User login
Thyroid Cancer: Incidence on the Rise
Detection of thyroid cancer is widespread, increasing by about 4.5% annually. In the past year, approximately 64,300 new cases were identified. An estimated one in 100 people will be diagnosed with thyroid cancer during their lifetime, making it the eighth most common cancer in the United States.1
Incidental thyroid nodules found on carotid ultrasounds and other neck imaging may account for much of the increase; evaluation of these “incidentalomas” may account for the doubling incidence of thyroid cancer cases. (For more on thyroid nodules, see “To Cut or Not to Cut?” Clinician Reviews. 2016;26[8]:34-36.) If this pace continues, thyroid cancer may become the third most common cancer among women in the US by 2019.2
RISK FACTORS
Generally, women are diagnosed with thyroid cancer more frequently than men.3 Other risk factors include
- Age (40 to 60 in women; 60 to 80 in men; median age at diagnosis, 51)
- Inherited conditions, such as multiple endocrine neoplasia (MEN) or familial medullary and nonmedullary thyroid carcinoma
- Other cancers, including breast cancer and familial adenomatous polyposis
- Iodine deficiency
- Radiation exposure, particularly head and neck radiation in childhood. This can be through treatment of acne, tinea capitis, enlarged tonsils, or adenoids (usually prior to 1960); treatment of lymphoma, Wilms tumor, or neuroblastoma; or proximity to Chernobyl in 1986.1,2
BIOPSY RECOMMENDATIONS
While thyroid nodules are fairly common, only 7% to 15% of nodules are found to be malignant.2 However, all patients presenting with a palpable thyroid nodule should undergo thyroid ultrasound for further evaluation.
According to American Thyroid Association guidelines, all nodules 2 cm or larger should be evaluated with fine needle aspiration (FNA) due to a concern for metastatic thyroid cancer in larger nodules.2 Some clinicians prefer to aspirate nodules 1 cm or larger. Nodules that are smaller than 2 cm with sonographic features suspicious for thyroid cancer (see Table 1) should be biopsied.

Nodules that are spongiform in appearance or are completely cystic with no solid components may be monitored without FNA.2
The FNA is typically performed by an endocrinologist under ultrasound guidance. No anesthetic is required, but a topical ethyl chloride spray can assist with patient comfort. Three to four passes are made into the nodule with a 27-gauge needle; most patients describe pressure or a pinching sensation, rather than pain, during the procedure. After the procedure, ice applied to the FNA area may help with patient comfort.
TYPES OF THYROID CANCER
Four possible types of thyroid cancer are identified on pathology after FNA: papillary, follicular, medullary, and anaplastic. Differentiated thyroid cancers, which encompass papillary and follicular cancers, are the most commonly diagnosed. Approximately 90% of thyroid cancers fall into this category.2
In most cases of differentiated thyroid cancer, patients can be treated with thyroidectomy alone if the cancer remains confined to the thyroid.2 Just over two-thirds of differentiated thyroid cancer cases are localized in the thyroid. The five-year survival rate for these patients is nearly 100%.1
About 27% of differentiated thyroid cancer is also found in neck lymph nodes; these patients may be treated with thyroidectomy and radioactive iodine.2 The five-year survival rate in these cases is nearly 98%.1 Chemotherapy is generally not needed for differentiated thyroid cancers.
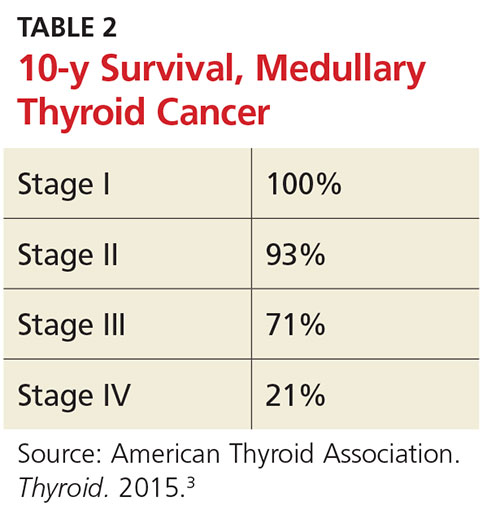
Medullary thyroid cancer (MTC) is diagnosed in up to 4% of thyroid cancer patients. Characterized by high levels of calcitonin, MTC can be genetically mediated or sporadic. MTC is associated with a variety of RET oncogene mutations; genetic testing of family members is recommended, as well as prophylactic thyroidectomy when high-risk RET oncogenes are detected.3
The 10-year survival prognosis for MTC patients varies according to stage at diagnosis (see Table 2). Up to 70% of patients with a palpable MTC nodule present with metastasis consistent with stage III or IV disease.3
Medullary thyroid cancer is treated with total thyroidectomy and cervical lymph node dissection. Radioactive iodine has not been proven effective for MTC patients, unless there is also papillary or follicular thyroid cancer present.3
Anaplastic thyroid cancer has the highest mortality rate of all types of thyroid cancer. Fortunately, it is relatively rare, occurring in only 1.7% of thyroid cancer patients. The one-year survival rate is 20%, with a median postdiagnosis survival prognosis of approximately five months. Anaplastic thyroid cancer is treated with total thyroidectomy and radical neck dissection when it is considered resectable. Metastatic lesions in the brain or spine are often indicators of unresectable disease. In some cases, external beam radiation therapy is used as palliative treatment.4
PEDIATRIC INCIDENCE
Thyroid cancer in children is rare, making up only 1.8% of all pediatric cancers diagnosed in the US annually. Patients are most often between ages 15 and 19, but it is possible for thyroid cancer to manifest in younger patients. Thyroid nodules are more likely to be malignant in children, with a greater incidence of metastatic disease at diagnosis. Prognosis is generally better in children than in adults, however, even with extensive disease.5
Children with prior history of other types of cancer treated with radiation, such as Hodgkin lymphoma or leukemia, are at increased risk for thyroid cancer and should be monitored.5 Children with a family history of MEN or MTC and evidence of RET oncogenes should be monitored starting as early as age 3 with thyroid exam, ultrasound, and measurement of calcitonin levels.3 Prophylactic thyroidectomy is an option in the first few months of life, depending on the presence of specific RET oncogenes.3
CHEMOTHERAPY
Chemotherapy may be helpful for metastatic medullary or anaplastic thyroid cancer, particularly in patients with unresectable disease. Though not usually curative, it may increase progression-free survival time. New chemotherapy agents approved for use in metastatic MTC include cabozantinib and vandetanib.3 Carboplatin, docetaxel, doxorubicin, and paclitaxel are used in treatment of anaplastic thyroid cancer.4
LONG-TERM PATIENT MANAGEMENT
After thyroidectomy and radioactive iodine treatment, follicular cell cancers (eg, papillary, follicular, anaplastic) are managed by following patients’ thyroid-stimulating hormone (TSH), thyroglobulin, and antithyroglobulin antibody levels. A cervical ultrasound is performed to detect possible disease in lymph nodes.2
Levothyroxine is dosed to suppress TSH below the recommended levels for hypothyroid patients in order to prevent disease recurrence. Low-risk patients may have TSH suppression below 1 to 2 mU/L, while high-risk patients may be managed with TSH levels below 0.1 mU/L.2
Lab levels should be checked annually and a cervical ultrasound performed at six to 12 months, then periodically thereafter depending on patient risk status.2 Patients with long-term TSH suppression must be monitored for atrial fibrillation and osteoporosis.
Patients who have been treated for medullary thyroid cancer require a different long-term management strategy. Patients should have ultrasound and measurement of TSH as well as calcitonin and carcinoembryonic antigen levels every six to 12 months.3 TSH suppression is not required; TSH may be maintained at typical euthyroid levels.
A FINAL THOUGHT
For clinicians, it’s easy to attempt to minimize thyroid cancer, since the disease is curable for most patients without the burden of chemotherapy and external radiation. However, for a patient, this is still a cancer diagnosis, with the accompanying surgery and required lifelong monitoring. It can be very disruptive to the lives of both patients and their families.
Support groups are available to help patients navigate their new reality. The Thyroid Cancer Survivors’ Association (www.thyca.org) has resources that may be beneficial to patients (and caregivers) as they learn how to live as a thyroid cancer survivor.
1. National Cancer Institute Surveillance, Epidemiology, and End Results Program. SEER stat fact sheets: thyroid cancer. http://seer.cancer.gov/statfacts/html/thyro.html. Accessed September 16, 2016.
2. Haugen BR, Alexander EK, Bible KC, et al; American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. 2015 American Thyroid Association guidelines for adult patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2016;26(1):1-133.
3. Wells SA Jr, Asa SL, Dralle H, et al; American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610.
4. Smallridge RC, Ain KB, Asa SL, et al; American Thyroid Association Anaplastic Thyroid Cancer Guidelines Taskforce. American Thyroid Association guidelines for the management of patients with anaplastic thyroid cancer. Thyroid. 2012;22(11):1104-1139.
5. Francis GL, Waguespack SG, Bauer AJ, et al; American Thyroid Association Guidelines Task Force. Management guidelines for children with thyroid nodules and differentiated thyroid cancer. Thyroid. 2015;25(7):716-759.
Clinician Reviews in partnership with
![]()
Melissa Murfin is an Assistant Professor and Academic Coordinator of the PA program at Elon University in North Carolina.
Clinician Reviews in partnership with
![]()
Melissa Murfin is an Assistant Professor and Academic Coordinator of the PA program at Elon University in North Carolina.
Clinician Reviews in partnership with
![]()
Melissa Murfin is an Assistant Professor and Academic Coordinator of the PA program at Elon University in North Carolina.
Detection of thyroid cancer is widespread, increasing by about 4.5% annually. In the past year, approximately 64,300 new cases were identified. An estimated one in 100 people will be diagnosed with thyroid cancer during their lifetime, making it the eighth most common cancer in the United States.1
Incidental thyroid nodules found on carotid ultrasounds and other neck imaging may account for much of the increase; evaluation of these “incidentalomas” may account for the doubling incidence of thyroid cancer cases. (For more on thyroid nodules, see “To Cut or Not to Cut?” Clinician Reviews. 2016;26[8]:34-36.) If this pace continues, thyroid cancer may become the third most common cancer among women in the US by 2019.2
RISK FACTORS
Generally, women are diagnosed with thyroid cancer more frequently than men.3 Other risk factors include
- Age (40 to 60 in women; 60 to 80 in men; median age at diagnosis, 51)
- Inherited conditions, such as multiple endocrine neoplasia (MEN) or familial medullary and nonmedullary thyroid carcinoma
- Other cancers, including breast cancer and familial adenomatous polyposis
- Iodine deficiency
- Radiation exposure, particularly head and neck radiation in childhood. This can be through treatment of acne, tinea capitis, enlarged tonsils, or adenoids (usually prior to 1960); treatment of lymphoma, Wilms tumor, or neuroblastoma; or proximity to Chernobyl in 1986.1,2
BIOPSY RECOMMENDATIONS
While thyroid nodules are fairly common, only 7% to 15% of nodules are found to be malignant.2 However, all patients presenting with a palpable thyroid nodule should undergo thyroid ultrasound for further evaluation.
According to American Thyroid Association guidelines, all nodules 2 cm or larger should be evaluated with fine needle aspiration (FNA) due to a concern for metastatic thyroid cancer in larger nodules.2 Some clinicians prefer to aspirate nodules 1 cm or larger. Nodules that are smaller than 2 cm with sonographic features suspicious for thyroid cancer (see Table 1) should be biopsied.
Nodules that are spongiform in appearance or are completely cystic with no solid components may be monitored without FNA.2
The FNA is typically performed by an endocrinologist under ultrasound guidance. No anesthetic is required, but a topical ethyl chloride spray can assist with patient comfort. Three to four passes are made into the nodule with a 27-gauge needle; most patients describe pressure or a pinching sensation, rather than pain, during the procedure. After the procedure, ice applied to the FNA area may help with patient comfort.
TYPES OF THYROID CANCER
Four possible types of thyroid cancer are identified on pathology after FNA: papillary, follicular, medullary, and anaplastic. Differentiated thyroid cancers, which encompass papillary and follicular cancers, are the most commonly diagnosed. Approximately 90% of thyroid cancers fall into this category.2
In most cases of differentiated thyroid cancer, patients can be treated with thyroidectomy alone if the cancer remains confined to the thyroid.2 Just over two-thirds of differentiated thyroid cancer cases are localized in the thyroid. The five-year survival rate for these patients is nearly 100%.1
About 27% of differentiated thyroid cancer is also found in neck lymph nodes; these patients may be treated with thyroidectomy and radioactive iodine.2 The five-year survival rate in these cases is nearly 98%.1 Chemotherapy is generally not needed for differentiated thyroid cancers.
Medullary thyroid cancer (MTC) is diagnosed in up to 4% of thyroid cancer patients. Characterized by high levels of calcitonin, MTC can be genetically mediated or sporadic. MTC is associated with a variety of RET oncogene mutations; genetic testing of family members is recommended, as well as prophylactic thyroidectomy when high-risk RET oncogenes are detected.3
The 10-year survival prognosis for MTC patients varies according to stage at diagnosis (see Table 2). Up to 70% of patients with a palpable MTC nodule present with metastasis consistent with stage III or IV disease.3
Medullary thyroid cancer is treated with total thyroidectomy and cervical lymph node dissection. Radioactive iodine has not been proven effective for MTC patients, unless there is also papillary or follicular thyroid cancer present.3
Anaplastic thyroid cancer has the highest mortality rate of all types of thyroid cancer. Fortunately, it is relatively rare, occurring in only 1.7% of thyroid cancer patients. The one-year survival rate is 20%, with a median postdiagnosis survival prognosis of approximately five months. Anaplastic thyroid cancer is treated with total thyroidectomy and radical neck dissection when it is considered resectable. Metastatic lesions in the brain or spine are often indicators of unresectable disease. In some cases, external beam radiation therapy is used as palliative treatment.4
PEDIATRIC INCIDENCE
Thyroid cancer in children is rare, making up only 1.8% of all pediatric cancers diagnosed in the US annually. Patients are most often between ages 15 and 19, but it is possible for thyroid cancer to manifest in younger patients. Thyroid nodules are more likely to be malignant in children, with a greater incidence of metastatic disease at diagnosis. Prognosis is generally better in children than in adults, however, even with extensive disease.5
Children with prior history of other types of cancer treated with radiation, such as Hodgkin lymphoma or leukemia, are at increased risk for thyroid cancer and should be monitored.5 Children with a family history of MEN or MTC and evidence of RET oncogenes should be monitored starting as early as age 3 with thyroid exam, ultrasound, and measurement of calcitonin levels.3 Prophylactic thyroidectomy is an option in the first few months of life, depending on the presence of specific RET oncogenes.3
CHEMOTHERAPY
Chemotherapy may be helpful for metastatic medullary or anaplastic thyroid cancer, particularly in patients with unresectable disease. Though not usually curative, it may increase progression-free survival time. New chemotherapy agents approved for use in metastatic MTC include cabozantinib and vandetanib.3 Carboplatin, docetaxel, doxorubicin, and paclitaxel are used in treatment of anaplastic thyroid cancer.4
LONG-TERM PATIENT MANAGEMENT
After thyroidectomy and radioactive iodine treatment, follicular cell cancers (eg, papillary, follicular, anaplastic) are managed by following patients’ thyroid-stimulating hormone (TSH), thyroglobulin, and antithyroglobulin antibody levels. A cervical ultrasound is performed to detect possible disease in lymph nodes.2
Levothyroxine is dosed to suppress TSH below the recommended levels for hypothyroid patients in order to prevent disease recurrence. Low-risk patients may have TSH suppression below 1 to 2 mU/L, while high-risk patients may be managed with TSH levels below 0.1 mU/L.2
Lab levels should be checked annually and a cervical ultrasound performed at six to 12 months, then periodically thereafter depending on patient risk status.2 Patients with long-term TSH suppression must be monitored for atrial fibrillation and osteoporosis.
Patients who have been treated for medullary thyroid cancer require a different long-term management strategy. Patients should have ultrasound and measurement of TSH as well as calcitonin and carcinoembryonic antigen levels every six to 12 months.3 TSH suppression is not required; TSH may be maintained at typical euthyroid levels.
A FINAL THOUGHT
For clinicians, it’s easy to attempt to minimize thyroid cancer, since the disease is curable for most patients without the burden of chemotherapy and external radiation. However, for a patient, this is still a cancer diagnosis, with the accompanying surgery and required lifelong monitoring. It can be very disruptive to the lives of both patients and their families.
Support groups are available to help patients navigate their new reality. The Thyroid Cancer Survivors’ Association (www.thyca.org) has resources that may be beneficial to patients (and caregivers) as they learn how to live as a thyroid cancer survivor.
Detection of thyroid cancer is widespread, increasing by about 4.5% annually. In the past year, approximately 64,300 new cases were identified. An estimated one in 100 people will be diagnosed with thyroid cancer during their lifetime, making it the eighth most common cancer in the United States.1
Incidental thyroid nodules found on carotid ultrasounds and other neck imaging may account for much of the increase; evaluation of these “incidentalomas” may account for the doubling incidence of thyroid cancer cases. (For more on thyroid nodules, see “To Cut or Not to Cut?” Clinician Reviews. 2016;26[8]:34-36.) If this pace continues, thyroid cancer may become the third most common cancer among women in the US by 2019.2
RISK FACTORS
Generally, women are diagnosed with thyroid cancer more frequently than men.3 Other risk factors include
- Age (40 to 60 in women; 60 to 80 in men; median age at diagnosis, 51)
- Inherited conditions, such as multiple endocrine neoplasia (MEN) or familial medullary and nonmedullary thyroid carcinoma
- Other cancers, including breast cancer and familial adenomatous polyposis
- Iodine deficiency
- Radiation exposure, particularly head and neck radiation in childhood. This can be through treatment of acne, tinea capitis, enlarged tonsils, or adenoids (usually prior to 1960); treatment of lymphoma, Wilms tumor, or neuroblastoma; or proximity to Chernobyl in 1986.1,2
BIOPSY RECOMMENDATIONS
While thyroid nodules are fairly common, only 7% to 15% of nodules are found to be malignant.2 However, all patients presenting with a palpable thyroid nodule should undergo thyroid ultrasound for further evaluation.
According to American Thyroid Association guidelines, all nodules 2 cm or larger should be evaluated with fine needle aspiration (FNA) due to a concern for metastatic thyroid cancer in larger nodules.2 Some clinicians prefer to aspirate nodules 1 cm or larger. Nodules that are smaller than 2 cm with sonographic features suspicious for thyroid cancer (see Table 1) should be biopsied.
Nodules that are spongiform in appearance or are completely cystic with no solid components may be monitored without FNA.2
The FNA is typically performed by an endocrinologist under ultrasound guidance. No anesthetic is required, but a topical ethyl chloride spray can assist with patient comfort. Three to four passes are made into the nodule with a 27-gauge needle; most patients describe pressure or a pinching sensation, rather than pain, during the procedure. After the procedure, ice applied to the FNA area may help with patient comfort.
TYPES OF THYROID CANCER
Four possible types of thyroid cancer are identified on pathology after FNA: papillary, follicular, medullary, and anaplastic. Differentiated thyroid cancers, which encompass papillary and follicular cancers, are the most commonly diagnosed. Approximately 90% of thyroid cancers fall into this category.2
In most cases of differentiated thyroid cancer, patients can be treated with thyroidectomy alone if the cancer remains confined to the thyroid.2 Just over two-thirds of differentiated thyroid cancer cases are localized in the thyroid. The five-year survival rate for these patients is nearly 100%.1
About 27% of differentiated thyroid cancer is also found in neck lymph nodes; these patients may be treated with thyroidectomy and radioactive iodine.2 The five-year survival rate in these cases is nearly 98%.1 Chemotherapy is generally not needed for differentiated thyroid cancers.
Medullary thyroid cancer (MTC) is diagnosed in up to 4% of thyroid cancer patients. Characterized by high levels of calcitonin, MTC can be genetically mediated or sporadic. MTC is associated with a variety of RET oncogene mutations; genetic testing of family members is recommended, as well as prophylactic thyroidectomy when high-risk RET oncogenes are detected.3
The 10-year survival prognosis for MTC patients varies according to stage at diagnosis (see Table 2). Up to 70% of patients with a palpable MTC nodule present with metastasis consistent with stage III or IV disease.3
Medullary thyroid cancer is treated with total thyroidectomy and cervical lymph node dissection. Radioactive iodine has not been proven effective for MTC patients, unless there is also papillary or follicular thyroid cancer present.3
Anaplastic thyroid cancer has the highest mortality rate of all types of thyroid cancer. Fortunately, it is relatively rare, occurring in only 1.7% of thyroid cancer patients. The one-year survival rate is 20%, with a median postdiagnosis survival prognosis of approximately five months. Anaplastic thyroid cancer is treated with total thyroidectomy and radical neck dissection when it is considered resectable. Metastatic lesions in the brain or spine are often indicators of unresectable disease. In some cases, external beam radiation therapy is used as palliative treatment.4
PEDIATRIC INCIDENCE
Thyroid cancer in children is rare, making up only 1.8% of all pediatric cancers diagnosed in the US annually. Patients are most often between ages 15 and 19, but it is possible for thyroid cancer to manifest in younger patients. Thyroid nodules are more likely to be malignant in children, with a greater incidence of metastatic disease at diagnosis. Prognosis is generally better in children than in adults, however, even with extensive disease.5
Children with prior history of other types of cancer treated with radiation, such as Hodgkin lymphoma or leukemia, are at increased risk for thyroid cancer and should be monitored.5 Children with a family history of MEN or MTC and evidence of RET oncogenes should be monitored starting as early as age 3 with thyroid exam, ultrasound, and measurement of calcitonin levels.3 Prophylactic thyroidectomy is an option in the first few months of life, depending on the presence of specific RET oncogenes.3
CHEMOTHERAPY
Chemotherapy may be helpful for metastatic medullary or anaplastic thyroid cancer, particularly in patients with unresectable disease. Though not usually curative, it may increase progression-free survival time. New chemotherapy agents approved for use in metastatic MTC include cabozantinib and vandetanib.3 Carboplatin, docetaxel, doxorubicin, and paclitaxel are used in treatment of anaplastic thyroid cancer.4
LONG-TERM PATIENT MANAGEMENT
After thyroidectomy and radioactive iodine treatment, follicular cell cancers (eg, papillary, follicular, anaplastic) are managed by following patients’ thyroid-stimulating hormone (TSH), thyroglobulin, and antithyroglobulin antibody levels. A cervical ultrasound is performed to detect possible disease in lymph nodes.2
Levothyroxine is dosed to suppress TSH below the recommended levels for hypothyroid patients in order to prevent disease recurrence. Low-risk patients may have TSH suppression below 1 to 2 mU/L, while high-risk patients may be managed with TSH levels below 0.1 mU/L.2
Lab levels should be checked annually and a cervical ultrasound performed at six to 12 months, then periodically thereafter depending on patient risk status.2 Patients with long-term TSH suppression must be monitored for atrial fibrillation and osteoporosis.
Patients who have been treated for medullary thyroid cancer require a different long-term management strategy. Patients should have ultrasound and measurement of TSH as well as calcitonin and carcinoembryonic antigen levels every six to 12 months.3 TSH suppression is not required; TSH may be maintained at typical euthyroid levels.
A FINAL THOUGHT
For clinicians, it’s easy to attempt to minimize thyroid cancer, since the disease is curable for most patients without the burden of chemotherapy and external radiation. However, for a patient, this is still a cancer diagnosis, with the accompanying surgery and required lifelong monitoring. It can be very disruptive to the lives of both patients and their families.
Support groups are available to help patients navigate their new reality. The Thyroid Cancer Survivors’ Association (www.thyca.org) has resources that may be beneficial to patients (and caregivers) as they learn how to live as a thyroid cancer survivor.
1. National Cancer Institute Surveillance, Epidemiology, and End Results Program. SEER stat fact sheets: thyroid cancer. http://seer.cancer.gov/statfacts/html/thyro.html. Accessed September 16, 2016.
2. Haugen BR, Alexander EK, Bible KC, et al; American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. 2015 American Thyroid Association guidelines for adult patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2016;26(1):1-133.
3. Wells SA Jr, Asa SL, Dralle H, et al; American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610.
4. Smallridge RC, Ain KB, Asa SL, et al; American Thyroid Association Anaplastic Thyroid Cancer Guidelines Taskforce. American Thyroid Association guidelines for the management of patients with anaplastic thyroid cancer. Thyroid. 2012;22(11):1104-1139.
5. Francis GL, Waguespack SG, Bauer AJ, et al; American Thyroid Association Guidelines Task Force. Management guidelines for children with thyroid nodules and differentiated thyroid cancer. Thyroid. 2015;25(7):716-759.
1. National Cancer Institute Surveillance, Epidemiology, and End Results Program. SEER stat fact sheets: thyroid cancer. http://seer.cancer.gov/statfacts/html/thyro.html. Accessed September 16, 2016.
2. Haugen BR, Alexander EK, Bible KC, et al; American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. 2015 American Thyroid Association guidelines for adult patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2016;26(1):1-133.
3. Wells SA Jr, Asa SL, Dralle H, et al; American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610.
4. Smallridge RC, Ain KB, Asa SL, et al; American Thyroid Association Anaplastic Thyroid Cancer Guidelines Taskforce. American Thyroid Association guidelines for the management of patients with anaplastic thyroid cancer. Thyroid. 2012;22(11):1104-1139.
5. Francis GL, Waguespack SG, Bauer AJ, et al; American Thyroid Association Guidelines Task Force. Management guidelines for children with thyroid nodules and differentiated thyroid cancer. Thyroid. 2015;25(7):716-759.
Sic transit gloria mundi
The email came with the words, “It is with sadness we report that Frank Moody died. …” I was instantly transported to the last time I saw the man and a flood of emotions swept over me. The name Frank Moody will ring a distant bell or none at all to some in our profession. Like many of the greats of surgery, he belongs to the ages.
I remember the first time I asked a student, “Who is Michael DeBakey?” I was dumbfounded to be greeted with a blank stare. How could a student of medicine not know of Dr. DeBakey? A few years later, the same question prompted a smart aleck reply that he was the man who invented DeBakey forceps. Well, of course he did invent the forceps, but to know nothing further of the man who was the world’s expert on ulcer disease in the 1940s, the progenitor of the National Medical Library, and among the foremost pioneers of heart surgery seemed beyond belief.

My mentor, Ernest Poulos, has long since left the active surgical scene. At times he would note the passing of one of his heroes like Carl Moyer (look it up!) and say, “Sic transit gloria mundi.” At 27 and anxious to get the right to cut into my fellow human beings, I would cock my head like a confounded puppy and wonder what that meant. I looked up the translation and meaning long ago, but now with age I understand the phrase in my bones.
I have long been a hanger-on at surgical meetings, hoping to meet those mighty figures that shaped surgical history. I saw W. Dean Warren once and had a very long hour with the great Mark Ravitch. Oliver Beahrs once performed magic tricks at a dinner I attended. At every surgical meeting there is an old guy (and now occasionally with the change in our profession, an elderly lady) getting on the bus to go to the reception or dinner dance. Often they are alone, their spouses having departed before them. As a young man, I wondered why the heck they came to the meetings. Just like every generation before, ours was eager to grab the reins, and in our ardor for future glory, we were polite but also restless for them to move aside. I hadn’t yet learned the importance of history and of listening.
What I missed while carousing with my young colleagues was an opportunity to hear history first hand and to learn that, what we thought was so cutting edge, these men and women had long ago considered. Many of our living legends imagined some of today’s innovations but they lacked the technology to bring their dreams to fruition, or time and age defeated them before they reached the final chapter of their research. It was when I was about 50 that I wised up and began seeking out living legends like Frank Moody and Frank Spencer.
In the case of Frank Moody, he was quite elderly when I first met him. For some reason, he knew who I was and shook my hand softly. I didn’t recognize him initially, but at the sound of his name, I knew I was in the presence of a major figure in 20th century gastrointestinal surgery. He had been at the University of California, San Francisco, during an historic time when George Sheldon, Donald Trunkey and other great surgeons trained there with J. Englebert Dunphy as their chief. Dr. Moody’s CV lists 141 articles in basic and clinical science that have had a profound impact on how we view the gastrointestinal tract. He was Chief at the University of Utah and the University of Alabama and finished his career as professor at the University of Texas-Houston. His awards and achievements were legion.
Parkinson’s had only recently really begun to affect him when I met him, and as the years went by his voice became so very faint that I had to lean in to hear him. We would sit together at the back of the dinner dance room so that we could hear each other. And while the other guests entertained themselves, Dr. Moody and I would discuss his life, scientific method and philosophy as well as his insights into his own case of Parkinsonism. I would see him at meetings, making his way slowly but steadily along a corridor while others briskly walked by, unaware that the man they just passed was among the most important surgical pioneers of our time. It was not sad that Dr. Moody was elderly and unrecognized, but that we younger surgeons missed knowing a great man in our tendency to rush past history.
History is not facts and dates, but rather, it is people and their lives. Yes, the history of our profession is embodied by pioneers like Frank Moody and the others I’ve mentioned.
We have many Fellows among us who are living history, still contributing – maybe not at the dais but at the dinner table, speaking softly and walking a bit slower than their juniors. Thanks to LaMar McGinnis who started it and Don Nakayama who continues it, the College has a History Community on the ACS Communities, an active Surgical History Group, and a will to acknowledge the history that lives and breathes among us. The Surgical History Group has organized a full program of events at the Clinical Congress and I hope many attendees take the opportunity to attend.
Take a moment at your next meeting or at the Clinical Congress and look for those historic surgeons still with us. Be smarter than I was at a young age and get to know them. You may learn something from them you can’t learn anyplace else.
Dr. Hughes is clinical professor in the department of surgery and director of medical education at the Kansas University School of Medicine, Salina Campus, and Co-Editor of ACS Surgery News.
The email came with the words, “It is with sadness we report that Frank Moody died. …” I was instantly transported to the last time I saw the man and a flood of emotions swept over me. The name Frank Moody will ring a distant bell or none at all to some in our profession. Like many of the greats of surgery, he belongs to the ages.
I remember the first time I asked a student, “Who is Michael DeBakey?” I was dumbfounded to be greeted with a blank stare. How could a student of medicine not know of Dr. DeBakey? A few years later, the same question prompted a smart aleck reply that he was the man who invented DeBakey forceps. Well, of course he did invent the forceps, but to know nothing further of the man who was the world’s expert on ulcer disease in the 1940s, the progenitor of the National Medical Library, and among the foremost pioneers of heart surgery seemed beyond belief.
My mentor, Ernest Poulos, has long since left the active surgical scene. At times he would note the passing of one of his heroes like Carl Moyer (look it up!) and say, “Sic transit gloria mundi.” At 27 and anxious to get the right to cut into my fellow human beings, I would cock my head like a confounded puppy and wonder what that meant. I looked up the translation and meaning long ago, but now with age I understand the phrase in my bones.
I have long been a hanger-on at surgical meetings, hoping to meet those mighty figures that shaped surgical history. I saw W. Dean Warren once and had a very long hour with the great Mark Ravitch. Oliver Beahrs once performed magic tricks at a dinner I attended. At every surgical meeting there is an old guy (and now occasionally with the change in our profession, an elderly lady) getting on the bus to go to the reception or dinner dance. Often they are alone, their spouses having departed before them. As a young man, I wondered why the heck they came to the meetings. Just like every generation before, ours was eager to grab the reins, and in our ardor for future glory, we were polite but also restless for them to move aside. I hadn’t yet learned the importance of history and of listening.
What I missed while carousing with my young colleagues was an opportunity to hear history first hand and to learn that, what we thought was so cutting edge, these men and women had long ago considered. Many of our living legends imagined some of today’s innovations but they lacked the technology to bring their dreams to fruition, or time and age defeated them before they reached the final chapter of their research. It was when I was about 50 that I wised up and began seeking out living legends like Frank Moody and Frank Spencer.
In the case of Frank Moody, he was quite elderly when I first met him. For some reason, he knew who I was and shook my hand softly. I didn’t recognize him initially, but at the sound of his name, I knew I was in the presence of a major figure in 20th century gastrointestinal surgery. He had been at the University of California, San Francisco, during an historic time when George Sheldon, Donald Trunkey and other great surgeons trained there with J. Englebert Dunphy as their chief. Dr. Moody’s CV lists 141 articles in basic and clinical science that have had a profound impact on how we view the gastrointestinal tract. He was Chief at the University of Utah and the University of Alabama and finished his career as professor at the University of Texas-Houston. His awards and achievements were legion.
Parkinson’s had only recently really begun to affect him when I met him, and as the years went by his voice became so very faint that I had to lean in to hear him. We would sit together at the back of the dinner dance room so that we could hear each other. And while the other guests entertained themselves, Dr. Moody and I would discuss his life, scientific method and philosophy as well as his insights into his own case of Parkinsonism. I would see him at meetings, making his way slowly but steadily along a corridor while others briskly walked by, unaware that the man they just passed was among the most important surgical pioneers of our time. It was not sad that Dr. Moody was elderly and unrecognized, but that we younger surgeons missed knowing a great man in our tendency to rush past history.
History is not facts and dates, but rather, it is people and their lives. Yes, the history of our profession is embodied by pioneers like Frank Moody and the others I’ve mentioned.
We have many Fellows among us who are living history, still contributing – maybe not at the dais but at the dinner table, speaking softly and walking a bit slower than their juniors. Thanks to LaMar McGinnis who started it and Don Nakayama who continues it, the College has a History Community on the ACS Communities, an active Surgical History Group, and a will to acknowledge the history that lives and breathes among us. The Surgical History Group has organized a full program of events at the Clinical Congress and I hope many attendees take the opportunity to attend.
Take a moment at your next meeting or at the Clinical Congress and look for those historic surgeons still with us. Be smarter than I was at a young age and get to know them. You may learn something from them you can’t learn anyplace else.
Dr. Hughes is clinical professor in the department of surgery and director of medical education at the Kansas University School of Medicine, Salina Campus, and Co-Editor of ACS Surgery News.
The email came with the words, “It is with sadness we report that Frank Moody died. …” I was instantly transported to the last time I saw the man and a flood of emotions swept over me. The name Frank Moody will ring a distant bell or none at all to some in our profession. Like many of the greats of surgery, he belongs to the ages.
I remember the first time I asked a student, “Who is Michael DeBakey?” I was dumbfounded to be greeted with a blank stare. How could a student of medicine not know of Dr. DeBakey? A few years later, the same question prompted a smart aleck reply that he was the man who invented DeBakey forceps. Well, of course he did invent the forceps, but to know nothing further of the man who was the world’s expert on ulcer disease in the 1940s, the progenitor of the National Medical Library, and among the foremost pioneers of heart surgery seemed beyond belief.
My mentor, Ernest Poulos, has long since left the active surgical scene. At times he would note the passing of one of his heroes like Carl Moyer (look it up!) and say, “Sic transit gloria mundi.” At 27 and anxious to get the right to cut into my fellow human beings, I would cock my head like a confounded puppy and wonder what that meant. I looked up the translation and meaning long ago, but now with age I understand the phrase in my bones.
I have long been a hanger-on at surgical meetings, hoping to meet those mighty figures that shaped surgical history. I saw W. Dean Warren once and had a very long hour with the great Mark Ravitch. Oliver Beahrs once performed magic tricks at a dinner I attended. At every surgical meeting there is an old guy (and now occasionally with the change in our profession, an elderly lady) getting on the bus to go to the reception or dinner dance. Often they are alone, their spouses having departed before them. As a young man, I wondered why the heck they came to the meetings. Just like every generation before, ours was eager to grab the reins, and in our ardor for future glory, we were polite but also restless for them to move aside. I hadn’t yet learned the importance of history and of listening.
What I missed while carousing with my young colleagues was an opportunity to hear history first hand and to learn that, what we thought was so cutting edge, these men and women had long ago considered. Many of our living legends imagined some of today’s innovations but they lacked the technology to bring their dreams to fruition, or time and age defeated them before they reached the final chapter of their research. It was when I was about 50 that I wised up and began seeking out living legends like Frank Moody and Frank Spencer.
In the case of Frank Moody, he was quite elderly when I first met him. For some reason, he knew who I was and shook my hand softly. I didn’t recognize him initially, but at the sound of his name, I knew I was in the presence of a major figure in 20th century gastrointestinal surgery. He had been at the University of California, San Francisco, during an historic time when George Sheldon, Donald Trunkey and other great surgeons trained there with J. Englebert Dunphy as their chief. Dr. Moody’s CV lists 141 articles in basic and clinical science that have had a profound impact on how we view the gastrointestinal tract. He was Chief at the University of Utah and the University of Alabama and finished his career as professor at the University of Texas-Houston. His awards and achievements were legion.
Parkinson’s had only recently really begun to affect him when I met him, and as the years went by his voice became so very faint that I had to lean in to hear him. We would sit together at the back of the dinner dance room so that we could hear each other. And while the other guests entertained themselves, Dr. Moody and I would discuss his life, scientific method and philosophy as well as his insights into his own case of Parkinsonism. I would see him at meetings, making his way slowly but steadily along a corridor while others briskly walked by, unaware that the man they just passed was among the most important surgical pioneers of our time. It was not sad that Dr. Moody was elderly and unrecognized, but that we younger surgeons missed knowing a great man in our tendency to rush past history.
History is not facts and dates, but rather, it is people and their lives. Yes, the history of our profession is embodied by pioneers like Frank Moody and the others I’ve mentioned.
We have many Fellows among us who are living history, still contributing – maybe not at the dais but at the dinner table, speaking softly and walking a bit slower than their juniors. Thanks to LaMar McGinnis who started it and Don Nakayama who continues it, the College has a History Community on the ACS Communities, an active Surgical History Group, and a will to acknowledge the history that lives and breathes among us. The Surgical History Group has organized a full program of events at the Clinical Congress and I hope many attendees take the opportunity to attend.
Take a moment at your next meeting or at the Clinical Congress and look for those historic surgeons still with us. Be smarter than I was at a young age and get to know them. You may learn something from them you can’t learn anyplace else.
Dr. Hughes is clinical professor in the department of surgery and director of medical education at the Kansas University School of Medicine, Salina Campus, and Co-Editor of ACS Surgery News.

Efficacy of Cladribine Tablets Continues After Conversion to MS
LONDON—Significant treatment effect versus placebo of cladribine tablets given to patients with clinically isolated syndrome during the initial treatment period continues to be observed in patients who convert to clinically definite multiple sclerosis (MS) and switch to treatment with a different disease-modifying drug (ie, subcutaneous interferon beta-1a), according to data presented at the 32nd Congress of the European Committee for Treatment and Research in MS (ECTRIMS).
Giancarlo Comi, MD, Professor of Neurology and Chairman of the Department of Neurology at Vita-Salute San Raffaele University in Milan, and colleagues reported that patients with clinically isolated syndrome who had been treated with cladribine tablets and who had converted to MS during the Oral Cladribine in Early MS (ORACLE-MS) initial treatment period had lower annualized relapse rates during the open-label maintenance period, relative to those patients who had received placebo during the ORACLE-MS initial treatment period.
The CLARITY (CLAdRIbine Tablets treating MS orallY) study in patients with active MS showed that annualized relapse rates and sustained disability worsening were reduced in patients treated with cladribine tablets annually for two years in short-duration courses, compared with placebo. The efficacy observed in the CLARITY study was maintained without further active treatment during the CLARITY extension study. In the ORACLE-MS study in patients with a first demyelinating event, cladribine tablets (3.5 mg/kg and 5.25 mg/kg) significantly reduced the risk of conversion to clinically definite MS, compared with placebo. If clinically definite MS occurred in the double-blinded, initial treatment period, patients were treated with subcutaneous interferon beta-1a in an open-label maintenance period.
The present study was designed to assess the annualized relapse rate during the ORACLE-MS open-label maintenance period in patients randomized to cladribine (3.5 mg/kg and 5.25 mg/kg) or placebo in the initial treatment period.
Similar to previous trials, participation in the ORACLE-MS open-label maintenance period was dependent upon the clinical course of the patient’s disease in the initial treatment period. Patients in ORACLE-MS who converted to clinically definite MS (according to Poser criteria) during the initial treatment period entered the open-label maintenance period and were treated with subcutaneous interferon beta-1a (titrated over four weeks up to the dose of 44 μg) administered three times per week.
A total of 109 patients in ORACLE-MS converted to clinically definite MS in the initial treatment period and received at least one dose of interferon beta-1a. The median time on interferon beta-1a was 56.0 weeks. Estimated annualized relapse rates in the open-label maintenance period were 0.14 for patients originally treated with cladribine 3.5 mg/kg (n = 25), 0.24 for patients originally treated with cladribine 5.25 mg/kg (n = 24), and 0.42 for patients who originally received placebo in the initial treatment period (n = 60).
According to the researchers, durable efficacy of cladribine tablets in ORACLE-MS into the open-label maintenance period is consistent with results of the CLARITY and CLARITY extension studies.
This study was sponsored by EMD Serono.
—Glenn S. Williams
Suggested Reading
Cook S, Vermersch P, Comi G, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Mult Scler. 2011;17(5):578-593.
Leist TP, Comi G, Cree BA, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. 2014;13(3):257-267.
LONDON—Significant treatment effect versus placebo of cladribine tablets given to patients with clinically isolated syndrome during the initial treatment period continues to be observed in patients who convert to clinically definite multiple sclerosis (MS) and switch to treatment with a different disease-modifying drug (ie, subcutaneous interferon beta-1a), according to data presented at the 32nd Congress of the European Committee for Treatment and Research in MS (ECTRIMS).
Giancarlo Comi, MD, Professor of Neurology and Chairman of the Department of Neurology at Vita-Salute San Raffaele University in Milan, and colleagues reported that patients with clinically isolated syndrome who had been treated with cladribine tablets and who had converted to MS during the Oral Cladribine in Early MS (ORACLE-MS) initial treatment period had lower annualized relapse rates during the open-label maintenance period, relative to those patients who had received placebo during the ORACLE-MS initial treatment period.
The CLARITY (CLAdRIbine Tablets treating MS orallY) study in patients with active MS showed that annualized relapse rates and sustained disability worsening were reduced in patients treated with cladribine tablets annually for two years in short-duration courses, compared with placebo. The efficacy observed in the CLARITY study was maintained without further active treatment during the CLARITY extension study. In the ORACLE-MS study in patients with a first demyelinating event, cladribine tablets (3.5 mg/kg and 5.25 mg/kg) significantly reduced the risk of conversion to clinically definite MS, compared with placebo. If clinically definite MS occurred in the double-blinded, initial treatment period, patients were treated with subcutaneous interferon beta-1a in an open-label maintenance period.
The present study was designed to assess the annualized relapse rate during the ORACLE-MS open-label maintenance period in patients randomized to cladribine (3.5 mg/kg and 5.25 mg/kg) or placebo in the initial treatment period.
Similar to previous trials, participation in the ORACLE-MS open-label maintenance period was dependent upon the clinical course of the patient’s disease in the initial treatment period. Patients in ORACLE-MS who converted to clinically definite MS (according to Poser criteria) during the initial treatment period entered the open-label maintenance period and were treated with subcutaneous interferon beta-1a (titrated over four weeks up to the dose of 44 μg) administered three times per week.
A total of 109 patients in ORACLE-MS converted to clinically definite MS in the initial treatment period and received at least one dose of interferon beta-1a. The median time on interferon beta-1a was 56.0 weeks. Estimated annualized relapse rates in the open-label maintenance period were 0.14 for patients originally treated with cladribine 3.5 mg/kg (n = 25), 0.24 for patients originally treated with cladribine 5.25 mg/kg (n = 24), and 0.42 for patients who originally received placebo in the initial treatment period (n = 60).
According to the researchers, durable efficacy of cladribine tablets in ORACLE-MS into the open-label maintenance period is consistent with results of the CLARITY and CLARITY extension studies.
This study was sponsored by EMD Serono.
—Glenn S. Williams
Suggested Reading
Cook S, Vermersch P, Comi G, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Mult Scler. 2011;17(5):578-593.
Leist TP, Comi G, Cree BA, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. 2014;13(3):257-267.
LONDON—Significant treatment effect versus placebo of cladribine tablets given to patients with clinically isolated syndrome during the initial treatment period continues to be observed in patients who convert to clinically definite multiple sclerosis (MS) and switch to treatment with a different disease-modifying drug (ie, subcutaneous interferon beta-1a), according to data presented at the 32nd Congress of the European Committee for Treatment and Research in MS (ECTRIMS).
Giancarlo Comi, MD, Professor of Neurology and Chairman of the Department of Neurology at Vita-Salute San Raffaele University in Milan, and colleagues reported that patients with clinically isolated syndrome who had been treated with cladribine tablets and who had converted to MS during the Oral Cladribine in Early MS (ORACLE-MS) initial treatment period had lower annualized relapse rates during the open-label maintenance period, relative to those patients who had received placebo during the ORACLE-MS initial treatment period.
The CLARITY (CLAdRIbine Tablets treating MS orallY) study in patients with active MS showed that annualized relapse rates and sustained disability worsening were reduced in patients treated with cladribine tablets annually for two years in short-duration courses, compared with placebo. The efficacy observed in the CLARITY study was maintained without further active treatment during the CLARITY extension study. In the ORACLE-MS study in patients with a first demyelinating event, cladribine tablets (3.5 mg/kg and 5.25 mg/kg) significantly reduced the risk of conversion to clinically definite MS, compared with placebo. If clinically definite MS occurred in the double-blinded, initial treatment period, patients were treated with subcutaneous interferon beta-1a in an open-label maintenance period.
The present study was designed to assess the annualized relapse rate during the ORACLE-MS open-label maintenance period in patients randomized to cladribine (3.5 mg/kg and 5.25 mg/kg) or placebo in the initial treatment period.
Similar to previous trials, participation in the ORACLE-MS open-label maintenance period was dependent upon the clinical course of the patient’s disease in the initial treatment period. Patients in ORACLE-MS who converted to clinically definite MS (according to Poser criteria) during the initial treatment period entered the open-label maintenance period and were treated with subcutaneous interferon beta-1a (titrated over four weeks up to the dose of 44 μg) administered three times per week.
A total of 109 patients in ORACLE-MS converted to clinically definite MS in the initial treatment period and received at least one dose of interferon beta-1a. The median time on interferon beta-1a was 56.0 weeks. Estimated annualized relapse rates in the open-label maintenance period were 0.14 for patients originally treated with cladribine 3.5 mg/kg (n = 25), 0.24 for patients originally treated with cladribine 5.25 mg/kg (n = 24), and 0.42 for patients who originally received placebo in the initial treatment period (n = 60).
According to the researchers, durable efficacy of cladribine tablets in ORACLE-MS into the open-label maintenance period is consistent with results of the CLARITY and CLARITY extension studies.
This study was sponsored by EMD Serono.
—Glenn S. Williams
Suggested Reading
Cook S, Vermersch P, Comi G, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Mult Scler. 2011;17(5):578-593.
Leist TP, Comi G, Cree BA, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. 2014;13(3):257-267.

ASPR Lends Support to New Screening Test for Zika
Reports in Brazil of Zika transmitted via blood transfusion call attention to the need for better ways to protect the blood supply. To that end, a blood screening test is getting a boost from the Office of the Assistant Secretary for Preparedness and Response (ASPR), with a $4.1 million agreement with Hologic, Inc., of Marlborough, Mass. This is the second screening test ASPR’s Biomedical Advanced Research and Development Authority (BARDA) is helping advance that may be used to test donated blood for Zika. Last April, ASPR announced support of a clinical study of a test developed by Roche Molecular Systems, Inc., of Branchburg, New Jersey.
Under the 1-year agreement, Hologic will advance development of its Procleix Zika Virus Assay, which is designed to detect Zika virus RNA in donated blood plasma up to 7 days post-infection. It runs on Hologic’s Panther automated system, which is already FDA cleared for some infectious disease in vitro diagnostic testing.
The contract could be extended to 18 months with an additional $6.2 million to support the clinical study to evaluate the sensitivity and specificity of the blood donation screening test in actual use, a necessary step before FDA approval.
The money is part of the $374 million HHS has repurposed for domestic Zika response and preparedness activities. BARDA has obligated $41.4 million of these “reprogrammed” funds to develop Zika vaccines, diagnostics, blood screening tests, and pathogen reduction technologies through private sector partners.
Reports in Brazil of Zika transmitted via blood transfusion call attention to the need for better ways to protect the blood supply. To that end, a blood screening test is getting a boost from the Office of the Assistant Secretary for Preparedness and Response (ASPR), with a $4.1 million agreement with Hologic, Inc., of Marlborough, Mass. This is the second screening test ASPR’s Biomedical Advanced Research and Development Authority (BARDA) is helping advance that may be used to test donated blood for Zika. Last April, ASPR announced support of a clinical study of a test developed by Roche Molecular Systems, Inc., of Branchburg, New Jersey.
Under the 1-year agreement, Hologic will advance development of its Procleix Zika Virus Assay, which is designed to detect Zika virus RNA in donated blood plasma up to 7 days post-infection. It runs on Hologic’s Panther automated system, which is already FDA cleared for some infectious disease in vitro diagnostic testing.
The contract could be extended to 18 months with an additional $6.2 million to support the clinical study to evaluate the sensitivity and specificity of the blood donation screening test in actual use, a necessary step before FDA approval.
The money is part of the $374 million HHS has repurposed for domestic Zika response and preparedness activities. BARDA has obligated $41.4 million of these “reprogrammed” funds to develop Zika vaccines, diagnostics, blood screening tests, and pathogen reduction technologies through private sector partners.
Reports in Brazil of Zika transmitted via blood transfusion call attention to the need for better ways to protect the blood supply. To that end, a blood screening test is getting a boost from the Office of the Assistant Secretary for Preparedness and Response (ASPR), with a $4.1 million agreement with Hologic, Inc., of Marlborough, Mass. This is the second screening test ASPR’s Biomedical Advanced Research and Development Authority (BARDA) is helping advance that may be used to test donated blood for Zika. Last April, ASPR announced support of a clinical study of a test developed by Roche Molecular Systems, Inc., of Branchburg, New Jersey.
Under the 1-year agreement, Hologic will advance development of its Procleix Zika Virus Assay, which is designed to detect Zika virus RNA in donated blood plasma up to 7 days post-infection. It runs on Hologic’s Panther automated system, which is already FDA cleared for some infectious disease in vitro diagnostic testing.
The contract could be extended to 18 months with an additional $6.2 million to support the clinical study to evaluate the sensitivity and specificity of the blood donation screening test in actual use, a necessary step before FDA approval.
The money is part of the $374 million HHS has repurposed for domestic Zika response and preparedness activities. BARDA has obligated $41.4 million of these “reprogrammed” funds to develop Zika vaccines, diagnostics, blood screening tests, and pathogen reduction technologies through private sector partners.
Real-World Safety and Effectiveness of Oral Anticoagulants for Afib
Clinical Question: Which oral anticoagulants are safest and most effective in nonvalvular atrial fibrillation?
Background: Use of direct oral anticoagulants (DOACs) has been increasing since their introduction and widespread marketing. While dosing is a challenge for warfarin, certain medical conditions limit the use of DOACs. Choosing the optimal oral anticoagulant is challenging with the increasing complexity of patients.
Study Design: Nationwide observational cohort study.
Setting: Three national Danish databases, from August 2011 to October 2015.
Synopsis: Authors reviewed data from 61,678 patients with nonvalvular atrial fibrillation who were new to oral anticoagulants. The study compared the efficacy, safety, and patient characteristics of DOACs and warfarin. Ischemic stroke, systemic embolism, and death were evaluated separately and as a composite measure of efficacy. Any bleeding, intracranial bleeding, and major bleeding were measured as safety outcomes. DOACs patients were younger and had lower CHA2DS2-VASc and HAS-BLED scores. No significant difference in risk of ischemic stroke was identified between DOACs and warfarin. Rivaroxaban was associated with lower rates of ischemic stroke and systemic embolism but had bleeding rates that were similar to warfarin. Any bleeding and major bleeding rates were lowest for dabigatran and apixaban. All-cause mortality was lowest in the dabigatran group and highest in the warfarin group.
Limitations were the retrospective, observational study design, with an average follow-up of only 1.9 years.
Bottom Line: All DOACs appear to be safer and more effective alternatives to warfarin. Oral anticoagulant selection needs to be based on individual patient clinical profile.
Citation: Larsen TB, Skjoth F, Nielsen PB, Kjaeldgaard JN, Lip GY. Comparative effectiveness and safety of non-vitamin K antagonist oral anticoagulants and warfarin in patients with atrial fibrillation: propensity weighted nationwide cohort study. BMJ. 2016;353:i3189.
Short Take
Mortality and Long-Acting Opiates
This retrospective cohort study raises questions about the safety of long-acting opioids for chronic noncancer pain. When compared with anticonvulsants or antidepressants, the adjusted hazard ratio was 1.64 for total mortality.
Citation: Ray W, Chung CP, Murray KT, Hall K, Stein CM. Prescription of long-acting opioids and mortality in patients with chronic noncancer pain. JAMA. 2016;315(22):2415-2423.
Clinical Question: Which oral anticoagulants are safest and most effective in nonvalvular atrial fibrillation?
Background: Use of direct oral anticoagulants (DOACs) has been increasing since their introduction and widespread marketing. While dosing is a challenge for warfarin, certain medical conditions limit the use of DOACs. Choosing the optimal oral anticoagulant is challenging with the increasing complexity of patients.
Study Design: Nationwide observational cohort study.
Setting: Three national Danish databases, from August 2011 to October 2015.
Synopsis: Authors reviewed data from 61,678 patients with nonvalvular atrial fibrillation who were new to oral anticoagulants. The study compared the efficacy, safety, and patient characteristics of DOACs and warfarin. Ischemic stroke, systemic embolism, and death were evaluated separately and as a composite measure of efficacy. Any bleeding, intracranial bleeding, and major bleeding were measured as safety outcomes. DOACs patients were younger and had lower CHA2DS2-VASc and HAS-BLED scores. No significant difference in risk of ischemic stroke was identified between DOACs and warfarin. Rivaroxaban was associated with lower rates of ischemic stroke and systemic embolism but had bleeding rates that were similar to warfarin. Any bleeding and major bleeding rates were lowest for dabigatran and apixaban. All-cause mortality was lowest in the dabigatran group and highest in the warfarin group.
Limitations were the retrospective, observational study design, with an average follow-up of only 1.9 years.
Bottom Line: All DOACs appear to be safer and more effective alternatives to warfarin. Oral anticoagulant selection needs to be based on individual patient clinical profile.
Citation: Larsen TB, Skjoth F, Nielsen PB, Kjaeldgaard JN, Lip GY. Comparative effectiveness and safety of non-vitamin K antagonist oral anticoagulants and warfarin in patients with atrial fibrillation: propensity weighted nationwide cohort study. BMJ. 2016;353:i3189.
Short Take
Mortality and Long-Acting Opiates
This retrospective cohort study raises questions about the safety of long-acting opioids for chronic noncancer pain. When compared with anticonvulsants or antidepressants, the adjusted hazard ratio was 1.64 for total mortality.
Citation: Ray W, Chung CP, Murray KT, Hall K, Stein CM. Prescription of long-acting opioids and mortality in patients with chronic noncancer pain. JAMA. 2016;315(22):2415-2423.
Clinical Question: Which oral anticoagulants are safest and most effective in nonvalvular atrial fibrillation?
Background: Use of direct oral anticoagulants (DOACs) has been increasing since their introduction and widespread marketing. While dosing is a challenge for warfarin, certain medical conditions limit the use of DOACs. Choosing the optimal oral anticoagulant is challenging with the increasing complexity of patients.
Study Design: Nationwide observational cohort study.
Setting: Three national Danish databases, from August 2011 to October 2015.
Synopsis: Authors reviewed data from 61,678 patients with nonvalvular atrial fibrillation who were new to oral anticoagulants. The study compared the efficacy, safety, and patient characteristics of DOACs and warfarin. Ischemic stroke, systemic embolism, and death were evaluated separately and as a composite measure of efficacy. Any bleeding, intracranial bleeding, and major bleeding were measured as safety outcomes. DOACs patients were younger and had lower CHA2DS2-VASc and HAS-BLED scores. No significant difference in risk of ischemic stroke was identified between DOACs and warfarin. Rivaroxaban was associated with lower rates of ischemic stroke and systemic embolism but had bleeding rates that were similar to warfarin. Any bleeding and major bleeding rates were lowest for dabigatran and apixaban. All-cause mortality was lowest in the dabigatran group and highest in the warfarin group.
Limitations were the retrospective, observational study design, with an average follow-up of only 1.9 years.
Bottom Line: All DOACs appear to be safer and more effective alternatives to warfarin. Oral anticoagulant selection needs to be based on individual patient clinical profile.
Citation: Larsen TB, Skjoth F, Nielsen PB, Kjaeldgaard JN, Lip GY. Comparative effectiveness and safety of non-vitamin K antagonist oral anticoagulants and warfarin in patients with atrial fibrillation: propensity weighted nationwide cohort study. BMJ. 2016;353:i3189.
Short Take
Mortality and Long-Acting Opiates
This retrospective cohort study raises questions about the safety of long-acting opioids for chronic noncancer pain. When compared with anticonvulsants or antidepressants, the adjusted hazard ratio was 1.64 for total mortality.
Citation: Ray W, Chung CP, Murray KT, Hall K, Stein CM. Prescription of long-acting opioids and mortality in patients with chronic noncancer pain. JAMA. 2016;315(22):2415-2423.

Prescribing Naloxone for Patients on Long-Term Opioid Therapy
Background: Unintentional opioid overdose is a major public health issue. Studies have shown that provision of naloxone to at-risk patients reduces mortality and improves survival. The CDC recommends considering naloxone prescription in high-risk patients. This study focused on patient education and prescription habits of providers rather than just making naloxone available.
Study Design: Non-randomized interventional study.
Setting: Six safety-net primary-care clinics in San Francisco.
Synopsis: The authors identified 1,985 adults on long-term opioid treatment, of which 759 were prescribed naloxone. Providers were encouraged to prescribe naloxone along with opioids. Patients were educated about use of the intranasal naloxone device. Outcomes included opioid-related emergency department visits and prescribed dosage. They noted that patients on a higher dose of opioids and with opioid-related ED visits in the prior 12 months were more likely to be prescribed naloxone. When compared to patients who were not prescribed naloxone, patients who received naloxone had 47% fewer ED visits per month in the first six months and 63% fewer ED visits over 12 months. Limitations include lack of randomization and being a single-center study.
Hospitalists can prioritize patients and consider providing naloxone prescription to reduce ED visits and perhaps readmissions. Further studies are needed focusing on patients who get discharged from the hospital.
Bottom Line: Naloxone prescription in patients on long-term opioid treatment may prevent opioid-related ED visits.
Citation: Coffin PO, Behar E, Rowe C, et al. Nonrandomized intervention study of naloxone coprescription for primary care patients receiving long-term opioid therapy for pain. Ann Intern Med. 2016;165(4):245-252.
Short Take
Mortality and Long-Acting Opiates
This retrospective cohort study raises questions about the safety of long-acting opioids for chronic noncancer pain. When compared with anticonvulsants or antidepressants, the adjusted hazard ratio was 1.64 for total mortality.
Citation: Ray W, Chung CP, Murray KT, Hall K, Stein CM. Prescription of long-acting opioids and mortality in patients with chronic noncancer pain. JAMA. 2016;315(22):2415-2423.
Background: Unintentional opioid overdose is a major public health issue. Studies have shown that provision of naloxone to at-risk patients reduces mortality and improves survival. The CDC recommends considering naloxone prescription in high-risk patients. This study focused on patient education and prescription habits of providers rather than just making naloxone available.
Study Design: Non-randomized interventional study.
Setting: Six safety-net primary-care clinics in San Francisco.
Synopsis: The authors identified 1,985 adults on long-term opioid treatment, of which 759 were prescribed naloxone. Providers were encouraged to prescribe naloxone along with opioids. Patients were educated about use of the intranasal naloxone device. Outcomes included opioid-related emergency department visits and prescribed dosage. They noted that patients on a higher dose of opioids and with opioid-related ED visits in the prior 12 months were more likely to be prescribed naloxone. When compared to patients who were not prescribed naloxone, patients who received naloxone had 47% fewer ED visits per month in the first six months and 63% fewer ED visits over 12 months. Limitations include lack of randomization and being a single-center study.
Hospitalists can prioritize patients and consider providing naloxone prescription to reduce ED visits and perhaps readmissions. Further studies are needed focusing on patients who get discharged from the hospital.
Bottom Line: Naloxone prescription in patients on long-term opioid treatment may prevent opioid-related ED visits.
Citation: Coffin PO, Behar E, Rowe C, et al. Nonrandomized intervention study of naloxone coprescription for primary care patients receiving long-term opioid therapy for pain. Ann Intern Med. 2016;165(4):245-252.
Short Take
Mortality and Long-Acting Opiates
This retrospective cohort study raises questions about the safety of long-acting opioids for chronic noncancer pain. When compared with anticonvulsants or antidepressants, the adjusted hazard ratio was 1.64 for total mortality.
Citation: Ray W, Chung CP, Murray KT, Hall K, Stein CM. Prescription of long-acting opioids and mortality in patients with chronic noncancer pain. JAMA. 2016;315(22):2415-2423.
Background: Unintentional opioid overdose is a major public health issue. Studies have shown that provision of naloxone to at-risk patients reduces mortality and improves survival. The CDC recommends considering naloxone prescription in high-risk patients. This study focused on patient education and prescription habits of providers rather than just making naloxone available.
Study Design: Non-randomized interventional study.
Setting: Six safety-net primary-care clinics in San Francisco.
Synopsis: The authors identified 1,985 adults on long-term opioid treatment, of which 759 were prescribed naloxone. Providers were encouraged to prescribe naloxone along with opioids. Patients were educated about use of the intranasal naloxone device. Outcomes included opioid-related emergency department visits and prescribed dosage. They noted that patients on a higher dose of opioids and with opioid-related ED visits in the prior 12 months were more likely to be prescribed naloxone. When compared to patients who were not prescribed naloxone, patients who received naloxone had 47% fewer ED visits per month in the first six months and 63% fewer ED visits over 12 months. Limitations include lack of randomization and being a single-center study.
Hospitalists can prioritize patients and consider providing naloxone prescription to reduce ED visits and perhaps readmissions. Further studies are needed focusing on patients who get discharged from the hospital.
Bottom Line: Naloxone prescription in patients on long-term opioid treatment may prevent opioid-related ED visits.
Citation: Coffin PO, Behar E, Rowe C, et al. Nonrandomized intervention study of naloxone coprescription for primary care patients receiving long-term opioid therapy for pain. Ann Intern Med. 2016;165(4):245-252.
Short Take
Mortality and Long-Acting Opiates
This retrospective cohort study raises questions about the safety of long-acting opioids for chronic noncancer pain. When compared with anticonvulsants or antidepressants, the adjusted hazard ratio was 1.64 for total mortality.
Citation: Ray W, Chung CP, Murray KT, Hall K, Stein CM. Prescription of long-acting opioids and mortality in patients with chronic noncancer pain. JAMA. 2016;315(22):2415-2423.
FDA grants priority review for MM drug

Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted priority review for part of a supplemental biologics license application (sBLA) for daratumumab (Darzalex®).
The priority review pertains to the use of daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat patients with multiple myeloma (MM) who have received at least 1 prior therapy.
To grant an application priority review, the FDA must believe the drug would provide a significant improvement in the treatment, diagnosis, or prevention of a serious condition.
The priority review designation means the FDA’s goal is to take action on an application within 6 months, rather than the 10 months typically taken for a standard review.
The FDA has assigned a Prescription Drug User Fee Act target date of February 17, 2017, to make a decision on daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone.
Standard review
The FDA has also granted a standard review period for part of the sBLA.
The standard review pertains to the use of daratumumab in combination with pomalidomide and dexamethasone to treat patients with relapsed or refractory MM who have received at least 2 prior therapies, including a proteasome inhibitor and an immunomodulatory agent.
The Prescription Drug User Fee Act date for the combination of daratumumab with pomalidomide and dexamethasone is June 17, 2017.
Trial data
The sBLA submission included data from a pair of phase 3 studies:
- The CASTOR study, in which researchers compared the combination of daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone in patients with relapsed or refractory MM
- The POLLUX study, in which researchers compared daratumumab in combination with lenalidomide and dexamethasone to lenalidomide and dexamethasone in patients with relapsed or refractory MM.
The sBLA submission also included data from a phase 1 study of daratumumab in combination with pomalidomide and dexamethasone in patients with relapsed or refractory MM.
About daratumumab
Daratumumab is a human IgG1k monoclonal antibody that binds to CD38, which is highly expressed on the surface of MM cells.
Daratumumab already has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
For more information on daratumumab, visit www.DARZALEX.com. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted priority review for part of a supplemental biologics license application (sBLA) for daratumumab (Darzalex®).
The priority review pertains to the use of daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat patients with multiple myeloma (MM) who have received at least 1 prior therapy.
To grant an application priority review, the FDA must believe the drug would provide a significant improvement in the treatment, diagnosis, or prevention of a serious condition.
The priority review designation means the FDA’s goal is to take action on an application within 6 months, rather than the 10 months typically taken for a standard review.
The FDA has assigned a Prescription Drug User Fee Act target date of February 17, 2017, to make a decision on daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone.
Standard review
The FDA has also granted a standard review period for part of the sBLA.
The standard review pertains to the use of daratumumab in combination with pomalidomide and dexamethasone to treat patients with relapsed or refractory MM who have received at least 2 prior therapies, including a proteasome inhibitor and an immunomodulatory agent.
The Prescription Drug User Fee Act date for the combination of daratumumab with pomalidomide and dexamethasone is June 17, 2017.
Trial data
The sBLA submission included data from a pair of phase 3 studies:
- The CASTOR study, in which researchers compared the combination of daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone in patients with relapsed or refractory MM
- The POLLUX study, in which researchers compared daratumumab in combination with lenalidomide and dexamethasone to lenalidomide and dexamethasone in patients with relapsed or refractory MM.
The sBLA submission also included data from a phase 1 study of daratumumab in combination with pomalidomide and dexamethasone in patients with relapsed or refractory MM.
About daratumumab
Daratumumab is a human IgG1k monoclonal antibody that binds to CD38, which is highly expressed on the surface of MM cells.
Daratumumab already has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
For more information on daratumumab, visit www.DARZALEX.com. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted priority review for part of a supplemental biologics license application (sBLA) for daratumumab (Darzalex®).
The priority review pertains to the use of daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat patients with multiple myeloma (MM) who have received at least 1 prior therapy.
To grant an application priority review, the FDA must believe the drug would provide a significant improvement in the treatment, diagnosis, or prevention of a serious condition.
The priority review designation means the FDA’s goal is to take action on an application within 6 months, rather than the 10 months typically taken for a standard review.
The FDA has assigned a Prescription Drug User Fee Act target date of February 17, 2017, to make a decision on daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone.
Standard review
The FDA has also granted a standard review period for part of the sBLA.
The standard review pertains to the use of daratumumab in combination with pomalidomide and dexamethasone to treat patients with relapsed or refractory MM who have received at least 2 prior therapies, including a proteasome inhibitor and an immunomodulatory agent.
The Prescription Drug User Fee Act date for the combination of daratumumab with pomalidomide and dexamethasone is June 17, 2017.
Trial data
The sBLA submission included data from a pair of phase 3 studies:
- The CASTOR study, in which researchers compared the combination of daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone in patients with relapsed or refractory MM
- The POLLUX study, in which researchers compared daratumumab in combination with lenalidomide and dexamethasone to lenalidomide and dexamethasone in patients with relapsed or refractory MM.
The sBLA submission also included data from a phase 1 study of daratumumab in combination with pomalidomide and dexamethasone in patients with relapsed or refractory MM.
About daratumumab
Daratumumab is a human IgG1k monoclonal antibody that binds to CD38, which is highly expressed on the surface of MM cells.
Daratumumab already has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
For more information on daratumumab, visit www.DARZALEX.com. ![]()
Temperature Extremes May Put Pregnancies at Risk
Very hot and very cold temperatures during pregnancy may increase the risk of preterm birth, say NIH researchers who analyzed records from 223,375 live births at 12 clinical centers.
They linked patients’ electronic records to hourly temperature records for the region surrounding each center. Because “hot” and “cold” can vary depending on the person and place, they defined cold and hot temperatures as below the 10th percentile and above the 90th percentile of average temperatures, respectively.
Related: Home-Visiting Program to Support Young Native American Families
Women who experienced extreme cold for the first 7 weeks of their pregnancies had a 20% higher risk of delivering before 34 weeks, a 9% higher risk of delivering between 34 and 36 weeks, and 3% higher risk of delivering in weeks 37 and 38.
But the researchers found more consistent associations with early delivery after exposure to extreme heat than extreme cold. Overall, exposure to extreme heat for the duration of pregnancy was associated with increases in risk for delivery at 34 weeks and 36 to 38 weeks by 6% to 21%. The researchers suggest that during cold spells, people are more likely to seek shelter and get warm—and may be more likely to just endure hot weather.
Related: Coordinating Better Care for Opioid-Addicted Women and Their Children
The researchers theorize that the stress of temperature extremes could hinder the development of the placenta or alter blood flow to the uterus, both of which could lead to early labor.
Very hot and very cold temperatures during pregnancy may increase the risk of preterm birth, say NIH researchers who analyzed records from 223,375 live births at 12 clinical centers.
They linked patients’ electronic records to hourly temperature records for the region surrounding each center. Because “hot” and “cold” can vary depending on the person and place, they defined cold and hot temperatures as below the 10th percentile and above the 90th percentile of average temperatures, respectively.
Related: Home-Visiting Program to Support Young Native American Families
Women who experienced extreme cold for the first 7 weeks of their pregnancies had a 20% higher risk of delivering before 34 weeks, a 9% higher risk of delivering between 34 and 36 weeks, and 3% higher risk of delivering in weeks 37 and 38.
But the researchers found more consistent associations with early delivery after exposure to extreme heat than extreme cold. Overall, exposure to extreme heat for the duration of pregnancy was associated with increases in risk for delivery at 34 weeks and 36 to 38 weeks by 6% to 21%. The researchers suggest that during cold spells, people are more likely to seek shelter and get warm—and may be more likely to just endure hot weather.
Related: Coordinating Better Care for Opioid-Addicted Women and Their Children
The researchers theorize that the stress of temperature extremes could hinder the development of the placenta or alter blood flow to the uterus, both of which could lead to early labor.
Very hot and very cold temperatures during pregnancy may increase the risk of preterm birth, say NIH researchers who analyzed records from 223,375 live births at 12 clinical centers.
They linked patients’ electronic records to hourly temperature records for the region surrounding each center. Because “hot” and “cold” can vary depending on the person and place, they defined cold and hot temperatures as below the 10th percentile and above the 90th percentile of average temperatures, respectively.
Related: Home-Visiting Program to Support Young Native American Families
Women who experienced extreme cold for the first 7 weeks of their pregnancies had a 20% higher risk of delivering before 34 weeks, a 9% higher risk of delivering between 34 and 36 weeks, and 3% higher risk of delivering in weeks 37 and 38.
But the researchers found more consistent associations with early delivery after exposure to extreme heat than extreme cold. Overall, exposure to extreme heat for the duration of pregnancy was associated with increases in risk for delivery at 34 weeks and 36 to 38 weeks by 6% to 21%. The researchers suggest that during cold spells, people are more likely to seek shelter and get warm—and may be more likely to just endure hot weather.
Related: Coordinating Better Care for Opioid-Addicted Women and Their Children
The researchers theorize that the stress of temperature extremes could hinder the development of the placenta or alter blood flow to the uterus, both of which could lead to early labor.
These Patients Knee’d Your Help
1. A 23-year-old man is brought in after being hit by a car. There is a moderate amount of soft tissue swelling around the knee, with limited flexion and extension due to pain. He can wiggle his toes, and there appears to be no neurovascular compromise.
Diagnosis: The image shows a comminuted and depressed fracture of the lateral tibial plateau. It is depressed approximately 6 to 7 mm. The patient was admitted, and orthopedic consultation was obtained. The patient subsequently underwent an open reduction and internal fixation of the fracture.
For more information, see “Clipped by an Oncoming Car.” Clinician Reviews. 2014;24(6):23,36.
2. A 20-year-old man presents after his car was broadsided by another vehicle. His air bag deployed, and the patient now complains of right-sided chest wall pain and right knee pain. Inspection of his right knee shows some joint deformity, with mild swelling and moderate tenderness. The patient is unable to perform flexion with his right knee. Good distal pulses are present, and sensation is intact.
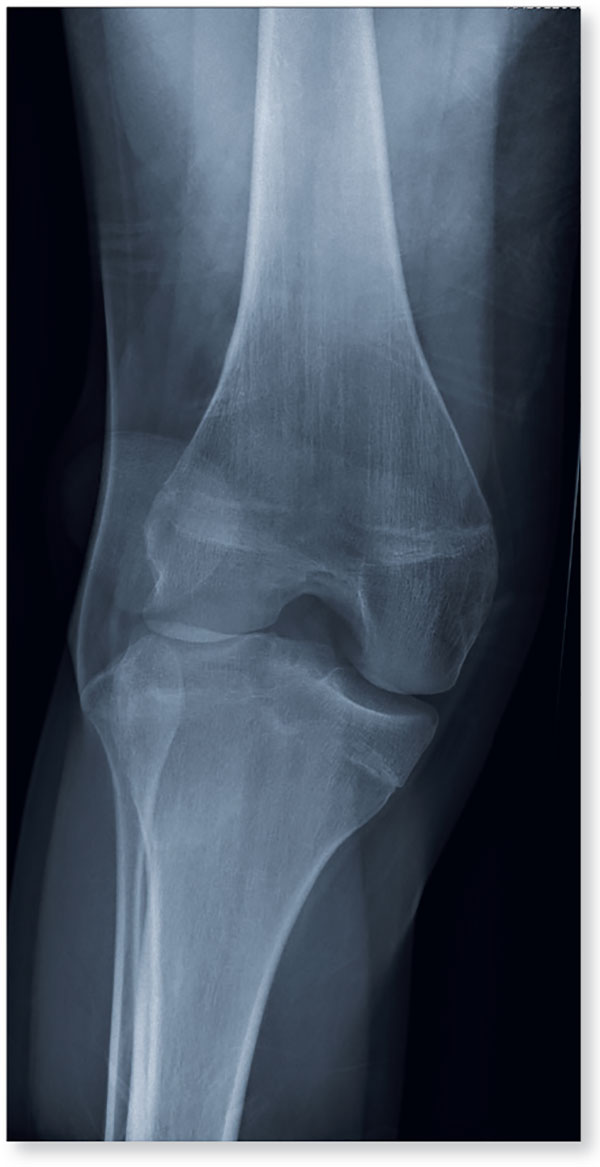
Diagnosis: The radiograph demonstrates lateral dislocation of the patella, with no evidence of an acute fracture in any surrounding bones. The patella was easily reduced in the emergency department, and the patient was placed in a knee immobilizer. Orthopedic consultation was obtained.
For more information, see “Chest Wall and Knee Pain Following Motor Vehicle Collision.” Clinician Reviews. 2013;23(1):8.
3. A 70-year-old woman presents for evaluation of right knee pain secondary to a fall. When she tripped and fell, all her weight landed on her right knee; she says it is now “extremely painful” to bear weight on that leg. Inspection of her right knee shows no obvious deformity, but a moderate amount of swelling and limited range of motion. She also has moderate tenderness circumferentially around the knee. There is additional swelling and mild bruising on both the medial and lateral aspects of the right ankle.

Diagnosis: The radiograph has several findings, one of which is a nondisplaced proximal fibula fracture. In addition, there is a moderate suprapatellar joint effusion. The patient also has fairly advanced tricompartment degenerative arthrosis. (To review, the tricompartment comprises all three anatomic areas of the knee: the patellofemoral, lateral tibiofemoral, and medial tibiofemoral joints.) The patient was placed in a knee immobilizer, and orthopedic evaluation was coordinated.
For more information, see “In Middle of Trip, Woman Falls.” Clinician Reviews. 2016;26(6):20,53.
4. A 28-year-old man is brought to you by EMS for evaluation after a motor vehicle accident. The patient was an unrestrained driver in a truck that went off the road into a ditch. The paramedics state that he was partially ejected, with his left leg caught in the window. He complains of back and left leg pain. Primary survey shows no obvious injury. Secondary survey reveals moderate swelling and decreased range of motion in the left knee. Good distal pulses are present.
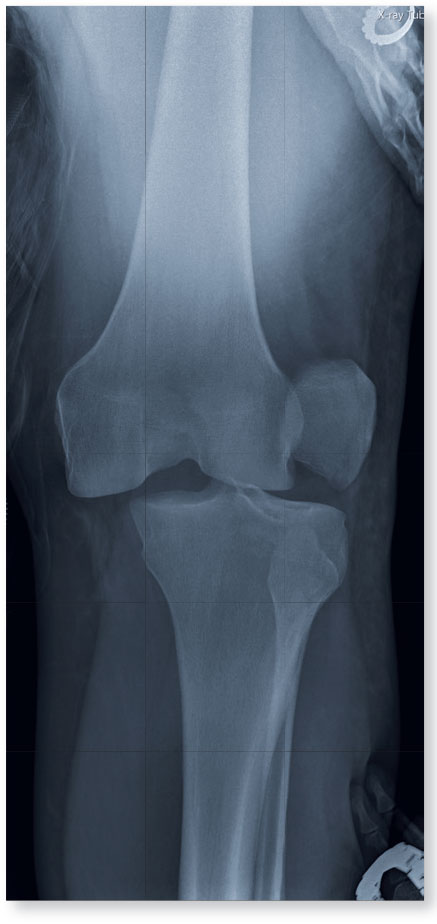
Diagnosis: The radiograph shows that the distal femur is medially dislocated relative to the tibial plateau. In addition, the patella is laterally dislocated. No obvious fractures are evident. Such injuries are typically associated with significant ligament injuries, especially of the medial collateral ligament (MCL), lateral collateral ligament (LCL), and anterior cruciate ligament (ACL). Orthopedics was consulted for reduction of the dislocation and further workup (including MRI of the knee).
For more information, see “Driver Partially Ejected From Vehicle.” Clinician Reviews. 2015;25(7):20,27.
1. A 23-year-old man is brought in after being hit by a car. There is a moderate amount of soft tissue swelling around the knee, with limited flexion and extension due to pain. He can wiggle his toes, and there appears to be no neurovascular compromise.
Diagnosis: The image shows a comminuted and depressed fracture of the lateral tibial plateau. It is depressed approximately 6 to 7 mm. The patient was admitted, and orthopedic consultation was obtained. The patient subsequently underwent an open reduction and internal fixation of the fracture.
For more information, see “Clipped by an Oncoming Car.” Clinician Reviews. 2014;24(6):23,36.
2. A 20-year-old man presents after his car was broadsided by another vehicle. His air bag deployed, and the patient now complains of right-sided chest wall pain and right knee pain. Inspection of his right knee shows some joint deformity, with mild swelling and moderate tenderness. The patient is unable to perform flexion with his right knee. Good distal pulses are present, and sensation is intact.
Diagnosis: The radiograph demonstrates lateral dislocation of the patella, with no evidence of an acute fracture in any surrounding bones. The patella was easily reduced in the emergency department, and the patient was placed in a knee immobilizer. Orthopedic consultation was obtained.
For more information, see “Chest Wall and Knee Pain Following Motor Vehicle Collision.” Clinician Reviews. 2013;23(1):8.
3. A 70-year-old woman presents for evaluation of right knee pain secondary to a fall. When she tripped and fell, all her weight landed on her right knee; she says it is now “extremely painful” to bear weight on that leg. Inspection of her right knee shows no obvious deformity, but a moderate amount of swelling and limited range of motion. She also has moderate tenderness circumferentially around the knee. There is additional swelling and mild bruising on both the medial and lateral aspects of the right ankle.
Diagnosis: The radiograph has several findings, one of which is a nondisplaced proximal fibula fracture. In addition, there is a moderate suprapatellar joint effusion. The patient also has fairly advanced tricompartment degenerative arthrosis. (To review, the tricompartment comprises all three anatomic areas of the knee: the patellofemoral, lateral tibiofemoral, and medial tibiofemoral joints.) The patient was placed in a knee immobilizer, and orthopedic evaluation was coordinated.
For more information, see “In Middle of Trip, Woman Falls.” Clinician Reviews. 2016;26(6):20,53.
4. A 28-year-old man is brought to you by EMS for evaluation after a motor vehicle accident. The patient was an unrestrained driver in a truck that went off the road into a ditch. The paramedics state that he was partially ejected, with his left leg caught in the window. He complains of back and left leg pain. Primary survey shows no obvious injury. Secondary survey reveals moderate swelling and decreased range of motion in the left knee. Good distal pulses are present.
Diagnosis: The radiograph shows that the distal femur is medially dislocated relative to the tibial plateau. In addition, the patella is laterally dislocated. No obvious fractures are evident. Such injuries are typically associated with significant ligament injuries, especially of the medial collateral ligament (MCL), lateral collateral ligament (LCL), and anterior cruciate ligament (ACL). Orthopedics was consulted for reduction of the dislocation and further workup (including MRI of the knee).
For more information, see “Driver Partially Ejected From Vehicle.” Clinician Reviews. 2015;25(7):20,27.
1. A 23-year-old man is brought in after being hit by a car. There is a moderate amount of soft tissue swelling around the knee, with limited flexion and extension due to pain. He can wiggle his toes, and there appears to be no neurovascular compromise.
Diagnosis: The image shows a comminuted and depressed fracture of the lateral tibial plateau. It is depressed approximately 6 to 7 mm. The patient was admitted, and orthopedic consultation was obtained. The patient subsequently underwent an open reduction and internal fixation of the fracture.
For more information, see “Clipped by an Oncoming Car.” Clinician Reviews. 2014;24(6):23,36.
2. A 20-year-old man presents after his car was broadsided by another vehicle. His air bag deployed, and the patient now complains of right-sided chest wall pain and right knee pain. Inspection of his right knee shows some joint deformity, with mild swelling and moderate tenderness. The patient is unable to perform flexion with his right knee. Good distal pulses are present, and sensation is intact.
Diagnosis: The radiograph demonstrates lateral dislocation of the patella, with no evidence of an acute fracture in any surrounding bones. The patella was easily reduced in the emergency department, and the patient was placed in a knee immobilizer. Orthopedic consultation was obtained.
For more information, see “Chest Wall and Knee Pain Following Motor Vehicle Collision.” Clinician Reviews. 2013;23(1):8.
3. A 70-year-old woman presents for evaluation of right knee pain secondary to a fall. When she tripped and fell, all her weight landed on her right knee; she says it is now “extremely painful” to bear weight on that leg. Inspection of her right knee shows no obvious deformity, but a moderate amount of swelling and limited range of motion. She also has moderate tenderness circumferentially around the knee. There is additional swelling and mild bruising on both the medial and lateral aspects of the right ankle.
Diagnosis: The radiograph has several findings, one of which is a nondisplaced proximal fibula fracture. In addition, there is a moderate suprapatellar joint effusion. The patient also has fairly advanced tricompartment degenerative arthrosis. (To review, the tricompartment comprises all three anatomic areas of the knee: the patellofemoral, lateral tibiofemoral, and medial tibiofemoral joints.) The patient was placed in a knee immobilizer, and orthopedic evaluation was coordinated.
For more information, see “In Middle of Trip, Woman Falls.” Clinician Reviews. 2016;26(6):20,53.
4. A 28-year-old man is brought to you by EMS for evaluation after a motor vehicle accident. The patient was an unrestrained driver in a truck that went off the road into a ditch. The paramedics state that he was partially ejected, with his left leg caught in the window. He complains of back and left leg pain. Primary survey shows no obvious injury. Secondary survey reveals moderate swelling and decreased range of motion in the left knee. Good distal pulses are present.
Diagnosis: The radiograph shows that the distal femur is medially dislocated relative to the tibial plateau. In addition, the patella is laterally dislocated. No obvious fractures are evident. Such injuries are typically associated with significant ligament injuries, especially of the medial collateral ligament (MCL), lateral collateral ligament (LCL), and anterior cruciate ligament (ACL). Orthopedics was consulted for reduction of the dislocation and further workup (including MRI of the knee).
For more information, see “Driver Partially Ejected From Vehicle.” Clinician Reviews. 2015;25(7):20,27.
Management of Relapsed and Refractory Multiple Myeloma
From the Division of Hematology and Oncology, University of North Carolina – Chapel Hill, Chapel Hill, NC (Dr. Reeves), and the Division of Cellular Therapy and Hematological Malignancies, Duke Cancer Institute, Durham, NC (Dr. Tuchman).
Abstract
- Objective: To review the management considerations in patients with relapsed and refractory multiple myeloma (RRMM).
- Methods: Review of the literature.
- Results: RRMM is a heterogeneous disease and numerous treatment regimens have been studied. Despite improvement in progression-free and overall survival in newly diagnosed multiple myeloma with current therapies, myeloma remains incurable and repeated relapses are inevitable. Relapses are often characterized by diminished response to chemotherapy (refractoriness) and duration of response.
- Conclusion: Management of RRMM should be individualized using both patient- and disease-related factors, given substantial heterogeneity in both. Further research regarding the optimal timing, regimen, and duration of treatment is warranted.
Although advancements in treating multiple myeloma (MM) have resulted in improved median survival from approximately 2 years in the 1990s to more recent estimates of over 6 years, the disease remains incurable [1–3]. Its overall course is generally defined by a series of increasingly short remissions and treatment-refractory relapses until eventual death due to MM occurs. Objective criteria for defining both relapsed and refractory MM have been published [4]. Briefly, relapsed myeloma is that which has been previously treated with some form of systemic therapy and which has recurred. That recurrence can be clinical (ie, the development of new or worsening signs or symptoms of active MM) and/or biochemical (ie, rising monoclonal MM proteins in the serum or urine). Refractory MM on the other hand refers to MM that is resistant to particular drugs, defined as MM that is nonresponsive to primary or salvage therapy, or MM that progresses within 60 days of the last therapy [4]. At any juncture during the course of relapsed MM, patients will have disease that is either sensitive or refractory to specific myeloma drugs. In this article, we discuss management of these often concurrent entities together as relapsed and refractory multiple myeloma (RRMM).
There are numerous treatment options for patients with RRMM—3 new drugs were approved in November 2015 alone. The abundance of available drugs leaves treating clinicians with a daunting task of sequencing therapies among several choices. The durability of response to treatment typically lessens with each disease relapse, such that the clinician needs to think of sequencing not just second-line therapy, but third- and fourth-line as well, further complicating the decision. In this review, we aim to help clinicians individualize treatment plans for patients with RRMM.
Case Studies
Patient A
A 62-year-old man with IgG-kappa MM was diagnosed 4 years ago during evaluation of a pathologic humeral fracture. The disease was prognostically standard risk, with revised International Staging System (RISS) stage I disease (beta-2 microglobulin 3.4 mcg/mL, albumin 4.1 g/dL, normal cytogenetics with 46,XY in 20 cells analyzed, and myeloma fluorescent in situ hybridization [FISH] panel showing t(11;14) but no del17p, t(14;16), t(14;20), or t(4;14)) [5], and normal blood counts, organ function, and lactate dehydrogenase (LDH) at diagnosis. He was treated with 5 cycles of standard lenalidomide, bortezomib, and dexamethasone followed by high-dose melphalan with autologous stem cell transplantation (ASCT) and then lenalidomide continuous maintenance. He achieved a stringent complete response (ie, complete disappearance of myeloma-derived monoclonal proteins in the serum and urine, a normal serum free light chain ratio, and undetectable monoclonal plasma cells on a bone marrow aspirate and biopsy) [4]. His MM was monitored every 2 to 3 months for disease progression and medication toxicity. At month 38, a monoclonal protein spike (M-spike) on serum protein electrophoresis (SPEP) remained undetectable, but serum kappa free light chain levels increased from 1.98 mg/dL to 8 mg/dL with stable lambda serum free light chains and a ratio that rose to 16, consistent with low-level biochemical recurrence. He had no evidence of end-organ damage and therefore was maintained on lenalidomide maintenance for the time being. Over the next 12 months, his kappa serum free light chain level continued to slowly rise, reaching 24 mg/dL, while the ratio rose to 50. There was still no detectable M-spike. He developed mild anemia during this time, with his hemoglobin dropping from a prior value of approximately 11 g/dL to 9.8 g/dL, though kidney function remained normal. A repeat bone marrow aspirate and biopsy revealed 20% kappa-restricted plasma cells.
Patient B
A 75-year-old woman with IgA-kappa MM was diagnosed after laboratory testing by her primary care physician incidentally showed an elevated serum total protein level. The MM was intermediate risk, with RISS stage II disease, and with mild renal impairment resulting in an estimated creatinine clearance of 45 mL/min that was felt to be due to MM. She was initially treated with bortezomib and dexamethasone but received only 2 cycles because she developed painful peripheral neuropathy secondary to bortezomib. Bortezomib was stopped and she was then treated with lenalidomide and dexamethasone for 4 cycles. She achieved a complete response and elected to stop treatment due to fatigue. Her fatigue did not improve off treatment. Six months after stopping therapy, an M-spike was detectable at 0.1 g/dL and she developed a new painful lytic lesion in the left humerus.
Patient C
A 59-year-old man with lambda free light chain MM was diagnosed when he presented with acute renal failure requiring dialysis. The disease was RISS-III at diagnosis (high risk), with the t(4;14) genetic abnormality in his MM cells detected on bone marrow aspirate, an abnormality that has been associated with poor prognosis MM [6–8]. The patient was treated with cyclophosphamide, bortezomib, and dexamethasone [9] for 6 cycles, at which point his disease was in a very good partial response (>90% reduction in M-spike) [4], and his renal function had recovered to a new baseline creatinine clearance of 45 mL/min. He then underwent ASCT after melphalan conditioning followed by bortezomib maintenance therapy every 2 weeks. Eight months after ASCT, his lambda free light chain level increased from 1.25 mg/dL to 45 mg/dL and the ratio increased from 4 to 22. Renal function was unchanged and there was stable anemia, with hemoglobin of 10.1 g/dL.
When should treatment for RRMM commence?
Patients with MM in remission are closely monitored, with clinical and laboratory examinations generally conducted every 1 to 3 months. The history is focused on MM-related symptoms such as increasing bone pain or weight loss, and symptoms of therapy-related toxicity such as fatigue, gastrointestinal distress, or peripheral neuropathy. Laboratory assessment typically includes blood counts and chemistry measurements, as well as measurements of MM-derived monoclonal proteins: SPEP, serum immunofixation (IFE), serum immunoglobulin free light chain measurements, and urine protein electrophoresis and immunofixation (UPEP/urine IFE) [10]. Progressive disease biochemically is defined as a 25% increase in M-spike (at least 0.5 g/dL if the M-spike is in serum or > 200 mg/24 hours if in urine), and/or a rise of greater than 10 mg/dL difference between the involved and uninvolved serum free light chains. Clinically progressive disease is denoted as new evidence of end-organ damage such as a new plasma-cytoma, unexplained hypercalcemia, or worsening anemia due to MM [4]. Many, if not most, patients will have biochemical recurrence identified by laboratory measurements ofmonoclonal proteins before clinical recurrence transpires.
The velocity of relapse can help guide decisions about when to reinitiate therapy. High-velocity disease relapse, meaning rapid rise in monoclonal proteins, is an indicator of more aggressive disease, and treatment should be initiated promptly before development of symptoms [11]. Conversely, low-level, indolent recurrence can often be followed with a “watch and wait” approach to determine how the myeloma will progress over time. Expert guidelines suggest that a monoclonal protein doubling time of 2 months may be an appropriate cutoff for determining high versus low velocity [12], although 2 months is not a firm rule and the decision of when to restart treatment for any given patient with asymptomatic biochemical recurrence should be individualized. Importantly, it is not clear that changing therapy at the time of biochemical recurrence, prior to clinical disease progression, improves outcomes, but clinicians are often nonetheless hesitant to hold therapy in the face of biochemically recurrent MM given the potential for complications, such as a pathologic fracture. In patients with biochemically recurrent MM for whom re-initiation of systemic anti-myeloma therapy is being deferred, one can consider re-initiation of zoledronic acid therapy, since in a randomized controlled trial, zoledronic acid commenced at the time of biochemical relapse resulted in fewer skeletal events as compared to placebo [13].
What disease factors should be considered in choosing treatment for RRMM?
MM exhibits genetic complexity, and prior treatments may result in clonal evolution of and selection for an initially nondominant, treatment-resistant clone [14,15]. This heterogeneity and selection pressure may explain why 3-drug regimens often outperform 1- or 2-drug regimens, why each remission is generally shorter than the last, and why patients who have enjoyed a long duration of response to one therapy and been off it for some length of time may again have a good response when re-treated with the same therapy at time of MM relapse. So how does one know if a new clone has emerged? While there is no standard for monitoring intra-clonal heterogeneity presently, changes in clinical phenotype likely correlate with evolving clones. Some such changes include free light chain escape (ie, MM that initially secreted an intact M-spike and then only secretes free light chain at relapse), new development of extramedullary disease (plasmacytomas outside of bone) in patients who previously had MM only in the bone marrow, and resistance of some sites to treatment while others respond (a mixed response). The former 2 phenotypes in particular portend poor prognosis and unsurprisingly they can be seen together [16–19]. Restaging, meaning a complete reassessment of MM disease status at the time of relapse, including bone marrow aspirate and biopsy, is beneficial to help guide therapy, as those with high-risk features including high ISS stage [20], high-risk cytogenetics, increased LDH, and extramedullary disease should be treated with triplet therapy when possible [11]. Repeat imaging should also be considered as a new baseline comparator. This can be done with standard x-rays, positron-emission tomography/computed tomography (PET-CT), or magnetic resonance imaging. PET-CT offers the advantage of showing active disease sites and the presence of extramedullary disease, although it exposes the patient to more radiation than the other methods.
In terms of using genetics to guide therapy decisions in RRMM, the presence of the del(17p) abnormality either by karyotyping or FISH portends high risk and pomalidomide in one study was shown to mitigate that risk [21]. How genetics and prognostic markers should dictate therapy selection in RRMM otherwise, however, is unclear and an area of active research efforts.
What patient factors should be considered in choosing treatment?
Given the relatively large selection of possible regimens for the treatment of RRMM, patient preference can be incorporated into regimen selection. Patients who have long commutes or who are trying to work may not be ideal candidates to receive carfilzomib-based regimens given the twice-per-week infusion schedule (though a once-a-week dosing schedule is being tested) [22]. Patients who have poor venous access may be good candidates for all-oral regimens. Prior treatment tolerability and side effects should also be considered. Patients who experienced significant peripheral neuropathy with bortezomib may have less neuropathy with carfilzomib. Those with renal failure may tolerate pomalidomide better than lenalidomide [23].
Patient age and functional status are important considerations in choosing a treatment regimen for RRMM. Very old patients (a subjective categorization to include patients > 80 years by chronologic or physiologic age), those with functional dependence, or patients harboring substantial medical comorbidities are at risk for therapy toxicity and so often warrant less intensive approaches [24]. Deciding which patients empirically warrant less dose-intensive approaches can be challenging, especially with the growing recognition that fit seniors can often tolerate and enjoy the benefits of full-dose approaches, including sometimes even ASCT. Geriatric assessment instruments that interrogate a variety of geriatric-relevant domains, such as number of falls, independence in activities of daily living, and polypharmacy, are being investigated as toxicity predictors and may help make those decisions in the future. Such instruments have been shown to predict chemotherapy toxicity in solid tumors [25,26] and preliminarily in MM [27], but they remain investigational. While no validated geriatric assessment instruments are currently available for routine clinical employment in MM, clinicians should consider the geriatric domains that these instruments assess when choosing among treatment options. Clinically, that often translates to choosing gentler regimens with likely better tolerability, albeit perhaps with less efficacy, for patients judged to be vulnerable to toxicity.
As part of therapy selection in RRMM, the clinician needs to consider if the patient is a candidate for ASCT. For patients who did not undergo ASCT as part of initial treatment, ASCT can be considered at the time of relapse. Ideally, all patients who could eventually undergo ASCT should have hematopoietic stem cells collected and stored at the time of first induction; however, collection after re-induction chemotherapy has been shown to be feasible [28,29]. ASCT for RRMM appears to be effective, although rigorous randomized comparisons of ASCT versus treatment purely with novel drugs are lacking [30–32]. For patients who did receive ASCT consolidation in the frontline, if a response is sustained for 18 months or greater, existing guidelines suggest that a second ASCT is likely worthwhile [29]. Whether the routine usage of maintenance therapies (low-dose, usually single drugs used to prolong duration of remission once remission is achieved) should change that 18-month cutoff is unclear, however, since maintenance “artificially” makes ASCT appear more effective by prolonging post-ASCT duration of remission. The “is it worth it” discussion is also largely subjective and hinges heavily on the patient’s experience with the first ASCT. In our practice, we often use 3 years as the cutoff for considering repeat ASCT in patients on maintenance therapy, meaning that if a patient underwent ASCT and received maintenance, a remission lasting more than 3 years means we consider ASCT as part of therapy for relapse.
Allogeneic stem cell transplantation (allo-SCT) is a treatment option for RRMM generally reserved for fit patients younger than 65 years [22,33]. The timing of allo-SCT is also controversial, with some reserving it as a last option given a historically high transplant-related mortality and improved progression-free survival but not necessarily overall survival benefit. A recent consensus statement has suggested allo-SCT be considered (preferentially in a clinical trial) for eligible patients with high-risk disease who relapse after primary treatment that included ASCT [29]. With the abundance of new treatment options in RRMM with reasonable toxicity profiles, it is not clear for whom and when allo-SCT is best considered.

Which regimen should be used to treat a first relapse?
Entry into a well-designed clinical trial for patients with RRMM should be considered for every patient since there is a lack of evidence to guide the best sequencing of chemotherapies [11]. Beyond that, the choice of therapy is based upon 2 main factors: the disease itself (eg, indolent, asymptomatic biochemical recurrence versus aggressive clinical recurrence with new fractures or extramedullary plasmacytomas), and the patient’s preferences and characteristics, such as age, performance status, comorbidities, and toxicities from prior therapies. In looking for the “best” re-induction regimen, it is tempting to compare the efficacy of regimens across trials, but such efforts are fraught given the significant heterogeneity of the patient populations between trials. As an example, comparing daratumumab + pomalidomide + dexamethasone (DPd) to daratumumab + lenalidomide + dexamethasone (DRd), one may conclude that DRd is superior, given an overall response rate of 88% in DRd versus 58% in DPd. However, the DPd trial included patients who were refractory to lenalidomide and bortezomib, while the DRd study required only treatment with one prior therapy [34,35].
For patients who enjoyed a long remission after any particular chemotherapy regimen with good tolerability and with indolent features at the time of relapse, re-treating with the same regimen can be considered, although nowadays with so many new and highly potent agents available such “backtracking” is less common and some studies suggest that employing new agents may be beneficial. As an example, in the randomized ENDEAVOR study of bortezomib + dexamethasone versus carfilzomib + dexamethasone in RRMM, 54% of patients had been exposed to bortezomib whereas virtually none had received carfilzomib prior to study enroll-ment. Among those patients with prior bortezomib exposure, median progression-free survival was 15.6 versus 8.1 months (hazard ratio 0.56, [95% confidence interval 0.44 to 0.73]) for carfilzomib versus bortezomib, respectively. Follow-up was too immature for definitive conclusions to be drawn about overall survival, but the substantial difference in progression-free survival provides a compelling argument for using carfilzomib instead of going back to bortezomib for patients with prior bortezomib exposure [36].
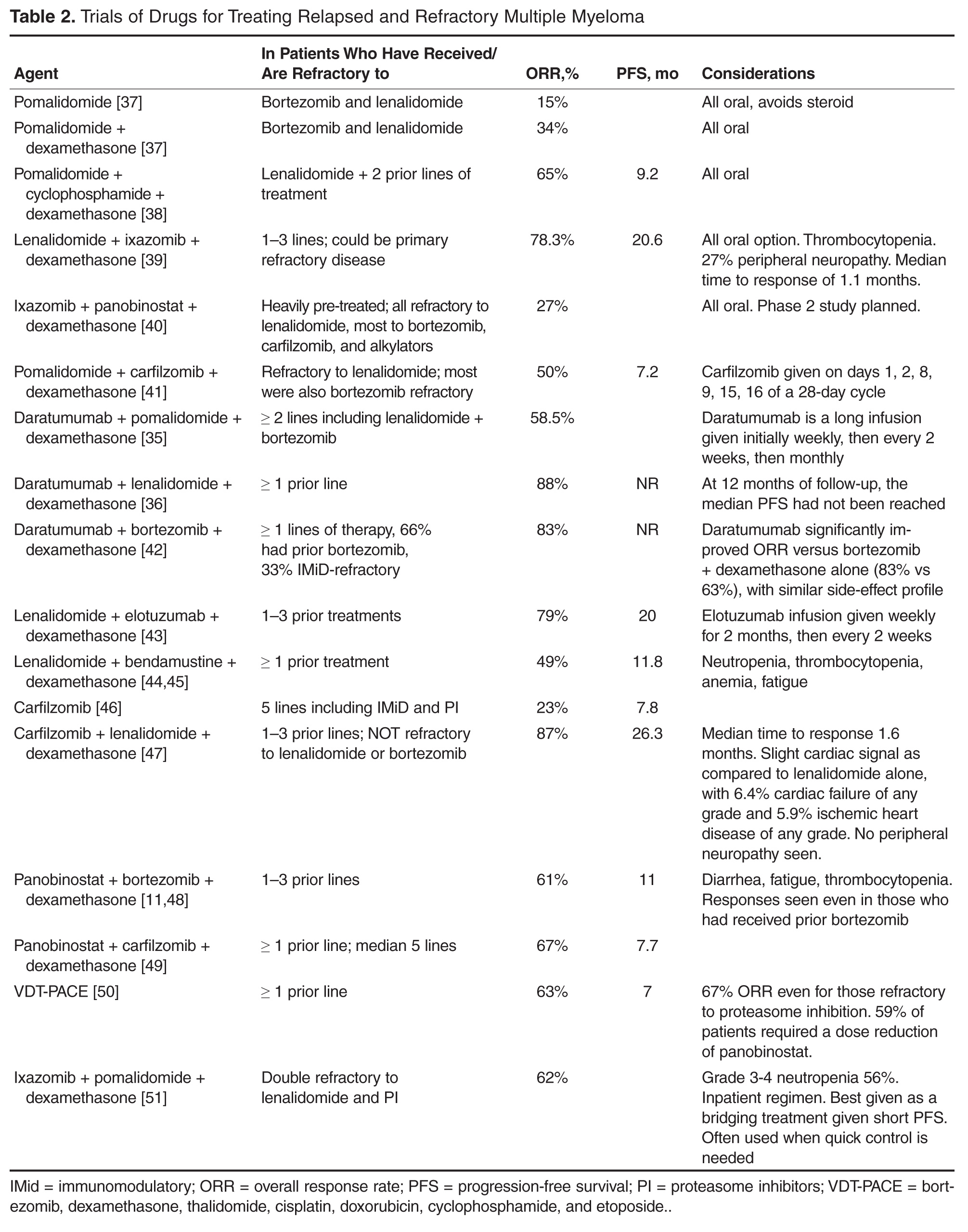
For patients who are fit and not very old, we generally employ triplet re-induction. For the large number of these patients who were previously exposed to both lenalidomide and bortezomib, including as part of a maintenance strategy, outside of clinical trials we routinely use carfilzomib + pomalidomide + dexamethasone [41]. For patients who are lenalidomide-naïve but bortezomib-exposed, we often employ carfilzomib + lenalidomide + dexamethasone based on the phase 3 ASPIRE trial, which showed a significantly improved progression-free survival with carfilzomib + lenalidomide + dexamethasone versus lenalidomide + dexamethasone [47]. For patients who have previously received lenalidomide but not bortezomib, we consider pomalidomide + bortezomib + dexamethasone [52]. These regimens take advantage of the arguably most potent, most proven drugs in treating RRMM, namely proteasome inhibitors (bortezomib and carfilzomib) and immunomodulatory agents (lenalidomide and pomalidomide).
For patients who are more vulnerable to toxicity due to advanced age or comorbidities, we consider less intensive regimens, including dose-reduced triplets or doublets. Patients who had received lenalidomide-based combinations but not bortezomib are considered for a bortezomib-based re-induction, including bortezomib + dexamethasone alone. In the case of someone who had initially received a bortezomib-based combination but no lenalidomide, the new drugs are viable options: ixazomib [53] or elotuzumab [43] can both be added to standard lenalidomide + dexamethasone, with expectations of increasing response rates and progression-free survival and an acceptably low increased risk of severe toxicity. Ixazomib + lenalidomide + dexamethasone also has the benefit of being all-oral. For patients with bortezomib- and lenalidomide-exposed RRMM, using carfilzomib [54] or pomalidomide [55] with dexamethasone is reasonable.
Once MM has progressed beyond the arguable “core drugs” of early-stage MM, namely lenalidomide, bortezomib, carfilzomib, and pomalidomide, off protocol we favor daratumumab monotherapy [56,57]. Other options include panobinostat (given usually with bortezomib) [58] and bendamustine [59], among others.
Can agents to which the MM was previously refractory be reused?
With the understanding that MM is not a disease defined by a single molecular mutation, but rather clones and subclones, it is reasonable to think that even treatments that have previously failed may be beneficial to patients if they have been off those treatments for some length of time and sensitive subclones reemerge. Additionally, combining the “failed” agent with a new drug may overcome the previously seen refractoriness, as in the case of panobinostat + bortezomib [48]. That said, given the multitude of new treatment options for RRMM and data from such trials as ENDEAVOR as mentioned, revisiting previously used drugs is probably best reserved for second or greater relapses.
What should be the duration of therapy for RRMM?
There is no evidence to guide duration of therapy in RRMM. Most patients with relapsed disease will be considered for continuous treatment until disease progression, which usually means treatment for 6 to 12 months with full-dose induction, often to maximal response, followed by transition to some form of lower-dose maintenance in which parts of a multi-drug regimen may be eliminated and/or the doses for the remaining drugs may be reduced. Patients with a slow-velocity relapse and no markers of high-risk disease may be suitable candidates for a defined course of treatment without maintenance therapy [11], but most patients nowadays remain on some form of maintenance for RRMM after achieving remission.
What supportive care is needed in RRMM?
Bone Health
Skeletal-related events, namely fractures, can be devastating in MM. Bisphosphonates have been shown to decrease such events in MM and zoledronic acid has shown a trend toward improved survival, perhaps related to its impact on the bone marrow microenvironment or direct toxicity to myeloma cells [60,61]. It is unclear whether bisphosphonates improve overall survival in the relapsed setting, although zoledronic acid has shown decreased skeletal-related events in the setting of biochemical-only disease progression [13,62]. In active RRMM, our general practice is to resume parenteral bisphosphonate therapy (either zoledronic acid or pamidronate in our U.S. practices) usually every 3 to 4 weeks, depending on the length of the chemotherapy cycle.
Supportive Care
RRMM is a complex disease in which patients often experience a multitude of symptoms and other complications as a result of the disease itself as well as therapy. Aggressive supportive care is of paramount importance. As examples, zoster prophylaxis is required for virtually all patients on proteasome inhibitors, anticoagulation/antiplatelet therapies should be considered for venous thrombotic event prophylaxis, and proton pump inhibitors may be appropriate for these patients who often have a real risk of peptic ulcer disease due to the use of corticosteroids, nonsteroidal anti-inflammatory drugs, and/or aspirin prophylaxis. Attention to dental health is important for patients on bisphosphonates to minimize the risk of osteonecrosis of the jaw. Nutritional problems should be monitored and can arise due to anorexia, dysgeusia, diarrhea, or constipation. Peripheral neuropathy is extremely common and support should be offered in the form of adjusting therapy to minimize risk of worsening it, analgesics if needed, assistive devices to aid in ambulation, and/or physical therapy. Depression and anxiety are understandably prevalent in patients with RRMM, who face an incurable disease that provides constant reminders of its presence due to symptoms, the need for daily pills, or frequent clinic visits for treatment and/or blood product transfusions [63]. Supporting a patient’s emotional health is a vital component of enhancing quality of life in RRMM.
Case Studies Continued
Patient A was noted to have biochemical progression initially, with relapse detectable only in serum free light chains. Treatment commenced at the time of worsening anemia. Notably, his disease originally secreted IgG-kappa and at relapse secreted kappa free light chain only; that is, he developed “light chain escape,” which signifies a high-risk disease and likely heralds clonal evolution [64]. He had excellent caregiver support and lived within 20 minutes of a treatment center. His performance status remained good at the time of relapse and he had normal organ function. He was treated with carfilzomib + pomalidomide + dexamethasone for 4 cycles, achieving a very good partial response. He then received a second ASCT with melphalan conditioning and again achieved stringent complete response. Indefinite maintenance therapy commenced with pomalidomide, and at 16 months post-ASCT he was doing well and still in remission.
Patient B was symptomatic at the time of disease progression. As her primary complaint was that of a painful humeral lytic lesion, she first underwent a course of palliative radiation, which alleviated her pain. She did not wish to restart systemic treatment and instead elected to watch her MM closely with her oncologist on a monthly basis. By 3 months, her M-spike had reached 0.6 g/dL and her serum creatinine had increased slightly, resulting in a creatinine clearance of 34 mL/min. She lived approximately 90 minutes from the closest treatment facility and found it difficult to come for visits more than once monthly. Her Eastern College Oncology Group (ECOG) performance status was 2. With her advanced age and frailty, she was not considered to be a good candidate for ASCT. She requested to go back on lenalidomide and decided with her oncologist to try ixazomib + lenalidomide + dexamethasone, with which she achieved a very good partial response. She had difficulty with myelosuppression with lenalidomide, which was dropped after 4 cycles, and she is planned for ixazomib maintenance until disease progression or drug intolerance. She receives monthly zoledronic acid to reduce the risk of fractures.
Patient C has high-risk disease as indicated by R-ISS III stage disease at diagnosis and progression only 8 months after ASCT and while on bortezomib maintenance therapy. Although he currently only has evidence of biochemical relapse, prompt initiation of treatment was warranted to prevent further renal compromise such as during his initial presentation [65]. Further, PET-CT showed the presence of extramedullary soft tissue disease, another high-risk feature. He was a robust patient with good social support and received carfilzomib + pomalidomide + dexamethasone re-induction. He was not considered for a second ASCT given his short duration of response. With his high-risk features of early relapse after ASCT, R-ISS III, and extramedullary disease, it was recommended that he continue triplet drug therapy until disease relapse or drug intolerance.
Ongoing and Future Trials
The management of RRMM will continue to evolve as paradigms for treating MM change and new treatment options become available. In particular, immunotherapies (ie, approaches that harness the immune system’s ability to fight cancer) are under exploration and some such drugs that are already FDA-approved in other diseases are being tested in MM. Chimeric antigen receptor-T cells (CAR-T), a form of cell-based immunotherapy, have generated tremendous excitement in acute lymphocytic leukemia [66] and are being tested in MM [67]. New analogs of old drugs may offer more effective, less toxic ways to control MM. The role of ASCT is being explored in randomized trials investigating whether ASCT should be pursued early or late in a patient’s MM course. These studies will no doubt further augment the armamentarium of anti-myeloma drugs that have already resulted in the increasingly longer survival we see today in this disease [3,68]. That said, MM remains incurable, and almost all patients who live long enough eventually relapse and die of MM. Hence, further research and progress are critical.
Summary
A well-designed clinical trial should be considered for all patients with RRMM, and in lieu of an available trial, regimen selection should be tailored upon disease and patient characteristics. Carfilzomib-based regimens are among the most popular at the time of first relapse currently based upon their efficacy in bortezomib-refractory cases and tolerability. Pomalidomide shows activity in lenalidomide-refractory patients. Due to intra-clonal heterogeneity, triplet regimens are preferred for fit patients, reserving doublet or monotherapy for those patients who are frail or who have an indolent disease relapse. Ongoing research will undoubtedly improve outcomes for RRMM, a disease for which the prognosis is far better than it formerly was, but which still has quite a bit of room for improvement.
Corresponding author: Brandi Reeves, MD, University of North Carolina – Chapel Hill, 170 Manning Dr., Physicians’ Office Building, CB 7305, Chapel Hill, NC 27599, brandi_reeves@med.unc.edu.
Financial disclosures: Dr. Tuchman reports the following: speakers’ bureau: Celgene, Takeda; consulting: Celgene, Takeda; research support: Celgene, Takeda, Novartis, Onyx.
1. Pulte D, Redaniel MT, Brenner H, et al. Recent improvement in survival of patients with multiple myeloma: variation by ethnicity. Leuk Lymphoma 2014;55:1083–9.
2. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 2014;28:1122–8.
3. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008;111:2516–20.
4. Rajkumar SV, Harousseau J-L, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 2011;117:4691–5.
5. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015;33:2863–9.
6. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood 2015;125:2095–100.
7. Karlin L, Soulier J, Chandesris O, et al. Clinical and biological features of t(4;14) multiple myeloma: a prospective study. Leuk Lymphoma 2011;52:238–46.
8. Moreau P, Cavo M, Sonneveld P, et al. Combination of international scoring system 3, high lactate dehydrogenase, and t(4;14) and/or del(17p) Identifies patients with multiple myeloma (MM) treated with front-line autologous stem-cell transplantation at high risk of early MM progression–related dDeath. J Clin Oncol 2014;32:2173–80.
9. Reeder CB, Reece DE, Kukreti V, et al. Long-term survival with cyclophosphamide, bortezomib and dexamethasone induction therapy in patients with newly diagnosed multiple myeloma. Br J Haematol 2014;167:563–5.
10. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016;17:e328–46.
11. Laubach J, Garderet L, Mahindra A, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia 2016;30:1005–17.
12. Palumbo A, Rajkumar SV, San Miguel JF, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not Eligible for standard autologous stem-cell transplantation. J Clin Oncol 2014;32:587–600.
13. Garcia-Sanz R, Oriol A, Moreno MJ, et al. Zoledronic acid as compared with observation in multiple myeloma patients at biochemical relapse: results of the randomized AZABACHE Spanish trial. Haematologica 2015;100:1207–13.
14. Bahlis NJ. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012;120:927–8.
15. Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012;120:1067–76.
16. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
17. Dawson MA, Patil S, Spencer A. Extramedullary relapse of multiple myeloma associated with a shift in secretion from intact immunoglobulin to light chains. Haematologica 2007;92:143–4.
18. Usmani SZ, Heuck C, Mitchell A, et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012;97:1761–7.
19. Varettoni M, Corso A, Pica G, et al. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: a longitudinal study on 1003 consecutive patients. Ann Oncol 2010;21:325–30.
20. Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol 2005;23:3412–20.
21. Leleu X, Karlin L, Macro M, et al. Pomalidomide plus low-dose dexamethasone in multiple myeloma with deletion 17p and/or translocation (4;14): IFM 2010-02 trial results. Blood 2015;125:1411–7.
22. Berenson JR, Cartmell A, Bessudo A, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood 2016 Jun 30;127:3360–8.
23. Richter J, Biran N, Duma N, et al. Safety and tolerability of pomalidomide-based regimens (pomalidomide-carfilzomib-dexamethasone with or without cyclophosphamide) in relapsed/refractory multiple myeloma and severe renal dysfunction: a case series. Hematol Oncol 2016 Mar 27. doi: 10.1002/hon.2290.
24. Wildes TM, Rosko A, Tuchman SA. Multiple myeloma in the older adult: better prospects, more challenges. J Clin Oncol 2014;32:2531–40.
25. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) score. Cancer 2012;118(13):3377-86.
26. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol 2011;29:3457–65.
27. Palumbo A, Bringhen S, Mateos M-V, et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood 2015;125:2068–74.
28. Lemieux E, Hulin C, Caillot D, et al. Autologous stem cell transplantation: an effective salvage therapy in multiple myeloma. Biol Blood Marrow Transplant 2013;19:445–9.
29. Giralt S, Garderet L, Durie B, et al. American Society of Blood and Marrow Transplantation, European Society of Blood and Marrow Transplantation, Blood and Marrow Transplant Clinical Trials Network, and International Myeloma Working Group Consensus Conference on Salvage Hematopoietic Cell Transplantation in Patients with Relapsed Multiple Myeloma. Biol Blood Marrow Transplant 2015;21:2039–51.
30. Cook G, Williams C, Brown JM, et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse [Intensive trial]): a randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:874–85.
31. Grövdal M, Nahi H, Gahrton G, et al. Autologous stem cell transplantation versus novel drugs or conventional chemotherapy for patients with relapsed multiple myeloma after previous ASCT. Bone Marrow Transplant 2015;50:808–12.
32. Singh Abbi KK, Zheng J, Devlin SM, et al. Second autologous stem cell transplant: an effective therapy for relapsed multiple myeloma. Biol Blood Marrow Transplant 2015;21:468–72.
33. Franssen LE, Raymakers RA, Buijs A, et al. Outcome of allogeneic transplantation in newly diagnosed and relapsed/refractory multiple myeloma: long-term follow-up in a single institution. European J Haematol 2016 Mar 29.
34. Chari AL, Sagar SA, Fay JW, et al. Open-label, multicenter, phase 1b study of daratumumab in combination with pomalidomide and dexamethasone in patients with at least 2 lines of prior therapy and relapsed or relapsed and refractory multiple myeloma. Blood 2015;126:508.
35. Plesner T, Gimsing P, Krejcik J, et al. Daratumumab in combination with lenalidomide and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: Updated results of a phase 1/2 study (GEN503). Blood 2015;126:507.
36. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016;17:27–38.
37. Richardson PG, Siegel D, Baz R, et al. Phase 1 study of pomalidomide MTD, safety, and efficacy in patients with refractory multiple myeloma who have received lenalidomide and bortezomib. Blood 2013;121:1961–7.
38. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 2014;123:1826–32.
39. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
40. Reu FJ, Valent J, Malek E, et al. A phase I study of ixazomib in combination with panobinostat and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood 2015;126:4221.
41. Shah JJ, Stadtmauer EA, Abonour R, et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood 2015;126:2284–90.
42. Palumbo A, Chanan-Khan AA, Weisel K, et al. Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study. J Clin Oncol 2016;34(suppl):abstr LBA4.
43. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015;373:621–31.
44. Lentzsch S, O’Sullivan A, Kennedy RC, et al. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: results of phase 1/2 open-label, dose escalation study. Blood 2012;119:4608–13.
45. Kumar SK, Krishnan A, LaPlant B, et al. Bendamustine, lenalidomide, and dexamethasone (BRD) is highly effective with durable responses in relapsed multiple myeloma. Am J Hematol 2015;90:1106–10.
46. Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012;120:2817–25.
47. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142–52.
48. San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncology 2014;15:1195–206.
49. Berdeja JG, Hart LL, Mace JR, et al. Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica 2015;100:670–6.
50. Buda G, Orciuolo E, Galimberti S, Ghio F, Petrini M. VTDPACE as salvage therapy for heavily pretreated MM patients. Blood 2013;122:5377.
51. Voorhees PM, Mulkey F, Hassoun H, et al. Alliance A061202. A phase I/II study of pomalidomide, dexamethasone and ixazomib versus pomalidomide and dexamethasone for patients with multiple myeloma refractory to lenalidomide and proteasome inhibitor based therapy: phase I results. Blood 2015;126:375.
52. Lacy MQ, LaPlant BR, Laumann KM, et al. Pomalidomide, bortezomib and dexamethasone (PVD) for patients with relapsed lenalidomide refractory multiple myeloma (MM). Blood 2014;124:304.
53. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
54. Vij R, Siegel DS, Jagannath S, et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol 2012;158:739–48.
55. Leleu X, Attal M, Arnulf B, et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide-refractory multiple myeloma: Intergroupe Francophone du Myelome 2009-02. Blood 2013;121:1968–75.
56. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016;387(10027):1551–60.
57. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015;373:1207–19.
58. Richardson PG, Schlossman RL, Alsina M, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013;122:2331–7.
59. Stöhr E, Schmeel FC, Schmeel LC, et al. Bendamustine in heavily pre-treated patients with relapsed or refractory multiple myeloma. J Cancer Res Clin Oncol 2015;141:2205–12.
60. Mhaskar R, Redzepovic J, Wheatley K, et al. Bisphosphonates in multiple myeloma: a network meta-analysis. Cochrane Database Syst Rev 2012(5):CD003188.
61. Dhodapkar MV, Singh J, Mehta J, et al. Anti-myeloma activity of pamidronate in vivo. Br J Haematol 1998;103:530–2.
62. Terpos E, Morgan G, Dimopoulos MA, et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J Clin Oncol 2013;31:2347–57.
63. Kiely F, Cran A, Finnerty D, O’Brien T. Self-reported quality of life and symptom burden in ambulatory patients with multiple myeloma on disease-modifying treatment. Am J Hosp Palliat Care 2016 May 2.
64. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
65. Ludwig H, Sonneveld P, Davies F, et al. European perspective on multiple myeloma treatment strategies in 2014. Oncologist 2014;19:829–44.
66. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17.
67. Garfall AL, Maus MV, Hwang W-T, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med 2015;373:1040–7.
68. Kumar SK, Dispenzieri A, Gertz MA, et al. Continued improvement in survival in multiple myeloma and the impact of novel agents. 54th ASH Annual Meeting Abstracts. Blood 2012;120(21):3972.
From the Division of Hematology and Oncology, University of North Carolina – Chapel Hill, Chapel Hill, NC (Dr. Reeves), and the Division of Cellular Therapy and Hematological Malignancies, Duke Cancer Institute, Durham, NC (Dr. Tuchman).
Abstract
- Objective: To review the management considerations in patients with relapsed and refractory multiple myeloma (RRMM).
- Methods: Review of the literature.
- Results: RRMM is a heterogeneous disease and numerous treatment regimens have been studied. Despite improvement in progression-free and overall survival in newly diagnosed multiple myeloma with current therapies, myeloma remains incurable and repeated relapses are inevitable. Relapses are often characterized by diminished response to chemotherapy (refractoriness) and duration of response.
- Conclusion: Management of RRMM should be individualized using both patient- and disease-related factors, given substantial heterogeneity in both. Further research regarding the optimal timing, regimen, and duration of treatment is warranted.
Although advancements in treating multiple myeloma (MM) have resulted in improved median survival from approximately 2 years in the 1990s to more recent estimates of over 6 years, the disease remains incurable [1–3]. Its overall course is generally defined by a series of increasingly short remissions and treatment-refractory relapses until eventual death due to MM occurs. Objective criteria for defining both relapsed and refractory MM have been published [4]. Briefly, relapsed myeloma is that which has been previously treated with some form of systemic therapy and which has recurred. That recurrence can be clinical (ie, the development of new or worsening signs or symptoms of active MM) and/or biochemical (ie, rising monoclonal MM proteins in the serum or urine). Refractory MM on the other hand refers to MM that is resistant to particular drugs, defined as MM that is nonresponsive to primary or salvage therapy, or MM that progresses within 60 days of the last therapy [4]. At any juncture during the course of relapsed MM, patients will have disease that is either sensitive or refractory to specific myeloma drugs. In this article, we discuss management of these often concurrent entities together as relapsed and refractory multiple myeloma (RRMM).
There are numerous treatment options for patients with RRMM—3 new drugs were approved in November 2015 alone. The abundance of available drugs leaves treating clinicians with a daunting task of sequencing therapies among several choices. The durability of response to treatment typically lessens with each disease relapse, such that the clinician needs to think of sequencing not just second-line therapy, but third- and fourth-line as well, further complicating the decision. In this review, we aim to help clinicians individualize treatment plans for patients with RRMM.
Case Studies
Patient A
A 62-year-old man with IgG-kappa MM was diagnosed 4 years ago during evaluation of a pathologic humeral fracture. The disease was prognostically standard risk, with revised International Staging System (RISS) stage I disease (beta-2 microglobulin 3.4 mcg/mL, albumin 4.1 g/dL, normal cytogenetics with 46,XY in 20 cells analyzed, and myeloma fluorescent in situ hybridization [FISH] panel showing t(11;14) but no del17p, t(14;16), t(14;20), or t(4;14)) [5], and normal blood counts, organ function, and lactate dehydrogenase (LDH) at diagnosis. He was treated with 5 cycles of standard lenalidomide, bortezomib, and dexamethasone followed by high-dose melphalan with autologous stem cell transplantation (ASCT) and then lenalidomide continuous maintenance. He achieved a stringent complete response (ie, complete disappearance of myeloma-derived monoclonal proteins in the serum and urine, a normal serum free light chain ratio, and undetectable monoclonal plasma cells on a bone marrow aspirate and biopsy) [4]. His MM was monitored every 2 to 3 months for disease progression and medication toxicity. At month 38, a monoclonal protein spike (M-spike) on serum protein electrophoresis (SPEP) remained undetectable, but serum kappa free light chain levels increased from 1.98 mg/dL to 8 mg/dL with stable lambda serum free light chains and a ratio that rose to 16, consistent with low-level biochemical recurrence. He had no evidence of end-organ damage and therefore was maintained on lenalidomide maintenance for the time being. Over the next 12 months, his kappa serum free light chain level continued to slowly rise, reaching 24 mg/dL, while the ratio rose to 50. There was still no detectable M-spike. He developed mild anemia during this time, with his hemoglobin dropping from a prior value of approximately 11 g/dL to 9.8 g/dL, though kidney function remained normal. A repeat bone marrow aspirate and biopsy revealed 20% kappa-restricted plasma cells.
Patient B
A 75-year-old woman with IgA-kappa MM was diagnosed after laboratory testing by her primary care physician incidentally showed an elevated serum total protein level. The MM was intermediate risk, with RISS stage II disease, and with mild renal impairment resulting in an estimated creatinine clearance of 45 mL/min that was felt to be due to MM. She was initially treated with bortezomib and dexamethasone but received only 2 cycles because she developed painful peripheral neuropathy secondary to bortezomib. Bortezomib was stopped and she was then treated with lenalidomide and dexamethasone for 4 cycles. She achieved a complete response and elected to stop treatment due to fatigue. Her fatigue did not improve off treatment. Six months after stopping therapy, an M-spike was detectable at 0.1 g/dL and she developed a new painful lytic lesion in the left humerus.
Patient C
A 59-year-old man with lambda free light chain MM was diagnosed when he presented with acute renal failure requiring dialysis. The disease was RISS-III at diagnosis (high risk), with the t(4;14) genetic abnormality in his MM cells detected on bone marrow aspirate, an abnormality that has been associated with poor prognosis MM [6–8]. The patient was treated with cyclophosphamide, bortezomib, and dexamethasone [9] for 6 cycles, at which point his disease was in a very good partial response (>90% reduction in M-spike) [4], and his renal function had recovered to a new baseline creatinine clearance of 45 mL/min. He then underwent ASCT after melphalan conditioning followed by bortezomib maintenance therapy every 2 weeks. Eight months after ASCT, his lambda free light chain level increased from 1.25 mg/dL to 45 mg/dL and the ratio increased from 4 to 22. Renal function was unchanged and there was stable anemia, with hemoglobin of 10.1 g/dL.
When should treatment for RRMM commence?
Patients with MM in remission are closely monitored, with clinical and laboratory examinations generally conducted every 1 to 3 months. The history is focused on MM-related symptoms such as increasing bone pain or weight loss, and symptoms of therapy-related toxicity such as fatigue, gastrointestinal distress, or peripheral neuropathy. Laboratory assessment typically includes blood counts and chemistry measurements, as well as measurements of MM-derived monoclonal proteins: SPEP, serum immunofixation (IFE), serum immunoglobulin free light chain measurements, and urine protein electrophoresis and immunofixation (UPEP/urine IFE) [10]. Progressive disease biochemically is defined as a 25% increase in M-spike (at least 0.5 g/dL if the M-spike is in serum or > 200 mg/24 hours if in urine), and/or a rise of greater than 10 mg/dL difference between the involved and uninvolved serum free light chains. Clinically progressive disease is denoted as new evidence of end-organ damage such as a new plasma-cytoma, unexplained hypercalcemia, or worsening anemia due to MM [4]. Many, if not most, patients will have biochemical recurrence identified by laboratory measurements ofmonoclonal proteins before clinical recurrence transpires.
The velocity of relapse can help guide decisions about when to reinitiate therapy. High-velocity disease relapse, meaning rapid rise in monoclonal proteins, is an indicator of more aggressive disease, and treatment should be initiated promptly before development of symptoms [11]. Conversely, low-level, indolent recurrence can often be followed with a “watch and wait” approach to determine how the myeloma will progress over time. Expert guidelines suggest that a monoclonal protein doubling time of 2 months may be an appropriate cutoff for determining high versus low velocity [12], although 2 months is not a firm rule and the decision of when to restart treatment for any given patient with asymptomatic biochemical recurrence should be individualized. Importantly, it is not clear that changing therapy at the time of biochemical recurrence, prior to clinical disease progression, improves outcomes, but clinicians are often nonetheless hesitant to hold therapy in the face of biochemically recurrent MM given the potential for complications, such as a pathologic fracture. In patients with biochemically recurrent MM for whom re-initiation of systemic anti-myeloma therapy is being deferred, one can consider re-initiation of zoledronic acid therapy, since in a randomized controlled trial, zoledronic acid commenced at the time of biochemical relapse resulted in fewer skeletal events as compared to placebo [13].
What disease factors should be considered in choosing treatment for RRMM?
MM exhibits genetic complexity, and prior treatments may result in clonal evolution of and selection for an initially nondominant, treatment-resistant clone [14,15]. This heterogeneity and selection pressure may explain why 3-drug regimens often outperform 1- or 2-drug regimens, why each remission is generally shorter than the last, and why patients who have enjoyed a long duration of response to one therapy and been off it for some length of time may again have a good response when re-treated with the same therapy at time of MM relapse. So how does one know if a new clone has emerged? While there is no standard for monitoring intra-clonal heterogeneity presently, changes in clinical phenotype likely correlate with evolving clones. Some such changes include free light chain escape (ie, MM that initially secreted an intact M-spike and then only secretes free light chain at relapse), new development of extramedullary disease (plasmacytomas outside of bone) in patients who previously had MM only in the bone marrow, and resistance of some sites to treatment while others respond (a mixed response). The former 2 phenotypes in particular portend poor prognosis and unsurprisingly they can be seen together [16–19]. Restaging, meaning a complete reassessment of MM disease status at the time of relapse, including bone marrow aspirate and biopsy, is beneficial to help guide therapy, as those with high-risk features including high ISS stage [20], high-risk cytogenetics, increased LDH, and extramedullary disease should be treated with triplet therapy when possible [11]. Repeat imaging should also be considered as a new baseline comparator. This can be done with standard x-rays, positron-emission tomography/computed tomography (PET-CT), or magnetic resonance imaging. PET-CT offers the advantage of showing active disease sites and the presence of extramedullary disease, although it exposes the patient to more radiation than the other methods.
In terms of using genetics to guide therapy decisions in RRMM, the presence of the del(17p) abnormality either by karyotyping or FISH portends high risk and pomalidomide in one study was shown to mitigate that risk [21]. How genetics and prognostic markers should dictate therapy selection in RRMM otherwise, however, is unclear and an area of active research efforts.
What patient factors should be considered in choosing treatment?
Given the relatively large selection of possible regimens for the treatment of RRMM, patient preference can be incorporated into regimen selection. Patients who have long commutes or who are trying to work may not be ideal candidates to receive carfilzomib-based regimens given the twice-per-week infusion schedule (though a once-a-week dosing schedule is being tested) [22]. Patients who have poor venous access may be good candidates for all-oral regimens. Prior treatment tolerability and side effects should also be considered. Patients who experienced significant peripheral neuropathy with bortezomib may have less neuropathy with carfilzomib. Those with renal failure may tolerate pomalidomide better than lenalidomide [23].
Patient age and functional status are important considerations in choosing a treatment regimen for RRMM. Very old patients (a subjective categorization to include patients > 80 years by chronologic or physiologic age), those with functional dependence, or patients harboring substantial medical comorbidities are at risk for therapy toxicity and so often warrant less intensive approaches [24]. Deciding which patients empirically warrant less dose-intensive approaches can be challenging, especially with the growing recognition that fit seniors can often tolerate and enjoy the benefits of full-dose approaches, including sometimes even ASCT. Geriatric assessment instruments that interrogate a variety of geriatric-relevant domains, such as number of falls, independence in activities of daily living, and polypharmacy, are being investigated as toxicity predictors and may help make those decisions in the future. Such instruments have been shown to predict chemotherapy toxicity in solid tumors [25,26] and preliminarily in MM [27], but they remain investigational. While no validated geriatric assessment instruments are currently available for routine clinical employment in MM, clinicians should consider the geriatric domains that these instruments assess when choosing among treatment options. Clinically, that often translates to choosing gentler regimens with likely better tolerability, albeit perhaps with less efficacy, for patients judged to be vulnerable to toxicity.
As part of therapy selection in RRMM, the clinician needs to consider if the patient is a candidate for ASCT. For patients who did not undergo ASCT as part of initial treatment, ASCT can be considered at the time of relapse. Ideally, all patients who could eventually undergo ASCT should have hematopoietic stem cells collected and stored at the time of first induction; however, collection after re-induction chemotherapy has been shown to be feasible [28,29]. ASCT for RRMM appears to be effective, although rigorous randomized comparisons of ASCT versus treatment purely with novel drugs are lacking [30–32]. For patients who did receive ASCT consolidation in the frontline, if a response is sustained for 18 months or greater, existing guidelines suggest that a second ASCT is likely worthwhile [29]. Whether the routine usage of maintenance therapies (low-dose, usually single drugs used to prolong duration of remission once remission is achieved) should change that 18-month cutoff is unclear, however, since maintenance “artificially” makes ASCT appear more effective by prolonging post-ASCT duration of remission. The “is it worth it” discussion is also largely subjective and hinges heavily on the patient’s experience with the first ASCT. In our practice, we often use 3 years as the cutoff for considering repeat ASCT in patients on maintenance therapy, meaning that if a patient underwent ASCT and received maintenance, a remission lasting more than 3 years means we consider ASCT as part of therapy for relapse.
Allogeneic stem cell transplantation (allo-SCT) is a treatment option for RRMM generally reserved for fit patients younger than 65 years [22,33]. The timing of allo-SCT is also controversial, with some reserving it as a last option given a historically high transplant-related mortality and improved progression-free survival but not necessarily overall survival benefit. A recent consensus statement has suggested allo-SCT be considered (preferentially in a clinical trial) for eligible patients with high-risk disease who relapse after primary treatment that included ASCT [29]. With the abundance of new treatment options in RRMM with reasonable toxicity profiles, it is not clear for whom and when allo-SCT is best considered.
Which regimen should be used to treat a first relapse?
Entry into a well-designed clinical trial for patients with RRMM should be considered for every patient since there is a lack of evidence to guide the best sequencing of chemotherapies [11]. Beyond that, the choice of therapy is based upon 2 main factors: the disease itself (eg, indolent, asymptomatic biochemical recurrence versus aggressive clinical recurrence with new fractures or extramedullary plasmacytomas), and the patient’s preferences and characteristics, such as age, performance status, comorbidities, and toxicities from prior therapies. In looking for the “best” re-induction regimen, it is tempting to compare the efficacy of regimens across trials, but such efforts are fraught given the significant heterogeneity of the patient populations between trials. As an example, comparing daratumumab + pomalidomide + dexamethasone (DPd) to daratumumab + lenalidomide + dexamethasone (DRd), one may conclude that DRd is superior, given an overall response rate of 88% in DRd versus 58% in DPd. However, the DPd trial included patients who were refractory to lenalidomide and bortezomib, while the DRd study required only treatment with one prior therapy [34,35].
For patients who enjoyed a long remission after any particular chemotherapy regimen with good tolerability and with indolent features at the time of relapse, re-treating with the same regimen can be considered, although nowadays with so many new and highly potent agents available such “backtracking” is less common and some studies suggest that employing new agents may be beneficial. As an example, in the randomized ENDEAVOR study of bortezomib + dexamethasone versus carfilzomib + dexamethasone in RRMM, 54% of patients had been exposed to bortezomib whereas virtually none had received carfilzomib prior to study enroll-ment. Among those patients with prior bortezomib exposure, median progression-free survival was 15.6 versus 8.1 months (hazard ratio 0.56, [95% confidence interval 0.44 to 0.73]) for carfilzomib versus bortezomib, respectively. Follow-up was too immature for definitive conclusions to be drawn about overall survival, but the substantial difference in progression-free survival provides a compelling argument for using carfilzomib instead of going back to bortezomib for patients with prior bortezomib exposure [36].
For patients who are fit and not very old, we generally employ triplet re-induction. For the large number of these patients who were previously exposed to both lenalidomide and bortezomib, including as part of a maintenance strategy, outside of clinical trials we routinely use carfilzomib + pomalidomide + dexamethasone [41]. For patients who are lenalidomide-naïve but bortezomib-exposed, we often employ carfilzomib + lenalidomide + dexamethasone based on the phase 3 ASPIRE trial, which showed a significantly improved progression-free survival with carfilzomib + lenalidomide + dexamethasone versus lenalidomide + dexamethasone [47]. For patients who have previously received lenalidomide but not bortezomib, we consider pomalidomide + bortezomib + dexamethasone [52]. These regimens take advantage of the arguably most potent, most proven drugs in treating RRMM, namely proteasome inhibitors (bortezomib and carfilzomib) and immunomodulatory agents (lenalidomide and pomalidomide).
For patients who are more vulnerable to toxicity due to advanced age or comorbidities, we consider less intensive regimens, including dose-reduced triplets or doublets. Patients who had received lenalidomide-based combinations but not bortezomib are considered for a bortezomib-based re-induction, including bortezomib + dexamethasone alone. In the case of someone who had initially received a bortezomib-based combination but no lenalidomide, the new drugs are viable options: ixazomib [53] or elotuzumab [43] can both be added to standard lenalidomide + dexamethasone, with expectations of increasing response rates and progression-free survival and an acceptably low increased risk of severe toxicity. Ixazomib + lenalidomide + dexamethasone also has the benefit of being all-oral. For patients with bortezomib- and lenalidomide-exposed RRMM, using carfilzomib [54] or pomalidomide [55] with dexamethasone is reasonable.
Once MM has progressed beyond the arguable “core drugs” of early-stage MM, namely lenalidomide, bortezomib, carfilzomib, and pomalidomide, off protocol we favor daratumumab monotherapy [56,57]. Other options include panobinostat (given usually with bortezomib) [58] and bendamustine [59], among others.
Can agents to which the MM was previously refractory be reused?
With the understanding that MM is not a disease defined by a single molecular mutation, but rather clones and subclones, it is reasonable to think that even treatments that have previously failed may be beneficial to patients if they have been off those treatments for some length of time and sensitive subclones reemerge. Additionally, combining the “failed” agent with a new drug may overcome the previously seen refractoriness, as in the case of panobinostat + bortezomib [48]. That said, given the multitude of new treatment options for RRMM and data from such trials as ENDEAVOR as mentioned, revisiting previously used drugs is probably best reserved for second or greater relapses.
What should be the duration of therapy for RRMM?
There is no evidence to guide duration of therapy in RRMM. Most patients with relapsed disease will be considered for continuous treatment until disease progression, which usually means treatment for 6 to 12 months with full-dose induction, often to maximal response, followed by transition to some form of lower-dose maintenance in which parts of a multi-drug regimen may be eliminated and/or the doses for the remaining drugs may be reduced. Patients with a slow-velocity relapse and no markers of high-risk disease may be suitable candidates for a defined course of treatment without maintenance therapy [11], but most patients nowadays remain on some form of maintenance for RRMM after achieving remission.
What supportive care is needed in RRMM?
Bone Health
Skeletal-related events, namely fractures, can be devastating in MM. Bisphosphonates have been shown to decrease such events in MM and zoledronic acid has shown a trend toward improved survival, perhaps related to its impact on the bone marrow microenvironment or direct toxicity to myeloma cells [60,61]. It is unclear whether bisphosphonates improve overall survival in the relapsed setting, although zoledronic acid has shown decreased skeletal-related events in the setting of biochemical-only disease progression [13,62]. In active RRMM, our general practice is to resume parenteral bisphosphonate therapy (either zoledronic acid or pamidronate in our U.S. practices) usually every 3 to 4 weeks, depending on the length of the chemotherapy cycle.
Supportive Care
RRMM is a complex disease in which patients often experience a multitude of symptoms and other complications as a result of the disease itself as well as therapy. Aggressive supportive care is of paramount importance. As examples, zoster prophylaxis is required for virtually all patients on proteasome inhibitors, anticoagulation/antiplatelet therapies should be considered for venous thrombotic event prophylaxis, and proton pump inhibitors may be appropriate for these patients who often have a real risk of peptic ulcer disease due to the use of corticosteroids, nonsteroidal anti-inflammatory drugs, and/or aspirin prophylaxis. Attention to dental health is important for patients on bisphosphonates to minimize the risk of osteonecrosis of the jaw. Nutritional problems should be monitored and can arise due to anorexia, dysgeusia, diarrhea, or constipation. Peripheral neuropathy is extremely common and support should be offered in the form of adjusting therapy to minimize risk of worsening it, analgesics if needed, assistive devices to aid in ambulation, and/or physical therapy. Depression and anxiety are understandably prevalent in patients with RRMM, who face an incurable disease that provides constant reminders of its presence due to symptoms, the need for daily pills, or frequent clinic visits for treatment and/or blood product transfusions [63]. Supporting a patient’s emotional health is a vital component of enhancing quality of life in RRMM.
Case Studies Continued
Patient A was noted to have biochemical progression initially, with relapse detectable only in serum free light chains. Treatment commenced at the time of worsening anemia. Notably, his disease originally secreted IgG-kappa and at relapse secreted kappa free light chain only; that is, he developed “light chain escape,” which signifies a high-risk disease and likely heralds clonal evolution [64]. He had excellent caregiver support and lived within 20 minutes of a treatment center. His performance status remained good at the time of relapse and he had normal organ function. He was treated with carfilzomib + pomalidomide + dexamethasone for 4 cycles, achieving a very good partial response. He then received a second ASCT with melphalan conditioning and again achieved stringent complete response. Indefinite maintenance therapy commenced with pomalidomide, and at 16 months post-ASCT he was doing well and still in remission.
Patient B was symptomatic at the time of disease progression. As her primary complaint was that of a painful humeral lytic lesion, she first underwent a course of palliative radiation, which alleviated her pain. She did not wish to restart systemic treatment and instead elected to watch her MM closely with her oncologist on a monthly basis. By 3 months, her M-spike had reached 0.6 g/dL and her serum creatinine had increased slightly, resulting in a creatinine clearance of 34 mL/min. She lived approximately 90 minutes from the closest treatment facility and found it difficult to come for visits more than once monthly. Her Eastern College Oncology Group (ECOG) performance status was 2. With her advanced age and frailty, she was not considered to be a good candidate for ASCT. She requested to go back on lenalidomide and decided with her oncologist to try ixazomib + lenalidomide + dexamethasone, with which she achieved a very good partial response. She had difficulty with myelosuppression with lenalidomide, which was dropped after 4 cycles, and she is planned for ixazomib maintenance until disease progression or drug intolerance. She receives monthly zoledronic acid to reduce the risk of fractures.
Patient C has high-risk disease as indicated by R-ISS III stage disease at diagnosis and progression only 8 months after ASCT and while on bortezomib maintenance therapy. Although he currently only has evidence of biochemical relapse, prompt initiation of treatment was warranted to prevent further renal compromise such as during his initial presentation [65]. Further, PET-CT showed the presence of extramedullary soft tissue disease, another high-risk feature. He was a robust patient with good social support and received carfilzomib + pomalidomide + dexamethasone re-induction. He was not considered for a second ASCT given his short duration of response. With his high-risk features of early relapse after ASCT, R-ISS III, and extramedullary disease, it was recommended that he continue triplet drug therapy until disease relapse or drug intolerance.
Ongoing and Future Trials
The management of RRMM will continue to evolve as paradigms for treating MM change and new treatment options become available. In particular, immunotherapies (ie, approaches that harness the immune system’s ability to fight cancer) are under exploration and some such drugs that are already FDA-approved in other diseases are being tested in MM. Chimeric antigen receptor-T cells (CAR-T), a form of cell-based immunotherapy, have generated tremendous excitement in acute lymphocytic leukemia [66] and are being tested in MM [67]. New analogs of old drugs may offer more effective, less toxic ways to control MM. The role of ASCT is being explored in randomized trials investigating whether ASCT should be pursued early or late in a patient’s MM course. These studies will no doubt further augment the armamentarium of anti-myeloma drugs that have already resulted in the increasingly longer survival we see today in this disease [3,68]. That said, MM remains incurable, and almost all patients who live long enough eventually relapse and die of MM. Hence, further research and progress are critical.
Summary
A well-designed clinical trial should be considered for all patients with RRMM, and in lieu of an available trial, regimen selection should be tailored upon disease and patient characteristics. Carfilzomib-based regimens are among the most popular at the time of first relapse currently based upon their efficacy in bortezomib-refractory cases and tolerability. Pomalidomide shows activity in lenalidomide-refractory patients. Due to intra-clonal heterogeneity, triplet regimens are preferred for fit patients, reserving doublet or monotherapy for those patients who are frail or who have an indolent disease relapse. Ongoing research will undoubtedly improve outcomes for RRMM, a disease for which the prognosis is far better than it formerly was, but which still has quite a bit of room for improvement.
Corresponding author: Brandi Reeves, MD, University of North Carolina – Chapel Hill, 170 Manning Dr., Physicians’ Office Building, CB 7305, Chapel Hill, NC 27599, brandi_reeves@med.unc.edu.
Financial disclosures: Dr. Tuchman reports the following: speakers’ bureau: Celgene, Takeda; consulting: Celgene, Takeda; research support: Celgene, Takeda, Novartis, Onyx.
From the Division of Hematology and Oncology, University of North Carolina – Chapel Hill, Chapel Hill, NC (Dr. Reeves), and the Division of Cellular Therapy and Hematological Malignancies, Duke Cancer Institute, Durham, NC (Dr. Tuchman).
Abstract
- Objective: To review the management considerations in patients with relapsed and refractory multiple myeloma (RRMM).
- Methods: Review of the literature.
- Results: RRMM is a heterogeneous disease and numerous treatment regimens have been studied. Despite improvement in progression-free and overall survival in newly diagnosed multiple myeloma with current therapies, myeloma remains incurable and repeated relapses are inevitable. Relapses are often characterized by diminished response to chemotherapy (refractoriness) and duration of response.
- Conclusion: Management of RRMM should be individualized using both patient- and disease-related factors, given substantial heterogeneity in both. Further research regarding the optimal timing, regimen, and duration of treatment is warranted.
Although advancements in treating multiple myeloma (MM) have resulted in improved median survival from approximately 2 years in the 1990s to more recent estimates of over 6 years, the disease remains incurable [1–3]. Its overall course is generally defined by a series of increasingly short remissions and treatment-refractory relapses until eventual death due to MM occurs. Objective criteria for defining both relapsed and refractory MM have been published [4]. Briefly, relapsed myeloma is that which has been previously treated with some form of systemic therapy and which has recurred. That recurrence can be clinical (ie, the development of new or worsening signs or symptoms of active MM) and/or biochemical (ie, rising monoclonal MM proteins in the serum or urine). Refractory MM on the other hand refers to MM that is resistant to particular drugs, defined as MM that is nonresponsive to primary or salvage therapy, or MM that progresses within 60 days of the last therapy [4]. At any juncture during the course of relapsed MM, patients will have disease that is either sensitive or refractory to specific myeloma drugs. In this article, we discuss management of these often concurrent entities together as relapsed and refractory multiple myeloma (RRMM).
There are numerous treatment options for patients with RRMM—3 new drugs were approved in November 2015 alone. The abundance of available drugs leaves treating clinicians with a daunting task of sequencing therapies among several choices. The durability of response to treatment typically lessens with each disease relapse, such that the clinician needs to think of sequencing not just second-line therapy, but third- and fourth-line as well, further complicating the decision. In this review, we aim to help clinicians individualize treatment plans for patients with RRMM.
Case Studies
Patient A
A 62-year-old man with IgG-kappa MM was diagnosed 4 years ago during evaluation of a pathologic humeral fracture. The disease was prognostically standard risk, with revised International Staging System (RISS) stage I disease (beta-2 microglobulin 3.4 mcg/mL, albumin 4.1 g/dL, normal cytogenetics with 46,XY in 20 cells analyzed, and myeloma fluorescent in situ hybridization [FISH] panel showing t(11;14) but no del17p, t(14;16), t(14;20), or t(4;14)) [5], and normal blood counts, organ function, and lactate dehydrogenase (LDH) at diagnosis. He was treated with 5 cycles of standard lenalidomide, bortezomib, and dexamethasone followed by high-dose melphalan with autologous stem cell transplantation (ASCT) and then lenalidomide continuous maintenance. He achieved a stringent complete response (ie, complete disappearance of myeloma-derived monoclonal proteins in the serum and urine, a normal serum free light chain ratio, and undetectable monoclonal plasma cells on a bone marrow aspirate and biopsy) [4]. His MM was monitored every 2 to 3 months for disease progression and medication toxicity. At month 38, a monoclonal protein spike (M-spike) on serum protein electrophoresis (SPEP) remained undetectable, but serum kappa free light chain levels increased from 1.98 mg/dL to 8 mg/dL with stable lambda serum free light chains and a ratio that rose to 16, consistent with low-level biochemical recurrence. He had no evidence of end-organ damage and therefore was maintained on lenalidomide maintenance for the time being. Over the next 12 months, his kappa serum free light chain level continued to slowly rise, reaching 24 mg/dL, while the ratio rose to 50. There was still no detectable M-spike. He developed mild anemia during this time, with his hemoglobin dropping from a prior value of approximately 11 g/dL to 9.8 g/dL, though kidney function remained normal. A repeat bone marrow aspirate and biopsy revealed 20% kappa-restricted plasma cells.
Patient B
A 75-year-old woman with IgA-kappa MM was diagnosed after laboratory testing by her primary care physician incidentally showed an elevated serum total protein level. The MM was intermediate risk, with RISS stage II disease, and with mild renal impairment resulting in an estimated creatinine clearance of 45 mL/min that was felt to be due to MM. She was initially treated with bortezomib and dexamethasone but received only 2 cycles because she developed painful peripheral neuropathy secondary to bortezomib. Bortezomib was stopped and she was then treated with lenalidomide and dexamethasone for 4 cycles. She achieved a complete response and elected to stop treatment due to fatigue. Her fatigue did not improve off treatment. Six months after stopping therapy, an M-spike was detectable at 0.1 g/dL and she developed a new painful lytic lesion in the left humerus.
Patient C
A 59-year-old man with lambda free light chain MM was diagnosed when he presented with acute renal failure requiring dialysis. The disease was RISS-III at diagnosis (high risk), with the t(4;14) genetic abnormality in his MM cells detected on bone marrow aspirate, an abnormality that has been associated with poor prognosis MM [6–8]. The patient was treated with cyclophosphamide, bortezomib, and dexamethasone [9] for 6 cycles, at which point his disease was in a very good partial response (>90% reduction in M-spike) [4], and his renal function had recovered to a new baseline creatinine clearance of 45 mL/min. He then underwent ASCT after melphalan conditioning followed by bortezomib maintenance therapy every 2 weeks. Eight months after ASCT, his lambda free light chain level increased from 1.25 mg/dL to 45 mg/dL and the ratio increased from 4 to 22. Renal function was unchanged and there was stable anemia, with hemoglobin of 10.1 g/dL.
When should treatment for RRMM commence?
Patients with MM in remission are closely monitored, with clinical and laboratory examinations generally conducted every 1 to 3 months. The history is focused on MM-related symptoms such as increasing bone pain or weight loss, and symptoms of therapy-related toxicity such as fatigue, gastrointestinal distress, or peripheral neuropathy. Laboratory assessment typically includes blood counts and chemistry measurements, as well as measurements of MM-derived monoclonal proteins: SPEP, serum immunofixation (IFE), serum immunoglobulin free light chain measurements, and urine protein electrophoresis and immunofixation (UPEP/urine IFE) [10]. Progressive disease biochemically is defined as a 25% increase in M-spike (at least 0.5 g/dL if the M-spike is in serum or > 200 mg/24 hours if in urine), and/or a rise of greater than 10 mg/dL difference between the involved and uninvolved serum free light chains. Clinically progressive disease is denoted as new evidence of end-organ damage such as a new plasma-cytoma, unexplained hypercalcemia, or worsening anemia due to MM [4]. Many, if not most, patients will have biochemical recurrence identified by laboratory measurements ofmonoclonal proteins before clinical recurrence transpires.
The velocity of relapse can help guide decisions about when to reinitiate therapy. High-velocity disease relapse, meaning rapid rise in monoclonal proteins, is an indicator of more aggressive disease, and treatment should be initiated promptly before development of symptoms [11]. Conversely, low-level, indolent recurrence can often be followed with a “watch and wait” approach to determine how the myeloma will progress over time. Expert guidelines suggest that a monoclonal protein doubling time of 2 months may be an appropriate cutoff for determining high versus low velocity [12], although 2 months is not a firm rule and the decision of when to restart treatment for any given patient with asymptomatic biochemical recurrence should be individualized. Importantly, it is not clear that changing therapy at the time of biochemical recurrence, prior to clinical disease progression, improves outcomes, but clinicians are often nonetheless hesitant to hold therapy in the face of biochemically recurrent MM given the potential for complications, such as a pathologic fracture. In patients with biochemically recurrent MM for whom re-initiation of systemic anti-myeloma therapy is being deferred, one can consider re-initiation of zoledronic acid therapy, since in a randomized controlled trial, zoledronic acid commenced at the time of biochemical relapse resulted in fewer skeletal events as compared to placebo [13].
What disease factors should be considered in choosing treatment for RRMM?
MM exhibits genetic complexity, and prior treatments may result in clonal evolution of and selection for an initially nondominant, treatment-resistant clone [14,15]. This heterogeneity and selection pressure may explain why 3-drug regimens often outperform 1- or 2-drug regimens, why each remission is generally shorter than the last, and why patients who have enjoyed a long duration of response to one therapy and been off it for some length of time may again have a good response when re-treated with the same therapy at time of MM relapse. So how does one know if a new clone has emerged? While there is no standard for monitoring intra-clonal heterogeneity presently, changes in clinical phenotype likely correlate with evolving clones. Some such changes include free light chain escape (ie, MM that initially secreted an intact M-spike and then only secretes free light chain at relapse), new development of extramedullary disease (plasmacytomas outside of bone) in patients who previously had MM only in the bone marrow, and resistance of some sites to treatment while others respond (a mixed response). The former 2 phenotypes in particular portend poor prognosis and unsurprisingly they can be seen together [16–19]. Restaging, meaning a complete reassessment of MM disease status at the time of relapse, including bone marrow aspirate and biopsy, is beneficial to help guide therapy, as those with high-risk features including high ISS stage [20], high-risk cytogenetics, increased LDH, and extramedullary disease should be treated with triplet therapy when possible [11]. Repeat imaging should also be considered as a new baseline comparator. This can be done with standard x-rays, positron-emission tomography/computed tomography (PET-CT), or magnetic resonance imaging. PET-CT offers the advantage of showing active disease sites and the presence of extramedullary disease, although it exposes the patient to more radiation than the other methods.
In terms of using genetics to guide therapy decisions in RRMM, the presence of the del(17p) abnormality either by karyotyping or FISH portends high risk and pomalidomide in one study was shown to mitigate that risk [21]. How genetics and prognostic markers should dictate therapy selection in RRMM otherwise, however, is unclear and an area of active research efforts.
What patient factors should be considered in choosing treatment?
Given the relatively large selection of possible regimens for the treatment of RRMM, patient preference can be incorporated into regimen selection. Patients who have long commutes or who are trying to work may not be ideal candidates to receive carfilzomib-based regimens given the twice-per-week infusion schedule (though a once-a-week dosing schedule is being tested) [22]. Patients who have poor venous access may be good candidates for all-oral regimens. Prior treatment tolerability and side effects should also be considered. Patients who experienced significant peripheral neuropathy with bortezomib may have less neuropathy with carfilzomib. Those with renal failure may tolerate pomalidomide better than lenalidomide [23].
Patient age and functional status are important considerations in choosing a treatment regimen for RRMM. Very old patients (a subjective categorization to include patients > 80 years by chronologic or physiologic age), those with functional dependence, or patients harboring substantial medical comorbidities are at risk for therapy toxicity and so often warrant less intensive approaches [24]. Deciding which patients empirically warrant less dose-intensive approaches can be challenging, especially with the growing recognition that fit seniors can often tolerate and enjoy the benefits of full-dose approaches, including sometimes even ASCT. Geriatric assessment instruments that interrogate a variety of geriatric-relevant domains, such as number of falls, independence in activities of daily living, and polypharmacy, are being investigated as toxicity predictors and may help make those decisions in the future. Such instruments have been shown to predict chemotherapy toxicity in solid tumors [25,26] and preliminarily in MM [27], but they remain investigational. While no validated geriatric assessment instruments are currently available for routine clinical employment in MM, clinicians should consider the geriatric domains that these instruments assess when choosing among treatment options. Clinically, that often translates to choosing gentler regimens with likely better tolerability, albeit perhaps with less efficacy, for patients judged to be vulnerable to toxicity.
As part of therapy selection in RRMM, the clinician needs to consider if the patient is a candidate for ASCT. For patients who did not undergo ASCT as part of initial treatment, ASCT can be considered at the time of relapse. Ideally, all patients who could eventually undergo ASCT should have hematopoietic stem cells collected and stored at the time of first induction; however, collection after re-induction chemotherapy has been shown to be feasible [28,29]. ASCT for RRMM appears to be effective, although rigorous randomized comparisons of ASCT versus treatment purely with novel drugs are lacking [30–32]. For patients who did receive ASCT consolidation in the frontline, if a response is sustained for 18 months or greater, existing guidelines suggest that a second ASCT is likely worthwhile [29]. Whether the routine usage of maintenance therapies (low-dose, usually single drugs used to prolong duration of remission once remission is achieved) should change that 18-month cutoff is unclear, however, since maintenance “artificially” makes ASCT appear more effective by prolonging post-ASCT duration of remission. The “is it worth it” discussion is also largely subjective and hinges heavily on the patient’s experience with the first ASCT. In our practice, we often use 3 years as the cutoff for considering repeat ASCT in patients on maintenance therapy, meaning that if a patient underwent ASCT and received maintenance, a remission lasting more than 3 years means we consider ASCT as part of therapy for relapse.
Allogeneic stem cell transplantation (allo-SCT) is a treatment option for RRMM generally reserved for fit patients younger than 65 years [22,33]. The timing of allo-SCT is also controversial, with some reserving it as a last option given a historically high transplant-related mortality and improved progression-free survival but not necessarily overall survival benefit. A recent consensus statement has suggested allo-SCT be considered (preferentially in a clinical trial) for eligible patients with high-risk disease who relapse after primary treatment that included ASCT [29]. With the abundance of new treatment options in RRMM with reasonable toxicity profiles, it is not clear for whom and when allo-SCT is best considered.
Which regimen should be used to treat a first relapse?
Entry into a well-designed clinical trial for patients with RRMM should be considered for every patient since there is a lack of evidence to guide the best sequencing of chemotherapies [11]. Beyond that, the choice of therapy is based upon 2 main factors: the disease itself (eg, indolent, asymptomatic biochemical recurrence versus aggressive clinical recurrence with new fractures or extramedullary plasmacytomas), and the patient’s preferences and characteristics, such as age, performance status, comorbidities, and toxicities from prior therapies. In looking for the “best” re-induction regimen, it is tempting to compare the efficacy of regimens across trials, but such efforts are fraught given the significant heterogeneity of the patient populations between trials. As an example, comparing daratumumab + pomalidomide + dexamethasone (DPd) to daratumumab + lenalidomide + dexamethasone (DRd), one may conclude that DRd is superior, given an overall response rate of 88% in DRd versus 58% in DPd. However, the DPd trial included patients who were refractory to lenalidomide and bortezomib, while the DRd study required only treatment with one prior therapy [34,35].
For patients who enjoyed a long remission after any particular chemotherapy regimen with good tolerability and with indolent features at the time of relapse, re-treating with the same regimen can be considered, although nowadays with so many new and highly potent agents available such “backtracking” is less common and some studies suggest that employing new agents may be beneficial. As an example, in the randomized ENDEAVOR study of bortezomib + dexamethasone versus carfilzomib + dexamethasone in RRMM, 54% of patients had been exposed to bortezomib whereas virtually none had received carfilzomib prior to study enroll-ment. Among those patients with prior bortezomib exposure, median progression-free survival was 15.6 versus 8.1 months (hazard ratio 0.56, [95% confidence interval 0.44 to 0.73]) for carfilzomib versus bortezomib, respectively. Follow-up was too immature for definitive conclusions to be drawn about overall survival, but the substantial difference in progression-free survival provides a compelling argument for using carfilzomib instead of going back to bortezomib for patients with prior bortezomib exposure [36].
For patients who are fit and not very old, we generally employ triplet re-induction. For the large number of these patients who were previously exposed to both lenalidomide and bortezomib, including as part of a maintenance strategy, outside of clinical trials we routinely use carfilzomib + pomalidomide + dexamethasone [41]. For patients who are lenalidomide-naïve but bortezomib-exposed, we often employ carfilzomib + lenalidomide + dexamethasone based on the phase 3 ASPIRE trial, which showed a significantly improved progression-free survival with carfilzomib + lenalidomide + dexamethasone versus lenalidomide + dexamethasone [47]. For patients who have previously received lenalidomide but not bortezomib, we consider pomalidomide + bortezomib + dexamethasone [52]. These regimens take advantage of the arguably most potent, most proven drugs in treating RRMM, namely proteasome inhibitors (bortezomib and carfilzomib) and immunomodulatory agents (lenalidomide and pomalidomide).
For patients who are more vulnerable to toxicity due to advanced age or comorbidities, we consider less intensive regimens, including dose-reduced triplets or doublets. Patients who had received lenalidomide-based combinations but not bortezomib are considered for a bortezomib-based re-induction, including bortezomib + dexamethasone alone. In the case of someone who had initially received a bortezomib-based combination but no lenalidomide, the new drugs are viable options: ixazomib [53] or elotuzumab [43] can both be added to standard lenalidomide + dexamethasone, with expectations of increasing response rates and progression-free survival and an acceptably low increased risk of severe toxicity. Ixazomib + lenalidomide + dexamethasone also has the benefit of being all-oral. For patients with bortezomib- and lenalidomide-exposed RRMM, using carfilzomib [54] or pomalidomide [55] with dexamethasone is reasonable.
Once MM has progressed beyond the arguable “core drugs” of early-stage MM, namely lenalidomide, bortezomib, carfilzomib, and pomalidomide, off protocol we favor daratumumab monotherapy [56,57]. Other options include panobinostat (given usually with bortezomib) [58] and bendamustine [59], among others.
Can agents to which the MM was previously refractory be reused?
With the understanding that MM is not a disease defined by a single molecular mutation, but rather clones and subclones, it is reasonable to think that even treatments that have previously failed may be beneficial to patients if they have been off those treatments for some length of time and sensitive subclones reemerge. Additionally, combining the “failed” agent with a new drug may overcome the previously seen refractoriness, as in the case of panobinostat + bortezomib [48]. That said, given the multitude of new treatment options for RRMM and data from such trials as ENDEAVOR as mentioned, revisiting previously used drugs is probably best reserved for second or greater relapses.
What should be the duration of therapy for RRMM?
There is no evidence to guide duration of therapy in RRMM. Most patients with relapsed disease will be considered for continuous treatment until disease progression, which usually means treatment for 6 to 12 months with full-dose induction, often to maximal response, followed by transition to some form of lower-dose maintenance in which parts of a multi-drug regimen may be eliminated and/or the doses for the remaining drugs may be reduced. Patients with a slow-velocity relapse and no markers of high-risk disease may be suitable candidates for a defined course of treatment without maintenance therapy [11], but most patients nowadays remain on some form of maintenance for RRMM after achieving remission.
What supportive care is needed in RRMM?
Bone Health
Skeletal-related events, namely fractures, can be devastating in MM. Bisphosphonates have been shown to decrease such events in MM and zoledronic acid has shown a trend toward improved survival, perhaps related to its impact on the bone marrow microenvironment or direct toxicity to myeloma cells [60,61]. It is unclear whether bisphosphonates improve overall survival in the relapsed setting, although zoledronic acid has shown decreased skeletal-related events in the setting of biochemical-only disease progression [13,62]. In active RRMM, our general practice is to resume parenteral bisphosphonate therapy (either zoledronic acid or pamidronate in our U.S. practices) usually every 3 to 4 weeks, depending on the length of the chemotherapy cycle.
Supportive Care
RRMM is a complex disease in which patients often experience a multitude of symptoms and other complications as a result of the disease itself as well as therapy. Aggressive supportive care is of paramount importance. As examples, zoster prophylaxis is required for virtually all patients on proteasome inhibitors, anticoagulation/antiplatelet therapies should be considered for venous thrombotic event prophylaxis, and proton pump inhibitors may be appropriate for these patients who often have a real risk of peptic ulcer disease due to the use of corticosteroids, nonsteroidal anti-inflammatory drugs, and/or aspirin prophylaxis. Attention to dental health is important for patients on bisphosphonates to minimize the risk of osteonecrosis of the jaw. Nutritional problems should be monitored and can arise due to anorexia, dysgeusia, diarrhea, or constipation. Peripheral neuropathy is extremely common and support should be offered in the form of adjusting therapy to minimize risk of worsening it, analgesics if needed, assistive devices to aid in ambulation, and/or physical therapy. Depression and anxiety are understandably prevalent in patients with RRMM, who face an incurable disease that provides constant reminders of its presence due to symptoms, the need for daily pills, or frequent clinic visits for treatment and/or blood product transfusions [63]. Supporting a patient’s emotional health is a vital component of enhancing quality of life in RRMM.
Case Studies Continued
Patient A was noted to have biochemical progression initially, with relapse detectable only in serum free light chains. Treatment commenced at the time of worsening anemia. Notably, his disease originally secreted IgG-kappa and at relapse secreted kappa free light chain only; that is, he developed “light chain escape,” which signifies a high-risk disease and likely heralds clonal evolution [64]. He had excellent caregiver support and lived within 20 minutes of a treatment center. His performance status remained good at the time of relapse and he had normal organ function. He was treated with carfilzomib + pomalidomide + dexamethasone for 4 cycles, achieving a very good partial response. He then received a second ASCT with melphalan conditioning and again achieved stringent complete response. Indefinite maintenance therapy commenced with pomalidomide, and at 16 months post-ASCT he was doing well and still in remission.
Patient B was symptomatic at the time of disease progression. As her primary complaint was that of a painful humeral lytic lesion, she first underwent a course of palliative radiation, which alleviated her pain. She did not wish to restart systemic treatment and instead elected to watch her MM closely with her oncologist on a monthly basis. By 3 months, her M-spike had reached 0.6 g/dL and her serum creatinine had increased slightly, resulting in a creatinine clearance of 34 mL/min. She lived approximately 90 minutes from the closest treatment facility and found it difficult to come for visits more than once monthly. Her Eastern College Oncology Group (ECOG) performance status was 2. With her advanced age and frailty, she was not considered to be a good candidate for ASCT. She requested to go back on lenalidomide and decided with her oncologist to try ixazomib + lenalidomide + dexamethasone, with which she achieved a very good partial response. She had difficulty with myelosuppression with lenalidomide, which was dropped after 4 cycles, and she is planned for ixazomib maintenance until disease progression or drug intolerance. She receives monthly zoledronic acid to reduce the risk of fractures.
Patient C has high-risk disease as indicated by R-ISS III stage disease at diagnosis and progression only 8 months after ASCT and while on bortezomib maintenance therapy. Although he currently only has evidence of biochemical relapse, prompt initiation of treatment was warranted to prevent further renal compromise such as during his initial presentation [65]. Further, PET-CT showed the presence of extramedullary soft tissue disease, another high-risk feature. He was a robust patient with good social support and received carfilzomib + pomalidomide + dexamethasone re-induction. He was not considered for a second ASCT given his short duration of response. With his high-risk features of early relapse after ASCT, R-ISS III, and extramedullary disease, it was recommended that he continue triplet drug therapy until disease relapse or drug intolerance.
Ongoing and Future Trials
The management of RRMM will continue to evolve as paradigms for treating MM change and new treatment options become available. In particular, immunotherapies (ie, approaches that harness the immune system’s ability to fight cancer) are under exploration and some such drugs that are already FDA-approved in other diseases are being tested in MM. Chimeric antigen receptor-T cells (CAR-T), a form of cell-based immunotherapy, have generated tremendous excitement in acute lymphocytic leukemia [66] and are being tested in MM [67]. New analogs of old drugs may offer more effective, less toxic ways to control MM. The role of ASCT is being explored in randomized trials investigating whether ASCT should be pursued early or late in a patient’s MM course. These studies will no doubt further augment the armamentarium of anti-myeloma drugs that have already resulted in the increasingly longer survival we see today in this disease [3,68]. That said, MM remains incurable, and almost all patients who live long enough eventually relapse and die of MM. Hence, further research and progress are critical.
Summary
A well-designed clinical trial should be considered for all patients with RRMM, and in lieu of an available trial, regimen selection should be tailored upon disease and patient characteristics. Carfilzomib-based regimens are among the most popular at the time of first relapse currently based upon their efficacy in bortezomib-refractory cases and tolerability. Pomalidomide shows activity in lenalidomide-refractory patients. Due to intra-clonal heterogeneity, triplet regimens are preferred for fit patients, reserving doublet or monotherapy for those patients who are frail or who have an indolent disease relapse. Ongoing research will undoubtedly improve outcomes for RRMM, a disease for which the prognosis is far better than it formerly was, but which still has quite a bit of room for improvement.
Corresponding author: Brandi Reeves, MD, University of North Carolina – Chapel Hill, 170 Manning Dr., Physicians’ Office Building, CB 7305, Chapel Hill, NC 27599, brandi_reeves@med.unc.edu.
Financial disclosures: Dr. Tuchman reports the following: speakers’ bureau: Celgene, Takeda; consulting: Celgene, Takeda; research support: Celgene, Takeda, Novartis, Onyx.
1. Pulte D, Redaniel MT, Brenner H, et al. Recent improvement in survival of patients with multiple myeloma: variation by ethnicity. Leuk Lymphoma 2014;55:1083–9.
2. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 2014;28:1122–8.
3. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008;111:2516–20.
4. Rajkumar SV, Harousseau J-L, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 2011;117:4691–5.
5. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015;33:2863–9.
6. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood 2015;125:2095–100.
7. Karlin L, Soulier J, Chandesris O, et al. Clinical and biological features of t(4;14) multiple myeloma: a prospective study. Leuk Lymphoma 2011;52:238–46.
8. Moreau P, Cavo M, Sonneveld P, et al. Combination of international scoring system 3, high lactate dehydrogenase, and t(4;14) and/or del(17p) Identifies patients with multiple myeloma (MM) treated with front-line autologous stem-cell transplantation at high risk of early MM progression–related dDeath. J Clin Oncol 2014;32:2173–80.
9. Reeder CB, Reece DE, Kukreti V, et al. Long-term survival with cyclophosphamide, bortezomib and dexamethasone induction therapy in patients with newly diagnosed multiple myeloma. Br J Haematol 2014;167:563–5.
10. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016;17:e328–46.
11. Laubach J, Garderet L, Mahindra A, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia 2016;30:1005–17.
12. Palumbo A, Rajkumar SV, San Miguel JF, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not Eligible for standard autologous stem-cell transplantation. J Clin Oncol 2014;32:587–600.
13. Garcia-Sanz R, Oriol A, Moreno MJ, et al. Zoledronic acid as compared with observation in multiple myeloma patients at biochemical relapse: results of the randomized AZABACHE Spanish trial. Haematologica 2015;100:1207–13.
14. Bahlis NJ. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012;120:927–8.
15. Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012;120:1067–76.
16. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
17. Dawson MA, Patil S, Spencer A. Extramedullary relapse of multiple myeloma associated with a shift in secretion from intact immunoglobulin to light chains. Haematologica 2007;92:143–4.
18. Usmani SZ, Heuck C, Mitchell A, et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012;97:1761–7.
19. Varettoni M, Corso A, Pica G, et al. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: a longitudinal study on 1003 consecutive patients. Ann Oncol 2010;21:325–30.
20. Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol 2005;23:3412–20.
21. Leleu X, Karlin L, Macro M, et al. Pomalidomide plus low-dose dexamethasone in multiple myeloma with deletion 17p and/or translocation (4;14): IFM 2010-02 trial results. Blood 2015;125:1411–7.
22. Berenson JR, Cartmell A, Bessudo A, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood 2016 Jun 30;127:3360–8.
23. Richter J, Biran N, Duma N, et al. Safety and tolerability of pomalidomide-based regimens (pomalidomide-carfilzomib-dexamethasone with or without cyclophosphamide) in relapsed/refractory multiple myeloma and severe renal dysfunction: a case series. Hematol Oncol 2016 Mar 27. doi: 10.1002/hon.2290.
24. Wildes TM, Rosko A, Tuchman SA. Multiple myeloma in the older adult: better prospects, more challenges. J Clin Oncol 2014;32:2531–40.
25. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) score. Cancer 2012;118(13):3377-86.
26. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol 2011;29:3457–65.
27. Palumbo A, Bringhen S, Mateos M-V, et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood 2015;125:2068–74.
28. Lemieux E, Hulin C, Caillot D, et al. Autologous stem cell transplantation: an effective salvage therapy in multiple myeloma. Biol Blood Marrow Transplant 2013;19:445–9.
29. Giralt S, Garderet L, Durie B, et al. American Society of Blood and Marrow Transplantation, European Society of Blood and Marrow Transplantation, Blood and Marrow Transplant Clinical Trials Network, and International Myeloma Working Group Consensus Conference on Salvage Hematopoietic Cell Transplantation in Patients with Relapsed Multiple Myeloma. Biol Blood Marrow Transplant 2015;21:2039–51.
30. Cook G, Williams C, Brown JM, et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse [Intensive trial]): a randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:874–85.
31. Grövdal M, Nahi H, Gahrton G, et al. Autologous stem cell transplantation versus novel drugs or conventional chemotherapy for patients with relapsed multiple myeloma after previous ASCT. Bone Marrow Transplant 2015;50:808–12.
32. Singh Abbi KK, Zheng J, Devlin SM, et al. Second autologous stem cell transplant: an effective therapy for relapsed multiple myeloma. Biol Blood Marrow Transplant 2015;21:468–72.
33. Franssen LE, Raymakers RA, Buijs A, et al. Outcome of allogeneic transplantation in newly diagnosed and relapsed/refractory multiple myeloma: long-term follow-up in a single institution. European J Haematol 2016 Mar 29.
34. Chari AL, Sagar SA, Fay JW, et al. Open-label, multicenter, phase 1b study of daratumumab in combination with pomalidomide and dexamethasone in patients with at least 2 lines of prior therapy and relapsed or relapsed and refractory multiple myeloma. Blood 2015;126:508.
35. Plesner T, Gimsing P, Krejcik J, et al. Daratumumab in combination with lenalidomide and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: Updated results of a phase 1/2 study (GEN503). Blood 2015;126:507.
36. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016;17:27–38.
37. Richardson PG, Siegel D, Baz R, et al. Phase 1 study of pomalidomide MTD, safety, and efficacy in patients with refractory multiple myeloma who have received lenalidomide and bortezomib. Blood 2013;121:1961–7.
38. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 2014;123:1826–32.
39. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
40. Reu FJ, Valent J, Malek E, et al. A phase I study of ixazomib in combination with panobinostat and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood 2015;126:4221.
41. Shah JJ, Stadtmauer EA, Abonour R, et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood 2015;126:2284–90.
42. Palumbo A, Chanan-Khan AA, Weisel K, et al. Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study. J Clin Oncol 2016;34(suppl):abstr LBA4.
43. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015;373:621–31.
44. Lentzsch S, O’Sullivan A, Kennedy RC, et al. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: results of phase 1/2 open-label, dose escalation study. Blood 2012;119:4608–13.
45. Kumar SK, Krishnan A, LaPlant B, et al. Bendamustine, lenalidomide, and dexamethasone (BRD) is highly effective with durable responses in relapsed multiple myeloma. Am J Hematol 2015;90:1106–10.
46. Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012;120:2817–25.
47. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142–52.
48. San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncology 2014;15:1195–206.
49. Berdeja JG, Hart LL, Mace JR, et al. Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica 2015;100:670–6.
50. Buda G, Orciuolo E, Galimberti S, Ghio F, Petrini M. VTDPACE as salvage therapy for heavily pretreated MM patients. Blood 2013;122:5377.
51. Voorhees PM, Mulkey F, Hassoun H, et al. Alliance A061202. A phase I/II study of pomalidomide, dexamethasone and ixazomib versus pomalidomide and dexamethasone for patients with multiple myeloma refractory to lenalidomide and proteasome inhibitor based therapy: phase I results. Blood 2015;126:375.
52. Lacy MQ, LaPlant BR, Laumann KM, et al. Pomalidomide, bortezomib and dexamethasone (PVD) for patients with relapsed lenalidomide refractory multiple myeloma (MM). Blood 2014;124:304.
53. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
54. Vij R, Siegel DS, Jagannath S, et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol 2012;158:739–48.
55. Leleu X, Attal M, Arnulf B, et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide-refractory multiple myeloma: Intergroupe Francophone du Myelome 2009-02. Blood 2013;121:1968–75.
56. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016;387(10027):1551–60.
57. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015;373:1207–19.
58. Richardson PG, Schlossman RL, Alsina M, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013;122:2331–7.
59. Stöhr E, Schmeel FC, Schmeel LC, et al. Bendamustine in heavily pre-treated patients with relapsed or refractory multiple myeloma. J Cancer Res Clin Oncol 2015;141:2205–12.
60. Mhaskar R, Redzepovic J, Wheatley K, et al. Bisphosphonates in multiple myeloma: a network meta-analysis. Cochrane Database Syst Rev 2012(5):CD003188.
61. Dhodapkar MV, Singh J, Mehta J, et al. Anti-myeloma activity of pamidronate in vivo. Br J Haematol 1998;103:530–2.
62. Terpos E, Morgan G, Dimopoulos MA, et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J Clin Oncol 2013;31:2347–57.
63. Kiely F, Cran A, Finnerty D, O’Brien T. Self-reported quality of life and symptom burden in ambulatory patients with multiple myeloma on disease-modifying treatment. Am J Hosp Palliat Care 2016 May 2.
64. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
65. Ludwig H, Sonneveld P, Davies F, et al. European perspective on multiple myeloma treatment strategies in 2014. Oncologist 2014;19:829–44.
66. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17.
67. Garfall AL, Maus MV, Hwang W-T, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med 2015;373:1040–7.
68. Kumar SK, Dispenzieri A, Gertz MA, et al. Continued improvement in survival in multiple myeloma and the impact of novel agents. 54th ASH Annual Meeting Abstracts. Blood 2012;120(21):3972.
1. Pulte D, Redaniel MT, Brenner H, et al. Recent improvement in survival of patients with multiple myeloma: variation by ethnicity. Leuk Lymphoma 2014;55:1083–9.
2. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 2014;28:1122–8.
3. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008;111:2516–20.
4. Rajkumar SV, Harousseau J-L, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 2011;117:4691–5.
5. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015;33:2863–9.
6. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood 2015;125:2095–100.
7. Karlin L, Soulier J, Chandesris O, et al. Clinical and biological features of t(4;14) multiple myeloma: a prospective study. Leuk Lymphoma 2011;52:238–46.
8. Moreau P, Cavo M, Sonneveld P, et al. Combination of international scoring system 3, high lactate dehydrogenase, and t(4;14) and/or del(17p) Identifies patients with multiple myeloma (MM) treated with front-line autologous stem-cell transplantation at high risk of early MM progression–related dDeath. J Clin Oncol 2014;32:2173–80.
9. Reeder CB, Reece DE, Kukreti V, et al. Long-term survival with cyclophosphamide, bortezomib and dexamethasone induction therapy in patients with newly diagnosed multiple myeloma. Br J Haematol 2014;167:563–5.
10. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016;17:e328–46.
11. Laubach J, Garderet L, Mahindra A, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia 2016;30:1005–17.
12. Palumbo A, Rajkumar SV, San Miguel JF, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not Eligible for standard autologous stem-cell transplantation. J Clin Oncol 2014;32:587–600.
13. Garcia-Sanz R, Oriol A, Moreno MJ, et al. Zoledronic acid as compared with observation in multiple myeloma patients at biochemical relapse: results of the randomized AZABACHE Spanish trial. Haematologica 2015;100:1207–13.
14. Bahlis NJ. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012;120:927–8.
15. Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012;120:1067–76.
16. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
17. Dawson MA, Patil S, Spencer A. Extramedullary relapse of multiple myeloma associated with a shift in secretion from intact immunoglobulin to light chains. Haematologica 2007;92:143–4.
18. Usmani SZ, Heuck C, Mitchell A, et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012;97:1761–7.
19. Varettoni M, Corso A, Pica G, et al. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: a longitudinal study on 1003 consecutive patients. Ann Oncol 2010;21:325–30.
20. Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol 2005;23:3412–20.
21. Leleu X, Karlin L, Macro M, et al. Pomalidomide plus low-dose dexamethasone in multiple myeloma with deletion 17p and/or translocation (4;14): IFM 2010-02 trial results. Blood 2015;125:1411–7.
22. Berenson JR, Cartmell A, Bessudo A, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood 2016 Jun 30;127:3360–8.
23. Richter J, Biran N, Duma N, et al. Safety and tolerability of pomalidomide-based regimens (pomalidomide-carfilzomib-dexamethasone with or without cyclophosphamide) in relapsed/refractory multiple myeloma and severe renal dysfunction: a case series. Hematol Oncol 2016 Mar 27. doi: 10.1002/hon.2290.
24. Wildes TM, Rosko A, Tuchman SA. Multiple myeloma in the older adult: better prospects, more challenges. J Clin Oncol 2014;32:2531–40.
25. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) score. Cancer 2012;118(13):3377-86.
26. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol 2011;29:3457–65.
27. Palumbo A, Bringhen S, Mateos M-V, et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood 2015;125:2068–74.
28. Lemieux E, Hulin C, Caillot D, et al. Autologous stem cell transplantation: an effective salvage therapy in multiple myeloma. Biol Blood Marrow Transplant 2013;19:445–9.
29. Giralt S, Garderet L, Durie B, et al. American Society of Blood and Marrow Transplantation, European Society of Blood and Marrow Transplantation, Blood and Marrow Transplant Clinical Trials Network, and International Myeloma Working Group Consensus Conference on Salvage Hematopoietic Cell Transplantation in Patients with Relapsed Multiple Myeloma. Biol Blood Marrow Transplant 2015;21:2039–51.
30. Cook G, Williams C, Brown JM, et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse [Intensive trial]): a randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:874–85.
31. Grövdal M, Nahi H, Gahrton G, et al. Autologous stem cell transplantation versus novel drugs or conventional chemotherapy for patients with relapsed multiple myeloma after previous ASCT. Bone Marrow Transplant 2015;50:808–12.
32. Singh Abbi KK, Zheng J, Devlin SM, et al. Second autologous stem cell transplant: an effective therapy for relapsed multiple myeloma. Biol Blood Marrow Transplant 2015;21:468–72.
33. Franssen LE, Raymakers RA, Buijs A, et al. Outcome of allogeneic transplantation in newly diagnosed and relapsed/refractory multiple myeloma: long-term follow-up in a single institution. European J Haematol 2016 Mar 29.
34. Chari AL, Sagar SA, Fay JW, et al. Open-label, multicenter, phase 1b study of daratumumab in combination with pomalidomide and dexamethasone in patients with at least 2 lines of prior therapy and relapsed or relapsed and refractory multiple myeloma. Blood 2015;126:508.
35. Plesner T, Gimsing P, Krejcik J, et al. Daratumumab in combination with lenalidomide and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: Updated results of a phase 1/2 study (GEN503). Blood 2015;126:507.
36. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016;17:27–38.
37. Richardson PG, Siegel D, Baz R, et al. Phase 1 study of pomalidomide MTD, safety, and efficacy in patients with refractory multiple myeloma who have received lenalidomide and bortezomib. Blood 2013;121:1961–7.
38. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 2014;123:1826–32.
39. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
40. Reu FJ, Valent J, Malek E, et al. A phase I study of ixazomib in combination with panobinostat and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood 2015;126:4221.
41. Shah JJ, Stadtmauer EA, Abonour R, et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood 2015;126:2284–90.
42. Palumbo A, Chanan-Khan AA, Weisel K, et al. Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study. J Clin Oncol 2016;34(suppl):abstr LBA4.
43. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015;373:621–31.
44. Lentzsch S, O’Sullivan A, Kennedy RC, et al. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: results of phase 1/2 open-label, dose escalation study. Blood 2012;119:4608–13.
45. Kumar SK, Krishnan A, LaPlant B, et al. Bendamustine, lenalidomide, and dexamethasone (BRD) is highly effective with durable responses in relapsed multiple myeloma. Am J Hematol 2015;90:1106–10.
46. Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012;120:2817–25.
47. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142–52.
48. San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncology 2014;15:1195–206.
49. Berdeja JG, Hart LL, Mace JR, et al. Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica 2015;100:670–6.
50. Buda G, Orciuolo E, Galimberti S, Ghio F, Petrini M. VTDPACE as salvage therapy for heavily pretreated MM patients. Blood 2013;122:5377.
51. Voorhees PM, Mulkey F, Hassoun H, et al. Alliance A061202. A phase I/II study of pomalidomide, dexamethasone and ixazomib versus pomalidomide and dexamethasone for patients with multiple myeloma refractory to lenalidomide and proteasome inhibitor based therapy: phase I results. Blood 2015;126:375.
52. Lacy MQ, LaPlant BR, Laumann KM, et al. Pomalidomide, bortezomib and dexamethasone (PVD) for patients with relapsed lenalidomide refractory multiple myeloma (MM). Blood 2014;124:304.
53. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016;374:1621–34.
54. Vij R, Siegel DS, Jagannath S, et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol 2012;158:739–48.
55. Leleu X, Attal M, Arnulf B, et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide-refractory multiple myeloma: Intergroupe Francophone du Myelome 2009-02. Blood 2013;121:1968–75.
56. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016;387(10027):1551–60.
57. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015;373:1207–19.
58. Richardson PG, Schlossman RL, Alsina M, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013;122:2331–7.
59. Stöhr E, Schmeel FC, Schmeel LC, et al. Bendamustine in heavily pre-treated patients with relapsed or refractory multiple myeloma. J Cancer Res Clin Oncol 2015;141:2205–12.
60. Mhaskar R, Redzepovic J, Wheatley K, et al. Bisphosphonates in multiple myeloma: a network meta-analysis. Cochrane Database Syst Rev 2012(5):CD003188.
61. Dhodapkar MV, Singh J, Mehta J, et al. Anti-myeloma activity of pamidronate in vivo. Br J Haematol 1998;103:530–2.
62. Terpos E, Morgan G, Dimopoulos MA, et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J Clin Oncol 2013;31:2347–57.
63. Kiely F, Cran A, Finnerty D, O’Brien T. Self-reported quality of life and symptom burden in ambulatory patients with multiple myeloma on disease-modifying treatment. Am J Hosp Palliat Care 2016 May 2.
64. Brioli A, Giles H, Pawlyn C, et al. Serum free immunoglobulin light chain evaluation as a marker of impact from intraclonal heterogeneity on myeloma outcome. Blood 2014;123:3414–9.
65. Ludwig H, Sonneveld P, Davies F, et al. European perspective on multiple myeloma treatment strategies in 2014. Oncologist 2014;19:829–44.
66. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17.
67. Garfall AL, Maus MV, Hwang W-T, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med 2015;373:1040–7.
68. Kumar SK, Dispenzieri A, Gertz MA, et al. Continued improvement in survival in multiple myeloma and the impact of novel agents. 54th ASH Annual Meeting Abstracts. Blood 2012;120(21):3972.