User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Acne Medications: What Factors Drive Variable Costs?


Gottron Papules Mimicking Dermatomyositis: An Unusual Manifestation of Systemic Lupus Erythematosus
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
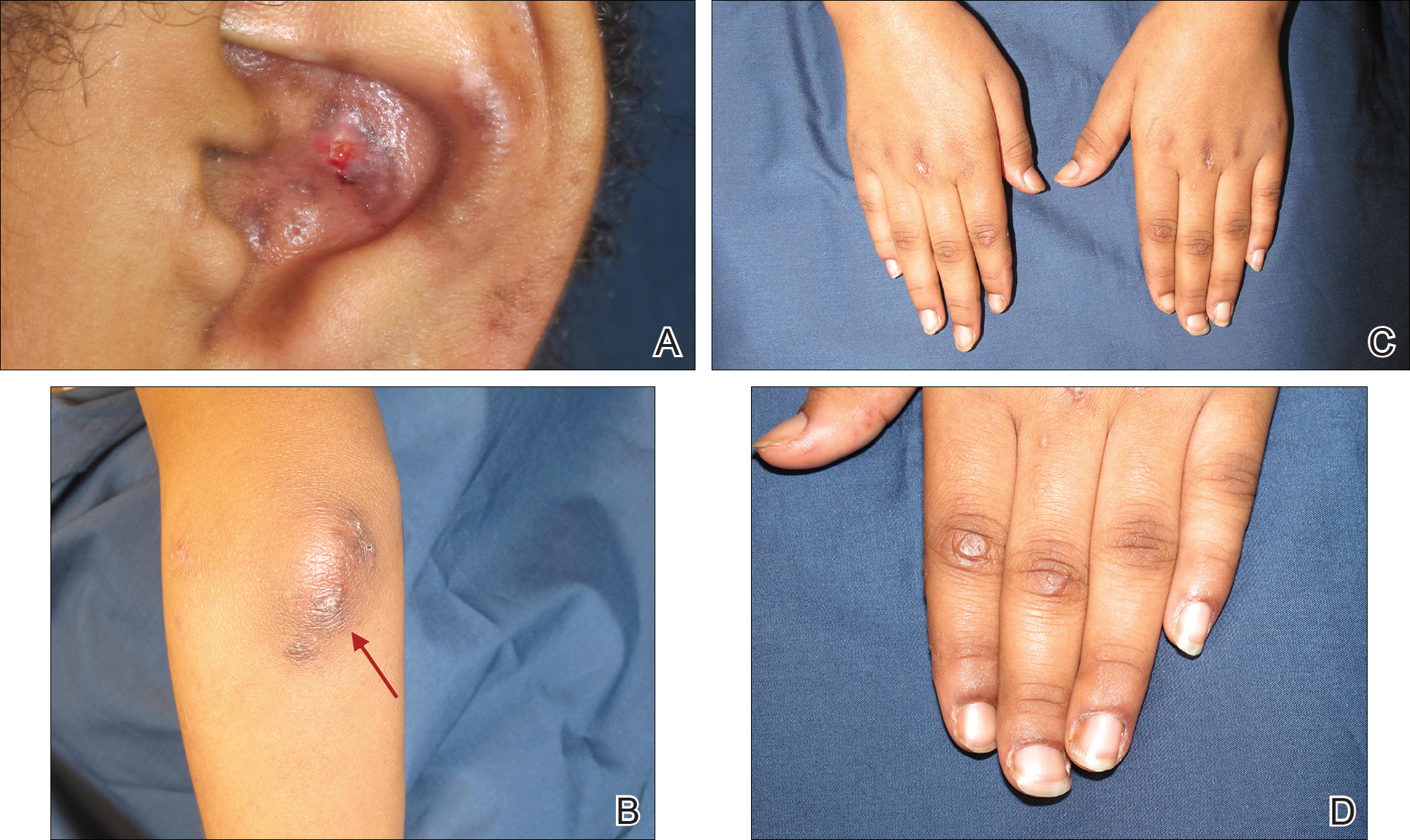
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
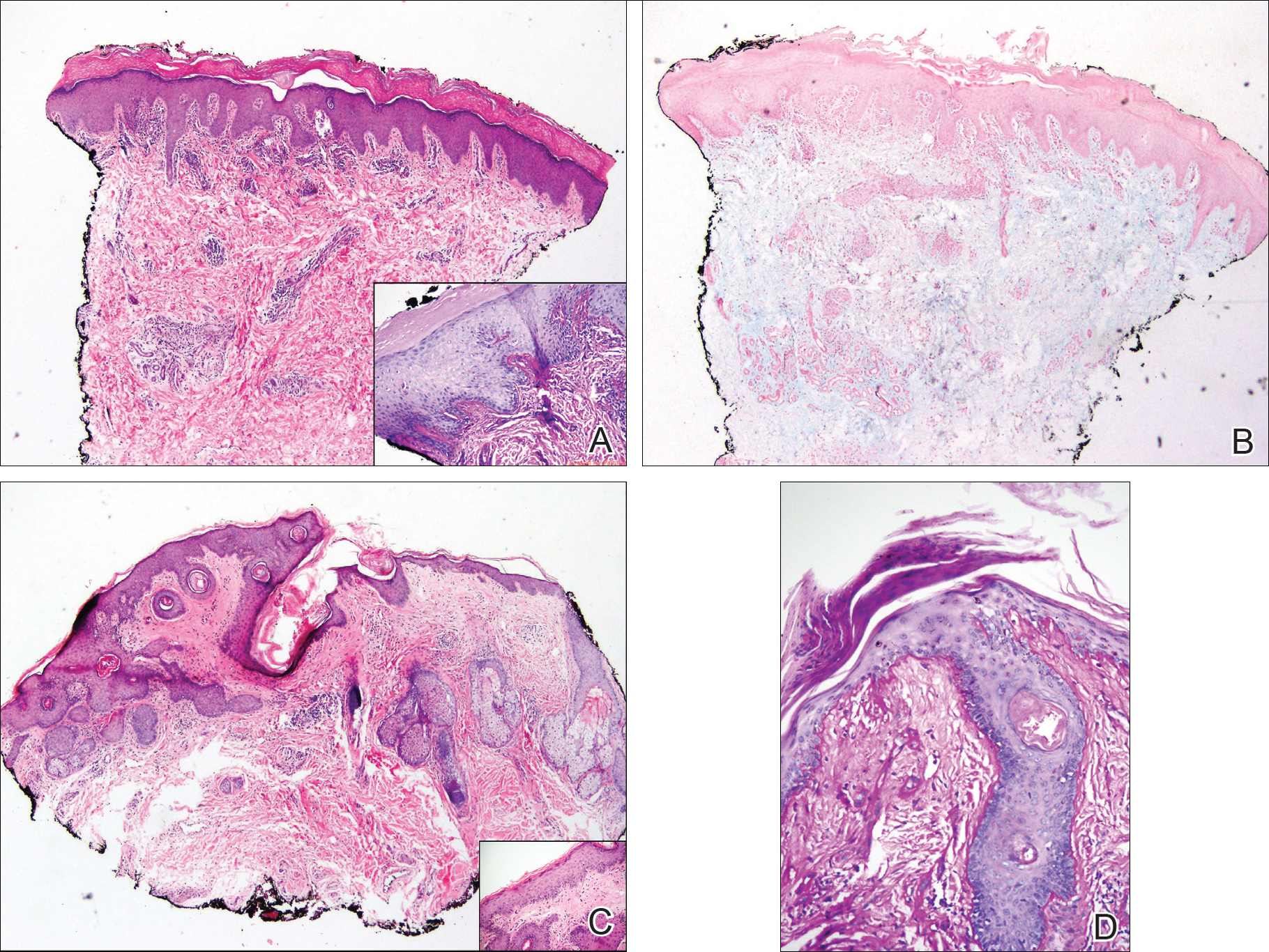
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
Practice Points
- Gottron-like papules can be a dermatologic presentation of lupus erythematosus.
- When present along with other findings of lupus erythematosus without any clinical manifestations of dermatomyositis, Gottron-like papules can be thought of as a manifestation of lupus erythematosus rather than dermatomyositis.

VIDEO: Look for Signs of Depression in Rosacea Patients
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Risk Stratification for Cellulitis Versus Noncellulitic Conditions of the Lower Extremity: A Retrospective Review of the NEW HAvUN Criteria
Cellulitis is defined as an acute or subacute, bacterial-induced inflammation of subcutaneous tissue that can extend superficially. The inciting incident often is assumed to be invasion of bacteria through loose connective tissue.1 Although cellulitis is bacterial in origin, it often is difficult to culture the offending microorganism from biopsy sites, swabs, or blood. Erythema, fever, induration, and tenderness are largely seen as clinical manifestations. Moderate and severe cases may be accompanied by fever, malaise, and leukocytosis. The lower extremity is the most common location of involvement (Figure 1), and usually a wound, ulcer, or interdigital superficial infection can be identified and implicated as the source of entry.

Effective treatment of cellulitis is necessary because complications such as abscesses, underlying fascia or muscle involvement, and septicemia can develop, leading to poor outcomes. Antibiotics should be administered intravenously in patients with suspected fascial involvement, septicemia, or dermal necrosis, or in those with an immunological comorbidity.2
The differential diagnosis of lower extremity cellulitis is wide due to the existence of several mimicking dermatologic conditions. These so-called pseudocellulitis conditions include stasis dermatitis, venous ulceration, acute lipodermatosclerosis, pigmented purpura, vasculopathy, contact dermatitis, adverse medication reaction, and arthropod bite. Stasis dermatitis and lipodermatosclerosis, both arising from venous insufficiency, are by far 2 of the most common skin conditions that imitate cellulitis.
Stasis dermatitis is a common condition in the United States and Europe, usually manifesting as a pigmented purpuric dermatosis on anterior tibial surfaces, around the ankle, or overlying dependent varicosities. Skin changes can include hyperpigmentation, edema, mild scaling, eczematous patches, and even ulceration.3
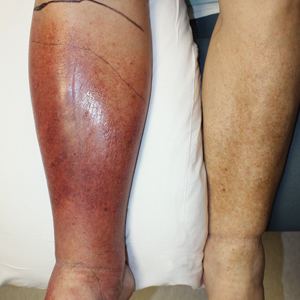
Lipodermatosclerosis is a disorder of progressive fibrosis of subcutaneous fat. It is more common in middle-aged women who have a high body mass index and a venous abnormality.4 This form of panniculitis typically affects the lower extremities bilaterally, manifesting as erythematous and indurated skin changes, sometimes described as inverted champagne bottles (Figure 2). At times, there can be accompanying painful ulceration on the erythematous areas, features that closely resemble cellulitis.5,6 Lipodermatosclerosis is commonly misdiagnosed as cellulitis, leading to inappropriate prescription of antibiotics.7

Distinguishing cellulitis from noncellulitic conditions of the lower extremity is paramount to effective patient management in the emergent setting. With a reported incidence of 24.6 per 100 person-years, cellulitis constitutes 1% to 14% of emergency department visits and 4% to 7% of hospital admissions.Therefore, prompt appropriate diagnosis and treatment can avoid life-threatening complications associated with infection such as sepsis, abscess, lymphangitis, and necrotizing fasciitis.8-11
It is estimated that 10% to 20% of patients who have been given a diagnosis of cellulitis do not actually have the disease.2,12 This discrepancy consumes a remarkable amount of hospital resources and can lead to inappropriate or excessive use of antibiotics.13 Although the true incidence of adverse antibiotic reactions is unknown, it is estimated that they are the cause of 3% to 6% of acute hospital admissions and occur in 10% to 15% of inpatients admitted for other primary reasons.14 These findings illustrate the potential for an increased risk for morbidity and increased length of stay for patients beginning an antibiotic regimen, especially when the agents are administered unnecessarily. In addition, inappropriate antibiotic use contributes to antibiotic resistance, which continues to be a major problem, especially in hospitalized patients.
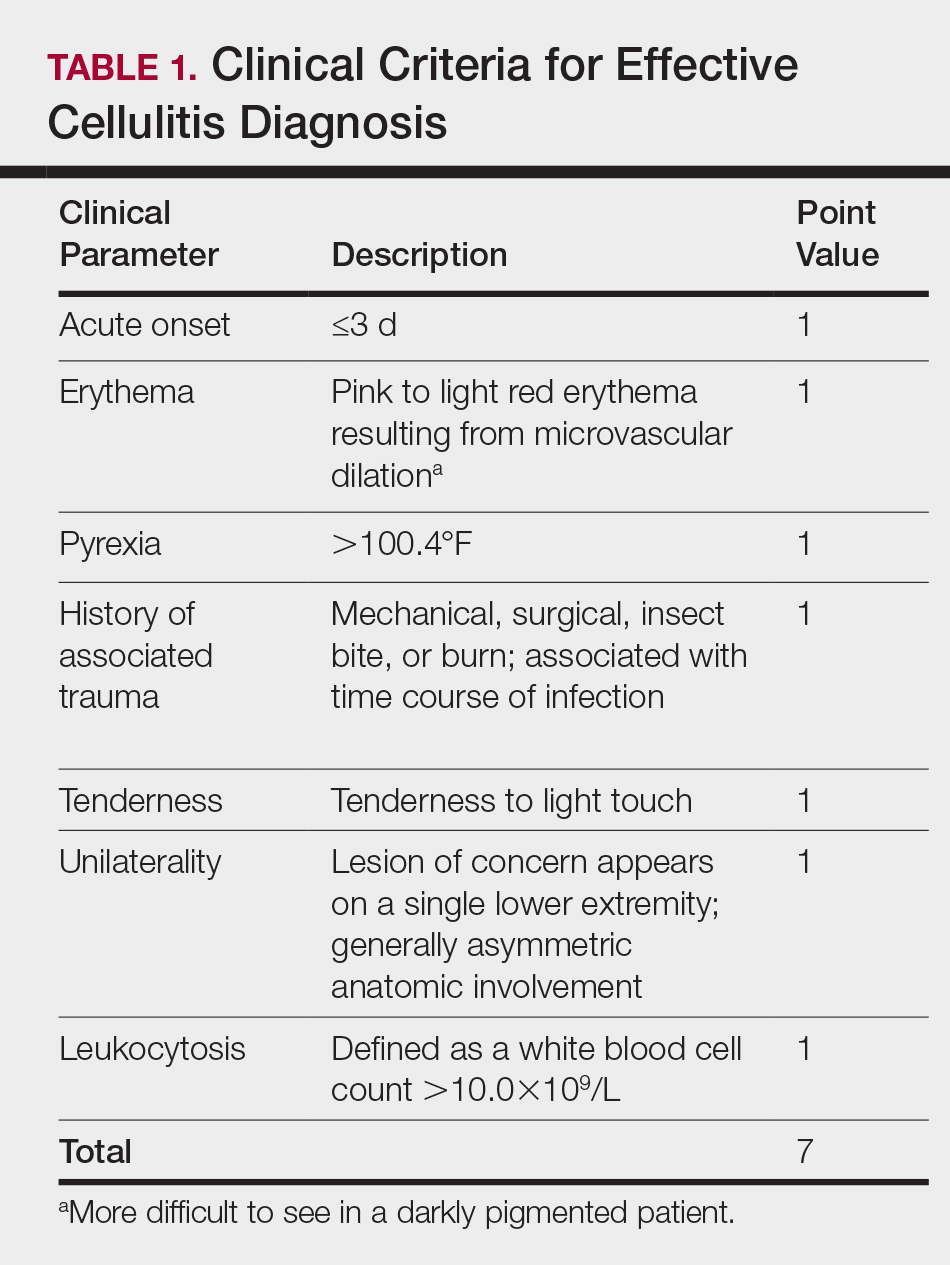
There is a lack of consensus in the literature about methods to risk stratify patients who present with acute dermatologic conditions that include and resemble cellulitis. We sought to identify clinical features based on available clinical literature-derived variables. We tested our scheme in a series of patients with a known diagnosis of cellulitis or other dermatologic pathology of the lower extremity to assess the validity of the following 7 clinical criteria: acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis.
Materials and Methods
This retrospective chart review was approved by the Yale University (New Haven, Connecticut) institutional review board (HIC#1409014533). Final diagnosis, demographic data, clinical manifestations, and relevant diagnostic laboratory values of 57 patients were obtained from a database in the dermatology department’s consultation log and electronic medical record database (December 2011 to December 2014). The presence of each clinical symptom—acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis—was assigned a score equal to 1; values were tallied to achieve a final score for each patient (Table 1). Patients who were seen initially as a consultation for possible cellulitis but given a final diagnosis of stasis dermatitis or lipodermatosclerosis were included (Table 2).

Clinical Criteria
The clinical criteria were developed based largely on clinical experience and relevant secondary literature.15-17 At the patient encounter, presence of each of the variables (Table 1) was assessed according to the following definitions:
- acute onset: within the prior 72 hours and more indicative of an acute infective process than a gradual and chronic consequence of venous stasis
- erythema: a subjective clinical marker for inflammation that can be associated with cellulitis, though darker, erythematous-appearing discolorations also can be seen in patients with chronic venous hypertension or valvular incompetence4,15
- pyrexia: body temperature greater than 100.4°F
- history of associated trauma: encompassing mechanical wounds, surgical incisions, burns, and insect bites that correlate closely to the time course of symptomatic development
- tenderness: tenderness to light touch, which may be more common in patients afflicted with cellulitis than in those with venous insufficiency
- unilaterality: a helpful distinguishing feature that points the diagnosis away from a dermatitislike clinical picture, especially because bilateral cellulitis is rare and regarded as a diagnostic pitfall18
- leukocytosis: white blood cell count greater than 10.0×109/L and is reasonably considered a cardinal metric of inflammatory processes, though it can be confounded by immunocompromise (low count) or steroid use (high count)
Statistical Analysis
Odds ratios (ORs) were calculated and χ2 analysis was performed for each presenting symptom using JMP 10.0 analytical software (SAS Institute Inc). Each patient was rated separately by means of the clinical feature–based scoring system for the calculation of a total score. After application of the score to the patient population, receiver operating characteristic curves were constructed to identify the optimal score threshold for discriminating cellulitis from dermatitis in this group. For each clinical feature, P<.05 was considered significant.
Results
Our cohort included 32 male and 25 female patients with a mean age of 63 and 61 years, respectively. The final clinical diagnosis of cellulitis was made in 20 patients (35%). An established diagnosis of cellulitis was assigned based on a dermatology evaluation located within our electronic medical record database (Table 2).
Each clinical parameter was evaluated separately for each patient; combined results are summarized in Table 3. Acute onset (≤3 days) was a clinical characteristic seen in 80% (16/20) of cellulitis cases and 22% (8/37) of noncellulitis cases (OR, 14.5; P<.001). Erythema had similar significance (OR, 10.3; prevalence, 95% [19/20] vs 65% [24/37]; P=.012). Pyrexia possessed an OR of 99.2 for cellulitis and was seen in 85% (17/20) of cellulitis cases and only 5% (2/37) of noncellulitis cases (P<.001).

A history of associated trauma had an OR of 36.0 for cellulitis, with 50% (10/20) and 3% (1/37) prevalence in cellulitis cases and noncellulitis cases, respectively (P<.001). Tenderness, documented in 90% (18/20) of cellulitis cases and 43% (16/37) of noncellulitis cases, had an OR of 11.8 (P<.001).
Unilaterality had 100% (20/20) prevalence in our cellulitis cohort and was the only characteristic within the algorithm that yielded an incalculable OR. Noncellulitis or stasis dermatitis of the lower extremity exhibited a unilateral lesion in 11 cases (30%), of which 1 case resulted from a unilateral tibial fracture. Leukocytosis was seen in 65% (13/20) of cellulitis cases and 8% (3/37) of noncellulitis cases, with an OR for cellulitis of 21.0 (P<.001).
All parameters were significant by χ2 analysis (Table 3).
Comment
We found that testing positive for 4 of 7 clinical criteria for assessing cellulitis was highly specific (95%) and sensitive (100%) for a diagnosis of cellulitis among its range of mimics (Figure 3). These cellulitis criteria can be remembered, with some modification, using NEW HAvUN as a mnemonic device (New onset,
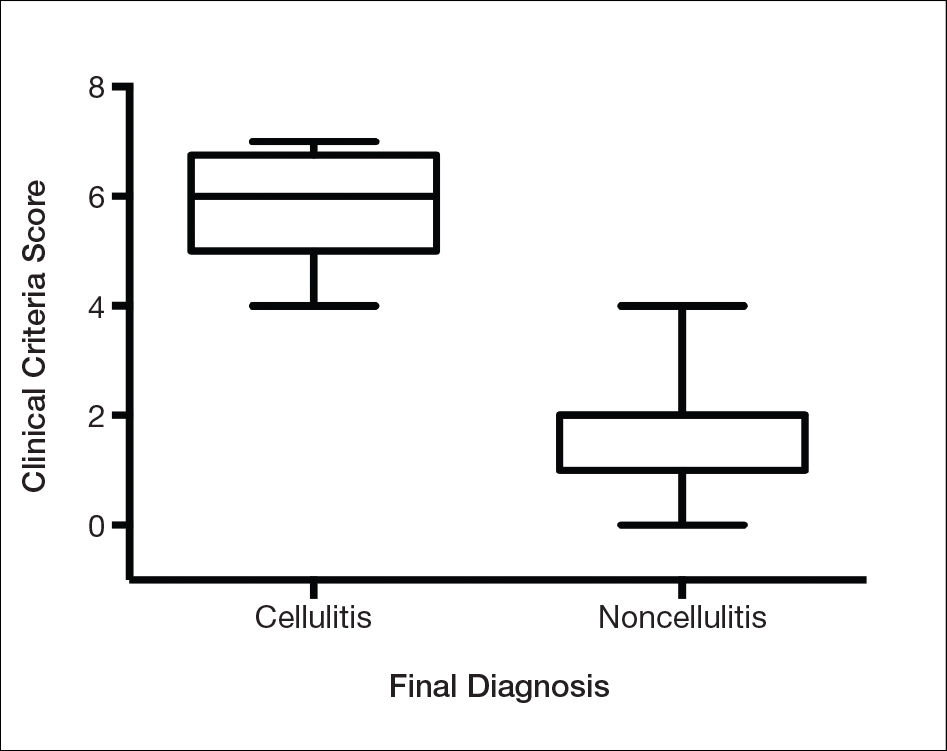
Consistent with the literature, pyrexia, history of associated trauma, and unilaterality also were predictors of cellulitis diagnosis. Unilaterality often is used as a diagnostic tool by dermatologist consultants when a patient lacks other criteria for cellulitis, so these findings are intuitive and consistent with our institutional experience. Interestingly, leukocytosis was seen in only 65% of cellulitis cases and 8% of noncellulitis cases and therefore might not serve as a sensitive independent predictor of a diagnosis of cellulitis, emphasizing the importance of the multifactorial scoring system we have put forward. Additionally, acuity of onset, erythema, and tenderness are not independently associated with cellulitis when assessing a patient because several of those findings are present in other dermatologic conditions of the lower extremity; when combined with the other criteria, however, these 3 findings can play a role in diagnosis.
Effective cellulitis diagnosis provides well-recognized challenges in the acute medical setting because many clinical mimics exist. The estimated rate of misdiagnosed cellulitis is certainly well-established: 30% to 75% in independent and multi-institutional studies. These studies also revealed that patients admitted for bilateral “cellulitis” overwhelmingly tended to be stasis clinical pictures.13,19
Cost implications from inappropriate diagnosis largely regard inappropriate antibiotic use and the potential for microbial resistance, with associated costs estimated to be more than $50 billion (2004 dollars).20,21 The true cost burden is extremely difficult to model or predict due to remarkable variations in the institutional misdiagnosis rate, prescribing pattern, and antibiotic cost and could represent avenues of further study. Misappropriation of antibiotics includes not only a monetary cost that encompasses all aspects of acute treatment and hospitalization but also an unquantifiable cost: human lives associated with the consequences of antibiotic resistance.
Conclusion
There is a lack of consensus or criteria for differentiating cellulitis from its most common clinical counterparts. Here, we propose a convenient clinical correlation system that we hope will lead to more efficient allocation of clinical resources, including antibiotics and hospital admissions, while lowering the incidence of adverse events and leading to better patient outcomes. We recognize that the small sample size of our study may limit broad application of these criteria, though we anticipate that further prospective studies can improve the diagnostic relevance and risk-assessment power of the NEW HAvUN criteria put forth here for assessing cellulitis in the acute medical setting.
Acknowledgement—Author H.H.E. recognizes the loving memory of Nadia Ezaldein for her profound influence on and motivation behind this research.
- Lep
pard BJ, Seal DV, Colman G, et al. The value of bacteriology and serology in the diagnosis of cellulitis and erysipelas. Br J Dermatol. 1985;112:559-567. - Hep
burn MJ, Dooley DP, Skidmore PJ, et al. Comparison of short-course (5 days) and standard (10 days) treatment for uncomplicated cellulitis. Arch Int Med. 2004;164:1669-1674. - Bergan JJ, Schmid-Schönbein GW, Smith PD, et al. Chronic venous disease. N Engl J Med. 2006;355:488-498.
- Bruc
e AJ, Bennett DD, Lohse CM, et al. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol. 2002;46:187-192. - Heym
ann WR. Lipodermatosclerosis. J Am Acad Dermatol. 2009;60:1022-1023. - Vesi
ć S, Vuković J, Medenica LJ, et al. Acute lipodermatosclerosis: an open clinical trial of stanozolol in patients unable to sustain compression therapy. Dermatol Online J. 2008;14:1. - Keller
EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleve Clin J Med. 2012;79:547-552. - Dong SL, Kelly KD, Oland RC, et al. ED management of cellulitis: a review of five urban centers. Am J Emerg Med. 2001;19:535-540.
- Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
- Manfredi R, Calza L, Chiodo F. Epidemiology and microbiology of cellulitis and bacterial soft tissue infection during HIV disease: a 10-year survey. J Cutan Pathol. 2002;29:168-172.
- Pascarella L, Schonbein GW, Bergan JJ. Microcirculation and venous ulcers: a review. Ann Vasc Surg. 2005;19:921-927.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician. 2003;67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J. 2011;17:1.
- Hay RJ, Adriaans BM. Bacterial infections. In: Thong BY, Tan TC. Epidemiology and risk factors for drug allergy. 8th ed. Br J Clin Pharmacol. 2011;71:684-700.
- Hay RJ, Adriaans BM. Bacterial infections. In: Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology. 8th ed. Hoboken, NJ: John Wiley & Sons, Inc; 2004:1345-1426.
- Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology In General Medicine. 7th ed. New York, NY: McGraw-Hill; 2003.
- Sommer LL, Reboli AC, Heymann WR. Bacterial infections. In: Bolognia J, Schaffer J, Cerroni L, et al. Dermatology. Vol 4. Philadelphia, PA: Elsevier Saunders; 2012:1462-1502.
- Cox NH. Management of lower leg cellulitis. Clin Med. 2002;2:23-27.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Pinder R, Sallis A, Berry D, et al. Behaviour change and antibiotic prescribing in healthcare settings: literature review and behavioural analysis. London, UK: Public Health England; February 2015. https://assets.publishing.service.gov.uk/government/
uploads/system/uploads/attachment_data/file/405031
/Behaviour_Change_for_Antibiotic_Prescribing_-_FINAL.pdf. Accessed May 7, 2018. - Smith R, Coast J. The true cost of antimicrobial resistance. BMJ. 2013;346:f1493.
Cellulitis is defined as an acute or subacute, bacterial-induced inflammation of subcutaneous tissue that can extend superficially. The inciting incident often is assumed to be invasion of bacteria through loose connective tissue.1 Although cellulitis is bacterial in origin, it often is difficult to culture the offending microorganism from biopsy sites, swabs, or blood. Erythema, fever, induration, and tenderness are largely seen as clinical manifestations. Moderate and severe cases may be accompanied by fever, malaise, and leukocytosis. The lower extremity is the most common location of involvement (Figure 1), and usually a wound, ulcer, or interdigital superficial infection can be identified and implicated as the source of entry.
Effective treatment of cellulitis is necessary because complications such as abscesses, underlying fascia or muscle involvement, and septicemia can develop, leading to poor outcomes. Antibiotics should be administered intravenously in patients with suspected fascial involvement, septicemia, or dermal necrosis, or in those with an immunological comorbidity.2
The differential diagnosis of lower extremity cellulitis is wide due to the existence of several mimicking dermatologic conditions. These so-called pseudocellulitis conditions include stasis dermatitis, venous ulceration, acute lipodermatosclerosis, pigmented purpura, vasculopathy, contact dermatitis, adverse medication reaction, and arthropod bite. Stasis dermatitis and lipodermatosclerosis, both arising from venous insufficiency, are by far 2 of the most common skin conditions that imitate cellulitis.
Stasis dermatitis is a common condition in the United States and Europe, usually manifesting as a pigmented purpuric dermatosis on anterior tibial surfaces, around the ankle, or overlying dependent varicosities. Skin changes can include hyperpigmentation, edema, mild scaling, eczematous patches, and even ulceration.3
Lipodermatosclerosis is a disorder of progressive fibrosis of subcutaneous fat. It is more common in middle-aged women who have a high body mass index and a venous abnormality.4 This form of panniculitis typically affects the lower extremities bilaterally, manifesting as erythematous and indurated skin changes, sometimes described as inverted champagne bottles (Figure 2). At times, there can be accompanying painful ulceration on the erythematous areas, features that closely resemble cellulitis.5,6 Lipodermatosclerosis is commonly misdiagnosed as cellulitis, leading to inappropriate prescription of antibiotics.7
Distinguishing cellulitis from noncellulitic conditions of the lower extremity is paramount to effective patient management in the emergent setting. With a reported incidence of 24.6 per 100 person-years, cellulitis constitutes 1% to 14% of emergency department visits and 4% to 7% of hospital admissions.Therefore, prompt appropriate diagnosis and treatment can avoid life-threatening complications associated with infection such as sepsis, abscess, lymphangitis, and necrotizing fasciitis.8-11
It is estimated that 10% to 20% of patients who have been given a diagnosis of cellulitis do not actually have the disease.2,12 This discrepancy consumes a remarkable amount of hospital resources and can lead to inappropriate or excessive use of antibiotics.13 Although the true incidence of adverse antibiotic reactions is unknown, it is estimated that they are the cause of 3% to 6% of acute hospital admissions and occur in 10% to 15% of inpatients admitted for other primary reasons.14 These findings illustrate the potential for an increased risk for morbidity and increased length of stay for patients beginning an antibiotic regimen, especially when the agents are administered unnecessarily. In addition, inappropriate antibiotic use contributes to antibiotic resistance, which continues to be a major problem, especially in hospitalized patients.
There is a lack of consensus in the literature about methods to risk stratify patients who present with acute dermatologic conditions that include and resemble cellulitis. We sought to identify clinical features based on available clinical literature-derived variables. We tested our scheme in a series of patients with a known diagnosis of cellulitis or other dermatologic pathology of the lower extremity to assess the validity of the following 7 clinical criteria: acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis.
Materials and Methods
This retrospective chart review was approved by the Yale University (New Haven, Connecticut) institutional review board (HIC#1409014533). Final diagnosis, demographic data, clinical manifestations, and relevant diagnostic laboratory values of 57 patients were obtained from a database in the dermatology department’s consultation log and electronic medical record database (December 2011 to December 2014). The presence of each clinical symptom—acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis—was assigned a score equal to 1; values were tallied to achieve a final score for each patient (Table 1). Patients who were seen initially as a consultation for possible cellulitis but given a final diagnosis of stasis dermatitis or lipodermatosclerosis were included (Table 2).
Clinical Criteria
The clinical criteria were developed based largely on clinical experience and relevant secondary literature.15-17 At the patient encounter, presence of each of the variables (Table 1) was assessed according to the following definitions:
- acute onset: within the prior 72 hours and more indicative of an acute infective process than a gradual and chronic consequence of venous stasis
- erythema: a subjective clinical marker for inflammation that can be associated with cellulitis, though darker, erythematous-appearing discolorations also can be seen in patients with chronic venous hypertension or valvular incompetence4,15
- pyrexia: body temperature greater than 100.4°F
- history of associated trauma: encompassing mechanical wounds, surgical incisions, burns, and insect bites that correlate closely to the time course of symptomatic development
- tenderness: tenderness to light touch, which may be more common in patients afflicted with cellulitis than in those with venous insufficiency
- unilaterality: a helpful distinguishing feature that points the diagnosis away from a dermatitislike clinical picture, especially because bilateral cellulitis is rare and regarded as a diagnostic pitfall18
- leukocytosis: white blood cell count greater than 10.0×109/L and is reasonably considered a cardinal metric of inflammatory processes, though it can be confounded by immunocompromise (low count) or steroid use (high count)
Statistical Analysis
Odds ratios (ORs) were calculated and χ2 analysis was performed for each presenting symptom using JMP 10.0 analytical software (SAS Institute Inc). Each patient was rated separately by means of the clinical feature–based scoring system for the calculation of a total score. After application of the score to the patient population, receiver operating characteristic curves were constructed to identify the optimal score threshold for discriminating cellulitis from dermatitis in this group. For each clinical feature, P<.05 was considered significant.
Results
Our cohort included 32 male and 25 female patients with a mean age of 63 and 61 years, respectively. The final clinical diagnosis of cellulitis was made in 20 patients (35%). An established diagnosis of cellulitis was assigned based on a dermatology evaluation located within our electronic medical record database (Table 2).
Each clinical parameter was evaluated separately for each patient; combined results are summarized in Table 3. Acute onset (≤3 days) was a clinical characteristic seen in 80% (16/20) of cellulitis cases and 22% (8/37) of noncellulitis cases (OR, 14.5; P<.001). Erythema had similar significance (OR, 10.3; prevalence, 95% [19/20] vs 65% [24/37]; P=.012). Pyrexia possessed an OR of 99.2 for cellulitis and was seen in 85% (17/20) of cellulitis cases and only 5% (2/37) of noncellulitis cases (P<.001).
A history of associated trauma had an OR of 36.0 for cellulitis, with 50% (10/20) and 3% (1/37) prevalence in cellulitis cases and noncellulitis cases, respectively (P<.001). Tenderness, documented in 90% (18/20) of cellulitis cases and 43% (16/37) of noncellulitis cases, had an OR of 11.8 (P<.001).
Unilaterality had 100% (20/20) prevalence in our cellulitis cohort and was the only characteristic within the algorithm that yielded an incalculable OR. Noncellulitis or stasis dermatitis of the lower extremity exhibited a unilateral lesion in 11 cases (30%), of which 1 case resulted from a unilateral tibial fracture. Leukocytosis was seen in 65% (13/20) of cellulitis cases and 8% (3/37) of noncellulitis cases, with an OR for cellulitis of 21.0 (P<.001).
All parameters were significant by χ2 analysis (Table 3).
Comment
We found that testing positive for 4 of 7 clinical criteria for assessing cellulitis was highly specific (95%) and sensitive (100%) for a diagnosis of cellulitis among its range of mimics (Figure 3). These cellulitis criteria can be remembered, with some modification, using NEW HAvUN as a mnemonic device (New onset,
Consistent with the literature, pyrexia, history of associated trauma, and unilaterality also were predictors of cellulitis diagnosis. Unilaterality often is used as a diagnostic tool by dermatologist consultants when a patient lacks other criteria for cellulitis, so these findings are intuitive and consistent with our institutional experience. Interestingly, leukocytosis was seen in only 65% of cellulitis cases and 8% of noncellulitis cases and therefore might not serve as a sensitive independent predictor of a diagnosis of cellulitis, emphasizing the importance of the multifactorial scoring system we have put forward. Additionally, acuity of onset, erythema, and tenderness are not independently associated with cellulitis when assessing a patient because several of those findings are present in other dermatologic conditions of the lower extremity; when combined with the other criteria, however, these 3 findings can play a role in diagnosis.
Effective cellulitis diagnosis provides well-recognized challenges in the acute medical setting because many clinical mimics exist. The estimated rate of misdiagnosed cellulitis is certainly well-established: 30% to 75% in independent and multi-institutional studies. These studies also revealed that patients admitted for bilateral “cellulitis” overwhelmingly tended to be stasis clinical pictures.13,19
Cost implications from inappropriate diagnosis largely regard inappropriate antibiotic use and the potential for microbial resistance, with associated costs estimated to be more than $50 billion (2004 dollars).20,21 The true cost burden is extremely difficult to model or predict due to remarkable variations in the institutional misdiagnosis rate, prescribing pattern, and antibiotic cost and could represent avenues of further study. Misappropriation of antibiotics includes not only a monetary cost that encompasses all aspects of acute treatment and hospitalization but also an unquantifiable cost: human lives associated with the consequences of antibiotic resistance.
Conclusion
There is a lack of consensus or criteria for differentiating cellulitis from its most common clinical counterparts. Here, we propose a convenient clinical correlation system that we hope will lead to more efficient allocation of clinical resources, including antibiotics and hospital admissions, while lowering the incidence of adverse events and leading to better patient outcomes. We recognize that the small sample size of our study may limit broad application of these criteria, though we anticipate that further prospective studies can improve the diagnostic relevance and risk-assessment power of the NEW HAvUN criteria put forth here for assessing cellulitis in the acute medical setting.
Acknowledgement—Author H.H.E. recognizes the loving memory of Nadia Ezaldein for her profound influence on and motivation behind this research.
Cellulitis is defined as an acute or subacute, bacterial-induced inflammation of subcutaneous tissue that can extend superficially. The inciting incident often is assumed to be invasion of bacteria through loose connective tissue.1 Although cellulitis is bacterial in origin, it often is difficult to culture the offending microorganism from biopsy sites, swabs, or blood. Erythema, fever, induration, and tenderness are largely seen as clinical manifestations. Moderate and severe cases may be accompanied by fever, malaise, and leukocytosis. The lower extremity is the most common location of involvement (Figure 1), and usually a wound, ulcer, or interdigital superficial infection can be identified and implicated as the source of entry.
Effective treatment of cellulitis is necessary because complications such as abscesses, underlying fascia or muscle involvement, and septicemia can develop, leading to poor outcomes. Antibiotics should be administered intravenously in patients with suspected fascial involvement, septicemia, or dermal necrosis, or in those with an immunological comorbidity.2
The differential diagnosis of lower extremity cellulitis is wide due to the existence of several mimicking dermatologic conditions. These so-called pseudocellulitis conditions include stasis dermatitis, venous ulceration, acute lipodermatosclerosis, pigmented purpura, vasculopathy, contact dermatitis, adverse medication reaction, and arthropod bite. Stasis dermatitis and lipodermatosclerosis, both arising from venous insufficiency, are by far 2 of the most common skin conditions that imitate cellulitis.
Stasis dermatitis is a common condition in the United States and Europe, usually manifesting as a pigmented purpuric dermatosis on anterior tibial surfaces, around the ankle, or overlying dependent varicosities. Skin changes can include hyperpigmentation, edema, mild scaling, eczematous patches, and even ulceration.3
Lipodermatosclerosis is a disorder of progressive fibrosis of subcutaneous fat. It is more common in middle-aged women who have a high body mass index and a venous abnormality.4 This form of panniculitis typically affects the lower extremities bilaterally, manifesting as erythematous and indurated skin changes, sometimes described as inverted champagne bottles (Figure 2). At times, there can be accompanying painful ulceration on the erythematous areas, features that closely resemble cellulitis.5,6 Lipodermatosclerosis is commonly misdiagnosed as cellulitis, leading to inappropriate prescription of antibiotics.7
Distinguishing cellulitis from noncellulitic conditions of the lower extremity is paramount to effective patient management in the emergent setting. With a reported incidence of 24.6 per 100 person-years, cellulitis constitutes 1% to 14% of emergency department visits and 4% to 7% of hospital admissions.Therefore, prompt appropriate diagnosis and treatment can avoid life-threatening complications associated with infection such as sepsis, abscess, lymphangitis, and necrotizing fasciitis.8-11
It is estimated that 10% to 20% of patients who have been given a diagnosis of cellulitis do not actually have the disease.2,12 This discrepancy consumes a remarkable amount of hospital resources and can lead to inappropriate or excessive use of antibiotics.13 Although the true incidence of adverse antibiotic reactions is unknown, it is estimated that they are the cause of 3% to 6% of acute hospital admissions and occur in 10% to 15% of inpatients admitted for other primary reasons.14 These findings illustrate the potential for an increased risk for morbidity and increased length of stay for patients beginning an antibiotic regimen, especially when the agents are administered unnecessarily. In addition, inappropriate antibiotic use contributes to antibiotic resistance, which continues to be a major problem, especially in hospitalized patients.
There is a lack of consensus in the literature about methods to risk stratify patients who present with acute dermatologic conditions that include and resemble cellulitis. We sought to identify clinical features based on available clinical literature-derived variables. We tested our scheme in a series of patients with a known diagnosis of cellulitis or other dermatologic pathology of the lower extremity to assess the validity of the following 7 clinical criteria: acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis.
Materials and Methods
This retrospective chart review was approved by the Yale University (New Haven, Connecticut) institutional review board (HIC#1409014533). Final diagnosis, demographic data, clinical manifestations, and relevant diagnostic laboratory values of 57 patients were obtained from a database in the dermatology department’s consultation log and electronic medical record database (December 2011 to December 2014). The presence of each clinical symptom—acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis—was assigned a score equal to 1; values were tallied to achieve a final score for each patient (Table 1). Patients who were seen initially as a consultation for possible cellulitis but given a final diagnosis of stasis dermatitis or lipodermatosclerosis were included (Table 2).
Clinical Criteria
The clinical criteria were developed based largely on clinical experience and relevant secondary literature.15-17 At the patient encounter, presence of each of the variables (Table 1) was assessed according to the following definitions:
- acute onset: within the prior 72 hours and more indicative of an acute infective process than a gradual and chronic consequence of venous stasis
- erythema: a subjective clinical marker for inflammation that can be associated with cellulitis, though darker, erythematous-appearing discolorations also can be seen in patients with chronic venous hypertension or valvular incompetence4,15
- pyrexia: body temperature greater than 100.4°F
- history of associated trauma: encompassing mechanical wounds, surgical incisions, burns, and insect bites that correlate closely to the time course of symptomatic development
- tenderness: tenderness to light touch, which may be more common in patients afflicted with cellulitis than in those with venous insufficiency
- unilaterality: a helpful distinguishing feature that points the diagnosis away from a dermatitislike clinical picture, especially because bilateral cellulitis is rare and regarded as a diagnostic pitfall18
- leukocytosis: white blood cell count greater than 10.0×109/L and is reasonably considered a cardinal metric of inflammatory processes, though it can be confounded by immunocompromise (low count) or steroid use (high count)
Statistical Analysis
Odds ratios (ORs) were calculated and χ2 analysis was performed for each presenting symptom using JMP 10.0 analytical software (SAS Institute Inc). Each patient was rated separately by means of the clinical feature–based scoring system for the calculation of a total score. After application of the score to the patient population, receiver operating characteristic curves were constructed to identify the optimal score threshold for discriminating cellulitis from dermatitis in this group. For each clinical feature, P<.05 was considered significant.
Results
Our cohort included 32 male and 25 female patients with a mean age of 63 and 61 years, respectively. The final clinical diagnosis of cellulitis was made in 20 patients (35%). An established diagnosis of cellulitis was assigned based on a dermatology evaluation located within our electronic medical record database (Table 2).
Each clinical parameter was evaluated separately for each patient; combined results are summarized in Table 3. Acute onset (≤3 days) was a clinical characteristic seen in 80% (16/20) of cellulitis cases and 22% (8/37) of noncellulitis cases (OR, 14.5; P<.001). Erythema had similar significance (OR, 10.3; prevalence, 95% [19/20] vs 65% [24/37]; P=.012). Pyrexia possessed an OR of 99.2 for cellulitis and was seen in 85% (17/20) of cellulitis cases and only 5% (2/37) of noncellulitis cases (P<.001).
A history of associated trauma had an OR of 36.0 for cellulitis, with 50% (10/20) and 3% (1/37) prevalence in cellulitis cases and noncellulitis cases, respectively (P<.001). Tenderness, documented in 90% (18/20) of cellulitis cases and 43% (16/37) of noncellulitis cases, had an OR of 11.8 (P<.001).
Unilaterality had 100% (20/20) prevalence in our cellulitis cohort and was the only characteristic within the algorithm that yielded an incalculable OR. Noncellulitis or stasis dermatitis of the lower extremity exhibited a unilateral lesion in 11 cases (30%), of which 1 case resulted from a unilateral tibial fracture. Leukocytosis was seen in 65% (13/20) of cellulitis cases and 8% (3/37) of noncellulitis cases, with an OR for cellulitis of 21.0 (P<.001).
All parameters were significant by χ2 analysis (Table 3).
Comment
We found that testing positive for 4 of 7 clinical criteria for assessing cellulitis was highly specific (95%) and sensitive (100%) for a diagnosis of cellulitis among its range of mimics (Figure 3). These cellulitis criteria can be remembered, with some modification, using NEW HAvUN as a mnemonic device (New onset,
Consistent with the literature, pyrexia, history of associated trauma, and unilaterality also were predictors of cellulitis diagnosis. Unilaterality often is used as a diagnostic tool by dermatologist consultants when a patient lacks other criteria for cellulitis, so these findings are intuitive and consistent with our institutional experience. Interestingly, leukocytosis was seen in only 65% of cellulitis cases and 8% of noncellulitis cases and therefore might not serve as a sensitive independent predictor of a diagnosis of cellulitis, emphasizing the importance of the multifactorial scoring system we have put forward. Additionally, acuity of onset, erythema, and tenderness are not independently associated with cellulitis when assessing a patient because several of those findings are present in other dermatologic conditions of the lower extremity; when combined with the other criteria, however, these 3 findings can play a role in diagnosis.
Effective cellulitis diagnosis provides well-recognized challenges in the acute medical setting because many clinical mimics exist. The estimated rate of misdiagnosed cellulitis is certainly well-established: 30% to 75% in independent and multi-institutional studies. These studies also revealed that patients admitted for bilateral “cellulitis” overwhelmingly tended to be stasis clinical pictures.13,19
Cost implications from inappropriate diagnosis largely regard inappropriate antibiotic use and the potential for microbial resistance, with associated costs estimated to be more than $50 billion (2004 dollars).20,21 The true cost burden is extremely difficult to model or predict due to remarkable variations in the institutional misdiagnosis rate, prescribing pattern, and antibiotic cost and could represent avenues of further study. Misappropriation of antibiotics includes not only a monetary cost that encompasses all aspects of acute treatment and hospitalization but also an unquantifiable cost: human lives associated with the consequences of antibiotic resistance.
Conclusion
There is a lack of consensus or criteria for differentiating cellulitis from its most common clinical counterparts. Here, we propose a convenient clinical correlation system that we hope will lead to more efficient allocation of clinical resources, including antibiotics and hospital admissions, while lowering the incidence of adverse events and leading to better patient outcomes. We recognize that the small sample size of our study may limit broad application of these criteria, though we anticipate that further prospective studies can improve the diagnostic relevance and risk-assessment power of the NEW HAvUN criteria put forth here for assessing cellulitis in the acute medical setting.
Acknowledgement—Author H.H.E. recognizes the loving memory of Nadia Ezaldein for her profound influence on and motivation behind this research.
- Lep
pard BJ, Seal DV, Colman G, et al. The value of bacteriology and serology in the diagnosis of cellulitis and erysipelas. Br J Dermatol. 1985;112:559-567. - Hep
burn MJ, Dooley DP, Skidmore PJ, et al. Comparison of short-course (5 days) and standard (10 days) treatment for uncomplicated cellulitis. Arch Int Med. 2004;164:1669-1674. - Bergan JJ, Schmid-Schönbein GW, Smith PD, et al. Chronic venous disease. N Engl J Med. 2006;355:488-498.
- Bruc
e AJ, Bennett DD, Lohse CM, et al. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol. 2002;46:187-192. - Heym
ann WR. Lipodermatosclerosis. J Am Acad Dermatol. 2009;60:1022-1023. - Vesi
ć S, Vuković J, Medenica LJ, et al. Acute lipodermatosclerosis: an open clinical trial of stanozolol in patients unable to sustain compression therapy. Dermatol Online J. 2008;14:1. - Keller
EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleve Clin J Med. 2012;79:547-552. - Dong SL, Kelly KD, Oland RC, et al. ED management of cellulitis: a review of five urban centers. Am J Emerg Med. 2001;19:535-540.
- Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
- Manfredi R, Calza L, Chiodo F. Epidemiology and microbiology of cellulitis and bacterial soft tissue infection during HIV disease: a 10-year survey. J Cutan Pathol. 2002;29:168-172.
- Pascarella L, Schonbein GW, Bergan JJ. Microcirculation and venous ulcers: a review. Ann Vasc Surg. 2005;19:921-927.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician. 2003;67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J. 2011;17:1.
- Hay RJ, Adriaans BM. Bacterial infections. In: Thong BY, Tan TC. Epidemiology and risk factors for drug allergy. 8th ed. Br J Clin Pharmacol. 2011;71:684-700.
- Hay RJ, Adriaans BM. Bacterial infections. In: Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology. 8th ed. Hoboken, NJ: John Wiley & Sons, Inc; 2004:1345-1426.
- Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology In General Medicine. 7th ed. New York, NY: McGraw-Hill; 2003.
- Sommer LL, Reboli AC, Heymann WR. Bacterial infections. In: Bolognia J, Schaffer J, Cerroni L, et al. Dermatology. Vol 4. Philadelphia, PA: Elsevier Saunders; 2012:1462-1502.
- Cox NH. Management of lower leg cellulitis. Clin Med. 2002;2:23-27.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Pinder R, Sallis A, Berry D, et al. Behaviour change and antibiotic prescribing in healthcare settings: literature review and behavioural analysis. London, UK: Public Health England; February 2015. https://assets.publishing.service.gov.uk/government/
uploads/system/uploads/attachment_data/file/405031
/Behaviour_Change_for_Antibiotic_Prescribing_-_FINAL.pdf. Accessed May 7, 2018. - Smith R, Coast J. The true cost of antimicrobial resistance. BMJ. 2013;346:f1493.
- Lep
pard BJ, Seal DV, Colman G, et al. The value of bacteriology and serology in the diagnosis of cellulitis and erysipelas. Br J Dermatol. 1985;112:559-567. - Hep
burn MJ, Dooley DP, Skidmore PJ, et al. Comparison of short-course (5 days) and standard (10 days) treatment for uncomplicated cellulitis. Arch Int Med. 2004;164:1669-1674. - Bergan JJ, Schmid-Schönbein GW, Smith PD, et al. Chronic venous disease. N Engl J Med. 2006;355:488-498.
- Bruc
e AJ, Bennett DD, Lohse CM, et al. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol. 2002;46:187-192. - Heym
ann WR. Lipodermatosclerosis. J Am Acad Dermatol. 2009;60:1022-1023. - Vesi
ć S, Vuković J, Medenica LJ, et al. Acute lipodermatosclerosis: an open clinical trial of stanozolol in patients unable to sustain compression therapy. Dermatol Online J. 2008;14:1. - Keller
EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleve Clin J Med. 2012;79:547-552. - Dong SL, Kelly KD, Oland RC, et al. ED management of cellulitis: a review of five urban centers. Am J Emerg Med. 2001;19:535-540.
- Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
- Manfredi R, Calza L, Chiodo F. Epidemiology and microbiology of cellulitis and bacterial soft tissue infection during HIV disease: a 10-year survey. J Cutan Pathol. 2002;29:168-172.
- Pascarella L, Schonbein GW, Bergan JJ. Microcirculation and venous ulcers: a review. Ann Vasc Surg. 2005;19:921-927.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician. 2003;67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J. 2011;17:1.
- Hay RJ, Adriaans BM. Bacterial infections. In: Thong BY, Tan TC. Epidemiology and risk factors for drug allergy. 8th ed. Br J Clin Pharmacol. 2011;71:684-700.
- Hay RJ, Adriaans BM. Bacterial infections. In: Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology. 8th ed. Hoboken, NJ: John Wiley & Sons, Inc; 2004:1345-1426.
- Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology In General Medicine. 7th ed. New York, NY: McGraw-Hill; 2003.
- Sommer LL, Reboli AC, Heymann WR. Bacterial infections. In: Bolognia J, Schaffer J, Cerroni L, et al. Dermatology. Vol 4. Philadelphia, PA: Elsevier Saunders; 2012:1462-1502.
- Cox NH. Management of lower leg cellulitis. Clin Med. 2002;2:23-27.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Pinder R, Sallis A, Berry D, et al. Behaviour change and antibiotic prescribing in healthcare settings: literature review and behavioural analysis. London, UK: Public Health England; February 2015. https://assets.publishing.service.gov.uk/government/
uploads/system/uploads/attachment_data/file/405031
/Behaviour_Change_for_Antibiotic_Prescribing_-_FINAL.pdf. Accessed May 7, 2018. - Smith R, Coast J. The true cost of antimicrobial resistance. BMJ. 2013;346:f1493.
Practice Points
- Distinguishing cellulitis from noncellulitic conditions of the lower extremity is paramount to effective patient management in the emergent setting, given that misdiagnosis consumes hospital resources and can lead to inappropriate or excessive use of antibiotics.
- We evaluated the specificity and sensitivity of the following 7 clinical criteria: acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis.
Hypopigmentation on the Ear
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
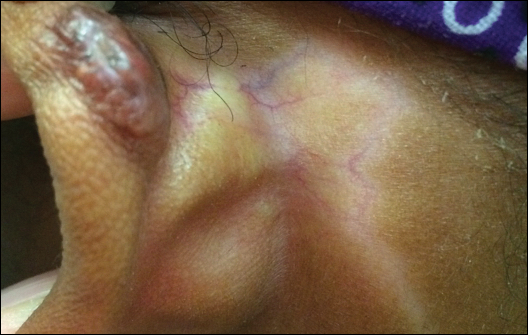
A 20-year-old black woman underwent multiple intralesional corticosteroid injections for treatment of a keloid on the superior aspect of the left helix and subsequently presented with a streak of atrophy and hypopigmentation in the postauricular region of unknown duration due to the lesion location.
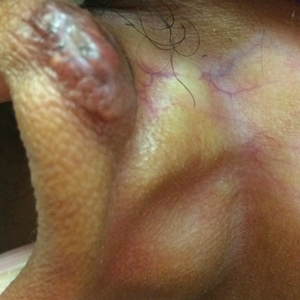
Ecthyma Gangrenosum Due to Pseudomonas fluorescens
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
...")
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
(H&E, original magnification ×100); biopsy from the periphery of the lesion showed leukocytoclastic vasculitis (B)...")
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
Practice Points
- Immunocompromised patients with a high exposure to agricultural products may be at increased risk for systemic infection by Pseudomonas fluorescens.
- Pseudomonas fluorescens is an opportunistic pathogen that can cause ecthyma gangrenosum, which necessitates rapid diagnosis and treatment to prevent mortality.
Human T-Lymphotropic Virus 1 Associated With Adult T-Cell Leukemia/Lymphoma
Adult T-cell leukemia/lymphoma (ATLL) is an uncommon neoplasm of mature T lymphocytes associated with infection by human T-lymphotropic virus 1 (HTLV-1),1-3 which is increasing in incidence in areas of the United States with large immigrant populations.4 Human T-lymphotrophic virus 1 infection is asymptomatic in most patients and has been associated with ATLL as well as tropical spastic paraparesis.5 We present a case of rapid-onset ATLL in an 82-year-old Japanese man who had immigrated to the United States.
Case Report
An 82-year-old Japanese man who had immigrated to the United States presented with papules and nodules on the neck, trunk, and arms of 4 weeks’ duration. Minimal pruritus was associated with the lesions, which were otherwise asymptomatic. The patient reported that he was generally healthy, and a review of systems was negative.
Physical examination revealed numerous erythematous and violaceous papules and nodules on the right side of the neck (Figure 1A), chest, back, abdomen, groin, left arm (Figure 1B), and medial thighs. Bilateral axillary and inguinal lymphadenopathy also was noted.
 and left arm (B) in a patient with adult T-cell leukemia/lymphoma.")
A biopsy from the abdomen revealed a dense, atypical, pandermal lymphoid infiltrate comprised of medium-sized lymphocytes with oval nuclei, fine chromatin, and pale cytoplasm (Figure 2). Mitotic figures and apoptotic cells also were observed. Immunostaining was strongly and diffusely positive for CD4 (Figure 3A), B-cell lymphoma 2 (Bcl-2)(Figure 3B), CD3, and programmed death 1, and was negative for CD8, CD10, CD20, CD30, and myeloperoxidase.
.")
(original magnification ×40) and B-cell lymphoma 2 (Bcl-2)(B)(original magnification ×40).")
A bone marrow biopsy revealed an atypical T-cell population on flow cytometry. Western blot analysis for HTLV-1 antibodies was positive. Complete blood cell count and complete metabolic panel were within reference range.
Clinical and histopathologic findings fit the diagnosis of ATLL. The patient was referred to hematology/oncology, but the rapid progression of lesions continued, and the patient died within 4 months of initial presentation.
Comment
Etiology
First described in 1977, ATLL is an uncommon neoplasm of mature T cells.6 The etiology is associated with infection by the retrovirus HTLV-1, which is endemic in Southern Japan, the Caribbean, Central and West Africa, and Central and South America, with increasing incidence in areas of the United States with large immigrant populations.7 The incidence of ATLL among all registered lymphoma cases from 2003 to 2008 in Japan was 8.3% compared to 0.2% in the United States.7
Transmission of HTLV-1
Human T-lymphotropic virus 1 is a retrovirus most commonly found in CD4+T cells and can be transmitted through breast milk, sexual intercourse, and blood exposure (eg, blood transfusion), with breastfeeding and blood exposure being the most common.8-10 Human T-lymphotrophic virus 1 has been described as the causative agent for 3 entities: (1) ATLL, (2) a nervous system degenerative disorder known as HTLV-1–associated myelopathy or tropical spastic paraparesis, and (3) HTLV-1 uveitis.5,11 It is thought that 10 to 20 million individuals worldwide are infected with HTLV-1.12
The evolution from infection with HTLV-1 to ATLL is thought to involve multiple steps.13,14 Those who contract the virus later in life rarely, if ever, develop ATLL, suggesting that this progression requires considerable time to evolve to carcinogenesis. More than 90% of those infected with HTLV-1 remain asymptomatic, while only 2% to 3% of women and 6% to 7% of men develop ATLL with a median incubation period greater than 15 to 20 years.7
Subtypes
Adult T-cell leukemia/lymphoma has been divided into 4 clinical subtypes based on clinical presentation and prognosis.15 The acute type is more aggressive and has a poorer prognosis, while the chronic and smoldering types have a more indolent course. The smoldering variant largely has only cutaneous involvement with less than 1% of the peripheral leukocytes being atypical lymphocytes.16 A cutaneous subtype in which few to no leukemic cells are present also has been described and may overlap with the smoldering variant.The cutaneous variant has been further classified into 2 subtypes, tumoral and erythematopapular, with the tumoral subtype carrying a worse prognosis.17,18 Clinically, 39% to 57% of ATLL cases have skin involvement, with nearly one-third reporting skin manifestations as the first symptom.19,20 The cutaneous manifestations vary greatly and may include papules, plaques, nodules, tumors, erythematous patches, or erythroderma.4,21 In addition to skin manifestations, most patients with acute ATLL demonstrate leukemia, lymphadenopathy, organomegaly, and hypercalcemia.22
Histopathology
Histologically, both the smoldering and chronic forms of tumoral or erythematopapular ATLL demonstrate a cutaneous, dermal, or subcutaneous infiltrate of small- to medium-sized CD4+ T cells with histiocytes and admixed granulomas.4 Epidermotropism and Pautrier microabscesses often are limited or absent but can be seen.
Differential Diagnosis
The differential diagnosis includes other small- or medium-sized T-cell lymphomas. The chronic and smoldering types can be difficult to distinguish from mycosis fungoides.
Treatment
Treatment decisions should be made based on the subclassification and prognostic factors at the time of diagnosis. High doses of interferon alfa and zidovudine may show some benefit, but many cases require multiagent chemotherapy.22 The only possible curative treatment is allogeneic stem cell transplant. Mogamulizumab, an antichemokine receptor 4 monoclonal antibody, has demonstrated some ATLL antitumor activity.24
- Uchiyama T, Yodoi J, Sagawa K, et al. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood. 1977;50:481-492.
- Poiesz BJ, Ruscetti FW, Gazdar AF, et al. Detection and isolation of type C retro-virus particles form fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A. 1980;77:7415-7419.
- Hinuma Y, Gotoh Y, Sugamura K, et al. A retrovirus associated with human adult T-cell leukemia: in vitro activation. Gan. 1982;73:341-344.
- Marchetti MA, Pulitzer MP, Myskowski PL, et al. Cutaneous manifestations of human T-cell lymphotropic virus type-1-associated adult T-cell leukemia/lymphoma: a single-center, retrospective study. J Am Acad Dermatol. 2015;72:293-301.
- Gessain A, Barin F, Vernant JC, et al. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet. 1985;2:407-410.
- Takatsuki K, Uchiyama T, Sagawa K, et al. Adult T cell leukemia in Japan. In: Seno S, Takasu F, Irino S, eds. Topics in Hematology. Amsterdam, Netherlands: Excerpta Medica; 1977:73-77.
- Yoshida N, Chihara D. Incidence of adult T-cell leukemia/lymphoma in nonendemic areas. Curr Treat Options Oncol. 2015;16:7.
- Tajima K, Tominaga S, Suchi T, et al. Epidemiological analysis of the distribution of antibody to adult T-cell leukemia-virus-associated antigen: possible horizontal transmission of adult T-cell leukemia virus. Gan. 1982;73:893-901.
- Kajiyama W, Kashiwagi S, Ikematsu H, et al. Intrafamilial transmission of adult T cell leukemia virus. J Infect Dis. 1986;154:851-857.
- Ichimaru M, Ikeda S, Kinoshita K, et al. Mother-to-child transmission of HTLV-1. Cancer Detect Prev. 1991;15:177-181.
- Lyra-da-Silva JO, de Mello Gonzaga YB, de Melo Espíndola O, et al. Adult t-cell leukemia/lymphoma: a case report of primary cutaneous tumoral type. Dermatol Pract Concept. 2012;2:202a03.
- Edlich RF, Arnette JA, Williams FM. Global epidemic of human T-cell lymphotropic virus type-I (HTLV-I). J Emerg Med. 2000;18:109-119.
- Magalhaes M, Oliveira PD, Bittencourt AL, et al. Microsatellite alterations are also present in the less aggressive types of adult T-cell leukemia-lymphoma. PLoS Negl Trop Dis. 2015;9:e0003403.
- Okamoto T, Ohno Y, Tsugane S, et al. Multi-step carcinogenesis model for adult T-cell leukemia. Jpn J Cancer Res. 1989;80:191-195.
- Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. Br J Haematol. 1991;79:428-437.
- Takahashi K, Tanaka T, Fujita M, et al. Cutaneous-type adult T-cell leukemia lymphoma. a unique clinical feature with monoclonal T-cell proliferation detected by Southern blot analysis Arch Dermatol. 1988;124:399-404.
- Amano M, Kurokawa M, Ogata K, et al. New entity, definition and diagnostic criteria of cutaneous adult T-cell leukemia/lymphoma: human T-lymphotropic virus type 1 proviral DNA load can distinguish between cutaneous and smoldering types. J Dermatol. 2008;35:270-275.
- Johno M, Ohishi M, Kojo Y, et al. Cutaneous manifestations of adult T-cell leukemia lymphoma. Gann Monogr Cancer Res. 1992;39:33-42.
- Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukemia-lymphoma: a report from the Lymphoma Study Group (1984-87). Br J Haematol. 1991;79:428-437.
- Levine PH, Manns A, Jaffe ES, et al. The effect of ethnic differences on the pattern of HTLV-I-associated T-cell leukemia/lymphoma (HATL) in the United States. Int J Cancer. 1994;56:177-181.
- Pezeshkpoor F, Yazdanpanah MJ, Shirdel A. Specific cutaneous manifestations in adult T-cell leukemia/lymphoma. Int J Dermatol. 2008;47:359-362.
- Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27:453-459.
- Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26:4124-4130.
- Ishida T, Joh T, Uike N, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012;30:837-842.
Adult T-cell leukemia/lymphoma (ATLL) is an uncommon neoplasm of mature T lymphocytes associated with infection by human T-lymphotropic virus 1 (HTLV-1),1-3 which is increasing in incidence in areas of the United States with large immigrant populations.4 Human T-lymphotrophic virus 1 infection is asymptomatic in most patients and has been associated with ATLL as well as tropical spastic paraparesis.5 We present a case of rapid-onset ATLL in an 82-year-old Japanese man who had immigrated to the United States.
Case Report
An 82-year-old Japanese man who had immigrated to the United States presented with papules and nodules on the neck, trunk, and arms of 4 weeks’ duration. Minimal pruritus was associated with the lesions, which were otherwise asymptomatic. The patient reported that he was generally healthy, and a review of systems was negative.
Physical examination revealed numerous erythematous and violaceous papules and nodules on the right side of the neck (Figure 1A), chest, back, abdomen, groin, left arm (Figure 1B), and medial thighs. Bilateral axillary and inguinal lymphadenopathy also was noted.
A biopsy from the abdomen revealed a dense, atypical, pandermal lymphoid infiltrate comprised of medium-sized lymphocytes with oval nuclei, fine chromatin, and pale cytoplasm (Figure 2). Mitotic figures and apoptotic cells also were observed. Immunostaining was strongly and diffusely positive for CD4 (Figure 3A), B-cell lymphoma 2 (Bcl-2)(Figure 3B), CD3, and programmed death 1, and was negative for CD8, CD10, CD20, CD30, and myeloperoxidase.
A bone marrow biopsy revealed an atypical T-cell population on flow cytometry. Western blot analysis for HTLV-1 antibodies was positive. Complete blood cell count and complete metabolic panel were within reference range.
Clinical and histopathologic findings fit the diagnosis of ATLL. The patient was referred to hematology/oncology, but the rapid progression of lesions continued, and the patient died within 4 months of initial presentation.
Comment
Etiology
First described in 1977, ATLL is an uncommon neoplasm of mature T cells.6 The etiology is associated with infection by the retrovirus HTLV-1, which is endemic in Southern Japan, the Caribbean, Central and West Africa, and Central and South America, with increasing incidence in areas of the United States with large immigrant populations.7 The incidence of ATLL among all registered lymphoma cases from 2003 to 2008 in Japan was 8.3% compared to 0.2% in the United States.7
Transmission of HTLV-1
Human T-lymphotropic virus 1 is a retrovirus most commonly found in CD4+T cells and can be transmitted through breast milk, sexual intercourse, and blood exposure (eg, blood transfusion), with breastfeeding and blood exposure being the most common.8-10 Human T-lymphotrophic virus 1 has been described as the causative agent for 3 entities: (1) ATLL, (2) a nervous system degenerative disorder known as HTLV-1–associated myelopathy or tropical spastic paraparesis, and (3) HTLV-1 uveitis.5,11 It is thought that 10 to 20 million individuals worldwide are infected with HTLV-1.12
The evolution from infection with HTLV-1 to ATLL is thought to involve multiple steps.13,14 Those who contract the virus later in life rarely, if ever, develop ATLL, suggesting that this progression requires considerable time to evolve to carcinogenesis. More than 90% of those infected with HTLV-1 remain asymptomatic, while only 2% to 3% of women and 6% to 7% of men develop ATLL with a median incubation period greater than 15 to 20 years.7
Subtypes
Adult T-cell leukemia/lymphoma has been divided into 4 clinical subtypes based on clinical presentation and prognosis.15 The acute type is more aggressive and has a poorer prognosis, while the chronic and smoldering types have a more indolent course. The smoldering variant largely has only cutaneous involvement with less than 1% of the peripheral leukocytes being atypical lymphocytes.16 A cutaneous subtype in which few to no leukemic cells are present also has been described and may overlap with the smoldering variant.The cutaneous variant has been further classified into 2 subtypes, tumoral and erythematopapular, with the tumoral subtype carrying a worse prognosis.17,18 Clinically, 39% to 57% of ATLL cases have skin involvement, with nearly one-third reporting skin manifestations as the first symptom.19,20 The cutaneous manifestations vary greatly and may include papules, plaques, nodules, tumors, erythematous patches, or erythroderma.4,21 In addition to skin manifestations, most patients with acute ATLL demonstrate leukemia, lymphadenopathy, organomegaly, and hypercalcemia.22
Histopathology
Histologically, both the smoldering and chronic forms of tumoral or erythematopapular ATLL demonstrate a cutaneous, dermal, or subcutaneous infiltrate of small- to medium-sized CD4+ T cells with histiocytes and admixed granulomas.4 Epidermotropism and Pautrier microabscesses often are limited or absent but can be seen.
Differential Diagnosis
The differential diagnosis includes other small- or medium-sized T-cell lymphomas. The chronic and smoldering types can be difficult to distinguish from mycosis fungoides.
Treatment
Treatment decisions should be made based on the subclassification and prognostic factors at the time of diagnosis. High doses of interferon alfa and zidovudine may show some benefit, but many cases require multiagent chemotherapy.22 The only possible curative treatment is allogeneic stem cell transplant. Mogamulizumab, an antichemokine receptor 4 monoclonal antibody, has demonstrated some ATLL antitumor activity.24
Adult T-cell leukemia/lymphoma (ATLL) is an uncommon neoplasm of mature T lymphocytes associated with infection by human T-lymphotropic virus 1 (HTLV-1),1-3 which is increasing in incidence in areas of the United States with large immigrant populations.4 Human T-lymphotrophic virus 1 infection is asymptomatic in most patients and has been associated with ATLL as well as tropical spastic paraparesis.5 We present a case of rapid-onset ATLL in an 82-year-old Japanese man who had immigrated to the United States.
Case Report
An 82-year-old Japanese man who had immigrated to the United States presented with papules and nodules on the neck, trunk, and arms of 4 weeks’ duration. Minimal pruritus was associated with the lesions, which were otherwise asymptomatic. The patient reported that he was generally healthy, and a review of systems was negative.
Physical examination revealed numerous erythematous and violaceous papules and nodules on the right side of the neck (Figure 1A), chest, back, abdomen, groin, left arm (Figure 1B), and medial thighs. Bilateral axillary and inguinal lymphadenopathy also was noted.
A biopsy from the abdomen revealed a dense, atypical, pandermal lymphoid infiltrate comprised of medium-sized lymphocytes with oval nuclei, fine chromatin, and pale cytoplasm (Figure 2). Mitotic figures and apoptotic cells also were observed. Immunostaining was strongly and diffusely positive for CD4 (Figure 3A), B-cell lymphoma 2 (Bcl-2)(Figure 3B), CD3, and programmed death 1, and was negative for CD8, CD10, CD20, CD30, and myeloperoxidase.
A bone marrow biopsy revealed an atypical T-cell population on flow cytometry. Western blot analysis for HTLV-1 antibodies was positive. Complete blood cell count and complete metabolic panel were within reference range.
Clinical and histopathologic findings fit the diagnosis of ATLL. The patient was referred to hematology/oncology, but the rapid progression of lesions continued, and the patient died within 4 months of initial presentation.
Comment
Etiology
First described in 1977, ATLL is an uncommon neoplasm of mature T cells.6 The etiology is associated with infection by the retrovirus HTLV-1, which is endemic in Southern Japan, the Caribbean, Central and West Africa, and Central and South America, with increasing incidence in areas of the United States with large immigrant populations.7 The incidence of ATLL among all registered lymphoma cases from 2003 to 2008 in Japan was 8.3% compared to 0.2% in the United States.7
Transmission of HTLV-1
Human T-lymphotropic virus 1 is a retrovirus most commonly found in CD4+T cells and can be transmitted through breast milk, sexual intercourse, and blood exposure (eg, blood transfusion), with breastfeeding and blood exposure being the most common.8-10 Human T-lymphotrophic virus 1 has been described as the causative agent for 3 entities: (1) ATLL, (2) a nervous system degenerative disorder known as HTLV-1–associated myelopathy or tropical spastic paraparesis, and (3) HTLV-1 uveitis.5,11 It is thought that 10 to 20 million individuals worldwide are infected with HTLV-1.12
The evolution from infection with HTLV-1 to ATLL is thought to involve multiple steps.13,14 Those who contract the virus later in life rarely, if ever, develop ATLL, suggesting that this progression requires considerable time to evolve to carcinogenesis. More than 90% of those infected with HTLV-1 remain asymptomatic, while only 2% to 3% of women and 6% to 7% of men develop ATLL with a median incubation period greater than 15 to 20 years.7
Subtypes
Adult T-cell leukemia/lymphoma has been divided into 4 clinical subtypes based on clinical presentation and prognosis.15 The acute type is more aggressive and has a poorer prognosis, while the chronic and smoldering types have a more indolent course. The smoldering variant largely has only cutaneous involvement with less than 1% of the peripheral leukocytes being atypical lymphocytes.16 A cutaneous subtype in which few to no leukemic cells are present also has been described and may overlap with the smoldering variant.The cutaneous variant has been further classified into 2 subtypes, tumoral and erythematopapular, with the tumoral subtype carrying a worse prognosis.17,18 Clinically, 39% to 57% of ATLL cases have skin involvement, with nearly one-third reporting skin manifestations as the first symptom.19,20 The cutaneous manifestations vary greatly and may include papules, plaques, nodules, tumors, erythematous patches, or erythroderma.4,21 In addition to skin manifestations, most patients with acute ATLL demonstrate leukemia, lymphadenopathy, organomegaly, and hypercalcemia.22
Histopathology
Histologically, both the smoldering and chronic forms of tumoral or erythematopapular ATLL demonstrate a cutaneous, dermal, or subcutaneous infiltrate of small- to medium-sized CD4+ T cells with histiocytes and admixed granulomas.4 Epidermotropism and Pautrier microabscesses often are limited or absent but can be seen.
Differential Diagnosis
The differential diagnosis includes other small- or medium-sized T-cell lymphomas. The chronic and smoldering types can be difficult to distinguish from mycosis fungoides.
Treatment
Treatment decisions should be made based on the subclassification and prognostic factors at the time of diagnosis. High doses of interferon alfa and zidovudine may show some benefit, but many cases require multiagent chemotherapy.22 The only possible curative treatment is allogeneic stem cell transplant. Mogamulizumab, an antichemokine receptor 4 monoclonal antibody, has demonstrated some ATLL antitumor activity.24
- Uchiyama T, Yodoi J, Sagawa K, et al. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood. 1977;50:481-492.
- Poiesz BJ, Ruscetti FW, Gazdar AF, et al. Detection and isolation of type C retro-virus particles form fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A. 1980;77:7415-7419.
- Hinuma Y, Gotoh Y, Sugamura K, et al. A retrovirus associated with human adult T-cell leukemia: in vitro activation. Gan. 1982;73:341-344.
- Marchetti MA, Pulitzer MP, Myskowski PL, et al. Cutaneous manifestations of human T-cell lymphotropic virus type-1-associated adult T-cell leukemia/lymphoma: a single-center, retrospective study. J Am Acad Dermatol. 2015;72:293-301.
- Gessain A, Barin F, Vernant JC, et al. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet. 1985;2:407-410.
- Takatsuki K, Uchiyama T, Sagawa K, et al. Adult T cell leukemia in Japan. In: Seno S, Takasu F, Irino S, eds. Topics in Hematology. Amsterdam, Netherlands: Excerpta Medica; 1977:73-77.
- Yoshida N, Chihara D. Incidence of adult T-cell leukemia/lymphoma in nonendemic areas. Curr Treat Options Oncol. 2015;16:7.
- Tajima K, Tominaga S, Suchi T, et al. Epidemiological analysis of the distribution of antibody to adult T-cell leukemia-virus-associated antigen: possible horizontal transmission of adult T-cell leukemia virus. Gan. 1982;73:893-901.
- Kajiyama W, Kashiwagi S, Ikematsu H, et al. Intrafamilial transmission of adult T cell leukemia virus. J Infect Dis. 1986;154:851-857.
- Ichimaru M, Ikeda S, Kinoshita K, et al. Mother-to-child transmission of HTLV-1. Cancer Detect Prev. 1991;15:177-181.
- Lyra-da-Silva JO, de Mello Gonzaga YB, de Melo Espíndola O, et al. Adult t-cell leukemia/lymphoma: a case report of primary cutaneous tumoral type. Dermatol Pract Concept. 2012;2:202a03.
- Edlich RF, Arnette JA, Williams FM. Global epidemic of human T-cell lymphotropic virus type-I (HTLV-I). J Emerg Med. 2000;18:109-119.
- Magalhaes M, Oliveira PD, Bittencourt AL, et al. Microsatellite alterations are also present in the less aggressive types of adult T-cell leukemia-lymphoma. PLoS Negl Trop Dis. 2015;9:e0003403.
- Okamoto T, Ohno Y, Tsugane S, et al. Multi-step carcinogenesis model for adult T-cell leukemia. Jpn J Cancer Res. 1989;80:191-195.
- Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. Br J Haematol. 1991;79:428-437.
- Takahashi K, Tanaka T, Fujita M, et al. Cutaneous-type adult T-cell leukemia lymphoma. a unique clinical feature with monoclonal T-cell proliferation detected by Southern blot analysis Arch Dermatol. 1988;124:399-404.
- Amano M, Kurokawa M, Ogata K, et al. New entity, definition and diagnostic criteria of cutaneous adult T-cell leukemia/lymphoma: human T-lymphotropic virus type 1 proviral DNA load can distinguish between cutaneous and smoldering types. J Dermatol. 2008;35:270-275.
- Johno M, Ohishi M, Kojo Y, et al. Cutaneous manifestations of adult T-cell leukemia lymphoma. Gann Monogr Cancer Res. 1992;39:33-42.
- Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukemia-lymphoma: a report from the Lymphoma Study Group (1984-87). Br J Haematol. 1991;79:428-437.
- Levine PH, Manns A, Jaffe ES, et al. The effect of ethnic differences on the pattern of HTLV-I-associated T-cell leukemia/lymphoma (HATL) in the United States. Int J Cancer. 1994;56:177-181.
- Pezeshkpoor F, Yazdanpanah MJ, Shirdel A. Specific cutaneous manifestations in adult T-cell leukemia/lymphoma. Int J Dermatol. 2008;47:359-362.
- Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27:453-459.
- Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26:4124-4130.
- Ishida T, Joh T, Uike N, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012;30:837-842.
- Uchiyama T, Yodoi J, Sagawa K, et al. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood. 1977;50:481-492.
- Poiesz BJ, Ruscetti FW, Gazdar AF, et al. Detection and isolation of type C retro-virus particles form fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A. 1980;77:7415-7419.
- Hinuma Y, Gotoh Y, Sugamura K, et al. A retrovirus associated with human adult T-cell leukemia: in vitro activation. Gan. 1982;73:341-344.
- Marchetti MA, Pulitzer MP, Myskowski PL, et al. Cutaneous manifestations of human T-cell lymphotropic virus type-1-associated adult T-cell leukemia/lymphoma: a single-center, retrospective study. J Am Acad Dermatol. 2015;72:293-301.
- Gessain A, Barin F, Vernant JC, et al. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet. 1985;2:407-410.
- Takatsuki K, Uchiyama T, Sagawa K, et al. Adult T cell leukemia in Japan. In: Seno S, Takasu F, Irino S, eds. Topics in Hematology. Amsterdam, Netherlands: Excerpta Medica; 1977:73-77.
- Yoshida N, Chihara D. Incidence of adult T-cell leukemia/lymphoma in nonendemic areas. Curr Treat Options Oncol. 2015;16:7.
- Tajima K, Tominaga S, Suchi T, et al. Epidemiological analysis of the distribution of antibody to adult T-cell leukemia-virus-associated antigen: possible horizontal transmission of adult T-cell leukemia virus. Gan. 1982;73:893-901.
- Kajiyama W, Kashiwagi S, Ikematsu H, et al. Intrafamilial transmission of adult T cell leukemia virus. J Infect Dis. 1986;154:851-857.
- Ichimaru M, Ikeda S, Kinoshita K, et al. Mother-to-child transmission of HTLV-1. Cancer Detect Prev. 1991;15:177-181.
- Lyra-da-Silva JO, de Mello Gonzaga YB, de Melo Espíndola O, et al. Adult t-cell leukemia/lymphoma: a case report of primary cutaneous tumoral type. Dermatol Pract Concept. 2012;2:202a03.
- Edlich RF, Arnette JA, Williams FM. Global epidemic of human T-cell lymphotropic virus type-I (HTLV-I). J Emerg Med. 2000;18:109-119.
- Magalhaes M, Oliveira PD, Bittencourt AL, et al. Microsatellite alterations are also present in the less aggressive types of adult T-cell leukemia-lymphoma. PLoS Negl Trop Dis. 2015;9:e0003403.
- Okamoto T, Ohno Y, Tsugane S, et al. Multi-step carcinogenesis model for adult T-cell leukemia. Jpn J Cancer Res. 1989;80:191-195.
- Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. Br J Haematol. 1991;79:428-437.
- Takahashi K, Tanaka T, Fujita M, et al. Cutaneous-type adult T-cell leukemia lymphoma. a unique clinical feature with monoclonal T-cell proliferation detected by Southern blot analysis Arch Dermatol. 1988;124:399-404.
- Amano M, Kurokawa M, Ogata K, et al. New entity, definition and diagnostic criteria of cutaneous adult T-cell leukemia/lymphoma: human T-lymphotropic virus type 1 proviral DNA load can distinguish between cutaneous and smoldering types. J Dermatol. 2008;35:270-275.
- Johno M, Ohishi M, Kojo Y, et al. Cutaneous manifestations of adult T-cell leukemia lymphoma. Gann Monogr Cancer Res. 1992;39:33-42.
- Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukemia-lymphoma: a report from the Lymphoma Study Group (1984-87). Br J Haematol. 1991;79:428-437.
- Levine PH, Manns A, Jaffe ES, et al. The effect of ethnic differences on the pattern of HTLV-I-associated T-cell leukemia/lymphoma (HATL) in the United States. Int J Cancer. 1994;56:177-181.
- Pezeshkpoor F, Yazdanpanah MJ, Shirdel A. Specific cutaneous manifestations in adult T-cell leukemia/lymphoma. Int J Dermatol. 2008;47:359-362.
- Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27:453-459.
- Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26:4124-4130.
- Ishida T, Joh T, Uike N, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012;30:837-842.
Practice Points
- Adult T-cell leukemia/lymphoma (ATLL) is an uncommon neoplasm of mature T lymphocytes associated with infection by human T-lymphotropic virus 1.
- In the United States, ATLL is increasing in incidence in areas with large immigrant populations.
- High suspicion and clinical features must be present to make the diagnosis of ATLL due to considerable histologic overlap with other cutaneous T-cell lymphomas.
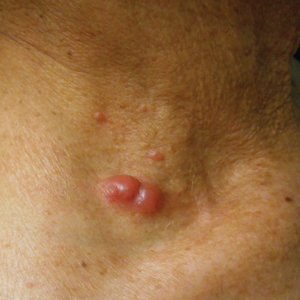
Debunking Atopic Dermatitis Myths: Can Adults Develop Eczema?
Myth: Atopic Dermatitis Does Not Start in Adulthood
Atopic dermatitis (AD) typically first appears in childhood and tends to disappear before puberty begins; however, some patients experience AD that persists into adulthood or occurs de novo. Bannister and Freeman coined the term adult-onset atopic dermatitis after reviewing 2604 cases of AD and noting that 243 patients (9%) were first diagnosed with AD at 20 years of age or older. Adult-onset AD may be its own subset of AD or childhood AD that was simply not diagnosed until adulthood or was forgotten by the patient.
Characteristically, AD presents in adults as inflammatory eczema with areas of lichenification. It could occur after a change in residence to a cold dry climate or exposure to central heating, as patients who grew up in warm, sunny, humid climates might not have had diagnosable AD in childhood or adolescence. The more common forms of adult-onset AD are hand and neck dermatitis, hand eczema, nummular eczema, or prurigo, while childhood AD often manifests in a flexural distribution. Because it is difficult to detect, adult-onset AD is diagnosed after ruling out other diseases. Diagnostic procedures, such as patch tests, skin prick tests, biopsies, or blood screenings, usually are necessary to rule out other diseases or types of eczema. Contact eczema is the first diagnostic sign of AD in adults.
Maintaining AD in the differential diagnosis for patients with clinical symptoms of pruritus and eczema is essential due to the quality of life impact of the condition. Sleep disturbance is common in adults with severe AD and treatment may help to improve sleep quality.
Hanifin suggested the following when assessing adults for AD:
- Verify diagnosis (not allergic contact dermatitis or psoriasis)
- Determine patient's history of allergies (eg, food allergy) or childhood eczema
- Obtain family history of eczema/allergies
- Evaluate if patient's occupation may impact condition (eg, contact with irritants or known contact allergens)
- Inquire about patient's childhood residence (eg, tropical climate)
Adult-onset AD is a recalcitrant condition that can be difficult to treat, and appropriately labeling/diagnosing the condition will lead to better management.
Expert Commentary
Whereas once it was the rite of the pediatric AD patient to outgrow disease, it has now become clear that resolution is not as hard a stop in atopic disease as expected. Adult-onset and persistent disease in AD is clearly a significant problem, especially in developed nations and carries a host of comorbidites. Time and enhanced research will hopefully identify interventions to reverse the trend towards persistence into adulthood.
—Nanette B. Silverberg, MD (New York, New York)
Bannister MJ, Freeman S. Adult-onset atopic dermatitis. Australas J Dermatol. 2000;41:225-228.
Hanifin JM. Adult-onset atopic dermatitis: fact or fancy? Dermatol Clin. 2017;35:299-302.
Silvestre Salvador JF, Romero-Pérez D, Encabo-Durán B. Atopic dermatitis in adults: a diagnostic challenge. J Investig Allergol Clin Immunol. 2017;27:78-88.
Myth: Atopic Dermatitis Does Not Start in Adulthood
Atopic dermatitis (AD) typically first appears in childhood and tends to disappear before puberty begins; however, some patients experience AD that persists into adulthood or occurs de novo. Bannister and Freeman coined the term adult-onset atopic dermatitis after reviewing 2604 cases of AD and noting that 243 patients (9%) were first diagnosed with AD at 20 years of age or older. Adult-onset AD may be its own subset of AD or childhood AD that was simply not diagnosed until adulthood or was forgotten by the patient.
Characteristically, AD presents in adults as inflammatory eczema with areas of lichenification. It could occur after a change in residence to a cold dry climate or exposure to central heating, as patients who grew up in warm, sunny, humid climates might not have had diagnosable AD in childhood or adolescence. The more common forms of adult-onset AD are hand and neck dermatitis, hand eczema, nummular eczema, or prurigo, while childhood AD often manifests in a flexural distribution. Because it is difficult to detect, adult-onset AD is diagnosed after ruling out other diseases. Diagnostic procedures, such as patch tests, skin prick tests, biopsies, or blood screenings, usually are necessary to rule out other diseases or types of eczema. Contact eczema is the first diagnostic sign of AD in adults.
Maintaining AD in the differential diagnosis for patients with clinical symptoms of pruritus and eczema is essential due to the quality of life impact of the condition. Sleep disturbance is common in adults with severe AD and treatment may help to improve sleep quality.
Hanifin suggested the following when assessing adults for AD:
- Verify diagnosis (not allergic contact dermatitis or psoriasis)
- Determine patient's history of allergies (eg, food allergy) or childhood eczema
- Obtain family history of eczema/allergies
- Evaluate if patient's occupation may impact condition (eg, contact with irritants or known contact allergens)
- Inquire about patient's childhood residence (eg, tropical climate)
Adult-onset AD is a recalcitrant condition that can be difficult to treat, and appropriately labeling/diagnosing the condition will lead to better management.
Expert Commentary
Whereas once it was the rite of the pediatric AD patient to outgrow disease, it has now become clear that resolution is not as hard a stop in atopic disease as expected. Adult-onset and persistent disease in AD is clearly a significant problem, especially in developed nations and carries a host of comorbidites. Time and enhanced research will hopefully identify interventions to reverse the trend towards persistence into adulthood.
—Nanette B. Silverberg, MD (New York, New York)
Myth: Atopic Dermatitis Does Not Start in Adulthood
Atopic dermatitis (AD) typically first appears in childhood and tends to disappear before puberty begins; however, some patients experience AD that persists into adulthood or occurs de novo. Bannister and Freeman coined the term adult-onset atopic dermatitis after reviewing 2604 cases of AD and noting that 243 patients (9%) were first diagnosed with AD at 20 years of age or older. Adult-onset AD may be its own subset of AD or childhood AD that was simply not diagnosed until adulthood or was forgotten by the patient.
Characteristically, AD presents in adults as inflammatory eczema with areas of lichenification. It could occur after a change in residence to a cold dry climate or exposure to central heating, as patients who grew up in warm, sunny, humid climates might not have had diagnosable AD in childhood or adolescence. The more common forms of adult-onset AD are hand and neck dermatitis, hand eczema, nummular eczema, or prurigo, while childhood AD often manifests in a flexural distribution. Because it is difficult to detect, adult-onset AD is diagnosed after ruling out other diseases. Diagnostic procedures, such as patch tests, skin prick tests, biopsies, or blood screenings, usually are necessary to rule out other diseases or types of eczema. Contact eczema is the first diagnostic sign of AD in adults.
Maintaining AD in the differential diagnosis for patients with clinical symptoms of pruritus and eczema is essential due to the quality of life impact of the condition. Sleep disturbance is common in adults with severe AD and treatment may help to improve sleep quality.
Hanifin suggested the following when assessing adults for AD:
- Verify diagnosis (not allergic contact dermatitis or psoriasis)
- Determine patient's history of allergies (eg, food allergy) or childhood eczema
- Obtain family history of eczema/allergies
- Evaluate if patient's occupation may impact condition (eg, contact with irritants or known contact allergens)
- Inquire about patient's childhood residence (eg, tropical climate)
Adult-onset AD is a recalcitrant condition that can be difficult to treat, and appropriately labeling/diagnosing the condition will lead to better management.
Expert Commentary
Whereas once it was the rite of the pediatric AD patient to outgrow disease, it has now become clear that resolution is not as hard a stop in atopic disease as expected. Adult-onset and persistent disease in AD is clearly a significant problem, especially in developed nations and carries a host of comorbidites. Time and enhanced research will hopefully identify interventions to reverse the trend towards persistence into adulthood.
—Nanette B. Silverberg, MD (New York, New York)
Bannister MJ, Freeman S. Adult-onset atopic dermatitis. Australas J Dermatol. 2000;41:225-228.
Hanifin JM. Adult-onset atopic dermatitis: fact or fancy? Dermatol Clin. 2017;35:299-302.
Silvestre Salvador JF, Romero-Pérez D, Encabo-Durán B. Atopic dermatitis in adults: a diagnostic challenge. J Investig Allergol Clin Immunol. 2017;27:78-88.
Bannister MJ, Freeman S. Adult-onset atopic dermatitis. Australas J Dermatol. 2000;41:225-228.
Hanifin JM. Adult-onset atopic dermatitis: fact or fancy? Dermatol Clin. 2017;35:299-302.
Silvestre Salvador JF, Romero-Pérez D, Encabo-Durán B. Atopic dermatitis in adults: a diagnostic challenge. J Investig Allergol Clin Immunol. 2017;27:78-88.

