativa treated with 3 courses of infliximab and died of metastatic squamous cell carcinoma. Dermatol Online J. 2014;20(3).
- Verdelli A, Antiga E, Bonciani D, et al. A fatal case of hidradenitis suppurativa associated with sepsis and squamous cell carcinoma. Int J Dermatol. 2016;55:E52-E53.
- Giesey R, Delost GR, Honaker J, et al. Metastatic squamous cell carcinoma in a patient treated with adalimumab for hidradenitis suppurativa. JAAD Case Rep. 2017;3:489-491.
- Roy C, Roy S, Ghazawi F, et al. Cutaneous squamous cell carcinoma arising in hidradenitis suppurativa: a case report. SAGE Open Med Case Rep. 2019;7:2050313X19847359.
- Lavogiez C, Delaporte E, Darras-Vercambre S, et al. Clinicopathological study of 13 cases of squamous cell carcinoma complicating hidradenitis suppurativa. Dermatology. 2010;220:147-153.
Practice Points
- Consider biopsy of representative lesions in men older than 20 years with moderate to severe disease of the groin and/or buttocks prior to initiation of tumor necrosis factor inhibitors.
- Consider more frequent clinical monitoring with a decrease in threshold to perform biopsy of any new or ulcerating lesions.
Dupilumab for the Treatment of Lichen Planus
To the Editor:
Lichen planus (LP) is an inflammatory mucocutaneous disorder that primarily affects adults aged 30 to 60 years.1 It can present across various regions such as the skin, scalp, oral cavity, genitalia, nails, and hair. It classically presents with pruritic, purple, polygonal papules or plaques. The proposed pathogenesis of this condition involves autoimmune destruction of epidermal basal keratinocytes.2 Management involves a stepwise approach, beginning with topical therapies such as corticosteroids and phototherapy and proceeding to systemic therapy including oral corticosteroids and retinoids. Additional medications with reported positive results include immunomodulators such as cyclosporine, tacrolimus, and mycophenolate mofetil.2-4 Dupilumab is a biologic immunomodulator and antagonist to the IL-4Rα on helper T cells (TH1). Although indicated for the treatment of moderate to severe atopic dermatitis, this medication’s immunomodulatory properties have been shown to aid various inflammatory cutaneous conditions, including prurigo nodularis.5-9 We present a case of dupilumab therapy for treatment-refractory LP.
A 52-year-old man presented with a new-onset progressive rash over the prior 6 months. He reported no history of atopic dermatitis. The patient described the rash as “severely pruritic” with a numeric rating scale itch intensity of 9/10 (0 being no itch; 10 being the worst itch imaginable). Physical examination revealed purple polygonal scaly papules on the arms, hands, legs, feet, chest, and back (Figure 1).
Three biopsies were taken, all indicative of lichenoid dermatitis consistent with LP. Rapid plasma reagin as well as HIV and hepatitis C virus serology tests were negative. Halobetasol ointment, tacrolimus ointment, and oral prednisone (28-day taper starting at 40 mg) all failed. Acitretin subsequently was initiated and failed to provide any benefit. The patient was unable to come to clinic 3 times a week for phototherapy due to his work schedule.
Due to the chronic, severe, and recalcitrant nature of his condition, as well as the lack of US Food and Drug Administration–approved treatments, the patient agreed to begin off-label treatment with dupilumab. Upon documentation, the patient’s primary diagnosis was listed as LP, clearly stating all commonly accepted treatments were attempted, except off-label therapy, and failed, and the plan was to treat him with dupilumab as if he had a severe form of atopic dermatitis. Dupilumab was approved with this documentation with a minimal co-pay, as the patient was on Medicaid. At 3-month follow-up (after 4 administrations of the medication), the patient showed remarkable improvement in appearance, and his numeric rating scale itch intensity score improved to 1/10.
Lichen planus is an immune-mediated, inflammatory condition that can affect the skin, hair, nails, and oral cavity. Although its etiology is not fully understood, research supports a primarily TH1 immunologic reaction.10 These T cells promote cytotoxic CD8 T-cell differentiation and migration, leading to subsequent destruction of epidermal basal keratinocytes. An important cytokine in this pathway—tumor necrosis factor α—stimulates a series of proinflammatory factors, including IL-1α, IL-8, and IL-6. IL-6 is of particular interest, as its elevation has been identified in the serum of patients with LP, with levels correlating to disease severity.11 This increase is thought to be multifactorial and a reliable predictor of disease activity.12,13 In addition to its proinflammatory role, IL-6 promotes the activity of IL-4, an essential cytokine in TH2 T-cell differentiation.
The TH2 pathway, enhanced by IL-6, increases the activity of downstream cytokines IL-4, IL-5, and IL-13. This pathway promotes IgE class switching and eosinophil maturation, pivotal factors in the development of atopic conditions such as allergic rhinitis, asthma, and atopic dermatitis. The role of IL-4 and TH2 cells in the pathogenesis of LP remains poorly understood.14 In prior basic laboratory studies, utilizing tissue sampling, RNA extraction, and real-time polymerase chain reaction assays, Yamauchi et al15 proposed that TH2-related chemokines played a pathogenic role in oral LP. Additional reports propose the pathogenic involvement of TH17, TH0, and TH2 T cells.16 These findings suggest that elevated IL-6 in those with LP may stimulate an increase in IL-4 and subsequent TH2 response. Dupilumab, a monoclonal antibody that targets IL-4Rα found on T cells, inhibits both IL-4 and IL-13 signaling, decreasing subsequent effector cell function.17,18 Several case reports have described dupilumab successfully treating various additional dermatoses, including prurigo nodularis, chronic pruritus, and bullous pemphigoid.5-9 Our case demonstrates an example of LP responsive to dupilumab. Our findings suggest that dupilumab interacts with the pathogenic cascade of LP, potentially implicating the role of TH2 in the pathophysiology of LP.
Treatment-refractory LP remains difficult to manage for both the patient and provider. Treatment regimens remain limited to small uncontrolled studies and case reports. Although primarily considered a TH1-mediated disease, the interplay of various alternative signaling pathways has been suggested. Our case of dupilumab-responsive LP suggests an underlying pathologic role of TH2-mediated activity. Dupilumab shows promise as an effective therapy for refractory LP, as evidenced by our patient’s remarkable response. Larger studies are warranted regarding the role of TH2-mediated inflammation and the use of dupilumab in LP.
- Cleach LL, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;266:723-732.
- Lehman, JS, Tollefson MM, Gibson LE. Lichen planus. Int J Dermatol. 2009;48:682-694.
- Frieling U, Bonsmann G, Schwarz T, et al. Treatment of severe lichen planus with mycophenolate mofetil. J Am Acad Dermatol. 2003;49:1063-1066.
- Cribier B, Frances C, Chosidow O. Treatment of lichen planus. an evidence-based medicine analysis of efficacy. Arch Dermatol. 1998;134:1521-1530.
- Calugareanu A, Jachiet C, Lepelletier C, et al. Dramatic improvement of generalized prurigo nodularis with dupilumab. J Eur Acad Dermatol Venereol. 2019;33:E303-E304.
- Kaye A, Gordon SC, Deverapalli SC, et al. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. 2018;154:1225-1226.
- Mollanazar NK, Qiu CC, Aldrich JL, et al. Use of dupilumab in HIV-positive patients: report of four cases. Br J Dermatol. 2019;181:1311-1312.
- Zhai LL, Savage KT, Qiu CC, et al. Chronic pruritus responding to dupilumab—a case series. Medicines (Basel). 2019;6:72.
- Mollanazar NK, Elgash M, Weaver L, et al. Reduced itch associated with dupilumab treatment in 4 patients with prurigo nodularis. JAMA Dermatol. 2019;155:121-122.
- Lodi G, Scully C, Carrozzo M, et al. Current controversies in oral lichen planus: report of an international consensus meeting. part 1. viral infections and etiopathogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:40-51.
- Yin M, Li G, Song H, et al. Identifying the association between interleukin-6 and lichen planus: a meta-analysis. Biomed Rep. 2017;6:571-575.
- Sun A, Chia JS, Chang YF, et al. Serum interleukin-6 level is a useful marker in evaluating therapeutic effects of levamisole and Chinese medicinal herbs on patients with oral lichen planus. J Oral Pathol Med. 2002;31:196-203.
- Rhodus NL, Cheng B, Bowles W, et al. Proinflammatory cytokine levels in saliva before and after treatment of (erosive) oral lichen planus with dexamethasone. Oral Dis. 2006;12:112-116.
- Carrozzo M. Understanding the pathobiology of oral lichen planus. Curr Oral Health Rep. 2014;1:173-179.
- Yamauchi M, Moriyama M, Hayashida JN, et al. Myeloid dendritic cells stimulated by thymic stromal lymphopoietin promote Th2 immune responses and the pathogenesis of oral lichen planus. Plos One. 2017:12:e0173017.
- Piccinni M-P, Lombardell L, Logidice F, et al. Potential pathogenetic role of Th17, Th0, and Th2 cells in erosive and reticular oral lichen planus. Oral Dis. 2013:20:212-218.
- Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. 2003;8:223-246.
- Noda S, Kruefer JG, Guttum-Yassky E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol. 2015;135:324-336.
To the Editor:
Lichen planus (LP) is an inflammatory mucocutaneous disorder that primarily affects adults aged 30 to 60 years.1 It can present across various regions such as the skin, scalp, oral cavity, genitalia, nails, and hair. It classically presents with pruritic, purple, polygonal papules or plaques. The proposed pathogenesis of this condition involves autoimmune destruction of epidermal basal keratinocytes.2 Management involves a stepwise approach, beginning with topical therapies such as corticosteroids and phototherapy and proceeding to systemic therapy including oral corticosteroids and retinoids. Additional medications with reported positive results include immunomodulators such as cyclosporine, tacrolimus, and mycophenolate mofetil.2-4 Dupilumab is a biologic immunomodulator and antagonist to the IL-4Rα on helper T cells (TH1). Although indicated for the treatment of moderate to severe atopic dermatitis, this medication’s immunomodulatory properties have been shown to aid various inflammatory cutaneous conditions, including prurigo nodularis.5-9 We present a case of dupilumab therapy for treatment-refractory LP.
A 52-year-old man presented with a new-onset progressive rash over the prior 6 months. He reported no history of atopic dermatitis. The patient described the rash as “severely pruritic” with a numeric rating scale itch intensity of 9/10 (0 being no itch; 10 being the worst itch imaginable). Physical examination revealed purple polygonal scaly papules on the arms, hands, legs, feet, chest, and back (Figure 1).
Three biopsies were taken, all indicative of lichenoid dermatitis consistent with LP. Rapid plasma reagin as well as HIV and hepatitis C virus serology tests were negative. Halobetasol ointment, tacrolimus ointment, and oral prednisone (28-day taper starting at 40 mg) all failed. Acitretin subsequently was initiated and failed to provide any benefit. The patient was unable to come to clinic 3 times a week for phototherapy due to his work schedule.
Due to the chronic, severe, and recalcitrant nature of his condition, as well as the lack of US Food and Drug Administration–approved treatments, the patient agreed to begin off-label treatment with dupilumab. Upon documentation, the patient’s primary diagnosis was listed as LP, clearly stating all commonly accepted treatments were attempted, except off-label therapy, and failed, and the plan was to treat him with dupilumab as if he had a severe form of atopic dermatitis. Dupilumab was approved with this documentation with a minimal co-pay, as the patient was on Medicaid. At 3-month follow-up (after 4 administrations of the medication), the patient showed remarkable improvement in appearance, and his numeric rating scale itch intensity score improved to 1/10.
Lichen planus is an immune-mediated, inflammatory condition that can affect the skin, hair, nails, and oral cavity. Although its etiology is not fully understood, research supports a primarily TH1 immunologic reaction.10 These T cells promote cytotoxic CD8 T-cell differentiation and migration, leading to subsequent destruction of epidermal basal keratinocytes. An important cytokine in this pathway—tumor necrosis factor α—stimulates a series of proinflammatory factors, including IL-1α, IL-8, and IL-6. IL-6 is of particular interest, as its elevation has been identified in the serum of patients with LP, with levels correlating to disease severity.11 This increase is thought to be multifactorial and a reliable predictor of disease activity.12,13 In addition to its proinflammatory role, IL-6 promotes the activity of IL-4, an essential cytokine in TH2 T-cell differentiation.
The TH2 pathway, enhanced by IL-6, increases the activity of downstream cytokines IL-4, IL-5, and IL-13. This pathway promotes IgE class switching and eosinophil maturation, pivotal factors in the development of atopic conditions such as allergic rhinitis, asthma, and atopic dermatitis. The role of IL-4 and TH2 cells in the pathogenesis of LP remains poorly understood.14 In prior basic laboratory studies, utilizing tissue sampling, RNA extraction, and real-time polymerase chain reaction assays, Yamauchi et al15 proposed that TH2-related chemokines played a pathogenic role in oral LP. Additional reports propose the pathogenic involvement of TH17, TH0, and TH2 T cells.16 These findings suggest that elevated IL-6 in those with LP may stimulate an increase in IL-4 and subsequent TH2 response. Dupilumab, a monoclonal antibody that targets IL-4Rα found on T cells, inhibits both IL-4 and IL-13 signaling, decreasing subsequent effector cell function.17,18 Several case reports have described dupilumab successfully treating various additional dermatoses, including prurigo nodularis, chronic pruritus, and bullous pemphigoid.5-9 Our case demonstrates an example of LP responsive to dupilumab. Our findings suggest that dupilumab interacts with the pathogenic cascade of LP, potentially implicating the role of TH2 in the pathophysiology of LP.
Treatment-refractory LP remains difficult to manage for both the patient and provider. Treatment regimens remain limited to small uncontrolled studies and case reports. Although primarily considered a TH1-mediated disease, the interplay of various alternative signaling pathways has been suggested. Our case of dupilumab-responsive LP suggests an underlying pathologic role of TH2-mediated activity. Dupilumab shows promise as an effective therapy for refractory LP, as evidenced by our patient’s remarkable response. Larger studies are warranted regarding the role of TH2-mediated inflammation and the use of dupilumab in LP.
To the Editor:
Lichen planus (LP) is an inflammatory mucocutaneous disorder that primarily affects adults aged 30 to 60 years.1 It can present across various regions such as the skin, scalp, oral cavity, genitalia, nails, and hair. It classically presents with pruritic, purple, polygonal papules or plaques. The proposed pathogenesis of this condition involves autoimmune destruction of epidermal basal keratinocytes.2 Management involves a stepwise approach, beginning with topical therapies such as corticosteroids and phototherapy and proceeding to systemic therapy including oral corticosteroids and retinoids. Additional medications with reported positive results include immunomodulators such as cyclosporine, tacrolimus, and mycophenolate mofetil.2-4 Dupilumab is a biologic immunomodulator and antagonist to the IL-4Rα on helper T cells (TH1). Although indicated for the treatment of moderate to severe atopic dermatitis, this medication’s immunomodulatory properties have been shown to aid various inflammatory cutaneous conditions, including prurigo nodularis.5-9 We present a case of dupilumab therapy for treatment-refractory LP.
A 52-year-old man presented with a new-onset progressive rash over the prior 6 months. He reported no history of atopic dermatitis. The patient described the rash as “severely pruritic” with a numeric rating scale itch intensity of 9/10 (0 being no itch; 10 being the worst itch imaginable). Physical examination revealed purple polygonal scaly papules on the arms, hands, legs, feet, chest, and back (Figure 1).
Three biopsies were taken, all indicative of lichenoid dermatitis consistent with LP. Rapid plasma reagin as well as HIV and hepatitis C virus serology tests were negative. Halobetasol ointment, tacrolimus ointment, and oral prednisone (28-day taper starting at 40 mg) all failed. Acitretin subsequently was initiated and failed to provide any benefit. The patient was unable to come to clinic 3 times a week for phototherapy due to his work schedule.
Due to the chronic, severe, and recalcitrant nature of his condition, as well as the lack of US Food and Drug Administration–approved treatments, the patient agreed to begin off-label treatment with dupilumab. Upon documentation, the patient’s primary diagnosis was listed as LP, clearly stating all commonly accepted treatments were attempted, except off-label therapy, and failed, and the plan was to treat him with dupilumab as if he had a severe form of atopic dermatitis. Dupilumab was approved with this documentation with a minimal co-pay, as the patient was on Medicaid. At 3-month follow-up (after 4 administrations of the medication), the patient showed remarkable improvement in appearance, and his numeric rating scale itch intensity score improved to 1/10.
Lichen planus is an immune-mediated, inflammatory condition that can affect the skin, hair, nails, and oral cavity. Although its etiology is not fully understood, research supports a primarily TH1 immunologic reaction.10 These T cells promote cytotoxic CD8 T-cell differentiation and migration, leading to subsequent destruction of epidermal basal keratinocytes. An important cytokine in this pathway—tumor necrosis factor α—stimulates a series of proinflammatory factors, including IL-1α, IL-8, and IL-6. IL-6 is of particular interest, as its elevation has been identified in the serum of patients with LP, with levels correlating to disease severity.11 This increase is thought to be multifactorial and a reliable predictor of disease activity.12,13 In addition to its proinflammatory role, IL-6 promotes the activity of IL-4, an essential cytokine in TH2 T-cell differentiation.
The TH2 pathway, enhanced by IL-6, increases the activity of downstream cytokines IL-4, IL-5, and IL-13. This pathway promotes IgE class switching and eosinophil maturation, pivotal factors in the development of atopic conditions such as allergic rhinitis, asthma, and atopic dermatitis. The role of IL-4 and TH2 cells in the pathogenesis of LP remains poorly understood.14 In prior basic laboratory studies, utilizing tissue sampling, RNA extraction, and real-time polymerase chain reaction assays, Yamauchi et al15 proposed that TH2-related chemokines played a pathogenic role in oral LP. Additional reports propose the pathogenic involvement of TH17, TH0, and TH2 T cells.16 These findings suggest that elevated IL-6 in those with LP may stimulate an increase in IL-4 and subsequent TH2 response. Dupilumab, a monoclonal antibody that targets IL-4Rα found on T cells, inhibits both IL-4 and IL-13 signaling, decreasing subsequent effector cell function.17,18 Several case reports have described dupilumab successfully treating various additional dermatoses, including prurigo nodularis, chronic pruritus, and bullous pemphigoid.5-9 Our case demonstrates an example of LP responsive to dupilumab. Our findings suggest that dupilumab interacts with the pathogenic cascade of LP, potentially implicating the role of TH2 in the pathophysiology of LP.
Treatment-refractory LP remains difficult to manage for both the patient and provider. Treatment regimens remain limited to small uncontrolled studies and case reports. Although primarily considered a TH1-mediated disease, the interplay of various alternative signaling pathways has been suggested. Our case of dupilumab-responsive LP suggests an underlying pathologic role of TH2-mediated activity. Dupilumab shows promise as an effective therapy for refractory LP, as evidenced by our patient’s remarkable response. Larger studies are warranted regarding the role of TH2-mediated inflammation and the use of dupilumab in LP.
- Cleach LL, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;266:723-732.
- Lehman, JS, Tollefson MM, Gibson LE. Lichen planus. Int J Dermatol. 2009;48:682-694.
- Frieling U, Bonsmann G, Schwarz T, et al. Treatment of severe lichen planus with mycophenolate mofetil. J Am Acad Dermatol. 2003;49:1063-1066.
- Cribier B, Frances C, Chosidow O. Treatment of lichen planus. an evidence-based medicine analysis of efficacy. Arch Dermatol. 1998;134:1521-1530.
- Calugareanu A, Jachiet C, Lepelletier C, et al. Dramatic improvement of generalized prurigo nodularis with dupilumab. J Eur Acad Dermatol Venereol. 2019;33:E303-E304.
- Kaye A, Gordon SC, Deverapalli SC, et al. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. 2018;154:1225-1226.
- Mollanazar NK, Qiu CC, Aldrich JL, et al. Use of dupilumab in HIV-positive patients: report of four cases. Br J Dermatol. 2019;181:1311-1312.
- Zhai LL, Savage KT, Qiu CC, et al. Chronic pruritus responding to dupilumab—a case series. Medicines (Basel). 2019;6:72.
- Mollanazar NK, Elgash M, Weaver L, et al. Reduced itch associated with dupilumab treatment in 4 patients with prurigo nodularis. JAMA Dermatol. 2019;155:121-122.
- Lodi G, Scully C, Carrozzo M, et al. Current controversies in oral lichen planus: report of an international consensus meeting. part 1. viral infections and etiopathogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:40-51.
- Yin M, Li G, Song H, et al. Identifying the association between interleukin-6 and lichen planus: a meta-analysis. Biomed Rep. 2017;6:571-575.
- Sun A, Chia JS, Chang YF, et al. Serum interleukin-6 level is a useful marker in evaluating therapeutic effects of levamisole and Chinese medicinal herbs on patients with oral lichen planus. J Oral Pathol Med. 2002;31:196-203.
- Rhodus NL, Cheng B, Bowles W, et al. Proinflammatory cytokine levels in saliva before and after treatment of (erosive) oral lichen planus with dexamethasone. Oral Dis. 2006;12:112-116.
- Carrozzo M. Understanding the pathobiology of oral lichen planus. Curr Oral Health Rep. 2014;1:173-179.
- Yamauchi M, Moriyama M, Hayashida JN, et al. Myeloid dendritic cells stimulated by thymic stromal lymphopoietin promote Th2 immune responses and the pathogenesis of oral lichen planus. Plos One. 2017:12:e0173017.
- Piccinni M-P, Lombardell L, Logidice F, et al. Potential pathogenetic role of Th17, Th0, and Th2 cells in erosive and reticular oral lichen planus. Oral Dis. 2013:20:212-218.
- Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. 2003;8:223-246.
- Noda S, Kruefer JG, Guttum-Yassky E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol. 2015;135:324-336.
- Cleach LL, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;266:723-732.
- Lehman, JS, Tollefson MM, Gibson LE. Lichen planus. Int J Dermatol. 2009;48:682-694.
- Frieling U, Bonsmann G, Schwarz T, et al. Treatment of severe lichen planus with mycophenolate mofetil. J Am Acad Dermatol. 2003;49:1063-1066.
- Cribier B, Frances C, Chosidow O. Treatment of lichen planus. an evidence-based medicine analysis of efficacy. Arch Dermatol. 1998;134:1521-1530.
- Calugareanu A, Jachiet C, Lepelletier C, et al. Dramatic improvement of generalized prurigo nodularis with dupilumab. J Eur Acad Dermatol Venereol. 2019;33:E303-E304.
- Kaye A, Gordon SC, Deverapalli SC, et al. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. 2018;154:1225-1226.
- Mollanazar NK, Qiu CC, Aldrich JL, et al. Use of dupilumab in HIV-positive patients: report of four cases. Br J Dermatol. 2019;181:1311-1312.
- Zhai LL, Savage KT, Qiu CC, et al. Chronic pruritus responding to dupilumab—a case series. Medicines (Basel). 2019;6:72.
- Mollanazar NK, Elgash M, Weaver L, et al. Reduced itch associated with dupilumab treatment in 4 patients with prurigo nodularis. JAMA Dermatol. 2019;155:121-122.
- Lodi G, Scully C, Carrozzo M, et al. Current controversies in oral lichen planus: report of an international consensus meeting. part 1. viral infections and etiopathogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:40-51.
- Yin M, Li G, Song H, et al. Identifying the association between interleukin-6 and lichen planus: a meta-analysis. Biomed Rep. 2017;6:571-575.
- Sun A, Chia JS, Chang YF, et al. Serum interleukin-6 level is a useful marker in evaluating therapeutic effects of levamisole and Chinese medicinal herbs on patients with oral lichen planus. J Oral Pathol Med. 2002;31:196-203.
- Rhodus NL, Cheng B, Bowles W, et al. Proinflammatory cytokine levels in saliva before and after treatment of (erosive) oral lichen planus with dexamethasone. Oral Dis. 2006;12:112-116.
- Carrozzo M. Understanding the pathobiology of oral lichen planus. Curr Oral Health Rep. 2014;1:173-179.
- Yamauchi M, Moriyama M, Hayashida JN, et al. Myeloid dendritic cells stimulated by thymic stromal lymphopoietin promote Th2 immune responses and the pathogenesis of oral lichen planus. Plos One. 2017:12:e0173017.
- Piccinni M-P, Lombardell L, Logidice F, et al. Potential pathogenetic role of Th17, Th0, and Th2 cells in erosive and reticular oral lichen planus. Oral Dis. 2013:20:212-218.
- Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. 2003;8:223-246.
- Noda S, Kruefer JG, Guttum-Yassky E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol. 2015;135:324-336.
Practice Points
- Lichen planus (LP) is an inflammatory mucocutaneous disorder that can present across various regions of the body with pruritic, purple, polygonal papules or plaques.
- The proposed pathogenesis of LP involves autoimmune destruction of epidermal basal keratinocytes.
- The immunomodulatory properties of dupilumab have been shown to aid various inflammatory cutaneous conditions.