User login
Crunch Time
Before his recent promotion, hospitalist Garth King, MD, medical director of the Schumacher Group at Southwest Medical Center in Lafayette, La., had hoped to add a fourth full-time doctor to his roster. The hiring made sense at the time. As recently as last summer, the group’s three full-time doctors were averaging 35 to 40 patient encounters a day, enough to warrant the additional hospitalist position. But the group’s census dropped 25% in the fourth quarter of 2008, and these days, Dr. King simply can’t justify the math to bring on another six-figure salary.
“Over the past two, three months, patient slowdown has pushed off the ability” to add staff, Dr. King explains. “We’ll wait until things ramp up again, once we get the numbers to where they were before.”
Dr. King could be in for a long wait. The fiscal meltdown that began in 2007 and last year mushroomed into a full-blown recession has taken hold in the world of hospital medicine. More and more, hospitals are reporting decreased revenues and increased levels of charity care. The result: Planned group expansions have been put on hold, open positions are going unfilled, and some hospitalists have been laid off.
Constituencies from health-system executives to rank-and-file hospitalists to economists remain cautious of acting too boldly before President Obama and the new Congress unveil much-anticipated changes to Medicare billing and reimbursement schedules. Aggressive reforms, such as extending health coverage to more than 45 million uninsured Americans, could swamp hospitals with new patients and lead to cuts in reimbursement rates. A more temperate approach by the new administration could leave a relative status quo.
Either way, hospital medicine has never endured a recession so deep that some healthcare economists liken it to the Great Depression, so the practical effect on the industry is difficult to forecast with accuracy. Hospitalists and their observers agree on one thing, though: This is the year hospital medicine will have to prove its worth more empirically than ever. Positive public relations and studies proclaiming reduced lengths of stay and sped-up emergency department throughput have given the industry a “rarified position as a specialty,” one researcher says. But in constrictive economic times, those figures likely will be revisited, says Mark Pauly, professor of healthcare management at The Wharton School at the University of Pennsylvania. “When revenues are falling, you go back and look at that evidence again,” Pauly says. “Is it really bulletproof?”
Still, the prognosis for economic health is not all bad. Many hospital medicine leaders think the concerns over whether chief financial officers will look to hospital contracts as places to cut spending might spawn improved coding and billing, create new partnerships between hospital medicine groups, and push new revenue streams, such as preoperative clinics or inpatient palliative-care initiatives. This also is a time for hospital groups to reaffirm to their respective C-suites—through a deft combination of data and intangible relationships—that they are an indispensable staffing measure that their respective institutions cannot do without.
“This is an opportunity for hospitalists,” says Joe Miller, SHM’s executive advisor to the CEO. “The problem is we’ve got young, inexperienced leaders. Can they see this? Can they recognize this and not see this as a challenge?”
Problem Identification
Hospitalist Marc Westle, DO, FACP, president and managing partner of Asheville Hospital Group in North Carolina, thinks tracking, collating, and reporting quantifiable metrics is the fastest way to convince hospital executives that hospitalists are not the place to cut spending. And when those same executives are looking at staff reductions—53% of hospitals already are cutting or considering cuts, according to the most recent American Hospital Association data—hospital medicine leaders need to be able to point to specific numbers to prove their worth. Detailed information on coding, cost of capture, revenue production, and patient referrals generated are data points that can strengthen a presentation, especially if an argument shows that revenue production and collection is maximized.
“Don’t leave money on the table,” Dr. Westle says. “Your billing department is only going to bill what your physicians tell them to bill. For hospital groups that have not mentioned both the upfront E&M coding by their doctors and the back-end billing efficiency, those are definite things they need to do, today or yesterday. That efficiency may not have hurt them before, but it could hurt them in the next 12 to 18 months.”
Dr. Westle and others also note that hospitalists have to see the economic downturn through the eyes of hospital executives—and the hospital’s bottom line. Recent AHA data show 29% of hospitals are reporting moderate decreases in admissions, and another 9% of hospitals categorize those drops as significant. More than 3 in 10 hospitals have reported a noticeable reduction in elective procedures.
Pauly, the Wharton professor, also cautions that a tightened economy might force primary-care physicians (PCPs) back into hospitals, taking away patients now in the hospitalists’ purview. Hospital medicine’s beginnings trace to those PCPs acquiescing hospital rounds to a new intermediary—hospitalists—in return for the ability to focus more on their daily practices, Pauly says. Ancillary benefits included not being on call 24 hours a day, seven days a week.
“A lower income may change that willingness,” Pauly notes. “Leisure is a luxury good, and if your revenue is falling, you may want to get that business back.”
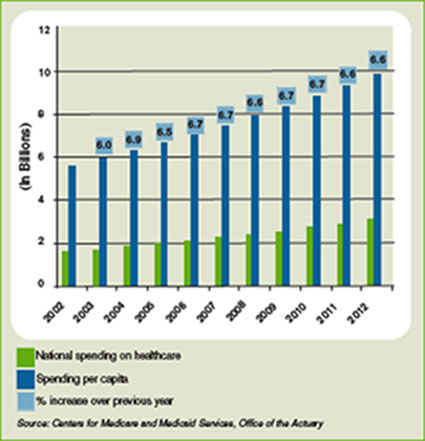
Another part of the equation is the rising tide of the uninsured, currently estimated at 47 million. It remains to be seen what effect the uninsured have on overall healthcare spending in the U.S., which is projected to rise 6.6% in 2009 to $2.6 trillion, according to the most recent data available from the Centers for Medicare and Medicaid Services (see Figure 1, below). Those projections, however, were released nearly a year ago, long before the severity of the current financial crisis became clear. Pauly says adjusted numbers released this winter, which assumedly will reflect the nation’s ever-deepening financial hole, should be a better gauge on whether healthcare remains as recession-proof as some think it is.
Hoangmai Pham, a senior health researcher at the Center for Studying Health System Change and lead author of a study on how the hospitalist model has focused attention on patient-care coordination, says changes at the federal level will be a driving factor in the strength of healthcare. Roughly 50% to 80% of a hospitalist’s annual salary comes from clinical billings, according to SHM data. The balance comes from hospital subsidies, in the form of annual contracts, monthly stipends, or pay-for-performance bonuses. Drastic changes in payment policy could have drastic implications on those subsidies.
“The ground is very fluid right now, in terms of where payment policy is going,” Pham says. “If I were hospitalists, part of the nervousness would be I’m not sure what direction things are heading in.”
Potential Solutions
Many hospitalists, however, see positives in the economic morass. Hospitalist Troy Ahlstrom, MD, is financial director of Hospitalists of Northwest Michigan in Traverse City, which serves roughly 400 beds at Munson Medical Center. He says groups can prove their worth by showing how they make it easier for other specialists—cardiologists, orthopedists, etc.—to perform the more lucrative procedures on which hospitals rely for higher reimbursements. Then, the group can negotiate for a piece of the savings under a pay-for-performance contracting model.
“What can we do to help make you more efficient, so you can do two extra surgical cases a week?” Dr. Ahlstrom says. “What if the hospital itself realizes a 15% increase in efficiency? Work out a deal that if we save you 15% … you give us a cut.”
Dr. Westle suggests analyzing cost accounting as another way to offer evidence of productivity. Paying overtime for back-office staff is ineffective if your salary overhead is greater than your billing collection. He also recommends a virtual office to employ billing specialists who work from home and doctors without off-site offices, eliminating real estate, utility, and infectious-waste-disposal costs that can cost private practices 55% to 60% in overhead costs.
Partnerships and reinvention are other avenues for cost-effectiveness. Some smaller, single-hospitalist groups might merge to cut costs through economies of scale, but SHM’s Miller thinks there is an opportunity to create a cooperative structure in which hospital medicine groups maintain individual identifies yet share certain functions, such as a common billing service.
Dr. King, the medical director who was promoted in November, is running a long-term acute-care center (LTAC) to help generate a parallel revenue stream. The center opened in May; during periods of decreased patient traffic at Southwest Medical Center, it affords his hospitalists another place to work, encountering patients and billing services. It also creates a referral stream for his hospital. He also has engaged in preliminary discussions for opening and staffing a pre-operative clinic or a wound-care clinic, but those discussions are in the early stages. King is sitting down with PCPs his group has little history with to increase referrals, and he is meeting with the hospital’s specialists to inform them that earlier consults with hospitalists could allow for streamlined service, shorter stays, and greater revenue.
“If you stop your research and development (R&D) during the hard times, 18 months from now, everyone else has stopped their R&D,” Dr. Westle says. “But if you continued your R&D, you’re 18 months ahead of everyone else.”
Dr. Westle refers to his method as “weaving yourself into the fabric of the health system.” Caring for the unassigned adult population and handling internal medicine assignments is low-hanging fruit for hospital medicine groups. He suggests creating programs to teach residents who are participating in hospital quality committees or other panels to deal with coding and billing, staffing LTACs, and improving the referral process for smaller, outlying hospitals to send patients to larger hospitals.
And, of course, there is relationship-building. Although tight economic times will require hospitalists to provide more quantitative data to prove their worth, the hospitalist model thrives on doctor-to-doctor interactions. Connections built with proceduralists and nurses, who can vouch for the value of a hospital medicine group, can mean a lot when hospital boards are searching for areas to trim costs.
“If you talk to seasoned directors of hospitalist programs, you win over hearts and minds one at a time,” Pham says. Most hospital executives “know there is value beyond that which can be proven on the balance sheet. It’s difficult to price things like convenience, satisfaction.”
What’s Next
Doug Cutler, MD, a hospitalist at Banner Sun City Hospitalists in Arizona, which serves 800-plus beds in two hospitals in Sun City and Sun City West, has watched economics change his situation. His old employer, Banner Health, recently merged its two hospitals with Sun Health. The new owners are learning how Cutler’s group works, talking to both hospitalists and other hospital staff. They have the group’s documentation to review, but individual doctors now have the opportunity to prove themselves as standouts.
“Find out the needs and service them the best you can,” Dr. Cutler says. “If it’s a throughput issue, work with them on that. Are you on committees? Are you on the quality committee? Pharmacy and therapeutics? Find what they need and fill a niche. Talk to medical directors, work with case managers. Don’t be the one that they hate to page; be the one they’re not worried about stopping in the hallways because you’re the go-to physician.”
SHM’s Miller agrees that individual hospitalists should take every opportunity to stand out. Whether it’s making sure hospital executives know your name, earning as many certifications as applicable, or applying for the society’s new Fellow in Hospital Medicine program, each doctor has to look at the economic crisis from a professional and personal viewpoint.
And while some could reason that Dr. Cutler’s situation—working for a hospital-owned group as opposed to a large multistate group or multispecialty practice—gives him more job security, he could argue the lack of negotiating leverage might give him less. But as he often tells his patients when they ask for a diagnosis: It’s hard to see 10 seconds into the future.
“I don’t want to think I’m sitting here fat, dumb, and happy and they won’t let me go,” Cutler says, knocking on wood. “I just don’t know. Every company … realistically needs to look at everything if they want to survive. We’re potentially on the chopping block, but do you lop off something that costs a million dollars when it saves you five? Are you willing to go back to a time before that?” TH
Richard Quinn is a freelance writer based in New Jersey.
Before his recent promotion, hospitalist Garth King, MD, medical director of the Schumacher Group at Southwest Medical Center in Lafayette, La., had hoped to add a fourth full-time doctor to his roster. The hiring made sense at the time. As recently as last summer, the group’s three full-time doctors were averaging 35 to 40 patient encounters a day, enough to warrant the additional hospitalist position. But the group’s census dropped 25% in the fourth quarter of 2008, and these days, Dr. King simply can’t justify the math to bring on another six-figure salary.
“Over the past two, three months, patient slowdown has pushed off the ability” to add staff, Dr. King explains. “We’ll wait until things ramp up again, once we get the numbers to where they were before.”
Dr. King could be in for a long wait. The fiscal meltdown that began in 2007 and last year mushroomed into a full-blown recession has taken hold in the world of hospital medicine. More and more, hospitals are reporting decreased revenues and increased levels of charity care. The result: Planned group expansions have been put on hold, open positions are going unfilled, and some hospitalists have been laid off.
Constituencies from health-system executives to rank-and-file hospitalists to economists remain cautious of acting too boldly before President Obama and the new Congress unveil much-anticipated changes to Medicare billing and reimbursement schedules. Aggressive reforms, such as extending health coverage to more than 45 million uninsured Americans, could swamp hospitals with new patients and lead to cuts in reimbursement rates. A more temperate approach by the new administration could leave a relative status quo.
Either way, hospital medicine has never endured a recession so deep that some healthcare economists liken it to the Great Depression, so the practical effect on the industry is difficult to forecast with accuracy. Hospitalists and their observers agree on one thing, though: This is the year hospital medicine will have to prove its worth more empirically than ever. Positive public relations and studies proclaiming reduced lengths of stay and sped-up emergency department throughput have given the industry a “rarified position as a specialty,” one researcher says. But in constrictive economic times, those figures likely will be revisited, says Mark Pauly, professor of healthcare management at The Wharton School at the University of Pennsylvania. “When revenues are falling, you go back and look at that evidence again,” Pauly says. “Is it really bulletproof?”
Still, the prognosis for economic health is not all bad. Many hospital medicine leaders think the concerns over whether chief financial officers will look to hospital contracts as places to cut spending might spawn improved coding and billing, create new partnerships between hospital medicine groups, and push new revenue streams, such as preoperative clinics or inpatient palliative-care initiatives. This also is a time for hospital groups to reaffirm to their respective C-suites—through a deft combination of data and intangible relationships—that they are an indispensable staffing measure that their respective institutions cannot do without.
“This is an opportunity for hospitalists,” says Joe Miller, SHM’s executive advisor to the CEO. “The problem is we’ve got young, inexperienced leaders. Can they see this? Can they recognize this and not see this as a challenge?”
Problem Identification
Hospitalist Marc Westle, DO, FACP, president and managing partner of Asheville Hospital Group in North Carolina, thinks tracking, collating, and reporting quantifiable metrics is the fastest way to convince hospital executives that hospitalists are not the place to cut spending. And when those same executives are looking at staff reductions—53% of hospitals already are cutting or considering cuts, according to the most recent American Hospital Association data—hospital medicine leaders need to be able to point to specific numbers to prove their worth. Detailed information on coding, cost of capture, revenue production, and patient referrals generated are data points that can strengthen a presentation, especially if an argument shows that revenue production and collection is maximized.
“Don’t leave money on the table,” Dr. Westle says. “Your billing department is only going to bill what your physicians tell them to bill. For hospital groups that have not mentioned both the upfront E&M coding by their doctors and the back-end billing efficiency, those are definite things they need to do, today or yesterday. That efficiency may not have hurt them before, but it could hurt them in the next 12 to 18 months.”
Dr. Westle and others also note that hospitalists have to see the economic downturn through the eyes of hospital executives—and the hospital’s bottom line. Recent AHA data show 29% of hospitals are reporting moderate decreases in admissions, and another 9% of hospitals categorize those drops as significant. More than 3 in 10 hospitals have reported a noticeable reduction in elective procedures.
Pauly, the Wharton professor, also cautions that a tightened economy might force primary-care physicians (PCPs) back into hospitals, taking away patients now in the hospitalists’ purview. Hospital medicine’s beginnings trace to those PCPs acquiescing hospital rounds to a new intermediary—hospitalists—in return for the ability to focus more on their daily practices, Pauly says. Ancillary benefits included not being on call 24 hours a day, seven days a week.
“A lower income may change that willingness,” Pauly notes. “Leisure is a luxury good, and if your revenue is falling, you may want to get that business back.”
Another part of the equation is the rising tide of the uninsured, currently estimated at 47 million. It remains to be seen what effect the uninsured have on overall healthcare spending in the U.S., which is projected to rise 6.6% in 2009 to $2.6 trillion, according to the most recent data available from the Centers for Medicare and Medicaid Services (see Figure 1, below). Those projections, however, were released nearly a year ago, long before the severity of the current financial crisis became clear. Pauly says adjusted numbers released this winter, which assumedly will reflect the nation’s ever-deepening financial hole, should be a better gauge on whether healthcare remains as recession-proof as some think it is.
Hoangmai Pham, a senior health researcher at the Center for Studying Health System Change and lead author of a study on how the hospitalist model has focused attention on patient-care coordination, says changes at the federal level will be a driving factor in the strength of healthcare. Roughly 50% to 80% of a hospitalist’s annual salary comes from clinical billings, according to SHM data. The balance comes from hospital subsidies, in the form of annual contracts, monthly stipends, or pay-for-performance bonuses. Drastic changes in payment policy could have drastic implications on those subsidies.
“The ground is very fluid right now, in terms of where payment policy is going,” Pham says. “If I were hospitalists, part of the nervousness would be I’m not sure what direction things are heading in.”
Potential Solutions
Many hospitalists, however, see positives in the economic morass. Hospitalist Troy Ahlstrom, MD, is financial director of Hospitalists of Northwest Michigan in Traverse City, which serves roughly 400 beds at Munson Medical Center. He says groups can prove their worth by showing how they make it easier for other specialists—cardiologists, orthopedists, etc.—to perform the more lucrative procedures on which hospitals rely for higher reimbursements. Then, the group can negotiate for a piece of the savings under a pay-for-performance contracting model.
“What can we do to help make you more efficient, so you can do two extra surgical cases a week?” Dr. Ahlstrom says. “What if the hospital itself realizes a 15% increase in efficiency? Work out a deal that if we save you 15% … you give us a cut.”
Dr. Westle suggests analyzing cost accounting as another way to offer evidence of productivity. Paying overtime for back-office staff is ineffective if your salary overhead is greater than your billing collection. He also recommends a virtual office to employ billing specialists who work from home and doctors without off-site offices, eliminating real estate, utility, and infectious-waste-disposal costs that can cost private practices 55% to 60% in overhead costs.
Partnerships and reinvention are other avenues for cost-effectiveness. Some smaller, single-hospitalist groups might merge to cut costs through economies of scale, but SHM’s Miller thinks there is an opportunity to create a cooperative structure in which hospital medicine groups maintain individual identifies yet share certain functions, such as a common billing service.
Dr. King, the medical director who was promoted in November, is running a long-term acute-care center (LTAC) to help generate a parallel revenue stream. The center opened in May; during periods of decreased patient traffic at Southwest Medical Center, it affords his hospitalists another place to work, encountering patients and billing services. It also creates a referral stream for his hospital. He also has engaged in preliminary discussions for opening and staffing a pre-operative clinic or a wound-care clinic, but those discussions are in the early stages. King is sitting down with PCPs his group has little history with to increase referrals, and he is meeting with the hospital’s specialists to inform them that earlier consults with hospitalists could allow for streamlined service, shorter stays, and greater revenue.
“If you stop your research and development (R&D) during the hard times, 18 months from now, everyone else has stopped their R&D,” Dr. Westle says. “But if you continued your R&D, you’re 18 months ahead of everyone else.”
Dr. Westle refers to his method as “weaving yourself into the fabric of the health system.” Caring for the unassigned adult population and handling internal medicine assignments is low-hanging fruit for hospital medicine groups. He suggests creating programs to teach residents who are participating in hospital quality committees or other panels to deal with coding and billing, staffing LTACs, and improving the referral process for smaller, outlying hospitals to send patients to larger hospitals.
And, of course, there is relationship-building. Although tight economic times will require hospitalists to provide more quantitative data to prove their worth, the hospitalist model thrives on doctor-to-doctor interactions. Connections built with proceduralists and nurses, who can vouch for the value of a hospital medicine group, can mean a lot when hospital boards are searching for areas to trim costs.
“If you talk to seasoned directors of hospitalist programs, you win over hearts and minds one at a time,” Pham says. Most hospital executives “know there is value beyond that which can be proven on the balance sheet. It’s difficult to price things like convenience, satisfaction.”
What’s Next
Doug Cutler, MD, a hospitalist at Banner Sun City Hospitalists in Arizona, which serves 800-plus beds in two hospitals in Sun City and Sun City West, has watched economics change his situation. His old employer, Banner Health, recently merged its two hospitals with Sun Health. The new owners are learning how Cutler’s group works, talking to both hospitalists and other hospital staff. They have the group’s documentation to review, but individual doctors now have the opportunity to prove themselves as standouts.
“Find out the needs and service them the best you can,” Dr. Cutler says. “If it’s a throughput issue, work with them on that. Are you on committees? Are you on the quality committee? Pharmacy and therapeutics? Find what they need and fill a niche. Talk to medical directors, work with case managers. Don’t be the one that they hate to page; be the one they’re not worried about stopping in the hallways because you’re the go-to physician.”
SHM’s Miller agrees that individual hospitalists should take every opportunity to stand out. Whether it’s making sure hospital executives know your name, earning as many certifications as applicable, or applying for the society’s new Fellow in Hospital Medicine program, each doctor has to look at the economic crisis from a professional and personal viewpoint.
And while some could reason that Dr. Cutler’s situation—working for a hospital-owned group as opposed to a large multistate group or multispecialty practice—gives him more job security, he could argue the lack of negotiating leverage might give him less. But as he often tells his patients when they ask for a diagnosis: It’s hard to see 10 seconds into the future.
“I don’t want to think I’m sitting here fat, dumb, and happy and they won’t let me go,” Cutler says, knocking on wood. “I just don’t know. Every company … realistically needs to look at everything if they want to survive. We’re potentially on the chopping block, but do you lop off something that costs a million dollars when it saves you five? Are you willing to go back to a time before that?” TH
Richard Quinn is a freelance writer based in New Jersey.
Before his recent promotion, hospitalist Garth King, MD, medical director of the Schumacher Group at Southwest Medical Center in Lafayette, La., had hoped to add a fourth full-time doctor to his roster. The hiring made sense at the time. As recently as last summer, the group’s three full-time doctors were averaging 35 to 40 patient encounters a day, enough to warrant the additional hospitalist position. But the group’s census dropped 25% in the fourth quarter of 2008, and these days, Dr. King simply can’t justify the math to bring on another six-figure salary.
“Over the past two, three months, patient slowdown has pushed off the ability” to add staff, Dr. King explains. “We’ll wait until things ramp up again, once we get the numbers to where they were before.”
Dr. King could be in for a long wait. The fiscal meltdown that began in 2007 and last year mushroomed into a full-blown recession has taken hold in the world of hospital medicine. More and more, hospitals are reporting decreased revenues and increased levels of charity care. The result: Planned group expansions have been put on hold, open positions are going unfilled, and some hospitalists have been laid off.
Constituencies from health-system executives to rank-and-file hospitalists to economists remain cautious of acting too boldly before President Obama and the new Congress unveil much-anticipated changes to Medicare billing and reimbursement schedules. Aggressive reforms, such as extending health coverage to more than 45 million uninsured Americans, could swamp hospitals with new patients and lead to cuts in reimbursement rates. A more temperate approach by the new administration could leave a relative status quo.
Either way, hospital medicine has never endured a recession so deep that some healthcare economists liken it to the Great Depression, so the practical effect on the industry is difficult to forecast with accuracy. Hospitalists and their observers agree on one thing, though: This is the year hospital medicine will have to prove its worth more empirically than ever. Positive public relations and studies proclaiming reduced lengths of stay and sped-up emergency department throughput have given the industry a “rarified position as a specialty,” one researcher says. But in constrictive economic times, those figures likely will be revisited, says Mark Pauly, professor of healthcare management at The Wharton School at the University of Pennsylvania. “When revenues are falling, you go back and look at that evidence again,” Pauly says. “Is it really bulletproof?”
Still, the prognosis for economic health is not all bad. Many hospital medicine leaders think the concerns over whether chief financial officers will look to hospital contracts as places to cut spending might spawn improved coding and billing, create new partnerships between hospital medicine groups, and push new revenue streams, such as preoperative clinics or inpatient palliative-care initiatives. This also is a time for hospital groups to reaffirm to their respective C-suites—through a deft combination of data and intangible relationships—that they are an indispensable staffing measure that their respective institutions cannot do without.
“This is an opportunity for hospitalists,” says Joe Miller, SHM’s executive advisor to the CEO. “The problem is we’ve got young, inexperienced leaders. Can they see this? Can they recognize this and not see this as a challenge?”
Problem Identification
Hospitalist Marc Westle, DO, FACP, president and managing partner of Asheville Hospital Group in North Carolina, thinks tracking, collating, and reporting quantifiable metrics is the fastest way to convince hospital executives that hospitalists are not the place to cut spending. And when those same executives are looking at staff reductions—53% of hospitals already are cutting or considering cuts, according to the most recent American Hospital Association data—hospital medicine leaders need to be able to point to specific numbers to prove their worth. Detailed information on coding, cost of capture, revenue production, and patient referrals generated are data points that can strengthen a presentation, especially if an argument shows that revenue production and collection is maximized.
“Don’t leave money on the table,” Dr. Westle says. “Your billing department is only going to bill what your physicians tell them to bill. For hospital groups that have not mentioned both the upfront E&M coding by their doctors and the back-end billing efficiency, those are definite things they need to do, today or yesterday. That efficiency may not have hurt them before, but it could hurt them in the next 12 to 18 months.”
Dr. Westle and others also note that hospitalists have to see the economic downturn through the eyes of hospital executives—and the hospital’s bottom line. Recent AHA data show 29% of hospitals are reporting moderate decreases in admissions, and another 9% of hospitals categorize those drops as significant. More than 3 in 10 hospitals have reported a noticeable reduction in elective procedures.
Pauly, the Wharton professor, also cautions that a tightened economy might force primary-care physicians (PCPs) back into hospitals, taking away patients now in the hospitalists’ purview. Hospital medicine’s beginnings trace to those PCPs acquiescing hospital rounds to a new intermediary—hospitalists—in return for the ability to focus more on their daily practices, Pauly says. Ancillary benefits included not being on call 24 hours a day, seven days a week.
“A lower income may change that willingness,” Pauly notes. “Leisure is a luxury good, and if your revenue is falling, you may want to get that business back.”
Another part of the equation is the rising tide of the uninsured, currently estimated at 47 million. It remains to be seen what effect the uninsured have on overall healthcare spending in the U.S., which is projected to rise 6.6% in 2009 to $2.6 trillion, according to the most recent data available from the Centers for Medicare and Medicaid Services (see Figure 1, below). Those projections, however, were released nearly a year ago, long before the severity of the current financial crisis became clear. Pauly says adjusted numbers released this winter, which assumedly will reflect the nation’s ever-deepening financial hole, should be a better gauge on whether healthcare remains as recession-proof as some think it is.
Hoangmai Pham, a senior health researcher at the Center for Studying Health System Change and lead author of a study on how the hospitalist model has focused attention on patient-care coordination, says changes at the federal level will be a driving factor in the strength of healthcare. Roughly 50% to 80% of a hospitalist’s annual salary comes from clinical billings, according to SHM data. The balance comes from hospital subsidies, in the form of annual contracts, monthly stipends, or pay-for-performance bonuses. Drastic changes in payment policy could have drastic implications on those subsidies.
“The ground is very fluid right now, in terms of where payment policy is going,” Pham says. “If I were hospitalists, part of the nervousness would be I’m not sure what direction things are heading in.”
Potential Solutions
Many hospitalists, however, see positives in the economic morass. Hospitalist Troy Ahlstrom, MD, is financial director of Hospitalists of Northwest Michigan in Traverse City, which serves roughly 400 beds at Munson Medical Center. He says groups can prove their worth by showing how they make it easier for other specialists—cardiologists, orthopedists, etc.—to perform the more lucrative procedures on which hospitals rely for higher reimbursements. Then, the group can negotiate for a piece of the savings under a pay-for-performance contracting model.
“What can we do to help make you more efficient, so you can do two extra surgical cases a week?” Dr. Ahlstrom says. “What if the hospital itself realizes a 15% increase in efficiency? Work out a deal that if we save you 15% … you give us a cut.”
Dr. Westle suggests analyzing cost accounting as another way to offer evidence of productivity. Paying overtime for back-office staff is ineffective if your salary overhead is greater than your billing collection. He also recommends a virtual office to employ billing specialists who work from home and doctors without off-site offices, eliminating real estate, utility, and infectious-waste-disposal costs that can cost private practices 55% to 60% in overhead costs.
Partnerships and reinvention are other avenues for cost-effectiveness. Some smaller, single-hospitalist groups might merge to cut costs through economies of scale, but SHM’s Miller thinks there is an opportunity to create a cooperative structure in which hospital medicine groups maintain individual identifies yet share certain functions, such as a common billing service.
Dr. King, the medical director who was promoted in November, is running a long-term acute-care center (LTAC) to help generate a parallel revenue stream. The center opened in May; during periods of decreased patient traffic at Southwest Medical Center, it affords his hospitalists another place to work, encountering patients and billing services. It also creates a referral stream for his hospital. He also has engaged in preliminary discussions for opening and staffing a pre-operative clinic or a wound-care clinic, but those discussions are in the early stages. King is sitting down with PCPs his group has little history with to increase referrals, and he is meeting with the hospital’s specialists to inform them that earlier consults with hospitalists could allow for streamlined service, shorter stays, and greater revenue.
“If you stop your research and development (R&D) during the hard times, 18 months from now, everyone else has stopped their R&D,” Dr. Westle says. “But if you continued your R&D, you’re 18 months ahead of everyone else.”
Dr. Westle refers to his method as “weaving yourself into the fabric of the health system.” Caring for the unassigned adult population and handling internal medicine assignments is low-hanging fruit for hospital medicine groups. He suggests creating programs to teach residents who are participating in hospital quality committees or other panels to deal with coding and billing, staffing LTACs, and improving the referral process for smaller, outlying hospitals to send patients to larger hospitals.
And, of course, there is relationship-building. Although tight economic times will require hospitalists to provide more quantitative data to prove their worth, the hospitalist model thrives on doctor-to-doctor interactions. Connections built with proceduralists and nurses, who can vouch for the value of a hospital medicine group, can mean a lot when hospital boards are searching for areas to trim costs.
“If you talk to seasoned directors of hospitalist programs, you win over hearts and minds one at a time,” Pham says. Most hospital executives “know there is value beyond that which can be proven on the balance sheet. It’s difficult to price things like convenience, satisfaction.”
What’s Next
Doug Cutler, MD, a hospitalist at Banner Sun City Hospitalists in Arizona, which serves 800-plus beds in two hospitals in Sun City and Sun City West, has watched economics change his situation. His old employer, Banner Health, recently merged its two hospitals with Sun Health. The new owners are learning how Cutler’s group works, talking to both hospitalists and other hospital staff. They have the group’s documentation to review, but individual doctors now have the opportunity to prove themselves as standouts.
“Find out the needs and service them the best you can,” Dr. Cutler says. “If it’s a throughput issue, work with them on that. Are you on committees? Are you on the quality committee? Pharmacy and therapeutics? Find what they need and fill a niche. Talk to medical directors, work with case managers. Don’t be the one that they hate to page; be the one they’re not worried about stopping in the hallways because you’re the go-to physician.”
SHM’s Miller agrees that individual hospitalists should take every opportunity to stand out. Whether it’s making sure hospital executives know your name, earning as many certifications as applicable, or applying for the society’s new Fellow in Hospital Medicine program, each doctor has to look at the economic crisis from a professional and personal viewpoint.
And while some could reason that Dr. Cutler’s situation—working for a hospital-owned group as opposed to a large multistate group or multispecialty practice—gives him more job security, he could argue the lack of negotiating leverage might give him less. But as he often tells his patients when they ask for a diagnosis: It’s hard to see 10 seconds into the future.
“I don’t want to think I’m sitting here fat, dumb, and happy and they won’t let me go,” Cutler says, knocking on wood. “I just don’t know. Every company … realistically needs to look at everything if they want to survive. We’re potentially on the chopping block, but do you lop off something that costs a million dollars when it saves you five? Are you willing to go back to a time before that?” TH
Richard Quinn is a freelance writer based in New Jersey.
System Overhaul
The global economy is on life support, unemployment is marching upward, wars rage on in Iraq and Afghanistan, and the federal deficit is approaching $1 trillion. By necessity, President Obama will push campaign promises to lower healthcare costs and provide affordable, accessible health insurance to all Americans to the end of his “to do” list, right?
Not necessarily.
“If we want to overcome our economic challenges, we must also finally address our healthcare challenge,” Obama said in a Dec. 11, 2008, speech in which he nominated former Sen. Tom Daschle (D-S.D.) to be his secretary of Health and Human Services and appointed him director of a new White House Office on Health Reform.
What this aggressive pursuit of healthcare change means for hospital medicine is still unclear, say health policy experts and hospitalists, because the Obama administration’s plan isn’t concrete and will change as it moves through Congress and the forums of public debate. Even so, some experts think an Obama healthcare overhaul would mean more revenue and information technology advancements for hospitals as well as significantly more patients as millions of newly insured Americans flood a system beset by a dwindling number of primary-care physicians.
For hospitalists and other physicians, the Obama plan could mean:
- Access to more information on what therapies work best for patients.
- A focus on preventative care.
- Greater emphasis on care-management programs and medical homes, especially for people with chronic conditions.
“He will lay out a bold vision on what he wants to do over time, and then he will enact it in several steps,” says Karen Davis, PhD, president of the Commonwealth Fund, a private healthcare research organization. “He’s certainly said it won’t be business as usual.”
Key Healthcare Officials in President Obama’s Administration

TOM DASCHLE
The secretary of Health and Human Services and director of a new White House Office on Health Reform is a former Democratic congressman and senator from South Dakota. He co-authored a book with Jeanne Lambrew this year, “Critical: What We Can Do About the Health-Care Crisis.” Obama has called the book “groundbreaking.” Prior to joining the Obama team, Daschle, 61, was a public policy adviser at Alston & Bird, a legal and lobbying firm. He remains a distinguished senior fellow at the Center for American Progress, where he’s pursued his interest in healthcare policy.

JEANNE LAMBREW
The deputy director of a new White House Office on Health Reform is an associate professor at the Lyndon B. Johnson School of Public Affairs at the University of Texas in Austin, and a senior fellow at the Center for American Progress. A co-author of the book Critical: What We Can Do About the Health-Care Crisis, Lambrew was a health policy advisor to President Clinton during his second term and helped establish the State Children’s Health Insurance Program.

PETER ORSZAG
The director of the Office of Management and Budget is a Princeton University graduate who has a Ph.D. from the London School of Economics. Prior to joining the Obama administration, Orszag, 39, directed the Con-gressional Budget Office and shifted a large part of its focus to healthcare issues. Orszag says the top fiscal threat facing the nation is escalating healthcare spending.
Sources: Obama-Biden transition Web site, www.change.gov; Center for American Progress; Congressional Quarterly
Right to Work
Obama says he will work immediately to expand eligibility for the State Children’s Health Insurance Program (SCHIP) and, in light of the recession, direct more federal money to states’ Medicaid programs, says Joseph Newhouse, PhD, a professor of health policy at Harvard University. Indeed, in the months before she was named deputy director of the White House’s new office on health reform, Jeanne Lambrew urged Congress to pass legislation that would boost federal funding for Medicaid and SCHIP.
Within the first few months of his administration, Obama also plans to push for investment in health information technology as a way to modernize the healthcare system and spur the economy, says Judy Feder, PhD, a professor and former dean of Georgetown University’s Public Policy Institute and a two-time Democratic congressional candidate who campaigned on a healthcare platform almost identical to the president’s.
Obama says he would like to direct $10 billion a year over the next five years to help the nation’s hospitals and healthcare providers install electronic billing and medical record systems.
“Somebody’s got to help set those up. We’ve got to buy computer systems and so forth. That’s an immediate boost to the economy…but it’s also laying the groundwork for reducing our healthcare costs over the long term,” Obama said in November upon naming Peter Orszag, an economist who regards rising healthcare spending as the nation’s top fiscal threat, director of the Office of Management and Budget.
Hospitals and hospitalists can benefit from IT advancements, but the technology should be slowly phased in to give users time to adjust, which may run counter to the quick economic stimulus Obama is trying to achieve, says David Meltzer, M.D., Ph.D., a professor in the department of medicine at the University of Chicago who has conducted considerable research in hospital medicine.
“The point is, health IT takes years to implement,” Dr. Meltzer says. “Just giving grants to buy and set up the equipment isn’t enough. You also want to give grants to prepare people on how to use it effectively.”
Dr. Meltzer is encouraged by Obama’s plan to create an independent, government-funded board charged with scientifically comparing the effectiveness of pharmaceutical drugs, medical devices, and procedures, and presenting the results to the medical community. He foresees hospitalists gaining opportunities to participate in clinical research as well as enroll patients in clinical trials.
“Over the long run, we’ll probably end up with therapies that will be better for patients and will control costs,” Dr. Meltzer says. “We spend a lot of money on things that don’t work or don’t work very well.”
Calling a comparative-effectiveness board “absolutely essential,” the Common-wealth Fund’s Davis says the U.S. has fallen far behind other countries in reviewing and rating therapies. Part of the reason is a fear that comparative effectiveness would stymie innovation and prevent doctors and patients from pursuing their choice of treatments, Dr. Meltzer says.
Opponents point to the book “Critical: What We Can Do About the Healthcare Crisis,” which Daschle and Lambrew co-wrote last year. In the book, Daschle advocates creating a federal health board outside the influence of Congress that would decide which procedures and therapies should be covered under public and private insurance plans. Obama has yet to support such a concept.
“There is that danger, but we live in an even more dangerous health system now,” says Dr. Meltzer, who predicts comparative-effectiveness legislation will advance this year. “I will be shocked and profoundly disappointed if we don’t see the legislation.”
Dr. Meltzer and other experts are less certain as to when Obama will move on other parts of his proposal, although Feder believes the president will try to create a national health-plan option and establish a national health insurance exchange, a kind of one-stop shop offering consumers health plans that would meet a minimum level of benefits, sometime in the next four years.
The national health-plan benefits could be similar to what federal employees receive, namely guaranteed health coverage and long-term care benefits, a wide variety of health plans to choose from, and insurability for pre-existing conditions. Private insurers would have to sell policies to everyone, regardless of pre-existing health conditions, and consumers who are unable to afford the premiums would be eligible for tax credits. The president’s plan stops short of requiring all Americans to have health insurance.
System Overload?
With more Americans insured, hospitals’ revenues will increase, according to Davis. Hospital patient loads—and hospitalists’ workloads—would increase, says Iris Mangulabnan, MD, a hospitalist at Covenant HealthCare in Saginaw, Mich.
“In the global scheme of things, if (Obama) is going to have insurance for about 45 million more patients, you’re going to see hospitals crammed with more people,” Dr. Mangulabnan says.
Adam Singer, MD, CEO of IPC: The Hospitalist Company, a national physician group practice based in California, says Obama’s plan has the potential to “overwhelm” the U.S. healthcare system. “Who’s going to take care of all these people?” he says.
Obama’s healthcare plan highlights preventive-care and disease management programs as ways to keep people out of hospitals and save money, but Dr. Mangulabnan says research has shown such initiatives aren’t always effective. “They hold a lot of conceptual promise, but I’m reminded of that fast-food commercial—you know, ‘Where’s the beef?’ ” Dr. Singer says.
Both doctors question how Obama’s healthcare plan, which would cost an estimated $75 billion a year when fully implemented, would be paid for. During his campaign, Obama talked about letting tax cuts expire for people making more than $250,000 a year and using that money for healthcare. But the economic crisis has forced the president to reconsider ending the tax cuts.
Cost is just one obstacle to Obama’s plan. Experts say the list also includes health insurers, pharmaceutical and medical product companies, doctors, congressional Republicans, an agenda full of other pressing problems, and change.
“It’s very difficult for a multitrillion-dollar industry to see the ground shift beneath it. It’s the known versus the unknown,” Davis says. “But I don’t see the economy as an obstacle. If anything, it increases the chance that healthcare will be addressed, because more people are being affected by problems in the system. The main thing that’s driving all of this is a feeling that it’s time.” TH
Lisa M. Ryan is a freelance writer based in New Jersey.
The global economy is on life support, unemployment is marching upward, wars rage on in Iraq and Afghanistan, and the federal deficit is approaching $1 trillion. By necessity, President Obama will push campaign promises to lower healthcare costs and provide affordable, accessible health insurance to all Americans to the end of his “to do” list, right?
Not necessarily.
“If we want to overcome our economic challenges, we must also finally address our healthcare challenge,” Obama said in a Dec. 11, 2008, speech in which he nominated former Sen. Tom Daschle (D-S.D.) to be his secretary of Health and Human Services and appointed him director of a new White House Office on Health Reform.
What this aggressive pursuit of healthcare change means for hospital medicine is still unclear, say health policy experts and hospitalists, because the Obama administration’s plan isn’t concrete and will change as it moves through Congress and the forums of public debate. Even so, some experts think an Obama healthcare overhaul would mean more revenue and information technology advancements for hospitals as well as significantly more patients as millions of newly insured Americans flood a system beset by a dwindling number of primary-care physicians.
For hospitalists and other physicians, the Obama plan could mean:
- Access to more information on what therapies work best for patients.
- A focus on preventative care.
- Greater emphasis on care-management programs and medical homes, especially for people with chronic conditions.
“He will lay out a bold vision on what he wants to do over time, and then he will enact it in several steps,” says Karen Davis, PhD, president of the Commonwealth Fund, a private healthcare research organization. “He’s certainly said it won’t be business as usual.”
Key Healthcare Officials in President Obama’s Administration
TOM DASCHLE
The secretary of Health and Human Services and director of a new White House Office on Health Reform is a former Democratic congressman and senator from South Dakota. He co-authored a book with Jeanne Lambrew this year, “Critical: What We Can Do About the Health-Care Crisis.” Obama has called the book “groundbreaking.” Prior to joining the Obama team, Daschle, 61, was a public policy adviser at Alston & Bird, a legal and lobbying firm. He remains a distinguished senior fellow at the Center for American Progress, where he’s pursued his interest in healthcare policy.
JEANNE LAMBREW
The deputy director of a new White House Office on Health Reform is an associate professor at the Lyndon B. Johnson School of Public Affairs at the University of Texas in Austin, and a senior fellow at the Center for American Progress. A co-author of the book Critical: What We Can Do About the Health-Care Crisis, Lambrew was a health policy advisor to President Clinton during his second term and helped establish the State Children’s Health Insurance Program.
PETER ORSZAG
The director of the Office of Management and Budget is a Princeton University graduate who has a Ph.D. from the London School of Economics. Prior to joining the Obama administration, Orszag, 39, directed the Con-gressional Budget Office and shifted a large part of its focus to healthcare issues. Orszag says the top fiscal threat facing the nation is escalating healthcare spending.
Sources: Obama-Biden transition Web site, www.change.gov; Center for American Progress; Congressional Quarterly
Right to Work
Obama says he will work immediately to expand eligibility for the State Children’s Health Insurance Program (SCHIP) and, in light of the recession, direct more federal money to states’ Medicaid programs, says Joseph Newhouse, PhD, a professor of health policy at Harvard University. Indeed, in the months before she was named deputy director of the White House’s new office on health reform, Jeanne Lambrew urged Congress to pass legislation that would boost federal funding for Medicaid and SCHIP.
Within the first few months of his administration, Obama also plans to push for investment in health information technology as a way to modernize the healthcare system and spur the economy, says Judy Feder, PhD, a professor and former dean of Georgetown University’s Public Policy Institute and a two-time Democratic congressional candidate who campaigned on a healthcare platform almost identical to the president’s.
Obama says he would like to direct $10 billion a year over the next five years to help the nation’s hospitals and healthcare providers install electronic billing and medical record systems.
“Somebody’s got to help set those up. We’ve got to buy computer systems and so forth. That’s an immediate boost to the economy…but it’s also laying the groundwork for reducing our healthcare costs over the long term,” Obama said in November upon naming Peter Orszag, an economist who regards rising healthcare spending as the nation’s top fiscal threat, director of the Office of Management and Budget.
Hospitals and hospitalists can benefit from IT advancements, but the technology should be slowly phased in to give users time to adjust, which may run counter to the quick economic stimulus Obama is trying to achieve, says David Meltzer, M.D., Ph.D., a professor in the department of medicine at the University of Chicago who has conducted considerable research in hospital medicine.
“The point is, health IT takes years to implement,” Dr. Meltzer says. “Just giving grants to buy and set up the equipment isn’t enough. You also want to give grants to prepare people on how to use it effectively.”
Dr. Meltzer is encouraged by Obama’s plan to create an independent, government-funded board charged with scientifically comparing the effectiveness of pharmaceutical drugs, medical devices, and procedures, and presenting the results to the medical community. He foresees hospitalists gaining opportunities to participate in clinical research as well as enroll patients in clinical trials.
“Over the long run, we’ll probably end up with therapies that will be better for patients and will control costs,” Dr. Meltzer says. “We spend a lot of money on things that don’t work or don’t work very well.”
Calling a comparative-effectiveness board “absolutely essential,” the Common-wealth Fund’s Davis says the U.S. has fallen far behind other countries in reviewing and rating therapies. Part of the reason is a fear that comparative effectiveness would stymie innovation and prevent doctors and patients from pursuing their choice of treatments, Dr. Meltzer says.
Opponents point to the book “Critical: What We Can Do About the Healthcare Crisis,” which Daschle and Lambrew co-wrote last year. In the book, Daschle advocates creating a federal health board outside the influence of Congress that would decide which procedures and therapies should be covered under public and private insurance plans. Obama has yet to support such a concept.
“There is that danger, but we live in an even more dangerous health system now,” says Dr. Meltzer, who predicts comparative-effectiveness legislation will advance this year. “I will be shocked and profoundly disappointed if we don’t see the legislation.”
Dr. Meltzer and other experts are less certain as to when Obama will move on other parts of his proposal, although Feder believes the president will try to create a national health-plan option and establish a national health insurance exchange, a kind of one-stop shop offering consumers health plans that would meet a minimum level of benefits, sometime in the next four years.
The national health-plan benefits could be similar to what federal employees receive, namely guaranteed health coverage and long-term care benefits, a wide variety of health plans to choose from, and insurability for pre-existing conditions. Private insurers would have to sell policies to everyone, regardless of pre-existing health conditions, and consumers who are unable to afford the premiums would be eligible for tax credits. The president’s plan stops short of requiring all Americans to have health insurance.
System Overload?
With more Americans insured, hospitals’ revenues will increase, according to Davis. Hospital patient loads—and hospitalists’ workloads—would increase, says Iris Mangulabnan, MD, a hospitalist at Covenant HealthCare in Saginaw, Mich.
“In the global scheme of things, if (Obama) is going to have insurance for about 45 million more patients, you’re going to see hospitals crammed with more people,” Dr. Mangulabnan says.
Adam Singer, MD, CEO of IPC: The Hospitalist Company, a national physician group practice based in California, says Obama’s plan has the potential to “overwhelm” the U.S. healthcare system. “Who’s going to take care of all these people?” he says.
Obama’s healthcare plan highlights preventive-care and disease management programs as ways to keep people out of hospitals and save money, but Dr. Mangulabnan says research has shown such initiatives aren’t always effective. “They hold a lot of conceptual promise, but I’m reminded of that fast-food commercial—you know, ‘Where’s the beef?’ ” Dr. Singer says.
Both doctors question how Obama’s healthcare plan, which would cost an estimated $75 billion a year when fully implemented, would be paid for. During his campaign, Obama talked about letting tax cuts expire for people making more than $250,000 a year and using that money for healthcare. But the economic crisis has forced the president to reconsider ending the tax cuts.
Cost is just one obstacle to Obama’s plan. Experts say the list also includes health insurers, pharmaceutical and medical product companies, doctors, congressional Republicans, an agenda full of other pressing problems, and change.
“It’s very difficult for a multitrillion-dollar industry to see the ground shift beneath it. It’s the known versus the unknown,” Davis says. “But I don’t see the economy as an obstacle. If anything, it increases the chance that healthcare will be addressed, because more people are being affected by problems in the system. The main thing that’s driving all of this is a feeling that it’s time.” TH
Lisa M. Ryan is a freelance writer based in New Jersey.
The global economy is on life support, unemployment is marching upward, wars rage on in Iraq and Afghanistan, and the federal deficit is approaching $1 trillion. By necessity, President Obama will push campaign promises to lower healthcare costs and provide affordable, accessible health insurance to all Americans to the end of his “to do” list, right?
Not necessarily.
“If we want to overcome our economic challenges, we must also finally address our healthcare challenge,” Obama said in a Dec. 11, 2008, speech in which he nominated former Sen. Tom Daschle (D-S.D.) to be his secretary of Health and Human Services and appointed him director of a new White House Office on Health Reform.
What this aggressive pursuit of healthcare change means for hospital medicine is still unclear, say health policy experts and hospitalists, because the Obama administration’s plan isn’t concrete and will change as it moves through Congress and the forums of public debate. Even so, some experts think an Obama healthcare overhaul would mean more revenue and information technology advancements for hospitals as well as significantly more patients as millions of newly insured Americans flood a system beset by a dwindling number of primary-care physicians.
For hospitalists and other physicians, the Obama plan could mean:
- Access to more information on what therapies work best for patients.
- A focus on preventative care.
- Greater emphasis on care-management programs and medical homes, especially for people with chronic conditions.
“He will lay out a bold vision on what he wants to do over time, and then he will enact it in several steps,” says Karen Davis, PhD, president of the Commonwealth Fund, a private healthcare research organization. “He’s certainly said it won’t be business as usual.”
Key Healthcare Officials in President Obama’s Administration
TOM DASCHLE
The secretary of Health and Human Services and director of a new White House Office on Health Reform is a former Democratic congressman and senator from South Dakota. He co-authored a book with Jeanne Lambrew this year, “Critical: What We Can Do About the Health-Care Crisis.” Obama has called the book “groundbreaking.” Prior to joining the Obama team, Daschle, 61, was a public policy adviser at Alston & Bird, a legal and lobbying firm. He remains a distinguished senior fellow at the Center for American Progress, where he’s pursued his interest in healthcare policy.
JEANNE LAMBREW
The deputy director of a new White House Office on Health Reform is an associate professor at the Lyndon B. Johnson School of Public Affairs at the University of Texas in Austin, and a senior fellow at the Center for American Progress. A co-author of the book Critical: What We Can Do About the Health-Care Crisis, Lambrew was a health policy advisor to President Clinton during his second term and helped establish the State Children’s Health Insurance Program.
PETER ORSZAG
The director of the Office of Management and Budget is a Princeton University graduate who has a Ph.D. from the London School of Economics. Prior to joining the Obama administration, Orszag, 39, directed the Con-gressional Budget Office and shifted a large part of its focus to healthcare issues. Orszag says the top fiscal threat facing the nation is escalating healthcare spending.
Sources: Obama-Biden transition Web site, www.change.gov; Center for American Progress; Congressional Quarterly
Right to Work
Obama says he will work immediately to expand eligibility for the State Children’s Health Insurance Program (SCHIP) and, in light of the recession, direct more federal money to states’ Medicaid programs, says Joseph Newhouse, PhD, a professor of health policy at Harvard University. Indeed, in the months before she was named deputy director of the White House’s new office on health reform, Jeanne Lambrew urged Congress to pass legislation that would boost federal funding for Medicaid and SCHIP.
Within the first few months of his administration, Obama also plans to push for investment in health information technology as a way to modernize the healthcare system and spur the economy, says Judy Feder, PhD, a professor and former dean of Georgetown University’s Public Policy Institute and a two-time Democratic congressional candidate who campaigned on a healthcare platform almost identical to the president’s.
Obama says he would like to direct $10 billion a year over the next five years to help the nation’s hospitals and healthcare providers install electronic billing and medical record systems.
“Somebody’s got to help set those up. We’ve got to buy computer systems and so forth. That’s an immediate boost to the economy…but it’s also laying the groundwork for reducing our healthcare costs over the long term,” Obama said in November upon naming Peter Orszag, an economist who regards rising healthcare spending as the nation’s top fiscal threat, director of the Office of Management and Budget.
Hospitals and hospitalists can benefit from IT advancements, but the technology should be slowly phased in to give users time to adjust, which may run counter to the quick economic stimulus Obama is trying to achieve, says David Meltzer, M.D., Ph.D., a professor in the department of medicine at the University of Chicago who has conducted considerable research in hospital medicine.
“The point is, health IT takes years to implement,” Dr. Meltzer says. “Just giving grants to buy and set up the equipment isn’t enough. You also want to give grants to prepare people on how to use it effectively.”
Dr. Meltzer is encouraged by Obama’s plan to create an independent, government-funded board charged with scientifically comparing the effectiveness of pharmaceutical drugs, medical devices, and procedures, and presenting the results to the medical community. He foresees hospitalists gaining opportunities to participate in clinical research as well as enroll patients in clinical trials.
“Over the long run, we’ll probably end up with therapies that will be better for patients and will control costs,” Dr. Meltzer says. “We spend a lot of money on things that don’t work or don’t work very well.”
Calling a comparative-effectiveness board “absolutely essential,” the Common-wealth Fund’s Davis says the U.S. has fallen far behind other countries in reviewing and rating therapies. Part of the reason is a fear that comparative effectiveness would stymie innovation and prevent doctors and patients from pursuing their choice of treatments, Dr. Meltzer says.
Opponents point to the book “Critical: What We Can Do About the Healthcare Crisis,” which Daschle and Lambrew co-wrote last year. In the book, Daschle advocates creating a federal health board outside the influence of Congress that would decide which procedures and therapies should be covered under public and private insurance plans. Obama has yet to support such a concept.
“There is that danger, but we live in an even more dangerous health system now,” says Dr. Meltzer, who predicts comparative-effectiveness legislation will advance this year. “I will be shocked and profoundly disappointed if we don’t see the legislation.”
Dr. Meltzer and other experts are less certain as to when Obama will move on other parts of his proposal, although Feder believes the president will try to create a national health-plan option and establish a national health insurance exchange, a kind of one-stop shop offering consumers health plans that would meet a minimum level of benefits, sometime in the next four years.
The national health-plan benefits could be similar to what federal employees receive, namely guaranteed health coverage and long-term care benefits, a wide variety of health plans to choose from, and insurability for pre-existing conditions. Private insurers would have to sell policies to everyone, regardless of pre-existing health conditions, and consumers who are unable to afford the premiums would be eligible for tax credits. The president’s plan stops short of requiring all Americans to have health insurance.
System Overload?
With more Americans insured, hospitals’ revenues will increase, according to Davis. Hospital patient loads—and hospitalists’ workloads—would increase, says Iris Mangulabnan, MD, a hospitalist at Covenant HealthCare in Saginaw, Mich.
“In the global scheme of things, if (Obama) is going to have insurance for about 45 million more patients, you’re going to see hospitals crammed with more people,” Dr. Mangulabnan says.
Adam Singer, MD, CEO of IPC: The Hospitalist Company, a national physician group practice based in California, says Obama’s plan has the potential to “overwhelm” the U.S. healthcare system. “Who’s going to take care of all these people?” he says.
Obama’s healthcare plan highlights preventive-care and disease management programs as ways to keep people out of hospitals and save money, but Dr. Mangulabnan says research has shown such initiatives aren’t always effective. “They hold a lot of conceptual promise, but I’m reminded of that fast-food commercial—you know, ‘Where’s the beef?’ ” Dr. Singer says.
Both doctors question how Obama’s healthcare plan, which would cost an estimated $75 billion a year when fully implemented, would be paid for. During his campaign, Obama talked about letting tax cuts expire for people making more than $250,000 a year and using that money for healthcare. But the economic crisis has forced the president to reconsider ending the tax cuts.
Cost is just one obstacle to Obama’s plan. Experts say the list also includes health insurers, pharmaceutical and medical product companies, doctors, congressional Republicans, an agenda full of other pressing problems, and change.
“It’s very difficult for a multitrillion-dollar industry to see the ground shift beneath it. It’s the known versus the unknown,” Davis says. “But I don’t see the economy as an obstacle. If anything, it increases the chance that healthcare will be addressed, because more people are being affected by problems in the system. The main thing that’s driving all of this is a feeling that it’s time.” TH
Lisa M. Ryan is a freelance writer based in New Jersey.
Perioperative Medicine Summit 2009
Summit Director:
Amir K. Jaffer, MD
Contents
Abstract 1: Pulmonary hypertension is an important predictor of perioperative outcomes in patients undergoing noncardiac surgery
Roop Kaw, MD; Esteban Walker, PhD; Vinay Pasupuleti, MD, PhD; Abhishek Deshpande, MD, PhD; Tarek Hamieh, MD; and Omar A. Minai, MD
Abstract 2: Analysis of administrative practices and residency training curricula in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 3: Is percent body fat a better predictor of surgical site infection risk than body mass index?
Emily Waisbren, BS; Angela M. Bader, MD, MPH; Heather Rosen, MD, MPH; Selwyn O. Rogers, Jr., MD, MPH; and Elof Eriksson, MD, PhD
Abstract 4: A nomogram for prediction of survival for patients undergoing elective major noncardiac surgery
Y. Olivia Xu-Cai, MD; and Michael W. Kattan, PhD
Abstract 5: Sustainability of an osteoporosis pathway
Catherine Gibb, MBBS, FRACP; Christopher Butcher, FRACS; Lesley Thomas, BNsg; and Jennifer Pink, BPharm
Abstract 6: Length of hospital stay is predicted by comorbidities
Catherine Gibb, MBBS, FRACP; and Professor Villis Marshall, FRACS
Abstract 7: Generalization of the POISE and Mangano studies on beta-blocker use in the perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; and Luc Lanthier, MD, MSc
Abstract 8: Impact of antihypertensive medication on perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; Danielle Pilon, MD, MSc; and Luc Lanthier, MD, MSc
Abstract 9: An analysis of preoperative testing protocols in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 10: Preoperative biomarkers of inflammation, ischemia, and heart failure and outcomes of vascular surgery
Matthew Griffee, MD; Ansgar Brambrink, MD, PhD; and Thomas Barrett, MD
Abstract 11: Alcohol-related predictors of postoperative delirium in major head and neck cancer surgery
Harrison Weed, MD; Summit Shah, BS; Xin He, PhD; Amit Agrawal, MD; Enver Ozer, MD; and David E. Schuller, MD
Abstract 12: Intraoperative coagulopathy: A low-volume treatment protocol that completely replaces fresh frozen plasma
Peter Kallas, MD; Mary Lou Green, MHS; and Anjali Desai, MD
Abstract 13: Is the Berlin Questionnaire an effective screening tool for obstructive sleep apnea in the preoperative total joint replacement population?
Peter Kallas, MD; Mark Schumacher; Mona Lazar, DO; and Anjali Desai, MD
Abstract 14: The impact of preoperative medical optimization on head and neck cancer surgery
Christopher Tan, MBBS; Catherine Gibb, MBBS, FRACP; and Suren Krishnan, MBBS, FRACS
Abstract 15: Reconceptualizing the preoperative process
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 16: Development of an electronic medical record smart set form to increase standardization, consistency, and compliance with ACC/AHA perioperative guidelines
Anitha Rajamanickam, MD; Ali Usmani, MD; Ajay Kumar, MD; and Brian Harte, MD
Abstract 17: Development of a perioperative electronic medical record research and quality improvement database
Anitha Rajamanickam, MD; Ali Usmani, MD; Feza Remzi, MD; Brian Harte, MD; and Ajay Kumar, MD
Abstract 18: An innovative perioperative/consultative curriculum for third-year internal medicine residents
Alex Rico, MD; Joshua Lenchus, DO; and Amir Jaffer, MD
Abstract 19: Preoperative medicine infobutton
Terrence J. Adam, MD, PhD
Abstract 20: Nurse practitioners: Bridging the gap in perioperative care
Sally Morgan, RN, MS, ANP-BC, ACNS-BC; and Angela Wright, RN, MSN, APRN, BC
Abstract 21: Intubation training of deploying far forward combat medical personnel with the video laryngoscope
Ben Boedeker, MD; Mary Barak-Bernhagen, BS; Kirsten Boedeker; and W. Bosseau Murray, MD
Abstract 22: The establishment of a perioperative skin integrity committee
Jeanne Lanchester, RN, MEd; Ann Leary, BSN, RNC; and Susan Vargas, AD, RN
Abstract 23: Development and implementation of a perianesthesia integrative care committee
Jeanne Lanchester, RN, MEd; Jeanette Cote, BWN, RN; Terri Jamros, RN; Charla Delillo, RN; Sherie Lavoie, BSN, RN; Jennifer Therminos, SN; Joan Compagnone, RN; and Nicole Engel, MSN, RN
Abstract 24: Development of a screening system to identify patients preoperatively who may benefit from a postoperative hospitalist consult
Elizabeth Marlow, MD, MA; and Chad Whelan, MD
Abstract 25: An algorithm for preoperative screening and management of sleep apnea: Have we created a monster?
Deborah C. Richman, MBChB, FFA(SA); Jorge M. Mallea, MD; Paul S. Richman, MD; and Pater S.A. Glass, MBChB
Abstract 26: Constructing a collaborative neuroscience hospitalist program
Rachel Thompson, MD; Christy Gilmore, MD; Kamal Ajam, MD; and Jennifer Thompson, MD
Abstract 27: The development of algorithms for preoperative management of antiplatelet and anticoagulation therapy in patients undergoing surgical or invasive procedures
Catherine McGowan, MSN, and Patricia Kidik, MSN
Abstract 28: Surgeon-initiated preoperative screening: A new approach
Christina Johnson, RN, PA-C; and Edward J. denBraven, CRNA
Abstract 29: A new process for ensuring the safety of patients having anesthesia outside of the operating room
Ellen Leary, MSN; Catherine McGowan, MSN; Kathleen McGrath, MSN; Sheila McCabe Hassan, MSN; and Theresa Kennedy, MSN
Abstract 30: Establishing a virtual preoperative evaluation clinic
Corey Zetterman, MD; Bobbie J. Sweitzer, MD; and Ben H. Boedeker, MD
Abstract 31: Perioperative hypoxemia and rhabdomyolysis in a medically complicated patient
Sarah Bodin, MD
Abstract 32: How soon is too soon? General anesthesia after coronary intervention with bare metal stents
Meghan Tadel, MD
Abstract 33: Can patients with critical aortic stenosis undergo noncardiac surgery without intervening aortic valve replacement?
M. Chadi Alraies, MD; Abdul Alraiyes, MD; Anitha Rajamanickam, MD; and Frank Michota, MD
Abstract 34: Is it safe to operate on cocaine-positive patients?
M. Chadi Alraies, MD; Abdul Hamid Alraiyes, MD; and Brian Harte, MD
Abstract 35: To intensive care or not?
Mona Lazar, DO; and Peter Kallas, MD
Abstract 36: Predicting surgical complications from liver disease
Mona Lazar, DO, and Peter Kallas, MD
Abstract 37: Preoperative coronary angiography: Friend or foe?
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 38: Heparin-induced thrombocytopenia with low molecular weight heparin after total knee replacement
Steven Cohn, MD
Abstract 39: Patient with Parkinson’s disease treated with implanted deep brain stimulators for laparotomy
Deborah C. Richman, MBChB, FFA(SA); Daryn H. Moller, MD; and Khoa N. Nguyen, MD
Abstract 40: Ethical dilemma in the preoperative assessment clinic: Can a patient refuse an indicated cardiac workup? Can we refuse to anesthetize?
Deborah C. Richman, MBChB, FFA(SA)
Abstract 41: Coronary artery bypass grafting as a precipitatin factor in diabetic ketoacidosis in type 2 diabetes
Vishal Sehgral, MD, and Abbas Kitabchi, MD
Summit Director:
Amir K. Jaffer, MD
Contents
Abstract 1: Pulmonary hypertension is an important predictor of perioperative outcomes in patients undergoing noncardiac surgery
Roop Kaw, MD; Esteban Walker, PhD; Vinay Pasupuleti, MD, PhD; Abhishek Deshpande, MD, PhD; Tarek Hamieh, MD; and Omar A. Minai, MD
Abstract 2: Analysis of administrative practices and residency training curricula in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 3: Is percent body fat a better predictor of surgical site infection risk than body mass index?
Emily Waisbren, BS; Angela M. Bader, MD, MPH; Heather Rosen, MD, MPH; Selwyn O. Rogers, Jr., MD, MPH; and Elof Eriksson, MD, PhD
Abstract 4: A nomogram for prediction of survival for patients undergoing elective major noncardiac surgery
Y. Olivia Xu-Cai, MD; and Michael W. Kattan, PhD
Abstract 5: Sustainability of an osteoporosis pathway
Catherine Gibb, MBBS, FRACP; Christopher Butcher, FRACS; Lesley Thomas, BNsg; and Jennifer Pink, BPharm
Abstract 6: Length of hospital stay is predicted by comorbidities
Catherine Gibb, MBBS, FRACP; and Professor Villis Marshall, FRACS
Abstract 7: Generalization of the POISE and Mangano studies on beta-blocker use in the perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; and Luc Lanthier, MD, MSc
Abstract 8: Impact of antihypertensive medication on perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; Danielle Pilon, MD, MSc; and Luc Lanthier, MD, MSc
Abstract 9: An analysis of preoperative testing protocols in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 10: Preoperative biomarkers of inflammation, ischemia, and heart failure and outcomes of vascular surgery
Matthew Griffee, MD; Ansgar Brambrink, MD, PhD; and Thomas Barrett, MD
Abstract 11: Alcohol-related predictors of postoperative delirium in major head and neck cancer surgery
Harrison Weed, MD; Summit Shah, BS; Xin He, PhD; Amit Agrawal, MD; Enver Ozer, MD; and David E. Schuller, MD
Abstract 12: Intraoperative coagulopathy: A low-volume treatment protocol that completely replaces fresh frozen plasma
Peter Kallas, MD; Mary Lou Green, MHS; and Anjali Desai, MD
Abstract 13: Is the Berlin Questionnaire an effective screening tool for obstructive sleep apnea in the preoperative total joint replacement population?
Peter Kallas, MD; Mark Schumacher; Mona Lazar, DO; and Anjali Desai, MD
Abstract 14: The impact of preoperative medical optimization on head and neck cancer surgery
Christopher Tan, MBBS; Catherine Gibb, MBBS, FRACP; and Suren Krishnan, MBBS, FRACS
Abstract 15: Reconceptualizing the preoperative process
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 16: Development of an electronic medical record smart set form to increase standardization, consistency, and compliance with ACC/AHA perioperative guidelines
Anitha Rajamanickam, MD; Ali Usmani, MD; Ajay Kumar, MD; and Brian Harte, MD
Abstract 17: Development of a perioperative electronic medical record research and quality improvement database
Anitha Rajamanickam, MD; Ali Usmani, MD; Feza Remzi, MD; Brian Harte, MD; and Ajay Kumar, MD
Abstract 18: An innovative perioperative/consultative curriculum for third-year internal medicine residents
Alex Rico, MD; Joshua Lenchus, DO; and Amir Jaffer, MD
Abstract 19: Preoperative medicine infobutton
Terrence J. Adam, MD, PhD
Abstract 20: Nurse practitioners: Bridging the gap in perioperative care
Sally Morgan, RN, MS, ANP-BC, ACNS-BC; and Angela Wright, RN, MSN, APRN, BC
Abstract 21: Intubation training of deploying far forward combat medical personnel with the video laryngoscope
Ben Boedeker, MD; Mary Barak-Bernhagen, BS; Kirsten Boedeker; and W. Bosseau Murray, MD
Abstract 22: The establishment of a perioperative skin integrity committee
Jeanne Lanchester, RN, MEd; Ann Leary, BSN, RNC; and Susan Vargas, AD, RN
Abstract 23: Development and implementation of a perianesthesia integrative care committee
Jeanne Lanchester, RN, MEd; Jeanette Cote, BWN, RN; Terri Jamros, RN; Charla Delillo, RN; Sherie Lavoie, BSN, RN; Jennifer Therminos, SN; Joan Compagnone, RN; and Nicole Engel, MSN, RN
Abstract 24: Development of a screening system to identify patients preoperatively who may benefit from a postoperative hospitalist consult
Elizabeth Marlow, MD, MA; and Chad Whelan, MD
Abstract 25: An algorithm for preoperative screening and management of sleep apnea: Have we created a monster?
Deborah C. Richman, MBChB, FFA(SA); Jorge M. Mallea, MD; Paul S. Richman, MD; and Pater S.A. Glass, MBChB
Abstract 26: Constructing a collaborative neuroscience hospitalist program
Rachel Thompson, MD; Christy Gilmore, MD; Kamal Ajam, MD; and Jennifer Thompson, MD
Abstract 27: The development of algorithms for preoperative management of antiplatelet and anticoagulation therapy in patients undergoing surgical or invasive procedures
Catherine McGowan, MSN, and Patricia Kidik, MSN
Abstract 28: Surgeon-initiated preoperative screening: A new approach
Christina Johnson, RN, PA-C; and Edward J. denBraven, CRNA
Abstract 29: A new process for ensuring the safety of patients having anesthesia outside of the operating room
Ellen Leary, MSN; Catherine McGowan, MSN; Kathleen McGrath, MSN; Sheila McCabe Hassan, MSN; and Theresa Kennedy, MSN
Abstract 30: Establishing a virtual preoperative evaluation clinic
Corey Zetterman, MD; Bobbie J. Sweitzer, MD; and Ben H. Boedeker, MD
Abstract 31: Perioperative hypoxemia and rhabdomyolysis in a medically complicated patient
Sarah Bodin, MD
Abstract 32: How soon is too soon? General anesthesia after coronary intervention with bare metal stents
Meghan Tadel, MD
Abstract 33: Can patients with critical aortic stenosis undergo noncardiac surgery without intervening aortic valve replacement?
M. Chadi Alraies, MD; Abdul Alraiyes, MD; Anitha Rajamanickam, MD; and Frank Michota, MD
Abstract 34: Is it safe to operate on cocaine-positive patients?
M. Chadi Alraies, MD; Abdul Hamid Alraiyes, MD; and Brian Harte, MD
Abstract 35: To intensive care or not?
Mona Lazar, DO; and Peter Kallas, MD
Abstract 36: Predicting surgical complications from liver disease
Mona Lazar, DO, and Peter Kallas, MD
Abstract 37: Preoperative coronary angiography: Friend or foe?
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 38: Heparin-induced thrombocytopenia with low molecular weight heparin after total knee replacement
Steven Cohn, MD
Abstract 39: Patient with Parkinson’s disease treated with implanted deep brain stimulators for laparotomy
Deborah C. Richman, MBChB, FFA(SA); Daryn H. Moller, MD; and Khoa N. Nguyen, MD
Abstract 40: Ethical dilemma in the preoperative assessment clinic: Can a patient refuse an indicated cardiac workup? Can we refuse to anesthetize?
Deborah C. Richman, MBChB, FFA(SA)
Abstract 41: Coronary artery bypass grafting as a precipitatin factor in diabetic ketoacidosis in type 2 diabetes
Vishal Sehgral, MD, and Abbas Kitabchi, MD
Summit Director:
Amir K. Jaffer, MD
Contents
Abstract 1: Pulmonary hypertension is an important predictor of perioperative outcomes in patients undergoing noncardiac surgery
Roop Kaw, MD; Esteban Walker, PhD; Vinay Pasupuleti, MD, PhD; Abhishek Deshpande, MD, PhD; Tarek Hamieh, MD; and Omar A. Minai, MD
Abstract 2: Analysis of administrative practices and residency training curricula in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 3: Is percent body fat a better predictor of surgical site infection risk than body mass index?
Emily Waisbren, BS; Angela M. Bader, MD, MPH; Heather Rosen, MD, MPH; Selwyn O. Rogers, Jr., MD, MPH; and Elof Eriksson, MD, PhD
Abstract 4: A nomogram for prediction of survival for patients undergoing elective major noncardiac surgery
Y. Olivia Xu-Cai, MD; and Michael W. Kattan, PhD
Abstract 5: Sustainability of an osteoporosis pathway
Catherine Gibb, MBBS, FRACP; Christopher Butcher, FRACS; Lesley Thomas, BNsg; and Jennifer Pink, BPharm
Abstract 6: Length of hospital stay is predicted by comorbidities
Catherine Gibb, MBBS, FRACP; and Professor Villis Marshall, FRACS
Abstract 7: Generalization of the POISE and Mangano studies on beta-blocker use in the perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; and Luc Lanthier, MD, MSc
Abstract 8: Impact of antihypertensive medication on perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; Danielle Pilon, MD, MSc; and Luc Lanthier, MD, MSc
Abstract 9: An analysis of preoperative testing protocols in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 10: Preoperative biomarkers of inflammation, ischemia, and heart failure and outcomes of vascular surgery
Matthew Griffee, MD; Ansgar Brambrink, MD, PhD; and Thomas Barrett, MD
Abstract 11: Alcohol-related predictors of postoperative delirium in major head and neck cancer surgery
Harrison Weed, MD; Summit Shah, BS; Xin He, PhD; Amit Agrawal, MD; Enver Ozer, MD; and David E. Schuller, MD
Abstract 12: Intraoperative coagulopathy: A low-volume treatment protocol that completely replaces fresh frozen plasma
Peter Kallas, MD; Mary Lou Green, MHS; and Anjali Desai, MD
Abstract 13: Is the Berlin Questionnaire an effective screening tool for obstructive sleep apnea in the preoperative total joint replacement population?
Peter Kallas, MD; Mark Schumacher; Mona Lazar, DO; and Anjali Desai, MD
Abstract 14: The impact of preoperative medical optimization on head and neck cancer surgery
Christopher Tan, MBBS; Catherine Gibb, MBBS, FRACP; and Suren Krishnan, MBBS, FRACS
Abstract 15: Reconceptualizing the preoperative process
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 16: Development of an electronic medical record smart set form to increase standardization, consistency, and compliance with ACC/AHA perioperative guidelines
Anitha Rajamanickam, MD; Ali Usmani, MD; Ajay Kumar, MD; and Brian Harte, MD
Abstract 17: Development of a perioperative electronic medical record research and quality improvement database
Anitha Rajamanickam, MD; Ali Usmani, MD; Feza Remzi, MD; Brian Harte, MD; and Ajay Kumar, MD
Abstract 18: An innovative perioperative/consultative curriculum for third-year internal medicine residents
Alex Rico, MD; Joshua Lenchus, DO; and Amir Jaffer, MD
Abstract 19: Preoperative medicine infobutton
Terrence J. Adam, MD, PhD
Abstract 20: Nurse practitioners: Bridging the gap in perioperative care
Sally Morgan, RN, MS, ANP-BC, ACNS-BC; and Angela Wright, RN, MSN, APRN, BC
Abstract 21: Intubation training of deploying far forward combat medical personnel with the video laryngoscope
Ben Boedeker, MD; Mary Barak-Bernhagen, BS; Kirsten Boedeker; and W. Bosseau Murray, MD
Abstract 22: The establishment of a perioperative skin integrity committee
Jeanne Lanchester, RN, MEd; Ann Leary, BSN, RNC; and Susan Vargas, AD, RN
Abstract 23: Development and implementation of a perianesthesia integrative care committee
Jeanne Lanchester, RN, MEd; Jeanette Cote, BWN, RN; Terri Jamros, RN; Charla Delillo, RN; Sherie Lavoie, BSN, RN; Jennifer Therminos, SN; Joan Compagnone, RN; and Nicole Engel, MSN, RN
Abstract 24: Development of a screening system to identify patients preoperatively who may benefit from a postoperative hospitalist consult
Elizabeth Marlow, MD, MA; and Chad Whelan, MD
Abstract 25: An algorithm for preoperative screening and management of sleep apnea: Have we created a monster?
Deborah C. Richman, MBChB, FFA(SA); Jorge M. Mallea, MD; Paul S. Richman, MD; and Pater S.A. Glass, MBChB
Abstract 26: Constructing a collaborative neuroscience hospitalist program
Rachel Thompson, MD; Christy Gilmore, MD; Kamal Ajam, MD; and Jennifer Thompson, MD
Abstract 27: The development of algorithms for preoperative management of antiplatelet and anticoagulation therapy in patients undergoing surgical or invasive procedures
Catherine McGowan, MSN, and Patricia Kidik, MSN
Abstract 28: Surgeon-initiated preoperative screening: A new approach
Christina Johnson, RN, PA-C; and Edward J. denBraven, CRNA
Abstract 29: A new process for ensuring the safety of patients having anesthesia outside of the operating room
Ellen Leary, MSN; Catherine McGowan, MSN; Kathleen McGrath, MSN; Sheila McCabe Hassan, MSN; and Theresa Kennedy, MSN
Abstract 30: Establishing a virtual preoperative evaluation clinic
Corey Zetterman, MD; Bobbie J. Sweitzer, MD; and Ben H. Boedeker, MD
Abstract 31: Perioperative hypoxemia and rhabdomyolysis in a medically complicated patient
Sarah Bodin, MD
Abstract 32: How soon is too soon? General anesthesia after coronary intervention with bare metal stents
Meghan Tadel, MD
Abstract 33: Can patients with critical aortic stenosis undergo noncardiac surgery without intervening aortic valve replacement?
M. Chadi Alraies, MD; Abdul Alraiyes, MD; Anitha Rajamanickam, MD; and Frank Michota, MD
Abstract 34: Is it safe to operate on cocaine-positive patients?
M. Chadi Alraies, MD; Abdul Hamid Alraiyes, MD; and Brian Harte, MD
Abstract 35: To intensive care or not?
Mona Lazar, DO; and Peter Kallas, MD
Abstract 36: Predicting surgical complications from liver disease
Mona Lazar, DO, and Peter Kallas, MD
Abstract 37: Preoperative coronary angiography: Friend or foe?
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 38: Heparin-induced thrombocytopenia with low molecular weight heparin after total knee replacement
Steven Cohn, MD
Abstract 39: Patient with Parkinson’s disease treated with implanted deep brain stimulators for laparotomy
Deborah C. Richman, MBChB, FFA(SA); Daryn H. Moller, MD; and Khoa N. Nguyen, MD
Abstract 40: Ethical dilemma in the preoperative assessment clinic: Can a patient refuse an indicated cardiac workup? Can we refuse to anesthetize?
Deborah C. Richman, MBChB, FFA(SA)
Abstract 41: Coronary artery bypass grafting as a precipitatin factor in diabetic ketoacidosis in type 2 diabetes
Vishal Sehgral, MD, and Abbas Kitabchi, MD
Lupus update: Perspective and clinical pearls
Many questions about systemic lupus erythematosus (SLE, lupus) remain unanswered. Why is this disease so difficult to diagnose even for rheumatologists? Why does lupus tend to develop in previously healthy young women? Why does the disease manifest in so many ways? Why are our current treatments suboptimal?
This article addresses these questions in a brief overview and update of SLE, with an emphasis on clinical pearls regarding prevention and treatment that are relevant to any physician who sees patients with this disease.
WOMEN AND MINORITIES ARE OVERREPRESENTED
Women have a much higher prevalence of almost all autoimmune diseases. SLE has a 12:1 female-to-male ratio during the ages of 15 to 45 years, but when disease develops in either children or the elderly, the female-to-male ratio is only 2:1.
African Americans, Asian Americans, and Hispanics have about a three to four times higher frequency of lupus than white non-Hispanics and often have more severe disease.
WHY IS SLE SO DIFFICULT TO DIAGNOSE?
SLE is frequently overlooked; patients spend an average of 4 years and see three physicians before the disease is correctly diagnosed. Part of the problem is that presentations of the disease vary so widely between patients and that signs and symptoms evolve over time. Often, physicians do not consider SLE in the differential diagnosis.
On the other hand, SLE is also often over-diagnosed. Narain et al1 evaluated 263 patients who had a presumptive diagnosis of SLE. Only about half of the patients had a confirmed diagnosis; about 5% had a different autoimmune disease, such as scleroderma, systemic sclerosis, Sjögren syndrome, or polymyositis; 5% had fibromyalgia; 29% tested positive for ANA but did not have an autoimmune disease; and 10% had a nonrheumatic disease, such as a hematologic malignancy with rheumatic disease manifestations. For patients referred by a community rheumatologist, the diagnostic accuracy was better, about 80%.
The traditional classification criteria for SLE2,3 are problematic. Some criteria are very specific for SLE but are not very sensitive—eg, anti-double-stranded DNA is present in about half of patients with SLE. Others tests, like ANA, are sensitive but not specific—although ANA is present in 95% of patients with SLE, the positive predictive value of the test for SLE for any given patient is only 11%.
Other criteria are highly subjective, including oral ulcers and photosensitivity. These signs may be present in normal individuals who get an occasional aphthous ulcer or who are fair-skinned and burn easily with prolonged sun exposure. It takes a trained clinician to distinguish these from the photosensitivity and oral ulcers associated with lupus.
Many diseases can mimic SLE
Fibromyalgia frequently presents in women and may include joint and muscle aches, fatigue, and occasionally a positive ANA. ANA may be seen in about 15% of healthy women.
Sjögren syndrome can also present with arthritis, fatigue, and a positive ANA; it is commonly overlooked because physicians do not often think to ask about the classic symptoms of dry eyes and dry mouth.
Dermatomyositis causes rashes that have many features in common with SLE. Even the skin biopsy is often indistinguishable from SLE.
Hematologic problems, such as idiopathic or thrombotic thrombocytopenic purpura, primary antiphospholipid syndrome, and hematologic neoplasms, can cause serologic changes, a positive ANA, and other manifestations seen in SLE.
Drug-induced lupus should always be considered in older patients presenting with SLE-like disease. Now with the use of minocycline (Minocin) and other related agents for the treatment of acne, we are seeing younger women with drug-induced lupus.
PATIENTS ASK ‘WHY ME?’
Lupus typically develops in a young woman who was previously healthy. Such patients inevitably wonder, why me?
Lupus is like a puzzle, with genetics, gender, and the environment being important pieces of the puzzle. If all the pieces come together, people develop defective immune regulation and a break in self-tolerance. Everyone generates antibodies to self, but these low-affinity, nonpathologic antibodies are inconsequential. In SLE, autoantibodies lead to the formation of immune complexes, complement activation, and tissue damage.
Genetics plays an important role
Genetics plays an important role but is clearly not the only determining factor. Clustering in families has been shown, although a patient with lupus is more likely to have a relative with another autoimmune disease, especially autoimmune thyroid disease, than with SLE. The likelihood of an identical twin of a patient with SLE having the disease is only 25% to 30%, and is only about 5% for a fraternal twin.
In the first few months of 2008, four major studies were published that shed light on the genetics of SLE.4–7 Together, the studies evaluated more than 5,000 patients with SLE using genome-wide association scans and identified areas of the genome that are frequently different in patients with lupus than in healthy controls. Three of the four studies identified the same genetic area as important and supported the concept that B cells and complement activation play important roles in the disease pathogenesis.
Although over 95% of cases of SLE cannot be attributed to a single gene, there are rare cases of lupus that may provide important clues to mechanisms of disease. For example a homozygous deficiency of C1q (an early component of complement) is extremely rare in lupus but is associated with the highest risk (nearly 90%) of developing the disease. Deficiencies in other components of the complement cascade also carry a high risk of disease development.
Investigators discovered that C1q plays an important role in clearing away apoptotic cellular debris. If a person is deficient in C1q, clearance of this debris is impaired. In a person genetically predisposed to getting lupus, the immune system now has an opportunity to react to self-antigens exposed during apoptosis that are not being cleared away.
Even though lupus cases cannot be explained by an absence of C1q, a defect in the clearance of apoptotic cells is a common, unifying feature of the disease.
Immune response is enhanced by environmental factors
Environmental factors, especially sun exposure, are also important. Following sunburn, skin cells undergo massive cell death, and patients with lupus have a huge release of self-antigens that can be recognized by the immune system. Sunburn is like having a booster vaccine of self-antigen to stimulate autoantibody production. Not only does the skin flare, but internal organs can also flare after intense sun exposure.
LUPUS SURVIVAL HAS IMPROVED; DISEASES OF AGING NOW A FOCUS
In 1950, only 50% of patients with SLE survived 5 years after diagnosis; now, thanks to better treatment and earlier diagnosis, 80% to 90% survive at least 10 years.
Early on, patients tend to die of active disease (manifestations of vasculitis, pulmonary hemorrhage, kidney problems) or infection. Over time, cardiovascular disease and osteoporosis become more of a problem. Patients also have a higher risk of cancer throughout life.
Lupus has an unpredictable course, with flares and remissions. But underlying the reversible inflammatory changes is irreversible organ damage caused by the disease itself and, possibly, by treatment. Preventing bone disease, heart disease, and cancer now play more prominent roles in managing SLE.
Increased bone disease
Fracture rates are higher than expected in women with lupus; Ramsey-Goldman et al8 calculated the rate as five times higher than in the general population. The increased risk of osteoporosis is partly due to treatment with corticosteroids, but it is also likely caused by inflammation from lupus. Even controlling for steroid use, increased bone loss is still evident in patients with SLE.
African American women with lupus are not exempt. Lee et al9 found that, after adjusting for body mass, steroid use, thyroid disease, and menopausal status, African American women with SLE had more than five times the risk of low bone mineral density in the spine than white women with the disease.
Increased cancer risk
Patients with SLE have an increased risk of hematologic cancer and possibly lung and hepatobiliary cancers.
Bernatsky et al10 evaluated cancer risk in an international cohort of patients with SLE from 23 sites. Among patients with SLE, for all cancers combined, the standardized incidence ratio was 1.2; for hematologic cancers the ratio was 2.8; and for non-Hodgkin lymphoma it was 2.4. Surprisingly, although SLE is primarily a disease of women, reproductive cancer rates in patients with SLE did not differ from background rates. Bernatsky et al did not compare rates of cervical cancer, as many cancer registries do not record it. However, studies from the National Institutes of Health indicate that cervical dysplasia is common in patients with lupus.
Other interesting findings included an increased risk of hepatobiliary cancer, especially among men with SLE. Lung cancers were also increased, which has been observed in patients with other autoimmune diseases such as scleroderma and polymyositis. Smoking is a strong predictor for developing autoimmune conditions and may play a role in the observed increased cancer risk.
Early and advanced cardiovascular disease
Patients with SLE are at high risk of atherosclerotic cardiovascular disease. At the University of Pittsburgh Medical Center from 1980 to 1993, we compared the incidence of myocardial infarction in nearly 500 women with SLE and more than 2,000 women of similar age in the Framingham Offspring Study. At ages 15 to 24, women with lupus had a rate of 6.33 per 1,000 person-years; at age 25 to 34, the rate was 3.66 per 1,000 person-years. None of the Framingham women in those age groups had events.
Women ages 35 to 44 with lupus had a risk of heart attack 50 times higher than women in the Framingham cohort, and women in older age groups had a risk 2.5 to 4 times higher.11
Subclinical cardiovascular disease is also increased in women with SLE. Asanuma et al12 used electron-beam computed tomography to screen for coronary artery calcification in 65 patients with SLE and 69 control subjects with no history of coronary artery disease matched for age, sex, and race. Calcification was present in 31% of patients with lupus vs 9% of controls (P = .002). Roman et al13 performed carotid ultrasonography on 197 patients with lupus and 197 matched controls and found more plaque in patients with lupus (37%) than in controls (15%, P < .001).
Other data also suggest that women with lupus have advanced cardiovascular disease and develop it early, with most studies finding the greatest relative risk of cardiovascular disease between ages 18 and 45 years.
Traditional risk factors for cardiovascular disease cannot fully explain the increased risk. Many patients with lupus have metabolic syndrome, hypertension, and renal disease, but even after adjusting for these risk factors, patients with lupus still have about a 7 to 10 times higher risk of nonfatal coronary heart disease and a 17 times higher risk of fatal coronary heart disease.14
Many investigators are now exploring the role of immune dysfunction and inflammation in cardiovascular disease. A number of biomarkers have been proposed for predicting risk of cardiovascular disease in the general population. The list includes many inflammatory factors that rheumatologists have been studying for decades, including myeloperoxidase, autoantibodies, inflammatory cytokines, tumor necrosis factor alpha, and adhesion molecules, many of which also play a role in autoimmunity.
In our patients with SLE, we found that about 90% had three or more modifiable cardiovascular risk factors that were not being addressed appropriately (unpublished data). Lipid management was the least often addressed by rheumatologists and primary caregivers.
There is reason to believe that lupus patients are a high-risk group that merit aggressive risk-factor management, but no formal recommendations can be made without clear evidence that this approach improves outcomes.
SLE INCREASES THE RISK OF ADVERSE PREGNANCY OUTCOMES
Women with SLE more commonly miscarry and deliver low-birth-weight babies than do other women. A history of renal disease is the factor most predictive of poor pregnancy outcome, and the presence of certain autoantibodies increases the risk of neonatal lupus.
We recommend that women with lupus have inactive disease for at least 6 months before becoming pregnant.
ORAL CONTRACEPTIVES AND HORMONE REPLACEMENT
Hormone replacement therapy and oral contraceptives do not increase the risk of significant disease activity flares in lupus. However, women with lupus have an increased risk of cardiovascular disease and thrombosis.
Buyon et al15 randomly assigned 351 menopausal women with inactive or stable active SLE to receive either hormone replacement therapy or placebo for 12 months. No significant increase in severe flares of the disease was observed, although the treatment group had a mild increase in minor flares.
Petri et al16 randomly assigned 183 women with inactive or stable active SLE to receive either combined oral contraceptives or placebo for 12 months and found similar rates of disease activity between the two groups.
A weakness of these trials is that women with antiphospholipid antibodies in high titers or who had previous thrombotic events were excluded.
TREATMENTS ON THE HORIZON?
In the past 50 years, only three drug treatments have been approved for lupus: corticosteroids, hydroxychloroquine, and aspirin. Fortunately, research in autoimmune diseases has rapidly expanded, and new drugs are on the horizon.
Mycophenolate mofetil (CellCept) may be a reasonable alternative to cyclophosphamide (Cytoxan) for lupus nephritis and may be appropriate as maintenance therapy after induction with cyclophosphamide.
Ginzler et al,17 in a randomized, open-label trial in 140 patients with active lupus nephritis, gave either oral mycophenolate mofetil (initial dosage 1,000 mg/day, increased to 3,000 mg/day) or monthly intravenous cyclophosphamide (0.5 g/m2, increased to 1.0 g/m2). Mycophenolate mofetil was more effective in inducing remission than cyclophosphamide and had a better safety profile.
The Aspreva Lupus Management Study was designed to assess the efficacy of oral mycophenolate mofetil compared with intravenous cyclophosphamide in the initial treatment of patients with active class III–V lupus nephritis and to assess the long-term efficacy of mycophenolate mofetil compared with azathioprine in maintaining remission and renal function. It was the largest study of mycophenolate mofetil in lupus nephritis to date. There were 370 patients with SLE enrolled. In the 24-week induction phase, patients were randomized to receive open-label mycophenolate mofetil with a target dose of 3 g/day or intravenous cyclophosphamide 0.5 to 1.0 g/m2 in monthly pulses. Both groups received prednisone. Response to treatment was defined as a decrease in proteinuria (as measured by the urinary protein-creatinine ratio) and improvement or stabilization in serum creatinine.
The results (presented at the American College of Rheumatology Meeting, November 6–11, 2007, in Boston, MA) showed that 104 (56%) of the 185 patients treated with mycophenolate mofetil responded, compared with 98 (53%) of the 185 patients treated with intravenous cyclophosphamide (P = .575). The study therefore did not meet its primary objective of showing a superior response rate with mycophenolate mofetil compared with cyclophosphamide. There was no difference in adverse events. It is this author’s opinion that having an agent that is at least as good as cyclphosphamide in treating lupus nephritis is a major step forward.
Mycophenolate mofetil can cause fetal harm and should not be used during pregnancy. It is recommended that the drug be stopped for 3 to 6 months before a woman tries to conceive.
New drugs target B cells
Many new drugs for lupus target B cells.
Rituximab (Rituxan) is a monoclonal antibody that depletes B cells by targeting the B-cell-specific antigen CD20. It has been studied for treating lupus in several open-label studies that altogether have included more than 400 patients.18–21 Regimens included either those used in oncology for treatment of lymphoma or those used in rheumatoid arthritis, coupled with high-dose corticosteroids and cyclophosphamide. In early studies, nearly 80% of treated patients entered at least partial remission, and 25% to 50% are still in remission more than 12 months later.
The first randomized controlled trial of rituximab vs placebo was recently completed and presented at the American College of Rheumatology meeting, October 24–28, 2008, in Boston, MA. The EXPLORER trial (sponsored by Genentech) included 257 patients with moderate to severe disease activity. The results showed that there was no difference in major or partial clinical response (based on a change in the British Isles Lupus Assessment Group index) in those on rituximab (28.4%) vs placebo (29.6%) at 12 months (P = .97). Overall, adverse events were balanced between the groups. It is this author’s opinion that the bar for “response” was set very high in this study, considering that all patients who entered were fairly sick and received significant doses of corticosteroids that were tapered over the course of the trial.
B-cell toleragens, which render B cells incapable of presenting specific antigens, are also of interest.
Other experimental drugs target the soluble cytokine BLyS, which normally binds to a B-cell receptor and prolongs B-cell survival. It may also be possible to inhibit costimulatory pathways (which are normally important for inducing activation, proliferation, and class-switching of B cells) with the use of cytotoxic T-lymphocyte-associated antigen 4 immunoglobulin inhibitor (CTLA4Ig) and anti-CD40 ligand.
The results of a 12-month exploratory, phase II trial of abatacept (Bristol-Myers Squibb) in patients with SLE and active polyarthritis, serositis, or discoid lesions were presented at the American College of Rheumatology meeting in 2008. The primary and secondary end points (based on an adjudicated British Isles Lupus Assessment Group index) were not met. There were no differences in adverse events. Post hoc analyses of other clinical end points and biomarkers suggested that abatacept may have benefit in lupus. Further studies are under way.
Downstream blockade may also be useful, with drugs that inhibit inflammatory cytokines, particularly interferon alfa. This is now being tested in clinical trials.
- Narain S, Richards HB, Satoh M, et al. Diagnostic accuracy for lupus and other systemic autoimmune diseases in the community setting. Arch Intern Med 2004; 164:2435–2441.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13 BLK and ITGAM ITGAX. N Engl J Med 2008; 358:900–909.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211–216.
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204–210.
- Nath SK, Han S, Kim Howard X, et al. A nonsynonymous functional variant in integrin alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008; 40:152–154.
- Ramsey-Goldman R, Dunn JE, Huang CF, et al. Frequency of fractures in women with systemic lupus erythematosus: comparison with United States population data. Arthritis Rheum 1999; 42:882–890.
- Lee C, Almagor O, Dunlop DD, et al. Association between African-American race/ethnicity and low bone mineral density in women with systemic lupus erythematosus. Arthritis Rheum 2007; 57:585–592.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005; 52:1481–1490.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol 1997; 145:408–415.
- Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2407–2415.
- Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2399–2406. Erratum in: N Engl J Med 2006; 355:1746.
- Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001; 44:2331–2337.
- Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005; 142:953–962.
- Petri M, Kim MY, Kalunian KC, et al; OC SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005; 353:2550–2558.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004; 50:3580–3590.
- Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose escalation trial of rituximab. Arthritis Rheum 2004; 50:2580–2589.
- Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum 2002; 46:2673–2677.
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 2006; 54:723–732.
Many questions about systemic lupus erythematosus (SLE, lupus) remain unanswered. Why is this disease so difficult to diagnose even for rheumatologists? Why does lupus tend to develop in previously healthy young women? Why does the disease manifest in so many ways? Why are our current treatments suboptimal?
This article addresses these questions in a brief overview and update of SLE, with an emphasis on clinical pearls regarding prevention and treatment that are relevant to any physician who sees patients with this disease.
WOMEN AND MINORITIES ARE OVERREPRESENTED
Women have a much higher prevalence of almost all autoimmune diseases. SLE has a 12:1 female-to-male ratio during the ages of 15 to 45 years, but when disease develops in either children or the elderly, the female-to-male ratio is only 2:1.
African Americans, Asian Americans, and Hispanics have about a three to four times higher frequency of lupus than white non-Hispanics and often have more severe disease.
WHY IS SLE SO DIFFICULT TO DIAGNOSE?
SLE is frequently overlooked; patients spend an average of 4 years and see three physicians before the disease is correctly diagnosed. Part of the problem is that presentations of the disease vary so widely between patients and that signs and symptoms evolve over time. Often, physicians do not consider SLE in the differential diagnosis.
On the other hand, SLE is also often over-diagnosed. Narain et al1 evaluated 263 patients who had a presumptive diagnosis of SLE. Only about half of the patients had a confirmed diagnosis; about 5% had a different autoimmune disease, such as scleroderma, systemic sclerosis, Sjögren syndrome, or polymyositis; 5% had fibromyalgia; 29% tested positive for ANA but did not have an autoimmune disease; and 10% had a nonrheumatic disease, such as a hematologic malignancy with rheumatic disease manifestations. For patients referred by a community rheumatologist, the diagnostic accuracy was better, about 80%.
The traditional classification criteria for SLE2,3 are problematic. Some criteria are very specific for SLE but are not very sensitive—eg, anti-double-stranded DNA is present in about half of patients with SLE. Others tests, like ANA, are sensitive but not specific—although ANA is present in 95% of patients with SLE, the positive predictive value of the test for SLE for any given patient is only 11%.
Other criteria are highly subjective, including oral ulcers and photosensitivity. These signs may be present in normal individuals who get an occasional aphthous ulcer or who are fair-skinned and burn easily with prolonged sun exposure. It takes a trained clinician to distinguish these from the photosensitivity and oral ulcers associated with lupus.
Many diseases can mimic SLE
Fibromyalgia frequently presents in women and may include joint and muscle aches, fatigue, and occasionally a positive ANA. ANA may be seen in about 15% of healthy women.
Sjögren syndrome can also present with arthritis, fatigue, and a positive ANA; it is commonly overlooked because physicians do not often think to ask about the classic symptoms of dry eyes and dry mouth.
Dermatomyositis causes rashes that have many features in common with SLE. Even the skin biopsy is often indistinguishable from SLE.
Hematologic problems, such as idiopathic or thrombotic thrombocytopenic purpura, primary antiphospholipid syndrome, and hematologic neoplasms, can cause serologic changes, a positive ANA, and other manifestations seen in SLE.
Drug-induced lupus should always be considered in older patients presenting with SLE-like disease. Now with the use of minocycline (Minocin) and other related agents for the treatment of acne, we are seeing younger women with drug-induced lupus.
PATIENTS ASK ‘WHY ME?’
Lupus typically develops in a young woman who was previously healthy. Such patients inevitably wonder, why me?
Lupus is like a puzzle, with genetics, gender, and the environment being important pieces of the puzzle. If all the pieces come together, people develop defective immune regulation and a break in self-tolerance. Everyone generates antibodies to self, but these low-affinity, nonpathologic antibodies are inconsequential. In SLE, autoantibodies lead to the formation of immune complexes, complement activation, and tissue damage.
Genetics plays an important role
Genetics plays an important role but is clearly not the only determining factor. Clustering in families has been shown, although a patient with lupus is more likely to have a relative with another autoimmune disease, especially autoimmune thyroid disease, than with SLE. The likelihood of an identical twin of a patient with SLE having the disease is only 25% to 30%, and is only about 5% for a fraternal twin.
In the first few months of 2008, four major studies were published that shed light on the genetics of SLE.4–7 Together, the studies evaluated more than 5,000 patients with SLE using genome-wide association scans and identified areas of the genome that are frequently different in patients with lupus than in healthy controls. Three of the four studies identified the same genetic area as important and supported the concept that B cells and complement activation play important roles in the disease pathogenesis.
Although over 95% of cases of SLE cannot be attributed to a single gene, there are rare cases of lupus that may provide important clues to mechanisms of disease. For example a homozygous deficiency of C1q (an early component of complement) is extremely rare in lupus but is associated with the highest risk (nearly 90%) of developing the disease. Deficiencies in other components of the complement cascade also carry a high risk of disease development.
Investigators discovered that C1q plays an important role in clearing away apoptotic cellular debris. If a person is deficient in C1q, clearance of this debris is impaired. In a person genetically predisposed to getting lupus, the immune system now has an opportunity to react to self-antigens exposed during apoptosis that are not being cleared away.
Even though lupus cases cannot be explained by an absence of C1q, a defect in the clearance of apoptotic cells is a common, unifying feature of the disease.
Immune response is enhanced by environmental factors
Environmental factors, especially sun exposure, are also important. Following sunburn, skin cells undergo massive cell death, and patients with lupus have a huge release of self-antigens that can be recognized by the immune system. Sunburn is like having a booster vaccine of self-antigen to stimulate autoantibody production. Not only does the skin flare, but internal organs can also flare after intense sun exposure.
LUPUS SURVIVAL HAS IMPROVED; DISEASES OF AGING NOW A FOCUS
In 1950, only 50% of patients with SLE survived 5 years after diagnosis; now, thanks to better treatment and earlier diagnosis, 80% to 90% survive at least 10 years.
Early on, patients tend to die of active disease (manifestations of vasculitis, pulmonary hemorrhage, kidney problems) or infection. Over time, cardiovascular disease and osteoporosis become more of a problem. Patients also have a higher risk of cancer throughout life.
Lupus has an unpredictable course, with flares and remissions. But underlying the reversible inflammatory changes is irreversible organ damage caused by the disease itself and, possibly, by treatment. Preventing bone disease, heart disease, and cancer now play more prominent roles in managing SLE.
Increased bone disease
Fracture rates are higher than expected in women with lupus; Ramsey-Goldman et al8 calculated the rate as five times higher than in the general population. The increased risk of osteoporosis is partly due to treatment with corticosteroids, but it is also likely caused by inflammation from lupus. Even controlling for steroid use, increased bone loss is still evident in patients with SLE.
African American women with lupus are not exempt. Lee et al9 found that, after adjusting for body mass, steroid use, thyroid disease, and menopausal status, African American women with SLE had more than five times the risk of low bone mineral density in the spine than white women with the disease.
Increased cancer risk
Patients with SLE have an increased risk of hematologic cancer and possibly lung and hepatobiliary cancers.
Bernatsky et al10 evaluated cancer risk in an international cohort of patients with SLE from 23 sites. Among patients with SLE, for all cancers combined, the standardized incidence ratio was 1.2; for hematologic cancers the ratio was 2.8; and for non-Hodgkin lymphoma it was 2.4. Surprisingly, although SLE is primarily a disease of women, reproductive cancer rates in patients with SLE did not differ from background rates. Bernatsky et al did not compare rates of cervical cancer, as many cancer registries do not record it. However, studies from the National Institutes of Health indicate that cervical dysplasia is common in patients with lupus.
Other interesting findings included an increased risk of hepatobiliary cancer, especially among men with SLE. Lung cancers were also increased, which has been observed in patients with other autoimmune diseases such as scleroderma and polymyositis. Smoking is a strong predictor for developing autoimmune conditions and may play a role in the observed increased cancer risk.
Early and advanced cardiovascular disease
Patients with SLE are at high risk of atherosclerotic cardiovascular disease. At the University of Pittsburgh Medical Center from 1980 to 1993, we compared the incidence of myocardial infarction in nearly 500 women with SLE and more than 2,000 women of similar age in the Framingham Offspring Study. At ages 15 to 24, women with lupus had a rate of 6.33 per 1,000 person-years; at age 25 to 34, the rate was 3.66 per 1,000 person-years. None of the Framingham women in those age groups had events.
Women ages 35 to 44 with lupus had a risk of heart attack 50 times higher than women in the Framingham cohort, and women in older age groups had a risk 2.5 to 4 times higher.11
Subclinical cardiovascular disease is also increased in women with SLE. Asanuma et al12 used electron-beam computed tomography to screen for coronary artery calcification in 65 patients with SLE and 69 control subjects with no history of coronary artery disease matched for age, sex, and race. Calcification was present in 31% of patients with lupus vs 9% of controls (P = .002). Roman et al13 performed carotid ultrasonography on 197 patients with lupus and 197 matched controls and found more plaque in patients with lupus (37%) than in controls (15%, P < .001).
Other data also suggest that women with lupus have advanced cardiovascular disease and develop it early, with most studies finding the greatest relative risk of cardiovascular disease between ages 18 and 45 years.
Traditional risk factors for cardiovascular disease cannot fully explain the increased risk. Many patients with lupus have metabolic syndrome, hypertension, and renal disease, but even after adjusting for these risk factors, patients with lupus still have about a 7 to 10 times higher risk of nonfatal coronary heart disease and a 17 times higher risk of fatal coronary heart disease.14
Many investigators are now exploring the role of immune dysfunction and inflammation in cardiovascular disease. A number of biomarkers have been proposed for predicting risk of cardiovascular disease in the general population. The list includes many inflammatory factors that rheumatologists have been studying for decades, including myeloperoxidase, autoantibodies, inflammatory cytokines, tumor necrosis factor alpha, and adhesion molecules, many of which also play a role in autoimmunity.
In our patients with SLE, we found that about 90% had three or more modifiable cardiovascular risk factors that were not being addressed appropriately (unpublished data). Lipid management was the least often addressed by rheumatologists and primary caregivers.
There is reason to believe that lupus patients are a high-risk group that merit aggressive risk-factor management, but no formal recommendations can be made without clear evidence that this approach improves outcomes.
SLE INCREASES THE RISK OF ADVERSE PREGNANCY OUTCOMES
Women with SLE more commonly miscarry and deliver low-birth-weight babies than do other women. A history of renal disease is the factor most predictive of poor pregnancy outcome, and the presence of certain autoantibodies increases the risk of neonatal lupus.
We recommend that women with lupus have inactive disease for at least 6 months before becoming pregnant.
ORAL CONTRACEPTIVES AND HORMONE REPLACEMENT
Hormone replacement therapy and oral contraceptives do not increase the risk of significant disease activity flares in lupus. However, women with lupus have an increased risk of cardiovascular disease and thrombosis.
Buyon et al15 randomly assigned 351 menopausal women with inactive or stable active SLE to receive either hormone replacement therapy or placebo for 12 months. No significant increase in severe flares of the disease was observed, although the treatment group had a mild increase in minor flares.
Petri et al16 randomly assigned 183 women with inactive or stable active SLE to receive either combined oral contraceptives or placebo for 12 months and found similar rates of disease activity between the two groups.
A weakness of these trials is that women with antiphospholipid antibodies in high titers or who had previous thrombotic events were excluded.
TREATMENTS ON THE HORIZON?
In the past 50 years, only three drug treatments have been approved for lupus: corticosteroids, hydroxychloroquine, and aspirin. Fortunately, research in autoimmune diseases has rapidly expanded, and new drugs are on the horizon.
Mycophenolate mofetil (CellCept) may be a reasonable alternative to cyclophosphamide (Cytoxan) for lupus nephritis and may be appropriate as maintenance therapy after induction with cyclophosphamide.
Ginzler et al,17 in a randomized, open-label trial in 140 patients with active lupus nephritis, gave either oral mycophenolate mofetil (initial dosage 1,000 mg/day, increased to 3,000 mg/day) or monthly intravenous cyclophosphamide (0.5 g/m2, increased to 1.0 g/m2). Mycophenolate mofetil was more effective in inducing remission than cyclophosphamide and had a better safety profile.
The Aspreva Lupus Management Study was designed to assess the efficacy of oral mycophenolate mofetil compared with intravenous cyclophosphamide in the initial treatment of patients with active class III–V lupus nephritis and to assess the long-term efficacy of mycophenolate mofetil compared with azathioprine in maintaining remission and renal function. It was the largest study of mycophenolate mofetil in lupus nephritis to date. There were 370 patients with SLE enrolled. In the 24-week induction phase, patients were randomized to receive open-label mycophenolate mofetil with a target dose of 3 g/day or intravenous cyclophosphamide 0.5 to 1.0 g/m2 in monthly pulses. Both groups received prednisone. Response to treatment was defined as a decrease in proteinuria (as measured by the urinary protein-creatinine ratio) and improvement or stabilization in serum creatinine.
The results (presented at the American College of Rheumatology Meeting, November 6–11, 2007, in Boston, MA) showed that 104 (56%) of the 185 patients treated with mycophenolate mofetil responded, compared with 98 (53%) of the 185 patients treated with intravenous cyclophosphamide (P = .575). The study therefore did not meet its primary objective of showing a superior response rate with mycophenolate mofetil compared with cyclophosphamide. There was no difference in adverse events. It is this author’s opinion that having an agent that is at least as good as cyclphosphamide in treating lupus nephritis is a major step forward.
Mycophenolate mofetil can cause fetal harm and should not be used during pregnancy. It is recommended that the drug be stopped for 3 to 6 months before a woman tries to conceive.
New drugs target B cells
Many new drugs for lupus target B cells.
Rituximab (Rituxan) is a monoclonal antibody that depletes B cells by targeting the B-cell-specific antigen CD20. It has been studied for treating lupus in several open-label studies that altogether have included more than 400 patients.18–21 Regimens included either those used in oncology for treatment of lymphoma or those used in rheumatoid arthritis, coupled with high-dose corticosteroids and cyclophosphamide. In early studies, nearly 80% of treated patients entered at least partial remission, and 25% to 50% are still in remission more than 12 months later.
The first randomized controlled trial of rituximab vs placebo was recently completed and presented at the American College of Rheumatology meeting, October 24–28, 2008, in Boston, MA. The EXPLORER trial (sponsored by Genentech) included 257 patients with moderate to severe disease activity. The results showed that there was no difference in major or partial clinical response (based on a change in the British Isles Lupus Assessment Group index) in those on rituximab (28.4%) vs placebo (29.6%) at 12 months (P = .97). Overall, adverse events were balanced between the groups. It is this author’s opinion that the bar for “response” was set very high in this study, considering that all patients who entered were fairly sick and received significant doses of corticosteroids that were tapered over the course of the trial.
B-cell toleragens, which render B cells incapable of presenting specific antigens, are also of interest.
Other experimental drugs target the soluble cytokine BLyS, which normally binds to a B-cell receptor and prolongs B-cell survival. It may also be possible to inhibit costimulatory pathways (which are normally important for inducing activation, proliferation, and class-switching of B cells) with the use of cytotoxic T-lymphocyte-associated antigen 4 immunoglobulin inhibitor (CTLA4Ig) and anti-CD40 ligand.
The results of a 12-month exploratory, phase II trial of abatacept (Bristol-Myers Squibb) in patients with SLE and active polyarthritis, serositis, or discoid lesions were presented at the American College of Rheumatology meeting in 2008. The primary and secondary end points (based on an adjudicated British Isles Lupus Assessment Group index) were not met. There were no differences in adverse events. Post hoc analyses of other clinical end points and biomarkers suggested that abatacept may have benefit in lupus. Further studies are under way.
Downstream blockade may also be useful, with drugs that inhibit inflammatory cytokines, particularly interferon alfa. This is now being tested in clinical trials.
Many questions about systemic lupus erythematosus (SLE, lupus) remain unanswered. Why is this disease so difficult to diagnose even for rheumatologists? Why does lupus tend to develop in previously healthy young women? Why does the disease manifest in so many ways? Why are our current treatments suboptimal?
This article addresses these questions in a brief overview and update of SLE, with an emphasis on clinical pearls regarding prevention and treatment that are relevant to any physician who sees patients with this disease.
WOMEN AND MINORITIES ARE OVERREPRESENTED
Women have a much higher prevalence of almost all autoimmune diseases. SLE has a 12:1 female-to-male ratio during the ages of 15 to 45 years, but when disease develops in either children or the elderly, the female-to-male ratio is only 2:1.
African Americans, Asian Americans, and Hispanics have about a three to four times higher frequency of lupus than white non-Hispanics and often have more severe disease.
WHY IS SLE SO DIFFICULT TO DIAGNOSE?
SLE is frequently overlooked; patients spend an average of 4 years and see three physicians before the disease is correctly diagnosed. Part of the problem is that presentations of the disease vary so widely between patients and that signs and symptoms evolve over time. Often, physicians do not consider SLE in the differential diagnosis.
On the other hand, SLE is also often over-diagnosed. Narain et al1 evaluated 263 patients who had a presumptive diagnosis of SLE. Only about half of the patients had a confirmed diagnosis; about 5% had a different autoimmune disease, such as scleroderma, systemic sclerosis, Sjögren syndrome, or polymyositis; 5% had fibromyalgia; 29% tested positive for ANA but did not have an autoimmune disease; and 10% had a nonrheumatic disease, such as a hematologic malignancy with rheumatic disease manifestations. For patients referred by a community rheumatologist, the diagnostic accuracy was better, about 80%.
The traditional classification criteria for SLE2,3 are problematic. Some criteria are very specific for SLE but are not very sensitive—eg, anti-double-stranded DNA is present in about half of patients with SLE. Others tests, like ANA, are sensitive but not specific—although ANA is present in 95% of patients with SLE, the positive predictive value of the test for SLE for any given patient is only 11%.
Other criteria are highly subjective, including oral ulcers and photosensitivity. These signs may be present in normal individuals who get an occasional aphthous ulcer or who are fair-skinned and burn easily with prolonged sun exposure. It takes a trained clinician to distinguish these from the photosensitivity and oral ulcers associated with lupus.
Many diseases can mimic SLE
Fibromyalgia frequently presents in women and may include joint and muscle aches, fatigue, and occasionally a positive ANA. ANA may be seen in about 15% of healthy women.
Sjögren syndrome can also present with arthritis, fatigue, and a positive ANA; it is commonly overlooked because physicians do not often think to ask about the classic symptoms of dry eyes and dry mouth.
Dermatomyositis causes rashes that have many features in common with SLE. Even the skin biopsy is often indistinguishable from SLE.
Hematologic problems, such as idiopathic or thrombotic thrombocytopenic purpura, primary antiphospholipid syndrome, and hematologic neoplasms, can cause serologic changes, a positive ANA, and other manifestations seen in SLE.
Drug-induced lupus should always be considered in older patients presenting with SLE-like disease. Now with the use of minocycline (Minocin) and other related agents for the treatment of acne, we are seeing younger women with drug-induced lupus.
PATIENTS ASK ‘WHY ME?’
Lupus typically develops in a young woman who was previously healthy. Such patients inevitably wonder, why me?
Lupus is like a puzzle, with genetics, gender, and the environment being important pieces of the puzzle. If all the pieces come together, people develop defective immune regulation and a break in self-tolerance. Everyone generates antibodies to self, but these low-affinity, nonpathologic antibodies are inconsequential. In SLE, autoantibodies lead to the formation of immune complexes, complement activation, and tissue damage.
Genetics plays an important role
Genetics plays an important role but is clearly not the only determining factor. Clustering in families has been shown, although a patient with lupus is more likely to have a relative with another autoimmune disease, especially autoimmune thyroid disease, than with SLE. The likelihood of an identical twin of a patient with SLE having the disease is only 25% to 30%, and is only about 5% for a fraternal twin.
In the first few months of 2008, four major studies were published that shed light on the genetics of SLE.4–7 Together, the studies evaluated more than 5,000 patients with SLE using genome-wide association scans and identified areas of the genome that are frequently different in patients with lupus than in healthy controls. Three of the four studies identified the same genetic area as important and supported the concept that B cells and complement activation play important roles in the disease pathogenesis.
Although over 95% of cases of SLE cannot be attributed to a single gene, there are rare cases of lupus that may provide important clues to mechanisms of disease. For example a homozygous deficiency of C1q (an early component of complement) is extremely rare in lupus but is associated with the highest risk (nearly 90%) of developing the disease. Deficiencies in other components of the complement cascade also carry a high risk of disease development.
Investigators discovered that C1q plays an important role in clearing away apoptotic cellular debris. If a person is deficient in C1q, clearance of this debris is impaired. In a person genetically predisposed to getting lupus, the immune system now has an opportunity to react to self-antigens exposed during apoptosis that are not being cleared away.
Even though lupus cases cannot be explained by an absence of C1q, a defect in the clearance of apoptotic cells is a common, unifying feature of the disease.
Immune response is enhanced by environmental factors
Environmental factors, especially sun exposure, are also important. Following sunburn, skin cells undergo massive cell death, and patients with lupus have a huge release of self-antigens that can be recognized by the immune system. Sunburn is like having a booster vaccine of self-antigen to stimulate autoantibody production. Not only does the skin flare, but internal organs can also flare after intense sun exposure.
LUPUS SURVIVAL HAS IMPROVED; DISEASES OF AGING NOW A FOCUS
In 1950, only 50% of patients with SLE survived 5 years after diagnosis; now, thanks to better treatment and earlier diagnosis, 80% to 90% survive at least 10 years.
Early on, patients tend to die of active disease (manifestations of vasculitis, pulmonary hemorrhage, kidney problems) or infection. Over time, cardiovascular disease and osteoporosis become more of a problem. Patients also have a higher risk of cancer throughout life.
Lupus has an unpredictable course, with flares and remissions. But underlying the reversible inflammatory changes is irreversible organ damage caused by the disease itself and, possibly, by treatment. Preventing bone disease, heart disease, and cancer now play more prominent roles in managing SLE.
Increased bone disease
Fracture rates are higher than expected in women with lupus; Ramsey-Goldman et al8 calculated the rate as five times higher than in the general population. The increased risk of osteoporosis is partly due to treatment with corticosteroids, but it is also likely caused by inflammation from lupus. Even controlling for steroid use, increased bone loss is still evident in patients with SLE.
African American women with lupus are not exempt. Lee et al9 found that, after adjusting for body mass, steroid use, thyroid disease, and menopausal status, African American women with SLE had more than five times the risk of low bone mineral density in the spine than white women with the disease.
Increased cancer risk
Patients with SLE have an increased risk of hematologic cancer and possibly lung and hepatobiliary cancers.
Bernatsky et al10 evaluated cancer risk in an international cohort of patients with SLE from 23 sites. Among patients with SLE, for all cancers combined, the standardized incidence ratio was 1.2; for hematologic cancers the ratio was 2.8; and for non-Hodgkin lymphoma it was 2.4. Surprisingly, although SLE is primarily a disease of women, reproductive cancer rates in patients with SLE did not differ from background rates. Bernatsky et al did not compare rates of cervical cancer, as many cancer registries do not record it. However, studies from the National Institutes of Health indicate that cervical dysplasia is common in patients with lupus.
Other interesting findings included an increased risk of hepatobiliary cancer, especially among men with SLE. Lung cancers were also increased, which has been observed in patients with other autoimmune diseases such as scleroderma and polymyositis. Smoking is a strong predictor for developing autoimmune conditions and may play a role in the observed increased cancer risk.
Early and advanced cardiovascular disease
Patients with SLE are at high risk of atherosclerotic cardiovascular disease. At the University of Pittsburgh Medical Center from 1980 to 1993, we compared the incidence of myocardial infarction in nearly 500 women with SLE and more than 2,000 women of similar age in the Framingham Offspring Study. At ages 15 to 24, women with lupus had a rate of 6.33 per 1,000 person-years; at age 25 to 34, the rate was 3.66 per 1,000 person-years. None of the Framingham women in those age groups had events.
Women ages 35 to 44 with lupus had a risk of heart attack 50 times higher than women in the Framingham cohort, and women in older age groups had a risk 2.5 to 4 times higher.11
Subclinical cardiovascular disease is also increased in women with SLE. Asanuma et al12 used electron-beam computed tomography to screen for coronary artery calcification in 65 patients with SLE and 69 control subjects with no history of coronary artery disease matched for age, sex, and race. Calcification was present in 31% of patients with lupus vs 9% of controls (P = .002). Roman et al13 performed carotid ultrasonography on 197 patients with lupus and 197 matched controls and found more plaque in patients with lupus (37%) than in controls (15%, P < .001).
Other data also suggest that women with lupus have advanced cardiovascular disease and develop it early, with most studies finding the greatest relative risk of cardiovascular disease between ages 18 and 45 years.
Traditional risk factors for cardiovascular disease cannot fully explain the increased risk. Many patients with lupus have metabolic syndrome, hypertension, and renal disease, but even after adjusting for these risk factors, patients with lupus still have about a 7 to 10 times higher risk of nonfatal coronary heart disease and a 17 times higher risk of fatal coronary heart disease.14
Many investigators are now exploring the role of immune dysfunction and inflammation in cardiovascular disease. A number of biomarkers have been proposed for predicting risk of cardiovascular disease in the general population. The list includes many inflammatory factors that rheumatologists have been studying for decades, including myeloperoxidase, autoantibodies, inflammatory cytokines, tumor necrosis factor alpha, and adhesion molecules, many of which also play a role in autoimmunity.
In our patients with SLE, we found that about 90% had three or more modifiable cardiovascular risk factors that were not being addressed appropriately (unpublished data). Lipid management was the least often addressed by rheumatologists and primary caregivers.
There is reason to believe that lupus patients are a high-risk group that merit aggressive risk-factor management, but no formal recommendations can be made without clear evidence that this approach improves outcomes.
SLE INCREASES THE RISK OF ADVERSE PREGNANCY OUTCOMES
Women with SLE more commonly miscarry and deliver low-birth-weight babies than do other women. A history of renal disease is the factor most predictive of poor pregnancy outcome, and the presence of certain autoantibodies increases the risk of neonatal lupus.
We recommend that women with lupus have inactive disease for at least 6 months before becoming pregnant.
ORAL CONTRACEPTIVES AND HORMONE REPLACEMENT
Hormone replacement therapy and oral contraceptives do not increase the risk of significant disease activity flares in lupus. However, women with lupus have an increased risk of cardiovascular disease and thrombosis.
Buyon et al15 randomly assigned 351 menopausal women with inactive or stable active SLE to receive either hormone replacement therapy or placebo for 12 months. No significant increase in severe flares of the disease was observed, although the treatment group had a mild increase in minor flares.
Petri et al16 randomly assigned 183 women with inactive or stable active SLE to receive either combined oral contraceptives or placebo for 12 months and found similar rates of disease activity between the two groups.
A weakness of these trials is that women with antiphospholipid antibodies in high titers or who had previous thrombotic events were excluded.
TREATMENTS ON THE HORIZON?
In the past 50 years, only three drug treatments have been approved for lupus: corticosteroids, hydroxychloroquine, and aspirin. Fortunately, research in autoimmune diseases has rapidly expanded, and new drugs are on the horizon.
Mycophenolate mofetil (CellCept) may be a reasonable alternative to cyclophosphamide (Cytoxan) for lupus nephritis and may be appropriate as maintenance therapy after induction with cyclophosphamide.
Ginzler et al,17 in a randomized, open-label trial in 140 patients with active lupus nephritis, gave either oral mycophenolate mofetil (initial dosage 1,000 mg/day, increased to 3,000 mg/day) or monthly intravenous cyclophosphamide (0.5 g/m2, increased to 1.0 g/m2). Mycophenolate mofetil was more effective in inducing remission than cyclophosphamide and had a better safety profile.
The Aspreva Lupus Management Study was designed to assess the efficacy of oral mycophenolate mofetil compared with intravenous cyclophosphamide in the initial treatment of patients with active class III–V lupus nephritis and to assess the long-term efficacy of mycophenolate mofetil compared with azathioprine in maintaining remission and renal function. It was the largest study of mycophenolate mofetil in lupus nephritis to date. There were 370 patients with SLE enrolled. In the 24-week induction phase, patients were randomized to receive open-label mycophenolate mofetil with a target dose of 3 g/day or intravenous cyclophosphamide 0.5 to 1.0 g/m2 in monthly pulses. Both groups received prednisone. Response to treatment was defined as a decrease in proteinuria (as measured by the urinary protein-creatinine ratio) and improvement or stabilization in serum creatinine.
The results (presented at the American College of Rheumatology Meeting, November 6–11, 2007, in Boston, MA) showed that 104 (56%) of the 185 patients treated with mycophenolate mofetil responded, compared with 98 (53%) of the 185 patients treated with intravenous cyclophosphamide (P = .575). The study therefore did not meet its primary objective of showing a superior response rate with mycophenolate mofetil compared with cyclophosphamide. There was no difference in adverse events. It is this author’s opinion that having an agent that is at least as good as cyclphosphamide in treating lupus nephritis is a major step forward.
Mycophenolate mofetil can cause fetal harm and should not be used during pregnancy. It is recommended that the drug be stopped for 3 to 6 months before a woman tries to conceive.
New drugs target B cells
Many new drugs for lupus target B cells.
Rituximab (Rituxan) is a monoclonal antibody that depletes B cells by targeting the B-cell-specific antigen CD20. It has been studied for treating lupus in several open-label studies that altogether have included more than 400 patients.18–21 Regimens included either those used in oncology for treatment of lymphoma or those used in rheumatoid arthritis, coupled with high-dose corticosteroids and cyclophosphamide. In early studies, nearly 80% of treated patients entered at least partial remission, and 25% to 50% are still in remission more than 12 months later.
The first randomized controlled trial of rituximab vs placebo was recently completed and presented at the American College of Rheumatology meeting, October 24–28, 2008, in Boston, MA. The EXPLORER trial (sponsored by Genentech) included 257 patients with moderate to severe disease activity. The results showed that there was no difference in major or partial clinical response (based on a change in the British Isles Lupus Assessment Group index) in those on rituximab (28.4%) vs placebo (29.6%) at 12 months (P = .97). Overall, adverse events were balanced between the groups. It is this author’s opinion that the bar for “response” was set very high in this study, considering that all patients who entered were fairly sick and received significant doses of corticosteroids that were tapered over the course of the trial.
B-cell toleragens, which render B cells incapable of presenting specific antigens, are also of interest.
Other experimental drugs target the soluble cytokine BLyS, which normally binds to a B-cell receptor and prolongs B-cell survival. It may also be possible to inhibit costimulatory pathways (which are normally important for inducing activation, proliferation, and class-switching of B cells) with the use of cytotoxic T-lymphocyte-associated antigen 4 immunoglobulin inhibitor (CTLA4Ig) and anti-CD40 ligand.
The results of a 12-month exploratory, phase II trial of abatacept (Bristol-Myers Squibb) in patients with SLE and active polyarthritis, serositis, or discoid lesions were presented at the American College of Rheumatology meeting in 2008. The primary and secondary end points (based on an adjudicated British Isles Lupus Assessment Group index) were not met. There were no differences in adverse events. Post hoc analyses of other clinical end points and biomarkers suggested that abatacept may have benefit in lupus. Further studies are under way.
Downstream blockade may also be useful, with drugs that inhibit inflammatory cytokines, particularly interferon alfa. This is now being tested in clinical trials.
- Narain S, Richards HB, Satoh M, et al. Diagnostic accuracy for lupus and other systemic autoimmune diseases in the community setting. Arch Intern Med 2004; 164:2435–2441.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13 BLK and ITGAM ITGAX. N Engl J Med 2008; 358:900–909.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211–216.
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204–210.
- Nath SK, Han S, Kim Howard X, et al. A nonsynonymous functional variant in integrin alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008; 40:152–154.
- Ramsey-Goldman R, Dunn JE, Huang CF, et al. Frequency of fractures in women with systemic lupus erythematosus: comparison with United States population data. Arthritis Rheum 1999; 42:882–890.
- Lee C, Almagor O, Dunlop DD, et al. Association between African-American race/ethnicity and low bone mineral density in women with systemic lupus erythematosus. Arthritis Rheum 2007; 57:585–592.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005; 52:1481–1490.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol 1997; 145:408–415.
- Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2407–2415.
- Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2399–2406. Erratum in: N Engl J Med 2006; 355:1746.
- Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001; 44:2331–2337.
- Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005; 142:953–962.
- Petri M, Kim MY, Kalunian KC, et al; OC SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005; 353:2550–2558.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004; 50:3580–3590.
- Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose escalation trial of rituximab. Arthritis Rheum 2004; 50:2580–2589.
- Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum 2002; 46:2673–2677.
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 2006; 54:723–732.
- Narain S, Richards HB, Satoh M, et al. Diagnostic accuracy for lupus and other systemic autoimmune diseases in the community setting. Arch Intern Med 2004; 164:2435–2441.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13 BLK and ITGAM ITGAX. N Engl J Med 2008; 358:900–909.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211–216.
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204–210.
- Nath SK, Han S, Kim Howard X, et al. A nonsynonymous functional variant in integrin alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008; 40:152–154.
- Ramsey-Goldman R, Dunn JE, Huang CF, et al. Frequency of fractures in women with systemic lupus erythematosus: comparison with United States population data. Arthritis Rheum 1999; 42:882–890.
- Lee C, Almagor O, Dunlop DD, et al. Association between African-American race/ethnicity and low bone mineral density in women with systemic lupus erythematosus. Arthritis Rheum 2007; 57:585–592.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005; 52:1481–1490.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol 1997; 145:408–415.
- Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2407–2415.
- Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2399–2406. Erratum in: N Engl J Med 2006; 355:1746.
- Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001; 44:2331–2337.
- Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005; 142:953–962.
- Petri M, Kim MY, Kalunian KC, et al; OC SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005; 353:2550–2558.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004; 50:3580–3590.
- Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose escalation trial of rituximab. Arthritis Rheum 2004; 50:2580–2589.
- Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum 2002; 46:2673–2677.
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 2006; 54:723–732.
KEY POINTS
- Lupus is often misdiagnosed. A person may be given a diagnosis based on a positive antinuclear antibody (ANA) test, a finding that alone is not sufficient to establish the diagnosis. In contrast, some patients with lupus may go several years and see numerous physicians before the proper diagnosis is made.
- One of the major mechanisms for lupus involves defective clearance of apoptotic cells, which act as a source of self-antigens. Because sun exposure can result in massive cell death of keratinocytes (skin cells), protection from the damaging effects of ultraviolet light plays an important role in the management of lupus.
- Patients at any age with SLE should have their modifiable cardiovascular risk factors managed.
- Drugs on the horizon for treating SLE inactivate B cells or their actions.
Role of barium esophagography in evaluating dysphagia
A 55-year-old woman presents with an intermittent sensation of food getting stuck in her mid to lower chest. The symptoms have occurred several times per year over the last 2 or 3 years and appear to be slowly worsening. She says she has no trouble swallowing liquids. She has a history of gastroesophageal reflux disease, for which she takes a proton pump inhibitor once a day. She says she has had no odynophagia, cough, regurgitation, or weight loss.
How should her symptoms best be evaluated?
DYSPHAGIA CAN BE OROPHARYNGEAL OR ESOPHAGEAL
Dysphagia is the subjective sensation of difficulty swallowing solids, liquids, or both. Symptoms can range from the inability to initiate a swallow to the sensation of esophageal obstruction. Other symptoms of esophageal disease may also be present, such as chest pain, heartburn, and regurgitation. There may also be nonesophageal symptoms related to the disease process causing the dysphagia.
Dysphagia can be separated into oropharyngeal and esophageal types.

Interestingly, many patients with symptoms of oropharyngeal dysphagia in fact have referred symptoms from primary esophageal dysphagia2; many patients with a distal mucosal ring describe a sense of something sticking in the cervical esophagus.
Esophageal dysphagia arises in the mid to distal esophagus or gastric cardia, and as a result, the symptoms are typically retrosternal.1 It can be caused by structural problems such as strictures, rings, webs, extrinsic compression, or a primary esophageal or gastroesophageal neoplasm, or by a primary motility abnormality such as achalasia (Table 1). Eosinophilic esophagitis is now a frequent cause of esophageal dysphagia, especially in white men.3
ESOPHAGOGRAPHY VS ENDOSCOPY IN EVALUATING DYSPHAGIA
Many gastroenterologists recommend endoscopy rather than barium esophagography as the initial examination in patients with dysphagia.4–8 Each test has certain advantages.
Advantages of endoscopy. Endoscopy is superior to esophagography in detecting milder grades of esophagitis. Further, interventions can be performed endoscopically (eg, dilation, biopsy, attachment of a wireless pH testing probe) that cannot be done during a radiographic procedure, and endoscopy does not expose the patient to radiation.
Advantages of esophagography. Endoscopy cannot detect evidence of gastroesophageal reflux disease unless mucosal injury is present. In dysphagia, the radiologic findings correlate well with endoscopic findings, including the detection of esophageal malignancy and moderate to severe esophagitis. Further, motility disorders can be detected with barium esophagography but not with endoscopy.9,10
Subtle abnormalities, especially rings and strictures, may be missed by narrow-diameter (9.8–10 mm) modern upper-endoscopic equipment. Further, esophagography is noninvasive, costs less, and may be more convenient (it does not require sedation and a chaperone for the patient after sedation). This examination also provides dynamic evaluation of the complex process of swallowing. Causes of dysphagia external to the esophagus can also be determined.
In view of the respective advantages and disadvantages of the two methods, we believe that in most instances barium esophagography should be the initial examination,1,9,11–15 and at our institution most patients presenting with dysphagia undergo barium esophagography before they undergo other examinations.14
OBTAIN A HISTORY BEFORE ORDERING ESOPHAGOGRAPHY
Before a barium examination of the esophagus is done, a focused medical history should be obtained, as it can guide the further workup as well as the esophageal study itself.
An attempt should be made to determine whether the dysphagia is oropharyngeal or if it is esophageal, as the former is generally best initially evaluated by a speech and language pathologist. Generally, the physician who orders the test judges whether the patient has oropharyngeal or esophageal dysphagia. Often, both an oropharyngeal examination, performed by a speech and language pathologist, and an esophageal examination, performed by a radiologist, are ordered.
Rapidly progressive symptoms, especially if accompanied by weight loss, should make one suspect cancer. Chronic symptoms usually point to gastroesophageal reflux disease or a motility disorder such as achalasia. Liquid dysphagia almost always means the patient has a motility disorder such as achalasia.
In view of the possibility of eosinophilic esophagitis, one should ask about food or seasonal allergies, especially in young patients with intermittent difficulty swallowing solids.3
BARIUM ESOPHAGOGRAPHY HAS EIGHT SEPARATE PHASES
Barium esophagography is tailored to the patient with dysphagia on the basis of his or her history. The standard examination is divided into eight separate phases (see below).14 Each phase addresses a specific question or questions concerning the structure and function of the esophagus.
At our institution, the first phase of the examination is determined by the presenting symptoms. If the patient has liquid dysphagia, we start with a timed barium swallow to assess esophageal emptying. If the patient does not have liquid dysphagia, we start with an air-contrast mucosal examination.
The patient must be cooperative and mobile to complete all phases of the examination.
Timed barium swallow to measure esophageal emptying
The timed barium swallow is an objective measure of esophageal emptying.16–18 This technique is essential in the initial evaluation of a patient with liquid dysphagia, a symptom common in patients with severe dysmotility, usually achalasia.
We use this examination in our patients with suspected or confirmed achalasia and to follow up patients who have been treated with pneumatic dilatation, botulinum toxin injection, and Heller myotomy.17,18 In addition, this timed test is an objective measure of emptying in patients who have undergone intervention but whose symptoms have not subjectively improved, and can suggest that further intervention may be required.
Air-contrast or mucosal phase
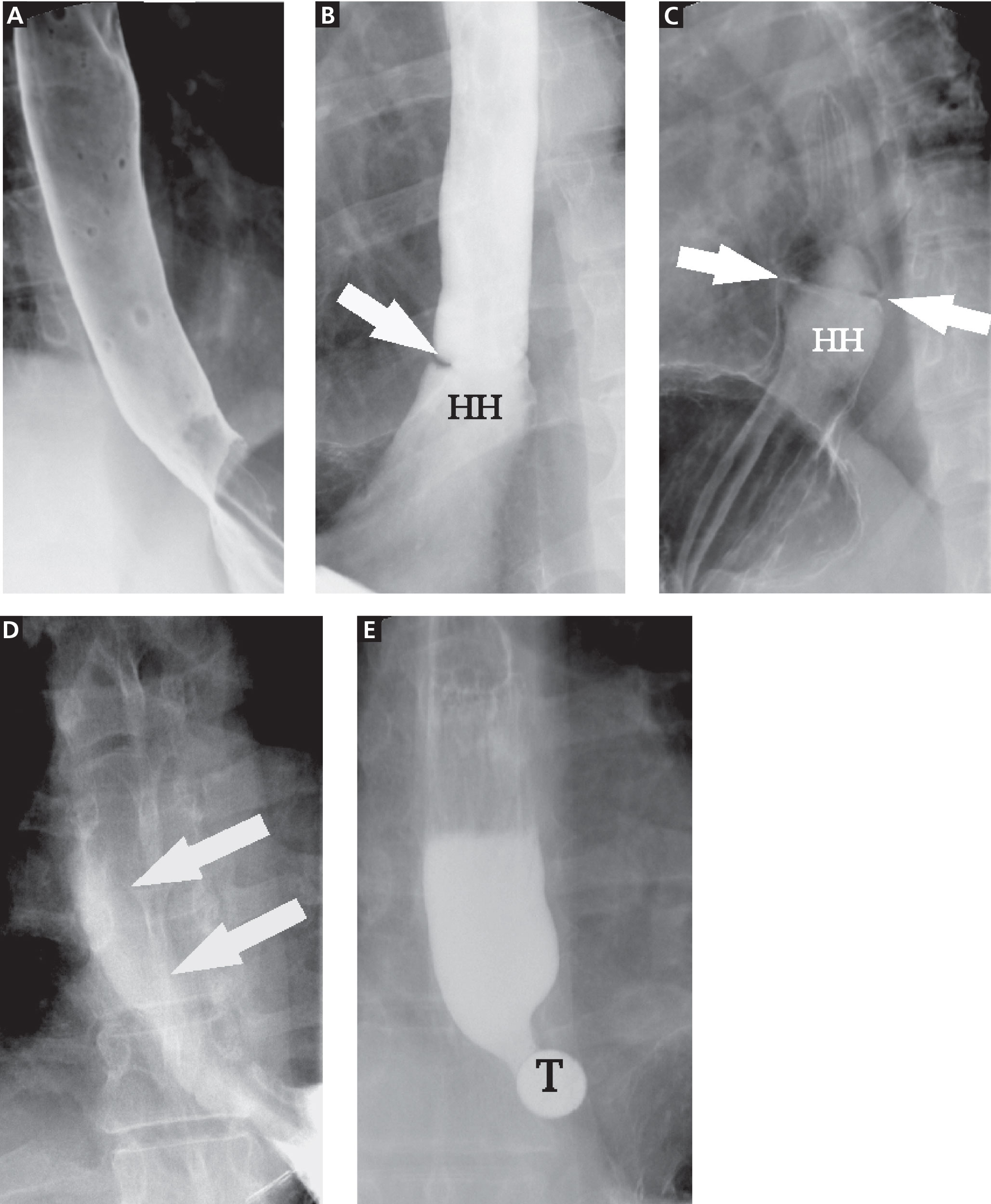
Although this phase is not as sensitive as endoscopy, it can detect masses, mucosal erosions, ulcers, and—most importantly in our experience—fixed hernias. Patients with a fixed hernia have a foreshortened esophagus, which is important to know about before repairing the hernia. Many esophageal surgeons believe that a foreshortened esophagus precludes a standard Nissen fundoplication and necessitates an esophageal lengthening procedure (ie, Collis gastroplasty with a Nissen fundoplication).14
Motility phase
The third phase examines esophageal motility. With the patient in a semiprone position, low-density barium is given in single swallows, separated by 25 to 30 seconds. The images are recorded on digital media to allow one to review them frame by frame.
The findings on this phase correlate well with those of manometry.19 This portion of the examination also uses impedance monitoring to assess bolus transfer, an aspect not evaluated by manometry.20,21 Impedance monitoring detects changes in resistance to current flow and correlates well with esophagraphic findings regarding bolus transfer.
While many patients with dysphagia also undergo esophageal manometry, the findings from this phase of the esophagographic examination may be the first indication of an esophageal motility disorder. In fact, this portion of the examination shows the distinct advantage of esophagography over endoscopy as the initial test in patients with dysphagia, as endoscopy may not identify patients with achalasia, especially early on.4
Single-contrast (full-column) phase to detect strictures, rings
The fourth phase of the esophagographic evaluation is the distended, single-contrast examination (Figure 2B). This is performed in the semiprone position with the patient rapidly drinking thin barium. It is done to detect esophageal strictures, rings, and contour abnormalities caused by extrinsic processes. Subtle abnormalities shown by this technique, including benign strictures and rings, are often not visualized with endoscopy.
Mucosal relief phase
The fifth phase is performed with a collapsed esophagus immediately after the distended, single-contrast phase, where spot films are taken of the barium-coated, collapsed esophagus (Figure 2C). This phase is used to evaluate thickened mucosal folds, a common finding in moderate to severe reflux esophagitis.
Reflux evaluation
Provocative maneuvers are used in the sixth phase to elicit gastroesophageal reflux (Figure 2D). With the patient supine, he or she is asked to roll side to side, do a Valsalva maneuver, and do a straight-leg raise. The patient then sips water in the supine position to assess for reflux (the water siphon test). If reflux is seen, the cause, the height of the reflux, and the duration of reflux retention are recorded.
Solid-bolus phase to assess strictures
In the seventh phase, the patient swallows a 13-mm barium tablet (Figure 2E). This allows one to assess the significance of a ring or stricture and to assess if dysphagia symptoms recur as a result of tablet obstruction. Subtle strictures that were not detected during the prior phases can also be detected using a tablet. If obstruction or impaired passage occurs, the site of obstruction and the presence or absence of symptoms are recorded.
Modified esophagography to assess the oropharynx
The final or eighth phase of barium esophagography is called “modified barium esophagography” or the modified barium swallow. However, it may be the first phase of the examination performed or the only portion of the examination performed, or it may not be performed at all.
Modified barium esophagography is used to define the anatomy of the oropharynx and to assess its function in swallowing.12 It may also guide rehabilitation strategies aimed at eliminating a patient’s swallowing symptoms.
Most patients referred for this test have sustained damage to the central nervous system or structures of the oropharynx, such as stroke or radiation therapy for laryngeal cancer. Many have difficulty in starting to swallow, aspirate when they try to swallow, or both.

The final esophagographic report should document the findings of each phase of the examination (Table 2).
WHAT HAPPENED TO OUR PATIENT?
Our patient underwent barium esophagography (Figure 2). A distal mucosal ring that transiently obstructed a 13-mm tablet was found. The patient underwent endoscopy and the ring was dilated. No biopsies were necessary.
- Levine MS, Rubesin SE. Radiologic investigation of dysphagia. AJR Am J Roentgenol 1990; 154:1157–1163.
- Smith DF, Ott DJ, Gelfand DW, Chen MY. Lower esophageal mucosal ring: correlation of referred symptoms with radiographic findings using a marshmallow bolus. AJR Am J Roentgenol 1998; 171:1361–1365.
- Furuta GT, Liacouras CA, Collins MH, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–1363.
- Spechler SJ. American Gastroenterological Association medical position statement on treatment of patients with dysphagia caused by benign disorders of the distal esophagus. Gastroenterology 1999; 117:229–233.
- American Society for Gastrointestinal Endoscopy. Appropriate use of gastrointestinal endoscopy. Gastrointest Endosc 2000; 52:831–837.
- Esfandyari T, Potter JW, Vaezi MF. Dysphagia: a cost analysis of the diagnostic approach. Am J Gastroenterol 2002; 97:2733–2737.
- Varadarajulu S, Eloubeidi MA, Patel RS, et al. The yield and the predictors of esophageal pathology when upper endoscopy is used for the initial evaluation of dysphagia. Gastrointest Endosc 2005; 61:804–808.
- Standards of Practice Committee. Role of endoscopy in the management of GERD. Gastrointest Endosc 2007; 66:219–224.
- Halpert RD, Feczko PJ, Spickler EM, Ackerman LV. Radiological assessment of dysphagia with endoscopic correlation. Radiology 1985; 157:599–602.
- Ott DJ. Gastroesophageal reflux disease. Radiol Clin North Am 1994; 32:1147–1166.
- Ekberg O, Pokieser P. Radiologic evaluation of the dysphagic patient. Eur Radiol 1997; 7:1285–1295.
- Logemann JA. Role of the modified barium swallow in management of patients with dysphagia. Otolaryngol Head Neck Surg 1997; 116:335–338.
- Baker ME, Rice TW. Radiologic evaluation of the esophagus: methods and value in motility disorders and GERD. Semin Thorac Cardiovasc Surg 2001; 13:201–225.
- Baker ME, Einstein DM, Herts BR, et al. Gastroesophageal reflux disease: integrating the barium esophagram before and after antire-flux surgery. Radiology 2007; 243:329–339.
- Levine MS, Rubesin SE, Laufer I. Barium esophagography: a study for all seasons. Clin Gastroenterol Hepatol 2008; 6:11–25.
- deOliveira JM, Birgisson S, Doinoff C, et al. Timed barium swallow: a simple technique for evaluating esophageal emptying in patients with achalasia. AJR Am J Roentgenol 1997; 169:473–479.
- Kostic SV, Rice TW, Baker ME, et al. Time barium esophagram: a simple physiologic assessment for achalasia. J Thorac Cardiovasc Surg 2000; 120:935–943.
- Vaezi MF, Baker ME, Achkar E, Richter JE. Timed barium oesophagram: better predictor of long term success after pneumatic dilation in achalasia than symptom assessment. Gut 2002; 50:765–770.
- Hewson EG, Ott DJ, Dalton CB, Chen YM, Wu WC, Richter JE. Manometry and radiology. Complementary studies in the assessment of esophageal motility disorders. Gastroenterology 1990; 98:626–632.
- Imam H, Shay S, Ali A, Baker M. Bolus transit patterns in healthy subjects: a study using simultaneous impedance monitoring, video-esophagram, and esophageal manometry. Am J Physiol Gastrointest Liver Physiol 2005;G1000–G1006.
- Imam H, Baker M, Shay S. Simultaneous barium esophagram, impedance monitoring and manometry in patients with dysphagia due to a tight fundoplication [abstract]. Gastroenterology 2004; 126:A-639.
A 55-year-old woman presents with an intermittent sensation of food getting stuck in her mid to lower chest. The symptoms have occurred several times per year over the last 2 or 3 years and appear to be slowly worsening. She says she has no trouble swallowing liquids. She has a history of gastroesophageal reflux disease, for which she takes a proton pump inhibitor once a day. She says she has had no odynophagia, cough, regurgitation, or weight loss.
How should her symptoms best be evaluated?
DYSPHAGIA CAN BE OROPHARYNGEAL OR ESOPHAGEAL
Dysphagia is the subjective sensation of difficulty swallowing solids, liquids, or both. Symptoms can range from the inability to initiate a swallow to the sensation of esophageal obstruction. Other symptoms of esophageal disease may also be present, such as chest pain, heartburn, and regurgitation. There may also be nonesophageal symptoms related to the disease process causing the dysphagia.
Dysphagia can be separated into oropharyngeal and esophageal types.
Interestingly, many patients with symptoms of oropharyngeal dysphagia in fact have referred symptoms from primary esophageal dysphagia2; many patients with a distal mucosal ring describe a sense of something sticking in the cervical esophagus.
Esophageal dysphagia arises in the mid to distal esophagus or gastric cardia, and as a result, the symptoms are typically retrosternal.1 It can be caused by structural problems such as strictures, rings, webs, extrinsic compression, or a primary esophageal or gastroesophageal neoplasm, or by a primary motility abnormality such as achalasia (Table 1). Eosinophilic esophagitis is now a frequent cause of esophageal dysphagia, especially in white men.3
ESOPHAGOGRAPHY VS ENDOSCOPY IN EVALUATING DYSPHAGIA
Many gastroenterologists recommend endoscopy rather than barium esophagography as the initial examination in patients with dysphagia.4–8 Each test has certain advantages.
Advantages of endoscopy. Endoscopy is superior to esophagography in detecting milder grades of esophagitis. Further, interventions can be performed endoscopically (eg, dilation, biopsy, attachment of a wireless pH testing probe) that cannot be done during a radiographic procedure, and endoscopy does not expose the patient to radiation.
Advantages of esophagography. Endoscopy cannot detect evidence of gastroesophageal reflux disease unless mucosal injury is present. In dysphagia, the radiologic findings correlate well with endoscopic findings, including the detection of esophageal malignancy and moderate to severe esophagitis. Further, motility disorders can be detected with barium esophagography but not with endoscopy.9,10
Subtle abnormalities, especially rings and strictures, may be missed by narrow-diameter (9.8–10 mm) modern upper-endoscopic equipment. Further, esophagography is noninvasive, costs less, and may be more convenient (it does not require sedation and a chaperone for the patient after sedation). This examination also provides dynamic evaluation of the complex process of swallowing. Causes of dysphagia external to the esophagus can also be determined.
In view of the respective advantages and disadvantages of the two methods, we believe that in most instances barium esophagography should be the initial examination,1,9,11–15 and at our institution most patients presenting with dysphagia undergo barium esophagography before they undergo other examinations.14
OBTAIN A HISTORY BEFORE ORDERING ESOPHAGOGRAPHY
Before a barium examination of the esophagus is done, a focused medical history should be obtained, as it can guide the further workup as well as the esophageal study itself.
An attempt should be made to determine whether the dysphagia is oropharyngeal or if it is esophageal, as the former is generally best initially evaluated by a speech and language pathologist. Generally, the physician who orders the test judges whether the patient has oropharyngeal or esophageal dysphagia. Often, both an oropharyngeal examination, performed by a speech and language pathologist, and an esophageal examination, performed by a radiologist, are ordered.
Rapidly progressive symptoms, especially if accompanied by weight loss, should make one suspect cancer. Chronic symptoms usually point to gastroesophageal reflux disease or a motility disorder such as achalasia. Liquid dysphagia almost always means the patient has a motility disorder such as achalasia.
In view of the possibility of eosinophilic esophagitis, one should ask about food or seasonal allergies, especially in young patients with intermittent difficulty swallowing solids.3
BARIUM ESOPHAGOGRAPHY HAS EIGHT SEPARATE PHASES
Barium esophagography is tailored to the patient with dysphagia on the basis of his or her history. The standard examination is divided into eight separate phases (see below).14 Each phase addresses a specific question or questions concerning the structure and function of the esophagus.
At our institution, the first phase of the examination is determined by the presenting symptoms. If the patient has liquid dysphagia, we start with a timed barium swallow to assess esophageal emptying. If the patient does not have liquid dysphagia, we start with an air-contrast mucosal examination.
The patient must be cooperative and mobile to complete all phases of the examination.
Timed barium swallow to measure esophageal emptying
The timed barium swallow is an objective measure of esophageal emptying.16–18 This technique is essential in the initial evaluation of a patient with liquid dysphagia, a symptom common in patients with severe dysmotility, usually achalasia.
We use this examination in our patients with suspected or confirmed achalasia and to follow up patients who have been treated with pneumatic dilatation, botulinum toxin injection, and Heller myotomy.17,18 In addition, this timed test is an objective measure of emptying in patients who have undergone intervention but whose symptoms have not subjectively improved, and can suggest that further intervention may be required.
Air-contrast or mucosal phase
Although this phase is not as sensitive as endoscopy, it can detect masses, mucosal erosions, ulcers, and—most importantly in our experience—fixed hernias. Patients with a fixed hernia have a foreshortened esophagus, which is important to know about before repairing the hernia. Many esophageal surgeons believe that a foreshortened esophagus precludes a standard Nissen fundoplication and necessitates an esophageal lengthening procedure (ie, Collis gastroplasty with a Nissen fundoplication).14
Motility phase
The third phase examines esophageal motility. With the patient in a semiprone position, low-density barium is given in single swallows, separated by 25 to 30 seconds. The images are recorded on digital media to allow one to review them frame by frame.
The findings on this phase correlate well with those of manometry.19 This portion of the examination also uses impedance monitoring to assess bolus transfer, an aspect not evaluated by manometry.20,21 Impedance monitoring detects changes in resistance to current flow and correlates well with esophagraphic findings regarding bolus transfer.
While many patients with dysphagia also undergo esophageal manometry, the findings from this phase of the esophagographic examination may be the first indication of an esophageal motility disorder. In fact, this portion of the examination shows the distinct advantage of esophagography over endoscopy as the initial test in patients with dysphagia, as endoscopy may not identify patients with achalasia, especially early on.4
Single-contrast (full-column) phase to detect strictures, rings
The fourth phase of the esophagographic evaluation is the distended, single-contrast examination (Figure 2B). This is performed in the semiprone position with the patient rapidly drinking thin barium. It is done to detect esophageal strictures, rings, and contour abnormalities caused by extrinsic processes. Subtle abnormalities shown by this technique, including benign strictures and rings, are often not visualized with endoscopy.
Mucosal relief phase
The fifth phase is performed with a collapsed esophagus immediately after the distended, single-contrast phase, where spot films are taken of the barium-coated, collapsed esophagus (Figure 2C). This phase is used to evaluate thickened mucosal folds, a common finding in moderate to severe reflux esophagitis.
Reflux evaluation
Provocative maneuvers are used in the sixth phase to elicit gastroesophageal reflux (Figure 2D). With the patient supine, he or she is asked to roll side to side, do a Valsalva maneuver, and do a straight-leg raise. The patient then sips water in the supine position to assess for reflux (the water siphon test). If reflux is seen, the cause, the height of the reflux, and the duration of reflux retention are recorded.
Solid-bolus phase to assess strictures
In the seventh phase, the patient swallows a 13-mm barium tablet (Figure 2E). This allows one to assess the significance of a ring or stricture and to assess if dysphagia symptoms recur as a result of tablet obstruction. Subtle strictures that were not detected during the prior phases can also be detected using a tablet. If obstruction or impaired passage occurs, the site of obstruction and the presence or absence of symptoms are recorded.
Modified esophagography to assess the oropharynx
The final or eighth phase of barium esophagography is called “modified barium esophagography” or the modified barium swallow. However, it may be the first phase of the examination performed or the only portion of the examination performed, or it may not be performed at all.
Modified barium esophagography is used to define the anatomy of the oropharynx and to assess its function in swallowing.12 It may also guide rehabilitation strategies aimed at eliminating a patient’s swallowing symptoms.
Most patients referred for this test have sustained damage to the central nervous system or structures of the oropharynx, such as stroke or radiation therapy for laryngeal cancer. Many have difficulty in starting to swallow, aspirate when they try to swallow, or both.
The final esophagographic report should document the findings of each phase of the examination (Table 2).
WHAT HAPPENED TO OUR PATIENT?
Our patient underwent barium esophagography (Figure 2). A distal mucosal ring that transiently obstructed a 13-mm tablet was found. The patient underwent endoscopy and the ring was dilated. No biopsies were necessary.
A 55-year-old woman presents with an intermittent sensation of food getting stuck in her mid to lower chest. The symptoms have occurred several times per year over the last 2 or 3 years and appear to be slowly worsening. She says she has no trouble swallowing liquids. She has a history of gastroesophageal reflux disease, for which she takes a proton pump inhibitor once a day. She says she has had no odynophagia, cough, regurgitation, or weight loss.
How should her symptoms best be evaluated?
DYSPHAGIA CAN BE OROPHARYNGEAL OR ESOPHAGEAL
Dysphagia is the subjective sensation of difficulty swallowing solids, liquids, or both. Symptoms can range from the inability to initiate a swallow to the sensation of esophageal obstruction. Other symptoms of esophageal disease may also be present, such as chest pain, heartburn, and regurgitation. There may also be nonesophageal symptoms related to the disease process causing the dysphagia.
Dysphagia can be separated into oropharyngeal and esophageal types.
Interestingly, many patients with symptoms of oropharyngeal dysphagia in fact have referred symptoms from primary esophageal dysphagia2; many patients with a distal mucosal ring describe a sense of something sticking in the cervical esophagus.
Esophageal dysphagia arises in the mid to distal esophagus or gastric cardia, and as a result, the symptoms are typically retrosternal.1 It can be caused by structural problems such as strictures, rings, webs, extrinsic compression, or a primary esophageal or gastroesophageal neoplasm, or by a primary motility abnormality such as achalasia (Table 1). Eosinophilic esophagitis is now a frequent cause of esophageal dysphagia, especially in white men.3
ESOPHAGOGRAPHY VS ENDOSCOPY IN EVALUATING DYSPHAGIA
Many gastroenterologists recommend endoscopy rather than barium esophagography as the initial examination in patients with dysphagia.4–8 Each test has certain advantages.
Advantages of endoscopy. Endoscopy is superior to esophagography in detecting milder grades of esophagitis. Further, interventions can be performed endoscopically (eg, dilation, biopsy, attachment of a wireless pH testing probe) that cannot be done during a radiographic procedure, and endoscopy does not expose the patient to radiation.
Advantages of esophagography. Endoscopy cannot detect evidence of gastroesophageal reflux disease unless mucosal injury is present. In dysphagia, the radiologic findings correlate well with endoscopic findings, including the detection of esophageal malignancy and moderate to severe esophagitis. Further, motility disorders can be detected with barium esophagography but not with endoscopy.9,10
Subtle abnormalities, especially rings and strictures, may be missed by narrow-diameter (9.8–10 mm) modern upper-endoscopic equipment. Further, esophagography is noninvasive, costs less, and may be more convenient (it does not require sedation and a chaperone for the patient after sedation). This examination also provides dynamic evaluation of the complex process of swallowing. Causes of dysphagia external to the esophagus can also be determined.
In view of the respective advantages and disadvantages of the two methods, we believe that in most instances barium esophagography should be the initial examination,1,9,11–15 and at our institution most patients presenting with dysphagia undergo barium esophagography before they undergo other examinations.14
OBTAIN A HISTORY BEFORE ORDERING ESOPHAGOGRAPHY
Before a barium examination of the esophagus is done, a focused medical history should be obtained, as it can guide the further workup as well as the esophageal study itself.
An attempt should be made to determine whether the dysphagia is oropharyngeal or if it is esophageal, as the former is generally best initially evaluated by a speech and language pathologist. Generally, the physician who orders the test judges whether the patient has oropharyngeal or esophageal dysphagia. Often, both an oropharyngeal examination, performed by a speech and language pathologist, and an esophageal examination, performed by a radiologist, are ordered.
Rapidly progressive symptoms, especially if accompanied by weight loss, should make one suspect cancer. Chronic symptoms usually point to gastroesophageal reflux disease or a motility disorder such as achalasia. Liquid dysphagia almost always means the patient has a motility disorder such as achalasia.
In view of the possibility of eosinophilic esophagitis, one should ask about food or seasonal allergies, especially in young patients with intermittent difficulty swallowing solids.3
BARIUM ESOPHAGOGRAPHY HAS EIGHT SEPARATE PHASES
Barium esophagography is tailored to the patient with dysphagia on the basis of his or her history. The standard examination is divided into eight separate phases (see below).14 Each phase addresses a specific question or questions concerning the structure and function of the esophagus.
At our institution, the first phase of the examination is determined by the presenting symptoms. If the patient has liquid dysphagia, we start with a timed barium swallow to assess esophageal emptying. If the patient does not have liquid dysphagia, we start with an air-contrast mucosal examination.
The patient must be cooperative and mobile to complete all phases of the examination.
Timed barium swallow to measure esophageal emptying
The timed barium swallow is an objective measure of esophageal emptying.16–18 This technique is essential in the initial evaluation of a patient with liquid dysphagia, a symptom common in patients with severe dysmotility, usually achalasia.
We use this examination in our patients with suspected or confirmed achalasia and to follow up patients who have been treated with pneumatic dilatation, botulinum toxin injection, and Heller myotomy.17,18 In addition, this timed test is an objective measure of emptying in patients who have undergone intervention but whose symptoms have not subjectively improved, and can suggest that further intervention may be required.
Air-contrast or mucosal phase
Although this phase is not as sensitive as endoscopy, it can detect masses, mucosal erosions, ulcers, and—most importantly in our experience—fixed hernias. Patients with a fixed hernia have a foreshortened esophagus, which is important to know about before repairing the hernia. Many esophageal surgeons believe that a foreshortened esophagus precludes a standard Nissen fundoplication and necessitates an esophageal lengthening procedure (ie, Collis gastroplasty with a Nissen fundoplication).14
Motility phase
The third phase examines esophageal motility. With the patient in a semiprone position, low-density barium is given in single swallows, separated by 25 to 30 seconds. The images are recorded on digital media to allow one to review them frame by frame.
The findings on this phase correlate well with those of manometry.19 This portion of the examination also uses impedance monitoring to assess bolus transfer, an aspect not evaluated by manometry.20,21 Impedance monitoring detects changes in resistance to current flow and correlates well with esophagraphic findings regarding bolus transfer.
While many patients with dysphagia also undergo esophageal manometry, the findings from this phase of the esophagographic examination may be the first indication of an esophageal motility disorder. In fact, this portion of the examination shows the distinct advantage of esophagography over endoscopy as the initial test in patients with dysphagia, as endoscopy may not identify patients with achalasia, especially early on.4
Single-contrast (full-column) phase to detect strictures, rings
The fourth phase of the esophagographic evaluation is the distended, single-contrast examination (Figure 2B). This is performed in the semiprone position with the patient rapidly drinking thin barium. It is done to detect esophageal strictures, rings, and contour abnormalities caused by extrinsic processes. Subtle abnormalities shown by this technique, including benign strictures and rings, are often not visualized with endoscopy.
Mucosal relief phase
The fifth phase is performed with a collapsed esophagus immediately after the distended, single-contrast phase, where spot films are taken of the barium-coated, collapsed esophagus (Figure 2C). This phase is used to evaluate thickened mucosal folds, a common finding in moderate to severe reflux esophagitis.
Reflux evaluation
Provocative maneuvers are used in the sixth phase to elicit gastroesophageal reflux (Figure 2D). With the patient supine, he or she is asked to roll side to side, do a Valsalva maneuver, and do a straight-leg raise. The patient then sips water in the supine position to assess for reflux (the water siphon test). If reflux is seen, the cause, the height of the reflux, and the duration of reflux retention are recorded.
Solid-bolus phase to assess strictures
In the seventh phase, the patient swallows a 13-mm barium tablet (Figure 2E). This allows one to assess the significance of a ring or stricture and to assess if dysphagia symptoms recur as a result of tablet obstruction. Subtle strictures that were not detected during the prior phases can also be detected using a tablet. If obstruction or impaired passage occurs, the site of obstruction and the presence or absence of symptoms are recorded.
Modified esophagography to assess the oropharynx
The final or eighth phase of barium esophagography is called “modified barium esophagography” or the modified barium swallow. However, it may be the first phase of the examination performed or the only portion of the examination performed, or it may not be performed at all.
Modified barium esophagography is used to define the anatomy of the oropharynx and to assess its function in swallowing.12 It may also guide rehabilitation strategies aimed at eliminating a patient’s swallowing symptoms.
Most patients referred for this test have sustained damage to the central nervous system or structures of the oropharynx, such as stroke or radiation therapy for laryngeal cancer. Many have difficulty in starting to swallow, aspirate when they try to swallow, or both.
The final esophagographic report should document the findings of each phase of the examination (Table 2).
WHAT HAPPENED TO OUR PATIENT?
Our patient underwent barium esophagography (Figure 2). A distal mucosal ring that transiently obstructed a 13-mm tablet was found. The patient underwent endoscopy and the ring was dilated. No biopsies were necessary.
- Levine MS, Rubesin SE. Radiologic investigation of dysphagia. AJR Am J Roentgenol 1990; 154:1157–1163.
- Smith DF, Ott DJ, Gelfand DW, Chen MY. Lower esophageal mucosal ring: correlation of referred symptoms with radiographic findings using a marshmallow bolus. AJR Am J Roentgenol 1998; 171:1361–1365.
- Furuta GT, Liacouras CA, Collins MH, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–1363.
- Spechler SJ. American Gastroenterological Association medical position statement on treatment of patients with dysphagia caused by benign disorders of the distal esophagus. Gastroenterology 1999; 117:229–233.
- American Society for Gastrointestinal Endoscopy. Appropriate use of gastrointestinal endoscopy. Gastrointest Endosc 2000; 52:831–837.
- Esfandyari T, Potter JW, Vaezi MF. Dysphagia: a cost analysis of the diagnostic approach. Am J Gastroenterol 2002; 97:2733–2737.
- Varadarajulu S, Eloubeidi MA, Patel RS, et al. The yield and the predictors of esophageal pathology when upper endoscopy is used for the initial evaluation of dysphagia. Gastrointest Endosc 2005; 61:804–808.
- Standards of Practice Committee. Role of endoscopy in the management of GERD. Gastrointest Endosc 2007; 66:219–224.
- Halpert RD, Feczko PJ, Spickler EM, Ackerman LV. Radiological assessment of dysphagia with endoscopic correlation. Radiology 1985; 157:599–602.
- Ott DJ. Gastroesophageal reflux disease. Radiol Clin North Am 1994; 32:1147–1166.
- Ekberg O, Pokieser P. Radiologic evaluation of the dysphagic patient. Eur Radiol 1997; 7:1285–1295.
- Logemann JA. Role of the modified barium swallow in management of patients with dysphagia. Otolaryngol Head Neck Surg 1997; 116:335–338.
- Baker ME, Rice TW. Radiologic evaluation of the esophagus: methods and value in motility disorders and GERD. Semin Thorac Cardiovasc Surg 2001; 13:201–225.
- Baker ME, Einstein DM, Herts BR, et al. Gastroesophageal reflux disease: integrating the barium esophagram before and after antire-flux surgery. Radiology 2007; 243:329–339.
- Levine MS, Rubesin SE, Laufer I. Barium esophagography: a study for all seasons. Clin Gastroenterol Hepatol 2008; 6:11–25.
- deOliveira JM, Birgisson S, Doinoff C, et al. Timed barium swallow: a simple technique for evaluating esophageal emptying in patients with achalasia. AJR Am J Roentgenol 1997; 169:473–479.
- Kostic SV, Rice TW, Baker ME, et al. Time barium esophagram: a simple physiologic assessment for achalasia. J Thorac Cardiovasc Surg 2000; 120:935–943.
- Vaezi MF, Baker ME, Achkar E, Richter JE. Timed barium oesophagram: better predictor of long term success after pneumatic dilation in achalasia than symptom assessment. Gut 2002; 50:765–770.
- Hewson EG, Ott DJ, Dalton CB, Chen YM, Wu WC, Richter JE. Manometry and radiology. Complementary studies in the assessment of esophageal motility disorders. Gastroenterology 1990; 98:626–632.
- Imam H, Shay S, Ali A, Baker M. Bolus transit patterns in healthy subjects: a study using simultaneous impedance monitoring, video-esophagram, and esophageal manometry. Am J Physiol Gastrointest Liver Physiol 2005;G1000–G1006.
- Imam H, Baker M, Shay S. Simultaneous barium esophagram, impedance monitoring and manometry in patients with dysphagia due to a tight fundoplication [abstract]. Gastroenterology 2004; 126:A-639.
- Levine MS, Rubesin SE. Radiologic investigation of dysphagia. AJR Am J Roentgenol 1990; 154:1157–1163.
- Smith DF, Ott DJ, Gelfand DW, Chen MY. Lower esophageal mucosal ring: correlation of referred symptoms with radiographic findings using a marshmallow bolus. AJR Am J Roentgenol 1998; 171:1361–1365.
- Furuta GT, Liacouras CA, Collins MH, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–1363.
- Spechler SJ. American Gastroenterological Association medical position statement on treatment of patients with dysphagia caused by benign disorders of the distal esophagus. Gastroenterology 1999; 117:229–233.
- American Society for Gastrointestinal Endoscopy. Appropriate use of gastrointestinal endoscopy. Gastrointest Endosc 2000; 52:831–837.
- Esfandyari T, Potter JW, Vaezi MF. Dysphagia: a cost analysis of the diagnostic approach. Am J Gastroenterol 2002; 97:2733–2737.
- Varadarajulu S, Eloubeidi MA, Patel RS, et al. The yield and the predictors of esophageal pathology when upper endoscopy is used for the initial evaluation of dysphagia. Gastrointest Endosc 2005; 61:804–808.
- Standards of Practice Committee. Role of endoscopy in the management of GERD. Gastrointest Endosc 2007; 66:219–224.
- Halpert RD, Feczko PJ, Spickler EM, Ackerman LV. Radiological assessment of dysphagia with endoscopic correlation. Radiology 1985; 157:599–602.
- Ott DJ. Gastroesophageal reflux disease. Radiol Clin North Am 1994; 32:1147–1166.
- Ekberg O, Pokieser P. Radiologic evaluation of the dysphagic patient. Eur Radiol 1997; 7:1285–1295.
- Logemann JA. Role of the modified barium swallow in management of patients with dysphagia. Otolaryngol Head Neck Surg 1997; 116:335–338.
- Baker ME, Rice TW. Radiologic evaluation of the esophagus: methods and value in motility disorders and GERD. Semin Thorac Cardiovasc Surg 2001; 13:201–225.
- Baker ME, Einstein DM, Herts BR, et al. Gastroesophageal reflux disease: integrating the barium esophagram before and after antire-flux surgery. Radiology 2007; 243:329–339.
- Levine MS, Rubesin SE, Laufer I. Barium esophagography: a study for all seasons. Clin Gastroenterol Hepatol 2008; 6:11–25.
- deOliveira JM, Birgisson S, Doinoff C, et al. Timed barium swallow: a simple technique for evaluating esophageal emptying in patients with achalasia. AJR Am J Roentgenol 1997; 169:473–479.
- Kostic SV, Rice TW, Baker ME, et al. Time barium esophagram: a simple physiologic assessment for achalasia. J Thorac Cardiovasc Surg 2000; 120:935–943.
- Vaezi MF, Baker ME, Achkar E, Richter JE. Timed barium oesophagram: better predictor of long term success after pneumatic dilation in achalasia than symptom assessment. Gut 2002; 50:765–770.
- Hewson EG, Ott DJ, Dalton CB, Chen YM, Wu WC, Richter JE. Manometry and radiology. Complementary studies in the assessment of esophageal motility disorders. Gastroenterology 1990; 98:626–632.
- Imam H, Shay S, Ali A, Baker M. Bolus transit patterns in healthy subjects: a study using simultaneous impedance monitoring, video-esophagram, and esophageal manometry. Am J Physiol Gastrointest Liver Physiol 2005;G1000–G1006.
- Imam H, Baker M, Shay S. Simultaneous barium esophagram, impedance monitoring and manometry in patients with dysphagia due to a tight fundoplication [abstract]. Gastroenterology 2004; 126:A-639.
KEY POINTS
- Dysphagia can be due to problems in the oropharynx and cervical esophagus or in the distal esophagus.
- Radiologic evaluation of dysphagia has distinct advantages over endoscopy, including its ability to diagnose both structural changes and motility disorders.
- A barium evaluation can include a modified barium-swallowing study to evaluate the oropharynx, barium esophagography to evaluate the esophagus, and a timed study to evaluate esophageal emptying.
- Often, the true cause of dysphagia is best approached with a combination of radiographic and endoscopic studies.
Autosomal dominant polycystic kidney disease: Emerging concepts of pathogenesis and new treatments
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
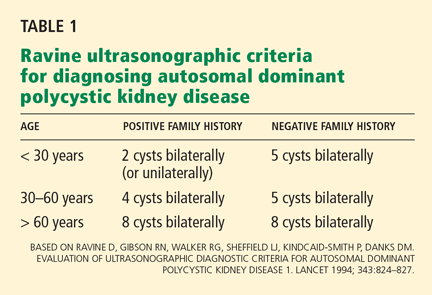
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
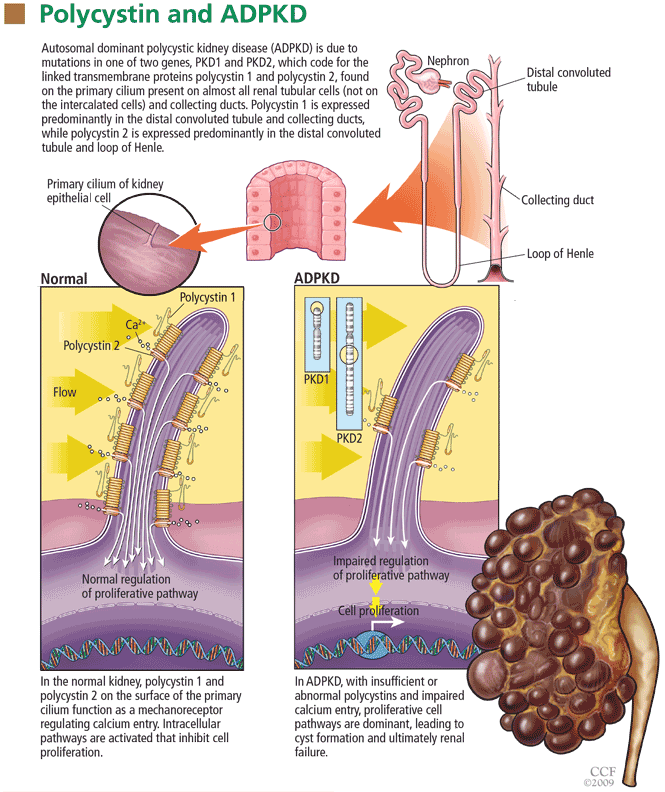
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
KEY POINTS
- In ADPKD the expanding cysts destroy normally functioning kidney tissue, causing hypertension, pain, and other complications, but renal function remains relatively stable until kidney volumes reach a critical size.
- Testing for genetic defects that cause ADPKD is available. The specific mutation involved (PKD1 or PKD2) affects the age of onset and therefore the rate of disease progression as well as the likelihood of cardiovascular complications. Other factors include somatic mutations (“second hits”) of the normal paired chromosome.
- Intracranial aneurysms are a key noncystic feature and may present with a very severe (“sentinel” or “thunderclap”) headache requiring immediate medical attention. Their occurrence is strongly influenced by family history.
- Basic research indicates that patients may be advised to increase their water intake, limit their sodium intake, and avoid caffeine and methylxanthine derivatives.
The battle of the clot

In this issue of the Journal we review two special situations in which low-molecular-weight heparins have special advantages. Babu and Carman discuss patients with cancer and thromboembolic disease. These patients are particularly difficult to manage since they tend to have recurrent thrombosis, sometimes even while on anticoagulant therapy, and they tend to have more bleeding complications from warfarin therapy. Inanition, drug interactions, and organ dysfunction make warfarin titration problematic, and the possibility of vascular metastases is always a concern. Low-molecular-weight heparins —which, unlike warfarin, work primarily by antagonizing factor Xa activity—have proven to be as effective as warfarin in reversing the many hypercoagulable effects of malignancy, although it wasn’t obvious at first that they would be.
Gibson and Powrie review the issues we face when pregnant patients need anticoagulation. While drug interactions and organ dysfunction are rarely problems in this setting, warfarin is teratogenic and is therefore strongly contraindicated early in pregnancy, and its peripartum use has been associated with bleeding complications. Furthermore, unfractionated heparin is associated with the development of osteoporosis, and it requires frequent injections. The low-molecular-weight heparins thus have a definite niche in the management of pregnant women, but with a caveat: dosing of these agents by weight alone in this setting is fraught with the potential for underdosing. Catastrophic outcomes have been reported in pregnant patients with older mechanical cardiac valves who were switched from warfarin to low-molecular-weight heparin therapy. Plus, if the patient is to receive neuraxial regional anesthesia, low-molecular-weight heparins should be discontinued at least 12 hours before catheter placement if prophylactic doses have been given, or 24 hours if therapeutic doses have been given.
Low-molecular-weight heparins have greatly enhanced our ability to treat thromboembolic disease. But, as the authors of these two papers discuss, many management nuances still must be noted.
In this issue of the Journal we review two special situations in which low-molecular-weight heparins have special advantages. Babu and Carman discuss patients with cancer and thromboembolic disease. These patients are particularly difficult to manage since they tend to have recurrent thrombosis, sometimes even while on anticoagulant therapy, and they tend to have more bleeding complications from warfarin therapy. Inanition, drug interactions, and organ dysfunction make warfarin titration problematic, and the possibility of vascular metastases is always a concern. Low-molecular-weight heparins —which, unlike warfarin, work primarily by antagonizing factor Xa activity—have proven to be as effective as warfarin in reversing the many hypercoagulable effects of malignancy, although it wasn’t obvious at first that they would be.
Gibson and Powrie review the issues we face when pregnant patients need anticoagulation. While drug interactions and organ dysfunction are rarely problems in this setting, warfarin is teratogenic and is therefore strongly contraindicated early in pregnancy, and its peripartum use has been associated with bleeding complications. Furthermore, unfractionated heparin is associated with the development of osteoporosis, and it requires frequent injections. The low-molecular-weight heparins thus have a definite niche in the management of pregnant women, but with a caveat: dosing of these agents by weight alone in this setting is fraught with the potential for underdosing. Catastrophic outcomes have been reported in pregnant patients with older mechanical cardiac valves who were switched from warfarin to low-molecular-weight heparin therapy. Plus, if the patient is to receive neuraxial regional anesthesia, low-molecular-weight heparins should be discontinued at least 12 hours before catheter placement if prophylactic doses have been given, or 24 hours if therapeutic doses have been given.
Low-molecular-weight heparins have greatly enhanced our ability to treat thromboembolic disease. But, as the authors of these two papers discuss, many management nuances still must be noted.
In this issue of the Journal we review two special situations in which low-molecular-weight heparins have special advantages. Babu and Carman discuss patients with cancer and thromboembolic disease. These patients are particularly difficult to manage since they tend to have recurrent thrombosis, sometimes even while on anticoagulant therapy, and they tend to have more bleeding complications from warfarin therapy. Inanition, drug interactions, and organ dysfunction make warfarin titration problematic, and the possibility of vascular metastases is always a concern. Low-molecular-weight heparins —which, unlike warfarin, work primarily by antagonizing factor Xa activity—have proven to be as effective as warfarin in reversing the many hypercoagulable effects of malignancy, although it wasn’t obvious at first that they would be.
Gibson and Powrie review the issues we face when pregnant patients need anticoagulation. While drug interactions and organ dysfunction are rarely problems in this setting, warfarin is teratogenic and is therefore strongly contraindicated early in pregnancy, and its peripartum use has been associated with bleeding complications. Furthermore, unfractionated heparin is associated with the development of osteoporosis, and it requires frequent injections. The low-molecular-weight heparins thus have a definite niche in the management of pregnant women, but with a caveat: dosing of these agents by weight alone in this setting is fraught with the potential for underdosing. Catastrophic outcomes have been reported in pregnant patients with older mechanical cardiac valves who were switched from warfarin to low-molecular-weight heparin therapy. Plus, if the patient is to receive neuraxial regional anesthesia, low-molecular-weight heparins should be discontinued at least 12 hours before catheter placement if prophylactic doses have been given, or 24 hours if therapeutic doses have been given.
Low-molecular-weight heparins have greatly enhanced our ability to treat thromboembolic disease. But, as the authors of these two papers discuss, many management nuances still must be noted.
Cancer and clots: All cases of venous thromboembolism are not treated the same
Venous thromboembolism (VTE) has various differing causes, so its treatment is not necessarily the same in all cases. Most cases of VTE are related to an easily identified risk factor. In patients with an apparently idiopathic event, identifying an underlying cause may alter therapy. In particular, identification of a malignancy may affect the choice of therapy and the duration of treatment.
In this review, we explore the role of cancer screening in patients with idiopathic VTE, then highlight the treatment for VTE in patients with cancer.
‘IDIOPATHIC’ VTE CAN BE DUE TO CANCER
Most patients with venous thrombosis have one of the components of Virchow’s triad: a hypercoagulable state, venous injury, or venous stasis. Those without identifiable risk factors for VTE are considered to have idiopathic VTE. In these patients, a search for a contributing factor may be indicated.
In 1861, the astute clinician Dr. Armand Trousseau noted a link between deep venous thrombosis and pancreatic cancer, stating that if cancer of an internal organ is suspected but the diagnosis cannot be verified, the diagnosis may be confirmed by the sudden, spontaneous appearance of thrombophlebitis in a large vein.1
Today, from 2% to 25% of patients with idiopathic VTE are found to have cancer within 24 months of the diagnosis of VTE.2–11 The goals of cancer screening in idiopathic VTE are to detect cancer at an early, treatable stage and to optimize the VTE therapy to decrease the risks of recurrence and anticoagulation-associated complications in patients who are found to have cancer. However, several questions must be considered first:
- What are the risks and costs of the screening?
- Will discovering the cancer sooner benefit the patient in terms of survival?
- If cancer is found, what are the possible complications or risks of the additional procedures, interventions, or treatments required?
- What is the psychological impact of the screening?
EVIDENCE SUPPORTING CANCER SCREENING AFTER IDIOPATHIC VTE
Piccioli et al12 recently performed a randomized, controlled trial comparing cancer-related death rates in 99 patients with idiopathic VTE screened for malignancy vs 102 patients with idiopathic VTE who were not screened.
The screened group underwent:
- Abdominal and pelvic ultrasonography and computed tomography (CT)
- Gastroscopy or double-contrast barium-swallow evaluation
- Colonoscopy or sigmoidoscopy followed by barium enema
- Testing for fecal occult blood
- Sputum cytology
- Measurement of carcinoembryonic antigen, alpha-fetoprotein, and cancer antigen 125.
- Mammography and Papanicolaou smears (women)
- Ultrasonography of the prostate and prostate-specific antigen testing (men).
Patients were followed for 2 years. The screening uncovered cancer in 13 patients. Cancer developed in one other patient in the screening group during follow-up; in the control group, 10 patients developed symptomatic cancer during follow-up. Overall, the time to cancer diagnosis was 11.6 months in the unscreened group vs 1 month in the screened group (P < .001). Nine of the 14 patients with cancer in the screened group had T1 or T2 disease without local or distant metastasis vs 2 of the 10 control patients with cancer (P = .047). Unfortunately, this study did not have adequate power to detect the effect of screening on survival.
Di Nisio et al13 used data from this trial to perform a decision analysis for cancer screening. They calculated that abdominal and pelvic CT, with or without mammography and with or without sputum cytologic testing, would cost the least per life-year gained and would harm the fewest number of patients. They also suggested that substituting CT of the chest for sputum cytology may provide additional diagnostic benefit.
However, this strategy has not been clinically tested. Given the limited number of patients and the short follow-up in this initial trial, larger trials are needed to look at the cost-effectiveness of this screening model and whether it increases survival.
Our recommendations
Because the data are limited, our approach to looking for an early, treatable malignancy in patients with idiopathic VTE follows the current consensus:
- A thorough history and physical, including an extensive review of systems
- Basic laboratory testing with a complete blood cell count, comprehensive metabolic profile, and urinalysis
- Chest radiography
- Other age- and sex-specific cancer screening tests.
Adding CT of the abdomen, pelvis, or chest to this evaluation may be considered. However, tumor marker testing, which typically has high false-positive rates, is not routinely warranted.13 Additional investigation should be considered if abnormalities are detected during the initial evaluation or in patients with recurrent VTE during therapy.
While this strategy may be most cost-effective, Monreal et al14 suggest that it may miss up to half of cancers ultimately discovered.
MANAGING VTE IN PATIENTS WITH KNOWN CANCER
Managing VTE is far more complex in cancer patients than in patients without cancer. Also, cancer patients with VTE have lower rates of survival than cancer patients without VTE and are at greater risk of adverse outcomes such as anticoagulant-associated bleeding and recurrent venous thrombotic events.15–17
Up to 21.5% of patients with VTE have another event within 5 years,18 but the risk is two to three times higher if they also have cancer.16,18 The risk of recurrence may be linked to the location of the thrombus and to the extent of the malignancy.
In one study, the 3-month rate of recurrence was up to 5.1% if the clot was in the popliteal vein, 5.3% if in the femoral vein, and 11.8% if in the iliac vein.19
Prandoni et al16 found that the risks of VTE recurrence and bleeding were higher in patients with extensive cancer than in those with less-extensive cancer. In this study, major bleeding was documented in 12.4% of patients with cancer vs 4.9% of patients without cancer. Compared with patients without cancer, the hazard ratio for a major bleeding event was 4.8 in patients with extensive cancer and 0.5 in patients with less-extensive cancer.
In addition, not all patients with bleeding had excessive levels of anticoagulation, and not all patients with recurrent events had subtherapeutic levels.16,17 Therefore, treatment of venous thrombosis in cancer patients requires a careful, individualized risk-to-benefit decision analysis.
ACUTE THERAPY FOR VTE: PARENTERAL AGENTS
Treatment in the first several hours or days after a thromboembolic event is with short-acting parenteral agents: unfractionated heparin; one of the low-molecular-weight heparins (LMWHs), ie, dalteparin (Fragmin), enoxaparin (Lovenox), or tinzaparin (Innohep); or fondaparinux (Arixtra).
Before starting anticoagulation, consider:
- Does the patient have severe chronic kidney disease (ie, a creatinine clearance < 30 mL/min)? If so, unfractionated heparin may be better than an LMWH or fondaparinux, which are cleared by the kidney.
- Does he or she need inpatient care? If not, LMWH therapy at home may be appropriate.
- Are there concerns about the ease of anticoagulation administration (ie, whether the patient can give the injections or have a family member do it), the cost of the drugs, or the ability to reverse the anticoagulant effect, if necessary? If so, unfractionated heparin may be more appropriate.
For acute treatment, the 2008 guidelines of the American College of Chest Physicians20 (ACCP) recommend using an LMWH in a weight-based dose; unfractionated heparin given intravenously; unfractionated heparin given subcutaneously with monitoring and dosing adjustments; unfractionated heparin given subcutaneously at a fixed dose; or fondaparinux (grade 1A recommendation). The 2007 National Comprehensive Cancer Network (NCCN) guidelines21 recommend an LMWH, fondaparinux, or unfractionated heparin. Treatment should start promptly after the diagnosis of VTE is confirmed. However, if VTE is strongly suspected and a delay in diagnostic testing is anticipated, therapy should be started while awaiting the test results.
LONG-TERM THERAPY: LMWH OR WARFARIN
The ACCP and the NCCN guidelines recommend LMWH monotherapy for extended treatment of VTE in patients with active malignancy, when appropriate.20,21 However, if long-term LMWH is not appropriate, then oral anticoagulation with a vitamin K antagonist, such as the coumarin derivative warfarin (Coumadin), is an alternative and should be started on the same day as the heparin. The heparin and the warfarin therapy must overlap for a minimum of 4 or 5 days and until a stable, therapeutic level of anticoagulation is achieved, ie, an international normalized ratio (INR) of 2 to 3 for 2 consecutive days.20
The duration of anticoagulant therapy depends on comorbidities and the patient’s underlying predisposition for VTE. In patients with limited disease, the guidelines recommend continuing anticoagulation for a minimum of 3 to 6 months for deep venous thrombosis and pulmonary embolism.20–21 Patients with active malignancy, ongoing treatment for the cancer, or continued risk factors may need indefinite treatment. In some circumstances, such as catheter-associated deep venous thrombosis, anticoagulation should continue for as long as the catheter is in place and for 1 to 3 months after its removal.21
WARFARIN CAN BE DIFFICULT TO USE
In 1954, the US Food and Drug Administration (FDA) approved the vitamin K antagonist warfarin for medical use in humans. Experience has shown it to be effective in preventing and treating VTE. However, it can be somewhat difficult to use, for several reasons:
- A narrow therapeutic window
- Genetic polymorphisms and variability in dose response
- Drug interactions and dietary considerations
- The need for laboratory monitoring and dose adjustment
- Patient noncompliance or miscommunication between the patient and physician.22
In cancer patients, the response to warfarin may be unpredictable because of poor nutrition, interactions with chemotherapy and antibiotics, and comorbid conditions.22 Furthermore, its onset of action can be delayed and its clearance may be prolonged, further increasing the risk of complications, especially in patients prone to developing chemotherapy-related anemia or thrombocytopenia.22 Bleeding risk is the highest in the first 3 months of therapy. In addition, the risk of bleeding is higher in older patients, women, and patients with a history of gastrointestinal bleeding, stroke, recent myocardial infarction, diabetes, renal insufficiency, malignancy, or anemia.23,24
ADVANTAGES AND DISADVANTAGES OF LMWH
The advantages of the LMWHs over unfractionated heparin include a lower risk of heparin-induced thrombocytopenia, greater bioavailability when given subcutaneously (which also permits once-daily or twice-daily dosing), and no need for laboratory monitoring in most patients. LMWHs have a short half-life, so omitting one or two doses will adequately interrupt therapy. Also, LMWHs have been shown to be as safe and effective as unfractionated heparin in treating VTE. They can be given safely at home, thus enhancing quality of life.25–31
On the other hand, these drugs cost more than unfractionated heparin or warfarin, their dosage must be adjusted in patients with renal insufficiency, their anticoagulant effect can be reversed only to a limited extent, and their dose must be adjusted according to weight in morbidly obese or in very thin patients.32,33
LMWHs are expensive, but may be worth it
As initial therapy, the LMWHs are cost-effective compared with unfractionated heparin in patients with VTE.34,35 However, they cost more with extended use. A cost-effectiveness analysis comparing 6 months of LMWH therapy to standard warfarin concluded that LMWH therapy was more costly.35 However, the impact of fewer hospitalizations, probably fewer bleeding complications, and better quality of life are difficult to analyze in this decision model and should also be considered when deciding about therapy for an individual patient.35
LMWHs are cleared by the kidney
All LMWHs are renally cleared, so patients with significant renal insufficiency (creatinine clearance < 30 mL/min) are at greater risk of bleeding complications. The rate below which clearance is impaired varies among the different LMWHs. Only enoxaparin has approved dosing regimens for use in patients with renal impairment.
If the patient has renal insufficiency, the ACCP guidelines suggest using unfractionated heparin, or if using LMWH, monitoring anti-factor Xa levels to avoid drug accumulation and increased bleeding risk.25 If bleeding occurs, LMWHs have limited reversibility with protamine sulfate, which is estimated to neutralize about 60% of the anti-factor Xa activity of LMWHs.25
Adjusting LMWHs for body weight
In the Registro Informatizado de la Enfermedad Tromboembólica (RIETE),33 patients weighing less than 50 kg had a higher risk of bleeding than patients weighing 50 to 100 kg, so in thinner patients the risk of bleeding from LMWH vs oral anticoagulation must be considered carefully and monitored prudently.
Although there is little evidence to suggest a higher bleeding risk in morbidly obese patients (> 150 kg), they may be at risk of subtherapeutic treatment, and monitoring with anti-factor Xa assays is recommended.25,32,33
LMWH VS WARFARIN FOR VTE IN CANCER PATIENTS
LMWHs are the first-line treatment for VTE in cancer patients.20,21 Several randomized controlled trials compared the efficacy of LMWH vs warfarin in patients with cancer.
Meyer et al36 randomized patients to receive either warfarin for 3 months at an INR between 2 and 3, or enoxaparin 1.5 mg/kg subcutaneously daily. Seventy-one patients received warfarin and 67 received enoxaparin. Fifteen (21%, 95% confidence interval [CI] 12%–32%) of the 71 patients assigned to warfarin experienced one major outcome event, defined as major bleeding or recurrent VTE, compared with 7 (10.5%) of the 67 patients assigned to receive enoxaparin (95% CI 4%–20%, P = .09). Six patients in the warfarin group died of bleeding vs none of the patients in the enoxaparin group. Overall, the warfarin group had a higher rate of bleeding, although this did not reach statistical significance. Despite weekly INR measurements, only 41% of the measured values were within the therapeutic range during the 3 months of treatment.36
Lee et al37 randomized cancer patients with deep venous thrombosis, pulmonary embolism, or both to receive 6 months of dalteparin alone, dosed at 200 IU/kg daily for 1 month, then decreased to 75% to 80% of the original dose (150 IU/kg) daily for the duration of therapy, or dalteparin followed by warfarin. During the 6-month follow-up, 17.4% of patients in the warfarin group had a recurrent thromboembolic event vs 8.8% in the dalteparin group (P = .0017). No statistically significant difference was noted in rates of major bleeding, minor bleeding, or death.37
Hull et al38 reported statistically significantly fewer episodes of recurrent VTE at 12 months in cancer patients treated with once-daily tinzaparin vs warfarin. In the tinzaparin group the recurrence rate was 7%, vs 16% in the warfarin group (P = .044). No difference in rates of bleeding or death were found.
Deitcher et al39 compared enoxaparin with long-term warfarin in 102 patients. While this trial did not have the power to detect clinical differences in recurrent thromboembolic events or bleeding complications, at 180 days they noted 97% compliance with once-daily or twice-daily enoxaparin therapy.
Noble and Finlay,40 in another small study, found LMWH therapy to be qualitatively more acceptable for palliative-care cancer patients than oral therapy.
In general, long-term therapy with once-daily or twice-daily LMWH is well tolerated. Currently, dalteparin is the only LMWH approved by the FDA for extended monotherapy in cancer-related VTE.
DO LMWHS AFFECT CANCER?
In vitro and animal studies indicate that LMWH may have antimetastatic and antiangiogenic properties.41–44
Altinbas et al45 reported significantly better chemotherapy-induced tumor response rates and survival rates in patients with small cell lung cancer randomized to receive combination chemotherapy plus prophylactic dalteparin 5,000 IU daily compared with combination chemotherapy alone. However, as provocative as these results may be, we need to test the effects of LWMHs on different cancer types in a prospective clinical trial. For now, this area remains controversial.
It has been suggested that anticoagulants may improve survival in patients with nonmetastatic cancer. Supporting this observation, a post hoc analysis of the trial by Lee et al37 found a statistically significantly lower cancer-specific mortality rate in nonmetastatic cancer patients treated with dalteparin vs oral therapy with a coumarin derivative. In patients without metastatic disease, the death rate at 12 months was 36% in patients treated with oral therapy vs 20% in patients treated with dalteparin (P = .03).46
These findings are consistent with those of the Fragmin Advanced Malignancy Outcome Study (FAMOUS),47 the first randomized, placebo-controlled trial of dalteparin 5,000 IU daily in patients with advanced solid tumors and without evidence of underlying thrombosis. Overall, dalteparin prophylaxis did not increase survival. However, in a subgroup of patients with a better prognosis and who were alive 17 months after diagnosis, survival was statistically significantly longer in patients treated with dalteparin.
Another small trial showed similar survival benefits in cancer patients without VTE.48 The results may suggest a long-term favorable effect of LMWH on tumor cell biology, which could translate into a favorable outcome in some patients. It is important to note, however, that not all trials have shown this same clinical benefit.49
In general, the growing body of laboratory and clinical data indicates that LMWHs may suppress tumor growth and metastasis. However, definitive conclusions about these effects are not yet possible because of variations in study design, tumor type, and patient populations. Further investigations into the role of LMWHs in the treatment of VTE and in cancer progression are ongoing.
THE EVIDENCE IN PERSPECTIVE
Illness and the recurrence of VTE in patients with cancer depend on the location and extent of the underlying cancer. Rates of death are higher in VTE patients with cancer than in VTE patients without cancer. Patients with limited or localized disease may not die of the cancer itself but of complications of acute pulmonary embolism. Therefore, it is important to recognize the different options for and the potential side effects of treating VTE.
If patients are hospitalized for an acute thromboembolic event and unfractionated heparin is chosen as the initial anticoagulant, using a weight-based nomogram has been shown to achieve therapeutic levels within 24 hours and reduce the rates of recurrence of thromboembolic events.50
Warfarin treatment may pose a particular challenge for both cancer patients and physicians, since multiple drug interactions, anorexia, and comorbid conditions contribute to an unpredictable response.
The risk of bleeding is higher in cancer patients than in the general population, and the decision to start anticoagulants should be based on an individualized risk-benefit profile. Several trials have shown LMWH to be more effective and safer than warfarin in cancer patients.
These considerations, along with the other advantages of LMWHs (ease of use, less need for laboratory monitoring, and better patient tolerance), make LMWHs a good choice for initial therapy. Extended LMWH therapy is currently favored for initial management in patients with cancer. Trials are under way to further assess the antitumor properties and potential survival benefit in patients with selected solid tumors.
- Aron E. The 100th anniversary of the death of A. Trousseau. Presse Med 1967; 75:1429–1430.
- Hettiarachchi RJ, Lok J, Prins MH, Büller HR, Prandoni P. Undiagnosed malignancy in patients with deep vein thrombosis: incidence, risk indicators, and diagnosis. Cancer 1998; 83:180–185.
- Baron JA, Gridley G, Weiderpass E, Nyrén O, Linet M. Venous thromboembolism and cancer. Lancet 1998; 351:1077–1080.
- Schulman S, Lindmarker P. Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. Duration of Anticoagulation Trial. N Engl J Med 2000; 342:1953–1958.
- Sørensen HT, Mellemkjaer L, Steffensen FH, Olsen JH, Nielsen GL. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998; 338:1169–1173.
- Monreal M, Lafoz E, Casals A, et al. Occult cancer in patients with deep venous thrombosis. A systematic approach. Cancer 1991; 67:541–545.
- Nordström M, Lindblad B, Anderson H, Bergqvist D, Kjellström T. Deep venous thrombosis and occult malignancy: an epidemiological study. BMJ 1994; 308:891–894.
- Prandoni P, Lensing AW, Büller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992; 327:1128–1133.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Fennerty T. Screening for cancer in venous thromboembolic disease. BMJ 2001; 323:704–705.
- Bastounis EA, Karayiannakis AJ, Makri GG, Alexiou D, Papalambros EL. The incidence of occult cancer in patients with deep venous thrombosis: a prospective study. J Intern Med 1996; 239:153–156.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Di Nisio M, Otten HM, Piccioli A, et al. Decision analysis for cancer screening in idiopathic venous thromboembolism. J Thromb Haemost 2005; 3:2391–2396.
- Monreal M, Lensing AW, Prins MH, et al. Screening for occult cancer in patients with acute deep vein thrombosis or pulmonary embolism. J Thromb Haemost 2004; 2:876–881.
- Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000; 343:1846–1850.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hutten BA, Prins MH, Gent M, Ginsberg J, Tijssen JG, Büller HR. Incidence of recurrent thromboembolic and bleeding complications among patients with venous thromboembolism in relation to both malignancy and achieved international normalized ratio: a retrospective analysis. J Clin Oncol 2000; 18:3078–3083.
- Hansson PO, Sörbo J, Eriksson H. Recurrent venous thromboembolism after deep vein thrombosis: incidence and risk factors. Arch Intern Med 2000; 160:769–774.
- Douketis JD, Crowther MA, Foster GA, Ginsberg JS. Does the location of thrombosis determine the risk of disease recurrence in patients with proximal deep vein thrombosis? Am J Med 2001; 110:515–519.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-based Clinical Practice Guidelines, 8th Edition. Chest 2008; 133 suppl 6:454S–545S.
- National Comprehensive Cancer Network. Venous Thromboembolic Disease Clinical Practice Guidelines in Oncology (V.1.2007). Available at www.nccn.org/professionals/physician_gls/PDF/vte.pdf. Accessed 01/02/2008.
- Ansell J, Hirsh J, Poller L, Bussey H, Jacobson A, Hylek E. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:204S–233S.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med 1999; 159:457–460.
- Hirsh J, Raschke R. Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic therapy. Chest 2004; 126 suppl 3:188S–203S.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Hettiarachchi RJ, Prins MH, Lensing AW, Büller HR. Low molecular weight heparin versus unfractionated heparin in the initial treatment of venous thromboembolism. Curr Opin Pulm Med 1998; 4:220–225.
- Gould MK, Dembitzer AD, Doyle RL, Hastie TJ, Garber AM. Low-molecular-weight heparins compared with unfractionated heparin for treatment of acute deep venous thrombosis. A meta-analysis of randomized, controlled trials. Ann Intern Med 1999; 130:800–809.
- Dolovich LR, Ginsberg JS, Douketis JD, Holbrook AM, Cheah G. A meta-analysis comparing low-molecular-weight heparins with un-fractionated heparin in the treatment of venous thromboembolism: examining some unanswered questions regarding location of treatment, product type, and dosing frequency. Arch Intern Med 2000; 160:181–188.
- van Dongen CJ, van den Belt AG, Prins MH, Lensing AW. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2004; 4:CD001100.
- Cook LM, Kahn SR, Goodwin J, Kovacs MJ. Frequency of renal impairment, advanced age, obesity, and cancer in venous thromboembolism patients in clinical practice. J Thromb Haemost 2007; 5:937–941.
- Barba R, Marco J, Martin-Alvarez H, et al. The influence of extreme body weight on clinical outcome of patients with venous thromboembolism: findings from a prospective registry (RIETE). J Thromb Haemost 2005; 3:856–862.
- Segal JB, Strieff MB, Hofmann LV, Thornton K, Bass EB. Management of venous thromboembolism: a systematic review for a practice guideline. Ann Intern Med 2007; 146:211–222.
- Aujesky D, Smith KJ, Cornuz J, Roberts MS. Cost-effectiveness of low-molecular-weight heparin for secondary prophylaxis of cancer-related venous thromboembolism. Thromb Haemost 2005; 93:592–599.
- Meyer G, Marjanovic Z, Valcke J, et al. Comparison of low-molecular-weight heparin and warfarin for the secondary prevention of venous thromboembolism in patients with cancer: a randomized controlled study. Arch Intern Med 2002; 162:1729–1735.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Deitcher SR, Kessler CM, Merli G, Rigas JR, Lyons RM, Fareed J ON-CENOX investigators. Secondary prevention of venous thromboembolic events in patients with active cancer: enoxaparin alone versus initial enoxaparin followed by warfarin for a 180-day period. Clin Appl Thromb Hemost 2006; 12:389–396.
- Noble SI, Finlay IG. Is long-term low-molecular-weight heparin acceptable to palliative care patients in the treatment of cancer related venous thromboembolism? A qualitative study. Palliat Med 2005; 19:197–201.
- Amirkhosravi A, Mousa SA, Amaya M, Francis JL. Antimetastatic effect of tinzaparin, a low-molecular-weight heparin. J Thromb Haemost 2003; 1:1972–1976.
- Kragh M, Binderup L, Vig Hjarnaa PJ, Bramm E, Johansen KB, Frimundt Petersen C. Non-anti-coagulant heparin inhibits metastasis but not primary tumor growth. Oncol Rep 2005; 14:99–104.
- Mousa SA, Mohamed S. Anti-angiogenic mechanisms and efficacy of the low molecular weight heparin, tinzaparin: anti-cancer efficacy. Oncol Rep 2004; 12:683–688.
- Bobek V, Kovarik J. Antitumor and antimetastatic effect of warfarin and heparins. Biomed Pharmacother 2004; 58:213–219.
- Altinbas M, Coskun HS, Er O, et al. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J Thromb Haemost 2004; 2:1266–1271.
- Lee AY, Rickles FR, Julian JA, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol 2005; 23:2123–2129.
- Kakkar AK, Levine MN, Kadziola Z, et al. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the Fragmin Advanced Malignancy Outcome Study (FAMOUS). J Clin Oncol 2004; 22:1944–1948.
- Klerk CP, Smorenburg SM, Otten HM, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol 2005; 23:2130–2135.
- Sideras K, Schaefer PL, Okuno SH, et al. Low-molecular-weight heparin in patients with advanced cancer: a phase 3 clinical trial. Mayo Clin Proc 2006; 81:758–767.
- Bernardi E, Piccioli A, Oliboni G, Zuin R, Girolami A, Prandoni P. Nomograms for the administration of unfractionated heparin in the initial treatment of acute thromboembolism—an overview. Thromb Haemost 2000; 84:22–26.
Venous thromboembolism (VTE) has various differing causes, so its treatment is not necessarily the same in all cases. Most cases of VTE are related to an easily identified risk factor. In patients with an apparently idiopathic event, identifying an underlying cause may alter therapy. In particular, identification of a malignancy may affect the choice of therapy and the duration of treatment.
In this review, we explore the role of cancer screening in patients with idiopathic VTE, then highlight the treatment for VTE in patients with cancer.
‘IDIOPATHIC’ VTE CAN BE DUE TO CANCER
Most patients with venous thrombosis have one of the components of Virchow’s triad: a hypercoagulable state, venous injury, or venous stasis. Those without identifiable risk factors for VTE are considered to have idiopathic VTE. In these patients, a search for a contributing factor may be indicated.
In 1861, the astute clinician Dr. Armand Trousseau noted a link between deep venous thrombosis and pancreatic cancer, stating that if cancer of an internal organ is suspected but the diagnosis cannot be verified, the diagnosis may be confirmed by the sudden, spontaneous appearance of thrombophlebitis in a large vein.1
Today, from 2% to 25% of patients with idiopathic VTE are found to have cancer within 24 months of the diagnosis of VTE.2–11 The goals of cancer screening in idiopathic VTE are to detect cancer at an early, treatable stage and to optimize the VTE therapy to decrease the risks of recurrence and anticoagulation-associated complications in patients who are found to have cancer. However, several questions must be considered first:
- What are the risks and costs of the screening?
- Will discovering the cancer sooner benefit the patient in terms of survival?
- If cancer is found, what are the possible complications or risks of the additional procedures, interventions, or treatments required?
- What is the psychological impact of the screening?
EVIDENCE SUPPORTING CANCER SCREENING AFTER IDIOPATHIC VTE
Piccioli et al12 recently performed a randomized, controlled trial comparing cancer-related death rates in 99 patients with idiopathic VTE screened for malignancy vs 102 patients with idiopathic VTE who were not screened.
The screened group underwent:
- Abdominal and pelvic ultrasonography and computed tomography (CT)
- Gastroscopy or double-contrast barium-swallow evaluation
- Colonoscopy or sigmoidoscopy followed by barium enema
- Testing for fecal occult blood
- Sputum cytology
- Measurement of carcinoembryonic antigen, alpha-fetoprotein, and cancer antigen 125.
- Mammography and Papanicolaou smears (women)
- Ultrasonography of the prostate and prostate-specific antigen testing (men).
Patients were followed for 2 years. The screening uncovered cancer in 13 patients. Cancer developed in one other patient in the screening group during follow-up; in the control group, 10 patients developed symptomatic cancer during follow-up. Overall, the time to cancer diagnosis was 11.6 months in the unscreened group vs 1 month in the screened group (P < .001). Nine of the 14 patients with cancer in the screened group had T1 or T2 disease without local or distant metastasis vs 2 of the 10 control patients with cancer (P = .047). Unfortunately, this study did not have adequate power to detect the effect of screening on survival.
Di Nisio et al13 used data from this trial to perform a decision analysis for cancer screening. They calculated that abdominal and pelvic CT, with or without mammography and with or without sputum cytologic testing, would cost the least per life-year gained and would harm the fewest number of patients. They also suggested that substituting CT of the chest for sputum cytology may provide additional diagnostic benefit.
However, this strategy has not been clinically tested. Given the limited number of patients and the short follow-up in this initial trial, larger trials are needed to look at the cost-effectiveness of this screening model and whether it increases survival.
Our recommendations
Because the data are limited, our approach to looking for an early, treatable malignancy in patients with idiopathic VTE follows the current consensus:
- A thorough history and physical, including an extensive review of systems
- Basic laboratory testing with a complete blood cell count, comprehensive metabolic profile, and urinalysis
- Chest radiography
- Other age- and sex-specific cancer screening tests.
Adding CT of the abdomen, pelvis, or chest to this evaluation may be considered. However, tumor marker testing, which typically has high false-positive rates, is not routinely warranted.13 Additional investigation should be considered if abnormalities are detected during the initial evaluation or in patients with recurrent VTE during therapy.
While this strategy may be most cost-effective, Monreal et al14 suggest that it may miss up to half of cancers ultimately discovered.
MANAGING VTE IN PATIENTS WITH KNOWN CANCER
Managing VTE is far more complex in cancer patients than in patients without cancer. Also, cancer patients with VTE have lower rates of survival than cancer patients without VTE and are at greater risk of adverse outcomes such as anticoagulant-associated bleeding and recurrent venous thrombotic events.15–17
Up to 21.5% of patients with VTE have another event within 5 years,18 but the risk is two to three times higher if they also have cancer.16,18 The risk of recurrence may be linked to the location of the thrombus and to the extent of the malignancy.
In one study, the 3-month rate of recurrence was up to 5.1% if the clot was in the popliteal vein, 5.3% if in the femoral vein, and 11.8% if in the iliac vein.19
Prandoni et al16 found that the risks of VTE recurrence and bleeding were higher in patients with extensive cancer than in those with less-extensive cancer. In this study, major bleeding was documented in 12.4% of patients with cancer vs 4.9% of patients without cancer. Compared with patients without cancer, the hazard ratio for a major bleeding event was 4.8 in patients with extensive cancer and 0.5 in patients with less-extensive cancer.
In addition, not all patients with bleeding had excessive levels of anticoagulation, and not all patients with recurrent events had subtherapeutic levels.16,17 Therefore, treatment of venous thrombosis in cancer patients requires a careful, individualized risk-to-benefit decision analysis.
ACUTE THERAPY FOR VTE: PARENTERAL AGENTS
Treatment in the first several hours or days after a thromboembolic event is with short-acting parenteral agents: unfractionated heparin; one of the low-molecular-weight heparins (LMWHs), ie, dalteparin (Fragmin), enoxaparin (Lovenox), or tinzaparin (Innohep); or fondaparinux (Arixtra).
Before starting anticoagulation, consider:
- Does the patient have severe chronic kidney disease (ie, a creatinine clearance < 30 mL/min)? If so, unfractionated heparin may be better than an LMWH or fondaparinux, which are cleared by the kidney.
- Does he or she need inpatient care? If not, LMWH therapy at home may be appropriate.
- Are there concerns about the ease of anticoagulation administration (ie, whether the patient can give the injections or have a family member do it), the cost of the drugs, or the ability to reverse the anticoagulant effect, if necessary? If so, unfractionated heparin may be more appropriate.
For acute treatment, the 2008 guidelines of the American College of Chest Physicians20 (ACCP) recommend using an LMWH in a weight-based dose; unfractionated heparin given intravenously; unfractionated heparin given subcutaneously with monitoring and dosing adjustments; unfractionated heparin given subcutaneously at a fixed dose; or fondaparinux (grade 1A recommendation). The 2007 National Comprehensive Cancer Network (NCCN) guidelines21 recommend an LMWH, fondaparinux, or unfractionated heparin. Treatment should start promptly after the diagnosis of VTE is confirmed. However, if VTE is strongly suspected and a delay in diagnostic testing is anticipated, therapy should be started while awaiting the test results.
LONG-TERM THERAPY: LMWH OR WARFARIN
The ACCP and the NCCN guidelines recommend LMWH monotherapy for extended treatment of VTE in patients with active malignancy, when appropriate.20,21 However, if long-term LMWH is not appropriate, then oral anticoagulation with a vitamin K antagonist, such as the coumarin derivative warfarin (Coumadin), is an alternative and should be started on the same day as the heparin. The heparin and the warfarin therapy must overlap for a minimum of 4 or 5 days and until a stable, therapeutic level of anticoagulation is achieved, ie, an international normalized ratio (INR) of 2 to 3 for 2 consecutive days.20
The duration of anticoagulant therapy depends on comorbidities and the patient’s underlying predisposition for VTE. In patients with limited disease, the guidelines recommend continuing anticoagulation for a minimum of 3 to 6 months for deep venous thrombosis and pulmonary embolism.20–21 Patients with active malignancy, ongoing treatment for the cancer, or continued risk factors may need indefinite treatment. In some circumstances, such as catheter-associated deep venous thrombosis, anticoagulation should continue for as long as the catheter is in place and for 1 to 3 months after its removal.21
WARFARIN CAN BE DIFFICULT TO USE
In 1954, the US Food and Drug Administration (FDA) approved the vitamin K antagonist warfarin for medical use in humans. Experience has shown it to be effective in preventing and treating VTE. However, it can be somewhat difficult to use, for several reasons:
- A narrow therapeutic window
- Genetic polymorphisms and variability in dose response
- Drug interactions and dietary considerations
- The need for laboratory monitoring and dose adjustment
- Patient noncompliance or miscommunication between the patient and physician.22
In cancer patients, the response to warfarin may be unpredictable because of poor nutrition, interactions with chemotherapy and antibiotics, and comorbid conditions.22 Furthermore, its onset of action can be delayed and its clearance may be prolonged, further increasing the risk of complications, especially in patients prone to developing chemotherapy-related anemia or thrombocytopenia.22 Bleeding risk is the highest in the first 3 months of therapy. In addition, the risk of bleeding is higher in older patients, women, and patients with a history of gastrointestinal bleeding, stroke, recent myocardial infarction, diabetes, renal insufficiency, malignancy, or anemia.23,24
ADVANTAGES AND DISADVANTAGES OF LMWH
The advantages of the LMWHs over unfractionated heparin include a lower risk of heparin-induced thrombocytopenia, greater bioavailability when given subcutaneously (which also permits once-daily or twice-daily dosing), and no need for laboratory monitoring in most patients. LMWHs have a short half-life, so omitting one or two doses will adequately interrupt therapy. Also, LMWHs have been shown to be as safe and effective as unfractionated heparin in treating VTE. They can be given safely at home, thus enhancing quality of life.25–31
On the other hand, these drugs cost more than unfractionated heparin or warfarin, their dosage must be adjusted in patients with renal insufficiency, their anticoagulant effect can be reversed only to a limited extent, and their dose must be adjusted according to weight in morbidly obese or in very thin patients.32,33
LMWHs are expensive, but may be worth it
As initial therapy, the LMWHs are cost-effective compared with unfractionated heparin in patients with VTE.34,35 However, they cost more with extended use. A cost-effectiveness analysis comparing 6 months of LMWH therapy to standard warfarin concluded that LMWH therapy was more costly.35 However, the impact of fewer hospitalizations, probably fewer bleeding complications, and better quality of life are difficult to analyze in this decision model and should also be considered when deciding about therapy for an individual patient.35
LMWHs are cleared by the kidney
All LMWHs are renally cleared, so patients with significant renal insufficiency (creatinine clearance < 30 mL/min) are at greater risk of bleeding complications. The rate below which clearance is impaired varies among the different LMWHs. Only enoxaparin has approved dosing regimens for use in patients with renal impairment.
If the patient has renal insufficiency, the ACCP guidelines suggest using unfractionated heparin, or if using LMWH, monitoring anti-factor Xa levels to avoid drug accumulation and increased bleeding risk.25 If bleeding occurs, LMWHs have limited reversibility with protamine sulfate, which is estimated to neutralize about 60% of the anti-factor Xa activity of LMWHs.25
Adjusting LMWHs for body weight
In the Registro Informatizado de la Enfermedad Tromboembólica (RIETE),33 patients weighing less than 50 kg had a higher risk of bleeding than patients weighing 50 to 100 kg, so in thinner patients the risk of bleeding from LMWH vs oral anticoagulation must be considered carefully and monitored prudently.
Although there is little evidence to suggest a higher bleeding risk in morbidly obese patients (> 150 kg), they may be at risk of subtherapeutic treatment, and monitoring with anti-factor Xa assays is recommended.25,32,33
LMWH VS WARFARIN FOR VTE IN CANCER PATIENTS
LMWHs are the first-line treatment for VTE in cancer patients.20,21 Several randomized controlled trials compared the efficacy of LMWH vs warfarin in patients with cancer.
Meyer et al36 randomized patients to receive either warfarin for 3 months at an INR between 2 and 3, or enoxaparin 1.5 mg/kg subcutaneously daily. Seventy-one patients received warfarin and 67 received enoxaparin. Fifteen (21%, 95% confidence interval [CI] 12%–32%) of the 71 patients assigned to warfarin experienced one major outcome event, defined as major bleeding or recurrent VTE, compared with 7 (10.5%) of the 67 patients assigned to receive enoxaparin (95% CI 4%–20%, P = .09). Six patients in the warfarin group died of bleeding vs none of the patients in the enoxaparin group. Overall, the warfarin group had a higher rate of bleeding, although this did not reach statistical significance. Despite weekly INR measurements, only 41% of the measured values were within the therapeutic range during the 3 months of treatment.36
Lee et al37 randomized cancer patients with deep venous thrombosis, pulmonary embolism, or both to receive 6 months of dalteparin alone, dosed at 200 IU/kg daily for 1 month, then decreased to 75% to 80% of the original dose (150 IU/kg) daily for the duration of therapy, or dalteparin followed by warfarin. During the 6-month follow-up, 17.4% of patients in the warfarin group had a recurrent thromboembolic event vs 8.8% in the dalteparin group (P = .0017). No statistically significant difference was noted in rates of major bleeding, minor bleeding, or death.37
Hull et al38 reported statistically significantly fewer episodes of recurrent VTE at 12 months in cancer patients treated with once-daily tinzaparin vs warfarin. In the tinzaparin group the recurrence rate was 7%, vs 16% in the warfarin group (P = .044). No difference in rates of bleeding or death were found.
Deitcher et al39 compared enoxaparin with long-term warfarin in 102 patients. While this trial did not have the power to detect clinical differences in recurrent thromboembolic events or bleeding complications, at 180 days they noted 97% compliance with once-daily or twice-daily enoxaparin therapy.
Noble and Finlay,40 in another small study, found LMWH therapy to be qualitatively more acceptable for palliative-care cancer patients than oral therapy.
In general, long-term therapy with once-daily or twice-daily LMWH is well tolerated. Currently, dalteparin is the only LMWH approved by the FDA for extended monotherapy in cancer-related VTE.
DO LMWHS AFFECT CANCER?
In vitro and animal studies indicate that LMWH may have antimetastatic and antiangiogenic properties.41–44
Altinbas et al45 reported significantly better chemotherapy-induced tumor response rates and survival rates in patients with small cell lung cancer randomized to receive combination chemotherapy plus prophylactic dalteparin 5,000 IU daily compared with combination chemotherapy alone. However, as provocative as these results may be, we need to test the effects of LWMHs on different cancer types in a prospective clinical trial. For now, this area remains controversial.
It has been suggested that anticoagulants may improve survival in patients with nonmetastatic cancer. Supporting this observation, a post hoc analysis of the trial by Lee et al37 found a statistically significantly lower cancer-specific mortality rate in nonmetastatic cancer patients treated with dalteparin vs oral therapy with a coumarin derivative. In patients without metastatic disease, the death rate at 12 months was 36% in patients treated with oral therapy vs 20% in patients treated with dalteparin (P = .03).46
These findings are consistent with those of the Fragmin Advanced Malignancy Outcome Study (FAMOUS),47 the first randomized, placebo-controlled trial of dalteparin 5,000 IU daily in patients with advanced solid tumors and without evidence of underlying thrombosis. Overall, dalteparin prophylaxis did not increase survival. However, in a subgroup of patients with a better prognosis and who were alive 17 months after diagnosis, survival was statistically significantly longer in patients treated with dalteparin.
Another small trial showed similar survival benefits in cancer patients without VTE.48 The results may suggest a long-term favorable effect of LMWH on tumor cell biology, which could translate into a favorable outcome in some patients. It is important to note, however, that not all trials have shown this same clinical benefit.49
In general, the growing body of laboratory and clinical data indicates that LMWHs may suppress tumor growth and metastasis. However, definitive conclusions about these effects are not yet possible because of variations in study design, tumor type, and patient populations. Further investigations into the role of LMWHs in the treatment of VTE and in cancer progression are ongoing.
THE EVIDENCE IN PERSPECTIVE
Illness and the recurrence of VTE in patients with cancer depend on the location and extent of the underlying cancer. Rates of death are higher in VTE patients with cancer than in VTE patients without cancer. Patients with limited or localized disease may not die of the cancer itself but of complications of acute pulmonary embolism. Therefore, it is important to recognize the different options for and the potential side effects of treating VTE.
If patients are hospitalized for an acute thromboembolic event and unfractionated heparin is chosen as the initial anticoagulant, using a weight-based nomogram has been shown to achieve therapeutic levels within 24 hours and reduce the rates of recurrence of thromboembolic events.50
Warfarin treatment may pose a particular challenge for both cancer patients and physicians, since multiple drug interactions, anorexia, and comorbid conditions contribute to an unpredictable response.
The risk of bleeding is higher in cancer patients than in the general population, and the decision to start anticoagulants should be based on an individualized risk-benefit profile. Several trials have shown LMWH to be more effective and safer than warfarin in cancer patients.
These considerations, along with the other advantages of LMWHs (ease of use, less need for laboratory monitoring, and better patient tolerance), make LMWHs a good choice for initial therapy. Extended LMWH therapy is currently favored for initial management in patients with cancer. Trials are under way to further assess the antitumor properties and potential survival benefit in patients with selected solid tumors.
Venous thromboembolism (VTE) has various differing causes, so its treatment is not necessarily the same in all cases. Most cases of VTE are related to an easily identified risk factor. In patients with an apparently idiopathic event, identifying an underlying cause may alter therapy. In particular, identification of a malignancy may affect the choice of therapy and the duration of treatment.
In this review, we explore the role of cancer screening in patients with idiopathic VTE, then highlight the treatment for VTE in patients with cancer.
‘IDIOPATHIC’ VTE CAN BE DUE TO CANCER
Most patients with venous thrombosis have one of the components of Virchow’s triad: a hypercoagulable state, venous injury, or venous stasis. Those without identifiable risk factors for VTE are considered to have idiopathic VTE. In these patients, a search for a contributing factor may be indicated.
In 1861, the astute clinician Dr. Armand Trousseau noted a link between deep venous thrombosis and pancreatic cancer, stating that if cancer of an internal organ is suspected but the diagnosis cannot be verified, the diagnosis may be confirmed by the sudden, spontaneous appearance of thrombophlebitis in a large vein.1
Today, from 2% to 25% of patients with idiopathic VTE are found to have cancer within 24 months of the diagnosis of VTE.2–11 The goals of cancer screening in idiopathic VTE are to detect cancer at an early, treatable stage and to optimize the VTE therapy to decrease the risks of recurrence and anticoagulation-associated complications in patients who are found to have cancer. However, several questions must be considered first:
- What are the risks and costs of the screening?
- Will discovering the cancer sooner benefit the patient in terms of survival?
- If cancer is found, what are the possible complications or risks of the additional procedures, interventions, or treatments required?
- What is the psychological impact of the screening?
EVIDENCE SUPPORTING CANCER SCREENING AFTER IDIOPATHIC VTE
Piccioli et al12 recently performed a randomized, controlled trial comparing cancer-related death rates in 99 patients with idiopathic VTE screened for malignancy vs 102 patients with idiopathic VTE who were not screened.
The screened group underwent:
- Abdominal and pelvic ultrasonography and computed tomography (CT)
- Gastroscopy or double-contrast barium-swallow evaluation
- Colonoscopy or sigmoidoscopy followed by barium enema
- Testing for fecal occult blood
- Sputum cytology
- Measurement of carcinoembryonic antigen, alpha-fetoprotein, and cancer antigen 125.
- Mammography and Papanicolaou smears (women)
- Ultrasonography of the prostate and prostate-specific antigen testing (men).
Patients were followed for 2 years. The screening uncovered cancer in 13 patients. Cancer developed in one other patient in the screening group during follow-up; in the control group, 10 patients developed symptomatic cancer during follow-up. Overall, the time to cancer diagnosis was 11.6 months in the unscreened group vs 1 month in the screened group (P < .001). Nine of the 14 patients with cancer in the screened group had T1 or T2 disease without local or distant metastasis vs 2 of the 10 control patients with cancer (P = .047). Unfortunately, this study did not have adequate power to detect the effect of screening on survival.
Di Nisio et al13 used data from this trial to perform a decision analysis for cancer screening. They calculated that abdominal and pelvic CT, with or without mammography and with or without sputum cytologic testing, would cost the least per life-year gained and would harm the fewest number of patients. They also suggested that substituting CT of the chest for sputum cytology may provide additional diagnostic benefit.
However, this strategy has not been clinically tested. Given the limited number of patients and the short follow-up in this initial trial, larger trials are needed to look at the cost-effectiveness of this screening model and whether it increases survival.
Our recommendations
Because the data are limited, our approach to looking for an early, treatable malignancy in patients with idiopathic VTE follows the current consensus:
- A thorough history and physical, including an extensive review of systems
- Basic laboratory testing with a complete blood cell count, comprehensive metabolic profile, and urinalysis
- Chest radiography
- Other age- and sex-specific cancer screening tests.
Adding CT of the abdomen, pelvis, or chest to this evaluation may be considered. However, tumor marker testing, which typically has high false-positive rates, is not routinely warranted.13 Additional investigation should be considered if abnormalities are detected during the initial evaluation or in patients with recurrent VTE during therapy.
While this strategy may be most cost-effective, Monreal et al14 suggest that it may miss up to half of cancers ultimately discovered.
MANAGING VTE IN PATIENTS WITH KNOWN CANCER
Managing VTE is far more complex in cancer patients than in patients without cancer. Also, cancer patients with VTE have lower rates of survival than cancer patients without VTE and are at greater risk of adverse outcomes such as anticoagulant-associated bleeding and recurrent venous thrombotic events.15–17
Up to 21.5% of patients with VTE have another event within 5 years,18 but the risk is two to three times higher if they also have cancer.16,18 The risk of recurrence may be linked to the location of the thrombus and to the extent of the malignancy.
In one study, the 3-month rate of recurrence was up to 5.1% if the clot was in the popliteal vein, 5.3% if in the femoral vein, and 11.8% if in the iliac vein.19
Prandoni et al16 found that the risks of VTE recurrence and bleeding were higher in patients with extensive cancer than in those with less-extensive cancer. In this study, major bleeding was documented in 12.4% of patients with cancer vs 4.9% of patients without cancer. Compared with patients without cancer, the hazard ratio for a major bleeding event was 4.8 in patients with extensive cancer and 0.5 in patients with less-extensive cancer.
In addition, not all patients with bleeding had excessive levels of anticoagulation, and not all patients with recurrent events had subtherapeutic levels.16,17 Therefore, treatment of venous thrombosis in cancer patients requires a careful, individualized risk-to-benefit decision analysis.
ACUTE THERAPY FOR VTE: PARENTERAL AGENTS
Treatment in the first several hours or days after a thromboembolic event is with short-acting parenteral agents: unfractionated heparin; one of the low-molecular-weight heparins (LMWHs), ie, dalteparin (Fragmin), enoxaparin (Lovenox), or tinzaparin (Innohep); or fondaparinux (Arixtra).
Before starting anticoagulation, consider:
- Does the patient have severe chronic kidney disease (ie, a creatinine clearance < 30 mL/min)? If so, unfractionated heparin may be better than an LMWH or fondaparinux, which are cleared by the kidney.
- Does he or she need inpatient care? If not, LMWH therapy at home may be appropriate.
- Are there concerns about the ease of anticoagulation administration (ie, whether the patient can give the injections or have a family member do it), the cost of the drugs, or the ability to reverse the anticoagulant effect, if necessary? If so, unfractionated heparin may be more appropriate.
For acute treatment, the 2008 guidelines of the American College of Chest Physicians20 (ACCP) recommend using an LMWH in a weight-based dose; unfractionated heparin given intravenously; unfractionated heparin given subcutaneously with monitoring and dosing adjustments; unfractionated heparin given subcutaneously at a fixed dose; or fondaparinux (grade 1A recommendation). The 2007 National Comprehensive Cancer Network (NCCN) guidelines21 recommend an LMWH, fondaparinux, or unfractionated heparin. Treatment should start promptly after the diagnosis of VTE is confirmed. However, if VTE is strongly suspected and a delay in diagnostic testing is anticipated, therapy should be started while awaiting the test results.
LONG-TERM THERAPY: LMWH OR WARFARIN
The ACCP and the NCCN guidelines recommend LMWH monotherapy for extended treatment of VTE in patients with active malignancy, when appropriate.20,21 However, if long-term LMWH is not appropriate, then oral anticoagulation with a vitamin K antagonist, such as the coumarin derivative warfarin (Coumadin), is an alternative and should be started on the same day as the heparin. The heparin and the warfarin therapy must overlap for a minimum of 4 or 5 days and until a stable, therapeutic level of anticoagulation is achieved, ie, an international normalized ratio (INR) of 2 to 3 for 2 consecutive days.20
The duration of anticoagulant therapy depends on comorbidities and the patient’s underlying predisposition for VTE. In patients with limited disease, the guidelines recommend continuing anticoagulation for a minimum of 3 to 6 months for deep venous thrombosis and pulmonary embolism.20–21 Patients with active malignancy, ongoing treatment for the cancer, or continued risk factors may need indefinite treatment. In some circumstances, such as catheter-associated deep venous thrombosis, anticoagulation should continue for as long as the catheter is in place and for 1 to 3 months after its removal.21
WARFARIN CAN BE DIFFICULT TO USE
In 1954, the US Food and Drug Administration (FDA) approved the vitamin K antagonist warfarin for medical use in humans. Experience has shown it to be effective in preventing and treating VTE. However, it can be somewhat difficult to use, for several reasons:
- A narrow therapeutic window
- Genetic polymorphisms and variability in dose response
- Drug interactions and dietary considerations
- The need for laboratory monitoring and dose adjustment
- Patient noncompliance or miscommunication between the patient and physician.22
In cancer patients, the response to warfarin may be unpredictable because of poor nutrition, interactions with chemotherapy and antibiotics, and comorbid conditions.22 Furthermore, its onset of action can be delayed and its clearance may be prolonged, further increasing the risk of complications, especially in patients prone to developing chemotherapy-related anemia or thrombocytopenia.22 Bleeding risk is the highest in the first 3 months of therapy. In addition, the risk of bleeding is higher in older patients, women, and patients with a history of gastrointestinal bleeding, stroke, recent myocardial infarction, diabetes, renal insufficiency, malignancy, or anemia.23,24
ADVANTAGES AND DISADVANTAGES OF LMWH
The advantages of the LMWHs over unfractionated heparin include a lower risk of heparin-induced thrombocytopenia, greater bioavailability when given subcutaneously (which also permits once-daily or twice-daily dosing), and no need for laboratory monitoring in most patients. LMWHs have a short half-life, so omitting one or two doses will adequately interrupt therapy. Also, LMWHs have been shown to be as safe and effective as unfractionated heparin in treating VTE. They can be given safely at home, thus enhancing quality of life.25–31
On the other hand, these drugs cost more than unfractionated heparin or warfarin, their dosage must be adjusted in patients with renal insufficiency, their anticoagulant effect can be reversed only to a limited extent, and their dose must be adjusted according to weight in morbidly obese or in very thin patients.32,33
LMWHs are expensive, but may be worth it
As initial therapy, the LMWHs are cost-effective compared with unfractionated heparin in patients with VTE.34,35 However, they cost more with extended use. A cost-effectiveness analysis comparing 6 months of LMWH therapy to standard warfarin concluded that LMWH therapy was more costly.35 However, the impact of fewer hospitalizations, probably fewer bleeding complications, and better quality of life are difficult to analyze in this decision model and should also be considered when deciding about therapy for an individual patient.35
LMWHs are cleared by the kidney
All LMWHs are renally cleared, so patients with significant renal insufficiency (creatinine clearance < 30 mL/min) are at greater risk of bleeding complications. The rate below which clearance is impaired varies among the different LMWHs. Only enoxaparin has approved dosing regimens for use in patients with renal impairment.
If the patient has renal insufficiency, the ACCP guidelines suggest using unfractionated heparin, or if using LMWH, monitoring anti-factor Xa levels to avoid drug accumulation and increased bleeding risk.25 If bleeding occurs, LMWHs have limited reversibility with protamine sulfate, which is estimated to neutralize about 60% of the anti-factor Xa activity of LMWHs.25
Adjusting LMWHs for body weight
In the Registro Informatizado de la Enfermedad Tromboembólica (RIETE),33 patients weighing less than 50 kg had a higher risk of bleeding than patients weighing 50 to 100 kg, so in thinner patients the risk of bleeding from LMWH vs oral anticoagulation must be considered carefully and monitored prudently.
Although there is little evidence to suggest a higher bleeding risk in morbidly obese patients (> 150 kg), they may be at risk of subtherapeutic treatment, and monitoring with anti-factor Xa assays is recommended.25,32,33
LMWH VS WARFARIN FOR VTE IN CANCER PATIENTS
LMWHs are the first-line treatment for VTE in cancer patients.20,21 Several randomized controlled trials compared the efficacy of LMWH vs warfarin in patients with cancer.
Meyer et al36 randomized patients to receive either warfarin for 3 months at an INR between 2 and 3, or enoxaparin 1.5 mg/kg subcutaneously daily. Seventy-one patients received warfarin and 67 received enoxaparin. Fifteen (21%, 95% confidence interval [CI] 12%–32%) of the 71 patients assigned to warfarin experienced one major outcome event, defined as major bleeding or recurrent VTE, compared with 7 (10.5%) of the 67 patients assigned to receive enoxaparin (95% CI 4%–20%, P = .09). Six patients in the warfarin group died of bleeding vs none of the patients in the enoxaparin group. Overall, the warfarin group had a higher rate of bleeding, although this did not reach statistical significance. Despite weekly INR measurements, only 41% of the measured values were within the therapeutic range during the 3 months of treatment.36
Lee et al37 randomized cancer patients with deep venous thrombosis, pulmonary embolism, or both to receive 6 months of dalteparin alone, dosed at 200 IU/kg daily for 1 month, then decreased to 75% to 80% of the original dose (150 IU/kg) daily for the duration of therapy, or dalteparin followed by warfarin. During the 6-month follow-up, 17.4% of patients in the warfarin group had a recurrent thromboembolic event vs 8.8% in the dalteparin group (P = .0017). No statistically significant difference was noted in rates of major bleeding, minor bleeding, or death.37
Hull et al38 reported statistically significantly fewer episodes of recurrent VTE at 12 months in cancer patients treated with once-daily tinzaparin vs warfarin. In the tinzaparin group the recurrence rate was 7%, vs 16% in the warfarin group (P = .044). No difference in rates of bleeding or death were found.
Deitcher et al39 compared enoxaparin with long-term warfarin in 102 patients. While this trial did not have the power to detect clinical differences in recurrent thromboembolic events or bleeding complications, at 180 days they noted 97% compliance with once-daily or twice-daily enoxaparin therapy.
Noble and Finlay,40 in another small study, found LMWH therapy to be qualitatively more acceptable for palliative-care cancer patients than oral therapy.
In general, long-term therapy with once-daily or twice-daily LMWH is well tolerated. Currently, dalteparin is the only LMWH approved by the FDA for extended monotherapy in cancer-related VTE.
DO LMWHS AFFECT CANCER?
In vitro and animal studies indicate that LMWH may have antimetastatic and antiangiogenic properties.41–44
Altinbas et al45 reported significantly better chemotherapy-induced tumor response rates and survival rates in patients with small cell lung cancer randomized to receive combination chemotherapy plus prophylactic dalteparin 5,000 IU daily compared with combination chemotherapy alone. However, as provocative as these results may be, we need to test the effects of LWMHs on different cancer types in a prospective clinical trial. For now, this area remains controversial.
It has been suggested that anticoagulants may improve survival in patients with nonmetastatic cancer. Supporting this observation, a post hoc analysis of the trial by Lee et al37 found a statistically significantly lower cancer-specific mortality rate in nonmetastatic cancer patients treated with dalteparin vs oral therapy with a coumarin derivative. In patients without metastatic disease, the death rate at 12 months was 36% in patients treated with oral therapy vs 20% in patients treated with dalteparin (P = .03).46
These findings are consistent with those of the Fragmin Advanced Malignancy Outcome Study (FAMOUS),47 the first randomized, placebo-controlled trial of dalteparin 5,000 IU daily in patients with advanced solid tumors and without evidence of underlying thrombosis. Overall, dalteparin prophylaxis did not increase survival. However, in a subgroup of patients with a better prognosis and who were alive 17 months after diagnosis, survival was statistically significantly longer in patients treated with dalteparin.
Another small trial showed similar survival benefits in cancer patients without VTE.48 The results may suggest a long-term favorable effect of LMWH on tumor cell biology, which could translate into a favorable outcome in some patients. It is important to note, however, that not all trials have shown this same clinical benefit.49
In general, the growing body of laboratory and clinical data indicates that LMWHs may suppress tumor growth and metastasis. However, definitive conclusions about these effects are not yet possible because of variations in study design, tumor type, and patient populations. Further investigations into the role of LMWHs in the treatment of VTE and in cancer progression are ongoing.
THE EVIDENCE IN PERSPECTIVE
Illness and the recurrence of VTE in patients with cancer depend on the location and extent of the underlying cancer. Rates of death are higher in VTE patients with cancer than in VTE patients without cancer. Patients with limited or localized disease may not die of the cancer itself but of complications of acute pulmonary embolism. Therefore, it is important to recognize the different options for and the potential side effects of treating VTE.
If patients are hospitalized for an acute thromboembolic event and unfractionated heparin is chosen as the initial anticoagulant, using a weight-based nomogram has been shown to achieve therapeutic levels within 24 hours and reduce the rates of recurrence of thromboembolic events.50
Warfarin treatment may pose a particular challenge for both cancer patients and physicians, since multiple drug interactions, anorexia, and comorbid conditions contribute to an unpredictable response.
The risk of bleeding is higher in cancer patients than in the general population, and the decision to start anticoagulants should be based on an individualized risk-benefit profile. Several trials have shown LMWH to be more effective and safer than warfarin in cancer patients.
These considerations, along with the other advantages of LMWHs (ease of use, less need for laboratory monitoring, and better patient tolerance), make LMWHs a good choice for initial therapy. Extended LMWH therapy is currently favored for initial management in patients with cancer. Trials are under way to further assess the antitumor properties and potential survival benefit in patients with selected solid tumors.
- Aron E. The 100th anniversary of the death of A. Trousseau. Presse Med 1967; 75:1429–1430.
- Hettiarachchi RJ, Lok J, Prins MH, Büller HR, Prandoni P. Undiagnosed malignancy in patients with deep vein thrombosis: incidence, risk indicators, and diagnosis. Cancer 1998; 83:180–185.
- Baron JA, Gridley G, Weiderpass E, Nyrén O, Linet M. Venous thromboembolism and cancer. Lancet 1998; 351:1077–1080.
- Schulman S, Lindmarker P. Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. Duration of Anticoagulation Trial. N Engl J Med 2000; 342:1953–1958.
- Sørensen HT, Mellemkjaer L, Steffensen FH, Olsen JH, Nielsen GL. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998; 338:1169–1173.
- Monreal M, Lafoz E, Casals A, et al. Occult cancer in patients with deep venous thrombosis. A systematic approach. Cancer 1991; 67:541–545.
- Nordström M, Lindblad B, Anderson H, Bergqvist D, Kjellström T. Deep venous thrombosis and occult malignancy: an epidemiological study. BMJ 1994; 308:891–894.
- Prandoni P, Lensing AW, Büller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992; 327:1128–1133.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Fennerty T. Screening for cancer in venous thromboembolic disease. BMJ 2001; 323:704–705.
- Bastounis EA, Karayiannakis AJ, Makri GG, Alexiou D, Papalambros EL. The incidence of occult cancer in patients with deep venous thrombosis: a prospective study. J Intern Med 1996; 239:153–156.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Di Nisio M, Otten HM, Piccioli A, et al. Decision analysis for cancer screening in idiopathic venous thromboembolism. J Thromb Haemost 2005; 3:2391–2396.
- Monreal M, Lensing AW, Prins MH, et al. Screening for occult cancer in patients with acute deep vein thrombosis or pulmonary embolism. J Thromb Haemost 2004; 2:876–881.
- Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000; 343:1846–1850.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hutten BA, Prins MH, Gent M, Ginsberg J, Tijssen JG, Büller HR. Incidence of recurrent thromboembolic and bleeding complications among patients with venous thromboembolism in relation to both malignancy and achieved international normalized ratio: a retrospective analysis. J Clin Oncol 2000; 18:3078–3083.
- Hansson PO, Sörbo J, Eriksson H. Recurrent venous thromboembolism after deep vein thrombosis: incidence and risk factors. Arch Intern Med 2000; 160:769–774.
- Douketis JD, Crowther MA, Foster GA, Ginsberg JS. Does the location of thrombosis determine the risk of disease recurrence in patients with proximal deep vein thrombosis? Am J Med 2001; 110:515–519.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-based Clinical Practice Guidelines, 8th Edition. Chest 2008; 133 suppl 6:454S–545S.
- National Comprehensive Cancer Network. Venous Thromboembolic Disease Clinical Practice Guidelines in Oncology (V.1.2007). Available at www.nccn.org/professionals/physician_gls/PDF/vte.pdf. Accessed 01/02/2008.
- Ansell J, Hirsh J, Poller L, Bussey H, Jacobson A, Hylek E. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:204S–233S.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med 1999; 159:457–460.
- Hirsh J, Raschke R. Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic therapy. Chest 2004; 126 suppl 3:188S–203S.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Hettiarachchi RJ, Prins MH, Lensing AW, Büller HR. Low molecular weight heparin versus unfractionated heparin in the initial treatment of venous thromboembolism. Curr Opin Pulm Med 1998; 4:220–225.
- Gould MK, Dembitzer AD, Doyle RL, Hastie TJ, Garber AM. Low-molecular-weight heparins compared with unfractionated heparin for treatment of acute deep venous thrombosis. A meta-analysis of randomized, controlled trials. Ann Intern Med 1999; 130:800–809.
- Dolovich LR, Ginsberg JS, Douketis JD, Holbrook AM, Cheah G. A meta-analysis comparing low-molecular-weight heparins with un-fractionated heparin in the treatment of venous thromboembolism: examining some unanswered questions regarding location of treatment, product type, and dosing frequency. Arch Intern Med 2000; 160:181–188.
- van Dongen CJ, van den Belt AG, Prins MH, Lensing AW. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2004; 4:CD001100.
- Cook LM, Kahn SR, Goodwin J, Kovacs MJ. Frequency of renal impairment, advanced age, obesity, and cancer in venous thromboembolism patients in clinical practice. J Thromb Haemost 2007; 5:937–941.
- Barba R, Marco J, Martin-Alvarez H, et al. The influence of extreme body weight on clinical outcome of patients with venous thromboembolism: findings from a prospective registry (RIETE). J Thromb Haemost 2005; 3:856–862.
- Segal JB, Strieff MB, Hofmann LV, Thornton K, Bass EB. Management of venous thromboembolism: a systematic review for a practice guideline. Ann Intern Med 2007; 146:211–222.
- Aujesky D, Smith KJ, Cornuz J, Roberts MS. Cost-effectiveness of low-molecular-weight heparin for secondary prophylaxis of cancer-related venous thromboembolism. Thromb Haemost 2005; 93:592–599.
- Meyer G, Marjanovic Z, Valcke J, et al. Comparison of low-molecular-weight heparin and warfarin for the secondary prevention of venous thromboembolism in patients with cancer: a randomized controlled study. Arch Intern Med 2002; 162:1729–1735.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Deitcher SR, Kessler CM, Merli G, Rigas JR, Lyons RM, Fareed J ON-CENOX investigators. Secondary prevention of venous thromboembolic events in patients with active cancer: enoxaparin alone versus initial enoxaparin followed by warfarin for a 180-day period. Clin Appl Thromb Hemost 2006; 12:389–396.
- Noble SI, Finlay IG. Is long-term low-molecular-weight heparin acceptable to palliative care patients in the treatment of cancer related venous thromboembolism? A qualitative study. Palliat Med 2005; 19:197–201.
- Amirkhosravi A, Mousa SA, Amaya M, Francis JL. Antimetastatic effect of tinzaparin, a low-molecular-weight heparin. J Thromb Haemost 2003; 1:1972–1976.
- Kragh M, Binderup L, Vig Hjarnaa PJ, Bramm E, Johansen KB, Frimundt Petersen C. Non-anti-coagulant heparin inhibits metastasis but not primary tumor growth. Oncol Rep 2005; 14:99–104.
- Mousa SA, Mohamed S. Anti-angiogenic mechanisms and efficacy of the low molecular weight heparin, tinzaparin: anti-cancer efficacy. Oncol Rep 2004; 12:683–688.
- Bobek V, Kovarik J. Antitumor and antimetastatic effect of warfarin and heparins. Biomed Pharmacother 2004; 58:213–219.
- Altinbas M, Coskun HS, Er O, et al. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J Thromb Haemost 2004; 2:1266–1271.
- Lee AY, Rickles FR, Julian JA, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol 2005; 23:2123–2129.
- Kakkar AK, Levine MN, Kadziola Z, et al. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the Fragmin Advanced Malignancy Outcome Study (FAMOUS). J Clin Oncol 2004; 22:1944–1948.
- Klerk CP, Smorenburg SM, Otten HM, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol 2005; 23:2130–2135.
- Sideras K, Schaefer PL, Okuno SH, et al. Low-molecular-weight heparin in patients with advanced cancer: a phase 3 clinical trial. Mayo Clin Proc 2006; 81:758–767.
- Bernardi E, Piccioli A, Oliboni G, Zuin R, Girolami A, Prandoni P. Nomograms for the administration of unfractionated heparin in the initial treatment of acute thromboembolism—an overview. Thromb Haemost 2000; 84:22–26.
- Aron E. The 100th anniversary of the death of A. Trousseau. Presse Med 1967; 75:1429–1430.
- Hettiarachchi RJ, Lok J, Prins MH, Büller HR, Prandoni P. Undiagnosed malignancy in patients with deep vein thrombosis: incidence, risk indicators, and diagnosis. Cancer 1998; 83:180–185.
- Baron JA, Gridley G, Weiderpass E, Nyrén O, Linet M. Venous thromboembolism and cancer. Lancet 1998; 351:1077–1080.
- Schulman S, Lindmarker P. Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. Duration of Anticoagulation Trial. N Engl J Med 2000; 342:1953–1958.
- Sørensen HT, Mellemkjaer L, Steffensen FH, Olsen JH, Nielsen GL. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998; 338:1169–1173.
- Monreal M, Lafoz E, Casals A, et al. Occult cancer in patients with deep venous thrombosis. A systematic approach. Cancer 1991; 67:541–545.
- Nordström M, Lindblad B, Anderson H, Bergqvist D, Kjellström T. Deep venous thrombosis and occult malignancy: an epidemiological study. BMJ 1994; 308:891–894.
- Prandoni P, Lensing AW, Büller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992; 327:1128–1133.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Fennerty T. Screening for cancer in venous thromboembolic disease. BMJ 2001; 323:704–705.
- Bastounis EA, Karayiannakis AJ, Makri GG, Alexiou D, Papalambros EL. The incidence of occult cancer in patients with deep venous thrombosis: a prospective study. J Intern Med 1996; 239:153–156.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Di Nisio M, Otten HM, Piccioli A, et al. Decision analysis for cancer screening in idiopathic venous thromboembolism. J Thromb Haemost 2005; 3:2391–2396.
- Monreal M, Lensing AW, Prins MH, et al. Screening for occult cancer in patients with acute deep vein thrombosis or pulmonary embolism. J Thromb Haemost 2004; 2:876–881.
- Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000; 343:1846–1850.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hutten BA, Prins MH, Gent M, Ginsberg J, Tijssen JG, Büller HR. Incidence of recurrent thromboembolic and bleeding complications among patients with venous thromboembolism in relation to both malignancy and achieved international normalized ratio: a retrospective analysis. J Clin Oncol 2000; 18:3078–3083.
- Hansson PO, Sörbo J, Eriksson H. Recurrent venous thromboembolism after deep vein thrombosis: incidence and risk factors. Arch Intern Med 2000; 160:769–774.
- Douketis JD, Crowther MA, Foster GA, Ginsberg JS. Does the location of thrombosis determine the risk of disease recurrence in patients with proximal deep vein thrombosis? Am J Med 2001; 110:515–519.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-based Clinical Practice Guidelines, 8th Edition. Chest 2008; 133 suppl 6:454S–545S.
- National Comprehensive Cancer Network. Venous Thromboembolic Disease Clinical Practice Guidelines in Oncology (V.1.2007). Available at www.nccn.org/professionals/physician_gls/PDF/vte.pdf. Accessed 01/02/2008.
- Ansell J, Hirsh J, Poller L, Bussey H, Jacobson A, Hylek E. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:204S–233S.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med 1999; 159:457–460.
- Hirsh J, Raschke R. Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic therapy. Chest 2004; 126 suppl 3:188S–203S.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Hettiarachchi RJ, Prins MH, Lensing AW, Büller HR. Low molecular weight heparin versus unfractionated heparin in the initial treatment of venous thromboembolism. Curr Opin Pulm Med 1998; 4:220–225.
- Gould MK, Dembitzer AD, Doyle RL, Hastie TJ, Garber AM. Low-molecular-weight heparins compared with unfractionated heparin for treatment of acute deep venous thrombosis. A meta-analysis of randomized, controlled trials. Ann Intern Med 1999; 130:800–809.
- Dolovich LR, Ginsberg JS, Douketis JD, Holbrook AM, Cheah G. A meta-analysis comparing low-molecular-weight heparins with un-fractionated heparin in the treatment of venous thromboembolism: examining some unanswered questions regarding location of treatment, product type, and dosing frequency. Arch Intern Med 2000; 160:181–188.
- van Dongen CJ, van den Belt AG, Prins MH, Lensing AW. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2004; 4:CD001100.
- Cook LM, Kahn SR, Goodwin J, Kovacs MJ. Frequency of renal impairment, advanced age, obesity, and cancer in venous thromboembolism patients in clinical practice. J Thromb Haemost 2007; 5:937–941.
- Barba R, Marco J, Martin-Alvarez H, et al. The influence of extreme body weight on clinical outcome of patients with venous thromboembolism: findings from a prospective registry (RIETE). J Thromb Haemost 2005; 3:856–862.
- Segal JB, Strieff MB, Hofmann LV, Thornton K, Bass EB. Management of venous thromboembolism: a systematic review for a practice guideline. Ann Intern Med 2007; 146:211–222.
- Aujesky D, Smith KJ, Cornuz J, Roberts MS. Cost-effectiveness of low-molecular-weight heparin for secondary prophylaxis of cancer-related venous thromboembolism. Thromb Haemost 2005; 93:592–599.
- Meyer G, Marjanovic Z, Valcke J, et al. Comparison of low-molecular-weight heparin and warfarin for the secondary prevention of venous thromboembolism in patients with cancer: a randomized controlled study. Arch Intern Med 2002; 162:1729–1735.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Deitcher SR, Kessler CM, Merli G, Rigas JR, Lyons RM, Fareed J ON-CENOX investigators. Secondary prevention of venous thromboembolic events in patients with active cancer: enoxaparin alone versus initial enoxaparin followed by warfarin for a 180-day period. Clin Appl Thromb Hemost 2006; 12:389–396.
- Noble SI, Finlay IG. Is long-term low-molecular-weight heparin acceptable to palliative care patients in the treatment of cancer related venous thromboembolism? A qualitative study. Palliat Med 2005; 19:197–201.
- Amirkhosravi A, Mousa SA, Amaya M, Francis JL. Antimetastatic effect of tinzaparin, a low-molecular-weight heparin. J Thromb Haemost 2003; 1:1972–1976.
- Kragh M, Binderup L, Vig Hjarnaa PJ, Bramm E, Johansen KB, Frimundt Petersen C. Non-anti-coagulant heparin inhibits metastasis but not primary tumor growth. Oncol Rep 2005; 14:99–104.
- Mousa SA, Mohamed S. Anti-angiogenic mechanisms and efficacy of the low molecular weight heparin, tinzaparin: anti-cancer efficacy. Oncol Rep 2004; 12:683–688.
- Bobek V, Kovarik J. Antitumor and antimetastatic effect of warfarin and heparins. Biomed Pharmacother 2004; 58:213–219.
- Altinbas M, Coskun HS, Er O, et al. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J Thromb Haemost 2004; 2:1266–1271.
- Lee AY, Rickles FR, Julian JA, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol 2005; 23:2123–2129.
- Kakkar AK, Levine MN, Kadziola Z, et al. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the Fragmin Advanced Malignancy Outcome Study (FAMOUS). J Clin Oncol 2004; 22:1944–1948.
- Klerk CP, Smorenburg SM, Otten HM, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol 2005; 23:2130–2135.
- Sideras K, Schaefer PL, Okuno SH, et al. Low-molecular-weight heparin in patients with advanced cancer: a phase 3 clinical trial. Mayo Clin Proc 2006; 81:758–767.
- Bernardi E, Piccioli A, Oliboni G, Zuin R, Girolami A, Prandoni P. Nomograms for the administration of unfractionated heparin in the initial treatment of acute thromboembolism—an overview. Thromb Haemost 2000; 84:22–26.
KEY POINTS
- We recommend judiciously screening for cancer with age- and sex-specific tests in patients with idiopathic VTE.
- Patients with VTE and cancer have a higher risk of both VTE recurrence and bleeding complications of anticoagulant therapy than do VTE patients without cancer.
- Either unfractionated heparin or a low-molecular-weight heparin (LMWH) should be started as soon as VTE is confirmed or even strongly suspected, while still awaiting confirmation.
- The current (grade 1A) recommendations for treating VTE in cancer patients are to use LMWH monotherapy for at least 3 to 6 months. Anticoagulation is necessary indefinitely when there is ongoing cancer treatment or persistent risk of VTE.
Anticoagulants and pregnancy: When are they safe?
Anticoagulation is essential in a wide variety of conditions in women of child-bearing age. Some, such as venous thromboembolism, occur more often during pregnancy. Others, such as recurrent fetal loss in the setting of antiphospholipid antibodies, are specific to pregnancy.
While anticoagulants are useful in many circumstances, their use during pregnancy increases the risk of hemorrhage and other adverse effects on the mother and the fetus. Treatment with anticoagulants during pregnancy must therefore be carefully considered, with judicious selection of the agent, and with reflection on the physiologic changes of pregnancy to ensure appropriate dosing. In this article, we review these issues.
WHY IS THROMBOTIC RISK HIGHER DURING PREGNANCY?
Venous thromboembolism is among the leading causes of maternal death in developed countries.1–3 Modern care has dramatically reduced the risk of maternal death from hemorrhage, infection, and hypertension, but rates of morbidity and death from thrombosis have remained stable or increased in recent years.4

- Much higher levels of fibrinogen and factors VII, VIII, IX, and X
- Lower levels of protein S and increased resistance to activated protein C
- Impaired fibrinolysis, due to inhibitors derived from the placenta.
Acquired antithrombin deficiency may also occur in high-proteinuric states such as nephrotic syndrome or preeclampsia, further increasing thrombotic risk. Pooling of venous blood, caused by progesterone-mediated venous dilation and compounded by compression of the inferior vena cava by the uterus in later pregnancy, also increases thrombotic risk. And endothelial disruption of the pelvic vessels may occur during delivery, particularly during cesarean section.
Additional factors that increase thrombotic risk include immobilization, such as bed rest for pregnancy complications; surgery, including cesarean section; ovarian hyperstimulation during gonadotropin use for in vitro fertilization; trauma; malignancy; and hereditary or acquired hypercoagulable states.6 These hypercoagulable states include deficiencies of antithrombin or the intrinsic anticoagulant proteins C or S; resistance to activated protein C, usually due to the factor V Leiden mutation; the PT20210A mutation of the prothrombin gene; hyperhomocystinemia due to mutation of the methyltetrahydrofolate reductase (MTHFR) gene; and the sustained presence of antiphospholipid antibodies, including lupus anticoagulant antibodies, sometimes also with moderately high titers of anticardiolipin or beta-2-glycoprotein I antibodies.
Other conditions that increase thrombotic risk include hyperemesis gravidarum, obesity, inflammatory bowel disease, infection, smoking, and indwelling intravenous catheters.6 Given the multitude of risk factors, pregnant women have a risk of thrombotic complications three to five times higher than nonpregnant women.7
HEPARIN USE DURING PREGNANCY
Low-molecular-weight heparins (LMWHs)8 and unfractionated heparin bind to anti-thrombin and thus change the shape of the antithrombin molecule, dramatically increasing its interaction with the clotting factors Xa and prothrombin (factor II). The enhanced clearance of these procoagulant proteins leads to the anticoagulant effect. Unfractionated heparin has roughly equivalent interaction with factors Xa and II and prolongs the activated partial thromboplastin time (aPTT), which is therefore used to monitor the intensity of anticoagulation.
LMWHs, on the other hand, interact relatively little with factor II and do not predictably prolong the aPTT. Monitoring their effect is therefore more difficult and requires direct measurement of anti-factor-Xa activity. This test is widely available, but it is time-consuming (it takes several hours and results may not be available within 24 hours if the test is requested “after hours”), and therefore it is of limited use in the acute clinical setting. While weight-based dosing of LMWHs is reliable and safe in nonpregnant patients, it has not yet been validated for pregnant women.
Unfractionated heparin has been used for decades for many indications during pregnancy. It is a large molecule, so it does not cross the placenta and thus, in contrast to the coumarin derivatives, does not cause teratogenesis or toxic fetal effects. Its main limitations in pregnancy are its inconvenient dosing (at least twice daily when given subcutaneously) and its potential maternal adverse effects (mainly osteoporosis and heparin-induced thrombocytopenia).
Over the last 10 years LMWHs have become the preferred anticoagulants for treating and preventing thromboembolism in all patients. They are equivalent or superior to unfractionated heparin in efficacy and safety in the initial treatment of acute deep venous thrombosis9,10 and pulmonary embolism11,12 outside of pregnancy. While comparative data are much less robust in pregnant patients, several series have confirmed the safety and efficacy of LMWHs in pregnancy.13–15 LMWHs do not cross the placenta15–17 and thus have a fetal safety profile equivalent to that of unfractionated heparin.
Pregnancy alters metabolism of LMWHs
The physiologic changes of pregnancy alter the metabolism of LMWH, resulting in lower peak levels and a higher rate of clearance,18,19 and so a pregnant woman may need higher doses or more frequent dosing.
Recent evidence suggests that thromboprophylaxis can be done with lower, fixed, once-daily doses of LMWH throughout pregnancy,20 although some clinicians still prefer twice-daily dosing (particularly during the latter half of pregnancy).
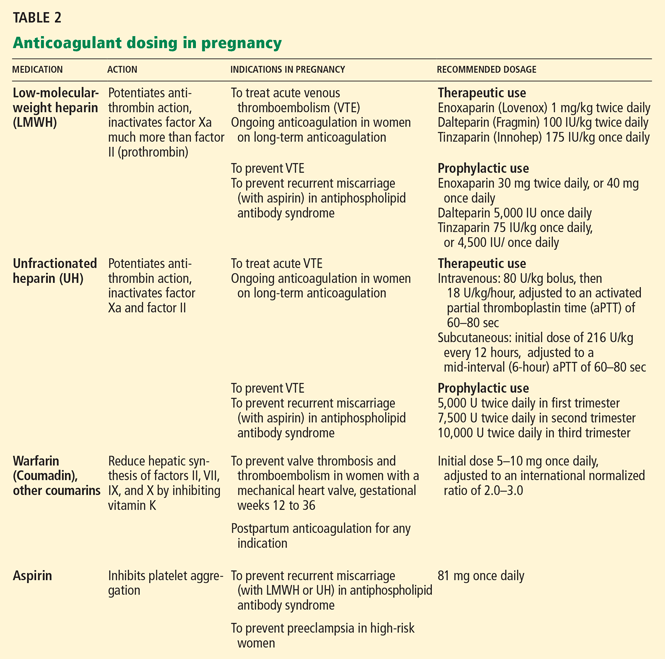
Pending more research on weight-based dosing of LMWH in pregnancy, anti-factor- Xa activity levels should be measured after treatment is started and every 1 to 3 months thereafter during pregnancy.21 Doses should be adjusted to keep the peak anti-Xa level (ie, 4 hours after the dose) at 0.5 to 1.2 U/mL.22
Heparin-induced thrombocytopenia
Type-2 heparin-induced thrombocytopenia is an uncommon but serious adverse effect of unfractionated heparin therapy (and, less commonly of LMWH), caused by heparin-dependent immunoglobulin G (IgG) antibodies that activate platelets via their Fc receptors, potentially precipitating life-threatening arterial or venous thrombosis.
In a trial in nonpregnant orthopedic patients,23 clinical heparin-induced thrombocytopenia occurred in 2.7% of patients receiving unfractionated heparin vs 0% of those receiving LMWH; heparin-dependent IgG was present in 7.8% vs 2.2%, respectively.
Fortunately, heparin-induced thrombocytopenia seems to be very rare in pregnancy: two recent prospective series evaluating prolonged LMWH use in pregnancy13,15 revealed no episodes of this disease. Nonetheless, it is reasonable to measure the platelet count once or twice weekly during the first few weeks of LMWH use and less often thereafter, unless symptoms of heparin-induced thrombocytopenia develop. In pregnant women with heparin-induced thrombocytopenia or heparin-related skin reactions, other anticoagulants must be considered24 (see discussion later).
Heparin-induced osteoporosis
Heparin-induced osteoporosis, a potential effect of prolonged heparin therapy, is of concern, given the prolonged duration and high doses of unfractionated heparin often needed to treat venous thromboembolism during pregnancy. Several studies found significant loss of bone mineral density in the proximal femur25 and lumbar spine26 during extended use of unfractionated heparin in pregnancy.
Fortunately, LMWH appears to be much safer with respect to bone loss. Three recent studies27–30 evaluated the use of LMWH for extended periods during pregnancy, and none found any greater loss of bone mineral density than that seen in normal pregnant controls. Giving supplemental calcium (1,000–1,500 mg/day) and vitamin D (400–1,000 IU/day) concomitantly with unfractionated heparin or LMWH in pregnancy is advisable to further reduce the risk.
Interrupt heparin to permit regional anesthesia
Heparin therapy should be temporarily stopped during the immediate peripartum interval to minimize the risk of hemorrhage and to permit regional anesthesia. Because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, many anesthetists will not perform neuraxial regional anesthesia in women who have recently received heparin.
Since unfractionated heparin has a relatively short duration of action, the American Society of Regional Anesthesia states that subcutaneous unfractionated heparin prophylaxis is not a contraindication to neuraxial regional anesthesia.31 However, LMWHs should be stopped for at least 12 to 24 hours before regional anesthesia can be considered safe. This issue is discussed in more detail in the section on peripartum and postpartum management of anticoagulation, below.
In summary, LMWH during pregnancy offers a number of advantages over unfractionated heparin: equivalent efficacy, once- or twice-daily dosing, lower risk of heparin-induced thrombocytopenia and osteoporosis, and less-intensive monitoring. Unfractionated heparin can be offered to women who cannot afford LMWH (which costs four to five times more), and it may be used peripartum to reduce hemorrhagic risk and to permit regional anesthesia.
COUMARINS
Coumarins are the mainstay of anticoagulant therapy in most nonpregnant women beyond the immediate thrombotic period.
Warfarin (Coumadin) is the most widely used coumarin because it has a predictable onset and duration of action and excellent bioavailability.32 Others, such as acenocoumarol (Sintrom) and phenprocoumon (Marcoumar), are used more outside the United States but can be ordered or brought into the United States.
Coumarins interfere with vitamin K metabolism, inhibiting the generation of vitamin-K-dependent procoagulant proteins (factors II, VII, IX, and X) and thereby preventing clotting. They also inhibit the formation of the vitamin-K-dependent intrinsic anticoagulant proteins C and S.
Major bleeding is the most significant side effect of coumarin therapy, occurring at a rate of 4% to 6% over 3 months when the prothrombin time is maintained at an international normalized ratio (INR) of 2 to 3,33 and more often if the INR is higher.
Other issues with warfarin are the effect of variations in dietary vitamin K intake on anticoagulation and potential drug interactions that may alter the anticoagulant effect. Thus, the INR needs to be monitored closely.
Risks to the fetus and the mother
Unlike the heparins, coumarins freely cross the placenta and thus pose a risk of teratogenicity. A cluster of fetal malformations including “warfarin embryopathy” (nasal bone hypoplasia and chondrodysplasia punctata) can occur when the drug is used between 6 and 12 weeks of gestation. Warfarin embryopathy may be avoided by stopping warfarin prior to 6 weeks from the onset of the last menstrual period (ie, 6-week “menstrual age” or 4-week gestational age34).
Later in pregnancy, warfarin is associated with potential fetal bleeding complications leading to central nervous system abnormalities, increased rates of intrauterine fetal death, and pregnancy loss. In pregnant women with mechanical cardiac valve prostheses who received oral anticoagulants throughout pregnancy, the incidence of congenital anomalies was 6.4% to 10.2%.35 Fetal demise (spontaneous abortion, stillbirth, neonatal death) was also very common (29.7% to 33.6% of pregnancies) in coumarin-treated women.
Severe maternal hemorrhage may also occur in pregnant women on oral anticoagulants, particularly those who remain fully anticoagulated around the time of labor and delivery.
General caveats to warfarin in pregnancy
Because of the many maternal and fetal concerns, oral anticoagulant use in pregnancy is largely restricted to women with older-generation prosthetic heart valves in whom the very high maternal thrombotic risk may outweigh the risk of maternal and fetal side effects.
While there are limited data on warfarin use in pregnant women with antiphospholipid syndrome,36 warfarin use in such patients should be considered only for those at highest risk and with careful informed consent. These issues are discussed further below in the section on mechanical heart valve prostheses.
ANTIPLATELET DRUGS
Aspirin is an antiplatelet agent rather than an anticoagulant. Although considered inadequate for preventing venous thrombosis in high-risk groups when used alone, aspirin can moderately reduce the risk of deep venous thrombosis and pulmonary embolism in nonpregnant patients.37 It also has a well-accepted role in preventing arterial thrombotic events, ie, coronary artery disease and stroke.38
Low-dose aspirin (≤ 100 mg/day) has been extensively evaluated during pregnancy39–41 and has been shown to be safe and effective in reducing the risk of preeclampsia in high-risk women39 and in treating women with antiphospholipid antibodies and recurrent pregnancy loss42 (in conjunction with prophylactic doses of heparin). Although higher doses of aspirin and other nonsteroidal anti-inflammatory drugs can be toxic to the fetus, low doses have been shown to be safe throughout pregnancy.43
Dipyridamole (Persantine) has been studied extensively in pregnancy, and while it appears to be safe, it has not found a well-defined therapeutic role.
Other antiplatelet drugs have been only rarely used, and data on their safety and efficacy during pregnancy are limited to case reports, for example, on ticlopidine44 (Ticlid) and clopidogrel45,46 (Plavix) given during pregnancy in women with cardiac disease. These drugs do not appear to be major teratogens or to cause specific fetal harm. Their use may be reasonable in some high-risk situations, such as recurrent thrombotic stroke despite aspirin therapy. They may be used alone or with other anticoagulants in women with a coronary or other vascular stent if fetal safety is uncertain or if there is an increased risk of maternal bleeding.
NEWER ANTICOAGULANTS
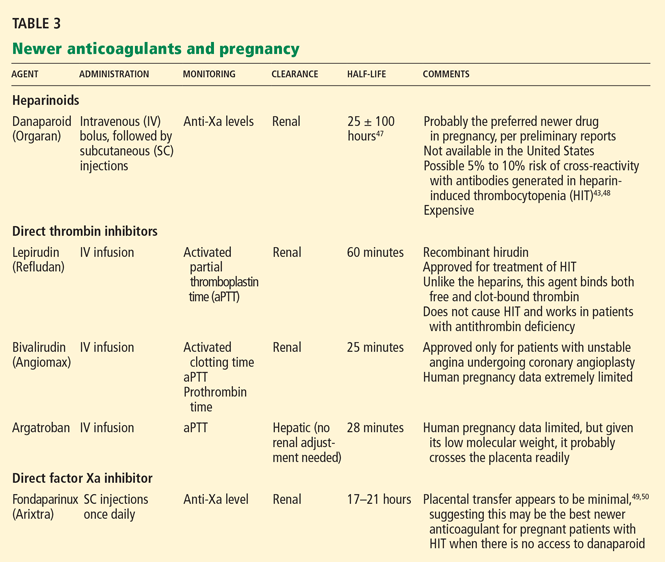
Danaparoid
The heparinoid danaparoid (Orgaran) is an LMWH, a combination of heparan, dermatan, and chondroitin sulfate. Since it is derived from heparin, in theory it can cross-react with antiheparin antibodies, but this is generally not a problem. Danaparoid inhibits factor Xa, and monitoring is via measurement of anti-factor-Xa activity levels. It has been shown to be safe and effective in nonpregnant patients with heparin-induced thrombocytopenia.51
Although no controlled study has been published on danaparoid in pregnancy, at least 51 pregnancies in 49 patients treated with danaparoid have been reported.52 Thirty-two of the patients received danaparoid because of heparin-induced thrombocytopenia and 19 because of heparin-induced skin intolerance. These reports suggest that danaparoid does not cross the placenta53 and that it may be effective and safe during pregnancy.54 For this reason, it is probably the preferred anticoagulant in pregnant patients with heparin-induced thrombocytopenia or other serious reactions to heparin.
Unfortunately, danaparoid has two major disadvantages. First, it has a prolonged half-life and no effective reversing agent, which makes its use problematic close to the time of delivery. Second, and perhaps more relevant to this discussion, it is not readily available in the United States; it was removed from the market by its manufacturer in April 2002 for business reasons rather than because of concerns over toxicity. It is still available in Canada and Europe, and it can be obtained in special circumstances in the United States via the US Food and Drug Administration (FDA); this may be worthwhile in pregnant patients who require a nonurgent alternative to heparin.
Direct thrombin inhibitors
Lepirudin (Refludan), bivalirudin (Angiomax), and argatroban are direct thrombin inhibitors and exert their anticoagulant effect independently of antithrombin. They are given by continuous intravenous infusion, and they have a very short half-life.
Lepirudin and argatroban are typically monitored via the aPTT. Bivalirudin can be monitored with the activated clotting time, partial thromboplastin time, or INR, depending on the circumstances. None of these agents generates or cross-reacts with antibodies generated in heparin-induced thrombocytopenia. None has an antidote, but the short half-life usually obviates the need for one.
Unfortunately, pregnancy data are very sparse for all three of these new agents. Argatroban has a low molecular weight and likely crosses the placenta. Also, because these agents are given intravenously, they are not practical for long-term use in pregnancy.
Fondaparinux
Fondaparinux (Arixtra), a direct factor Xa inhibitor, binds to antithrombin, causing an irreversible conformational change that increases antithrombin’s ability to inactivate factor Xa (as do the heparins). It has no effect on factor IIa (thrombin) and does not predictably affect the aPTT. Its half-life is 17 hours, and no agent is known to reverse its anticoagulant effect, although some experts would recommend a trial of high-dose recombinant factor VIIa (Novo-Seven) in uncontrolled hemorrhage.
While not FDA-approved for treating heparin-induced thrombocytopenia, it has been used for this in some patients.55–58 Animal studies and in vitro human placental perfusion studies suggest that fondaparinux does not cross the placenta in significant amounts.49 Since danaparoid is not available in the United States, fondaparinux would likely be the first choice among the newer anticoagulants when treating heparin-induced thrombocytopenia in pregnancy.
INDICATIONS FOR ANTICOAGULANTS DURING PREGNANCY
Acute deep venous thrombosis and pulmonary embolism
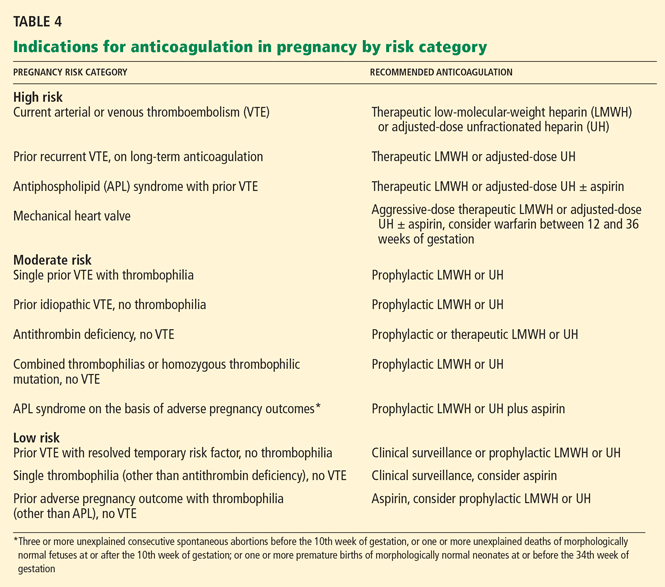
Anticoagulant therapy should begin as full doses of either LMWH or intravenous unfractionated heparin. We prefer starting with LMWH, as it can be started rapidly with less need for nursing care (eg, no need to start and maintain an intravenous line and monitor the aPTT) and has excellent safety. If LMWH is selected, initial dosing should be based on the current weight (Table 2). Subsequent monitoring of the peak anti-factor-Xa activity levels (ie, 4 hours after the dose) is recommended, with the first level drawn in the first few days of treatment, and repeat levels every 1 to 3 months for the rest of treatment. As mentioned earlier, weight-based dosing has not been systematically evaluated in pregnancy.
If unfractionated heparin is the initial agent, it should be given as a bolus followed by a continuous infusion, ideally utilizing a weight-based nomogram to estimate required doses, with adjustment of the infusion rate to maintain the aPTT at 1.5 to 2.5 times the baseline value (obtained during pregnancy). After several days, the heparin may be switched to LMWH in therapeutic doses (Table 2).
Alternatively, in women approaching term or who cannot afford LMWH, anticoagulation may be continued as adjusted-dose subcutaneous unfractionated heparin, ie, two or three large daily doses of subcutaneous heparin to provide therapeutic levels of anticoagulation. The starting dose can be calculated as the total units of heparin required to maintain full anticoagulation intravenously over 24 hours, given as two or three divided doses (Table 2). The aPTT at the mid-dosing interval (eg, 6 hours after the subcutaneous dose during every-12-hour dosing) should be monitored and the dose adjusted to maintain the aPTT at 1.5 to 2.5 times the baseline value.
A therapeutic level of anticoagulation should be maintained for at least 3 months after an acute thrombotic event during pregnancy, though many physicians prefer to continue full anticoagulation for a total of 6 months. Beyond this interval, if the woman is still pregnant, the anticoagulation may be reduced in intensity, perhaps even to a prophylactic level for the duration of the pregnancy (see discussion below on prior venous thromboembolic events) (Table 2). Peripartum and postpartum anticoagulation are discussed further below.
PRIOR VENOUS THROMBOEMBOLIC EVENT
While all pregnant women are at higher risk of venous thrombosis, the overall incidence of thromboembolism is only about one event per 1,000 pregnancies. Routine thromboprophylaxis in all pregnant women is therefore not justified. However, women who have previously had a venous thromboembolic event are at a substantially higher risk of recurrent thrombosis and should be considered for thromboprophylaxis in all subsequent high-risk situations, including pregnancy.
For women on indefinite therapeutic anticoagulation (ie, because of recurrent thrombosis), full therapeutic anticoagulation with LMWH or adjusted-dose unfractionated heparin should be maintained throughout pregnancy, as described above.
Which other women should receive prophylactic anticoagulation is a topic of ongoing debate and controversy.
How great is the risk of recurrent thromboembolism?
A small observational study59 examined the risk of recurrent venous thromboembolism during subsequent pregnancies in women with a prior thrombotic event. Anticoagulation was withheld during the antepartum period and restarted briefly after delivery. Among the 125 women enrolled, recurrent venous thromboembolism occurred in 4.8%, with half of the events occurring during the antepartum period. Among those with underlying thrombophilia, the rate of recurrent venous thromboembolism was 13% (95% confidence interval [CI] 1.7%–40.5%) to 20% (95% CI 2.5%–56.5%), and those with a prior idiopathic clot without thrombophilia had an event rate of 7.7% (95% CI 0.01%–25.1%). The subgroup with a prior reversible risk factor (at the time of their initial venous thromboembolic event) and without detectable thrombophilia had no recurrent events.
This study suggests that women with prior venous thromboembolism and thrombophilia or a prior idiopathic thrombotic event are at a substantial risk of recurrent thrombotic events during pregnancy. And other data confirm the high risk of recurrent venous thromboembolism in thrombophilic pregnant women.60 These women should all be offered active antepartum and postpartum thromboprophylaxis with LMWH or unfractionated heparin (Tables 2 and 4). Women without thrombophilia but with a history of venous thromboembolism related to pregnancy or oral contraceptive use also have a substantial risk of recurrent venous thrombosis and should be offered antepartum and postpartum thromboprophylaxis.61 In contrast, women with a prior “secondary” clot, no thrombophilia, and no additional current risk factors (Table 1) appear to be at low risk of recurrent venous thromboembolism.
The risks should be discussed with these women, with an option for close clinical surveillance during pregnancy (Table 4), but with a low threshold to investigate any worrisome symptoms. Such women may also elect to take LMWH or unfractionated heparin during pregnancy.
Which heparin to use?
Prophylactic anticoagulation during pregnancy can be with either LMWH or unfractionated heparin. For most women this involves “prophylactic” dosing with the goal of maintaining a mid-interval anti-factor-Xa activity level of approximately 0.05 to 0.2 U/mL. Thromboprophylaxis with LMWH can be with lower, fixed, once-daily doses throughout pregnancy20 (Table 2), although some clinicians still prefer twice-daily dosing. The heparin should be started as soon as pregnancy is confirmed, as the pregnancy-associated increase in thrombotic risk begins by the middle of the first trimester.
To maintain effective prophylactic levels, the dose of unfractionated heparin should be increased sequentially over the trimesters62,63: approximately 5,000 units subcutaneously twice daily in the first trimester, then 7,500 units twice daily in the second trimester, and 10,000 units twice daily in the third trimester for a woman of average size.
When to add low-dose aspirin
Women with antiphospholipid antibodies, particularly those with prior recurrent pregnancy loss or fetal demise, should receive aspirin 81 mg/day in addition to heparin.39 The aspirin may be started prior to conception or when pregnancy is confirmed.
Other measures
Women on anticoagulant therapy who are at risk of recurrent venous thromboembolism should be encouraged to wear elastic compression stockings. Intermittent pneumatic compression of the legs via automated devices may be considered for women hospitalized for any reason or on bedrest.
Whichever measures are used, a high index of suspicion and a low threshold for investigating for recurrent thrombosis should be maintained throughout pregnancy and the puerperium.
PERIPARTUM AND POSTPARTUM MANAGEMENT OF ANTICOAGULATION
Heparin therapy must be interrupted temporarily during the immediate peripartum interval to minimize the risk of hemorrhage and to allow for the option of regional anesthesia. As mentioned earlier, because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, the American Society of Regional Anesthesia guidelines advise waiting to insert the needle at least 10 to 12 hours after the last prophylactic dose of LMWH, and at least 24 hours after the last therapeutic dose.31
The guidelines state that neuraxial anesthesia is not contraindicated in patients on prophylactic unfractionated heparin.31
To facilitate use of regional anesthesia in these women, therefore, options include:
- Electively stopping LMWH 24 hours before planned induction of labor
- Electively stopping prophylactic-dose LMWH or unfractionated heparin at about 38 weeks of gestation, to await spontaneous labor, or
- Switching therapeutic or prophylactic LMWH to unfractionated heparin at about 36 weeks of gestation, with instructions to discontinue the injections in the earliest stages of spontaneous labor. This aims to shorten the heparin-free period required before neuraxial anesthesia while minimizing maternal thrombotic risk.
Additional advantages to using unfractionated heparin peripartum include the option of obtaining a rapid aPTT measurement to confirm the absence of a significant ongoing heparin effect prior to regional anesthesia or delivery, and the ability to completely reverse the heparin effect with protamine sulfate if major bleeding occurs. LMWHs are only partially reversible.64
Interrupting anticoagulation after an initial thrombotic event
If therapeutic anticoagulation must be interrupted for labor within 1 month of the initial thrombotic event, the risk of recurrent thrombotic complications is high65; these women must be observed very carefully and may benefit from intravenous heparin before and after delivery. They may even merit placement of a temporary vena cava filter (particularly if less than 2 weeks have elapsed since the venous thromboembolic event and in women with a large deep venous clot burden), a procedure that has been used safely but little studied in pregnant women.66
Fluoroscopic guidance may be needed for filter placement. This exposes the fetus to radiation, but the low-level exposure at this late gestational age is unlikely to pose a significant risk. The filter may be removed within 1 to 2 weeks postpartum, assuming there are no ongoing contraindications to anticoagulation.
In the rare woman with antithrombin deficiency and a recent or prior thrombotic event, giving antithrombin concentrate during the peripartum (heparin-free) interval has been described and may be considered under the guidance of a hematologist.67
Ongoing anticoagulation is essential postpartum, as the puerperium is the period of highest day-to-day risk of thromboembolic events: about one-third of pregnancy-associated events occur during these 6 to 12 weeks.2 Heparin should be resumed 6 to 12 hours after delivery, once hemostasis is confirmed.
Options for women requiring ongoing therapeutic anticoagulation include intravenous heparin started without a bolus, to minimize bleeding risk, with aPTT measured 12 hours later, or an initial prophylactic dose of LMWH 6 to 12 hours postpartum, with therapeutic dosing resumed on postpartum day 1. If prophylactic dosing is desired, unfractionated heparin or LMWH may be given subcutaneously starting at about 6 hours postpartum.
Warfarin in the puerperium
Women may subsequently be maintained on either LMWH or unfractionated heparin, or switched to an oral anticoagulant such as warfarin. Although warfarin may appear in minute amounts in breast milk, it has not been associated with adverse events in newborns and is considered compatible with breastfeeding.68 Heparin should be continued during the initial days of warfarin therapy, until the INR is at a therapeutic level for 24 hours. Some physicians prefer to delay warfarin for several days, giving LMWH alone in the immediate postpartum period, to allow wound-healing and to reduce bleeding risk.
Postpartum, anticoagulation should be continued for at least 6 to 12 weeks, at which point the physiologic changes in the coagulation system related to pregnancy will have returned to normal.
THROMBOPHILIA WITHOUT A PREVIOUS THROMBOEMBOLIC EVENT
Over the last 5 to 10 years, practitioners have been seeing many more young women with genetic or acquired thrombophilias who have never had a venous thromboembolic event. Physicians must advise these women about their risk of thromboembolic events during pregnancy and about the appropriateness of anticoagulant use.
Thrombophilias are often detected in women who develop venous thrombosis during pregnancy,69–71 but they are also very common in the general population (around 15%). While women with thrombophilia are at above-average risk of venous thromboembolism during pregnancy, the magnitude of risk in an individual patient is often difficult to estimate.
Data suggest that some types of thrombophilia confer greater thrombotic risk than others. McColl et al72 derived risk estimates for a primary event in women with several of the disorders: 0.23% in women heterozygous for the factor V Leiden mutation, 0.88% in women with protein C deficiency, and 2.4% to 35.7% in women with antithrombin deficiency. A case-control study70 found that all thrombophilic states were more common in women with pregnancy-associated venous thromboembolism than in healthy pregnant controls, except those with the MTHFR mutation and protein S deficiency. The estimated risk during pregnancy was 0.03% in women with no defect, 0.1% in women with protein C deficiency, 0.25% in women with the factor V Leiden mutation, 0.4% in those with antithrombin deficiency, 0.5% in those with the prothrombin gene mutation, and 4.6% in those with both factor V Leiden and prothrombin gene mutations.
Routine anticoagulation not advised in pregnant thrombophilic women
Because the risk of a primary venous thromboembolic event is less than 1% for most thrombophilic women, routine anticoagulant therapy does not seem prudent for this indication. Given the low absolute risk of venous thromboembolism, the cost and potential side effects of anticoagulant use are difficult to justify.
The women who seem at higher risk and in whom anticoagulation should be considered include those with antithrombin deficiency; those with high-titer anticardiolipin antibodies or a lupus anticoagulant antibody (treat with heparin and low-dose aspirin); those with combined thrombophilic defects or who are homozygotes for the factor V Leiden or prothrombin gene mutations; and those with multiple other current risk factors for venous thromboembolism (Table 1).
Since anticoagulants for primary prevention of adverse pregnancy outcomes in thrombophilic women have not yet been shown to have a definitive benefit, they are not recommended for this purpose.
ADVERSE PREGNANCY OUTCOMES IN WOMEN WITH THROMBOPHILIAS
Women with antiphospholipid antibodies and a previous poor obstetric outcome are clearly at increased risk of recurrent adverse pregnancy outcomes such as recurrent spontaneous abortion, unexplained fetal death, placental insufficiency, and early or severe preeclampsia. In such women who have both antiphospholipid antibodies and a history of venous thromboembolism or adverse pregnancy outcome, treatment during subsequent pregnancy with low-dose aspirin and prophylactic-dose LMWH or unfractionated heparin improves pregnancy outcomes.36–42 Women with antiphospholipid antibodies without previous thrombosis or pregnancy complications may also be at increased risk, but it is unclear whether thromboprophylaxis improves their outcomes.
Recent epidemiologic data reveal that women with other thrombophilic conditions also are at increased risk of early, severe preeclampsia73 as well as other pregnancy complications, including recurrent pregnancy loss, placental abruption, fetal growth restriction, and stillbirth.74 A recent meta-analysis75 looked at individual thrombophilias and found that factor V Leiden and prothrombin gene mutations were associated with recurrent fetal loss, stillbirth, and preeclampsia; that protein S deficiency was associated with recurrent fetal loss and stillbirth; that antiphospholipid antibodies were associated with recurrent pregnancy loss, preeclampsia, and intrauterine growth restriction; that the MTHFR mutation (homozygous) was associated with preeclampsia; and that protein C and antithrombin deficiencies were not significantly associated with adverse pregnancy outcomes. Data were scant for some of the rarer thrombophilias.75
Several recent small studies76–78 suggest that anticoagulants may improve pregnancy outcomes in women with genetic thrombophilias and recurrent pregnancy loss. These findings have not yet been confirmed in high-quality clinical trials, but such trials are under way. It is still unclear whether anticoagulants also reduce the risk of other adverse pregnancy outcomes associated with thrombophilias.
The current American College of Chest Physicians guidelines recommend testing of women with adverse pregnancy outcomes (recurrent pregnancy loss, prior severe or recurrent preeclampsia, abruptions, or otherwise unexplained intrauterine death) for congenital thrombophilias and antiphospholipid antibodies, and offering treatment to such women, if thrombophilic, with low-dose aspirin plus prophylactic heparin (unfractionated or LMWH).22 The authors of the guidelines admit that the evidence for this recommendation is weak, but they argue that the heparin will also serve as thromboprophylaxis in this high-risk group. Hopefully, the randomized clinical trials currently under way will provide clearer guidance regarding the most appropriate therapy in this difficult clinical situation.
MECHANICAL HEART VALVES
Internists may occasionally encounter a woman with a mechanical heart valve prosthesis who is either pregnant or is planning a pregnancy and therefore needs advice regarding optimal anticoagulant management. This should generally be undertaken in a multi-disciplinary fashion, with input from cardiology, hematology, and maternal-fetal medicine. The substantial maternal and fetal risks and the lack of definitive data on which to base treatment decisions make it a treacherous and stressful undertaking. Nonetheless, all internists should have a basic understanding of the complex issues regarding this management.
Outside of pregnancy, oral anticoagulants are the mainstay of therapy for patients with mechanical heart valves. Unfortunately, as discussed above, the use of these agents during pregnancy carries a risk of teratogenicity and toxic fetal effects and increases the risk of pregnancy loss and maternal hemorrhage. Heparins have been used in this setting for many years, but data on their efficacy and safety are very limited, and there are numerous reports of catastrophic maternal thrombotic complications.79,80
A systematic review of anticoagulation in pregnant women with prosthetic heart valves34 found very limited data on heparin use throughout pregnancy. Women maintained on warfarin vs heparin between pregnancy weeks 6 and 12 had higher rates of congenital anomalies (6.4% with warfarin vs 3.4% with heparin) and total fetal wastage (33.6% vs 26.5%). The warfarin group had fewer maternal thromboembolic complications (3.9% vs 9.2%), however, and a slightly lower rate of maternal death (1.8% vs 4.2%). Most of the women had higher-risk older-generation valves in the mitral position.
Recent data on LMWH consist mainly of case reports and case series,81 with a likely bias to publication of worse outcomes. Controlled trials in this area will be difficult to conduct. Still, aggressive anticoagulation with LMWH or unfractionated heparin, with close monitoring of the intensity of anticoagulation, may be safe and effective for pregnant women with newer-generation mechanical heart valves.82 A recent consensus statement22 suggested several regimens for pregnant women with mechanical heart valves:
- Twice-daily LMWH throughout pregnancy, with the dose adjusted either by weight, or to keep the 4-hour postinjection anti-factor-Xa activity level around 1.0 to 1.2 U/mL
- Aggressive adjusted-dose unfractionated heparin throughout pregnancy, given subcutaneously every 12 hours and adjusted to keep the mid-interval aPTT at least twice the control value or to attain a mid-interval anti-factor-Xa activity level of 0.35 to 0.70 U/mL
- Unfractionated heparin or LMWH (as above) until gestation week 13, then warfarin until the middle of the third trimester, and then heparin again.22
The authors also recommended adding low-dose aspirin (75–162 mg/day) in high-risk women.22
These options all seem reasonable, given our current knowledge, though warfarin use during pregnancy should be restricted to very-high-risk situations, such as women with older-generation mitral prostheses. LM-WHs may become the preferred therapy for this indication once further controlled data regarding their efficacy and safety become available.
- Chang J, Elam-Evans LD, Berg CJ, et al. Pregnancy-related mortality surveillance-United States, 1991–1999. MMWR Surveill Summ 2003; 52:1–8.
- Lewis G, Drife JO, Clutton-Brock T, et al. Why Mothers Die, 2000–2002. The Sixth Report of the Confidential Enquiries into Maternal Deaths in the United Kingdom. London: RCOG Press, 2004.
- Health Canada. Special Report on Maternal Mortality and Severe Morbidity in Canada—Enhanced Surveillance: The Path to Prevention. Ottawa: Minister of Public Works and Government Services Canada, 2004. www.phac-aspc.gc.ca/rhs-ssg/srmm-rsmm/page1-eng.php. Accessed 11/26/2008.
- Stein PD, Hull RD, Kayali F, et al. Venous thromboembolism in pregnancy: 21-year trends. Am J Med 2004; 117:121–125.
- Greer IA. Thrombosis in pregnancy: maternal and fetal issues. Lancet 1999; 353:1258–1265.
- Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet 1999; 353:1167–1173.
- Gherman RB, Goodwin TM, Leung B, et al. Incidence, clinical characteristics, and timing of objectively diagnosed venous thromboembolism during pregnancy. Obstet Gynecol 1999; 94:730–734.
- Weitz JI. Low-molecular-weight heparins. N Engl J Med 1997; 337:688–698.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Simonneau G, Sors H, Charbonnier B, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for acute pulmonary embolism. The THESEE Study Group. Tinzaparine ou Heparine Standard: Evaluations dans l’Embolie Pulmonaire. N Engl J Med 1997; 337:663–669.
- Hull RD, Raskob GE, Brant RF, et al. Low-molecular-weight heparin vs heparin in the treatment of patients with pulmonary embolism. American-Canadian Thrombosis Study Group. Arch Intern Med 2000; 160:229–236.
- Sanson BJ, Lensing AW, Prins MH, et al. Safety of low-molecular-weight heparin in pregnancy: a systematic review. Thromb Haemost 1999; 81:668–672.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005; 106:401–407.
- Melissari E, Parker CJ, Wilson NV, et al. Use of low molecular weight heparin in pregnancy. Thromb Haemost 1992; 68:652–656.
- Forestier F, Daffos F, Capella-Pavlovsky M. Low molecular weight heparin (PK 10169) does not cross the placenta during the second trimester of pregnancy study by direct fetal blood sampling under ultrasound. Thromb Res 1984; 34:557–560.
- Forestier F, Daffos F, Rainaut M, Toulemonde F. Low molecular weight heparin (CY 216) does not cross the placenta during the third trimester of pregnancy. Thromb Haemost 1987; 57:234.
- Barbour LA, Oja JL, Schultz LK. A prospective trial that demonstrates that dalteparin requirements increase in pregnancy to maintain therapeutic levels of anticoagulation. Am J Obstet Gynecol 2004; 191:1024–1029.
- Smith MP, Norris LA, Steer PJ, Savidge GF, Bonnar J. Tinzaparin sodium for thrombosis treatment and prevention during pregnancy. Am J Obstet Gynecol 2004; 190:495–501.
- Ellison J, Walker ID, Greer IA. Antenatal use of enoxaparin for prevention and treatment of thromboembolism in pregnancy. BJOG 2000; 107:1116–1121.
- Sarig G, Brenner B. Monitoring of low molecular weight heparin (LMWH) in pregnancy. Thromb Res 2005; 115 suppl 1:84–86.
- Bates SM, Greer IA, Hirsh J, Ginsberg JS. Use of antithrombotic agents during pregnancy: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:627S–644S.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995; 332:1330–1335.
- Hassell K. The management of patients with heparin-induced thrombocytopenia who require anticoagulant therapy. Chest 2005; 127 suppl 2:1S–8S.
- Barbour LA, Kick SD, Steiner JF, et al. A prospective study of heparin-induced osteoporosis in pregnancy using bone densitometry. Am J Obst Gynecol 1994; 170:862–869.
- Douketis JD, Ginsberg JS, Burrows RF, Duku EK, Webber CE, Brill-Edwards P. The effects of long-term heparin therapy during pregnancy on bone density. A prospective matched cohort study. Thromb Haemost 1996; 75:254–257.
- Pettila V, Leinonen P, Markkola A, Hiilesmaa V, Kaaja R. Postpartum bone mineral density in women treated for thromboprophylaxis with unfractionated heparin or LMW heparin. Thromb Haemost 2002; 87:182–186.
- Carlin AJ, Farquharson RG, Quenby SM, Topping J, Fraser WD. Prospective observational study of bone mineral density during pregnancy: low molecular weight heparin versus control. Hum Reprod 2004; 19:1211–1214.
- Casele HL, Laifer SA. Prospective evaluation of bone density in pregnant women receiving the low molecular weight heparin enoxaparin sodium. J Matern Fetal Med 2000; 9:122–125.
- Casele H, Haney EI, James A, Rosene-Montella K, Carson M. Bone density changes in women who receive thromboprophylaxis in pregnancy. Am J Obstet Gynecol 2006; 195:1109–1113.
- Horlocker TT, Wedel DJ, Benzon H, et al. Regional anesthesia in the anticoagulated patient: defining the risks (the second ASRA Consensus Conference on Neuraxial Anesthesia and Anticoagulation). Reg Anesth Pain Med 2003; 28:172–197.
- Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 2001; 119 suppl 1:8S–21S.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:287S–310S.
- Holmes LB. Teratogen-induced limb defects. Am J Med Genet 2002; 112:297–303.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Pauzner R, Dulitzki M, Langevitz P, Livneh A, Kenett R, Many A. Low molecular weight heparin and warfarin in the treatment of patients with antiphospholipid syndrome during pregnancy. Thromb Haemost 2001; 86:1379–1384.
- Pulmonary Embolism Prevention (PEP) Trial Collaborative Group. Prevention of pulmonary embolism and deep vein thrombosis with low dose aspirin: Pulmonary Embolism Prevention (PEP) trial. Lancet 2000; 355:1295–1302.
- Patrono C, Coller B, FitzGerald GA, Hirsh J, Roth G. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:234S–264S.
- Duley L, Henderson-Smart DJ, Knight M, King JF. Antiplatelet agents for preventing preeclampsia and its complications. Cochrane Database Syst Rev. 2004; ( 1):CD004659.
- Coomarasamy A, Honest H, Papaioannou S, Gee H, Khan KS. Aspirin for prevention of preeclampsia in women with historical risk factors: a systematic review. Obstet Gynecol 2003; 101:1319–1332.
- Caritis SN, Sibai BM, Hauth J, et al, and the National Institute of Child Health and Human Development Network of Maternal Fetal Medicine Units. Low-dose aspirin to prevent preeclampsia in women at high risk. N Engl J Med 1998; 338:701–705.
- Rai R, Cohen H, Dave M, Regan L. Randomised controlled trial of aspirin and aspirin plus heparin in pregnant women with recurrent miscarriage associated with phospholipid antibodies (or antiphospholipid antibodies). BMJ 1997; 314:253–257.
- Kozer E, Nikfar S, Costei A, Boskovic R, Nulman I, Koren G. Aspirin consumption during the first trimester of pregnancy and congenital anomalies: a meta-analysis. Am J Obstet Gynecol 2002; 187:1623–1630.
- Sebastian C, Scherlag M, Kugelmass A, Schechter E. Primary stent implantation for acute myocardial infarction during pregnancy: use of abciximab, ticlopidine, and aspirin. Cathet Cardiovasc Diagn 1998; 45:275–249.
- Wilson AM, Boyle AJ, Fox P. Management of ischaemic heart disease in women of child-bearing age. Intern Med J 2004; 34:694–697.
- Klinzing P, Markert UR, Liesaus K, Peiker G. Case report: successful pregnancy and delivery after myocardial infarction and essential thrombocythemia treated with clopidogrel. Clin Exp Obstet Gynecol 2001; 28:215–216.
- Danhof M, de Boer A, Magnani HN, Stiekema JC. Pharmacokinetic considerations on Orgaran (Org 10172) therapy. Haemostasis 1992; 22:73–84.
- Tardy-Poncet B, Tardy B, Reynaud J, et al. Efficacy and safety of danaparoid sodium (ORG 10172) in critically ill patients with heparin-associated thrombocytopenia. Chest 1999; 115:1616–1620.
- Lagrange F, Vergnes C, Brun JL, et al. Absence of placental transfer of pentasaccharide (fondaparinux, Arixtra) in the dually perfused human cotyledon in vitro. Thromb Haemost 2002; 87:831–835.
- Dempfle CE. Minor transplacental passge of fondapinux in vivo. N Engl J Med 2004; 350:1914.
- Magnani HN. Heparin-induced thrombocytopenia (HIT): an overview of 230 patients treated with orgaran (Org 10172). Thromb Haemost 1993; 70:554–561.
- Lindhoff-Last E, Kreutzenbeck HJ, Magnani HN. Treatment of 51 pregnancies with danaparoid because of heparin intolerance. Thromb Haemost 2005; 93:63–69.
- Greinacher A, Eckhardt T, Mussmann J, Mueller-Eckhardt C. Pregnancy complicated by heparin associated thrombocytopenia: management by a prospectively in vitro selected heparinoid (Org 10172). Thromb Res 1993; 71:123–126.
- Schindewolf M, Mosch G, Bauersachs RM, Lindhoff-Last E. Safe anticoagulation with danaparoid in pregnancy and lactation. Thromb Haemost 2004; 92:211.
- Harenberg J. Treatment of a woman with lupus and thromboembolism and cutaneous intolerance to heparins using fondaparinux during pregnancy. Thromb Res 2007; 119:385–388.
- Wijesiriwardana A, Lees DA, Lush C. Fondaparinux as anticoagulant in a pregnant woman with heparin allergy. Blood Coagul Fibrinolysis 2006; 17:147–149.
- Mazzolai L, Hohlfeld P, Spertini F, Hayoz D, Schapira M, Duchosal MA. Fondaparinux is a safe alternative in case of heparin intolerance during pregnancy. Blood 2006; 108:1569–1570.
- Hawkins D, Evans J. Minimizing the risk of heparin-induced osteoporosis during pregnancy. Expert Opin Drug Saf 2005; 4:583–590.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of clot in this pregnancy study group. N Engl J Med 2000; 343:1439–1444.
- Martinelli I, Legnani C, Bucciarelli P, Grandone E, De Stefano V, Mannucci PM. Risk of pregnancy-related venous thrombosis in carriers of severe inherited thrombophilia. Thromb Haemost 2001; 86:800–803.
- De Stefano V, Martinelli I, Rossi E, Battaglioli T, Za T, Mannucci PM, Leone G. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006; 135:386–391.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995; 173:1869–1873.
- Ensom MH, Stephenson MD. Pharmacokinetics of low molecular weight heparin and unfractionated heparin in pregnancy. J Soc Gynecol Investig 2004; 11:377–383.
- Crowther MA, Berry LR, Monagle PT, Chan AK. Mechanisms responsible for the failure of protamine to inactivate low-molecular-weight heparin. Br J Haematol 2002; 116:178–186.
- Kearon C, Hirsh J. Management of anticoagulation before and after elective surgery. N Engl J Med 1997; 336:1506–1511.
- Thomas LA, Summers RR, Cardwell MS. Use of Greenfield filters in pregnant women at risk for pulmonary embolism. South Med J 1997; 90:215–217.
- Maclean PS, Tait RC. Hereditary and acquired antithrombin deficiency: epidemiology, pathogenesis and treatment options. Drugs 2007; 67:1429–1440.
- Information from LactMed: http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?LACT, LactMed Record Number: 279. Accessed 11/26/2008.
- Gerhardt A, Scharf RE, Beckmann MW, et al. Prothrombin and factor V mutations in women with a history of thrombosis during pregnancy and the puerperium. N Engl J Med 2000; 342:374–380.
- Hirsch DR, Mikkola KM, Marks PW, et al. Pulmonary embolism and deep venous thrombosis during pregnancy or oral contraceptive use: prevalence of factor V Leiden. Am Heart J 1996; 131:1145–1148.
- Dizon-Townson DS, Nelson LM, Jang H, Varner MW, Ward K. The incidence of the factor V Leiden mutation in an obstetric population and its relationship to deep vein thrombosis. Am J Obstet Gynecol 1997; 176:883–886.
- McColl MD, Ramsay JE, Tait RC, et al. Risk factors for pregnancy associated venous thromboembolism. Thromb Haemost 1997; 78:1183–1188.
- Kupferminc MJ, Fait G, Many A, Gordon D, Eldor A, Lessing JB. Severe preeclampsia and high frequency of genetic thrombophilic mutations. Obstet Gynecol 2000; 96:45–49.
- Kupferminc MJ, Eldor A, Steinman N, et al. Increased frequency of genetic thrombophilia in women with complications of pregnancy. N Engl J Med 1999; 340:9–13.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006; 132:171–196.
- Brenner B, Hoffman R, Blumenfeld Z, Weiner Z, Younis JS. Gestational outcome in thrombophilic women with recurrent pregnancy loss treated by enoxaparin. Thromb Haemost 2000; 83:693–697.
- Carp H, Dolitzky M, Inbal A. Thromboprophylaxis improves the live birth rate in women with consecutive recurrent miscarriages and hereditary thrombophilia. J Thromb Haemost 2003; 1:433–438.
- Gris JC, Mercier E, Quere I, et al. Low-molecular-weight heparin versus low-dose aspirin in women with one fetal loss and a constitutional thrombophilic disorder. Blood 2004; 103:3695–3699.
- Salazar E, Izaguirre R, Verdejo J, Mutchinick O. Failure of adjusted doses of subcutaneous heparin to prevent thromboembolic phenomena in pregnant patients with mechanical cardiac valve prostheses. J Am Coll Cardiol 1996; 27:1698–1703.
- Iturbe-Alessio I, Fonseca MC, Mutchinik O, Santos MA, Zajarias A, Salazar E. Risks of anticoagulant therapy in pregnant women with artificial heart valves. N Engl J Med 1986; 315:1390–1393.
- Rowan JA, McCowan LM, Raudkivi PJ, North RA. Enoxaparin treatment in women with mechanical heart valves during pregnancy. Am J Obstet Gynecol 2001; 185:633–637.
- Oran B, Lee-Parritz A, Ansell J. Low molecular weight heparin for the prophylaxis of thromboembolism in women with prosthetic mechanical heart valves during pregnancy. Thromb Haemost 2004; 92:747–751.
Anticoagulation is essential in a wide variety of conditions in women of child-bearing age. Some, such as venous thromboembolism, occur more often during pregnancy. Others, such as recurrent fetal loss in the setting of antiphospholipid antibodies, are specific to pregnancy.
While anticoagulants are useful in many circumstances, their use during pregnancy increases the risk of hemorrhage and other adverse effects on the mother and the fetus. Treatment with anticoagulants during pregnancy must therefore be carefully considered, with judicious selection of the agent, and with reflection on the physiologic changes of pregnancy to ensure appropriate dosing. In this article, we review these issues.
WHY IS THROMBOTIC RISK HIGHER DURING PREGNANCY?
Venous thromboembolism is among the leading causes of maternal death in developed countries.1–3 Modern care has dramatically reduced the risk of maternal death from hemorrhage, infection, and hypertension, but rates of morbidity and death from thrombosis have remained stable or increased in recent years.4
- Much higher levels of fibrinogen and factors VII, VIII, IX, and X
- Lower levels of protein S and increased resistance to activated protein C
- Impaired fibrinolysis, due to inhibitors derived from the placenta.
Acquired antithrombin deficiency may also occur in high-proteinuric states such as nephrotic syndrome or preeclampsia, further increasing thrombotic risk. Pooling of venous blood, caused by progesterone-mediated venous dilation and compounded by compression of the inferior vena cava by the uterus in later pregnancy, also increases thrombotic risk. And endothelial disruption of the pelvic vessels may occur during delivery, particularly during cesarean section.
Additional factors that increase thrombotic risk include immobilization, such as bed rest for pregnancy complications; surgery, including cesarean section; ovarian hyperstimulation during gonadotropin use for in vitro fertilization; trauma; malignancy; and hereditary or acquired hypercoagulable states.6 These hypercoagulable states include deficiencies of antithrombin or the intrinsic anticoagulant proteins C or S; resistance to activated protein C, usually due to the factor V Leiden mutation; the PT20210A mutation of the prothrombin gene; hyperhomocystinemia due to mutation of the methyltetrahydrofolate reductase (MTHFR) gene; and the sustained presence of antiphospholipid antibodies, including lupus anticoagulant antibodies, sometimes also with moderately high titers of anticardiolipin or beta-2-glycoprotein I antibodies.
Other conditions that increase thrombotic risk include hyperemesis gravidarum, obesity, inflammatory bowel disease, infection, smoking, and indwelling intravenous catheters.6 Given the multitude of risk factors, pregnant women have a risk of thrombotic complications three to five times higher than nonpregnant women.7
HEPARIN USE DURING PREGNANCY
Low-molecular-weight heparins (LMWHs)8 and unfractionated heparin bind to anti-thrombin and thus change the shape of the antithrombin molecule, dramatically increasing its interaction with the clotting factors Xa and prothrombin (factor II). The enhanced clearance of these procoagulant proteins leads to the anticoagulant effect. Unfractionated heparin has roughly equivalent interaction with factors Xa and II and prolongs the activated partial thromboplastin time (aPTT), which is therefore used to monitor the intensity of anticoagulation.
LMWHs, on the other hand, interact relatively little with factor II and do not predictably prolong the aPTT. Monitoring their effect is therefore more difficult and requires direct measurement of anti-factor-Xa activity. This test is widely available, but it is time-consuming (it takes several hours and results may not be available within 24 hours if the test is requested “after hours”), and therefore it is of limited use in the acute clinical setting. While weight-based dosing of LMWHs is reliable and safe in nonpregnant patients, it has not yet been validated for pregnant women.
Unfractionated heparin has been used for decades for many indications during pregnancy. It is a large molecule, so it does not cross the placenta and thus, in contrast to the coumarin derivatives, does not cause teratogenesis or toxic fetal effects. Its main limitations in pregnancy are its inconvenient dosing (at least twice daily when given subcutaneously) and its potential maternal adverse effects (mainly osteoporosis and heparin-induced thrombocytopenia).
Over the last 10 years LMWHs have become the preferred anticoagulants for treating and preventing thromboembolism in all patients. They are equivalent or superior to unfractionated heparin in efficacy and safety in the initial treatment of acute deep venous thrombosis9,10 and pulmonary embolism11,12 outside of pregnancy. While comparative data are much less robust in pregnant patients, several series have confirmed the safety and efficacy of LMWHs in pregnancy.13–15 LMWHs do not cross the placenta15–17 and thus have a fetal safety profile equivalent to that of unfractionated heparin.
Pregnancy alters metabolism of LMWHs
The physiologic changes of pregnancy alter the metabolism of LMWH, resulting in lower peak levels and a higher rate of clearance,18,19 and so a pregnant woman may need higher doses or more frequent dosing.
Recent evidence suggests that thromboprophylaxis can be done with lower, fixed, once-daily doses of LMWH throughout pregnancy,20 although some clinicians still prefer twice-daily dosing (particularly during the latter half of pregnancy).
Pending more research on weight-based dosing of LMWH in pregnancy, anti-factor- Xa activity levels should be measured after treatment is started and every 1 to 3 months thereafter during pregnancy.21 Doses should be adjusted to keep the peak anti-Xa level (ie, 4 hours after the dose) at 0.5 to 1.2 U/mL.22
Heparin-induced thrombocytopenia
Type-2 heparin-induced thrombocytopenia is an uncommon but serious adverse effect of unfractionated heparin therapy (and, less commonly of LMWH), caused by heparin-dependent immunoglobulin G (IgG) antibodies that activate platelets via their Fc receptors, potentially precipitating life-threatening arterial or venous thrombosis.
In a trial in nonpregnant orthopedic patients,23 clinical heparin-induced thrombocytopenia occurred in 2.7% of patients receiving unfractionated heparin vs 0% of those receiving LMWH; heparin-dependent IgG was present in 7.8% vs 2.2%, respectively.
Fortunately, heparin-induced thrombocytopenia seems to be very rare in pregnancy: two recent prospective series evaluating prolonged LMWH use in pregnancy13,15 revealed no episodes of this disease. Nonetheless, it is reasonable to measure the platelet count once or twice weekly during the first few weeks of LMWH use and less often thereafter, unless symptoms of heparin-induced thrombocytopenia develop. In pregnant women with heparin-induced thrombocytopenia or heparin-related skin reactions, other anticoagulants must be considered24 (see discussion later).
Heparin-induced osteoporosis
Heparin-induced osteoporosis, a potential effect of prolonged heparin therapy, is of concern, given the prolonged duration and high doses of unfractionated heparin often needed to treat venous thromboembolism during pregnancy. Several studies found significant loss of bone mineral density in the proximal femur25 and lumbar spine26 during extended use of unfractionated heparin in pregnancy.
Fortunately, LMWH appears to be much safer with respect to bone loss. Three recent studies27–30 evaluated the use of LMWH for extended periods during pregnancy, and none found any greater loss of bone mineral density than that seen in normal pregnant controls. Giving supplemental calcium (1,000–1,500 mg/day) and vitamin D (400–1,000 IU/day) concomitantly with unfractionated heparin or LMWH in pregnancy is advisable to further reduce the risk.
Interrupt heparin to permit regional anesthesia
Heparin therapy should be temporarily stopped during the immediate peripartum interval to minimize the risk of hemorrhage and to permit regional anesthesia. Because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, many anesthetists will not perform neuraxial regional anesthesia in women who have recently received heparin.
Since unfractionated heparin has a relatively short duration of action, the American Society of Regional Anesthesia states that subcutaneous unfractionated heparin prophylaxis is not a contraindication to neuraxial regional anesthesia.31 However, LMWHs should be stopped for at least 12 to 24 hours before regional anesthesia can be considered safe. This issue is discussed in more detail in the section on peripartum and postpartum management of anticoagulation, below.
In summary, LMWH during pregnancy offers a number of advantages over unfractionated heparin: equivalent efficacy, once- or twice-daily dosing, lower risk of heparin-induced thrombocytopenia and osteoporosis, and less-intensive monitoring. Unfractionated heparin can be offered to women who cannot afford LMWH (which costs four to five times more), and it may be used peripartum to reduce hemorrhagic risk and to permit regional anesthesia.
COUMARINS
Coumarins are the mainstay of anticoagulant therapy in most nonpregnant women beyond the immediate thrombotic period.
Warfarin (Coumadin) is the most widely used coumarin because it has a predictable onset and duration of action and excellent bioavailability.32 Others, such as acenocoumarol (Sintrom) and phenprocoumon (Marcoumar), are used more outside the United States but can be ordered or brought into the United States.
Coumarins interfere with vitamin K metabolism, inhibiting the generation of vitamin-K-dependent procoagulant proteins (factors II, VII, IX, and X) and thereby preventing clotting. They also inhibit the formation of the vitamin-K-dependent intrinsic anticoagulant proteins C and S.
Major bleeding is the most significant side effect of coumarin therapy, occurring at a rate of 4% to 6% over 3 months when the prothrombin time is maintained at an international normalized ratio (INR) of 2 to 3,33 and more often if the INR is higher.
Other issues with warfarin are the effect of variations in dietary vitamin K intake on anticoagulation and potential drug interactions that may alter the anticoagulant effect. Thus, the INR needs to be monitored closely.
Risks to the fetus and the mother
Unlike the heparins, coumarins freely cross the placenta and thus pose a risk of teratogenicity. A cluster of fetal malformations including “warfarin embryopathy” (nasal bone hypoplasia and chondrodysplasia punctata) can occur when the drug is used between 6 and 12 weeks of gestation. Warfarin embryopathy may be avoided by stopping warfarin prior to 6 weeks from the onset of the last menstrual period (ie, 6-week “menstrual age” or 4-week gestational age34).
Later in pregnancy, warfarin is associated with potential fetal bleeding complications leading to central nervous system abnormalities, increased rates of intrauterine fetal death, and pregnancy loss. In pregnant women with mechanical cardiac valve prostheses who received oral anticoagulants throughout pregnancy, the incidence of congenital anomalies was 6.4% to 10.2%.35 Fetal demise (spontaneous abortion, stillbirth, neonatal death) was also very common (29.7% to 33.6% of pregnancies) in coumarin-treated women.
Severe maternal hemorrhage may also occur in pregnant women on oral anticoagulants, particularly those who remain fully anticoagulated around the time of labor and delivery.
General caveats to warfarin in pregnancy
Because of the many maternal and fetal concerns, oral anticoagulant use in pregnancy is largely restricted to women with older-generation prosthetic heart valves in whom the very high maternal thrombotic risk may outweigh the risk of maternal and fetal side effects.
While there are limited data on warfarin use in pregnant women with antiphospholipid syndrome,36 warfarin use in such patients should be considered only for those at highest risk and with careful informed consent. These issues are discussed further below in the section on mechanical heart valve prostheses.
ANTIPLATELET DRUGS
Aspirin is an antiplatelet agent rather than an anticoagulant. Although considered inadequate for preventing venous thrombosis in high-risk groups when used alone, aspirin can moderately reduce the risk of deep venous thrombosis and pulmonary embolism in nonpregnant patients.37 It also has a well-accepted role in preventing arterial thrombotic events, ie, coronary artery disease and stroke.38
Low-dose aspirin (≤ 100 mg/day) has been extensively evaluated during pregnancy39–41 and has been shown to be safe and effective in reducing the risk of preeclampsia in high-risk women39 and in treating women with antiphospholipid antibodies and recurrent pregnancy loss42 (in conjunction with prophylactic doses of heparin). Although higher doses of aspirin and other nonsteroidal anti-inflammatory drugs can be toxic to the fetus, low doses have been shown to be safe throughout pregnancy.43
Dipyridamole (Persantine) has been studied extensively in pregnancy, and while it appears to be safe, it has not found a well-defined therapeutic role.
Other antiplatelet drugs have been only rarely used, and data on their safety and efficacy during pregnancy are limited to case reports, for example, on ticlopidine44 (Ticlid) and clopidogrel45,46 (Plavix) given during pregnancy in women with cardiac disease. These drugs do not appear to be major teratogens or to cause specific fetal harm. Their use may be reasonable in some high-risk situations, such as recurrent thrombotic stroke despite aspirin therapy. They may be used alone or with other anticoagulants in women with a coronary or other vascular stent if fetal safety is uncertain or if there is an increased risk of maternal bleeding.
NEWER ANTICOAGULANTS
Danaparoid
The heparinoid danaparoid (Orgaran) is an LMWH, a combination of heparan, dermatan, and chondroitin sulfate. Since it is derived from heparin, in theory it can cross-react with antiheparin antibodies, but this is generally not a problem. Danaparoid inhibits factor Xa, and monitoring is via measurement of anti-factor-Xa activity levels. It has been shown to be safe and effective in nonpregnant patients with heparin-induced thrombocytopenia.51
Although no controlled study has been published on danaparoid in pregnancy, at least 51 pregnancies in 49 patients treated with danaparoid have been reported.52 Thirty-two of the patients received danaparoid because of heparin-induced thrombocytopenia and 19 because of heparin-induced skin intolerance. These reports suggest that danaparoid does not cross the placenta53 and that it may be effective and safe during pregnancy.54 For this reason, it is probably the preferred anticoagulant in pregnant patients with heparin-induced thrombocytopenia or other serious reactions to heparin.
Unfortunately, danaparoid has two major disadvantages. First, it has a prolonged half-life and no effective reversing agent, which makes its use problematic close to the time of delivery. Second, and perhaps more relevant to this discussion, it is not readily available in the United States; it was removed from the market by its manufacturer in April 2002 for business reasons rather than because of concerns over toxicity. It is still available in Canada and Europe, and it can be obtained in special circumstances in the United States via the US Food and Drug Administration (FDA); this may be worthwhile in pregnant patients who require a nonurgent alternative to heparin.
Direct thrombin inhibitors
Lepirudin (Refludan), bivalirudin (Angiomax), and argatroban are direct thrombin inhibitors and exert their anticoagulant effect independently of antithrombin. They are given by continuous intravenous infusion, and they have a very short half-life.
Lepirudin and argatroban are typically monitored via the aPTT. Bivalirudin can be monitored with the activated clotting time, partial thromboplastin time, or INR, depending on the circumstances. None of these agents generates or cross-reacts with antibodies generated in heparin-induced thrombocytopenia. None has an antidote, but the short half-life usually obviates the need for one.
Unfortunately, pregnancy data are very sparse for all three of these new agents. Argatroban has a low molecular weight and likely crosses the placenta. Also, because these agents are given intravenously, they are not practical for long-term use in pregnancy.
Fondaparinux
Fondaparinux (Arixtra), a direct factor Xa inhibitor, binds to antithrombin, causing an irreversible conformational change that increases antithrombin’s ability to inactivate factor Xa (as do the heparins). It has no effect on factor IIa (thrombin) and does not predictably affect the aPTT. Its half-life is 17 hours, and no agent is known to reverse its anticoagulant effect, although some experts would recommend a trial of high-dose recombinant factor VIIa (Novo-Seven) in uncontrolled hemorrhage.
While not FDA-approved for treating heparin-induced thrombocytopenia, it has been used for this in some patients.55–58 Animal studies and in vitro human placental perfusion studies suggest that fondaparinux does not cross the placenta in significant amounts.49 Since danaparoid is not available in the United States, fondaparinux would likely be the first choice among the newer anticoagulants when treating heparin-induced thrombocytopenia in pregnancy.
INDICATIONS FOR ANTICOAGULANTS DURING PREGNANCY
Acute deep venous thrombosis and pulmonary embolism
Anticoagulant therapy should begin as full doses of either LMWH or intravenous unfractionated heparin. We prefer starting with LMWH, as it can be started rapidly with less need for nursing care (eg, no need to start and maintain an intravenous line and monitor the aPTT) and has excellent safety. If LMWH is selected, initial dosing should be based on the current weight (Table 2). Subsequent monitoring of the peak anti-factor-Xa activity levels (ie, 4 hours after the dose) is recommended, with the first level drawn in the first few days of treatment, and repeat levels every 1 to 3 months for the rest of treatment. As mentioned earlier, weight-based dosing has not been systematically evaluated in pregnancy.
If unfractionated heparin is the initial agent, it should be given as a bolus followed by a continuous infusion, ideally utilizing a weight-based nomogram to estimate required doses, with adjustment of the infusion rate to maintain the aPTT at 1.5 to 2.5 times the baseline value (obtained during pregnancy). After several days, the heparin may be switched to LMWH in therapeutic doses (Table 2).
Alternatively, in women approaching term or who cannot afford LMWH, anticoagulation may be continued as adjusted-dose subcutaneous unfractionated heparin, ie, two or three large daily doses of subcutaneous heparin to provide therapeutic levels of anticoagulation. The starting dose can be calculated as the total units of heparin required to maintain full anticoagulation intravenously over 24 hours, given as two or three divided doses (Table 2). The aPTT at the mid-dosing interval (eg, 6 hours after the subcutaneous dose during every-12-hour dosing) should be monitored and the dose adjusted to maintain the aPTT at 1.5 to 2.5 times the baseline value.
A therapeutic level of anticoagulation should be maintained for at least 3 months after an acute thrombotic event during pregnancy, though many physicians prefer to continue full anticoagulation for a total of 6 months. Beyond this interval, if the woman is still pregnant, the anticoagulation may be reduced in intensity, perhaps even to a prophylactic level for the duration of the pregnancy (see discussion below on prior venous thromboembolic events) (Table 2). Peripartum and postpartum anticoagulation are discussed further below.
PRIOR VENOUS THROMBOEMBOLIC EVENT
While all pregnant women are at higher risk of venous thrombosis, the overall incidence of thromboembolism is only about one event per 1,000 pregnancies. Routine thromboprophylaxis in all pregnant women is therefore not justified. However, women who have previously had a venous thromboembolic event are at a substantially higher risk of recurrent thrombosis and should be considered for thromboprophylaxis in all subsequent high-risk situations, including pregnancy.
For women on indefinite therapeutic anticoagulation (ie, because of recurrent thrombosis), full therapeutic anticoagulation with LMWH or adjusted-dose unfractionated heparin should be maintained throughout pregnancy, as described above.
Which other women should receive prophylactic anticoagulation is a topic of ongoing debate and controversy.
How great is the risk of recurrent thromboembolism?
A small observational study59 examined the risk of recurrent venous thromboembolism during subsequent pregnancies in women with a prior thrombotic event. Anticoagulation was withheld during the antepartum period and restarted briefly after delivery. Among the 125 women enrolled, recurrent venous thromboembolism occurred in 4.8%, with half of the events occurring during the antepartum period. Among those with underlying thrombophilia, the rate of recurrent venous thromboembolism was 13% (95% confidence interval [CI] 1.7%–40.5%) to 20% (95% CI 2.5%–56.5%), and those with a prior idiopathic clot without thrombophilia had an event rate of 7.7% (95% CI 0.01%–25.1%). The subgroup with a prior reversible risk factor (at the time of their initial venous thromboembolic event) and without detectable thrombophilia had no recurrent events.
This study suggests that women with prior venous thromboembolism and thrombophilia or a prior idiopathic thrombotic event are at a substantial risk of recurrent thrombotic events during pregnancy. And other data confirm the high risk of recurrent venous thromboembolism in thrombophilic pregnant women.60 These women should all be offered active antepartum and postpartum thromboprophylaxis with LMWH or unfractionated heparin (Tables 2 and 4). Women without thrombophilia but with a history of venous thromboembolism related to pregnancy or oral contraceptive use also have a substantial risk of recurrent venous thrombosis and should be offered antepartum and postpartum thromboprophylaxis.61 In contrast, women with a prior “secondary” clot, no thrombophilia, and no additional current risk factors (Table 1) appear to be at low risk of recurrent venous thromboembolism.
The risks should be discussed with these women, with an option for close clinical surveillance during pregnancy (Table 4), but with a low threshold to investigate any worrisome symptoms. Such women may also elect to take LMWH or unfractionated heparin during pregnancy.
Which heparin to use?
Prophylactic anticoagulation during pregnancy can be with either LMWH or unfractionated heparin. For most women this involves “prophylactic” dosing with the goal of maintaining a mid-interval anti-factor-Xa activity level of approximately 0.05 to 0.2 U/mL. Thromboprophylaxis with LMWH can be with lower, fixed, once-daily doses throughout pregnancy20 (Table 2), although some clinicians still prefer twice-daily dosing. The heparin should be started as soon as pregnancy is confirmed, as the pregnancy-associated increase in thrombotic risk begins by the middle of the first trimester.
To maintain effective prophylactic levels, the dose of unfractionated heparin should be increased sequentially over the trimesters62,63: approximately 5,000 units subcutaneously twice daily in the first trimester, then 7,500 units twice daily in the second trimester, and 10,000 units twice daily in the third trimester for a woman of average size.
When to add low-dose aspirin
Women with antiphospholipid antibodies, particularly those with prior recurrent pregnancy loss or fetal demise, should receive aspirin 81 mg/day in addition to heparin.39 The aspirin may be started prior to conception or when pregnancy is confirmed.
Other measures
Women on anticoagulant therapy who are at risk of recurrent venous thromboembolism should be encouraged to wear elastic compression stockings. Intermittent pneumatic compression of the legs via automated devices may be considered for women hospitalized for any reason or on bedrest.
Whichever measures are used, a high index of suspicion and a low threshold for investigating for recurrent thrombosis should be maintained throughout pregnancy and the puerperium.
PERIPARTUM AND POSTPARTUM MANAGEMENT OF ANTICOAGULATION
Heparin therapy must be interrupted temporarily during the immediate peripartum interval to minimize the risk of hemorrhage and to allow for the option of regional anesthesia. As mentioned earlier, because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, the American Society of Regional Anesthesia guidelines advise waiting to insert the needle at least 10 to 12 hours after the last prophylactic dose of LMWH, and at least 24 hours after the last therapeutic dose.31
The guidelines state that neuraxial anesthesia is not contraindicated in patients on prophylactic unfractionated heparin.31
To facilitate use of regional anesthesia in these women, therefore, options include:
- Electively stopping LMWH 24 hours before planned induction of labor
- Electively stopping prophylactic-dose LMWH or unfractionated heparin at about 38 weeks of gestation, to await spontaneous labor, or
- Switching therapeutic or prophylactic LMWH to unfractionated heparin at about 36 weeks of gestation, with instructions to discontinue the injections in the earliest stages of spontaneous labor. This aims to shorten the heparin-free period required before neuraxial anesthesia while minimizing maternal thrombotic risk.
Additional advantages to using unfractionated heparin peripartum include the option of obtaining a rapid aPTT measurement to confirm the absence of a significant ongoing heparin effect prior to regional anesthesia or delivery, and the ability to completely reverse the heparin effect with protamine sulfate if major bleeding occurs. LMWHs are only partially reversible.64
Interrupting anticoagulation after an initial thrombotic event
If therapeutic anticoagulation must be interrupted for labor within 1 month of the initial thrombotic event, the risk of recurrent thrombotic complications is high65; these women must be observed very carefully and may benefit from intravenous heparin before and after delivery. They may even merit placement of a temporary vena cava filter (particularly if less than 2 weeks have elapsed since the venous thromboembolic event and in women with a large deep venous clot burden), a procedure that has been used safely but little studied in pregnant women.66
Fluoroscopic guidance may be needed for filter placement. This exposes the fetus to radiation, but the low-level exposure at this late gestational age is unlikely to pose a significant risk. The filter may be removed within 1 to 2 weeks postpartum, assuming there are no ongoing contraindications to anticoagulation.
In the rare woman with antithrombin deficiency and a recent or prior thrombotic event, giving antithrombin concentrate during the peripartum (heparin-free) interval has been described and may be considered under the guidance of a hematologist.67
Ongoing anticoagulation is essential postpartum, as the puerperium is the period of highest day-to-day risk of thromboembolic events: about one-third of pregnancy-associated events occur during these 6 to 12 weeks.2 Heparin should be resumed 6 to 12 hours after delivery, once hemostasis is confirmed.
Options for women requiring ongoing therapeutic anticoagulation include intravenous heparin started without a bolus, to minimize bleeding risk, with aPTT measured 12 hours later, or an initial prophylactic dose of LMWH 6 to 12 hours postpartum, with therapeutic dosing resumed on postpartum day 1. If prophylactic dosing is desired, unfractionated heparin or LMWH may be given subcutaneously starting at about 6 hours postpartum.
Warfarin in the puerperium
Women may subsequently be maintained on either LMWH or unfractionated heparin, or switched to an oral anticoagulant such as warfarin. Although warfarin may appear in minute amounts in breast milk, it has not been associated with adverse events in newborns and is considered compatible with breastfeeding.68 Heparin should be continued during the initial days of warfarin therapy, until the INR is at a therapeutic level for 24 hours. Some physicians prefer to delay warfarin for several days, giving LMWH alone in the immediate postpartum period, to allow wound-healing and to reduce bleeding risk.
Postpartum, anticoagulation should be continued for at least 6 to 12 weeks, at which point the physiologic changes in the coagulation system related to pregnancy will have returned to normal.
THROMBOPHILIA WITHOUT A PREVIOUS THROMBOEMBOLIC EVENT
Over the last 5 to 10 years, practitioners have been seeing many more young women with genetic or acquired thrombophilias who have never had a venous thromboembolic event. Physicians must advise these women about their risk of thromboembolic events during pregnancy and about the appropriateness of anticoagulant use.
Thrombophilias are often detected in women who develop venous thrombosis during pregnancy,69–71 but they are also very common in the general population (around 15%). While women with thrombophilia are at above-average risk of venous thromboembolism during pregnancy, the magnitude of risk in an individual patient is often difficult to estimate.
Data suggest that some types of thrombophilia confer greater thrombotic risk than others. McColl et al72 derived risk estimates for a primary event in women with several of the disorders: 0.23% in women heterozygous for the factor V Leiden mutation, 0.88% in women with protein C deficiency, and 2.4% to 35.7% in women with antithrombin deficiency. A case-control study70 found that all thrombophilic states were more common in women with pregnancy-associated venous thromboembolism than in healthy pregnant controls, except those with the MTHFR mutation and protein S deficiency. The estimated risk during pregnancy was 0.03% in women with no defect, 0.1% in women with protein C deficiency, 0.25% in women with the factor V Leiden mutation, 0.4% in those with antithrombin deficiency, 0.5% in those with the prothrombin gene mutation, and 4.6% in those with both factor V Leiden and prothrombin gene mutations.
Routine anticoagulation not advised in pregnant thrombophilic women
Because the risk of a primary venous thromboembolic event is less than 1% for most thrombophilic women, routine anticoagulant therapy does not seem prudent for this indication. Given the low absolute risk of venous thromboembolism, the cost and potential side effects of anticoagulant use are difficult to justify.
The women who seem at higher risk and in whom anticoagulation should be considered include those with antithrombin deficiency; those with high-titer anticardiolipin antibodies or a lupus anticoagulant antibody (treat with heparin and low-dose aspirin); those with combined thrombophilic defects or who are homozygotes for the factor V Leiden or prothrombin gene mutations; and those with multiple other current risk factors for venous thromboembolism (Table 1).
Since anticoagulants for primary prevention of adverse pregnancy outcomes in thrombophilic women have not yet been shown to have a definitive benefit, they are not recommended for this purpose.
ADVERSE PREGNANCY OUTCOMES IN WOMEN WITH THROMBOPHILIAS
Women with antiphospholipid antibodies and a previous poor obstetric outcome are clearly at increased risk of recurrent adverse pregnancy outcomes such as recurrent spontaneous abortion, unexplained fetal death, placental insufficiency, and early or severe preeclampsia. In such women who have both antiphospholipid antibodies and a history of venous thromboembolism or adverse pregnancy outcome, treatment during subsequent pregnancy with low-dose aspirin and prophylactic-dose LMWH or unfractionated heparin improves pregnancy outcomes.36–42 Women with antiphospholipid antibodies without previous thrombosis or pregnancy complications may also be at increased risk, but it is unclear whether thromboprophylaxis improves their outcomes.
Recent epidemiologic data reveal that women with other thrombophilic conditions also are at increased risk of early, severe preeclampsia73 as well as other pregnancy complications, including recurrent pregnancy loss, placental abruption, fetal growth restriction, and stillbirth.74 A recent meta-analysis75 looked at individual thrombophilias and found that factor V Leiden and prothrombin gene mutations were associated with recurrent fetal loss, stillbirth, and preeclampsia; that protein S deficiency was associated with recurrent fetal loss and stillbirth; that antiphospholipid antibodies were associated with recurrent pregnancy loss, preeclampsia, and intrauterine growth restriction; that the MTHFR mutation (homozygous) was associated with preeclampsia; and that protein C and antithrombin deficiencies were not significantly associated with adverse pregnancy outcomes. Data were scant for some of the rarer thrombophilias.75
Several recent small studies76–78 suggest that anticoagulants may improve pregnancy outcomes in women with genetic thrombophilias and recurrent pregnancy loss. These findings have not yet been confirmed in high-quality clinical trials, but such trials are under way. It is still unclear whether anticoagulants also reduce the risk of other adverse pregnancy outcomes associated with thrombophilias.
The current American College of Chest Physicians guidelines recommend testing of women with adverse pregnancy outcomes (recurrent pregnancy loss, prior severe or recurrent preeclampsia, abruptions, or otherwise unexplained intrauterine death) for congenital thrombophilias and antiphospholipid antibodies, and offering treatment to such women, if thrombophilic, with low-dose aspirin plus prophylactic heparin (unfractionated or LMWH).22 The authors of the guidelines admit that the evidence for this recommendation is weak, but they argue that the heparin will also serve as thromboprophylaxis in this high-risk group. Hopefully, the randomized clinical trials currently under way will provide clearer guidance regarding the most appropriate therapy in this difficult clinical situation.
MECHANICAL HEART VALVES
Internists may occasionally encounter a woman with a mechanical heart valve prosthesis who is either pregnant or is planning a pregnancy and therefore needs advice regarding optimal anticoagulant management. This should generally be undertaken in a multi-disciplinary fashion, with input from cardiology, hematology, and maternal-fetal medicine. The substantial maternal and fetal risks and the lack of definitive data on which to base treatment decisions make it a treacherous and stressful undertaking. Nonetheless, all internists should have a basic understanding of the complex issues regarding this management.
Outside of pregnancy, oral anticoagulants are the mainstay of therapy for patients with mechanical heart valves. Unfortunately, as discussed above, the use of these agents during pregnancy carries a risk of teratogenicity and toxic fetal effects and increases the risk of pregnancy loss and maternal hemorrhage. Heparins have been used in this setting for many years, but data on their efficacy and safety are very limited, and there are numerous reports of catastrophic maternal thrombotic complications.79,80
A systematic review of anticoagulation in pregnant women with prosthetic heart valves34 found very limited data on heparin use throughout pregnancy. Women maintained on warfarin vs heparin between pregnancy weeks 6 and 12 had higher rates of congenital anomalies (6.4% with warfarin vs 3.4% with heparin) and total fetal wastage (33.6% vs 26.5%). The warfarin group had fewer maternal thromboembolic complications (3.9% vs 9.2%), however, and a slightly lower rate of maternal death (1.8% vs 4.2%). Most of the women had higher-risk older-generation valves in the mitral position.
Recent data on LMWH consist mainly of case reports and case series,81 with a likely bias to publication of worse outcomes. Controlled trials in this area will be difficult to conduct. Still, aggressive anticoagulation with LMWH or unfractionated heparin, with close monitoring of the intensity of anticoagulation, may be safe and effective for pregnant women with newer-generation mechanical heart valves.82 A recent consensus statement22 suggested several regimens for pregnant women with mechanical heart valves:
- Twice-daily LMWH throughout pregnancy, with the dose adjusted either by weight, or to keep the 4-hour postinjection anti-factor-Xa activity level around 1.0 to 1.2 U/mL
- Aggressive adjusted-dose unfractionated heparin throughout pregnancy, given subcutaneously every 12 hours and adjusted to keep the mid-interval aPTT at least twice the control value or to attain a mid-interval anti-factor-Xa activity level of 0.35 to 0.70 U/mL
- Unfractionated heparin or LMWH (as above) until gestation week 13, then warfarin until the middle of the third trimester, and then heparin again.22
The authors also recommended adding low-dose aspirin (75–162 mg/day) in high-risk women.22
These options all seem reasonable, given our current knowledge, though warfarin use during pregnancy should be restricted to very-high-risk situations, such as women with older-generation mitral prostheses. LM-WHs may become the preferred therapy for this indication once further controlled data regarding their efficacy and safety become available.
Anticoagulation is essential in a wide variety of conditions in women of child-bearing age. Some, such as venous thromboembolism, occur more often during pregnancy. Others, such as recurrent fetal loss in the setting of antiphospholipid antibodies, are specific to pregnancy.
While anticoagulants are useful in many circumstances, their use during pregnancy increases the risk of hemorrhage and other adverse effects on the mother and the fetus. Treatment with anticoagulants during pregnancy must therefore be carefully considered, with judicious selection of the agent, and with reflection on the physiologic changes of pregnancy to ensure appropriate dosing. In this article, we review these issues.
WHY IS THROMBOTIC RISK HIGHER DURING PREGNANCY?
Venous thromboembolism is among the leading causes of maternal death in developed countries.1–3 Modern care has dramatically reduced the risk of maternal death from hemorrhage, infection, and hypertension, but rates of morbidity and death from thrombosis have remained stable or increased in recent years.4
- Much higher levels of fibrinogen and factors VII, VIII, IX, and X
- Lower levels of protein S and increased resistance to activated protein C
- Impaired fibrinolysis, due to inhibitors derived from the placenta.
Acquired antithrombin deficiency may also occur in high-proteinuric states such as nephrotic syndrome or preeclampsia, further increasing thrombotic risk. Pooling of venous blood, caused by progesterone-mediated venous dilation and compounded by compression of the inferior vena cava by the uterus in later pregnancy, also increases thrombotic risk. And endothelial disruption of the pelvic vessels may occur during delivery, particularly during cesarean section.
Additional factors that increase thrombotic risk include immobilization, such as bed rest for pregnancy complications; surgery, including cesarean section; ovarian hyperstimulation during gonadotropin use for in vitro fertilization; trauma; malignancy; and hereditary or acquired hypercoagulable states.6 These hypercoagulable states include deficiencies of antithrombin or the intrinsic anticoagulant proteins C or S; resistance to activated protein C, usually due to the factor V Leiden mutation; the PT20210A mutation of the prothrombin gene; hyperhomocystinemia due to mutation of the methyltetrahydrofolate reductase (MTHFR) gene; and the sustained presence of antiphospholipid antibodies, including lupus anticoagulant antibodies, sometimes also with moderately high titers of anticardiolipin or beta-2-glycoprotein I antibodies.
Other conditions that increase thrombotic risk include hyperemesis gravidarum, obesity, inflammatory bowel disease, infection, smoking, and indwelling intravenous catheters.6 Given the multitude of risk factors, pregnant women have a risk of thrombotic complications three to five times higher than nonpregnant women.7
HEPARIN USE DURING PREGNANCY
Low-molecular-weight heparins (LMWHs)8 and unfractionated heparin bind to anti-thrombin and thus change the shape of the antithrombin molecule, dramatically increasing its interaction with the clotting factors Xa and prothrombin (factor II). The enhanced clearance of these procoagulant proteins leads to the anticoagulant effect. Unfractionated heparin has roughly equivalent interaction with factors Xa and II and prolongs the activated partial thromboplastin time (aPTT), which is therefore used to monitor the intensity of anticoagulation.
LMWHs, on the other hand, interact relatively little with factor II and do not predictably prolong the aPTT. Monitoring their effect is therefore more difficult and requires direct measurement of anti-factor-Xa activity. This test is widely available, but it is time-consuming (it takes several hours and results may not be available within 24 hours if the test is requested “after hours”), and therefore it is of limited use in the acute clinical setting. While weight-based dosing of LMWHs is reliable and safe in nonpregnant patients, it has not yet been validated for pregnant women.
Unfractionated heparin has been used for decades for many indications during pregnancy. It is a large molecule, so it does not cross the placenta and thus, in contrast to the coumarin derivatives, does not cause teratogenesis or toxic fetal effects. Its main limitations in pregnancy are its inconvenient dosing (at least twice daily when given subcutaneously) and its potential maternal adverse effects (mainly osteoporosis and heparin-induced thrombocytopenia).
Over the last 10 years LMWHs have become the preferred anticoagulants for treating and preventing thromboembolism in all patients. They are equivalent or superior to unfractionated heparin in efficacy and safety in the initial treatment of acute deep venous thrombosis9,10 and pulmonary embolism11,12 outside of pregnancy. While comparative data are much less robust in pregnant patients, several series have confirmed the safety and efficacy of LMWHs in pregnancy.13–15 LMWHs do not cross the placenta15–17 and thus have a fetal safety profile equivalent to that of unfractionated heparin.
Pregnancy alters metabolism of LMWHs
The physiologic changes of pregnancy alter the metabolism of LMWH, resulting in lower peak levels and a higher rate of clearance,18,19 and so a pregnant woman may need higher doses or more frequent dosing.
Recent evidence suggests that thromboprophylaxis can be done with lower, fixed, once-daily doses of LMWH throughout pregnancy,20 although some clinicians still prefer twice-daily dosing (particularly during the latter half of pregnancy).
Pending more research on weight-based dosing of LMWH in pregnancy, anti-factor- Xa activity levels should be measured after treatment is started and every 1 to 3 months thereafter during pregnancy.21 Doses should be adjusted to keep the peak anti-Xa level (ie, 4 hours after the dose) at 0.5 to 1.2 U/mL.22
Heparin-induced thrombocytopenia
Type-2 heparin-induced thrombocytopenia is an uncommon but serious adverse effect of unfractionated heparin therapy (and, less commonly of LMWH), caused by heparin-dependent immunoglobulin G (IgG) antibodies that activate platelets via their Fc receptors, potentially precipitating life-threatening arterial or venous thrombosis.
In a trial in nonpregnant orthopedic patients,23 clinical heparin-induced thrombocytopenia occurred in 2.7% of patients receiving unfractionated heparin vs 0% of those receiving LMWH; heparin-dependent IgG was present in 7.8% vs 2.2%, respectively.
Fortunately, heparin-induced thrombocytopenia seems to be very rare in pregnancy: two recent prospective series evaluating prolonged LMWH use in pregnancy13,15 revealed no episodes of this disease. Nonetheless, it is reasonable to measure the platelet count once or twice weekly during the first few weeks of LMWH use and less often thereafter, unless symptoms of heparin-induced thrombocytopenia develop. In pregnant women with heparin-induced thrombocytopenia or heparin-related skin reactions, other anticoagulants must be considered24 (see discussion later).
Heparin-induced osteoporosis
Heparin-induced osteoporosis, a potential effect of prolonged heparin therapy, is of concern, given the prolonged duration and high doses of unfractionated heparin often needed to treat venous thromboembolism during pregnancy. Several studies found significant loss of bone mineral density in the proximal femur25 and lumbar spine26 during extended use of unfractionated heparin in pregnancy.
Fortunately, LMWH appears to be much safer with respect to bone loss. Three recent studies27–30 evaluated the use of LMWH for extended periods during pregnancy, and none found any greater loss of bone mineral density than that seen in normal pregnant controls. Giving supplemental calcium (1,000–1,500 mg/day) and vitamin D (400–1,000 IU/day) concomitantly with unfractionated heparin or LMWH in pregnancy is advisable to further reduce the risk.
Interrupt heparin to permit regional anesthesia
Heparin therapy should be temporarily stopped during the immediate peripartum interval to minimize the risk of hemorrhage and to permit regional anesthesia. Because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, many anesthetists will not perform neuraxial regional anesthesia in women who have recently received heparin.
Since unfractionated heparin has a relatively short duration of action, the American Society of Regional Anesthesia states that subcutaneous unfractionated heparin prophylaxis is not a contraindication to neuraxial regional anesthesia.31 However, LMWHs should be stopped for at least 12 to 24 hours before regional anesthesia can be considered safe. This issue is discussed in more detail in the section on peripartum and postpartum management of anticoagulation, below.
In summary, LMWH during pregnancy offers a number of advantages over unfractionated heparin: equivalent efficacy, once- or twice-daily dosing, lower risk of heparin-induced thrombocytopenia and osteoporosis, and less-intensive monitoring. Unfractionated heparin can be offered to women who cannot afford LMWH (which costs four to five times more), and it may be used peripartum to reduce hemorrhagic risk and to permit regional anesthesia.
COUMARINS
Coumarins are the mainstay of anticoagulant therapy in most nonpregnant women beyond the immediate thrombotic period.
Warfarin (Coumadin) is the most widely used coumarin because it has a predictable onset and duration of action and excellent bioavailability.32 Others, such as acenocoumarol (Sintrom) and phenprocoumon (Marcoumar), are used more outside the United States but can be ordered or brought into the United States.
Coumarins interfere with vitamin K metabolism, inhibiting the generation of vitamin-K-dependent procoagulant proteins (factors II, VII, IX, and X) and thereby preventing clotting. They also inhibit the formation of the vitamin-K-dependent intrinsic anticoagulant proteins C and S.
Major bleeding is the most significant side effect of coumarin therapy, occurring at a rate of 4% to 6% over 3 months when the prothrombin time is maintained at an international normalized ratio (INR) of 2 to 3,33 and more often if the INR is higher.
Other issues with warfarin are the effect of variations in dietary vitamin K intake on anticoagulation and potential drug interactions that may alter the anticoagulant effect. Thus, the INR needs to be monitored closely.
Risks to the fetus and the mother
Unlike the heparins, coumarins freely cross the placenta and thus pose a risk of teratogenicity. A cluster of fetal malformations including “warfarin embryopathy” (nasal bone hypoplasia and chondrodysplasia punctata) can occur when the drug is used between 6 and 12 weeks of gestation. Warfarin embryopathy may be avoided by stopping warfarin prior to 6 weeks from the onset of the last menstrual period (ie, 6-week “menstrual age” or 4-week gestational age34).
Later in pregnancy, warfarin is associated with potential fetal bleeding complications leading to central nervous system abnormalities, increased rates of intrauterine fetal death, and pregnancy loss. In pregnant women with mechanical cardiac valve prostheses who received oral anticoagulants throughout pregnancy, the incidence of congenital anomalies was 6.4% to 10.2%.35 Fetal demise (spontaneous abortion, stillbirth, neonatal death) was also very common (29.7% to 33.6% of pregnancies) in coumarin-treated women.
Severe maternal hemorrhage may also occur in pregnant women on oral anticoagulants, particularly those who remain fully anticoagulated around the time of labor and delivery.
General caveats to warfarin in pregnancy
Because of the many maternal and fetal concerns, oral anticoagulant use in pregnancy is largely restricted to women with older-generation prosthetic heart valves in whom the very high maternal thrombotic risk may outweigh the risk of maternal and fetal side effects.
While there are limited data on warfarin use in pregnant women with antiphospholipid syndrome,36 warfarin use in such patients should be considered only for those at highest risk and with careful informed consent. These issues are discussed further below in the section on mechanical heart valve prostheses.
ANTIPLATELET DRUGS
Aspirin is an antiplatelet agent rather than an anticoagulant. Although considered inadequate for preventing venous thrombosis in high-risk groups when used alone, aspirin can moderately reduce the risk of deep venous thrombosis and pulmonary embolism in nonpregnant patients.37 It also has a well-accepted role in preventing arterial thrombotic events, ie, coronary artery disease and stroke.38
Low-dose aspirin (≤ 100 mg/day) has been extensively evaluated during pregnancy39–41 and has been shown to be safe and effective in reducing the risk of preeclampsia in high-risk women39 and in treating women with antiphospholipid antibodies and recurrent pregnancy loss42 (in conjunction with prophylactic doses of heparin). Although higher doses of aspirin and other nonsteroidal anti-inflammatory drugs can be toxic to the fetus, low doses have been shown to be safe throughout pregnancy.43
Dipyridamole (Persantine) has been studied extensively in pregnancy, and while it appears to be safe, it has not found a well-defined therapeutic role.
Other antiplatelet drugs have been only rarely used, and data on their safety and efficacy during pregnancy are limited to case reports, for example, on ticlopidine44 (Ticlid) and clopidogrel45,46 (Plavix) given during pregnancy in women with cardiac disease. These drugs do not appear to be major teratogens or to cause specific fetal harm. Their use may be reasonable in some high-risk situations, such as recurrent thrombotic stroke despite aspirin therapy. They may be used alone or with other anticoagulants in women with a coronary or other vascular stent if fetal safety is uncertain or if there is an increased risk of maternal bleeding.
NEWER ANTICOAGULANTS
Danaparoid
The heparinoid danaparoid (Orgaran) is an LMWH, a combination of heparan, dermatan, and chondroitin sulfate. Since it is derived from heparin, in theory it can cross-react with antiheparin antibodies, but this is generally not a problem. Danaparoid inhibits factor Xa, and monitoring is via measurement of anti-factor-Xa activity levels. It has been shown to be safe and effective in nonpregnant patients with heparin-induced thrombocytopenia.51
Although no controlled study has been published on danaparoid in pregnancy, at least 51 pregnancies in 49 patients treated with danaparoid have been reported.52 Thirty-two of the patients received danaparoid because of heparin-induced thrombocytopenia and 19 because of heparin-induced skin intolerance. These reports suggest that danaparoid does not cross the placenta53 and that it may be effective and safe during pregnancy.54 For this reason, it is probably the preferred anticoagulant in pregnant patients with heparin-induced thrombocytopenia or other serious reactions to heparin.
Unfortunately, danaparoid has two major disadvantages. First, it has a prolonged half-life and no effective reversing agent, which makes its use problematic close to the time of delivery. Second, and perhaps more relevant to this discussion, it is not readily available in the United States; it was removed from the market by its manufacturer in April 2002 for business reasons rather than because of concerns over toxicity. It is still available in Canada and Europe, and it can be obtained in special circumstances in the United States via the US Food and Drug Administration (FDA); this may be worthwhile in pregnant patients who require a nonurgent alternative to heparin.
Direct thrombin inhibitors
Lepirudin (Refludan), bivalirudin (Angiomax), and argatroban are direct thrombin inhibitors and exert their anticoagulant effect independently of antithrombin. They are given by continuous intravenous infusion, and they have a very short half-life.
Lepirudin and argatroban are typically monitored via the aPTT. Bivalirudin can be monitored with the activated clotting time, partial thromboplastin time, or INR, depending on the circumstances. None of these agents generates or cross-reacts with antibodies generated in heparin-induced thrombocytopenia. None has an antidote, but the short half-life usually obviates the need for one.
Unfortunately, pregnancy data are very sparse for all three of these new agents. Argatroban has a low molecular weight and likely crosses the placenta. Also, because these agents are given intravenously, they are not practical for long-term use in pregnancy.
Fondaparinux
Fondaparinux (Arixtra), a direct factor Xa inhibitor, binds to antithrombin, causing an irreversible conformational change that increases antithrombin’s ability to inactivate factor Xa (as do the heparins). It has no effect on factor IIa (thrombin) and does not predictably affect the aPTT. Its half-life is 17 hours, and no agent is known to reverse its anticoagulant effect, although some experts would recommend a trial of high-dose recombinant factor VIIa (Novo-Seven) in uncontrolled hemorrhage.
While not FDA-approved for treating heparin-induced thrombocytopenia, it has been used for this in some patients.55–58 Animal studies and in vitro human placental perfusion studies suggest that fondaparinux does not cross the placenta in significant amounts.49 Since danaparoid is not available in the United States, fondaparinux would likely be the first choice among the newer anticoagulants when treating heparin-induced thrombocytopenia in pregnancy.
INDICATIONS FOR ANTICOAGULANTS DURING PREGNANCY
Acute deep venous thrombosis and pulmonary embolism
Anticoagulant therapy should begin as full doses of either LMWH or intravenous unfractionated heparin. We prefer starting with LMWH, as it can be started rapidly with less need for nursing care (eg, no need to start and maintain an intravenous line and monitor the aPTT) and has excellent safety. If LMWH is selected, initial dosing should be based on the current weight (Table 2). Subsequent monitoring of the peak anti-factor-Xa activity levels (ie, 4 hours after the dose) is recommended, with the first level drawn in the first few days of treatment, and repeat levels every 1 to 3 months for the rest of treatment. As mentioned earlier, weight-based dosing has not been systematically evaluated in pregnancy.
If unfractionated heparin is the initial agent, it should be given as a bolus followed by a continuous infusion, ideally utilizing a weight-based nomogram to estimate required doses, with adjustment of the infusion rate to maintain the aPTT at 1.5 to 2.5 times the baseline value (obtained during pregnancy). After several days, the heparin may be switched to LMWH in therapeutic doses (Table 2).
Alternatively, in women approaching term or who cannot afford LMWH, anticoagulation may be continued as adjusted-dose subcutaneous unfractionated heparin, ie, two or three large daily doses of subcutaneous heparin to provide therapeutic levels of anticoagulation. The starting dose can be calculated as the total units of heparin required to maintain full anticoagulation intravenously over 24 hours, given as two or three divided doses (Table 2). The aPTT at the mid-dosing interval (eg, 6 hours after the subcutaneous dose during every-12-hour dosing) should be monitored and the dose adjusted to maintain the aPTT at 1.5 to 2.5 times the baseline value.
A therapeutic level of anticoagulation should be maintained for at least 3 months after an acute thrombotic event during pregnancy, though many physicians prefer to continue full anticoagulation for a total of 6 months. Beyond this interval, if the woman is still pregnant, the anticoagulation may be reduced in intensity, perhaps even to a prophylactic level for the duration of the pregnancy (see discussion below on prior venous thromboembolic events) (Table 2). Peripartum and postpartum anticoagulation are discussed further below.
PRIOR VENOUS THROMBOEMBOLIC EVENT
While all pregnant women are at higher risk of venous thrombosis, the overall incidence of thromboembolism is only about one event per 1,000 pregnancies. Routine thromboprophylaxis in all pregnant women is therefore not justified. However, women who have previously had a venous thromboembolic event are at a substantially higher risk of recurrent thrombosis and should be considered for thromboprophylaxis in all subsequent high-risk situations, including pregnancy.
For women on indefinite therapeutic anticoagulation (ie, because of recurrent thrombosis), full therapeutic anticoagulation with LMWH or adjusted-dose unfractionated heparin should be maintained throughout pregnancy, as described above.
Which other women should receive prophylactic anticoagulation is a topic of ongoing debate and controversy.
How great is the risk of recurrent thromboembolism?
A small observational study59 examined the risk of recurrent venous thromboembolism during subsequent pregnancies in women with a prior thrombotic event. Anticoagulation was withheld during the antepartum period and restarted briefly after delivery. Among the 125 women enrolled, recurrent venous thromboembolism occurred in 4.8%, with half of the events occurring during the antepartum period. Among those with underlying thrombophilia, the rate of recurrent venous thromboembolism was 13% (95% confidence interval [CI] 1.7%–40.5%) to 20% (95% CI 2.5%–56.5%), and those with a prior idiopathic clot without thrombophilia had an event rate of 7.7% (95% CI 0.01%–25.1%). The subgroup with a prior reversible risk factor (at the time of their initial venous thromboembolic event) and without detectable thrombophilia had no recurrent events.
This study suggests that women with prior venous thromboembolism and thrombophilia or a prior idiopathic thrombotic event are at a substantial risk of recurrent thrombotic events during pregnancy. And other data confirm the high risk of recurrent venous thromboembolism in thrombophilic pregnant women.60 These women should all be offered active antepartum and postpartum thromboprophylaxis with LMWH or unfractionated heparin (Tables 2 and 4). Women without thrombophilia but with a history of venous thromboembolism related to pregnancy or oral contraceptive use also have a substantial risk of recurrent venous thrombosis and should be offered antepartum and postpartum thromboprophylaxis.61 In contrast, women with a prior “secondary” clot, no thrombophilia, and no additional current risk factors (Table 1) appear to be at low risk of recurrent venous thromboembolism.
The risks should be discussed with these women, with an option for close clinical surveillance during pregnancy (Table 4), but with a low threshold to investigate any worrisome symptoms. Such women may also elect to take LMWH or unfractionated heparin during pregnancy.
Which heparin to use?
Prophylactic anticoagulation during pregnancy can be with either LMWH or unfractionated heparin. For most women this involves “prophylactic” dosing with the goal of maintaining a mid-interval anti-factor-Xa activity level of approximately 0.05 to 0.2 U/mL. Thromboprophylaxis with LMWH can be with lower, fixed, once-daily doses throughout pregnancy20 (Table 2), although some clinicians still prefer twice-daily dosing. The heparin should be started as soon as pregnancy is confirmed, as the pregnancy-associated increase in thrombotic risk begins by the middle of the first trimester.
To maintain effective prophylactic levels, the dose of unfractionated heparin should be increased sequentially over the trimesters62,63: approximately 5,000 units subcutaneously twice daily in the first trimester, then 7,500 units twice daily in the second trimester, and 10,000 units twice daily in the third trimester for a woman of average size.
When to add low-dose aspirin
Women with antiphospholipid antibodies, particularly those with prior recurrent pregnancy loss or fetal demise, should receive aspirin 81 mg/day in addition to heparin.39 The aspirin may be started prior to conception or when pregnancy is confirmed.
Other measures
Women on anticoagulant therapy who are at risk of recurrent venous thromboembolism should be encouraged to wear elastic compression stockings. Intermittent pneumatic compression of the legs via automated devices may be considered for women hospitalized for any reason or on bedrest.
Whichever measures are used, a high index of suspicion and a low threshold for investigating for recurrent thrombosis should be maintained throughout pregnancy and the puerperium.
PERIPARTUM AND POSTPARTUM MANAGEMENT OF ANTICOAGULATION
Heparin therapy must be interrupted temporarily during the immediate peripartum interval to minimize the risk of hemorrhage and to allow for the option of regional anesthesia. As mentioned earlier, because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, the American Society of Regional Anesthesia guidelines advise waiting to insert the needle at least 10 to 12 hours after the last prophylactic dose of LMWH, and at least 24 hours after the last therapeutic dose.31
The guidelines state that neuraxial anesthesia is not contraindicated in patients on prophylactic unfractionated heparin.31
To facilitate use of regional anesthesia in these women, therefore, options include:
- Electively stopping LMWH 24 hours before planned induction of labor
- Electively stopping prophylactic-dose LMWH or unfractionated heparin at about 38 weeks of gestation, to await spontaneous labor, or
- Switching therapeutic or prophylactic LMWH to unfractionated heparin at about 36 weeks of gestation, with instructions to discontinue the injections in the earliest stages of spontaneous labor. This aims to shorten the heparin-free period required before neuraxial anesthesia while minimizing maternal thrombotic risk.
Additional advantages to using unfractionated heparin peripartum include the option of obtaining a rapid aPTT measurement to confirm the absence of a significant ongoing heparin effect prior to regional anesthesia or delivery, and the ability to completely reverse the heparin effect with protamine sulfate if major bleeding occurs. LMWHs are only partially reversible.64
Interrupting anticoagulation after an initial thrombotic event
If therapeutic anticoagulation must be interrupted for labor within 1 month of the initial thrombotic event, the risk of recurrent thrombotic complications is high65; these women must be observed very carefully and may benefit from intravenous heparin before and after delivery. They may even merit placement of a temporary vena cava filter (particularly if less than 2 weeks have elapsed since the venous thromboembolic event and in women with a large deep venous clot burden), a procedure that has been used safely but little studied in pregnant women.66
Fluoroscopic guidance may be needed for filter placement. This exposes the fetus to radiation, but the low-level exposure at this late gestational age is unlikely to pose a significant risk. The filter may be removed within 1 to 2 weeks postpartum, assuming there are no ongoing contraindications to anticoagulation.
In the rare woman with antithrombin deficiency and a recent or prior thrombotic event, giving antithrombin concentrate during the peripartum (heparin-free) interval has been described and may be considered under the guidance of a hematologist.67
Ongoing anticoagulation is essential postpartum, as the puerperium is the period of highest day-to-day risk of thromboembolic events: about one-third of pregnancy-associated events occur during these 6 to 12 weeks.2 Heparin should be resumed 6 to 12 hours after delivery, once hemostasis is confirmed.
Options for women requiring ongoing therapeutic anticoagulation include intravenous heparin started without a bolus, to minimize bleeding risk, with aPTT measured 12 hours later, or an initial prophylactic dose of LMWH 6 to 12 hours postpartum, with therapeutic dosing resumed on postpartum day 1. If prophylactic dosing is desired, unfractionated heparin or LMWH may be given subcutaneously starting at about 6 hours postpartum.
Warfarin in the puerperium
Women may subsequently be maintained on either LMWH or unfractionated heparin, or switched to an oral anticoagulant such as warfarin. Although warfarin may appear in minute amounts in breast milk, it has not been associated with adverse events in newborns and is considered compatible with breastfeeding.68 Heparin should be continued during the initial days of warfarin therapy, until the INR is at a therapeutic level for 24 hours. Some physicians prefer to delay warfarin for several days, giving LMWH alone in the immediate postpartum period, to allow wound-healing and to reduce bleeding risk.
Postpartum, anticoagulation should be continued for at least 6 to 12 weeks, at which point the physiologic changes in the coagulation system related to pregnancy will have returned to normal.
THROMBOPHILIA WITHOUT A PREVIOUS THROMBOEMBOLIC EVENT
Over the last 5 to 10 years, practitioners have been seeing many more young women with genetic or acquired thrombophilias who have never had a venous thromboembolic event. Physicians must advise these women about their risk of thromboembolic events during pregnancy and about the appropriateness of anticoagulant use.
Thrombophilias are often detected in women who develop venous thrombosis during pregnancy,69–71 but they are also very common in the general population (around 15%). While women with thrombophilia are at above-average risk of venous thromboembolism during pregnancy, the magnitude of risk in an individual patient is often difficult to estimate.
Data suggest that some types of thrombophilia confer greater thrombotic risk than others. McColl et al72 derived risk estimates for a primary event in women with several of the disorders: 0.23% in women heterozygous for the factor V Leiden mutation, 0.88% in women with protein C deficiency, and 2.4% to 35.7% in women with antithrombin deficiency. A case-control study70 found that all thrombophilic states were more common in women with pregnancy-associated venous thromboembolism than in healthy pregnant controls, except those with the MTHFR mutation and protein S deficiency. The estimated risk during pregnancy was 0.03% in women with no defect, 0.1% in women with protein C deficiency, 0.25% in women with the factor V Leiden mutation, 0.4% in those with antithrombin deficiency, 0.5% in those with the prothrombin gene mutation, and 4.6% in those with both factor V Leiden and prothrombin gene mutations.
Routine anticoagulation not advised in pregnant thrombophilic women
Because the risk of a primary venous thromboembolic event is less than 1% for most thrombophilic women, routine anticoagulant therapy does not seem prudent for this indication. Given the low absolute risk of venous thromboembolism, the cost and potential side effects of anticoagulant use are difficult to justify.
The women who seem at higher risk and in whom anticoagulation should be considered include those with antithrombin deficiency; those with high-titer anticardiolipin antibodies or a lupus anticoagulant antibody (treat with heparin and low-dose aspirin); those with combined thrombophilic defects or who are homozygotes for the factor V Leiden or prothrombin gene mutations; and those with multiple other current risk factors for venous thromboembolism (Table 1).
Since anticoagulants for primary prevention of adverse pregnancy outcomes in thrombophilic women have not yet been shown to have a definitive benefit, they are not recommended for this purpose.
ADVERSE PREGNANCY OUTCOMES IN WOMEN WITH THROMBOPHILIAS
Women with antiphospholipid antibodies and a previous poor obstetric outcome are clearly at increased risk of recurrent adverse pregnancy outcomes such as recurrent spontaneous abortion, unexplained fetal death, placental insufficiency, and early or severe preeclampsia. In such women who have both antiphospholipid antibodies and a history of venous thromboembolism or adverse pregnancy outcome, treatment during subsequent pregnancy with low-dose aspirin and prophylactic-dose LMWH or unfractionated heparin improves pregnancy outcomes.36–42 Women with antiphospholipid antibodies without previous thrombosis or pregnancy complications may also be at increased risk, but it is unclear whether thromboprophylaxis improves their outcomes.
Recent epidemiologic data reveal that women with other thrombophilic conditions also are at increased risk of early, severe preeclampsia73 as well as other pregnancy complications, including recurrent pregnancy loss, placental abruption, fetal growth restriction, and stillbirth.74 A recent meta-analysis75 looked at individual thrombophilias and found that factor V Leiden and prothrombin gene mutations were associated with recurrent fetal loss, stillbirth, and preeclampsia; that protein S deficiency was associated with recurrent fetal loss and stillbirth; that antiphospholipid antibodies were associated with recurrent pregnancy loss, preeclampsia, and intrauterine growth restriction; that the MTHFR mutation (homozygous) was associated with preeclampsia; and that protein C and antithrombin deficiencies were not significantly associated with adverse pregnancy outcomes. Data were scant for some of the rarer thrombophilias.75
Several recent small studies76–78 suggest that anticoagulants may improve pregnancy outcomes in women with genetic thrombophilias and recurrent pregnancy loss. These findings have not yet been confirmed in high-quality clinical trials, but such trials are under way. It is still unclear whether anticoagulants also reduce the risk of other adverse pregnancy outcomes associated with thrombophilias.
The current American College of Chest Physicians guidelines recommend testing of women with adverse pregnancy outcomes (recurrent pregnancy loss, prior severe or recurrent preeclampsia, abruptions, or otherwise unexplained intrauterine death) for congenital thrombophilias and antiphospholipid antibodies, and offering treatment to such women, if thrombophilic, with low-dose aspirin plus prophylactic heparin (unfractionated or LMWH).22 The authors of the guidelines admit that the evidence for this recommendation is weak, but they argue that the heparin will also serve as thromboprophylaxis in this high-risk group. Hopefully, the randomized clinical trials currently under way will provide clearer guidance regarding the most appropriate therapy in this difficult clinical situation.
MECHANICAL HEART VALVES
Internists may occasionally encounter a woman with a mechanical heart valve prosthesis who is either pregnant or is planning a pregnancy and therefore needs advice regarding optimal anticoagulant management. This should generally be undertaken in a multi-disciplinary fashion, with input from cardiology, hematology, and maternal-fetal medicine. The substantial maternal and fetal risks and the lack of definitive data on which to base treatment decisions make it a treacherous and stressful undertaking. Nonetheless, all internists should have a basic understanding of the complex issues regarding this management.
Outside of pregnancy, oral anticoagulants are the mainstay of therapy for patients with mechanical heart valves. Unfortunately, as discussed above, the use of these agents during pregnancy carries a risk of teratogenicity and toxic fetal effects and increases the risk of pregnancy loss and maternal hemorrhage. Heparins have been used in this setting for many years, but data on their efficacy and safety are very limited, and there are numerous reports of catastrophic maternal thrombotic complications.79,80
A systematic review of anticoagulation in pregnant women with prosthetic heart valves34 found very limited data on heparin use throughout pregnancy. Women maintained on warfarin vs heparin between pregnancy weeks 6 and 12 had higher rates of congenital anomalies (6.4% with warfarin vs 3.4% with heparin) and total fetal wastage (33.6% vs 26.5%). The warfarin group had fewer maternal thromboembolic complications (3.9% vs 9.2%), however, and a slightly lower rate of maternal death (1.8% vs 4.2%). Most of the women had higher-risk older-generation valves in the mitral position.
Recent data on LMWH consist mainly of case reports and case series,81 with a likely bias to publication of worse outcomes. Controlled trials in this area will be difficult to conduct. Still, aggressive anticoagulation with LMWH or unfractionated heparin, with close monitoring of the intensity of anticoagulation, may be safe and effective for pregnant women with newer-generation mechanical heart valves.82 A recent consensus statement22 suggested several regimens for pregnant women with mechanical heart valves:
- Twice-daily LMWH throughout pregnancy, with the dose adjusted either by weight, or to keep the 4-hour postinjection anti-factor-Xa activity level around 1.0 to 1.2 U/mL
- Aggressive adjusted-dose unfractionated heparin throughout pregnancy, given subcutaneously every 12 hours and adjusted to keep the mid-interval aPTT at least twice the control value or to attain a mid-interval anti-factor-Xa activity level of 0.35 to 0.70 U/mL
- Unfractionated heparin or LMWH (as above) until gestation week 13, then warfarin until the middle of the third trimester, and then heparin again.22
The authors also recommended adding low-dose aspirin (75–162 mg/day) in high-risk women.22
These options all seem reasonable, given our current knowledge, though warfarin use during pregnancy should be restricted to very-high-risk situations, such as women with older-generation mitral prostheses. LM-WHs may become the preferred therapy for this indication once further controlled data regarding their efficacy and safety become available.
- Chang J, Elam-Evans LD, Berg CJ, et al. Pregnancy-related mortality surveillance-United States, 1991–1999. MMWR Surveill Summ 2003; 52:1–8.
- Lewis G, Drife JO, Clutton-Brock T, et al. Why Mothers Die, 2000–2002. The Sixth Report of the Confidential Enquiries into Maternal Deaths in the United Kingdom. London: RCOG Press, 2004.
- Health Canada. Special Report on Maternal Mortality and Severe Morbidity in Canada—Enhanced Surveillance: The Path to Prevention. Ottawa: Minister of Public Works and Government Services Canada, 2004. www.phac-aspc.gc.ca/rhs-ssg/srmm-rsmm/page1-eng.php. Accessed 11/26/2008.
- Stein PD, Hull RD, Kayali F, et al. Venous thromboembolism in pregnancy: 21-year trends. Am J Med 2004; 117:121–125.
- Greer IA. Thrombosis in pregnancy: maternal and fetal issues. Lancet 1999; 353:1258–1265.
- Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet 1999; 353:1167–1173.
- Gherman RB, Goodwin TM, Leung B, et al. Incidence, clinical characteristics, and timing of objectively diagnosed venous thromboembolism during pregnancy. Obstet Gynecol 1999; 94:730–734.
- Weitz JI. Low-molecular-weight heparins. N Engl J Med 1997; 337:688–698.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Simonneau G, Sors H, Charbonnier B, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for acute pulmonary embolism. The THESEE Study Group. Tinzaparine ou Heparine Standard: Evaluations dans l’Embolie Pulmonaire. N Engl J Med 1997; 337:663–669.
- Hull RD, Raskob GE, Brant RF, et al. Low-molecular-weight heparin vs heparin in the treatment of patients with pulmonary embolism. American-Canadian Thrombosis Study Group. Arch Intern Med 2000; 160:229–236.
- Sanson BJ, Lensing AW, Prins MH, et al. Safety of low-molecular-weight heparin in pregnancy: a systematic review. Thromb Haemost 1999; 81:668–672.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005; 106:401–407.
- Melissari E, Parker CJ, Wilson NV, et al. Use of low molecular weight heparin in pregnancy. Thromb Haemost 1992; 68:652–656.
- Forestier F, Daffos F, Capella-Pavlovsky M. Low molecular weight heparin (PK 10169) does not cross the placenta during the second trimester of pregnancy study by direct fetal blood sampling under ultrasound. Thromb Res 1984; 34:557–560.
- Forestier F, Daffos F, Rainaut M, Toulemonde F. Low molecular weight heparin (CY 216) does not cross the placenta during the third trimester of pregnancy. Thromb Haemost 1987; 57:234.
- Barbour LA, Oja JL, Schultz LK. A prospective trial that demonstrates that dalteparin requirements increase in pregnancy to maintain therapeutic levels of anticoagulation. Am J Obstet Gynecol 2004; 191:1024–1029.
- Smith MP, Norris LA, Steer PJ, Savidge GF, Bonnar J. Tinzaparin sodium for thrombosis treatment and prevention during pregnancy. Am J Obstet Gynecol 2004; 190:495–501.
- Ellison J, Walker ID, Greer IA. Antenatal use of enoxaparin for prevention and treatment of thromboembolism in pregnancy. BJOG 2000; 107:1116–1121.
- Sarig G, Brenner B. Monitoring of low molecular weight heparin (LMWH) in pregnancy. Thromb Res 2005; 115 suppl 1:84–86.
- Bates SM, Greer IA, Hirsh J, Ginsberg JS. Use of antithrombotic agents during pregnancy: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:627S–644S.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995; 332:1330–1335.
- Hassell K. The management of patients with heparin-induced thrombocytopenia who require anticoagulant therapy. Chest 2005; 127 suppl 2:1S–8S.
- Barbour LA, Kick SD, Steiner JF, et al. A prospective study of heparin-induced osteoporosis in pregnancy using bone densitometry. Am J Obst Gynecol 1994; 170:862–869.
- Douketis JD, Ginsberg JS, Burrows RF, Duku EK, Webber CE, Brill-Edwards P. The effects of long-term heparin therapy during pregnancy on bone density. A prospective matched cohort study. Thromb Haemost 1996; 75:254–257.
- Pettila V, Leinonen P, Markkola A, Hiilesmaa V, Kaaja R. Postpartum bone mineral density in women treated for thromboprophylaxis with unfractionated heparin or LMW heparin. Thromb Haemost 2002; 87:182–186.
- Carlin AJ, Farquharson RG, Quenby SM, Topping J, Fraser WD. Prospective observational study of bone mineral density during pregnancy: low molecular weight heparin versus control. Hum Reprod 2004; 19:1211–1214.
- Casele HL, Laifer SA. Prospective evaluation of bone density in pregnant women receiving the low molecular weight heparin enoxaparin sodium. J Matern Fetal Med 2000; 9:122–125.
- Casele H, Haney EI, James A, Rosene-Montella K, Carson M. Bone density changes in women who receive thromboprophylaxis in pregnancy. Am J Obstet Gynecol 2006; 195:1109–1113.
- Horlocker TT, Wedel DJ, Benzon H, et al. Regional anesthesia in the anticoagulated patient: defining the risks (the second ASRA Consensus Conference on Neuraxial Anesthesia and Anticoagulation). Reg Anesth Pain Med 2003; 28:172–197.
- Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 2001; 119 suppl 1:8S–21S.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:287S–310S.
- Holmes LB. Teratogen-induced limb defects. Am J Med Genet 2002; 112:297–303.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Pauzner R, Dulitzki M, Langevitz P, Livneh A, Kenett R, Many A. Low molecular weight heparin and warfarin in the treatment of patients with antiphospholipid syndrome during pregnancy. Thromb Haemost 2001; 86:1379–1384.
- Pulmonary Embolism Prevention (PEP) Trial Collaborative Group. Prevention of pulmonary embolism and deep vein thrombosis with low dose aspirin: Pulmonary Embolism Prevention (PEP) trial. Lancet 2000; 355:1295–1302.
- Patrono C, Coller B, FitzGerald GA, Hirsh J, Roth G. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:234S–264S.
- Duley L, Henderson-Smart DJ, Knight M, King JF. Antiplatelet agents for preventing preeclampsia and its complications. Cochrane Database Syst Rev. 2004; ( 1):CD004659.
- Coomarasamy A, Honest H, Papaioannou S, Gee H, Khan KS. Aspirin for prevention of preeclampsia in women with historical risk factors: a systematic review. Obstet Gynecol 2003; 101:1319–1332.
- Caritis SN, Sibai BM, Hauth J, et al, and the National Institute of Child Health and Human Development Network of Maternal Fetal Medicine Units. Low-dose aspirin to prevent preeclampsia in women at high risk. N Engl J Med 1998; 338:701–705.
- Rai R, Cohen H, Dave M, Regan L. Randomised controlled trial of aspirin and aspirin plus heparin in pregnant women with recurrent miscarriage associated with phospholipid antibodies (or antiphospholipid antibodies). BMJ 1997; 314:253–257.
- Kozer E, Nikfar S, Costei A, Boskovic R, Nulman I, Koren G. Aspirin consumption during the first trimester of pregnancy and congenital anomalies: a meta-analysis. Am J Obstet Gynecol 2002; 187:1623–1630.
- Sebastian C, Scherlag M, Kugelmass A, Schechter E. Primary stent implantation for acute myocardial infarction during pregnancy: use of abciximab, ticlopidine, and aspirin. Cathet Cardiovasc Diagn 1998; 45:275–249.
- Wilson AM, Boyle AJ, Fox P. Management of ischaemic heart disease in women of child-bearing age. Intern Med J 2004; 34:694–697.
- Klinzing P, Markert UR, Liesaus K, Peiker G. Case report: successful pregnancy and delivery after myocardial infarction and essential thrombocythemia treated with clopidogrel. Clin Exp Obstet Gynecol 2001; 28:215–216.
- Danhof M, de Boer A, Magnani HN, Stiekema JC. Pharmacokinetic considerations on Orgaran (Org 10172) therapy. Haemostasis 1992; 22:73–84.
- Tardy-Poncet B, Tardy B, Reynaud J, et al. Efficacy and safety of danaparoid sodium (ORG 10172) in critically ill patients with heparin-associated thrombocytopenia. Chest 1999; 115:1616–1620.
- Lagrange F, Vergnes C, Brun JL, et al. Absence of placental transfer of pentasaccharide (fondaparinux, Arixtra) in the dually perfused human cotyledon in vitro. Thromb Haemost 2002; 87:831–835.
- Dempfle CE. Minor transplacental passge of fondapinux in vivo. N Engl J Med 2004; 350:1914.
- Magnani HN. Heparin-induced thrombocytopenia (HIT): an overview of 230 patients treated with orgaran (Org 10172). Thromb Haemost 1993; 70:554–561.
- Lindhoff-Last E, Kreutzenbeck HJ, Magnani HN. Treatment of 51 pregnancies with danaparoid because of heparin intolerance. Thromb Haemost 2005; 93:63–69.
- Greinacher A, Eckhardt T, Mussmann J, Mueller-Eckhardt C. Pregnancy complicated by heparin associated thrombocytopenia: management by a prospectively in vitro selected heparinoid (Org 10172). Thromb Res 1993; 71:123–126.
- Schindewolf M, Mosch G, Bauersachs RM, Lindhoff-Last E. Safe anticoagulation with danaparoid in pregnancy and lactation. Thromb Haemost 2004; 92:211.
- Harenberg J. Treatment of a woman with lupus and thromboembolism and cutaneous intolerance to heparins using fondaparinux during pregnancy. Thromb Res 2007; 119:385–388.
- Wijesiriwardana A, Lees DA, Lush C. Fondaparinux as anticoagulant in a pregnant woman with heparin allergy. Blood Coagul Fibrinolysis 2006; 17:147–149.
- Mazzolai L, Hohlfeld P, Spertini F, Hayoz D, Schapira M, Duchosal MA. Fondaparinux is a safe alternative in case of heparin intolerance during pregnancy. Blood 2006; 108:1569–1570.
- Hawkins D, Evans J. Minimizing the risk of heparin-induced osteoporosis during pregnancy. Expert Opin Drug Saf 2005; 4:583–590.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of clot in this pregnancy study group. N Engl J Med 2000; 343:1439–1444.
- Martinelli I, Legnani C, Bucciarelli P, Grandone E, De Stefano V, Mannucci PM. Risk of pregnancy-related venous thrombosis in carriers of severe inherited thrombophilia. Thromb Haemost 2001; 86:800–803.
- De Stefano V, Martinelli I, Rossi E, Battaglioli T, Za T, Mannucci PM, Leone G. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006; 135:386–391.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995; 173:1869–1873.
- Ensom MH, Stephenson MD. Pharmacokinetics of low molecular weight heparin and unfractionated heparin in pregnancy. J Soc Gynecol Investig 2004; 11:377–383.
- Crowther MA, Berry LR, Monagle PT, Chan AK. Mechanisms responsible for the failure of protamine to inactivate low-molecular-weight heparin. Br J Haematol 2002; 116:178–186.
- Kearon C, Hirsh J. Management of anticoagulation before and after elective surgery. N Engl J Med 1997; 336:1506–1511.
- Thomas LA, Summers RR, Cardwell MS. Use of Greenfield filters in pregnant women at risk for pulmonary embolism. South Med J 1997; 90:215–217.
- Maclean PS, Tait RC. Hereditary and acquired antithrombin deficiency: epidemiology, pathogenesis and treatment options. Drugs 2007; 67:1429–1440.
- Information from LactMed: http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?LACT, LactMed Record Number: 279. Accessed 11/26/2008.
- Gerhardt A, Scharf RE, Beckmann MW, et al. Prothrombin and factor V mutations in women with a history of thrombosis during pregnancy and the puerperium. N Engl J Med 2000; 342:374–380.
- Hirsch DR, Mikkola KM, Marks PW, et al. Pulmonary embolism and deep venous thrombosis during pregnancy or oral contraceptive use: prevalence of factor V Leiden. Am Heart J 1996; 131:1145–1148.
- Dizon-Townson DS, Nelson LM, Jang H, Varner MW, Ward K. The incidence of the factor V Leiden mutation in an obstetric population and its relationship to deep vein thrombosis. Am J Obstet Gynecol 1997; 176:883–886.
- McColl MD, Ramsay JE, Tait RC, et al. Risk factors for pregnancy associated venous thromboembolism. Thromb Haemost 1997; 78:1183–1188.
- Kupferminc MJ, Fait G, Many A, Gordon D, Eldor A, Lessing JB. Severe preeclampsia and high frequency of genetic thrombophilic mutations. Obstet Gynecol 2000; 96:45–49.
- Kupferminc MJ, Eldor A, Steinman N, et al. Increased frequency of genetic thrombophilia in women with complications of pregnancy. N Engl J Med 1999; 340:9–13.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006; 132:171–196.
- Brenner B, Hoffman R, Blumenfeld Z, Weiner Z, Younis JS. Gestational outcome in thrombophilic women with recurrent pregnancy loss treated by enoxaparin. Thromb Haemost 2000; 83:693–697.
- Carp H, Dolitzky M, Inbal A. Thromboprophylaxis improves the live birth rate in women with consecutive recurrent miscarriages and hereditary thrombophilia. J Thromb Haemost 2003; 1:433–438.
- Gris JC, Mercier E, Quere I, et al. Low-molecular-weight heparin versus low-dose aspirin in women with one fetal loss and a constitutional thrombophilic disorder. Blood 2004; 103:3695–3699.
- Salazar E, Izaguirre R, Verdejo J, Mutchinick O. Failure of adjusted doses of subcutaneous heparin to prevent thromboembolic phenomena in pregnant patients with mechanical cardiac valve prostheses. J Am Coll Cardiol 1996; 27:1698–1703.
- Iturbe-Alessio I, Fonseca MC, Mutchinik O, Santos MA, Zajarias A, Salazar E. Risks of anticoagulant therapy in pregnant women with artificial heart valves. N Engl J Med 1986; 315:1390–1393.
- Rowan JA, McCowan LM, Raudkivi PJ, North RA. Enoxaparin treatment in women with mechanical heart valves during pregnancy. Am J Obstet Gynecol 2001; 185:633–637.
- Oran B, Lee-Parritz A, Ansell J. Low molecular weight heparin for the prophylaxis of thromboembolism in women with prosthetic mechanical heart valves during pregnancy. Thromb Haemost 2004; 92:747–751.
- Chang J, Elam-Evans LD, Berg CJ, et al. Pregnancy-related mortality surveillance-United States, 1991–1999. MMWR Surveill Summ 2003; 52:1–8.
- Lewis G, Drife JO, Clutton-Brock T, et al. Why Mothers Die, 2000–2002. The Sixth Report of the Confidential Enquiries into Maternal Deaths in the United Kingdom. London: RCOG Press, 2004.
- Health Canada. Special Report on Maternal Mortality and Severe Morbidity in Canada—Enhanced Surveillance: The Path to Prevention. Ottawa: Minister of Public Works and Government Services Canada, 2004. www.phac-aspc.gc.ca/rhs-ssg/srmm-rsmm/page1-eng.php. Accessed 11/26/2008.
- Stein PD, Hull RD, Kayali F, et al. Venous thromboembolism in pregnancy: 21-year trends. Am J Med 2004; 117:121–125.
- Greer IA. Thrombosis in pregnancy: maternal and fetal issues. Lancet 1999; 353:1258–1265.
- Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet 1999; 353:1167–1173.
- Gherman RB, Goodwin TM, Leung B, et al. Incidence, clinical characteristics, and timing of objectively diagnosed venous thromboembolism during pregnancy. Obstet Gynecol 1999; 94:730–734.
- Weitz JI. Low-molecular-weight heparins. N Engl J Med 1997; 337:688–698.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Simonneau G, Sors H, Charbonnier B, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for acute pulmonary embolism. The THESEE Study Group. Tinzaparine ou Heparine Standard: Evaluations dans l’Embolie Pulmonaire. N Engl J Med 1997; 337:663–669.
- Hull RD, Raskob GE, Brant RF, et al. Low-molecular-weight heparin vs heparin in the treatment of patients with pulmonary embolism. American-Canadian Thrombosis Study Group. Arch Intern Med 2000; 160:229–236.
- Sanson BJ, Lensing AW, Prins MH, et al. Safety of low-molecular-weight heparin in pregnancy: a systematic review. Thromb Haemost 1999; 81:668–672.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005; 106:401–407.
- Melissari E, Parker CJ, Wilson NV, et al. Use of low molecular weight heparin in pregnancy. Thromb Haemost 1992; 68:652–656.
- Forestier F, Daffos F, Capella-Pavlovsky M. Low molecular weight heparin (PK 10169) does not cross the placenta during the second trimester of pregnancy study by direct fetal blood sampling under ultrasound. Thromb Res 1984; 34:557–560.
- Forestier F, Daffos F, Rainaut M, Toulemonde F. Low molecular weight heparin (CY 216) does not cross the placenta during the third trimester of pregnancy. Thromb Haemost 1987; 57:234.
- Barbour LA, Oja JL, Schultz LK. A prospective trial that demonstrates that dalteparin requirements increase in pregnancy to maintain therapeutic levels of anticoagulation. Am J Obstet Gynecol 2004; 191:1024–1029.
- Smith MP, Norris LA, Steer PJ, Savidge GF, Bonnar J. Tinzaparin sodium for thrombosis treatment and prevention during pregnancy. Am J Obstet Gynecol 2004; 190:495–501.
- Ellison J, Walker ID, Greer IA. Antenatal use of enoxaparin for prevention and treatment of thromboembolism in pregnancy. BJOG 2000; 107:1116–1121.
- Sarig G, Brenner B. Monitoring of low molecular weight heparin (LMWH) in pregnancy. Thromb Res 2005; 115 suppl 1:84–86.
- Bates SM, Greer IA, Hirsh J, Ginsberg JS. Use of antithrombotic agents during pregnancy: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:627S–644S.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995; 332:1330–1335.
- Hassell K. The management of patients with heparin-induced thrombocytopenia who require anticoagulant therapy. Chest 2005; 127 suppl 2:1S–8S.
- Barbour LA, Kick SD, Steiner JF, et al. A prospective study of heparin-induced osteoporosis in pregnancy using bone densitometry. Am J Obst Gynecol 1994; 170:862–869.
- Douketis JD, Ginsberg JS, Burrows RF, Duku EK, Webber CE, Brill-Edwards P. The effects of long-term heparin therapy during pregnancy on bone density. A prospective matched cohort study. Thromb Haemost 1996; 75:254–257.
- Pettila V, Leinonen P, Markkola A, Hiilesmaa V, Kaaja R. Postpartum bone mineral density in women treated for thromboprophylaxis with unfractionated heparin or LMW heparin. Thromb Haemost 2002; 87:182–186.
- Carlin AJ, Farquharson RG, Quenby SM, Topping J, Fraser WD. Prospective observational study of bone mineral density during pregnancy: low molecular weight heparin versus control. Hum Reprod 2004; 19:1211–1214.
- Casele HL, Laifer SA. Prospective evaluation of bone density in pregnant women receiving the low molecular weight heparin enoxaparin sodium. J Matern Fetal Med 2000; 9:122–125.
- Casele H, Haney EI, James A, Rosene-Montella K, Carson M. Bone density changes in women who receive thromboprophylaxis in pregnancy. Am J Obstet Gynecol 2006; 195:1109–1113.
- Horlocker TT, Wedel DJ, Benzon H, et al. Regional anesthesia in the anticoagulated patient: defining the risks (the second ASRA Consensus Conference on Neuraxial Anesthesia and Anticoagulation). Reg Anesth Pain Med 2003; 28:172–197.
- Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 2001; 119 suppl 1:8S–21S.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:287S–310S.
- Holmes LB. Teratogen-induced limb defects. Am J Med Genet 2002; 112:297–303.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Pauzner R, Dulitzki M, Langevitz P, Livneh A, Kenett R, Many A. Low molecular weight heparin and warfarin in the treatment of patients with antiphospholipid syndrome during pregnancy. Thromb Haemost 2001; 86:1379–1384.
- Pulmonary Embolism Prevention (PEP) Trial Collaborative Group. Prevention of pulmonary embolism and deep vein thrombosis with low dose aspirin: Pulmonary Embolism Prevention (PEP) trial. Lancet 2000; 355:1295–1302.
- Patrono C, Coller B, FitzGerald GA, Hirsh J, Roth G. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:234S–264S.
- Duley L, Henderson-Smart DJ, Knight M, King JF. Antiplatelet agents for preventing preeclampsia and its complications. Cochrane Database Syst Rev. 2004; ( 1):CD004659.
- Coomarasamy A, Honest H, Papaioannou S, Gee H, Khan KS. Aspirin for prevention of preeclampsia in women with historical risk factors: a systematic review. Obstet Gynecol 2003; 101:1319–1332.
- Caritis SN, Sibai BM, Hauth J, et al, and the National Institute of Child Health and Human Development Network of Maternal Fetal Medicine Units. Low-dose aspirin to prevent preeclampsia in women at high risk. N Engl J Med 1998; 338:701–705.
- Rai R, Cohen H, Dave M, Regan L. Randomised controlled trial of aspirin and aspirin plus heparin in pregnant women with recurrent miscarriage associated with phospholipid antibodies (or antiphospholipid antibodies). BMJ 1997; 314:253–257.
- Kozer E, Nikfar S, Costei A, Boskovic R, Nulman I, Koren G. Aspirin consumption during the first trimester of pregnancy and congenital anomalies: a meta-analysis. Am J Obstet Gynecol 2002; 187:1623–1630.
- Sebastian C, Scherlag M, Kugelmass A, Schechter E. Primary stent implantation for acute myocardial infarction during pregnancy: use of abciximab, ticlopidine, and aspirin. Cathet Cardiovasc Diagn 1998; 45:275–249.
- Wilson AM, Boyle AJ, Fox P. Management of ischaemic heart disease in women of child-bearing age. Intern Med J 2004; 34:694–697.
- Klinzing P, Markert UR, Liesaus K, Peiker G. Case report: successful pregnancy and delivery after myocardial infarction and essential thrombocythemia treated with clopidogrel. Clin Exp Obstet Gynecol 2001; 28:215–216.
- Danhof M, de Boer A, Magnani HN, Stiekema JC. Pharmacokinetic considerations on Orgaran (Org 10172) therapy. Haemostasis 1992; 22:73–84.
- Tardy-Poncet B, Tardy B, Reynaud J, et al. Efficacy and safety of danaparoid sodium (ORG 10172) in critically ill patients with heparin-associated thrombocytopenia. Chest 1999; 115:1616–1620.
- Lagrange F, Vergnes C, Brun JL, et al. Absence of placental transfer of pentasaccharide (fondaparinux, Arixtra) in the dually perfused human cotyledon in vitro. Thromb Haemost 2002; 87:831–835.
- Dempfle CE. Minor transplacental passge of fondapinux in vivo. N Engl J Med 2004; 350:1914.
- Magnani HN. Heparin-induced thrombocytopenia (HIT): an overview of 230 patients treated with orgaran (Org 10172). Thromb Haemost 1993; 70:554–561.
- Lindhoff-Last E, Kreutzenbeck HJ, Magnani HN. Treatment of 51 pregnancies with danaparoid because of heparin intolerance. Thromb Haemost 2005; 93:63–69.
- Greinacher A, Eckhardt T, Mussmann J, Mueller-Eckhardt C. Pregnancy complicated by heparin associated thrombocytopenia: management by a prospectively in vitro selected heparinoid (Org 10172). Thromb Res 1993; 71:123–126.
- Schindewolf M, Mosch G, Bauersachs RM, Lindhoff-Last E. Safe anticoagulation with danaparoid in pregnancy and lactation. Thromb Haemost 2004; 92:211.
- Harenberg J. Treatment of a woman with lupus and thromboembolism and cutaneous intolerance to heparins using fondaparinux during pregnancy. Thromb Res 2007; 119:385–388.
- Wijesiriwardana A, Lees DA, Lush C. Fondaparinux as anticoagulant in a pregnant woman with heparin allergy. Blood Coagul Fibrinolysis 2006; 17:147–149.
- Mazzolai L, Hohlfeld P, Spertini F, Hayoz D, Schapira M, Duchosal MA. Fondaparinux is a safe alternative in case of heparin intolerance during pregnancy. Blood 2006; 108:1569–1570.
- Hawkins D, Evans J. Minimizing the risk of heparin-induced osteoporosis during pregnancy. Expert Opin Drug Saf 2005; 4:583–590.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of clot in this pregnancy study group. N Engl J Med 2000; 343:1439–1444.
- Martinelli I, Legnani C, Bucciarelli P, Grandone E, De Stefano V, Mannucci PM. Risk of pregnancy-related venous thrombosis in carriers of severe inherited thrombophilia. Thromb Haemost 2001; 86:800–803.
- De Stefano V, Martinelli I, Rossi E, Battaglioli T, Za T, Mannucci PM, Leone G. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006; 135:386–391.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995; 173:1869–1873.
- Ensom MH, Stephenson MD. Pharmacokinetics of low molecular weight heparin and unfractionated heparin in pregnancy. J Soc Gynecol Investig 2004; 11:377–383.
- Crowther MA, Berry LR, Monagle PT, Chan AK. Mechanisms responsible for the failure of protamine to inactivate low-molecular-weight heparin. Br J Haematol 2002; 116:178–186.
- Kearon C, Hirsh J. Management of anticoagulation before and after elective surgery. N Engl J Med 1997; 336:1506–1511.
- Thomas LA, Summers RR, Cardwell MS. Use of Greenfield filters in pregnant women at risk for pulmonary embolism. South Med J 1997; 90:215–217.
- Maclean PS, Tait RC. Hereditary and acquired antithrombin deficiency: epidemiology, pathogenesis and treatment options. Drugs 2007; 67:1429–1440.
- Information from LactMed: http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?LACT, LactMed Record Number: 279. Accessed 11/26/2008.
- Gerhardt A, Scharf RE, Beckmann MW, et al. Prothrombin and factor V mutations in women with a history of thrombosis during pregnancy and the puerperium. N Engl J Med 2000; 342:374–380.
- Hirsch DR, Mikkola KM, Marks PW, et al. Pulmonary embolism and deep venous thrombosis during pregnancy or oral contraceptive use: prevalence of factor V Leiden. Am Heart J 1996; 131:1145–1148.
- Dizon-Townson DS, Nelson LM, Jang H, Varner MW, Ward K. The incidence of the factor V Leiden mutation in an obstetric population and its relationship to deep vein thrombosis. Am J Obstet Gynecol 1997; 176:883–886.
- McColl MD, Ramsay JE, Tait RC, et al. Risk factors for pregnancy associated venous thromboembolism. Thromb Haemost 1997; 78:1183–1188.
- Kupferminc MJ, Fait G, Many A, Gordon D, Eldor A, Lessing JB. Severe preeclampsia and high frequency of genetic thrombophilic mutations. Obstet Gynecol 2000; 96:45–49.
- Kupferminc MJ, Eldor A, Steinman N, et al. Increased frequency of genetic thrombophilia in women with complications of pregnancy. N Engl J Med 1999; 340:9–13.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006; 132:171–196.
- Brenner B, Hoffman R, Blumenfeld Z, Weiner Z, Younis JS. Gestational outcome in thrombophilic women with recurrent pregnancy loss treated by enoxaparin. Thromb Haemost 2000; 83:693–697.
- Carp H, Dolitzky M, Inbal A. Thromboprophylaxis improves the live birth rate in women with consecutive recurrent miscarriages and hereditary thrombophilia. J Thromb Haemost 2003; 1:433–438.
- Gris JC, Mercier E, Quere I, et al. Low-molecular-weight heparin versus low-dose aspirin in women with one fetal loss and a constitutional thrombophilic disorder. Blood 2004; 103:3695–3699.
- Salazar E, Izaguirre R, Verdejo J, Mutchinick O. Failure of adjusted doses of subcutaneous heparin to prevent thromboembolic phenomena in pregnant patients with mechanical cardiac valve prostheses. J Am Coll Cardiol 1996; 27:1698–1703.
- Iturbe-Alessio I, Fonseca MC, Mutchinik O, Santos MA, Zajarias A, Salazar E. Risks of anticoagulant therapy in pregnant women with artificial heart valves. N Engl J Med 1986; 315:1390–1393.
- Rowan JA, McCowan LM, Raudkivi PJ, North RA. Enoxaparin treatment in women with mechanical heart valves during pregnancy. Am J Obstet Gynecol 2001; 185:633–637.
- Oran B, Lee-Parritz A, Ansell J. Low molecular weight heparin for the prophylaxis of thromboembolism in women with prosthetic mechanical heart valves during pregnancy. Thromb Haemost 2004; 92:747–751.
KEY POINTS
- Pregnancy is a hypercoagulable state. Thrombotic risk in an individual pregnancy depends on many maternal and situational factors.
- When indicated, careful anticoagulation can proceed with minimal risk to the mother and fetus.
- Heparins, especially LMWHs, are the main anticoagulants used in pregnancy. Dosing depends on the clinical indications and on the agent selected.
- If anticoagulation is absolutely necessary and LMWH is contraindicated, a newer, alternative anticoagulant should be considered.
- Warfarin should not be used in pregnancy in any but the highest-risk situations.