User login
Hepatocellular carcinoma: Options for diagnosing and managing a deadly disease
Hepatocellular carcinoma (HCC) is a common cause of death worldwide. However, it can be detected early in high-risk individuals by using effective screening strategies, resulting in the ability to provide curative treatment.
Here, we review the risk factors for HCC, strategies for surveillance and diagnosis, and therapies that can be used.
EPIDEMIOLOGY
HCC is the most common primary malignancy of the liver. Overall, it is the fifth most common type of cancer in men and the seventh most common in women.1
Cirrhosis is present in 80% to 90% of patients with HCC.
Male sex. The male-to-female ratio is from 2:1 to 4:1, depending on the region.2 In the United States, the overall male-to-female ratio has been reported2 as 2.4:1. In another report,3 the incidence rate of HCC per 100,000 person-years was 3.7 for men and 2.0 for women.
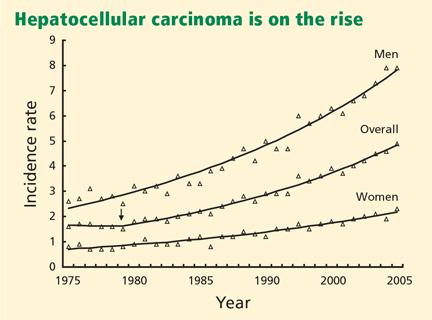
Geographic areas with a high incidence of HCC include sub-Saharan Africa and eastern Asia, whereas Canada and the United States are low-incidence areas. The difference has been because of a lower prevalence of hepatitis B virus infection in North America. However, recent data show a downward trend in incidence of HCC in eastern Asia and an upward trend in North America (Figure 1).3,4
Viral hepatitis (ie, hepatitis B or hepatitis C) is the main risk factor for cirrhosis and HCC.
Diabetes mellitus can predispose to nonalcoholic steatohepatitis, which can subsequently progress to cirrhosis. Thus, it increases the risk of HCC.
Obesity increases the risk of death from liver cancer, with obese people (body mass index ≥ 30 kg/m2) having a higher HCC-related death rate than leaner individuals.5 And as obesity becomes more prevalent, the number of deaths from HCC could increase.
Other diseases that predispose to HCC include alcohol abuse, hereditary hemochromatosis, alpha-1-antitrypsin deficiency, and glycogen storage disease.
SURVEILLANCE OF PATIENTS AT RISK
Patients at high risk of developing liver cancer require frequent screening (Table 1).

Patients with cirrhosis. Sarasin et al6 calculated that surveillance is cost-effective and increases the odds of survival in patients with cirrhosis if the incidence of HCC exceeds 1.5% per year (which it does). In view of this finding, all patients with cirrhosis should be screened every 6 months, irrespective of the cause of the cirrhosis.
Hepatitis B carriers. Surveillance is also indicated in some hepatitis B carriers (Table 1), eg, those with a family history of HCC in a first-degree relative (an independent risk factor for developing the disease in this group).7 Also, Africans with hepatitis B tend to develop HCC early in life.8 Though it has been recommended that surveillance be started at a younger age in these patients,9 the age at which it should begin has not been clearly established. In addition, it is not clear if black people born outside Africa are at higher risk.
Benefit of surveillance
HCC surveillance has shown to lower the death rate. A randomized controlled trial in China compared screening (with abdominal ultrasonography and alpha-fetoprotein levels) vs no screening in patients with hepatitis B. It showed that screening led to a 37% decrease in the death rate.12 Studies have also established that patients with early-stage HCC have a better survival rate than patients with more-advanced disease.10,11 This survival benefit is largely explained by the availability of effective treatments for early-stage cancer, including liver transplantation. Therefore, early-stage asymptomatic patients diagnosed by a surveillance program should have a better survival rate than symptomatic patients.
Surveillance methods
The tests most often used in surveillance for HCC are serum alpha-fetoprotein levels and liver ultrasonography.
Serum alpha-fetoprotein levels by themselves have not been shown to be useful, whereas the combination of alpha-fetoprotein levels and ultrasonography has been shown to reduce the death rate when used for surveillance in a randomized trial.12 A 2012 study reported that the combination of alpha-fetoprotein testing and ultrasonography had a higher sensitivity (90%) than ultrasonography alone (58%), but at the expense of a lower specificity.13
Alpha-fetoprotein has a low sensitivity (ie, 54%) for HCC.14 Tumor size is one of the factors limiting the sensitivity of alpha-fetoprotein, 14 and this would imply that this test may not be helpful in detecting HCC at an early stage. Alpha-fetoprotein L3, an isoform of alpha-fetoprotein, may be helpful in patients with alpha-fetoprotein levels in the intermediate range, and it is currently being studied.
Liver ultrasonography is operator-dependent, and it may not be as accurate in overweight or obese people.
Computed tomography (CT) and magnetic resonance imaging (MRI) are not recommended for surveillance. Serial CT poses risks of radiation-induced damage, contrast-related anaphylaxis, and renal failure, and MRI is not cost-effective and can also lead to gadolinium-induced nephrogenic systemic fibrosis in patients with renal failure.
Currently, the American Association for the Study of Liver Diseases9 recommends ultrasonography only, every 6 months, for surveillance for HCC. However, it may be premature to conclude that alpha-fetoprotein measurement is no longer required for surveillance, and if new data emerge that support its role, it may be reincorporated into the guidelines.
DIAGNOSING HEPATOCELLULAR CARCINOMA
Lesions larger than 1 cm on ultrasonography
The finding of a liver lesion larger than 1 cm on ultrasonography during surveillance warrants further testing.
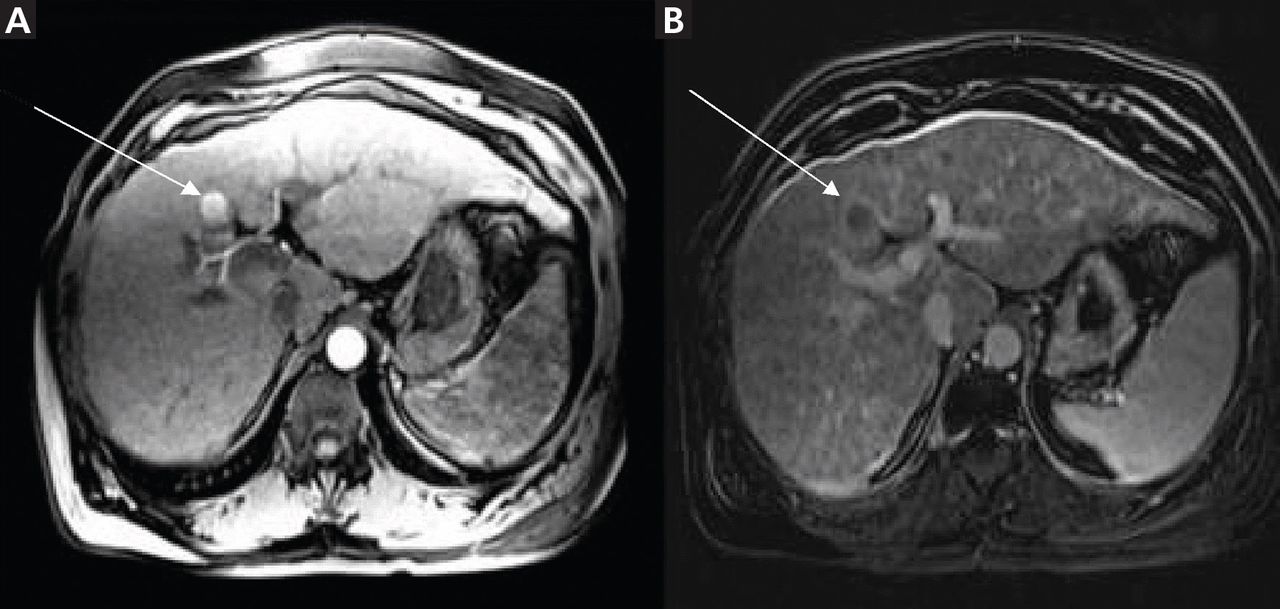
Noninvasive testing with four-phase multidetector CT or dynamic contrast-enhanced MRI is the next step. Typical findings on either of these imaging studies are sufficient to make a diagnosis of HCC, as they have a high specificity and positive predictive value.15 Arterial hyperenhancement with a venous-phase or delayed-phase washout of contrast medium confirms the diagnosis (Figure 2).9 If one of the two imaging studies is typical for HCC, liver biopsy is not needed.
Other imaging studies, including contrast-enhanced ultrasonography, have not been shown to be specific for this diagnosis.16
Liver biopsy is indicated in patients in whom the imaging findings are atypical for HCC.9,17 Biopsy has very good sensitivity and specificity for cancer, but false-negative findings do occur.18 Therefore, a negative biopsy does not entirely exclude HCC. In this situation, patients should be followed by serial ultrasonography, and any further growth or change in character should be reevaluated.
Lesions smaller than 1 cm
For lesions smaller than 1 cm, the incidence of HCC is low, and currently available diagnostic tests are not reliable.15,19 Lesions of this size should be followed by serial ultrasonography every 3 to 4 months until they either enlarge to greater than 1 cm or remain stable at 2 years.9 If they remain stable at the end of 2 years, regular surveillance ultrasonography once every 6 months can be continued.
CURATIVE AND PALLIATIVE THERAPIES

Therapies for HCC (Table 2) can be divided into two categories: curative and palliative.
Curative treatments include surgical resection, liver transplantation, and radiofrequency ablation. All other treatments are palliative, including transarterial chemoembolization and medical therapy with sorafenib.
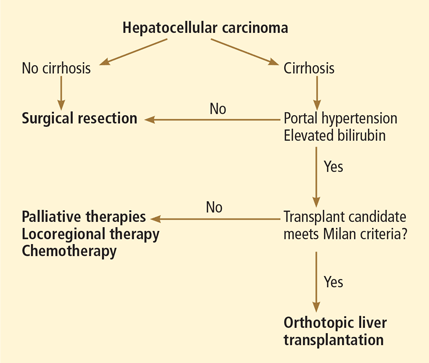
The choice of treatment depends on the characteristics of the tumor, the degree of liver dysfunction, and the patient’s current level of function. The Barcelona Clinic Liver Cancer classification is widely used in making these decisions, as it incorporates both clinical features and tumor stage.9 Figure 3 shows a simplified management algorithm.
SURGICAL RESECTION
Surgical resection is the preferred treatment for patients who have a solitary HCC lesion without cirrhosis.9 It is also indicated in patients with well-compensated cirrhosis who have normal portal pressure, a normal serum bilirubin level, and a platelet count greater than 100 × 109/L.20,21 In such patients, the 5-year survival rate is about 74%, compared with 25% in patients with portal hypertension and serum bilirubin levels higher than 1 mg/dL.21
Surgical resection is not recommended for patients with decompensated cirrhosis, as it can worsen liver function postoperatively and increase the risk of death.19,20 In Western countries, where cirrhosis from hepatitis C is the commonest cause of HCC, most patients have poorly preserved hepatic function at the time of diagnosis, leaving only a minority of patients as candidates for surgical resection.
After surgical resection of HCC, the recurrence rate can be as high as 70% to 80% at 5 years.22,23 Studies have consistently found larger tumor size and vascular invasion to be factors that predict recurrence.24,25 Vascular invasion was also found to predict poor survival after recurrence.24 Studies have so far not shown any conclusive benefit from post-surgical adjuvant chemotherapy in reducing the rate of recurrence of HCC.26,27
How to treat recurrent HCC after surgical resection has not been clearly established. Radiofrequency ablation, transarterial chemoembolization, repeat resection, and liver transplantation have all improved survival when used alone or in combination.28 However, randomized controlled trials are needed to establish the effective treatment strategy and the benefit of multimodal treatment of recurrent HCC.
LIVER TRANSPLANTATION
Orthotopic liver transplantation is the preferred treatment for patients with HCC complicated by cirrhosis and portal hypertension. It has the advantage not only of being potentially curative, but also of overcoming liver cirrhosis by replacing the liver.
To qualify for liver transplantation, patients must meet the Milan criteria (ie, have a single nodule less than 5 cm in diameter or up to three nodules, with the largest being less than 3 cm in diameter, with no evidence of vascular invasion or distant metastasis). These patients have an expected 4-year survival rate of 85% and a recurrence-free survival rate of 92% after transplantation, compared with 50% and 59%, respectively, in patients whose tumors exceeded these criteria.29
Some believe that the Milan criteria are too restrictive and could be expanded. Yao et al at the University of California-San Francisco30 reported that patients with HCC meeting the criteria of having a solitary tumor smaller than 6.5 cm or having up to three nodules, with the largest smaller than 4.5 cm, and total tumor diameter less than 8 cm, had survival rates of 90% at 1 year and 75.2% at 5 years after liver transplantation, compared with 50% at 1 year for patients with tumors exceeding these limits. (These have come to be known as the UCSF criteria.) However, the United Network for Organ Sharing (UNOS) has not adopted these expanded criteria. UNOS has a point system for allocating livers for transplant called the Model for End-Stage Liver Disease (MELD). Patients who meet the Milan criteria receive extra points, putting them higher on the transplant list. This allows for early transplantation, thus reducing tumor progression and dropout from the transplant list. UNOS allocates a MELD score of 22 to all patients who meet the Milan criteria, and the score is further adjusted once every 3 months to reflect a 10% increase in the mortality rate. However, patients who have a single lesion smaller than 2 cm and are candidates for liver transplantation are not assigned additional MELD points per UNOS policy, as the risk of tumor progression beyond the Milan criteria in these patients is deemed to be low.
Therapies while awaiting transplantation
Even if they receive additional MELD points to give them priority on the waiting list, patients face a considerable wait before transplantation because of the limited availability of donor organs. In the interim, they have a risk of tumor progression beyond the Milan criteria and subsequent dropout from the transplant list.31 Patients on the waiting list may therefore undergo a locoregional therapy such as transarterial chemoembolization or radiofrequency ablation as bridging therapy.
These therapies have been shown to decrease dropout from the waiting list.31 A prospective study showed that in 48 patients who underwent transarterial chemoembolization while awaiting liver transplantation, none had tumor progression, and 41 did receive a transplant, with excellent posttransplantation survival rates.32 Similarly, radioembolization using yttrium-90-labeled microspheres or radiofrequency ablation while on the waiting list has been shown to significantly decrease the rate of dropout, with good posttransplantation outcomes.33,34
However, in spite of these benefits, these bridging therapies do not increase survival rates after transplantation. It is also unclear whether they are useful in regions with short waiting times for liver transplantation.
Adjuvant systemic chemotherapy has not been shown to improve survival in patients undergoing liver transplantation. For example, in a randomized controlled trial of doxorubicin given before, during, and after surgery, the survival rate at 5 years was 38% with doxorubicin and 40% without.35
ABLATIVE LOCOREGIONAL THERAPIES
Locoregional therapies play an important role in managing HCC. They are classified as ablative and perfusion-based.
Ablative locoregional therapies include chemical modalities such as percutaneous ethanol injection; thermal therapies such as radiofrequency ablation, microwave ablation, laser ablation, and cryotherapy; and newer methods such as irreversible electroporation and light-activated drug therapy. Of these, radiofrequency ablation is the most widely used.
Radiofrequency ablation
Radiofrequency ablation induces thermal injury, resulting in tumor necrosis. It can be used as an alternative to surgery in patients who have a single HCC lesion less than 3 to 5 cm in diameter, confined to the liver, and in a site amenable to this procedure and who have a reasonable coagulation profile. The procedure can be performed percutaneously or via laparoscopy.
Radiofrequency ablation is contraindicated in patients with decompensated cirrhosis, Child-Pugh class C cirrhosis (the most severe category), vascular or bile duct invasion, extrahepatic disease, or lesions that are not accessible or are adjacent to structures such as the gall bladder, bowel, stomach, or diaphragm.
Radiofrequency ablation has been compared with surgical resection in patients who had small tumors. Though a randomized controlled trial did not show any difference between the two treatment groups in terms of survival at 5 years and recurrence rates,36 a meta-analysis showed that overall survival rates at 3 years and 5 years were significantly higher with surgical resection than with radiofrequency ablation.37 Patients also had a higher rate of local recurrence with radiofrequency ablation than with surgical resection.37 In addition, radiofrequency ablation has been shown to be effective only in small tumors and does not perform as well in lesions larger than 2 or 3 cm.
Thus, based on current evidence, surgical resection is preferable to radiofrequency ablation as first-line treatment. The latter, however, is also used as a bridging therapy in patients awaiting liver transplantation.
Percutaneous ethanol injection
Percutaneous ethanol injection is used less frequently than radiofrequency ablation, as studies have shown the latter to be superior in regard to local recurrence-free survival rates.38 However, percutaneous ethanol injection is used instead of radiofrequency ablation in a small number of patients, when the lesion is very close to organs such as the bile duct (which could be damaged by radiofrequency ablation) or the large vessels (which may make radiofrequency ablation less effective, since heat may dissipate as a result of excessive blood flow in this region).
Microwave ablation
Microwave ablation is an emerging therapy for HCC. Its advantage over radiofrequency ablation is that its use is not limited by blood vessels in close proximity to the ablation site.
Earlier studies did not show microwave ablation to be superior to radiofrequency ablation.39,40 However, current studies involving newer techniques of microwave ablation are more promising.41
PERFUSION-BASED LOCOREGIONAL THERAPIES
Perfusion-based locoregional therapies deliver embolic particles, chemotherapeutic agents, or radioactive materials into the artery feeding the tumor. The portal blood flow allows for preservation of vital liver tissue during arterial embolization of liver tumors. Perfusionbased therapies include transarterial chemoembolization, transarterial chemoembolization with doxorubicin-eluting beads (DEB-TACE), “bland” embolization, and radioembolization.
Transarterial chemoembolization
Transarterial chemoembolization is a minimally invasive procedure in which the hepatic artery is cannulated through a percutaneous puncture, the branches of the hepatic artery supplying the tumor are identified, and then embolic particles and chemotherapeutic agents are injected. This serves a dual purpose: it embolizes the feeding vessel that supplies the tumor, causing tumor necrosis, and it focuses the chemotherapy on the tumor and thus minimizes the systemic effects of the chemotherapeutic agent.
This therapy is contraindicated in patients with portal vein thrombosis, advanced liver dysfunction, or a transjugular intrahepatic portosystemic shunt. Side effects of the procedure include a postembolization syndrome of abdominal pain and fever (occurring in about 50% of patients from ischemic injury to the liver), hepatic abscesses, injury to the hepatic artery, development of ascites, liver dysfunction, and contrast-induced renal failure.
In addition to bridging patients to liver transplantation, transarterial chemoembolization is recommended as palliative treatment to prolong survival in patients with HCC who are not candidates for liver transplantation, surgical resection, or radiofrequency ablation.9,42 Patients who have Child-Pugh grade A or B cirrhosis but do not have main portal vein thrombosis or extrahepatic spread are candidates for this therapy. Patients such as these who undergo this therapy have a better survival rate at 2 years compared with untreated patients.43,44
Transarterial chemoembolization has also been used to reduce the size of (ie, to “downstage”) tumors that are outside the Milan criteria in patients who are otherwise candidates for liver transplantation. It induces tumor necrosis and has been shown to decrease the tumor size in a selected group of patients and to bring them within the Milan criteria, thus potentially enabling them to be put on the transplant list.45 Studies have shown that patients who receive a transplant after successful down-staging may achieve a 5-year survival rate comparable with that of patients who were initially within the Milan criteria and received a transplant without the need for down-staging.45 However, factors that predict successful down-staging have not been clearly established.
Newer techniques have been developed. A randomized controlled trial found transarterial chemoembolization with doxorubicin-eluting beads to be safer and better tolerated than conventional transarterial chemembolization.46
Bland embolization is transarterial embolization without chemotherapeutic agents and is performed in patients with significant liver dysfunction who might not tolerate chemotherapy. The benefits of this approach are yet to be determined.
Radioembolization
Radioembolization with yttrium-90 microspheres has recently been introduced as an alternative to transarterial chemoembolization, especially in patients with portal vein thrombosis, a portocaval shunt, or a transjugular intrahepatic portosystemic shunt.
In observational studies, radioembolization was as effective as transarterial chemoembolization, with a similar survival benefit.47 However, significant pulmonary shunting must be ruled out before radioembolization, as it would lead to radiation-induced pulmonary disease. Randomized controlled trials are under way to compare the efficacy of the two methods.
CHEMOTHERAPY
Sorafenib
Sorafenib is an oral antiangiogenic agent. A kinase inhibitor, it interacts with multiple intracellular and cell-surface kinases, including vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and Raf proto-oncogene, inhibiting tumor cell proliferation and angiogenesis.
Sorafenib has been shown to prolong survival in patients with advanced-stage HCC.48 A randomized placebo-controlled trial in patients with Child-Pugh grade A cirrhosis and advanced HCC who had not received chemotherapy showed that sorafenib increased the life expectancy by nearly 3 months compared with placebo.47 Sorafenib therapy is very expensive, but it is usually covered by insurance.
Sorafenib is recommended in patients who have advanced HCC with vascular invasion, extrahepatic dissemination, or minimal constitutional symptoms. It is not recommended for patients with severe advanced liver disease who have moderate to severe tumor-related constitutional symptoms or Child-Pugh grade C cirrhosis, or for patients with a life expectancy of less than 3 months.
The most common side effects of sorafenib are diarrhea, weight loss, and skin reactions on the hands and feet. These commonly lead to decreased tolerability and dose reductions.47 Doses should be adjusted on the basis of the bilirubin and albumin levels.49
Other chemotherapeutic agents
Several molecular targeted agents are undergoing clinical trials for the treatment of HCC. These include bevacizumab, erlotinib, brivanib, and ramucirumab. Chemotherapeutic agents such as doxorubicin and everolimus are also being studied.
PALLIATIVE TREATMENT
Patients with end-stage HCC with moderate to severe constitutional symptoms, extrahepatic disease progression, and decompensated liver disease have a survival of less than 3 months and are treated for pain and symptom control.9
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010; 127:2893–2917.
- El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132:2557–2576.
- El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012; 142:1264–1273.e1.
- Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 2009; 27:1485–1491.
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med 2003; 348:1625–1638.
- Sarasin FP, Giostra E, Hadengue A. Cost-effectiveness of screening for detection of small hepatocellular carcinoma in western patients with Child-Pugh class A cirrhosis. Am J Med 1996; 101:422–434.
- Yu MW, Chang HC, Liaw YF, et al. Familial risk of hepatocellular carcinoma among chronic hepatitis B carriers and their relatives. J Natl Cancer Inst 2000; 92:1159–1164.
- Kew MC, Macerollo P. Effect of age on the etiologic role of the hepatitis B virus in hepatocellular carcinoma in blacks. Gastroenterology 1988; 94:439–442.
- Bruix J, Sherman M; American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology 2011; 53:1020–1022.
- Bruix J, Llovet JM. Major achievements in hepatocellular carcinoma. Lancet 2009; 373:614–616.
- Gómez-Rodríguez R, Romero-Gutiérrez M, Artaza-Varasa T, et al. The value of the Barcelona Clinic Liver Cancer and alpha-fetoprotein in the prognosis of hepatocellular carcinoma. Rev Esp Enferm Dig 2012; 104:298–304.
- Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol 2004; 130:417–422.
- Giannini EG, Erroi V, Trevisani F. Effectiveness of a-fetoprotein for hepatocellular carcinoma surveillance: the return of the living-dead? Expert Rev Gastroenterol Hepatol 2012; 6:441–444.
- Farinati F, Marino D, De Giorgio M, et al. Diagnostic and prognostic role of alpha-fetoprotein in hepatocellular carcinoma: both or neither? Am J Gastroenterol 2006; 101:524–532.
- Forner A, Vilana R, Ayuso C, et al. Diagnosis of hepatic nodules 20 mm or smaller in cirrhosis: prospective validation of the noninvasive diagnostic criteria for hepatocellular carcinoma. Hepatology 2008; 47:97–104.
- Vilana R, Forner A, Bianchi L, et al. Intrahepatic peripheral cholangiocarcinoma in cirrhosis patients may display a vascular pattern similar to hepatocellular carcinoma on contrast-enhanced ultrasound. Hepatology 2010; 51:2020–2029.
- Kojiro M. Pathological diagnosis at early stage: reaching international consensus. Oncology 2010; 78(suppl 1):31–35.
- Schölmerich J, Schacherer D. Diagnostic biopsy for hepatocellular carcinoma in cirrhosis: useful, necessary, dangerous, or academic sport? Gut 2004; 53:1224–1226.
- Durand F, Regimbeau JM, Belghiti J, et al. Assessment of the benefits and risks of percutaneous biopsy before surgical resection of hepatocellular carcinoma. J Hepatol 2001; 35:254–258.
- Bruix J, Castells A, Bosch J, et al. Surgical resection of hepatocellular carcinoma in cirrhotic patients: prognostic value of preoperative portal pressure. Gastroenterology 1996; 111:1018–1022.
- Llovet JM, Fuster J, Bruix J. Intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: resection versus transplantation. Hepatology 1999; 30:1434–1440.
- Nagasue N, Uchida M, Makino Y, et al. Incidence and factors associated with intrahepatic recurrence following resection of hepatocellular carcinoma. Gastroenterology 1993; 105:488–494.
- Arii S, Tanaka J, Yamazoe Y, et al. Predictive factors for intrahepatic recurrence of hepatocellular carcinoma after partial hepatectomy. Cancer 1992; 69:913–919.
- Cha C, Fong Y, Jarnagin WR, Blumgart LH, DeMatteo RP. Predictors and patterns of recurrence after resection of hepatocellular carcinoma. J Am Coll Surg 2003; 197:753–758.
- Shah SA, Cleary SP, Wei AC, et al. Recurrence after liver resection for hepatocellular carcinoma: risk factors, treatment, and outcomes. Surgery 2007; 141:330–339.
- Kohno H, Nagasue N, Hayashi T, et al. Postoperative adjuvant chemotherapy after radical hepatic resection for hepatocellular carcinoma (HCC). Hepatogastroenterology 1996; 43:1405–1409.
- Ono T, Nagasue N, Kohno H, et al. Adjuvant chemotherapy with epirubicin and carmofur after radical resection of hepatocellular carcinoma: a prospective randomized study. Semin Oncol 1997; 24(suppl 6):S6–25.
- Poon RT, Fan ST, Lo CM, Liu CL, Wong J. Intrahepatic recurrence after curative resection of hepatocellular carcinoma: Long-term results of treatment and prognostic factors. Ann Surg 1999; 229:216–222.
- Mazzaferro V, Regalia E, Doci R, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med 1996; 334:693–699.
- Yao FY, Ferrell L, Bass NM, et al. Liver transplantation for hepatocellular carcinoma: expansion of the tumor size limits does not adversely impact survival. Hepatology 2001; 33:1394–1403.
- Majno P, Lencioni R, Mornex F, Girard N, Poon RT, Cherqui D. Is the treatment of hepatocellular carcinoma on the waiting list necessary? Liver Transpl 2011; 17(suppl 2):S98–S108.
- Graziadei IW, Sandmueller H, Waldenberger P, et al. Chemoembolization followed by liver transplantation for hepatocellular carcinoma impedes tumor progression while on the waiting list and leads to excellent outcome. Liver Transpl 2003; 9:557–563.
- Kulik LM, Atassi B, van Holsbeeck L, et al. Yttrium-90 microspheres (TheraSphere) treatment of unresectable hepatocellular carcinoma: downstaging to resection, RFA and bridge to transplantation. J Surg Oncol 2006; 94:572–586.
- Lu DS, Yu NC, Raman SS, et al. Percutaneous radiofrequency ablation of hepatocellular carcinoma as a bridge to liver transplantation. Hepatology 2005; 41:1130–1137.
- Pokorny H, Gnant M, Rasoul-Rockenschaub S, et al. Does additional doxorubicin chemotherapy improve outcome in patients with hepatocellular carcinoma treated by liver transplantation? Am J Transplant 2005; 5:788–794.
- Feng K, Yan J, Li X, et al. A randomized controlled trial of radiofrequency ablation and surgical resection in the treatment of small hepatocellular carcinoma. J Hepatol 2012; 57:794–802.
- Zhou Y, Zhao Y, Li B, et al. Meta-analysis of radiofrequency ablation versus hepatic resection for small hepatocellular carcinoma. BMC Gastroenterol 2010; 10:78.
- Lencioni RA, Allgaier HP, Cioni D, et al. Small hepatocellular carcinoma in cirrhosis: Randomized comparison of radiofrequency thermal ablation versus percutaneous ethanol injection. Radiology 2003; 228:235–240.
- Ohmoto K, Yoshioka N, Tomiyama Y, et al. Comparison of therapeutic effects between radiofrequency ablation and percutaneous microwave coagulation therapy for small hepatocellular carcinomas. J Gastroenterol Hepatol 2009; 24:223–227.
- Shibata T, Iimuro Y, Yamamoto Y, et al. Small hepatocellular carcinoma: comparison of radiofrequency ablation and percutaneous microwave coagulation therapy. Radiology 2002; 223:331–337.
- Qian GJ, Wang N, Shen Q, et al. Efficacy of microwave versus radiofrequency ablation for treatment of small hepatocellular carcinoma: Experimental and clinical studies. Eur Radiol 2012; 22:1983–1990.
- Burrel M, Reig M, Forner A, et al. Survival of patients with hepatocellular carcinoma treated by transarterial chemoembolisation (TACE) using drug eluting beads. Implications for clinical practice and trial design. J Hepatol 2012; 56:1330–1335.
- Cammà C, Schepis F, Orlando A, et al. Transarterial chemoembolization for unresectable hepatocellular carcinoma: meta-analysis of randomized controlled trials. Radiology 2002; 224:47–54.
- Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 2003; 37:429–442.
- Yao FY, Kerlan RK, Hirose R, et al. Excellent outcome following down-staging of hepatocellular carcinoma prior to liver transplantation: an intention-to-treat analysis. Hepatology 2008; 48:819–827.
- Ferrer Puchol MD, la Parra C, Esteban E, et al. Comparison of doxorubicin-eluting bead transarterial chemoembolization (DEBTACE) with conventional transarterial chemoembolization (TACE) for the treatment of hepatocellular carcinoma (article in Spanish). Radiologia 2011; 53:246–253.
- Salem R, Lewandowski RJ, Kulik L, et al. Radioembolization results in longer time-to-progression and reduced toxicity compared with chemoembolization in patients with hepatocellular carcinoma. Gastroenterology 2011; 140:497–507.e2.
- Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359:378–390.
- Miller AA, Murry DJ, Owzar K, et al. Phase I and pharmacokinetic study of sorafenib in patients with hepatic or renal dysfunction: CALGB 60301. J Clin Oncol 2009; 27:1800–1805.
Hepatocellular carcinoma (HCC) is a common cause of death worldwide. However, it can be detected early in high-risk individuals by using effective screening strategies, resulting in the ability to provide curative treatment.
Here, we review the risk factors for HCC, strategies for surveillance and diagnosis, and therapies that can be used.
EPIDEMIOLOGY
HCC is the most common primary malignancy of the liver. Overall, it is the fifth most common type of cancer in men and the seventh most common in women.1
Cirrhosis is present in 80% to 90% of patients with HCC.
Male sex. The male-to-female ratio is from 2:1 to 4:1, depending on the region.2 In the United States, the overall male-to-female ratio has been reported2 as 2.4:1. In another report,3 the incidence rate of HCC per 100,000 person-years was 3.7 for men and 2.0 for women.
Geographic areas with a high incidence of HCC include sub-Saharan Africa and eastern Asia, whereas Canada and the United States are low-incidence areas. The difference has been because of a lower prevalence of hepatitis B virus infection in North America. However, recent data show a downward trend in incidence of HCC in eastern Asia and an upward trend in North America (Figure 1).3,4
Viral hepatitis (ie, hepatitis B or hepatitis C) is the main risk factor for cirrhosis and HCC.
Diabetes mellitus can predispose to nonalcoholic steatohepatitis, which can subsequently progress to cirrhosis. Thus, it increases the risk of HCC.
Obesity increases the risk of death from liver cancer, with obese people (body mass index ≥ 30 kg/m2) having a higher HCC-related death rate than leaner individuals.5 And as obesity becomes more prevalent, the number of deaths from HCC could increase.
Other diseases that predispose to HCC include alcohol abuse, hereditary hemochromatosis, alpha-1-antitrypsin deficiency, and glycogen storage disease.
SURVEILLANCE OF PATIENTS AT RISK
Patients at high risk of developing liver cancer require frequent screening (Table 1).
Patients with cirrhosis. Sarasin et al6 calculated that surveillance is cost-effective and increases the odds of survival in patients with cirrhosis if the incidence of HCC exceeds 1.5% per year (which it does). In view of this finding, all patients with cirrhosis should be screened every 6 months, irrespective of the cause of the cirrhosis.
Hepatitis B carriers. Surveillance is also indicated in some hepatitis B carriers (Table 1), eg, those with a family history of HCC in a first-degree relative (an independent risk factor for developing the disease in this group).7 Also, Africans with hepatitis B tend to develop HCC early in life.8 Though it has been recommended that surveillance be started at a younger age in these patients,9 the age at which it should begin has not been clearly established. In addition, it is not clear if black people born outside Africa are at higher risk.
Benefit of surveillance
HCC surveillance has shown to lower the death rate. A randomized controlled trial in China compared screening (with abdominal ultrasonography and alpha-fetoprotein levels) vs no screening in patients with hepatitis B. It showed that screening led to a 37% decrease in the death rate.12 Studies have also established that patients with early-stage HCC have a better survival rate than patients with more-advanced disease.10,11 This survival benefit is largely explained by the availability of effective treatments for early-stage cancer, including liver transplantation. Therefore, early-stage asymptomatic patients diagnosed by a surveillance program should have a better survival rate than symptomatic patients.
Surveillance methods
The tests most often used in surveillance for HCC are serum alpha-fetoprotein levels and liver ultrasonography.
Serum alpha-fetoprotein levels by themselves have not been shown to be useful, whereas the combination of alpha-fetoprotein levels and ultrasonography has been shown to reduce the death rate when used for surveillance in a randomized trial.12 A 2012 study reported that the combination of alpha-fetoprotein testing and ultrasonography had a higher sensitivity (90%) than ultrasonography alone (58%), but at the expense of a lower specificity.13
Alpha-fetoprotein has a low sensitivity (ie, 54%) for HCC.14 Tumor size is one of the factors limiting the sensitivity of alpha-fetoprotein, 14 and this would imply that this test may not be helpful in detecting HCC at an early stage. Alpha-fetoprotein L3, an isoform of alpha-fetoprotein, may be helpful in patients with alpha-fetoprotein levels in the intermediate range, and it is currently being studied.
Liver ultrasonography is operator-dependent, and it may not be as accurate in overweight or obese people.
Computed tomography (CT) and magnetic resonance imaging (MRI) are not recommended for surveillance. Serial CT poses risks of radiation-induced damage, contrast-related anaphylaxis, and renal failure, and MRI is not cost-effective and can also lead to gadolinium-induced nephrogenic systemic fibrosis in patients with renal failure.
Currently, the American Association for the Study of Liver Diseases9 recommends ultrasonography only, every 6 months, for surveillance for HCC. However, it may be premature to conclude that alpha-fetoprotein measurement is no longer required for surveillance, and if new data emerge that support its role, it may be reincorporated into the guidelines.
DIAGNOSING HEPATOCELLULAR CARCINOMA
Lesions larger than 1 cm on ultrasonography
The finding of a liver lesion larger than 1 cm on ultrasonography during surveillance warrants further testing.
Noninvasive testing with four-phase multidetector CT or dynamic contrast-enhanced MRI is the next step. Typical findings on either of these imaging studies are sufficient to make a diagnosis of HCC, as they have a high specificity and positive predictive value.15 Arterial hyperenhancement with a venous-phase or delayed-phase washout of contrast medium confirms the diagnosis (Figure 2).9 If one of the two imaging studies is typical for HCC, liver biopsy is not needed.
Other imaging studies, including contrast-enhanced ultrasonography, have not been shown to be specific for this diagnosis.16
Liver biopsy is indicated in patients in whom the imaging findings are atypical for HCC.9,17 Biopsy has very good sensitivity and specificity for cancer, but false-negative findings do occur.18 Therefore, a negative biopsy does not entirely exclude HCC. In this situation, patients should be followed by serial ultrasonography, and any further growth or change in character should be reevaluated.
Lesions smaller than 1 cm
For lesions smaller than 1 cm, the incidence of HCC is low, and currently available diagnostic tests are not reliable.15,19 Lesions of this size should be followed by serial ultrasonography every 3 to 4 months until they either enlarge to greater than 1 cm or remain stable at 2 years.9 If they remain stable at the end of 2 years, regular surveillance ultrasonography once every 6 months can be continued.
CURATIVE AND PALLIATIVE THERAPIES
Therapies for HCC (Table 2) can be divided into two categories: curative and palliative.
Curative treatments include surgical resection, liver transplantation, and radiofrequency ablation. All other treatments are palliative, including transarterial chemoembolization and medical therapy with sorafenib.
The choice of treatment depends on the characteristics of the tumor, the degree of liver dysfunction, and the patient’s current level of function. The Barcelona Clinic Liver Cancer classification is widely used in making these decisions, as it incorporates both clinical features and tumor stage.9 Figure 3 shows a simplified management algorithm.
SURGICAL RESECTION
Surgical resection is the preferred treatment for patients who have a solitary HCC lesion without cirrhosis.9 It is also indicated in patients with well-compensated cirrhosis who have normal portal pressure, a normal serum bilirubin level, and a platelet count greater than 100 × 109/L.20,21 In such patients, the 5-year survival rate is about 74%, compared with 25% in patients with portal hypertension and serum bilirubin levels higher than 1 mg/dL.21
Surgical resection is not recommended for patients with decompensated cirrhosis, as it can worsen liver function postoperatively and increase the risk of death.19,20 In Western countries, where cirrhosis from hepatitis C is the commonest cause of HCC, most patients have poorly preserved hepatic function at the time of diagnosis, leaving only a minority of patients as candidates for surgical resection.
After surgical resection of HCC, the recurrence rate can be as high as 70% to 80% at 5 years.22,23 Studies have consistently found larger tumor size and vascular invasion to be factors that predict recurrence.24,25 Vascular invasion was also found to predict poor survival after recurrence.24 Studies have so far not shown any conclusive benefit from post-surgical adjuvant chemotherapy in reducing the rate of recurrence of HCC.26,27
How to treat recurrent HCC after surgical resection has not been clearly established. Radiofrequency ablation, transarterial chemoembolization, repeat resection, and liver transplantation have all improved survival when used alone or in combination.28 However, randomized controlled trials are needed to establish the effective treatment strategy and the benefit of multimodal treatment of recurrent HCC.
LIVER TRANSPLANTATION
Orthotopic liver transplantation is the preferred treatment for patients with HCC complicated by cirrhosis and portal hypertension. It has the advantage not only of being potentially curative, but also of overcoming liver cirrhosis by replacing the liver.
To qualify for liver transplantation, patients must meet the Milan criteria (ie, have a single nodule less than 5 cm in diameter or up to three nodules, with the largest being less than 3 cm in diameter, with no evidence of vascular invasion or distant metastasis). These patients have an expected 4-year survival rate of 85% and a recurrence-free survival rate of 92% after transplantation, compared with 50% and 59%, respectively, in patients whose tumors exceeded these criteria.29
Some believe that the Milan criteria are too restrictive and could be expanded. Yao et al at the University of California-San Francisco30 reported that patients with HCC meeting the criteria of having a solitary tumor smaller than 6.5 cm or having up to three nodules, with the largest smaller than 4.5 cm, and total tumor diameter less than 8 cm, had survival rates of 90% at 1 year and 75.2% at 5 years after liver transplantation, compared with 50% at 1 year for patients with tumors exceeding these limits. (These have come to be known as the UCSF criteria.) However, the United Network for Organ Sharing (UNOS) has not adopted these expanded criteria. UNOS has a point system for allocating livers for transplant called the Model for End-Stage Liver Disease (MELD). Patients who meet the Milan criteria receive extra points, putting them higher on the transplant list. This allows for early transplantation, thus reducing tumor progression and dropout from the transplant list. UNOS allocates a MELD score of 22 to all patients who meet the Milan criteria, and the score is further adjusted once every 3 months to reflect a 10% increase in the mortality rate. However, patients who have a single lesion smaller than 2 cm and are candidates for liver transplantation are not assigned additional MELD points per UNOS policy, as the risk of tumor progression beyond the Milan criteria in these patients is deemed to be low.
Therapies while awaiting transplantation
Even if they receive additional MELD points to give them priority on the waiting list, patients face a considerable wait before transplantation because of the limited availability of donor organs. In the interim, they have a risk of tumor progression beyond the Milan criteria and subsequent dropout from the transplant list.31 Patients on the waiting list may therefore undergo a locoregional therapy such as transarterial chemoembolization or radiofrequency ablation as bridging therapy.
These therapies have been shown to decrease dropout from the waiting list.31 A prospective study showed that in 48 patients who underwent transarterial chemoembolization while awaiting liver transplantation, none had tumor progression, and 41 did receive a transplant, with excellent posttransplantation survival rates.32 Similarly, radioembolization using yttrium-90-labeled microspheres or radiofrequency ablation while on the waiting list has been shown to significantly decrease the rate of dropout, with good posttransplantation outcomes.33,34
However, in spite of these benefits, these bridging therapies do not increase survival rates after transplantation. It is also unclear whether they are useful in regions with short waiting times for liver transplantation.
Adjuvant systemic chemotherapy has not been shown to improve survival in patients undergoing liver transplantation. For example, in a randomized controlled trial of doxorubicin given before, during, and after surgery, the survival rate at 5 years was 38% with doxorubicin and 40% without.35
ABLATIVE LOCOREGIONAL THERAPIES
Locoregional therapies play an important role in managing HCC. They are classified as ablative and perfusion-based.
Ablative locoregional therapies include chemical modalities such as percutaneous ethanol injection; thermal therapies such as radiofrequency ablation, microwave ablation, laser ablation, and cryotherapy; and newer methods such as irreversible electroporation and light-activated drug therapy. Of these, radiofrequency ablation is the most widely used.
Radiofrequency ablation
Radiofrequency ablation induces thermal injury, resulting in tumor necrosis. It can be used as an alternative to surgery in patients who have a single HCC lesion less than 3 to 5 cm in diameter, confined to the liver, and in a site amenable to this procedure and who have a reasonable coagulation profile. The procedure can be performed percutaneously or via laparoscopy.
Radiofrequency ablation is contraindicated in patients with decompensated cirrhosis, Child-Pugh class C cirrhosis (the most severe category), vascular or bile duct invasion, extrahepatic disease, or lesions that are not accessible or are adjacent to structures such as the gall bladder, bowel, stomach, or diaphragm.
Radiofrequency ablation has been compared with surgical resection in patients who had small tumors. Though a randomized controlled trial did not show any difference between the two treatment groups in terms of survival at 5 years and recurrence rates,36 a meta-analysis showed that overall survival rates at 3 years and 5 years were significantly higher with surgical resection than with radiofrequency ablation.37 Patients also had a higher rate of local recurrence with radiofrequency ablation than with surgical resection.37 In addition, radiofrequency ablation has been shown to be effective only in small tumors and does not perform as well in lesions larger than 2 or 3 cm.
Thus, based on current evidence, surgical resection is preferable to radiofrequency ablation as first-line treatment. The latter, however, is also used as a bridging therapy in patients awaiting liver transplantation.
Percutaneous ethanol injection
Percutaneous ethanol injection is used less frequently than radiofrequency ablation, as studies have shown the latter to be superior in regard to local recurrence-free survival rates.38 However, percutaneous ethanol injection is used instead of radiofrequency ablation in a small number of patients, when the lesion is very close to organs such as the bile duct (which could be damaged by radiofrequency ablation) or the large vessels (which may make radiofrequency ablation less effective, since heat may dissipate as a result of excessive blood flow in this region).
Microwave ablation
Microwave ablation is an emerging therapy for HCC. Its advantage over radiofrequency ablation is that its use is not limited by blood vessels in close proximity to the ablation site.
Earlier studies did not show microwave ablation to be superior to radiofrequency ablation.39,40 However, current studies involving newer techniques of microwave ablation are more promising.41
PERFUSION-BASED LOCOREGIONAL THERAPIES
Perfusion-based locoregional therapies deliver embolic particles, chemotherapeutic agents, or radioactive materials into the artery feeding the tumor. The portal blood flow allows for preservation of vital liver tissue during arterial embolization of liver tumors. Perfusionbased therapies include transarterial chemoembolization, transarterial chemoembolization with doxorubicin-eluting beads (DEB-TACE), “bland” embolization, and radioembolization.
Transarterial chemoembolization
Transarterial chemoembolization is a minimally invasive procedure in which the hepatic artery is cannulated through a percutaneous puncture, the branches of the hepatic artery supplying the tumor are identified, and then embolic particles and chemotherapeutic agents are injected. This serves a dual purpose: it embolizes the feeding vessel that supplies the tumor, causing tumor necrosis, and it focuses the chemotherapy on the tumor and thus minimizes the systemic effects of the chemotherapeutic agent.
This therapy is contraindicated in patients with portal vein thrombosis, advanced liver dysfunction, or a transjugular intrahepatic portosystemic shunt. Side effects of the procedure include a postembolization syndrome of abdominal pain and fever (occurring in about 50% of patients from ischemic injury to the liver), hepatic abscesses, injury to the hepatic artery, development of ascites, liver dysfunction, and contrast-induced renal failure.
In addition to bridging patients to liver transplantation, transarterial chemoembolization is recommended as palliative treatment to prolong survival in patients with HCC who are not candidates for liver transplantation, surgical resection, or radiofrequency ablation.9,42 Patients who have Child-Pugh grade A or B cirrhosis but do not have main portal vein thrombosis or extrahepatic spread are candidates for this therapy. Patients such as these who undergo this therapy have a better survival rate at 2 years compared with untreated patients.43,44
Transarterial chemoembolization has also been used to reduce the size of (ie, to “downstage”) tumors that are outside the Milan criteria in patients who are otherwise candidates for liver transplantation. It induces tumor necrosis and has been shown to decrease the tumor size in a selected group of patients and to bring them within the Milan criteria, thus potentially enabling them to be put on the transplant list.45 Studies have shown that patients who receive a transplant after successful down-staging may achieve a 5-year survival rate comparable with that of patients who were initially within the Milan criteria and received a transplant without the need for down-staging.45 However, factors that predict successful down-staging have not been clearly established.
Newer techniques have been developed. A randomized controlled trial found transarterial chemoembolization with doxorubicin-eluting beads to be safer and better tolerated than conventional transarterial chemembolization.46
Bland embolization is transarterial embolization without chemotherapeutic agents and is performed in patients with significant liver dysfunction who might not tolerate chemotherapy. The benefits of this approach are yet to be determined.
Radioembolization
Radioembolization with yttrium-90 microspheres has recently been introduced as an alternative to transarterial chemoembolization, especially in patients with portal vein thrombosis, a portocaval shunt, or a transjugular intrahepatic portosystemic shunt.
In observational studies, radioembolization was as effective as transarterial chemoembolization, with a similar survival benefit.47 However, significant pulmonary shunting must be ruled out before radioembolization, as it would lead to radiation-induced pulmonary disease. Randomized controlled trials are under way to compare the efficacy of the two methods.
CHEMOTHERAPY
Sorafenib
Sorafenib is an oral antiangiogenic agent. A kinase inhibitor, it interacts with multiple intracellular and cell-surface kinases, including vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and Raf proto-oncogene, inhibiting tumor cell proliferation and angiogenesis.
Sorafenib has been shown to prolong survival in patients with advanced-stage HCC.48 A randomized placebo-controlled trial in patients with Child-Pugh grade A cirrhosis and advanced HCC who had not received chemotherapy showed that sorafenib increased the life expectancy by nearly 3 months compared with placebo.47 Sorafenib therapy is very expensive, but it is usually covered by insurance.
Sorafenib is recommended in patients who have advanced HCC with vascular invasion, extrahepatic dissemination, or minimal constitutional symptoms. It is not recommended for patients with severe advanced liver disease who have moderate to severe tumor-related constitutional symptoms or Child-Pugh grade C cirrhosis, or for patients with a life expectancy of less than 3 months.
The most common side effects of sorafenib are diarrhea, weight loss, and skin reactions on the hands and feet. These commonly lead to decreased tolerability and dose reductions.47 Doses should be adjusted on the basis of the bilirubin and albumin levels.49
Other chemotherapeutic agents
Several molecular targeted agents are undergoing clinical trials for the treatment of HCC. These include bevacizumab, erlotinib, brivanib, and ramucirumab. Chemotherapeutic agents such as doxorubicin and everolimus are also being studied.
PALLIATIVE TREATMENT
Patients with end-stage HCC with moderate to severe constitutional symptoms, extrahepatic disease progression, and decompensated liver disease have a survival of less than 3 months and are treated for pain and symptom control.9
Hepatocellular carcinoma (HCC) is a common cause of death worldwide. However, it can be detected early in high-risk individuals by using effective screening strategies, resulting in the ability to provide curative treatment.
Here, we review the risk factors for HCC, strategies for surveillance and diagnosis, and therapies that can be used.
EPIDEMIOLOGY
HCC is the most common primary malignancy of the liver. Overall, it is the fifth most common type of cancer in men and the seventh most common in women.1
Cirrhosis is present in 80% to 90% of patients with HCC.
Male sex. The male-to-female ratio is from 2:1 to 4:1, depending on the region.2 In the United States, the overall male-to-female ratio has been reported2 as 2.4:1. In another report,3 the incidence rate of HCC per 100,000 person-years was 3.7 for men and 2.0 for women.
Geographic areas with a high incidence of HCC include sub-Saharan Africa and eastern Asia, whereas Canada and the United States are low-incidence areas. The difference has been because of a lower prevalence of hepatitis B virus infection in North America. However, recent data show a downward trend in incidence of HCC in eastern Asia and an upward trend in North America (Figure 1).3,4
Viral hepatitis (ie, hepatitis B or hepatitis C) is the main risk factor for cirrhosis and HCC.
Diabetes mellitus can predispose to nonalcoholic steatohepatitis, which can subsequently progress to cirrhosis. Thus, it increases the risk of HCC.
Obesity increases the risk of death from liver cancer, with obese people (body mass index ≥ 30 kg/m2) having a higher HCC-related death rate than leaner individuals.5 And as obesity becomes more prevalent, the number of deaths from HCC could increase.
Other diseases that predispose to HCC include alcohol abuse, hereditary hemochromatosis, alpha-1-antitrypsin deficiency, and glycogen storage disease.
SURVEILLANCE OF PATIENTS AT RISK
Patients at high risk of developing liver cancer require frequent screening (Table 1).
Patients with cirrhosis. Sarasin et al6 calculated that surveillance is cost-effective and increases the odds of survival in patients with cirrhosis if the incidence of HCC exceeds 1.5% per year (which it does). In view of this finding, all patients with cirrhosis should be screened every 6 months, irrespective of the cause of the cirrhosis.
Hepatitis B carriers. Surveillance is also indicated in some hepatitis B carriers (Table 1), eg, those with a family history of HCC in a first-degree relative (an independent risk factor for developing the disease in this group).7 Also, Africans with hepatitis B tend to develop HCC early in life.8 Though it has been recommended that surveillance be started at a younger age in these patients,9 the age at which it should begin has not been clearly established. In addition, it is not clear if black people born outside Africa are at higher risk.
Benefit of surveillance
HCC surveillance has shown to lower the death rate. A randomized controlled trial in China compared screening (with abdominal ultrasonography and alpha-fetoprotein levels) vs no screening in patients with hepatitis B. It showed that screening led to a 37% decrease in the death rate.12 Studies have also established that patients with early-stage HCC have a better survival rate than patients with more-advanced disease.10,11 This survival benefit is largely explained by the availability of effective treatments for early-stage cancer, including liver transplantation. Therefore, early-stage asymptomatic patients diagnosed by a surveillance program should have a better survival rate than symptomatic patients.
Surveillance methods
The tests most often used in surveillance for HCC are serum alpha-fetoprotein levels and liver ultrasonography.
Serum alpha-fetoprotein levels by themselves have not been shown to be useful, whereas the combination of alpha-fetoprotein levels and ultrasonography has been shown to reduce the death rate when used for surveillance in a randomized trial.12 A 2012 study reported that the combination of alpha-fetoprotein testing and ultrasonography had a higher sensitivity (90%) than ultrasonography alone (58%), but at the expense of a lower specificity.13
Alpha-fetoprotein has a low sensitivity (ie, 54%) for HCC.14 Tumor size is one of the factors limiting the sensitivity of alpha-fetoprotein, 14 and this would imply that this test may not be helpful in detecting HCC at an early stage. Alpha-fetoprotein L3, an isoform of alpha-fetoprotein, may be helpful in patients with alpha-fetoprotein levels in the intermediate range, and it is currently being studied.
Liver ultrasonography is operator-dependent, and it may not be as accurate in overweight or obese people.
Computed tomography (CT) and magnetic resonance imaging (MRI) are not recommended for surveillance. Serial CT poses risks of radiation-induced damage, contrast-related anaphylaxis, and renal failure, and MRI is not cost-effective and can also lead to gadolinium-induced nephrogenic systemic fibrosis in patients with renal failure.
Currently, the American Association for the Study of Liver Diseases9 recommends ultrasonography only, every 6 months, for surveillance for HCC. However, it may be premature to conclude that alpha-fetoprotein measurement is no longer required for surveillance, and if new data emerge that support its role, it may be reincorporated into the guidelines.
DIAGNOSING HEPATOCELLULAR CARCINOMA
Lesions larger than 1 cm on ultrasonography
The finding of a liver lesion larger than 1 cm on ultrasonography during surveillance warrants further testing.
Noninvasive testing with four-phase multidetector CT or dynamic contrast-enhanced MRI is the next step. Typical findings on either of these imaging studies are sufficient to make a diagnosis of HCC, as they have a high specificity and positive predictive value.15 Arterial hyperenhancement with a venous-phase or delayed-phase washout of contrast medium confirms the diagnosis (Figure 2).9 If one of the two imaging studies is typical for HCC, liver biopsy is not needed.
Other imaging studies, including contrast-enhanced ultrasonography, have not been shown to be specific for this diagnosis.16
Liver biopsy is indicated in patients in whom the imaging findings are atypical for HCC.9,17 Biopsy has very good sensitivity and specificity for cancer, but false-negative findings do occur.18 Therefore, a negative biopsy does not entirely exclude HCC. In this situation, patients should be followed by serial ultrasonography, and any further growth or change in character should be reevaluated.
Lesions smaller than 1 cm
For lesions smaller than 1 cm, the incidence of HCC is low, and currently available diagnostic tests are not reliable.15,19 Lesions of this size should be followed by serial ultrasonography every 3 to 4 months until they either enlarge to greater than 1 cm or remain stable at 2 years.9 If they remain stable at the end of 2 years, regular surveillance ultrasonography once every 6 months can be continued.
CURATIVE AND PALLIATIVE THERAPIES
Therapies for HCC (Table 2) can be divided into two categories: curative and palliative.
Curative treatments include surgical resection, liver transplantation, and radiofrequency ablation. All other treatments are palliative, including transarterial chemoembolization and medical therapy with sorafenib.
The choice of treatment depends on the characteristics of the tumor, the degree of liver dysfunction, and the patient’s current level of function. The Barcelona Clinic Liver Cancer classification is widely used in making these decisions, as it incorporates both clinical features and tumor stage.9 Figure 3 shows a simplified management algorithm.
SURGICAL RESECTION
Surgical resection is the preferred treatment for patients who have a solitary HCC lesion without cirrhosis.9 It is also indicated in patients with well-compensated cirrhosis who have normal portal pressure, a normal serum bilirubin level, and a platelet count greater than 100 × 109/L.20,21 In such patients, the 5-year survival rate is about 74%, compared with 25% in patients with portal hypertension and serum bilirubin levels higher than 1 mg/dL.21
Surgical resection is not recommended for patients with decompensated cirrhosis, as it can worsen liver function postoperatively and increase the risk of death.19,20 In Western countries, where cirrhosis from hepatitis C is the commonest cause of HCC, most patients have poorly preserved hepatic function at the time of diagnosis, leaving only a minority of patients as candidates for surgical resection.
After surgical resection of HCC, the recurrence rate can be as high as 70% to 80% at 5 years.22,23 Studies have consistently found larger tumor size and vascular invasion to be factors that predict recurrence.24,25 Vascular invasion was also found to predict poor survival after recurrence.24 Studies have so far not shown any conclusive benefit from post-surgical adjuvant chemotherapy in reducing the rate of recurrence of HCC.26,27
How to treat recurrent HCC after surgical resection has not been clearly established. Radiofrequency ablation, transarterial chemoembolization, repeat resection, and liver transplantation have all improved survival when used alone or in combination.28 However, randomized controlled trials are needed to establish the effective treatment strategy and the benefit of multimodal treatment of recurrent HCC.
LIVER TRANSPLANTATION
Orthotopic liver transplantation is the preferred treatment for patients with HCC complicated by cirrhosis and portal hypertension. It has the advantage not only of being potentially curative, but also of overcoming liver cirrhosis by replacing the liver.
To qualify for liver transplantation, patients must meet the Milan criteria (ie, have a single nodule less than 5 cm in diameter or up to three nodules, with the largest being less than 3 cm in diameter, with no evidence of vascular invasion or distant metastasis). These patients have an expected 4-year survival rate of 85% and a recurrence-free survival rate of 92% after transplantation, compared with 50% and 59%, respectively, in patients whose tumors exceeded these criteria.29
Some believe that the Milan criteria are too restrictive and could be expanded. Yao et al at the University of California-San Francisco30 reported that patients with HCC meeting the criteria of having a solitary tumor smaller than 6.5 cm or having up to three nodules, with the largest smaller than 4.5 cm, and total tumor diameter less than 8 cm, had survival rates of 90% at 1 year and 75.2% at 5 years after liver transplantation, compared with 50% at 1 year for patients with tumors exceeding these limits. (These have come to be known as the UCSF criteria.) However, the United Network for Organ Sharing (UNOS) has not adopted these expanded criteria. UNOS has a point system for allocating livers for transplant called the Model for End-Stage Liver Disease (MELD). Patients who meet the Milan criteria receive extra points, putting them higher on the transplant list. This allows for early transplantation, thus reducing tumor progression and dropout from the transplant list. UNOS allocates a MELD score of 22 to all patients who meet the Milan criteria, and the score is further adjusted once every 3 months to reflect a 10% increase in the mortality rate. However, patients who have a single lesion smaller than 2 cm and are candidates for liver transplantation are not assigned additional MELD points per UNOS policy, as the risk of tumor progression beyond the Milan criteria in these patients is deemed to be low.
Therapies while awaiting transplantation
Even if they receive additional MELD points to give them priority on the waiting list, patients face a considerable wait before transplantation because of the limited availability of donor organs. In the interim, they have a risk of tumor progression beyond the Milan criteria and subsequent dropout from the transplant list.31 Patients on the waiting list may therefore undergo a locoregional therapy such as transarterial chemoembolization or radiofrequency ablation as bridging therapy.
These therapies have been shown to decrease dropout from the waiting list.31 A prospective study showed that in 48 patients who underwent transarterial chemoembolization while awaiting liver transplantation, none had tumor progression, and 41 did receive a transplant, with excellent posttransplantation survival rates.32 Similarly, radioembolization using yttrium-90-labeled microspheres or radiofrequency ablation while on the waiting list has been shown to significantly decrease the rate of dropout, with good posttransplantation outcomes.33,34
However, in spite of these benefits, these bridging therapies do not increase survival rates after transplantation. It is also unclear whether they are useful in regions with short waiting times for liver transplantation.
Adjuvant systemic chemotherapy has not been shown to improve survival in patients undergoing liver transplantation. For example, in a randomized controlled trial of doxorubicin given before, during, and after surgery, the survival rate at 5 years was 38% with doxorubicin and 40% without.35
ABLATIVE LOCOREGIONAL THERAPIES
Locoregional therapies play an important role in managing HCC. They are classified as ablative and perfusion-based.
Ablative locoregional therapies include chemical modalities such as percutaneous ethanol injection; thermal therapies such as radiofrequency ablation, microwave ablation, laser ablation, and cryotherapy; and newer methods such as irreversible electroporation and light-activated drug therapy. Of these, radiofrequency ablation is the most widely used.
Radiofrequency ablation
Radiofrequency ablation induces thermal injury, resulting in tumor necrosis. It can be used as an alternative to surgery in patients who have a single HCC lesion less than 3 to 5 cm in diameter, confined to the liver, and in a site amenable to this procedure and who have a reasonable coagulation profile. The procedure can be performed percutaneously or via laparoscopy.
Radiofrequency ablation is contraindicated in patients with decompensated cirrhosis, Child-Pugh class C cirrhosis (the most severe category), vascular or bile duct invasion, extrahepatic disease, or lesions that are not accessible or are adjacent to structures such as the gall bladder, bowel, stomach, or diaphragm.
Radiofrequency ablation has been compared with surgical resection in patients who had small tumors. Though a randomized controlled trial did not show any difference between the two treatment groups in terms of survival at 5 years and recurrence rates,36 a meta-analysis showed that overall survival rates at 3 years and 5 years were significantly higher with surgical resection than with radiofrequency ablation.37 Patients also had a higher rate of local recurrence with radiofrequency ablation than with surgical resection.37 In addition, radiofrequency ablation has been shown to be effective only in small tumors and does not perform as well in lesions larger than 2 or 3 cm.
Thus, based on current evidence, surgical resection is preferable to radiofrequency ablation as first-line treatment. The latter, however, is also used as a bridging therapy in patients awaiting liver transplantation.
Percutaneous ethanol injection
Percutaneous ethanol injection is used less frequently than radiofrequency ablation, as studies have shown the latter to be superior in regard to local recurrence-free survival rates.38 However, percutaneous ethanol injection is used instead of radiofrequency ablation in a small number of patients, when the lesion is very close to organs such as the bile duct (which could be damaged by radiofrequency ablation) or the large vessels (which may make radiofrequency ablation less effective, since heat may dissipate as a result of excessive blood flow in this region).
Microwave ablation
Microwave ablation is an emerging therapy for HCC. Its advantage over radiofrequency ablation is that its use is not limited by blood vessels in close proximity to the ablation site.
Earlier studies did not show microwave ablation to be superior to radiofrequency ablation.39,40 However, current studies involving newer techniques of microwave ablation are more promising.41
PERFUSION-BASED LOCOREGIONAL THERAPIES
Perfusion-based locoregional therapies deliver embolic particles, chemotherapeutic agents, or radioactive materials into the artery feeding the tumor. The portal blood flow allows for preservation of vital liver tissue during arterial embolization of liver tumors. Perfusionbased therapies include transarterial chemoembolization, transarterial chemoembolization with doxorubicin-eluting beads (DEB-TACE), “bland” embolization, and radioembolization.
Transarterial chemoembolization
Transarterial chemoembolization is a minimally invasive procedure in which the hepatic artery is cannulated through a percutaneous puncture, the branches of the hepatic artery supplying the tumor are identified, and then embolic particles and chemotherapeutic agents are injected. This serves a dual purpose: it embolizes the feeding vessel that supplies the tumor, causing tumor necrosis, and it focuses the chemotherapy on the tumor and thus minimizes the systemic effects of the chemotherapeutic agent.
This therapy is contraindicated in patients with portal vein thrombosis, advanced liver dysfunction, or a transjugular intrahepatic portosystemic shunt. Side effects of the procedure include a postembolization syndrome of abdominal pain and fever (occurring in about 50% of patients from ischemic injury to the liver), hepatic abscesses, injury to the hepatic artery, development of ascites, liver dysfunction, and contrast-induced renal failure.
In addition to bridging patients to liver transplantation, transarterial chemoembolization is recommended as palliative treatment to prolong survival in patients with HCC who are not candidates for liver transplantation, surgical resection, or radiofrequency ablation.9,42 Patients who have Child-Pugh grade A or B cirrhosis but do not have main portal vein thrombosis or extrahepatic spread are candidates for this therapy. Patients such as these who undergo this therapy have a better survival rate at 2 years compared with untreated patients.43,44
Transarterial chemoembolization has also been used to reduce the size of (ie, to “downstage”) tumors that are outside the Milan criteria in patients who are otherwise candidates for liver transplantation. It induces tumor necrosis and has been shown to decrease the tumor size in a selected group of patients and to bring them within the Milan criteria, thus potentially enabling them to be put on the transplant list.45 Studies have shown that patients who receive a transplant after successful down-staging may achieve a 5-year survival rate comparable with that of patients who were initially within the Milan criteria and received a transplant without the need for down-staging.45 However, factors that predict successful down-staging have not been clearly established.
Newer techniques have been developed. A randomized controlled trial found transarterial chemoembolization with doxorubicin-eluting beads to be safer and better tolerated than conventional transarterial chemembolization.46
Bland embolization is transarterial embolization without chemotherapeutic agents and is performed in patients with significant liver dysfunction who might not tolerate chemotherapy. The benefits of this approach are yet to be determined.
Radioembolization
Radioembolization with yttrium-90 microspheres has recently been introduced as an alternative to transarterial chemoembolization, especially in patients with portal vein thrombosis, a portocaval shunt, or a transjugular intrahepatic portosystemic shunt.
In observational studies, radioembolization was as effective as transarterial chemoembolization, with a similar survival benefit.47 However, significant pulmonary shunting must be ruled out before radioembolization, as it would lead to radiation-induced pulmonary disease. Randomized controlled trials are under way to compare the efficacy of the two methods.
CHEMOTHERAPY
Sorafenib
Sorafenib is an oral antiangiogenic agent. A kinase inhibitor, it interacts with multiple intracellular and cell-surface kinases, including vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and Raf proto-oncogene, inhibiting tumor cell proliferation and angiogenesis.
Sorafenib has been shown to prolong survival in patients with advanced-stage HCC.48 A randomized placebo-controlled trial in patients with Child-Pugh grade A cirrhosis and advanced HCC who had not received chemotherapy showed that sorafenib increased the life expectancy by nearly 3 months compared with placebo.47 Sorafenib therapy is very expensive, but it is usually covered by insurance.
Sorafenib is recommended in patients who have advanced HCC with vascular invasion, extrahepatic dissemination, or minimal constitutional symptoms. It is not recommended for patients with severe advanced liver disease who have moderate to severe tumor-related constitutional symptoms or Child-Pugh grade C cirrhosis, or for patients with a life expectancy of less than 3 months.
The most common side effects of sorafenib are diarrhea, weight loss, and skin reactions on the hands and feet. These commonly lead to decreased tolerability and dose reductions.47 Doses should be adjusted on the basis of the bilirubin and albumin levels.49
Other chemotherapeutic agents
Several molecular targeted agents are undergoing clinical trials for the treatment of HCC. These include bevacizumab, erlotinib, brivanib, and ramucirumab. Chemotherapeutic agents such as doxorubicin and everolimus are also being studied.
PALLIATIVE TREATMENT
Patients with end-stage HCC with moderate to severe constitutional symptoms, extrahepatic disease progression, and decompensated liver disease have a survival of less than 3 months and are treated for pain and symptom control.9
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010; 127:2893–2917.
- El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132:2557–2576.
- El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012; 142:1264–1273.e1.
- Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 2009; 27:1485–1491.
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med 2003; 348:1625–1638.
- Sarasin FP, Giostra E, Hadengue A. Cost-effectiveness of screening for detection of small hepatocellular carcinoma in western patients with Child-Pugh class A cirrhosis. Am J Med 1996; 101:422–434.
- Yu MW, Chang HC, Liaw YF, et al. Familial risk of hepatocellular carcinoma among chronic hepatitis B carriers and their relatives. J Natl Cancer Inst 2000; 92:1159–1164.
- Kew MC, Macerollo P. Effect of age on the etiologic role of the hepatitis B virus in hepatocellular carcinoma in blacks. Gastroenterology 1988; 94:439–442.
- Bruix J, Sherman M; American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology 2011; 53:1020–1022.
- Bruix J, Llovet JM. Major achievements in hepatocellular carcinoma. Lancet 2009; 373:614–616.
- Gómez-Rodríguez R, Romero-Gutiérrez M, Artaza-Varasa T, et al. The value of the Barcelona Clinic Liver Cancer and alpha-fetoprotein in the prognosis of hepatocellular carcinoma. Rev Esp Enferm Dig 2012; 104:298–304.
- Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol 2004; 130:417–422.
- Giannini EG, Erroi V, Trevisani F. Effectiveness of a-fetoprotein for hepatocellular carcinoma surveillance: the return of the living-dead? Expert Rev Gastroenterol Hepatol 2012; 6:441–444.
- Farinati F, Marino D, De Giorgio M, et al. Diagnostic and prognostic role of alpha-fetoprotein in hepatocellular carcinoma: both or neither? Am J Gastroenterol 2006; 101:524–532.
- Forner A, Vilana R, Ayuso C, et al. Diagnosis of hepatic nodules 20 mm or smaller in cirrhosis: prospective validation of the noninvasive diagnostic criteria for hepatocellular carcinoma. Hepatology 2008; 47:97–104.
- Vilana R, Forner A, Bianchi L, et al. Intrahepatic peripheral cholangiocarcinoma in cirrhosis patients may display a vascular pattern similar to hepatocellular carcinoma on contrast-enhanced ultrasound. Hepatology 2010; 51:2020–2029.
- Kojiro M. Pathological diagnosis at early stage: reaching international consensus. Oncology 2010; 78(suppl 1):31–35.
- Schölmerich J, Schacherer D. Diagnostic biopsy for hepatocellular carcinoma in cirrhosis: useful, necessary, dangerous, or academic sport? Gut 2004; 53:1224–1226.
- Durand F, Regimbeau JM, Belghiti J, et al. Assessment of the benefits and risks of percutaneous biopsy before surgical resection of hepatocellular carcinoma. J Hepatol 2001; 35:254–258.
- Bruix J, Castells A, Bosch J, et al. Surgical resection of hepatocellular carcinoma in cirrhotic patients: prognostic value of preoperative portal pressure. Gastroenterology 1996; 111:1018–1022.
- Llovet JM, Fuster J, Bruix J. Intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: resection versus transplantation. Hepatology 1999; 30:1434–1440.
- Nagasue N, Uchida M, Makino Y, et al. Incidence and factors associated with intrahepatic recurrence following resection of hepatocellular carcinoma. Gastroenterology 1993; 105:488–494.
- Arii S, Tanaka J, Yamazoe Y, et al. Predictive factors for intrahepatic recurrence of hepatocellular carcinoma after partial hepatectomy. Cancer 1992; 69:913–919.
- Cha C, Fong Y, Jarnagin WR, Blumgart LH, DeMatteo RP. Predictors and patterns of recurrence after resection of hepatocellular carcinoma. J Am Coll Surg 2003; 197:753–758.
- Shah SA, Cleary SP, Wei AC, et al. Recurrence after liver resection for hepatocellular carcinoma: risk factors, treatment, and outcomes. Surgery 2007; 141:330–339.
- Kohno H, Nagasue N, Hayashi T, et al. Postoperative adjuvant chemotherapy after radical hepatic resection for hepatocellular carcinoma (HCC). Hepatogastroenterology 1996; 43:1405–1409.
- Ono T, Nagasue N, Kohno H, et al. Adjuvant chemotherapy with epirubicin and carmofur after radical resection of hepatocellular carcinoma: a prospective randomized study. Semin Oncol 1997; 24(suppl 6):S6–25.
- Poon RT, Fan ST, Lo CM, Liu CL, Wong J. Intrahepatic recurrence after curative resection of hepatocellular carcinoma: Long-term results of treatment and prognostic factors. Ann Surg 1999; 229:216–222.
- Mazzaferro V, Regalia E, Doci R, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med 1996; 334:693–699.
- Yao FY, Ferrell L, Bass NM, et al. Liver transplantation for hepatocellular carcinoma: expansion of the tumor size limits does not adversely impact survival. Hepatology 2001; 33:1394–1403.
- Majno P, Lencioni R, Mornex F, Girard N, Poon RT, Cherqui D. Is the treatment of hepatocellular carcinoma on the waiting list necessary? Liver Transpl 2011; 17(suppl 2):S98–S108.
- Graziadei IW, Sandmueller H, Waldenberger P, et al. Chemoembolization followed by liver transplantation for hepatocellular carcinoma impedes tumor progression while on the waiting list and leads to excellent outcome. Liver Transpl 2003; 9:557–563.
- Kulik LM, Atassi B, van Holsbeeck L, et al. Yttrium-90 microspheres (TheraSphere) treatment of unresectable hepatocellular carcinoma: downstaging to resection, RFA and bridge to transplantation. J Surg Oncol 2006; 94:572–586.
- Lu DS, Yu NC, Raman SS, et al. Percutaneous radiofrequency ablation of hepatocellular carcinoma as a bridge to liver transplantation. Hepatology 2005; 41:1130–1137.
- Pokorny H, Gnant M, Rasoul-Rockenschaub S, et al. Does additional doxorubicin chemotherapy improve outcome in patients with hepatocellular carcinoma treated by liver transplantation? Am J Transplant 2005; 5:788–794.
- Feng K, Yan J, Li X, et al. A randomized controlled trial of radiofrequency ablation and surgical resection in the treatment of small hepatocellular carcinoma. J Hepatol 2012; 57:794–802.
- Zhou Y, Zhao Y, Li B, et al. Meta-analysis of radiofrequency ablation versus hepatic resection for small hepatocellular carcinoma. BMC Gastroenterol 2010; 10:78.
- Lencioni RA, Allgaier HP, Cioni D, et al. Small hepatocellular carcinoma in cirrhosis: Randomized comparison of radiofrequency thermal ablation versus percutaneous ethanol injection. Radiology 2003; 228:235–240.
- Ohmoto K, Yoshioka N, Tomiyama Y, et al. Comparison of therapeutic effects between radiofrequency ablation and percutaneous microwave coagulation therapy for small hepatocellular carcinomas. J Gastroenterol Hepatol 2009; 24:223–227.
- Shibata T, Iimuro Y, Yamamoto Y, et al. Small hepatocellular carcinoma: comparison of radiofrequency ablation and percutaneous microwave coagulation therapy. Radiology 2002; 223:331–337.
- Qian GJ, Wang N, Shen Q, et al. Efficacy of microwave versus radiofrequency ablation for treatment of small hepatocellular carcinoma: Experimental and clinical studies. Eur Radiol 2012; 22:1983–1990.
- Burrel M, Reig M, Forner A, et al. Survival of patients with hepatocellular carcinoma treated by transarterial chemoembolisation (TACE) using drug eluting beads. Implications for clinical practice and trial design. J Hepatol 2012; 56:1330–1335.
- Cammà C, Schepis F, Orlando A, et al. Transarterial chemoembolization for unresectable hepatocellular carcinoma: meta-analysis of randomized controlled trials. Radiology 2002; 224:47–54.
- Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 2003; 37:429–442.
- Yao FY, Kerlan RK, Hirose R, et al. Excellent outcome following down-staging of hepatocellular carcinoma prior to liver transplantation: an intention-to-treat analysis. Hepatology 2008; 48:819–827.
- Ferrer Puchol MD, la Parra C, Esteban E, et al. Comparison of doxorubicin-eluting bead transarterial chemoembolization (DEBTACE) with conventional transarterial chemoembolization (TACE) for the treatment of hepatocellular carcinoma (article in Spanish). Radiologia 2011; 53:246–253.
- Salem R, Lewandowski RJ, Kulik L, et al. Radioembolization results in longer time-to-progression and reduced toxicity compared with chemoembolization in patients with hepatocellular carcinoma. Gastroenterology 2011; 140:497–507.e2.
- Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359:378–390.
- Miller AA, Murry DJ, Owzar K, et al. Phase I and pharmacokinetic study of sorafenib in patients with hepatic or renal dysfunction: CALGB 60301. J Clin Oncol 2009; 27:1800–1805.
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010; 127:2893–2917.
- El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132:2557–2576.
- El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012; 142:1264–1273.e1.
- Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 2009; 27:1485–1491.
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med 2003; 348:1625–1638.
- Sarasin FP, Giostra E, Hadengue A. Cost-effectiveness of screening for detection of small hepatocellular carcinoma in western patients with Child-Pugh class A cirrhosis. Am J Med 1996; 101:422–434.
- Yu MW, Chang HC, Liaw YF, et al. Familial risk of hepatocellular carcinoma among chronic hepatitis B carriers and their relatives. J Natl Cancer Inst 2000; 92:1159–1164.
- Kew MC, Macerollo P. Effect of age on the etiologic role of the hepatitis B virus in hepatocellular carcinoma in blacks. Gastroenterology 1988; 94:439–442.
- Bruix J, Sherman M; American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology 2011; 53:1020–1022.
- Bruix J, Llovet JM. Major achievements in hepatocellular carcinoma. Lancet 2009; 373:614–616.
- Gómez-Rodríguez R, Romero-Gutiérrez M, Artaza-Varasa T, et al. The value of the Barcelona Clinic Liver Cancer and alpha-fetoprotein in the prognosis of hepatocellular carcinoma. Rev Esp Enferm Dig 2012; 104:298–304.
- Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol 2004; 130:417–422.
- Giannini EG, Erroi V, Trevisani F. Effectiveness of a-fetoprotein for hepatocellular carcinoma surveillance: the return of the living-dead? Expert Rev Gastroenterol Hepatol 2012; 6:441–444.
- Farinati F, Marino D, De Giorgio M, et al. Diagnostic and prognostic role of alpha-fetoprotein in hepatocellular carcinoma: both or neither? Am J Gastroenterol 2006; 101:524–532.
- Forner A, Vilana R, Ayuso C, et al. Diagnosis of hepatic nodules 20 mm or smaller in cirrhosis: prospective validation of the noninvasive diagnostic criteria for hepatocellular carcinoma. Hepatology 2008; 47:97–104.
- Vilana R, Forner A, Bianchi L, et al. Intrahepatic peripheral cholangiocarcinoma in cirrhosis patients may display a vascular pattern similar to hepatocellular carcinoma on contrast-enhanced ultrasound. Hepatology 2010; 51:2020–2029.
- Kojiro M. Pathological diagnosis at early stage: reaching international consensus. Oncology 2010; 78(suppl 1):31–35.
- Schölmerich J, Schacherer D. Diagnostic biopsy for hepatocellular carcinoma in cirrhosis: useful, necessary, dangerous, or academic sport? Gut 2004; 53:1224–1226.
- Durand F, Regimbeau JM, Belghiti J, et al. Assessment of the benefits and risks of percutaneous biopsy before surgical resection of hepatocellular carcinoma. J Hepatol 2001; 35:254–258.
- Bruix J, Castells A, Bosch J, et al. Surgical resection of hepatocellular carcinoma in cirrhotic patients: prognostic value of preoperative portal pressure. Gastroenterology 1996; 111:1018–1022.
- Llovet JM, Fuster J, Bruix J. Intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: resection versus transplantation. Hepatology 1999; 30:1434–1440.
- Nagasue N, Uchida M, Makino Y, et al. Incidence and factors associated with intrahepatic recurrence following resection of hepatocellular carcinoma. Gastroenterology 1993; 105:488–494.
- Arii S, Tanaka J, Yamazoe Y, et al. Predictive factors for intrahepatic recurrence of hepatocellular carcinoma after partial hepatectomy. Cancer 1992; 69:913–919.
- Cha C, Fong Y, Jarnagin WR, Blumgart LH, DeMatteo RP. Predictors and patterns of recurrence after resection of hepatocellular carcinoma. J Am Coll Surg 2003; 197:753–758.
- Shah SA, Cleary SP, Wei AC, et al. Recurrence after liver resection for hepatocellular carcinoma: risk factors, treatment, and outcomes. Surgery 2007; 141:330–339.
- Kohno H, Nagasue N, Hayashi T, et al. Postoperative adjuvant chemotherapy after radical hepatic resection for hepatocellular carcinoma (HCC). Hepatogastroenterology 1996; 43:1405–1409.
- Ono T, Nagasue N, Kohno H, et al. Adjuvant chemotherapy with epirubicin and carmofur after radical resection of hepatocellular carcinoma: a prospective randomized study. Semin Oncol 1997; 24(suppl 6):S6–25.
- Poon RT, Fan ST, Lo CM, Liu CL, Wong J. Intrahepatic recurrence after curative resection of hepatocellular carcinoma: Long-term results of treatment and prognostic factors. Ann Surg 1999; 229:216–222.
- Mazzaferro V, Regalia E, Doci R, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med 1996; 334:693–699.
- Yao FY, Ferrell L, Bass NM, et al. Liver transplantation for hepatocellular carcinoma: expansion of the tumor size limits does not adversely impact survival. Hepatology 2001; 33:1394–1403.
- Majno P, Lencioni R, Mornex F, Girard N, Poon RT, Cherqui D. Is the treatment of hepatocellular carcinoma on the waiting list necessary? Liver Transpl 2011; 17(suppl 2):S98–S108.
- Graziadei IW, Sandmueller H, Waldenberger P, et al. Chemoembolization followed by liver transplantation for hepatocellular carcinoma impedes tumor progression while on the waiting list and leads to excellent outcome. Liver Transpl 2003; 9:557–563.
- Kulik LM, Atassi B, van Holsbeeck L, et al. Yttrium-90 microspheres (TheraSphere) treatment of unresectable hepatocellular carcinoma: downstaging to resection, RFA and bridge to transplantation. J Surg Oncol 2006; 94:572–586.
- Lu DS, Yu NC, Raman SS, et al. Percutaneous radiofrequency ablation of hepatocellular carcinoma as a bridge to liver transplantation. Hepatology 2005; 41:1130–1137.
- Pokorny H, Gnant M, Rasoul-Rockenschaub S, et al. Does additional doxorubicin chemotherapy improve outcome in patients with hepatocellular carcinoma treated by liver transplantation? Am J Transplant 2005; 5:788–794.
- Feng K, Yan J, Li X, et al. A randomized controlled trial of radiofrequency ablation and surgical resection in the treatment of small hepatocellular carcinoma. J Hepatol 2012; 57:794–802.
- Zhou Y, Zhao Y, Li B, et al. Meta-analysis of radiofrequency ablation versus hepatic resection for small hepatocellular carcinoma. BMC Gastroenterol 2010; 10:78.
- Lencioni RA, Allgaier HP, Cioni D, et al. Small hepatocellular carcinoma in cirrhosis: Randomized comparison of radiofrequency thermal ablation versus percutaneous ethanol injection. Radiology 2003; 228:235–240.
- Ohmoto K, Yoshioka N, Tomiyama Y, et al. Comparison of therapeutic effects between radiofrequency ablation and percutaneous microwave coagulation therapy for small hepatocellular carcinomas. J Gastroenterol Hepatol 2009; 24:223–227.
- Shibata T, Iimuro Y, Yamamoto Y, et al. Small hepatocellular carcinoma: comparison of radiofrequency ablation and percutaneous microwave coagulation therapy. Radiology 2002; 223:331–337.
- Qian GJ, Wang N, Shen Q, et al. Efficacy of microwave versus radiofrequency ablation for treatment of small hepatocellular carcinoma: Experimental and clinical studies. Eur Radiol 2012; 22:1983–1990.
- Burrel M, Reig M, Forner A, et al. Survival of patients with hepatocellular carcinoma treated by transarterial chemoembolisation (TACE) using drug eluting beads. Implications for clinical practice and trial design. J Hepatol 2012; 56:1330–1335.
- Cammà C, Schepis F, Orlando A, et al. Transarterial chemoembolization for unresectable hepatocellular carcinoma: meta-analysis of randomized controlled trials. Radiology 2002; 224:47–54.
- Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 2003; 37:429–442.
- Yao FY, Kerlan RK, Hirose R, et al. Excellent outcome following down-staging of hepatocellular carcinoma prior to liver transplantation: an intention-to-treat analysis. Hepatology 2008; 48:819–827.
- Ferrer Puchol MD, la Parra C, Esteban E, et al. Comparison of doxorubicin-eluting bead transarterial chemoembolization (DEBTACE) with conventional transarterial chemoembolization (TACE) for the treatment of hepatocellular carcinoma (article in Spanish). Radiologia 2011; 53:246–253.
- Salem R, Lewandowski RJ, Kulik L, et al. Radioembolization results in longer time-to-progression and reduced toxicity compared with chemoembolization in patients with hepatocellular carcinoma. Gastroenterology 2011; 140:497–507.e2.
- Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359:378–390.
- Miller AA, Murry DJ, Owzar K, et al. Phase I and pharmacokinetic study of sorafenib in patients with hepatic or renal dysfunction: CALGB 60301. J Clin Oncol 2009; 27:1800–1805.
KEY POINTS
- Surveillance for HCC is indicated in all patients with cirrhosis, regardless of the cause of the cirrhosis.
- Liver biopsy is not needed to make the diagnosis if the findings on four-phase multidetector computed tomography or dynamic contrast-enhanced magnetic resonance imaging are typical of HCC (arterial hyperenhancement with venous-phase or delayed-phase washout).
- Many treatments are available, including surgical resection, liver transplantation, ablative therapy, perfusion-based therapy, chemotherapy, and palliative therapy.

The jugular venous pressure revisited
In this age of technological marvels, it is easy to become so reliant on them as to neglect the value of bedside physical signs. Yet these signs provide information that adds no cost, is immediately available, and can be repeated at will.
Few physical findings are as useful but as undervalued as is the estimation of the jugular venous pressure. Unfortunately, many practitioners at many levels of seniority and experience do not measure it correctly, leading to a vicious circle of unreliable information, lack of confidence, and underuse. Another reason for its underuse is that the jugular venous pressure does not correlate precisely with the right atrial pressure, as we will see below.
In this review, we will attempt to clarify physiologic principles and describe technical details. Much of this is simple but, as always, the devil is in the details.
ANATOMIC CONSIDERATIONS
Think of the systemic veins as a soft-walled and mildly distensible reservoir with fingerlike projections, analogous to a partially fluidfilled surgical glove.1 In a semi-upright position, the venous system is partially filled with blood and is collapsed above the level that this blood reaches up to.
Blood is constantly flowing in and out of this reservoir, flowing in by venous return and flowing out by the pumping action of the right side of the heart. The volume in the venous reservoir and hence the pressure are normally maintained by the variability of right ventricular stroke volume in accordance with the Frank-Starling law. Excess volume and pressure indicate failure of this homeostatic mechanism.
The internal jugular veins, being continuous with the superior vena cava, provide a visible measure of the degree to which the systemic venous reservoir is filled, a manometer that reflects the pressure in the right atrium—at least in theory.2 Thus, the vertical height above the right atrium to which they are distended and above which they are in a collapsed state should reflect the right atrial pressure.
(In fact, the jugular venous pressure may underestimate the right atrial pressure, for reasons still not understood. This will be discussed below.)
In a healthy person, the visible jugular veins are fully collapsed when the person is standing and are often distended to a variable degree when the person is supine. Selecting an appropriate intermediate position permits the top of the column (the meniscus) to become visible in the neck between the clavicle and the mandible.
DISCREPANCY BETWEEN JUGULAR VENOUS AND RIGHT ATRIAL PRESSURE
Several reports have indicated that the jugular venous pressure may underestimate the right atrial pressure. Deol et al3 confirmed this, while establishing an excellent correlation between the level of venous collapse (observed on ultrasonography) and the jugular venous pressure. The difference between the right atrial pressure and the jugular venous pressure tended to be greater at higher venous pressures.3
Most people have a valve near the termination of the internal jugular vein, with variable competence. Inhibition of reflux of blood from the superior vena cava into the internal jugular vein by this valve is the most plausible cause of this disparity.4
The failure of the jugular venous pressure to correlate with the right atrial pressure has been cited by some as a reason to doubt the value of a sign that cardiologists have long relied on. How do we reconcile this apparent paradox? Careful review of the literature that has demonstrated this lack of correlation reveals the following:
- When unequal, the jugular venous pressure always underestimates the right atrial pressure.
- The lack of correlation is less evident at lower venous pressures.
This indicates the following:
- In the presence of congestive heart failure, the right atrial pressure is at least as high and perhaps higher than the jugular venous pressure. Hence, if the jugular venous pressure is high, further treatment, especially diuresis, is needed.
- A jugular venous pressure of zero implies a euvolemic state.
Thus, the jugular venous pressure provides excellent guidance when administering diuresis in congestive heart failure. These deductions obviously require the clinical judgment that the elevated right atrial pressure and jugular venous pressure do indeed reflect elevation of pulmonary capillary wedge pressure rather than other conditions discussed later in this article.
WHICH REFERENCE POINT TO USE?
The two points that can be used as references above which the jugular venous pressure is expressed are the center of the right atrium and the sternal angle. While the former may reflect physiology, the latter is preferred, as it is always visible and has the added advantage of being close to the upper limit of normal, which is about 3 cm above this level.
The difference in height between these two reference points has often been quoted as 5 cm, but this is an underestimate in the body positions used in examination.5 Seth et al6 found a mean of 8 cm at 30° elevation, 9.7 cm at 45°, and 9.8 cm at 60°. The difference also varied between patients, being larger in association with smoking, older age, large body mass index, and large anterior-posterior diameter. These factors should be considered when trying to evaluate the significance of a particular jugular venous pressure.
The junction of the midaxillary line and the fourth left intercostal space (“the phlebostatic point”) has been recommended as a reference point by some, as it is level with the mid-right atrium. However, using the phlebostatic point as a reference position is cumbersome and results in a valid measurement only with the patient in the supine position.7
TECHNIQUE IS VITAL
Close adherence to technical details is vital in reliably and reproducibly measuring the pressure in the internal jugular veins (Figure 1).
The right side is usually observed first, as it is the side on which the examiner usually stands. Using the right side also avoids the rare occurrence of external compression of the left brachiocephalic vein.
Head and shoulders
The sternocleidomastoid muscle lies anterior to each internal jugular vein.8 When tense, it impedes good observation. Shortening, and hence relaxing, this muscle permits the meniscus to be observed. Correct positioning is achieved by:
- Placing a folded pillow behind the patient’s head
- Keeping the shoulders on the mattress
- Turning the head away and elevating the jaw, both slightly; this is often best achieved by gentle pressure of the palm of the observer's hand on the patient's forehead.
Degree of head elevation
Although the proper degree of head elevation is sometimes said to be between 30° and 60°, these numbers are approximate. The correct angle is that which brings the venous meniscus into the window of visibility in the neck between the clavicle and mandible.
Lighting
Shining a flashlight tangentially to the skin is often helpful, casting shadows that improve the visibility of vein motion. Dimming the room lighting may further enhance this effect. Directing a light perpendicular to the skin is not helpful.
Also check the external jugular vein
Checking the external jugular vein can help establish that the jugular venous pressure is normal. If the vein is initially collapsed, light finger pressure at the base of the neck will distend it. If the distention rapidly clears after release of this pressure, the jugular venous pressure is not elevated. However, if external jugular venous distention persists, this does not prove true jugular venous pressure elevation, since it may reflect external compression of the vein by the cervical fascia or delayed blood flow caused by sclerotic venous valves.9 In these instances, the internal jugular pulsation level must be sought.
Jugular venous collapse with inspiration
Collapse of the inferior vena cava with forced inspiration is routinely evaluated during echocardiography as a way to estimate right atrial pressure. This finding has been extrapolated to the jugular veins, wherein the absence of venous collapse during vigorous inspiration or sniffing indicates elevated central venous pressures.10
Distinguishing venous from arterial pulsation
Features indicating venous rather than arterial pulsation were listed by Wood more than 50 years ago11 and are still relevant today. These include internal jugular pulsation that:
- Is soft, diffuse, undulant
- Is not palpable
- Has two crests and two troughs per cardiac cycle
- Has crests that do not coincide with the palpated carotid pulse (exceptions may be seen with the systolic timing of the v wave of tricuspid regurgitation)
- Has higher pressure in expiration, lower in inspiration (exceptions may be seen when Kussmaul physiology is present)
- Has pressure that rises with abdominal pressure
- Is obliterated by light pressure at the base of the neck.
In addition to the above criteria, a wave whose movement is predominantly a descent is nearly always venous.
Abdominojugular reflex
Firm, steady pressure over the abdomen will often result in a small rise in jugular venous pressure. In healthy people, this normalizes in a few seconds, even while manual pressure is maintained. Persistence of jugular venous pressure elevation beyond 10 seconds, followed by an abrupt fall upon withdrawal of manual pressure, is abnormal. This finding has implications similar to those of an elevated baseline jugular venous pressure.
SIGNIFICANCE OF JUGULAR VENOUS PRESSURE ELEVATION
Elevated jugular venous pressure is a manifestation of abnormal right heart dynamics, mostly commonly reflecting elevated pulmonary capillary wedge pressure from left heart failure.12 This usually implies fluid overload, indicating the need for diuresis.
Exceptions to this therapeutic implication include the presence of a primary right heart condition, pericardial disease, certain arrhythmias, and conditions that elevate intrathoracic pressure. These will be discussed below. One important example is the acute jugular venous pressure elevation seen in right ventricular infarction, in which the high venous pressure is compensatory and its reduction can produce hypotension and shock.13
Primary right heart conditions also include right-sided valvular disease, cor pulmonale (including pulmonary embolism and pulmonary hypertension), and the compressive effect of pericardial tamponade or constriction. A normal or near-normal jugular venous pressure significantly decreases the likelihood of significant constriction or of tamponade of a degree necessitating urgent pericardiocentesis.14
SPECIAL CIRCUMSTANCES
Presence of an intravenous line in the neck
An intravenous line in the neck will often prevent observation of the jugular venous pressure. A simple measure can often compensate for this. If the venous line can be temporarily disconnected, the central venous pressure can be measured directly. Using sterile technique, the line can be flushed with saline and aspirated to bring blood into the transparent tubing. Leaving the proximal end open to the air, and alternately raising and lowering it to confirm free flow, the level to which the blood rises can be easily observed. Observing small cardiac and respiratory variations of the meniscus confirms free communication with the central veins. Attaching the line to a transducer is another option, but this may be time-consuming, and establishing an accurate zero point is often difficult.
The previously described discrepancy between jugular venous pressure and central venous pressure has to be considered when drawing conclusions from this measurement.
Intrathoracic pressure elevators
Positive pressure ventilation will elevate intrathoracic pressure (including right atrial pressure) and hence the jugular venous pressure, making interpretation difficult.15 Large pleural effusions or pneumothorax may have a similar effect.16
Superior vena cava syndrome
Markedly elevated jugular venous pressure is here associated with absent or very diminished pulsation, as the caval obstruction has eliminated free communication with the right atrium.17 Associated facial plethora and edema, papilledema, and superficial venous distention over the chest wall will often confirm this diagnosis.
THE WAVEFORM
While the main purpose of viewing the neck veins is to establish the mean pressure, useful information can often be obtained by assessing the waveform. Abnormalities reflect arrhythmias, right heart hemodynamics, or pericardial disease.18 Changes may be subtle and difficult to detect, but some patterns can be quite readily appreciated (Figure 2). A limited selection follows.
Arrhythmias
Cannon a waves. These intermittent sharp positive deflections in the venous pulse represent right atrial contraction against a closed tricuspid valve. They are most commonly associated with premature ventricular complexes, but they occur in other conditions in which atrial and ventricular beating are dissociated, including complete heart block, atrioventricular dissociation, and electronic ventricular pacing.19–21
Repetitive cannon waves. These may be seen with atrioventricular junctional tachycardia or ventricular tachycardia with 1:1 retrograde ventriculoatrial conduction in which the tricuspid valve is closed to every atrial beat.
Fine rapid regular pulsation may be seen in atrial flutter and may be a useful clue in distinguishing this from sinus rhythm when there is 4:1 atrioventricular conduction and a normal ventricular rate.
Abnormal right heart hemodynamics
Large v waves (Lancisi sign). These surges, replacing the usual x descent in systole, are seen in tricuspid insufficiency when the right atrium and its venous attachments are not protected from the right ventricular systolic pressure.22 High right ventricular pressure will obviously enhance this systolic surge.
Large a waves. These reflect resistance to right atrial outflow and may be seen when right ventricular compliance is reduced by hypertrophy from chronic pressure overload or in tricuspid stenosis.23
Pericardial disease
Kussmaul sign is the paradoxical increase in jugular venous pressure with inspiration, observed in conditions associated with limited filling of the right ventricle. It is typically associated with constrictive pericarditis, although it occurs in only a minority of people with this condition.24 It may also be seen in restrictive cardiomyopathy, massive pulmonary embolism, right ventricular infarction, and tricuspid stenosis.25
Diaphragmatic descent during inspiration increases intra-abdominal pressure and decreases intrathoracic pressure. The resulting increased gradient between the abdomen and thorax enhances venous return from splanchnic vessels, which in the setting of a noncompliant right ventricle may result in increased right atrial (and, hence, jugular venous) pressure.26
It is important to point out that the Kussmaul sign does not occur with cardiac tamponade in the absence of associated pericardial constriction.
Exaggerated y descent is typically seen in pericardial constriction, in which the high pressure of the v wave falls rapidly at the onset of diastole, given initial minimal right ventricular resistance. Flow is abruptly stopped when the intrapericardial space is filled.
- Sherwood L. Human Physiology: From Cells to Systems. 8th ed. Belmont, CA: Brooks/Cole; 2012.
- Constant J. Using internal jugular pulsations as a manometer for right atrial pressure measurements. Cardiology 2000; 93:26–30.
- Deol GR, Collett N, Ashby A, Schmidt GA. Ultrasound accurately reflects the jugular venous examination but underestimates central venous pressure. Chest 2011; 139:95–100.
- Wu X, Studer W, Erb T, Skarvan K, Seeberger MD. Competence of the internal jugular vein valve is damaged by cannulation and catheterization of the internal jugular vein. Anesthesiology 2000; 93:319–324.
- Ramana RK, Sanagala T, Lichtenberg R. A new angle on the angle of Louis. Congest Heart Fail 2006; 12:196–199.
- Seth R, Magner P, Matzinger F, van Walraven C. How far is the sternal angle from the mid-right atrium? J Gen Intern Med 2002; 17:852–856.
- Kee LL, Simonson JS, Stotts NA, Skov P, Schiller NB. Echocardiographic determination of valid zero reference levels in supine and lateral positions. Am J Crit Care 1993; 2:72–80.
- Park SY, Kim MJ, Kim MG, et al. Changes in the relationship between the right internal jugular vein and an anatomical landmark after head rotation. Korean J Anesthesiol 2011; 61:107–111.
- Sankoff J, Zidulka A. Non-invasive method for the rapid assessment of central venous pressure: description and validation by a single examiner. West J Emerg Med 2008; 9:201–205.
- Conn RD, O’Keefe JH. Simplified evaluation of the jugular venous pressure: significance of inspiratory collapse of jugular veins. Mo Med 2012; 109:150–152.
- Wood PH. Diseases of the Heart and Circulation. 2nd ed. Philadelphia, PA: Lippincott; 1956.
- Drazner MH, Brown RN, Kaiser PA, et al. Relationship of right- and left-sided filling pressures in patients with advanced heart failure: a 14-year multi-institutional analysis. J Heart Lung Transplant 2012; 31:67–72.
- Clark G, Strauss HD, Roberts R. Dobutamine vs furosemide in the treatment of cardiac failure due to right ventricular infarction. Chest 1980; 77:220–223.
- Roy CL, Minor MA, Brookhart MA, Choudhry NK. Does this patient with a pericardial effusion have cardiac tamponade? JAMA 2007; 297:1810–1818.
- Zhou Q, Xiao W, An E, Zhou H, Yan M. Effects of four different positive airway pressures on right internal jugular vein catheterisation. Eur J Anaesthesiol 2012; 29:223–228.
- Jolobe OM. Disproportionate elevation of jugular venous pressure in pleural effusion. Br J Hosp Med (Lond) 2011; 72:582–585.
- Seo M, Shin WJ, Jun IG. Central venous catheter-related superior vena cava syndrome following renal transplantation—a case report. Korean J Anesthesiol 2012; 63:550–554.
- Applefeld MM. The jugular venous pressure and pulse contour. In:Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd ed. Boston, MA: Butterworths; 1990.
- El Gamal MI, Van Gelder LM. Chronic ventricular pacing with ventriculo-atrial conduction versus atrial pacing in three patients with symptomatic sinus bradycardia. Pacing Clin Electrophysiol 1981; 4:100–105.
- Berman ND, Waxman MB. Cannon waves with A-V association. Am Heart J 1976; 91:643–644.
- Luisada AA, Singhal A, Kim K. The jugular and hepatic tracings in normal subjects and in conduction defects. Acta Cardiol 1983; 38:405–424.
- Miller MJ, McKay RG, Ferguson JJ, et al. Right atrial pressure-volume relationships in tricuspid regurgitation. Circulation 1986; 73:799–808.
- Wooley CF, Fontana ME, Kilman JW, Ryan JM. Tricuspid stenosis. Atrial systolic murmur, tricuspid opening snap, and right atrial pressure pulse. Am J Med 1985; 78:375–384.
- McGee SR. Evidence-Based Physical Diagnosis. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
- Mittal SR, Garg S, Lalgarhia M. Jugular venous pressure and pulse wave form in the diagnosis of right ventricular infarction. Int J Cardiol 1996; 53:253–256.
- Bilchick KC, Wise RA. Paradoxical physical findings described by Kussmaul: pulsus paradoxus and Kussmaul’s sign. Lancet 2002; 359:1940–1942.
In this age of technological marvels, it is easy to become so reliant on them as to neglect the value of bedside physical signs. Yet these signs provide information that adds no cost, is immediately available, and can be repeated at will.
Few physical findings are as useful but as undervalued as is the estimation of the jugular venous pressure. Unfortunately, many practitioners at many levels of seniority and experience do not measure it correctly, leading to a vicious circle of unreliable information, lack of confidence, and underuse. Another reason for its underuse is that the jugular venous pressure does not correlate precisely with the right atrial pressure, as we will see below.
In this review, we will attempt to clarify physiologic principles and describe technical details. Much of this is simple but, as always, the devil is in the details.
ANATOMIC CONSIDERATIONS
Think of the systemic veins as a soft-walled and mildly distensible reservoir with fingerlike projections, analogous to a partially fluidfilled surgical glove.1 In a semi-upright position, the venous system is partially filled with blood and is collapsed above the level that this blood reaches up to.
Blood is constantly flowing in and out of this reservoir, flowing in by venous return and flowing out by the pumping action of the right side of the heart. The volume in the venous reservoir and hence the pressure are normally maintained by the variability of right ventricular stroke volume in accordance with the Frank-Starling law. Excess volume and pressure indicate failure of this homeostatic mechanism.
The internal jugular veins, being continuous with the superior vena cava, provide a visible measure of the degree to which the systemic venous reservoir is filled, a manometer that reflects the pressure in the right atrium—at least in theory.2 Thus, the vertical height above the right atrium to which they are distended and above which they are in a collapsed state should reflect the right atrial pressure.
(In fact, the jugular venous pressure may underestimate the right atrial pressure, for reasons still not understood. This will be discussed below.)
In a healthy person, the visible jugular veins are fully collapsed when the person is standing and are often distended to a variable degree when the person is supine. Selecting an appropriate intermediate position permits the top of the column (the meniscus) to become visible in the neck between the clavicle and the mandible.
DISCREPANCY BETWEEN JUGULAR VENOUS AND RIGHT ATRIAL PRESSURE
Several reports have indicated that the jugular venous pressure may underestimate the right atrial pressure. Deol et al3 confirmed this, while establishing an excellent correlation between the level of venous collapse (observed on ultrasonography) and the jugular venous pressure. The difference between the right atrial pressure and the jugular venous pressure tended to be greater at higher venous pressures.3
Most people have a valve near the termination of the internal jugular vein, with variable competence. Inhibition of reflux of blood from the superior vena cava into the internal jugular vein by this valve is the most plausible cause of this disparity.4
The failure of the jugular venous pressure to correlate with the right atrial pressure has been cited by some as a reason to doubt the value of a sign that cardiologists have long relied on. How do we reconcile this apparent paradox? Careful review of the literature that has demonstrated this lack of correlation reveals the following:
- When unequal, the jugular venous pressure always underestimates the right atrial pressure.
- The lack of correlation is less evident at lower venous pressures.
This indicates the following:
- In the presence of congestive heart failure, the right atrial pressure is at least as high and perhaps higher than the jugular venous pressure. Hence, if the jugular venous pressure is high, further treatment, especially diuresis, is needed.
- A jugular venous pressure of zero implies a euvolemic state.
Thus, the jugular venous pressure provides excellent guidance when administering diuresis in congestive heart failure. These deductions obviously require the clinical judgment that the elevated right atrial pressure and jugular venous pressure do indeed reflect elevation of pulmonary capillary wedge pressure rather than other conditions discussed later in this article.
WHICH REFERENCE POINT TO USE?
The two points that can be used as references above which the jugular venous pressure is expressed are the center of the right atrium and the sternal angle. While the former may reflect physiology, the latter is preferred, as it is always visible and has the added advantage of being close to the upper limit of normal, which is about 3 cm above this level.
The difference in height between these two reference points has often been quoted as 5 cm, but this is an underestimate in the body positions used in examination.5 Seth et al6 found a mean of 8 cm at 30° elevation, 9.7 cm at 45°, and 9.8 cm at 60°. The difference also varied between patients, being larger in association with smoking, older age, large body mass index, and large anterior-posterior diameter. These factors should be considered when trying to evaluate the significance of a particular jugular venous pressure.
The junction of the midaxillary line and the fourth left intercostal space (“the phlebostatic point”) has been recommended as a reference point by some, as it is level with the mid-right atrium. However, using the phlebostatic point as a reference position is cumbersome and results in a valid measurement only with the patient in the supine position.7
TECHNIQUE IS VITAL
Close adherence to technical details is vital in reliably and reproducibly measuring the pressure in the internal jugular veins (Figure 1).
The right side is usually observed first, as it is the side on which the examiner usually stands. Using the right side also avoids the rare occurrence of external compression of the left brachiocephalic vein.
Head and shoulders
The sternocleidomastoid muscle lies anterior to each internal jugular vein.8 When tense, it impedes good observation. Shortening, and hence relaxing, this muscle permits the meniscus to be observed. Correct positioning is achieved by:
- Placing a folded pillow behind the patient’s head
- Keeping the shoulders on the mattress
- Turning the head away and elevating the jaw, both slightly; this is often best achieved by gentle pressure of the palm of the observer's hand on the patient's forehead.
Degree of head elevation
Although the proper degree of head elevation is sometimes said to be between 30° and 60°, these numbers are approximate. The correct angle is that which brings the venous meniscus into the window of visibility in the neck between the clavicle and mandible.
Lighting
Shining a flashlight tangentially to the skin is often helpful, casting shadows that improve the visibility of vein motion. Dimming the room lighting may further enhance this effect. Directing a light perpendicular to the skin is not helpful.
Also check the external jugular vein
Checking the external jugular vein can help establish that the jugular venous pressure is normal. If the vein is initially collapsed, light finger pressure at the base of the neck will distend it. If the distention rapidly clears after release of this pressure, the jugular venous pressure is not elevated. However, if external jugular venous distention persists, this does not prove true jugular venous pressure elevation, since it may reflect external compression of the vein by the cervical fascia or delayed blood flow caused by sclerotic venous valves.9 In these instances, the internal jugular pulsation level must be sought.
Jugular venous collapse with inspiration
Collapse of the inferior vena cava with forced inspiration is routinely evaluated during echocardiography as a way to estimate right atrial pressure. This finding has been extrapolated to the jugular veins, wherein the absence of venous collapse during vigorous inspiration or sniffing indicates elevated central venous pressures.10
Distinguishing venous from arterial pulsation
Features indicating venous rather than arterial pulsation were listed by Wood more than 50 years ago11 and are still relevant today. These include internal jugular pulsation that:
- Is soft, diffuse, undulant
- Is not palpable
- Has two crests and two troughs per cardiac cycle
- Has crests that do not coincide with the palpated carotid pulse (exceptions may be seen with the systolic timing of the v wave of tricuspid regurgitation)
- Has higher pressure in expiration, lower in inspiration (exceptions may be seen when Kussmaul physiology is present)
- Has pressure that rises with abdominal pressure
- Is obliterated by light pressure at the base of the neck.
In addition to the above criteria, a wave whose movement is predominantly a descent is nearly always venous.
Abdominojugular reflex
Firm, steady pressure over the abdomen will often result in a small rise in jugular venous pressure. In healthy people, this normalizes in a few seconds, even while manual pressure is maintained. Persistence of jugular venous pressure elevation beyond 10 seconds, followed by an abrupt fall upon withdrawal of manual pressure, is abnormal. This finding has implications similar to those of an elevated baseline jugular venous pressure.
SIGNIFICANCE OF JUGULAR VENOUS PRESSURE ELEVATION
Elevated jugular venous pressure is a manifestation of abnormal right heart dynamics, mostly commonly reflecting elevated pulmonary capillary wedge pressure from left heart failure.12 This usually implies fluid overload, indicating the need for diuresis.
Exceptions to this therapeutic implication include the presence of a primary right heart condition, pericardial disease, certain arrhythmias, and conditions that elevate intrathoracic pressure. These will be discussed below. One important example is the acute jugular venous pressure elevation seen in right ventricular infarction, in which the high venous pressure is compensatory and its reduction can produce hypotension and shock.13
Primary right heart conditions also include right-sided valvular disease, cor pulmonale (including pulmonary embolism and pulmonary hypertension), and the compressive effect of pericardial tamponade or constriction. A normal or near-normal jugular venous pressure significantly decreases the likelihood of significant constriction or of tamponade of a degree necessitating urgent pericardiocentesis.14
SPECIAL CIRCUMSTANCES
Presence of an intravenous line in the neck
An intravenous line in the neck will often prevent observation of the jugular venous pressure. A simple measure can often compensate for this. If the venous line can be temporarily disconnected, the central venous pressure can be measured directly. Using sterile technique, the line can be flushed with saline and aspirated to bring blood into the transparent tubing. Leaving the proximal end open to the air, and alternately raising and lowering it to confirm free flow, the level to which the blood rises can be easily observed. Observing small cardiac and respiratory variations of the meniscus confirms free communication with the central veins. Attaching the line to a transducer is another option, but this may be time-consuming, and establishing an accurate zero point is often difficult.
The previously described discrepancy between jugular venous pressure and central venous pressure has to be considered when drawing conclusions from this measurement.
Intrathoracic pressure elevators
Positive pressure ventilation will elevate intrathoracic pressure (including right atrial pressure) and hence the jugular venous pressure, making interpretation difficult.15 Large pleural effusions or pneumothorax may have a similar effect.16
Superior vena cava syndrome
Markedly elevated jugular venous pressure is here associated with absent or very diminished pulsation, as the caval obstruction has eliminated free communication with the right atrium.17 Associated facial plethora and edema, papilledema, and superficial venous distention over the chest wall will often confirm this diagnosis.
THE WAVEFORM
While the main purpose of viewing the neck veins is to establish the mean pressure, useful information can often be obtained by assessing the waveform. Abnormalities reflect arrhythmias, right heart hemodynamics, or pericardial disease.18 Changes may be subtle and difficult to detect, but some patterns can be quite readily appreciated (Figure 2). A limited selection follows.
Arrhythmias
Cannon a waves. These intermittent sharp positive deflections in the venous pulse represent right atrial contraction against a closed tricuspid valve. They are most commonly associated with premature ventricular complexes, but they occur in other conditions in which atrial and ventricular beating are dissociated, including complete heart block, atrioventricular dissociation, and electronic ventricular pacing.19–21
Repetitive cannon waves. These may be seen with atrioventricular junctional tachycardia or ventricular tachycardia with 1:1 retrograde ventriculoatrial conduction in which the tricuspid valve is closed to every atrial beat.
Fine rapid regular pulsation may be seen in atrial flutter and may be a useful clue in distinguishing this from sinus rhythm when there is 4:1 atrioventricular conduction and a normal ventricular rate.
Abnormal right heart hemodynamics
Large v waves (Lancisi sign). These surges, replacing the usual x descent in systole, are seen in tricuspid insufficiency when the right atrium and its venous attachments are not protected from the right ventricular systolic pressure.22 High right ventricular pressure will obviously enhance this systolic surge.
Large a waves. These reflect resistance to right atrial outflow and may be seen when right ventricular compliance is reduced by hypertrophy from chronic pressure overload or in tricuspid stenosis.23
Pericardial disease
Kussmaul sign is the paradoxical increase in jugular venous pressure with inspiration, observed in conditions associated with limited filling of the right ventricle. It is typically associated with constrictive pericarditis, although it occurs in only a minority of people with this condition.24 It may also be seen in restrictive cardiomyopathy, massive pulmonary embolism, right ventricular infarction, and tricuspid stenosis.25
Diaphragmatic descent during inspiration increases intra-abdominal pressure and decreases intrathoracic pressure. The resulting increased gradient between the abdomen and thorax enhances venous return from splanchnic vessels, which in the setting of a noncompliant right ventricle may result in increased right atrial (and, hence, jugular venous) pressure.26
It is important to point out that the Kussmaul sign does not occur with cardiac tamponade in the absence of associated pericardial constriction.
Exaggerated y descent is typically seen in pericardial constriction, in which the high pressure of the v wave falls rapidly at the onset of diastole, given initial minimal right ventricular resistance. Flow is abruptly stopped when the intrapericardial space is filled.
In this age of technological marvels, it is easy to become so reliant on them as to neglect the value of bedside physical signs. Yet these signs provide information that adds no cost, is immediately available, and can be repeated at will.
Few physical findings are as useful but as undervalued as is the estimation of the jugular venous pressure. Unfortunately, many practitioners at many levels of seniority and experience do not measure it correctly, leading to a vicious circle of unreliable information, lack of confidence, and underuse. Another reason for its underuse is that the jugular venous pressure does not correlate precisely with the right atrial pressure, as we will see below.
In this review, we will attempt to clarify physiologic principles and describe technical details. Much of this is simple but, as always, the devil is in the details.
ANATOMIC CONSIDERATIONS
Think of the systemic veins as a soft-walled and mildly distensible reservoir with fingerlike projections, analogous to a partially fluidfilled surgical glove.1 In a semi-upright position, the venous system is partially filled with blood and is collapsed above the level that this blood reaches up to.
Blood is constantly flowing in and out of this reservoir, flowing in by venous return and flowing out by the pumping action of the right side of the heart. The volume in the venous reservoir and hence the pressure are normally maintained by the variability of right ventricular stroke volume in accordance with the Frank-Starling law. Excess volume and pressure indicate failure of this homeostatic mechanism.
The internal jugular veins, being continuous with the superior vena cava, provide a visible measure of the degree to which the systemic venous reservoir is filled, a manometer that reflects the pressure in the right atrium—at least in theory.2 Thus, the vertical height above the right atrium to which they are distended and above which they are in a collapsed state should reflect the right atrial pressure.
(In fact, the jugular venous pressure may underestimate the right atrial pressure, for reasons still not understood. This will be discussed below.)
In a healthy person, the visible jugular veins are fully collapsed when the person is standing and are often distended to a variable degree when the person is supine. Selecting an appropriate intermediate position permits the top of the column (the meniscus) to become visible in the neck between the clavicle and the mandible.
DISCREPANCY BETWEEN JUGULAR VENOUS AND RIGHT ATRIAL PRESSURE
Several reports have indicated that the jugular venous pressure may underestimate the right atrial pressure. Deol et al3 confirmed this, while establishing an excellent correlation between the level of venous collapse (observed on ultrasonography) and the jugular venous pressure. The difference between the right atrial pressure and the jugular venous pressure tended to be greater at higher venous pressures.3
Most people have a valve near the termination of the internal jugular vein, with variable competence. Inhibition of reflux of blood from the superior vena cava into the internal jugular vein by this valve is the most plausible cause of this disparity.4
The failure of the jugular venous pressure to correlate with the right atrial pressure has been cited by some as a reason to doubt the value of a sign that cardiologists have long relied on. How do we reconcile this apparent paradox? Careful review of the literature that has demonstrated this lack of correlation reveals the following:
- When unequal, the jugular venous pressure always underestimates the right atrial pressure.
- The lack of correlation is less evident at lower venous pressures.
This indicates the following:
- In the presence of congestive heart failure, the right atrial pressure is at least as high and perhaps higher than the jugular venous pressure. Hence, if the jugular venous pressure is high, further treatment, especially diuresis, is needed.
- A jugular venous pressure of zero implies a euvolemic state.
Thus, the jugular venous pressure provides excellent guidance when administering diuresis in congestive heart failure. These deductions obviously require the clinical judgment that the elevated right atrial pressure and jugular venous pressure do indeed reflect elevation of pulmonary capillary wedge pressure rather than other conditions discussed later in this article.
WHICH REFERENCE POINT TO USE?
The two points that can be used as references above which the jugular venous pressure is expressed are the center of the right atrium and the sternal angle. While the former may reflect physiology, the latter is preferred, as it is always visible and has the added advantage of being close to the upper limit of normal, which is about 3 cm above this level.
The difference in height between these two reference points has often been quoted as 5 cm, but this is an underestimate in the body positions used in examination.5 Seth et al6 found a mean of 8 cm at 30° elevation, 9.7 cm at 45°, and 9.8 cm at 60°. The difference also varied between patients, being larger in association with smoking, older age, large body mass index, and large anterior-posterior diameter. These factors should be considered when trying to evaluate the significance of a particular jugular venous pressure.
The junction of the midaxillary line and the fourth left intercostal space (“the phlebostatic point”) has been recommended as a reference point by some, as it is level with the mid-right atrium. However, using the phlebostatic point as a reference position is cumbersome and results in a valid measurement only with the patient in the supine position.7
TECHNIQUE IS VITAL
Close adherence to technical details is vital in reliably and reproducibly measuring the pressure in the internal jugular veins (Figure 1).
The right side is usually observed first, as it is the side on which the examiner usually stands. Using the right side also avoids the rare occurrence of external compression of the left brachiocephalic vein.
Head and shoulders
The sternocleidomastoid muscle lies anterior to each internal jugular vein.8 When tense, it impedes good observation. Shortening, and hence relaxing, this muscle permits the meniscus to be observed. Correct positioning is achieved by:
- Placing a folded pillow behind the patient’s head
- Keeping the shoulders on the mattress
- Turning the head away and elevating the jaw, both slightly; this is often best achieved by gentle pressure of the palm of the observer's hand on the patient's forehead.
Degree of head elevation
Although the proper degree of head elevation is sometimes said to be between 30° and 60°, these numbers are approximate. The correct angle is that which brings the venous meniscus into the window of visibility in the neck between the clavicle and mandible.
Lighting
Shining a flashlight tangentially to the skin is often helpful, casting shadows that improve the visibility of vein motion. Dimming the room lighting may further enhance this effect. Directing a light perpendicular to the skin is not helpful.
Also check the external jugular vein
Checking the external jugular vein can help establish that the jugular venous pressure is normal. If the vein is initially collapsed, light finger pressure at the base of the neck will distend it. If the distention rapidly clears after release of this pressure, the jugular venous pressure is not elevated. However, if external jugular venous distention persists, this does not prove true jugular venous pressure elevation, since it may reflect external compression of the vein by the cervical fascia or delayed blood flow caused by sclerotic venous valves.9 In these instances, the internal jugular pulsation level must be sought.
Jugular venous collapse with inspiration
Collapse of the inferior vena cava with forced inspiration is routinely evaluated during echocardiography as a way to estimate right atrial pressure. This finding has been extrapolated to the jugular veins, wherein the absence of venous collapse during vigorous inspiration or sniffing indicates elevated central venous pressures.10
Distinguishing venous from arterial pulsation
Features indicating venous rather than arterial pulsation were listed by Wood more than 50 years ago11 and are still relevant today. These include internal jugular pulsation that:
- Is soft, diffuse, undulant
- Is not palpable
- Has two crests and two troughs per cardiac cycle
- Has crests that do not coincide with the palpated carotid pulse (exceptions may be seen with the systolic timing of the v wave of tricuspid regurgitation)
- Has higher pressure in expiration, lower in inspiration (exceptions may be seen when Kussmaul physiology is present)
- Has pressure that rises with abdominal pressure
- Is obliterated by light pressure at the base of the neck.
In addition to the above criteria, a wave whose movement is predominantly a descent is nearly always venous.
Abdominojugular reflex
Firm, steady pressure over the abdomen will often result in a small rise in jugular venous pressure. In healthy people, this normalizes in a few seconds, even while manual pressure is maintained. Persistence of jugular venous pressure elevation beyond 10 seconds, followed by an abrupt fall upon withdrawal of manual pressure, is abnormal. This finding has implications similar to those of an elevated baseline jugular venous pressure.
SIGNIFICANCE OF JUGULAR VENOUS PRESSURE ELEVATION
Elevated jugular venous pressure is a manifestation of abnormal right heart dynamics, mostly commonly reflecting elevated pulmonary capillary wedge pressure from left heart failure.12 This usually implies fluid overload, indicating the need for diuresis.
Exceptions to this therapeutic implication include the presence of a primary right heart condition, pericardial disease, certain arrhythmias, and conditions that elevate intrathoracic pressure. These will be discussed below. One important example is the acute jugular venous pressure elevation seen in right ventricular infarction, in which the high venous pressure is compensatory and its reduction can produce hypotension and shock.13
Primary right heart conditions also include right-sided valvular disease, cor pulmonale (including pulmonary embolism and pulmonary hypertension), and the compressive effect of pericardial tamponade or constriction. A normal or near-normal jugular venous pressure significantly decreases the likelihood of significant constriction or of tamponade of a degree necessitating urgent pericardiocentesis.14
SPECIAL CIRCUMSTANCES
Presence of an intravenous line in the neck
An intravenous line in the neck will often prevent observation of the jugular venous pressure. A simple measure can often compensate for this. If the venous line can be temporarily disconnected, the central venous pressure can be measured directly. Using sterile technique, the line can be flushed with saline and aspirated to bring blood into the transparent tubing. Leaving the proximal end open to the air, and alternately raising and lowering it to confirm free flow, the level to which the blood rises can be easily observed. Observing small cardiac and respiratory variations of the meniscus confirms free communication with the central veins. Attaching the line to a transducer is another option, but this may be time-consuming, and establishing an accurate zero point is often difficult.
The previously described discrepancy between jugular venous pressure and central venous pressure has to be considered when drawing conclusions from this measurement.
Intrathoracic pressure elevators
Positive pressure ventilation will elevate intrathoracic pressure (including right atrial pressure) and hence the jugular venous pressure, making interpretation difficult.15 Large pleural effusions or pneumothorax may have a similar effect.16
Superior vena cava syndrome
Markedly elevated jugular venous pressure is here associated with absent or very diminished pulsation, as the caval obstruction has eliminated free communication with the right atrium.17 Associated facial plethora and edema, papilledema, and superficial venous distention over the chest wall will often confirm this diagnosis.
THE WAVEFORM
While the main purpose of viewing the neck veins is to establish the mean pressure, useful information can often be obtained by assessing the waveform. Abnormalities reflect arrhythmias, right heart hemodynamics, or pericardial disease.18 Changes may be subtle and difficult to detect, but some patterns can be quite readily appreciated (Figure 2). A limited selection follows.
Arrhythmias
Cannon a waves. These intermittent sharp positive deflections in the venous pulse represent right atrial contraction against a closed tricuspid valve. They are most commonly associated with premature ventricular complexes, but they occur in other conditions in which atrial and ventricular beating are dissociated, including complete heart block, atrioventricular dissociation, and electronic ventricular pacing.19–21
Repetitive cannon waves. These may be seen with atrioventricular junctional tachycardia or ventricular tachycardia with 1:1 retrograde ventriculoatrial conduction in which the tricuspid valve is closed to every atrial beat.
Fine rapid regular pulsation may be seen in atrial flutter and may be a useful clue in distinguishing this from sinus rhythm when there is 4:1 atrioventricular conduction and a normal ventricular rate.
Abnormal right heart hemodynamics
Large v waves (Lancisi sign). These surges, replacing the usual x descent in systole, are seen in tricuspid insufficiency when the right atrium and its venous attachments are not protected from the right ventricular systolic pressure.22 High right ventricular pressure will obviously enhance this systolic surge.
Large a waves. These reflect resistance to right atrial outflow and may be seen when right ventricular compliance is reduced by hypertrophy from chronic pressure overload or in tricuspid stenosis.23
Pericardial disease
Kussmaul sign is the paradoxical increase in jugular venous pressure with inspiration, observed in conditions associated with limited filling of the right ventricle. It is typically associated with constrictive pericarditis, although it occurs in only a minority of people with this condition.24 It may also be seen in restrictive cardiomyopathy, massive pulmonary embolism, right ventricular infarction, and tricuspid stenosis.25
Diaphragmatic descent during inspiration increases intra-abdominal pressure and decreases intrathoracic pressure. The resulting increased gradient between the abdomen and thorax enhances venous return from splanchnic vessels, which in the setting of a noncompliant right ventricle may result in increased right atrial (and, hence, jugular venous) pressure.26
It is important to point out that the Kussmaul sign does not occur with cardiac tamponade in the absence of associated pericardial constriction.
Exaggerated y descent is typically seen in pericardial constriction, in which the high pressure of the v wave falls rapidly at the onset of diastole, given initial minimal right ventricular resistance. Flow is abruptly stopped when the intrapericardial space is filled.
- Sherwood L. Human Physiology: From Cells to Systems. 8th ed. Belmont, CA: Brooks/Cole; 2012.
- Constant J. Using internal jugular pulsations as a manometer for right atrial pressure measurements. Cardiology 2000; 93:26–30.
- Deol GR, Collett N, Ashby A, Schmidt GA. Ultrasound accurately reflects the jugular venous examination but underestimates central venous pressure. Chest 2011; 139:95–100.
- Wu X, Studer W, Erb T, Skarvan K, Seeberger MD. Competence of the internal jugular vein valve is damaged by cannulation and catheterization of the internal jugular vein. Anesthesiology 2000; 93:319–324.
- Ramana RK, Sanagala T, Lichtenberg R. A new angle on the angle of Louis. Congest Heart Fail 2006; 12:196–199.
- Seth R, Magner P, Matzinger F, van Walraven C. How far is the sternal angle from the mid-right atrium? J Gen Intern Med 2002; 17:852–856.
- Kee LL, Simonson JS, Stotts NA, Skov P, Schiller NB. Echocardiographic determination of valid zero reference levels in supine and lateral positions. Am J Crit Care 1993; 2:72–80.
- Park SY, Kim MJ, Kim MG, et al. Changes in the relationship between the right internal jugular vein and an anatomical landmark after head rotation. Korean J Anesthesiol 2011; 61:107–111.
- Sankoff J, Zidulka A. Non-invasive method for the rapid assessment of central venous pressure: description and validation by a single examiner. West J Emerg Med 2008; 9:201–205.
- Conn RD, O’Keefe JH. Simplified evaluation of the jugular venous pressure: significance of inspiratory collapse of jugular veins. Mo Med 2012; 109:150–152.
- Wood PH. Diseases of the Heart and Circulation. 2nd ed. Philadelphia, PA: Lippincott; 1956.
- Drazner MH, Brown RN, Kaiser PA, et al. Relationship of right- and left-sided filling pressures in patients with advanced heart failure: a 14-year multi-institutional analysis. J Heart Lung Transplant 2012; 31:67–72.
- Clark G, Strauss HD, Roberts R. Dobutamine vs furosemide in the treatment of cardiac failure due to right ventricular infarction. Chest 1980; 77:220–223.
- Roy CL, Minor MA, Brookhart MA, Choudhry NK. Does this patient with a pericardial effusion have cardiac tamponade? JAMA 2007; 297:1810–1818.
- Zhou Q, Xiao W, An E, Zhou H, Yan M. Effects of four different positive airway pressures on right internal jugular vein catheterisation. Eur J Anaesthesiol 2012; 29:223–228.
- Jolobe OM. Disproportionate elevation of jugular venous pressure in pleural effusion. Br J Hosp Med (Lond) 2011; 72:582–585.
- Seo M, Shin WJ, Jun IG. Central venous catheter-related superior vena cava syndrome following renal transplantation—a case report. Korean J Anesthesiol 2012; 63:550–554.
- Applefeld MM. The jugular venous pressure and pulse contour. In:Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd ed. Boston, MA: Butterworths; 1990.
- El Gamal MI, Van Gelder LM. Chronic ventricular pacing with ventriculo-atrial conduction versus atrial pacing in three patients with symptomatic sinus bradycardia. Pacing Clin Electrophysiol 1981; 4:100–105.
- Berman ND, Waxman MB. Cannon waves with A-V association. Am Heart J 1976; 91:643–644.
- Luisada AA, Singhal A, Kim K. The jugular and hepatic tracings in normal subjects and in conduction defects. Acta Cardiol 1983; 38:405–424.
- Miller MJ, McKay RG, Ferguson JJ, et al. Right atrial pressure-volume relationships in tricuspid regurgitation. Circulation 1986; 73:799–808.
- Wooley CF, Fontana ME, Kilman JW, Ryan JM. Tricuspid stenosis. Atrial systolic murmur, tricuspid opening snap, and right atrial pressure pulse. Am J Med 1985; 78:375–384.
- McGee SR. Evidence-Based Physical Diagnosis. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
- Mittal SR, Garg S, Lalgarhia M. Jugular venous pressure and pulse wave form in the diagnosis of right ventricular infarction. Int J Cardiol 1996; 53:253–256.
- Bilchick KC, Wise RA. Paradoxical physical findings described by Kussmaul: pulsus paradoxus and Kussmaul’s sign. Lancet 2002; 359:1940–1942.
- Sherwood L. Human Physiology: From Cells to Systems. 8th ed. Belmont, CA: Brooks/Cole; 2012.
- Constant J. Using internal jugular pulsations as a manometer for right atrial pressure measurements. Cardiology 2000; 93:26–30.
- Deol GR, Collett N, Ashby A, Schmidt GA. Ultrasound accurately reflects the jugular venous examination but underestimates central venous pressure. Chest 2011; 139:95–100.
- Wu X, Studer W, Erb T, Skarvan K, Seeberger MD. Competence of the internal jugular vein valve is damaged by cannulation and catheterization of the internal jugular vein. Anesthesiology 2000; 93:319–324.
- Ramana RK, Sanagala T, Lichtenberg R. A new angle on the angle of Louis. Congest Heart Fail 2006; 12:196–199.
- Seth R, Magner P, Matzinger F, van Walraven C. How far is the sternal angle from the mid-right atrium? J Gen Intern Med 2002; 17:852–856.
- Kee LL, Simonson JS, Stotts NA, Skov P, Schiller NB. Echocardiographic determination of valid zero reference levels in supine and lateral positions. Am J Crit Care 1993; 2:72–80.
- Park SY, Kim MJ, Kim MG, et al. Changes in the relationship between the right internal jugular vein and an anatomical landmark after head rotation. Korean J Anesthesiol 2011; 61:107–111.
- Sankoff J, Zidulka A. Non-invasive method for the rapid assessment of central venous pressure: description and validation by a single examiner. West J Emerg Med 2008; 9:201–205.
- Conn RD, O’Keefe JH. Simplified evaluation of the jugular venous pressure: significance of inspiratory collapse of jugular veins. Mo Med 2012; 109:150–152.
- Wood PH. Diseases of the Heart and Circulation. 2nd ed. Philadelphia, PA: Lippincott; 1956.
- Drazner MH, Brown RN, Kaiser PA, et al. Relationship of right- and left-sided filling pressures in patients with advanced heart failure: a 14-year multi-institutional analysis. J Heart Lung Transplant 2012; 31:67–72.
- Clark G, Strauss HD, Roberts R. Dobutamine vs furosemide in the treatment of cardiac failure due to right ventricular infarction. Chest 1980; 77:220–223.
- Roy CL, Minor MA, Brookhart MA, Choudhry NK. Does this patient with a pericardial effusion have cardiac tamponade? JAMA 2007; 297:1810–1818.
- Zhou Q, Xiao W, An E, Zhou H, Yan M. Effects of four different positive airway pressures on right internal jugular vein catheterisation. Eur J Anaesthesiol 2012; 29:223–228.
- Jolobe OM. Disproportionate elevation of jugular venous pressure in pleural effusion. Br J Hosp Med (Lond) 2011; 72:582–585.
- Seo M, Shin WJ, Jun IG. Central venous catheter-related superior vena cava syndrome following renal transplantation—a case report. Korean J Anesthesiol 2012; 63:550–554.
- Applefeld MM. The jugular venous pressure and pulse contour. In:Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd ed. Boston, MA: Butterworths; 1990.
- El Gamal MI, Van Gelder LM. Chronic ventricular pacing with ventriculo-atrial conduction versus atrial pacing in three patients with symptomatic sinus bradycardia. Pacing Clin Electrophysiol 1981; 4:100–105.
- Berman ND, Waxman MB. Cannon waves with A-V association. Am Heart J 1976; 91:643–644.
- Luisada AA, Singhal A, Kim K. The jugular and hepatic tracings in normal subjects and in conduction defects. Acta Cardiol 1983; 38:405–424.
- Miller MJ, McKay RG, Ferguson JJ, et al. Right atrial pressure-volume relationships in tricuspid regurgitation. Circulation 1986; 73:799–808.
- Wooley CF, Fontana ME, Kilman JW, Ryan JM. Tricuspid stenosis. Atrial systolic murmur, tricuspid opening snap, and right atrial pressure pulse. Am J Med 1985; 78:375–384.
- McGee SR. Evidence-Based Physical Diagnosis. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
- Mittal SR, Garg S, Lalgarhia M. Jugular venous pressure and pulse wave form in the diagnosis of right ventricular infarction. Int J Cardiol 1996; 53:253–256.
- Bilchick KC, Wise RA. Paradoxical physical findings described by Kussmaul: pulsus paradoxus and Kussmaul’s sign. Lancet 2002; 359:1940–1942.
KEY POINTS
- If the jugular venous pressure differs from the true right atrial pressure, the jugular venous pressure is always the lower value.
- The jugular venous pressure is useful to observe when diagnosing congestive heart failure and when considering the need for or the adequacy of diuresis.
- The jugular venous wave form is more difficult to observe than its elevation but can yield useful information in the assessment of certain arrhythmias, right-heart conditions, and pericardial disease.
More than skin-deep
A 68-year-old man presented for evaluation of a diffuse rash with mucosal involvement. During the past 9 months, he had had oral ulcers, a truncal rash, and blistering lesions on his hands with fingernail erosion, and all of these had been getting worse (Figure 1). He also reported cycles of fever and a weight loss of 50 lb.
Computed tomography revealed diffuse lymphadenopathy, including a bulky 18-cm mediastinal mass. Lymph node biopsy confirmed follicular non-Hodgkin lymphoma.
On serum enzyme-linked immunosorbent assay (ELISA), the desmoglein 1 antibody titer was 62.4 U (< 14 is negative, > 20 is positive) and the desmoglein 3 antibody titer was 36.0 U (< 9 is negative, > 20 is positive). Indirect immunofluorescence testing on monkey esophagus substrate was reactive.
ELISA detected no immunoglobulin G (IgG) reactivity against the bullous pemphigoid autoantigens BP180 and BP230. Direct immunofluorescence testing revealed strong IgG and C3 deposition on epithelial cell surfaces, and indirect immunofluorescence on rat bladder substrate was positive, suggesting the diagnosis of paraneoplastic pemphigus.
The patient was treated with dexamethasone and bendamustine-rituximab. Five months later, desmoglein 1 and 3 antibody titers were measured again and were within normal limits. His lesions had improved significantly (Figure 2), but he had endured multiple medical setbacks, including Pseudomonas peritonitis, fistulizing cytomegalovirus perianal ulceration, septic shock secondary to fulminant Clostridium difficile colitis, and toxic megacolon necessitating total colectomy with end-ileostomy.
PARANEOPLASTIC PEMPHIGUS
Paraneoplastic pemphigus usually occurs in the presence of underlying lymphoproliferative neoplasm, most often non-Hodgkin lymphoma, chronic lymphocytic leukemia, or Castleman disease. It may also occur with epithelial carcinoma, mesenchymal sarcoma, and, rarely, malignant melanoma.1 The association between malignancy and autoimmune mucocutaneous disease was first described in 1990.2 Autoantibodies against periplakin, envoplakin, and desmoglein 3 are often present and may play a role in pathogenesis.
Paraneoplastic pemphigus associated with malignancy portends a poor prognosis. Although historically reported to have an overall death rate ranging from 75% to 90%, with a mean survival of less than 1 year, a recent retrospective cohort has shown slightly improved outcomes, with 49% of patients remaining alive at 1 year and 38% alive at 5 years.3 The most common causes of death are sepsis, respiratory failure, and the underlying malignancy. Bronchiolitis obliterans may occur late in the disease course and is an ominous prognostic factor, with a death rate of 41% after a median interval of 13 months.4
Immunosuppressive drugs are often used to control the disease. First-line treatment is with high-dose steroids. Immunosuppressives such as azathioprine and cyclosporin may be used in conjunction to reduce the steroid dose that is required and to limit the adverse effects of steroid therapy. Treating the underlying malignancy may decrease autoantibody production and lead to clinical improvement.
- Kaplan I, Hodak E, Ackerman L, Mimouni D, Anhalt GJ, Calderon S. Neoplasms associated with paraneoplastic pemphigus: a review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol 2004; 40:553–562.
- Anhalt GJ, Kim SC, Stanley JR, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med 1990; 323:1729–1735.
- Leger S, Picard D, Ingen-Housz-Oro S, et al. Prognostic factors of paraneoplastic pemphigus. Arch Dermatol 2012; 148:1165–1172.
- Maldonado F, Pittelkow MR, Ryu JH. Constrictive bronchiolitis associated with pareaneoplastic autoimmune multi-organ syndrome. Respirology 2009; 14:129–133.
A 68-year-old man presented for evaluation of a diffuse rash with mucosal involvement. During the past 9 months, he had had oral ulcers, a truncal rash, and blistering lesions on his hands with fingernail erosion, and all of these had been getting worse (Figure 1). He also reported cycles of fever and a weight loss of 50 lb.
Computed tomography revealed diffuse lymphadenopathy, including a bulky 18-cm mediastinal mass. Lymph node biopsy confirmed follicular non-Hodgkin lymphoma.
On serum enzyme-linked immunosorbent assay (ELISA), the desmoglein 1 antibody titer was 62.4 U (< 14 is negative, > 20 is positive) and the desmoglein 3 antibody titer was 36.0 U (< 9 is negative, > 20 is positive). Indirect immunofluorescence testing on monkey esophagus substrate was reactive.
ELISA detected no immunoglobulin G (IgG) reactivity against the bullous pemphigoid autoantigens BP180 and BP230. Direct immunofluorescence testing revealed strong IgG and C3 deposition on epithelial cell surfaces, and indirect immunofluorescence on rat bladder substrate was positive, suggesting the diagnosis of paraneoplastic pemphigus.
The patient was treated with dexamethasone and bendamustine-rituximab. Five months later, desmoglein 1 and 3 antibody titers were measured again and were within normal limits. His lesions had improved significantly (Figure 2), but he had endured multiple medical setbacks, including Pseudomonas peritonitis, fistulizing cytomegalovirus perianal ulceration, septic shock secondary to fulminant Clostridium difficile colitis, and toxic megacolon necessitating total colectomy with end-ileostomy.
PARANEOPLASTIC PEMPHIGUS
Paraneoplastic pemphigus usually occurs in the presence of underlying lymphoproliferative neoplasm, most often non-Hodgkin lymphoma, chronic lymphocytic leukemia, or Castleman disease. It may also occur with epithelial carcinoma, mesenchymal sarcoma, and, rarely, malignant melanoma.1 The association between malignancy and autoimmune mucocutaneous disease was first described in 1990.2 Autoantibodies against periplakin, envoplakin, and desmoglein 3 are often present and may play a role in pathogenesis.
Paraneoplastic pemphigus associated with malignancy portends a poor prognosis. Although historically reported to have an overall death rate ranging from 75% to 90%, with a mean survival of less than 1 year, a recent retrospective cohort has shown slightly improved outcomes, with 49% of patients remaining alive at 1 year and 38% alive at 5 years.3 The most common causes of death are sepsis, respiratory failure, and the underlying malignancy. Bronchiolitis obliterans may occur late in the disease course and is an ominous prognostic factor, with a death rate of 41% after a median interval of 13 months.4
Immunosuppressive drugs are often used to control the disease. First-line treatment is with high-dose steroids. Immunosuppressives such as azathioprine and cyclosporin may be used in conjunction to reduce the steroid dose that is required and to limit the adverse effects of steroid therapy. Treating the underlying malignancy may decrease autoantibody production and lead to clinical improvement.
A 68-year-old man presented for evaluation of a diffuse rash with mucosal involvement. During the past 9 months, he had had oral ulcers, a truncal rash, and blistering lesions on his hands with fingernail erosion, and all of these had been getting worse (Figure 1). He also reported cycles of fever and a weight loss of 50 lb.
Computed tomography revealed diffuse lymphadenopathy, including a bulky 18-cm mediastinal mass. Lymph node biopsy confirmed follicular non-Hodgkin lymphoma.
On serum enzyme-linked immunosorbent assay (ELISA), the desmoglein 1 antibody titer was 62.4 U (< 14 is negative, > 20 is positive) and the desmoglein 3 antibody titer was 36.0 U (< 9 is negative, > 20 is positive). Indirect immunofluorescence testing on monkey esophagus substrate was reactive.
ELISA detected no immunoglobulin G (IgG) reactivity against the bullous pemphigoid autoantigens BP180 and BP230. Direct immunofluorescence testing revealed strong IgG and C3 deposition on epithelial cell surfaces, and indirect immunofluorescence on rat bladder substrate was positive, suggesting the diagnosis of paraneoplastic pemphigus.
The patient was treated with dexamethasone and bendamustine-rituximab. Five months later, desmoglein 1 and 3 antibody titers were measured again and were within normal limits. His lesions had improved significantly (Figure 2), but he had endured multiple medical setbacks, including Pseudomonas peritonitis, fistulizing cytomegalovirus perianal ulceration, septic shock secondary to fulminant Clostridium difficile colitis, and toxic megacolon necessitating total colectomy with end-ileostomy.
PARANEOPLASTIC PEMPHIGUS
Paraneoplastic pemphigus usually occurs in the presence of underlying lymphoproliferative neoplasm, most often non-Hodgkin lymphoma, chronic lymphocytic leukemia, or Castleman disease. It may also occur with epithelial carcinoma, mesenchymal sarcoma, and, rarely, malignant melanoma.1 The association between malignancy and autoimmune mucocutaneous disease was first described in 1990.2 Autoantibodies against periplakin, envoplakin, and desmoglein 3 are often present and may play a role in pathogenesis.
Paraneoplastic pemphigus associated with malignancy portends a poor prognosis. Although historically reported to have an overall death rate ranging from 75% to 90%, with a mean survival of less than 1 year, a recent retrospective cohort has shown slightly improved outcomes, with 49% of patients remaining alive at 1 year and 38% alive at 5 years.3 The most common causes of death are sepsis, respiratory failure, and the underlying malignancy. Bronchiolitis obliterans may occur late in the disease course and is an ominous prognostic factor, with a death rate of 41% after a median interval of 13 months.4
Immunosuppressive drugs are often used to control the disease. First-line treatment is with high-dose steroids. Immunosuppressives such as azathioprine and cyclosporin may be used in conjunction to reduce the steroid dose that is required and to limit the adverse effects of steroid therapy. Treating the underlying malignancy may decrease autoantibody production and lead to clinical improvement.
- Kaplan I, Hodak E, Ackerman L, Mimouni D, Anhalt GJ, Calderon S. Neoplasms associated with paraneoplastic pemphigus: a review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol 2004; 40:553–562.
- Anhalt GJ, Kim SC, Stanley JR, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med 1990; 323:1729–1735.
- Leger S, Picard D, Ingen-Housz-Oro S, et al. Prognostic factors of paraneoplastic pemphigus. Arch Dermatol 2012; 148:1165–1172.
- Maldonado F, Pittelkow MR, Ryu JH. Constrictive bronchiolitis associated with pareaneoplastic autoimmune multi-organ syndrome. Respirology 2009; 14:129–133.
- Kaplan I, Hodak E, Ackerman L, Mimouni D, Anhalt GJ, Calderon S. Neoplasms associated with paraneoplastic pemphigus: a review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol 2004; 40:553–562.
- Anhalt GJ, Kim SC, Stanley JR, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med 1990; 323:1729–1735.
- Leger S, Picard D, Ingen-Housz-Oro S, et al. Prognostic factors of paraneoplastic pemphigus. Arch Dermatol 2012; 148:1165–1172.
- Maldonado F, Pittelkow MR, Ryu JH. Constrictive bronchiolitis associated with pareaneoplastic autoimmune multi-organ syndrome. Respirology 2009; 14:129–133.
A practical approach to prescribing antidepressants
With the variety of drugs available for treating depression, choosing one can be daunting. Different agents have characteristics that may make them a better choice for different types of patients, but even so, treating any kind of mental illness often requires an element of trial and error.
Primary care providers are on the frontline of treating mental illness, often evaluating patients before they are seen by a psychiatrist. The purpose of this article is to provide insight into the art of prescribing antidepressants in the primary care setting. We will discuss common patient presentations, including depressed patients without other medical comorbidities as well as those with common comorbidities, with our recommendations for first-line treatment.
We hope our recommendations will help you to navigate the uncertainty more confidently, resulting in more efficient and tailored treatment for your patients.
BASELINE TESTING
When starting a patient on antidepressant drug therapy, we recommend obtaining a set of baseline laboratory tests to rule out underlying medical conditions that may be contributing to the patient’s depression or that may preclude the use of a given drug. (For example, elevation of liver enzymes may preclude the use of duloxetine.) Tests should include:
- A complete blood cell count
- A complete metabolic panel
- A thyroid-stimulating hormone level.
Electrocardiography may also be useful, as some antidepressants can prolong the QT interval or elevate the blood levels of other drugs with this effect.
GENERAL TREATMENT CONSIDERATIONS
There are several classes of antidepressants, and each class has a number of agents. Research has found little difference in efficacy among agents. So to simplify choosing which one to use, we recommend becoming comfortable with an agent from each class, ie:
- A selective serotonin reuptake inhibitor (SSRI)
- A selective serotonin-norepinephrine reuptake inhibitor (SNRI)
- A tricyclic antidepressant (TCA)
- A monoamine oxidase (MAO) inhibitor.
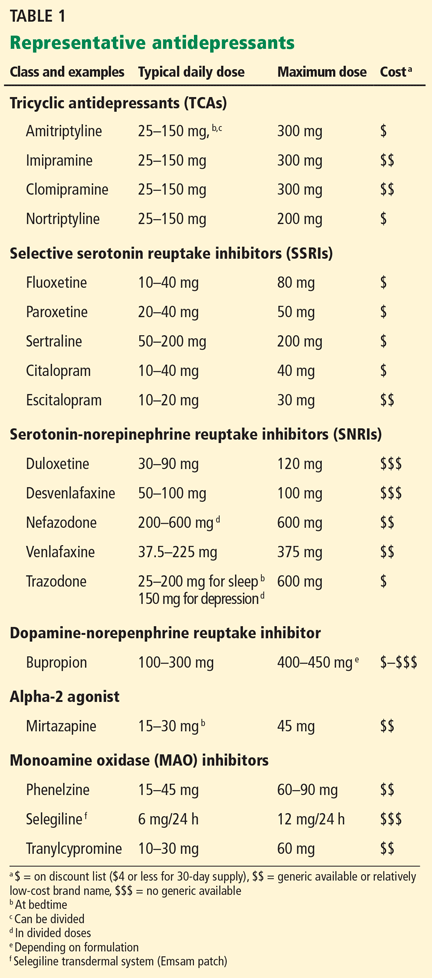
Each class includes generic agents, many of which are on the discount lists of retail pharmacies. Table 1 shows representative drugs from each class, with their relative costs.
Start low and go slow. In general, when starting an antidepressant, consider starting at half the normal dose, titrating upward as tolerated about every 14 days. This approach can minimize side effects. For example, if prescribing fluoxetine, start with 10 mg and titrate every 2 weeks based on tolerance and patient response. That said, each patient may respond differently, requiring perhaps a lower starting dose or a longer titration schedule.
Anticipate side effects. Most of the side effects of an antidepressant drug can be explained by its mechanism of action. Although side effects should certainly be considered when choosing an agent, patients can be reassured that most are transient and benign. A detailed discussion of side effects of antidepressant drugs is beyond the scope of this article, but a review by Khawam et al1 was published earlier in this journal.
Reassess. If after 4 to 6 weeks the patient has had little or no response, it is reasonable to switch agents. For a patient who was on an SSRI, the change can be to another SSRI or to an SNRI. However, if two SSRIs have already failed, then choose an SNRI. Agents are commonly cross-tapered during the switch to avoid abrupt cessation of one drug or the increased risk of adverse events such as cytochrome P450 interactions, serotonin syndrome, or hypertensive crisis (when switching to an MAO inhibitor).
Beware of interactions. All SSRIs and SNRIs are metabolized through the P450 system in the liver and therefore have the potential for drug-drug interactions. Care must be taken when giving these agents together with drugs whose metabolism can be altered by P450 inhibition. For TCAs, blood levels can be checked if there is concern about toxicity; however, dosing is not strictly based on this level. Great care should be taken if a TCA is given together with an SNRI or an SSRI, as the TCA blood level can become significantly elevated. This may result in QT interval prolongation, as mentioned earlier.
Refer. Referral to a psychiatrist is appropriate for patients for whom multiple classes have failed, for patients who have another psychiatric comorbidity (such as psychosis, hypomania, or mania), or for patients who may need hospitalization. Referral is also appropriate if the physician is concerned about suicide risk.
PATIENTS WITH MAJOR DEPRESSION ONLY
For a patient presenting with depression but no other significant medical comorbidity, the first-line therapy is often an SSRI. Several generic SSRIs are available, and some are on the discount lists at retail pharmacies.
Symptoms should start to improve in about 2 weeks, and the optimal response should be achieved in 4 to 6 weeks of treatment. If this does not occur, consider either adding an augmenting agent or switching to a different antidepressant.
PATIENTS WITH CHRONIC PAIN

Chronic pain and depression often go hand in hand and can potentiate each other. When considering an antidepressant in a patient who has both conditions, the SNRIs and TCAs are typically preferred. Some SNRIs, namely duloxetine and milnacipran, are approved for certain chronic pain conditions, such as fibromyalgia. SNRIs are frequently used off-label for other chronic pain conditions such as headache and neuropathic pain.2
TCAs such as amitriptyline, nortriptyline, and doxepin are also often used in patients with chronic pain. These agents, like the SNRIs, inhibit the reuptake of serotonin and norepinephrine and are used off-label for neuropathic pain,3,4 migraine, interstitial cystitis,5 and other pain conditions.6–9
For TCAs and SNRIs, the effective dose range for chronic pain overlaps that for depression. However, TCAs are often given at lower doses to patients without depression. We recommend starting at a low dose and slowly titrating upward to an effective dose. SNRIs are often preferred over TCAs because they do not have anticholinergic side effects and because an overdose is much less likely to be lethal.
PATIENTS WITH SEXUAL DYSFUNCTION
One of the more commonly reported side effects of antidepressants is sexual dysfunction, generally in the form of delayed orgasm or decreased libido.10 Typically, these complaints are attributed to SSRIs and SNRIs; however, TCAs and MAO inhibitors have also been associated wth sexual dysfunction.
Both erectile dysfunction and priapism have been linked to certain antidepressants. In particular, trazodone is a known cause of priapism. Even if using low doses for sleep, male patients should be made aware of this adverse effect.
Switching from one agent to another in the same class is not likely to improve sexual side effects. In particular, all the SSRIs are similar in their likelihood of causing sexual dysfunction. In a patient taking an SSRI who experiences this side effect, switching to bupropion11 or mirtazapine12 can be quite useful. Bupropion acts primarily on dopamine and norepinephrine, whereas mirtazapine acts on serotonin and norepinephrine but in a different manner from SSRIs and SNRIs.
Adjunctive treatment such as a cholinergic agonist, yohimbine (contraindicated with MAO inhibitors), a serotonergic agent (eg, buspirone), or a drug that acts on nitric oxide (eg, sildenafil, tadalafil) may have some utility but is often ineffective. Dose reduction, if possible, can be of value.
PATIENTS WITH ANXIETY
Many antidepressants are also approved for anxiety disorders, and still more are used off-label for this purpose. Anxiety and depression often occur together, so being able to treat both conditions with one drug can be quite useful.13 In general, the antidepressant effects are seen at lower doses of SSRIs and SNRIs, whereas more of the anxiolytic effects are seen at higher doses, particularly for obsessive-compulsive disorder.14
First-line treatment would be an SSRI or SNRI. Most anxiety disorders respond to either class, but there are some more-specific recommendations. SSRIs are best studied in panic disorder, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Fluoxetine, citalopram, escitalopram, and sertraline15 can all be effective in both major depressive disorder and generalized anxiety disorder. Panic disorder also tends to respond well to SSRIs. SNRIs have been evaluated primarily in generalized anxiety disorder but may also be useful in many of the other conditions.
Additionally, mirtazapine (used off-label)12 and the TCAs16–18 can help treat anxiety. Clomipramine is used to treat obsessive-compulsive disorder.19 These drugs are especially useful for nighttime anxiety, as they can aid sleep. Of note, the anxiolytic effect of mirtazapine may be greater at higher doses.
MAO inhibitors often go unused because of the dietary and medication restrictions involved. However, very refractory cases of certain anxiety disorders may respond preferentially to these agents.
Bupropion tends to be more activating than other antidepressants, so is often avoided in anxious patients. However, some research suggests this is not always necessary.20 If the anxiety is secondary to depression, it will often improve significantly with this agent.
When starting or increasing the dose of an antidepressant, patients may experience increased anxiety or feel “jittery.” This feeling usually passes within the first week of treatment, and it is important to inform patients about this effect. “Start low and go slow” in patients with significant comorbid anxiety. Temporarily using a benzodiazepine such as clonazepam may make the transition more tolerable.
PATIENTS WITH CHRONIC FATIGUE SYNDROME OR FIBROMYALGIA
Increasing recognition of both chronic fatigue syndrome and fibromyalgia has led to more proactive treatment for these disorders. Depression can go hand in hand with these disorders, and certain antidepressants, namely the SNRIs, can be useful in this population.
More data exist for the treatment of fibromyalgia. Both duloxetine and milnacipran are approved by the US Food and Drug Administration (FDA) for the treatment of fibromyalgia.21 Venlafaxine is also used off-label for this purpose. SSRIs such as fluoxetine and citalopram have had mixed results.21–23 TCAs have been used with some success; however, their side effects and lethal potential are often limiting.21,24,25 A recent study in Spain also suggested there may be benefit from using MAO inhibitors for fibromyalgia, but data are quite limited.26
The data for treating chronic fatigue syndrome with SSRIs, SNRIs, or MAO inhibitors are conflicting.27–29 However, managing the co-existing depression may provide some relief in and of itself.
PATIENTS WITH FREQUENT INSOMNIA
Insomnia can be a symptom of depression, but it can also be a side effect of certain antidepressants. The SSRIs and SNRIs can disrupt sleep patterns in some patients by shortening the rapid-eye-movement (REM) stage.30,31
In patients with severe insomnia, it may be best to first recommend taking the antidepressant in the morning if they notice worsening sleep after initiating treatment. Patients can be told with any antidepressant, “If it makes you tired, take it at night, and if it wakes you up, take it in the morning.” Of note, a recent South African study suggested that escitalopram may be able to improve sleep.32
If that does not solve the problem, there are other options. For instance, mirtazapine, particularly in doses of 15 mg or 30 mg, aids depression and insomnia. At higher doses (45 mg), the sleep-aiding effect may be reduced. Low doses of TCAs, particularly doxepin, maprotiline (technically speaking, a tetracyclic antidepressant), amitriptyline, and nortriptyline can be effective sleep aids. These agents may be used as an adjunct to another antidepressant to enhance sleep and mood. However, the TCAs also shorten the REM stage of sleep.33
The previously mentioned drug interactions with SSRIs and SNRIs also need to be considered. Caution should be used when discontinuing these medications, as patients may experience rebound symptoms in the form of much more vivid dreams. MAO inhibitors may worsen insomnia because they suppress REM sleep.34
Trazodone is another agent that at lower doses (25–150 mg) can be an effective, nonaddicting sleep aid. When used as an antidepressant, it is generally prescribed at higher doses (300–400 mg), but its sedating effects can be quite limiting at these levels. It is important to remember the possibility of priapism in male patients.
GERIATRIC PATIENTS
Old age brings its own set of concerns when treating depression. Elderly patients are more susceptible to potential bradycardia caused by SSRIs. The TCAs have the more worrisome cardiac side effect of QTc prolongation. TCAs can slow cognitive function, whereas the SSRIs, bupropion, and the SNRIs tend not to affect cognition. Escitalopram and duloxetine have been suggested to be particularly effective in the elderly.35,36 A study from the Netherlands linked SSRIs with increased risk of falling in geriatric patients with dementia.37 Constipation, which could lead to ileus, is increased with TCAs and certain other agents (ie, paroxetine) in the geriatric population.
Mirtazapine is often very useful in elderly patients for many reasons: it treats both anxiety and depression, stimulates appetite and weight gain, can help with nausea, and is an effective sleep aid. Concerns about weight, appetite, and sleep are particularly common in the elderly, whereas younger patients can be less tolerant of drugs that make them gain weight and sleep more. Normal age-related changes to the sleep cycle contribute to decreased satisfaction with sleep as we age. In addition, depression often further impairs sleep. So, in the elderly, optimizing sleep is key. Research has also shown mirtazapine to be effective in patients with both Alzheimer dementia and depression.38
DIABETIC PATIENTS
One of the more worrisome side effects of psychiatric medications in diabetic patients is weight gain. Certain antidepressants have a greater propensity for weight gain and should likely be avoided as first-line treatments in this population.12 Typically, these agents include those that have more antihistamine action such as paroxetine and the TCAs. These agents also may lead to constipation, which could potentially worsen gastroparesis. Mirtazapine and the MAO inhibitors are also known to cause weight gain.
Bupropion and nefazodone are the most weight-neutral of all antidepressants. Nefazodone has fallen out of favor because of its potential to cause fulminant liver failure in rare cases. However, it remains a reasonable option for patients with comorbid anxiety and depression who have significant weight gain with other agents.
SSRIs and MAO inhibitors may improve or be neutral toward glucose metabolism, and some data suggest that SNRIs may impair this process.39
PATIENTS WITH CARDIAC CONDITIONS
Major depression often coexists with cardiac conditions. In particular, many patients develop depression after suffering a myocardial infarction, and increasingly they are being treated for it.40 Treatment in this situation is appropriate, since depression, if untreated, can increase the risk of recurrence of myocardial infarction.41
However, there are many concerns that accompany treating depression in cardiac patients. Therefore, a baseline electrocardiogram should be obtained before starting an antidepressant.
TCAs and tetracyclic agents have a tendency to prolong the QTc interval and potentiate ventricular arrhythmias,42 so it may be prudent to avoid these in patients at risk. These agents can also significantly increase the pulse rate. This tachycardia increases the risk of angina or myocardial infarction from the anticholinergic effects of these drugs.
In February 2013, the FDA issued a warning about possible arrhythmias with citalopram at doses greater than 40 mg in adult patients43; however, research has suggested citalopram is effective in treating depression in cardiac patients.44 Research has not shown an increase in efficacy at doses greater than 40 mg daily, so we recommend following the black-box warning.
TCAs and MAO inhibitors can also cause orthostatic hypotension. On the other hand, consuming large amounts of tyramine, in foods such as aged cheese, can precipitate a hypertensive crisis in patients taking MAO inhibitors.
Which antidepressants tend to be safer in cardiac patients? Sertraline has been shown to be safe in congestive heart failure and coronary artery disease,45–47 but the SSRIs are typically safe. Fluoxetine has shown efficacy in patients who have had a myocardial infarction.48 Mirtazapine has also been shown to be efficacious in cardiac patients.49 Nefazodone, mirtazapine, bupropion, SSRIs, and SNRIs have little or no tendency toward orthostatic hypotension.
- Khawam EA, Laurencic G, Malone DA. Side effects of antidepressants: an overview. Cleve Clin J Med 2006; 73:351–361.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab Res Rev 2008; 24(suppl 1):S52–S57.
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81:1372–1373.
- Tanenberg RJ, Irving GA, Risser RC, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86:615–626.
- Hertle L, van Ophoven A. Long-term results of amitriptyline treatment for interstitial cystitis. Aktuelle Urol 2010; 41(suppl 1):S61–S65.
- Nguyen TM, Eslick GD. Systematic review: the treatment of noncardiac chest pain with antidepressants. Aliment Pharmacol Ther 2012; 35:493–500.
- Lee H, Kim JH, Min BH, et al. Efficacy of venlafaxine for symptomatic relief in young adult patients with functional chest pain: a randomized, double-blind, placebo-controlled, crossover trial. Am J Gastroenterol 2010; 105:1504–1512.
- Varia I, Logue E, O’Connor C, et al. Randomized trial of sertraline in patients with unexplained chest pain of noncardiac origin. Am Heart J 2000; 140:367–372.
- Doraiswamy PM, Varia I, Hellegers C, et al. A randomized controlled trial of paroxetine for noncardiac chest pain. Psychopharmacol Bull 2006; 39:15–24.
- Clayton AH. Understanding antidepressant mechanism of action and its effect on efficacy and safety. J Clin Psychiatry 2012; 73:e11.
- Gartlehner G, Hansen RA, Morgan LC, et al. Second-generation antidepressants in the pharmacologic treatment of adult depression: an update of the 2007 comparative effectiveness review (Internet). Rockville (MD): Agency for Healthcare Research and Quality (US); 2011 Dec. Comparative Effectiveness Reviews, No. 46. http://www.ncbi.nlm.nih.gov/books/NBK83442/. Accessed February 27, 2013.
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev 2011; 12:CD006528.
- Hofmeijer-Sevink MK, Batelaan NM, van Megen HJ, et al. Clinical relevance of comorbidity in anxiety disorders: a report from the Netherlands Study of Depression and Anxiety (NESDA). J Affect Disord 2012; 137:106–112.
- Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci 2011; 13:423–437.
- Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol 2009; 24:43–60.
- Huh J, Goebert D, Takeshita J, Lu BY, Kang M. Treatment of generalized anxiety disorder: a comprehensive review of the literature for psychopharmacologic alternatives to newer antidepressants and benzodiazepines. Prim Care Companion CNS Disord 2011; 13: 4088/PCC.08r00709blu.
- Rickels K, Downing R, Schweizer E, Hassman H. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry 1993; 50:884–895.
- Uher R, Maier W, Hauser J, et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259.
- Kellner M. Drug treatment of obsessive-compulsive disorder. Dialogues Clin Neurosci 2010; 12:187–197.
- Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology 2001; 25:131–138.
- Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol 2011; 25:285–297.
- Wolfe F, Cathey MA, Hawley DJ. A double-blind placebo controlled trial of fluoxetine in fibromyalgia. Scand J Rheumatol 1994; 23:255–259.
- Arnold LM, Hess EV, Hudson JI, Welge JA, Berno SE, Keck PE. A randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med 2002; 112:191–197.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41:104–113.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Tort S, Urrútia G, Nishishinya MB, Walitt B. Monoamine oxidase inhibitors (MAOIs) for fibromyalgia syndrome. Cochrane Database Syst Rev 2012; 4:CD009807.
- Vercoulen JH, Swanink CM, Zitman FG, et al. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996; 347:858–861.
- Natelson BH, Cheu J, Pareja J, Ellis SP, Policastro T, Findley TW. Randomized, double blind, controlled placebo-phase in trial of low dose phenelzine in the chronic fatigue syndrome. Psychopharmacology (Berl) 1996; 124:226–230.
- Reid S, Chalder T, Cleare A, Hotopf M, Wessely S. Chronic fatigue syndrome. BMJ 2000; 320:292–296.
- Kupfer DJ, Spiker DG, Coble PA, Neil JF, Ulrich R, Shaw DH. Sleep and treatment prediction in endogenous depression. Am J Psychiatry 1981; 138:429–434.
- Argyropoulos SV, Hicks JA, Nash JR, et al. Redistribution of slow wave activity of sleep during pharmacological treatment of depression with paroxetine but not with nefazodone. J Sleep Res 2009; 18:342–348.
- Stein DJ, Lopez AG. Effects of escitalopram on sleep problems in patients with major depression or generalized anxiety disorder. Adv Ther 2011; 28:1021–1037.
- Ehlers CL, Havstad JW, Kupfer DJ. Estimation of the time course of slow-wave sleep over the night in depressed patients: effects of clomipramine and clinical response. Biol Psychiatry 1996; 39:171–181.
- Landolt HP, Raimo EB, Schnierow BJ, Kelsoe JR, Rapaport MH, Gillin JC. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch Gen Psychiatry 2001; 58:268–276.
- Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res 2011; 39:1946–1953.
- Dolder C, Nelson M, Stump A. Pharmacological and clinical profile of newer antidepressants: implications for the treatment of elderly patients. Drugs Aging 2010; 27:625–640.
- Sterke CS, Ziere G, van Beeck EF, Looman CW, van der Cammen TJ. Dose-response relationship between selective serotonin re-uptake inhibitors and injurious falls: a study in nursing home residents with dementia. Br J Clin Pharmacol 2012; 73:812–820.
- Raji MA, Brady SR. Mirtazapine for treatment of depression and comorbidities in Alzheimer disease. Ann Pharmacother 2001; 35:1024–1027.
- Hennings JM, Schaaf L, Fulda S. Glucose metabolism and antidepressant medication. Curr Pharm Des 2012; 18:5900–5919.
- Czarny MJ, Arthurs E, Coffie DF, et al. Prevalence of antidepressant prescription or use in patients with acute coronary syndrome: a systematic review. PLoS One 2011; 6:e27671.
- Zuidersma M, Ormel J, Conradi HJ, de Jonge P. An increase in depressive symptoms after myocardial infarction predicts new cardiac events irrespective of depressive symptoms before myocardial infarction. Psychol Med 2012; 42:683–693.
- van Noord C, Straus SM, Sturkenboom MC, et al. Psychotropic drugs associated with corrected QT interval prolongation. J Clin Psychopharmacol 2009; 29:9–15.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: abnormal heart rhythms associated with high doses of Celexa (citalopram hydrobromide). http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed August 25, 2013.
- Lespérance F, Frasure-Smith N, Koszycki D, et al; CREATE Investigators. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- O’Connor CM, Jiang W, Kuchibhatla M, et al; SADHART-CHF Investigators. Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Glassman AH, O’Connor CM, Califf RM, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Swenson JR, O’Connor CM, Barton D, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol 2003; 92:1271–1276.
- Strik JJ, Honig A, Lousberg R, et al. Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med 2000; 62:783–789.
- Honig A, Kuyper AM, Schene AH, et al; MIND-IT investigators. Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med 2007; 69:606–613.
With the variety of drugs available for treating depression, choosing one can be daunting. Different agents have characteristics that may make them a better choice for different types of patients, but even so, treating any kind of mental illness often requires an element of trial and error.
Primary care providers are on the frontline of treating mental illness, often evaluating patients before they are seen by a psychiatrist. The purpose of this article is to provide insight into the art of prescribing antidepressants in the primary care setting. We will discuss common patient presentations, including depressed patients without other medical comorbidities as well as those with common comorbidities, with our recommendations for first-line treatment.
We hope our recommendations will help you to navigate the uncertainty more confidently, resulting in more efficient and tailored treatment for your patients.
BASELINE TESTING
When starting a patient on antidepressant drug therapy, we recommend obtaining a set of baseline laboratory tests to rule out underlying medical conditions that may be contributing to the patient’s depression or that may preclude the use of a given drug. (For example, elevation of liver enzymes may preclude the use of duloxetine.) Tests should include:
- A complete blood cell count
- A complete metabolic panel
- A thyroid-stimulating hormone level.
Electrocardiography may also be useful, as some antidepressants can prolong the QT interval or elevate the blood levels of other drugs with this effect.
GENERAL TREATMENT CONSIDERATIONS
There are several classes of antidepressants, and each class has a number of agents. Research has found little difference in efficacy among agents. So to simplify choosing which one to use, we recommend becoming comfortable with an agent from each class, ie:
- A selective serotonin reuptake inhibitor (SSRI)
- A selective serotonin-norepinephrine reuptake inhibitor (SNRI)
- A tricyclic antidepressant (TCA)
- A monoamine oxidase (MAO) inhibitor.
Each class includes generic agents, many of which are on the discount lists of retail pharmacies. Table 1 shows representative drugs from each class, with their relative costs.
Start low and go slow. In general, when starting an antidepressant, consider starting at half the normal dose, titrating upward as tolerated about every 14 days. This approach can minimize side effects. For example, if prescribing fluoxetine, start with 10 mg and titrate every 2 weeks based on tolerance and patient response. That said, each patient may respond differently, requiring perhaps a lower starting dose or a longer titration schedule.
Anticipate side effects. Most of the side effects of an antidepressant drug can be explained by its mechanism of action. Although side effects should certainly be considered when choosing an agent, patients can be reassured that most are transient and benign. A detailed discussion of side effects of antidepressant drugs is beyond the scope of this article, but a review by Khawam et al1 was published earlier in this journal.
Reassess. If after 4 to 6 weeks the patient has had little or no response, it is reasonable to switch agents. For a patient who was on an SSRI, the change can be to another SSRI or to an SNRI. However, if two SSRIs have already failed, then choose an SNRI. Agents are commonly cross-tapered during the switch to avoid abrupt cessation of one drug or the increased risk of adverse events such as cytochrome P450 interactions, serotonin syndrome, or hypertensive crisis (when switching to an MAO inhibitor).
Beware of interactions. All SSRIs and SNRIs are metabolized through the P450 system in the liver and therefore have the potential for drug-drug interactions. Care must be taken when giving these agents together with drugs whose metabolism can be altered by P450 inhibition. For TCAs, blood levels can be checked if there is concern about toxicity; however, dosing is not strictly based on this level. Great care should be taken if a TCA is given together with an SNRI or an SSRI, as the TCA blood level can become significantly elevated. This may result in QT interval prolongation, as mentioned earlier.
Refer. Referral to a psychiatrist is appropriate for patients for whom multiple classes have failed, for patients who have another psychiatric comorbidity (such as psychosis, hypomania, or mania), or for patients who may need hospitalization. Referral is also appropriate if the physician is concerned about suicide risk.
PATIENTS WITH MAJOR DEPRESSION ONLY
For a patient presenting with depression but no other significant medical comorbidity, the first-line therapy is often an SSRI. Several generic SSRIs are available, and some are on the discount lists at retail pharmacies.
Symptoms should start to improve in about 2 weeks, and the optimal response should be achieved in 4 to 6 weeks of treatment. If this does not occur, consider either adding an augmenting agent or switching to a different antidepressant.
PATIENTS WITH CHRONIC PAIN
Chronic pain and depression often go hand in hand and can potentiate each other. When considering an antidepressant in a patient who has both conditions, the SNRIs and TCAs are typically preferred. Some SNRIs, namely duloxetine and milnacipran, are approved for certain chronic pain conditions, such as fibromyalgia. SNRIs are frequently used off-label for other chronic pain conditions such as headache and neuropathic pain.2
TCAs such as amitriptyline, nortriptyline, and doxepin are also often used in patients with chronic pain. These agents, like the SNRIs, inhibit the reuptake of serotonin and norepinephrine and are used off-label for neuropathic pain,3,4 migraine, interstitial cystitis,5 and other pain conditions.6–9
For TCAs and SNRIs, the effective dose range for chronic pain overlaps that for depression. However, TCAs are often given at lower doses to patients without depression. We recommend starting at a low dose and slowly titrating upward to an effective dose. SNRIs are often preferred over TCAs because they do not have anticholinergic side effects and because an overdose is much less likely to be lethal.
PATIENTS WITH SEXUAL DYSFUNCTION
One of the more commonly reported side effects of antidepressants is sexual dysfunction, generally in the form of delayed orgasm or decreased libido.10 Typically, these complaints are attributed to SSRIs and SNRIs; however, TCAs and MAO inhibitors have also been associated wth sexual dysfunction.
Both erectile dysfunction and priapism have been linked to certain antidepressants. In particular, trazodone is a known cause of priapism. Even if using low doses for sleep, male patients should be made aware of this adverse effect.
Switching from one agent to another in the same class is not likely to improve sexual side effects. In particular, all the SSRIs are similar in their likelihood of causing sexual dysfunction. In a patient taking an SSRI who experiences this side effect, switching to bupropion11 or mirtazapine12 can be quite useful. Bupropion acts primarily on dopamine and norepinephrine, whereas mirtazapine acts on serotonin and norepinephrine but in a different manner from SSRIs and SNRIs.
Adjunctive treatment such as a cholinergic agonist, yohimbine (contraindicated with MAO inhibitors), a serotonergic agent (eg, buspirone), or a drug that acts on nitric oxide (eg, sildenafil, tadalafil) may have some utility but is often ineffective. Dose reduction, if possible, can be of value.
PATIENTS WITH ANXIETY
Many antidepressants are also approved for anxiety disorders, and still more are used off-label for this purpose. Anxiety and depression often occur together, so being able to treat both conditions with one drug can be quite useful.13 In general, the antidepressant effects are seen at lower doses of SSRIs and SNRIs, whereas more of the anxiolytic effects are seen at higher doses, particularly for obsessive-compulsive disorder.14
First-line treatment would be an SSRI or SNRI. Most anxiety disorders respond to either class, but there are some more-specific recommendations. SSRIs are best studied in panic disorder, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Fluoxetine, citalopram, escitalopram, and sertraline15 can all be effective in both major depressive disorder and generalized anxiety disorder. Panic disorder also tends to respond well to SSRIs. SNRIs have been evaluated primarily in generalized anxiety disorder but may also be useful in many of the other conditions.
Additionally, mirtazapine (used off-label)12 and the TCAs16–18 can help treat anxiety. Clomipramine is used to treat obsessive-compulsive disorder.19 These drugs are especially useful for nighttime anxiety, as they can aid sleep. Of note, the anxiolytic effect of mirtazapine may be greater at higher doses.
MAO inhibitors often go unused because of the dietary and medication restrictions involved. However, very refractory cases of certain anxiety disorders may respond preferentially to these agents.
Bupropion tends to be more activating than other antidepressants, so is often avoided in anxious patients. However, some research suggests this is not always necessary.20 If the anxiety is secondary to depression, it will often improve significantly with this agent.
When starting or increasing the dose of an antidepressant, patients may experience increased anxiety or feel “jittery.” This feeling usually passes within the first week of treatment, and it is important to inform patients about this effect. “Start low and go slow” in patients with significant comorbid anxiety. Temporarily using a benzodiazepine such as clonazepam may make the transition more tolerable.
PATIENTS WITH CHRONIC FATIGUE SYNDROME OR FIBROMYALGIA
Increasing recognition of both chronic fatigue syndrome and fibromyalgia has led to more proactive treatment for these disorders. Depression can go hand in hand with these disorders, and certain antidepressants, namely the SNRIs, can be useful in this population.
More data exist for the treatment of fibromyalgia. Both duloxetine and milnacipran are approved by the US Food and Drug Administration (FDA) for the treatment of fibromyalgia.21 Venlafaxine is also used off-label for this purpose. SSRIs such as fluoxetine and citalopram have had mixed results.21–23 TCAs have been used with some success; however, their side effects and lethal potential are often limiting.21,24,25 A recent study in Spain also suggested there may be benefit from using MAO inhibitors for fibromyalgia, but data are quite limited.26
The data for treating chronic fatigue syndrome with SSRIs, SNRIs, or MAO inhibitors are conflicting.27–29 However, managing the co-existing depression may provide some relief in and of itself.
PATIENTS WITH FREQUENT INSOMNIA
Insomnia can be a symptom of depression, but it can also be a side effect of certain antidepressants. The SSRIs and SNRIs can disrupt sleep patterns in some patients by shortening the rapid-eye-movement (REM) stage.30,31
In patients with severe insomnia, it may be best to first recommend taking the antidepressant in the morning if they notice worsening sleep after initiating treatment. Patients can be told with any antidepressant, “If it makes you tired, take it at night, and if it wakes you up, take it in the morning.” Of note, a recent South African study suggested that escitalopram may be able to improve sleep.32
If that does not solve the problem, there are other options. For instance, mirtazapine, particularly in doses of 15 mg or 30 mg, aids depression and insomnia. At higher doses (45 mg), the sleep-aiding effect may be reduced. Low doses of TCAs, particularly doxepin, maprotiline (technically speaking, a tetracyclic antidepressant), amitriptyline, and nortriptyline can be effective sleep aids. These agents may be used as an adjunct to another antidepressant to enhance sleep and mood. However, the TCAs also shorten the REM stage of sleep.33
The previously mentioned drug interactions with SSRIs and SNRIs also need to be considered. Caution should be used when discontinuing these medications, as patients may experience rebound symptoms in the form of much more vivid dreams. MAO inhibitors may worsen insomnia because they suppress REM sleep.34
Trazodone is another agent that at lower doses (25–150 mg) can be an effective, nonaddicting sleep aid. When used as an antidepressant, it is generally prescribed at higher doses (300–400 mg), but its sedating effects can be quite limiting at these levels. It is important to remember the possibility of priapism in male patients.
GERIATRIC PATIENTS
Old age brings its own set of concerns when treating depression. Elderly patients are more susceptible to potential bradycardia caused by SSRIs. The TCAs have the more worrisome cardiac side effect of QTc prolongation. TCAs can slow cognitive function, whereas the SSRIs, bupropion, and the SNRIs tend not to affect cognition. Escitalopram and duloxetine have been suggested to be particularly effective in the elderly.35,36 A study from the Netherlands linked SSRIs with increased risk of falling in geriatric patients with dementia.37 Constipation, which could lead to ileus, is increased with TCAs and certain other agents (ie, paroxetine) in the geriatric population.
Mirtazapine is often very useful in elderly patients for many reasons: it treats both anxiety and depression, stimulates appetite and weight gain, can help with nausea, and is an effective sleep aid. Concerns about weight, appetite, and sleep are particularly common in the elderly, whereas younger patients can be less tolerant of drugs that make them gain weight and sleep more. Normal age-related changes to the sleep cycle contribute to decreased satisfaction with sleep as we age. In addition, depression often further impairs sleep. So, in the elderly, optimizing sleep is key. Research has also shown mirtazapine to be effective in patients with both Alzheimer dementia and depression.38
DIABETIC PATIENTS
One of the more worrisome side effects of psychiatric medications in diabetic patients is weight gain. Certain antidepressants have a greater propensity for weight gain and should likely be avoided as first-line treatments in this population.12 Typically, these agents include those that have more antihistamine action such as paroxetine and the TCAs. These agents also may lead to constipation, which could potentially worsen gastroparesis. Mirtazapine and the MAO inhibitors are also known to cause weight gain.
Bupropion and nefazodone are the most weight-neutral of all antidepressants. Nefazodone has fallen out of favor because of its potential to cause fulminant liver failure in rare cases. However, it remains a reasonable option for patients with comorbid anxiety and depression who have significant weight gain with other agents.
SSRIs and MAO inhibitors may improve or be neutral toward glucose metabolism, and some data suggest that SNRIs may impair this process.39
PATIENTS WITH CARDIAC CONDITIONS
Major depression often coexists with cardiac conditions. In particular, many patients develop depression after suffering a myocardial infarction, and increasingly they are being treated for it.40 Treatment in this situation is appropriate, since depression, if untreated, can increase the risk of recurrence of myocardial infarction.41
However, there are many concerns that accompany treating depression in cardiac patients. Therefore, a baseline electrocardiogram should be obtained before starting an antidepressant.
TCAs and tetracyclic agents have a tendency to prolong the QTc interval and potentiate ventricular arrhythmias,42 so it may be prudent to avoid these in patients at risk. These agents can also significantly increase the pulse rate. This tachycardia increases the risk of angina or myocardial infarction from the anticholinergic effects of these drugs.
In February 2013, the FDA issued a warning about possible arrhythmias with citalopram at doses greater than 40 mg in adult patients43; however, research has suggested citalopram is effective in treating depression in cardiac patients.44 Research has not shown an increase in efficacy at doses greater than 40 mg daily, so we recommend following the black-box warning.
TCAs and MAO inhibitors can also cause orthostatic hypotension. On the other hand, consuming large amounts of tyramine, in foods such as aged cheese, can precipitate a hypertensive crisis in patients taking MAO inhibitors.
Which antidepressants tend to be safer in cardiac patients? Sertraline has been shown to be safe in congestive heart failure and coronary artery disease,45–47 but the SSRIs are typically safe. Fluoxetine has shown efficacy in patients who have had a myocardial infarction.48 Mirtazapine has also been shown to be efficacious in cardiac patients.49 Nefazodone, mirtazapine, bupropion, SSRIs, and SNRIs have little or no tendency toward orthostatic hypotension.
With the variety of drugs available for treating depression, choosing one can be daunting. Different agents have characteristics that may make them a better choice for different types of patients, but even so, treating any kind of mental illness often requires an element of trial and error.
Primary care providers are on the frontline of treating mental illness, often evaluating patients before they are seen by a psychiatrist. The purpose of this article is to provide insight into the art of prescribing antidepressants in the primary care setting. We will discuss common patient presentations, including depressed patients without other medical comorbidities as well as those with common comorbidities, with our recommendations for first-line treatment.
We hope our recommendations will help you to navigate the uncertainty more confidently, resulting in more efficient and tailored treatment for your patients.
BASELINE TESTING
When starting a patient on antidepressant drug therapy, we recommend obtaining a set of baseline laboratory tests to rule out underlying medical conditions that may be contributing to the patient’s depression or that may preclude the use of a given drug. (For example, elevation of liver enzymes may preclude the use of duloxetine.) Tests should include:
- A complete blood cell count
- A complete metabolic panel
- A thyroid-stimulating hormone level.
Electrocardiography may also be useful, as some antidepressants can prolong the QT interval or elevate the blood levels of other drugs with this effect.
GENERAL TREATMENT CONSIDERATIONS
There are several classes of antidepressants, and each class has a number of agents. Research has found little difference in efficacy among agents. So to simplify choosing which one to use, we recommend becoming comfortable with an agent from each class, ie:
- A selective serotonin reuptake inhibitor (SSRI)
- A selective serotonin-norepinephrine reuptake inhibitor (SNRI)
- A tricyclic antidepressant (TCA)
- A monoamine oxidase (MAO) inhibitor.
Each class includes generic agents, many of which are on the discount lists of retail pharmacies. Table 1 shows representative drugs from each class, with their relative costs.
Start low and go slow. In general, when starting an antidepressant, consider starting at half the normal dose, titrating upward as tolerated about every 14 days. This approach can minimize side effects. For example, if prescribing fluoxetine, start with 10 mg and titrate every 2 weeks based on tolerance and patient response. That said, each patient may respond differently, requiring perhaps a lower starting dose or a longer titration schedule.
Anticipate side effects. Most of the side effects of an antidepressant drug can be explained by its mechanism of action. Although side effects should certainly be considered when choosing an agent, patients can be reassured that most are transient and benign. A detailed discussion of side effects of antidepressant drugs is beyond the scope of this article, but a review by Khawam et al1 was published earlier in this journal.
Reassess. If after 4 to 6 weeks the patient has had little or no response, it is reasonable to switch agents. For a patient who was on an SSRI, the change can be to another SSRI or to an SNRI. However, if two SSRIs have already failed, then choose an SNRI. Agents are commonly cross-tapered during the switch to avoid abrupt cessation of one drug or the increased risk of adverse events such as cytochrome P450 interactions, serotonin syndrome, or hypertensive crisis (when switching to an MAO inhibitor).
Beware of interactions. All SSRIs and SNRIs are metabolized through the P450 system in the liver and therefore have the potential for drug-drug interactions. Care must be taken when giving these agents together with drugs whose metabolism can be altered by P450 inhibition. For TCAs, blood levels can be checked if there is concern about toxicity; however, dosing is not strictly based on this level. Great care should be taken if a TCA is given together with an SNRI or an SSRI, as the TCA blood level can become significantly elevated. This may result in QT interval prolongation, as mentioned earlier.
Refer. Referral to a psychiatrist is appropriate for patients for whom multiple classes have failed, for patients who have another psychiatric comorbidity (such as psychosis, hypomania, or mania), or for patients who may need hospitalization. Referral is also appropriate if the physician is concerned about suicide risk.
PATIENTS WITH MAJOR DEPRESSION ONLY
For a patient presenting with depression but no other significant medical comorbidity, the first-line therapy is often an SSRI. Several generic SSRIs are available, and some are on the discount lists at retail pharmacies.
Symptoms should start to improve in about 2 weeks, and the optimal response should be achieved in 4 to 6 weeks of treatment. If this does not occur, consider either adding an augmenting agent or switching to a different antidepressant.
PATIENTS WITH CHRONIC PAIN
Chronic pain and depression often go hand in hand and can potentiate each other. When considering an antidepressant in a patient who has both conditions, the SNRIs and TCAs are typically preferred. Some SNRIs, namely duloxetine and milnacipran, are approved for certain chronic pain conditions, such as fibromyalgia. SNRIs are frequently used off-label for other chronic pain conditions such as headache and neuropathic pain.2
TCAs such as amitriptyline, nortriptyline, and doxepin are also often used in patients with chronic pain. These agents, like the SNRIs, inhibit the reuptake of serotonin and norepinephrine and are used off-label for neuropathic pain,3,4 migraine, interstitial cystitis,5 and other pain conditions.6–9
For TCAs and SNRIs, the effective dose range for chronic pain overlaps that for depression. However, TCAs are often given at lower doses to patients without depression. We recommend starting at a low dose and slowly titrating upward to an effective dose. SNRIs are often preferred over TCAs because they do not have anticholinergic side effects and because an overdose is much less likely to be lethal.
PATIENTS WITH SEXUAL DYSFUNCTION
One of the more commonly reported side effects of antidepressants is sexual dysfunction, generally in the form of delayed orgasm or decreased libido.10 Typically, these complaints are attributed to SSRIs and SNRIs; however, TCAs and MAO inhibitors have also been associated wth sexual dysfunction.
Both erectile dysfunction and priapism have been linked to certain antidepressants. In particular, trazodone is a known cause of priapism. Even if using low doses for sleep, male patients should be made aware of this adverse effect.
Switching from one agent to another in the same class is not likely to improve sexual side effects. In particular, all the SSRIs are similar in their likelihood of causing sexual dysfunction. In a patient taking an SSRI who experiences this side effect, switching to bupropion11 or mirtazapine12 can be quite useful. Bupropion acts primarily on dopamine and norepinephrine, whereas mirtazapine acts on serotonin and norepinephrine but in a different manner from SSRIs and SNRIs.
Adjunctive treatment such as a cholinergic agonist, yohimbine (contraindicated with MAO inhibitors), a serotonergic agent (eg, buspirone), or a drug that acts on nitric oxide (eg, sildenafil, tadalafil) may have some utility but is often ineffective. Dose reduction, if possible, can be of value.
PATIENTS WITH ANXIETY
Many antidepressants are also approved for anxiety disorders, and still more are used off-label for this purpose. Anxiety and depression often occur together, so being able to treat both conditions with one drug can be quite useful.13 In general, the antidepressant effects are seen at lower doses of SSRIs and SNRIs, whereas more of the anxiolytic effects are seen at higher doses, particularly for obsessive-compulsive disorder.14
First-line treatment would be an SSRI or SNRI. Most anxiety disorders respond to either class, but there are some more-specific recommendations. SSRIs are best studied in panic disorder, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Fluoxetine, citalopram, escitalopram, and sertraline15 can all be effective in both major depressive disorder and generalized anxiety disorder. Panic disorder also tends to respond well to SSRIs. SNRIs have been evaluated primarily in generalized anxiety disorder but may also be useful in many of the other conditions.
Additionally, mirtazapine (used off-label)12 and the TCAs16–18 can help treat anxiety. Clomipramine is used to treat obsessive-compulsive disorder.19 These drugs are especially useful for nighttime anxiety, as they can aid sleep. Of note, the anxiolytic effect of mirtazapine may be greater at higher doses.
MAO inhibitors often go unused because of the dietary and medication restrictions involved. However, very refractory cases of certain anxiety disorders may respond preferentially to these agents.
Bupropion tends to be more activating than other antidepressants, so is often avoided in anxious patients. However, some research suggests this is not always necessary.20 If the anxiety is secondary to depression, it will often improve significantly with this agent.
When starting or increasing the dose of an antidepressant, patients may experience increased anxiety or feel “jittery.” This feeling usually passes within the first week of treatment, and it is important to inform patients about this effect. “Start low and go slow” in patients with significant comorbid anxiety. Temporarily using a benzodiazepine such as clonazepam may make the transition more tolerable.
PATIENTS WITH CHRONIC FATIGUE SYNDROME OR FIBROMYALGIA
Increasing recognition of both chronic fatigue syndrome and fibromyalgia has led to more proactive treatment for these disorders. Depression can go hand in hand with these disorders, and certain antidepressants, namely the SNRIs, can be useful in this population.
More data exist for the treatment of fibromyalgia. Both duloxetine and milnacipran are approved by the US Food and Drug Administration (FDA) for the treatment of fibromyalgia.21 Venlafaxine is also used off-label for this purpose. SSRIs such as fluoxetine and citalopram have had mixed results.21–23 TCAs have been used with some success; however, their side effects and lethal potential are often limiting.21,24,25 A recent study in Spain also suggested there may be benefit from using MAO inhibitors for fibromyalgia, but data are quite limited.26
The data for treating chronic fatigue syndrome with SSRIs, SNRIs, or MAO inhibitors are conflicting.27–29 However, managing the co-existing depression may provide some relief in and of itself.
PATIENTS WITH FREQUENT INSOMNIA
Insomnia can be a symptom of depression, but it can also be a side effect of certain antidepressants. The SSRIs and SNRIs can disrupt sleep patterns in some patients by shortening the rapid-eye-movement (REM) stage.30,31
In patients with severe insomnia, it may be best to first recommend taking the antidepressant in the morning if they notice worsening sleep after initiating treatment. Patients can be told with any antidepressant, “If it makes you tired, take it at night, and if it wakes you up, take it in the morning.” Of note, a recent South African study suggested that escitalopram may be able to improve sleep.32
If that does not solve the problem, there are other options. For instance, mirtazapine, particularly in doses of 15 mg or 30 mg, aids depression and insomnia. At higher doses (45 mg), the sleep-aiding effect may be reduced. Low doses of TCAs, particularly doxepin, maprotiline (technically speaking, a tetracyclic antidepressant), amitriptyline, and nortriptyline can be effective sleep aids. These agents may be used as an adjunct to another antidepressant to enhance sleep and mood. However, the TCAs also shorten the REM stage of sleep.33
The previously mentioned drug interactions with SSRIs and SNRIs also need to be considered. Caution should be used when discontinuing these medications, as patients may experience rebound symptoms in the form of much more vivid dreams. MAO inhibitors may worsen insomnia because they suppress REM sleep.34
Trazodone is another agent that at lower doses (25–150 mg) can be an effective, nonaddicting sleep aid. When used as an antidepressant, it is generally prescribed at higher doses (300–400 mg), but its sedating effects can be quite limiting at these levels. It is important to remember the possibility of priapism in male patients.
GERIATRIC PATIENTS
Old age brings its own set of concerns when treating depression. Elderly patients are more susceptible to potential bradycardia caused by SSRIs. The TCAs have the more worrisome cardiac side effect of QTc prolongation. TCAs can slow cognitive function, whereas the SSRIs, bupropion, and the SNRIs tend not to affect cognition. Escitalopram and duloxetine have been suggested to be particularly effective in the elderly.35,36 A study from the Netherlands linked SSRIs with increased risk of falling in geriatric patients with dementia.37 Constipation, which could lead to ileus, is increased with TCAs and certain other agents (ie, paroxetine) in the geriatric population.
Mirtazapine is often very useful in elderly patients for many reasons: it treats both anxiety and depression, stimulates appetite and weight gain, can help with nausea, and is an effective sleep aid. Concerns about weight, appetite, and sleep are particularly common in the elderly, whereas younger patients can be less tolerant of drugs that make them gain weight and sleep more. Normal age-related changes to the sleep cycle contribute to decreased satisfaction with sleep as we age. In addition, depression often further impairs sleep. So, in the elderly, optimizing sleep is key. Research has also shown mirtazapine to be effective in patients with both Alzheimer dementia and depression.38
DIABETIC PATIENTS
One of the more worrisome side effects of psychiatric medications in diabetic patients is weight gain. Certain antidepressants have a greater propensity for weight gain and should likely be avoided as first-line treatments in this population.12 Typically, these agents include those that have more antihistamine action such as paroxetine and the TCAs. These agents also may lead to constipation, which could potentially worsen gastroparesis. Mirtazapine and the MAO inhibitors are also known to cause weight gain.
Bupropion and nefazodone are the most weight-neutral of all antidepressants. Nefazodone has fallen out of favor because of its potential to cause fulminant liver failure in rare cases. However, it remains a reasonable option for patients with comorbid anxiety and depression who have significant weight gain with other agents.
SSRIs and MAO inhibitors may improve or be neutral toward glucose metabolism, and some data suggest that SNRIs may impair this process.39
PATIENTS WITH CARDIAC CONDITIONS
Major depression often coexists with cardiac conditions. In particular, many patients develop depression after suffering a myocardial infarction, and increasingly they are being treated for it.40 Treatment in this situation is appropriate, since depression, if untreated, can increase the risk of recurrence of myocardial infarction.41
However, there are many concerns that accompany treating depression in cardiac patients. Therefore, a baseline electrocardiogram should be obtained before starting an antidepressant.
TCAs and tetracyclic agents have a tendency to prolong the QTc interval and potentiate ventricular arrhythmias,42 so it may be prudent to avoid these in patients at risk. These agents can also significantly increase the pulse rate. This tachycardia increases the risk of angina or myocardial infarction from the anticholinergic effects of these drugs.
In February 2013, the FDA issued a warning about possible arrhythmias with citalopram at doses greater than 40 mg in adult patients43; however, research has suggested citalopram is effective in treating depression in cardiac patients.44 Research has not shown an increase in efficacy at doses greater than 40 mg daily, so we recommend following the black-box warning.
TCAs and MAO inhibitors can also cause orthostatic hypotension. On the other hand, consuming large amounts of tyramine, in foods such as aged cheese, can precipitate a hypertensive crisis in patients taking MAO inhibitors.
Which antidepressants tend to be safer in cardiac patients? Sertraline has been shown to be safe in congestive heart failure and coronary artery disease,45–47 but the SSRIs are typically safe. Fluoxetine has shown efficacy in patients who have had a myocardial infarction.48 Mirtazapine has also been shown to be efficacious in cardiac patients.49 Nefazodone, mirtazapine, bupropion, SSRIs, and SNRIs have little or no tendency toward orthostatic hypotension.
- Khawam EA, Laurencic G, Malone DA. Side effects of antidepressants: an overview. Cleve Clin J Med 2006; 73:351–361.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab Res Rev 2008; 24(suppl 1):S52–S57.
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81:1372–1373.
- Tanenberg RJ, Irving GA, Risser RC, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86:615–626.
- Hertle L, van Ophoven A. Long-term results of amitriptyline treatment for interstitial cystitis. Aktuelle Urol 2010; 41(suppl 1):S61–S65.
- Nguyen TM, Eslick GD. Systematic review: the treatment of noncardiac chest pain with antidepressants. Aliment Pharmacol Ther 2012; 35:493–500.
- Lee H, Kim JH, Min BH, et al. Efficacy of venlafaxine for symptomatic relief in young adult patients with functional chest pain: a randomized, double-blind, placebo-controlled, crossover trial. Am J Gastroenterol 2010; 105:1504–1512.
- Varia I, Logue E, O’Connor C, et al. Randomized trial of sertraline in patients with unexplained chest pain of noncardiac origin. Am Heart J 2000; 140:367–372.
- Doraiswamy PM, Varia I, Hellegers C, et al. A randomized controlled trial of paroxetine for noncardiac chest pain. Psychopharmacol Bull 2006; 39:15–24.
- Clayton AH. Understanding antidepressant mechanism of action and its effect on efficacy and safety. J Clin Psychiatry 2012; 73:e11.
- Gartlehner G, Hansen RA, Morgan LC, et al. Second-generation antidepressants in the pharmacologic treatment of adult depression: an update of the 2007 comparative effectiveness review (Internet). Rockville (MD): Agency for Healthcare Research and Quality (US); 2011 Dec. Comparative Effectiveness Reviews, No. 46. http://www.ncbi.nlm.nih.gov/books/NBK83442/. Accessed February 27, 2013.
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev 2011; 12:CD006528.
- Hofmeijer-Sevink MK, Batelaan NM, van Megen HJ, et al. Clinical relevance of comorbidity in anxiety disorders: a report from the Netherlands Study of Depression and Anxiety (NESDA). J Affect Disord 2012; 137:106–112.
- Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci 2011; 13:423–437.
- Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol 2009; 24:43–60.
- Huh J, Goebert D, Takeshita J, Lu BY, Kang M. Treatment of generalized anxiety disorder: a comprehensive review of the literature for psychopharmacologic alternatives to newer antidepressants and benzodiazepines. Prim Care Companion CNS Disord 2011; 13: 4088/PCC.08r00709blu.
- Rickels K, Downing R, Schweizer E, Hassman H. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry 1993; 50:884–895.
- Uher R, Maier W, Hauser J, et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259.
- Kellner M. Drug treatment of obsessive-compulsive disorder. Dialogues Clin Neurosci 2010; 12:187–197.
- Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology 2001; 25:131–138.
- Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol 2011; 25:285–297.
- Wolfe F, Cathey MA, Hawley DJ. A double-blind placebo controlled trial of fluoxetine in fibromyalgia. Scand J Rheumatol 1994; 23:255–259.
- Arnold LM, Hess EV, Hudson JI, Welge JA, Berno SE, Keck PE. A randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med 2002; 112:191–197.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41:104–113.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Tort S, Urrútia G, Nishishinya MB, Walitt B. Monoamine oxidase inhibitors (MAOIs) for fibromyalgia syndrome. Cochrane Database Syst Rev 2012; 4:CD009807.
- Vercoulen JH, Swanink CM, Zitman FG, et al. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996; 347:858–861.
- Natelson BH, Cheu J, Pareja J, Ellis SP, Policastro T, Findley TW. Randomized, double blind, controlled placebo-phase in trial of low dose phenelzine in the chronic fatigue syndrome. Psychopharmacology (Berl) 1996; 124:226–230.
- Reid S, Chalder T, Cleare A, Hotopf M, Wessely S. Chronic fatigue syndrome. BMJ 2000; 320:292–296.
- Kupfer DJ, Spiker DG, Coble PA, Neil JF, Ulrich R, Shaw DH. Sleep and treatment prediction in endogenous depression. Am J Psychiatry 1981; 138:429–434.
- Argyropoulos SV, Hicks JA, Nash JR, et al. Redistribution of slow wave activity of sleep during pharmacological treatment of depression with paroxetine but not with nefazodone. J Sleep Res 2009; 18:342–348.
- Stein DJ, Lopez AG. Effects of escitalopram on sleep problems in patients with major depression or generalized anxiety disorder. Adv Ther 2011; 28:1021–1037.
- Ehlers CL, Havstad JW, Kupfer DJ. Estimation of the time course of slow-wave sleep over the night in depressed patients: effects of clomipramine and clinical response. Biol Psychiatry 1996; 39:171–181.
- Landolt HP, Raimo EB, Schnierow BJ, Kelsoe JR, Rapaport MH, Gillin JC. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch Gen Psychiatry 2001; 58:268–276.
- Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res 2011; 39:1946–1953.
- Dolder C, Nelson M, Stump A. Pharmacological and clinical profile of newer antidepressants: implications for the treatment of elderly patients. Drugs Aging 2010; 27:625–640.
- Sterke CS, Ziere G, van Beeck EF, Looman CW, van der Cammen TJ. Dose-response relationship between selective serotonin re-uptake inhibitors and injurious falls: a study in nursing home residents with dementia. Br J Clin Pharmacol 2012; 73:812–820.
- Raji MA, Brady SR. Mirtazapine for treatment of depression and comorbidities in Alzheimer disease. Ann Pharmacother 2001; 35:1024–1027.
- Hennings JM, Schaaf L, Fulda S. Glucose metabolism and antidepressant medication. Curr Pharm Des 2012; 18:5900–5919.
- Czarny MJ, Arthurs E, Coffie DF, et al. Prevalence of antidepressant prescription or use in patients with acute coronary syndrome: a systematic review. PLoS One 2011; 6:e27671.
- Zuidersma M, Ormel J, Conradi HJ, de Jonge P. An increase in depressive symptoms after myocardial infarction predicts new cardiac events irrespective of depressive symptoms before myocardial infarction. Psychol Med 2012; 42:683–693.
- van Noord C, Straus SM, Sturkenboom MC, et al. Psychotropic drugs associated with corrected QT interval prolongation. J Clin Psychopharmacol 2009; 29:9–15.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: abnormal heart rhythms associated with high doses of Celexa (citalopram hydrobromide). http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed August 25, 2013.
- Lespérance F, Frasure-Smith N, Koszycki D, et al; CREATE Investigators. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- O’Connor CM, Jiang W, Kuchibhatla M, et al; SADHART-CHF Investigators. Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Glassman AH, O’Connor CM, Califf RM, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Swenson JR, O’Connor CM, Barton D, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol 2003; 92:1271–1276.
- Strik JJ, Honig A, Lousberg R, et al. Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med 2000; 62:783–789.
- Honig A, Kuyper AM, Schene AH, et al; MIND-IT investigators. Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med 2007; 69:606–613.
- Khawam EA, Laurencic G, Malone DA. Side effects of antidepressants: an overview. Cleve Clin J Med 2006; 73:351–361.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab Res Rev 2008; 24(suppl 1):S52–S57.
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81:1372–1373.
- Tanenberg RJ, Irving GA, Risser RC, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86:615–626.
- Hertle L, van Ophoven A. Long-term results of amitriptyline treatment for interstitial cystitis. Aktuelle Urol 2010; 41(suppl 1):S61–S65.
- Nguyen TM, Eslick GD. Systematic review: the treatment of noncardiac chest pain with antidepressants. Aliment Pharmacol Ther 2012; 35:493–500.
- Lee H, Kim JH, Min BH, et al. Efficacy of venlafaxine for symptomatic relief in young adult patients with functional chest pain: a randomized, double-blind, placebo-controlled, crossover trial. Am J Gastroenterol 2010; 105:1504–1512.
- Varia I, Logue E, O’Connor C, et al. Randomized trial of sertraline in patients with unexplained chest pain of noncardiac origin. Am Heart J 2000; 140:367–372.
- Doraiswamy PM, Varia I, Hellegers C, et al. A randomized controlled trial of paroxetine for noncardiac chest pain. Psychopharmacol Bull 2006; 39:15–24.
- Clayton AH. Understanding antidepressant mechanism of action and its effect on efficacy and safety. J Clin Psychiatry 2012; 73:e11.
- Gartlehner G, Hansen RA, Morgan LC, et al. Second-generation antidepressants in the pharmacologic treatment of adult depression: an update of the 2007 comparative effectiveness review (Internet). Rockville (MD): Agency for Healthcare Research and Quality (US); 2011 Dec. Comparative Effectiveness Reviews, No. 46. http://www.ncbi.nlm.nih.gov/books/NBK83442/. Accessed February 27, 2013.
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev 2011; 12:CD006528.
- Hofmeijer-Sevink MK, Batelaan NM, van Megen HJ, et al. Clinical relevance of comorbidity in anxiety disorders: a report from the Netherlands Study of Depression and Anxiety (NESDA). J Affect Disord 2012; 137:106–112.
- Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci 2011; 13:423–437.
- Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol 2009; 24:43–60.
- Huh J, Goebert D, Takeshita J, Lu BY, Kang M. Treatment of generalized anxiety disorder: a comprehensive review of the literature for psychopharmacologic alternatives to newer antidepressants and benzodiazepines. Prim Care Companion CNS Disord 2011; 13: 4088/PCC.08r00709blu.
- Rickels K, Downing R, Schweizer E, Hassman H. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry 1993; 50:884–895.
- Uher R, Maier W, Hauser J, et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259.
- Kellner M. Drug treatment of obsessive-compulsive disorder. Dialogues Clin Neurosci 2010; 12:187–197.
- Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology 2001; 25:131–138.
- Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol 2011; 25:285–297.
- Wolfe F, Cathey MA, Hawley DJ. A double-blind placebo controlled trial of fluoxetine in fibromyalgia. Scand J Rheumatol 1994; 23:255–259.
- Arnold LM, Hess EV, Hudson JI, Welge JA, Berno SE, Keck PE. A randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med 2002; 112:191–197.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41:104–113.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Tort S, Urrútia G, Nishishinya MB, Walitt B. Monoamine oxidase inhibitors (MAOIs) for fibromyalgia syndrome. Cochrane Database Syst Rev 2012; 4:CD009807.
- Vercoulen JH, Swanink CM, Zitman FG, et al. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996; 347:858–861.
- Natelson BH, Cheu J, Pareja J, Ellis SP, Policastro T, Findley TW. Randomized, double blind, controlled placebo-phase in trial of low dose phenelzine in the chronic fatigue syndrome. Psychopharmacology (Berl) 1996; 124:226–230.
- Reid S, Chalder T, Cleare A, Hotopf M, Wessely S. Chronic fatigue syndrome. BMJ 2000; 320:292–296.
- Kupfer DJ, Spiker DG, Coble PA, Neil JF, Ulrich R, Shaw DH. Sleep and treatment prediction in endogenous depression. Am J Psychiatry 1981; 138:429–434.
- Argyropoulos SV, Hicks JA, Nash JR, et al. Redistribution of slow wave activity of sleep during pharmacological treatment of depression with paroxetine but not with nefazodone. J Sleep Res 2009; 18:342–348.
- Stein DJ, Lopez AG. Effects of escitalopram on sleep problems in patients with major depression or generalized anxiety disorder. Adv Ther 2011; 28:1021–1037.
- Ehlers CL, Havstad JW, Kupfer DJ. Estimation of the time course of slow-wave sleep over the night in depressed patients: effects of clomipramine and clinical response. Biol Psychiatry 1996; 39:171–181.
- Landolt HP, Raimo EB, Schnierow BJ, Kelsoe JR, Rapaport MH, Gillin JC. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch Gen Psychiatry 2001; 58:268–276.
- Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res 2011; 39:1946–1953.
- Dolder C, Nelson M, Stump A. Pharmacological and clinical profile of newer antidepressants: implications for the treatment of elderly patients. Drugs Aging 2010; 27:625–640.
- Sterke CS, Ziere G, van Beeck EF, Looman CW, van der Cammen TJ. Dose-response relationship between selective serotonin re-uptake inhibitors and injurious falls: a study in nursing home residents with dementia. Br J Clin Pharmacol 2012; 73:812–820.
- Raji MA, Brady SR. Mirtazapine for treatment of depression and comorbidities in Alzheimer disease. Ann Pharmacother 2001; 35:1024–1027.
- Hennings JM, Schaaf L, Fulda S. Glucose metabolism and antidepressant medication. Curr Pharm Des 2012; 18:5900–5919.
- Czarny MJ, Arthurs E, Coffie DF, et al. Prevalence of antidepressant prescription or use in patients with acute coronary syndrome: a systematic review. PLoS One 2011; 6:e27671.
- Zuidersma M, Ormel J, Conradi HJ, de Jonge P. An increase in depressive symptoms after myocardial infarction predicts new cardiac events irrespective of depressive symptoms before myocardial infarction. Psychol Med 2012; 42:683–693.
- van Noord C, Straus SM, Sturkenboom MC, et al. Psychotropic drugs associated with corrected QT interval prolongation. J Clin Psychopharmacol 2009; 29:9–15.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: abnormal heart rhythms associated with high doses of Celexa (citalopram hydrobromide). http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed August 25, 2013.
- Lespérance F, Frasure-Smith N, Koszycki D, et al; CREATE Investigators. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- O’Connor CM, Jiang W, Kuchibhatla M, et al; SADHART-CHF Investigators. Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Glassman AH, O’Connor CM, Califf RM, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Swenson JR, O’Connor CM, Barton D, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol 2003; 92:1271–1276.
- Strik JJ, Honig A, Lousberg R, et al. Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med 2000; 62:783–789.
- Honig A, Kuyper AM, Schene AH, et al; MIND-IT investigators. Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med 2007; 69:606–613.
KEY POINTS
- We suggest that clinicians become familiar with one drug from each class of antidepressants.
- Many antidepressants are also approved for conditions other than depression, and for patients who have both depression and one or more of these comcomitant conditions, these drugs can have a “two-for-one” benefit.
- Adverse effects of an antidepressant are usually predictable on the basis of the drug’s mechanism of action.
Peripheral opacity on plain chest radiography
An 82-year-old woman was admitted to the hospital with dyspnea and chest discomfort over the past 24 hours. She was known to have paroxysmal atrial fibrillation and was taking warfarin, but that had been stopped 2 weeks earlier because of an acute ischemic stroke.
At the time of admission, she had no fever, cough, orthopnea, or leg swelling. Her physical activity was restricted, with residual right-sided weakness after her stroke. Her heart rate was 125 bpm; her oxygen saturation level was 98% on 2 L of oxygen per minute via nasal cannula. She had an irregularly irregular rhythm, a jugular venous pressure of 7 cm H2O, and no cardiac murmurs. Lung sounds were reduced at the bases, with faint crepitations.
Her hemoglobin concentration and white blood cell count were normal. Her brain-natriuretic peptide level was elevated at 2,648 pg/mL (reference range < 167), but cardiac enzyme levels were normal.
Electrocardiography showed atrial fibrillation with rapid ventricular response.
Plain chest radiography showed a 3-cm wedge-shaped opacity in the right mid-thorax (Figure 1), a finding known as the Hampton hump—a sign of pulmonary infarction caused by embolism.
Contrast-enhanced computed tomography (CT) of the chest showed acute thromboembolism in the right interlobar artery and wedge-shaped consolidation in the right-middle lobe (Figure 2), indicating pulmonary infarction.
Brain CT showed a stable infarction. Anticoagulation was restarted, and the patient was discharged in stable condition.
THE HAMPTON HUMP IN PULMONARY EMBOLISM
Because the lungs have a dual blood supply, pulmonary infarction is seen in only a minority of cases of pulmonary embolism. Infarction is more common in patients with peripheral pulmonary embolism, owing to the rapid inflow of bronchial blood, and in patients with medical comorbidities such as heart failure and chronic lung disease.2
The Hampton hump, first described by Aubrey Otis Hampton in 1940, is a peripheral (pleural-based) opacity that represents alveolar hemorrhage from underlying pulmonary infarction. It is one of several radiographic features that have been associated with pulmonary embolism; another is the Westermark sign, indicating oligemia.3
Worsley et al4 examined the diagnostic value of these radiographic features and found that the Hampton hump had a sensitivity of 22% and a specificity of 82% for detecting pulmonary embolism in the right hemithorax, and 24% and 82%, respectively, in the left hemithorax. The prevalence of pleural-based opacities was not significantly different in patients with or without pulmonary embolism. The authors concluded that chest radiography has limited diagnostic value in excluding or diagnosing pulmonary embolism.
In contrast, computed tomographic pulmonary angiography is the first-line imaging test in patients with suspected pulmonary embolism, because of its high sensitivity and specificity.1
We were not specifically looking for a pulmonary embolism when we found this new opacity on our patient’s radiograph, but this prompted further imaging, which led to the diagnosis. Although a near-normal chest radiograph is the most common radiologic finding in pulmonary embolism, this case shows how careful observation can detect unusual signs.
- Mos IC, Klok FA, Kroft LJ, de Roos A, Huisman MV. Imaging tests in the diagnosis of pulmonary embolism. Semin Respir Crit Care Med 2012; 33:138–143.
- Cha SI, Shin KM, Lee J, et al. Clinical relevance of pulmonary infarction in patients with pulmonary embolism. Thromb Res 2012; 130:e1–e5.
- Algın O, GÖkalp G, Topal U. Signs in chest imaging. Diagn Interv Radiol 2011; 17:18–29.
- Worsley DF, Alavi A, Aronchick JM, Chen JT, Greenspan RH, Ravin CE. Chest radiographic findings in patients with acute pulmonary embolism: observations from the PIOPED study. Radiology 1993; 189:133–136.
An 82-year-old woman was admitted to the hospital with dyspnea and chest discomfort over the past 24 hours. She was known to have paroxysmal atrial fibrillation and was taking warfarin, but that had been stopped 2 weeks earlier because of an acute ischemic stroke.
At the time of admission, she had no fever, cough, orthopnea, or leg swelling. Her physical activity was restricted, with residual right-sided weakness after her stroke. Her heart rate was 125 bpm; her oxygen saturation level was 98% on 2 L of oxygen per minute via nasal cannula. She had an irregularly irregular rhythm, a jugular venous pressure of 7 cm H2O, and no cardiac murmurs. Lung sounds were reduced at the bases, with faint crepitations.
Her hemoglobin concentration and white blood cell count were normal. Her brain-natriuretic peptide level was elevated at 2,648 pg/mL (reference range < 167), but cardiac enzyme levels were normal.
Electrocardiography showed atrial fibrillation with rapid ventricular response.
Plain chest radiography showed a 3-cm wedge-shaped opacity in the right mid-thorax (Figure 1), a finding known as the Hampton hump—a sign of pulmonary infarction caused by embolism.
Contrast-enhanced computed tomography (CT) of the chest showed acute thromboembolism in the right interlobar artery and wedge-shaped consolidation in the right-middle lobe (Figure 2), indicating pulmonary infarction.
Brain CT showed a stable infarction. Anticoagulation was restarted, and the patient was discharged in stable condition.
THE HAMPTON HUMP IN PULMONARY EMBOLISM
Because the lungs have a dual blood supply, pulmonary infarction is seen in only a minority of cases of pulmonary embolism. Infarction is more common in patients with peripheral pulmonary embolism, owing to the rapid inflow of bronchial blood, and in patients with medical comorbidities such as heart failure and chronic lung disease.2
The Hampton hump, first described by Aubrey Otis Hampton in 1940, is a peripheral (pleural-based) opacity that represents alveolar hemorrhage from underlying pulmonary infarction. It is one of several radiographic features that have been associated with pulmonary embolism; another is the Westermark sign, indicating oligemia.3
Worsley et al4 examined the diagnostic value of these radiographic features and found that the Hampton hump had a sensitivity of 22% and a specificity of 82% for detecting pulmonary embolism in the right hemithorax, and 24% and 82%, respectively, in the left hemithorax. The prevalence of pleural-based opacities was not significantly different in patients with or without pulmonary embolism. The authors concluded that chest radiography has limited diagnostic value in excluding or diagnosing pulmonary embolism.
In contrast, computed tomographic pulmonary angiography is the first-line imaging test in patients with suspected pulmonary embolism, because of its high sensitivity and specificity.1
We were not specifically looking for a pulmonary embolism when we found this new opacity on our patient’s radiograph, but this prompted further imaging, which led to the diagnosis. Although a near-normal chest radiograph is the most common radiologic finding in pulmonary embolism, this case shows how careful observation can detect unusual signs.
An 82-year-old woman was admitted to the hospital with dyspnea and chest discomfort over the past 24 hours. She was known to have paroxysmal atrial fibrillation and was taking warfarin, but that had been stopped 2 weeks earlier because of an acute ischemic stroke.
At the time of admission, she had no fever, cough, orthopnea, or leg swelling. Her physical activity was restricted, with residual right-sided weakness after her stroke. Her heart rate was 125 bpm; her oxygen saturation level was 98% on 2 L of oxygen per minute via nasal cannula. She had an irregularly irregular rhythm, a jugular venous pressure of 7 cm H2O, and no cardiac murmurs. Lung sounds were reduced at the bases, with faint crepitations.
Her hemoglobin concentration and white blood cell count were normal. Her brain-natriuretic peptide level was elevated at 2,648 pg/mL (reference range < 167), but cardiac enzyme levels were normal.
Electrocardiography showed atrial fibrillation with rapid ventricular response.
Plain chest radiography showed a 3-cm wedge-shaped opacity in the right mid-thorax (Figure 1), a finding known as the Hampton hump—a sign of pulmonary infarction caused by embolism.
Contrast-enhanced computed tomography (CT) of the chest showed acute thromboembolism in the right interlobar artery and wedge-shaped consolidation in the right-middle lobe (Figure 2), indicating pulmonary infarction.
Brain CT showed a stable infarction. Anticoagulation was restarted, and the patient was discharged in stable condition.
THE HAMPTON HUMP IN PULMONARY EMBOLISM
Because the lungs have a dual blood supply, pulmonary infarction is seen in only a minority of cases of pulmonary embolism. Infarction is more common in patients with peripheral pulmonary embolism, owing to the rapid inflow of bronchial blood, and in patients with medical comorbidities such as heart failure and chronic lung disease.2
The Hampton hump, first described by Aubrey Otis Hampton in 1940, is a peripheral (pleural-based) opacity that represents alveolar hemorrhage from underlying pulmonary infarction. It is one of several radiographic features that have been associated with pulmonary embolism; another is the Westermark sign, indicating oligemia.3
Worsley et al4 examined the diagnostic value of these radiographic features and found that the Hampton hump had a sensitivity of 22% and a specificity of 82% for detecting pulmonary embolism in the right hemithorax, and 24% and 82%, respectively, in the left hemithorax. The prevalence of pleural-based opacities was not significantly different in patients with or without pulmonary embolism. The authors concluded that chest radiography has limited diagnostic value in excluding or diagnosing pulmonary embolism.
In contrast, computed tomographic pulmonary angiography is the first-line imaging test in patients with suspected pulmonary embolism, because of its high sensitivity and specificity.1
We were not specifically looking for a pulmonary embolism when we found this new opacity on our patient’s radiograph, but this prompted further imaging, which led to the diagnosis. Although a near-normal chest radiograph is the most common radiologic finding in pulmonary embolism, this case shows how careful observation can detect unusual signs.
- Mos IC, Klok FA, Kroft LJ, de Roos A, Huisman MV. Imaging tests in the diagnosis of pulmonary embolism. Semin Respir Crit Care Med 2012; 33:138–143.
- Cha SI, Shin KM, Lee J, et al. Clinical relevance of pulmonary infarction in patients with pulmonary embolism. Thromb Res 2012; 130:e1–e5.
- Algın O, GÖkalp G, Topal U. Signs in chest imaging. Diagn Interv Radiol 2011; 17:18–29.
- Worsley DF, Alavi A, Aronchick JM, Chen JT, Greenspan RH, Ravin CE. Chest radiographic findings in patients with acute pulmonary embolism: observations from the PIOPED study. Radiology 1993; 189:133–136.
- Mos IC, Klok FA, Kroft LJ, de Roos A, Huisman MV. Imaging tests in the diagnosis of pulmonary embolism. Semin Respir Crit Care Med 2012; 33:138–143.
- Cha SI, Shin KM, Lee J, et al. Clinical relevance of pulmonary infarction in patients with pulmonary embolism. Thromb Res 2012; 130:e1–e5.
- Algın O, GÖkalp G, Topal U. Signs in chest imaging. Diagn Interv Radiol 2011; 17:18–29.
- Worsley DF, Alavi A, Aronchick JM, Chen JT, Greenspan RH, Ravin CE. Chest radiographic findings in patients with acute pulmonary embolism: observations from the PIOPED study. Radiology 1993; 189:133–136.
The overdiagnosis of pneumonia
Pneumonia was once considered the “old man’s friend,” but in the modern world, has it become the physician’s?
The definition of pneumonia has increasingly been stretched, and physicians occasionally make the diagnosis without canonical signs or symptoms, or even with negative chest radiography. The hallmark of overdiagnosis is identifying illness for which treatment is not needed or is not helpful, and some cases of pneumonia likely fit this description. Empirical evidence over the last 3 decades shows a sustained increase in the diagnosis of pneumonia, but little evidence of a decrease in the rates of pneumonia morbidity and mortality. The central problem with pneumonia is one common to many diagnoses, such as pulmonary embolism, coronary artery disease, and infectious conditions—diagnostic criteria remain divorced from outcomes data. Linking the two has the potential to improve the evidence base of medicine.
Like many long-recognized diagnoses, pneumonia lacks a standardized definition. Most physicians believe that although fever, cough, sputum production, dyspnea, and pleurisy are hallmark symptoms, confirmatory chest radiography is needed to cement the diagnosis.1 But what if a patient has only a fever, cough, and infiltrate? What if the infiltrate is not visible on radiography, but only on computed tomography (CT)? And what if the patient has a cough but is afebrile and has nonspecific findings on CT?
THE RATE OF HOSPITAL ADMISSIONS FOR PNEUMONIA IS RISING
In current clinical practice, any or all of the above cases are called pneumonia. The pneumonia label, once applied, justifies the use of antibiotics, which patients or physicians may overtly desire. One prospective observational study of six hospitals found that 21% of patients admitted with pneumonia and 43% of those treated as outpatients had negative chest radiographs.2 Empirical evidence suggests that the incidence of these “soft” diagnoses may be growing in number.
In the United States, hospitalizations with discharge codes listing pneumonia increased 20% from the late 1980s to the early 2000s.3 The rates of hospitalization for the 10 other most frequent causes of admission did not change significantly over this same period, suggesting a selective increase in hospital admissions for pneumonia.
This focus on pneumonia would be justified if it led to a proportionate benefit for pneumonia outcomes. However, in the same data set, the risk of death from pneumonia did not improve more than that from the other 10 common conditions—all improved similarly—and the rate of discharge from the hospital to a long-term care facility was unchanged. We are hospitalizing more patients with pneumonia, but this has not improved outcomes beyond global trends in mortality.
Data from England suggest that overdiagnosis may be a worldwide phenomenon. Between 1997 and 2005, hospitalization rates in England for pneumonia, adjusted for age, increased 34% from 1.48 to 1.98 per 1,000 persons.4 The 30-day in-hospital death rate for pneumonia remained about the same over this period. In the absence of a paradigm-shifting technology, one that would alter hospitalization practices, or an environmental cause of increased incidence—and with pneumonia there has been neither—the most likely explanation for these documented trends is that hospitals are admitting patients with pneumonia that is less severe.
Finally, data from the 2000s that at first seemed to reverse the trend of increasing hospitalizations for pneumonia have been reanalyzed to account for alternative coding.5 For instance, a pneumonia admission may be coded with respiratory failure as the primary diagnosis and pneumonia as the secondary diagnosis. Examining data from large populations from 2002 to 2009, and correcting as such, shows that the incidence of pneumonia has reached a plateau or has declined only slightly from the elevated rates of the early 2000s. The death rate remains unchanged.
PNEUMONIA: A DIAGNOSIS IN THE EYE OF THE BEHOLDER
Apparently, when it comes to pneumonia, the diagnosis is in the eye of the beholder. Different physicians have different thresholds for applying the label. In the wake of quality efforts to ensure that emergency physicians deliver antibiotics within 4 hours, emergency doctors have been shown to have worse accuracy in diagnosing pneumonia.1 But worse accuracy compared with what standard?
In an investigation by Welker et al,1 the standard definition of pneumonia was based on the one favored by the US Food and Drug Administration for clinical trials. Patients had to have all of the following:
- A new or increasing infiltrate on radiography or CT
- A fever, an elevated white blood cell count, or a shift to immature polymorphonuclear leukocytes
- At least two signs or symptoms of the condition (eg, cough, dyspnea, egophany).
Although this definition is reasonable and ensures homogeneity in clinical trials, it is not steadfastly adhered to in clinical practice and has never been shown to cleanly delineate a population that benefits from antibiotics.
Another challenge to devising a perfect definition of pneumonia is the lack of a pathologic gold standard. Based on a review of 17,340 Medicare patients hospitalized for community-acquired pneumonia, microbial confirmation is often of little assistance, and a probable pathogen is identified in only 7.6% of cases.6
RATES OF OUTPATIENT DIAGNOSIS ARE LIKELY SIMILAR
Thus far, we have examined trends in inpatient diagnosis but not those of outpatient diagnosis. There is no well-done observational study that documents outpatient trends, but there is little reason to suppose the trends are different. Risk-scoring systems in pneumonia, such as the PORT7 and the CURB-65,8 have been designed to decrease unnecessary inpatient admissions, but they do not lend clarity to the diagnosis itself.
The central problem with pneumonia, as with many long-recognized clinical conditions, is that the diagnosis is separated from the treatment. In other words, although physicians are confident that antibiotics benefit patients who have what Sir William Osler would have called pneumonia (elevated white blood cell count, fever, cough, dyspnea, pleurisy, egophany, lobular infiltrate), we don’t know whether the treatment benefits patients whose pneumonia would have been unrecognizable decades ago (with cough, low-grade fever, and infiltrate on CT alone). Improvements in imaging may exacerbate the problem. In this sense, pneumonia exists on a spectrum, as do many medical diagnoses. Not all cases are equally severe, and some may not deserve to be labeled as pneumonia.
No randomized trial has compared antibiotics against supportive care in pneumonia, and, likely, no such trial is needed for clear cases. However, with the growing number of soft diagnoses, randomized trials are desperately needed to delineate where harms outweigh benefits, and where the fuzzy edge of the pneumonia diagnosis must end. And as is always the case with studies that challenge a standard of care, null results should prompt further trials.
WELL-DESIGNED TRIALS COULD END THE UNCERTAINTY
In the next few years, clinical trials, rationally planned, may end most of the uncertainty regarding pneumonia.
Existing observational data may be used to identify groups of patients who, in today’s world, are diagnosed with pneumonia but who do exceptionally well (eg, younger patients with fewer comorbidities, who present with low-grade fever but no signs of consolidation on physical examination, and with dubious results on chest radiography). These are patients for whom equipoise exists, and randomized trials should compare a strategy of antibiotics with a strategy of best supportive care. Trials should be powered for patient-centered outcomes, such as the duration and the complications of illness. The death rate should be scrupulously recorded.
Patients whose pneumonia would have been unrecognizable decades ago should be another target population for the trials I propose.
In a short time, pneumonia may become synonymous with a set of factors for lung infection that predict who will benefit from antibiotics, and who can be safely followed. Already, we are moving toward this standard in other diseases.9 For pulmonary embolism, ongoing trials are testing if anticoagulation can be safely omitted in patients with subsegmental clots (clinicaltrials.gov identifier NCT01455818). Such trials are, at last, translating old diagnoses into the language of evidence-based medicine.
For patients with pneumonia who are not hospitalized, the current outpatient therapy is based on data from studies that show a low rate of failure with empiric treatment based on consideration of the common pathogens for this condition, with few patients subsequently requiring hospitalization. Today, this reasoning is inadequate. The basis for any therapy must be proven benefit for patients with a defined condition compared with a lesser strategy. Data already demonstrate that a short course of antibiotics is no worse than a long course for many hospitalized and outpatients with pneumonia,10,11 but many other patients may require no treatment at all. The time has come to find out.
- Welker JA, Huston M, McCue JD. Antibiotic timing and errors in diagnosing pneumonia. Arch Intern Med 2008; 168:351–356.
- Marrie TJ, Huang JQ. Low-risk patients admitted with community-acquired pneumonia. Am J Med 2005; 118:1357–1363.
- Fry AM, Shay DK, Holman RC, Curns AT, Anderson LJ. Trends in hospitalizations for pneumonia among persons aged 65 years or older in the United States, 1988–2002. JAMA 2005; 294:2712–2719.
- Trotter CL, Stuart JM, George R, Miller E. Increasing hospital admissions for pneumonia, England. Emerg Infect Dis 2008; 14:727–733.
- Lindenauer PK, Lagu T, Shieh MS, Pekow PS, Rothberg MB. Association of diagnostic coding with trends in hospitalizations and mortality of patients with pneumonia, 2003–2009. JAMA 2012; 307:1405–1413.
- Bartlett JG. Diagnostic tests for agents of community-acquired pneumonia. Clin Infect Dis 2011; 52(suppl 4):S296–S304.
- Fine MJ, Auble TE, Yealy DM, et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N Engl J Med 1997; 336:242–250.
- Lim W, van der Eerden MM, Laing R, et al. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax 2003; 58:377–382.
- Prasad V, Rho J, Cifu A. The diagnosis and treatment of pulmonary embolism: a metaphor for medicine in the evidence-based medicine era. Arch Intern Med 2012; 172:955–958.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. A proposed solution for indiscriminate antibiotic prescription. Am J Respir Crit Care Med 2000; 162:505–511.
- Li JZ, Winston LG, Moore DH, Bent S. Efficacy of short-course antibiotic regimens for community-acquired pneumonia: a meta-analysis. Am J Med 2007; 120:783–790.
Pneumonia was once considered the “old man’s friend,” but in the modern world, has it become the physician’s?
The definition of pneumonia has increasingly been stretched, and physicians occasionally make the diagnosis without canonical signs or symptoms, or even with negative chest radiography. The hallmark of overdiagnosis is identifying illness for which treatment is not needed or is not helpful, and some cases of pneumonia likely fit this description. Empirical evidence over the last 3 decades shows a sustained increase in the diagnosis of pneumonia, but little evidence of a decrease in the rates of pneumonia morbidity and mortality. The central problem with pneumonia is one common to many diagnoses, such as pulmonary embolism, coronary artery disease, and infectious conditions—diagnostic criteria remain divorced from outcomes data. Linking the two has the potential to improve the evidence base of medicine.
Like many long-recognized diagnoses, pneumonia lacks a standardized definition. Most physicians believe that although fever, cough, sputum production, dyspnea, and pleurisy are hallmark symptoms, confirmatory chest radiography is needed to cement the diagnosis.1 But what if a patient has only a fever, cough, and infiltrate? What if the infiltrate is not visible on radiography, but only on computed tomography (CT)? And what if the patient has a cough but is afebrile and has nonspecific findings on CT?
THE RATE OF HOSPITAL ADMISSIONS FOR PNEUMONIA IS RISING
In current clinical practice, any or all of the above cases are called pneumonia. The pneumonia label, once applied, justifies the use of antibiotics, which patients or physicians may overtly desire. One prospective observational study of six hospitals found that 21% of patients admitted with pneumonia and 43% of those treated as outpatients had negative chest radiographs.2 Empirical evidence suggests that the incidence of these “soft” diagnoses may be growing in number.
In the United States, hospitalizations with discharge codes listing pneumonia increased 20% from the late 1980s to the early 2000s.3 The rates of hospitalization for the 10 other most frequent causes of admission did not change significantly over this same period, suggesting a selective increase in hospital admissions for pneumonia.
This focus on pneumonia would be justified if it led to a proportionate benefit for pneumonia outcomes. However, in the same data set, the risk of death from pneumonia did not improve more than that from the other 10 common conditions—all improved similarly—and the rate of discharge from the hospital to a long-term care facility was unchanged. We are hospitalizing more patients with pneumonia, but this has not improved outcomes beyond global trends in mortality.
Data from England suggest that overdiagnosis may be a worldwide phenomenon. Between 1997 and 2005, hospitalization rates in England for pneumonia, adjusted for age, increased 34% from 1.48 to 1.98 per 1,000 persons.4 The 30-day in-hospital death rate for pneumonia remained about the same over this period. In the absence of a paradigm-shifting technology, one that would alter hospitalization practices, or an environmental cause of increased incidence—and with pneumonia there has been neither—the most likely explanation for these documented trends is that hospitals are admitting patients with pneumonia that is less severe.
Finally, data from the 2000s that at first seemed to reverse the trend of increasing hospitalizations for pneumonia have been reanalyzed to account for alternative coding.5 For instance, a pneumonia admission may be coded with respiratory failure as the primary diagnosis and pneumonia as the secondary diagnosis. Examining data from large populations from 2002 to 2009, and correcting as such, shows that the incidence of pneumonia has reached a plateau or has declined only slightly from the elevated rates of the early 2000s. The death rate remains unchanged.
PNEUMONIA: A DIAGNOSIS IN THE EYE OF THE BEHOLDER
Apparently, when it comes to pneumonia, the diagnosis is in the eye of the beholder. Different physicians have different thresholds for applying the label. In the wake of quality efforts to ensure that emergency physicians deliver antibiotics within 4 hours, emergency doctors have been shown to have worse accuracy in diagnosing pneumonia.1 But worse accuracy compared with what standard?
In an investigation by Welker et al,1 the standard definition of pneumonia was based on the one favored by the US Food and Drug Administration for clinical trials. Patients had to have all of the following:
- A new or increasing infiltrate on radiography or CT
- A fever, an elevated white blood cell count, or a shift to immature polymorphonuclear leukocytes
- At least two signs or symptoms of the condition (eg, cough, dyspnea, egophany).
Although this definition is reasonable and ensures homogeneity in clinical trials, it is not steadfastly adhered to in clinical practice and has never been shown to cleanly delineate a population that benefits from antibiotics.
Another challenge to devising a perfect definition of pneumonia is the lack of a pathologic gold standard. Based on a review of 17,340 Medicare patients hospitalized for community-acquired pneumonia, microbial confirmation is often of little assistance, and a probable pathogen is identified in only 7.6% of cases.6
RATES OF OUTPATIENT DIAGNOSIS ARE LIKELY SIMILAR
Thus far, we have examined trends in inpatient diagnosis but not those of outpatient diagnosis. There is no well-done observational study that documents outpatient trends, but there is little reason to suppose the trends are different. Risk-scoring systems in pneumonia, such as the PORT7 and the CURB-65,8 have been designed to decrease unnecessary inpatient admissions, but they do not lend clarity to the diagnosis itself.
The central problem with pneumonia, as with many long-recognized clinical conditions, is that the diagnosis is separated from the treatment. In other words, although physicians are confident that antibiotics benefit patients who have what Sir William Osler would have called pneumonia (elevated white blood cell count, fever, cough, dyspnea, pleurisy, egophany, lobular infiltrate), we don’t know whether the treatment benefits patients whose pneumonia would have been unrecognizable decades ago (with cough, low-grade fever, and infiltrate on CT alone). Improvements in imaging may exacerbate the problem. In this sense, pneumonia exists on a spectrum, as do many medical diagnoses. Not all cases are equally severe, and some may not deserve to be labeled as pneumonia.
No randomized trial has compared antibiotics against supportive care in pneumonia, and, likely, no such trial is needed for clear cases. However, with the growing number of soft diagnoses, randomized trials are desperately needed to delineate where harms outweigh benefits, and where the fuzzy edge of the pneumonia diagnosis must end. And as is always the case with studies that challenge a standard of care, null results should prompt further trials.
WELL-DESIGNED TRIALS COULD END THE UNCERTAINTY
In the next few years, clinical trials, rationally planned, may end most of the uncertainty regarding pneumonia.
Existing observational data may be used to identify groups of patients who, in today’s world, are diagnosed with pneumonia but who do exceptionally well (eg, younger patients with fewer comorbidities, who present with low-grade fever but no signs of consolidation on physical examination, and with dubious results on chest radiography). These are patients for whom equipoise exists, and randomized trials should compare a strategy of antibiotics with a strategy of best supportive care. Trials should be powered for patient-centered outcomes, such as the duration and the complications of illness. The death rate should be scrupulously recorded.
Patients whose pneumonia would have been unrecognizable decades ago should be another target population for the trials I propose.
In a short time, pneumonia may become synonymous with a set of factors for lung infection that predict who will benefit from antibiotics, and who can be safely followed. Already, we are moving toward this standard in other diseases.9 For pulmonary embolism, ongoing trials are testing if anticoagulation can be safely omitted in patients with subsegmental clots (clinicaltrials.gov identifier NCT01455818). Such trials are, at last, translating old diagnoses into the language of evidence-based medicine.
For patients with pneumonia who are not hospitalized, the current outpatient therapy is based on data from studies that show a low rate of failure with empiric treatment based on consideration of the common pathogens for this condition, with few patients subsequently requiring hospitalization. Today, this reasoning is inadequate. The basis for any therapy must be proven benefit for patients with a defined condition compared with a lesser strategy. Data already demonstrate that a short course of antibiotics is no worse than a long course for many hospitalized and outpatients with pneumonia,10,11 but many other patients may require no treatment at all. The time has come to find out.
Pneumonia was once considered the “old man’s friend,” but in the modern world, has it become the physician’s?
The definition of pneumonia has increasingly been stretched, and physicians occasionally make the diagnosis without canonical signs or symptoms, or even with negative chest radiography. The hallmark of overdiagnosis is identifying illness for which treatment is not needed or is not helpful, and some cases of pneumonia likely fit this description. Empirical evidence over the last 3 decades shows a sustained increase in the diagnosis of pneumonia, but little evidence of a decrease in the rates of pneumonia morbidity and mortality. The central problem with pneumonia is one common to many diagnoses, such as pulmonary embolism, coronary artery disease, and infectious conditions—diagnostic criteria remain divorced from outcomes data. Linking the two has the potential to improve the evidence base of medicine.
Like many long-recognized diagnoses, pneumonia lacks a standardized definition. Most physicians believe that although fever, cough, sputum production, dyspnea, and pleurisy are hallmark symptoms, confirmatory chest radiography is needed to cement the diagnosis.1 But what if a patient has only a fever, cough, and infiltrate? What if the infiltrate is not visible on radiography, but only on computed tomography (CT)? And what if the patient has a cough but is afebrile and has nonspecific findings on CT?
THE RATE OF HOSPITAL ADMISSIONS FOR PNEUMONIA IS RISING
In current clinical practice, any or all of the above cases are called pneumonia. The pneumonia label, once applied, justifies the use of antibiotics, which patients or physicians may overtly desire. One prospective observational study of six hospitals found that 21% of patients admitted with pneumonia and 43% of those treated as outpatients had negative chest radiographs.2 Empirical evidence suggests that the incidence of these “soft” diagnoses may be growing in number.
In the United States, hospitalizations with discharge codes listing pneumonia increased 20% from the late 1980s to the early 2000s.3 The rates of hospitalization for the 10 other most frequent causes of admission did not change significantly over this same period, suggesting a selective increase in hospital admissions for pneumonia.
This focus on pneumonia would be justified if it led to a proportionate benefit for pneumonia outcomes. However, in the same data set, the risk of death from pneumonia did not improve more than that from the other 10 common conditions—all improved similarly—and the rate of discharge from the hospital to a long-term care facility was unchanged. We are hospitalizing more patients with pneumonia, but this has not improved outcomes beyond global trends in mortality.
Data from England suggest that overdiagnosis may be a worldwide phenomenon. Between 1997 and 2005, hospitalization rates in England for pneumonia, adjusted for age, increased 34% from 1.48 to 1.98 per 1,000 persons.4 The 30-day in-hospital death rate for pneumonia remained about the same over this period. In the absence of a paradigm-shifting technology, one that would alter hospitalization practices, or an environmental cause of increased incidence—and with pneumonia there has been neither—the most likely explanation for these documented trends is that hospitals are admitting patients with pneumonia that is less severe.
Finally, data from the 2000s that at first seemed to reverse the trend of increasing hospitalizations for pneumonia have been reanalyzed to account for alternative coding.5 For instance, a pneumonia admission may be coded with respiratory failure as the primary diagnosis and pneumonia as the secondary diagnosis. Examining data from large populations from 2002 to 2009, and correcting as such, shows that the incidence of pneumonia has reached a plateau or has declined only slightly from the elevated rates of the early 2000s. The death rate remains unchanged.
PNEUMONIA: A DIAGNOSIS IN THE EYE OF THE BEHOLDER
Apparently, when it comes to pneumonia, the diagnosis is in the eye of the beholder. Different physicians have different thresholds for applying the label. In the wake of quality efforts to ensure that emergency physicians deliver antibiotics within 4 hours, emergency doctors have been shown to have worse accuracy in diagnosing pneumonia.1 But worse accuracy compared with what standard?
In an investigation by Welker et al,1 the standard definition of pneumonia was based on the one favored by the US Food and Drug Administration for clinical trials. Patients had to have all of the following:
- A new or increasing infiltrate on radiography or CT
- A fever, an elevated white blood cell count, or a shift to immature polymorphonuclear leukocytes
- At least two signs or symptoms of the condition (eg, cough, dyspnea, egophany).
Although this definition is reasonable and ensures homogeneity in clinical trials, it is not steadfastly adhered to in clinical practice and has never been shown to cleanly delineate a population that benefits from antibiotics.
Another challenge to devising a perfect definition of pneumonia is the lack of a pathologic gold standard. Based on a review of 17,340 Medicare patients hospitalized for community-acquired pneumonia, microbial confirmation is often of little assistance, and a probable pathogen is identified in only 7.6% of cases.6
RATES OF OUTPATIENT DIAGNOSIS ARE LIKELY SIMILAR
Thus far, we have examined trends in inpatient diagnosis but not those of outpatient diagnosis. There is no well-done observational study that documents outpatient trends, but there is little reason to suppose the trends are different. Risk-scoring systems in pneumonia, such as the PORT7 and the CURB-65,8 have been designed to decrease unnecessary inpatient admissions, but they do not lend clarity to the diagnosis itself.
The central problem with pneumonia, as with many long-recognized clinical conditions, is that the diagnosis is separated from the treatment. In other words, although physicians are confident that antibiotics benefit patients who have what Sir William Osler would have called pneumonia (elevated white blood cell count, fever, cough, dyspnea, pleurisy, egophany, lobular infiltrate), we don’t know whether the treatment benefits patients whose pneumonia would have been unrecognizable decades ago (with cough, low-grade fever, and infiltrate on CT alone). Improvements in imaging may exacerbate the problem. In this sense, pneumonia exists on a spectrum, as do many medical diagnoses. Not all cases are equally severe, and some may not deserve to be labeled as pneumonia.
No randomized trial has compared antibiotics against supportive care in pneumonia, and, likely, no such trial is needed for clear cases. However, with the growing number of soft diagnoses, randomized trials are desperately needed to delineate where harms outweigh benefits, and where the fuzzy edge of the pneumonia diagnosis must end. And as is always the case with studies that challenge a standard of care, null results should prompt further trials.
WELL-DESIGNED TRIALS COULD END THE UNCERTAINTY
In the next few years, clinical trials, rationally planned, may end most of the uncertainty regarding pneumonia.
Existing observational data may be used to identify groups of patients who, in today’s world, are diagnosed with pneumonia but who do exceptionally well (eg, younger patients with fewer comorbidities, who present with low-grade fever but no signs of consolidation on physical examination, and with dubious results on chest radiography). These are patients for whom equipoise exists, and randomized trials should compare a strategy of antibiotics with a strategy of best supportive care. Trials should be powered for patient-centered outcomes, such as the duration and the complications of illness. The death rate should be scrupulously recorded.
Patients whose pneumonia would have been unrecognizable decades ago should be another target population for the trials I propose.
In a short time, pneumonia may become synonymous with a set of factors for lung infection that predict who will benefit from antibiotics, and who can be safely followed. Already, we are moving toward this standard in other diseases.9 For pulmonary embolism, ongoing trials are testing if anticoagulation can be safely omitted in patients with subsegmental clots (clinicaltrials.gov identifier NCT01455818). Such trials are, at last, translating old diagnoses into the language of evidence-based medicine.
For patients with pneumonia who are not hospitalized, the current outpatient therapy is based on data from studies that show a low rate of failure with empiric treatment based on consideration of the common pathogens for this condition, with few patients subsequently requiring hospitalization. Today, this reasoning is inadequate. The basis for any therapy must be proven benefit for patients with a defined condition compared with a lesser strategy. Data already demonstrate that a short course of antibiotics is no worse than a long course for many hospitalized and outpatients with pneumonia,10,11 but many other patients may require no treatment at all. The time has come to find out.
- Welker JA, Huston M, McCue JD. Antibiotic timing and errors in diagnosing pneumonia. Arch Intern Med 2008; 168:351–356.
- Marrie TJ, Huang JQ. Low-risk patients admitted with community-acquired pneumonia. Am J Med 2005; 118:1357–1363.
- Fry AM, Shay DK, Holman RC, Curns AT, Anderson LJ. Trends in hospitalizations for pneumonia among persons aged 65 years or older in the United States, 1988–2002. JAMA 2005; 294:2712–2719.
- Trotter CL, Stuart JM, George R, Miller E. Increasing hospital admissions for pneumonia, England. Emerg Infect Dis 2008; 14:727–733.
- Lindenauer PK, Lagu T, Shieh MS, Pekow PS, Rothberg MB. Association of diagnostic coding with trends in hospitalizations and mortality of patients with pneumonia, 2003–2009. JAMA 2012; 307:1405–1413.
- Bartlett JG. Diagnostic tests for agents of community-acquired pneumonia. Clin Infect Dis 2011; 52(suppl 4):S296–S304.
- Fine MJ, Auble TE, Yealy DM, et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N Engl J Med 1997; 336:242–250.
- Lim W, van der Eerden MM, Laing R, et al. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax 2003; 58:377–382.
- Prasad V, Rho J, Cifu A. The diagnosis and treatment of pulmonary embolism: a metaphor for medicine in the evidence-based medicine era. Arch Intern Med 2012; 172:955–958.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. A proposed solution for indiscriminate antibiotic prescription. Am J Respir Crit Care Med 2000; 162:505–511.
- Li JZ, Winston LG, Moore DH, Bent S. Efficacy of short-course antibiotic regimens for community-acquired pneumonia: a meta-analysis. Am J Med 2007; 120:783–790.
- Welker JA, Huston M, McCue JD. Antibiotic timing and errors in diagnosing pneumonia. Arch Intern Med 2008; 168:351–356.
- Marrie TJ, Huang JQ. Low-risk patients admitted with community-acquired pneumonia. Am J Med 2005; 118:1357–1363.
- Fry AM, Shay DK, Holman RC, Curns AT, Anderson LJ. Trends in hospitalizations for pneumonia among persons aged 65 years or older in the United States, 1988–2002. JAMA 2005; 294:2712–2719.
- Trotter CL, Stuart JM, George R, Miller E. Increasing hospital admissions for pneumonia, England. Emerg Infect Dis 2008; 14:727–733.
- Lindenauer PK, Lagu T, Shieh MS, Pekow PS, Rothberg MB. Association of diagnostic coding with trends in hospitalizations and mortality of patients with pneumonia, 2003–2009. JAMA 2012; 307:1405–1413.
- Bartlett JG. Diagnostic tests for agents of community-acquired pneumonia. Clin Infect Dis 2011; 52(suppl 4):S296–S304.
- Fine MJ, Auble TE, Yealy DM, et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N Engl J Med 1997; 336:242–250.
- Lim W, van der Eerden MM, Laing R, et al. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax 2003; 58:377–382.
- Prasad V, Rho J, Cifu A. The diagnosis and treatment of pulmonary embolism: a metaphor for medicine in the evidence-based medicine era. Arch Intern Med 2012; 172:955–958.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. A proposed solution for indiscriminate antibiotic prescription. Am J Respir Crit Care Med 2000; 162:505–511.
- Li JZ, Winston LG, Moore DH, Bent S. Efficacy of short-course antibiotic regimens for community-acquired pneumonia: a meta-analysis. Am J Med 2007; 120:783–790.
Another perspective: Reducing the overtreatment of pneumonia
In his commentary in this issue, Dr. Vinay Prasad provides a well-supported perspective on the overdiagnosis of pneumonia.
Although I agree that there can be a tendency to overdiagnose pneumonia, we must not overlook the fact that pneumonia is still a leading cause of death in the United States.
The number of cases of invasive pneumococcal disease (mostly bacteremic pneumonia) in people over age 65 has increased over the past decade.1 This increase is not the result of overdiagnosis, since the diagnosis relies on the well-established US Centers for Disease Control and Prevention (CDC) surveillance system, which requires a positive culture from a sterile site. However, to an extent, the increase can be explained by the increasing age of our population and by the associated comorbidities.2 These comorbidities increase the predisposition to and the severity of pneumonia and adversely affect the outcome—which may also explain why we have seen no significant decrease in the death rate for patients admitted to the hospital.
In addition, a 2012 report3 that drew data from a variety of sources, including the CDC, projected that between 2004 and 2040, the US population would increase by 38% and at the same time pneumococcal pneumonia hospitalizations would increase by 96%, since population growth is fastest in older age groups, who have the highest rates of disease.3
Thus, I believe that pneumonia will continue to be a big problem and that we should pursue efforts to prevent it (including vaccinating more people against it) and to better manage it.
I agree that “over-calling” acute respiratory infections as pneumonia is often the result of imprecise diagnosis. Clinical criteria, even a combination of symptoms (cough) and signs (fever, tachycardia, and crackles), are not reliably predictive when using chest radiography as the standard.4 This can lead to the overuse of antimicrobials.
The Centers for Medicare and Medicaid Services used to call for starting the first dose of antibiotics within a specified time (at one time it was 4 hours, subsequently it became 6 hours) of presentation to the hospital with community-acquired pneumonia. Although the actual effect was uncertain, many feared that this performance measure would unintentionally lead to the indiscriminate use of antimicrobials to achieve high rates of compliance in patients who have little evidence of pneumonia, in order to not miss a possible case.5,6 However, it is important to be aware that this measure was retired in 2012 and is no longer in effect.
WE NEED BETTER ASSESSMENTS AND TREATMENTS
We need better ways to assess and manage patients with pneumonia. I believe part of the solution to better assessment lies in improved diagnostic tools, and these are now becoming available.
As Dr. Prasad notes, the causative pathogen is rarely identified using standard diagnostic methods. However, polymerase chain reaction testing and measurement of the biomarker procalcitonin may improve our diagnostic accuracy and, hopefully, lead to better outcomes.7 The results of these tests can be rapidly available and thus may aid the point-of-care decision to treat or not to treat with antimicrobials and to allow for therapy to be started within an acceptable period. In addition, procalcitonin testing has been shown to help differentiate viral from bacterial causes of respiratory tract infection.
Dr. Prasad states that no randomized trial has compared antibiotics with supportive care in pneumonia. Although this is true for recent trials, historical studies demonstrate that antibiotics reduce death rates in patients with pneumonia.8 Indeed, these are the basis for the recent changes in the US Food and Drug Administration guidance for clinical trials of pneumonia.9
Treatment is best when it is directed at the pathogen, but there is little consensus on the practicality of achieving this goal at the primary point of care. A study funded by the National Institutes of Health is about to start enrollment to compare the effect of narrow-spectrum therapy vs the standard of care based on rapid diagnostics.10 Identification of a specific pathogen will allow directed therapy without the need for a broad-spectrum empiric regimen. In contrast, finding a viral cause without an accompanying bacterial cause will prevent the unnecessary use of antibacterials in many cases. A significant percent of cases of pneumonia in adults are caused by viruses alone.6
Thus, the question will not be, “Should we treat community-acquired pneumonia with antibacterials” but rather, “What is the optimal treatment for pneumonia with a defined cause?” This is a major change from an empirical broad-spectrum regimen (treat all likely pathogens) to a more specific approach that has several potential benefits, including better patient outcomes and less emergence of resistance. To paraphrase Dr. Prasad, the time has come to find this out.
- US Centers for Disease Control and Prevention. Active bacterial core surveillance (ABCs) http://www.cdc.gov/abcs/reports-findings/surv-reports.html. Accessed June 20, 2013.
- Fry AM, Shay DK, Holman RC, Curns AT, Anderson LJ. Trends in hospitalizations for pneumonia among persons aged 65 years or older in the United States, 1988–2002. JAMA 2005; 294:2712–2719.
- Wroe PC, Finkelstein JA, Ray GT, et al. Aging population and future burden of pneumococcal pneumonia in the United States. J Infect Dis 2012; 205:1589–1592.
- Metlay JP, Fine MJ. Testing strategies in the initial management of patients with community-acquired pneumonia. Ann Intern Med 2003; 138:109–118.
- File TM, Solomkin JS, Cosgrove SE. Strategies for improving antimicrobial use and the role of antimicrobial stewardship programs. Clin Infect Dis 2011; 53(suppl 1):S15–S22.
- File TM, Gross PA. Performance measurement in community-acquired pneumonia: consequences intended and unintended. Clin Infect Dis 2007; 44:942–944.
- File TM. New diagnostic tests for pneumonia: what is their role in clinical practice? Clin Chest Med 2011; 32:417–430.
- Spellberg B, Talbot GH, Brass EP, Bradley JS, Boucher HW, Gilbert DN; Infectious Diseases Society of America. Position paper: recommended design features of future clinical trials of antibacterial agents for community-acquired pneumonia. Clin Infect Dis 2008; 47(suppl 3):S249–S265.
- Division of Drug Information. Guidance for industry. Community-acquired bacterial pneumonia: Developing drugs for treatment. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm123686.pdf. Accessed August 4, 2013.
- US National Institutes of Health. Microbiology testing with the aim of directed antimicrobial therapy for CAP (NIHCAP). Department of Microbiology Infectious Diseases Protocol 10-0061. http://clinicaltrials.gov/ct2/show/NCT01662258?term=community+acquired+pneumonia&cond=%22Pneumonia%22&titles=microbiology+testing+with+the+aim+of+directed+antimicrobial+therapy&rank=1. Accessed August 4, 2013.
In his commentary in this issue, Dr. Vinay Prasad provides a well-supported perspective on the overdiagnosis of pneumonia.
Although I agree that there can be a tendency to overdiagnose pneumonia, we must not overlook the fact that pneumonia is still a leading cause of death in the United States.
The number of cases of invasive pneumococcal disease (mostly bacteremic pneumonia) in people over age 65 has increased over the past decade.1 This increase is not the result of overdiagnosis, since the diagnosis relies on the well-established US Centers for Disease Control and Prevention (CDC) surveillance system, which requires a positive culture from a sterile site. However, to an extent, the increase can be explained by the increasing age of our population and by the associated comorbidities.2 These comorbidities increase the predisposition to and the severity of pneumonia and adversely affect the outcome—which may also explain why we have seen no significant decrease in the death rate for patients admitted to the hospital.
In addition, a 2012 report3 that drew data from a variety of sources, including the CDC, projected that between 2004 and 2040, the US population would increase by 38% and at the same time pneumococcal pneumonia hospitalizations would increase by 96%, since population growth is fastest in older age groups, who have the highest rates of disease.3
Thus, I believe that pneumonia will continue to be a big problem and that we should pursue efforts to prevent it (including vaccinating more people against it) and to better manage it.
I agree that “over-calling” acute respiratory infections as pneumonia is often the result of imprecise diagnosis. Clinical criteria, even a combination of symptoms (cough) and signs (fever, tachycardia, and crackles), are not reliably predictive when using chest radiography as the standard.4 This can lead to the overuse of antimicrobials.
The Centers for Medicare and Medicaid Services used to call for starting the first dose of antibiotics within a specified time (at one time it was 4 hours, subsequently it became 6 hours) of presentation to the hospital with community-acquired pneumonia. Although the actual effect was uncertain, many feared that this performance measure would unintentionally lead to the indiscriminate use of antimicrobials to achieve high rates of compliance in patients who have little evidence of pneumonia, in order to not miss a possible case.5,6 However, it is important to be aware that this measure was retired in 2012 and is no longer in effect.
WE NEED BETTER ASSESSMENTS AND TREATMENTS
We need better ways to assess and manage patients with pneumonia. I believe part of the solution to better assessment lies in improved diagnostic tools, and these are now becoming available.
As Dr. Prasad notes, the causative pathogen is rarely identified using standard diagnostic methods. However, polymerase chain reaction testing and measurement of the biomarker procalcitonin may improve our diagnostic accuracy and, hopefully, lead to better outcomes.7 The results of these tests can be rapidly available and thus may aid the point-of-care decision to treat or not to treat with antimicrobials and to allow for therapy to be started within an acceptable period. In addition, procalcitonin testing has been shown to help differentiate viral from bacterial causes of respiratory tract infection.
Dr. Prasad states that no randomized trial has compared antibiotics with supportive care in pneumonia. Although this is true for recent trials, historical studies demonstrate that antibiotics reduce death rates in patients with pneumonia.8 Indeed, these are the basis for the recent changes in the US Food and Drug Administration guidance for clinical trials of pneumonia.9
Treatment is best when it is directed at the pathogen, but there is little consensus on the practicality of achieving this goal at the primary point of care. A study funded by the National Institutes of Health is about to start enrollment to compare the effect of narrow-spectrum therapy vs the standard of care based on rapid diagnostics.10 Identification of a specific pathogen will allow directed therapy without the need for a broad-spectrum empiric regimen. In contrast, finding a viral cause without an accompanying bacterial cause will prevent the unnecessary use of antibacterials in many cases. A significant percent of cases of pneumonia in adults are caused by viruses alone.6
Thus, the question will not be, “Should we treat community-acquired pneumonia with antibacterials” but rather, “What is the optimal treatment for pneumonia with a defined cause?” This is a major change from an empirical broad-spectrum regimen (treat all likely pathogens) to a more specific approach that has several potential benefits, including better patient outcomes and less emergence of resistance. To paraphrase Dr. Prasad, the time has come to find this out.
In his commentary in this issue, Dr. Vinay Prasad provides a well-supported perspective on the overdiagnosis of pneumonia.
Although I agree that there can be a tendency to overdiagnose pneumonia, we must not overlook the fact that pneumonia is still a leading cause of death in the United States.
The number of cases of invasive pneumococcal disease (mostly bacteremic pneumonia) in people over age 65 has increased over the past decade.1 This increase is not the result of overdiagnosis, since the diagnosis relies on the well-established US Centers for Disease Control and Prevention (CDC) surveillance system, which requires a positive culture from a sterile site. However, to an extent, the increase can be explained by the increasing age of our population and by the associated comorbidities.2 These comorbidities increase the predisposition to and the severity of pneumonia and adversely affect the outcome—which may also explain why we have seen no significant decrease in the death rate for patients admitted to the hospital.
In addition, a 2012 report3 that drew data from a variety of sources, including the CDC, projected that between 2004 and 2040, the US population would increase by 38% and at the same time pneumococcal pneumonia hospitalizations would increase by 96%, since population growth is fastest in older age groups, who have the highest rates of disease.3
Thus, I believe that pneumonia will continue to be a big problem and that we should pursue efforts to prevent it (including vaccinating more people against it) and to better manage it.
I agree that “over-calling” acute respiratory infections as pneumonia is often the result of imprecise diagnosis. Clinical criteria, even a combination of symptoms (cough) and signs (fever, tachycardia, and crackles), are not reliably predictive when using chest radiography as the standard.4 This can lead to the overuse of antimicrobials.
The Centers for Medicare and Medicaid Services used to call for starting the first dose of antibiotics within a specified time (at one time it was 4 hours, subsequently it became 6 hours) of presentation to the hospital with community-acquired pneumonia. Although the actual effect was uncertain, many feared that this performance measure would unintentionally lead to the indiscriminate use of antimicrobials to achieve high rates of compliance in patients who have little evidence of pneumonia, in order to not miss a possible case.5,6 However, it is important to be aware that this measure was retired in 2012 and is no longer in effect.
WE NEED BETTER ASSESSMENTS AND TREATMENTS
We need better ways to assess and manage patients with pneumonia. I believe part of the solution to better assessment lies in improved diagnostic tools, and these are now becoming available.
As Dr. Prasad notes, the causative pathogen is rarely identified using standard diagnostic methods. However, polymerase chain reaction testing and measurement of the biomarker procalcitonin may improve our diagnostic accuracy and, hopefully, lead to better outcomes.7 The results of these tests can be rapidly available and thus may aid the point-of-care decision to treat or not to treat with antimicrobials and to allow for therapy to be started within an acceptable period. In addition, procalcitonin testing has been shown to help differentiate viral from bacterial causes of respiratory tract infection.
Dr. Prasad states that no randomized trial has compared antibiotics with supportive care in pneumonia. Although this is true for recent trials, historical studies demonstrate that antibiotics reduce death rates in patients with pneumonia.8 Indeed, these are the basis for the recent changes in the US Food and Drug Administration guidance for clinical trials of pneumonia.9
Treatment is best when it is directed at the pathogen, but there is little consensus on the practicality of achieving this goal at the primary point of care. A study funded by the National Institutes of Health is about to start enrollment to compare the effect of narrow-spectrum therapy vs the standard of care based on rapid diagnostics.10 Identification of a specific pathogen will allow directed therapy without the need for a broad-spectrum empiric regimen. In contrast, finding a viral cause without an accompanying bacterial cause will prevent the unnecessary use of antibacterials in many cases. A significant percent of cases of pneumonia in adults are caused by viruses alone.6
Thus, the question will not be, “Should we treat community-acquired pneumonia with antibacterials” but rather, “What is the optimal treatment for pneumonia with a defined cause?” This is a major change from an empirical broad-spectrum regimen (treat all likely pathogens) to a more specific approach that has several potential benefits, including better patient outcomes and less emergence of resistance. To paraphrase Dr. Prasad, the time has come to find this out.
- US Centers for Disease Control and Prevention. Active bacterial core surveillance (ABCs) http://www.cdc.gov/abcs/reports-findings/surv-reports.html. Accessed June 20, 2013.
- Fry AM, Shay DK, Holman RC, Curns AT, Anderson LJ. Trends in hospitalizations for pneumonia among persons aged 65 years or older in the United States, 1988–2002. JAMA 2005; 294:2712–2719.
- Wroe PC, Finkelstein JA, Ray GT, et al. Aging population and future burden of pneumococcal pneumonia in the United States. J Infect Dis 2012; 205:1589–1592.
- Metlay JP, Fine MJ. Testing strategies in the initial management of patients with community-acquired pneumonia. Ann Intern Med 2003; 138:109–118.
- File TM, Solomkin JS, Cosgrove SE. Strategies for improving antimicrobial use and the role of antimicrobial stewardship programs. Clin Infect Dis 2011; 53(suppl 1):S15–S22.
- File TM, Gross PA. Performance measurement in community-acquired pneumonia: consequences intended and unintended. Clin Infect Dis 2007; 44:942–944.
- File TM. New diagnostic tests for pneumonia: what is their role in clinical practice? Clin Chest Med 2011; 32:417–430.
- Spellberg B, Talbot GH, Brass EP, Bradley JS, Boucher HW, Gilbert DN; Infectious Diseases Society of America. Position paper: recommended design features of future clinical trials of antibacterial agents for community-acquired pneumonia. Clin Infect Dis 2008; 47(suppl 3):S249–S265.
- Division of Drug Information. Guidance for industry. Community-acquired bacterial pneumonia: Developing drugs for treatment. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm123686.pdf. Accessed August 4, 2013.
- US National Institutes of Health. Microbiology testing with the aim of directed antimicrobial therapy for CAP (NIHCAP). Department of Microbiology Infectious Diseases Protocol 10-0061. http://clinicaltrials.gov/ct2/show/NCT01662258?term=community+acquired+pneumonia&cond=%22Pneumonia%22&titles=microbiology+testing+with+the+aim+of+directed+antimicrobial+therapy&rank=1. Accessed August 4, 2013.
- US Centers for Disease Control and Prevention. Active bacterial core surveillance (ABCs) http://www.cdc.gov/abcs/reports-findings/surv-reports.html. Accessed June 20, 2013.
- Fry AM, Shay DK, Holman RC, Curns AT, Anderson LJ. Trends in hospitalizations for pneumonia among persons aged 65 years or older in the United States, 1988–2002. JAMA 2005; 294:2712–2719.
- Wroe PC, Finkelstein JA, Ray GT, et al. Aging population and future burden of pneumococcal pneumonia in the United States. J Infect Dis 2012; 205:1589–1592.
- Metlay JP, Fine MJ. Testing strategies in the initial management of patients with community-acquired pneumonia. Ann Intern Med 2003; 138:109–118.
- File TM, Solomkin JS, Cosgrove SE. Strategies for improving antimicrobial use and the role of antimicrobial stewardship programs. Clin Infect Dis 2011; 53(suppl 1):S15–S22.
- File TM, Gross PA. Performance measurement in community-acquired pneumonia: consequences intended and unintended. Clin Infect Dis 2007; 44:942–944.
- File TM. New diagnostic tests for pneumonia: what is their role in clinical practice? Clin Chest Med 2011; 32:417–430.
- Spellberg B, Talbot GH, Brass EP, Bradley JS, Boucher HW, Gilbert DN; Infectious Diseases Society of America. Position paper: recommended design features of future clinical trials of antibacterial agents for community-acquired pneumonia. Clin Infect Dis 2008; 47(suppl 3):S249–S265.
- Division of Drug Information. Guidance for industry. Community-acquired bacterial pneumonia: Developing drugs for treatment. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm123686.pdf. Accessed August 4, 2013.
- US National Institutes of Health. Microbiology testing with the aim of directed antimicrobial therapy for CAP (NIHCAP). Department of Microbiology Infectious Diseases Protocol 10-0061. http://clinicaltrials.gov/ct2/show/NCT01662258?term=community+acquired+pneumonia&cond=%22Pneumonia%22&titles=microbiology+testing+with+the+aim+of+directed+antimicrobial+therapy&rank=1. Accessed August 4, 2013.
Is anticoagulation appropriate for all patients with portal vein thrombosis?
No. in general, the decision to treat portal vein thrombosis with anticoagulant drugs is complex and depends on whether the thrombosis is acute or chronic, and whether the cause is a local factor, cirrhosis of the liver, or a systemic condition (Table 1). A “one-size-fits-all” approach should be avoided (Figure 1).
ACUTE PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS

No randomized controlled trial has yet evaluated anticoagulation in acute portal vein thrombosis. But a prospective study published in 2010 showed that the portal vein and its left or right branch were patent in 39% of anticoagulated patients (vs 13% initially), the splenic vein in 80% (vs 57% initially), and the superior mesenteric vein in 73% (vs 42% initially).1 Further, there appears to be a 20% reduction in the overall mortality rate associated with anticoagulation for acute portal vein thrombosis in retrospective studies.2
In the absence of contraindications, anticoagulation with heparin or low-molecular-weight heparin is recommended, with complete bridging to oral anticoagulation with a vitamin K antagonist. Anticoagulation should be continued for at least 3 months, and indefinitely in patients with permanent hypercoaguable risk factors.3
CHRONIC PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
All patients with chronic portal vein thrombosis should undergo esophagogastroduodenoscopy to evaluate for varices. Patients with large varices should be treated orally with a nonselective beta-adrenergic blocker or endoscopically. Though no prospective study has validated this practice, a retrospective analysis showed a decreased risk of first or recurrent bleeding.4
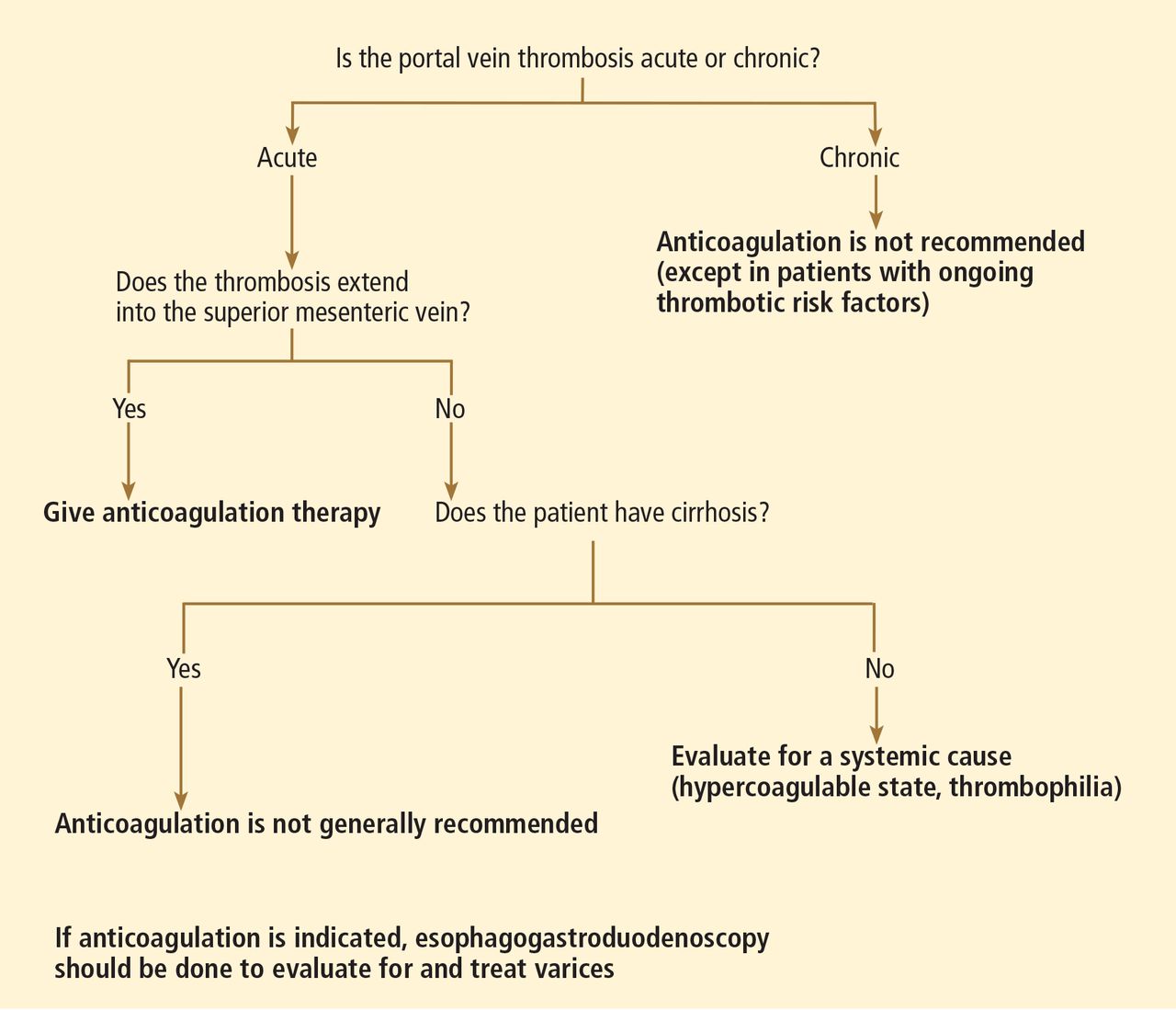
In 2007, a retrospective study showed a lower rate of death in patients with portomesenteric venous thrombosis treated with an oral vitamin K antagonist.5 Patients with chronic portal vein thrombosis with ongoing thrombotic risk factors should be treated with long-term anticoagulation after screening for varices, and if varices are present, primary prophylaxis should be started.3 With this approach, less than 5% of patients died from classic complications of portal vein thrombosis at 5 years of follow-up.4
ACUTE OR CHRONIC PORTAL VEIN THROMBOSIS WITH CIRRHOSIS
Portal vein thrombosis is common in patients with underlying cirrhosis. The risk in patients with cirrhosis significantly increases as liver function worsens. In patients with well-compensated cirrhosis, the risk is less than 1% vs 8% to 25% in those with advanced cirrhosis.6
In patients awaiting liver transplantation, a large retrospective study7 showed that the rate of partial or complete recanalization of the splanchnic veins was significantly higher in those who received anticoagulation (8 of 19) than in those who did not (0 of 10, P = .002). The rate of survival was significantly lower in those who had complete thrombotic obstruction of the portal vein at the time of surgery (P = .04). However, there was no difference in survival rates between those with partial obstruction who received anticoagulation and those with a patent portal vein.7
A later retrospective study8 showed no significant benefit in the rate of transplantation-free survival or survival after liver transplantation in patients with or without chronic portal vein thrombosis.8
Unfortunately, we have no data from prospective controlled trials and only limited data from retrospective studies to make a strong recommendation for or against anticoagulation in either acute and chronic portal vein thrombosis associated with cirrhosis. As such, each case must be evaluated on an individual basis in association with expert consultation.
In our experience, the risk of bleeding in patients with liver cirrhosis is substantial because of the decreased synthesis of coagulation factors and the presence of varices, whereas the efficacy and the benefits of recanalizing the portal vein in asymptomatic patients with liver cirrhosis and portal vein thrombosis are unknown. Therefore, unless the thrombosis extends into the mesenteric vein, thus posing a risk of mesenteric ischemia, we do not generally recommend anticoagulation in asymptomatic portal vein thrombosis in patients with cirrhosis.
- Plessier A, Darwish-Murad S, Hernandez-Guerra M, et al; European Network for Vascular Disorders of the Liver (EN-Vie). Acute portal vein thrombosis unrelated to cirrhosis: a prospective multicenter follow-up study. Hepatology 2010; 51:210–218.
- Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med 2001; 345:1683–1688.
- de Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut 2005; 54:691–697.
- John BV, Konjeti VR, Aggarwal A, et al. The impact of portal vein thrombosis (PVT) on cirrhotics awaiting liver transplantation (abstract). Hepatology 2010; 52(suppl1):888A–889A.
No. in general, the decision to treat portal vein thrombosis with anticoagulant drugs is complex and depends on whether the thrombosis is acute or chronic, and whether the cause is a local factor, cirrhosis of the liver, or a systemic condition (Table 1). A “one-size-fits-all” approach should be avoided (Figure 1).
ACUTE PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
No randomized controlled trial has yet evaluated anticoagulation in acute portal vein thrombosis. But a prospective study published in 2010 showed that the portal vein and its left or right branch were patent in 39% of anticoagulated patients (vs 13% initially), the splenic vein in 80% (vs 57% initially), and the superior mesenteric vein in 73% (vs 42% initially).1 Further, there appears to be a 20% reduction in the overall mortality rate associated with anticoagulation for acute portal vein thrombosis in retrospective studies.2
In the absence of contraindications, anticoagulation with heparin or low-molecular-weight heparin is recommended, with complete bridging to oral anticoagulation with a vitamin K antagonist. Anticoagulation should be continued for at least 3 months, and indefinitely in patients with permanent hypercoaguable risk factors.3
CHRONIC PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
All patients with chronic portal vein thrombosis should undergo esophagogastroduodenoscopy to evaluate for varices. Patients with large varices should be treated orally with a nonselective beta-adrenergic blocker or endoscopically. Though no prospective study has validated this practice, a retrospective analysis showed a decreased risk of first or recurrent bleeding.4
In 2007, a retrospective study showed a lower rate of death in patients with portomesenteric venous thrombosis treated with an oral vitamin K antagonist.5 Patients with chronic portal vein thrombosis with ongoing thrombotic risk factors should be treated with long-term anticoagulation after screening for varices, and if varices are present, primary prophylaxis should be started.3 With this approach, less than 5% of patients died from classic complications of portal vein thrombosis at 5 years of follow-up.4
ACUTE OR CHRONIC PORTAL VEIN THROMBOSIS WITH CIRRHOSIS
Portal vein thrombosis is common in patients with underlying cirrhosis. The risk in patients with cirrhosis significantly increases as liver function worsens. In patients with well-compensated cirrhosis, the risk is less than 1% vs 8% to 25% in those with advanced cirrhosis.6
In patients awaiting liver transplantation, a large retrospective study7 showed that the rate of partial or complete recanalization of the splanchnic veins was significantly higher in those who received anticoagulation (8 of 19) than in those who did not (0 of 10, P = .002). The rate of survival was significantly lower in those who had complete thrombotic obstruction of the portal vein at the time of surgery (P = .04). However, there was no difference in survival rates between those with partial obstruction who received anticoagulation and those with a patent portal vein.7
A later retrospective study8 showed no significant benefit in the rate of transplantation-free survival or survival after liver transplantation in patients with or without chronic portal vein thrombosis.8
Unfortunately, we have no data from prospective controlled trials and only limited data from retrospective studies to make a strong recommendation for or against anticoagulation in either acute and chronic portal vein thrombosis associated with cirrhosis. As such, each case must be evaluated on an individual basis in association with expert consultation.
In our experience, the risk of bleeding in patients with liver cirrhosis is substantial because of the decreased synthesis of coagulation factors and the presence of varices, whereas the efficacy and the benefits of recanalizing the portal vein in asymptomatic patients with liver cirrhosis and portal vein thrombosis are unknown. Therefore, unless the thrombosis extends into the mesenteric vein, thus posing a risk of mesenteric ischemia, we do not generally recommend anticoagulation in asymptomatic portal vein thrombosis in patients with cirrhosis.
No. in general, the decision to treat portal vein thrombosis with anticoagulant drugs is complex and depends on whether the thrombosis is acute or chronic, and whether the cause is a local factor, cirrhosis of the liver, or a systemic condition (Table 1). A “one-size-fits-all” approach should be avoided (Figure 1).
ACUTE PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
No randomized controlled trial has yet evaluated anticoagulation in acute portal vein thrombosis. But a prospective study published in 2010 showed that the portal vein and its left or right branch were patent in 39% of anticoagulated patients (vs 13% initially), the splenic vein in 80% (vs 57% initially), and the superior mesenteric vein in 73% (vs 42% initially).1 Further, there appears to be a 20% reduction in the overall mortality rate associated with anticoagulation for acute portal vein thrombosis in retrospective studies.2
In the absence of contraindications, anticoagulation with heparin or low-molecular-weight heparin is recommended, with complete bridging to oral anticoagulation with a vitamin K antagonist. Anticoagulation should be continued for at least 3 months, and indefinitely in patients with permanent hypercoaguable risk factors.3
CHRONIC PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
All patients with chronic portal vein thrombosis should undergo esophagogastroduodenoscopy to evaluate for varices. Patients with large varices should be treated orally with a nonselective beta-adrenergic blocker or endoscopically. Though no prospective study has validated this practice, a retrospective analysis showed a decreased risk of first or recurrent bleeding.4
In 2007, a retrospective study showed a lower rate of death in patients with portomesenteric venous thrombosis treated with an oral vitamin K antagonist.5 Patients with chronic portal vein thrombosis with ongoing thrombotic risk factors should be treated with long-term anticoagulation after screening for varices, and if varices are present, primary prophylaxis should be started.3 With this approach, less than 5% of patients died from classic complications of portal vein thrombosis at 5 years of follow-up.4
ACUTE OR CHRONIC PORTAL VEIN THROMBOSIS WITH CIRRHOSIS
Portal vein thrombosis is common in patients with underlying cirrhosis. The risk in patients with cirrhosis significantly increases as liver function worsens. In patients with well-compensated cirrhosis, the risk is less than 1% vs 8% to 25% in those with advanced cirrhosis.6
In patients awaiting liver transplantation, a large retrospective study7 showed that the rate of partial or complete recanalization of the splanchnic veins was significantly higher in those who received anticoagulation (8 of 19) than in those who did not (0 of 10, P = .002). The rate of survival was significantly lower in those who had complete thrombotic obstruction of the portal vein at the time of surgery (P = .04). However, there was no difference in survival rates between those with partial obstruction who received anticoagulation and those with a patent portal vein.7
A later retrospective study8 showed no significant benefit in the rate of transplantation-free survival or survival after liver transplantation in patients with or without chronic portal vein thrombosis.8
Unfortunately, we have no data from prospective controlled trials and only limited data from retrospective studies to make a strong recommendation for or against anticoagulation in either acute and chronic portal vein thrombosis associated with cirrhosis. As such, each case must be evaluated on an individual basis in association with expert consultation.
In our experience, the risk of bleeding in patients with liver cirrhosis is substantial because of the decreased synthesis of coagulation factors and the presence of varices, whereas the efficacy and the benefits of recanalizing the portal vein in asymptomatic patients with liver cirrhosis and portal vein thrombosis are unknown. Therefore, unless the thrombosis extends into the mesenteric vein, thus posing a risk of mesenteric ischemia, we do not generally recommend anticoagulation in asymptomatic portal vein thrombosis in patients with cirrhosis.
- Plessier A, Darwish-Murad S, Hernandez-Guerra M, et al; European Network for Vascular Disorders of the Liver (EN-Vie). Acute portal vein thrombosis unrelated to cirrhosis: a prospective multicenter follow-up study. Hepatology 2010; 51:210–218.
- Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med 2001; 345:1683–1688.
- de Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut 2005; 54:691–697.
- John BV, Konjeti VR, Aggarwal A, et al. The impact of portal vein thrombosis (PVT) on cirrhotics awaiting liver transplantation (abstract). Hepatology 2010; 52(suppl1):888A–889A.
- Plessier A, Darwish-Murad S, Hernandez-Guerra M, et al; European Network for Vascular Disorders of the Liver (EN-Vie). Acute portal vein thrombosis unrelated to cirrhosis: a prospective multicenter follow-up study. Hepatology 2010; 51:210–218.
- Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med 2001; 345:1683–1688.
- de Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut 2005; 54:691–697.
- John BV, Konjeti VR, Aggarwal A, et al. The impact of portal vein thrombosis (PVT) on cirrhotics awaiting liver transplantation (abstract). Hepatology 2010; 52(suppl1):888A–889A.
An uncommon syndrome makes us reflect on our approach to diagnosis

In this issue of the Journal, Dr. Soumya Chatterjee and colleagues discuss the antisynthetase syndrome. Although uncommon, this syndrome is important for internists and subspecialists to be aware of. Patients present in several different ways, and potentially life-threatening organ involvement may initially not be recognized or may not be linked with other components of the syndrome, such as involvement of the lungs, muscles, heart, and esophagus and fever.
I am currently on our inpatient rheumatology consultation service, and so I am reminded daily of the challenges hospitalists and subspecialists confront in ordering tests while trying to balance limiting length of stay with cost-efficiency and the desire to obtain a correct diagnosis. And I am repeatedly sensitized to several common test-ordering pitfalls intrinsic to the evaluation of patients with multisystem disease, including myositis. Most have a shared theme—limited time is spent in thoughtful reflection before ordering.
Patients with myositis rarely present with the textbook description of proximal muscle weakness. They describe fatigue, malaise, and sometimes a generalized sense of weakness. It is the probing questioning of their functional capacity and focused examination that reveal that the weakness is characterized by difficulty getting up off the floor, out of a low chair, or off the toilet. Then, with further questioning, some patients note that their fatigue and tiredness may also include getting winded easily with exertion, such as when climbing stairs, thus raising the question of cardiac dysfunction, pulmonary hypertension, or interstitial lung disease.
The responses to those probing questions and the subsequent examination should transform the interpretation of elevated aminotransferase levels (“liver tests”: AST and ALT) from liver disease into suspicion of muscle disease and the appropriate ordering of the creatine kinase level (avoiding liver imaging and hepatology consultation). The carefully repeated and now focused neurologic examination distinguishes the initial “poor cooperation” from the proximal weakness of myopathy. The probing interview leads to the performance of a focused physical examination that frames the appropriate interpretation of the routinely obtained “admission lab studies”!
The thoughtful history and examination are the basic stuff of clinical medicine that can easily be pushed aside by any of us as we deal with the tensions of high-volume, “high-throughput” medical care. It is a low-resistance path from hearing the symptom of fatigue with elevated “liver enzymes” to immediately checking ferritin, ceruloplasmin, and a hepatitis screen in preparation for getting a liver biopsy. It is easy to go through the motions without reflection. Easy, but sometimes wrong. And it is just as easy (but likely to be costly and unhelpful) to identify a patient prematurely with “possible autoimmune disease” and to immediately order a panoply of antinuclear and autoimmune serologies, including the Jo-1 autoantibody test.
As Dr. Chatterjee et al point out, we must continuously reflect on our diagnoses, for even after we navigate the pitfalls and avoid missing the diagnosis of myositis, if we don’t continuously assess all the patient’s symptoms, repeat the examination in a directed manner, and then look for circulating Jo-1 antibody when appropriate, we may well miss the opportunity to recognize that our patient’s ongoing fatigue with exertion is a reflection of the well-described association of myositis with interstitial lung disease (which may warrant a change in therapy), and not steroid myopathy or just poor conditioning.
Alternatively, in evaluating a patient who describes a year of feeling tired, suffering generalized muscle pains with low-grade fevers with temperatures of 99.8°F, and total exhaustion for 3 days after cleaning the oven, testing for antinuclear antibodies, extractable nuclear antigen antibodies, and a “vasculitis panel” in anticipation of a rheumatology consultation is not likely to be useful therapeutically or diagnostically.
Despite the daily pressures, we need to keep ourselves grounded in the fundamentals of clinical care: careful listening, purposeful examination, and directed use of laboratory tests and imaging. The downstream consequences of ordering tests for the sake of efficient throughput are quite real, and thoughtful test ordering is one step toward quality care, as well as cost-effective care.
In future months, the Journal will delve more deeply into test ordering when, in a joint effort with the American College of Physicians, we will be discussing the use and misuse of specific tests.
In this issue of the Journal, Dr. Soumya Chatterjee and colleagues discuss the antisynthetase syndrome. Although uncommon, this syndrome is important for internists and subspecialists to be aware of. Patients present in several different ways, and potentially life-threatening organ involvement may initially not be recognized or may not be linked with other components of the syndrome, such as involvement of the lungs, muscles, heart, and esophagus and fever.
I am currently on our inpatient rheumatology consultation service, and so I am reminded daily of the challenges hospitalists and subspecialists confront in ordering tests while trying to balance limiting length of stay with cost-efficiency and the desire to obtain a correct diagnosis. And I am repeatedly sensitized to several common test-ordering pitfalls intrinsic to the evaluation of patients with multisystem disease, including myositis. Most have a shared theme—limited time is spent in thoughtful reflection before ordering.
Patients with myositis rarely present with the textbook description of proximal muscle weakness. They describe fatigue, malaise, and sometimes a generalized sense of weakness. It is the probing questioning of their functional capacity and focused examination that reveal that the weakness is characterized by difficulty getting up off the floor, out of a low chair, or off the toilet. Then, with further questioning, some patients note that their fatigue and tiredness may also include getting winded easily with exertion, such as when climbing stairs, thus raising the question of cardiac dysfunction, pulmonary hypertension, or interstitial lung disease.
The responses to those probing questions and the subsequent examination should transform the interpretation of elevated aminotransferase levels (“liver tests”: AST and ALT) from liver disease into suspicion of muscle disease and the appropriate ordering of the creatine kinase level (avoiding liver imaging and hepatology consultation). The carefully repeated and now focused neurologic examination distinguishes the initial “poor cooperation” from the proximal weakness of myopathy. The probing interview leads to the performance of a focused physical examination that frames the appropriate interpretation of the routinely obtained “admission lab studies”!
The thoughtful history and examination are the basic stuff of clinical medicine that can easily be pushed aside by any of us as we deal with the tensions of high-volume, “high-throughput” medical care. It is a low-resistance path from hearing the symptom of fatigue with elevated “liver enzymes” to immediately checking ferritin, ceruloplasmin, and a hepatitis screen in preparation for getting a liver biopsy. It is easy to go through the motions without reflection. Easy, but sometimes wrong. And it is just as easy (but likely to be costly and unhelpful) to identify a patient prematurely with “possible autoimmune disease” and to immediately order a panoply of antinuclear and autoimmune serologies, including the Jo-1 autoantibody test.
As Dr. Chatterjee et al point out, we must continuously reflect on our diagnoses, for even after we navigate the pitfalls and avoid missing the diagnosis of myositis, if we don’t continuously assess all the patient’s symptoms, repeat the examination in a directed manner, and then look for circulating Jo-1 antibody when appropriate, we may well miss the opportunity to recognize that our patient’s ongoing fatigue with exertion is a reflection of the well-described association of myositis with interstitial lung disease (which may warrant a change in therapy), and not steroid myopathy or just poor conditioning.
Alternatively, in evaluating a patient who describes a year of feeling tired, suffering generalized muscle pains with low-grade fevers with temperatures of 99.8°F, and total exhaustion for 3 days after cleaning the oven, testing for antinuclear antibodies, extractable nuclear antigen antibodies, and a “vasculitis panel” in anticipation of a rheumatology consultation is not likely to be useful therapeutically or diagnostically.
Despite the daily pressures, we need to keep ourselves grounded in the fundamentals of clinical care: careful listening, purposeful examination, and directed use of laboratory tests and imaging. The downstream consequences of ordering tests for the sake of efficient throughput are quite real, and thoughtful test ordering is one step toward quality care, as well as cost-effective care.
In future months, the Journal will delve more deeply into test ordering when, in a joint effort with the American College of Physicians, we will be discussing the use and misuse of specific tests.
In this issue of the Journal, Dr. Soumya Chatterjee and colleagues discuss the antisynthetase syndrome. Although uncommon, this syndrome is important for internists and subspecialists to be aware of. Patients present in several different ways, and potentially life-threatening organ involvement may initially not be recognized or may not be linked with other components of the syndrome, such as involvement of the lungs, muscles, heart, and esophagus and fever.
I am currently on our inpatient rheumatology consultation service, and so I am reminded daily of the challenges hospitalists and subspecialists confront in ordering tests while trying to balance limiting length of stay with cost-efficiency and the desire to obtain a correct diagnosis. And I am repeatedly sensitized to several common test-ordering pitfalls intrinsic to the evaluation of patients with multisystem disease, including myositis. Most have a shared theme—limited time is spent in thoughtful reflection before ordering.
Patients with myositis rarely present with the textbook description of proximal muscle weakness. They describe fatigue, malaise, and sometimes a generalized sense of weakness. It is the probing questioning of their functional capacity and focused examination that reveal that the weakness is characterized by difficulty getting up off the floor, out of a low chair, or off the toilet. Then, with further questioning, some patients note that their fatigue and tiredness may also include getting winded easily with exertion, such as when climbing stairs, thus raising the question of cardiac dysfunction, pulmonary hypertension, or interstitial lung disease.
The responses to those probing questions and the subsequent examination should transform the interpretation of elevated aminotransferase levels (“liver tests”: AST and ALT) from liver disease into suspicion of muscle disease and the appropriate ordering of the creatine kinase level (avoiding liver imaging and hepatology consultation). The carefully repeated and now focused neurologic examination distinguishes the initial “poor cooperation” from the proximal weakness of myopathy. The probing interview leads to the performance of a focused physical examination that frames the appropriate interpretation of the routinely obtained “admission lab studies”!
The thoughtful history and examination are the basic stuff of clinical medicine that can easily be pushed aside by any of us as we deal with the tensions of high-volume, “high-throughput” medical care. It is a low-resistance path from hearing the symptom of fatigue with elevated “liver enzymes” to immediately checking ferritin, ceruloplasmin, and a hepatitis screen in preparation for getting a liver biopsy. It is easy to go through the motions without reflection. Easy, but sometimes wrong. And it is just as easy (but likely to be costly and unhelpful) to identify a patient prematurely with “possible autoimmune disease” and to immediately order a panoply of antinuclear and autoimmune serologies, including the Jo-1 autoantibody test.
As Dr. Chatterjee et al point out, we must continuously reflect on our diagnoses, for even after we navigate the pitfalls and avoid missing the diagnosis of myositis, if we don’t continuously assess all the patient’s symptoms, repeat the examination in a directed manner, and then look for circulating Jo-1 antibody when appropriate, we may well miss the opportunity to recognize that our patient’s ongoing fatigue with exertion is a reflection of the well-described association of myositis with interstitial lung disease (which may warrant a change in therapy), and not steroid myopathy or just poor conditioning.
Alternatively, in evaluating a patient who describes a year of feeling tired, suffering generalized muscle pains with low-grade fevers with temperatures of 99.8°F, and total exhaustion for 3 days after cleaning the oven, testing for antinuclear antibodies, extractable nuclear antigen antibodies, and a “vasculitis panel” in anticipation of a rheumatology consultation is not likely to be useful therapeutically or diagnostically.
Despite the daily pressures, we need to keep ourselves grounded in the fundamentals of clinical care: careful listening, purposeful examination, and directed use of laboratory tests and imaging. The downstream consequences of ordering tests for the sake of efficient throughput are quite real, and thoughtful test ordering is one step toward quality care, as well as cost-effective care.
In future months, the Journal will delve more deeply into test ordering when, in a joint effort with the American College of Physicians, we will be discussing the use and misuse of specific tests.
Antisynthetase syndrome: Not just an inflammatory myopathy
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
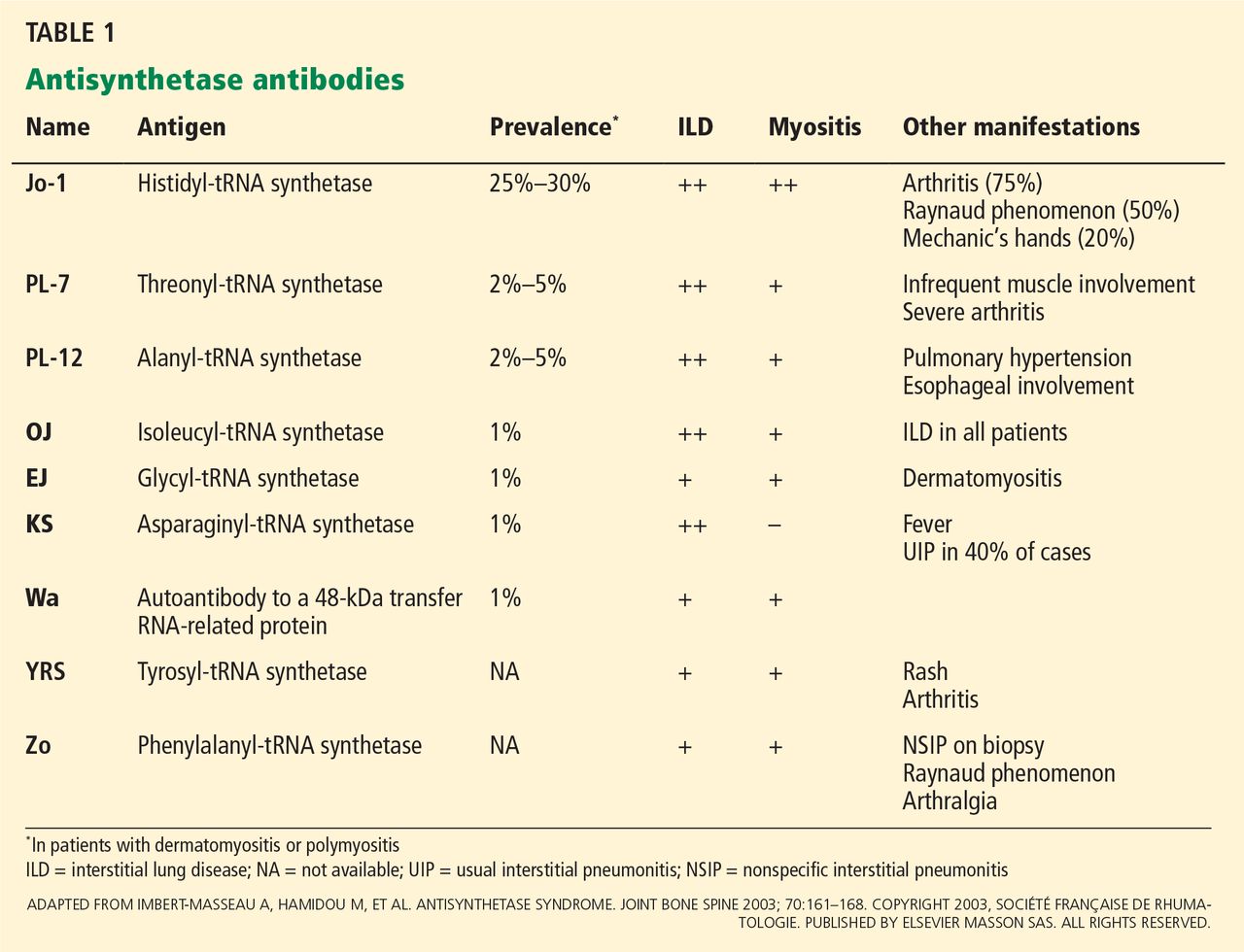
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
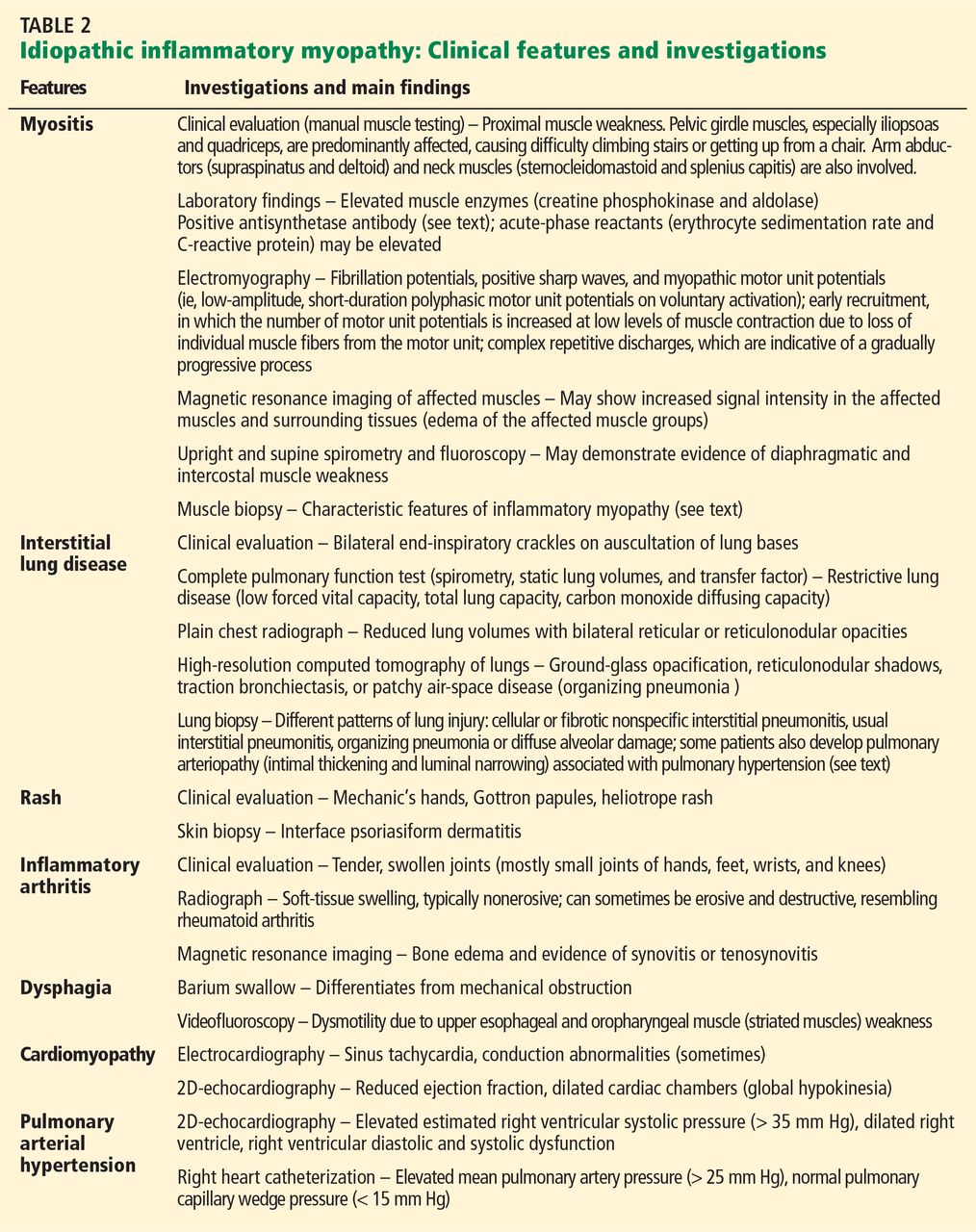
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
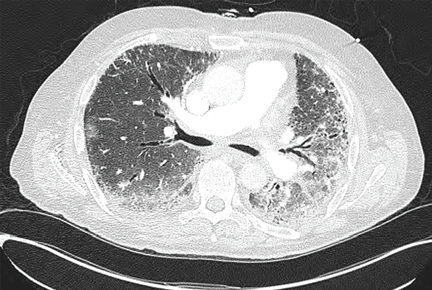
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.
Mechanic’s hands
In about 30% of patients, the skin of the tips and margins of the fingers becomes thickened, hyperkeratotic, and fissured, the appearance of which is classically described as mechanic’s hands. It is a common manifestation of antisynthetase syndrome and is particularly prominent on the radial side of the index fingers (Figure 1). Biopsy of affected skin shows an interface psoriasiform dermatitis.19 In addition, some dermatomyositis patients with Gottron papules and a heliotrope rash have antisynthetase antibodies.
Raynaud phenomenon
Raynaud phenomenon develops in about 40% of patients. Some have nailfold capillary abnormalities.20 However, persistent or severe digital ischemia leading to digital ulceration or infarction is uncommon.21
Inflammatory arthritis
Arthralgias and arthritis are common (50%), the most common form being a symmetric polyarthritis of the small joints of the hands and feet. It is typically nonerosive but can sometimes be erosive and destructive.20
Because inflammatory arthritis mimics rheumatoid arthritis, antisynthetase syndrome should be considered in rheumatoid factor-negative patients presenting with polyarthritis.
ASSOCIATION WITH MALIGNANCY
Traditional teaching has been that antisynthetase antibody is protective against an underlying malignancy.22,23 However, several recently published case studies have reported various malignancies occurring within 6 to 12 months of the diagnosis of antisynthetase syndrome.7,24 The debate as to whether these are chance associations or causal (a paraneoplastic phenomenon) has not been resolved at this time.24
It is now recommended that patients with antisynthetase syndrome be screened for malignancies as appropriate for the patient’s age and sex. Screening should include a careful history and physical examination, complete blood cell count, comprehensive metabolic panel, chest radiography, mammography, and a gynecologic examination for women.25 If abnormalities are found, a more thorough evaluation for cancer is appropriate.
DIAGNOSIS
Muscle enzyme levels are often elevated
Muscle enzymes (creatine kinase and aldolase) are often elevated. Serum creatine kinase levels can range between 5 to 50 times the upper limit of normal. In an established case, creatine kinase levels along with careful manual muscle strength testing may help evaluate myositis activity. However, in chronic and advanced disease, creatine kinase may be within the normal range despite active myositis, partly because of extensive loss of muscle mass. In myositis, it may be prudent to check both creatine kinase and aldolase; sometimes only serum aldolase level rises, when immune-mediated injury predominantly affects the early regenerative myocytes.26
Judicious use of autoantibody testing
The characteristic clinical presentation is the initial clue to the diagnosis of antisynthetase syndrome, which is then supported by serologic testing.
Injudicious testing for a long list of antibodies should be avoided, as the cost is considerable and it does not influence further management. However, ordering an anti-Jo-1 antibody test in the correct clinical setting is appropriate, as it has high specificity,27,28 and thus can help establish or refute the clinical suspicion of antisynthetase syndrome.
Screening pulmonary function testing and thoracic high-resolution computed tomography for all patients with polymyositis or dermatomyositis is not considered “standard of care” and will likely not be reimbursed by third-party payers. However, in a patient with symptoms and signs of myositis, the presence of an antisynthetase antibody should prompt screening for occult interstitial lung disease, even in the absence of symptoms. As lung disease ultimately determines the prognosis in antisynthetase syndrome, early diagnosis and management is the key. Therefore, these tests would likely be approved to establish the diagnosis of interstitial lung disease and evaluate its severity.
If a myositis patient is also found to have interstitial lung disease or develops mechanic’s hands, the likely diagnosis is antisynthetase syndrome, which can be confirmed by serologic testing for antisynthetase antibodies. Interstitial lung disease in antisynthetase syndrome is often from “nonspecific interstitial pneumonitis”; therefore, medications tested and proven effective for this condition should be approved and reimbursed by payers.29–32
The coexistence of myositis and interstitial lung disease increases the sensitivity of anti-Jo-1 antibody.11 Thus, the clinician can have more confidence in early recognition and initiation of aggressive but targeted disease-modifying therapy.
Various methods can be used for detecting antisynthetase antibodies, with comparable results.28 Anti-Jo-1 antibody testing costs about $140. If that test is negative and antisynthetase syndrome is still suspected, then testing for the non-Jo-1 antisynthetase antibodies may be justified (Table 1). Though the cost of this panel of autoantibodies is about $300, it helps to confirm the diagnosis, and it influences the choice of second-line immunosuppressive agents such as tacrolimus29 and rituximab32 in patients resistant to conventional immunosuppressive agents such as azathioprine and methotrexate.
Often, anti-Ro52 SS-A antibodies are present concurrently in patients with anti-Jo-1 syndrome.33 In observational studies in patients with anti-Jo-1 antibody-associated interstitial lung disease, coexistence of anti-Ro52 SS-A antibody tended to predict a worse pulmonary outcome than in those with anti-Jo-1 antibody alone.34,35
Electromyography
Electromyography not only helps differentiate between myopathic and neuropathic weakness, but it may also support the diagnosis of “inflammatory” myopathy as suggested by prominent muscle membrane irritability (fibrillations, positive sharp waves) and abnormal motor unit action potentials (spontaneous activity showing small, short, polyphasic potentials and early recruitment). However, the findings can be nonspecific, and may even be normal in 10% to 15% of patients.36 Electromyographic abnormalities are most consistently observed in weak proximal muscles, and electromyography is also helpful in selecting a muscle for biopsy. Although no single electromyographic pattern is considered diagnostic for inflammatory myopathy, abnormalities are present in around 90% of patients (Table 2).3
Magnetic resonance imaging
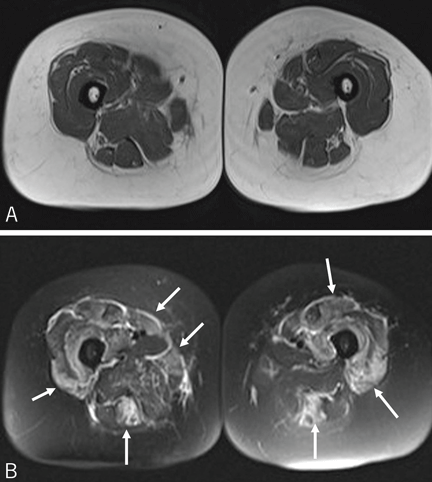
Magnetic resonance imaging may show increased signal intensity in the affected muscles and surrounding tissues (Figure 3).37 Because it lacks sensitivity and specificity, magnetic resonance imaging is not helpful in diagnosing the disease. However, in the correct clinical setting, it may be used to guide muscle biopsy, and it can help in monitoring the disease progress.38
Muscle histopathology
Muscle biopsy, though often helpful, is not always diagnostic, and antisynthetase syndrome should still be suspected in the right clinical context, even in the absence of characteristic pathologic changes.
Biopsy of sites recently studied by electromyography should be avoided, and if the patient has undergone electromyography recently, the contralateral side should be selected for biopsy.
Reports of histopathologic findings in muscle biopsies in patients with antisynthetase syndrome document inflammatory myopathic features (Figure 4). In a series of patients with anti-Jo-1 syndrome, inflammation was noted in all cases, predominantly perimysial in location, with occasional endomysial and perivascular inflammation.39 Many of the inflammatory cells seen were macrophages and lymphocytes, in contrast to the predominantly lymphocytic infiltrates described in classic polymyositis and dermatomyositis. Perifascicular atrophy, similar to what is seen in dermatomyositis, was encountered; however, vascular changes, typical of dermatomyositis, were absent. Occasional degenerating and regenerating muscle fibers were also observed in most cases. Additionally, a characteristic perimysial connective tissue fragmentation was described, a feature less often seen in classic polymyositis and dermatomyositis.39
Pulmonary function testing
If antisynthetase syndrome is suspected or diagnosed, baseline pulmonary function testing (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease (reduced forced vital capacity and carbon monoxide diffusion capacity), and the diagnosis will be substantiated on thoracic high-resolution computed tomography. Respiratory muscle weakness can be detected with upright and supine spirometry.40 Weakness of these muscles contributes to shortness of breath, and patients may need ventilatory support.
Thoracic high-resolution computed tomography
Different patterns of lung injury can be seen in antisynthetase syndrome. Diffuse ground-glass opacification may suggest a nonspecific interstitial pneumonitis pattern, which is the most common form of interstitial lung disease (Figure 2), whereas coarse reticulation or honeycombing correlates with a usual interstitial pneumonitis pattern. Patchy consolidation or air-space disease can occur if cryptogenic organizing pneumonia is the predominant pattern of lung injury.
Swallowing evaluation
A comprehensive swallowing evaluation by a speech therapist may be necessary for evaluation of dysphagia (from oropharyngeal and striated esophageal muscle weakness) and determination of aspiration risk (Table 2).
Lung histopathology
If necessary, a surgical lung biopsy is needed to document the pathologic pattern of injury, including the amount of fibrosis in the lung. Historically, in idiopathic inflammatory myopathy patients in general, this has taken the form of usual interstitial pneumonia, organizing pneumonia, or diffuse alveolar damage.41 With the emergence of the definition of nonspecific interstitial pneumonitis and fibrosis as a documented and accepted pattern, more studies have found this to be the most common pattern of lung injury.16 It is characterized by diffuse involvement of the lung by an interstitial chronic inflammatory infiltrate, a cellular type of nonspecific interstitial pneumonitis that progresses in a uniform pattern to a fibrotic type (Figure 5). This form of fibrosis rarely results in significant remodeling, so-called honeycomb changes. In addition, anti-Jo-1 antibody patients may also have an increased incidence of acute lung injury, including acute diffuse alveolar damage that is often superimposed on the underlying chronic lung disease.42
In patients with pulmonary hypertension, histopathologic studies of the muscular pulmonary arteries often show moderate intimal fibroplasia, suggesting that a pulmonary arteriopathy with intimal thickening and luminal narrowing develops in some of these patients (Figure 5), independent of chronic hypoxic pulmonary vasoconstriction or vascular obstruction due to its entrapment within fibrotic lung tissue.17
TREATMENT
Glucocorticoids are the mainstay
Glucocorticoids are considered the mainstay of treatment. Patients should be advised that long-term use of glucocorticoids is necessary, though the response is variable. It is also important to discuss possible side effects of long-term glucocorticoid use.
Standard practice is to initiate treatment with high doses for the first 4 to 6 weeks to achieve disease control, followed by a slow taper over the next 9 to 12 months to the lowest effective dose to maintain remission. If the patient is profoundly weak, especially with respiratory muscle weakness or significant dysphagia and aspiration risk, hospital admission for intravenous methylprednisolone 1,000 mg daily for 3 to 5 days may be necessary. Otherwise, oral prednisone 1 mg/kg/day would be the usual starting dose.
If the patient’s muscle strength initially improves and then declines weeks to months later despite adequate therapy, glucocorticoid-induced myopathy should be suspected, especially if the muscle enzymes are within the reference range. This is more likely to occur if high-dose prednisone is continued for more than 6 to 8 weeks.
Improvement in muscle strength, which can take several weeks to several months, is a more reliable indicator of response to therapy than the serum creatine kinase level, which may take much longer to normalize. Relying on normalization of the creatine kinase level alone may lead to unnecessary prolongation of high-dose glucocorticoid therapy. It may take several months for the muscle enzymes to normalize, and there is usually a time lag between normalization of muscle enzymes and complete recovery of muscle strength.
Long-term use of high-dose prednisone leads to glucocorticoid-induced osteoporosis. Therefore, patients should receive osteoporosis prophylaxis including antiresorptive therapy with a bisphosphonate. In addition, prophylaxis against Pneumocystis jirovecii is indicated for patients treated with high-dose glucocorticoids.
Additional immunosuppressive agents
Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine and methotrexate are often required, both as glucocorticoid-sparing agents and to achieve adequate disease control.10 Addition of such agents from the outset is particularly necessary in patients with profound muscle weakness or those who have concomitant symptomatic interstitial lung disease.
No randomized controlled trial comparing azathioprine and methotrexate has been conducted to date. Therefore, the choice is based on patient preference, presence of coexisting interstitial lung disease or liver disease, commitment to limit alcohol consumption, and thiopurine methyltransferase status. Most patients need prolonged therapy.
In a randomized clinical trial, concomitant therapy with prednisone and azathioprine resulted in better functional outcomes and a significantly lower prednisone dose requirement for maintenance therapy at 3 years than with prednisone alone.43,44 Although no such randomized study has been conducted using methotrexate, several retrospective studies have demonstrated 70% to 80% response rates, including those for whom monotherapy with glucocorticoids had failed.45,46 The combination of methotrexate and azathioprine may be beneficial in patients who previously had inadequate responses to either of these agents alone.47
For severe pulmonary involvement associated with antisynthetase syndrome, monthly intravenous infusion of cyclophosphamide has been shown to be effective.48,49
Some recent studies established the role of tacrolimus in the treatment of both interstitial lung disease and myositis associated with antisynthetase syndrome.29 Cyclosporine has also been successfully used in a case of interstitial lung disease associated with anti Jo-1 syndrome.30
Rituximab, a monoclonal antibody to Blymphocyte antigen CD20, can also be used successfully in refractory disease,31 including refractory interstitial lung disease.32
In an open-label prospective study, polymyositis refractory to glucocorticoids and multiple conventional immunosuppressive agents responded well to high-dose intravenous immune globulin in the short term.50 However, the antisynthetase antibody status in this cohort was unknown; therefore, no definite conclusion could be drawn about the efficacy of intravenous immune globulin specifically in antisynthetase syndrome.
General measures
In patients with profound muscle weakness, physical therapy and rehabilitation should begin early. The goal is to reduce further muscle wasting from disuse and prevent muscle contractures. Patients with oropharyngeal and esophageal dysmotility should be advised about aspiration precautions and may need a swallow evaluation by a speech therapist; some may need temporary parenteral hyper-alimentation or J-tube insertion.
PROGNOSIS
If skeletal muscle involvement is the sole manifestation of antisynthetase syndrome, patients usually respond to glucocorticoids and immunosuppressive therapy and do fairly well. However, the outcome is not so promising when patients also develop interstitial lung disease, and the severity and type of lung injury usually determine the prognosis. As expected, patients with a progressive course of interstitial lung disease fare poorly, whereas those with a nonprogressive course tend to do relatively better. Older age at onset (> 60 years), presence of a malignancy, and a negative antinuclear antibody test are associated with a poor prognosis.7
Acknowledgment: The authors are grateful to Dr. Stephen Hatem, MD, staff radiologist, musculoskeletal radiology, Cleveland Clinic Imaging Institute, for help in the preparation of the magnetic resonance images. We also thank Dr. Steven Shook, MD, staff neurologist, Cleveland Clinic Neurological Institute, for help in summarizing the EMG findings.
- Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13:175–181.
- Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum 1980; 23:881–888.
- Nagaraju K, Lundberg IE. Inflammatory diseases of muscle and other myopathies. In:Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. Philadelphia, PA: Saunders; 2008:1353–1380.
- Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60:116–123.
- Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum 1996; 39:292–296.
- Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum 1996; 39:1507–1518.
- Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with antisynthetase autoantibodies. Intern Med J 2011; 41:674–679.
- Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 1996; 26:459–467.
- Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39:233–241.
- Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53–59.
- Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol 2000; 12:475–481.
- Ancuta CM, Ancuta E, Chirieac RM. Aminoacyl-tRNA synthetases in idiopathic inflammatory myopathies: an update on immunopathogenic significance, clinical and therapeutic implications. In:Gran JT, editor. Idiopathic Inflammatory Myopathies - Recent Developments. Rijeka, Croatia: InTech; 2011:77–90.
- Kajihara M, Kuwana M, Tokuda H, et al. Myositis and interstitial lung disease associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27:2707–2710.
- Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54:2004–2009.
- Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep 2003; 5:425–430.
- Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23:874–880.
- Chatterjee S, Farver C. Severe pulmonary hypertension in anti-Jo-1 syndrome. Arthritis Care Res (Hoboken) 2010; 62:425–429.
- Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus 2009; 18:1006–1010.
- Bugatti L, De Angelis R, Filosa G, Salaffi F. Bilateral, asymptomatic scaly and fissured cutaneous lesions of the fingers in a patient presenting with myositis. Indian J Dermatol Venereol Leprol 2005; 71:137–138.
- Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010; 16:307–312.
- Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum 1992; 35:449–456.
- Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70:360–374.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol 2001; 144:825–831.
- Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol 2008; 35:169–171.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol 2010; 22:627–632.
- Casciola-Rosen L, Hall JC, Mammen AL, Christopher-Stine L, Rosen A. Isolated elevation of aldolase in the serum of myositis patients: a potential biomarker of damaged early regenerating muscle cells. Clin Exp Rheumatol 2012; 30:548–553.
- Shovman O, Gilburd B, Barzilai O, et al. Evaluation of the BioPlex 2200 ANA screen: analysis of 510 healthy subjects: incidence of natural/predictive autoantibodies. Ann N Y Acad Sci 2005; 1050:380–388.
- Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity 2005; 38:73–78.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Jankowska M, Butto B, Debska-Slizien A, Rutkowski B. Beneficial effect of treatment with cyclosporin A in a case of refractory antisynthetase syndrome. Rheumatol Int 2007; 27:775–780.
- Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 2012; 42:e4–e7.
- Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106:581–587.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32–40.
- La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39:249–253.
- Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29:989–994.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255–286.
- Reimers CD, Finkenstaedt M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 9:475–485.
- O’Connell MJ. Whole-body MR imaging in the diagnosis of polymyositis. Am J Roentgenol 2002; 179:967–971.
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472–478.
- Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil 2001; 82:123–128.
- Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(suppl 1):513S–519S.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162:2213–2217.
- Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 1980; 92:365–369.
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum 1981; 24:45–48.
- Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182–189.
- Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379–387.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998; 41:392–399.
- Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46:124–130.
- al-Janadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/dermatomyositis. J Rheumatol 1989; 16:1592–1596.
- Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum 2002; 46:467–474.
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.
Mechanic’s hands
In about 30% of patients, the skin of the tips and margins of the fingers becomes thickened, hyperkeratotic, and fissured, the appearance of which is classically described as mechanic’s hands. It is a common manifestation of antisynthetase syndrome and is particularly prominent on the radial side of the index fingers (Figure 1). Biopsy of affected skin shows an interface psoriasiform dermatitis.19 In addition, some dermatomyositis patients with Gottron papules and a heliotrope rash have antisynthetase antibodies.
Raynaud phenomenon
Raynaud phenomenon develops in about 40% of patients. Some have nailfold capillary abnormalities.20 However, persistent or severe digital ischemia leading to digital ulceration or infarction is uncommon.21
Inflammatory arthritis
Arthralgias and arthritis are common (50%), the most common form being a symmetric polyarthritis of the small joints of the hands and feet. It is typically nonerosive but can sometimes be erosive and destructive.20
Because inflammatory arthritis mimics rheumatoid arthritis, antisynthetase syndrome should be considered in rheumatoid factor-negative patients presenting with polyarthritis.
ASSOCIATION WITH MALIGNANCY
Traditional teaching has been that antisynthetase antibody is protective against an underlying malignancy.22,23 However, several recently published case studies have reported various malignancies occurring within 6 to 12 months of the diagnosis of antisynthetase syndrome.7,24 The debate as to whether these are chance associations or causal (a paraneoplastic phenomenon) has not been resolved at this time.24
It is now recommended that patients with antisynthetase syndrome be screened for malignancies as appropriate for the patient’s age and sex. Screening should include a careful history and physical examination, complete blood cell count, comprehensive metabolic panel, chest radiography, mammography, and a gynecologic examination for women.25 If abnormalities are found, a more thorough evaluation for cancer is appropriate.
DIAGNOSIS
Muscle enzyme levels are often elevated
Muscle enzymes (creatine kinase and aldolase) are often elevated. Serum creatine kinase levels can range between 5 to 50 times the upper limit of normal. In an established case, creatine kinase levels along with careful manual muscle strength testing may help evaluate myositis activity. However, in chronic and advanced disease, creatine kinase may be within the normal range despite active myositis, partly because of extensive loss of muscle mass. In myositis, it may be prudent to check both creatine kinase and aldolase; sometimes only serum aldolase level rises, when immune-mediated injury predominantly affects the early regenerative myocytes.26
Judicious use of autoantibody testing
The characteristic clinical presentation is the initial clue to the diagnosis of antisynthetase syndrome, which is then supported by serologic testing.
Injudicious testing for a long list of antibodies should be avoided, as the cost is considerable and it does not influence further management. However, ordering an anti-Jo-1 antibody test in the correct clinical setting is appropriate, as it has high specificity,27,28 and thus can help establish or refute the clinical suspicion of antisynthetase syndrome.
Screening pulmonary function testing and thoracic high-resolution computed tomography for all patients with polymyositis or dermatomyositis is not considered “standard of care” and will likely not be reimbursed by third-party payers. However, in a patient with symptoms and signs of myositis, the presence of an antisynthetase antibody should prompt screening for occult interstitial lung disease, even in the absence of symptoms. As lung disease ultimately determines the prognosis in antisynthetase syndrome, early diagnosis and management is the key. Therefore, these tests would likely be approved to establish the diagnosis of interstitial lung disease and evaluate its severity.
If a myositis patient is also found to have interstitial lung disease or develops mechanic’s hands, the likely diagnosis is antisynthetase syndrome, which can be confirmed by serologic testing for antisynthetase antibodies. Interstitial lung disease in antisynthetase syndrome is often from “nonspecific interstitial pneumonitis”; therefore, medications tested and proven effective for this condition should be approved and reimbursed by payers.29–32
The coexistence of myositis and interstitial lung disease increases the sensitivity of anti-Jo-1 antibody.11 Thus, the clinician can have more confidence in early recognition and initiation of aggressive but targeted disease-modifying therapy.
Various methods can be used for detecting antisynthetase antibodies, with comparable results.28 Anti-Jo-1 antibody testing costs about $140. If that test is negative and antisynthetase syndrome is still suspected, then testing for the non-Jo-1 antisynthetase antibodies may be justified (Table 1). Though the cost of this panel of autoantibodies is about $300, it helps to confirm the diagnosis, and it influences the choice of second-line immunosuppressive agents such as tacrolimus29 and rituximab32 in patients resistant to conventional immunosuppressive agents such as azathioprine and methotrexate.
Often, anti-Ro52 SS-A antibodies are present concurrently in patients with anti-Jo-1 syndrome.33 In observational studies in patients with anti-Jo-1 antibody-associated interstitial lung disease, coexistence of anti-Ro52 SS-A antibody tended to predict a worse pulmonary outcome than in those with anti-Jo-1 antibody alone.34,35
Electromyography
Electromyography not only helps differentiate between myopathic and neuropathic weakness, but it may also support the diagnosis of “inflammatory” myopathy as suggested by prominent muscle membrane irritability (fibrillations, positive sharp waves) and abnormal motor unit action potentials (spontaneous activity showing small, short, polyphasic potentials and early recruitment). However, the findings can be nonspecific, and may even be normal in 10% to 15% of patients.36 Electromyographic abnormalities are most consistently observed in weak proximal muscles, and electromyography is also helpful in selecting a muscle for biopsy. Although no single electromyographic pattern is considered diagnostic for inflammatory myopathy, abnormalities are present in around 90% of patients (Table 2).3
Magnetic resonance imaging
Magnetic resonance imaging may show increased signal intensity in the affected muscles and surrounding tissues (Figure 3).37 Because it lacks sensitivity and specificity, magnetic resonance imaging is not helpful in diagnosing the disease. However, in the correct clinical setting, it may be used to guide muscle biopsy, and it can help in monitoring the disease progress.38
Muscle histopathology
Muscle biopsy, though often helpful, is not always diagnostic, and antisynthetase syndrome should still be suspected in the right clinical context, even in the absence of characteristic pathologic changes.
Biopsy of sites recently studied by electromyography should be avoided, and if the patient has undergone electromyography recently, the contralateral side should be selected for biopsy.
Reports of histopathologic findings in muscle biopsies in patients with antisynthetase syndrome document inflammatory myopathic features (Figure 4). In a series of patients with anti-Jo-1 syndrome, inflammation was noted in all cases, predominantly perimysial in location, with occasional endomysial and perivascular inflammation.39 Many of the inflammatory cells seen were macrophages and lymphocytes, in contrast to the predominantly lymphocytic infiltrates described in classic polymyositis and dermatomyositis. Perifascicular atrophy, similar to what is seen in dermatomyositis, was encountered; however, vascular changes, typical of dermatomyositis, were absent. Occasional degenerating and regenerating muscle fibers were also observed in most cases. Additionally, a characteristic perimysial connective tissue fragmentation was described, a feature less often seen in classic polymyositis and dermatomyositis.39
Pulmonary function testing
If antisynthetase syndrome is suspected or diagnosed, baseline pulmonary function testing (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease (reduced forced vital capacity and carbon monoxide diffusion capacity), and the diagnosis will be substantiated on thoracic high-resolution computed tomography. Respiratory muscle weakness can be detected with upright and supine spirometry.40 Weakness of these muscles contributes to shortness of breath, and patients may need ventilatory support.
Thoracic high-resolution computed tomography
Different patterns of lung injury can be seen in antisynthetase syndrome. Diffuse ground-glass opacification may suggest a nonspecific interstitial pneumonitis pattern, which is the most common form of interstitial lung disease (Figure 2), whereas coarse reticulation or honeycombing correlates with a usual interstitial pneumonitis pattern. Patchy consolidation or air-space disease can occur if cryptogenic organizing pneumonia is the predominant pattern of lung injury.
Swallowing evaluation
A comprehensive swallowing evaluation by a speech therapist may be necessary for evaluation of dysphagia (from oropharyngeal and striated esophageal muscle weakness) and determination of aspiration risk (Table 2).
Lung histopathology
If necessary, a surgical lung biopsy is needed to document the pathologic pattern of injury, including the amount of fibrosis in the lung. Historically, in idiopathic inflammatory myopathy patients in general, this has taken the form of usual interstitial pneumonia, organizing pneumonia, or diffuse alveolar damage.41 With the emergence of the definition of nonspecific interstitial pneumonitis and fibrosis as a documented and accepted pattern, more studies have found this to be the most common pattern of lung injury.16 It is characterized by diffuse involvement of the lung by an interstitial chronic inflammatory infiltrate, a cellular type of nonspecific interstitial pneumonitis that progresses in a uniform pattern to a fibrotic type (Figure 5). This form of fibrosis rarely results in significant remodeling, so-called honeycomb changes. In addition, anti-Jo-1 antibody patients may also have an increased incidence of acute lung injury, including acute diffuse alveolar damage that is often superimposed on the underlying chronic lung disease.42
In patients with pulmonary hypertension, histopathologic studies of the muscular pulmonary arteries often show moderate intimal fibroplasia, suggesting that a pulmonary arteriopathy with intimal thickening and luminal narrowing develops in some of these patients (Figure 5), independent of chronic hypoxic pulmonary vasoconstriction or vascular obstruction due to its entrapment within fibrotic lung tissue.17
TREATMENT
Glucocorticoids are the mainstay
Glucocorticoids are considered the mainstay of treatment. Patients should be advised that long-term use of glucocorticoids is necessary, though the response is variable. It is also important to discuss possible side effects of long-term glucocorticoid use.
Standard practice is to initiate treatment with high doses for the first 4 to 6 weeks to achieve disease control, followed by a slow taper over the next 9 to 12 months to the lowest effective dose to maintain remission. If the patient is profoundly weak, especially with respiratory muscle weakness or significant dysphagia and aspiration risk, hospital admission for intravenous methylprednisolone 1,000 mg daily for 3 to 5 days may be necessary. Otherwise, oral prednisone 1 mg/kg/day would be the usual starting dose.
If the patient’s muscle strength initially improves and then declines weeks to months later despite adequate therapy, glucocorticoid-induced myopathy should be suspected, especially if the muscle enzymes are within the reference range. This is more likely to occur if high-dose prednisone is continued for more than 6 to 8 weeks.
Improvement in muscle strength, which can take several weeks to several months, is a more reliable indicator of response to therapy than the serum creatine kinase level, which may take much longer to normalize. Relying on normalization of the creatine kinase level alone may lead to unnecessary prolongation of high-dose glucocorticoid therapy. It may take several months for the muscle enzymes to normalize, and there is usually a time lag between normalization of muscle enzymes and complete recovery of muscle strength.
Long-term use of high-dose prednisone leads to glucocorticoid-induced osteoporosis. Therefore, patients should receive osteoporosis prophylaxis including antiresorptive therapy with a bisphosphonate. In addition, prophylaxis against Pneumocystis jirovecii is indicated for patients treated with high-dose glucocorticoids.
Additional immunosuppressive agents
Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine and methotrexate are often required, both as glucocorticoid-sparing agents and to achieve adequate disease control.10 Addition of such agents from the outset is particularly necessary in patients with profound muscle weakness or those who have concomitant symptomatic interstitial lung disease.
No randomized controlled trial comparing azathioprine and methotrexate has been conducted to date. Therefore, the choice is based on patient preference, presence of coexisting interstitial lung disease or liver disease, commitment to limit alcohol consumption, and thiopurine methyltransferase status. Most patients need prolonged therapy.
In a randomized clinical trial, concomitant therapy with prednisone and azathioprine resulted in better functional outcomes and a significantly lower prednisone dose requirement for maintenance therapy at 3 years than with prednisone alone.43,44 Although no such randomized study has been conducted using methotrexate, several retrospective studies have demonstrated 70% to 80% response rates, including those for whom monotherapy with glucocorticoids had failed.45,46 The combination of methotrexate and azathioprine may be beneficial in patients who previously had inadequate responses to either of these agents alone.47
For severe pulmonary involvement associated with antisynthetase syndrome, monthly intravenous infusion of cyclophosphamide has been shown to be effective.48,49
Some recent studies established the role of tacrolimus in the treatment of both interstitial lung disease and myositis associated with antisynthetase syndrome.29 Cyclosporine has also been successfully used in a case of interstitial lung disease associated with anti Jo-1 syndrome.30
Rituximab, a monoclonal antibody to Blymphocyte antigen CD20, can also be used successfully in refractory disease,31 including refractory interstitial lung disease.32
In an open-label prospective study, polymyositis refractory to glucocorticoids and multiple conventional immunosuppressive agents responded well to high-dose intravenous immune globulin in the short term.50 However, the antisynthetase antibody status in this cohort was unknown; therefore, no definite conclusion could be drawn about the efficacy of intravenous immune globulin specifically in antisynthetase syndrome.
General measures
In patients with profound muscle weakness, physical therapy and rehabilitation should begin early. The goal is to reduce further muscle wasting from disuse and prevent muscle contractures. Patients with oropharyngeal and esophageal dysmotility should be advised about aspiration precautions and may need a swallow evaluation by a speech therapist; some may need temporary parenteral hyper-alimentation or J-tube insertion.
PROGNOSIS
If skeletal muscle involvement is the sole manifestation of antisynthetase syndrome, patients usually respond to glucocorticoids and immunosuppressive therapy and do fairly well. However, the outcome is not so promising when patients also develop interstitial lung disease, and the severity and type of lung injury usually determine the prognosis. As expected, patients with a progressive course of interstitial lung disease fare poorly, whereas those with a nonprogressive course tend to do relatively better. Older age at onset (> 60 years), presence of a malignancy, and a negative antinuclear antibody test are associated with a poor prognosis.7
Acknowledgment: The authors are grateful to Dr. Stephen Hatem, MD, staff radiologist, musculoskeletal radiology, Cleveland Clinic Imaging Institute, for help in the preparation of the magnetic resonance images. We also thank Dr. Steven Shook, MD, staff neurologist, Cleveland Clinic Neurological Institute, for help in summarizing the EMG findings.
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.
Mechanic’s hands
In about 30% of patients, the skin of the tips and margins of the fingers becomes thickened, hyperkeratotic, and fissured, the appearance of which is classically described as mechanic’s hands. It is a common manifestation of antisynthetase syndrome and is particularly prominent on the radial side of the index fingers (Figure 1). Biopsy of affected skin shows an interface psoriasiform dermatitis.19 In addition, some dermatomyositis patients with Gottron papules and a heliotrope rash have antisynthetase antibodies.
Raynaud phenomenon
Raynaud phenomenon develops in about 40% of patients. Some have nailfold capillary abnormalities.20 However, persistent or severe digital ischemia leading to digital ulceration or infarction is uncommon.21
Inflammatory arthritis
Arthralgias and arthritis are common (50%), the most common form being a symmetric polyarthritis of the small joints of the hands and feet. It is typically nonerosive but can sometimes be erosive and destructive.20
Because inflammatory arthritis mimics rheumatoid arthritis, antisynthetase syndrome should be considered in rheumatoid factor-negative patients presenting with polyarthritis.
ASSOCIATION WITH MALIGNANCY
Traditional teaching has been that antisynthetase antibody is protective against an underlying malignancy.22,23 However, several recently published case studies have reported various malignancies occurring within 6 to 12 months of the diagnosis of antisynthetase syndrome.7,24 The debate as to whether these are chance associations or causal (a paraneoplastic phenomenon) has not been resolved at this time.24
It is now recommended that patients with antisynthetase syndrome be screened for malignancies as appropriate for the patient’s age and sex. Screening should include a careful history and physical examination, complete blood cell count, comprehensive metabolic panel, chest radiography, mammography, and a gynecologic examination for women.25 If abnormalities are found, a more thorough evaluation for cancer is appropriate.
DIAGNOSIS
Muscle enzyme levels are often elevated
Muscle enzymes (creatine kinase and aldolase) are often elevated. Serum creatine kinase levels can range between 5 to 50 times the upper limit of normal. In an established case, creatine kinase levels along with careful manual muscle strength testing may help evaluate myositis activity. However, in chronic and advanced disease, creatine kinase may be within the normal range despite active myositis, partly because of extensive loss of muscle mass. In myositis, it may be prudent to check both creatine kinase and aldolase; sometimes only serum aldolase level rises, when immune-mediated injury predominantly affects the early regenerative myocytes.26
Judicious use of autoantibody testing
The characteristic clinical presentation is the initial clue to the diagnosis of antisynthetase syndrome, which is then supported by serologic testing.
Injudicious testing for a long list of antibodies should be avoided, as the cost is considerable and it does not influence further management. However, ordering an anti-Jo-1 antibody test in the correct clinical setting is appropriate, as it has high specificity,27,28 and thus can help establish or refute the clinical suspicion of antisynthetase syndrome.
Screening pulmonary function testing and thoracic high-resolution computed tomography for all patients with polymyositis or dermatomyositis is not considered “standard of care” and will likely not be reimbursed by third-party payers. However, in a patient with symptoms and signs of myositis, the presence of an antisynthetase antibody should prompt screening for occult interstitial lung disease, even in the absence of symptoms. As lung disease ultimately determines the prognosis in antisynthetase syndrome, early diagnosis and management is the key. Therefore, these tests would likely be approved to establish the diagnosis of interstitial lung disease and evaluate its severity.
If a myositis patient is also found to have interstitial lung disease or develops mechanic’s hands, the likely diagnosis is antisynthetase syndrome, which can be confirmed by serologic testing for antisynthetase antibodies. Interstitial lung disease in antisynthetase syndrome is often from “nonspecific interstitial pneumonitis”; therefore, medications tested and proven effective for this condition should be approved and reimbursed by payers.29–32
The coexistence of myositis and interstitial lung disease increases the sensitivity of anti-Jo-1 antibody.11 Thus, the clinician can have more confidence in early recognition and initiation of aggressive but targeted disease-modifying therapy.
Various methods can be used for detecting antisynthetase antibodies, with comparable results.28 Anti-Jo-1 antibody testing costs about $140. If that test is negative and antisynthetase syndrome is still suspected, then testing for the non-Jo-1 antisynthetase antibodies may be justified (Table 1). Though the cost of this panel of autoantibodies is about $300, it helps to confirm the diagnosis, and it influences the choice of second-line immunosuppressive agents such as tacrolimus29 and rituximab32 in patients resistant to conventional immunosuppressive agents such as azathioprine and methotrexate.
Often, anti-Ro52 SS-A antibodies are present concurrently in patients with anti-Jo-1 syndrome.33 In observational studies in patients with anti-Jo-1 antibody-associated interstitial lung disease, coexistence of anti-Ro52 SS-A antibody tended to predict a worse pulmonary outcome than in those with anti-Jo-1 antibody alone.34,35
Electromyography
Electromyography not only helps differentiate between myopathic and neuropathic weakness, but it may also support the diagnosis of “inflammatory” myopathy as suggested by prominent muscle membrane irritability (fibrillations, positive sharp waves) and abnormal motor unit action potentials (spontaneous activity showing small, short, polyphasic potentials and early recruitment). However, the findings can be nonspecific, and may even be normal in 10% to 15% of patients.36 Electromyographic abnormalities are most consistently observed in weak proximal muscles, and electromyography is also helpful in selecting a muscle for biopsy. Although no single electromyographic pattern is considered diagnostic for inflammatory myopathy, abnormalities are present in around 90% of patients (Table 2).3
Magnetic resonance imaging
Magnetic resonance imaging may show increased signal intensity in the affected muscles and surrounding tissues (Figure 3).37 Because it lacks sensitivity and specificity, magnetic resonance imaging is not helpful in diagnosing the disease. However, in the correct clinical setting, it may be used to guide muscle biopsy, and it can help in monitoring the disease progress.38
Muscle histopathology
Muscle biopsy, though often helpful, is not always diagnostic, and antisynthetase syndrome should still be suspected in the right clinical context, even in the absence of characteristic pathologic changes.
Biopsy of sites recently studied by electromyography should be avoided, and if the patient has undergone electromyography recently, the contralateral side should be selected for biopsy.
Reports of histopathologic findings in muscle biopsies in patients with antisynthetase syndrome document inflammatory myopathic features (Figure 4). In a series of patients with anti-Jo-1 syndrome, inflammation was noted in all cases, predominantly perimysial in location, with occasional endomysial and perivascular inflammation.39 Many of the inflammatory cells seen were macrophages and lymphocytes, in contrast to the predominantly lymphocytic infiltrates described in classic polymyositis and dermatomyositis. Perifascicular atrophy, similar to what is seen in dermatomyositis, was encountered; however, vascular changes, typical of dermatomyositis, were absent. Occasional degenerating and regenerating muscle fibers were also observed in most cases. Additionally, a characteristic perimysial connective tissue fragmentation was described, a feature less often seen in classic polymyositis and dermatomyositis.39
Pulmonary function testing
If antisynthetase syndrome is suspected or diagnosed, baseline pulmonary function testing (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease (reduced forced vital capacity and carbon monoxide diffusion capacity), and the diagnosis will be substantiated on thoracic high-resolution computed tomography. Respiratory muscle weakness can be detected with upright and supine spirometry.40 Weakness of these muscles contributes to shortness of breath, and patients may need ventilatory support.
Thoracic high-resolution computed tomography
Different patterns of lung injury can be seen in antisynthetase syndrome. Diffuse ground-glass opacification may suggest a nonspecific interstitial pneumonitis pattern, which is the most common form of interstitial lung disease (Figure 2), whereas coarse reticulation or honeycombing correlates with a usual interstitial pneumonitis pattern. Patchy consolidation or air-space disease can occur if cryptogenic organizing pneumonia is the predominant pattern of lung injury.
Swallowing evaluation
A comprehensive swallowing evaluation by a speech therapist may be necessary for evaluation of dysphagia (from oropharyngeal and striated esophageal muscle weakness) and determination of aspiration risk (Table 2).
Lung histopathology
If necessary, a surgical lung biopsy is needed to document the pathologic pattern of injury, including the amount of fibrosis in the lung. Historically, in idiopathic inflammatory myopathy patients in general, this has taken the form of usual interstitial pneumonia, organizing pneumonia, or diffuse alveolar damage.41 With the emergence of the definition of nonspecific interstitial pneumonitis and fibrosis as a documented and accepted pattern, more studies have found this to be the most common pattern of lung injury.16 It is characterized by diffuse involvement of the lung by an interstitial chronic inflammatory infiltrate, a cellular type of nonspecific interstitial pneumonitis that progresses in a uniform pattern to a fibrotic type (Figure 5). This form of fibrosis rarely results in significant remodeling, so-called honeycomb changes. In addition, anti-Jo-1 antibody patients may also have an increased incidence of acute lung injury, including acute diffuse alveolar damage that is often superimposed on the underlying chronic lung disease.42
In patients with pulmonary hypertension, histopathologic studies of the muscular pulmonary arteries often show moderate intimal fibroplasia, suggesting that a pulmonary arteriopathy with intimal thickening and luminal narrowing develops in some of these patients (Figure 5), independent of chronic hypoxic pulmonary vasoconstriction or vascular obstruction due to its entrapment within fibrotic lung tissue.17
TREATMENT
Glucocorticoids are the mainstay
Glucocorticoids are considered the mainstay of treatment. Patients should be advised that long-term use of glucocorticoids is necessary, though the response is variable. It is also important to discuss possible side effects of long-term glucocorticoid use.
Standard practice is to initiate treatment with high doses for the first 4 to 6 weeks to achieve disease control, followed by a slow taper over the next 9 to 12 months to the lowest effective dose to maintain remission. If the patient is profoundly weak, especially with respiratory muscle weakness or significant dysphagia and aspiration risk, hospital admission for intravenous methylprednisolone 1,000 mg daily for 3 to 5 days may be necessary. Otherwise, oral prednisone 1 mg/kg/day would be the usual starting dose.
If the patient’s muscle strength initially improves and then declines weeks to months later despite adequate therapy, glucocorticoid-induced myopathy should be suspected, especially if the muscle enzymes are within the reference range. This is more likely to occur if high-dose prednisone is continued for more than 6 to 8 weeks.
Improvement in muscle strength, which can take several weeks to several months, is a more reliable indicator of response to therapy than the serum creatine kinase level, which may take much longer to normalize. Relying on normalization of the creatine kinase level alone may lead to unnecessary prolongation of high-dose glucocorticoid therapy. It may take several months for the muscle enzymes to normalize, and there is usually a time lag between normalization of muscle enzymes and complete recovery of muscle strength.
Long-term use of high-dose prednisone leads to glucocorticoid-induced osteoporosis. Therefore, patients should receive osteoporosis prophylaxis including antiresorptive therapy with a bisphosphonate. In addition, prophylaxis against Pneumocystis jirovecii is indicated for patients treated with high-dose glucocorticoids.
Additional immunosuppressive agents
Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine and methotrexate are often required, both as glucocorticoid-sparing agents and to achieve adequate disease control.10 Addition of such agents from the outset is particularly necessary in patients with profound muscle weakness or those who have concomitant symptomatic interstitial lung disease.
No randomized controlled trial comparing azathioprine and methotrexate has been conducted to date. Therefore, the choice is based on patient preference, presence of coexisting interstitial lung disease or liver disease, commitment to limit alcohol consumption, and thiopurine methyltransferase status. Most patients need prolonged therapy.
In a randomized clinical trial, concomitant therapy with prednisone and azathioprine resulted in better functional outcomes and a significantly lower prednisone dose requirement for maintenance therapy at 3 years than with prednisone alone.43,44 Although no such randomized study has been conducted using methotrexate, several retrospective studies have demonstrated 70% to 80% response rates, including those for whom monotherapy with glucocorticoids had failed.45,46 The combination of methotrexate and azathioprine may be beneficial in patients who previously had inadequate responses to either of these agents alone.47
For severe pulmonary involvement associated with antisynthetase syndrome, monthly intravenous infusion of cyclophosphamide has been shown to be effective.48,49
Some recent studies established the role of tacrolimus in the treatment of both interstitial lung disease and myositis associated with antisynthetase syndrome.29 Cyclosporine has also been successfully used in a case of interstitial lung disease associated with anti Jo-1 syndrome.30
Rituximab, a monoclonal antibody to Blymphocyte antigen CD20, can also be used successfully in refractory disease,31 including refractory interstitial lung disease.32
In an open-label prospective study, polymyositis refractory to glucocorticoids and multiple conventional immunosuppressive agents responded well to high-dose intravenous immune globulin in the short term.50 However, the antisynthetase antibody status in this cohort was unknown; therefore, no definite conclusion could be drawn about the efficacy of intravenous immune globulin specifically in antisynthetase syndrome.
General measures
In patients with profound muscle weakness, physical therapy and rehabilitation should begin early. The goal is to reduce further muscle wasting from disuse and prevent muscle contractures. Patients with oropharyngeal and esophageal dysmotility should be advised about aspiration precautions and may need a swallow evaluation by a speech therapist; some may need temporary parenteral hyper-alimentation or J-tube insertion.
PROGNOSIS
If skeletal muscle involvement is the sole manifestation of antisynthetase syndrome, patients usually respond to glucocorticoids and immunosuppressive therapy and do fairly well. However, the outcome is not so promising when patients also develop interstitial lung disease, and the severity and type of lung injury usually determine the prognosis. As expected, patients with a progressive course of interstitial lung disease fare poorly, whereas those with a nonprogressive course tend to do relatively better. Older age at onset (> 60 years), presence of a malignancy, and a negative antinuclear antibody test are associated with a poor prognosis.7
Acknowledgment: The authors are grateful to Dr. Stephen Hatem, MD, staff radiologist, musculoskeletal radiology, Cleveland Clinic Imaging Institute, for help in the preparation of the magnetic resonance images. We also thank Dr. Steven Shook, MD, staff neurologist, Cleveland Clinic Neurological Institute, for help in summarizing the EMG findings.
- Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13:175–181.
- Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum 1980; 23:881–888.
- Nagaraju K, Lundberg IE. Inflammatory diseases of muscle and other myopathies. In:Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. Philadelphia, PA: Saunders; 2008:1353–1380.
- Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60:116–123.
- Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum 1996; 39:292–296.
- Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum 1996; 39:1507–1518.
- Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with antisynthetase autoantibodies. Intern Med J 2011; 41:674–679.
- Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 1996; 26:459–467.
- Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39:233–241.
- Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53–59.
- Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol 2000; 12:475–481.
- Ancuta CM, Ancuta E, Chirieac RM. Aminoacyl-tRNA synthetases in idiopathic inflammatory myopathies: an update on immunopathogenic significance, clinical and therapeutic implications. In:Gran JT, editor. Idiopathic Inflammatory Myopathies - Recent Developments. Rijeka, Croatia: InTech; 2011:77–90.
- Kajihara M, Kuwana M, Tokuda H, et al. Myositis and interstitial lung disease associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27:2707–2710.
- Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54:2004–2009.
- Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep 2003; 5:425–430.
- Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23:874–880.
- Chatterjee S, Farver C. Severe pulmonary hypertension in anti-Jo-1 syndrome. Arthritis Care Res (Hoboken) 2010; 62:425–429.
- Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus 2009; 18:1006–1010.
- Bugatti L, De Angelis R, Filosa G, Salaffi F. Bilateral, asymptomatic scaly and fissured cutaneous lesions of the fingers in a patient presenting with myositis. Indian J Dermatol Venereol Leprol 2005; 71:137–138.
- Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010; 16:307–312.
- Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum 1992; 35:449–456.
- Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70:360–374.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol 2001; 144:825–831.
- Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol 2008; 35:169–171.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol 2010; 22:627–632.
- Casciola-Rosen L, Hall JC, Mammen AL, Christopher-Stine L, Rosen A. Isolated elevation of aldolase in the serum of myositis patients: a potential biomarker of damaged early regenerating muscle cells. Clin Exp Rheumatol 2012; 30:548–553.
- Shovman O, Gilburd B, Barzilai O, et al. Evaluation of the BioPlex 2200 ANA screen: analysis of 510 healthy subjects: incidence of natural/predictive autoantibodies. Ann N Y Acad Sci 2005; 1050:380–388.
- Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity 2005; 38:73–78.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Jankowska M, Butto B, Debska-Slizien A, Rutkowski B. Beneficial effect of treatment with cyclosporin A in a case of refractory antisynthetase syndrome. Rheumatol Int 2007; 27:775–780.
- Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 2012; 42:e4–e7.
- Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106:581–587.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32–40.
- La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39:249–253.
- Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29:989–994.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255–286.
- Reimers CD, Finkenstaedt M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 9:475–485.
- O’Connell MJ. Whole-body MR imaging in the diagnosis of polymyositis. Am J Roentgenol 2002; 179:967–971.
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472–478.
- Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil 2001; 82:123–128.
- Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(suppl 1):513S–519S.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162:2213–2217.
- Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 1980; 92:365–369.
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum 1981; 24:45–48.
- Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182–189.
- Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379–387.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998; 41:392–399.
- Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46:124–130.
- al-Janadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/dermatomyositis. J Rheumatol 1989; 16:1592–1596.
- Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum 2002; 46:467–474.
- Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13:175–181.
- Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum 1980; 23:881–888.
- Nagaraju K, Lundberg IE. Inflammatory diseases of muscle and other myopathies. In:Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. Philadelphia, PA: Saunders; 2008:1353–1380.
- Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60:116–123.
- Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum 1996; 39:292–296.
- Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum 1996; 39:1507–1518.
- Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with antisynthetase autoantibodies. Intern Med J 2011; 41:674–679.
- Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 1996; 26:459–467.
- Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39:233–241.
- Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53–59.
- Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol 2000; 12:475–481.
- Ancuta CM, Ancuta E, Chirieac RM. Aminoacyl-tRNA synthetases in idiopathic inflammatory myopathies: an update on immunopathogenic significance, clinical and therapeutic implications. In:Gran JT, editor. Idiopathic Inflammatory Myopathies - Recent Developments. Rijeka, Croatia: InTech; 2011:77–90.
- Kajihara M, Kuwana M, Tokuda H, et al. Myositis and interstitial lung disease associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27:2707–2710.
- Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54:2004–2009.
- Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep 2003; 5:425–430.
- Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23:874–880.
- Chatterjee S, Farver C. Severe pulmonary hypertension in anti-Jo-1 syndrome. Arthritis Care Res (Hoboken) 2010; 62:425–429.
- Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus 2009; 18:1006–1010.
- Bugatti L, De Angelis R, Filosa G, Salaffi F. Bilateral, asymptomatic scaly and fissured cutaneous lesions of the fingers in a patient presenting with myositis. Indian J Dermatol Venereol Leprol 2005; 71:137–138.
- Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010; 16:307–312.
- Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum 1992; 35:449–456.
- Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70:360–374.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol 2001; 144:825–831.
- Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol 2008; 35:169–171.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol 2010; 22:627–632.
- Casciola-Rosen L, Hall JC, Mammen AL, Christopher-Stine L, Rosen A. Isolated elevation of aldolase in the serum of myositis patients: a potential biomarker of damaged early regenerating muscle cells. Clin Exp Rheumatol 2012; 30:548–553.
- Shovman O, Gilburd B, Barzilai O, et al. Evaluation of the BioPlex 2200 ANA screen: analysis of 510 healthy subjects: incidence of natural/predictive autoantibodies. Ann N Y Acad Sci 2005; 1050:380–388.
- Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity 2005; 38:73–78.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Jankowska M, Butto B, Debska-Slizien A, Rutkowski B. Beneficial effect of treatment with cyclosporin A in a case of refractory antisynthetase syndrome. Rheumatol Int 2007; 27:775–780.
- Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 2012; 42:e4–e7.
- Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106:581–587.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32–40.
- La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39:249–253.
- Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29:989–994.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255–286.
- Reimers CD, Finkenstaedt M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 9:475–485.
- O’Connell MJ. Whole-body MR imaging in the diagnosis of polymyositis. Am J Roentgenol 2002; 179:967–971.
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472–478.
- Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil 2001; 82:123–128.
- Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(suppl 1):513S–519S.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162:2213–2217.
- Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 1980; 92:365–369.
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum 1981; 24:45–48.
- Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182–189.
- Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379–387.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998; 41:392–399.
- Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46:124–130.
- al-Janadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/dermatomyositis. J Rheumatol 1989; 16:1592–1596.
- Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum 2002; 46:467–474.
KEY POINTS
- Antisynthetase syndrome can present with a wide variety of clinical manifestations, including myositis and interstitial lung disease.
- The type and severity of interstitial lung disease usually determine the long-term outcome.
- In the appropriate clinical setting, the diagnosis is usually confirmed by the detection of antibodies to various aminoacyl-transfer RNA synthetases, anti-Jo-1 antibody being the most common.
- Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine or methotrexate are often required as steroid-sparing agents and also to achieve disease control.
- In the case of severe pulmonary involvement, cyclophosphamide is recommended.