User login
Lung cancer screening: What to expect
Lung cancer can sometimes be detected with a low-dose screening CT (computed tomography) scan. During the scan, you will lie down in a donut-like structure while x-rays are passed through your body. Computers then use these x-rays to produce images of the inside of your body. The scan does not hurt and takes only a few seconds to complete.
Benefits of screening
Screening for lung cancer with a chest CT scan has been shown to lower your chance of dying from lung cancer by 20%. That means that for every 5 people who would have died from lung cancer without screening, 1 of these 5 will not.
Downside of screening
False alarms. Screening for lung cancer with a chest CT has been shown to find a small spot or spots (called lung nodules) in the lungs of at least one-quarter of everyone who gets the CT scan. Only 3 or 4 out of 100 of the lung nodules found are cancer, while the rest are small scars that will never affect your health.
For many of the small lung nodules found, there is no way to tell without additional tests if they are a small scar or a lung cancer. These tests usually include CT scans done over time to see if the lung nodule grows. If the lung nodule is large enough, a biopsy may also be required. Therefore, many people who have a lung cancer screening CT scan and do not have lung cancer will have additional tests performed.
The physician who ordered the screening test will be able to advise you about how the lung nodule should be evaluated. He or she may choose to have you visit a lung nodule clinic for advice as well.
Cost. Most insurance programs do not currently cover the cost of a lung cancer screening chest CT, but they usually do cover the evaluation of any abnormal findings.
Quit smoking!
If you currently smoke, you can lower your risk of dying from lung cancer by quitting smoking. The amount your risk will be lowered by quitting smoking is greater than the amount your risk will be lowered by being screened with a CT scan. If you smoke, try to quit. Talk to your doctor about the best strategies for quitting.
This information is provided by your physician and the Cleveland Clinic Journal of Medicine. It is not designed to replace a physician’s medical assessment and judgment.
This page may be reproduced noncommercially to share with patients. Any other reproduction is subject to Cleveland Clinic Journal of Medicine approval. Bulk color reprints are available by calling 216-444-2661.
For patient information on hundreds of health topics, see the Patient Education and Health Information web site, www.clevelandclinic.org/health
Lung cancer can sometimes be detected with a low-dose screening CT (computed tomography) scan. During the scan, you will lie down in a donut-like structure while x-rays are passed through your body. Computers then use these x-rays to produce images of the inside of your body. The scan does not hurt and takes only a few seconds to complete.
Benefits of screening
Screening for lung cancer with a chest CT scan has been shown to lower your chance of dying from lung cancer by 20%. That means that for every 5 people who would have died from lung cancer without screening, 1 of these 5 will not.
Downside of screening
False alarms. Screening for lung cancer with a chest CT has been shown to find a small spot or spots (called lung nodules) in the lungs of at least one-quarter of everyone who gets the CT scan. Only 3 or 4 out of 100 of the lung nodules found are cancer, while the rest are small scars that will never affect your health.
For many of the small lung nodules found, there is no way to tell without additional tests if they are a small scar or a lung cancer. These tests usually include CT scans done over time to see if the lung nodule grows. If the lung nodule is large enough, a biopsy may also be required. Therefore, many people who have a lung cancer screening CT scan and do not have lung cancer will have additional tests performed.
The physician who ordered the screening test will be able to advise you about how the lung nodule should be evaluated. He or she may choose to have you visit a lung nodule clinic for advice as well.
Cost. Most insurance programs do not currently cover the cost of a lung cancer screening chest CT, but they usually do cover the evaluation of any abnormal findings.
Quit smoking!
If you currently smoke, you can lower your risk of dying from lung cancer by quitting smoking. The amount your risk will be lowered by quitting smoking is greater than the amount your risk will be lowered by being screened with a CT scan. If you smoke, try to quit. Talk to your doctor about the best strategies for quitting.
This information is provided by your physician and the Cleveland Clinic Journal of Medicine. It is not designed to replace a physician’s medical assessment and judgment.
This page may be reproduced noncommercially to share with patients. Any other reproduction is subject to Cleveland Clinic Journal of Medicine approval. Bulk color reprints are available by calling 216-444-2661.
For patient information on hundreds of health topics, see the Patient Education and Health Information web site, www.clevelandclinic.org/health
Lung cancer can sometimes be detected with a low-dose screening CT (computed tomography) scan. During the scan, you will lie down in a donut-like structure while x-rays are passed through your body. Computers then use these x-rays to produce images of the inside of your body. The scan does not hurt and takes only a few seconds to complete.
Benefits of screening
Screening for lung cancer with a chest CT scan has been shown to lower your chance of dying from lung cancer by 20%. That means that for every 5 people who would have died from lung cancer without screening, 1 of these 5 will not.
Downside of screening
False alarms. Screening for lung cancer with a chest CT has been shown to find a small spot or spots (called lung nodules) in the lungs of at least one-quarter of everyone who gets the CT scan. Only 3 or 4 out of 100 of the lung nodules found are cancer, while the rest are small scars that will never affect your health.
For many of the small lung nodules found, there is no way to tell without additional tests if they are a small scar or a lung cancer. These tests usually include CT scans done over time to see if the lung nodule grows. If the lung nodule is large enough, a biopsy may also be required. Therefore, many people who have a lung cancer screening CT scan and do not have lung cancer will have additional tests performed.
The physician who ordered the screening test will be able to advise you about how the lung nodule should be evaluated. He or she may choose to have you visit a lung nodule clinic for advice as well.
Cost. Most insurance programs do not currently cover the cost of a lung cancer screening chest CT, but they usually do cover the evaluation of any abnormal findings.
Quit smoking!
If you currently smoke, you can lower your risk of dying from lung cancer by quitting smoking. The amount your risk will be lowered by quitting smoking is greater than the amount your risk will be lowered by being screened with a CT scan. If you smoke, try to quit. Talk to your doctor about the best strategies for quitting.
This information is provided by your physician and the Cleveland Clinic Journal of Medicine. It is not designed to replace a physician’s medical assessment and judgment.
This page may be reproduced noncommercially to share with patients. Any other reproduction is subject to Cleveland Clinic Journal of Medicine approval. Bulk color reprints are available by calling 216-444-2661.
For patient information on hundreds of health topics, see the Patient Education and Health Information web site, www.clevelandclinic.org/health
Family history: Still relevant in the genomics era
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
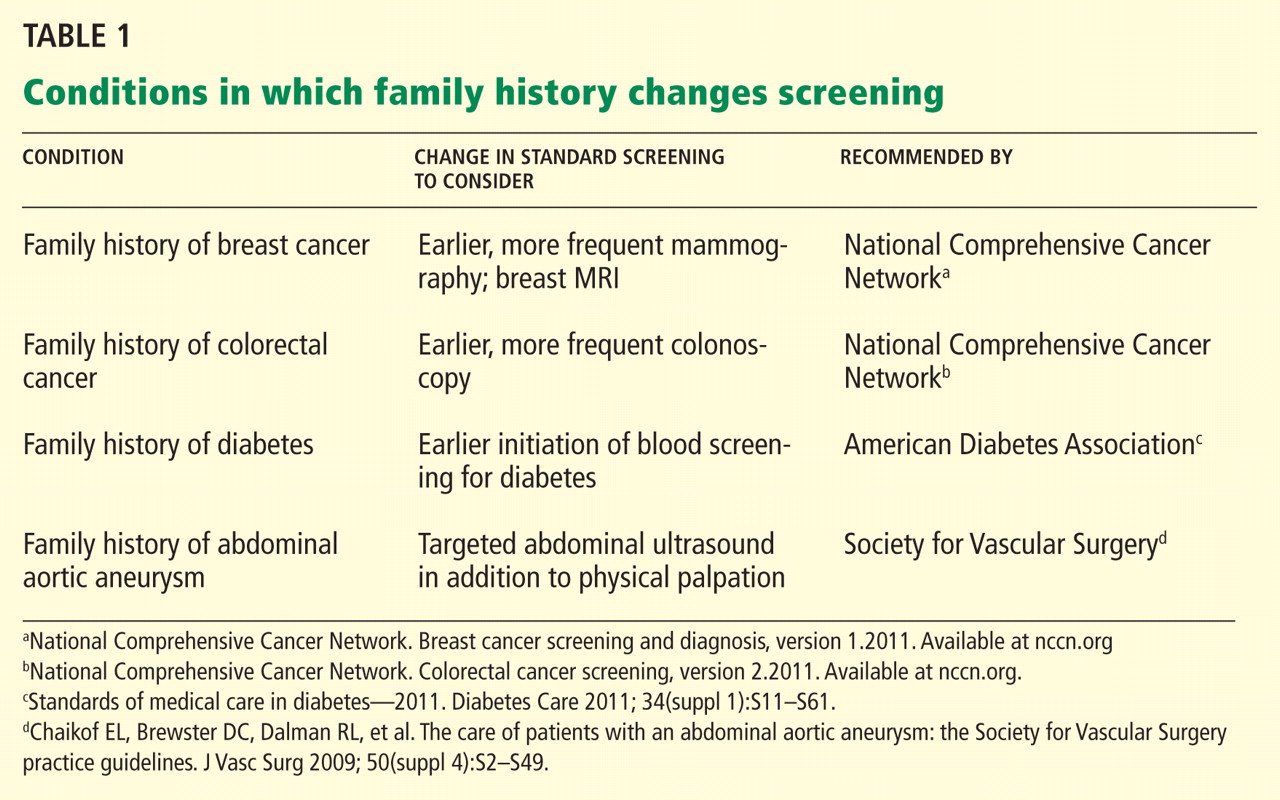
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
KEY POINTS
- The family history is an underused tool for predicting the risk of disease and for personalizing preventive care.
- Barriers to the appropriate collection and use of the family history include concerns over the reliability of patient reporting, a lack of time and reimbursement, and provider knowledge gaps.
- Use of family history to inform genetic testing for hereditary cancer syndromes has been shown to improve outcomes and may reduce overall health care costs.
- Future solutions need to focus on creating time-effective ways to collect and analyze the family history, and on developing innovative methods of educating medical providers at all levels of training as to how to apply the family history in clinical practice.
Personalizing patient care
The concept and promise of personalized health care have been anticipated for decades. Yet in its breadth and in the way we would like it practiced, it is in its infancy.
Personalized health care aims to individualize care by integrating a person’s unique clinical, molecular (ie, genetic, genomic), and environmental information. Applied not only when the patient is sick but also when he or she is well, it builds on and enhances our current standards of care.
As Sir William Osler recognized more than a century ago, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”1
PREDICTING THE RISK OF DISEASE
For years, we have attempted to predict and stratify the risk of disease, and the Human Genome Project has given us a new set of tools to help understand the complexity of disease and its variability.
We know and have known for decades—and in some cultures, for centuries—that family history is the most clinically validated tool for predicting the risk of disease.
Nevertheless, evidence suggests that we physicians are not collecting adequate family histories, and because of this, we are missing opportunities to intervene and prevent diseases predicted by the family history. The current standard of care is to use family medical histories to hone genetic differential diagnoses, and based on the differential diagnoses, to target specific genes to test in the setting of genetic counseling. Current genetic testing is used for molecular diagnoses and predictive testing so that gene-specific clinical management can be subsequently tailored.
PREDICTING RESPONSE TO TREATMENT
The use of personalized health care to predict response to treatment is a novel and constantly evolving practice.
ABO blood typing is a form of genetics-based personalization of safe transfusion that dates back to World War II.
A prominent, recent success story is in cancer treatment. For example, the American Society of Clinical Oncology now recommends that tumors from patients with node-negative, estrogen-receptor-positive breast cancer be evaluated with the Oncotype DX assay.2 This test measures the expression of 21 genes, and the score obtained identifies patients most likely to benefit from adjuvant chemotherapy. A similar 12-gene expression signature has been developed for colon cancer, and others have been developed for hematologic cancers.2,3 As with other new but apparently valid tests, the risk scores derived are sensitive at the extremes but ambiguous in the mid-ranges. We anticipate many more developments in this field.
In the field of pharmacogenomics, there is evidence to suggest that prior knowledge of CYP2C9 and VKORC1 genotypes enhances outcomes for patients starting treatment with warfarin. The US Food and Drug Administration revised the label on warfarin in February 2010, suggesting that genotypes be taken into consideration when the drug is prescribed.4
However, clinicians have been slow to adopt genotype testing when prescribing warfarin. Some cite the paucity of large, randomized, controlled trials demonstrating clinical utility of genotype-informed prescribing. Others cite concern that warfarin will soon become obsolete with the arrival of newer anticoagulants (such as factor X inhibitors) that do not carry warfarin’s adverse effects, and these genotypes will therefore become moot. Perhaps, as we move forward and new drugs are developed, companion genotype tests could be developed at the same time to be used with them.5
IMPROVING CARE, SAVING MONEY, AND EMPOWERING PATIENTS
The goal of personalized health care, by customizing treatments (medication types and dosages) and preventive strategies, is to optimize medical care and improve outcomes for each patient. It could improve the quality of care by targeting interventions and reducing adverse events, topics that are important to all of us in the current environment of health care reform.
A personalized approach might also, in the long run, decrease the cost of health care by driving appropriate utilization of resources.
Lastly, the true value of personalized health care may be in its potential to improve patient satisfaction and to empower our patients to work with us towards better health.
WE LAUNCH A NEW SERIES
To keep physicians up-to-date on progress in personalized health care, the Cleveland Clinic Journal of Medicine will present a series of articles on the topic. The series, to run once a quarter, begins in this issue, on page 331, with an article on the importance of the family history as a piece of genetic information that can help to predict the risk of disease and inform preventive care plans. Future topics will include the role of genetics and genomics in personalized care of patients with breast and colorectal cancers; the genetic counselor as a part of the health care team; pharmacogenomics; and ethical, legal, and societal considerations.
Our goal in this series is to provide practical information to help our readers incorporate personalized approaches into daily practice. In addition, as patients become more interested in and informed about personalized health care, we hope this information will help clinicians to effectively coach them about its potential benefits and risks. We also hope this information will enable our readers to ask the right questions so that patient and health care provider can work together to help the patient grow old gracefully.
As the series unfolds, we ask you to send us feedback and to suggest other topics in personalized health care you would like us to cover in this series.
- Osler W. Aequanimitas, With Other Addresses to Medical Students, Nurses and Practitioners of Medicine. 2nd edition. Philadelphia, PA: P. Blakiston’s Sone & Co, 1906:348.
- McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med 2011; 364:340–350.
- Eng C. Microenvironmental protection in diffuse large-B-cell lymphoma. N Engl J Med 2008; 359:2379–2381.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med 2010; 363:301–304.
The concept and promise of personalized health care have been anticipated for decades. Yet in its breadth and in the way we would like it practiced, it is in its infancy.
Personalized health care aims to individualize care by integrating a person’s unique clinical, molecular (ie, genetic, genomic), and environmental information. Applied not only when the patient is sick but also when he or she is well, it builds on and enhances our current standards of care.
As Sir William Osler recognized more than a century ago, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”1
PREDICTING THE RISK OF DISEASE
For years, we have attempted to predict and stratify the risk of disease, and the Human Genome Project has given us a new set of tools to help understand the complexity of disease and its variability.
We know and have known for decades—and in some cultures, for centuries—that family history is the most clinically validated tool for predicting the risk of disease.
Nevertheless, evidence suggests that we physicians are not collecting adequate family histories, and because of this, we are missing opportunities to intervene and prevent diseases predicted by the family history. The current standard of care is to use family medical histories to hone genetic differential diagnoses, and based on the differential diagnoses, to target specific genes to test in the setting of genetic counseling. Current genetic testing is used for molecular diagnoses and predictive testing so that gene-specific clinical management can be subsequently tailored.
PREDICTING RESPONSE TO TREATMENT
The use of personalized health care to predict response to treatment is a novel and constantly evolving practice.
ABO blood typing is a form of genetics-based personalization of safe transfusion that dates back to World War II.
A prominent, recent success story is in cancer treatment. For example, the American Society of Clinical Oncology now recommends that tumors from patients with node-negative, estrogen-receptor-positive breast cancer be evaluated with the Oncotype DX assay.2 This test measures the expression of 21 genes, and the score obtained identifies patients most likely to benefit from adjuvant chemotherapy. A similar 12-gene expression signature has been developed for colon cancer, and others have been developed for hematologic cancers.2,3 As with other new but apparently valid tests, the risk scores derived are sensitive at the extremes but ambiguous in the mid-ranges. We anticipate many more developments in this field.
In the field of pharmacogenomics, there is evidence to suggest that prior knowledge of CYP2C9 and VKORC1 genotypes enhances outcomes for patients starting treatment with warfarin. The US Food and Drug Administration revised the label on warfarin in February 2010, suggesting that genotypes be taken into consideration when the drug is prescribed.4
However, clinicians have been slow to adopt genotype testing when prescribing warfarin. Some cite the paucity of large, randomized, controlled trials demonstrating clinical utility of genotype-informed prescribing. Others cite concern that warfarin will soon become obsolete with the arrival of newer anticoagulants (such as factor X inhibitors) that do not carry warfarin’s adverse effects, and these genotypes will therefore become moot. Perhaps, as we move forward and new drugs are developed, companion genotype tests could be developed at the same time to be used with them.5
IMPROVING CARE, SAVING MONEY, AND EMPOWERING PATIENTS
The goal of personalized health care, by customizing treatments (medication types and dosages) and preventive strategies, is to optimize medical care and improve outcomes for each patient. It could improve the quality of care by targeting interventions and reducing adverse events, topics that are important to all of us in the current environment of health care reform.
A personalized approach might also, in the long run, decrease the cost of health care by driving appropriate utilization of resources.
Lastly, the true value of personalized health care may be in its potential to improve patient satisfaction and to empower our patients to work with us towards better health.
WE LAUNCH A NEW SERIES
To keep physicians up-to-date on progress in personalized health care, the Cleveland Clinic Journal of Medicine will present a series of articles on the topic. The series, to run once a quarter, begins in this issue, on page 331, with an article on the importance of the family history as a piece of genetic information that can help to predict the risk of disease and inform preventive care plans. Future topics will include the role of genetics and genomics in personalized care of patients with breast and colorectal cancers; the genetic counselor as a part of the health care team; pharmacogenomics; and ethical, legal, and societal considerations.
Our goal in this series is to provide practical information to help our readers incorporate personalized approaches into daily practice. In addition, as patients become more interested in and informed about personalized health care, we hope this information will help clinicians to effectively coach them about its potential benefits and risks. We also hope this information will enable our readers to ask the right questions so that patient and health care provider can work together to help the patient grow old gracefully.
As the series unfolds, we ask you to send us feedback and to suggest other topics in personalized health care you would like us to cover in this series.
The concept and promise of personalized health care have been anticipated for decades. Yet in its breadth and in the way we would like it practiced, it is in its infancy.
Personalized health care aims to individualize care by integrating a person’s unique clinical, molecular (ie, genetic, genomic), and environmental information. Applied not only when the patient is sick but also when he or she is well, it builds on and enhances our current standards of care.
As Sir William Osler recognized more than a century ago, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”1
PREDICTING THE RISK OF DISEASE
For years, we have attempted to predict and stratify the risk of disease, and the Human Genome Project has given us a new set of tools to help understand the complexity of disease and its variability.
We know and have known for decades—and in some cultures, for centuries—that family history is the most clinically validated tool for predicting the risk of disease.
Nevertheless, evidence suggests that we physicians are not collecting adequate family histories, and because of this, we are missing opportunities to intervene and prevent diseases predicted by the family history. The current standard of care is to use family medical histories to hone genetic differential diagnoses, and based on the differential diagnoses, to target specific genes to test in the setting of genetic counseling. Current genetic testing is used for molecular diagnoses and predictive testing so that gene-specific clinical management can be subsequently tailored.
PREDICTING RESPONSE TO TREATMENT
The use of personalized health care to predict response to treatment is a novel and constantly evolving practice.
ABO blood typing is a form of genetics-based personalization of safe transfusion that dates back to World War II.
A prominent, recent success story is in cancer treatment. For example, the American Society of Clinical Oncology now recommends that tumors from patients with node-negative, estrogen-receptor-positive breast cancer be evaluated with the Oncotype DX assay.2 This test measures the expression of 21 genes, and the score obtained identifies patients most likely to benefit from adjuvant chemotherapy. A similar 12-gene expression signature has been developed for colon cancer, and others have been developed for hematologic cancers.2,3 As with other new but apparently valid tests, the risk scores derived are sensitive at the extremes but ambiguous in the mid-ranges. We anticipate many more developments in this field.
In the field of pharmacogenomics, there is evidence to suggest that prior knowledge of CYP2C9 and VKORC1 genotypes enhances outcomes for patients starting treatment with warfarin. The US Food and Drug Administration revised the label on warfarin in February 2010, suggesting that genotypes be taken into consideration when the drug is prescribed.4
However, clinicians have been slow to adopt genotype testing when prescribing warfarin. Some cite the paucity of large, randomized, controlled trials demonstrating clinical utility of genotype-informed prescribing. Others cite concern that warfarin will soon become obsolete with the arrival of newer anticoagulants (such as factor X inhibitors) that do not carry warfarin’s adverse effects, and these genotypes will therefore become moot. Perhaps, as we move forward and new drugs are developed, companion genotype tests could be developed at the same time to be used with them.5
IMPROVING CARE, SAVING MONEY, AND EMPOWERING PATIENTS
The goal of personalized health care, by customizing treatments (medication types and dosages) and preventive strategies, is to optimize medical care and improve outcomes for each patient. It could improve the quality of care by targeting interventions and reducing adverse events, topics that are important to all of us in the current environment of health care reform.
A personalized approach might also, in the long run, decrease the cost of health care by driving appropriate utilization of resources.
Lastly, the true value of personalized health care may be in its potential to improve patient satisfaction and to empower our patients to work with us towards better health.
WE LAUNCH A NEW SERIES
To keep physicians up-to-date on progress in personalized health care, the Cleveland Clinic Journal of Medicine will present a series of articles on the topic. The series, to run once a quarter, begins in this issue, on page 331, with an article on the importance of the family history as a piece of genetic information that can help to predict the risk of disease and inform preventive care plans. Future topics will include the role of genetics and genomics in personalized care of patients with breast and colorectal cancers; the genetic counselor as a part of the health care team; pharmacogenomics; and ethical, legal, and societal considerations.
Our goal in this series is to provide practical information to help our readers incorporate personalized approaches into daily practice. In addition, as patients become more interested in and informed about personalized health care, we hope this information will help clinicians to effectively coach them about its potential benefits and risks. We also hope this information will enable our readers to ask the right questions so that patient and health care provider can work together to help the patient grow old gracefully.
As the series unfolds, we ask you to send us feedback and to suggest other topics in personalized health care you would like us to cover in this series.
- Osler W. Aequanimitas, With Other Addresses to Medical Students, Nurses and Practitioners of Medicine. 2nd edition. Philadelphia, PA: P. Blakiston’s Sone & Co, 1906:348.
- McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med 2011; 364:340–350.
- Eng C. Microenvironmental protection in diffuse large-B-cell lymphoma. N Engl J Med 2008; 359:2379–2381.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med 2010; 363:301–304.
- Osler W. Aequanimitas, With Other Addresses to Medical Students, Nurses and Practitioners of Medicine. 2nd edition. Philadelphia, PA: P. Blakiston’s Sone & Co, 1906:348.
- McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med 2011; 364:340–350.
- Eng C. Microenvironmental protection in diffuse large-B-cell lymphoma. N Engl J Med 2008; 359:2379–2381.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med 2010; 363:301–304.
Disparities in prostate cancer in African American men: What primary care physicians can do
Prostate cancer is the most common cancer affecting American men. In 2010, an estimated 217,730 men were diagnosed with it and 32,050 died of it.1 African American men are disproportionately affected, with a prostate cancer incidence two-thirds higher than whites and a mortality rate twice as high.1 Owing to such disparities, the life expectancy of African Americans is several years shorter than that of non-Hispanic whites.2
For the primary care provider, who is often the first access point for health care in the United States, it is important to understand what mechanisms may underlie these differences and what can be done to narrow the gap.3
WHAT IS THE CAUSE OF THESE DIFFERENCES?
Many studies have looked into the causes of the higher incidence of prostate cancer in African American men and their higher mortality rate from it. The disparity may be due to a variety of factors, some socioeconomic and some biologic.
Poorer access to care, or lower-quality care?
A study of US servicemen who had equal access to care showed that African American men had a higher rate of prostate cancer regardless of access to care and socioeconomic status.4
However, the 2002 Institute of Medicine report, Unequal Treatment: Confronting Racial and Ethnic Disparities in Health Care, found evidence that racial and ethnic minorities tend to receive lower-quality health care than whites, “even when access-related factors, such as patients’ insurance status and income, are controlled.”5
Genetic predisposition?
Some have proposed that the disparity may be a function of genetic predisposition.
Evidence of a genetic component to the high incidence and mortality rate in African American men comes from epidemiologic studies of men with similar genetic backgrounds. For example, men in Nigeria and Ghana also have a high incidence of prostate cancer, as do men of African descent in the Caribbean islands and in the United Kingdom.6
Chromosome 8q24 variants have been shown in several studies to be associated with prostate cancer risk and are more common in African American men.7–10 Some studies have also shown a higher rate of variations in cell apoptosis genes such as BCL211 and tumor-suppression genes such as EphB2 in African American men.12
These findings suggest that genetic differences may contribute to the higher prostate cancer incidence and mortality rate seen in African American men.
More-aggressive cancer, or later detection?
Not only do African American men tend to have a higher incidence of prostate cancer, they also tend to have more-aggressive disease (ie, a higher pathologic grade) at the time of diagnosis, which may contribute to the disparity in mortality rates.13–19
Initially, there was some controversy as to whether this observation is a result of genetic and biologic factors that may predispose African American men to more-aggressive disease, or if it is due to inadequate screening and delayed presentation. However, a body of evidence supports the contention that prostate cancer is more aggressive in African American men.
For example, a study of autopsy data from men who died of prostate cancer at ages 20 to 49 showed that the age of onset of prostate cancer was similar between African American and white men.20 The Surveillance Epidemiology and End Results (SEER) database showed that African American men had a higher incidence of metastatic disease across all age groups.20 A similar study conducted 10 years later confirmed that rates of subclinical prostate cancer in African American and white men do not differ by race at the early ages, but that advanced or metastatic disease occurred nearly four times as frequently in African American men.21
Another study examined prostate biopsies from African American men and found that their tumors expressed higher levels of biomarkers, suggesting they had more-aggressive disease.22
SCREENING FOR PROSTATE CANCER
Serum prostate-specific antigen (PSA) testing has become the method of choice for prostate cancer screening. However, PSA screening in asymptomatic men is under debate, because it can lead to overdetection and subsequent overtreatment of indolent disease.23
Several recent studies showed differing results from prostate cancer screening.
The US Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial found that the mortality rate was no lower with combined PSA screening and digital rectal examination during a median follow-up of 11 years than in a control group that had a lower rate of screening.24 However, further analysis of these data, with stratifying by comorbidities, showed that PSA screening in young and healthy men reduces the risk of death from prostate cancer, with minimal overtreatment.25
The European Randomized Study of Screening for Prostate Cancer found a statistically significant 20% reduction in deaths from prostate cancer with PSA screening, but that it was necessary to treat 48 men in order to save one life.26
Another study, published in 2010, showed that regular PSA screening reduced the rate of prostate cancer mortality by half over 14 years.27
African American men generally present with disease that is more advanced than in white men.28 This historically has been attributed to the fact that African Americans have been less likely to be screened for prostate cancer, though recent data indicate the gap is lessening.29–31 A cross-sectional study from the Texas Medical Center showed that 54.4% of African American men had received PSA screening, compared with 63.2% of white men.32
Another study showed that African Americans were more likely to have had a longer interval between PSA screenings before diagnosis, and that a longer PSA screening interval was associated with greater odds of having advanced disease at diagnosis.33 However, when the researchers controlled for the PSA screening interval, they found that African Americans had the same odds of being diagnosed with advanced prostate cancer as white patients did. They concluded that more frequent or systematic PSA screening may reduce the racial differences in cancer stage at diagnosis and in deaths.
Reasons for the disparities in screening
Many reasons have been proposed to explain why African Americans receive less screening, including poor communication between physicians and minority patients due to lack of cultural competency among physicians, lack of health insurance (and poor access to quality care as a result), and deficiency of knowledge about screening. Though awareness is rising, many African Americans are unaware of early detection methods for prostate cancer (eg, PSA testing), and other barriers such as cost and transportation exist that may prevent African American men from being screened.34,35
As gatekeepers, primary care physicians are in a position to address these shortcomings in patient education and to enhance the physician-patient relationship.36
Black men have higher PSA levels, with or without cancer
Physicians must also be aware of racial differences in PSA levels and realize that the predictive value of PSA in the diagnosis of prostate cancer may differ between African Americans and whites.
Black men, with or without prostate cancer, have been found to have higher PSA levels. Kyle and colleagues37 found that African American men without prostate cancer had significantly higher mean PSA levels than white men across all age groups. Furthermore, Vijayakumar et al38 found that African Americans with newly diagnosed localized prostate cancer had higher serum PSA levels than whites at diagnosis.
Although PSA cutoff levels have not been officially modified according to race, primary care physicians should have a lower threshold for referring African American men who have a suspiciously high PSA level for further urologic evaluation. Close partnership between the internist, family practitioner, and urologist will aid in the optimal use of PSA testing for the early detection of prostate cancer.
When to start PSA screening? How often to screen?
The age at which African American men should begin to have their PSA levels checked (with or without a digital rectal examination) continues to debated. However, the American Cancer Society39 recommends that African American men who have a father or brother who had prostate cancer before age 65 should begin having discussions with their physician on this topic and, with their informed consent, screening at age 45.
The frequency of PSA screening depends on the individual’s PSA level. The National Comprehensive Cancer Network40 recommends that men at high risk be offered a baseline PSA measurement and digital rectal examination at age 40 and, if the PSA level is higher than 1 ng/mL, that they be offered annual follow-ups. If the PSA level is less than 1 ng/mL, they recommend screening again at age 45. Risk factors for prostate cancer include family history as well as African American race.41
How should PSA levels be interpreted?
Interpreting PSA results is important in detecting prostate cancer at early stages.
At first, we believed the normal range of PSA for all men was 4.0 ng/mL or less. However, the American Urological Association now recognizes that the normal PSA range, in addition to varying along racial lines, also is age-dependent.42 The Cleveland Clinic Minority Men's Health Center's suggested normal ranges of PSA in African American men are:
- Age 40–49: ≤ 2.5 ng/mL
- Age 50–59: ≤ 3.0 ng/mL
- Age 60–69: ≤ 3.5 ng/mL
- Age 70–79: ≤ 4.5 ng/mL
- Age > 80: ≤ 5.0 ng/mL.
Remember that an elevated PSA does not necessarily signify prostate cancer, and that these are reference ranges only and may vary in individual men.
SURVIVAL AFTER DIAGNOSIS
African American men with prostate cancer have significantly higher mortality rates than white men. The possible causes of worse outcomes are many, and there have been many studies that attempted to address this disparity. The question of a more biologically aggressive cancer was previously discussed, but additional factors such as socioeconomic factors, comorbidities, and treatment received have also been studied, and data are mixed.43–45
In a large SEER database review, once confounding variables of socioeconomic status, cancer stage, and treatment received were eliminated, African Americans had similar stage-for-stage survival from prostate cancer.46 Another study found, in 2,046 men, that differences in socioeconomic status explained the difference in mortality rates between white and black patients.47
However, other studies that adjusted for socioeconomic status as well as patient and tumor characteristics found that African American and Hispanic men were more likely to die of prostate cancer than white men.48
Do African American men receive less-aggressive care?
Studies have also determined that there may be differences in treatments offered to patients, which in turn negatively affect survival.28,49–53 Potentially curative local therapies (including radical surgery or radiation) may be recommended less often to black men because of major comorbidities or socioeconomic considerations.49–52
Additionally, potential metastatic disease may be identified in a less timely and accurate manner, as African American men are less likely to undergo pelvic lymph node dissection. This was associated with worse survival in men with poorly differentiated prostate cancer.53
However, returning to the possibility that prostate cancer is biologically more aggressive in African American men, some studies have shown that even after adjusting for treatment, African Americans continue to have worse survival rates.54,55 One study in men with stage T1 to T3 prostate cancer who chose brachytherapy for treatment reported that after adjusting for PSA, clinical stage, socioeconomic status, and comorbidities, African American and Hispanic race were associated with higher all-cause mortality rates.55
Equal care, equal outcomes?
In total, these results suggest that factors unrelated to tumor biology may be additional reasons for the poorer survival rates in African American men with prostate cancer. More favorable survival outcomes for African Americans with localized disease may be achieved with uniform assignment of treatment.
Fowler and Terrell56 reviewed the outcomes of 148 black and 209 white men with localized prostate cancer treated with surgery or radiation therapy over an 11-year period at a Veterans Administration hospital. Not surprisingly, the black men presented more often with advanced disease. However, survival outcomes were equivalent between whites and blacks when treatment was assigned in a uniform manner without regard to race. After a median follow-up of 96 months, there were no significant differences in all-cause, cause-specific, metastasis-free, clinical disease-free, or PSA recurrence-free survival rates in 109 black and 167 white men with low-stage cancer treated with surgery or radiation therapy or in 39 black and 42 white men with high-stage disease treated with radiotherapy.56
Similarly, Tewari et al57 studied a cohort of 402 African American and 642 white men, all of whom underwent radical prostatectomy for clinically localized prostate cancer. They were followed for PSA recurrence to determine if race-specific differences in PSA doubling time or histopathologic variables might account for the higher mortality rate in black men. While there were race-specific differences in baseline serum PSA and incidence of high-grade prostatic intraepithelial neoplasia, race was not an independent risk factor for biochemical recurrence. Instead, other variables such as the Gleason pathology score, bilateral cancers, and margin positivity were independently associated with biochemical recurrence.
Furthermore, researchers at Louisiana State University58 retrospectively analyzed data from 205 men of different races with early-stage prostate cancer. The African American men had a higher serum PSA level, suggesting more advanced disease or greater tumor burden at presentation, but no statistically significant differences were found among the pretreatment biopsy variables, including prostate volume (measured by ultrasonography), Gleason score, millimeters of cancer within the biopsy specimen, and percentage of cancer within the biopsy specimen. After treatment, there were no significant differences in survival outcomes along racial lines, leading the authors to conclude that early detection and treatment of prostate cancer in African Americans would be the best approach to lowering mortality rates.
Taken together, these data suggest that if localized prostate cancer is treated adequately and appropriately, African American patients may have improved survival rates.
DIETARY AND LIFESTYLE FACTORS
The incidence of prostate cancer is increasing in other countries where Western diets and lifestyles have been adopted,59,60 suggesting that nutritional factors may also contribute partly to prostate carcinogenesis. Culture- and race-specific differences in diet may play an important role in prostate cancer risk in certain racial minorities. Many aspects of diet and nutrition have been studied for their impact on prostate cancer.
Dietary risk factors
Too much red meat and processed meat? Although some have suggested that diets high in red and processed meats may lead to a higher risk of prostate cancer, a meta-analysis showed no association.61,62
Too much calcium? The European Prospective Investigation Into Cancer and Nutrition study found that high dietary intake of dairy protein and calcium from dairy products was associated with a higher risk of prostate cancer.63 A cohort study in the United States had similar findings with regard to calcium.64 However, the higher risk of prostate cancer was associated with consumption of 2,000 mg or more of calcium per day, which was consumed by only 2% of the study’s cohort and, as the study’s authors reported, fewer than 1% of US men. As such, only a small population of American men seem to be exposing themselves to a higher risk of prostate cancer by high calcium consumption.
High fat intake? Certain fatty acids have been implicated in general tumor genesis, and that risk has been extrapolated to prostate cancer.65 For example, high fat intake and obesity are associated with increased levels of insulin-like growth factor 1, which in turn has been shown to correlate with a significantly elevated risk of prostate cancer.63,65
Obesity has been shown to increase the risk of more-aggressive prostate cancer, but not of less-aggressive tumors.66 Moreover, men who lost weight had a lower risk of prostate cancer than those who maintained their weight over 10 years.66 Obesity may be particularly risky for African American men, in whom it was found to be associated with shorter biochemical relapse-free survival, whereas it was not an independent risk factor in white men.67
Preventive dietary agents have been elusive
Unfortunately, despite attempts to identify preventive dietary agents, none has yet been confirmed.
No benefit from selenium or vitamin E. The Selenium and Vitamin E Cancer Prevention Trial was discontinued, as there was no evidence that either agent prevented prostate cancer in relatively healthy men.68
Vitamin D? It has been suggested that lower levels of vitamin D could contribute to the higher rates of prostate cancer in African Americans, as vitamin D deficiency is more common in African Americans.69 However, several meta-analyses have shown no association between vitamin D and prostate cancer.70–72
Soy? Attempts at correlating the relatively low incidence of prostate cancer in Asians have revealed that high soy intake may be protective. Asians consume more soy than Americans do (100 vs 3 mg/day), and soy isoflavones such as genistein, glycitein, and daidzein lower the incidence of prostate cancer in laboratory mice.73
Other lifestyle factors
Other lifestyle factors have also been analyzed to see if they contribute to prostate cancer.
Pollution. Some studies have suggested that the etiology of prostate cancer may lie in environmental exposures to pesticides,74 metal industrial facilities,75 and urban living.76
Smoking. Watters et al77 found that current and former cigarette smokers were actually at a lower risk of being diagnosed with non-advanced prostate cancer, but current smokers were at higher risk of dying from prostate cancer.
Physical activity. A prospective study of lifetime physical activity of more than 45,000 men found that men who were not sedentary during work and who walked or bicycled more than 30 minutes per day during adult life had an approximately 20% lower incidence of prostate cancer.78
In sum, primary care providers who are generally promoting healthy lifestyles can point to a reduction in risk for prostate cancer as yet another benefit to a low-fat diet, a healthy body mass index, and daily exercise.
HOW PRIMARY CARE PHYSICIANS CAN HELP CLOSE THE GAP
Primary care physicians serve as the first point of health access for many in the United States today.
The diagnosis of prostate cancer is made more frequently in African American men than in other American men, often at a higher pathological grade, and with a worse mortality rate. Primary care physicians can help improve these statistics. Interventions targeting overall health, such as promotion of a healthy diet, could be established at primary care visits and could also reduce the incidence of prostate cancer in African American men. Patient education regarding prostate cancer screening, the impact of family history, and the rate of PSA screening could be improved.
Primary care physicians serve a vital role in health education and prostate cancer screening, and therefore they begin the process in potentially reducing the impact of prostate cancer in African American men. The racial disparity seen in prostate cancer may begin to be minimized with primary care physicians and specialists working together to ensure that all men receive appropriate treatment.
- Altekruse SF, Kosary CL, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2007, National Cancer Institute. Bethesda, MD. http://seer.cancer.gov/csr/1975_2007/, based on November 2009 SEER data submission, posted to the SEER web site, 2010. Accessed April 2, 2011.
- Arias E. United States life tables, 2007. National vital statistics reports; vol 59 no 9. Hyattsville, MD: National Center for Health Statistics. 2011.
- Klein JB, Nguyen CT, Saffore L, Modlin C, Modlin CS. Racial disparities in urologic health care. J Natl Med Assoc 2010; 102:108–117.
- Wells TS, Bukowinski AT, Smith TC, et al. Racial differences in prostate cancer risk remain among US servicemen with equal access to care. Prostate 2010; 70:727–734.
- Smedley BD, Stith AY, Nelson AR, editors. Unequal Treatment: Confronting Racial and Ethnic Disparities in Health Care. Institute of Medicine. National Academy Press; 2002.
- Odedina FT, Akinremi TO, Chinegwundoh F, et al. Prostate cancer disparities in black men of African descent: a comparative literature review of prostate cancer burden among black men in the United States, Caribbean, United Kingdom, and West Africa. Infect Agent Cancer 2009; 4(suppl 1):S2.
- Okobia MN, Zmuda JM, Ferrell RE, Patrick AL, Bunker CH. Chromosome 8q24 variants are associated with prostate cancer risk in a high risk population of African ancestry. Prostate 2011; 71:1054–1063.
- Haiman CA, Chen GK, Blot WJ, et al. Characterizing genetic risk at known prostate cancer susceptibility loci in African Americans. PLoS Genet 2011; 7:e1001387.
- Freedman ML, Haiman CA, Patterson N, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci U S A 2006; 103:14068–14073.
- Chang BL, Isaacs SD, Wiley KE, et al. Genome-wide screen for prostate cancer susceptibility genes in men with clinically significant disease. Prostate 2005; 64:356–361.
- Hatcher D, Daniels G, Osman I, Lee P. Molecular mechanisms involving prostate cancer racial disparity. Am J Transl Res 2009; 1:235–248.
- Robbins CM, Hooker S, Kittles RA, Carpten JD. EphB2 SNPs and sporadic prostate cancer risk in African American men. PLoS One 2011; 6:e19494.
- American Cancer Society. Cancer Facts & Figures for African Americans 2009–2010. http://www.cancer.org/acs/groups/content/@nho/documents/document/cffaa20092010pdf.pdf. Accessed April 2, 2012.
- Ayanian JZ, Udvarhelyi IS, Gatsonis CA, Pashos CL, Epstein AM. Racial differences in the use of revascularization procedures after coronary angiography. JAMA 1993; 269:2642–2646.
- Fine MJ, Ibrahim SA, Thomas SB. The role of race and genetics in health disparities research. Am J Public Health 2005; 95:2125–2128.
- Horner RD, Oddone EZ, Matchar DB. Theories explaining racial differences in the utilization of diagnostic and therapeutic procedures for cerebrovascular disease. Milbank Q 1995; 73:443–462.
- Juckett G. Cross-cultural medicine. Am Fam Physician 2005; 72:2267–2274.
- Ndubuisi SC, Kofie VY, Andoh JY, Schwartz EM. Black-white differences in the stage at presentation of prostate cancer in the District of Columbia. Urology 1995; 46:71–77.
- Misra-Hebert AD. Physician cultural competence: cross-cultural communication improves care. Cleve Clin J Med 2003; 70:289,293,296–298.
- Powell I, Sakr W, Weiss L, et al. Prostate cancer is biologically more aggressive among African Americans than Caucasian men under age 70: hypothesis supported by autopsy and SEER data. Program and abstracts from the American Urological Association 95th Annual Meeting; April 29–May 4, 2000: Atlanta, GA.
- Powell IJ, Bock CH, Ruterbusch JJ, Sakr W. Evidence supports a faster growth rate and/or earlier transformation to clinically significant prostate cancer in black than in white American men, and influences racial progression and mortality disparity. J Urol 2010; 183:1792–1796.
- Kim HS, Moreira DM, Jayachandran J, et al. Prostate biopsies from black men express higher levels of aggressive disease biomarkers than prostate biopsies from white men. Prostate Cancer Prostatic Dis 2011; 14:262–265.
- Duffy MJ. Prostate-specific antigen: does the current evidence support its use in prostate cancer screening? Ann Clin Biochem 2011; 48:310–316.
- Andriole GL, Crawford ED, Grubb RL, et al; PLCO Project Team. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med 2009; 360:1310–1319.
- Crawford ED, Grubb R, Black A, et al. Comorbidity and mortality results from a randomized prostate cancer screening trial. J Clin Oncol 2011; 29:355–361.
- Schröder FH, Hugosson J, Roobol MJ, et al; ERSPC Investigators. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 2009; 360:1320–1328.
- Hugosson J, Carlsson S, Aus G, et al. Mortality results from the Göteborg randomised population-based prostate-cancer screening trial. Lancet Oncol 2010; 11:725–732.
- Chornokur G, Dalton K, Borysova ME, Kumar NB. Disparities at presentation, diagnosis, treatment, and survival in African American men, affected by prostate cancer. Prostate 2011; 71:985–997.
- Boyd MD, Weinrich SP, Weinrich M, Norton A. Obstacles to prostate cancer screening in African-American men. J Natl Black Nurses Assoc 2001; 12:1–5.
- Freedland SJ, Isaacs WB. Explaining racial differences in prostate cancer in the United States: sociology or biology? Prostate 2005; 62:243–252.
- Ross LE, Berkowitz Z, Ekwueme DU. Use of the prostate-specific antigen test among U.S. men: findings from the 2005 National Health Interview Survey. Cancer Epidemiol Biomarkers Prev 2008; 17:636–644.
- Hosain GM, Sanderson M, Du XL, Chan W, Strom SS. Racial/ethnic differences in predictors of PSA screening in a tri-ethnic population. Cent Eur J Public Health 2011; 19:30–34.
- Carpenter WR, Howard DL, Taylor YJ, Ross LE, Wobker SE, Godley PA. Racial differences in PSA screening interval and stage at diagnosis. Cancer Causes Control 2010; 21:1071–1080.
- Betancourt JR, Maina AW. The Institute of Medicine report “Unequal Treatment”: implications for academic health centers. Mt Sinai J Med 2004; 71:314–321.
- Patel K, Kenerson D, Wang H, et al. Factors influencing prostate cancer screening in low-income African Americans in Tennessee. J Health Care Poor Underserved 2010; 21(suppl 1):114–126.
- Modlin CS. Culture, race, and disparities in health care. Cleve Clin J Med 2003; 70:283–288.
- Kyle C, Ewing T, Wu XC, et al. Statewide analysis of serum prostate specific antigen levels in Louisiana men without prostate cancer. J La State Med Soc 2004; 156:319–323.
- Vijayakumar S, Winter K, Sause W, et al. Prostate-specific antigen levels are higher in African-American than in white patients in a multicenter registration study: results of RTOG 94-12. Int J Radiat Oncol Biol Phys 1998; 40:17–25.
- Chang BL, Spangler E, Gallagher S, et al. Validation of genome-wide prostate cancer associations in men of African descent. Cancer Epidemiol Biomarkers Prev 2011; 20:23–32.
- National Comprehensive Cancer Network (NCCN). NCCN Stresses Importance of PSA Testing in High-Risk Men. http://www.nccn.org/about/news/newsinfo.asp?NewsID=218. Accessed April 2, 2012.
- National Cancer Institute. Prostate-Specific Antigen (PSA) Test. http://www.cancer.gov/cancertopics/factsheet/detection/PSA. Accessed April 2, 2012.
- Duggan D, Zheng SL, Knowlton M, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. Natl Cancer Inst 2007; 99:1836–1844.
- Grossfeld GD, Latini DM, Downs T, Lubeck DP, Mehta SS, Carroll PR. Is ethnicity an independent predictor of prostate cancer recurrence after radical prostatectomy? J Urol 2002; 168:2510–2515.
- Hoffman RM, Harlan LC, Klabunde CN, et al. Racial differences in initial treatment for clinically localized prostate cancer. Results from the prostate cancer outcomes study. J Gen Intern Med 2003; 18:845–853.
- Polednak AP. Prostate cancer treatment in black and white men: the need to consider both stage at diagnosis and socioeconomic status. J Natl Med Assoc 1998; 90:101–104.
- Merrill RM, Lyon JL. Explaining the difference in prostate cancer mortality rates between white and black men in the United States. Urology 2000; 55:730–735.
- Tewari AK, Gold HT, Demers RY, et al. Effect of socioeconomic factors on long-term mortality in men with clinically localized prostate cancer. Urology 2009; 73:624–630.
- White A, Coker AL, Du XL, Eggleston KS, Williams M. Racial/ethnic disparities in survival among men diagnosed with prostate cancer in Texas. Cancer 2011; 117:1080–1088.
- Moses KA, Paciorek AT, Penson DF, Carroll PR, Master VA. Impact of ethnicity on primary treatment choice and mortality in men with prostate cancer: data from CaPSURE. J Clin Oncol 2010; 28:1069–1074.
- Demers RY, Tiwari A, Wei J, Weiss LK, Severson RK, Montie J. Trends in the utilization of androgen-deprivation therapy for patients with prostate carcinoma suggest an effect on mortality. Cancer 2001; 92:2309–2317.
- Hsing AW, Chokkalingam AP. Prostate cancer epidemiology. Front Biosci 2006; 11:1388–1413.
- Schwartz K, Powell IJ, Underwood W, George J, Yee C, Banerjee M. Interplay of race, socioeconomic status, and treatment on survival of patients with prostate cancer. Urology 2009; 74:1296–1302.
- Hayn MH, Orom H, Shavers VL, et al. Racial/ethnic differences in receipt of pelvic lymph node dissection among men with localized/regional prostate cancer. Cancer 2011. [Epub ahead of print]
- Du XL, Lin CC, Johnson NJ, Altekruse S. Effects of individual-level socioeconomic factors on racial disparities in cancer treatment and survival: findings from the National Longitudinal Mortality Study, 1979–2003. Cancer 2011; 117:3242–3251.
- Winkfield KM, Chen MH, Dosoretz DE, et al. Race and survival following brachytherapy-based treatment for men with localized or locally advanced adenocarcinoma of the prostate. Int J Radiat Oncol Biol Phys 20115; 81:e345–e350.
- Fowler JE, Terrell F. Survival in blacks and whites after treatment for localized prostate cancer. J Urol 1996; 156:133–136.
- Tewari A, Horninger W, Badani KK, et al. Racial differences in serum prostate-specific antigen (PSA) doubling time, histopathological variables and long-term PSA recurrence between African-American and white American men undergoing radical prostatectomy for clinically localized prostate cancer. BJU Int 2005; 96:29–33.
- Bozeman C, Williams BJ, Whatley T, Crow A, Eastham J. Clinical and biopsy specimen features in black and white men with clinically localized prostate cancer. South Med J 2000; 93:400–402.
- Delongchamps NB, Singh A, Haas GP. Epidemiology of prostate cancer in Africa: another step in the understanding of the disease? Curr Probl Cancer 2007; 31:226–236.
- Quinn M, Babb P. Patterns and trends in prostate cancer incidence, survival, prevalence and mortality. Part I: international comparisons. BJU Int 2002; 90:162–173.
- Muller DC, Severi G, Baglietto L, et al. Dietary patterns and prostate cancer risk. Cancer Epidemiol Biomarkers Prev 2009; 18:3126–3129.
- Alexander DD, Mink PJ, Cushing CA, Sceurman B. A review and meta-analysis of prospective studies of red and processed meat intake and prostate cancer. Nutr J 2010; 9:50.
- Gonzalez CA, Riboli E. Diet and cancer prevention: contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Eur J Cancer 2010; 46:2555–2562.
- Rodriguez C, McCullough ML, Mondul AM, et al. Calcium, dairy products, and risk of prostate cancer in a prospective cohort of United States men. Cancer Epidemiol Biomarkers Prev 2003; 12:597–603.
- McCarty MF. Mortality from Western cancers rose dramatically among African-Americans during the 20th century: are dietary animal products to blame? Med Hypotheses 2001; 57:169–174.
- Rodriguez C, Freedland SJ, Deka A, et al. Body mass index, weight change, and risk of prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol Biomarkers Prev 2007; 16:63–69.
- Spangler E, Zeigler-Johnson CM, Coomes M, Malkowicz SB, Wein A, Rebbeck TR. Association of obesity with tumor characteristics and treatment failure of prostate cancer in African-American and European American men. J Urol 2007; 178:1939–1944.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Oakley-Girvan I, Feldman D, Eccleshall TR, et al. Risk of early-onset prostate cancer in relation to germ line polymorphisms of the vitamin D receptor. Cancer Epidemiol Biomarkers Prev 2004; 13:1325–1330.
- Gilbert R, Martin RM, Beynon R, et al. Associations of circulating and dietary vitamin D with prostate cancer risk: a systematic review and dose-response meta-analysis. Cancer Causes Control 2011; 22:319–340.
- Gandini S, Boniol M, Haukka J, et al. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. Int J Cancer 2011; 128:1414–1424.
- Yin L, Raum E, Haug U, Arndt V, Brenner H. Meta-analysis of longitudinal studies: serum vitamin D and prostate cancer risk. Cancer Epidemiol 2009; 33:435–445.
- McCormick DL, Johnson WD, Bosland MC, Lubet RA, Steele VE. Chemoprevention of rat prostate carcinogenesis by soy isoflavones and by Bowman-Birk inhibitor. Nutr Cancer 2007; 57:184–193.
- Belpomme D, Irigaray P, Ossondo M, Vacque D, Martin M. Prostate cancer as an environmental disease: an ecological study in the French Caribbean islands, Martinique and Guadeloupe. Int J Oncol 2009; 34:1037–1044.
- Ramis R, Diggle P, Cambra K, López-Abente G. Prostate cancer and industrial pollution. Risk around putative focus in a multi-source scenario. Environ Int 2011; 37:577–585.
- Dey S, Zhang Z, Hablas A, et al. Geographic patterns of cancer in the population-based registry of Egypt: possible links to environmental exposures. Cancer Epidemiol 2011; 35:254–264.
- Watters JL, Park Y, Hollenbeck A, Schatzkin A, Albanes D. Cigarette smoking and prostate cancer in a prospective US cohort study. Cancer Epidemiol Biomarkers Prev 2009; 18:2427–2435.
- Orsini N, Bellocco R, Bottai M, et al. A prospective study of lifetime physical activity and prostate cancer incidence and mortality. Br J Cancer 2009; 101:1932–1938.
Prostate cancer is the most common cancer affecting American men. In 2010, an estimated 217,730 men were diagnosed with it and 32,050 died of it.1 African American men are disproportionately affected, with a prostate cancer incidence two-thirds higher than whites and a mortality rate twice as high.1 Owing to such disparities, the life expectancy of African Americans is several years shorter than that of non-Hispanic whites.2
For the primary care provider, who is often the first access point for health care in the United States, it is important to understand what mechanisms may underlie these differences and what can be done to narrow the gap.3
WHAT IS THE CAUSE OF THESE DIFFERENCES?
Many studies have looked into the causes of the higher incidence of prostate cancer in African American men and their higher mortality rate from it. The disparity may be due to a variety of factors, some socioeconomic and some biologic.
Poorer access to care, or lower-quality care?
A study of US servicemen who had equal access to care showed that African American men had a higher rate of prostate cancer regardless of access to care and socioeconomic status.4
However, the 2002 Institute of Medicine report, Unequal Treatment: Confronting Racial and Ethnic Disparities in Health Care, found evidence that racial and ethnic minorities tend to receive lower-quality health care than whites, “even when access-related factors, such as patients’ insurance status and income, are controlled.”5
Genetic predisposition?
Some have proposed that the disparity may be a function of genetic predisposition.
Evidence of a genetic component to the high incidence and mortality rate in African American men comes from epidemiologic studies of men with similar genetic backgrounds. For example, men in Nigeria and Ghana also have a high incidence of prostate cancer, as do men of African descent in the Caribbean islands and in the United Kingdom.6
Chromosome 8q24 variants have been shown in several studies to be associated with prostate cancer risk and are more common in African American men.7–10 Some studies have also shown a higher rate of variations in cell apoptosis genes such as BCL211 and tumor-suppression genes such as EphB2 in African American men.12
These findings suggest that genetic differences may contribute to the higher prostate cancer incidence and mortality rate seen in African American men.
More-aggressive cancer, or later detection?
Not only do African American men tend to have a higher incidence of prostate cancer, they also tend to have more-aggressive disease (ie, a higher pathologic grade) at the time of diagnosis, which may contribute to the disparity in mortality rates.13–19
Initially, there was some controversy as to whether this observation is a result of genetic and biologic factors that may predispose African American men to more-aggressive disease, or if it is due to inadequate screening and delayed presentation. However, a body of evidence supports the contention that prostate cancer is more aggressive in African American men.
For example, a study of autopsy data from men who died of prostate cancer at ages 20 to 49 showed that the age of onset of prostate cancer was similar between African American and white men.20 The Surveillance Epidemiology and End Results (SEER) database showed that African American men had a higher incidence of metastatic disease across all age groups.20 A similar study conducted 10 years later confirmed that rates of subclinical prostate cancer in African American and white men do not differ by race at the early ages, but that advanced or metastatic disease occurred nearly four times as frequently in African American men.21
Another study examined prostate biopsies from African American men and found that their tumors expressed higher levels of biomarkers, suggesting they had more-aggressive disease.22
SCREENING FOR PROSTATE CANCER
Serum prostate-specific antigen (PSA) testing has become the method of choice for prostate cancer screening. However, PSA screening in asymptomatic men is under debate, because it can lead to overdetection and subsequent overtreatment of indolent disease.23
Several recent studies showed differing results from prostate cancer screening.
The US Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial found that the mortality rate was no lower with combined PSA screening and digital rectal examination during a median follow-up of 11 years than in a control group that had a lower rate of screening.24 However, further analysis of these data, with stratifying by comorbidities, showed that PSA screening in young and healthy men reduces the risk of death from prostate cancer, with minimal overtreatment.25
The European Randomized Study of Screening for Prostate Cancer found a statistically significant 20% reduction in deaths from prostate cancer with PSA screening, but that it was necessary to treat 48 men in order to save one life.26
Another study, published in 2010, showed that regular PSA screening reduced the rate of prostate cancer mortality by half over 14 years.27
African American men generally present with disease that is more advanced than in white men.28 This historically has been attributed to the fact that African Americans have been less likely to be screened for prostate cancer, though recent data indicate the gap is lessening.29–31 A cross-sectional study from the Texas Medical Center showed that 54.4% of African American men had received PSA screening, compared with 63.2% of white men.32
Another study showed that African Americans were more likely to have had a longer interval between PSA screenings before diagnosis, and that a longer PSA screening interval was associated with greater odds of having advanced disease at diagnosis.33 However, when the researchers controlled for the PSA screening interval, they found that African Americans had the same odds of being diagnosed with advanced prostate cancer as white patients did. They concluded that more frequent or systematic PSA screening may reduce the racial differences in cancer stage at diagnosis and in deaths.
Reasons for the disparities in screening
Many reasons have been proposed to explain why African Americans receive less screening, including poor communication between physicians and minority patients due to lack of cultural competency among physicians, lack of health insurance (and poor access to quality care as a result), and deficiency of knowledge about screening. Though awareness is rising, many African Americans are unaware of early detection methods for prostate cancer (eg, PSA testing), and other barriers such as cost and transportation exist that may prevent African American men from being screened.34,35
As gatekeepers, primary care physicians are in a position to address these shortcomings in patient education and to enhance the physician-patient relationship.36
Black men have higher PSA levels, with or without cancer
Physicians must also be aware of racial differences in PSA levels and realize that the predictive value of PSA in the diagnosis of prostate cancer may differ between African Americans and whites.
Black men, with or without prostate cancer, have been found to have higher PSA levels. Kyle and colleagues37 found that African American men without prostate cancer had significantly higher mean PSA levels than white men across all age groups. Furthermore, Vijayakumar et al38 found that African Americans with newly diagnosed localized prostate cancer had higher serum PSA levels than whites at diagnosis.
Although PSA cutoff levels have not been officially modified according to race, primary care physicians should have a lower threshold for referring African American men who have a suspiciously high PSA level for further urologic evaluation. Close partnership between the internist, family practitioner, and urologist will aid in the optimal use of PSA testing for the early detection of prostate cancer.
When to start PSA screening? How often to screen?
The age at which African American men should begin to have their PSA levels checked (with or without a digital rectal examination) continues to debated. However, the American Cancer Society39 recommends that African American men who have a father or brother who had prostate cancer before age 65 should begin having discussions with their physician on this topic and, with their informed consent, screening at age 45.
The frequency of PSA screening depends on the individual’s PSA level. The National Comprehensive Cancer Network40 recommends that men at high risk be offered a baseline PSA measurement and digital rectal examination at age 40 and, if the PSA level is higher than 1 ng/mL, that they be offered annual follow-ups. If the PSA level is less than 1 ng/mL, they recommend screening again at age 45. Risk factors for prostate cancer include family history as well as African American race.41
How should PSA levels be interpreted?
Interpreting PSA results is important in detecting prostate cancer at early stages.
At first, we believed the normal range of PSA for all men was 4.0 ng/mL or less. However, the American Urological Association now recognizes that the normal PSA range, in addition to varying along racial lines, also is age-dependent.42 The Cleveland Clinic Minority Men's Health Center's suggested normal ranges of PSA in African American men are:
- Age 40–49: ≤ 2.5 ng/mL
- Age 50–59: ≤ 3.0 ng/mL
- Age 60–69: ≤ 3.5 ng/mL
- Age 70–79: ≤ 4.5 ng/mL
- Age > 80: ≤ 5.0 ng/mL.
Remember that an elevated PSA does not necessarily signify prostate cancer, and that these are reference ranges only and may vary in individual men.
SURVIVAL AFTER DIAGNOSIS
African American men with prostate cancer have significantly higher mortality rates than white men. The possible causes of worse outcomes are many, and there have been many studies that attempted to address this disparity. The question of a more biologically aggressive cancer was previously discussed, but additional factors such as socioeconomic factors, comorbidities, and treatment received have also been studied, and data are mixed.43–45
In a large SEER database review, once confounding variables of socioeconomic status, cancer stage, and treatment received were eliminated, African Americans had similar stage-for-stage survival from prostate cancer.46 Another study found, in 2,046 men, that differences in socioeconomic status explained the difference in mortality rates between white and black patients.47
However, other studies that adjusted for socioeconomic status as well as patient and tumor characteristics found that African American and Hispanic men were more likely to die of prostate cancer than white men.48
Do African American men receive less-aggressive care?
Studies have also determined that there may be differences in treatments offered to patients, which in turn negatively affect survival.28,49–53 Potentially curative local therapies (including radical surgery or radiation) may be recommended less often to black men because of major comorbidities or socioeconomic considerations.49–52
Additionally, potential metastatic disease may be identified in a less timely and accurate manner, as African American men are less likely to undergo pelvic lymph node dissection. This was associated with worse survival in men with poorly differentiated prostate cancer.53
However, returning to the possibility that prostate cancer is biologically more aggressive in African American men, some studies have shown that even after adjusting for treatment, African Americans continue to have worse survival rates.54,55 One study in men with stage T1 to T3 prostate cancer who chose brachytherapy for treatment reported that after adjusting for PSA, clinical stage, socioeconomic status, and comorbidities, African American and Hispanic race were associated with higher all-cause mortality rates.55
Equal care, equal outcomes?
In total, these results suggest that factors unrelated to tumor biology may be additional reasons for the poorer survival rates in African American men with prostate cancer. More favorable survival outcomes for African Americans with localized disease may be achieved with uniform assignment of treatment.
Fowler and Terrell56 reviewed the outcomes of 148 black and 209 white men with localized prostate cancer treated with surgery or radiation therapy over an 11-year period at a Veterans Administration hospital. Not surprisingly, the black men presented more often with advanced disease. However, survival outcomes were equivalent between whites and blacks when treatment was assigned in a uniform manner without regard to race. After a median follow-up of 96 months, there were no significant differences in all-cause, cause-specific, metastasis-free, clinical disease-free, or PSA recurrence-free survival rates in 109 black and 167 white men with low-stage cancer treated with surgery or radiation therapy or in 39 black and 42 white men with high-stage disease treated with radiotherapy.56
Similarly, Tewari et al57 studied a cohort of 402 African American and 642 white men, all of whom underwent radical prostatectomy for clinically localized prostate cancer. They were followed for PSA recurrence to determine if race-specific differences in PSA doubling time or histopathologic variables might account for the higher mortality rate in black men. While there were race-specific differences in baseline serum PSA and incidence of high-grade prostatic intraepithelial neoplasia, race was not an independent risk factor for biochemical recurrence. Instead, other variables such as the Gleason pathology score, bilateral cancers, and margin positivity were independently associated with biochemical recurrence.
Furthermore, researchers at Louisiana State University58 retrospectively analyzed data from 205 men of different races with early-stage prostate cancer. The African American men had a higher serum PSA level, suggesting more advanced disease or greater tumor burden at presentation, but no statistically significant differences were found among the pretreatment biopsy variables, including prostate volume (measured by ultrasonography), Gleason score, millimeters of cancer within the biopsy specimen, and percentage of cancer within the biopsy specimen. After treatment, there were no significant differences in survival outcomes along racial lines, leading the authors to conclude that early detection and treatment of prostate cancer in African Americans would be the best approach to lowering mortality rates.
Taken together, these data suggest that if localized prostate cancer is treated adequately and appropriately, African American patients may have improved survival rates.
DIETARY AND LIFESTYLE FACTORS
The incidence of prostate cancer is increasing in other countries where Western diets and lifestyles have been adopted,59,60 suggesting that nutritional factors may also contribute partly to prostate carcinogenesis. Culture- and race-specific differences in diet may play an important role in prostate cancer risk in certain racial minorities. Many aspects of diet and nutrition have been studied for their impact on prostate cancer.
Dietary risk factors
Too much red meat and processed meat? Although some have suggested that diets high in red and processed meats may lead to a higher risk of prostate cancer, a meta-analysis showed no association.61,62
Too much calcium? The European Prospective Investigation Into Cancer and Nutrition study found that high dietary intake of dairy protein and calcium from dairy products was associated with a higher risk of prostate cancer.63 A cohort study in the United States had similar findings with regard to calcium.64 However, the higher risk of prostate cancer was associated with consumption of 2,000 mg or more of calcium per day, which was consumed by only 2% of the study’s cohort and, as the study’s authors reported, fewer than 1% of US men. As such, only a small population of American men seem to be exposing themselves to a higher risk of prostate cancer by high calcium consumption.
High fat intake? Certain fatty acids have been implicated in general tumor genesis, and that risk has been extrapolated to prostate cancer.65 For example, high fat intake and obesity are associated with increased levels of insulin-like growth factor 1, which in turn has been shown to correlate with a significantly elevated risk of prostate cancer.63,65
Obesity has been shown to increase the risk of more-aggressive prostate cancer, but not of less-aggressive tumors.66 Moreover, men who lost weight had a lower risk of prostate cancer than those who maintained their weight over 10 years.66 Obesity may be particularly risky for African American men, in whom it was found to be associated with shorter biochemical relapse-free survival, whereas it was not an independent risk factor in white men.67
Preventive dietary agents have been elusive
Unfortunately, despite attempts to identify preventive dietary agents, none has yet been confirmed.
No benefit from selenium or vitamin E. The Selenium and Vitamin E Cancer Prevention Trial was discontinued, as there was no evidence that either agent prevented prostate cancer in relatively healthy men.68
Vitamin D? It has been suggested that lower levels of vitamin D could contribute to the higher rates of prostate cancer in African Americans, as vitamin D deficiency is more common in African Americans.69 However, several meta-analyses have shown no association between vitamin D and prostate cancer.70–72
Soy? Attempts at correlating the relatively low incidence of prostate cancer in Asians have revealed that high soy intake may be protective. Asians consume more soy than Americans do (100 vs 3 mg/day), and soy isoflavones such as genistein, glycitein, and daidzein lower the incidence of prostate cancer in laboratory mice.73
Other lifestyle factors
Other lifestyle factors have also been analyzed to see if they contribute to prostate cancer.
Pollution. Some studies have suggested that the etiology of prostate cancer may lie in environmental exposures to pesticides,74 metal industrial facilities,75 and urban living.76
Smoking. Watters et al77 found that current and former cigarette smokers were actually at a lower risk of being diagnosed with non-advanced prostate cancer, but current smokers were at higher risk of dying from prostate cancer.
Physical activity. A prospective study of lifetime physical activity of more than 45,000 men found that men who were not sedentary during work and who walked or bicycled more than 30 minutes per day during adult life had an approximately 20% lower incidence of prostate cancer.78
In sum, primary care providers who are generally promoting healthy lifestyles can point to a reduction in risk for prostate cancer as yet another benefit to a low-fat diet, a healthy body mass index, and daily exercise.
HOW PRIMARY CARE PHYSICIANS CAN HELP CLOSE THE GAP
Primary care physicians serve as the first point of health access for many in the United States today.
The diagnosis of prostate cancer is made more frequently in African American men than in other American men, often at a higher pathological grade, and with a worse mortality rate. Primary care physicians can help improve these statistics. Interventions targeting overall health, such as promotion of a healthy diet, could be established at primary care visits and could also reduce the incidence of prostate cancer in African American men. Patient education regarding prostate cancer screening, the impact of family history, and the rate of PSA screening could be improved.
Primary care physicians serve a vital role in health education and prostate cancer screening, and therefore they begin the process in potentially reducing the impact of prostate cancer in African American men. The racial disparity seen in prostate cancer may begin to be minimized with primary care physicians and specialists working together to ensure that all men receive appropriate treatment.
Prostate cancer is the most common cancer affecting American men. In 2010, an estimated 217,730 men were diagnosed with it and 32,050 died of it.1 African American men are disproportionately affected, with a prostate cancer incidence two-thirds higher than whites and a mortality rate twice as high.1 Owing to such disparities, the life expectancy of African Americans is several years shorter than that of non-Hispanic whites.2
For the primary care provider, who is often the first access point for health care in the United States, it is important to understand what mechanisms may underlie these differences and what can be done to narrow the gap.3
WHAT IS THE CAUSE OF THESE DIFFERENCES?
Many studies have looked into the causes of the higher incidence of prostate cancer in African American men and their higher mortality rate from it. The disparity may be due to a variety of factors, some socioeconomic and some biologic.
Poorer access to care, or lower-quality care?
A study of US servicemen who had equal access to care showed that African American men had a higher rate of prostate cancer regardless of access to care and socioeconomic status.4
However, the 2002 Institute of Medicine report, Unequal Treatment: Confronting Racial and Ethnic Disparities in Health Care, found evidence that racial and ethnic minorities tend to receive lower-quality health care than whites, “even when access-related factors, such as patients’ insurance status and income, are controlled.”5
Genetic predisposition?
Some have proposed that the disparity may be a function of genetic predisposition.
Evidence of a genetic component to the high incidence and mortality rate in African American men comes from epidemiologic studies of men with similar genetic backgrounds. For example, men in Nigeria and Ghana also have a high incidence of prostate cancer, as do men of African descent in the Caribbean islands and in the United Kingdom.6
Chromosome 8q24 variants have been shown in several studies to be associated with prostate cancer risk and are more common in African American men.7–10 Some studies have also shown a higher rate of variations in cell apoptosis genes such as BCL211 and tumor-suppression genes such as EphB2 in African American men.12
These findings suggest that genetic differences may contribute to the higher prostate cancer incidence and mortality rate seen in African American men.
More-aggressive cancer, or later detection?
Not only do African American men tend to have a higher incidence of prostate cancer, they also tend to have more-aggressive disease (ie, a higher pathologic grade) at the time of diagnosis, which may contribute to the disparity in mortality rates.13–19
Initially, there was some controversy as to whether this observation is a result of genetic and biologic factors that may predispose African American men to more-aggressive disease, or if it is due to inadequate screening and delayed presentation. However, a body of evidence supports the contention that prostate cancer is more aggressive in African American men.
For example, a study of autopsy data from men who died of prostate cancer at ages 20 to 49 showed that the age of onset of prostate cancer was similar between African American and white men.20 The Surveillance Epidemiology and End Results (SEER) database showed that African American men had a higher incidence of metastatic disease across all age groups.20 A similar study conducted 10 years later confirmed that rates of subclinical prostate cancer in African American and white men do not differ by race at the early ages, but that advanced or metastatic disease occurred nearly four times as frequently in African American men.21
Another study examined prostate biopsies from African American men and found that their tumors expressed higher levels of biomarkers, suggesting they had more-aggressive disease.22
SCREENING FOR PROSTATE CANCER
Serum prostate-specific antigen (PSA) testing has become the method of choice for prostate cancer screening. However, PSA screening in asymptomatic men is under debate, because it can lead to overdetection and subsequent overtreatment of indolent disease.23
Several recent studies showed differing results from prostate cancer screening.
The US Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial found that the mortality rate was no lower with combined PSA screening and digital rectal examination during a median follow-up of 11 years than in a control group that had a lower rate of screening.24 However, further analysis of these data, with stratifying by comorbidities, showed that PSA screening in young and healthy men reduces the risk of death from prostate cancer, with minimal overtreatment.25
The European Randomized Study of Screening for Prostate Cancer found a statistically significant 20% reduction in deaths from prostate cancer with PSA screening, but that it was necessary to treat 48 men in order to save one life.26
Another study, published in 2010, showed that regular PSA screening reduced the rate of prostate cancer mortality by half over 14 years.27
African American men generally present with disease that is more advanced than in white men.28 This historically has been attributed to the fact that African Americans have been less likely to be screened for prostate cancer, though recent data indicate the gap is lessening.29–31 A cross-sectional study from the Texas Medical Center showed that 54.4% of African American men had received PSA screening, compared with 63.2% of white men.32
Another study showed that African Americans were more likely to have had a longer interval between PSA screenings before diagnosis, and that a longer PSA screening interval was associated with greater odds of having advanced disease at diagnosis.33 However, when the researchers controlled for the PSA screening interval, they found that African Americans had the same odds of being diagnosed with advanced prostate cancer as white patients did. They concluded that more frequent or systematic PSA screening may reduce the racial differences in cancer stage at diagnosis and in deaths.
Reasons for the disparities in screening
Many reasons have been proposed to explain why African Americans receive less screening, including poor communication between physicians and minority patients due to lack of cultural competency among physicians, lack of health insurance (and poor access to quality care as a result), and deficiency of knowledge about screening. Though awareness is rising, many African Americans are unaware of early detection methods for prostate cancer (eg, PSA testing), and other barriers such as cost and transportation exist that may prevent African American men from being screened.34,35
As gatekeepers, primary care physicians are in a position to address these shortcomings in patient education and to enhance the physician-patient relationship.36
Black men have higher PSA levels, with or without cancer
Physicians must also be aware of racial differences in PSA levels and realize that the predictive value of PSA in the diagnosis of prostate cancer may differ between African Americans and whites.
Black men, with or without prostate cancer, have been found to have higher PSA levels. Kyle and colleagues37 found that African American men without prostate cancer had significantly higher mean PSA levels than white men across all age groups. Furthermore, Vijayakumar et al38 found that African Americans with newly diagnosed localized prostate cancer had higher serum PSA levels than whites at diagnosis.
Although PSA cutoff levels have not been officially modified according to race, primary care physicians should have a lower threshold for referring African American men who have a suspiciously high PSA level for further urologic evaluation. Close partnership between the internist, family practitioner, and urologist will aid in the optimal use of PSA testing for the early detection of prostate cancer.
When to start PSA screening? How often to screen?
The age at which African American men should begin to have their PSA levels checked (with or without a digital rectal examination) continues to debated. However, the American Cancer Society39 recommends that African American men who have a father or brother who had prostate cancer before age 65 should begin having discussions with their physician on this topic and, with their informed consent, screening at age 45.
The frequency of PSA screening depends on the individual’s PSA level. The National Comprehensive Cancer Network40 recommends that men at high risk be offered a baseline PSA measurement and digital rectal examination at age 40 and, if the PSA level is higher than 1 ng/mL, that they be offered annual follow-ups. If the PSA level is less than 1 ng/mL, they recommend screening again at age 45. Risk factors for prostate cancer include family history as well as African American race.41
How should PSA levels be interpreted?
Interpreting PSA results is important in detecting prostate cancer at early stages.
At first, we believed the normal range of PSA for all men was 4.0 ng/mL or less. However, the American Urological Association now recognizes that the normal PSA range, in addition to varying along racial lines, also is age-dependent.42 The Cleveland Clinic Minority Men's Health Center's suggested normal ranges of PSA in African American men are:
- Age 40–49: ≤ 2.5 ng/mL
- Age 50–59: ≤ 3.0 ng/mL
- Age 60–69: ≤ 3.5 ng/mL
- Age 70–79: ≤ 4.5 ng/mL
- Age > 80: ≤ 5.0 ng/mL.
Remember that an elevated PSA does not necessarily signify prostate cancer, and that these are reference ranges only and may vary in individual men.
SURVIVAL AFTER DIAGNOSIS
African American men with prostate cancer have significantly higher mortality rates than white men. The possible causes of worse outcomes are many, and there have been many studies that attempted to address this disparity. The question of a more biologically aggressive cancer was previously discussed, but additional factors such as socioeconomic factors, comorbidities, and treatment received have also been studied, and data are mixed.43–45
In a large SEER database review, once confounding variables of socioeconomic status, cancer stage, and treatment received were eliminated, African Americans had similar stage-for-stage survival from prostate cancer.46 Another study found, in 2,046 men, that differences in socioeconomic status explained the difference in mortality rates between white and black patients.47
However, other studies that adjusted for socioeconomic status as well as patient and tumor characteristics found that African American and Hispanic men were more likely to die of prostate cancer than white men.48
Do African American men receive less-aggressive care?
Studies have also determined that there may be differences in treatments offered to patients, which in turn negatively affect survival.28,49–53 Potentially curative local therapies (including radical surgery or radiation) may be recommended less often to black men because of major comorbidities or socioeconomic considerations.49–52
Additionally, potential metastatic disease may be identified in a less timely and accurate manner, as African American men are less likely to undergo pelvic lymph node dissection. This was associated with worse survival in men with poorly differentiated prostate cancer.53
However, returning to the possibility that prostate cancer is biologically more aggressive in African American men, some studies have shown that even after adjusting for treatment, African Americans continue to have worse survival rates.54,55 One study in men with stage T1 to T3 prostate cancer who chose brachytherapy for treatment reported that after adjusting for PSA, clinical stage, socioeconomic status, and comorbidities, African American and Hispanic race were associated with higher all-cause mortality rates.55
Equal care, equal outcomes?
In total, these results suggest that factors unrelated to tumor biology may be additional reasons for the poorer survival rates in African American men with prostate cancer. More favorable survival outcomes for African Americans with localized disease may be achieved with uniform assignment of treatment.
Fowler and Terrell56 reviewed the outcomes of 148 black and 209 white men with localized prostate cancer treated with surgery or radiation therapy over an 11-year period at a Veterans Administration hospital. Not surprisingly, the black men presented more often with advanced disease. However, survival outcomes were equivalent between whites and blacks when treatment was assigned in a uniform manner without regard to race. After a median follow-up of 96 months, there were no significant differences in all-cause, cause-specific, metastasis-free, clinical disease-free, or PSA recurrence-free survival rates in 109 black and 167 white men with low-stage cancer treated with surgery or radiation therapy or in 39 black and 42 white men with high-stage disease treated with radiotherapy.56
Similarly, Tewari et al57 studied a cohort of 402 African American and 642 white men, all of whom underwent radical prostatectomy for clinically localized prostate cancer. They were followed for PSA recurrence to determine if race-specific differences in PSA doubling time or histopathologic variables might account for the higher mortality rate in black men. While there were race-specific differences in baseline serum PSA and incidence of high-grade prostatic intraepithelial neoplasia, race was not an independent risk factor for biochemical recurrence. Instead, other variables such as the Gleason pathology score, bilateral cancers, and margin positivity were independently associated with biochemical recurrence.
Furthermore, researchers at Louisiana State University58 retrospectively analyzed data from 205 men of different races with early-stage prostate cancer. The African American men had a higher serum PSA level, suggesting more advanced disease or greater tumor burden at presentation, but no statistically significant differences were found among the pretreatment biopsy variables, including prostate volume (measured by ultrasonography), Gleason score, millimeters of cancer within the biopsy specimen, and percentage of cancer within the biopsy specimen. After treatment, there were no significant differences in survival outcomes along racial lines, leading the authors to conclude that early detection and treatment of prostate cancer in African Americans would be the best approach to lowering mortality rates.
Taken together, these data suggest that if localized prostate cancer is treated adequately and appropriately, African American patients may have improved survival rates.
DIETARY AND LIFESTYLE FACTORS
The incidence of prostate cancer is increasing in other countries where Western diets and lifestyles have been adopted,59,60 suggesting that nutritional factors may also contribute partly to prostate carcinogenesis. Culture- and race-specific differences in diet may play an important role in prostate cancer risk in certain racial minorities. Many aspects of diet and nutrition have been studied for their impact on prostate cancer.
Dietary risk factors
Too much red meat and processed meat? Although some have suggested that diets high in red and processed meats may lead to a higher risk of prostate cancer, a meta-analysis showed no association.61,62
Too much calcium? The European Prospective Investigation Into Cancer and Nutrition study found that high dietary intake of dairy protein and calcium from dairy products was associated with a higher risk of prostate cancer.63 A cohort study in the United States had similar findings with regard to calcium.64 However, the higher risk of prostate cancer was associated with consumption of 2,000 mg or more of calcium per day, which was consumed by only 2% of the study’s cohort and, as the study’s authors reported, fewer than 1% of US men. As such, only a small population of American men seem to be exposing themselves to a higher risk of prostate cancer by high calcium consumption.
High fat intake? Certain fatty acids have been implicated in general tumor genesis, and that risk has been extrapolated to prostate cancer.65 For example, high fat intake and obesity are associated with increased levels of insulin-like growth factor 1, which in turn has been shown to correlate with a significantly elevated risk of prostate cancer.63,65
Obesity has been shown to increase the risk of more-aggressive prostate cancer, but not of less-aggressive tumors.66 Moreover, men who lost weight had a lower risk of prostate cancer than those who maintained their weight over 10 years.66 Obesity may be particularly risky for African American men, in whom it was found to be associated with shorter biochemical relapse-free survival, whereas it was not an independent risk factor in white men.67
Preventive dietary agents have been elusive
Unfortunately, despite attempts to identify preventive dietary agents, none has yet been confirmed.
No benefit from selenium or vitamin E. The Selenium and Vitamin E Cancer Prevention Trial was discontinued, as there was no evidence that either agent prevented prostate cancer in relatively healthy men.68
Vitamin D? It has been suggested that lower levels of vitamin D could contribute to the higher rates of prostate cancer in African Americans, as vitamin D deficiency is more common in African Americans.69 However, several meta-analyses have shown no association between vitamin D and prostate cancer.70–72
Soy? Attempts at correlating the relatively low incidence of prostate cancer in Asians have revealed that high soy intake may be protective. Asians consume more soy than Americans do (100 vs 3 mg/day), and soy isoflavones such as genistein, glycitein, and daidzein lower the incidence of prostate cancer in laboratory mice.73
Other lifestyle factors
Other lifestyle factors have also been analyzed to see if they contribute to prostate cancer.
Pollution. Some studies have suggested that the etiology of prostate cancer may lie in environmental exposures to pesticides,74 metal industrial facilities,75 and urban living.76
Smoking. Watters et al77 found that current and former cigarette smokers were actually at a lower risk of being diagnosed with non-advanced prostate cancer, but current smokers were at higher risk of dying from prostate cancer.
Physical activity. A prospective study of lifetime physical activity of more than 45,000 men found that men who were not sedentary during work and who walked or bicycled more than 30 minutes per day during adult life had an approximately 20% lower incidence of prostate cancer.78
In sum, primary care providers who are generally promoting healthy lifestyles can point to a reduction in risk for prostate cancer as yet another benefit to a low-fat diet, a healthy body mass index, and daily exercise.
HOW PRIMARY CARE PHYSICIANS CAN HELP CLOSE THE GAP
Primary care physicians serve as the first point of health access for many in the United States today.
The diagnosis of prostate cancer is made more frequently in African American men than in other American men, often at a higher pathological grade, and with a worse mortality rate. Primary care physicians can help improve these statistics. Interventions targeting overall health, such as promotion of a healthy diet, could be established at primary care visits and could also reduce the incidence of prostate cancer in African American men. Patient education regarding prostate cancer screening, the impact of family history, and the rate of PSA screening could be improved.
Primary care physicians serve a vital role in health education and prostate cancer screening, and therefore they begin the process in potentially reducing the impact of prostate cancer in African American men. The racial disparity seen in prostate cancer may begin to be minimized with primary care physicians and specialists working together to ensure that all men receive appropriate treatment.
- Altekruse SF, Kosary CL, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2007, National Cancer Institute. Bethesda, MD. http://seer.cancer.gov/csr/1975_2007/, based on November 2009 SEER data submission, posted to the SEER web site, 2010. Accessed April 2, 2011.
- Arias E. United States life tables, 2007. National vital statistics reports; vol 59 no 9. Hyattsville, MD: National Center for Health Statistics. 2011.
- Klein JB, Nguyen CT, Saffore L, Modlin C, Modlin CS. Racial disparities in urologic health care. J Natl Med Assoc 2010; 102:108–117.
- Wells TS, Bukowinski AT, Smith TC, et al. Racial differences in prostate cancer risk remain among US servicemen with equal access to care. Prostate 2010; 70:727–734.
- Smedley BD, Stith AY, Nelson AR, editors. Unequal Treatment: Confronting Racial and Ethnic Disparities in Health Care. Institute of Medicine. National Academy Press; 2002.
- Odedina FT, Akinremi TO, Chinegwundoh F, et al. Prostate cancer disparities in black men of African descent: a comparative literature review of prostate cancer burden among black men in the United States, Caribbean, United Kingdom, and West Africa. Infect Agent Cancer 2009; 4(suppl 1):S2.
- Okobia MN, Zmuda JM, Ferrell RE, Patrick AL, Bunker CH. Chromosome 8q24 variants are associated with prostate cancer risk in a high risk population of African ancestry. Prostate 2011; 71:1054–1063.
- Haiman CA, Chen GK, Blot WJ, et al. Characterizing genetic risk at known prostate cancer susceptibility loci in African Americans. PLoS Genet 2011; 7:e1001387.
- Freedman ML, Haiman CA, Patterson N, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci U S A 2006; 103:14068–14073.
- Chang BL, Isaacs SD, Wiley KE, et al. Genome-wide screen for prostate cancer susceptibility genes in men with clinically significant disease. Prostate 2005; 64:356–361.
- Hatcher D, Daniels G, Osman I, Lee P. Molecular mechanisms involving prostate cancer racial disparity. Am J Transl Res 2009; 1:235–248.
- Robbins CM, Hooker S, Kittles RA, Carpten JD. EphB2 SNPs and sporadic prostate cancer risk in African American men. PLoS One 2011; 6:e19494.
- American Cancer Society. Cancer Facts & Figures for African Americans 2009–2010. http://www.cancer.org/acs/groups/content/@nho/documents/document/cffaa20092010pdf.pdf. Accessed April 2, 2012.
- Ayanian JZ, Udvarhelyi IS, Gatsonis CA, Pashos CL, Epstein AM. Racial differences in the use of revascularization procedures after coronary angiography. JAMA 1993; 269:2642–2646.
- Fine MJ, Ibrahim SA, Thomas SB. The role of race and genetics in health disparities research. Am J Public Health 2005; 95:2125–2128.
- Horner RD, Oddone EZ, Matchar DB. Theories explaining racial differences in the utilization of diagnostic and therapeutic procedures for cerebrovascular disease. Milbank Q 1995; 73:443–462.
- Juckett G. Cross-cultural medicine. Am Fam Physician 2005; 72:2267–2274.
- Ndubuisi SC, Kofie VY, Andoh JY, Schwartz EM. Black-white differences in the stage at presentation of prostate cancer in the District of Columbia. Urology 1995; 46:71–77.
- Misra-Hebert AD. Physician cultural competence: cross-cultural communication improves care. Cleve Clin J Med 2003; 70:289,293,296–298.
- Powell I, Sakr W, Weiss L, et al. Prostate cancer is biologically more aggressive among African Americans than Caucasian men under age 70: hypothesis supported by autopsy and SEER data. Program and abstracts from the American Urological Association 95th Annual Meeting; April 29–May 4, 2000: Atlanta, GA.
- Powell IJ, Bock CH, Ruterbusch JJ, Sakr W. Evidence supports a faster growth rate and/or earlier transformation to clinically significant prostate cancer in black than in white American men, and influences racial progression and mortality disparity. J Urol 2010; 183:1792–1796.
- Kim HS, Moreira DM, Jayachandran J, et al. Prostate biopsies from black men express higher levels of aggressive disease biomarkers than prostate biopsies from white men. Prostate Cancer Prostatic Dis 2011; 14:262–265.
- Duffy MJ. Prostate-specific antigen: does the current evidence support its use in prostate cancer screening? Ann Clin Biochem 2011; 48:310–316.
- Andriole GL, Crawford ED, Grubb RL, et al; PLCO Project Team. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med 2009; 360:1310–1319.
- Crawford ED, Grubb R, Black A, et al. Comorbidity and mortality results from a randomized prostate cancer screening trial. J Clin Oncol 2011; 29:355–361.
- Schröder FH, Hugosson J, Roobol MJ, et al; ERSPC Investigators. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 2009; 360:1320–1328.
- Hugosson J, Carlsson S, Aus G, et al. Mortality results from the Göteborg randomised population-based prostate-cancer screening trial. Lancet Oncol 2010; 11:725–732.
- Chornokur G, Dalton K, Borysova ME, Kumar NB. Disparities at presentation, diagnosis, treatment, and survival in African American men, affected by prostate cancer. Prostate 2011; 71:985–997.
- Boyd MD, Weinrich SP, Weinrich M, Norton A. Obstacles to prostate cancer screening in African-American men. J Natl Black Nurses Assoc 2001; 12:1–5.
- Freedland SJ, Isaacs WB. Explaining racial differences in prostate cancer in the United States: sociology or biology? Prostate 2005; 62:243–252.
- Ross LE, Berkowitz Z, Ekwueme DU. Use of the prostate-specific antigen test among U.S. men: findings from the 2005 National Health Interview Survey. Cancer Epidemiol Biomarkers Prev 2008; 17:636–644.
- Hosain GM, Sanderson M, Du XL, Chan W, Strom SS. Racial/ethnic differences in predictors of PSA screening in a tri-ethnic population. Cent Eur J Public Health 2011; 19:30–34.
- Carpenter WR, Howard DL, Taylor YJ, Ross LE, Wobker SE, Godley PA. Racial differences in PSA screening interval and stage at diagnosis. Cancer Causes Control 2010; 21:1071–1080.
- Betancourt JR, Maina AW. The Institute of Medicine report “Unequal Treatment”: implications for academic health centers. Mt Sinai J Med 2004; 71:314–321.
- Patel K, Kenerson D, Wang H, et al. Factors influencing prostate cancer screening in low-income African Americans in Tennessee. J Health Care Poor Underserved 2010; 21(suppl 1):114–126.
- Modlin CS. Culture, race, and disparities in health care. Cleve Clin J Med 2003; 70:283–288.
- Kyle C, Ewing T, Wu XC, et al. Statewide analysis of serum prostate specific antigen levels in Louisiana men without prostate cancer. J La State Med Soc 2004; 156:319–323.
- Vijayakumar S, Winter K, Sause W, et al. Prostate-specific antigen levels are higher in African-American than in white patients in a multicenter registration study: results of RTOG 94-12. Int J Radiat Oncol Biol Phys 1998; 40:17–25.
- Chang BL, Spangler E, Gallagher S, et al. Validation of genome-wide prostate cancer associations in men of African descent. Cancer Epidemiol Biomarkers Prev 2011; 20:23–32.
- National Comprehensive Cancer Network (NCCN). NCCN Stresses Importance of PSA Testing in High-Risk Men. http://www.nccn.org/about/news/newsinfo.asp?NewsID=218. Accessed April 2, 2012.
- National Cancer Institute. Prostate-Specific Antigen (PSA) Test. http://www.cancer.gov/cancertopics/factsheet/detection/PSA. Accessed April 2, 2012.
- Duggan D, Zheng SL, Knowlton M, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. Natl Cancer Inst 2007; 99:1836–1844.
- Grossfeld GD, Latini DM, Downs T, Lubeck DP, Mehta SS, Carroll PR. Is ethnicity an independent predictor of prostate cancer recurrence after radical prostatectomy? J Urol 2002; 168:2510–2515.
- Hoffman RM, Harlan LC, Klabunde CN, et al. Racial differences in initial treatment for clinically localized prostate cancer. Results from the prostate cancer outcomes study. J Gen Intern Med 2003; 18:845–853.
- Polednak AP. Prostate cancer treatment in black and white men: the need to consider both stage at diagnosis and socioeconomic status. J Natl Med Assoc 1998; 90:101–104.
- Merrill RM, Lyon JL. Explaining the difference in prostate cancer mortality rates between white and black men in the United States. Urology 2000; 55:730–735.
- Tewari AK, Gold HT, Demers RY, et al. Effect of socioeconomic factors on long-term mortality in men with clinically localized prostate cancer. Urology 2009; 73:624–630.
- White A, Coker AL, Du XL, Eggleston KS, Williams M. Racial/ethnic disparities in survival among men diagnosed with prostate cancer in Texas. Cancer 2011; 117:1080–1088.
- Moses KA, Paciorek AT, Penson DF, Carroll PR, Master VA. Impact of ethnicity on primary treatment choice and mortality in men with prostate cancer: data from CaPSURE. J Clin Oncol 2010; 28:1069–1074.
- Demers RY, Tiwari A, Wei J, Weiss LK, Severson RK, Montie J. Trends in the utilization of androgen-deprivation therapy for patients with prostate carcinoma suggest an effect on mortality. Cancer 2001; 92:2309–2317.
- Hsing AW, Chokkalingam AP. Prostate cancer epidemiology. Front Biosci 2006; 11:1388–1413.
- Schwartz K, Powell IJ, Underwood W, George J, Yee C, Banerjee M. Interplay of race, socioeconomic status, and treatment on survival of patients with prostate cancer. Urology 2009; 74:1296–1302.
- Hayn MH, Orom H, Shavers VL, et al. Racial/ethnic differences in receipt of pelvic lymph node dissection among men with localized/regional prostate cancer. Cancer 2011. [Epub ahead of print]
- Du XL, Lin CC, Johnson NJ, Altekruse S. Effects of individual-level socioeconomic factors on racial disparities in cancer treatment and survival: findings from the National Longitudinal Mortality Study, 1979–2003. Cancer 2011; 117:3242–3251.
- Winkfield KM, Chen MH, Dosoretz DE, et al. Race and survival following brachytherapy-based treatment for men with localized or locally advanced adenocarcinoma of the prostate. Int J Radiat Oncol Biol Phys 20115; 81:e345–e350.
- Fowler JE, Terrell F. Survival in blacks and whites after treatment for localized prostate cancer. J Urol 1996; 156:133–136.
- Tewari A, Horninger W, Badani KK, et al. Racial differences in serum prostate-specific antigen (PSA) doubling time, histopathological variables and long-term PSA recurrence between African-American and white American men undergoing radical prostatectomy for clinically localized prostate cancer. BJU Int 2005; 96:29–33.
- Bozeman C, Williams BJ, Whatley T, Crow A, Eastham J. Clinical and biopsy specimen features in black and white men with clinically localized prostate cancer. South Med J 2000; 93:400–402.
- Delongchamps NB, Singh A, Haas GP. Epidemiology of prostate cancer in Africa: another step in the understanding of the disease? Curr Probl Cancer 2007; 31:226–236.
- Quinn M, Babb P. Patterns and trends in prostate cancer incidence, survival, prevalence and mortality. Part I: international comparisons. BJU Int 2002; 90:162–173.
- Muller DC, Severi G, Baglietto L, et al. Dietary patterns and prostate cancer risk. Cancer Epidemiol Biomarkers Prev 2009; 18:3126–3129.
- Alexander DD, Mink PJ, Cushing CA, Sceurman B. A review and meta-analysis of prospective studies of red and processed meat intake and prostate cancer. Nutr J 2010; 9:50.
- Gonzalez CA, Riboli E. Diet and cancer prevention: contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Eur J Cancer 2010; 46:2555–2562.
- Rodriguez C, McCullough ML, Mondul AM, et al. Calcium, dairy products, and risk of prostate cancer in a prospective cohort of United States men. Cancer Epidemiol Biomarkers Prev 2003; 12:597–603.
- McCarty MF. Mortality from Western cancers rose dramatically among African-Americans during the 20th century: are dietary animal products to blame? Med Hypotheses 2001; 57:169–174.
- Rodriguez C, Freedland SJ, Deka A, et al. Body mass index, weight change, and risk of prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol Biomarkers Prev 2007; 16:63–69.
- Spangler E, Zeigler-Johnson CM, Coomes M, Malkowicz SB, Wein A, Rebbeck TR. Association of obesity with tumor characteristics and treatment failure of prostate cancer in African-American and European American men. J Urol 2007; 178:1939–1944.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Oakley-Girvan I, Feldman D, Eccleshall TR, et al. Risk of early-onset prostate cancer in relation to germ line polymorphisms of the vitamin D receptor. Cancer Epidemiol Biomarkers Prev 2004; 13:1325–1330.
- Gilbert R, Martin RM, Beynon R, et al. Associations of circulating and dietary vitamin D with prostate cancer risk: a systematic review and dose-response meta-analysis. Cancer Causes Control 2011; 22:319–340.
- Gandini S, Boniol M, Haukka J, et al. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. Int J Cancer 2011; 128:1414–1424.
- Yin L, Raum E, Haug U, Arndt V, Brenner H. Meta-analysis of longitudinal studies: serum vitamin D and prostate cancer risk. Cancer Epidemiol 2009; 33:435–445.
- McCormick DL, Johnson WD, Bosland MC, Lubet RA, Steele VE. Chemoprevention of rat prostate carcinogenesis by soy isoflavones and by Bowman-Birk inhibitor. Nutr Cancer 2007; 57:184–193.
- Belpomme D, Irigaray P, Ossondo M, Vacque D, Martin M. Prostate cancer as an environmental disease: an ecological study in the French Caribbean islands, Martinique and Guadeloupe. Int J Oncol 2009; 34:1037–1044.
- Ramis R, Diggle P, Cambra K, López-Abente G. Prostate cancer and industrial pollution. Risk around putative focus in a multi-source scenario. Environ Int 2011; 37:577–585.
- Dey S, Zhang Z, Hablas A, et al. Geographic patterns of cancer in the population-based registry of Egypt: possible links to environmental exposures. Cancer Epidemiol 2011; 35:254–264.
- Watters JL, Park Y, Hollenbeck A, Schatzkin A, Albanes D. Cigarette smoking and prostate cancer in a prospective US cohort study. Cancer Epidemiol Biomarkers Prev 2009; 18:2427–2435.
- Orsini N, Bellocco R, Bottai M, et al. A prospective study of lifetime physical activity and prostate cancer incidence and mortality. Br J Cancer 2009; 101:1932–1938.
- Altekruse SF, Kosary CL, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2007, National Cancer Institute. Bethesda, MD. http://seer.cancer.gov/csr/1975_2007/, based on November 2009 SEER data submission, posted to the SEER web site, 2010. Accessed April 2, 2011.
- Arias E. United States life tables, 2007. National vital statistics reports; vol 59 no 9. Hyattsville, MD: National Center for Health Statistics. 2011.
- Klein JB, Nguyen CT, Saffore L, Modlin C, Modlin CS. Racial disparities in urologic health care. J Natl Med Assoc 2010; 102:108–117.
- Wells TS, Bukowinski AT, Smith TC, et al. Racial differences in prostate cancer risk remain among US servicemen with equal access to care. Prostate 2010; 70:727–734.
- Smedley BD, Stith AY, Nelson AR, editors. Unequal Treatment: Confronting Racial and Ethnic Disparities in Health Care. Institute of Medicine. National Academy Press; 2002.
- Odedina FT, Akinremi TO, Chinegwundoh F, et al. Prostate cancer disparities in black men of African descent: a comparative literature review of prostate cancer burden among black men in the United States, Caribbean, United Kingdom, and West Africa. Infect Agent Cancer 2009; 4(suppl 1):S2.
- Okobia MN, Zmuda JM, Ferrell RE, Patrick AL, Bunker CH. Chromosome 8q24 variants are associated with prostate cancer risk in a high risk population of African ancestry. Prostate 2011; 71:1054–1063.
- Haiman CA, Chen GK, Blot WJ, et al. Characterizing genetic risk at known prostate cancer susceptibility loci in African Americans. PLoS Genet 2011; 7:e1001387.
- Freedman ML, Haiman CA, Patterson N, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci U S A 2006; 103:14068–14073.
- Chang BL, Isaacs SD, Wiley KE, et al. Genome-wide screen for prostate cancer susceptibility genes in men with clinically significant disease. Prostate 2005; 64:356–361.
- Hatcher D, Daniels G, Osman I, Lee P. Molecular mechanisms involving prostate cancer racial disparity. Am J Transl Res 2009; 1:235–248.
- Robbins CM, Hooker S, Kittles RA, Carpten JD. EphB2 SNPs and sporadic prostate cancer risk in African American men. PLoS One 2011; 6:e19494.
- American Cancer Society. Cancer Facts & Figures for African Americans 2009–2010. http://www.cancer.org/acs/groups/content/@nho/documents/document/cffaa20092010pdf.pdf. Accessed April 2, 2012.
- Ayanian JZ, Udvarhelyi IS, Gatsonis CA, Pashos CL, Epstein AM. Racial differences in the use of revascularization procedures after coronary angiography. JAMA 1993; 269:2642–2646.
- Fine MJ, Ibrahim SA, Thomas SB. The role of race and genetics in health disparities research. Am J Public Health 2005; 95:2125–2128.
- Horner RD, Oddone EZ, Matchar DB. Theories explaining racial differences in the utilization of diagnostic and therapeutic procedures for cerebrovascular disease. Milbank Q 1995; 73:443–462.
- Juckett G. Cross-cultural medicine. Am Fam Physician 2005; 72:2267–2274.
- Ndubuisi SC, Kofie VY, Andoh JY, Schwartz EM. Black-white differences in the stage at presentation of prostate cancer in the District of Columbia. Urology 1995; 46:71–77.
- Misra-Hebert AD. Physician cultural competence: cross-cultural communication improves care. Cleve Clin J Med 2003; 70:289,293,296–298.
- Powell I, Sakr W, Weiss L, et al. Prostate cancer is biologically more aggressive among African Americans than Caucasian men under age 70: hypothesis supported by autopsy and SEER data. Program and abstracts from the American Urological Association 95th Annual Meeting; April 29–May 4, 2000: Atlanta, GA.
- Powell IJ, Bock CH, Ruterbusch JJ, Sakr W. Evidence supports a faster growth rate and/or earlier transformation to clinically significant prostate cancer in black than in white American men, and influences racial progression and mortality disparity. J Urol 2010; 183:1792–1796.
- Kim HS, Moreira DM, Jayachandran J, et al. Prostate biopsies from black men express higher levels of aggressive disease biomarkers than prostate biopsies from white men. Prostate Cancer Prostatic Dis 2011; 14:262–265.
- Duffy MJ. Prostate-specific antigen: does the current evidence support its use in prostate cancer screening? Ann Clin Biochem 2011; 48:310–316.
- Andriole GL, Crawford ED, Grubb RL, et al; PLCO Project Team. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med 2009; 360:1310–1319.
- Crawford ED, Grubb R, Black A, et al. Comorbidity and mortality results from a randomized prostate cancer screening trial. J Clin Oncol 2011; 29:355–361.
- Schröder FH, Hugosson J, Roobol MJ, et al; ERSPC Investigators. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 2009; 360:1320–1328.
- Hugosson J, Carlsson S, Aus G, et al. Mortality results from the Göteborg randomised population-based prostate-cancer screening trial. Lancet Oncol 2010; 11:725–732.
- Chornokur G, Dalton K, Borysova ME, Kumar NB. Disparities at presentation, diagnosis, treatment, and survival in African American men, affected by prostate cancer. Prostate 2011; 71:985–997.
- Boyd MD, Weinrich SP, Weinrich M, Norton A. Obstacles to prostate cancer screening in African-American men. J Natl Black Nurses Assoc 2001; 12:1–5.
- Freedland SJ, Isaacs WB. Explaining racial differences in prostate cancer in the United States: sociology or biology? Prostate 2005; 62:243–252.
- Ross LE, Berkowitz Z, Ekwueme DU. Use of the prostate-specific antigen test among U.S. men: findings from the 2005 National Health Interview Survey. Cancer Epidemiol Biomarkers Prev 2008; 17:636–644.
- Hosain GM, Sanderson M, Du XL, Chan W, Strom SS. Racial/ethnic differences in predictors of PSA screening in a tri-ethnic population. Cent Eur J Public Health 2011; 19:30–34.
- Carpenter WR, Howard DL, Taylor YJ, Ross LE, Wobker SE, Godley PA. Racial differences in PSA screening interval and stage at diagnosis. Cancer Causes Control 2010; 21:1071–1080.
- Betancourt JR, Maina AW. The Institute of Medicine report “Unequal Treatment”: implications for academic health centers. Mt Sinai J Med 2004; 71:314–321.
- Patel K, Kenerson D, Wang H, et al. Factors influencing prostate cancer screening in low-income African Americans in Tennessee. J Health Care Poor Underserved 2010; 21(suppl 1):114–126.
- Modlin CS. Culture, race, and disparities in health care. Cleve Clin J Med 2003; 70:283–288.
- Kyle C, Ewing T, Wu XC, et al. Statewide analysis of serum prostate specific antigen levels in Louisiana men without prostate cancer. J La State Med Soc 2004; 156:319–323.
- Vijayakumar S, Winter K, Sause W, et al. Prostate-specific antigen levels are higher in African-American than in white patients in a multicenter registration study: results of RTOG 94-12. Int J Radiat Oncol Biol Phys 1998; 40:17–25.
- Chang BL, Spangler E, Gallagher S, et al. Validation of genome-wide prostate cancer associations in men of African descent. Cancer Epidemiol Biomarkers Prev 2011; 20:23–32.
- National Comprehensive Cancer Network (NCCN). NCCN Stresses Importance of PSA Testing in High-Risk Men. http://www.nccn.org/about/news/newsinfo.asp?NewsID=218. Accessed April 2, 2012.
- National Cancer Institute. Prostate-Specific Antigen (PSA) Test. http://www.cancer.gov/cancertopics/factsheet/detection/PSA. Accessed April 2, 2012.
- Duggan D, Zheng SL, Knowlton M, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. Natl Cancer Inst 2007; 99:1836–1844.
- Grossfeld GD, Latini DM, Downs T, Lubeck DP, Mehta SS, Carroll PR. Is ethnicity an independent predictor of prostate cancer recurrence after radical prostatectomy? J Urol 2002; 168:2510–2515.
- Hoffman RM, Harlan LC, Klabunde CN, et al. Racial differences in initial treatment for clinically localized prostate cancer. Results from the prostate cancer outcomes study. J Gen Intern Med 2003; 18:845–853.
- Polednak AP. Prostate cancer treatment in black and white men: the need to consider both stage at diagnosis and socioeconomic status. J Natl Med Assoc 1998; 90:101–104.
- Merrill RM, Lyon JL. Explaining the difference in prostate cancer mortality rates between white and black men in the United States. Urology 2000; 55:730–735.
- Tewari AK, Gold HT, Demers RY, et al. Effect of socioeconomic factors on long-term mortality in men with clinically localized prostate cancer. Urology 2009; 73:624–630.
- White A, Coker AL, Du XL, Eggleston KS, Williams M. Racial/ethnic disparities in survival among men diagnosed with prostate cancer in Texas. Cancer 2011; 117:1080–1088.
- Moses KA, Paciorek AT, Penson DF, Carroll PR, Master VA. Impact of ethnicity on primary treatment choice and mortality in men with prostate cancer: data from CaPSURE. J Clin Oncol 2010; 28:1069–1074.
- Demers RY, Tiwari A, Wei J, Weiss LK, Severson RK, Montie J. Trends in the utilization of androgen-deprivation therapy for patients with prostate carcinoma suggest an effect on mortality. Cancer 2001; 92:2309–2317.
- Hsing AW, Chokkalingam AP. Prostate cancer epidemiology. Front Biosci 2006; 11:1388–1413.
- Schwartz K, Powell IJ, Underwood W, George J, Yee C, Banerjee M. Interplay of race, socioeconomic status, and treatment on survival of patients with prostate cancer. Urology 2009; 74:1296–1302.
- Hayn MH, Orom H, Shavers VL, et al. Racial/ethnic differences in receipt of pelvic lymph node dissection among men with localized/regional prostate cancer. Cancer 2011. [Epub ahead of print]
- Du XL, Lin CC, Johnson NJ, Altekruse S. Effects of individual-level socioeconomic factors on racial disparities in cancer treatment and survival: findings from the National Longitudinal Mortality Study, 1979–2003. Cancer 2011; 117:3242–3251.
- Winkfield KM, Chen MH, Dosoretz DE, et al. Race and survival following brachytherapy-based treatment for men with localized or locally advanced adenocarcinoma of the prostate. Int J Radiat Oncol Biol Phys 20115; 81:e345–e350.
- Fowler JE, Terrell F. Survival in blacks and whites after treatment for localized prostate cancer. J Urol 1996; 156:133–136.
- Tewari A, Horninger W, Badani KK, et al. Racial differences in serum prostate-specific antigen (PSA) doubling time, histopathological variables and long-term PSA recurrence between African-American and white American men undergoing radical prostatectomy for clinically localized prostate cancer. BJU Int 2005; 96:29–33.
- Bozeman C, Williams BJ, Whatley T, Crow A, Eastham J. Clinical and biopsy specimen features in black and white men with clinically localized prostate cancer. South Med J 2000; 93:400–402.
- Delongchamps NB, Singh A, Haas GP. Epidemiology of prostate cancer in Africa: another step in the understanding of the disease? Curr Probl Cancer 2007; 31:226–236.
- Quinn M, Babb P. Patterns and trends in prostate cancer incidence, survival, prevalence and mortality. Part I: international comparisons. BJU Int 2002; 90:162–173.
- Muller DC, Severi G, Baglietto L, et al. Dietary patterns and prostate cancer risk. Cancer Epidemiol Biomarkers Prev 2009; 18:3126–3129.
- Alexander DD, Mink PJ, Cushing CA, Sceurman B. A review and meta-analysis of prospective studies of red and processed meat intake and prostate cancer. Nutr J 2010; 9:50.
- Gonzalez CA, Riboli E. Diet and cancer prevention: contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Eur J Cancer 2010; 46:2555–2562.
- Rodriguez C, McCullough ML, Mondul AM, et al. Calcium, dairy products, and risk of prostate cancer in a prospective cohort of United States men. Cancer Epidemiol Biomarkers Prev 2003; 12:597–603.
- McCarty MF. Mortality from Western cancers rose dramatically among African-Americans during the 20th century: are dietary animal products to blame? Med Hypotheses 2001; 57:169–174.
- Rodriguez C, Freedland SJ, Deka A, et al. Body mass index, weight change, and risk of prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol Biomarkers Prev 2007; 16:63–69.
- Spangler E, Zeigler-Johnson CM, Coomes M, Malkowicz SB, Wein A, Rebbeck TR. Association of obesity with tumor characteristics and treatment failure of prostate cancer in African-American and European American men. J Urol 2007; 178:1939–1944.
- Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009; 301:39–51.
- Oakley-Girvan I, Feldman D, Eccleshall TR, et al. Risk of early-onset prostate cancer in relation to germ line polymorphisms of the vitamin D receptor. Cancer Epidemiol Biomarkers Prev 2004; 13:1325–1330.
- Gilbert R, Martin RM, Beynon R, et al. Associations of circulating and dietary vitamin D with prostate cancer risk: a systematic review and dose-response meta-analysis. Cancer Causes Control 2011; 22:319–340.
- Gandini S, Boniol M, Haukka J, et al. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. Int J Cancer 2011; 128:1414–1424.
- Yin L, Raum E, Haug U, Arndt V, Brenner H. Meta-analysis of longitudinal studies: serum vitamin D and prostate cancer risk. Cancer Epidemiol 2009; 33:435–445.
- McCormick DL, Johnson WD, Bosland MC, Lubet RA, Steele VE. Chemoprevention of rat prostate carcinogenesis by soy isoflavones and by Bowman-Birk inhibitor. Nutr Cancer 2007; 57:184–193.
- Belpomme D, Irigaray P, Ossondo M, Vacque D, Martin M. Prostate cancer as an environmental disease: an ecological study in the French Caribbean islands, Martinique and Guadeloupe. Int J Oncol 2009; 34:1037–1044.
- Ramis R, Diggle P, Cambra K, López-Abente G. Prostate cancer and industrial pollution. Risk around putative focus in a multi-source scenario. Environ Int 2011; 37:577–585.
- Dey S, Zhang Z, Hablas A, et al. Geographic patterns of cancer in the population-based registry of Egypt: possible links to environmental exposures. Cancer Epidemiol 2011; 35:254–264.
- Watters JL, Park Y, Hollenbeck A, Schatzkin A, Albanes D. Cigarette smoking and prostate cancer in a prospective US cohort study. Cancer Epidemiol Biomarkers Prev 2009; 18:2427–2435.
- Orsini N, Bellocco R, Bottai M, et al. A prospective study of lifetime physical activity and prostate cancer incidence and mortality. Br J Cancer 2009; 101:1932–1938.
KEY POINTS
- African American men have the dual disadvantages of being less likely to receive adequate care and also, possibly, of having biological differences that make them more prone to prostate cancer and more-aggressive cancer.
- Prostate-specific antigen (PSA) cutoff levels have not been officially modified according to race, but we believe primary care physicians should have a lower threshold for referring African American men who have a suspiciously high PSA level for further urologic evaluation.
- A healthy lifestyle, with a low-fat diet, healthy body mass index, and daily exercise, may decrease the risk of prostate cancer, among other benefits.
- Primary care physicians, who are often the gatekeepers to care, play a key role in educating and screening their patients.
Hyperpigmentation and hypotension
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Initial laboratory values are as follows:
- White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
- Mild anemia, with a hemoglobin of 116 g/L (140–175)
- Activated partial thromboplastin time 59.9 sec (23.0–32.4)
- Serum sodium 135 mmol/L (136–142)
- Serum potassium 4.6 mmol/L (3.5–5.0)
- Aspartate aminotransferase 58 U/L (10–30)
- Alanine aminotransferase 16 U/L (10–40)
- Alkaline phosphatase 328 U/L (30–120)
- Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
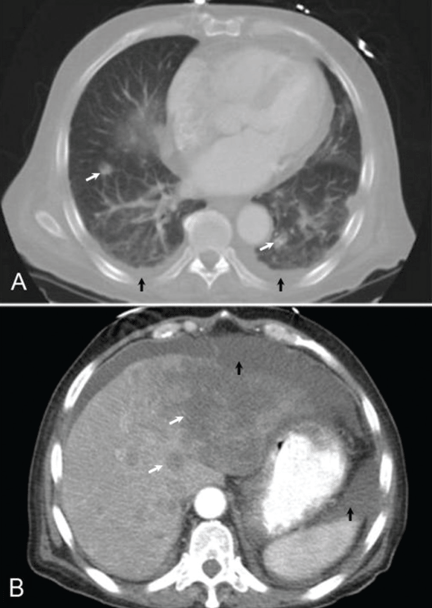
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
- Serum parathyroid-hormone-related protein
- Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
- Serum thyrotropin level
- Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
- Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
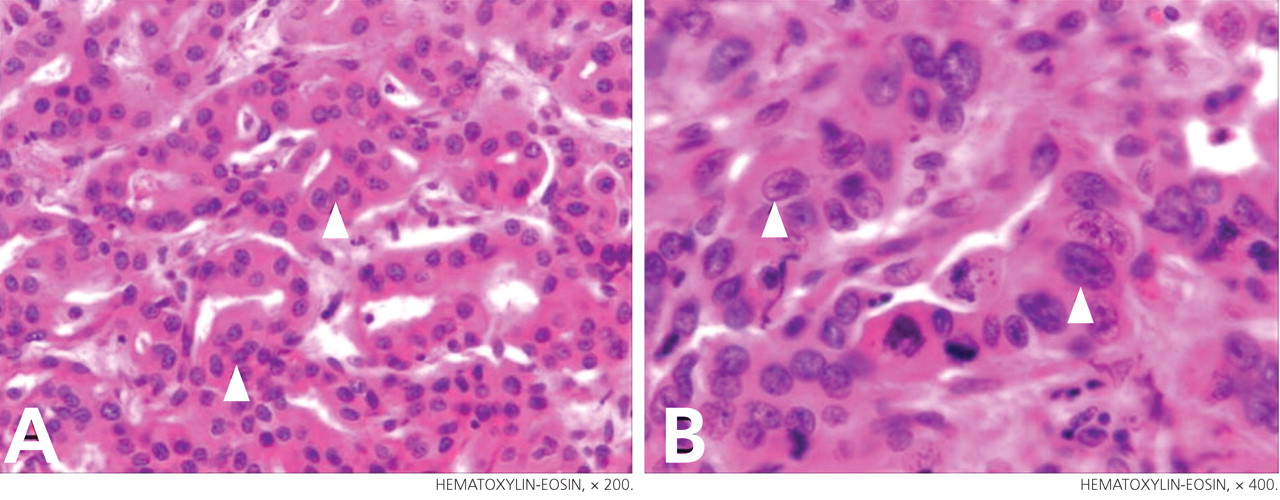
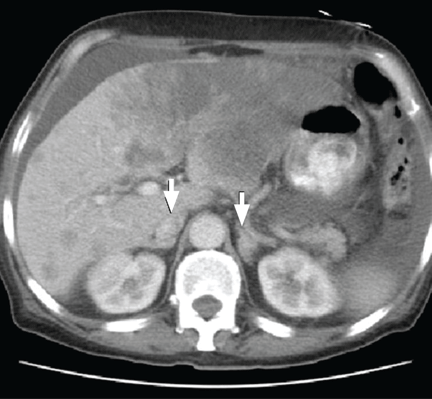
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
- Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol 2009; 160:233–237.
- Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335:1206–1212.
- Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer 1987; 60:103–107.
- Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev 1949; 29:132.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun 1995; 8:121–130.
- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol 2000; 143:91–97.
- Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer 1990; 65:177–179.
- Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet 1981; 152:607–610.
- Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J 1978; 1:1591–1592.
- Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer 1984; 54:552–557.
- Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol 2010; 2010. pii:749239.
- Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J 2008; 32:1047–1052.
- Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J 2009; 102:665–667.
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Initial laboratory values are as follows:
- White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
- Mild anemia, with a hemoglobin of 116 g/L (140–175)
- Activated partial thromboplastin time 59.9 sec (23.0–32.4)
- Serum sodium 135 mmol/L (136–142)
- Serum potassium 4.6 mmol/L (3.5–5.0)
- Aspartate aminotransferase 58 U/L (10–30)
- Alanine aminotransferase 16 U/L (10–40)
- Alkaline phosphatase 328 U/L (30–120)
- Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
- Serum parathyroid-hormone-related protein
- Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
- Serum thyrotropin level
- Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
- Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Initial laboratory values are as follows:
- White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
- Mild anemia, with a hemoglobin of 116 g/L (140–175)
- Activated partial thromboplastin time 59.9 sec (23.0–32.4)
- Serum sodium 135 mmol/L (136–142)
- Serum potassium 4.6 mmol/L (3.5–5.0)
- Aspartate aminotransferase 58 U/L (10–30)
- Alanine aminotransferase 16 U/L (10–40)
- Alkaline phosphatase 328 U/L (30–120)
- Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
- Serum parathyroid-hormone-related protein
- Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
- Serum thyrotropin level
- Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
- Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
- Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol 2009; 160:233–237.
- Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335:1206–1212.
- Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer 1987; 60:103–107.
- Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev 1949; 29:132.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun 1995; 8:121–130.
- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol 2000; 143:91–97.
- Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer 1990; 65:177–179.
- Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet 1981; 152:607–610.
- Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J 1978; 1:1591–1592.
- Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer 1984; 54:552–557.
- Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol 2010; 2010. pii:749239.
- Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J 2008; 32:1047–1052.
- Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J 2009; 102:665–667.
- Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol 2009; 160:233–237.
- Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335:1206–1212.
- Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer 1987; 60:103–107.
- Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev 1949; 29:132.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun 1995; 8:121–130.
- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol 2000; 143:91–97.
- Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer 1990; 65:177–179.
- Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet 1981; 152:607–610.
- Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J 1978; 1:1591–1592.
- Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer 1984; 54:552–557.
- Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol 2010; 2010. pii:749239.
- Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J 2008; 32:1047–1052.
- Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J 2009; 102:665–667.
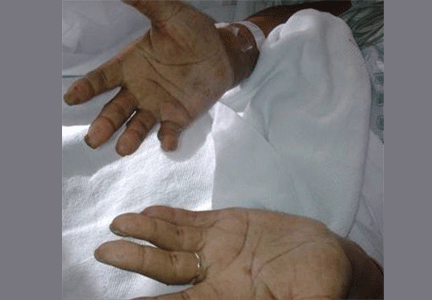
Lung cancer screening: One step forward

Screening seems to be such an easy concept: look for cancer before it is symptomatic, find it at an early stage, and treat it. We should be more able to cure cancer if it is found during screening, or at least to significantly prolong the patient’s survival by slowing the cancer’s growth and metastasis. But exactly which screening strategies save lives (and what level of efficacy is cost-effective and risk-acceptable to society and individuals) has turned out to be difficult to prove in clinical trials.
For screening to be efficacious, the test must be able to detect cancer at a stage at which early treatment makes a difference. Herein lie two challenges. A person with a cancer that grows so slowly that early treatment may not make a survival difference will not benefit from screening, and neither will someone with cancer that is so aggressive that early treatment will not significantly slow its malignant outcome. The first scenario is called “overdiagnosis”—a diagnosis made during screening that may not affect the prognosis but can lead to significant anxiety as well as additional testing and treatments, with associated costs. This has yet to be fully addressed in lung cancer screening using repeated CT imaging, but it has been discussed in breast and prostate screening.
Other challenges include how individual physicians will implement a successful lung screening program, which is more complex than yearly mammography, requiring consecutive yearly CT screening with tracking of specific results and incidental findings. How will screening be limited to appropriate patients, as dictated by trial results? Will CT review be as successful in the community as it was in trial centers of excellence? Since smoking (an act of personal choice) is the major risk factor that warrants screening, who should bear the cost?
Then there are potential unintended consequences. What if lung cancer screening makes current smokers more complacent about continuing to smoke? We must increase our educational efforts on smoking cessation, efforts that I sense are having a disappointingly limited impact on the younger generation.
Screening seems to be such an easy concept: look for cancer before it is symptomatic, find it at an early stage, and treat it. We should be more able to cure cancer if it is found during screening, or at least to significantly prolong the patient’s survival by slowing the cancer’s growth and metastasis. But exactly which screening strategies save lives (and what level of efficacy is cost-effective and risk-acceptable to society and individuals) has turned out to be difficult to prove in clinical trials.
For screening to be efficacious, the test must be able to detect cancer at a stage at which early treatment makes a difference. Herein lie two challenges. A person with a cancer that grows so slowly that early treatment may not make a survival difference will not benefit from screening, and neither will someone with cancer that is so aggressive that early treatment will not significantly slow its malignant outcome. The first scenario is called “overdiagnosis”—a diagnosis made during screening that may not affect the prognosis but can lead to significant anxiety as well as additional testing and treatments, with associated costs. This has yet to be fully addressed in lung cancer screening using repeated CT imaging, but it has been discussed in breast and prostate screening.
Other challenges include how individual physicians will implement a successful lung screening program, which is more complex than yearly mammography, requiring consecutive yearly CT screening with tracking of specific results and incidental findings. How will screening be limited to appropriate patients, as dictated by trial results? Will CT review be as successful in the community as it was in trial centers of excellence? Since smoking (an act of personal choice) is the major risk factor that warrants screening, who should bear the cost?
Then there are potential unintended consequences. What if lung cancer screening makes current smokers more complacent about continuing to smoke? We must increase our educational efforts on smoking cessation, efforts that I sense are having a disappointingly limited impact on the younger generation.
Screening seems to be such an easy concept: look for cancer before it is symptomatic, find it at an early stage, and treat it. We should be more able to cure cancer if it is found during screening, or at least to significantly prolong the patient’s survival by slowing the cancer’s growth and metastasis. But exactly which screening strategies save lives (and what level of efficacy is cost-effective and risk-acceptable to society and individuals) has turned out to be difficult to prove in clinical trials.
For screening to be efficacious, the test must be able to detect cancer at a stage at which early treatment makes a difference. Herein lie two challenges. A person with a cancer that grows so slowly that early treatment may not make a survival difference will not benefit from screening, and neither will someone with cancer that is so aggressive that early treatment will not significantly slow its malignant outcome. The first scenario is called “overdiagnosis”—a diagnosis made during screening that may not affect the prognosis but can lead to significant anxiety as well as additional testing and treatments, with associated costs. This has yet to be fully addressed in lung cancer screening using repeated CT imaging, but it has been discussed in breast and prostate screening.
Other challenges include how individual physicians will implement a successful lung screening program, which is more complex than yearly mammography, requiring consecutive yearly CT screening with tracking of specific results and incidental findings. How will screening be limited to appropriate patients, as dictated by trial results? Will CT review be as successful in the community as it was in trial centers of excellence? Since smoking (an act of personal choice) is the major risk factor that warrants screening, who should bear the cost?
Then there are potential unintended consequences. What if lung cancer screening makes current smokers more complacent about continuing to smoke? We must increase our educational efforts on smoking cessation, efforts that I sense are having a disappointingly limited impact on the younger generation.
Geriatrics update 2012: What parts of our practice to change, what to ‘think about’
A number of new studies and guidelines published over the last few years are changing the way we treat older patients. This article summarizes these recent developments in a variety of areas—from prevention of falls to targets for hypertension therapy—relevant to the treatment of geriatric patients.
A MULTICOMPONENT APPROACH TO PREVENTING FALS
The American Geriatrics Society and British Geriatrics Society’s 2010 Clinical Practice Guideline for Prevention of Falls in Older Persons1 has added an important new element since the 2001 guideline: in addition to asking older patients about a fall, clinicians should also ask whether a gait or balance problem has developed.
A complete falls evaluation and multicomponent intervention is indicated for patients who in the past year or since the previous visit have had one fall with an injury or more than one fall, or for patients who report or have been diagnosed with a gait or balance problem. A falls risk assessment is not indicated for a patient with no gait or balance problem and who has had only one noninjurious fall in the previous year that did not require medical attention.
The multicomponent evaluation detailed in the guideline is very thorough and comprises more elements than can be done in a follow-up office visit. In addition to the relevant medical history, physical examination, and cognitive and functional assessment, the fall-risk evaluation includes a falls history, medication review, visual acuity testing, gait and balance assessment, postural and heart-rate evaluation, examination of the feet and footwear, and, if appropriate, a referral for home assessment of environmental hazards.
Intervention consists of many aspects
Of the interventions, exercise has the strongest correlation with falls prevention, and a prescription should include exercises for balance, gait, and strength. Tai chi is specifically recommended.
Medications should be reduced or withdrawn. The previous guideline recommended reducing medications for patients taking four or more medications, but the current guideline applies to everyone.
First cataract removal is associated with reducing the risk of falls.
Postural hypotension should be treated if present.
Vitamin D at 800 U per day is recommended for all elderly people at risk. For elderly people in long-term care, giving vitamin D for proven or suspected deficiency is by itself correlated with risk reduction.
Interventions that by themselves are not associated with risk reduction include education (eg, providing a handout on preventing falls) and having vision checked. For adults who are cognitively impaired, there is insufficient evidence that even the multicomponent intervention helps prevent falls.
CALCIUM AND VITAMIN D MAY NOT BE HARMLESS

Calcium supplements: A cause of heart attack?
Questions have arisen in recent studies about the potential risks of calcium supplementation.
A meta-analysis of 11 trials with nearly 12,000 participants found that the risk of myocardial infarction was significantly higher in people taking calcium supplementation (relative risk 1.27; 95% confidence interval [CI] 1.01–1.59, P = .038).2 Patients were predominantly postmenopausal women and were followed for a mean of 4 years. The incidence of stroke and death were also higher in people who took calcium, but the differences did not reach statistical significance. The dosages were primarily 1,000 mg per day (range 600 mg to 2 g). Risk was independent of age, sex, and type of supplement.
The authors concluded (somewhat provocatively, because only the risk of myocardial infarction reached statistical significance) that if 1,000 people were treated with calcium supplementation for 5 years, 26 fractures would be prevented but 14 myocardial infarctions, 10 strokes, and 13 deaths would be caused.

Comments. These data suggest that physicians may wish to prescribe calcium to supplement (not replace) dietary calcium to help patients reach but not exceed current guidelines for total calcium intake for age and sex. They may also want to advise the patient to take the calcium supplement separately from medications, as indicated in Table 2.
Benefits of vitamin D may depend on dosing
Studies show that the risk of hip fracture can be reduced with modest daily vitamin D supplementation, up to 800 U daily, regardless of calcium intake.3 Some vitamin D dosing regimens, however, may also entail risk.
Sanders et al4 randomized women age 70 and older to receive an annual injection of a high dose of vitamin D (500,000 U) or placebo for 3 to 5 years. Women in the vitamin D group had 15% more falls and 25% more fractures than those in the placebo group. The once-yearly dose of 500,000 U equates to 1,370 U/day, which is not much higher than the recommended daily dosage. The median baseline serum level was 49 nmol/L and reached 120 nmol/L at 30 days in the treatment group, which was not in the toxic range.
Comments. This study cautions physicians against giving large doses of vitamin D at long intervals. Future studies should focus on long-term clinical outcomes of falls and fractures for dosing regimens currently in practice, such as 50,000 units weekly or monthly.
BISPHOSPHONATES AND NONTRAUMATIC THICK BONE FRACTURES
Bisphosphonates have been regarded as the best drugs for preventing hip fracture. But in 2010, the US Food and Drug Administration (FDA) issued a warning that bisphosphonates have been associated with “atypical” femoral fractures. The atypical fracture pattern is a clean break through the thick bone of the shaft that occurs after minimal or no trauma.5 This pattern contrasts with the splintering “typical” fracture in the proximal femur in osteoporotic bone, usually after a fall.
Another characteristic of the atypical fractures is a higher incidence of postoperative complications requiring revision surgery. In more than 14,000 women in secondary analyses of three large randomized bisphosphonate trials, 12 fractures in 10 patients were found that were classified as atypical, averaging to an incidence of 2.3 per 10,000 patient-years.6
A population-based, nested case-control study7 using Canadian pharmacy records evaluated more than 200,000 women at least 68 years old who received bisphosphonate therapy. Of these, 716 (0.35%) sustained an atypical femoral fracture and 9,723 (4.7%) had a typical osteoporotic femoral fracture. Comparing the duration of bisphosphonate use between the two groups, the authors found that the risk of an atypical fracture increased with years of usage (at 5 years or more, the adjusted odds ratio was 2.74, 95% CI 1.25–6.02), but the risk of a typical fracture decreased (at 5 years or more, the adjusted odds ratio was 0.76, 95% CI 0.63–0.93). The study suggests that for every 100 hip fractures that bisphosphonate therapy prevents, it causes one atypical hip fracture.
Comments. These studies have caused some experts to advocate periodic bisphosphonate “vacations,”8 but for how long remains an open question because the risk of a typical fracture will increase. It is possible that a biomarker can help establish the best course, but that has yet to be determined.
DENOSUMAB: A NEW DRUG FOR OSTEOPOROSIS WITH A BIG PRICE TAG
Denosumab (Prolia, Xgeva), a newly available injectable drug, is a monoclonal antibody member of the tumor necrosis factor super-family.9 It is FDA-approved for osteoporosis in postmenopausal women at a dosage of 60 mg every 6 months and for skeletal metastases from solid tumors (120 mg every 4 weeks). It is also being used off-label for skeletal protection in women taking aromatase inhibitors and for men with androgen deficiency.
This drug is expensive, costing $850 per 60-mg dose wholesale, and no data are yet available on its long-term effects.
Since the drug is not cleared via renal mechanisms, there is some hope that it can be used to treat osteoporosis in patients with advanced chronic kidney disease, since bisphosphonates are contraindicated in those with an estimated glomerular filtration rate (GFR) less than 30 to 35 mL/min. However, the major study of denosumab to date, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) study, had no patients with stage 5 chronic kidney disease (GFR < 15 mL/min/1.73 m2 or on dialysis), and too few with stage 4 chronic kidney disease (GFR 15–29) to demonstrate either the safety or efficacy of denosumab in patients with advanced chronic kidney disease.10
HYPERTENSION TREATMENT
A secondary analysis of a recent large hypertension study confirmed the benefits of antihypertensive therapy in very old adults and suggested new targets for systolic and diastolic blood pressures.11,12
The Systolic Hypertension in the Elderly Program (SHEP) trial,13 the Systolic Hypertension in Europe (Syst-Eur) trial,14 and the Hypertension in the Very Elderly Trial (HYVET)15 are the major, randomized, placebo-controlled antihypertensive trials in older adults. They all showed a reduction in the risk of stroke and cardiovascular events. The diuretic studies (SHEP and HYVET)13,15 also showed a lower risk of heart failure and death.
Most recently, secondary analysis of the International Verapamil-Trandolapril (INVEST) study11,12 showed that adults in the oldest groups (age 70–79 and 80 and older), experienced a greater risk of adverse cardiovascular outcomes if systolic blood pressure was lowered to below about 130 mm Hg. As diastolic blood pressure was lowered to about the 65–70 mm Hg range, all age groups in the study experienced an increased risk of cardiovascular events. These results confirm the findings of a secondary analysis of the SHEP trial,16 showing an increased risk of cardiovascular events when diastolic pressure was lowered to below approximately 65 mm Hg.
These studies have been incorporated into 75 pages of the 2011 Expert Consensus Document on Hypertension in the Elderly issued by the American College of Cardiology Foundation and the American Heart Association.17 In a nutshell, the guidelines suggest that older adults less than 80 years of age be treated comparably to middle-aged adults. However, for adults age 80 and older:
- A target for systolic blood pressure of 140 to 145 mm Hg “can be acceptable.”
- Initiating treatment with monotherapy (with a low-dose thiazide, calcium channel blocker, or renin-angiotensin-aldosterone system drug) is reasonable. A second drug may be added if needed.
- Patients should be monitored for “excessive” orthostasis.
- Systolic blood pressure lower than 130 mm Hg and diastolic blood pressure lower than 65 mm Hg should be avoided.
TRANSCATHETER AORTIC VALVE IMPLANTATION APPROVED BY THE FDA
An estimated 2% to 9% of the elderly have aortic stenosis. Aortic valve replacement reduces mortality rates and improves function in all age groups, including octogenarians. Those with asymptomatic aortic stenosis tend to decline very quickly once they develop heart failure, syncope, or angina. Aortic valve replacement has been shown to put people back on the course they were on before they became symptomatic.
Transcatheter self-expanding transaortic valve implantation was approved by the FDA in November 2011. The procedure does not require open surgery and involves angioplasty of the old valve, with the new valve being passed into place through a catheter and expanded. Access is either transfemoral or transapical.
Transaortic valve implantation has been rapidly adopted in Europe since 2002 without any randomized control trials. The Placement of Aortic Transcatheter Valves (PARTNER) trial18 in 2011 was the first randomized trial of this therapy. It was conducted at 25 centers, with nearly 700 patients with severe aortic stenosis randomized to undergo either transcatheter aortic valve replacement with a balloon-expandable valve (244 via the transfemoral and 104 via the transapical approach) or surgical replacement. The mean age of the patients was 84 years, and the Society of Thoracic Surgeons mean score was 12%, indicating high perioperative risk.
At 30 days after the procedure, the rates of death were 3.4% with transcatheter implantation and 6.5% with surgical replacement (P = .07). At 1 year, the rates were 24.2% and 26.8%, respectively (P = 0.44, and P = .001 for noninferiority). However, the rate of major stroke was higher in the transcatheter implantation group: 3.8% vs 2.1% in the surgical group (P = .20) at 1 month and 5.1% vs 2.4% (P = .07) at 1 year. Vascular complications were significantly more frequent in the transcatheter implantation group, and the new onset of atrial fibrillation and major bleeding were significantly higher in the surgical group.
Patients in the transcatheter implantation group had a significantly shorter length of stay in the intensive care unit and a shorter index hospitalization. At 30 days, the transcatheter group also had a significant improvement in New York Heart Association functional status and a better 6-minute walk performance, although at 1 year, these measures were similar between the two groups and were greatly improved over baseline. Quality of life, measured using the Kansas City Cardiomyopathy Questionnaire, was higher both at 6 months and at 1 year in the transcatheter implantation group compared with those who underwent the open surgical procedure.19
Comments. The higher risk of stroke with the transcatheter implantation procedure remains a concern. More evaluation is also needed with respect to function and cognition in the very elderly, and of efficacy and safety in higher- and lower-risk patients.
DEPRESSION CAN BE EFFECTIVELY TREATED WITH MEDICATION
Many placebo-controlled trials have demonstrated the effectiveness of treating depression with medications in elderly people who are cognitively intact and living in the community. A Cochrane Review20 found that in placebo-controlled trials, the number needed to treat to produce one recovery with tricyclic antidepressants, selective serotonin reuptake inhibitors, and monoamine oxidase inhibitors was less than 10 for each of the drug classes.
Since the newer drugs appear to be safer and to have fewer adverse effects than the older drugs, more older adults have been treated with antidepressants, including patients with comorbidities such as dementia that were exclusion criteria in early studies. For example, the number of older adults treated with antidepressants has increased 25% since 1992; at the same time the number being referred for cognitive-based therapies has been reduced by 43%.21 Similar trends are apparent in elderly people in long-term care. In 1999, about one-third of people in long-term care were diagnosed with depression; in 2007 more than one-half were.22
Treating depression is less effective when dementia is present
Up to half of adults age 85 and older living in the community may have dementia. In long-term care facilities, most residents likely have some cognitive impairment or are diagnosed with dementia. Many of these are also taking antidepressive agents.
A review of studies in the Medline and Cochrane registries found seven trials that treated 330 patients with antidepressants for combined depression and dementia. Efficacy was not confirmed.23
After this study was published, Banerjee et al24 treated 218 patients who had depression and dementia in nine centers in the United Kingdom. Patients received sertraline (Zoloft), mirtazapine (Remeron), or placebo. Reductions in depression scores at 13 weeks and at 39 weeks did not differ between the groups, and adverse events were more frequent in the treatment groups than in the placebo groups.
Comments. The poor performance of antidepressants in patients with dementia may be due to misdiagnosis, such as mistaking apathy for depression.25 It is also possible that better criteria than we have now are needed to diagnose depression in patients with dementia, or that current outcome measures are not sensitive for depression when dementia is present.
It may also be unsafe to treat older adults long-term with antidepressive agents. For example, although selective serotonin reuptake inhibitors, the most commonly prescribed antidepressive agents, are considered safe, their side effects are numerous and include sexual dysfunction, bleeding (due to platelet dysfunction), hyponatremia, early weight loss, tremor (mostly with paroxetine [Paxil]), sedation, apathy (especially with high doses), loose stools (with sertraline), urinary incontinence, falls, bone loss, and QTc prolongation.
Citalopram: Maximum dosage in elderly
In August 2011, an FDA Safety Communication was issued for citalopram (Celexa), stating that the daily dose should not exceed 40 mg in the general population and should not exceed 20 mg in patients age 60 and older. The dose should also not exceed 20 mg for a patient at any age who has hepatic impairment, who is known to be a poor metabolizer of CYP 2C19, or who takes cimetidine (Tagamet), since that drug inhibits the metabolism of citalopram at the CYP 2C19 enzyme site.
Although the FDA warning specifically mentions only cimetidine, physicians may have concerns about other drugs that inhibit CYP 2C19, such as proton pump inhibitors (eg, omeprazole [Prilosec]) when taken concomitantly with citalopram. Also, escitalopram (Lexapro) and sertraline are quite similar to citalopram; although they were not mentioned in the FDA Safety Communication, higher doses of these drugs may put patients at similar risk.
ALZHEIMER DISEASE: NEED TO BETTER IDENTIFY PEOPLE AT RISK
The definition of dementia is essentially the presence of a cognitive problem that affects the ability to function. For people with Alzheimer disease, impairment of cognitive performance precedes functional decline. Those with a cognitive deficit who still function well have, by definition, mild cognitive impairment (MCI). Although MCI could be caused by a variety of vascular and other neurologic processes, the most common cause of MCI in the United States is Alzheimer disease.
Unfortunately, the population with MCI currently enrolled in clinical trials to reduce the risk of progression to Alzheimer disease is heterogeneous. Many study participants may never get dementia, and others may have had the pathology present for decades and are progressing rapidly. Imaging and biomarkers are emerging as good indicators that predict progression and could help to better define populations for clinical trials.26
Studies now indicate that people with MCI that is ultimately due to Alzheimer disease are likely to have amyloid beta peptide 42 evident in the cerebrospinal fluid 10 to 20 years before symptoms arise. At the same time, amyloid is also likely to be evident in the brain with amyloid-imaging positron emission tomography (PET). Some time later, abnormalities in metabolism are also evident on fluorodeoxyglucose (FDG) PET, as are changes such as reduced hippocampal volume on magnetic resonance imaging (MRI).
The 1984 criteria for diagnosing MCI due to Alzheimer disease were recently revised to incorporate the evolving availability of biomarkers.27,28 The diagnosis of MCI itself is still based on clinical ascertainment including history, physical examination, and cognitive testing. It requires diagnosis of a cognitive decline from a prior level but maintenance of activities of daily living with no or minimal assistance. This diagnosis is certainly challenging since it requires ascertainment of a prior level of function and corroboration, when feasible, with an informant. Blood tests and imaging, which are readily available, constitute an important part of the assessment.
Attributing the MCI to Alzheimer disease requires consistency of the disease course—a gradual decline in Alzheimer disease, rather than a stroke, head injury, neurologic disease such as Parkinson disease, or mixed causes.
Knowledge of genetic factors, such as the presence of a mutation in APP, PS1, or PS2, can be predictive with young patients. The presence of one or two 34 alleles in the apolipoprotein E (APOE) gene is the only genetic variant broadly accepted as increasing the risk for late-onset Alzheimer dementia, whereas the 32 allele decreases risk.
Refining the risk attribution to Alzheimer disease requires biomarkers, currently available only in research settings:
- High likelihood—amyloid beta peptide detected by PET or cerebrospinal fluid analysis and evidence of neuronal degeneration or injury (elevated tau in the cerebrospinal fluid, decreased FDG uptake on PET, and atrophy evident by structural MRI)
- Intermediate likelihood—presence of amyloid beta peptide or evidence of neuronal degeneration or injury
- Unlikely—biomarkers tested and negative
- No comment—biomarkers not tested or reporting is indeterminate.
Comments. There is significant potential for misunderstanding the new definition for MCI. Patients who are concerned about their memory may request biomarker testing in an effort to determine if they currently have or will acquire Alzheimer disease. Doctors may be tempted to refer patients for biomarker testing (via imaging or lumbar puncture) to “screen” for MCI or Alzheimer disease.
It should be emphasized that MCI itself is still a clinical diagnosis, with the challenges noted above of determining whether there has been a cognitive decline from a prior level of function but preservation of activities of daily living. The biomarkers are not proposed to diagnose MCI, but only to help identify the subset of MCI patients most likely to progress rapidly to Alzheimer disease.
At present, the best use of biomarker testing is to aid research by identifying high-risk people among those with MCI who enroll in prospective trials for testing interventions to reduce the progression of Alzheimer disease.
- Panel on Prevention of Falls in Older Persons, American Geriatrics Society and British Geriatrics Society. J Am Geriatr Soc 2011; 59:148–157.
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: metaanalysis. BMJ 2010; 341:c3691.
- Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009; 169:551–561.
- Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women. A randomized controlled trial. JAMA 2010; 303:1815–1822.
- Kuehn BM. Prolonged bisphosphonate use linked to rare fractures, esophageal cancer. JAMA 2010; 304:2114–2115.
- Black DM, Kelly MP, Genant HK, et al; Fracture Intervention Trial Steering Committee; HORIZON Pivotal Fracture Trial Steering Committee. Bisphosphonates and fractures of the subtrochanteric or diaphyseal femur. N Engl J Med 2010; 362:1761–1771.
- Park-Wyllie LY, Mamdani MM, Juurlink DN, et al. Bisphosphonate use and the risk of subtrochanteric or femoral shaft fractures in older women. JAMA 2011; 305:783–789.
- Ott SM. What is the optimal duration of bisphosphonate therapy? Cleve Clin J Med 2011; 78:619–630.
- Cummings SR, San Martin J, McClung MR, et al; for the FREEDOM trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Pepine CJ, Handberg EM, Cooper-Dehoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Denardo SJ, Gong Y, Nichols WW, et al. Blood pressure and outcomes in very old hypertensive coronary artery disease patients: an INVEST substudy. Am J Med 2010; 123:719–726.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al; for the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension (erratum published in Lancet 1997; 350:1636). Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; for the HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Somes G, Pahor M, Shorr R, Cushman WC, Applegate WB. The role of diastolic blood pressure when treating isolated systolic hypertension. Arch Intern Med 1999; 159:2004–2009.
- Aronow WS, Fleg JL, Pepine CJ, et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly. J Am Coll Cardiol 2011; 57:2037–2114.
- Smith CR, Leon MB, Mack MJ, et al; PARTNER Trial Investigators. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med 2011; 364:2187–2198.
- Reynolds MR, Magnuson EA, Lei Y, et al; Placement of Aortic Transcatheter Valves (PARTNER) Investigators. Health-related quality of life after transcatheter aortic valve replacement in inoperable patients with severe aortic stenosis. Circulation 2011; 124:1964–1972.
- Wilson K, Mottram P, Sivanranthan A, Nightingale A. Antidepressant versus placebo for depressed elderly. Cochrane Database Syst Rev 2001;(2):CD000561.
- Akincigil A, Olfson M, Walkup JT, et al. Diagnosis and treatment of depression in older community-dwelling adults: 1992–2005. J Am Geriatr Soc 2011; 59:1042–1051.
- Gaboda D, Lucas J, Siegel M, Kalay E, Crystal S. No longer undertreated? Depression diagnosis and antidepressant therapy in elderly long-stay nursing home residents, 1999 to 2007. J Am Geriatr Soc 2011; 59:673–680.
- Nelson JC, Devanand DP. A systematic review and meta-analysis of placebo-controlled antidepressant studies in peoloe with depression and dementia. J Am Geriatr Soc 2011; 59:577–585.
- Banerjee S, Hellier J, Dewey M, et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial. Lancet 2011; 378:403–411.
- Landes AM, Sperry SD, Strauss ME, Geldmacher DS. Apathy in Alzheimer’s disease. J Am Geriatr Soc 2001; 49:1700–1707.
- Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010; 9:1118–1127.
- Daviglus ML, Bell CC, Berrettini W, et al. National Institutes of Health State-of-the-Science Conference statement: preventing Alzheimer disease and cognitive decline. Ann Intern Med 2010; 153:176–181.
- McKhann GM, Knopman DS, Chertkow H, et al The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263–269.
A number of new studies and guidelines published over the last few years are changing the way we treat older patients. This article summarizes these recent developments in a variety of areas—from prevention of falls to targets for hypertension therapy—relevant to the treatment of geriatric patients.
A MULTICOMPONENT APPROACH TO PREVENTING FALS
The American Geriatrics Society and British Geriatrics Society’s 2010 Clinical Practice Guideline for Prevention of Falls in Older Persons1 has added an important new element since the 2001 guideline: in addition to asking older patients about a fall, clinicians should also ask whether a gait or balance problem has developed.
A complete falls evaluation and multicomponent intervention is indicated for patients who in the past year or since the previous visit have had one fall with an injury or more than one fall, or for patients who report or have been diagnosed with a gait or balance problem. A falls risk assessment is not indicated for a patient with no gait or balance problem and who has had only one noninjurious fall in the previous year that did not require medical attention.
The multicomponent evaluation detailed in the guideline is very thorough and comprises more elements than can be done in a follow-up office visit. In addition to the relevant medical history, physical examination, and cognitive and functional assessment, the fall-risk evaluation includes a falls history, medication review, visual acuity testing, gait and balance assessment, postural and heart-rate evaluation, examination of the feet and footwear, and, if appropriate, a referral for home assessment of environmental hazards.
Intervention consists of many aspects
Of the interventions, exercise has the strongest correlation with falls prevention, and a prescription should include exercises for balance, gait, and strength. Tai chi is specifically recommended.
Medications should be reduced or withdrawn. The previous guideline recommended reducing medications for patients taking four or more medications, but the current guideline applies to everyone.
First cataract removal is associated with reducing the risk of falls.
Postural hypotension should be treated if present.
Vitamin D at 800 U per day is recommended for all elderly people at risk. For elderly people in long-term care, giving vitamin D for proven or suspected deficiency is by itself correlated with risk reduction.
Interventions that by themselves are not associated with risk reduction include education (eg, providing a handout on preventing falls) and having vision checked. For adults who are cognitively impaired, there is insufficient evidence that even the multicomponent intervention helps prevent falls.
CALCIUM AND VITAMIN D MAY NOT BE HARMLESS
Calcium supplements: A cause of heart attack?
Questions have arisen in recent studies about the potential risks of calcium supplementation.
A meta-analysis of 11 trials with nearly 12,000 participants found that the risk of myocardial infarction was significantly higher in people taking calcium supplementation (relative risk 1.27; 95% confidence interval [CI] 1.01–1.59, P = .038).2 Patients were predominantly postmenopausal women and were followed for a mean of 4 years. The incidence of stroke and death were also higher in people who took calcium, but the differences did not reach statistical significance. The dosages were primarily 1,000 mg per day (range 600 mg to 2 g). Risk was independent of age, sex, and type of supplement.
The authors concluded (somewhat provocatively, because only the risk of myocardial infarction reached statistical significance) that if 1,000 people were treated with calcium supplementation for 5 years, 26 fractures would be prevented but 14 myocardial infarctions, 10 strokes, and 13 deaths would be caused.
Comments. These data suggest that physicians may wish to prescribe calcium to supplement (not replace) dietary calcium to help patients reach but not exceed current guidelines for total calcium intake for age and sex. They may also want to advise the patient to take the calcium supplement separately from medications, as indicated in Table 2.
Benefits of vitamin D may depend on dosing
Studies show that the risk of hip fracture can be reduced with modest daily vitamin D supplementation, up to 800 U daily, regardless of calcium intake.3 Some vitamin D dosing regimens, however, may also entail risk.
Sanders et al4 randomized women age 70 and older to receive an annual injection of a high dose of vitamin D (500,000 U) or placebo for 3 to 5 years. Women in the vitamin D group had 15% more falls and 25% more fractures than those in the placebo group. The once-yearly dose of 500,000 U equates to 1,370 U/day, which is not much higher than the recommended daily dosage. The median baseline serum level was 49 nmol/L and reached 120 nmol/L at 30 days in the treatment group, which was not in the toxic range.
Comments. This study cautions physicians against giving large doses of vitamin D at long intervals. Future studies should focus on long-term clinical outcomes of falls and fractures for dosing regimens currently in practice, such as 50,000 units weekly or monthly.
BISPHOSPHONATES AND NONTRAUMATIC THICK BONE FRACTURES
Bisphosphonates have been regarded as the best drugs for preventing hip fracture. But in 2010, the US Food and Drug Administration (FDA) issued a warning that bisphosphonates have been associated with “atypical” femoral fractures. The atypical fracture pattern is a clean break through the thick bone of the shaft that occurs after minimal or no trauma.5 This pattern contrasts with the splintering “typical” fracture in the proximal femur in osteoporotic bone, usually after a fall.
Another characteristic of the atypical fractures is a higher incidence of postoperative complications requiring revision surgery. In more than 14,000 women in secondary analyses of three large randomized bisphosphonate trials, 12 fractures in 10 patients were found that were classified as atypical, averaging to an incidence of 2.3 per 10,000 patient-years.6
A population-based, nested case-control study7 using Canadian pharmacy records evaluated more than 200,000 women at least 68 years old who received bisphosphonate therapy. Of these, 716 (0.35%) sustained an atypical femoral fracture and 9,723 (4.7%) had a typical osteoporotic femoral fracture. Comparing the duration of bisphosphonate use between the two groups, the authors found that the risk of an atypical fracture increased with years of usage (at 5 years or more, the adjusted odds ratio was 2.74, 95% CI 1.25–6.02), but the risk of a typical fracture decreased (at 5 years or more, the adjusted odds ratio was 0.76, 95% CI 0.63–0.93). The study suggests that for every 100 hip fractures that bisphosphonate therapy prevents, it causes one atypical hip fracture.
Comments. These studies have caused some experts to advocate periodic bisphosphonate “vacations,”8 but for how long remains an open question because the risk of a typical fracture will increase. It is possible that a biomarker can help establish the best course, but that has yet to be determined.
DENOSUMAB: A NEW DRUG FOR OSTEOPOROSIS WITH A BIG PRICE TAG
Denosumab (Prolia, Xgeva), a newly available injectable drug, is a monoclonal antibody member of the tumor necrosis factor super-family.9 It is FDA-approved for osteoporosis in postmenopausal women at a dosage of 60 mg every 6 months and for skeletal metastases from solid tumors (120 mg every 4 weeks). It is also being used off-label for skeletal protection in women taking aromatase inhibitors and for men with androgen deficiency.
This drug is expensive, costing $850 per 60-mg dose wholesale, and no data are yet available on its long-term effects.
Since the drug is not cleared via renal mechanisms, there is some hope that it can be used to treat osteoporosis in patients with advanced chronic kidney disease, since bisphosphonates are contraindicated in those with an estimated glomerular filtration rate (GFR) less than 30 to 35 mL/min. However, the major study of denosumab to date, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) study, had no patients with stage 5 chronic kidney disease (GFR < 15 mL/min/1.73 m2 or on dialysis), and too few with stage 4 chronic kidney disease (GFR 15–29) to demonstrate either the safety or efficacy of denosumab in patients with advanced chronic kidney disease.10
HYPERTENSION TREATMENT
A secondary analysis of a recent large hypertension study confirmed the benefits of antihypertensive therapy in very old adults and suggested new targets for systolic and diastolic blood pressures.11,12
The Systolic Hypertension in the Elderly Program (SHEP) trial,13 the Systolic Hypertension in Europe (Syst-Eur) trial,14 and the Hypertension in the Very Elderly Trial (HYVET)15 are the major, randomized, placebo-controlled antihypertensive trials in older adults. They all showed a reduction in the risk of stroke and cardiovascular events. The diuretic studies (SHEP and HYVET)13,15 also showed a lower risk of heart failure and death.
Most recently, secondary analysis of the International Verapamil-Trandolapril (INVEST) study11,12 showed that adults in the oldest groups (age 70–79 and 80 and older), experienced a greater risk of adverse cardiovascular outcomes if systolic blood pressure was lowered to below about 130 mm Hg. As diastolic blood pressure was lowered to about the 65–70 mm Hg range, all age groups in the study experienced an increased risk of cardiovascular events. These results confirm the findings of a secondary analysis of the SHEP trial,16 showing an increased risk of cardiovascular events when diastolic pressure was lowered to below approximately 65 mm Hg.
These studies have been incorporated into 75 pages of the 2011 Expert Consensus Document on Hypertension in the Elderly issued by the American College of Cardiology Foundation and the American Heart Association.17 In a nutshell, the guidelines suggest that older adults less than 80 years of age be treated comparably to middle-aged adults. However, for adults age 80 and older:
- A target for systolic blood pressure of 140 to 145 mm Hg “can be acceptable.”
- Initiating treatment with monotherapy (with a low-dose thiazide, calcium channel blocker, or renin-angiotensin-aldosterone system drug) is reasonable. A second drug may be added if needed.
- Patients should be monitored for “excessive” orthostasis.
- Systolic blood pressure lower than 130 mm Hg and diastolic blood pressure lower than 65 mm Hg should be avoided.
TRANSCATHETER AORTIC VALVE IMPLANTATION APPROVED BY THE FDA
An estimated 2% to 9% of the elderly have aortic stenosis. Aortic valve replacement reduces mortality rates and improves function in all age groups, including octogenarians. Those with asymptomatic aortic stenosis tend to decline very quickly once they develop heart failure, syncope, or angina. Aortic valve replacement has been shown to put people back on the course they were on before they became symptomatic.
Transcatheter self-expanding transaortic valve implantation was approved by the FDA in November 2011. The procedure does not require open surgery and involves angioplasty of the old valve, with the new valve being passed into place through a catheter and expanded. Access is either transfemoral or transapical.
Transaortic valve implantation has been rapidly adopted in Europe since 2002 without any randomized control trials. The Placement of Aortic Transcatheter Valves (PARTNER) trial18 in 2011 was the first randomized trial of this therapy. It was conducted at 25 centers, with nearly 700 patients with severe aortic stenosis randomized to undergo either transcatheter aortic valve replacement with a balloon-expandable valve (244 via the transfemoral and 104 via the transapical approach) or surgical replacement. The mean age of the patients was 84 years, and the Society of Thoracic Surgeons mean score was 12%, indicating high perioperative risk.
At 30 days after the procedure, the rates of death were 3.4% with transcatheter implantation and 6.5% with surgical replacement (P = .07). At 1 year, the rates were 24.2% and 26.8%, respectively (P = 0.44, and P = .001 for noninferiority). However, the rate of major stroke was higher in the transcatheter implantation group: 3.8% vs 2.1% in the surgical group (P = .20) at 1 month and 5.1% vs 2.4% (P = .07) at 1 year. Vascular complications were significantly more frequent in the transcatheter implantation group, and the new onset of atrial fibrillation and major bleeding were significantly higher in the surgical group.
Patients in the transcatheter implantation group had a significantly shorter length of stay in the intensive care unit and a shorter index hospitalization. At 30 days, the transcatheter group also had a significant improvement in New York Heart Association functional status and a better 6-minute walk performance, although at 1 year, these measures were similar between the two groups and were greatly improved over baseline. Quality of life, measured using the Kansas City Cardiomyopathy Questionnaire, was higher both at 6 months and at 1 year in the transcatheter implantation group compared with those who underwent the open surgical procedure.19
Comments. The higher risk of stroke with the transcatheter implantation procedure remains a concern. More evaluation is also needed with respect to function and cognition in the very elderly, and of efficacy and safety in higher- and lower-risk patients.
DEPRESSION CAN BE EFFECTIVELY TREATED WITH MEDICATION
Many placebo-controlled trials have demonstrated the effectiveness of treating depression with medications in elderly people who are cognitively intact and living in the community. A Cochrane Review20 found that in placebo-controlled trials, the number needed to treat to produce one recovery with tricyclic antidepressants, selective serotonin reuptake inhibitors, and monoamine oxidase inhibitors was less than 10 for each of the drug classes.
Since the newer drugs appear to be safer and to have fewer adverse effects than the older drugs, more older adults have been treated with antidepressants, including patients with comorbidities such as dementia that were exclusion criteria in early studies. For example, the number of older adults treated with antidepressants has increased 25% since 1992; at the same time the number being referred for cognitive-based therapies has been reduced by 43%.21 Similar trends are apparent in elderly people in long-term care. In 1999, about one-third of people in long-term care were diagnosed with depression; in 2007 more than one-half were.22
Treating depression is less effective when dementia is present
Up to half of adults age 85 and older living in the community may have dementia. In long-term care facilities, most residents likely have some cognitive impairment or are diagnosed with dementia. Many of these are also taking antidepressive agents.
A review of studies in the Medline and Cochrane registries found seven trials that treated 330 patients with antidepressants for combined depression and dementia. Efficacy was not confirmed.23
After this study was published, Banerjee et al24 treated 218 patients who had depression and dementia in nine centers in the United Kingdom. Patients received sertraline (Zoloft), mirtazapine (Remeron), or placebo. Reductions in depression scores at 13 weeks and at 39 weeks did not differ between the groups, and adverse events were more frequent in the treatment groups than in the placebo groups.
Comments. The poor performance of antidepressants in patients with dementia may be due to misdiagnosis, such as mistaking apathy for depression.25 It is also possible that better criteria than we have now are needed to diagnose depression in patients with dementia, or that current outcome measures are not sensitive for depression when dementia is present.
It may also be unsafe to treat older adults long-term with antidepressive agents. For example, although selective serotonin reuptake inhibitors, the most commonly prescribed antidepressive agents, are considered safe, their side effects are numerous and include sexual dysfunction, bleeding (due to platelet dysfunction), hyponatremia, early weight loss, tremor (mostly with paroxetine [Paxil]), sedation, apathy (especially with high doses), loose stools (with sertraline), urinary incontinence, falls, bone loss, and QTc prolongation.
Citalopram: Maximum dosage in elderly
In August 2011, an FDA Safety Communication was issued for citalopram (Celexa), stating that the daily dose should not exceed 40 mg in the general population and should not exceed 20 mg in patients age 60 and older. The dose should also not exceed 20 mg for a patient at any age who has hepatic impairment, who is known to be a poor metabolizer of CYP 2C19, or who takes cimetidine (Tagamet), since that drug inhibits the metabolism of citalopram at the CYP 2C19 enzyme site.
Although the FDA warning specifically mentions only cimetidine, physicians may have concerns about other drugs that inhibit CYP 2C19, such as proton pump inhibitors (eg, omeprazole [Prilosec]) when taken concomitantly with citalopram. Also, escitalopram (Lexapro) and sertraline are quite similar to citalopram; although they were not mentioned in the FDA Safety Communication, higher doses of these drugs may put patients at similar risk.
ALZHEIMER DISEASE: NEED TO BETTER IDENTIFY PEOPLE AT RISK
The definition of dementia is essentially the presence of a cognitive problem that affects the ability to function. For people with Alzheimer disease, impairment of cognitive performance precedes functional decline. Those with a cognitive deficit who still function well have, by definition, mild cognitive impairment (MCI). Although MCI could be caused by a variety of vascular and other neurologic processes, the most common cause of MCI in the United States is Alzheimer disease.
Unfortunately, the population with MCI currently enrolled in clinical trials to reduce the risk of progression to Alzheimer disease is heterogeneous. Many study participants may never get dementia, and others may have had the pathology present for decades and are progressing rapidly. Imaging and biomarkers are emerging as good indicators that predict progression and could help to better define populations for clinical trials.26
Studies now indicate that people with MCI that is ultimately due to Alzheimer disease are likely to have amyloid beta peptide 42 evident in the cerebrospinal fluid 10 to 20 years before symptoms arise. At the same time, amyloid is also likely to be evident in the brain with amyloid-imaging positron emission tomography (PET). Some time later, abnormalities in metabolism are also evident on fluorodeoxyglucose (FDG) PET, as are changes such as reduced hippocampal volume on magnetic resonance imaging (MRI).
The 1984 criteria for diagnosing MCI due to Alzheimer disease were recently revised to incorporate the evolving availability of biomarkers.27,28 The diagnosis of MCI itself is still based on clinical ascertainment including history, physical examination, and cognitive testing. It requires diagnosis of a cognitive decline from a prior level but maintenance of activities of daily living with no or minimal assistance. This diagnosis is certainly challenging since it requires ascertainment of a prior level of function and corroboration, when feasible, with an informant. Blood tests and imaging, which are readily available, constitute an important part of the assessment.
Attributing the MCI to Alzheimer disease requires consistency of the disease course—a gradual decline in Alzheimer disease, rather than a stroke, head injury, neurologic disease such as Parkinson disease, or mixed causes.
Knowledge of genetic factors, such as the presence of a mutation in APP, PS1, or PS2, can be predictive with young patients. The presence of one or two 34 alleles in the apolipoprotein E (APOE) gene is the only genetic variant broadly accepted as increasing the risk for late-onset Alzheimer dementia, whereas the 32 allele decreases risk.
Refining the risk attribution to Alzheimer disease requires biomarkers, currently available only in research settings:
- High likelihood—amyloid beta peptide detected by PET or cerebrospinal fluid analysis and evidence of neuronal degeneration or injury (elevated tau in the cerebrospinal fluid, decreased FDG uptake on PET, and atrophy evident by structural MRI)
- Intermediate likelihood—presence of amyloid beta peptide or evidence of neuronal degeneration or injury
- Unlikely—biomarkers tested and negative
- No comment—biomarkers not tested or reporting is indeterminate.
Comments. There is significant potential for misunderstanding the new definition for MCI. Patients who are concerned about their memory may request biomarker testing in an effort to determine if they currently have or will acquire Alzheimer disease. Doctors may be tempted to refer patients for biomarker testing (via imaging or lumbar puncture) to “screen” for MCI or Alzheimer disease.
It should be emphasized that MCI itself is still a clinical diagnosis, with the challenges noted above of determining whether there has been a cognitive decline from a prior level of function but preservation of activities of daily living. The biomarkers are not proposed to diagnose MCI, but only to help identify the subset of MCI patients most likely to progress rapidly to Alzheimer disease.
At present, the best use of biomarker testing is to aid research by identifying high-risk people among those with MCI who enroll in prospective trials for testing interventions to reduce the progression of Alzheimer disease.
A number of new studies and guidelines published over the last few years are changing the way we treat older patients. This article summarizes these recent developments in a variety of areas—from prevention of falls to targets for hypertension therapy—relevant to the treatment of geriatric patients.
A MULTICOMPONENT APPROACH TO PREVENTING FALS
The American Geriatrics Society and British Geriatrics Society’s 2010 Clinical Practice Guideline for Prevention of Falls in Older Persons1 has added an important new element since the 2001 guideline: in addition to asking older patients about a fall, clinicians should also ask whether a gait or balance problem has developed.
A complete falls evaluation and multicomponent intervention is indicated for patients who in the past year or since the previous visit have had one fall with an injury or more than one fall, or for patients who report or have been diagnosed with a gait or balance problem. A falls risk assessment is not indicated for a patient with no gait or balance problem and who has had only one noninjurious fall in the previous year that did not require medical attention.
The multicomponent evaluation detailed in the guideline is very thorough and comprises more elements than can be done in a follow-up office visit. In addition to the relevant medical history, physical examination, and cognitive and functional assessment, the fall-risk evaluation includes a falls history, medication review, visual acuity testing, gait and balance assessment, postural and heart-rate evaluation, examination of the feet and footwear, and, if appropriate, a referral for home assessment of environmental hazards.
Intervention consists of many aspects
Of the interventions, exercise has the strongest correlation with falls prevention, and a prescription should include exercises for balance, gait, and strength. Tai chi is specifically recommended.
Medications should be reduced or withdrawn. The previous guideline recommended reducing medications for patients taking four or more medications, but the current guideline applies to everyone.
First cataract removal is associated with reducing the risk of falls.
Postural hypotension should be treated if present.
Vitamin D at 800 U per day is recommended for all elderly people at risk. For elderly people in long-term care, giving vitamin D for proven or suspected deficiency is by itself correlated with risk reduction.
Interventions that by themselves are not associated with risk reduction include education (eg, providing a handout on preventing falls) and having vision checked. For adults who are cognitively impaired, there is insufficient evidence that even the multicomponent intervention helps prevent falls.
CALCIUM AND VITAMIN D MAY NOT BE HARMLESS
Calcium supplements: A cause of heart attack?
Questions have arisen in recent studies about the potential risks of calcium supplementation.
A meta-analysis of 11 trials with nearly 12,000 participants found that the risk of myocardial infarction was significantly higher in people taking calcium supplementation (relative risk 1.27; 95% confidence interval [CI] 1.01–1.59, P = .038).2 Patients were predominantly postmenopausal women and were followed for a mean of 4 years. The incidence of stroke and death were also higher in people who took calcium, but the differences did not reach statistical significance. The dosages were primarily 1,000 mg per day (range 600 mg to 2 g). Risk was independent of age, sex, and type of supplement.
The authors concluded (somewhat provocatively, because only the risk of myocardial infarction reached statistical significance) that if 1,000 people were treated with calcium supplementation for 5 years, 26 fractures would be prevented but 14 myocardial infarctions, 10 strokes, and 13 deaths would be caused.
Comments. These data suggest that physicians may wish to prescribe calcium to supplement (not replace) dietary calcium to help patients reach but not exceed current guidelines for total calcium intake for age and sex. They may also want to advise the patient to take the calcium supplement separately from medications, as indicated in Table 2.
Benefits of vitamin D may depend on dosing
Studies show that the risk of hip fracture can be reduced with modest daily vitamin D supplementation, up to 800 U daily, regardless of calcium intake.3 Some vitamin D dosing regimens, however, may also entail risk.
Sanders et al4 randomized women age 70 and older to receive an annual injection of a high dose of vitamin D (500,000 U) or placebo for 3 to 5 years. Women in the vitamin D group had 15% more falls and 25% more fractures than those in the placebo group. The once-yearly dose of 500,000 U equates to 1,370 U/day, which is not much higher than the recommended daily dosage. The median baseline serum level was 49 nmol/L and reached 120 nmol/L at 30 days in the treatment group, which was not in the toxic range.
Comments. This study cautions physicians against giving large doses of vitamin D at long intervals. Future studies should focus on long-term clinical outcomes of falls and fractures for dosing regimens currently in practice, such as 50,000 units weekly or monthly.
BISPHOSPHONATES AND NONTRAUMATIC THICK BONE FRACTURES
Bisphosphonates have been regarded as the best drugs for preventing hip fracture. But in 2010, the US Food and Drug Administration (FDA) issued a warning that bisphosphonates have been associated with “atypical” femoral fractures. The atypical fracture pattern is a clean break through the thick bone of the shaft that occurs after minimal or no trauma.5 This pattern contrasts with the splintering “typical” fracture in the proximal femur in osteoporotic bone, usually after a fall.
Another characteristic of the atypical fractures is a higher incidence of postoperative complications requiring revision surgery. In more than 14,000 women in secondary analyses of three large randomized bisphosphonate trials, 12 fractures in 10 patients were found that were classified as atypical, averaging to an incidence of 2.3 per 10,000 patient-years.6
A population-based, nested case-control study7 using Canadian pharmacy records evaluated more than 200,000 women at least 68 years old who received bisphosphonate therapy. Of these, 716 (0.35%) sustained an atypical femoral fracture and 9,723 (4.7%) had a typical osteoporotic femoral fracture. Comparing the duration of bisphosphonate use between the two groups, the authors found that the risk of an atypical fracture increased with years of usage (at 5 years or more, the adjusted odds ratio was 2.74, 95% CI 1.25–6.02), but the risk of a typical fracture decreased (at 5 years or more, the adjusted odds ratio was 0.76, 95% CI 0.63–0.93). The study suggests that for every 100 hip fractures that bisphosphonate therapy prevents, it causes one atypical hip fracture.
Comments. These studies have caused some experts to advocate periodic bisphosphonate “vacations,”8 but for how long remains an open question because the risk of a typical fracture will increase. It is possible that a biomarker can help establish the best course, but that has yet to be determined.
DENOSUMAB: A NEW DRUG FOR OSTEOPOROSIS WITH A BIG PRICE TAG
Denosumab (Prolia, Xgeva), a newly available injectable drug, is a monoclonal antibody member of the tumor necrosis factor super-family.9 It is FDA-approved for osteoporosis in postmenopausal women at a dosage of 60 mg every 6 months and for skeletal metastases from solid tumors (120 mg every 4 weeks). It is also being used off-label for skeletal protection in women taking aromatase inhibitors and for men with androgen deficiency.
This drug is expensive, costing $850 per 60-mg dose wholesale, and no data are yet available on its long-term effects.
Since the drug is not cleared via renal mechanisms, there is some hope that it can be used to treat osteoporosis in patients with advanced chronic kidney disease, since bisphosphonates are contraindicated in those with an estimated glomerular filtration rate (GFR) less than 30 to 35 mL/min. However, the major study of denosumab to date, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) study, had no patients with stage 5 chronic kidney disease (GFR < 15 mL/min/1.73 m2 or on dialysis), and too few with stage 4 chronic kidney disease (GFR 15–29) to demonstrate either the safety or efficacy of denosumab in patients with advanced chronic kidney disease.10
HYPERTENSION TREATMENT
A secondary analysis of a recent large hypertension study confirmed the benefits of antihypertensive therapy in very old adults and suggested new targets for systolic and diastolic blood pressures.11,12
The Systolic Hypertension in the Elderly Program (SHEP) trial,13 the Systolic Hypertension in Europe (Syst-Eur) trial,14 and the Hypertension in the Very Elderly Trial (HYVET)15 are the major, randomized, placebo-controlled antihypertensive trials in older adults. They all showed a reduction in the risk of stroke and cardiovascular events. The diuretic studies (SHEP and HYVET)13,15 also showed a lower risk of heart failure and death.
Most recently, secondary analysis of the International Verapamil-Trandolapril (INVEST) study11,12 showed that adults in the oldest groups (age 70–79 and 80 and older), experienced a greater risk of adverse cardiovascular outcomes if systolic blood pressure was lowered to below about 130 mm Hg. As diastolic blood pressure was lowered to about the 65–70 mm Hg range, all age groups in the study experienced an increased risk of cardiovascular events. These results confirm the findings of a secondary analysis of the SHEP trial,16 showing an increased risk of cardiovascular events when diastolic pressure was lowered to below approximately 65 mm Hg.
These studies have been incorporated into 75 pages of the 2011 Expert Consensus Document on Hypertension in the Elderly issued by the American College of Cardiology Foundation and the American Heart Association.17 In a nutshell, the guidelines suggest that older adults less than 80 years of age be treated comparably to middle-aged adults. However, for adults age 80 and older:
- A target for systolic blood pressure of 140 to 145 mm Hg “can be acceptable.”
- Initiating treatment with monotherapy (with a low-dose thiazide, calcium channel blocker, or renin-angiotensin-aldosterone system drug) is reasonable. A second drug may be added if needed.
- Patients should be monitored for “excessive” orthostasis.
- Systolic blood pressure lower than 130 mm Hg and diastolic blood pressure lower than 65 mm Hg should be avoided.
TRANSCATHETER AORTIC VALVE IMPLANTATION APPROVED BY THE FDA
An estimated 2% to 9% of the elderly have aortic stenosis. Aortic valve replacement reduces mortality rates and improves function in all age groups, including octogenarians. Those with asymptomatic aortic stenosis tend to decline very quickly once they develop heart failure, syncope, or angina. Aortic valve replacement has been shown to put people back on the course they were on before they became symptomatic.
Transcatheter self-expanding transaortic valve implantation was approved by the FDA in November 2011. The procedure does not require open surgery and involves angioplasty of the old valve, with the new valve being passed into place through a catheter and expanded. Access is either transfemoral or transapical.
Transaortic valve implantation has been rapidly adopted in Europe since 2002 without any randomized control trials. The Placement of Aortic Transcatheter Valves (PARTNER) trial18 in 2011 was the first randomized trial of this therapy. It was conducted at 25 centers, with nearly 700 patients with severe aortic stenosis randomized to undergo either transcatheter aortic valve replacement with a balloon-expandable valve (244 via the transfemoral and 104 via the transapical approach) or surgical replacement. The mean age of the patients was 84 years, and the Society of Thoracic Surgeons mean score was 12%, indicating high perioperative risk.
At 30 days after the procedure, the rates of death were 3.4% with transcatheter implantation and 6.5% with surgical replacement (P = .07). At 1 year, the rates were 24.2% and 26.8%, respectively (P = 0.44, and P = .001 for noninferiority). However, the rate of major stroke was higher in the transcatheter implantation group: 3.8% vs 2.1% in the surgical group (P = .20) at 1 month and 5.1% vs 2.4% (P = .07) at 1 year. Vascular complications were significantly more frequent in the transcatheter implantation group, and the new onset of atrial fibrillation and major bleeding were significantly higher in the surgical group.
Patients in the transcatheter implantation group had a significantly shorter length of stay in the intensive care unit and a shorter index hospitalization. At 30 days, the transcatheter group also had a significant improvement in New York Heart Association functional status and a better 6-minute walk performance, although at 1 year, these measures were similar between the two groups and were greatly improved over baseline. Quality of life, measured using the Kansas City Cardiomyopathy Questionnaire, was higher both at 6 months and at 1 year in the transcatheter implantation group compared with those who underwent the open surgical procedure.19
Comments. The higher risk of stroke with the transcatheter implantation procedure remains a concern. More evaluation is also needed with respect to function and cognition in the very elderly, and of efficacy and safety in higher- and lower-risk patients.
DEPRESSION CAN BE EFFECTIVELY TREATED WITH MEDICATION
Many placebo-controlled trials have demonstrated the effectiveness of treating depression with medications in elderly people who are cognitively intact and living in the community. A Cochrane Review20 found that in placebo-controlled trials, the number needed to treat to produce one recovery with tricyclic antidepressants, selective serotonin reuptake inhibitors, and monoamine oxidase inhibitors was less than 10 for each of the drug classes.
Since the newer drugs appear to be safer and to have fewer adverse effects than the older drugs, more older adults have been treated with antidepressants, including patients with comorbidities such as dementia that were exclusion criteria in early studies. For example, the number of older adults treated with antidepressants has increased 25% since 1992; at the same time the number being referred for cognitive-based therapies has been reduced by 43%.21 Similar trends are apparent in elderly people in long-term care. In 1999, about one-third of people in long-term care were diagnosed with depression; in 2007 more than one-half were.22
Treating depression is less effective when dementia is present
Up to half of adults age 85 and older living in the community may have dementia. In long-term care facilities, most residents likely have some cognitive impairment or are diagnosed with dementia. Many of these are also taking antidepressive agents.
A review of studies in the Medline and Cochrane registries found seven trials that treated 330 patients with antidepressants for combined depression and dementia. Efficacy was not confirmed.23
After this study was published, Banerjee et al24 treated 218 patients who had depression and dementia in nine centers in the United Kingdom. Patients received sertraline (Zoloft), mirtazapine (Remeron), or placebo. Reductions in depression scores at 13 weeks and at 39 weeks did not differ between the groups, and adverse events were more frequent in the treatment groups than in the placebo groups.
Comments. The poor performance of antidepressants in patients with dementia may be due to misdiagnosis, such as mistaking apathy for depression.25 It is also possible that better criteria than we have now are needed to diagnose depression in patients with dementia, or that current outcome measures are not sensitive for depression when dementia is present.
It may also be unsafe to treat older adults long-term with antidepressive agents. For example, although selective serotonin reuptake inhibitors, the most commonly prescribed antidepressive agents, are considered safe, their side effects are numerous and include sexual dysfunction, bleeding (due to platelet dysfunction), hyponatremia, early weight loss, tremor (mostly with paroxetine [Paxil]), sedation, apathy (especially with high doses), loose stools (with sertraline), urinary incontinence, falls, bone loss, and QTc prolongation.
Citalopram: Maximum dosage in elderly
In August 2011, an FDA Safety Communication was issued for citalopram (Celexa), stating that the daily dose should not exceed 40 mg in the general population and should not exceed 20 mg in patients age 60 and older. The dose should also not exceed 20 mg for a patient at any age who has hepatic impairment, who is known to be a poor metabolizer of CYP 2C19, or who takes cimetidine (Tagamet), since that drug inhibits the metabolism of citalopram at the CYP 2C19 enzyme site.
Although the FDA warning specifically mentions only cimetidine, physicians may have concerns about other drugs that inhibit CYP 2C19, such as proton pump inhibitors (eg, omeprazole [Prilosec]) when taken concomitantly with citalopram. Also, escitalopram (Lexapro) and sertraline are quite similar to citalopram; although they were not mentioned in the FDA Safety Communication, higher doses of these drugs may put patients at similar risk.
ALZHEIMER DISEASE: NEED TO BETTER IDENTIFY PEOPLE AT RISK
The definition of dementia is essentially the presence of a cognitive problem that affects the ability to function. For people with Alzheimer disease, impairment of cognitive performance precedes functional decline. Those with a cognitive deficit who still function well have, by definition, mild cognitive impairment (MCI). Although MCI could be caused by a variety of vascular and other neurologic processes, the most common cause of MCI in the United States is Alzheimer disease.
Unfortunately, the population with MCI currently enrolled in clinical trials to reduce the risk of progression to Alzheimer disease is heterogeneous. Many study participants may never get dementia, and others may have had the pathology present for decades and are progressing rapidly. Imaging and biomarkers are emerging as good indicators that predict progression and could help to better define populations for clinical trials.26
Studies now indicate that people with MCI that is ultimately due to Alzheimer disease are likely to have amyloid beta peptide 42 evident in the cerebrospinal fluid 10 to 20 years before symptoms arise. At the same time, amyloid is also likely to be evident in the brain with amyloid-imaging positron emission tomography (PET). Some time later, abnormalities in metabolism are also evident on fluorodeoxyglucose (FDG) PET, as are changes such as reduced hippocampal volume on magnetic resonance imaging (MRI).
The 1984 criteria for diagnosing MCI due to Alzheimer disease were recently revised to incorporate the evolving availability of biomarkers.27,28 The diagnosis of MCI itself is still based on clinical ascertainment including history, physical examination, and cognitive testing. It requires diagnosis of a cognitive decline from a prior level but maintenance of activities of daily living with no or minimal assistance. This diagnosis is certainly challenging since it requires ascertainment of a prior level of function and corroboration, when feasible, with an informant. Blood tests and imaging, which are readily available, constitute an important part of the assessment.
Attributing the MCI to Alzheimer disease requires consistency of the disease course—a gradual decline in Alzheimer disease, rather than a stroke, head injury, neurologic disease such as Parkinson disease, or mixed causes.
Knowledge of genetic factors, such as the presence of a mutation in APP, PS1, or PS2, can be predictive with young patients. The presence of one or two 34 alleles in the apolipoprotein E (APOE) gene is the only genetic variant broadly accepted as increasing the risk for late-onset Alzheimer dementia, whereas the 32 allele decreases risk.
Refining the risk attribution to Alzheimer disease requires biomarkers, currently available only in research settings:
- High likelihood—amyloid beta peptide detected by PET or cerebrospinal fluid analysis and evidence of neuronal degeneration or injury (elevated tau in the cerebrospinal fluid, decreased FDG uptake on PET, and atrophy evident by structural MRI)
- Intermediate likelihood—presence of amyloid beta peptide or evidence of neuronal degeneration or injury
- Unlikely—biomarkers tested and negative
- No comment—biomarkers not tested or reporting is indeterminate.
Comments. There is significant potential for misunderstanding the new definition for MCI. Patients who are concerned about their memory may request biomarker testing in an effort to determine if they currently have or will acquire Alzheimer disease. Doctors may be tempted to refer patients for biomarker testing (via imaging or lumbar puncture) to “screen” for MCI or Alzheimer disease.
It should be emphasized that MCI itself is still a clinical diagnosis, with the challenges noted above of determining whether there has been a cognitive decline from a prior level of function but preservation of activities of daily living. The biomarkers are not proposed to diagnose MCI, but only to help identify the subset of MCI patients most likely to progress rapidly to Alzheimer disease.
At present, the best use of biomarker testing is to aid research by identifying high-risk people among those with MCI who enroll in prospective trials for testing interventions to reduce the progression of Alzheimer disease.
- Panel on Prevention of Falls in Older Persons, American Geriatrics Society and British Geriatrics Society. J Am Geriatr Soc 2011; 59:148–157.
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: metaanalysis. BMJ 2010; 341:c3691.
- Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009; 169:551–561.
- Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women. A randomized controlled trial. JAMA 2010; 303:1815–1822.
- Kuehn BM. Prolonged bisphosphonate use linked to rare fractures, esophageal cancer. JAMA 2010; 304:2114–2115.
- Black DM, Kelly MP, Genant HK, et al; Fracture Intervention Trial Steering Committee; HORIZON Pivotal Fracture Trial Steering Committee. Bisphosphonates and fractures of the subtrochanteric or diaphyseal femur. N Engl J Med 2010; 362:1761–1771.
- Park-Wyllie LY, Mamdani MM, Juurlink DN, et al. Bisphosphonate use and the risk of subtrochanteric or femoral shaft fractures in older women. JAMA 2011; 305:783–789.
- Ott SM. What is the optimal duration of bisphosphonate therapy? Cleve Clin J Med 2011; 78:619–630.
- Cummings SR, San Martin J, McClung MR, et al; for the FREEDOM trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Pepine CJ, Handberg EM, Cooper-Dehoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Denardo SJ, Gong Y, Nichols WW, et al. Blood pressure and outcomes in very old hypertensive coronary artery disease patients: an INVEST substudy. Am J Med 2010; 123:719–726.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al; for the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension (erratum published in Lancet 1997; 350:1636). Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; for the HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Somes G, Pahor M, Shorr R, Cushman WC, Applegate WB. The role of diastolic blood pressure when treating isolated systolic hypertension. Arch Intern Med 1999; 159:2004–2009.
- Aronow WS, Fleg JL, Pepine CJ, et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly. J Am Coll Cardiol 2011; 57:2037–2114.
- Smith CR, Leon MB, Mack MJ, et al; PARTNER Trial Investigators. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med 2011; 364:2187–2198.
- Reynolds MR, Magnuson EA, Lei Y, et al; Placement of Aortic Transcatheter Valves (PARTNER) Investigators. Health-related quality of life after transcatheter aortic valve replacement in inoperable patients with severe aortic stenosis. Circulation 2011; 124:1964–1972.
- Wilson K, Mottram P, Sivanranthan A, Nightingale A. Antidepressant versus placebo for depressed elderly. Cochrane Database Syst Rev 2001;(2):CD000561.
- Akincigil A, Olfson M, Walkup JT, et al. Diagnosis and treatment of depression in older community-dwelling adults: 1992–2005. J Am Geriatr Soc 2011; 59:1042–1051.
- Gaboda D, Lucas J, Siegel M, Kalay E, Crystal S. No longer undertreated? Depression diagnosis and antidepressant therapy in elderly long-stay nursing home residents, 1999 to 2007. J Am Geriatr Soc 2011; 59:673–680.
- Nelson JC, Devanand DP. A systematic review and meta-analysis of placebo-controlled antidepressant studies in peoloe with depression and dementia. J Am Geriatr Soc 2011; 59:577–585.
- Banerjee S, Hellier J, Dewey M, et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial. Lancet 2011; 378:403–411.
- Landes AM, Sperry SD, Strauss ME, Geldmacher DS. Apathy in Alzheimer’s disease. J Am Geriatr Soc 2001; 49:1700–1707.
- Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010; 9:1118–1127.
- Daviglus ML, Bell CC, Berrettini W, et al. National Institutes of Health State-of-the-Science Conference statement: preventing Alzheimer disease and cognitive decline. Ann Intern Med 2010; 153:176–181.
- McKhann GM, Knopman DS, Chertkow H, et al The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263–269.
- Panel on Prevention of Falls in Older Persons, American Geriatrics Society and British Geriatrics Society. J Am Geriatr Soc 2011; 59:148–157.
- Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: metaanalysis. BMJ 2010; 341:c3691.
- Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009; 169:551–561.
- Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women. A randomized controlled trial. JAMA 2010; 303:1815–1822.
- Kuehn BM. Prolonged bisphosphonate use linked to rare fractures, esophageal cancer. JAMA 2010; 304:2114–2115.
- Black DM, Kelly MP, Genant HK, et al; Fracture Intervention Trial Steering Committee; HORIZON Pivotal Fracture Trial Steering Committee. Bisphosphonates and fractures of the subtrochanteric or diaphyseal femur. N Engl J Med 2010; 362:1761–1771.
- Park-Wyllie LY, Mamdani MM, Juurlink DN, et al. Bisphosphonate use and the risk of subtrochanteric or femoral shaft fractures in older women. JAMA 2011; 305:783–789.
- Ott SM. What is the optimal duration of bisphosphonate therapy? Cleve Clin J Med 2011; 78:619–630.
- Cummings SR, San Martin J, McClung MR, et al; for the FREEDOM trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Pepine CJ, Handberg EM, Cooper-Dehoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Denardo SJ, Gong Y, Nichols WW, et al. Blood pressure and outcomes in very old hypertensive coronary artery disease patients: an INVEST substudy. Am J Med 2010; 123:719–726.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al; for the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension (erratum published in Lancet 1997; 350:1636). Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; for the HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- Somes G, Pahor M, Shorr R, Cushman WC, Applegate WB. The role of diastolic blood pressure when treating isolated systolic hypertension. Arch Intern Med 1999; 159:2004–2009.
- Aronow WS, Fleg JL, Pepine CJ, et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly. J Am Coll Cardiol 2011; 57:2037–2114.
- Smith CR, Leon MB, Mack MJ, et al; PARTNER Trial Investigators. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N Engl J Med 2011; 364:2187–2198.
- Reynolds MR, Magnuson EA, Lei Y, et al; Placement of Aortic Transcatheter Valves (PARTNER) Investigators. Health-related quality of life after transcatheter aortic valve replacement in inoperable patients with severe aortic stenosis. Circulation 2011; 124:1964–1972.
- Wilson K, Mottram P, Sivanranthan A, Nightingale A. Antidepressant versus placebo for depressed elderly. Cochrane Database Syst Rev 2001;(2):CD000561.
- Akincigil A, Olfson M, Walkup JT, et al. Diagnosis and treatment of depression in older community-dwelling adults: 1992–2005. J Am Geriatr Soc 2011; 59:1042–1051.
- Gaboda D, Lucas J, Siegel M, Kalay E, Crystal S. No longer undertreated? Depression diagnosis and antidepressant therapy in elderly long-stay nursing home residents, 1999 to 2007. J Am Geriatr Soc 2011; 59:673–680.
- Nelson JC, Devanand DP. A systematic review and meta-analysis of placebo-controlled antidepressant studies in peoloe with depression and dementia. J Am Geriatr Soc 2011; 59:577–585.
- Banerjee S, Hellier J, Dewey M, et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial. Lancet 2011; 378:403–411.
- Landes AM, Sperry SD, Strauss ME, Geldmacher DS. Apathy in Alzheimer’s disease. J Am Geriatr Soc 2001; 49:1700–1707.
- Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010; 9:1118–1127.
- Daviglus ML, Bell CC, Berrettini W, et al. National Institutes of Health State-of-the-Science Conference statement: preventing Alzheimer disease and cognitive decline. Ann Intern Med 2010; 153:176–181.
- McKhann GM, Knopman DS, Chertkow H, et al The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:263–269.
KEY POINTS
- To prevent falls, patients should be asked not only about recent falls but about balance. Referral for a multicomponent falls evaluation should be considered.
- For patients age 80 and older, a target systolic blood pressure of 140 to 145 mm Hg is acceptable, and blood pressure below 130 mm Hg systolic and 65 mm Hg diastolic should be avoided.
- The dosage of the antidepressant citalopram (Celexa) should not exceed 40 mg per day in the general population and 20 mg in patients age 60 and older.
- Calcium supplementation may increase the risk of myocardial infarction and stroke. A large annual dose of vitamin D appears harmful, raising questions about the long-term safety of large doses given weekly or monthly.
The rationale for, and design of, a lung cancer screening program
In 2011, two papers were published that will shape the way we think about lung cancer screening for years to come.
See related patient information sheet
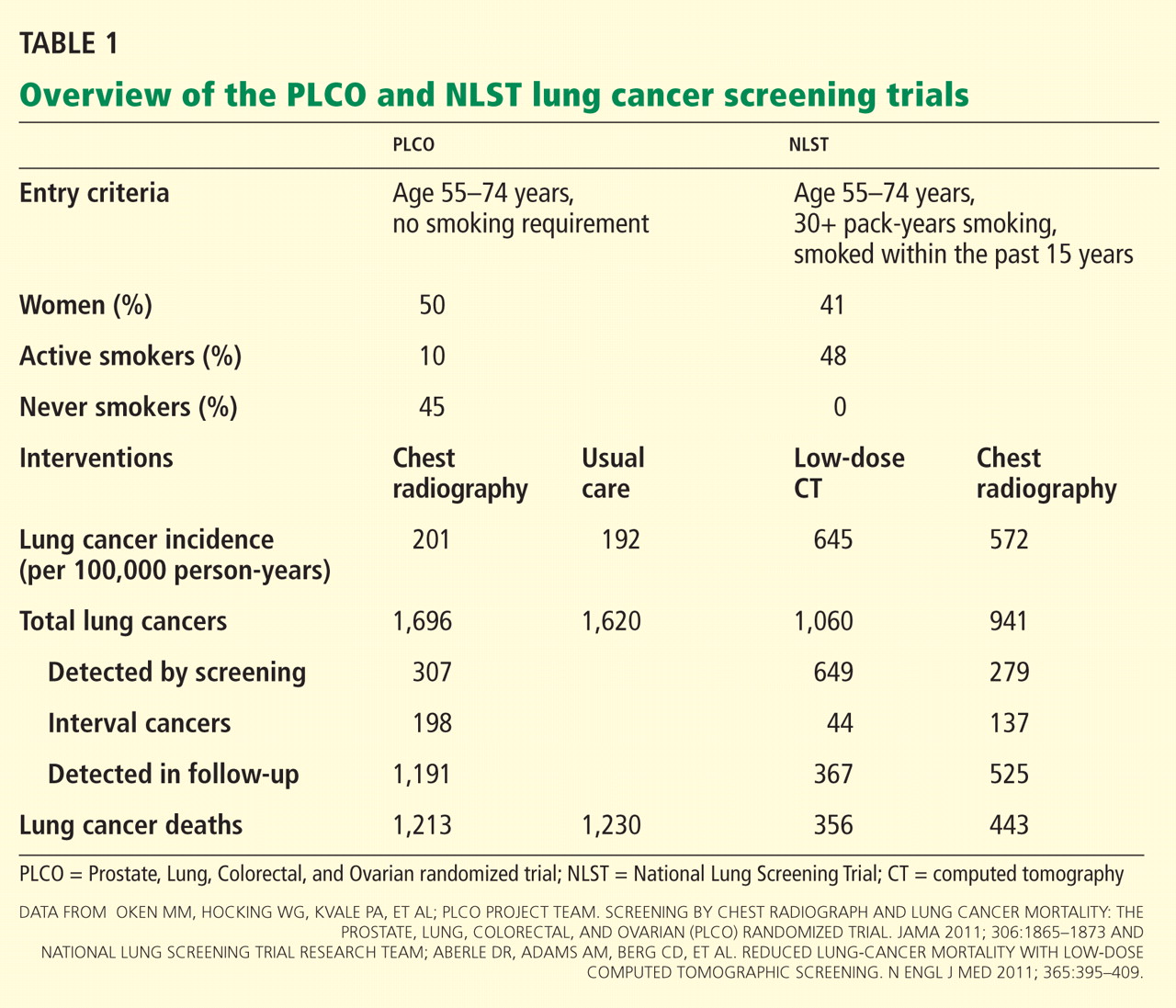
While the ability to screen for lung cancer is a major positive change, it also raises many thorny questions, such as who should be screened, how often should they be screened, and how should we respond when a nodule is detected.
To answer some of these questions, we will outline how Cleveland Clinic has structured its lung cancer screening program, and the rationale we used for making pragmatic patient-care decisions within this program. We will conclude with our thoughts about the potential evolution of lung cancer screening programs.
THE 40-YEAR QUEST FOR EFFECTIVE LUNG CANCER SCREENING
Lung cancer kills more people in the United States than the next four most lethal types of cancer combined.3 It is curable if found early in its course. Unfortunately, most people who develop lung cancer feel no symptoms when it is early in its course, and therefore it is too often diagnosed at a late stage. Treatment for late-stage lung cancer is effective, but it is rarely curative.
Screening refers to testing people at risk of developing a disease before its symptoms or signs have appeared. The goal of screening is to reduce the disease-specific mortality rate. For this to happen, the disease must be detectable in a preclinical form, and treatment must be more successful when applied early. Ideally, the screening test should pose little risk to the patient, be sensitive for detecting the disease early in its course, give few false-positive results, be acceptable to the patient, and be relatively inexpensive to the health system.
Over the past 4 decades, a large volume of research has been done in the hope of proving that conventional radiography or CT could be an effective screening test for lung cancer.4,5
Cohort studies (ie, in which all the patients were screened) of radiography or CT have shown a longer survival from the time of lung cancer diagnosis than would be expected without screening. These studies were not designed to prove a reduction in the lung cancer-specific mortality rate.
Controlled trials (in which half the patients received the screening and the other half did not) of chest radiography have been interpreted as not showing a reduction in lung cancer mortality rates, though debate about the interpretation of these trials persisted until this past year. Biases inherent in using duration of survival rather than the mortality rate as an end point have been suggested as the reason for the apparent benefit in survival without a reduction in the mortality rate.
Controlled trials of CT screening were started nearly a decade ago. Until 2011, the results of these trials were not mature enough to comment on.
THE PROSTATE, LUNG, COLORECTAL, AND OVARIAN TRIAL
The lung cancer screening portion of the PLCO trial aimed to determine the effect of screening chest radiography on lung cancer-specific mortality rates.1
In this trial, 154,901 people were randomized to undergo either posteroanterior chest radiography every year for 4 years or usual care, ie, no lung cancer screening. Participants were men and women age 55 to 74 with no history of prostate, lung, colorectal, or ovarian cancer. They did not need to be a smoker to participate. Those who had never smoked and who were randomized to the screening group received only 3 years of testing. All were followed for 13 years or until the conclusion of the study (8 years after the final participant was enrolled). About half were women, and nearly two-thirds were age 55 through 64. Only 10% were current smokers, while a full 45% had never smoked.
Results. Adherence to screening in the screening group ranged from 79% to 86.6% over the years of screening, and 11% of the usual-care group was estimated to have undergone screening chest radiography.
Cumulative lung cancer incidence rates were 201 per 100,000 person-years in the screening group and 192 in the usual-care group.
In the screening group, there were a total of 1,696 lung cancers during the entire study. Of these, 307 (18%) were detected by screening, 198 (12%) were interval cancers (diagnosed during the screening period but not by the screening test), and the remainder were diagnosed after the screening period during the years of follow-up. In the screening group, the cancers detected by screening were more likely to be adenocarcinomas and less likely to be small-cell carcinomas than those not detected by screening. Also in the screening group, the cancers detected by screening were more likely to be stage I (50%) than those not detected by screening.
The cumulative number of deaths from lung cancer was slightly but not significantly lower in the screening group from years 4 through 11. However, by the end of follow-up, the number of lung cancer deaths was equal between the groups (1,213 in the screening group vs 1,230 in the usual-care group). The cumulative overall mortality rate was also similar between the groups. For the subgroup who would have qualified for the NLST (see below), the lung cancer mortality rate was statistically similar between the two groups.
Comments. The results of the PLCO screening trial will be interpreted as the final word in lung cancer screening with standard chest radiography. The conclusion is that annual screening with chest radiography does not reduce lung cancer mortality rates and thus should not be performed in this context.
THE NATIONAL LUNG SCREENING TRIAL
The NLST aimed to determine if screening with low-dose chest CT could reduce lung cancer mortality rates.2
This controlled trial enrolled 53,454 people, who were randomized to undergo either low-dose chest CT or posteroanterior chest radiography at baseline and then yearly for 2 years.
Participants were men and women age 55 to 74 with at least 30 pack-years of cigarette smoking. If they had quit smoking, they had to have quit within the past 15 years. All were followed until study conclusion (median 6.5 years, maximum 7.4). About 41% were women, and nearly three-quarters were age 55 through 64. More than 48% were current smokers, with the rest being former smokers.
Results. Adherence to screening was 95% in the CT group and 93% in the radiography group, with a 4.3% annual rate of CT outside the study during the screening phase.
Cumulative lung cancer incidence rates were 645 per 100,000 person-years in the CT group and 572 in the radiography group.
In the CT group there were a total of 1,060 lung cancers during the entire study. Of these, 649 (61%) were detected by screening, 44 (4%) were interval cancers, and the rest were diagnosed after the screening period during follow-up.
In the chest radiography group, there were a total of 941 lung cancers during the entire study. Of these, 279 (30%) were detected by screening, 137 (15%) were interval cancers, and the rest were diagnosed after the screening period. Within the CT group, the cancers detected by screening were more likely to be adenocarcinomas and less likely to be small-cell carcinomas than those not detected by screening. Also within the CT group, the cancers detected by screening were more likely to be stage I (63%) than those not detected by screening.
The cumulative number of deaths from lung cancer was 443 in the radiography group, but only 356 in the CT group—20.0% lower (P =.004). The cumulative overall mortality rate was 6.7% lower in the CT group (P = .02).
Comments. The results of the NLST provide the first evidence that lung cancer mortality rates can be reduced by screening. Though many questions remain, the conclusions of this study are that screening a well-defined high-risk group with low-dose CT reduces the rate of death from lung cancer.
REMAINING CHALLENGES
The NLST showed that lung cancer screening with low-dose CT can meet the most important criterion for a successful screening program, ie, a reduction in the disease-specific mortality rate. Many challenges remain in meeting the other criteria for a successful or ideal screening program (low risk, few false-positive results, acceptability to the patient, and affordability). The issues with low-dose CT-based screening that challenge these ideals are outlined in this section.
Lung nodules: Benign or malignant?
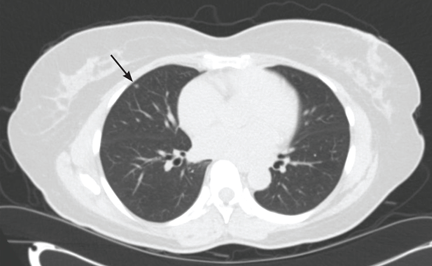
A meta-analysis of CT screening studies found that for every 1,000 people screened at baseline, 9 were found to have stage I non-small-cell lung cancer, 235 had false-positive nodules, and 4 underwent thoracotomy for benign lesions.6
The NLST results were similar. In this trial, only nodules that were 4 mm or greater in diameter were reported. Using these criteria, over 27% of all study participants were found to have a lung nodule on CT at baseline and at year 1. The rate fell to nearly 17% at year 2, as nodules present from baseline were not reported. Of all the lung nodules detected, only 3.6% were ultimately proven to represent lung cancer.2
Many issues with small lung nodules need to be considered. The nodules are difficult to find, with highly variable reporting even by expert radiologists.7 They are difficult to measure accurately and thus are difficult to assess for growth.8 Adjunctive imaging and nonsurgical biopsy have a low yield for small nodules.9–11 Follow-up of these lung nodules includes additional imaging and nonsurgical and surgical biopsy procedures, adding expense to the program and risk to the patient. Finally, knowing that they have a lung nodule makes patients feel anxious and thus negatively affects their quality of life.12,13
Radiation exposure: How great is the risk?
There is a great deal of concern about radiation exposure from medical imaging, as many people receive a substantial amount of radiation each year from medical testing.14 A single low-dose scan with chest CT delivers a whole-body effective dose of about 1.5 mSv—less than one-fifth of the radiation dose of a typical diagnostic CT scan.
Many have tried to estimate the consequences of radiation exposure from low-dose CT screening. All estimates are extrapolations from unrelated radiation exposures. The increase in risk of death ranged from 0.01% to a few percent,15 and the increase in cancers was as high as 1.8% over a 25-year screening period.16 In general, the risks are felt to be very low but not negligible.
Cost-effectiveness is unknown
The cost-effectiveness of lung cancer screening is also unknown. Many highly variable estimates have been published.17–20 The studies have differed in the perspective taken, the costs of testing assumed, and the rounds of screening included. The most cost-effective estimates are in populations with the highest risk of cancer, in programs that achieve the greatest reduction in mortality rate, and in programs that lead to high rates of smoking cessation.
Screening in the real world as opposed to a clinical trial may involve different risks, benefits, and costs. Compliance with screening and with nodule management algorithms may be lower outside of a study. One study suggested that those at highest risk of developing lung cancer would be the least likely to enroll in a screening program and the least likely to accept curative-intent surgery for screening-detected cancer.21
We expect that the NLST data will be analyzed for cost-effectiveness. This should provide the most accurate estimates for the group that was studied.
WE SET OUT TO DESIGN A SCREENING PROGRAM
With the evidence supporting a reduction in the rate of lung cancer mortality, and knowing the remaining challenges, we set out to provide a lung cancer screening program within Cleveland Clinic. In the design of our program, we considered several questions, outlined below.
Who should be offered low-dose CT screening?
The results of the NLST led to a great deal of excitement about lung cancer screening in both the medical community and the general public. The positive side of this publicity is that lung cancer is receiving attention that may lead to support for further advances. The negative side is that many patients who may seek out lung cancer screening are not at high enough risk of lung cancer to clearly benefit from it.
In the NLST, a very high-risk cohort was studied, as defined by clinical variables (age 55 to 74, at least 30 pack-years of smoking, and if a former smoker, had quit within the past 15 years). In this high-risk group, 320 patients needed to be screened (with three yearly chest CT scans) for one life to be saved from lung cancer, and only 3.6% of all lung nodules found (4 mm or larger) were actually lung cancer. In a group at lower risk, the number that needed to be screened to save one life would be higher, and the percentage of lung nodules that truly were lung cancer would be lower. This would lead to higher risks and costs related to screening, without a proven benefit to members of the lower-risk group.
The risk of the NLST cohort developing lung cancer was approximately 0.6% per year. Lung cancer risk-prediction models have been developed and published. Up to 2011, the three most commonly used models had only moderate accuracy at predicting risk.22–25 In 2011 a risk model based on the PLCO cohort was developed and published.26 This model seemed to be more accurate but perhaps a bit harder to apply in practice.
We discussed whether using a validated risk predictor with a target of 0.6% per year (ie, the risk in the NLST trial) would be an adequate means of deciding on candidacy for lung cancer screening or if we should strictly adhere to the inclusion criteria of the NLST cohort. We feel that the NLST cohort is the only group with true evidence of benefit (a reduction in the lung cancer-specific mortality rate). Thus, for our program’s entry criteria, we decided to use the same clinical predictors used for entry in the NLST.
How will the right patients get scheduled for low-dose screening CT?
Patients who enter the lung cancer screening program from our health system will require a physician’s order.
We are fortunate to have an electronic medical record in place. We have created an order set within the electronic record for low-dose chest CT. The order will eventually be able to be entered as “CT lung screening w/o” (ie, without contrast).
For patients from outside of our health system who would like to enter the lung cancer screening program, the entry criteria will be the same (see above). We will ask for the name of the patient’s primary care practitioner. If the patient does not have one, a member of our Respiratory Institute will see and enroll the patient.
How often should patients be screened, and for how many years?
Unfortunately, questions about the frequency of screening and how many years it should continue remain unanswered.
In the NLST, a similar number of early-stage lung cancers were detected during each of the three screening rounds. In both the NLST and PLCO trials, differences in the mortality rate curves began to narrow during the observation period, when active screening was no longer occurring. Thus, it is possible that a longer duration of screening could lead to a further reduction in mortality rates. Others have questioned whether a similar benefit, with less cost and risk, could be obtained by screening every 2 years.
The large amount of data obtained from the NLST and other CT-based studies is being reviewed so that models can be developed to help answer these questions. For now, we suggest at least three yearly CT screenings, with the hope that we will have clearer answers to these questions over time.
How will low-dose CT be performed and interpreted?
The parameters for low-dose CT were very tightly controlled and monitored during the NLST. This quality-control effort, designed to improve consistency across sites and to minimize risk to patients, should be carried into lung cancer screening programs.
Our program will closely mimic the CT performance criteria used in the NLST (tube current-time product 40 mAs for all patients, field of view lungs only, lung kernel images 3 mm at 1.5-mm intervals, and soft-tissue kernel images 5 mm at 2.5-mm intervals).27 In the initial phase of the program, all screening scans will be performed at Cleveland Clinic’s main imaging facility.
Small lung nodules remain quite challenging to detect and measure. To minimize variability in scan interpretation, the NLST readers were all expertly trained radiologists. Despite this, much variability was noted in the number of nodules detected, their measured size, and the follow-up recommendations. All of the screening CT images for our program will be interpreted by board-certified radiologists with expertise in chest imaging.
Other screening studies have included novel imaging assessment in their testing algorithms, particularly volumetric analysis of lung nodules.28 These tools may prove to assist in nodule detection, measurement, and management over time. At this point, we do not think they have been studied and standardized enough to include them in a standard-of-care screening program. We hope that they will evolve to the point of clinical utility in the near future.
Lung cancer screening is not currently covered by most insurers, including Medicare, although one major insurer has recently started to cover it. We expect decisions on coverage from other insurers in the next 12 months. In the meantime, we offer a low-dose screening chest CT to our patients for $125, which includes the radiologist’s fee for interpreting the scan.
Smoking cessation
The NLST showed that low-dose CT screening can reduce lung cancer mortality rates by 20% in a high-risk group. A 50-year-old active smoker who quits smoking reduces his or her risk of dying of lung cancer by more than 50%.29 Entry into a lung cancer screening program provides an opportunity for education and assistance with tobacco dependency.
At Cleveland Clinic, we have an active Tobacco Treatment Center within our Wellness Institute. All lung cancer screening participants who are identified as active smokers will be given a program brochure and will be offered a consult in the program.
What do we identify as a lung nodule, and how should they be managed?
Studies of CT-based screening have highlighted the tremendous number of lung nodules that are identified and the low likelihood of malignancy in those that are less than 1 cm in diameter. Many screening studies define a positive result as a lung nodule above a particular size. The NLST used 4 mm or greater as the cutoff. The lower the cutoff, the greater the number of nodules found, and the lower the overall likelihood of malignancy in the nodules.
Studies in which annual CT screening was the intervention are able to use size criteria in part because the study design ensures another CT will be performed 12 months later. Current nodule management guidelines suggest 12-month CT follow-up of incidentally discovered lung nodules, 4 mm or smaller, in at-risk patients.30 In a screening program, particularly one for which the patient must pay, the 12-month screening CT cannot be guaranteed. This makes it more difficult to ignore the smallest nodules identified on CT screening. Given this, we will be reporting all lung nodules identified, regardless of size on the initial screening.
Most studies of CT screening have reported any new nodule identified in subsequent screening rounds regardless of size. Though it is intuitive that a new nodule would have a high likelihood of malignancy in a high-risk cohort, malignancy rates have been reported to be as low as 1% for new nodules. As with the initial round of screening, we will report all new lung nodules identified in subsequent screening rounds.

The recommendations for the evaluation of lung nodules, both within the report and at the lung nodule clinic, are in keeping with currently available guidelines, such as those from the Fleischner Society30 and the American College of Chest Physicians.31 For incidentally discovered lung nodules in patients at high risk, the Fleischner Society recommendations are as follows30:
- For nodules 4 mm or smaller, follow-up in 12 months; if no growth, then no further follow-up
- For nodules 4 to 6 mm, follow-up at 6 to 12 months, then 18 to 24 months if no growth
- For nodules 6 to 8 mm, follow-up at 3 to 6 months, then 9 to 12 months, then 24 months if no growth
- For nodules 8 mm or larger, follow-up at 3, 9, and 24 months, or positron emission tomography, or biopsy, or both.
If the nodule is large enough or is deemed to be of high enough risk, adjuvant testing with diagnostic imaging, guided bronchoscopy, transthoracic needle aspiration, or minimally invasive resection will be offered. All patients with nodules believed to require biopsy will be discussed at our multidisciplinary lung cancer tumor board before biopsy.
How do we make practitioners and patients aware of the program and its indications, risks, and benefits?
Education will be the key to having lung cancer screening adopted as the standard of care, to lung cancer screening being provided within a well-designed and capable system, and to ensuring that patients have realistic expectations about screening. Articles such as this and grand rounds presentations within our health system will help provide education to our colleagues. Broader marketing campaigns will be considered in the future once demand and system capabilities are clearly identified. A patient information brochure will be provided at the time of the screening test (see the patient information sheet that accompanies this article).
How do we help to advance best practice?
As excited as we are that low-dose CT-based lung cancer screening has been proven to reduce lung cancer mortality rates, it is clear that there is a lot of room to improve the programs that are developed based on current data.
Advances in our ability to accurately predict an individual’s risk of developing lung cancer will allow us to offer screening to those it is most likely to benefit.
Advances in smoking cessation and chemoprevention will help to minimize the number of lung cancers that develop.
Advances in our ability to determine the nature of lung nodules will allow us to accelerate treatment of very early lung cancer while minimizing additional testing on benign nodules; advances in our ability to treat localized and advanced disease will improve the outcome for those identified as having lung cancer.
To help move the science of screening forward, we will develop a screening program registry that can be populated from the order set and the templated report. The registry can be used to ensure appropriate patient care, while studying relevant epidemiologic, quality, and cost-related questions.
We hope to assess novel imaging software capable of assisting with the detection and characterization of lung nodules.
We have an active biomarker development program to assess the ability of breath and blood-based biomarkers to identify those at risk of developing lung cancer; to assist with the management of screening-detected lung nodules; to assist with the diagnosis of early stage lung cancer; and to characterize the nature of the cancers identified. Accurate biomarkers could lead to further decreases in mortality rates while reducing the risks and costs of a screening program.
We have strong surgical, medical, and radiation oncology programs, actively pursuing advances in minimally invasive resection procedures and ablative and targeted therapies.
ENTERING A NEW ERA
We are entering a new era of lung cancer screening. The NLST has shown that lung cancer morality rates can be reduced through low-dose CT screening in a high-risk population. Many challenges remain, such as managing the nodules that are discovered, determining if the program is cost-effective, and minimizing radiation exposure. These need to be considered when designing a lung cancer screening program. Advances over time will help us optimize the programs that are developed.
- Oken MM, Hocking WG, Kvale PA, et al; PLCO Project Team. Screening by chest radiograph and lung cancer mortality: the Prostate, Lung, Colorectal, and Ovarian (PLCO) randomized trial. JAMA 2011; 306:1865–1873.
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365:395–409.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Mazzone PJ, Mekhail T. Lung cancer screening. Curr Oncol Rep 2007; 9:265–274.
- Mazzone PJ. Lung cancer screening: an update, discussion, and look ahead. Curr Oncol Rep 2010; 12:226–234.
- Gopal M, Abdullah SE, Grady JJ, Goodwin JS. Screening for lung cancer with low-dose computed tomography: a systematic review and meta-analysis of the baseline findings of randomized controlled trials. J Thorac Oncol 2010; 5:1233–1239.
- Gierada DS, Pilgram TK, Ford M, et al. Lung cancer: interobserver agreement on interpretation of pulmonary findings at low-dose CT screening. Radiology 2008; 246:265–272.
- Singh S, Pinsky P, Fineberg NS, et al. Evaluation of reader variability in the interpretation of follow-up CT scans at lung cancer screening. Radiology 2011; 259:263–270.
- Lindell RM, Hartman TE, Swensen SJ, et al. Lung cancer screening experience: a retrospective review of PET in 22 non-small cell lung carcinomas detected on screening chest CT in a high-risk population. AJR Am J Roentgenol 2005; 185:126–131.
- Baaklini WA, Reinoso MA, Gorin AB, Sharafkaneh A, Manian P. Diagnostic yield of fiberoptic bronchoscopy in evaluating solitary pulmonary nodules. Chest 2000; 117:1049–1054.
- Kothary N, Lock L, Sze DY, Hofmann LV. Computed tomography-guided percutaneous needle biopsy of pulmonary nodules: impact of nodule size on diagnostic accuracy. Clin Lung Cancer 2009; 10:360–363.
- van den Bergh KA, Essink-Bot ML, Borsboom GJ, et al. Short-term health-related quality of life consequences in a lung cancer CT screening trial (NELSON). Br J Cancer 2010; 102:27–34.
- Lemonnier I, Baumann C, Jolly D, et al. Solitary pulmonary nodules: consequences for patient quality of life. Qual Life Res 2011; 20:101–109.
- Fazel R, Krumholz HM, Wang Y, et al. Exposure to low-dose ionizing radiation from medical imaging procedures. N Engl J Med 2009; 361:849–857.
- Buls N, de Mey J, Covens P, Stadnik T. Health screening with CT: prospective assessment of radiation dose and associated detriment. JBR-BTR 2005; 88:12–16.
- Brenner DJ. Radiation risks potentially associated with low-dose CT screening of adult smokers for lung cancer. Radiology 2004; 231:440–445.
- Mahadevia PJ, Fleisher LA, Frick KD, Eng J, Goodman SN, Powe NR. Lung cancer screening with helical computed tomography in older adult smokers: a decision and cost-effectiveness analysis. JAMA 2003; 289:313–322.
- Wisnivesky JP, Mushlin AI, Sicherman N, Henschke C. The cost-effectiveness of low-dose CT screening for lung cancer: preliminary results of baseline screening. Chest 2003; 124:614–621.
- Manser R, Dalton A, Carter R, Byrnes G, Elwood M, Campbell DA. Cost-effectiveness analysis of screening for lung cancer with low dose spiral CT (computed tomography) in the Australian setting. Lung Cancer 2005; 48:171–185.
- McMahon PM, Kong CY, Bouzan C, et al. Cost-effectiveness of computed tomography screening for lung cancer in the United States. J Thorac Oncol 2011; 6:1841–1848.
- Silvestri GA, Nietert PJ, Zoller J, Carter C, Bradford D. Attitudes towards screening for lung cancer among smokers and their nonsmoking counterparts. Thorax 2007; 62:126–130.
- Bach PB, Kattan MW, Thornquist MD, et al. Variations in lung cancer risk among smokers. J Natl Cancer Inst 2003; 95:470–478.
- Spitz MR, Hong WK, Amos CI, et al. A risk model for prediction of lung cancer. J Natl Cancer Inst 2007; 99:715–726.
- Cassidy A, Myles JP, van Tongeren M, et al. The LLP risk model: an individual risk prediction model for lung cancer. Br J Cancer 2008; 98:270–276.
- D’Amelio AM, Cassidy A, Asomaning K, et al. Comparison of discriminatory power and accuracy of three lung cancer risk models. Br J Cancer 2010; 103:423–429.
- Tammemagi CM, Pinsky PF, Caporaso NE, et al. Lung cancer risk prediction: Prostate, Lung, Colorectal And Ovarian Cancer Screening Trial models and validation. J Natl Cancer Inst 2011; 103:1058–1068.
- National Lung Screening Trial Research Team; Aberle DR, Berg CD, Black WC, et al. The National Lung Screening Trial: overview and study design. Radiology 2011; 258:243–253.
- van Klaveren RJ, Oudkerk M, Prokop M, et al. Management of lung nodules detected by volume CT scanning. N Engl J Med 2009; 361:2221–2229.
- Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ 2000; 321:323–329.
- MacMahon H, Austin JH, Gamsu G, et al; Fleischner Society. Guidelines for management of small pulmonary nodules detected on CT scans: a statement from the Fleischner Society. Radiology 2005; 237:395–400.
- Gould MK, Fletcher J, Iannettoni MD, et al; American College of Chest Physicians. Evaluation of patients with pulmonary nodules: when is it lung cancer?: ACCP evidence-based clinical practice guidelines (2nd edition). Chest 2007; 132(suppl 3):108S–130S.
In 2011, two papers were published that will shape the way we think about lung cancer screening for years to come.
See related patient information sheet
While the ability to screen for lung cancer is a major positive change, it also raises many thorny questions, such as who should be screened, how often should they be screened, and how should we respond when a nodule is detected.
To answer some of these questions, we will outline how Cleveland Clinic has structured its lung cancer screening program, and the rationale we used for making pragmatic patient-care decisions within this program. We will conclude with our thoughts about the potential evolution of lung cancer screening programs.
THE 40-YEAR QUEST FOR EFFECTIVE LUNG CANCER SCREENING
Lung cancer kills more people in the United States than the next four most lethal types of cancer combined.3 It is curable if found early in its course. Unfortunately, most people who develop lung cancer feel no symptoms when it is early in its course, and therefore it is too often diagnosed at a late stage. Treatment for late-stage lung cancer is effective, but it is rarely curative.
Screening refers to testing people at risk of developing a disease before its symptoms or signs have appeared. The goal of screening is to reduce the disease-specific mortality rate. For this to happen, the disease must be detectable in a preclinical form, and treatment must be more successful when applied early. Ideally, the screening test should pose little risk to the patient, be sensitive for detecting the disease early in its course, give few false-positive results, be acceptable to the patient, and be relatively inexpensive to the health system.
Over the past 4 decades, a large volume of research has been done in the hope of proving that conventional radiography or CT could be an effective screening test for lung cancer.4,5
Cohort studies (ie, in which all the patients were screened) of radiography or CT have shown a longer survival from the time of lung cancer diagnosis than would be expected without screening. These studies were not designed to prove a reduction in the lung cancer-specific mortality rate.
Controlled trials (in which half the patients received the screening and the other half did not) of chest radiography have been interpreted as not showing a reduction in lung cancer mortality rates, though debate about the interpretation of these trials persisted until this past year. Biases inherent in using duration of survival rather than the mortality rate as an end point have been suggested as the reason for the apparent benefit in survival without a reduction in the mortality rate.
Controlled trials of CT screening were started nearly a decade ago. Until 2011, the results of these trials were not mature enough to comment on.
THE PROSTATE, LUNG, COLORECTAL, AND OVARIAN TRIAL
The lung cancer screening portion of the PLCO trial aimed to determine the effect of screening chest radiography on lung cancer-specific mortality rates.1
In this trial, 154,901 people were randomized to undergo either posteroanterior chest radiography every year for 4 years or usual care, ie, no lung cancer screening. Participants were men and women age 55 to 74 with no history of prostate, lung, colorectal, or ovarian cancer. They did not need to be a smoker to participate. Those who had never smoked and who were randomized to the screening group received only 3 years of testing. All were followed for 13 years or until the conclusion of the study (8 years after the final participant was enrolled). About half were women, and nearly two-thirds were age 55 through 64. Only 10% were current smokers, while a full 45% had never smoked.
Results. Adherence to screening in the screening group ranged from 79% to 86.6% over the years of screening, and 11% of the usual-care group was estimated to have undergone screening chest radiography.
Cumulative lung cancer incidence rates were 201 per 100,000 person-years in the screening group and 192 in the usual-care group.
In the screening group, there were a total of 1,696 lung cancers during the entire study. Of these, 307 (18%) were detected by screening, 198 (12%) were interval cancers (diagnosed during the screening period but not by the screening test), and the remainder were diagnosed after the screening period during the years of follow-up. In the screening group, the cancers detected by screening were more likely to be adenocarcinomas and less likely to be small-cell carcinomas than those not detected by screening. Also in the screening group, the cancers detected by screening were more likely to be stage I (50%) than those not detected by screening.
The cumulative number of deaths from lung cancer was slightly but not significantly lower in the screening group from years 4 through 11. However, by the end of follow-up, the number of lung cancer deaths was equal between the groups (1,213 in the screening group vs 1,230 in the usual-care group). The cumulative overall mortality rate was also similar between the groups. For the subgroup who would have qualified for the NLST (see below), the lung cancer mortality rate was statistically similar between the two groups.
Comments. The results of the PLCO screening trial will be interpreted as the final word in lung cancer screening with standard chest radiography. The conclusion is that annual screening with chest radiography does not reduce lung cancer mortality rates and thus should not be performed in this context.
THE NATIONAL LUNG SCREENING TRIAL
The NLST aimed to determine if screening with low-dose chest CT could reduce lung cancer mortality rates.2
This controlled trial enrolled 53,454 people, who were randomized to undergo either low-dose chest CT or posteroanterior chest radiography at baseline and then yearly for 2 years.
Participants were men and women age 55 to 74 with at least 30 pack-years of cigarette smoking. If they had quit smoking, they had to have quit within the past 15 years. All were followed until study conclusion (median 6.5 years, maximum 7.4). About 41% were women, and nearly three-quarters were age 55 through 64. More than 48% were current smokers, with the rest being former smokers.
Results. Adherence to screening was 95% in the CT group and 93% in the radiography group, with a 4.3% annual rate of CT outside the study during the screening phase.
Cumulative lung cancer incidence rates were 645 per 100,000 person-years in the CT group and 572 in the radiography group.
In the CT group there were a total of 1,060 lung cancers during the entire study. Of these, 649 (61%) were detected by screening, 44 (4%) were interval cancers, and the rest were diagnosed after the screening period during follow-up.
In the chest radiography group, there were a total of 941 lung cancers during the entire study. Of these, 279 (30%) were detected by screening, 137 (15%) were interval cancers, and the rest were diagnosed after the screening period. Within the CT group, the cancers detected by screening were more likely to be adenocarcinomas and less likely to be small-cell carcinomas than those not detected by screening. Also within the CT group, the cancers detected by screening were more likely to be stage I (63%) than those not detected by screening.
The cumulative number of deaths from lung cancer was 443 in the radiography group, but only 356 in the CT group—20.0% lower (P =.004). The cumulative overall mortality rate was 6.7% lower in the CT group (P = .02).
Comments. The results of the NLST provide the first evidence that lung cancer mortality rates can be reduced by screening. Though many questions remain, the conclusions of this study are that screening a well-defined high-risk group with low-dose CT reduces the rate of death from lung cancer.
REMAINING CHALLENGES
The NLST showed that lung cancer screening with low-dose CT can meet the most important criterion for a successful screening program, ie, a reduction in the disease-specific mortality rate. Many challenges remain in meeting the other criteria for a successful or ideal screening program (low risk, few false-positive results, acceptability to the patient, and affordability). The issues with low-dose CT-based screening that challenge these ideals are outlined in this section.
Lung nodules: Benign or malignant?
A meta-analysis of CT screening studies found that for every 1,000 people screened at baseline, 9 were found to have stage I non-small-cell lung cancer, 235 had false-positive nodules, and 4 underwent thoracotomy for benign lesions.6
The NLST results were similar. In this trial, only nodules that were 4 mm or greater in diameter were reported. Using these criteria, over 27% of all study participants were found to have a lung nodule on CT at baseline and at year 1. The rate fell to nearly 17% at year 2, as nodules present from baseline were not reported. Of all the lung nodules detected, only 3.6% were ultimately proven to represent lung cancer.2
Many issues with small lung nodules need to be considered. The nodules are difficult to find, with highly variable reporting even by expert radiologists.7 They are difficult to measure accurately and thus are difficult to assess for growth.8 Adjunctive imaging and nonsurgical biopsy have a low yield for small nodules.9–11 Follow-up of these lung nodules includes additional imaging and nonsurgical and surgical biopsy procedures, adding expense to the program and risk to the patient. Finally, knowing that they have a lung nodule makes patients feel anxious and thus negatively affects their quality of life.12,13
Radiation exposure: How great is the risk?
There is a great deal of concern about radiation exposure from medical imaging, as many people receive a substantial amount of radiation each year from medical testing.14 A single low-dose scan with chest CT delivers a whole-body effective dose of about 1.5 mSv—less than one-fifth of the radiation dose of a typical diagnostic CT scan.
Many have tried to estimate the consequences of radiation exposure from low-dose CT screening. All estimates are extrapolations from unrelated radiation exposures. The increase in risk of death ranged from 0.01% to a few percent,15 and the increase in cancers was as high as 1.8% over a 25-year screening period.16 In general, the risks are felt to be very low but not negligible.
Cost-effectiveness is unknown
The cost-effectiveness of lung cancer screening is also unknown. Many highly variable estimates have been published.17–20 The studies have differed in the perspective taken, the costs of testing assumed, and the rounds of screening included. The most cost-effective estimates are in populations with the highest risk of cancer, in programs that achieve the greatest reduction in mortality rate, and in programs that lead to high rates of smoking cessation.
Screening in the real world as opposed to a clinical trial may involve different risks, benefits, and costs. Compliance with screening and with nodule management algorithms may be lower outside of a study. One study suggested that those at highest risk of developing lung cancer would be the least likely to enroll in a screening program and the least likely to accept curative-intent surgery for screening-detected cancer.21
We expect that the NLST data will be analyzed for cost-effectiveness. This should provide the most accurate estimates for the group that was studied.
WE SET OUT TO DESIGN A SCREENING PROGRAM
With the evidence supporting a reduction in the rate of lung cancer mortality, and knowing the remaining challenges, we set out to provide a lung cancer screening program within Cleveland Clinic. In the design of our program, we considered several questions, outlined below.
Who should be offered low-dose CT screening?
The results of the NLST led to a great deal of excitement about lung cancer screening in both the medical community and the general public. The positive side of this publicity is that lung cancer is receiving attention that may lead to support for further advances. The negative side is that many patients who may seek out lung cancer screening are not at high enough risk of lung cancer to clearly benefit from it.
In the NLST, a very high-risk cohort was studied, as defined by clinical variables (age 55 to 74, at least 30 pack-years of smoking, and if a former smoker, had quit within the past 15 years). In this high-risk group, 320 patients needed to be screened (with three yearly chest CT scans) for one life to be saved from lung cancer, and only 3.6% of all lung nodules found (4 mm or larger) were actually lung cancer. In a group at lower risk, the number that needed to be screened to save one life would be higher, and the percentage of lung nodules that truly were lung cancer would be lower. This would lead to higher risks and costs related to screening, without a proven benefit to members of the lower-risk group.
The risk of the NLST cohort developing lung cancer was approximately 0.6% per year. Lung cancer risk-prediction models have been developed and published. Up to 2011, the three most commonly used models had only moderate accuracy at predicting risk.22–25 In 2011 a risk model based on the PLCO cohort was developed and published.26 This model seemed to be more accurate but perhaps a bit harder to apply in practice.
We discussed whether using a validated risk predictor with a target of 0.6% per year (ie, the risk in the NLST trial) would be an adequate means of deciding on candidacy for lung cancer screening or if we should strictly adhere to the inclusion criteria of the NLST cohort. We feel that the NLST cohort is the only group with true evidence of benefit (a reduction in the lung cancer-specific mortality rate). Thus, for our program’s entry criteria, we decided to use the same clinical predictors used for entry in the NLST.
How will the right patients get scheduled for low-dose screening CT?
Patients who enter the lung cancer screening program from our health system will require a physician’s order.
We are fortunate to have an electronic medical record in place. We have created an order set within the electronic record for low-dose chest CT. The order will eventually be able to be entered as “CT lung screening w/o” (ie, without contrast).
For patients from outside of our health system who would like to enter the lung cancer screening program, the entry criteria will be the same (see above). We will ask for the name of the patient’s primary care practitioner. If the patient does not have one, a member of our Respiratory Institute will see and enroll the patient.
How often should patients be screened, and for how many years?
Unfortunately, questions about the frequency of screening and how many years it should continue remain unanswered.
In the NLST, a similar number of early-stage lung cancers were detected during each of the three screening rounds. In both the NLST and PLCO trials, differences in the mortality rate curves began to narrow during the observation period, when active screening was no longer occurring. Thus, it is possible that a longer duration of screening could lead to a further reduction in mortality rates. Others have questioned whether a similar benefit, with less cost and risk, could be obtained by screening every 2 years.
The large amount of data obtained from the NLST and other CT-based studies is being reviewed so that models can be developed to help answer these questions. For now, we suggest at least three yearly CT screenings, with the hope that we will have clearer answers to these questions over time.
How will low-dose CT be performed and interpreted?
The parameters for low-dose CT were very tightly controlled and monitored during the NLST. This quality-control effort, designed to improve consistency across sites and to minimize risk to patients, should be carried into lung cancer screening programs.
Our program will closely mimic the CT performance criteria used in the NLST (tube current-time product 40 mAs for all patients, field of view lungs only, lung kernel images 3 mm at 1.5-mm intervals, and soft-tissue kernel images 5 mm at 2.5-mm intervals).27 In the initial phase of the program, all screening scans will be performed at Cleveland Clinic’s main imaging facility.
Small lung nodules remain quite challenging to detect and measure. To minimize variability in scan interpretation, the NLST readers were all expertly trained radiologists. Despite this, much variability was noted in the number of nodules detected, their measured size, and the follow-up recommendations. All of the screening CT images for our program will be interpreted by board-certified radiologists with expertise in chest imaging.
Other screening studies have included novel imaging assessment in their testing algorithms, particularly volumetric analysis of lung nodules.28 These tools may prove to assist in nodule detection, measurement, and management over time. At this point, we do not think they have been studied and standardized enough to include them in a standard-of-care screening program. We hope that they will evolve to the point of clinical utility in the near future.
Lung cancer screening is not currently covered by most insurers, including Medicare, although one major insurer has recently started to cover it. We expect decisions on coverage from other insurers in the next 12 months. In the meantime, we offer a low-dose screening chest CT to our patients for $125, which includes the radiologist’s fee for interpreting the scan.
Smoking cessation
The NLST showed that low-dose CT screening can reduce lung cancer mortality rates by 20% in a high-risk group. A 50-year-old active smoker who quits smoking reduces his or her risk of dying of lung cancer by more than 50%.29 Entry into a lung cancer screening program provides an opportunity for education and assistance with tobacco dependency.
At Cleveland Clinic, we have an active Tobacco Treatment Center within our Wellness Institute. All lung cancer screening participants who are identified as active smokers will be given a program brochure and will be offered a consult in the program.
What do we identify as a lung nodule, and how should they be managed?
Studies of CT-based screening have highlighted the tremendous number of lung nodules that are identified and the low likelihood of malignancy in those that are less than 1 cm in diameter. Many screening studies define a positive result as a lung nodule above a particular size. The NLST used 4 mm or greater as the cutoff. The lower the cutoff, the greater the number of nodules found, and the lower the overall likelihood of malignancy in the nodules.
Studies in which annual CT screening was the intervention are able to use size criteria in part because the study design ensures another CT will be performed 12 months later. Current nodule management guidelines suggest 12-month CT follow-up of incidentally discovered lung nodules, 4 mm or smaller, in at-risk patients.30 In a screening program, particularly one for which the patient must pay, the 12-month screening CT cannot be guaranteed. This makes it more difficult to ignore the smallest nodules identified on CT screening. Given this, we will be reporting all lung nodules identified, regardless of size on the initial screening.
Most studies of CT screening have reported any new nodule identified in subsequent screening rounds regardless of size. Though it is intuitive that a new nodule would have a high likelihood of malignancy in a high-risk cohort, malignancy rates have been reported to be as low as 1% for new nodules. As with the initial round of screening, we will report all new lung nodules identified in subsequent screening rounds.
The recommendations for the evaluation of lung nodules, both within the report and at the lung nodule clinic, are in keeping with currently available guidelines, such as those from the Fleischner Society30 and the American College of Chest Physicians.31 For incidentally discovered lung nodules in patients at high risk, the Fleischner Society recommendations are as follows30:
- For nodules 4 mm or smaller, follow-up in 12 months; if no growth, then no further follow-up
- For nodules 4 to 6 mm, follow-up at 6 to 12 months, then 18 to 24 months if no growth
- For nodules 6 to 8 mm, follow-up at 3 to 6 months, then 9 to 12 months, then 24 months if no growth
- For nodules 8 mm or larger, follow-up at 3, 9, and 24 months, or positron emission tomography, or biopsy, or both.
If the nodule is large enough or is deemed to be of high enough risk, adjuvant testing with diagnostic imaging, guided bronchoscopy, transthoracic needle aspiration, or minimally invasive resection will be offered. All patients with nodules believed to require biopsy will be discussed at our multidisciplinary lung cancer tumor board before biopsy.
How do we make practitioners and patients aware of the program and its indications, risks, and benefits?
Education will be the key to having lung cancer screening adopted as the standard of care, to lung cancer screening being provided within a well-designed and capable system, and to ensuring that patients have realistic expectations about screening. Articles such as this and grand rounds presentations within our health system will help provide education to our colleagues. Broader marketing campaigns will be considered in the future once demand and system capabilities are clearly identified. A patient information brochure will be provided at the time of the screening test (see the patient information sheet that accompanies this article).
How do we help to advance best practice?
As excited as we are that low-dose CT-based lung cancer screening has been proven to reduce lung cancer mortality rates, it is clear that there is a lot of room to improve the programs that are developed based on current data.
Advances in our ability to accurately predict an individual’s risk of developing lung cancer will allow us to offer screening to those it is most likely to benefit.
Advances in smoking cessation and chemoprevention will help to minimize the number of lung cancers that develop.
Advances in our ability to determine the nature of lung nodules will allow us to accelerate treatment of very early lung cancer while minimizing additional testing on benign nodules; advances in our ability to treat localized and advanced disease will improve the outcome for those identified as having lung cancer.
To help move the science of screening forward, we will develop a screening program registry that can be populated from the order set and the templated report. The registry can be used to ensure appropriate patient care, while studying relevant epidemiologic, quality, and cost-related questions.
We hope to assess novel imaging software capable of assisting with the detection and characterization of lung nodules.
We have an active biomarker development program to assess the ability of breath and blood-based biomarkers to identify those at risk of developing lung cancer; to assist with the management of screening-detected lung nodules; to assist with the diagnosis of early stage lung cancer; and to characterize the nature of the cancers identified. Accurate biomarkers could lead to further decreases in mortality rates while reducing the risks and costs of a screening program.
We have strong surgical, medical, and radiation oncology programs, actively pursuing advances in minimally invasive resection procedures and ablative and targeted therapies.
ENTERING A NEW ERA
We are entering a new era of lung cancer screening. The NLST has shown that lung cancer morality rates can be reduced through low-dose CT screening in a high-risk population. Many challenges remain, such as managing the nodules that are discovered, determining if the program is cost-effective, and minimizing radiation exposure. These need to be considered when designing a lung cancer screening program. Advances over time will help us optimize the programs that are developed.
In 2011, two papers were published that will shape the way we think about lung cancer screening for years to come.
See related patient information sheet
While the ability to screen for lung cancer is a major positive change, it also raises many thorny questions, such as who should be screened, how often should they be screened, and how should we respond when a nodule is detected.
To answer some of these questions, we will outline how Cleveland Clinic has structured its lung cancer screening program, and the rationale we used for making pragmatic patient-care decisions within this program. We will conclude with our thoughts about the potential evolution of lung cancer screening programs.
THE 40-YEAR QUEST FOR EFFECTIVE LUNG CANCER SCREENING
Lung cancer kills more people in the United States than the next four most lethal types of cancer combined.3 It is curable if found early in its course. Unfortunately, most people who develop lung cancer feel no symptoms when it is early in its course, and therefore it is too often diagnosed at a late stage. Treatment for late-stage lung cancer is effective, but it is rarely curative.
Screening refers to testing people at risk of developing a disease before its symptoms or signs have appeared. The goal of screening is to reduce the disease-specific mortality rate. For this to happen, the disease must be detectable in a preclinical form, and treatment must be more successful when applied early. Ideally, the screening test should pose little risk to the patient, be sensitive for detecting the disease early in its course, give few false-positive results, be acceptable to the patient, and be relatively inexpensive to the health system.
Over the past 4 decades, a large volume of research has been done in the hope of proving that conventional radiography or CT could be an effective screening test for lung cancer.4,5
Cohort studies (ie, in which all the patients were screened) of radiography or CT have shown a longer survival from the time of lung cancer diagnosis than would be expected without screening. These studies were not designed to prove a reduction in the lung cancer-specific mortality rate.
Controlled trials (in which half the patients received the screening and the other half did not) of chest radiography have been interpreted as not showing a reduction in lung cancer mortality rates, though debate about the interpretation of these trials persisted until this past year. Biases inherent in using duration of survival rather than the mortality rate as an end point have been suggested as the reason for the apparent benefit in survival without a reduction in the mortality rate.
Controlled trials of CT screening were started nearly a decade ago. Until 2011, the results of these trials were not mature enough to comment on.
THE PROSTATE, LUNG, COLORECTAL, AND OVARIAN TRIAL
The lung cancer screening portion of the PLCO trial aimed to determine the effect of screening chest radiography on lung cancer-specific mortality rates.1
In this trial, 154,901 people were randomized to undergo either posteroanterior chest radiography every year for 4 years or usual care, ie, no lung cancer screening. Participants were men and women age 55 to 74 with no history of prostate, lung, colorectal, or ovarian cancer. They did not need to be a smoker to participate. Those who had never smoked and who were randomized to the screening group received only 3 years of testing. All were followed for 13 years or until the conclusion of the study (8 years after the final participant was enrolled). About half were women, and nearly two-thirds were age 55 through 64. Only 10% were current smokers, while a full 45% had never smoked.
Results. Adherence to screening in the screening group ranged from 79% to 86.6% over the years of screening, and 11% of the usual-care group was estimated to have undergone screening chest radiography.
Cumulative lung cancer incidence rates were 201 per 100,000 person-years in the screening group and 192 in the usual-care group.
In the screening group, there were a total of 1,696 lung cancers during the entire study. Of these, 307 (18%) were detected by screening, 198 (12%) were interval cancers (diagnosed during the screening period but not by the screening test), and the remainder were diagnosed after the screening period during the years of follow-up. In the screening group, the cancers detected by screening were more likely to be adenocarcinomas and less likely to be small-cell carcinomas than those not detected by screening. Also in the screening group, the cancers detected by screening were more likely to be stage I (50%) than those not detected by screening.
The cumulative number of deaths from lung cancer was slightly but not significantly lower in the screening group from years 4 through 11. However, by the end of follow-up, the number of lung cancer deaths was equal between the groups (1,213 in the screening group vs 1,230 in the usual-care group). The cumulative overall mortality rate was also similar between the groups. For the subgroup who would have qualified for the NLST (see below), the lung cancer mortality rate was statistically similar between the two groups.
Comments. The results of the PLCO screening trial will be interpreted as the final word in lung cancer screening with standard chest radiography. The conclusion is that annual screening with chest radiography does not reduce lung cancer mortality rates and thus should not be performed in this context.
THE NATIONAL LUNG SCREENING TRIAL
The NLST aimed to determine if screening with low-dose chest CT could reduce lung cancer mortality rates.2
This controlled trial enrolled 53,454 people, who were randomized to undergo either low-dose chest CT or posteroanterior chest radiography at baseline and then yearly for 2 years.
Participants were men and women age 55 to 74 with at least 30 pack-years of cigarette smoking. If they had quit smoking, they had to have quit within the past 15 years. All were followed until study conclusion (median 6.5 years, maximum 7.4). About 41% were women, and nearly three-quarters were age 55 through 64. More than 48% were current smokers, with the rest being former smokers.
Results. Adherence to screening was 95% in the CT group and 93% in the radiography group, with a 4.3% annual rate of CT outside the study during the screening phase.
Cumulative lung cancer incidence rates were 645 per 100,000 person-years in the CT group and 572 in the radiography group.
In the CT group there were a total of 1,060 lung cancers during the entire study. Of these, 649 (61%) were detected by screening, 44 (4%) were interval cancers, and the rest were diagnosed after the screening period during follow-up.
In the chest radiography group, there were a total of 941 lung cancers during the entire study. Of these, 279 (30%) were detected by screening, 137 (15%) were interval cancers, and the rest were diagnosed after the screening period. Within the CT group, the cancers detected by screening were more likely to be adenocarcinomas and less likely to be small-cell carcinomas than those not detected by screening. Also within the CT group, the cancers detected by screening were more likely to be stage I (63%) than those not detected by screening.
The cumulative number of deaths from lung cancer was 443 in the radiography group, but only 356 in the CT group—20.0% lower (P =.004). The cumulative overall mortality rate was 6.7% lower in the CT group (P = .02).
Comments. The results of the NLST provide the first evidence that lung cancer mortality rates can be reduced by screening. Though many questions remain, the conclusions of this study are that screening a well-defined high-risk group with low-dose CT reduces the rate of death from lung cancer.
REMAINING CHALLENGES
The NLST showed that lung cancer screening with low-dose CT can meet the most important criterion for a successful screening program, ie, a reduction in the disease-specific mortality rate. Many challenges remain in meeting the other criteria for a successful or ideal screening program (low risk, few false-positive results, acceptability to the patient, and affordability). The issues with low-dose CT-based screening that challenge these ideals are outlined in this section.
Lung nodules: Benign or malignant?
A meta-analysis of CT screening studies found that for every 1,000 people screened at baseline, 9 were found to have stage I non-small-cell lung cancer, 235 had false-positive nodules, and 4 underwent thoracotomy for benign lesions.6
The NLST results were similar. In this trial, only nodules that were 4 mm or greater in diameter were reported. Using these criteria, over 27% of all study participants were found to have a lung nodule on CT at baseline and at year 1. The rate fell to nearly 17% at year 2, as nodules present from baseline were not reported. Of all the lung nodules detected, only 3.6% were ultimately proven to represent lung cancer.2
Many issues with small lung nodules need to be considered. The nodules are difficult to find, with highly variable reporting even by expert radiologists.7 They are difficult to measure accurately and thus are difficult to assess for growth.8 Adjunctive imaging and nonsurgical biopsy have a low yield for small nodules.9–11 Follow-up of these lung nodules includes additional imaging and nonsurgical and surgical biopsy procedures, adding expense to the program and risk to the patient. Finally, knowing that they have a lung nodule makes patients feel anxious and thus negatively affects their quality of life.12,13
Radiation exposure: How great is the risk?
There is a great deal of concern about radiation exposure from medical imaging, as many people receive a substantial amount of radiation each year from medical testing.14 A single low-dose scan with chest CT delivers a whole-body effective dose of about 1.5 mSv—less than one-fifth of the radiation dose of a typical diagnostic CT scan.
Many have tried to estimate the consequences of radiation exposure from low-dose CT screening. All estimates are extrapolations from unrelated radiation exposures. The increase in risk of death ranged from 0.01% to a few percent,15 and the increase in cancers was as high as 1.8% over a 25-year screening period.16 In general, the risks are felt to be very low but not negligible.
Cost-effectiveness is unknown
The cost-effectiveness of lung cancer screening is also unknown. Many highly variable estimates have been published.17–20 The studies have differed in the perspective taken, the costs of testing assumed, and the rounds of screening included. The most cost-effective estimates are in populations with the highest risk of cancer, in programs that achieve the greatest reduction in mortality rate, and in programs that lead to high rates of smoking cessation.
Screening in the real world as opposed to a clinical trial may involve different risks, benefits, and costs. Compliance with screening and with nodule management algorithms may be lower outside of a study. One study suggested that those at highest risk of developing lung cancer would be the least likely to enroll in a screening program and the least likely to accept curative-intent surgery for screening-detected cancer.21
We expect that the NLST data will be analyzed for cost-effectiveness. This should provide the most accurate estimates for the group that was studied.
WE SET OUT TO DESIGN A SCREENING PROGRAM
With the evidence supporting a reduction in the rate of lung cancer mortality, and knowing the remaining challenges, we set out to provide a lung cancer screening program within Cleveland Clinic. In the design of our program, we considered several questions, outlined below.
Who should be offered low-dose CT screening?
The results of the NLST led to a great deal of excitement about lung cancer screening in both the medical community and the general public. The positive side of this publicity is that lung cancer is receiving attention that may lead to support for further advances. The negative side is that many patients who may seek out lung cancer screening are not at high enough risk of lung cancer to clearly benefit from it.
In the NLST, a very high-risk cohort was studied, as defined by clinical variables (age 55 to 74, at least 30 pack-years of smoking, and if a former smoker, had quit within the past 15 years). In this high-risk group, 320 patients needed to be screened (with three yearly chest CT scans) for one life to be saved from lung cancer, and only 3.6% of all lung nodules found (4 mm or larger) were actually lung cancer. In a group at lower risk, the number that needed to be screened to save one life would be higher, and the percentage of lung nodules that truly were lung cancer would be lower. This would lead to higher risks and costs related to screening, without a proven benefit to members of the lower-risk group.
The risk of the NLST cohort developing lung cancer was approximately 0.6% per year. Lung cancer risk-prediction models have been developed and published. Up to 2011, the three most commonly used models had only moderate accuracy at predicting risk.22–25 In 2011 a risk model based on the PLCO cohort was developed and published.26 This model seemed to be more accurate but perhaps a bit harder to apply in practice.
We discussed whether using a validated risk predictor with a target of 0.6% per year (ie, the risk in the NLST trial) would be an adequate means of deciding on candidacy for lung cancer screening or if we should strictly adhere to the inclusion criteria of the NLST cohort. We feel that the NLST cohort is the only group with true evidence of benefit (a reduction in the lung cancer-specific mortality rate). Thus, for our program’s entry criteria, we decided to use the same clinical predictors used for entry in the NLST.
How will the right patients get scheduled for low-dose screening CT?
Patients who enter the lung cancer screening program from our health system will require a physician’s order.
We are fortunate to have an electronic medical record in place. We have created an order set within the electronic record for low-dose chest CT. The order will eventually be able to be entered as “CT lung screening w/o” (ie, without contrast).
For patients from outside of our health system who would like to enter the lung cancer screening program, the entry criteria will be the same (see above). We will ask for the name of the patient’s primary care practitioner. If the patient does not have one, a member of our Respiratory Institute will see and enroll the patient.
How often should patients be screened, and for how many years?
Unfortunately, questions about the frequency of screening and how many years it should continue remain unanswered.
In the NLST, a similar number of early-stage lung cancers were detected during each of the three screening rounds. In both the NLST and PLCO trials, differences in the mortality rate curves began to narrow during the observation period, when active screening was no longer occurring. Thus, it is possible that a longer duration of screening could lead to a further reduction in mortality rates. Others have questioned whether a similar benefit, with less cost and risk, could be obtained by screening every 2 years.
The large amount of data obtained from the NLST and other CT-based studies is being reviewed so that models can be developed to help answer these questions. For now, we suggest at least three yearly CT screenings, with the hope that we will have clearer answers to these questions over time.
How will low-dose CT be performed and interpreted?
The parameters for low-dose CT were very tightly controlled and monitored during the NLST. This quality-control effort, designed to improve consistency across sites and to minimize risk to patients, should be carried into lung cancer screening programs.
Our program will closely mimic the CT performance criteria used in the NLST (tube current-time product 40 mAs for all patients, field of view lungs only, lung kernel images 3 mm at 1.5-mm intervals, and soft-tissue kernel images 5 mm at 2.5-mm intervals).27 In the initial phase of the program, all screening scans will be performed at Cleveland Clinic’s main imaging facility.
Small lung nodules remain quite challenging to detect and measure. To minimize variability in scan interpretation, the NLST readers were all expertly trained radiologists. Despite this, much variability was noted in the number of nodules detected, their measured size, and the follow-up recommendations. All of the screening CT images for our program will be interpreted by board-certified radiologists with expertise in chest imaging.
Other screening studies have included novel imaging assessment in their testing algorithms, particularly volumetric analysis of lung nodules.28 These tools may prove to assist in nodule detection, measurement, and management over time. At this point, we do not think they have been studied and standardized enough to include them in a standard-of-care screening program. We hope that they will evolve to the point of clinical utility in the near future.
Lung cancer screening is not currently covered by most insurers, including Medicare, although one major insurer has recently started to cover it. We expect decisions on coverage from other insurers in the next 12 months. In the meantime, we offer a low-dose screening chest CT to our patients for $125, which includes the radiologist’s fee for interpreting the scan.
Smoking cessation
The NLST showed that low-dose CT screening can reduce lung cancer mortality rates by 20% in a high-risk group. A 50-year-old active smoker who quits smoking reduces his or her risk of dying of lung cancer by more than 50%.29 Entry into a lung cancer screening program provides an opportunity for education and assistance with tobacco dependency.
At Cleveland Clinic, we have an active Tobacco Treatment Center within our Wellness Institute. All lung cancer screening participants who are identified as active smokers will be given a program brochure and will be offered a consult in the program.
What do we identify as a lung nodule, and how should they be managed?
Studies of CT-based screening have highlighted the tremendous number of lung nodules that are identified and the low likelihood of malignancy in those that are less than 1 cm in diameter. Many screening studies define a positive result as a lung nodule above a particular size. The NLST used 4 mm or greater as the cutoff. The lower the cutoff, the greater the number of nodules found, and the lower the overall likelihood of malignancy in the nodules.
Studies in which annual CT screening was the intervention are able to use size criteria in part because the study design ensures another CT will be performed 12 months later. Current nodule management guidelines suggest 12-month CT follow-up of incidentally discovered lung nodules, 4 mm or smaller, in at-risk patients.30 In a screening program, particularly one for which the patient must pay, the 12-month screening CT cannot be guaranteed. This makes it more difficult to ignore the smallest nodules identified on CT screening. Given this, we will be reporting all lung nodules identified, regardless of size on the initial screening.
Most studies of CT screening have reported any new nodule identified in subsequent screening rounds regardless of size. Though it is intuitive that a new nodule would have a high likelihood of malignancy in a high-risk cohort, malignancy rates have been reported to be as low as 1% for new nodules. As with the initial round of screening, we will report all new lung nodules identified in subsequent screening rounds.
The recommendations for the evaluation of lung nodules, both within the report and at the lung nodule clinic, are in keeping with currently available guidelines, such as those from the Fleischner Society30 and the American College of Chest Physicians.31 For incidentally discovered lung nodules in patients at high risk, the Fleischner Society recommendations are as follows30:
- For nodules 4 mm or smaller, follow-up in 12 months; if no growth, then no further follow-up
- For nodules 4 to 6 mm, follow-up at 6 to 12 months, then 18 to 24 months if no growth
- For nodules 6 to 8 mm, follow-up at 3 to 6 months, then 9 to 12 months, then 24 months if no growth
- For nodules 8 mm or larger, follow-up at 3, 9, and 24 months, or positron emission tomography, or biopsy, or both.
If the nodule is large enough or is deemed to be of high enough risk, adjuvant testing with diagnostic imaging, guided bronchoscopy, transthoracic needle aspiration, or minimally invasive resection will be offered. All patients with nodules believed to require biopsy will be discussed at our multidisciplinary lung cancer tumor board before biopsy.
How do we make practitioners and patients aware of the program and its indications, risks, and benefits?
Education will be the key to having lung cancer screening adopted as the standard of care, to lung cancer screening being provided within a well-designed and capable system, and to ensuring that patients have realistic expectations about screening. Articles such as this and grand rounds presentations within our health system will help provide education to our colleagues. Broader marketing campaigns will be considered in the future once demand and system capabilities are clearly identified. A patient information brochure will be provided at the time of the screening test (see the patient information sheet that accompanies this article).
How do we help to advance best practice?
As excited as we are that low-dose CT-based lung cancer screening has been proven to reduce lung cancer mortality rates, it is clear that there is a lot of room to improve the programs that are developed based on current data.
Advances in our ability to accurately predict an individual’s risk of developing lung cancer will allow us to offer screening to those it is most likely to benefit.
Advances in smoking cessation and chemoprevention will help to minimize the number of lung cancers that develop.
Advances in our ability to determine the nature of lung nodules will allow us to accelerate treatment of very early lung cancer while minimizing additional testing on benign nodules; advances in our ability to treat localized and advanced disease will improve the outcome for those identified as having lung cancer.
To help move the science of screening forward, we will develop a screening program registry that can be populated from the order set and the templated report. The registry can be used to ensure appropriate patient care, while studying relevant epidemiologic, quality, and cost-related questions.
We hope to assess novel imaging software capable of assisting with the detection and characterization of lung nodules.
We have an active biomarker development program to assess the ability of breath and blood-based biomarkers to identify those at risk of developing lung cancer; to assist with the management of screening-detected lung nodules; to assist with the diagnosis of early stage lung cancer; and to characterize the nature of the cancers identified. Accurate biomarkers could lead to further decreases in mortality rates while reducing the risks and costs of a screening program.
We have strong surgical, medical, and radiation oncology programs, actively pursuing advances in minimally invasive resection procedures and ablative and targeted therapies.
ENTERING A NEW ERA
We are entering a new era of lung cancer screening. The NLST has shown that lung cancer morality rates can be reduced through low-dose CT screening in a high-risk population. Many challenges remain, such as managing the nodules that are discovered, determining if the program is cost-effective, and minimizing radiation exposure. These need to be considered when designing a lung cancer screening program. Advances over time will help us optimize the programs that are developed.
- Oken MM, Hocking WG, Kvale PA, et al; PLCO Project Team. Screening by chest radiograph and lung cancer mortality: the Prostate, Lung, Colorectal, and Ovarian (PLCO) randomized trial. JAMA 2011; 306:1865–1873.
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365:395–409.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Mazzone PJ, Mekhail T. Lung cancer screening. Curr Oncol Rep 2007; 9:265–274.
- Mazzone PJ. Lung cancer screening: an update, discussion, and look ahead. Curr Oncol Rep 2010; 12:226–234.
- Gopal M, Abdullah SE, Grady JJ, Goodwin JS. Screening for lung cancer with low-dose computed tomography: a systematic review and meta-analysis of the baseline findings of randomized controlled trials. J Thorac Oncol 2010; 5:1233–1239.
- Gierada DS, Pilgram TK, Ford M, et al. Lung cancer: interobserver agreement on interpretation of pulmonary findings at low-dose CT screening. Radiology 2008; 246:265–272.
- Singh S, Pinsky P, Fineberg NS, et al. Evaluation of reader variability in the interpretation of follow-up CT scans at lung cancer screening. Radiology 2011; 259:263–270.
- Lindell RM, Hartman TE, Swensen SJ, et al. Lung cancer screening experience: a retrospective review of PET in 22 non-small cell lung carcinomas detected on screening chest CT in a high-risk population. AJR Am J Roentgenol 2005; 185:126–131.
- Baaklini WA, Reinoso MA, Gorin AB, Sharafkaneh A, Manian P. Diagnostic yield of fiberoptic bronchoscopy in evaluating solitary pulmonary nodules. Chest 2000; 117:1049–1054.
- Kothary N, Lock L, Sze DY, Hofmann LV. Computed tomography-guided percutaneous needle biopsy of pulmonary nodules: impact of nodule size on diagnostic accuracy. Clin Lung Cancer 2009; 10:360–363.
- van den Bergh KA, Essink-Bot ML, Borsboom GJ, et al. Short-term health-related quality of life consequences in a lung cancer CT screening trial (NELSON). Br J Cancer 2010; 102:27–34.
- Lemonnier I, Baumann C, Jolly D, et al. Solitary pulmonary nodules: consequences for patient quality of life. Qual Life Res 2011; 20:101–109.
- Fazel R, Krumholz HM, Wang Y, et al. Exposure to low-dose ionizing radiation from medical imaging procedures. N Engl J Med 2009; 361:849–857.
- Buls N, de Mey J, Covens P, Stadnik T. Health screening with CT: prospective assessment of radiation dose and associated detriment. JBR-BTR 2005; 88:12–16.
- Brenner DJ. Radiation risks potentially associated with low-dose CT screening of adult smokers for lung cancer. Radiology 2004; 231:440–445.
- Mahadevia PJ, Fleisher LA, Frick KD, Eng J, Goodman SN, Powe NR. Lung cancer screening with helical computed tomography in older adult smokers: a decision and cost-effectiveness analysis. JAMA 2003; 289:313–322.
- Wisnivesky JP, Mushlin AI, Sicherman N, Henschke C. The cost-effectiveness of low-dose CT screening for lung cancer: preliminary results of baseline screening. Chest 2003; 124:614–621.
- Manser R, Dalton A, Carter R, Byrnes G, Elwood M, Campbell DA. Cost-effectiveness analysis of screening for lung cancer with low dose spiral CT (computed tomography) in the Australian setting. Lung Cancer 2005; 48:171–185.
- McMahon PM, Kong CY, Bouzan C, et al. Cost-effectiveness of computed tomography screening for lung cancer in the United States. J Thorac Oncol 2011; 6:1841–1848.
- Silvestri GA, Nietert PJ, Zoller J, Carter C, Bradford D. Attitudes towards screening for lung cancer among smokers and their nonsmoking counterparts. Thorax 2007; 62:126–130.
- Bach PB, Kattan MW, Thornquist MD, et al. Variations in lung cancer risk among smokers. J Natl Cancer Inst 2003; 95:470–478.
- Spitz MR, Hong WK, Amos CI, et al. A risk model for prediction of lung cancer. J Natl Cancer Inst 2007; 99:715–726.
- Cassidy A, Myles JP, van Tongeren M, et al. The LLP risk model: an individual risk prediction model for lung cancer. Br J Cancer 2008; 98:270–276.
- D’Amelio AM, Cassidy A, Asomaning K, et al. Comparison of discriminatory power and accuracy of three lung cancer risk models. Br J Cancer 2010; 103:423–429.
- Tammemagi CM, Pinsky PF, Caporaso NE, et al. Lung cancer risk prediction: Prostate, Lung, Colorectal And Ovarian Cancer Screening Trial models and validation. J Natl Cancer Inst 2011; 103:1058–1068.
- National Lung Screening Trial Research Team; Aberle DR, Berg CD, Black WC, et al. The National Lung Screening Trial: overview and study design. Radiology 2011; 258:243–253.
- van Klaveren RJ, Oudkerk M, Prokop M, et al. Management of lung nodules detected by volume CT scanning. N Engl J Med 2009; 361:2221–2229.
- Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ 2000; 321:323–329.
- MacMahon H, Austin JH, Gamsu G, et al; Fleischner Society. Guidelines for management of small pulmonary nodules detected on CT scans: a statement from the Fleischner Society. Radiology 2005; 237:395–400.
- Gould MK, Fletcher J, Iannettoni MD, et al; American College of Chest Physicians. Evaluation of patients with pulmonary nodules: when is it lung cancer?: ACCP evidence-based clinical practice guidelines (2nd edition). Chest 2007; 132(suppl 3):108S–130S.
- Oken MM, Hocking WG, Kvale PA, et al; PLCO Project Team. Screening by chest radiograph and lung cancer mortality: the Prostate, Lung, Colorectal, and Ovarian (PLCO) randomized trial. JAMA 2011; 306:1865–1873.
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365:395–409.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Mazzone PJ, Mekhail T. Lung cancer screening. Curr Oncol Rep 2007; 9:265–274.
- Mazzone PJ. Lung cancer screening: an update, discussion, and look ahead. Curr Oncol Rep 2010; 12:226–234.
- Gopal M, Abdullah SE, Grady JJ, Goodwin JS. Screening for lung cancer with low-dose computed tomography: a systematic review and meta-analysis of the baseline findings of randomized controlled trials. J Thorac Oncol 2010; 5:1233–1239.
- Gierada DS, Pilgram TK, Ford M, et al. Lung cancer: interobserver agreement on interpretation of pulmonary findings at low-dose CT screening. Radiology 2008; 246:265–272.
- Singh S, Pinsky P, Fineberg NS, et al. Evaluation of reader variability in the interpretation of follow-up CT scans at lung cancer screening. Radiology 2011; 259:263–270.
- Lindell RM, Hartman TE, Swensen SJ, et al. Lung cancer screening experience: a retrospective review of PET in 22 non-small cell lung carcinomas detected on screening chest CT in a high-risk population. AJR Am J Roentgenol 2005; 185:126–131.
- Baaklini WA, Reinoso MA, Gorin AB, Sharafkaneh A, Manian P. Diagnostic yield of fiberoptic bronchoscopy in evaluating solitary pulmonary nodules. Chest 2000; 117:1049–1054.
- Kothary N, Lock L, Sze DY, Hofmann LV. Computed tomography-guided percutaneous needle biopsy of pulmonary nodules: impact of nodule size on diagnostic accuracy. Clin Lung Cancer 2009; 10:360–363.
- van den Bergh KA, Essink-Bot ML, Borsboom GJ, et al. Short-term health-related quality of life consequences in a lung cancer CT screening trial (NELSON). Br J Cancer 2010; 102:27–34.
- Lemonnier I, Baumann C, Jolly D, et al. Solitary pulmonary nodules: consequences for patient quality of life. Qual Life Res 2011; 20:101–109.
- Fazel R, Krumholz HM, Wang Y, et al. Exposure to low-dose ionizing radiation from medical imaging procedures. N Engl J Med 2009; 361:849–857.
- Buls N, de Mey J, Covens P, Stadnik T. Health screening with CT: prospective assessment of radiation dose and associated detriment. JBR-BTR 2005; 88:12–16.
- Brenner DJ. Radiation risks potentially associated with low-dose CT screening of adult smokers for lung cancer. Radiology 2004; 231:440–445.
- Mahadevia PJ, Fleisher LA, Frick KD, Eng J, Goodman SN, Powe NR. Lung cancer screening with helical computed tomography in older adult smokers: a decision and cost-effectiveness analysis. JAMA 2003; 289:313–322.
- Wisnivesky JP, Mushlin AI, Sicherman N, Henschke C. The cost-effectiveness of low-dose CT screening for lung cancer: preliminary results of baseline screening. Chest 2003; 124:614–621.
- Manser R, Dalton A, Carter R, Byrnes G, Elwood M, Campbell DA. Cost-effectiveness analysis of screening for lung cancer with low dose spiral CT (computed tomography) in the Australian setting. Lung Cancer 2005; 48:171–185.
- McMahon PM, Kong CY, Bouzan C, et al. Cost-effectiveness of computed tomography screening for lung cancer in the United States. J Thorac Oncol 2011; 6:1841–1848.
- Silvestri GA, Nietert PJ, Zoller J, Carter C, Bradford D. Attitudes towards screening for lung cancer among smokers and their nonsmoking counterparts. Thorax 2007; 62:126–130.
- Bach PB, Kattan MW, Thornquist MD, et al. Variations in lung cancer risk among smokers. J Natl Cancer Inst 2003; 95:470–478.
- Spitz MR, Hong WK, Amos CI, et al. A risk model for prediction of lung cancer. J Natl Cancer Inst 2007; 99:715–726.
- Cassidy A, Myles JP, van Tongeren M, et al. The LLP risk model: an individual risk prediction model for lung cancer. Br J Cancer 2008; 98:270–276.
- D’Amelio AM, Cassidy A, Asomaning K, et al. Comparison of discriminatory power and accuracy of three lung cancer risk models. Br J Cancer 2010; 103:423–429.
- Tammemagi CM, Pinsky PF, Caporaso NE, et al. Lung cancer risk prediction: Prostate, Lung, Colorectal And Ovarian Cancer Screening Trial models and validation. J Natl Cancer Inst 2011; 103:1058–1068.
- National Lung Screening Trial Research Team; Aberle DR, Berg CD, Black WC, et al. The National Lung Screening Trial: overview and study design. Radiology 2011; 258:243–253.
- van Klaveren RJ, Oudkerk M, Prokop M, et al. Management of lung nodules detected by volume CT scanning. N Engl J Med 2009; 361:2221–2229.
- Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ 2000; 321:323–329.
- MacMahon H, Austin JH, Gamsu G, et al; Fleischner Society. Guidelines for management of small pulmonary nodules detected on CT scans: a statement from the Fleischner Society. Radiology 2005; 237:395–400.
- Gould MK, Fletcher J, Iannettoni MD, et al; American College of Chest Physicians. Evaluation of patients with pulmonary nodules: when is it lung cancer?: ACCP evidence-based clinical practice guidelines (2nd edition). Chest 2007; 132(suppl 3):108S–130S.
KEY POINTS
- The NLST documented a 20% reduction in the rate of death from lung cancer with low-dose CT screening compared with chest radiography screening (number needed to treat = 320). This was in a population at high risk (age 55–74 with a smoking history of at least 30 pack-years, at least some of it within the past 15 years).
- CT screening detects many lung nodules, of which only a few (3.6% in the NLST) prove to be cancer.
- In view of the positive results of the NLST, Cleveland Clinic has begun a lung cancer screening program, using the same entry criteria as those in the NLST.
- Of possibly greater impact than detecting lung cancer will be the opportunity to promote smoking cessation.
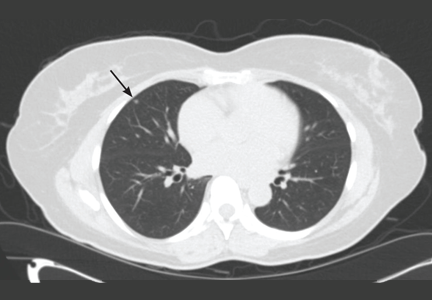
VA's Project Reach Contest to Help Homeless Find Services; DoD Changes Sexual Assault Response; Faster Claims Processing Now Available; NAMI Statement on Afghanistan Tragedy; Mentors Prove Helpful to African American Vets With Diabetes; Veterans With PTSD
Grand Rounds: Man, 62, With New-Onset Atrial Fibrillation
A 62-year-old black nursing home resident was transported to the hospital emergency department with fever of 102°F, new-onset atrial fibrillation (A-fib), and dementia. His medical history was significant for hypertension and multiple strokes.
His inpatient work-up for A-fib and dementia revealed a thyroid-stimulating hormone (TSH) level below 0.005 µIU/mL (normal range, 0.3 to 3.0 µIU/mL). Results of thyroid function testing (TFT) revealed a triiodothyronine (T3) level within normal range but a free thyroxine (T4) level of 2.9 ng/dL (normal range, 0.7 to 1.5 ng/dL) and a total T4 of 17.8 µg/dL (normal, 4.5 to 12.0 µg/dL). The abnormal TSH and T4 levels were considered suggestive of a thyrotoxic state, warranting an endocrinology consult. Cardiology was consulted regarding new-onset A-fib.
During history taking, the patient denied any shortness of breath, cough, palpitations, heat intolerance, anxiety, tremors, insomnia, dysphagia, diarrhea, dysuria, weight loss, or recent ingestion of iodine-containing medications or supplements.
On examination, the patient was febrile, with a blood pressure of 106/71 mm Hg; pulse, 74 beats/min; respiratory rate, 20 breaths/min; and O2 saturation, 98% to 99% on room air. ECG showed a normal sinus rhythm and a ventricular rate of 64 beats/min.
The patient's weight was 58.9 kg, and his height, 63" (BMI, 22.8). The patient had no skin changes, and his mucous membranes were slightly moist. The patient's head was atraumatic and normocephalic. His extraocular movements were intact, and his pupils were equal, round, and reactive to light, with nonicteric sclera. There was no proptosis or ophthalmoplegia. The patient's neck was supple, with no jugular venous distension, tracheal deviation, or thyromegaly.
The cardiovascular exam revealed an irregular heartbeat, and repeat ECG showed A-fib with a ventricular rate of 151 beats/min (see Figure 1). The patient's chest was clear, with no wheezing or rhonchi. The abdomen was soft and slightly obese, and bowel sounds were present. The neurologic examination revealed no hyperreflexia. The patient's mental status was altered at times and he was alert, awake, and oriented to others. His speech was slightly slow, and some left-sided weakness was noted.
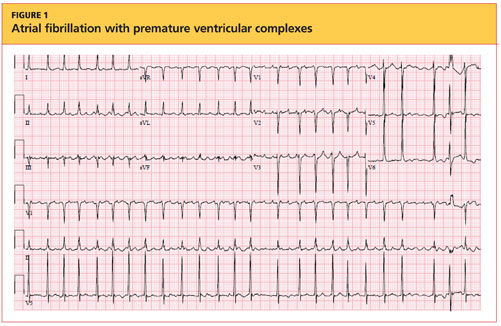
As recommended during the endocrinology consult, the patient underwent an I-123 sodium iodide thyroid scan, which showed faint uptake at the base of the neck, slightly to the left of midline; and a 24-hour radioactive iodide uptake (RAIU), which measured 2.8% (normal range, 8% to 35%).
The patient's chest X-ray showed a right tracheal deviation not previously noted on physical examination (see Figure 2); the possible cause of a thyroid mass was considered. Subsequent ultrasonography of the thyroid revealed generally normal dimensions and parenchymal echogenicity. However, a large complex mass was detected, arising from the inferior pole of the thyroid and displacing the trachea toward the right (see Figure 3). According to the radiologist's notes, the mass contained both solid and cystic elements, scattered calcifications, and foci of flow on color Doppler. It measured about 6 cm in the largest (transverse) dimension. A 2.0-mm nodule was noted in the isthmus, slightly to the right of midline, consistent with multinodular goiter.
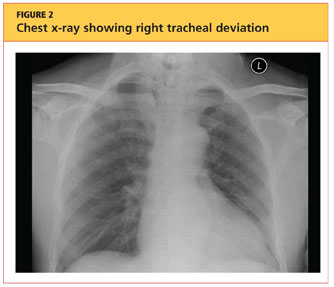
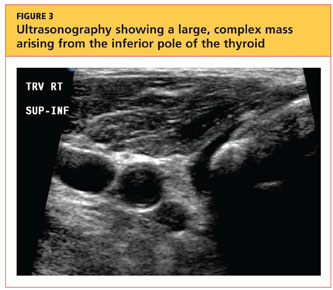
Following the cardiology consult, a diltiazem drip was initiated, but the patient was later optimized on flecainide for rhythm control and metoprolol for rate control. He was also initially anticoagulated using a heparin drip and bridged to warfarin, with target international normalized ratio (INR) between 2.0 and 3.0. Echocardiography revealed normal systolic function with ejection fraction of 55%, left ventricular hypertrophy, pulmonary artery systolic pressure of 35 mm Hg, and no pericardial effusions or valvular disease.
Regarding the patient's unexplained fever, results of chest imaging were negative for signs of pneumonia or atelectasis, which might have suggested a pulmonary cause. Urinalysis results were normal. Complete blood count showed no leukocytosis. The patient's fever subsided within 48 hours.
The differential diagnosis included Graves' disease, toxic multinodular goiter, Jod-Basedow syndrome, and subacute thyroiditis.
Graves' disease, an autoimmune disease with an unknown trigger, is the most common cause of hyperthyroidism. In affected patients, the thyroid gland overproduces thyroid hormones, leading to thyrotoxicosis. Thyrotoxicosis can result in multiple clinical signs and symptoms, including Graves' ophthalmopathy, pretibial myxedema, and goiter; TFT results typically include elevated T3 and T4 and low TSH.1-5 In the case patient (who had no history of thyroid disease, nor clinical signs or symptoms of Graves' disease), low uptake of iodine on thyroid scan precluded this diagnosis.
Toxic multinodular goiter, the second most common cause of hyperthyroidism, can be responsible for A-fib, tachycardia, and congestive heart failure.6,7 Iodine deficiency causes enlargement of the thyroid gland, where numerous nodules can develop, as seen in the case patient. These nodules can function independently, sometimes producing excess thyroid hormone; this leads to hyperplasia of the thyroid gland, resulting in a nontoxic multinodular goiter. From this goiter, a toxic multinodular goiter can emerge insidiously. However, in this condition, RAIU typically exceeds 30%; in the case patient, low 24-hour RAIU (2.8%) and the absence of functioning nodules on scanning made it possible to rule out this diagnosis.
Jod-Basedow syndrome refers to hyperthyroidism that develops as a result of administration of iodide, either as a dietary supplement or as IV contrast medium, or as an adverse effect of the antiarrhythmic drug amiodarone. This phenomenon is usually seen in a patient with endemic goiter.8-11 The relatively limited nature of the case patient's goiter and absence of a precipitating exposure to iodine made this diagnosis highly unlikely.
Subacute thyroiditis is a condition to which the patient's abnormal TFT results could reasonably be attributed. The patient had a substernal multinodular goiter that could not be palpated on physical examination, but it was visualized in the extended lower neck during thyroid scintigraphy.3 RAIU was minimal—a typical finding in this disorder,6 as TSH is suppressed by leakage of the excessive amounts of thyroid hormone. A tentative diagnosis of subacute thyroiditis was made.
As subacute thyroiditis is a self-limiting disorder, the patient was not started on any medications for hyperthyroidism but was advised to follow up with his primary care provider or an endocrinologist for repeat TFT and for fine-needle aspiration biopsy of the large thyroid nodule (a complex mass, containing cystic elements and calcifications, with a potential for malignancy) to rule out thyroid cancer.
Repeat ECG before discharge showed normal sinus rhythm with a ventricular rate of 74 beats/min. The patient was alert, awake, and oriented at discharge. He was continued on flecainide, metoprolol, and warfarin and advised to follow up with his primary care provider regarding his target INR.
DISCUSSION
The incidence of subacute thyroiditis, according to findings reported in 2003 from the Rochester Epidemiology Project in Olmsted County, Minnesota,12 is 12.1 cases per 100,000/year, with a higher incidence in women than men. It is most common in young adults and decreases with advancing age. Coxsackie virus, adenovirus, mumps, echovirus, influenza, and Epstein-Barr virus have been implicated in the disorder.12,13
Subacute thyroiditis is associated with a triphasic clinical course of hyperthyroidism, then hypothyroidism, then a return to normal thyroid function—as was seen in the case patient. Onset of subacute thyroiditis has been associated with recent viral infection, which may serve as a precipitant. The cause of this patient's high fever was never identified; thus, the etiology may have been viral.
The initial high thyroid hormone levels result from inflammation of thyroid tissue and release of preformed thyroid hormone into the circulation.6 At this point, TSH is suppressed and patients have very low RAIU, as was true in the case patient.
The condition is self-limiting and does not require treatment in the majority of patients, as TFT results return to normal levels within about two months.6 Patients can appear extremely ill due to thyrotoxicosis from subacute thyroiditis, but this usually lasts no longer than six to eight weeks.3 Subacute thyroiditis can be associated with atrial arrhythmia or heart failure.14,15
PATIENT OUTCOME
New-onset A-fib was attributed to the patient's thyrotoxicosis, which in turn was caused by subacute thyroiditis. He had a multinodular goiter, although he had not received any iodine supplements or IV contrast. As in most cases of subacute thyroiditis, no precipitating event was identified. However, given this patient's residence in a nursing facility and presentation with a high fever with no identifiable cause, a viral etiology for his subacute thyroiditis is possible.6
The patient's dementia may have been secondary to acute thyrotoxicosis, as his mental state improved during the hospital stay. His vitamin B12, folate, and A1C levels were within normal range. CT of the head showed multiple chronic infarcts and cerebral atrophy, and MRI of the brain indicated microvascular ischemic disease.
The patient was readmitted one month later for an episode of near-syncope (which, it was concluded, was a vasovagal episode). At that time, his TSH was found normal at 1.350 µIU/mL. Flecainide and metoprolol were discontinued; he was started on diltiazem for continued rate and rhythm control (as recommended by cardiology) and continued on warfarin.
CONCLUSION
In this case, subacute thyroiditis was most likely caused by a viral infection that led to destruction of the normal thyroid follicles and release of their preformed thyroid hormone into the circulation; this in turn led to sudden-onset A-fib. The diagnosis of subacute thyroiditis was suggested based on the abnormalities seen in this patient's TFT results, coupled with the suppressed RAIU—a typical finding in this disease.
Because subacute thyroiditis is a self-limiting condition, there is no role for antithyroid medication. Instead, treatment should be focused on relieving the patient's symptoms, such as ß-blockade or calcium channel blockers for tachycardia and corticosteroids or NSAIDs for neck pain.
REFERENCES
1. Weetman AP. Graves' disease. N Engl J Med. 2000;343(17):1236-1248.
2. Delgado Hurtado JJ, Pineda M. Images in medicine: Graves' disease. N Engl J Med. 2011; 364(20):1955.
3. Al-Sharif AA, Abujbara MA, Chiacchio S, et al. Contribution of radioiodine uptake measurement and thyroid scintigraphy to the differential diagnosis of thyrotoxicosis. Hell J Nucl Med. 2010;13(2):132-137.
4. Buccelletti F, Carroccia A, Marsiliani D, et al. Utility of routine thyroid-stimulating hormone determination in new-onset atrial fibrillation in the ED. Am J Emerg Med. 2011;29(9):1158-1162.
5. Ross DS. Radioiodine therapy for hyperthyroidism. N Engl J Med. 2011;364(6):542-550.
6. Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011;17(3):456-520.
7. Erickson D, Gharib H, Li H, van Heerden JA. Treatment of patients with toxic multinodular goiter. Thyroid. 1998;8(4):277-282.
8. Basaria S, Cooper DS. Amiodarone and the thyroid. Am J Med. 2005;118(7):706-714.
9. Bogazzi F, Bartalena L, Martino E. Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2010;95(6):2529-2535.
10. El-Shirbiny AM, Stavrou SS, Dnistrian A, et al. Jod-Basedow syndrome following oral iodine and radioiodinated-antibody administration. J Nucl Med. 1997;38(11):1816-1817.
11. Stanbury JB, Ermans AE, Bourdoux P, et al. Iodine-induced hyperthyroidism: occurrence and epidemiology. Thyroid. 1998;8(1):83-100.
12. Fatourechi V, Aniszewski JP, Fatourechi GZ, et al. Clinical features and outcome of subacute thyroiditis in an incidence cohort: Olmsted County, Minnesota, study. J Clin Endocrinol Metab. 2003;88(5):2100-2105.
13. Golden SH, Robinson KA, Saldanha I, et al. Clinical review: prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab. 2009;94(6):1853-1878.
14. Volpé R. The management of subacute (DeQuervain's) thyroiditis. Thyroid. 1993;3(3):253-255.
15. Lee SL. Subacute thyroiditis (2009). http://emedicine.medscape.com/article/125648-overview. Accessed April 17, 2012.
A 62-year-old black nursing home resident was transported to the hospital emergency department with fever of 102°F, new-onset atrial fibrillation (A-fib), and dementia. His medical history was significant for hypertension and multiple strokes.
His inpatient work-up for A-fib and dementia revealed a thyroid-stimulating hormone (TSH) level below 0.005 µIU/mL (normal range, 0.3 to 3.0 µIU/mL). Results of thyroid function testing (TFT) revealed a triiodothyronine (T3) level within normal range but a free thyroxine (T4) level of 2.9 ng/dL (normal range, 0.7 to 1.5 ng/dL) and a total T4 of 17.8 µg/dL (normal, 4.5 to 12.0 µg/dL). The abnormal TSH and T4 levels were considered suggestive of a thyrotoxic state, warranting an endocrinology consult. Cardiology was consulted regarding new-onset A-fib.
During history taking, the patient denied any shortness of breath, cough, palpitations, heat intolerance, anxiety, tremors, insomnia, dysphagia, diarrhea, dysuria, weight loss, or recent ingestion of iodine-containing medications or supplements.
On examination, the patient was febrile, with a blood pressure of 106/71 mm Hg; pulse, 74 beats/min; respiratory rate, 20 breaths/min; and O2 saturation, 98% to 99% on room air. ECG showed a normal sinus rhythm and a ventricular rate of 64 beats/min.
The patient's weight was 58.9 kg, and his height, 63" (BMI, 22.8). The patient had no skin changes, and his mucous membranes were slightly moist. The patient's head was atraumatic and normocephalic. His extraocular movements were intact, and his pupils were equal, round, and reactive to light, with nonicteric sclera. There was no proptosis or ophthalmoplegia. The patient's neck was supple, with no jugular venous distension, tracheal deviation, or thyromegaly.
The cardiovascular exam revealed an irregular heartbeat, and repeat ECG showed A-fib with a ventricular rate of 151 beats/min (see Figure 1). The patient's chest was clear, with no wheezing or rhonchi. The abdomen was soft and slightly obese, and bowel sounds were present. The neurologic examination revealed no hyperreflexia. The patient's mental status was altered at times and he was alert, awake, and oriented to others. His speech was slightly slow, and some left-sided weakness was noted.
As recommended during the endocrinology consult, the patient underwent an I-123 sodium iodide thyroid scan, which showed faint uptake at the base of the neck, slightly to the left of midline; and a 24-hour radioactive iodide uptake (RAIU), which measured 2.8% (normal range, 8% to 35%).
The patient's chest X-ray showed a right tracheal deviation not previously noted on physical examination (see Figure 2); the possible cause of a thyroid mass was considered. Subsequent ultrasonography of the thyroid revealed generally normal dimensions and parenchymal echogenicity. However, a large complex mass was detected, arising from the inferior pole of the thyroid and displacing the trachea toward the right (see Figure 3). According to the radiologist's notes, the mass contained both solid and cystic elements, scattered calcifications, and foci of flow on color Doppler. It measured about 6 cm in the largest (transverse) dimension. A 2.0-mm nodule was noted in the isthmus, slightly to the right of midline, consistent with multinodular goiter.
Following the cardiology consult, a diltiazem drip was initiated, but the patient was later optimized on flecainide for rhythm control and metoprolol for rate control. He was also initially anticoagulated using a heparin drip and bridged to warfarin, with target international normalized ratio (INR) between 2.0 and 3.0. Echocardiography revealed normal systolic function with ejection fraction of 55%, left ventricular hypertrophy, pulmonary artery systolic pressure of 35 mm Hg, and no pericardial effusions or valvular disease.
Regarding the patient's unexplained fever, results of chest imaging were negative for signs of pneumonia or atelectasis, which might have suggested a pulmonary cause. Urinalysis results were normal. Complete blood count showed no leukocytosis. The patient's fever subsided within 48 hours.
The differential diagnosis included Graves' disease, toxic multinodular goiter, Jod-Basedow syndrome, and subacute thyroiditis.
Graves' disease, an autoimmune disease with an unknown trigger, is the most common cause of hyperthyroidism. In affected patients, the thyroid gland overproduces thyroid hormones, leading to thyrotoxicosis. Thyrotoxicosis can result in multiple clinical signs and symptoms, including Graves' ophthalmopathy, pretibial myxedema, and goiter; TFT results typically include elevated T3 and T4 and low TSH.1-5 In the case patient (who had no history of thyroid disease, nor clinical signs or symptoms of Graves' disease), low uptake of iodine on thyroid scan precluded this diagnosis.
Toxic multinodular goiter, the second most common cause of hyperthyroidism, can be responsible for A-fib, tachycardia, and congestive heart failure.6,7 Iodine deficiency causes enlargement of the thyroid gland, where numerous nodules can develop, as seen in the case patient. These nodules can function independently, sometimes producing excess thyroid hormone; this leads to hyperplasia of the thyroid gland, resulting in a nontoxic multinodular goiter. From this goiter, a toxic multinodular goiter can emerge insidiously. However, in this condition, RAIU typically exceeds 30%; in the case patient, low 24-hour RAIU (2.8%) and the absence of functioning nodules on scanning made it possible to rule out this diagnosis.
Jod-Basedow syndrome refers to hyperthyroidism that develops as a result of administration of iodide, either as a dietary supplement or as IV contrast medium, or as an adverse effect of the antiarrhythmic drug amiodarone. This phenomenon is usually seen in a patient with endemic goiter.8-11 The relatively limited nature of the case patient's goiter and absence of a precipitating exposure to iodine made this diagnosis highly unlikely.
Subacute thyroiditis is a condition to which the patient's abnormal TFT results could reasonably be attributed. The patient had a substernal multinodular goiter that could not be palpated on physical examination, but it was visualized in the extended lower neck during thyroid scintigraphy.3 RAIU was minimal—a typical finding in this disorder,6 as TSH is suppressed by leakage of the excessive amounts of thyroid hormone. A tentative diagnosis of subacute thyroiditis was made.
As subacute thyroiditis is a self-limiting disorder, the patient was not started on any medications for hyperthyroidism but was advised to follow up with his primary care provider or an endocrinologist for repeat TFT and for fine-needle aspiration biopsy of the large thyroid nodule (a complex mass, containing cystic elements and calcifications, with a potential for malignancy) to rule out thyroid cancer.
Repeat ECG before discharge showed normal sinus rhythm with a ventricular rate of 74 beats/min. The patient was alert, awake, and oriented at discharge. He was continued on flecainide, metoprolol, and warfarin and advised to follow up with his primary care provider regarding his target INR.
DISCUSSION
The incidence of subacute thyroiditis, according to findings reported in 2003 from the Rochester Epidemiology Project in Olmsted County, Minnesota,12 is 12.1 cases per 100,000/year, with a higher incidence in women than men. It is most common in young adults and decreases with advancing age. Coxsackie virus, adenovirus, mumps, echovirus, influenza, and Epstein-Barr virus have been implicated in the disorder.12,13
Subacute thyroiditis is associated with a triphasic clinical course of hyperthyroidism, then hypothyroidism, then a return to normal thyroid function—as was seen in the case patient. Onset of subacute thyroiditis has been associated with recent viral infection, which may serve as a precipitant. The cause of this patient's high fever was never identified; thus, the etiology may have been viral.
The initial high thyroid hormone levels result from inflammation of thyroid tissue and release of preformed thyroid hormone into the circulation.6 At this point, TSH is suppressed and patients have very low RAIU, as was true in the case patient.
The condition is self-limiting and does not require treatment in the majority of patients, as TFT results return to normal levels within about two months.6 Patients can appear extremely ill due to thyrotoxicosis from subacute thyroiditis, but this usually lasts no longer than six to eight weeks.3 Subacute thyroiditis can be associated with atrial arrhythmia or heart failure.14,15
PATIENT OUTCOME
New-onset A-fib was attributed to the patient's thyrotoxicosis, which in turn was caused by subacute thyroiditis. He had a multinodular goiter, although he had not received any iodine supplements or IV contrast. As in most cases of subacute thyroiditis, no precipitating event was identified. However, given this patient's residence in a nursing facility and presentation with a high fever with no identifiable cause, a viral etiology for his subacute thyroiditis is possible.6
The patient's dementia may have been secondary to acute thyrotoxicosis, as his mental state improved during the hospital stay. His vitamin B12, folate, and A1C levels were within normal range. CT of the head showed multiple chronic infarcts and cerebral atrophy, and MRI of the brain indicated microvascular ischemic disease.
The patient was readmitted one month later for an episode of near-syncope (which, it was concluded, was a vasovagal episode). At that time, his TSH was found normal at 1.350 µIU/mL. Flecainide and metoprolol were discontinued; he was started on diltiazem for continued rate and rhythm control (as recommended by cardiology) and continued on warfarin.
CONCLUSION
In this case, subacute thyroiditis was most likely caused by a viral infection that led to destruction of the normal thyroid follicles and release of their preformed thyroid hormone into the circulation; this in turn led to sudden-onset A-fib. The diagnosis of subacute thyroiditis was suggested based on the abnormalities seen in this patient's TFT results, coupled with the suppressed RAIU—a typical finding in this disease.
Because subacute thyroiditis is a self-limiting condition, there is no role for antithyroid medication. Instead, treatment should be focused on relieving the patient's symptoms, such as ß-blockade or calcium channel blockers for tachycardia and corticosteroids or NSAIDs for neck pain.
REFERENCES
1. Weetman AP. Graves' disease. N Engl J Med. 2000;343(17):1236-1248.
2. Delgado Hurtado JJ, Pineda M. Images in medicine: Graves' disease. N Engl J Med. 2011; 364(20):1955.
3. Al-Sharif AA, Abujbara MA, Chiacchio S, et al. Contribution of radioiodine uptake measurement and thyroid scintigraphy to the differential diagnosis of thyrotoxicosis. Hell J Nucl Med. 2010;13(2):132-137.
4. Buccelletti F, Carroccia A, Marsiliani D, et al. Utility of routine thyroid-stimulating hormone determination in new-onset atrial fibrillation in the ED. Am J Emerg Med. 2011;29(9):1158-1162.
5. Ross DS. Radioiodine therapy for hyperthyroidism. N Engl J Med. 2011;364(6):542-550.
6. Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011;17(3):456-520.
7. Erickson D, Gharib H, Li H, van Heerden JA. Treatment of patients with toxic multinodular goiter. Thyroid. 1998;8(4):277-282.
8. Basaria S, Cooper DS. Amiodarone and the thyroid. Am J Med. 2005;118(7):706-714.
9. Bogazzi F, Bartalena L, Martino E. Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2010;95(6):2529-2535.
10. El-Shirbiny AM, Stavrou SS, Dnistrian A, et al. Jod-Basedow syndrome following oral iodine and radioiodinated-antibody administration. J Nucl Med. 1997;38(11):1816-1817.
11. Stanbury JB, Ermans AE, Bourdoux P, et al. Iodine-induced hyperthyroidism: occurrence and epidemiology. Thyroid. 1998;8(1):83-100.
12. Fatourechi V, Aniszewski JP, Fatourechi GZ, et al. Clinical features and outcome of subacute thyroiditis in an incidence cohort: Olmsted County, Minnesota, study. J Clin Endocrinol Metab. 2003;88(5):2100-2105.
13. Golden SH, Robinson KA, Saldanha I, et al. Clinical review: prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab. 2009;94(6):1853-1878.
14. Volpé R. The management of subacute (DeQuervain's) thyroiditis. Thyroid. 1993;3(3):253-255.
15. Lee SL. Subacute thyroiditis (2009). http://emedicine.medscape.com/article/125648-overview. Accessed April 17, 2012.
A 62-year-old black nursing home resident was transported to the hospital emergency department with fever of 102°F, new-onset atrial fibrillation (A-fib), and dementia. His medical history was significant for hypertension and multiple strokes.
His inpatient work-up for A-fib and dementia revealed a thyroid-stimulating hormone (TSH) level below 0.005 µIU/mL (normal range, 0.3 to 3.0 µIU/mL). Results of thyroid function testing (TFT) revealed a triiodothyronine (T3) level within normal range but a free thyroxine (T4) level of 2.9 ng/dL (normal range, 0.7 to 1.5 ng/dL) and a total T4 of 17.8 µg/dL (normal, 4.5 to 12.0 µg/dL). The abnormal TSH and T4 levels were considered suggestive of a thyrotoxic state, warranting an endocrinology consult. Cardiology was consulted regarding new-onset A-fib.
During history taking, the patient denied any shortness of breath, cough, palpitations, heat intolerance, anxiety, tremors, insomnia, dysphagia, diarrhea, dysuria, weight loss, or recent ingestion of iodine-containing medications or supplements.
On examination, the patient was febrile, with a blood pressure of 106/71 mm Hg; pulse, 74 beats/min; respiratory rate, 20 breaths/min; and O2 saturation, 98% to 99% on room air. ECG showed a normal sinus rhythm and a ventricular rate of 64 beats/min.
The patient's weight was 58.9 kg, and his height, 63" (BMI, 22.8). The patient had no skin changes, and his mucous membranes were slightly moist. The patient's head was atraumatic and normocephalic. His extraocular movements were intact, and his pupils were equal, round, and reactive to light, with nonicteric sclera. There was no proptosis or ophthalmoplegia. The patient's neck was supple, with no jugular venous distension, tracheal deviation, or thyromegaly.
The cardiovascular exam revealed an irregular heartbeat, and repeat ECG showed A-fib with a ventricular rate of 151 beats/min (see Figure 1). The patient's chest was clear, with no wheezing or rhonchi. The abdomen was soft and slightly obese, and bowel sounds were present. The neurologic examination revealed no hyperreflexia. The patient's mental status was altered at times and he was alert, awake, and oriented to others. His speech was slightly slow, and some left-sided weakness was noted.
As recommended during the endocrinology consult, the patient underwent an I-123 sodium iodide thyroid scan, which showed faint uptake at the base of the neck, slightly to the left of midline; and a 24-hour radioactive iodide uptake (RAIU), which measured 2.8% (normal range, 8% to 35%).
The patient's chest X-ray showed a right tracheal deviation not previously noted on physical examination (see Figure 2); the possible cause of a thyroid mass was considered. Subsequent ultrasonography of the thyroid revealed generally normal dimensions and parenchymal echogenicity. However, a large complex mass was detected, arising from the inferior pole of the thyroid and displacing the trachea toward the right (see Figure 3). According to the radiologist's notes, the mass contained both solid and cystic elements, scattered calcifications, and foci of flow on color Doppler. It measured about 6 cm in the largest (transverse) dimension. A 2.0-mm nodule was noted in the isthmus, slightly to the right of midline, consistent with multinodular goiter.
Following the cardiology consult, a diltiazem drip was initiated, but the patient was later optimized on flecainide for rhythm control and metoprolol for rate control. He was also initially anticoagulated using a heparin drip and bridged to warfarin, with target international normalized ratio (INR) between 2.0 and 3.0. Echocardiography revealed normal systolic function with ejection fraction of 55%, left ventricular hypertrophy, pulmonary artery systolic pressure of 35 mm Hg, and no pericardial effusions or valvular disease.
Regarding the patient's unexplained fever, results of chest imaging were negative for signs of pneumonia or atelectasis, which might have suggested a pulmonary cause. Urinalysis results were normal. Complete blood count showed no leukocytosis. The patient's fever subsided within 48 hours.
The differential diagnosis included Graves' disease, toxic multinodular goiter, Jod-Basedow syndrome, and subacute thyroiditis.
Graves' disease, an autoimmune disease with an unknown trigger, is the most common cause of hyperthyroidism. In affected patients, the thyroid gland overproduces thyroid hormones, leading to thyrotoxicosis. Thyrotoxicosis can result in multiple clinical signs and symptoms, including Graves' ophthalmopathy, pretibial myxedema, and goiter; TFT results typically include elevated T3 and T4 and low TSH.1-5 In the case patient (who had no history of thyroid disease, nor clinical signs or symptoms of Graves' disease), low uptake of iodine on thyroid scan precluded this diagnosis.
Toxic multinodular goiter, the second most common cause of hyperthyroidism, can be responsible for A-fib, tachycardia, and congestive heart failure.6,7 Iodine deficiency causes enlargement of the thyroid gland, where numerous nodules can develop, as seen in the case patient. These nodules can function independently, sometimes producing excess thyroid hormone; this leads to hyperplasia of the thyroid gland, resulting in a nontoxic multinodular goiter. From this goiter, a toxic multinodular goiter can emerge insidiously. However, in this condition, RAIU typically exceeds 30%; in the case patient, low 24-hour RAIU (2.8%) and the absence of functioning nodules on scanning made it possible to rule out this diagnosis.
Jod-Basedow syndrome refers to hyperthyroidism that develops as a result of administration of iodide, either as a dietary supplement or as IV contrast medium, or as an adverse effect of the antiarrhythmic drug amiodarone. This phenomenon is usually seen in a patient with endemic goiter.8-11 The relatively limited nature of the case patient's goiter and absence of a precipitating exposure to iodine made this diagnosis highly unlikely.
Subacute thyroiditis is a condition to which the patient's abnormal TFT results could reasonably be attributed. The patient had a substernal multinodular goiter that could not be palpated on physical examination, but it was visualized in the extended lower neck during thyroid scintigraphy.3 RAIU was minimal—a typical finding in this disorder,6 as TSH is suppressed by leakage of the excessive amounts of thyroid hormone. A tentative diagnosis of subacute thyroiditis was made.
As subacute thyroiditis is a self-limiting disorder, the patient was not started on any medications for hyperthyroidism but was advised to follow up with his primary care provider or an endocrinologist for repeat TFT and for fine-needle aspiration biopsy of the large thyroid nodule (a complex mass, containing cystic elements and calcifications, with a potential for malignancy) to rule out thyroid cancer.
Repeat ECG before discharge showed normal sinus rhythm with a ventricular rate of 74 beats/min. The patient was alert, awake, and oriented at discharge. He was continued on flecainide, metoprolol, and warfarin and advised to follow up with his primary care provider regarding his target INR.
DISCUSSION
The incidence of subacute thyroiditis, according to findings reported in 2003 from the Rochester Epidemiology Project in Olmsted County, Minnesota,12 is 12.1 cases per 100,000/year, with a higher incidence in women than men. It is most common in young adults and decreases with advancing age. Coxsackie virus, adenovirus, mumps, echovirus, influenza, and Epstein-Barr virus have been implicated in the disorder.12,13
Subacute thyroiditis is associated with a triphasic clinical course of hyperthyroidism, then hypothyroidism, then a return to normal thyroid function—as was seen in the case patient. Onset of subacute thyroiditis has been associated with recent viral infection, which may serve as a precipitant. The cause of this patient's high fever was never identified; thus, the etiology may have been viral.
The initial high thyroid hormone levels result from inflammation of thyroid tissue and release of preformed thyroid hormone into the circulation.6 At this point, TSH is suppressed and patients have very low RAIU, as was true in the case patient.
The condition is self-limiting and does not require treatment in the majority of patients, as TFT results return to normal levels within about two months.6 Patients can appear extremely ill due to thyrotoxicosis from subacute thyroiditis, but this usually lasts no longer than six to eight weeks.3 Subacute thyroiditis can be associated with atrial arrhythmia or heart failure.14,15
PATIENT OUTCOME
New-onset A-fib was attributed to the patient's thyrotoxicosis, which in turn was caused by subacute thyroiditis. He had a multinodular goiter, although he had not received any iodine supplements or IV contrast. As in most cases of subacute thyroiditis, no precipitating event was identified. However, given this patient's residence in a nursing facility and presentation with a high fever with no identifiable cause, a viral etiology for his subacute thyroiditis is possible.6
The patient's dementia may have been secondary to acute thyrotoxicosis, as his mental state improved during the hospital stay. His vitamin B12, folate, and A1C levels were within normal range. CT of the head showed multiple chronic infarcts and cerebral atrophy, and MRI of the brain indicated microvascular ischemic disease.
The patient was readmitted one month later for an episode of near-syncope (which, it was concluded, was a vasovagal episode). At that time, his TSH was found normal at 1.350 µIU/mL. Flecainide and metoprolol were discontinued; he was started on diltiazem for continued rate and rhythm control (as recommended by cardiology) and continued on warfarin.
CONCLUSION
In this case, subacute thyroiditis was most likely caused by a viral infection that led to destruction of the normal thyroid follicles and release of their preformed thyroid hormone into the circulation; this in turn led to sudden-onset A-fib. The diagnosis of subacute thyroiditis was suggested based on the abnormalities seen in this patient's TFT results, coupled with the suppressed RAIU—a typical finding in this disease.
Because subacute thyroiditis is a self-limiting condition, there is no role for antithyroid medication. Instead, treatment should be focused on relieving the patient's symptoms, such as ß-blockade or calcium channel blockers for tachycardia and corticosteroids or NSAIDs for neck pain.
REFERENCES
1. Weetman AP. Graves' disease. N Engl J Med. 2000;343(17):1236-1248.
2. Delgado Hurtado JJ, Pineda M. Images in medicine: Graves' disease. N Engl J Med. 2011; 364(20):1955.
3. Al-Sharif AA, Abujbara MA, Chiacchio S, et al. Contribution of radioiodine uptake measurement and thyroid scintigraphy to the differential diagnosis of thyrotoxicosis. Hell J Nucl Med. 2010;13(2):132-137.
4. Buccelletti F, Carroccia A, Marsiliani D, et al. Utility of routine thyroid-stimulating hormone determination in new-onset atrial fibrillation in the ED. Am J Emerg Med. 2011;29(9):1158-1162.
5. Ross DS. Radioiodine therapy for hyperthyroidism. N Engl J Med. 2011;364(6):542-550.
6. Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011;17(3):456-520.
7. Erickson D, Gharib H, Li H, van Heerden JA. Treatment of patients with toxic multinodular goiter. Thyroid. 1998;8(4):277-282.
8. Basaria S, Cooper DS. Amiodarone and the thyroid. Am J Med. 2005;118(7):706-714.
9. Bogazzi F, Bartalena L, Martino E. Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2010;95(6):2529-2535.
10. El-Shirbiny AM, Stavrou SS, Dnistrian A, et al. Jod-Basedow syndrome following oral iodine and radioiodinated-antibody administration. J Nucl Med. 1997;38(11):1816-1817.
11. Stanbury JB, Ermans AE, Bourdoux P, et al. Iodine-induced hyperthyroidism: occurrence and epidemiology. Thyroid. 1998;8(1):83-100.
12. Fatourechi V, Aniszewski JP, Fatourechi GZ, et al. Clinical features and outcome of subacute thyroiditis in an incidence cohort: Olmsted County, Minnesota, study. J Clin Endocrinol Metab. 2003;88(5):2100-2105.
13. Golden SH, Robinson KA, Saldanha I, et al. Clinical review: prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab. 2009;94(6):1853-1878.
14. Volpé R. The management of subacute (DeQuervain's) thyroiditis. Thyroid. 1993;3(3):253-255.
15. Lee SL. Subacute thyroiditis (2009). http://emedicine.medscape.com/article/125648-overview. Accessed April 17, 2012.