User login
Supreme Court decisions in 2017 that affected your practice
Despite being short-handed (there were only 8 justices for most of the Term), the United States Supreme Court decided a number of important cases during its most recent Term, which concluded on June 27, 2017. Among the 69 cases, several are of particular interest to ObGyns.

1. Arbitration in health care
In Kindred Nursing Centers v Clark, the Court decided an important case involving arbitration in health care.1
At stake. The families of 2 people who died after being in a long-term care facility filed lawsuits against the facility, claiming personal injury, violations of Kentucky statutes regarding long-term care facilities, and wrongful death. However, during admission to the facility, the patients (technically, their agents under a power of attorney) signed an agreement that any disputes would be taken to arbitration. The facility successfully had the lawsuits dismissed.
Final ruling. The Supreme Court agreed that the case had to go to arbitration rather than to court, even though the arbitration clause violated state law. The Federal Arbitration Act (FAA) preempts state law. The Court has been very aggressive in enforcing arbitration agreements and striking down state laws that are inconsistent with the FAA. This case emphasizes that the FAA applies in the health care context.
The case suggests both a warning and an opportunity for health care providers. The warning is that arbitration clauses will be enforced; thoughtlessly entering into arbitration for future disputes may be dangerous. Among other things, the decision of arbitrators is essentially unreviewable. Appellate courts review the decisions of lower courts, but there is no such review in arbitration. Furthermore, arbitration may be stacked in favor of commercial entities that often use arbitrators.
The opportunity for health care providers lies in that it may be possible to include arbitration clauses in agreements with patients. This should be considered only after obtaining legal advice. The agreements should, for example, be consistent with the obligations to patients (in the case of the Kentucky facility, it made clear that accepting the arbitration agreement was not necessary in order to receive care or be admitted to the facility). Because arbitration agreements are becoming ubiquitous and rigorously enforced by federal courts, arbitration is bound to have an important function in health care.
2. Pharmaceuticals
Biologics and biosimilars
Biologics play an important role in health care. Eight of the top 10 selling drugs in 2016 were biologics.2 The case of Sandoz v Amgen involved biosimilar pharmaceuticals, essentially the generics of biologic drugs.3
At stake. While biologics hold great promise in medicine, they are generally very expensive. Just as with generics, brand-name companies (generally referred to as “reference” biologics) want to keep biosimilars off the market for as long as possible, thereby extending the advantages of monopolistic pricing. This Term the Supreme Court considered the statutory rules for licensing biosimilar drugs.
Final ruling. The Court’s decision will allow biosimilar companies to speed up the licensing process by at least 180 days. This is a modest win for patients and their physicians, but the legal issues around biosimilars will need additional attention.
Class action suits
In another case, the Court made it more difficult to file class action suits against pharmaceutical companies in state courts.4 Although this is a fairly technical decision, it is likely to have a significant impact in pharmaceutical liability by limiting classactions.

3. The travel ban
The American College of Obstetricians and Gynecologists joined other medical organizations in an amicus curiae (friend of the court) brief to challenge President Trump’s “travel ban.”5
At stake. The brief argued that the United States “relies upon a significant number of health professionals and scientists who have entered the country through the immigration system.”5
Final ruling. The Court allowed most of the travel ban to stay in place, but did permit entry into the United States by foreign nationals “with a close familial relationship,” or pre-existing ties to US businesses or institutions (such as students who have been admitted to American colleges, workers who have accepted US employment, or lecturers invited to address American audiences).6 Following the Term, the Administration issued a different travel ban, so the issue was taken off the Court’s calendar for the moment. There undoubtedly will be additional chapters to come.

4. Birth certificates and same-sex marriage
In Pavan v Smith, the legal question concerned whether married same-sex couples may have both parents listed on the birth certificate of children born during the marriage.7 Two same-sex couples conceived children through anonymous sperm donation and gave birth in Arkansas. The Department of Health in Arkansas issued birth certificates listing the mother’s name, but refused to list the spouse on the birth certificate.
At stake. The couples brought suit claiming a constitutional right to have both parents listed. In particular, they noted that under Arkansas law, the woman who gives birth is deemed to be the mother. When the woman is married, the husband’s name is “entered on the certificate as the father of the child.”8 The same-sex parents argued that a 2015 decision of the Supreme Court, which held that the Constitution requires states to recognize same-sex marriages, made it clear that same-sex couples should have the benefits of marriage.9 Eventually the case wound its way to the Supreme Court.
Final ruling. The Court held that if the state ordinarily lists the names of both husband and wife on such certificates, then same-sex couples are entitled to have birth certificates listing both parents. The Court noted that laws are unconstitutional if they treat same-sex couples differently than opposite-sex couples. Based on this principle, the Court held that parental birth certificate registration is part of the “constellations of benefits” linked to marriage that the Constitution affords same-sex couples. This ruling applies as a matter of constitutional right in all states.
Read about more interesting Supreme Court decisions

5. Sexual offenders and social media
States struggle to protect children from convicted sex offenders. North Carolina, for example, made it a felony for sex offenders (who had completed their sentences) to use social media sites that “permit minor children to become members or create and maintain personal web pages.”10
At stake. In Packingham v North Carolina, the Court was asked to decide whether this statute violates the First Amendment (free speech) rights of sex offenders.11
Final ruling. The Court held that the North Carolina limitation on sex offenders’ use of social media was too broad. It noted the wide range of political, employment, news, personal, commercial, and religious websites that are off limits to sex offenders under the statute—hardly narrowly tailored. It suggested, however, that it probably would be constitutional for a state to prohibit sex offenders “from engaging in conduct that often presages a sexual crime, like contacting a minor or using a website to gather information about a minor.”11
It was important in this case that the defendant had already served his entire sentence and was “no longer subject to the supervision of the criminal justice system.”11 If he had still been in prison, the state could limit or prohibit his Internet use. Even if he had been on probation or parole (under the supervision of the criminal justice system) the restrictions may well have been permitted. In addition, the state could impose new, narrowly tailored restrictions.
This case is also a reminder that ObGyns are very important in the efforts to eliminate child sexual abuse. All states have laws that require the reporting of known or suspected sexual abuse. In addition to complying with the law, such reports are often critical to discovering and ending the abuse.

6. Transgender rights
The Court had accepted a “transgender bathroom case” in Gloucester County School Board v G.G.12
At stake. This case essentially challenged the Obama Administration’s requirement that schools allow transgender students to use the restrooms in which they feel most comfortable. It was one of the most anticipated cases of the Term, but it essentially disappeared. Following the presidential election, the Department of Education rescinded the earlier guidance on which the case was based.
Final ruling. The Court returned the case to the Fourth Circuit for reconsideration. This issue, however, may reappear before the Court in the form of a claim that the states must provide this accommodation as a matter of federal statutory right, or even Equal Protection.
- In an important First Amendment decision, the Court held that it is a violation of the Freedom of Religion to deny a church-related school access to generally available state grant funds solely because of its religious status (in this case the program funded playground surfacing grants).1
- In several cases, it was apparent that the Court is uncomfortable with the way death penalty cases are handled in some states.2
- Juries may be questioned about racial bias that was expressed during jury deliberations--a substantial change for many courts.3
- The failure of the Patent and Trademark Office (PTO) to register the trademark for the band "The Slants" was a First Amendment violation. One reason that this case was watched was because of the effort of the PTO to deregister the trademark of the Washington Redskins.4
- The Court considered 9 cases involving revoking citizenship, deportation, and cross-border liability (an extraordinary number). Two cases that could change the nature and process of deportation were held over to the next Term for reargument.
- Individualized educational plans under the federal Individuals with Disabilities Education Act (IDEA) must target more than trivial progress for the students.5
References
- Trinity Lutheran Church of Columbia, Inc. v Comer, 582 US 15 577 (2017).
- McAllister S. Death-penalty symposium: A court increasingly uncomfortable with the death penalty. SCOTUSblog.com. http://www.scotusblog.com/2017/06/death-penalty-symposium-court-increasingly-uncomfortable-death-penalty/. Published June 29, 2017. Accessed November 2, 2017.
- Pena-Rodriguez v Colorado, 580 US 15 606 (2017).
- Matal v Tam, 582 US 15 1293 (2017).
- Endrew F v Douglas County School District, RE-1, 580 US 15 827 (2017).
Summary of the Term
The Term was notable for the level of agreement. With 69 decided cases, 41 (69%) were unanimous. In 59 cases (85%), there was a strong consensus, with no more than 2 justices dissenting. Only 7 decisions (10%) were 5 to 4. Justice Kennedy was, as usual, the deciding vote in most of the close cases. He voted in the majority in 97% of the decisions. Justice Gorsuch took the place of Justice Scalia (who passed away in February 2016), so arguably the Court is ideologically close to where it has been for a number of years. Despite rumors that Justice Kennedy would announce his resignation from the Court, neither he nor any other justice has left. The Supreme Court began its new Term on October 2, 2017, with a full complement of 9 justices.
What’s to come
The Court will add cases through much of its new Term, but it has already accepted cases dealing with arbitration agreements (again); public employees’ union dues; immigration (again); the privacy of information held by mobile phone companies; a constitutional challenge to political gerrymandering; bakeries and gay-marriage ceremonies; whistleblowers and Dodd-Frank regulations; sports gambling and the NCAA; and more.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Kindred Nursing Centers, LP v Clark, 581 US 16 32 (2017).
- Anderson L. Looking Ahead: Pharma Projections for 2016 - & Beyond. Perma.cc Website. . Reviewed March 30, 2017. Accessed November 2, 2017.
- Sandoz Inc v Amgen Inc, 581 US 15 1039 1195 (2017).
- Bristol-Myers Squibb Co v Superior Court of California, San Francisco County, 582 US 16 466 (2017).
- Trinity FR, Sterling AM, Rogaczewski JD, et al. Motion for Leave to File and Brief for the Association of American Medical Colleges and Others as Amici Curiae Supporting Respondents. SCOTUSblog. http://www.scotusblog.com/wp-content/uploads/2017/06/16-1436-ac-AAMC-supporting-respondents.pdf. Accessed November 2, 2017.
- Donald J. Trump, President of the United States v International Refugee Assistance Project, 582 US 16 1436 (2017).
- Pavan v Smith, 582 US 16 992 (2017).
- Arkansas Code, §20 18 401(f)(1) (2014).
- Obergefell v Hodges, 576 US ___ (2015).
- NC Gen. Stat. Ann. §§14-202.5(a),(e).
- Packingham v North Carolina, 582 US 15 1194 (2017).
- Gloucester County School Board v G.G. SCOTUSblog. http://www.scotusblog.com/case-files/cases/gloucester-county-school-board-v-g-g/. Published March 6, 2017. Accessed November 2, 2017.
In this column, medical and legal experts and educators provide clear takeaways for your practice.

Mr. Smith is Professor Emeritus of Law and Dean Emeritus at California Western School of Law, San Diego, California. He is an

Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the
The authors report no financial relationships relevant to this article.
In this column, medical and legal experts and educators provide clear takeaways for your practice.
Mr. Smith is Professor Emeritus of Law and Dean Emeritus at California Western School of Law, San Diego, California. He is an
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the
The authors report no financial relationships relevant to this article.
In this column, medical and legal experts and educators provide clear takeaways for your practice.
Mr. Smith is Professor Emeritus of Law and Dean Emeritus at California Western School of Law, San Diego, California. He is an
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the
The authors report no financial relationships relevant to this article.
Despite being short-handed (there were only 8 justices for most of the Term), the United States Supreme Court decided a number of important cases during its most recent Term, which concluded on June 27, 2017. Among the 69 cases, several are of particular interest to ObGyns.
1. Arbitration in health care
In Kindred Nursing Centers v Clark, the Court decided an important case involving arbitration in health care.1
At stake. The families of 2 people who died after being in a long-term care facility filed lawsuits against the facility, claiming personal injury, violations of Kentucky statutes regarding long-term care facilities, and wrongful death. However, during admission to the facility, the patients (technically, their agents under a power of attorney) signed an agreement that any disputes would be taken to arbitration. The facility successfully had the lawsuits dismissed.
Final ruling. The Supreme Court agreed that the case had to go to arbitration rather than to court, even though the arbitration clause violated state law. The Federal Arbitration Act (FAA) preempts state law. The Court has been very aggressive in enforcing arbitration agreements and striking down state laws that are inconsistent with the FAA. This case emphasizes that the FAA applies in the health care context.
The case suggests both a warning and an opportunity for health care providers. The warning is that arbitration clauses will be enforced; thoughtlessly entering into arbitration for future disputes may be dangerous. Among other things, the decision of arbitrators is essentially unreviewable. Appellate courts review the decisions of lower courts, but there is no such review in arbitration. Furthermore, arbitration may be stacked in favor of commercial entities that often use arbitrators.
The opportunity for health care providers lies in that it may be possible to include arbitration clauses in agreements with patients. This should be considered only after obtaining legal advice. The agreements should, for example, be consistent with the obligations to patients (in the case of the Kentucky facility, it made clear that accepting the arbitration agreement was not necessary in order to receive care or be admitted to the facility). Because arbitration agreements are becoming ubiquitous and rigorously enforced by federal courts, arbitration is bound to have an important function in health care.
2. Pharmaceuticals
Biologics and biosimilars
Biologics play an important role in health care. Eight of the top 10 selling drugs in 2016 were biologics.2 The case of Sandoz v Amgen involved biosimilar pharmaceuticals, essentially the generics of biologic drugs.3
At stake. While biologics hold great promise in medicine, they are generally very expensive. Just as with generics, brand-name companies (generally referred to as “reference” biologics) want to keep biosimilars off the market for as long as possible, thereby extending the advantages of monopolistic pricing. This Term the Supreme Court considered the statutory rules for licensing biosimilar drugs.
Final ruling. The Court’s decision will allow biosimilar companies to speed up the licensing process by at least 180 days. This is a modest win for patients and their physicians, but the legal issues around biosimilars will need additional attention.
Class action suits
In another case, the Court made it more difficult to file class action suits against pharmaceutical companies in state courts.4 Although this is a fairly technical decision, it is likely to have a significant impact in pharmaceutical liability by limiting classactions.
3. The travel ban
The American College of Obstetricians and Gynecologists joined other medical organizations in an amicus curiae (friend of the court) brief to challenge President Trump’s “travel ban.”5
At stake. The brief argued that the United States “relies upon a significant number of health professionals and scientists who have entered the country through the immigration system.”5
Final ruling. The Court allowed most of the travel ban to stay in place, but did permit entry into the United States by foreign nationals “with a close familial relationship,” or pre-existing ties to US businesses or institutions (such as students who have been admitted to American colleges, workers who have accepted US employment, or lecturers invited to address American audiences).6 Following the Term, the Administration issued a different travel ban, so the issue was taken off the Court’s calendar for the moment. There undoubtedly will be additional chapters to come.
4. Birth certificates and same-sex marriage
In Pavan v Smith, the legal question concerned whether married same-sex couples may have both parents listed on the birth certificate of children born during the marriage.7 Two same-sex couples conceived children through anonymous sperm donation and gave birth in Arkansas. The Department of Health in Arkansas issued birth certificates listing the mother’s name, but refused to list the spouse on the birth certificate.
At stake. The couples brought suit claiming a constitutional right to have both parents listed. In particular, they noted that under Arkansas law, the woman who gives birth is deemed to be the mother. When the woman is married, the husband’s name is “entered on the certificate as the father of the child.”8 The same-sex parents argued that a 2015 decision of the Supreme Court, which held that the Constitution requires states to recognize same-sex marriages, made it clear that same-sex couples should have the benefits of marriage.9 Eventually the case wound its way to the Supreme Court.
Final ruling. The Court held that if the state ordinarily lists the names of both husband and wife on such certificates, then same-sex couples are entitled to have birth certificates listing both parents. The Court noted that laws are unconstitutional if they treat same-sex couples differently than opposite-sex couples. Based on this principle, the Court held that parental birth certificate registration is part of the “constellations of benefits” linked to marriage that the Constitution affords same-sex couples. This ruling applies as a matter of constitutional right in all states.
Read about more interesting Supreme Court decisions
5. Sexual offenders and social media
States struggle to protect children from convicted sex offenders. North Carolina, for example, made it a felony for sex offenders (who had completed their sentences) to use social media sites that “permit minor children to become members or create and maintain personal web pages.”10
At stake. In Packingham v North Carolina, the Court was asked to decide whether this statute violates the First Amendment (free speech) rights of sex offenders.11
Final ruling. The Court held that the North Carolina limitation on sex offenders’ use of social media was too broad. It noted the wide range of political, employment, news, personal, commercial, and religious websites that are off limits to sex offenders under the statute—hardly narrowly tailored. It suggested, however, that it probably would be constitutional for a state to prohibit sex offenders “from engaging in conduct that often presages a sexual crime, like contacting a minor or using a website to gather information about a minor.”11
It was important in this case that the defendant had already served his entire sentence and was “no longer subject to the supervision of the criminal justice system.”11 If he had still been in prison, the state could limit or prohibit his Internet use. Even if he had been on probation or parole (under the supervision of the criminal justice system) the restrictions may well have been permitted. In addition, the state could impose new, narrowly tailored restrictions.
This case is also a reminder that ObGyns are very important in the efforts to eliminate child sexual abuse. All states have laws that require the reporting of known or suspected sexual abuse. In addition to complying with the law, such reports are often critical to discovering and ending the abuse.
6. Transgender rights
The Court had accepted a “transgender bathroom case” in Gloucester County School Board v G.G.12
At stake. This case essentially challenged the Obama Administration’s requirement that schools allow transgender students to use the restrooms in which they feel most comfortable. It was one of the most anticipated cases of the Term, but it essentially disappeared. Following the presidential election, the Department of Education rescinded the earlier guidance on which the case was based.
Final ruling. The Court returned the case to the Fourth Circuit for reconsideration. This issue, however, may reappear before the Court in the form of a claim that the states must provide this accommodation as a matter of federal statutory right, or even Equal Protection.
- In an important First Amendment decision, the Court held that it is a violation of the Freedom of Religion to deny a church-related school access to generally available state grant funds solely because of its religious status (in this case the program funded playground surfacing grants).1
- In several cases, it was apparent that the Court is uncomfortable with the way death penalty cases are handled in some states.2
- Juries may be questioned about racial bias that was expressed during jury deliberations--a substantial change for many courts.3
- The failure of the Patent and Trademark Office (PTO) to register the trademark for the band "The Slants" was a First Amendment violation. One reason that this case was watched was because of the effort of the PTO to deregister the trademark of the Washington Redskins.4
- The Court considered 9 cases involving revoking citizenship, deportation, and cross-border liability (an extraordinary number). Two cases that could change the nature and process of deportation were held over to the next Term for reargument.
- Individualized educational plans under the federal Individuals with Disabilities Education Act (IDEA) must target more than trivial progress for the students.5
References
- Trinity Lutheran Church of Columbia, Inc. v Comer, 582 US 15 577 (2017).
- McAllister S. Death-penalty symposium: A court increasingly uncomfortable with the death penalty. SCOTUSblog.com. http://www.scotusblog.com/2017/06/death-penalty-symposium-court-increasingly-uncomfortable-death-penalty/. Published June 29, 2017. Accessed November 2, 2017.
- Pena-Rodriguez v Colorado, 580 US 15 606 (2017).
- Matal v Tam, 582 US 15 1293 (2017).
- Endrew F v Douglas County School District, RE-1, 580 US 15 827 (2017).
Summary of the Term
The Term was notable for the level of agreement. With 69 decided cases, 41 (69%) were unanimous. In 59 cases (85%), there was a strong consensus, with no more than 2 justices dissenting. Only 7 decisions (10%) were 5 to 4. Justice Kennedy was, as usual, the deciding vote in most of the close cases. He voted in the majority in 97% of the decisions. Justice Gorsuch took the place of Justice Scalia (who passed away in February 2016), so arguably the Court is ideologically close to where it has been for a number of years. Despite rumors that Justice Kennedy would announce his resignation from the Court, neither he nor any other justice has left. The Supreme Court began its new Term on October 2, 2017, with a full complement of 9 justices.
What’s to come
The Court will add cases through much of its new Term, but it has already accepted cases dealing with arbitration agreements (again); public employees’ union dues; immigration (again); the privacy of information held by mobile phone companies; a constitutional challenge to political gerrymandering; bakeries and gay-marriage ceremonies; whistleblowers and Dodd-Frank regulations; sports gambling and the NCAA; and more.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
Despite being short-handed (there were only 8 justices for most of the Term), the United States Supreme Court decided a number of important cases during its most recent Term, which concluded on June 27, 2017. Among the 69 cases, several are of particular interest to ObGyns.
1. Arbitration in health care
In Kindred Nursing Centers v Clark, the Court decided an important case involving arbitration in health care.1
At stake. The families of 2 people who died after being in a long-term care facility filed lawsuits against the facility, claiming personal injury, violations of Kentucky statutes regarding long-term care facilities, and wrongful death. However, during admission to the facility, the patients (technically, their agents under a power of attorney) signed an agreement that any disputes would be taken to arbitration. The facility successfully had the lawsuits dismissed.
Final ruling. The Supreme Court agreed that the case had to go to arbitration rather than to court, even though the arbitration clause violated state law. The Federal Arbitration Act (FAA) preempts state law. The Court has been very aggressive in enforcing arbitration agreements and striking down state laws that are inconsistent with the FAA. This case emphasizes that the FAA applies in the health care context.
The case suggests both a warning and an opportunity for health care providers. The warning is that arbitration clauses will be enforced; thoughtlessly entering into arbitration for future disputes may be dangerous. Among other things, the decision of arbitrators is essentially unreviewable. Appellate courts review the decisions of lower courts, but there is no such review in arbitration. Furthermore, arbitration may be stacked in favor of commercial entities that often use arbitrators.
The opportunity for health care providers lies in that it may be possible to include arbitration clauses in agreements with patients. This should be considered only after obtaining legal advice. The agreements should, for example, be consistent with the obligations to patients (in the case of the Kentucky facility, it made clear that accepting the arbitration agreement was not necessary in order to receive care or be admitted to the facility). Because arbitration agreements are becoming ubiquitous and rigorously enforced by federal courts, arbitration is bound to have an important function in health care.
2. Pharmaceuticals
Biologics and biosimilars
Biologics play an important role in health care. Eight of the top 10 selling drugs in 2016 were biologics.2 The case of Sandoz v Amgen involved biosimilar pharmaceuticals, essentially the generics of biologic drugs.3
At stake. While biologics hold great promise in medicine, they are generally very expensive. Just as with generics, brand-name companies (generally referred to as “reference” biologics) want to keep biosimilars off the market for as long as possible, thereby extending the advantages of monopolistic pricing. This Term the Supreme Court considered the statutory rules for licensing biosimilar drugs.
Final ruling. The Court’s decision will allow biosimilar companies to speed up the licensing process by at least 180 days. This is a modest win for patients and their physicians, but the legal issues around biosimilars will need additional attention.
Class action suits
In another case, the Court made it more difficult to file class action suits against pharmaceutical companies in state courts.4 Although this is a fairly technical decision, it is likely to have a significant impact in pharmaceutical liability by limiting classactions.
3. The travel ban
The American College of Obstetricians and Gynecologists joined other medical organizations in an amicus curiae (friend of the court) brief to challenge President Trump’s “travel ban.”5
At stake. The brief argued that the United States “relies upon a significant number of health professionals and scientists who have entered the country through the immigration system.”5
Final ruling. The Court allowed most of the travel ban to stay in place, but did permit entry into the United States by foreign nationals “with a close familial relationship,” or pre-existing ties to US businesses or institutions (such as students who have been admitted to American colleges, workers who have accepted US employment, or lecturers invited to address American audiences).6 Following the Term, the Administration issued a different travel ban, so the issue was taken off the Court’s calendar for the moment. There undoubtedly will be additional chapters to come.
4. Birth certificates and same-sex marriage
In Pavan v Smith, the legal question concerned whether married same-sex couples may have both parents listed on the birth certificate of children born during the marriage.7 Two same-sex couples conceived children through anonymous sperm donation and gave birth in Arkansas. The Department of Health in Arkansas issued birth certificates listing the mother’s name, but refused to list the spouse on the birth certificate.
At stake. The couples brought suit claiming a constitutional right to have both parents listed. In particular, they noted that under Arkansas law, the woman who gives birth is deemed to be the mother. When the woman is married, the husband’s name is “entered on the certificate as the father of the child.”8 The same-sex parents argued that a 2015 decision of the Supreme Court, which held that the Constitution requires states to recognize same-sex marriages, made it clear that same-sex couples should have the benefits of marriage.9 Eventually the case wound its way to the Supreme Court.
Final ruling. The Court held that if the state ordinarily lists the names of both husband and wife on such certificates, then same-sex couples are entitled to have birth certificates listing both parents. The Court noted that laws are unconstitutional if they treat same-sex couples differently than opposite-sex couples. Based on this principle, the Court held that parental birth certificate registration is part of the “constellations of benefits” linked to marriage that the Constitution affords same-sex couples. This ruling applies as a matter of constitutional right in all states.
Read about more interesting Supreme Court decisions
5. Sexual offenders and social media
States struggle to protect children from convicted sex offenders. North Carolina, for example, made it a felony for sex offenders (who had completed their sentences) to use social media sites that “permit minor children to become members or create and maintain personal web pages.”10
At stake. In Packingham v North Carolina, the Court was asked to decide whether this statute violates the First Amendment (free speech) rights of sex offenders.11
Final ruling. The Court held that the North Carolina limitation on sex offenders’ use of social media was too broad. It noted the wide range of political, employment, news, personal, commercial, and religious websites that are off limits to sex offenders under the statute—hardly narrowly tailored. It suggested, however, that it probably would be constitutional for a state to prohibit sex offenders “from engaging in conduct that often presages a sexual crime, like contacting a minor or using a website to gather information about a minor.”11
It was important in this case that the defendant had already served his entire sentence and was “no longer subject to the supervision of the criminal justice system.”11 If he had still been in prison, the state could limit or prohibit his Internet use. Even if he had been on probation or parole (under the supervision of the criminal justice system) the restrictions may well have been permitted. In addition, the state could impose new, narrowly tailored restrictions.
This case is also a reminder that ObGyns are very important in the efforts to eliminate child sexual abuse. All states have laws that require the reporting of known or suspected sexual abuse. In addition to complying with the law, such reports are often critical to discovering and ending the abuse.
6. Transgender rights
The Court had accepted a “transgender bathroom case” in Gloucester County School Board v G.G.12
At stake. This case essentially challenged the Obama Administration’s requirement that schools allow transgender students to use the restrooms in which they feel most comfortable. It was one of the most anticipated cases of the Term, but it essentially disappeared. Following the presidential election, the Department of Education rescinded the earlier guidance on which the case was based.
Final ruling. The Court returned the case to the Fourth Circuit for reconsideration. This issue, however, may reappear before the Court in the form of a claim that the states must provide this accommodation as a matter of federal statutory right, or even Equal Protection.
- In an important First Amendment decision, the Court held that it is a violation of the Freedom of Religion to deny a church-related school access to generally available state grant funds solely because of its religious status (in this case the program funded playground surfacing grants).1
- In several cases, it was apparent that the Court is uncomfortable with the way death penalty cases are handled in some states.2
- Juries may be questioned about racial bias that was expressed during jury deliberations--a substantial change for many courts.3
- The failure of the Patent and Trademark Office (PTO) to register the trademark for the band "The Slants" was a First Amendment violation. One reason that this case was watched was because of the effort of the PTO to deregister the trademark of the Washington Redskins.4
- The Court considered 9 cases involving revoking citizenship, deportation, and cross-border liability (an extraordinary number). Two cases that could change the nature and process of deportation were held over to the next Term for reargument.
- Individualized educational plans under the federal Individuals with Disabilities Education Act (IDEA) must target more than trivial progress for the students.5
References
- Trinity Lutheran Church of Columbia, Inc. v Comer, 582 US 15 577 (2017).
- McAllister S. Death-penalty symposium: A court increasingly uncomfortable with the death penalty. SCOTUSblog.com. http://www.scotusblog.com/2017/06/death-penalty-symposium-court-increasingly-uncomfortable-death-penalty/. Published June 29, 2017. Accessed November 2, 2017.
- Pena-Rodriguez v Colorado, 580 US 15 606 (2017).
- Matal v Tam, 582 US 15 1293 (2017).
- Endrew F v Douglas County School District, RE-1, 580 US 15 827 (2017).
Summary of the Term
The Term was notable for the level of agreement. With 69 decided cases, 41 (69%) were unanimous. In 59 cases (85%), there was a strong consensus, with no more than 2 justices dissenting. Only 7 decisions (10%) were 5 to 4. Justice Kennedy was, as usual, the deciding vote in most of the close cases. He voted in the majority in 97% of the decisions. Justice Gorsuch took the place of Justice Scalia (who passed away in February 2016), so arguably the Court is ideologically close to where it has been for a number of years. Despite rumors that Justice Kennedy would announce his resignation from the Court, neither he nor any other justice has left. The Supreme Court began its new Term on October 2, 2017, with a full complement of 9 justices.
What’s to come
The Court will add cases through much of its new Term, but it has already accepted cases dealing with arbitration agreements (again); public employees’ union dues; immigration (again); the privacy of information held by mobile phone companies; a constitutional challenge to political gerrymandering; bakeries and gay-marriage ceremonies; whistleblowers and Dodd-Frank regulations; sports gambling and the NCAA; and more.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Kindred Nursing Centers, LP v Clark, 581 US 16 32 (2017).
- Anderson L. Looking Ahead: Pharma Projections for 2016 - & Beyond. Perma.cc Website. . Reviewed March 30, 2017. Accessed November 2, 2017.
- Sandoz Inc v Amgen Inc, 581 US 15 1039 1195 (2017).
- Bristol-Myers Squibb Co v Superior Court of California, San Francisco County, 582 US 16 466 (2017).
- Trinity FR, Sterling AM, Rogaczewski JD, et al. Motion for Leave to File and Brief for the Association of American Medical Colleges and Others as Amici Curiae Supporting Respondents. SCOTUSblog. http://www.scotusblog.com/wp-content/uploads/2017/06/16-1436-ac-AAMC-supporting-respondents.pdf. Accessed November 2, 2017.
- Donald J. Trump, President of the United States v International Refugee Assistance Project, 582 US 16 1436 (2017).
- Pavan v Smith, 582 US 16 992 (2017).
- Arkansas Code, §20 18 401(f)(1) (2014).
- Obergefell v Hodges, 576 US ___ (2015).
- NC Gen. Stat. Ann. §§14-202.5(a),(e).
- Packingham v North Carolina, 582 US 15 1194 (2017).
- Gloucester County School Board v G.G. SCOTUSblog. http://www.scotusblog.com/case-files/cases/gloucester-county-school-board-v-g-g/. Published March 6, 2017. Accessed November 2, 2017.
- Kindred Nursing Centers, LP v Clark, 581 US 16 32 (2017).
- Anderson L. Looking Ahead: Pharma Projections for 2016 - & Beyond. Perma.cc Website. . Reviewed March 30, 2017. Accessed November 2, 2017.
- Sandoz Inc v Amgen Inc, 581 US 15 1039 1195 (2017).
- Bristol-Myers Squibb Co v Superior Court of California, San Francisco County, 582 US 16 466 (2017).
- Trinity FR, Sterling AM, Rogaczewski JD, et al. Motion for Leave to File and Brief for the Association of American Medical Colleges and Others as Amici Curiae Supporting Respondents. SCOTUSblog. http://www.scotusblog.com/wp-content/uploads/2017/06/16-1436-ac-AAMC-supporting-respondents.pdf. Accessed November 2, 2017.
- Donald J. Trump, President of the United States v International Refugee Assistance Project, 582 US 16 1436 (2017).
- Pavan v Smith, 582 US 16 992 (2017).
- Arkansas Code, §20 18 401(f)(1) (2014).
- Obergefell v Hodges, 576 US ___ (2015).
- NC Gen. Stat. Ann. §§14-202.5(a),(e).
- Packingham v North Carolina, 582 US 15 1194 (2017).
- Gloucester County School Board v G.G. SCOTUSblog. http://www.scotusblog.com/case-files/cases/gloucester-county-school-board-v-g-g/. Published March 6, 2017. Accessed November 2, 2017.

Is Change in Personality an Early Sign of Dementia?
Evidence does not support the hypothesis that change in personality is a marker for preclinical mild cognitive impairment or dementia, according to research published online ahead of print September 20 in JAMA Psychiatry. Patients who subsequently develop dementia, however, appear to score higher on neuroticism and lower on conscientiousness and extraversion than people who remain cognitively healthy.
“These findings suggest that tracking change in self-rated personality as an early indicator of dementia is unlikely to be fruitful, while a single assessment provides reliable information on the personality traits that increase resilience (eg, conscientiousness) or vulnerability (eg, neuroticism) to clinical dementia,” said Antonio Terracciano, PhD, Associate Professor of Geriatrics at Florida State University College of Medicine in Tallahassee. 
Changes in behavior and personality are one criterion for the diagnosis of dementia. It is uncertain whether such changes begin before the clinical onset of the disease, however. Prospective studies of this question have either not assessed all five major dimensions of personality or administered assessments too close in time to the diagnosis of dementia.
Examining Change in Personality Traits
Dr. Terracciano and colleagues conducted a study to determine whether increases in neuroticism, declines in conscientiousness, and changes in other personality traits occur before the onset of mild cognitive impairment or dementia. They examined data for a cohort of 2,046 community-dwelling older adults who participated in the Baltimore Longitudinal Study of Aging. In the latter study, participants with no cognitive impairment underwent periodic personality and clinical assessments between 1980 and July 13, 2016. Follow-up lasted for as long as 36 years.
Participants completed the self-report version of the Revised NEO Personality Inventory (NEO-PI-R), or an earlier version, in 1980 and 1986. The NEO-PI-R is a 240-item questionnaire that assesses 30 facets of personality, including six for each of the following five major dimensions: neuroticism, extraversion, openness, agreeableness, and conscientiousness. In addition, participants were evaluated at enrollment for history of neurologic or cerebrovascular disease and for impairment of cognitive or behavioral functioning. Follow-up evaluations included a neuropsychologic battery and clinical examination, including an informant-and-participant-structured interview.
Patients With Dementia Had Higher Neuroticism Scores
Participants’ mean age at baseline was 62. In all, 931 participants (46%) were women. The population sample included 374 blacks (18%), 1,582 whites (77%), and 55 Asians or Pacific Islanders (3%). During the study, 104 participants (5%) developed mild cognitive impairment, and 255 (13%) developed all-cause dementia, including 194 (10%) who developed Alzheimer’s disease.
Change in personality before the onset of mild cognitive impairment or dementia was not significantly different between participants who remained unimpaired and those who developed Alzheimer’s disease. Change in personality for participants who developed mild cognitive impairment and all-cause dementia also was similar to that in unimpaired participants. Neuroticism and conscientiousness did not change significantly as disease onset approached.
One limitation of this study was that the population sample was selective and highly educated, said the authors. Another limitation was that the unimpaired participants were relatively young and could develop dementia in the future. “More research is needed on personality and Alzheimer’s disease biomarkers and how personality may increase resilience against neuropathology and forestall the emergence of clinical dementia,” said Dr. Terracciano.
—Erica Tricarico
Suggested Reading
Terracciano A, An Y, Sutin AR, et al. Personality change in the preclinical phase of Alzheimer disease. JAMA Psychiatry. 2017 Sep 20 [Epub ahead of print].
Evidence does not support the hypothesis that change in personality is a marker for preclinical mild cognitive impairment or dementia, according to research published online ahead of print September 20 in JAMA Psychiatry. Patients who subsequently develop dementia, however, appear to score higher on neuroticism and lower on conscientiousness and extraversion than people who remain cognitively healthy.
“These findings suggest that tracking change in self-rated personality as an early indicator of dementia is unlikely to be fruitful, while a single assessment provides reliable information on the personality traits that increase resilience (eg, conscientiousness) or vulnerability (eg, neuroticism) to clinical dementia,” said Antonio Terracciano, PhD, Associate Professor of Geriatrics at Florida State University College of Medicine in Tallahassee.
Changes in behavior and personality are one criterion for the diagnosis of dementia. It is uncertain whether such changes begin before the clinical onset of the disease, however. Prospective studies of this question have either not assessed all five major dimensions of personality or administered assessments too close in time to the diagnosis of dementia.
Examining Change in Personality Traits
Dr. Terracciano and colleagues conducted a study to determine whether increases in neuroticism, declines in conscientiousness, and changes in other personality traits occur before the onset of mild cognitive impairment or dementia. They examined data for a cohort of 2,046 community-dwelling older adults who participated in the Baltimore Longitudinal Study of Aging. In the latter study, participants with no cognitive impairment underwent periodic personality and clinical assessments between 1980 and July 13, 2016. Follow-up lasted for as long as 36 years.
Participants completed the self-report version of the Revised NEO Personality Inventory (NEO-PI-R), or an earlier version, in 1980 and 1986. The NEO-PI-R is a 240-item questionnaire that assesses 30 facets of personality, including six for each of the following five major dimensions: neuroticism, extraversion, openness, agreeableness, and conscientiousness. In addition, participants were evaluated at enrollment for history of neurologic or cerebrovascular disease and for impairment of cognitive or behavioral functioning. Follow-up evaluations included a neuropsychologic battery and clinical examination, including an informant-and-participant-structured interview.
Patients With Dementia Had Higher Neuroticism Scores
Participants’ mean age at baseline was 62. In all, 931 participants (46%) were women. The population sample included 374 blacks (18%), 1,582 whites (77%), and 55 Asians or Pacific Islanders (3%). During the study, 104 participants (5%) developed mild cognitive impairment, and 255 (13%) developed all-cause dementia, including 194 (10%) who developed Alzheimer’s disease.
Change in personality before the onset of mild cognitive impairment or dementia was not significantly different between participants who remained unimpaired and those who developed Alzheimer’s disease. Change in personality for participants who developed mild cognitive impairment and all-cause dementia also was similar to that in unimpaired participants. Neuroticism and conscientiousness did not change significantly as disease onset approached.
One limitation of this study was that the population sample was selective and highly educated, said the authors. Another limitation was that the unimpaired participants were relatively young and could develop dementia in the future. “More research is needed on personality and Alzheimer’s disease biomarkers and how personality may increase resilience against neuropathology and forestall the emergence of clinical dementia,” said Dr. Terracciano.
—Erica Tricarico
Suggested Reading
Terracciano A, An Y, Sutin AR, et al. Personality change in the preclinical phase of Alzheimer disease. JAMA Psychiatry. 2017 Sep 20 [Epub ahead of print].
Evidence does not support the hypothesis that change in personality is a marker for preclinical mild cognitive impairment or dementia, according to research published online ahead of print September 20 in JAMA Psychiatry. Patients who subsequently develop dementia, however, appear to score higher on neuroticism and lower on conscientiousness and extraversion than people who remain cognitively healthy.
“These findings suggest that tracking change in self-rated personality as an early indicator of dementia is unlikely to be fruitful, while a single assessment provides reliable information on the personality traits that increase resilience (eg, conscientiousness) or vulnerability (eg, neuroticism) to clinical dementia,” said Antonio Terracciano, PhD, Associate Professor of Geriatrics at Florida State University College of Medicine in Tallahassee.
Changes in behavior and personality are one criterion for the diagnosis of dementia. It is uncertain whether such changes begin before the clinical onset of the disease, however. Prospective studies of this question have either not assessed all five major dimensions of personality or administered assessments too close in time to the diagnosis of dementia.
Examining Change in Personality Traits
Dr. Terracciano and colleagues conducted a study to determine whether increases in neuroticism, declines in conscientiousness, and changes in other personality traits occur before the onset of mild cognitive impairment or dementia. They examined data for a cohort of 2,046 community-dwelling older adults who participated in the Baltimore Longitudinal Study of Aging. In the latter study, participants with no cognitive impairment underwent periodic personality and clinical assessments between 1980 and July 13, 2016. Follow-up lasted for as long as 36 years.
Participants completed the self-report version of the Revised NEO Personality Inventory (NEO-PI-R), or an earlier version, in 1980 and 1986. The NEO-PI-R is a 240-item questionnaire that assesses 30 facets of personality, including six for each of the following five major dimensions: neuroticism, extraversion, openness, agreeableness, and conscientiousness. In addition, participants were evaluated at enrollment for history of neurologic or cerebrovascular disease and for impairment of cognitive or behavioral functioning. Follow-up evaluations included a neuropsychologic battery and clinical examination, including an informant-and-participant-structured interview.
Patients With Dementia Had Higher Neuroticism Scores
Participants’ mean age at baseline was 62. In all, 931 participants (46%) were women. The population sample included 374 blacks (18%), 1,582 whites (77%), and 55 Asians or Pacific Islanders (3%). During the study, 104 participants (5%) developed mild cognitive impairment, and 255 (13%) developed all-cause dementia, including 194 (10%) who developed Alzheimer’s disease.
Change in personality before the onset of mild cognitive impairment or dementia was not significantly different between participants who remained unimpaired and those who developed Alzheimer’s disease. Change in personality for participants who developed mild cognitive impairment and all-cause dementia also was similar to that in unimpaired participants. Neuroticism and conscientiousness did not change significantly as disease onset approached.
One limitation of this study was that the population sample was selective and highly educated, said the authors. Another limitation was that the unimpaired participants were relatively young and could develop dementia in the future. “More research is needed on personality and Alzheimer’s disease biomarkers and how personality may increase resilience against neuropathology and forestall the emergence of clinical dementia,” said Dr. Terracciano.
—Erica Tricarico
Suggested Reading
Terracciano A, An Y, Sutin AR, et al. Personality change in the preclinical phase of Alzheimer disease. JAMA Psychiatry. 2017 Sep 20 [Epub ahead of print].
2017 Update on bone health
Bone health remains one of the most important health care concerns in the United States today. In 2004, the Surgeon General released a report on bone health and osteoporosis. According to the report’s introduction:
This first-ever Surgeon General’s Report on bone health and osteoporosis illustrates the large burden that bone disease places on our Nation and its citizens. Like other chronic diseases that disproportionately affect the elderly, the prevalence of bone disease and fractures is projected to increase markedly as the population ages. If these predictions come true, bone disease and fractures will have a tremendous negative impact on the future well-being of Americans. But as this report makes clear, they need not come true: by working together we can change the picture of aging in America. Osteoporosis and fractures…no longer should be thought of as an inevitable part of growing old. By focusing on prevention and lifestyle changes, including physical activity and nutrition, as well as early diagnosis and appropriate treatment, Americans can avoid much of the damaging impact of bone disease.1
Related article:
2016 Update on bone health
Although men also experience osteoporosis as they age, in women the rapid loss of bone at menopause makes their disease burden much greater. As women’s health care providers, we stand at the front line for preventing, diagnosing, and treating osteoporosis to reduce the impact of this disease. In this Update I focus on important information that has emerged in the past year.
Read about new ACP guidelines to assess fracture risk
Guidelines for therapy: How to assess fracture risk and when to treat
American College of Obstetricians and Gynecologists Committee on Practice Bulletins--Gynecology. ACOG Practice Bulletin No. 129: Osteoporosis. Obstet Gynecol. 2012;120(3):718-734.
Qaseem A, Forciea MA, McLean RM, Denberg TD; Clinical Guidelines Committee of the American College of Physicians. Treatment of low bone density or osteoporosis to prevent fractures in men and women: a clinical practice guideline update from the American College of Physicians. Ann Intern Med. 2017;166(11):818-839.
A crucial component for good bone health maintenance and osteoporotic fracture prevention is understanding the current guidelines for therapy. The most recent practice bulletin of the American College of Obstetricians and Gynecologists (ACOG) on osteoporosis was published in 2012. ACOG states that treatment be recommended for women who have a bone mineral density (BMD) T-score of -2.5 or lower.
For women in the low bone mass category (T-score between -1 and -2.5), use of the Fracture Risk Assessment Tool (FRAX) calculator can assist in making an informed treatment decision.2 Based on the FRAX calculator, women who have a 10-year risk of major osteoporotic fracture of 20% or greater, or a risk of hip fracture of 3% or greater, are candidates for pharmacologic therapy.
Women who have experienced a low-trauma fracture (especially of the vertebra or hip) also are candidates for treatment, even in the absence of osteoporosis on a dual-energy x-ray absorptiometry (DXA) report.
Related article:
Women’s Preventive Services Initiative Guidelines provide consensus for practicing ObGyns
Updated recommendations from the ACP
The 2017 guideline published by the American College of Physicians (ACP), whose target audience is "all clinicians," recommends that, for women who have known osteoporosis, clinicians offer pharmacologic treatment with alendronate, risedronate, zoledronic acid, or denosumab to reduce the risk for hip and vertebral fractures.
In addition, the ACP recommends that clinicians make the decision whether or not to treat osteopenic women 65 years of age or older who are at a high risk for fracture based on a discussion of patient preferences, fracture risk profile, and benefits, harms, and costs of medications. This may seem somewhat contradictory to ACOG's guidance vis-a-vis women younger than 65 years of age.
The ACP further states that given the limited evidence supporting the benefit of treatment, the balance of benefits and harms in treating osteopenic women is most favorable when the risk for fracture is high. Women younger than 65 years with osteopenia and women older than 65 years with mild osteopenia (T-score between -1.0 and -1.5) will benefit less than women who are 65 years of age or older with severe osteopenia (T-score <-2.0).
Risk factors and risk assessment tools
Clinicians can use their own judgment based on risk factors for fracture (lower body weight, smoking, weight loss, family history of fractures, decreased physical activity, alcohol or caffeine use, low calcium and vitamin D intake, corticosteroid use), or they can use a risk assessment tool. Several risk assessment tools, such as the FRAX calculator mentioned earlier, are available to predict fracture risk among untreated people with low bone density. Although the FRAX calculator is widely used, there is no evidence from randomized controlled trials demonstrating a benefit of fracture reduction when FRAX scores are used in treatment decision making.
Duration of therapy. The ACP recommends that clinicians treat osteoporotic women with pharmacologic therapy for 5 years. Bone density monitoring is not recommended during the 5-year treatment period for osteoporosis in women; current evidence does not show any benefit for bone density monitoring during treatment.
Moderate-quality evidence demonstrated that women treated with antiresorptive therapies (including bisphosphonates, raloxifene, and teriparatide) benefited from reduced fractures, even if no increase in BMD occurred or if BMD decreased.
As before, all women with osteoporosis or a previous low-trauma fracture should be treated. Use of the FRAX calculator should involve clinician judgment, and other risk factors should be taken into account. For most women, treatment should be continued for 5 years. There is no benefit in continued bone mass assessment (DXA testing) while a patient is on pharmacologic therapy.
Read about fracture risk after stopping HT
Another WHI update: No increase in fractures after stopping HT
Watts NB, Cauley JA, Jackson RD, et al; Women's Health Initiative Investigators. No increase in fractures after stopping hormone therapy: results from the Women's Health Initiative. J Clin Endocrinol Metab. 2017;102(1):302-308.
The analysis and reanalysis of the Women's Health Initiative (WHI) trial data seems never-ending, yet the article by Watts and colleagues is important. Although the WHI hormone therapy (HT) trials showed that treatment protects against hip and total fractures, a later observational report suggested loss of benefit and rebound increased risk after HT was discontinued.3 The purpose of the Watts' study was to examine fractures after stopping HT.
Related article:
Did long-term follow-up of WHI participants reveal any mortality increase among women who received HT?
Details of the study
Two placebo-controlled randomized trials served as the study setting. The study included WHI participants (n = 15,187) who continued to take active HT or placebo through the intervention period and who did not take HT in the postintervention period. The trial interventions included conjugated equine estrogen (CEE) plus medroxyprogesterone acetate (MPA) for women with natural menopause and CEE alone for women with prior hysterectomy. The investigators recorded total fractures and hip fractures through 5 years after HT discontinuation.
Findings on fractures. Hip fractures occurred infrequently, with approximately 2.5 per 1,000 person-years. This finding was similar between trials and in former HT users and placebo groups.
No difference was found in total fractures in the CEE plus MPA trial for former HT users compared with former placebo users (28.9 per 1,000 person-years and 29.9 per 1,000 person-years, respectively; hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.87-1.09; P = .63). In the CEE-alone trial, however, total fractures were higher in former placebo users (36.9 per 1,000 person-years) compared with the former active-treatment group (31.1 per 1,000 person-years). This finding suggests a residual benefit of CEE in reducing total fractures (HR, 0.85; 95% CI, 0.73-0.98; P = .03).
Investigators' takeaway. The authors concluded that, after discontinuing HT, there was no evidence of increased fracture risk (sustained or transient) in former HT users compared with former placebo users. In the CEE-alone trial, there was a residual benefit for total fracture reduction in former HT users compared with placebo users.
Gynecologists have long believed that on stopping HT, the loss of bone mass will follow at the same rate as it would at natural menopause. These WHI trials demonstrate, however, that through 5 years, women who stopped HT had no increase in hip or total fractures, and hysterectomized women who stopped estrogen therapy actually had fewer fractures than the placebo group. Keep in mind that this large cohort was not chosen based on risk of osteoporotic fractures. In fact, baseline bone mass was not even measured in these women, making the results even more "real world."
Read about reassessing FRAX scores
A new look at fracture risk assessment scores
Gourlay ML, Overman RA, Fine JP, et al; Women's Health Initiative Investigators. Time to clinically relevant fracture risk scores in postmenopausal women. Am J Med. 2017;130:862.e15-e23.
Jiang X, Gruner M, Trémollieres F, et al. Diagnostic accuracy of FRAX in predicting the 10-year risk of osteoporotic fractures using the USA treatment thresholds: a systematic review and meta-analysis. Bone. 2017;99:20-25.
The FRAX score has become a popular form of triage for women who do not yet meet the bone mass criteria of osteoporosis. Current practice guidelines recommend use of fracture risk scores for screening and pharmacologic therapeutic decision making. Some newer data, however, may give rise to questions about its utility, especially in younger women.
Fracture risk analysis in a large postmenopausal population
Gourlay and colleagues conducted a retrospective competing risk analysis of new occurrence of treatment-level and screening-level fracture risk scores. Study participants were postmenopausal women aged 50 years and older who had not previously received pharmacologic treatment and had not had a first hip or clinical vertebral facture.
Details of the study
In 54,280 postmenopausal women aged 50 to 64 years who did not have a bone mineral density test, the time for 10% to develop a treatment-level FRAX score could not be estimated accurately because the incidence of treatment-level scores was rare.
A total of 6,096 women had FRAX scores calculated with bone mineral density testing. In this group, the estimated unadjusted time to treatment-level FRAX scores was 7.6 years (95% CI, 6.6-8.7) for those aged 65 to 69, and 5.1 years (95% CI, 3.5-7.5) for women aged 75 to 79 at baseline.
Of 17,967 women aged 50 to 64 who had a screening-level FRAX at baseline, 100 (0.6%) experienced a hip or clinical vertebral fracture by age 65 years.
Age is key factor. Gourlay and colleagues concluded that postmenopausal women who had subthreshold fracture risk scores at baseline would be unlikely to develop a treatment-level FRAX score between ages 50 and 64. The increased incidence of treatment-level fracture risk scores, osteoporosis, and major osteoporotic fracture after age 65, however, supports more frequent consideration of FRAX assessment and bone mineral density testing.
Related article:
2015 Update on osteoporosis
Meta-analysis of FRAX tool accuracy
In another study, Jiang and colleagues conducted a systematic review and meta-analysis to determine how the FRAX score performed in predicting the 10-year risk of major osteoporotic fractures and hip fractures. The investigators used the US treatment thresholds.
Details of the study
Seven studies (n = 57,027) were analyzed to assess the diagnostic accuracy of FRAX in predicting major osteoporotic fractures; 20% was used as the 10-year fracture risk threshold for intervention. The mean sensitivity and specificity, along with their 95% CIs, were 10.25% (3.76%-25.06%) and 97.02% (91.17%-99.03%), respectively.
For hip fracture prediction, 6 studies (n = 50,944) were analyzed, and 3% was used as the 10-year fracture risk threshold. The mean sensitivity and specificity, along with their 95% CIs, were 45.70% (24.88%-68.13%) and 84.70% (76.41%-90.44%), respectively.
Predictive value of FRAX. The authors concluded that, using the 10-year intervention thresholds of 20% for major osteoporotic fracture and 3% for hip fracture, FRAX performed better in identifying individuals who will not have a major osteoporotic fracture or hip fracture within 10 years than in identifying those who will experience a fracture. A substantial number of those who developed fractures, especially major osteoporotic fracture within 10 years of follow up, were missed by the baseline FRAX assessment.
Increasing age is still arguably among the most important factors for decreasing bone health. Older women are more likely to develop treatment-level FRAX scores more quickly than younger women. In addition, the FRAX tool is better in predicting which women will not develop a fracture in the next 10 years than in predicting those who will experience a fracture.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- United States Office of the Surgeon General. Bone health and osteoporosis: a report of the Surgeon General. Rockville, Maryland: Office of the Surgeon General (US); 2004. https://www.ncbi.nlm.nih.gov/books/NBK45513/. Accessed November 6, 2017.
- Centre for Metabolic Bone Diseases, University of Sheffield, United Kingdom. FRAX Fracture Risk Assessment Tool website. www.sheffield.ac.uk/FRAX. Accessed November 6, 2017.
- Yates J, Barrett-Connor E, Barlas S, Chen YT, Miller PD, Siris ES. Rapid loss of hip fracture protection after estrogen cessation: evidence from the National Osteoporosis Risk Assessment. Obstet Gynecol. 2004;103(3):440–446.

Dr. Goldstein is Professor in the Department of Obstetrics and Gynecology, New York
University School of Medicine, and Director of Gynecologic Ultrasound and Co-
Director of Bone Densitometry and Body Composition at New York University Medical
Center in New York, New York. He serves on the OBG Management Board of Editors.
The author reports no financial relationships relevant to this article
Dr. Goldstein is Professor in the Department of Obstetrics and Gynecology, New York
University School of Medicine, and Director of Gynecologic Ultrasound and Co-
Director of Bone Densitometry and Body Composition at New York University Medical
Center in New York, New York. He serves on the OBG Management Board of Editors.
The author reports no financial relationships relevant to this article
Dr. Goldstein is Professor in the Department of Obstetrics and Gynecology, New York
University School of Medicine, and Director of Gynecologic Ultrasound and Co-
Director of Bone Densitometry and Body Composition at New York University Medical
Center in New York, New York. He serves on the OBG Management Board of Editors.
The author reports no financial relationships relevant to this article
Bone health remains one of the most important health care concerns in the United States today. In 2004, the Surgeon General released a report on bone health and osteoporosis. According to the report’s introduction:
This first-ever Surgeon General’s Report on bone health and osteoporosis illustrates the large burden that bone disease places on our Nation and its citizens. Like other chronic diseases that disproportionately affect the elderly, the prevalence of bone disease and fractures is projected to increase markedly as the population ages. If these predictions come true, bone disease and fractures will have a tremendous negative impact on the future well-being of Americans. But as this report makes clear, they need not come true: by working together we can change the picture of aging in America. Osteoporosis and fractures…no longer should be thought of as an inevitable part of growing old. By focusing on prevention and lifestyle changes, including physical activity and nutrition, as well as early diagnosis and appropriate treatment, Americans can avoid much of the damaging impact of bone disease.1
Related article:
2016 Update on bone health
Although men also experience osteoporosis as they age, in women the rapid loss of bone at menopause makes their disease burden much greater. As women’s health care providers, we stand at the front line for preventing, diagnosing, and treating osteoporosis to reduce the impact of this disease. In this Update I focus on important information that has emerged in the past year.
Read about new ACP guidelines to assess fracture risk
Guidelines for therapy: How to assess fracture risk and when to treat
American College of Obstetricians and Gynecologists Committee on Practice Bulletins--Gynecology. ACOG Practice Bulletin No. 129: Osteoporosis. Obstet Gynecol. 2012;120(3):718-734.
Qaseem A, Forciea MA, McLean RM, Denberg TD; Clinical Guidelines Committee of the American College of Physicians. Treatment of low bone density or osteoporosis to prevent fractures in men and women: a clinical practice guideline update from the American College of Physicians. Ann Intern Med. 2017;166(11):818-839.
A crucial component for good bone health maintenance and osteoporotic fracture prevention is understanding the current guidelines for therapy. The most recent practice bulletin of the American College of Obstetricians and Gynecologists (ACOG) on osteoporosis was published in 2012. ACOG states that treatment be recommended for women who have a bone mineral density (BMD) T-score of -2.5 or lower.
For women in the low bone mass category (T-score between -1 and -2.5), use of the Fracture Risk Assessment Tool (FRAX) calculator can assist in making an informed treatment decision.2 Based on the FRAX calculator, women who have a 10-year risk of major osteoporotic fracture of 20% or greater, or a risk of hip fracture of 3% or greater, are candidates for pharmacologic therapy.
Women who have experienced a low-trauma fracture (especially of the vertebra or hip) also are candidates for treatment, even in the absence of osteoporosis on a dual-energy x-ray absorptiometry (DXA) report.
Related article:
Women’s Preventive Services Initiative Guidelines provide consensus for practicing ObGyns
Updated recommendations from the ACP
The 2017 guideline published by the American College of Physicians (ACP), whose target audience is "all clinicians," recommends that, for women who have known osteoporosis, clinicians offer pharmacologic treatment with alendronate, risedronate, zoledronic acid, or denosumab to reduce the risk for hip and vertebral fractures.
In addition, the ACP recommends that clinicians make the decision whether or not to treat osteopenic women 65 years of age or older who are at a high risk for fracture based on a discussion of patient preferences, fracture risk profile, and benefits, harms, and costs of medications. This may seem somewhat contradictory to ACOG's guidance vis-a-vis women younger than 65 years of age.
The ACP further states that given the limited evidence supporting the benefit of treatment, the balance of benefits and harms in treating osteopenic women is most favorable when the risk for fracture is high. Women younger than 65 years with osteopenia and women older than 65 years with mild osteopenia (T-score between -1.0 and -1.5) will benefit less than women who are 65 years of age or older with severe osteopenia (T-score <-2.0).
Risk factors and risk assessment tools
Clinicians can use their own judgment based on risk factors for fracture (lower body weight, smoking, weight loss, family history of fractures, decreased physical activity, alcohol or caffeine use, low calcium and vitamin D intake, corticosteroid use), or they can use a risk assessment tool. Several risk assessment tools, such as the FRAX calculator mentioned earlier, are available to predict fracture risk among untreated people with low bone density. Although the FRAX calculator is widely used, there is no evidence from randomized controlled trials demonstrating a benefit of fracture reduction when FRAX scores are used in treatment decision making.
Duration of therapy. The ACP recommends that clinicians treat osteoporotic women with pharmacologic therapy for 5 years. Bone density monitoring is not recommended during the 5-year treatment period for osteoporosis in women; current evidence does not show any benefit for bone density monitoring during treatment.
Moderate-quality evidence demonstrated that women treated with antiresorptive therapies (including bisphosphonates, raloxifene, and teriparatide) benefited from reduced fractures, even if no increase in BMD occurred or if BMD decreased.
As before, all women with osteoporosis or a previous low-trauma fracture should be treated. Use of the FRAX calculator should involve clinician judgment, and other risk factors should be taken into account. For most women, treatment should be continued for 5 years. There is no benefit in continued bone mass assessment (DXA testing) while a patient is on pharmacologic therapy.
Read about fracture risk after stopping HT
Another WHI update: No increase in fractures after stopping HT
Watts NB, Cauley JA, Jackson RD, et al; Women's Health Initiative Investigators. No increase in fractures after stopping hormone therapy: results from the Women's Health Initiative. J Clin Endocrinol Metab. 2017;102(1):302-308.
The analysis and reanalysis of the Women's Health Initiative (WHI) trial data seems never-ending, yet the article by Watts and colleagues is important. Although the WHI hormone therapy (HT) trials showed that treatment protects against hip and total fractures, a later observational report suggested loss of benefit and rebound increased risk after HT was discontinued.3 The purpose of the Watts' study was to examine fractures after stopping HT.
Related article:
Did long-term follow-up of WHI participants reveal any mortality increase among women who received HT?
Details of the study
Two placebo-controlled randomized trials served as the study setting. The study included WHI participants (n = 15,187) who continued to take active HT or placebo through the intervention period and who did not take HT in the postintervention period. The trial interventions included conjugated equine estrogen (CEE) plus medroxyprogesterone acetate (MPA) for women with natural menopause and CEE alone for women with prior hysterectomy. The investigators recorded total fractures and hip fractures through 5 years after HT discontinuation.
Findings on fractures. Hip fractures occurred infrequently, with approximately 2.5 per 1,000 person-years. This finding was similar between trials and in former HT users and placebo groups.
No difference was found in total fractures in the CEE plus MPA trial for former HT users compared with former placebo users (28.9 per 1,000 person-years and 29.9 per 1,000 person-years, respectively; hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.87-1.09; P = .63). In the CEE-alone trial, however, total fractures were higher in former placebo users (36.9 per 1,000 person-years) compared with the former active-treatment group (31.1 per 1,000 person-years). This finding suggests a residual benefit of CEE in reducing total fractures (HR, 0.85; 95% CI, 0.73-0.98; P = .03).
Investigators' takeaway. The authors concluded that, after discontinuing HT, there was no evidence of increased fracture risk (sustained or transient) in former HT users compared with former placebo users. In the CEE-alone trial, there was a residual benefit for total fracture reduction in former HT users compared with placebo users.
Gynecologists have long believed that on stopping HT, the loss of bone mass will follow at the same rate as it would at natural menopause. These WHI trials demonstrate, however, that through 5 years, women who stopped HT had no increase in hip or total fractures, and hysterectomized women who stopped estrogen therapy actually had fewer fractures than the placebo group. Keep in mind that this large cohort was not chosen based on risk of osteoporotic fractures. In fact, baseline bone mass was not even measured in these women, making the results even more "real world."
Read about reassessing FRAX scores
A new look at fracture risk assessment scores
Gourlay ML, Overman RA, Fine JP, et al; Women's Health Initiative Investigators. Time to clinically relevant fracture risk scores in postmenopausal women. Am J Med. 2017;130:862.e15-e23.
Jiang X, Gruner M, Trémollieres F, et al. Diagnostic accuracy of FRAX in predicting the 10-year risk of osteoporotic fractures using the USA treatment thresholds: a systematic review and meta-analysis. Bone. 2017;99:20-25.
The FRAX score has become a popular form of triage for women who do not yet meet the bone mass criteria of osteoporosis. Current practice guidelines recommend use of fracture risk scores for screening and pharmacologic therapeutic decision making. Some newer data, however, may give rise to questions about its utility, especially in younger women.
Fracture risk analysis in a large postmenopausal population
Gourlay and colleagues conducted a retrospective competing risk analysis of new occurrence of treatment-level and screening-level fracture risk scores. Study participants were postmenopausal women aged 50 years and older who had not previously received pharmacologic treatment and had not had a first hip or clinical vertebral facture.
Details of the study
In 54,280 postmenopausal women aged 50 to 64 years who did not have a bone mineral density test, the time for 10% to develop a treatment-level FRAX score could not be estimated accurately because the incidence of treatment-level scores was rare.
A total of 6,096 women had FRAX scores calculated with bone mineral density testing. In this group, the estimated unadjusted time to treatment-level FRAX scores was 7.6 years (95% CI, 6.6-8.7) for those aged 65 to 69, and 5.1 years (95% CI, 3.5-7.5) for women aged 75 to 79 at baseline.
Of 17,967 women aged 50 to 64 who had a screening-level FRAX at baseline, 100 (0.6%) experienced a hip or clinical vertebral fracture by age 65 years.
Age is key factor. Gourlay and colleagues concluded that postmenopausal women who had subthreshold fracture risk scores at baseline would be unlikely to develop a treatment-level FRAX score between ages 50 and 64. The increased incidence of treatment-level fracture risk scores, osteoporosis, and major osteoporotic fracture after age 65, however, supports more frequent consideration of FRAX assessment and bone mineral density testing.
Related article:
2015 Update on osteoporosis
Meta-analysis of FRAX tool accuracy
In another study, Jiang and colleagues conducted a systematic review and meta-analysis to determine how the FRAX score performed in predicting the 10-year risk of major osteoporotic fractures and hip fractures. The investigators used the US treatment thresholds.
Details of the study
Seven studies (n = 57,027) were analyzed to assess the diagnostic accuracy of FRAX in predicting major osteoporotic fractures; 20% was used as the 10-year fracture risk threshold for intervention. The mean sensitivity and specificity, along with their 95% CIs, were 10.25% (3.76%-25.06%) and 97.02% (91.17%-99.03%), respectively.
For hip fracture prediction, 6 studies (n = 50,944) were analyzed, and 3% was used as the 10-year fracture risk threshold. The mean sensitivity and specificity, along with their 95% CIs, were 45.70% (24.88%-68.13%) and 84.70% (76.41%-90.44%), respectively.
Predictive value of FRAX. The authors concluded that, using the 10-year intervention thresholds of 20% for major osteoporotic fracture and 3% for hip fracture, FRAX performed better in identifying individuals who will not have a major osteoporotic fracture or hip fracture within 10 years than in identifying those who will experience a fracture. A substantial number of those who developed fractures, especially major osteoporotic fracture within 10 years of follow up, were missed by the baseline FRAX assessment.
Increasing age is still arguably among the most important factors for decreasing bone health. Older women are more likely to develop treatment-level FRAX scores more quickly than younger women. In addition, the FRAX tool is better in predicting which women will not develop a fracture in the next 10 years than in predicting those who will experience a fracture.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
Bone health remains one of the most important health care concerns in the United States today. In 2004, the Surgeon General released a report on bone health and osteoporosis. According to the report’s introduction:
This first-ever Surgeon General’s Report on bone health and osteoporosis illustrates the large burden that bone disease places on our Nation and its citizens. Like other chronic diseases that disproportionately affect the elderly, the prevalence of bone disease and fractures is projected to increase markedly as the population ages. If these predictions come true, bone disease and fractures will have a tremendous negative impact on the future well-being of Americans. But as this report makes clear, they need not come true: by working together we can change the picture of aging in America. Osteoporosis and fractures…no longer should be thought of as an inevitable part of growing old. By focusing on prevention and lifestyle changes, including physical activity and nutrition, as well as early diagnosis and appropriate treatment, Americans can avoid much of the damaging impact of bone disease.1
Related article:
2016 Update on bone health
Although men also experience osteoporosis as they age, in women the rapid loss of bone at menopause makes their disease burden much greater. As women’s health care providers, we stand at the front line for preventing, diagnosing, and treating osteoporosis to reduce the impact of this disease. In this Update I focus on important information that has emerged in the past year.
Read about new ACP guidelines to assess fracture risk
Guidelines for therapy: How to assess fracture risk and when to treat
American College of Obstetricians and Gynecologists Committee on Practice Bulletins--Gynecology. ACOG Practice Bulletin No. 129: Osteoporosis. Obstet Gynecol. 2012;120(3):718-734.
Qaseem A, Forciea MA, McLean RM, Denberg TD; Clinical Guidelines Committee of the American College of Physicians. Treatment of low bone density or osteoporosis to prevent fractures in men and women: a clinical practice guideline update from the American College of Physicians. Ann Intern Med. 2017;166(11):818-839.
A crucial component for good bone health maintenance and osteoporotic fracture prevention is understanding the current guidelines for therapy. The most recent practice bulletin of the American College of Obstetricians and Gynecologists (ACOG) on osteoporosis was published in 2012. ACOG states that treatment be recommended for women who have a bone mineral density (BMD) T-score of -2.5 or lower.
For women in the low bone mass category (T-score between -1 and -2.5), use of the Fracture Risk Assessment Tool (FRAX) calculator can assist in making an informed treatment decision.2 Based on the FRAX calculator, women who have a 10-year risk of major osteoporotic fracture of 20% or greater, or a risk of hip fracture of 3% or greater, are candidates for pharmacologic therapy.
Women who have experienced a low-trauma fracture (especially of the vertebra or hip) also are candidates for treatment, even in the absence of osteoporosis on a dual-energy x-ray absorptiometry (DXA) report.
Related article:
Women’s Preventive Services Initiative Guidelines provide consensus for practicing ObGyns
Updated recommendations from the ACP
The 2017 guideline published by the American College of Physicians (ACP), whose target audience is "all clinicians," recommends that, for women who have known osteoporosis, clinicians offer pharmacologic treatment with alendronate, risedronate, zoledronic acid, or denosumab to reduce the risk for hip and vertebral fractures.
In addition, the ACP recommends that clinicians make the decision whether or not to treat osteopenic women 65 years of age or older who are at a high risk for fracture based on a discussion of patient preferences, fracture risk profile, and benefits, harms, and costs of medications. This may seem somewhat contradictory to ACOG's guidance vis-a-vis women younger than 65 years of age.
The ACP further states that given the limited evidence supporting the benefit of treatment, the balance of benefits and harms in treating osteopenic women is most favorable when the risk for fracture is high. Women younger than 65 years with osteopenia and women older than 65 years with mild osteopenia (T-score between -1.0 and -1.5) will benefit less than women who are 65 years of age or older with severe osteopenia (T-score <-2.0).
Risk factors and risk assessment tools
Clinicians can use their own judgment based on risk factors for fracture (lower body weight, smoking, weight loss, family history of fractures, decreased physical activity, alcohol or caffeine use, low calcium and vitamin D intake, corticosteroid use), or they can use a risk assessment tool. Several risk assessment tools, such as the FRAX calculator mentioned earlier, are available to predict fracture risk among untreated people with low bone density. Although the FRAX calculator is widely used, there is no evidence from randomized controlled trials demonstrating a benefit of fracture reduction when FRAX scores are used in treatment decision making.
Duration of therapy. The ACP recommends that clinicians treat osteoporotic women with pharmacologic therapy for 5 years. Bone density monitoring is not recommended during the 5-year treatment period for osteoporosis in women; current evidence does not show any benefit for bone density monitoring during treatment.
Moderate-quality evidence demonstrated that women treated with antiresorptive therapies (including bisphosphonates, raloxifene, and teriparatide) benefited from reduced fractures, even if no increase in BMD occurred or if BMD decreased.
As before, all women with osteoporosis or a previous low-trauma fracture should be treated. Use of the FRAX calculator should involve clinician judgment, and other risk factors should be taken into account. For most women, treatment should be continued for 5 years. There is no benefit in continued bone mass assessment (DXA testing) while a patient is on pharmacologic therapy.
Read about fracture risk after stopping HT
Another WHI update: No increase in fractures after stopping HT
Watts NB, Cauley JA, Jackson RD, et al; Women's Health Initiative Investigators. No increase in fractures after stopping hormone therapy: results from the Women's Health Initiative. J Clin Endocrinol Metab. 2017;102(1):302-308.
The analysis and reanalysis of the Women's Health Initiative (WHI) trial data seems never-ending, yet the article by Watts and colleagues is important. Although the WHI hormone therapy (HT) trials showed that treatment protects against hip and total fractures, a later observational report suggested loss of benefit and rebound increased risk after HT was discontinued.3 The purpose of the Watts' study was to examine fractures after stopping HT.
Related article:
Did long-term follow-up of WHI participants reveal any mortality increase among women who received HT?
Details of the study
Two placebo-controlled randomized trials served as the study setting. The study included WHI participants (n = 15,187) who continued to take active HT or placebo through the intervention period and who did not take HT in the postintervention period. The trial interventions included conjugated equine estrogen (CEE) plus medroxyprogesterone acetate (MPA) for women with natural menopause and CEE alone for women with prior hysterectomy. The investigators recorded total fractures and hip fractures through 5 years after HT discontinuation.
Findings on fractures. Hip fractures occurred infrequently, with approximately 2.5 per 1,000 person-years. This finding was similar between trials and in former HT users and placebo groups.
No difference was found in total fractures in the CEE plus MPA trial for former HT users compared with former placebo users (28.9 per 1,000 person-years and 29.9 per 1,000 person-years, respectively; hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.87-1.09; P = .63). In the CEE-alone trial, however, total fractures were higher in former placebo users (36.9 per 1,000 person-years) compared with the former active-treatment group (31.1 per 1,000 person-years). This finding suggests a residual benefit of CEE in reducing total fractures (HR, 0.85; 95% CI, 0.73-0.98; P = .03).
Investigators' takeaway. The authors concluded that, after discontinuing HT, there was no evidence of increased fracture risk (sustained or transient) in former HT users compared with former placebo users. In the CEE-alone trial, there was a residual benefit for total fracture reduction in former HT users compared with placebo users.
Gynecologists have long believed that on stopping HT, the loss of bone mass will follow at the same rate as it would at natural menopause. These WHI trials demonstrate, however, that through 5 years, women who stopped HT had no increase in hip or total fractures, and hysterectomized women who stopped estrogen therapy actually had fewer fractures than the placebo group. Keep in mind that this large cohort was not chosen based on risk of osteoporotic fractures. In fact, baseline bone mass was not even measured in these women, making the results even more "real world."
Read about reassessing FRAX scores
A new look at fracture risk assessment scores
Gourlay ML, Overman RA, Fine JP, et al; Women's Health Initiative Investigators. Time to clinically relevant fracture risk scores in postmenopausal women. Am J Med. 2017;130:862.e15-e23.
Jiang X, Gruner M, Trémollieres F, et al. Diagnostic accuracy of FRAX in predicting the 10-year risk of osteoporotic fractures using the USA treatment thresholds: a systematic review and meta-analysis. Bone. 2017;99:20-25.
The FRAX score has become a popular form of triage for women who do not yet meet the bone mass criteria of osteoporosis. Current practice guidelines recommend use of fracture risk scores for screening and pharmacologic therapeutic decision making. Some newer data, however, may give rise to questions about its utility, especially in younger women.
Fracture risk analysis in a large postmenopausal population
Gourlay and colleagues conducted a retrospective competing risk analysis of new occurrence of treatment-level and screening-level fracture risk scores. Study participants were postmenopausal women aged 50 years and older who had not previously received pharmacologic treatment and had not had a first hip or clinical vertebral facture.
Details of the study
In 54,280 postmenopausal women aged 50 to 64 years who did not have a bone mineral density test, the time for 10% to develop a treatment-level FRAX score could not be estimated accurately because the incidence of treatment-level scores was rare.
A total of 6,096 women had FRAX scores calculated with bone mineral density testing. In this group, the estimated unadjusted time to treatment-level FRAX scores was 7.6 years (95% CI, 6.6-8.7) for those aged 65 to 69, and 5.1 years (95% CI, 3.5-7.5) for women aged 75 to 79 at baseline.
Of 17,967 women aged 50 to 64 who had a screening-level FRAX at baseline, 100 (0.6%) experienced a hip or clinical vertebral fracture by age 65 years.
Age is key factor. Gourlay and colleagues concluded that postmenopausal women who had subthreshold fracture risk scores at baseline would be unlikely to develop a treatment-level FRAX score between ages 50 and 64. The increased incidence of treatment-level fracture risk scores, osteoporosis, and major osteoporotic fracture after age 65, however, supports more frequent consideration of FRAX assessment and bone mineral density testing.
Related article:
2015 Update on osteoporosis
Meta-analysis of FRAX tool accuracy
In another study, Jiang and colleagues conducted a systematic review and meta-analysis to determine how the FRAX score performed in predicting the 10-year risk of major osteoporotic fractures and hip fractures. The investigators used the US treatment thresholds.
Details of the study
Seven studies (n = 57,027) were analyzed to assess the diagnostic accuracy of FRAX in predicting major osteoporotic fractures; 20% was used as the 10-year fracture risk threshold for intervention. The mean sensitivity and specificity, along with their 95% CIs, were 10.25% (3.76%-25.06%) and 97.02% (91.17%-99.03%), respectively.
For hip fracture prediction, 6 studies (n = 50,944) were analyzed, and 3% was used as the 10-year fracture risk threshold. The mean sensitivity and specificity, along with their 95% CIs, were 45.70% (24.88%-68.13%) and 84.70% (76.41%-90.44%), respectively.
Predictive value of FRAX. The authors concluded that, using the 10-year intervention thresholds of 20% for major osteoporotic fracture and 3% for hip fracture, FRAX performed better in identifying individuals who will not have a major osteoporotic fracture or hip fracture within 10 years than in identifying those who will experience a fracture. A substantial number of those who developed fractures, especially major osteoporotic fracture within 10 years of follow up, were missed by the baseline FRAX assessment.
Increasing age is still arguably among the most important factors for decreasing bone health. Older women are more likely to develop treatment-level FRAX scores more quickly than younger women. In addition, the FRAX tool is better in predicting which women will not develop a fracture in the next 10 years than in predicting those who will experience a fracture.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- United States Office of the Surgeon General. Bone health and osteoporosis: a report of the Surgeon General. Rockville, Maryland: Office of the Surgeon General (US); 2004. https://www.ncbi.nlm.nih.gov/books/NBK45513/. Accessed November 6, 2017.
- Centre for Metabolic Bone Diseases, University of Sheffield, United Kingdom. FRAX Fracture Risk Assessment Tool website. www.sheffield.ac.uk/FRAX. Accessed November 6, 2017.
- Yates J, Barrett-Connor E, Barlas S, Chen YT, Miller PD, Siris ES. Rapid loss of hip fracture protection after estrogen cessation: evidence from the National Osteoporosis Risk Assessment. Obstet Gynecol. 2004;103(3):440–446.
- United States Office of the Surgeon General. Bone health and osteoporosis: a report of the Surgeon General. Rockville, Maryland: Office of the Surgeon General (US); 2004. https://www.ncbi.nlm.nih.gov/books/NBK45513/. Accessed November 6, 2017.
- Centre for Metabolic Bone Diseases, University of Sheffield, United Kingdom. FRAX Fracture Risk Assessment Tool website. www.sheffield.ac.uk/FRAX. Accessed November 6, 2017.
- Yates J, Barrett-Connor E, Barlas S, Chen YT, Miller PD, Siris ES. Rapid loss of hip fracture protection after estrogen cessation: evidence from the National Osteoporosis Risk Assessment. Obstet Gynecol. 2004;103(3):440–446.
Recognize and treat iron deficiency anemia in pregnant women
All mammalian life is dependent on a continuous supply of molecular oxygen. Molecular oxygen is carried to cells by noncovalent binding to the iron moiety in the hemoglobin of red blood cells. It is utilized within cells by noncovalent binding to the iron moiety in various microsomal and mitochondrial proteins, including myoglobin and cytochromes. Consequently, to efficiently utilize molecular oxygen all mammalian life is dependent on an adequate supply of iron. Surprisingly, in an era of high technology precision medicine, many pregnant women are iron deficient, anemic, and not receiving adequate iron supplementation.
Iron deficiency is prevalent in women and pregnant women
Women often become iron deficient because of pregnancy or heavy menstrual bleeding. During pregnancy, maternal iron is provided to supply the needs of the fetus and placenta. Additional iron is needed to expand maternal red blood cell volume and replace iron lost due to bleeding at delivery. In the National Health and Nutrition Examination Survey (NHANES) of 1988–1994, 11% of women aged 16 to 49 years were iron deficient. By contrast, less than 1% of men aged 16 to 49 years were iron deficient.1
In a NHANES study from 1999–2006, risk factors for iron deficiency included multiparity, current pregnancy, and regular menstrual cycles. Use of hormonal contraception reduced the rate of iron deficiency.2 Using the same data, the prevalences of iron deficiency during the first, second, and third trimesters of pregnancy were reported to be 7%, 14%, and 30%, respectively.3 In addition to pregnancy and menstrual bleeding there are many other medical problems that may contribute to iron deficiency, including Helicobacter pylori (H pylori) infection, gastritis, celiac disease, and bariatric surgery.
Iron deficiency anemia may be associated with adverse pregnancy outcomes
In a retrospective study of 75,660 singleton pregnancies, 7,977 women were diagnosed with iron deficiency anemia when they were admitted for delivery. Compared with pregnant women without iron deficiency, the presence of iron deficiency increased the risk of:
- blood transfusion (odds ratio [OR], 5.48; 95% confidence interval [CI], 4.57–6.58)
- preterm delivery (OR, 1.54; 95% CI, 1.36–1.76)
- cesarean delivery (OR, 1.30; 95% CI, 1.13–1.49)
- 5-minute Apgar score <7 (OR, 2.21; 95% CI, 1.84–2.64)
- intensive care unit (ICU) admission (OR, 1.28; 95% CI, 1.20–1.39).4
In a systematic review and meta-analysis of 26 studies, maternal anemia (mostly iron deficiency anemia) was associated with a higher risk of low birth weight (relative risk [RR], 1.31; 95% CI, 1.13–1.51), preterm birth (RR, 1.63; 95% CI, 1.33–2.01), perinatal mortality (RR, 1.51; 95% CI, 1.30–1.76), and neonatal mortality (RR, 2.72; 95% CI, 1.19–6.25).5
In a clinical trial, pregnant women were randomly assigned to receive folic acid alone; folic acid plus iron supplements; or 15 vitamins and minerals, including folic acid and iron. At delivery, women in the iron-folic acid and the 15 vitamin and minerals groups had higher hemoglobin concentrations than the folic acid monotherapy group. Among 4,697 live births, women in the iron-folic acid group had significantly fewer preterm births (<34 weeks’ gestation) than the folic acid group (RR, 0.50; 95% CI, 0.27–0.94; P = .031).6 Data from additional randomized trials are needed to further clarify the effect of iron supplementation on obstetric outcomes.
Related article:
Treating polycystic ovary syndrome: Start using dual medical therapy
The diagnosis of iron deficiency is optimized by measuring serum ferritin
Serum ferritin measurement is an excellent test of iron deficiency. We recommend that all pregnant women have serum ferritin measured at the first prenatal visit and at the beginning of the third trimester to assess maternal iron stores. In pregnancy, the Centers for Disease Control and Prevention and the World Health Organization define anemia as a hemoglobin level of less than 11 g/dL or hematocrit less than 33% in the first and third trimesters. If a pregnant woman is not anemic, a serum ferritin level less than 15 ng/mL indicates iron deficiency.7 Some experts believe that in pregnant women who are not anemic, a serum ferritin level between 15 and 30 ng/mL may also indicate iron deficiency.8 If the pregnant woman is anemic and does not have another cause of the anemia, a serum ferritin level less than 40 ng/mL is indicative of iron deficiency.7
Ferritin is an acute phase reactant and levels may be falsely elevated due to chronic or acute inflammation, liver disease, renal failure, metabolic syndrome, or malignancy. Some women with iron deficiency due to bariatric surgery or malabsorption also have vitamin B12 and, less commonly, folate deficiency, which can contribute to the development of anemia (see “Diagnosis of anemia, iron deficiency, and iron deficiency anemia in pregnancy.”) Clinicians are often advised that a mean corpuscular volume demonstrating microcytosis is the “best test” to assess a patient for iron deficiency. However, reduced iron availability and low ferritin precede microcytosis. Hence microcytosis is a lagging measure and iron deficiency is diagnosed at an earlier stage by ferritin.
Requirements for a diagnosis of anemia in pregnancy
The American College of Obstetricians and Gynecologists recommends obtaining a hemoglobin and hematocrit test at the first prenatal visit and at the beginning of the third trimester of pregnancy.1
If the hemoglobin concentration is less than 11 g/dL, or hematocrit is less than 33%, anemia is present.2,3
If anemia is diagnosed, additional testing to investigate potential causes of anemia includes hemoglobin electrophoresis and measurement of vitamin B12 and folate levels. Many obstetricians perform hemoglobin electrophoresis on all their pregnant patients as part of the routine prenatal screen.
Requirements for a diagnosis of iron deficiency in pregnancy
We recommend obtaining a ferritin measurement at the first prenatal visit and at the beginning of the third trimester.
In pregnant women with anemia, iron deficiency is present if the ferritin is less than 40 ng/mL.
If a pregnant woman is not anemic, iron deficiency is present if the ferritin is less than 15 ng/mL.4
Requirements for a diagnosis of iron deficiency anemia
Hemoglobin concentration less than 11 g/dL, or hematocrit less than 33% (diagnosis of anemia).
PLUS
Ferritin less than 40 ng/mL (diagnosis of iron deficiency in an anemic woman)
PLUS
Evaluation for other known major causes of anemia, including blood loss, hemolysis, bone marrow disease, medications that suppress bone marrow function, kidney disease, malignancy, hemoglobinopathy, and vitamin B12 or folate deficiency.
References
- Guidelines for Perinatal Care. 8th ed. Washington DC: American Academy of Pediatrics, American College of Obstetricians and Gynecologists;2017.
- Centers for Disease Control and Prevention. CDC criteria for anemia in children and childbearing-aged women. MMWR Morb Mortal Wkly Rep. 1989;38(22):400-404.
- World Health Organization. Iron deficiency anaemia: assessment, prevention and control. A guide for programme managers. World Health Organization: Geneva, Switzerland; 2001. http://www.who.int/nutrition/publications/en/ida_assessment_prevention_control.pdf. Accessed November 8, 2017.
- Guyatt GH, Oxman AD, Ali M, Willan A, McIlroy W, Patterson C. Laboratory diagnosis of iron-deficiency: an overview. J Gen Intern Med. 1992;7(2):145-153.
Dietary iron
Iron in food is present in heme (meat, poultry, fish) and non-heme forms (grains, plant food, supplements). Heme iron is better absorbed than non-heme iron. Foods rich in non-heme iron include spinach, lentils, prune juice, dried prunes, and fortified cereals. Absorption of non-heme iron can be increased by vitamin C or vitamin C–rich foods (broccoli, bell peppers, cantaloupe, grapefruit, oranges, strawberries, and tomatoes). Absorption of non-heme iron is reduced by consumption of dairy products, coffee, tea, and chocolate.
Oral iron treatment
Oral iron is an effective treatment for iron deficiency9,10 and is inexpensive, safe, and widely available. The CDC recommends that all pregnant women take a 30 mg/day iron supplement, unless they have hemochromatosis.11 For women with a low ferritin level and anemia, iron supplementation should be increased to 30 to 120 mg daily.11 Not all prenatal vitamins contain iron; those that do typically contain 17 to 28 mg of elemental iron per dose.
Many pregnant women taking oral iron, especially at doses greater than 30 mg daily, have gastrointestinal side effects, which cause them to discontinue the iron therapy.12 Taking iron supplementation on an intermittent basis may help to reduce gastrointestinal side effects and improve iron stores.13
In the past, a standard approach to the treatment of iron deficiency anemia was oral ferrous sulfate 325 mg (65 mg elemental iron) spaced in 3 doses each day for a total daily dose of 195 mg elemental iron. However, recent absorption studies concluded that maximal absorption of iron occurs with a dose in the range of 40 to 80 mg of elemental iron daily. Greater doses do not result in more iron absorption and are associated with more side effects.14,15 (See “Start using alternate-day oral iron dosing, and stop using daily iron dosing.”)
Recent research reports alternate-day oral iron dosing compared with daily oral iron dosing results in higher absorption of iron.
Details of the study
A total of 40 iron deficient women (mean serum ferritin level, 14 ng/mL) were randomly assigned to receive a daily dose of 60 mg of elemental iron (325 mg of ferrous sulfate) for 14 days or an alternate-day dose of 60 mg for 28 days. A small amount of radioactive iron was added to the oral medication to assess iron absorption. The primary outcome was fractional and total iron absorption, calculated by measuring radioactive iron in circulating red blood cells 14 days after the final oral iron dose.
Alternate-day iron dosing, compared with daily dosing, resulted in a higher fraction of the iron dose being absorbed (22% vs 16%; P = .0013). In addition, alternate-day iron dosing resulted in greater cumulative total iron absorption (175 mg vs 131 mg; P = .001). Nausea was reported less frequently by women in the alternate-day dosing group (11%) than in the daily iron dose group (29%).
The investigators concluded that prescribing iron as a single alternate-day
dose may be a superior dosing regimen compared with daily dosing.
Reference
- Stoffel NU, Cercamondi CI, Brittenham G, et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials. Lancet Haematol. 2017;4(11):e524–e533.
Oral iron should not be taken in close approximation to the consumption of milk, cereals, tea, coffee, eggs, or calcium supplements. The absorption of oral iron is enhanced by the consumption of orange juice or 250 mg of vitamin C. Gastrointestinal side effects include nausea, flatulence, constipation, diarrhea, epigastric distress, and vomiting. If gastrointestinal side effects occur, interventions that might improve tolerability include: reduce the dose of iron or administer intermittently or use a low dose of oral iron, where dosing can be more easily titrated.
We re-check ferritin and hemoglobin levels 2 to 4 weeks after initiation of oral iron therapy and expect to see a hemoglobin rise of 1 g/dL if the therapy is effective.
Intravenous iron treatment
For women with iron deficiency anemia who cannot tolerate oral iron or in whom oral iron treatment has not resolved their anemia, intravenous (IV) iron treatment may be an optimal approach. Women in the third trimester of pregnancy with iron deficiency anemia have very little time to consume sufficient quantities of oral iron in food and supplements to restore their deficiency and reverse their anemia. Consequently, treatment with IV iron may be especially appropriate for women with iron deficiency anemia in the third trimester of pregnancy. Prior gastric surgery, including gastric bypass, results in reduced gastric acid production and causes severe impairment of intestinal absorption of iron. Patients with malabsorption syndromes, including celiac disease, also may have limited absorption of oral iron. These populations of pregnant women may particularly benefit from the use of IV iron. In pregnant women IV iron has fewer gastrointestinal side effects than oral iron.16
Many severely iron deficient patients need 1,000 mg of iron to resolve their deficit. In order to avoid giving multiple standard doses (200 mg per infusion, with 5 infusions over many days), some centers have explored the use of 1 large dose of IV iron (1,000 mg of low molecular weight iron dextran administered over 1 hour) (INFeD, Watson Pharma).17–19 This is not a regimen that is specifically approved by the US Food and Drug Administration. An alternative regimen is to administer 750 mg of ferrous carboxymaltose (Injectafer, Luitpold Pharmaceuticals) over 15 minutes, which is an FDA-approved regimen.18 Many hematologists prefer to administer multiple smaller doses of iron. For example, in our practice, pregnant women are commonly treated with IV iron sucrose (300 mg) every 2 weeks for 3 doses. To increase access of pregnant women to IV iron treatment, obstetricians need to work with hematologists and infusion centers to create collaborative protocols to expeditiously treat women in the third trimester.
There is an epidemic of iron deficiency in pregnant women in the United States. In an era of high technology medicine, it is surprising that iron deficiency remains an unsolved obstetric problem in our country.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Looker AC, Dallman PR, Carroll MD, Gunter EW, Johnson CL. Prevalence of iron deficiency in the United States. JAMA. 1997;277(12):973–976.
- Miller EM. Iron status and reproduction in US women: National Health and Nutrition Examination Survey 1999–2006. PLoS One. 2014;9(11):e112216.
- Mei Z, Cogswell ME, Looker AC, et al. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999–2006. Am J Clin Nutr. 2011;93(6):1312–1320.
- Drukker L, Hants Y, Farkash R, Ruchlemer R, Samueloff A, Grisaru-Granovsky S. Iron deficiency anemia at admission for labor and delivery is associated with an increased risk for Cesarean section and adverse maternal and neonatal outcomes. Transfusion. 2015;55(12):2799–2806.
- Rahmann MM, Abe SK, Rahman MS, et al. Maternal anemia and risk of adverse birth and health outcomes in low- and middle-income countries: systematic review and meta-analysis. Am J Clin Nutr. 2016;103(2):495–504.
- Zeng L, Dibley MJ, Cheng Y, et al. Impact of micronutrient supplementation during pregnancy on birth weight, duration of gestation, and perinatal mortality in rural western China: double blind cluster randomised controlled trial. BMJ. 2008;337:a2001.
- Guyatt GH, Oxman AD, Ali M, Willan A, McIlroy W, Patterson C. Laboratory diagnosis of iron-deficiency: an overview. J Gen Intern Med. 1992;7(2):145–153.
- van den Broek NR, Letsky EA, White SA, Shenkin A. Iron status in pregnant women: which measurements are valid? Br J Haematol. 1998;103(3):817–824.
- Peña-Rosas JP, De-Regil LM, Garcia-Casal MN, Dowswell T. Daily oral iron supplementation during pregnancy. Cochrane Database Syst Rev. 2015(7);CD004736.
- Cantor AG, Bougatsos C, Dana T, Blazina I, McDonagh M. Routine iron supplementation and screening for iron deficiency anemia in pregnancy: a systematic review for the US Preventive Services Task Force. Ann Intern Med. 2015;162(8):566–576.
- Centers for Disease Control and Prevention. Recommendations to prevent and control iron deficiency in the United States. MMWR Recomm Rep. 1998;47(RR-3):1–29.
- Tolkien Z, Stecher L, Mander AP, Pereira DI, Powell JJ. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10(2):e0117383.
- Peña-Rosas JP, De-Regil LM, Gomez Malave H, Flores-Urrutia MC, Dowswell T. Intermittent oral iron supplementation during pregnancy. Cochrane Database Syst Rev. 2015(10);CD009997.
- Moretti D, Goede JS, Zeder C, et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015;126(17):1981–1989.
- Schrier SL. So you know how to treat iron deficiency anemia. Blood. 2015;126(17):1971.
- Breymann C, Milman N, Mezzacasa A, Bernard R, Dudenhausen J; FER-ASAP investigators. Ferric carboxymaltose vs oral iron in the treatment of pregnant women with iron deficiency anemia: an international, open-label, randomized controlled trial (FER-ASAP). J Perinatal Med. 2017;45(4):443–453.
- Auerbach M, Pappadakis JA, Bahrain H, Auerbach SA, Ballard H, Dahl NV. Safety and efficacy of rapidly administered (one hour) one gram of low molecular weight iron dextran (INFeD) for the treatment of iron deficient anemia. Am J Hematol. 2011;86(10):860–862.
- Auerbach M, Adamson JW. How we diagnose and treat iron deficiency anemia. Am J Hematol. 2016;91(1):31–38.
- Wong L, Smith S, Gilstrop M, et al. Safety and efficacy of rapid (1,000 mg in 1 hr) intravenous iron dextran for treatment of maternal iron deficient anemia of pregnancy. Am J Hematol. 2016;91(6):590–593.

Dr. Schantz-Dunn is Instructor, Department of Obstetrics, Gynecology, and Reproductive Biology Brigham and Women's Hospital and Harvard Medical School Boston, Massachusetts.

Dr. Barbieri is Editor in Chief, OBG Management, and Chair, Department of Obstetrics and Gynecology, Brigham and Women's Hospital, and Kate Macy Ladd Professor of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School.
The authors report no financial relationships relevant to this article.
Dr. Schantz-Dunn is Instructor, Department of Obstetrics, Gynecology, and Reproductive Biology Brigham and Women's Hospital and Harvard Medical School Boston, Massachusetts.
Dr. Barbieri is Editor in Chief, OBG Management, and Chair, Department of Obstetrics and Gynecology, Brigham and Women's Hospital, and Kate Macy Ladd Professor of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School.
The authors report no financial relationships relevant to this article.
Dr. Schantz-Dunn is Instructor, Department of Obstetrics, Gynecology, and Reproductive Biology Brigham and Women's Hospital and Harvard Medical School Boston, Massachusetts.
Dr. Barbieri is Editor in Chief, OBG Management, and Chair, Department of Obstetrics and Gynecology, Brigham and Women's Hospital, and Kate Macy Ladd Professor of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School.
The authors report no financial relationships relevant to this article.
All mammalian life is dependent on a continuous supply of molecular oxygen. Molecular oxygen is carried to cells by noncovalent binding to the iron moiety in the hemoglobin of red blood cells. It is utilized within cells by noncovalent binding to the iron moiety in various microsomal and mitochondrial proteins, including myoglobin and cytochromes. Consequently, to efficiently utilize molecular oxygen all mammalian life is dependent on an adequate supply of iron. Surprisingly, in an era of high technology precision medicine, many pregnant women are iron deficient, anemic, and not receiving adequate iron supplementation.
Iron deficiency is prevalent in women and pregnant women
Women often become iron deficient because of pregnancy or heavy menstrual bleeding. During pregnancy, maternal iron is provided to supply the needs of the fetus and placenta. Additional iron is needed to expand maternal red blood cell volume and replace iron lost due to bleeding at delivery. In the National Health and Nutrition Examination Survey (NHANES) of 1988–1994, 11% of women aged 16 to 49 years were iron deficient. By contrast, less than 1% of men aged 16 to 49 years were iron deficient.1
In a NHANES study from 1999–2006, risk factors for iron deficiency included multiparity, current pregnancy, and regular menstrual cycles. Use of hormonal contraception reduced the rate of iron deficiency.2 Using the same data, the prevalences of iron deficiency during the first, second, and third trimesters of pregnancy were reported to be 7%, 14%, and 30%, respectively.3 In addition to pregnancy and menstrual bleeding there are many other medical problems that may contribute to iron deficiency, including Helicobacter pylori (H pylori) infection, gastritis, celiac disease, and bariatric surgery.
Iron deficiency anemia may be associated with adverse pregnancy outcomes
In a retrospective study of 75,660 singleton pregnancies, 7,977 women were diagnosed with iron deficiency anemia when they were admitted for delivery. Compared with pregnant women without iron deficiency, the presence of iron deficiency increased the risk of:
- blood transfusion (odds ratio [OR], 5.48; 95% confidence interval [CI], 4.57–6.58)
- preterm delivery (OR, 1.54; 95% CI, 1.36–1.76)
- cesarean delivery (OR, 1.30; 95% CI, 1.13–1.49)
- 5-minute Apgar score <7 (OR, 2.21; 95% CI, 1.84–2.64)
- intensive care unit (ICU) admission (OR, 1.28; 95% CI, 1.20–1.39).4
In a systematic review and meta-analysis of 26 studies, maternal anemia (mostly iron deficiency anemia) was associated with a higher risk of low birth weight (relative risk [RR], 1.31; 95% CI, 1.13–1.51), preterm birth (RR, 1.63; 95% CI, 1.33–2.01), perinatal mortality (RR, 1.51; 95% CI, 1.30–1.76), and neonatal mortality (RR, 2.72; 95% CI, 1.19–6.25).5
In a clinical trial, pregnant women were randomly assigned to receive folic acid alone; folic acid plus iron supplements; or 15 vitamins and minerals, including folic acid and iron. At delivery, women in the iron-folic acid and the 15 vitamin and minerals groups had higher hemoglobin concentrations than the folic acid monotherapy group. Among 4,697 live births, women in the iron-folic acid group had significantly fewer preterm births (<34 weeks’ gestation) than the folic acid group (RR, 0.50; 95% CI, 0.27–0.94; P = .031).6 Data from additional randomized trials are needed to further clarify the effect of iron supplementation on obstetric outcomes.
Related article:
Treating polycystic ovary syndrome: Start using dual medical therapy
The diagnosis of iron deficiency is optimized by measuring serum ferritin
Serum ferritin measurement is an excellent test of iron deficiency. We recommend that all pregnant women have serum ferritin measured at the first prenatal visit and at the beginning of the third trimester to assess maternal iron stores. In pregnancy, the Centers for Disease Control and Prevention and the World Health Organization define anemia as a hemoglobin level of less than 11 g/dL or hematocrit less than 33% in the first and third trimesters. If a pregnant woman is not anemic, a serum ferritin level less than 15 ng/mL indicates iron deficiency.7 Some experts believe that in pregnant women who are not anemic, a serum ferritin level between 15 and 30 ng/mL may also indicate iron deficiency.8 If the pregnant woman is anemic and does not have another cause of the anemia, a serum ferritin level less than 40 ng/mL is indicative of iron deficiency.7
Ferritin is an acute phase reactant and levels may be falsely elevated due to chronic or acute inflammation, liver disease, renal failure, metabolic syndrome, or malignancy. Some women with iron deficiency due to bariatric surgery or malabsorption also have vitamin B12 and, less commonly, folate deficiency, which can contribute to the development of anemia (see “Diagnosis of anemia, iron deficiency, and iron deficiency anemia in pregnancy.”) Clinicians are often advised that a mean corpuscular volume demonstrating microcytosis is the “best test” to assess a patient for iron deficiency. However, reduced iron availability and low ferritin precede microcytosis. Hence microcytosis is a lagging measure and iron deficiency is diagnosed at an earlier stage by ferritin.
Requirements for a diagnosis of anemia in pregnancy
The American College of Obstetricians and Gynecologists recommends obtaining a hemoglobin and hematocrit test at the first prenatal visit and at the beginning of the third trimester of pregnancy.1
If the hemoglobin concentration is less than 11 g/dL, or hematocrit is less than 33%, anemia is present.2,3
If anemia is diagnosed, additional testing to investigate potential causes of anemia includes hemoglobin electrophoresis and measurement of vitamin B12 and folate levels. Many obstetricians perform hemoglobin electrophoresis on all their pregnant patients as part of the routine prenatal screen.
Requirements for a diagnosis of iron deficiency in pregnancy
We recommend obtaining a ferritin measurement at the first prenatal visit and at the beginning of the third trimester.
In pregnant women with anemia, iron deficiency is present if the ferritin is less than 40 ng/mL.
If a pregnant woman is not anemic, iron deficiency is present if the ferritin is less than 15 ng/mL.4
Requirements for a diagnosis of iron deficiency anemia
Hemoglobin concentration less than 11 g/dL, or hematocrit less than 33% (diagnosis of anemia).
PLUS
Ferritin less than 40 ng/mL (diagnosis of iron deficiency in an anemic woman)
PLUS
Evaluation for other known major causes of anemia, including blood loss, hemolysis, bone marrow disease, medications that suppress bone marrow function, kidney disease, malignancy, hemoglobinopathy, and vitamin B12 or folate deficiency.
References
- Guidelines for Perinatal Care. 8th ed. Washington DC: American Academy of Pediatrics, American College of Obstetricians and Gynecologists;2017.
- Centers for Disease Control and Prevention. CDC criteria for anemia in children and childbearing-aged women. MMWR Morb Mortal Wkly Rep. 1989;38(22):400-404.
- World Health Organization. Iron deficiency anaemia: assessment, prevention and control. A guide for programme managers. World Health Organization: Geneva, Switzerland; 2001. http://www.who.int/nutrition/publications/en/ida_assessment_prevention_control.pdf. Accessed November 8, 2017.
- Guyatt GH, Oxman AD, Ali M, Willan A, McIlroy W, Patterson C. Laboratory diagnosis of iron-deficiency: an overview. J Gen Intern Med. 1992;7(2):145-153.
Dietary iron
Iron in food is present in heme (meat, poultry, fish) and non-heme forms (grains, plant food, supplements). Heme iron is better absorbed than non-heme iron. Foods rich in non-heme iron include spinach, lentils, prune juice, dried prunes, and fortified cereals. Absorption of non-heme iron can be increased by vitamin C or vitamin C–rich foods (broccoli, bell peppers, cantaloupe, grapefruit, oranges, strawberries, and tomatoes). Absorption of non-heme iron is reduced by consumption of dairy products, coffee, tea, and chocolate.
Oral iron treatment
Oral iron is an effective treatment for iron deficiency9,10 and is inexpensive, safe, and widely available. The CDC recommends that all pregnant women take a 30 mg/day iron supplement, unless they have hemochromatosis.11 For women with a low ferritin level and anemia, iron supplementation should be increased to 30 to 120 mg daily.11 Not all prenatal vitamins contain iron; those that do typically contain 17 to 28 mg of elemental iron per dose.
Many pregnant women taking oral iron, especially at doses greater than 30 mg daily, have gastrointestinal side effects, which cause them to discontinue the iron therapy.12 Taking iron supplementation on an intermittent basis may help to reduce gastrointestinal side effects and improve iron stores.13
In the past, a standard approach to the treatment of iron deficiency anemia was oral ferrous sulfate 325 mg (65 mg elemental iron) spaced in 3 doses each day for a total daily dose of 195 mg elemental iron. However, recent absorption studies concluded that maximal absorption of iron occurs with a dose in the range of 40 to 80 mg of elemental iron daily. Greater doses do not result in more iron absorption and are associated with more side effects.14,15 (See “Start using alternate-day oral iron dosing, and stop using daily iron dosing.”)
Recent research reports alternate-day oral iron dosing compared with daily oral iron dosing results in higher absorption of iron.
Details of the study
A total of 40 iron deficient women (mean serum ferritin level, 14 ng/mL) were randomly assigned to receive a daily dose of 60 mg of elemental iron (325 mg of ferrous sulfate) for 14 days or an alternate-day dose of 60 mg for 28 days. A small amount of radioactive iron was added to the oral medication to assess iron absorption. The primary outcome was fractional and total iron absorption, calculated by measuring radioactive iron in circulating red blood cells 14 days after the final oral iron dose.
Alternate-day iron dosing, compared with daily dosing, resulted in a higher fraction of the iron dose being absorbed (22% vs 16%; P = .0013). In addition, alternate-day iron dosing resulted in greater cumulative total iron absorption (175 mg vs 131 mg; P = .001). Nausea was reported less frequently by women in the alternate-day dosing group (11%) than in the daily iron dose group (29%).
The investigators concluded that prescribing iron as a single alternate-day
dose may be a superior dosing regimen compared with daily dosing.
Reference
- Stoffel NU, Cercamondi CI, Brittenham G, et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials. Lancet Haematol. 2017;4(11):e524–e533.
Oral iron should not be taken in close approximation to the consumption of milk, cereals, tea, coffee, eggs, or calcium supplements. The absorption of oral iron is enhanced by the consumption of orange juice or 250 mg of vitamin C. Gastrointestinal side effects include nausea, flatulence, constipation, diarrhea, epigastric distress, and vomiting. If gastrointestinal side effects occur, interventions that might improve tolerability include: reduce the dose of iron or administer intermittently or use a low dose of oral iron, where dosing can be more easily titrated.
We re-check ferritin and hemoglobin levels 2 to 4 weeks after initiation of oral iron therapy and expect to see a hemoglobin rise of 1 g/dL if the therapy is effective.
Intravenous iron treatment
For women with iron deficiency anemia who cannot tolerate oral iron or in whom oral iron treatment has not resolved their anemia, intravenous (IV) iron treatment may be an optimal approach. Women in the third trimester of pregnancy with iron deficiency anemia have very little time to consume sufficient quantities of oral iron in food and supplements to restore their deficiency and reverse their anemia. Consequently, treatment with IV iron may be especially appropriate for women with iron deficiency anemia in the third trimester of pregnancy. Prior gastric surgery, including gastric bypass, results in reduced gastric acid production and causes severe impairment of intestinal absorption of iron. Patients with malabsorption syndromes, including celiac disease, also may have limited absorption of oral iron. These populations of pregnant women may particularly benefit from the use of IV iron. In pregnant women IV iron has fewer gastrointestinal side effects than oral iron.16
Many severely iron deficient patients need 1,000 mg of iron to resolve their deficit. In order to avoid giving multiple standard doses (200 mg per infusion, with 5 infusions over many days), some centers have explored the use of 1 large dose of IV iron (1,000 mg of low molecular weight iron dextran administered over 1 hour) (INFeD, Watson Pharma).17–19 This is not a regimen that is specifically approved by the US Food and Drug Administration. An alternative regimen is to administer 750 mg of ferrous carboxymaltose (Injectafer, Luitpold Pharmaceuticals) over 15 minutes, which is an FDA-approved regimen.18 Many hematologists prefer to administer multiple smaller doses of iron. For example, in our practice, pregnant women are commonly treated with IV iron sucrose (300 mg) every 2 weeks for 3 doses. To increase access of pregnant women to IV iron treatment, obstetricians need to work with hematologists and infusion centers to create collaborative protocols to expeditiously treat women in the third trimester.
There is an epidemic of iron deficiency in pregnant women in the United States. In an era of high technology medicine, it is surprising that iron deficiency remains an unsolved obstetric problem in our country.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
All mammalian life is dependent on a continuous supply of molecular oxygen. Molecular oxygen is carried to cells by noncovalent binding to the iron moiety in the hemoglobin of red blood cells. It is utilized within cells by noncovalent binding to the iron moiety in various microsomal and mitochondrial proteins, including myoglobin and cytochromes. Consequently, to efficiently utilize molecular oxygen all mammalian life is dependent on an adequate supply of iron. Surprisingly, in an era of high technology precision medicine, many pregnant women are iron deficient, anemic, and not receiving adequate iron supplementation.
Iron deficiency is prevalent in women and pregnant women
Women often become iron deficient because of pregnancy or heavy menstrual bleeding. During pregnancy, maternal iron is provided to supply the needs of the fetus and placenta. Additional iron is needed to expand maternal red blood cell volume and replace iron lost due to bleeding at delivery. In the National Health and Nutrition Examination Survey (NHANES) of 1988–1994, 11% of women aged 16 to 49 years were iron deficient. By contrast, less than 1% of men aged 16 to 49 years were iron deficient.1
In a NHANES study from 1999–2006, risk factors for iron deficiency included multiparity, current pregnancy, and regular menstrual cycles. Use of hormonal contraception reduced the rate of iron deficiency.2 Using the same data, the prevalences of iron deficiency during the first, second, and third trimesters of pregnancy were reported to be 7%, 14%, and 30%, respectively.3 In addition to pregnancy and menstrual bleeding there are many other medical problems that may contribute to iron deficiency, including Helicobacter pylori (H pylori) infection, gastritis, celiac disease, and bariatric surgery.
Iron deficiency anemia may be associated with adverse pregnancy outcomes
In a retrospective study of 75,660 singleton pregnancies, 7,977 women were diagnosed with iron deficiency anemia when they were admitted for delivery. Compared with pregnant women without iron deficiency, the presence of iron deficiency increased the risk of:
- blood transfusion (odds ratio [OR], 5.48; 95% confidence interval [CI], 4.57–6.58)
- preterm delivery (OR, 1.54; 95% CI, 1.36–1.76)
- cesarean delivery (OR, 1.30; 95% CI, 1.13–1.49)
- 5-minute Apgar score <7 (OR, 2.21; 95% CI, 1.84–2.64)
- intensive care unit (ICU) admission (OR, 1.28; 95% CI, 1.20–1.39).4
In a systematic review and meta-analysis of 26 studies, maternal anemia (mostly iron deficiency anemia) was associated with a higher risk of low birth weight (relative risk [RR], 1.31; 95% CI, 1.13–1.51), preterm birth (RR, 1.63; 95% CI, 1.33–2.01), perinatal mortality (RR, 1.51; 95% CI, 1.30–1.76), and neonatal mortality (RR, 2.72; 95% CI, 1.19–6.25).5
In a clinical trial, pregnant women were randomly assigned to receive folic acid alone; folic acid plus iron supplements; or 15 vitamins and minerals, including folic acid and iron. At delivery, women in the iron-folic acid and the 15 vitamin and minerals groups had higher hemoglobin concentrations than the folic acid monotherapy group. Among 4,697 live births, women in the iron-folic acid group had significantly fewer preterm births (<34 weeks’ gestation) than the folic acid group (RR, 0.50; 95% CI, 0.27–0.94; P = .031).6 Data from additional randomized trials are needed to further clarify the effect of iron supplementation on obstetric outcomes.
Related article:
Treating polycystic ovary syndrome: Start using dual medical therapy
The diagnosis of iron deficiency is optimized by measuring serum ferritin
Serum ferritin measurement is an excellent test of iron deficiency. We recommend that all pregnant women have serum ferritin measured at the first prenatal visit and at the beginning of the third trimester to assess maternal iron stores. In pregnancy, the Centers for Disease Control and Prevention and the World Health Organization define anemia as a hemoglobin level of less than 11 g/dL or hematocrit less than 33% in the first and third trimesters. If a pregnant woman is not anemic, a serum ferritin level less than 15 ng/mL indicates iron deficiency.7 Some experts believe that in pregnant women who are not anemic, a serum ferritin level between 15 and 30 ng/mL may also indicate iron deficiency.8 If the pregnant woman is anemic and does not have another cause of the anemia, a serum ferritin level less than 40 ng/mL is indicative of iron deficiency.7
Ferritin is an acute phase reactant and levels may be falsely elevated due to chronic or acute inflammation, liver disease, renal failure, metabolic syndrome, or malignancy. Some women with iron deficiency due to bariatric surgery or malabsorption also have vitamin B12 and, less commonly, folate deficiency, which can contribute to the development of anemia (see “Diagnosis of anemia, iron deficiency, and iron deficiency anemia in pregnancy.”) Clinicians are often advised that a mean corpuscular volume demonstrating microcytosis is the “best test” to assess a patient for iron deficiency. However, reduced iron availability and low ferritin precede microcytosis. Hence microcytosis is a lagging measure and iron deficiency is diagnosed at an earlier stage by ferritin.
Requirements for a diagnosis of anemia in pregnancy
The American College of Obstetricians and Gynecologists recommends obtaining a hemoglobin and hematocrit test at the first prenatal visit and at the beginning of the third trimester of pregnancy.1
If the hemoglobin concentration is less than 11 g/dL, or hematocrit is less than 33%, anemia is present.2,3
If anemia is diagnosed, additional testing to investigate potential causes of anemia includes hemoglobin electrophoresis and measurement of vitamin B12 and folate levels. Many obstetricians perform hemoglobin electrophoresis on all their pregnant patients as part of the routine prenatal screen.
Requirements for a diagnosis of iron deficiency in pregnancy
We recommend obtaining a ferritin measurement at the first prenatal visit and at the beginning of the third trimester.
In pregnant women with anemia, iron deficiency is present if the ferritin is less than 40 ng/mL.
If a pregnant woman is not anemic, iron deficiency is present if the ferritin is less than 15 ng/mL.4
Requirements for a diagnosis of iron deficiency anemia
Hemoglobin concentration less than 11 g/dL, or hematocrit less than 33% (diagnosis of anemia).
PLUS
Ferritin less than 40 ng/mL (diagnosis of iron deficiency in an anemic woman)
PLUS
Evaluation for other known major causes of anemia, including blood loss, hemolysis, bone marrow disease, medications that suppress bone marrow function, kidney disease, malignancy, hemoglobinopathy, and vitamin B12 or folate deficiency.
References
- Guidelines for Perinatal Care. 8th ed. Washington DC: American Academy of Pediatrics, American College of Obstetricians and Gynecologists;2017.
- Centers for Disease Control and Prevention. CDC criteria for anemia in children and childbearing-aged women. MMWR Morb Mortal Wkly Rep. 1989;38(22):400-404.
- World Health Organization. Iron deficiency anaemia: assessment, prevention and control. A guide for programme managers. World Health Organization: Geneva, Switzerland; 2001. http://www.who.int/nutrition/publications/en/ida_assessment_prevention_control.pdf. Accessed November 8, 2017.
- Guyatt GH, Oxman AD, Ali M, Willan A, McIlroy W, Patterson C. Laboratory diagnosis of iron-deficiency: an overview. J Gen Intern Med. 1992;7(2):145-153.
Dietary iron
Iron in food is present in heme (meat, poultry, fish) and non-heme forms (grains, plant food, supplements). Heme iron is better absorbed than non-heme iron. Foods rich in non-heme iron include spinach, lentils, prune juice, dried prunes, and fortified cereals. Absorption of non-heme iron can be increased by vitamin C or vitamin C–rich foods (broccoli, bell peppers, cantaloupe, grapefruit, oranges, strawberries, and tomatoes). Absorption of non-heme iron is reduced by consumption of dairy products, coffee, tea, and chocolate.
Oral iron treatment
Oral iron is an effective treatment for iron deficiency9,10 and is inexpensive, safe, and widely available. The CDC recommends that all pregnant women take a 30 mg/day iron supplement, unless they have hemochromatosis.11 For women with a low ferritin level and anemia, iron supplementation should be increased to 30 to 120 mg daily.11 Not all prenatal vitamins contain iron; those that do typically contain 17 to 28 mg of elemental iron per dose.
Many pregnant women taking oral iron, especially at doses greater than 30 mg daily, have gastrointestinal side effects, which cause them to discontinue the iron therapy.12 Taking iron supplementation on an intermittent basis may help to reduce gastrointestinal side effects and improve iron stores.13
In the past, a standard approach to the treatment of iron deficiency anemia was oral ferrous sulfate 325 mg (65 mg elemental iron) spaced in 3 doses each day for a total daily dose of 195 mg elemental iron. However, recent absorption studies concluded that maximal absorption of iron occurs with a dose in the range of 40 to 80 mg of elemental iron daily. Greater doses do not result in more iron absorption and are associated with more side effects.14,15 (See “Start using alternate-day oral iron dosing, and stop using daily iron dosing.”)
Recent research reports alternate-day oral iron dosing compared with daily oral iron dosing results in higher absorption of iron.
Details of the study
A total of 40 iron deficient women (mean serum ferritin level, 14 ng/mL) were randomly assigned to receive a daily dose of 60 mg of elemental iron (325 mg of ferrous sulfate) for 14 days or an alternate-day dose of 60 mg for 28 days. A small amount of radioactive iron was added to the oral medication to assess iron absorption. The primary outcome was fractional and total iron absorption, calculated by measuring radioactive iron in circulating red blood cells 14 days after the final oral iron dose.
Alternate-day iron dosing, compared with daily dosing, resulted in a higher fraction of the iron dose being absorbed (22% vs 16%; P = .0013). In addition, alternate-day iron dosing resulted in greater cumulative total iron absorption (175 mg vs 131 mg; P = .001). Nausea was reported less frequently by women in the alternate-day dosing group (11%) than in the daily iron dose group (29%).
The investigators concluded that prescribing iron as a single alternate-day
dose may be a superior dosing regimen compared with daily dosing.
Reference
- Stoffel NU, Cercamondi CI, Brittenham G, et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials. Lancet Haematol. 2017;4(11):e524–e533.
Oral iron should not be taken in close approximation to the consumption of milk, cereals, tea, coffee, eggs, or calcium supplements. The absorption of oral iron is enhanced by the consumption of orange juice or 250 mg of vitamin C. Gastrointestinal side effects include nausea, flatulence, constipation, diarrhea, epigastric distress, and vomiting. If gastrointestinal side effects occur, interventions that might improve tolerability include: reduce the dose of iron or administer intermittently or use a low dose of oral iron, where dosing can be more easily titrated.
We re-check ferritin and hemoglobin levels 2 to 4 weeks after initiation of oral iron therapy and expect to see a hemoglobin rise of 1 g/dL if the therapy is effective.
Intravenous iron treatment
For women with iron deficiency anemia who cannot tolerate oral iron or in whom oral iron treatment has not resolved their anemia, intravenous (IV) iron treatment may be an optimal approach. Women in the third trimester of pregnancy with iron deficiency anemia have very little time to consume sufficient quantities of oral iron in food and supplements to restore their deficiency and reverse their anemia. Consequently, treatment with IV iron may be especially appropriate for women with iron deficiency anemia in the third trimester of pregnancy. Prior gastric surgery, including gastric bypass, results in reduced gastric acid production and causes severe impairment of intestinal absorption of iron. Patients with malabsorption syndromes, including celiac disease, also may have limited absorption of oral iron. These populations of pregnant women may particularly benefit from the use of IV iron. In pregnant women IV iron has fewer gastrointestinal side effects than oral iron.16
Many severely iron deficient patients need 1,000 mg of iron to resolve their deficit. In order to avoid giving multiple standard doses (200 mg per infusion, with 5 infusions over many days), some centers have explored the use of 1 large dose of IV iron (1,000 mg of low molecular weight iron dextran administered over 1 hour) (INFeD, Watson Pharma).17–19 This is not a regimen that is specifically approved by the US Food and Drug Administration. An alternative regimen is to administer 750 mg of ferrous carboxymaltose (Injectafer, Luitpold Pharmaceuticals) over 15 minutes, which is an FDA-approved regimen.18 Many hematologists prefer to administer multiple smaller doses of iron. For example, in our practice, pregnant women are commonly treated with IV iron sucrose (300 mg) every 2 weeks for 3 doses. To increase access of pregnant women to IV iron treatment, obstetricians need to work with hematologists and infusion centers to create collaborative protocols to expeditiously treat women in the third trimester.
There is an epidemic of iron deficiency in pregnant women in the United States. In an era of high technology medicine, it is surprising that iron deficiency remains an unsolved obstetric problem in our country.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Looker AC, Dallman PR, Carroll MD, Gunter EW, Johnson CL. Prevalence of iron deficiency in the United States. JAMA. 1997;277(12):973–976.
- Miller EM. Iron status and reproduction in US women: National Health and Nutrition Examination Survey 1999–2006. PLoS One. 2014;9(11):e112216.
- Mei Z, Cogswell ME, Looker AC, et al. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999–2006. Am J Clin Nutr. 2011;93(6):1312–1320.
- Drukker L, Hants Y, Farkash R, Ruchlemer R, Samueloff A, Grisaru-Granovsky S. Iron deficiency anemia at admission for labor and delivery is associated with an increased risk for Cesarean section and adverse maternal and neonatal outcomes. Transfusion. 2015;55(12):2799–2806.
- Rahmann MM, Abe SK, Rahman MS, et al. Maternal anemia and risk of adverse birth and health outcomes in low- and middle-income countries: systematic review and meta-analysis. Am J Clin Nutr. 2016;103(2):495–504.
- Zeng L, Dibley MJ, Cheng Y, et al. Impact of micronutrient supplementation during pregnancy on birth weight, duration of gestation, and perinatal mortality in rural western China: double blind cluster randomised controlled trial. BMJ. 2008;337:a2001.
- Guyatt GH, Oxman AD, Ali M, Willan A, McIlroy W, Patterson C. Laboratory diagnosis of iron-deficiency: an overview. J Gen Intern Med. 1992;7(2):145–153.
- van den Broek NR, Letsky EA, White SA, Shenkin A. Iron status in pregnant women: which measurements are valid? Br J Haematol. 1998;103(3):817–824.
- Peña-Rosas JP, De-Regil LM, Garcia-Casal MN, Dowswell T. Daily oral iron supplementation during pregnancy. Cochrane Database Syst Rev. 2015(7);CD004736.
- Cantor AG, Bougatsos C, Dana T, Blazina I, McDonagh M. Routine iron supplementation and screening for iron deficiency anemia in pregnancy: a systematic review for the US Preventive Services Task Force. Ann Intern Med. 2015;162(8):566–576.
- Centers for Disease Control and Prevention. Recommendations to prevent and control iron deficiency in the United States. MMWR Recomm Rep. 1998;47(RR-3):1–29.
- Tolkien Z, Stecher L, Mander AP, Pereira DI, Powell JJ. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10(2):e0117383.
- Peña-Rosas JP, De-Regil LM, Gomez Malave H, Flores-Urrutia MC, Dowswell T. Intermittent oral iron supplementation during pregnancy. Cochrane Database Syst Rev. 2015(10);CD009997.
- Moretti D, Goede JS, Zeder C, et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015;126(17):1981–1989.
- Schrier SL. So you know how to treat iron deficiency anemia. Blood. 2015;126(17):1971.
- Breymann C, Milman N, Mezzacasa A, Bernard R, Dudenhausen J; FER-ASAP investigators. Ferric carboxymaltose vs oral iron in the treatment of pregnant women with iron deficiency anemia: an international, open-label, randomized controlled trial (FER-ASAP). J Perinatal Med. 2017;45(4):443–453.
- Auerbach M, Pappadakis JA, Bahrain H, Auerbach SA, Ballard H, Dahl NV. Safety and efficacy of rapidly administered (one hour) one gram of low molecular weight iron dextran (INFeD) for the treatment of iron deficient anemia. Am J Hematol. 2011;86(10):860–862.
- Auerbach M, Adamson JW. How we diagnose and treat iron deficiency anemia. Am J Hematol. 2016;91(1):31–38.
- Wong L, Smith S, Gilstrop M, et al. Safety and efficacy of rapid (1,000 mg in 1 hr) intravenous iron dextran for treatment of maternal iron deficient anemia of pregnancy. Am J Hematol. 2016;91(6):590–593.
- Looker AC, Dallman PR, Carroll MD, Gunter EW, Johnson CL. Prevalence of iron deficiency in the United States. JAMA. 1997;277(12):973–976.
- Miller EM. Iron status and reproduction in US women: National Health and Nutrition Examination Survey 1999–2006. PLoS One. 2014;9(11):e112216.
- Mei Z, Cogswell ME, Looker AC, et al. Assessment of iron status in US pregnant women from the National Health and Nutrition Examination Survey (NHANES), 1999–2006. Am J Clin Nutr. 2011;93(6):1312–1320.
- Drukker L, Hants Y, Farkash R, Ruchlemer R, Samueloff A, Grisaru-Granovsky S. Iron deficiency anemia at admission for labor and delivery is associated with an increased risk for Cesarean section and adverse maternal and neonatal outcomes. Transfusion. 2015;55(12):2799–2806.
- Rahmann MM, Abe SK, Rahman MS, et al. Maternal anemia and risk of adverse birth and health outcomes in low- and middle-income countries: systematic review and meta-analysis. Am J Clin Nutr. 2016;103(2):495–504.
- Zeng L, Dibley MJ, Cheng Y, et al. Impact of micronutrient supplementation during pregnancy on birth weight, duration of gestation, and perinatal mortality in rural western China: double blind cluster randomised controlled trial. BMJ. 2008;337:a2001.
- Guyatt GH, Oxman AD, Ali M, Willan A, McIlroy W, Patterson C. Laboratory diagnosis of iron-deficiency: an overview. J Gen Intern Med. 1992;7(2):145–153.
- van den Broek NR, Letsky EA, White SA, Shenkin A. Iron status in pregnant women: which measurements are valid? Br J Haematol. 1998;103(3):817–824.
- Peña-Rosas JP, De-Regil LM, Garcia-Casal MN, Dowswell T. Daily oral iron supplementation during pregnancy. Cochrane Database Syst Rev. 2015(7);CD004736.
- Cantor AG, Bougatsos C, Dana T, Blazina I, McDonagh M. Routine iron supplementation and screening for iron deficiency anemia in pregnancy: a systematic review for the US Preventive Services Task Force. Ann Intern Med. 2015;162(8):566–576.
- Centers for Disease Control and Prevention. Recommendations to prevent and control iron deficiency in the United States. MMWR Recomm Rep. 1998;47(RR-3):1–29.
- Tolkien Z, Stecher L, Mander AP, Pereira DI, Powell JJ. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10(2):e0117383.
- Peña-Rosas JP, De-Regil LM, Gomez Malave H, Flores-Urrutia MC, Dowswell T. Intermittent oral iron supplementation during pregnancy. Cochrane Database Syst Rev. 2015(10);CD009997.
- Moretti D, Goede JS, Zeder C, et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015;126(17):1981–1989.
- Schrier SL. So you know how to treat iron deficiency anemia. Blood. 2015;126(17):1971.
- Breymann C, Milman N, Mezzacasa A, Bernard R, Dudenhausen J; FER-ASAP investigators. Ferric carboxymaltose vs oral iron in the treatment of pregnant women with iron deficiency anemia: an international, open-label, randomized controlled trial (FER-ASAP). J Perinatal Med. 2017;45(4):443–453.
- Auerbach M, Pappadakis JA, Bahrain H, Auerbach SA, Ballard H, Dahl NV. Safety and efficacy of rapidly administered (one hour) one gram of low molecular weight iron dextran (INFeD) for the treatment of iron deficient anemia. Am J Hematol. 2011;86(10):860–862.
- Auerbach M, Adamson JW. How we diagnose and treat iron deficiency anemia. Am J Hematol. 2016;91(1):31–38.
- Wong L, Smith S, Gilstrop M, et al. Safety and efficacy of rapid (1,000 mg in 1 hr) intravenous iron dextran for treatment of maternal iron deficient anemia of pregnancy. Am J Hematol. 2016;91(6):590–593.
The importance of weight management and exercise: Practical advice for your patients
Over the past 3 decades, the prevalence of overweight and obesity has increased dramatically in the United States. A study published in 2016 showed the age-adjusted prevalence of obesity in 2013–2014 was 35% among men and 40.4% among women.1 It comes as no surprise that increased reliance on inexpensive fast foods coupled with progressively more sedentary lifestyles have been implicated as causative factors.2
With the rise in obesity also has come an attendant rise in related chronic diseases, such as type 2 diabetes mellitus and cardiovascular disease. Women who are obese are also at risk for certain women’s health conditions, such as polycystic ovary syndrome, breast cancer, and endometrial cancer.
It is clear that curbing this public health crisis will require concerted efforts from individuals, clinicians, and policy makers, as well as changes in societal norms.
Linda D. Bradley, MD: I think it is important for us not to lecture our patients. I could list all of the things that patients should or could do to prevent or even reverse disease states, in terms of eating right and exercising, but I think motivational interviewing is a more productive approach to elicit and evoke change (see “Principles and practice of motivational interviewing”). I used to preach to my patients. I would say, “You know, if you stay at this weight, you’re going to get diabetes, you’re going to increase your breast cancer risk, you’re going to have abnormal bleeding, you’re not going to be able to get pregnant,” and so on. It is easy to slip into that in the 7 minutes that you have with your patient, but to me, that is not the right way.
With motivational interviewing, our interactions with patients are shaped by:
- asking
- advising
- assisting
- arranging.
We begin by asking permission: “Do you mind if we talk about your weight?” or “Can we talk about your level of exercise?” Once the patient has granted permission, we ask open-ended questions and use reflective listening: “What I hear you saying is that you are concerned you will not be able to lose the weight,” or “It sounds like you don’t like to exercise, but you are worried about the health consequences of that.”
Utilizing motivational interviewing to help patients identify thoughts and feelings that contribute to unhealthy behaviors--and replacing those thoughts and feelings with new thought patterns that aid in behavior change--has been shown to be an effective and efficient facilitator for change. By incorporating the following principles of motivational interviewing into practice, clinicians can have an important impact on the prevention or management of serious diseases in women1:
- Express empathy and avoid arguments. "I know it has been difficult for you to take the first step to losing weight. That is something that is difficult for a lot of my patients. How can I help you take that first step?"
- Develop discrepancies to help the patient understand the difference between her behavior and her goals. "You have said that you would like to lose some weight. I think you know that exercise would help with that. Why do you think it has been hard for you to start exercising more?"
- Roll with resistance and provide personalized feedback to help the patient find ways to succeed. "What I hear you saying is your work schedule does not allow you time to work out at the gym. What about walking during lunch breaks or taking the stairs instead of the elevator--is that something you think you can commit to doing?"
- Support self-efficacy and elicit self-motivation. "What would you like to see differently about your health? What makes you think you need to change? What happens if you don't change?"
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243-246.
I find these skills useful for addressing anything from smoking to drinking to weight management to excessive shopping—any extreme behavior that is affecting a patient negatively. When a patient is not ready to talk about her clinical problems or make changes, I let her know my door is always open to her and that I have many resources available to help her when she is ready (TABLE).4 In those cases, I might say something like, “I have many patients who really don’t want to talk about this when I first ask them, but I just want you to know, Mrs. Jones, that I want you to succeed and I want you to be healthy. We have a team approach to taking care of all of you, and when you are ready, we are here to help.”

Related article:
2017 Update on fertility: Effects of obesity on reproduction
It is important to provide practical advice to patients—including how much to exercise, the importance of keeping a food journal, and determining a goal for slow, safe weight loss—and provide resources as necessary (such as for Weight Watchers, nutrition, and dieticians). Each day we have more than 30 opportunities to select foods to eat, drink, or purchase. Have a plan and advise your patients do the same. Recommend patients cook their own meals. Suggest weight loss apps. Counsel them to celebrate successes, find a buddy (for social support), practice positive self-talk (positive language), and plan for challenges (travel, parties, working late) and setbacks, which do not need to become a fall. Find an activity or exercise that the patient enjoys and tell them to seek professional help if needed.
Read about how to educate your patients on wellness.
Dr. Bradley: About 86% of the health care dollars spent in the United States are due to chronic diseases, and chronic diseases are the leading cause of death and disability in the country.5 The most common chronic diseases—cardiovascular disease, hypertension, type 2 diabetes, colon cancer, depression, dementia, cognitive problems, higher rates of fractures—all have been associated, at least in part, with unhealthy food choices and lack of exercise. That applies to breast cancer, too.
The good news is, we can prevent and even reverse disease. As Hippocrates said, let food be thy medicine and medicine be thy food. We have all seen success stories where consistent exercise and dietary changes definitely change the paradigm for what the disease state represents. A multiplicity of factors affect poor health—noncompliance, obesity, smoking—but when we begin to make consistent, healthy changes with diet and exercise, this creates a sort of domino effect.
In the book Us! Our Life. Our Health. Our Legacy,1 co-authored by Dr. Bradley and her colleague, Margaret L. McKenzie, MD, the authors highlight the 10 healthiest behaviors to bring about youthfulness and robust health:
- Walk at least 30-45 minutes per day most days of the week.
- Engage in resistance training 2-3 days per week.
- Eat a primarily plant-based diet made up of a variety of whole foods.
- Do not smoke.
- Maintain a waist line that measures less than half your height.
- Drink alcohol only in moderation.
- Get 7-8 hours of sleep most nights.
- Forgive.
- Have gratitude.
- Believe in something greater than yourself.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
Dr. Bradley: I think we need to get to the root cause of these clinical problems and provide the resources and support that patients need to reverse or even prevent these diseases. Clinicians need to become more aware—be an example and a role model. Our patients are watching us as much as we are watching them. Together, we can form good partnerships in order to promote better health.
Dr. Bradley: I think when you are about to be a change agent for your body and become what I call the best version of yourself, you can have these great ideas, but you need to turn those ideas into actions and make them consistent. And we know that is difficult to do, so I do try to have patients write down specific goals, their plan for achieving them, and list the reasons why it is important for them to reach their goals. That gives them something tangible to look at when the going gets tough. It is also important to work into the contract ways to reward positive behaviors when goals are met, and to plan for challenges and setbacks and how to get back on track.
I also encourage patients to document their progress and learn how to make quick adjustments when necessary to get back on track. Another important element involves setting milestones—by what date are you going to reach this goal? Like any other contract, I have my patients date and sign their wellness contracts. I also encourage them to visualize what their new self is going to look like, how they will feel when they reach their goal, what they will wear, and what activities they will engage in.
Related article:
Obesity medicine: How to incorporate it into your practice
Dr. Bradley: I do, but the amount of nutrition education that most of us get in medical school is minimal to nonexistent and not practical. As physicians, we know that food is health, exercise is fitness, and that our patients need both of them. We also know that we did not get this information in school and that our education was more about treating disease than preventing disease. Many of us were not trained in robot surgery either, because it did not exist. So what did we do? We took classes, attended lectures, read books, and learned. We can do the same with wellness. There are many courses around the country. We have to begin to relearn and reteach ourselves about health, nutrition, and exercise and then pass that information on to our patients—be a resource and a guide. We should be able to write a prescription for health as quickly as we can write a prescription for insulin or a statin.
I also bring up portion distortion with my patients. The National Institutes of Health has resources on their website (https://www.nhlbi.nih.gov/health/educational /wecan/eat-right/portion-distortion.html) that include great visuals that show portion sizes 20 years ago and what they are now. For instance, 20 years ago a bagel was 3 inches and 140 calories; today’s bagel is 6 inches and 350 calories (plus whatever toppings are added). I tell that to my patients and then explain how much more exercise is needed to burn off just that 1 bagel.
Related article:
How to help your patients control gestational weight gain
Dr. Bradley: They may not know that term directly, but I think people understand that you have the potential to pass on poor lifestyle and/or health issues related to how things are when you are in utero and later in life. It gets back to letting people know to be healthy in pregnancy and even pre-pregnancy, and that includes one’s emotional state, physical state, and spiritual state. We are what we are in our mother’s womb. Getting the best start in life starts with a healthy mom, healthy dad, and a healthy environment.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in obesity among adults in the United States, 2005 to 2014. JAMA.2016;315(21):2284Arial–2291.
- Sturm R, An R. Obesity and economic environments. CA Cancer J Clin. 2014;64(5):337Arial–350.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243–246.
- Centers for Disease Control and Prevention. Chronic disease overview. https://www.cdc.gov/chronicdisease/overview/index.htm. Updated June 28, 2017. Accessed November 3, 2017.

Dr. Bradley is Professor of Surgery and Vice Chair of Obstetrics and Gynecology at the Women’s Health Institute, and Director, Center for Menstrual Disorders, Fibroids, and Hysteroscopic Services, Cleveland Clinic, Cleveland, Ohio. Dr. Bradley serves on the OBG
Dr. Bradley reports that she has received research or grant support from and is a consultant and speaker for Bayer, is a speaker for Smith & Nephew and Teva, serves on the scientific advisory board for Boston Scientific, is a consultant for Karl Storz, and has received royalties from UpToDate and Elsevier.
Dr. Bradley is Professor of Surgery and Vice Chair of Obstetrics and Gynecology at the Women’s Health Institute, and Director, Center for Menstrual Disorders, Fibroids, and Hysteroscopic Services, Cleveland Clinic, Cleveland, Ohio. Dr. Bradley serves on the OBG
Dr. Bradley reports that she has received research or grant support from and is a consultant and speaker for Bayer, is a speaker for Smith & Nephew and Teva, serves on the scientific advisory board for Boston Scientific, is a consultant for Karl Storz, and has received royalties from UpToDate and Elsevier.
Dr. Bradley is Professor of Surgery and Vice Chair of Obstetrics and Gynecology at the Women’s Health Institute, and Director, Center for Menstrual Disorders, Fibroids, and Hysteroscopic Services, Cleveland Clinic, Cleveland, Ohio. Dr. Bradley serves on the OBG
Dr. Bradley reports that she has received research or grant support from and is a consultant and speaker for Bayer, is a speaker for Smith & Nephew and Teva, serves on the scientific advisory board for Boston Scientific, is a consultant for Karl Storz, and has received royalties from UpToDate and Elsevier.
Over the past 3 decades, the prevalence of overweight and obesity has increased dramatically in the United States. A study published in 2016 showed the age-adjusted prevalence of obesity in 2013–2014 was 35% among men and 40.4% among women.1 It comes as no surprise that increased reliance on inexpensive fast foods coupled with progressively more sedentary lifestyles have been implicated as causative factors.2
With the rise in obesity also has come an attendant rise in related chronic diseases, such as type 2 diabetes mellitus and cardiovascular disease. Women who are obese are also at risk for certain women’s health conditions, such as polycystic ovary syndrome, breast cancer, and endometrial cancer.
It is clear that curbing this public health crisis will require concerted efforts from individuals, clinicians, and policy makers, as well as changes in societal norms.
Linda D. Bradley, MD: I think it is important for us not to lecture our patients. I could list all of the things that patients should or could do to prevent or even reverse disease states, in terms of eating right and exercising, but I think motivational interviewing is a more productive approach to elicit and evoke change (see “Principles and practice of motivational interviewing”). I used to preach to my patients. I would say, “You know, if you stay at this weight, you’re going to get diabetes, you’re going to increase your breast cancer risk, you’re going to have abnormal bleeding, you’re not going to be able to get pregnant,” and so on. It is easy to slip into that in the 7 minutes that you have with your patient, but to me, that is not the right way.
With motivational interviewing, our interactions with patients are shaped by:
- asking
- advising
- assisting
- arranging.
We begin by asking permission: “Do you mind if we talk about your weight?” or “Can we talk about your level of exercise?” Once the patient has granted permission, we ask open-ended questions and use reflective listening: “What I hear you saying is that you are concerned you will not be able to lose the weight,” or “It sounds like you don’t like to exercise, but you are worried about the health consequences of that.”
Utilizing motivational interviewing to help patients identify thoughts and feelings that contribute to unhealthy behaviors--and replacing those thoughts and feelings with new thought patterns that aid in behavior change--has been shown to be an effective and efficient facilitator for change. By incorporating the following principles of motivational interviewing into practice, clinicians can have an important impact on the prevention or management of serious diseases in women1:
- Express empathy and avoid arguments. "I know it has been difficult for you to take the first step to losing weight. That is something that is difficult for a lot of my patients. How can I help you take that first step?"
- Develop discrepancies to help the patient understand the difference between her behavior and her goals. "You have said that you would like to lose some weight. I think you know that exercise would help with that. Why do you think it has been hard for you to start exercising more?"
- Roll with resistance and provide personalized feedback to help the patient find ways to succeed. "What I hear you saying is your work schedule does not allow you time to work out at the gym. What about walking during lunch breaks or taking the stairs instead of the elevator--is that something you think you can commit to doing?"
- Support self-efficacy and elicit self-motivation. "What would you like to see differently about your health? What makes you think you need to change? What happens if you don't change?"
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243-246.
I find these skills useful for addressing anything from smoking to drinking to weight management to excessive shopping—any extreme behavior that is affecting a patient negatively. When a patient is not ready to talk about her clinical problems or make changes, I let her know my door is always open to her and that I have many resources available to help her when she is ready (TABLE).4 In those cases, I might say something like, “I have many patients who really don’t want to talk about this when I first ask them, but I just want you to know, Mrs. Jones, that I want you to succeed and I want you to be healthy. We have a team approach to taking care of all of you, and when you are ready, we are here to help.”
Related article:
2017 Update on fertility: Effects of obesity on reproduction
It is important to provide practical advice to patients—including how much to exercise, the importance of keeping a food journal, and determining a goal for slow, safe weight loss—and provide resources as necessary (such as for Weight Watchers, nutrition, and dieticians). Each day we have more than 30 opportunities to select foods to eat, drink, or purchase. Have a plan and advise your patients do the same. Recommend patients cook their own meals. Suggest weight loss apps. Counsel them to celebrate successes, find a buddy (for social support), practice positive self-talk (positive language), and plan for challenges (travel, parties, working late) and setbacks, which do not need to become a fall. Find an activity or exercise that the patient enjoys and tell them to seek professional help if needed.
Read about how to educate your patients on wellness.
Dr. Bradley: About 86% of the health care dollars spent in the United States are due to chronic diseases, and chronic diseases are the leading cause of death and disability in the country.5 The most common chronic diseases—cardiovascular disease, hypertension, type 2 diabetes, colon cancer, depression, dementia, cognitive problems, higher rates of fractures—all have been associated, at least in part, with unhealthy food choices and lack of exercise. That applies to breast cancer, too.
The good news is, we can prevent and even reverse disease. As Hippocrates said, let food be thy medicine and medicine be thy food. We have all seen success stories where consistent exercise and dietary changes definitely change the paradigm for what the disease state represents. A multiplicity of factors affect poor health—noncompliance, obesity, smoking—but when we begin to make consistent, healthy changes with diet and exercise, this creates a sort of domino effect.
In the book Us! Our Life. Our Health. Our Legacy,1 co-authored by Dr. Bradley and her colleague, Margaret L. McKenzie, MD, the authors highlight the 10 healthiest behaviors to bring about youthfulness and robust health:
- Walk at least 30-45 minutes per day most days of the week.
- Engage in resistance training 2-3 days per week.
- Eat a primarily plant-based diet made up of a variety of whole foods.
- Do not smoke.
- Maintain a waist line that measures less than half your height.
- Drink alcohol only in moderation.
- Get 7-8 hours of sleep most nights.
- Forgive.
- Have gratitude.
- Believe in something greater than yourself.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
Dr. Bradley: I think we need to get to the root cause of these clinical problems and provide the resources and support that patients need to reverse or even prevent these diseases. Clinicians need to become more aware—be an example and a role model. Our patients are watching us as much as we are watching them. Together, we can form good partnerships in order to promote better health.
Dr. Bradley: I think when you are about to be a change agent for your body and become what I call the best version of yourself, you can have these great ideas, but you need to turn those ideas into actions and make them consistent. And we know that is difficult to do, so I do try to have patients write down specific goals, their plan for achieving them, and list the reasons why it is important for them to reach their goals. That gives them something tangible to look at when the going gets tough. It is also important to work into the contract ways to reward positive behaviors when goals are met, and to plan for challenges and setbacks and how to get back on track.
I also encourage patients to document their progress and learn how to make quick adjustments when necessary to get back on track. Another important element involves setting milestones—by what date are you going to reach this goal? Like any other contract, I have my patients date and sign their wellness contracts. I also encourage them to visualize what their new self is going to look like, how they will feel when they reach their goal, what they will wear, and what activities they will engage in.
Related article:
Obesity medicine: How to incorporate it into your practice
Dr. Bradley: I do, but the amount of nutrition education that most of us get in medical school is minimal to nonexistent and not practical. As physicians, we know that food is health, exercise is fitness, and that our patients need both of them. We also know that we did not get this information in school and that our education was more about treating disease than preventing disease. Many of us were not trained in robot surgery either, because it did not exist. So what did we do? We took classes, attended lectures, read books, and learned. We can do the same with wellness. There are many courses around the country. We have to begin to relearn and reteach ourselves about health, nutrition, and exercise and then pass that information on to our patients—be a resource and a guide. We should be able to write a prescription for health as quickly as we can write a prescription for insulin or a statin.
I also bring up portion distortion with my patients. The National Institutes of Health has resources on their website (https://www.nhlbi.nih.gov/health/educational /wecan/eat-right/portion-distortion.html) that include great visuals that show portion sizes 20 years ago and what they are now. For instance, 20 years ago a bagel was 3 inches and 140 calories; today’s bagel is 6 inches and 350 calories (plus whatever toppings are added). I tell that to my patients and then explain how much more exercise is needed to burn off just that 1 bagel.
Related article:
How to help your patients control gestational weight gain
Dr. Bradley: They may not know that term directly, but I think people understand that you have the potential to pass on poor lifestyle and/or health issues related to how things are when you are in utero and later in life. It gets back to letting people know to be healthy in pregnancy and even pre-pregnancy, and that includes one’s emotional state, physical state, and spiritual state. We are what we are in our mother’s womb. Getting the best start in life starts with a healthy mom, healthy dad, and a healthy environment.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
Over the past 3 decades, the prevalence of overweight and obesity has increased dramatically in the United States. A study published in 2016 showed the age-adjusted prevalence of obesity in 2013–2014 was 35% among men and 40.4% among women.1 It comes as no surprise that increased reliance on inexpensive fast foods coupled with progressively more sedentary lifestyles have been implicated as causative factors.2
With the rise in obesity also has come an attendant rise in related chronic diseases, such as type 2 diabetes mellitus and cardiovascular disease. Women who are obese are also at risk for certain women’s health conditions, such as polycystic ovary syndrome, breast cancer, and endometrial cancer.
It is clear that curbing this public health crisis will require concerted efforts from individuals, clinicians, and policy makers, as well as changes in societal norms.
Linda D. Bradley, MD: I think it is important for us not to lecture our patients. I could list all of the things that patients should or could do to prevent or even reverse disease states, in terms of eating right and exercising, but I think motivational interviewing is a more productive approach to elicit and evoke change (see “Principles and practice of motivational interviewing”). I used to preach to my patients. I would say, “You know, if you stay at this weight, you’re going to get diabetes, you’re going to increase your breast cancer risk, you’re going to have abnormal bleeding, you’re not going to be able to get pregnant,” and so on. It is easy to slip into that in the 7 minutes that you have with your patient, but to me, that is not the right way.
With motivational interviewing, our interactions with patients are shaped by:
- asking
- advising
- assisting
- arranging.
We begin by asking permission: “Do you mind if we talk about your weight?” or “Can we talk about your level of exercise?” Once the patient has granted permission, we ask open-ended questions and use reflective listening: “What I hear you saying is that you are concerned you will not be able to lose the weight,” or “It sounds like you don’t like to exercise, but you are worried about the health consequences of that.”
Utilizing motivational interviewing to help patients identify thoughts and feelings that contribute to unhealthy behaviors--and replacing those thoughts and feelings with new thought patterns that aid in behavior change--has been shown to be an effective and efficient facilitator for change. By incorporating the following principles of motivational interviewing into practice, clinicians can have an important impact on the prevention or management of serious diseases in women1:
- Express empathy and avoid arguments. "I know it has been difficult for you to take the first step to losing weight. That is something that is difficult for a lot of my patients. How can I help you take that first step?"
- Develop discrepancies to help the patient understand the difference between her behavior and her goals. "You have said that you would like to lose some weight. I think you know that exercise would help with that. Why do you think it has been hard for you to start exercising more?"
- Roll with resistance and provide personalized feedback to help the patient find ways to succeed. "What I hear you saying is your work schedule does not allow you time to work out at the gym. What about walking during lunch breaks or taking the stairs instead of the elevator--is that something you think you can commit to doing?"
- Support self-efficacy and elicit self-motivation. "What would you like to see differently about your health? What makes you think you need to change? What happens if you don't change?"
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243-246.
I find these skills useful for addressing anything from smoking to drinking to weight management to excessive shopping—any extreme behavior that is affecting a patient negatively. When a patient is not ready to talk about her clinical problems or make changes, I let her know my door is always open to her and that I have many resources available to help her when she is ready (TABLE).4 In those cases, I might say something like, “I have many patients who really don’t want to talk about this when I first ask them, but I just want you to know, Mrs. Jones, that I want you to succeed and I want you to be healthy. We have a team approach to taking care of all of you, and when you are ready, we are here to help.”
Related article:
2017 Update on fertility: Effects of obesity on reproduction
It is important to provide practical advice to patients—including how much to exercise, the importance of keeping a food journal, and determining a goal for slow, safe weight loss—and provide resources as necessary (such as for Weight Watchers, nutrition, and dieticians). Each day we have more than 30 opportunities to select foods to eat, drink, or purchase. Have a plan and advise your patients do the same. Recommend patients cook their own meals. Suggest weight loss apps. Counsel them to celebrate successes, find a buddy (for social support), practice positive self-talk (positive language), and plan for challenges (travel, parties, working late) and setbacks, which do not need to become a fall. Find an activity or exercise that the patient enjoys and tell them to seek professional help if needed.
Read about how to educate your patients on wellness.
Dr. Bradley: About 86% of the health care dollars spent in the United States are due to chronic diseases, and chronic diseases are the leading cause of death and disability in the country.5 The most common chronic diseases—cardiovascular disease, hypertension, type 2 diabetes, colon cancer, depression, dementia, cognitive problems, higher rates of fractures—all have been associated, at least in part, with unhealthy food choices and lack of exercise. That applies to breast cancer, too.
The good news is, we can prevent and even reverse disease. As Hippocrates said, let food be thy medicine and medicine be thy food. We have all seen success stories where consistent exercise and dietary changes definitely change the paradigm for what the disease state represents. A multiplicity of factors affect poor health—noncompliance, obesity, smoking—but when we begin to make consistent, healthy changes with diet and exercise, this creates a sort of domino effect.
In the book Us! Our Life. Our Health. Our Legacy,1 co-authored by Dr. Bradley and her colleague, Margaret L. McKenzie, MD, the authors highlight the 10 healthiest behaviors to bring about youthfulness and robust health:
- Walk at least 30-45 minutes per day most days of the week.
- Engage in resistance training 2-3 days per week.
- Eat a primarily plant-based diet made up of a variety of whole foods.
- Do not smoke.
- Maintain a waist line that measures less than half your height.
- Drink alcohol only in moderation.
- Get 7-8 hours of sleep most nights.
- Forgive.
- Have gratitude.
- Believe in something greater than yourself.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
Dr. Bradley: I think we need to get to the root cause of these clinical problems and provide the resources and support that patients need to reverse or even prevent these diseases. Clinicians need to become more aware—be an example and a role model. Our patients are watching us as much as we are watching them. Together, we can form good partnerships in order to promote better health.
Dr. Bradley: I think when you are about to be a change agent for your body and become what I call the best version of yourself, you can have these great ideas, but you need to turn those ideas into actions and make them consistent. And we know that is difficult to do, so I do try to have patients write down specific goals, their plan for achieving them, and list the reasons why it is important for them to reach their goals. That gives them something tangible to look at when the going gets tough. It is also important to work into the contract ways to reward positive behaviors when goals are met, and to plan for challenges and setbacks and how to get back on track.
I also encourage patients to document their progress and learn how to make quick adjustments when necessary to get back on track. Another important element involves setting milestones—by what date are you going to reach this goal? Like any other contract, I have my patients date and sign their wellness contracts. I also encourage them to visualize what their new self is going to look like, how they will feel when they reach their goal, what they will wear, and what activities they will engage in.
Related article:
Obesity medicine: How to incorporate it into your practice
Dr. Bradley: I do, but the amount of nutrition education that most of us get in medical school is minimal to nonexistent and not practical. As physicians, we know that food is health, exercise is fitness, and that our patients need both of them. We also know that we did not get this information in school and that our education was more about treating disease than preventing disease. Many of us were not trained in robot surgery either, because it did not exist. So what did we do? We took classes, attended lectures, read books, and learned. We can do the same with wellness. There are many courses around the country. We have to begin to relearn and reteach ourselves about health, nutrition, and exercise and then pass that information on to our patients—be a resource and a guide. We should be able to write a prescription for health as quickly as we can write a prescription for insulin or a statin.
I also bring up portion distortion with my patients. The National Institutes of Health has resources on their website (https://www.nhlbi.nih.gov/health/educational /wecan/eat-right/portion-distortion.html) that include great visuals that show portion sizes 20 years ago and what they are now. For instance, 20 years ago a bagel was 3 inches and 140 calories; today’s bagel is 6 inches and 350 calories (plus whatever toppings are added). I tell that to my patients and then explain how much more exercise is needed to burn off just that 1 bagel.
Related article:
How to help your patients control gestational weight gain
Dr. Bradley: They may not know that term directly, but I think people understand that you have the potential to pass on poor lifestyle and/or health issues related to how things are when you are in utero and later in life. It gets back to letting people know to be healthy in pregnancy and even pre-pregnancy, and that includes one’s emotional state, physical state, and spiritual state. We are what we are in our mother’s womb. Getting the best start in life starts with a healthy mom, healthy dad, and a healthy environment.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in obesity among adults in the United States, 2005 to 2014. JAMA.2016;315(21):2284Arial–2291.
- Sturm R, An R. Obesity and economic environments. CA Cancer J Clin. 2014;64(5):337Arial–350.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243–246.
- Centers for Disease Control and Prevention. Chronic disease overview. https://www.cdc.gov/chronicdisease/overview/index.htm. Updated June 28, 2017. Accessed November 3, 2017.
- Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in obesity among adults in the United States, 2005 to 2014. JAMA.2016;315(21):2284Arial–2291.
- Sturm R, An R. Obesity and economic environments. CA Cancer J Clin. 2014;64(5):337Arial–350.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243–246.
- Centers for Disease Control and Prevention. Chronic disease overview. https://www.cdc.gov/chronicdisease/overview/index.htm. Updated June 28, 2017. Accessed November 3, 2017.

Luxury drug treatment centers: Close scrutiny advised
About 2.5 million people received mental and/or substance use disorder treatment last year, according to the Substance Abuse and Mental Health Services Administration.1 Of those, a small but significant percentage chose support from a luxury type facility. This article intends to provide clinicians with guidance in addressing medically supervised luxury detox and/or rehabilitation programs for drug and alcohol abuse.
We recommend that individuals entering substance abuse services pursue these treatments with “eyes wide open.” A vast amount of literature indicates a rise in programs designed to attract vulnerable clients seeking treatment.2 They offer an array of luxury services such as equine, massage, and yoga therapy, as well as holistic approaches. These services are all packaged in a five-star hotel–like environment in a desirable area (by the sea, mountains, etc.).
The question is whether a $50,000-per-month treatment center is better at keeping its clients in remission than a facility that charges a fraction of the price per month. We believe that success rates may be less a function of financial cost and more a function of evidenced-based treatment strategies that are specific to recovery.
We would like to advise individuals to look for the following elements when reviewing a potential treatment center:
• Accepted by major insurance companies such as Blue Cross/Blue Shield and United Healthcare, to name a few.
• No cookie-cutter approaches: Programs allowing for inter-individual flexibility regarding length of stay (not specifically 21, 30, or 45 days), as well as flexibility of services.
• Group therapy should have no more than 15 clients. Some individuals may not be appropriate for group therapy or may have a strong aversion to this modality.
• Licensed and/or certified staff (not peer coaching and/or paraprofessionals alone).
• Minimum of 20 hours per week, per client, of clinically supervised evidence-based methods, techniques, and/or practices including individual counseling, group therapies, and family involvement.
• If the length of stay is longer, fewer staff members may be more appropriate.
• Availability of higher level of professional staff, psychologists, psychiatrists, and other physicians to address all comorbid concerns.
• Adequate aftercare treatment.
After reviewing social media that rate treatment facilities, one common thread we noticed was the total absence of aftercare services.3 Aftercare services were something that clients may not think of at the initial intake.
References
1 Substance Abuse and Mental Health Services Administration (SAMHSA). “Behavioral Health Treatments and Services.”
2 The Verge, Sept. 6, 2017
3 Rehabs.com
4. The New York Times, Sept. 14, 2017
Dr. Lesser is executive director of RANE, Medical & Mental Health, in New York City. In his recent positions as medical director for New York City and State, he was instrumental in developing and implementing nationally recognized emergency and response programs. Dr. Brenner is chief of Behavioral Health Service Line for Catholic Health Services of Long Island, in New York. He is a clinical professor of psychiatry for the State University of New York, Brooklyn, and medical director and CEO of Neurobehavioral Research Inc. Dr. Ferber is a licensed psychologist in New York and California. He has been the director of Behavioral Health Central Intake at Catholic Health Services of Long Island for the last 2 years. Dr. Ferber specializes in addiction treatment. Ms. Howard is a psychologist in training with specialization in clinical psychology. She currently treats numerous dual-diagnosed patients with comorbidities on an acute psychiatric ward.
About 2.5 million people received mental and/or substance use disorder treatment last year, according to the Substance Abuse and Mental Health Services Administration.1 Of those, a small but significant percentage chose support from a luxury type facility. This article intends to provide clinicians with guidance in addressing medically supervised luxury detox and/or rehabilitation programs for drug and alcohol abuse.
We recommend that individuals entering substance abuse services pursue these treatments with “eyes wide open.” A vast amount of literature indicates a rise in programs designed to attract vulnerable clients seeking treatment.2 They offer an array of luxury services such as equine, massage, and yoga therapy, as well as holistic approaches. These services are all packaged in a five-star hotel–like environment in a desirable area (by the sea, mountains, etc.).
The question is whether a $50,000-per-month treatment center is better at keeping its clients in remission than a facility that charges a fraction of the price per month. We believe that success rates may be less a function of financial cost and more a function of evidenced-based treatment strategies that are specific to recovery.
We would like to advise individuals to look for the following elements when reviewing a potential treatment center:
• Accepted by major insurance companies such as Blue Cross/Blue Shield and United Healthcare, to name a few.
• No cookie-cutter approaches: Programs allowing for inter-individual flexibility regarding length of stay (not specifically 21, 30, or 45 days), as well as flexibility of services.
• Group therapy should have no more than 15 clients. Some individuals may not be appropriate for group therapy or may have a strong aversion to this modality.
• Licensed and/or certified staff (not peer coaching and/or paraprofessionals alone).
• Minimum of 20 hours per week, per client, of clinically supervised evidence-based methods, techniques, and/or practices including individual counseling, group therapies, and family involvement.
• If the length of stay is longer, fewer staff members may be more appropriate.
• Availability of higher level of professional staff, psychologists, psychiatrists, and other physicians to address all comorbid concerns.
• Adequate aftercare treatment.
After reviewing social media that rate treatment facilities, one common thread we noticed was the total absence of aftercare services.3 Aftercare services were something that clients may not think of at the initial intake.
References
1 Substance Abuse and Mental Health Services Administration (SAMHSA). “Behavioral Health Treatments and Services.”
2 The Verge, Sept. 6, 2017
3 Rehabs.com
4. The New York Times, Sept. 14, 2017
Dr. Lesser is executive director of RANE, Medical & Mental Health, in New York City. In his recent positions as medical director for New York City and State, he was instrumental in developing and implementing nationally recognized emergency and response programs. Dr. Brenner is chief of Behavioral Health Service Line for Catholic Health Services of Long Island, in New York. He is a clinical professor of psychiatry for the State University of New York, Brooklyn, and medical director and CEO of Neurobehavioral Research Inc. Dr. Ferber is a licensed psychologist in New York and California. He has been the director of Behavioral Health Central Intake at Catholic Health Services of Long Island for the last 2 years. Dr. Ferber specializes in addiction treatment. Ms. Howard is a psychologist in training with specialization in clinical psychology. She currently treats numerous dual-diagnosed patients with comorbidities on an acute psychiatric ward.
About 2.5 million people received mental and/or substance use disorder treatment last year, according to the Substance Abuse and Mental Health Services Administration.1 Of those, a small but significant percentage chose support from a luxury type facility. This article intends to provide clinicians with guidance in addressing medically supervised luxury detox and/or rehabilitation programs for drug and alcohol abuse.
We recommend that individuals entering substance abuse services pursue these treatments with “eyes wide open.” A vast amount of literature indicates a rise in programs designed to attract vulnerable clients seeking treatment.2 They offer an array of luxury services such as equine, massage, and yoga therapy, as well as holistic approaches. These services are all packaged in a five-star hotel–like environment in a desirable area (by the sea, mountains, etc.).
The question is whether a $50,000-per-month treatment center is better at keeping its clients in remission than a facility that charges a fraction of the price per month. We believe that success rates may be less a function of financial cost and more a function of evidenced-based treatment strategies that are specific to recovery.
We would like to advise individuals to look for the following elements when reviewing a potential treatment center:
• Accepted by major insurance companies such as Blue Cross/Blue Shield and United Healthcare, to name a few.
• No cookie-cutter approaches: Programs allowing for inter-individual flexibility regarding length of stay (not specifically 21, 30, or 45 days), as well as flexibility of services.
• Group therapy should have no more than 15 clients. Some individuals may not be appropriate for group therapy or may have a strong aversion to this modality.
• Licensed and/or certified staff (not peer coaching and/or paraprofessionals alone).
• Minimum of 20 hours per week, per client, of clinically supervised evidence-based methods, techniques, and/or practices including individual counseling, group therapies, and family involvement.
• If the length of stay is longer, fewer staff members may be more appropriate.
• Availability of higher level of professional staff, psychologists, psychiatrists, and other physicians to address all comorbid concerns.
• Adequate aftercare treatment.
After reviewing social media that rate treatment facilities, one common thread we noticed was the total absence of aftercare services.3 Aftercare services were something that clients may not think of at the initial intake.
References
1 Substance Abuse and Mental Health Services Administration (SAMHSA). “Behavioral Health Treatments and Services.”
2 The Verge, Sept. 6, 2017
3 Rehabs.com
4. The New York Times, Sept. 14, 2017
Dr. Lesser is executive director of RANE, Medical & Mental Health, in New York City. In his recent positions as medical director for New York City and State, he was instrumental in developing and implementing nationally recognized emergency and response programs. Dr. Brenner is chief of Behavioral Health Service Line for Catholic Health Services of Long Island, in New York. He is a clinical professor of psychiatry for the State University of New York, Brooklyn, and medical director and CEO of Neurobehavioral Research Inc. Dr. Ferber is a licensed psychologist in New York and California. He has been the director of Behavioral Health Central Intake at Catholic Health Services of Long Island for the last 2 years. Dr. Ferber specializes in addiction treatment. Ms. Howard is a psychologist in training with specialization in clinical psychology. She currently treats numerous dual-diagnosed patients with comorbidities on an acute psychiatric ward.
Are fewer nonpregnant women seeing ObGyns?
EXPERT COMMENTARY
Health care services for women are fragmented due to multiple types of providers who offer a variety of care. Simon and Uddin’s recent research analysis indicates that the percentage of nonpregnant women who visit a general ObGyn, whether alone or in combination with an internist, family physician, or general practitioner, has declined.
Details of the study
The authors used data from the National Health Interview Survey of a representative sample of US women age 18 or older. They sought to identify whether the women saw or talked to a physician who either specialized in women’s health (presumably an ObGyn) or treated a variety of illnesses during the previous 12 months.
While the percentage of women who saw a general physician remained essentially the same (70%–74%), it declined for seeing an ObGyn from 45% to 41% between 2003 and 2007, and from 42% to 38% between 2011 and 2015. Furthermore, the percentage of women who saw both an ObGyn and a general physician declined from a peak of 35% in 2003 to 30% in 2015.
Study strengths and weaknesses
The data used in this study were from a nationally representative, cross-sectional, multistage sample, population health survey conducted by the Centers for Disease Control and Prevention. The study period was sufficient to draw conclusions.
From my perspective, the study had 2 major limitations: 1) only physicians, not mid-level providers, were included in the analysis, and 2) no breakdown of the women’s age groups was provided.
Many ObGyn offices employ nurse practitioners and midwives, and these providers’ roles are increasingly important for improving frontline access to care and different levels of care. Women aged 19 to 39 seek almost all their health care from ObGyns or family physicians, and significant sharing of care exists across these provider groups.1 Women aged 45 to 64 are more likely to obtain care exclusively at the offices of family physicians or general internists than at those of ObGyns.2 Most ObGyns are engaged to some degree with women aged 65 years or older, especially for preventive care, disease screening and early detection, and urogenital conditions.3
The decline in the percentage of women seeking care from ObGyns is likely related to the patient's age, reason for seeking care, and access to care. The US population of adult women, especially those who are beyond the reproductive years, is rising in relation to the number of physicians in general ObGyn practice. Providing a team-based collaborative model of care should allow for improved access and value. Defining the roles of what constitutes evidence-based care also will impact when a person needs to see a women's health care specialist. Geographic distribution of ObGyns in relation to the patient population will invariably impact on the percentage of women who seek care at the office of an ObGyn alone, in combination with another general physician, or not at all. Given the overlap in care provided at more than one physician's office, continued surveillance is needed to minimize redundant costs and optimize resource utilization. I look forward to what unfolds over the next 15 years.
-- William F. Rayburn, MD, MBA
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Petterson SM, Bazemore AW, Phillips RL, Rayburn WF. Trends in office-based care for reproductive-aged women according to physician specialty: a ten-year study. J Womens Health (Larchmt). 2014;23(12):1021–1026.
- Raffoul MC, Petterson SM, Rayburn WF, Wingrove P, Bazemore AW. Office visits for women aged 45–64 years according to physician specialties. J Womens Health (Larchmt). 2016:25(12):1231–1236.
- Rayburn WF, Raglan GB, Herman CJ, Schulkin J. A survey of obstetrician-gynecologists regarding their care of women 65 years or older. J Geriatr Med Gerontol. 2015;1:2-5.
EXPERT COMMENTARY
Health care services for women are fragmented due to multiple types of providers who offer a variety of care. Simon and Uddin’s recent research analysis indicates that the percentage of nonpregnant women who visit a general ObGyn, whether alone or in combination with an internist, family physician, or general practitioner, has declined.
Details of the study
The authors used data from the National Health Interview Survey of a representative sample of US women age 18 or older. They sought to identify whether the women saw or talked to a physician who either specialized in women’s health (presumably an ObGyn) or treated a variety of illnesses during the previous 12 months.
While the percentage of women who saw a general physician remained essentially the same (70%–74%), it declined for seeing an ObGyn from 45% to 41% between 2003 and 2007, and from 42% to 38% between 2011 and 2015. Furthermore, the percentage of women who saw both an ObGyn and a general physician declined from a peak of 35% in 2003 to 30% in 2015.
Study strengths and weaknesses
The data used in this study were from a nationally representative, cross-sectional, multistage sample, population health survey conducted by the Centers for Disease Control and Prevention. The study period was sufficient to draw conclusions.
From my perspective, the study had 2 major limitations: 1) only physicians, not mid-level providers, were included in the analysis, and 2) no breakdown of the women’s age groups was provided.
Many ObGyn offices employ nurse practitioners and midwives, and these providers’ roles are increasingly important for improving frontline access to care and different levels of care. Women aged 19 to 39 seek almost all their health care from ObGyns or family physicians, and significant sharing of care exists across these provider groups.1 Women aged 45 to 64 are more likely to obtain care exclusively at the offices of family physicians or general internists than at those of ObGyns.2 Most ObGyns are engaged to some degree with women aged 65 years or older, especially for preventive care, disease screening and early detection, and urogenital conditions.3
The decline in the percentage of women seeking care from ObGyns is likely related to the patient's age, reason for seeking care, and access to care. The US population of adult women, especially those who are beyond the reproductive years, is rising in relation to the number of physicians in general ObGyn practice. Providing a team-based collaborative model of care should allow for improved access and value. Defining the roles of what constitutes evidence-based care also will impact when a person needs to see a women's health care specialist. Geographic distribution of ObGyns in relation to the patient population will invariably impact on the percentage of women who seek care at the office of an ObGyn alone, in combination with another general physician, or not at all. Given the overlap in care provided at more than one physician's office, continued surveillance is needed to minimize redundant costs and optimize resource utilization. I look forward to what unfolds over the next 15 years.
-- William F. Rayburn, MD, MBA
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
EXPERT COMMENTARY
Health care services for women are fragmented due to multiple types of providers who offer a variety of care. Simon and Uddin’s recent research analysis indicates that the percentage of nonpregnant women who visit a general ObGyn, whether alone or in combination with an internist, family physician, or general practitioner, has declined.
Details of the study
The authors used data from the National Health Interview Survey of a representative sample of US women age 18 or older. They sought to identify whether the women saw or talked to a physician who either specialized in women’s health (presumably an ObGyn) or treated a variety of illnesses during the previous 12 months.
While the percentage of women who saw a general physician remained essentially the same (70%–74%), it declined for seeing an ObGyn from 45% to 41% between 2003 and 2007, and from 42% to 38% between 2011 and 2015. Furthermore, the percentage of women who saw both an ObGyn and a general physician declined from a peak of 35% in 2003 to 30% in 2015.
Study strengths and weaknesses
The data used in this study were from a nationally representative, cross-sectional, multistage sample, population health survey conducted by the Centers for Disease Control and Prevention. The study period was sufficient to draw conclusions.
From my perspective, the study had 2 major limitations: 1) only physicians, not mid-level providers, were included in the analysis, and 2) no breakdown of the women’s age groups was provided.
Many ObGyn offices employ nurse practitioners and midwives, and these providers’ roles are increasingly important for improving frontline access to care and different levels of care. Women aged 19 to 39 seek almost all their health care from ObGyns or family physicians, and significant sharing of care exists across these provider groups.1 Women aged 45 to 64 are more likely to obtain care exclusively at the offices of family physicians or general internists than at those of ObGyns.2 Most ObGyns are engaged to some degree with women aged 65 years or older, especially for preventive care, disease screening and early detection, and urogenital conditions.3
The decline in the percentage of women seeking care from ObGyns is likely related to the patient's age, reason for seeking care, and access to care. The US population of adult women, especially those who are beyond the reproductive years, is rising in relation to the number of physicians in general ObGyn practice. Providing a team-based collaborative model of care should allow for improved access and value. Defining the roles of what constitutes evidence-based care also will impact when a person needs to see a women's health care specialist. Geographic distribution of ObGyns in relation to the patient population will invariably impact on the percentage of women who seek care at the office of an ObGyn alone, in combination with another general physician, or not at all. Given the overlap in care provided at more than one physician's office, continued surveillance is needed to minimize redundant costs and optimize resource utilization. I look forward to what unfolds over the next 15 years.
-- William F. Rayburn, MD, MBA
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Petterson SM, Bazemore AW, Phillips RL, Rayburn WF. Trends in office-based care for reproductive-aged women according to physician specialty: a ten-year study. J Womens Health (Larchmt). 2014;23(12):1021–1026.
- Raffoul MC, Petterson SM, Rayburn WF, Wingrove P, Bazemore AW. Office visits for women aged 45–64 years according to physician specialties. J Womens Health (Larchmt). 2016:25(12):1231–1236.
- Rayburn WF, Raglan GB, Herman CJ, Schulkin J. A survey of obstetrician-gynecologists regarding their care of women 65 years or older. J Geriatr Med Gerontol. 2015;1:2-5.
- Petterson SM, Bazemore AW, Phillips RL, Rayburn WF. Trends in office-based care for reproductive-aged women according to physician specialty: a ten-year study. J Womens Health (Larchmt). 2014;23(12):1021–1026.
- Raffoul MC, Petterson SM, Rayburn WF, Wingrove P, Bazemore AW. Office visits for women aged 45–64 years according to physician specialties. J Womens Health (Larchmt). 2016:25(12):1231–1236.
- Rayburn WF, Raglan GB, Herman CJ, Schulkin J. A survey of obstetrician-gynecologists regarding their care of women 65 years or older. J Geriatr Med Gerontol. 2015;1:2-5.
Shulkin: VA Focused on Transparency and Modernization
After a concerted effort, the VA is now approaching its goal of 100% treatment for patients infected with hepatitis C virus (HCV), VA Secretary David J. Shulkin, MD noted at a speech at the AMSUS Annual Meeting. More than 100,000 veterans have been treated for HCV infection, and 90% have been cured. According to Dr. Shulkin, the VA also has seen significant drops in measures of opioid use and homelessness among veterans. Although the VA is no longer focused on completely ending veteran homelessness, the rate has dropped 50% since 2010, and 3 states (Virginia, Connecticut, and Delaware) have eliminated homelessness completely.
According to Dr. Shulkin, the VA currently is the only health care system that publishes its wait times, quality scores, and disciplinary actions taken against employees. That level of transparency is not only unusual in health care systems, but also unprecedented in the federal government, Dr. Shulkin noted. “I am the only cabinet member that publishes my travel schedule,” Dr. Shulkin told the VA, DoD, and PHS audience.
In addition to promoting current VA progress, Dr. Shulkin also outlined his priorities for the VA moving forward. The priorities included:
- Greater Choice. Dr. Shulkin maintained that the VA should not only provide more options for veterans seeking health care, but also facilitate their ability to make better health care decisions. Choice for veterans will mean being able to access care both inside and outside the VA system.
- Modernize the VA. The signature part of VA’s modernization effort has been its decision to adopt the Cerner/GENESIS electronic health record (EHR) system, but it also includes the VA decision to close 1,100 facilities and focus its resources on the remaining facilities. Dr. Shulkin hopes that the combined power of the VA and DoD on a single EHR system will force EHR providers to improve interoperability. In addition, a new program will allow PHS officers to serve in the VA, bringing in much needed public health experience.
- Improve the Timeliness of Services. Currently, 97% of VA appointments are completed within 30 days, 86% are within a week, and 21% are same day. Improving those metrics will require the VA to “double down on technology,” Dr. Shulkin insisted. The VA now sees 700,000 veterans via telehealth.
- Focus Resources More Efficiently. The VA can and should be “world class” in areas where it matters most, Dr. Shulkin argued, including areas like prosthetics, traumatic brain injury, posttraumatic stress disorder, and spinal cord injury. Better coordination with the DoD also will help both health care systems to improve efficiency. A “Caregivers Moonshot” initiative also will allow more veterans to remain at home at end of life.
- Suicide Prevention. While suicide is a public health crisis across the U.S., it is especially acute amongst veterans. According to Dr. Shulkin, reducing veteran suicide will require action both inside and outside the VA. The VA needs to connect with community mental health providers and provide more services to other than honorable discharges.
After a concerted effort, the VA is now approaching its goal of 100% treatment for patients infected with hepatitis C virus (HCV), VA Secretary David J. Shulkin, MD noted at a speech at the AMSUS Annual Meeting. More than 100,000 veterans have been treated for HCV infection, and 90% have been cured. According to Dr. Shulkin, the VA also has seen significant drops in measures of opioid use and homelessness among veterans. Although the VA is no longer focused on completely ending veteran homelessness, the rate has dropped 50% since 2010, and 3 states (Virginia, Connecticut, and Delaware) have eliminated homelessness completely.
According to Dr. Shulkin, the VA currently is the only health care system that publishes its wait times, quality scores, and disciplinary actions taken against employees. That level of transparency is not only unusual in health care systems, but also unprecedented in the federal government, Dr. Shulkin noted. “I am the only cabinet member that publishes my travel schedule,” Dr. Shulkin told the VA, DoD, and PHS audience.
In addition to promoting current VA progress, Dr. Shulkin also outlined his priorities for the VA moving forward. The priorities included:
- Greater Choice. Dr. Shulkin maintained that the VA should not only provide more options for veterans seeking health care, but also facilitate their ability to make better health care decisions. Choice for veterans will mean being able to access care both inside and outside the VA system.
- Modernize the VA. The signature part of VA’s modernization effort has been its decision to adopt the Cerner/GENESIS electronic health record (EHR) system, but it also includes the VA decision to close 1,100 facilities and focus its resources on the remaining facilities. Dr. Shulkin hopes that the combined power of the VA and DoD on a single EHR system will force EHR providers to improve interoperability. In addition, a new program will allow PHS officers to serve in the VA, bringing in much needed public health experience.
- Improve the Timeliness of Services. Currently, 97% of VA appointments are completed within 30 days, 86% are within a week, and 21% are same day. Improving those metrics will require the VA to “double down on technology,” Dr. Shulkin insisted. The VA now sees 700,000 veterans via telehealth.
- Focus Resources More Efficiently. The VA can and should be “world class” in areas where it matters most, Dr. Shulkin argued, including areas like prosthetics, traumatic brain injury, posttraumatic stress disorder, and spinal cord injury. Better coordination with the DoD also will help both health care systems to improve efficiency. A “Caregivers Moonshot” initiative also will allow more veterans to remain at home at end of life.
- Suicide Prevention. While suicide is a public health crisis across the U.S., it is especially acute amongst veterans. According to Dr. Shulkin, reducing veteran suicide will require action both inside and outside the VA. The VA needs to connect with community mental health providers and provide more services to other than honorable discharges.
After a concerted effort, the VA is now approaching its goal of 100% treatment for patients infected with hepatitis C virus (HCV), VA Secretary David J. Shulkin, MD noted at a speech at the AMSUS Annual Meeting. More than 100,000 veterans have been treated for HCV infection, and 90% have been cured. According to Dr. Shulkin, the VA also has seen significant drops in measures of opioid use and homelessness among veterans. Although the VA is no longer focused on completely ending veteran homelessness, the rate has dropped 50% since 2010, and 3 states (Virginia, Connecticut, and Delaware) have eliminated homelessness completely.
According to Dr. Shulkin, the VA currently is the only health care system that publishes its wait times, quality scores, and disciplinary actions taken against employees. That level of transparency is not only unusual in health care systems, but also unprecedented in the federal government, Dr. Shulkin noted. “I am the only cabinet member that publishes my travel schedule,” Dr. Shulkin told the VA, DoD, and PHS audience.
In addition to promoting current VA progress, Dr. Shulkin also outlined his priorities for the VA moving forward. The priorities included:
- Greater Choice. Dr. Shulkin maintained that the VA should not only provide more options for veterans seeking health care, but also facilitate their ability to make better health care decisions. Choice for veterans will mean being able to access care both inside and outside the VA system.
- Modernize the VA. The signature part of VA’s modernization effort has been its decision to adopt the Cerner/GENESIS electronic health record (EHR) system, but it also includes the VA decision to close 1,100 facilities and focus its resources on the remaining facilities. Dr. Shulkin hopes that the combined power of the VA and DoD on a single EHR system will force EHR providers to improve interoperability. In addition, a new program will allow PHS officers to serve in the VA, bringing in much needed public health experience.
- Improve the Timeliness of Services. Currently, 97% of VA appointments are completed within 30 days, 86% are within a week, and 21% are same day. Improving those metrics will require the VA to “double down on technology,” Dr. Shulkin insisted. The VA now sees 700,000 veterans via telehealth.
- Focus Resources More Efficiently. The VA can and should be “world class” in areas where it matters most, Dr. Shulkin argued, including areas like prosthetics, traumatic brain injury, posttraumatic stress disorder, and spinal cord injury. Better coordination with the DoD also will help both health care systems to improve efficiency. A “Caregivers Moonshot” initiative also will allow more veterans to remain at home at end of life.
- Suicide Prevention. While suicide is a public health crisis across the U.S., it is especially acute amongst veterans. According to Dr. Shulkin, reducing veteran suicide will require action both inside and outside the VA. The VA needs to connect with community mental health providers and provide more services to other than honorable discharges.
Immune Thrombocytopenia
Introduction
Immune thrombocytopenia (ITP) is a common acquired autoimmune disease characterized by low platelet counts and an increased risk of bleeding. The incidence of ITP is approximately 3.3 per 100,000 adults.1 There is considerable controversy about all aspects of the disease, with little “hard” data on which to base decisions given the lack of randomized clinical trials to address most clinical questions. This article reviews the presentation and diagnosis of ITP and its treatment options and discusses management of ITP in specific clinical situations.
Pathogenesis and Epidemiology
ITP is caused by autoantibodies binding to platelet surface proteins, most often to the platelet receptor GP IIb/IIIa.2-4 These antibody-coated platelets then bind to Fc receptors in macrophages and are removed from circulation. The initiating event in ITP is unknown. It is speculated that the patient responds to a viral or bacterial infection by creating antibodies which cross-react with the platelet receptors. Continued exposure to platelets perpetuates the immune response. ITP that occurs in childhood appears to be an acute response to viral infection and usually resolves. ITP in adults may occur in any age group but is seen especially in young women.
Despite the increased platelet destruction that occurs in ITP, the production of new platelets often is not significantly increased. This is most likely due to lack of an increase in thrombopoietin, the predominant platelet growth factor.5
It had been thought that most adult patients who present with ITP go on to have a chronic course, but more recent studies have shown this is not the case. In modern series the percentage of patients who are “cured” with steroids ranges from 30% to 70%.6–9 In addition, it has been appreciated that even in patients with modest thrombocytopenia, no therapy is required if the platelet count remains higher than 30 × 103/µL. However, this leaves a considerable number of patients who will require chronic therapy.
Clinical Presentation
Presentation can range from a symptomatic patient with low platelets found on a routine blood count to a patient with massive bleeding. Typically, patients first present with petechiae (small bruises 1 mm in size) on the shins. True petechiae are seen only in severe thrombocytopenia. Patients will also report frequent bruising and bleeding from the gums. Patients with very low platelet counts will notice “wet purpura,” which is characterized by blood-filled bullae in the oral cavity. Life-threatening bleeding is a very unusual presenting sign unless other problems (trauma, ulcers) are present. The physical examination is only remarkable for stigmata of bleeding such as the petechiae. The presence of splenomegaly or lymphadenopathy weighs strongly against a diagnosis of ITP. Many patients with ITP will note fatigue when their platelets counts are lower.10
Diagnosis
Extremely low platelet counts with a normal blood smear and an otherwise healthy patient are diagnostic of ITP. The platelet count cutoff for considering ITP is 100 × 103/µL as the majority of patients with counts in the 100 to 150 × 103/µL range will not develop greater thrombocytopenia.11 Also, the platelet count decreases with age (9 × 103/µL per decade in one study), and this also needs to be factored into the evaluation.12 The finding of relatives with ITP should raise suspicion for congenital thrombocytopenia.13 One should question the patient carefully about drug exposure (see Drug-Induced Thrombocytopenia), especially about over-the-counter medicines, “natural” remedies, or recreational drugs.
There is no laboratory test that rules in ITP; rather, it is a diagnosis of exclusion. The blood smear should be carefully examined for evidence of microangiopathic hemolytic anemias (schistocytes), bone marrow disease (blasts, teardrop cells), or any other evidence of a primary bone marrow disease. In ITP, the platelets can be larger than normal, but finding some platelets the size of red cells should raise the issue of congenital thrombocytopenia.14 Pseudo-thrombocytopenia, which is the clumping of platelets due to a reaction to the EDTA anticoagulant in the tube, should be excluded. The diagnosis is established by drawing the blood in a citrated (blue-top) tube to perform the platelet count. There is no role for antiplatelet antibody assay because this test lacks sensitivity and specificity. In a patient without a history of autoimmune disease or symptoms, empiric testing for autoimmune disease is not recommended.
Patients who present with ITP should be tested for both HIV and hepatitis C infection.15,16 These are the most common viral causes of secondary ITP, and both have prognostic and treatment implications. Some authorities also recommend checking thyroid function as hypothyroidism can present or aggravate the thrombocytopenia.
The role of bone marrow examination is controversial.17 Patients with a classic presentation of ITP (young woman, normal blood smear) do not require a bone marrow exam before therapy is initiated, although patients who do not respond to initial therapy should have a bone marrow aspiration. The rare entity amegakaryocytic thrombocytopenia can present with a clinical picture similar to that of ITP, but amegakaryocytic thrombocytopenia will not respond to steroids. Bone marrow aspiration reveals the absence of megakaryocytes in this entity. It is rare, however, that another hematologic disease is diagnosed in patients with a classic clinical presentation of ITP.
In the future, measurement of thrombopoietin and reticulated platelets may provide clues to the diagnosis.4 Patients with ITP paradoxically have normal or only mildly elevated thrombopoietin levels. The finding of a significantly elevated thrombopoietin level should lead to questioning of the diagnosis. One can also measure “reticulated platelets,” which are analogous to red cell reticulocytes. Patients with ITP (or any platelet destructive disorders) will have high levels of reticulated platelets. These tests are not recommended for routine evaluation, but may be helpful in difficult cases.
Treatment
In general, therapy in ITP should be guided by the patient’s signs of bleeding and not by unquestioning adherence to measuring platelet levels,15 as patients tolerate thrombocytopenia well. It is unusual to have life-threatening bleeding with platelet counts greater than 5 × 103/µL in the absence of mechanical lesions. Despite the low platelet count in patients with ITP, the overall mortality is estimated to be only 0.3% to 1.3%.18 It is sobering that in one study the rate of death from infections was twice as high as that from bleeding.19 Rare patients will have antibodies that interfere with the function of the platelet, and these patients can have profound bleeding with only modestly lowered platelet counts.20 A suggested cut-off for treating newly diagnosed patients is 30 × 103/µL.21
Initial Therapy
The primary therapy of ITP is glucocorticoids, either prednisone or dexamethasone. In the past prednisone at a dose of 60 to 80 mg/day was started at the time of diagnosis (Table 1). 
For rapid induction of a response, there are 2 options. A single dose of intravenous immune globulin (IVIG) at 1 g/kg or intravenous anti-D immunoglobulin (anti-D) at 50 to 75 µg/kg can induce a response in more than 80% of patients in 24 to 48 hours.21,24 IVIG has several drawbacks. It can cause aseptic meningitis, and in patients with vascular disease the increased viscosity can induce ischemia. There is also a considerable fluid load delivered with the IVIG, and it needs to be given over several hours.
The use of anti-D is limited to Rh-positive patients who have not had a splenectomy. It should not be used in patients who are Coombs positive due to the risk of provoking more hemolysis. Rarely anti-D has been reported to cause a severe hemolytic disseminated intravascular coagulation syndrome (1:20,000 patients), which has led to restrictions in its use.25 Although the drug can be rapidly given over 15 minutes, due to these concerns current recommendations are now to observe patients for 8 hours after their dose and to perform a urine dipstick test for blood at 2, 4, and 8 hours. Concerns about this rare but serious side effect have led to a dramatic decrease in the use of anti-D.
For patients who are severely thrombocytopenic and do not respond to initial therapy, there are 2 options for raising the platelet counts. One is to use a combination of IVIG, methylprednisolone, vincristine, and/or anti-D.26 The combination of IVIG and anti-D may be synergistic since these agents block different Fc receptors. A response of 71% has been reported for this 3- or 4-drug combination in a series of 35 patients.26 The other option is to treat with a continuous infusion of platelets (1 unit over 6 hours) and IVIG 1 g/kg for 24 hours. Response rates of 62.7% have been reported with this combination, and this rapid rise in platelets can allow time for other therapies to take effect.27,28
Patients with severe thrombocytopenia who relapse with reduction of steroids or who do not respond to steroids have several options for further management. Repeated doses of IVIG can transiently raise the platelet count, and some patients may only need several courses of therapy over the course of many months. One study showed that 60% of patients could delay or defer therapy by receiving multiple doses of anti-D. However, 30% of patients did eventually receive splenectomy and 20% of patients required ongoing therapy with anti-D.29 In a randomized trial comparing early use of anti-D to steroids to avoid splenectomy, there was no difference in splenectomy rate (38% versus 42%).30 Finally, an option as mentioned above is to try a 6-month course of pulse dexamethasone 40 mg/day for 4 days, repeated every 28 days.
Options for Refractory ITP
There are multiple options for patients who do not respond to initial ITP therapies. These can be divided into several broad groups: curative therapies (splenectomy and rituximab), thrombopoietin receptor agonists, and anecdotal therapies.
Splenectomy
In patients with severe thrombocytopenia who do not respond or who relapse with lower doses of prednisone, splenectomy should be strongly considered. Splenectomy will induce a good response in 60% to 70% of patients and is durable in most patients. In 2 recently published reviews of splenectomy, the complete response rate was 67% and the total response rate was 88% to 90%%.8,31 Between 15% and 28% of patients relapsed over 5 years, with most recurrences occurring in the first 2 years. Splenectomy carries a short-term surgical risk, and the life-long risk of increased susceptibility to overwhelming sepsis is discussed below. However, the absolute magnitude of these risks is low and is often lower than the risks of continued prednisone therapy or of continued cytotoxic therapy.
Timing of splenectomy depends on the patient’s presentation. Most patients should be given a 6-month trial of steroids or other therapies before proceeding to splenectomy.31 However, patients who persist with severe thrombocytopenia despite initial therapies or who are suffering intolerable side effects from therapy should be considered sooner for splenectomy.31 In the George review, multiple factors such as responding to IVIG were found not to be predictive of response to splenectomy.8
Method of splenectomy appears not to matter.21 Rates of finding accessory spleens are just as high or higher with laparoscopic splenectomy and the patient can recover faster. In patients who are severely thrombocytopenic, open splenectomy can allow for quicker control of the vascular access of the spleen.
Rates of splenectomy in recent years have decreased for many reasons,32 including the acceptance of lower platelet counts in asymptomatic patients and the availability of alternative therapies such as rituximab. In addition, despite abundant data for good outcomes, there is a concern that splenectomy responses are not durable. Although splenectomy will not cure every patient with ITP, splenectomy is the therapy with the most patients, the longest follow-up, and the most consistent rate of cure, and it should be discussed with every ITP patient who does not respond to initial therapy and needs further treatment.
The risk of overwhelming sepsis varies by indications for splenectomy but appears to be about 1%.33,34 The use of pneumococcal vaccine and recognition of this syndrome have helped reduce the risk. Asplenic patients need to be counseled about the risk of overwhelming infections, should be vaccinated for pneumococcus, meningococcus, and Haemophilus influenzae, and should wear an ID bracelet.35–37 Patients previously vaccinated for pneumococcus should be re-vaccinated every 3 to 5 years. The role of prophylactic antibiotics in adults is controversial, but patients under the age of 18 should be on penicillin VK 250 mg orally twice daily.
Rituximab
Rituximab has been shown to be very active in ITP. Most studies used the standard dose of 375 mg/m2 weekly for 4 weeks, but other studies have shown that 1000 mg twice 14 days apart (ie, on days 1 and 15) resulted in the same response rate and may be more convenient for patients.38,39 The response time can vary, with patients either showing a rapid response or requiring up to 8 weeks for their counts to go up. Although experience is limited, the response seems to be durable, especially in those patients whose counts rise higher than 150 × 103/µL; in patients who relapse, a response can be re-induced with a repeat course. Overall the response rate for rituximab is about 60%, but only approximately 20% to 40% of patients will remain in long-term remission.40–42 There is no evidence yet that “maintenance” therapy or monitoring CD19/CD20 cells can help further the duration of remission.
Whether to give rituximab pre- or post-splenectomy is also uncertain. An advantage of presplenectomy rituximab is that many patients will achieve remission, delaying the need for surgery. Also, rituximab is a good option for patients whose medical conditions put them at high risk for complications with splenectomy. However, it is unknown whether rituximab poses any long-term risks, while the long-term risks of splenectomy are well-defined. Rituximab is the only curative option left for patients who have failed splenectomy and is a reasonable option for these patients.
There is an intriguing trial in which patients were randomly assigned to dexamethasone alone versus dexamethasone plus rituximab upon presentation with ITP; those who were refractory to dexamethasone alone received salvage therapy with dexamethasone plus rituximab.43 The dexamethasone plus rituximab group had an overall higher rate of sustained remission at 6 months than the dexamethasone group, 63% versus 36%. Interestingly, patients who failed their first course of dexamethasone but then were “salvaged” with dexamethasone/rituximab had a similar overall response rate of 56%, suggesting that saving the addition of rituximab for steroid failures may be an effective option.
Although not “chemotherapy,” rituximab is not without risks. Patients can develop infusion reactions, which can be severe in 1% to 2% of patients. In a meta-analysis the fatal reaction rate was 2.9%.40 Patients with chronic hepatitis B infections can experience reactivation with rituximab, and thus all patients should be screened before treatment. Finally, the very rare but devastating complication of progressive multifocal leukoencephalopathy has been reported.
Thrombopoietin Receptor Agonists
Although patients with ITP have low platelet counts, studies starting with Dameshek have shown that these patients also have reduced production of platelets.44 Despite the very low circulating platelet count, levels of the platelet growth factor thrombopoietin (TPO) are not raised.45 Seminal studies with recombinant TPO in the 1990s showed that ITP patients responded to thrombopoietin-stimulating protein, but the formation of anti-TPO antibodies halted trials with the first generation of these agents. Two TPO receptor agonists (TPO-RA) are approved for use in patients with ITP.
Romiplostim. Romiplostim is a peptibody, a combination of a peptide that binds and stimulates the TPO receptor and an Fc domain to extend its half-life.46 It is administered in a weekly subcutaneous dose starting at 1 to 3 µg/kg. Use of romiplostim in ITP patients produces a response rate of 80% to 88%, with 87% of patients being able to wean off or decrease other anti-ITP medications.47 In a long-term extension study, the response was again high at 87%.48 These studies have also shown a reduced incidence of bleeding.
The major side effect of romiplostim seen in clinical trials was marrow reticulin formation, which occurred in up to 5.6% of patients.47,48 The clinical course in these patients is the development of anemia and a myelophthisic blood smear with teardrop cells and nucleated red cells. These changes appear to reverse with cessation of the drug. The bone marrow shows increased reticulin formation but rarely, if ever, shows the collagen deposition seen with primary myelofibrosis.
Thrombosis has also been seen, with a rate of 0.08 to 0.1 cases per 100 patient-weeks,49 but it remains unclear if this is due to the drug, part of the natural history of ITP, or expected complications in older patients undergoing any type of medical therapy. Surprisingly, despite the low platelet counts, patients with ITP in one study had double the risk of venous thrombosis, demonstrating that ITP itself can be a risk factor for thrombosis.50 These trials have shown no long-term concerns for other clinical problems such as liver disease.
Eltrombopag. The other available TPO-RA is eltrombopag,51 an oral agent that stimulates the TPO receptor by binding the transmembrane domain and activating it. The drug is given orally starting at 50 mg/day (25 mg for patients of Asian ancestry or with liver disease) and can be dose escalated to 75 mg/day. The drug needs to be taken on an empty stomach. Eltrombopag has been shown to be effective in chronic ITP, with response rates of 59% to 80% and reduction in use of rescue medications.47,51,52 As with romiplostim, the incidence of bleeding was also decreased with eltrombopag in these trials.47,51
Clinical trials demonstrated that eltrombopag shares with romiplostim the risk for marrow fibrosis. A side effect unique to eltrombopag observed in these trials was a 3% to 7% incidence of elevated liver function tests.21,52 These abnormal findings appeared to resolve in most patients, but liver function tests need to be monitored in patients receiving eltrombopag.
Clinical use. The clearest indication for the use of TPO-RAs is in patients who have failed several therapies and remain symptomatic or are on intolerable doses of other medications such as prednisone. The clear benefits are their relative safety and high rates of success. The main drawback of TPO-RAs is the need for continuing therapy as the platelet count will return to baseline shortly after these agents are stopped. Currently there is no clear indication for one medication over the other. The advantages of romiplostim are great flexibility in dosing (1–10 µg/kg week) and no concerns about drug interaction. The current drawback of romiplostim is the Food and Drug Administration’s requirement for patients to receive the drug from a clinic and not at home. Eltrombopag offers the advantage of oral use, but it has a limited dose range and potential for drug interactions. Both agents have been associated with marrow reticulin formation, although in clinical use this risk appears to be very low.53
Other Options
In the literature there are numerous options for the treatment of ITP.54,55 Most of these studies are anecdotal, enrolled small number of patients, and sometimes included patients with mild thrombocytopenia, but these therapeutic options can be tried in patients who are refractory to standard therapies and have bleeding. The agents with the greatest amount of supporting data are danazol, vincristine, azathioprine, cyclophosphamide, and fostamatinib.
Danazol 200 mg 4 times daily is thought to downregulate the macrophage Fc receptor. The onset of action may be delayed and a therapeutic trial of up to 4 to 6 months is advised. Danazol is very effective in patients with antiphospholipid antibody syndrome who develop ITP and may be more effective in premenopausal women.56 Once a response is seen, danazol should be continued for 6 months and then an attempt to wean the patient off the agent should be made. A partial response can be seen in 70% to 90% of patients, but a complete response is rare.54
Vincristine 1.4 mg/m2 weekly has a low response rate, but if a response is going to occur, it will occur rapidly within 2 weeks. Thus, a prolonged trial of vincristine is not needed; if no platelet rise is seen in several weeks, the drug should be stopped. Again, partial responses are more common than complete response—50% to 63% versus 0% to 6%.54Azathioprine 150 mg orally daily, like danazol, demonstrates a delayed response and requires several months to assess for response. However, 19% to 25% of patients may have a complete response.54 It has been reported that the related agent mycophenolate 1000 mg twice daily is also effective in ITP.57
Cyclophosphamide 1 g/m2 intravenously repeated every 28 days has been reported to have a response rate of up to 40%.58 Although considered more aggressive, this is a standard immunosuppressive dose and should be considered in patients with very low platelet counts. Patients who have not responded to single-agent cyclophosphamide may respond to multi-agent chemotherapy with agents such as etoposide and vincristine plus cyclophosphamide.59
Fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, is currently under investigation for the treatment of ITP.60 This agent prevents phagocytosis of antibody-coated platelets by macrophages. In early studies fostamatinib has been well tolerated at a dose of 150 mg twice daily, with 75% of patients showing a response. Large phase 3 trials are underway, and if the earlier promising results hold up fostamatinib may be a novel option for refractory patients.
A Practical Approach to Refractory ITP
One approach is to divide patients into bleeders, or those with either very low platelet counts (< 5 × 103/µL) or who have had significant bleeding in the past, and nonbleeders, or those with platelet counts above 5 × 103/µL and no history of severe bleeding. Bleeders who do not respond adequately to splenectomy should first start with rituximab since it is not cytotoxic and is the only other “curative” therapy (Table 2). 
Nonbleeders should be tried on danazol and other relatively safe agents. If this fails, rituximab or TPO-RAs can be considered. Before one considers cytotoxic therapy, the risk of the therapy must be weighed against the risk posed by the thrombocytopenia. The mortality from ITP is fairly low (5%) and is restricted to patients with severe disease. Patients with only moderate thrombocytopenia and no bleeding are better served with conservative management. There is little justification for the use of continuous steroid therapy in this group of patients given the long-term risks of this therapy.
Special Situations
Surgery
Patients with ITP who need surgery either for splenectomy or for other reasons should have their platelet counts raised to a level greater than 20 to 30 × 103/µL before surgery. Most patients with ITP have increased platelet function and will not have excessive bleeding with these platelet counts. For patients with platelet counts below this level, an infusion of immune globulin or anti-D may rapidly increase the platelet counts. If the surgery is elective, short-term use of TPO-RAs to raise the counts can also be considered.
Pregnancy
Up to 10% of pregnant women will develop low platelet counts during their pregnancy.61,62 The most common etiology is gestational thrombocytopenia, which is an exaggeration of the lowered platelet count seen in pregnancy. Counts may fall as low as 50 × 103/µL at the time of delivery. No therapy is required as the fetus is not affected and the mother does not have an increased risk of bleeding. Pregnancy complications such as HELLP syndrome and thrombotic microangiopathies also present with low platelet counts, but these can be diagnosed by history.61,63
Women with ITP can either develop the disease during pregnancy or have a worsening of the symptoms.64 Counts often drop dramatically during the first trimester. Early management should be conservative with low doses of prednisone to keep the count above 10 × 103/µL.21 Immunoglobulin is also effective,65 but there are rare reports of pulmonary edema. Rarely patients who are refractory will require splenectomy, which may be safely performed in the second trimester. For delivery the count should be greater than 30 × 103/µL and for an epidural greater than 50 × 103/µL.64 There are reports of the use of TPO-RAs in pregnancy, and this can be considered for refractory cases.66
Most controversy centers on management of the delivery. In the past it was feared that fetal thrombocytopenia could lead to intracranial hemorrhage, and Caesarean section was always recommended. It now appears that most cases of intracranial hemorrhage were due to alloimmune thrombocytopenia and not ITP. Furthermore, the nadir of the baby’s platelet count is not at birth but several days after. It appears the safest course is to proceed with a vaginal or C-section delivery determined by obstetrical indications and then immediately check the baby’s platelet count. If the platelet count is low in the neonate, immunoglobulin will raise the count. Since the neonatal thrombocytopenia is due to passive transfer of maternal antibody, the platelet destruction will abate in 4 to 6 weeks.
Pediatric Patients
The incidence of ITP in children is 2.2 to 5.3 per 100,000 children.1 There are several distinct differences in pediatric ITP. Most cases will resolve in weeks, with only a minority of patients transforming into chronic ITP (5%–10%). Also, the rates of serious bleeding are lower in children than in adults, with intracranial hemorrhage rates of 0.1% to 0.5% being seen.67 For most patients with no or mild bleeding, management now is observation alone regardless of platelet count because it is felt that the risks of therapies are higher than the risk of bleeding.21 For patients with bleeding, IVIG, anti-D, or a short course of steroids can be used. Given the risk of overwhelming sepsis, splenectomy is often deferred as long as possible. Rituximab is increasingly being used in children due to concerns about use of agents such a cyclophosphamide or azathioprine in children.68 Abundant data on use of TPO-RAs in children showing high response rates and safety support their use, and these should be considered in refractory ITP before any cytotoxic agent.69–71
Helicobacter Pylori Infection
There has been much interest in the relationship between H. pylori and ITP.16,72,73H. pylori infections have been associated with a variety of autoimmune diseases, and there is a confusing literature on this infection and ITP. Several meta-analyses have shown that eradication of H. pylori will result in an ITP response rate of 20% to 30%, but responses curiously appear to be limited to certain geographic areas such as Japan and Italy but not the United States. In patients with recalcitrant ITP, especially in geographic areas with high incidence, it may be worthwhile to check for H. pylori infection and treat accordingly if positive.
Drug-Induced Thrombocytopenia
Patients with drug-induced thrombocytopenia present with very low (< 10 × 103/µL) platelet counts 1 to 3 weeks after starting a new medication.74–76 In patients with a possible drug-induced thrombocytopenia, the primary therapy is to stop the suspect drug.77 If there are multiple new medications, the best approach is to stop any drug that has been strongly associated with thrombocytopenia (Table 3).74,78,79
Immune globulin, corticosteroids, or intravenous anti‑D have been suggested as useful in drug‑related thrombocytopenia. However, since most of these thrombocytopenic patients recover when the agent is cleared from the body, this therapy is probably not necessary and withholding treatment avoids exposing the patients to the adverse events associated with further therapy.
Evans Syndrome
Evans syndrome is defined as the combination of autoimmune hemolytic anemia (AIHA) and ITP.80,81 These cytopenias can present simultaneously or sequentially. Patients with Evans syndrome are thought to have a more severe disease process, to be more prone to bleeding, and to be more difficult to treat, but the rarity of this syndrome makes this hard to quantify.
The classic clinical presentation of Evans syndrome is severe anemia and thrombocytopenia. Children with Evans syndrome often have complex immunodeficiencies such as autoimmune lymphoproliferative syndrome.82,83 In adults, Evans syndrome most often complicates other autoimmune diseases such as lupus. There are increasing reports of Evans syndrome occurring as a complication of T-cell lymphomas. Often the autoimmune disease can predate the lymphoma diagnosis by months or even years.
In theory the diagnostic approach is straightforward by showing a Coombs-positive hemolytic anemia in the setting of a clinical diagnosis of immune thrombocytopenia. The blood smear will show spherocytes and a diminished platelet count. The presence of other abnormal red cell forms should raise the possibility of an alternative diagnosis. It is unclear how vigorously one should search for other underlying diseases. Many patients will already have the diagnosis of an underlying autoimmune disease. The presence of lymphadenopathy should raise concern for lymphoma.
Initial therapy is high-dose steroids (2 mg/kg/day). IVIG should be added if severe thrombocytopenia is present. Patients who cannot be weaned off prednisone or relapse after prednisone should be considered for splenectomy, although these patients are at higher risk of relapsing.80 Increasingly rituximab is being used with success.84,85 For patients who fail splenectomy and rituximab, aggressive immunosuppression should be considered. Increasing data support the benefits of sirolimus, and this should be considered for refractory patients.86 For patients with Evans syndrome due to underlying lymphoma, antineoplastic therapy often results in prompt resolution of the symptoms. Recurrence of the autoimmune cytopenias often heralds relapse.
1. Terrell DR, Beebe LA, Vesely SK, et al. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol 2010;85:174–80.
2. McMillan R, Lopez-Dee J, Bowditch R. Clonal restriction of platelet-associated anti-GPIIb/IIIa autoantibodies in patients with chronic ITP. Thromb Haemost 2001;85:821–3.
3. Aster RH, George JN, McMillan R, Ganguly P. Workshop on autoimmune (idiopathic) thrombocytopenic purpura: Pathogenesis and new approaches to therapy. Am J Hematol 1998;58:231–4.
4. Toltl LJ, Arnold DM. Pathophysiology and management of chronic immune thrombocytopenia: focusing on what matters. Br J Haematol 2011;152:52–60.
5. Kuter DJ, Gernsheimer TB. Thrombopoietin and platelet production in chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1193–211.
6. Pamuk GE, Pamuk ON, Baslar Z, et al. Overview of 321 patients with idiopathic thrombocytopenic purpura. Retrospective analysis of the clinical features and response to therapy. Ann Hematol 2002;81:436–40.
7. Stasi R, Stipa E, Masi M, et al. Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 1995;98:436–42.
8. Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004;104:2623–34.
9. Matschke J, Muller-Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment-naive adult patients with immune thrombocytopenia: EIS 2002 study. Acta Haematol 2016;136:101–7.
10. Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011;86:420–9.
11. Stasi R, Amadori S, Osborn J, et al. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006;3:e24.
12. Biino G, Balduini CL, Casula L, et al. Analysis of 12,517 inhabitants of a Sardinian geographic isolate reveals that predispositions to thrombocytopenia and thrombocytosis are inherited traits. Haematologica 2011;96:96–101.
13. Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 2004;103:390–8.
14. Geddis AE, Balduini CL. Diagnosis of immune thrombocytopenic purpura in children. Curr Opin Hematol 2007;14:520–5.
15. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010;115:168–86.
16. Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1275–97.
17. Jubelirer SJ, Harpold R. The role of the bone marrow examination in the diagnosis of immune thrombocytopenic purpura: case series and literature review. Clin Appl Thromb Hemost 2002;8:73–6.
18. George JN. Management of patients with refractory immune thrombocytopenic purpura. J Thromb Haemost 2006;4:1664–72.
19. Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001;97:2549–54.
20. McMillan R, Bowditch RD, Tani P, et al. A non-thrombocytopenic bleeding disorder due to an IgG4- kappa anti-GPIIb/IIIa autoantibody. Br J Haematol 1996;95:747–9.
21. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011;117:4190–207.22. Mazzucconi MG, Fazi P, Bernasconi S, et al. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007;109:1401–7.
23. Wei Y, Ji XB, Wang YW, et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. Blood 2016;127:296–302.
24. Newman GC, Novoa MV, Fodero EM, et al. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001;112:1076–8.
25. Gaines AR. Acute onset hemoglobinemia and/or hemoglobinuria and sequelae following Rho(D) immune globulin intravenous administration in immune thrombocytopenic purpura patients. Blood 2000;95:2523–9.
26. Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007;110:3526–31.
27. Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008;83:122–5.
28. Olson SR, Chu C, Shatzel JJ, Deloughery TG. The “platelet boilermaker”: A treatment protocol to rapidly increase platelets in patients with immune-mediated thrombocytopenia. Am J Hematol 2016;91:E330–1.
29. Cooper N, Woloski BM, Fodero EM, et al. Does treatment with intermittent infusions of intravenous anti-D allow a proportion of adults with recently diagnosed immune thrombocytopenic purpura to avoid splenectomy? Blood 2002;99:1922–7.
30. George JN, Raskob GE, Vesely SK, et al. Initial management of immune thrombocytopenic purpura in adults: a randomized controlled trial comparing intermittent anti-D with routine care. Am J Hematol 2003;74:161–9.
31. Mikhael J, Northridge K, Lindquist K, et al. Short-term and long-term failure of laparoscopic splenectomy in adult immune thrombocytopenic purpura patients: a systematic review. Am J Hematol 2009;84:743–8.
32. Palandri F, Polverelli N, Sollazzo D, et al. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am J Hematol 2016;91:E267–72.
33. Landgren O, Bjorkholm M, Konradsen HB, et al. A prospective study on antibody response to repeated vaccinations with pneumococcal capsular polysaccharide in splenectomized individuals with special reference to Hodgkin’s lymphoma. J Intern Med 2004;255:664–73.
34. Bisharat N, Omari H, Lavi I, Raz R. Risk of infection and death among post-splenectomy patients. J Infect 2001;43:182–6.
35. Mileno MD, Bia FJ. The compromised traveler. Infect Dis Clin North Am 1998;12:369–412.
36. Guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Working Party of the British Committee for Standards in Haematology Clinical Haematology Task Force. BMJ 1996;312:430–4.
37. Ericsson CD. Travellers with pre-existing medical conditions. Int J Antimicrob Agents 2003;21:181–8.
38. Tran H, Brighton T, Grigg A, et al. A multi-centre, single-arm, open-label study evaluating the safety and efficacy of fixed dose rituximab in patients with refractory, relapsed or chronic idiopathic thrombocytopenic purpura (R-ITP1000 study). Br J Haematol 2014;167:243–51.
39. Mahevas M, Ebbo M, Audia S, et al. Efficacy and safety of rituximab given at 1,000 mg on days 1 and 15 compared to the standard regimen to treat adult immune thrombocytopenia. Am J Hematol 2013;88:858–61.
40. Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007;146:25–33.
41. Khellaf M, Charles-Nelson A, Fain O, et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: results from a prospective registry including 248 patients. Blood 2014;124:3228–36.
42. Ghanima W, Khelif A, Waage A, et al. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015;385:1653–61.
43. Zaja F, Baccarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010;115:2755–62.
44. Dameshek W, Miller EB. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood 1946;1:27–50.
45. Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment of thrombocytopenia. Annu Rev Med 2009;60:193–206.
46. Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006;355:1672–81.
47. Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:641–8.
48. Bussel JB, Kuter DJ, Pullarkat V, et al. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009;113:2161–71.
49. Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010;8:1372–82.
50. Severinsen MT, Engebjerg MC, Farkas DK, et al. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol 2011;152:360–2.
51. Bussel JB, Cheng G, Saleh MN, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med 2007;357:2237–47.
52. Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011;377:393–402.
53. Brynes RK, Orazi A, Theodore D, et al. Evaluation of bone marrow reticulin in patients with chronic immune thrombocytopenia treated with eltrombopag: Data from the EXTEND study. Am J Hematol 2015;90:598–601.
54. George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000;37:290–8.
55. McMillan R. Therapy for adults with refractory chronic immune thrombocytopenic purpura. Ann Intern Med 1997;126:307–14.
56. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Successful therapy with danazol in refractory autoimmune thrombocytopenia associated with rheumatic diseases. Br J Rheumatol 1997;36:1095–9.
57. Provan D, Moss AJ, Newland AC, Bussel JB. Efficacy of mycophenolate mofetil as single-agent therapy for refractory immune thrombocytopenic purpura. Am J Hematol 2006;81:19–25.
58. Reiner A, Gernsheimer T, Slichter SJ. Pulse cyclophosphamide therapy for refractory autoimmune thrombocytopenic purpura. Blood 1995;85:351–8.
59. Figueroa M, Gehlsen J, Hammond D, et al. Combination chemotherapy in refractory immune thrombocytopenic purpura. N Engl J Med 1993;328:1226–9.
60. Newland A, Lee EJ, McDonald V, Bussel JB. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy 2 Oct 2017.
61. McCrae KR. Thrombocytopenia in pregnancy. Hematology Am Soc Hematol Educ Program 2010;2010:397–402.
62. Gernsheimer T, McCrae KR. Immune thrombocytopenic purpura in pregnancy. Curr Opin Hematol 2007;14:574–80.
63. DeLoughery TG. Critical care clotting catastrophies. Crit Care Clin 2005;21:531–62.
64. Stavrou E, McCrae KR. Immune thrombocytopenia in pregnancy. Hematol Oncol Clin North Am 2009;23:1299–316.
65. Sun D, Shehata N, Ye XY, et al. Corticosteroids compared with intravenous immunoglobulin for the treatment of immune thrombocytopenia in pregnancy. Blood 2016;128:1329–35.
66. Kong Z, Qin P, Xiao S, et al. A novel recombinant human thrombopoietin therapy for the management of immune thrombocytopenia in pregnancy. Blood 2017;130:1097–103.
67. Psaila B, Petrovic A, Page LK, et al. Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood 2009;114:4777–83.
68. Journeycake JM. Childhood immune thrombocytopenia: role of rituximab, recombinant thrombopoietin, and other new therapeutics. Hematology Am Soc Hematol Educ Program 2012;2012:444–9.
69. Zhang J, Liang Y, Ai Y, et al. Thrombopoietin-receptor agonists for children with immune thrombocytopenia: a systematic review. Expert Opin Pharmacother 2017;18:1543–51.
70. Tarantino MD, Bussel JB, Blanchette VS, et al. Romiplostim in children with immune thrombocytopenia: a phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016;388:45–54.71. Grainger JD, Locatelli F, Chotsampancharoen T, et al. Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet 2015;386:1649–58.
72. Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009;113:1231–40.
73. Arnold DM, Bernotas A, Nazi I, et al. Platelet count response to H. pylori treatment in patients with immune thrombocytopenic purpura with and without H. pylori infection: a systematic review. Haematologica 2009;94:850–6.
74. Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med 2007;357:580–7.
75. Reese JA, Li X, Hauben M, et al. Identifying drugs that cause acute thrombocytopenia: an analysis using 3 distinct methods. Blood 2010;116:2127–33.
76. Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis and management. J Thromb Haemost 2009;7:911–8.
77. Zondor SD, George JN, Medina PJ. Treatment of drug-induced thrombocytopenia. Expert Opin Drug Saf 2002;1:173–80.
78. George JN, Raskob GE, Shah SR, et al. Drug-induced thrombocytopenia: A systematic review of published case reports. Ann Intern Med 1998;129:886–90.
79. Green D, Hougie C, Kazmier FJ, et al. Report of the working party on acquired inhibitors of coagulation: studies of the “lupus” anticoagulant. Thromb Haemost 1983;49:144–6.
80. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood 2009;114:3167–72.
81. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology 2008;13:356–60.
82. Notarangelo LD. Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematology Am Soc Hematol Educ Program 2009:139–43.
83. Martinez-Valdez L, Deya-Martinez A, Giner MT, et al. Evans syndrome as first manifestation of primary immunodeficiency in clinical practice. J Pediatr Hematol Oncol 2017;39:490–4.
84. Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003;78:1340–6.
85. Mantadakis E, Danilatou V, Stiakaki E, Kalmanti M. Rituximab for refractory Evans syndrome and other immune-mediated hematologic diseases. Am J Hematol 2004;77:303–10.
86. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol 2017;39:420–4.
Introduction
Immune thrombocytopenia (ITP) is a common acquired autoimmune disease characterized by low platelet counts and an increased risk of bleeding. The incidence of ITP is approximately 3.3 per 100,000 adults.1 There is considerable controversy about all aspects of the disease, with little “hard” data on which to base decisions given the lack of randomized clinical trials to address most clinical questions. This article reviews the presentation and diagnosis of ITP and its treatment options and discusses management of ITP in specific clinical situations.
Pathogenesis and Epidemiology
ITP is caused by autoantibodies binding to platelet surface proteins, most often to the platelet receptor GP IIb/IIIa.2-4 These antibody-coated platelets then bind to Fc receptors in macrophages and are removed from circulation. The initiating event in ITP is unknown. It is speculated that the patient responds to a viral or bacterial infection by creating antibodies which cross-react with the platelet receptors. Continued exposure to platelets perpetuates the immune response. ITP that occurs in childhood appears to be an acute response to viral infection and usually resolves. ITP in adults may occur in any age group but is seen especially in young women.
Despite the increased platelet destruction that occurs in ITP, the production of new platelets often is not significantly increased. This is most likely due to lack of an increase in thrombopoietin, the predominant platelet growth factor.5
It had been thought that most adult patients who present with ITP go on to have a chronic course, but more recent studies have shown this is not the case. In modern series the percentage of patients who are “cured” with steroids ranges from 30% to 70%.6–9 In addition, it has been appreciated that even in patients with modest thrombocytopenia, no therapy is required if the platelet count remains higher than 30 × 103/µL. However, this leaves a considerable number of patients who will require chronic therapy.
Clinical Presentation
Presentation can range from a symptomatic patient with low platelets found on a routine blood count to a patient with massive bleeding. Typically, patients first present with petechiae (small bruises 1 mm in size) on the shins. True petechiae are seen only in severe thrombocytopenia. Patients will also report frequent bruising and bleeding from the gums. Patients with very low platelet counts will notice “wet purpura,” which is characterized by blood-filled bullae in the oral cavity. Life-threatening bleeding is a very unusual presenting sign unless other problems (trauma, ulcers) are present. The physical examination is only remarkable for stigmata of bleeding such as the petechiae. The presence of splenomegaly or lymphadenopathy weighs strongly against a diagnosis of ITP. Many patients with ITP will note fatigue when their platelets counts are lower.10
Diagnosis
Extremely low platelet counts with a normal blood smear and an otherwise healthy patient are diagnostic of ITP. The platelet count cutoff for considering ITP is 100 × 103/µL as the majority of patients with counts in the 100 to 150 × 103/µL range will not develop greater thrombocytopenia.11 Also, the platelet count decreases with age (9 × 103/µL per decade in one study), and this also needs to be factored into the evaluation.12 The finding of relatives with ITP should raise suspicion for congenital thrombocytopenia.13 One should question the patient carefully about drug exposure (see Drug-Induced Thrombocytopenia), especially about over-the-counter medicines, “natural” remedies, or recreational drugs.
There is no laboratory test that rules in ITP; rather, it is a diagnosis of exclusion. The blood smear should be carefully examined for evidence of microangiopathic hemolytic anemias (schistocytes), bone marrow disease (blasts, teardrop cells), or any other evidence of a primary bone marrow disease. In ITP, the platelets can be larger than normal, but finding some platelets the size of red cells should raise the issue of congenital thrombocytopenia.14 Pseudo-thrombocytopenia, which is the clumping of platelets due to a reaction to the EDTA anticoagulant in the tube, should be excluded. The diagnosis is established by drawing the blood in a citrated (blue-top) tube to perform the platelet count. There is no role for antiplatelet antibody assay because this test lacks sensitivity and specificity. In a patient without a history of autoimmune disease or symptoms, empiric testing for autoimmune disease is not recommended.
Patients who present with ITP should be tested for both HIV and hepatitis C infection.15,16 These are the most common viral causes of secondary ITP, and both have prognostic and treatment implications. Some authorities also recommend checking thyroid function as hypothyroidism can present or aggravate the thrombocytopenia.
The role of bone marrow examination is controversial.17 Patients with a classic presentation of ITP (young woman, normal blood smear) do not require a bone marrow exam before therapy is initiated, although patients who do not respond to initial therapy should have a bone marrow aspiration. The rare entity amegakaryocytic thrombocytopenia can present with a clinical picture similar to that of ITP, but amegakaryocytic thrombocytopenia will not respond to steroids. Bone marrow aspiration reveals the absence of megakaryocytes in this entity. It is rare, however, that another hematologic disease is diagnosed in patients with a classic clinical presentation of ITP.
In the future, measurement of thrombopoietin and reticulated platelets may provide clues to the diagnosis.4 Patients with ITP paradoxically have normal or only mildly elevated thrombopoietin levels. The finding of a significantly elevated thrombopoietin level should lead to questioning of the diagnosis. One can also measure “reticulated platelets,” which are analogous to red cell reticulocytes. Patients with ITP (or any platelet destructive disorders) will have high levels of reticulated platelets. These tests are not recommended for routine evaluation, but may be helpful in difficult cases.
Treatment
In general, therapy in ITP should be guided by the patient’s signs of bleeding and not by unquestioning adherence to measuring platelet levels,15 as patients tolerate thrombocytopenia well. It is unusual to have life-threatening bleeding with platelet counts greater than 5 × 103/µL in the absence of mechanical lesions. Despite the low platelet count in patients with ITP, the overall mortality is estimated to be only 0.3% to 1.3%.18 It is sobering that in one study the rate of death from infections was twice as high as that from bleeding.19 Rare patients will have antibodies that interfere with the function of the platelet, and these patients can have profound bleeding with only modestly lowered platelet counts.20 A suggested cut-off for treating newly diagnosed patients is 30 × 103/µL.21
Initial Therapy
The primary therapy of ITP is glucocorticoids, either prednisone or dexamethasone. In the past prednisone at a dose of 60 to 80 mg/day was started at the time of diagnosis (Table 1).
For rapid induction of a response, there are 2 options. A single dose of intravenous immune globulin (IVIG) at 1 g/kg or intravenous anti-D immunoglobulin (anti-D) at 50 to 75 µg/kg can induce a response in more than 80% of patients in 24 to 48 hours.21,24 IVIG has several drawbacks. It can cause aseptic meningitis, and in patients with vascular disease the increased viscosity can induce ischemia. There is also a considerable fluid load delivered with the IVIG, and it needs to be given over several hours.
The use of anti-D is limited to Rh-positive patients who have not had a splenectomy. It should not be used in patients who are Coombs positive due to the risk of provoking more hemolysis. Rarely anti-D has been reported to cause a severe hemolytic disseminated intravascular coagulation syndrome (1:20,000 patients), which has led to restrictions in its use.25 Although the drug can be rapidly given over 15 minutes, due to these concerns current recommendations are now to observe patients for 8 hours after their dose and to perform a urine dipstick test for blood at 2, 4, and 8 hours. Concerns about this rare but serious side effect have led to a dramatic decrease in the use of anti-D.
For patients who are severely thrombocytopenic and do not respond to initial therapy, there are 2 options for raising the platelet counts. One is to use a combination of IVIG, methylprednisolone, vincristine, and/or anti-D.26 The combination of IVIG and anti-D may be synergistic since these agents block different Fc receptors. A response of 71% has been reported for this 3- or 4-drug combination in a series of 35 patients.26 The other option is to treat with a continuous infusion of platelets (1 unit over 6 hours) and IVIG 1 g/kg for 24 hours. Response rates of 62.7% have been reported with this combination, and this rapid rise in platelets can allow time for other therapies to take effect.27,28
Patients with severe thrombocytopenia who relapse with reduction of steroids or who do not respond to steroids have several options for further management. Repeated doses of IVIG can transiently raise the platelet count, and some patients may only need several courses of therapy over the course of many months. One study showed that 60% of patients could delay or defer therapy by receiving multiple doses of anti-D. However, 30% of patients did eventually receive splenectomy and 20% of patients required ongoing therapy with anti-D.29 In a randomized trial comparing early use of anti-D to steroids to avoid splenectomy, there was no difference in splenectomy rate (38% versus 42%).30 Finally, an option as mentioned above is to try a 6-month course of pulse dexamethasone 40 mg/day for 4 days, repeated every 28 days.
Options for Refractory ITP
There are multiple options for patients who do not respond to initial ITP therapies. These can be divided into several broad groups: curative therapies (splenectomy and rituximab), thrombopoietin receptor agonists, and anecdotal therapies.
Splenectomy
In patients with severe thrombocytopenia who do not respond or who relapse with lower doses of prednisone, splenectomy should be strongly considered. Splenectomy will induce a good response in 60% to 70% of patients and is durable in most patients. In 2 recently published reviews of splenectomy, the complete response rate was 67% and the total response rate was 88% to 90%%.8,31 Between 15% and 28% of patients relapsed over 5 years, with most recurrences occurring in the first 2 years. Splenectomy carries a short-term surgical risk, and the life-long risk of increased susceptibility to overwhelming sepsis is discussed below. However, the absolute magnitude of these risks is low and is often lower than the risks of continued prednisone therapy or of continued cytotoxic therapy.
Timing of splenectomy depends on the patient’s presentation. Most patients should be given a 6-month trial of steroids or other therapies before proceeding to splenectomy.31 However, patients who persist with severe thrombocytopenia despite initial therapies or who are suffering intolerable side effects from therapy should be considered sooner for splenectomy.31 In the George review, multiple factors such as responding to IVIG were found not to be predictive of response to splenectomy.8
Method of splenectomy appears not to matter.21 Rates of finding accessory spleens are just as high or higher with laparoscopic splenectomy and the patient can recover faster. In patients who are severely thrombocytopenic, open splenectomy can allow for quicker control of the vascular access of the spleen.
Rates of splenectomy in recent years have decreased for many reasons,32 including the acceptance of lower platelet counts in asymptomatic patients and the availability of alternative therapies such as rituximab. In addition, despite abundant data for good outcomes, there is a concern that splenectomy responses are not durable. Although splenectomy will not cure every patient with ITP, splenectomy is the therapy with the most patients, the longest follow-up, and the most consistent rate of cure, and it should be discussed with every ITP patient who does not respond to initial therapy and needs further treatment.
The risk of overwhelming sepsis varies by indications for splenectomy but appears to be about 1%.33,34 The use of pneumococcal vaccine and recognition of this syndrome have helped reduce the risk. Asplenic patients need to be counseled about the risk of overwhelming infections, should be vaccinated for pneumococcus, meningococcus, and Haemophilus influenzae, and should wear an ID bracelet.35–37 Patients previously vaccinated for pneumococcus should be re-vaccinated every 3 to 5 years. The role of prophylactic antibiotics in adults is controversial, but patients under the age of 18 should be on penicillin VK 250 mg orally twice daily.
Rituximab
Rituximab has been shown to be very active in ITP. Most studies used the standard dose of 375 mg/m2 weekly for 4 weeks, but other studies have shown that 1000 mg twice 14 days apart (ie, on days 1 and 15) resulted in the same response rate and may be more convenient for patients.38,39 The response time can vary, with patients either showing a rapid response or requiring up to 8 weeks for their counts to go up. Although experience is limited, the response seems to be durable, especially in those patients whose counts rise higher than 150 × 103/µL; in patients who relapse, a response can be re-induced with a repeat course. Overall the response rate for rituximab is about 60%, but only approximately 20% to 40% of patients will remain in long-term remission.40–42 There is no evidence yet that “maintenance” therapy or monitoring CD19/CD20 cells can help further the duration of remission.
Whether to give rituximab pre- or post-splenectomy is also uncertain. An advantage of presplenectomy rituximab is that many patients will achieve remission, delaying the need for surgery. Also, rituximab is a good option for patients whose medical conditions put them at high risk for complications with splenectomy. However, it is unknown whether rituximab poses any long-term risks, while the long-term risks of splenectomy are well-defined. Rituximab is the only curative option left for patients who have failed splenectomy and is a reasonable option for these patients.
There is an intriguing trial in which patients were randomly assigned to dexamethasone alone versus dexamethasone plus rituximab upon presentation with ITP; those who were refractory to dexamethasone alone received salvage therapy with dexamethasone plus rituximab.43 The dexamethasone plus rituximab group had an overall higher rate of sustained remission at 6 months than the dexamethasone group, 63% versus 36%. Interestingly, patients who failed their first course of dexamethasone but then were “salvaged” with dexamethasone/rituximab had a similar overall response rate of 56%, suggesting that saving the addition of rituximab for steroid failures may be an effective option.
Although not “chemotherapy,” rituximab is not without risks. Patients can develop infusion reactions, which can be severe in 1% to 2% of patients. In a meta-analysis the fatal reaction rate was 2.9%.40 Patients with chronic hepatitis B infections can experience reactivation with rituximab, and thus all patients should be screened before treatment. Finally, the very rare but devastating complication of progressive multifocal leukoencephalopathy has been reported.
Thrombopoietin Receptor Agonists
Although patients with ITP have low platelet counts, studies starting with Dameshek have shown that these patients also have reduced production of platelets.44 Despite the very low circulating platelet count, levels of the platelet growth factor thrombopoietin (TPO) are not raised.45 Seminal studies with recombinant TPO in the 1990s showed that ITP patients responded to thrombopoietin-stimulating protein, but the formation of anti-TPO antibodies halted trials with the first generation of these agents. Two TPO receptor agonists (TPO-RA) are approved for use in patients with ITP.
Romiplostim. Romiplostim is a peptibody, a combination of a peptide that binds and stimulates the TPO receptor and an Fc domain to extend its half-life.46 It is administered in a weekly subcutaneous dose starting at 1 to 3 µg/kg. Use of romiplostim in ITP patients produces a response rate of 80% to 88%, with 87% of patients being able to wean off or decrease other anti-ITP medications.47 In a long-term extension study, the response was again high at 87%.48 These studies have also shown a reduced incidence of bleeding.
The major side effect of romiplostim seen in clinical trials was marrow reticulin formation, which occurred in up to 5.6% of patients.47,48 The clinical course in these patients is the development of anemia and a myelophthisic blood smear with teardrop cells and nucleated red cells. These changes appear to reverse with cessation of the drug. The bone marrow shows increased reticulin formation but rarely, if ever, shows the collagen deposition seen with primary myelofibrosis.
Thrombosis has also been seen, with a rate of 0.08 to 0.1 cases per 100 patient-weeks,49 but it remains unclear if this is due to the drug, part of the natural history of ITP, or expected complications in older patients undergoing any type of medical therapy. Surprisingly, despite the low platelet counts, patients with ITP in one study had double the risk of venous thrombosis, demonstrating that ITP itself can be a risk factor for thrombosis.50 These trials have shown no long-term concerns for other clinical problems such as liver disease.
Eltrombopag. The other available TPO-RA is eltrombopag,51 an oral agent that stimulates the TPO receptor by binding the transmembrane domain and activating it. The drug is given orally starting at 50 mg/day (25 mg for patients of Asian ancestry or with liver disease) and can be dose escalated to 75 mg/day. The drug needs to be taken on an empty stomach. Eltrombopag has been shown to be effective in chronic ITP, with response rates of 59% to 80% and reduction in use of rescue medications.47,51,52 As with romiplostim, the incidence of bleeding was also decreased with eltrombopag in these trials.47,51
Clinical trials demonstrated that eltrombopag shares with romiplostim the risk for marrow fibrosis. A side effect unique to eltrombopag observed in these trials was a 3% to 7% incidence of elevated liver function tests.21,52 These abnormal findings appeared to resolve in most patients, but liver function tests need to be monitored in patients receiving eltrombopag.
Clinical use. The clearest indication for the use of TPO-RAs is in patients who have failed several therapies and remain symptomatic or are on intolerable doses of other medications such as prednisone. The clear benefits are their relative safety and high rates of success. The main drawback of TPO-RAs is the need for continuing therapy as the platelet count will return to baseline shortly after these agents are stopped. Currently there is no clear indication for one medication over the other. The advantages of romiplostim are great flexibility in dosing (1–10 µg/kg week) and no concerns about drug interaction. The current drawback of romiplostim is the Food and Drug Administration’s requirement for patients to receive the drug from a clinic and not at home. Eltrombopag offers the advantage of oral use, but it has a limited dose range and potential for drug interactions. Both agents have been associated with marrow reticulin formation, although in clinical use this risk appears to be very low.53
Other Options
In the literature there are numerous options for the treatment of ITP.54,55 Most of these studies are anecdotal, enrolled small number of patients, and sometimes included patients with mild thrombocytopenia, but these therapeutic options can be tried in patients who are refractory to standard therapies and have bleeding. The agents with the greatest amount of supporting data are danazol, vincristine, azathioprine, cyclophosphamide, and fostamatinib.
Danazol 200 mg 4 times daily is thought to downregulate the macrophage Fc receptor. The onset of action may be delayed and a therapeutic trial of up to 4 to 6 months is advised. Danazol is very effective in patients with antiphospholipid antibody syndrome who develop ITP and may be more effective in premenopausal women.56 Once a response is seen, danazol should be continued for 6 months and then an attempt to wean the patient off the agent should be made. A partial response can be seen in 70% to 90% of patients, but a complete response is rare.54
Vincristine 1.4 mg/m2 weekly has a low response rate, but if a response is going to occur, it will occur rapidly within 2 weeks. Thus, a prolonged trial of vincristine is not needed; if no platelet rise is seen in several weeks, the drug should be stopped. Again, partial responses are more common than complete response—50% to 63% versus 0% to 6%.54Azathioprine 150 mg orally daily, like danazol, demonstrates a delayed response and requires several months to assess for response. However, 19% to 25% of patients may have a complete response.54 It has been reported that the related agent mycophenolate 1000 mg twice daily is also effective in ITP.57
Cyclophosphamide 1 g/m2 intravenously repeated every 28 days has been reported to have a response rate of up to 40%.58 Although considered more aggressive, this is a standard immunosuppressive dose and should be considered in patients with very low platelet counts. Patients who have not responded to single-agent cyclophosphamide may respond to multi-agent chemotherapy with agents such as etoposide and vincristine plus cyclophosphamide.59
Fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, is currently under investigation for the treatment of ITP.60 This agent prevents phagocytosis of antibody-coated platelets by macrophages. In early studies fostamatinib has been well tolerated at a dose of 150 mg twice daily, with 75% of patients showing a response. Large phase 3 trials are underway, and if the earlier promising results hold up fostamatinib may be a novel option for refractory patients.
A Practical Approach to Refractory ITP
One approach is to divide patients into bleeders, or those with either very low platelet counts (< 5 × 103/µL) or who have had significant bleeding in the past, and nonbleeders, or those with platelet counts above 5 × 103/µL and no history of severe bleeding. Bleeders who do not respond adequately to splenectomy should first start with rituximab since it is not cytotoxic and is the only other “curative” therapy (Table 2).
Nonbleeders should be tried on danazol and other relatively safe agents. If this fails, rituximab or TPO-RAs can be considered. Before one considers cytotoxic therapy, the risk of the therapy must be weighed against the risk posed by the thrombocytopenia. The mortality from ITP is fairly low (5%) and is restricted to patients with severe disease. Patients with only moderate thrombocytopenia and no bleeding are better served with conservative management. There is little justification for the use of continuous steroid therapy in this group of patients given the long-term risks of this therapy.
Special Situations
Surgery
Patients with ITP who need surgery either for splenectomy or for other reasons should have their platelet counts raised to a level greater than 20 to 30 × 103/µL before surgery. Most patients with ITP have increased platelet function and will not have excessive bleeding with these platelet counts. For patients with platelet counts below this level, an infusion of immune globulin or anti-D may rapidly increase the platelet counts. If the surgery is elective, short-term use of TPO-RAs to raise the counts can also be considered.
Pregnancy
Up to 10% of pregnant women will develop low platelet counts during their pregnancy.61,62 The most common etiology is gestational thrombocytopenia, which is an exaggeration of the lowered platelet count seen in pregnancy. Counts may fall as low as 50 × 103/µL at the time of delivery. No therapy is required as the fetus is not affected and the mother does not have an increased risk of bleeding. Pregnancy complications such as HELLP syndrome and thrombotic microangiopathies also present with low platelet counts, but these can be diagnosed by history.61,63
Women with ITP can either develop the disease during pregnancy or have a worsening of the symptoms.64 Counts often drop dramatically during the first trimester. Early management should be conservative with low doses of prednisone to keep the count above 10 × 103/µL.21 Immunoglobulin is also effective,65 but there are rare reports of pulmonary edema. Rarely patients who are refractory will require splenectomy, which may be safely performed in the second trimester. For delivery the count should be greater than 30 × 103/µL and for an epidural greater than 50 × 103/µL.64 There are reports of the use of TPO-RAs in pregnancy, and this can be considered for refractory cases.66
Most controversy centers on management of the delivery. In the past it was feared that fetal thrombocytopenia could lead to intracranial hemorrhage, and Caesarean section was always recommended. It now appears that most cases of intracranial hemorrhage were due to alloimmune thrombocytopenia and not ITP. Furthermore, the nadir of the baby’s platelet count is not at birth but several days after. It appears the safest course is to proceed with a vaginal or C-section delivery determined by obstetrical indications and then immediately check the baby’s platelet count. If the platelet count is low in the neonate, immunoglobulin will raise the count. Since the neonatal thrombocytopenia is due to passive transfer of maternal antibody, the platelet destruction will abate in 4 to 6 weeks.
Pediatric Patients
The incidence of ITP in children is 2.2 to 5.3 per 100,000 children.1 There are several distinct differences in pediatric ITP. Most cases will resolve in weeks, with only a minority of patients transforming into chronic ITP (5%–10%). Also, the rates of serious bleeding are lower in children than in adults, with intracranial hemorrhage rates of 0.1% to 0.5% being seen.67 For most patients with no or mild bleeding, management now is observation alone regardless of platelet count because it is felt that the risks of therapies are higher than the risk of bleeding.21 For patients with bleeding, IVIG, anti-D, or a short course of steroids can be used. Given the risk of overwhelming sepsis, splenectomy is often deferred as long as possible. Rituximab is increasingly being used in children due to concerns about use of agents such a cyclophosphamide or azathioprine in children.68 Abundant data on use of TPO-RAs in children showing high response rates and safety support their use, and these should be considered in refractory ITP before any cytotoxic agent.69–71
Helicobacter Pylori Infection
There has been much interest in the relationship between H. pylori and ITP.16,72,73H. pylori infections have been associated with a variety of autoimmune diseases, and there is a confusing literature on this infection and ITP. Several meta-analyses have shown that eradication of H. pylori will result in an ITP response rate of 20% to 30%, but responses curiously appear to be limited to certain geographic areas such as Japan and Italy but not the United States. In patients with recalcitrant ITP, especially in geographic areas with high incidence, it may be worthwhile to check for H. pylori infection and treat accordingly if positive.
Drug-Induced Thrombocytopenia
Patients with drug-induced thrombocytopenia present with very low (< 10 × 103/µL) platelet counts 1 to 3 weeks after starting a new medication.74–76 In patients with a possible drug-induced thrombocytopenia, the primary therapy is to stop the suspect drug.77 If there are multiple new medications, the best approach is to stop any drug that has been strongly associated with thrombocytopenia (Table 3).74,78,79
Immune globulin, corticosteroids, or intravenous anti‑D have been suggested as useful in drug‑related thrombocytopenia. However, since most of these thrombocytopenic patients recover when the agent is cleared from the body, this therapy is probably not necessary and withholding treatment avoids exposing the patients to the adverse events associated with further therapy.
Evans Syndrome
Evans syndrome is defined as the combination of autoimmune hemolytic anemia (AIHA) and ITP.80,81 These cytopenias can present simultaneously or sequentially. Patients with Evans syndrome are thought to have a more severe disease process, to be more prone to bleeding, and to be more difficult to treat, but the rarity of this syndrome makes this hard to quantify.
The classic clinical presentation of Evans syndrome is severe anemia and thrombocytopenia. Children with Evans syndrome often have complex immunodeficiencies such as autoimmune lymphoproliferative syndrome.82,83 In adults, Evans syndrome most often complicates other autoimmune diseases such as lupus. There are increasing reports of Evans syndrome occurring as a complication of T-cell lymphomas. Often the autoimmune disease can predate the lymphoma diagnosis by months or even years.
In theory the diagnostic approach is straightforward by showing a Coombs-positive hemolytic anemia in the setting of a clinical diagnosis of immune thrombocytopenia. The blood smear will show spherocytes and a diminished platelet count. The presence of other abnormal red cell forms should raise the possibility of an alternative diagnosis. It is unclear how vigorously one should search for other underlying diseases. Many patients will already have the diagnosis of an underlying autoimmune disease. The presence of lymphadenopathy should raise concern for lymphoma.
Initial therapy is high-dose steroids (2 mg/kg/day). IVIG should be added if severe thrombocytopenia is present. Patients who cannot be weaned off prednisone or relapse after prednisone should be considered for splenectomy, although these patients are at higher risk of relapsing.80 Increasingly rituximab is being used with success.84,85 For patients who fail splenectomy and rituximab, aggressive immunosuppression should be considered. Increasing data support the benefits of sirolimus, and this should be considered for refractory patients.86 For patients with Evans syndrome due to underlying lymphoma, antineoplastic therapy often results in prompt resolution of the symptoms. Recurrence of the autoimmune cytopenias often heralds relapse.
Introduction
Immune thrombocytopenia (ITP) is a common acquired autoimmune disease characterized by low platelet counts and an increased risk of bleeding. The incidence of ITP is approximately 3.3 per 100,000 adults.1 There is considerable controversy about all aspects of the disease, with little “hard” data on which to base decisions given the lack of randomized clinical trials to address most clinical questions. This article reviews the presentation and diagnosis of ITP and its treatment options and discusses management of ITP in specific clinical situations.
Pathogenesis and Epidemiology
ITP is caused by autoantibodies binding to platelet surface proteins, most often to the platelet receptor GP IIb/IIIa.2-4 These antibody-coated platelets then bind to Fc receptors in macrophages and are removed from circulation. The initiating event in ITP is unknown. It is speculated that the patient responds to a viral or bacterial infection by creating antibodies which cross-react with the platelet receptors. Continued exposure to platelets perpetuates the immune response. ITP that occurs in childhood appears to be an acute response to viral infection and usually resolves. ITP in adults may occur in any age group but is seen especially in young women.
Despite the increased platelet destruction that occurs in ITP, the production of new platelets often is not significantly increased. This is most likely due to lack of an increase in thrombopoietin, the predominant platelet growth factor.5
It had been thought that most adult patients who present with ITP go on to have a chronic course, but more recent studies have shown this is not the case. In modern series the percentage of patients who are “cured” with steroids ranges from 30% to 70%.6–9 In addition, it has been appreciated that even in patients with modest thrombocytopenia, no therapy is required if the platelet count remains higher than 30 × 103/µL. However, this leaves a considerable number of patients who will require chronic therapy.
Clinical Presentation
Presentation can range from a symptomatic patient with low platelets found on a routine blood count to a patient with massive bleeding. Typically, patients first present with petechiae (small bruises 1 mm in size) on the shins. True petechiae are seen only in severe thrombocytopenia. Patients will also report frequent bruising and bleeding from the gums. Patients with very low platelet counts will notice “wet purpura,” which is characterized by blood-filled bullae in the oral cavity. Life-threatening bleeding is a very unusual presenting sign unless other problems (trauma, ulcers) are present. The physical examination is only remarkable for stigmata of bleeding such as the petechiae. The presence of splenomegaly or lymphadenopathy weighs strongly against a diagnosis of ITP. Many patients with ITP will note fatigue when their platelets counts are lower.10
Diagnosis
Extremely low platelet counts with a normal blood smear and an otherwise healthy patient are diagnostic of ITP. The platelet count cutoff for considering ITP is 100 × 103/µL as the majority of patients with counts in the 100 to 150 × 103/µL range will not develop greater thrombocytopenia.11 Also, the platelet count decreases with age (9 × 103/µL per decade in one study), and this also needs to be factored into the evaluation.12 The finding of relatives with ITP should raise suspicion for congenital thrombocytopenia.13 One should question the patient carefully about drug exposure (see Drug-Induced Thrombocytopenia), especially about over-the-counter medicines, “natural” remedies, or recreational drugs.
There is no laboratory test that rules in ITP; rather, it is a diagnosis of exclusion. The blood smear should be carefully examined for evidence of microangiopathic hemolytic anemias (schistocytes), bone marrow disease (blasts, teardrop cells), or any other evidence of a primary bone marrow disease. In ITP, the platelets can be larger than normal, but finding some platelets the size of red cells should raise the issue of congenital thrombocytopenia.14 Pseudo-thrombocytopenia, which is the clumping of platelets due to a reaction to the EDTA anticoagulant in the tube, should be excluded. The diagnosis is established by drawing the blood in a citrated (blue-top) tube to perform the platelet count. There is no role for antiplatelet antibody assay because this test lacks sensitivity and specificity. In a patient without a history of autoimmune disease or symptoms, empiric testing for autoimmune disease is not recommended.
Patients who present with ITP should be tested for both HIV and hepatitis C infection.15,16 These are the most common viral causes of secondary ITP, and both have prognostic and treatment implications. Some authorities also recommend checking thyroid function as hypothyroidism can present or aggravate the thrombocytopenia.
The role of bone marrow examination is controversial.17 Patients with a classic presentation of ITP (young woman, normal blood smear) do not require a bone marrow exam before therapy is initiated, although patients who do not respond to initial therapy should have a bone marrow aspiration. The rare entity amegakaryocytic thrombocytopenia can present with a clinical picture similar to that of ITP, but amegakaryocytic thrombocytopenia will not respond to steroids. Bone marrow aspiration reveals the absence of megakaryocytes in this entity. It is rare, however, that another hematologic disease is diagnosed in patients with a classic clinical presentation of ITP.
In the future, measurement of thrombopoietin and reticulated platelets may provide clues to the diagnosis.4 Patients with ITP paradoxically have normal or only mildly elevated thrombopoietin levels. The finding of a significantly elevated thrombopoietin level should lead to questioning of the diagnosis. One can also measure “reticulated platelets,” which are analogous to red cell reticulocytes. Patients with ITP (or any platelet destructive disorders) will have high levels of reticulated platelets. These tests are not recommended for routine evaluation, but may be helpful in difficult cases.
Treatment
In general, therapy in ITP should be guided by the patient’s signs of bleeding and not by unquestioning adherence to measuring platelet levels,15 as patients tolerate thrombocytopenia well. It is unusual to have life-threatening bleeding with platelet counts greater than 5 × 103/µL in the absence of mechanical lesions. Despite the low platelet count in patients with ITP, the overall mortality is estimated to be only 0.3% to 1.3%.18 It is sobering that in one study the rate of death from infections was twice as high as that from bleeding.19 Rare patients will have antibodies that interfere with the function of the platelet, and these patients can have profound bleeding with only modestly lowered platelet counts.20 A suggested cut-off for treating newly diagnosed patients is 30 × 103/µL.21
Initial Therapy
The primary therapy of ITP is glucocorticoids, either prednisone or dexamethasone. In the past prednisone at a dose of 60 to 80 mg/day was started at the time of diagnosis (Table 1).
For rapid induction of a response, there are 2 options. A single dose of intravenous immune globulin (IVIG) at 1 g/kg or intravenous anti-D immunoglobulin (anti-D) at 50 to 75 µg/kg can induce a response in more than 80% of patients in 24 to 48 hours.21,24 IVIG has several drawbacks. It can cause aseptic meningitis, and in patients with vascular disease the increased viscosity can induce ischemia. There is also a considerable fluid load delivered with the IVIG, and it needs to be given over several hours.
The use of anti-D is limited to Rh-positive patients who have not had a splenectomy. It should not be used in patients who are Coombs positive due to the risk of provoking more hemolysis. Rarely anti-D has been reported to cause a severe hemolytic disseminated intravascular coagulation syndrome (1:20,000 patients), which has led to restrictions in its use.25 Although the drug can be rapidly given over 15 minutes, due to these concerns current recommendations are now to observe patients for 8 hours after their dose and to perform a urine dipstick test for blood at 2, 4, and 8 hours. Concerns about this rare but serious side effect have led to a dramatic decrease in the use of anti-D.
For patients who are severely thrombocytopenic and do not respond to initial therapy, there are 2 options for raising the platelet counts. One is to use a combination of IVIG, methylprednisolone, vincristine, and/or anti-D.26 The combination of IVIG and anti-D may be synergistic since these agents block different Fc receptors. A response of 71% has been reported for this 3- or 4-drug combination in a series of 35 patients.26 The other option is to treat with a continuous infusion of platelets (1 unit over 6 hours) and IVIG 1 g/kg for 24 hours. Response rates of 62.7% have been reported with this combination, and this rapid rise in platelets can allow time for other therapies to take effect.27,28
Patients with severe thrombocytopenia who relapse with reduction of steroids or who do not respond to steroids have several options for further management. Repeated doses of IVIG can transiently raise the platelet count, and some patients may only need several courses of therapy over the course of many months. One study showed that 60% of patients could delay or defer therapy by receiving multiple doses of anti-D. However, 30% of patients did eventually receive splenectomy and 20% of patients required ongoing therapy with anti-D.29 In a randomized trial comparing early use of anti-D to steroids to avoid splenectomy, there was no difference in splenectomy rate (38% versus 42%).30 Finally, an option as mentioned above is to try a 6-month course of pulse dexamethasone 40 mg/day for 4 days, repeated every 28 days.
Options for Refractory ITP
There are multiple options for patients who do not respond to initial ITP therapies. These can be divided into several broad groups: curative therapies (splenectomy and rituximab), thrombopoietin receptor agonists, and anecdotal therapies.
Splenectomy
In patients with severe thrombocytopenia who do not respond or who relapse with lower doses of prednisone, splenectomy should be strongly considered. Splenectomy will induce a good response in 60% to 70% of patients and is durable in most patients. In 2 recently published reviews of splenectomy, the complete response rate was 67% and the total response rate was 88% to 90%%.8,31 Between 15% and 28% of patients relapsed over 5 years, with most recurrences occurring in the first 2 years. Splenectomy carries a short-term surgical risk, and the life-long risk of increased susceptibility to overwhelming sepsis is discussed below. However, the absolute magnitude of these risks is low and is often lower than the risks of continued prednisone therapy or of continued cytotoxic therapy.
Timing of splenectomy depends on the patient’s presentation. Most patients should be given a 6-month trial of steroids or other therapies before proceeding to splenectomy.31 However, patients who persist with severe thrombocytopenia despite initial therapies or who are suffering intolerable side effects from therapy should be considered sooner for splenectomy.31 In the George review, multiple factors such as responding to IVIG were found not to be predictive of response to splenectomy.8
Method of splenectomy appears not to matter.21 Rates of finding accessory spleens are just as high or higher with laparoscopic splenectomy and the patient can recover faster. In patients who are severely thrombocytopenic, open splenectomy can allow for quicker control of the vascular access of the spleen.
Rates of splenectomy in recent years have decreased for many reasons,32 including the acceptance of lower platelet counts in asymptomatic patients and the availability of alternative therapies such as rituximab. In addition, despite abundant data for good outcomes, there is a concern that splenectomy responses are not durable. Although splenectomy will not cure every patient with ITP, splenectomy is the therapy with the most patients, the longest follow-up, and the most consistent rate of cure, and it should be discussed with every ITP patient who does not respond to initial therapy and needs further treatment.
The risk of overwhelming sepsis varies by indications for splenectomy but appears to be about 1%.33,34 The use of pneumococcal vaccine and recognition of this syndrome have helped reduce the risk. Asplenic patients need to be counseled about the risk of overwhelming infections, should be vaccinated for pneumococcus, meningococcus, and Haemophilus influenzae, and should wear an ID bracelet.35–37 Patients previously vaccinated for pneumococcus should be re-vaccinated every 3 to 5 years. The role of prophylactic antibiotics in adults is controversial, but patients under the age of 18 should be on penicillin VK 250 mg orally twice daily.
Rituximab
Rituximab has been shown to be very active in ITP. Most studies used the standard dose of 375 mg/m2 weekly for 4 weeks, but other studies have shown that 1000 mg twice 14 days apart (ie, on days 1 and 15) resulted in the same response rate and may be more convenient for patients.38,39 The response time can vary, with patients either showing a rapid response or requiring up to 8 weeks for their counts to go up. Although experience is limited, the response seems to be durable, especially in those patients whose counts rise higher than 150 × 103/µL; in patients who relapse, a response can be re-induced with a repeat course. Overall the response rate for rituximab is about 60%, but only approximately 20% to 40% of patients will remain in long-term remission.40–42 There is no evidence yet that “maintenance” therapy or monitoring CD19/CD20 cells can help further the duration of remission.
Whether to give rituximab pre- or post-splenectomy is also uncertain. An advantage of presplenectomy rituximab is that many patients will achieve remission, delaying the need for surgery. Also, rituximab is a good option for patients whose medical conditions put them at high risk for complications with splenectomy. However, it is unknown whether rituximab poses any long-term risks, while the long-term risks of splenectomy are well-defined. Rituximab is the only curative option left for patients who have failed splenectomy and is a reasonable option for these patients.
There is an intriguing trial in which patients were randomly assigned to dexamethasone alone versus dexamethasone plus rituximab upon presentation with ITP; those who were refractory to dexamethasone alone received salvage therapy with dexamethasone plus rituximab.43 The dexamethasone plus rituximab group had an overall higher rate of sustained remission at 6 months than the dexamethasone group, 63% versus 36%. Interestingly, patients who failed their first course of dexamethasone but then were “salvaged” with dexamethasone/rituximab had a similar overall response rate of 56%, suggesting that saving the addition of rituximab for steroid failures may be an effective option.
Although not “chemotherapy,” rituximab is not without risks. Patients can develop infusion reactions, which can be severe in 1% to 2% of patients. In a meta-analysis the fatal reaction rate was 2.9%.40 Patients with chronic hepatitis B infections can experience reactivation with rituximab, and thus all patients should be screened before treatment. Finally, the very rare but devastating complication of progressive multifocal leukoencephalopathy has been reported.
Thrombopoietin Receptor Agonists
Although patients with ITP have low platelet counts, studies starting with Dameshek have shown that these patients also have reduced production of platelets.44 Despite the very low circulating platelet count, levels of the platelet growth factor thrombopoietin (TPO) are not raised.45 Seminal studies with recombinant TPO in the 1990s showed that ITP patients responded to thrombopoietin-stimulating protein, but the formation of anti-TPO antibodies halted trials with the first generation of these agents. Two TPO receptor agonists (TPO-RA) are approved for use in patients with ITP.
Romiplostim. Romiplostim is a peptibody, a combination of a peptide that binds and stimulates the TPO receptor and an Fc domain to extend its half-life.46 It is administered in a weekly subcutaneous dose starting at 1 to 3 µg/kg. Use of romiplostim in ITP patients produces a response rate of 80% to 88%, with 87% of patients being able to wean off or decrease other anti-ITP medications.47 In a long-term extension study, the response was again high at 87%.48 These studies have also shown a reduced incidence of bleeding.
The major side effect of romiplostim seen in clinical trials was marrow reticulin formation, which occurred in up to 5.6% of patients.47,48 The clinical course in these patients is the development of anemia and a myelophthisic blood smear with teardrop cells and nucleated red cells. These changes appear to reverse with cessation of the drug. The bone marrow shows increased reticulin formation but rarely, if ever, shows the collagen deposition seen with primary myelofibrosis.
Thrombosis has also been seen, with a rate of 0.08 to 0.1 cases per 100 patient-weeks,49 but it remains unclear if this is due to the drug, part of the natural history of ITP, or expected complications in older patients undergoing any type of medical therapy. Surprisingly, despite the low platelet counts, patients with ITP in one study had double the risk of venous thrombosis, demonstrating that ITP itself can be a risk factor for thrombosis.50 These trials have shown no long-term concerns for other clinical problems such as liver disease.
Eltrombopag. The other available TPO-RA is eltrombopag,51 an oral agent that stimulates the TPO receptor by binding the transmembrane domain and activating it. The drug is given orally starting at 50 mg/day (25 mg for patients of Asian ancestry or with liver disease) and can be dose escalated to 75 mg/day. The drug needs to be taken on an empty stomach. Eltrombopag has been shown to be effective in chronic ITP, with response rates of 59% to 80% and reduction in use of rescue medications.47,51,52 As with romiplostim, the incidence of bleeding was also decreased with eltrombopag in these trials.47,51
Clinical trials demonstrated that eltrombopag shares with romiplostim the risk for marrow fibrosis. A side effect unique to eltrombopag observed in these trials was a 3% to 7% incidence of elevated liver function tests.21,52 These abnormal findings appeared to resolve in most patients, but liver function tests need to be monitored in patients receiving eltrombopag.
Clinical use. The clearest indication for the use of TPO-RAs is in patients who have failed several therapies and remain symptomatic or are on intolerable doses of other medications such as prednisone. The clear benefits are their relative safety and high rates of success. The main drawback of TPO-RAs is the need for continuing therapy as the platelet count will return to baseline shortly after these agents are stopped. Currently there is no clear indication for one medication over the other. The advantages of romiplostim are great flexibility in dosing (1–10 µg/kg week) and no concerns about drug interaction. The current drawback of romiplostim is the Food and Drug Administration’s requirement for patients to receive the drug from a clinic and not at home. Eltrombopag offers the advantage of oral use, but it has a limited dose range and potential for drug interactions. Both agents have been associated with marrow reticulin formation, although in clinical use this risk appears to be very low.53
Other Options
In the literature there are numerous options for the treatment of ITP.54,55 Most of these studies are anecdotal, enrolled small number of patients, and sometimes included patients with mild thrombocytopenia, but these therapeutic options can be tried in patients who are refractory to standard therapies and have bleeding. The agents with the greatest amount of supporting data are danazol, vincristine, azathioprine, cyclophosphamide, and fostamatinib.
Danazol 200 mg 4 times daily is thought to downregulate the macrophage Fc receptor. The onset of action may be delayed and a therapeutic trial of up to 4 to 6 months is advised. Danazol is very effective in patients with antiphospholipid antibody syndrome who develop ITP and may be more effective in premenopausal women.56 Once a response is seen, danazol should be continued for 6 months and then an attempt to wean the patient off the agent should be made. A partial response can be seen in 70% to 90% of patients, but a complete response is rare.54
Vincristine 1.4 mg/m2 weekly has a low response rate, but if a response is going to occur, it will occur rapidly within 2 weeks. Thus, a prolonged trial of vincristine is not needed; if no platelet rise is seen in several weeks, the drug should be stopped. Again, partial responses are more common than complete response—50% to 63% versus 0% to 6%.54Azathioprine 150 mg orally daily, like danazol, demonstrates a delayed response and requires several months to assess for response. However, 19% to 25% of patients may have a complete response.54 It has been reported that the related agent mycophenolate 1000 mg twice daily is also effective in ITP.57
Cyclophosphamide 1 g/m2 intravenously repeated every 28 days has been reported to have a response rate of up to 40%.58 Although considered more aggressive, this is a standard immunosuppressive dose and should be considered in patients with very low platelet counts. Patients who have not responded to single-agent cyclophosphamide may respond to multi-agent chemotherapy with agents such as etoposide and vincristine plus cyclophosphamide.59
Fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, is currently under investigation for the treatment of ITP.60 This agent prevents phagocytosis of antibody-coated platelets by macrophages. In early studies fostamatinib has been well tolerated at a dose of 150 mg twice daily, with 75% of patients showing a response. Large phase 3 trials are underway, and if the earlier promising results hold up fostamatinib may be a novel option for refractory patients.
A Practical Approach to Refractory ITP
One approach is to divide patients into bleeders, or those with either very low platelet counts (< 5 × 103/µL) or who have had significant bleeding in the past, and nonbleeders, or those with platelet counts above 5 × 103/µL and no history of severe bleeding. Bleeders who do not respond adequately to splenectomy should first start with rituximab since it is not cytotoxic and is the only other “curative” therapy (Table 2).
Nonbleeders should be tried on danazol and other relatively safe agents. If this fails, rituximab or TPO-RAs can be considered. Before one considers cytotoxic therapy, the risk of the therapy must be weighed against the risk posed by the thrombocytopenia. The mortality from ITP is fairly low (5%) and is restricted to patients with severe disease. Patients with only moderate thrombocytopenia and no bleeding are better served with conservative management. There is little justification for the use of continuous steroid therapy in this group of patients given the long-term risks of this therapy.
Special Situations
Surgery
Patients with ITP who need surgery either for splenectomy or for other reasons should have their platelet counts raised to a level greater than 20 to 30 × 103/µL before surgery. Most patients with ITP have increased platelet function and will not have excessive bleeding with these platelet counts. For patients with platelet counts below this level, an infusion of immune globulin or anti-D may rapidly increase the platelet counts. If the surgery is elective, short-term use of TPO-RAs to raise the counts can also be considered.
Pregnancy
Up to 10% of pregnant women will develop low platelet counts during their pregnancy.61,62 The most common etiology is gestational thrombocytopenia, which is an exaggeration of the lowered platelet count seen in pregnancy. Counts may fall as low as 50 × 103/µL at the time of delivery. No therapy is required as the fetus is not affected and the mother does not have an increased risk of bleeding. Pregnancy complications such as HELLP syndrome and thrombotic microangiopathies also present with low platelet counts, but these can be diagnosed by history.61,63
Women with ITP can either develop the disease during pregnancy or have a worsening of the symptoms.64 Counts often drop dramatically during the first trimester. Early management should be conservative with low doses of prednisone to keep the count above 10 × 103/µL.21 Immunoglobulin is also effective,65 but there are rare reports of pulmonary edema. Rarely patients who are refractory will require splenectomy, which may be safely performed in the second trimester. For delivery the count should be greater than 30 × 103/µL and for an epidural greater than 50 × 103/µL.64 There are reports of the use of TPO-RAs in pregnancy, and this can be considered for refractory cases.66
Most controversy centers on management of the delivery. In the past it was feared that fetal thrombocytopenia could lead to intracranial hemorrhage, and Caesarean section was always recommended. It now appears that most cases of intracranial hemorrhage were due to alloimmune thrombocytopenia and not ITP. Furthermore, the nadir of the baby’s platelet count is not at birth but several days after. It appears the safest course is to proceed with a vaginal or C-section delivery determined by obstetrical indications and then immediately check the baby’s platelet count. If the platelet count is low in the neonate, immunoglobulin will raise the count. Since the neonatal thrombocytopenia is due to passive transfer of maternal antibody, the platelet destruction will abate in 4 to 6 weeks.
Pediatric Patients
The incidence of ITP in children is 2.2 to 5.3 per 100,000 children.1 There are several distinct differences in pediatric ITP. Most cases will resolve in weeks, with only a minority of patients transforming into chronic ITP (5%–10%). Also, the rates of serious bleeding are lower in children than in adults, with intracranial hemorrhage rates of 0.1% to 0.5% being seen.67 For most patients with no or mild bleeding, management now is observation alone regardless of platelet count because it is felt that the risks of therapies are higher than the risk of bleeding.21 For patients with bleeding, IVIG, anti-D, or a short course of steroids can be used. Given the risk of overwhelming sepsis, splenectomy is often deferred as long as possible. Rituximab is increasingly being used in children due to concerns about use of agents such a cyclophosphamide or azathioprine in children.68 Abundant data on use of TPO-RAs in children showing high response rates and safety support their use, and these should be considered in refractory ITP before any cytotoxic agent.69–71
Helicobacter Pylori Infection
There has been much interest in the relationship between H. pylori and ITP.16,72,73H. pylori infections have been associated with a variety of autoimmune diseases, and there is a confusing literature on this infection and ITP. Several meta-analyses have shown that eradication of H. pylori will result in an ITP response rate of 20% to 30%, but responses curiously appear to be limited to certain geographic areas such as Japan and Italy but not the United States. In patients with recalcitrant ITP, especially in geographic areas with high incidence, it may be worthwhile to check for H. pylori infection and treat accordingly if positive.
Drug-Induced Thrombocytopenia
Patients with drug-induced thrombocytopenia present with very low (< 10 × 103/µL) platelet counts 1 to 3 weeks after starting a new medication.74–76 In patients with a possible drug-induced thrombocytopenia, the primary therapy is to stop the suspect drug.77 If there are multiple new medications, the best approach is to stop any drug that has been strongly associated with thrombocytopenia (Table 3).74,78,79
Immune globulin, corticosteroids, or intravenous anti‑D have been suggested as useful in drug‑related thrombocytopenia. However, since most of these thrombocytopenic patients recover when the agent is cleared from the body, this therapy is probably not necessary and withholding treatment avoids exposing the patients to the adverse events associated with further therapy.
Evans Syndrome
Evans syndrome is defined as the combination of autoimmune hemolytic anemia (AIHA) and ITP.80,81 These cytopenias can present simultaneously or sequentially. Patients with Evans syndrome are thought to have a more severe disease process, to be more prone to bleeding, and to be more difficult to treat, but the rarity of this syndrome makes this hard to quantify.
The classic clinical presentation of Evans syndrome is severe anemia and thrombocytopenia. Children with Evans syndrome often have complex immunodeficiencies such as autoimmune lymphoproliferative syndrome.82,83 In adults, Evans syndrome most often complicates other autoimmune diseases such as lupus. There are increasing reports of Evans syndrome occurring as a complication of T-cell lymphomas. Often the autoimmune disease can predate the lymphoma diagnosis by months or even years.
In theory the diagnostic approach is straightforward by showing a Coombs-positive hemolytic anemia in the setting of a clinical diagnosis of immune thrombocytopenia. The blood smear will show spherocytes and a diminished platelet count. The presence of other abnormal red cell forms should raise the possibility of an alternative diagnosis. It is unclear how vigorously one should search for other underlying diseases. Many patients will already have the diagnosis of an underlying autoimmune disease. The presence of lymphadenopathy should raise concern for lymphoma.
Initial therapy is high-dose steroids (2 mg/kg/day). IVIG should be added if severe thrombocytopenia is present. Patients who cannot be weaned off prednisone or relapse after prednisone should be considered for splenectomy, although these patients are at higher risk of relapsing.80 Increasingly rituximab is being used with success.84,85 For patients who fail splenectomy and rituximab, aggressive immunosuppression should be considered. Increasing data support the benefits of sirolimus, and this should be considered for refractory patients.86 For patients with Evans syndrome due to underlying lymphoma, antineoplastic therapy often results in prompt resolution of the symptoms. Recurrence of the autoimmune cytopenias often heralds relapse.
1. Terrell DR, Beebe LA, Vesely SK, et al. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol 2010;85:174–80.
2. McMillan R, Lopez-Dee J, Bowditch R. Clonal restriction of platelet-associated anti-GPIIb/IIIa autoantibodies in patients with chronic ITP. Thromb Haemost 2001;85:821–3.
3. Aster RH, George JN, McMillan R, Ganguly P. Workshop on autoimmune (idiopathic) thrombocytopenic purpura: Pathogenesis and new approaches to therapy. Am J Hematol 1998;58:231–4.
4. Toltl LJ, Arnold DM. Pathophysiology and management of chronic immune thrombocytopenia: focusing on what matters. Br J Haematol 2011;152:52–60.
5. Kuter DJ, Gernsheimer TB. Thrombopoietin and platelet production in chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1193–211.
6. Pamuk GE, Pamuk ON, Baslar Z, et al. Overview of 321 patients with idiopathic thrombocytopenic purpura. Retrospective analysis of the clinical features and response to therapy. Ann Hematol 2002;81:436–40.
7. Stasi R, Stipa E, Masi M, et al. Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 1995;98:436–42.
8. Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004;104:2623–34.
9. Matschke J, Muller-Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment-naive adult patients with immune thrombocytopenia: EIS 2002 study. Acta Haematol 2016;136:101–7.
10. Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011;86:420–9.
11. Stasi R, Amadori S, Osborn J, et al. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006;3:e24.
12. Biino G, Balduini CL, Casula L, et al. Analysis of 12,517 inhabitants of a Sardinian geographic isolate reveals that predispositions to thrombocytopenia and thrombocytosis are inherited traits. Haematologica 2011;96:96–101.
13. Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 2004;103:390–8.
14. Geddis AE, Balduini CL. Diagnosis of immune thrombocytopenic purpura in children. Curr Opin Hematol 2007;14:520–5.
15. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010;115:168–86.
16. Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1275–97.
17. Jubelirer SJ, Harpold R. The role of the bone marrow examination in the diagnosis of immune thrombocytopenic purpura: case series and literature review. Clin Appl Thromb Hemost 2002;8:73–6.
18. George JN. Management of patients with refractory immune thrombocytopenic purpura. J Thromb Haemost 2006;4:1664–72.
19. Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001;97:2549–54.
20. McMillan R, Bowditch RD, Tani P, et al. A non-thrombocytopenic bleeding disorder due to an IgG4- kappa anti-GPIIb/IIIa autoantibody. Br J Haematol 1996;95:747–9.
21. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011;117:4190–207.22. Mazzucconi MG, Fazi P, Bernasconi S, et al. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007;109:1401–7.
23. Wei Y, Ji XB, Wang YW, et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. Blood 2016;127:296–302.
24. Newman GC, Novoa MV, Fodero EM, et al. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001;112:1076–8.
25. Gaines AR. Acute onset hemoglobinemia and/or hemoglobinuria and sequelae following Rho(D) immune globulin intravenous administration in immune thrombocytopenic purpura patients. Blood 2000;95:2523–9.
26. Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007;110:3526–31.
27. Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008;83:122–5.
28. Olson SR, Chu C, Shatzel JJ, Deloughery TG. The “platelet boilermaker”: A treatment protocol to rapidly increase platelets in patients with immune-mediated thrombocytopenia. Am J Hematol 2016;91:E330–1.
29. Cooper N, Woloski BM, Fodero EM, et al. Does treatment with intermittent infusions of intravenous anti-D allow a proportion of adults with recently diagnosed immune thrombocytopenic purpura to avoid splenectomy? Blood 2002;99:1922–7.
30. George JN, Raskob GE, Vesely SK, et al. Initial management of immune thrombocytopenic purpura in adults: a randomized controlled trial comparing intermittent anti-D with routine care. Am J Hematol 2003;74:161–9.
31. Mikhael J, Northridge K, Lindquist K, et al. Short-term and long-term failure of laparoscopic splenectomy in adult immune thrombocytopenic purpura patients: a systematic review. Am J Hematol 2009;84:743–8.
32. Palandri F, Polverelli N, Sollazzo D, et al. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am J Hematol 2016;91:E267–72.
33. Landgren O, Bjorkholm M, Konradsen HB, et al. A prospective study on antibody response to repeated vaccinations with pneumococcal capsular polysaccharide in splenectomized individuals with special reference to Hodgkin’s lymphoma. J Intern Med 2004;255:664–73.
34. Bisharat N, Omari H, Lavi I, Raz R. Risk of infection and death among post-splenectomy patients. J Infect 2001;43:182–6.
35. Mileno MD, Bia FJ. The compromised traveler. Infect Dis Clin North Am 1998;12:369–412.
36. Guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Working Party of the British Committee for Standards in Haematology Clinical Haematology Task Force. BMJ 1996;312:430–4.
37. Ericsson CD. Travellers with pre-existing medical conditions. Int J Antimicrob Agents 2003;21:181–8.
38. Tran H, Brighton T, Grigg A, et al. A multi-centre, single-arm, open-label study evaluating the safety and efficacy of fixed dose rituximab in patients with refractory, relapsed or chronic idiopathic thrombocytopenic purpura (R-ITP1000 study). Br J Haematol 2014;167:243–51.
39. Mahevas M, Ebbo M, Audia S, et al. Efficacy and safety of rituximab given at 1,000 mg on days 1 and 15 compared to the standard regimen to treat adult immune thrombocytopenia. Am J Hematol 2013;88:858–61.
40. Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007;146:25–33.
41. Khellaf M, Charles-Nelson A, Fain O, et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: results from a prospective registry including 248 patients. Blood 2014;124:3228–36.
42. Ghanima W, Khelif A, Waage A, et al. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015;385:1653–61.
43. Zaja F, Baccarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010;115:2755–62.
44. Dameshek W, Miller EB. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood 1946;1:27–50.
45. Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment of thrombocytopenia. Annu Rev Med 2009;60:193–206.
46. Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006;355:1672–81.
47. Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:641–8.
48. Bussel JB, Kuter DJ, Pullarkat V, et al. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009;113:2161–71.
49. Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010;8:1372–82.
50. Severinsen MT, Engebjerg MC, Farkas DK, et al. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol 2011;152:360–2.
51. Bussel JB, Cheng G, Saleh MN, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med 2007;357:2237–47.
52. Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011;377:393–402.
53. Brynes RK, Orazi A, Theodore D, et al. Evaluation of bone marrow reticulin in patients with chronic immune thrombocytopenia treated with eltrombopag: Data from the EXTEND study. Am J Hematol 2015;90:598–601.
54. George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000;37:290–8.
55. McMillan R. Therapy for adults with refractory chronic immune thrombocytopenic purpura. Ann Intern Med 1997;126:307–14.
56. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Successful therapy with danazol in refractory autoimmune thrombocytopenia associated with rheumatic diseases. Br J Rheumatol 1997;36:1095–9.
57. Provan D, Moss AJ, Newland AC, Bussel JB. Efficacy of mycophenolate mofetil as single-agent therapy for refractory immune thrombocytopenic purpura. Am J Hematol 2006;81:19–25.
58. Reiner A, Gernsheimer T, Slichter SJ. Pulse cyclophosphamide therapy for refractory autoimmune thrombocytopenic purpura. Blood 1995;85:351–8.
59. Figueroa M, Gehlsen J, Hammond D, et al. Combination chemotherapy in refractory immune thrombocytopenic purpura. N Engl J Med 1993;328:1226–9.
60. Newland A, Lee EJ, McDonald V, Bussel JB. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy 2 Oct 2017.
61. McCrae KR. Thrombocytopenia in pregnancy. Hematology Am Soc Hematol Educ Program 2010;2010:397–402.
62. Gernsheimer T, McCrae KR. Immune thrombocytopenic purpura in pregnancy. Curr Opin Hematol 2007;14:574–80.
63. DeLoughery TG. Critical care clotting catastrophies. Crit Care Clin 2005;21:531–62.
64. Stavrou E, McCrae KR. Immune thrombocytopenia in pregnancy. Hematol Oncol Clin North Am 2009;23:1299–316.
65. Sun D, Shehata N, Ye XY, et al. Corticosteroids compared with intravenous immunoglobulin for the treatment of immune thrombocytopenia in pregnancy. Blood 2016;128:1329–35.
66. Kong Z, Qin P, Xiao S, et al. A novel recombinant human thrombopoietin therapy for the management of immune thrombocytopenia in pregnancy. Blood 2017;130:1097–103.
67. Psaila B, Petrovic A, Page LK, et al. Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood 2009;114:4777–83.
68. Journeycake JM. Childhood immune thrombocytopenia: role of rituximab, recombinant thrombopoietin, and other new therapeutics. Hematology Am Soc Hematol Educ Program 2012;2012:444–9.
69. Zhang J, Liang Y, Ai Y, et al. Thrombopoietin-receptor agonists for children with immune thrombocytopenia: a systematic review. Expert Opin Pharmacother 2017;18:1543–51.
70. Tarantino MD, Bussel JB, Blanchette VS, et al. Romiplostim in children with immune thrombocytopenia: a phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016;388:45–54.71. Grainger JD, Locatelli F, Chotsampancharoen T, et al. Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet 2015;386:1649–58.
72. Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009;113:1231–40.
73. Arnold DM, Bernotas A, Nazi I, et al. Platelet count response to H. pylori treatment in patients with immune thrombocytopenic purpura with and without H. pylori infection: a systematic review. Haematologica 2009;94:850–6.
74. Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med 2007;357:580–7.
75. Reese JA, Li X, Hauben M, et al. Identifying drugs that cause acute thrombocytopenia: an analysis using 3 distinct methods. Blood 2010;116:2127–33.
76. Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis and management. J Thromb Haemost 2009;7:911–8.
77. Zondor SD, George JN, Medina PJ. Treatment of drug-induced thrombocytopenia. Expert Opin Drug Saf 2002;1:173–80.
78. George JN, Raskob GE, Shah SR, et al. Drug-induced thrombocytopenia: A systematic review of published case reports. Ann Intern Med 1998;129:886–90.
79. Green D, Hougie C, Kazmier FJ, et al. Report of the working party on acquired inhibitors of coagulation: studies of the “lupus” anticoagulant. Thromb Haemost 1983;49:144–6.
80. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood 2009;114:3167–72.
81. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology 2008;13:356–60.
82. Notarangelo LD. Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematology Am Soc Hematol Educ Program 2009:139–43.
83. Martinez-Valdez L, Deya-Martinez A, Giner MT, et al. Evans syndrome as first manifestation of primary immunodeficiency in clinical practice. J Pediatr Hematol Oncol 2017;39:490–4.
84. Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003;78:1340–6.
85. Mantadakis E, Danilatou V, Stiakaki E, Kalmanti M. Rituximab for refractory Evans syndrome and other immune-mediated hematologic diseases. Am J Hematol 2004;77:303–10.
86. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol 2017;39:420–4.
1. Terrell DR, Beebe LA, Vesely SK, et al. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol 2010;85:174–80.
2. McMillan R, Lopez-Dee J, Bowditch R. Clonal restriction of platelet-associated anti-GPIIb/IIIa autoantibodies in patients with chronic ITP. Thromb Haemost 2001;85:821–3.
3. Aster RH, George JN, McMillan R, Ganguly P. Workshop on autoimmune (idiopathic) thrombocytopenic purpura: Pathogenesis and new approaches to therapy. Am J Hematol 1998;58:231–4.
4. Toltl LJ, Arnold DM. Pathophysiology and management of chronic immune thrombocytopenia: focusing on what matters. Br J Haematol 2011;152:52–60.
5. Kuter DJ, Gernsheimer TB. Thrombopoietin and platelet production in chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1193–211.
6. Pamuk GE, Pamuk ON, Baslar Z, et al. Overview of 321 patients with idiopathic thrombocytopenic purpura. Retrospective analysis of the clinical features and response to therapy. Ann Hematol 2002;81:436–40.
7. Stasi R, Stipa E, Masi M, et al. Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 1995;98:436–42.
8. Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004;104:2623–34.
9. Matschke J, Muller-Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment-naive adult patients with immune thrombocytopenia: EIS 2002 study. Acta Haematol 2016;136:101–7.
10. Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011;86:420–9.
11. Stasi R, Amadori S, Osborn J, et al. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006;3:e24.
12. Biino G, Balduini CL, Casula L, et al. Analysis of 12,517 inhabitants of a Sardinian geographic isolate reveals that predispositions to thrombocytopenia and thrombocytosis are inherited traits. Haematologica 2011;96:96–101.
13. Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 2004;103:390–8.
14. Geddis AE, Balduini CL. Diagnosis of immune thrombocytopenic purpura in children. Curr Opin Hematol 2007;14:520–5.
15. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010;115:168–86.
16. Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1275–97.
17. Jubelirer SJ, Harpold R. The role of the bone marrow examination in the diagnosis of immune thrombocytopenic purpura: case series and literature review. Clin Appl Thromb Hemost 2002;8:73–6.
18. George JN. Management of patients with refractory immune thrombocytopenic purpura. J Thromb Haemost 2006;4:1664–72.
19. Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001;97:2549–54.
20. McMillan R, Bowditch RD, Tani P, et al. A non-thrombocytopenic bleeding disorder due to an IgG4- kappa anti-GPIIb/IIIa autoantibody. Br J Haematol 1996;95:747–9.
21. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011;117:4190–207.22. Mazzucconi MG, Fazi P, Bernasconi S, et al. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007;109:1401–7.
23. Wei Y, Ji XB, Wang YW, et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. Blood 2016;127:296–302.
24. Newman GC, Novoa MV, Fodero EM, et al. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001;112:1076–8.
25. Gaines AR. Acute onset hemoglobinemia and/or hemoglobinuria and sequelae following Rho(D) immune globulin intravenous administration in immune thrombocytopenic purpura patients. Blood 2000;95:2523–9.
26. Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007;110:3526–31.
27. Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008;83:122–5.
28. Olson SR, Chu C, Shatzel JJ, Deloughery TG. The “platelet boilermaker”: A treatment protocol to rapidly increase platelets in patients with immune-mediated thrombocytopenia. Am J Hematol 2016;91:E330–1.
29. Cooper N, Woloski BM, Fodero EM, et al. Does treatment with intermittent infusions of intravenous anti-D allow a proportion of adults with recently diagnosed immune thrombocytopenic purpura to avoid splenectomy? Blood 2002;99:1922–7.
30. George JN, Raskob GE, Vesely SK, et al. Initial management of immune thrombocytopenic purpura in adults: a randomized controlled trial comparing intermittent anti-D with routine care. Am J Hematol 2003;74:161–9.
31. Mikhael J, Northridge K, Lindquist K, et al. Short-term and long-term failure of laparoscopic splenectomy in adult immune thrombocytopenic purpura patients: a systematic review. Am J Hematol 2009;84:743–8.
32. Palandri F, Polverelli N, Sollazzo D, et al. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am J Hematol 2016;91:E267–72.
33. Landgren O, Bjorkholm M, Konradsen HB, et al. A prospective study on antibody response to repeated vaccinations with pneumococcal capsular polysaccharide in splenectomized individuals with special reference to Hodgkin’s lymphoma. J Intern Med 2004;255:664–73.
34. Bisharat N, Omari H, Lavi I, Raz R. Risk of infection and death among post-splenectomy patients. J Infect 2001;43:182–6.
35. Mileno MD, Bia FJ. The compromised traveler. Infect Dis Clin North Am 1998;12:369–412.
36. Guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Working Party of the British Committee for Standards in Haematology Clinical Haematology Task Force. BMJ 1996;312:430–4.
37. Ericsson CD. Travellers with pre-existing medical conditions. Int J Antimicrob Agents 2003;21:181–8.
38. Tran H, Brighton T, Grigg A, et al. A multi-centre, single-arm, open-label study evaluating the safety and efficacy of fixed dose rituximab in patients with refractory, relapsed or chronic idiopathic thrombocytopenic purpura (R-ITP1000 study). Br J Haematol 2014;167:243–51.
39. Mahevas M, Ebbo M, Audia S, et al. Efficacy and safety of rituximab given at 1,000 mg on days 1 and 15 compared to the standard regimen to treat adult immune thrombocytopenia. Am J Hematol 2013;88:858–61.
40. Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007;146:25–33.
41. Khellaf M, Charles-Nelson A, Fain O, et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: results from a prospective registry including 248 patients. Blood 2014;124:3228–36.
42. Ghanima W, Khelif A, Waage A, et al. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015;385:1653–61.
43. Zaja F, Baccarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010;115:2755–62.
44. Dameshek W, Miller EB. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood 1946;1:27–50.
45. Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment of thrombocytopenia. Annu Rev Med 2009;60:193–206.
46. Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006;355:1672–81.
47. Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:641–8.
48. Bussel JB, Kuter DJ, Pullarkat V, et al. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009;113:2161–71.
49. Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010;8:1372–82.
50. Severinsen MT, Engebjerg MC, Farkas DK, et al. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol 2011;152:360–2.
51. Bussel JB, Cheng G, Saleh MN, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med 2007;357:2237–47.
52. Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011;377:393–402.
53. Brynes RK, Orazi A, Theodore D, et al. Evaluation of bone marrow reticulin in patients with chronic immune thrombocytopenia treated with eltrombopag: Data from the EXTEND study. Am J Hematol 2015;90:598–601.
54. George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000;37:290–8.
55. McMillan R. Therapy for adults with refractory chronic immune thrombocytopenic purpura. Ann Intern Med 1997;126:307–14.
56. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Successful therapy with danazol in refractory autoimmune thrombocytopenia associated with rheumatic diseases. Br J Rheumatol 1997;36:1095–9.
57. Provan D, Moss AJ, Newland AC, Bussel JB. Efficacy of mycophenolate mofetil as single-agent therapy for refractory immune thrombocytopenic purpura. Am J Hematol 2006;81:19–25.
58. Reiner A, Gernsheimer T, Slichter SJ. Pulse cyclophosphamide therapy for refractory autoimmune thrombocytopenic purpura. Blood 1995;85:351–8.
59. Figueroa M, Gehlsen J, Hammond D, et al. Combination chemotherapy in refractory immune thrombocytopenic purpura. N Engl J Med 1993;328:1226–9.
60. Newland A, Lee EJ, McDonald V, Bussel JB. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy 2 Oct 2017.
61. McCrae KR. Thrombocytopenia in pregnancy. Hematology Am Soc Hematol Educ Program 2010;2010:397–402.
62. Gernsheimer T, McCrae KR. Immune thrombocytopenic purpura in pregnancy. Curr Opin Hematol 2007;14:574–80.
63. DeLoughery TG. Critical care clotting catastrophies. Crit Care Clin 2005;21:531–62.
64. Stavrou E, McCrae KR. Immune thrombocytopenia in pregnancy. Hematol Oncol Clin North Am 2009;23:1299–316.
65. Sun D, Shehata N, Ye XY, et al. Corticosteroids compared with intravenous immunoglobulin for the treatment of immune thrombocytopenia in pregnancy. Blood 2016;128:1329–35.
66. Kong Z, Qin P, Xiao S, et al. A novel recombinant human thrombopoietin therapy for the management of immune thrombocytopenia in pregnancy. Blood 2017;130:1097–103.
67. Psaila B, Petrovic A, Page LK, et al. Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood 2009;114:4777–83.
68. Journeycake JM. Childhood immune thrombocytopenia: role of rituximab, recombinant thrombopoietin, and other new therapeutics. Hematology Am Soc Hematol Educ Program 2012;2012:444–9.
69. Zhang J, Liang Y, Ai Y, et al. Thrombopoietin-receptor agonists for children with immune thrombocytopenia: a systematic review. Expert Opin Pharmacother 2017;18:1543–51.
70. Tarantino MD, Bussel JB, Blanchette VS, et al. Romiplostim in children with immune thrombocytopenia: a phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016;388:45–54.71. Grainger JD, Locatelli F, Chotsampancharoen T, et al. Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet 2015;386:1649–58.
72. Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009;113:1231–40.
73. Arnold DM, Bernotas A, Nazi I, et al. Platelet count response to H. pylori treatment in patients with immune thrombocytopenic purpura with and without H. pylori infection: a systematic review. Haematologica 2009;94:850–6.
74. Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med 2007;357:580–7.
75. Reese JA, Li X, Hauben M, et al. Identifying drugs that cause acute thrombocytopenia: an analysis using 3 distinct methods. Blood 2010;116:2127–33.
76. Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis and management. J Thromb Haemost 2009;7:911–8.
77. Zondor SD, George JN, Medina PJ. Treatment of drug-induced thrombocytopenia. Expert Opin Drug Saf 2002;1:173–80.
78. George JN, Raskob GE, Shah SR, et al. Drug-induced thrombocytopenia: A systematic review of published case reports. Ann Intern Med 1998;129:886–90.
79. Green D, Hougie C, Kazmier FJ, et al. Report of the working party on acquired inhibitors of coagulation: studies of the “lupus” anticoagulant. Thromb Haemost 1983;49:144–6.
80. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood 2009;114:3167–72.
81. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology 2008;13:356–60.
82. Notarangelo LD. Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematology Am Soc Hematol Educ Program 2009:139–43.
83. Martinez-Valdez L, Deya-Martinez A, Giner MT, et al. Evans syndrome as first manifestation of primary immunodeficiency in clinical practice. J Pediatr Hematol Oncol 2017;39:490–4.
84. Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003;78:1340–6.
85. Mantadakis E, Danilatou V, Stiakaki E, Kalmanti M. Rituximab for refractory Evans syndrome and other immune-mediated hematologic diseases. Am J Hematol 2004;77:303–10.
86. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol 2017;39:420–4.
Hairy Cell Leukemia
Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14 
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.
While BRAF V600E is identified in nearly all cases of cHCL, it is rare in vHCL.18 The variant type of HCL was classified as a distinct clinical entity in 2008 and can now often be distinguished from cHCL on the basis of BRAF mutational status, among other differences. Interestingly, in the rare cases of BRAF V600E–negative cHCL, other mutations in BRAF or downstream targets as well as aberrant activation of the RAF-MEK-ERK signaling cascade are observed, indicating that this pathway is critical in HCL and may still represent a viable therapeutic target. Expression of the IGHV4-34 immunoglobulin rearrangement, while more common in vHCL, has also been identified in 10% of cHCL cases and appears to confer poor prognosis.19 Other mutated genes that have been identified in HCL include CDKN1B, TP53, U2AF1, ARID1A, EZH2, and KDM6A.20
Classic HCL is characterized by the immunophenotypic expression of CD11c, CD25, CD103, and CD123, with kappa or lambda light chain restriction indicating clonality; HCL B cells are generally negative for CD5, CD10, CD23, CD27, and CD79b. In contrast, vHCL often lacks expression of CD25 and CD123.18 The B-cell receptor (BCR) is expressed on hairy cells and its activation promotes proliferation and survival in vitro.21 The role of BCR signaling in B-cell malignancies is increasingly recognized, and therapies that target the BCR and associated signaling molecules offer an attractive treatment strategy.22 HCL B cells also typically express CD19, CD20, CD22, CD79a, CD200, CD1d, and annexin A1. Tartrate-resistant acid phosphatase (TRAP) positivity by immunohistochemistry is a hallmark of cHCL. Interestingly, changes to the patient’s original immunophenotype have been observed following treatment and upon disease recurrence, highlighting the importance of tracking immunophenotype throughout the course of disease.
Diagnosis
Prior to the advent of annual screening evaluations with routine examination of complete blood counts (CBC), patients were most often diagnosed with HCL when they presented with symptoms of the disease such as splenomegaly, infections, or complications of anemia or thrombocytopenia.23 In the current era, patients are more likely to be incidentally diagnosed when they are found to have an abnormal value on a CBC. Any blood lineage may be affected and patients may have pancytopenia or isolated cytopenias. Of note, monocytopenia is a common finding in cHCL that is not entirely understood. The cells typical of cHCL do not usually circulate in the peripheral blood, but if present would appear as mature lymphocytes with villous cytoplasmic projections, pale blue cytoplasm, and reniform nuclei with open chromatin (Figure).9 Even if the morphologic examination is highly suggestive of HCL, additional testing is required to differentiate between cHCL, vHCL, and other hematologic malignancies which may also have cytoplasmic projections. A complete assessment of the immunophenotype, molecular profile, and cytogenetic features is required to arrive at this diagnosis.
The international Hairy Cell Leukemia Foundation recently published consensus guidelines for the diagnosis and treatment of HCL.24 These guidelines recommend that patients undergo examination of the peripheral blood for morphology and immunophenotyping and further recommend obtaining bone marrow core and aspirate biopsy samples for immunophenotyping via immunohistochemical staining and flow cytometry. The characteristic immunophenotype of cHCL is a population of monoclonal B lymphocytes which co-express CD19, CD20, CD11c, CD25, CD103, and CD123. Variant HCL is characterized by a very similar immunophenotype but is usually negative for CD25 and CD123. It is notable that CD25 positivity may be lost following treatment, and the absence of this marker should not be used as the sole basis of a cHCL versus vHCL diagnosis. Because marrow fibrosis in HCL may prevent a marrow aspirate from being obtained, many of the key diagnostic studies are performed on the core biopsy, including morphological evaluation and immunohistochemical stains such as CD20 (a pan-B cell antigen), annexin-1 (an anti-inflammatory protein expressed only in cHCL), and VE1 (a BRAF V600E stain).
As noted above, recurrent cytogenetic abnormalities have now been identified that may inform the diagnosis or prognosis of HCL. Next-generation sequencing and other testing of the genetic landscape are taking on a larger role in subtype differentiation, and it is likely that future guidelines will recommend evaluation for significant mutations. Given that BRAF V600E mutation status is a key feature of cHCL and is absent in vHCL, it is important to perform this testing at the time of diagnosis whenever possible. The mutation may be detected via VE1 immunohistochemical staining, allele-specific polymerase chain reaction, or next-generation sequencing. Other less sensitive tests exist but are utilized less frequently.
Minimal Residual Disease
There is currently no accepted standard for minimal residual disease (MRD) monitoring in HCL. While detection of MRD has been clearly associated with increased risk of disease progression, cHCL cells typically do not circulate in the peripheral blood, limiting the use of peripheral blood immunophenotyping for quantitative MRD assessment. For quantitative monitoring of marrow involvement by HCL, immunohistochemical staining of the bone marrow core biopsy is usually required. Staining may be performed for CD20, or, in patients who have received anti-CD20 therapy, DBA.44, VE-1, or CD79a. There is currently not a consensus regarding what level of disease involvement constitutes MRD. One group studied this issue and found that relapse could be predicted by evaluating MRD by percentage of positive cells in the marrow by immunohistochemical staining, with less than 1% involvement having the lowest risk for disease relapse and greater than 5% having the highest risk for disease relapse.25 A recent study evaluated MRD patterns in the peripheral blood of 32 cHCL patients who had completed frontline therapy. This group performed flow cytometry on the peripheral blood of patients at 1, 3, 6, and 12 months following therapy. All patients had achieved a complete response with initial therapy and peripheral blood MRD negativity at the completion of therapy. At a median follow-up of 100 months post therapy, 5 patients converted from peripheral blood–MRD negative to peripheral blood–MRD positive, and 6 patients developed overt disease progression. In all patients who progressed, progression was preceded by an increase in detectable peripheral blood MRD cells.26 Although larger studies are needed, peripheral blood flow cytometric monitoring for MRD may be a useful adjunct to predict ongoing response or impending relapse. In addition, newer, more sensitive methods of disease monitoring may ultimately supplant flow cytometry.
Risk Stratification
Although much progress has been made in the risk stratification profiling of hematologic malignancies in general, HCL has unfortunately lagged behind in this effort. The most recent risk stratification analysis was performed in 1982 by Jansen and colleagues.27 This group of researchers performed a retrospective analysis of 391 HCL patients treated at 22 centers. One of the central questions in their analysis was survival time from diagnosis in patients who had not yet undergone splenectomy (a standard treatment at the time). This group consisted of a total of 154 patients. As this study predated modern pathological and molecular testing, clinical and laboratory features were examined, and these mostly consisted of physical exam findings and analysis of the peripheral blood. This group found that several factors influenced the survival of these patients, including duration of symptoms prior to diagnosis, the degree of splenomegaly, hemoglobin level, and number of hairy cells in the peripheral blood. However, because of interobserver variation for the majority of these variables, only hemoglobin and spleen size were included in the proportional hazard model. Using only these 2 variables, the authors were able to determine 3 clinical stages for HCL (Table 1). The stages were found to correlate with median survival: patients with stage 1 disease had a median survival not reached at 72 months, but patients with stage 2 disease had a median survival of 18 months, which decreased to only 12 months in patients with stage 3 disease. 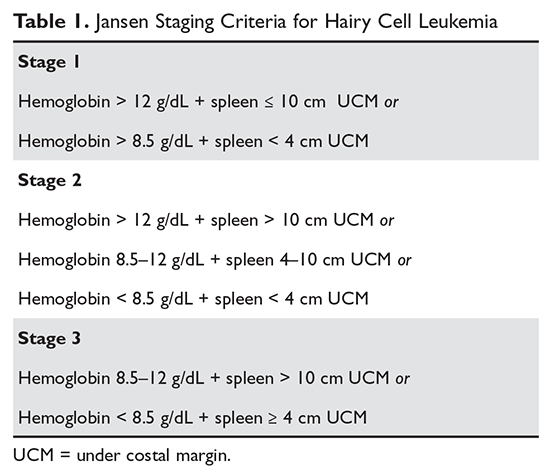
Because the majority of patients with HCL in the modern era will be diagnosed prior to reaching stage 3, a risk stratification system incorporating clinical features, laboratory parameters, and molecular and genetic testing is of considerable interest and is a subject of ongoing research. Ultimately, the goal will be to identify patients at higher risk of early relapse so that more intensive therapies can be applied to initial treatment that will result in longer treatment-free intervals.
Treatment
Because there is no curative treatment for either cHCL or vHCL outside allogeneic transplantation, and it is not clear that early treatment leads to better outcomes in HCL, patients do not always receive treatment at the time of diagnosis or relapse. The general consensus is that patients should be treated if there is a declining trend in hematologic parameters or they experience symptoms from the disease.24 Current consensus guidelines recommend treatment when any of the following hematologic parameters are met: hemoglobin less than 11 g/dL, platelet count less than 100 × 103/µL, or absolute neutrophil count less than 1000/µL.24 These parameters are surrogate markers that indicate compromised bone marrow function. Cytopenias may also be caused by splenomegaly, and symptomatic splenomegaly with or without cytopenias is an indication for treatment. A small number of patients with HCL (approximately 10%) do not require immediate therapy after diagnosis and are monitored by their provider until treatment is indicated.
First-Line Therapy
Despite advances in targeted therapies for HCL, because no treatment has been shown to extend the treatment-free interval longer than chemotherapy, treatment with a purine nucleoside analog is usually the recommended first-line therapy. This includes either cladribine or pentostatin. Both agents appear to be equally effective, and the choice of therapy is determined by the treating physician based on his or her experience. Cladribine administration has been studied using a number of different schedules and routes: intravenous continuous infusion (0.1 mg/kg) for 7 days, intravenous infusion (0.14 mg/kg/day) over 2 hours on a 5-day regimen, or alternatively subcutaneously (0.1–0.14 mg/kg/day) on a once-per-day or once-per-week regimen (Table 2).28,29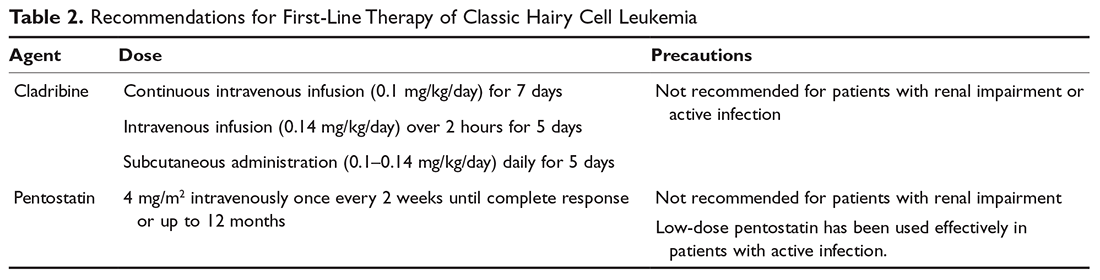
Unlike cHCL, vHCL remains difficult to treat and early disease progression is common. The best outcomes have been seen in patients who have received combination chemo-immunotherapy such as purine nucleoside analog therapy plus rituximab or bendamustine plus rituximab.31 One pilot study of bendamustine plus rituximab in 12 patients found an overall response rate of 100%, with the majority of patients achieving a complete response.31 For patients who achieved a complete response, the median duration of response had not been reached, but patients achieving only a partial response had a median duration of response of only 20 months, indicating there is a subgroup of patients who will require a different treatment approach.32 A randomized phase 2 trial of rituximab with either pentostatin or bendamustine is ongoing.33
Assessment of Response
Response assessment involves physical examination for estimation of spleen size, assessment of hematologic parameters, and a bone marrow biopsy for evaluation of marrow response. It is recommended that the bone marrow biopsy be performed 4 to 6 months following cladribine administration, or after completion of 12 doses of pentostatin. Detailed response assessment criteria are shown in Table 3. 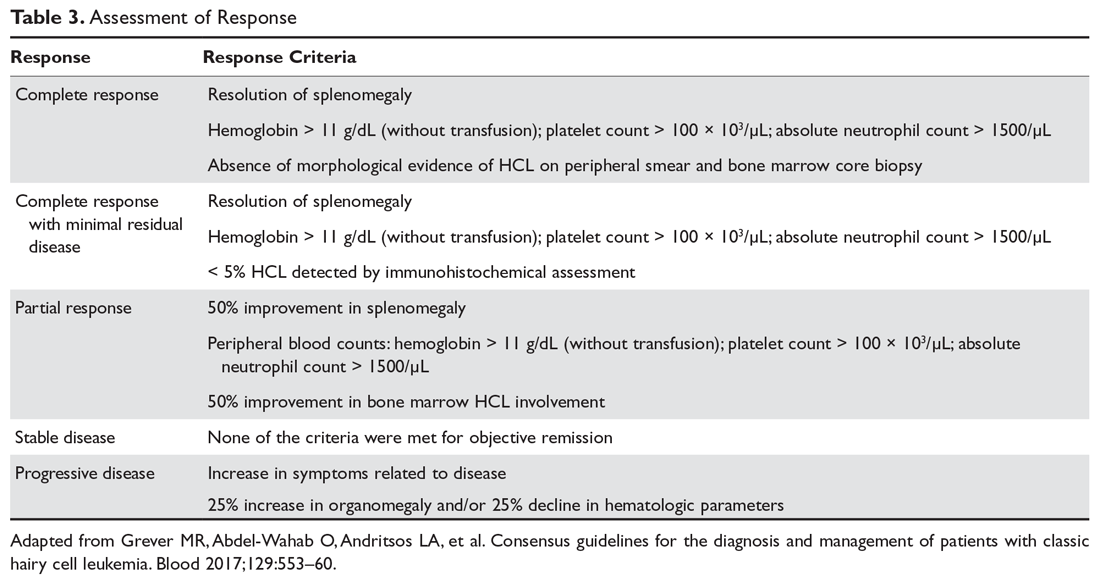
Second-Line Therapy
Although the majority of patients treated with purine analogs will achieve durable remissions, approximately 40% of patients will eventually require second-line therapy. Criteria for treatment at relapse are the same as the criteria for initial therapy, including symptomatic disease or progressive anemia, thrombocytopenia, or neutropenia. The choice of treatment is based on clinical parameters and the duration of the previous remission. If the initial remission was longer than 65 months and the patient is eligible to receive chemotherapy, re-treatment with initial therapy is recommended. For a remission between 24 and 65 months, re-treatment with a purine analog combined with an anti-CD20 monoclonal antibody may be considered.34 If the first remission is shorter than 24 months, confirmation of the original diagnosis as well as consideration for testing for additional mutations with therapeutic targets (BRAF V600E, MAP2K1) should be considered before a treatment decision is made. For these patients, alternative therapies, including investigational agents, should be considered.24
Monoclonal antibody therapy has been studied in both the up-front setting and in relapsed or refractory HCL.35 An initial study of 15 patients with relapsed HCL found an overall response rate of 80%, with 8 patients achieving a complete response. A subsequent study of 26 patients who relapsed after cladribine therapy found an overall response rate of 80%, with a complete response rate of 32%. Median relapse-free survival was 27 months.36 Ravandi and others studied rituximab in the up-front setting in combination with cladribine, and found an overall response rate of 100%, including in patients with vHCL. At the time of publication of the study results, the median survival had not been reached.37 As has been seen with other lymphoid malignancies, concurrent therapy with rituximab appears to enhance the activity of the agent with which it is combined. While its use in the up-front setting remains an area of active investigation, there is a clear role for chemo-immunotherapy in the relapsed setting.
In patients with cHCL, excellent results including complete remissions have been reported with the use of BRAF inhibitors, both as a single agent and when combined with anti-CD20 therapy. The 2 commercially available BRAF inhibitors are vemurafenib and dabrafenib, and both have been tested in relapsed cHCL.38,39 The first study of vemurafenib was reported by Tiacci and colleagues, who found an overall response rate of 96% after a median of 8 weeks and a 100% response rate after a median of 12 weeks, with complete response rates up to 42%.38 The median relapse-free survival was 23 months (decreasing to only 6 months in patients who achieved only a partial remission), indicating that these agents will likely need to be administered in combination with other effective therapies with non-overlapping toxicities. Vemurafenib has been administered concurrently with rituximab, and preliminary results of this combination therapy showed early rates of complete responses.40 Dabrafenib has been reported for use as a single agent in cHCL and clinical trials are underway evaluating its efficacy when administered with trametinib, a MEK inhibitor.39,41 Of note, patients receiving BRAF inhibitors frequently develop cutaneous complications of RAF inhibition including cutaneous squamous cell carcinomas and keratoacanthomas, and close dermatologic surveillance is required.
Variant HCL does not harbor the BRAF V600E mutation, but up to half of patients have been found to have mutations of MAP2K1, which upregulates MEK1 expression.42 Trametinib is approved by the US Food and Drug Administration for the treatment of patients with melanoma at a dose of 2 mg orally daily, and has been successfully used to treat 1 patient with vHCL.43 Further evaluation of this targeted therapy is underway.
Ibrutinib, a Bruton tyrosine kinase inhibitor, and moxetumomab pasudotox, an immunotoxin conjugate, are currently being studied in National Institutes of Health–sponsored multi-institutional trials for patients with HCL. Ibrutinib is administered orally at 420 mg per day until relapse.44 Moxetumomab pasudotox was tested at different doses between 5 and 50 μg/kg intravenously every other day for 3 doses for up to 16 cycles unless they experienced disease progression or developed neutralizing antibodies.45 Both agents have been shown to have significant activity in cHCL and vHCL and will likely be included in the treatment armamentarium once trials are completed. Second-line therapy options are summarized in Table 4.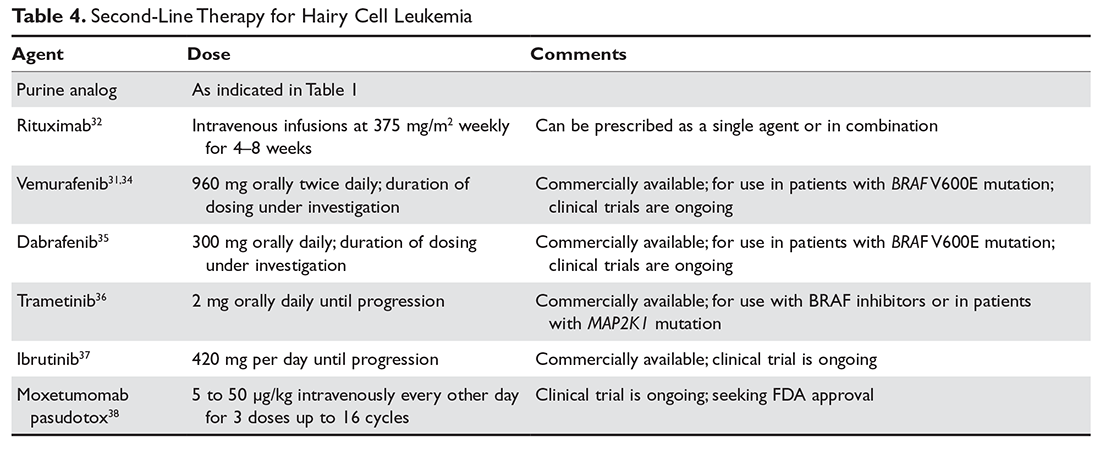
Complications and Supportive Care
The complications of HCL may be separated into the pre-, intra-, and post-treatment periods. At the time of diagnosis and prior to the initiation of therapy, marrow infiltration by HCL frequently leads to cytopenias which cause symptomatic anemia, infection, and/or bleeding complications. Many patients develop splenomegaly, which may further lower the blood counts and which is experienced as abdominal fullness or distention, with early satiety leading to weight loss. Patients may also experience constitutional symptoms with fatigue, fevers in the absence of infection, and unintentional weight loss even without splenomegaly.
For patients who initiate therapy with purine nucleoside analogs, the early part of treatment is associated with the greatest risk of morbidity and mortality. Chemotherapy leads to both immunosuppression (altered cellular immunity) as well as myelosuppression. Thus, patients who are already in need of treatment because of disease-related cytopenias will experience an abrupt and sometimes significant decline in the peripheral blood counts. The treatment period prior to recovery of neutrophils requires the greatest vigilance. Because patients are profoundly immunocompromised, febrile neutropenia is a common complication leading to hospital admission and the cause is often difficult to identify. Treatment with broad-spectrum antibiotics, investigation for opportunistic and viral infections, and considerations for antifungal prophylaxis or therapy are required in this setting. It is recommended that all patients treated with purine nucleoside analogs receive prophylactic antimicrobials for herpes simplex virus and varicella zoster virus, as well as prophylaxis against Pneumocystis jirovecii. Unfortunately, growth factor support has not proven successful in this patient population but is not contraindicated.46
Following successful completion of therapy, patients may remain functionally immunocompromised for a significant period of time even with a normal neutrophil count. Monitoring of the CD4 count may help to determine when prophylactic antimicrobials may be discontinued. A CD4 count greater than 200 cells/µL is generally considered to be adequate for prevention of opportunistic infections. Although immunizations have not been well studied in HCL, it is recommended that patients receive annual influenza immunizations as well as age-appropriate immunizations against Streptococcus pneumoniae and other infectious illnesses as indicated. Live viral vaccines such as the currently available herpes zoster vaccine can lead to infections in this patient population and are not recommended.
Like many hematologic malignancies, HCL may be associated with comorbid conditions related to immune dysfunction. There is a known association with an increased risk of second primary malignancies, which may predate the diagnosis of HCL.47 Therefore, it is recommended that patients continue annual cancer screenings as well as undergo prompt evaluation for potential symptoms of second malignancies. In addition, it is thought that there may be an increased risk for autoimmune disorders such as inflammatory arthritis or immune-mediated cytopenias. One case-control study found a possible association between autoimmune diseases and HCL, noting that at times these diseases are diagnosed concurrently.48 However, because of the rarity of the disease it has been difficult to quantify these associated conditions in a systematic way. There is currently an international patient data registry under development for the systematic study of HCL and its complications which may answer many of these questions.
Survivorship and quality of life are important considerations in chronic diseases. It is not uncommon for patients to develop anxiety related to the trauma of diagnosis and treatment, especially when intensive care has been required. Patients may have lingering fears regarding concerns of developing infections due to exposure to ill persons or fears regarding risk of relapse and need for re-treatment. A proactive approach with partnership with psychosocial oncology may be of benefit, especially when symptoms of post-traumatic stress disorder are evident.
Conclusion
HCL is a rare, chronic lymphoid malignancy that is now subclassified into classic and variant HCL. Further investigations into the disease subtypes will allow more precise disease definitions, and these studies are underway. Renewed efforts toward updated risk stratification and clinical staging systems will be important aspects of these investigations. Refinements in treatment and supportive care have resulted in greatly improved overall survival, which has translated into larger numbers of people living with HCL. However, new treatment paradigms for vHCL are needed as the progression-free survival in this disease remains significantly lower than that of cHCL. Future efforts toward understanding survivorship issues and management of long-term treatment and disease-related complications will be critical for ensuring good quality of life for patients living with HCL.
1. Teras LR, Desantis DE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2008.
3. Yetgin S, Olcay L, Yenicesu I, et al. Relapse in hairy cell leukemia due to isolated nodular skin infiltration. Pediatr Hematol Oncol 2001;18:415–7.
4. Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:175–9.
5. Veterans and agent orange: update 2014. Mil Med 2017;182:1619–20.
6. Villemagne B, Bay JO, Tournilhac O, et al. Two new cases of familial hairy cell leukemia associated with HLA haplotypes A2, B7, Bw4, Bw6. Leuk Lymphoma 2005;46:243–5.
7. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol 2013;163:407–9.
8. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958;13:609–30.
9. Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966;27:199–211.
10. Polliack A, Tadmor T. Surface topography of hairy cell leukemia cells compared to other leukemias as seen by scanning electron microscopy. Leuk Lymphoma 2011;52 Suppl 2:14–7.
11. Miranda RN, Cousar JB, Hammer RD, et al. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol 1999;30:306–12.
12. Vanhentenrijk V, Tierens A, Wlodarska I, et al. V(H) gene analysis of hairy cell leukemia reveals a homogeneous mutation status and suggests its marginal zone B-cell origin. Leukemia 2004;18:1729–32.
13. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59–68.
14. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med 2014;6:238ra71.
15. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
16. Kamiguti AS, Harris RJ, Slupsky JR, et al. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003;22:2272–84.
17. Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: From the laboratory to clinical trials. Crit Rev Oncol Hematol 2014;90:220–32.
18. Shao H, Calvo KR, Gronborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res 2013;37:401–9.
19. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330–2.
20. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014;15:187–209.
21. Sivina M, Kreitman RJ, Arons E, et al. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014;166:177–88.
22. Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood 2015;126:842–50.
23. Andritsos LA, Grever MR. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:166–74.
24. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017;129:553–60.
25. Mhawech-Fauceglia P, Oberholzer M, Aschenafi S, et al. Potential predictive patterns of minimal residual disease detected by immunohistochemistry on bone marrow biopsy specimens during a long-term follow-up in patients treated with cladribine for hairy cell leukemia. Arch Pathol Lab Med 2006;130:374–7.
26. Ortiz-Maldonado V, Villamor N, Baumann T, et al., Is there a role for minimal residual disease monitoring in the management of patients with hairy-cell leukaemia? Br J Haematol 2017 Aug 18.
27. Jansen J, Hermans J. Clinical staging system for hairy-cell leukemia. Blood 1982;60:571–7.
28. Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583–90.
29. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
30. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974–82.
31. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res 2013;19:6873–81.
32. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res 2013;19:6313–21.
33. Randomized phase II trial of rituximab with either pentostatin or bendamustine for multiply relapsed or refractory hairy cell leukemia. 2017 [cited 2017 Oct 26]; NCT01059786. https://clinicaltrials.gov/ct2/show/NCT01059786.
34. Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52 Suppl 2:75–8.
35. Thomas DA, O’Brien S, Bueso-Ramos C, et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003;102:3906–11.
36. Zenhäusern R, Simcock M, Gratwohl A, et al. Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008;93(9):1426–8.
37. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
38. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47.
39. Blachly JS, Lozanski G, Lucas DM, et al. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw 2015;13:9–13.
40. Tiacci E, De Carolis L, Zaja F, et al. Vemurafenib plus rituximab in hairy cell leukemia: a promisingchemotherapy-free regimen for relapsed or refractory patients. Blood 2016;128:1.
41. A phase II, open-label study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib. 2017 [cited 2017 Oct 26]; NCT02034110. https://clinicaltrials.gov/ct2/show/NCT02034110.
42. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014;46:8–10.
43. Andritsos LA, Grieselhuber NR, Anghelina M, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma 2017 Sep 18:1–4.
44. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–506.
45. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30:1822–8.
46. Saven A, Burian C, Adusumalli J, Koziol JA. Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood 1999;93:2471–7.
47. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014;166:390–400.
48. Anderson LA, Engels EA. Autoimmune conditions and hairy cell leukemia: an exploratory case-control study. J Hematol Oncol 2010;3:35.
Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.
While BRAF V600E is identified in nearly all cases of cHCL, it is rare in vHCL.18 The variant type of HCL was classified as a distinct clinical entity in 2008 and can now often be distinguished from cHCL on the basis of BRAF mutational status, among other differences. Interestingly, in the rare cases of BRAF V600E–negative cHCL, other mutations in BRAF or downstream targets as well as aberrant activation of the RAF-MEK-ERK signaling cascade are observed, indicating that this pathway is critical in HCL and may still represent a viable therapeutic target. Expression of the IGHV4-34 immunoglobulin rearrangement, while more common in vHCL, has also been identified in 10% of cHCL cases and appears to confer poor prognosis.19 Other mutated genes that have been identified in HCL include CDKN1B, TP53, U2AF1, ARID1A, EZH2, and KDM6A.20
Classic HCL is characterized by the immunophenotypic expression of CD11c, CD25, CD103, and CD123, with kappa or lambda light chain restriction indicating clonality; HCL B cells are generally negative for CD5, CD10, CD23, CD27, and CD79b. In contrast, vHCL often lacks expression of CD25 and CD123.18 The B-cell receptor (BCR) is expressed on hairy cells and its activation promotes proliferation and survival in vitro.21 The role of BCR signaling in B-cell malignancies is increasingly recognized, and therapies that target the BCR and associated signaling molecules offer an attractive treatment strategy.22 HCL B cells also typically express CD19, CD20, CD22, CD79a, CD200, CD1d, and annexin A1. Tartrate-resistant acid phosphatase (TRAP) positivity by immunohistochemistry is a hallmark of cHCL. Interestingly, changes to the patient’s original immunophenotype have been observed following treatment and upon disease recurrence, highlighting the importance of tracking immunophenotype throughout the course of disease.
Diagnosis
Prior to the advent of annual screening evaluations with routine examination of complete blood counts (CBC), patients were most often diagnosed with HCL when they presented with symptoms of the disease such as splenomegaly, infections, or complications of anemia or thrombocytopenia.23 In the current era, patients are more likely to be incidentally diagnosed when they are found to have an abnormal value on a CBC. Any blood lineage may be affected and patients may have pancytopenia or isolated cytopenias. Of note, monocytopenia is a common finding in cHCL that is not entirely understood. The cells typical of cHCL do not usually circulate in the peripheral blood, but if present would appear as mature lymphocytes with villous cytoplasmic projections, pale blue cytoplasm, and reniform nuclei with open chromatin (Figure).9 Even if the morphologic examination is highly suggestive of HCL, additional testing is required to differentiate between cHCL, vHCL, and other hematologic malignancies which may also have cytoplasmic projections. A complete assessment of the immunophenotype, molecular profile, and cytogenetic features is required to arrive at this diagnosis.
The international Hairy Cell Leukemia Foundation recently published consensus guidelines for the diagnosis and treatment of HCL.24 These guidelines recommend that patients undergo examination of the peripheral blood for morphology and immunophenotyping and further recommend obtaining bone marrow core and aspirate biopsy samples for immunophenotyping via immunohistochemical staining and flow cytometry. The characteristic immunophenotype of cHCL is a population of monoclonal B lymphocytes which co-express CD19, CD20, CD11c, CD25, CD103, and CD123. Variant HCL is characterized by a very similar immunophenotype but is usually negative for CD25 and CD123. It is notable that CD25 positivity may be lost following treatment, and the absence of this marker should not be used as the sole basis of a cHCL versus vHCL diagnosis. Because marrow fibrosis in HCL may prevent a marrow aspirate from being obtained, many of the key diagnostic studies are performed on the core biopsy, including morphological evaluation and immunohistochemical stains such as CD20 (a pan-B cell antigen), annexin-1 (an anti-inflammatory protein expressed only in cHCL), and VE1 (a BRAF V600E stain).
As noted above, recurrent cytogenetic abnormalities have now been identified that may inform the diagnosis or prognosis of HCL. Next-generation sequencing and other testing of the genetic landscape are taking on a larger role in subtype differentiation, and it is likely that future guidelines will recommend evaluation for significant mutations. Given that BRAF V600E mutation status is a key feature of cHCL and is absent in vHCL, it is important to perform this testing at the time of diagnosis whenever possible. The mutation may be detected via VE1 immunohistochemical staining, allele-specific polymerase chain reaction, or next-generation sequencing. Other less sensitive tests exist but are utilized less frequently.
Minimal Residual Disease
There is currently no accepted standard for minimal residual disease (MRD) monitoring in HCL. While detection of MRD has been clearly associated with increased risk of disease progression, cHCL cells typically do not circulate in the peripheral blood, limiting the use of peripheral blood immunophenotyping for quantitative MRD assessment. For quantitative monitoring of marrow involvement by HCL, immunohistochemical staining of the bone marrow core biopsy is usually required. Staining may be performed for CD20, or, in patients who have received anti-CD20 therapy, DBA.44, VE-1, or CD79a. There is currently not a consensus regarding what level of disease involvement constitutes MRD. One group studied this issue and found that relapse could be predicted by evaluating MRD by percentage of positive cells in the marrow by immunohistochemical staining, with less than 1% involvement having the lowest risk for disease relapse and greater than 5% having the highest risk for disease relapse.25 A recent study evaluated MRD patterns in the peripheral blood of 32 cHCL patients who had completed frontline therapy. This group performed flow cytometry on the peripheral blood of patients at 1, 3, 6, and 12 months following therapy. All patients had achieved a complete response with initial therapy and peripheral blood MRD negativity at the completion of therapy. At a median follow-up of 100 months post therapy, 5 patients converted from peripheral blood–MRD negative to peripheral blood–MRD positive, and 6 patients developed overt disease progression. In all patients who progressed, progression was preceded by an increase in detectable peripheral blood MRD cells.26 Although larger studies are needed, peripheral blood flow cytometric monitoring for MRD may be a useful adjunct to predict ongoing response or impending relapse. In addition, newer, more sensitive methods of disease monitoring may ultimately supplant flow cytometry.
Risk Stratification
Although much progress has been made in the risk stratification profiling of hematologic malignancies in general, HCL has unfortunately lagged behind in this effort. The most recent risk stratification analysis was performed in 1982 by Jansen and colleagues.27 This group of researchers performed a retrospective analysis of 391 HCL patients treated at 22 centers. One of the central questions in their analysis was survival time from diagnosis in patients who had not yet undergone splenectomy (a standard treatment at the time). This group consisted of a total of 154 patients. As this study predated modern pathological and molecular testing, clinical and laboratory features were examined, and these mostly consisted of physical exam findings and analysis of the peripheral blood. This group found that several factors influenced the survival of these patients, including duration of symptoms prior to diagnosis, the degree of splenomegaly, hemoglobin level, and number of hairy cells in the peripheral blood. However, because of interobserver variation for the majority of these variables, only hemoglobin and spleen size were included in the proportional hazard model. Using only these 2 variables, the authors were able to determine 3 clinical stages for HCL (Table 1). The stages were found to correlate with median survival: patients with stage 1 disease had a median survival not reached at 72 months, but patients with stage 2 disease had a median survival of 18 months, which decreased to only 12 months in patients with stage 3 disease.
Because the majority of patients with HCL in the modern era will be diagnosed prior to reaching stage 3, a risk stratification system incorporating clinical features, laboratory parameters, and molecular and genetic testing is of considerable interest and is a subject of ongoing research. Ultimately, the goal will be to identify patients at higher risk of early relapse so that more intensive therapies can be applied to initial treatment that will result in longer treatment-free intervals.
Treatment
Because there is no curative treatment for either cHCL or vHCL outside allogeneic transplantation, and it is not clear that early treatment leads to better outcomes in HCL, patients do not always receive treatment at the time of diagnosis or relapse. The general consensus is that patients should be treated if there is a declining trend in hematologic parameters or they experience symptoms from the disease.24 Current consensus guidelines recommend treatment when any of the following hematologic parameters are met: hemoglobin less than 11 g/dL, platelet count less than 100 × 103/µL, or absolute neutrophil count less than 1000/µL.24 These parameters are surrogate markers that indicate compromised bone marrow function. Cytopenias may also be caused by splenomegaly, and symptomatic splenomegaly with or without cytopenias is an indication for treatment. A small number of patients with HCL (approximately 10%) do not require immediate therapy after diagnosis and are monitored by their provider until treatment is indicated.
First-Line Therapy
Despite advances in targeted therapies for HCL, because no treatment has been shown to extend the treatment-free interval longer than chemotherapy, treatment with a purine nucleoside analog is usually the recommended first-line therapy. This includes either cladribine or pentostatin. Both agents appear to be equally effective, and the choice of therapy is determined by the treating physician based on his or her experience. Cladribine administration has been studied using a number of different schedules and routes: intravenous continuous infusion (0.1 mg/kg) for 7 days, intravenous infusion (0.14 mg/kg/day) over 2 hours on a 5-day regimen, or alternatively subcutaneously (0.1–0.14 mg/kg/day) on a once-per-day or once-per-week regimen (Table 2).28,29
Unlike cHCL, vHCL remains difficult to treat and early disease progression is common. The best outcomes have been seen in patients who have received combination chemo-immunotherapy such as purine nucleoside analog therapy plus rituximab or bendamustine plus rituximab.31 One pilot study of bendamustine plus rituximab in 12 patients found an overall response rate of 100%, with the majority of patients achieving a complete response.31 For patients who achieved a complete response, the median duration of response had not been reached, but patients achieving only a partial response had a median duration of response of only 20 months, indicating there is a subgroup of patients who will require a different treatment approach.32 A randomized phase 2 trial of rituximab with either pentostatin or bendamustine is ongoing.33
Assessment of Response
Response assessment involves physical examination for estimation of spleen size, assessment of hematologic parameters, and a bone marrow biopsy for evaluation of marrow response. It is recommended that the bone marrow biopsy be performed 4 to 6 months following cladribine administration, or after completion of 12 doses of pentostatin. Detailed response assessment criteria are shown in Table 3.
Second-Line Therapy
Although the majority of patients treated with purine analogs will achieve durable remissions, approximately 40% of patients will eventually require second-line therapy. Criteria for treatment at relapse are the same as the criteria for initial therapy, including symptomatic disease or progressive anemia, thrombocytopenia, or neutropenia. The choice of treatment is based on clinical parameters and the duration of the previous remission. If the initial remission was longer than 65 months and the patient is eligible to receive chemotherapy, re-treatment with initial therapy is recommended. For a remission between 24 and 65 months, re-treatment with a purine analog combined with an anti-CD20 monoclonal antibody may be considered.34 If the first remission is shorter than 24 months, confirmation of the original diagnosis as well as consideration for testing for additional mutations with therapeutic targets (BRAF V600E, MAP2K1) should be considered before a treatment decision is made. For these patients, alternative therapies, including investigational agents, should be considered.24
Monoclonal antibody therapy has been studied in both the up-front setting and in relapsed or refractory HCL.35 An initial study of 15 patients with relapsed HCL found an overall response rate of 80%, with 8 patients achieving a complete response. A subsequent study of 26 patients who relapsed after cladribine therapy found an overall response rate of 80%, with a complete response rate of 32%. Median relapse-free survival was 27 months.36 Ravandi and others studied rituximab in the up-front setting in combination with cladribine, and found an overall response rate of 100%, including in patients with vHCL. At the time of publication of the study results, the median survival had not been reached.37 As has been seen with other lymphoid malignancies, concurrent therapy with rituximab appears to enhance the activity of the agent with which it is combined. While its use in the up-front setting remains an area of active investigation, there is a clear role for chemo-immunotherapy in the relapsed setting.
In patients with cHCL, excellent results including complete remissions have been reported with the use of BRAF inhibitors, both as a single agent and when combined with anti-CD20 therapy. The 2 commercially available BRAF inhibitors are vemurafenib and dabrafenib, and both have been tested in relapsed cHCL.38,39 The first study of vemurafenib was reported by Tiacci and colleagues, who found an overall response rate of 96% after a median of 8 weeks and a 100% response rate after a median of 12 weeks, with complete response rates up to 42%.38 The median relapse-free survival was 23 months (decreasing to only 6 months in patients who achieved only a partial remission), indicating that these agents will likely need to be administered in combination with other effective therapies with non-overlapping toxicities. Vemurafenib has been administered concurrently with rituximab, and preliminary results of this combination therapy showed early rates of complete responses.40 Dabrafenib has been reported for use as a single agent in cHCL and clinical trials are underway evaluating its efficacy when administered with trametinib, a MEK inhibitor.39,41 Of note, patients receiving BRAF inhibitors frequently develop cutaneous complications of RAF inhibition including cutaneous squamous cell carcinomas and keratoacanthomas, and close dermatologic surveillance is required.
Variant HCL does not harbor the BRAF V600E mutation, but up to half of patients have been found to have mutations of MAP2K1, which upregulates MEK1 expression.42 Trametinib is approved by the US Food and Drug Administration for the treatment of patients with melanoma at a dose of 2 mg orally daily, and has been successfully used to treat 1 patient with vHCL.43 Further evaluation of this targeted therapy is underway.
Ibrutinib, a Bruton tyrosine kinase inhibitor, and moxetumomab pasudotox, an immunotoxin conjugate, are currently being studied in National Institutes of Health–sponsored multi-institutional trials for patients with HCL. Ibrutinib is administered orally at 420 mg per day until relapse.44 Moxetumomab pasudotox was tested at different doses between 5 and 50 μg/kg intravenously every other day for 3 doses for up to 16 cycles unless they experienced disease progression or developed neutralizing antibodies.45 Both agents have been shown to have significant activity in cHCL and vHCL and will likely be included in the treatment armamentarium once trials are completed. Second-line therapy options are summarized in Table 4.
Complications and Supportive Care
The complications of HCL may be separated into the pre-, intra-, and post-treatment periods. At the time of diagnosis and prior to the initiation of therapy, marrow infiltration by HCL frequently leads to cytopenias which cause symptomatic anemia, infection, and/or bleeding complications. Many patients develop splenomegaly, which may further lower the blood counts and which is experienced as abdominal fullness or distention, with early satiety leading to weight loss. Patients may also experience constitutional symptoms with fatigue, fevers in the absence of infection, and unintentional weight loss even without splenomegaly.
For patients who initiate therapy with purine nucleoside analogs, the early part of treatment is associated with the greatest risk of morbidity and mortality. Chemotherapy leads to both immunosuppression (altered cellular immunity) as well as myelosuppression. Thus, patients who are already in need of treatment because of disease-related cytopenias will experience an abrupt and sometimes significant decline in the peripheral blood counts. The treatment period prior to recovery of neutrophils requires the greatest vigilance. Because patients are profoundly immunocompromised, febrile neutropenia is a common complication leading to hospital admission and the cause is often difficult to identify. Treatment with broad-spectrum antibiotics, investigation for opportunistic and viral infections, and considerations for antifungal prophylaxis or therapy are required in this setting. It is recommended that all patients treated with purine nucleoside analogs receive prophylactic antimicrobials for herpes simplex virus and varicella zoster virus, as well as prophylaxis against Pneumocystis jirovecii. Unfortunately, growth factor support has not proven successful in this patient population but is not contraindicated.46
Following successful completion of therapy, patients may remain functionally immunocompromised for a significant period of time even with a normal neutrophil count. Monitoring of the CD4 count may help to determine when prophylactic antimicrobials may be discontinued. A CD4 count greater than 200 cells/µL is generally considered to be adequate for prevention of opportunistic infections. Although immunizations have not been well studied in HCL, it is recommended that patients receive annual influenza immunizations as well as age-appropriate immunizations against Streptococcus pneumoniae and other infectious illnesses as indicated. Live viral vaccines such as the currently available herpes zoster vaccine can lead to infections in this patient population and are not recommended.
Like many hematologic malignancies, HCL may be associated with comorbid conditions related to immune dysfunction. There is a known association with an increased risk of second primary malignancies, which may predate the diagnosis of HCL.47 Therefore, it is recommended that patients continue annual cancer screenings as well as undergo prompt evaluation for potential symptoms of second malignancies. In addition, it is thought that there may be an increased risk for autoimmune disorders such as inflammatory arthritis or immune-mediated cytopenias. One case-control study found a possible association between autoimmune diseases and HCL, noting that at times these diseases are diagnosed concurrently.48 However, because of the rarity of the disease it has been difficult to quantify these associated conditions in a systematic way. There is currently an international patient data registry under development for the systematic study of HCL and its complications which may answer many of these questions.
Survivorship and quality of life are important considerations in chronic diseases. It is not uncommon for patients to develop anxiety related to the trauma of diagnosis and treatment, especially when intensive care has been required. Patients may have lingering fears regarding concerns of developing infections due to exposure to ill persons or fears regarding risk of relapse and need for re-treatment. A proactive approach with partnership with psychosocial oncology may be of benefit, especially when symptoms of post-traumatic stress disorder are evident.
Conclusion
HCL is a rare, chronic lymphoid malignancy that is now subclassified into classic and variant HCL. Further investigations into the disease subtypes will allow more precise disease definitions, and these studies are underway. Renewed efforts toward updated risk stratification and clinical staging systems will be important aspects of these investigations. Refinements in treatment and supportive care have resulted in greatly improved overall survival, which has translated into larger numbers of people living with HCL. However, new treatment paradigms for vHCL are needed as the progression-free survival in this disease remains significantly lower than that of cHCL. Future efforts toward understanding survivorship issues and management of long-term treatment and disease-related complications will be critical for ensuring good quality of life for patients living with HCL.
Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.
While BRAF V600E is identified in nearly all cases of cHCL, it is rare in vHCL.18 The variant type of HCL was classified as a distinct clinical entity in 2008 and can now often be distinguished from cHCL on the basis of BRAF mutational status, among other differences. Interestingly, in the rare cases of BRAF V600E–negative cHCL, other mutations in BRAF or downstream targets as well as aberrant activation of the RAF-MEK-ERK signaling cascade are observed, indicating that this pathway is critical in HCL and may still represent a viable therapeutic target. Expression of the IGHV4-34 immunoglobulin rearrangement, while more common in vHCL, has also been identified in 10% of cHCL cases and appears to confer poor prognosis.19 Other mutated genes that have been identified in HCL include CDKN1B, TP53, U2AF1, ARID1A, EZH2, and KDM6A.20
Classic HCL is characterized by the immunophenotypic expression of CD11c, CD25, CD103, and CD123, with kappa or lambda light chain restriction indicating clonality; HCL B cells are generally negative for CD5, CD10, CD23, CD27, and CD79b. In contrast, vHCL often lacks expression of CD25 and CD123.18 The B-cell receptor (BCR) is expressed on hairy cells and its activation promotes proliferation and survival in vitro.21 The role of BCR signaling in B-cell malignancies is increasingly recognized, and therapies that target the BCR and associated signaling molecules offer an attractive treatment strategy.22 HCL B cells also typically express CD19, CD20, CD22, CD79a, CD200, CD1d, and annexin A1. Tartrate-resistant acid phosphatase (TRAP) positivity by immunohistochemistry is a hallmark of cHCL. Interestingly, changes to the patient’s original immunophenotype have been observed following treatment and upon disease recurrence, highlighting the importance of tracking immunophenotype throughout the course of disease.
Diagnosis
Prior to the advent of annual screening evaluations with routine examination of complete blood counts (CBC), patients were most often diagnosed with HCL when they presented with symptoms of the disease such as splenomegaly, infections, or complications of anemia or thrombocytopenia.23 In the current era, patients are more likely to be incidentally diagnosed when they are found to have an abnormal value on a CBC. Any blood lineage may be affected and patients may have pancytopenia or isolated cytopenias. Of note, monocytopenia is a common finding in cHCL that is not entirely understood. The cells typical of cHCL do not usually circulate in the peripheral blood, but if present would appear as mature lymphocytes with villous cytoplasmic projections, pale blue cytoplasm, and reniform nuclei with open chromatin (Figure).9 Even if the morphologic examination is highly suggestive of HCL, additional testing is required to differentiate between cHCL, vHCL, and other hematologic malignancies which may also have cytoplasmic projections. A complete assessment of the immunophenotype, molecular profile, and cytogenetic features is required to arrive at this diagnosis.
The international Hairy Cell Leukemia Foundation recently published consensus guidelines for the diagnosis and treatment of HCL.24 These guidelines recommend that patients undergo examination of the peripheral blood for morphology and immunophenotyping and further recommend obtaining bone marrow core and aspirate biopsy samples for immunophenotyping via immunohistochemical staining and flow cytometry. The characteristic immunophenotype of cHCL is a population of monoclonal B lymphocytes which co-express CD19, CD20, CD11c, CD25, CD103, and CD123. Variant HCL is characterized by a very similar immunophenotype but is usually negative for CD25 and CD123. It is notable that CD25 positivity may be lost following treatment, and the absence of this marker should not be used as the sole basis of a cHCL versus vHCL diagnosis. Because marrow fibrosis in HCL may prevent a marrow aspirate from being obtained, many of the key diagnostic studies are performed on the core biopsy, including morphological evaluation and immunohistochemical stains such as CD20 (a pan-B cell antigen), annexin-1 (an anti-inflammatory protein expressed only in cHCL), and VE1 (a BRAF V600E stain).
As noted above, recurrent cytogenetic abnormalities have now been identified that may inform the diagnosis or prognosis of HCL. Next-generation sequencing and other testing of the genetic landscape are taking on a larger role in subtype differentiation, and it is likely that future guidelines will recommend evaluation for significant mutations. Given that BRAF V600E mutation status is a key feature of cHCL and is absent in vHCL, it is important to perform this testing at the time of diagnosis whenever possible. The mutation may be detected via VE1 immunohistochemical staining, allele-specific polymerase chain reaction, or next-generation sequencing. Other less sensitive tests exist but are utilized less frequently.
Minimal Residual Disease
There is currently no accepted standard for minimal residual disease (MRD) monitoring in HCL. While detection of MRD has been clearly associated with increased risk of disease progression, cHCL cells typically do not circulate in the peripheral blood, limiting the use of peripheral blood immunophenotyping for quantitative MRD assessment. For quantitative monitoring of marrow involvement by HCL, immunohistochemical staining of the bone marrow core biopsy is usually required. Staining may be performed for CD20, or, in patients who have received anti-CD20 therapy, DBA.44, VE-1, or CD79a. There is currently not a consensus regarding what level of disease involvement constitutes MRD. One group studied this issue and found that relapse could be predicted by evaluating MRD by percentage of positive cells in the marrow by immunohistochemical staining, with less than 1% involvement having the lowest risk for disease relapse and greater than 5% having the highest risk for disease relapse.25 A recent study evaluated MRD patterns in the peripheral blood of 32 cHCL patients who had completed frontline therapy. This group performed flow cytometry on the peripheral blood of patients at 1, 3, 6, and 12 months following therapy. All patients had achieved a complete response with initial therapy and peripheral blood MRD negativity at the completion of therapy. At a median follow-up of 100 months post therapy, 5 patients converted from peripheral blood–MRD negative to peripheral blood–MRD positive, and 6 patients developed overt disease progression. In all patients who progressed, progression was preceded by an increase in detectable peripheral blood MRD cells.26 Although larger studies are needed, peripheral blood flow cytometric monitoring for MRD may be a useful adjunct to predict ongoing response or impending relapse. In addition, newer, more sensitive methods of disease monitoring may ultimately supplant flow cytometry.
Risk Stratification
Although much progress has been made in the risk stratification profiling of hematologic malignancies in general, HCL has unfortunately lagged behind in this effort. The most recent risk stratification analysis was performed in 1982 by Jansen and colleagues.27 This group of researchers performed a retrospective analysis of 391 HCL patients treated at 22 centers. One of the central questions in their analysis was survival time from diagnosis in patients who had not yet undergone splenectomy (a standard treatment at the time). This group consisted of a total of 154 patients. As this study predated modern pathological and molecular testing, clinical and laboratory features were examined, and these mostly consisted of physical exam findings and analysis of the peripheral blood. This group found that several factors influenced the survival of these patients, including duration of symptoms prior to diagnosis, the degree of splenomegaly, hemoglobin level, and number of hairy cells in the peripheral blood. However, because of interobserver variation for the majority of these variables, only hemoglobin and spleen size were included in the proportional hazard model. Using only these 2 variables, the authors were able to determine 3 clinical stages for HCL (Table 1). The stages were found to correlate with median survival: patients with stage 1 disease had a median survival not reached at 72 months, but patients with stage 2 disease had a median survival of 18 months, which decreased to only 12 months in patients with stage 3 disease.
Because the majority of patients with HCL in the modern era will be diagnosed prior to reaching stage 3, a risk stratification system incorporating clinical features, laboratory parameters, and molecular and genetic testing is of considerable interest and is a subject of ongoing research. Ultimately, the goal will be to identify patients at higher risk of early relapse so that more intensive therapies can be applied to initial treatment that will result in longer treatment-free intervals.
Treatment
Because there is no curative treatment for either cHCL or vHCL outside allogeneic transplantation, and it is not clear that early treatment leads to better outcomes in HCL, patients do not always receive treatment at the time of diagnosis or relapse. The general consensus is that patients should be treated if there is a declining trend in hematologic parameters or they experience symptoms from the disease.24 Current consensus guidelines recommend treatment when any of the following hematologic parameters are met: hemoglobin less than 11 g/dL, platelet count less than 100 × 103/µL, or absolute neutrophil count less than 1000/µL.24 These parameters are surrogate markers that indicate compromised bone marrow function. Cytopenias may also be caused by splenomegaly, and symptomatic splenomegaly with or without cytopenias is an indication for treatment. A small number of patients with HCL (approximately 10%) do not require immediate therapy after diagnosis and are monitored by their provider until treatment is indicated.
First-Line Therapy
Despite advances in targeted therapies for HCL, because no treatment has been shown to extend the treatment-free interval longer than chemotherapy, treatment with a purine nucleoside analog is usually the recommended first-line therapy. This includes either cladribine or pentostatin. Both agents appear to be equally effective, and the choice of therapy is determined by the treating physician based on his or her experience. Cladribine administration has been studied using a number of different schedules and routes: intravenous continuous infusion (0.1 mg/kg) for 7 days, intravenous infusion (0.14 mg/kg/day) over 2 hours on a 5-day regimen, or alternatively subcutaneously (0.1–0.14 mg/kg/day) on a once-per-day or once-per-week regimen (Table 2).28,29
Unlike cHCL, vHCL remains difficult to treat and early disease progression is common. The best outcomes have been seen in patients who have received combination chemo-immunotherapy such as purine nucleoside analog therapy plus rituximab or bendamustine plus rituximab.31 One pilot study of bendamustine plus rituximab in 12 patients found an overall response rate of 100%, with the majority of patients achieving a complete response.31 For patients who achieved a complete response, the median duration of response had not been reached, but patients achieving only a partial response had a median duration of response of only 20 months, indicating there is a subgroup of patients who will require a different treatment approach.32 A randomized phase 2 trial of rituximab with either pentostatin or bendamustine is ongoing.33
Assessment of Response
Response assessment involves physical examination for estimation of spleen size, assessment of hematologic parameters, and a bone marrow biopsy for evaluation of marrow response. It is recommended that the bone marrow biopsy be performed 4 to 6 months following cladribine administration, or after completion of 12 doses of pentostatin. Detailed response assessment criteria are shown in Table 3.
Second-Line Therapy
Although the majority of patients treated with purine analogs will achieve durable remissions, approximately 40% of patients will eventually require second-line therapy. Criteria for treatment at relapse are the same as the criteria for initial therapy, including symptomatic disease or progressive anemia, thrombocytopenia, or neutropenia. The choice of treatment is based on clinical parameters and the duration of the previous remission. If the initial remission was longer than 65 months and the patient is eligible to receive chemotherapy, re-treatment with initial therapy is recommended. For a remission between 24 and 65 months, re-treatment with a purine analog combined with an anti-CD20 monoclonal antibody may be considered.34 If the first remission is shorter than 24 months, confirmation of the original diagnosis as well as consideration for testing for additional mutations with therapeutic targets (BRAF V600E, MAP2K1) should be considered before a treatment decision is made. For these patients, alternative therapies, including investigational agents, should be considered.24
Monoclonal antibody therapy has been studied in both the up-front setting and in relapsed or refractory HCL.35 An initial study of 15 patients with relapsed HCL found an overall response rate of 80%, with 8 patients achieving a complete response. A subsequent study of 26 patients who relapsed after cladribine therapy found an overall response rate of 80%, with a complete response rate of 32%. Median relapse-free survival was 27 months.36 Ravandi and others studied rituximab in the up-front setting in combination with cladribine, and found an overall response rate of 100%, including in patients with vHCL. At the time of publication of the study results, the median survival had not been reached.37 As has been seen with other lymphoid malignancies, concurrent therapy with rituximab appears to enhance the activity of the agent with which it is combined. While its use in the up-front setting remains an area of active investigation, there is a clear role for chemo-immunotherapy in the relapsed setting.
In patients with cHCL, excellent results including complete remissions have been reported with the use of BRAF inhibitors, both as a single agent and when combined with anti-CD20 therapy. The 2 commercially available BRAF inhibitors are vemurafenib and dabrafenib, and both have been tested in relapsed cHCL.38,39 The first study of vemurafenib was reported by Tiacci and colleagues, who found an overall response rate of 96% after a median of 8 weeks and a 100% response rate after a median of 12 weeks, with complete response rates up to 42%.38 The median relapse-free survival was 23 months (decreasing to only 6 months in patients who achieved only a partial remission), indicating that these agents will likely need to be administered in combination with other effective therapies with non-overlapping toxicities. Vemurafenib has been administered concurrently with rituximab, and preliminary results of this combination therapy showed early rates of complete responses.40 Dabrafenib has been reported for use as a single agent in cHCL and clinical trials are underway evaluating its efficacy when administered with trametinib, a MEK inhibitor.39,41 Of note, patients receiving BRAF inhibitors frequently develop cutaneous complications of RAF inhibition including cutaneous squamous cell carcinomas and keratoacanthomas, and close dermatologic surveillance is required.
Variant HCL does not harbor the BRAF V600E mutation, but up to half of patients have been found to have mutations of MAP2K1, which upregulates MEK1 expression.42 Trametinib is approved by the US Food and Drug Administration for the treatment of patients with melanoma at a dose of 2 mg orally daily, and has been successfully used to treat 1 patient with vHCL.43 Further evaluation of this targeted therapy is underway.
Ibrutinib, a Bruton tyrosine kinase inhibitor, and moxetumomab pasudotox, an immunotoxin conjugate, are currently being studied in National Institutes of Health–sponsored multi-institutional trials for patients with HCL. Ibrutinib is administered orally at 420 mg per day until relapse.44 Moxetumomab pasudotox was tested at different doses between 5 and 50 μg/kg intravenously every other day for 3 doses for up to 16 cycles unless they experienced disease progression or developed neutralizing antibodies.45 Both agents have been shown to have significant activity in cHCL and vHCL and will likely be included in the treatment armamentarium once trials are completed. Second-line therapy options are summarized in Table 4.
Complications and Supportive Care
The complications of HCL may be separated into the pre-, intra-, and post-treatment periods. At the time of diagnosis and prior to the initiation of therapy, marrow infiltration by HCL frequently leads to cytopenias which cause symptomatic anemia, infection, and/or bleeding complications. Many patients develop splenomegaly, which may further lower the blood counts and which is experienced as abdominal fullness or distention, with early satiety leading to weight loss. Patients may also experience constitutional symptoms with fatigue, fevers in the absence of infection, and unintentional weight loss even without splenomegaly.
For patients who initiate therapy with purine nucleoside analogs, the early part of treatment is associated with the greatest risk of morbidity and mortality. Chemotherapy leads to both immunosuppression (altered cellular immunity) as well as myelosuppression. Thus, patients who are already in need of treatment because of disease-related cytopenias will experience an abrupt and sometimes significant decline in the peripheral blood counts. The treatment period prior to recovery of neutrophils requires the greatest vigilance. Because patients are profoundly immunocompromised, febrile neutropenia is a common complication leading to hospital admission and the cause is often difficult to identify. Treatment with broad-spectrum antibiotics, investigation for opportunistic and viral infections, and considerations for antifungal prophylaxis or therapy are required in this setting. It is recommended that all patients treated with purine nucleoside analogs receive prophylactic antimicrobials for herpes simplex virus and varicella zoster virus, as well as prophylaxis against Pneumocystis jirovecii. Unfortunately, growth factor support has not proven successful in this patient population but is not contraindicated.46
Following successful completion of therapy, patients may remain functionally immunocompromised for a significant period of time even with a normal neutrophil count. Monitoring of the CD4 count may help to determine when prophylactic antimicrobials may be discontinued. A CD4 count greater than 200 cells/µL is generally considered to be adequate for prevention of opportunistic infections. Although immunizations have not been well studied in HCL, it is recommended that patients receive annual influenza immunizations as well as age-appropriate immunizations against Streptococcus pneumoniae and other infectious illnesses as indicated. Live viral vaccines such as the currently available herpes zoster vaccine can lead to infections in this patient population and are not recommended.
Like many hematologic malignancies, HCL may be associated with comorbid conditions related to immune dysfunction. There is a known association with an increased risk of second primary malignancies, which may predate the diagnosis of HCL.47 Therefore, it is recommended that patients continue annual cancer screenings as well as undergo prompt evaluation for potential symptoms of second malignancies. In addition, it is thought that there may be an increased risk for autoimmune disorders such as inflammatory arthritis or immune-mediated cytopenias. One case-control study found a possible association between autoimmune diseases and HCL, noting that at times these diseases are diagnosed concurrently.48 However, because of the rarity of the disease it has been difficult to quantify these associated conditions in a systematic way. There is currently an international patient data registry under development for the systematic study of HCL and its complications which may answer many of these questions.
Survivorship and quality of life are important considerations in chronic diseases. It is not uncommon for patients to develop anxiety related to the trauma of diagnosis and treatment, especially when intensive care has been required. Patients may have lingering fears regarding concerns of developing infections due to exposure to ill persons or fears regarding risk of relapse and need for re-treatment. A proactive approach with partnership with psychosocial oncology may be of benefit, especially when symptoms of post-traumatic stress disorder are evident.
Conclusion
HCL is a rare, chronic lymphoid malignancy that is now subclassified into classic and variant HCL. Further investigations into the disease subtypes will allow more precise disease definitions, and these studies are underway. Renewed efforts toward updated risk stratification and clinical staging systems will be important aspects of these investigations. Refinements in treatment and supportive care have resulted in greatly improved overall survival, which has translated into larger numbers of people living with HCL. However, new treatment paradigms for vHCL are needed as the progression-free survival in this disease remains significantly lower than that of cHCL. Future efforts toward understanding survivorship issues and management of long-term treatment and disease-related complications will be critical for ensuring good quality of life for patients living with HCL.
1. Teras LR, Desantis DE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2008.
3. Yetgin S, Olcay L, Yenicesu I, et al. Relapse in hairy cell leukemia due to isolated nodular skin infiltration. Pediatr Hematol Oncol 2001;18:415–7.
4. Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:175–9.
5. Veterans and agent orange: update 2014. Mil Med 2017;182:1619–20.
6. Villemagne B, Bay JO, Tournilhac O, et al. Two new cases of familial hairy cell leukemia associated with HLA haplotypes A2, B7, Bw4, Bw6. Leuk Lymphoma 2005;46:243–5.
7. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol 2013;163:407–9.
8. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958;13:609–30.
9. Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966;27:199–211.
10. Polliack A, Tadmor T. Surface topography of hairy cell leukemia cells compared to other leukemias as seen by scanning electron microscopy. Leuk Lymphoma 2011;52 Suppl 2:14–7.
11. Miranda RN, Cousar JB, Hammer RD, et al. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol 1999;30:306–12.
12. Vanhentenrijk V, Tierens A, Wlodarska I, et al. V(H) gene analysis of hairy cell leukemia reveals a homogeneous mutation status and suggests its marginal zone B-cell origin. Leukemia 2004;18:1729–32.
13. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59–68.
14. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med 2014;6:238ra71.
15. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
16. Kamiguti AS, Harris RJ, Slupsky JR, et al. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003;22:2272–84.
17. Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: From the laboratory to clinical trials. Crit Rev Oncol Hematol 2014;90:220–32.
18. Shao H, Calvo KR, Gronborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res 2013;37:401–9.
19. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330–2.
20. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014;15:187–209.
21. Sivina M, Kreitman RJ, Arons E, et al. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014;166:177–88.
22. Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood 2015;126:842–50.
23. Andritsos LA, Grever MR. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:166–74.
24. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017;129:553–60.
25. Mhawech-Fauceglia P, Oberholzer M, Aschenafi S, et al. Potential predictive patterns of minimal residual disease detected by immunohistochemistry on bone marrow biopsy specimens during a long-term follow-up in patients treated with cladribine for hairy cell leukemia. Arch Pathol Lab Med 2006;130:374–7.
26. Ortiz-Maldonado V, Villamor N, Baumann T, et al., Is there a role for minimal residual disease monitoring in the management of patients with hairy-cell leukaemia? Br J Haematol 2017 Aug 18.
27. Jansen J, Hermans J. Clinical staging system for hairy-cell leukemia. Blood 1982;60:571–7.
28. Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583–90.
29. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
30. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974–82.
31. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res 2013;19:6873–81.
32. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res 2013;19:6313–21.
33. Randomized phase II trial of rituximab with either pentostatin or bendamustine for multiply relapsed or refractory hairy cell leukemia. 2017 [cited 2017 Oct 26]; NCT01059786. https://clinicaltrials.gov/ct2/show/NCT01059786.
34. Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52 Suppl 2:75–8.
35. Thomas DA, O’Brien S, Bueso-Ramos C, et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003;102:3906–11.
36. Zenhäusern R, Simcock M, Gratwohl A, et al. Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008;93(9):1426–8.
37. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
38. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47.
39. Blachly JS, Lozanski G, Lucas DM, et al. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw 2015;13:9–13.
40. Tiacci E, De Carolis L, Zaja F, et al. Vemurafenib plus rituximab in hairy cell leukemia: a promisingchemotherapy-free regimen for relapsed or refractory patients. Blood 2016;128:1.
41. A phase II, open-label study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib. 2017 [cited 2017 Oct 26]; NCT02034110. https://clinicaltrials.gov/ct2/show/NCT02034110.
42. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014;46:8–10.
43. Andritsos LA, Grieselhuber NR, Anghelina M, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma 2017 Sep 18:1–4.
44. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–506.
45. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30:1822–8.
46. Saven A, Burian C, Adusumalli J, Koziol JA. Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood 1999;93:2471–7.
47. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014;166:390–400.
48. Anderson LA, Engels EA. Autoimmune conditions and hairy cell leukemia: an exploratory case-control study. J Hematol Oncol 2010;3:35.
1. Teras LR, Desantis DE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2008.
3. Yetgin S, Olcay L, Yenicesu I, et al. Relapse in hairy cell leukemia due to isolated nodular skin infiltration. Pediatr Hematol Oncol 2001;18:415–7.
4. Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:175–9.
5. Veterans and agent orange: update 2014. Mil Med 2017;182:1619–20.
6. Villemagne B, Bay JO, Tournilhac O, et al. Two new cases of familial hairy cell leukemia associated with HLA haplotypes A2, B7, Bw4, Bw6. Leuk Lymphoma 2005;46:243–5.
7. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol 2013;163:407–9.
8. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958;13:609–30.
9. Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966;27:199–211.
10. Polliack A, Tadmor T. Surface topography of hairy cell leukemia cells compared to other leukemias as seen by scanning electron microscopy. Leuk Lymphoma 2011;52 Suppl 2:14–7.
11. Miranda RN, Cousar JB, Hammer RD, et al. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol 1999;30:306–12.
12. Vanhentenrijk V, Tierens A, Wlodarska I, et al. V(H) gene analysis of hairy cell leukemia reveals a homogeneous mutation status and suggests its marginal zone B-cell origin. Leukemia 2004;18:1729–32.
13. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59–68.
14. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med 2014;6:238ra71.
15. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
16. Kamiguti AS, Harris RJ, Slupsky JR, et al. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003;22:2272–84.
17. Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: From the laboratory to clinical trials. Crit Rev Oncol Hematol 2014;90:220–32.
18. Shao H, Calvo KR, Gronborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res 2013;37:401–9.
19. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330–2.
20. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014;15:187–209.
21. Sivina M, Kreitman RJ, Arons E, et al. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014;166:177–88.
22. Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood 2015;126:842–50.
23. Andritsos LA, Grever MR. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:166–74.
24. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017;129:553–60.
25. Mhawech-Fauceglia P, Oberholzer M, Aschenafi S, et al. Potential predictive patterns of minimal residual disease detected by immunohistochemistry on bone marrow biopsy specimens during a long-term follow-up in patients treated with cladribine for hairy cell leukemia. Arch Pathol Lab Med 2006;130:374–7.
26. Ortiz-Maldonado V, Villamor N, Baumann T, et al., Is there a role for minimal residual disease monitoring in the management of patients with hairy-cell leukaemia? Br J Haematol 2017 Aug 18.
27. Jansen J, Hermans J. Clinical staging system for hairy-cell leukemia. Blood 1982;60:571–7.
28. Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583–90.
29. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
30. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974–82.
31. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res 2013;19:6873–81.
32. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res 2013;19:6313–21.
33. Randomized phase II trial of rituximab with either pentostatin or bendamustine for multiply relapsed or refractory hairy cell leukemia. 2017 [cited 2017 Oct 26]; NCT01059786. https://clinicaltrials.gov/ct2/show/NCT01059786.
34. Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52 Suppl 2:75–8.
35. Thomas DA, O’Brien S, Bueso-Ramos C, et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003;102:3906–11.
36. Zenhäusern R, Simcock M, Gratwohl A, et al. Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008;93(9):1426–8.
37. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
38. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47.
39. Blachly JS, Lozanski G, Lucas DM, et al. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw 2015;13:9–13.
40. Tiacci E, De Carolis L, Zaja F, et al. Vemurafenib plus rituximab in hairy cell leukemia: a promisingchemotherapy-free regimen for relapsed or refractory patients. Blood 2016;128:1.
41. A phase II, open-label study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib. 2017 [cited 2017 Oct 26]; NCT02034110. https://clinicaltrials.gov/ct2/show/NCT02034110.
42. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014;46:8–10.
43. Andritsos LA, Grieselhuber NR, Anghelina M, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma 2017 Sep 18:1–4.
44. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–506.
45. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30:1822–8.
46. Saven A, Burian C, Adusumalli J, Koziol JA. Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood 1999;93:2471–7.
47. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014;166:390–400.
48. Anderson LA, Engels EA. Autoimmune conditions and hairy cell leukemia: an exploratory case-control study. J Hematol Oncol 2010;3:35.