User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Pediatric Subungual Exostosis
Exostosis is a type of benign bone tumor in which trabecular (spongy) bone overgrows its normal border in a nodular pattern. 1,2 Histologically, it usually is surrounded by a fibrocartilaginous cap. 3 It is most commonly found on the lateral or medial aspect of the foot and is thought to be caused by trauma, either physical pressure or infection. 4 When this lesion is found under the nail bed, it is termed subungual exostosis ( Dupuytren exostosis ) . 3 Sequelae of a subungual exostosis include nail dystrophy and lifting of the nail away from the toe, in addition to infection and possible loss of the toenail (onycholysis). There are only 2 genetic conditions related to exostosis: hereditary multiple exostosis and multiple exostoses-mental retardation syndrome.
An exostosis may appear to be a wart on first inspection. It may present similar to osteochondromas, and the only way to get a true diagnosis is by biopsy of the lesion. The treatment for an exostosis is surgery. The surgeon must remove the lesion at the base of the bone from which it grows to prevent recurrence of the lesion.5
Because exostosis may cause nail bed disruption, the differential diagnosis may include nail deformities, such as traumatic onycholysis, onychogryphosis, verrucae, subungual infection, or nail trauma.6,7
Case Report
A 7-year-old boy presented with changes of the right great toenail over the last 4 months. The patient noted that the affected nail was discolored, dystrophic, painful, and thickened. He did not recall prior trauma to the affected nail, and his mother stated that the lesion was growing and becoming more painful with a throbbing sensation at times. He described the pain as stabbing, which was exacerbated while walking and playing sports. Neither the patient nor his family had ever had any similar condition. He was not taking any medications, only a daily multivitamin. He had a history of eczematous dermatitis and keratosis pilaris without any other medical illnesses. He had a family history of psoriasis; however, no prior instances of exostosis had been reported. He had no medication allergies.
A full-body cutaneous and nail examination showed a well-developed, well-nourished boy who was in no acute distress. A firm, subungual, pink, pearly,hyperkeratotic nodule was appreciated on the right great toe (Figure 1). The lesion was tender to palpation. The rest of the examination and review of systems were normal.
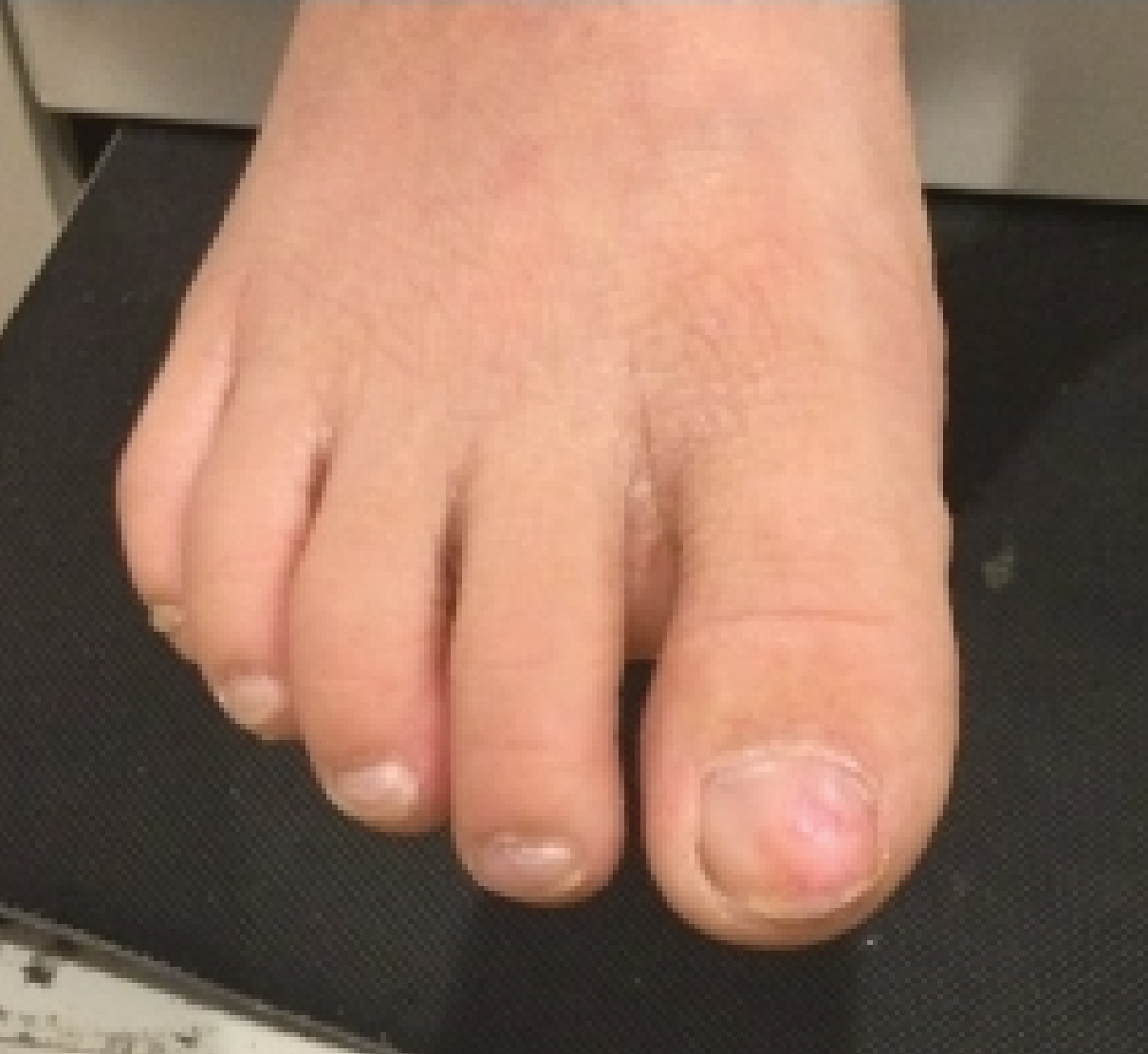
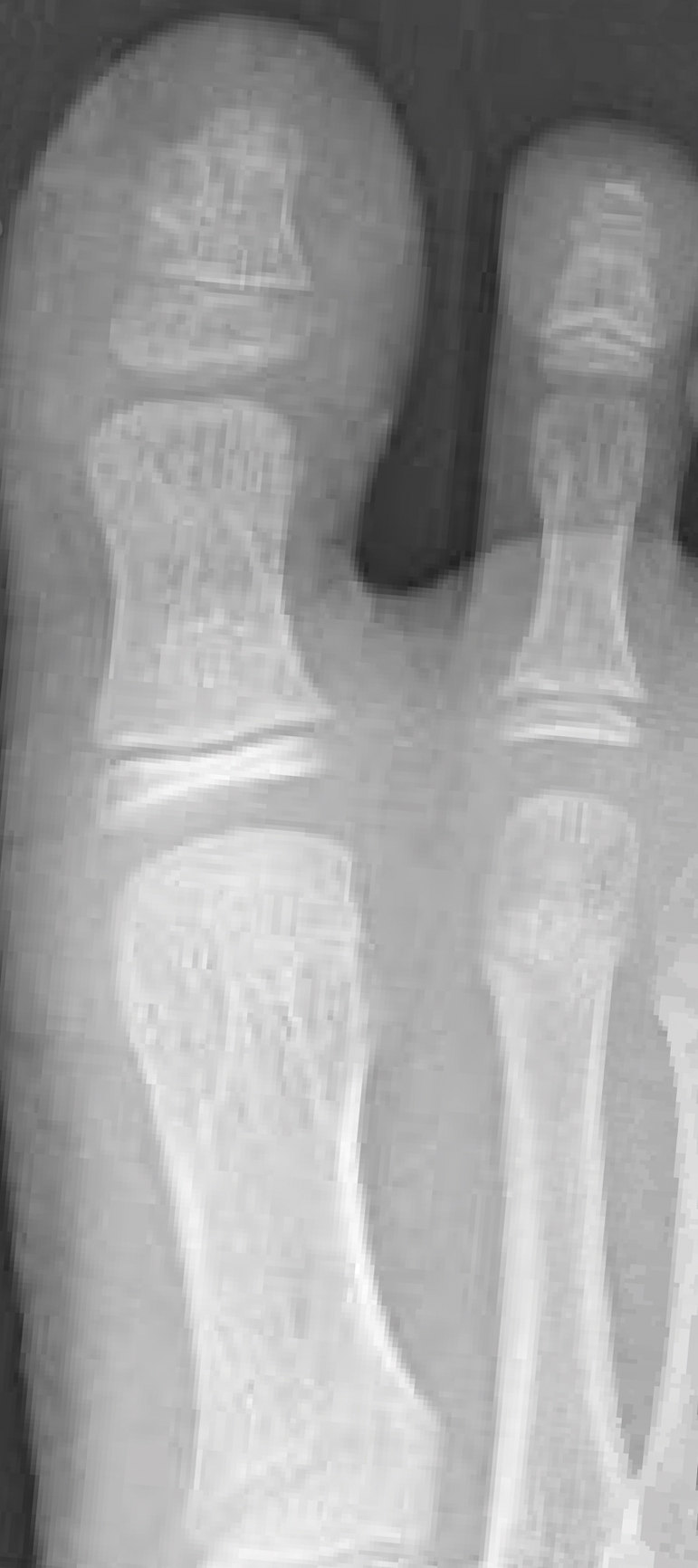
From the clinical findings, a differential diagnosis of glomus tumor, hemangioma, and infection was considered. Periodic acid–Schiff stain was negative, which ruled out fungal infection. Nail avulsion and a shave biopsy were performed under general anesthesia. There was an exostosis arising from the dorsal aspect of the great toe measuring approximately 5 mm in width at the base and approximately 1 mm in height, which endorsed a diagnosis of distal phalanx subungual exostosis. A postsurgery radiograph (Figure 2) showed residual bone below the level of shave removal at the nail bed.
Comment
Exostosis is most commonly found on the lateral or medial aspect of the hallux (great toe) in patients younger than 18 years.8 Diagnosis often is obvious, even without a radiograph or biopsy, because the exostosis comes out from under the tip of the nail. Our case was interesting because the patient was a child, and the exostosis did not lift the nail or extrude from the distal tip of the nail bed. Evidence suggests that a greater-than-expected genetic influence contributes to an exostosis, though further investigation is needed to determine all of the causes and risk factors for subungual bony exostosis. Timely diagnosis and treatment are essential to the prevention of sequelae of the disease, such as toe infection or chronic pain.
- de Palma L, Gigante A, Specchia N. Subungual exostosis of the foot. Foot Ankle Int. 1996;17:758-763. doi:10.1177/107110079601701208
- Multhopp-Stephens H, Walling AK. Subungual (Dupuytren’s) exostosis. J Pediatr Orthop. 1995;15:582-584. doi:10.1097/01241398-199509000-00006
- Davis DA, Cohen PR. Subungual exostosis: case report and review of the literature. Pediatr Dermatol. 1996;13:212-218.
- Guarneri C, Guarneri F, Risitano G, et al. Solitary asymptomatic nodule of the great toe. Int J Dermatol. 2005;44:245-247.
- Letts M, Davidson D, Nizalik E. Subungual exostosis: diagnosis and treatment in children. J Trauma. 1998;44:346-349.
- Hoy NY, Leung AKC, Metelitsa AI, et al. New concepts in median nail dystrophy, onychomycosis, and hand, foot, and mouth disease nail pathology. ISRN Dermatol. 2012;2012:680163.
- Rich P, Scher RK. Examination of the nail and work-up of nail conditions. In: Rich P, Scher RK, eds. An Atlas of Diseases of the Nail. Parthenon Publishing; 2003.
- DaCambra MP, Gupta SK, Ferri-de-Barros F. Subungual exostosis of the toes: a systematic review. Clin Orthop Relat Res. 2014;472:1251-1259. doi:10.1007/s11999-013-3345-4
Exostosis is a type of benign bone tumor in which trabecular (spongy) bone overgrows its normal border in a nodular pattern. 1,2 Histologically, it usually is surrounded by a fibrocartilaginous cap. 3 It is most commonly found on the lateral or medial aspect of the foot and is thought to be caused by trauma, either physical pressure or infection. 4 When this lesion is found under the nail bed, it is termed subungual exostosis ( Dupuytren exostosis ) . 3 Sequelae of a subungual exostosis include nail dystrophy and lifting of the nail away from the toe, in addition to infection and possible loss of the toenail (onycholysis). There are only 2 genetic conditions related to exostosis: hereditary multiple exostosis and multiple exostoses-mental retardation syndrome.
An exostosis may appear to be a wart on first inspection. It may present similar to osteochondromas, and the only way to get a true diagnosis is by biopsy of the lesion. The treatment for an exostosis is surgery. The surgeon must remove the lesion at the base of the bone from which it grows to prevent recurrence of the lesion.5
Because exostosis may cause nail bed disruption, the differential diagnosis may include nail deformities, such as traumatic onycholysis, onychogryphosis, verrucae, subungual infection, or nail trauma.6,7
Case Report
A 7-year-old boy presented with changes of the right great toenail over the last 4 months. The patient noted that the affected nail was discolored, dystrophic, painful, and thickened. He did not recall prior trauma to the affected nail, and his mother stated that the lesion was growing and becoming more painful with a throbbing sensation at times. He described the pain as stabbing, which was exacerbated while walking and playing sports. Neither the patient nor his family had ever had any similar condition. He was not taking any medications, only a daily multivitamin. He had a history of eczematous dermatitis and keratosis pilaris without any other medical illnesses. He had a family history of psoriasis; however, no prior instances of exostosis had been reported. He had no medication allergies.
A full-body cutaneous and nail examination showed a well-developed, well-nourished boy who was in no acute distress. A firm, subungual, pink, pearly,hyperkeratotic nodule was appreciated on the right great toe (Figure 1). The lesion was tender to palpation. The rest of the examination and review of systems were normal.
From the clinical findings, a differential diagnosis of glomus tumor, hemangioma, and infection was considered. Periodic acid–Schiff stain was negative, which ruled out fungal infection. Nail avulsion and a shave biopsy were performed under general anesthesia. There was an exostosis arising from the dorsal aspect of the great toe measuring approximately 5 mm in width at the base and approximately 1 mm in height, which endorsed a diagnosis of distal phalanx subungual exostosis. A postsurgery radiograph (Figure 2) showed residual bone below the level of shave removal at the nail bed.
Comment
Exostosis is most commonly found on the lateral or medial aspect of the hallux (great toe) in patients younger than 18 years.8 Diagnosis often is obvious, even without a radiograph or biopsy, because the exostosis comes out from under the tip of the nail. Our case was interesting because the patient was a child, and the exostosis did not lift the nail or extrude from the distal tip of the nail bed. Evidence suggests that a greater-than-expected genetic influence contributes to an exostosis, though further investigation is needed to determine all of the causes and risk factors for subungual bony exostosis. Timely diagnosis and treatment are essential to the prevention of sequelae of the disease, such as toe infection or chronic pain.
Exostosis is a type of benign bone tumor in which trabecular (spongy) bone overgrows its normal border in a nodular pattern. 1,2 Histologically, it usually is surrounded by a fibrocartilaginous cap. 3 It is most commonly found on the lateral or medial aspect of the foot and is thought to be caused by trauma, either physical pressure or infection. 4 When this lesion is found under the nail bed, it is termed subungual exostosis ( Dupuytren exostosis ) . 3 Sequelae of a subungual exostosis include nail dystrophy and lifting of the nail away from the toe, in addition to infection and possible loss of the toenail (onycholysis). There are only 2 genetic conditions related to exostosis: hereditary multiple exostosis and multiple exostoses-mental retardation syndrome.
An exostosis may appear to be a wart on first inspection. It may present similar to osteochondromas, and the only way to get a true diagnosis is by biopsy of the lesion. The treatment for an exostosis is surgery. The surgeon must remove the lesion at the base of the bone from which it grows to prevent recurrence of the lesion.5
Because exostosis may cause nail bed disruption, the differential diagnosis may include nail deformities, such as traumatic onycholysis, onychogryphosis, verrucae, subungual infection, or nail trauma.6,7
Case Report
A 7-year-old boy presented with changes of the right great toenail over the last 4 months. The patient noted that the affected nail was discolored, dystrophic, painful, and thickened. He did not recall prior trauma to the affected nail, and his mother stated that the lesion was growing and becoming more painful with a throbbing sensation at times. He described the pain as stabbing, which was exacerbated while walking and playing sports. Neither the patient nor his family had ever had any similar condition. He was not taking any medications, only a daily multivitamin. He had a history of eczematous dermatitis and keratosis pilaris without any other medical illnesses. He had a family history of psoriasis; however, no prior instances of exostosis had been reported. He had no medication allergies.
A full-body cutaneous and nail examination showed a well-developed, well-nourished boy who was in no acute distress. A firm, subungual, pink, pearly,hyperkeratotic nodule was appreciated on the right great toe (Figure 1). The lesion was tender to palpation. The rest of the examination and review of systems were normal.
From the clinical findings, a differential diagnosis of glomus tumor, hemangioma, and infection was considered. Periodic acid–Schiff stain was negative, which ruled out fungal infection. Nail avulsion and a shave biopsy were performed under general anesthesia. There was an exostosis arising from the dorsal aspect of the great toe measuring approximately 5 mm in width at the base and approximately 1 mm in height, which endorsed a diagnosis of distal phalanx subungual exostosis. A postsurgery radiograph (Figure 2) showed residual bone below the level of shave removal at the nail bed.
Comment
Exostosis is most commonly found on the lateral or medial aspect of the hallux (great toe) in patients younger than 18 years.8 Diagnosis often is obvious, even without a radiograph or biopsy, because the exostosis comes out from under the tip of the nail. Our case was interesting because the patient was a child, and the exostosis did not lift the nail or extrude from the distal tip of the nail bed. Evidence suggests that a greater-than-expected genetic influence contributes to an exostosis, though further investigation is needed to determine all of the causes and risk factors for subungual bony exostosis. Timely diagnosis and treatment are essential to the prevention of sequelae of the disease, such as toe infection or chronic pain.
- de Palma L, Gigante A, Specchia N. Subungual exostosis of the foot. Foot Ankle Int. 1996;17:758-763. doi:10.1177/107110079601701208
- Multhopp-Stephens H, Walling AK. Subungual (Dupuytren’s) exostosis. J Pediatr Orthop. 1995;15:582-584. doi:10.1097/01241398-199509000-00006
- Davis DA, Cohen PR. Subungual exostosis: case report and review of the literature. Pediatr Dermatol. 1996;13:212-218.
- Guarneri C, Guarneri F, Risitano G, et al. Solitary asymptomatic nodule of the great toe. Int J Dermatol. 2005;44:245-247.
- Letts M, Davidson D, Nizalik E. Subungual exostosis: diagnosis and treatment in children. J Trauma. 1998;44:346-349.
- Hoy NY, Leung AKC, Metelitsa AI, et al. New concepts in median nail dystrophy, onychomycosis, and hand, foot, and mouth disease nail pathology. ISRN Dermatol. 2012;2012:680163.
- Rich P, Scher RK. Examination of the nail and work-up of nail conditions. In: Rich P, Scher RK, eds. An Atlas of Diseases of the Nail. Parthenon Publishing; 2003.
- DaCambra MP, Gupta SK, Ferri-de-Barros F. Subungual exostosis of the toes: a systematic review. Clin Orthop Relat Res. 2014;472:1251-1259. doi:10.1007/s11999-013-3345-4
- de Palma L, Gigante A, Specchia N. Subungual exostosis of the foot. Foot Ankle Int. 1996;17:758-763. doi:10.1177/107110079601701208
- Multhopp-Stephens H, Walling AK. Subungual (Dupuytren’s) exostosis. J Pediatr Orthop. 1995;15:582-584. doi:10.1097/01241398-199509000-00006
- Davis DA, Cohen PR. Subungual exostosis: case report and review of the literature. Pediatr Dermatol. 1996;13:212-218.
- Guarneri C, Guarneri F, Risitano G, et al. Solitary asymptomatic nodule of the great toe. Int J Dermatol. 2005;44:245-247.
- Letts M, Davidson D, Nizalik E. Subungual exostosis: diagnosis and treatment in children. J Trauma. 1998;44:346-349.
- Hoy NY, Leung AKC, Metelitsa AI, et al. New concepts in median nail dystrophy, onychomycosis, and hand, foot, and mouth disease nail pathology. ISRN Dermatol. 2012;2012:680163.
- Rich P, Scher RK. Examination of the nail and work-up of nail conditions. In: Rich P, Scher RK, eds. An Atlas of Diseases of the Nail. Parthenon Publishing; 2003.
- DaCambra MP, Gupta SK, Ferri-de-Barros F. Subungual exostosis of the toes: a systematic review. Clin Orthop Relat Res. 2014;472:1251-1259. doi:10.1007/s11999-013-3345-4
Practice Points
- Nail dystrophy can have a variety of causes, most commonly trauma, onychomycosis, verrucae, or subungual exostosis.
- Exostosis is a benign osteochondral tumor commonly found on the lateral or medial aspect of the hallux (great toe) in pediatric and young adult patients.
- A radiograph can be used as a preliminary tool for diagnosis, but subungual exostosis must be confirmed by biopsy or tissue histology at the time of excision.
Phototoxicity Secondary to Home Fireplace Exposure After Photodynamic Therapy for Actinic Keratosis
To the Editor:
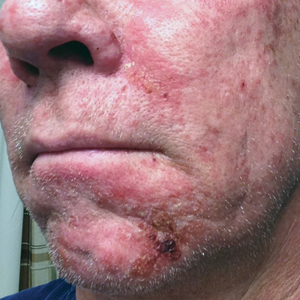
Photodynamic therapy (PDT) is a US Food and Drug Administration–approved treatment for actinic keratosis (AK). It also commonly is administered off label for basal cell carcinoma, Bowen disease, photoaging, and acne vulgaris and is being investigated for other applications.1,2 In the context of treating AK, the mechanism employed in PDT most commonly involves the application of exogenous aminolevulinic acid (ALA), which is metabolized to the endogenous photosensitizer protoporphyrin IX (PpIX) in skin cells by enzymes in the heme biosynthetic pathway.3 The preferential uptake of ALA and conversion to PpIX is due to the altered and increased permeability of abnormal keratin layers of aging, sun-damaged cells, and skin tumors. Selectivity of ALA also occurs due to the preferential intracellular accumulation of PpIX in proliferating, relatively iron–deficient, precancerous and cancerous cells. The therapeutic effect is achieved with light exposure to blue light wavelength at 417 nm and corresponds to the excitation peak of PpIX,4 which activates PpIX and forms reactive oxygen species in the presence of oxygen that ultimately cause cell necrosis and apoptosis.5 Because it takes approximately 24 hours for PpIX to be completely metabolized from the skin, patients are counseled to avoid sun or artificial light exposure in the first 24 hours post-PDT, regardless of the indication, to avoid a severe phototoxic reaction.3,6,7 Although it is normal and desirable for patients to experience some form of a phototoxic reaction, which may include erythema, edema, crusting, vesiculation, or erosion in most patients, these types of reactions most often are secondary to the intended exposure and incidental natural or artificial light exposures.6 We report a case of a severe phototoxic reaction in which a patient experienced painful erythema and purulence on the left side of the chin after being within an arm’s length of a flame in a fireplace following PDT treatment.
A 59-year-old man presented to our dermatology clinic for his second of 3 PDT sessions to treat AKs on the face. He had a history of a basal cell carcinoma on the left nasolabial fold that previously was treated with Mohs micrographic surgery and melanoma on the left ear that was previously treated with excision. The patient received the initial PDT session 1 month prior and experienced a mild reaction with minimal redness and peeling that resolved in 4 to 5 days. For the second treatment, per standard protocol at our clinic, ALA was applied to the face, after which the patient incubated for 1 hour prior to blue light exposure (mean [SD] peak output of 417 [5] nm for 1000 seconds and 10 J/cm2).
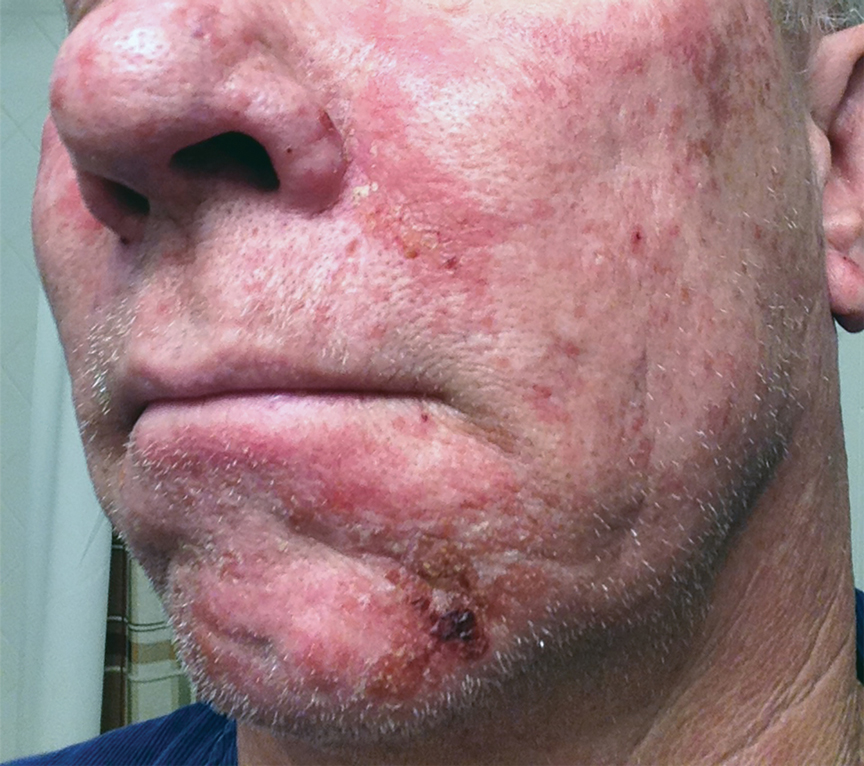
After blue light exposure, broad-spectrum sunscreen (sun protection factor 47) was applied to our patient’s face, and he wore a wide-brimmed hat upon leaving the clinic and walking to his car. Similar to the first PDT session 1 month prior, he experienced minimal pain immediately after treatment. Once home and approximately 4 to 5 hours after PDT, he tended to a fire using his left hand and leaned into the fireplace with the left side of his face, which was within an arm’s length of the flames. Although his skin did not come in direct contact with the flames, the brief 2- to 3-minute exposure to the flame’s light and heat produced an immediate intense burning pain that the patient likened to the pain of blue light exposure. Within 24 hours, he developed a severe inflammatory reaction that included erythema, edema, desquamation, and pustules on the left side of the chin and cheek that produced a purulent discharge (Figure). The purulence resolved the next day; however, the other clinical manifestations persisted for 1 week. Despite the discomfort and symptoms, our patient did not seek medical attention and instead managed his symptoms conservatively with cold compresses. Although he noticed an overall subjective improvement in the appearance of his face after this second treatment, he received a third treatment with PDT approximately 1 month later, which resulted in a response that was similar to his first visit.
Photodynamic therapy is an increasingly accepted treatment modality for a plethora of benign and malignant dermatologic conditions. Although blue and red light are the most common light sources utilized with PDT because their wavelengths (404–420 nm and 635 nm, respectively) correspond to the excitation peaks of photosensitizers, alternative light sources increasingly are being explored. There is increasing interest in utilizing infrared (IR) light sources (700–1,000,000 nm) to penetrate deeper into the skin in the treatment of precancerous and cancerous lesions. Exposure to IR radiation is known to raise skin temperature via inside-out dermal water absorption and is thought to be useful in PDT-ALA by promoting ALA penetration and its conversion to PpIX.8 In a randomized controlled trial by Giehl et al,9 visible light plus water-filtered IR-A light was shown to produce considerably less pain in ALA-PDT compared to placebo, though efficacy was not statistically affected. There are burgeoning trials examining the role of IR in treating dermatologic conditions such as acne, but research is still needed on ALA-PDT activated by IR radiation to target AKs.
Although the PDT side-effect profile of phototoxicity, dyspigmentation, and hypersensitivity is well documented, phototoxicity secondary to flame exposure is rare. In our patient, the synergistic effect of light and heat produced an exuberant phototoxic reaction. As the applications for PDT continue to broaden, this case may represent the importance of addressing additional precautions, such as avoiding open flames in the house or while camping, in the PDT aftercare instructions to maximize patient safety.
- Fritsch C, Ruzicka T. Fluorescence diagnosis and photodynamic therapy in dermatology from experimental state to clinic standard methods. J Environ Pathol Toxicol Oncol. 2006;25:425-439.
- Lang K, Schulte KW, Ruzicka T, et al. Aminolevulinic acid (Levulan)in photodynamic therapy of actinic keratoses. Skin Therapy Lett. 2001;6:1-2, 5.
- Kennedy JC, Pottier RH. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J Photochem Photobiol B. 1992;14:275-292.
- Wan MT, Lin JY. Current evidence and applications of photodynamic therapy in dermatology. Clin Cosmet Investig Dermatol. 2014;7:145-163.
- Gad F, Viau G, Boushira M, et al. Photodynamic therapy with 5-aminolevulinic acid induces apoptosis and caspase activation in malignant T cells. J Cutan Med Surg. 2001;5:8-13.
- Piacquadio DJ, Chen DM, Farber HF, et al. Photodynamic therapy with aminolevulinic acid topical solution and visible blue light in the treatment of multiple actinic keratoses of the face and scalp: investigator-blinded, phase 3, multicenter trials. Arch Dermatol. 2004;140:41-46.
- Rhodes LE, Tsoukas MM, Anderson RR, et al. Iontophoretic delivery of ALA provides a quantitative model for ALA pharmacokinetics and PpIX phototoxicity in human skin. J Invest Dermatol. 1997;108:87-91.
- Dover JS, Phillips TJ, Arndt KA. Cutaneous effects and therapeutic uses of heat with emphasis on infrared radiation. J Am Acad Dermatol. 1989;20(2, pt 1):278-286.
- Giehl KA, Kriz M, Grahovac M, et al. A controlled trial of photodynamic therapy of actinic keratosis comparing different red light sources. Eur J Dermatol. 2014;24:335-341.
To the Editor:
Photodynamic therapy (PDT) is a US Food and Drug Administration–approved treatment for actinic keratosis (AK). It also commonly is administered off label for basal cell carcinoma, Bowen disease, photoaging, and acne vulgaris and is being investigated for other applications.1,2 In the context of treating AK, the mechanism employed in PDT most commonly involves the application of exogenous aminolevulinic acid (ALA), which is metabolized to the endogenous photosensitizer protoporphyrin IX (PpIX) in skin cells by enzymes in the heme biosynthetic pathway.3 The preferential uptake of ALA and conversion to PpIX is due to the altered and increased permeability of abnormal keratin layers of aging, sun-damaged cells, and skin tumors. Selectivity of ALA also occurs due to the preferential intracellular accumulation of PpIX in proliferating, relatively iron–deficient, precancerous and cancerous cells. The therapeutic effect is achieved with light exposure to blue light wavelength at 417 nm and corresponds to the excitation peak of PpIX,4 which activates PpIX and forms reactive oxygen species in the presence of oxygen that ultimately cause cell necrosis and apoptosis.5 Because it takes approximately 24 hours for PpIX to be completely metabolized from the skin, patients are counseled to avoid sun or artificial light exposure in the first 24 hours post-PDT, regardless of the indication, to avoid a severe phototoxic reaction.3,6,7 Although it is normal and desirable for patients to experience some form of a phototoxic reaction, which may include erythema, edema, crusting, vesiculation, or erosion in most patients, these types of reactions most often are secondary to the intended exposure and incidental natural or artificial light exposures.6 We report a case of a severe phototoxic reaction in which a patient experienced painful erythema and purulence on the left side of the chin after being within an arm’s length of a flame in a fireplace following PDT treatment.
A 59-year-old man presented to our dermatology clinic for his second of 3 PDT sessions to treat AKs on the face. He had a history of a basal cell carcinoma on the left nasolabial fold that previously was treated with Mohs micrographic surgery and melanoma on the left ear that was previously treated with excision. The patient received the initial PDT session 1 month prior and experienced a mild reaction with minimal redness and peeling that resolved in 4 to 5 days. For the second treatment, per standard protocol at our clinic, ALA was applied to the face, after which the patient incubated for 1 hour prior to blue light exposure (mean [SD] peak output of 417 [5] nm for 1000 seconds and 10 J/cm2).
After blue light exposure, broad-spectrum sunscreen (sun protection factor 47) was applied to our patient’s face, and he wore a wide-brimmed hat upon leaving the clinic and walking to his car. Similar to the first PDT session 1 month prior, he experienced minimal pain immediately after treatment. Once home and approximately 4 to 5 hours after PDT, he tended to a fire using his left hand and leaned into the fireplace with the left side of his face, which was within an arm’s length of the flames. Although his skin did not come in direct contact with the flames, the brief 2- to 3-minute exposure to the flame’s light and heat produced an immediate intense burning pain that the patient likened to the pain of blue light exposure. Within 24 hours, he developed a severe inflammatory reaction that included erythema, edema, desquamation, and pustules on the left side of the chin and cheek that produced a purulent discharge (Figure). The purulence resolved the next day; however, the other clinical manifestations persisted for 1 week. Despite the discomfort and symptoms, our patient did not seek medical attention and instead managed his symptoms conservatively with cold compresses. Although he noticed an overall subjective improvement in the appearance of his face after this second treatment, he received a third treatment with PDT approximately 1 month later, which resulted in a response that was similar to his first visit.
Photodynamic therapy is an increasingly accepted treatment modality for a plethora of benign and malignant dermatologic conditions. Although blue and red light are the most common light sources utilized with PDT because their wavelengths (404–420 nm and 635 nm, respectively) correspond to the excitation peaks of photosensitizers, alternative light sources increasingly are being explored. There is increasing interest in utilizing infrared (IR) light sources (700–1,000,000 nm) to penetrate deeper into the skin in the treatment of precancerous and cancerous lesions. Exposure to IR radiation is known to raise skin temperature via inside-out dermal water absorption and is thought to be useful in PDT-ALA by promoting ALA penetration and its conversion to PpIX.8 In a randomized controlled trial by Giehl et al,9 visible light plus water-filtered IR-A light was shown to produce considerably less pain in ALA-PDT compared to placebo, though efficacy was not statistically affected. There are burgeoning trials examining the role of IR in treating dermatologic conditions such as acne, but research is still needed on ALA-PDT activated by IR radiation to target AKs.
Although the PDT side-effect profile of phototoxicity, dyspigmentation, and hypersensitivity is well documented, phototoxicity secondary to flame exposure is rare. In our patient, the synergistic effect of light and heat produced an exuberant phototoxic reaction. As the applications for PDT continue to broaden, this case may represent the importance of addressing additional precautions, such as avoiding open flames in the house or while camping, in the PDT aftercare instructions to maximize patient safety.
To the Editor:
Photodynamic therapy (PDT) is a US Food and Drug Administration–approved treatment for actinic keratosis (AK). It also commonly is administered off label for basal cell carcinoma, Bowen disease, photoaging, and acne vulgaris and is being investigated for other applications.1,2 In the context of treating AK, the mechanism employed in PDT most commonly involves the application of exogenous aminolevulinic acid (ALA), which is metabolized to the endogenous photosensitizer protoporphyrin IX (PpIX) in skin cells by enzymes in the heme biosynthetic pathway.3 The preferential uptake of ALA and conversion to PpIX is due to the altered and increased permeability of abnormal keratin layers of aging, sun-damaged cells, and skin tumors. Selectivity of ALA also occurs due to the preferential intracellular accumulation of PpIX in proliferating, relatively iron–deficient, precancerous and cancerous cells. The therapeutic effect is achieved with light exposure to blue light wavelength at 417 nm and corresponds to the excitation peak of PpIX,4 which activates PpIX and forms reactive oxygen species in the presence of oxygen that ultimately cause cell necrosis and apoptosis.5 Because it takes approximately 24 hours for PpIX to be completely metabolized from the skin, patients are counseled to avoid sun or artificial light exposure in the first 24 hours post-PDT, regardless of the indication, to avoid a severe phototoxic reaction.3,6,7 Although it is normal and desirable for patients to experience some form of a phototoxic reaction, which may include erythema, edema, crusting, vesiculation, or erosion in most patients, these types of reactions most often are secondary to the intended exposure and incidental natural or artificial light exposures.6 We report a case of a severe phototoxic reaction in which a patient experienced painful erythema and purulence on the left side of the chin after being within an arm’s length of a flame in a fireplace following PDT treatment.
A 59-year-old man presented to our dermatology clinic for his second of 3 PDT sessions to treat AKs on the face. He had a history of a basal cell carcinoma on the left nasolabial fold that previously was treated with Mohs micrographic surgery and melanoma on the left ear that was previously treated with excision. The patient received the initial PDT session 1 month prior and experienced a mild reaction with minimal redness and peeling that resolved in 4 to 5 days. For the second treatment, per standard protocol at our clinic, ALA was applied to the face, after which the patient incubated for 1 hour prior to blue light exposure (mean [SD] peak output of 417 [5] nm for 1000 seconds and 10 J/cm2).
After blue light exposure, broad-spectrum sunscreen (sun protection factor 47) was applied to our patient’s face, and he wore a wide-brimmed hat upon leaving the clinic and walking to his car. Similar to the first PDT session 1 month prior, he experienced minimal pain immediately after treatment. Once home and approximately 4 to 5 hours after PDT, he tended to a fire using his left hand and leaned into the fireplace with the left side of his face, which was within an arm’s length of the flames. Although his skin did not come in direct contact with the flames, the brief 2- to 3-minute exposure to the flame’s light and heat produced an immediate intense burning pain that the patient likened to the pain of blue light exposure. Within 24 hours, he developed a severe inflammatory reaction that included erythema, edema, desquamation, and pustules on the left side of the chin and cheek that produced a purulent discharge (Figure). The purulence resolved the next day; however, the other clinical manifestations persisted for 1 week. Despite the discomfort and symptoms, our patient did not seek medical attention and instead managed his symptoms conservatively with cold compresses. Although he noticed an overall subjective improvement in the appearance of his face after this second treatment, he received a third treatment with PDT approximately 1 month later, which resulted in a response that was similar to his first visit.
Photodynamic therapy is an increasingly accepted treatment modality for a plethora of benign and malignant dermatologic conditions. Although blue and red light are the most common light sources utilized with PDT because their wavelengths (404–420 nm and 635 nm, respectively) correspond to the excitation peaks of photosensitizers, alternative light sources increasingly are being explored. There is increasing interest in utilizing infrared (IR) light sources (700–1,000,000 nm) to penetrate deeper into the skin in the treatment of precancerous and cancerous lesions. Exposure to IR radiation is known to raise skin temperature via inside-out dermal water absorption and is thought to be useful in PDT-ALA by promoting ALA penetration and its conversion to PpIX.8 In a randomized controlled trial by Giehl et al,9 visible light plus water-filtered IR-A light was shown to produce considerably less pain in ALA-PDT compared to placebo, though efficacy was not statistically affected. There are burgeoning trials examining the role of IR in treating dermatologic conditions such as acne, but research is still needed on ALA-PDT activated by IR radiation to target AKs.
Although the PDT side-effect profile of phototoxicity, dyspigmentation, and hypersensitivity is well documented, phototoxicity secondary to flame exposure is rare. In our patient, the synergistic effect of light and heat produced an exuberant phototoxic reaction. As the applications for PDT continue to broaden, this case may represent the importance of addressing additional precautions, such as avoiding open flames in the house or while camping, in the PDT aftercare instructions to maximize patient safety.
- Fritsch C, Ruzicka T. Fluorescence diagnosis and photodynamic therapy in dermatology from experimental state to clinic standard methods. J Environ Pathol Toxicol Oncol. 2006;25:425-439.
- Lang K, Schulte KW, Ruzicka T, et al. Aminolevulinic acid (Levulan)in photodynamic therapy of actinic keratoses. Skin Therapy Lett. 2001;6:1-2, 5.
- Kennedy JC, Pottier RH. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J Photochem Photobiol B. 1992;14:275-292.
- Wan MT, Lin JY. Current evidence and applications of photodynamic therapy in dermatology. Clin Cosmet Investig Dermatol. 2014;7:145-163.
- Gad F, Viau G, Boushira M, et al. Photodynamic therapy with 5-aminolevulinic acid induces apoptosis and caspase activation in malignant T cells. J Cutan Med Surg. 2001;5:8-13.
- Piacquadio DJ, Chen DM, Farber HF, et al. Photodynamic therapy with aminolevulinic acid topical solution and visible blue light in the treatment of multiple actinic keratoses of the face and scalp: investigator-blinded, phase 3, multicenter trials. Arch Dermatol. 2004;140:41-46.
- Rhodes LE, Tsoukas MM, Anderson RR, et al. Iontophoretic delivery of ALA provides a quantitative model for ALA pharmacokinetics and PpIX phototoxicity in human skin. J Invest Dermatol. 1997;108:87-91.
- Dover JS, Phillips TJ, Arndt KA. Cutaneous effects and therapeutic uses of heat with emphasis on infrared radiation. J Am Acad Dermatol. 1989;20(2, pt 1):278-286.
- Giehl KA, Kriz M, Grahovac M, et al. A controlled trial of photodynamic therapy of actinic keratosis comparing different red light sources. Eur J Dermatol. 2014;24:335-341.
- Fritsch C, Ruzicka T. Fluorescence diagnosis and photodynamic therapy in dermatology from experimental state to clinic standard methods. J Environ Pathol Toxicol Oncol. 2006;25:425-439.
- Lang K, Schulte KW, Ruzicka T, et al. Aminolevulinic acid (Levulan)in photodynamic therapy of actinic keratoses. Skin Therapy Lett. 2001;6:1-2, 5.
- Kennedy JC, Pottier RH. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J Photochem Photobiol B. 1992;14:275-292.
- Wan MT, Lin JY. Current evidence and applications of photodynamic therapy in dermatology. Clin Cosmet Investig Dermatol. 2014;7:145-163.
- Gad F, Viau G, Boushira M, et al. Photodynamic therapy with 5-aminolevulinic acid induces apoptosis and caspase activation in malignant T cells. J Cutan Med Surg. 2001;5:8-13.
- Piacquadio DJ, Chen DM, Farber HF, et al. Photodynamic therapy with aminolevulinic acid topical solution and visible blue light in the treatment of multiple actinic keratoses of the face and scalp: investigator-blinded, phase 3, multicenter trials. Arch Dermatol. 2004;140:41-46.
- Rhodes LE, Tsoukas MM, Anderson RR, et al. Iontophoretic delivery of ALA provides a quantitative model for ALA pharmacokinetics and PpIX phototoxicity in human skin. J Invest Dermatol. 1997;108:87-91.
- Dover JS, Phillips TJ, Arndt KA. Cutaneous effects and therapeutic uses of heat with emphasis on infrared radiation. J Am Acad Dermatol. 1989;20(2, pt 1):278-286.
- Giehl KA, Kriz M, Grahovac M, et al. A controlled trial of photodynamic therapy of actinic keratosis comparing different red light sources. Eur J Dermatol. 2014;24:335-341.
Practice Points
- As the applications of photodynamic therapy (PDT) in dermatology continue to expand, it is imperative for providers and patients alike to be knowledgeable with aftercare instructions and potential adverse effects.
- Avoid open flames in the house or while camping following PDT to maximize patient safety and prevent phototoxicity.
Early Pilomatrix Carcinoma: A Case Report With Emphasis on Molecular Pathology and Review of the Literature
Pilomatrix carcinoma is a rare adnexal tumor with origin from the germinative matrical cells of the hair follicle. Clinically, it presents as a solitary lesion commonly found in the head and neck region as well as the upper back. The tumors cannot be distinguished by their clinical appearance only and frequently are mistaken for cysts. Histopathologic examination provides the definitive diagnosis in most cases. These carcinomas are aggressive neoplasms with a high probability of local recurrence and distant metastasis. Assessment of the Wnt signaling pathway components such as β-catenin, lymphoid enhancer-binding factor 1 (LEF-1), and caudal-related homeobox transcription factor 2 (CDX-2) potentially can be used for diagnostic purposes and targeted therapy.
We report a rare and unique case of early pilomatrix carcinoma with intralesional melanocytes. We review the molecular pathology and pathogenesis of these carcinomas as well as the significance of early diagnosis.
Case Report
A 73-year-old man with a history of extensive sun exposure presented with a 1-cm, raised, rapidly growing, slightly irregular, purple lesion on the right forearm of 3 months’ duration with tendency to bleed. He did not have a history of skin cancers and was otherwise healthy. Excision was recommended due to the progressive and rapid growth of the lesion.
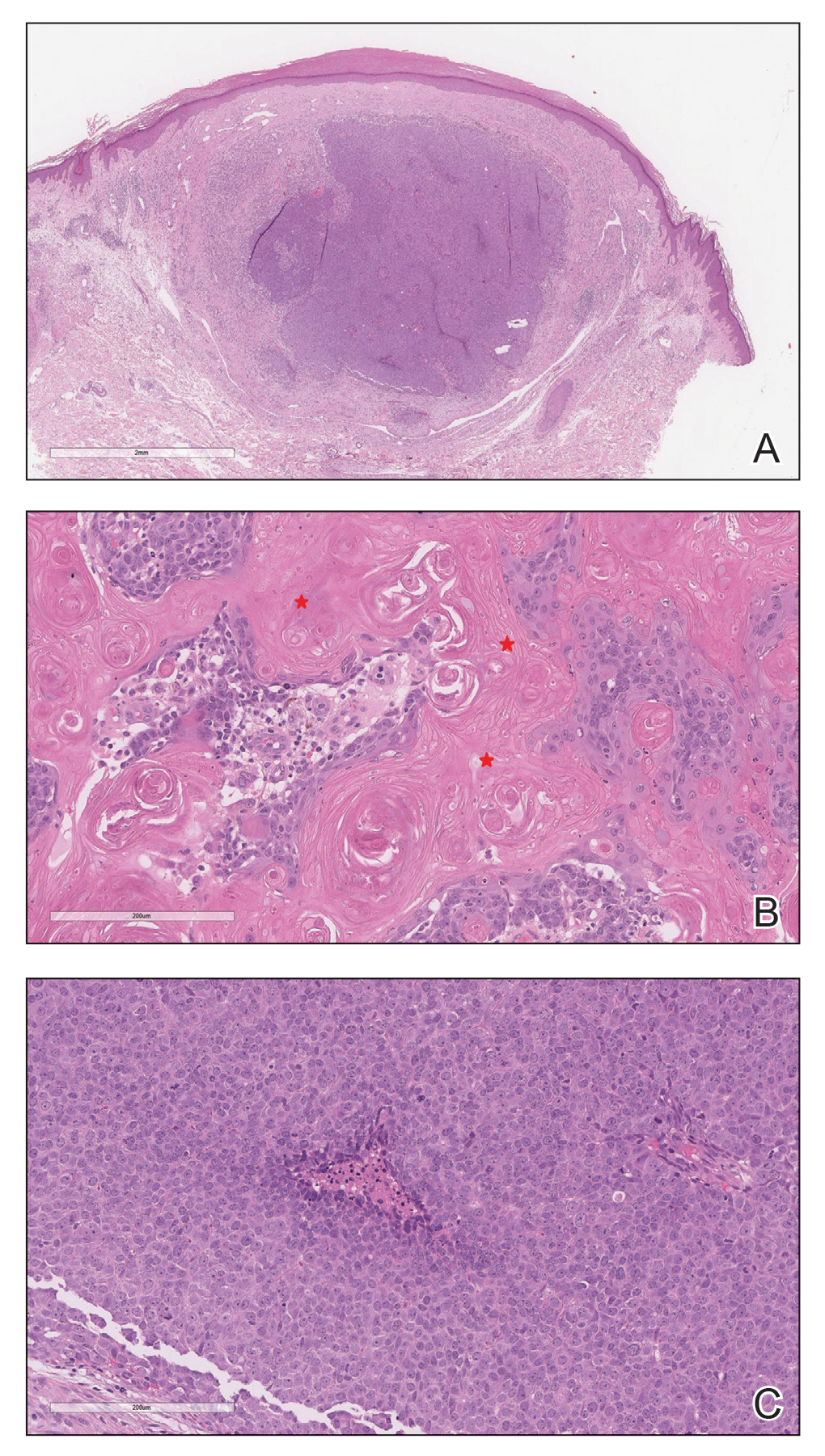
Histopathologic Findings—Gross examination revealed a 0.9×0.7-cm, raised, slightly irregular lesion located 1 mm away from the closest peripheral margin. Histologically, the lesion was a relatively circumscribed, dermal-based basaloid neoplasm with slightly ill-defined edges involving the superficial and deep dermis (Figure 1A). The neoplasm was formed predominantly of sheets of basaloid cells and small nests of ghost cells, in addition to some squamoid and transitional cells (Figure 1B). The basaloid cells exhibited severe nuclear atypia, pleomorphism, increased nuclear to cytoplasmic ratio (Figure 1C), minimal to moderate amounts of eosinophilic cytoplasm, enlarged nuclei, prominent nucleoli, and coarse chromatin pattern. Abundant mitotic activity and apoptotic bodies were present as well as focal area of central necrosis (Figure 1C). Also, melanophages and a multinucleated giant cell reaction was noted. Elastic trichrome special stain highlighted focal infiltration of the neoplastic cells into the adjacent desmoplastic stroma. Melanin stain was negative for melanin pigment within the neoplasm. Given the presence of severely atypical basaloid cells along with ghost cells indicating matrical differentiation, a diagnosis of pilomatrix carcinoma was rendered.
Immunohistochemistry—The neoplastic cells were diffusely positive for p63, CDX-2 (Figure 2A), β-catenin (Figure 2B), and CD10 (Figure 2C), and focally and weakly positive for cytokeratin (CK) 5, BerEP4 (staining the tumor periphery), androgen receptor, and CK18 (a low-molecular-weight keratin). They were negative for monoclonal carcinoembryonic antigen, epithelial membrane antigen, CK7, CK20, CD34, SOX-10, CD56, synaptophysin, and chromogranin. Cytokeratin 14 was positive in the squamoid cells but negative in the basaloid cells. SOX-10 and melanoma cocktail immunostains demonstrated few intralesional dendritic melanocytes.

Comment
Pilomatrix carcinoma is a rare malignant cutaneous adnexal neoplasm with origin from the germinative matrix of the hair bulb region of hair follicles. Pilomatrix carcinoma was first reported in 1980.1,2 These tumors are characterized by rapid growth and aggressive behavior. Their benign counterpart, pilomatrixoma, is a slow-growing, dermal or subcutaneous tumor that rarely recurs after complete excision.
As with pilomatrixoma, pilomatrix carcinomas are asymptomatic and present as solitary dermal or subcutaneous masses3,4 that most commonly are found in the posterior neck, upper back, and preauricular regions of middle-aged or elderly adults with male predominance.5 They range in size from 0.5 to 20 cm with a mean of 4 cm that is slightly larger than pilomatrixoma. Pilomatrix carcinomas predominantly are firm tumors with or without cystic components, and they exhibit a high probability of recurrence and have risk for distant metastasis.6-15
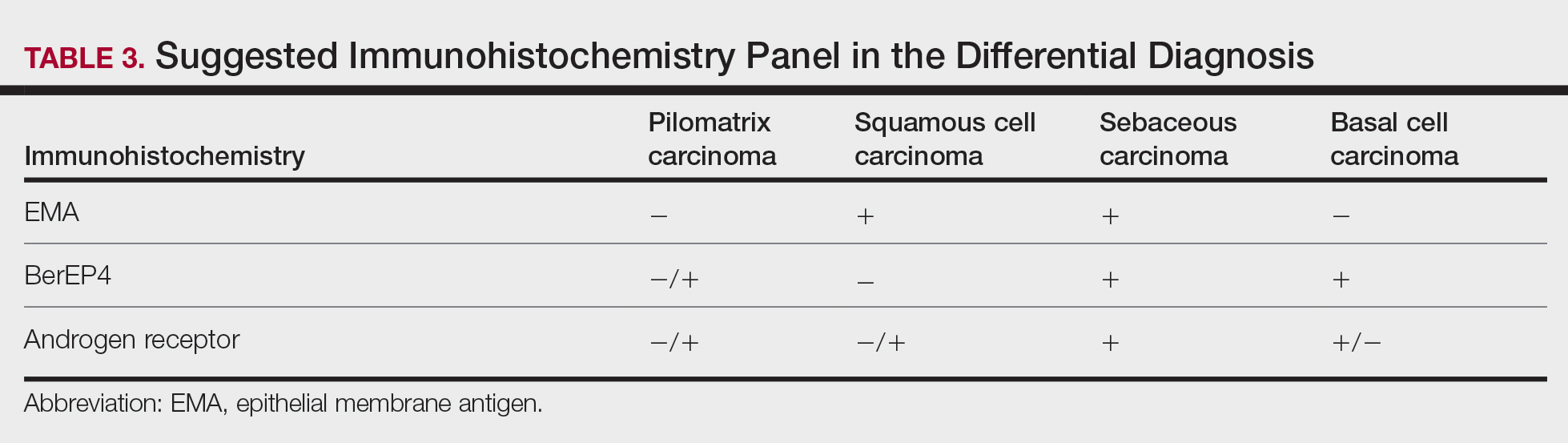
The differential diagnosis includes epidermal cysts, pilomatrixoma, basal cell carcinoma with matrical differentiation, trichoblastoma/trichoblastic carcinoma, and trichilemmal carcinoma. Pilomatrix carcinomas frequently are mistaken for epidermal cysts on clinical examination. Such a distinction can be easily resolved by histopathologic evaluation. The more challenging differential diagnosis is with pilomatrixoma. Histologically, pilomatrixomas consist of a distinct population of cells including basaloid, squamoid, transitional, and shadow cells in variable proportions. The basaloid cells transition to shadow cells in an organized zonal fashion.16 Compared to pilomatrixomas, pilomatrix carcinomas often show predominance of the basaloid cells; marked cytologic atypia and pleomorphism; numerous mitotic figures; deep infiltrative pattern into subcutaneous fat, fascia, and skeletal muscle; stromal desmoplasia; necrosis; and neurovascular invasion (Tables 1 and 2). Furthermore, the shadow cells tend to form a small nested pattern in pilomatrix carcinoma instead of the flat sheetlike pattern usually observed in pilomatrixoma.16 Basal cell carcinoma with matrical differentiation can pose a diagnostic challenge in the differential diagnosis; basal cell carcinoma usually exhibits a peripheral palisade of the basaloid cells accompanied by retraction spaces separating the tumor from the stroma. Trichoblastoma/trichoblastic carcinoma with matrical differentiation can be distinguished by its exuberant stroma, prominent primitive hair follicles, and papillary mesenchymal bodies. Trichilemmal carcinomas are recognized by their connection to the overlying epidermis, peripheral palisading, and presence of clear cells, while pilomatrix carcinoma lacks connection to the surface epithelium.

Immunohistochemical stains have little to no role in the differential diagnosis, and morphology is the mainstay in making the diagnosis. Rarely, pilomatrix carcinoma can be confused with poorly differentiated sebaceous carcinoma and poorly differentiated squamous cell carcinoma. Although careful scrutiny of the histologic features may help identify mature sebocytes in sebaceous carcinoma, evidence of keratinization in squamous cell carcinoma and ghost cells in pilomatrix carcinoma, using a panel of immunohistochemical stains can be helpful in reaching the final diagnosis (Table 3).

The development of hair matrix tumors have been known to harbor mutations in exon 3 of the catenin beta-1 gene, CTNNB1, that encodes for β-catenin, a downstream effector in the Wnt signaling pathway responsible for differentiation, proliferation, and adhesion of epithelial stem cells.17-21 In a study conducted by Kazakov et al,22 DNA was extracted from 86 lesions: 4 were pilomatrixomas and 1 was a pilomatrix carcinoma. A polymerase chain reaction assay revealed 8 pathogenic variants of the β-catenin gene. D32Y (CTNNB1):c.94G>T (p.Asp32Tyr) and G34R (CTNNB1):c.100G>C (p.Gly34Arg) were the mutations present in pilomatrixoma and pilomatrix carcinoma, respectively.22 In addition, there are several proteins that are part of the Wnt pathway in addition to β-catenin—LEF-1 and CDX-2.
Tumminello and Hosler23 found that pilomatrixomas and pilomatrix carcinomas were positive for CDX-2, β-catenin, and LEF-1 by immunohistochemistry. These downstream molecules in the Wnt signaling pathway could have the potential to be used as diagnostic and prognostic markers.2,13,15,23
Although the pathogenesis is unclear, there are 2 possible mechanisms by which pilomatrix carcinomas develop. They can either arise as de novo tumors, or it is possible that initial mutations in β-catenin result in the formation of pilomatrixomas at an early age that may undergo malignant transformation in elderly patients over time with additional mutations.2
Our case was strongly and diffusely positive for β-catenin in a nuclear and cytoplasmic pattern and CDX-2 in a nuclear pattern, supporting the role of the Wnt signaling pathway in such tumors. Furthermore, our case demonstrated the presence of few intralesional normal dendritic melanocytes, a rare finding1,24,25 but not unexpected, as melanocytes normally are present within the hair follicle matrix.
Pilomatrix carcinomas are aggressive tumors with a high risk for local recurrence and tendency for metastasis. In a study of 13 cases of pilomatrix carcinomas, Herrmann et al13 found that metastasis was significantly associated with local tumor recurrence (P<.0413). They concluded that the combination of overall high local recurrence and metastatic rates of pilomatrix carcinoma as well as documented tumor-related deaths would warrant continued patient follow-up, especially for recurrent tumors.13 Rapid growth of a tumor, either de novo or following several months of stable size, should alert physicians to perform a diagnostic biopsy.
Management options of pilomatrix carcinoma include surgery or radiation with close follow-up. The most widely reported treatment of pilomatrix carcinoma is wide local excision with histologically confirmed clear margins. Mohs micrographic surgery is an excellent treatment option.2,13-15 Adjuvant radiation therapy may be necessary following excision. Currently there is no consensus on surgical management, and standard excisional margins have not been defined.26 Jones et al2 concluded that complete excision with wide margins likely is curative, with decreased rates of recurrence, and better awareness of this carcinoma would lead to appropriate treatment while avoiding unnecessary diagnostic tests.2
Conclusion
We report an exceptionally unique case of early pilomatrix carcinoma with a discussion on the pathogenesis and molecular pathology of hair matrix tumors. A large cohort of patients with longer follow-up periods and better molecular characterization is essential in drawing accurate information about their prognosis, identifying molecular markers that can be used as therapeutic targets, and determining ideal management strategy.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38.
- Forbis R, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606.
- Elder D, Elenitsas R, Ragsdale BD. Tumors of epidermal appendages. In: Elder D, Elenitsas R, Jaworsky C, eds. Lever’s Histopathology of the Skin. 8th ed. Lippincott Raven; 1997:757-759.
- Aherne NJ, Fitzpatrick DA, Gibbons D, et al. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol. 2008;3:47.
- Papadakis M, de Bree E, Floros N, et al. Pilomatrix carcinoma: more malignant biological behavior than was considered in the past. Mol Clin Oncol. 2017;6:415-418.
- LeBoit PE, Parslow TG, Choy SH. Hair matrix differentiation: occurrence in lesions other than pilomatricoma. Am J Dermatopathol. 1987;9:399-405.
- Campoy F, Stiefel P, Stiefel E, et al. Pilomatrix carcinoma: role played by MR imaging. Neuroradiology. 1989;31:196-198.
- Tateyama H, Eimoto T, Tada T, et al. Malignant pilomatricoma: an immunohistochemical study with antihair keratin antibody. Cancer. 1992;69:127-132.
- O’Donovan DG, Freemont AJ, Adams JE, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1993;23:385-386.
- Cross P, Richmond I, Wells S, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1994;24:499-500.
- Niedermeyer HP, Peris K, Höfler H. Pilomatrix carcinoma with multiple visceral metastases: report of a case. Cancer. 1996;77:1311-1314.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Xing L, Marzolf SA, Vandergriff T, et al. Facial pilomatrix carcinomas treated with Mohs micrographic surgery. JAAD Case Rep. 2018;4:253-255.
- Fernandez-Flores A, Cassarino DS. Sarcomatoid pilomatrix carcinoma. J Cutan Pathol. 2018;45:508-514.
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498.
- Chan E, Gat U, McNiff JM, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410-413.
- Huelsken J, Vogel R, Erdmann B, et al. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533-545.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225-229.
- Durand M, Moles J. Beta-catenin mutations in a common skin cancer: pilomatricoma. Bull Cancer. 1999;86:725-726.
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Kazakov DV, Sima R, Vanecek T, et al. Mutations in exon 3 of the CTNNB1 gene (β-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol. 2009;31:248-255.
- Tumminello K, Hosler GA. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J Cutan Pathol. 2018;45:318-324.
- Rodic´ N, Taube JM, Manson P, et al Locally invasive dermal squamomelanocytic tumor with matrical differentiation: a peculiar case with review of the literature. Am J Dermatopathol. 2013;35:E72-E76.
- Perez C, Debbaneh M, Cassarino D. Preference for the term pilomatrical carcinoma with melanocytic hyperplasia: letter to the editor. J Cutan Pathol. 2017;44:655-657.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
Pilomatrix carcinoma is a rare adnexal tumor with origin from the germinative matrical cells of the hair follicle. Clinically, it presents as a solitary lesion commonly found in the head and neck region as well as the upper back. The tumors cannot be distinguished by their clinical appearance only and frequently are mistaken for cysts. Histopathologic examination provides the definitive diagnosis in most cases. These carcinomas are aggressive neoplasms with a high probability of local recurrence and distant metastasis. Assessment of the Wnt signaling pathway components such as β-catenin, lymphoid enhancer-binding factor 1 (LEF-1), and caudal-related homeobox transcription factor 2 (CDX-2) potentially can be used for diagnostic purposes and targeted therapy.
We report a rare and unique case of early pilomatrix carcinoma with intralesional melanocytes. We review the molecular pathology and pathogenesis of these carcinomas as well as the significance of early diagnosis.
Case Report
A 73-year-old man with a history of extensive sun exposure presented with a 1-cm, raised, rapidly growing, slightly irregular, purple lesion on the right forearm of 3 months’ duration with tendency to bleed. He did not have a history of skin cancers and was otherwise healthy. Excision was recommended due to the progressive and rapid growth of the lesion.
Histopathologic Findings—Gross examination revealed a 0.9×0.7-cm, raised, slightly irregular lesion located 1 mm away from the closest peripheral margin. Histologically, the lesion was a relatively circumscribed, dermal-based basaloid neoplasm with slightly ill-defined edges involving the superficial and deep dermis (Figure 1A). The neoplasm was formed predominantly of sheets of basaloid cells and small nests of ghost cells, in addition to some squamoid and transitional cells (Figure 1B). The basaloid cells exhibited severe nuclear atypia, pleomorphism, increased nuclear to cytoplasmic ratio (Figure 1C), minimal to moderate amounts of eosinophilic cytoplasm, enlarged nuclei, prominent nucleoli, and coarse chromatin pattern. Abundant mitotic activity and apoptotic bodies were present as well as focal area of central necrosis (Figure 1C). Also, melanophages and a multinucleated giant cell reaction was noted. Elastic trichrome special stain highlighted focal infiltration of the neoplastic cells into the adjacent desmoplastic stroma. Melanin stain was negative for melanin pigment within the neoplasm. Given the presence of severely atypical basaloid cells along with ghost cells indicating matrical differentiation, a diagnosis of pilomatrix carcinoma was rendered.
Immunohistochemistry—The neoplastic cells were diffusely positive for p63, CDX-2 (Figure 2A), β-catenin (Figure 2B), and CD10 (Figure 2C), and focally and weakly positive for cytokeratin (CK) 5, BerEP4 (staining the tumor periphery), androgen receptor, and CK18 (a low-molecular-weight keratin). They were negative for monoclonal carcinoembryonic antigen, epithelial membrane antigen, CK7, CK20, CD34, SOX-10, CD56, synaptophysin, and chromogranin. Cytokeratin 14 was positive in the squamoid cells but negative in the basaloid cells. SOX-10 and melanoma cocktail immunostains demonstrated few intralesional dendritic melanocytes.
Comment
Pilomatrix carcinoma is a rare malignant cutaneous adnexal neoplasm with origin from the germinative matrix of the hair bulb region of hair follicles. Pilomatrix carcinoma was first reported in 1980.1,2 These tumors are characterized by rapid growth and aggressive behavior. Their benign counterpart, pilomatrixoma, is a slow-growing, dermal or subcutaneous tumor that rarely recurs after complete excision.
As with pilomatrixoma, pilomatrix carcinomas are asymptomatic and present as solitary dermal or subcutaneous masses3,4 that most commonly are found in the posterior neck, upper back, and preauricular regions of middle-aged or elderly adults with male predominance.5 They range in size from 0.5 to 20 cm with a mean of 4 cm that is slightly larger than pilomatrixoma. Pilomatrix carcinomas predominantly are firm tumors with or without cystic components, and they exhibit a high probability of recurrence and have risk for distant metastasis.6-15
The differential diagnosis includes epidermal cysts, pilomatrixoma, basal cell carcinoma with matrical differentiation, trichoblastoma/trichoblastic carcinoma, and trichilemmal carcinoma. Pilomatrix carcinomas frequently are mistaken for epidermal cysts on clinical examination. Such a distinction can be easily resolved by histopathologic evaluation. The more challenging differential diagnosis is with pilomatrixoma. Histologically, pilomatrixomas consist of a distinct population of cells including basaloid, squamoid, transitional, and shadow cells in variable proportions. The basaloid cells transition to shadow cells in an organized zonal fashion.16 Compared to pilomatrixomas, pilomatrix carcinomas often show predominance of the basaloid cells; marked cytologic atypia and pleomorphism; numerous mitotic figures; deep infiltrative pattern into subcutaneous fat, fascia, and skeletal muscle; stromal desmoplasia; necrosis; and neurovascular invasion (Tables 1 and 2). Furthermore, the shadow cells tend to form a small nested pattern in pilomatrix carcinoma instead of the flat sheetlike pattern usually observed in pilomatrixoma.16 Basal cell carcinoma with matrical differentiation can pose a diagnostic challenge in the differential diagnosis; basal cell carcinoma usually exhibits a peripheral palisade of the basaloid cells accompanied by retraction spaces separating the tumor from the stroma. Trichoblastoma/trichoblastic carcinoma with matrical differentiation can be distinguished by its exuberant stroma, prominent primitive hair follicles, and papillary mesenchymal bodies. Trichilemmal carcinomas are recognized by their connection to the overlying epidermis, peripheral palisading, and presence of clear cells, while pilomatrix carcinoma lacks connection to the surface epithelium.
Immunohistochemical stains have little to no role in the differential diagnosis, and morphology is the mainstay in making the diagnosis. Rarely, pilomatrix carcinoma can be confused with poorly differentiated sebaceous carcinoma and poorly differentiated squamous cell carcinoma. Although careful scrutiny of the histologic features may help identify mature sebocytes in sebaceous carcinoma, evidence of keratinization in squamous cell carcinoma and ghost cells in pilomatrix carcinoma, using a panel of immunohistochemical stains can be helpful in reaching the final diagnosis (Table 3).
The development of hair matrix tumors have been known to harbor mutations in exon 3 of the catenin beta-1 gene, CTNNB1, that encodes for β-catenin, a downstream effector in the Wnt signaling pathway responsible for differentiation, proliferation, and adhesion of epithelial stem cells.17-21 In a study conducted by Kazakov et al,22 DNA was extracted from 86 lesions: 4 were pilomatrixomas and 1 was a pilomatrix carcinoma. A polymerase chain reaction assay revealed 8 pathogenic variants of the β-catenin gene. D32Y (CTNNB1):c.94G>T (p.Asp32Tyr) and G34R (CTNNB1):c.100G>C (p.Gly34Arg) were the mutations present in pilomatrixoma and pilomatrix carcinoma, respectively.22 In addition, there are several proteins that are part of the Wnt pathway in addition to β-catenin—LEF-1 and CDX-2.
Tumminello and Hosler23 found that pilomatrixomas and pilomatrix carcinomas were positive for CDX-2, β-catenin, and LEF-1 by immunohistochemistry. These downstream molecules in the Wnt signaling pathway could have the potential to be used as diagnostic and prognostic markers.2,13,15,23
Although the pathogenesis is unclear, there are 2 possible mechanisms by which pilomatrix carcinomas develop. They can either arise as de novo tumors, or it is possible that initial mutations in β-catenin result in the formation of pilomatrixomas at an early age that may undergo malignant transformation in elderly patients over time with additional mutations.2
Our case was strongly and diffusely positive for β-catenin in a nuclear and cytoplasmic pattern and CDX-2 in a nuclear pattern, supporting the role of the Wnt signaling pathway in such tumors. Furthermore, our case demonstrated the presence of few intralesional normal dendritic melanocytes, a rare finding1,24,25 but not unexpected, as melanocytes normally are present within the hair follicle matrix.
Pilomatrix carcinomas are aggressive tumors with a high risk for local recurrence and tendency for metastasis. In a study of 13 cases of pilomatrix carcinomas, Herrmann et al13 found that metastasis was significantly associated with local tumor recurrence (P<.0413). They concluded that the combination of overall high local recurrence and metastatic rates of pilomatrix carcinoma as well as documented tumor-related deaths would warrant continued patient follow-up, especially for recurrent tumors.13 Rapid growth of a tumor, either de novo or following several months of stable size, should alert physicians to perform a diagnostic biopsy.
Management options of pilomatrix carcinoma include surgery or radiation with close follow-up. The most widely reported treatment of pilomatrix carcinoma is wide local excision with histologically confirmed clear margins. Mohs micrographic surgery is an excellent treatment option.2,13-15 Adjuvant radiation therapy may be necessary following excision. Currently there is no consensus on surgical management, and standard excisional margins have not been defined.26 Jones et al2 concluded that complete excision with wide margins likely is curative, with decreased rates of recurrence, and better awareness of this carcinoma would lead to appropriate treatment while avoiding unnecessary diagnostic tests.2
Conclusion
We report an exceptionally unique case of early pilomatrix carcinoma with a discussion on the pathogenesis and molecular pathology of hair matrix tumors. A large cohort of patients with longer follow-up periods and better molecular characterization is essential in drawing accurate information about their prognosis, identifying molecular markers that can be used as therapeutic targets, and determining ideal management strategy.
Pilomatrix carcinoma is a rare adnexal tumor with origin from the germinative matrical cells of the hair follicle. Clinically, it presents as a solitary lesion commonly found in the head and neck region as well as the upper back. The tumors cannot be distinguished by their clinical appearance only and frequently are mistaken for cysts. Histopathologic examination provides the definitive diagnosis in most cases. These carcinomas are aggressive neoplasms with a high probability of local recurrence and distant metastasis. Assessment of the Wnt signaling pathway components such as β-catenin, lymphoid enhancer-binding factor 1 (LEF-1), and caudal-related homeobox transcription factor 2 (CDX-2) potentially can be used for diagnostic purposes and targeted therapy.
We report a rare and unique case of early pilomatrix carcinoma with intralesional melanocytes. We review the molecular pathology and pathogenesis of these carcinomas as well as the significance of early diagnosis.
Case Report
A 73-year-old man with a history of extensive sun exposure presented with a 1-cm, raised, rapidly growing, slightly irregular, purple lesion on the right forearm of 3 months’ duration with tendency to bleed. He did not have a history of skin cancers and was otherwise healthy. Excision was recommended due to the progressive and rapid growth of the lesion.
Histopathologic Findings—Gross examination revealed a 0.9×0.7-cm, raised, slightly irregular lesion located 1 mm away from the closest peripheral margin. Histologically, the lesion was a relatively circumscribed, dermal-based basaloid neoplasm with slightly ill-defined edges involving the superficial and deep dermis (Figure 1A). The neoplasm was formed predominantly of sheets of basaloid cells and small nests of ghost cells, in addition to some squamoid and transitional cells (Figure 1B). The basaloid cells exhibited severe nuclear atypia, pleomorphism, increased nuclear to cytoplasmic ratio (Figure 1C), minimal to moderate amounts of eosinophilic cytoplasm, enlarged nuclei, prominent nucleoli, and coarse chromatin pattern. Abundant mitotic activity and apoptotic bodies were present as well as focal area of central necrosis (Figure 1C). Also, melanophages and a multinucleated giant cell reaction was noted. Elastic trichrome special stain highlighted focal infiltration of the neoplastic cells into the adjacent desmoplastic stroma. Melanin stain was negative for melanin pigment within the neoplasm. Given the presence of severely atypical basaloid cells along with ghost cells indicating matrical differentiation, a diagnosis of pilomatrix carcinoma was rendered.
Immunohistochemistry—The neoplastic cells were diffusely positive for p63, CDX-2 (Figure 2A), β-catenin (Figure 2B), and CD10 (Figure 2C), and focally and weakly positive for cytokeratin (CK) 5, BerEP4 (staining the tumor periphery), androgen receptor, and CK18 (a low-molecular-weight keratin). They were negative for monoclonal carcinoembryonic antigen, epithelial membrane antigen, CK7, CK20, CD34, SOX-10, CD56, synaptophysin, and chromogranin. Cytokeratin 14 was positive in the squamoid cells but negative in the basaloid cells. SOX-10 and melanoma cocktail immunostains demonstrated few intralesional dendritic melanocytes.
Comment
Pilomatrix carcinoma is a rare malignant cutaneous adnexal neoplasm with origin from the germinative matrix of the hair bulb region of hair follicles. Pilomatrix carcinoma was first reported in 1980.1,2 These tumors are characterized by rapid growth and aggressive behavior. Their benign counterpart, pilomatrixoma, is a slow-growing, dermal or subcutaneous tumor that rarely recurs after complete excision.
As with pilomatrixoma, pilomatrix carcinomas are asymptomatic and present as solitary dermal or subcutaneous masses3,4 that most commonly are found in the posterior neck, upper back, and preauricular regions of middle-aged or elderly adults with male predominance.5 They range in size from 0.5 to 20 cm with a mean of 4 cm that is slightly larger than pilomatrixoma. Pilomatrix carcinomas predominantly are firm tumors with or without cystic components, and they exhibit a high probability of recurrence and have risk for distant metastasis.6-15
The differential diagnosis includes epidermal cysts, pilomatrixoma, basal cell carcinoma with matrical differentiation, trichoblastoma/trichoblastic carcinoma, and trichilemmal carcinoma. Pilomatrix carcinomas frequently are mistaken for epidermal cysts on clinical examination. Such a distinction can be easily resolved by histopathologic evaluation. The more challenging differential diagnosis is with pilomatrixoma. Histologically, pilomatrixomas consist of a distinct population of cells including basaloid, squamoid, transitional, and shadow cells in variable proportions. The basaloid cells transition to shadow cells in an organized zonal fashion.16 Compared to pilomatrixomas, pilomatrix carcinomas often show predominance of the basaloid cells; marked cytologic atypia and pleomorphism; numerous mitotic figures; deep infiltrative pattern into subcutaneous fat, fascia, and skeletal muscle; stromal desmoplasia; necrosis; and neurovascular invasion (Tables 1 and 2). Furthermore, the shadow cells tend to form a small nested pattern in pilomatrix carcinoma instead of the flat sheetlike pattern usually observed in pilomatrixoma.16 Basal cell carcinoma with matrical differentiation can pose a diagnostic challenge in the differential diagnosis; basal cell carcinoma usually exhibits a peripheral palisade of the basaloid cells accompanied by retraction spaces separating the tumor from the stroma. Trichoblastoma/trichoblastic carcinoma with matrical differentiation can be distinguished by its exuberant stroma, prominent primitive hair follicles, and papillary mesenchymal bodies. Trichilemmal carcinomas are recognized by their connection to the overlying epidermis, peripheral palisading, and presence of clear cells, while pilomatrix carcinoma lacks connection to the surface epithelium.
Immunohistochemical stains have little to no role in the differential diagnosis, and morphology is the mainstay in making the diagnosis. Rarely, pilomatrix carcinoma can be confused with poorly differentiated sebaceous carcinoma and poorly differentiated squamous cell carcinoma. Although careful scrutiny of the histologic features may help identify mature sebocytes in sebaceous carcinoma, evidence of keratinization in squamous cell carcinoma and ghost cells in pilomatrix carcinoma, using a panel of immunohistochemical stains can be helpful in reaching the final diagnosis (Table 3).
The development of hair matrix tumors have been known to harbor mutations in exon 3 of the catenin beta-1 gene, CTNNB1, that encodes for β-catenin, a downstream effector in the Wnt signaling pathway responsible for differentiation, proliferation, and adhesion of epithelial stem cells.17-21 In a study conducted by Kazakov et al,22 DNA was extracted from 86 lesions: 4 were pilomatrixomas and 1 was a pilomatrix carcinoma. A polymerase chain reaction assay revealed 8 pathogenic variants of the β-catenin gene. D32Y (CTNNB1):c.94G>T (p.Asp32Tyr) and G34R (CTNNB1):c.100G>C (p.Gly34Arg) were the mutations present in pilomatrixoma and pilomatrix carcinoma, respectively.22 In addition, there are several proteins that are part of the Wnt pathway in addition to β-catenin—LEF-1 and CDX-2.
Tumminello and Hosler23 found that pilomatrixomas and pilomatrix carcinomas were positive for CDX-2, β-catenin, and LEF-1 by immunohistochemistry. These downstream molecules in the Wnt signaling pathway could have the potential to be used as diagnostic and prognostic markers.2,13,15,23
Although the pathogenesis is unclear, there are 2 possible mechanisms by which pilomatrix carcinomas develop. They can either arise as de novo tumors, or it is possible that initial mutations in β-catenin result in the formation of pilomatrixomas at an early age that may undergo malignant transformation in elderly patients over time with additional mutations.2
Our case was strongly and diffusely positive for β-catenin in a nuclear and cytoplasmic pattern and CDX-2 in a nuclear pattern, supporting the role of the Wnt signaling pathway in such tumors. Furthermore, our case demonstrated the presence of few intralesional normal dendritic melanocytes, a rare finding1,24,25 but not unexpected, as melanocytes normally are present within the hair follicle matrix.
Pilomatrix carcinomas are aggressive tumors with a high risk for local recurrence and tendency for metastasis. In a study of 13 cases of pilomatrix carcinomas, Herrmann et al13 found that metastasis was significantly associated with local tumor recurrence (P<.0413). They concluded that the combination of overall high local recurrence and metastatic rates of pilomatrix carcinoma as well as documented tumor-related deaths would warrant continued patient follow-up, especially for recurrent tumors.13 Rapid growth of a tumor, either de novo or following several months of stable size, should alert physicians to perform a diagnostic biopsy.
Management options of pilomatrix carcinoma include surgery or radiation with close follow-up. The most widely reported treatment of pilomatrix carcinoma is wide local excision with histologically confirmed clear margins. Mohs micrographic surgery is an excellent treatment option.2,13-15 Adjuvant radiation therapy may be necessary following excision. Currently there is no consensus on surgical management, and standard excisional margins have not been defined.26 Jones et al2 concluded that complete excision with wide margins likely is curative, with decreased rates of recurrence, and better awareness of this carcinoma would lead to appropriate treatment while avoiding unnecessary diagnostic tests.2
Conclusion
We report an exceptionally unique case of early pilomatrix carcinoma with a discussion on the pathogenesis and molecular pathology of hair matrix tumors. A large cohort of patients with longer follow-up periods and better molecular characterization is essential in drawing accurate information about their prognosis, identifying molecular markers that can be used as therapeutic targets, and determining ideal management strategy.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38.
- Forbis R, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606.
- Elder D, Elenitsas R, Ragsdale BD. Tumors of epidermal appendages. In: Elder D, Elenitsas R, Jaworsky C, eds. Lever’s Histopathology of the Skin. 8th ed. Lippincott Raven; 1997:757-759.
- Aherne NJ, Fitzpatrick DA, Gibbons D, et al. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol. 2008;3:47.
- Papadakis M, de Bree E, Floros N, et al. Pilomatrix carcinoma: more malignant biological behavior than was considered in the past. Mol Clin Oncol. 2017;6:415-418.
- LeBoit PE, Parslow TG, Choy SH. Hair matrix differentiation: occurrence in lesions other than pilomatricoma. Am J Dermatopathol. 1987;9:399-405.
- Campoy F, Stiefel P, Stiefel E, et al. Pilomatrix carcinoma: role played by MR imaging. Neuroradiology. 1989;31:196-198.
- Tateyama H, Eimoto T, Tada T, et al. Malignant pilomatricoma: an immunohistochemical study with antihair keratin antibody. Cancer. 1992;69:127-132.
- O’Donovan DG, Freemont AJ, Adams JE, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1993;23:385-386.
- Cross P, Richmond I, Wells S, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1994;24:499-500.
- Niedermeyer HP, Peris K, Höfler H. Pilomatrix carcinoma with multiple visceral metastases: report of a case. Cancer. 1996;77:1311-1314.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Xing L, Marzolf SA, Vandergriff T, et al. Facial pilomatrix carcinomas treated with Mohs micrographic surgery. JAAD Case Rep. 2018;4:253-255.
- Fernandez-Flores A, Cassarino DS. Sarcomatoid pilomatrix carcinoma. J Cutan Pathol. 2018;45:508-514.
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498.
- Chan E, Gat U, McNiff JM, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410-413.
- Huelsken J, Vogel R, Erdmann B, et al. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533-545.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225-229.
- Durand M, Moles J. Beta-catenin mutations in a common skin cancer: pilomatricoma. Bull Cancer. 1999;86:725-726.
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Kazakov DV, Sima R, Vanecek T, et al. Mutations in exon 3 of the CTNNB1 gene (β-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol. 2009;31:248-255.
- Tumminello K, Hosler GA. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J Cutan Pathol. 2018;45:318-324.
- Rodic´ N, Taube JM, Manson P, et al Locally invasive dermal squamomelanocytic tumor with matrical differentiation: a peculiar case with review of the literature. Am J Dermatopathol. 2013;35:E72-E76.
- Perez C, Debbaneh M, Cassarino D. Preference for the term pilomatrical carcinoma with melanocytic hyperplasia: letter to the editor. J Cutan Pathol. 2017;44:655-657.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38.
- Forbis R, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606.
- Elder D, Elenitsas R, Ragsdale BD. Tumors of epidermal appendages. In: Elder D, Elenitsas R, Jaworsky C, eds. Lever’s Histopathology of the Skin. 8th ed. Lippincott Raven; 1997:757-759.
- Aherne NJ, Fitzpatrick DA, Gibbons D, et al. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol. 2008;3:47.
- Papadakis M, de Bree E, Floros N, et al. Pilomatrix carcinoma: more malignant biological behavior than was considered in the past. Mol Clin Oncol. 2017;6:415-418.
- LeBoit PE, Parslow TG, Choy SH. Hair matrix differentiation: occurrence in lesions other than pilomatricoma. Am J Dermatopathol. 1987;9:399-405.
- Campoy F, Stiefel P, Stiefel E, et al. Pilomatrix carcinoma: role played by MR imaging. Neuroradiology. 1989;31:196-198.
- Tateyama H, Eimoto T, Tada T, et al. Malignant pilomatricoma: an immunohistochemical study with antihair keratin antibody. Cancer. 1992;69:127-132.
- O’Donovan DG, Freemont AJ, Adams JE, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1993;23:385-386.
- Cross P, Richmond I, Wells S, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1994;24:499-500.
- Niedermeyer HP, Peris K, Höfler H. Pilomatrix carcinoma with multiple visceral metastases: report of a case. Cancer. 1996;77:1311-1314.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Xing L, Marzolf SA, Vandergriff T, et al. Facial pilomatrix carcinomas treated with Mohs micrographic surgery. JAAD Case Rep. 2018;4:253-255.
- Fernandez-Flores A, Cassarino DS. Sarcomatoid pilomatrix carcinoma. J Cutan Pathol. 2018;45:508-514.
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498.
- Chan E, Gat U, McNiff JM, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410-413.
- Huelsken J, Vogel R, Erdmann B, et al. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533-545.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225-229.
- Durand M, Moles J. Beta-catenin mutations in a common skin cancer: pilomatricoma. Bull Cancer. 1999;86:725-726.
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Kazakov DV, Sima R, Vanecek T, et al. Mutations in exon 3 of the CTNNB1 gene (β-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol. 2009;31:248-255.
- Tumminello K, Hosler GA. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J Cutan Pathol. 2018;45:318-324.
- Rodic´ N, Taube JM, Manson P, et al Locally invasive dermal squamomelanocytic tumor with matrical differentiation: a peculiar case with review of the literature. Am J Dermatopathol. 2013;35:E72-E76.
- Perez C, Debbaneh M, Cassarino D. Preference for the term pilomatrical carcinoma with melanocytic hyperplasia: letter to the editor. J Cutan Pathol. 2017;44:655-657.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
Practice Points
- Clinicians and pathologists should be aware of pilomatrix carcinoma to facilitate early detection.
- Early diagnosis and prompt treatment of pilomatrix carcinoma is crucial in lowering recurrence rate and avoiding a poor outcome.
- Caudal-related homeobox transcription factor 2 and β-catenin components of the Wnt signaling pathway play an important role in the pathogenesis of pilomatrix carcinoma.
- Although controversial, wide local excision is the treatment of choice for pilomatrix carcinoma.

TANS Syndrome: Tanorexia, Anorexia, and Nonmelanoma Skin Cancer
The term tanorexia describes compulsive use of a tanning bed, a disorder often identified in White patients. This compulsion is driven by underlying psychological distress that typically correlates with another psychiatric disorder, such as anxiety, body dysmorphic disorder, or an eating disorder. 1 Severe anorexia combined with excessive indoor tanning led to a notable burden of cutaneous squamous cell carcinomas (SCCs) and keratoacanthomas in one of our patients. We discuss the management and approach to patient care in this difficult situation, which we have coined TANS syndrome (for T anorexia, A norexia, and N onmelanoma s kin cancer).
A Patient With TANS Syndrome
A 35-year-old cachectic woman, who appeared much older than her chronologic age, presented for management of numerous painful bleeding skin lesions. Diffuse, erythematous, tender nodules with central keratotic cores, some several centimeters in diameter, were scattered on the abdomen, chest, and extremities (Figure 1); similar lesions were noted on the neck (Figure 2). Numerous erythematous scaly papules and plaques consistent with actinic keratoses were noted throughout the body.
The patient reported that the cutaneous SCCs presented over the last few years, whereas her eating disorder began in adolescence and persisted despite multiple intensive outpatient and inpatient programs. The patient adamantly refused repeat hospitalization, against repeated suggestions by health care providers and her family. Comorbidities related to her anorexia included severe renal insufficiency, iron deficiency anemia, hypertriglyceridemia, kwashiorkor, and pellagra.
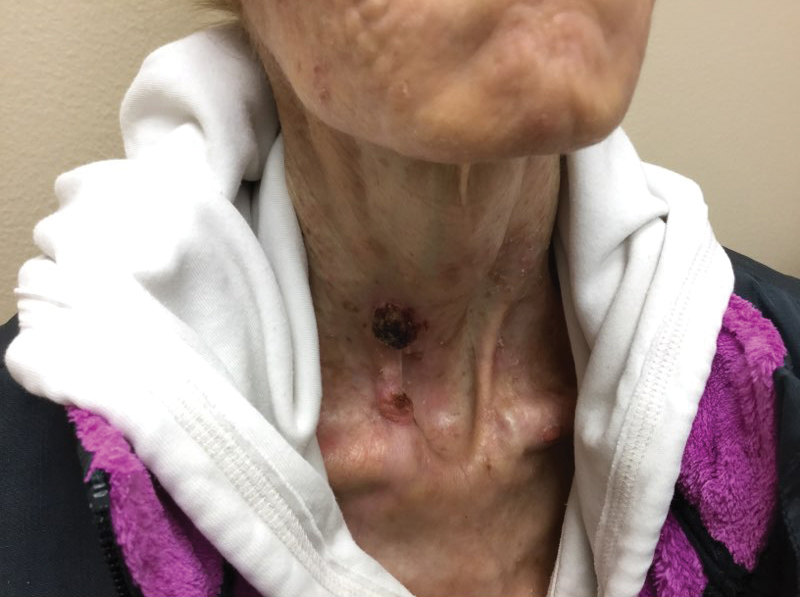
Within the last year, the patient had several biopsies showing SCC, keratoacanthoma type. The largest tumors had been treated by Mohs micrographic surgery, excision, and electrodesiccation or curettage. Adjuvant therapy over the last 2 years consisted of tazarotene cream 0.1%, imiquimod cream 5%, oral nicotinamide 500 mg twice daily, and acitretin 10 to 20 mg daily. Human papillomavirus 9-valent vaccine, recombinant, also had been tried as a chemopreventive and treatment, based on a published report of 2 patients in whom keratinocytic carcinomas decreased after such vaccination.2 The dose of acitretin was kept low because of the patient’s severe renal insufficiency and lack of supporting data for its use in this setting. Despite these modalities, our patient continued to develop new cutaneous SCCs.
We considered starting intralesional methotrexate but deferred this course of action, given the patient’s deteriorating renal function. Our plan was to initiate intralesional 5-fluorouracil; however, the patient was admitted to the hospital and subsequently died due to cardiovascular complications of anorexia.
UV Radiation in the Setting of Immune Compromise
Habitual tanning bed use has been recognized as a psychologic addiction.3,4 After exposure to UV radiation, damaged DNA upregulates pro-opiomelanocortin, which posttranslationally generates β-endorphins to elevate mood.3,5
Tanning beds deliver a higher dose of UVA radiation than UVB radiation and cause darkening of pigmentation by oxidation of preformed melanin and redistribution of melanosomes.3 UVA radiation (320–400 nm) emitted from a tanning bed is 10- to 15-times higher than the radiation emitted by the midday sun and causes DNA damage through generation of reactive oxygen species. UVA penetrates the dermis; its harmful effect on DNA contributes to the pathogenesis of melanoma.
UVB radiation (290–320 nm) is mainly restricted to the epidermis and is largely responsible for erythema of the skin. UVB specifically causes direct damage to DNA by forming pyrimidine dimers, superficially causing sunburn. Excessive exposure to UVB radiation increases the risk for nonmelanoma skin cancer.6
Severe starvation and chronic malnutrition, as seen in anorexia nervosa, also are known to lead to immunosuppression.7 Exposure to UV radiation has been shown to impair the function of antigen-presenting cells, cytokines, and suppressor T cells, and is classified as a Group 1 carcinogen by the World Health Organization.3,8 Combining a compromised immune system in anorexia with DNA damage from frequent indoor tanning provides a dangerous milieu for carcinogenesis.8 Without immune surveillance, as occurs with adequate nutrition, treatment of cutaneous SCC is, at best, challenging.
Primary care physicians, dermatologists, psychiatrists, nutritionists, and public health officials should educate high-risk patients to prevent TANS syndrome.
- Petit A, Karila L, Chalmin F, et al. Phenomenology and psychopathology of excessive indoor tanning. Int J Dermatol. 2014;53:664-672. doi:10.1111/ijd.12336
- Nichols AJ, Allen AH, Shareef S, et al. Association of human papillomavirus vaccine with the development of keratinocyte carcinomas. JAMA Dermatol. 2017;153:571-574. doi:10.1001/jamadermatol.2016.5703
- Madigan LM, Lim HW. Tanning beds: impact on health, and recent regulations. Clin Dermatol. 2016;34:640-648. doi:10.1016/j.clindermatol.2016.05.016
- Schwebel DC. Adolescent tanning, disordered eating, and risk taking. J Dev Behav Pediatr. 2014;35:225-227. doi:10.1097/DBP.0000000000000045
- Friedman B, English JC 3rd, Ferris LK. Indoor tanning, skin cancer and the young female patient: a review of the literature. J Pediatr Adolesc Gynecol. 2015;28:275-283. doi:10.1016/j.jpag.2014.07.015
- Armstrong BK, Kricker A. Epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8-18. doi:10.1016/s1011-1344(01)00198-1
- Hanachi M, Bohem V, Bemer P, et al. Negative role of malnutrition in cell-mediated immune response: Pneumocystis jirovecii pneumonia (PCP) in a severely malnourished, HIV-negative patient with anorexia nervosa. Clin Nutr ESPEN. 2018;25:163-165. doi:10.1016/j.clnesp.2018.03.121
- Schwarz T, Beissert S. Milestones in photoimmunology. J Invest Dermatol. 2013;133:E7-E10. doi:10.1038/skinbio.2013.177
The term tanorexia describes compulsive use of a tanning bed, a disorder often identified in White patients. This compulsion is driven by underlying psychological distress that typically correlates with another psychiatric disorder, such as anxiety, body dysmorphic disorder, or an eating disorder. 1 Severe anorexia combined with excessive indoor tanning led to a notable burden of cutaneous squamous cell carcinomas (SCCs) and keratoacanthomas in one of our patients. We discuss the management and approach to patient care in this difficult situation, which we have coined TANS syndrome (for T anorexia, A norexia, and N onmelanoma s kin cancer).
A Patient With TANS Syndrome
A 35-year-old cachectic woman, who appeared much older than her chronologic age, presented for management of numerous painful bleeding skin lesions. Diffuse, erythematous, tender nodules with central keratotic cores, some several centimeters in diameter, were scattered on the abdomen, chest, and extremities (Figure 1); similar lesions were noted on the neck (Figure 2). Numerous erythematous scaly papules and plaques consistent with actinic keratoses were noted throughout the body.
The patient reported that the cutaneous SCCs presented over the last few years, whereas her eating disorder began in adolescence and persisted despite multiple intensive outpatient and inpatient programs. The patient adamantly refused repeat hospitalization, against repeated suggestions by health care providers and her family. Comorbidities related to her anorexia included severe renal insufficiency, iron deficiency anemia, hypertriglyceridemia, kwashiorkor, and pellagra.
Within the last year, the patient had several biopsies showing SCC, keratoacanthoma type. The largest tumors had been treated by Mohs micrographic surgery, excision, and electrodesiccation or curettage. Adjuvant therapy over the last 2 years consisted of tazarotene cream 0.1%, imiquimod cream 5%, oral nicotinamide 500 mg twice daily, and acitretin 10 to 20 mg daily. Human papillomavirus 9-valent vaccine, recombinant, also had been tried as a chemopreventive and treatment, based on a published report of 2 patients in whom keratinocytic carcinomas decreased after such vaccination.2 The dose of acitretin was kept low because of the patient’s severe renal insufficiency and lack of supporting data for its use in this setting. Despite these modalities, our patient continued to develop new cutaneous SCCs.
We considered starting intralesional methotrexate but deferred this course of action, given the patient’s deteriorating renal function. Our plan was to initiate intralesional 5-fluorouracil; however, the patient was admitted to the hospital and subsequently died due to cardiovascular complications of anorexia.
UV Radiation in the Setting of Immune Compromise
Habitual tanning bed use has been recognized as a psychologic addiction.3,4 After exposure to UV radiation, damaged DNA upregulates pro-opiomelanocortin, which posttranslationally generates β-endorphins to elevate mood.3,5
Tanning beds deliver a higher dose of UVA radiation than UVB radiation and cause darkening of pigmentation by oxidation of preformed melanin and redistribution of melanosomes.3 UVA radiation (320–400 nm) emitted from a tanning bed is 10- to 15-times higher than the radiation emitted by the midday sun and causes DNA damage through generation of reactive oxygen species. UVA penetrates the dermis; its harmful effect on DNA contributes to the pathogenesis of melanoma.
UVB radiation (290–320 nm) is mainly restricted to the epidermis and is largely responsible for erythema of the skin. UVB specifically causes direct damage to DNA by forming pyrimidine dimers, superficially causing sunburn. Excessive exposure to UVB radiation increases the risk for nonmelanoma skin cancer.6
Severe starvation and chronic malnutrition, as seen in anorexia nervosa, also are known to lead to immunosuppression.7 Exposure to UV radiation has been shown to impair the function of antigen-presenting cells, cytokines, and suppressor T cells, and is classified as a Group 1 carcinogen by the World Health Organization.3,8 Combining a compromised immune system in anorexia with DNA damage from frequent indoor tanning provides a dangerous milieu for carcinogenesis.8 Without immune surveillance, as occurs with adequate nutrition, treatment of cutaneous SCC is, at best, challenging.
Primary care physicians, dermatologists, psychiatrists, nutritionists, and public health officials should educate high-risk patients to prevent TANS syndrome.
The term tanorexia describes compulsive use of a tanning bed, a disorder often identified in White patients. This compulsion is driven by underlying psychological distress that typically correlates with another psychiatric disorder, such as anxiety, body dysmorphic disorder, or an eating disorder. 1 Severe anorexia combined with excessive indoor tanning led to a notable burden of cutaneous squamous cell carcinomas (SCCs) and keratoacanthomas in one of our patients. We discuss the management and approach to patient care in this difficult situation, which we have coined TANS syndrome (for T anorexia, A norexia, and N onmelanoma s kin cancer).
A Patient With TANS Syndrome
A 35-year-old cachectic woman, who appeared much older than her chronologic age, presented for management of numerous painful bleeding skin lesions. Diffuse, erythematous, tender nodules with central keratotic cores, some several centimeters in diameter, were scattered on the abdomen, chest, and extremities (Figure 1); similar lesions were noted on the neck (Figure 2). Numerous erythematous scaly papules and plaques consistent with actinic keratoses were noted throughout the body.
The patient reported that the cutaneous SCCs presented over the last few years, whereas her eating disorder began in adolescence and persisted despite multiple intensive outpatient and inpatient programs. The patient adamantly refused repeat hospitalization, against repeated suggestions by health care providers and her family. Comorbidities related to her anorexia included severe renal insufficiency, iron deficiency anemia, hypertriglyceridemia, kwashiorkor, and pellagra.
Within the last year, the patient had several biopsies showing SCC, keratoacanthoma type. The largest tumors had been treated by Mohs micrographic surgery, excision, and electrodesiccation or curettage. Adjuvant therapy over the last 2 years consisted of tazarotene cream 0.1%, imiquimod cream 5%, oral nicotinamide 500 mg twice daily, and acitretin 10 to 20 mg daily. Human papillomavirus 9-valent vaccine, recombinant, also had been tried as a chemopreventive and treatment, based on a published report of 2 patients in whom keratinocytic carcinomas decreased after such vaccination.2 The dose of acitretin was kept low because of the patient’s severe renal insufficiency and lack of supporting data for its use in this setting. Despite these modalities, our patient continued to develop new cutaneous SCCs.
We considered starting intralesional methotrexate but deferred this course of action, given the patient’s deteriorating renal function. Our plan was to initiate intralesional 5-fluorouracil; however, the patient was admitted to the hospital and subsequently died due to cardiovascular complications of anorexia.
UV Radiation in the Setting of Immune Compromise
Habitual tanning bed use has been recognized as a psychologic addiction.3,4 After exposure to UV radiation, damaged DNA upregulates pro-opiomelanocortin, which posttranslationally generates β-endorphins to elevate mood.3,5
Tanning beds deliver a higher dose of UVA radiation than UVB radiation and cause darkening of pigmentation by oxidation of preformed melanin and redistribution of melanosomes.3 UVA radiation (320–400 nm) emitted from a tanning bed is 10- to 15-times higher than the radiation emitted by the midday sun and causes DNA damage through generation of reactive oxygen species. UVA penetrates the dermis; its harmful effect on DNA contributes to the pathogenesis of melanoma.
UVB radiation (290–320 nm) is mainly restricted to the epidermis and is largely responsible for erythema of the skin. UVB specifically causes direct damage to DNA by forming pyrimidine dimers, superficially causing sunburn. Excessive exposure to UVB radiation increases the risk for nonmelanoma skin cancer.6
Severe starvation and chronic malnutrition, as seen in anorexia nervosa, also are known to lead to immunosuppression.7 Exposure to UV radiation has been shown to impair the function of antigen-presenting cells, cytokines, and suppressor T cells, and is classified as a Group 1 carcinogen by the World Health Organization.3,8 Combining a compromised immune system in anorexia with DNA damage from frequent indoor tanning provides a dangerous milieu for carcinogenesis.8 Without immune surveillance, as occurs with adequate nutrition, treatment of cutaneous SCC is, at best, challenging.
Primary care physicians, dermatologists, psychiatrists, nutritionists, and public health officials should educate high-risk patients to prevent TANS syndrome.
- Petit A, Karila L, Chalmin F, et al. Phenomenology and psychopathology of excessive indoor tanning. Int J Dermatol. 2014;53:664-672. doi:10.1111/ijd.12336
- Nichols AJ, Allen AH, Shareef S, et al. Association of human papillomavirus vaccine with the development of keratinocyte carcinomas. JAMA Dermatol. 2017;153:571-574. doi:10.1001/jamadermatol.2016.5703
- Madigan LM, Lim HW. Tanning beds: impact on health, and recent regulations. Clin Dermatol. 2016;34:640-648. doi:10.1016/j.clindermatol.2016.05.016
- Schwebel DC. Adolescent tanning, disordered eating, and risk taking. J Dev Behav Pediatr. 2014;35:225-227. doi:10.1097/DBP.0000000000000045
- Friedman B, English JC 3rd, Ferris LK. Indoor tanning, skin cancer and the young female patient: a review of the literature. J Pediatr Adolesc Gynecol. 2015;28:275-283. doi:10.1016/j.jpag.2014.07.015
- Armstrong BK, Kricker A. Epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8-18. doi:10.1016/s1011-1344(01)00198-1
- Hanachi M, Bohem V, Bemer P, et al. Negative role of malnutrition in cell-mediated immune response: Pneumocystis jirovecii pneumonia (PCP) in a severely malnourished, HIV-negative patient with anorexia nervosa. Clin Nutr ESPEN. 2018;25:163-165. doi:10.1016/j.clnesp.2018.03.121
- Schwarz T, Beissert S. Milestones in photoimmunology. J Invest Dermatol. 2013;133:E7-E10. doi:10.1038/skinbio.2013.177
- Petit A, Karila L, Chalmin F, et al. Phenomenology and psychopathology of excessive indoor tanning. Int J Dermatol. 2014;53:664-672. doi:10.1111/ijd.12336
- Nichols AJ, Allen AH, Shareef S, et al. Association of human papillomavirus vaccine with the development of keratinocyte carcinomas. JAMA Dermatol. 2017;153:571-574. doi:10.1001/jamadermatol.2016.5703
- Madigan LM, Lim HW. Tanning beds: impact on health, and recent regulations. Clin Dermatol. 2016;34:640-648. doi:10.1016/j.clindermatol.2016.05.016
- Schwebel DC. Adolescent tanning, disordered eating, and risk taking. J Dev Behav Pediatr. 2014;35:225-227. doi:10.1097/DBP.0000000000000045
- Friedman B, English JC 3rd, Ferris LK. Indoor tanning, skin cancer and the young female patient: a review of the literature. J Pediatr Adolesc Gynecol. 2015;28:275-283. doi:10.1016/j.jpag.2014.07.015
- Armstrong BK, Kricker A. Epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8-18. doi:10.1016/s1011-1344(01)00198-1
- Hanachi M, Bohem V, Bemer P, et al. Negative role of malnutrition in cell-mediated immune response: Pneumocystis jirovecii pneumonia (PCP) in a severely malnourished, HIV-negative patient with anorexia nervosa. Clin Nutr ESPEN. 2018;25:163-165. doi:10.1016/j.clnesp.2018.03.121
- Schwarz T, Beissert S. Milestones in photoimmunology. J Invest Dermatol. 2013;133:E7-E10. doi:10.1038/skinbio.2013.177
Practice Points
- Primary care physicians, dermatologists, psychiatrists, nutritionists, and public health officials should educate high-risk patients to prevent TANS syndrome.
- Combining a compromised immune system in anorexia with DNA damage from frequent indoor tanning provides a dangerous milieu for carcinogenesis.
- Comorbidities related to TANS syndrome make it challenging to effectively treat cutaneous squamous cell carcinoma.
Ulcer on the Leg
The Diagnosis: Calcinosis Cutis Due to Systemic Sclerosis Sine Scleroderma
Laboratory evaluation was notable for high titers of antinuclear antibodies (>1/320; reference range, 0–1/80) and positive anticentromere antibodies. There were no other relevant laboratory findings; phosphocalcic metabolism was within normal limits, and urinary sediment was normal. Biopsy of the edge of the ulcer revealed basophilic material compatible with calcium deposits. In a 3D volume rendering reconstruction from the lower limb scanner, grouped calcifications were observed in subcutaneous cellular tissue near the ulcer (Figure). The patient had a restrictive ventilatory pattern observed in a pulmonary function test. An esophageal motility study was normal.
The patient was diagnosed with systemic sclerosis sine scleroderma (ssSSc) type II because she met the 4 criteria established by Poormoghim et al1 : (1) Raynaud phenomenon or a peripheral vascular equivalent (ie, digital pitting scars, digital-tip ulcers, digital-tip gangrene, abnormal nail fold capillaries); (2) positive antinuclear antibodies; (3) distal esophageal hypomotility, small bowel hypomotility, pulmonary interstitial fibrosis, primary pulmonary arterial hypertension (without fibrosis), cardiac involvement typical of scleroderma, or renal failure; and (4) no other defined connective tissue or other disease as a cause of the prior conditions.
Systemic sclerosis is a chronic disease characterized by progressive fibrosis of the skin and other internal organs—especially the lungs, kidneys, digestive tract, and heart—as well as generalized vascular dysfunction. Cutaneous induration is its hallmark; however, up to 10% of affected patients have ssSSc.2 This entity is characterized by the total or partial absence of cutaneous manifestations of systemic sclerosis with the occurrence of internal organ involvement and serologic abnormalities. There are 3 types of ssSSc depending on the grade of skin involvement. Type I is characterized by the lack of any typical cutaneous stigmata of the disease. Type II is without sclerodactyly but can coexist with other cutaneous findings such as calcifications, telangiectases, or pitting scars. Type III is characterized clinically by internal organ involvement, typical of systemic sclerosis, that has appeared before skin changes.2
An abnormal deposit of calcium in the cutaneous and subcutaneous tissue is called calcinosis cutis. There are 5 subtypes of calcinosis cutis: dystrophic, metastatic, idiopathic, iatrogenic, and calciphylaxis. Dystrophic skin calcifications may appear in patients with connective tissue diseases such as dermatomyositis or systemic sclerosis.3 Up to 25% of patients with systemic sclerosis can develop calcinosis cutis due to local tissue damage, with normal phosphocalcic metabolism.3
Calcinosis cutis is more common in patients with systemic sclerosis and positive anticentromere antibodies.4 The calcifications usually are located in areas that are subject to repeated trauma, such as the fingers or arms, though other locations have been described such as cervical, paraspinal, or on the hips.5,6 Our patient developed calcifications on both legs, which represent atypical areas for this process.
Dermatomyositis also can present with calcinosis cutis. There are 4 patterns of calcification: superficial nodulelike calcified masses; deep calcified masses; deep sheetlike calcifications within the fascial planes; and a rare, diffuse, superficial lacy and reticular calcification that involves almost the entire body surface area.7 Patients with calcinosis cutis secondary to dermatomyositis usually develop proximal muscle weakness, high titers of creatine kinase, heliotrope rash, or interstitial lung disease with specific antibodies.
Calciphylaxis is a serious disorder involving the calcification of dermal and subcutaneous arterioles and capillaries. It presents with painful cutaneous areas of necrosis.
Venous ulcers also can present with secondary dystrophic calcification due to local tissue damage. These patients usually have cutaneous signs of chronic venous insufficiency. Our patient denied prior trauma to the area; therefore, a traumatic ulcer with secondary calcification was ruled out.
The most concerning complication of calcinosis cutis is the development of ulcers, which occurred in 154 of 316 calcinoses (48.7%) in patients with systemic sclerosis and secondary calcifications.8 These ulcers can cause disabling pain or become superinfected, as in our patient.
There currently is no drug capable of removing dystrophic calcifications, but diltiazem, minocycline, or colchicine can reduce their size and prevent their progression. In the event of neurologic compromise or intractable pain, the treatment of choice is surgical removal of the calcification.9 Curettage, intralesional sodium thiosulfate, and intravenous sodium thiosulfate also have been suggested as therapeutic options.10 Antibiotic treatment was carried out in our patient, which controlled the superinfection of the ulcers. Diltiazem also was started, with stabilization of the calcium deposits without a reduction in their size.
There are few studies evaluating the presence of nondigital ulcers in patients with systemic sclerosis. Shanmugam et al11 calculated a 4% (N=249) prevalence of ulcers in the lower limbs of systemic sclerosis patients. In a study by Bohelay et al12 of 45 patients, the estimated prevalence of lower limb ulcers was 12.8%, and the etiologies consisted of 22 cases of venous insufficiency (49%), 21 cases of ischemic causes (47%), and 2 cases of other causes (4%).
We present the case of a woman with ssSSc who developed dystrophic calcinosis cutis in atypical areas with secondary ulceration and superinfection. The skin usually plays a key role in the diagnosis of systemic sclerosis, as sclerodactyly and the characteristic generalized skin induration stand out in affected individuals. Although our patient was diagnosed with ssSSc, her skin manifestations also were crucial for the diagnosis, as she had ulcers on the lower limbs.
- Poormoghim H, Lucas M, Fertig N, et al. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum. 2000;43:444-451.
- Kucharz EJ, Kopec´-Me˛ drek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med. 2017;26:875-880.
- Valenzuela A, Baron M, Herrick AL, et al. Calcinosis is associated with digital ulcers and osteoporosis in patients with systemic sclerosis: a scleroderma clinical trials consortium study. Semin Arthritis Rheum. 2016;46:344-349.
- D’Aoust J, Hudson M, Tatibouet S, et al. Clinical and serologic correlates of antiPM/Scl antibodies in systemic sclerosis: a multicenter study of 763 patients. Arthritis Rheum. 2014;66:1608-1615.
- Contreras I, Sallés M, Mínguez S, et al. Hard paracervical tumor in a patient with limited systemic sclerosis. Rheumatol Clin. 2014; 10:336-337.
- Meriglier E, Lafourcade F, Gombert B, et al. Giant calcinosis revealing systemic sclerosis. Int J Rheum Dis. 2019;22:1787-1788.
- Chung CH. Calcinosis universalis in juvenile dermatomyositis [published online September 24, 2020]. Chonnam Med J. 2020;56:212-213.
- Bartoli F, Fiori G, Braschi F, et al. Calcinosis in systemic sclerosis: subsets, distribution and complications [published online May 30, 2016]. Rheumatology (Oxford). 2016;55:1610-1614.
- Jung H, Lee D, Cho J, et al. Surgical treatment of extensive tumoral calcinosis associated with systemic sclerosis. Korean J Thorac Cardiovasc Surg. 2015;48:151-154.
- Badawi AH, Patel V, Warner AE, et al. Dystrophic calcinosis cutis: treatment with intravenous sodium thiosulfate. Cutis. 2020;106:E15-E17.
- Shanmugam V, Price P, Attinger C, et al. Lower extremity ulcers in systemic sclerosis: features and response to therapy [published online August 18, 2010]. Int J Rheumatol. doi:10.1155/2010/747946
- Bohelay G, Blaise S, Levy P, et al. Lower-limb ulcers in systemic sclerosis: a multicentre retrospective case-control study. Acta Derm Venereol. 2018;98:677-682.
The Diagnosis: Calcinosis Cutis Due to Systemic Sclerosis Sine Scleroderma
Laboratory evaluation was notable for high titers of antinuclear antibodies (>1/320; reference range, 0–1/80) and positive anticentromere antibodies. There were no other relevant laboratory findings; phosphocalcic metabolism was within normal limits, and urinary sediment was normal. Biopsy of the edge of the ulcer revealed basophilic material compatible with calcium deposits. In a 3D volume rendering reconstruction from the lower limb scanner, grouped calcifications were observed in subcutaneous cellular tissue near the ulcer (Figure). The patient had a restrictive ventilatory pattern observed in a pulmonary function test. An esophageal motility study was normal.
The patient was diagnosed with systemic sclerosis sine scleroderma (ssSSc) type II because she met the 4 criteria established by Poormoghim et al1 : (1) Raynaud phenomenon or a peripheral vascular equivalent (ie, digital pitting scars, digital-tip ulcers, digital-tip gangrene, abnormal nail fold capillaries); (2) positive antinuclear antibodies; (3) distal esophageal hypomotility, small bowel hypomotility, pulmonary interstitial fibrosis, primary pulmonary arterial hypertension (without fibrosis), cardiac involvement typical of scleroderma, or renal failure; and (4) no other defined connective tissue or other disease as a cause of the prior conditions.
Systemic sclerosis is a chronic disease characterized by progressive fibrosis of the skin and other internal organs—especially the lungs, kidneys, digestive tract, and heart—as well as generalized vascular dysfunction. Cutaneous induration is its hallmark; however, up to 10% of affected patients have ssSSc.2 This entity is characterized by the total or partial absence of cutaneous manifestations of systemic sclerosis with the occurrence of internal organ involvement and serologic abnormalities. There are 3 types of ssSSc depending on the grade of skin involvement. Type I is characterized by the lack of any typical cutaneous stigmata of the disease. Type II is without sclerodactyly but can coexist with other cutaneous findings such as calcifications, telangiectases, or pitting scars. Type III is characterized clinically by internal organ involvement, typical of systemic sclerosis, that has appeared before skin changes.2
An abnormal deposit of calcium in the cutaneous and subcutaneous tissue is called calcinosis cutis. There are 5 subtypes of calcinosis cutis: dystrophic, metastatic, idiopathic, iatrogenic, and calciphylaxis. Dystrophic skin calcifications may appear in patients with connective tissue diseases such as dermatomyositis or systemic sclerosis.3 Up to 25% of patients with systemic sclerosis can develop calcinosis cutis due to local tissue damage, with normal phosphocalcic metabolism.3
Calcinosis cutis is more common in patients with systemic sclerosis and positive anticentromere antibodies.4 The calcifications usually are located in areas that are subject to repeated trauma, such as the fingers or arms, though other locations have been described such as cervical, paraspinal, or on the hips.5,6 Our patient developed calcifications on both legs, which represent atypical areas for this process.
Dermatomyositis also can present with calcinosis cutis. There are 4 patterns of calcification: superficial nodulelike calcified masses; deep calcified masses; deep sheetlike calcifications within the fascial planes; and a rare, diffuse, superficial lacy and reticular calcification that involves almost the entire body surface area.7 Patients with calcinosis cutis secondary to dermatomyositis usually develop proximal muscle weakness, high titers of creatine kinase, heliotrope rash, or interstitial lung disease with specific antibodies.
Calciphylaxis is a serious disorder involving the calcification of dermal and subcutaneous arterioles and capillaries. It presents with painful cutaneous areas of necrosis.
Venous ulcers also can present with secondary dystrophic calcification due to local tissue damage. These patients usually have cutaneous signs of chronic venous insufficiency. Our patient denied prior trauma to the area; therefore, a traumatic ulcer with secondary calcification was ruled out.
The most concerning complication of calcinosis cutis is the development of ulcers, which occurred in 154 of 316 calcinoses (48.7%) in patients with systemic sclerosis and secondary calcifications.8 These ulcers can cause disabling pain or become superinfected, as in our patient.
There currently is no drug capable of removing dystrophic calcifications, but diltiazem, minocycline, or colchicine can reduce their size and prevent their progression. In the event of neurologic compromise or intractable pain, the treatment of choice is surgical removal of the calcification.9 Curettage, intralesional sodium thiosulfate, and intravenous sodium thiosulfate also have been suggested as therapeutic options.10 Antibiotic treatment was carried out in our patient, which controlled the superinfection of the ulcers. Diltiazem also was started, with stabilization of the calcium deposits without a reduction in their size.
There are few studies evaluating the presence of nondigital ulcers in patients with systemic sclerosis. Shanmugam et al11 calculated a 4% (N=249) prevalence of ulcers in the lower limbs of systemic sclerosis patients. In a study by Bohelay et al12 of 45 patients, the estimated prevalence of lower limb ulcers was 12.8%, and the etiologies consisted of 22 cases of venous insufficiency (49%), 21 cases of ischemic causes (47%), and 2 cases of other causes (4%).
We present the case of a woman with ssSSc who developed dystrophic calcinosis cutis in atypical areas with secondary ulceration and superinfection. The skin usually plays a key role in the diagnosis of systemic sclerosis, as sclerodactyly and the characteristic generalized skin induration stand out in affected individuals. Although our patient was diagnosed with ssSSc, her skin manifestations also were crucial for the diagnosis, as she had ulcers on the lower limbs.
The Diagnosis: Calcinosis Cutis Due to Systemic Sclerosis Sine Scleroderma
Laboratory evaluation was notable for high titers of antinuclear antibodies (>1/320; reference range, 0–1/80) and positive anticentromere antibodies. There were no other relevant laboratory findings; phosphocalcic metabolism was within normal limits, and urinary sediment was normal. Biopsy of the edge of the ulcer revealed basophilic material compatible with calcium deposits. In a 3D volume rendering reconstruction from the lower limb scanner, grouped calcifications were observed in subcutaneous cellular tissue near the ulcer (Figure). The patient had a restrictive ventilatory pattern observed in a pulmonary function test. An esophageal motility study was normal.
The patient was diagnosed with systemic sclerosis sine scleroderma (ssSSc) type II because she met the 4 criteria established by Poormoghim et al1 : (1) Raynaud phenomenon or a peripheral vascular equivalent (ie, digital pitting scars, digital-tip ulcers, digital-tip gangrene, abnormal nail fold capillaries); (2) positive antinuclear antibodies; (3) distal esophageal hypomotility, small bowel hypomotility, pulmonary interstitial fibrosis, primary pulmonary arterial hypertension (without fibrosis), cardiac involvement typical of scleroderma, or renal failure; and (4) no other defined connective tissue or other disease as a cause of the prior conditions.
Systemic sclerosis is a chronic disease characterized by progressive fibrosis of the skin and other internal organs—especially the lungs, kidneys, digestive tract, and heart—as well as generalized vascular dysfunction. Cutaneous induration is its hallmark; however, up to 10% of affected patients have ssSSc.2 This entity is characterized by the total or partial absence of cutaneous manifestations of systemic sclerosis with the occurrence of internal organ involvement and serologic abnormalities. There are 3 types of ssSSc depending on the grade of skin involvement. Type I is characterized by the lack of any typical cutaneous stigmata of the disease. Type II is without sclerodactyly but can coexist with other cutaneous findings such as calcifications, telangiectases, or pitting scars. Type III is characterized clinically by internal organ involvement, typical of systemic sclerosis, that has appeared before skin changes.2
An abnormal deposit of calcium in the cutaneous and subcutaneous tissue is called calcinosis cutis. There are 5 subtypes of calcinosis cutis: dystrophic, metastatic, idiopathic, iatrogenic, and calciphylaxis. Dystrophic skin calcifications may appear in patients with connective tissue diseases such as dermatomyositis or systemic sclerosis.3 Up to 25% of patients with systemic sclerosis can develop calcinosis cutis due to local tissue damage, with normal phosphocalcic metabolism.3
Calcinosis cutis is more common in patients with systemic sclerosis and positive anticentromere antibodies.4 The calcifications usually are located in areas that are subject to repeated trauma, such as the fingers or arms, though other locations have been described such as cervical, paraspinal, or on the hips.5,6 Our patient developed calcifications on both legs, which represent atypical areas for this process.
Dermatomyositis also can present with calcinosis cutis. There are 4 patterns of calcification: superficial nodulelike calcified masses; deep calcified masses; deep sheetlike calcifications within the fascial planes; and a rare, diffuse, superficial lacy and reticular calcification that involves almost the entire body surface area.7 Patients with calcinosis cutis secondary to dermatomyositis usually develop proximal muscle weakness, high titers of creatine kinase, heliotrope rash, or interstitial lung disease with specific antibodies.
Calciphylaxis is a serious disorder involving the calcification of dermal and subcutaneous arterioles and capillaries. It presents with painful cutaneous areas of necrosis.
Venous ulcers also can present with secondary dystrophic calcification due to local tissue damage. These patients usually have cutaneous signs of chronic venous insufficiency. Our patient denied prior trauma to the area; therefore, a traumatic ulcer with secondary calcification was ruled out.
The most concerning complication of calcinosis cutis is the development of ulcers, which occurred in 154 of 316 calcinoses (48.7%) in patients with systemic sclerosis and secondary calcifications.8 These ulcers can cause disabling pain or become superinfected, as in our patient.
There currently is no drug capable of removing dystrophic calcifications, but diltiazem, minocycline, or colchicine can reduce their size and prevent their progression. In the event of neurologic compromise or intractable pain, the treatment of choice is surgical removal of the calcification.9 Curettage, intralesional sodium thiosulfate, and intravenous sodium thiosulfate also have been suggested as therapeutic options.10 Antibiotic treatment was carried out in our patient, which controlled the superinfection of the ulcers. Diltiazem also was started, with stabilization of the calcium deposits without a reduction in their size.
There are few studies evaluating the presence of nondigital ulcers in patients with systemic sclerosis. Shanmugam et al11 calculated a 4% (N=249) prevalence of ulcers in the lower limbs of systemic sclerosis patients. In a study by Bohelay et al12 of 45 patients, the estimated prevalence of lower limb ulcers was 12.8%, and the etiologies consisted of 22 cases of venous insufficiency (49%), 21 cases of ischemic causes (47%), and 2 cases of other causes (4%).
We present the case of a woman with ssSSc who developed dystrophic calcinosis cutis in atypical areas with secondary ulceration and superinfection. The skin usually plays a key role in the diagnosis of systemic sclerosis, as sclerodactyly and the characteristic generalized skin induration stand out in affected individuals. Although our patient was diagnosed with ssSSc, her skin manifestations also were crucial for the diagnosis, as she had ulcers on the lower limbs.
- Poormoghim H, Lucas M, Fertig N, et al. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum. 2000;43:444-451.
- Kucharz EJ, Kopec´-Me˛ drek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med. 2017;26:875-880.
- Valenzuela A, Baron M, Herrick AL, et al. Calcinosis is associated with digital ulcers and osteoporosis in patients with systemic sclerosis: a scleroderma clinical trials consortium study. Semin Arthritis Rheum. 2016;46:344-349.
- D’Aoust J, Hudson M, Tatibouet S, et al. Clinical and serologic correlates of antiPM/Scl antibodies in systemic sclerosis: a multicenter study of 763 patients. Arthritis Rheum. 2014;66:1608-1615.
- Contreras I, Sallés M, Mínguez S, et al. Hard paracervical tumor in a patient with limited systemic sclerosis. Rheumatol Clin. 2014; 10:336-337.
- Meriglier E, Lafourcade F, Gombert B, et al. Giant calcinosis revealing systemic sclerosis. Int J Rheum Dis. 2019;22:1787-1788.
- Chung CH. Calcinosis universalis in juvenile dermatomyositis [published online September 24, 2020]. Chonnam Med J. 2020;56:212-213.
- Bartoli F, Fiori G, Braschi F, et al. Calcinosis in systemic sclerosis: subsets, distribution and complications [published online May 30, 2016]. Rheumatology (Oxford). 2016;55:1610-1614.
- Jung H, Lee D, Cho J, et al. Surgical treatment of extensive tumoral calcinosis associated with systemic sclerosis. Korean J Thorac Cardiovasc Surg. 2015;48:151-154.
- Badawi AH, Patel V, Warner AE, et al. Dystrophic calcinosis cutis: treatment with intravenous sodium thiosulfate. Cutis. 2020;106:E15-E17.
- Shanmugam V, Price P, Attinger C, et al. Lower extremity ulcers in systemic sclerosis: features and response to therapy [published online August 18, 2010]. Int J Rheumatol. doi:10.1155/2010/747946
- Bohelay G, Blaise S, Levy P, et al. Lower-limb ulcers in systemic sclerosis: a multicentre retrospective case-control study. Acta Derm Venereol. 2018;98:677-682.
- Poormoghim H, Lucas M, Fertig N, et al. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum. 2000;43:444-451.
- Kucharz EJ, Kopec´-Me˛ drek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med. 2017;26:875-880.
- Valenzuela A, Baron M, Herrick AL, et al. Calcinosis is associated with digital ulcers and osteoporosis in patients with systemic sclerosis: a scleroderma clinical trials consortium study. Semin Arthritis Rheum. 2016;46:344-349.
- D’Aoust J, Hudson M, Tatibouet S, et al. Clinical and serologic correlates of antiPM/Scl antibodies in systemic sclerosis: a multicenter study of 763 patients. Arthritis Rheum. 2014;66:1608-1615.
- Contreras I, Sallés M, Mínguez S, et al. Hard paracervical tumor in a patient with limited systemic sclerosis. Rheumatol Clin. 2014; 10:336-337.
- Meriglier E, Lafourcade F, Gombert B, et al. Giant calcinosis revealing systemic sclerosis. Int J Rheum Dis. 2019;22:1787-1788.
- Chung CH. Calcinosis universalis in juvenile dermatomyositis [published online September 24, 2020]. Chonnam Med J. 2020;56:212-213.
- Bartoli F, Fiori G, Braschi F, et al. Calcinosis in systemic sclerosis: subsets, distribution and complications [published online May 30, 2016]. Rheumatology (Oxford). 2016;55:1610-1614.
- Jung H, Lee D, Cho J, et al. Surgical treatment of extensive tumoral calcinosis associated with systemic sclerosis. Korean J Thorac Cardiovasc Surg. 2015;48:151-154.
- Badawi AH, Patel V, Warner AE, et al. Dystrophic calcinosis cutis: treatment with intravenous sodium thiosulfate. Cutis. 2020;106:E15-E17.
- Shanmugam V, Price P, Attinger C, et al. Lower extremity ulcers in systemic sclerosis: features and response to therapy [published online August 18, 2010]. Int J Rheumatol. doi:10.1155/2010/747946
- Bohelay G, Blaise S, Levy P, et al. Lower-limb ulcers in systemic sclerosis: a multicentre retrospective case-control study. Acta Derm Venereol. 2018;98:677-682.
A 49-year-old woman with type 2 diabetes mellitus, morbid obesity, pulmonary fibrosis, and pulmonary arterial hypertension presented to our hospital with an ulcer on the left leg of unknown etiology that was superinfected by multidrug-resistant Klebsiella according to bacterial culture. She had an axillary temperature of 38.6 °C. She underwent amputation of the second and third toes on the left foot 5 years prior to presentation due to distal necrotic ulcers of ischemic origin. Physical examination revealed an 8×2-cm deep ulcer with abrupt edges on the left leg with fibrin and a purulent exudate. Deep palpation of the perilesional skin revealed indurated subcutaneous nodules. She also had scars on the fingertips of both hands with no induration on the rest of the skin surface. Capillaroscopy showed no pathologic findings. Blood cultures were performed, and she was admitted to the hospital for intravenous antibiotic therapy. During ulcer debridement, some solid whitish material was released.
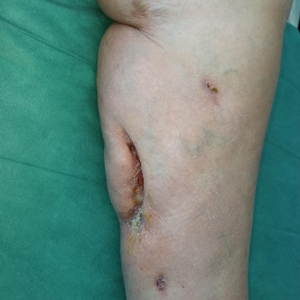
Molluscum Contagiosum Superimposed on Lymphangioma Circumscriptum
To the Editor:
Lymphangioma circumscriptum (LC) is a benign malformation of the lymphatic system.1 It is postulated to arise from abnormal lymphatic cisterns, and it grows separately from the normal lymphatic system. These cisterns are connected to malformed dermal lymphatic channels, and the contraction of smooth muscles lining cisterns will cause dilatation of connected lymphatic channels in the papillary dermis due to back pressure,1,2 which causes a classic LC manifestation characterized by multiple translucent, sometimes red-brown, small vesicles grouped together. Lymphangioma circumscriptum can be difficult to differentiate from molluscum contagiosum (MC) due to the similar morphology.1 We present a notable case of MC superimposed on LC.
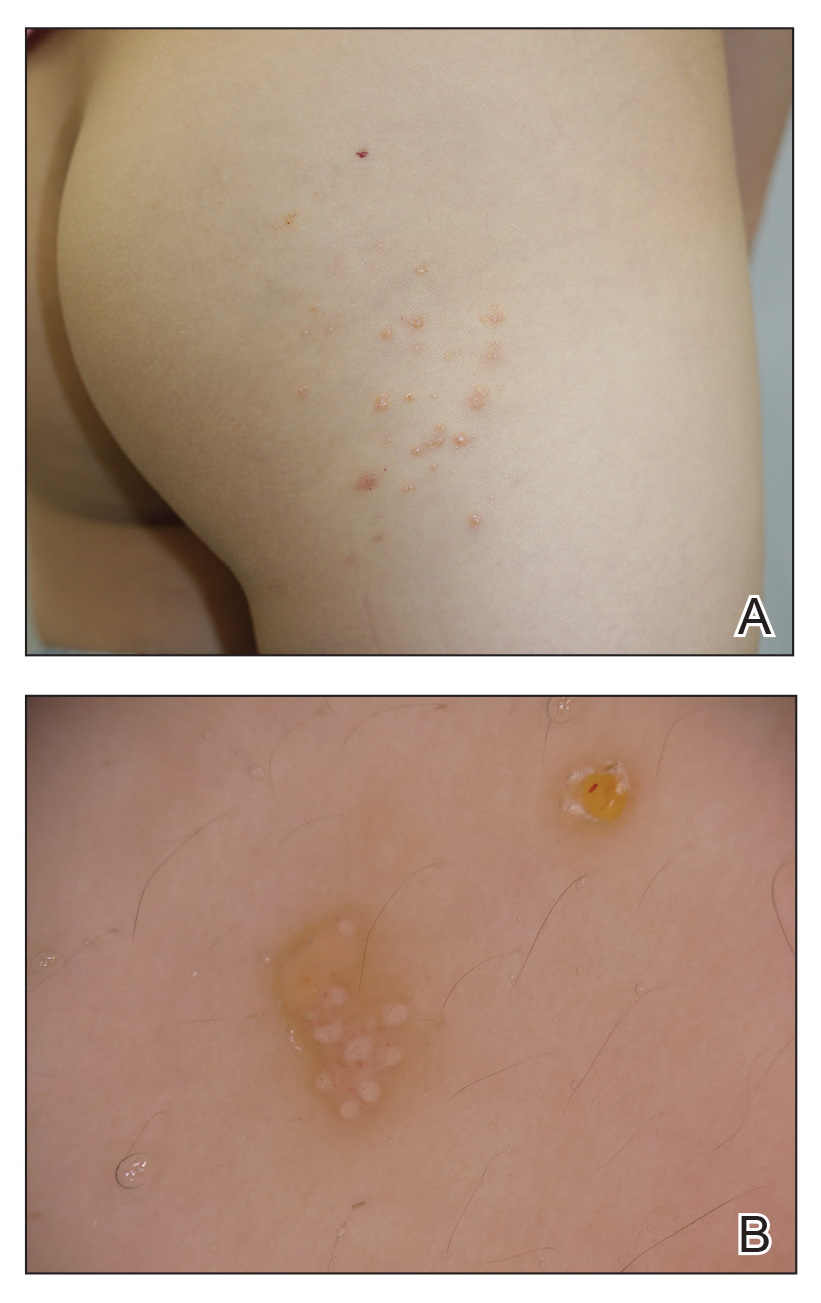
A 6-year-old girl presented with multiple grouped, clear, vesicular papules on the right buttock of 18 months’ duration. Some of the papules showed tiny whitish pearl-like particles on the top (Figure 1). Similar lesions were not present elsewhere on the body. She had no underlying disease and did not have a history of procedure, edema, or malformation of the lower extremities. Histopathology from one of the lesions showed dilated cystic lymphatic spaces in the papillary dermis lined with flattened endothelium and cup-shaped downward proliferation of the epidermis with presence of large intracytoplasmic inclusion bodies—features of both LC and MC (Figure 2). We waited 4 additional months for the MC lesions to self-resolve, but they persisted. The patient’s mother strongly requested for their removal, and the residual MC lesions were carefully removed by CO2 laser. To prevent unnecessary physical damage to underlying LC lesions and minimize scarring, we opted to use the CO2 laser and not simple curettage. She currently is under periodic observation with no signs of clinical recurrence of MC, but the LC lesions naturally persisted.
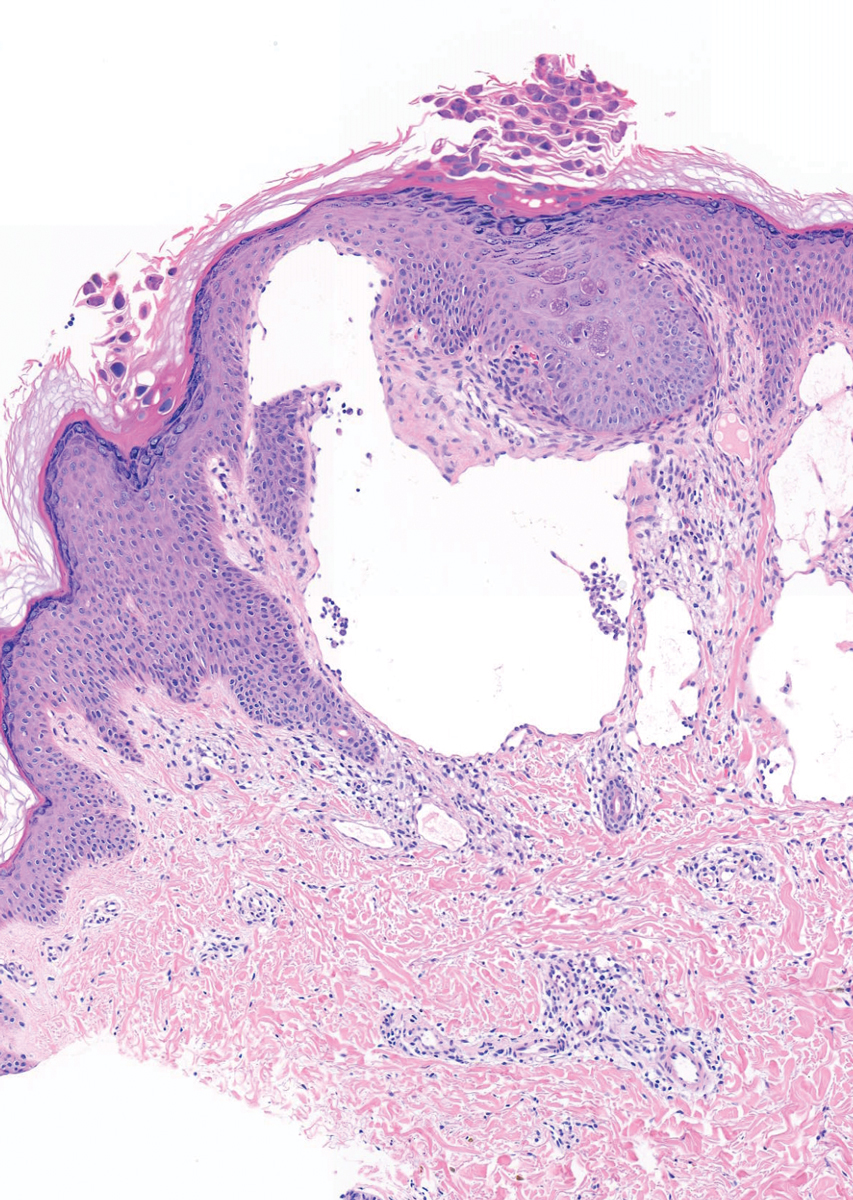
Due to its vesicular and sometimes warty appearance, LC can sometimes be hard to differentiate from MC. In one report, a vesicular plaquelike lesion on the trunk initially was misdiagnosed and treated as MC but was histologically confirmed as LC several years later.3 Our case demonstrates the coexistence of MC and LC. Although this phenomenon may be coincidental, we have not noticed any additional MC lesions on the body and MC only existed over the LC lesions, implying a possible pathophysiologic relationship. It is unlikely that MC might have preceded the development of LC. Although acquired LC exists, it has mostly been reported in the genital region of patients with conditions leading to lymphatic obstruction such as surgery, radiation therapy, malignancy, or serious infections.4 Because our patient developed lesions at an early age without any remarkable medical history, it is likely that she had congenital LC that was secondarily infected by the MC virus. Vesicular lesions in LC are known to rupture easily and may serve as a vulnerable entry site for pathogens. Subsequent secondary bacterial infections are common, with Staphylococcus aureus being the most prominent entity.1 However, secondary viral infection rarely is reported. It is possible that the abnormally dilated lymphatic channels of LC that lack communication with the normal lymphatic system have contributed to an LC site-specific vulnerability to MC virus. Further studies and subsequent reports are required to confirm this hypothesis.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111/j.1365-4632.2009.04226.x
- Fatima S, Uddin N, Idrees R, et al. Lymphangioma circumscriptum: clinicopathological spectrum of 29 cases. J Coll Physicians Surg Pak. 2015;25:658-661. doi:09.2015/JCPSP.658661
- Patel GA, Siperstein RD, Ragi G, Schwartz RA. Zosteriform lymphangioma circumscriptum. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:179-182.
- Chang MB, Newman CC, Davis MD, et al. Acquired lymphangiectasia (lymphangioma circumscriptum) of the vulva: clinicopathologic study of 11 patients from a single institution and 67 from the literature. Int J Dermatol. 2016;55:E482-E487. doi:10.1111/ijd.13264
To the Editor:
Lymphangioma circumscriptum (LC) is a benign malformation of the lymphatic system.1 It is postulated to arise from abnormal lymphatic cisterns, and it grows separately from the normal lymphatic system. These cisterns are connected to malformed dermal lymphatic channels, and the contraction of smooth muscles lining cisterns will cause dilatation of connected lymphatic channels in the papillary dermis due to back pressure,1,2 which causes a classic LC manifestation characterized by multiple translucent, sometimes red-brown, small vesicles grouped together. Lymphangioma circumscriptum can be difficult to differentiate from molluscum contagiosum (MC) due to the similar morphology.1 We present a notable case of MC superimposed on LC.
A 6-year-old girl presented with multiple grouped, clear, vesicular papules on the right buttock of 18 months’ duration. Some of the papules showed tiny whitish pearl-like particles on the top (Figure 1). Similar lesions were not present elsewhere on the body. She had no underlying disease and did not have a history of procedure, edema, or malformation of the lower extremities. Histopathology from one of the lesions showed dilated cystic lymphatic spaces in the papillary dermis lined with flattened endothelium and cup-shaped downward proliferation of the epidermis with presence of large intracytoplasmic inclusion bodies—features of both LC and MC (Figure 2). We waited 4 additional months for the MC lesions to self-resolve, but they persisted. The patient’s mother strongly requested for their removal, and the residual MC lesions were carefully removed by CO2 laser. To prevent unnecessary physical damage to underlying LC lesions and minimize scarring, we opted to use the CO2 laser and not simple curettage. She currently is under periodic observation with no signs of clinical recurrence of MC, but the LC lesions naturally persisted.
Due to its vesicular and sometimes warty appearance, LC can sometimes be hard to differentiate from MC. In one report, a vesicular plaquelike lesion on the trunk initially was misdiagnosed and treated as MC but was histologically confirmed as LC several years later.3 Our case demonstrates the coexistence of MC and LC. Although this phenomenon may be coincidental, we have not noticed any additional MC lesions on the body and MC only existed over the LC lesions, implying a possible pathophysiologic relationship. It is unlikely that MC might have preceded the development of LC. Although acquired LC exists, it has mostly been reported in the genital region of patients with conditions leading to lymphatic obstruction such as surgery, radiation therapy, malignancy, or serious infections.4 Because our patient developed lesions at an early age without any remarkable medical history, it is likely that she had congenital LC that was secondarily infected by the MC virus. Vesicular lesions in LC are known to rupture easily and may serve as a vulnerable entry site for pathogens. Subsequent secondary bacterial infections are common, with Staphylococcus aureus being the most prominent entity.1 However, secondary viral infection rarely is reported. It is possible that the abnormally dilated lymphatic channels of LC that lack communication with the normal lymphatic system have contributed to an LC site-specific vulnerability to MC virus. Further studies and subsequent reports are required to confirm this hypothesis.
To the Editor:
Lymphangioma circumscriptum (LC) is a benign malformation of the lymphatic system.1 It is postulated to arise from abnormal lymphatic cisterns, and it grows separately from the normal lymphatic system. These cisterns are connected to malformed dermal lymphatic channels, and the contraction of smooth muscles lining cisterns will cause dilatation of connected lymphatic channels in the papillary dermis due to back pressure,1,2 which causes a classic LC manifestation characterized by multiple translucent, sometimes red-brown, small vesicles grouped together. Lymphangioma circumscriptum can be difficult to differentiate from molluscum contagiosum (MC) due to the similar morphology.1 We present a notable case of MC superimposed on LC.
A 6-year-old girl presented with multiple grouped, clear, vesicular papules on the right buttock of 18 months’ duration. Some of the papules showed tiny whitish pearl-like particles on the top (Figure 1). Similar lesions were not present elsewhere on the body. She had no underlying disease and did not have a history of procedure, edema, or malformation of the lower extremities. Histopathology from one of the lesions showed dilated cystic lymphatic spaces in the papillary dermis lined with flattened endothelium and cup-shaped downward proliferation of the epidermis with presence of large intracytoplasmic inclusion bodies—features of both LC and MC (Figure 2). We waited 4 additional months for the MC lesions to self-resolve, but they persisted. The patient’s mother strongly requested for their removal, and the residual MC lesions were carefully removed by CO2 laser. To prevent unnecessary physical damage to underlying LC lesions and minimize scarring, we opted to use the CO2 laser and not simple curettage. She currently is under periodic observation with no signs of clinical recurrence of MC, but the LC lesions naturally persisted.
Due to its vesicular and sometimes warty appearance, LC can sometimes be hard to differentiate from MC. In one report, a vesicular plaquelike lesion on the trunk initially was misdiagnosed and treated as MC but was histologically confirmed as LC several years later.3 Our case demonstrates the coexistence of MC and LC. Although this phenomenon may be coincidental, we have not noticed any additional MC lesions on the body and MC only existed over the LC lesions, implying a possible pathophysiologic relationship. It is unlikely that MC might have preceded the development of LC. Although acquired LC exists, it has mostly been reported in the genital region of patients with conditions leading to lymphatic obstruction such as surgery, radiation therapy, malignancy, or serious infections.4 Because our patient developed lesions at an early age without any remarkable medical history, it is likely that she had congenital LC that was secondarily infected by the MC virus. Vesicular lesions in LC are known to rupture easily and may serve as a vulnerable entry site for pathogens. Subsequent secondary bacterial infections are common, with Staphylococcus aureus being the most prominent entity.1 However, secondary viral infection rarely is reported. It is possible that the abnormally dilated lymphatic channels of LC that lack communication with the normal lymphatic system have contributed to an LC site-specific vulnerability to MC virus. Further studies and subsequent reports are required to confirm this hypothesis.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111/j.1365-4632.2009.04226.x
- Fatima S, Uddin N, Idrees R, et al. Lymphangioma circumscriptum: clinicopathological spectrum of 29 cases. J Coll Physicians Surg Pak. 2015;25:658-661. doi:09.2015/JCPSP.658661
- Patel GA, Siperstein RD, Ragi G, Schwartz RA. Zosteriform lymphangioma circumscriptum. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:179-182.
- Chang MB, Newman CC, Davis MD, et al. Acquired lymphangiectasia (lymphangioma circumscriptum) of the vulva: clinicopathologic study of 11 patients from a single institution and 67 from the literature. Int J Dermatol. 2016;55:E482-E487. doi:10.1111/ijd.13264
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111/j.1365-4632.2009.04226.x
- Fatima S, Uddin N, Idrees R, et al. Lymphangioma circumscriptum: clinicopathological spectrum of 29 cases. J Coll Physicians Surg Pak. 2015;25:658-661. doi:09.2015/JCPSP.658661
- Patel GA, Siperstein RD, Ragi G, Schwartz RA. Zosteriform lymphangioma circumscriptum. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:179-182.
- Chang MB, Newman CC, Davis MD, et al. Acquired lymphangiectasia (lymphangioma circumscriptum) of the vulva: clinicopathologic study of 11 patients from a single institution and 67 from the literature. Int J Dermatol. 2016;55:E482-E487. doi:10.1111/ijd.13264
Practice Points
- Lymphangioma circumscriptum (LC) is a benign malformation of the lymphatic system that can be misdiagnosed as molluscum contagiosum (MC).
- Secondary infection of LC is common, with Staphylococcus aureus being the most common entity, but MC virus also can be secondarily infected.
Enoxaparin-Induced Hemorrhagic Bullae at Sites of Trauma and Endothelial Pathology
To the Editor:
A 67-year-old man with diabetes mellitus was admitted to the hospital for exacerbation of congestive heart failure and atrial flutter with rapid ventricular response. He subsequently developed a non-ST segment elevation myocardial infarction and was started on subcutaneous enoxaparin 110 mg twice daily. On day 9 of hospitalization, small “blood blisters” on the legs were noted by the nurse, and dermatology was consulted.
Physical examination revealed tense hemorrhagic bullae with erythematous haloes scattered over the arms and legs and to a lesser extent on the trunk. The bullae were most concentrated at the surrounding subcutaneous injection sites of insulin and enoxaparin with secondary bruising (Figure 1). The lesions also were present on the legs, where pitting edema and capillaritis also were appreciated (Figure 2).
Laboratory workup for heparin-induced thrombocytopenia was negative. A diagnosis of enoxaparin-associated hemorrhagic bullae was made. Biopsy was recommended, but the patient declined based on anecdotal reports that the bullae typically self-resolve.
The enoxaparin was discontinued 7 days after the dermatology consultation, and the patient was transitioned to apixaban. A review of the medical record during the dermatology consultation revealed he had been on aspirin (81–385 mg/d) for 13 years prior to admission and had received prophylactic enoxaparin (40 mg/d) while hospitalized 2 and 7 years prior to the current episode of hemorrhagic bullae.
The patient declined outpatient dermatology follow-up; however, his cardiologist noted that the skin lesions had resolved at a 3-week postdischarge appointment. Approximately 5 months after discharge, the patient was re-treated by the cardiologist with enoxaparin 110 mg twice daily for 3 days to bridge to warfarin after he developed a deep vein thrombosis while taking apixaban. He did not develop hemorrhagic bullae upon retreatment with enoxaparin.
Heparin-induced hemorrhagic bullous dermatosis (HBD) has been associated with administration of both unfractionated and low-molecular-weight heparin.1 The condition typically develops 5 to 21 days after initiation of heparin as asymptomatic, purple-to-black bullae, sometimes with an erythematous halo.2,3 The arms and legs are the most common location, but the exact pathogenesis of the lesions remains unknown.3,4 Most cases resolve within weeks of discontinuing heparin, although some reports have suggested that discontinuation is unnecessary.3,4
Histopathologic analysis shows intraepidermal or subepidermal bullae with red blood cells and fibrin in the absence of vasculitis and intravascular thrombi.1,4 Immunofluorescence studies are negative.3 In a comprehensive review of HBD, the investigators hypothesized that the pathogenesis may be related to noninflammatory to pauci-inflammatory activation of basement membrane zone proteases or possibly epithelial or endothelial fragility in conjunction with trauma that causes disruption of the vascular endothelium (eg, subcutaneous injections, vasculitis).4
Our case is of particular interest because the bullae were strikingly limited to sites of subcutaneous injection and surrounding areas along with coexistent endothelial pathology on the lower legs (capillaritis and pitting edema). These clinical observations support trauma from the injections and altered endothelia as pathogenetic factors in HBD.
Of interest, our patient had 2 prior hospitalizations during which he received prophylactic enoxaparin and did not develop hemorrhagic bullae. Furthermore, repeat exposure to therapeutic dosing of enoxaparin with a shorter duration did not result in recurrence of HBD. This suggests that heparin dosing and duration of therapy also might be involved in the development of HBD.
Our hope is that future reports of HBD will address the presence or absence of coexistent cutaneous pathology, such as edema, stasis dermatitis, bruising, and capillaritis, along with heparin dosing, duration, and prior exposure to heparin treatment so that risk factors and pathogenesis can be further investigated. We also agree with Snow et al4 that HBD should be included as an outcome in future trials of heparin therapy.
- Komforti MK, Bressler ES, Selim MA, et al. A rare cutaneous manifestation of hemorrhagic bullae to low-molecular-weight heparin and fondaparinux: report of two cases: letter to the editor. J Cutan Pathol. 2017;44:104-106. doi:10.1111/cup.12821
- Peña ZG, Suszko JW, Morrison LH. Hemorrhagic bullae in a 73-year-old man. JAMA Dermatol. 2013;149:871-872. doi:10.1001/jamadermatol.2013.3364a
- Gouveia AI, Lopes L, Soares-Almeida L, et al. Bullous hemorrhagic dermatosis induced by enoxaparin. Cutan Ocul Toxicol. 2016;35:160-162. doi:10.3109/15569527.2015.1041033
- Snow SC, Pearson DR, Fathi R, et al. Heparin‐induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398. doi:10.1111/ced.13327
To the Editor:
A 67-year-old man with diabetes mellitus was admitted to the hospital for exacerbation of congestive heart failure and atrial flutter with rapid ventricular response. He subsequently developed a non-ST segment elevation myocardial infarction and was started on subcutaneous enoxaparin 110 mg twice daily. On day 9 of hospitalization, small “blood blisters” on the legs were noted by the nurse, and dermatology was consulted.
Physical examination revealed tense hemorrhagic bullae with erythematous haloes scattered over the arms and legs and to a lesser extent on the trunk. The bullae were most concentrated at the surrounding subcutaneous injection sites of insulin and enoxaparin with secondary bruising (Figure 1). The lesions also were present on the legs, where pitting edema and capillaritis also were appreciated (Figure 2).
Laboratory workup for heparin-induced thrombocytopenia was negative. A diagnosis of enoxaparin-associated hemorrhagic bullae was made. Biopsy was recommended, but the patient declined based on anecdotal reports that the bullae typically self-resolve.
The enoxaparin was discontinued 7 days after the dermatology consultation, and the patient was transitioned to apixaban. A review of the medical record during the dermatology consultation revealed he had been on aspirin (81–385 mg/d) for 13 years prior to admission and had received prophylactic enoxaparin (40 mg/d) while hospitalized 2 and 7 years prior to the current episode of hemorrhagic bullae.
The patient declined outpatient dermatology follow-up; however, his cardiologist noted that the skin lesions had resolved at a 3-week postdischarge appointment. Approximately 5 months after discharge, the patient was re-treated by the cardiologist with enoxaparin 110 mg twice daily for 3 days to bridge to warfarin after he developed a deep vein thrombosis while taking apixaban. He did not develop hemorrhagic bullae upon retreatment with enoxaparin.
Heparin-induced hemorrhagic bullous dermatosis (HBD) has been associated with administration of both unfractionated and low-molecular-weight heparin.1 The condition typically develops 5 to 21 days after initiation of heparin as asymptomatic, purple-to-black bullae, sometimes with an erythematous halo.2,3 The arms and legs are the most common location, but the exact pathogenesis of the lesions remains unknown.3,4 Most cases resolve within weeks of discontinuing heparin, although some reports have suggested that discontinuation is unnecessary.3,4
Histopathologic analysis shows intraepidermal or subepidermal bullae with red blood cells and fibrin in the absence of vasculitis and intravascular thrombi.1,4 Immunofluorescence studies are negative.3 In a comprehensive review of HBD, the investigators hypothesized that the pathogenesis may be related to noninflammatory to pauci-inflammatory activation of basement membrane zone proteases or possibly epithelial or endothelial fragility in conjunction with trauma that causes disruption of the vascular endothelium (eg, subcutaneous injections, vasculitis).4
Our case is of particular interest because the bullae were strikingly limited to sites of subcutaneous injection and surrounding areas along with coexistent endothelial pathology on the lower legs (capillaritis and pitting edema). These clinical observations support trauma from the injections and altered endothelia as pathogenetic factors in HBD.
Of interest, our patient had 2 prior hospitalizations during which he received prophylactic enoxaparin and did not develop hemorrhagic bullae. Furthermore, repeat exposure to therapeutic dosing of enoxaparin with a shorter duration did not result in recurrence of HBD. This suggests that heparin dosing and duration of therapy also might be involved in the development of HBD.
Our hope is that future reports of HBD will address the presence or absence of coexistent cutaneous pathology, such as edema, stasis dermatitis, bruising, and capillaritis, along with heparin dosing, duration, and prior exposure to heparin treatment so that risk factors and pathogenesis can be further investigated. We also agree with Snow et al4 that HBD should be included as an outcome in future trials of heparin therapy.
To the Editor:
A 67-year-old man with diabetes mellitus was admitted to the hospital for exacerbation of congestive heart failure and atrial flutter with rapid ventricular response. He subsequently developed a non-ST segment elevation myocardial infarction and was started on subcutaneous enoxaparin 110 mg twice daily. On day 9 of hospitalization, small “blood blisters” on the legs were noted by the nurse, and dermatology was consulted.
Physical examination revealed tense hemorrhagic bullae with erythematous haloes scattered over the arms and legs and to a lesser extent on the trunk. The bullae were most concentrated at the surrounding subcutaneous injection sites of insulin and enoxaparin with secondary bruising (Figure 1). The lesions also were present on the legs, where pitting edema and capillaritis also were appreciated (Figure 2).
Laboratory workup for heparin-induced thrombocytopenia was negative. A diagnosis of enoxaparin-associated hemorrhagic bullae was made. Biopsy was recommended, but the patient declined based on anecdotal reports that the bullae typically self-resolve.
The enoxaparin was discontinued 7 days after the dermatology consultation, and the patient was transitioned to apixaban. A review of the medical record during the dermatology consultation revealed he had been on aspirin (81–385 mg/d) for 13 years prior to admission and had received prophylactic enoxaparin (40 mg/d) while hospitalized 2 and 7 years prior to the current episode of hemorrhagic bullae.
The patient declined outpatient dermatology follow-up; however, his cardiologist noted that the skin lesions had resolved at a 3-week postdischarge appointment. Approximately 5 months after discharge, the patient was re-treated by the cardiologist with enoxaparin 110 mg twice daily for 3 days to bridge to warfarin after he developed a deep vein thrombosis while taking apixaban. He did not develop hemorrhagic bullae upon retreatment with enoxaparin.
Heparin-induced hemorrhagic bullous dermatosis (HBD) has been associated with administration of both unfractionated and low-molecular-weight heparin.1 The condition typically develops 5 to 21 days after initiation of heparin as asymptomatic, purple-to-black bullae, sometimes with an erythematous halo.2,3 The arms and legs are the most common location, but the exact pathogenesis of the lesions remains unknown.3,4 Most cases resolve within weeks of discontinuing heparin, although some reports have suggested that discontinuation is unnecessary.3,4
Histopathologic analysis shows intraepidermal or subepidermal bullae with red blood cells and fibrin in the absence of vasculitis and intravascular thrombi.1,4 Immunofluorescence studies are negative.3 In a comprehensive review of HBD, the investigators hypothesized that the pathogenesis may be related to noninflammatory to pauci-inflammatory activation of basement membrane zone proteases or possibly epithelial or endothelial fragility in conjunction with trauma that causes disruption of the vascular endothelium (eg, subcutaneous injections, vasculitis).4
Our case is of particular interest because the bullae were strikingly limited to sites of subcutaneous injection and surrounding areas along with coexistent endothelial pathology on the lower legs (capillaritis and pitting edema). These clinical observations support trauma from the injections and altered endothelia as pathogenetic factors in HBD.
Of interest, our patient had 2 prior hospitalizations during which he received prophylactic enoxaparin and did not develop hemorrhagic bullae. Furthermore, repeat exposure to therapeutic dosing of enoxaparin with a shorter duration did not result in recurrence of HBD. This suggests that heparin dosing and duration of therapy also might be involved in the development of HBD.
Our hope is that future reports of HBD will address the presence or absence of coexistent cutaneous pathology, such as edema, stasis dermatitis, bruising, and capillaritis, along with heparin dosing, duration, and prior exposure to heparin treatment so that risk factors and pathogenesis can be further investigated. We also agree with Snow et al4 that HBD should be included as an outcome in future trials of heparin therapy.
- Komforti MK, Bressler ES, Selim MA, et al. A rare cutaneous manifestation of hemorrhagic bullae to low-molecular-weight heparin and fondaparinux: report of two cases: letter to the editor. J Cutan Pathol. 2017;44:104-106. doi:10.1111/cup.12821
- Peña ZG, Suszko JW, Morrison LH. Hemorrhagic bullae in a 73-year-old man. JAMA Dermatol. 2013;149:871-872. doi:10.1001/jamadermatol.2013.3364a
- Gouveia AI, Lopes L, Soares-Almeida L, et al. Bullous hemorrhagic dermatosis induced by enoxaparin. Cutan Ocul Toxicol. 2016;35:160-162. doi:10.3109/15569527.2015.1041033
- Snow SC, Pearson DR, Fathi R, et al. Heparin‐induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398. doi:10.1111/ced.13327
- Komforti MK, Bressler ES, Selim MA, et al. A rare cutaneous manifestation of hemorrhagic bullae to low-molecular-weight heparin and fondaparinux: report of two cases: letter to the editor. J Cutan Pathol. 2017;44:104-106. doi:10.1111/cup.12821
- Peña ZG, Suszko JW, Morrison LH. Hemorrhagic bullae in a 73-year-old man. JAMA Dermatol. 2013;149:871-872. doi:10.1001/jamadermatol.2013.3364a
- Gouveia AI, Lopes L, Soares-Almeida L, et al. Bullous hemorrhagic dermatosis induced by enoxaparin. Cutan Ocul Toxicol. 2016;35:160-162. doi:10.3109/15569527.2015.1041033
- Snow SC, Pearson DR, Fathi R, et al. Heparin‐induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398. doi:10.1111/ced.13327
Granulomatous Facial Dermatoses
Cutaneous granulomatous diseases encompass many entities that are skin-limited or systemic. The prototypical cutaneous granuloma is a painless, rounded, well-defined, red-pink or flesh-colored papule1 and is smooth, owing to minimal epidermal involvement. Examples of conditions that present with such lesions include granulomatous periorificial dermatitis (GPD), granulomatous rosacea (GR), lupus miliaris disseminatus faciei (LMDF), and papular sarcoidosis. These entities commonly are seen on the face and can be a source of distress to patients when they are extensive. Several reports have raised the possibility that these conditions lie on a spectrum.2-4 We present 2 cases of patients with facial papular granulomas, discuss potential causes of the lesions, review historical aspects from the literature, and highlight the challenges that these lesions can pose to the clinician.
Case Reports
Patient 1—A 10-year-old Ethiopian girl with a history of atopic dermatitis presented with a facial rash of 4 months’ duration. Her pediatrician initially treated the rash as pityriasis alba and prescribed hydrocortisone cream. Two months into treatment, the patient developed an otherwise asymptomatic, unilateral, papular dermatosis on the right cheek. She subsequently was switched to treatment with benzoyl peroxide and topical clindamycin, which she had been using for 2 months with no improvement at the time of the current presentation. The lesions then spread bilaterally and periorally.
At the current presentation, physical examination demonstrated fine, diffuse, follicular-based, flesh-colored papules over both cheeks, the right side of the nose, and the perioral region (Figure 1). A biopsy of a papular lesion from the right cheek revealed well-formed, noncaseating granulomas in the superficial and mid dermis with an associated lymphocytic infiltrate (Figure 2). No organisms were identified on acid-fast, Fite, or periodic acid–Schiff staining. A tuberculin skin test was negative. A chest radiograph showed small calcified hilar lymph nodes bilaterally. Pulmonary function tests were unremarkable. Calcium and angiotensin-converting enzyme levels were normal.
The patient denied any fever, chills, hemoptysis, cough, dyspnea, lymphadenopathy, scleral or conjunctival pain or erythema, visual disturbances, or arthralgias. Hydroxychloroquine 200 mg twice daily was started with minimal improvement after 5 months. Methotrexate 20 mg once weekly was then added. Topical fluocinonide 0.05% also was started at this time, as the patient had required several prednisone tapers over the past 3 months for symptomatic relief. The lesions improved minimally after 5 more months of treatment, at which time she had developed inflammatory papules, pustules, and open comedones in the same areas as well as the glabella.
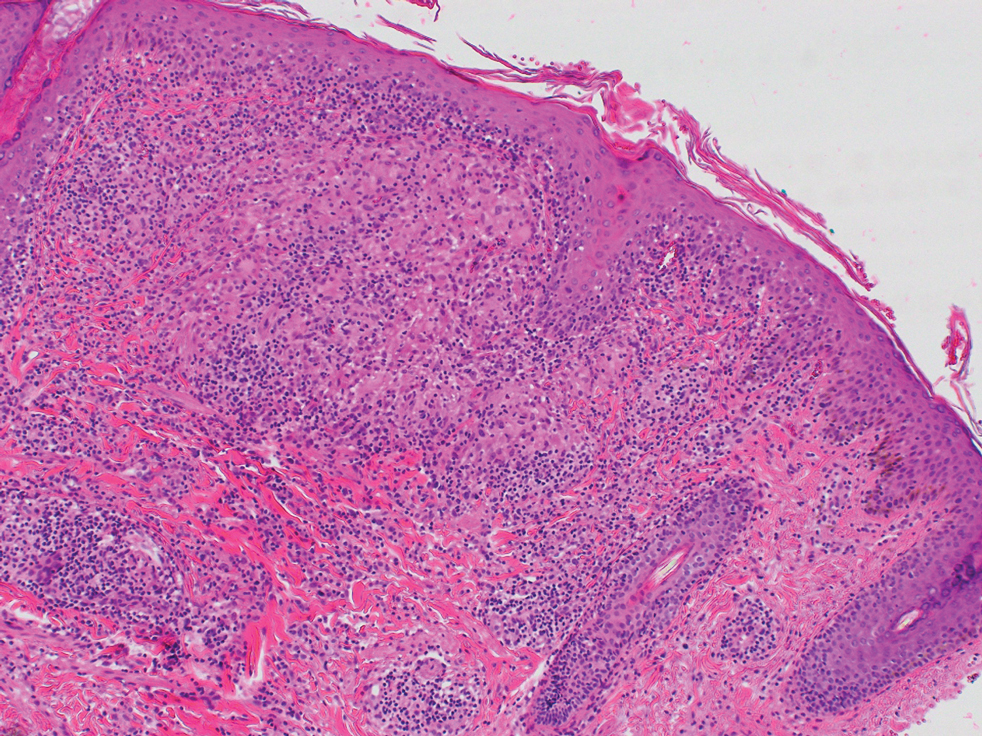
Repeat biopsy of a papular lesion demonstrated noncaseating granulomas and an associated chronic lymphocytic infiltrate in a follicular and perifollicular distribution (Figure 3). Biopsy of a pustule demonstrated acute Demodex folliculitis. Fluocinonide was stopped, and anti-mite therapy with ivermectin, permethrin cream 5%, and selenium sulfide lotion 2.5% was started, with good response from the pustular lesions.
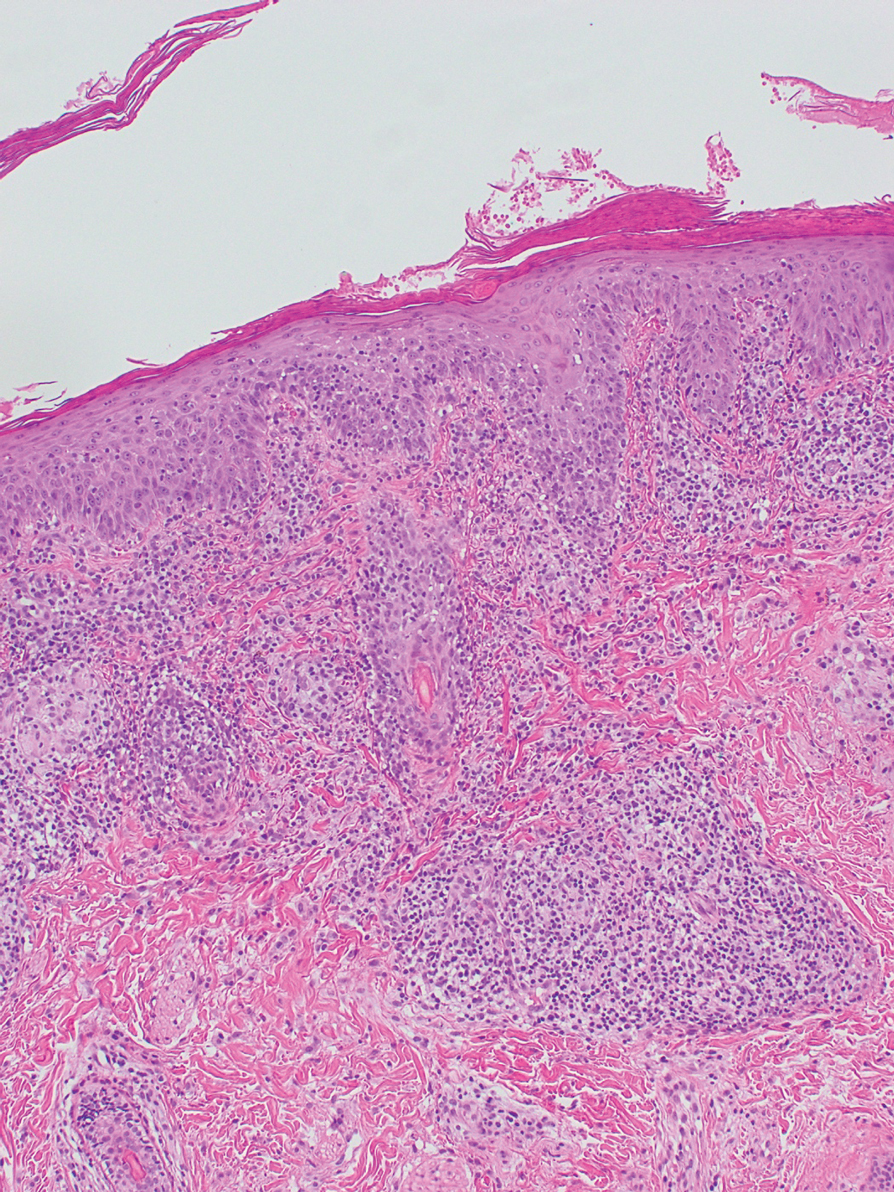
The patient continued taking methotrexate 20 mg once weekly during this time, with improvement in the papular lesions. She discontinued methotrexate after 12 months with complete resolution. At follow-up 12 months after stopping the methotrexate (roughly 2 years after initial presentation), she showed sustained resolution, with small pitted scars on both cheeks and the nasal tip.
Patient 2—A 33-year-old Ethiopian woman presented with a facial rash of 15 years’ duration. The lesions had been accumulating slowly and were asymptomatic. Physical examination revealed multiple follicular-based, flesh-colored, and erythematous papules on the cheeks, chin, perioral area, and forehead (Figure 4). There were no pustules or telangiectasias. Treatment with tretinoin cream 0.05% for 6 months offered minimal relief.
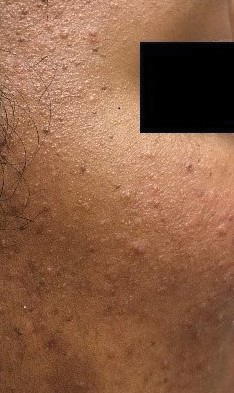
Biopsy of a papule from the left mandible showed superficial vascular telangiectasias, noncaseating granulomas comprising epithelioid histiocytes and lymphocytes in the superficial dermis, and a perifollicular lymphocytic infiltrate (Figure 5). No organisms were identified on Fite or Gomori methenamine silver staining.
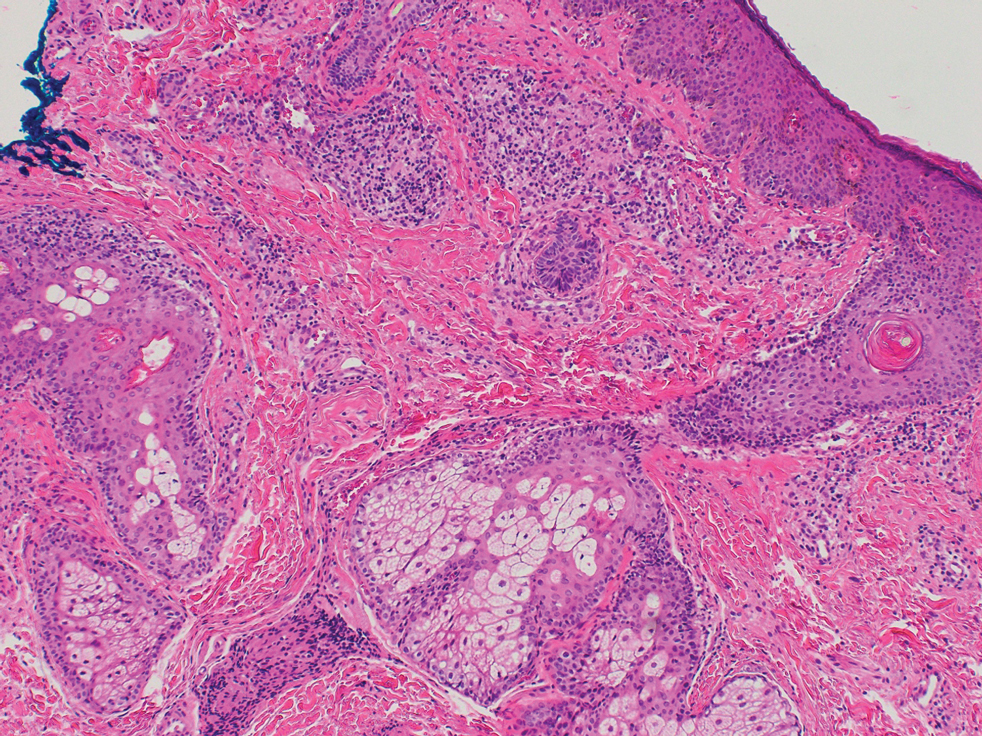
Comment
The first step in differentiating cutaneous granulomatous lesions should be to distinguish infectious from noninfectious causes.1 Noninfectious cutaneous granulomas can appear nearly anywhere; however, certain processes have a predilection for the face, including GPD, GR, LMDF, and papular sarcoidosis.5-7 These conditions generally present with papular granulomas with features as described above.
Granulomatous Periorificial Dermatitis—In 1970, Gianotti and colleagues8 briefly described the first possible cases of GPD in 5 children. The eruption comprised numerous yellow, dome-shaped papules in a mostly perioral distribution. Tuberculin and the Kveim tests were nonreactive; histopathology was described as sarcoid-type and not necessarily follicular or perifollicular.8 In 1974, Marten et al9 described 22 Afro-Caribbean children with flesh-colored, papular eruptions on the face that did not show histologic granulomatous changes but were morphologically similar to the reports by Gianotti et al.8 By 1989, Frieden and colleagues10 described this facial eruption as “granulomatous perioral dermatitis in children”. Additionally, the investigators observed granulomatous infiltrates in a perifollicular distribution and suggested follicular disruption as a possible cause. It was clear from the case discussions that these eruptions were not uncommonly diagnosed as papular sarcoidosis.10 The following year, Williams et al11 reported 5 cases of similar papular eruptions in 5 Afro-Caribbean children, coining the term facial Afro-Caribbean eruption.11 Knautz and Lesher12 referred to this entity as “childhood GPD” in 1996 to avoid limiting the diagnosis to Afro-Caribbean patients and to a perioral distribution; this is the most popular current terminology.12 Since then, reports of extrafacial involvement and disease in adults have been published.13,14
Granulomatous periorificial dermatitis often is seen in the perinasal, periocular, and perioral regions of the face.2 It is associated with topical steroid exposure.5 Histologically, noncaseating granulomas around the upper half of undisrupted hair follicles with a lymphocytic infiltrate are typical.13 Treatment should begin with cessation of any topical steroids; first-line agents are oral tetracycline or macrolide antibiotics.5 These agents can be used alone or in combination with topical erythromycin, metronidazole, or sulfur-based lotions.13 Rarely, GPD presents extrafacially.13 Even so, it usually resolves within 2 weeks to 6 months, especially with therapy; scarring is unusual.5,13,15
Granulomatous Rosacea—A report in the early 20th century described patients with tuberculoid granulomas resembling papular rosacea; the initial belief was that this finding represented a rosacealike tuberculid eruption.5 However, this belief was questioned by Snapp,16 among others, who demonstrated near universal lack of reactivity to tuberculin among 20 of these patients in 1949; more recent evidence has substantiated these findings.17 Still, Snapp16 postulated that these rosacealike granulomatous lesions were distinct from classic rosacea because they lacked vascular symptoms and pustules and were recalcitrant to rosacea treatment modalities.
In 1970, Mullanax and colleagues18 introduced the term granulomatous rosacea, reiterating that this entity was not tuberculous. They documented papulopustular lesions as well as telangiectasias, raising the possibility that GR does overlap with acne rosacea. More recent studies have established the current theory that GR is a histologic variant of acne rosacea because, in addition to typical granulomatous papules, its microscopic features can be seen across subtypes of acne rosacea.19,20
Various causes have been proposed for GR. Demodex mites have been reported in association with GR for nearly 30 years.19,20 In the past 10 years, molecular studies have started to define the role of metalloproteinases, UV radiation, and cutaneous peptides in the pathogenesis of acne rosacea and GR.21,22
Granulomatous rosacea typically is seen in middle-aged women.20,23 Hallmarks of rosacea, such as facial erythema, flushing, telangiectasias, pustules, and rhinophyma, are not always present in GR.5,20,23 Lesions usually are distributed around the central face, although extension to the cheeks, total facial involvement, and extrafacial lesions are possible.5,20 Histologically, perifollicular and follicular-based noncaseating granulomas with dilatation of the dermal papillary vasculature are seen.17,23 As a whole, rosacea is comparatively uncommon in dark-skinned patients; when it does occur, GR is a frequent presentation.24
First-line treatment for GR is tetracycline antibiotics.5 Unresponsive cases have been treated—largely anecdotally—with topical modalities (eg, metronidazole, steroids, immunomodulators), systemic agents (eg, dapsone, erythromycin, isotretinoin), and other therapies.5 Granulomatous rosacea tends to have a chronic course.5,23
Lupus Miliaris Disseminatus Faciei—Classic LMDF demonstrates caseating perifollicular granulomas histologically.6,17,25 Lesions tend to appear on the central face, particularly the eyelids, and can be seen extrafacially.3,6,25,26 Although LMDF originally was categorized as a tuberculid eruption, this no longer is thought to be the case.27 It is now regarded by some as a variant of GR25; however, LMDF responds poorly to tetracyclines, is more common in males, and lacks rosacealike vascular abnormalities, leading some to question this association.3,6,17 In the past 20 years, some have proposed renaming LMDF to better reflect its clinical course and to consider it independent of tuberculosis and GR.28 It usually resolves spontaneously after 1 to 3 years, leaving pitted scars.3,6
Papular Sarcoidosis—The first potential documented case of sarcoidosis was by Hutchinson29 in 1869 in a patient seen in London. The author labeled purple plaques on the index patient’s legs and hands as “livid papillary psoriasis.” In 1889, Besnier30 described a patient with violaceous swellings on the nose, ears, and fingers, which he called “lupus pernio”; his contemporary, Tenneson,31 published a case of lupus pernio and described its histologic profile as comprising epithelioid cells and giant cells. It was not until 1899 that the term sarkoid was used to describe these cutaneous lesions by Boeck,32 who thought they were reminiscent of sarcoma. In 1915, Kuznitsky and Bittorf33 described a patient with cutaneous lesions histologically consistent with Boeck’s sarkoid but additionally with hilar lymphadenopathy and pulmonary infiltrates. Around 1916 or 1917, Schaumann34 described patients with cutaneous lesions and additionally with involvement of pulmonary, osseous, hepatosplenic, and tonsillar tissue. These reports are among the first to recognize the multisystemic nature of sarcoidosis. The first possible case of childhood sarcoidosis might have been reported by Osler35 in the United States in 1898.
In the past century or so, an ongoing effort by researchers has focused on identifying etiologic triggers for sarcoidosis. Microbial agents have been considered in this role, with Mycobacterium and Propionibacterium organisms the most intensively studied; the possibility that foreign material contributes to the formation of granulomas also has been raised.36 Current models of the pathogenesis of sarcoidosis involve an interplay between the immune system in genetically predisposed patients and an infection that leads to a hyperimmune type 1 T–helper cell response that clears the infection but not antigens generated by the microbes and the acute host response, including proteins such as serum amyloid A and vimentin.36,37 These antigens aggregate and serve as a nidus for granuloma formation and maintenance long after infection has resolved.
Cutaneous lesions of sarcoidosis include macules, papules, plaques, and lupus pernio, as well as lesions arising within scars or tattoos, with many less common presentations.7,38 Papular sarcoidosis is common on the face but also can involve the extremities.4,7 Strictly, at least 2 organ systems must be involved to diagnose sarcoidosis, but this is debatable.4,7 Among 41 patients with cutaneous sarcoidosis, 24 (58.5%) had systemic disease; cutaneous lesions were the presenting sign in 87.5% (21/24) of patients.38 Histologic analysis, regardless of the lesion, usually shows noncaseating so-called “naked” granulomas, which have minimal lymphocytic infiltrate associated with the epithelioid histiocytes.38,39 Perifollicular granulomas are possible but unusual.40
Treatment depends on the extent of cutaneous and systemic involvement. Pharmacotherapeutic modalities include topical steroids, immunomodulators, and retinoids; systemic immunomodulators and immunosuppressants; and biologic agents.7 Isolated cutaneous sarcoidosis, particularly the papular variant, usually is associated with acute disease lasting less than 2 years, with resolution of skin lesions.7,38 That said, a recent report suggested that cutaneous sarcoidosis can progress to multisystemic disease as long as 7 years after the initial diagnosis.41
Clinical and Histologic Overlap—Despite this categorization of noninfectious facial granulomatous conditions, each has some clinical and histologic overlap with the others, which must be considered when encountering a granulomatous facial dermatosis. Both GPD and GR tend to present with lesions near the eyes, mouth, and nose, although GR can extend to lateral aspects of the face, below the mandible, and the forehead and has different demographic features.15,20,23 Granulomas in both GPD and GR generally are noncaseating and form in a follicular or perifollicular distribution within the dermis.2,15,23 Lupus miliaris disseminatus faciei and GR share a similar facial distribution in some cases.17,20 Even papular cutaneous sarcoidosis has masqueraded as GR clinically and histologically.4
Diagnostic and Treatment Difficulty—Our cases illustrate the range of difficulty in evaluating and managing patients with facial papular granulomas. On one hand, our adult patient’s clinical and histologic findings were highly consistent with GR; on the other hand, our younger patient had clinicopathologic features of both sarcoidosis and GPD at varying times. Both conditions are more common in dark-skinned patients.11,42
Juvenile sarcoidosis is comparatively rare, with a reported annual incidence of 0.22 to 0.27 for every 100,000 children younger than 15 years; however, juvenile sarcoidosis commonly presents around 8 to 15 years of age.43
It is unusual for sarcoid granulomas to be isolated to the skin, much less to the face.4,7,43,44 Patient 1 initially presented in this manner and lacked convincing laboratory or radiographic evidence of systemic sarcoidosis. Bilateral hilar calcifications in sarcoidosis are more typical among adults after 5 to 20 years; there were no signs or symptoms of active infection that could account for the pulmonary and cutaneous lesions.45
The presence of perifollicular granulomas with associated lymphocytic infiltrates on repeat biopsy, coupled with the use of topical steroids, made it difficult to rule out a contribution by GPD to her clinical course. That her lesions resolved with pitted scarring while she was taking methotrexate and after topical steroids had been stopped could be the result of successful management or spontaneous resolution of her dermatosis; both papular sarcoidosis and GPD tend to have a self-limited course.7,13
Conclusion
We present 2 cases of papular facial granulomas in patients with similar skin types who had different clinical courses. Evaluation of such lesions remains challenging given the similarity between specific entities that present in this manner. Certainly, it is reasonable to consider a spectrum upon which all of these conditions fall, in light of the findings of these cases and those reported previously.
- Beretta-Piccoli BT, Mainetti C, Peeters M-A, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146. doi:10.1007/s12016-017-8666-8
- Lucas CR, Korman NJ, Gilliam AC. Granulomatous periorificial dermatitis: a variant of granulomatous rosacea in children? J Cutan Med Surg. 2009;13:115-118. doi:10.2310/7750.2008.07088
- van de Scheur MR, van der Waal RIF, Starink TM. Lupus miliaris disseminatus faciei: a distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120-123. doi:10.1159/000068457
- Simonart T, Lowy M, Rasquin F, et al. Overlap of sarcoidosis and rosacea. Dermatology. 1997;194:416-418. doi:10.1159/000246165
- Lee GL, Zirwas MJ. Granulomatous rosacea and periorificial dermatitis: controversies and review of management. Dermatol Clin. 2015;33:447-455. doi:10.1016/j.det.2015.03.009
- Michaels JD, Cook-Norris RH, Lehman JS, et al. Adult with papular eruption of the central aspect of the face. J Am Acad Dermatol. 2014;71:410-412. doi:10.1016/j.jaad.2012.06.039
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;38:685-702. doi:10.1016/j.ccm.2015.08.010
- Gianotti F, Ermacora E, Benelli MG, et al. Particulière dermatite peri-orale infantile. observations sur 5 cas. Bull Soc Fr Dermatol Syphiligr. 1970;77:341.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual papular and acneiform facial eruption in the negro child. Br J Dermatol. 1974;91:435-438. doi:10.1111/j.1365-2133.1974.tb13083.x
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Williams HC, Ashworth J, Pembroke AC, et al. FACE—facial Afro-Caribbean childhood eruption. Clin Exp Dermatol. 1990;15:163-166. doi:10.1111/j.1365-2230.1990.tb02063.x
- Knautz MA, Lesher JL Jr. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134. doi:10.1111/j.1525-1470.1996.tb01419.x
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358. doi:10.1001/archderm.138.10.1354
- Vincenzi C, Parente G, Tosti A. Perioral granulomatous dermatitis: two cases treated with clarithromycin. J Dermatol Treat. 2000;11:57-61.
- Kim YJ, Shin JW, Lee JS, et al. Childhood granulomatous periorificial dermatitis. Ann Dermatol. 2011;23:386-388. doi:10.5021/ad.2011.23.3.386
- Snapp RH. Lewandowsky’s rosacea-like eruption; a clinical study. J Invest Dermatol. 1949;13:175-190. doi:10.1038/jid.1949.86
- Chougule A, Chatterjee D, Sethi S, et al. Granulomatous rosacea versus lupus miliaris disseminatus faciei—2 faces of facial granulomatous disorder: a clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819-823. doi:10.1097/DAD.0000000000001243
- Mullanax MG, Kierland RR. Granulomatous rosacea. Arch Dermatol. 1970;101:206-211.
- Sánchez JL, Berlingeri-Ramos AC, Dueño DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6-9. doi:10.1097/DAD.0b013e31815bc191
- Helm KF, Menz J, Gibson LE, et al. A clinical and histopathologic study of granulomatous rosacea. J Am Acad Dermatol. 1991;25:1038-1043. doi:10.1016/0190-9622(91)70304-k
- Kanada KN, Nakatsuji T, Gallo RL. Doxycycline indirectly inhibits proteolytic activation of tryptic kallikrein-related peptidases and activation of cathelicidin. J Invest Dermatol. 2012;132:1435-1442. doi:10.1038/jid.2012.14
- Jang YH, Sim JH, Kang HY, et al. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol. 2011;25:544-548. doi:10.1111/j.1468-3083.2010.03825.x
- Khokhar O, Khachemoune A. A case of granulomatous rosacea: sorting granulomatous rosacea from other granulomatous diseases that affect the face. Dermatol Online J. 2004;10:6.
- Rosen T, Stone MS. Acne rosacea in blacks. J Am Acad Dermatol. 1987;17:70-73. doi:10.1016/s0190-9622(87)70173-x
- Adams AK, Davis JL, Davis MDP, et al. What is your diagnosis? granulomatous rosacea (lupus miliaris disseminatus faciei, acne agminata). Cutis. 2008;82:103-112.
- Shitara A. Lupus miliaris disseminatus faciei. Int J Dermatol. 1984;23:542-544. doi:10.1111/j.1365-4362.1984.tb04206.x
- Hodak E, Trattner A, Feuerman H, et al. Lupus miliaris disseminatus faciei—the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614-619. doi: 10.1111/j.1365-2133.1997.tb03797.x
- Skowron F, Causeret AS, Pabion C, et al. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution. Dermatology. 2000;201:287-289. doi:10.1159/000051539
- Hutchinson J. Case of livid papillary psoriasis. In: London J, Churchill A, eds. Illustrations of Clinical Surgery. J&A Churchill; 1877:42-43.
- Besnier E. Lupus pernio of the face [in French]. Ann Dermatol Syphiligr (Paris). 1889;10:33-36.
- Tenneson H. Lupus pernio. Ann Dermatol Syphiligr (Paris). 1889;10:333-336.
- Boeck C. Multiple benign sarkoid of the skin [in Norwegian]. Norsk Mag Laegevidensk. 1899;14:1321-1334.
- Kuznitsky E, Bittorf A. Sarkoid mit beteiligung innerer organe. Münch Med Wochenschr. 1915;62:1349-1353.
- Schaumann J. Etude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. Ann Dermatol Syphiligr. 1916-1917;6:357-373.
- Osler W. On chronic symmetrical enlargement of the salivary and lacrimal glands. Am J Med Sci. 1898;115:27-30.
- Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49:6-18. doi:10.1007/s12016-015-8481-z
- Eberhardt C, Thillai M, Parker R, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12:e0170285. doi:10.1371/journal.pone.0170285
- Esteves TC, Aparicio G, Ferrer B, et al. Prognostic value of skin lesions in sarcoidosis: clinical and histopathological clues. Eur J Dermatol. 2015;25:556-562. doi:10.1684/ejd.2015.2666
- Cardoso JC, Cravo M, Reis JP, et al. Cutaneous sarcoidosis: a histopathological study. J Eur Acad Dermatol Venereol. 2009;23:678-682. doi:10.1111/j.1468-3083.2009.03153.x
- Mangas C, Fernández-Figueras M-T, Fité E, et al. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772-777. doi:10.1111/j.1600-0560.2006.00563.x
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis. clinical spectrum and histological analysis of 40 cases. Int J Dermatol. 2019;58:178-184. doi: 10.1111/ijd.14218
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16. doi:10.1186/1546-0096-6-16
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87:871-878. doi:10.1080/08035259875001366244. A, H, Yapıcı I. Isolated cutaneous sarcoidosis. Arch Bronconeumol. 2016;52:220.
- Scadding JG. The late stages of pulmonary sarcoidosis. Postgrad Med J. 1970;46:530-536. doi:10.1136/pgmj.46.538.530
Cutaneous granulomatous diseases encompass many entities that are skin-limited or systemic. The prototypical cutaneous granuloma is a painless, rounded, well-defined, red-pink or flesh-colored papule1 and is smooth, owing to minimal epidermal involvement. Examples of conditions that present with such lesions include granulomatous periorificial dermatitis (GPD), granulomatous rosacea (GR), lupus miliaris disseminatus faciei (LMDF), and papular sarcoidosis. These entities commonly are seen on the face and can be a source of distress to patients when they are extensive. Several reports have raised the possibility that these conditions lie on a spectrum.2-4 We present 2 cases of patients with facial papular granulomas, discuss potential causes of the lesions, review historical aspects from the literature, and highlight the challenges that these lesions can pose to the clinician.
Case Reports
Patient 1—A 10-year-old Ethiopian girl with a history of atopic dermatitis presented with a facial rash of 4 months’ duration. Her pediatrician initially treated the rash as pityriasis alba and prescribed hydrocortisone cream. Two months into treatment, the patient developed an otherwise asymptomatic, unilateral, papular dermatosis on the right cheek. She subsequently was switched to treatment with benzoyl peroxide and topical clindamycin, which she had been using for 2 months with no improvement at the time of the current presentation. The lesions then spread bilaterally and periorally.
At the current presentation, physical examination demonstrated fine, diffuse, follicular-based, flesh-colored papules over both cheeks, the right side of the nose, and the perioral region (Figure 1). A biopsy of a papular lesion from the right cheek revealed well-formed, noncaseating granulomas in the superficial and mid dermis with an associated lymphocytic infiltrate (Figure 2). No organisms were identified on acid-fast, Fite, or periodic acid–Schiff staining. A tuberculin skin test was negative. A chest radiograph showed small calcified hilar lymph nodes bilaterally. Pulmonary function tests were unremarkable. Calcium and angiotensin-converting enzyme levels were normal.
The patient denied any fever, chills, hemoptysis, cough, dyspnea, lymphadenopathy, scleral or conjunctival pain or erythema, visual disturbances, or arthralgias. Hydroxychloroquine 200 mg twice daily was started with minimal improvement after 5 months. Methotrexate 20 mg once weekly was then added. Topical fluocinonide 0.05% also was started at this time, as the patient had required several prednisone tapers over the past 3 months for symptomatic relief. The lesions improved minimally after 5 more months of treatment, at which time she had developed inflammatory papules, pustules, and open comedones in the same areas as well as the glabella.
Repeat biopsy of a papular lesion demonstrated noncaseating granulomas and an associated chronic lymphocytic infiltrate in a follicular and perifollicular distribution (Figure 3). Biopsy of a pustule demonstrated acute Demodex folliculitis. Fluocinonide was stopped, and anti-mite therapy with ivermectin, permethrin cream 5%, and selenium sulfide lotion 2.5% was started, with good response from the pustular lesions.
The patient continued taking methotrexate 20 mg once weekly during this time, with improvement in the papular lesions. She discontinued methotrexate after 12 months with complete resolution. At follow-up 12 months after stopping the methotrexate (roughly 2 years after initial presentation), she showed sustained resolution, with small pitted scars on both cheeks and the nasal tip.
Patient 2—A 33-year-old Ethiopian woman presented with a facial rash of 15 years’ duration. The lesions had been accumulating slowly and were asymptomatic. Physical examination revealed multiple follicular-based, flesh-colored, and erythematous papules on the cheeks, chin, perioral area, and forehead (Figure 4). There were no pustules or telangiectasias. Treatment with tretinoin cream 0.05% for 6 months offered minimal relief.
Biopsy of a papule from the left mandible showed superficial vascular telangiectasias, noncaseating granulomas comprising epithelioid histiocytes and lymphocytes in the superficial dermis, and a perifollicular lymphocytic infiltrate (Figure 5). No organisms were identified on Fite or Gomori methenamine silver staining.
Comment
The first step in differentiating cutaneous granulomatous lesions should be to distinguish infectious from noninfectious causes.1 Noninfectious cutaneous granulomas can appear nearly anywhere; however, certain processes have a predilection for the face, including GPD, GR, LMDF, and papular sarcoidosis.5-7 These conditions generally present with papular granulomas with features as described above.
Granulomatous Periorificial Dermatitis—In 1970, Gianotti and colleagues8 briefly described the first possible cases of GPD in 5 children. The eruption comprised numerous yellow, dome-shaped papules in a mostly perioral distribution. Tuberculin and the Kveim tests were nonreactive; histopathology was described as sarcoid-type and not necessarily follicular or perifollicular.8 In 1974, Marten et al9 described 22 Afro-Caribbean children with flesh-colored, papular eruptions on the face that did not show histologic granulomatous changes but were morphologically similar to the reports by Gianotti et al.8 By 1989, Frieden and colleagues10 described this facial eruption as “granulomatous perioral dermatitis in children”. Additionally, the investigators observed granulomatous infiltrates in a perifollicular distribution and suggested follicular disruption as a possible cause. It was clear from the case discussions that these eruptions were not uncommonly diagnosed as papular sarcoidosis.10 The following year, Williams et al11 reported 5 cases of similar papular eruptions in 5 Afro-Caribbean children, coining the term facial Afro-Caribbean eruption.11 Knautz and Lesher12 referred to this entity as “childhood GPD” in 1996 to avoid limiting the diagnosis to Afro-Caribbean patients and to a perioral distribution; this is the most popular current terminology.12 Since then, reports of extrafacial involvement and disease in adults have been published.13,14
Granulomatous periorificial dermatitis often is seen in the perinasal, periocular, and perioral regions of the face.2 It is associated with topical steroid exposure.5 Histologically, noncaseating granulomas around the upper half of undisrupted hair follicles with a lymphocytic infiltrate are typical.13 Treatment should begin with cessation of any topical steroids; first-line agents are oral tetracycline or macrolide antibiotics.5 These agents can be used alone or in combination with topical erythromycin, metronidazole, or sulfur-based lotions.13 Rarely, GPD presents extrafacially.13 Even so, it usually resolves within 2 weeks to 6 months, especially with therapy; scarring is unusual.5,13,15
Granulomatous Rosacea—A report in the early 20th century described patients with tuberculoid granulomas resembling papular rosacea; the initial belief was that this finding represented a rosacealike tuberculid eruption.5 However, this belief was questioned by Snapp,16 among others, who demonstrated near universal lack of reactivity to tuberculin among 20 of these patients in 1949; more recent evidence has substantiated these findings.17 Still, Snapp16 postulated that these rosacealike granulomatous lesions were distinct from classic rosacea because they lacked vascular symptoms and pustules and were recalcitrant to rosacea treatment modalities.
In 1970, Mullanax and colleagues18 introduced the term granulomatous rosacea, reiterating that this entity was not tuberculous. They documented papulopustular lesions as well as telangiectasias, raising the possibility that GR does overlap with acne rosacea. More recent studies have established the current theory that GR is a histologic variant of acne rosacea because, in addition to typical granulomatous papules, its microscopic features can be seen across subtypes of acne rosacea.19,20
Various causes have been proposed for GR. Demodex mites have been reported in association with GR for nearly 30 years.19,20 In the past 10 years, molecular studies have started to define the role of metalloproteinases, UV radiation, and cutaneous peptides in the pathogenesis of acne rosacea and GR.21,22
Granulomatous rosacea typically is seen in middle-aged women.20,23 Hallmarks of rosacea, such as facial erythema, flushing, telangiectasias, pustules, and rhinophyma, are not always present in GR.5,20,23 Lesions usually are distributed around the central face, although extension to the cheeks, total facial involvement, and extrafacial lesions are possible.5,20 Histologically, perifollicular and follicular-based noncaseating granulomas with dilatation of the dermal papillary vasculature are seen.17,23 As a whole, rosacea is comparatively uncommon in dark-skinned patients; when it does occur, GR is a frequent presentation.24
First-line treatment for GR is tetracycline antibiotics.5 Unresponsive cases have been treated—largely anecdotally—with topical modalities (eg, metronidazole, steroids, immunomodulators), systemic agents (eg, dapsone, erythromycin, isotretinoin), and other therapies.5 Granulomatous rosacea tends to have a chronic course.5,23
Lupus Miliaris Disseminatus Faciei—Classic LMDF demonstrates caseating perifollicular granulomas histologically.6,17,25 Lesions tend to appear on the central face, particularly the eyelids, and can be seen extrafacially.3,6,25,26 Although LMDF originally was categorized as a tuberculid eruption, this no longer is thought to be the case.27 It is now regarded by some as a variant of GR25; however, LMDF responds poorly to tetracyclines, is more common in males, and lacks rosacealike vascular abnormalities, leading some to question this association.3,6,17 In the past 20 years, some have proposed renaming LMDF to better reflect its clinical course and to consider it independent of tuberculosis and GR.28 It usually resolves spontaneously after 1 to 3 years, leaving pitted scars.3,6
Papular Sarcoidosis—The first potential documented case of sarcoidosis was by Hutchinson29 in 1869 in a patient seen in London. The author labeled purple plaques on the index patient’s legs and hands as “livid papillary psoriasis.” In 1889, Besnier30 described a patient with violaceous swellings on the nose, ears, and fingers, which he called “lupus pernio”; his contemporary, Tenneson,31 published a case of lupus pernio and described its histologic profile as comprising epithelioid cells and giant cells. It was not until 1899 that the term sarkoid was used to describe these cutaneous lesions by Boeck,32 who thought they were reminiscent of sarcoma. In 1915, Kuznitsky and Bittorf33 described a patient with cutaneous lesions histologically consistent with Boeck’s sarkoid but additionally with hilar lymphadenopathy and pulmonary infiltrates. Around 1916 or 1917, Schaumann34 described patients with cutaneous lesions and additionally with involvement of pulmonary, osseous, hepatosplenic, and tonsillar tissue. These reports are among the first to recognize the multisystemic nature of sarcoidosis. The first possible case of childhood sarcoidosis might have been reported by Osler35 in the United States in 1898.
In the past century or so, an ongoing effort by researchers has focused on identifying etiologic triggers for sarcoidosis. Microbial agents have been considered in this role, with Mycobacterium and Propionibacterium organisms the most intensively studied; the possibility that foreign material contributes to the formation of granulomas also has been raised.36 Current models of the pathogenesis of sarcoidosis involve an interplay between the immune system in genetically predisposed patients and an infection that leads to a hyperimmune type 1 T–helper cell response that clears the infection but not antigens generated by the microbes and the acute host response, including proteins such as serum amyloid A and vimentin.36,37 These antigens aggregate and serve as a nidus for granuloma formation and maintenance long after infection has resolved.
Cutaneous lesions of sarcoidosis include macules, papules, plaques, and lupus pernio, as well as lesions arising within scars or tattoos, with many less common presentations.7,38 Papular sarcoidosis is common on the face but also can involve the extremities.4,7 Strictly, at least 2 organ systems must be involved to diagnose sarcoidosis, but this is debatable.4,7 Among 41 patients with cutaneous sarcoidosis, 24 (58.5%) had systemic disease; cutaneous lesions were the presenting sign in 87.5% (21/24) of patients.38 Histologic analysis, regardless of the lesion, usually shows noncaseating so-called “naked” granulomas, which have minimal lymphocytic infiltrate associated with the epithelioid histiocytes.38,39 Perifollicular granulomas are possible but unusual.40
Treatment depends on the extent of cutaneous and systemic involvement. Pharmacotherapeutic modalities include topical steroids, immunomodulators, and retinoids; systemic immunomodulators and immunosuppressants; and biologic agents.7 Isolated cutaneous sarcoidosis, particularly the papular variant, usually is associated with acute disease lasting less than 2 years, with resolution of skin lesions.7,38 That said, a recent report suggested that cutaneous sarcoidosis can progress to multisystemic disease as long as 7 years after the initial diagnosis.41
Clinical and Histologic Overlap—Despite this categorization of noninfectious facial granulomatous conditions, each has some clinical and histologic overlap with the others, which must be considered when encountering a granulomatous facial dermatosis. Both GPD and GR tend to present with lesions near the eyes, mouth, and nose, although GR can extend to lateral aspects of the face, below the mandible, and the forehead and has different demographic features.15,20,23 Granulomas in both GPD and GR generally are noncaseating and form in a follicular or perifollicular distribution within the dermis.2,15,23 Lupus miliaris disseminatus faciei and GR share a similar facial distribution in some cases.17,20 Even papular cutaneous sarcoidosis has masqueraded as GR clinically and histologically.4
Diagnostic and Treatment Difficulty—Our cases illustrate the range of difficulty in evaluating and managing patients with facial papular granulomas. On one hand, our adult patient’s clinical and histologic findings were highly consistent with GR; on the other hand, our younger patient had clinicopathologic features of both sarcoidosis and GPD at varying times. Both conditions are more common in dark-skinned patients.11,42
Juvenile sarcoidosis is comparatively rare, with a reported annual incidence of 0.22 to 0.27 for every 100,000 children younger than 15 years; however, juvenile sarcoidosis commonly presents around 8 to 15 years of age.43
It is unusual for sarcoid granulomas to be isolated to the skin, much less to the face.4,7,43,44 Patient 1 initially presented in this manner and lacked convincing laboratory or radiographic evidence of systemic sarcoidosis. Bilateral hilar calcifications in sarcoidosis are more typical among adults after 5 to 20 years; there were no signs or symptoms of active infection that could account for the pulmonary and cutaneous lesions.45
The presence of perifollicular granulomas with associated lymphocytic infiltrates on repeat biopsy, coupled with the use of topical steroids, made it difficult to rule out a contribution by GPD to her clinical course. That her lesions resolved with pitted scarring while she was taking methotrexate and after topical steroids had been stopped could be the result of successful management or spontaneous resolution of her dermatosis; both papular sarcoidosis and GPD tend to have a self-limited course.7,13
Conclusion
We present 2 cases of papular facial granulomas in patients with similar skin types who had different clinical courses. Evaluation of such lesions remains challenging given the similarity between specific entities that present in this manner. Certainly, it is reasonable to consider a spectrum upon which all of these conditions fall, in light of the findings of these cases and those reported previously.
Cutaneous granulomatous diseases encompass many entities that are skin-limited or systemic. The prototypical cutaneous granuloma is a painless, rounded, well-defined, red-pink or flesh-colored papule1 and is smooth, owing to minimal epidermal involvement. Examples of conditions that present with such lesions include granulomatous periorificial dermatitis (GPD), granulomatous rosacea (GR), lupus miliaris disseminatus faciei (LMDF), and papular sarcoidosis. These entities commonly are seen on the face and can be a source of distress to patients when they are extensive. Several reports have raised the possibility that these conditions lie on a spectrum.2-4 We present 2 cases of patients with facial papular granulomas, discuss potential causes of the lesions, review historical aspects from the literature, and highlight the challenges that these lesions can pose to the clinician.
Case Reports
Patient 1—A 10-year-old Ethiopian girl with a history of atopic dermatitis presented with a facial rash of 4 months’ duration. Her pediatrician initially treated the rash as pityriasis alba and prescribed hydrocortisone cream. Two months into treatment, the patient developed an otherwise asymptomatic, unilateral, papular dermatosis on the right cheek. She subsequently was switched to treatment with benzoyl peroxide and topical clindamycin, which she had been using for 2 months with no improvement at the time of the current presentation. The lesions then spread bilaterally and periorally.
At the current presentation, physical examination demonstrated fine, diffuse, follicular-based, flesh-colored papules over both cheeks, the right side of the nose, and the perioral region (Figure 1). A biopsy of a papular lesion from the right cheek revealed well-formed, noncaseating granulomas in the superficial and mid dermis with an associated lymphocytic infiltrate (Figure 2). No organisms were identified on acid-fast, Fite, or periodic acid–Schiff staining. A tuberculin skin test was negative. A chest radiograph showed small calcified hilar lymph nodes bilaterally. Pulmonary function tests were unremarkable. Calcium and angiotensin-converting enzyme levels were normal.
The patient denied any fever, chills, hemoptysis, cough, dyspnea, lymphadenopathy, scleral or conjunctival pain or erythema, visual disturbances, or arthralgias. Hydroxychloroquine 200 mg twice daily was started with minimal improvement after 5 months. Methotrexate 20 mg once weekly was then added. Topical fluocinonide 0.05% also was started at this time, as the patient had required several prednisone tapers over the past 3 months for symptomatic relief. The lesions improved minimally after 5 more months of treatment, at which time she had developed inflammatory papules, pustules, and open comedones in the same areas as well as the glabella.
Repeat biopsy of a papular lesion demonstrated noncaseating granulomas and an associated chronic lymphocytic infiltrate in a follicular and perifollicular distribution (Figure 3). Biopsy of a pustule demonstrated acute Demodex folliculitis. Fluocinonide was stopped, and anti-mite therapy with ivermectin, permethrin cream 5%, and selenium sulfide lotion 2.5% was started, with good response from the pustular lesions.
The patient continued taking methotrexate 20 mg once weekly during this time, with improvement in the papular lesions. She discontinued methotrexate after 12 months with complete resolution. At follow-up 12 months after stopping the methotrexate (roughly 2 years after initial presentation), she showed sustained resolution, with small pitted scars on both cheeks and the nasal tip.
Patient 2—A 33-year-old Ethiopian woman presented with a facial rash of 15 years’ duration. The lesions had been accumulating slowly and were asymptomatic. Physical examination revealed multiple follicular-based, flesh-colored, and erythematous papules on the cheeks, chin, perioral area, and forehead (Figure 4). There were no pustules or telangiectasias. Treatment with tretinoin cream 0.05% for 6 months offered minimal relief.
Biopsy of a papule from the left mandible showed superficial vascular telangiectasias, noncaseating granulomas comprising epithelioid histiocytes and lymphocytes in the superficial dermis, and a perifollicular lymphocytic infiltrate (Figure 5). No organisms were identified on Fite or Gomori methenamine silver staining.
Comment
The first step in differentiating cutaneous granulomatous lesions should be to distinguish infectious from noninfectious causes.1 Noninfectious cutaneous granulomas can appear nearly anywhere; however, certain processes have a predilection for the face, including GPD, GR, LMDF, and papular sarcoidosis.5-7 These conditions generally present with papular granulomas with features as described above.
Granulomatous Periorificial Dermatitis—In 1970, Gianotti and colleagues8 briefly described the first possible cases of GPD in 5 children. The eruption comprised numerous yellow, dome-shaped papules in a mostly perioral distribution. Tuberculin and the Kveim tests were nonreactive; histopathology was described as sarcoid-type and not necessarily follicular or perifollicular.8 In 1974, Marten et al9 described 22 Afro-Caribbean children with flesh-colored, papular eruptions on the face that did not show histologic granulomatous changes but were morphologically similar to the reports by Gianotti et al.8 By 1989, Frieden and colleagues10 described this facial eruption as “granulomatous perioral dermatitis in children”. Additionally, the investigators observed granulomatous infiltrates in a perifollicular distribution and suggested follicular disruption as a possible cause. It was clear from the case discussions that these eruptions were not uncommonly diagnosed as papular sarcoidosis.10 The following year, Williams et al11 reported 5 cases of similar papular eruptions in 5 Afro-Caribbean children, coining the term facial Afro-Caribbean eruption.11 Knautz and Lesher12 referred to this entity as “childhood GPD” in 1996 to avoid limiting the diagnosis to Afro-Caribbean patients and to a perioral distribution; this is the most popular current terminology.12 Since then, reports of extrafacial involvement and disease in adults have been published.13,14
Granulomatous periorificial dermatitis often is seen in the perinasal, periocular, and perioral regions of the face.2 It is associated with topical steroid exposure.5 Histologically, noncaseating granulomas around the upper half of undisrupted hair follicles with a lymphocytic infiltrate are typical.13 Treatment should begin with cessation of any topical steroids; first-line agents are oral tetracycline or macrolide antibiotics.5 These agents can be used alone or in combination with topical erythromycin, metronidazole, or sulfur-based lotions.13 Rarely, GPD presents extrafacially.13 Even so, it usually resolves within 2 weeks to 6 months, especially with therapy; scarring is unusual.5,13,15
Granulomatous Rosacea—A report in the early 20th century described patients with tuberculoid granulomas resembling papular rosacea; the initial belief was that this finding represented a rosacealike tuberculid eruption.5 However, this belief was questioned by Snapp,16 among others, who demonstrated near universal lack of reactivity to tuberculin among 20 of these patients in 1949; more recent evidence has substantiated these findings.17 Still, Snapp16 postulated that these rosacealike granulomatous lesions were distinct from classic rosacea because they lacked vascular symptoms and pustules and were recalcitrant to rosacea treatment modalities.
In 1970, Mullanax and colleagues18 introduced the term granulomatous rosacea, reiterating that this entity was not tuberculous. They documented papulopustular lesions as well as telangiectasias, raising the possibility that GR does overlap with acne rosacea. More recent studies have established the current theory that GR is a histologic variant of acne rosacea because, in addition to typical granulomatous papules, its microscopic features can be seen across subtypes of acne rosacea.19,20
Various causes have been proposed for GR. Demodex mites have been reported in association with GR for nearly 30 years.19,20 In the past 10 years, molecular studies have started to define the role of metalloproteinases, UV radiation, and cutaneous peptides in the pathogenesis of acne rosacea and GR.21,22
Granulomatous rosacea typically is seen in middle-aged women.20,23 Hallmarks of rosacea, such as facial erythema, flushing, telangiectasias, pustules, and rhinophyma, are not always present in GR.5,20,23 Lesions usually are distributed around the central face, although extension to the cheeks, total facial involvement, and extrafacial lesions are possible.5,20 Histologically, perifollicular and follicular-based noncaseating granulomas with dilatation of the dermal papillary vasculature are seen.17,23 As a whole, rosacea is comparatively uncommon in dark-skinned patients; when it does occur, GR is a frequent presentation.24
First-line treatment for GR is tetracycline antibiotics.5 Unresponsive cases have been treated—largely anecdotally—with topical modalities (eg, metronidazole, steroids, immunomodulators), systemic agents (eg, dapsone, erythromycin, isotretinoin), and other therapies.5 Granulomatous rosacea tends to have a chronic course.5,23
Lupus Miliaris Disseminatus Faciei—Classic LMDF demonstrates caseating perifollicular granulomas histologically.6,17,25 Lesions tend to appear on the central face, particularly the eyelids, and can be seen extrafacially.3,6,25,26 Although LMDF originally was categorized as a tuberculid eruption, this no longer is thought to be the case.27 It is now regarded by some as a variant of GR25; however, LMDF responds poorly to tetracyclines, is more common in males, and lacks rosacealike vascular abnormalities, leading some to question this association.3,6,17 In the past 20 years, some have proposed renaming LMDF to better reflect its clinical course and to consider it independent of tuberculosis and GR.28 It usually resolves spontaneously after 1 to 3 years, leaving pitted scars.3,6
Papular Sarcoidosis—The first potential documented case of sarcoidosis was by Hutchinson29 in 1869 in a patient seen in London. The author labeled purple plaques on the index patient’s legs and hands as “livid papillary psoriasis.” In 1889, Besnier30 described a patient with violaceous swellings on the nose, ears, and fingers, which he called “lupus pernio”; his contemporary, Tenneson,31 published a case of lupus pernio and described its histologic profile as comprising epithelioid cells and giant cells. It was not until 1899 that the term sarkoid was used to describe these cutaneous lesions by Boeck,32 who thought they were reminiscent of sarcoma. In 1915, Kuznitsky and Bittorf33 described a patient with cutaneous lesions histologically consistent with Boeck’s sarkoid but additionally with hilar lymphadenopathy and pulmonary infiltrates. Around 1916 or 1917, Schaumann34 described patients with cutaneous lesions and additionally with involvement of pulmonary, osseous, hepatosplenic, and tonsillar tissue. These reports are among the first to recognize the multisystemic nature of sarcoidosis. The first possible case of childhood sarcoidosis might have been reported by Osler35 in the United States in 1898.
In the past century or so, an ongoing effort by researchers has focused on identifying etiologic triggers for sarcoidosis. Microbial agents have been considered in this role, with Mycobacterium and Propionibacterium organisms the most intensively studied; the possibility that foreign material contributes to the formation of granulomas also has been raised.36 Current models of the pathogenesis of sarcoidosis involve an interplay between the immune system in genetically predisposed patients and an infection that leads to a hyperimmune type 1 T–helper cell response that clears the infection but not antigens generated by the microbes and the acute host response, including proteins such as serum amyloid A and vimentin.36,37 These antigens aggregate and serve as a nidus for granuloma formation and maintenance long after infection has resolved.
Cutaneous lesions of sarcoidosis include macules, papules, plaques, and lupus pernio, as well as lesions arising within scars or tattoos, with many less common presentations.7,38 Papular sarcoidosis is common on the face but also can involve the extremities.4,7 Strictly, at least 2 organ systems must be involved to diagnose sarcoidosis, but this is debatable.4,7 Among 41 patients with cutaneous sarcoidosis, 24 (58.5%) had systemic disease; cutaneous lesions were the presenting sign in 87.5% (21/24) of patients.38 Histologic analysis, regardless of the lesion, usually shows noncaseating so-called “naked” granulomas, which have minimal lymphocytic infiltrate associated with the epithelioid histiocytes.38,39 Perifollicular granulomas are possible but unusual.40
Treatment depends on the extent of cutaneous and systemic involvement. Pharmacotherapeutic modalities include topical steroids, immunomodulators, and retinoids; systemic immunomodulators and immunosuppressants; and biologic agents.7 Isolated cutaneous sarcoidosis, particularly the papular variant, usually is associated with acute disease lasting less than 2 years, with resolution of skin lesions.7,38 That said, a recent report suggested that cutaneous sarcoidosis can progress to multisystemic disease as long as 7 years after the initial diagnosis.41
Clinical and Histologic Overlap—Despite this categorization of noninfectious facial granulomatous conditions, each has some clinical and histologic overlap with the others, which must be considered when encountering a granulomatous facial dermatosis. Both GPD and GR tend to present with lesions near the eyes, mouth, and nose, although GR can extend to lateral aspects of the face, below the mandible, and the forehead and has different demographic features.15,20,23 Granulomas in both GPD and GR generally are noncaseating and form in a follicular or perifollicular distribution within the dermis.2,15,23 Lupus miliaris disseminatus faciei and GR share a similar facial distribution in some cases.17,20 Even papular cutaneous sarcoidosis has masqueraded as GR clinically and histologically.4
Diagnostic and Treatment Difficulty—Our cases illustrate the range of difficulty in evaluating and managing patients with facial papular granulomas. On one hand, our adult patient’s clinical and histologic findings were highly consistent with GR; on the other hand, our younger patient had clinicopathologic features of both sarcoidosis and GPD at varying times. Both conditions are more common in dark-skinned patients.11,42
Juvenile sarcoidosis is comparatively rare, with a reported annual incidence of 0.22 to 0.27 for every 100,000 children younger than 15 years; however, juvenile sarcoidosis commonly presents around 8 to 15 years of age.43
It is unusual for sarcoid granulomas to be isolated to the skin, much less to the face.4,7,43,44 Patient 1 initially presented in this manner and lacked convincing laboratory or radiographic evidence of systemic sarcoidosis. Bilateral hilar calcifications in sarcoidosis are more typical among adults after 5 to 20 years; there were no signs or symptoms of active infection that could account for the pulmonary and cutaneous lesions.45
The presence of perifollicular granulomas with associated lymphocytic infiltrates on repeat biopsy, coupled with the use of topical steroids, made it difficult to rule out a contribution by GPD to her clinical course. That her lesions resolved with pitted scarring while she was taking methotrexate and after topical steroids had been stopped could be the result of successful management or spontaneous resolution of her dermatosis; both papular sarcoidosis and GPD tend to have a self-limited course.7,13
Conclusion
We present 2 cases of papular facial granulomas in patients with similar skin types who had different clinical courses. Evaluation of such lesions remains challenging given the similarity between specific entities that present in this manner. Certainly, it is reasonable to consider a spectrum upon which all of these conditions fall, in light of the findings of these cases and those reported previously.
- Beretta-Piccoli BT, Mainetti C, Peeters M-A, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146. doi:10.1007/s12016-017-8666-8
- Lucas CR, Korman NJ, Gilliam AC. Granulomatous periorificial dermatitis: a variant of granulomatous rosacea in children? J Cutan Med Surg. 2009;13:115-118. doi:10.2310/7750.2008.07088
- van de Scheur MR, van der Waal RIF, Starink TM. Lupus miliaris disseminatus faciei: a distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120-123. doi:10.1159/000068457
- Simonart T, Lowy M, Rasquin F, et al. Overlap of sarcoidosis and rosacea. Dermatology. 1997;194:416-418. doi:10.1159/000246165
- Lee GL, Zirwas MJ. Granulomatous rosacea and periorificial dermatitis: controversies and review of management. Dermatol Clin. 2015;33:447-455. doi:10.1016/j.det.2015.03.009
- Michaels JD, Cook-Norris RH, Lehman JS, et al. Adult with papular eruption of the central aspect of the face. J Am Acad Dermatol. 2014;71:410-412. doi:10.1016/j.jaad.2012.06.039
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;38:685-702. doi:10.1016/j.ccm.2015.08.010
- Gianotti F, Ermacora E, Benelli MG, et al. Particulière dermatite peri-orale infantile. observations sur 5 cas. Bull Soc Fr Dermatol Syphiligr. 1970;77:341.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual papular and acneiform facial eruption in the negro child. Br J Dermatol. 1974;91:435-438. doi:10.1111/j.1365-2133.1974.tb13083.x
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Williams HC, Ashworth J, Pembroke AC, et al. FACE—facial Afro-Caribbean childhood eruption. Clin Exp Dermatol. 1990;15:163-166. doi:10.1111/j.1365-2230.1990.tb02063.x
- Knautz MA, Lesher JL Jr. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134. doi:10.1111/j.1525-1470.1996.tb01419.x
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358. doi:10.1001/archderm.138.10.1354
- Vincenzi C, Parente G, Tosti A. Perioral granulomatous dermatitis: two cases treated with clarithromycin. J Dermatol Treat. 2000;11:57-61.
- Kim YJ, Shin JW, Lee JS, et al. Childhood granulomatous periorificial dermatitis. Ann Dermatol. 2011;23:386-388. doi:10.5021/ad.2011.23.3.386
- Snapp RH. Lewandowsky’s rosacea-like eruption; a clinical study. J Invest Dermatol. 1949;13:175-190. doi:10.1038/jid.1949.86
- Chougule A, Chatterjee D, Sethi S, et al. Granulomatous rosacea versus lupus miliaris disseminatus faciei—2 faces of facial granulomatous disorder: a clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819-823. doi:10.1097/DAD.0000000000001243
- Mullanax MG, Kierland RR. Granulomatous rosacea. Arch Dermatol. 1970;101:206-211.
- Sánchez JL, Berlingeri-Ramos AC, Dueño DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6-9. doi:10.1097/DAD.0b013e31815bc191
- Helm KF, Menz J, Gibson LE, et al. A clinical and histopathologic study of granulomatous rosacea. J Am Acad Dermatol. 1991;25:1038-1043. doi:10.1016/0190-9622(91)70304-k
- Kanada KN, Nakatsuji T, Gallo RL. Doxycycline indirectly inhibits proteolytic activation of tryptic kallikrein-related peptidases and activation of cathelicidin. J Invest Dermatol. 2012;132:1435-1442. doi:10.1038/jid.2012.14
- Jang YH, Sim JH, Kang HY, et al. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol. 2011;25:544-548. doi:10.1111/j.1468-3083.2010.03825.x
- Khokhar O, Khachemoune A. A case of granulomatous rosacea: sorting granulomatous rosacea from other granulomatous diseases that affect the face. Dermatol Online J. 2004;10:6.
- Rosen T, Stone MS. Acne rosacea in blacks. J Am Acad Dermatol. 1987;17:70-73. doi:10.1016/s0190-9622(87)70173-x
- Adams AK, Davis JL, Davis MDP, et al. What is your diagnosis? granulomatous rosacea (lupus miliaris disseminatus faciei, acne agminata). Cutis. 2008;82:103-112.
- Shitara A. Lupus miliaris disseminatus faciei. Int J Dermatol. 1984;23:542-544. doi:10.1111/j.1365-4362.1984.tb04206.x
- Hodak E, Trattner A, Feuerman H, et al. Lupus miliaris disseminatus faciei—the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614-619. doi: 10.1111/j.1365-2133.1997.tb03797.x
- Skowron F, Causeret AS, Pabion C, et al. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution. Dermatology. 2000;201:287-289. doi:10.1159/000051539
- Hutchinson J. Case of livid papillary psoriasis. In: London J, Churchill A, eds. Illustrations of Clinical Surgery. J&A Churchill; 1877:42-43.
- Besnier E. Lupus pernio of the face [in French]. Ann Dermatol Syphiligr (Paris). 1889;10:33-36.
- Tenneson H. Lupus pernio. Ann Dermatol Syphiligr (Paris). 1889;10:333-336.
- Boeck C. Multiple benign sarkoid of the skin [in Norwegian]. Norsk Mag Laegevidensk. 1899;14:1321-1334.
- Kuznitsky E, Bittorf A. Sarkoid mit beteiligung innerer organe. Münch Med Wochenschr. 1915;62:1349-1353.
- Schaumann J. Etude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. Ann Dermatol Syphiligr. 1916-1917;6:357-373.
- Osler W. On chronic symmetrical enlargement of the salivary and lacrimal glands. Am J Med Sci. 1898;115:27-30.
- Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49:6-18. doi:10.1007/s12016-015-8481-z
- Eberhardt C, Thillai M, Parker R, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12:e0170285. doi:10.1371/journal.pone.0170285
- Esteves TC, Aparicio G, Ferrer B, et al. Prognostic value of skin lesions in sarcoidosis: clinical and histopathological clues. Eur J Dermatol. 2015;25:556-562. doi:10.1684/ejd.2015.2666
- Cardoso JC, Cravo M, Reis JP, et al. Cutaneous sarcoidosis: a histopathological study. J Eur Acad Dermatol Venereol. 2009;23:678-682. doi:10.1111/j.1468-3083.2009.03153.x
- Mangas C, Fernández-Figueras M-T, Fité E, et al. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772-777. doi:10.1111/j.1600-0560.2006.00563.x
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis. clinical spectrum and histological analysis of 40 cases. Int J Dermatol. 2019;58:178-184. doi: 10.1111/ijd.14218
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16. doi:10.1186/1546-0096-6-16
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87:871-878. doi:10.1080/08035259875001366244. A, H, Yapıcı I. Isolated cutaneous sarcoidosis. Arch Bronconeumol. 2016;52:220.
- Scadding JG. The late stages of pulmonary sarcoidosis. Postgrad Med J. 1970;46:530-536. doi:10.1136/pgmj.46.538.530
- Beretta-Piccoli BT, Mainetti C, Peeters M-A, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146. doi:10.1007/s12016-017-8666-8
- Lucas CR, Korman NJ, Gilliam AC. Granulomatous periorificial dermatitis: a variant of granulomatous rosacea in children? J Cutan Med Surg. 2009;13:115-118. doi:10.2310/7750.2008.07088
- van de Scheur MR, van der Waal RIF, Starink TM. Lupus miliaris disseminatus faciei: a distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120-123. doi:10.1159/000068457
- Simonart T, Lowy M, Rasquin F, et al. Overlap of sarcoidosis and rosacea. Dermatology. 1997;194:416-418. doi:10.1159/000246165
- Lee GL, Zirwas MJ. Granulomatous rosacea and periorificial dermatitis: controversies and review of management. Dermatol Clin. 2015;33:447-455. doi:10.1016/j.det.2015.03.009
- Michaels JD, Cook-Norris RH, Lehman JS, et al. Adult with papular eruption of the central aspect of the face. J Am Acad Dermatol. 2014;71:410-412. doi:10.1016/j.jaad.2012.06.039
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;38:685-702. doi:10.1016/j.ccm.2015.08.010
- Gianotti F, Ermacora E, Benelli MG, et al. Particulière dermatite peri-orale infantile. observations sur 5 cas. Bull Soc Fr Dermatol Syphiligr. 1970;77:341.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual papular and acneiform facial eruption in the negro child. Br J Dermatol. 1974;91:435-438. doi:10.1111/j.1365-2133.1974.tb13083.x
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Williams HC, Ashworth J, Pembroke AC, et al. FACE—facial Afro-Caribbean childhood eruption. Clin Exp Dermatol. 1990;15:163-166. doi:10.1111/j.1365-2230.1990.tb02063.x
- Knautz MA, Lesher JL Jr. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134. doi:10.1111/j.1525-1470.1996.tb01419.x
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358. doi:10.1001/archderm.138.10.1354
- Vincenzi C, Parente G, Tosti A. Perioral granulomatous dermatitis: two cases treated with clarithromycin. J Dermatol Treat. 2000;11:57-61.
- Kim YJ, Shin JW, Lee JS, et al. Childhood granulomatous periorificial dermatitis. Ann Dermatol. 2011;23:386-388. doi:10.5021/ad.2011.23.3.386
- Snapp RH. Lewandowsky’s rosacea-like eruption; a clinical study. J Invest Dermatol. 1949;13:175-190. doi:10.1038/jid.1949.86
- Chougule A, Chatterjee D, Sethi S, et al. Granulomatous rosacea versus lupus miliaris disseminatus faciei—2 faces of facial granulomatous disorder: a clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819-823. doi:10.1097/DAD.0000000000001243
- Mullanax MG, Kierland RR. Granulomatous rosacea. Arch Dermatol. 1970;101:206-211.
- Sánchez JL, Berlingeri-Ramos AC, Dueño DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6-9. doi:10.1097/DAD.0b013e31815bc191
- Helm KF, Menz J, Gibson LE, et al. A clinical and histopathologic study of granulomatous rosacea. J Am Acad Dermatol. 1991;25:1038-1043. doi:10.1016/0190-9622(91)70304-k
- Kanada KN, Nakatsuji T, Gallo RL. Doxycycline indirectly inhibits proteolytic activation of tryptic kallikrein-related peptidases and activation of cathelicidin. J Invest Dermatol. 2012;132:1435-1442. doi:10.1038/jid.2012.14
- Jang YH, Sim JH, Kang HY, et al. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol. 2011;25:544-548. doi:10.1111/j.1468-3083.2010.03825.x
- Khokhar O, Khachemoune A. A case of granulomatous rosacea: sorting granulomatous rosacea from other granulomatous diseases that affect the face. Dermatol Online J. 2004;10:6.
- Rosen T, Stone MS. Acne rosacea in blacks. J Am Acad Dermatol. 1987;17:70-73. doi:10.1016/s0190-9622(87)70173-x
- Adams AK, Davis JL, Davis MDP, et al. What is your diagnosis? granulomatous rosacea (lupus miliaris disseminatus faciei, acne agminata). Cutis. 2008;82:103-112.
- Shitara A. Lupus miliaris disseminatus faciei. Int J Dermatol. 1984;23:542-544. doi:10.1111/j.1365-4362.1984.tb04206.x
- Hodak E, Trattner A, Feuerman H, et al. Lupus miliaris disseminatus faciei—the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614-619. doi: 10.1111/j.1365-2133.1997.tb03797.x
- Skowron F, Causeret AS, Pabion C, et al. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution. Dermatology. 2000;201:287-289. doi:10.1159/000051539
- Hutchinson J. Case of livid papillary psoriasis. In: London J, Churchill A, eds. Illustrations of Clinical Surgery. J&A Churchill; 1877:42-43.
- Besnier E. Lupus pernio of the face [in French]. Ann Dermatol Syphiligr (Paris). 1889;10:33-36.
- Tenneson H. Lupus pernio. Ann Dermatol Syphiligr (Paris). 1889;10:333-336.
- Boeck C. Multiple benign sarkoid of the skin [in Norwegian]. Norsk Mag Laegevidensk. 1899;14:1321-1334.
- Kuznitsky E, Bittorf A. Sarkoid mit beteiligung innerer organe. Münch Med Wochenschr. 1915;62:1349-1353.
- Schaumann J. Etude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. Ann Dermatol Syphiligr. 1916-1917;6:357-373.
- Osler W. On chronic symmetrical enlargement of the salivary and lacrimal glands. Am J Med Sci. 1898;115:27-30.
- Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49:6-18. doi:10.1007/s12016-015-8481-z
- Eberhardt C, Thillai M, Parker R, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12:e0170285. doi:10.1371/journal.pone.0170285
- Esteves TC, Aparicio G, Ferrer B, et al. Prognostic value of skin lesions in sarcoidosis: clinical and histopathological clues. Eur J Dermatol. 2015;25:556-562. doi:10.1684/ejd.2015.2666
- Cardoso JC, Cravo M, Reis JP, et al. Cutaneous sarcoidosis: a histopathological study. J Eur Acad Dermatol Venereol. 2009;23:678-682. doi:10.1111/j.1468-3083.2009.03153.x
- Mangas C, Fernández-Figueras M-T, Fité E, et al. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772-777. doi:10.1111/j.1600-0560.2006.00563.x
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis. clinical spectrum and histological analysis of 40 cases. Int J Dermatol. 2019;58:178-184. doi: 10.1111/ijd.14218
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16. doi:10.1186/1546-0096-6-16
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87:871-878. doi:10.1080/08035259875001366244. A, H, Yapıcı I. Isolated cutaneous sarcoidosis. Arch Bronconeumol. 2016;52:220.
- Scadding JG. The late stages of pulmonary sarcoidosis. Postgrad Med J. 1970;46:530-536. doi:10.1136/pgmj.46.538.530
Practice Points
- Dermatologists should be aware that noninfectious granulomatous dermatosis of the face can be caused by granulomatous periorificial dermatitis, granulomatous rosacea, lupus miliaris disseminatus faciei, and papular sarcoidosis.
- These conditions lie on a spectrum, suggested by their historical description and clinical and histological features.
- Because their clinical courses can vary considerably from patient to patient, a thorough effort should be made to differentiate these conditions.
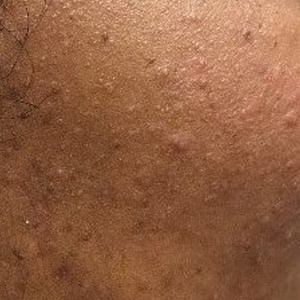
Flesh-Colored Papule in the Nose of a Child
The Diagnosis: Striated Muscle Hamartoma
Histopathologic evaluation revealed a dome-shaped papule with a center composed of mature striated muscle bundles, vellus hairs, sebaceous lobules, and nerve twigs (Figure) consistent with a diagnosis of striated muscle hamartoma (SMH).
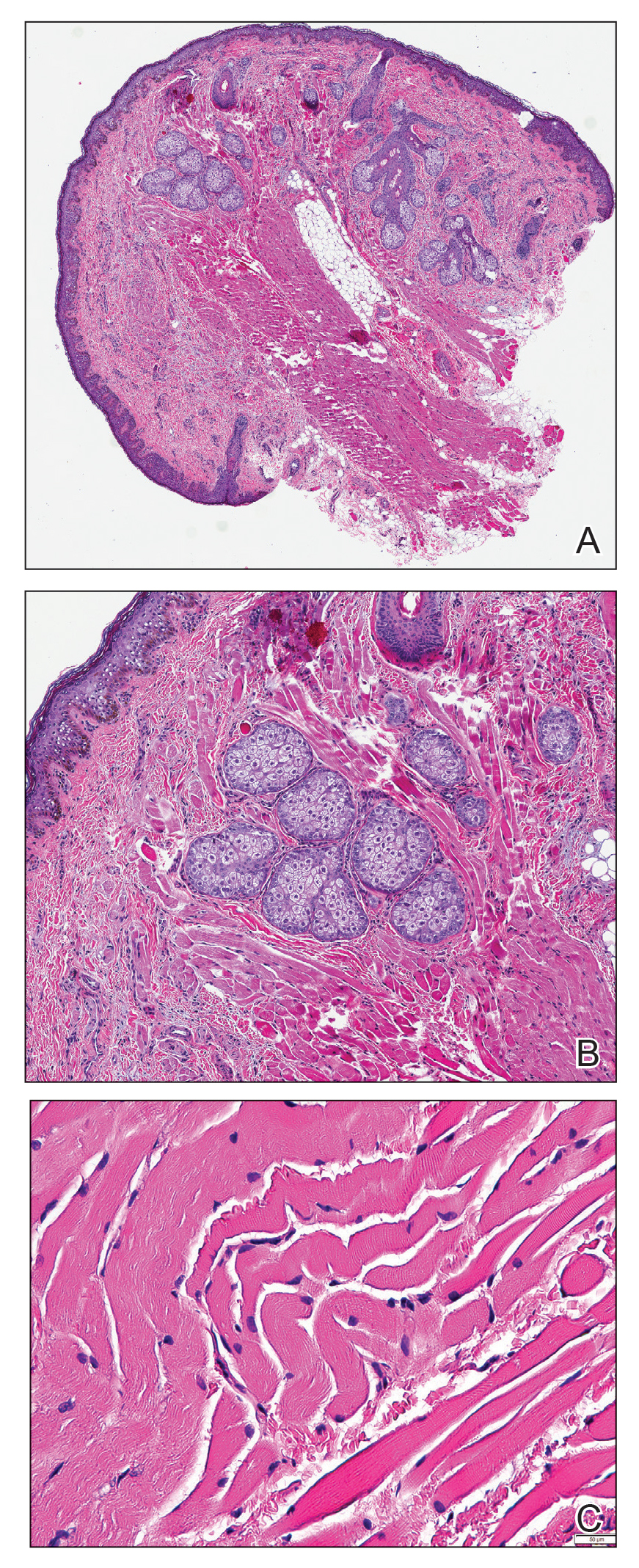
Striated muscle hamartoma was first described in 1986 by Hendrick et al1 with 2 cases in neonates. Biopsies of the lesions taken from the upper lip and sternum showed a characteristic histology consisting of dermal striated muscle fibers and nerve bundles in the central core of the papules associated with a marked number of adnexa. In 1989, the diagnosis of rhabdomyomatous mesenchymal hamartoma was described, which showed similar findings.2 Cases reported since these entities were discovered have used the terms striated muscle hamartoma and rhabdomyomatous mesenchymal hamartoma interchangeably.3
Most commonly found on the head and neck, SMH has now been observed in diverse locations including the sternum, hallux, vagina, and oral cavity.1-15 Many reported cases describe lesions around or in the nose.4,7,8 Multiple congenital anomalies have been described alongside SMH and may be associated with this entity including amniotic bands, cleft lip and palate, coloboma, and Delleman syndrome.1,3,4 Almost all of the lesions present as a sessile or pedunculated papule, polyp, nodule, or plaque measuring from 0.3 cm up to 4.9 cm and typically are present since birth.3,5,15 However, there are a few cases of lesions presenting in adults with no prior history.5,6,15
Microscopically, SMH is defined by a dermal lesion with a core comprised of mature skeletal muscle admixed with adipose tissue, adnexa, nerve bundles, and fibrovascular tissue.1 There are other entities that should be considered before making the diagnosis of SMH. Other hamartomas such as accessory tragus, connective tissue nevus, fibrous hamartoma of infancy, and nevus lipomatosis may present similarly; however, these lesions classically lack skeletal muscle. Benign triton tumors, or neuromuscular hamartomas, are rare lesions composed of skeletal muscle and abundant, intimately associated neural tissue. Neuromuscular hamartomas frequently involve large nerves.16 Rhabdomyomas also should be considered. Adult rhabdomyomas are composed of eosinophilic polygonal cells with granular cytoplasm and occasional cross-striations. Fetal rhabdomyomas have multiple histologic types and are defined by a variable myxoid stroma, eosinophilic spindled cells, and rhabdomyocytes in various stages of maturity. Genital rhabdomyomas histopathologically appear similar to fetal rhabdomyomas but are confined to the genital region. The skeletal muscle present in rhabdomyomas typically is less differentiated.17 TMature skeletal bundles should be a dominant component of the lesion before diagnosing SMH.
Typically presenting as congenital lesions in the head and neck region, papules with a dermal core of mature skeletal muscle associated with adnexa and nerve twigs should prompt consideration of a diagnosis of SMH or rhabdomyomatous mesenchymal hamartoma. These lesions are benign and usually are cured with complete excision.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol. 1986;3:153-157.
- Mills AE. Rhabdomyomatous mesenchymal hamartoma of skin. Am J Dermatopathol. 1989;1:58-63.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: an unusual dermal entity with a report of two cases and a review of the literature. J Cutan Pathol. 2002;29:238-243.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman’s syndrome. J Cutan Pathol. 1994;21:40-46.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005;21:185-188.
- Harris MA, Dutton JJ, Proia AD. Striated muscle hamartoma of the eyelid in an adult woman. Ophthalmic Plast Reconstr Surg. 2008;24:492-494.
- Nakanishi H, Hashimoto I, Takiwaki H, et al. Striated muscle hamartoma of the nostril. J Dermatol. 1995;22:504-507.
- Farris PE, Manning S, Veatch F. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol. 1994;16:73-75.
- Grilli R, Escalonilla P, Soriano ML, et al. The so-called striated muscle hamartoma is a hamartoma of cutaneous adnexa and mesenchyme, but not of striated muscle. Acta Derm Venereol. 1998;78:390.
- Sampat K, Cheesman E, Siminas S. Perianal rhabdomyomatous mesenchymal hamartoma. Ann R Coll Surg Engl. 2017;99:E193-E195.
- Brinster NK, Farmer ER. Rhabdomyomatous mesenchymal hamartoma presenting on a digit. J Cutan Pathol. 2009;36:61-63.
- Han SH, Song HJ, Hong WK, et al. Rhabdomyomatous mesenchymal hamartoma of the vagina. Pediatr Dermatol. 2009;26:753-755.
- De la Sotta P, Salomone C, González S. Rhabdomyomatous (mesenchymal) hamartoma of the tongue: report of a case. J Oral Pathol Med. 2007;36:58-59.
- Magro G, Di Benedetto A, Sanges G, et al. Rhabdomyomatous mesenchymal hamartoma of oral cavity: an unusual location for such a rare lesion. Virchows Arch. 2005;446:346-347.
- Wang Y, Zhao H, Yue X, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a big subcutaneous mass on the neck: a case report. J Med Case Rep. 2014;8:410.
- Amita K, Shankar SV, Nischal KC, et al. Benign triton tumor: a rare entity in head and neck region. Korean J Pathol. 2013;47:74-76.
- Walsh S, Hurt M. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32:485-491.
The Diagnosis: Striated Muscle Hamartoma
Histopathologic evaluation revealed a dome-shaped papule with a center composed of mature striated muscle bundles, vellus hairs, sebaceous lobules, and nerve twigs (Figure) consistent with a diagnosis of striated muscle hamartoma (SMH).
Striated muscle hamartoma was first described in 1986 by Hendrick et al1 with 2 cases in neonates. Biopsies of the lesions taken from the upper lip and sternum showed a characteristic histology consisting of dermal striated muscle fibers and nerve bundles in the central core of the papules associated with a marked number of adnexa. In 1989, the diagnosis of rhabdomyomatous mesenchymal hamartoma was described, which showed similar findings.2 Cases reported since these entities were discovered have used the terms striated muscle hamartoma and rhabdomyomatous mesenchymal hamartoma interchangeably.3
Most commonly found on the head and neck, SMH has now been observed in diverse locations including the sternum, hallux, vagina, and oral cavity.1-15 Many reported cases describe lesions around or in the nose.4,7,8 Multiple congenital anomalies have been described alongside SMH and may be associated with this entity including amniotic bands, cleft lip and palate, coloboma, and Delleman syndrome.1,3,4 Almost all of the lesions present as a sessile or pedunculated papule, polyp, nodule, or plaque measuring from 0.3 cm up to 4.9 cm and typically are present since birth.3,5,15 However, there are a few cases of lesions presenting in adults with no prior history.5,6,15
Microscopically, SMH is defined by a dermal lesion with a core comprised of mature skeletal muscle admixed with adipose tissue, adnexa, nerve bundles, and fibrovascular tissue.1 There are other entities that should be considered before making the diagnosis of SMH. Other hamartomas such as accessory tragus, connective tissue nevus, fibrous hamartoma of infancy, and nevus lipomatosis may present similarly; however, these lesions classically lack skeletal muscle. Benign triton tumors, or neuromuscular hamartomas, are rare lesions composed of skeletal muscle and abundant, intimately associated neural tissue. Neuromuscular hamartomas frequently involve large nerves.16 Rhabdomyomas also should be considered. Adult rhabdomyomas are composed of eosinophilic polygonal cells with granular cytoplasm and occasional cross-striations. Fetal rhabdomyomas have multiple histologic types and are defined by a variable myxoid stroma, eosinophilic spindled cells, and rhabdomyocytes in various stages of maturity. Genital rhabdomyomas histopathologically appear similar to fetal rhabdomyomas but are confined to the genital region. The skeletal muscle present in rhabdomyomas typically is less differentiated.17 TMature skeletal bundles should be a dominant component of the lesion before diagnosing SMH.
Typically presenting as congenital lesions in the head and neck region, papules with a dermal core of mature skeletal muscle associated with adnexa and nerve twigs should prompt consideration of a diagnosis of SMH or rhabdomyomatous mesenchymal hamartoma. These lesions are benign and usually are cured with complete excision.
The Diagnosis: Striated Muscle Hamartoma
Histopathologic evaluation revealed a dome-shaped papule with a center composed of mature striated muscle bundles, vellus hairs, sebaceous lobules, and nerve twigs (Figure) consistent with a diagnosis of striated muscle hamartoma (SMH).
Striated muscle hamartoma was first described in 1986 by Hendrick et al1 with 2 cases in neonates. Biopsies of the lesions taken from the upper lip and sternum showed a characteristic histology consisting of dermal striated muscle fibers and nerve bundles in the central core of the papules associated with a marked number of adnexa. In 1989, the diagnosis of rhabdomyomatous mesenchymal hamartoma was described, which showed similar findings.2 Cases reported since these entities were discovered have used the terms striated muscle hamartoma and rhabdomyomatous mesenchymal hamartoma interchangeably.3
Most commonly found on the head and neck, SMH has now been observed in diverse locations including the sternum, hallux, vagina, and oral cavity.1-15 Many reported cases describe lesions around or in the nose.4,7,8 Multiple congenital anomalies have been described alongside SMH and may be associated with this entity including amniotic bands, cleft lip and palate, coloboma, and Delleman syndrome.1,3,4 Almost all of the lesions present as a sessile or pedunculated papule, polyp, nodule, or plaque measuring from 0.3 cm up to 4.9 cm and typically are present since birth.3,5,15 However, there are a few cases of lesions presenting in adults with no prior history.5,6,15
Microscopically, SMH is defined by a dermal lesion with a core comprised of mature skeletal muscle admixed with adipose tissue, adnexa, nerve bundles, and fibrovascular tissue.1 There are other entities that should be considered before making the diagnosis of SMH. Other hamartomas such as accessory tragus, connective tissue nevus, fibrous hamartoma of infancy, and nevus lipomatosis may present similarly; however, these lesions classically lack skeletal muscle. Benign triton tumors, or neuromuscular hamartomas, are rare lesions composed of skeletal muscle and abundant, intimately associated neural tissue. Neuromuscular hamartomas frequently involve large nerves.16 Rhabdomyomas also should be considered. Adult rhabdomyomas are composed of eosinophilic polygonal cells with granular cytoplasm and occasional cross-striations. Fetal rhabdomyomas have multiple histologic types and are defined by a variable myxoid stroma, eosinophilic spindled cells, and rhabdomyocytes in various stages of maturity. Genital rhabdomyomas histopathologically appear similar to fetal rhabdomyomas but are confined to the genital region. The skeletal muscle present in rhabdomyomas typically is less differentiated.17 TMature skeletal bundles should be a dominant component of the lesion before diagnosing SMH.
Typically presenting as congenital lesions in the head and neck region, papules with a dermal core of mature skeletal muscle associated with adnexa and nerve twigs should prompt consideration of a diagnosis of SMH or rhabdomyomatous mesenchymal hamartoma. These lesions are benign and usually are cured with complete excision.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol. 1986;3:153-157.
- Mills AE. Rhabdomyomatous mesenchymal hamartoma of skin. Am J Dermatopathol. 1989;1:58-63.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: an unusual dermal entity with a report of two cases and a review of the literature. J Cutan Pathol. 2002;29:238-243.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman’s syndrome. J Cutan Pathol. 1994;21:40-46.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005;21:185-188.
- Harris MA, Dutton JJ, Proia AD. Striated muscle hamartoma of the eyelid in an adult woman. Ophthalmic Plast Reconstr Surg. 2008;24:492-494.
- Nakanishi H, Hashimoto I, Takiwaki H, et al. Striated muscle hamartoma of the nostril. J Dermatol. 1995;22:504-507.
- Farris PE, Manning S, Veatch F. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol. 1994;16:73-75.
- Grilli R, Escalonilla P, Soriano ML, et al. The so-called striated muscle hamartoma is a hamartoma of cutaneous adnexa and mesenchyme, but not of striated muscle. Acta Derm Venereol. 1998;78:390.
- Sampat K, Cheesman E, Siminas S. Perianal rhabdomyomatous mesenchymal hamartoma. Ann R Coll Surg Engl. 2017;99:E193-E195.
- Brinster NK, Farmer ER. Rhabdomyomatous mesenchymal hamartoma presenting on a digit. J Cutan Pathol. 2009;36:61-63.
- Han SH, Song HJ, Hong WK, et al. Rhabdomyomatous mesenchymal hamartoma of the vagina. Pediatr Dermatol. 2009;26:753-755.
- De la Sotta P, Salomone C, González S. Rhabdomyomatous (mesenchymal) hamartoma of the tongue: report of a case. J Oral Pathol Med. 2007;36:58-59.
- Magro G, Di Benedetto A, Sanges G, et al. Rhabdomyomatous mesenchymal hamartoma of oral cavity: an unusual location for such a rare lesion. Virchows Arch. 2005;446:346-347.
- Wang Y, Zhao H, Yue X, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a big subcutaneous mass on the neck: a case report. J Med Case Rep. 2014;8:410.
- Amita K, Shankar SV, Nischal KC, et al. Benign triton tumor: a rare entity in head and neck region. Korean J Pathol. 2013;47:74-76.
- Walsh S, Hurt M. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32:485-491.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol. 1986;3:153-157.
- Mills AE. Rhabdomyomatous mesenchymal hamartoma of skin. Am J Dermatopathol. 1989;1:58-63.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: an unusual dermal entity with a report of two cases and a review of the literature. J Cutan Pathol. 2002;29:238-243.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman’s syndrome. J Cutan Pathol. 1994;21:40-46.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005;21:185-188.
- Harris MA, Dutton JJ, Proia AD. Striated muscle hamartoma of the eyelid in an adult woman. Ophthalmic Plast Reconstr Surg. 2008;24:492-494.
- Nakanishi H, Hashimoto I, Takiwaki H, et al. Striated muscle hamartoma of the nostril. J Dermatol. 1995;22:504-507.
- Farris PE, Manning S, Veatch F. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol. 1994;16:73-75.
- Grilli R, Escalonilla P, Soriano ML, et al. The so-called striated muscle hamartoma is a hamartoma of cutaneous adnexa and mesenchyme, but not of striated muscle. Acta Derm Venereol. 1998;78:390.
- Sampat K, Cheesman E, Siminas S. Perianal rhabdomyomatous mesenchymal hamartoma. Ann R Coll Surg Engl. 2017;99:E193-E195.
- Brinster NK, Farmer ER. Rhabdomyomatous mesenchymal hamartoma presenting on a digit. J Cutan Pathol. 2009;36:61-63.
- Han SH, Song HJ, Hong WK, et al. Rhabdomyomatous mesenchymal hamartoma of the vagina. Pediatr Dermatol. 2009;26:753-755.
- De la Sotta P, Salomone C, González S. Rhabdomyomatous (mesenchymal) hamartoma of the tongue: report of a case. J Oral Pathol Med. 2007;36:58-59.
- Magro G, Di Benedetto A, Sanges G, et al. Rhabdomyomatous mesenchymal hamartoma of oral cavity: an unusual location for such a rare lesion. Virchows Arch. 2005;446:346-347.
- Wang Y, Zhao H, Yue X, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a big subcutaneous mass on the neck: a case report. J Med Case Rep. 2014;8:410.
- Amita K, Shankar SV, Nischal KC, et al. Benign triton tumor: a rare entity in head and neck region. Korean J Pathol. 2013;47:74-76.
- Walsh S, Hurt M. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32:485-491.
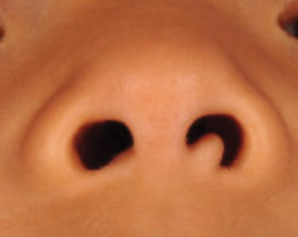
A 4-year-old girl presented to our clinic with an asymptomatic flesh-colored papule in the left nostril. The lesion had been present since birth and grew in relation to the patient with no rapid changes. There had been no pigmentation changes and no bleeding, pain, or itching. The patient’s birth and developmental history were normal. Physical examination revealed a singular, 10×5-mm, flesh-colored, pedunculated mass on the left nasal sill. There were no additional lesions present. An excisional biopsy was performed and submitted for pathologic diagnosis.
