User login
Anti-CD22 CAR T-cells shift ALL into complete remission
SAN DIEGO – When one CAR stops one working, try another: chimeric antigen receptor (CAR) T-cell therapy for children and young adults with acute lymphoblastic leukemia is driving forward with a novel anti-CD22 target that in an early dose-finding trial has induced complete remissions in some patients with relapsed or refractory disease, including patients previously treated with anti-CD19 CAR-T therapy.
In the first-in-humans trial, CAR T-cell therapy directed against CD22 was shown to be safe and was associated with minimal residual disease (MRD)-negative complete remissions in eight of 10 children and young adults with relapsed/refractory B-precursor acute lymphoblastic leukemia treated at the highest dose level.
“This is the first successful salvage CAR therapy for CD19-negative B-[lineage] ALL,” said co-principal investigator Terry J. Fry, MD, from the Center for Cancer Research at the National Cancer Institute in Bethesda, Md.
Preliminary experience with anti-CD22 immunotherapy suggests that it is comparable in potency to anti-CD19 CAR, and investigators are exploring the possibility that the two chimeric antigen targets could be combined for greater efficacy, he said during a briefing at the annual meeting of the American Society of Hematology.
Tough target
As reported previously from the 2013 ASH annual meeting, anti-CD19 CAR T cells induced complete responses in 10 of 16 children and young adults with relapsed/refractory ALL, and in a second study, CD19-targeted T cells induced complete molecular responses in 12 of 16 adults with B-lineage ALL refractory to chemotherapy.
In current phase 2 trials, anti-CD19 CAR-T therapy is associated with complete remission rates of 80% to 90% of those treated.
However, “we’re learning now that one of the limitations of this approach is the loss of CD19 expression occurring in a substantial number of patients, although it has not been systematically analyzed,” Dr. Fry said.
CD22, an antigen restricted to B-lineage cells, is a promising alternative to CD19 as a target, but finding just the right anti-CD22 CAR was tricky, Dr. Fry said in an interview. The investigators found that many candidate antigens bound well to T cells but had no efficacy, and it took several years of trying before they identified the current version of the antigen
In the phase I trial, the investigators enrolled 16 children and young adults (ages 7 to 22 years) with relapsed/refractory CD22-positive hematologic malignancies. All patients had previously undergone at least one allogeneic stem cell transplant, 11 had previously received anti-CD19 CAR-T cell therapy, and 9 were CD19-negative or had reduced CD19 expression on ALL cells.
The patients underwent peripheral blood mononuclear cells (PBMCs) collected through autologous leukapheresis. The cells were then enriched and expanded, and transduced with a lentiviral vector containing an anti-CD22 CAR for 7 to 10 days, allowing the cells to identify and bind to CD22 expressed on ALL blasts.
The patients then underwent lymphodepletion with fludarabine, and cyclophosphamide, and received infusions of the transduced T-cells at one of three dose levels, starting at 3 x 105 transduced T-cells per recipient weight in kilograms (DL-1), 1 x 106/kg (DL-2), and 3 x 106/kg (DL-3).
The complete remission rate at DL-2 and -3 combined was 80%, with the cytokine-release syndrome (CRS) at a maximum of grade 2.
As noted before, three of the remissions were comparatively durable, with one lasting more than a year.
There were no dose-limiting toxicities at DL-2, and grade 4 hypoxia at DL-3 was seen in one patient.There was one death from sepsis and multi-organ failure in one patient in an expansion cohort. There have been no cases of severe neurotoxicity thus far.
In five patients who experienced relapse, one treated at DL-1 had a loss of CAR cells, and four had changes in CD22 expression, primarily a decrease in site density that may cause the CD22 expression to fall below the threshold for CAR activity, Dr. Fry said.
“At least in our eyes, this may not be best used as a salvage therapy, but we’re beginning to think about how this should be included with CD19 in the upfront CAR treatment,” he said.
The study was funded by the National Institutes of Health with support from Lentigen and Juno Therapeutics. Dr. Fry reported no relevant disclosures.
SAN DIEGO – When one CAR stops one working, try another: chimeric antigen receptor (CAR) T-cell therapy for children and young adults with acute lymphoblastic leukemia is driving forward with a novel anti-CD22 target that in an early dose-finding trial has induced complete remissions in some patients with relapsed or refractory disease, including patients previously treated with anti-CD19 CAR-T therapy.
In the first-in-humans trial, CAR T-cell therapy directed against CD22 was shown to be safe and was associated with minimal residual disease (MRD)-negative complete remissions in eight of 10 children and young adults with relapsed/refractory B-precursor acute lymphoblastic leukemia treated at the highest dose level.
“This is the first successful salvage CAR therapy for CD19-negative B-[lineage] ALL,” said co-principal investigator Terry J. Fry, MD, from the Center for Cancer Research at the National Cancer Institute in Bethesda, Md.
Preliminary experience with anti-CD22 immunotherapy suggests that it is comparable in potency to anti-CD19 CAR, and investigators are exploring the possibility that the two chimeric antigen targets could be combined for greater efficacy, he said during a briefing at the annual meeting of the American Society of Hematology.
Tough target
As reported previously from the 2013 ASH annual meeting, anti-CD19 CAR T cells induced complete responses in 10 of 16 children and young adults with relapsed/refractory ALL, and in a second study, CD19-targeted T cells induced complete molecular responses in 12 of 16 adults with B-lineage ALL refractory to chemotherapy.
In current phase 2 trials, anti-CD19 CAR-T therapy is associated with complete remission rates of 80% to 90% of those treated.
However, “we’re learning now that one of the limitations of this approach is the loss of CD19 expression occurring in a substantial number of patients, although it has not been systematically analyzed,” Dr. Fry said.
CD22, an antigen restricted to B-lineage cells, is a promising alternative to CD19 as a target, but finding just the right anti-CD22 CAR was tricky, Dr. Fry said in an interview. The investigators found that many candidate antigens bound well to T cells but had no efficacy, and it took several years of trying before they identified the current version of the antigen
In the phase I trial, the investigators enrolled 16 children and young adults (ages 7 to 22 years) with relapsed/refractory CD22-positive hematologic malignancies. All patients had previously undergone at least one allogeneic stem cell transplant, 11 had previously received anti-CD19 CAR-T cell therapy, and 9 were CD19-negative or had reduced CD19 expression on ALL cells.
The patients underwent peripheral blood mononuclear cells (PBMCs) collected through autologous leukapheresis. The cells were then enriched and expanded, and transduced with a lentiviral vector containing an anti-CD22 CAR for 7 to 10 days, allowing the cells to identify and bind to CD22 expressed on ALL blasts.
The patients then underwent lymphodepletion with fludarabine, and cyclophosphamide, and received infusions of the transduced T-cells at one of three dose levels, starting at 3 x 105 transduced T-cells per recipient weight in kilograms (DL-1), 1 x 106/kg (DL-2), and 3 x 106/kg (DL-3).
The complete remission rate at DL-2 and -3 combined was 80%, with the cytokine-release syndrome (CRS) at a maximum of grade 2.
As noted before, three of the remissions were comparatively durable, with one lasting more than a year.
There were no dose-limiting toxicities at DL-2, and grade 4 hypoxia at DL-3 was seen in one patient.There was one death from sepsis and multi-organ failure in one patient in an expansion cohort. There have been no cases of severe neurotoxicity thus far.
In five patients who experienced relapse, one treated at DL-1 had a loss of CAR cells, and four had changes in CD22 expression, primarily a decrease in site density that may cause the CD22 expression to fall below the threshold for CAR activity, Dr. Fry said.
“At least in our eyes, this may not be best used as a salvage therapy, but we’re beginning to think about how this should be included with CD19 in the upfront CAR treatment,” he said.
The study was funded by the National Institutes of Health with support from Lentigen and Juno Therapeutics. Dr. Fry reported no relevant disclosures.
SAN DIEGO – When one CAR stops one working, try another: chimeric antigen receptor (CAR) T-cell therapy for children and young adults with acute lymphoblastic leukemia is driving forward with a novel anti-CD22 target that in an early dose-finding trial has induced complete remissions in some patients with relapsed or refractory disease, including patients previously treated with anti-CD19 CAR-T therapy.
In the first-in-humans trial, CAR T-cell therapy directed against CD22 was shown to be safe and was associated with minimal residual disease (MRD)-negative complete remissions in eight of 10 children and young adults with relapsed/refractory B-precursor acute lymphoblastic leukemia treated at the highest dose level.
“This is the first successful salvage CAR therapy for CD19-negative B-[lineage] ALL,” said co-principal investigator Terry J. Fry, MD, from the Center for Cancer Research at the National Cancer Institute in Bethesda, Md.
Preliminary experience with anti-CD22 immunotherapy suggests that it is comparable in potency to anti-CD19 CAR, and investigators are exploring the possibility that the two chimeric antigen targets could be combined for greater efficacy, he said during a briefing at the annual meeting of the American Society of Hematology.
Tough target
As reported previously from the 2013 ASH annual meeting, anti-CD19 CAR T cells induced complete responses in 10 of 16 children and young adults with relapsed/refractory ALL, and in a second study, CD19-targeted T cells induced complete molecular responses in 12 of 16 adults with B-lineage ALL refractory to chemotherapy.
In current phase 2 trials, anti-CD19 CAR-T therapy is associated with complete remission rates of 80% to 90% of those treated.
However, “we’re learning now that one of the limitations of this approach is the loss of CD19 expression occurring in a substantial number of patients, although it has not been systematically analyzed,” Dr. Fry said.
CD22, an antigen restricted to B-lineage cells, is a promising alternative to CD19 as a target, but finding just the right anti-CD22 CAR was tricky, Dr. Fry said in an interview. The investigators found that many candidate antigens bound well to T cells but had no efficacy, and it took several years of trying before they identified the current version of the antigen
In the phase I trial, the investigators enrolled 16 children and young adults (ages 7 to 22 years) with relapsed/refractory CD22-positive hematologic malignancies. All patients had previously undergone at least one allogeneic stem cell transplant, 11 had previously received anti-CD19 CAR-T cell therapy, and 9 were CD19-negative or had reduced CD19 expression on ALL cells.
The patients underwent peripheral blood mononuclear cells (PBMCs) collected through autologous leukapheresis. The cells were then enriched and expanded, and transduced with a lentiviral vector containing an anti-CD22 CAR for 7 to 10 days, allowing the cells to identify and bind to CD22 expressed on ALL blasts.
The patients then underwent lymphodepletion with fludarabine, and cyclophosphamide, and received infusions of the transduced T-cells at one of three dose levels, starting at 3 x 105 transduced T-cells per recipient weight in kilograms (DL-1), 1 x 106/kg (DL-2), and 3 x 106/kg (DL-3).
The complete remission rate at DL-2 and -3 combined was 80%, with the cytokine-release syndrome (CRS) at a maximum of grade 2.
As noted before, three of the remissions were comparatively durable, with one lasting more than a year.
There were no dose-limiting toxicities at DL-2, and grade 4 hypoxia at DL-3 was seen in one patient.There was one death from sepsis and multi-organ failure in one patient in an expansion cohort. There have been no cases of severe neurotoxicity thus far.
In five patients who experienced relapse, one treated at DL-1 had a loss of CAR cells, and four had changes in CD22 expression, primarily a decrease in site density that may cause the CD22 expression to fall below the threshold for CAR activity, Dr. Fry said.
“At least in our eyes, this may not be best used as a salvage therapy, but we’re beginning to think about how this should be included with CD19 in the upfront CAR treatment,” he said.
The study was funded by the National Institutes of Health with support from Lentigen and Juno Therapeutics. Dr. Fry reported no relevant disclosures.
FROM ASH 2016
Key clinical point: CAR T-cell therapy with an anti-CD22 antigen induced complete, MRD-negative remissions in children/young adults with acute lymphoblastic leukemia.
Major finding: The complete remission rate among patients treated at the two highest dose levels was 80%.
Data source: Phase 1 dose-finding trial in 16 children/young adults with relapsed/refractory ALL or diffuse large B-cell lymphoma.
Disclosures The study was funded by the National Institutes of Health with support from Lentigen and Juno Therapeutics. Dr. Fry reported no relevant disclosures views
Malaria elimination in sub-Saharan Africa is possible, study suggests

Kariba area of southern Zambia
where malaria elimination
programs are underway.
Photo from Milen Nikolov
Malaria elimination in historically high transmission areas like southern Africa is possible with tools that are already available, according to new research.
The study suggests that high levels of vector control are key, and mass drug campaigns cannot make much of an impact without proper vector control.
Milen Nikolov, of the Institute for Disease Modeling in Bellevue, Washington, and colleagues reported these findings in PLOS Computational Biology.
The researchers said the Lake Kariba region of Southern Province, Zambia, is part of a multi-country malaria elimination effort. However, elimination in this area is challenging because villages with high and low malaria burden are interconnected through human travel.
With this in mind, the researchers combined a mathematical model of malaria transmission with field data from Zambia to test a variety of strategies for eliminating malaria in the Lake Kariba region.
The team used detailed spatial surveillance data from field studies—including household locations, climate, clinical malaria incidence, prevalence of malaria infections, and bednet usage rates—to construct a model of interconnected villages, then tested a variety of intervention scenarios to see which ones could lead to elimination.
The results indicate that elimination requires high, yet realistic, levels of vector control. And mass drug campaigns deployed to kill parasites in the human population can boost the chances of achieving elimination as long as vector control is well-implemented.
The researchers said this work suggests that elimination programs in sub-Saharan Africa should focus on how to achieve and maintain excellent coverage of vector control measures rather than spending resources on mass drug campaigns that are predicted to have little effect without well-implemented vector control already in place.
Human movement within the region should be targeted to achieve elimination, as should the importation of infections from outside the region. This is because both impact the likelihood of achieving elimination and understanding regional movement patterns can help guide strategies on targeting specific groups of at-risk people.
While no sub-Saharan African country has yet eliminated malaria, the researchers predict that regional malaria elimination is within reach with current tools, provided the efficacy and operational efficiency attained in southern Zambia can be extended and targeted to other key areas. ![]()
Kariba area of southern Zambia
where malaria elimination
programs are underway.
Photo from Milen Nikolov
Malaria elimination in historically high transmission areas like southern Africa is possible with tools that are already available, according to new research.
The study suggests that high levels of vector control are key, and mass drug campaigns cannot make much of an impact without proper vector control.
Milen Nikolov, of the Institute for Disease Modeling in Bellevue, Washington, and colleagues reported these findings in PLOS Computational Biology.
The researchers said the Lake Kariba region of Southern Province, Zambia, is part of a multi-country malaria elimination effort. However, elimination in this area is challenging because villages with high and low malaria burden are interconnected through human travel.
With this in mind, the researchers combined a mathematical model of malaria transmission with field data from Zambia to test a variety of strategies for eliminating malaria in the Lake Kariba region.
The team used detailed spatial surveillance data from field studies—including household locations, climate, clinical malaria incidence, prevalence of malaria infections, and bednet usage rates—to construct a model of interconnected villages, then tested a variety of intervention scenarios to see which ones could lead to elimination.
The results indicate that elimination requires high, yet realistic, levels of vector control. And mass drug campaigns deployed to kill parasites in the human population can boost the chances of achieving elimination as long as vector control is well-implemented.
The researchers said this work suggests that elimination programs in sub-Saharan Africa should focus on how to achieve and maintain excellent coverage of vector control measures rather than spending resources on mass drug campaigns that are predicted to have little effect without well-implemented vector control already in place.
Human movement within the region should be targeted to achieve elimination, as should the importation of infections from outside the region. This is because both impact the likelihood of achieving elimination and understanding regional movement patterns can help guide strategies on targeting specific groups of at-risk people.
While no sub-Saharan African country has yet eliminated malaria, the researchers predict that regional malaria elimination is within reach with current tools, provided the efficacy and operational efficiency attained in southern Zambia can be extended and targeted to other key areas. ![]()
Kariba area of southern Zambia
where malaria elimination
programs are underway.
Photo from Milen Nikolov
Malaria elimination in historically high transmission areas like southern Africa is possible with tools that are already available, according to new research.
The study suggests that high levels of vector control are key, and mass drug campaigns cannot make much of an impact without proper vector control.
Milen Nikolov, of the Institute for Disease Modeling in Bellevue, Washington, and colleagues reported these findings in PLOS Computational Biology.
The researchers said the Lake Kariba region of Southern Province, Zambia, is part of a multi-country malaria elimination effort. However, elimination in this area is challenging because villages with high and low malaria burden are interconnected through human travel.
With this in mind, the researchers combined a mathematical model of malaria transmission with field data from Zambia to test a variety of strategies for eliminating malaria in the Lake Kariba region.
The team used detailed spatial surveillance data from field studies—including household locations, climate, clinical malaria incidence, prevalence of malaria infections, and bednet usage rates—to construct a model of interconnected villages, then tested a variety of intervention scenarios to see which ones could lead to elimination.
The results indicate that elimination requires high, yet realistic, levels of vector control. And mass drug campaigns deployed to kill parasites in the human population can boost the chances of achieving elimination as long as vector control is well-implemented.
The researchers said this work suggests that elimination programs in sub-Saharan Africa should focus on how to achieve and maintain excellent coverage of vector control measures rather than spending resources on mass drug campaigns that are predicted to have little effect without well-implemented vector control already in place.
Human movement within the region should be targeted to achieve elimination, as should the importation of infections from outside the region. This is because both impact the likelihood of achieving elimination and understanding regional movement patterns can help guide strategies on targeting specific groups of at-risk people.
While no sub-Saharan African country has yet eliminated malaria, the researchers predict that regional malaria elimination is within reach with current tools, provided the efficacy and operational efficiency attained in southern Zambia can be extended and targeted to other key areas. ![]()
VIDEO: Anti-CD22 CAR for R/R ALL impresses in early trial
SAN DIEGO – In a first-in-humans trial, chimeric antigen receptor (CAR) T-cell therapy directed against CD22 was shown to be safe and was associated with minimal residual disease (MRD)–negative complete remissions in 8 of 10 children and young adults with relapsed/refractory B-precursor acute lymphoblastic leukemia treated at the highest dose levels. One patient remains in remission more than 1 year of treatment, one had a 6-month remission, and one had a remission lasting 3 months.
In a video interview, co-principal investigator Terry J. Fry, MD, of the Center for Cancer Research at the National Cancer Institute in Bethesda, Md., discusses the rationale behind using an alternative antigen target in salvage therapy for ALL, and the potential for combining antigen targets to treat patients with relapsed/refractory ALL.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – In a first-in-humans trial, chimeric antigen receptor (CAR) T-cell therapy directed against CD22 was shown to be safe and was associated with minimal residual disease (MRD)–negative complete remissions in 8 of 10 children and young adults with relapsed/refractory B-precursor acute lymphoblastic leukemia treated at the highest dose levels. One patient remains in remission more than 1 year of treatment, one had a 6-month remission, and one had a remission lasting 3 months.
In a video interview, co-principal investigator Terry J. Fry, MD, of the Center for Cancer Research at the National Cancer Institute in Bethesda, Md., discusses the rationale behind using an alternative antigen target in salvage therapy for ALL, and the potential for combining antigen targets to treat patients with relapsed/refractory ALL.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – In a first-in-humans trial, chimeric antigen receptor (CAR) T-cell therapy directed against CD22 was shown to be safe and was associated with minimal residual disease (MRD)–negative complete remissions in 8 of 10 children and young adults with relapsed/refractory B-precursor acute lymphoblastic leukemia treated at the highest dose levels. One patient remains in remission more than 1 year of treatment, one had a 6-month remission, and one had a remission lasting 3 months.
In a video interview, co-principal investigator Terry J. Fry, MD, of the Center for Cancer Research at the National Cancer Institute in Bethesda, Md., discusses the rationale behind using an alternative antigen target in salvage therapy for ALL, and the potential for combining antigen targets to treat patients with relapsed/refractory ALL.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2016

ASH: Hemophilia B gene therapy posts strong update
SAN DIEGO – Patients with hemophilia B who received a single 1-hour infusion of the gene transfer therapy SPK-9001 achieved steady-state factor IX activity levels averaging 28% and persisting over 1,650 cumulative days of observation, according to updated results from a phase I/II trial.
All nine patients treated to date have exceeded the 12% steady-state factor IX activity level typically needed to prevent breakthrough bleeds, Katherine A. High, MD, said during a press briefing at the annual meeting of the American Society of Hematology. One patient infused himself once with factor IX after developing a suspected ankle bleed 2 days after treatment, Dr. High and her associates reported in the accompanying abstract.
This therapy works at a lower dose than previous factor IX gene transfer products and therefore has not caused the hepatotoxicity that halted their development, according to Dr. High, president and chief scientific officer of Spark Therapeutics, which makes SPK-9001. Two of nine patients developed an immune response to the viral capsid in the product, with a corresponding drop in factor IX activity levels, but the immune response was halted by tapering doses of corticosteroids, and patients maintained sufficient levels of factor IX activity to prevent breakthrough bleeds or the need for replacement factor, she said.
Because the virus capsid breaks down over time, a transient immune response to it “is not really a safety issue, but is an efficacy issue,” Dr. High emphasized. “If it is not caught in time, and patients are not given steroids promptly, they can lose the donated gene. Therefore, quick recognition is key.” Patients who develop an immune response to the viral capsid show sharp declines in factor IX activity levels, rises in baseline AST and ALT, and mononuclear cell reactivity, she explained during an interview.
The current standard of care for hemophilia B involves the cost and treatment burden of intravenous factor IX injections given one to three times weekly. Previous work evaluated factor IX gene transfer mediated by adeno-associated virus, but long-term factor IX activity levels did not reach the trough levels typically achieved with long-acting factor IX prophylaxis. Simply escalating the vector dose did not work because the viral capsid triggered immune-mediated hepatotoxicity, Dr. High noted.
To develop a more efficient product that works at lower doses, she and her associates created a recombinant vector containing a bioengineered adeno-associated virus capsid and a DNA sequence with a promoter designed to drive hepatic expression of a highly active variant of factor IX. To test the product, researchers in Mississippi, Pennsylvania, and California enrolled men aged 18-52 years with a confirmed diagnosis of hemophilia B (no more than 2 IU/dL or 2% endogenous factor IX) who had received at least 50 days of exposure to factor IX products and averaged at least four bleeding events per year requiring factor IX treatment or prophylaxis. Patients had no measurable inhibitory antibodies but otherwise represented the “general hemophilia B population,” Dr. High said. Five of nine patients had multiple target joints, liver disease associated with hepatitis C virus infection, or both. Each patient received a 1-hour infusion of 5 x 1011 vector genomes per body weight and was followed for 7-52 weeks.
Among seven patients who, by Nov. 30, 2016, had surpassed the 12 weeks needed to reach steady state factor IX expression levels, median steady-state level was 30% (range, 13%-38%), Dr. High reported. “Now we can give one quarter the dose [of adeno-associated virus vector] that was given before, and its driving factor IX expression levels five to eight times higher,” she concluded. Results for the first seven treated patients prompted Food and Drug Administration to give the product orphan drug designation in July 2016. Plans for phase III trials are underway, and researchers also are planning to investigate this approach to gene therapy in hemophilia A, Dr. High said.
Spark Therapeutics Inc. and Pfizer sponsored the study. Dr. High is president and chief scientific officer of Spark. Dr. George had no relevant financial disclosures.
SAN DIEGO – Patients with hemophilia B who received a single 1-hour infusion of the gene transfer therapy SPK-9001 achieved steady-state factor IX activity levels averaging 28% and persisting over 1,650 cumulative days of observation, according to updated results from a phase I/II trial.
All nine patients treated to date have exceeded the 12% steady-state factor IX activity level typically needed to prevent breakthrough bleeds, Katherine A. High, MD, said during a press briefing at the annual meeting of the American Society of Hematology. One patient infused himself once with factor IX after developing a suspected ankle bleed 2 days after treatment, Dr. High and her associates reported in the accompanying abstract.
This therapy works at a lower dose than previous factor IX gene transfer products and therefore has not caused the hepatotoxicity that halted their development, according to Dr. High, president and chief scientific officer of Spark Therapeutics, which makes SPK-9001. Two of nine patients developed an immune response to the viral capsid in the product, with a corresponding drop in factor IX activity levels, but the immune response was halted by tapering doses of corticosteroids, and patients maintained sufficient levels of factor IX activity to prevent breakthrough bleeds or the need for replacement factor, she said.
Because the virus capsid breaks down over time, a transient immune response to it “is not really a safety issue, but is an efficacy issue,” Dr. High emphasized. “If it is not caught in time, and patients are not given steroids promptly, they can lose the donated gene. Therefore, quick recognition is key.” Patients who develop an immune response to the viral capsid show sharp declines in factor IX activity levels, rises in baseline AST and ALT, and mononuclear cell reactivity, she explained during an interview.
The current standard of care for hemophilia B involves the cost and treatment burden of intravenous factor IX injections given one to three times weekly. Previous work evaluated factor IX gene transfer mediated by adeno-associated virus, but long-term factor IX activity levels did not reach the trough levels typically achieved with long-acting factor IX prophylaxis. Simply escalating the vector dose did not work because the viral capsid triggered immune-mediated hepatotoxicity, Dr. High noted.
To develop a more efficient product that works at lower doses, she and her associates created a recombinant vector containing a bioengineered adeno-associated virus capsid and a DNA sequence with a promoter designed to drive hepatic expression of a highly active variant of factor IX. To test the product, researchers in Mississippi, Pennsylvania, and California enrolled men aged 18-52 years with a confirmed diagnosis of hemophilia B (no more than 2 IU/dL or 2% endogenous factor IX) who had received at least 50 days of exposure to factor IX products and averaged at least four bleeding events per year requiring factor IX treatment or prophylaxis. Patients had no measurable inhibitory antibodies but otherwise represented the “general hemophilia B population,” Dr. High said. Five of nine patients had multiple target joints, liver disease associated with hepatitis C virus infection, or both. Each patient received a 1-hour infusion of 5 x 1011 vector genomes per body weight and was followed for 7-52 weeks.
Among seven patients who, by Nov. 30, 2016, had surpassed the 12 weeks needed to reach steady state factor IX expression levels, median steady-state level was 30% (range, 13%-38%), Dr. High reported. “Now we can give one quarter the dose [of adeno-associated virus vector] that was given before, and its driving factor IX expression levels five to eight times higher,” she concluded. Results for the first seven treated patients prompted Food and Drug Administration to give the product orphan drug designation in July 2016. Plans for phase III trials are underway, and researchers also are planning to investigate this approach to gene therapy in hemophilia A, Dr. High said.
Spark Therapeutics Inc. and Pfizer sponsored the study. Dr. High is president and chief scientific officer of Spark. Dr. George had no relevant financial disclosures.
SAN DIEGO – Patients with hemophilia B who received a single 1-hour infusion of the gene transfer therapy SPK-9001 achieved steady-state factor IX activity levels averaging 28% and persisting over 1,650 cumulative days of observation, according to updated results from a phase I/II trial.
All nine patients treated to date have exceeded the 12% steady-state factor IX activity level typically needed to prevent breakthrough bleeds, Katherine A. High, MD, said during a press briefing at the annual meeting of the American Society of Hematology. One patient infused himself once with factor IX after developing a suspected ankle bleed 2 days after treatment, Dr. High and her associates reported in the accompanying abstract.
This therapy works at a lower dose than previous factor IX gene transfer products and therefore has not caused the hepatotoxicity that halted their development, according to Dr. High, president and chief scientific officer of Spark Therapeutics, which makes SPK-9001. Two of nine patients developed an immune response to the viral capsid in the product, with a corresponding drop in factor IX activity levels, but the immune response was halted by tapering doses of corticosteroids, and patients maintained sufficient levels of factor IX activity to prevent breakthrough bleeds or the need for replacement factor, she said.
Because the virus capsid breaks down over time, a transient immune response to it “is not really a safety issue, but is an efficacy issue,” Dr. High emphasized. “If it is not caught in time, and patients are not given steroids promptly, they can lose the donated gene. Therefore, quick recognition is key.” Patients who develop an immune response to the viral capsid show sharp declines in factor IX activity levels, rises in baseline AST and ALT, and mononuclear cell reactivity, she explained during an interview.
The current standard of care for hemophilia B involves the cost and treatment burden of intravenous factor IX injections given one to three times weekly. Previous work evaluated factor IX gene transfer mediated by adeno-associated virus, but long-term factor IX activity levels did not reach the trough levels typically achieved with long-acting factor IX prophylaxis. Simply escalating the vector dose did not work because the viral capsid triggered immune-mediated hepatotoxicity, Dr. High noted.
To develop a more efficient product that works at lower doses, she and her associates created a recombinant vector containing a bioengineered adeno-associated virus capsid and a DNA sequence with a promoter designed to drive hepatic expression of a highly active variant of factor IX. To test the product, researchers in Mississippi, Pennsylvania, and California enrolled men aged 18-52 years with a confirmed diagnosis of hemophilia B (no more than 2 IU/dL or 2% endogenous factor IX) who had received at least 50 days of exposure to factor IX products and averaged at least four bleeding events per year requiring factor IX treatment or prophylaxis. Patients had no measurable inhibitory antibodies but otherwise represented the “general hemophilia B population,” Dr. High said. Five of nine patients had multiple target joints, liver disease associated with hepatitis C virus infection, or both. Each patient received a 1-hour infusion of 5 x 1011 vector genomes per body weight and was followed for 7-52 weeks.
Among seven patients who, by Nov. 30, 2016, had surpassed the 12 weeks needed to reach steady state factor IX expression levels, median steady-state level was 30% (range, 13%-38%), Dr. High reported. “Now we can give one quarter the dose [of adeno-associated virus vector] that was given before, and its driving factor IX expression levels five to eight times higher,” she concluded. Results for the first seven treated patients prompted Food and Drug Administration to give the product orphan drug designation in July 2016. Plans for phase III trials are underway, and researchers also are planning to investigate this approach to gene therapy in hemophilia A, Dr. High said.
Spark Therapeutics Inc. and Pfizer sponsored the study. Dr. High is president and chief scientific officer of Spark. Dr. George had no relevant financial disclosures.
AT ASH 2016
Key clinical point: Gene therapy with SPK-9001 continues to post strong results in patients with moderate to severe hemophilia B.
Major finding: As of Nov. 30, median steady-state factor IX levels were 30% (range, 13% to 38%). Two of nine patients developed an immune response to the adeno-associated virus capsid that appears to have been halted with tapering doses of corticosteroids.
Data source: An ongoing phase I/II trial of SPK-9001, dosed at 5 x 1011 vector genomes (vg)/kg body weight.
Disclosures: Spark Therapeutics Inc. and Pfizer sponsored the work. Dr. High is president and chief scientific officer of Spark. Dr. George had no relevant financial disclosures.
VIDEO: Hemophilia B gene therapy maintains factor IX levels averaging 28%
SAN DIEGO – Patients with hemophilia B who received a single infusion of the gene transfer therapy SPK-9001 achieved steady-state factor IX activity levels averaging 28% and persisting over 1,650 cumulative days of observation, according to updated results from a phase I/II trial.
All nine patients treated to date have exceeded the steady-state factor IX activity level typically needed to prevent breakthrough bleeds, Katherine A. High, MD, reported at the American Society of Hematology. There have been no confirmed bleeds, all patients remain off prophylactic factor IX, none have developed factor IX inhibitory antibodies, and Enzyme-Linked ImmunoSpot testing has uncovered no evidence of emergent reactivity to the gene product. Two patients developed an immune response to the viral capsid in the product, with a corresponding drop in factor IX activity levels. Tapering doses of corticosteroids halted the immune response and patients maintained sufficient levels of factor IX activity to prevent breakthrough bleeds or the need for replacement factor.
Spark Therapeutics Inc. and Pfizer sponsored the work. Dr. High is president and chief scientific officer of Spark. She discussed the trial in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – Patients with hemophilia B who received a single infusion of the gene transfer therapy SPK-9001 achieved steady-state factor IX activity levels averaging 28% and persisting over 1,650 cumulative days of observation, according to updated results from a phase I/II trial.
All nine patients treated to date have exceeded the steady-state factor IX activity level typically needed to prevent breakthrough bleeds, Katherine A. High, MD, reported at the American Society of Hematology. There have been no confirmed bleeds, all patients remain off prophylactic factor IX, none have developed factor IX inhibitory antibodies, and Enzyme-Linked ImmunoSpot testing has uncovered no evidence of emergent reactivity to the gene product. Two patients developed an immune response to the viral capsid in the product, with a corresponding drop in factor IX activity levels. Tapering doses of corticosteroids halted the immune response and patients maintained sufficient levels of factor IX activity to prevent breakthrough bleeds or the need for replacement factor.
Spark Therapeutics Inc. and Pfizer sponsored the work. Dr. High is president and chief scientific officer of Spark. She discussed the trial in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – Patients with hemophilia B who received a single infusion of the gene transfer therapy SPK-9001 achieved steady-state factor IX activity levels averaging 28% and persisting over 1,650 cumulative days of observation, according to updated results from a phase I/II trial.
All nine patients treated to date have exceeded the steady-state factor IX activity level typically needed to prevent breakthrough bleeds, Katherine A. High, MD, reported at the American Society of Hematology. There have been no confirmed bleeds, all patients remain off prophylactic factor IX, none have developed factor IX inhibitory antibodies, and Enzyme-Linked ImmunoSpot testing has uncovered no evidence of emergent reactivity to the gene product. Two patients developed an immune response to the viral capsid in the product, with a corresponding drop in factor IX activity levels. Tapering doses of corticosteroids halted the immune response and patients maintained sufficient levels of factor IX activity to prevent breakthrough bleeds or the need for replacement factor.
Spark Therapeutics Inc. and Pfizer sponsored the work. Dr. High is president and chief scientific officer of Spark. She discussed the trial in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2016

ASH: Novel microcapsules show promise in hemophilia A with inhibitory antibodies
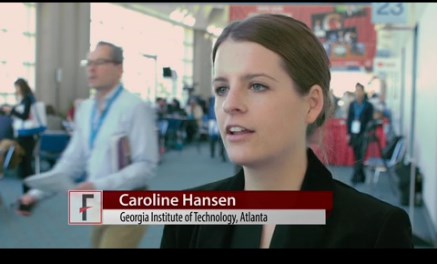
SAN DIEGO – Novel microcapsules loaded with factor VIII outperformed systemic factor VIII infusions in a model of hemophilia A with inhibitory antibodies, Caroline E. Hansen reported at the annual meeting of the American Society of Hematology.
“This is a completely new paradigm that uses platelet biomechanics to target and deliver a drug,” Ms. Hansen said at a press briefing.
To create the microcapsules, the investigators deposited alternatingly charged layers of polyelectrolytes, poly-L-lysine, and poly-L-glutamic acid onto a calcium carbonate core covered with factor VIII and dextran. They added fibrinogen to the final polyelectrolyte layer and then chelated out the innermost core, leaving the dextran layer as a shield between factor VIII and the outside of the microcapsule. Initial in vitro experiments showed that the microcapsules adhered to platelets and were incorporated into fibrin networks when platelets were activated, Ms. Hansen reported. Because the microcapsules only ruptured upon platelet contraction, factor VIII was only delivered to actively forming clots as intended, she added.
As a next step, the researchers perfused recalcified whole blood and platelet-poor plasma into a collagen and tissue factor patch designed to mimic vascular injury, and then measured fibrin fluorescence on the patch. Microcapsules lacking dextran, fibrinogen, or loaded factor VIII did not work – a treated sample and a phosphate-buffered saline (PBS) control yielded statistically similar fibrin production. However, complete microcapsules loaded with 0.01 U/mL factor VIII produced four times more fibrin than systemic infusion of 0.05 U/mL factor VIII.
“These were really promising results but we want to take a step back and see if a clot would form in the presence of inhibitory antibodies,” Ms. Hansen said. Accordingly, they added factor VIII inhibitory antibody 2-76 into blood samples from healthy donors. The microcapsules triggered 2.7 times more fibrin production in this setting than systemic treatment did (P less than .05). “This increased efficacy is likely due to the microcapsule shielding effect on factor VIII, preventing exposure to inhibitory antibodies,” Ms. Hansen and her associates concluded in their abstract.
The investigators are now studying the extent to which the microcapsules induce thrombin production, and how agents such as blebbistatin, ROCK, and myosin affect platelet contraction force and the efficiency of the microcapsule.
Ms. Hansen had no disclosures.
SAN DIEGO – Novel microcapsules loaded with factor VIII outperformed systemic factor VIII infusions in a model of hemophilia A with inhibitory antibodies, Caroline E. Hansen reported at the annual meeting of the American Society of Hematology.
“This is a completely new paradigm that uses platelet biomechanics to target and deliver a drug,” Ms. Hansen said at a press briefing.
To create the microcapsules, the investigators deposited alternatingly charged layers of polyelectrolytes, poly-L-lysine, and poly-L-glutamic acid onto a calcium carbonate core covered with factor VIII and dextran. They added fibrinogen to the final polyelectrolyte layer and then chelated out the innermost core, leaving the dextran layer as a shield between factor VIII and the outside of the microcapsule. Initial in vitro experiments showed that the microcapsules adhered to platelets and were incorporated into fibrin networks when platelets were activated, Ms. Hansen reported. Because the microcapsules only ruptured upon platelet contraction, factor VIII was only delivered to actively forming clots as intended, she added.
As a next step, the researchers perfused recalcified whole blood and platelet-poor plasma into a collagen and tissue factor patch designed to mimic vascular injury, and then measured fibrin fluorescence on the patch. Microcapsules lacking dextran, fibrinogen, or loaded factor VIII did not work – a treated sample and a phosphate-buffered saline (PBS) control yielded statistically similar fibrin production. However, complete microcapsules loaded with 0.01 U/mL factor VIII produced four times more fibrin than systemic infusion of 0.05 U/mL factor VIII.
“These were really promising results but we want to take a step back and see if a clot would form in the presence of inhibitory antibodies,” Ms. Hansen said. Accordingly, they added factor VIII inhibitory antibody 2-76 into blood samples from healthy donors. The microcapsules triggered 2.7 times more fibrin production in this setting than systemic treatment did (P less than .05). “This increased efficacy is likely due to the microcapsule shielding effect on factor VIII, preventing exposure to inhibitory antibodies,” Ms. Hansen and her associates concluded in their abstract.
The investigators are now studying the extent to which the microcapsules induce thrombin production, and how agents such as blebbistatin, ROCK, and myosin affect platelet contraction force and the efficiency of the microcapsule.
Ms. Hansen had no disclosures.
SAN DIEGO – Novel microcapsules loaded with factor VIII outperformed systemic factor VIII infusions in a model of hemophilia A with inhibitory antibodies, Caroline E. Hansen reported at the annual meeting of the American Society of Hematology.
“This is a completely new paradigm that uses platelet biomechanics to target and deliver a drug,” Ms. Hansen said at a press briefing.
To create the microcapsules, the investigators deposited alternatingly charged layers of polyelectrolytes, poly-L-lysine, and poly-L-glutamic acid onto a calcium carbonate core covered with factor VIII and dextran. They added fibrinogen to the final polyelectrolyte layer and then chelated out the innermost core, leaving the dextran layer as a shield between factor VIII and the outside of the microcapsule. Initial in vitro experiments showed that the microcapsules adhered to platelets and were incorporated into fibrin networks when platelets were activated, Ms. Hansen reported. Because the microcapsules only ruptured upon platelet contraction, factor VIII was only delivered to actively forming clots as intended, she added.
As a next step, the researchers perfused recalcified whole blood and platelet-poor plasma into a collagen and tissue factor patch designed to mimic vascular injury, and then measured fibrin fluorescence on the patch. Microcapsules lacking dextran, fibrinogen, or loaded factor VIII did not work – a treated sample and a phosphate-buffered saline (PBS) control yielded statistically similar fibrin production. However, complete microcapsules loaded with 0.01 U/mL factor VIII produced four times more fibrin than systemic infusion of 0.05 U/mL factor VIII.
“These were really promising results but we want to take a step back and see if a clot would form in the presence of inhibitory antibodies,” Ms. Hansen said. Accordingly, they added factor VIII inhibitory antibody 2-76 into blood samples from healthy donors. The microcapsules triggered 2.7 times more fibrin production in this setting than systemic treatment did (P less than .05). “This increased efficacy is likely due to the microcapsule shielding effect on factor VIII, preventing exposure to inhibitory antibodies,” Ms. Hansen and her associates concluded in their abstract.
The investigators are now studying the extent to which the microcapsules induce thrombin production, and how agents such as blebbistatin, ROCK, and myosin affect platelet contraction force and the efficiency of the microcapsule.
Ms. Hansen had no disclosures.
AT ASH 2016
Key clinical point: Novel microcapsules loaded with factor VIII outperformed systemic factor VIII infusions in an in vitro model of hemophilia A with inhibitory antibodies.
Major finding: In an in vitro model of this disease state, the microcapsules triggered 2.7 times more fibrin production than systemic treatment with factor VIII (P less than .05).
Data source: A multicenter laboratory study.
Disclosures: Ms. Hansen had no relevant financial disclosures.
VIDEO: Novel microcapsules show promise in hemophilia A with inhibitory antibodies
SAN DIEGO – Novel microcapsules loaded with factor VIII outperformed systemic factor VIII infusions in models of hemophilia A with inhibitory antibodies, Caroline E. Hansen reported at the annual meeting of the American Society of Hematology.
“This is a completely new paradigm that uses platelet biomechanics to target and deliver a drug,” said Ms. Hansen of Georgia Institute of Technology, Atlanta.
The microcapsules are designed to mechanically shield factor VIII from the immune system. When they reached a modeled site of vascular injury, they contracted and released factor VIII. Initial work showed that this approach triggered significantly more fibrin production in a developing clot than did systemic infusions of factor VIII.
Ms. Hansen had no disclosures. She discussed the findings in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – Novel microcapsules loaded with factor VIII outperformed systemic factor VIII infusions in models of hemophilia A with inhibitory antibodies, Caroline E. Hansen reported at the annual meeting of the American Society of Hematology.
“This is a completely new paradigm that uses platelet biomechanics to target and deliver a drug,” said Ms. Hansen of Georgia Institute of Technology, Atlanta.
The microcapsules are designed to mechanically shield factor VIII from the immune system. When they reached a modeled site of vascular injury, they contracted and released factor VIII. Initial work showed that this approach triggered significantly more fibrin production in a developing clot than did systemic infusions of factor VIII.
Ms. Hansen had no disclosures. She discussed the findings in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – Novel microcapsules loaded with factor VIII outperformed systemic factor VIII infusions in models of hemophilia A with inhibitory antibodies, Caroline E. Hansen reported at the annual meeting of the American Society of Hematology.
“This is a completely new paradigm that uses platelet biomechanics to target and deliver a drug,” said Ms. Hansen of Georgia Institute of Technology, Atlanta.
The microcapsules are designed to mechanically shield factor VIII from the immune system. When they reached a modeled site of vascular injury, they contracted and released factor VIII. Initial work showed that this approach triggered significantly more fibrin production in a developing clot than did systemic infusions of factor VIII.
Ms. Hansen had no disclosures. She discussed the findings in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2016
Key clinical point: Novel microcapsules loaded with factor VIII outperformed systemic factor VIII infusions in an in vitro model of hemophilia A with inhibitory antibodies.
Major finding: In an in vitro model, the microcapsules triggered 2.7 times more fibrin production than systemic treatment with factor VIII (P less than .05).
Data source: A multicenter laboratory study.
Disclosures: Ms. Hansen had no relevant financial disclosures.
Phase II trial: Drug reduces sickle cell ‘pain crises’
An industry-funded phase II trial has shown that high doses of the experimental drug crizanlizumab significantly reduced the number of dangerous “pain crises” in subjects with sickle cell disease.
The median per-year rate of pain crises was 45.3% lower among those who took the high dose of crizanlizumab, compared with the placebo group (P = .01) More than a third of the subjects who took the high dose reported no pain crises during the treatment phase, more than double the rate among the placebo group.
The trial findings were released at the annual meeting of the American Society of Hematology and published simultaneously in the New England Journal of Medicine (doi: 10.1056/NEJMoa1611770).
The American Society of Hematology estimates that 70,000-100,000 people in the United States have sickle cell anemia and some patients are treated with hydroxyurea (Hydrea) are available. According to background material provided in the trial report, however, hydroxyurea has limited value, and some patients still face the prospect of pain crises which can lead to end-organ damage, and early death.
The SUSTAIN trial focuses on pain crises, also known as vaso-occlusive and sickle cell crises, which can occur without warning when sickle cells block blood flow and decrease oxygen delivery.
Researchers led by Kenneth I. Ataga, MB, of the University of North Carolina, Chapel Hill, recruited 198 subjects who had sickle cell disease and who had experienced 2-10 pain crises related to their condition over the past year. They randomly assigned 67 subjects to receive a low 2.5-mg/kg dose of crizanlizumab (also known as SelG1), 66 to a high 5.0-mg/kg dose, and 65 to a placebo. Crizanlizumab is an antibody against the molecule P-selectin, whose up-regulation in certain cells and platelets is thought to contribute to vaso-occlusion and sickle cell pain crises.
All the doses were administered intravenously 14 times over a year at sites in Brazil, the United States, and Jamaica. Risk groups for sickle cell include people of African and South American descent, among groups.
The first two doses were loading doses given at 2-week intervals, and the rest were given at 4-week intervals.
Subjects were aged 16-63 years; the median age was 29 for the two crizanlizumab groups and 26 for the placebo group. The percentage of black subjects ranged from 90% to 94% in each group, and the percentage of female subjects ranged from 52% to 58%.
Some subjects, but not all, were taking hydroxyurea. If they were taking the drug, they needed to have been on it for at least 6 months prior to the trial, and at least the last 3 months at a steady dose. Those who didn’t take hydroxyurea weren’t allowed to start taking it.
The researchers found that the median number of pain crises per year was 1.63 in the high-dose group, 2.01 in the low-dose group, and 2.98 in the placebo group. That translates to a 45.3% lower rate for the high-dose group than placebo (P = .01) and a 32.6% lower rate for low-dose than placebo (P = .18).
A total of 36% of the subjects in the high-dose group had no pain crises during the treatment phase, compared with 18% and 17% in the low-dose and placebo groups, respectively.
In a per-protocol analysis of 125 subjects, the researchers found similar numbers for median pain crises and no pain crises with one exception: The rate of annual pain crises was only 8.3% lower for the low-dose group than the placebo (P = .13).
Overall, the researchers wrote, the rates of adverse and serious adverse events were “similar” among all the subjects regardless of their randomized group.
Five patients died during the trial: two from the high dose group, one in the low dose group, and two in the placebo group. Among serious adverse events, pyrexia and pneumonia occurred more frequently in at least one of the crizanlizumab groups than in the placebo group, but their levels were low at zero to three cases of each event in the three groups.
The researchers noted that they didn’t detect any antibody response against crizanlizumab. However, “longer follow-up and monitoring are necessary to ensure that late neutralizing antibodies do not emerge that might limit the ability to administer crizanlizumab on a long-term basis.”
The study was funded by Selexys Pharmaceuticals, which received grants from the National Heart, Lung, and Blood Institute and the Food and Drug Administration’s Orphan Products Grant Program. Dr. Ataga reports personal fees from Selexys Pharmaceuticals. The other authors report various disclosures or none. The complete list of disclosures is available at NEJM.org.
An industry-funded phase II trial has shown that high doses of the experimental drug crizanlizumab significantly reduced the number of dangerous “pain crises” in subjects with sickle cell disease.
The median per-year rate of pain crises was 45.3% lower among those who took the high dose of crizanlizumab, compared with the placebo group (P = .01) More than a third of the subjects who took the high dose reported no pain crises during the treatment phase, more than double the rate among the placebo group.
The trial findings were released at the annual meeting of the American Society of Hematology and published simultaneously in the New England Journal of Medicine (doi: 10.1056/NEJMoa1611770).
The American Society of Hematology estimates that 70,000-100,000 people in the United States have sickle cell anemia and some patients are treated with hydroxyurea (Hydrea) are available. According to background material provided in the trial report, however, hydroxyurea has limited value, and some patients still face the prospect of pain crises which can lead to end-organ damage, and early death.
The SUSTAIN trial focuses on pain crises, also known as vaso-occlusive and sickle cell crises, which can occur without warning when sickle cells block blood flow and decrease oxygen delivery.
Researchers led by Kenneth I. Ataga, MB, of the University of North Carolina, Chapel Hill, recruited 198 subjects who had sickle cell disease and who had experienced 2-10 pain crises related to their condition over the past year. They randomly assigned 67 subjects to receive a low 2.5-mg/kg dose of crizanlizumab (also known as SelG1), 66 to a high 5.0-mg/kg dose, and 65 to a placebo. Crizanlizumab is an antibody against the molecule P-selectin, whose up-regulation in certain cells and platelets is thought to contribute to vaso-occlusion and sickle cell pain crises.
All the doses were administered intravenously 14 times over a year at sites in Brazil, the United States, and Jamaica. Risk groups for sickle cell include people of African and South American descent, among groups.
The first two doses were loading doses given at 2-week intervals, and the rest were given at 4-week intervals.
Subjects were aged 16-63 years; the median age was 29 for the two crizanlizumab groups and 26 for the placebo group. The percentage of black subjects ranged from 90% to 94% in each group, and the percentage of female subjects ranged from 52% to 58%.
Some subjects, but not all, were taking hydroxyurea. If they were taking the drug, they needed to have been on it for at least 6 months prior to the trial, and at least the last 3 months at a steady dose. Those who didn’t take hydroxyurea weren’t allowed to start taking it.
The researchers found that the median number of pain crises per year was 1.63 in the high-dose group, 2.01 in the low-dose group, and 2.98 in the placebo group. That translates to a 45.3% lower rate for the high-dose group than placebo (P = .01) and a 32.6% lower rate for low-dose than placebo (P = .18).
A total of 36% of the subjects in the high-dose group had no pain crises during the treatment phase, compared with 18% and 17% in the low-dose and placebo groups, respectively.
In a per-protocol analysis of 125 subjects, the researchers found similar numbers for median pain crises and no pain crises with one exception: The rate of annual pain crises was only 8.3% lower for the low-dose group than the placebo (P = .13).
Overall, the researchers wrote, the rates of adverse and serious adverse events were “similar” among all the subjects regardless of their randomized group.
Five patients died during the trial: two from the high dose group, one in the low dose group, and two in the placebo group. Among serious adverse events, pyrexia and pneumonia occurred more frequently in at least one of the crizanlizumab groups than in the placebo group, but their levels were low at zero to three cases of each event in the three groups.
The researchers noted that they didn’t detect any antibody response against crizanlizumab. However, “longer follow-up and monitoring are necessary to ensure that late neutralizing antibodies do not emerge that might limit the ability to administer crizanlizumab on a long-term basis.”
The study was funded by Selexys Pharmaceuticals, which received grants from the National Heart, Lung, and Blood Institute and the Food and Drug Administration’s Orphan Products Grant Program. Dr. Ataga reports personal fees from Selexys Pharmaceuticals. The other authors report various disclosures or none. The complete list of disclosures is available at NEJM.org.
An industry-funded phase II trial has shown that high doses of the experimental drug crizanlizumab significantly reduced the number of dangerous “pain crises” in subjects with sickle cell disease.
The median per-year rate of pain crises was 45.3% lower among those who took the high dose of crizanlizumab, compared with the placebo group (P = .01) More than a third of the subjects who took the high dose reported no pain crises during the treatment phase, more than double the rate among the placebo group.
The trial findings were released at the annual meeting of the American Society of Hematology and published simultaneously in the New England Journal of Medicine (doi: 10.1056/NEJMoa1611770).
The American Society of Hematology estimates that 70,000-100,000 people in the United States have sickle cell anemia and some patients are treated with hydroxyurea (Hydrea) are available. According to background material provided in the trial report, however, hydroxyurea has limited value, and some patients still face the prospect of pain crises which can lead to end-organ damage, and early death.
The SUSTAIN trial focuses on pain crises, also known as vaso-occlusive and sickle cell crises, which can occur without warning when sickle cells block blood flow and decrease oxygen delivery.
Researchers led by Kenneth I. Ataga, MB, of the University of North Carolina, Chapel Hill, recruited 198 subjects who had sickle cell disease and who had experienced 2-10 pain crises related to their condition over the past year. They randomly assigned 67 subjects to receive a low 2.5-mg/kg dose of crizanlizumab (also known as SelG1), 66 to a high 5.0-mg/kg dose, and 65 to a placebo. Crizanlizumab is an antibody against the molecule P-selectin, whose up-regulation in certain cells and platelets is thought to contribute to vaso-occlusion and sickle cell pain crises.
All the doses were administered intravenously 14 times over a year at sites in Brazil, the United States, and Jamaica. Risk groups for sickle cell include people of African and South American descent, among groups.
The first two doses were loading doses given at 2-week intervals, and the rest were given at 4-week intervals.
Subjects were aged 16-63 years; the median age was 29 for the two crizanlizumab groups and 26 for the placebo group. The percentage of black subjects ranged from 90% to 94% in each group, and the percentage of female subjects ranged from 52% to 58%.
Some subjects, but not all, were taking hydroxyurea. If they were taking the drug, they needed to have been on it for at least 6 months prior to the trial, and at least the last 3 months at a steady dose. Those who didn’t take hydroxyurea weren’t allowed to start taking it.
The researchers found that the median number of pain crises per year was 1.63 in the high-dose group, 2.01 in the low-dose group, and 2.98 in the placebo group. That translates to a 45.3% lower rate for the high-dose group than placebo (P = .01) and a 32.6% lower rate for low-dose than placebo (P = .18).
A total of 36% of the subjects in the high-dose group had no pain crises during the treatment phase, compared with 18% and 17% in the low-dose and placebo groups, respectively.
In a per-protocol analysis of 125 subjects, the researchers found similar numbers for median pain crises and no pain crises with one exception: The rate of annual pain crises was only 8.3% lower for the low-dose group than the placebo (P = .13).
Overall, the researchers wrote, the rates of adverse and serious adverse events were “similar” among all the subjects regardless of their randomized group.
Five patients died during the trial: two from the high dose group, one in the low dose group, and two in the placebo group. Among serious adverse events, pyrexia and pneumonia occurred more frequently in at least one of the crizanlizumab groups than in the placebo group, but their levels were low at zero to three cases of each event in the three groups.
The researchers noted that they didn’t detect any antibody response against crizanlizumab. However, “longer follow-up and monitoring are necessary to ensure that late neutralizing antibodies do not emerge that might limit the ability to administer crizanlizumab on a long-term basis.”
The study was funded by Selexys Pharmaceuticals, which received grants from the National Heart, Lung, and Blood Institute and the Food and Drug Administration’s Orphan Products Grant Program. Dr. Ataga reports personal fees from Selexys Pharmaceuticals. The other authors report various disclosures or none. The complete list of disclosures is available at NEJM.org.
FROM ASH 2016
Key clinical point: High-dose crizanlizumab significantly lowers, but does not eliminate, dangerous ‘pain crises’ that strike sickle cell patients.
Major finding: Patients who took high-dose crizanlizumab had a median of 1.63 pain crises a year versus 2.98 for the placebo group. (P = .01)
Data source: A phase II, 12-month, multicenter, double-blind, randomized, placebo-controlled study of 198 patients with sickle cell disease; 129 subjects completed the trial.
Disclosures: The study was funded by Selexys Pharmaceuticals, which received grants from the National Heart, Lung, and Blood Institute and the FDA’s Orphan Products Grant Program. Dr. Ataga reports personal fees from Selexys Pharmaceuticals. The other authors report various disclosures or none. The complete list of disclosures is available at NEJM.org.
Empagliflozin first antidiabetes drug to gain cardioprotective indication
Empagliflozin is the first antidiabetes medication to be approved for reducing the risk of cardiovascular death in patients with type 2 diabetes and concomitant cardiovascular disease.
The Food and Drug Administration granted the new indication based on the EMPA-REG OUTCOME study, a postmarketing analysis that found that empagliflozin (Jardiance, Boehringer-Ingelheim) reduced the risk of cardiovascular death by 38% when added to standard-of-care type 2 diabetes therapy.
When empagliflozin was approved for type 2 diabetes in 2014, the FDA required an additional postmarketing study to examine its cardiovascular safety. The 48-month, open-label EMPA-REG enrolled more than 7,000 patients who had type 2 diabetes and a high risk of cardiovascular disease.
The study’s big surprise, however, was not empagliflozin’s safety, but its striking cardioprotective qualities. It reduced by 14% the risk of the primary endpoint, a composite of cardiovascular death, nonfatal stroke, and nonfatal myocardial infarction (N Engl J Med. 2015;373:2117-28).
When examined as individual outcomes in a secondary analysis, empagliflozin significantly reduced the risk of cardiovascular death by 38%. However, risk reductions on the other endpoints were not significant. Nevertheless, experts called empagliflozin’s cardiovascular benefit a potential game-changer for the clinical challenge of managing patients with both disorders.
But the drug barely squeaked by its June FDA approval hearing for the cardioprotective indication, receiving a split 12-11 endorsement from the Endocrinologic and Metabolic Drugs Advisory Committee. The major sticking point was that EMPA-REG was a test of empagliflozin’s cardiovascular safety, not its efficacy, and that cardiovascular death was not a prespecified endpoint.
Although there were no significant cardiovascular safety issues, empagliflozin has been associated with hypotension, serious urinary tract infection, acute kidney injury, and genital infections.
“Cardiovascular disease is a leading cause of death in adults with type 2 diabetes mellitus,” Jean-Marc Guettier, MD, director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research, wrote in a press statement. “Availability of antidiabetes therapies that can help people live longer by reducing the risk of cardiovascular death is an important advance for adults with type 2 diabetes.”
msullivan@frontlinemedcom.com
On Twitter @alz_gal
Empagliflozin is the first antidiabetes medication to be approved for reducing the risk of cardiovascular death in patients with type 2 diabetes and concomitant cardiovascular disease.
The Food and Drug Administration granted the new indication based on the EMPA-REG OUTCOME study, a postmarketing analysis that found that empagliflozin (Jardiance, Boehringer-Ingelheim) reduced the risk of cardiovascular death by 38% when added to standard-of-care type 2 diabetes therapy.
When empagliflozin was approved for type 2 diabetes in 2014, the FDA required an additional postmarketing study to examine its cardiovascular safety. The 48-month, open-label EMPA-REG enrolled more than 7,000 patients who had type 2 diabetes and a high risk of cardiovascular disease.
The study’s big surprise, however, was not empagliflozin’s safety, but its striking cardioprotective qualities. It reduced by 14% the risk of the primary endpoint, a composite of cardiovascular death, nonfatal stroke, and nonfatal myocardial infarction (N Engl J Med. 2015;373:2117-28).
When examined as individual outcomes in a secondary analysis, empagliflozin significantly reduced the risk of cardiovascular death by 38%. However, risk reductions on the other endpoints were not significant. Nevertheless, experts called empagliflozin’s cardiovascular benefit a potential game-changer for the clinical challenge of managing patients with both disorders.
But the drug barely squeaked by its June FDA approval hearing for the cardioprotective indication, receiving a split 12-11 endorsement from the Endocrinologic and Metabolic Drugs Advisory Committee. The major sticking point was that EMPA-REG was a test of empagliflozin’s cardiovascular safety, not its efficacy, and that cardiovascular death was not a prespecified endpoint.
Although there were no significant cardiovascular safety issues, empagliflozin has been associated with hypotension, serious urinary tract infection, acute kidney injury, and genital infections.
“Cardiovascular disease is a leading cause of death in adults with type 2 diabetes mellitus,” Jean-Marc Guettier, MD, director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research, wrote in a press statement. “Availability of antidiabetes therapies that can help people live longer by reducing the risk of cardiovascular death is an important advance for adults with type 2 diabetes.”
msullivan@frontlinemedcom.com
On Twitter @alz_gal
Empagliflozin is the first antidiabetes medication to be approved for reducing the risk of cardiovascular death in patients with type 2 diabetes and concomitant cardiovascular disease.
The Food and Drug Administration granted the new indication based on the EMPA-REG OUTCOME study, a postmarketing analysis that found that empagliflozin (Jardiance, Boehringer-Ingelheim) reduced the risk of cardiovascular death by 38% when added to standard-of-care type 2 diabetes therapy.
When empagliflozin was approved for type 2 diabetes in 2014, the FDA required an additional postmarketing study to examine its cardiovascular safety. The 48-month, open-label EMPA-REG enrolled more than 7,000 patients who had type 2 diabetes and a high risk of cardiovascular disease.
The study’s big surprise, however, was not empagliflozin’s safety, but its striking cardioprotective qualities. It reduced by 14% the risk of the primary endpoint, a composite of cardiovascular death, nonfatal stroke, and nonfatal myocardial infarction (N Engl J Med. 2015;373:2117-28).
When examined as individual outcomes in a secondary analysis, empagliflozin significantly reduced the risk of cardiovascular death by 38%. However, risk reductions on the other endpoints were not significant. Nevertheless, experts called empagliflozin’s cardiovascular benefit a potential game-changer for the clinical challenge of managing patients with both disorders.
But the drug barely squeaked by its June FDA approval hearing for the cardioprotective indication, receiving a split 12-11 endorsement from the Endocrinologic and Metabolic Drugs Advisory Committee. The major sticking point was that EMPA-REG was a test of empagliflozin’s cardiovascular safety, not its efficacy, and that cardiovascular death was not a prespecified endpoint.
Although there were no significant cardiovascular safety issues, empagliflozin has been associated with hypotension, serious urinary tract infection, acute kidney injury, and genital infections.
“Cardiovascular disease is a leading cause of death in adults with type 2 diabetes mellitus,” Jean-Marc Guettier, MD, director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research, wrote in a press statement. “Availability of antidiabetes therapies that can help people live longer by reducing the risk of cardiovascular death is an important advance for adults with type 2 diabetes.”
msullivan@frontlinemedcom.com
On Twitter @alz_gal
U.S. okay looms for third drug-coated PAD balloon
WASHINGTON – Good pivotal-trial performance of a drug-coated balloon for treating superficial femoral and popliteal artery stenoses raised the prospect that it might soon be the third drug-coated balloon on the U.S. market, creating an opportunity for lower prices and competitive improvements for an increasingly used device.
“Having another drug-coated balloon would be useful for several reasons,” commented William A. Gray, MD, during the Transcatheter Cardiovascular Therapeutics annual meeting. The competition should mean lower cost, and accumulating reports on performance might identify a specific drug-coated balloon as most effective. Drug-coated balloons for peripheral artery stenoses “have been introduced over the past 2 years, with a significant increase in use during that time. It’s still not a majority of patients, but it’s increasing,” said Dr. Gray, chief of the division of cardiovascular disease at Main Line Health and president of Main Line Health’s Lankenau Heart Institute in Wynnewood, Pa.
The ILLUMENATE pivotal trial enrolled 300 patients at 43 centers in the United States and Europe. Patients had Rutherford 2, 3 or 4 disease, and averaged about 69 years old. More than 60% had class 3 disease and another 30% had class 2 disease.
The study’s primary safety endpoint was freedom from device- or procedure-related death to 30 days, and freedom from clinically drived target lesion revascularization at 12 months, a 92% rate in the 200 patients who had PTA with the Stellarex drug-coated balloon and 83% in the 100 controls who had PTA with an uncoated balloon. This statistically significant eight percentage point difference met the prespecified criteria for safety superiority.
The two drug-coated balloons already approved for U.S. use are the Lutonix and the IN.PACT Admiral.
“All the drug-coated balloons have worked well. It’s pretty exciting to see them work. It will be interesting to compare them against each other. We need side-by-side comparisons,” commented Craig M. Walker, MD, an interventional cardiologist in Houma, La. and a discussant for Dr. Lyden’s report.
The ILLUMENATE Pivotal trial was funded by Spectranetics, the company that is developing the Stellarex drug-coated balloon. Dr. Lyden has been a consultant to Spectranetics and to Biomet, Endologix, and TVA Medical. He received research support from Spectranetics and several other companies. Dr. Gray has been a consultant to Abbott Vascular, Boston Scientific, Cook, Medtronic, and Shockwave. He has received research support from Gore and Intact Vascular. Dr. Walker has been a consultant to Spectranetics as well as to Abbott Vascular, Bard, Boston Scientific, Cook, Gore, and Medtronic.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
It’s good to have competition among various models of drug-eluting balloons because it will help drive costs down and help drive additional improvements in device design. We win by having a third good drug-coated balloon option available.
Drug-coated balloons are increasingly used in routine U.S. practice. A recent report showed that one of the drug-coated balloons already on the U.S. market outperformed balloon angioplasty out to 3 years of follow-up. Drug-coated balloons hold an advantage over stents by leaving nothing behind. Another attraction of drug-coated balloons is that they can potentially be used as an adjunct to additional interventions for complex lesions, such as atherectomy.
So far, we have not seen a clear winner for safety and efficacy among the two drug-coated balloons already on the U.S. market and this new drug-coated balloon, which may soon be the third option for U.S. practice. But there is no single class effect from these drug-coated balloons; they must be evaluated individually.
D. Christopher Metzger, MD, is an interventional cardiologist and director of cardiac and peripheral vascular catheterization labs at the Wellmont CVA Heart Institute in Kingsport, Tenn. He has been a consultant to and received honoraria from Abbott Vascular, Bard, and Medtronic. He made these comments in an interview.
It’s good to have competition among various models of drug-eluting balloons because it will help drive costs down and help drive additional improvements in device design. We win by having a third good drug-coated balloon option available.
Drug-coated balloons are increasingly used in routine U.S. practice. A recent report showed that one of the drug-coated balloons already on the U.S. market outperformed balloon angioplasty out to 3 years of follow-up. Drug-coated balloons hold an advantage over stents by leaving nothing behind. Another attraction of drug-coated balloons is that they can potentially be used as an adjunct to additional interventions for complex lesions, such as atherectomy.
So far, we have not seen a clear winner for safety and efficacy among the two drug-coated balloons already on the U.S. market and this new drug-coated balloon, which may soon be the third option for U.S. practice. But there is no single class effect from these drug-coated balloons; they must be evaluated individually.
D. Christopher Metzger, MD, is an interventional cardiologist and director of cardiac and peripheral vascular catheterization labs at the Wellmont CVA Heart Institute in Kingsport, Tenn. He has been a consultant to and received honoraria from Abbott Vascular, Bard, and Medtronic. He made these comments in an interview.
It’s good to have competition among various models of drug-eluting balloons because it will help drive costs down and help drive additional improvements in device design. We win by having a third good drug-coated balloon option available.
Drug-coated balloons are increasingly used in routine U.S. practice. A recent report showed that one of the drug-coated balloons already on the U.S. market outperformed balloon angioplasty out to 3 years of follow-up. Drug-coated balloons hold an advantage over stents by leaving nothing behind. Another attraction of drug-coated balloons is that they can potentially be used as an adjunct to additional interventions for complex lesions, such as atherectomy.
So far, we have not seen a clear winner for safety and efficacy among the two drug-coated balloons already on the U.S. market and this new drug-coated balloon, which may soon be the third option for U.S. practice. But there is no single class effect from these drug-coated balloons; they must be evaluated individually.
D. Christopher Metzger, MD, is an interventional cardiologist and director of cardiac and peripheral vascular catheterization labs at the Wellmont CVA Heart Institute in Kingsport, Tenn. He has been a consultant to and received honoraria from Abbott Vascular, Bard, and Medtronic. He made these comments in an interview.
WASHINGTON – Good pivotal-trial performance of a drug-coated balloon for treating superficial femoral and popliteal artery stenoses raised the prospect that it might soon be the third drug-coated balloon on the U.S. market, creating an opportunity for lower prices and competitive improvements for an increasingly used device.
“Having another drug-coated balloon would be useful for several reasons,” commented William A. Gray, MD, during the Transcatheter Cardiovascular Therapeutics annual meeting. The competition should mean lower cost, and accumulating reports on performance might identify a specific drug-coated balloon as most effective. Drug-coated balloons for peripheral artery stenoses “have been introduced over the past 2 years, with a significant increase in use during that time. It’s still not a majority of patients, but it’s increasing,” said Dr. Gray, chief of the division of cardiovascular disease at Main Line Health and president of Main Line Health’s Lankenau Heart Institute in Wynnewood, Pa.
The ILLUMENATE pivotal trial enrolled 300 patients at 43 centers in the United States and Europe. Patients had Rutherford 2, 3 or 4 disease, and averaged about 69 years old. More than 60% had class 3 disease and another 30% had class 2 disease.
The study’s primary safety endpoint was freedom from device- or procedure-related death to 30 days, and freedom from clinically drived target lesion revascularization at 12 months, a 92% rate in the 200 patients who had PTA with the Stellarex drug-coated balloon and 83% in the 100 controls who had PTA with an uncoated balloon. This statistically significant eight percentage point difference met the prespecified criteria for safety superiority.
The two drug-coated balloons already approved for U.S. use are the Lutonix and the IN.PACT Admiral.
“All the drug-coated balloons have worked well. It’s pretty exciting to see them work. It will be interesting to compare them against each other. We need side-by-side comparisons,” commented Craig M. Walker, MD, an interventional cardiologist in Houma, La. and a discussant for Dr. Lyden’s report.
The ILLUMENATE Pivotal trial was funded by Spectranetics, the company that is developing the Stellarex drug-coated balloon. Dr. Lyden has been a consultant to Spectranetics and to Biomet, Endologix, and TVA Medical. He received research support from Spectranetics and several other companies. Dr. Gray has been a consultant to Abbott Vascular, Boston Scientific, Cook, Medtronic, and Shockwave. He has received research support from Gore and Intact Vascular. Dr. Walker has been a consultant to Spectranetics as well as to Abbott Vascular, Bard, Boston Scientific, Cook, Gore, and Medtronic.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
WASHINGTON – Good pivotal-trial performance of a drug-coated balloon for treating superficial femoral and popliteal artery stenoses raised the prospect that it might soon be the third drug-coated balloon on the U.S. market, creating an opportunity for lower prices and competitive improvements for an increasingly used device.
“Having another drug-coated balloon would be useful for several reasons,” commented William A. Gray, MD, during the Transcatheter Cardiovascular Therapeutics annual meeting. The competition should mean lower cost, and accumulating reports on performance might identify a specific drug-coated balloon as most effective. Drug-coated balloons for peripheral artery stenoses “have been introduced over the past 2 years, with a significant increase in use during that time. It’s still not a majority of patients, but it’s increasing,” said Dr. Gray, chief of the division of cardiovascular disease at Main Line Health and president of Main Line Health’s Lankenau Heart Institute in Wynnewood, Pa.
The ILLUMENATE pivotal trial enrolled 300 patients at 43 centers in the United States and Europe. Patients had Rutherford 2, 3 or 4 disease, and averaged about 69 years old. More than 60% had class 3 disease and another 30% had class 2 disease.
The study’s primary safety endpoint was freedom from device- or procedure-related death to 30 days, and freedom from clinically drived target lesion revascularization at 12 months, a 92% rate in the 200 patients who had PTA with the Stellarex drug-coated balloon and 83% in the 100 controls who had PTA with an uncoated balloon. This statistically significant eight percentage point difference met the prespecified criteria for safety superiority.
The two drug-coated balloons already approved for U.S. use are the Lutonix and the IN.PACT Admiral.
“All the drug-coated balloons have worked well. It’s pretty exciting to see them work. It will be interesting to compare them against each other. We need side-by-side comparisons,” commented Craig M. Walker, MD, an interventional cardiologist in Houma, La. and a discussant for Dr. Lyden’s report.
The ILLUMENATE Pivotal trial was funded by Spectranetics, the company that is developing the Stellarex drug-coated balloon. Dr. Lyden has been a consultant to Spectranetics and to Biomet, Endologix, and TVA Medical. He received research support from Spectranetics and several other companies. Dr. Gray has been a consultant to Abbott Vascular, Boston Scientific, Cook, Medtronic, and Shockwave. He has received research support from Gore and Intact Vascular. Dr. Walker has been a consultant to Spectranetics as well as to Abbott Vascular, Bard, Boston Scientific, Cook, Gore, and Medtronic.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
Key clinical point:
Major finding: The primary efficacy endpoint occurred in 76% of patients in the drug-coated balloon arm and 58% of controls.
Data source: The ILLUMENATE pivotal trial, which enrolled 300 patients at 63 U.S. and European centers.
Disclosures: The ILLUMENATE pivotal trial was funded by Spectranetics, the company that is developing the Stellarex drug-coated balloon. Dr. Lyden has been a consultant to Spectranetics and to Biomet, Endologix, and TVA Medical. He received research support from Spectranetics and several other companies. Dr. Gray has been a consultant to Abbott Vascular, Boston Scientific, Cook, Medtronic, and Shockwave. He has received research support from Gore and Intact Vascular. Dr. Walker has been a consultant to Spectranetics as well as to Abbott Vascular, Bard, Boston Scientific, Cook, Gore, and Medtronic.