User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Umbilicated Neoplasm on the Chest
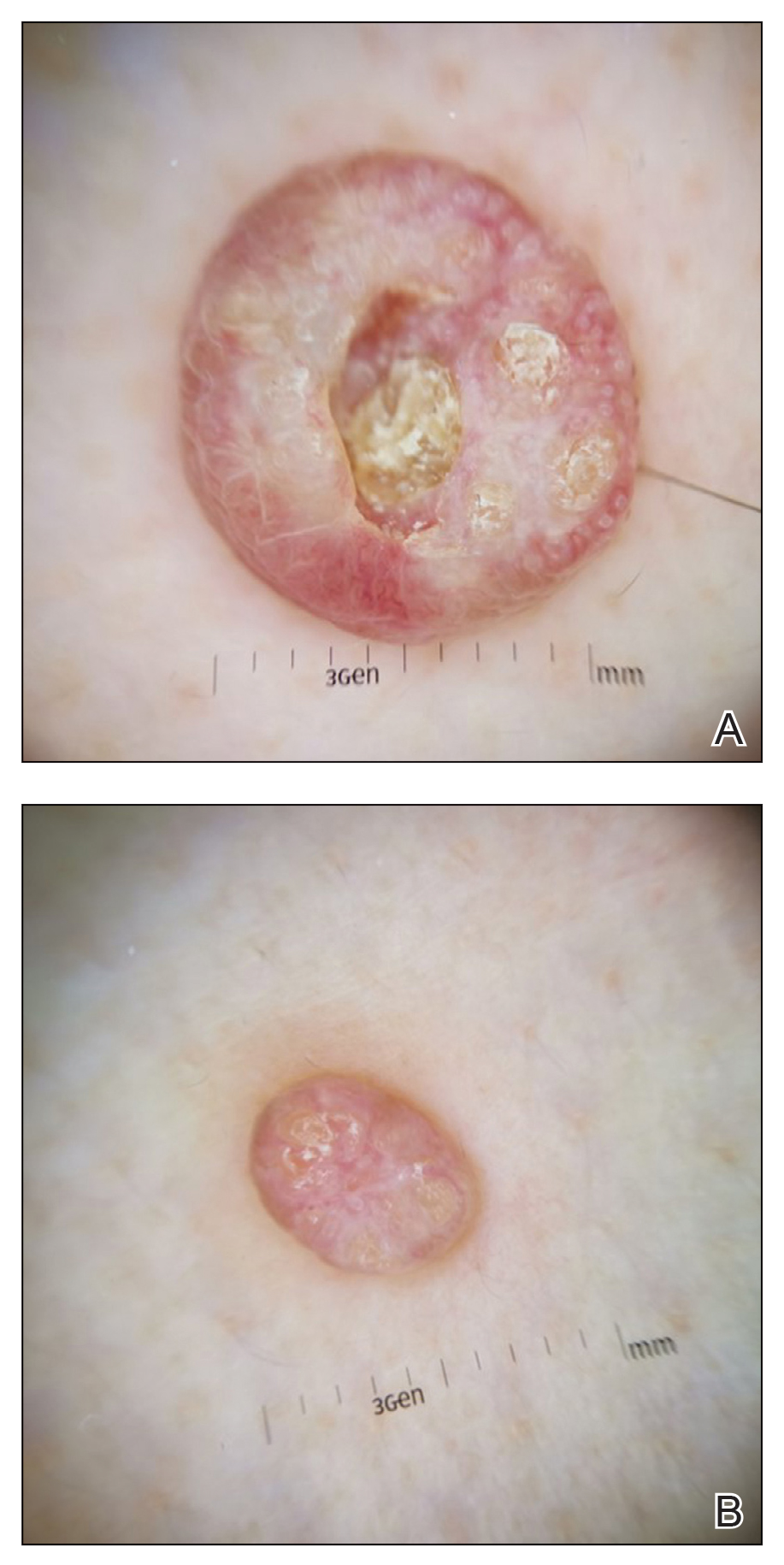
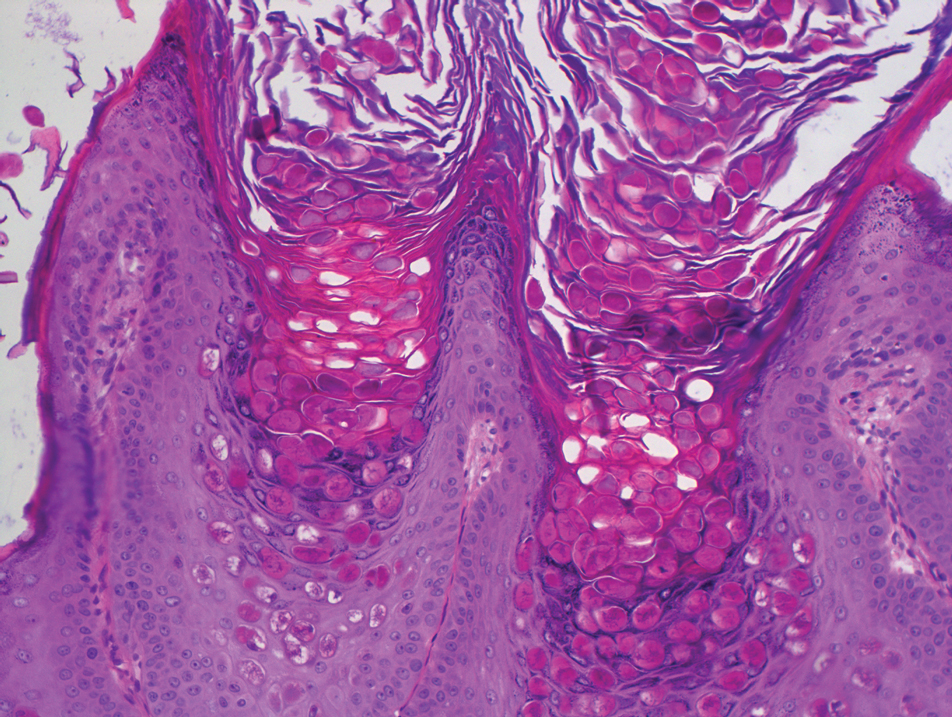
Dermoscopy showed polylobular, whitish yellow, amorphous structures at the center of the lesion surrounded by a crown of vessels (Figure 1). Histopathology revealed hyperplastic crateriform lesions containing large eosinophilic intracytoplasmic inclusion bodies within keratinocytes (Figure 2). At follow-up 2 weeks after the biopsy, the patient presented with approximately 20 more reddish papules of varying sizes on the abdomen and back that presented as dome-shaped papules and had a typical umbilicated center. The clinical manifestations, dermoscopy, and pathology findings were consistent with molluscum contagiosum (MC).
Molluscum contagiosum was first described in 1814. It is a benign cutaneous infectious disease caused by a double-stranded DNA virus of the poxvirus family. Molluscum contagiosum lesions usually manifest clinically as dome-shaped, flesh-colored or translucent, umbilicated papules measuring 1 to 5 mm in diameter that are commonly distributed over the face, trunk, and extremities and usually are self-limiting.1
Giant MC is rare and can be seen either in patients on immunosuppressive therapy or in those with diseases that can cause immunosuppression, such as human immunodeficiency virus, leukemia, atopic dermatitis, Wiskott-Aldrich syndrome, and sarcoidosis. In these instances, MC often is greater than 1 cm in diameter. Atypical variants may have an eczematous presentation or a lesion with secondary abscess formation and also can be spread widely over the body.2 Due to these atypical appearances and large dimensions in immunocompromised patients, other dermatologic diseases should be considered in the differential diagnosis, such as basal cell carcinoma, keratoacanthoma, squamous cell carcinoma, cutaneous horn, cutaneous cryptococcosis, histoplasmosis, and xanthomatosis.3
In our patient, the differential diagnosis included keratoacanthoma, which may present as a solitary, discrete, round to oval, flesh-colored, umbilicated nodule with a central keratin-filled crater and has a rapid clinical evolution, usually regressing within 4 to 6 months.
Squamous cell carcinoma may appear as scaly red patches, open sores, warts, or elevated growths with a central depression and may crust or bleed. Basal cell carcinoma typically may appear as a dome-shaped skin nodule with visible blood vessels or sometimes presents as a red patch similar to eczema. Xanthomatosis often appears as yellow to orange, mostly asymptomatic, supple patches or plaques, usually with sharp and distinctive edges.
Ancillary diagnostic modalities such as dermoscopy may be used to improve diagnostic accuracy. The best known capillaroscopic feature of MC is the peripheral crown of vessels in a radial distribution. A study of 258 MC lesions highlighted that crown and crown plus radial arrangements are the most common vascular structure patterns under dermoscopy. In addition, polylobular amorphous white structures in the center of the lesions tend to be a feature of larger MC papules.4 Histologically, MC shows lobulated crateriform lesions, thickening of the epidermis into the dermis, and the typical appearance of large eosinophilic intracytoplasmic inclusion bodies within keratinocytes.5
There are several treatment options available for MC. Common modalities include liquid nitrogen cryospray, curettage, and electrocauterization. In immunocompromised patients, MC lesions usually are resistant to ordinary therapy. The efficacy of topical agents such as imiquimod, which can induce high levels of IFN-α and other cytokines, has been demonstrated in these patients.6 Cidofovir, a nucleoside analog that has potent antiviral properties, also can be included as a therapeutic option.3 Our patient’s largest MC lesion was treated with surgical excision, the 2 large lesions on the left side of the chest with cryotherapy, and the other small lesions with curettage.
- Hanson D, Diven DG. Molluscum contagiosum. Dermatol Online J. 2003;9:2.
- Singh S, Swain M, Shukla S, et al. An unusual presentation of giant molluscum contagiosum diagnosed on cytology. Diagn Cytopathol. 2018;46:794-796.
- Mansur AT, Goktay F, Gunduz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Ku SH, Cho EB, Park EJ, et al. Dermoscopic features of molluscum contagiosum based on white structures and their correlation with histopathological findings. Clin Exp Dermatol. 2015;40:208-210.
- Trčko K, Hošnjak L, Kušar B, et al. Clinical, histopathological, and virological evaluation of 203 patients with a clinical diagnosis of molluscum contagiosum [published online November 12, 2018]. Open Forum Infect Dis. 2018;5.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2010;31:452-453.
Dermoscopy showed polylobular, whitish yellow, amorphous structures at the center of the lesion surrounded by a crown of vessels (Figure 1). Histopathology revealed hyperplastic crateriform lesions containing large eosinophilic intracytoplasmic inclusion bodies within keratinocytes (Figure 2). At follow-up 2 weeks after the biopsy, the patient presented with approximately 20 more reddish papules of varying sizes on the abdomen and back that presented as dome-shaped papules and had a typical umbilicated center. The clinical manifestations, dermoscopy, and pathology findings were consistent with molluscum contagiosum (MC).
Molluscum contagiosum was first described in 1814. It is a benign cutaneous infectious disease caused by a double-stranded DNA virus of the poxvirus family. Molluscum contagiosum lesions usually manifest clinically as dome-shaped, flesh-colored or translucent, umbilicated papules measuring 1 to 5 mm in diameter that are commonly distributed over the face, trunk, and extremities and usually are self-limiting.1
Giant MC is rare and can be seen either in patients on immunosuppressive therapy or in those with diseases that can cause immunosuppression, such as human immunodeficiency virus, leukemia, atopic dermatitis, Wiskott-Aldrich syndrome, and sarcoidosis. In these instances, MC often is greater than 1 cm in diameter. Atypical variants may have an eczematous presentation or a lesion with secondary abscess formation and also can be spread widely over the body.2 Due to these atypical appearances and large dimensions in immunocompromised patients, other dermatologic diseases should be considered in the differential diagnosis, such as basal cell carcinoma, keratoacanthoma, squamous cell carcinoma, cutaneous horn, cutaneous cryptococcosis, histoplasmosis, and xanthomatosis.3
In our patient, the differential diagnosis included keratoacanthoma, which may present as a solitary, discrete, round to oval, flesh-colored, umbilicated nodule with a central keratin-filled crater and has a rapid clinical evolution, usually regressing within 4 to 6 months.
Squamous cell carcinoma may appear as scaly red patches, open sores, warts, or elevated growths with a central depression and may crust or bleed. Basal cell carcinoma typically may appear as a dome-shaped skin nodule with visible blood vessels or sometimes presents as a red patch similar to eczema. Xanthomatosis often appears as yellow to orange, mostly asymptomatic, supple patches or plaques, usually with sharp and distinctive edges.
Ancillary diagnostic modalities such as dermoscopy may be used to improve diagnostic accuracy. The best known capillaroscopic feature of MC is the peripheral crown of vessels in a radial distribution. A study of 258 MC lesions highlighted that crown and crown plus radial arrangements are the most common vascular structure patterns under dermoscopy. In addition, polylobular amorphous white structures in the center of the lesions tend to be a feature of larger MC papules.4 Histologically, MC shows lobulated crateriform lesions, thickening of the epidermis into the dermis, and the typical appearance of large eosinophilic intracytoplasmic inclusion bodies within keratinocytes.5
There are several treatment options available for MC. Common modalities include liquid nitrogen cryospray, curettage, and electrocauterization. In immunocompromised patients, MC lesions usually are resistant to ordinary therapy. The efficacy of topical agents such as imiquimod, which can induce high levels of IFN-α and other cytokines, has been demonstrated in these patients.6 Cidofovir, a nucleoside analog that has potent antiviral properties, also can be included as a therapeutic option.3 Our patient’s largest MC lesion was treated with surgical excision, the 2 large lesions on the left side of the chest with cryotherapy, and the other small lesions with curettage.
Dermoscopy showed polylobular, whitish yellow, amorphous structures at the center of the lesion surrounded by a crown of vessels (Figure 1). Histopathology revealed hyperplastic crateriform lesions containing large eosinophilic intracytoplasmic inclusion bodies within keratinocytes (Figure 2). At follow-up 2 weeks after the biopsy, the patient presented with approximately 20 more reddish papules of varying sizes on the abdomen and back that presented as dome-shaped papules and had a typical umbilicated center. The clinical manifestations, dermoscopy, and pathology findings were consistent with molluscum contagiosum (MC).
Molluscum contagiosum was first described in 1814. It is a benign cutaneous infectious disease caused by a double-stranded DNA virus of the poxvirus family. Molluscum contagiosum lesions usually manifest clinically as dome-shaped, flesh-colored or translucent, umbilicated papules measuring 1 to 5 mm in diameter that are commonly distributed over the face, trunk, and extremities and usually are self-limiting.1
Giant MC is rare and can be seen either in patients on immunosuppressive therapy or in those with diseases that can cause immunosuppression, such as human immunodeficiency virus, leukemia, atopic dermatitis, Wiskott-Aldrich syndrome, and sarcoidosis. In these instances, MC often is greater than 1 cm in diameter. Atypical variants may have an eczematous presentation or a lesion with secondary abscess formation and also can be spread widely over the body.2 Due to these atypical appearances and large dimensions in immunocompromised patients, other dermatologic diseases should be considered in the differential diagnosis, such as basal cell carcinoma, keratoacanthoma, squamous cell carcinoma, cutaneous horn, cutaneous cryptococcosis, histoplasmosis, and xanthomatosis.3
In our patient, the differential diagnosis included keratoacanthoma, which may present as a solitary, discrete, round to oval, flesh-colored, umbilicated nodule with a central keratin-filled crater and has a rapid clinical evolution, usually regressing within 4 to 6 months.
Squamous cell carcinoma may appear as scaly red patches, open sores, warts, or elevated growths with a central depression and may crust or bleed. Basal cell carcinoma typically may appear as a dome-shaped skin nodule with visible blood vessels or sometimes presents as a red patch similar to eczema. Xanthomatosis often appears as yellow to orange, mostly asymptomatic, supple patches or plaques, usually with sharp and distinctive edges.
Ancillary diagnostic modalities such as dermoscopy may be used to improve diagnostic accuracy. The best known capillaroscopic feature of MC is the peripheral crown of vessels in a radial distribution. A study of 258 MC lesions highlighted that crown and crown plus radial arrangements are the most common vascular structure patterns under dermoscopy. In addition, polylobular amorphous white structures in the center of the lesions tend to be a feature of larger MC papules.4 Histologically, MC shows lobulated crateriform lesions, thickening of the epidermis into the dermis, and the typical appearance of large eosinophilic intracytoplasmic inclusion bodies within keratinocytes.5
There are several treatment options available for MC. Common modalities include liquid nitrogen cryospray, curettage, and electrocauterization. In immunocompromised patients, MC lesions usually are resistant to ordinary therapy. The efficacy of topical agents such as imiquimod, which can induce high levels of IFN-α and other cytokines, has been demonstrated in these patients.6 Cidofovir, a nucleoside analog that has potent antiviral properties, also can be included as a therapeutic option.3 Our patient’s largest MC lesion was treated with surgical excision, the 2 large lesions on the left side of the chest with cryotherapy, and the other small lesions with curettage.
- Hanson D, Diven DG. Molluscum contagiosum. Dermatol Online J. 2003;9:2.
- Singh S, Swain M, Shukla S, et al. An unusual presentation of giant molluscum contagiosum diagnosed on cytology. Diagn Cytopathol. 2018;46:794-796.
- Mansur AT, Goktay F, Gunduz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Ku SH, Cho EB, Park EJ, et al. Dermoscopic features of molluscum contagiosum based on white structures and their correlation with histopathological findings. Clin Exp Dermatol. 2015;40:208-210.
- Trčko K, Hošnjak L, Kušar B, et al. Clinical, histopathological, and virological evaluation of 203 patients with a clinical diagnosis of molluscum contagiosum [published online November 12, 2018]. Open Forum Infect Dis. 2018;5.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2010;31:452-453.
- Hanson D, Diven DG. Molluscum contagiosum. Dermatol Online J. 2003;9:2.
- Singh S, Swain M, Shukla S, et al. An unusual presentation of giant molluscum contagiosum diagnosed on cytology. Diagn Cytopathol. 2018;46:794-796.
- Mansur AT, Goktay F, Gunduz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Ku SH, Cho EB, Park EJ, et al. Dermoscopic features of molluscum contagiosum based on white structures and their correlation with histopathological findings. Clin Exp Dermatol. 2015;40:208-210.
- Trčko K, Hošnjak L, Kušar B, et al. Clinical, histopathological, and virological evaluation of 203 patients with a clinical diagnosis of molluscum contagiosum [published online November 12, 2018]. Open Forum Infect Dis. 2018;5.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2010;31:452-453.
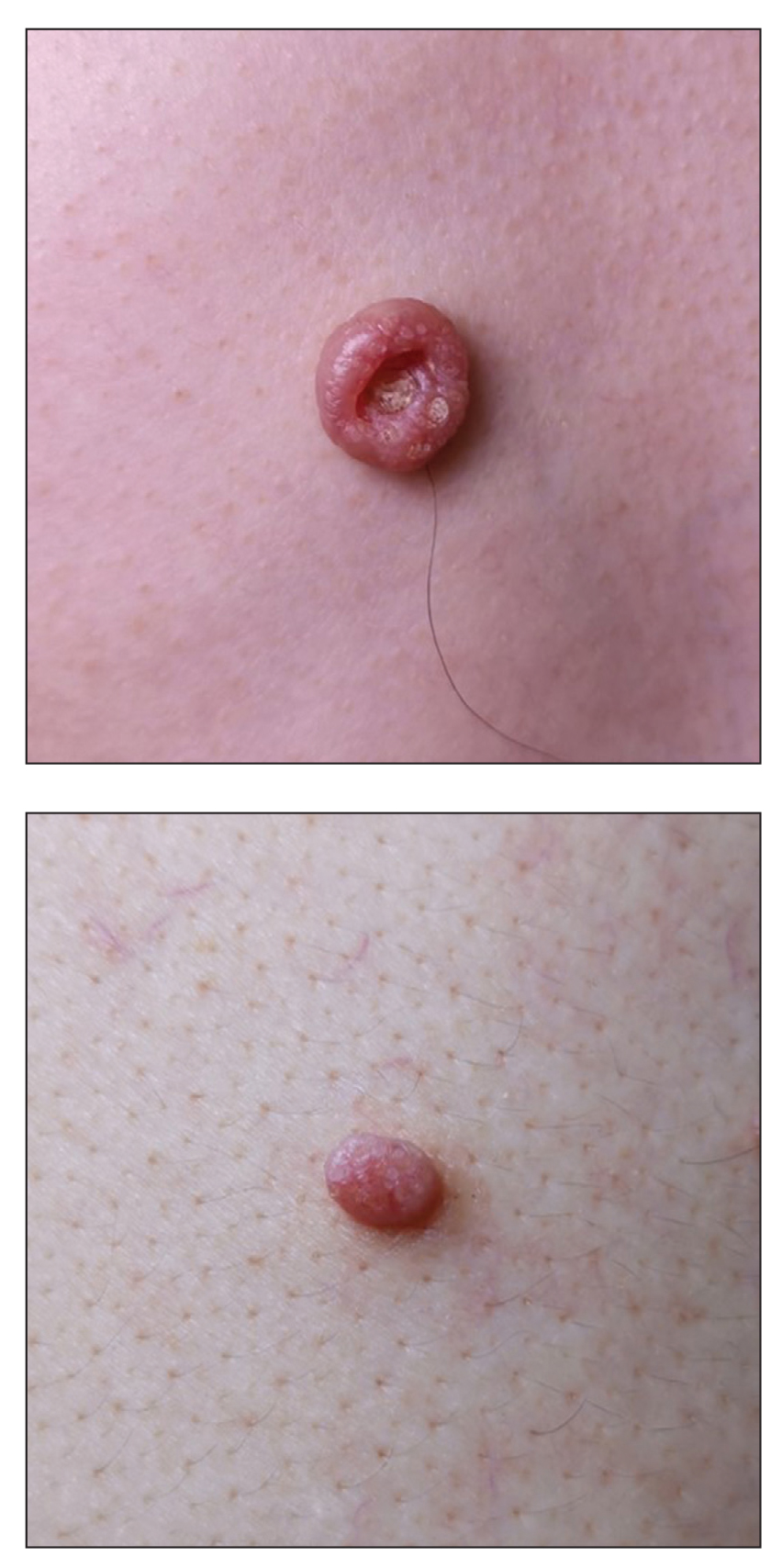
A 49-year-old man presented with a slow-growing mass on the chest of 1 year’s duration. The neoplasm started as a small papule that gradually increased in size. The patient denied pain, itching, bleeding, or discharge. He had a history of end-stage renal disease with a kidney transplant 8 years prior. His medication history included long-term use of oral tacrolimus, mycophenolate mofetil, and prednisone. Physical examination revealed a yellowish red, exogenous, pedunculated neoplasm on the right side of the chest measuring 1 cm in diameter with an umbilicated center and keratotic material (top). There were 2 more yellowish red papules on the left side of the chest measuring 0.5 cm in diameter without an umbilicated center (bottom). Dermoscopy and a biopsy were performed.
Recurrent Cutaneous Exophiala Phaeohyphomycosis in an Immunosuppressed Patient
To the Editor:
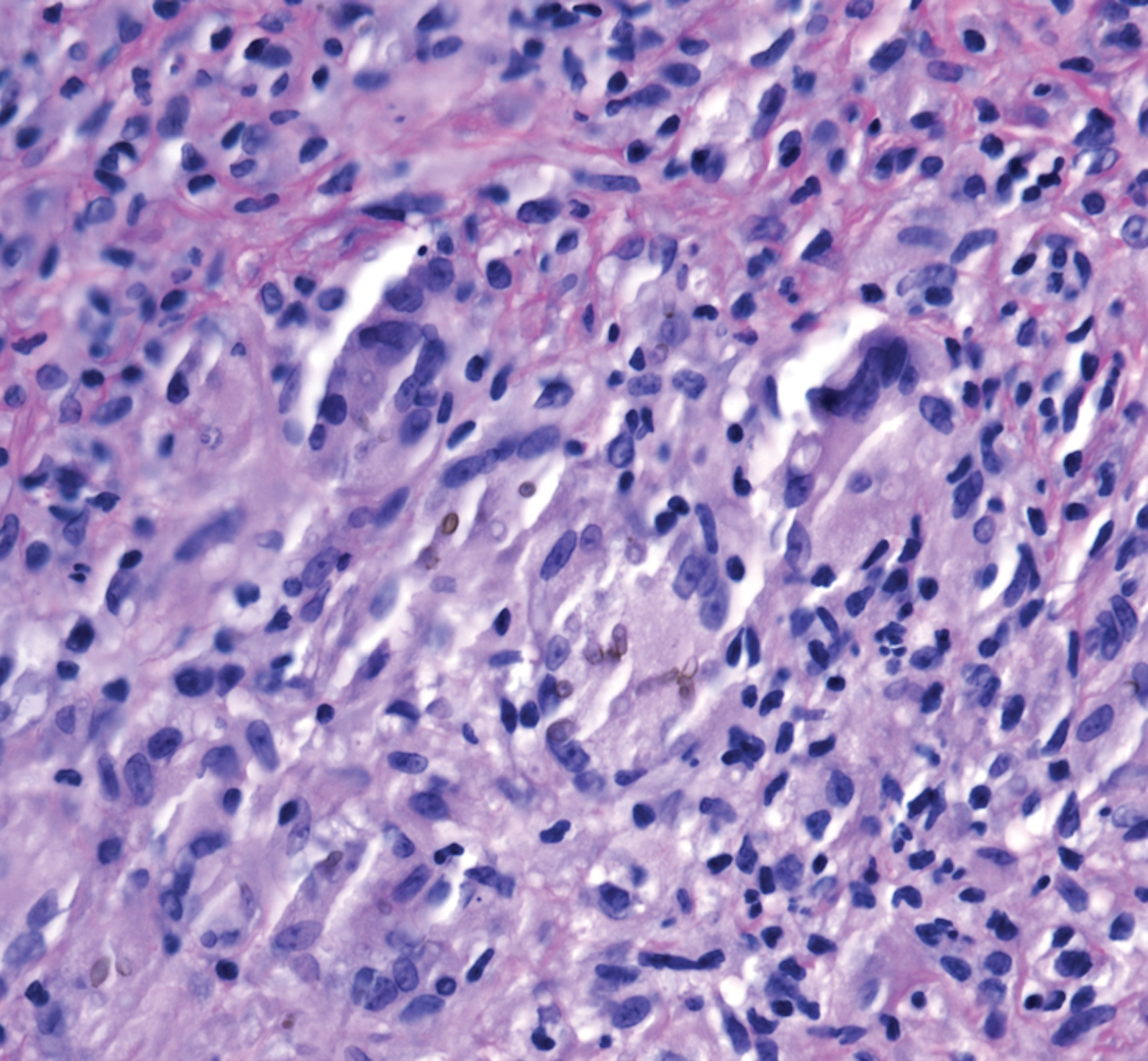
A 73-year-old man presented with a 2.5-cm, recurrent, fluctuant, multiloculated nodule on the left forearm. The lesion was nontender with occasional chalky, white to yellow discharge from multiple sinus tracts. He was otherwise well appearing without signs of systemic infection. He reported similar lesions in slightly different anatomic locations on the left forearm both 7 and 4 years prior to the current presentation. In both instances, the nodules were excised at an outside hospital without any additional treatment. Histopathology of the excised tissue from both prior occasions demonstrated brown septate hyphae surrounded by suppurative and granulomatous inflammation consistent with dematiaceous fungal infection of the dermis (Figures 1 and 2); the organisms were highlighted with periodic acid–Schiff stain.

The patient’s medical history was notable for advanced heart failure with an ejection fraction of 25% and autosomal-dominant polycystic kidney disease. He received an orthotopic kidney transplant 17 years prior to the current presentation. Medications included tacrolimus, mycophenolate mofetil, and prednisone. He denied any trauma or notable exposures to vegetation, and his travel history was unremarkable. A review of systems was negative.
At the current presentation, a sterile fungal culture was performed and found positive for Exophiala species, while bacterial and mycobacterial cultures were negative. A diagnosis of phaeohyphomycosis was made, and he was scheduled for re-excision. Out of concern for interactions with his immunosuppressive regimen, he chose to forgo any systemic antifungal therapy. He died from hospital-acquired pneumonia and volume overload unresponsive to diuretics or dialysis.
Phaeohyphomycosis is a rare fungal infection caused by several genera of dematiaceous fungi that are characterized by the presence of melaninlike cell wall pigments thought to locally hinder immune clearance by scavenging phagocyte-derived free radicals. These fungi are ubiquitous in soil and vegetation and usually penetrate the skin at sites of minor trauma.1 Phaeohyphomycosis typically affects immunosuppressed hosts, and its incidence among organ transplant recipients currently is 9%.2 The incidence in this population has been rising, however, as recent advances in immunosuppressive therapies have increased posttransplant survival.3
Subcutaneous phaeohyphomycosis can present with nodules, cysts, tumors, and/or verrucous plaques, and the diagnosis almost always requires clinicopathologic correlation.3 Rapid diagnosis can be made when septate brown hyphae and/or yeast forms are observed on hematoxylin and eosin stain. Rarely, patients present with disseminated infection, characterized by fungemia; central nervous system involvement; and/or infection of multiple deep structures including the eyes, lungs, bones, and sinuses.4 The risk for dissemination from the skin likely is related to the culprit organism’s genus; Lomentospora, Cladophialophora, and Verruconis often are associated with dissemination, while Alternaria, Exophiala, and Fonsecaea typically remain confined to the skin and subcutis.5 Due to this difference and its potential to impact management, obtaining a tissue fungal culture is advisable when phaeohyphomycosis is suspected.
There is no standard treatment of phaeohyphomycosis. Regimens typically consist of excision and prolonged courses of azole therapy, though excision alone with close follow-up may be a reasonable alternative.6 The latter is a particularly important consideration when managing phaeohyphomycosis in organ transplant recipients, as azoles are known cytochrome P450 3A4 inhibitors that can affect serum levels of common immunosuppressive medications including calcineurin inhibitors and mammalian target of rapamycin inhibitors.3 Local recurrence is common regardless of whether azole therapy is pursued,7 and dissemination of localized Exophiala infections is exceedingly rare.8 There is a strong argument to be made for our patient’s decision to forgo antifungal therapy.
This case underscores the difficulty inherent to eradicating local subcutaneous Exophiala phaeohyphomycosis while providing reassurance that with treatment, the risk of life-threatening complications is low. Obtaining tissue for both hematoxylin and eosin stain and sterile culture is crucial to ensuring prompt diagnosis and tailoring the optimal treatment and surveillance strategy to the culprit organism. To avoid delays in diagnosis and treatment, it is important for clinicians to consider phaeohyphomycosis in the differential diagnosis for recurrent nodulocystic lesions in immunosuppressed patients and to recognize that presentations may span many years.
- Bhardwaj S, Capoor MR, Kolte S, et al. Phaeohyphomycosis due to Exophiala jeanselmei: an emerging pathogen in India—case report and review. Mycopathologia. 2016;181:279-284.
- Isa-Isa R, Garcia C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Tirico MCCP, Neto CF, Cruz LL, et al. Clinical spectrum of phaeohyphomycosis in solid organ transplant recipients. JAAD Case Rep. 2016;2:465-469.
- Revankar SG, Patterson JE, Sutton DA, et al. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin Infect Dis. 2002;34:467-476.
- Revankar SG, Baddley JW, Chen SC-A, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200.
- Oberlin KE, Nichols AJ, Rosa R, et al. Phaeohyphomycosis due to Exophiala infections in solid organ transplant recipients: case report and literature review [published online June 26, 2017]. Transpl Infect Dis. 2017;19. doi:10.1111/tid.12723.
- Shirbur S, Telkar S, Goudar B, et al. Recurrent phaeohyphomycosis: a case report. J Clin Diagn Res. 2013;7:2015-2016.
- Li D-M, Li R-Y, de Hoog GS, et al. Fatal Exophiala infections in China, with a report of seven cases. Mycoses. 2011;54:E136-E142.
To the Editor:
A 73-year-old man presented with a 2.5-cm, recurrent, fluctuant, multiloculated nodule on the left forearm. The lesion was nontender with occasional chalky, white to yellow discharge from multiple sinus tracts. He was otherwise well appearing without signs of systemic infection. He reported similar lesions in slightly different anatomic locations on the left forearm both 7 and 4 years prior to the current presentation. In both instances, the nodules were excised at an outside hospital without any additional treatment. Histopathology of the excised tissue from both prior occasions demonstrated brown septate hyphae surrounded by suppurative and granulomatous inflammation consistent with dematiaceous fungal infection of the dermis (Figures 1 and 2); the organisms were highlighted with periodic acid–Schiff stain.
The patient’s medical history was notable for advanced heart failure with an ejection fraction of 25% and autosomal-dominant polycystic kidney disease. He received an orthotopic kidney transplant 17 years prior to the current presentation. Medications included tacrolimus, mycophenolate mofetil, and prednisone. He denied any trauma or notable exposures to vegetation, and his travel history was unremarkable. A review of systems was negative.
At the current presentation, a sterile fungal culture was performed and found positive for Exophiala species, while bacterial and mycobacterial cultures were negative. A diagnosis of phaeohyphomycosis was made, and he was scheduled for re-excision. Out of concern for interactions with his immunosuppressive regimen, he chose to forgo any systemic antifungal therapy. He died from hospital-acquired pneumonia and volume overload unresponsive to diuretics or dialysis.
Phaeohyphomycosis is a rare fungal infection caused by several genera of dematiaceous fungi that are characterized by the presence of melaninlike cell wall pigments thought to locally hinder immune clearance by scavenging phagocyte-derived free radicals. These fungi are ubiquitous in soil and vegetation and usually penetrate the skin at sites of minor trauma.1 Phaeohyphomycosis typically affects immunosuppressed hosts, and its incidence among organ transplant recipients currently is 9%.2 The incidence in this population has been rising, however, as recent advances in immunosuppressive therapies have increased posttransplant survival.3
Subcutaneous phaeohyphomycosis can present with nodules, cysts, tumors, and/or verrucous plaques, and the diagnosis almost always requires clinicopathologic correlation.3 Rapid diagnosis can be made when septate brown hyphae and/or yeast forms are observed on hematoxylin and eosin stain. Rarely, patients present with disseminated infection, characterized by fungemia; central nervous system involvement; and/or infection of multiple deep structures including the eyes, lungs, bones, and sinuses.4 The risk for dissemination from the skin likely is related to the culprit organism’s genus; Lomentospora, Cladophialophora, and Verruconis often are associated with dissemination, while Alternaria, Exophiala, and Fonsecaea typically remain confined to the skin and subcutis.5 Due to this difference and its potential to impact management, obtaining a tissue fungal culture is advisable when phaeohyphomycosis is suspected.
There is no standard treatment of phaeohyphomycosis. Regimens typically consist of excision and prolonged courses of azole therapy, though excision alone with close follow-up may be a reasonable alternative.6 The latter is a particularly important consideration when managing phaeohyphomycosis in organ transplant recipients, as azoles are known cytochrome P450 3A4 inhibitors that can affect serum levels of common immunosuppressive medications including calcineurin inhibitors and mammalian target of rapamycin inhibitors.3 Local recurrence is common regardless of whether azole therapy is pursued,7 and dissemination of localized Exophiala infections is exceedingly rare.8 There is a strong argument to be made for our patient’s decision to forgo antifungal therapy.
This case underscores the difficulty inherent to eradicating local subcutaneous Exophiala phaeohyphomycosis while providing reassurance that with treatment, the risk of life-threatening complications is low. Obtaining tissue for both hematoxylin and eosin stain and sterile culture is crucial to ensuring prompt diagnosis and tailoring the optimal treatment and surveillance strategy to the culprit organism. To avoid delays in diagnosis and treatment, it is important for clinicians to consider phaeohyphomycosis in the differential diagnosis for recurrent nodulocystic lesions in immunosuppressed patients and to recognize that presentations may span many years.
To the Editor:
A 73-year-old man presented with a 2.5-cm, recurrent, fluctuant, multiloculated nodule on the left forearm. The lesion was nontender with occasional chalky, white to yellow discharge from multiple sinus tracts. He was otherwise well appearing without signs of systemic infection. He reported similar lesions in slightly different anatomic locations on the left forearm both 7 and 4 years prior to the current presentation. In both instances, the nodules were excised at an outside hospital without any additional treatment. Histopathology of the excised tissue from both prior occasions demonstrated brown septate hyphae surrounded by suppurative and granulomatous inflammation consistent with dematiaceous fungal infection of the dermis (Figures 1 and 2); the organisms were highlighted with periodic acid–Schiff stain.
The patient’s medical history was notable for advanced heart failure with an ejection fraction of 25% and autosomal-dominant polycystic kidney disease. He received an orthotopic kidney transplant 17 years prior to the current presentation. Medications included tacrolimus, mycophenolate mofetil, and prednisone. He denied any trauma or notable exposures to vegetation, and his travel history was unremarkable. A review of systems was negative.
At the current presentation, a sterile fungal culture was performed and found positive for Exophiala species, while bacterial and mycobacterial cultures were negative. A diagnosis of phaeohyphomycosis was made, and he was scheduled for re-excision. Out of concern for interactions with his immunosuppressive regimen, he chose to forgo any systemic antifungal therapy. He died from hospital-acquired pneumonia and volume overload unresponsive to diuretics or dialysis.
Phaeohyphomycosis is a rare fungal infection caused by several genera of dematiaceous fungi that are characterized by the presence of melaninlike cell wall pigments thought to locally hinder immune clearance by scavenging phagocyte-derived free radicals. These fungi are ubiquitous in soil and vegetation and usually penetrate the skin at sites of minor trauma.1 Phaeohyphomycosis typically affects immunosuppressed hosts, and its incidence among organ transplant recipients currently is 9%.2 The incidence in this population has been rising, however, as recent advances in immunosuppressive therapies have increased posttransplant survival.3
Subcutaneous phaeohyphomycosis can present with nodules, cysts, tumors, and/or verrucous plaques, and the diagnosis almost always requires clinicopathologic correlation.3 Rapid diagnosis can be made when septate brown hyphae and/or yeast forms are observed on hematoxylin and eosin stain. Rarely, patients present with disseminated infection, characterized by fungemia; central nervous system involvement; and/or infection of multiple deep structures including the eyes, lungs, bones, and sinuses.4 The risk for dissemination from the skin likely is related to the culprit organism’s genus; Lomentospora, Cladophialophora, and Verruconis often are associated with dissemination, while Alternaria, Exophiala, and Fonsecaea typically remain confined to the skin and subcutis.5 Due to this difference and its potential to impact management, obtaining a tissue fungal culture is advisable when phaeohyphomycosis is suspected.
There is no standard treatment of phaeohyphomycosis. Regimens typically consist of excision and prolonged courses of azole therapy, though excision alone with close follow-up may be a reasonable alternative.6 The latter is a particularly important consideration when managing phaeohyphomycosis in organ transplant recipients, as azoles are known cytochrome P450 3A4 inhibitors that can affect serum levels of common immunosuppressive medications including calcineurin inhibitors and mammalian target of rapamycin inhibitors.3 Local recurrence is common regardless of whether azole therapy is pursued,7 and dissemination of localized Exophiala infections is exceedingly rare.8 There is a strong argument to be made for our patient’s decision to forgo antifungal therapy.
This case underscores the difficulty inherent to eradicating local subcutaneous Exophiala phaeohyphomycosis while providing reassurance that with treatment, the risk of life-threatening complications is low. Obtaining tissue for both hematoxylin and eosin stain and sterile culture is crucial to ensuring prompt diagnosis and tailoring the optimal treatment and surveillance strategy to the culprit organism. To avoid delays in diagnosis and treatment, it is important for clinicians to consider phaeohyphomycosis in the differential diagnosis for recurrent nodulocystic lesions in immunosuppressed patients and to recognize that presentations may span many years.
- Bhardwaj S, Capoor MR, Kolte S, et al. Phaeohyphomycosis due to Exophiala jeanselmei: an emerging pathogen in India—case report and review. Mycopathologia. 2016;181:279-284.
- Isa-Isa R, Garcia C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Tirico MCCP, Neto CF, Cruz LL, et al. Clinical spectrum of phaeohyphomycosis in solid organ transplant recipients. JAAD Case Rep. 2016;2:465-469.
- Revankar SG, Patterson JE, Sutton DA, et al. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin Infect Dis. 2002;34:467-476.
- Revankar SG, Baddley JW, Chen SC-A, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200.
- Oberlin KE, Nichols AJ, Rosa R, et al. Phaeohyphomycosis due to Exophiala infections in solid organ transplant recipients: case report and literature review [published online June 26, 2017]. Transpl Infect Dis. 2017;19. doi:10.1111/tid.12723.
- Shirbur S, Telkar S, Goudar B, et al. Recurrent phaeohyphomycosis: a case report. J Clin Diagn Res. 2013;7:2015-2016.
- Li D-M, Li R-Y, de Hoog GS, et al. Fatal Exophiala infections in China, with a report of seven cases. Mycoses. 2011;54:E136-E142.
- Bhardwaj S, Capoor MR, Kolte S, et al. Phaeohyphomycosis due to Exophiala jeanselmei: an emerging pathogen in India—case report and review. Mycopathologia. 2016;181:279-284.
- Isa-Isa R, Garcia C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Tirico MCCP, Neto CF, Cruz LL, et al. Clinical spectrum of phaeohyphomycosis in solid organ transplant recipients. JAAD Case Rep. 2016;2:465-469.
- Revankar SG, Patterson JE, Sutton DA, et al. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin Infect Dis. 2002;34:467-476.
- Revankar SG, Baddley JW, Chen SC-A, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200.
- Oberlin KE, Nichols AJ, Rosa R, et al. Phaeohyphomycosis due to Exophiala infections in solid organ transplant recipients: case report and literature review [published online June 26, 2017]. Transpl Infect Dis. 2017;19. doi:10.1111/tid.12723.
- Shirbur S, Telkar S, Goudar B, et al. Recurrent phaeohyphomycosis: a case report. J Clin Diagn Res. 2013;7:2015-2016.
- Li D-M, Li R-Y, de Hoog GS, et al. Fatal Exophiala infections in China, with a report of seven cases. Mycoses. 2011;54:E136-E142.
Practice Points
- Phaeohyphomycosis is an infection with dematiaceous fungi that most commonly affects immunosuppressed patients.
- Subcutaneous phaeohyphomycosis may present with nodulocystic lesions that recur over the course of years.
- Tissue fungal culture should be obtained when the diagnosis is suspected, as the risk for dissemination is related to the culprit organism.
- Surgical excision with close follow-up may be an appropriate management strategy for patients on immunosuppressive medications to avoid interactions with azole therapy.
Scalp Arteriovenous Fistula With Intracranial Communication
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
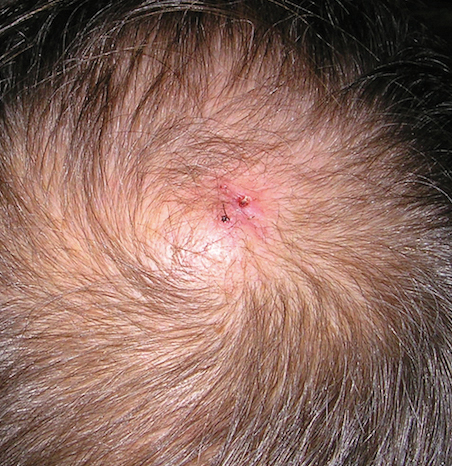
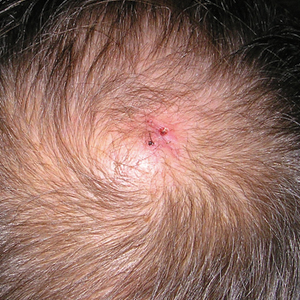
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
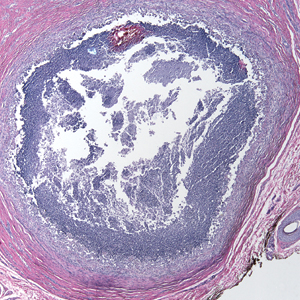
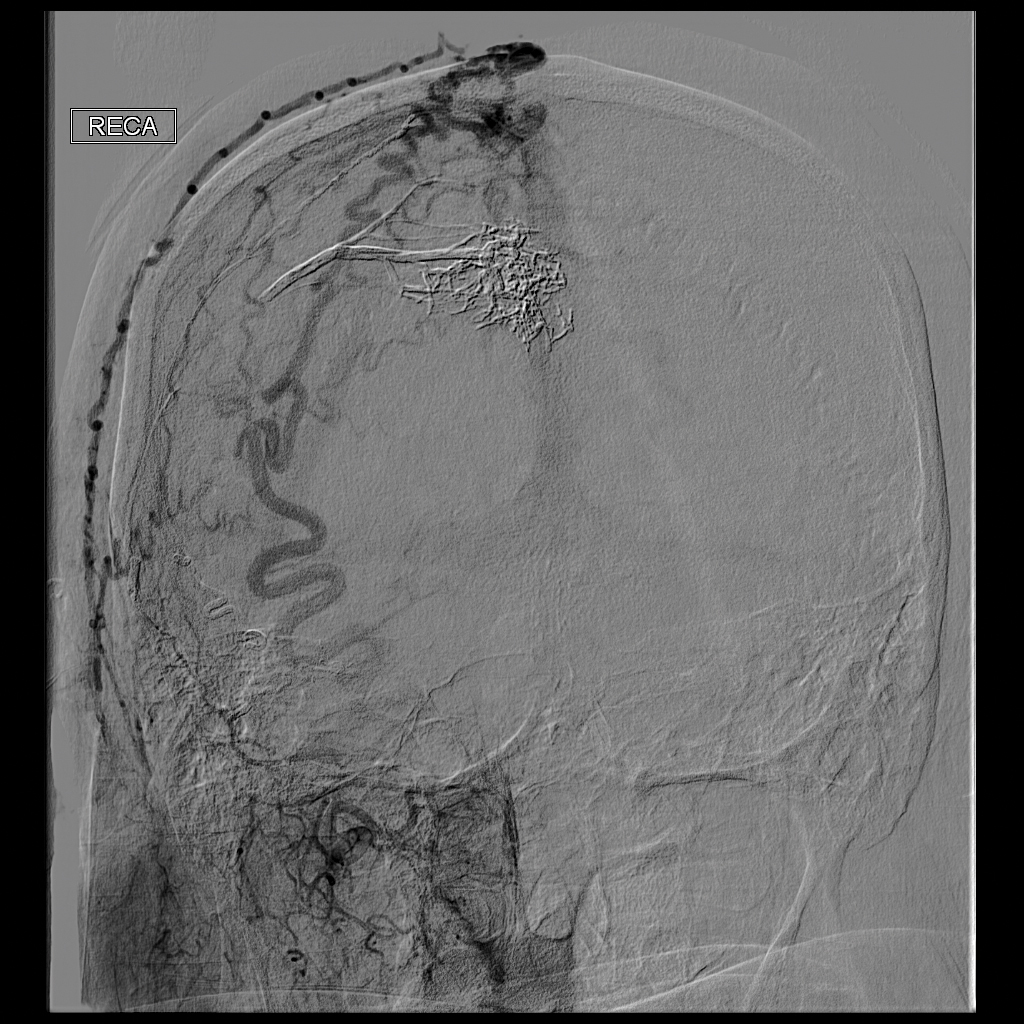
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
Practice Points
- Scalp arteriovenous fistulas may be traumatic or spontaneous and present as either an innocuous-looking scalp nodule or as a progressively enlarging pulsatile mass on the scalp.
- Clinical detection followed by appropriate imaging and referral to neurosurgery or interventional neuroradiology is vital to patient safety.
Nail Unit Squamous Cell Carcinoma: Updates on Diagnosis, Surgical Approach, and the Use of Mohs Micrographic Surgery
Nail unit squamous cell carcinoma (NSCC) is a malignant neoplasm that can arise from any part of the nail unit. Diagnosis often is delayed due to its clinical presentation mimicking benign conditions such as onychomycosis, warts, and paronychia. Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion. It is imperative for dermatologists who are early in their training to recognize this entity and refer for treatment. Many approaches have been used to treat NSCC, including wide local excision, digital amputation, cryotherapy, topical modalities, and recently Mohs micrographic surgery (MMS). This article provides an overview of the clinical presentation and diagnosis of NSCC, the role of human papillomavirus (HPV) in NSCC pathogenesis, and the evidence supporting surgical management.
NSCC Clinical Presentation and Diagnosis
Nail unit squamous cell carcinoma is a malignant neoplasm that can arise from any part of the nail unit including the nail bed, matrix, groove, and nail fold.1 Although NSCC is the most common malignant nail neoplasm, its diagnosis often is delayed partly due to the clinical presentation of NSCC mimicking benign conditions such as onychomycosis, warts, and paronychia.2,3 Nail unit SCC most commonly is mistaken for verruca vulgaris, and thus it is important to exclude malignancy in nonresolving verrucae of the fingernails or toenails. Another reason for a delay in the diagnosis is the painless and often asymptomatic presentation of this tumor, which keeps patients from seeking care.4 While evaluating a subungual lesion, dermatologists should keep in mind red flags that would prompt a biopsy to rule out NSCC (Table 1), including chronic nonhealing lesions, nail plate nodularity, known history of infection with HPV types 16 and 18, history of radiation or arsenic exposure, and immunosuppression. Table 2 lists the differential diagnosis of a persisting or nonhealing subungual tumor.


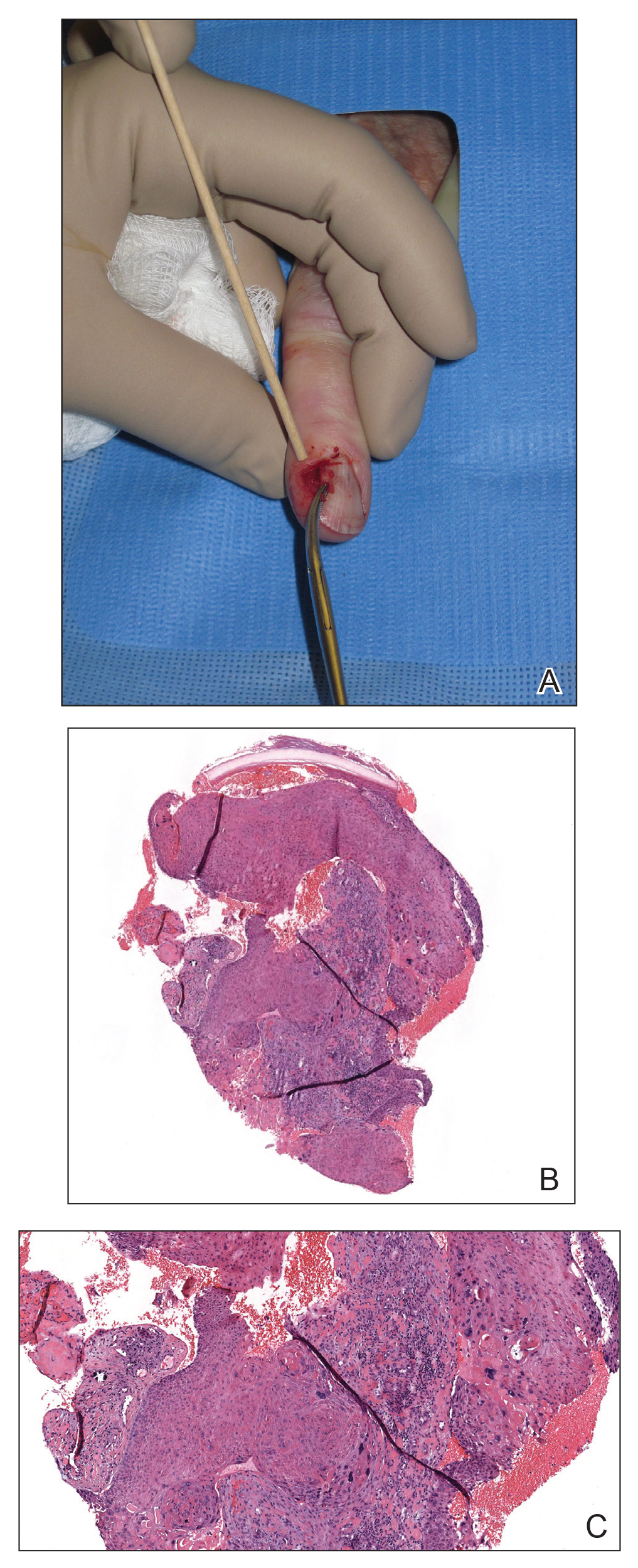
Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion.5 Based on several reports, NSCC more commonly is found in middle-aged and older individuals, has a male predilection, and more often is seen on fingernails than toenails.1,2,6 Figure A shows an example of the clinical presentation of NSCC affecting the right thumb.
Although there often is a delay in the presentation and biopsy of NSCC, no correlation has been observed between time to biopsy and rate of disease invasion and recurrence.7 Nevertheless, Starace et al7 noted that a low threshold for biopsy of nail unit lesions is necessary. It is recommended to perform a deep shave or a nail matrix biopsy, especially if matrical involvement is suspected.8 Patients should be closely followed after a diagnosis of NSCC is made, especially if they are immunocompromised or have genetic skin cancer syndromes, as multiple NSCCs can occur in the same individual.9 For instance, one report discussed a patient with xeroderma pigmentosum who developed 3 separate NSCCs. Interestingly, in this patient, the authors suspected HPV as a cause for the field cancerization, as 2 of 3 NSCCs were noted on initial histopathology to have arisen from verrucae.10
Histologic Features
A biopsy from an NSCC tumor shows features similar to cutaneous SCC in the affected areas (ie, nail bed, nail matrix, nail groove, nail fold). Characteristic histologic findings include tongues or whorls of atypical squamous epithelium that invade deeply into the dermis.11 The cells appear as atypical keratinocytes, exhibit distinct intracellular bridges, and possess hyperchromatic and pleomorphic nuclei with dyskeratosis and keratin pearls within the dermis.12 Immunoperoxidase staining for cytokeratin AE1/AE3 can be helpful to confirm the diagnosis and assess whether the depth of invasion involves the bone.13 Figures B and C demonstrate the histopathology of NSCC biopsied from the tumor shown in Figure A.
Role of HPV in NSCC Pathogenesis
There is no clear pathogenic etiology for NSCC; however, there have been some reports of HPV as a risk factor. Shimizu et al14 reviewed 136 cases of HPV-associated NSCC and found that half of the cases were associated with high-risk HPV. They also found that 24% of the patients with NSCC had a history of other HPV-associated diseases. As such, the authors hypothesized that there is a possibility for genitodigital HPV transmission and that NSCC could be a reservoir for sexually transmitted high-risk HPV.14 Other risk factors are radiation exposure, chemical insult, and chronic trauma.15 The higher propensity for fingernails likely is reflective of the role of UV light exposure and infection with HPV in the development of these tumors.14,15
Several nonsurgical approaches have been suggested to treat NSCC, including topical agents, cryotherapy, CO2 laser, and photodynamic therapy.3,16 Unfortunately, there are no large case series to demonstrate the cure rate or effectiveness of these methods.17 In one study, the authors did not recommend use of photodynamic therapy or topical modalities such as imiquimod cream 5% or fluorouracil cream 5% as first-line treatments of NSCC due to the difficulty in ensuring complete treatment of the sulci of the lateral and proximal nail folds.18
More evidence in the literature supports surgical approaches, including wide local excision, MMS, and digital amputation. Clinicians should consider relapse rates and the impact on digital functioning when choosing a surgical approach.
For wide local excisions, the most common approach is en bloc excision of the nail unit including the lateral nail folds, the proximal nail fold, and the distal nail fold. The excision starts with a transverse incision on the base of the distal phalanx, which is then prolonged laterally and distally to the distal nail fold down to the bone. After the incision is made to the depth of the bone, the matrical horns are destroyed by electrocoagulation, and the defect is closed either by a full-thickness skin graft or secondary intent.19
Topin-Ruiz et al19 followed patients with biopsy-proven NSCC without bone invasion who underwent en bloc excision followed by full-thickness skin graft. In their consecutive series of 55 patients with 5 years of follow-up, the rate of recurrence was only 4%. There was a low rate of complications including graft infection, delayed wound healing, and severe pain in a small percentage of patients. They also reported a high patient satisfaction rate.19 Due to the low recurrence rate, this study suggested that total excision of the nail unit followed by a full-thickness skin graft is a safe and efficient treatment of NSCC without bone involvement. Similarly, in another case series, wide local excision of the entire nail apparatus had a relapse rate of only 5%, in contrast to partial excision of the nail unit with a relapse of 56%.20 These studies suggest that wide nail unit excision is an acceptable and effective approach; however, in cases in which invasion cannot be ruled out, histologic clearance would be a reasonable approach.21 As such, several case series demonstrated the merits of MMS for NSCC. de Berker et al22 reported 8 patients with NSCC treated using slow MMS and showed tumor clearance after a mean of 3 stages over a mean period of 6.9 days. In all cases, the wounds were allowed to heal by secondary intention, and the distal phalanx was preserved. During a mean follow-up period of 3.1 years, no recurrence was seen, and involved digits remained functional.22
Other studies tested the efficacy of MMS for NSCC. Young et al23 reported the outcomes of 14 NSCC cases treated with MMS. In their case series, they found that the mean number of MMS surgical stages required to achieve histologic clearance was 2, while the mean number of tissue sections was 4.23 All cases were allowed to heal by secondary intent with excellent outcomes, except for 1 patient who received primary closure of a small defect. They reported a 78% cure rate with an average time to recurrence of 47 months.23 In a series of 42 cases of NSCC treated with MMS, Gou et al17 noted a cure rate close to 93%. In their study, recurrences were observed in only 3 patients (7.1%). These recurrent cases were then successfully treated with another round of MMS.17 This study’s cure rate was comparable to the cure rate of MMS for SCC in other cutaneous areas. Goldminz and Bennett24 demonstrated a cure rate of 92% in their case series of 25 patients. Two patients developed recurrent disease and were treated again with MMS resulting in no subsequent recurrence. In this study, the authors allowed all defects to heal by secondary intention and found that there were excellent cosmetic and functional outcomes.24 Dika et al25 evaluated the long-term effectiveness of MMS in the treatment of NSCC, in particular its ability to reduce the number of digital amputations. Fifteen patients diagnosed with NSCC were treated with MMS as the first-line surgical approach and were followed for 2 to 5 years. They found that in utilizing MMS, they were able to avoid amputations in 13 of 15 cases with no recurrence in any of these tumors. Two cases, however, still required amputation of the distal phalanx.25
Although these studies suggest that MMS achieves a high cure rate ranging from 78% to 93%, it is not yet clear in the literature whether MMS is superior to wide local excision. More studies and clinical trials comparing these 2 surgical approaches should be performed to identify which surgical approach would be the gold standard for NSCC and which select cases would benefit from MMS as first-line treatment.
Final Thoughts
Nail unit SCC is one of the most common nail unit malignancies and can mimic several benign entities. Dermatologists who are early in their training should consider biopsy of subungual lesions with certain red flags (Table 1). It is important to diagnose NSCC for early intervention. Referral for wide local excision or MMS would be ideal. There are data in the literature supporting both surgical approaches as being effective; however, there are no trials comparing both approaches. Distal amputation should be considered as a last resort when wide local excision is not reasonable or when MMS fails to achieve clear margins, thereby reducing unnecessary amputations and patient morbidity.17
- Dika E, Starace M, Patrizi A, et al. Squamous cell carcinoma of the nail unit: a clinical histopathologic study and a proposal for classification. Dermatol Surg. 2019;45:365-370.
- Lee TM, Jo G, Kim M, et al. Squamous cell carcinoma of the nail unit: a retrospective review of 19 cases in Asia and comparative review of Western literature. Int J Dermatol. 2019;58:428-432.
- Tambe SA, Patil PD, Saple DG, et al. Squamous cell carcinoma of the nail bed: the great mimicker. J Cutan Aesthet Surg. 2017;10:59-60.
- Perrin C. Tumors of the nail unit. a review. part II: acquired localized longitudinal pachyonychia and masked nail tumors. Am J Dermatopathol. 2013;35:693-712.
- Li PF, Zhu N, Lu H. Squamous cell carcinoma of the nail bed: a case report. World J Clin Cases. 2019;7:3590-3594.
- Kaul S, Singal A, Grover C, et al. Clinical and histological spectrum of nail psoriasis: a cross-sectional study. J Cutan Pathol. 2018;45:824-830.
- Starace M, Alessandrini A, Dika E, et al. Squamous cell carcinoma of the nail unit. Dermatol Pract Concept. 2018;8:238-244.
- Kelly KJ, Kalani AD, Storrs S, et al. Subungual squamous cell carcinoma of the toe: working toward a standardized therapeutic approach. J Surg Educ. 2008;65:297-301.
- Ormerod E, De Berker D. Nail unit squamous cell carcinoma in people with immunosuppression. Br J Dermatol. 2015;173:701-712.
- Ventéjou S, Bagny K, Waldmeyer J, et al. Skin cancers in patients of skin phototype V or VI with xeroderma pigmentosum type C (XP-C): a retrospective study. Ann Dermatol Venereol. 2019;146:192-203.
- Mikhail GR. Subungual epidermoid carcinoma. J Am Acad Dermatol. 1984;11:291-298.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Kurokawa I, Senba Y, Kakeda M, et al. Cytokeratin expression in subungual squamous cell carcinoma. J Int Med Res. 2006;34:441-443.
- Shimizu A, Kuriyama Y, Hasegawa M, et al. Nail squamous cell carcinoma: a hidden high-risk human papillomavirus reservoir for sexually transmitted infections. J Am Acad Dermatol. 2019;81:1358-1370.
- Tang N, Maloney ME, Clark AH, et al. A retrospective study of nail squamous cell carcinoma at 2 institutions. Dermatol Surg. 2016;42(suppl 1):S8-S17.
- An Q, Zheng S, Zhang L, et al. Subungual squamous cell carcinoma treated by topical photodynamic therapy. Chin Med J (Engl). 2020;133:881-882.
- Gou D, Nijhawan RI, Srivastava D. Mohs micrographic surgery as the standard of care for nail unit squamous cell carcinoma. Dermatol Surg. 2020;46:725-732.
- Dika E, Fanti PA, Patrizi A, et al. Mohs surgery for squamous cell carcinoma of the nail unit: 10 years of experience. Dermatol Surg. 2015;41:1015-1019.
- Topin-Ruiz S, Surinach C, Dalle S, et al. Surgical treatment of subungual squamous cell carcinoma by wide excision of the nail unit and skin graft reconstruction: an evaluation of treatment efficiency and outcomes. JAMA Dermatol. 2017;153:442-448.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Zaiac MN, Weiss E. Mohs micrographic surgery of the nail unit and squamous cell carcinoma. Dermatol Surg. 2001;27:246-251.
- de Berker DA, Dahl MG, Malcolm AJ, et al. Micrographic surgery for subungual squamous cell carcinoma. Br J Plast Surg. 1996;49:414-419.
- Young LC, Tuxen AJ, Goodman G. Mohs’ micrographic surgery as treatment for squamous dysplasia of the nail unit. Australas J Dermatol. 2012;53:123-127.
- Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
- Dika E, Piraccini BM, Balestri R, et al. Mohs surgery for squamous cell carcinoma of the nail: report of 15 cases. our experience and a long-term follow-up. Br J Dermatol. 2012;167:1310-1314.
Nail unit squamous cell carcinoma (NSCC) is a malignant neoplasm that can arise from any part of the nail unit. Diagnosis often is delayed due to its clinical presentation mimicking benign conditions such as onychomycosis, warts, and paronychia. Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion. It is imperative for dermatologists who are early in their training to recognize this entity and refer for treatment. Many approaches have been used to treat NSCC, including wide local excision, digital amputation, cryotherapy, topical modalities, and recently Mohs micrographic surgery (MMS). This article provides an overview of the clinical presentation and diagnosis of NSCC, the role of human papillomavirus (HPV) in NSCC pathogenesis, and the evidence supporting surgical management.
NSCC Clinical Presentation and Diagnosis
Nail unit squamous cell carcinoma is a malignant neoplasm that can arise from any part of the nail unit including the nail bed, matrix, groove, and nail fold.1 Although NSCC is the most common malignant nail neoplasm, its diagnosis often is delayed partly due to the clinical presentation of NSCC mimicking benign conditions such as onychomycosis, warts, and paronychia.2,3 Nail unit SCC most commonly is mistaken for verruca vulgaris, and thus it is important to exclude malignancy in nonresolving verrucae of the fingernails or toenails. Another reason for a delay in the diagnosis is the painless and often asymptomatic presentation of this tumor, which keeps patients from seeking care.4 While evaluating a subungual lesion, dermatologists should keep in mind red flags that would prompt a biopsy to rule out NSCC (Table 1), including chronic nonhealing lesions, nail plate nodularity, known history of infection with HPV types 16 and 18, history of radiation or arsenic exposure, and immunosuppression. Table 2 lists the differential diagnosis of a persisting or nonhealing subungual tumor.
Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion.5 Based on several reports, NSCC more commonly is found in middle-aged and older individuals, has a male predilection, and more often is seen on fingernails than toenails.1,2,6 Figure A shows an example of the clinical presentation of NSCC affecting the right thumb.
Although there often is a delay in the presentation and biopsy of NSCC, no correlation has been observed between time to biopsy and rate of disease invasion and recurrence.7 Nevertheless, Starace et al7 noted that a low threshold for biopsy of nail unit lesions is necessary. It is recommended to perform a deep shave or a nail matrix biopsy, especially if matrical involvement is suspected.8 Patients should be closely followed after a diagnosis of NSCC is made, especially if they are immunocompromised or have genetic skin cancer syndromes, as multiple NSCCs can occur in the same individual.9 For instance, one report discussed a patient with xeroderma pigmentosum who developed 3 separate NSCCs. Interestingly, in this patient, the authors suspected HPV as a cause for the field cancerization, as 2 of 3 NSCCs were noted on initial histopathology to have arisen from verrucae.10
Histologic Features
A biopsy from an NSCC tumor shows features similar to cutaneous SCC in the affected areas (ie, nail bed, nail matrix, nail groove, nail fold). Characteristic histologic findings include tongues or whorls of atypical squamous epithelium that invade deeply into the dermis.11 The cells appear as atypical keratinocytes, exhibit distinct intracellular bridges, and possess hyperchromatic and pleomorphic nuclei with dyskeratosis and keratin pearls within the dermis.12 Immunoperoxidase staining for cytokeratin AE1/AE3 can be helpful to confirm the diagnosis and assess whether the depth of invasion involves the bone.13 Figures B and C demonstrate the histopathology of NSCC biopsied from the tumor shown in Figure A.
Role of HPV in NSCC Pathogenesis
There is no clear pathogenic etiology for NSCC; however, there have been some reports of HPV as a risk factor. Shimizu et al14 reviewed 136 cases of HPV-associated NSCC and found that half of the cases were associated with high-risk HPV. They also found that 24% of the patients with NSCC had a history of other HPV-associated diseases. As such, the authors hypothesized that there is a possibility for genitodigital HPV transmission and that NSCC could be a reservoir for sexually transmitted high-risk HPV.14 Other risk factors are radiation exposure, chemical insult, and chronic trauma.15 The higher propensity for fingernails likely is reflective of the role of UV light exposure and infection with HPV in the development of these tumors.14,15
Several nonsurgical approaches have been suggested to treat NSCC, including topical agents, cryotherapy, CO2 laser, and photodynamic therapy.3,16 Unfortunately, there are no large case series to demonstrate the cure rate or effectiveness of these methods.17 In one study, the authors did not recommend use of photodynamic therapy or topical modalities such as imiquimod cream 5% or fluorouracil cream 5% as first-line treatments of NSCC due to the difficulty in ensuring complete treatment of the sulci of the lateral and proximal nail folds.18
More evidence in the literature supports surgical approaches, including wide local excision, MMS, and digital amputation. Clinicians should consider relapse rates and the impact on digital functioning when choosing a surgical approach.
For wide local excisions, the most common approach is en bloc excision of the nail unit including the lateral nail folds, the proximal nail fold, and the distal nail fold. The excision starts with a transverse incision on the base of the distal phalanx, which is then prolonged laterally and distally to the distal nail fold down to the bone. After the incision is made to the depth of the bone, the matrical horns are destroyed by electrocoagulation, and the defect is closed either by a full-thickness skin graft or secondary intent.19
Topin-Ruiz et al19 followed patients with biopsy-proven NSCC without bone invasion who underwent en bloc excision followed by full-thickness skin graft. In their consecutive series of 55 patients with 5 years of follow-up, the rate of recurrence was only 4%. There was a low rate of complications including graft infection, delayed wound healing, and severe pain in a small percentage of patients. They also reported a high patient satisfaction rate.19 Due to the low recurrence rate, this study suggested that total excision of the nail unit followed by a full-thickness skin graft is a safe and efficient treatment of NSCC without bone involvement. Similarly, in another case series, wide local excision of the entire nail apparatus had a relapse rate of only 5%, in contrast to partial excision of the nail unit with a relapse of 56%.20 These studies suggest that wide nail unit excision is an acceptable and effective approach; however, in cases in which invasion cannot be ruled out, histologic clearance would be a reasonable approach.21 As such, several case series demonstrated the merits of MMS for NSCC. de Berker et al22 reported 8 patients with NSCC treated using slow MMS and showed tumor clearance after a mean of 3 stages over a mean period of 6.9 days. In all cases, the wounds were allowed to heal by secondary intention, and the distal phalanx was preserved. During a mean follow-up period of 3.1 years, no recurrence was seen, and involved digits remained functional.22
Other studies tested the efficacy of MMS for NSCC. Young et al23 reported the outcomes of 14 NSCC cases treated with MMS. In their case series, they found that the mean number of MMS surgical stages required to achieve histologic clearance was 2, while the mean number of tissue sections was 4.23 All cases were allowed to heal by secondary intent with excellent outcomes, except for 1 patient who received primary closure of a small defect. They reported a 78% cure rate with an average time to recurrence of 47 months.23 In a series of 42 cases of NSCC treated with MMS, Gou et al17 noted a cure rate close to 93%. In their study, recurrences were observed in only 3 patients (7.1%). These recurrent cases were then successfully treated with another round of MMS.17 This study’s cure rate was comparable to the cure rate of MMS for SCC in other cutaneous areas. Goldminz and Bennett24 demonstrated a cure rate of 92% in their case series of 25 patients. Two patients developed recurrent disease and were treated again with MMS resulting in no subsequent recurrence. In this study, the authors allowed all defects to heal by secondary intention and found that there were excellent cosmetic and functional outcomes.24 Dika et al25 evaluated the long-term effectiveness of MMS in the treatment of NSCC, in particular its ability to reduce the number of digital amputations. Fifteen patients diagnosed with NSCC were treated with MMS as the first-line surgical approach and were followed for 2 to 5 years. They found that in utilizing MMS, they were able to avoid amputations in 13 of 15 cases with no recurrence in any of these tumors. Two cases, however, still required amputation of the distal phalanx.25
Although these studies suggest that MMS achieves a high cure rate ranging from 78% to 93%, it is not yet clear in the literature whether MMS is superior to wide local excision. More studies and clinical trials comparing these 2 surgical approaches should be performed to identify which surgical approach would be the gold standard for NSCC and which select cases would benefit from MMS as first-line treatment.
Final Thoughts
Nail unit SCC is one of the most common nail unit malignancies and can mimic several benign entities. Dermatologists who are early in their training should consider biopsy of subungual lesions with certain red flags (Table 1). It is important to diagnose NSCC for early intervention. Referral for wide local excision or MMS would be ideal. There are data in the literature supporting both surgical approaches as being effective; however, there are no trials comparing both approaches. Distal amputation should be considered as a last resort when wide local excision is not reasonable or when MMS fails to achieve clear margins, thereby reducing unnecessary amputations and patient morbidity.17
Nail unit squamous cell carcinoma (NSCC) is a malignant neoplasm that can arise from any part of the nail unit. Diagnosis often is delayed due to its clinical presentation mimicking benign conditions such as onychomycosis, warts, and paronychia. Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion. It is imperative for dermatologists who are early in their training to recognize this entity and refer for treatment. Many approaches have been used to treat NSCC, including wide local excision, digital amputation, cryotherapy, topical modalities, and recently Mohs micrographic surgery (MMS). This article provides an overview of the clinical presentation and diagnosis of NSCC, the role of human papillomavirus (HPV) in NSCC pathogenesis, and the evidence supporting surgical management.
NSCC Clinical Presentation and Diagnosis
Nail unit squamous cell carcinoma is a malignant neoplasm that can arise from any part of the nail unit including the nail bed, matrix, groove, and nail fold.1 Although NSCC is the most common malignant nail neoplasm, its diagnosis often is delayed partly due to the clinical presentation of NSCC mimicking benign conditions such as onychomycosis, warts, and paronychia.2,3 Nail unit SCC most commonly is mistaken for verruca vulgaris, and thus it is important to exclude malignancy in nonresolving verrucae of the fingernails or toenails. Another reason for a delay in the diagnosis is the painless and often asymptomatic presentation of this tumor, which keeps patients from seeking care.4 While evaluating a subungual lesion, dermatologists should keep in mind red flags that would prompt a biopsy to rule out NSCC (Table 1), including chronic nonhealing lesions, nail plate nodularity, known history of infection with HPV types 16 and 18, history of radiation or arsenic exposure, and immunosuppression. Table 2 lists the differential diagnosis of a persisting or nonhealing subungual tumor.
Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion.5 Based on several reports, NSCC more commonly is found in middle-aged and older individuals, has a male predilection, and more often is seen on fingernails than toenails.1,2,6 Figure A shows an example of the clinical presentation of NSCC affecting the right thumb.
Although there often is a delay in the presentation and biopsy of NSCC, no correlation has been observed between time to biopsy and rate of disease invasion and recurrence.7 Nevertheless, Starace et al7 noted that a low threshold for biopsy of nail unit lesions is necessary. It is recommended to perform a deep shave or a nail matrix biopsy, especially if matrical involvement is suspected.8 Patients should be closely followed after a diagnosis of NSCC is made, especially if they are immunocompromised or have genetic skin cancer syndromes, as multiple NSCCs can occur in the same individual.9 For instance, one report discussed a patient with xeroderma pigmentosum who developed 3 separate NSCCs. Interestingly, in this patient, the authors suspected HPV as a cause for the field cancerization, as 2 of 3 NSCCs were noted on initial histopathology to have arisen from verrucae.10
Histologic Features
A biopsy from an NSCC tumor shows features similar to cutaneous SCC in the affected areas (ie, nail bed, nail matrix, nail groove, nail fold). Characteristic histologic findings include tongues or whorls of atypical squamous epithelium that invade deeply into the dermis.11 The cells appear as atypical keratinocytes, exhibit distinct intracellular bridges, and possess hyperchromatic and pleomorphic nuclei with dyskeratosis and keratin pearls within the dermis.12 Immunoperoxidase staining for cytokeratin AE1/AE3 can be helpful to confirm the diagnosis and assess whether the depth of invasion involves the bone.13 Figures B and C demonstrate the histopathology of NSCC biopsied from the tumor shown in Figure A.
Role of HPV in NSCC Pathogenesis
There is no clear pathogenic etiology for NSCC; however, there have been some reports of HPV as a risk factor. Shimizu et al14 reviewed 136 cases of HPV-associated NSCC and found that half of the cases were associated with high-risk HPV. They also found that 24% of the patients with NSCC had a history of other HPV-associated diseases. As such, the authors hypothesized that there is a possibility for genitodigital HPV transmission and that NSCC could be a reservoir for sexually transmitted high-risk HPV.14 Other risk factors are radiation exposure, chemical insult, and chronic trauma.15 The higher propensity for fingernails likely is reflective of the role of UV light exposure and infection with HPV in the development of these tumors.14,15
Several nonsurgical approaches have been suggested to treat NSCC, including topical agents, cryotherapy, CO2 laser, and photodynamic therapy.3,16 Unfortunately, there are no large case series to demonstrate the cure rate or effectiveness of these methods.17 In one study, the authors did not recommend use of photodynamic therapy or topical modalities such as imiquimod cream 5% or fluorouracil cream 5% as first-line treatments of NSCC due to the difficulty in ensuring complete treatment of the sulci of the lateral and proximal nail folds.18
More evidence in the literature supports surgical approaches, including wide local excision, MMS, and digital amputation. Clinicians should consider relapse rates and the impact on digital functioning when choosing a surgical approach.
For wide local excisions, the most common approach is en bloc excision of the nail unit including the lateral nail folds, the proximal nail fold, and the distal nail fold. The excision starts with a transverse incision on the base of the distal phalanx, which is then prolonged laterally and distally to the distal nail fold down to the bone. After the incision is made to the depth of the bone, the matrical horns are destroyed by electrocoagulation, and the defect is closed either by a full-thickness skin graft or secondary intent.19
Topin-Ruiz et al19 followed patients with biopsy-proven NSCC without bone invasion who underwent en bloc excision followed by full-thickness skin graft. In their consecutive series of 55 patients with 5 years of follow-up, the rate of recurrence was only 4%. There was a low rate of complications including graft infection, delayed wound healing, and severe pain in a small percentage of patients. They also reported a high patient satisfaction rate.19 Due to the low recurrence rate, this study suggested that total excision of the nail unit followed by a full-thickness skin graft is a safe and efficient treatment of NSCC without bone involvement. Similarly, in another case series, wide local excision of the entire nail apparatus had a relapse rate of only 5%, in contrast to partial excision of the nail unit with a relapse of 56%.20 These studies suggest that wide nail unit excision is an acceptable and effective approach; however, in cases in which invasion cannot be ruled out, histologic clearance would be a reasonable approach.21 As such, several case series demonstrated the merits of MMS for NSCC. de Berker et al22 reported 8 patients with NSCC treated using slow MMS and showed tumor clearance after a mean of 3 stages over a mean period of 6.9 days. In all cases, the wounds were allowed to heal by secondary intention, and the distal phalanx was preserved. During a mean follow-up period of 3.1 years, no recurrence was seen, and involved digits remained functional.22
Other studies tested the efficacy of MMS for NSCC. Young et al23 reported the outcomes of 14 NSCC cases treated with MMS. In their case series, they found that the mean number of MMS surgical stages required to achieve histologic clearance was 2, while the mean number of tissue sections was 4.23 All cases were allowed to heal by secondary intent with excellent outcomes, except for 1 patient who received primary closure of a small defect. They reported a 78% cure rate with an average time to recurrence of 47 months.23 In a series of 42 cases of NSCC treated with MMS, Gou et al17 noted a cure rate close to 93%. In their study, recurrences were observed in only 3 patients (7.1%). These recurrent cases were then successfully treated with another round of MMS.17 This study’s cure rate was comparable to the cure rate of MMS for SCC in other cutaneous areas. Goldminz and Bennett24 demonstrated a cure rate of 92% in their case series of 25 patients. Two patients developed recurrent disease and were treated again with MMS resulting in no subsequent recurrence. In this study, the authors allowed all defects to heal by secondary intention and found that there were excellent cosmetic and functional outcomes.24 Dika et al25 evaluated the long-term effectiveness of MMS in the treatment of NSCC, in particular its ability to reduce the number of digital amputations. Fifteen patients diagnosed with NSCC were treated with MMS as the first-line surgical approach and were followed for 2 to 5 years. They found that in utilizing MMS, they were able to avoid amputations in 13 of 15 cases with no recurrence in any of these tumors. Two cases, however, still required amputation of the distal phalanx.25
Although these studies suggest that MMS achieves a high cure rate ranging from 78% to 93%, it is not yet clear in the literature whether MMS is superior to wide local excision. More studies and clinical trials comparing these 2 surgical approaches should be performed to identify which surgical approach would be the gold standard for NSCC and which select cases would benefit from MMS as first-line treatment.
Final Thoughts
Nail unit SCC is one of the most common nail unit malignancies and can mimic several benign entities. Dermatologists who are early in their training should consider biopsy of subungual lesions with certain red flags (Table 1). It is important to diagnose NSCC for early intervention. Referral for wide local excision or MMS would be ideal. There are data in the literature supporting both surgical approaches as being effective; however, there are no trials comparing both approaches. Distal amputation should be considered as a last resort when wide local excision is not reasonable or when MMS fails to achieve clear margins, thereby reducing unnecessary amputations and patient morbidity.17
- Dika E, Starace M, Patrizi A, et al. Squamous cell carcinoma of the nail unit: a clinical histopathologic study and a proposal for classification. Dermatol Surg. 2019;45:365-370.
- Lee TM, Jo G, Kim M, et al. Squamous cell carcinoma of the nail unit: a retrospective review of 19 cases in Asia and comparative review of Western literature. Int J Dermatol. 2019;58:428-432.
- Tambe SA, Patil PD, Saple DG, et al. Squamous cell carcinoma of the nail bed: the great mimicker. J Cutan Aesthet Surg. 2017;10:59-60.
- Perrin C. Tumors of the nail unit. a review. part II: acquired localized longitudinal pachyonychia and masked nail tumors. Am J Dermatopathol. 2013;35:693-712.
- Li PF, Zhu N, Lu H. Squamous cell carcinoma of the nail bed: a case report. World J Clin Cases. 2019;7:3590-3594.
- Kaul S, Singal A, Grover C, et al. Clinical and histological spectrum of nail psoriasis: a cross-sectional study. J Cutan Pathol. 2018;45:824-830.
- Starace M, Alessandrini A, Dika E, et al. Squamous cell carcinoma of the nail unit. Dermatol Pract Concept. 2018;8:238-244.
- Kelly KJ, Kalani AD, Storrs S, et al. Subungual squamous cell carcinoma of the toe: working toward a standardized therapeutic approach. J Surg Educ. 2008;65:297-301.
- Ormerod E, De Berker D. Nail unit squamous cell carcinoma in people with immunosuppression. Br J Dermatol. 2015;173:701-712.
- Ventéjou S, Bagny K, Waldmeyer J, et al. Skin cancers in patients of skin phototype V or VI with xeroderma pigmentosum type C (XP-C): a retrospective study. Ann Dermatol Venereol. 2019;146:192-203.
- Mikhail GR. Subungual epidermoid carcinoma. J Am Acad Dermatol. 1984;11:291-298.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Kurokawa I, Senba Y, Kakeda M, et al. Cytokeratin expression in subungual squamous cell carcinoma. J Int Med Res. 2006;34:441-443.
- Shimizu A, Kuriyama Y, Hasegawa M, et al. Nail squamous cell carcinoma: a hidden high-risk human papillomavirus reservoir for sexually transmitted infections. J Am Acad Dermatol. 2019;81:1358-1370.
- Tang N, Maloney ME, Clark AH, et al. A retrospective study of nail squamous cell carcinoma at 2 institutions. Dermatol Surg. 2016;42(suppl 1):S8-S17.
- An Q, Zheng S, Zhang L, et al. Subungual squamous cell carcinoma treated by topical photodynamic therapy. Chin Med J (Engl). 2020;133:881-882.
- Gou D, Nijhawan RI, Srivastava D. Mohs micrographic surgery as the standard of care for nail unit squamous cell carcinoma. Dermatol Surg. 2020;46:725-732.
- Dika E, Fanti PA, Patrizi A, et al. Mohs surgery for squamous cell carcinoma of the nail unit: 10 years of experience. Dermatol Surg. 2015;41:1015-1019.
- Topin-Ruiz S, Surinach C, Dalle S, et al. Surgical treatment of subungual squamous cell carcinoma by wide excision of the nail unit and skin graft reconstruction: an evaluation of treatment efficiency and outcomes. JAMA Dermatol. 2017;153:442-448.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Zaiac MN, Weiss E. Mohs micrographic surgery of the nail unit and squamous cell carcinoma. Dermatol Surg. 2001;27:246-251.
- de Berker DA, Dahl MG, Malcolm AJ, et al. Micrographic surgery for subungual squamous cell carcinoma. Br J Plast Surg. 1996;49:414-419.
- Young LC, Tuxen AJ, Goodman G. Mohs’ micrographic surgery as treatment for squamous dysplasia of the nail unit. Australas J Dermatol. 2012;53:123-127.
- Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
- Dika E, Piraccini BM, Balestri R, et al. Mohs surgery for squamous cell carcinoma of the nail: report of 15 cases. our experience and a long-term follow-up. Br J Dermatol. 2012;167:1310-1314.
- Dika E, Starace M, Patrizi A, et al. Squamous cell carcinoma of the nail unit: a clinical histopathologic study and a proposal for classification. Dermatol Surg. 2019;45:365-370.
- Lee TM, Jo G, Kim M, et al. Squamous cell carcinoma of the nail unit: a retrospective review of 19 cases in Asia and comparative review of Western literature. Int J Dermatol. 2019;58:428-432.
- Tambe SA, Patil PD, Saple DG, et al. Squamous cell carcinoma of the nail bed: the great mimicker. J Cutan Aesthet Surg. 2017;10:59-60.
- Perrin C. Tumors of the nail unit. a review. part II: acquired localized longitudinal pachyonychia and masked nail tumors. Am J Dermatopathol. 2013;35:693-712.
- Li PF, Zhu N, Lu H. Squamous cell carcinoma of the nail bed: a case report. World J Clin Cases. 2019;7:3590-3594.
- Kaul S, Singal A, Grover C, et al. Clinical and histological spectrum of nail psoriasis: a cross-sectional study. J Cutan Pathol. 2018;45:824-830.
- Starace M, Alessandrini A, Dika E, et al. Squamous cell carcinoma of the nail unit. Dermatol Pract Concept. 2018;8:238-244.
- Kelly KJ, Kalani AD, Storrs S, et al. Subungual squamous cell carcinoma of the toe: working toward a standardized therapeutic approach. J Surg Educ. 2008;65:297-301.
- Ormerod E, De Berker D. Nail unit squamous cell carcinoma in people with immunosuppression. Br J Dermatol. 2015;173:701-712.
- Ventéjou S, Bagny K, Waldmeyer J, et al. Skin cancers in patients of skin phototype V or VI with xeroderma pigmentosum type C (XP-C): a retrospective study. Ann Dermatol Venereol. 2019;146:192-203.
- Mikhail GR. Subungual epidermoid carcinoma. J Am Acad Dermatol. 1984;11:291-298.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Kurokawa I, Senba Y, Kakeda M, et al. Cytokeratin expression in subungual squamous cell carcinoma. J Int Med Res. 2006;34:441-443.
- Shimizu A, Kuriyama Y, Hasegawa M, et al. Nail squamous cell carcinoma: a hidden high-risk human papillomavirus reservoir for sexually transmitted infections. J Am Acad Dermatol. 2019;81:1358-1370.
- Tang N, Maloney ME, Clark AH, et al. A retrospective study of nail squamous cell carcinoma at 2 institutions. Dermatol Surg. 2016;42(suppl 1):S8-S17.
- An Q, Zheng S, Zhang L, et al. Subungual squamous cell carcinoma treated by topical photodynamic therapy. Chin Med J (Engl). 2020;133:881-882.
- Gou D, Nijhawan RI, Srivastava D. Mohs micrographic surgery as the standard of care for nail unit squamous cell carcinoma. Dermatol Surg. 2020;46:725-732.
- Dika E, Fanti PA, Patrizi A, et al. Mohs surgery for squamous cell carcinoma of the nail unit: 10 years of experience. Dermatol Surg. 2015;41:1015-1019.
- Topin-Ruiz S, Surinach C, Dalle S, et al. Surgical treatment of subungual squamous cell carcinoma by wide excision of the nail unit and skin graft reconstruction: an evaluation of treatment efficiency and outcomes. JAMA Dermatol. 2017;153:442-448.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Zaiac MN, Weiss E. Mohs micrographic surgery of the nail unit and squamous cell carcinoma. Dermatol Surg. 2001;27:246-251.
- de Berker DA, Dahl MG, Malcolm AJ, et al. Micrographic surgery for subungual squamous cell carcinoma. Br J Plast Surg. 1996;49:414-419.
- Young LC, Tuxen AJ, Goodman G. Mohs’ micrographic surgery as treatment for squamous dysplasia of the nail unit. Australas J Dermatol. 2012;53:123-127.
- Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
- Dika E, Piraccini BM, Balestri R, et al. Mohs surgery for squamous cell carcinoma of the nail: report of 15 cases. our experience and a long-term follow-up. Br J Dermatol. 2012;167:1310-1314.
Resident Pearls
- The diagnosis of nail unit squamous cell carcinoma often is delayed due to its clinical presentation, which frequently mimics benign nail conditions.
- Treatment includes wide local excision, Mohs micrographic surgery, digital amputation, cryotherapy, and topical modalities.

Oral Hairy Leukoplakia Associated With the Use of Adalimumab
To the Editor:
Oral hairy leukoplakia (OHL) is an Epstein-Barr virus (EBV)–mediated mucocutaneous disease that often involves the lingual epithelium. The lateral portions of the tongue are the most commonly affected sites. The lesions often are described as asymptomatic, white, corrugated patches or plaques that are unable to be scraped off.1 Oral hairy leukoplakia was first identified in 1984 and was considered to be associated with AIDS.2 An association between the presence of OHL and the degree of immunosuppression as well as the severity of human immunodeficiency virus (HIV) has been reported.3 Although OHL initially was considered to be pathognomonic for HIV, it has since been described in multiple other immunosuppressive conditions.4 Numerous medical conditions and combinations of immunosuppressive medications have been associated with OHL in patients who were HIV negative.5
Adalimumab is an injectable human IgG1 recombinant antibody to tumor necrosis factor α (TNF-α).6 It currently is approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn disease, ulcerative colitis, noninfectious uveitis, hidradenitis suppurativa, and plaque psoriasis.7 We report a case of OHL associated with the use of adalimumab.
A 47-year-old woman initially presented with chronic plaque-type psoriasis. Her medical history was notable for bipolar disorder, migraines, hypertension, and tobacco use. The patient’s psoriasis initially was well controlled on a regimen of topical steroids and methotrexate; however, methotrexate was stopped after 2.5 years due to a mildly elevated alanine aminotransferase level, as well as an abnormal liver biopsy showing mildly active (grade 1 of 3) steatohepatitis with portal chronic inflammation, pericellular fibrosis, and portal and focal periportal fibrosis (stage 1-2 of 4). The patient and her dermatologist were uncomfortable continuing methotrexate with these findings. After baseline screening including a negative purified protein derivative skin test, adalimumab was initiated. A loading dose of 80 mg subcutaneously (SQ) was given, followed by adalimumab 40 mg SQ 1 week later and 40 mg every other week as maintenance.
The patient’s psoriasis was well controlled with adalimumab for 22 months, but she then developed a thin white plaque on the right lateral tongue (Figure 1). An incisional biopsy of the tongue performed by an oral surgeon revealed hyperkeratosis with Candida colonization and viral cytopathic effect (Figure 2). An EBV DNA in situ hybridization stain revealed focal positivity within these cells (Figure 3), leading to a diagnosis of OHL. Laboratory evaluation demonstrated a normal complete blood cell count with differential and liver panel as well as a negative HIV test. The patient otherwise felt well and denied fevers, lymphadenopathy, and weight loss.
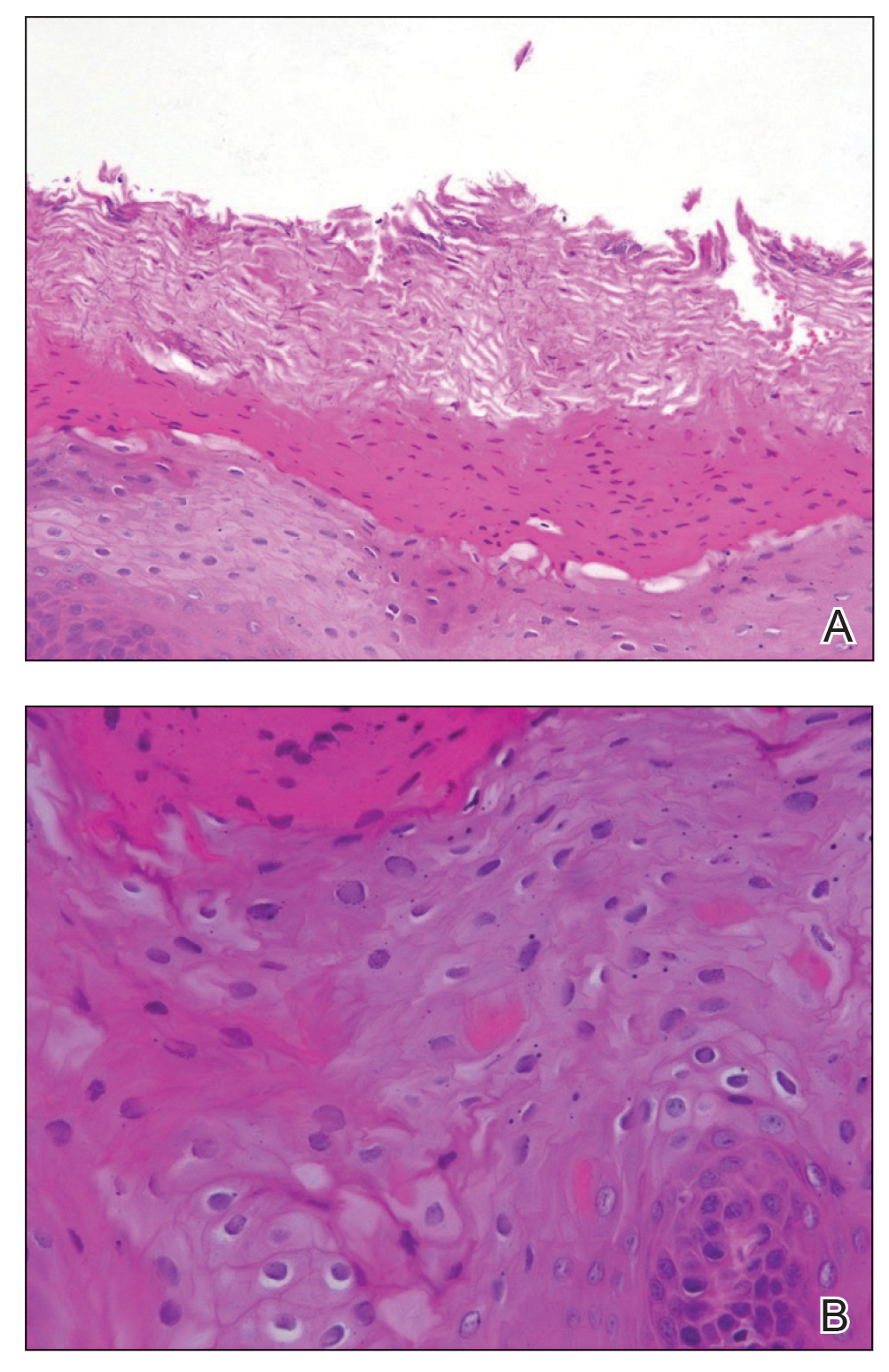
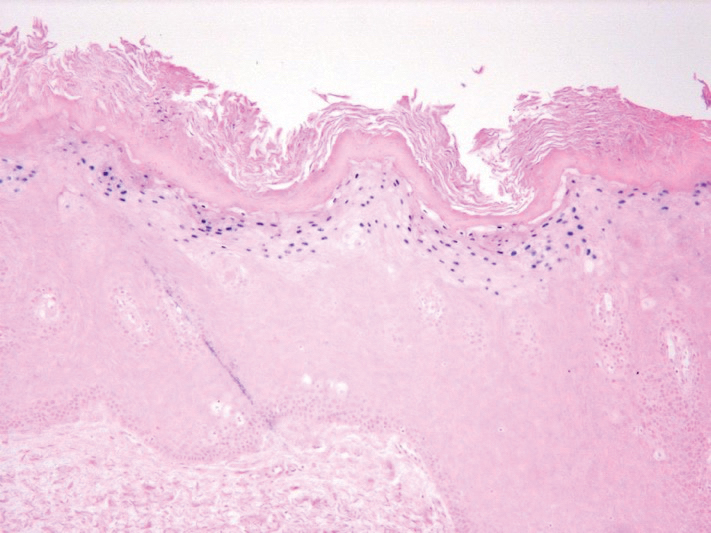
We consulted with an infectious disease and immunodeficiency specialist regarding the patient’s case. Before conducting further evaluation beyond HIV screening for immunodeficiency states, adalimumab was discontinued to see if the OHL would spontaneously resolve. Three months after discontinuation of adalimumab, the white plaque on the right lateral tongue was notably improved. The OHL continued to disappear and was completely resolved 1 year after discontinuation of adalimumab. The patient’s psoriasis had subsequently remained well controlled with diet and weight loss, smoking cessation, topical steroids, and apremilast without any recurrence of the OHL.
Oral hairy leukoplakia is associated with upregulated EBV replication and EBV-encoded proteins such as latent membrane protein 1.2 It often presents as white or gray patches on the lateral lingual margins with prominent folds and/or projections, giving a shaggy appearance. Oral hairy leukoplakia often is specific for HIV infection and rarely is associated with other immunodeficiencies.2 Prasad and Bilodeau5 performed a literature review of medical conditions and immunosuppressive medications associated with OHL in patients without HIV. Allogeneic transplant was associated with the highest incidence of OHL in HIV-negative patients (59.2% [45/76]).5 Various combinations of immunosuppressive medications (eg, prednisone, cyclosporine, azathioprine) also may be implicated in cases of HIV-negative patients with OHL. A case of OHL also has been reported with long-standing use of inhaled corticosteroids in an immunocompetent, HIV-negative patient.6 Another case was reported with long-term use of the aromatic antiepileptic lamotrigine, which resolved once stopping the medication.8 Although EBV is an oncovirus and has been associated with lymphoproliferative disorders and nasopharyngeal carcinoma, OHL is not considered to be a premalignant lesion.7 Despite the strong association between OHL and HIV, our patient was HIV negative. The only immunocompromising factor in our patient was the use of adalimumab to treat psoriasis. We did not conduct further testing for immunodeficiency states because the OHL spontaneously resolved when the adalimumab was discontinued.
PubMed and Ovid searches of articles indexed for MEDLINE using the terms adalimumab and oral hairy leukoplakia as well as TNF-alpha inhibitor and oral hairy leukoplakia with humans and English language as limitations revealed that no cases have been reported in the literature demonstrating an association between OHL and adalimumab or any other TNF-α inhibitor. However, Cetkovska et al9 reported a case of EBV hepatitis and subsequently chronic hepatitis as a complication of infliximab used for the treatment of chronic psoriasis. Because TNF-α and IFN-γ play an important role in controlling viral infections, there is an increased risk for reactivating a viral illness when depleting TNF through pharmacologic measures (ie, adalimumab, infliximab).8 Another case of EBV-associated plasmablastic lymphoma was reported after 1 year of adalimumab use in a patient with Crohn disease. The plasmablastic lymphoma resolved after 4 rounds of chemotherapy.10
The only contraindication for the use of adalimumab is a known hypersensitivity to the drug. Relative contraindications for use of adalimumab include active tuberculosis, demyelinating disease, hematologic diseases (ie, thrombocytopenia, pancytopenia), lymphoma, hepatitis C, and hepatitis B.11 The most common adverse effect of adalimumab is an injection-site reaction. Additional reported adverse effects of TNF-α inhibitors as a class are lymphoma, melanoma, nonmelanoma skin cancer, reactivation of latent tuberculosis, congestive heart failure, autoimmunity, and hematologic toxicity.11
This case demonstrates an association between adalimumab and OHL in an HIV-negative patient. Although the mechanism behind OHL and immunosuppression remains to be elucidated, this association is important to keep in mind when using adalimumab or other TNF-α inhibitors for the treatment of psoriasis or other medical conditions.
- Triantos D, Porter SR, Scully C, et al. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Clin Infect Dis. 1997;25:1392-1396.
- Greenspan D, Greenspan JS, Conant M, et al. Oral “hairy” leucoplakia in male homosexuals: evidence of association with both papillomavirus and a herpes-group virus. Lancet. 1984;2:831-834.
- Glick M, Muzyka BC, Lurie D, et al. Oral manifestations associated with HIV-related disease as marks for immune suppression and AIDS. Oral Surg Oral Med Oral Pathol. 1994;77:344-349.
- Chambers AE, Conn B, Pemberton M, et al. Twenty-first-century oral hair leukoplakia—a non-HIV-associated entity. Oral Surg Oral Med Oral Patho Oral Radiol. 2015;119:326-332.
- Prasad JL, Bilodeau EA. Oral hairy leukoplakia in patients without HIV: presentation of 2 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:E151-E160.
- Moffat M, Jauhar S, Jones ME, et al. Oral hairy leukoplakia in an HIV-negative, immunocompetent patient. Oral Biosci Med. 2005;2:282-284.
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67:396-403.
- Gordins P, Sloan P, Spickett GP, et al. Oral hairy leukoplakia in a patient on long-term anticonvulsant treatment with lamotrigine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:E17-E23.
- Cetkovska P, Lomicova I, Mukensnabl P, et al. Anti-tumour necrosis factor treatment of severe psoriasis complicated by Epstein-Barr virus hepatitis and subsequently by chronic hepatitis. Dermatol Ther. 2015;28:369-372.
- Liu L, Charabaty A, Ozdemirli M. EBV-associated plasmablastic lymphoma in a patient with Crohn’s disease after adalimumab treatment. J Crohns Colitis. 2013;7:E118-E119.
- Humira [package insert]. North Chicago, IL: AbbVie Inc; 2018.
To the Editor:
Oral hairy leukoplakia (OHL) is an Epstein-Barr virus (EBV)–mediated mucocutaneous disease that often involves the lingual epithelium. The lateral portions of the tongue are the most commonly affected sites. The lesions often are described as asymptomatic, white, corrugated patches or plaques that are unable to be scraped off.1 Oral hairy leukoplakia was first identified in 1984 and was considered to be associated with AIDS.2 An association between the presence of OHL and the degree of immunosuppression as well as the severity of human immunodeficiency virus (HIV) has been reported.3 Although OHL initially was considered to be pathognomonic for HIV, it has since been described in multiple other immunosuppressive conditions.4 Numerous medical conditions and combinations of immunosuppressive medications have been associated with OHL in patients who were HIV negative.5
Adalimumab is an injectable human IgG1 recombinant antibody to tumor necrosis factor α (TNF-α).6 It currently is approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn disease, ulcerative colitis, noninfectious uveitis, hidradenitis suppurativa, and plaque psoriasis.7 We report a case of OHL associated with the use of adalimumab.
A 47-year-old woman initially presented with chronic plaque-type psoriasis. Her medical history was notable for bipolar disorder, migraines, hypertension, and tobacco use. The patient’s psoriasis initially was well controlled on a regimen of topical steroids and methotrexate; however, methotrexate was stopped after 2.5 years due to a mildly elevated alanine aminotransferase level, as well as an abnormal liver biopsy showing mildly active (grade 1 of 3) steatohepatitis with portal chronic inflammation, pericellular fibrosis, and portal and focal periportal fibrosis (stage 1-2 of 4). The patient and her dermatologist were uncomfortable continuing methotrexate with these findings. After baseline screening including a negative purified protein derivative skin test, adalimumab was initiated. A loading dose of 80 mg subcutaneously (SQ) was given, followed by adalimumab 40 mg SQ 1 week later and 40 mg every other week as maintenance.
The patient’s psoriasis was well controlled with adalimumab for 22 months, but she then developed a thin white plaque on the right lateral tongue (Figure 1). An incisional biopsy of the tongue performed by an oral surgeon revealed hyperkeratosis with Candida colonization and viral cytopathic effect (Figure 2). An EBV DNA in situ hybridization stain revealed focal positivity within these cells (Figure 3), leading to a diagnosis of OHL. Laboratory evaluation demonstrated a normal complete blood cell count with differential and liver panel as well as a negative HIV test. The patient otherwise felt well and denied fevers, lymphadenopathy, and weight loss.
We consulted with an infectious disease and immunodeficiency specialist regarding the patient’s case. Before conducting further evaluation beyond HIV screening for immunodeficiency states, adalimumab was discontinued to see if the OHL would spontaneously resolve. Three months after discontinuation of adalimumab, the white plaque on the right lateral tongue was notably improved. The OHL continued to disappear and was completely resolved 1 year after discontinuation of adalimumab. The patient’s psoriasis had subsequently remained well controlled with diet and weight loss, smoking cessation, topical steroids, and apremilast without any recurrence of the OHL.
Oral hairy leukoplakia is associated with upregulated EBV replication and EBV-encoded proteins such as latent membrane protein 1.2 It often presents as white or gray patches on the lateral lingual margins with prominent folds and/or projections, giving a shaggy appearance. Oral hairy leukoplakia often is specific for HIV infection and rarely is associated with other immunodeficiencies.2 Prasad and Bilodeau5 performed a literature review of medical conditions and immunosuppressive medications associated with OHL in patients without HIV. Allogeneic transplant was associated with the highest incidence of OHL in HIV-negative patients (59.2% [45/76]).5 Various combinations of immunosuppressive medications (eg, prednisone, cyclosporine, azathioprine) also may be implicated in cases of HIV-negative patients with OHL. A case of OHL also has been reported with long-standing use of inhaled corticosteroids in an immunocompetent, HIV-negative patient.6 Another case was reported with long-term use of the aromatic antiepileptic lamotrigine, which resolved once stopping the medication.8 Although EBV is an oncovirus and has been associated with lymphoproliferative disorders and nasopharyngeal carcinoma, OHL is not considered to be a premalignant lesion.7 Despite the strong association between OHL and HIV, our patient was HIV negative. The only immunocompromising factor in our patient was the use of adalimumab to treat psoriasis. We did not conduct further testing for immunodeficiency states because the OHL spontaneously resolved when the adalimumab was discontinued.
PubMed and Ovid searches of articles indexed for MEDLINE using the terms adalimumab and oral hairy leukoplakia as well as TNF-alpha inhibitor and oral hairy leukoplakia with humans and English language as limitations revealed that no cases have been reported in the literature demonstrating an association between OHL and adalimumab or any other TNF-α inhibitor. However, Cetkovska et al9 reported a case of EBV hepatitis and subsequently chronic hepatitis as a complication of infliximab used for the treatment of chronic psoriasis. Because TNF-α and IFN-γ play an important role in controlling viral infections, there is an increased risk for reactivating a viral illness when depleting TNF through pharmacologic measures (ie, adalimumab, infliximab).8 Another case of EBV-associated plasmablastic lymphoma was reported after 1 year of adalimumab use in a patient with Crohn disease. The plasmablastic lymphoma resolved after 4 rounds of chemotherapy.10
The only contraindication for the use of adalimumab is a known hypersensitivity to the drug. Relative contraindications for use of adalimumab include active tuberculosis, demyelinating disease, hematologic diseases (ie, thrombocytopenia, pancytopenia), lymphoma, hepatitis C, and hepatitis B.11 The most common adverse effect of adalimumab is an injection-site reaction. Additional reported adverse effects of TNF-α inhibitors as a class are lymphoma, melanoma, nonmelanoma skin cancer, reactivation of latent tuberculosis, congestive heart failure, autoimmunity, and hematologic toxicity.11
This case demonstrates an association between adalimumab and OHL in an HIV-negative patient. Although the mechanism behind OHL and immunosuppression remains to be elucidated, this association is important to keep in mind when using adalimumab or other TNF-α inhibitors for the treatment of psoriasis or other medical conditions.
To the Editor:
Oral hairy leukoplakia (OHL) is an Epstein-Barr virus (EBV)–mediated mucocutaneous disease that often involves the lingual epithelium. The lateral portions of the tongue are the most commonly affected sites. The lesions often are described as asymptomatic, white, corrugated patches or plaques that are unable to be scraped off.1 Oral hairy leukoplakia was first identified in 1984 and was considered to be associated with AIDS.2 An association between the presence of OHL and the degree of immunosuppression as well as the severity of human immunodeficiency virus (HIV) has been reported.3 Although OHL initially was considered to be pathognomonic for HIV, it has since been described in multiple other immunosuppressive conditions.4 Numerous medical conditions and combinations of immunosuppressive medications have been associated with OHL in patients who were HIV negative.5
Adalimumab is an injectable human IgG1 recombinant antibody to tumor necrosis factor α (TNF-α).6 It currently is approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn disease, ulcerative colitis, noninfectious uveitis, hidradenitis suppurativa, and plaque psoriasis.7 We report a case of OHL associated with the use of adalimumab.
A 47-year-old woman initially presented with chronic plaque-type psoriasis. Her medical history was notable for bipolar disorder, migraines, hypertension, and tobacco use. The patient’s psoriasis initially was well controlled on a regimen of topical steroids and methotrexate; however, methotrexate was stopped after 2.5 years due to a mildly elevated alanine aminotransferase level, as well as an abnormal liver biopsy showing mildly active (grade 1 of 3) steatohepatitis with portal chronic inflammation, pericellular fibrosis, and portal and focal periportal fibrosis (stage 1-2 of 4). The patient and her dermatologist were uncomfortable continuing methotrexate with these findings. After baseline screening including a negative purified protein derivative skin test, adalimumab was initiated. A loading dose of 80 mg subcutaneously (SQ) was given, followed by adalimumab 40 mg SQ 1 week later and 40 mg every other week as maintenance.
The patient’s psoriasis was well controlled with adalimumab for 22 months, but she then developed a thin white plaque on the right lateral tongue (Figure 1). An incisional biopsy of the tongue performed by an oral surgeon revealed hyperkeratosis with Candida colonization and viral cytopathic effect (Figure 2). An EBV DNA in situ hybridization stain revealed focal positivity within these cells (Figure 3), leading to a diagnosis of OHL. Laboratory evaluation demonstrated a normal complete blood cell count with differential and liver panel as well as a negative HIV test. The patient otherwise felt well and denied fevers, lymphadenopathy, and weight loss.
We consulted with an infectious disease and immunodeficiency specialist regarding the patient’s case. Before conducting further evaluation beyond HIV screening for immunodeficiency states, adalimumab was discontinued to see if the OHL would spontaneously resolve. Three months after discontinuation of adalimumab, the white plaque on the right lateral tongue was notably improved. The OHL continued to disappear and was completely resolved 1 year after discontinuation of adalimumab. The patient’s psoriasis had subsequently remained well controlled with diet and weight loss, smoking cessation, topical steroids, and apremilast without any recurrence of the OHL.
Oral hairy leukoplakia is associated with upregulated EBV replication and EBV-encoded proteins such as latent membrane protein 1.2 It often presents as white or gray patches on the lateral lingual margins with prominent folds and/or projections, giving a shaggy appearance. Oral hairy leukoplakia often is specific for HIV infection and rarely is associated with other immunodeficiencies.2 Prasad and Bilodeau5 performed a literature review of medical conditions and immunosuppressive medications associated with OHL in patients without HIV. Allogeneic transplant was associated with the highest incidence of OHL in HIV-negative patients (59.2% [45/76]).5 Various combinations of immunosuppressive medications (eg, prednisone, cyclosporine, azathioprine) also may be implicated in cases of HIV-negative patients with OHL. A case of OHL also has been reported with long-standing use of inhaled corticosteroids in an immunocompetent, HIV-negative patient.6 Another case was reported with long-term use of the aromatic antiepileptic lamotrigine, which resolved once stopping the medication.8 Although EBV is an oncovirus and has been associated with lymphoproliferative disorders and nasopharyngeal carcinoma, OHL is not considered to be a premalignant lesion.7 Despite the strong association between OHL and HIV, our patient was HIV negative. The only immunocompromising factor in our patient was the use of adalimumab to treat psoriasis. We did not conduct further testing for immunodeficiency states because the OHL spontaneously resolved when the adalimumab was discontinued.
PubMed and Ovid searches of articles indexed for MEDLINE using the terms adalimumab and oral hairy leukoplakia as well as TNF-alpha inhibitor and oral hairy leukoplakia with humans and English language as limitations revealed that no cases have been reported in the literature demonstrating an association between OHL and adalimumab or any other TNF-α inhibitor. However, Cetkovska et al9 reported a case of EBV hepatitis and subsequently chronic hepatitis as a complication of infliximab used for the treatment of chronic psoriasis. Because TNF-α and IFN-γ play an important role in controlling viral infections, there is an increased risk for reactivating a viral illness when depleting TNF through pharmacologic measures (ie, adalimumab, infliximab).8 Another case of EBV-associated plasmablastic lymphoma was reported after 1 year of adalimumab use in a patient with Crohn disease. The plasmablastic lymphoma resolved after 4 rounds of chemotherapy.10
The only contraindication for the use of adalimumab is a known hypersensitivity to the drug. Relative contraindications for use of adalimumab include active tuberculosis, demyelinating disease, hematologic diseases (ie, thrombocytopenia, pancytopenia), lymphoma, hepatitis C, and hepatitis B.11 The most common adverse effect of adalimumab is an injection-site reaction. Additional reported adverse effects of TNF-α inhibitors as a class are lymphoma, melanoma, nonmelanoma skin cancer, reactivation of latent tuberculosis, congestive heart failure, autoimmunity, and hematologic toxicity.11
This case demonstrates an association between adalimumab and OHL in an HIV-negative patient. Although the mechanism behind OHL and immunosuppression remains to be elucidated, this association is important to keep in mind when using adalimumab or other TNF-α inhibitors for the treatment of psoriasis or other medical conditions.
- Triantos D, Porter SR, Scully C, et al. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Clin Infect Dis. 1997;25:1392-1396.
- Greenspan D, Greenspan JS, Conant M, et al. Oral “hairy” leucoplakia in male homosexuals: evidence of association with both papillomavirus and a herpes-group virus. Lancet. 1984;2:831-834.
- Glick M, Muzyka BC, Lurie D, et al. Oral manifestations associated with HIV-related disease as marks for immune suppression and AIDS. Oral Surg Oral Med Oral Pathol. 1994;77:344-349.
- Chambers AE, Conn B, Pemberton M, et al. Twenty-first-century oral hair leukoplakia—a non-HIV-associated entity. Oral Surg Oral Med Oral Patho Oral Radiol. 2015;119:326-332.
- Prasad JL, Bilodeau EA. Oral hairy leukoplakia in patients without HIV: presentation of 2 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:E151-E160.
- Moffat M, Jauhar S, Jones ME, et al. Oral hairy leukoplakia in an HIV-negative, immunocompetent patient. Oral Biosci Med. 2005;2:282-284.
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67:396-403.
- Gordins P, Sloan P, Spickett GP, et al. Oral hairy leukoplakia in a patient on long-term anticonvulsant treatment with lamotrigine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:E17-E23.
- Cetkovska P, Lomicova I, Mukensnabl P, et al. Anti-tumour necrosis factor treatment of severe psoriasis complicated by Epstein-Barr virus hepatitis and subsequently by chronic hepatitis. Dermatol Ther. 2015;28:369-372.
- Liu L, Charabaty A, Ozdemirli M. EBV-associated plasmablastic lymphoma in a patient with Crohn’s disease after adalimumab treatment. J Crohns Colitis. 2013;7:E118-E119.
- Humira [package insert]. North Chicago, IL: AbbVie Inc; 2018.
- Triantos D, Porter SR, Scully C, et al. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Clin Infect Dis. 1997;25:1392-1396.
- Greenspan D, Greenspan JS, Conant M, et al. Oral “hairy” leucoplakia in male homosexuals: evidence of association with both papillomavirus and a herpes-group virus. Lancet. 1984;2:831-834.
- Glick M, Muzyka BC, Lurie D, et al. Oral manifestations associated with HIV-related disease as marks for immune suppression and AIDS. Oral Surg Oral Med Oral Pathol. 1994;77:344-349.
- Chambers AE, Conn B, Pemberton M, et al. Twenty-first-century oral hair leukoplakia—a non-HIV-associated entity. Oral Surg Oral Med Oral Patho Oral Radiol. 2015;119:326-332.
- Prasad JL, Bilodeau EA. Oral hairy leukoplakia in patients without HIV: presentation of 2 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:E151-E160.
- Moffat M, Jauhar S, Jones ME, et al. Oral hairy leukoplakia in an HIV-negative, immunocompetent patient. Oral Biosci Med. 2005;2:282-284.
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67:396-403.
- Gordins P, Sloan P, Spickett GP, et al. Oral hairy leukoplakia in a patient on long-term anticonvulsant treatment with lamotrigine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:E17-E23.
- Cetkovska P, Lomicova I, Mukensnabl P, et al. Anti-tumour necrosis factor treatment of severe psoriasis complicated by Epstein-Barr virus hepatitis and subsequently by chronic hepatitis. Dermatol Ther. 2015;28:369-372.
- Liu L, Charabaty A, Ozdemirli M. EBV-associated plasmablastic lymphoma in a patient with Crohn’s disease after adalimumab treatment. J Crohns Colitis. 2013;7:E118-E119.
- Humira [package insert]. North Chicago, IL: AbbVie Inc; 2018.
Practice Points
- Workup for new-onset oral hairy leukoplakia should include a comprehensive medication history.
- Oral hairy leukoplakia is an uncommon side effect of adalimumab.
Painful Papules on the Arms
The Diagnosis: Piloleiomyoma
Leiomyoma cutis, also known as cutaneous leiomyoma, is a benign smooth muscle tumor first described in 1854.1 Cutaneous leiomyoma is comprised of 3 distinct types that depend on the origin of smooth muscle tumor: piloleiomyoma (arrector pili muscle), angioleiomyoma (tunica media of arteries/veins), and genital leiomyoma (dartos muscle of the scrotum and labia majora, erectile muscle of nipple).2 It affects both sexes equally, though some reports have noted an increased prevalence in females. Piloleiomyomas commonly present on the extensor surfaces of the extremities (solitary) and trunk (multiple).1 Tumors most often present as firm flesh-colored or pink-brown papulonodules. They can be linear, dermatomal, segmental, or diffuse, and often are painful. Clinical differential diagnosis for painful skin tumors is aided by the acronym "BLEND AN EGG": blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor.3 For isolated lesions, surgical excision is the treatment of choice. For numerous lesions in which excision would not be feasible, intralesional corticosteroids, medications (eg, calcium channel blockers, alpha blockers, nitroglycerin), and botulinum toxin have been used for pain relief.4
Notably, multiple cutaneous leiomyomas can be seen in association with uterine leiomyomas in Reed syndrome due to an autosomal-dominant or de novo mutation in the fumarate hydratase gene, FH. Reed syndrome is associated with a lifetime risk for renal cell carcinoma (hereditary leiomyomatosis and renal cell cancer) in 15% of cases with FH mutations.5 In our patient, both immunohistochemical staining and blood testing for FH were performed. Immunohistochemistry revealed notably diminished staining with only weak patchy granular cytoplasmic staining present (Figure 1). Genetic testing revealed heterozygosity for a pathogenic variant of the FH gene, consistent with a diagnosis of Reed syndrome.
Histologically, the differential diagnosis includes other spindle cell tumors, such as dermatofibroma, neurofibroma, and dermatomyofibroma. The histologic appearance varies depending on the type, with piloleiomyoma typically located within the reticular dermis with possible subcutaneous extension. Fascicles of eosinophilic smooth muscle cells in an interlacing arrangement often ramify between neighboring dermal collagen; these smooth muscle cells contain cigar-shaped, blunt-ended nuclei with a perinuclear clear vacuole. Marked epidermal hyperplasia is possible.6 A close association with a nearby hair follicle frequently is noted. Although differentiated smooth muscle cells usually are evident on hematoxylin and eosin, positive staining for smooth muscle actin (SMA) and desmin can aid in diagnosis.7 Immunohistochemical staining for FH has proven to be highly specific (97.6%) with moderate sensitivity (70.0%).8 Angioleiomyomas appear as well-demarcated dermal to subcutaneous tumors composed of smooth muscle cells surrounding thick-walled vaculature.9 Scrotal and vulvar leiomyomas are composed of eosinophilic spindle cells, though vulvar leiomyomas have shown epithelioid differentiation.10 Nipple leiomyomas appear similar to piloleiomyomas on histology with interlacing smooth muscle fiber bundles.
Eccrine spiradenoma is a relatively uncommon adnexal tumor derived from eccrine sweat glands. It most often presents as a small, painful or tender, intradermal nodule (or rarely as nodules) on the head or ventral trunk.11 There is no sexual predilection. It affects adults at any age but most often from 15 to 35 years. Although rare, malignant transformation is possible. Histologically, eccrine spiradenomas appear as a well-demarcated dermal tumor composed of bland basaloid cells with minimal cytoplasm, often with numerous admixed lymphocytes and variably prominent vasculature (Figure 2). Eosinophilic basement membrane material can be seen within or surrounding the nodules of tumor cells. Multiple spiradenomas can occur in the setting of Brooke-Spiegler syndrome, which is an autosomal-dominant disorder due to an inherited mutation in the CYLD gene. Spiradenomas are benign neoplasms, and surgical excision with clear margins is the treatment of choice.12
Dermatofibroma, also known as cutaneous benign fibrous histiocytoma, is a firm, flesh-colored papule or nodule that most often presents on the lower extremities. It typically is seen in women aged 20 to 40 years.13 The etiology is uncertain, and dermatofibromas often spontaneously develop, though there are inconsistent reports of development with local trauma including insect bites and puncture wounds. The dimple sign refers to skin dimpling with lateral pressure.13 Most commonly, dermatofibromas consist of a dermal proliferation of bland fibroblastic cells with entrapment of dermal collagen bundles at the periphery of the tumors (Figure 3). The fibroblastic cells often are paler and less eosinophilic than smooth muscle cells seen in cutaneous leiomyomas, with tapered nuclei that lack a perinuclear vacuole. Admixed histocytes and other inflammatory cells often are present. Overlying epidermal hyperplasia and/or hyperpigmentation also may be present. Numerous histologic variants have been described, including cellular, epithelioid, aneurysmal, atypical, and hemosiderotic types.14 Immunohistochemical stains may show patchy positive staining for SMA, but h-caldesmon and desmin typically are negative.
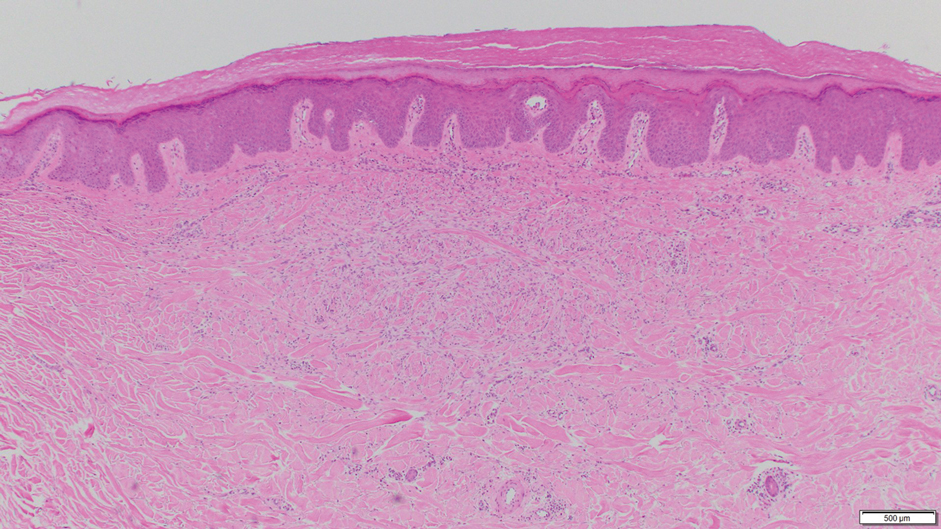
Neurofibroma is a tumor derived from neuromesenchymal tissue with nerve axons. They form through neuromesenchyme (eg, Schwann cells, mast cells, perineural cells, endoneural fibroblast) proliferation. Solitary neurofibromas occur most commonly in adults and have no gender predilection. The most common presentation is an asymptomatic, solitary, soft, flesh-colored papulonodule.15 Clinical variants include pigmented, diffuse, and plexiform, with plexiform neurofibromas almost always being consistent with a diagnosis of neurofibromatosis type 1. Histologically, neurofibromas present as dermal or subcutaneous nodules composed of randomly arranged spindle cells with wavy tapered nuclei within a loose collagenous stroma (Figure 4).16 The spindle cells in neurofibromas will stain positively for S-100 protein and SOX-10 and negatively for SMA and desmin.

Angiolipoma is a benign tumor composed of adipocytes that also contains vasculature.17 The majority of cases are of unknown etiology, though familial cases have been described. They typically present as multiple painful or tender (differentiating from lipomas) subcutaneous swellings over the forearms in individuals aged 20 to 30 years.18 On histopathology, angiolipomas appear as well-circumscribed subcutaneous tumors containing mature adipocytes intermixed with small capillary vessels, some of which contain luminal fibrin thrombi (Figure 5).
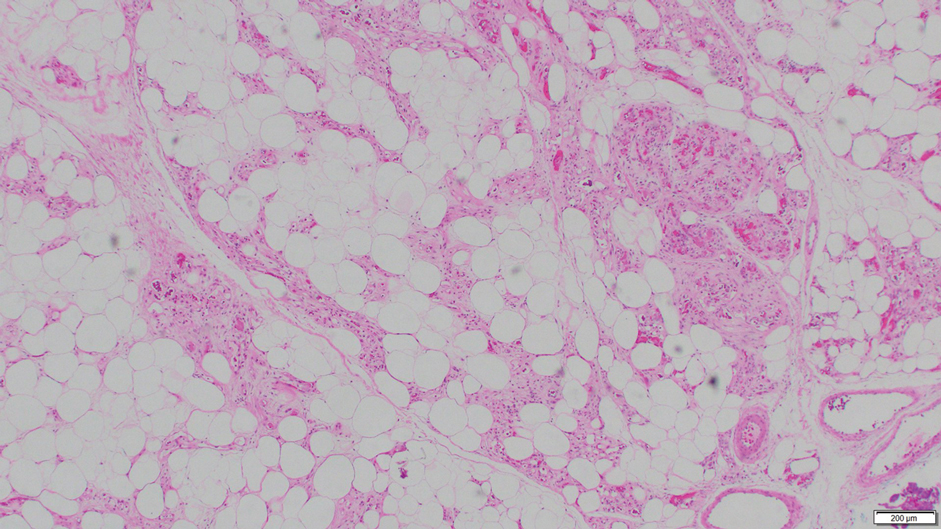
- Malik K, Patel P, Chen J, et al. Leiomyoma cutis: a focused review on presentation, management, and association with malignancy. Am J Clin Dermatol. 2015;16:35-46.
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Delfino S, Toto V, Brunetti B, et al. Recurrent atypical eccrine spiradenoma of the forehead. In Vivo. 2008;22:821-823.
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Choi JH, Ro JY. Cutaneous spindle cell neoplasms: pattern-based diagnostic approach. Arch Pathol Lab Med. 2018;142:958-972.
- Carter CS, Skala SL, Chinnaiyan AM, et al. Immunohistochemical characterization of fumarate hydratase (FH) and succinate dehydrogenase (SDH) in cutaneous leiomyomas for detection of familial cancer syndromes. Am J Surg Pathol. 2017;41:801-809.
- Kanitakis J. Angioleiomyoma of the auricle: an unusual tumor on a rare location. Case Rep Otolaryngol. 2017;2017:1-3.
- Tavassoli FA, Norris HJ. Smooth muscle tumors of the vulva. Obstet Gynecol. 1979;53:213-217.
- Phukan J, Sinha A, Pal S. Fine needle aspiration cytology of eccrine spiradenoma of back: report of a rare case. J Lab Physicians. 2014;6:130.
- Zheng Y, Tian Q, Wang J, et al. Differential diagnosis of eccrine spiradenoma: a case report. Exp Ther Med. 2014;8:1097-1101.
- Bandyopadhyay MR, Besra M, Dutta S, et al. Dermatofibroma: atypical presentations. Indian J Dermatol. 2016;61:121.
- Commons JD, Parish L, Yazdanian S, et al. Dermatofibroma: a curious tumor. Skinmed. 2012;10:268-270.
- Lee YB, Lee JI, Park HJ, et al. Solitary neurofibromas: does an uncommon site exist? Ann Dermatol. 2012;24:101-102.
- Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018;91:S5-S13.
- Howard WR. Angiolipoma. Arch Dermatol. 1960;82:924.
- Ghosh S, Haldar BA. Multiple angiolipomas. Indian J Dermatol Venereol Leprol. 1990;56:143-144.
The Diagnosis: Piloleiomyoma
Leiomyoma cutis, also known as cutaneous leiomyoma, is a benign smooth muscle tumor first described in 1854.1 Cutaneous leiomyoma is comprised of 3 distinct types that depend on the origin of smooth muscle tumor: piloleiomyoma (arrector pili muscle), angioleiomyoma (tunica media of arteries/veins), and genital leiomyoma (dartos muscle of the scrotum and labia majora, erectile muscle of nipple).2 It affects both sexes equally, though some reports have noted an increased prevalence in females. Piloleiomyomas commonly present on the extensor surfaces of the extremities (solitary) and trunk (multiple).1 Tumors most often present as firm flesh-colored or pink-brown papulonodules. They can be linear, dermatomal, segmental, or diffuse, and often are painful. Clinical differential diagnosis for painful skin tumors is aided by the acronym "BLEND AN EGG": blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor.3 For isolated lesions, surgical excision is the treatment of choice. For numerous lesions in which excision would not be feasible, intralesional corticosteroids, medications (eg, calcium channel blockers, alpha blockers, nitroglycerin), and botulinum toxin have been used for pain relief.4
Notably, multiple cutaneous leiomyomas can be seen in association with uterine leiomyomas in Reed syndrome due to an autosomal-dominant or de novo mutation in the fumarate hydratase gene, FH. Reed syndrome is associated with a lifetime risk for renal cell carcinoma (hereditary leiomyomatosis and renal cell cancer) in 15% of cases with FH mutations.5 In our patient, both immunohistochemical staining and blood testing for FH were performed. Immunohistochemistry revealed notably diminished staining with only weak patchy granular cytoplasmic staining present (Figure 1). Genetic testing revealed heterozygosity for a pathogenic variant of the FH gene, consistent with a diagnosis of Reed syndrome.
Histologically, the differential diagnosis includes other spindle cell tumors, such as dermatofibroma, neurofibroma, and dermatomyofibroma. The histologic appearance varies depending on the type, with piloleiomyoma typically located within the reticular dermis with possible subcutaneous extension. Fascicles of eosinophilic smooth muscle cells in an interlacing arrangement often ramify between neighboring dermal collagen; these smooth muscle cells contain cigar-shaped, blunt-ended nuclei with a perinuclear clear vacuole. Marked epidermal hyperplasia is possible.6 A close association with a nearby hair follicle frequently is noted. Although differentiated smooth muscle cells usually are evident on hematoxylin and eosin, positive staining for smooth muscle actin (SMA) and desmin can aid in diagnosis.7 Immunohistochemical staining for FH has proven to be highly specific (97.6%) with moderate sensitivity (70.0%).8 Angioleiomyomas appear as well-demarcated dermal to subcutaneous tumors composed of smooth muscle cells surrounding thick-walled vaculature.9 Scrotal and vulvar leiomyomas are composed of eosinophilic spindle cells, though vulvar leiomyomas have shown epithelioid differentiation.10 Nipple leiomyomas appear similar to piloleiomyomas on histology with interlacing smooth muscle fiber bundles.
Eccrine spiradenoma is a relatively uncommon adnexal tumor derived from eccrine sweat glands. It most often presents as a small, painful or tender, intradermal nodule (or rarely as nodules) on the head or ventral trunk.11 There is no sexual predilection. It affects adults at any age but most often from 15 to 35 years. Although rare, malignant transformation is possible. Histologically, eccrine spiradenomas appear as a well-demarcated dermal tumor composed of bland basaloid cells with minimal cytoplasm, often with numerous admixed lymphocytes and variably prominent vasculature (Figure 2). Eosinophilic basement membrane material can be seen within or surrounding the nodules of tumor cells. Multiple spiradenomas can occur in the setting of Brooke-Spiegler syndrome, which is an autosomal-dominant disorder due to an inherited mutation in the CYLD gene. Spiradenomas are benign neoplasms, and surgical excision with clear margins is the treatment of choice.12
Dermatofibroma, also known as cutaneous benign fibrous histiocytoma, is a firm, flesh-colored papule or nodule that most often presents on the lower extremities. It typically is seen in women aged 20 to 40 years.13 The etiology is uncertain, and dermatofibromas often spontaneously develop, though there are inconsistent reports of development with local trauma including insect bites and puncture wounds. The dimple sign refers to skin dimpling with lateral pressure.13 Most commonly, dermatofibromas consist of a dermal proliferation of bland fibroblastic cells with entrapment of dermal collagen bundles at the periphery of the tumors (Figure 3). The fibroblastic cells often are paler and less eosinophilic than smooth muscle cells seen in cutaneous leiomyomas, with tapered nuclei that lack a perinuclear vacuole. Admixed histocytes and other inflammatory cells often are present. Overlying epidermal hyperplasia and/or hyperpigmentation also may be present. Numerous histologic variants have been described, including cellular, epithelioid, aneurysmal, atypical, and hemosiderotic types.14 Immunohistochemical stains may show patchy positive staining for SMA, but h-caldesmon and desmin typically are negative.
Neurofibroma is a tumor derived from neuromesenchymal tissue with nerve axons. They form through neuromesenchyme (eg, Schwann cells, mast cells, perineural cells, endoneural fibroblast) proliferation. Solitary neurofibromas occur most commonly in adults and have no gender predilection. The most common presentation is an asymptomatic, solitary, soft, flesh-colored papulonodule.15 Clinical variants include pigmented, diffuse, and plexiform, with plexiform neurofibromas almost always being consistent with a diagnosis of neurofibromatosis type 1. Histologically, neurofibromas present as dermal or subcutaneous nodules composed of randomly arranged spindle cells with wavy tapered nuclei within a loose collagenous stroma (Figure 4).16 The spindle cells in neurofibromas will stain positively for S-100 protein and SOX-10 and negatively for SMA and desmin.
Angiolipoma is a benign tumor composed of adipocytes that also contains vasculature.17 The majority of cases are of unknown etiology, though familial cases have been described. They typically present as multiple painful or tender (differentiating from lipomas) subcutaneous swellings over the forearms in individuals aged 20 to 30 years.18 On histopathology, angiolipomas appear as well-circumscribed subcutaneous tumors containing mature adipocytes intermixed with small capillary vessels, some of which contain luminal fibrin thrombi (Figure 5).
The Diagnosis: Piloleiomyoma
Leiomyoma cutis, also known as cutaneous leiomyoma, is a benign smooth muscle tumor first described in 1854.1 Cutaneous leiomyoma is comprised of 3 distinct types that depend on the origin of smooth muscle tumor: piloleiomyoma (arrector pili muscle), angioleiomyoma (tunica media of arteries/veins), and genital leiomyoma (dartos muscle of the scrotum and labia majora, erectile muscle of nipple).2 It affects both sexes equally, though some reports have noted an increased prevalence in females. Piloleiomyomas commonly present on the extensor surfaces of the extremities (solitary) and trunk (multiple).1 Tumors most often present as firm flesh-colored or pink-brown papulonodules. They can be linear, dermatomal, segmental, or diffuse, and often are painful. Clinical differential diagnosis for painful skin tumors is aided by the acronym "BLEND AN EGG": blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor.3 For isolated lesions, surgical excision is the treatment of choice. For numerous lesions in which excision would not be feasible, intralesional corticosteroids, medications (eg, calcium channel blockers, alpha blockers, nitroglycerin), and botulinum toxin have been used for pain relief.4
Notably, multiple cutaneous leiomyomas can be seen in association with uterine leiomyomas in Reed syndrome due to an autosomal-dominant or de novo mutation in the fumarate hydratase gene, FH. Reed syndrome is associated with a lifetime risk for renal cell carcinoma (hereditary leiomyomatosis and renal cell cancer) in 15% of cases with FH mutations.5 In our patient, both immunohistochemical staining and blood testing for FH were performed. Immunohistochemistry revealed notably diminished staining with only weak patchy granular cytoplasmic staining present (Figure 1). Genetic testing revealed heterozygosity for a pathogenic variant of the FH gene, consistent with a diagnosis of Reed syndrome.
Histologically, the differential diagnosis includes other spindle cell tumors, such as dermatofibroma, neurofibroma, and dermatomyofibroma. The histologic appearance varies depending on the type, with piloleiomyoma typically located within the reticular dermis with possible subcutaneous extension. Fascicles of eosinophilic smooth muscle cells in an interlacing arrangement often ramify between neighboring dermal collagen; these smooth muscle cells contain cigar-shaped, blunt-ended nuclei with a perinuclear clear vacuole. Marked epidermal hyperplasia is possible.6 A close association with a nearby hair follicle frequently is noted. Although differentiated smooth muscle cells usually are evident on hematoxylin and eosin, positive staining for smooth muscle actin (SMA) and desmin can aid in diagnosis.7 Immunohistochemical staining for FH has proven to be highly specific (97.6%) with moderate sensitivity (70.0%).8 Angioleiomyomas appear as well-demarcated dermal to subcutaneous tumors composed of smooth muscle cells surrounding thick-walled vaculature.9 Scrotal and vulvar leiomyomas are composed of eosinophilic spindle cells, though vulvar leiomyomas have shown epithelioid differentiation.10 Nipple leiomyomas appear similar to piloleiomyomas on histology with interlacing smooth muscle fiber bundles.
Eccrine spiradenoma is a relatively uncommon adnexal tumor derived from eccrine sweat glands. It most often presents as a small, painful or tender, intradermal nodule (or rarely as nodules) on the head or ventral trunk.11 There is no sexual predilection. It affects adults at any age but most often from 15 to 35 years. Although rare, malignant transformation is possible. Histologically, eccrine spiradenomas appear as a well-demarcated dermal tumor composed of bland basaloid cells with minimal cytoplasm, often with numerous admixed lymphocytes and variably prominent vasculature (Figure 2). Eosinophilic basement membrane material can be seen within or surrounding the nodules of tumor cells. Multiple spiradenomas can occur in the setting of Brooke-Spiegler syndrome, which is an autosomal-dominant disorder due to an inherited mutation in the CYLD gene. Spiradenomas are benign neoplasms, and surgical excision with clear margins is the treatment of choice.12
Dermatofibroma, also known as cutaneous benign fibrous histiocytoma, is a firm, flesh-colored papule or nodule that most often presents on the lower extremities. It typically is seen in women aged 20 to 40 years.13 The etiology is uncertain, and dermatofibromas often spontaneously develop, though there are inconsistent reports of development with local trauma including insect bites and puncture wounds. The dimple sign refers to skin dimpling with lateral pressure.13 Most commonly, dermatofibromas consist of a dermal proliferation of bland fibroblastic cells with entrapment of dermal collagen bundles at the periphery of the tumors (Figure 3). The fibroblastic cells often are paler and less eosinophilic than smooth muscle cells seen in cutaneous leiomyomas, with tapered nuclei that lack a perinuclear vacuole. Admixed histocytes and other inflammatory cells often are present. Overlying epidermal hyperplasia and/or hyperpigmentation also may be present. Numerous histologic variants have been described, including cellular, epithelioid, aneurysmal, atypical, and hemosiderotic types.14 Immunohistochemical stains may show patchy positive staining for SMA, but h-caldesmon and desmin typically are negative.
Neurofibroma is a tumor derived from neuromesenchymal tissue with nerve axons. They form through neuromesenchyme (eg, Schwann cells, mast cells, perineural cells, endoneural fibroblast) proliferation. Solitary neurofibromas occur most commonly in adults and have no gender predilection. The most common presentation is an asymptomatic, solitary, soft, flesh-colored papulonodule.15 Clinical variants include pigmented, diffuse, and plexiform, with plexiform neurofibromas almost always being consistent with a diagnosis of neurofibromatosis type 1. Histologically, neurofibromas present as dermal or subcutaneous nodules composed of randomly arranged spindle cells with wavy tapered nuclei within a loose collagenous stroma (Figure 4).16 The spindle cells in neurofibromas will stain positively for S-100 protein and SOX-10 and negatively for SMA and desmin.
Angiolipoma is a benign tumor composed of adipocytes that also contains vasculature.17 The majority of cases are of unknown etiology, though familial cases have been described. They typically present as multiple painful or tender (differentiating from lipomas) subcutaneous swellings over the forearms in individuals aged 20 to 30 years.18 On histopathology, angiolipomas appear as well-circumscribed subcutaneous tumors containing mature adipocytes intermixed with small capillary vessels, some of which contain luminal fibrin thrombi (Figure 5).
- Malik K, Patel P, Chen J, et al. Leiomyoma cutis: a focused review on presentation, management, and association with malignancy. Am J Clin Dermatol. 2015;16:35-46.
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Delfino S, Toto V, Brunetti B, et al. Recurrent atypical eccrine spiradenoma of the forehead. In Vivo. 2008;22:821-823.
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Choi JH, Ro JY. Cutaneous spindle cell neoplasms: pattern-based diagnostic approach. Arch Pathol Lab Med. 2018;142:958-972.
- Carter CS, Skala SL, Chinnaiyan AM, et al. Immunohistochemical characterization of fumarate hydratase (FH) and succinate dehydrogenase (SDH) in cutaneous leiomyomas for detection of familial cancer syndromes. Am J Surg Pathol. 2017;41:801-809.
- Kanitakis J. Angioleiomyoma of the auricle: an unusual tumor on a rare location. Case Rep Otolaryngol. 2017;2017:1-3.
- Tavassoli FA, Norris HJ. Smooth muscle tumors of the vulva. Obstet Gynecol. 1979;53:213-217.
- Phukan J, Sinha A, Pal S. Fine needle aspiration cytology of eccrine spiradenoma of back: report of a rare case. J Lab Physicians. 2014;6:130.
- Zheng Y, Tian Q, Wang J, et al. Differential diagnosis of eccrine spiradenoma: a case report. Exp Ther Med. 2014;8:1097-1101.
- Bandyopadhyay MR, Besra M, Dutta S, et al. Dermatofibroma: atypical presentations. Indian J Dermatol. 2016;61:121.
- Commons JD, Parish L, Yazdanian S, et al. Dermatofibroma: a curious tumor. Skinmed. 2012;10:268-270.
- Lee YB, Lee JI, Park HJ, et al. Solitary neurofibromas: does an uncommon site exist? Ann Dermatol. 2012;24:101-102.
- Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018;91:S5-S13.
- Howard WR. Angiolipoma. Arch Dermatol. 1960;82:924.
- Ghosh S, Haldar BA. Multiple angiolipomas. Indian J Dermatol Venereol Leprol. 1990;56:143-144.
- Malik K, Patel P, Chen J, et al. Leiomyoma cutis: a focused review on presentation, management, and association with malignancy. Am J Clin Dermatol. 2015;16:35-46.
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Delfino S, Toto V, Brunetti B, et al. Recurrent atypical eccrine spiradenoma of the forehead. In Vivo. 2008;22:821-823.
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Choi JH, Ro JY. Cutaneous spindle cell neoplasms: pattern-based diagnostic approach. Arch Pathol Lab Med. 2018;142:958-972.
- Carter CS, Skala SL, Chinnaiyan AM, et al. Immunohistochemical characterization of fumarate hydratase (FH) and succinate dehydrogenase (SDH) in cutaneous leiomyomas for detection of familial cancer syndromes. Am J Surg Pathol. 2017;41:801-809.
- Kanitakis J. Angioleiomyoma of the auricle: an unusual tumor on a rare location. Case Rep Otolaryngol. 2017;2017:1-3.
- Tavassoli FA, Norris HJ. Smooth muscle tumors of the vulva. Obstet Gynecol. 1979;53:213-217.
- Phukan J, Sinha A, Pal S. Fine needle aspiration cytology of eccrine spiradenoma of back: report of a rare case. J Lab Physicians. 2014;6:130.
- Zheng Y, Tian Q, Wang J, et al. Differential diagnosis of eccrine spiradenoma: a case report. Exp Ther Med. 2014;8:1097-1101.
- Bandyopadhyay MR, Besra M, Dutta S, et al. Dermatofibroma: atypical presentations. Indian J Dermatol. 2016;61:121.
- Commons JD, Parish L, Yazdanian S, et al. Dermatofibroma: a curious tumor. Skinmed. 2012;10:268-270.
- Lee YB, Lee JI, Park HJ, et al. Solitary neurofibromas: does an uncommon site exist? Ann Dermatol. 2012;24:101-102.
- Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018;91:S5-S13.
- Howard WR. Angiolipoma. Arch Dermatol. 1960;82:924.
- Ghosh S, Haldar BA. Multiple angiolipomas. Indian J Dermatol Venereol Leprol. 1990;56:143-144.
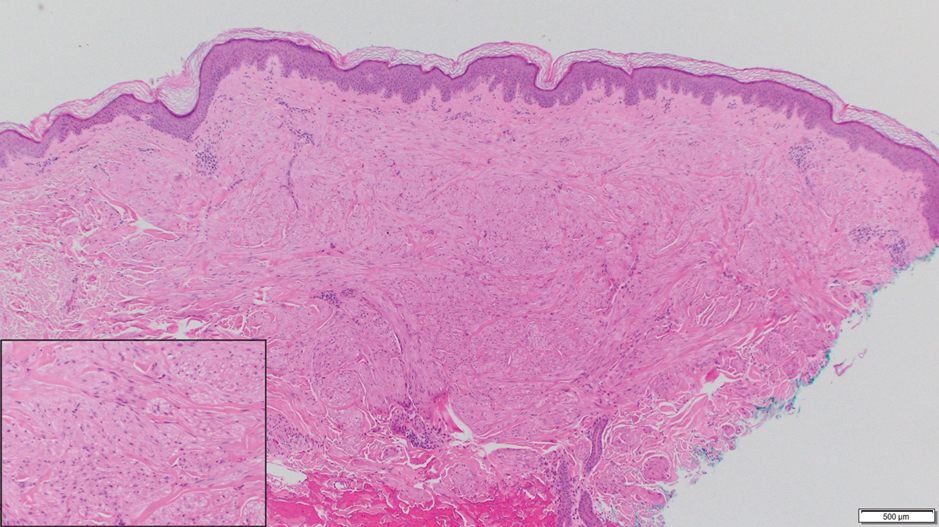
A 36-year-old woman presented with multiple new-onset, firm, tender, subcutaneous papules and nodules involving the upper arms and shoulders.
Sparse Hair on the Scalp
The Diagnosis: Monilethrix
Trichoscopy showed a beaded appearance of the hair shafts (Figure, A). Light microscopy demonstrated normal medullated nodes of hair coupled with internodal, thin, nonmedullated hair at regular intervals (Figure, B). Clinical and trichoscopic findings led to a diagnosis of monilethrix.
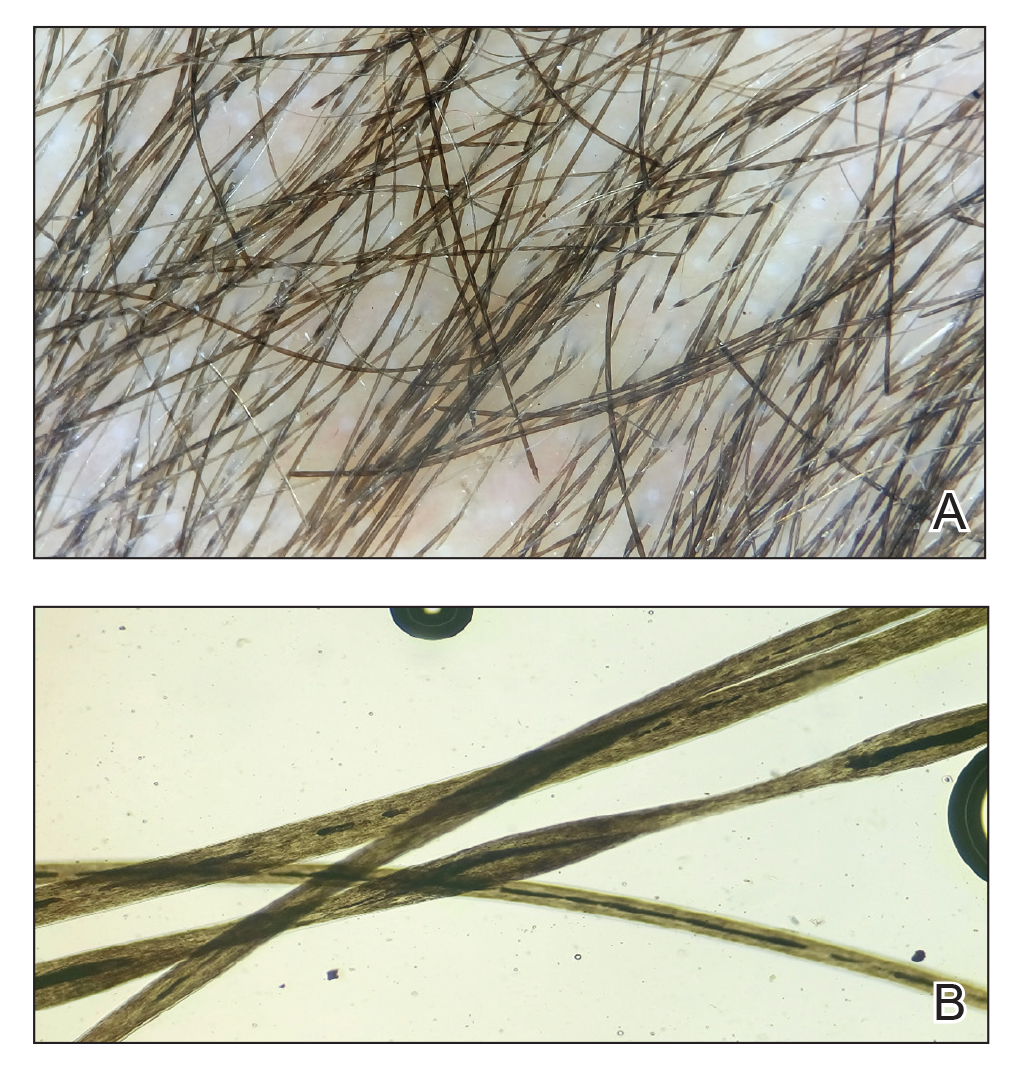
Monilethrix is a genetic hair disorder characterized by regular periodic thinning of the hair shafts, giving the strands a beaded appearance. The hair tends to break at these constricted parts, resulting in short hairs. Nodosities represent the normal hair shaft, whereas the constricted points are the site of the defect. The hair tends to be normal at birth and then becomes short, fragile, and brittle within months, leading to hypotrichosis, particularly on the occipital scalp.1 Monilethrix also may involve the eyebrows and eyelashes in addition to scalp hair. Follicular hyperkeratotic papules with perifollicular erythema frequently are noted on the occipital area. Monilethrix can be inherited in an autosomal-dominant fashion with mutations involving KRT81, KRT83, and KRT86, which code for the type II hair keratins Hb1, Hb3, and Hb6, respectively. The autosomal-recessive form is caused by mutations in the DSG4 gene, coding for the desmoglein 4 protein.2 Trichoscopy or light microscopy is essential to establish a diagnosis of monilethrix. Trichoscopy is an easy and rapid tool that is utilized to illustrate the beaded appearance of the hair shafts.3 Light microscopy shows the distinctive nodes that are medullated, with a normal hair diameter alternating with the internodes, or constrictions, that are nonmedullated and represent the sites of fracture.1 Monilethrix can improve by puberty. There is no definitive treatment; however, some patients show considerable improvement on minoxidil.4 Treatment with minoxidil was initiated in this patient; however, she was lost to follow-up.
Genetic hair disorders are rare and can be an isolated phenomenon or part of concurrent genetic syndromes. Therefore, thorough clinical examination of other ectodermal structures such as the nails and teeth is crucial as well as obtaining a detailed family history and review of systems to exclude other syndromes.2 Hypotrichosis simplex is characterized by hair loss exclusively on the scalp, sparing other ectodermal structures and with no systemic abnormalities. Ectodermal dysplasia is a heterogeneous group of disorders affecting not only the hair but also the teeth, nails, and sweat glands.2 Pili torti is another rare genetic hair disorder that is characterized by twisting of the hair fiber on its own axis. It presents clinically as sparse, depigmented, lusterless hair that is easily broken. Light microscopy demonstrates twists of hair at irregular intervals. Pili annulati is characterized by bright and dark bands when viewed with reflected light. Unlike monilethrix, there is no fragility, and the hair can grow long.5
- Mirmirani P, Huang KP, Price VH. A practical, algorithmic approach to diagnosing hair shaft disorders. Int J Dermatol. 2011;50:1-12.
- Ahmed A, Almohanna H, Griggs J, et al. Genetic hair disorders: a review. Dermatol Ther. 2019;9:421-448.
- Liu C-I, Hsu C-H. Rapid diagnosis of monilethrix using dermoscopy. Br J Dermatol. 2008;159:741-743.
- Rossi A, Iorio A, Fortuna MC, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol. 2011;24:239-242.
- Singh G, Miteva M. Prognosis and management of congenital hair shaft disorders with fragility—part I. Pediatr Dermatol. 2016;33:473-480.
The Diagnosis: Monilethrix
Trichoscopy showed a beaded appearance of the hair shafts (Figure, A). Light microscopy demonstrated normal medullated nodes of hair coupled with internodal, thin, nonmedullated hair at regular intervals (Figure, B). Clinical and trichoscopic findings led to a diagnosis of monilethrix.
Monilethrix is a genetic hair disorder characterized by regular periodic thinning of the hair shafts, giving the strands a beaded appearance. The hair tends to break at these constricted parts, resulting in short hairs. Nodosities represent the normal hair shaft, whereas the constricted points are the site of the defect. The hair tends to be normal at birth and then becomes short, fragile, and brittle within months, leading to hypotrichosis, particularly on the occipital scalp.1 Monilethrix also may involve the eyebrows and eyelashes in addition to scalp hair. Follicular hyperkeratotic papules with perifollicular erythema frequently are noted on the occipital area. Monilethrix can be inherited in an autosomal-dominant fashion with mutations involving KRT81, KRT83, and KRT86, which code for the type II hair keratins Hb1, Hb3, and Hb6, respectively. The autosomal-recessive form is caused by mutations in the DSG4 gene, coding for the desmoglein 4 protein.2 Trichoscopy or light microscopy is essential to establish a diagnosis of monilethrix. Trichoscopy is an easy and rapid tool that is utilized to illustrate the beaded appearance of the hair shafts.3 Light microscopy shows the distinctive nodes that are medullated, with a normal hair diameter alternating with the internodes, or constrictions, that are nonmedullated and represent the sites of fracture.1 Monilethrix can improve by puberty. There is no definitive treatment; however, some patients show considerable improvement on minoxidil.4 Treatment with minoxidil was initiated in this patient; however, she was lost to follow-up.
Genetic hair disorders are rare and can be an isolated phenomenon or part of concurrent genetic syndromes. Therefore, thorough clinical examination of other ectodermal structures such as the nails and teeth is crucial as well as obtaining a detailed family history and review of systems to exclude other syndromes.2 Hypotrichosis simplex is characterized by hair loss exclusively on the scalp, sparing other ectodermal structures and with no systemic abnormalities. Ectodermal dysplasia is a heterogeneous group of disorders affecting not only the hair but also the teeth, nails, and sweat glands.2 Pili torti is another rare genetic hair disorder that is characterized by twisting of the hair fiber on its own axis. It presents clinically as sparse, depigmented, lusterless hair that is easily broken. Light microscopy demonstrates twists of hair at irregular intervals. Pili annulati is characterized by bright and dark bands when viewed with reflected light. Unlike monilethrix, there is no fragility, and the hair can grow long.5
The Diagnosis: Monilethrix
Trichoscopy showed a beaded appearance of the hair shafts (Figure, A). Light microscopy demonstrated normal medullated nodes of hair coupled with internodal, thin, nonmedullated hair at regular intervals (Figure, B). Clinical and trichoscopic findings led to a diagnosis of monilethrix.
Monilethrix is a genetic hair disorder characterized by regular periodic thinning of the hair shafts, giving the strands a beaded appearance. The hair tends to break at these constricted parts, resulting in short hairs. Nodosities represent the normal hair shaft, whereas the constricted points are the site of the defect. The hair tends to be normal at birth and then becomes short, fragile, and brittle within months, leading to hypotrichosis, particularly on the occipital scalp.1 Monilethrix also may involve the eyebrows and eyelashes in addition to scalp hair. Follicular hyperkeratotic papules with perifollicular erythema frequently are noted on the occipital area. Monilethrix can be inherited in an autosomal-dominant fashion with mutations involving KRT81, KRT83, and KRT86, which code for the type II hair keratins Hb1, Hb3, and Hb6, respectively. The autosomal-recessive form is caused by mutations in the DSG4 gene, coding for the desmoglein 4 protein.2 Trichoscopy or light microscopy is essential to establish a diagnosis of monilethrix. Trichoscopy is an easy and rapid tool that is utilized to illustrate the beaded appearance of the hair shafts.3 Light microscopy shows the distinctive nodes that are medullated, with a normal hair diameter alternating with the internodes, or constrictions, that are nonmedullated and represent the sites of fracture.1 Monilethrix can improve by puberty. There is no definitive treatment; however, some patients show considerable improvement on minoxidil.4 Treatment with minoxidil was initiated in this patient; however, she was lost to follow-up.
Genetic hair disorders are rare and can be an isolated phenomenon or part of concurrent genetic syndromes. Therefore, thorough clinical examination of other ectodermal structures such as the nails and teeth is crucial as well as obtaining a detailed family history and review of systems to exclude other syndromes.2 Hypotrichosis simplex is characterized by hair loss exclusively on the scalp, sparing other ectodermal structures and with no systemic abnormalities. Ectodermal dysplasia is a heterogeneous group of disorders affecting not only the hair but also the teeth, nails, and sweat glands.2 Pili torti is another rare genetic hair disorder that is characterized by twisting of the hair fiber on its own axis. It presents clinically as sparse, depigmented, lusterless hair that is easily broken. Light microscopy demonstrates twists of hair at irregular intervals. Pili annulati is characterized by bright and dark bands when viewed with reflected light. Unlike monilethrix, there is no fragility, and the hair can grow long.5
- Mirmirani P, Huang KP, Price VH. A practical, algorithmic approach to diagnosing hair shaft disorders. Int J Dermatol. 2011;50:1-12.
- Ahmed A, Almohanna H, Griggs J, et al. Genetic hair disorders: a review. Dermatol Ther. 2019;9:421-448.
- Liu C-I, Hsu C-H. Rapid diagnosis of monilethrix using dermoscopy. Br J Dermatol. 2008;159:741-743.
- Rossi A, Iorio A, Fortuna MC, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol. 2011;24:239-242.
- Singh G, Miteva M. Prognosis and management of congenital hair shaft disorders with fragility—part I. Pediatr Dermatol. 2016;33:473-480.
- Mirmirani P, Huang KP, Price VH. A practical, algorithmic approach to diagnosing hair shaft disorders. Int J Dermatol. 2011;50:1-12.
- Ahmed A, Almohanna H, Griggs J, et al. Genetic hair disorders: a review. Dermatol Ther. 2019;9:421-448.
- Liu C-I, Hsu C-H. Rapid diagnosis of monilethrix using dermoscopy. Br J Dermatol. 2008;159:741-743.
- Rossi A, Iorio A, Fortuna MC, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol. 2011;24:239-242.
- Singh G, Miteva M. Prognosis and management of congenital hair shaft disorders with fragility—part I. Pediatr Dermatol. 2016;33:473-480.
A 5-year-old girl presented to our clinic with sparse scalp hair. Her mother reported thinning of the hair and breakage that appeared shortly after birth. She also reported that the patient’s hair was dull, dry, and unable to be grown long. The patient was otherwise healthy. She was born to nonconsanguineous parents, and her family history was unremarkable. Physical examination revealed dry, brittle, and short hairs. The hair was sparser on the occipital area of the scalp, and multiple keratotic papules were noted in this area. No abnormalities were detected on the teeth or nails, and a review of systems was unremarkable. Trichoscopy and light microscopy were performed.
Pediatric Procedural Dermatology
Performing dermatologic procedures in infants, children, and teenagers presents many unique challenges. There may be unique diagnoses, different instruments, differences in skin biology, or different approaches to pain management and anesthesia; the inclusion of a third party (caregivers) in decision processes; or a need to assess maturity level or to optimize outcomes over the patient’s lifetime. The field of pediatric procedural dermatology is broad. This article reviews some of the more common procedures performed by pediatric dermatologists and some of the more common ethical and quality-of-life (QOL) considerations one might face in procedural pediatric dermatology. (The textbook Procedural Pediatric Dermatology1 offers a thorough discussion of this topic.)
Quality of Life
More often than not, procedures are performed in pediatric dermatology to improve QOL rather than to prevent morbidity or mortality. In the case of many self-limited conditions, such as ingrown nails or pyogenic granulomas, it is clear that intervention will improve the patient’s QOL. In the case of warts and molluscum contagiosum, emotional, social, and cultural considerations play a large role in determining whether an intervention will improve QOL. Finally, some conditions, such as genodermatoses, giant congenital melanocytic nevi, and large vascular malformations, may be associated with additional systemic symptoms and may not have good treatment options for cure. In these cases, procedural interventions will result in a mixture of positive and negative QOL outcomes that can occur at the same time.
Bemmels et al2 published a qualitative study that provides a good foundation for understanding the positive and negative effects of procedural interventions on children and teenagers. In their study, children and teenagers who underwent reconstructive surgery for craniofacial differences noted improved self-esteem and reduced stigmatization. However, they also experienced negative outcomes, including an addiction to attaining a perfect surgical face, missing school for treatments, difficulty adjusting to an evolving appearance, anxiety related to not knowing when treatments will end, and experiencing stigma related to undergoing surgery.2 Thus, a comprehensive plan for the management of children who need ongoing procedures should include some level of psychosocial support. Two good references on supporting young patients with visible differences include CBT for Appearance Anxiety: Psychosocial Interventions for Anxiety Due to Visible Difference3 and Reaching Teens: Strength-Based, Trauma-Sensitive, Resilience-Building Communication Strategies Rooted in Positive Youth Development.4
Ethics
Ethical decisions in pediatric procedural dermatology differ from adult dermatology in 3 major ways: (1) the involvement of a third party (ie, parents or legal guardians), (2) the need to assess the maturity of the patient, and (3) the need to know local laws in the jurisdiction in which care is being provided. Ethical dilemmas occur when the desires of the child, parents/guardians, and dermatologist are not in alignment. In these cases, it is important to be prepared with a moral or ethical framework to guide decision-making when conflicts occur. Two great resources are the best interest standard5 and the publication entitled, “Informed Consent in Decision-making in Pediatric Practice,” from the American Academy of Pediatrics.6
In pediatrics, it often is better to conceptualize medical decision-making as a combination of informed permission and assent of the patient rather than informed consent. Informed permission describes how a parent or surrogate makes decisions for the child or adolescent and is similar to informed consent. A parent’s informed permission may be in conflict with a child’s wishes, but it is assumed that the parent is acting in the best interest of the child. Assent of the patient is the process of obtaining a minor’s agreement to undergo an intervention even though he/she may lack legal authority or decision-making capacity to provide standard informed consent. It is important to respect the child’s right to assent to interventions to the extent that their maturity level permits to develop trust with the dermatologist and medical encounters in general.
These differences emphasize an active process in which the patient, caregiver, and physician are all involved in the health care process and allow for increasing inclusion of the child as is developmentally appropriate. In the end, however, parents have the legal authority to give or withhold permission for a procedure.7 When this conflicts with a child’s dissent, the dermatologist will need to objectively explore the reasons for the conflict and decide if a procedure is not in the child’s best interests. If a mutual understanding cannot be reached between the dermatologist and parents, obtaining a second opinion is a good option.8
Common Diagnoses
The most common diagnoses unique to procedural pediatric dermatology include congenital melanocytic nevi, vascular anomalies, midline lesions, epidermal nevi, and pilomatricomas. Prior to intervening on these lesions, it is important to consider evaluating for associated diseases.
Congenital Melanocytic Nevi
Nevus Outreach has published best practices for the management of congenital melanocytic nevi.9 In newborns with a congenital melanocytic nevus greater than 3 cm in diameter or more than 20 satellite lesions, it is recommended that magnetic resonance imaging (MRI) of the brain and spine with and without gadolinium contrast be obtained before 6 months of age. Within the first 6 months of life, these children also should see ophthalmologists, neurologists, pediatric dermatologists, and plastic surgeons. These early referrals will help to establish a baseline for the patient and plan for possible interventions, if needed. Additionally, before 3 years of age, every child should be referred to psychology, even if he/she is asymptomatic.10
Vascular Anomalies
Prior to intervening on a vascular anomaly, it is important to accurately classify the lesion. Once the lesion is classified, an evaluation and treatment plan can be developed. The International Society for the Study of Vascular Anomalies has published a detailed classification guide that is a useful starting point in the management of vascular anomalies.11 Once a diagnosis is confirmed, further evaluation may include imaging, specialty referrals, genetic testing, biopsy, or blood tests, and a pediatric dermatologist usually helps to coordinate the care of patients with complex vascular anomalies.
Midline Lesions
Certain lesions in the midline may have a higher risk for neural tube dysraphism, and imaging should be performed prior to any procedural intervention.12 Midline cutaneous findings that are highly likely to be associated with dysraphism are lipomas, acrochordons, pseudotails, true tails, aplasia cutis congenita, congenital scars, dermoid cysts, dermoid sinuses, and infantile hemangiomas that are greater than 2.5 cm in diameter. An MRI should be performed for all high-risk lesions. Intermediate-risk lesions are atypical dimples (>5 mm in diameter or >2.5 cm from the anal verge), hemangiomas less than 2.5 cm in diameter, and hypertrichosis. An ultrasound can screen for spinal dysraphism in these cases as long as imaging is performed prior to 6 months of age. If the child is older than 6 months, an MRI should be performed. Low-risk lesions that do not require imaging are simple dimples, hyperpigmentation, hypopigmentation, melanocytic nevi, port-wine stains, and telangiectases.
Epidermal Nevi
Children with epidermal nevi should have a complete physical examination, focusing on the skeletal system, central nervous system, and eyes. There are no specifically recommended imaging studies or referrals; however, several diagnostic clues can aid in the diagnosis of an epidermal nevus syndrome13:
• Schimmelpenning syndrome: extensive nevus sebaceous and bowing or pain in the legs after 2 years of age
• Phacomatosis pigmentokeratotica: nevus sebaceous and nevus spilus
• Nevus comedonicus syndrome: ipsilateral cataract
• Angora hair nevus syndrome: soft white hair within the nevus
• Becker nevus syndrome: breast hypoplasia
• Proteus syndrome: cerebriform plantar changes
• PIK3CA-related overgrowth spectrum: lipomas, macrodactyly, and/or vascular malformations
• Congenital hemidysplasia with ichthyosiform erythroderma and limb defects: inflammatory epidermal nevi, lateralization, ptychotropism, and ipsilateral limb defects
• Conradi-Hünermann-Happle syndrome: scaly red epidermal nevi without hair follicles and asymmetric limb shortening
Pilomatricomas
In addition to the tent sign—an angulated shape can be appreciated by stretching the skin overlying pilomatricomas—diagnosis of pilomatricoma can be confirmed by transillumination with an otoscope. In this case, a dark shadow typically is cast distal to where the otoscope touches the skin.14 In the case of multiple lesions, the patient should be evaluated for signs of myotonic dystrophy, Turner syndrome, and Gardner syndrome.15
Common Procedures
Pulsed Dye Laser
The pulsed dye laser is the most common laser used for red-colored lesions such as port-wine stains, facial telangiectases, and superficial hemangiomas. It also can be used to treat erythematous scars, verrucae, and psoriasis. In large vascular lesions, it typically is employed at 0.45 to 10 milliseconds every 4 to 6 weeks for 10 or more treatments. Port-wine stains preferably are treated within the first few months of life to provide the most fading without the need for general anesthesia.16 On the other hand, systemic therapy with propranolol is preferred over lasers for infantile hemangiomas.17
Long-Pulsed Alexandrite Laser (755 nm)
The alexandrite laser often is used to treat deeper vascular lesions such as venous lakes and hypertrophic port-wine stains. The operator needs to be cautious, as this laser has a higher incidence of scarring at the settings used to treat vascular lesions (typically fluences around 60–85 J/cm2).18 It also may be used for hair reduction in disorders with hypertrichosis or hidradenitis suppurativa.19
Long-Pulsed Nd:YAG Laser
The long-pulsed Nd:YAG laser also can be used to treat deep vascular lesions and remove unwanted hair. Because of its low window of safety in the treatment of vascular lesions, the alexandrite laser usually is preferred. However, it is the preferred laser for treatment of unwanted hair and hidradenitis suppurativa in darker skin types. It often provides a 50% reduction in hair density after 9 treatments.20
Quality-Switched Lasers
Pigment granules in melanosomes and tattoo particles are targeted with quality-switched (QS) lasers. Typically, a device will contain a combination of QS 532-nm potassium-titanyl-phosphate (KTP) lasers, QS 1064-nm Nd:YAG lasers, and QS 755-nm alexandrite lasers in 1 machine. In general, shorter wavelengths are used to treat epidermal lesions such as ephelides, lentigines, and café-au-lait macules. Longer wavelengths are used to treat deeper lesions such as nevus of Ota. A 2017 review suggested that café-au-lait macules with ragged borders (so-called coast of Maine borders) may respond well to QS lasers.21
Ablative Lasers
The 10,600-nm
Fractionated Lasers
Fractionated lasers can be nonablative (several devices are available in the 1410- to 1927-nm range) or ablative (CO2 or erbium:YAG). In pediatrics, they are usually used to treat burn scars, traumatic scars, and mild to moderate acne scarring.22 The most common side effects from fractionated lasers are prolonged erythema or hyperpigmentation. In addition, it typically takes at least 3 treatments to notice improvements.
Excisions
Pediatric procedural dermatologists remove a variety of unique lesions through excision. A few tips are provided for some of the more common lesions that may be excised in children.
Accessory Tragi
Prior to excising an accessory tragus, the surgeon should consider documenting a facial nerve examination, as accessory tragi can be associated with complete or partial facial nerve dysfunction. Additionally, there usually is an underlying cartilage structure present within the tragus. The cartilage stalk also should be addressed during the excision to avoid a continued palpable deformity after excision.
Dermoid Cysts
Dermoid cysts are the most commonly diagnosed benign orbital lesion in children.23 Exophytic periorbital lesions, which extend outside the orbital rim, can be removed through an infrabrow incision. Endophytic periorbital lesions, which are inside the orbital rim, should be removed through a crease incision. Midline lesions may have an intracranial extension and should be imaged through MRI and/or a computed tomography.24 Because dermoid cysts usually are located below the orbicularis oculi muscle, the muscle should be fixed with a suture prior to closing with skin sutures.
Pilomatricomas
Typically, a linear incision is made overlying the lesion, and then the underlying tumor is removed with sharp or blunt dissection. However, if the overlying skin has been stretched thin, a lenticular excision that includes the thinned skin may improve cosmesis.
Congenital Nevi
Large congenital nevi typically are removed through staged excisions. Lower extremity lesions are best removed before 10 months of age or before walking begins to minimize wound tension. However, if the procedure is not performed in infancy, it is best to wait until walking becomes stable.25 In older children, it is advisable to splint the affected lower extremity for 2 weeks to prevent dehiscence. The interval between excisions typically is 4 to 6 weeks for small lesions and 3 months for larger nevi.
Conclusion
Procedural pediatric dermatology is a broad and emerging field. As this article highlights, children are not small versions of adults and have unique biology, diseases, therapies, social situations, and ethical challenges from adults. This article provides a superficial overview of some of the more common issues faced by pediatric dermatologists and providers who perform procedures on infants, children, and teenagers. Readers who are interested in obtaining a more in-depth understanding of procedural pediatric dermatology should look at Procedural Pediatric Dermatology,1 the first textbook to provide expert opinion and evidence-based information on procedural management of pediatric skin conditions.
- Krakowski AC. Procedural Pediatric Dermatology. Phialdelphia, PA: Wolters Kluwer; 2011.
- Bemmels H, Biesecker B, Schmidt J, et al. Psychological and social factors in undergoing reconstructive surgery among individuals with craniofacial conditions: an exploratory study. Cleft Palate Craniofac J. 2013;50:158-167.
- Clarke A, Thompson AR, Jenkinson E, et al. CBT for Appearance Anxiety: Psychosocial Interventions for Anxiety Due to Visible Difference. Chichester, West Sussex: Wiley-Blackwell; 2013.
- Ginsburg KR, Ramirez McClain ZB, eds. Reaching Teens: Strength-Based, Trauma-Sensitive, Resilience-Building Communication Strategies Rooted in Positive Youth Development. 2nd ed. Itasca, IL: American Academy of Pediatrics; 2020.
- Kopelman LM. The best interests standard for incompetent or incapacitated patients of all ages. J Law Med Ethics. 2007;35:187-196.
- Katz AL, Webb SA; Committee on Bioethics. Informed consent in decision-making in pediatric practice. Pediatrics. 2016;138:e20161485. doi:10.1542/peds.2016-1485.
- Michon K. Emancipation of minors. NOLO website. https://www.nolo.com/legal-encyclopedia/emancipation-of-minors-32237.html. Accessed October 14, 2020.
- Cobb C, Bercovitch L. Ethical dilemmas. In: Krakowski AC, ed. Procedural Pediatric Dermatology. Philadelphia, PA: Wolters Kluwer; 2021:7-10.
- Nevus Outreach, Inc., releases best practice guidelines [news release]. Bartlesville, OK: Nevus Outreach Inc; July 7, 2018. https://www.nevus.org/matrices/page_file_download.php?id=239. Accessed October 14, 2020.
- 10. Masnari O, Neuhaus K, Aegerter T, et al. Predictors of health-related quality of life and psychological adjustment in children and adolescents with congenital melanocytic nevi: analysis of parent reports. J Pediatr Psychol. 2019;44:714-725.
- ISSVA classification for vascular anomalies. International Society for the Study of Vascular Anomalies website. https://www.issva.org/UserFiles/file/ISSVA-Classification-2018.pdf. Approved April 2014. Revised May 2018. Accessed October 14, 2020.
- Sewell MJ, Chiu YE, Drolet BA. Neural tube dysraphism: review of cutaneous markers and imaging. Pediatr Dermatol. 2015;32:161-170.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Berreto-Chang OL, Gorell ES, Yamaguma MA, et al. Diagnosis of pilomatricoma using an otoscope. Pediatr Dermatol. 2010;27:554-557.
- Danielson-Cohen A, Lin SJ, Hughes CA, et al. Head and neck pilomatrixoma in children. Arch Otolaryngol Head Neck Surg. 2001;127:1481-1483.
- Jeon H, Bernstein LJ, Belkin DA, et al. Pulsed dye laser treatment of port-wine stains in infancy without the need for general anesthesia. JAMA Dermatol. 2019;155:435-441.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:e20183475. doi:10.1542/peds.2018-3475.
- Tierney EP, Hanke CW. Alexandrite laser for the treatment of port wine stains refractory to pulsed dye laser. Dermatol Surg. 2011;37:1268-1278.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2019;81:76-90.
- Rao K, Sankar TK. Long-pulsed Nd:YAG laser-assisted hair removal in Fitzpatrick skin types IV-VI. Lasers Med Sci. 2011;26:623-626.
- Belkin DA, Neckman JP, Jeon H, et al. Response to laser treatment of café au lait macules based on morphologic features. JAMA Dermatol. 2017;153:1158-1161.
- Kelly K, Lehmer L. Laser surgery. In: Krakowski AC, ed. Procedural Pediatric Dermatology. Philadelphia, PA: Wolters Kluwer; 2021:92-106.
- Eldesouky MA, Elbakary MA. Orbital dermoid cyst: classification and its impact on surgical management. Semin Ophthalmol. 2018;33:170-174.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Metz BJ. Procedural pediatric dermatology. Dermatol Clin. 2013;31:337-346.
Performing dermatologic procedures in infants, children, and teenagers presents many unique challenges. There may be unique diagnoses, different instruments, differences in skin biology, or different approaches to pain management and anesthesia; the inclusion of a third party (caregivers) in decision processes; or a need to assess maturity level or to optimize outcomes over the patient’s lifetime. The field of pediatric procedural dermatology is broad. This article reviews some of the more common procedures performed by pediatric dermatologists and some of the more common ethical and quality-of-life (QOL) considerations one might face in procedural pediatric dermatology. (The textbook Procedural Pediatric Dermatology1 offers a thorough discussion of this topic.)
Quality of Life
More often than not, procedures are performed in pediatric dermatology to improve QOL rather than to prevent morbidity or mortality. In the case of many self-limited conditions, such as ingrown nails or pyogenic granulomas, it is clear that intervention will improve the patient’s QOL. In the case of warts and molluscum contagiosum, emotional, social, and cultural considerations play a large role in determining whether an intervention will improve QOL. Finally, some conditions, such as genodermatoses, giant congenital melanocytic nevi, and large vascular malformations, may be associated with additional systemic symptoms and may not have good treatment options for cure. In these cases, procedural interventions will result in a mixture of positive and negative QOL outcomes that can occur at the same time.
Bemmels et al2 published a qualitative study that provides a good foundation for understanding the positive and negative effects of procedural interventions on children and teenagers. In their study, children and teenagers who underwent reconstructive surgery for craniofacial differences noted improved self-esteem and reduced stigmatization. However, they also experienced negative outcomes, including an addiction to attaining a perfect surgical face, missing school for treatments, difficulty adjusting to an evolving appearance, anxiety related to not knowing when treatments will end, and experiencing stigma related to undergoing surgery.2 Thus, a comprehensive plan for the management of children who need ongoing procedures should include some level of psychosocial support. Two good references on supporting young patients with visible differences include CBT for Appearance Anxiety: Psychosocial Interventions for Anxiety Due to Visible Difference3 and Reaching Teens: Strength-Based, Trauma-Sensitive, Resilience-Building Communication Strategies Rooted in Positive Youth Development.4
Ethics
Ethical decisions in pediatric procedural dermatology differ from adult dermatology in 3 major ways: (1) the involvement of a third party (ie, parents or legal guardians), (2) the need to assess the maturity of the patient, and (3) the need to know local laws in the jurisdiction in which care is being provided. Ethical dilemmas occur when the desires of the child, parents/guardians, and dermatologist are not in alignment. In these cases, it is important to be prepared with a moral or ethical framework to guide decision-making when conflicts occur. Two great resources are the best interest standard5 and the publication entitled, “Informed Consent in Decision-making in Pediatric Practice,” from the American Academy of Pediatrics.6
In pediatrics, it often is better to conceptualize medical decision-making as a combination of informed permission and assent of the patient rather than informed consent. Informed permission describes how a parent or surrogate makes decisions for the child or adolescent and is similar to informed consent. A parent’s informed permission may be in conflict with a child’s wishes, but it is assumed that the parent is acting in the best interest of the child. Assent of the patient is the process of obtaining a minor’s agreement to undergo an intervention even though he/she may lack legal authority or decision-making capacity to provide standard informed consent. It is important to respect the child’s right to assent to interventions to the extent that their maturity level permits to develop trust with the dermatologist and medical encounters in general.
These differences emphasize an active process in which the patient, caregiver, and physician are all involved in the health care process and allow for increasing inclusion of the child as is developmentally appropriate. In the end, however, parents have the legal authority to give or withhold permission for a procedure.7 When this conflicts with a child’s dissent, the dermatologist will need to objectively explore the reasons for the conflict and decide if a procedure is not in the child’s best interests. If a mutual understanding cannot be reached between the dermatologist and parents, obtaining a second opinion is a good option.8
Common Diagnoses
The most common diagnoses unique to procedural pediatric dermatology include congenital melanocytic nevi, vascular anomalies, midline lesions, epidermal nevi, and pilomatricomas. Prior to intervening on these lesions, it is important to consider evaluating for associated diseases.
Congenital Melanocytic Nevi
Nevus Outreach has published best practices for the management of congenital melanocytic nevi.9 In newborns with a congenital melanocytic nevus greater than 3 cm in diameter or more than 20 satellite lesions, it is recommended that magnetic resonance imaging (MRI) of the brain and spine with and without gadolinium contrast be obtained before 6 months of age. Within the first 6 months of life, these children also should see ophthalmologists, neurologists, pediatric dermatologists, and plastic surgeons. These early referrals will help to establish a baseline for the patient and plan for possible interventions, if needed. Additionally, before 3 years of age, every child should be referred to psychology, even if he/she is asymptomatic.10
Vascular Anomalies
Prior to intervening on a vascular anomaly, it is important to accurately classify the lesion. Once the lesion is classified, an evaluation and treatment plan can be developed. The International Society for the Study of Vascular Anomalies has published a detailed classification guide that is a useful starting point in the management of vascular anomalies.11 Once a diagnosis is confirmed, further evaluation may include imaging, specialty referrals, genetic testing, biopsy, or blood tests, and a pediatric dermatologist usually helps to coordinate the care of patients with complex vascular anomalies.
Midline Lesions
Certain lesions in the midline may have a higher risk for neural tube dysraphism, and imaging should be performed prior to any procedural intervention.12 Midline cutaneous findings that are highly likely to be associated with dysraphism are lipomas, acrochordons, pseudotails, true tails, aplasia cutis congenita, congenital scars, dermoid cysts, dermoid sinuses, and infantile hemangiomas that are greater than 2.5 cm in diameter. An MRI should be performed for all high-risk lesions. Intermediate-risk lesions are atypical dimples (>5 mm in diameter or >2.5 cm from the anal verge), hemangiomas less than 2.5 cm in diameter, and hypertrichosis. An ultrasound can screen for spinal dysraphism in these cases as long as imaging is performed prior to 6 months of age. If the child is older than 6 months, an MRI should be performed. Low-risk lesions that do not require imaging are simple dimples, hyperpigmentation, hypopigmentation, melanocytic nevi, port-wine stains, and telangiectases.
Epidermal Nevi
Children with epidermal nevi should have a complete physical examination, focusing on the skeletal system, central nervous system, and eyes. There are no specifically recommended imaging studies or referrals; however, several diagnostic clues can aid in the diagnosis of an epidermal nevus syndrome13:
• Schimmelpenning syndrome: extensive nevus sebaceous and bowing or pain in the legs after 2 years of age
• Phacomatosis pigmentokeratotica: nevus sebaceous and nevus spilus
• Nevus comedonicus syndrome: ipsilateral cataract
• Angora hair nevus syndrome: soft white hair within the nevus
• Becker nevus syndrome: breast hypoplasia
• Proteus syndrome: cerebriform plantar changes
• PIK3CA-related overgrowth spectrum: lipomas, macrodactyly, and/or vascular malformations
• Congenital hemidysplasia with ichthyosiform erythroderma and limb defects: inflammatory epidermal nevi, lateralization, ptychotropism, and ipsilateral limb defects
• Conradi-Hünermann-Happle syndrome: scaly red epidermal nevi without hair follicles and asymmetric limb shortening
Pilomatricomas
In addition to the tent sign—an angulated shape can be appreciated by stretching the skin overlying pilomatricomas—diagnosis of pilomatricoma can be confirmed by transillumination with an otoscope. In this case, a dark shadow typically is cast distal to where the otoscope touches the skin.14 In the case of multiple lesions, the patient should be evaluated for signs of myotonic dystrophy, Turner syndrome, and Gardner syndrome.15
Common Procedures
Pulsed Dye Laser
The pulsed dye laser is the most common laser used for red-colored lesions such as port-wine stains, facial telangiectases, and superficial hemangiomas. It also can be used to treat erythematous scars, verrucae, and psoriasis. In large vascular lesions, it typically is employed at 0.45 to 10 milliseconds every 4 to 6 weeks for 10 or more treatments. Port-wine stains preferably are treated within the first few months of life to provide the most fading without the need for general anesthesia.16 On the other hand, systemic therapy with propranolol is preferred over lasers for infantile hemangiomas.17
Long-Pulsed Alexandrite Laser (755 nm)
The alexandrite laser often is used to treat deeper vascular lesions such as venous lakes and hypertrophic port-wine stains. The operator needs to be cautious, as this laser has a higher incidence of scarring at the settings used to treat vascular lesions (typically fluences around 60–85 J/cm2).18 It also may be used for hair reduction in disorders with hypertrichosis or hidradenitis suppurativa.19
Long-Pulsed Nd:YAG Laser
The long-pulsed Nd:YAG laser also can be used to treat deep vascular lesions and remove unwanted hair. Because of its low window of safety in the treatment of vascular lesions, the alexandrite laser usually is preferred. However, it is the preferred laser for treatment of unwanted hair and hidradenitis suppurativa in darker skin types. It often provides a 50% reduction in hair density after 9 treatments.20
Quality-Switched Lasers
Pigment granules in melanosomes and tattoo particles are targeted with quality-switched (QS) lasers. Typically, a device will contain a combination of QS 532-nm potassium-titanyl-phosphate (KTP) lasers, QS 1064-nm Nd:YAG lasers, and QS 755-nm alexandrite lasers in 1 machine. In general, shorter wavelengths are used to treat epidermal lesions such as ephelides, lentigines, and café-au-lait macules. Longer wavelengths are used to treat deeper lesions such as nevus of Ota. A 2017 review suggested that café-au-lait macules with ragged borders (so-called coast of Maine borders) may respond well to QS lasers.21
Ablative Lasers
The 10,600-nm
Fractionated Lasers
Fractionated lasers can be nonablative (several devices are available in the 1410- to 1927-nm range) or ablative (CO2 or erbium:YAG). In pediatrics, they are usually used to treat burn scars, traumatic scars, and mild to moderate acne scarring.22 The most common side effects from fractionated lasers are prolonged erythema or hyperpigmentation. In addition, it typically takes at least 3 treatments to notice improvements.
Excisions
Pediatric procedural dermatologists remove a variety of unique lesions through excision. A few tips are provided for some of the more common lesions that may be excised in children.
Accessory Tragi
Prior to excising an accessory tragus, the surgeon should consider documenting a facial nerve examination, as accessory tragi can be associated with complete or partial facial nerve dysfunction. Additionally, there usually is an underlying cartilage structure present within the tragus. The cartilage stalk also should be addressed during the excision to avoid a continued palpable deformity after excision.
Dermoid Cysts
Dermoid cysts are the most commonly diagnosed benign orbital lesion in children.23 Exophytic periorbital lesions, which extend outside the orbital rim, can be removed through an infrabrow incision. Endophytic periorbital lesions, which are inside the orbital rim, should be removed through a crease incision. Midline lesions may have an intracranial extension and should be imaged through MRI and/or a computed tomography.24 Because dermoid cysts usually are located below the orbicularis oculi muscle, the muscle should be fixed with a suture prior to closing with skin sutures.
Pilomatricomas
Typically, a linear incision is made overlying the lesion, and then the underlying tumor is removed with sharp or blunt dissection. However, if the overlying skin has been stretched thin, a lenticular excision that includes the thinned skin may improve cosmesis.
Congenital Nevi
Large congenital nevi typically are removed through staged excisions. Lower extremity lesions are best removed before 10 months of age or before walking begins to minimize wound tension. However, if the procedure is not performed in infancy, it is best to wait until walking becomes stable.25 In older children, it is advisable to splint the affected lower extremity for 2 weeks to prevent dehiscence. The interval between excisions typically is 4 to 6 weeks for small lesions and 3 months for larger nevi.
Conclusion
Procedural pediatric dermatology is a broad and emerging field. As this article highlights, children are not small versions of adults and have unique biology, diseases, therapies, social situations, and ethical challenges from adults. This article provides a superficial overview of some of the more common issues faced by pediatric dermatologists and providers who perform procedures on infants, children, and teenagers. Readers who are interested in obtaining a more in-depth understanding of procedural pediatric dermatology should look at Procedural Pediatric Dermatology,1 the first textbook to provide expert opinion and evidence-based information on procedural management of pediatric skin conditions.
Performing dermatologic procedures in infants, children, and teenagers presents many unique challenges. There may be unique diagnoses, different instruments, differences in skin biology, or different approaches to pain management and anesthesia; the inclusion of a third party (caregivers) in decision processes; or a need to assess maturity level or to optimize outcomes over the patient’s lifetime. The field of pediatric procedural dermatology is broad. This article reviews some of the more common procedures performed by pediatric dermatologists and some of the more common ethical and quality-of-life (QOL) considerations one might face in procedural pediatric dermatology. (The textbook Procedural Pediatric Dermatology1 offers a thorough discussion of this topic.)
Quality of Life
More often than not, procedures are performed in pediatric dermatology to improve QOL rather than to prevent morbidity or mortality. In the case of many self-limited conditions, such as ingrown nails or pyogenic granulomas, it is clear that intervention will improve the patient’s QOL. In the case of warts and molluscum contagiosum, emotional, social, and cultural considerations play a large role in determining whether an intervention will improve QOL. Finally, some conditions, such as genodermatoses, giant congenital melanocytic nevi, and large vascular malformations, may be associated with additional systemic symptoms and may not have good treatment options for cure. In these cases, procedural interventions will result in a mixture of positive and negative QOL outcomes that can occur at the same time.
Bemmels et al2 published a qualitative study that provides a good foundation for understanding the positive and negative effects of procedural interventions on children and teenagers. In their study, children and teenagers who underwent reconstructive surgery for craniofacial differences noted improved self-esteem and reduced stigmatization. However, they also experienced negative outcomes, including an addiction to attaining a perfect surgical face, missing school for treatments, difficulty adjusting to an evolving appearance, anxiety related to not knowing when treatments will end, and experiencing stigma related to undergoing surgery.2 Thus, a comprehensive plan for the management of children who need ongoing procedures should include some level of psychosocial support. Two good references on supporting young patients with visible differences include CBT for Appearance Anxiety: Psychosocial Interventions for Anxiety Due to Visible Difference3 and Reaching Teens: Strength-Based, Trauma-Sensitive, Resilience-Building Communication Strategies Rooted in Positive Youth Development.4
Ethics
Ethical decisions in pediatric procedural dermatology differ from adult dermatology in 3 major ways: (1) the involvement of a third party (ie, parents or legal guardians), (2) the need to assess the maturity of the patient, and (3) the need to know local laws in the jurisdiction in which care is being provided. Ethical dilemmas occur when the desires of the child, parents/guardians, and dermatologist are not in alignment. In these cases, it is important to be prepared with a moral or ethical framework to guide decision-making when conflicts occur. Two great resources are the best interest standard5 and the publication entitled, “Informed Consent in Decision-making in Pediatric Practice,” from the American Academy of Pediatrics.6
In pediatrics, it often is better to conceptualize medical decision-making as a combination of informed permission and assent of the patient rather than informed consent. Informed permission describes how a parent or surrogate makes decisions for the child or adolescent and is similar to informed consent. A parent’s informed permission may be in conflict with a child’s wishes, but it is assumed that the parent is acting in the best interest of the child. Assent of the patient is the process of obtaining a minor’s agreement to undergo an intervention even though he/she may lack legal authority or decision-making capacity to provide standard informed consent. It is important to respect the child’s right to assent to interventions to the extent that their maturity level permits to develop trust with the dermatologist and medical encounters in general.
These differences emphasize an active process in which the patient, caregiver, and physician are all involved in the health care process and allow for increasing inclusion of the child as is developmentally appropriate. In the end, however, parents have the legal authority to give or withhold permission for a procedure.7 When this conflicts with a child’s dissent, the dermatologist will need to objectively explore the reasons for the conflict and decide if a procedure is not in the child’s best interests. If a mutual understanding cannot be reached between the dermatologist and parents, obtaining a second opinion is a good option.8
Common Diagnoses
The most common diagnoses unique to procedural pediatric dermatology include congenital melanocytic nevi, vascular anomalies, midline lesions, epidermal nevi, and pilomatricomas. Prior to intervening on these lesions, it is important to consider evaluating for associated diseases.
Congenital Melanocytic Nevi
Nevus Outreach has published best practices for the management of congenital melanocytic nevi.9 In newborns with a congenital melanocytic nevus greater than 3 cm in diameter or more than 20 satellite lesions, it is recommended that magnetic resonance imaging (MRI) of the brain and spine with and without gadolinium contrast be obtained before 6 months of age. Within the first 6 months of life, these children also should see ophthalmologists, neurologists, pediatric dermatologists, and plastic surgeons. These early referrals will help to establish a baseline for the patient and plan for possible interventions, if needed. Additionally, before 3 years of age, every child should be referred to psychology, even if he/she is asymptomatic.10
Vascular Anomalies
Prior to intervening on a vascular anomaly, it is important to accurately classify the lesion. Once the lesion is classified, an evaluation and treatment plan can be developed. The International Society for the Study of Vascular Anomalies has published a detailed classification guide that is a useful starting point in the management of vascular anomalies.11 Once a diagnosis is confirmed, further evaluation may include imaging, specialty referrals, genetic testing, biopsy, or blood tests, and a pediatric dermatologist usually helps to coordinate the care of patients with complex vascular anomalies.
Midline Lesions
Certain lesions in the midline may have a higher risk for neural tube dysraphism, and imaging should be performed prior to any procedural intervention.12 Midline cutaneous findings that are highly likely to be associated with dysraphism are lipomas, acrochordons, pseudotails, true tails, aplasia cutis congenita, congenital scars, dermoid cysts, dermoid sinuses, and infantile hemangiomas that are greater than 2.5 cm in diameter. An MRI should be performed for all high-risk lesions. Intermediate-risk lesions are atypical dimples (>5 mm in diameter or >2.5 cm from the anal verge), hemangiomas less than 2.5 cm in diameter, and hypertrichosis. An ultrasound can screen for spinal dysraphism in these cases as long as imaging is performed prior to 6 months of age. If the child is older than 6 months, an MRI should be performed. Low-risk lesions that do not require imaging are simple dimples, hyperpigmentation, hypopigmentation, melanocytic nevi, port-wine stains, and telangiectases.
Epidermal Nevi
Children with epidermal nevi should have a complete physical examination, focusing on the skeletal system, central nervous system, and eyes. There are no specifically recommended imaging studies or referrals; however, several diagnostic clues can aid in the diagnosis of an epidermal nevus syndrome13:
• Schimmelpenning syndrome: extensive nevus sebaceous and bowing or pain in the legs after 2 years of age
• Phacomatosis pigmentokeratotica: nevus sebaceous and nevus spilus
• Nevus comedonicus syndrome: ipsilateral cataract
• Angora hair nevus syndrome: soft white hair within the nevus
• Becker nevus syndrome: breast hypoplasia
• Proteus syndrome: cerebriform plantar changes
• PIK3CA-related overgrowth spectrum: lipomas, macrodactyly, and/or vascular malformations
• Congenital hemidysplasia with ichthyosiform erythroderma and limb defects: inflammatory epidermal nevi, lateralization, ptychotropism, and ipsilateral limb defects
• Conradi-Hünermann-Happle syndrome: scaly red epidermal nevi without hair follicles and asymmetric limb shortening
Pilomatricomas
In addition to the tent sign—an angulated shape can be appreciated by stretching the skin overlying pilomatricomas—diagnosis of pilomatricoma can be confirmed by transillumination with an otoscope. In this case, a dark shadow typically is cast distal to where the otoscope touches the skin.14 In the case of multiple lesions, the patient should be evaluated for signs of myotonic dystrophy, Turner syndrome, and Gardner syndrome.15
Common Procedures
Pulsed Dye Laser
The pulsed dye laser is the most common laser used for red-colored lesions such as port-wine stains, facial telangiectases, and superficial hemangiomas. It also can be used to treat erythematous scars, verrucae, and psoriasis. In large vascular lesions, it typically is employed at 0.45 to 10 milliseconds every 4 to 6 weeks for 10 or more treatments. Port-wine stains preferably are treated within the first few months of life to provide the most fading without the need for general anesthesia.16 On the other hand, systemic therapy with propranolol is preferred over lasers for infantile hemangiomas.17
Long-Pulsed Alexandrite Laser (755 nm)
The alexandrite laser often is used to treat deeper vascular lesions such as venous lakes and hypertrophic port-wine stains. The operator needs to be cautious, as this laser has a higher incidence of scarring at the settings used to treat vascular lesions (typically fluences around 60–85 J/cm2).18 It also may be used for hair reduction in disorders with hypertrichosis or hidradenitis suppurativa.19
Long-Pulsed Nd:YAG Laser
The long-pulsed Nd:YAG laser also can be used to treat deep vascular lesions and remove unwanted hair. Because of its low window of safety in the treatment of vascular lesions, the alexandrite laser usually is preferred. However, it is the preferred laser for treatment of unwanted hair and hidradenitis suppurativa in darker skin types. It often provides a 50% reduction in hair density after 9 treatments.20
Quality-Switched Lasers
Pigment granules in melanosomes and tattoo particles are targeted with quality-switched (QS) lasers. Typically, a device will contain a combination of QS 532-nm potassium-titanyl-phosphate (KTP) lasers, QS 1064-nm Nd:YAG lasers, and QS 755-nm alexandrite lasers in 1 machine. In general, shorter wavelengths are used to treat epidermal lesions such as ephelides, lentigines, and café-au-lait macules. Longer wavelengths are used to treat deeper lesions such as nevus of Ota. A 2017 review suggested that café-au-lait macules with ragged borders (so-called coast of Maine borders) may respond well to QS lasers.21
Ablative Lasers
The 10,600-nm
Fractionated Lasers
Fractionated lasers can be nonablative (several devices are available in the 1410- to 1927-nm range) or ablative (CO2 or erbium:YAG). In pediatrics, they are usually used to treat burn scars, traumatic scars, and mild to moderate acne scarring.22 The most common side effects from fractionated lasers are prolonged erythema or hyperpigmentation. In addition, it typically takes at least 3 treatments to notice improvements.
Excisions
Pediatric procedural dermatologists remove a variety of unique lesions through excision. A few tips are provided for some of the more common lesions that may be excised in children.
Accessory Tragi
Prior to excising an accessory tragus, the surgeon should consider documenting a facial nerve examination, as accessory tragi can be associated with complete or partial facial nerve dysfunction. Additionally, there usually is an underlying cartilage structure present within the tragus. The cartilage stalk also should be addressed during the excision to avoid a continued palpable deformity after excision.
Dermoid Cysts
Dermoid cysts are the most commonly diagnosed benign orbital lesion in children.23 Exophytic periorbital lesions, which extend outside the orbital rim, can be removed through an infrabrow incision. Endophytic periorbital lesions, which are inside the orbital rim, should be removed through a crease incision. Midline lesions may have an intracranial extension and should be imaged through MRI and/or a computed tomography.24 Because dermoid cysts usually are located below the orbicularis oculi muscle, the muscle should be fixed with a suture prior to closing with skin sutures.
Pilomatricomas
Typically, a linear incision is made overlying the lesion, and then the underlying tumor is removed with sharp or blunt dissection. However, if the overlying skin has been stretched thin, a lenticular excision that includes the thinned skin may improve cosmesis.
Congenital Nevi
Large congenital nevi typically are removed through staged excisions. Lower extremity lesions are best removed before 10 months of age or before walking begins to minimize wound tension. However, if the procedure is not performed in infancy, it is best to wait until walking becomes stable.25 In older children, it is advisable to splint the affected lower extremity for 2 weeks to prevent dehiscence. The interval between excisions typically is 4 to 6 weeks for small lesions and 3 months for larger nevi.
Conclusion
Procedural pediatric dermatology is a broad and emerging field. As this article highlights, children are not small versions of adults and have unique biology, diseases, therapies, social situations, and ethical challenges from adults. This article provides a superficial overview of some of the more common issues faced by pediatric dermatologists and providers who perform procedures on infants, children, and teenagers. Readers who are interested in obtaining a more in-depth understanding of procedural pediatric dermatology should look at Procedural Pediatric Dermatology,1 the first textbook to provide expert opinion and evidence-based information on procedural management of pediatric skin conditions.
- Krakowski AC. Procedural Pediatric Dermatology. Phialdelphia, PA: Wolters Kluwer; 2011.
- Bemmels H, Biesecker B, Schmidt J, et al. Psychological and social factors in undergoing reconstructive surgery among individuals with craniofacial conditions: an exploratory study. Cleft Palate Craniofac J. 2013;50:158-167.
- Clarke A, Thompson AR, Jenkinson E, et al. CBT for Appearance Anxiety: Psychosocial Interventions for Anxiety Due to Visible Difference. Chichester, West Sussex: Wiley-Blackwell; 2013.
- Ginsburg KR, Ramirez McClain ZB, eds. Reaching Teens: Strength-Based, Trauma-Sensitive, Resilience-Building Communication Strategies Rooted in Positive Youth Development. 2nd ed. Itasca, IL: American Academy of Pediatrics; 2020.
- Kopelman LM. The best interests standard for incompetent or incapacitated patients of all ages. J Law Med Ethics. 2007;35:187-196.
- Katz AL, Webb SA; Committee on Bioethics. Informed consent in decision-making in pediatric practice. Pediatrics. 2016;138:e20161485. doi:10.1542/peds.2016-1485.
- Michon K. Emancipation of minors. NOLO website. https://www.nolo.com/legal-encyclopedia/emancipation-of-minors-32237.html. Accessed October 14, 2020.
- Cobb C, Bercovitch L. Ethical dilemmas. In: Krakowski AC, ed. Procedural Pediatric Dermatology. Philadelphia, PA: Wolters Kluwer; 2021:7-10.
- Nevus Outreach, Inc., releases best practice guidelines [news release]. Bartlesville, OK: Nevus Outreach Inc; July 7, 2018. https://www.nevus.org/matrices/page_file_download.php?id=239. Accessed October 14, 2020.
- 10. Masnari O, Neuhaus K, Aegerter T, et al. Predictors of health-related quality of life and psychological adjustment in children and adolescents with congenital melanocytic nevi: analysis of parent reports. J Pediatr Psychol. 2019;44:714-725.
- ISSVA classification for vascular anomalies. International Society for the Study of Vascular Anomalies website. https://www.issva.org/UserFiles/file/ISSVA-Classification-2018.pdf. Approved April 2014. Revised May 2018. Accessed October 14, 2020.
- Sewell MJ, Chiu YE, Drolet BA. Neural tube dysraphism: review of cutaneous markers and imaging. Pediatr Dermatol. 2015;32:161-170.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Berreto-Chang OL, Gorell ES, Yamaguma MA, et al. Diagnosis of pilomatricoma using an otoscope. Pediatr Dermatol. 2010;27:554-557.
- Danielson-Cohen A, Lin SJ, Hughes CA, et al. Head and neck pilomatrixoma in children. Arch Otolaryngol Head Neck Surg. 2001;127:1481-1483.
- Jeon H, Bernstein LJ, Belkin DA, et al. Pulsed dye laser treatment of port-wine stains in infancy without the need for general anesthesia. JAMA Dermatol. 2019;155:435-441.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:e20183475. doi:10.1542/peds.2018-3475.
- Tierney EP, Hanke CW. Alexandrite laser for the treatment of port wine stains refractory to pulsed dye laser. Dermatol Surg. 2011;37:1268-1278.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2019;81:76-90.
- Rao K, Sankar TK. Long-pulsed Nd:YAG laser-assisted hair removal in Fitzpatrick skin types IV-VI. Lasers Med Sci. 2011;26:623-626.
- Belkin DA, Neckman JP, Jeon H, et al. Response to laser treatment of café au lait macules based on morphologic features. JAMA Dermatol. 2017;153:1158-1161.
- Kelly K, Lehmer L. Laser surgery. In: Krakowski AC, ed. Procedural Pediatric Dermatology. Philadelphia, PA: Wolters Kluwer; 2021:92-106.
- Eldesouky MA, Elbakary MA. Orbital dermoid cyst: classification and its impact on surgical management. Semin Ophthalmol. 2018;33:170-174.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Metz BJ. Procedural pediatric dermatology. Dermatol Clin. 2013;31:337-346.
- Krakowski AC. Procedural Pediatric Dermatology. Phialdelphia, PA: Wolters Kluwer; 2011.
- Bemmels H, Biesecker B, Schmidt J, et al. Psychological and social factors in undergoing reconstructive surgery among individuals with craniofacial conditions: an exploratory study. Cleft Palate Craniofac J. 2013;50:158-167.
- Clarke A, Thompson AR, Jenkinson E, et al. CBT for Appearance Anxiety: Psychosocial Interventions for Anxiety Due to Visible Difference. Chichester, West Sussex: Wiley-Blackwell; 2013.
- Ginsburg KR, Ramirez McClain ZB, eds. Reaching Teens: Strength-Based, Trauma-Sensitive, Resilience-Building Communication Strategies Rooted in Positive Youth Development. 2nd ed. Itasca, IL: American Academy of Pediatrics; 2020.
- Kopelman LM. The best interests standard for incompetent or incapacitated patients of all ages. J Law Med Ethics. 2007;35:187-196.
- Katz AL, Webb SA; Committee on Bioethics. Informed consent in decision-making in pediatric practice. Pediatrics. 2016;138:e20161485. doi:10.1542/peds.2016-1485.
- Michon K. Emancipation of minors. NOLO website. https://www.nolo.com/legal-encyclopedia/emancipation-of-minors-32237.html. Accessed October 14, 2020.
- Cobb C, Bercovitch L. Ethical dilemmas. In: Krakowski AC, ed. Procedural Pediatric Dermatology. Philadelphia, PA: Wolters Kluwer; 2021:7-10.
- Nevus Outreach, Inc., releases best practice guidelines [news release]. Bartlesville, OK: Nevus Outreach Inc; July 7, 2018. https://www.nevus.org/matrices/page_file_download.php?id=239. Accessed October 14, 2020.
- 10. Masnari O, Neuhaus K, Aegerter T, et al. Predictors of health-related quality of life and psychological adjustment in children and adolescents with congenital melanocytic nevi: analysis of parent reports. J Pediatr Psychol. 2019;44:714-725.
- ISSVA classification for vascular anomalies. International Society for the Study of Vascular Anomalies website. https://www.issva.org/UserFiles/file/ISSVA-Classification-2018.pdf. Approved April 2014. Revised May 2018. Accessed October 14, 2020.
- Sewell MJ, Chiu YE, Drolet BA. Neural tube dysraphism: review of cutaneous markers and imaging. Pediatr Dermatol. 2015;32:161-170.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Berreto-Chang OL, Gorell ES, Yamaguma MA, et al. Diagnosis of pilomatricoma using an otoscope. Pediatr Dermatol. 2010;27:554-557.
- Danielson-Cohen A, Lin SJ, Hughes CA, et al. Head and neck pilomatrixoma in children. Arch Otolaryngol Head Neck Surg. 2001;127:1481-1483.
- Jeon H, Bernstein LJ, Belkin DA, et al. Pulsed dye laser treatment of port-wine stains in infancy without the need for general anesthesia. JAMA Dermatol. 2019;155:435-441.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:e20183475. doi:10.1542/peds.2018-3475.
- Tierney EP, Hanke CW. Alexandrite laser for the treatment of port wine stains refractory to pulsed dye laser. Dermatol Surg. 2011;37:1268-1278.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2019;81:76-90.
- Rao K, Sankar TK. Long-pulsed Nd:YAG laser-assisted hair removal in Fitzpatrick skin types IV-VI. Lasers Med Sci. 2011;26:623-626.
- Belkin DA, Neckman JP, Jeon H, et al. Response to laser treatment of café au lait macules based on morphologic features. JAMA Dermatol. 2017;153:1158-1161.
- Kelly K, Lehmer L. Laser surgery. In: Krakowski AC, ed. Procedural Pediatric Dermatology. Philadelphia, PA: Wolters Kluwer; 2021:92-106.
- Eldesouky MA, Elbakary MA. Orbital dermoid cyst: classification and its impact on surgical management. Semin Ophthalmol. 2018;33:170-174.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Metz BJ. Procedural pediatric dermatology. Dermatol Clin. 2013;31:337-346.
Practice Points
- Children who require repetitive laser or surgical procedures over time benefit from regular monitoring of psychosocial needs.
- The informed consent process for children differs from adult procedural dermatology and should be adjusted to the maturity level of the patient.
- Common diagnoses unique to procedural pediatric dermatology that may require additional investigation include congenital melanocytic nevi, vascular anomalies, epidermal nevi, and midline lesions.
- Specific measures can be performed to improve outcomes when removing accessory tragi, dermoid cysts, pilomatricomas, and congenital nevi.
Bonds and Bridges: The Role of Social Capital in Building a More Diverse Dermatology Workforce
As our specialty seeks to address its lack of racial diversity, many dermatologists have answered recent calls to action.1,2 As we work toward dismantling systemic issues that have created pervasive inequality in our residency application review and interview processes, consideration also should be given to psychosocial issues that underrepresented-in-medicine (UIM) students face before their applications come to our attention. In this article, we explore how potential differences in the social capital of UIM and other disadvantaged dermatology residency applicants contribute to persistent homogeneity among dermatology training programs and the workforce.
The Theory of Capital
The concepts of economic, social, and cultural capital originate from the writings of social theorist Pierre Bourdieu.3 All 3 forms of capital are interconnected, and they relate to each other in ways that often facilitate social division and inequality. Economic capital denotes an individual’s economic resources or wealth, while cultural capital refers to the knowledge, behaviors, and skills that demonstrate his/her economic class (eg, communication style, table manners).3 Social capital refers to an individual’s interpersonal connections in personal and professional settings and can be subdivided into 3 categories: bonds, bridges, and linkages.4,5 Herein, we will focus on bonds and bridges.
It has been suggested that bonds are important for “getting by,” while bridges are critical for “getting ahead.”5 Bonds refer to close relationships within a community of people with shared characteristics, such as racial/ethnic identity and culture, access to information, and resources (eg, family, friends). These bonds provide trust, safety, and financial and emotional support; however, they are considered to be inward-looking and can promote exclusion and homogeneity.5
On the other hand, bridges refer to social relationships that extend outward beyond one’s close circle of family and friends to other people with shared interests and goals who may have different social or cultural identities (eg, professional colleagues). These bridges are considered to be outward-looking and provide many benefits to individuals and society. They link diverse individuals, which tends to increase tolerance and disrupt stereotypes, and they facilitate the sharing of ideas, information, and innovation. Additionally, bridges between individuals from different networks facilitate access to increased resources and opportunities for all parties.5
The 3 forms of capital are inextricably linked. For example, with economic capital, a child’s family can purchase access to a prestigious private high school, where he/she will gain valuable social capital through bridges with other students and their families. At this school, the child also will accumulate cultural capital that increases his/her sense of belonging in these circles. Subsequently, both the social and cultural capital accumulated at this private high school can be exchanged for economic capital via social networks, skills, values, and behaviors that facilitate entry into higher education and professional training. As such, these 3 forms of capital work together to continue social/class divisions, hierarchies, and ultimately inequality.
Impact of Social Capital in Pursuing a Medical Career
For medical students whose bonds (ie, close family, friends) include physicians or other health care professionals, the journey to studying medicine and entering their chosen specialty will be facilitated by financial security, valuable “inside information” about the application process, study skills, and even clinical guidance. Additionally, these students will have access to professional networks for mentorship, shadowing experiences, and other potential advantages. Furthermore, social capital is associated with higher self-esteem,6 which likely improves academic performance and wards off imposter syndrome in these students.
For medical students from lower socioeconomic status backgrounds or those whose inner circles do not include physicians or other health care professionals, accumulating the social and cultural capital needed to successfully navigate a medical career is more difficult. Although they may receive support and encouragement from family and friends, they will not have access to the same valuable information and connections that facilitate success; rather, they will have a further distance to travel, and this distance should be acknowledged in the residency application review process.
Acquiring Social Capital as a UIM Student
Despite the benefits of social and cultural capital, acquiring them takes a toll. For those UIM students who start life from a disadvantaged place, the accumulation of social capital does not come easily; rather, it demands effort and time that has the potential to detract from a student’s focus on the academic demands of medical education.7 Programs that attempt to improve disadvantaged students’ access to credible information, role models, and mentors can help lift some of the burden from the individual student’s shoulders. For example, studies have demonstrated the benefits of harnessing technology to enhance mentorship programs that increase social capital of disadvantaged populations.8-11 This approach already is in progress, bolstered by advances made in digital communications during the coronavirus disease 2019 pandemic.12 Student-led networking groups that connect remotely have been shown to build social capital bonds and bridges that facilitate collaborative learning, relationship building, and information sharing.8-11 There are existing online UIM student networks that individual dermatologists, institutions, and national organizations can partner with to facilitate the construction of bridges between these UIM student groups and dermatologists who can provide accurate, high-yield information and professional networking; however, one limitation of this suggestion is the disparate access to technology in the UIM community.
Final Thoughts
It is important to note that assumptions should not be made about the level of economic, social, or cultural capital an individual possesses based on his/her race or ethnicity. Instead, mentors should attempt to be available to a diverse pool of students; take the time to get to know these students; and then provide the types of mentorship, information, exposure, and networking that each individual student needs. Another approach is to make a concerted effort to ensure that all students receive the same amount and quality of information about medical education and our specialty regardless of their level of economic, cultural, or social capital. Moreover, beyond the promotion of diversity through increasing numbers of UIM applicants, we should seek to reshape our specialty into a space that does not require students to subdue their existing diverse forms of capital but rather to bring these different perspectives and lived experiences to the table.13
- Bray JK, McMichael AJ, Huang WW, et al. Publication rates on the topic of racial and ethnic diversity in dermatology versus other specialties. Dermatol Online J. 2020;26:7.
- Pritchett EN, Pandya AG, Ferguson NN, et al. Diversity in dermatology: roadmap for improvement. J Am Acad Dermatol. 2018;79:337-341.
- Bourdieu P. The forms of capital. In: Richardson J, ed. Handbook of Theory and Research for the Sociology of Education. Westport, CT: Greenwood; 1986:241-258.
- Granovetter MS. The strength of weak ties. Am J Sociol. 1973;78:1360-1380.
- Putnam RD. Bowling alone: America’s declining social capital. J Democracy. 1995;6:65-78.
- Han S. Longitudinal association between social capital and self-esteem: a matter of context. Psychiatry Research. 2015;226:340-346.
- Kirschling JM. Building social capital: leading and leveraging constituencies outside the college. J Nurs Educ. 2004;43:517-519.
- Radlick RL, Svedberg P, Nygren JM, et al. Digitally enhanced mentoring for immigrant youth social capital: protocol for a mixed methods pilot study and a randomized controlled trial [published online March 17, 2020]. JMIR Research Protocols. doi:10.2196/16472.
- Koh LC, Walker R, Wollersheim D, et al. I think someone is walking with me: the use of mobile phone for social capital development among women in four refugee communities. Int J Migration Health Social Care. 2018;14:411-424.
- Hartley A, Kassam AA. Social networking for learning in higher education: capitalising on social capital. ResearchGate website.https://www.researchgate.net/publication/311097860_Social_Networking_for_Learning_in_Higher_Education_Capitalising_on_Social_Capital. Published November 2016. Accessed October 19, 2020.
- Zalon ML. Using technology to build community in professional associations. J Contin Educ Nurs. 2008;39:235-240.
- Stewart CR, Chernoff KA, Wildman HF, et al. Recommendations for medical student preparedness and equity for dermatology residency applications during the COVID-19 pandemic. J Am Acad Dermatol. 2020;83:E225-E226.
- Brosnan C, Southgate E, Outram S, et al. Experiences of medical students who are first in family to attend university. Med Educ. 2016;50:842-851.
As our specialty seeks to address its lack of racial diversity, many dermatologists have answered recent calls to action.1,2 As we work toward dismantling systemic issues that have created pervasive inequality in our residency application review and interview processes, consideration also should be given to psychosocial issues that underrepresented-in-medicine (UIM) students face before their applications come to our attention. In this article, we explore how potential differences in the social capital of UIM and other disadvantaged dermatology residency applicants contribute to persistent homogeneity among dermatology training programs and the workforce.
The Theory of Capital
The concepts of economic, social, and cultural capital originate from the writings of social theorist Pierre Bourdieu.3 All 3 forms of capital are interconnected, and they relate to each other in ways that often facilitate social division and inequality. Economic capital denotes an individual’s economic resources or wealth, while cultural capital refers to the knowledge, behaviors, and skills that demonstrate his/her economic class (eg, communication style, table manners).3 Social capital refers to an individual’s interpersonal connections in personal and professional settings and can be subdivided into 3 categories: bonds, bridges, and linkages.4,5 Herein, we will focus on bonds and bridges.
It has been suggested that bonds are important for “getting by,” while bridges are critical for “getting ahead.”5 Bonds refer to close relationships within a community of people with shared characteristics, such as racial/ethnic identity and culture, access to information, and resources (eg, family, friends). These bonds provide trust, safety, and financial and emotional support; however, they are considered to be inward-looking and can promote exclusion and homogeneity.5
On the other hand, bridges refer to social relationships that extend outward beyond one’s close circle of family and friends to other people with shared interests and goals who may have different social or cultural identities (eg, professional colleagues). These bridges are considered to be outward-looking and provide many benefits to individuals and society. They link diverse individuals, which tends to increase tolerance and disrupt stereotypes, and they facilitate the sharing of ideas, information, and innovation. Additionally, bridges between individuals from different networks facilitate access to increased resources and opportunities for all parties.5
The 3 forms of capital are inextricably linked. For example, with economic capital, a child’s family can purchase access to a prestigious private high school, where he/she will gain valuable social capital through bridges with other students and their families. At this school, the child also will accumulate cultural capital that increases his/her sense of belonging in these circles. Subsequently, both the social and cultural capital accumulated at this private high school can be exchanged for economic capital via social networks, skills, values, and behaviors that facilitate entry into higher education and professional training. As such, these 3 forms of capital work together to continue social/class divisions, hierarchies, and ultimately inequality.
Impact of Social Capital in Pursuing a Medical Career
For medical students whose bonds (ie, close family, friends) include physicians or other health care professionals, the journey to studying medicine and entering their chosen specialty will be facilitated by financial security, valuable “inside information” about the application process, study skills, and even clinical guidance. Additionally, these students will have access to professional networks for mentorship, shadowing experiences, and other potential advantages. Furthermore, social capital is associated with higher self-esteem,6 which likely improves academic performance and wards off imposter syndrome in these students.
For medical students from lower socioeconomic status backgrounds or those whose inner circles do not include physicians or other health care professionals, accumulating the social and cultural capital needed to successfully navigate a medical career is more difficult. Although they may receive support and encouragement from family and friends, they will not have access to the same valuable information and connections that facilitate success; rather, they will have a further distance to travel, and this distance should be acknowledged in the residency application review process.
Acquiring Social Capital as a UIM Student
Despite the benefits of social and cultural capital, acquiring them takes a toll. For those UIM students who start life from a disadvantaged place, the accumulation of social capital does not come easily; rather, it demands effort and time that has the potential to detract from a student’s focus on the academic demands of medical education.7 Programs that attempt to improve disadvantaged students’ access to credible information, role models, and mentors can help lift some of the burden from the individual student’s shoulders. For example, studies have demonstrated the benefits of harnessing technology to enhance mentorship programs that increase social capital of disadvantaged populations.8-11 This approach already is in progress, bolstered by advances made in digital communications during the coronavirus disease 2019 pandemic.12 Student-led networking groups that connect remotely have been shown to build social capital bonds and bridges that facilitate collaborative learning, relationship building, and information sharing.8-11 There are existing online UIM student networks that individual dermatologists, institutions, and national organizations can partner with to facilitate the construction of bridges between these UIM student groups and dermatologists who can provide accurate, high-yield information and professional networking; however, one limitation of this suggestion is the disparate access to technology in the UIM community.
Final Thoughts
It is important to note that assumptions should not be made about the level of economic, social, or cultural capital an individual possesses based on his/her race or ethnicity. Instead, mentors should attempt to be available to a diverse pool of students; take the time to get to know these students; and then provide the types of mentorship, information, exposure, and networking that each individual student needs. Another approach is to make a concerted effort to ensure that all students receive the same amount and quality of information about medical education and our specialty regardless of their level of economic, cultural, or social capital. Moreover, beyond the promotion of diversity through increasing numbers of UIM applicants, we should seek to reshape our specialty into a space that does not require students to subdue their existing diverse forms of capital but rather to bring these different perspectives and lived experiences to the table.13
As our specialty seeks to address its lack of racial diversity, many dermatologists have answered recent calls to action.1,2 As we work toward dismantling systemic issues that have created pervasive inequality in our residency application review and interview processes, consideration also should be given to psychosocial issues that underrepresented-in-medicine (UIM) students face before their applications come to our attention. In this article, we explore how potential differences in the social capital of UIM and other disadvantaged dermatology residency applicants contribute to persistent homogeneity among dermatology training programs and the workforce.
The Theory of Capital
The concepts of economic, social, and cultural capital originate from the writings of social theorist Pierre Bourdieu.3 All 3 forms of capital are interconnected, and they relate to each other in ways that often facilitate social division and inequality. Economic capital denotes an individual’s economic resources or wealth, while cultural capital refers to the knowledge, behaviors, and skills that demonstrate his/her economic class (eg, communication style, table manners).3 Social capital refers to an individual’s interpersonal connections in personal and professional settings and can be subdivided into 3 categories: bonds, bridges, and linkages.4,5 Herein, we will focus on bonds and bridges.
It has been suggested that bonds are important for “getting by,” while bridges are critical for “getting ahead.”5 Bonds refer to close relationships within a community of people with shared characteristics, such as racial/ethnic identity and culture, access to information, and resources (eg, family, friends). These bonds provide trust, safety, and financial and emotional support; however, they are considered to be inward-looking and can promote exclusion and homogeneity.5
On the other hand, bridges refer to social relationships that extend outward beyond one’s close circle of family and friends to other people with shared interests and goals who may have different social or cultural identities (eg, professional colleagues). These bridges are considered to be outward-looking and provide many benefits to individuals and society. They link diverse individuals, which tends to increase tolerance and disrupt stereotypes, and they facilitate the sharing of ideas, information, and innovation. Additionally, bridges between individuals from different networks facilitate access to increased resources and opportunities for all parties.5
The 3 forms of capital are inextricably linked. For example, with economic capital, a child’s family can purchase access to a prestigious private high school, where he/she will gain valuable social capital through bridges with other students and their families. At this school, the child also will accumulate cultural capital that increases his/her sense of belonging in these circles. Subsequently, both the social and cultural capital accumulated at this private high school can be exchanged for economic capital via social networks, skills, values, and behaviors that facilitate entry into higher education and professional training. As such, these 3 forms of capital work together to continue social/class divisions, hierarchies, and ultimately inequality.
Impact of Social Capital in Pursuing a Medical Career
For medical students whose bonds (ie, close family, friends) include physicians or other health care professionals, the journey to studying medicine and entering their chosen specialty will be facilitated by financial security, valuable “inside information” about the application process, study skills, and even clinical guidance. Additionally, these students will have access to professional networks for mentorship, shadowing experiences, and other potential advantages. Furthermore, social capital is associated with higher self-esteem,6 which likely improves academic performance and wards off imposter syndrome in these students.
For medical students from lower socioeconomic status backgrounds or those whose inner circles do not include physicians or other health care professionals, accumulating the social and cultural capital needed to successfully navigate a medical career is more difficult. Although they may receive support and encouragement from family and friends, they will not have access to the same valuable information and connections that facilitate success; rather, they will have a further distance to travel, and this distance should be acknowledged in the residency application review process.
Acquiring Social Capital as a UIM Student
Despite the benefits of social and cultural capital, acquiring them takes a toll. For those UIM students who start life from a disadvantaged place, the accumulation of social capital does not come easily; rather, it demands effort and time that has the potential to detract from a student’s focus on the academic demands of medical education.7 Programs that attempt to improve disadvantaged students’ access to credible information, role models, and mentors can help lift some of the burden from the individual student’s shoulders. For example, studies have demonstrated the benefits of harnessing technology to enhance mentorship programs that increase social capital of disadvantaged populations.8-11 This approach already is in progress, bolstered by advances made in digital communications during the coronavirus disease 2019 pandemic.12 Student-led networking groups that connect remotely have been shown to build social capital bonds and bridges that facilitate collaborative learning, relationship building, and information sharing.8-11 There are existing online UIM student networks that individual dermatologists, institutions, and national organizations can partner with to facilitate the construction of bridges between these UIM student groups and dermatologists who can provide accurate, high-yield information and professional networking; however, one limitation of this suggestion is the disparate access to technology in the UIM community.
Final Thoughts
It is important to note that assumptions should not be made about the level of economic, social, or cultural capital an individual possesses based on his/her race or ethnicity. Instead, mentors should attempt to be available to a diverse pool of students; take the time to get to know these students; and then provide the types of mentorship, information, exposure, and networking that each individual student needs. Another approach is to make a concerted effort to ensure that all students receive the same amount and quality of information about medical education and our specialty regardless of their level of economic, cultural, or social capital. Moreover, beyond the promotion of diversity through increasing numbers of UIM applicants, we should seek to reshape our specialty into a space that does not require students to subdue their existing diverse forms of capital but rather to bring these different perspectives and lived experiences to the table.13
- Bray JK, McMichael AJ, Huang WW, et al. Publication rates on the topic of racial and ethnic diversity in dermatology versus other specialties. Dermatol Online J. 2020;26:7.
- Pritchett EN, Pandya AG, Ferguson NN, et al. Diversity in dermatology: roadmap for improvement. J Am Acad Dermatol. 2018;79:337-341.
- Bourdieu P. The forms of capital. In: Richardson J, ed. Handbook of Theory and Research for the Sociology of Education. Westport, CT: Greenwood; 1986:241-258.
- Granovetter MS. The strength of weak ties. Am J Sociol. 1973;78:1360-1380.
- Putnam RD. Bowling alone: America’s declining social capital. J Democracy. 1995;6:65-78.
- Han S. Longitudinal association between social capital and self-esteem: a matter of context. Psychiatry Research. 2015;226:340-346.
- Kirschling JM. Building social capital: leading and leveraging constituencies outside the college. J Nurs Educ. 2004;43:517-519.
- Radlick RL, Svedberg P, Nygren JM, et al. Digitally enhanced mentoring for immigrant youth social capital: protocol for a mixed methods pilot study and a randomized controlled trial [published online March 17, 2020]. JMIR Research Protocols. doi:10.2196/16472.
- Koh LC, Walker R, Wollersheim D, et al. I think someone is walking with me: the use of mobile phone for social capital development among women in four refugee communities. Int J Migration Health Social Care. 2018;14:411-424.
- Hartley A, Kassam AA. Social networking for learning in higher education: capitalising on social capital. ResearchGate website.https://www.researchgate.net/publication/311097860_Social_Networking_for_Learning_in_Higher_Education_Capitalising_on_Social_Capital. Published November 2016. Accessed October 19, 2020.
- Zalon ML. Using technology to build community in professional associations. J Contin Educ Nurs. 2008;39:235-240.
- Stewart CR, Chernoff KA, Wildman HF, et al. Recommendations for medical student preparedness and equity for dermatology residency applications during the COVID-19 pandemic. J Am Acad Dermatol. 2020;83:E225-E226.
- Brosnan C, Southgate E, Outram S, et al. Experiences of medical students who are first in family to attend university. Med Educ. 2016;50:842-851.
- Bray JK, McMichael AJ, Huang WW, et al. Publication rates on the topic of racial and ethnic diversity in dermatology versus other specialties. Dermatol Online J. 2020;26:7.
- Pritchett EN, Pandya AG, Ferguson NN, et al. Diversity in dermatology: roadmap for improvement. J Am Acad Dermatol. 2018;79:337-341.
- Bourdieu P. The forms of capital. In: Richardson J, ed. Handbook of Theory and Research for the Sociology of Education. Westport, CT: Greenwood; 1986:241-258.
- Granovetter MS. The strength of weak ties. Am J Sociol. 1973;78:1360-1380.
- Putnam RD. Bowling alone: America’s declining social capital. J Democracy. 1995;6:65-78.
- Han S. Longitudinal association between social capital and self-esteem: a matter of context. Psychiatry Research. 2015;226:340-346.
- Kirschling JM. Building social capital: leading and leveraging constituencies outside the college. J Nurs Educ. 2004;43:517-519.
- Radlick RL, Svedberg P, Nygren JM, et al. Digitally enhanced mentoring for immigrant youth social capital: protocol for a mixed methods pilot study and a randomized controlled trial [published online March 17, 2020]. JMIR Research Protocols. doi:10.2196/16472.
- Koh LC, Walker R, Wollersheim D, et al. I think someone is walking with me: the use of mobile phone for social capital development among women in four refugee communities. Int J Migration Health Social Care. 2018;14:411-424.
- Hartley A, Kassam AA. Social networking for learning in higher education: capitalising on social capital. ResearchGate website.https://www.researchgate.net/publication/311097860_Social_Networking_for_Learning_in_Higher_Education_Capitalising_on_Social_Capital. Published November 2016. Accessed October 19, 2020.
- Zalon ML. Using technology to build community in professional associations. J Contin Educ Nurs. 2008;39:235-240.
- Stewart CR, Chernoff KA, Wildman HF, et al. Recommendations for medical student preparedness and equity for dermatology residency applications during the COVID-19 pandemic. J Am Acad Dermatol. 2020;83:E225-E226.
- Brosnan C, Southgate E, Outram S, et al. Experiences of medical students who are first in family to attend university. Med Educ. 2016;50:842-851.
Practice Points
- Achieving diversity in the field of dermatology will require a concerted effort to equalize access to mentorship, information, exposure, and networking for students of all backgrounds.
- Valuing diverse forms of capital in applicants ultimately will strengthen the dermatology workforce through inclusion of various lived experiences and perspectives.

Translating the 2019 AAD-NPF Guidelines of Care for the Management of Psoriasis in Pediatric Patients
In November 2019, the American Academy of Dermatology (AAD) and the National Psoriasis Foundation (NPF) released their first set of recommendations for the management of pediatric psoriasis.1 The pediatric guidelines discuss methods of quantifying disease severity in children, triggers and comorbidities, and the efficacy and safety of various therapeutic agents. This review aims to discuss, in a condensed form, special considerations unique to the management of children with psoriasis as presented in the guidelines as well as grade A– and grade B–level treatment recommendations (Table).
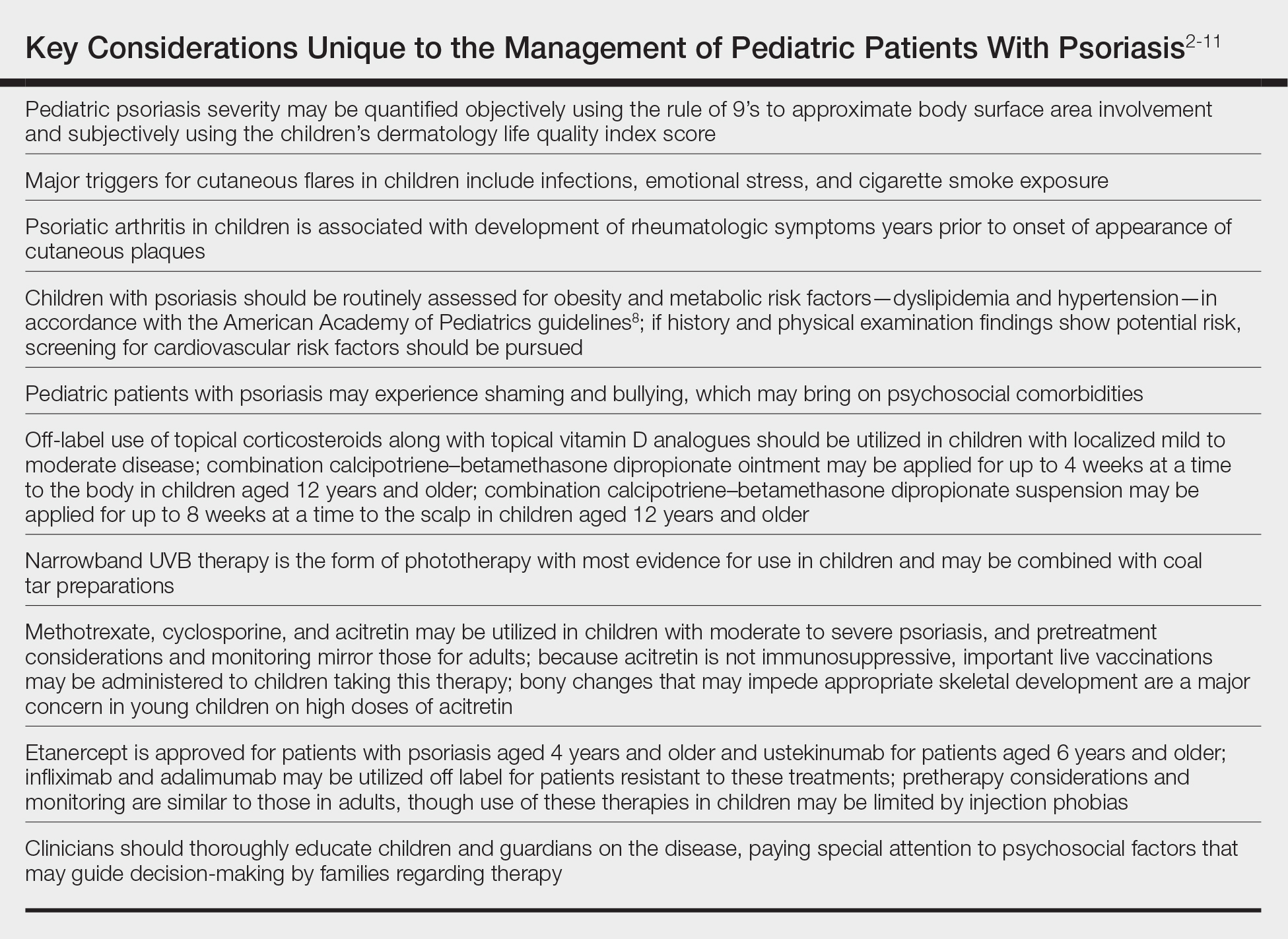
Quantifying Psoriasis Severity in Children
Percentage body surface area (BSA) involvement is the most common mode of grading psoriasis severity, with less than 3% BSA involvement being considered mild, 3% to 10% BSA moderate, and more than 10% severe disease. In children, the standard method of measuring BSA is the rule of 9’s: the head and each arm make up 9% of the total BSA, each leg and the front and back of the torso respectively each make up 18%, and the genitalia make up 1%. It also is important to consider impact on quality of life, which may be remarkable in spite of limited BSA involvement. The children’s dermatology life quality index score may be utilized in combination with affected BSA to determine the burden of psoriasis in context of impact on daily life. This metric is available in both written and cartoon form, and it consists of 10 questions that include variables such as severity of itch, impact on social life, and effects on sleep. Most notably, this tool incorporates pruritus,2 which generally is addressed inadequately in pediatric psoriasis.
Triggers and Comorbidities in Pediatric Patients
In children, it is important to identify and eliminate modifiable factors that may prompt psoriasis flares. Infections, particularly group A beta-hemolytic streptococcal infections, are a major trigger in neonates and infants. Other exacerbating factors in children include emotional stress, secondhand cigarette smoke, Kawasaki disease, and withdrawal from systemic corticosteroids.
Psoriatic arthritis (PsA) is a burdensome comorbidity affecting children with psoriasis. The prevalence of joint disease is 15-times greater in children with psoriasis vs those without,3 and 80% of children with PsA develop rheumatologic symptoms, which typically include oligoarticular disease and dactylitis in infants and girls and enthesitis and axial joint involvement in boys and older children, years prior to the onset of cutaneous disease.4 Uveitis often occurs in children with psoriasis and PsA but not in those with isolated cutaneous disease.
Compared to unaffected children, pediatric patients with psoriasis have greater prevalence of metabolic and cardiovascular risk factors during childhood, including central obesity, hypertension, hypertriglyceridemia, hypercholesterolemia, insulin resistance, atherosclerosis, arrythmia, and valvular heart disease. Family history of obesity increases the risk for early-onset development of cutaneous lesions,5,6 and weight reduction may alleviate severity of psoriasis lesions.7 In the United States, many of the metabolic associations observed are particularly robust in Black and Hispanic children vs those of other races. Furthermore, the prevalence of inflammatory bowel disease is 3- to 4-times higher in children with psoriasis compared to those without.
As with other cutaneous diseases, it is important to be aware of social and mental health concerns in children with psoriasis. The majority of pediatric patients with psoriasis experience name-calling, shaming, or bullying, and many have concerns from skin shedding and malodor. Independent risk for depression after the onset of psoriasis is high. Affected older children and adolescents are at increased risk for alcohol and drug abuse as well as eating disorders.
Despite these identified comorbidities, there are no unique screening recommendations for arthritis, ophthalmologic disease, metabolic disease, cardiovascular disease, gastrointestinal tract disease, or mental health issues in children with psoriasis. Rather, these patients should be monitored according to the American Academy of Pediatrics or American Diabetes Association guidelines for all pediatric patients.8,9 Nonetheless, educating patients and guardians about these potential issues may be warranted.
Topical Therapies
For children with mild to moderate psoriasis, topical therapies are first line. Despite being off label, topical corticosteroids are the mainstay of therapy for localized psoriatic plaques in children. Topical vitamin D analogues—calcitriol and calcipotriol/calcipotriene—are highly effective and well tolerated, and they frequently are used in combination with topical corticosteroids. Topical calcineurin inhibitors, namely tacrolimus, also are used off label but are considered first line for sensitive regions of the skin in children, including the face, genitalia, and body folds. There currently is limited evidence for supporting the use of the topical vitamin A analogue tazarotene in children with psoriasis, though some consider its off-label use effective for pediatric nail psoriasis. It also may be used as an adjunct to topical corticosteroids to minimize irritation.
Although there is no gold standard topical regimen, combination therapy with a high-potency topical steroid and topical vitamin D analogue commonly is used to minimize steroid-induced side effects. For the first 2 weeks of treatment, they each may be applied once daily or mixed together and applied twice daily. For subsequent maintenance, topical calcipotriene may be applied on weekdays and topical steroids only on weekends. Combination calcipotriol–betamethasone dipropionate also is available as cream, ointment, foam, and suspension vehicles for use on the body and scalp in children aged 12 years and older. Tacrolimus ointment 0.1% may be applied in a thin layer up to twice daily. Concurrent emollient use also is recommended with these therapies.
Health care providers should educate patients and guardians about the potential side effects of topical therapies. They also should provide explicit instructions for amount, site, frequency, and duration of application. Topical corticosteroids commonly result in burning on application and may potentially cause skin thinning and striae with overuse. Topical vitamin D analogues may result in local irritation that may be improved by concurrent emollient use, and they generally should be avoided on sensitive sites. Topical calcineurin inhibitors are associated with burning, stinging, and pruritus, and the US Food and Drug Administration has issued a black-box warning related to risk for lymphoma with their chronic intermittent use. However, it was based on rare reports of lymphoma in transplant patients taking oral calcineurin inhibitors; no clinical trials to date in humans have demonstrated an increased risk for malignancy with topical calcineurin inhibitors.10 Tazarotene should be used cautiously in females of childbearing age given its teratogenic potential.
Children younger than 7 years are especially prone to suppression of the hypothalamic-pituitary-adrenal axis from topical corticosteroid therapy and theoretically hypercalcemia and hypervitaminosis D from topical vitamin D analogues, as their high BSA-to-volume ratio increases potential for systemic absorption. Children should avoid occlusive application of topical vitamin D analogues to large areas of the skin. Monitoring of vitamin D metabolites in the serum may be considered if calcipotriene or calcipotriol application to a large BSA is warranted.
Light-Based Therapy
In children with widespread psoriasis or those refractory to topical therapy, phototherapy may be considered. Narrowband UVB (311- to 313-nm wavelength) therapy is considered a first-line form of phototherapy in pediatric psoriasis. Mineral oil or emollient pretreatment to affected areas may augment the efficacy of UV-based treatments.11 Excimer laser and UVA also may be efficacious, though evidence is limited in children. Treatment is recommended to start at 3 days a week, and once improvement is seen, the frequency can be decreased to 2 days a week. Once desired clearance is achieved, maintenance therapy can be continued at even longer intervals. Adjunctive use of tar preparations may potentiate the efficacy of phototherapy, though there is a theoretical increased risk for carcinogenicity with prolonged use of coal tar. Side effects of phototherapy include erythema, blistering hyperpigmentation, and pruritus. Psoralen is contraindicated in children younger than 12 years. All forms of phototherapy are contraindicated in children with generalized erythroderma and cutaneous cancer syndromes. Other important pediatric-specific considerations include anxiety that may be provoked by UV light machines and inconvenience of frequent appointments.
Nonbiologic Systemic Therapies
Systemic therapies may be considered in children with recalcitrant, widespread, or rapidly progressing psoriasis, particularly if the disease is accompanied by severe emotional and psychological burden. These drugs, which include methotrexate, cyclosporine, and acitretin (see eTable for recommended dosing), are advantageous in that they may be combined with other therapies; however, they have potential for dangerous toxicities.
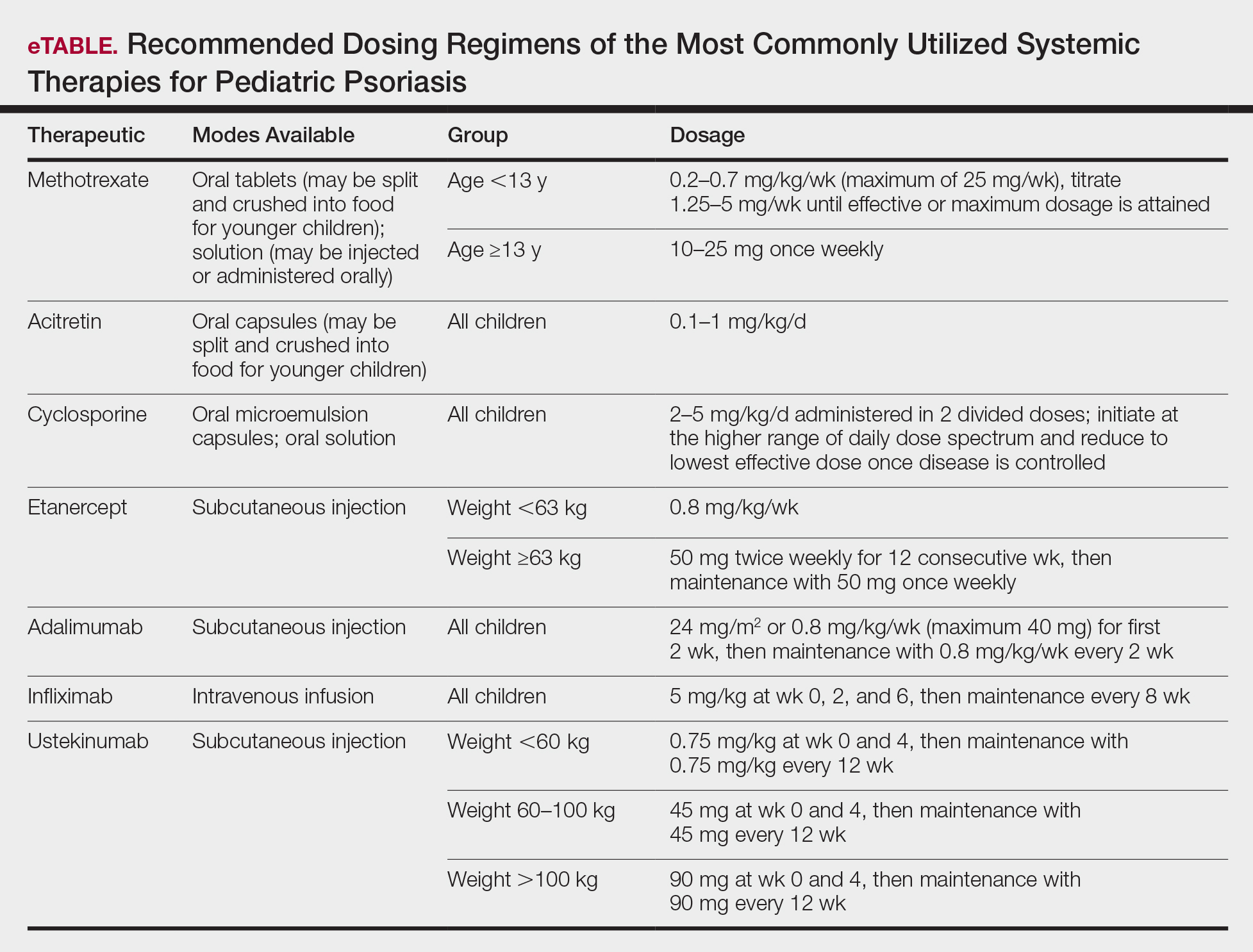
Methotrexate is the most frequently utilized systemic therapy for psoriasis worldwide in children because of its low cost, once-weekly dosing, and the substantial amount of long-term efficacy and safety data available in the pediatric population. It is slow acting initially but has excellent long-term efficacy for nearly every subtype of psoriasis. The most common side effect of methotrexate is gastrointestinal tract intolerance. Nonetheless, adverse events are rare in children without prior history, with 1 large study (N=289) reporting no adverse events in more than 90% of patients aged 9 to 14 years treated with methotrexate.12 Current guidelines recommend monitoring for bone marrow suppression and elevated transaminase levels 4 to 6 days after initiating treatment.1 The absolute contraindications for methotrexate are pregnancy and liver disease, and caution should be taken in children with metabolic risk factors. Adolescents must be counseled regarding the elevated risk for hepatotoxicity associated with alcohol ingestion. Methotrexate therapy also requires 1 mg folic acid supplementation 6 to 7 days a week, which decreases the risk for developing folic acid deficiency and may decrease gastrointestinal tract intolerance and hepatic side effects that may result from therapy.
Cyclosporine is an effective and well-tolerated option for rapid control of severe psoriasis in children. It is useful for various types of psoriasis but generally is reserved for more severe subtypes, such as generalized pustular psoriasis, erythrodermic psoriasis, and uncontrolled plaque psoriasis. Long-term use of cyclosporine may result in renal toxicity and hypertension, and this therapy is absolutely contraindicated in children with kidney disease or hypertension at baseline. It is strongly recommended to evaluate blood pressure every week for the first month of therapy and at every subsequent follow-up visit, which may occur at variable intervals based on the judgement of the provider. Evaluation before and during treatment with cyclosporine also should include a complete blood cell count, complete metabolic panel, and lipid panel.
Systemic retinoids have a unique advantage over methotrexate and cyclosporine in that they are not immunosuppressive and therefore are not contraindicated in children who are very young or immunosuppressed. Children receiving systemic retinoids also can receive routine live vaccines—measles-mumps-rubella, varicella zoster, and rotavirus—that are contraindicated with other systemic therapies. Acitretin is particularly effective in pediatric patients with diffuse guttate psoriasis, pustular psoriasis, and palmoplantar psoriasis. Narrowband UVB therapy has been shown to augment the effectiveness of acitretin in children, which may allow for reduced acitretin dosing. Pustular psoriasis may respond as quickly as 3 weeks after initiation, whereas it may take 2 to 3 months before improvement is noticed in plaque psoriasis. Side effects of retinoids include skin dryness, hyperlipidemia, and gastrointestinal tract upset. The most severe long-term concern is skeletal toxicity, including premature epiphyseal closure, hyperostosis, periosteal bone formation, and decreased bone mineral density.1 Vitamin A derivatives also are known teratogens and should be avoided in females of childbearing potential. Lipids and transaminases should be monitored routinely, and screening for depression and psychiatric symptoms should be performed frequently.1
When utilizing systemic therapies, the objective should be to control the disease, maintain stability, and ultimately taper to the lowest effective dose or transition to a topical therapy, if feasible. Although no particular systemic therapy is recommended as first line for children with psoriasis, it is important to consider comorbidities, contraindications, monitoring frequency, mode of administration (injectable therapies elicit more psychological trauma in children than oral therapies), and expense when determining the best choice.
Biologics
Biologic agents are associated with very high to total psoriatic plaque clearance rates and require infrequent dosing and monitoring. However, their use may be limited by cost and injection phobias in children as well as limited evidence for their efficacy and safety in pediatric psoriasis. Several studies have established the safety and effectiveness of biologics in children with plaque psoriasis (see eTable for recommended dosing), whereas the evidence supporting their use in treating pustular and erythrodermic variants are limited to case reports and case series. The tumor necrosis factor α (TNF-α) inhibitor etanercept has been approved for use in children aged 4 years and older, and the IL-12/IL-23 inhibitor ustekinumab is approved in children aged 6 years and older. Other TNF-α inhibitors, namely infliximab and adalimumab, commonly are utilized off label for pediatric psoriasis. The most common side effect of biologic therapies in pediatric patients is injection-site reactions.1 Prior to initiating therapy, children must undergo tuberculosis screening either by purified protein derivative testing or IFN-γ release assay. Testing should be repeated annually in individuals taking TNF-α inhibitors, though the utility of repeat testing when taking biologics in other classes is not clear. High-risk patients also should be screened for human immunodeficiency virus and hepatitis. Follow-up frequency may range from every 3 months to annually, based on judgement of the provider. In children who develop loss of response to biologics, methotrexate can be added to the regimen to attenuate formation of efficacy-reducing antidrug antibodies.
Final Thoughts
When managing children with psoriasis, it is important for dermatologists to appropriately educate guardians and children on the disease course, as well as consider the psychological, emotional, social, and financial factors that may direct decision-making regarding optimal therapeutics. Dermatologists should consider collaboration with the child’s primary care physician and other specialists to ensure that all needs are met.
These guidelines provide a framework agreed upon by numerous experts in pediatric psoriasis, but they are limited by gaps in the research. There still is much to be learned regarding the pathophysiology of psoriasis; the risk for developing comorbidities during adulthood; and the efficacy and safety of certain therapeutics, particularly biologics, in pediatric patients with psoriasis.
- Menter A, Cordoro KM, Davis DMR, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management and treatment of psoriasis in pediatric patients [published online November 5, 2019]. J Am Acad Dermatol. 2020;82:161-201.
- Lewis-Jones MS, Finlay AY. The Children’s Dermatology Life Quality Index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132:942-949.
- Augustin M, Radtke MA, Glaeske G, et al. Epidemiology and comorbidity in children with psoriasis and atopic eczema. Dermatology. 2015;231:35-40.
- Osier E, Wang AS, Tollefson MM, et al. Pediatric psoriasis comorbidity screening guidelines. JAMA Dermatol. 2017;153:698-704.
- Boccardi D, Menni S, La Vecchia C, et al. Overweight and childhood psoriasis. Br J Dermatol. 2009;161:484-486.
- Becker L, Tom WL, Eshagh K, et al. Excess adiposity preceding pediatric psoriasis. JAMA Dermatol. 2014;150:573-574.
- Alotaibi HA. Effects of weight loss on psoriasis: a review of clinical trials. Cureus. 2018;10:E3491.
- Guidelines summaries—American Academy of Pediatrics. Guideline Central
website. https://www.guidelinecentral.com/summaries/organizations/american-academy-of-pediatrics/2019. Accessed October 27, 2020. - Standards of Medical Care in Diabetes. American Diabetes Association website. https://care.diabetesjournals.org/content/43/Supplement_1. Published January 1, 2020. Accessed May 8, 2020.
- Siegfried EC, Jaworski JC, Hebert AA. Topical calcineurin inhibitors and lymphoma risk: evidence update with implications for daily practice. Am J Clin Dermatol. 2013;14:163-178.
- Jain VK, Bansal A, Aggarwal K, et al. Enhanced response of childhood psoriasis to narrow-band UV-B phototherapy with preirradiation use of mineral oil. Pediatr Dermatol. 2008;25:559-564.
- Ergun T, Seckin Gencosmanoglu D, Alpsoy E, et al. Efficacy, safety and drug survival of conventional agents in pediatric psoriasis: a multicenter, cohort study. J Dermatol. 2017;44:630-634.
In November 2019, the American Academy of Dermatology (AAD) and the National Psoriasis Foundation (NPF) released their first set of recommendations for the management of pediatric psoriasis.1 The pediatric guidelines discuss methods of quantifying disease severity in children, triggers and comorbidities, and the efficacy and safety of various therapeutic agents. This review aims to discuss, in a condensed form, special considerations unique to the management of children with psoriasis as presented in the guidelines as well as grade A– and grade B–level treatment recommendations (Table).
Quantifying Psoriasis Severity in Children
Percentage body surface area (BSA) involvement is the most common mode of grading psoriasis severity, with less than 3% BSA involvement being considered mild, 3% to 10% BSA moderate, and more than 10% severe disease. In children, the standard method of measuring BSA is the rule of 9’s: the head and each arm make up 9% of the total BSA, each leg and the front and back of the torso respectively each make up 18%, and the genitalia make up 1%. It also is important to consider impact on quality of life, which may be remarkable in spite of limited BSA involvement. The children’s dermatology life quality index score may be utilized in combination with affected BSA to determine the burden of psoriasis in context of impact on daily life. This metric is available in both written and cartoon form, and it consists of 10 questions that include variables such as severity of itch, impact on social life, and effects on sleep. Most notably, this tool incorporates pruritus,2 which generally is addressed inadequately in pediatric psoriasis.
Triggers and Comorbidities in Pediatric Patients
In children, it is important to identify and eliminate modifiable factors that may prompt psoriasis flares. Infections, particularly group A beta-hemolytic streptococcal infections, are a major trigger in neonates and infants. Other exacerbating factors in children include emotional stress, secondhand cigarette smoke, Kawasaki disease, and withdrawal from systemic corticosteroids.
Psoriatic arthritis (PsA) is a burdensome comorbidity affecting children with psoriasis. The prevalence of joint disease is 15-times greater in children with psoriasis vs those without,3 and 80% of children with PsA develop rheumatologic symptoms, which typically include oligoarticular disease and dactylitis in infants and girls and enthesitis and axial joint involvement in boys and older children, years prior to the onset of cutaneous disease.4 Uveitis often occurs in children with psoriasis and PsA but not in those with isolated cutaneous disease.
Compared to unaffected children, pediatric patients with psoriasis have greater prevalence of metabolic and cardiovascular risk factors during childhood, including central obesity, hypertension, hypertriglyceridemia, hypercholesterolemia, insulin resistance, atherosclerosis, arrythmia, and valvular heart disease. Family history of obesity increases the risk for early-onset development of cutaneous lesions,5,6 and weight reduction may alleviate severity of psoriasis lesions.7 In the United States, many of the metabolic associations observed are particularly robust in Black and Hispanic children vs those of other races. Furthermore, the prevalence of inflammatory bowel disease is 3- to 4-times higher in children with psoriasis compared to those without.
As with other cutaneous diseases, it is important to be aware of social and mental health concerns in children with psoriasis. The majority of pediatric patients with psoriasis experience name-calling, shaming, or bullying, and many have concerns from skin shedding and malodor. Independent risk for depression after the onset of psoriasis is high. Affected older children and adolescents are at increased risk for alcohol and drug abuse as well as eating disorders.
Despite these identified comorbidities, there are no unique screening recommendations for arthritis, ophthalmologic disease, metabolic disease, cardiovascular disease, gastrointestinal tract disease, or mental health issues in children with psoriasis. Rather, these patients should be monitored according to the American Academy of Pediatrics or American Diabetes Association guidelines for all pediatric patients.8,9 Nonetheless, educating patients and guardians about these potential issues may be warranted.
Topical Therapies
For children with mild to moderate psoriasis, topical therapies are first line. Despite being off label, topical corticosteroids are the mainstay of therapy for localized psoriatic plaques in children. Topical vitamin D analogues—calcitriol and calcipotriol/calcipotriene—are highly effective and well tolerated, and they frequently are used in combination with topical corticosteroids. Topical calcineurin inhibitors, namely tacrolimus, also are used off label but are considered first line for sensitive regions of the skin in children, including the face, genitalia, and body folds. There currently is limited evidence for supporting the use of the topical vitamin A analogue tazarotene in children with psoriasis, though some consider its off-label use effective for pediatric nail psoriasis. It also may be used as an adjunct to topical corticosteroids to minimize irritation.
Although there is no gold standard topical regimen, combination therapy with a high-potency topical steroid and topical vitamin D analogue commonly is used to minimize steroid-induced side effects. For the first 2 weeks of treatment, they each may be applied once daily or mixed together and applied twice daily. For subsequent maintenance, topical calcipotriene may be applied on weekdays and topical steroids only on weekends. Combination calcipotriol–betamethasone dipropionate also is available as cream, ointment, foam, and suspension vehicles for use on the body and scalp in children aged 12 years and older. Tacrolimus ointment 0.1% may be applied in a thin layer up to twice daily. Concurrent emollient use also is recommended with these therapies.
Health care providers should educate patients and guardians about the potential side effects of topical therapies. They also should provide explicit instructions for amount, site, frequency, and duration of application. Topical corticosteroids commonly result in burning on application and may potentially cause skin thinning and striae with overuse. Topical vitamin D analogues may result in local irritation that may be improved by concurrent emollient use, and they generally should be avoided on sensitive sites. Topical calcineurin inhibitors are associated with burning, stinging, and pruritus, and the US Food and Drug Administration has issued a black-box warning related to risk for lymphoma with their chronic intermittent use. However, it was based on rare reports of lymphoma in transplant patients taking oral calcineurin inhibitors; no clinical trials to date in humans have demonstrated an increased risk for malignancy with topical calcineurin inhibitors.10 Tazarotene should be used cautiously in females of childbearing age given its teratogenic potential.
Children younger than 7 years are especially prone to suppression of the hypothalamic-pituitary-adrenal axis from topical corticosteroid therapy and theoretically hypercalcemia and hypervitaminosis D from topical vitamin D analogues, as their high BSA-to-volume ratio increases potential for systemic absorption. Children should avoid occlusive application of topical vitamin D analogues to large areas of the skin. Monitoring of vitamin D metabolites in the serum may be considered if calcipotriene or calcipotriol application to a large BSA is warranted.
Light-Based Therapy
In children with widespread psoriasis or those refractory to topical therapy, phototherapy may be considered. Narrowband UVB (311- to 313-nm wavelength) therapy is considered a first-line form of phototherapy in pediatric psoriasis. Mineral oil or emollient pretreatment to affected areas may augment the efficacy of UV-based treatments.11 Excimer laser and UVA also may be efficacious, though evidence is limited in children. Treatment is recommended to start at 3 days a week, and once improvement is seen, the frequency can be decreased to 2 days a week. Once desired clearance is achieved, maintenance therapy can be continued at even longer intervals. Adjunctive use of tar preparations may potentiate the efficacy of phototherapy, though there is a theoretical increased risk for carcinogenicity with prolonged use of coal tar. Side effects of phototherapy include erythema, blistering hyperpigmentation, and pruritus. Psoralen is contraindicated in children younger than 12 years. All forms of phototherapy are contraindicated in children with generalized erythroderma and cutaneous cancer syndromes. Other important pediatric-specific considerations include anxiety that may be provoked by UV light machines and inconvenience of frequent appointments.
Nonbiologic Systemic Therapies
Systemic therapies may be considered in children with recalcitrant, widespread, or rapidly progressing psoriasis, particularly if the disease is accompanied by severe emotional and psychological burden. These drugs, which include methotrexate, cyclosporine, and acitretin (see eTable for recommended dosing), are advantageous in that they may be combined with other therapies; however, they have potential for dangerous toxicities.
Methotrexate is the most frequently utilized systemic therapy for psoriasis worldwide in children because of its low cost, once-weekly dosing, and the substantial amount of long-term efficacy and safety data available in the pediatric population. It is slow acting initially but has excellent long-term efficacy for nearly every subtype of psoriasis. The most common side effect of methotrexate is gastrointestinal tract intolerance. Nonetheless, adverse events are rare in children without prior history, with 1 large study (N=289) reporting no adverse events in more than 90% of patients aged 9 to 14 years treated with methotrexate.12 Current guidelines recommend monitoring for bone marrow suppression and elevated transaminase levels 4 to 6 days after initiating treatment.1 The absolute contraindications for methotrexate are pregnancy and liver disease, and caution should be taken in children with metabolic risk factors. Adolescents must be counseled regarding the elevated risk for hepatotoxicity associated with alcohol ingestion. Methotrexate therapy also requires 1 mg folic acid supplementation 6 to 7 days a week, which decreases the risk for developing folic acid deficiency and may decrease gastrointestinal tract intolerance and hepatic side effects that may result from therapy.
Cyclosporine is an effective and well-tolerated option for rapid control of severe psoriasis in children. It is useful for various types of psoriasis but generally is reserved for more severe subtypes, such as generalized pustular psoriasis, erythrodermic psoriasis, and uncontrolled plaque psoriasis. Long-term use of cyclosporine may result in renal toxicity and hypertension, and this therapy is absolutely contraindicated in children with kidney disease or hypertension at baseline. It is strongly recommended to evaluate blood pressure every week for the first month of therapy and at every subsequent follow-up visit, which may occur at variable intervals based on the judgement of the provider. Evaluation before and during treatment with cyclosporine also should include a complete blood cell count, complete metabolic panel, and lipid panel.
Systemic retinoids have a unique advantage over methotrexate and cyclosporine in that they are not immunosuppressive and therefore are not contraindicated in children who are very young or immunosuppressed. Children receiving systemic retinoids also can receive routine live vaccines—measles-mumps-rubella, varicella zoster, and rotavirus—that are contraindicated with other systemic therapies. Acitretin is particularly effective in pediatric patients with diffuse guttate psoriasis, pustular psoriasis, and palmoplantar psoriasis. Narrowband UVB therapy has been shown to augment the effectiveness of acitretin in children, which may allow for reduced acitretin dosing. Pustular psoriasis may respond as quickly as 3 weeks after initiation, whereas it may take 2 to 3 months before improvement is noticed in plaque psoriasis. Side effects of retinoids include skin dryness, hyperlipidemia, and gastrointestinal tract upset. The most severe long-term concern is skeletal toxicity, including premature epiphyseal closure, hyperostosis, periosteal bone formation, and decreased bone mineral density.1 Vitamin A derivatives also are known teratogens and should be avoided in females of childbearing potential. Lipids and transaminases should be monitored routinely, and screening for depression and psychiatric symptoms should be performed frequently.1
When utilizing systemic therapies, the objective should be to control the disease, maintain stability, and ultimately taper to the lowest effective dose or transition to a topical therapy, if feasible. Although no particular systemic therapy is recommended as first line for children with psoriasis, it is important to consider comorbidities, contraindications, monitoring frequency, mode of administration (injectable therapies elicit more psychological trauma in children than oral therapies), and expense when determining the best choice.
Biologics
Biologic agents are associated with very high to total psoriatic plaque clearance rates and require infrequent dosing and monitoring. However, their use may be limited by cost and injection phobias in children as well as limited evidence for their efficacy and safety in pediatric psoriasis. Several studies have established the safety and effectiveness of biologics in children with plaque psoriasis (see eTable for recommended dosing), whereas the evidence supporting their use in treating pustular and erythrodermic variants are limited to case reports and case series. The tumor necrosis factor α (TNF-α) inhibitor etanercept has been approved for use in children aged 4 years and older, and the IL-12/IL-23 inhibitor ustekinumab is approved in children aged 6 years and older. Other TNF-α inhibitors, namely infliximab and adalimumab, commonly are utilized off label for pediatric psoriasis. The most common side effect of biologic therapies in pediatric patients is injection-site reactions.1 Prior to initiating therapy, children must undergo tuberculosis screening either by purified protein derivative testing or IFN-γ release assay. Testing should be repeated annually in individuals taking TNF-α inhibitors, though the utility of repeat testing when taking biologics in other classes is not clear. High-risk patients also should be screened for human immunodeficiency virus and hepatitis. Follow-up frequency may range from every 3 months to annually, based on judgement of the provider. In children who develop loss of response to biologics, methotrexate can be added to the regimen to attenuate formation of efficacy-reducing antidrug antibodies.
Final Thoughts
When managing children with psoriasis, it is important for dermatologists to appropriately educate guardians and children on the disease course, as well as consider the psychological, emotional, social, and financial factors that may direct decision-making regarding optimal therapeutics. Dermatologists should consider collaboration with the child’s primary care physician and other specialists to ensure that all needs are met.
These guidelines provide a framework agreed upon by numerous experts in pediatric psoriasis, but they are limited by gaps in the research. There still is much to be learned regarding the pathophysiology of psoriasis; the risk for developing comorbidities during adulthood; and the efficacy and safety of certain therapeutics, particularly biologics, in pediatric patients with psoriasis.
In November 2019, the American Academy of Dermatology (AAD) and the National Psoriasis Foundation (NPF) released their first set of recommendations for the management of pediatric psoriasis.1 The pediatric guidelines discuss methods of quantifying disease severity in children, triggers and comorbidities, and the efficacy and safety of various therapeutic agents. This review aims to discuss, in a condensed form, special considerations unique to the management of children with psoriasis as presented in the guidelines as well as grade A– and grade B–level treatment recommendations (Table).
Quantifying Psoriasis Severity in Children
Percentage body surface area (BSA) involvement is the most common mode of grading psoriasis severity, with less than 3% BSA involvement being considered mild, 3% to 10% BSA moderate, and more than 10% severe disease. In children, the standard method of measuring BSA is the rule of 9’s: the head and each arm make up 9% of the total BSA, each leg and the front and back of the torso respectively each make up 18%, and the genitalia make up 1%. It also is important to consider impact on quality of life, which may be remarkable in spite of limited BSA involvement. The children’s dermatology life quality index score may be utilized in combination with affected BSA to determine the burden of psoriasis in context of impact on daily life. This metric is available in both written and cartoon form, and it consists of 10 questions that include variables such as severity of itch, impact on social life, and effects on sleep. Most notably, this tool incorporates pruritus,2 which generally is addressed inadequately in pediatric psoriasis.
Triggers and Comorbidities in Pediatric Patients
In children, it is important to identify and eliminate modifiable factors that may prompt psoriasis flares. Infections, particularly group A beta-hemolytic streptococcal infections, are a major trigger in neonates and infants. Other exacerbating factors in children include emotional stress, secondhand cigarette smoke, Kawasaki disease, and withdrawal from systemic corticosteroids.
Psoriatic arthritis (PsA) is a burdensome comorbidity affecting children with psoriasis. The prevalence of joint disease is 15-times greater in children with psoriasis vs those without,3 and 80% of children with PsA develop rheumatologic symptoms, which typically include oligoarticular disease and dactylitis in infants and girls and enthesitis and axial joint involvement in boys and older children, years prior to the onset of cutaneous disease.4 Uveitis often occurs in children with psoriasis and PsA but not in those with isolated cutaneous disease.
Compared to unaffected children, pediatric patients with psoriasis have greater prevalence of metabolic and cardiovascular risk factors during childhood, including central obesity, hypertension, hypertriglyceridemia, hypercholesterolemia, insulin resistance, atherosclerosis, arrythmia, and valvular heart disease. Family history of obesity increases the risk for early-onset development of cutaneous lesions,5,6 and weight reduction may alleviate severity of psoriasis lesions.7 In the United States, many of the metabolic associations observed are particularly robust in Black and Hispanic children vs those of other races. Furthermore, the prevalence of inflammatory bowel disease is 3- to 4-times higher in children with psoriasis compared to those without.
As with other cutaneous diseases, it is important to be aware of social and mental health concerns in children with psoriasis. The majority of pediatric patients with psoriasis experience name-calling, shaming, or bullying, and many have concerns from skin shedding and malodor. Independent risk for depression after the onset of psoriasis is high. Affected older children and adolescents are at increased risk for alcohol and drug abuse as well as eating disorders.
Despite these identified comorbidities, there are no unique screening recommendations for arthritis, ophthalmologic disease, metabolic disease, cardiovascular disease, gastrointestinal tract disease, or mental health issues in children with psoriasis. Rather, these patients should be monitored according to the American Academy of Pediatrics or American Diabetes Association guidelines for all pediatric patients.8,9 Nonetheless, educating patients and guardians about these potential issues may be warranted.
Topical Therapies
For children with mild to moderate psoriasis, topical therapies are first line. Despite being off label, topical corticosteroids are the mainstay of therapy for localized psoriatic plaques in children. Topical vitamin D analogues—calcitriol and calcipotriol/calcipotriene—are highly effective and well tolerated, and they frequently are used in combination with topical corticosteroids. Topical calcineurin inhibitors, namely tacrolimus, also are used off label but are considered first line for sensitive regions of the skin in children, including the face, genitalia, and body folds. There currently is limited evidence for supporting the use of the topical vitamin A analogue tazarotene in children with psoriasis, though some consider its off-label use effective for pediatric nail psoriasis. It also may be used as an adjunct to topical corticosteroids to minimize irritation.
Although there is no gold standard topical regimen, combination therapy with a high-potency topical steroid and topical vitamin D analogue commonly is used to minimize steroid-induced side effects. For the first 2 weeks of treatment, they each may be applied once daily or mixed together and applied twice daily. For subsequent maintenance, topical calcipotriene may be applied on weekdays and topical steroids only on weekends. Combination calcipotriol–betamethasone dipropionate also is available as cream, ointment, foam, and suspension vehicles for use on the body and scalp in children aged 12 years and older. Tacrolimus ointment 0.1% may be applied in a thin layer up to twice daily. Concurrent emollient use also is recommended with these therapies.
Health care providers should educate patients and guardians about the potential side effects of topical therapies. They also should provide explicit instructions for amount, site, frequency, and duration of application. Topical corticosteroids commonly result in burning on application and may potentially cause skin thinning and striae with overuse. Topical vitamin D analogues may result in local irritation that may be improved by concurrent emollient use, and they generally should be avoided on sensitive sites. Topical calcineurin inhibitors are associated with burning, stinging, and pruritus, and the US Food and Drug Administration has issued a black-box warning related to risk for lymphoma with their chronic intermittent use. However, it was based on rare reports of lymphoma in transplant patients taking oral calcineurin inhibitors; no clinical trials to date in humans have demonstrated an increased risk for malignancy with topical calcineurin inhibitors.10 Tazarotene should be used cautiously in females of childbearing age given its teratogenic potential.
Children younger than 7 years are especially prone to suppression of the hypothalamic-pituitary-adrenal axis from topical corticosteroid therapy and theoretically hypercalcemia and hypervitaminosis D from topical vitamin D analogues, as their high BSA-to-volume ratio increases potential for systemic absorption. Children should avoid occlusive application of topical vitamin D analogues to large areas of the skin. Monitoring of vitamin D metabolites in the serum may be considered if calcipotriene or calcipotriol application to a large BSA is warranted.
Light-Based Therapy
In children with widespread psoriasis or those refractory to topical therapy, phototherapy may be considered. Narrowband UVB (311- to 313-nm wavelength) therapy is considered a first-line form of phototherapy in pediatric psoriasis. Mineral oil or emollient pretreatment to affected areas may augment the efficacy of UV-based treatments.11 Excimer laser and UVA also may be efficacious, though evidence is limited in children. Treatment is recommended to start at 3 days a week, and once improvement is seen, the frequency can be decreased to 2 days a week. Once desired clearance is achieved, maintenance therapy can be continued at even longer intervals. Adjunctive use of tar preparations may potentiate the efficacy of phototherapy, though there is a theoretical increased risk for carcinogenicity with prolonged use of coal tar. Side effects of phototherapy include erythema, blistering hyperpigmentation, and pruritus. Psoralen is contraindicated in children younger than 12 years. All forms of phototherapy are contraindicated in children with generalized erythroderma and cutaneous cancer syndromes. Other important pediatric-specific considerations include anxiety that may be provoked by UV light machines and inconvenience of frequent appointments.
Nonbiologic Systemic Therapies
Systemic therapies may be considered in children with recalcitrant, widespread, or rapidly progressing psoriasis, particularly if the disease is accompanied by severe emotional and psychological burden. These drugs, which include methotrexate, cyclosporine, and acitretin (see eTable for recommended dosing), are advantageous in that they may be combined with other therapies; however, they have potential for dangerous toxicities.
Methotrexate is the most frequently utilized systemic therapy for psoriasis worldwide in children because of its low cost, once-weekly dosing, and the substantial amount of long-term efficacy and safety data available in the pediatric population. It is slow acting initially but has excellent long-term efficacy for nearly every subtype of psoriasis. The most common side effect of methotrexate is gastrointestinal tract intolerance. Nonetheless, adverse events are rare in children without prior history, with 1 large study (N=289) reporting no adverse events in more than 90% of patients aged 9 to 14 years treated with methotrexate.12 Current guidelines recommend monitoring for bone marrow suppression and elevated transaminase levels 4 to 6 days after initiating treatment.1 The absolute contraindications for methotrexate are pregnancy and liver disease, and caution should be taken in children with metabolic risk factors. Adolescents must be counseled regarding the elevated risk for hepatotoxicity associated with alcohol ingestion. Methotrexate therapy also requires 1 mg folic acid supplementation 6 to 7 days a week, which decreases the risk for developing folic acid deficiency and may decrease gastrointestinal tract intolerance and hepatic side effects that may result from therapy.
Cyclosporine is an effective and well-tolerated option for rapid control of severe psoriasis in children. It is useful for various types of psoriasis but generally is reserved for more severe subtypes, such as generalized pustular psoriasis, erythrodermic psoriasis, and uncontrolled plaque psoriasis. Long-term use of cyclosporine may result in renal toxicity and hypertension, and this therapy is absolutely contraindicated in children with kidney disease or hypertension at baseline. It is strongly recommended to evaluate blood pressure every week for the first month of therapy and at every subsequent follow-up visit, which may occur at variable intervals based on the judgement of the provider. Evaluation before and during treatment with cyclosporine also should include a complete blood cell count, complete metabolic panel, and lipid panel.
Systemic retinoids have a unique advantage over methotrexate and cyclosporine in that they are not immunosuppressive and therefore are not contraindicated in children who are very young or immunosuppressed. Children receiving systemic retinoids also can receive routine live vaccines—measles-mumps-rubella, varicella zoster, and rotavirus—that are contraindicated with other systemic therapies. Acitretin is particularly effective in pediatric patients with diffuse guttate psoriasis, pustular psoriasis, and palmoplantar psoriasis. Narrowband UVB therapy has been shown to augment the effectiveness of acitretin in children, which may allow for reduced acitretin dosing. Pustular psoriasis may respond as quickly as 3 weeks after initiation, whereas it may take 2 to 3 months before improvement is noticed in plaque psoriasis. Side effects of retinoids include skin dryness, hyperlipidemia, and gastrointestinal tract upset. The most severe long-term concern is skeletal toxicity, including premature epiphyseal closure, hyperostosis, periosteal bone formation, and decreased bone mineral density.1 Vitamin A derivatives also are known teratogens and should be avoided in females of childbearing potential. Lipids and transaminases should be monitored routinely, and screening for depression and psychiatric symptoms should be performed frequently.1
When utilizing systemic therapies, the objective should be to control the disease, maintain stability, and ultimately taper to the lowest effective dose or transition to a topical therapy, if feasible. Although no particular systemic therapy is recommended as first line for children with psoriasis, it is important to consider comorbidities, contraindications, monitoring frequency, mode of administration (injectable therapies elicit more psychological trauma in children than oral therapies), and expense when determining the best choice.
Biologics
Biologic agents are associated with very high to total psoriatic plaque clearance rates and require infrequent dosing and monitoring. However, their use may be limited by cost and injection phobias in children as well as limited evidence for their efficacy and safety in pediatric psoriasis. Several studies have established the safety and effectiveness of biologics in children with plaque psoriasis (see eTable for recommended dosing), whereas the evidence supporting their use in treating pustular and erythrodermic variants are limited to case reports and case series. The tumor necrosis factor α (TNF-α) inhibitor etanercept has been approved for use in children aged 4 years and older, and the IL-12/IL-23 inhibitor ustekinumab is approved in children aged 6 years and older. Other TNF-α inhibitors, namely infliximab and adalimumab, commonly are utilized off label for pediatric psoriasis. The most common side effect of biologic therapies in pediatric patients is injection-site reactions.1 Prior to initiating therapy, children must undergo tuberculosis screening either by purified protein derivative testing or IFN-γ release assay. Testing should be repeated annually in individuals taking TNF-α inhibitors, though the utility of repeat testing when taking biologics in other classes is not clear. High-risk patients also should be screened for human immunodeficiency virus and hepatitis. Follow-up frequency may range from every 3 months to annually, based on judgement of the provider. In children who develop loss of response to biologics, methotrexate can be added to the regimen to attenuate formation of efficacy-reducing antidrug antibodies.
Final Thoughts
When managing children with psoriasis, it is important for dermatologists to appropriately educate guardians and children on the disease course, as well as consider the psychological, emotional, social, and financial factors that may direct decision-making regarding optimal therapeutics. Dermatologists should consider collaboration with the child’s primary care physician and other specialists to ensure that all needs are met.
These guidelines provide a framework agreed upon by numerous experts in pediatric psoriasis, but they are limited by gaps in the research. There still is much to be learned regarding the pathophysiology of psoriasis; the risk for developing comorbidities during adulthood; and the efficacy and safety of certain therapeutics, particularly biologics, in pediatric patients with psoriasis.
- Menter A, Cordoro KM, Davis DMR, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management and treatment of psoriasis in pediatric patients [published online November 5, 2019]. J Am Acad Dermatol. 2020;82:161-201.
- Lewis-Jones MS, Finlay AY. The Children’s Dermatology Life Quality Index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132:942-949.
- Augustin M, Radtke MA, Glaeske G, et al. Epidemiology and comorbidity in children with psoriasis and atopic eczema. Dermatology. 2015;231:35-40.
- Osier E, Wang AS, Tollefson MM, et al. Pediatric psoriasis comorbidity screening guidelines. JAMA Dermatol. 2017;153:698-704.
- Boccardi D, Menni S, La Vecchia C, et al. Overweight and childhood psoriasis. Br J Dermatol. 2009;161:484-486.
- Becker L, Tom WL, Eshagh K, et al. Excess adiposity preceding pediatric psoriasis. JAMA Dermatol. 2014;150:573-574.
- Alotaibi HA. Effects of weight loss on psoriasis: a review of clinical trials. Cureus. 2018;10:E3491.
- Guidelines summaries—American Academy of Pediatrics. Guideline Central
website. https://www.guidelinecentral.com/summaries/organizations/american-academy-of-pediatrics/2019. Accessed October 27, 2020. - Standards of Medical Care in Diabetes. American Diabetes Association website. https://care.diabetesjournals.org/content/43/Supplement_1. Published January 1, 2020. Accessed May 8, 2020.
- Siegfried EC, Jaworski JC, Hebert AA. Topical calcineurin inhibitors and lymphoma risk: evidence update with implications for daily practice. Am J Clin Dermatol. 2013;14:163-178.
- Jain VK, Bansal A, Aggarwal K, et al. Enhanced response of childhood psoriasis to narrow-band UV-B phototherapy with preirradiation use of mineral oil. Pediatr Dermatol. 2008;25:559-564.
- Ergun T, Seckin Gencosmanoglu D, Alpsoy E, et al. Efficacy, safety and drug survival of conventional agents in pediatric psoriasis: a multicenter, cohort study. J Dermatol. 2017;44:630-634.
- Menter A, Cordoro KM, Davis DMR, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management and treatment of psoriasis in pediatric patients [published online November 5, 2019]. J Am Acad Dermatol. 2020;82:161-201.
- Lewis-Jones MS, Finlay AY. The Children’s Dermatology Life Quality Index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132:942-949.
- Augustin M, Radtke MA, Glaeske G, et al. Epidemiology and comorbidity in children with psoriasis and atopic eczema. Dermatology. 2015;231:35-40.
- Osier E, Wang AS, Tollefson MM, et al. Pediatric psoriasis comorbidity screening guidelines. JAMA Dermatol. 2017;153:698-704.
- Boccardi D, Menni S, La Vecchia C, et al. Overweight and childhood psoriasis. Br J Dermatol. 2009;161:484-486.
- Becker L, Tom WL, Eshagh K, et al. Excess adiposity preceding pediatric psoriasis. JAMA Dermatol. 2014;150:573-574.
- Alotaibi HA. Effects of weight loss on psoriasis: a review of clinical trials. Cureus. 2018;10:E3491.
- Guidelines summaries—American Academy of Pediatrics. Guideline Central
website. https://www.guidelinecentral.com/summaries/organizations/american-academy-of-pediatrics/2019. Accessed October 27, 2020. - Standards of Medical Care in Diabetes. American Diabetes Association website. https://care.diabetesjournals.org/content/43/Supplement_1. Published January 1, 2020. Accessed May 8, 2020.
- Siegfried EC, Jaworski JC, Hebert AA. Topical calcineurin inhibitors and lymphoma risk: evidence update with implications for daily practice. Am J Clin Dermatol. 2013;14:163-178.
- Jain VK, Bansal A, Aggarwal K, et al. Enhanced response of childhood psoriasis to narrow-band UV-B phototherapy with preirradiation use of mineral oil. Pediatr Dermatol. 2008;25:559-564.
- Ergun T, Seckin Gencosmanoglu D, Alpsoy E, et al. Efficacy, safety and drug survival of conventional agents in pediatric psoriasis: a multicenter, cohort study. J Dermatol. 2017;44:630-634.
Practice Points
- For children, several environmental factors may prompt psoriasis flares, and it is critical to identify and eliminate these triggers.
- Although the use of biologics may be limited by cost and injection phobias in children, they may be an appropriate option for children with moderate to severe psoriasis when other therapies have failed. A growing body of literature is establishing the safety and effectiveness of biologics in children.
- Clinicians should thoroughly educate parents/ guardians on the course of psoriasis and treatment options as well as pay special attention to treatment goals and psychosocial factors that may guide decision-making regarding therapy.
