User login
MOC: ACOG’s role in developing a solution to the heated controversy

The American Board of Medical Specialties (ABMS) has decided to trade the phrase “maintenance of certification” (MOC) for “continuing board certification,” a seemingly minor change that has an important backstory. This is the story of how the physician community flexed its collective muscle and how the American College of Obstetricians and Gynecologists (ACOG) helped broker an important détente and pathway in a highly contentious issue.
Founded in 1933 as a nonprofit organization dedicated to maintaining high uniform standards among physicians, the ABMS and many of its specialty boards have found themselves, for more than a decade, under heavy fire from physicians (especially family physicians, internists, and surgeons), their 24 subspecialties, and the state medical societies representing them.
The ObGyn experience with the American Board of Obstetrics and Gynecology (ABOG), however, is better for a number of reasons. Historically, ABOG and ACOG have worked closely together, which is an anomaly among boards as many boards have an arms-length or even an antagonistic relationship with their specialty society.
The discussion below outlines physician concerns with the ABMS and related boards and describes efforts to address and rebuild the continuing board certification process.
Direct and indirect costs
Physicians are very concerned with the costs involved in MOC. Measurable costs include testing fees, while indirect costs include time, stress, travel to test centers, and threats to livelihood for failing a high-stakes examination. Physicians want the high-stakes exam eliminated.
Relevance to practice
Physicians often feel that the MOC has little relevance to their practice, which fuels a sense of resentment toward boards that they believe are dominated by physicians who no longer practice. Subspecialists feel farther away from general practice and the base exams. Generalists feel that the exams miss the points of their daily practice.
Lack of data to show improved quality of care
Physicians want to know that the MOC is worth their time, effort, and money because it improves patient care. To date, however, empirical or clinical data on patient outcomes are absent or ambiguous; most studies lack high-level data or do not investigate the MOC requirements. Physicians want to know what the best MOC practices are, what improves care, and that practices that make no difference will be discarded. In addition, they want timely knowledge alerts when evidence changes.
Relationship to licensing, employment, privileging, credentialing, and reimbursement
Hospitals, insurers, and states increasingly—and inappropriately—use board certification as the primary (sometimes only) default measure of a physician’s fitness for patient care. Physicians without board certification often are denied hospital privileges, inclusion in insurance panels, and even medical licenses. This changes certification from a voluntary physician self-improvement exercise into a can’t-earn-a-living-without-it cudgel.
Variation
Boards vary significantly in their MOC requirements and costs. The importance of an equal standard across all boards is a clear theme among physician concerns.
Role and authority of the ABMS and related boards
Many physicians are frustrated with the perceived autocratic nature of their boards—boards that lack transparency, do not solicit or allow input from practicing physicians, and are unresponsive to physician concerns.
According to Susan Ramin, MD, ABOG Associate Executive Director, ABOG is leading in a number of these areas, including:
- rapidly disseminating clinical information on emerging topics, such as Zika virus infection and opioid misuse
- offering physician choice of testing categories
- exempting high scorers from the secured written exam, which saved physicians a total of $881,000 in exam fees
- crediting physicians for what they already are doing, including serving on maternal mortality review committees, participating in registries, and participating in the Alliance for Innovation on Maternal Health (AIM)
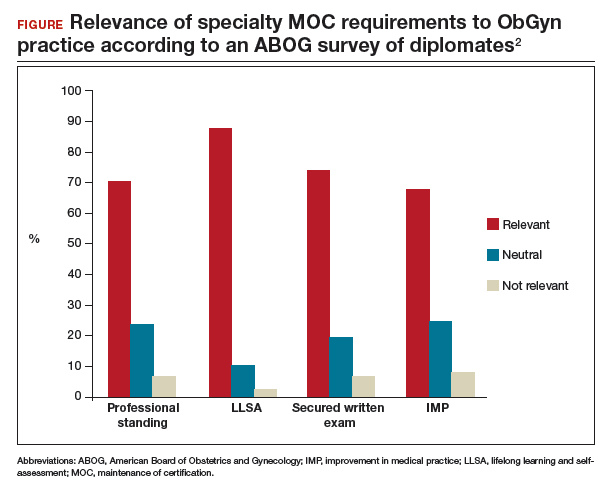
- providing Lifelong Learning and Self-Assessment (LLSA) articles that, according to 90% of diplomates surveyed, are beneficial to their clinical practice (FIGURE).1,2
Our colleague physicians are not so lucky. In a 2015 New England Journal of Medicine Perspective, one physician called out the American Board of Internal Medicine as “a private, self-appointed certifying organization,” a not-for-profit organization that has “grown into a $55-million-per-year business.”3 He concluded that “many physicians are waking up to the fact that our profession is increasingly controlled by people not directly involved in patient care who have lost contact with the realities of day-to-day clinical practice.”3
State and society responses to MOC requirements
Frustration with an inability to resolve these concerns has grown steadily, bubbling over into state governments. The American Medical Association developed “model state legislation intended to prohibit hospitals, health care insurers, and state boards of medicine and osteopathic medicine from requiring participation in MOC processes as a condition of credentialing, privileging, insurance panel participation, licensure, or licensure renewal.”4
Some states are proposing or have enacted legislation that prohibits the use of MOC as a criterion for licensure, privileging, employment, reimbursement, and/or insurance panel participation. Eight states (Arizona, Georgia, Kentucky, Maryland, Maine, Missouri, Oklahoma, Tennessee) have enacted laws to prohibit the use of MOC for initial and renewal licensure decisions. Many states are actively considering MOC-related legislation, including Alaska, Florida, Iowa, Indiana, Maryland, Massachusetts, Michigan, Missouri, New Hampshire, New York, Ohio, Oklahoma, Rhode Island, South Carolina, Tennessee, Utah, Washington, and Wisconsin.
Legislation is not the only outlet for physician frustration. Some medical specialty societies are considering dropping board certification as a membership requirement; physicians are exploring developing alternative boards; and some physicians are defying the board certification requirement altogether, with thousands signing anti-MOC petitions.
ACOG asserts importance of maintaining self-regulation
While other specialties are actively advocating state legislation, ACOG and ABOG have worked together to oppose state legislation, believing that physician self-regulation is paramount. In fact, in 2017, ACOG and ABOG issued a joint statement urging state lawmakers to “not interfere with our decades of successful self-regulation and to realize that each medical society has its own experience with its MOC program.”5
Negotiations lead to new initiative
This brings us to an interesting situation. ACOG’s Executive Vice President and CEO Hal Lawrence III, MD, was tapped (in his position as Chair of the Specialty Society CEO Consortium) to represent physician specialties in negotiations and discussions with the boards, which were represented by Lois Nora, MD, JD, President and CEO of the ABMS, and state medical societies, represented by Donald Palmisano Jr, JD, Executive Director and CEO of the Medical Association of Georgia. Many state medical societies, boards, and physician specialty organizations participated in these meetings.
Throughout months of debate, Dr. Lawrence urged his colleagues to stay at the table and do the hard work of reaching an agreement, rather than ask politicians to solve medicine’s problems. This approach was leveraged by the serious efforts and threats of state legislation, which brought the boards to the table. In August 2017, 41 state medical societies and 33 national medical specialty societies wrote to Dr. Nora expressing their concerns that “professional self-regulation is under attack. Concerns regarding the usefulness of the high-stakes exam, the exorbitant costs of the MOC process, and the lack of transparent communication from the certifying boards have led to damaging the MOC brand, and creating state-based attacks on the MOC process.”6
In December 2017, Dr. Lawrence and Mr. Palmisano led a meeting of principals from the national medical specialty societies and state medical societies with leaders of ABMS and 8 specialty boards, including ABOG, an opportunity to secure meaningful change. Dr. Lawrence began by stressing that the interests of physicians and patients would be best served by all parties coming together and collaborating on a meaningful solution, to repair trust and preserve physician self-regulation.
Dr. Ramin presented ABOG’s approach to continuous certification, lifelong learning, and self-assessment. The American Board of Urology and the American Board of Psychiatry and Neurology indicated that they were basing important changes in their MOC process on ABOG’s work, including using 5 modules (1 general and 4 specific to the physician’s practice) and multiple open-book mini-exams based on selected journal articles as an alternative to the 10-year MOC exam.
The Vision Initiative. At that meeting and others, the ABMS and other boards heard physicians’ candid and sometimes blunt concerns. Dr. Nora spoke to the recently announced Continuing Board Certification: Vision for the Future program, also known as the “Vision Initiative,” a process designed to fundamentally rebuild the continuing certification process with input and guidance from practicing physicians. Physician response seemed uniform: Seeing is believing.
Importantly, all participants at the December meeting agreed to work together to rebuild trust and ensure professionalism and professional self-regulation, reflected in this Statement of Shared Purpose:
ABMS certifying boards and national medical specialty societies will collaborate to resolve differences in the process of ongoing certification and to fulfill the principles of professional self-regulation, achieving appropriate standardization, and assuring that ongoing certification is relevant to the practices of physicians without undue burden. Furthermore, the boards and societies, and their organizations (ABMS and CMSS [Council of Medical Specialty Societies]), will undertake necessary changes in a timely manner, and will commit to ongoing communication with state medical associations to solicit their input.4
Two ObGyns participating in the Vision Initiative are Haywood Brown, MD, ACOG’s Immediate Past President, and George Wendel, MD, ABOG’s Executive Director. The Vision Initiative is composed of 3 parts. Part 1, Organization, is complete. The committee is currently working on part 2, Envisioning the Future, an information-gathering component that includes physician surveys, hearings, open solicited input, and identifying new and better approaches. After the final report is delivered to the ABMS in February 2019, part 3, Implementation, will begin.
The Vision Initiative offers physicians an important opportunity to help shape the future of continuing education and certification. ObGyns and other physicians should consider reviewing and commenting on the draft report, due in November, during the public comment period. Visit https://visioninitiative.org for more information and to sign up for email updates.

Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
- American Board of Obstetrics and Gynecology. From pilot to permanent: ABOG's program offering an innovative pathway integrating lifelong learning and self-assessment and external assessment is approved. https://www.abog.org/new/ABOG_PilotToPermanent.aspx. Accessed July 6, 2018.
- Ramin S. American Board of Obstetrics and Gynecology MOC program. PowerPoint presentation; December 4, 2017.
- Teirstein PS. Boarded to death--why maintenance of certification is bad for doctors and patients. N Engl J Med. 2015;372(2):106-108.
- AMA Council on Medical Education. Executive summary. 2017. https://www.ama-assn.org/sites/default/files/media-browser/public/council-on-med-ed/a18-cme-02.pdf. Accessed July 6, 2018.
- American College of Obstetricians and Gynecologists. ACOG-ABOG joint statement: political interference in physician maintenance of skills threatens women's health care. https://www.acog.org/-/media/Departments/State-Legislative-Activities/2017ACOG-ABMS-MOC-Statement.pdf?dmc=1&ts=20180706T1615538746. Accessed July 6, 2018.
- Letter to Lois Nora, MD, JD. August 18, 2017. https://www.mainemed.com/sites/default/files/content/MOC%20Letter%20082117.pdf. Accessed July 6, 2018.

Ms. DiVenere is Officer, Government and Political Affairs, at the American College of Obstetricians and Gynecologists in Washington, DC. She is an OBG Management Contributing Editor.
The author reports no financial relationships relevant to this article.
Ms. DiVenere is Officer, Government and Political Affairs, at the American College of Obstetricians and Gynecologists in Washington, DC. She is an OBG Management Contributing Editor.
The author reports no financial relationships relevant to this article.
Ms. DiVenere is Officer, Government and Political Affairs, at the American College of Obstetricians and Gynecologists in Washington, DC. She is an OBG Management Contributing Editor.
The author reports no financial relationships relevant to this article.
The American Board of Medical Specialties (ABMS) has decided to trade the phrase “maintenance of certification” (MOC) for “continuing board certification,” a seemingly minor change that has an important backstory. This is the story of how the physician community flexed its collective muscle and how the American College of Obstetricians and Gynecologists (ACOG) helped broker an important détente and pathway in a highly contentious issue.
Founded in 1933 as a nonprofit organization dedicated to maintaining high uniform standards among physicians, the ABMS and many of its specialty boards have found themselves, for more than a decade, under heavy fire from physicians (especially family physicians, internists, and surgeons), their 24 subspecialties, and the state medical societies representing them.
The ObGyn experience with the American Board of Obstetrics and Gynecology (ABOG), however, is better for a number of reasons. Historically, ABOG and ACOG have worked closely together, which is an anomaly among boards as many boards have an arms-length or even an antagonistic relationship with their specialty society.
The discussion below outlines physician concerns with the ABMS and related boards and describes efforts to address and rebuild the continuing board certification process.
Direct and indirect costs
Physicians are very concerned with the costs involved in MOC. Measurable costs include testing fees, while indirect costs include time, stress, travel to test centers, and threats to livelihood for failing a high-stakes examination. Physicians want the high-stakes exam eliminated.
Relevance to practice
Physicians often feel that the MOC has little relevance to their practice, which fuels a sense of resentment toward boards that they believe are dominated by physicians who no longer practice. Subspecialists feel farther away from general practice and the base exams. Generalists feel that the exams miss the points of their daily practice.
Lack of data to show improved quality of care
Physicians want to know that the MOC is worth their time, effort, and money because it improves patient care. To date, however, empirical or clinical data on patient outcomes are absent or ambiguous; most studies lack high-level data or do not investigate the MOC requirements. Physicians want to know what the best MOC practices are, what improves care, and that practices that make no difference will be discarded. In addition, they want timely knowledge alerts when evidence changes.
Relationship to licensing, employment, privileging, credentialing, and reimbursement
Hospitals, insurers, and states increasingly—and inappropriately—use board certification as the primary (sometimes only) default measure of a physician’s fitness for patient care. Physicians without board certification often are denied hospital privileges, inclusion in insurance panels, and even medical licenses. This changes certification from a voluntary physician self-improvement exercise into a can’t-earn-a-living-without-it cudgel.
Variation
Boards vary significantly in their MOC requirements and costs. The importance of an equal standard across all boards is a clear theme among physician concerns.
Role and authority of the ABMS and related boards
Many physicians are frustrated with the perceived autocratic nature of their boards—boards that lack transparency, do not solicit or allow input from practicing physicians, and are unresponsive to physician concerns.
According to Susan Ramin, MD, ABOG Associate Executive Director, ABOG is leading in a number of these areas, including:
- rapidly disseminating clinical information on emerging topics, such as Zika virus infection and opioid misuse
- offering physician choice of testing categories
- exempting high scorers from the secured written exam, which saved physicians a total of $881,000 in exam fees
- crediting physicians for what they already are doing, including serving on maternal mortality review committees, participating in registries, and participating in the Alliance for Innovation on Maternal Health (AIM)
- providing Lifelong Learning and Self-Assessment (LLSA) articles that, according to 90% of diplomates surveyed, are beneficial to their clinical practice (FIGURE).1,2
Our colleague physicians are not so lucky. In a 2015 New England Journal of Medicine Perspective, one physician called out the American Board of Internal Medicine as “a private, self-appointed certifying organization,” a not-for-profit organization that has “grown into a $55-million-per-year business.”3 He concluded that “many physicians are waking up to the fact that our profession is increasingly controlled by people not directly involved in patient care who have lost contact with the realities of day-to-day clinical practice.”3
State and society responses to MOC requirements
Frustration with an inability to resolve these concerns has grown steadily, bubbling over into state governments. The American Medical Association developed “model state legislation intended to prohibit hospitals, health care insurers, and state boards of medicine and osteopathic medicine from requiring participation in MOC processes as a condition of credentialing, privileging, insurance panel participation, licensure, or licensure renewal.”4
Some states are proposing or have enacted legislation that prohibits the use of MOC as a criterion for licensure, privileging, employment, reimbursement, and/or insurance panel participation. Eight states (Arizona, Georgia, Kentucky, Maryland, Maine, Missouri, Oklahoma, Tennessee) have enacted laws to prohibit the use of MOC for initial and renewal licensure decisions. Many states are actively considering MOC-related legislation, including Alaska, Florida, Iowa, Indiana, Maryland, Massachusetts, Michigan, Missouri, New Hampshire, New York, Ohio, Oklahoma, Rhode Island, South Carolina, Tennessee, Utah, Washington, and Wisconsin.
Legislation is not the only outlet for physician frustration. Some medical specialty societies are considering dropping board certification as a membership requirement; physicians are exploring developing alternative boards; and some physicians are defying the board certification requirement altogether, with thousands signing anti-MOC petitions.
ACOG asserts importance of maintaining self-regulation
While other specialties are actively advocating state legislation, ACOG and ABOG have worked together to oppose state legislation, believing that physician self-regulation is paramount. In fact, in 2017, ACOG and ABOG issued a joint statement urging state lawmakers to “not interfere with our decades of successful self-regulation and to realize that each medical society has its own experience with its MOC program.”5
Negotiations lead to new initiative
This brings us to an interesting situation. ACOG’s Executive Vice President and CEO Hal Lawrence III, MD, was tapped (in his position as Chair of the Specialty Society CEO Consortium) to represent physician specialties in negotiations and discussions with the boards, which were represented by Lois Nora, MD, JD, President and CEO of the ABMS, and state medical societies, represented by Donald Palmisano Jr, JD, Executive Director and CEO of the Medical Association of Georgia. Many state medical societies, boards, and physician specialty organizations participated in these meetings.
Throughout months of debate, Dr. Lawrence urged his colleagues to stay at the table and do the hard work of reaching an agreement, rather than ask politicians to solve medicine’s problems. This approach was leveraged by the serious efforts and threats of state legislation, which brought the boards to the table. In August 2017, 41 state medical societies and 33 national medical specialty societies wrote to Dr. Nora expressing their concerns that “professional self-regulation is under attack. Concerns regarding the usefulness of the high-stakes exam, the exorbitant costs of the MOC process, and the lack of transparent communication from the certifying boards have led to damaging the MOC brand, and creating state-based attacks on the MOC process.”6
In December 2017, Dr. Lawrence and Mr. Palmisano led a meeting of principals from the national medical specialty societies and state medical societies with leaders of ABMS and 8 specialty boards, including ABOG, an opportunity to secure meaningful change. Dr. Lawrence began by stressing that the interests of physicians and patients would be best served by all parties coming together and collaborating on a meaningful solution, to repair trust and preserve physician self-regulation.
Dr. Ramin presented ABOG’s approach to continuous certification, lifelong learning, and self-assessment. The American Board of Urology and the American Board of Psychiatry and Neurology indicated that they were basing important changes in their MOC process on ABOG’s work, including using 5 modules (1 general and 4 specific to the physician’s practice) and multiple open-book mini-exams based on selected journal articles as an alternative to the 10-year MOC exam.
The Vision Initiative. At that meeting and others, the ABMS and other boards heard physicians’ candid and sometimes blunt concerns. Dr. Nora spoke to the recently announced Continuing Board Certification: Vision for the Future program, also known as the “Vision Initiative,” a process designed to fundamentally rebuild the continuing certification process with input and guidance from practicing physicians. Physician response seemed uniform: Seeing is believing.
Importantly, all participants at the December meeting agreed to work together to rebuild trust and ensure professionalism and professional self-regulation, reflected in this Statement of Shared Purpose:
ABMS certifying boards and national medical specialty societies will collaborate to resolve differences in the process of ongoing certification and to fulfill the principles of professional self-regulation, achieving appropriate standardization, and assuring that ongoing certification is relevant to the practices of physicians without undue burden. Furthermore, the boards and societies, and their organizations (ABMS and CMSS [Council of Medical Specialty Societies]), will undertake necessary changes in a timely manner, and will commit to ongoing communication with state medical associations to solicit their input.4
Two ObGyns participating in the Vision Initiative are Haywood Brown, MD, ACOG’s Immediate Past President, and George Wendel, MD, ABOG’s Executive Director. The Vision Initiative is composed of 3 parts. Part 1, Organization, is complete. The committee is currently working on part 2, Envisioning the Future, an information-gathering component that includes physician surveys, hearings, open solicited input, and identifying new and better approaches. After the final report is delivered to the ABMS in February 2019, part 3, Implementation, will begin.
The Vision Initiative offers physicians an important opportunity to help shape the future of continuing education and certification. ObGyns and other physicians should consider reviewing and commenting on the draft report, due in November, during the public comment period. Visit https://visioninitiative.org for more information and to sign up for email updates.
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
The American Board of Medical Specialties (ABMS) has decided to trade the phrase “maintenance of certification” (MOC) for “continuing board certification,” a seemingly minor change that has an important backstory. This is the story of how the physician community flexed its collective muscle and how the American College of Obstetricians and Gynecologists (ACOG) helped broker an important détente and pathway in a highly contentious issue.
Founded in 1933 as a nonprofit organization dedicated to maintaining high uniform standards among physicians, the ABMS and many of its specialty boards have found themselves, for more than a decade, under heavy fire from physicians (especially family physicians, internists, and surgeons), their 24 subspecialties, and the state medical societies representing them.
The ObGyn experience with the American Board of Obstetrics and Gynecology (ABOG), however, is better for a number of reasons. Historically, ABOG and ACOG have worked closely together, which is an anomaly among boards as many boards have an arms-length or even an antagonistic relationship with their specialty society.
The discussion below outlines physician concerns with the ABMS and related boards and describes efforts to address and rebuild the continuing board certification process.
Direct and indirect costs
Physicians are very concerned with the costs involved in MOC. Measurable costs include testing fees, while indirect costs include time, stress, travel to test centers, and threats to livelihood for failing a high-stakes examination. Physicians want the high-stakes exam eliminated.
Relevance to practice
Physicians often feel that the MOC has little relevance to their practice, which fuels a sense of resentment toward boards that they believe are dominated by physicians who no longer practice. Subspecialists feel farther away from general practice and the base exams. Generalists feel that the exams miss the points of their daily practice.
Lack of data to show improved quality of care
Physicians want to know that the MOC is worth their time, effort, and money because it improves patient care. To date, however, empirical or clinical data on patient outcomes are absent or ambiguous; most studies lack high-level data or do not investigate the MOC requirements. Physicians want to know what the best MOC practices are, what improves care, and that practices that make no difference will be discarded. In addition, they want timely knowledge alerts when evidence changes.
Relationship to licensing, employment, privileging, credentialing, and reimbursement
Hospitals, insurers, and states increasingly—and inappropriately—use board certification as the primary (sometimes only) default measure of a physician’s fitness for patient care. Physicians without board certification often are denied hospital privileges, inclusion in insurance panels, and even medical licenses. This changes certification from a voluntary physician self-improvement exercise into a can’t-earn-a-living-without-it cudgel.
Variation
Boards vary significantly in their MOC requirements and costs. The importance of an equal standard across all boards is a clear theme among physician concerns.
Role and authority of the ABMS and related boards
Many physicians are frustrated with the perceived autocratic nature of their boards—boards that lack transparency, do not solicit or allow input from practicing physicians, and are unresponsive to physician concerns.
According to Susan Ramin, MD, ABOG Associate Executive Director, ABOG is leading in a number of these areas, including:
- rapidly disseminating clinical information on emerging topics, such as Zika virus infection and opioid misuse
- offering physician choice of testing categories
- exempting high scorers from the secured written exam, which saved physicians a total of $881,000 in exam fees
- crediting physicians for what they already are doing, including serving on maternal mortality review committees, participating in registries, and participating in the Alliance for Innovation on Maternal Health (AIM)
- providing Lifelong Learning and Self-Assessment (LLSA) articles that, according to 90% of diplomates surveyed, are beneficial to their clinical practice (FIGURE).1,2
Our colleague physicians are not so lucky. In a 2015 New England Journal of Medicine Perspective, one physician called out the American Board of Internal Medicine as “a private, self-appointed certifying organization,” a not-for-profit organization that has “grown into a $55-million-per-year business.”3 He concluded that “many physicians are waking up to the fact that our profession is increasingly controlled by people not directly involved in patient care who have lost contact with the realities of day-to-day clinical practice.”3
State and society responses to MOC requirements
Frustration with an inability to resolve these concerns has grown steadily, bubbling over into state governments. The American Medical Association developed “model state legislation intended to prohibit hospitals, health care insurers, and state boards of medicine and osteopathic medicine from requiring participation in MOC processes as a condition of credentialing, privileging, insurance panel participation, licensure, or licensure renewal.”4
Some states are proposing or have enacted legislation that prohibits the use of MOC as a criterion for licensure, privileging, employment, reimbursement, and/or insurance panel participation. Eight states (Arizona, Georgia, Kentucky, Maryland, Maine, Missouri, Oklahoma, Tennessee) have enacted laws to prohibit the use of MOC for initial and renewal licensure decisions. Many states are actively considering MOC-related legislation, including Alaska, Florida, Iowa, Indiana, Maryland, Massachusetts, Michigan, Missouri, New Hampshire, New York, Ohio, Oklahoma, Rhode Island, South Carolina, Tennessee, Utah, Washington, and Wisconsin.
Legislation is not the only outlet for physician frustration. Some medical specialty societies are considering dropping board certification as a membership requirement; physicians are exploring developing alternative boards; and some physicians are defying the board certification requirement altogether, with thousands signing anti-MOC petitions.
ACOG asserts importance of maintaining self-regulation
While other specialties are actively advocating state legislation, ACOG and ABOG have worked together to oppose state legislation, believing that physician self-regulation is paramount. In fact, in 2017, ACOG and ABOG issued a joint statement urging state lawmakers to “not interfere with our decades of successful self-regulation and to realize that each medical society has its own experience with its MOC program.”5
Negotiations lead to new initiative
This brings us to an interesting situation. ACOG’s Executive Vice President and CEO Hal Lawrence III, MD, was tapped (in his position as Chair of the Specialty Society CEO Consortium) to represent physician specialties in negotiations and discussions with the boards, which were represented by Lois Nora, MD, JD, President and CEO of the ABMS, and state medical societies, represented by Donald Palmisano Jr, JD, Executive Director and CEO of the Medical Association of Georgia. Many state medical societies, boards, and physician specialty organizations participated in these meetings.
Throughout months of debate, Dr. Lawrence urged his colleagues to stay at the table and do the hard work of reaching an agreement, rather than ask politicians to solve medicine’s problems. This approach was leveraged by the serious efforts and threats of state legislation, which brought the boards to the table. In August 2017, 41 state medical societies and 33 national medical specialty societies wrote to Dr. Nora expressing their concerns that “professional self-regulation is under attack. Concerns regarding the usefulness of the high-stakes exam, the exorbitant costs of the MOC process, and the lack of transparent communication from the certifying boards have led to damaging the MOC brand, and creating state-based attacks on the MOC process.”6
In December 2017, Dr. Lawrence and Mr. Palmisano led a meeting of principals from the national medical specialty societies and state medical societies with leaders of ABMS and 8 specialty boards, including ABOG, an opportunity to secure meaningful change. Dr. Lawrence began by stressing that the interests of physicians and patients would be best served by all parties coming together and collaborating on a meaningful solution, to repair trust and preserve physician self-regulation.
Dr. Ramin presented ABOG’s approach to continuous certification, lifelong learning, and self-assessment. The American Board of Urology and the American Board of Psychiatry and Neurology indicated that they were basing important changes in their MOC process on ABOG’s work, including using 5 modules (1 general and 4 specific to the physician’s practice) and multiple open-book mini-exams based on selected journal articles as an alternative to the 10-year MOC exam.
The Vision Initiative. At that meeting and others, the ABMS and other boards heard physicians’ candid and sometimes blunt concerns. Dr. Nora spoke to the recently announced Continuing Board Certification: Vision for the Future program, also known as the “Vision Initiative,” a process designed to fundamentally rebuild the continuing certification process with input and guidance from practicing physicians. Physician response seemed uniform: Seeing is believing.
Importantly, all participants at the December meeting agreed to work together to rebuild trust and ensure professionalism and professional self-regulation, reflected in this Statement of Shared Purpose:
ABMS certifying boards and national medical specialty societies will collaborate to resolve differences in the process of ongoing certification and to fulfill the principles of professional self-regulation, achieving appropriate standardization, and assuring that ongoing certification is relevant to the practices of physicians without undue burden. Furthermore, the boards and societies, and their organizations (ABMS and CMSS [Council of Medical Specialty Societies]), will undertake necessary changes in a timely manner, and will commit to ongoing communication with state medical associations to solicit their input.4
Two ObGyns participating in the Vision Initiative are Haywood Brown, MD, ACOG’s Immediate Past President, and George Wendel, MD, ABOG’s Executive Director. The Vision Initiative is composed of 3 parts. Part 1, Organization, is complete. The committee is currently working on part 2, Envisioning the Future, an information-gathering component that includes physician surveys, hearings, open solicited input, and identifying new and better approaches. After the final report is delivered to the ABMS in February 2019, part 3, Implementation, will begin.
The Vision Initiative offers physicians an important opportunity to help shape the future of continuing education and certification. ObGyns and other physicians should consider reviewing and commenting on the draft report, due in November, during the public comment period. Visit https://visioninitiative.org for more information and to sign up for email updates.
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
- American Board of Obstetrics and Gynecology. From pilot to permanent: ABOG's program offering an innovative pathway integrating lifelong learning and self-assessment and external assessment is approved. https://www.abog.org/new/ABOG_PilotToPermanent.aspx. Accessed July 6, 2018.
- Ramin S. American Board of Obstetrics and Gynecology MOC program. PowerPoint presentation; December 4, 2017.
- Teirstein PS. Boarded to death--why maintenance of certification is bad for doctors and patients. N Engl J Med. 2015;372(2):106-108.
- AMA Council on Medical Education. Executive summary. 2017. https://www.ama-assn.org/sites/default/files/media-browser/public/council-on-med-ed/a18-cme-02.pdf. Accessed July 6, 2018.
- American College of Obstetricians and Gynecologists. ACOG-ABOG joint statement: political interference in physician maintenance of skills threatens women's health care. https://www.acog.org/-/media/Departments/State-Legislative-Activities/2017ACOG-ABMS-MOC-Statement.pdf?dmc=1&ts=20180706T1615538746. Accessed July 6, 2018.
- Letter to Lois Nora, MD, JD. August 18, 2017. https://www.mainemed.com/sites/default/files/content/MOC%20Letter%20082117.pdf. Accessed July 6, 2018.
- American Board of Obstetrics and Gynecology. From pilot to permanent: ABOG's program offering an innovative pathway integrating lifelong learning and self-assessment and external assessment is approved. https://www.abog.org/new/ABOG_PilotToPermanent.aspx. Accessed July 6, 2018.
- Ramin S. American Board of Obstetrics and Gynecology MOC program. PowerPoint presentation; December 4, 2017.
- Teirstein PS. Boarded to death--why maintenance of certification is bad for doctors and patients. N Engl J Med. 2015;372(2):106-108.
- AMA Council on Medical Education. Executive summary. 2017. https://www.ama-assn.org/sites/default/files/media-browser/public/council-on-med-ed/a18-cme-02.pdf. Accessed July 6, 2018.
- American College of Obstetricians and Gynecologists. ACOG-ABOG joint statement: political interference in physician maintenance of skills threatens women's health care. https://www.acog.org/-/media/Departments/State-Legislative-Activities/2017ACOG-ABMS-MOC-Statement.pdf?dmc=1&ts=20180706T1615538746. Accessed July 6, 2018.
- Letter to Lois Nora, MD, JD. August 18, 2017. https://www.mainemed.com/sites/default/files/content/MOC%20Letter%20082117.pdf. Accessed July 6, 2018.

CDC apps specific for ObGyns
The Centers for Disease Control and Prevention (CDC) is a US federal agency under the Department of Health and Human Services. It is the nation’s leading public health institute. Its main goal is to save lives and protect people from health, safety, and security threats. The CDC website lists 25 no-cost applications that the agency has developed: https://www.cdc.gov/mobile/mobileapp.html.
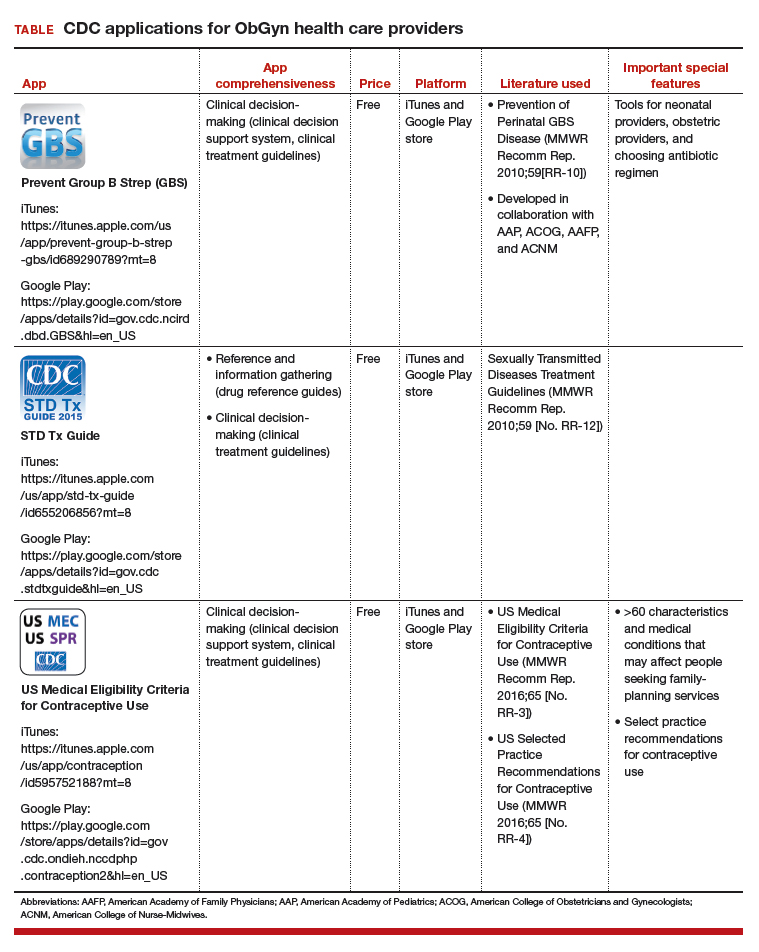
This review will focus on 3 CDC apps (Table) that I feel are useful to ObGyn health care providers: Prevent Group B Strep (GBS), STD Tx Guide, and US Medical Eligibility Criteria for Contraceptive Use. In fact, in an evaluation of contraception apps for providers of family planning services, US Medical Eligibility Criteria for Contraceptive Use was one of the highest scoring apps.1 I will evaluate each app by a shortened version of the APPLICATIONS scoring system, APPLI (app comprehensiveness, price, platform, literature use, and important special features).2 I commend the CDC for developing these useful tools to assist health care providers.
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
- Perry R, Lunde B, Chen KT. An evaluation of contraception mobile applications for providers of family planning services. Contraception. 2016;93(6):539-544.
- Chyjek K, Farag S, Chen KT. Rating pregnancy wheel applications using the APPLICATIONS scoring system. Obstet Gynecol. 2015;125(6):1478-1483.

Dr. Chen is Professor of Obstetrics, Gynecology, and Reproductive Science and Medical Education,Vice-Chair of Ob-Gyn Education for the Mount Sinai Health System, Icahn School of Medicine, Mount Sinai, New York, New York. She is an OBG Management Contributing Editor.
The author reports being an advisory board member and receiving royalties from UpToDate, Inc.
Dr. Chen is Professor of Obstetrics, Gynecology, and Reproductive Science and Medical Education,Vice-Chair of Ob-Gyn Education for the Mount Sinai Health System, Icahn School of Medicine, Mount Sinai, New York, New York. She is an OBG Management Contributing Editor.
The author reports being an advisory board member and receiving royalties from UpToDate, Inc.
Dr. Chen is Professor of Obstetrics, Gynecology, and Reproductive Science and Medical Education,Vice-Chair of Ob-Gyn Education for the Mount Sinai Health System, Icahn School of Medicine, Mount Sinai, New York, New York. She is an OBG Management Contributing Editor.
The author reports being an advisory board member and receiving royalties from UpToDate, Inc.
The Centers for Disease Control and Prevention (CDC) is a US federal agency under the Department of Health and Human Services. It is the nation’s leading public health institute. Its main goal is to save lives and protect people from health, safety, and security threats. The CDC website lists 25 no-cost applications that the agency has developed: https://www.cdc.gov/mobile/mobileapp.html.
This review will focus on 3 CDC apps (Table) that I feel are useful to ObGyn health care providers: Prevent Group B Strep (GBS), STD Tx Guide, and US Medical Eligibility Criteria for Contraceptive Use. In fact, in an evaluation of contraception apps for providers of family planning services, US Medical Eligibility Criteria for Contraceptive Use was one of the highest scoring apps.1 I will evaluate each app by a shortened version of the APPLICATIONS scoring system, APPLI (app comprehensiveness, price, platform, literature use, and important special features).2 I commend the CDC for developing these useful tools to assist health care providers.
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
The Centers for Disease Control and Prevention (CDC) is a US federal agency under the Department of Health and Human Services. It is the nation’s leading public health institute. Its main goal is to save lives and protect people from health, safety, and security threats. The CDC website lists 25 no-cost applications that the agency has developed: https://www.cdc.gov/mobile/mobileapp.html.
This review will focus on 3 CDC apps (Table) that I feel are useful to ObGyn health care providers: Prevent Group B Strep (GBS), STD Tx Guide, and US Medical Eligibility Criteria for Contraceptive Use. In fact, in an evaluation of contraception apps for providers of family planning services, US Medical Eligibility Criteria for Contraceptive Use was one of the highest scoring apps.1 I will evaluate each app by a shortened version of the APPLICATIONS scoring system, APPLI (app comprehensiveness, price, platform, literature use, and important special features).2 I commend the CDC for developing these useful tools to assist health care providers.
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
- Perry R, Lunde B, Chen KT. An evaluation of contraception mobile applications for providers of family planning services. Contraception. 2016;93(6):539-544.
- Chyjek K, Farag S, Chen KT. Rating pregnancy wheel applications using the APPLICATIONS scoring system. Obstet Gynecol. 2015;125(6):1478-1483.
- Perry R, Lunde B, Chen KT. An evaluation of contraception mobile applications for providers of family planning services. Contraception. 2016;93(6):539-544.
- Chyjek K, Farag S, Chen KT. Rating pregnancy wheel applications using the APPLICATIONS scoring system. Obstet Gynecol. 2015;125(6):1478-1483.
IN THIS ARTICLE
- Details on recommended apps

Liver enzymes: No trivial elevations, even if asymptomatic
Elevated levels of circulating enzymes that are frequently of hepatic origin (aminotransferases and alkaline phosphatase) and bilirubin in the absence of symptoms are common in clinical practice. A dogmatic but true statement holds that there are no trivial elevations in these substances. All persistent elevations of liver enzymes need a methodical evaluation and an appropriate working diagnosis.1
Here, we outline a framework for the workup and treatment of common causes of liver enzyme elevations.
PATTERN OF ELEVATION: CHOLESTATIC OR HEPATOCELLULAR

Based on the pattern of elevation, causes of elevated liver enzymes can be sorted into disorders of cholestasis and disorders of hepatocellular injury (Table 1).1
Cholestatic disorders tend to cause elevations in alkaline phosphatase, bilirubin, and gamma-glutamyl transferase (GGT).
Hepatocellular injury raises levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST).
HOW SHOULD ABNORMAL RESULTS BE EVALUATED?
When approaching liver enzyme elevations, the clinician should develop a working differential diagnosis based on the medical and social history and physical examination.
Think about alcohol, drugs, and fat
The most common causes of liver enzyme elevation are alcohol toxicity, medication overdose, and fatty liver disease.
Alcohol intake should be ascertained. “Significant” consumption is defined as more than 21 drinks per week in men or more than 14 drinks per week in women, over a period of at least 2 years.2
The exact pathogenesis of alcoholic hepatitis is incompletely understood, but alcohol is primarily metabolized by the liver, and damage likely occurs during metabolism of the ingested alcohol. AST elevations tend to be higher than ALT elevations; the reason is ascribed to hepatic deficiency of pyridoxal 5´-phosphate, a cofactor of the enzymatic activity of ALT, which leads to a lesser increase in ALT than in AST.
Alcoholic liver disease can be difficult to diagnose, as many people are initially reluctant to fully disclose how much they drink, but it should be suspected when the ratio of AST to ALT is 2 or greater.
In a classic study, a ratio greater than 2 was found in 70% of patients with alcoholic hepatitis and cirrhosis, compared with 26% of patients with postnecrotic cirrhosis, 8% with chronic hepatitis, 4% with viral hepatitis, and none with obstructive jaundice.3 Importantly, the disorder is often correctable if the patient is able to remain abstinent from alcohol over time.

A detailed medication history is important and should focus especially on recently added medications, dosage changes, medication overuse, and use of nonprescription drugs and herbal supplements. Common medications that affect liver enzyme levels include statins, which cause hepatic dysfunction primarily during the first 3 months of therapy, nonsteroidal anti-inflammatory drugs, antiepileptic drugs, antibiotics, anabolic steroids, and acetaminophen (Table 2).1 Use of illicit drugs and herbal remedies should be discussed, as they may cause toxin-mediated hepatitis.
Although inflammation from drug toxicity will resolve if the offending agent is discontinued, complete recovery may take weeks to months.4
A pertinent social history includes exposure to environmental hepatotoxins such as amatoxin (contained in some wild mushrooms) and occupational hazards (eg, vinyl chloride). Risk factors for viral hepatitis should be evaluated, including intravenous drug use, blood transfusions, unprotected sexual contact, organ transplant, perinatal transmission, and a history of work in healthcare facilities or travel to regions in which hepatitis A or E is endemic.
The medical and family history should include details of associated conditions, such as:
- Right heart failure (a cause of congestive hepatopathy)
- Metabolic syndrome (associated with fatty liver disease)
- Inflammatory bowel disease and primary sclerosing cholangitis
- Early-onset emphysema and alpha-1 antitrypsin deficiency.
The physical examination should be thorough, with emphasis on the abdomen, and search for stigmata of advanced liver disease such as hepatomegaly, splenomegaly, ascites, edema, spider angiomata, jaundice, and asterixis. Any patient with evidence of chronic liver disease should be referred to a subspecialist for further evaluation.
Further diagnostic workup
Abnormal liver enzyme findings or physical examination findings should direct the subsequent diagnostic workup with laboratory testing and imaging.5
For cholestasis. If laboratory data are consistent with cholestasis or abnormal bile flow, it should be further characterized as extrahepatic or intrahepatic. Common causes of extrahepatic cholestasis include biliary tree obstruction due to stones or malignancy, often visualized as intraductal biliary dilation on ultrasonography of the right upper quadrant. Common causes of intrahepatic cholestasis include viral and alcoholic hepatitis, nonalcoholic steatohepatitis, certain drugs and toxins such as alkylated steroids and herbal medications, infiltrative diseases such as amyloid, sarcoid, lymphoma, and tuberculosis, and primary biliary cholangitis.
Abnormal findings on ultrasonography should be further pursued with advanced imaging, ie, computed tomography or magnetic resonance cholangiopancreatography (MRCP). The confirmation of a lesion on imaging is often followed by endoscopic retrograde cholangiopancreatography (ERCP) in an attempt to obtain biopsy samples, remove obstructions, and place therapeutic stents. In instances when endoscopic attempts fail to relieve the obstruction, surgical referral may be appropriate.
For nonhepatobiliary problems. Depending on clinical presentation, it may also be important to consider nonhepatobiliary causes of elevated liver enzymes.
Alkaline phosphatase is found in many other tissue types, including bone, kidney, and the placenta, and can be elevated during pregnancy, adolescence, and even after fatty meals due to intestinal release.6 After screening for the aforementioned physiologic conditions, isolated elevated alkaline phosphatase should be further evaluated by obtaining GGT or 5-nucleotidase levels, which are more specifically of hepatic origin. If these levels are within normal limits, further evaluation for conditions of bone growth and cellular turnover such as Paget disease, hyperparathyroidism, and malignancy should be considered. Specifically, Stauffer syndrome should be considered when there is a paraneoplastic rise in the alkaline phosphatase level in the setting of renal cell carcinoma without liver metastases.
AST and ALT levels may also be elevated in clinical situations and syndromes unrelated to liver disease. Rhabdomyolysis, for instance, may be associated with elevations of AST in more than 90% of cases, and ALT in more than 75%.7 Markers of muscle injury including serum creatine kinase should be obtained in the setting of heat stroke, muscle weakness, strenuous activity, or seizures, as related elevations in AST and ALT may not always be clinically indicative of liver injury.
Given the many conditions that may cause elevated liver enzymes, evaluation and treatment should focus on identifying and removing offending agents and targeting the underlying process with appropriate medical therapy.
FATTY LIVER
With rates of obesity and type 2 diabetes on the rise in the general population, identifying and treating nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) require increased awareness and close coordination between primary care providers and subspecialists.
According to current estimates, up to one-third of the US population (100 million people) may have NAFLD, and 1% to 3% of the population (4–6 million people) likely have NASH, defined as steatosis with inflammation. Development of NASH places patients at a significantly higher risk of fibrosis, hepatocellular injury, and cancer.8
NAFLD is more common in men than in women. It is present in around 80% to 90% of obese adults, two-thirds of adults with type 2 diabetes, and many people with hyperlipidemia. It is also becoming more common in children, with 40% to 70% of obese children likely having some element of NAFLD.
Diagnosis of fatty liver
Although liver enzymes are more likely to be abnormal in individuals with NAFLD, many individuals with underlying NAFLD may have normal laboratory evaluations. ALT may be elevated in only up to 20% of cases and does not likely correlate with the level of underlying liver damage, although increasing GGT may serve as a marker of fibrosis over time.9–11 In contrast to alcohol injury, however, the AST-ALT ratio is usually less than 1.0.
Noninvasive tools for diagnosing NAFLD include the NAFLD fibrosis score, which incorporates age, hyperglycemia, body mass index, platelet count, albumin level, and AST-ALT ratio. This and related scoring algorithms may be useful in differentiating patients with minimal fibrosis from those with advanced fibrosis.12,13
Ultrasonography is a first-line diagnostic test for steatosis, although it may demonstrate fatty infiltration only around 60% of the time. Computed tomography and magnetic resonance imaging are more sensitive, but costlier. Transient elastography (FibroScan; Echosens, Paris, France) has become more popular and has been shown to correlate with findings on liver biopsy in diagnosing or excluding advanced liver fibrosis.14,15
The gold standard for diagnosing NAFLD and NASH is identifying fat-laden hepatocytes or portal inflammation on biopsy; however, biopsy is generally reserved for cases in which the diagnosis remains uncertain.
Behavioral treatment
The primary treatment for NAFLD consists of behavioral modification including weight loss, exercise, and adherence to a low-fat diet, in addition to tight glycemic control and treatment of any underlying lipid abnormalities. Studies have shown that a reduction of 7% to 10% of body weight is associated with a decrease in the inflammation of NAFLD, though no strict guidelines have been established.16
Given the prevalence of NAFLD and the need for longitudinal treatment, primary care physicians will play a significant role in long-term monitoring and management of patients with fatty liver disease.
OTHER DISORDERS OF LIVER FUNCTION
Hereditary hemochromatosis
Hereditary hemochromatosis is the most common inherited liver disorder in adults of European descent,17 and can be effectively treated if discovered early. But its clinical diagnosis can be challenging, as many patients have no symptoms at presentation despite abnormal liver enzyme levels. Early symptoms may include severe fatigue, arthralgias, and, in men, impotence, before the appearance of the classic triad of “bronze diabetes” with cirrhosis, diabetes, and darkening of the skin.18
If hemochromatosis is suspected, laboratory tests should include a calculation of percent transferrin saturation, with saturation greater than 45% warranting serum ferritin measurement to evaluate for iron overload (ferritin > 200–300 ng/mL in men, > 150–200 ng/mL in women).19 If iron overload is confirmed, referral to a gastroenterologist is recommended.
Genetic evaluation is often pursued, but patients may ultimately require liver biopsy regardless of the findings, as some patients homozygous for the HFE mutation C282Y may not have clinical hemochromatosis, whereas others with hereditary hemochromatosis may not have the HFE mutation.
Therapeutic phlebotomy is the treatment of choice, and most patients tolerate it well.
Chronic hepatitis B virus and hepatitis C virus infections
Chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are common in the United States, with HBV affecting more than 1 million people and HCV affecting an estimated 3.5 million.
Chronic HCV infection. Direct-acting antiviral drugs have revolutionized HCV treatment and have led to a sustained viral response and presumed cure at 12 weeks in more than 95% of cases across all HCV genotypes.20 Given the recent development of effective and well-tolerated treatments, primary care physicians have assumed a pivotal role in screening for HCV.
The American Association for the Study of Liver Diseases and the Infectious Diseases Society of America21 recommend screening for HCV in people who have risk factors for it, ie:
- HCV exposure
- HIV infection
- Behavioral or environmental risks for contracting the virus such as intravenous drug use or incarceration
- Birth between 1945 and 1965 (one-time testing).
If HCV antibody screening is positive, HCV RNA should be obtained to quantify the viral load and confirm active infection, and genotype testing should be performed to guide treatment. Among the 6 most common HCV genotypes, genotype 1 is the most common in North America, accounting for over 70% of cases in the United States.
Although recommendations and therapies are constantly evolving, the selection of a treatment regimen and the duration of therapy are determined by viral genotype, history of prior treatment, stage of liver fibrosis, potential drug interactions, and frequently, medication cost and insurance coverage.
HBV infection. The treatment for acute HBV infection is generally supportive, though viral suppression with tenofovir or entecavir may be required for those who develop coagulopathy, bilirubinemia, or liver failure. Treatment of chronic HBV infection may not be required and is generally considered for those with elevated ALT, high viral load, or evidence of liver fibrosis on noninvasive measurements such as transient elastography.
Autoimmune hepatitis
Autoimmune causes of liver enzyme elevations should also be considered during initial screening. Positive antinuclear antibody and positive antismooth muscle antibody tests are common in cases of autoimmune hepatitis.22 Autoimmune hepatitis affects women more often than men, with a ratio of 4:1. The peaks of incidence occur during adolescence and between ages 30 and 45.23
Primary biliary cholangitis
Additionally, an elevated alkaline phosphatase level should raise concern for underlying primary biliary cholangitis (formerly called primary biliary cirrhosis), an autoimmune disorder that affects the small and medium intrahepatic bile ducts. Diagnosis of primary biliary cholangitis can be assisted by a positive test for antimitochondrial antibody, present in almost 90% of patients.24
Primary sclerosing cholangitis
Elevated alkaline phosphatase is also the hallmark of primary sclerosing cholangitis, which is associated with inflammatory bowel disease.25 Primary sclerosing cholangitis is characterized by inflammation and fibrosis of the intrahepatic and extrahepatic bile ducts, which are visualized on MRCP and confirmed by biopsy if needed.
REFERRAL
Subspecialty referral should be considered if the cause remains ambiguous or unknown, if there is concern for a rare hepatic disorder such as an autoimmune condition, Wilson disease, or alpha-1 antitrypsin deficiency, or if there is evidence of advanced or chronic liver disease.
Primary care physicians are at the forefront of detecting and diagnosing liver disease, and close coordination with subspecialists will remain crucial in delivering patient care.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77(3):195–204. doi:10.3949/ccjm.77a.09064
- Chalasani N, Younossi Z, Lavine JE, et al; American Gastroenterological Association; American Association for the Study of Liver Diseases; American College of Gastroenterology. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012; 142(7):1592–1609. doi:10.1053/j.gastro.2012.04.001
- Cohen JA, Kaplan MM. The SGOT/SGPT ratio—an indicator of alcoholic liver disease. Dig Dis Sci 1979; 24(11):835–838. pmid:520102
- Kaplan MM. Alanine aminotransferase levels: what’s normal? Ann Intern Med 2002; 137(1):49-51. pmid:12093245
- Pratt DS, Kaplan MM. Evaluation of abnormal liver enzyme results in asymptomatic patients. N Engl J Med 2000; 342(17):1266–1271. doi:10.1056/NEJM200004273421707
- Sharma U, Pal D, Prasad R. Alkaline phosphatase: an overview. Indian J Clin Biochem 2014; 29(3):269–278. doi:10.1007/s12291-013-0408-y
- Weibrecht K, Dayno M, Darling C, Bird SB. Liver aminotransferases are elevated with rhabdomyolysis in the absence of significant liver injury. J Med Toxicol 2010; 6(3):294–300. doi:10.1007/s13181-010-0075-9
- Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis 2010; 28(1):155–161. doi:10.1159/000282080
- Adams LA, Feldstein AE. Non-invasive diagnosis of nonalcoholic fatty liver and nonalcoholic steatohepatitis. J Dig Dis 2011; 12(1):10–16. doi:10.1111/j.1751-2980.2010.00471.x
- Fracanzani AL, Valenti L, Bugianesi E, et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: a role for insulin resistance and diabetes. Hepatology 2008; 48(3):792–798. doi:10.1002/hep.22429
- Tahan V, Canbakan B, Balci H, et al. Serum gamma-glutamyltranspeptidase distinguishes non-alcoholic fatty liver disease at high risk. Hepatogastroenterolgoy 2008; 55(85):1433-1438. pmid:18795706
- McPherson S, Stewart S, Henderson E, Burt AD, Day CP. Simple non-invasive fibrosis scoring systems can reliably exclude advanced fibrosis in patients with non-alcoholic fatty liver disease. Gut 2010; 59(9):1265–1269. doi:10.1136/gut.2010.216077
- Angulo P, Hui JM, Marchesini G, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007; 45(4):846–854. doi:10.1002/hep.21496
- Petta S, Vanni E, Bugianesi E, et al. The combination of liver stiffness measurement and NAFLD fibrosis score improves the noninvasive diagnostic accuracy for severe liver fibrosis in patients with nonalcoholic fatty liver disease. Liver Int 2015; 35(5):1566–1573. doi:10.1111/liv.12584
- Hashemi SA, Alavian SM, Gholami-Fesharaki M. Assessment of transient elastography (FibroScan) for diagnosis of fibrosis in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Caspian J Intern Med 2016; 7(4):242–252. pmid:27999641
- Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010; 51(1):121–129. doi:10.1002/hep.23276
- Adams PH, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med 2005; 352(17):1769-1778. doi:10.1056/NEJMoa041534
- Brissot P, de Bels F. Current approaches to the management of hemochromatosis. Hematology Am Soc Hematol Educ Program 2006; 2006(1):36–41. doi:10.1182/asheducation-2006.1.36
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54(1):328–343. doi:10.1002/hep.24330
- Weiler N, Zeuzem S, Welker MW. Concise review: interferon-free treatment of hepatitis C virus-associated cirrhosis and liver graft infection. World J Gastroenterol 2016; 22(41):9044–9056. doi:10.3748/wjg.v22.i41.9044
- American Association for the Study of Liver Disease, Infectious Diseases Society of America. HCV guidance: recommendations for testing, managing, and treating hepatitis C. www.hcvguidelines.org. Accessed July 16, 2018.
- Manns MP, Czaja AJ, Gorham JD, et al; American Association for the Study of Liver Diseases. Diagnosis and management of autoimmune hepatitis. Hepatology 2010; 51(6):2193–2213. doi:10.1002/hep.23584
- Liberal R, Krawitt EL, Vierling JM, Manns MP, Mieli-Vergani G, Vergani D. Cutting edge issues in autoimmune hepatitis. J Autoimmun 2016; 75:6–19. doi:10.1016/j.jaut.2016.07.005
- Mousa HS, Carbone M, Malinverno F, Ronca V, Gershwin ME, Invernizzi P. Novel therapeutics for primary biliary cholangitis: Toward a disease-stage-based approach. Autoimmun Rev 2016; 15(9):870–876. doi:10.1016/j.autrev.2016.07.003
- de Vries AB, Janse M, Blokzijl H, Weersma RK. Distinctive inflammatory bowel disease phenotype in primary sclerosing cholangitis. World J Gastroenterol 2015; 21(6):1956–1971. doi:10.3748/wjg.v21.i6.1956
Elevated levels of circulating enzymes that are frequently of hepatic origin (aminotransferases and alkaline phosphatase) and bilirubin in the absence of symptoms are common in clinical practice. A dogmatic but true statement holds that there are no trivial elevations in these substances. All persistent elevations of liver enzymes need a methodical evaluation and an appropriate working diagnosis.1
Here, we outline a framework for the workup and treatment of common causes of liver enzyme elevations.
PATTERN OF ELEVATION: CHOLESTATIC OR HEPATOCELLULAR
Based on the pattern of elevation, causes of elevated liver enzymes can be sorted into disorders of cholestasis and disorders of hepatocellular injury (Table 1).1
Cholestatic disorders tend to cause elevations in alkaline phosphatase, bilirubin, and gamma-glutamyl transferase (GGT).
Hepatocellular injury raises levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST).
HOW SHOULD ABNORMAL RESULTS BE EVALUATED?
When approaching liver enzyme elevations, the clinician should develop a working differential diagnosis based on the medical and social history and physical examination.
Think about alcohol, drugs, and fat
The most common causes of liver enzyme elevation are alcohol toxicity, medication overdose, and fatty liver disease.
Alcohol intake should be ascertained. “Significant” consumption is defined as more than 21 drinks per week in men or more than 14 drinks per week in women, over a period of at least 2 years.2
The exact pathogenesis of alcoholic hepatitis is incompletely understood, but alcohol is primarily metabolized by the liver, and damage likely occurs during metabolism of the ingested alcohol. AST elevations tend to be higher than ALT elevations; the reason is ascribed to hepatic deficiency of pyridoxal 5´-phosphate, a cofactor of the enzymatic activity of ALT, which leads to a lesser increase in ALT than in AST.
Alcoholic liver disease can be difficult to diagnose, as many people are initially reluctant to fully disclose how much they drink, but it should be suspected when the ratio of AST to ALT is 2 or greater.
In a classic study, a ratio greater than 2 was found in 70% of patients with alcoholic hepatitis and cirrhosis, compared with 26% of patients with postnecrotic cirrhosis, 8% with chronic hepatitis, 4% with viral hepatitis, and none with obstructive jaundice.3 Importantly, the disorder is often correctable if the patient is able to remain abstinent from alcohol over time.
A detailed medication history is important and should focus especially on recently added medications, dosage changes, medication overuse, and use of nonprescription drugs and herbal supplements. Common medications that affect liver enzyme levels include statins, which cause hepatic dysfunction primarily during the first 3 months of therapy, nonsteroidal anti-inflammatory drugs, antiepileptic drugs, antibiotics, anabolic steroids, and acetaminophen (Table 2).1 Use of illicit drugs and herbal remedies should be discussed, as they may cause toxin-mediated hepatitis.
Although inflammation from drug toxicity will resolve if the offending agent is discontinued, complete recovery may take weeks to months.4
A pertinent social history includes exposure to environmental hepatotoxins such as amatoxin (contained in some wild mushrooms) and occupational hazards (eg, vinyl chloride). Risk factors for viral hepatitis should be evaluated, including intravenous drug use, blood transfusions, unprotected sexual contact, organ transplant, perinatal transmission, and a history of work in healthcare facilities or travel to regions in which hepatitis A or E is endemic.
The medical and family history should include details of associated conditions, such as:
- Right heart failure (a cause of congestive hepatopathy)
- Metabolic syndrome (associated with fatty liver disease)
- Inflammatory bowel disease and primary sclerosing cholangitis
- Early-onset emphysema and alpha-1 antitrypsin deficiency.
The physical examination should be thorough, with emphasis on the abdomen, and search for stigmata of advanced liver disease such as hepatomegaly, splenomegaly, ascites, edema, spider angiomata, jaundice, and asterixis. Any patient with evidence of chronic liver disease should be referred to a subspecialist for further evaluation.
Further diagnostic workup
Abnormal liver enzyme findings or physical examination findings should direct the subsequent diagnostic workup with laboratory testing and imaging.5
For cholestasis. If laboratory data are consistent with cholestasis or abnormal bile flow, it should be further characterized as extrahepatic or intrahepatic. Common causes of extrahepatic cholestasis include biliary tree obstruction due to stones or malignancy, often visualized as intraductal biliary dilation on ultrasonography of the right upper quadrant. Common causes of intrahepatic cholestasis include viral and alcoholic hepatitis, nonalcoholic steatohepatitis, certain drugs and toxins such as alkylated steroids and herbal medications, infiltrative diseases such as amyloid, sarcoid, lymphoma, and tuberculosis, and primary biliary cholangitis.
Abnormal findings on ultrasonography should be further pursued with advanced imaging, ie, computed tomography or magnetic resonance cholangiopancreatography (MRCP). The confirmation of a lesion on imaging is often followed by endoscopic retrograde cholangiopancreatography (ERCP) in an attempt to obtain biopsy samples, remove obstructions, and place therapeutic stents. In instances when endoscopic attempts fail to relieve the obstruction, surgical referral may be appropriate.
For nonhepatobiliary problems. Depending on clinical presentation, it may also be important to consider nonhepatobiliary causes of elevated liver enzymes.
Alkaline phosphatase is found in many other tissue types, including bone, kidney, and the placenta, and can be elevated during pregnancy, adolescence, and even after fatty meals due to intestinal release.6 After screening for the aforementioned physiologic conditions, isolated elevated alkaline phosphatase should be further evaluated by obtaining GGT or 5-nucleotidase levels, which are more specifically of hepatic origin. If these levels are within normal limits, further evaluation for conditions of bone growth and cellular turnover such as Paget disease, hyperparathyroidism, and malignancy should be considered. Specifically, Stauffer syndrome should be considered when there is a paraneoplastic rise in the alkaline phosphatase level in the setting of renal cell carcinoma without liver metastases.
AST and ALT levels may also be elevated in clinical situations and syndromes unrelated to liver disease. Rhabdomyolysis, for instance, may be associated with elevations of AST in more than 90% of cases, and ALT in more than 75%.7 Markers of muscle injury including serum creatine kinase should be obtained in the setting of heat stroke, muscle weakness, strenuous activity, or seizures, as related elevations in AST and ALT may not always be clinically indicative of liver injury.
Given the many conditions that may cause elevated liver enzymes, evaluation and treatment should focus on identifying and removing offending agents and targeting the underlying process with appropriate medical therapy.
FATTY LIVER
With rates of obesity and type 2 diabetes on the rise in the general population, identifying and treating nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) require increased awareness and close coordination between primary care providers and subspecialists.
According to current estimates, up to one-third of the US population (100 million people) may have NAFLD, and 1% to 3% of the population (4–6 million people) likely have NASH, defined as steatosis with inflammation. Development of NASH places patients at a significantly higher risk of fibrosis, hepatocellular injury, and cancer.8
NAFLD is more common in men than in women. It is present in around 80% to 90% of obese adults, two-thirds of adults with type 2 diabetes, and many people with hyperlipidemia. It is also becoming more common in children, with 40% to 70% of obese children likely having some element of NAFLD.
Diagnosis of fatty liver
Although liver enzymes are more likely to be abnormal in individuals with NAFLD, many individuals with underlying NAFLD may have normal laboratory evaluations. ALT may be elevated in only up to 20% of cases and does not likely correlate with the level of underlying liver damage, although increasing GGT may serve as a marker of fibrosis over time.9–11 In contrast to alcohol injury, however, the AST-ALT ratio is usually less than 1.0.
Noninvasive tools for diagnosing NAFLD include the NAFLD fibrosis score, which incorporates age, hyperglycemia, body mass index, platelet count, albumin level, and AST-ALT ratio. This and related scoring algorithms may be useful in differentiating patients with minimal fibrosis from those with advanced fibrosis.12,13
Ultrasonography is a first-line diagnostic test for steatosis, although it may demonstrate fatty infiltration only around 60% of the time. Computed tomography and magnetic resonance imaging are more sensitive, but costlier. Transient elastography (FibroScan; Echosens, Paris, France) has become more popular and has been shown to correlate with findings on liver biopsy in diagnosing or excluding advanced liver fibrosis.14,15
The gold standard for diagnosing NAFLD and NASH is identifying fat-laden hepatocytes or portal inflammation on biopsy; however, biopsy is generally reserved for cases in which the diagnosis remains uncertain.
Behavioral treatment
The primary treatment for NAFLD consists of behavioral modification including weight loss, exercise, and adherence to a low-fat diet, in addition to tight glycemic control and treatment of any underlying lipid abnormalities. Studies have shown that a reduction of 7% to 10% of body weight is associated with a decrease in the inflammation of NAFLD, though no strict guidelines have been established.16
Given the prevalence of NAFLD and the need for longitudinal treatment, primary care physicians will play a significant role in long-term monitoring and management of patients with fatty liver disease.
OTHER DISORDERS OF LIVER FUNCTION
Hereditary hemochromatosis
Hereditary hemochromatosis is the most common inherited liver disorder in adults of European descent,17 and can be effectively treated if discovered early. But its clinical diagnosis can be challenging, as many patients have no symptoms at presentation despite abnormal liver enzyme levels. Early symptoms may include severe fatigue, arthralgias, and, in men, impotence, before the appearance of the classic triad of “bronze diabetes” with cirrhosis, diabetes, and darkening of the skin.18
If hemochromatosis is suspected, laboratory tests should include a calculation of percent transferrin saturation, with saturation greater than 45% warranting serum ferritin measurement to evaluate for iron overload (ferritin > 200–300 ng/mL in men, > 150–200 ng/mL in women).19 If iron overload is confirmed, referral to a gastroenterologist is recommended.
Genetic evaluation is often pursued, but patients may ultimately require liver biopsy regardless of the findings, as some patients homozygous for the HFE mutation C282Y may not have clinical hemochromatosis, whereas others with hereditary hemochromatosis may not have the HFE mutation.
Therapeutic phlebotomy is the treatment of choice, and most patients tolerate it well.
Chronic hepatitis B virus and hepatitis C virus infections
Chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are common in the United States, with HBV affecting more than 1 million people and HCV affecting an estimated 3.5 million.
Chronic HCV infection. Direct-acting antiviral drugs have revolutionized HCV treatment and have led to a sustained viral response and presumed cure at 12 weeks in more than 95% of cases across all HCV genotypes.20 Given the recent development of effective and well-tolerated treatments, primary care physicians have assumed a pivotal role in screening for HCV.
The American Association for the Study of Liver Diseases and the Infectious Diseases Society of America21 recommend screening for HCV in people who have risk factors for it, ie:
- HCV exposure
- HIV infection
- Behavioral or environmental risks for contracting the virus such as intravenous drug use or incarceration
- Birth between 1945 and 1965 (one-time testing).
If HCV antibody screening is positive, HCV RNA should be obtained to quantify the viral load and confirm active infection, and genotype testing should be performed to guide treatment. Among the 6 most common HCV genotypes, genotype 1 is the most common in North America, accounting for over 70% of cases in the United States.
Although recommendations and therapies are constantly evolving, the selection of a treatment regimen and the duration of therapy are determined by viral genotype, history of prior treatment, stage of liver fibrosis, potential drug interactions, and frequently, medication cost and insurance coverage.
HBV infection. The treatment for acute HBV infection is generally supportive, though viral suppression with tenofovir or entecavir may be required for those who develop coagulopathy, bilirubinemia, or liver failure. Treatment of chronic HBV infection may not be required and is generally considered for those with elevated ALT, high viral load, or evidence of liver fibrosis on noninvasive measurements such as transient elastography.
Autoimmune hepatitis
Autoimmune causes of liver enzyme elevations should also be considered during initial screening. Positive antinuclear antibody and positive antismooth muscle antibody tests are common in cases of autoimmune hepatitis.22 Autoimmune hepatitis affects women more often than men, with a ratio of 4:1. The peaks of incidence occur during adolescence and between ages 30 and 45.23
Primary biliary cholangitis
Additionally, an elevated alkaline phosphatase level should raise concern for underlying primary biliary cholangitis (formerly called primary biliary cirrhosis), an autoimmune disorder that affects the small and medium intrahepatic bile ducts. Diagnosis of primary biliary cholangitis can be assisted by a positive test for antimitochondrial antibody, present in almost 90% of patients.24
Primary sclerosing cholangitis
Elevated alkaline phosphatase is also the hallmark of primary sclerosing cholangitis, which is associated with inflammatory bowel disease.25 Primary sclerosing cholangitis is characterized by inflammation and fibrosis of the intrahepatic and extrahepatic bile ducts, which are visualized on MRCP and confirmed by biopsy if needed.
REFERRAL
Subspecialty referral should be considered if the cause remains ambiguous or unknown, if there is concern for a rare hepatic disorder such as an autoimmune condition, Wilson disease, or alpha-1 antitrypsin deficiency, or if there is evidence of advanced or chronic liver disease.
Primary care physicians are at the forefront of detecting and diagnosing liver disease, and close coordination with subspecialists will remain crucial in delivering patient care.
Elevated levels of circulating enzymes that are frequently of hepatic origin (aminotransferases and alkaline phosphatase) and bilirubin in the absence of symptoms are common in clinical practice. A dogmatic but true statement holds that there are no trivial elevations in these substances. All persistent elevations of liver enzymes need a methodical evaluation and an appropriate working diagnosis.1
Here, we outline a framework for the workup and treatment of common causes of liver enzyme elevations.
PATTERN OF ELEVATION: CHOLESTATIC OR HEPATOCELLULAR
Based on the pattern of elevation, causes of elevated liver enzymes can be sorted into disorders of cholestasis and disorders of hepatocellular injury (Table 1).1
Cholestatic disorders tend to cause elevations in alkaline phosphatase, bilirubin, and gamma-glutamyl transferase (GGT).
Hepatocellular injury raises levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST).
HOW SHOULD ABNORMAL RESULTS BE EVALUATED?
When approaching liver enzyme elevations, the clinician should develop a working differential diagnosis based on the medical and social history and physical examination.
Think about alcohol, drugs, and fat
The most common causes of liver enzyme elevation are alcohol toxicity, medication overdose, and fatty liver disease.
Alcohol intake should be ascertained. “Significant” consumption is defined as more than 21 drinks per week in men or more than 14 drinks per week in women, over a period of at least 2 years.2
The exact pathogenesis of alcoholic hepatitis is incompletely understood, but alcohol is primarily metabolized by the liver, and damage likely occurs during metabolism of the ingested alcohol. AST elevations tend to be higher than ALT elevations; the reason is ascribed to hepatic deficiency of pyridoxal 5´-phosphate, a cofactor of the enzymatic activity of ALT, which leads to a lesser increase in ALT than in AST.
Alcoholic liver disease can be difficult to diagnose, as many people are initially reluctant to fully disclose how much they drink, but it should be suspected when the ratio of AST to ALT is 2 or greater.
In a classic study, a ratio greater than 2 was found in 70% of patients with alcoholic hepatitis and cirrhosis, compared with 26% of patients with postnecrotic cirrhosis, 8% with chronic hepatitis, 4% with viral hepatitis, and none with obstructive jaundice.3 Importantly, the disorder is often correctable if the patient is able to remain abstinent from alcohol over time.
A detailed medication history is important and should focus especially on recently added medications, dosage changes, medication overuse, and use of nonprescription drugs and herbal supplements. Common medications that affect liver enzyme levels include statins, which cause hepatic dysfunction primarily during the first 3 months of therapy, nonsteroidal anti-inflammatory drugs, antiepileptic drugs, antibiotics, anabolic steroids, and acetaminophen (Table 2).1 Use of illicit drugs and herbal remedies should be discussed, as they may cause toxin-mediated hepatitis.
Although inflammation from drug toxicity will resolve if the offending agent is discontinued, complete recovery may take weeks to months.4
A pertinent social history includes exposure to environmental hepatotoxins such as amatoxin (contained in some wild mushrooms) and occupational hazards (eg, vinyl chloride). Risk factors for viral hepatitis should be evaluated, including intravenous drug use, blood transfusions, unprotected sexual contact, organ transplant, perinatal transmission, and a history of work in healthcare facilities or travel to regions in which hepatitis A or E is endemic.
The medical and family history should include details of associated conditions, such as:
- Right heart failure (a cause of congestive hepatopathy)
- Metabolic syndrome (associated with fatty liver disease)
- Inflammatory bowel disease and primary sclerosing cholangitis
- Early-onset emphysema and alpha-1 antitrypsin deficiency.
The physical examination should be thorough, with emphasis on the abdomen, and search for stigmata of advanced liver disease such as hepatomegaly, splenomegaly, ascites, edema, spider angiomata, jaundice, and asterixis. Any patient with evidence of chronic liver disease should be referred to a subspecialist for further evaluation.
Further diagnostic workup
Abnormal liver enzyme findings or physical examination findings should direct the subsequent diagnostic workup with laboratory testing and imaging.5
For cholestasis. If laboratory data are consistent with cholestasis or abnormal bile flow, it should be further characterized as extrahepatic or intrahepatic. Common causes of extrahepatic cholestasis include biliary tree obstruction due to stones or malignancy, often visualized as intraductal biliary dilation on ultrasonography of the right upper quadrant. Common causes of intrahepatic cholestasis include viral and alcoholic hepatitis, nonalcoholic steatohepatitis, certain drugs and toxins such as alkylated steroids and herbal medications, infiltrative diseases such as amyloid, sarcoid, lymphoma, and tuberculosis, and primary biliary cholangitis.
Abnormal findings on ultrasonography should be further pursued with advanced imaging, ie, computed tomography or magnetic resonance cholangiopancreatography (MRCP). The confirmation of a lesion on imaging is often followed by endoscopic retrograde cholangiopancreatography (ERCP) in an attempt to obtain biopsy samples, remove obstructions, and place therapeutic stents. In instances when endoscopic attempts fail to relieve the obstruction, surgical referral may be appropriate.
For nonhepatobiliary problems. Depending on clinical presentation, it may also be important to consider nonhepatobiliary causes of elevated liver enzymes.
Alkaline phosphatase is found in many other tissue types, including bone, kidney, and the placenta, and can be elevated during pregnancy, adolescence, and even after fatty meals due to intestinal release.6 After screening for the aforementioned physiologic conditions, isolated elevated alkaline phosphatase should be further evaluated by obtaining GGT or 5-nucleotidase levels, which are more specifically of hepatic origin. If these levels are within normal limits, further evaluation for conditions of bone growth and cellular turnover such as Paget disease, hyperparathyroidism, and malignancy should be considered. Specifically, Stauffer syndrome should be considered when there is a paraneoplastic rise in the alkaline phosphatase level in the setting of renal cell carcinoma without liver metastases.
AST and ALT levels may also be elevated in clinical situations and syndromes unrelated to liver disease. Rhabdomyolysis, for instance, may be associated with elevations of AST in more than 90% of cases, and ALT in more than 75%.7 Markers of muscle injury including serum creatine kinase should be obtained in the setting of heat stroke, muscle weakness, strenuous activity, or seizures, as related elevations in AST and ALT may not always be clinically indicative of liver injury.
Given the many conditions that may cause elevated liver enzymes, evaluation and treatment should focus on identifying and removing offending agents and targeting the underlying process with appropriate medical therapy.
FATTY LIVER
With rates of obesity and type 2 diabetes on the rise in the general population, identifying and treating nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) require increased awareness and close coordination between primary care providers and subspecialists.
According to current estimates, up to one-third of the US population (100 million people) may have NAFLD, and 1% to 3% of the population (4–6 million people) likely have NASH, defined as steatosis with inflammation. Development of NASH places patients at a significantly higher risk of fibrosis, hepatocellular injury, and cancer.8
NAFLD is more common in men than in women. It is present in around 80% to 90% of obese adults, two-thirds of adults with type 2 diabetes, and many people with hyperlipidemia. It is also becoming more common in children, with 40% to 70% of obese children likely having some element of NAFLD.
Diagnosis of fatty liver
Although liver enzymes are more likely to be abnormal in individuals with NAFLD, many individuals with underlying NAFLD may have normal laboratory evaluations. ALT may be elevated in only up to 20% of cases and does not likely correlate with the level of underlying liver damage, although increasing GGT may serve as a marker of fibrosis over time.9–11 In contrast to alcohol injury, however, the AST-ALT ratio is usually less than 1.0.
Noninvasive tools for diagnosing NAFLD include the NAFLD fibrosis score, which incorporates age, hyperglycemia, body mass index, platelet count, albumin level, and AST-ALT ratio. This and related scoring algorithms may be useful in differentiating patients with minimal fibrosis from those with advanced fibrosis.12,13
Ultrasonography is a first-line diagnostic test for steatosis, although it may demonstrate fatty infiltration only around 60% of the time. Computed tomography and magnetic resonance imaging are more sensitive, but costlier. Transient elastography (FibroScan; Echosens, Paris, France) has become more popular and has been shown to correlate with findings on liver biopsy in diagnosing or excluding advanced liver fibrosis.14,15
The gold standard for diagnosing NAFLD and NASH is identifying fat-laden hepatocytes or portal inflammation on biopsy; however, biopsy is generally reserved for cases in which the diagnosis remains uncertain.
Behavioral treatment
The primary treatment for NAFLD consists of behavioral modification including weight loss, exercise, and adherence to a low-fat diet, in addition to tight glycemic control and treatment of any underlying lipid abnormalities. Studies have shown that a reduction of 7% to 10% of body weight is associated with a decrease in the inflammation of NAFLD, though no strict guidelines have been established.16
Given the prevalence of NAFLD and the need for longitudinal treatment, primary care physicians will play a significant role in long-term monitoring and management of patients with fatty liver disease.
OTHER DISORDERS OF LIVER FUNCTION
Hereditary hemochromatosis
Hereditary hemochromatosis is the most common inherited liver disorder in adults of European descent,17 and can be effectively treated if discovered early. But its clinical diagnosis can be challenging, as many patients have no symptoms at presentation despite abnormal liver enzyme levels. Early symptoms may include severe fatigue, arthralgias, and, in men, impotence, before the appearance of the classic triad of “bronze diabetes” with cirrhosis, diabetes, and darkening of the skin.18
If hemochromatosis is suspected, laboratory tests should include a calculation of percent transferrin saturation, with saturation greater than 45% warranting serum ferritin measurement to evaluate for iron overload (ferritin > 200–300 ng/mL in men, > 150–200 ng/mL in women).19 If iron overload is confirmed, referral to a gastroenterologist is recommended.
Genetic evaluation is often pursued, but patients may ultimately require liver biopsy regardless of the findings, as some patients homozygous for the HFE mutation C282Y may not have clinical hemochromatosis, whereas others with hereditary hemochromatosis may not have the HFE mutation.
Therapeutic phlebotomy is the treatment of choice, and most patients tolerate it well.
Chronic hepatitis B virus and hepatitis C virus infections
Chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are common in the United States, with HBV affecting more than 1 million people and HCV affecting an estimated 3.5 million.
Chronic HCV infection. Direct-acting antiviral drugs have revolutionized HCV treatment and have led to a sustained viral response and presumed cure at 12 weeks in more than 95% of cases across all HCV genotypes.20 Given the recent development of effective and well-tolerated treatments, primary care physicians have assumed a pivotal role in screening for HCV.
The American Association for the Study of Liver Diseases and the Infectious Diseases Society of America21 recommend screening for HCV in people who have risk factors for it, ie:
- HCV exposure
- HIV infection
- Behavioral or environmental risks for contracting the virus such as intravenous drug use or incarceration
- Birth between 1945 and 1965 (one-time testing).
If HCV antibody screening is positive, HCV RNA should be obtained to quantify the viral load and confirm active infection, and genotype testing should be performed to guide treatment. Among the 6 most common HCV genotypes, genotype 1 is the most common in North America, accounting for over 70% of cases in the United States.
Although recommendations and therapies are constantly evolving, the selection of a treatment regimen and the duration of therapy are determined by viral genotype, history of prior treatment, stage of liver fibrosis, potential drug interactions, and frequently, medication cost and insurance coverage.
HBV infection. The treatment for acute HBV infection is generally supportive, though viral suppression with tenofovir or entecavir may be required for those who develop coagulopathy, bilirubinemia, or liver failure. Treatment of chronic HBV infection may not be required and is generally considered for those with elevated ALT, high viral load, or evidence of liver fibrosis on noninvasive measurements such as transient elastography.
Autoimmune hepatitis
Autoimmune causes of liver enzyme elevations should also be considered during initial screening. Positive antinuclear antibody and positive antismooth muscle antibody tests are common in cases of autoimmune hepatitis.22 Autoimmune hepatitis affects women more often than men, with a ratio of 4:1. The peaks of incidence occur during adolescence and between ages 30 and 45.23
Primary biliary cholangitis
Additionally, an elevated alkaline phosphatase level should raise concern for underlying primary biliary cholangitis (formerly called primary biliary cirrhosis), an autoimmune disorder that affects the small and medium intrahepatic bile ducts. Diagnosis of primary biliary cholangitis can be assisted by a positive test for antimitochondrial antibody, present in almost 90% of patients.24
Primary sclerosing cholangitis
Elevated alkaline phosphatase is also the hallmark of primary sclerosing cholangitis, which is associated with inflammatory bowel disease.25 Primary sclerosing cholangitis is characterized by inflammation and fibrosis of the intrahepatic and extrahepatic bile ducts, which are visualized on MRCP and confirmed by biopsy if needed.
REFERRAL
Subspecialty referral should be considered if the cause remains ambiguous or unknown, if there is concern for a rare hepatic disorder such as an autoimmune condition, Wilson disease, or alpha-1 antitrypsin deficiency, or if there is evidence of advanced or chronic liver disease.
Primary care physicians are at the forefront of detecting and diagnosing liver disease, and close coordination with subspecialists will remain crucial in delivering patient care.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77(3):195–204. doi:10.3949/ccjm.77a.09064
- Chalasani N, Younossi Z, Lavine JE, et al; American Gastroenterological Association; American Association for the Study of Liver Diseases; American College of Gastroenterology. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012; 142(7):1592–1609. doi:10.1053/j.gastro.2012.04.001
- Cohen JA, Kaplan MM. The SGOT/SGPT ratio—an indicator of alcoholic liver disease. Dig Dis Sci 1979; 24(11):835–838. pmid:520102
- Kaplan MM. Alanine aminotransferase levels: what’s normal? Ann Intern Med 2002; 137(1):49-51. pmid:12093245
- Pratt DS, Kaplan MM. Evaluation of abnormal liver enzyme results in asymptomatic patients. N Engl J Med 2000; 342(17):1266–1271. doi:10.1056/NEJM200004273421707
- Sharma U, Pal D, Prasad R. Alkaline phosphatase: an overview. Indian J Clin Biochem 2014; 29(3):269–278. doi:10.1007/s12291-013-0408-y
- Weibrecht K, Dayno M, Darling C, Bird SB. Liver aminotransferases are elevated with rhabdomyolysis in the absence of significant liver injury. J Med Toxicol 2010; 6(3):294–300. doi:10.1007/s13181-010-0075-9
- Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis 2010; 28(1):155–161. doi:10.1159/000282080
- Adams LA, Feldstein AE. Non-invasive diagnosis of nonalcoholic fatty liver and nonalcoholic steatohepatitis. J Dig Dis 2011; 12(1):10–16. doi:10.1111/j.1751-2980.2010.00471.x
- Fracanzani AL, Valenti L, Bugianesi E, et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: a role for insulin resistance and diabetes. Hepatology 2008; 48(3):792–798. doi:10.1002/hep.22429
- Tahan V, Canbakan B, Balci H, et al. Serum gamma-glutamyltranspeptidase distinguishes non-alcoholic fatty liver disease at high risk. Hepatogastroenterolgoy 2008; 55(85):1433-1438. pmid:18795706
- McPherson S, Stewart S, Henderson E, Burt AD, Day CP. Simple non-invasive fibrosis scoring systems can reliably exclude advanced fibrosis in patients with non-alcoholic fatty liver disease. Gut 2010; 59(9):1265–1269. doi:10.1136/gut.2010.216077
- Angulo P, Hui JM, Marchesini G, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007; 45(4):846–854. doi:10.1002/hep.21496
- Petta S, Vanni E, Bugianesi E, et al. The combination of liver stiffness measurement and NAFLD fibrosis score improves the noninvasive diagnostic accuracy for severe liver fibrosis in patients with nonalcoholic fatty liver disease. Liver Int 2015; 35(5):1566–1573. doi:10.1111/liv.12584
- Hashemi SA, Alavian SM, Gholami-Fesharaki M. Assessment of transient elastography (FibroScan) for diagnosis of fibrosis in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Caspian J Intern Med 2016; 7(4):242–252. pmid:27999641
- Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010; 51(1):121–129. doi:10.1002/hep.23276
- Adams PH, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med 2005; 352(17):1769-1778. doi:10.1056/NEJMoa041534
- Brissot P, de Bels F. Current approaches to the management of hemochromatosis. Hematology Am Soc Hematol Educ Program 2006; 2006(1):36–41. doi:10.1182/asheducation-2006.1.36
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54(1):328–343. doi:10.1002/hep.24330
- Weiler N, Zeuzem S, Welker MW. Concise review: interferon-free treatment of hepatitis C virus-associated cirrhosis and liver graft infection. World J Gastroenterol 2016; 22(41):9044–9056. doi:10.3748/wjg.v22.i41.9044
- American Association for the Study of Liver Disease, Infectious Diseases Society of America. HCV guidance: recommendations for testing, managing, and treating hepatitis C. www.hcvguidelines.org. Accessed July 16, 2018.
- Manns MP, Czaja AJ, Gorham JD, et al; American Association for the Study of Liver Diseases. Diagnosis and management of autoimmune hepatitis. Hepatology 2010; 51(6):2193–2213. doi:10.1002/hep.23584
- Liberal R, Krawitt EL, Vierling JM, Manns MP, Mieli-Vergani G, Vergani D. Cutting edge issues in autoimmune hepatitis. J Autoimmun 2016; 75:6–19. doi:10.1016/j.jaut.2016.07.005
- Mousa HS, Carbone M, Malinverno F, Ronca V, Gershwin ME, Invernizzi P. Novel therapeutics for primary biliary cholangitis: Toward a disease-stage-based approach. Autoimmun Rev 2016; 15(9):870–876. doi:10.1016/j.autrev.2016.07.003
- de Vries AB, Janse M, Blokzijl H, Weersma RK. Distinctive inflammatory bowel disease phenotype in primary sclerosing cholangitis. World J Gastroenterol 2015; 21(6):1956–1971. doi:10.3748/wjg.v21.i6.1956
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77(3):195–204. doi:10.3949/ccjm.77a.09064
- Chalasani N, Younossi Z, Lavine JE, et al; American Gastroenterological Association; American Association for the Study of Liver Diseases; American College of Gastroenterology. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012; 142(7):1592–1609. doi:10.1053/j.gastro.2012.04.001
- Cohen JA, Kaplan MM. The SGOT/SGPT ratio—an indicator of alcoholic liver disease. Dig Dis Sci 1979; 24(11):835–838. pmid:520102
- Kaplan MM. Alanine aminotransferase levels: what’s normal? Ann Intern Med 2002; 137(1):49-51. pmid:12093245
- Pratt DS, Kaplan MM. Evaluation of abnormal liver enzyme results in asymptomatic patients. N Engl J Med 2000; 342(17):1266–1271. doi:10.1056/NEJM200004273421707
- Sharma U, Pal D, Prasad R. Alkaline phosphatase: an overview. Indian J Clin Biochem 2014; 29(3):269–278. doi:10.1007/s12291-013-0408-y
- Weibrecht K, Dayno M, Darling C, Bird SB. Liver aminotransferases are elevated with rhabdomyolysis in the absence of significant liver injury. J Med Toxicol 2010; 6(3):294–300. doi:10.1007/s13181-010-0075-9
- Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis 2010; 28(1):155–161. doi:10.1159/000282080
- Adams LA, Feldstein AE. Non-invasive diagnosis of nonalcoholic fatty liver and nonalcoholic steatohepatitis. J Dig Dis 2011; 12(1):10–16. doi:10.1111/j.1751-2980.2010.00471.x
- Fracanzani AL, Valenti L, Bugianesi E, et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: a role for insulin resistance and diabetes. Hepatology 2008; 48(3):792–798. doi:10.1002/hep.22429
- Tahan V, Canbakan B, Balci H, et al. Serum gamma-glutamyltranspeptidase distinguishes non-alcoholic fatty liver disease at high risk. Hepatogastroenterolgoy 2008; 55(85):1433-1438. pmid:18795706
- McPherson S, Stewart S, Henderson E, Burt AD, Day CP. Simple non-invasive fibrosis scoring systems can reliably exclude advanced fibrosis in patients with non-alcoholic fatty liver disease. Gut 2010; 59(9):1265–1269. doi:10.1136/gut.2010.216077
- Angulo P, Hui JM, Marchesini G, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007; 45(4):846–854. doi:10.1002/hep.21496
- Petta S, Vanni E, Bugianesi E, et al. The combination of liver stiffness measurement and NAFLD fibrosis score improves the noninvasive diagnostic accuracy for severe liver fibrosis in patients with nonalcoholic fatty liver disease. Liver Int 2015; 35(5):1566–1573. doi:10.1111/liv.12584
- Hashemi SA, Alavian SM, Gholami-Fesharaki M. Assessment of transient elastography (FibroScan) for diagnosis of fibrosis in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Caspian J Intern Med 2016; 7(4):242–252. pmid:27999641
- Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010; 51(1):121–129. doi:10.1002/hep.23276
- Adams PH, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med 2005; 352(17):1769-1778. doi:10.1056/NEJMoa041534
- Brissot P, de Bels F. Current approaches to the management of hemochromatosis. Hematology Am Soc Hematol Educ Program 2006; 2006(1):36–41. doi:10.1182/asheducation-2006.1.36
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54(1):328–343. doi:10.1002/hep.24330
- Weiler N, Zeuzem S, Welker MW. Concise review: interferon-free treatment of hepatitis C virus-associated cirrhosis and liver graft infection. World J Gastroenterol 2016; 22(41):9044–9056. doi:10.3748/wjg.v22.i41.9044
- American Association for the Study of Liver Disease, Infectious Diseases Society of America. HCV guidance: recommendations for testing, managing, and treating hepatitis C. www.hcvguidelines.org. Accessed July 16, 2018.
- Manns MP, Czaja AJ, Gorham JD, et al; American Association for the Study of Liver Diseases. Diagnosis and management of autoimmune hepatitis. Hepatology 2010; 51(6):2193–2213. doi:10.1002/hep.23584
- Liberal R, Krawitt EL, Vierling JM, Manns MP, Mieli-Vergani G, Vergani D. Cutting edge issues in autoimmune hepatitis. J Autoimmun 2016; 75:6–19. doi:10.1016/j.jaut.2016.07.005
- Mousa HS, Carbone M, Malinverno F, Ronca V, Gershwin ME, Invernizzi P. Novel therapeutics for primary biliary cholangitis: Toward a disease-stage-based approach. Autoimmun Rev 2016; 15(9):870–876. doi:10.1016/j.autrev.2016.07.003
- de Vries AB, Janse M, Blokzijl H, Weersma RK. Distinctive inflammatory bowel disease phenotype in primary sclerosing cholangitis. World J Gastroenterol 2015; 21(6):1956–1971. doi:10.3748/wjg.v21.i6.1956
KEY POINTS
- Disorders of hepatocellular injury tend to elevate levels of aminotransferases, whereas cholestatic disorders cause elevations of alkaline phosphatase and bilirubin.
- The three most common causes of liver enzyme elevation are alcohol toxicity, medication overdose, and fatty liver disease.
- Other disorders of liver dysfunction include hereditary hemochromatosis, viral hepatitis, autoimmune hepatitis, primary biliary cholangitis, primary sclerosing cholangitis, and alpha-1 antitrypsin disease.
- Nonhepatic causes of elevated “liver enzymes” also need to be considered. For instance, rhabdomyolysis causes elevations in aminotransferase levels.

The bias of word choice and the interpretation of laboratory tests
In the current sociopolitical environment in the United States, the slogan “words matter” has become a battle cry for several groups and causes, emphasizing that our choice of words can influence the way we assess a specific person or situation. We are not immune to the subliminal bias of words, even as we evaluate such seemingly objective components of clinical management as laboratory test results.
Several years ago, I was supervising teaching rounds on a general medicine service. It was the first rounds of the month, and the patients were relatively new to the residents and totally unknown to me. One patient was an elderly man with weight loss, fatigue, weakness, and a history of excessive alcohol ingestion. His family had corroborated the last detail, but he had stopped drinking a long time before his admission. He had normal creatinine, minimal anemia, and markedly elevated and unexplained “liver function tests.” Liver biopsy was planned.
As we entered his room, we saw a gaunt man struggle to rise from the bedside chair to get back into bed. He rocked several times and then pushed himself up from the chair using his arms. Then, after a few short steps, he plopped back into bed and greeted us. His breakfast tray was untouched at the bedside. I introduced myself, we chatted for a short while as I examined him in front of our team, and we left.
In the hallway I asked, “Who would like to get an additional blood test before we do a liver biopsy?” Without waiting for a response I asked a second question, “What exactly are liver function tests?”
Words do matter, and they influence the way we analyze clinical scenarios. It could be argued that a complete and careful history would have established that our patient’s fatigue and weakness were due to proximal muscle weakness and not general asthenia, and that detailed questioning would have revealed that his weight loss was mainly from difficulty in swallowing without a sense of choking and coughing. But faced with an elderly man, a likely explanation for liver disease, and markedly elevated aspartate and alanine aminotransferase (AST and ALT) levels, there was premature closure of the diagnosis, and the decision was made to obtain a liver biopsy—which our hepatology consultants surely would not have done. I believe that a major contributor to the premature diagnosis was the choice of the words “liver function tests” and the default assumption that elevated serum levels of these enzymes always reflect liver disease.
Aminotransferases are fairly ubiquitous, likely present in various concentrations in all cells in our body. AST exists in mitochondrial and cytosolic forms, and ALT in the cytosol. The concentration of ALT is higher in the liver than in other organs, and its enzymatic activity is suppressed by hepatic exposure to alcohol. Both enzymes are present in muscle, and although AST is more abundant in cells other than hepatocytes, the longer serum half-life of ALT may result in roughly equal serum levels in the setting of chronic muscle injury such as myositis (the true diagnosis in our weak patient).
While a meticulous history and examination would indeed have led to the diagnosis of muscle disease in this man, they alone could not have determined whether he had coexistent liver and muscle disease. And this is a real challenge when acute muscle toxicity and liver toxicity are equally possible (eg, statin or immune checkpoint autoimmune tissue damage, or after significant trauma).
There are many nuances in the interpretation of even the most common laboratory tests. In this issue of the Journal, Agganis et al discuss liver enzymes (a term slightly more acceptable to me than liver function tests). In future issues, we will address the interpretation of other laboratory tests.
In the current sociopolitical environment in the United States, the slogan “words matter” has become a battle cry for several groups and causes, emphasizing that our choice of words can influence the way we assess a specific person or situation. We are not immune to the subliminal bias of words, even as we evaluate such seemingly objective components of clinical management as laboratory test results.
Several years ago, I was supervising teaching rounds on a general medicine service. It was the first rounds of the month, and the patients were relatively new to the residents and totally unknown to me. One patient was an elderly man with weight loss, fatigue, weakness, and a history of excessive alcohol ingestion. His family had corroborated the last detail, but he had stopped drinking a long time before his admission. He had normal creatinine, minimal anemia, and markedly elevated and unexplained “liver function tests.” Liver biopsy was planned.
As we entered his room, we saw a gaunt man struggle to rise from the bedside chair to get back into bed. He rocked several times and then pushed himself up from the chair using his arms. Then, after a few short steps, he plopped back into bed and greeted us. His breakfast tray was untouched at the bedside. I introduced myself, we chatted for a short while as I examined him in front of our team, and we left.
In the hallway I asked, “Who would like to get an additional blood test before we do a liver biopsy?” Without waiting for a response I asked a second question, “What exactly are liver function tests?”
Words do matter, and they influence the way we analyze clinical scenarios. It could be argued that a complete and careful history would have established that our patient’s fatigue and weakness were due to proximal muscle weakness and not general asthenia, and that detailed questioning would have revealed that his weight loss was mainly from difficulty in swallowing without a sense of choking and coughing. But faced with an elderly man, a likely explanation for liver disease, and markedly elevated aspartate and alanine aminotransferase (AST and ALT) levels, there was premature closure of the diagnosis, and the decision was made to obtain a liver biopsy—which our hepatology consultants surely would not have done. I believe that a major contributor to the premature diagnosis was the choice of the words “liver function tests” and the default assumption that elevated serum levels of these enzymes always reflect liver disease.
Aminotransferases are fairly ubiquitous, likely present in various concentrations in all cells in our body. AST exists in mitochondrial and cytosolic forms, and ALT in the cytosol. The concentration of ALT is higher in the liver than in other organs, and its enzymatic activity is suppressed by hepatic exposure to alcohol. Both enzymes are present in muscle, and although AST is more abundant in cells other than hepatocytes, the longer serum half-life of ALT may result in roughly equal serum levels in the setting of chronic muscle injury such as myositis (the true diagnosis in our weak patient).
While a meticulous history and examination would indeed have led to the diagnosis of muscle disease in this man, they alone could not have determined whether he had coexistent liver and muscle disease. And this is a real challenge when acute muscle toxicity and liver toxicity are equally possible (eg, statin or immune checkpoint autoimmune tissue damage, or after significant trauma).
There are many nuances in the interpretation of even the most common laboratory tests. In this issue of the Journal, Agganis et al discuss liver enzymes (a term slightly more acceptable to me than liver function tests). In future issues, we will address the interpretation of other laboratory tests.
In the current sociopolitical environment in the United States, the slogan “words matter” has become a battle cry for several groups and causes, emphasizing that our choice of words can influence the way we assess a specific person or situation. We are not immune to the subliminal bias of words, even as we evaluate such seemingly objective components of clinical management as laboratory test results.
Several years ago, I was supervising teaching rounds on a general medicine service. It was the first rounds of the month, and the patients were relatively new to the residents and totally unknown to me. One patient was an elderly man with weight loss, fatigue, weakness, and a history of excessive alcohol ingestion. His family had corroborated the last detail, but he had stopped drinking a long time before his admission. He had normal creatinine, minimal anemia, and markedly elevated and unexplained “liver function tests.” Liver biopsy was planned.
As we entered his room, we saw a gaunt man struggle to rise from the bedside chair to get back into bed. He rocked several times and then pushed himself up from the chair using his arms. Then, after a few short steps, he plopped back into bed and greeted us. His breakfast tray was untouched at the bedside. I introduced myself, we chatted for a short while as I examined him in front of our team, and we left.
In the hallway I asked, “Who would like to get an additional blood test before we do a liver biopsy?” Without waiting for a response I asked a second question, “What exactly are liver function tests?”
Words do matter, and they influence the way we analyze clinical scenarios. It could be argued that a complete and careful history would have established that our patient’s fatigue and weakness were due to proximal muscle weakness and not general asthenia, and that detailed questioning would have revealed that his weight loss was mainly from difficulty in swallowing without a sense of choking and coughing. But faced with an elderly man, a likely explanation for liver disease, and markedly elevated aspartate and alanine aminotransferase (AST and ALT) levels, there was premature closure of the diagnosis, and the decision was made to obtain a liver biopsy—which our hepatology consultants surely would not have done. I believe that a major contributor to the premature diagnosis was the choice of the words “liver function tests” and the default assumption that elevated serum levels of these enzymes always reflect liver disease.
Aminotransferases are fairly ubiquitous, likely present in various concentrations in all cells in our body. AST exists in mitochondrial and cytosolic forms, and ALT in the cytosol. The concentration of ALT is higher in the liver than in other organs, and its enzymatic activity is suppressed by hepatic exposure to alcohol. Both enzymes are present in muscle, and although AST is more abundant in cells other than hepatocytes, the longer serum half-life of ALT may result in roughly equal serum levels in the setting of chronic muscle injury such as myositis (the true diagnosis in our weak patient).
While a meticulous history and examination would indeed have led to the diagnosis of muscle disease in this man, they alone could not have determined whether he had coexistent liver and muscle disease. And this is a real challenge when acute muscle toxicity and liver toxicity are equally possible (eg, statin or immune checkpoint autoimmune tissue damage, or after significant trauma).
There are many nuances in the interpretation of even the most common laboratory tests. In this issue of the Journal, Agganis et al discuss liver enzymes (a term slightly more acceptable to me than liver function tests). In future issues, we will address the interpretation of other laboratory tests.

Phosphorus binders: The new and the old, and how to choose
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
See related editorial and article
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
- Phosphorus balance
- The interplay of hormones, including fibroblast growth factor 23 (FGF23)
- The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.

An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7


Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
- Decreased calcitriol due to its inhibition by FGF239
- Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
- Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).

As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.

Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.

Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
- Complicated diabetes mellitus
- Vascular or valvular calcification
- Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
- Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
- Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
- Lederer E. Regulation of serum phosphate. J Physiol 2014; 592(18):3985–3995. doi:10.1113/jphysiol.2014.273979
- Lederer E. Renal phosphate transporters. Curr Opin Nephrol Hypertens 2014; 23(5):502–506. doi:10.1097/MNH.0000000000000053
- Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
- Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
- Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
- Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
- Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
- Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
- Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
- Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
- Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
- Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
- Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
- Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
- Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
- Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
- Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
- National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
- Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
- Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
- Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
- Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
- Braunlin W, Zhorov E, Guo A, et al. Bile acid binding to sevelamer HCl. Kidney Int 2002; 62(2):611–619. doi:10.1046/j.1523-1755.2002.00459.x
- Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
- Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
- Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
- Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
- Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
- Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
- Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
- Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
- Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
- Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
- Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
- Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
- Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
- Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
- Suzuki NT. Hyperphosphatemia in nondialyzed TPN patients. JPEN J Parenter Enteral Nutr 1987; 11(5):512. doi:10.1177/0148607187011005512
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
See related editorial and article
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
- Phosphorus balance
- The interplay of hormones, including fibroblast growth factor 23 (FGF23)
- The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
- Decreased calcitriol due to its inhibition by FGF239
- Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
- Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
- Complicated diabetes mellitus
- Vascular or valvular calcification
- Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
See related editorial and article
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
- Phosphorus balance
- The interplay of hormones, including fibroblast growth factor 23 (FGF23)
- The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
- Decreased calcitriol due to its inhibition by FGF239
- Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
- Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
- Complicated diabetes mellitus
- Vascular or valvular calcification
- Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
- Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
- Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
- Lederer E. Regulation of serum phosphate. J Physiol 2014; 592(18):3985–3995. doi:10.1113/jphysiol.2014.273979
- Lederer E. Renal phosphate transporters. Curr Opin Nephrol Hypertens 2014; 23(5):502–506. doi:10.1097/MNH.0000000000000053
- Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
- Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
- Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
- Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
- Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
- Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
- Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
- Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
- Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
- Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
- Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
- Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
- Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
- Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
- Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
- National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
- Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
- Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
- Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
- Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
- Braunlin W, Zhorov E, Guo A, et al. Bile acid binding to sevelamer HCl. Kidney Int 2002; 62(2):611–619. doi:10.1046/j.1523-1755.2002.00459.x
- Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
- Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
- Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
- Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
- Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
- Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
- Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
- Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
- Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
- Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
- Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
- Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
- Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
- Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
- Suzuki NT. Hyperphosphatemia in nondialyzed TPN patients. JPEN J Parenter Enteral Nutr 1987; 11(5):512. doi:10.1177/0148607187011005512
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
- Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
- Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
- Lederer E. Regulation of serum phosphate. J Physiol 2014; 592(18):3985–3995. doi:10.1113/jphysiol.2014.273979
- Lederer E. Renal phosphate transporters. Curr Opin Nephrol Hypertens 2014; 23(5):502–506. doi:10.1097/MNH.0000000000000053
- Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
- Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
- Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
- Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
- Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
- Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
- Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
- Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
- Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
- Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
- Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
- Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
- Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
- Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
- Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
- National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
- Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
- Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
- Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
- Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
- Braunlin W, Zhorov E, Guo A, et al. Bile acid binding to sevelamer HCl. Kidney Int 2002; 62(2):611–619. doi:10.1046/j.1523-1755.2002.00459.x
- Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
- Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
- Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
- Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
- Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
- Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
- Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
- Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
- Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
- Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
- Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
- Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
- Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
- Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
- Suzuki NT. Hyperphosphatemia in nondialyzed TPN patients. JPEN J Parenter Enteral Nutr 1987; 11(5):512. doi:10.1177/0148607187011005512
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
KEY POINTS
- Serum phosphorus is maintained within normal levels in a tightly regulated system involving interplay between organs, hormones, diet, and other factors.
- Dietary phosphorus comes mainly from protein, so restricting phosphorus without introducing protein deficiency is difficult. Food with a low phosphorus-to-protein ratio and plant-based sources of protein may be preferable.
- Although dialysis removes phosphorus, it usually does not remove enough, and many patients require phosphorus-binding drugs.
- Selection of an appropriate binder should consider serum calcium levels, pill burden, serum iron stores, and cost.
Phosphorus in kidney disease: Culprit or bystander?
Phosphorus is essential for life. However, both low and high levels of phosphorus in the body have consequences, and its concentration in the blood is tightly regulated through dietary absorption, bone flux, and renal excretion and is influenced by calcitriol (1,25 hydroxyvitamin D3), parathyroid hormone, and fibroblast growth factor 23 (FGF23).
See related articles by M. Shetty and A. Sekar
Sekar et al,1 in this issue of the Journal, provide an extensive review of the pathophysiology of phosphorus metabolism and strategies to control phosphorus levels in patients with hyperphosphatemia and end-stage kidney disease.
PHOSPHORUS OR PHOSPHATE?
What's in a name? That which we call a rose
By any other word would smell as sweet.
—Shakespeare, Romeo and Juliet
The terms phosphate and phosphorus are often used interchangeably, though most writers still prefer phosphate over phosphorus.
The serum concentrations of phosphate and phosphorus are the same when expressed in millimoles per liter, as every mole of phosphate contains 1 mole of phosphorus, but not the same when expressed in milligrams per deciliter.2 The molecular weight of phosphorus is 30.97, whereas the molecular weight of the phosphate ion (PO43–) is 94.97—more than 3 times higher. Therefore, using these terms interchangeably in this context can lead to numerical error.3
Phosphorus, being highly reactive, does not exist by itself in nature and is typically present as phosphates in biologic systems. When describing phosphorus metabolism, the term phosphates should ideally be used because phosphates are the actual participants in the bodily processes. But in the clinical laboratory, all methods that measure serum phosphorus in fact measure inorganic phosphate and are expressed in terms of milligrams of phosphorus per deciliter rather than milligrams of phosphate per deciliter, and using these 2 terms interchangeably in clinical practice should not be of concern.4
THE PROBLEM
US adults typically ingest 1,200 mg of phosphorus each day, and about 60% to 70% of the ingested phosphorus is absorbed both by passive paracellular diffusion via tight junctions and by active transcellular transport via sodium-phosphate cotransport. The kidneys must excrete the same amount daily to maintain a steady state. As kidney function declines, phosphorus accumulates in the blood, leading to hyperphosphatemia.
Hyperphosphatemia is often asymptomatic, but it can cause generalized itching, red eyes, and adverse effects on the bone and parathyroid glands. Higher serum phosphorus levels have been shown to be associated with vascular calcification,5 cardiovascular events, and higher all-cause mortality rates in the general population,6 in patients with diabetes,7 and in those with chronic kidney disease.8 This association between higher serum phosphorus levels and the all-cause mortality rate led to the assumption that lowering serum phosphorus levels in these patients could reduce the rates of cardiovascular events and death, and to efforts to correct hyperphosphatemia.
Research into FGF23 continues, especially its role in cardiovascular complications of chronic kidney disease, as both phosphorus and FGF23 levels are elevated in chronic kidney disease and are implicated in poor clinical outcomes in these patients. However, both FGF23 and parathyroid hormone levels rise early in the course of kidney disease, long before overt hyperphosphatemia develops. Further, FGF23 rises earlier than parathyroid hormone and has been found to be an independent risk factor for cardiovascular events and death from any cause in end-stage kidney disease.9
Whether hyperphosphatemia is the culprit or merely an epiphenomenon of metabolic complications of chronic kidney disease is still unclear, as more molecules are being identified in the complex process of cardiovascular calcification.10
However, one thing is clear: vascular calcification is not just a simple precipitation of calcium and phosphorus. Instead, it is an active process that involves many regulators of mineral metabolism.10 The complex nature of this process is likely one of the reasons that evidence is conflicting11 about the benefits of phosphorus binders in terms of cardiovascular events or all-cause mortality in these patients.
STRATEGIES TO CONTROL HYPERPHOSPHATEMIA
Reducing intake
Dietary phosphorus restriction is the first step in controlling serum phosphorus. But reducing phosphorus intake while otherwise trying to optimize the nutritional status can be challenging.
The recommended daily protein intake is 1.0 to 1.2 g/kg. But phosphorus is typically found in foods rich in proteins, and restricting protein severely can compromise nutritional status and may be as bad as elevated phosphate levels in terms of outcomes.
Although plant-based foods contain more phosphate per gram of protein (ie, they have a higher ratio of phosphorus to protein) than animal-based foods, the bioavailability of phosphorus from plant foods is lower. Phosphorus in plant-based foods is mainly in the form of phytate. Humans cannot hydrolyze phytate because we lack the phytase enzyme; hence, the phosphorus in plant-based foods is not well absorbed. Therefore, a vegetarian diet may be preferable and beneficial in patients with chronic kidney disease. A small study in humans showed that a vegetarian diet resulted in lower serum phosphorus and FGF23 levels, but the study was limited by its small sample size.12
Patients should be advised to avoid foods that have a high phosphate content, such as processed foods, fast foods, and cola beverages, which often have phosphate-based food additives.
Further, one should be cautious about using supplements with healthy-sounding names. A case in point is “vitamin water”: 12 oz of this fruit punch-flavored beverage contains 392 mg of phosphorus,13 and this alone would require 12 to 15 phosphate binder tablets to bind its phosphorus content.
In addition, many prescription drugs have significant amounts of phosphorus, and this is often unrecognized.
Sherman et al14 reviewed 200 of the most commonly prescribed drugs in dialysis patients and found that 23 (11.5%) of the drug labels listed phosphorus-containing ingredients, but the actual amount of phosphorus was not listed. The phosphorus content ranged from 1.4 mg (clonidine 0.2 mg, Blue Point Laboratories, Dublin, Ireland) to 111.5 mg (paroxetine 40 mg, GlaxoSmith Kline, Philadelphia, PA). The phosphorus content was inconsistent and varied with the dose of the agent, type of formulation (tablet or syrup), branded or generic formulation, and manufacturer.
Branded lisinopril (Merck, Kenilworth, NJ) had 21.4 mg of phosphorus per 10-mg dose, while a generic product (Blue Point Laboratories, Dublin, Ireland) had 32.6 mg. Different brands of generic amlodipine 10 mg varied in their phosphorus content from 8.6 mg (Lupin Pharmaceuticals, Mumbai, India) to 27.8 mg (Greenstone LLC, Peapack, NJ) to 40.1 mg (Qualitest Pharmaceuticals, Huntsville, AL. Rena-Vite (Cypress Pharmaceuticals, Madison, MS), a multivitamin marketed to patients with kidney disease, had 37.7 mg of phosphorus per tablet. Thus, just to bind the phosphorus content of these 3 tablets (lisinopril, amlodipine, and Rena-Vite), a patient could need at least 3 to 4 extra doses of phosphate binder.
The phosphate content of medications should be considered when prescribing. For example, Reno Caps (Nnodum Pharmaceuticals, Cincinnati, OH), another vitamin supplement, has only 1.7 mg of phosphorus per tablet and should be considered, especially in patients with poorly controlled serum phosphorus levels. However, the challenge is that medication labels do not provide the phosphorus content.
Reducing phosphorus absorption

Although these agents reduce serum phosphorus and help reduce symptoms, an important quality-of-life measure, it is uncertain whether they improve clinical outcomes.11 To date, no specific phosphorus binder offers a survival benefit over placebo.11
Based on the limited and conflicting evidence, the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, recently updated, suggest that oral phosphorus binders should be used in patients with hyperphosphatemia to lower serum phosphorus levels toward the normal range.15 They further recommend not exceeding 1,500 mg of elemental calcium per day if a calcium-based binder is used, and they recommend avoiding calcium-based binders in patients with hypercalcemia, adynamic bone disease, or vascular calcification.
Phosphorus binders may account for up to 50% of the daily pill burden and may contribute to poor medication adherence.16 Dialysis patients need to take a lot of these drugs: by weight, 5 to 6 pounds per year.
These drugs can bind and interfere with the absorption of other vital medications and so should be taken with meals and separately from other medications.

Removing phosphorus
Removal of phosphorus by adequate dialysis or kidney transplant is the final strategy.
New agents under study
To improve phosphorus control, other agents that inhibit absorption of phosphate are being investigated.
Nicotinamide reduces expression of the sodium-phosphorus cotransporter NTP2b. Its use in combination with a low-phosphorus diet and phosphorus binders may maximize reductions in phosphorus absorption and is being studied in the CKD Optimal Management With Binders and Nicotinamide (COMBINE) study.
Tenapanor, an inhibitor of the sodium-hydrogen transporter NHE3, has been shown in animal studies to increase fecal phosphate excretion and decrease urinary phosphate excretion17 but requires further evaluation.
- Sekar A, Kaur T, Nally JV Jr, Rincon-Choles H, Jolly S, Nakhoul G. Phosphorus binders: the new and the old, and how to choose. Cleve Clin J Med 2018; 85(8):629–638. doi:10.3949/ccjm.85a.17054
- Young DS. "Phosphorus" or "phosphate." Ann Intern Med 1980; 93(4):631. pmid:7436198
- Bartter FC. Reporting of phosphate and phosphorus plasma values. Am J Med 1981; 71(5):848. pmid:7304659.
- Iheagwara OS, Ing TS, Kjellstrand CM, Lew SQ. Phosphorus, phosphorous, and phosphate. Hemodial Int 2013; 17(4):479–482. doi:10.1111/hdi.12010
- Adeney KL, Siscovick DS, Ix JH, et al. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 2009; 20(2):381–387. doi:10.1681/ASN.2008040349
- Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 2007; 167(9):879–885. doi:10.1001/archinte.167.9.879
- Chonchol M, Dale R, Schrier RW, Estacio R. Serum phosphorus and cardiovascular mortality in type 2 diabetes. Am J Med 2009; 122(4):380–386. doi:10.1016/j.amjmed.2008.09.039
- Covic A, Kothawala P, Bernal M, Robbins S, Chalian A, Goldsmith D. Systematic review of the evidence underlying the association between mineral metabolism disturbances and risk of all-cause mortality, cardiovascular mortality and cardiovascular events in chronic kidney disease. Nephrol Dial Transplant 2009; 24(5):1506–1523. doi:10.1093/ndt/gfn613
- Gutiérrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359(6):584–592. doi:10.1056/NEJMoa0706130
- Lullo LD, Barbera V, Bellasi A, et al. Vascular and valvular calcifications in chronic kidney disease: an update. EMJ Nephrol 2016; 4(1):84–91. https://pdfs.semanticscholar.org/150f/c7b5dfe671c9b61e4c76d54b7d713b60ba6a.pdf. Accesssed June 5, 2018.
- Palmer SC, Gardner S, Tonelli M, et al. Phosphate-binding agents in adults with CKD: a network meta-analysis of randomized trials. Am J Kidney Dis 2016; 68(5):691–702. doi:10.1053/j.ajkd.2016.05.015
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
- Moser M, White K, Henry B, et al. Phosphorus content of popular beverages. Am J Kidney Dis 2015; 65(6):969–971. doi:10.1053/j.ajkd.2015.02.330
- Sherman RA, Ravella S, Kapoian T. A dearth of data: the problem of phosphorus in prescription medications. Kidney Int 2015; 87(6):1097–1099. doi:10.1038/ki.2015.67
- KDIGO 2017 clinical practice guideline update for diagnosis, evaluation, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Supplements 2017; 7(1 suppl): 1–59. www.kisupplements.org/article/S2157-1716(17)30001-1/pdf. Accessed June 5, 2018.
- Fissell RB, Karaboyas A, Bieber BA, et al. Phosphate binder pill burden, patient-reported non-adherence, and mineral bone disorder markers: findings from the DOPPS. Hemodial Int 2016; 20(1):38–49. doi:10.1111/hdi.12315
- Labonté ED, Carreras CW, Leadbetter MR, et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol 2015; 26(5):1138–1149. doi:10.1681/ASN.2014030317
Phosphorus is essential for life. However, both low and high levels of phosphorus in the body have consequences, and its concentration in the blood is tightly regulated through dietary absorption, bone flux, and renal excretion and is influenced by calcitriol (1,25 hydroxyvitamin D3), parathyroid hormone, and fibroblast growth factor 23 (FGF23).
See related articles by M. Shetty and A. Sekar
Sekar et al,1 in this issue of the Journal, provide an extensive review of the pathophysiology of phosphorus metabolism and strategies to control phosphorus levels in patients with hyperphosphatemia and end-stage kidney disease.
PHOSPHORUS OR PHOSPHATE?
What's in a name? That which we call a rose
By any other word would smell as sweet.
—Shakespeare, Romeo and Juliet
The terms phosphate and phosphorus are often used interchangeably, though most writers still prefer phosphate over phosphorus.
The serum concentrations of phosphate and phosphorus are the same when expressed in millimoles per liter, as every mole of phosphate contains 1 mole of phosphorus, but not the same when expressed in milligrams per deciliter.2 The molecular weight of phosphorus is 30.97, whereas the molecular weight of the phosphate ion (PO43–) is 94.97—more than 3 times higher. Therefore, using these terms interchangeably in this context can lead to numerical error.3
Phosphorus, being highly reactive, does not exist by itself in nature and is typically present as phosphates in biologic systems. When describing phosphorus metabolism, the term phosphates should ideally be used because phosphates are the actual participants in the bodily processes. But in the clinical laboratory, all methods that measure serum phosphorus in fact measure inorganic phosphate and are expressed in terms of milligrams of phosphorus per deciliter rather than milligrams of phosphate per deciliter, and using these 2 terms interchangeably in clinical practice should not be of concern.4
THE PROBLEM
US adults typically ingest 1,200 mg of phosphorus each day, and about 60% to 70% of the ingested phosphorus is absorbed both by passive paracellular diffusion via tight junctions and by active transcellular transport via sodium-phosphate cotransport. The kidneys must excrete the same amount daily to maintain a steady state. As kidney function declines, phosphorus accumulates in the blood, leading to hyperphosphatemia.
Hyperphosphatemia is often asymptomatic, but it can cause generalized itching, red eyes, and adverse effects on the bone and parathyroid glands. Higher serum phosphorus levels have been shown to be associated with vascular calcification,5 cardiovascular events, and higher all-cause mortality rates in the general population,6 in patients with diabetes,7 and in those with chronic kidney disease.8 This association between higher serum phosphorus levels and the all-cause mortality rate led to the assumption that lowering serum phosphorus levels in these patients could reduce the rates of cardiovascular events and death, and to efforts to correct hyperphosphatemia.
Research into FGF23 continues, especially its role in cardiovascular complications of chronic kidney disease, as both phosphorus and FGF23 levels are elevated in chronic kidney disease and are implicated in poor clinical outcomes in these patients. However, both FGF23 and parathyroid hormone levels rise early in the course of kidney disease, long before overt hyperphosphatemia develops. Further, FGF23 rises earlier than parathyroid hormone and has been found to be an independent risk factor for cardiovascular events and death from any cause in end-stage kidney disease.9
Whether hyperphosphatemia is the culprit or merely an epiphenomenon of metabolic complications of chronic kidney disease is still unclear, as more molecules are being identified in the complex process of cardiovascular calcification.10
However, one thing is clear: vascular calcification is not just a simple precipitation of calcium and phosphorus. Instead, it is an active process that involves many regulators of mineral metabolism.10 The complex nature of this process is likely one of the reasons that evidence is conflicting11 about the benefits of phosphorus binders in terms of cardiovascular events or all-cause mortality in these patients.
STRATEGIES TO CONTROL HYPERPHOSPHATEMIA
Reducing intake
Dietary phosphorus restriction is the first step in controlling serum phosphorus. But reducing phosphorus intake while otherwise trying to optimize the nutritional status can be challenging.
The recommended daily protein intake is 1.0 to 1.2 g/kg. But phosphorus is typically found in foods rich in proteins, and restricting protein severely can compromise nutritional status and may be as bad as elevated phosphate levels in terms of outcomes.
Although plant-based foods contain more phosphate per gram of protein (ie, they have a higher ratio of phosphorus to protein) than animal-based foods, the bioavailability of phosphorus from plant foods is lower. Phosphorus in plant-based foods is mainly in the form of phytate. Humans cannot hydrolyze phytate because we lack the phytase enzyme; hence, the phosphorus in plant-based foods is not well absorbed. Therefore, a vegetarian diet may be preferable and beneficial in patients with chronic kidney disease. A small study in humans showed that a vegetarian diet resulted in lower serum phosphorus and FGF23 levels, but the study was limited by its small sample size.12
Patients should be advised to avoid foods that have a high phosphate content, such as processed foods, fast foods, and cola beverages, which often have phosphate-based food additives.
Further, one should be cautious about using supplements with healthy-sounding names. A case in point is “vitamin water”: 12 oz of this fruit punch-flavored beverage contains 392 mg of phosphorus,13 and this alone would require 12 to 15 phosphate binder tablets to bind its phosphorus content.
In addition, many prescription drugs have significant amounts of phosphorus, and this is often unrecognized.
Sherman et al14 reviewed 200 of the most commonly prescribed drugs in dialysis patients and found that 23 (11.5%) of the drug labels listed phosphorus-containing ingredients, but the actual amount of phosphorus was not listed. The phosphorus content ranged from 1.4 mg (clonidine 0.2 mg, Blue Point Laboratories, Dublin, Ireland) to 111.5 mg (paroxetine 40 mg, GlaxoSmith Kline, Philadelphia, PA). The phosphorus content was inconsistent and varied with the dose of the agent, type of formulation (tablet or syrup), branded or generic formulation, and manufacturer.
Branded lisinopril (Merck, Kenilworth, NJ) had 21.4 mg of phosphorus per 10-mg dose, while a generic product (Blue Point Laboratories, Dublin, Ireland) had 32.6 mg. Different brands of generic amlodipine 10 mg varied in their phosphorus content from 8.6 mg (Lupin Pharmaceuticals, Mumbai, India) to 27.8 mg (Greenstone LLC, Peapack, NJ) to 40.1 mg (Qualitest Pharmaceuticals, Huntsville, AL. Rena-Vite (Cypress Pharmaceuticals, Madison, MS), a multivitamin marketed to patients with kidney disease, had 37.7 mg of phosphorus per tablet. Thus, just to bind the phosphorus content of these 3 tablets (lisinopril, amlodipine, and Rena-Vite), a patient could need at least 3 to 4 extra doses of phosphate binder.
The phosphate content of medications should be considered when prescribing. For example, Reno Caps (Nnodum Pharmaceuticals, Cincinnati, OH), another vitamin supplement, has only 1.7 mg of phosphorus per tablet and should be considered, especially in patients with poorly controlled serum phosphorus levels. However, the challenge is that medication labels do not provide the phosphorus content.
Reducing phosphorus absorption
Although these agents reduce serum phosphorus and help reduce symptoms, an important quality-of-life measure, it is uncertain whether they improve clinical outcomes.11 To date, no specific phosphorus binder offers a survival benefit over placebo.11
Based on the limited and conflicting evidence, the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, recently updated, suggest that oral phosphorus binders should be used in patients with hyperphosphatemia to lower serum phosphorus levels toward the normal range.15 They further recommend not exceeding 1,500 mg of elemental calcium per day if a calcium-based binder is used, and they recommend avoiding calcium-based binders in patients with hypercalcemia, adynamic bone disease, or vascular calcification.
Phosphorus binders may account for up to 50% of the daily pill burden and may contribute to poor medication adherence.16 Dialysis patients need to take a lot of these drugs: by weight, 5 to 6 pounds per year.
These drugs can bind and interfere with the absorption of other vital medications and so should be taken with meals and separately from other medications.
Removing phosphorus
Removal of phosphorus by adequate dialysis or kidney transplant is the final strategy.
New agents under study
To improve phosphorus control, other agents that inhibit absorption of phosphate are being investigated.
Nicotinamide reduces expression of the sodium-phosphorus cotransporter NTP2b. Its use in combination with a low-phosphorus diet and phosphorus binders may maximize reductions in phosphorus absorption and is being studied in the CKD Optimal Management With Binders and Nicotinamide (COMBINE) study.
Tenapanor, an inhibitor of the sodium-hydrogen transporter NHE3, has been shown in animal studies to increase fecal phosphate excretion and decrease urinary phosphate excretion17 but requires further evaluation.
Phosphorus is essential for life. However, both low and high levels of phosphorus in the body have consequences, and its concentration in the blood is tightly regulated through dietary absorption, bone flux, and renal excretion and is influenced by calcitriol (1,25 hydroxyvitamin D3), parathyroid hormone, and fibroblast growth factor 23 (FGF23).
See related articles by M. Shetty and A. Sekar
Sekar et al,1 in this issue of the Journal, provide an extensive review of the pathophysiology of phosphorus metabolism and strategies to control phosphorus levels in patients with hyperphosphatemia and end-stage kidney disease.
PHOSPHORUS OR PHOSPHATE?
What's in a name? That which we call a rose
By any other word would smell as sweet.
—Shakespeare, Romeo and Juliet
The terms phosphate and phosphorus are often used interchangeably, though most writers still prefer phosphate over phosphorus.
The serum concentrations of phosphate and phosphorus are the same when expressed in millimoles per liter, as every mole of phosphate contains 1 mole of phosphorus, but not the same when expressed in milligrams per deciliter.2 The molecular weight of phosphorus is 30.97, whereas the molecular weight of the phosphate ion (PO43–) is 94.97—more than 3 times higher. Therefore, using these terms interchangeably in this context can lead to numerical error.3
Phosphorus, being highly reactive, does not exist by itself in nature and is typically present as phosphates in biologic systems. When describing phosphorus metabolism, the term phosphates should ideally be used because phosphates are the actual participants in the bodily processes. But in the clinical laboratory, all methods that measure serum phosphorus in fact measure inorganic phosphate and are expressed in terms of milligrams of phosphorus per deciliter rather than milligrams of phosphate per deciliter, and using these 2 terms interchangeably in clinical practice should not be of concern.4
THE PROBLEM
US adults typically ingest 1,200 mg of phosphorus each day, and about 60% to 70% of the ingested phosphorus is absorbed both by passive paracellular diffusion via tight junctions and by active transcellular transport via sodium-phosphate cotransport. The kidneys must excrete the same amount daily to maintain a steady state. As kidney function declines, phosphorus accumulates in the blood, leading to hyperphosphatemia.
Hyperphosphatemia is often asymptomatic, but it can cause generalized itching, red eyes, and adverse effects on the bone and parathyroid glands. Higher serum phosphorus levels have been shown to be associated with vascular calcification,5 cardiovascular events, and higher all-cause mortality rates in the general population,6 in patients with diabetes,7 and in those with chronic kidney disease.8 This association between higher serum phosphorus levels and the all-cause mortality rate led to the assumption that lowering serum phosphorus levels in these patients could reduce the rates of cardiovascular events and death, and to efforts to correct hyperphosphatemia.
Research into FGF23 continues, especially its role in cardiovascular complications of chronic kidney disease, as both phosphorus and FGF23 levels are elevated in chronic kidney disease and are implicated in poor clinical outcomes in these patients. However, both FGF23 and parathyroid hormone levels rise early in the course of kidney disease, long before overt hyperphosphatemia develops. Further, FGF23 rises earlier than parathyroid hormone and has been found to be an independent risk factor for cardiovascular events and death from any cause in end-stage kidney disease.9
Whether hyperphosphatemia is the culprit or merely an epiphenomenon of metabolic complications of chronic kidney disease is still unclear, as more molecules are being identified in the complex process of cardiovascular calcification.10
However, one thing is clear: vascular calcification is not just a simple precipitation of calcium and phosphorus. Instead, it is an active process that involves many regulators of mineral metabolism.10 The complex nature of this process is likely one of the reasons that evidence is conflicting11 about the benefits of phosphorus binders in terms of cardiovascular events or all-cause mortality in these patients.
STRATEGIES TO CONTROL HYPERPHOSPHATEMIA
Reducing intake
Dietary phosphorus restriction is the first step in controlling serum phosphorus. But reducing phosphorus intake while otherwise trying to optimize the nutritional status can be challenging.
The recommended daily protein intake is 1.0 to 1.2 g/kg. But phosphorus is typically found in foods rich in proteins, and restricting protein severely can compromise nutritional status and may be as bad as elevated phosphate levels in terms of outcomes.
Although plant-based foods contain more phosphate per gram of protein (ie, they have a higher ratio of phosphorus to protein) than animal-based foods, the bioavailability of phosphorus from plant foods is lower. Phosphorus in plant-based foods is mainly in the form of phytate. Humans cannot hydrolyze phytate because we lack the phytase enzyme; hence, the phosphorus in plant-based foods is not well absorbed. Therefore, a vegetarian diet may be preferable and beneficial in patients with chronic kidney disease. A small study in humans showed that a vegetarian diet resulted in lower serum phosphorus and FGF23 levels, but the study was limited by its small sample size.12
Patients should be advised to avoid foods that have a high phosphate content, such as processed foods, fast foods, and cola beverages, which often have phosphate-based food additives.
Further, one should be cautious about using supplements with healthy-sounding names. A case in point is “vitamin water”: 12 oz of this fruit punch-flavored beverage contains 392 mg of phosphorus,13 and this alone would require 12 to 15 phosphate binder tablets to bind its phosphorus content.
In addition, many prescription drugs have significant amounts of phosphorus, and this is often unrecognized.
Sherman et al14 reviewed 200 of the most commonly prescribed drugs in dialysis patients and found that 23 (11.5%) of the drug labels listed phosphorus-containing ingredients, but the actual amount of phosphorus was not listed. The phosphorus content ranged from 1.4 mg (clonidine 0.2 mg, Blue Point Laboratories, Dublin, Ireland) to 111.5 mg (paroxetine 40 mg, GlaxoSmith Kline, Philadelphia, PA). The phosphorus content was inconsistent and varied with the dose of the agent, type of formulation (tablet or syrup), branded or generic formulation, and manufacturer.
Branded lisinopril (Merck, Kenilworth, NJ) had 21.4 mg of phosphorus per 10-mg dose, while a generic product (Blue Point Laboratories, Dublin, Ireland) had 32.6 mg. Different brands of generic amlodipine 10 mg varied in their phosphorus content from 8.6 mg (Lupin Pharmaceuticals, Mumbai, India) to 27.8 mg (Greenstone LLC, Peapack, NJ) to 40.1 mg (Qualitest Pharmaceuticals, Huntsville, AL. Rena-Vite (Cypress Pharmaceuticals, Madison, MS), a multivitamin marketed to patients with kidney disease, had 37.7 mg of phosphorus per tablet. Thus, just to bind the phosphorus content of these 3 tablets (lisinopril, amlodipine, and Rena-Vite), a patient could need at least 3 to 4 extra doses of phosphate binder.
The phosphate content of medications should be considered when prescribing. For example, Reno Caps (Nnodum Pharmaceuticals, Cincinnati, OH), another vitamin supplement, has only 1.7 mg of phosphorus per tablet and should be considered, especially in patients with poorly controlled serum phosphorus levels. However, the challenge is that medication labels do not provide the phosphorus content.
Reducing phosphorus absorption
Although these agents reduce serum phosphorus and help reduce symptoms, an important quality-of-life measure, it is uncertain whether they improve clinical outcomes.11 To date, no specific phosphorus binder offers a survival benefit over placebo.11
Based on the limited and conflicting evidence, the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, recently updated, suggest that oral phosphorus binders should be used in patients with hyperphosphatemia to lower serum phosphorus levels toward the normal range.15 They further recommend not exceeding 1,500 mg of elemental calcium per day if a calcium-based binder is used, and they recommend avoiding calcium-based binders in patients with hypercalcemia, adynamic bone disease, or vascular calcification.
Phosphorus binders may account for up to 50% of the daily pill burden and may contribute to poor medication adherence.16 Dialysis patients need to take a lot of these drugs: by weight, 5 to 6 pounds per year.
These drugs can bind and interfere with the absorption of other vital medications and so should be taken with meals and separately from other medications.
Removing phosphorus
Removal of phosphorus by adequate dialysis or kidney transplant is the final strategy.
New agents under study
To improve phosphorus control, other agents that inhibit absorption of phosphate are being investigated.
Nicotinamide reduces expression of the sodium-phosphorus cotransporter NTP2b. Its use in combination with a low-phosphorus diet and phosphorus binders may maximize reductions in phosphorus absorption and is being studied in the CKD Optimal Management With Binders and Nicotinamide (COMBINE) study.
Tenapanor, an inhibitor of the sodium-hydrogen transporter NHE3, has been shown in animal studies to increase fecal phosphate excretion and decrease urinary phosphate excretion17 but requires further evaluation.
- Sekar A, Kaur T, Nally JV Jr, Rincon-Choles H, Jolly S, Nakhoul G. Phosphorus binders: the new and the old, and how to choose. Cleve Clin J Med 2018; 85(8):629–638. doi:10.3949/ccjm.85a.17054
- Young DS. "Phosphorus" or "phosphate." Ann Intern Med 1980; 93(4):631. pmid:7436198
- Bartter FC. Reporting of phosphate and phosphorus plasma values. Am J Med 1981; 71(5):848. pmid:7304659.
- Iheagwara OS, Ing TS, Kjellstrand CM, Lew SQ. Phosphorus, phosphorous, and phosphate. Hemodial Int 2013; 17(4):479–482. doi:10.1111/hdi.12010
- Adeney KL, Siscovick DS, Ix JH, et al. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 2009; 20(2):381–387. doi:10.1681/ASN.2008040349
- Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 2007; 167(9):879–885. doi:10.1001/archinte.167.9.879
- Chonchol M, Dale R, Schrier RW, Estacio R. Serum phosphorus and cardiovascular mortality in type 2 diabetes. Am J Med 2009; 122(4):380–386. doi:10.1016/j.amjmed.2008.09.039
- Covic A, Kothawala P, Bernal M, Robbins S, Chalian A, Goldsmith D. Systematic review of the evidence underlying the association between mineral metabolism disturbances and risk of all-cause mortality, cardiovascular mortality and cardiovascular events in chronic kidney disease. Nephrol Dial Transplant 2009; 24(5):1506–1523. doi:10.1093/ndt/gfn613
- Gutiérrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359(6):584–592. doi:10.1056/NEJMoa0706130
- Lullo LD, Barbera V, Bellasi A, et al. Vascular and valvular calcifications in chronic kidney disease: an update. EMJ Nephrol 2016; 4(1):84–91. https://pdfs.semanticscholar.org/150f/c7b5dfe671c9b61e4c76d54b7d713b60ba6a.pdf. Accesssed June 5, 2018.
- Palmer SC, Gardner S, Tonelli M, et al. Phosphate-binding agents in adults with CKD: a network meta-analysis of randomized trials. Am J Kidney Dis 2016; 68(5):691–702. doi:10.1053/j.ajkd.2016.05.015
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
- Moser M, White K, Henry B, et al. Phosphorus content of popular beverages. Am J Kidney Dis 2015; 65(6):969–971. doi:10.1053/j.ajkd.2015.02.330
- Sherman RA, Ravella S, Kapoian T. A dearth of data: the problem of phosphorus in prescription medications. Kidney Int 2015; 87(6):1097–1099. doi:10.1038/ki.2015.67
- KDIGO 2017 clinical practice guideline update for diagnosis, evaluation, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Supplements 2017; 7(1 suppl): 1–59. www.kisupplements.org/article/S2157-1716(17)30001-1/pdf. Accessed June 5, 2018.
- Fissell RB, Karaboyas A, Bieber BA, et al. Phosphate binder pill burden, patient-reported non-adherence, and mineral bone disorder markers: findings from the DOPPS. Hemodial Int 2016; 20(1):38–49. doi:10.1111/hdi.12315
- Labonté ED, Carreras CW, Leadbetter MR, et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol 2015; 26(5):1138–1149. doi:10.1681/ASN.2014030317
- Sekar A, Kaur T, Nally JV Jr, Rincon-Choles H, Jolly S, Nakhoul G. Phosphorus binders: the new and the old, and how to choose. Cleve Clin J Med 2018; 85(8):629–638. doi:10.3949/ccjm.85a.17054
- Young DS. "Phosphorus" or "phosphate." Ann Intern Med 1980; 93(4):631. pmid:7436198
- Bartter FC. Reporting of phosphate and phosphorus plasma values. Am J Med 1981; 71(5):848. pmid:7304659.
- Iheagwara OS, Ing TS, Kjellstrand CM, Lew SQ. Phosphorus, phosphorous, and phosphate. Hemodial Int 2013; 17(4):479–482. doi:10.1111/hdi.12010
- Adeney KL, Siscovick DS, Ix JH, et al. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 2009; 20(2):381–387. doi:10.1681/ASN.2008040349
- Dhingra R, Sullivan LM, Fox CS, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 2007; 167(9):879–885. doi:10.1001/archinte.167.9.879
- Chonchol M, Dale R, Schrier RW, Estacio R. Serum phosphorus and cardiovascular mortality in type 2 diabetes. Am J Med 2009; 122(4):380–386. doi:10.1016/j.amjmed.2008.09.039
- Covic A, Kothawala P, Bernal M, Robbins S, Chalian A, Goldsmith D. Systematic review of the evidence underlying the association between mineral metabolism disturbances and risk of all-cause mortality, cardiovascular mortality and cardiovascular events in chronic kidney disease. Nephrol Dial Transplant 2009; 24(5):1506–1523. doi:10.1093/ndt/gfn613
- Gutiérrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359(6):584–592. doi:10.1056/NEJMoa0706130
- Lullo LD, Barbera V, Bellasi A, et al. Vascular and valvular calcifications in chronic kidney disease: an update. EMJ Nephrol 2016; 4(1):84–91. https://pdfs.semanticscholar.org/150f/c7b5dfe671c9b61e4c76d54b7d713b60ba6a.pdf. Accesssed June 5, 2018.
- Palmer SC, Gardner S, Tonelli M, et al. Phosphate-binding agents in adults with CKD: a network meta-analysis of randomized trials. Am J Kidney Dis 2016; 68(5):691–702. doi:10.1053/j.ajkd.2016.05.015
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
- Moser M, White K, Henry B, et al. Phosphorus content of popular beverages. Am J Kidney Dis 2015; 65(6):969–971. doi:10.1053/j.ajkd.2015.02.330
- Sherman RA, Ravella S, Kapoian T. A dearth of data: the problem of phosphorus in prescription medications. Kidney Int 2015; 87(6):1097–1099. doi:10.1038/ki.2015.67
- KDIGO 2017 clinical practice guideline update for diagnosis, evaluation, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Supplements 2017; 7(1 suppl): 1–59. www.kisupplements.org/article/S2157-1716(17)30001-1/pdf. Accessed June 5, 2018.
- Fissell RB, Karaboyas A, Bieber BA, et al. Phosphate binder pill burden, patient-reported non-adherence, and mineral bone disorder markers: findings from the DOPPS. Hemodial Int 2016; 20(1):38–49. doi:10.1111/hdi.12315
- Labonté ED, Carreras CW, Leadbetter MR, et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol 2015; 26(5):1138–1149. doi:10.1681/ASN.2014030317
Calcific uremic arteriolopathy
A 51-year-old man with end-stage renal disease, on peritoneal dialysis for the past 4 years, presented to the emergency department with severe pain in both legs. The pain had started 2 months previously and had progressively worsened. After multiple admissions in the past for hyperkalemia and volume overload due to noncompliance, he had been advised to switch to hemodialysis.
See related article and editorial

Laboratory analysis revealed the following values:
- Serum creatinine 12.62 mg/dL (reference range 0.73–1.22)
- Blood urea nitrogen 159 mg/dL (9–24)
- Serum calcium corrected for serum albumin 8.1 mg/dL (8.4–10.0)
- Serum phosphorus 10.6 mg/dL (2.7–4.8).
His history of end-stage renal disease, failure of peritoneal dialysis, high calcium-phosphorus product (8.1 mg/dL × 10.6 mg/dL = 85.9 mg2/dL2, reference range ≤ 55), and characteristic physical findings led to the diagnosis of calcific uremic arteriolopathy.
CALCIFIC UREMIC ARTERIOLOPATHY
Calcific uremic arteriolopathy or “calciphylaxis,” seen most often in patients with end-stage renal disease, is caused by calcium deposition in the media of the dermo-hypodermic arterioles, leading to infarction of adjacent tissue.1–3 A high calcium-phosphorus product (> 55) has been implicated in its development; however, the calcium-phosphorus product can be normal despite hyperphosphatemia, which itself may promote ectopic calcification.
Early ischemic manifestations include livedo reticularis and painful retiform purpura on the thighs and other areas of high adiposity. Lesions evolve into violaceous plaquelike subcutaneous nodules that can infarct, become necrotic, ulcerate, and become infected. Punch biopsy demonstrating arteriolar calcification, subintimal fibrosis, and thrombosis confirms the diagnosis.
Differential diagnosis
Warfarin necrosis can cause large, irregular, bloody bullae that ulcerate and turn into eschar that may resemble lesions of calcific uremic arteriolopathy. Our patient, however, had no exposure to warfarin.
Pemphigus foliaceus, an immunoglobulin G4-mediated autoimmune disorder targeted against desmoglein-1, leads to the formation of fragile blisters that easily rupture when rubbed (Nikolsky sign). Lesions evolve into scaling, crusty erosions on an erythematous base. With tender blisters and lack of mucous membrane involvement, pemphigus foliaceus shares similarities with calcific uremic arteriolopathy, but the presence of necrotic eschar surrounded by violaceous plaques in our patient made it an unlikely diagnosis.
Cryofibrinogenemia. In the right clinical scenario, ie, in a patient with vasculitis, malignancy, infection, cryoglobulinemia, or collagen diseases, cryofibrinogen-mediated cold-induced occlusive lesions may mimic calcific uremic arteriolopathy, with painful or pruritic erythema, purpura, livedo reticularis, necrosis, and ulceration.4 Our patient had no color changes with exposure to cold, nor any history of Raynaud phenomenon or joint pain, making the diagnosis of cryofibrinogenemia less likely.
Nephrogenic systemic fibrosis. Gadolinium contrast medium in magnetic resonance imaging can cause nephrogenic systemic fibrosis, characterized by erythematous papules that coalesce into brawny plaques with surrounding woody induration, which may resemble lesions of calcific uremic arteriolopathy.5 However, our patient had not been exposed to gadolinium.
Management
Management is multidisciplinary and includes the following1:
- Hemodialysis, modified to optimize calcium balance2
- Intravenous sodium thiosulfate: the exact mechanism of action remains unclear, but it is thought to play a role in chelating calcium from tissue deposits, thus decreasing pain and promoting regression of skin lesions3
- Wound care, including chemical debridement agents, negative-pressure wound therapy, and surgical debridement for infected wounds6
- Pain management with opioid analgesics.
The patient was treated with all these measures. However, he died of sudden cardiac arrest during the same admission.
- Weenig RH, Sewell LD, Davis MD, McCarthy JT, Pittelkow MR. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol 2007; 56(4):569–579. doi:10.1016/j.jaad.2006.08.065
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66(1):133–146. doi:10.1053/j.ajkd.2015.01.034
- Janigan DT, Hirsch DJ, Klassen GA, MacDonald AS. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis 2000; 35(4):588–597. pmid:10739777
- Michaud M, Pourrat J. Cryofibrinogenemia. J Clin Rheumatol 2013; 19(3):142–148. doi:10.1097/RHU.0b013e318289e06e
- Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol 2006; 18(6):614–617. doi:10.1097/01.bor.0000245725.94887.8d
- Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, eds. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill Professional; 2003:558–562.
A 51-year-old man with end-stage renal disease, on peritoneal dialysis for the past 4 years, presented to the emergency department with severe pain in both legs. The pain had started 2 months previously and had progressively worsened. After multiple admissions in the past for hyperkalemia and volume overload due to noncompliance, he had been advised to switch to hemodialysis.
See related article and editorial
Laboratory analysis revealed the following values:
- Serum creatinine 12.62 mg/dL (reference range 0.73–1.22)
- Blood urea nitrogen 159 mg/dL (9–24)
- Serum calcium corrected for serum albumin 8.1 mg/dL (8.4–10.0)
- Serum phosphorus 10.6 mg/dL (2.7–4.8).
His history of end-stage renal disease, failure of peritoneal dialysis, high calcium-phosphorus product (8.1 mg/dL × 10.6 mg/dL = 85.9 mg2/dL2, reference range ≤ 55), and characteristic physical findings led to the diagnosis of calcific uremic arteriolopathy.
CALCIFIC UREMIC ARTERIOLOPATHY
Calcific uremic arteriolopathy or “calciphylaxis,” seen most often in patients with end-stage renal disease, is caused by calcium deposition in the media of the dermo-hypodermic arterioles, leading to infarction of adjacent tissue.1–3 A high calcium-phosphorus product (> 55) has been implicated in its development; however, the calcium-phosphorus product can be normal despite hyperphosphatemia, which itself may promote ectopic calcification.
Early ischemic manifestations include livedo reticularis and painful retiform purpura on the thighs and other areas of high adiposity. Lesions evolve into violaceous plaquelike subcutaneous nodules that can infarct, become necrotic, ulcerate, and become infected. Punch biopsy demonstrating arteriolar calcification, subintimal fibrosis, and thrombosis confirms the diagnosis.
Differential diagnosis
Warfarin necrosis can cause large, irregular, bloody bullae that ulcerate and turn into eschar that may resemble lesions of calcific uremic arteriolopathy. Our patient, however, had no exposure to warfarin.
Pemphigus foliaceus, an immunoglobulin G4-mediated autoimmune disorder targeted against desmoglein-1, leads to the formation of fragile blisters that easily rupture when rubbed (Nikolsky sign). Lesions evolve into scaling, crusty erosions on an erythematous base. With tender blisters and lack of mucous membrane involvement, pemphigus foliaceus shares similarities with calcific uremic arteriolopathy, but the presence of necrotic eschar surrounded by violaceous plaques in our patient made it an unlikely diagnosis.
Cryofibrinogenemia. In the right clinical scenario, ie, in a patient with vasculitis, malignancy, infection, cryoglobulinemia, or collagen diseases, cryofibrinogen-mediated cold-induced occlusive lesions may mimic calcific uremic arteriolopathy, with painful or pruritic erythema, purpura, livedo reticularis, necrosis, and ulceration.4 Our patient had no color changes with exposure to cold, nor any history of Raynaud phenomenon or joint pain, making the diagnosis of cryofibrinogenemia less likely.
Nephrogenic systemic fibrosis. Gadolinium contrast medium in magnetic resonance imaging can cause nephrogenic systemic fibrosis, characterized by erythematous papules that coalesce into brawny plaques with surrounding woody induration, which may resemble lesions of calcific uremic arteriolopathy.5 However, our patient had not been exposed to gadolinium.
Management
Management is multidisciplinary and includes the following1:
- Hemodialysis, modified to optimize calcium balance2
- Intravenous sodium thiosulfate: the exact mechanism of action remains unclear, but it is thought to play a role in chelating calcium from tissue deposits, thus decreasing pain and promoting regression of skin lesions3
- Wound care, including chemical debridement agents, negative-pressure wound therapy, and surgical debridement for infected wounds6
- Pain management with opioid analgesics.
The patient was treated with all these measures. However, he died of sudden cardiac arrest during the same admission.
A 51-year-old man with end-stage renal disease, on peritoneal dialysis for the past 4 years, presented to the emergency department with severe pain in both legs. The pain had started 2 months previously and had progressively worsened. After multiple admissions in the past for hyperkalemia and volume overload due to noncompliance, he had been advised to switch to hemodialysis.
See related article and editorial
Laboratory analysis revealed the following values:
- Serum creatinine 12.62 mg/dL (reference range 0.73–1.22)
- Blood urea nitrogen 159 mg/dL (9–24)
- Serum calcium corrected for serum albumin 8.1 mg/dL (8.4–10.0)
- Serum phosphorus 10.6 mg/dL (2.7–4.8).
His history of end-stage renal disease, failure of peritoneal dialysis, high calcium-phosphorus product (8.1 mg/dL × 10.6 mg/dL = 85.9 mg2/dL2, reference range ≤ 55), and characteristic physical findings led to the diagnosis of calcific uremic arteriolopathy.
CALCIFIC UREMIC ARTERIOLOPATHY
Calcific uremic arteriolopathy or “calciphylaxis,” seen most often in patients with end-stage renal disease, is caused by calcium deposition in the media of the dermo-hypodermic arterioles, leading to infarction of adjacent tissue.1–3 A high calcium-phosphorus product (> 55) has been implicated in its development; however, the calcium-phosphorus product can be normal despite hyperphosphatemia, which itself may promote ectopic calcification.
Early ischemic manifestations include livedo reticularis and painful retiform purpura on the thighs and other areas of high adiposity. Lesions evolve into violaceous plaquelike subcutaneous nodules that can infarct, become necrotic, ulcerate, and become infected. Punch biopsy demonstrating arteriolar calcification, subintimal fibrosis, and thrombosis confirms the diagnosis.
Differential diagnosis
Warfarin necrosis can cause large, irregular, bloody bullae that ulcerate and turn into eschar that may resemble lesions of calcific uremic arteriolopathy. Our patient, however, had no exposure to warfarin.
Pemphigus foliaceus, an immunoglobulin G4-mediated autoimmune disorder targeted against desmoglein-1, leads to the formation of fragile blisters that easily rupture when rubbed (Nikolsky sign). Lesions evolve into scaling, crusty erosions on an erythematous base. With tender blisters and lack of mucous membrane involvement, pemphigus foliaceus shares similarities with calcific uremic arteriolopathy, but the presence of necrotic eschar surrounded by violaceous plaques in our patient made it an unlikely diagnosis.
Cryofibrinogenemia. In the right clinical scenario, ie, in a patient with vasculitis, malignancy, infection, cryoglobulinemia, or collagen diseases, cryofibrinogen-mediated cold-induced occlusive lesions may mimic calcific uremic arteriolopathy, with painful or pruritic erythema, purpura, livedo reticularis, necrosis, and ulceration.4 Our patient had no color changes with exposure to cold, nor any history of Raynaud phenomenon or joint pain, making the diagnosis of cryofibrinogenemia less likely.
Nephrogenic systemic fibrosis. Gadolinium contrast medium in magnetic resonance imaging can cause nephrogenic systemic fibrosis, characterized by erythematous papules that coalesce into brawny plaques with surrounding woody induration, which may resemble lesions of calcific uremic arteriolopathy.5 However, our patient had not been exposed to gadolinium.
Management
Management is multidisciplinary and includes the following1:
- Hemodialysis, modified to optimize calcium balance2
- Intravenous sodium thiosulfate: the exact mechanism of action remains unclear, but it is thought to play a role in chelating calcium from tissue deposits, thus decreasing pain and promoting regression of skin lesions3
- Wound care, including chemical debridement agents, negative-pressure wound therapy, and surgical debridement for infected wounds6
- Pain management with opioid analgesics.
The patient was treated with all these measures. However, he died of sudden cardiac arrest during the same admission.
- Weenig RH, Sewell LD, Davis MD, McCarthy JT, Pittelkow MR. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol 2007; 56(4):569–579. doi:10.1016/j.jaad.2006.08.065
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66(1):133–146. doi:10.1053/j.ajkd.2015.01.034
- Janigan DT, Hirsch DJ, Klassen GA, MacDonald AS. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis 2000; 35(4):588–597. pmid:10739777
- Michaud M, Pourrat J. Cryofibrinogenemia. J Clin Rheumatol 2013; 19(3):142–148. doi:10.1097/RHU.0b013e318289e06e
- Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol 2006; 18(6):614–617. doi:10.1097/01.bor.0000245725.94887.8d
- Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, eds. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill Professional; 2003:558–562.
- Weenig RH, Sewell LD, Davis MD, McCarthy JT, Pittelkow MR. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol 2007; 56(4):569–579. doi:10.1016/j.jaad.2006.08.065
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66(1):133–146. doi:10.1053/j.ajkd.2015.01.034
- Janigan DT, Hirsch DJ, Klassen GA, MacDonald AS. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis 2000; 35(4):588–597. pmid:10739777
- Michaud M, Pourrat J. Cryofibrinogenemia. J Clin Rheumatol 2013; 19(3):142–148. doi:10.1097/RHU.0b013e318289e06e
- Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol 2006; 18(6):614–617. doi:10.1097/01.bor.0000245725.94887.8d
- Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, eds. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill Professional; 2003:558–562.
Diagnosing and treating bipolar disorder in primary care
Patients presenting with depression commonly have undiagnosed bipolar depression,1–7 that is, depression with shifts to periods of mania. During manic or hypomanic episodes, people feel energetic, need little sleep, and are often happy and charming.8 But too much of a good thing can also wreak havoc on their life.
Bipolar depression (ie, depression in patients with a diagnosis of bipolar disorder) is treated differently from unipolar depression,3,9–13 making it especially important that clinicians recognize if a patient who presents with depression has a history of (hypo)manic symptoms.
CASE 1: THE IMPULSIVE NURSE
A 32-year-old nurse presents to her primary care provider with depressed mood. She reports having had a single depressive episode when she was a college freshman. Her family history includes depression, bipolar disorder, and schizophrenia, and her paternal grandfather and a maternal aunt committed suicide. Upon questioning, she reveals that in the past, she has had 3 episodes lasting several weeks and characterized by insubordinate behavior at work, irritability, high energy, and decreased need for sleep. She regrets impulsive sexual and financial decisions that she made during these episodes and recently filed for personal bankruptcy. For the past month, her mood has been persistently low, with reduced sleep, appetite, energy, and concentration, and with passive thoughts of suicide.
A CAREFUL HISTORY IS CRITICAL
This case illustrates many typical features of bipolar depression that are revealed only by taking a thorough history. Although the patient is high-functioning, having attained a professional career, she has serious problems with sexual and financial impulsivity and at her job. She has a strong family history of mood disorder. And she describes episodes of depression and mania in the past.
Starts in young adulthood, strong heritability
Bipolar disorder can be a devastating condition with lifelong consequences,14–20 especially as it typically starts when patients are getting an education or embarking on a career. It usually first manifests in the late teenage years and progresses in the patient’s early 20s.21,22 The first hospitalization can occur soon thereafter.23,24
Bipolar disorder is one of the most heritable conditions in psychiatry, and about 13% of children who have an afflicted parent develop it.25 In identical twins, the concordance is about 50% to 75%, indicating the importance of genetics and environmental factors.26,27
Associated with migraine, other conditions

The disorder is associated with a variety of conditions (Table 1).28,29 Some conditions (eg, thyroid disease) can cause mood cycling, and some (eg, sexually transmitted infections, accidents) are the consequences of the lifestyle that may accompany mania. For unknown reasons, migraine is highly associated with bipolar disorder.
DEPRESSION AND MANIA: TWO SIDES OF THE SAME COIN
Symptoms of depression and mania are frequently viewed as opposite mood states, though many times patients report a mixture of them.17,30–35 For both states, the features of a distinct change from the patient’s normal condition and the sustained nature of the symptoms are important diagnostically and indicate a likely underlying biological cause.
Major depressive disorder: Slowing down
The American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5),8 defines major depressive disorder as having either depressed mood or markedly diminished pleasure in most activities for most days during at least 2 weeks.
In addition, at least 4 of the following must be present during the same period:
- Appetite disturbance
- Sleep disturbance
- Motor retardation or agitation
- Lack of energy
- Feelings of worthlessness or excessive guilt
- Decreased concentration
- Recurrent thoughts of death or suicide.
An estimated 20% of the population experience a major depressive episode over their lifetime. A surprisingly high proportion of people with depression—30% to 40%—also have had subthreshold symptoms of mania (symptoms not meeting the criteria for hypomania or mania in terms of number of symptoms or duration).21,22 Because of these odds, it is important to suspect bipolar disorder even in patients who present with depression but who may not yet have manifested episodes of mania or hypomania.
Mood disorders can be regarded as falling into a spectrum, ranging from unipolar or “pure” major depression without any features of hypomania to major depression and severe mania.17,31–36
Mania: Speeding up
The DSM-5 defines mania as the presence of persistently elevated, expansive, or irritable mood with increased activity for more than 1 week. In addition, at least 3 of the following features must be present, with impaired functioning (4 features are required if mood is only irritable)8:
- Inflated self-esteem or grandiosity
- Decreased need for sleep
- Pressured speech
- Racing thoughts
- Distractibility
- Excessive involvement in pleasurable, high-risk activities.
Hypomania: No functional impairment
Hypomania is a less severe condition, in which the abnormally elevated mood is of shorter duration (4–7 days) and meets the other criteria for mania but without significant functional impairment. People may, in fact, be very functional and productive during hypomanic episodes.8
CLASSIFYING BIPOLAR DISORDER
Bipolar disorder is categorized according to severity.24,37,38 The most severe form, bipolar I disorder, is marked by major depression and manic episodes. It affects up to 1.5% of the US population, with equal proportions of men and women.39 Bipolar II disorder is less severe. It affects 0.8% to 1.6% of the US population, predominantly women.21,40 In bipolar II disorder, depression is more prominent, with episodes of hypomania.
Subthreshold bipolar disorders are characterized by episodic symptoms that do not meet the threshold for depression or hypomania; the symptoms are fewer or of shorter duration. These minor types of bipolar disorder affect up to 6% of the US population.17
Other conditions within the spectrum of bipolar and depressive disorders include medication- and substance-induced mania, agitated or anxious depression, and mixed states.31,34–36
DISTINGUISHING UNIPOLAR FROM BIPOLAR DEPRESSION
Considerable research has focused on finding a clear-cut clinical or biological feature to differentiate unipolar from bipolar depression, but so far none has been discovered. Distinguishing the two conditions still depends on clinical judgment. There are important reasons to identify the distinction between unipolar depression and bipolar disorder.
Prognosis differs. Bipolar disorder tends to be a more severe condition. Young people, who may initially present with only mild symptoms of mania, may develop serious episodes over the years. People may lose their savings, their marriage, and their career during a manic episode. The more critical the occupation (eg, doctor, pilot), the greater the potential consequences of impaired judgment brought on by even mild hypomania.14–20
Treatment differs. Typical antidepressants given for depression can trigger a manic episode in patients with bipolar depression, with devastating consequences. Atypical neuroleptic drugs used to treat bipolar disorder can also have serious effects (eg, metabolic and neurologic effects, including irreversible tardive dyskinesia).3,13,40–43
Despite the good reasons to do so, many doctors (including some psychiatrists) do not ask their patients about a propensity to mania or hypomania.4–6 More stigma is attached to the diagnosis of bipolar disorder than to depression44–47; once it is in the medical record, the patient may have problems with employment and obtaining medical insurance.17,44 The old term “manic-depressive” is often associated in the public mind with a person on the streets displaying severely psychotic behavior; the condition is now understood to consist of a spectrum from mild to more severe illness.
Clinical indicators of bipolarity
There are many indicators that a person who presents with depression may be on the bipolar spectrum, but this is not always easily identified.48–53
History of hypomanic symptoms or subthreshold manic symptoms. Although directly asking the patient about the defining symptoms (eg, “Have you ever had episodes of being ‘hyper’ or too happy?”) may help elicit the diagnosis, many patients with bipolar disorder only report depression, as it is psychically painful. In contrast, hypomania and even mania can be perceived as positive, as patients may have less insight into the abnormality of the condition and feel that they are functioning extremely well.
Early age of onset of a mood disorder, such as severe depression in childhood or early adulthood, points toward bipolar disorder. Diagnosing mood disorders in childhood is difficult, as children are less able to recognize or verbalize many of their symptoms.
Postpartum mood disorder, particularly with psychotic symptoms, indicates a strong possibility of a diagnosis of bipolar disorder.
Drug-induced mania, hypomania, and periods of hyperactivity are key features of bipolar disorder. If asked, patients may report feeling a “buzz” when taking an antidepressant.
Erratic patterns in work and relationships are a red flag and are viewed as “soft signs” of bipolar depression. Akiskal54 described the “rule of three” that should make one consider bipolar disorder: for example, three failed marriages, three current jobs or frequent job changes, three distinct professions practiced at the same time, and simultaneously dating three people. Such features indicate both the hyperfunctioning and the disruptive aspects of mania.
Family history of bipolar disorder or severe psychiatric illness is a very important clue. A more subtle clue described by Akiskal54 may be that several members of the family are very high-functioning in several different fields: eg, one may be a highly accomplished doctor, another a famous lawyer, and another a prominent politician. Or several members of the family may have erratic patterns of work and relationships. However, these subtle clues have been derived from clinical experiences and have not been validated in large-scale studies.
CASE 2: THE FRIENDLY SURGEON
Dr. Z is a prominent surgical subspecialist who is part of a small group practice. His wife has become increasingly worried about his behavior changes at home, including sleeping only a few hours a night, spending sprees, and binge drinking. He reluctantly agrees to an outpatient psychiatric evaluation if she attends with him. He creates a disturbance in the waiting room by shaking everyone’s hands and trying to hug all the women. During his examination, he is loud and expansive, denying he has any problems and describing himself as “the greatest doctor in the world.” The psychiatrist recommends hospitalization, but Dr. Z refuses and becomes belligerent. He announces that he just needs a career change and that he will fly to Mexico to open a bar.
This case, from the Texas Medical Association Archives,55 is not unusual. In addition to many characteristics discussed above, this case is typical in that the spouse brought the patient in, reflecting that the patient lacked insight that his behavior was abnormal. The disinhibition (hugging women), grandiosity, and unrealistic plans are also typical.
DIFFERENTIAL DIAGNOSIS OF BIPOLAR DEPRESSION

Anxiety disorders may be associated with dissociative speech or racing thoughts, which can be confused with bipolar illness. Personality disorders (eg, borderline, narcissistic, sociopathic) can involve a tumultuous and impulsive lifestyle resembling episodes of depression and mania. Schizoaffective illness has features of schizophrenia and bipolar disorder.
It is also possible that, despite what may look like mild features of bipolar disorder, there is no psychiatric condition. Some people with mild mania—often successful professionals or politicians—have high energy and can function very well with only a few hours of sleep. Similarly, depressive symptoms for short periods of time can be adaptive, such as in the face of a serious setback when extreme reflection and a period of inactivity can be useful, leading to subsequent reorganization.
A psychiatric diagnosis is usually made only when there is an abnormality, ie, the behavior is beyond normal limits, the person cannot control his or her symptoms, or social or occupational functioning is impaired.
SCREENING INSTRUMENTS
A few tools help determine the likelihood of bipolar disorder.
The Patient Health Questionnaire (PHQ-9)59,60 is a good 9-item screening tool for depression.
The Mood Disorder Questionnaire60 is specific for bipolar disorder, and like the PHQ-9, it is a patient-reported, short questionnaire that is available free online. The Mood Disorder Questionnaire asks about the symptoms of mania in a yes-no format. The result is positive if all of the following are present:
- A “yes” response to 7 of the 13 features
- Several features occur simultaneously
- The features are a moderate or serious problem.
Unlike most screening instruments, the Mood Disorder Questionnaire is more specific than sensitive. It is 93% specific for bipolar disorder in patients treated for depression in a primary care setting, but only 58% sensitive.61–63
WHEN TO REFER TO PSYCHIATRY
Patients suspected of having bipolar disorder or who have been previously diagnosed with it should be referred to a psychiatrist if they have certain features, including:
- Bipolar I disorder
- Psychotic symptoms
- Suicide risk or in danger of harming others
- Significantly impaired functioning
- Unclear diagnosis.
CASE 3: A TELEVISION ANCHOR’S DREAM TURNS TO NIGHTMARE
According to a famous news anchor’s autobiography,64 the steroids prescribed for her hives “revved her up.” The next course left her depressed. Antidepressant medications propelled her into a manic state, and she was soon planning a book, a television show, and a magazine all at once. During that time, she bought a cottage online. Her shyness evaporated at parties. “I was suddenly the equal of my high-energy friends who move fast and talk fast and loud,” she wrote. “I told everyone that I could understand why men felt like they could run the world, because I felt like that. This was a new me, and I liked her!”64 She was soon diagnosed with bipolar disorder and admitted to a psychiatric clinic.
TREAT WITH ANTIDEPRESSANTS, MOOD STABILIZERS
In general, acute bipolar disorder should be treated with a combination of an antidepressant and a mood stabilizer, and possibly an antipsychotic drug. An antidepressant should not be used alone, particularly with patients with a diagnosis of bipolar I disorder, because of the risk of triggering mania or the risk of faster cycling between mania and depression.13
Mood stabilizers include lithium, lamotrigine, and valproate. Each can prevent episodes of depression and mania. Lithium, which has been used as a mood stabilizer for 60 years, is specific for bipolar disorder, and it remains the best mood stabilizer treatment.
Antidepressants. The first-line antidepressant medication is bupropion, which is thought to be less likely to precipitate a manic episode,65 though all antidepressants have been associated with this side effect in patients with bipolar disorder. Other antidepressants—for example, selective serotonin reuptake inhibitors such as fluoxetine and dual reuptake inhibitors such as venlafaxine and duloxetine—can also be used. The precipitation of mania and possible increased mood cycling was first described with tricylic antidepressants, so drugs of this class should be used with caution.
Neuroleptic drugs such as aripiprazole, quetiapine, and lurasidone may be the easiest drugs to use, as they have antidepressant effects and can also prevent the occurrence of mania. These medications are frequently classified as mood stabilizers. However, they may not have true mood stabilizing properties such as that of lithium. Importantly, their use tends to entail significant metabolic problems and can lead to hyperlipidemia and diabetes. In addition, Parkinson disease-like symptoms— and in some cases irreversible involuntary movements of the mouth and tongue, as well as the body (tardive dyskinesia)—are important possible side effects.

All psychiatric medications have potential side effects (Table 3). Newer antidepressants and neuroleptics may have fewer side effects than older medications but are not more effective.
Should milder forms of bipolar depression be treated?
A dilemma is whether we should treat milder forms of bipolar depression, such as bipolar II depression, depression with subthreshold hypomania symptoms, or depression in persons with a strong family history of bipolar disorder.
Many doctors are justifiably reluctant to prescribe antidepressants for depression because of the risk of triggering mania. Although mood stabilizers such as lithium would counteract possible mania emergence, physicians often do not prescribe them because of inexperience and fear of risks and possible side effects. Patients are likewise resistant because they feel that use of mood stabilizers is tantamount to being told they are “manic-depressive,” with its associated stigma.
Overuse of atypical neuroleptics such as aripiprazole, quetiapine, and olanzapine has led to an awareness of metabolic syndrome and tardive dyskinesia, also making doctors cautious about using these drugs.
Answer: Yes, but treat with caution
Not treating depression consigns a patient to suffer with untreated depression, sometimes for years. Outcomes for patients with depression and bipolar disorder are often poor because the conditions are not recognized, and even when the conditions are recognized, doctors and patients may be reluctant to medicate appropriately. Medications should be used as needed to treat depression, but with an awareness of the possible side effects and with close patient monitoring.
A truly sustained manic state (unlike the brief euphoria brought on by some drugs) is not actually so easy to induce. In an unpublished Cleveland Clinic study, we monitored peaks of hypomanic symptoms in young patients (ages 15–30) during antidepressant treatment without mood stabilizers. About 30% to 40% of patients had subthreshold manic symptoms or a family history of bipolar disorder; 3 patients out of 51 developed hypomania leading to a change of diagnosis to bipolar disorder. Even in patients who had no risk factors for bipolar disorder, 2 out of 53 converted to a bipolar diagnosis. So conversion rates in patients with subthreshold bipolar disorder seem to be low, and the disorder can be identified early by monitoring the patient closely.
NONPHARMACOLOGIC TREATMENTS FOR DEPRESSION
Psychotherapy is indicated for all patients on medications for depression, as both pharmacologic and nonpharmacologic treatments are more effective when combined.66 Other treatments include transcranial magnetic stimulation, electroconvulsive therapy, light therapy, and exercise. Having a consistent daily routine, particularly regarding the sleep-wake schedule, is also helpful, and patients should be educated about its importance.
- Cicchetti D, Toth SL. The development of depression in children and adolescents. Am Psychol 1998; 53(2):221–241. doi:10.1037/0003-066X.53.2.221
- Glick ID. Undiagnosed bipolar disorder: new syndromes and new treatments. Prim Care Companion J Clin Psychiatry 2004;6(1):27–33. pmid:15486598
- Ghaemi SN, Boiman EE, Goodwin FK. Diagnosing bipolar disorder and the effect of antidepressants: a naturalistic study. J Clin Psychiatry 2000; 61(10):804–808. pmid:11078046
- Singh T, Rajput M. Misdiagnosis of bipolar disorder. Psychiatry (Edgmont) 2006; 3(10):57–63. pmid: 20877548
- Lish JD, Dime-Meenan S, Whybrow PC, Price RA, Hirschfeld RM. The National Depressive and Manic-depressive Association (DMDA) survey of bipolar members. J Affect Disord 1994; 31(4):281–294. pmid:7989643
- Howes OD, Falkenberg I. Early detection and intervention in bipolar affective disorder: targeting the development of the disorder. Curr Psychiatry Rep 2011; 13(6):493–499. pmid:21850462
- Ghaemi SN, Sachs GS, Chiou AM, Pandurangi AK, Goodwin K. Is bipolar disorder still underdiagnosed? Are antidepressants overutilized? J Affect Disord 1999; 52(1–3):135–144. pmid:10357026
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-5. Washington, DC: American Psychiatric Publishing, 2013.
- Hlastala SA, Frank E, Mallinger AG, Thase ME, Ritenour AM, Kupfer DJ. Bipolar depression: an underestimated treatment challenge. Depress Anxiety 1997; 5(2):73–83. pmid:9262937
- Smith DJ, Craddock N. Unipolar and bipolar depression: different or the same? Br J Psychiatry 2011; 199(4):272–274. doi:10.1192/bjp.bp.111.092726
- Viktorin A, Lichtenstein P, Thase ME, et al. The risk of switch to mania in patients with bipolar disorder during treatment with an antidepressant alone and in combination with a mood stabilizer. Am J Psychiatry 2014; 171(10):1067–1073. doi:10.1176/appi.ajp.2014.13111501
- Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med 2007; 356(17):1711–1722. doi:10.1056/NEJMoa064135
- American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry 2002; 159(4 suppl):1–50. pmid:11958165
- Leonpacher AK, Liebers D, Pirooznia M, et al. Distinguishing bipolar from unipolar depression: the importance of clinical symptoms and illness features. Psychol Med 2015; 45(11):2437–2446. doi:10.1017/S0033291715000446
- Angst J, Gamma A, Benazzi F, Ajdacic V, Eich D, Rössler W. Toward a re-definition of subthreshold bipolarity: epidemiology and proposed criteria for bipolar-II, minor bipolar disorders and hypomania. J Affect Disord 2003; 73(1–2):133–146. pmid:12507746
- Faravelli C, Rosi S, Alessandra Scarpato M, Lampronti L, Amedei SG, Rana N. Threshold and subthreshold bipolar disorders in the Sesto Fiorentino Study. J Affect Disord 2006; 94(1–3):111–119. pmid:16701902
- Judd LL, Akiskal HS. The prevalence and disability of bipolar spectrum disorders in the US population: re-analysis of the ECA database taking into account subthreshold cases. J Affect Disord 2003; 73(1–2):123–131. pmid:12507745
- Park Y-M, Lee B-H. Treatment response in relation to subthreshold bipolarity in patients with major depressive disorder receiving antidepressant monotherapy: a post hoc data analysis (KOMDD study). Neuropsychiatr Dis Treat 2016; 12:1221–1227. doi:10.2147/NDT.S104188
- Perlis RH, Uher R, Ostacher M, Goldberg JF, et al. Association between bipolar spectrum features and treatment outcomes in outpatients with major depressive disorder. Arch Gen Psychiatry 2011; 68(4):351–360. doi:10.1001/archgenpsychiatry.2010.179
- Dudek D, Siwek M, Zielin´ska D, Jaeschke R, Rybakowski J. Diagnostic conversions from major depressive disorder into bipolar disorder in an outpatient setting: results of a retrospective chart review. J Affect Disord 2013; 144(1–2):112–115. doi:10.1016/j.jad.2012.06.014
- Angst J, Cui L, Swendsen J, et al. Major depressive disorder with subthreshold bipolarity in the National Comorbidity Survey Replication. Am J Psychiatry 2010; 167(10):1194–1201. doi:10.1176/appi.ajp.2010.09071011
- Zimmermann P, Brückl T, Nocon A, et al. Heterogeneity of DSM-IV major depressive disorder as a consequence of subthreshold bipolarity. Arch Gen Psychiatry 2009; 66(12):1341–1352. doi:10.1001/archgenpsychiatry.2009.158
- Patel NC, DelBello MP, Keck PE, Strakowski SM. Phenomenology associated with age at onset in patients with bipolar disorder at their first psychiatric hospitalization. Bipolar Disord 2006; 8(1):91–94. pmid:16411986
- Hirschfeld RMA, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the National Depressive and Manic-Depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry 2003; 64(2):161–174. pmid:12633125
- Craddock N, Jones I. Genetics of bipolar disorder. J Med Genet 1999; 36(8):585–594. pmid:10465107
- Griswold KS, Pessar LF. Management of bipolar disorder. Am Fam Physician 2000; 62(6):1343–1358. pmid:11011863
- Kerner B. Genetics of bipolar disorder. Appl Clin Genet 2014; 7:33–42. doi:10.2147/TACG.S39297
- Scheffer RE, Linden S. Concurrent medical conditions with pediatric bipolar disorder. Curr Opin Psychiatry 2007; 20(4):398–401. doi:10.1097/YCO.0b013e3281a305c3
- Carney CP, Jones LE. Medical comorbidity in women and men with bipolar disorders: a population-based controlled study. Psychosom Med 2006;68(5):684–691. doi:10.1097/01.psy.0000237316.09601.88
- Cassano GB, Akiskal HS, Savino M, Musetti L, Perugi G. Proposed subtypes of bipolar II and related disorders: with hypomanic episodes (or cyclothymia) and with hyperthymic temperament. J Affect Disord 1992; 26(2):127–140. pmid:1447430
- Akiskal HS, Mallya G. Criteria for the “soft” bipolar spectrum: treatment implications. Psychopharmacol Bull 1987; 23(1):68–73. pmid:3602332
- Fiedorowicz JG, Endicott J, Leon AC, Solomon DA, Keller MB, Coryell WH. Subthreshold hypomanic symptoms in progression from unipolar major depression to bipolar disorder. Am J Psychiatry 2011; 168(1):40–48. doi:10.1176/appi.ajp.2010.10030328
- Maj M, Pirozzi R, Magliano L, Fiorillo A, Bartoli L. Agitated “unipolar” major depression: prevalence, phenomenology, and outcome. J Clin Psychiatry 2006; 67(5):712–719. pmid:16841620
- Akiskal HS. The bipolar spectrum: new concepts in classification and diagnosis. In: Psychiatry Update: the American Psychiatric Association Annual Review. Washington, DC: American Psychiatric Press; 1983:271–292.
- Akiskal HS, Pinto O. The evolving bipolar spectrum. Prototypes I, II, III, and IV. Psychiatr Clin North Am 1999; 22(3):517–534. pmid:10550853
- Cassano GB, Savino M, Perugi G, Musetti L, Akiskal HS. Major depressive episode: unipolar and bipolar II. Encephale 1992 Jan;18 Spec No:15–18. pmid:1600898
- Müller-Oerlinghausen B, Berghöfer A, Bauer M. Bipolar disorder. Lancet 2002; 359(9302):241–247. pmid:11812578
- Hirschfeld RMA, Calabrese JR, Weissman MM, et al. Screening for bipolar disorder in the community. J Clin Psychiatry 2003; 64(1):53–59. pmid:12590624
- Blanco C, Compton WM, Saha TD, et al. Epidemiology of DSM-5 bipolar I disorder: results from the National Epidemiologic survey on Alcohol and Related Conditions—III. J Psychiatr Res 2017; 84:310–317. doi:10.1016/j.jpsychires.2016.10.003
- McGirr A, Vöhringer PA, Ghaemi SN, Lam RW, Yatham LN. Safety and efficacy of adjunctive second-generation antidepressant therapy with a mood stabiliser or an atypical antipsychotic in acute bipolar depression: a systematic review and meta-analysis of randomised placebo-controlled trials. Lancet Psychiatry 2016; 3(12):1138–1146. doi:10.1016/S2215-0366(16)30264-4
- Gijsman HJ, Geddes JR, Rendell JM, Nolen WA, Goodwin GM. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry 2004; 161(9):1537–1547. doi:10.1176/appi.ajp.161.9.1537
- Sidor MM, Macqueen GM. Antidepressants for the acute treatment of bipolar depression: a systematic review and meta-analysis. J Clin Psychiatry 2011; 72(2):156–167. doi:10.4088/JCP.09r05385gre
- Liu B, Zhang Y, Fang H, Liu J, Liu T, Li L. Efficacy and safety of long-term antidepressant treatment for bipolar disorders - A meta-analysis of randomized controlled trials. J Affect Disord 2017; 223(139):41–48. doi:10.1016/j.jad.2017.07.023
- Krupa T, Kirsh B, Cockburn L, Gewurtz R. Understanding the stigma of mental illness in employment. Work 2009; 33(4):413–425. doi:10.3233/WOR-2009-0890
- Hawke LD, Parikh SV, Michalak EE. Stigma and bipolar disorder: a review of the literature. J Affect Disord 2013; 150(2):181–191. doi:10.1016/j.jad.2013.05.030
- Cerit C, Filizer A, Tural Ü, Tufan AE. Stigma: a core factor on predicting functionality in bipolar disorder. Compr Psychiatry 2012; 53(5):484–489. doi:10.1016/j.comppsych.2011.08.010
- O’Donnell L, Himle JA, Ryan K, et al. Social aspects of the workplace among individuals with bipolar disorder. J Soc Social Work Res 2017; 8(3):379–398. doi:10.1086/693163
- Akiskal HS, Maser JD, Zeller PJ, et al. Switching from “unipolar” to bipolar II. An 11-year prospective study of clinical and temperamental predictors in 559 patients. Arch Gen Psychiatry 1995; 52(2):114–123. pmid:7848047
- Kroon JS, Wohlfarth TD, Dieleman J, et al. Incidence rates and risk factors of bipolar disorder in the general population: a population-based cohort study. Bipolar Disord 2013; 15(3):306–313. doi:10.1111/bdi.12058
- Fiedorowicz JG, Endicott J, Leon AC, Solomon DA, Keller MB, Coryell WH. Subthreshold hypomanic symptoms in progression from unipolar major depression to bipolar disorder. Am J Psychiatry 2011; 168(1):40–48. doi:10.1176/appi.ajp.2010.10030328
- Akiskal HS, Walker P, Puzantian VR, King D, Rosenthal TL, Dranon M. Bipolar outcome in the course of depressive illness. Phenomenologic, familial, and pharmacologic predictors. J Affect Disord 1983; 5(2):115–128. pmid:6222091
- Strober M, Carlson G. Bipolar illness in adolescents with major depression: clinical, genetic, and psychopharmacologic predictors in a three- to four-year prospective follow-up investigation. Arch Gen Psychiatry 1982; 39(5):549–555. pmid:7092488
- Goldberg JF, Harrow M, Whiteside JE. Risk for bipolar illness in patients initially hospitalized for unipolar depression. Am J Psychiatry 2001; 158(8):1265–1270. pmid:11481161
- Akiskal HS. Searching for behavioral indicators of bipolar II in patients presenting with major depressive episodes: the “red sign,” the “rule of three” and other biographic signs of temperamental extravagance, activation and hypomania. J Affect Disord 2005; 84(2–3):279–290. pmid:15708427
- Texas Medical Association. Mood disorders in physicians. www.texmed.org/Template.aspx?id=6833. Accessed June 7, 2018.
- Hirschfeld RM. Differential diagnosis of bipolar disorder and major depressive disorder. J Affect Disord 2014;169(suppl 1):S12–S16. doi:10.1016/S0165-0327(14)70004-7
- Dunner DL. Differential diagnosis of bipolar disorder. J Clin Psychopharmacol 1992; 12(1suppl):7S–12S. pmid:1541721
- Peet M, Peters S. Drug-induced mania. Drug Saf 1995; 12(2):146–153. pmid:7766338
- Spitzer RL, Kroenke K, Williams JBW. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA 1999; 282(18):1737–1744. pmid:10568646
- Kroenke K, Spitzer RL, Williams JBW. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001; 16(9):606–613. pmid:11556941
- Hirschfeld RMMA, Williams JBBW, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: the Mood Disorder Questionnaire. Am J Psychiatry 2000; 157(11)1873–1875. doi:10.1176/appi.ajp.157.11.1873
- Hirschfeld RMA. The Mood Disorder Questionnaire: a simple, patient-rated screening instrument for bipolar disorder. Prim Care Companion J Clin Psychiatry 2002; 4(1):9–11. pmid: 15014728
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293(8):956–963. doi:10.1001/jama.293.8.956
- Pauley J. Skywriting: A Life Out of the Blue. New York: Random House, 2004.
- Goren JL, Levin GM. Mania with bupropion: a dose-related phenomenon? Ann Pharmacother 2000; 34(5):619–621. doi:10.1345/aph.19313
- Swann AC. Long-term treatment in bipolar disorder. J Clin Psychiatry 2005; 66(suppl 1):7–12. pmid:15693746
Patients presenting with depression commonly have undiagnosed bipolar depression,1–7 that is, depression with shifts to periods of mania. During manic or hypomanic episodes, people feel energetic, need little sleep, and are often happy and charming.8 But too much of a good thing can also wreak havoc on their life.
Bipolar depression (ie, depression in patients with a diagnosis of bipolar disorder) is treated differently from unipolar depression,3,9–13 making it especially important that clinicians recognize if a patient who presents with depression has a history of (hypo)manic symptoms.
CASE 1: THE IMPULSIVE NURSE
A 32-year-old nurse presents to her primary care provider with depressed mood. She reports having had a single depressive episode when she was a college freshman. Her family history includes depression, bipolar disorder, and schizophrenia, and her paternal grandfather and a maternal aunt committed suicide. Upon questioning, she reveals that in the past, she has had 3 episodes lasting several weeks and characterized by insubordinate behavior at work, irritability, high energy, and decreased need for sleep. She regrets impulsive sexual and financial decisions that she made during these episodes and recently filed for personal bankruptcy. For the past month, her mood has been persistently low, with reduced sleep, appetite, energy, and concentration, and with passive thoughts of suicide.
A CAREFUL HISTORY IS CRITICAL
This case illustrates many typical features of bipolar depression that are revealed only by taking a thorough history. Although the patient is high-functioning, having attained a professional career, she has serious problems with sexual and financial impulsivity and at her job. She has a strong family history of mood disorder. And she describes episodes of depression and mania in the past.
Starts in young adulthood, strong heritability
Bipolar disorder can be a devastating condition with lifelong consequences,14–20 especially as it typically starts when patients are getting an education or embarking on a career. It usually first manifests in the late teenage years and progresses in the patient’s early 20s.21,22 The first hospitalization can occur soon thereafter.23,24
Bipolar disorder is one of the most heritable conditions in psychiatry, and about 13% of children who have an afflicted parent develop it.25 In identical twins, the concordance is about 50% to 75%, indicating the importance of genetics and environmental factors.26,27
Associated with migraine, other conditions
The disorder is associated with a variety of conditions (Table 1).28,29 Some conditions (eg, thyroid disease) can cause mood cycling, and some (eg, sexually transmitted infections, accidents) are the consequences of the lifestyle that may accompany mania. For unknown reasons, migraine is highly associated with bipolar disorder.
DEPRESSION AND MANIA: TWO SIDES OF THE SAME COIN
Symptoms of depression and mania are frequently viewed as opposite mood states, though many times patients report a mixture of them.17,30–35 For both states, the features of a distinct change from the patient’s normal condition and the sustained nature of the symptoms are important diagnostically and indicate a likely underlying biological cause.
Major depressive disorder: Slowing down
The American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5),8 defines major depressive disorder as having either depressed mood or markedly diminished pleasure in most activities for most days during at least 2 weeks.
In addition, at least 4 of the following must be present during the same period:
- Appetite disturbance
- Sleep disturbance
- Motor retardation or agitation
- Lack of energy
- Feelings of worthlessness or excessive guilt
- Decreased concentration
- Recurrent thoughts of death or suicide.
An estimated 20% of the population experience a major depressive episode over their lifetime. A surprisingly high proportion of people with depression—30% to 40%—also have had subthreshold symptoms of mania (symptoms not meeting the criteria for hypomania or mania in terms of number of symptoms or duration).21,22 Because of these odds, it is important to suspect bipolar disorder even in patients who present with depression but who may not yet have manifested episodes of mania or hypomania.
Mood disorders can be regarded as falling into a spectrum, ranging from unipolar or “pure” major depression without any features of hypomania to major depression and severe mania.17,31–36
Mania: Speeding up
The DSM-5 defines mania as the presence of persistently elevated, expansive, or irritable mood with increased activity for more than 1 week. In addition, at least 3 of the following features must be present, with impaired functioning (4 features are required if mood is only irritable)8:
- Inflated self-esteem or grandiosity
- Decreased need for sleep
- Pressured speech
- Racing thoughts
- Distractibility
- Excessive involvement in pleasurable, high-risk activities.
Hypomania: No functional impairment
Hypomania is a less severe condition, in which the abnormally elevated mood is of shorter duration (4–7 days) and meets the other criteria for mania but without significant functional impairment. People may, in fact, be very functional and productive during hypomanic episodes.8
CLASSIFYING BIPOLAR DISORDER
Bipolar disorder is categorized according to severity.24,37,38 The most severe form, bipolar I disorder, is marked by major depression and manic episodes. It affects up to 1.5% of the US population, with equal proportions of men and women.39 Bipolar II disorder is less severe. It affects 0.8% to 1.6% of the US population, predominantly women.21,40 In bipolar II disorder, depression is more prominent, with episodes of hypomania.
Subthreshold bipolar disorders are characterized by episodic symptoms that do not meet the threshold for depression or hypomania; the symptoms are fewer or of shorter duration. These minor types of bipolar disorder affect up to 6% of the US population.17
Other conditions within the spectrum of bipolar and depressive disorders include medication- and substance-induced mania, agitated or anxious depression, and mixed states.31,34–36
DISTINGUISHING UNIPOLAR FROM BIPOLAR DEPRESSION
Considerable research has focused on finding a clear-cut clinical or biological feature to differentiate unipolar from bipolar depression, but so far none has been discovered. Distinguishing the two conditions still depends on clinical judgment. There are important reasons to identify the distinction between unipolar depression and bipolar disorder.
Prognosis differs. Bipolar disorder tends to be a more severe condition. Young people, who may initially present with only mild symptoms of mania, may develop serious episodes over the years. People may lose their savings, their marriage, and their career during a manic episode. The more critical the occupation (eg, doctor, pilot), the greater the potential consequences of impaired judgment brought on by even mild hypomania.14–20
Treatment differs. Typical antidepressants given for depression can trigger a manic episode in patients with bipolar depression, with devastating consequences. Atypical neuroleptic drugs used to treat bipolar disorder can also have serious effects (eg, metabolic and neurologic effects, including irreversible tardive dyskinesia).3,13,40–43
Despite the good reasons to do so, many doctors (including some psychiatrists) do not ask their patients about a propensity to mania or hypomania.4–6 More stigma is attached to the diagnosis of bipolar disorder than to depression44–47; once it is in the medical record, the patient may have problems with employment and obtaining medical insurance.17,44 The old term “manic-depressive” is often associated in the public mind with a person on the streets displaying severely psychotic behavior; the condition is now understood to consist of a spectrum from mild to more severe illness.
Clinical indicators of bipolarity
There are many indicators that a person who presents with depression may be on the bipolar spectrum, but this is not always easily identified.48–53
History of hypomanic symptoms or subthreshold manic symptoms. Although directly asking the patient about the defining symptoms (eg, “Have you ever had episodes of being ‘hyper’ or too happy?”) may help elicit the diagnosis, many patients with bipolar disorder only report depression, as it is psychically painful. In contrast, hypomania and even mania can be perceived as positive, as patients may have less insight into the abnormality of the condition and feel that they are functioning extremely well.
Early age of onset of a mood disorder, such as severe depression in childhood or early adulthood, points toward bipolar disorder. Diagnosing mood disorders in childhood is difficult, as children are less able to recognize or verbalize many of their symptoms.
Postpartum mood disorder, particularly with psychotic symptoms, indicates a strong possibility of a diagnosis of bipolar disorder.
Drug-induced mania, hypomania, and periods of hyperactivity are key features of bipolar disorder. If asked, patients may report feeling a “buzz” when taking an antidepressant.
Erratic patterns in work and relationships are a red flag and are viewed as “soft signs” of bipolar depression. Akiskal54 described the “rule of three” that should make one consider bipolar disorder: for example, three failed marriages, three current jobs or frequent job changes, three distinct professions practiced at the same time, and simultaneously dating three people. Such features indicate both the hyperfunctioning and the disruptive aspects of mania.
Family history of bipolar disorder or severe psychiatric illness is a very important clue. A more subtle clue described by Akiskal54 may be that several members of the family are very high-functioning in several different fields: eg, one may be a highly accomplished doctor, another a famous lawyer, and another a prominent politician. Or several members of the family may have erratic patterns of work and relationships. However, these subtle clues have been derived from clinical experiences and have not been validated in large-scale studies.
CASE 2: THE FRIENDLY SURGEON
Dr. Z is a prominent surgical subspecialist who is part of a small group practice. His wife has become increasingly worried about his behavior changes at home, including sleeping only a few hours a night, spending sprees, and binge drinking. He reluctantly agrees to an outpatient psychiatric evaluation if she attends with him. He creates a disturbance in the waiting room by shaking everyone’s hands and trying to hug all the women. During his examination, he is loud and expansive, denying he has any problems and describing himself as “the greatest doctor in the world.” The psychiatrist recommends hospitalization, but Dr. Z refuses and becomes belligerent. He announces that he just needs a career change and that he will fly to Mexico to open a bar.
This case, from the Texas Medical Association Archives,55 is not unusual. In addition to many characteristics discussed above, this case is typical in that the spouse brought the patient in, reflecting that the patient lacked insight that his behavior was abnormal. The disinhibition (hugging women), grandiosity, and unrealistic plans are also typical.
DIFFERENTIAL DIAGNOSIS OF BIPOLAR DEPRESSION
Anxiety disorders may be associated with dissociative speech or racing thoughts, which can be confused with bipolar illness. Personality disorders (eg, borderline, narcissistic, sociopathic) can involve a tumultuous and impulsive lifestyle resembling episodes of depression and mania. Schizoaffective illness has features of schizophrenia and bipolar disorder.
It is also possible that, despite what may look like mild features of bipolar disorder, there is no psychiatric condition. Some people with mild mania—often successful professionals or politicians—have high energy and can function very well with only a few hours of sleep. Similarly, depressive symptoms for short periods of time can be adaptive, such as in the face of a serious setback when extreme reflection and a period of inactivity can be useful, leading to subsequent reorganization.
A psychiatric diagnosis is usually made only when there is an abnormality, ie, the behavior is beyond normal limits, the person cannot control his or her symptoms, or social or occupational functioning is impaired.
SCREENING INSTRUMENTS
A few tools help determine the likelihood of bipolar disorder.
The Patient Health Questionnaire (PHQ-9)59,60 is a good 9-item screening tool for depression.
The Mood Disorder Questionnaire60 is specific for bipolar disorder, and like the PHQ-9, it is a patient-reported, short questionnaire that is available free online. The Mood Disorder Questionnaire asks about the symptoms of mania in a yes-no format. The result is positive if all of the following are present:
- A “yes” response to 7 of the 13 features
- Several features occur simultaneously
- The features are a moderate or serious problem.
Unlike most screening instruments, the Mood Disorder Questionnaire is more specific than sensitive. It is 93% specific for bipolar disorder in patients treated for depression in a primary care setting, but only 58% sensitive.61–63
WHEN TO REFER TO PSYCHIATRY
Patients suspected of having bipolar disorder or who have been previously diagnosed with it should be referred to a psychiatrist if they have certain features, including:
- Bipolar I disorder
- Psychotic symptoms
- Suicide risk or in danger of harming others
- Significantly impaired functioning
- Unclear diagnosis.
CASE 3: A TELEVISION ANCHOR’S DREAM TURNS TO NIGHTMARE
According to a famous news anchor’s autobiography,64 the steroids prescribed for her hives “revved her up.” The next course left her depressed. Antidepressant medications propelled her into a manic state, and she was soon planning a book, a television show, and a magazine all at once. During that time, she bought a cottage online. Her shyness evaporated at parties. “I was suddenly the equal of my high-energy friends who move fast and talk fast and loud,” she wrote. “I told everyone that I could understand why men felt like they could run the world, because I felt like that. This was a new me, and I liked her!”64 She was soon diagnosed with bipolar disorder and admitted to a psychiatric clinic.
TREAT WITH ANTIDEPRESSANTS, MOOD STABILIZERS
In general, acute bipolar disorder should be treated with a combination of an antidepressant and a mood stabilizer, and possibly an antipsychotic drug. An antidepressant should not be used alone, particularly with patients with a diagnosis of bipolar I disorder, because of the risk of triggering mania or the risk of faster cycling between mania and depression.13
Mood stabilizers include lithium, lamotrigine, and valproate. Each can prevent episodes of depression and mania. Lithium, which has been used as a mood stabilizer for 60 years, is specific for bipolar disorder, and it remains the best mood stabilizer treatment.
Antidepressants. The first-line antidepressant medication is bupropion, which is thought to be less likely to precipitate a manic episode,65 though all antidepressants have been associated with this side effect in patients with bipolar disorder. Other antidepressants—for example, selective serotonin reuptake inhibitors such as fluoxetine and dual reuptake inhibitors such as venlafaxine and duloxetine—can also be used. The precipitation of mania and possible increased mood cycling was first described with tricylic antidepressants, so drugs of this class should be used with caution.
Neuroleptic drugs such as aripiprazole, quetiapine, and lurasidone may be the easiest drugs to use, as they have antidepressant effects and can also prevent the occurrence of mania. These medications are frequently classified as mood stabilizers. However, they may not have true mood stabilizing properties such as that of lithium. Importantly, their use tends to entail significant metabolic problems and can lead to hyperlipidemia and diabetes. In addition, Parkinson disease-like symptoms— and in some cases irreversible involuntary movements of the mouth and tongue, as well as the body (tardive dyskinesia)—are important possible side effects.
All psychiatric medications have potential side effects (Table 3). Newer antidepressants and neuroleptics may have fewer side effects than older medications but are not more effective.
Should milder forms of bipolar depression be treated?
A dilemma is whether we should treat milder forms of bipolar depression, such as bipolar II depression, depression with subthreshold hypomania symptoms, or depression in persons with a strong family history of bipolar disorder.
Many doctors are justifiably reluctant to prescribe antidepressants for depression because of the risk of triggering mania. Although mood stabilizers such as lithium would counteract possible mania emergence, physicians often do not prescribe them because of inexperience and fear of risks and possible side effects. Patients are likewise resistant because they feel that use of mood stabilizers is tantamount to being told they are “manic-depressive,” with its associated stigma.
Overuse of atypical neuroleptics such as aripiprazole, quetiapine, and olanzapine has led to an awareness of metabolic syndrome and tardive dyskinesia, also making doctors cautious about using these drugs.
Answer: Yes, but treat with caution
Not treating depression consigns a patient to suffer with untreated depression, sometimes for years. Outcomes for patients with depression and bipolar disorder are often poor because the conditions are not recognized, and even when the conditions are recognized, doctors and patients may be reluctant to medicate appropriately. Medications should be used as needed to treat depression, but with an awareness of the possible side effects and with close patient monitoring.
A truly sustained manic state (unlike the brief euphoria brought on by some drugs) is not actually so easy to induce. In an unpublished Cleveland Clinic study, we monitored peaks of hypomanic symptoms in young patients (ages 15–30) during antidepressant treatment without mood stabilizers. About 30% to 40% of patients had subthreshold manic symptoms or a family history of bipolar disorder; 3 patients out of 51 developed hypomania leading to a change of diagnosis to bipolar disorder. Even in patients who had no risk factors for bipolar disorder, 2 out of 53 converted to a bipolar diagnosis. So conversion rates in patients with subthreshold bipolar disorder seem to be low, and the disorder can be identified early by monitoring the patient closely.
NONPHARMACOLOGIC TREATMENTS FOR DEPRESSION
Psychotherapy is indicated for all patients on medications for depression, as both pharmacologic and nonpharmacologic treatments are more effective when combined.66 Other treatments include transcranial magnetic stimulation, electroconvulsive therapy, light therapy, and exercise. Having a consistent daily routine, particularly regarding the sleep-wake schedule, is also helpful, and patients should be educated about its importance.
Patients presenting with depression commonly have undiagnosed bipolar depression,1–7 that is, depression with shifts to periods of mania. During manic or hypomanic episodes, people feel energetic, need little sleep, and are often happy and charming.8 But too much of a good thing can also wreak havoc on their life.
Bipolar depression (ie, depression in patients with a diagnosis of bipolar disorder) is treated differently from unipolar depression,3,9–13 making it especially important that clinicians recognize if a patient who presents with depression has a history of (hypo)manic symptoms.
CASE 1: THE IMPULSIVE NURSE
A 32-year-old nurse presents to her primary care provider with depressed mood. She reports having had a single depressive episode when she was a college freshman. Her family history includes depression, bipolar disorder, and schizophrenia, and her paternal grandfather and a maternal aunt committed suicide. Upon questioning, she reveals that in the past, she has had 3 episodes lasting several weeks and characterized by insubordinate behavior at work, irritability, high energy, and decreased need for sleep. She regrets impulsive sexual and financial decisions that she made during these episodes and recently filed for personal bankruptcy. For the past month, her mood has been persistently low, with reduced sleep, appetite, energy, and concentration, and with passive thoughts of suicide.
A CAREFUL HISTORY IS CRITICAL
This case illustrates many typical features of bipolar depression that are revealed only by taking a thorough history. Although the patient is high-functioning, having attained a professional career, she has serious problems with sexual and financial impulsivity and at her job. She has a strong family history of mood disorder. And she describes episodes of depression and mania in the past.
Starts in young adulthood, strong heritability
Bipolar disorder can be a devastating condition with lifelong consequences,14–20 especially as it typically starts when patients are getting an education or embarking on a career. It usually first manifests in the late teenage years and progresses in the patient’s early 20s.21,22 The first hospitalization can occur soon thereafter.23,24
Bipolar disorder is one of the most heritable conditions in psychiatry, and about 13% of children who have an afflicted parent develop it.25 In identical twins, the concordance is about 50% to 75%, indicating the importance of genetics and environmental factors.26,27
Associated with migraine, other conditions
The disorder is associated with a variety of conditions (Table 1).28,29 Some conditions (eg, thyroid disease) can cause mood cycling, and some (eg, sexually transmitted infections, accidents) are the consequences of the lifestyle that may accompany mania. For unknown reasons, migraine is highly associated with bipolar disorder.
DEPRESSION AND MANIA: TWO SIDES OF THE SAME COIN
Symptoms of depression and mania are frequently viewed as opposite mood states, though many times patients report a mixture of them.17,30–35 For both states, the features of a distinct change from the patient’s normal condition and the sustained nature of the symptoms are important diagnostically and indicate a likely underlying biological cause.
Major depressive disorder: Slowing down
The American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5),8 defines major depressive disorder as having either depressed mood or markedly diminished pleasure in most activities for most days during at least 2 weeks.
In addition, at least 4 of the following must be present during the same period:
- Appetite disturbance
- Sleep disturbance
- Motor retardation or agitation
- Lack of energy
- Feelings of worthlessness or excessive guilt
- Decreased concentration
- Recurrent thoughts of death or suicide.
An estimated 20% of the population experience a major depressive episode over their lifetime. A surprisingly high proportion of people with depression—30% to 40%—also have had subthreshold symptoms of mania (symptoms not meeting the criteria for hypomania or mania in terms of number of symptoms or duration).21,22 Because of these odds, it is important to suspect bipolar disorder even in patients who present with depression but who may not yet have manifested episodes of mania or hypomania.
Mood disorders can be regarded as falling into a spectrum, ranging from unipolar or “pure” major depression without any features of hypomania to major depression and severe mania.17,31–36
Mania: Speeding up
The DSM-5 defines mania as the presence of persistently elevated, expansive, or irritable mood with increased activity for more than 1 week. In addition, at least 3 of the following features must be present, with impaired functioning (4 features are required if mood is only irritable)8:
- Inflated self-esteem or grandiosity
- Decreased need for sleep
- Pressured speech
- Racing thoughts
- Distractibility
- Excessive involvement in pleasurable, high-risk activities.
Hypomania: No functional impairment
Hypomania is a less severe condition, in which the abnormally elevated mood is of shorter duration (4–7 days) and meets the other criteria for mania but without significant functional impairment. People may, in fact, be very functional and productive during hypomanic episodes.8
CLASSIFYING BIPOLAR DISORDER
Bipolar disorder is categorized according to severity.24,37,38 The most severe form, bipolar I disorder, is marked by major depression and manic episodes. It affects up to 1.5% of the US population, with equal proportions of men and women.39 Bipolar II disorder is less severe. It affects 0.8% to 1.6% of the US population, predominantly women.21,40 In bipolar II disorder, depression is more prominent, with episodes of hypomania.
Subthreshold bipolar disorders are characterized by episodic symptoms that do not meet the threshold for depression or hypomania; the symptoms are fewer or of shorter duration. These minor types of bipolar disorder affect up to 6% of the US population.17
Other conditions within the spectrum of bipolar and depressive disorders include medication- and substance-induced mania, agitated or anxious depression, and mixed states.31,34–36
DISTINGUISHING UNIPOLAR FROM BIPOLAR DEPRESSION
Considerable research has focused on finding a clear-cut clinical or biological feature to differentiate unipolar from bipolar depression, but so far none has been discovered. Distinguishing the two conditions still depends on clinical judgment. There are important reasons to identify the distinction between unipolar depression and bipolar disorder.
Prognosis differs. Bipolar disorder tends to be a more severe condition. Young people, who may initially present with only mild symptoms of mania, may develop serious episodes over the years. People may lose their savings, their marriage, and their career during a manic episode. The more critical the occupation (eg, doctor, pilot), the greater the potential consequences of impaired judgment brought on by even mild hypomania.14–20
Treatment differs. Typical antidepressants given for depression can trigger a manic episode in patients with bipolar depression, with devastating consequences. Atypical neuroleptic drugs used to treat bipolar disorder can also have serious effects (eg, metabolic and neurologic effects, including irreversible tardive dyskinesia).3,13,40–43
Despite the good reasons to do so, many doctors (including some psychiatrists) do not ask their patients about a propensity to mania or hypomania.4–6 More stigma is attached to the diagnosis of bipolar disorder than to depression44–47; once it is in the medical record, the patient may have problems with employment and obtaining medical insurance.17,44 The old term “manic-depressive” is often associated in the public mind with a person on the streets displaying severely psychotic behavior; the condition is now understood to consist of a spectrum from mild to more severe illness.
Clinical indicators of bipolarity
There are many indicators that a person who presents with depression may be on the bipolar spectrum, but this is not always easily identified.48–53
History of hypomanic symptoms or subthreshold manic symptoms. Although directly asking the patient about the defining symptoms (eg, “Have you ever had episodes of being ‘hyper’ or too happy?”) may help elicit the diagnosis, many patients with bipolar disorder only report depression, as it is psychically painful. In contrast, hypomania and even mania can be perceived as positive, as patients may have less insight into the abnormality of the condition and feel that they are functioning extremely well.
Early age of onset of a mood disorder, such as severe depression in childhood or early adulthood, points toward bipolar disorder. Diagnosing mood disorders in childhood is difficult, as children are less able to recognize or verbalize many of their symptoms.
Postpartum mood disorder, particularly with psychotic symptoms, indicates a strong possibility of a diagnosis of bipolar disorder.
Drug-induced mania, hypomania, and periods of hyperactivity are key features of bipolar disorder. If asked, patients may report feeling a “buzz” when taking an antidepressant.
Erratic patterns in work and relationships are a red flag and are viewed as “soft signs” of bipolar depression. Akiskal54 described the “rule of three” that should make one consider bipolar disorder: for example, three failed marriages, three current jobs or frequent job changes, three distinct professions practiced at the same time, and simultaneously dating three people. Such features indicate both the hyperfunctioning and the disruptive aspects of mania.
Family history of bipolar disorder or severe psychiatric illness is a very important clue. A more subtle clue described by Akiskal54 may be that several members of the family are very high-functioning in several different fields: eg, one may be a highly accomplished doctor, another a famous lawyer, and another a prominent politician. Or several members of the family may have erratic patterns of work and relationships. However, these subtle clues have been derived from clinical experiences and have not been validated in large-scale studies.
CASE 2: THE FRIENDLY SURGEON
Dr. Z is a prominent surgical subspecialist who is part of a small group practice. His wife has become increasingly worried about his behavior changes at home, including sleeping only a few hours a night, spending sprees, and binge drinking. He reluctantly agrees to an outpatient psychiatric evaluation if she attends with him. He creates a disturbance in the waiting room by shaking everyone’s hands and trying to hug all the women. During his examination, he is loud and expansive, denying he has any problems and describing himself as “the greatest doctor in the world.” The psychiatrist recommends hospitalization, but Dr. Z refuses and becomes belligerent. He announces that he just needs a career change and that he will fly to Mexico to open a bar.
This case, from the Texas Medical Association Archives,55 is not unusual. In addition to many characteristics discussed above, this case is typical in that the spouse brought the patient in, reflecting that the patient lacked insight that his behavior was abnormal. The disinhibition (hugging women), grandiosity, and unrealistic plans are also typical.
DIFFERENTIAL DIAGNOSIS OF BIPOLAR DEPRESSION
Anxiety disorders may be associated with dissociative speech or racing thoughts, which can be confused with bipolar illness. Personality disorders (eg, borderline, narcissistic, sociopathic) can involve a tumultuous and impulsive lifestyle resembling episodes of depression and mania. Schizoaffective illness has features of schizophrenia and bipolar disorder.
It is also possible that, despite what may look like mild features of bipolar disorder, there is no psychiatric condition. Some people with mild mania—often successful professionals or politicians—have high energy and can function very well with only a few hours of sleep. Similarly, depressive symptoms for short periods of time can be adaptive, such as in the face of a serious setback when extreme reflection and a period of inactivity can be useful, leading to subsequent reorganization.
A psychiatric diagnosis is usually made only when there is an abnormality, ie, the behavior is beyond normal limits, the person cannot control his or her symptoms, or social or occupational functioning is impaired.
SCREENING INSTRUMENTS
A few tools help determine the likelihood of bipolar disorder.
The Patient Health Questionnaire (PHQ-9)59,60 is a good 9-item screening tool for depression.
The Mood Disorder Questionnaire60 is specific for bipolar disorder, and like the PHQ-9, it is a patient-reported, short questionnaire that is available free online. The Mood Disorder Questionnaire asks about the symptoms of mania in a yes-no format. The result is positive if all of the following are present:
- A “yes” response to 7 of the 13 features
- Several features occur simultaneously
- The features are a moderate or serious problem.
Unlike most screening instruments, the Mood Disorder Questionnaire is more specific than sensitive. It is 93% specific for bipolar disorder in patients treated for depression in a primary care setting, but only 58% sensitive.61–63
WHEN TO REFER TO PSYCHIATRY
Patients suspected of having bipolar disorder or who have been previously diagnosed with it should be referred to a psychiatrist if they have certain features, including:
- Bipolar I disorder
- Psychotic symptoms
- Suicide risk or in danger of harming others
- Significantly impaired functioning
- Unclear diagnosis.
CASE 3: A TELEVISION ANCHOR’S DREAM TURNS TO NIGHTMARE
According to a famous news anchor’s autobiography,64 the steroids prescribed for her hives “revved her up.” The next course left her depressed. Antidepressant medications propelled her into a manic state, and she was soon planning a book, a television show, and a magazine all at once. During that time, she bought a cottage online. Her shyness evaporated at parties. “I was suddenly the equal of my high-energy friends who move fast and talk fast and loud,” she wrote. “I told everyone that I could understand why men felt like they could run the world, because I felt like that. This was a new me, and I liked her!”64 She was soon diagnosed with bipolar disorder and admitted to a psychiatric clinic.
TREAT WITH ANTIDEPRESSANTS, MOOD STABILIZERS
In general, acute bipolar disorder should be treated with a combination of an antidepressant and a mood stabilizer, and possibly an antipsychotic drug. An antidepressant should not be used alone, particularly with patients with a diagnosis of bipolar I disorder, because of the risk of triggering mania or the risk of faster cycling between mania and depression.13
Mood stabilizers include lithium, lamotrigine, and valproate. Each can prevent episodes of depression and mania. Lithium, which has been used as a mood stabilizer for 60 years, is specific for bipolar disorder, and it remains the best mood stabilizer treatment.
Antidepressants. The first-line antidepressant medication is bupropion, which is thought to be less likely to precipitate a manic episode,65 though all antidepressants have been associated with this side effect in patients with bipolar disorder. Other antidepressants—for example, selective serotonin reuptake inhibitors such as fluoxetine and dual reuptake inhibitors such as venlafaxine and duloxetine—can also be used. The precipitation of mania and possible increased mood cycling was first described with tricylic antidepressants, so drugs of this class should be used with caution.
Neuroleptic drugs such as aripiprazole, quetiapine, and lurasidone may be the easiest drugs to use, as they have antidepressant effects and can also prevent the occurrence of mania. These medications are frequently classified as mood stabilizers. However, they may not have true mood stabilizing properties such as that of lithium. Importantly, their use tends to entail significant metabolic problems and can lead to hyperlipidemia and diabetes. In addition, Parkinson disease-like symptoms— and in some cases irreversible involuntary movements of the mouth and tongue, as well as the body (tardive dyskinesia)—are important possible side effects.
All psychiatric medications have potential side effects (Table 3). Newer antidepressants and neuroleptics may have fewer side effects than older medications but are not more effective.
Should milder forms of bipolar depression be treated?
A dilemma is whether we should treat milder forms of bipolar depression, such as bipolar II depression, depression with subthreshold hypomania symptoms, or depression in persons with a strong family history of bipolar disorder.
Many doctors are justifiably reluctant to prescribe antidepressants for depression because of the risk of triggering mania. Although mood stabilizers such as lithium would counteract possible mania emergence, physicians often do not prescribe them because of inexperience and fear of risks and possible side effects. Patients are likewise resistant because they feel that use of mood stabilizers is tantamount to being told they are “manic-depressive,” with its associated stigma.
Overuse of atypical neuroleptics such as aripiprazole, quetiapine, and olanzapine has led to an awareness of metabolic syndrome and tardive dyskinesia, also making doctors cautious about using these drugs.
Answer: Yes, but treat with caution
Not treating depression consigns a patient to suffer with untreated depression, sometimes for years. Outcomes for patients with depression and bipolar disorder are often poor because the conditions are not recognized, and even when the conditions are recognized, doctors and patients may be reluctant to medicate appropriately. Medications should be used as needed to treat depression, but with an awareness of the possible side effects and with close patient monitoring.
A truly sustained manic state (unlike the brief euphoria brought on by some drugs) is not actually so easy to induce. In an unpublished Cleveland Clinic study, we monitored peaks of hypomanic symptoms in young patients (ages 15–30) during antidepressant treatment without mood stabilizers. About 30% to 40% of patients had subthreshold manic symptoms or a family history of bipolar disorder; 3 patients out of 51 developed hypomania leading to a change of diagnosis to bipolar disorder. Even in patients who had no risk factors for bipolar disorder, 2 out of 53 converted to a bipolar diagnosis. So conversion rates in patients with subthreshold bipolar disorder seem to be low, and the disorder can be identified early by monitoring the patient closely.
NONPHARMACOLOGIC TREATMENTS FOR DEPRESSION
Psychotherapy is indicated for all patients on medications for depression, as both pharmacologic and nonpharmacologic treatments are more effective when combined.66 Other treatments include transcranial magnetic stimulation, electroconvulsive therapy, light therapy, and exercise. Having a consistent daily routine, particularly regarding the sleep-wake schedule, is also helpful, and patients should be educated about its importance.
- Cicchetti D, Toth SL. The development of depression in children and adolescents. Am Psychol 1998; 53(2):221–241. doi:10.1037/0003-066X.53.2.221
- Glick ID. Undiagnosed bipolar disorder: new syndromes and new treatments. Prim Care Companion J Clin Psychiatry 2004;6(1):27–33. pmid:15486598
- Ghaemi SN, Boiman EE, Goodwin FK. Diagnosing bipolar disorder and the effect of antidepressants: a naturalistic study. J Clin Psychiatry 2000; 61(10):804–808. pmid:11078046
- Singh T, Rajput M. Misdiagnosis of bipolar disorder. Psychiatry (Edgmont) 2006; 3(10):57–63. pmid: 20877548
- Lish JD, Dime-Meenan S, Whybrow PC, Price RA, Hirschfeld RM. The National Depressive and Manic-depressive Association (DMDA) survey of bipolar members. J Affect Disord 1994; 31(4):281–294. pmid:7989643
- Howes OD, Falkenberg I. Early detection and intervention in bipolar affective disorder: targeting the development of the disorder. Curr Psychiatry Rep 2011; 13(6):493–499. pmid:21850462
- Ghaemi SN, Sachs GS, Chiou AM, Pandurangi AK, Goodwin K. Is bipolar disorder still underdiagnosed? Are antidepressants overutilized? J Affect Disord 1999; 52(1–3):135–144. pmid:10357026
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-5. Washington, DC: American Psychiatric Publishing, 2013.
- Hlastala SA, Frank E, Mallinger AG, Thase ME, Ritenour AM, Kupfer DJ. Bipolar depression: an underestimated treatment challenge. Depress Anxiety 1997; 5(2):73–83. pmid:9262937
- Smith DJ, Craddock N. Unipolar and bipolar depression: different or the same? Br J Psychiatry 2011; 199(4):272–274. doi:10.1192/bjp.bp.111.092726
- Viktorin A, Lichtenstein P, Thase ME, et al. The risk of switch to mania in patients with bipolar disorder during treatment with an antidepressant alone and in combination with a mood stabilizer. Am J Psychiatry 2014; 171(10):1067–1073. doi:10.1176/appi.ajp.2014.13111501
- Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med 2007; 356(17):1711–1722. doi:10.1056/NEJMoa064135
- American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry 2002; 159(4 suppl):1–50. pmid:11958165
- Leonpacher AK, Liebers D, Pirooznia M, et al. Distinguishing bipolar from unipolar depression: the importance of clinical symptoms and illness features. Psychol Med 2015; 45(11):2437–2446. doi:10.1017/S0033291715000446
- Angst J, Gamma A, Benazzi F, Ajdacic V, Eich D, Rössler W. Toward a re-definition of subthreshold bipolarity: epidemiology and proposed criteria for bipolar-II, minor bipolar disorders and hypomania. J Affect Disord 2003; 73(1–2):133–146. pmid:12507746
- Faravelli C, Rosi S, Alessandra Scarpato M, Lampronti L, Amedei SG, Rana N. Threshold and subthreshold bipolar disorders in the Sesto Fiorentino Study. J Affect Disord 2006; 94(1–3):111–119. pmid:16701902
- Judd LL, Akiskal HS. The prevalence and disability of bipolar spectrum disorders in the US population: re-analysis of the ECA database taking into account subthreshold cases. J Affect Disord 2003; 73(1–2):123–131. pmid:12507745
- Park Y-M, Lee B-H. Treatment response in relation to subthreshold bipolarity in patients with major depressive disorder receiving antidepressant monotherapy: a post hoc data analysis (KOMDD study). Neuropsychiatr Dis Treat 2016; 12:1221–1227. doi:10.2147/NDT.S104188
- Perlis RH, Uher R, Ostacher M, Goldberg JF, et al. Association between bipolar spectrum features and treatment outcomes in outpatients with major depressive disorder. Arch Gen Psychiatry 2011; 68(4):351–360. doi:10.1001/archgenpsychiatry.2010.179
- Dudek D, Siwek M, Zielin´ska D, Jaeschke R, Rybakowski J. Diagnostic conversions from major depressive disorder into bipolar disorder in an outpatient setting: results of a retrospective chart review. J Affect Disord 2013; 144(1–2):112–115. doi:10.1016/j.jad.2012.06.014
- Angst J, Cui L, Swendsen J, et al. Major depressive disorder with subthreshold bipolarity in the National Comorbidity Survey Replication. Am J Psychiatry 2010; 167(10):1194–1201. doi:10.1176/appi.ajp.2010.09071011
- Zimmermann P, Brückl T, Nocon A, et al. Heterogeneity of DSM-IV major depressive disorder as a consequence of subthreshold bipolarity. Arch Gen Psychiatry 2009; 66(12):1341–1352. doi:10.1001/archgenpsychiatry.2009.158
- Patel NC, DelBello MP, Keck PE, Strakowski SM. Phenomenology associated with age at onset in patients with bipolar disorder at their first psychiatric hospitalization. Bipolar Disord 2006; 8(1):91–94. pmid:16411986
- Hirschfeld RMA, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the National Depressive and Manic-Depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry 2003; 64(2):161–174. pmid:12633125
- Craddock N, Jones I. Genetics of bipolar disorder. J Med Genet 1999; 36(8):585–594. pmid:10465107
- Griswold KS, Pessar LF. Management of bipolar disorder. Am Fam Physician 2000; 62(6):1343–1358. pmid:11011863
- Kerner B. Genetics of bipolar disorder. Appl Clin Genet 2014; 7:33–42. doi:10.2147/TACG.S39297
- Scheffer RE, Linden S. Concurrent medical conditions with pediatric bipolar disorder. Curr Opin Psychiatry 2007; 20(4):398–401. doi:10.1097/YCO.0b013e3281a305c3
- Carney CP, Jones LE. Medical comorbidity in women and men with bipolar disorders: a population-based controlled study. Psychosom Med 2006;68(5):684–691. doi:10.1097/01.psy.0000237316.09601.88
- Cassano GB, Akiskal HS, Savino M, Musetti L, Perugi G. Proposed subtypes of bipolar II and related disorders: with hypomanic episodes (or cyclothymia) and with hyperthymic temperament. J Affect Disord 1992; 26(2):127–140. pmid:1447430
- Akiskal HS, Mallya G. Criteria for the “soft” bipolar spectrum: treatment implications. Psychopharmacol Bull 1987; 23(1):68–73. pmid:3602332
- Fiedorowicz JG, Endicott J, Leon AC, Solomon DA, Keller MB, Coryell WH. Subthreshold hypomanic symptoms in progression from unipolar major depression to bipolar disorder. Am J Psychiatry 2011; 168(1):40–48. doi:10.1176/appi.ajp.2010.10030328
- Maj M, Pirozzi R, Magliano L, Fiorillo A, Bartoli L. Agitated “unipolar” major depression: prevalence, phenomenology, and outcome. J Clin Psychiatry 2006; 67(5):712–719. pmid:16841620
- Akiskal HS. The bipolar spectrum: new concepts in classification and diagnosis. In: Psychiatry Update: the American Psychiatric Association Annual Review. Washington, DC: American Psychiatric Press; 1983:271–292.
- Akiskal HS, Pinto O. The evolving bipolar spectrum. Prototypes I, II, III, and IV. Psychiatr Clin North Am 1999; 22(3):517–534. pmid:10550853
- Cassano GB, Savino M, Perugi G, Musetti L, Akiskal HS. Major depressive episode: unipolar and bipolar II. Encephale 1992 Jan;18 Spec No:15–18. pmid:1600898
- Müller-Oerlinghausen B, Berghöfer A, Bauer M. Bipolar disorder. Lancet 2002; 359(9302):241–247. pmid:11812578
- Hirschfeld RMA, Calabrese JR, Weissman MM, et al. Screening for bipolar disorder in the community. J Clin Psychiatry 2003; 64(1):53–59. pmid:12590624
- Blanco C, Compton WM, Saha TD, et al. Epidemiology of DSM-5 bipolar I disorder: results from the National Epidemiologic survey on Alcohol and Related Conditions—III. J Psychiatr Res 2017; 84:310–317. doi:10.1016/j.jpsychires.2016.10.003
- McGirr A, Vöhringer PA, Ghaemi SN, Lam RW, Yatham LN. Safety and efficacy of adjunctive second-generation antidepressant therapy with a mood stabiliser or an atypical antipsychotic in acute bipolar depression: a systematic review and meta-analysis of randomised placebo-controlled trials. Lancet Psychiatry 2016; 3(12):1138–1146. doi:10.1016/S2215-0366(16)30264-4
- Gijsman HJ, Geddes JR, Rendell JM, Nolen WA, Goodwin GM. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry 2004; 161(9):1537–1547. doi:10.1176/appi.ajp.161.9.1537
- Sidor MM, Macqueen GM. Antidepressants for the acute treatment of bipolar depression: a systematic review and meta-analysis. J Clin Psychiatry 2011; 72(2):156–167. doi:10.4088/JCP.09r05385gre
- Liu B, Zhang Y, Fang H, Liu J, Liu T, Li L. Efficacy and safety of long-term antidepressant treatment for bipolar disorders - A meta-analysis of randomized controlled trials. J Affect Disord 2017; 223(139):41–48. doi:10.1016/j.jad.2017.07.023
- Krupa T, Kirsh B, Cockburn L, Gewurtz R. Understanding the stigma of mental illness in employment. Work 2009; 33(4):413–425. doi:10.3233/WOR-2009-0890
- Hawke LD, Parikh SV, Michalak EE. Stigma and bipolar disorder: a review of the literature. J Affect Disord 2013; 150(2):181–191. doi:10.1016/j.jad.2013.05.030
- Cerit C, Filizer A, Tural Ü, Tufan AE. Stigma: a core factor on predicting functionality in bipolar disorder. Compr Psychiatry 2012; 53(5):484–489. doi:10.1016/j.comppsych.2011.08.010
- O’Donnell L, Himle JA, Ryan K, et al. Social aspects of the workplace among individuals with bipolar disorder. J Soc Social Work Res 2017; 8(3):379–398. doi:10.1086/693163
- Akiskal HS, Maser JD, Zeller PJ, et al. Switching from “unipolar” to bipolar II. An 11-year prospective study of clinical and temperamental predictors in 559 patients. Arch Gen Psychiatry 1995; 52(2):114–123. pmid:7848047
- Kroon JS, Wohlfarth TD, Dieleman J, et al. Incidence rates and risk factors of bipolar disorder in the general population: a population-based cohort study. Bipolar Disord 2013; 15(3):306–313. doi:10.1111/bdi.12058
- Fiedorowicz JG, Endicott J, Leon AC, Solomon DA, Keller MB, Coryell WH. Subthreshold hypomanic symptoms in progression from unipolar major depression to bipolar disorder. Am J Psychiatry 2011; 168(1):40–48. doi:10.1176/appi.ajp.2010.10030328
- Akiskal HS, Walker P, Puzantian VR, King D, Rosenthal TL, Dranon M. Bipolar outcome in the course of depressive illness. Phenomenologic, familial, and pharmacologic predictors. J Affect Disord 1983; 5(2):115–128. pmid:6222091
- Strober M, Carlson G. Bipolar illness in adolescents with major depression: clinical, genetic, and psychopharmacologic predictors in a three- to four-year prospective follow-up investigation. Arch Gen Psychiatry 1982; 39(5):549–555. pmid:7092488
- Goldberg JF, Harrow M, Whiteside JE. Risk for bipolar illness in patients initially hospitalized for unipolar depression. Am J Psychiatry 2001; 158(8):1265–1270. pmid:11481161
- Akiskal HS. Searching for behavioral indicators of bipolar II in patients presenting with major depressive episodes: the “red sign,” the “rule of three” and other biographic signs of temperamental extravagance, activation and hypomania. J Affect Disord 2005; 84(2–3):279–290. pmid:15708427
- Texas Medical Association. Mood disorders in physicians. www.texmed.org/Template.aspx?id=6833. Accessed June 7, 2018.
- Hirschfeld RM. Differential diagnosis of bipolar disorder and major depressive disorder. J Affect Disord 2014;169(suppl 1):S12–S16. doi:10.1016/S0165-0327(14)70004-7
- Dunner DL. Differential diagnosis of bipolar disorder. J Clin Psychopharmacol 1992; 12(1suppl):7S–12S. pmid:1541721
- Peet M, Peters S. Drug-induced mania. Drug Saf 1995; 12(2):146–153. pmid:7766338
- Spitzer RL, Kroenke K, Williams JBW. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA 1999; 282(18):1737–1744. pmid:10568646
- Kroenke K, Spitzer RL, Williams JBW. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001; 16(9):606–613. pmid:11556941
- Hirschfeld RMMA, Williams JBBW, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: the Mood Disorder Questionnaire. Am J Psychiatry 2000; 157(11)1873–1875. doi:10.1176/appi.ajp.157.11.1873
- Hirschfeld RMA. The Mood Disorder Questionnaire: a simple, patient-rated screening instrument for bipolar disorder. Prim Care Companion J Clin Psychiatry 2002; 4(1):9–11. pmid: 15014728
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293(8):956–963. doi:10.1001/jama.293.8.956
- Pauley J. Skywriting: A Life Out of the Blue. New York: Random House, 2004.
- Goren JL, Levin GM. Mania with bupropion: a dose-related phenomenon? Ann Pharmacother 2000; 34(5):619–621. doi:10.1345/aph.19313
- Swann AC. Long-term treatment in bipolar disorder. J Clin Psychiatry 2005; 66(suppl 1):7–12. pmid:15693746
- Cicchetti D, Toth SL. The development of depression in children and adolescents. Am Psychol 1998; 53(2):221–241. doi:10.1037/0003-066X.53.2.221
- Glick ID. Undiagnosed bipolar disorder: new syndromes and new treatments. Prim Care Companion J Clin Psychiatry 2004;6(1):27–33. pmid:15486598
- Ghaemi SN, Boiman EE, Goodwin FK. Diagnosing bipolar disorder and the effect of antidepressants: a naturalistic study. J Clin Psychiatry 2000; 61(10):804–808. pmid:11078046
- Singh T, Rajput M. Misdiagnosis of bipolar disorder. Psychiatry (Edgmont) 2006; 3(10):57–63. pmid: 20877548
- Lish JD, Dime-Meenan S, Whybrow PC, Price RA, Hirschfeld RM. The National Depressive and Manic-depressive Association (DMDA) survey of bipolar members. J Affect Disord 1994; 31(4):281–294. pmid:7989643
- Howes OD, Falkenberg I. Early detection and intervention in bipolar affective disorder: targeting the development of the disorder. Curr Psychiatry Rep 2011; 13(6):493–499. pmid:21850462
- Ghaemi SN, Sachs GS, Chiou AM, Pandurangi AK, Goodwin K. Is bipolar disorder still underdiagnosed? Are antidepressants overutilized? J Affect Disord 1999; 52(1–3):135–144. pmid:10357026
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-5. Washington, DC: American Psychiatric Publishing, 2013.
- Hlastala SA, Frank E, Mallinger AG, Thase ME, Ritenour AM, Kupfer DJ. Bipolar depression: an underestimated treatment challenge. Depress Anxiety 1997; 5(2):73–83. pmid:9262937
- Smith DJ, Craddock N. Unipolar and bipolar depression: different or the same? Br J Psychiatry 2011; 199(4):272–274. doi:10.1192/bjp.bp.111.092726
- Viktorin A, Lichtenstein P, Thase ME, et al. The risk of switch to mania in patients with bipolar disorder during treatment with an antidepressant alone and in combination with a mood stabilizer. Am J Psychiatry 2014; 171(10):1067–1073. doi:10.1176/appi.ajp.2014.13111501
- Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med 2007; 356(17):1711–1722. doi:10.1056/NEJMoa064135
- American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry 2002; 159(4 suppl):1–50. pmid:11958165
- Leonpacher AK, Liebers D, Pirooznia M, et al. Distinguishing bipolar from unipolar depression: the importance of clinical symptoms and illness features. Psychol Med 2015; 45(11):2437–2446. doi:10.1017/S0033291715000446
- Angst J, Gamma A, Benazzi F, Ajdacic V, Eich D, Rössler W. Toward a re-definition of subthreshold bipolarity: epidemiology and proposed criteria for bipolar-II, minor bipolar disorders and hypomania. J Affect Disord 2003; 73(1–2):133–146. pmid:12507746
- Faravelli C, Rosi S, Alessandra Scarpato M, Lampronti L, Amedei SG, Rana N. Threshold and subthreshold bipolar disorders in the Sesto Fiorentino Study. J Affect Disord 2006; 94(1–3):111–119. pmid:16701902
- Judd LL, Akiskal HS. The prevalence and disability of bipolar spectrum disorders in the US population: re-analysis of the ECA database taking into account subthreshold cases. J Affect Disord 2003; 73(1–2):123–131. pmid:12507745
- Park Y-M, Lee B-H. Treatment response in relation to subthreshold bipolarity in patients with major depressive disorder receiving antidepressant monotherapy: a post hoc data analysis (KOMDD study). Neuropsychiatr Dis Treat 2016; 12:1221–1227. doi:10.2147/NDT.S104188
- Perlis RH, Uher R, Ostacher M, Goldberg JF, et al. Association between bipolar spectrum features and treatment outcomes in outpatients with major depressive disorder. Arch Gen Psychiatry 2011; 68(4):351–360. doi:10.1001/archgenpsychiatry.2010.179
- Dudek D, Siwek M, Zielin´ska D, Jaeschke R, Rybakowski J. Diagnostic conversions from major depressive disorder into bipolar disorder in an outpatient setting: results of a retrospective chart review. J Affect Disord 2013; 144(1–2):112–115. doi:10.1016/j.jad.2012.06.014
- Angst J, Cui L, Swendsen J, et al. Major depressive disorder with subthreshold bipolarity in the National Comorbidity Survey Replication. Am J Psychiatry 2010; 167(10):1194–1201. doi:10.1176/appi.ajp.2010.09071011
- Zimmermann P, Brückl T, Nocon A, et al. Heterogeneity of DSM-IV major depressive disorder as a consequence of subthreshold bipolarity. Arch Gen Psychiatry 2009; 66(12):1341–1352. doi:10.1001/archgenpsychiatry.2009.158
- Patel NC, DelBello MP, Keck PE, Strakowski SM. Phenomenology associated with age at onset in patients with bipolar disorder at their first psychiatric hospitalization. Bipolar Disord 2006; 8(1):91–94. pmid:16411986
- Hirschfeld RMA, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the National Depressive and Manic-Depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry 2003; 64(2):161–174. pmid:12633125
- Craddock N, Jones I. Genetics of bipolar disorder. J Med Genet 1999; 36(8):585–594. pmid:10465107
- Griswold KS, Pessar LF. Management of bipolar disorder. Am Fam Physician 2000; 62(6):1343–1358. pmid:11011863
- Kerner B. Genetics of bipolar disorder. Appl Clin Genet 2014; 7:33–42. doi:10.2147/TACG.S39297
- Scheffer RE, Linden S. Concurrent medical conditions with pediatric bipolar disorder. Curr Opin Psychiatry 2007; 20(4):398–401. doi:10.1097/YCO.0b013e3281a305c3
- Carney CP, Jones LE. Medical comorbidity in women and men with bipolar disorders: a population-based controlled study. Psychosom Med 2006;68(5):684–691. doi:10.1097/01.psy.0000237316.09601.88
- Cassano GB, Akiskal HS, Savino M, Musetti L, Perugi G. Proposed subtypes of bipolar II and related disorders: with hypomanic episodes (or cyclothymia) and with hyperthymic temperament. J Affect Disord 1992; 26(2):127–140. pmid:1447430
- Akiskal HS, Mallya G. Criteria for the “soft” bipolar spectrum: treatment implications. Psychopharmacol Bull 1987; 23(1):68–73. pmid:3602332
- Fiedorowicz JG, Endicott J, Leon AC, Solomon DA, Keller MB, Coryell WH. Subthreshold hypomanic symptoms in progression from unipolar major depression to bipolar disorder. Am J Psychiatry 2011; 168(1):40–48. doi:10.1176/appi.ajp.2010.10030328
- Maj M, Pirozzi R, Magliano L, Fiorillo A, Bartoli L. Agitated “unipolar” major depression: prevalence, phenomenology, and outcome. J Clin Psychiatry 2006; 67(5):712–719. pmid:16841620
- Akiskal HS. The bipolar spectrum: new concepts in classification and diagnosis. In: Psychiatry Update: the American Psychiatric Association Annual Review. Washington, DC: American Psychiatric Press; 1983:271–292.
- Akiskal HS, Pinto O. The evolving bipolar spectrum. Prototypes I, II, III, and IV. Psychiatr Clin North Am 1999; 22(3):517–534. pmid:10550853
- Cassano GB, Savino M, Perugi G, Musetti L, Akiskal HS. Major depressive episode: unipolar and bipolar II. Encephale 1992 Jan;18 Spec No:15–18. pmid:1600898
- Müller-Oerlinghausen B, Berghöfer A, Bauer M. Bipolar disorder. Lancet 2002; 359(9302):241–247. pmid:11812578
- Hirschfeld RMA, Calabrese JR, Weissman MM, et al. Screening for bipolar disorder in the community. J Clin Psychiatry 2003; 64(1):53–59. pmid:12590624
- Blanco C, Compton WM, Saha TD, et al. Epidemiology of DSM-5 bipolar I disorder: results from the National Epidemiologic survey on Alcohol and Related Conditions—III. J Psychiatr Res 2017; 84:310–317. doi:10.1016/j.jpsychires.2016.10.003
- McGirr A, Vöhringer PA, Ghaemi SN, Lam RW, Yatham LN. Safety and efficacy of adjunctive second-generation antidepressant therapy with a mood stabiliser or an atypical antipsychotic in acute bipolar depression: a systematic review and meta-analysis of randomised placebo-controlled trials. Lancet Psychiatry 2016; 3(12):1138–1146. doi:10.1016/S2215-0366(16)30264-4
- Gijsman HJ, Geddes JR, Rendell JM, Nolen WA, Goodwin GM. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry 2004; 161(9):1537–1547. doi:10.1176/appi.ajp.161.9.1537
- Sidor MM, Macqueen GM. Antidepressants for the acute treatment of bipolar depression: a systematic review and meta-analysis. J Clin Psychiatry 2011; 72(2):156–167. doi:10.4088/JCP.09r05385gre
- Liu B, Zhang Y, Fang H, Liu J, Liu T, Li L. Efficacy and safety of long-term antidepressant treatment for bipolar disorders - A meta-analysis of randomized controlled trials. J Affect Disord 2017; 223(139):41–48. doi:10.1016/j.jad.2017.07.023
- Krupa T, Kirsh B, Cockburn L, Gewurtz R. Understanding the stigma of mental illness in employment. Work 2009; 33(4):413–425. doi:10.3233/WOR-2009-0890
- Hawke LD, Parikh SV, Michalak EE. Stigma and bipolar disorder: a review of the literature. J Affect Disord 2013; 150(2):181–191. doi:10.1016/j.jad.2013.05.030
- Cerit C, Filizer A, Tural Ü, Tufan AE. Stigma: a core factor on predicting functionality in bipolar disorder. Compr Psychiatry 2012; 53(5):484–489. doi:10.1016/j.comppsych.2011.08.010
- O’Donnell L, Himle JA, Ryan K, et al. Social aspects of the workplace among individuals with bipolar disorder. J Soc Social Work Res 2017; 8(3):379–398. doi:10.1086/693163
- Akiskal HS, Maser JD, Zeller PJ, et al. Switching from “unipolar” to bipolar II. An 11-year prospective study of clinical and temperamental predictors in 559 patients. Arch Gen Psychiatry 1995; 52(2):114–123. pmid:7848047
- Kroon JS, Wohlfarth TD, Dieleman J, et al. Incidence rates and risk factors of bipolar disorder in the general population: a population-based cohort study. Bipolar Disord 2013; 15(3):306–313. doi:10.1111/bdi.12058
- Fiedorowicz JG, Endicott J, Leon AC, Solomon DA, Keller MB, Coryell WH. Subthreshold hypomanic symptoms in progression from unipolar major depression to bipolar disorder. Am J Psychiatry 2011; 168(1):40–48. doi:10.1176/appi.ajp.2010.10030328
- Akiskal HS, Walker P, Puzantian VR, King D, Rosenthal TL, Dranon M. Bipolar outcome in the course of depressive illness. Phenomenologic, familial, and pharmacologic predictors. J Affect Disord 1983; 5(2):115–128. pmid:6222091
- Strober M, Carlson G. Bipolar illness in adolescents with major depression: clinical, genetic, and psychopharmacologic predictors in a three- to four-year prospective follow-up investigation. Arch Gen Psychiatry 1982; 39(5):549–555. pmid:7092488
- Goldberg JF, Harrow M, Whiteside JE. Risk for bipolar illness in patients initially hospitalized for unipolar depression. Am J Psychiatry 2001; 158(8):1265–1270. pmid:11481161
- Akiskal HS. Searching for behavioral indicators of bipolar II in patients presenting with major depressive episodes: the “red sign,” the “rule of three” and other biographic signs of temperamental extravagance, activation and hypomania. J Affect Disord 2005; 84(2–3):279–290. pmid:15708427
- Texas Medical Association. Mood disorders in physicians. www.texmed.org/Template.aspx?id=6833. Accessed June 7, 2018.
- Hirschfeld RM. Differential diagnosis of bipolar disorder and major depressive disorder. J Affect Disord 2014;169(suppl 1):S12–S16. doi:10.1016/S0165-0327(14)70004-7
- Dunner DL. Differential diagnosis of bipolar disorder. J Clin Psychopharmacol 1992; 12(1suppl):7S–12S. pmid:1541721
- Peet M, Peters S. Drug-induced mania. Drug Saf 1995; 12(2):146–153. pmid:7766338
- Spitzer RL, Kroenke K, Williams JBW. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA 1999; 282(18):1737–1744. pmid:10568646
- Kroenke K, Spitzer RL, Williams JBW. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001; 16(9):606–613. pmid:11556941
- Hirschfeld RMMA, Williams JBBW, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: the Mood Disorder Questionnaire. Am J Psychiatry 2000; 157(11)1873–1875. doi:10.1176/appi.ajp.157.11.1873
- Hirschfeld RMA. The Mood Disorder Questionnaire: a simple, patient-rated screening instrument for bipolar disorder. Prim Care Companion J Clin Psychiatry 2002; 4(1):9–11. pmid: 15014728
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293(8):956–963. doi:10.1001/jama.293.8.956
- Pauley J. Skywriting: A Life Out of the Blue. New York: Random House, 2004.
- Goren JL, Levin GM. Mania with bupropion: a dose-related phenomenon? Ann Pharmacother 2000; 34(5):619–621. doi:10.1345/aph.19313
- Swann AC. Long-term treatment in bipolar disorder. J Clin Psychiatry 2005; 66(suppl 1):7–12. pmid:15693746
KEY POINTS
- Bipolar depression in its manifest and subthreshold forms is nearly as prevalent as unipolar depression and often occurs in successful professionals.
- A manic or hypomanic episode can make a patient highly productive, but it can also be severely disruptive, leading to loss of job, marriage, and financial savings.
- Identifying bipolar depression depends on asking about bipolar symptoms, using screening instruments, and being aware of clues from the patient’s history.
- A major depressive episode in patients with a history of mania or hypomania should be treated with a combination of an antidepressant and a mood stabilizer or a mood stabilizer alone.

Which patients with a parapneumonic effusion need a chest tube?
Hospitalized patients with pneumonia who develop a complicated parapneumonic effusion or empyema need to undergo chest tube placement.
WHAT IS PARAPNEUMONIC EFFUSION?
Parapneumonic effusion is a pleural effusion that forms concurrently with bacterial or viral pneumonia. Up to 40% of patients hospitalized with pneumonia develop a parapneumonic effusion.1 The effusion progresses through a continuum of 3 stages: uncomplicated, complicated, and empyema.
Uncomplicated parapneumonic effusion is an exudative effusion without bacteria or pus that is caused by movement of fluid and neutrophils into the pleural space. Pneumonia itself causes an increase in interstitial fluid and capillary leakage. The effusion becomes complicated as a result of bacteria invading the pleural space, causing a further increase in neutrophils in the pleural fluid. Empyema is defined as the presence of frank pus in the pleural space.
CLINICAL SIGNIFICANCE
According to the US Centers for Disease Control and Prevention, pneumonia accounts for 674,000 emergency department visits each year; of the patients hospitalized, up to 40% develop a parapneumonic effusion.2 The only study done on rates of death associated with parapneumonic effusion showed that, compared with patients with no effusion, the risk of death was 3.7 times higher with a unilateral effusion and 6.5 times higher with bilateral effusions.3
INITIAL EVALUATION
The initial evaluation of suspected parapneumonic effusion should include chest radiography with lateral or decubitus views, followed by thoracentesis if indicated. If thoracentesis is performed, the fluid should be tested as follows:
- Gram stain
- Appropriate cultures based on clinical scenario (eg, aerobic, anaerobic, fungal)
- Total protein in pleural fluid and serum
- Lactate dehydrogenase (LDH) in pleural fluid and serum
- Glucose
- pH.
CLASSIFICATION OF EFFUSIONS
When pleural fluid is obtained, the total protein and LDH levels are used to categorize the effusion as either transudative or exudative based on the Light criteria.4 An effusion is confirmed as exudative when 1 of the following 3 criteria is met:
- The ratio of pleural fluid protein to serum protein is greater than 0.5
- The ratio of pleural fluid LDH to serum LDH is greater than 0.6
- The pleural fluid LDH is greater than two-thirds the upper limit of normal for the serum LDH.

Category 1 effusions are defined as free- flowing fluid with a thickness of less than 10 mm on any imaging modality. Thoracentesis for pleural fluid analysis is not required. The prognosis is very good.
Category 2 effusions are defined as free- flowing fluid with a thickness greater than 10 mm and less than 50% of the hemithorax. Thoracentesis is typically done because of the size of the effusion, but Gram stain and culture of the pleural fluid are usually negative, and the pH is at least 7.2. The prognosis is good.
Category 3 effusions are considered complicated because the anatomy of the pleural space becomes altered or because bacteria have invaded the pleural space. The effusion is larger than 50% of the hemithorax or is loculated, or the parietal pleura is thickened. Since the bacteria have invaded the pleural space, Gram stain or culture of pleural fluid may be positive, the pleural fluid pH may be less than 7.2, or the glucose level of the fluid may be less than 60 mg/dL. The prognosis for category 3 is poor.
Category 4 effusions are defined as empyema. The only characteristic that separates this from category 3 is frank pus in the pleural space. The prognosis is very poor.
TO PLACE A CHEST TUBE OR NOT
For category 1 or 2 effusions, treatment with antibiotics alone is typically enough. Category 3 effusions usually do not respond to antibiotics alone and may require complete drainage of the fluid with or without a chest tube depending on whether loculations are present, as loculations are difficult to drain with a chest tube. Category 4 effusions require both antibiotics and chest tube placement.
WHAT TYPE OF CHEST TUBE?
Studies have shown that small-bore chest tubes (< 20 F) are as efficacious as larger tubes (≥ 20 F) for the treatment of complicated parapneumonic effusion and empyema.6,7 Studies have also shown that the size of the tube makes no difference in the time needed to drain the effusion, the length of hospital stay, or the complication rate.8,9 Based on these studies, a small-bore chest tube should be placed first when clinically appropriate. When a chest tube is placed for empyema, computed tomography should be performed within 24 hours to confirm proper tube placement.
ADVANCED THERAPIES FOR EMPYEMA
Empyema treatment fails when antibiotic coverage is inadequate or when a loculation is not drained appropriately. Options if treatment fails include instillation of fibrinolytics into the pleural space, video-assisted thorascopic surgery, and decortication.
The role of fibrinolytics has not been well-established, but fibrinolytics should be considered in loculated effusions or empyema, or if drainage of the effusion slows.10 Video-assisted thorascopic surgery is reserved for effusions that are incompletely drained with a chest tube with or without fibrinolytics; studies have shown shorter hospital length of stay and higher treatment efficacy when this is performed earlier for loculated effusions.11 Decortication is reserved for symptomatic patients who have a thickened pleura more than 6 months after the initial infection.12 Timing for each of these procedures is not clearly defined and so must be individualized.
TAKE-AWAY POINTS
- Parapneumonic effusion occurs concurrently with pneumonia and with a high frequency.1
- Effusions are associated with an increased risk of death.3
- Categorizing the effusion helps guide treatment.
- Chest tubes should be placed for some cases of complicated effusion and for all cases of empyema.
- A small-bore chest tube (< 20 F) should be tried first.
- Light RW. Parapneumonic effusions and empyema. Proc Am Thorac Soc 2006; 3(1):75–80. doi:10.1513/pats.200510-113JH
- US Department of Health and Human Services; Centers for Disease Control and Prevention; National Center for Health Statistics. National hospital ambulatory medical care survey: 2013 emergency department summary tables. www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf.
- Hasley PB, Albaum MN, Li YH, et al. Do pulmonary radiographic findings at presentation predict mortality in patients with community-acquired pneumonia? Arch Intern Med 1996; 156(19):2206–2212. doi:10.1001/archinte.1996.00440180068008
- Light RW, Macgregor MI, Luchsinger PC, Ball WC. Pleural effusions: the diagnostic separation of transudates and exudates. Ann Intern Med 1972; 77(4):507–513. doi:10.7326/0003-4819-77-4-507
- Colice GL, Curtis A, Deslauriers J, et al. Medical and surgical treatment of parapneumonic effusions: an evidence-based guideline. Chest 2000; 118(4):1158–1171. doi:10.1378/CHEST.118.4.1158
- Ali I, Unruh H. Management of empyema thoracis. Ann Thorac Surg 1990; 50(3):355–359. doi:10.1016/0003-4975(90)90474-K
- Ashbaugh DG. Empyema thoracis. Factors influencing morbidity and mortality. Chest 1991; 99(5):1162–1165. doi:10.1378/CHEST.99.5.1162
- Cooke DT, David EA. Large-bore and small-bore chest tubes: types, function, and placement. Thorac Surg Clin 2013; 23(1):17–24. doi:10.1016/j.thorsurg.2012.10.006
- Halifax RJ, Psallidas I, Rahman NM. Chest drain size: the debate continues. Curr Pulmonol Rep 2017; 6(1):26–29. doi:10.1007/s13665-017-0162-3
- Maskell NA, Davies CW, Nunn AJ, et al; First Multicenter Intrapleural Sepsis Trial (MIST1) Group. UK controlled trial of intrapleural streptokinase for pleural infection. N Engl J Med 2005; 352(9):865–874. doi:10.1056/NEJMoa042473
- Wait MA, Sharma S, Hohn J, Dal Nogare A. A randomized trial of empyema therapy. Chest 1997; 111(6):1548–1551. doi:10.1378/chest.111.6.1548
- Rzyman W, Skokowski J, Romanowicz G, Lass P, Dziadziuszko R. Decortication in chronic pleural empyema—effect on lung function. Eur J Cardiothorac Surg 2002; 21(3):502–507. doi:10.1016/S1010-7940(01)01167-8
Hospitalized patients with pneumonia who develop a complicated parapneumonic effusion or empyema need to undergo chest tube placement.
WHAT IS PARAPNEUMONIC EFFUSION?
Parapneumonic effusion is a pleural effusion that forms concurrently with bacterial or viral pneumonia. Up to 40% of patients hospitalized with pneumonia develop a parapneumonic effusion.1 The effusion progresses through a continuum of 3 stages: uncomplicated, complicated, and empyema.
Uncomplicated parapneumonic effusion is an exudative effusion without bacteria or pus that is caused by movement of fluid and neutrophils into the pleural space. Pneumonia itself causes an increase in interstitial fluid and capillary leakage. The effusion becomes complicated as a result of bacteria invading the pleural space, causing a further increase in neutrophils in the pleural fluid. Empyema is defined as the presence of frank pus in the pleural space.
CLINICAL SIGNIFICANCE
According to the US Centers for Disease Control and Prevention, pneumonia accounts for 674,000 emergency department visits each year; of the patients hospitalized, up to 40% develop a parapneumonic effusion.2 The only study done on rates of death associated with parapneumonic effusion showed that, compared with patients with no effusion, the risk of death was 3.7 times higher with a unilateral effusion and 6.5 times higher with bilateral effusions.3
INITIAL EVALUATION
The initial evaluation of suspected parapneumonic effusion should include chest radiography with lateral or decubitus views, followed by thoracentesis if indicated. If thoracentesis is performed, the fluid should be tested as follows:
- Gram stain
- Appropriate cultures based on clinical scenario (eg, aerobic, anaerobic, fungal)
- Total protein in pleural fluid and serum
- Lactate dehydrogenase (LDH) in pleural fluid and serum
- Glucose
- pH.
CLASSIFICATION OF EFFUSIONS
When pleural fluid is obtained, the total protein and LDH levels are used to categorize the effusion as either transudative or exudative based on the Light criteria.4 An effusion is confirmed as exudative when 1 of the following 3 criteria is met:
- The ratio of pleural fluid protein to serum protein is greater than 0.5
- The ratio of pleural fluid LDH to serum LDH is greater than 0.6
- The pleural fluid LDH is greater than two-thirds the upper limit of normal for the serum LDH.
Category 1 effusions are defined as free- flowing fluid with a thickness of less than 10 mm on any imaging modality. Thoracentesis for pleural fluid analysis is not required. The prognosis is very good.
Category 2 effusions are defined as free- flowing fluid with a thickness greater than 10 mm and less than 50% of the hemithorax. Thoracentesis is typically done because of the size of the effusion, but Gram stain and culture of the pleural fluid are usually negative, and the pH is at least 7.2. The prognosis is good.
Category 3 effusions are considered complicated because the anatomy of the pleural space becomes altered or because bacteria have invaded the pleural space. The effusion is larger than 50% of the hemithorax or is loculated, or the parietal pleura is thickened. Since the bacteria have invaded the pleural space, Gram stain or culture of pleural fluid may be positive, the pleural fluid pH may be less than 7.2, or the glucose level of the fluid may be less than 60 mg/dL. The prognosis for category 3 is poor.
Category 4 effusions are defined as empyema. The only characteristic that separates this from category 3 is frank pus in the pleural space. The prognosis is very poor.
TO PLACE A CHEST TUBE OR NOT
For category 1 or 2 effusions, treatment with antibiotics alone is typically enough. Category 3 effusions usually do not respond to antibiotics alone and may require complete drainage of the fluid with or without a chest tube depending on whether loculations are present, as loculations are difficult to drain with a chest tube. Category 4 effusions require both antibiotics and chest tube placement.
WHAT TYPE OF CHEST TUBE?
Studies have shown that small-bore chest tubes (< 20 F) are as efficacious as larger tubes (≥ 20 F) for the treatment of complicated parapneumonic effusion and empyema.6,7 Studies have also shown that the size of the tube makes no difference in the time needed to drain the effusion, the length of hospital stay, or the complication rate.8,9 Based on these studies, a small-bore chest tube should be placed first when clinically appropriate. When a chest tube is placed for empyema, computed tomography should be performed within 24 hours to confirm proper tube placement.
ADVANCED THERAPIES FOR EMPYEMA
Empyema treatment fails when antibiotic coverage is inadequate or when a loculation is not drained appropriately. Options if treatment fails include instillation of fibrinolytics into the pleural space, video-assisted thorascopic surgery, and decortication.
The role of fibrinolytics has not been well-established, but fibrinolytics should be considered in loculated effusions or empyema, or if drainage of the effusion slows.10 Video-assisted thorascopic surgery is reserved for effusions that are incompletely drained with a chest tube with or without fibrinolytics; studies have shown shorter hospital length of stay and higher treatment efficacy when this is performed earlier for loculated effusions.11 Decortication is reserved for symptomatic patients who have a thickened pleura more than 6 months after the initial infection.12 Timing for each of these procedures is not clearly defined and so must be individualized.
TAKE-AWAY POINTS
- Parapneumonic effusion occurs concurrently with pneumonia and with a high frequency.1
- Effusions are associated with an increased risk of death.3
- Categorizing the effusion helps guide treatment.
- Chest tubes should be placed for some cases of complicated effusion and for all cases of empyema.
- A small-bore chest tube (< 20 F) should be tried first.
Hospitalized patients with pneumonia who develop a complicated parapneumonic effusion or empyema need to undergo chest tube placement.
WHAT IS PARAPNEUMONIC EFFUSION?
Parapneumonic effusion is a pleural effusion that forms concurrently with bacterial or viral pneumonia. Up to 40% of patients hospitalized with pneumonia develop a parapneumonic effusion.1 The effusion progresses through a continuum of 3 stages: uncomplicated, complicated, and empyema.
Uncomplicated parapneumonic effusion is an exudative effusion without bacteria or pus that is caused by movement of fluid and neutrophils into the pleural space. Pneumonia itself causes an increase in interstitial fluid and capillary leakage. The effusion becomes complicated as a result of bacteria invading the pleural space, causing a further increase in neutrophils in the pleural fluid. Empyema is defined as the presence of frank pus in the pleural space.
CLINICAL SIGNIFICANCE
According to the US Centers for Disease Control and Prevention, pneumonia accounts for 674,000 emergency department visits each year; of the patients hospitalized, up to 40% develop a parapneumonic effusion.2 The only study done on rates of death associated with parapneumonic effusion showed that, compared with patients with no effusion, the risk of death was 3.7 times higher with a unilateral effusion and 6.5 times higher with bilateral effusions.3
INITIAL EVALUATION
The initial evaluation of suspected parapneumonic effusion should include chest radiography with lateral or decubitus views, followed by thoracentesis if indicated. If thoracentesis is performed, the fluid should be tested as follows:
- Gram stain
- Appropriate cultures based on clinical scenario (eg, aerobic, anaerobic, fungal)
- Total protein in pleural fluid and serum
- Lactate dehydrogenase (LDH) in pleural fluid and serum
- Glucose
- pH.
CLASSIFICATION OF EFFUSIONS
When pleural fluid is obtained, the total protein and LDH levels are used to categorize the effusion as either transudative or exudative based on the Light criteria.4 An effusion is confirmed as exudative when 1 of the following 3 criteria is met:
- The ratio of pleural fluid protein to serum protein is greater than 0.5
- The ratio of pleural fluid LDH to serum LDH is greater than 0.6
- The pleural fluid LDH is greater than two-thirds the upper limit of normal for the serum LDH.
Category 1 effusions are defined as free- flowing fluid with a thickness of less than 10 mm on any imaging modality. Thoracentesis for pleural fluid analysis is not required. The prognosis is very good.
Category 2 effusions are defined as free- flowing fluid with a thickness greater than 10 mm and less than 50% of the hemithorax. Thoracentesis is typically done because of the size of the effusion, but Gram stain and culture of the pleural fluid are usually negative, and the pH is at least 7.2. The prognosis is good.
Category 3 effusions are considered complicated because the anatomy of the pleural space becomes altered or because bacteria have invaded the pleural space. The effusion is larger than 50% of the hemithorax or is loculated, or the parietal pleura is thickened. Since the bacteria have invaded the pleural space, Gram stain or culture of pleural fluid may be positive, the pleural fluid pH may be less than 7.2, or the glucose level of the fluid may be less than 60 mg/dL. The prognosis for category 3 is poor.
Category 4 effusions are defined as empyema. The only characteristic that separates this from category 3 is frank pus in the pleural space. The prognosis is very poor.
TO PLACE A CHEST TUBE OR NOT
For category 1 or 2 effusions, treatment with antibiotics alone is typically enough. Category 3 effusions usually do not respond to antibiotics alone and may require complete drainage of the fluid with or without a chest tube depending on whether loculations are present, as loculations are difficult to drain with a chest tube. Category 4 effusions require both antibiotics and chest tube placement.
WHAT TYPE OF CHEST TUBE?
Studies have shown that small-bore chest tubes (< 20 F) are as efficacious as larger tubes (≥ 20 F) for the treatment of complicated parapneumonic effusion and empyema.6,7 Studies have also shown that the size of the tube makes no difference in the time needed to drain the effusion, the length of hospital stay, or the complication rate.8,9 Based on these studies, a small-bore chest tube should be placed first when clinically appropriate. When a chest tube is placed for empyema, computed tomography should be performed within 24 hours to confirm proper tube placement.
ADVANCED THERAPIES FOR EMPYEMA
Empyema treatment fails when antibiotic coverage is inadequate or when a loculation is not drained appropriately. Options if treatment fails include instillation of fibrinolytics into the pleural space, video-assisted thorascopic surgery, and decortication.
The role of fibrinolytics has not been well-established, but fibrinolytics should be considered in loculated effusions or empyema, or if drainage of the effusion slows.10 Video-assisted thorascopic surgery is reserved for effusions that are incompletely drained with a chest tube with or without fibrinolytics; studies have shown shorter hospital length of stay and higher treatment efficacy when this is performed earlier for loculated effusions.11 Decortication is reserved for symptomatic patients who have a thickened pleura more than 6 months after the initial infection.12 Timing for each of these procedures is not clearly defined and so must be individualized.
TAKE-AWAY POINTS
- Parapneumonic effusion occurs concurrently with pneumonia and with a high frequency.1
- Effusions are associated with an increased risk of death.3
- Categorizing the effusion helps guide treatment.
- Chest tubes should be placed for some cases of complicated effusion and for all cases of empyema.
- A small-bore chest tube (< 20 F) should be tried first.
- Light RW. Parapneumonic effusions and empyema. Proc Am Thorac Soc 2006; 3(1):75–80. doi:10.1513/pats.200510-113JH
- US Department of Health and Human Services; Centers for Disease Control and Prevention; National Center for Health Statistics. National hospital ambulatory medical care survey: 2013 emergency department summary tables. www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf.
- Hasley PB, Albaum MN, Li YH, et al. Do pulmonary radiographic findings at presentation predict mortality in patients with community-acquired pneumonia? Arch Intern Med 1996; 156(19):2206–2212. doi:10.1001/archinte.1996.00440180068008
- Light RW, Macgregor MI, Luchsinger PC, Ball WC. Pleural effusions: the diagnostic separation of transudates and exudates. Ann Intern Med 1972; 77(4):507–513. doi:10.7326/0003-4819-77-4-507
- Colice GL, Curtis A, Deslauriers J, et al. Medical and surgical treatment of parapneumonic effusions: an evidence-based guideline. Chest 2000; 118(4):1158–1171. doi:10.1378/CHEST.118.4.1158
- Ali I, Unruh H. Management of empyema thoracis. Ann Thorac Surg 1990; 50(3):355–359. doi:10.1016/0003-4975(90)90474-K
- Ashbaugh DG. Empyema thoracis. Factors influencing morbidity and mortality. Chest 1991; 99(5):1162–1165. doi:10.1378/CHEST.99.5.1162
- Cooke DT, David EA. Large-bore and small-bore chest tubes: types, function, and placement. Thorac Surg Clin 2013; 23(1):17–24. doi:10.1016/j.thorsurg.2012.10.006
- Halifax RJ, Psallidas I, Rahman NM. Chest drain size: the debate continues. Curr Pulmonol Rep 2017; 6(1):26–29. doi:10.1007/s13665-017-0162-3
- Maskell NA, Davies CW, Nunn AJ, et al; First Multicenter Intrapleural Sepsis Trial (MIST1) Group. UK controlled trial of intrapleural streptokinase for pleural infection. N Engl J Med 2005; 352(9):865–874. doi:10.1056/NEJMoa042473
- Wait MA, Sharma S, Hohn J, Dal Nogare A. A randomized trial of empyema therapy. Chest 1997; 111(6):1548–1551. doi:10.1378/chest.111.6.1548
- Rzyman W, Skokowski J, Romanowicz G, Lass P, Dziadziuszko R. Decortication in chronic pleural empyema—effect on lung function. Eur J Cardiothorac Surg 2002; 21(3):502–507. doi:10.1016/S1010-7940(01)01167-8
- Light RW. Parapneumonic effusions and empyema. Proc Am Thorac Soc 2006; 3(1):75–80. doi:10.1513/pats.200510-113JH
- US Department of Health and Human Services; Centers for Disease Control and Prevention; National Center for Health Statistics. National hospital ambulatory medical care survey: 2013 emergency department summary tables. www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf.
- Hasley PB, Albaum MN, Li YH, et al. Do pulmonary radiographic findings at presentation predict mortality in patients with community-acquired pneumonia? Arch Intern Med 1996; 156(19):2206–2212. doi:10.1001/archinte.1996.00440180068008
- Light RW, Macgregor MI, Luchsinger PC, Ball WC. Pleural effusions: the diagnostic separation of transudates and exudates. Ann Intern Med 1972; 77(4):507–513. doi:10.7326/0003-4819-77-4-507
- Colice GL, Curtis A, Deslauriers J, et al. Medical and surgical treatment of parapneumonic effusions: an evidence-based guideline. Chest 2000; 118(4):1158–1171. doi:10.1378/CHEST.118.4.1158
- Ali I, Unruh H. Management of empyema thoracis. Ann Thorac Surg 1990; 50(3):355–359. doi:10.1016/0003-4975(90)90474-K
- Ashbaugh DG. Empyema thoracis. Factors influencing morbidity and mortality. Chest 1991; 99(5):1162–1165. doi:10.1378/CHEST.99.5.1162
- Cooke DT, David EA. Large-bore and small-bore chest tubes: types, function, and placement. Thorac Surg Clin 2013; 23(1):17–24. doi:10.1016/j.thorsurg.2012.10.006
- Halifax RJ, Psallidas I, Rahman NM. Chest drain size: the debate continues. Curr Pulmonol Rep 2017; 6(1):26–29. doi:10.1007/s13665-017-0162-3
- Maskell NA, Davies CW, Nunn AJ, et al; First Multicenter Intrapleural Sepsis Trial (MIST1) Group. UK controlled trial of intrapleural streptokinase for pleural infection. N Engl J Med 2005; 352(9):865–874. doi:10.1056/NEJMoa042473
- Wait MA, Sharma S, Hohn J, Dal Nogare A. A randomized trial of empyema therapy. Chest 1997; 111(6):1548–1551. doi:10.1378/chest.111.6.1548
- Rzyman W, Skokowski J, Romanowicz G, Lass P, Dziadziuszko R. Decortication in chronic pleural empyema—effect on lung function. Eur J Cardiothorac Surg 2002; 21(3):502–507. doi:10.1016/S1010-7940(01)01167-8
Palmoplantar exanthema and liver dysfunction

A 51-year-old man with type 2 diabetes was referred to our hospital because of liver dysfunction and nonpruritic exanthema, with papulosquamous, scaly, papular and macular lesions on his trunk and extremities, including his palms (Figure 1) and soles. Also noted were tiny grayish mucus patches on the oral mucosa. Axillary and inguinal superficial lymph nodes were palpable.
Laboratory testing revealed elevated serum levels of markers of liver disease, ie:
- Total bilirubin 9.8 mg/dL (reference range 0.2–1.3)
- Direct bilirubin 8.0 mg/dL (< 0.2)
- Aspartate aminotransferase 57 IU/L (13–35)
- Alanine aminotransferase 90 IU/L (10–54)
- Alkaline phosphatase 4,446 IU/L (36–108).
Possible causes of liver dysfunction such as legal and illicit drugs, alcohol abuse, obstructive biliary tract or liver disease, viral hepatitis, and primary biliary cirrhosis were ruled out by history, serologic testing, abdominal ultrasonography, and computed tomography.
Secondary syphilis was suspected in view of the characteristic distribution of exanthema involving the trunk and extremities, especially the palms and soles. On questioning, the patient admitted to having had unprotected sex with a female sex worker, which also raised the probability of syphilis infection.
The rapid plasma reagin test was positive at a titer of 1:16, and the Treponema pallidum agglutination test was positive at a signal-to-cutoff ratio of 27.02. Antibody testing for human immunodeficiency virus (HIV) was negative.
The patient was started on penicillin G, but 4 hours later, he developed a fever with a temperature of 100.2°F (37.9°C), which was assumed to be a Jarisch-Herxheimer reaction. The fever resolved by the next morning without further treatment.
His course was otherwise uneventful. The exanthema resolved within 3 months, and his liver function returned to normal. Five months later, the rapid plasma reagin test was repeated on an outpatient basis, and the result was normal.
SYPHILIS IS NOT A DISEASE OF THE PAST
Syphilis is caused by T pallidum and is mainly transmitted by sexual contact.1
The incidence of syphilis has substantially increased in recent years in Japan2,3 and worldwide.4 The typical patient is a young man who has sex with men, is infected with HIV, and has a history of syphilis infection.3 However, the rapid increase in syphilis infections in Japan in recent years is largely because of an increase in heterosexual transmission.3
Infectious in its early stages
Syphilis is potentially infectious in its early (primary, secondary, and early latent) stages.1,5 The secondary stage generally begins 6 to 8 weeks after the primary infection1 and presents with diverse symptoms, including arthralgia, condylomata lata, generalized lymphadenopathy, maculopapular and papulosquamous exanthema, myalgia, and pharyngitis.1
Liver dysfunction in secondary syphilis
Liver dysfunction is common in secondary syphilis, occurring in 25% to 50% of cases.5 The liver enzyme pattern in most cases is a disproportionate increase in the alkaline phosphatase level compared with modest elevations of aminotransferases and bilirubin.2,5 However, some cases may show predominant hepatocellular damage (with prominent elevations in aminotransferase levels), and others may present with severe cholestasis (with prominent elevations in alkaline phosphatase and bilirubin) or even fulminant hepatic failure.2,5
The diagnostic criteria for syphilitic hepatitis are abnormal liver enzyme levels, serologic evidence of syphilis in conjunction with acute clinical presentation of secondary syphilis, exclusion of alternative causes of liver dysfunction, and prompt recovery of liver function after antimicrobial therapy.2,5
Pathogenic mechanisms in syphilitic hepatitis include direct portal venous inoculation and immune complex-mediated disease.2 However, direct hepatotoxicity of the microorganism seems unlikely, as spirochetes are rarely detected in liver specimens.2,5
Jarisch-Herxheimer reaction
The Jarisch-Herxheimer reaction is an acute febrile illness during the first 24 hours of antimicrobial treatment.1,6 It is assumed to be due to lysis of large numbers of spirochetes, releasing lipopolysaccharides (endotoxins) that further incite the release of a range of cytokines, resulting in symptoms such as fever, chills, myalgias, headache, tachycardia, hyperventilation, vasodilation with flushing, and mild hypotension.6,7
The frequency of Jarisch-Herxheimer reaction in syphilis and other spirochetal infections has varied widely in different reports.8 It is common in primary and secondary syphilis but usually does not occur in latent syphilis.6
Consider the diagnosis
Physicians should consider secondary syphilis in patients who present with characteristic generalized reddish macules and papules with papulosquamous lesions, including on the palms and soles as in our patient, and also in patients who have had unprotected sexual contact. Syphilis is not a disease of the past.
Acknowledgment: The authors thank Dr. Joel Branch, Shonan Kamakura General Hospital, Japan, for his editorial assistance.
- Mattei PL, Beachkofsky TM, Gilson RT, Wisco OJ. Syphilis: a reemerging infection. Am Fam Physician 2012; 86(5):433–440. pmid:22963062
- Miura H, Nakano M, Ryu T, Kitamura S, Suzaki A. A case of syphilis presenting with initial syphilitic hepatitis and serological recurrence with cerebrospinal abnormality. Intern Med 2010; 49(14):1377–1381. pmid:20647651
- Nishijima T, Teruya K, Shibata S, et al. Incidence and risk factors for incident syphilis among HIV-1-infected men who have sex with men in a large urban HIV clinic in Tokyo, 2008-2015. PLoS One 2016; 11(12):e0168642. doi:10.1371/journal.pone.0168642
- US Preventive Services Task Force (USPSTF), Bibbins-Domingo K, Grossman DC, Curry SJ, et al. Screening for syphilis infection in nonpregnant adults and adolescents: US Preventive Services Task Force Recommendation Statement. JAMA 2016; 315(21):2321–2327. doi:10.1001/jama.2016.5824
- Aggarwal A, Sharma V, Vaiphei K, Duseja A, Chawla YK. An unusual cause of cholestatic hepatitis: syphilis. Dig Dis Sci 2013; 58(10):3049–3051. doi:10.1007/s10620-013-2581-5
- Belum GR, Belum VR, Chaitanya Arudra SK, Reddy BS. The Jarisch-Herxheimer reaction: revisited. Travel Med Infect Dis 2013; 11(4):231–237. doi:10.1016/j.tmaid.2013.04.001
- Nau R, Eiffert H. Modulation of release of proinflammatory bacterial compounds by antibacterials: potential impact on course of inflammation and outcome in sepsis and meningitis. Clin Microbiol Rev 2002; 15(1):95–110. pmid:11781269
- Butler T. The Jarisch-Herxheimer reaction after antibiotic treatment of spirochetal infections: a review of recent cases and our understanding of pathogenesis. Am J Trop Med Hyg 2017; 96(1):46–52. doi:10.4269/ajtmh.16-0434
A 51-year-old man with type 2 diabetes was referred to our hospital because of liver dysfunction and nonpruritic exanthema, with papulosquamous, scaly, papular and macular lesions on his trunk and extremities, including his palms (Figure 1) and soles. Also noted were tiny grayish mucus patches on the oral mucosa. Axillary and inguinal superficial lymph nodes were palpable.
Laboratory testing revealed elevated serum levels of markers of liver disease, ie:
- Total bilirubin 9.8 mg/dL (reference range 0.2–1.3)
- Direct bilirubin 8.0 mg/dL (< 0.2)
- Aspartate aminotransferase 57 IU/L (13–35)
- Alanine aminotransferase 90 IU/L (10–54)
- Alkaline phosphatase 4,446 IU/L (36–108).
Possible causes of liver dysfunction such as legal and illicit drugs, alcohol abuse, obstructive biliary tract or liver disease, viral hepatitis, and primary biliary cirrhosis were ruled out by history, serologic testing, abdominal ultrasonography, and computed tomography.
Secondary syphilis was suspected in view of the characteristic distribution of exanthema involving the trunk and extremities, especially the palms and soles. On questioning, the patient admitted to having had unprotected sex with a female sex worker, which also raised the probability of syphilis infection.
The rapid plasma reagin test was positive at a titer of 1:16, and the Treponema pallidum agglutination test was positive at a signal-to-cutoff ratio of 27.02. Antibody testing for human immunodeficiency virus (HIV) was negative.
The patient was started on penicillin G, but 4 hours later, he developed a fever with a temperature of 100.2°F (37.9°C), which was assumed to be a Jarisch-Herxheimer reaction. The fever resolved by the next morning without further treatment.
His course was otherwise uneventful. The exanthema resolved within 3 months, and his liver function returned to normal. Five months later, the rapid plasma reagin test was repeated on an outpatient basis, and the result was normal.
SYPHILIS IS NOT A DISEASE OF THE PAST
Syphilis is caused by T pallidum and is mainly transmitted by sexual contact.1
The incidence of syphilis has substantially increased in recent years in Japan2,3 and worldwide.4 The typical patient is a young man who has sex with men, is infected with HIV, and has a history of syphilis infection.3 However, the rapid increase in syphilis infections in Japan in recent years is largely because of an increase in heterosexual transmission.3
Infectious in its early stages
Syphilis is potentially infectious in its early (primary, secondary, and early latent) stages.1,5 The secondary stage generally begins 6 to 8 weeks after the primary infection1 and presents with diverse symptoms, including arthralgia, condylomata lata, generalized lymphadenopathy, maculopapular and papulosquamous exanthema, myalgia, and pharyngitis.1
Liver dysfunction in secondary syphilis
Liver dysfunction is common in secondary syphilis, occurring in 25% to 50% of cases.5 The liver enzyme pattern in most cases is a disproportionate increase in the alkaline phosphatase level compared with modest elevations of aminotransferases and bilirubin.2,5 However, some cases may show predominant hepatocellular damage (with prominent elevations in aminotransferase levels), and others may present with severe cholestasis (with prominent elevations in alkaline phosphatase and bilirubin) or even fulminant hepatic failure.2,5
The diagnostic criteria for syphilitic hepatitis are abnormal liver enzyme levels, serologic evidence of syphilis in conjunction with acute clinical presentation of secondary syphilis, exclusion of alternative causes of liver dysfunction, and prompt recovery of liver function after antimicrobial therapy.2,5
Pathogenic mechanisms in syphilitic hepatitis include direct portal venous inoculation and immune complex-mediated disease.2 However, direct hepatotoxicity of the microorganism seems unlikely, as spirochetes are rarely detected in liver specimens.2,5
Jarisch-Herxheimer reaction
The Jarisch-Herxheimer reaction is an acute febrile illness during the first 24 hours of antimicrobial treatment.1,6 It is assumed to be due to lysis of large numbers of spirochetes, releasing lipopolysaccharides (endotoxins) that further incite the release of a range of cytokines, resulting in symptoms such as fever, chills, myalgias, headache, tachycardia, hyperventilation, vasodilation with flushing, and mild hypotension.6,7
The frequency of Jarisch-Herxheimer reaction in syphilis and other spirochetal infections has varied widely in different reports.8 It is common in primary and secondary syphilis but usually does not occur in latent syphilis.6
Consider the diagnosis
Physicians should consider secondary syphilis in patients who present with characteristic generalized reddish macules and papules with papulosquamous lesions, including on the palms and soles as in our patient, and also in patients who have had unprotected sexual contact. Syphilis is not a disease of the past.
Acknowledgment: The authors thank Dr. Joel Branch, Shonan Kamakura General Hospital, Japan, for his editorial assistance.
A 51-year-old man with type 2 diabetes was referred to our hospital because of liver dysfunction and nonpruritic exanthema, with papulosquamous, scaly, papular and macular lesions on his trunk and extremities, including his palms (Figure 1) and soles. Also noted were tiny grayish mucus patches on the oral mucosa. Axillary and inguinal superficial lymph nodes were palpable.
Laboratory testing revealed elevated serum levels of markers of liver disease, ie:
- Total bilirubin 9.8 mg/dL (reference range 0.2–1.3)
- Direct bilirubin 8.0 mg/dL (< 0.2)
- Aspartate aminotransferase 57 IU/L (13–35)
- Alanine aminotransferase 90 IU/L (10–54)
- Alkaline phosphatase 4,446 IU/L (36–108).
Possible causes of liver dysfunction such as legal and illicit drugs, alcohol abuse, obstructive biliary tract or liver disease, viral hepatitis, and primary biliary cirrhosis were ruled out by history, serologic testing, abdominal ultrasonography, and computed tomography.
Secondary syphilis was suspected in view of the characteristic distribution of exanthema involving the trunk and extremities, especially the palms and soles. On questioning, the patient admitted to having had unprotected sex with a female sex worker, which also raised the probability of syphilis infection.
The rapid plasma reagin test was positive at a titer of 1:16, and the Treponema pallidum agglutination test was positive at a signal-to-cutoff ratio of 27.02. Antibody testing for human immunodeficiency virus (HIV) was negative.
The patient was started on penicillin G, but 4 hours later, he developed a fever with a temperature of 100.2°F (37.9°C), which was assumed to be a Jarisch-Herxheimer reaction. The fever resolved by the next morning without further treatment.
His course was otherwise uneventful. The exanthema resolved within 3 months, and his liver function returned to normal. Five months later, the rapid plasma reagin test was repeated on an outpatient basis, and the result was normal.
SYPHILIS IS NOT A DISEASE OF THE PAST
Syphilis is caused by T pallidum and is mainly transmitted by sexual contact.1
The incidence of syphilis has substantially increased in recent years in Japan2,3 and worldwide.4 The typical patient is a young man who has sex with men, is infected with HIV, and has a history of syphilis infection.3 However, the rapid increase in syphilis infections in Japan in recent years is largely because of an increase in heterosexual transmission.3
Infectious in its early stages
Syphilis is potentially infectious in its early (primary, secondary, and early latent) stages.1,5 The secondary stage generally begins 6 to 8 weeks after the primary infection1 and presents with diverse symptoms, including arthralgia, condylomata lata, generalized lymphadenopathy, maculopapular and papulosquamous exanthema, myalgia, and pharyngitis.1
Liver dysfunction in secondary syphilis
Liver dysfunction is common in secondary syphilis, occurring in 25% to 50% of cases.5 The liver enzyme pattern in most cases is a disproportionate increase in the alkaline phosphatase level compared with modest elevations of aminotransferases and bilirubin.2,5 However, some cases may show predominant hepatocellular damage (with prominent elevations in aminotransferase levels), and others may present with severe cholestasis (with prominent elevations in alkaline phosphatase and bilirubin) or even fulminant hepatic failure.2,5
The diagnostic criteria for syphilitic hepatitis are abnormal liver enzyme levels, serologic evidence of syphilis in conjunction with acute clinical presentation of secondary syphilis, exclusion of alternative causes of liver dysfunction, and prompt recovery of liver function after antimicrobial therapy.2,5
Pathogenic mechanisms in syphilitic hepatitis include direct portal venous inoculation and immune complex-mediated disease.2 However, direct hepatotoxicity of the microorganism seems unlikely, as spirochetes are rarely detected in liver specimens.2,5
Jarisch-Herxheimer reaction
The Jarisch-Herxheimer reaction is an acute febrile illness during the first 24 hours of antimicrobial treatment.1,6 It is assumed to be due to lysis of large numbers of spirochetes, releasing lipopolysaccharides (endotoxins) that further incite the release of a range of cytokines, resulting in symptoms such as fever, chills, myalgias, headache, tachycardia, hyperventilation, vasodilation with flushing, and mild hypotension.6,7
The frequency of Jarisch-Herxheimer reaction in syphilis and other spirochetal infections has varied widely in different reports.8 It is common in primary and secondary syphilis but usually does not occur in latent syphilis.6
Consider the diagnosis
Physicians should consider secondary syphilis in patients who present with characteristic generalized reddish macules and papules with papulosquamous lesions, including on the palms and soles as in our patient, and also in patients who have had unprotected sexual contact. Syphilis is not a disease of the past.
Acknowledgment: The authors thank Dr. Joel Branch, Shonan Kamakura General Hospital, Japan, for his editorial assistance.
- Mattei PL, Beachkofsky TM, Gilson RT, Wisco OJ. Syphilis: a reemerging infection. Am Fam Physician 2012; 86(5):433–440. pmid:22963062
- Miura H, Nakano M, Ryu T, Kitamura S, Suzaki A. A case of syphilis presenting with initial syphilitic hepatitis and serological recurrence with cerebrospinal abnormality. Intern Med 2010; 49(14):1377–1381. pmid:20647651
- Nishijima T, Teruya K, Shibata S, et al. Incidence and risk factors for incident syphilis among HIV-1-infected men who have sex with men in a large urban HIV clinic in Tokyo, 2008-2015. PLoS One 2016; 11(12):e0168642. doi:10.1371/journal.pone.0168642
- US Preventive Services Task Force (USPSTF), Bibbins-Domingo K, Grossman DC, Curry SJ, et al. Screening for syphilis infection in nonpregnant adults and adolescents: US Preventive Services Task Force Recommendation Statement. JAMA 2016; 315(21):2321–2327. doi:10.1001/jama.2016.5824
- Aggarwal A, Sharma V, Vaiphei K, Duseja A, Chawla YK. An unusual cause of cholestatic hepatitis: syphilis. Dig Dis Sci 2013; 58(10):3049–3051. doi:10.1007/s10620-013-2581-5
- Belum GR, Belum VR, Chaitanya Arudra SK, Reddy BS. The Jarisch-Herxheimer reaction: revisited. Travel Med Infect Dis 2013; 11(4):231–237. doi:10.1016/j.tmaid.2013.04.001
- Nau R, Eiffert H. Modulation of release of proinflammatory bacterial compounds by antibacterials: potential impact on course of inflammation and outcome in sepsis and meningitis. Clin Microbiol Rev 2002; 15(1):95–110. pmid:11781269
- Butler T. The Jarisch-Herxheimer reaction after antibiotic treatment of spirochetal infections: a review of recent cases and our understanding of pathogenesis. Am J Trop Med Hyg 2017; 96(1):46–52. doi:10.4269/ajtmh.16-0434
- Mattei PL, Beachkofsky TM, Gilson RT, Wisco OJ. Syphilis: a reemerging infection. Am Fam Physician 2012; 86(5):433–440. pmid:22963062
- Miura H, Nakano M, Ryu T, Kitamura S, Suzaki A. A case of syphilis presenting with initial syphilitic hepatitis and serological recurrence with cerebrospinal abnormality. Intern Med 2010; 49(14):1377–1381. pmid:20647651
- Nishijima T, Teruya K, Shibata S, et al. Incidence and risk factors for incident syphilis among HIV-1-infected men who have sex with men in a large urban HIV clinic in Tokyo, 2008-2015. PLoS One 2016; 11(12):e0168642. doi:10.1371/journal.pone.0168642
- US Preventive Services Task Force (USPSTF), Bibbins-Domingo K, Grossman DC, Curry SJ, et al. Screening for syphilis infection in nonpregnant adults and adolescents: US Preventive Services Task Force Recommendation Statement. JAMA 2016; 315(21):2321–2327. doi:10.1001/jama.2016.5824
- Aggarwal A, Sharma V, Vaiphei K, Duseja A, Chawla YK. An unusual cause of cholestatic hepatitis: syphilis. Dig Dis Sci 2013; 58(10):3049–3051. doi:10.1007/s10620-013-2581-5
- Belum GR, Belum VR, Chaitanya Arudra SK, Reddy BS. The Jarisch-Herxheimer reaction: revisited. Travel Med Infect Dis 2013; 11(4):231–237. doi:10.1016/j.tmaid.2013.04.001
- Nau R, Eiffert H. Modulation of release of proinflammatory bacterial compounds by antibacterials: potential impact on course of inflammation and outcome in sepsis and meningitis. Clin Microbiol Rev 2002; 15(1):95–110. pmid:11781269
- Butler T. The Jarisch-Herxheimer reaction after antibiotic treatment of spirochetal infections: a review of recent cases and our understanding of pathogenesis. Am J Trop Med Hyg 2017; 96(1):46–52. doi:10.4269/ajtmh.16-0434