User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Tumor Necrosis Factor Inhibitors May Reduce Cardiovascular Morbidity in Patients With Psoriasis
The connection between psoriasis and increased major adverse cardiovascular events (MACEs) has been well studied. 1,2 Although treatment of psoriasis can improve skin and joint symptoms, it is less clear whether therapies may mitigate the increased risk for cardiovascular comorbidities. Tumor necrosis factor (TNF) inhibitors in particular have been studied with great interest given the role of TNF in vascular and metabolic functions. 3 Using a retrospective cohort design, Wu and colleagues 4 examined if treatment with TNF inhibitors in patients with psoriasis would be associated with a lower risk for MACEs compared to phototherapy. Results suggested a significantly lower hazard of MACEs in patients using TNF inhibitors vs patients treated with phototherapy (adjusted hazard ratio, 0.77; P = .046). Moreover, based on these findings, they calculated that treating approximately 161 patients with TNF inhibitors rather than phototherapy would result in 1 less MACE per year overall. 4
Patients with psoriasis have been shown to have a greater noncalcified coronary plaque burden and prevalence of high-risk plaque compared to healthy patients.5 Lerman and colleagues5 measured the coronary plaque burden of 105 patients with psoriasis and 25 healthy volunteers using coronary computed tomography angiography. Although the patients were on average 10 years younger and had lower cardiovascular risk as measured by traditional risk scores, patients with psoriasis were found to have a greater noncalcified coronary plaque burden compared to 100 patients with hyperlipidemia. This burden was associated with an increased prevalence of high-risk plaques. Furthermore, in patients followed for 1 year, improvements in psoriasis severity were associated with reductions in noncalcified coronary plaque burden, though this finding was across all treatment modalities. However, there was no significant difference in calcified coronary plaque burden associated with reduced psoriasis severity.5
Moreover, Pina et al6 conducted a prospective study evaluating the use of the TNF inhibitor adalimumab to improve endothelial function and arterial stiffness in patients with moderate to severe psoriasis. Among 29 patients, they found a significant improvement in endothelial function as measured by flow-mediated dilatation after 6 months of adalimumab therapy, with a mean increase from 6.19% to 7.46% (P=.008). They also reported decreases in arterial stiffness by pulse wave velocity (P=.03). Despite a small sample size, these findings provide 2 potential mechanisms by which TNF inhibitor therapy may reduce the risk for cardiovascular events.6
A retrospective cohort study evaluating data from the Kaiser Permanente Southern California health plan assessed whether TNF inhibitor therapy was associated with a lower risk for MACE in patients with psoriasis.7 A total of 18,194 patients were included; of these, 1463 received TNF inhibitor therapy for at least 2 months. After controlling for other variables, including age at psoriasis diagnosis, sex, race/ethnicity, and other cardiovascular risk factors (eg, history of smoking or alcohol use; use of clopidogrel, antihypertensive agents, antihyperlipidemics, or anticoagulants), patients in the TNF inhibitor cohort demonstrated a significantly lower MACE hazard ratio compared to patients treated with topicals (hazard ratio, 0.80; 95% confidence interval, 0.66-0.98; P<.05).7
Conversely, a randomized, placebo-controlled trial of 107 patients found no difference in vascular inflammation of the ascending aorta and the carotids after 16 weeks of adalimumab treatment vs placebo. In this study, however, most patients had only moderate psoriasis based on a mean psoriasis area and severity index score of 9.8.8 Given studies finding higher risk burden in patients with more severe skin disease,2 it is possible that the effect of TNF inhibitor therapy may not be as pronounced in patients with less skin involvement. There was a significant effect on C-reactive protein levels in patients receiving TNF inhibitor therapy compared to placebo at 16 weeks (P=.012), suggesting TNF does play some role in systemic inflammation, and it is possible it may exert cardiovascular effects through a mechanism other than vascular inflammation.8
A second double-blind, randomized trial reported similar results.9 Among 97 patients randomized to receive adalimumab, placebo, or phototherapy, no significant difference in vascular inflammation was found after 12 weeks of therapy. In contrast, levels of C-reactive protein, IL-6, and glycoprotein acetylation were markedly reduced. The authors also reported adverse effects of adalimumab therapy on lipid metabolism with reduced cholesterol efflux capacity, a marker of ability of high-density-lipoprotein particles to perform reverse cholesterol transport, and high-density-lipoprotein particles, suggesting these effects may counteract some of the anti-inflammatory effects of TNF inhibitors.9
A growing body of data regarding the effect of TNF inhibitors on cardiovascular morbidity in patients with psoriasis is being collected, but no strong conclusions can be made. Given the disconnect of findings across these studies, it is possible that we have yet to elucidate the full mechanism by which TNF inhibitors may affect cardiovascular health. However, there may be additional confounding factors or patient characteristics at play. More large, prospective, randomized, controlled studies are needed to further understand this relationship.
- Ogdie A, Yu Y, Haynes K, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2015;74:326-332.
- Ahlehoff O, Gislason GH, Charlot M, et al. Psoriasis is associated with clinically significant cardiovascular risk: a Danish nationwide cohort study. J Intern Med. 2011;270:147-157.
- Kölliker Frers RA, Bisoendial RJ, Montoya SF, et al. Psoriasis and cardiovascular risk: immune-mediated crosstalk between metabolic, vascular, and autoimmune inflammation. Int J Cardiol Metab Endocr. 2015;6:43-54.
- Wu JJ, Sundaram M, Cloutier M, et al. The risk of cardiovascular events in psoriasis patients treated with tumor necrosis factor-α inhibitors versus phototherapy: an observational cohort study. J Am Acad Dermatol. 2018;79:60-68.
- Lerman JB, Joshi AA, Chaturvedi A, et al. Coronary plaque characterization in psoriasis reveals high-risk features that improve after treatment in a prospective observational study. Circulation. 2017;136:263-276.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-α therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6 month prospective study. J Dermatol. 2016;43:1267-1272.
- Wu JJ, Joshi AA, Reddy SP, et al. Anti-inflammatory therapy with tumor necrosis factor inhibitors is associated with reduced risk of major adverse cardiovascular events in psoriasis [published online March 24, 2018]. J Eur Acad Dermatol Venereol. doi:10.1111/jdv.14951.
- Bissonnette R, Harel F, Krueger JG, et al. TNF-α antagonist and vascular inflammation patients with psoriasis vulgaris: a randomized placebo-controlled study. J Invest Dermatol. 2017;137:1638-1645 .
- Mehta NN, Shin DB, Joshi AA, et al. Effect of 2 psoriasis treatments on vascular inflammation and novel inflammatory cardiovascular biomarkers: a randomized placebo-controlled trial. Circ Cardiovasc Imaging. 2018;11:e007394.
The connection between psoriasis and increased major adverse cardiovascular events (MACEs) has been well studied. 1,2 Although treatment of psoriasis can improve skin and joint symptoms, it is less clear whether therapies may mitigate the increased risk for cardiovascular comorbidities. Tumor necrosis factor (TNF) inhibitors in particular have been studied with great interest given the role of TNF in vascular and metabolic functions. 3 Using a retrospective cohort design, Wu and colleagues 4 examined if treatment with TNF inhibitors in patients with psoriasis would be associated with a lower risk for MACEs compared to phototherapy. Results suggested a significantly lower hazard of MACEs in patients using TNF inhibitors vs patients treated with phototherapy (adjusted hazard ratio, 0.77; P = .046). Moreover, based on these findings, they calculated that treating approximately 161 patients with TNF inhibitors rather than phototherapy would result in 1 less MACE per year overall. 4
Patients with psoriasis have been shown to have a greater noncalcified coronary plaque burden and prevalence of high-risk plaque compared to healthy patients.5 Lerman and colleagues5 measured the coronary plaque burden of 105 patients with psoriasis and 25 healthy volunteers using coronary computed tomography angiography. Although the patients were on average 10 years younger and had lower cardiovascular risk as measured by traditional risk scores, patients with psoriasis were found to have a greater noncalcified coronary plaque burden compared to 100 patients with hyperlipidemia. This burden was associated with an increased prevalence of high-risk plaques. Furthermore, in patients followed for 1 year, improvements in psoriasis severity were associated with reductions in noncalcified coronary plaque burden, though this finding was across all treatment modalities. However, there was no significant difference in calcified coronary plaque burden associated with reduced psoriasis severity.5
Moreover, Pina et al6 conducted a prospective study evaluating the use of the TNF inhibitor adalimumab to improve endothelial function and arterial stiffness in patients with moderate to severe psoriasis. Among 29 patients, they found a significant improvement in endothelial function as measured by flow-mediated dilatation after 6 months of adalimumab therapy, with a mean increase from 6.19% to 7.46% (P=.008). They also reported decreases in arterial stiffness by pulse wave velocity (P=.03). Despite a small sample size, these findings provide 2 potential mechanisms by which TNF inhibitor therapy may reduce the risk for cardiovascular events.6
A retrospective cohort study evaluating data from the Kaiser Permanente Southern California health plan assessed whether TNF inhibitor therapy was associated with a lower risk for MACE in patients with psoriasis.7 A total of 18,194 patients were included; of these, 1463 received TNF inhibitor therapy for at least 2 months. After controlling for other variables, including age at psoriasis diagnosis, sex, race/ethnicity, and other cardiovascular risk factors (eg, history of smoking or alcohol use; use of clopidogrel, antihypertensive agents, antihyperlipidemics, or anticoagulants), patients in the TNF inhibitor cohort demonstrated a significantly lower MACE hazard ratio compared to patients treated with topicals (hazard ratio, 0.80; 95% confidence interval, 0.66-0.98; P<.05).7
Conversely, a randomized, placebo-controlled trial of 107 patients found no difference in vascular inflammation of the ascending aorta and the carotids after 16 weeks of adalimumab treatment vs placebo. In this study, however, most patients had only moderate psoriasis based on a mean psoriasis area and severity index score of 9.8.8 Given studies finding higher risk burden in patients with more severe skin disease,2 it is possible that the effect of TNF inhibitor therapy may not be as pronounced in patients with less skin involvement. There was a significant effect on C-reactive protein levels in patients receiving TNF inhibitor therapy compared to placebo at 16 weeks (P=.012), suggesting TNF does play some role in systemic inflammation, and it is possible it may exert cardiovascular effects through a mechanism other than vascular inflammation.8
A second double-blind, randomized trial reported similar results.9 Among 97 patients randomized to receive adalimumab, placebo, or phototherapy, no significant difference in vascular inflammation was found after 12 weeks of therapy. In contrast, levels of C-reactive protein, IL-6, and glycoprotein acetylation were markedly reduced. The authors also reported adverse effects of adalimumab therapy on lipid metabolism with reduced cholesterol efflux capacity, a marker of ability of high-density-lipoprotein particles to perform reverse cholesterol transport, and high-density-lipoprotein particles, suggesting these effects may counteract some of the anti-inflammatory effects of TNF inhibitors.9
A growing body of data regarding the effect of TNF inhibitors on cardiovascular morbidity in patients with psoriasis is being collected, but no strong conclusions can be made. Given the disconnect of findings across these studies, it is possible that we have yet to elucidate the full mechanism by which TNF inhibitors may affect cardiovascular health. However, there may be additional confounding factors or patient characteristics at play. More large, prospective, randomized, controlled studies are needed to further understand this relationship.
The connection between psoriasis and increased major adverse cardiovascular events (MACEs) has been well studied. 1,2 Although treatment of psoriasis can improve skin and joint symptoms, it is less clear whether therapies may mitigate the increased risk for cardiovascular comorbidities. Tumor necrosis factor (TNF) inhibitors in particular have been studied with great interest given the role of TNF in vascular and metabolic functions. 3 Using a retrospective cohort design, Wu and colleagues 4 examined if treatment with TNF inhibitors in patients with psoriasis would be associated with a lower risk for MACEs compared to phototherapy. Results suggested a significantly lower hazard of MACEs in patients using TNF inhibitors vs patients treated with phototherapy (adjusted hazard ratio, 0.77; P = .046). Moreover, based on these findings, they calculated that treating approximately 161 patients with TNF inhibitors rather than phototherapy would result in 1 less MACE per year overall. 4
Patients with psoriasis have been shown to have a greater noncalcified coronary plaque burden and prevalence of high-risk plaque compared to healthy patients.5 Lerman and colleagues5 measured the coronary plaque burden of 105 patients with psoriasis and 25 healthy volunteers using coronary computed tomography angiography. Although the patients were on average 10 years younger and had lower cardiovascular risk as measured by traditional risk scores, patients with psoriasis were found to have a greater noncalcified coronary plaque burden compared to 100 patients with hyperlipidemia. This burden was associated with an increased prevalence of high-risk plaques. Furthermore, in patients followed for 1 year, improvements in psoriasis severity were associated with reductions in noncalcified coronary plaque burden, though this finding was across all treatment modalities. However, there was no significant difference in calcified coronary plaque burden associated with reduced psoriasis severity.5
Moreover, Pina et al6 conducted a prospective study evaluating the use of the TNF inhibitor adalimumab to improve endothelial function and arterial stiffness in patients with moderate to severe psoriasis. Among 29 patients, they found a significant improvement in endothelial function as measured by flow-mediated dilatation after 6 months of adalimumab therapy, with a mean increase from 6.19% to 7.46% (P=.008). They also reported decreases in arterial stiffness by pulse wave velocity (P=.03). Despite a small sample size, these findings provide 2 potential mechanisms by which TNF inhibitor therapy may reduce the risk for cardiovascular events.6
A retrospective cohort study evaluating data from the Kaiser Permanente Southern California health plan assessed whether TNF inhibitor therapy was associated with a lower risk for MACE in patients with psoriasis.7 A total of 18,194 patients were included; of these, 1463 received TNF inhibitor therapy for at least 2 months. After controlling for other variables, including age at psoriasis diagnosis, sex, race/ethnicity, and other cardiovascular risk factors (eg, history of smoking or alcohol use; use of clopidogrel, antihypertensive agents, antihyperlipidemics, or anticoagulants), patients in the TNF inhibitor cohort demonstrated a significantly lower MACE hazard ratio compared to patients treated with topicals (hazard ratio, 0.80; 95% confidence interval, 0.66-0.98; P<.05).7
Conversely, a randomized, placebo-controlled trial of 107 patients found no difference in vascular inflammation of the ascending aorta and the carotids after 16 weeks of adalimumab treatment vs placebo. In this study, however, most patients had only moderate psoriasis based on a mean psoriasis area and severity index score of 9.8.8 Given studies finding higher risk burden in patients with more severe skin disease,2 it is possible that the effect of TNF inhibitor therapy may not be as pronounced in patients with less skin involvement. There was a significant effect on C-reactive protein levels in patients receiving TNF inhibitor therapy compared to placebo at 16 weeks (P=.012), suggesting TNF does play some role in systemic inflammation, and it is possible it may exert cardiovascular effects through a mechanism other than vascular inflammation.8
A second double-blind, randomized trial reported similar results.9 Among 97 patients randomized to receive adalimumab, placebo, or phototherapy, no significant difference in vascular inflammation was found after 12 weeks of therapy. In contrast, levels of C-reactive protein, IL-6, and glycoprotein acetylation were markedly reduced. The authors also reported adverse effects of adalimumab therapy on lipid metabolism with reduced cholesterol efflux capacity, a marker of ability of high-density-lipoprotein particles to perform reverse cholesterol transport, and high-density-lipoprotein particles, suggesting these effects may counteract some of the anti-inflammatory effects of TNF inhibitors.9
A growing body of data regarding the effect of TNF inhibitors on cardiovascular morbidity in patients with psoriasis is being collected, but no strong conclusions can be made. Given the disconnect of findings across these studies, it is possible that we have yet to elucidate the full mechanism by which TNF inhibitors may affect cardiovascular health. However, there may be additional confounding factors or patient characteristics at play. More large, prospective, randomized, controlled studies are needed to further understand this relationship.
- Ogdie A, Yu Y, Haynes K, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2015;74:326-332.
- Ahlehoff O, Gislason GH, Charlot M, et al. Psoriasis is associated with clinically significant cardiovascular risk: a Danish nationwide cohort study. J Intern Med. 2011;270:147-157.
- Kölliker Frers RA, Bisoendial RJ, Montoya SF, et al. Psoriasis and cardiovascular risk: immune-mediated crosstalk between metabolic, vascular, and autoimmune inflammation. Int J Cardiol Metab Endocr. 2015;6:43-54.
- Wu JJ, Sundaram M, Cloutier M, et al. The risk of cardiovascular events in psoriasis patients treated with tumor necrosis factor-α inhibitors versus phototherapy: an observational cohort study. J Am Acad Dermatol. 2018;79:60-68.
- Lerman JB, Joshi AA, Chaturvedi A, et al. Coronary plaque characterization in psoriasis reveals high-risk features that improve after treatment in a prospective observational study. Circulation. 2017;136:263-276.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-α therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6 month prospective study. J Dermatol. 2016;43:1267-1272.
- Wu JJ, Joshi AA, Reddy SP, et al. Anti-inflammatory therapy with tumor necrosis factor inhibitors is associated with reduced risk of major adverse cardiovascular events in psoriasis [published online March 24, 2018]. J Eur Acad Dermatol Venereol. doi:10.1111/jdv.14951.
- Bissonnette R, Harel F, Krueger JG, et al. TNF-α antagonist and vascular inflammation patients with psoriasis vulgaris: a randomized placebo-controlled study. J Invest Dermatol. 2017;137:1638-1645 .
- Mehta NN, Shin DB, Joshi AA, et al. Effect of 2 psoriasis treatments on vascular inflammation and novel inflammatory cardiovascular biomarkers: a randomized placebo-controlled trial. Circ Cardiovasc Imaging. 2018;11:e007394.
- Ogdie A, Yu Y, Haynes K, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2015;74:326-332.
- Ahlehoff O, Gislason GH, Charlot M, et al. Psoriasis is associated with clinically significant cardiovascular risk: a Danish nationwide cohort study. J Intern Med. 2011;270:147-157.
- Kölliker Frers RA, Bisoendial RJ, Montoya SF, et al. Psoriasis and cardiovascular risk: immune-mediated crosstalk between metabolic, vascular, and autoimmune inflammation. Int J Cardiol Metab Endocr. 2015;6:43-54.
- Wu JJ, Sundaram M, Cloutier M, et al. The risk of cardiovascular events in psoriasis patients treated with tumor necrosis factor-α inhibitors versus phototherapy: an observational cohort study. J Am Acad Dermatol. 2018;79:60-68.
- Lerman JB, Joshi AA, Chaturvedi A, et al. Coronary plaque characterization in psoriasis reveals high-risk features that improve after treatment in a prospective observational study. Circulation. 2017;136:263-276.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-α therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6 month prospective study. J Dermatol. 2016;43:1267-1272.
- Wu JJ, Joshi AA, Reddy SP, et al. Anti-inflammatory therapy with tumor necrosis factor inhibitors is associated with reduced risk of major adverse cardiovascular events in psoriasis [published online March 24, 2018]. J Eur Acad Dermatol Venereol. doi:10.1111/jdv.14951.
- Bissonnette R, Harel F, Krueger JG, et al. TNF-α antagonist and vascular inflammation patients with psoriasis vulgaris: a randomized placebo-controlled study. J Invest Dermatol. 2017;137:1638-1645 .
- Mehta NN, Shin DB, Joshi AA, et al. Effect of 2 psoriasis treatments on vascular inflammation and novel inflammatory cardiovascular biomarkers: a randomized placebo-controlled trial. Circ Cardiovasc Imaging. 2018;11:e007394.
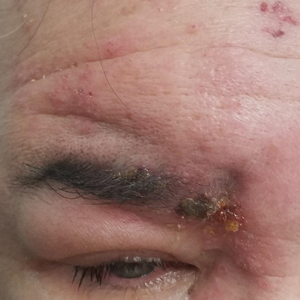
Periorbital Swelling and Rash Following Trauma
The Diagnosis: Herpes Zoster Opthalmicus
Due to the potential concern of vision loss, the patient was directed to a local emergency department for immediate ophthalmologic evaluation. He was diagnosed with herpes zoster ophthalmicus (HZO) and treated with oral acyclovir and prednisone. The rash and periorbital swelling resolved within 2 weeks of treatment, and he remained asymptomatic at follow-up 3 months later.
Herpes zoster ophthalmicus presents with an erythematous and vesicular rash in the distribution of cranial nerve V1. The herpetiform grouping of lesions on the forehead is diagnostic of HZO. Varicella-zoster virus (VZV) infection presents in 2 distinct forms. Primary infection (commonly known as chickenpox) presents clinically as a vesicular rash usually located on the face, arms, and trunk. Although the initial presentation usually occurs in childhood and is self-limited, the virus becomes latent in the dorsal root ganglia of sensory neurons. Varicella-zoster virus may become reactivated later in life and is termed herpes zoster (commonly known as shingles). It most often presents as a painful vesicular rash that may later form pustules.
Zoster outbreaks typically do not cross the midline but may in disseminated disease. Patients may experience a prodrome in the form of pain or less commonly pruritus or paresthesia along the dermatome between 1 and 10 days before the rash appears. Triggers for herpes zoster include illness, medications, malnutrition, surgery, or the natural decline in immune function due to aging. Trauma is another important precipitating event for VZV reactivation; one case-control study showed that zoster patients were 3.4 times more likely than controls to have had trauma the week prior.1 Patients with cranial zoster are more than 25 times more likely to have experienced trauma in the preceding week. Local trauma may predispose these patients to VZV reactivation by stimulating local sensory nerves or by disrupting local cutaneous immunity.2
Herpes zoster ophthalmicus occurs when zoster presents in the ophthalmic division of the fifth cranial nerve. It is a serious, vision-threatening condition with a presentation that can include conjunctivitis, scleritis, keratitis, optic neuritis, exophthalmos, lid retraction, ptosis, and extraocular muscle palsies. Treatment includes antiviral medication (eg, acyclovir, valacyclovir, famciclovir) and prompt ophthalmologic consultation due to potential vision-threatening complications, such as acute retinal necrosis. Acute pain control may be necessary with nonsteroidal anti-inflammatory drugs, opioids, steroids, tricyclic antidepressants, or anticonvulsants.3 Wet-to-dry dressings with sterile saline or Burow solution and/or calamine lotion can provide symptomatic relief of itching.
Periorbital and preseptal cellulitis typically present with more erythema of the skin surrounding the eye and without the accompanying rash. Periorbital cellulitis is the more serious infection and may be clinically distinguished by the presence of pain with extraocular muscle movement. Contact dermatitis and pemphigus vulgaris are possibilities, but both were less likely than HZO in this case presentation given the distribution of the rash and the patient history. Contact dermatitis typically presents with no prodrome with a main concern of pruritus. Pemphigus vulgaris nearly always includes involvement of the oral mucous membranes.
- Goh CL, Khoo L. A retrospective study of the clinical presentation and outcome of herpes zoster in a tertiary dermatology outpatient referral clinic. Int J Dermatol. 1997;36:667-672.
- Zhang JX, Joesoef RM, Bialek S, et al. Association of physical trauma with risk of herpes zoster among Medicare beneficiaries in the United States. J Infect Dis. 2013;207:1007-1011.
- Rousseau A, Bourcier T, Colin J, et al. Herpes zoster ophthalmicus--diagnosis and management. US Ophthalm Rev. 2013;6:119-124.
The Diagnosis: Herpes Zoster Opthalmicus
Due to the potential concern of vision loss, the patient was directed to a local emergency department for immediate ophthalmologic evaluation. He was diagnosed with herpes zoster ophthalmicus (HZO) and treated with oral acyclovir and prednisone. The rash and periorbital swelling resolved within 2 weeks of treatment, and he remained asymptomatic at follow-up 3 months later.
Herpes zoster ophthalmicus presents with an erythematous and vesicular rash in the distribution of cranial nerve V1. The herpetiform grouping of lesions on the forehead is diagnostic of HZO. Varicella-zoster virus (VZV) infection presents in 2 distinct forms. Primary infection (commonly known as chickenpox) presents clinically as a vesicular rash usually located on the face, arms, and trunk. Although the initial presentation usually occurs in childhood and is self-limited, the virus becomes latent in the dorsal root ganglia of sensory neurons. Varicella-zoster virus may become reactivated later in life and is termed herpes zoster (commonly known as shingles). It most often presents as a painful vesicular rash that may later form pustules.
Zoster outbreaks typically do not cross the midline but may in disseminated disease. Patients may experience a prodrome in the form of pain or less commonly pruritus or paresthesia along the dermatome between 1 and 10 days before the rash appears. Triggers for herpes zoster include illness, medications, malnutrition, surgery, or the natural decline in immune function due to aging. Trauma is another important precipitating event for VZV reactivation; one case-control study showed that zoster patients were 3.4 times more likely than controls to have had trauma the week prior.1 Patients with cranial zoster are more than 25 times more likely to have experienced trauma in the preceding week. Local trauma may predispose these patients to VZV reactivation by stimulating local sensory nerves or by disrupting local cutaneous immunity.2
Herpes zoster ophthalmicus occurs when zoster presents in the ophthalmic division of the fifth cranial nerve. It is a serious, vision-threatening condition with a presentation that can include conjunctivitis, scleritis, keratitis, optic neuritis, exophthalmos, lid retraction, ptosis, and extraocular muscle palsies. Treatment includes antiviral medication (eg, acyclovir, valacyclovir, famciclovir) and prompt ophthalmologic consultation due to potential vision-threatening complications, such as acute retinal necrosis. Acute pain control may be necessary with nonsteroidal anti-inflammatory drugs, opioids, steroids, tricyclic antidepressants, or anticonvulsants.3 Wet-to-dry dressings with sterile saline or Burow solution and/or calamine lotion can provide symptomatic relief of itching.
Periorbital and preseptal cellulitis typically present with more erythema of the skin surrounding the eye and without the accompanying rash. Periorbital cellulitis is the more serious infection and may be clinically distinguished by the presence of pain with extraocular muscle movement. Contact dermatitis and pemphigus vulgaris are possibilities, but both were less likely than HZO in this case presentation given the distribution of the rash and the patient history. Contact dermatitis typically presents with no prodrome with a main concern of pruritus. Pemphigus vulgaris nearly always includes involvement of the oral mucous membranes.
The Diagnosis: Herpes Zoster Opthalmicus
Due to the potential concern of vision loss, the patient was directed to a local emergency department for immediate ophthalmologic evaluation. He was diagnosed with herpes zoster ophthalmicus (HZO) and treated with oral acyclovir and prednisone. The rash and periorbital swelling resolved within 2 weeks of treatment, and he remained asymptomatic at follow-up 3 months later.
Herpes zoster ophthalmicus presents with an erythematous and vesicular rash in the distribution of cranial nerve V1. The herpetiform grouping of lesions on the forehead is diagnostic of HZO. Varicella-zoster virus (VZV) infection presents in 2 distinct forms. Primary infection (commonly known as chickenpox) presents clinically as a vesicular rash usually located on the face, arms, and trunk. Although the initial presentation usually occurs in childhood and is self-limited, the virus becomes latent in the dorsal root ganglia of sensory neurons. Varicella-zoster virus may become reactivated later in life and is termed herpes zoster (commonly known as shingles). It most often presents as a painful vesicular rash that may later form pustules.
Zoster outbreaks typically do not cross the midline but may in disseminated disease. Patients may experience a prodrome in the form of pain or less commonly pruritus or paresthesia along the dermatome between 1 and 10 days before the rash appears. Triggers for herpes zoster include illness, medications, malnutrition, surgery, or the natural decline in immune function due to aging. Trauma is another important precipitating event for VZV reactivation; one case-control study showed that zoster patients were 3.4 times more likely than controls to have had trauma the week prior.1 Patients with cranial zoster are more than 25 times more likely to have experienced trauma in the preceding week. Local trauma may predispose these patients to VZV reactivation by stimulating local sensory nerves or by disrupting local cutaneous immunity.2
Herpes zoster ophthalmicus occurs when zoster presents in the ophthalmic division of the fifth cranial nerve. It is a serious, vision-threatening condition with a presentation that can include conjunctivitis, scleritis, keratitis, optic neuritis, exophthalmos, lid retraction, ptosis, and extraocular muscle palsies. Treatment includes antiviral medication (eg, acyclovir, valacyclovir, famciclovir) and prompt ophthalmologic consultation due to potential vision-threatening complications, such as acute retinal necrosis. Acute pain control may be necessary with nonsteroidal anti-inflammatory drugs, opioids, steroids, tricyclic antidepressants, or anticonvulsants.3 Wet-to-dry dressings with sterile saline or Burow solution and/or calamine lotion can provide symptomatic relief of itching.
Periorbital and preseptal cellulitis typically present with more erythema of the skin surrounding the eye and without the accompanying rash. Periorbital cellulitis is the more serious infection and may be clinically distinguished by the presence of pain with extraocular muscle movement. Contact dermatitis and pemphigus vulgaris are possibilities, but both were less likely than HZO in this case presentation given the distribution of the rash and the patient history. Contact dermatitis typically presents with no prodrome with a main concern of pruritus. Pemphigus vulgaris nearly always includes involvement of the oral mucous membranes.
- Goh CL, Khoo L. A retrospective study of the clinical presentation and outcome of herpes zoster in a tertiary dermatology outpatient referral clinic. Int J Dermatol. 1997;36:667-672.
- Zhang JX, Joesoef RM, Bialek S, et al. Association of physical trauma with risk of herpes zoster among Medicare beneficiaries in the United States. J Infect Dis. 2013;207:1007-1011.
- Rousseau A, Bourcier T, Colin J, et al. Herpes zoster ophthalmicus--diagnosis and management. US Ophthalm Rev. 2013;6:119-124.
- Goh CL, Khoo L. A retrospective study of the clinical presentation and outcome of herpes zoster in a tertiary dermatology outpatient referral clinic. Int J Dermatol. 1997;36:667-672.
- Zhang JX, Joesoef RM, Bialek S, et al. Association of physical trauma with risk of herpes zoster among Medicare beneficiaries in the United States. J Infect Dis. 2013;207:1007-1011.
- Rousseau A, Bourcier T, Colin J, et al. Herpes zoster ophthalmicus--diagnosis and management. US Ophthalm Rev. 2013;6:119-124.
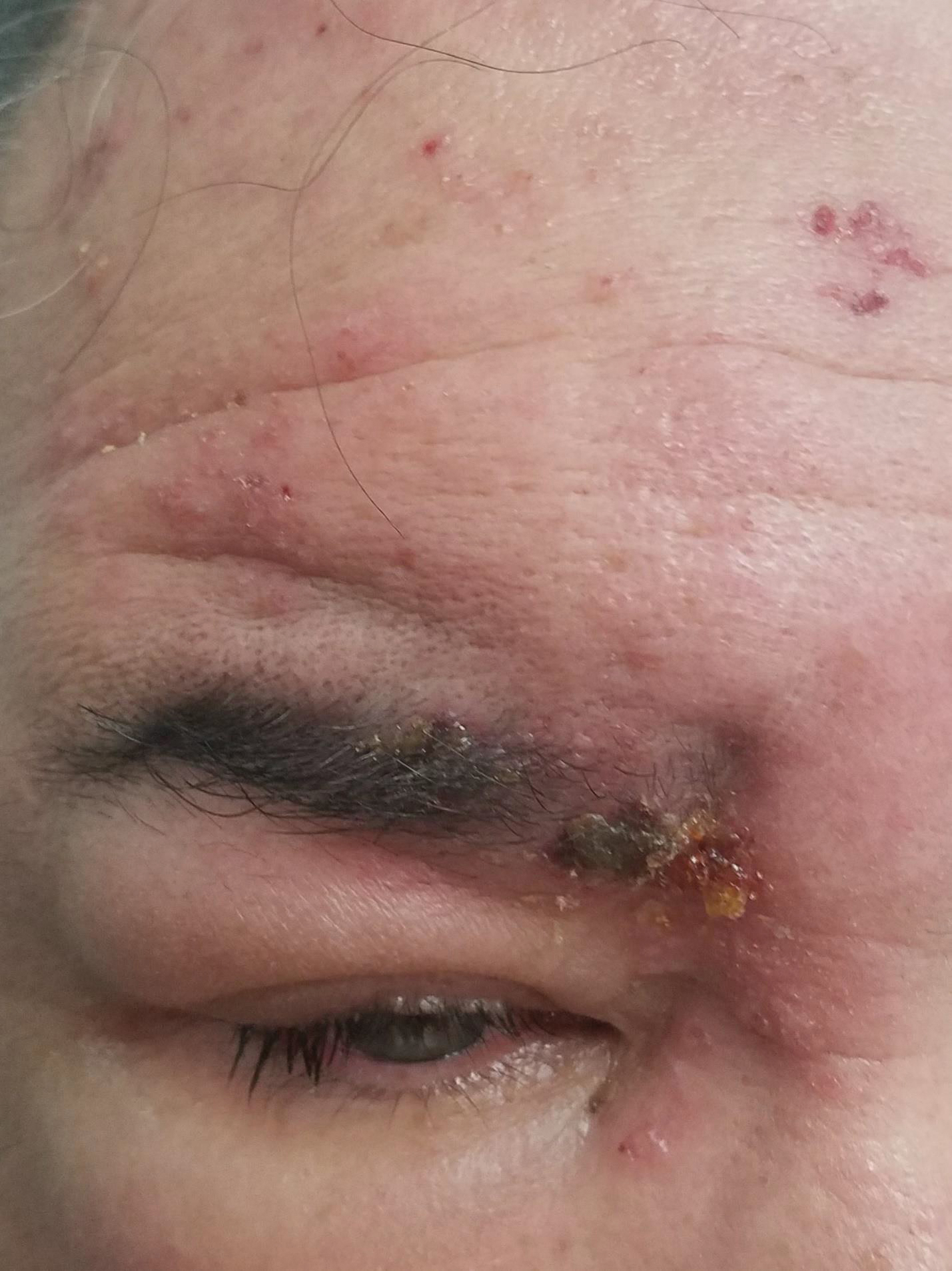
A 56-year-old man presented to an urgent care clinic with right periorbital swelling. He reported hitting his head on the door to a storage unit 2 days prior but did not lose consciousness. The swelling presented 2 days later. He reported mild headache and swelling around the right eye that coincided with an uncomfortable rash on the face and scalp. He also reported visual disruption due to the swelling but denied any eye pain, discharge from the eye, or painful eye movements. He had no lesions on the lips or inside the mouth. He denied any history of skin conditions. He further denied fever, joint pain, or any other systemic symptoms. His chronic medical conditions included diabetes mellitus, hypertension, and hyperlipidemia that were stable on metformin, carvedilol, amlodipine, enalapril, and simvastatin, which he had taken for several years. He had not started any new medications, and there were no recent changes in the dosing of his medications.
Infographic: Applications for the Ketogenic Diet in Dermatology
This infographic is available in the PDF above.
This infographic is available in the PDF above.
This infographic is available in the PDF above.

Subungual Hemorrhage From an Epidermal Growth Factor Receptor Inhibitor
To the Editor:
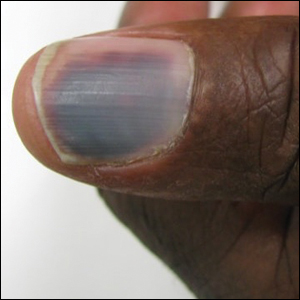
The epidermal growth factor receptor (EGFR) signaling pathway plays a role in the differentiation, proliferation, and survival of several cell types.1 Erlotinib is an EGFR inhibitor that targets aberrant cells that overexpress this receptor and has been used in the treatment of various solid malignant tumors.2,3 Common dermatologic side effects associated with EGFR inhibitors include papulopustular rash, xeroderma, and paronychia.2,3 We present a unique finding of subungual hemorrhage of the thumbnails in a patient taking erlotinib.
A 50-year-old man presented with acute-onset tenderness and discoloration of the thumbnails of 1 week’s duration. There was no preceding trauma or history of similar symptoms. His medical history was notable for recurrent lung adenocarcinoma with EGFR L858R mutation. Erlotinib therapy was initiated 5 weeks prior to symptom onset. He developed notable xeroderma of the palms and soles that preceded nail changes by a few days. He completed treatment with carboplatin and pemetrexed 16 months prior to relapse after paclitaxel failed due to a severe allergic reaction. There were no nail symptoms during that time. The patient did not have a documented coagulation disorder and was not on any known medications that would predispose him to bleeding. Physical examination demonstrated subungual hemorrhage of the thumbnails with tenderness on palpation (Figure). There was no evidence of periungual changes or nail plate abnormality. All other nails appeared normal. Laboratory test results showed normal platelets. Supportive therapeutic measures were recommended, and the patient was advised to avoid trauma to the nails.
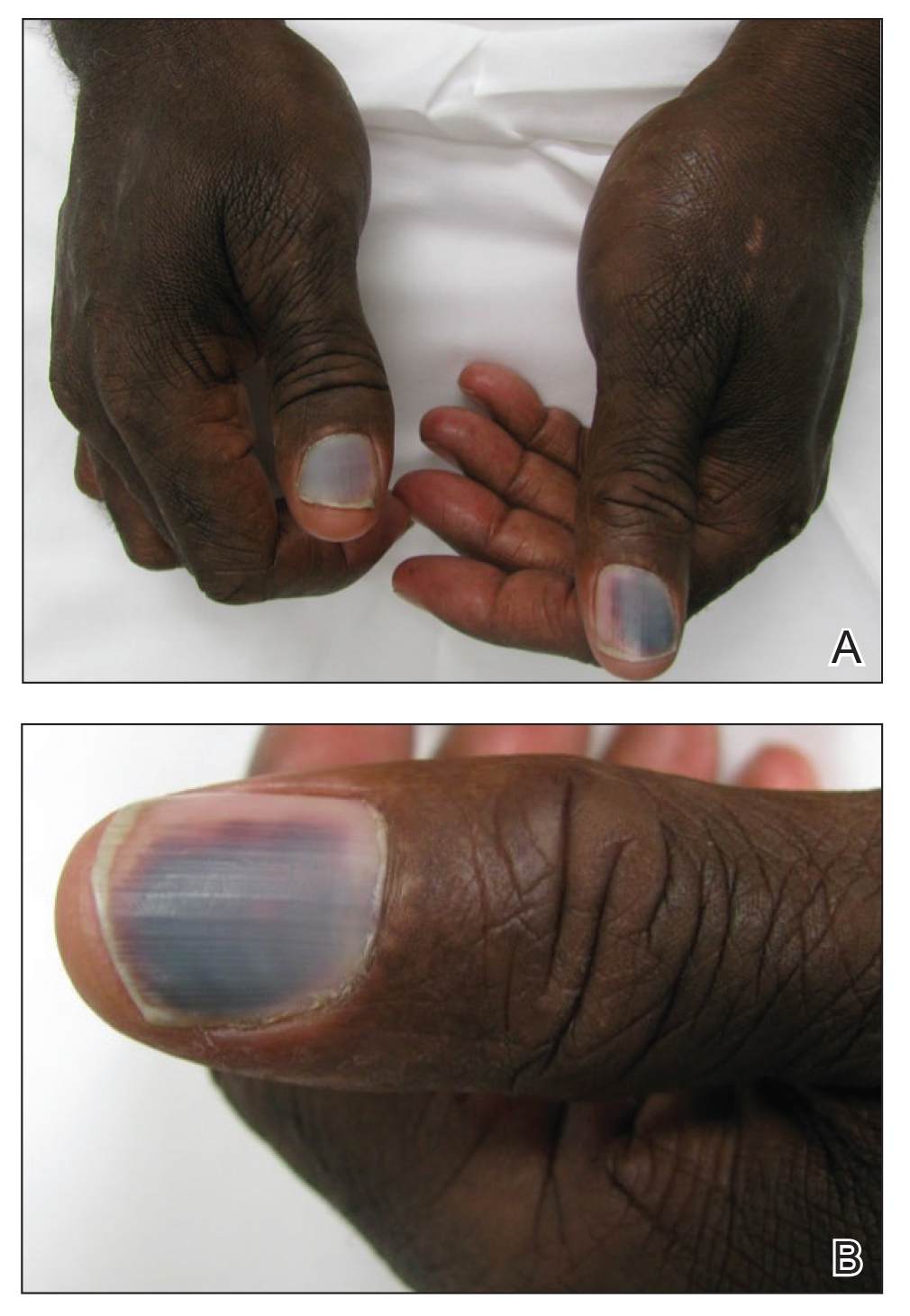
Nail toxicities reported with EGFR inhibitors include paronychia, periungual pyogenic granulomas, and ingrown nails.1-3 Inflammation of the nail bed also can lead to secondary nail changes, such as onychodystrophy or onycholysis.2 Subungual hemorrhage has been reported as a side effect of taxanes, anticoagulants, anthracyclines, anti-inflammatory agents, and retinoids.4,5
The pathogenesis of nail toxicity secondary to EGFR inhibitors is not entirely clear. Symptoms commonly occur several weeks to months after therapy initiation.6 Epidermal growth factor receptor inhibitors disrupt proliferation and promote apoptosis of keratinocytes that is thought to enhance fragility of the periungual skin and nail plate.1,3 Under the influence of EGFR inhibition, a proinflammatory microenvironment in the skin is created through a type I interferon response leading to tissue damage.7 These changes may predispose patients to develop subungual hemorrhage in response to repeated nail microtrauma. Subungual asymptomatic splinter hemorrhage is a nail finding described in patients treated with the multikinase inhibitors sorafenib and sunitinib. Splinter hemorrhages of the nails are thought to be secondary to capillary microinjuries of the digits that cannot be repaired due to inhibition of vascular EGFRs.4
The time course of erlotinib administration and the simultaneous onset of xeroderma, a known side effect of the drug, in our patient are consistent with other cases.6 Subungual hemorrhage, which the patient reported observing only days after the onset of xeroderma, provides increased support that the anti-EGFR medication was likely responsible for both side effects concurrently. Bilateral involvement of the thumbs makes trauma as an inciting event unlikely.
Incidence of nail changes secondary to anti-EGFR drugs are likely underestimated and underreported.3 Subungual hemorrhage should be considered as an additional, less common nail side effect of EGFR inhibitors that clinicians and patients may encounter. Improved awareness and understanding of nail toxicities associated with EGFR inhibitors may offer better insight into the pathogenesis of these side effects and management options.
- Piraccini BM, Alessandrini A. Drug-related nail disease. Clin Dermatol. 2013;31:618-626.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol. 2013;69:463-472.
- Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;333-337.
- Garden BC, Wu S, Lacouture ME. The risk of nail changes with epidermal growth factor receptor inhibitors: a systematic review of the literature and meta-analysis. J Am Acad Dermatol. 2012;67:400-408.
- Fox LP. Nail toxicity associated with epidermal growth factor receptor inhibitor therapy. J Am Acad Dermatol. 2007;56:460-465.
- Chen KL, Lin CC, Cho YT, et al. Comparison of skin toxic effects associated with gefitinib, erlotinib or afatinib treatment for non-small cell lung cancer. JAMA Dermatol. 2016;152:340-342.
- Lulli D, Carbone ML, Pastore S. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget. 2016;7:47777-47793.
To the Editor:
The epidermal growth factor receptor (EGFR) signaling pathway plays a role in the differentiation, proliferation, and survival of several cell types.1 Erlotinib is an EGFR inhibitor that targets aberrant cells that overexpress this receptor and has been used in the treatment of various solid malignant tumors.2,3 Common dermatologic side effects associated with EGFR inhibitors include papulopustular rash, xeroderma, and paronychia.2,3 We present a unique finding of subungual hemorrhage of the thumbnails in a patient taking erlotinib.
A 50-year-old man presented with acute-onset tenderness and discoloration of the thumbnails of 1 week’s duration. There was no preceding trauma or history of similar symptoms. His medical history was notable for recurrent lung adenocarcinoma with EGFR L858R mutation. Erlotinib therapy was initiated 5 weeks prior to symptom onset. He developed notable xeroderma of the palms and soles that preceded nail changes by a few days. He completed treatment with carboplatin and pemetrexed 16 months prior to relapse after paclitaxel failed due to a severe allergic reaction. There were no nail symptoms during that time. The patient did not have a documented coagulation disorder and was not on any known medications that would predispose him to bleeding. Physical examination demonstrated subungual hemorrhage of the thumbnails with tenderness on palpation (Figure). There was no evidence of periungual changes or nail plate abnormality. All other nails appeared normal. Laboratory test results showed normal platelets. Supportive therapeutic measures were recommended, and the patient was advised to avoid trauma to the nails.
Nail toxicities reported with EGFR inhibitors include paronychia, periungual pyogenic granulomas, and ingrown nails.1-3 Inflammation of the nail bed also can lead to secondary nail changes, such as onychodystrophy or onycholysis.2 Subungual hemorrhage has been reported as a side effect of taxanes, anticoagulants, anthracyclines, anti-inflammatory agents, and retinoids.4,5
The pathogenesis of nail toxicity secondary to EGFR inhibitors is not entirely clear. Symptoms commonly occur several weeks to months after therapy initiation.6 Epidermal growth factor receptor inhibitors disrupt proliferation and promote apoptosis of keratinocytes that is thought to enhance fragility of the periungual skin and nail plate.1,3 Under the influence of EGFR inhibition, a proinflammatory microenvironment in the skin is created through a type I interferon response leading to tissue damage.7 These changes may predispose patients to develop subungual hemorrhage in response to repeated nail microtrauma. Subungual asymptomatic splinter hemorrhage is a nail finding described in patients treated with the multikinase inhibitors sorafenib and sunitinib. Splinter hemorrhages of the nails are thought to be secondary to capillary microinjuries of the digits that cannot be repaired due to inhibition of vascular EGFRs.4
The time course of erlotinib administration and the simultaneous onset of xeroderma, a known side effect of the drug, in our patient are consistent with other cases.6 Subungual hemorrhage, which the patient reported observing only days after the onset of xeroderma, provides increased support that the anti-EGFR medication was likely responsible for both side effects concurrently. Bilateral involvement of the thumbs makes trauma as an inciting event unlikely.
Incidence of nail changes secondary to anti-EGFR drugs are likely underestimated and underreported.3 Subungual hemorrhage should be considered as an additional, less common nail side effect of EGFR inhibitors that clinicians and patients may encounter. Improved awareness and understanding of nail toxicities associated with EGFR inhibitors may offer better insight into the pathogenesis of these side effects and management options.
To the Editor:
The epidermal growth factor receptor (EGFR) signaling pathway plays a role in the differentiation, proliferation, and survival of several cell types.1 Erlotinib is an EGFR inhibitor that targets aberrant cells that overexpress this receptor and has been used in the treatment of various solid malignant tumors.2,3 Common dermatologic side effects associated with EGFR inhibitors include papulopustular rash, xeroderma, and paronychia.2,3 We present a unique finding of subungual hemorrhage of the thumbnails in a patient taking erlotinib.
A 50-year-old man presented with acute-onset tenderness and discoloration of the thumbnails of 1 week’s duration. There was no preceding trauma or history of similar symptoms. His medical history was notable for recurrent lung adenocarcinoma with EGFR L858R mutation. Erlotinib therapy was initiated 5 weeks prior to symptom onset. He developed notable xeroderma of the palms and soles that preceded nail changes by a few days. He completed treatment with carboplatin and pemetrexed 16 months prior to relapse after paclitaxel failed due to a severe allergic reaction. There were no nail symptoms during that time. The patient did not have a documented coagulation disorder and was not on any known medications that would predispose him to bleeding. Physical examination demonstrated subungual hemorrhage of the thumbnails with tenderness on palpation (Figure). There was no evidence of periungual changes or nail plate abnormality. All other nails appeared normal. Laboratory test results showed normal platelets. Supportive therapeutic measures were recommended, and the patient was advised to avoid trauma to the nails.
Nail toxicities reported with EGFR inhibitors include paronychia, periungual pyogenic granulomas, and ingrown nails.1-3 Inflammation of the nail bed also can lead to secondary nail changes, such as onychodystrophy or onycholysis.2 Subungual hemorrhage has been reported as a side effect of taxanes, anticoagulants, anthracyclines, anti-inflammatory agents, and retinoids.4,5
The pathogenesis of nail toxicity secondary to EGFR inhibitors is not entirely clear. Symptoms commonly occur several weeks to months after therapy initiation.6 Epidermal growth factor receptor inhibitors disrupt proliferation and promote apoptosis of keratinocytes that is thought to enhance fragility of the periungual skin and nail plate.1,3 Under the influence of EGFR inhibition, a proinflammatory microenvironment in the skin is created through a type I interferon response leading to tissue damage.7 These changes may predispose patients to develop subungual hemorrhage in response to repeated nail microtrauma. Subungual asymptomatic splinter hemorrhage is a nail finding described in patients treated with the multikinase inhibitors sorafenib and sunitinib. Splinter hemorrhages of the nails are thought to be secondary to capillary microinjuries of the digits that cannot be repaired due to inhibition of vascular EGFRs.4
The time course of erlotinib administration and the simultaneous onset of xeroderma, a known side effect of the drug, in our patient are consistent with other cases.6 Subungual hemorrhage, which the patient reported observing only days after the onset of xeroderma, provides increased support that the anti-EGFR medication was likely responsible for both side effects concurrently. Bilateral involvement of the thumbs makes trauma as an inciting event unlikely.
Incidence of nail changes secondary to anti-EGFR drugs are likely underestimated and underreported.3 Subungual hemorrhage should be considered as an additional, less common nail side effect of EGFR inhibitors that clinicians and patients may encounter. Improved awareness and understanding of nail toxicities associated with EGFR inhibitors may offer better insight into the pathogenesis of these side effects and management options.
- Piraccini BM, Alessandrini A. Drug-related nail disease. Clin Dermatol. 2013;31:618-626.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol. 2013;69:463-472.
- Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;333-337.
- Garden BC, Wu S, Lacouture ME. The risk of nail changes with epidermal growth factor receptor inhibitors: a systematic review of the literature and meta-analysis. J Am Acad Dermatol. 2012;67:400-408.
- Fox LP. Nail toxicity associated with epidermal growth factor receptor inhibitor therapy. J Am Acad Dermatol. 2007;56:460-465.
- Chen KL, Lin CC, Cho YT, et al. Comparison of skin toxic effects associated with gefitinib, erlotinib or afatinib treatment for non-small cell lung cancer. JAMA Dermatol. 2016;152:340-342.
- Lulli D, Carbone ML, Pastore S. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget. 2016;7:47777-47793.
- Piraccini BM, Alessandrini A. Drug-related nail disease. Clin Dermatol. 2013;31:618-626.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol. 2013;69:463-472.
- Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;333-337.
- Garden BC, Wu S, Lacouture ME. The risk of nail changes with epidermal growth factor receptor inhibitors: a systematic review of the literature and meta-analysis. J Am Acad Dermatol. 2012;67:400-408.
- Fox LP. Nail toxicity associated with epidermal growth factor receptor inhibitor therapy. J Am Acad Dermatol. 2007;56:460-465.
- Chen KL, Lin CC, Cho YT, et al. Comparison of skin toxic effects associated with gefitinib, erlotinib or afatinib treatment for non-small cell lung cancer. JAMA Dermatol. 2016;152:340-342.
- Lulli D, Carbone ML, Pastore S. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget. 2016;7:47777-47793.
Practice Points
- Subungual hemorrhage is a potential adverse side effect of epidermal growth factor receptor inhibitors.
- Epidermal growth factor receptor inhibition may lead to enhanced fragility of the periungual skin and nail plate as well as a proinflammatory microenvironment in the skin, predisposing patients to nail toxicity.
Nonuremic Calciphylaxis Triggered by Rapid Weight Loss and Hypotension
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
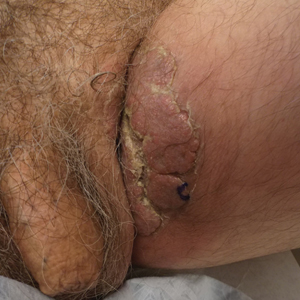
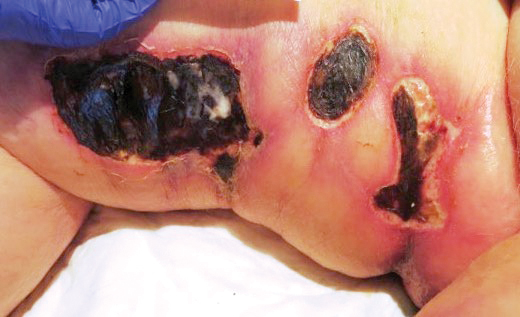
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).

Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
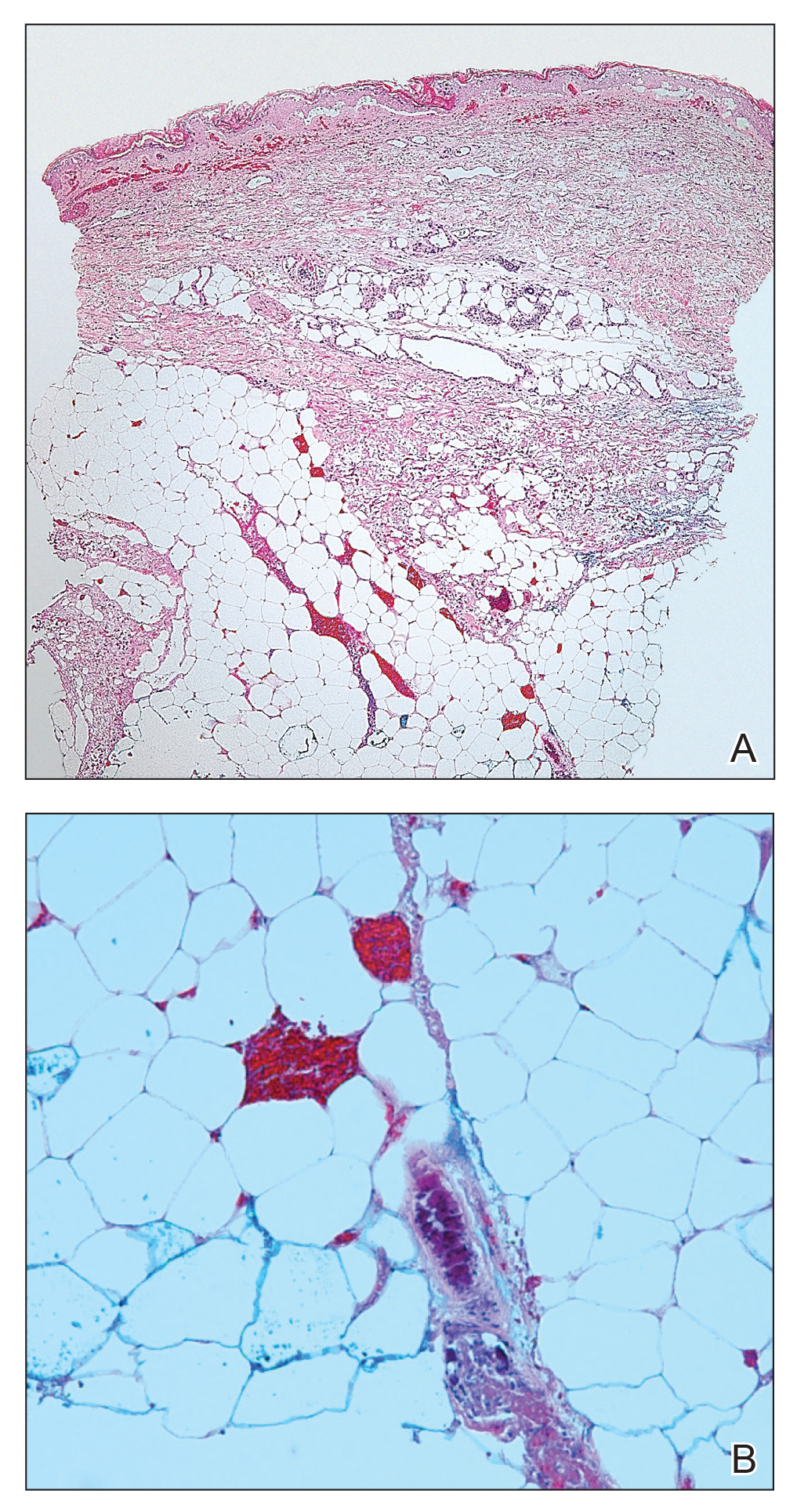
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Practice Points
- Calciphylaxis is a potentially fatal disease caused by metastatic calcification of cutaneous small- and medium-sized blood vessels leading to ischemia and necrosis.
- Calciphylaxis most commonly is seen in patients with renal disease requiring dialysis, but it also may be triggered by nonuremic causes in patients with known risk factors for calciphylaxis.
- Risk factors for calciphylaxis include female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.
- The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature.
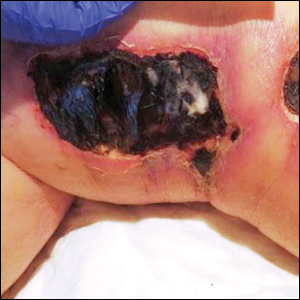
The Lowdown on Low-Dose Naltrexone
Low-dose naltrexone (LDN) has shown efficacy in off-label treatment of a variety of inflammatory diseases ranging from Crohn disease to multiple sclerosis.1 There are limited data about the use of LDN in dermatology, but reports regarding how it works as an anti-inflammatory agent have been published.1,2
Naltrexone is an opioid receptor antagonist that originally was approved by the US Food and Drug Administration to treat addiction to alcohol, opiates, and heroin.2 The dose of naltrexone to treat addiction ranges from 50 to 100 mg/d, and at these levels the effects of opioids are blocked for 24 hours; however, the dosing for LDN is much lower, ranging from 1.5 to 4.5 mg/d.3 At this low dose, naltrexone partially binds to various opioid receptors, leading to a temporary blockade.4 One of the downstream effects of this opioid receptor blockade is a paradoxical increase in endogenous endorphins.3
In addition to opioid blockage, lower doses of naltrexone have anti-inflammatory effects by inhibiting nonopioid receptors. Naltrexone blocks toll-like receptor 4, which is found on keratinocytes and also on macrophages such as microglia.5 These macrophages also contain inflammatory compounds such as tumor necrosis factor α and IL-6. Low-dose naltrexone can suppress levels of these inflammatory markers. It is important to note that these anti-inflammatory effects have not been observed at the standard higher doses of naltrexone.1
When to Use
Low-dose naltrexone is a treatment option for inflammatory dermatologic conditions. A recent review of the literature outlined the use of LDN in a variety of inflammatory skin conditions. Improvement was noted in patients with Hailey-Hailey disease, lichen planopilaris, and various types of pruritus (ie, aquagenic, cholestatic, uremic, atopic dermatitis related).3 A case report of LDN successfully treating a patient with psoriasis also has been published.6 We often use LDN at the University of Wisconsin (Madison, Wisconsin) to treat patients with psoriasis. Ekelem et al3 also discussed patients with skin conditions that either had no response or worsened with naltrexone treatment, including various types of pruritus (ie, uremic, mycosis fungoides related, other causes of pruritus). Importantly, in the majority of cases without an improved response, the dose used was 50 mg/d.3 Higher doses of naltrexone are not known to have anti-inflammatory effects.
Low-dose naltrexone can be considered as a treatment option in patients with contraindications to other systemic anti-inflammatory treatments; for example, patients with a history of malignancy may prefer to avoid treatment with biologic agents. Low-dose naltrexone also can be considered as a treatment option in patients who are uncomfortable with the side-effect profiles of other systemic anti-inflammatory treatments, such as the risk for leukemias and lymphomas associated with biologic agents, the risk for liver toxicity with methotrexate, or the risk for hyperlipidemia with acitretin.
How to Monitor
The following monitoring information is adapted from the practice of Apple Bodemer, MD, a board-certified dermatologist at the University of Wisconsin (Madison, Wisconsin) who also is fellowship trained in integrative medicine.
There is a paucity of published data about LDN dosing for inflammatory skin diseases. However, prescribers should be aware that LDN can alter thyroid hormone levels, especially in patients with autoimmune thyroid disease. If a thyroid-stimulating hormone (TSH) level within reference range has not been noted in the last year, consider screening with a TSH test and also assessing for a personal or family history of thyroid disease. If the TSH level is within reference range, there generally is no need to monitor while treating with LDN. Consider checking TSH levels every 4 months in patients with thyroid disease while they are on LDN therapy and be sure to educate them about symptoms of hyperthyroidism.
Side Effects
Low-dose naltrexone has a minimal side-effect profile with self-limited side effects that often resolve within approximately 1 week. One of the most commonly reported side effects is sleep disturbance with vivid dreams, which has been reported in 37% of participants.1 If your patients experience this side effect, you can reassure them that it improves with time. You also can switch to morning dosing to try and alleviate sleep disturbances at night. Another possible side effect is gastrointestinal tract upset. Importantly, there is no known abuse potential for LDN.1 To stop LDN, patients should be stable for 6 to 12 months, and there is no need to wean them off it.
Cost and Availability
Because use of LDN in dermatology is considered off label and it is not approved by the US Food and Drug Administration to treat any medical conditions, it must be prescribed through a compounding pharmacy, usually without insurance coverage. The monthly cost is approximately $30 depending on the pharmacy (unpublished data), which may be cost prohibitive for patients, so it is important to counsel them about price
Final Thoughts
Low-dose naltrexone is an alternative treatment option that can be considered in patients with inflammatory skin diseases. It has a favorable side-effect profile, especially compared to other systemic anti-inflammatory agents; however, additional studies are needed to learn more about its safety and efficacy. If patients ask you about LDN, the information provided here can guide you with how it works and how to prescribe it.
- Younger J, Parkitny L, McLain D. The use of low-dose naltrexone (LDN) as a novel anti-inflammatory treatment for chronic pain. Clin Rheumatol. 2014;33:451-459.
- Brown N, Panksepp J. Low-dose naltrexone for disease prevention and quality of life. Med Hypotheses. 2009;72:333-337.
- Ekelem C, Juhasz M, Khera P, et al. Utility of naltrexone treatment for chronic inflammatory dermatologic conditions: a systematic review. JAMA Dermatol. 2019;155:229-236.
- Bihari B. Efficacy of low dose naltrexone as an immune stabilizing agent for the treatment of HIV/AIDS. AIDS Patient Care. 1995;9:3.
- Lee B, Elston DM. The uses of naltrexone in dermatologic conditions [published online December 21, 2018]. J Am Acad Dermatol. 2019;80:1746-1752.
- Bridgman AC, Kirchhof MG. Treatment of psoriasis vulgaris using low-dose naltrexone. JAAD Case Rep. 2018;4:827-829.
Low-dose naltrexone (LDN) has shown efficacy in off-label treatment of a variety of inflammatory diseases ranging from Crohn disease to multiple sclerosis.1 There are limited data about the use of LDN in dermatology, but reports regarding how it works as an anti-inflammatory agent have been published.1,2
Naltrexone is an opioid receptor antagonist that originally was approved by the US Food and Drug Administration to treat addiction to alcohol, opiates, and heroin.2 The dose of naltrexone to treat addiction ranges from 50 to 100 mg/d, and at these levels the effects of opioids are blocked for 24 hours; however, the dosing for LDN is much lower, ranging from 1.5 to 4.5 mg/d.3 At this low dose, naltrexone partially binds to various opioid receptors, leading to a temporary blockade.4 One of the downstream effects of this opioid receptor blockade is a paradoxical increase in endogenous endorphins.3
In addition to opioid blockage, lower doses of naltrexone have anti-inflammatory effects by inhibiting nonopioid receptors. Naltrexone blocks toll-like receptor 4, which is found on keratinocytes and also on macrophages such as microglia.5 These macrophages also contain inflammatory compounds such as tumor necrosis factor α and IL-6. Low-dose naltrexone can suppress levels of these inflammatory markers. It is important to note that these anti-inflammatory effects have not been observed at the standard higher doses of naltrexone.1
When to Use
Low-dose naltrexone is a treatment option for inflammatory dermatologic conditions. A recent review of the literature outlined the use of LDN in a variety of inflammatory skin conditions. Improvement was noted in patients with Hailey-Hailey disease, lichen planopilaris, and various types of pruritus (ie, aquagenic, cholestatic, uremic, atopic dermatitis related).3 A case report of LDN successfully treating a patient with psoriasis also has been published.6 We often use LDN at the University of Wisconsin (Madison, Wisconsin) to treat patients with psoriasis. Ekelem et al3 also discussed patients with skin conditions that either had no response or worsened with naltrexone treatment, including various types of pruritus (ie, uremic, mycosis fungoides related, other causes of pruritus). Importantly, in the majority of cases without an improved response, the dose used was 50 mg/d.3 Higher doses of naltrexone are not known to have anti-inflammatory effects.
Low-dose naltrexone can be considered as a treatment option in patients with contraindications to other systemic anti-inflammatory treatments; for example, patients with a history of malignancy may prefer to avoid treatment with biologic agents. Low-dose naltrexone also can be considered as a treatment option in patients who are uncomfortable with the side-effect profiles of other systemic anti-inflammatory treatments, such as the risk for leukemias and lymphomas associated with biologic agents, the risk for liver toxicity with methotrexate, or the risk for hyperlipidemia with acitretin.
How to Monitor
The following monitoring information is adapted from the practice of Apple Bodemer, MD, a board-certified dermatologist at the University of Wisconsin (Madison, Wisconsin) who also is fellowship trained in integrative medicine.
There is a paucity of published data about LDN dosing for inflammatory skin diseases. However, prescribers should be aware that LDN can alter thyroid hormone levels, especially in patients with autoimmune thyroid disease. If a thyroid-stimulating hormone (TSH) level within reference range has not been noted in the last year, consider screening with a TSH test and also assessing for a personal or family history of thyroid disease. If the TSH level is within reference range, there generally is no need to monitor while treating with LDN. Consider checking TSH levels every 4 months in patients with thyroid disease while they are on LDN therapy and be sure to educate them about symptoms of hyperthyroidism.
Side Effects
Low-dose naltrexone has a minimal side-effect profile with self-limited side effects that often resolve within approximately 1 week. One of the most commonly reported side effects is sleep disturbance with vivid dreams, which has been reported in 37% of participants.1 If your patients experience this side effect, you can reassure them that it improves with time. You also can switch to morning dosing to try and alleviate sleep disturbances at night. Another possible side effect is gastrointestinal tract upset. Importantly, there is no known abuse potential for LDN.1 To stop LDN, patients should be stable for 6 to 12 months, and there is no need to wean them off it.
Cost and Availability
Because use of LDN in dermatology is considered off label and it is not approved by the US Food and Drug Administration to treat any medical conditions, it must be prescribed through a compounding pharmacy, usually without insurance coverage. The monthly cost is approximately $30 depending on the pharmacy (unpublished data), which may be cost prohibitive for patients, so it is important to counsel them about price
Final Thoughts
Low-dose naltrexone is an alternative treatment option that can be considered in patients with inflammatory skin diseases. It has a favorable side-effect profile, especially compared to other systemic anti-inflammatory agents; however, additional studies are needed to learn more about its safety and efficacy. If patients ask you about LDN, the information provided here can guide you with how it works and how to prescribe it.
Low-dose naltrexone (LDN) has shown efficacy in off-label treatment of a variety of inflammatory diseases ranging from Crohn disease to multiple sclerosis.1 There are limited data about the use of LDN in dermatology, but reports regarding how it works as an anti-inflammatory agent have been published.1,2
Naltrexone is an opioid receptor antagonist that originally was approved by the US Food and Drug Administration to treat addiction to alcohol, opiates, and heroin.2 The dose of naltrexone to treat addiction ranges from 50 to 100 mg/d, and at these levels the effects of opioids are blocked for 24 hours; however, the dosing for LDN is much lower, ranging from 1.5 to 4.5 mg/d.3 At this low dose, naltrexone partially binds to various opioid receptors, leading to a temporary blockade.4 One of the downstream effects of this opioid receptor blockade is a paradoxical increase in endogenous endorphins.3
In addition to opioid blockage, lower doses of naltrexone have anti-inflammatory effects by inhibiting nonopioid receptors. Naltrexone blocks toll-like receptor 4, which is found on keratinocytes and also on macrophages such as microglia.5 These macrophages also contain inflammatory compounds such as tumor necrosis factor α and IL-6. Low-dose naltrexone can suppress levels of these inflammatory markers. It is important to note that these anti-inflammatory effects have not been observed at the standard higher doses of naltrexone.1
When to Use
Low-dose naltrexone is a treatment option for inflammatory dermatologic conditions. A recent review of the literature outlined the use of LDN in a variety of inflammatory skin conditions. Improvement was noted in patients with Hailey-Hailey disease, lichen planopilaris, and various types of pruritus (ie, aquagenic, cholestatic, uremic, atopic dermatitis related).3 A case report of LDN successfully treating a patient with psoriasis also has been published.6 We often use LDN at the University of Wisconsin (Madison, Wisconsin) to treat patients with psoriasis. Ekelem et al3 also discussed patients with skin conditions that either had no response or worsened with naltrexone treatment, including various types of pruritus (ie, uremic, mycosis fungoides related, other causes of pruritus). Importantly, in the majority of cases without an improved response, the dose used was 50 mg/d.3 Higher doses of naltrexone are not known to have anti-inflammatory effects.
Low-dose naltrexone can be considered as a treatment option in patients with contraindications to other systemic anti-inflammatory treatments; for example, patients with a history of malignancy may prefer to avoid treatment with biologic agents. Low-dose naltrexone also can be considered as a treatment option in patients who are uncomfortable with the side-effect profiles of other systemic anti-inflammatory treatments, such as the risk for leukemias and lymphomas associated with biologic agents, the risk for liver toxicity with methotrexate, or the risk for hyperlipidemia with acitretin.
How to Monitor
The following monitoring information is adapted from the practice of Apple Bodemer, MD, a board-certified dermatologist at the University of Wisconsin (Madison, Wisconsin) who also is fellowship trained in integrative medicine.
There is a paucity of published data about LDN dosing for inflammatory skin diseases. However, prescribers should be aware that LDN can alter thyroid hormone levels, especially in patients with autoimmune thyroid disease. If a thyroid-stimulating hormone (TSH) level within reference range has not been noted in the last year, consider screening with a TSH test and also assessing for a personal or family history of thyroid disease. If the TSH level is within reference range, there generally is no need to monitor while treating with LDN. Consider checking TSH levels every 4 months in patients with thyroid disease while they are on LDN therapy and be sure to educate them about symptoms of hyperthyroidism.
Side Effects
Low-dose naltrexone has a minimal side-effect profile with self-limited side effects that often resolve within approximately 1 week. One of the most commonly reported side effects is sleep disturbance with vivid dreams, which has been reported in 37% of participants.1 If your patients experience this side effect, you can reassure them that it improves with time. You also can switch to morning dosing to try and alleviate sleep disturbances at night. Another possible side effect is gastrointestinal tract upset. Importantly, there is no known abuse potential for LDN.1 To stop LDN, patients should be stable for 6 to 12 months, and there is no need to wean them off it.
Cost and Availability
Because use of LDN in dermatology is considered off label and it is not approved by the US Food and Drug Administration to treat any medical conditions, it must be prescribed through a compounding pharmacy, usually without insurance coverage. The monthly cost is approximately $30 depending on the pharmacy (unpublished data), which may be cost prohibitive for patients, so it is important to counsel them about price
Final Thoughts
Low-dose naltrexone is an alternative treatment option that can be considered in patients with inflammatory skin diseases. It has a favorable side-effect profile, especially compared to other systemic anti-inflammatory agents; however, additional studies are needed to learn more about its safety and efficacy. If patients ask you about LDN, the information provided here can guide you with how it works and how to prescribe it.
- Younger J, Parkitny L, McLain D. The use of low-dose naltrexone (LDN) as a novel anti-inflammatory treatment for chronic pain. Clin Rheumatol. 2014;33:451-459.
- Brown N, Panksepp J. Low-dose naltrexone for disease prevention and quality of life. Med Hypotheses. 2009;72:333-337.
- Ekelem C, Juhasz M, Khera P, et al. Utility of naltrexone treatment for chronic inflammatory dermatologic conditions: a systematic review. JAMA Dermatol. 2019;155:229-236.
- Bihari B. Efficacy of low dose naltrexone as an immune stabilizing agent for the treatment of HIV/AIDS. AIDS Patient Care. 1995;9:3.
- Lee B, Elston DM. The uses of naltrexone in dermatologic conditions [published online December 21, 2018]. J Am Acad Dermatol. 2019;80:1746-1752.
- Bridgman AC, Kirchhof MG. Treatment of psoriasis vulgaris using low-dose naltrexone. JAAD Case Rep. 2018;4:827-829.
- Younger J, Parkitny L, McLain D. The use of low-dose naltrexone (LDN) as a novel anti-inflammatory treatment for chronic pain. Clin Rheumatol. 2014;33:451-459.
- Brown N, Panksepp J. Low-dose naltrexone for disease prevention and quality of life. Med Hypotheses. 2009;72:333-337.
- Ekelem C, Juhasz M, Khera P, et al. Utility of naltrexone treatment for chronic inflammatory dermatologic conditions: a systematic review. JAMA Dermatol. 2019;155:229-236.
- Bihari B. Efficacy of low dose naltrexone as an immune stabilizing agent for the treatment of HIV/AIDS. AIDS Patient Care. 1995;9:3.
- Lee B, Elston DM. The uses of naltrexone in dermatologic conditions [published online December 21, 2018]. J Am Acad Dermatol. 2019;80:1746-1752.
- Bridgman AC, Kirchhof MG. Treatment of psoriasis vulgaris using low-dose naltrexone. JAAD Case Rep. 2018;4:827-829.
Resident Pearl
- Low-dose naltrexone is an alternative antiinflammatory treatment to consider in patients with inflammatory skin diseases, with a minimal side-effect profile.

Friable Scalp Nodule
The Diagnosis: Adnexal Neoplasm Arising in a Nevus Sebaceus
Biopsy of the lesion showed a proliferation of basaloid-appearing cells with focal ductal differentiation and ulceration consistent with poroma (Figure 1). Due to the superficial nature of the biopsy, the pathologist recommended excision to ensure complete removal and to rule out a well-differentiated porocarcinoma. Excision of the lesion showed large basaloid aggregates with a hypercellular stroma and a surrounding papillomatous epidermis with well-developed sebaceous lobules consistent with a trichoblastoma and a nevus sebaceus, respectively (Figure 2). There also was evidence of poroma; however, there were no findings concerning for porocarcinoma, which could lead to metastasis (Figure 3).
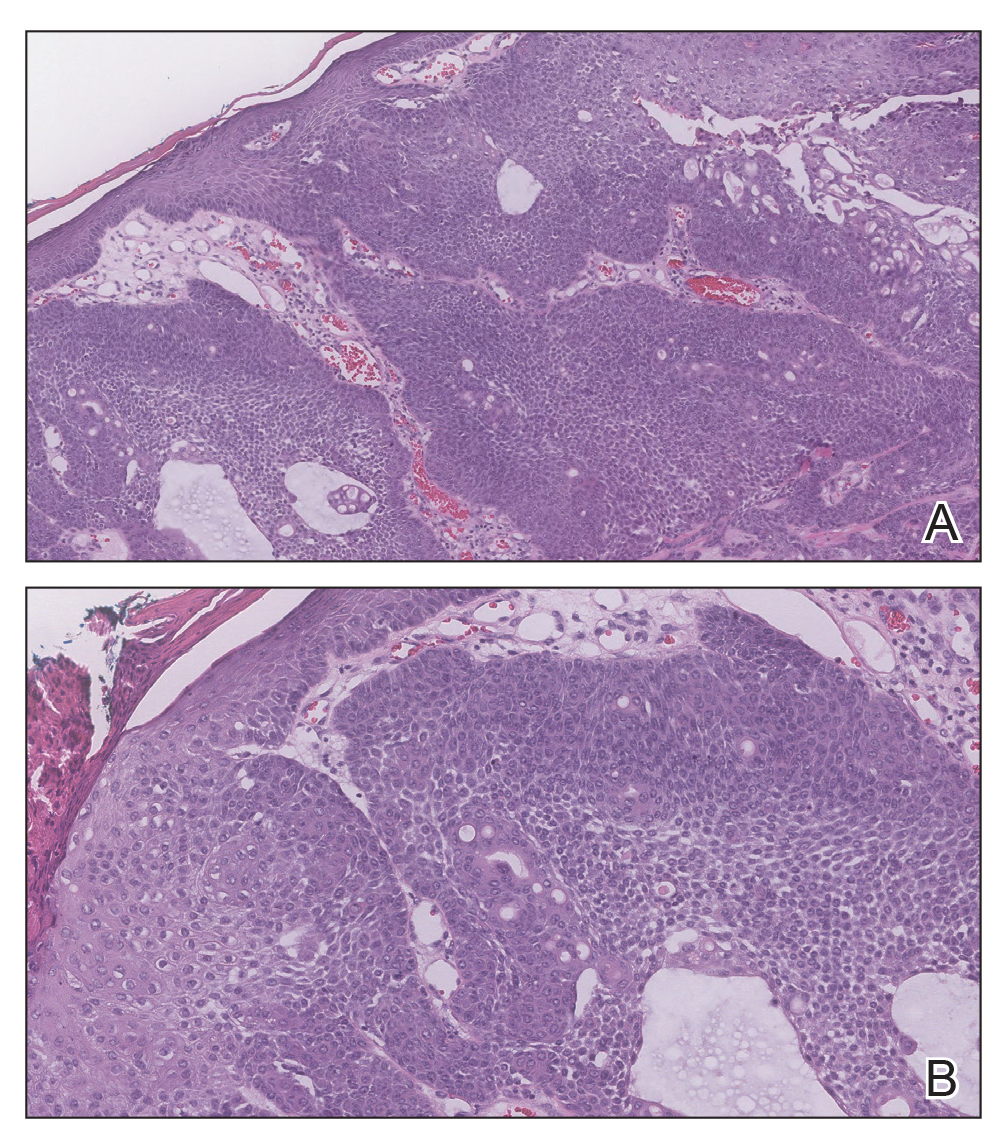
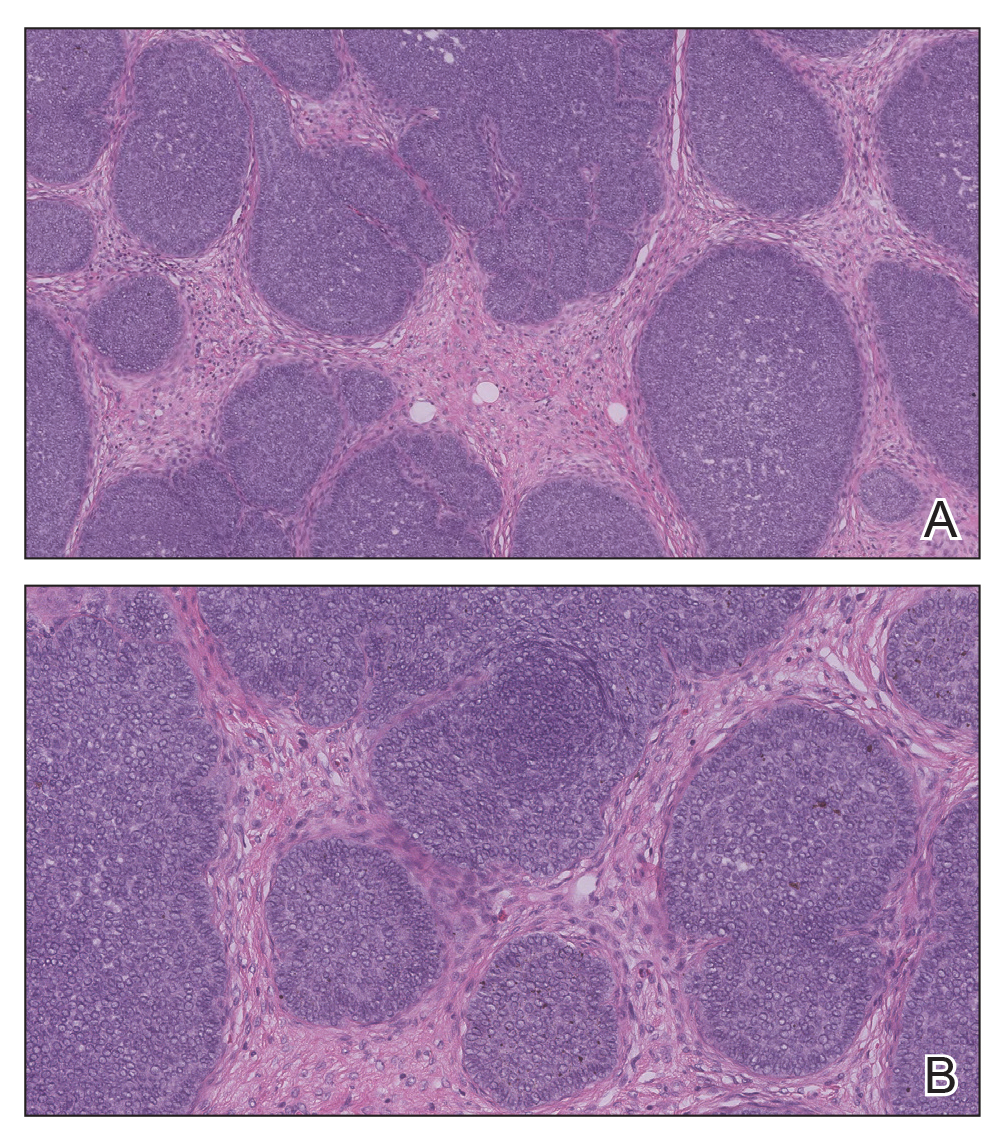
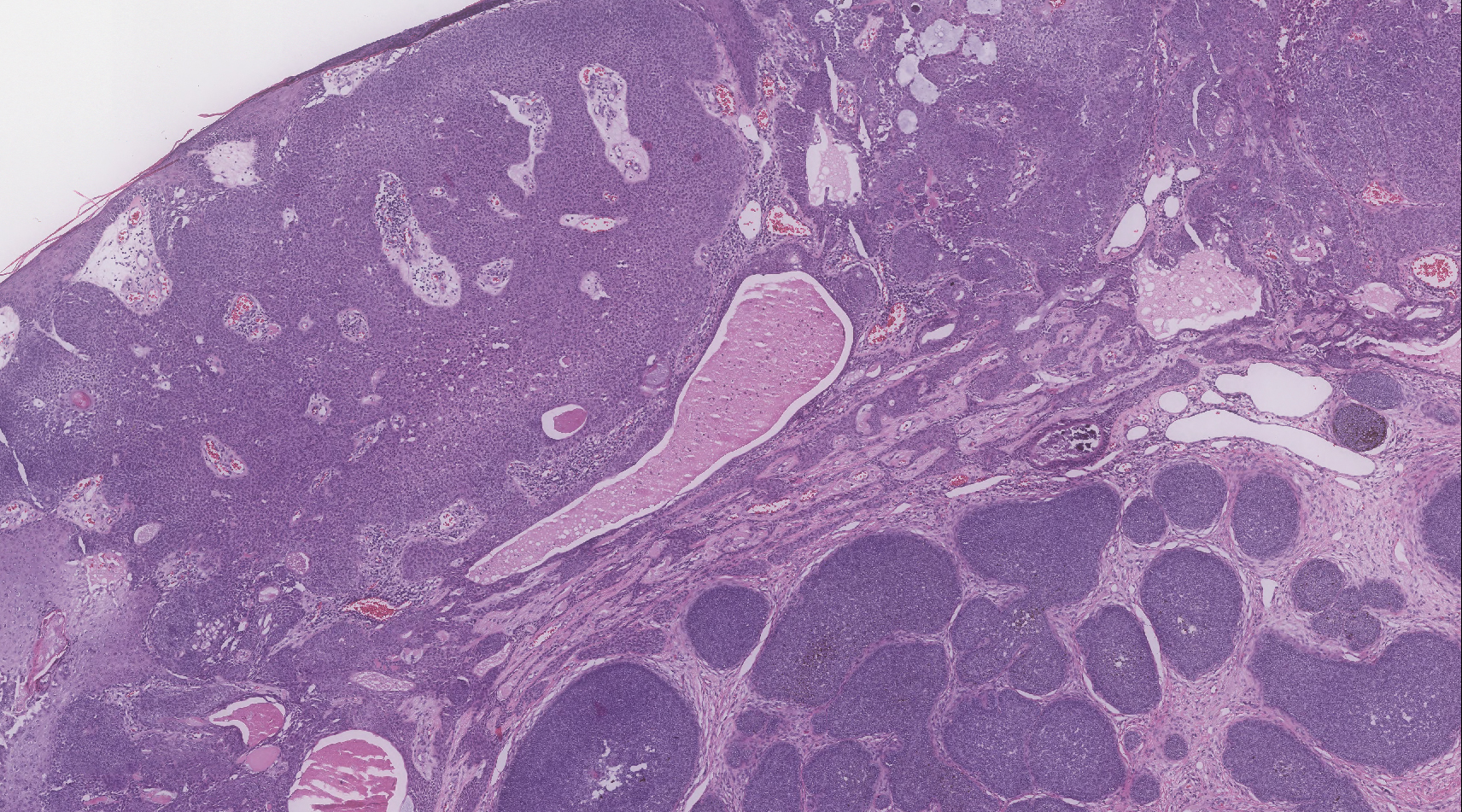
Nevus sebaceus is a benign, hamartomatous, congenital growth that occurs in approximately 1% of patients presenting to dermatology offices. It usually presents as a single asymptomatic plaque on the scalp (62.5%) or face (24.5%) that changes in morphology over its lifetime.1,2 In children, a nevus manifests as a yellowish, smooth, waxy skin lesion. As the sebaceous glands become more developed during adolescence, the lesion takes on more of a verrucous appearance and also can darken.
Although nevus sebaceus is benign, it may give rise to both benign and malignant neoplasms. In a 2014 study of 707 cases of nevus sebaceus, 21.4% developed secondary neoplasms, 88% of which were benign.2 The origins of these neoplasms can be epithelial, sebaceous, apocrine, and/or follicular. The 3 most common secondary neoplasms found in nevus sebaceus are trichoblastoma (34.7%), syringocystadenoma papilliferum (24.7%), and apocrine/eccrine adenoma (10%), all of which are benign.2 Trichoblastomas represent a type of hair follicle tumor. Malignant lesions manifest in approximately 2.5% of cases, with basal cell carcinoma (BCC) being the most common (5.3% of all neoplasms), followed by squamous cell carcinoma (2.7% of all neoplasms).2 Differentiating BCC from trichoblastoma can be difficult, but histologically BCCs usually have tumor stromal clefting while trichoblastomas do not.3 The incidence of secondary tumors in nevus sebaceus displays a strong correlation with age; thus, the highest proportion of neoplasms occur in adults.
Treatment of nevus sebaceus depends on the patient's age. In children, because of the low probability of secondary neoplasms, observation in lieu of surgical excision is a common approach. In adults, the approach typically is surgical excision or close follow-up, as there is a concern for secondary neoplasm and the potential for malignant degeneration.
A nevus sebaceus leading to 2 or more tumors within the same lesion is rare (seen in only 4.2% of lesions). The most common combination is trichoblastoma with syringocystadenoma papilliferum (0.6% of all cases).2 Poromas represent sweat gland tumors that usually appear on the soles (65%) or palms (10%).4 It is uncommon for these neoplasms to manifest on the scalp or within a nevus sebaceus. Three independent studies (N=596; N=707; N=450) did not report any occurrences of eccrine poroma.1,2,5 Eccrine poroma in conjunction with nodular trichoblastoma arising in a nevus sebaceus is unusual, and definitive excision should be strongly considered because of the possibility to develop a porocarcinoma.6
Atypical fibroxanthoma presents on sun-exposed areas as an exophytic nodule or plaque that frequently ulcerates. Pathology of this tumor shows a spindled cell proliferation that can stain positively for CD10 and procollagen 1. Basal cell carcinoma presents as a pearly papule or nodule displaying basaloid-appearing aggregates with tumor stromal clefting and can stain with Ber-EP4. Cylindromas typically present on the scalp as large rubbery-appearing plaques and nodules. Cylindromas usually present as a solitary tumor, but in the familial form there can be clusters of multiple nodules. Metastatic renal cell carcinoma frequently appears as a bleeding nodule on the scalp in patients with known renal cell cancer or as the initial presentation.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(pt 1):263-268.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Wang E, Lee JS, Kazakov DV. A rare combination of sebaceoma with carcinomatous change (sebaceous carcinoma), trichoblastoma, and poroma arising from a nevus sebaceus. J Cutan Pathol. 2013;40:676-682.
- Bae MI, Cho TH, Shin MK, et al. An unusual clinical presentation of eccrine poroma occurring on the auricle. Indian J Dermatol. 2015;60:523.
- Hsu MC, Liau JY, Hong JL, et al. Secondary neoplasms arising from nevus sebaceus: a retrospective study of 450 cases in Taiwan. J Dermatol. 2016;43:175-180.
- Takhan II, Domingo J. Metastasizing eccrine porocarcinoma developing in a sebaceous nevus of Jadassohn. report of a case. Arch Dermatol. 1985;121:413-415.
The Diagnosis: Adnexal Neoplasm Arising in a Nevus Sebaceus
Biopsy of the lesion showed a proliferation of basaloid-appearing cells with focal ductal differentiation and ulceration consistent with poroma (Figure 1). Due to the superficial nature of the biopsy, the pathologist recommended excision to ensure complete removal and to rule out a well-differentiated porocarcinoma. Excision of the lesion showed large basaloid aggregates with a hypercellular stroma and a surrounding papillomatous epidermis with well-developed sebaceous lobules consistent with a trichoblastoma and a nevus sebaceus, respectively (Figure 2). There also was evidence of poroma; however, there were no findings concerning for porocarcinoma, which could lead to metastasis (Figure 3).
Nevus sebaceus is a benign, hamartomatous, congenital growth that occurs in approximately 1% of patients presenting to dermatology offices. It usually presents as a single asymptomatic plaque on the scalp (62.5%) or face (24.5%) that changes in morphology over its lifetime.1,2 In children, a nevus manifests as a yellowish, smooth, waxy skin lesion. As the sebaceous glands become more developed during adolescence, the lesion takes on more of a verrucous appearance and also can darken.
Although nevus sebaceus is benign, it may give rise to both benign and malignant neoplasms. In a 2014 study of 707 cases of nevus sebaceus, 21.4% developed secondary neoplasms, 88% of which were benign.2 The origins of these neoplasms can be epithelial, sebaceous, apocrine, and/or follicular. The 3 most common secondary neoplasms found in nevus sebaceus are trichoblastoma (34.7%), syringocystadenoma papilliferum (24.7%), and apocrine/eccrine adenoma (10%), all of which are benign.2 Trichoblastomas represent a type of hair follicle tumor. Malignant lesions manifest in approximately 2.5% of cases, with basal cell carcinoma (BCC) being the most common (5.3% of all neoplasms), followed by squamous cell carcinoma (2.7% of all neoplasms).2 Differentiating BCC from trichoblastoma can be difficult, but histologically BCCs usually have tumor stromal clefting while trichoblastomas do not.3 The incidence of secondary tumors in nevus sebaceus displays a strong correlation with age; thus, the highest proportion of neoplasms occur in adults.
Treatment of nevus sebaceus depends on the patient's age. In children, because of the low probability of secondary neoplasms, observation in lieu of surgical excision is a common approach. In adults, the approach typically is surgical excision or close follow-up, as there is a concern for secondary neoplasm and the potential for malignant degeneration.
A nevus sebaceus leading to 2 or more tumors within the same lesion is rare (seen in only 4.2% of lesions). The most common combination is trichoblastoma with syringocystadenoma papilliferum (0.6% of all cases).2 Poromas represent sweat gland tumors that usually appear on the soles (65%) or palms (10%).4 It is uncommon for these neoplasms to manifest on the scalp or within a nevus sebaceus. Three independent studies (N=596; N=707; N=450) did not report any occurrences of eccrine poroma.1,2,5 Eccrine poroma in conjunction with nodular trichoblastoma arising in a nevus sebaceus is unusual, and definitive excision should be strongly considered because of the possibility to develop a porocarcinoma.6
Atypical fibroxanthoma presents on sun-exposed areas as an exophytic nodule or plaque that frequently ulcerates. Pathology of this tumor shows a spindled cell proliferation that can stain positively for CD10 and procollagen 1. Basal cell carcinoma presents as a pearly papule or nodule displaying basaloid-appearing aggregates with tumor stromal clefting and can stain with Ber-EP4. Cylindromas typically present on the scalp as large rubbery-appearing plaques and nodules. Cylindromas usually present as a solitary tumor, but in the familial form there can be clusters of multiple nodules. Metastatic renal cell carcinoma frequently appears as a bleeding nodule on the scalp in patients with known renal cell cancer or as the initial presentation.
The Diagnosis: Adnexal Neoplasm Arising in a Nevus Sebaceus
Biopsy of the lesion showed a proliferation of basaloid-appearing cells with focal ductal differentiation and ulceration consistent with poroma (Figure 1). Due to the superficial nature of the biopsy, the pathologist recommended excision to ensure complete removal and to rule out a well-differentiated porocarcinoma. Excision of the lesion showed large basaloid aggregates with a hypercellular stroma and a surrounding papillomatous epidermis with well-developed sebaceous lobules consistent with a trichoblastoma and a nevus sebaceus, respectively (Figure 2). There also was evidence of poroma; however, there were no findings concerning for porocarcinoma, which could lead to metastasis (Figure 3).
Nevus sebaceus is a benign, hamartomatous, congenital growth that occurs in approximately 1% of patients presenting to dermatology offices. It usually presents as a single asymptomatic plaque on the scalp (62.5%) or face (24.5%) that changes in morphology over its lifetime.1,2 In children, a nevus manifests as a yellowish, smooth, waxy skin lesion. As the sebaceous glands become more developed during adolescence, the lesion takes on more of a verrucous appearance and also can darken.
Although nevus sebaceus is benign, it may give rise to both benign and malignant neoplasms. In a 2014 study of 707 cases of nevus sebaceus, 21.4% developed secondary neoplasms, 88% of which were benign.2 The origins of these neoplasms can be epithelial, sebaceous, apocrine, and/or follicular. The 3 most common secondary neoplasms found in nevus sebaceus are trichoblastoma (34.7%), syringocystadenoma papilliferum (24.7%), and apocrine/eccrine adenoma (10%), all of which are benign.2 Trichoblastomas represent a type of hair follicle tumor. Malignant lesions manifest in approximately 2.5% of cases, with basal cell carcinoma (BCC) being the most common (5.3% of all neoplasms), followed by squamous cell carcinoma (2.7% of all neoplasms).2 Differentiating BCC from trichoblastoma can be difficult, but histologically BCCs usually have tumor stromal clefting while trichoblastomas do not.3 The incidence of secondary tumors in nevus sebaceus displays a strong correlation with age; thus, the highest proportion of neoplasms occur in adults.
Treatment of nevus sebaceus depends on the patient's age. In children, because of the low probability of secondary neoplasms, observation in lieu of surgical excision is a common approach. In adults, the approach typically is surgical excision or close follow-up, as there is a concern for secondary neoplasm and the potential for malignant degeneration.
A nevus sebaceus leading to 2 or more tumors within the same lesion is rare (seen in only 4.2% of lesions). The most common combination is trichoblastoma with syringocystadenoma papilliferum (0.6% of all cases).2 Poromas represent sweat gland tumors that usually appear on the soles (65%) or palms (10%).4 It is uncommon for these neoplasms to manifest on the scalp or within a nevus sebaceus. Three independent studies (N=596; N=707; N=450) did not report any occurrences of eccrine poroma.1,2,5 Eccrine poroma in conjunction with nodular trichoblastoma arising in a nevus sebaceus is unusual, and definitive excision should be strongly considered because of the possibility to develop a porocarcinoma.6
Atypical fibroxanthoma presents on sun-exposed areas as an exophytic nodule or plaque that frequently ulcerates. Pathology of this tumor shows a spindled cell proliferation that can stain positively for CD10 and procollagen 1. Basal cell carcinoma presents as a pearly papule or nodule displaying basaloid-appearing aggregates with tumor stromal clefting and can stain with Ber-EP4. Cylindromas typically present on the scalp as large rubbery-appearing plaques and nodules. Cylindromas usually present as a solitary tumor, but in the familial form there can be clusters of multiple nodules. Metastatic renal cell carcinoma frequently appears as a bleeding nodule on the scalp in patients with known renal cell cancer or as the initial presentation.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(pt 1):263-268.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Wang E, Lee JS, Kazakov DV. A rare combination of sebaceoma with carcinomatous change (sebaceous carcinoma), trichoblastoma, and poroma arising from a nevus sebaceus. J Cutan Pathol. 2013;40:676-682.
- Bae MI, Cho TH, Shin MK, et al. An unusual clinical presentation of eccrine poroma occurring on the auricle. Indian J Dermatol. 2015;60:523.
- Hsu MC, Liau JY, Hong JL, et al. Secondary neoplasms arising from nevus sebaceus: a retrospective study of 450 cases in Taiwan. J Dermatol. 2016;43:175-180.
- Takhan II, Domingo J. Metastasizing eccrine porocarcinoma developing in a sebaceous nevus of Jadassohn. report of a case. Arch Dermatol. 1985;121:413-415.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(pt 1):263-268.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Wang E, Lee JS, Kazakov DV. A rare combination of sebaceoma with carcinomatous change (sebaceous carcinoma), trichoblastoma, and poroma arising from a nevus sebaceus. J Cutan Pathol. 2013;40:676-682.
- Bae MI, Cho TH, Shin MK, et al. An unusual clinical presentation of eccrine poroma occurring on the auricle. Indian J Dermatol. 2015;60:523.
- Hsu MC, Liau JY, Hong JL, et al. Secondary neoplasms arising from nevus sebaceus: a retrospective study of 450 cases in Taiwan. J Dermatol. 2016;43:175-180.
- Takhan II, Domingo J. Metastasizing eccrine porocarcinoma developing in a sebaceous nevus of Jadassohn. report of a case. Arch Dermatol. 1985;121:413-415.
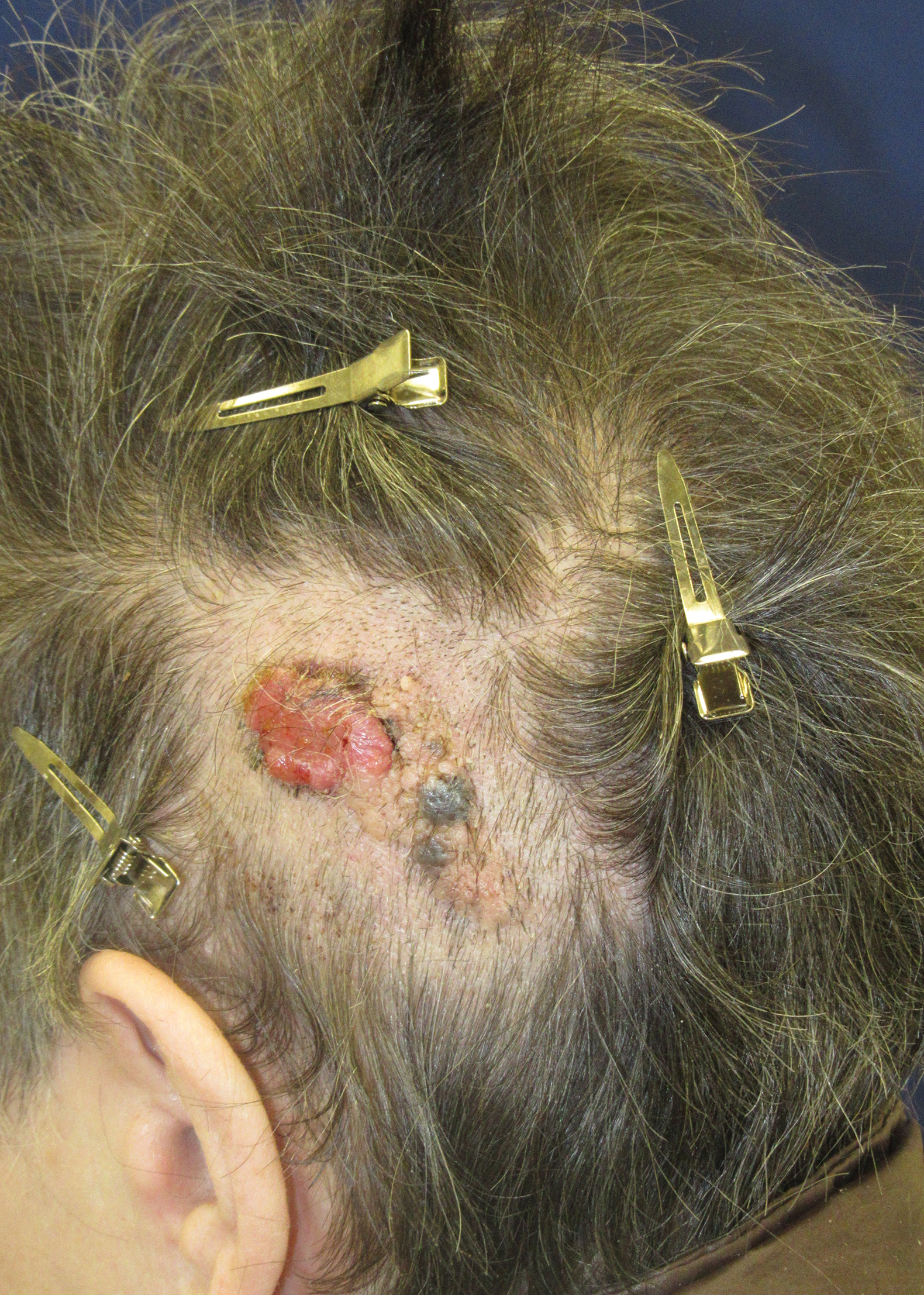
A 75-year-old woman presented with an enlarging plaque on the scalp of 5 years' duration. Physical examination revealed a 5.6.2 ×2.9-cm, tan-colored, verrucous plaque with an overlying pink friable nodule on the left occipital scalp. The lesion was not painful or pruritic, and the patient did not have any constitutional symptoms such as fever, night sweats, or weight loss. The patient denied prior tanning bed use and reported intermittent sun exposure over her lifetime. She denied any prior surgical intervention. There was no family history of similar lesions.
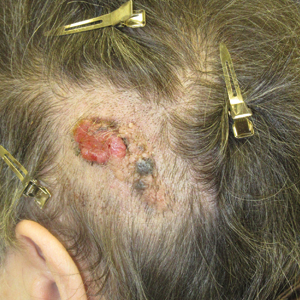
Scleromyxedema in a Patient With Thyroid Disease: An Atypical Case or a Case for Revised Criteria?
Scleromyxedema (SM) is a generalized papular and sclerodermoid form of lichen myxedematosus (LM), commonly referred to as papular mucinosis. It is a rare progressive disease of unknown etiology with systemic manifestations that cause serious morbidity and mortality. Diagnostic criteria were initially created by Montgomery and Underwood1 in 1953 and revised by Rongioletti and Rebora2 in 2001 as follows: (1) generalized papular and sclerodermoid eruption; (2) histologic triad of mucin deposition, fibroblast proliferation, and fibrosis; (3) monoclonal gammopathy; and (4) absence of thyroid disease. There are several reports of LM in association with hypothyroidism, most of which can be characterized as atypical.3-8 We present a case of SM in a patient with Hashimoto thyroiditis and propose that the presence of thyroid disease should not preclude the diagnosis of SM.
Case Report
A 44-year-old woman presented with a progressive eruption of thickened skin and papules spanning many months. The papules ranged from flesh colored to erythematous and covered more than 80% of the body surface area, most notably involving the face, neck, ears, arms, chest, abdomen, and thighs (Figures 1A and 2A). Review of systems was notable for pruritus, muscle pain but no weakness, dysphagia, and constipation. Her medical history included childhood atopic dermatitis and Hashimoto thyroiditis. Hypothyroidism was diagnosed with support of a thyroid ultrasound and thyroid peroxidase antibodies. It was treated with oral levothyroxine for 2 years prior to the skin eruption. Thyroid biopsy was not performed. Her thyroid-stimulating hormone levels notably fluctuated in the year prior to presentation despite close clinical and laboratory monitoring by an endocrinologist. Laboratory results are summarized in Table 1. Both skin and muscle9 biopsies were consistent with SM (Figure 3) and are summarized in Table 1.
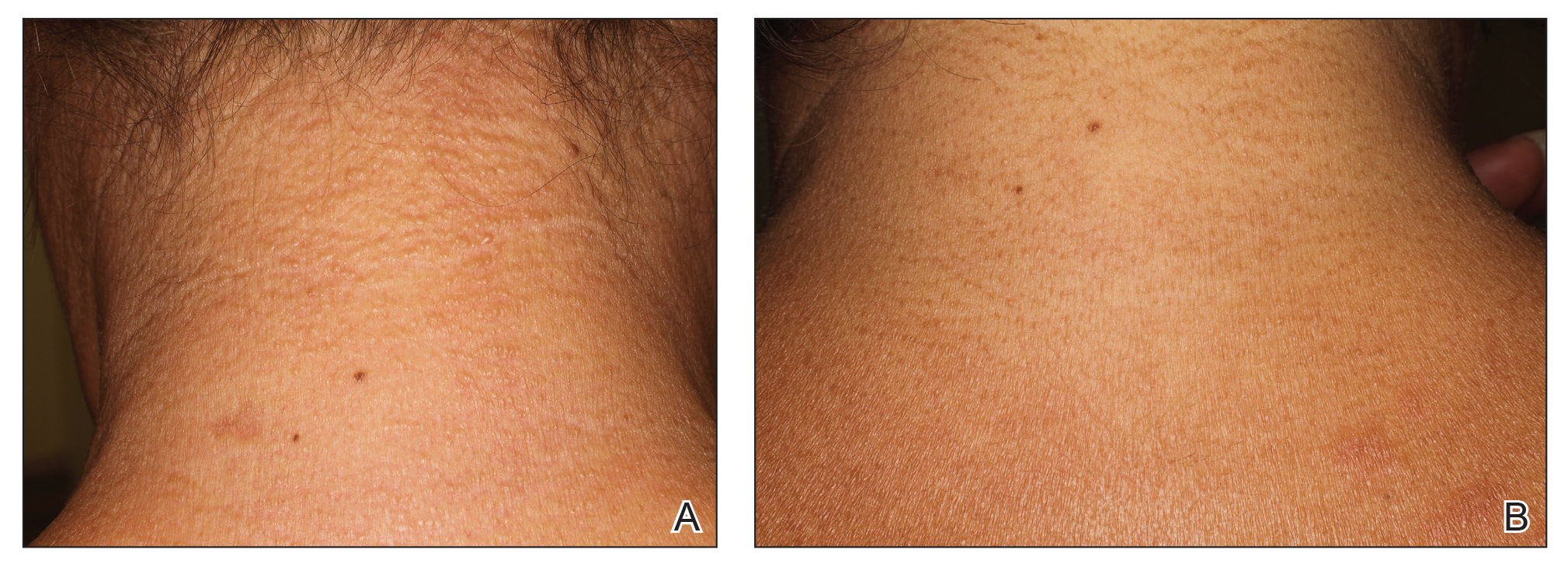

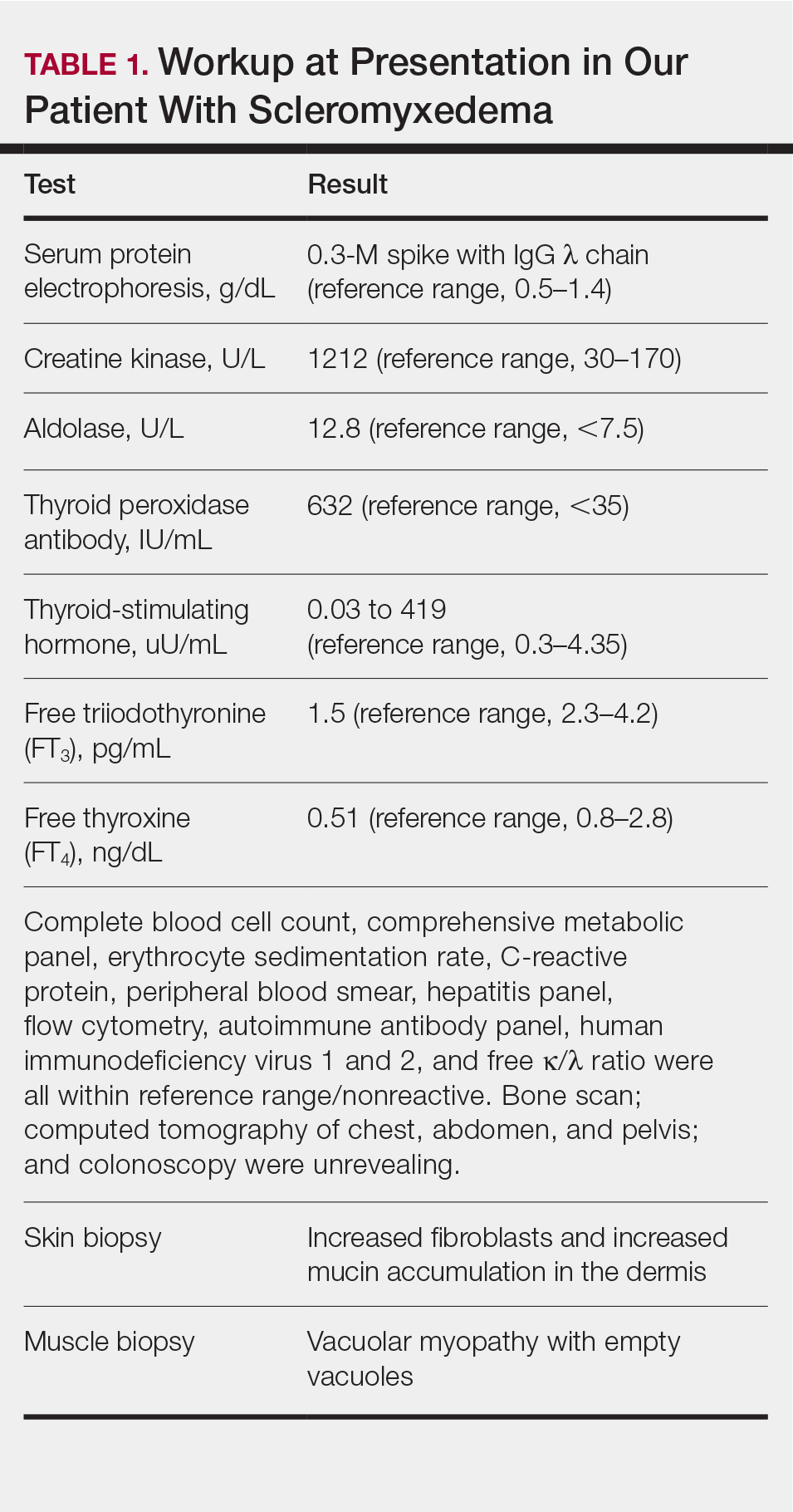
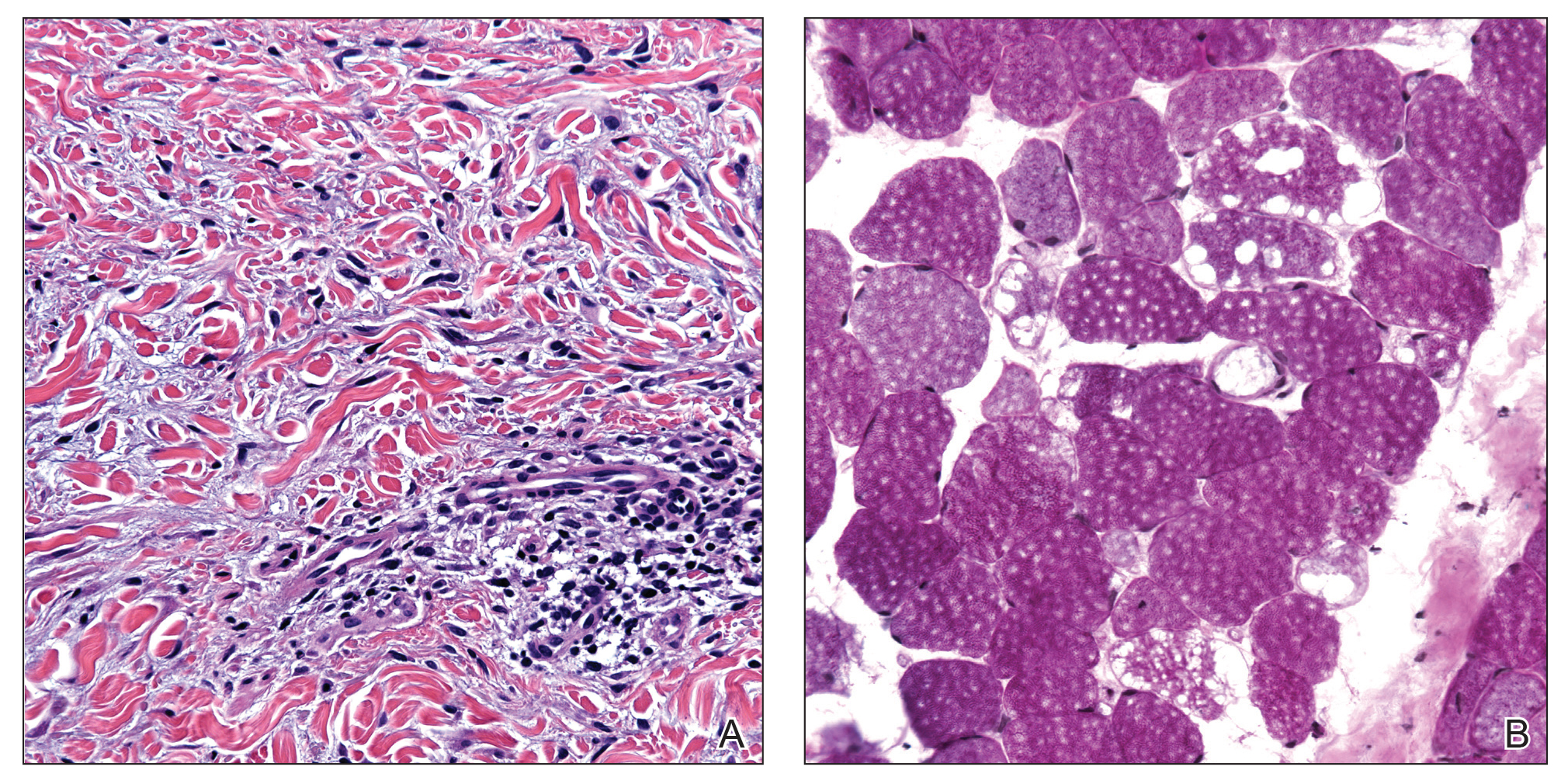
Shortly after presentation to our clinic the patient developed acute concerns of confusion and muscle weakness. She was admitted for further inpatient management due to concern for dermato-neuro syndrome, a rare but potentially fatal decline in neurological status that can progress to coma and death, rather than myxedema coma. On admission, a thyroid function test showed subclinical hypothyroidism with a thyroid-stimulating hormone level of 6.35 uU/mL (reference range, 0.3–4.35 uU/mL) and free thyroxine (FT4) level of 1.5 ng/dL (reference range, 0.8–2.8 ng/dL). While hospitalized she was started on intravenous levothyroxine, systemic steroids, and a course of intravenous immunoglobulin (IVIg) treatment consisting of 2 g/kg divided over 5 days. On this regimen, her mental status quickly returned to baseline and other symptoms improved, including the skin eruption (Figures 1B and 2B). She has been maintained on lenalidomide 25 mg/d for the first 3 weeks of each month as well as monthly IVIg infusions. Her thyroid levels have persistently fluctuated despite intramuscular levothyroxine dosing, but her skin has remained clear with continued SM-directed therapy.
Comment
Classification
Lichen myxedematosus is differentiated into localized and generalized forms. The former is limited to the skin and lacks monoclonal gammopathy. The latter, also known as SM, is associated with monoclonal gammopathy and systemic symptoms. Atypical LM is an umbrella term for intermediate cases.
Clinical Presentation
Skin manifestations of SM are described as 1- to 3-mm, firm, waxy, dome-shaped papules that commonly affect the hands, forearms, face, neck, trunk, and thighs. The surrounding skin may be reddish brown and edematous with evidence of skin thickening. Extracutaneous manifestations in SM are numerous and unpredictable. Any organ system can be involved, but gastrointestinal, rheumatologic, pulmonary, and cardiovascular complications are most common.10 A comprehensive multidisciplinary evaluation is necessary based on clinical symptoms and laboratory findings.
Management
Many treatments have been proposed for SM in case reports and case series. Prior treatments have had little success. Most recently, in one of the largest case series on SM, Rongioletti et al10 demonstrated IVIg to be a safe and effective treatment modality.
Differential Diagnosis
An important differential diagnosis is generalized myxedema, which is seen in long-standing hypothyroidism and may present with cutaneous mucinosis and systemic symptoms that resemble SM. Hypothyroid myxedema is associated with a widespread slowing of the body’s metabolic processes and deposition of mucin in various organs, including the skin, creating a generalized nonpitting edema. Classic clinical signs include macroglossia, periorbital puffiness, thick lips, and acral swelling. The skin tends to be cold, dry, and pale. Hair is characterized as being coarse, dry, and brittle with diffuse partial alopecia. Histologically, there is hyperkeratosis with follicular plugging and diffuse mucin and edema splaying between collagen fibers spanning the entire dermis.11 In contradistinction with SM, there is no fibroblast proliferation. The treatment is thyroid replacement therapy. Hyperthyroidism has distinct clinical and histologic changes. Clinically, there is moist and smooth skin with soft, fine, and sometimes alopecic hair. Graves disease, the most common cause of hyperthyroidism, is further characterized by Graves ophthalmopathy and pretibial myxedema, or pink to brown, raised, firm, indurated, asymmetric plaques most commonly affecting the shins. Histologically there is increased mucin in the lower to mid dermis without fibroblast proliferation. The epidermis can be hyperkeratotic, which will clinically correlate with verrucous lesions.12
Hypothyroid encephalopathy is a rare disorder that can cause a change in mental status. It is a steroid-responsive autoimmune process characterized by encephalopathy that is associated with cognitive impairment and psychiatric features. It is a diagnosis of exclusion and should be suspected in women with a history of autoimmune disease, especially antithyroid peroxidase antibodies, a negative infectious workup, and encephalitis with behavioral changes. Although typically highly responsive to systemic steroids, IVIg also has shown efficacy.13
Presence of Thyroid Disease
According to a PubMed search of articles indexed for MEDLINE using the terms scleromyxedema and lichen myxedematosus, there are 7 cases in the literature that potentially describe LM associated with hypothyroidism (Table 2).3-8 The majority of these cases lack monoclonal gammopathy; improved with thyroid replacement therapy; or had severely atypical clinical presentations, rendering them cases of atypical LM or atypical thyroid dermopathy.3-6 Macnab and Kenny7 presented a case of subclinical hypothyroidism with a generalized papular eruption, monoclonal gammopathy, and consistent histologic changes that responded to IVIg therapy. These findings are suggestive of SM, but limited to the current diagnostic criteria, the patient was diagnosed with atypical LM.7 Shenoy et al8 described 2 cases of LM with hypothyroidism. One patient had biopsy-proven SM that was responsive to IVIg as well as Hashimoto thyroiditis with delayed onset of monoclonal gammopathy. The second patient had a medical history of hypothyroidism and Hodgkin lymphoma with active rheumatoid arthritis and biopsy-proven LM that was responsive to systemic steroids.8
Current literature states that thyroid disorder precludes the diagnosis of SM. However, historic literature would suggest otherwise. Because of inconsistent reports and theories regarding the pathogenesis of various sclerodermoid and mucin deposition diseases, in 1953 Montgomery and Underwood1 sought to differentiate LM from scleroderma and generalized myxedema. They stressed clinical appearance and proposed diagnostic criteria for LM as generalized papular mucinosis in which “[n]o relation to disturbance of the thyroid or other endocrine glands is apparent,” whereas generalized myxedema was defined as a “[t]rue cutaneous myxedema, with diffuse edema and the usual commonly recognized changes” in patients with endocrine abnormalities.1 With this classification, the authors made a clear distinction between mucinosis caused by thyroid abnormalities and LM, which is not caused by a thyroid disorder. Since this original description was published, associations with monoclonal gammopathy and fibroblast proliferation have been made, ultimately culminating into the current 2001 criteria that incorporate the absence of thyroid disease.2

Conclusion
We believe our case is consistent with the classification initially proposed by Montgomery and Underwood1 and is strengthened with the more recent associations with monoclonal gammopathy and specific histopathologic findings. Although there is no definitive way to rule out myxedema coma or Hashimoto encephalopathy to describe our patient’s transient neurologic decline, her clinical symptoms, laboratory findings, and biopsy results all supported the diagnosis of SM. Furthermore, her response to SM-directed therapy, despite fluctuating thyroid function test results, also supported the diagnosis. In the setting of cutaneous mucinosis with conflicting findings for hypothyroid myxedema, LM should be ruled out. Given the features presented in this report and others, diagnostic criteria should allow for SM and thyroid dysfunction to be concurrent diagnoses. Most importantly, we believe it is essential to identify and diagnose SM in a timely manner to facilitate SM-directed therapy, namely IVIg, to potentially minimize the disease’s notable morbidity and mortality.
- Montgomery H, Underwood LJ. Lichen myxedematosus; differentiation from cutaneous myxedemas or mucoid states. J Invest Dermatol. 1953;20:213-236.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Archibald GC, Calvert HT. Hypothyroidsm and lichen myxedematosus. Arch Dermatol. 1977;113:684.
- Schaeffer D, Bruce S, Rosen T. Cutaneous mucinosis associated with thyroid dysfunction. Cutis. 1983;11:449-456.
- Martin-Ezquerra G, Sanchez-Regaña M, Massana-Gil J, et al. Papular mucinosis associated with subclinical hypothyroidism: improvement with thyroxine therapy. J Eur Acad Dermatol Venereol. 2006;20:1340-1341.
- Volpato MB, Jaime TJ, Proença MP, et al. Papular mucinosis associated with hypothyroidism. An Bras Dermatol. 2010;85:89-92.
- Macnab M, Kenny P. Successful intravenous immunoglobulin treatment of atypical lichen myxedematosus associated with hypothyroidism and central nervous system. involvement: case report and discussion of the literature. J Cutan Med Surg. 2013;17:69-73.
- Shenoy A, Steixner J, Beltrani V, et al. Discrete papular lichen myxedematosus and scleromyxedema with hypothyroidism: a report of two cases. Case Rep Dermatol. 2019;11:64-70.
- Helfrich DJ, Walker ER, Martinez AJ, et al. Scleromyxedema myopathy: case report and review of the literature. Arthritis Rheum. 1988;31:1437-1441.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Jackson EM, English JC 3rd. Diffuse cutaneous mucinoses. Dermatol Clin. 2002;20:493-501.
- Leonhardt JM, Heymann WR. Thyroid disease and the skin. Dermatol Clin. 2002;20:473-481.
- Zhou JY, Xu B, Lopes J, et al. Hashimoto encephalopathy: literature review. Acta Neurol Scand. 2017;135:285-290.
Scleromyxedema (SM) is a generalized papular and sclerodermoid form of lichen myxedematosus (LM), commonly referred to as papular mucinosis. It is a rare progressive disease of unknown etiology with systemic manifestations that cause serious morbidity and mortality. Diagnostic criteria were initially created by Montgomery and Underwood1 in 1953 and revised by Rongioletti and Rebora2 in 2001 as follows: (1) generalized papular and sclerodermoid eruption; (2) histologic triad of mucin deposition, fibroblast proliferation, and fibrosis; (3) monoclonal gammopathy; and (4) absence of thyroid disease. There are several reports of LM in association with hypothyroidism, most of which can be characterized as atypical.3-8 We present a case of SM in a patient with Hashimoto thyroiditis and propose that the presence of thyroid disease should not preclude the diagnosis of SM.
Case Report
A 44-year-old woman presented with a progressive eruption of thickened skin and papules spanning many months. The papules ranged from flesh colored to erythematous and covered more than 80% of the body surface area, most notably involving the face, neck, ears, arms, chest, abdomen, and thighs (Figures 1A and 2A). Review of systems was notable for pruritus, muscle pain but no weakness, dysphagia, and constipation. Her medical history included childhood atopic dermatitis and Hashimoto thyroiditis. Hypothyroidism was diagnosed with support of a thyroid ultrasound and thyroid peroxidase antibodies. It was treated with oral levothyroxine for 2 years prior to the skin eruption. Thyroid biopsy was not performed. Her thyroid-stimulating hormone levels notably fluctuated in the year prior to presentation despite close clinical and laboratory monitoring by an endocrinologist. Laboratory results are summarized in Table 1. Both skin and muscle9 biopsies were consistent with SM (Figure 3) and are summarized in Table 1.
Shortly after presentation to our clinic the patient developed acute concerns of confusion and muscle weakness. She was admitted for further inpatient management due to concern for dermato-neuro syndrome, a rare but potentially fatal decline in neurological status that can progress to coma and death, rather than myxedema coma. On admission, a thyroid function test showed subclinical hypothyroidism with a thyroid-stimulating hormone level of 6.35 uU/mL (reference range, 0.3–4.35 uU/mL) and free thyroxine (FT4) level of 1.5 ng/dL (reference range, 0.8–2.8 ng/dL). While hospitalized she was started on intravenous levothyroxine, systemic steroids, and a course of intravenous immunoglobulin (IVIg) treatment consisting of 2 g/kg divided over 5 days. On this regimen, her mental status quickly returned to baseline and other symptoms improved, including the skin eruption (Figures 1B and 2B). She has been maintained on lenalidomide 25 mg/d for the first 3 weeks of each month as well as monthly IVIg infusions. Her thyroid levels have persistently fluctuated despite intramuscular levothyroxine dosing, but her skin has remained clear with continued SM-directed therapy.
Comment
Classification
Lichen myxedematosus is differentiated into localized and generalized forms. The former is limited to the skin and lacks monoclonal gammopathy. The latter, also known as SM, is associated with monoclonal gammopathy and systemic symptoms. Atypical LM is an umbrella term for intermediate cases.
Clinical Presentation
Skin manifestations of SM are described as 1- to 3-mm, firm, waxy, dome-shaped papules that commonly affect the hands, forearms, face, neck, trunk, and thighs. The surrounding skin may be reddish brown and edematous with evidence of skin thickening. Extracutaneous manifestations in SM are numerous and unpredictable. Any organ system can be involved, but gastrointestinal, rheumatologic, pulmonary, and cardiovascular complications are most common.10 A comprehensive multidisciplinary evaluation is necessary based on clinical symptoms and laboratory findings.
Management
Many treatments have been proposed for SM in case reports and case series. Prior treatments have had little success. Most recently, in one of the largest case series on SM, Rongioletti et al10 demonstrated IVIg to be a safe and effective treatment modality.
Differential Diagnosis
An important differential diagnosis is generalized myxedema, which is seen in long-standing hypothyroidism and may present with cutaneous mucinosis and systemic symptoms that resemble SM. Hypothyroid myxedema is associated with a widespread slowing of the body’s metabolic processes and deposition of mucin in various organs, including the skin, creating a generalized nonpitting edema. Classic clinical signs include macroglossia, periorbital puffiness, thick lips, and acral swelling. The skin tends to be cold, dry, and pale. Hair is characterized as being coarse, dry, and brittle with diffuse partial alopecia. Histologically, there is hyperkeratosis with follicular plugging and diffuse mucin and edema splaying between collagen fibers spanning the entire dermis.11 In contradistinction with SM, there is no fibroblast proliferation. The treatment is thyroid replacement therapy. Hyperthyroidism has distinct clinical and histologic changes. Clinically, there is moist and smooth skin with soft, fine, and sometimes alopecic hair. Graves disease, the most common cause of hyperthyroidism, is further characterized by Graves ophthalmopathy and pretibial myxedema, or pink to brown, raised, firm, indurated, asymmetric plaques most commonly affecting the shins. Histologically there is increased mucin in the lower to mid dermis without fibroblast proliferation. The epidermis can be hyperkeratotic, which will clinically correlate with verrucous lesions.12
Hypothyroid encephalopathy is a rare disorder that can cause a change in mental status. It is a steroid-responsive autoimmune process characterized by encephalopathy that is associated with cognitive impairment and psychiatric features. It is a diagnosis of exclusion and should be suspected in women with a history of autoimmune disease, especially antithyroid peroxidase antibodies, a negative infectious workup, and encephalitis with behavioral changes. Although typically highly responsive to systemic steroids, IVIg also has shown efficacy.13
Presence of Thyroid Disease
According to a PubMed search of articles indexed for MEDLINE using the terms scleromyxedema and lichen myxedematosus, there are 7 cases in the literature that potentially describe LM associated with hypothyroidism (Table 2).3-8 The majority of these cases lack monoclonal gammopathy; improved with thyroid replacement therapy; or had severely atypical clinical presentations, rendering them cases of atypical LM or atypical thyroid dermopathy.3-6 Macnab and Kenny7 presented a case of subclinical hypothyroidism with a generalized papular eruption, monoclonal gammopathy, and consistent histologic changes that responded to IVIg therapy. These findings are suggestive of SM, but limited to the current diagnostic criteria, the patient was diagnosed with atypical LM.7 Shenoy et al8 described 2 cases of LM with hypothyroidism. One patient had biopsy-proven SM that was responsive to IVIg as well as Hashimoto thyroiditis with delayed onset of monoclonal gammopathy. The second patient had a medical history of hypothyroidism and Hodgkin lymphoma with active rheumatoid arthritis and biopsy-proven LM that was responsive to systemic steroids.8
Current literature states that thyroid disorder precludes the diagnosis of SM. However, historic literature would suggest otherwise. Because of inconsistent reports and theories regarding the pathogenesis of various sclerodermoid and mucin deposition diseases, in 1953 Montgomery and Underwood1 sought to differentiate LM from scleroderma and generalized myxedema. They stressed clinical appearance and proposed diagnostic criteria for LM as generalized papular mucinosis in which “[n]o relation to disturbance of the thyroid or other endocrine glands is apparent,” whereas generalized myxedema was defined as a “[t]rue cutaneous myxedema, with diffuse edema and the usual commonly recognized changes” in patients with endocrine abnormalities.1 With this classification, the authors made a clear distinction between mucinosis caused by thyroid abnormalities and LM, which is not caused by a thyroid disorder. Since this original description was published, associations with monoclonal gammopathy and fibroblast proliferation have been made, ultimately culminating into the current 2001 criteria that incorporate the absence of thyroid disease.2
Conclusion
We believe our case is consistent with the classification initially proposed by Montgomery and Underwood1 and is strengthened with the more recent associations with monoclonal gammopathy and specific histopathologic findings. Although there is no definitive way to rule out myxedema coma or Hashimoto encephalopathy to describe our patient’s transient neurologic decline, her clinical symptoms, laboratory findings, and biopsy results all supported the diagnosis of SM. Furthermore, her response to SM-directed therapy, despite fluctuating thyroid function test results, also supported the diagnosis. In the setting of cutaneous mucinosis with conflicting findings for hypothyroid myxedema, LM should be ruled out. Given the features presented in this report and others, diagnostic criteria should allow for SM and thyroid dysfunction to be concurrent diagnoses. Most importantly, we believe it is essential to identify and diagnose SM in a timely manner to facilitate SM-directed therapy, namely IVIg, to potentially minimize the disease’s notable morbidity and mortality.
Scleromyxedema (SM) is a generalized papular and sclerodermoid form of lichen myxedematosus (LM), commonly referred to as papular mucinosis. It is a rare progressive disease of unknown etiology with systemic manifestations that cause serious morbidity and mortality. Diagnostic criteria were initially created by Montgomery and Underwood1 in 1953 and revised by Rongioletti and Rebora2 in 2001 as follows: (1) generalized papular and sclerodermoid eruption; (2) histologic triad of mucin deposition, fibroblast proliferation, and fibrosis; (3) monoclonal gammopathy; and (4) absence of thyroid disease. There are several reports of LM in association with hypothyroidism, most of which can be characterized as atypical.3-8 We present a case of SM in a patient with Hashimoto thyroiditis and propose that the presence of thyroid disease should not preclude the diagnosis of SM.
Case Report
A 44-year-old woman presented with a progressive eruption of thickened skin and papules spanning many months. The papules ranged from flesh colored to erythematous and covered more than 80% of the body surface area, most notably involving the face, neck, ears, arms, chest, abdomen, and thighs (Figures 1A and 2A). Review of systems was notable for pruritus, muscle pain but no weakness, dysphagia, and constipation. Her medical history included childhood atopic dermatitis and Hashimoto thyroiditis. Hypothyroidism was diagnosed with support of a thyroid ultrasound and thyroid peroxidase antibodies. It was treated with oral levothyroxine for 2 years prior to the skin eruption. Thyroid biopsy was not performed. Her thyroid-stimulating hormone levels notably fluctuated in the year prior to presentation despite close clinical and laboratory monitoring by an endocrinologist. Laboratory results are summarized in Table 1. Both skin and muscle9 biopsies were consistent with SM (Figure 3) and are summarized in Table 1.
Shortly after presentation to our clinic the patient developed acute concerns of confusion and muscle weakness. She was admitted for further inpatient management due to concern for dermato-neuro syndrome, a rare but potentially fatal decline in neurological status that can progress to coma and death, rather than myxedema coma. On admission, a thyroid function test showed subclinical hypothyroidism with a thyroid-stimulating hormone level of 6.35 uU/mL (reference range, 0.3–4.35 uU/mL) and free thyroxine (FT4) level of 1.5 ng/dL (reference range, 0.8–2.8 ng/dL). While hospitalized she was started on intravenous levothyroxine, systemic steroids, and a course of intravenous immunoglobulin (IVIg) treatment consisting of 2 g/kg divided over 5 days. On this regimen, her mental status quickly returned to baseline and other symptoms improved, including the skin eruption (Figures 1B and 2B). She has been maintained on lenalidomide 25 mg/d for the first 3 weeks of each month as well as monthly IVIg infusions. Her thyroid levels have persistently fluctuated despite intramuscular levothyroxine dosing, but her skin has remained clear with continued SM-directed therapy.
Comment
Classification
Lichen myxedematosus is differentiated into localized and generalized forms. The former is limited to the skin and lacks monoclonal gammopathy. The latter, also known as SM, is associated with monoclonal gammopathy and systemic symptoms. Atypical LM is an umbrella term for intermediate cases.
Clinical Presentation
Skin manifestations of SM are described as 1- to 3-mm, firm, waxy, dome-shaped papules that commonly affect the hands, forearms, face, neck, trunk, and thighs. The surrounding skin may be reddish brown and edematous with evidence of skin thickening. Extracutaneous manifestations in SM are numerous and unpredictable. Any organ system can be involved, but gastrointestinal, rheumatologic, pulmonary, and cardiovascular complications are most common.10 A comprehensive multidisciplinary evaluation is necessary based on clinical symptoms and laboratory findings.
Management
Many treatments have been proposed for SM in case reports and case series. Prior treatments have had little success. Most recently, in one of the largest case series on SM, Rongioletti et al10 demonstrated IVIg to be a safe and effective treatment modality.
Differential Diagnosis
An important differential diagnosis is generalized myxedema, which is seen in long-standing hypothyroidism and may present with cutaneous mucinosis and systemic symptoms that resemble SM. Hypothyroid myxedema is associated with a widespread slowing of the body’s metabolic processes and deposition of mucin in various organs, including the skin, creating a generalized nonpitting edema. Classic clinical signs include macroglossia, periorbital puffiness, thick lips, and acral swelling. The skin tends to be cold, dry, and pale. Hair is characterized as being coarse, dry, and brittle with diffuse partial alopecia. Histologically, there is hyperkeratosis with follicular plugging and diffuse mucin and edema splaying between collagen fibers spanning the entire dermis.11 In contradistinction with SM, there is no fibroblast proliferation. The treatment is thyroid replacement therapy. Hyperthyroidism has distinct clinical and histologic changes. Clinically, there is moist and smooth skin with soft, fine, and sometimes alopecic hair. Graves disease, the most common cause of hyperthyroidism, is further characterized by Graves ophthalmopathy and pretibial myxedema, or pink to brown, raised, firm, indurated, asymmetric plaques most commonly affecting the shins. Histologically there is increased mucin in the lower to mid dermis without fibroblast proliferation. The epidermis can be hyperkeratotic, which will clinically correlate with verrucous lesions.12
Hypothyroid encephalopathy is a rare disorder that can cause a change in mental status. It is a steroid-responsive autoimmune process characterized by encephalopathy that is associated with cognitive impairment and psychiatric features. It is a diagnosis of exclusion and should be suspected in women with a history of autoimmune disease, especially antithyroid peroxidase antibodies, a negative infectious workup, and encephalitis with behavioral changes. Although typically highly responsive to systemic steroids, IVIg also has shown efficacy.13
Presence of Thyroid Disease
According to a PubMed search of articles indexed for MEDLINE using the terms scleromyxedema and lichen myxedematosus, there are 7 cases in the literature that potentially describe LM associated with hypothyroidism (Table 2).3-8 The majority of these cases lack monoclonal gammopathy; improved with thyroid replacement therapy; or had severely atypical clinical presentations, rendering them cases of atypical LM or atypical thyroid dermopathy.3-6 Macnab and Kenny7 presented a case of subclinical hypothyroidism with a generalized papular eruption, monoclonal gammopathy, and consistent histologic changes that responded to IVIg therapy. These findings are suggestive of SM, but limited to the current diagnostic criteria, the patient was diagnosed with atypical LM.7 Shenoy et al8 described 2 cases of LM with hypothyroidism. One patient had biopsy-proven SM that was responsive to IVIg as well as Hashimoto thyroiditis with delayed onset of monoclonal gammopathy. The second patient had a medical history of hypothyroidism and Hodgkin lymphoma with active rheumatoid arthritis and biopsy-proven LM that was responsive to systemic steroids.8
Current literature states that thyroid disorder precludes the diagnosis of SM. However, historic literature would suggest otherwise. Because of inconsistent reports and theories regarding the pathogenesis of various sclerodermoid and mucin deposition diseases, in 1953 Montgomery and Underwood1 sought to differentiate LM from scleroderma and generalized myxedema. They stressed clinical appearance and proposed diagnostic criteria for LM as generalized papular mucinosis in which “[n]o relation to disturbance of the thyroid or other endocrine glands is apparent,” whereas generalized myxedema was defined as a “[t]rue cutaneous myxedema, with diffuse edema and the usual commonly recognized changes” in patients with endocrine abnormalities.1 With this classification, the authors made a clear distinction between mucinosis caused by thyroid abnormalities and LM, which is not caused by a thyroid disorder. Since this original description was published, associations with monoclonal gammopathy and fibroblast proliferation have been made, ultimately culminating into the current 2001 criteria that incorporate the absence of thyroid disease.2
Conclusion
We believe our case is consistent with the classification initially proposed by Montgomery and Underwood1 and is strengthened with the more recent associations with monoclonal gammopathy and specific histopathologic findings. Although there is no definitive way to rule out myxedema coma or Hashimoto encephalopathy to describe our patient’s transient neurologic decline, her clinical symptoms, laboratory findings, and biopsy results all supported the diagnosis of SM. Furthermore, her response to SM-directed therapy, despite fluctuating thyroid function test results, also supported the diagnosis. In the setting of cutaneous mucinosis with conflicting findings for hypothyroid myxedema, LM should be ruled out. Given the features presented in this report and others, diagnostic criteria should allow for SM and thyroid dysfunction to be concurrent diagnoses. Most importantly, we believe it is essential to identify and diagnose SM in a timely manner to facilitate SM-directed therapy, namely IVIg, to potentially minimize the disease’s notable morbidity and mortality.
- Montgomery H, Underwood LJ. Lichen myxedematosus; differentiation from cutaneous myxedemas or mucoid states. J Invest Dermatol. 1953;20:213-236.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Archibald GC, Calvert HT. Hypothyroidsm and lichen myxedematosus. Arch Dermatol. 1977;113:684.
- Schaeffer D, Bruce S, Rosen T. Cutaneous mucinosis associated with thyroid dysfunction. Cutis. 1983;11:449-456.
- Martin-Ezquerra G, Sanchez-Regaña M, Massana-Gil J, et al. Papular mucinosis associated with subclinical hypothyroidism: improvement with thyroxine therapy. J Eur Acad Dermatol Venereol. 2006;20:1340-1341.
- Volpato MB, Jaime TJ, Proença MP, et al. Papular mucinosis associated with hypothyroidism. An Bras Dermatol. 2010;85:89-92.
- Macnab M, Kenny P. Successful intravenous immunoglobulin treatment of atypical lichen myxedematosus associated with hypothyroidism and central nervous system. involvement: case report and discussion of the literature. J Cutan Med Surg. 2013;17:69-73.
- Shenoy A, Steixner J, Beltrani V, et al. Discrete papular lichen myxedematosus and scleromyxedema with hypothyroidism: a report of two cases. Case Rep Dermatol. 2019;11:64-70.
- Helfrich DJ, Walker ER, Martinez AJ, et al. Scleromyxedema myopathy: case report and review of the literature. Arthritis Rheum. 1988;31:1437-1441.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Jackson EM, English JC 3rd. Diffuse cutaneous mucinoses. Dermatol Clin. 2002;20:493-501.
- Leonhardt JM, Heymann WR. Thyroid disease and the skin. Dermatol Clin. 2002;20:473-481.
- Zhou JY, Xu B, Lopes J, et al. Hashimoto encephalopathy: literature review. Acta Neurol Scand. 2017;135:285-290.
- Montgomery H, Underwood LJ. Lichen myxedematosus; differentiation from cutaneous myxedemas or mucoid states. J Invest Dermatol. 1953;20:213-236.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Archibald GC, Calvert HT. Hypothyroidsm and lichen myxedematosus. Arch Dermatol. 1977;113:684.
- Schaeffer D, Bruce S, Rosen T. Cutaneous mucinosis associated with thyroid dysfunction. Cutis. 1983;11:449-456.
- Martin-Ezquerra G, Sanchez-Regaña M, Massana-Gil J, et al. Papular mucinosis associated with subclinical hypothyroidism: improvement with thyroxine therapy. J Eur Acad Dermatol Venereol. 2006;20:1340-1341.
- Volpato MB, Jaime TJ, Proença MP, et al. Papular mucinosis associated with hypothyroidism. An Bras Dermatol. 2010;85:89-92.
- Macnab M, Kenny P. Successful intravenous immunoglobulin treatment of atypical lichen myxedematosus associated with hypothyroidism and central nervous system. involvement: case report and discussion of the literature. J Cutan Med Surg. 2013;17:69-73.
- Shenoy A, Steixner J, Beltrani V, et al. Discrete papular lichen myxedematosus and scleromyxedema with hypothyroidism: a report of two cases. Case Rep Dermatol. 2019;11:64-70.
- Helfrich DJ, Walker ER, Martinez AJ, et al. Scleromyxedema myopathy: case report and review of the literature. Arthritis Rheum. 1988;31:1437-1441.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Jackson EM, English JC 3rd. Diffuse cutaneous mucinoses. Dermatol Clin. 2002;20:493-501.
- Leonhardt JM, Heymann WR. Thyroid disease and the skin. Dermatol Clin. 2002;20:473-481.
- Zhou JY, Xu B, Lopes J, et al. Hashimoto encephalopathy: literature review. Acta Neurol Scand. 2017;135:285-290.
Practice Points
- Scleromyxedema (SM) is progressive disease of unknown etiology with unpredictable behavior.
- Systemic manifestations associated with SM can cause serious morbidity and mortality.
- Intravenous immunoglobulin is the most effective treatment modality in SM.
- The presence of thyroid disease should not preclude the diagnosis of SM.
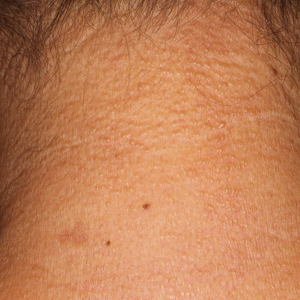
Cutaneous Pemphigus Vegetans Co-occurring With Oral Pemphigus Vulgaris
To the Editor:
A 74-year-old man with a history of colon cancer and no history of sexually transmitted diseases presented with tender, moist, vegetating, and verrucous plaques localized to the inguinal creases and behind the scrotum of 3 weeks’ duration (Figure 1). The patient recently had taken lisinopril prescribed by his primary care physician for a couple of years for hypertension before switching to losartan prior to the current presentation. He later noticed the groin eruptions. He also noticed white tongue plaques temporally associated with the groin plaques and a long history of recurrent oral ulcerations. Prior to being seen in our clinic, outside physicians cultured methicillin-sensitive Staphylococcus aureus from the groin plaques and treated him with oral clindamycin, cephalexin, and topical mupirocin without a clinical response.
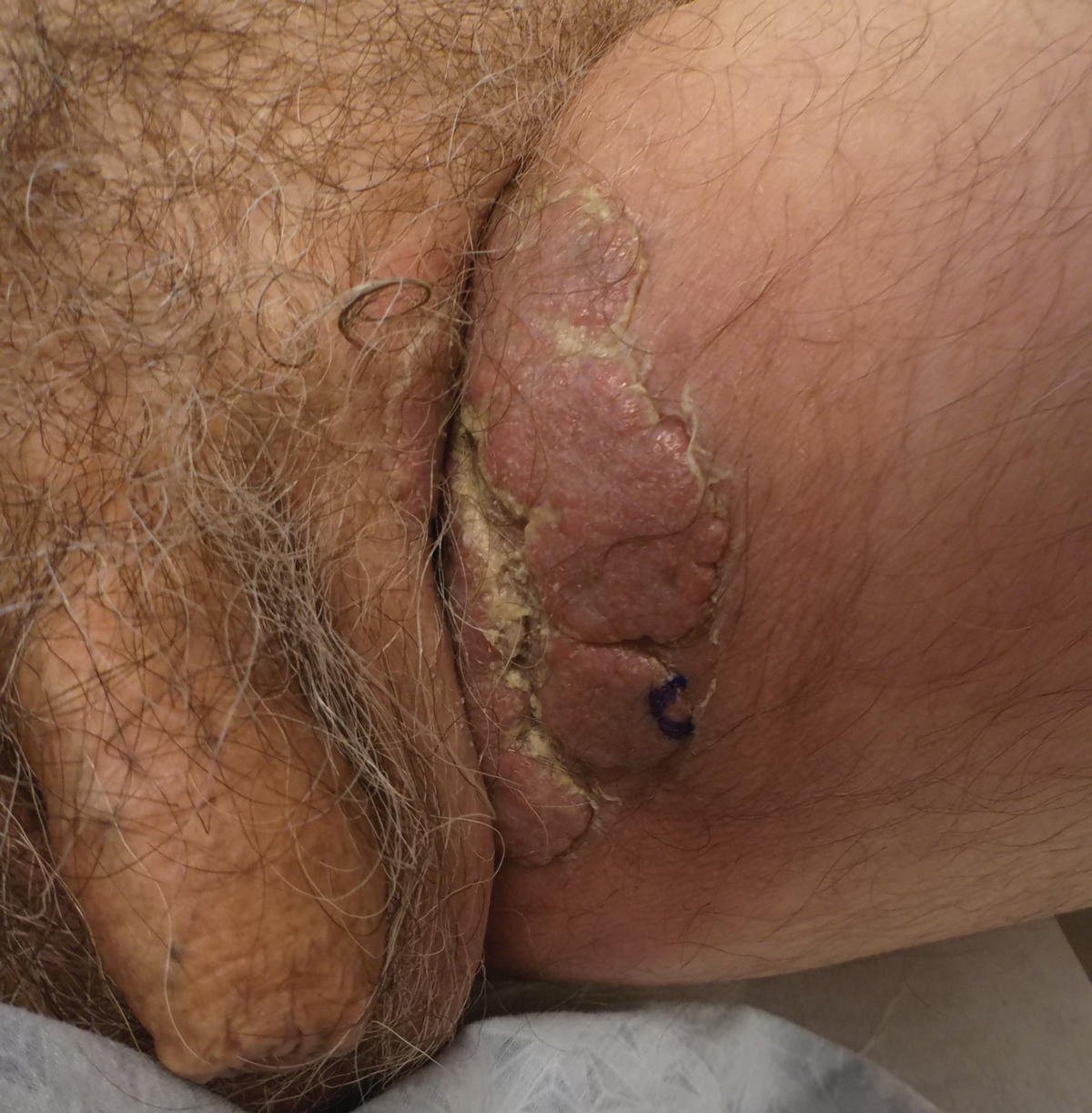
Our differential diagnosis included condyloma acuminata, condyloma lata, and cutaneous pemphigus vegetans. Laboratory testing revealed a nonreactive rapid plasma reagin test and peripheral eosinophilia of 14.9% (reference range, 0%–6%). Biopsy of a left groin plaque revealed epidermal hyperplasia with spongiosis and an eosinophilic-rich infiltrate on hematoxylin and eosin staining (Figure 2A), and direct immunofluorescence revealed diffuse epidermal intercellular IgG deposits (Figure 2B). The patient’s clinical and histologic presentation was consistent with cutaneous pemphigus vegetans. Biopsy of an oral ulcer revealed denuded acantholytic mucositis with eosinophilic-rich submucosal infiltrate and fibrosis (Figure 3A). Direct immunofluorescence was positive for lacelike intercellular staining for IgG and C3 within the squamous epithelium (Figure 3B). Together the clinical and histologic findings were consistent with oral pemphigus vulgaris.
The patient initially was started on oral minocycline 100 mg twice daily and mometasone furoate cream 0.1% twice daily to affected groin areas. With these interventions, the groin plaques almost completely resolved after several months, leaving only residual hyperpigmentation (Figure 4). The oral pemphigus vulgaris initially was treated with dexamethasone 0.5 mg/5 mL solution 2 to 3 times daily, but the lesions were refractory to this approach and also did not improve after the losartan was discontinued for several months. As such, mycophenolate mofetil was started. He was titrated to the lowest effective dose and showed near-complete resolution with 500 mg 3 times daily.
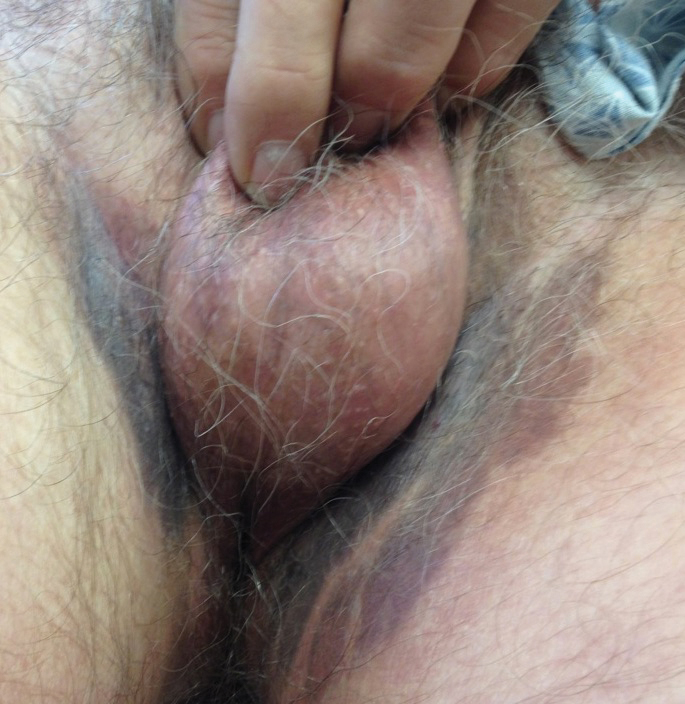
Cutaneous pemphigus vegetans, a rare variant of pemphigus vulgaris, is characterized by vegetating plaques commonly localized to the skin folds, scalp, face, and mucous membranes.1 Involvement of the oral mucosa occurs in a majority of cases. Although our patient had oral ulcerations, he did not have characteristic cerebriform changes of the dorsal tongue or associated verrucous hyperkeratotic lesions involving the buccal mucosa, hard and soft palate, or vermilion border of the lips that typically are seen in pemphigus vegetans.2-5 Subsequent biopsy of the oral mucosa confirmed oral pemphigus vulgaris in our patient.
This case presentation of co-occurring cutaneous pemphigus vegetans and oral pemphigus vulgaris is uncommonly reported in the literature. Although the etiology of this co-occurrence is not clear, it could represent a form of epitope spread, with the mechanism similar to that proposed for the progression of pemphigus vulgaris from the mucosal to the mucocutaneous stage by Chan et al,6 who suggested that an autoimmune reaction against specific desmoglein 3 epitopes (an important protein component for desmosomes and the autoantigen in pemphigus vulgaris) on mucosal membranes could induce local damage. These injuries could then expose the autoreactive immune cells to a secondary desmoglein 3 epitope present in the skin, leading to the development of cutaneous lesions.6 Salato et al7 also supported this idea of intramolecular epitope spread in pemphigus vulgaris, explaining that at various stages of the disease (mucosal and mucocutaneous), the antibodies have “different tissue-binding patterns and pathogenic activities, suggesting that they may recognize distinct epitopes.” This concept of epitope spread from the oral mucosal form to the cutaneous form of pemphigus vulgaris also could help explain our patient’s presentation, as he had a long history of recurrent oral ulcerations prior to developing the vegetating cutaneous plaques of cutaneous pemphigus vegetans.
We also appreciate that either the cutaneous pemphigus vegetans or oral pemphigus vulgaris could have been drug induced in our case. Captopril has been reported to cause pemphigus vulgaris,8 so it is conceivable that the related medication lisinopril was the culprit in our case. A prior case report described an elderly man who developed lisinopril-induced pemphigus foliaceus; however, there was no oral involvement in this case and no further blister formation within 48 hours of discontinuing lisinopril.9 An additional case report implicated lisinopril in the development of a bullous eruption on the oral mucosa in a female patient, though direct and indirect immunofluorescence did not reveal the autoantibodies that typically are seen in pemphigus vulgaris.10 Our patient’s blood eosinophilia also could support an adverse drug reaction. Our patient’s losartan was discontinued for several months without respite of the oral ulcerations and thus was restarted. The cutaneous pemphigus vegetans continues to be in remission and was unaffected by restarting the losartan, making it a less likely culprit for his presentation.
We identified another case in the literature in which an individual with a history of colon cancer was diagnosed with cutaneous pemphigus vegetans.11 As such, we considered a possible link between the 2 diagnoses; however, the temporal disconnect between both conditions in our patient makes this less likely, unlike the other reported case in which the internal neoplasm and pemphigus vegetans appeared nearly simultaneously.11
Finally, our case supports a combination of topical steroids and minocycline for treatment of cutaneous pemphigus vegetans.
Our case demonstrates the importance of considering cutaneous pemphigus vegetans in the differential diagnosis, despite its rarity, when patients present with vegetating plaques. In addition, although oral involvement is common with this condition, if the patient’s oral lesions do not fit the characteristic oral findings seen in pemphigus vegetans, alternative diagnoses should be considered.
- de Almeida HL Jr, Neugebauer MG, Guarenti IM, et al. Pemphigus vegetans associated with verrucous lesions: expanding a phenotype. Clinics (Sao Paulo). 2006;61:279-282.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Markopoulos AK, Antoniades DZ, Zaraboukas T. Pemphigus vegetans of the oral cavity. Int J Dermatol. 2006;45:425-428.
- Woo TY, Solomon AR, Fairley JA. Pemphigus vegetans limited to the lips and oral mucosa. Arch Dermatol. 1985;121:271-272.
- Yuen KL, Yau KC. An old gentleman with vegetative plaques and erosions: a case of pemphigus vegetans. Hong Kong J Dermatol Venereol. 2012;20:179-182.
- Chan LS, Vanderlugt CJ, Hashimoto T, et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol. 1998;110:103-109.
- Salato VK, Hacker-Foegen MK, Lazarova Z, et al. Role of intramolecular epitope spreading in pemphigus vulgaris. Clin Immunol. 2005;116:54-64.
- Dashore A, Choudhary SD. Captopril induced pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 1987;53:293-294.
- Patterson CR, Davies MG. Pemphigus foliaceus: an adverse reaction to lisinopril. J Dermatolog Treat. 2004;15:60-62.
- Baričević M, Mravak Stipeti´c M, Situm M, et al. Oral bullous eruption after taking lisinopril—case report and literature review. Wien Klin Wochenschr. 2013;125:408-411.
- Torres T, Ferreira M, Sanches M, et al. Pemphigus vegetans in a patient with colonic cancer. Indian J Dermatol Venereol Leprol. 2009;75:603-605.
To the Editor:
A 74-year-old man with a history of colon cancer and no history of sexually transmitted diseases presented with tender, moist, vegetating, and verrucous plaques localized to the inguinal creases and behind the scrotum of 3 weeks’ duration (Figure 1). The patient recently had taken lisinopril prescribed by his primary care physician for a couple of years for hypertension before switching to losartan prior to the current presentation. He later noticed the groin eruptions. He also noticed white tongue plaques temporally associated with the groin plaques and a long history of recurrent oral ulcerations. Prior to being seen in our clinic, outside physicians cultured methicillin-sensitive Staphylococcus aureus from the groin plaques and treated him with oral clindamycin, cephalexin, and topical mupirocin without a clinical response.
Our differential diagnosis included condyloma acuminata, condyloma lata, and cutaneous pemphigus vegetans. Laboratory testing revealed a nonreactive rapid plasma reagin test and peripheral eosinophilia of 14.9% (reference range, 0%–6%). Biopsy of a left groin plaque revealed epidermal hyperplasia with spongiosis and an eosinophilic-rich infiltrate on hematoxylin and eosin staining (Figure 2A), and direct immunofluorescence revealed diffuse epidermal intercellular IgG deposits (Figure 2B). The patient’s clinical and histologic presentation was consistent with cutaneous pemphigus vegetans. Biopsy of an oral ulcer revealed denuded acantholytic mucositis with eosinophilic-rich submucosal infiltrate and fibrosis (Figure 3A). Direct immunofluorescence was positive for lacelike intercellular staining for IgG and C3 within the squamous epithelium (Figure 3B). Together the clinical and histologic findings were consistent with oral pemphigus vulgaris.
The patient initially was started on oral minocycline 100 mg twice daily and mometasone furoate cream 0.1% twice daily to affected groin areas. With these interventions, the groin plaques almost completely resolved after several months, leaving only residual hyperpigmentation (Figure 4). The oral pemphigus vulgaris initially was treated with dexamethasone 0.5 mg/5 mL solution 2 to 3 times daily, but the lesions were refractory to this approach and also did not improve after the losartan was discontinued for several months. As such, mycophenolate mofetil was started. He was titrated to the lowest effective dose and showed near-complete resolution with 500 mg 3 times daily.
Cutaneous pemphigus vegetans, a rare variant of pemphigus vulgaris, is characterized by vegetating plaques commonly localized to the skin folds, scalp, face, and mucous membranes.1 Involvement of the oral mucosa occurs in a majority of cases. Although our patient had oral ulcerations, he did not have characteristic cerebriform changes of the dorsal tongue or associated verrucous hyperkeratotic lesions involving the buccal mucosa, hard and soft palate, or vermilion border of the lips that typically are seen in pemphigus vegetans.2-5 Subsequent biopsy of the oral mucosa confirmed oral pemphigus vulgaris in our patient.
This case presentation of co-occurring cutaneous pemphigus vegetans and oral pemphigus vulgaris is uncommonly reported in the literature. Although the etiology of this co-occurrence is not clear, it could represent a form of epitope spread, with the mechanism similar to that proposed for the progression of pemphigus vulgaris from the mucosal to the mucocutaneous stage by Chan et al,6 who suggested that an autoimmune reaction against specific desmoglein 3 epitopes (an important protein component for desmosomes and the autoantigen in pemphigus vulgaris) on mucosal membranes could induce local damage. These injuries could then expose the autoreactive immune cells to a secondary desmoglein 3 epitope present in the skin, leading to the development of cutaneous lesions.6 Salato et al7 also supported this idea of intramolecular epitope spread in pemphigus vulgaris, explaining that at various stages of the disease (mucosal and mucocutaneous), the antibodies have “different tissue-binding patterns and pathogenic activities, suggesting that they may recognize distinct epitopes.” This concept of epitope spread from the oral mucosal form to the cutaneous form of pemphigus vulgaris also could help explain our patient’s presentation, as he had a long history of recurrent oral ulcerations prior to developing the vegetating cutaneous plaques of cutaneous pemphigus vegetans.
We also appreciate that either the cutaneous pemphigus vegetans or oral pemphigus vulgaris could have been drug induced in our case. Captopril has been reported to cause pemphigus vulgaris,8 so it is conceivable that the related medication lisinopril was the culprit in our case. A prior case report described an elderly man who developed lisinopril-induced pemphigus foliaceus; however, there was no oral involvement in this case and no further blister formation within 48 hours of discontinuing lisinopril.9 An additional case report implicated lisinopril in the development of a bullous eruption on the oral mucosa in a female patient, though direct and indirect immunofluorescence did not reveal the autoantibodies that typically are seen in pemphigus vulgaris.10 Our patient’s blood eosinophilia also could support an adverse drug reaction. Our patient’s losartan was discontinued for several months without respite of the oral ulcerations and thus was restarted. The cutaneous pemphigus vegetans continues to be in remission and was unaffected by restarting the losartan, making it a less likely culprit for his presentation.
We identified another case in the literature in which an individual with a history of colon cancer was diagnosed with cutaneous pemphigus vegetans.11 As such, we considered a possible link between the 2 diagnoses; however, the temporal disconnect between both conditions in our patient makes this less likely, unlike the other reported case in which the internal neoplasm and pemphigus vegetans appeared nearly simultaneously.11
Finally, our case supports a combination of topical steroids and minocycline for treatment of cutaneous pemphigus vegetans.
Our case demonstrates the importance of considering cutaneous pemphigus vegetans in the differential diagnosis, despite its rarity, when patients present with vegetating plaques. In addition, although oral involvement is common with this condition, if the patient’s oral lesions do not fit the characteristic oral findings seen in pemphigus vegetans, alternative diagnoses should be considered.
To the Editor:
A 74-year-old man with a history of colon cancer and no history of sexually transmitted diseases presented with tender, moist, vegetating, and verrucous plaques localized to the inguinal creases and behind the scrotum of 3 weeks’ duration (Figure 1). The patient recently had taken lisinopril prescribed by his primary care physician for a couple of years for hypertension before switching to losartan prior to the current presentation. He later noticed the groin eruptions. He also noticed white tongue plaques temporally associated with the groin plaques and a long history of recurrent oral ulcerations. Prior to being seen in our clinic, outside physicians cultured methicillin-sensitive Staphylococcus aureus from the groin plaques and treated him with oral clindamycin, cephalexin, and topical mupirocin without a clinical response.
Our differential diagnosis included condyloma acuminata, condyloma lata, and cutaneous pemphigus vegetans. Laboratory testing revealed a nonreactive rapid plasma reagin test and peripheral eosinophilia of 14.9% (reference range, 0%–6%). Biopsy of a left groin plaque revealed epidermal hyperplasia with spongiosis and an eosinophilic-rich infiltrate on hematoxylin and eosin staining (Figure 2A), and direct immunofluorescence revealed diffuse epidermal intercellular IgG deposits (Figure 2B). The patient’s clinical and histologic presentation was consistent with cutaneous pemphigus vegetans. Biopsy of an oral ulcer revealed denuded acantholytic mucositis with eosinophilic-rich submucosal infiltrate and fibrosis (Figure 3A). Direct immunofluorescence was positive for lacelike intercellular staining for IgG and C3 within the squamous epithelium (Figure 3B). Together the clinical and histologic findings were consistent with oral pemphigus vulgaris.
The patient initially was started on oral minocycline 100 mg twice daily and mometasone furoate cream 0.1% twice daily to affected groin areas. With these interventions, the groin plaques almost completely resolved after several months, leaving only residual hyperpigmentation (Figure 4). The oral pemphigus vulgaris initially was treated with dexamethasone 0.5 mg/5 mL solution 2 to 3 times daily, but the lesions were refractory to this approach and also did not improve after the losartan was discontinued for several months. As such, mycophenolate mofetil was started. He was titrated to the lowest effective dose and showed near-complete resolution with 500 mg 3 times daily.
Cutaneous pemphigus vegetans, a rare variant of pemphigus vulgaris, is characterized by vegetating plaques commonly localized to the skin folds, scalp, face, and mucous membranes.1 Involvement of the oral mucosa occurs in a majority of cases. Although our patient had oral ulcerations, he did not have characteristic cerebriform changes of the dorsal tongue or associated verrucous hyperkeratotic lesions involving the buccal mucosa, hard and soft palate, or vermilion border of the lips that typically are seen in pemphigus vegetans.2-5 Subsequent biopsy of the oral mucosa confirmed oral pemphigus vulgaris in our patient.
This case presentation of co-occurring cutaneous pemphigus vegetans and oral pemphigus vulgaris is uncommonly reported in the literature. Although the etiology of this co-occurrence is not clear, it could represent a form of epitope spread, with the mechanism similar to that proposed for the progression of pemphigus vulgaris from the mucosal to the mucocutaneous stage by Chan et al,6 who suggested that an autoimmune reaction against specific desmoglein 3 epitopes (an important protein component for desmosomes and the autoantigen in pemphigus vulgaris) on mucosal membranes could induce local damage. These injuries could then expose the autoreactive immune cells to a secondary desmoglein 3 epitope present in the skin, leading to the development of cutaneous lesions.6 Salato et al7 also supported this idea of intramolecular epitope spread in pemphigus vulgaris, explaining that at various stages of the disease (mucosal and mucocutaneous), the antibodies have “different tissue-binding patterns and pathogenic activities, suggesting that they may recognize distinct epitopes.” This concept of epitope spread from the oral mucosal form to the cutaneous form of pemphigus vulgaris also could help explain our patient’s presentation, as he had a long history of recurrent oral ulcerations prior to developing the vegetating cutaneous plaques of cutaneous pemphigus vegetans.
We also appreciate that either the cutaneous pemphigus vegetans or oral pemphigus vulgaris could have been drug induced in our case. Captopril has been reported to cause pemphigus vulgaris,8 so it is conceivable that the related medication lisinopril was the culprit in our case. A prior case report described an elderly man who developed lisinopril-induced pemphigus foliaceus; however, there was no oral involvement in this case and no further blister formation within 48 hours of discontinuing lisinopril.9 An additional case report implicated lisinopril in the development of a bullous eruption on the oral mucosa in a female patient, though direct and indirect immunofluorescence did not reveal the autoantibodies that typically are seen in pemphigus vulgaris.10 Our patient’s blood eosinophilia also could support an adverse drug reaction. Our patient’s losartan was discontinued for several months without respite of the oral ulcerations and thus was restarted. The cutaneous pemphigus vegetans continues to be in remission and was unaffected by restarting the losartan, making it a less likely culprit for his presentation.
We identified another case in the literature in which an individual with a history of colon cancer was diagnosed with cutaneous pemphigus vegetans.11 As such, we considered a possible link between the 2 diagnoses; however, the temporal disconnect between both conditions in our patient makes this less likely, unlike the other reported case in which the internal neoplasm and pemphigus vegetans appeared nearly simultaneously.11
Finally, our case supports a combination of topical steroids and minocycline for treatment of cutaneous pemphigus vegetans.
Our case demonstrates the importance of considering cutaneous pemphigus vegetans in the differential diagnosis, despite its rarity, when patients present with vegetating plaques. In addition, although oral involvement is common with this condition, if the patient’s oral lesions do not fit the characteristic oral findings seen in pemphigus vegetans, alternative diagnoses should be considered.
- de Almeida HL Jr, Neugebauer MG, Guarenti IM, et al. Pemphigus vegetans associated with verrucous lesions: expanding a phenotype. Clinics (Sao Paulo). 2006;61:279-282.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Markopoulos AK, Antoniades DZ, Zaraboukas T. Pemphigus vegetans of the oral cavity. Int J Dermatol. 2006;45:425-428.
- Woo TY, Solomon AR, Fairley JA. Pemphigus vegetans limited to the lips and oral mucosa. Arch Dermatol. 1985;121:271-272.
- Yuen KL, Yau KC. An old gentleman with vegetative plaques and erosions: a case of pemphigus vegetans. Hong Kong J Dermatol Venereol. 2012;20:179-182.
- Chan LS, Vanderlugt CJ, Hashimoto T, et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol. 1998;110:103-109.
- Salato VK, Hacker-Foegen MK, Lazarova Z, et al. Role of intramolecular epitope spreading in pemphigus vulgaris. Clin Immunol. 2005;116:54-64.
- Dashore A, Choudhary SD. Captopril induced pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 1987;53:293-294.
- Patterson CR, Davies MG. Pemphigus foliaceus: an adverse reaction to lisinopril. J Dermatolog Treat. 2004;15:60-62.
- Baričević M, Mravak Stipeti´c M, Situm M, et al. Oral bullous eruption after taking lisinopril—case report and literature review. Wien Klin Wochenschr. 2013;125:408-411.
- Torres T, Ferreira M, Sanches M, et al. Pemphigus vegetans in a patient with colonic cancer. Indian J Dermatol Venereol Leprol. 2009;75:603-605.
- de Almeida HL Jr, Neugebauer MG, Guarenti IM, et al. Pemphigus vegetans associated with verrucous lesions: expanding a phenotype. Clinics (Sao Paulo). 2006;61:279-282.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Markopoulos AK, Antoniades DZ, Zaraboukas T. Pemphigus vegetans of the oral cavity. Int J Dermatol. 2006;45:425-428.
- Woo TY, Solomon AR, Fairley JA. Pemphigus vegetans limited to the lips and oral mucosa. Arch Dermatol. 1985;121:271-272.
- Yuen KL, Yau KC. An old gentleman with vegetative plaques and erosions: a case of pemphigus vegetans. Hong Kong J Dermatol Venereol. 2012;20:179-182.
- Chan LS, Vanderlugt CJ, Hashimoto T, et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol. 1998;110:103-109.
- Salato VK, Hacker-Foegen MK, Lazarova Z, et al. Role of intramolecular epitope spreading in pemphigus vulgaris. Clin Immunol. 2005;116:54-64.
- Dashore A, Choudhary SD. Captopril induced pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 1987;53:293-294.
- Patterson CR, Davies MG. Pemphigus foliaceus: an adverse reaction to lisinopril. J Dermatolog Treat. 2004;15:60-62.
- Baričević M, Mravak Stipeti´c M, Situm M, et al. Oral bullous eruption after taking lisinopril—case report and literature review. Wien Klin Wochenschr. 2013;125:408-411.
- Torres T, Ferreira M, Sanches M, et al. Pemphigus vegetans in a patient with colonic cancer. Indian J Dermatol Venereol Leprol. 2009;75:603-605.
Practice Points
- Recognize the clinical and histologic features of pemphigus vegetans, a rare variant of pemphigus vulgaris.
- Consider mechanisms of co-occurring cutaneous pemphigus vegetans and oral pemphigus vulgaris.