User login
Complete Heart Block in a Patient With Metastatic Papillary Thyroid Carcinoma
A 74-year-old woman presented with a 2-day history of exertional dyspnea and palpitations. Her past medical history was significant for metastatic papillary thyroid carcinoma treated with total thyroidectomy and radioactive iodine ablation with levothyroxine for chronic suppressive therapy.
On examination, the patient was afebrile with an oxygen saturation of 98% on room air, heart rate of 92 beats/min, and blood pressure of 100/54 mm Hg. There was trace bilateral lower extremity edema, and her cardiopulmonary examination was unremarkable. The laboratory studies showed a white blood cell count of 24,300/µL (3,400-9,800); platelets 86,000/µL (142,000-362,000); thyroid stimulating hormone 0.009 mlU/L (0.4-4.1); free T4 2.07 ng/dL (0.8-2.0); thyroglobulin antibody titer < 1:10 (< 1:160); thyroid microsomal antibody titer < 1:100 (< 1:1600); and thyroglobulin 17.9 ng/mL (2.0-35.0). Her initial troponin T was undetectable.
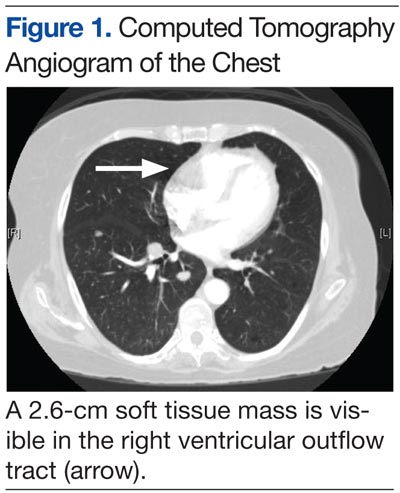
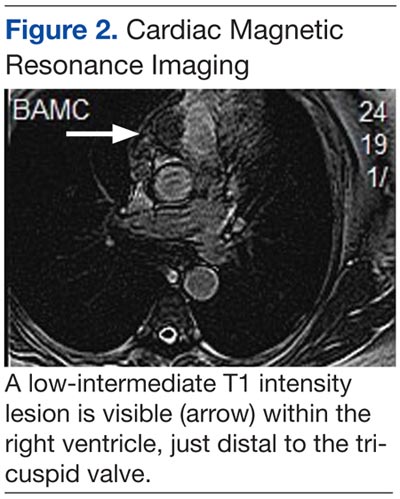
An electrocardiogram showed a first-degree atrioventricular block and subsequently a new intermittent third-degree atrioventricular block. A computed tomography angiogram (Figure 1) and cardiac magnetic resonance imaging (Figure 2) revealed a 2.6-cm soft tissue mass in the right ventricular outflow tract along with multiple pulmonary emboli and previously diagnosed pulmonary metastases. A positron emission tomography (PET) scan (not shown) revealed a 3.5-cm PET-avid lesion within the right ventricular outflow tract.
- What is your diagnosis?
- How would you treat this patient?
[Click through to the next page to see the answer.]
Our Treatment
Diagnosis and Discussion
This patient experienced complete heart block due to a cardiac tumor from papillary thyroid carcinoma metastasis. Complete heart block is not an unprecedented symptom of metastatic disease, but to our knowledge this is the first reported case of heart block secondary to metastatic papillary thyroid cancer.1 In general, metastatic cardiac tumors, usually associated with cancers of the breast and lung, melanoma, and lymphoma, are more common than are primary cardiac tumors and are often asymptomatic and discovered mostly postmortem.2,3 The frequency of thyroid metastasis to the heart has been reported to be as low as 0% to 2%, and a review of the literature demonstrated only 13 total cases in the past 30 years.
Theoretical mechanisms for invasion into the heart include lymphatic spread, hematogenous dissemination, or direct right ventricular invasion from the thoracic duct. It has been suggested that the lower blood flow to the myocardium (240 mL/min) relative to bone (600 mL/min) or the brain (750 mL/min) is the reason for a lower likelihood of cardiac involvement in metastatic disease.3 Given the findings in this case, evidence of cardiac conduction abnormalities in the setting of papillary thyroid cancer should raise suspicion for cardiac metastatic disease.
Case Outcome
In this patient, a permanent pacemaker was implanted for high-grade atrioventricular block, with resolution of the palpitations. The pulmonary emboli were concomitantly treated with enoxaparin, and the patient was discharged to a rehabilitation facility. Her prognosis was extremely poor given that survival with cardiac metastasis from any type of cancer is limited to a few weeks to months.3 She was to be reevaluated for experimental chemotherapy after reconditioning. However, not long after discharge she was readmitted in respiratory failure and died.
Acknowledgments
We would like to thank Dr. Kevin Steel, Lt Col, USAF, MC, imaging cardiologist at the Brooke Army Medical Center for his time and effort in accessing and preparing the CT and MRI images for this article.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect the official policy or position of Federal Practitioner, Frontline Medical Communications Inc., Brooke Army Medical Center, the U.S. Army Medical Department, the U.S. Army Office of the Surgeon General, the Department of the Army, Department of Defense, the U.S. Government, or any other of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Conley M, Hawkins K, Ririe D. Complete heart block and cardiac tamponade secondary to Merkel cell carcinoma cardiac metastases. South Med J. 2006;99(1):74-78.
2. Pascale P, Prior JO, Carron PN, Pruvot E, Muller O. Haemoptysis and complete atrioventricular block. Eur Heart J. 2008;29(11):1396.
3. Giuffrida D, Gharib H. Cardiac metastasis from primary anaplastic thyroid carcinoma: Report of three cases and a review of the literature. Endocr Relat Cancer. 2001;8(1):71-73.
A 74-year-old woman presented with a 2-day history of exertional dyspnea and palpitations. Her past medical history was significant for metastatic papillary thyroid carcinoma treated with total thyroidectomy and radioactive iodine ablation with levothyroxine for chronic suppressive therapy.
On examination, the patient was afebrile with an oxygen saturation of 98% on room air, heart rate of 92 beats/min, and blood pressure of 100/54 mm Hg. There was trace bilateral lower extremity edema, and her cardiopulmonary examination was unremarkable. The laboratory studies showed a white blood cell count of 24,300/µL (3,400-9,800); platelets 86,000/µL (142,000-362,000); thyroid stimulating hormone 0.009 mlU/L (0.4-4.1); free T4 2.07 ng/dL (0.8-2.0); thyroglobulin antibody titer < 1:10 (< 1:160); thyroid microsomal antibody titer < 1:100 (< 1:1600); and thyroglobulin 17.9 ng/mL (2.0-35.0). Her initial troponin T was undetectable.
An electrocardiogram showed a first-degree atrioventricular block and subsequently a new intermittent third-degree atrioventricular block. A computed tomography angiogram (Figure 1) and cardiac magnetic resonance imaging (Figure 2) revealed a 2.6-cm soft tissue mass in the right ventricular outflow tract along with multiple pulmonary emboli and previously diagnosed pulmonary metastases. A positron emission tomography (PET) scan (not shown) revealed a 3.5-cm PET-avid lesion within the right ventricular outflow tract.
- What is your diagnosis?
- How would you treat this patient?
[Click through to the next page to see the answer.]
Our Treatment
Diagnosis and Discussion
This patient experienced complete heart block due to a cardiac tumor from papillary thyroid carcinoma metastasis. Complete heart block is not an unprecedented symptom of metastatic disease, but to our knowledge this is the first reported case of heart block secondary to metastatic papillary thyroid cancer.1 In general, metastatic cardiac tumors, usually associated with cancers of the breast and lung, melanoma, and lymphoma, are more common than are primary cardiac tumors and are often asymptomatic and discovered mostly postmortem.2,3 The frequency of thyroid metastasis to the heart has been reported to be as low as 0% to 2%, and a review of the literature demonstrated only 13 total cases in the past 30 years.
Theoretical mechanisms for invasion into the heart include lymphatic spread, hematogenous dissemination, or direct right ventricular invasion from the thoracic duct. It has been suggested that the lower blood flow to the myocardium (240 mL/min) relative to bone (600 mL/min) or the brain (750 mL/min) is the reason for a lower likelihood of cardiac involvement in metastatic disease.3 Given the findings in this case, evidence of cardiac conduction abnormalities in the setting of papillary thyroid cancer should raise suspicion for cardiac metastatic disease.
Case Outcome
In this patient, a permanent pacemaker was implanted for high-grade atrioventricular block, with resolution of the palpitations. The pulmonary emboli were concomitantly treated with enoxaparin, and the patient was discharged to a rehabilitation facility. Her prognosis was extremely poor given that survival with cardiac metastasis from any type of cancer is limited to a few weeks to months.3 She was to be reevaluated for experimental chemotherapy after reconditioning. However, not long after discharge she was readmitted in respiratory failure and died.
Acknowledgments
We would like to thank Dr. Kevin Steel, Lt Col, USAF, MC, imaging cardiologist at the Brooke Army Medical Center for his time and effort in accessing and preparing the CT and MRI images for this article.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect the official policy or position of Federal Practitioner, Frontline Medical Communications Inc., Brooke Army Medical Center, the U.S. Army Medical Department, the U.S. Army Office of the Surgeon General, the Department of the Army, Department of Defense, the U.S. Government, or any other of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
A 74-year-old woman presented with a 2-day history of exertional dyspnea and palpitations. Her past medical history was significant for metastatic papillary thyroid carcinoma treated with total thyroidectomy and radioactive iodine ablation with levothyroxine for chronic suppressive therapy.
On examination, the patient was afebrile with an oxygen saturation of 98% on room air, heart rate of 92 beats/min, and blood pressure of 100/54 mm Hg. There was trace bilateral lower extremity edema, and her cardiopulmonary examination was unremarkable. The laboratory studies showed a white blood cell count of 24,300/µL (3,400-9,800); platelets 86,000/µL (142,000-362,000); thyroid stimulating hormone 0.009 mlU/L (0.4-4.1); free T4 2.07 ng/dL (0.8-2.0); thyroglobulin antibody titer < 1:10 (< 1:160); thyroid microsomal antibody titer < 1:100 (< 1:1600); and thyroglobulin 17.9 ng/mL (2.0-35.0). Her initial troponin T was undetectable.
An electrocardiogram showed a first-degree atrioventricular block and subsequently a new intermittent third-degree atrioventricular block. A computed tomography angiogram (Figure 1) and cardiac magnetic resonance imaging (Figure 2) revealed a 2.6-cm soft tissue mass in the right ventricular outflow tract along with multiple pulmonary emboli and previously diagnosed pulmonary metastases. A positron emission tomography (PET) scan (not shown) revealed a 3.5-cm PET-avid lesion within the right ventricular outflow tract.
- What is your diagnosis?
- How would you treat this patient?
[Click through to the next page to see the answer.]
Our Treatment
Diagnosis and Discussion
This patient experienced complete heart block due to a cardiac tumor from papillary thyroid carcinoma metastasis. Complete heart block is not an unprecedented symptom of metastatic disease, but to our knowledge this is the first reported case of heart block secondary to metastatic papillary thyroid cancer.1 In general, metastatic cardiac tumors, usually associated with cancers of the breast and lung, melanoma, and lymphoma, are more common than are primary cardiac tumors and are often asymptomatic and discovered mostly postmortem.2,3 The frequency of thyroid metastasis to the heart has been reported to be as low as 0% to 2%, and a review of the literature demonstrated only 13 total cases in the past 30 years.
Theoretical mechanisms for invasion into the heart include lymphatic spread, hematogenous dissemination, or direct right ventricular invasion from the thoracic duct. It has been suggested that the lower blood flow to the myocardium (240 mL/min) relative to bone (600 mL/min) or the brain (750 mL/min) is the reason for a lower likelihood of cardiac involvement in metastatic disease.3 Given the findings in this case, evidence of cardiac conduction abnormalities in the setting of papillary thyroid cancer should raise suspicion for cardiac metastatic disease.
Case Outcome
In this patient, a permanent pacemaker was implanted for high-grade atrioventricular block, with resolution of the palpitations. The pulmonary emboli were concomitantly treated with enoxaparin, and the patient was discharged to a rehabilitation facility. Her prognosis was extremely poor given that survival with cardiac metastasis from any type of cancer is limited to a few weeks to months.3 She was to be reevaluated for experimental chemotherapy after reconditioning. However, not long after discharge she was readmitted in respiratory failure and died.
Acknowledgments
We would like to thank Dr. Kevin Steel, Lt Col, USAF, MC, imaging cardiologist at the Brooke Army Medical Center for his time and effort in accessing and preparing the CT and MRI images for this article.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect the official policy or position of Federal Practitioner, Frontline Medical Communications Inc., Brooke Army Medical Center, the U.S. Army Medical Department, the U.S. Army Office of the Surgeon General, the Department of the Army, Department of Defense, the U.S. Government, or any other of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Conley M, Hawkins K, Ririe D. Complete heart block and cardiac tamponade secondary to Merkel cell carcinoma cardiac metastases. South Med J. 2006;99(1):74-78.
2. Pascale P, Prior JO, Carron PN, Pruvot E, Muller O. Haemoptysis and complete atrioventricular block. Eur Heart J. 2008;29(11):1396.
3. Giuffrida D, Gharib H. Cardiac metastasis from primary anaplastic thyroid carcinoma: Report of three cases and a review of the literature. Endocr Relat Cancer. 2001;8(1):71-73.
1. Conley M, Hawkins K, Ririe D. Complete heart block and cardiac tamponade secondary to Merkel cell carcinoma cardiac metastases. South Med J. 2006;99(1):74-78.
2. Pascale P, Prior JO, Carron PN, Pruvot E, Muller O. Haemoptysis and complete atrioventricular block. Eur Heart J. 2008;29(11):1396.
3. Giuffrida D, Gharib H. Cardiac metastasis from primary anaplastic thyroid carcinoma: Report of three cases and a review of the literature. Endocr Relat Cancer. 2001;8(1):71-73.
Herpes Esophagitis in the Setting of Immunosuppression From Pemphigus Vulgaris Therapy
Pemphigus vulgaris (PV) is a chronic autoimmune intraepithelial bullous disease caused by pathogenic IgG antibodies at the intraepidermal cell-surface proteins desmoglein 1 (DSG1) and desmoglein 3 (DSG3), which are members of the cadherin superfamily of desmosomal proteins and are involved in keratinocyte adhesion. Autoantibody binding to these molecules leads to the loss of cell-cell adhesion in the epithelial suprabasilar layer, producing flaccid blisters on an erythematous base with a positive Nikolsky sign.1 The blisters frequently rupture, leaving painful nonscarring erosions with the potential for secondary infection.
The clinical phenotype of PV is directly related to the autoantibody profile. Clinically, PV often is mucosal dominant on presentation with painful oropharyngeal involvement and associated IgG antibodies against DSG3. Progression to cutaneous disease, such as on the scalp or axillae, is accompanied by a shift in IgG antibodies against both DSG1 and DSG3.2,3
Combination therapy with prednisone and mycophenolate mofetil (MMF) has proven to be an effective method of controlling the signs and symptoms of PV4; however, the immunosuppressive effects of these medications put the patient at risk for a host of opportunistic infections. Herpes simplex virus (HSV) has been associated with PV lesions of the oral mucosa, though a clear-cut relationship between these 2 entities has yet to be established.5 Herpes simplex virus has likewise been confirmed in therapy-resistant exacerbations of PV.6 Herpes esophagitis is a rare consequence of treatment with prednisone and MMF that is primarily encountered in patients with a history of solid organ transplantation7 and rarely has been reported in PV patients undergoing therapeutic immunosuppression.
Acute odynophagia in patients undergoing systemic treatment of active PV warrants prompt endoscopic evaluation to rule out esophageal pemphigus or superinfection. We report the case of a 35-year-old man with stable but poorly controlled PV who was undergoing systemic treatment and experienced rapid deterioration due to herpes esophagitis from immunosuppression.
Case Report
A 35-year-old man was referred to our clinic for evaluation of blisters on the scalp, oral mucosa, and proximal upper and lower extremities of 4 months’ duration. A biopsy performed by his primary care physician within a month of onset of symptoms was reportedly suggestive of PV; although no direct immunofluorescence had been performed, serum indirect immunofluorescence was highly positive for IgG antibodies toward DSG3 and to a lesser extent DSG1. The blisters failed to improve with a 2-week prednisone taper completed 1 month prior to presentation. The patient was not currently taking any other medications. He had a remote history of fever blisters but no other dermatologic issues.
Initial examination revealed flaccid bullae on an erythematous base involving the posterior scalp as well as tender white erosions to shallow ulcers on the tongue and hard and soft palates. A Tzanck smear (modified Wright-Giemsa stain) of these erosions confirmed acantholytic mucosal cells. Punch biopsies of lesional and perilesional skin from the scalp were obtained for histopathologic confirmation and immunofluorescence. An acantholytic dermatosis with a tombstone pattern along the basement membrane was present on hematoxylin and eosin staining, and direct immunofluorescence was positive for IgG and C3 in an intraepidermal lacelike pattern, confirming a diagnosis of PV.
Despite starting an oral regimen of high-dose corticosteroids (prednisone 80 mg once daily), no improvement was noted at 2-week follow-up. He had developed flaccid blisters on the left axillae and mildly worsened oral erosions. He also reported moderate difficulty eating due to pain with swallowing. Mycophenolate mofetil (500 mg twice daily) was added as combination therapy with the prednisone.
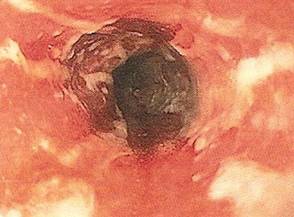
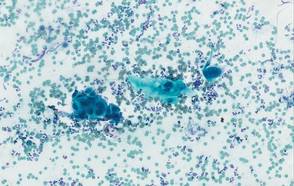
One week later, the patient was unable to eat or drink due to worsening odynophagia. He was admitted as an inpatient for treatment with intravenous methylprednisolone (120 mg every 8 hours) and MMF (1000 mg daily). The gastroenterology department was consulted and an esophagogastroduodenoscopy revealed diffuse areas of denuded and friable mucosa with an overlay of white exudate (Figure 1). Cytology performed on esophageal brushings revealed viral cytopathic changes confirming herpes esophagitis (Figure 2). No esophageal viral cultures were taken. The patient was started on intravenous acyclovir (800 mg 4 times daily), leading to rapid resolution of the odynophagia. He was discharged after 4 days with a course of oral acyclovir (400 mg 4 times daily for 14 days). Tzanck smears and HSV cultures of oral lesions performed immediately following discharge were negative. Combination therapy with MMF (500 mg twice daily) and a slow taper of prednisone (down to 5 mg once daily) was continued past 1 year without flare of his cutaneous disease.
Comment
Although PV may have been considered a fatal disease at one time, treatment with systemic steroids has made it a manageable, albeit relapsing, condition. The development of corticosteroid-sparing, adjuvant immunosuppressives such as MMF has allowed for the more aggressive treatment of this disease with fewer steroid-related side effects.4,8,9 As seen in solid organ transplant recipients who often utilize combination therapy, the use of adjuvant immunosuppressives is associated with potential complications including bone marrow suppression and an increased risk for infections.7,10
Odynophagia is among the potential complications in patients with PV and has a wide differential diagnosis. Mucosal lesions of PV previously have been associated with HSV colonization, though a causal relationship has not been corroborated.5 Herpes simplex virus is more often detected in PV patients being treated with immunosuppressive agents than in nontreated patient groups.11 Recalcitrant or suddenly exacerbated oral mucosal lesions of PV under appropriate therapy may therefore be the result of HSV superinfection, which has been deferentially referred to as pemphigus herpeticum.12 Esophageal mucosal involvement by PV also may be more common than previously thought and should be suspected in patients with active oral disease.13 Esophagitis secondary to medications or various opportunistic organisms such as Candida, cytomegalovirus, or HSV also should be ruled out in patients taking immunosuppressives.5,10
Herpes esophagitis primarily occurs in immunocompromised hosts and is well documented in the literature regarding treatment with MMF and prednisone following renal and cardiac transplantation.10 Prednisone therapy in patients with chronic obstructive pulmonary disease also has been implicated.14 Reactivation of latent HSV resulting from immunosuppression is most often described, though primary infection also is possible.15 Patients typically present with acute odynophagia progressing to dysphagia, with complications ranging from sequelae of poor oral intake to esophageal perforation and hemorrhage, but the course generally is self-limited if immune function is promptly restored. Intravenous acyclovir has been known to hasten the recovery process and improve symptoms.16 Characteristic findings on esophagogastroduodenoscopy in combination with tissue biopsy, viral culture, and/or polymerase chain reaction aid in the diagnosis of herpes esophagitis.15,16 Our patient had a grossly abnormal esophagogastroduodenoscopy with positive cytology; however, no further diagnostic workup was performed. The cytologic findings and the rapid symptomatic improvement following the initiation of acyclovir helped support HSV as the etiology.
Conclusion
We present a case of herpes esophagitis that complicated the treatment of PV with MMF and prednisone. A diagnosis of herpes esophagitis must be ruled out in patients with PV who are undergoing therapeutic immunosuppression and present with an acute episode of odynophagia that is resistant to upscaling of therapy.
- Mustasim DF, Bilic M, Hawayek LH, et al. Immunobullous diseases. J Am Acad Dermatol. 2005;52:1029-1043.
- Amagai M, Tsunoda K, Zillikens D, et al. The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J Am Acad Dermatol. 1999;40(2, pt 1):167-170.
- Sirois DA, Fatahzadeh M, Roth R, et al. Diagnostic patterns and delays in pemphigus vulgaris: experience from 99 patients. Arch Dermatol. 2000;136:1569-1570.
- Strowd LC, Taylor SL, Jorizzo JL, et al. Therapeutic ladder for pemphigus vulgaris: emphasis on achieving complete remission. J Am Acad Dermatol. 2011;64:490-494.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Hale EK, Bystryn JC. Atypical herpes simplex can mimic a flare of disease activity in patients with pemphigus vulgaris. J Eur Acad Dermatol Venereol. 1999;13:221-223.
- Smak Gregoor PJ, van Gelder T, van Riemsdijk-van Overbeeke IC, et al. Unusual presentation of herpes virus infections in renal transplant recipients exposed to high mycophenolic acid plasma concentrations. Transpl Infect Dis. 2003;5:79-83.
- Beissert S, Mimouni D, Kanwar AJ, et al. Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo-controlled trial. J Invest Dermatol. 2010;130:2041-2048.
- Yeh SW, Sami N, Ahmed RA. Treatment of pemphigus vulgaris: current and emerging options. Am J Clin Dermatol. 2005;6:327-342.
- Eisen HJ, Kobashigawa J, Keogh A, et al. Three-year results of a randomized, double-blind, controlled trial of mycophenolate mofetil versus azathioprine in cardiac transplant recipients. J Heart Lung Transplant. 2005;24:517-525.
- Marzano AV, Tourlaki A, Merlo V, et al. Herpes simplex virus infection and pemphigus. Int J Immunopathol Pharmacol. 2009;22:781-786.
- Feldmeyer L, Trüeb RM, French LE, et al. Pitfall: pemphigus herpeticatus should not be confounded with resistant pemphigus vulgaris. J Dermatolog Treat. 2010;21:311-313.
- Rao PN, Samarth A, Aurangabadkar SJ, et al. Study of upper gastrointestinal tract involvement in pemphigus by esophago-gastro-duodenoscopy. Indian J Dermatol Venereol Leprol. 2006;72:421-424.
- Wiest PM, Flanigan T, Salata RA, et al. Serious infectious complications of corticosteroid therapy for COPD. Chest. 1989;95:1180-1184.
- Lee B, Caddy G. A rare cause of dysphagia: herpes simplex esophagitis. World J Gastroenterol. 2007;13:2756-2757.
- Robertson AG, Dunn LJ, Immanuel A, et al. An unusual presentation of herpes simplex esophagitis: a nonhealing “peptic” ulcer. Endoscopy. 2009;41(suppl 2):E213.
Pemphigus vulgaris (PV) is a chronic autoimmune intraepithelial bullous disease caused by pathogenic IgG antibodies at the intraepidermal cell-surface proteins desmoglein 1 (DSG1) and desmoglein 3 (DSG3), which are members of the cadherin superfamily of desmosomal proteins and are involved in keratinocyte adhesion. Autoantibody binding to these molecules leads to the loss of cell-cell adhesion in the epithelial suprabasilar layer, producing flaccid blisters on an erythematous base with a positive Nikolsky sign.1 The blisters frequently rupture, leaving painful nonscarring erosions with the potential for secondary infection.
The clinical phenotype of PV is directly related to the autoantibody profile. Clinically, PV often is mucosal dominant on presentation with painful oropharyngeal involvement and associated IgG antibodies against DSG3. Progression to cutaneous disease, such as on the scalp or axillae, is accompanied by a shift in IgG antibodies against both DSG1 and DSG3.2,3
Combination therapy with prednisone and mycophenolate mofetil (MMF) has proven to be an effective method of controlling the signs and symptoms of PV4; however, the immunosuppressive effects of these medications put the patient at risk for a host of opportunistic infections. Herpes simplex virus (HSV) has been associated with PV lesions of the oral mucosa, though a clear-cut relationship between these 2 entities has yet to be established.5 Herpes simplex virus has likewise been confirmed in therapy-resistant exacerbations of PV.6 Herpes esophagitis is a rare consequence of treatment with prednisone and MMF that is primarily encountered in patients with a history of solid organ transplantation7 and rarely has been reported in PV patients undergoing therapeutic immunosuppression.
Acute odynophagia in patients undergoing systemic treatment of active PV warrants prompt endoscopic evaluation to rule out esophageal pemphigus or superinfection. We report the case of a 35-year-old man with stable but poorly controlled PV who was undergoing systemic treatment and experienced rapid deterioration due to herpes esophagitis from immunosuppression.
Case Report
A 35-year-old man was referred to our clinic for evaluation of blisters on the scalp, oral mucosa, and proximal upper and lower extremities of 4 months’ duration. A biopsy performed by his primary care physician within a month of onset of symptoms was reportedly suggestive of PV; although no direct immunofluorescence had been performed, serum indirect immunofluorescence was highly positive for IgG antibodies toward DSG3 and to a lesser extent DSG1. The blisters failed to improve with a 2-week prednisone taper completed 1 month prior to presentation. The patient was not currently taking any other medications. He had a remote history of fever blisters but no other dermatologic issues.
Initial examination revealed flaccid bullae on an erythematous base involving the posterior scalp as well as tender white erosions to shallow ulcers on the tongue and hard and soft palates. A Tzanck smear (modified Wright-Giemsa stain) of these erosions confirmed acantholytic mucosal cells. Punch biopsies of lesional and perilesional skin from the scalp were obtained for histopathologic confirmation and immunofluorescence. An acantholytic dermatosis with a tombstone pattern along the basement membrane was present on hematoxylin and eosin staining, and direct immunofluorescence was positive for IgG and C3 in an intraepidermal lacelike pattern, confirming a diagnosis of PV.
Despite starting an oral regimen of high-dose corticosteroids (prednisone 80 mg once daily), no improvement was noted at 2-week follow-up. He had developed flaccid blisters on the left axillae and mildly worsened oral erosions. He also reported moderate difficulty eating due to pain with swallowing. Mycophenolate mofetil (500 mg twice daily) was added as combination therapy with the prednisone.
One week later, the patient was unable to eat or drink due to worsening odynophagia. He was admitted as an inpatient for treatment with intravenous methylprednisolone (120 mg every 8 hours) and MMF (1000 mg daily). The gastroenterology department was consulted and an esophagogastroduodenoscopy revealed diffuse areas of denuded and friable mucosa with an overlay of white exudate (Figure 1). Cytology performed on esophageal brushings revealed viral cytopathic changes confirming herpes esophagitis (Figure 2). No esophageal viral cultures were taken. The patient was started on intravenous acyclovir (800 mg 4 times daily), leading to rapid resolution of the odynophagia. He was discharged after 4 days with a course of oral acyclovir (400 mg 4 times daily for 14 days). Tzanck smears and HSV cultures of oral lesions performed immediately following discharge were negative. Combination therapy with MMF (500 mg twice daily) and a slow taper of prednisone (down to 5 mg once daily) was continued past 1 year without flare of his cutaneous disease.
Comment
Although PV may have been considered a fatal disease at one time, treatment with systemic steroids has made it a manageable, albeit relapsing, condition. The development of corticosteroid-sparing, adjuvant immunosuppressives such as MMF has allowed for the more aggressive treatment of this disease with fewer steroid-related side effects.4,8,9 As seen in solid organ transplant recipients who often utilize combination therapy, the use of adjuvant immunosuppressives is associated with potential complications including bone marrow suppression and an increased risk for infections.7,10
Odynophagia is among the potential complications in patients with PV and has a wide differential diagnosis. Mucosal lesions of PV previously have been associated with HSV colonization, though a causal relationship has not been corroborated.5 Herpes simplex virus is more often detected in PV patients being treated with immunosuppressive agents than in nontreated patient groups.11 Recalcitrant or suddenly exacerbated oral mucosal lesions of PV under appropriate therapy may therefore be the result of HSV superinfection, which has been deferentially referred to as pemphigus herpeticum.12 Esophageal mucosal involvement by PV also may be more common than previously thought and should be suspected in patients with active oral disease.13 Esophagitis secondary to medications or various opportunistic organisms such as Candida, cytomegalovirus, or HSV also should be ruled out in patients taking immunosuppressives.5,10
Herpes esophagitis primarily occurs in immunocompromised hosts and is well documented in the literature regarding treatment with MMF and prednisone following renal and cardiac transplantation.10 Prednisone therapy in patients with chronic obstructive pulmonary disease also has been implicated.14 Reactivation of latent HSV resulting from immunosuppression is most often described, though primary infection also is possible.15 Patients typically present with acute odynophagia progressing to dysphagia, with complications ranging from sequelae of poor oral intake to esophageal perforation and hemorrhage, but the course generally is self-limited if immune function is promptly restored. Intravenous acyclovir has been known to hasten the recovery process and improve symptoms.16 Characteristic findings on esophagogastroduodenoscopy in combination with tissue biopsy, viral culture, and/or polymerase chain reaction aid in the diagnosis of herpes esophagitis.15,16 Our patient had a grossly abnormal esophagogastroduodenoscopy with positive cytology; however, no further diagnostic workup was performed. The cytologic findings and the rapid symptomatic improvement following the initiation of acyclovir helped support HSV as the etiology.
Conclusion
We present a case of herpes esophagitis that complicated the treatment of PV with MMF and prednisone. A diagnosis of herpes esophagitis must be ruled out in patients with PV who are undergoing therapeutic immunosuppression and present with an acute episode of odynophagia that is resistant to upscaling of therapy.
Pemphigus vulgaris (PV) is a chronic autoimmune intraepithelial bullous disease caused by pathogenic IgG antibodies at the intraepidermal cell-surface proteins desmoglein 1 (DSG1) and desmoglein 3 (DSG3), which are members of the cadherin superfamily of desmosomal proteins and are involved in keratinocyte adhesion. Autoantibody binding to these molecules leads to the loss of cell-cell adhesion in the epithelial suprabasilar layer, producing flaccid blisters on an erythematous base with a positive Nikolsky sign.1 The blisters frequently rupture, leaving painful nonscarring erosions with the potential for secondary infection.
The clinical phenotype of PV is directly related to the autoantibody profile. Clinically, PV often is mucosal dominant on presentation with painful oropharyngeal involvement and associated IgG antibodies against DSG3. Progression to cutaneous disease, such as on the scalp or axillae, is accompanied by a shift in IgG antibodies against both DSG1 and DSG3.2,3
Combination therapy with prednisone and mycophenolate mofetil (MMF) has proven to be an effective method of controlling the signs and symptoms of PV4; however, the immunosuppressive effects of these medications put the patient at risk for a host of opportunistic infections. Herpes simplex virus (HSV) has been associated with PV lesions of the oral mucosa, though a clear-cut relationship between these 2 entities has yet to be established.5 Herpes simplex virus has likewise been confirmed in therapy-resistant exacerbations of PV.6 Herpes esophagitis is a rare consequence of treatment with prednisone and MMF that is primarily encountered in patients with a history of solid organ transplantation7 and rarely has been reported in PV patients undergoing therapeutic immunosuppression.
Acute odynophagia in patients undergoing systemic treatment of active PV warrants prompt endoscopic evaluation to rule out esophageal pemphigus or superinfection. We report the case of a 35-year-old man with stable but poorly controlled PV who was undergoing systemic treatment and experienced rapid deterioration due to herpes esophagitis from immunosuppression.
Case Report
A 35-year-old man was referred to our clinic for evaluation of blisters on the scalp, oral mucosa, and proximal upper and lower extremities of 4 months’ duration. A biopsy performed by his primary care physician within a month of onset of symptoms was reportedly suggestive of PV; although no direct immunofluorescence had been performed, serum indirect immunofluorescence was highly positive for IgG antibodies toward DSG3 and to a lesser extent DSG1. The blisters failed to improve with a 2-week prednisone taper completed 1 month prior to presentation. The patient was not currently taking any other medications. He had a remote history of fever blisters but no other dermatologic issues.
Initial examination revealed flaccid bullae on an erythematous base involving the posterior scalp as well as tender white erosions to shallow ulcers on the tongue and hard and soft palates. A Tzanck smear (modified Wright-Giemsa stain) of these erosions confirmed acantholytic mucosal cells. Punch biopsies of lesional and perilesional skin from the scalp were obtained for histopathologic confirmation and immunofluorescence. An acantholytic dermatosis with a tombstone pattern along the basement membrane was present on hematoxylin and eosin staining, and direct immunofluorescence was positive for IgG and C3 in an intraepidermal lacelike pattern, confirming a diagnosis of PV.
Despite starting an oral regimen of high-dose corticosteroids (prednisone 80 mg once daily), no improvement was noted at 2-week follow-up. He had developed flaccid blisters on the left axillae and mildly worsened oral erosions. He also reported moderate difficulty eating due to pain with swallowing. Mycophenolate mofetil (500 mg twice daily) was added as combination therapy with the prednisone.
One week later, the patient was unable to eat or drink due to worsening odynophagia. He was admitted as an inpatient for treatment with intravenous methylprednisolone (120 mg every 8 hours) and MMF (1000 mg daily). The gastroenterology department was consulted and an esophagogastroduodenoscopy revealed diffuse areas of denuded and friable mucosa with an overlay of white exudate (Figure 1). Cytology performed on esophageal brushings revealed viral cytopathic changes confirming herpes esophagitis (Figure 2). No esophageal viral cultures were taken. The patient was started on intravenous acyclovir (800 mg 4 times daily), leading to rapid resolution of the odynophagia. He was discharged after 4 days with a course of oral acyclovir (400 mg 4 times daily for 14 days). Tzanck smears and HSV cultures of oral lesions performed immediately following discharge were negative. Combination therapy with MMF (500 mg twice daily) and a slow taper of prednisone (down to 5 mg once daily) was continued past 1 year without flare of his cutaneous disease.
Comment
Although PV may have been considered a fatal disease at one time, treatment with systemic steroids has made it a manageable, albeit relapsing, condition. The development of corticosteroid-sparing, adjuvant immunosuppressives such as MMF has allowed for the more aggressive treatment of this disease with fewer steroid-related side effects.4,8,9 As seen in solid organ transplant recipients who often utilize combination therapy, the use of adjuvant immunosuppressives is associated with potential complications including bone marrow suppression and an increased risk for infections.7,10
Odynophagia is among the potential complications in patients with PV and has a wide differential diagnosis. Mucosal lesions of PV previously have been associated with HSV colonization, though a causal relationship has not been corroborated.5 Herpes simplex virus is more often detected in PV patients being treated with immunosuppressive agents than in nontreated patient groups.11 Recalcitrant or suddenly exacerbated oral mucosal lesions of PV under appropriate therapy may therefore be the result of HSV superinfection, which has been deferentially referred to as pemphigus herpeticum.12 Esophageal mucosal involvement by PV also may be more common than previously thought and should be suspected in patients with active oral disease.13 Esophagitis secondary to medications or various opportunistic organisms such as Candida, cytomegalovirus, or HSV also should be ruled out in patients taking immunosuppressives.5,10
Herpes esophagitis primarily occurs in immunocompromised hosts and is well documented in the literature regarding treatment with MMF and prednisone following renal and cardiac transplantation.10 Prednisone therapy in patients with chronic obstructive pulmonary disease also has been implicated.14 Reactivation of latent HSV resulting from immunosuppression is most often described, though primary infection also is possible.15 Patients typically present with acute odynophagia progressing to dysphagia, with complications ranging from sequelae of poor oral intake to esophageal perforation and hemorrhage, but the course generally is self-limited if immune function is promptly restored. Intravenous acyclovir has been known to hasten the recovery process and improve symptoms.16 Characteristic findings on esophagogastroduodenoscopy in combination with tissue biopsy, viral culture, and/or polymerase chain reaction aid in the diagnosis of herpes esophagitis.15,16 Our patient had a grossly abnormal esophagogastroduodenoscopy with positive cytology; however, no further diagnostic workup was performed. The cytologic findings and the rapid symptomatic improvement following the initiation of acyclovir helped support HSV as the etiology.
Conclusion
We present a case of herpes esophagitis that complicated the treatment of PV with MMF and prednisone. A diagnosis of herpes esophagitis must be ruled out in patients with PV who are undergoing therapeutic immunosuppression and present with an acute episode of odynophagia that is resistant to upscaling of therapy.
- Mustasim DF, Bilic M, Hawayek LH, et al. Immunobullous diseases. J Am Acad Dermatol. 2005;52:1029-1043.
- Amagai M, Tsunoda K, Zillikens D, et al. The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J Am Acad Dermatol. 1999;40(2, pt 1):167-170.
- Sirois DA, Fatahzadeh M, Roth R, et al. Diagnostic patterns and delays in pemphigus vulgaris: experience from 99 patients. Arch Dermatol. 2000;136:1569-1570.
- Strowd LC, Taylor SL, Jorizzo JL, et al. Therapeutic ladder for pemphigus vulgaris: emphasis on achieving complete remission. J Am Acad Dermatol. 2011;64:490-494.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Hale EK, Bystryn JC. Atypical herpes simplex can mimic a flare of disease activity in patients with pemphigus vulgaris. J Eur Acad Dermatol Venereol. 1999;13:221-223.
- Smak Gregoor PJ, van Gelder T, van Riemsdijk-van Overbeeke IC, et al. Unusual presentation of herpes virus infections in renal transplant recipients exposed to high mycophenolic acid plasma concentrations. Transpl Infect Dis. 2003;5:79-83.
- Beissert S, Mimouni D, Kanwar AJ, et al. Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo-controlled trial. J Invest Dermatol. 2010;130:2041-2048.
- Yeh SW, Sami N, Ahmed RA. Treatment of pemphigus vulgaris: current and emerging options. Am J Clin Dermatol. 2005;6:327-342.
- Eisen HJ, Kobashigawa J, Keogh A, et al. Three-year results of a randomized, double-blind, controlled trial of mycophenolate mofetil versus azathioprine in cardiac transplant recipients. J Heart Lung Transplant. 2005;24:517-525.
- Marzano AV, Tourlaki A, Merlo V, et al. Herpes simplex virus infection and pemphigus. Int J Immunopathol Pharmacol. 2009;22:781-786.
- Feldmeyer L, Trüeb RM, French LE, et al. Pitfall: pemphigus herpeticatus should not be confounded with resistant pemphigus vulgaris. J Dermatolog Treat. 2010;21:311-313.
- Rao PN, Samarth A, Aurangabadkar SJ, et al. Study of upper gastrointestinal tract involvement in pemphigus by esophago-gastro-duodenoscopy. Indian J Dermatol Venereol Leprol. 2006;72:421-424.
- Wiest PM, Flanigan T, Salata RA, et al. Serious infectious complications of corticosteroid therapy for COPD. Chest. 1989;95:1180-1184.
- Lee B, Caddy G. A rare cause of dysphagia: herpes simplex esophagitis. World J Gastroenterol. 2007;13:2756-2757.
- Robertson AG, Dunn LJ, Immanuel A, et al. An unusual presentation of herpes simplex esophagitis: a nonhealing “peptic” ulcer. Endoscopy. 2009;41(suppl 2):E213.
- Mustasim DF, Bilic M, Hawayek LH, et al. Immunobullous diseases. J Am Acad Dermatol. 2005;52:1029-1043.
- Amagai M, Tsunoda K, Zillikens D, et al. The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J Am Acad Dermatol. 1999;40(2, pt 1):167-170.
- Sirois DA, Fatahzadeh M, Roth R, et al. Diagnostic patterns and delays in pemphigus vulgaris: experience from 99 patients. Arch Dermatol. 2000;136:1569-1570.
- Strowd LC, Taylor SL, Jorizzo JL, et al. Therapeutic ladder for pemphigus vulgaris: emphasis on achieving complete remission. J Am Acad Dermatol. 2011;64:490-494.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Hale EK, Bystryn JC. Atypical herpes simplex can mimic a flare of disease activity in patients with pemphigus vulgaris. J Eur Acad Dermatol Venereol. 1999;13:221-223.
- Smak Gregoor PJ, van Gelder T, van Riemsdijk-van Overbeeke IC, et al. Unusual presentation of herpes virus infections in renal transplant recipients exposed to high mycophenolic acid plasma concentrations. Transpl Infect Dis. 2003;5:79-83.
- Beissert S, Mimouni D, Kanwar AJ, et al. Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo-controlled trial. J Invest Dermatol. 2010;130:2041-2048.
- Yeh SW, Sami N, Ahmed RA. Treatment of pemphigus vulgaris: current and emerging options. Am J Clin Dermatol. 2005;6:327-342.
- Eisen HJ, Kobashigawa J, Keogh A, et al. Three-year results of a randomized, double-blind, controlled trial of mycophenolate mofetil versus azathioprine in cardiac transplant recipients. J Heart Lung Transplant. 2005;24:517-525.
- Marzano AV, Tourlaki A, Merlo V, et al. Herpes simplex virus infection and pemphigus. Int J Immunopathol Pharmacol. 2009;22:781-786.
- Feldmeyer L, Trüeb RM, French LE, et al. Pitfall: pemphigus herpeticatus should not be confounded with resistant pemphigus vulgaris. J Dermatolog Treat. 2010;21:311-313.
- Rao PN, Samarth A, Aurangabadkar SJ, et al. Study of upper gastrointestinal tract involvement in pemphigus by esophago-gastro-duodenoscopy. Indian J Dermatol Venereol Leprol. 2006;72:421-424.
- Wiest PM, Flanigan T, Salata RA, et al. Serious infectious complications of corticosteroid therapy for COPD. Chest. 1989;95:1180-1184.
- Lee B, Caddy G. A rare cause of dysphagia: herpes simplex esophagitis. World J Gastroenterol. 2007;13:2756-2757.
- Robertson AG, Dunn LJ, Immanuel A, et al. An unusual presentation of herpes simplex esophagitis: a nonhealing “peptic” ulcer. Endoscopy. 2009;41(suppl 2):E213.
Practice Points
- Pemphigus vulgaris (PV) often requires therapeutic immunosuppression for disease control.
- Acute odynophagia in the setting of systemic immunosuppression for PV requires endoscopic evaluation.
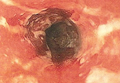
Solitary Morphea Profunda Following Trauma Sustained in an Automobile Accident
Case Report
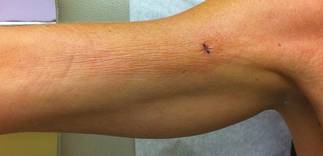
A 50-year-old white woman presented to our clinic for evaluation of what she described as a “very hard red line” on the right upper arm. The lesion had developed suddenly overnight. Several months prior to presentation the patient sustained trauma to the same area in a car accident and she thought the lesion might be related to the resulting nerve damage. Initially she presented to her primary care physician who used ultrasonography of the area to rule out muscle or bone involvement. The patient presented to our dermatology clinic 2 months later with an 18×4-cm, brownish, rectangular, sclerotic, bound-down, hypertrophic plaque that started on the right mid forearm and extended to the right shoulder (Figure 1). Her medical history was notable for high blood pressure, which was controlled with valsartan.
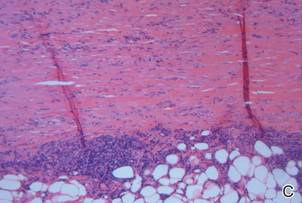
A review of systems was unremarkable. Physical examination revealed a well-developed, well-nourished woman. Examination of the right arm revealed no motion restriction (muscle strength, 5/5) and no pain; however, she described a burning sensation at the site of the lesion. She reported no allergies. A 4-mm punch biopsy was performed and laboratory tests were ordered including an antinuclear antibody (ANA) test with reflex, double-stranded DNA test, DNA antitopoisomerase antibodies test, and Lyme titers (IgM and IgG). Initially, the patient was treated with calcipotriene 0.005%–betamethasone dipropionate 0.064% ointment twice daily; she also was treated empirically for Lyme disease with doxycycline 50 mg twice daily. All laboratory tests were within reference range, and a punch biopsy revealed markedly thickened fibrous septa within the subcutaneous fat. At the edge of the septa there were nodular aggregates of lymphocytes. Due to clinical presentation, laboratory data, and histopathology, solitary morphea profunda (SMP) was diagnosed.
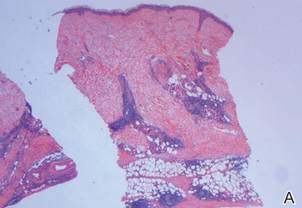
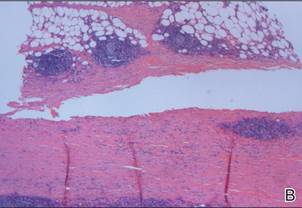
Following histopathologic examination (Figure 2), the patient was instructed to continue treatment with calcipotriene–betamethasone dipropionate as well as doxycycline. A trial of prednisone and/or hydroxychloroquine also was considered pending her response to the initial treatment. At approximately 1-month follow-up, remarkable improvement of the lesion was noted.
Comment
|
There is limited literature available about the diagnosis and treatment of SMP. Our case prompted us to further examine the data to emphasize the necessity of greater research surrounding SMP.
Classification of SMP
Morphea is a localized form of scleroderma, an inflammatory disease that primarily affects the dermis but can extend down to the bone and also can limit motion. There are several types of morphea that are classified according to the extent, depth, and distribution of the lesions, including plaque, generalized, bullous, linear (including morphea en coup de sabre), guttate, nodular, and deep morphea.1,2 Other subtypes have been described including subcutaneous morphea, eosinophilic fasciitis (EF), pansclerotic morphea, and morphea profunda.3 Linear and deep morphea are characterized by involvement of the deep dermis, subcutaneous tissue, fascia, and/or superficial muscle.2,4
In 1981, Su and Person5 first described morphea profunda (MP). In their study, 22 of 23 patients presented with generalized MP. One patient developed a single lesion,5 which ultimately was classified as SMP by Whittaker et al6 in 1989.
Epidemiology
Morphea profunda occurs more frequently in females than in males, with sclerosis manifesting over a period of several months.7 In 2004, Azad et al4 suggested that only 9 cases of SMP had been reported in the literature. Although there is insignificant data to determine the epidemiology of SMP, the authors concluded that it most commonly affects middle-aged individuals with equal sex distribution.4 The single plaque in patients with SMP most commonly presents on the shoulder, back, or neck or in the paraspinal area.
Etiology
Because of the limited amount of literature on MP, a definitive etiology is unknown, but investigators have cited many possible causes. Genetic, autoimmune, hormonal, traumatic,8 vaccination,2,8 radiation,9 viral, neurogenic, and vascular factors all have been implicated,10 as well as infectious agents such as Borrelia burgdorferi in the United States,11,12Borrelia afzelii in Europe,2 and Borrelia garinii in Japan.2 Because our patient experienced a traumatic episode several months prior to presentation, it is important to investigate trauma as a likely etiology. Furthermore, traumatic events have been reported in 23% of children with linear morphea.13
Diagnostic Studies
Morphea profunda is diagnosed clinically and skin biopsy can be used for confirmation. Biopsy requires deep excision down to the muscle, which can aid in determining if the fascia is incorporated. Elevated levels of IgG and IgM have been detected in deep and linear morphea and are known to correlate with disease activity and the development of joint contractures in linear morphea.2 Serum procollagen type I has been considered by some as a useful indicator of disease severity.14 Elevated serum levels of antifibrillin-1 antibodies also have been demonstrated in patients with localized scleroderma (LS).15 Radiography and magnetic resonance imaging can be used for monitoring and analyzing lesion depth. Furthermore, magnetic resonance imaging can be used to differentiate MP from EF.2
The presence of ANAs in LS is controversial. According to Nguyen et al,2 ANAs are present in approximately 46% to 80% of patients with morphea, with a higher prevalence in patients with generalized, linear, and deep subtypes. However, Savoia et al16 found that patients with morphea typically do not present with ANAs; rather ANAs usually are found in patients with EF.
Pathogenesis
After the inflammatory phase in LS, fibrillar collagen types I and III accumulate, causing dermal fibrosis. The extracellular matrix increases due to the activation of connective tissue growth factor, transforming growth factor β (TGF-β), TGF-β receptors, IL-4, and several other cytokines.17 The TGF-β receptors combine with the connective tissue growth factor released by fibroblasts to create an autocrine production loop that causes fibroblast and matrix production.17 As the inflammation progresses to sclerosis, the CD34 count decreases.18
Physical Findings
In patients with MP, lesions manifest as thickened taut skin with deep, solitary, and sclerotic indurated plaques. Clinically, plaques are mildly inflamed, hyperpigmented, symmetric, and somewhat ill defined, and the skin feels thickened and bound to the underlying fascia and muscle. Plaques usually are smooth and shiny, but areas of both dermal and subcutaneous atrophy may be present, particularly in chronic lesions.19 Morphea profunda also can be described as having a cobblestone or pseudocellulite appearance. The groove sign is used to describe a depression along the course of a vein and/or between muscle groups. Both clinical presentations may manifest later in the course of disease.2
Histopathology
Su and Person5 described 3 main characteristics of MP that stand out histopathologically. First, there is thickening and hyalinization of collagen bundles in the deep dermis, subcutis, and fascia that are prominent between the junction of the dermis and subcutaneous fat. There also are fewer sebaceous glands and hair follicles. Second, MP presents with an increased inflammatory cell infiltrate composed mainly of lymphocytes located around small blood vessels and the interstitium. In some patients, the lymphocytes consist predominantly of collections of plasma cells. Third, MP contains deposits of mucin in deep portions of the dermis with occasional eosinophils and mast cells. The presence of eosinophils allows EF to be a part of this spectrum and to be included as a differential diagnosis.5 Eosinophilic fasciitis has a similar presentation to MP because the fibrosis affects the dermis, subcutaneous fat, and underlying structures.20 Although EF presents with the histopathologic characteristic of fascial fibrosis, a clear distinction between EF and morphea has not been established in the literature. Some authors classify EF as a variant of morphea, whereas others consider it as its own entity. We believe EF is its own entity. Eosinophilic fasciitis can be distinguished from morphea because 60% to 80% of patients with EF have peripheral eosinophilia and 20% to 70% of patients with EF have hypergammaglobulinemia. Additionally, morphea does not present as symmetrically or abruptly as EF.21
Treatment
To date, there is conflicting literature regarding the treatment regimen for MP. There is controversy regarding whether MP responds to corticosteroids.19 Different treatment regimens have been discussed for LS, but there is a lack of reports specifically describing therapies for MP and SMP. Because MP and SMP fall under the umbrella of LS, many investigators have reported using the following treatment regimens for patients with MP and SMP: bosentan,22 D-penicillamine,23 phototherapy,24-26 retinoids,26 oral steroids,27 methotrexate,27-29 vitamin D3 (oral calcitriol),30,31 cyclosporine,32 mycophenolate mofetil,33 and extracorporeal photochemotherapy.34
Falanga and Medsger23 reported 64% (7/11) treatment success with D-penicillamine in patients who exhibited severe LS. Psoralen plus UVA,24 methoxsalen, and UVA1 therapy are widely used in the treatment of LS.25 Kreuter et al25 advocated for phototherapy as the first approach in the management of LS after reporting improvement in all participants in their study (N=64), 2 participants with deep morphea while the rest exhibited other forms of morphea. Ozdemir et al26 proposed that retinoic acid combined with psoralen plus UVA is a good treatment choice for plaque-type LS; however, UVA only has the ability to target the epidermis and dermis, which may not be useful for deep forms of morphea.
Several studies have shown positive results in patients treated with methylprednisolone combined with low-dose methotrexate sodium.27-29 Kreuter et al30 and Elst et al31 proposed that calcitriol is effective in treating LS, whereas Hulshof et al35 indicated that it is not. It should be noted that none of these studies specifically mentioned MP. Martini et al33 demonstrated success with mycophenolate mofetil in the treatment of 10 LS patients who were resistant to methotrexate sodium and corticosteroids. Although none of the participants in the study had MP, 2 patients had disabling pansclerotic morphea, 3 had generalized morphea, and 5 had linear scleroderma (morphea en coup de sabre) affecting the limbs (n=2) and face (n=3).33 Because there is no established therapy or consensus for the treatment of MP, we have found success in starting with corticosteroids and then trying alternative therapies.
Prognosis
Morphea has transitioned into systemic scleroderma in a small number of reported cases.10,16,20,36 Therefore, patient follow-up is imperative to consistently identify systemic evolution. Although visceral complications are rare in the setting of LS, associated clinical findings have been reported, including arthralgia, arthritis, contractures, and carpal tunnel syndrome, as well as pulmonary, esophageal, and cardiac abnormalities.7,34
Conclusion
The morphologic features observed in our patient appear to correspond most closely to the type of lesion described by Su and Person5 and Whittaker et al.6 Although our case was clinically difficult to distinguish from linear morphea, the histology suggested SMP over other causes. If our patient’s SMP progressed to the joints, physical therapy would be needed to maintain range of motion and function of the extremities,2 and mandatory long-term follow-up would be required due to the risk for relapse after discontinuation of therapy. Our case highlights the inherent difficulties in the treatment of MP. Due to limited reports of SMP and MP in the literature as well as the conflicting views regarding effective and appropriate treatment options, additional investigation of these conditions and therapeutic options are necessary to further understand this debilitating condition.
1. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
2. Nguyen JV, Werth VP, Fett N. Morphea treatment & management. Medscape Web site. http://emedicine.medscape.com/article/1065782-treatment. Updated July 21, 2014. Accessed December 16, 2104.
3. Melani L, Cardinali C, Giomi B, et al. Case study: periodic follow-up is necessary in morphea profunda to identify systemic evolution. Skinmed. 2005;4:188-190.
4. Azad J, Dawn G, Shaffrali FC, et al. Does solitary morphoea profunda progress? Clin Exp Dermatol. 2004;29:25-27.
5. Su WP, Person JR. Morphea profunda. a new concept and a histopathologic study of 23 cases. Am J Dermatopathol. 1981;3:251-260.
6. Whittaker SJ, Smith NP, Jones RR. Solitary morphoea profunda. Br J Dermatol. 1989;120:431-440.
7. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
8. Torrelo A, Suárez J, Colmenero I, et al. Deep morphea after vaccination in two young children. Pediatr Dermatol. 2006;23:484-487.
9. Kreft B, Wohlrab J, Radant K, et al. Unrecognized radiation-induced localized scleroderma: a cause of postoperative wound-healing disorder [published online ahead of print June 22, 2009]. Clin Exp Dermatol. 2009;34:e383-e384.
10. Braun-Falco O, Plewig G, Wolff HH, et al, eds. Dermatology. Berlin, Germany: Springer; 2002.
11. Prinz JC, Kutasi Z, Weisenseel P, et al. “Borrelia-associated early-onset morphea”: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? results of a cohort analysis and presentation of three cases [published online ahead of print November 20, 2008]. J Am Acad Dermatol. 2009;60:248-255.
12. Wojas-Pelc A, Wielowieyska-Szybińska D, Kiełtyka A. Presence of the antinuclear antibodies and antibodies to Borrelia burgdorferi among patients with morphea en plaque, deep linear scleroderma and atrophoderma Pasini-Pierini [in Polish]. Przegl Lek. 2002;59:898-902.
13. Falanga V, Medsger TA Jr, Reichlin M, et al. Linear scleroderma. clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med. 1986;104:849-857.
14. Kikuchi K, Sato S, Kadono T, et al. Serum concentration of procollagen type I carboxyterminal propeptide in localized scleroderma. Arch Dermatol. 1994;130:1269-1272.
15. Arnett FC, Tan FK, Uziel Y, et al. Autoantibodies to the extracellular matrix microfibrillar protein, fibrilin 1, in patients with localized scleroderma. Arthritis Rheum. 1999;42:2656-2659.
16. Savoia P, Zaccagna A, Bernengo MG. Guess what? inflammatory disseminated morphea profunda. Eur J Dermatol. 1999;9:654-656.
17. Igarashi A, Nashiro K, Kikuchi K, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J Invest Dermatol. 1996;106:729-733.
18. Gilmour TK, Wilkinson B, Breit SN, et al. Analysis of dendritic cell populations using a revised histological staging of morphoea. Br J Dermatol. 2000;143:1183-1192.
19. Sayama K, Chen M, Shiraishi S, et al. Morphea profunda. Int J Dermatol. 1991;30:873-875.
20. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
21. Bischoff L, Derk CT. Eosinophilic fasciitis: demographics disease pattern and response to treatment: report of 12 cases and review of the literature. Int J Dermatol. 2008;47:29-35.
22. Roldan R, Morote G, Castro Mdel C, et al. Efficacy of bosentan in treatment of unresponsive cutaneous ulceration in disabling pansclerotic morphea in children. J Rheumatol. 2006;33:2538-2540.
23. Falanga V, Medsger TA Jr. D-penicillamine in the treatment of localized scleroderma. Arch Dermatol. 1990;126:609-612.
24. Breuckmann F, Gambichler T, Altmeyer P, et al. UVA/UVA1 phototherapy and PUVA photochemotherapy in connective tissue diseases and related disorders: a research based review. BMC Dermatol. 2004;4:11.
25. Kreuter A, Hyun J, Stücker M, et al. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma [published online ahead of print January 30, 2006]. J Am Acad Dermatol. 2006;54:440-447.
26. Ozdemir M, Engin B, Toy H, et al. Treatment of plaque-type localized scleroderma with retinoic acid and ultraviolet A plus the photosensitizer psoralen: a case series. J Eur Acad Dermatol Venereol. 2008;22:519-521.
27. Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141:847-852.
28. Kroft EB, Creemers MC, van den Hoogen FH, et al. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study [published online ahead of print February 4, 2009]. Br J Dermatol. 2009;160:1075-1082.
29. Weibel L, Sampaio MC, Visentin MT, et al. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. 2006;155:1013-1020.
30. Kreuter A, Gambichler T, Avermaete A, et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet A1 phototherapy in childhood morphea. Pediatr Dermatol. 2001;18:241-245.
31. Elst EF, Van Suijlekom-Smit LW, Oranje AP. Treatment of linear scleroderma with oral 1,25-dihydroxy vitamin D3 (calcitriol) in seven children. Pediatr Dermatol. 1999;16:53-58.
32. Crespo MP, Mas IB, Díaz JM, et al. Rapid response to cyclosporine and maintenance with methotrexate in linear scleroderma in a young girl. Pediatr Dermatol. 2009;26:118-120.
33. Martini G, Ramanan AV, Falcini F, et al. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil [published online ahead of print August 27, 2009]. Rheumatology (Oxford). 2009;48:1410-1413.
34. Neustadter JH, Samarin F, Carlson KR, et al. Extracorporeal photochemotherapy for generalized deep morphea. Arch Dermatol. 2009;145:127-130.
35. Hulshof MM, Bouwes Bavinck JN, Bergman W, et al. Double-blind, placebo-controlled study of oral calcitriol for the treatment of localized and systemic scleroderma. J Am Acad Dermatol. 2000;43:1017-1023.
36. Toledano C, Rabhi S, Kettaneh A, et al. Localized scleroderma: a series of 52 patients [published online ahead of print September 17, 2008]. Eur J Intern Med. 2009;20:331-336.
Case Report
A 50-year-old white woman presented to our clinic for evaluation of what she described as a “very hard red line” on the right upper arm. The lesion had developed suddenly overnight. Several months prior to presentation the patient sustained trauma to the same area in a car accident and she thought the lesion might be related to the resulting nerve damage. Initially she presented to her primary care physician who used ultrasonography of the area to rule out muscle or bone involvement. The patient presented to our dermatology clinic 2 months later with an 18×4-cm, brownish, rectangular, sclerotic, bound-down, hypertrophic plaque that started on the right mid forearm and extended to the right shoulder (Figure 1). Her medical history was notable for high blood pressure, which was controlled with valsartan.
A review of systems was unremarkable. Physical examination revealed a well-developed, well-nourished woman. Examination of the right arm revealed no motion restriction (muscle strength, 5/5) and no pain; however, she described a burning sensation at the site of the lesion. She reported no allergies. A 4-mm punch biopsy was performed and laboratory tests were ordered including an antinuclear antibody (ANA) test with reflex, double-stranded DNA test, DNA antitopoisomerase antibodies test, and Lyme titers (IgM and IgG). Initially, the patient was treated with calcipotriene 0.005%–betamethasone dipropionate 0.064% ointment twice daily; she also was treated empirically for Lyme disease with doxycycline 50 mg twice daily. All laboratory tests were within reference range, and a punch biopsy revealed markedly thickened fibrous septa within the subcutaneous fat. At the edge of the septa there were nodular aggregates of lymphocytes. Due to clinical presentation, laboratory data, and histopathology, solitary morphea profunda (SMP) was diagnosed.
Following histopathologic examination (Figure 2), the patient was instructed to continue treatment with calcipotriene–betamethasone dipropionate as well as doxycycline. A trial of prednisone and/or hydroxychloroquine also was considered pending her response to the initial treatment. At approximately 1-month follow-up, remarkable improvement of the lesion was noted.
Comment
|
There is limited literature available about the diagnosis and treatment of SMP. Our case prompted us to further examine the data to emphasize the necessity of greater research surrounding SMP.
Classification of SMP
Morphea is a localized form of scleroderma, an inflammatory disease that primarily affects the dermis but can extend down to the bone and also can limit motion. There are several types of morphea that are classified according to the extent, depth, and distribution of the lesions, including plaque, generalized, bullous, linear (including morphea en coup de sabre), guttate, nodular, and deep morphea.1,2 Other subtypes have been described including subcutaneous morphea, eosinophilic fasciitis (EF), pansclerotic morphea, and morphea profunda.3 Linear and deep morphea are characterized by involvement of the deep dermis, subcutaneous tissue, fascia, and/or superficial muscle.2,4
In 1981, Su and Person5 first described morphea profunda (MP). In their study, 22 of 23 patients presented with generalized MP. One patient developed a single lesion,5 which ultimately was classified as SMP by Whittaker et al6 in 1989.
Epidemiology
Morphea profunda occurs more frequently in females than in males, with sclerosis manifesting over a period of several months.7 In 2004, Azad et al4 suggested that only 9 cases of SMP had been reported in the literature. Although there is insignificant data to determine the epidemiology of SMP, the authors concluded that it most commonly affects middle-aged individuals with equal sex distribution.4 The single plaque in patients with SMP most commonly presents on the shoulder, back, or neck or in the paraspinal area.
Etiology
Because of the limited amount of literature on MP, a definitive etiology is unknown, but investigators have cited many possible causes. Genetic, autoimmune, hormonal, traumatic,8 vaccination,2,8 radiation,9 viral, neurogenic, and vascular factors all have been implicated,10 as well as infectious agents such as Borrelia burgdorferi in the United States,11,12Borrelia afzelii in Europe,2 and Borrelia garinii in Japan.2 Because our patient experienced a traumatic episode several months prior to presentation, it is important to investigate trauma as a likely etiology. Furthermore, traumatic events have been reported in 23% of children with linear morphea.13
Diagnostic Studies
Morphea profunda is diagnosed clinically and skin biopsy can be used for confirmation. Biopsy requires deep excision down to the muscle, which can aid in determining if the fascia is incorporated. Elevated levels of IgG and IgM have been detected in deep and linear morphea and are known to correlate with disease activity and the development of joint contractures in linear morphea.2 Serum procollagen type I has been considered by some as a useful indicator of disease severity.14 Elevated serum levels of antifibrillin-1 antibodies also have been demonstrated in patients with localized scleroderma (LS).15 Radiography and magnetic resonance imaging can be used for monitoring and analyzing lesion depth. Furthermore, magnetic resonance imaging can be used to differentiate MP from EF.2
The presence of ANAs in LS is controversial. According to Nguyen et al,2 ANAs are present in approximately 46% to 80% of patients with morphea, with a higher prevalence in patients with generalized, linear, and deep subtypes. However, Savoia et al16 found that patients with morphea typically do not present with ANAs; rather ANAs usually are found in patients with EF.
Pathogenesis
After the inflammatory phase in LS, fibrillar collagen types I and III accumulate, causing dermal fibrosis. The extracellular matrix increases due to the activation of connective tissue growth factor, transforming growth factor β (TGF-β), TGF-β receptors, IL-4, and several other cytokines.17 The TGF-β receptors combine with the connective tissue growth factor released by fibroblasts to create an autocrine production loop that causes fibroblast and matrix production.17 As the inflammation progresses to sclerosis, the CD34 count decreases.18
Physical Findings
In patients with MP, lesions manifest as thickened taut skin with deep, solitary, and sclerotic indurated plaques. Clinically, plaques are mildly inflamed, hyperpigmented, symmetric, and somewhat ill defined, and the skin feels thickened and bound to the underlying fascia and muscle. Plaques usually are smooth and shiny, but areas of both dermal and subcutaneous atrophy may be present, particularly in chronic lesions.19 Morphea profunda also can be described as having a cobblestone or pseudocellulite appearance. The groove sign is used to describe a depression along the course of a vein and/or between muscle groups. Both clinical presentations may manifest later in the course of disease.2
Histopathology
Su and Person5 described 3 main characteristics of MP that stand out histopathologically. First, there is thickening and hyalinization of collagen bundles in the deep dermis, subcutis, and fascia that are prominent between the junction of the dermis and subcutaneous fat. There also are fewer sebaceous glands and hair follicles. Second, MP presents with an increased inflammatory cell infiltrate composed mainly of lymphocytes located around small blood vessels and the interstitium. In some patients, the lymphocytes consist predominantly of collections of plasma cells. Third, MP contains deposits of mucin in deep portions of the dermis with occasional eosinophils and mast cells. The presence of eosinophils allows EF to be a part of this spectrum and to be included as a differential diagnosis.5 Eosinophilic fasciitis has a similar presentation to MP because the fibrosis affects the dermis, subcutaneous fat, and underlying structures.20 Although EF presents with the histopathologic characteristic of fascial fibrosis, a clear distinction between EF and morphea has not been established in the literature. Some authors classify EF as a variant of morphea, whereas others consider it as its own entity. We believe EF is its own entity. Eosinophilic fasciitis can be distinguished from morphea because 60% to 80% of patients with EF have peripheral eosinophilia and 20% to 70% of patients with EF have hypergammaglobulinemia. Additionally, morphea does not present as symmetrically or abruptly as EF.21
Treatment
To date, there is conflicting literature regarding the treatment regimen for MP. There is controversy regarding whether MP responds to corticosteroids.19 Different treatment regimens have been discussed for LS, but there is a lack of reports specifically describing therapies for MP and SMP. Because MP and SMP fall under the umbrella of LS, many investigators have reported using the following treatment regimens for patients with MP and SMP: bosentan,22 D-penicillamine,23 phototherapy,24-26 retinoids,26 oral steroids,27 methotrexate,27-29 vitamin D3 (oral calcitriol),30,31 cyclosporine,32 mycophenolate mofetil,33 and extracorporeal photochemotherapy.34
Falanga and Medsger23 reported 64% (7/11) treatment success with D-penicillamine in patients who exhibited severe LS. Psoralen plus UVA,24 methoxsalen, and UVA1 therapy are widely used in the treatment of LS.25 Kreuter et al25 advocated for phototherapy as the first approach in the management of LS after reporting improvement in all participants in their study (N=64), 2 participants with deep morphea while the rest exhibited other forms of morphea. Ozdemir et al26 proposed that retinoic acid combined with psoralen plus UVA is a good treatment choice for plaque-type LS; however, UVA only has the ability to target the epidermis and dermis, which may not be useful for deep forms of morphea.
Several studies have shown positive results in patients treated with methylprednisolone combined with low-dose methotrexate sodium.27-29 Kreuter et al30 and Elst et al31 proposed that calcitriol is effective in treating LS, whereas Hulshof et al35 indicated that it is not. It should be noted that none of these studies specifically mentioned MP. Martini et al33 demonstrated success with mycophenolate mofetil in the treatment of 10 LS patients who were resistant to methotrexate sodium and corticosteroids. Although none of the participants in the study had MP, 2 patients had disabling pansclerotic morphea, 3 had generalized morphea, and 5 had linear scleroderma (morphea en coup de sabre) affecting the limbs (n=2) and face (n=3).33 Because there is no established therapy or consensus for the treatment of MP, we have found success in starting with corticosteroids and then trying alternative therapies.
Prognosis
Morphea has transitioned into systemic scleroderma in a small number of reported cases.10,16,20,36 Therefore, patient follow-up is imperative to consistently identify systemic evolution. Although visceral complications are rare in the setting of LS, associated clinical findings have been reported, including arthralgia, arthritis, contractures, and carpal tunnel syndrome, as well as pulmonary, esophageal, and cardiac abnormalities.7,34
Conclusion
The morphologic features observed in our patient appear to correspond most closely to the type of lesion described by Su and Person5 and Whittaker et al.6 Although our case was clinically difficult to distinguish from linear morphea, the histology suggested SMP over other causes. If our patient’s SMP progressed to the joints, physical therapy would be needed to maintain range of motion and function of the extremities,2 and mandatory long-term follow-up would be required due to the risk for relapse after discontinuation of therapy. Our case highlights the inherent difficulties in the treatment of MP. Due to limited reports of SMP and MP in the literature as well as the conflicting views regarding effective and appropriate treatment options, additional investigation of these conditions and therapeutic options are necessary to further understand this debilitating condition.
Case Report
A 50-year-old white woman presented to our clinic for evaluation of what she described as a “very hard red line” on the right upper arm. The lesion had developed suddenly overnight. Several months prior to presentation the patient sustained trauma to the same area in a car accident and she thought the lesion might be related to the resulting nerve damage. Initially she presented to her primary care physician who used ultrasonography of the area to rule out muscle or bone involvement. The patient presented to our dermatology clinic 2 months later with an 18×4-cm, brownish, rectangular, sclerotic, bound-down, hypertrophic plaque that started on the right mid forearm and extended to the right shoulder (Figure 1). Her medical history was notable for high blood pressure, which was controlled with valsartan.
A review of systems was unremarkable. Physical examination revealed a well-developed, well-nourished woman. Examination of the right arm revealed no motion restriction (muscle strength, 5/5) and no pain; however, she described a burning sensation at the site of the lesion. She reported no allergies. A 4-mm punch biopsy was performed and laboratory tests were ordered including an antinuclear antibody (ANA) test with reflex, double-stranded DNA test, DNA antitopoisomerase antibodies test, and Lyme titers (IgM and IgG). Initially, the patient was treated with calcipotriene 0.005%–betamethasone dipropionate 0.064% ointment twice daily; she also was treated empirically for Lyme disease with doxycycline 50 mg twice daily. All laboratory tests were within reference range, and a punch biopsy revealed markedly thickened fibrous septa within the subcutaneous fat. At the edge of the septa there were nodular aggregates of lymphocytes. Due to clinical presentation, laboratory data, and histopathology, solitary morphea profunda (SMP) was diagnosed.
Following histopathologic examination (Figure 2), the patient was instructed to continue treatment with calcipotriene–betamethasone dipropionate as well as doxycycline. A trial of prednisone and/or hydroxychloroquine also was considered pending her response to the initial treatment. At approximately 1-month follow-up, remarkable improvement of the lesion was noted.
Comment
|
There is limited literature available about the diagnosis and treatment of SMP. Our case prompted us to further examine the data to emphasize the necessity of greater research surrounding SMP.
Classification of SMP
Morphea is a localized form of scleroderma, an inflammatory disease that primarily affects the dermis but can extend down to the bone and also can limit motion. There are several types of morphea that are classified according to the extent, depth, and distribution of the lesions, including plaque, generalized, bullous, linear (including morphea en coup de sabre), guttate, nodular, and deep morphea.1,2 Other subtypes have been described including subcutaneous morphea, eosinophilic fasciitis (EF), pansclerotic morphea, and morphea profunda.3 Linear and deep morphea are characterized by involvement of the deep dermis, subcutaneous tissue, fascia, and/or superficial muscle.2,4
In 1981, Su and Person5 first described morphea profunda (MP). In their study, 22 of 23 patients presented with generalized MP. One patient developed a single lesion,5 which ultimately was classified as SMP by Whittaker et al6 in 1989.
Epidemiology
Morphea profunda occurs more frequently in females than in males, with sclerosis manifesting over a period of several months.7 In 2004, Azad et al4 suggested that only 9 cases of SMP had been reported in the literature. Although there is insignificant data to determine the epidemiology of SMP, the authors concluded that it most commonly affects middle-aged individuals with equal sex distribution.4 The single plaque in patients with SMP most commonly presents on the shoulder, back, or neck or in the paraspinal area.
Etiology
Because of the limited amount of literature on MP, a definitive etiology is unknown, but investigators have cited many possible causes. Genetic, autoimmune, hormonal, traumatic,8 vaccination,2,8 radiation,9 viral, neurogenic, and vascular factors all have been implicated,10 as well as infectious agents such as Borrelia burgdorferi in the United States,11,12Borrelia afzelii in Europe,2 and Borrelia garinii in Japan.2 Because our patient experienced a traumatic episode several months prior to presentation, it is important to investigate trauma as a likely etiology. Furthermore, traumatic events have been reported in 23% of children with linear morphea.13
Diagnostic Studies
Morphea profunda is diagnosed clinically and skin biopsy can be used for confirmation. Biopsy requires deep excision down to the muscle, which can aid in determining if the fascia is incorporated. Elevated levels of IgG and IgM have been detected in deep and linear morphea and are known to correlate with disease activity and the development of joint contractures in linear morphea.2 Serum procollagen type I has been considered by some as a useful indicator of disease severity.14 Elevated serum levels of antifibrillin-1 antibodies also have been demonstrated in patients with localized scleroderma (LS).15 Radiography and magnetic resonance imaging can be used for monitoring and analyzing lesion depth. Furthermore, magnetic resonance imaging can be used to differentiate MP from EF.2
The presence of ANAs in LS is controversial. According to Nguyen et al,2 ANAs are present in approximately 46% to 80% of patients with morphea, with a higher prevalence in patients with generalized, linear, and deep subtypes. However, Savoia et al16 found that patients with morphea typically do not present with ANAs; rather ANAs usually are found in patients with EF.
Pathogenesis
After the inflammatory phase in LS, fibrillar collagen types I and III accumulate, causing dermal fibrosis. The extracellular matrix increases due to the activation of connective tissue growth factor, transforming growth factor β (TGF-β), TGF-β receptors, IL-4, and several other cytokines.17 The TGF-β receptors combine with the connective tissue growth factor released by fibroblasts to create an autocrine production loop that causes fibroblast and matrix production.17 As the inflammation progresses to sclerosis, the CD34 count decreases.18
Physical Findings
In patients with MP, lesions manifest as thickened taut skin with deep, solitary, and sclerotic indurated plaques. Clinically, plaques are mildly inflamed, hyperpigmented, symmetric, and somewhat ill defined, and the skin feels thickened and bound to the underlying fascia and muscle. Plaques usually are smooth and shiny, but areas of both dermal and subcutaneous atrophy may be present, particularly in chronic lesions.19 Morphea profunda also can be described as having a cobblestone or pseudocellulite appearance. The groove sign is used to describe a depression along the course of a vein and/or between muscle groups. Both clinical presentations may manifest later in the course of disease.2
Histopathology
Su and Person5 described 3 main characteristics of MP that stand out histopathologically. First, there is thickening and hyalinization of collagen bundles in the deep dermis, subcutis, and fascia that are prominent between the junction of the dermis and subcutaneous fat. There also are fewer sebaceous glands and hair follicles. Second, MP presents with an increased inflammatory cell infiltrate composed mainly of lymphocytes located around small blood vessels and the interstitium. In some patients, the lymphocytes consist predominantly of collections of plasma cells. Third, MP contains deposits of mucin in deep portions of the dermis with occasional eosinophils and mast cells. The presence of eosinophils allows EF to be a part of this spectrum and to be included as a differential diagnosis.5 Eosinophilic fasciitis has a similar presentation to MP because the fibrosis affects the dermis, subcutaneous fat, and underlying structures.20 Although EF presents with the histopathologic characteristic of fascial fibrosis, a clear distinction between EF and morphea has not been established in the literature. Some authors classify EF as a variant of morphea, whereas others consider it as its own entity. We believe EF is its own entity. Eosinophilic fasciitis can be distinguished from morphea because 60% to 80% of patients with EF have peripheral eosinophilia and 20% to 70% of patients with EF have hypergammaglobulinemia. Additionally, morphea does not present as symmetrically or abruptly as EF.21
Treatment
To date, there is conflicting literature regarding the treatment regimen for MP. There is controversy regarding whether MP responds to corticosteroids.19 Different treatment regimens have been discussed for LS, but there is a lack of reports specifically describing therapies for MP and SMP. Because MP and SMP fall under the umbrella of LS, many investigators have reported using the following treatment regimens for patients with MP and SMP: bosentan,22 D-penicillamine,23 phototherapy,24-26 retinoids,26 oral steroids,27 methotrexate,27-29 vitamin D3 (oral calcitriol),30,31 cyclosporine,32 mycophenolate mofetil,33 and extracorporeal photochemotherapy.34
Falanga and Medsger23 reported 64% (7/11) treatment success with D-penicillamine in patients who exhibited severe LS. Psoralen plus UVA,24 methoxsalen, and UVA1 therapy are widely used in the treatment of LS.25 Kreuter et al25 advocated for phototherapy as the first approach in the management of LS after reporting improvement in all participants in their study (N=64), 2 participants with deep morphea while the rest exhibited other forms of morphea. Ozdemir et al26 proposed that retinoic acid combined with psoralen plus UVA is a good treatment choice for plaque-type LS; however, UVA only has the ability to target the epidermis and dermis, which may not be useful for deep forms of morphea.
Several studies have shown positive results in patients treated with methylprednisolone combined with low-dose methotrexate sodium.27-29 Kreuter et al30 and Elst et al31 proposed that calcitriol is effective in treating LS, whereas Hulshof et al35 indicated that it is not. It should be noted that none of these studies specifically mentioned MP. Martini et al33 demonstrated success with mycophenolate mofetil in the treatment of 10 LS patients who were resistant to methotrexate sodium and corticosteroids. Although none of the participants in the study had MP, 2 patients had disabling pansclerotic morphea, 3 had generalized morphea, and 5 had linear scleroderma (morphea en coup de sabre) affecting the limbs (n=2) and face (n=3).33 Because there is no established therapy or consensus for the treatment of MP, we have found success in starting with corticosteroids and then trying alternative therapies.
Prognosis
Morphea has transitioned into systemic scleroderma in a small number of reported cases.10,16,20,36 Therefore, patient follow-up is imperative to consistently identify systemic evolution. Although visceral complications are rare in the setting of LS, associated clinical findings have been reported, including arthralgia, arthritis, contractures, and carpal tunnel syndrome, as well as pulmonary, esophageal, and cardiac abnormalities.7,34
Conclusion
The morphologic features observed in our patient appear to correspond most closely to the type of lesion described by Su and Person5 and Whittaker et al.6 Although our case was clinically difficult to distinguish from linear morphea, the histology suggested SMP over other causes. If our patient’s SMP progressed to the joints, physical therapy would be needed to maintain range of motion and function of the extremities,2 and mandatory long-term follow-up would be required due to the risk for relapse after discontinuation of therapy. Our case highlights the inherent difficulties in the treatment of MP. Due to limited reports of SMP and MP in the literature as well as the conflicting views regarding effective and appropriate treatment options, additional investigation of these conditions and therapeutic options are necessary to further understand this debilitating condition.
1. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
2. Nguyen JV, Werth VP, Fett N. Morphea treatment & management. Medscape Web site. http://emedicine.medscape.com/article/1065782-treatment. Updated July 21, 2014. Accessed December 16, 2104.
3. Melani L, Cardinali C, Giomi B, et al. Case study: periodic follow-up is necessary in morphea profunda to identify systemic evolution. Skinmed. 2005;4:188-190.
4. Azad J, Dawn G, Shaffrali FC, et al. Does solitary morphoea profunda progress? Clin Exp Dermatol. 2004;29:25-27.
5. Su WP, Person JR. Morphea profunda. a new concept and a histopathologic study of 23 cases. Am J Dermatopathol. 1981;3:251-260.
6. Whittaker SJ, Smith NP, Jones RR. Solitary morphoea profunda. Br J Dermatol. 1989;120:431-440.
7. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
8. Torrelo A, Suárez J, Colmenero I, et al. Deep morphea after vaccination in two young children. Pediatr Dermatol. 2006;23:484-487.
9. Kreft B, Wohlrab J, Radant K, et al. Unrecognized radiation-induced localized scleroderma: a cause of postoperative wound-healing disorder [published online ahead of print June 22, 2009]. Clin Exp Dermatol. 2009;34:e383-e384.
10. Braun-Falco O, Plewig G, Wolff HH, et al, eds. Dermatology. Berlin, Germany: Springer; 2002.
11. Prinz JC, Kutasi Z, Weisenseel P, et al. “Borrelia-associated early-onset morphea”: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? results of a cohort analysis and presentation of three cases [published online ahead of print November 20, 2008]. J Am Acad Dermatol. 2009;60:248-255.
12. Wojas-Pelc A, Wielowieyska-Szybińska D, Kiełtyka A. Presence of the antinuclear antibodies and antibodies to Borrelia burgdorferi among patients with morphea en plaque, deep linear scleroderma and atrophoderma Pasini-Pierini [in Polish]. Przegl Lek. 2002;59:898-902.
13. Falanga V, Medsger TA Jr, Reichlin M, et al. Linear scleroderma. clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med. 1986;104:849-857.
14. Kikuchi K, Sato S, Kadono T, et al. Serum concentration of procollagen type I carboxyterminal propeptide in localized scleroderma. Arch Dermatol. 1994;130:1269-1272.
15. Arnett FC, Tan FK, Uziel Y, et al. Autoantibodies to the extracellular matrix microfibrillar protein, fibrilin 1, in patients with localized scleroderma. Arthritis Rheum. 1999;42:2656-2659.
16. Savoia P, Zaccagna A, Bernengo MG. Guess what? inflammatory disseminated morphea profunda. Eur J Dermatol. 1999;9:654-656.
17. Igarashi A, Nashiro K, Kikuchi K, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J Invest Dermatol. 1996;106:729-733.
18. Gilmour TK, Wilkinson B, Breit SN, et al. Analysis of dendritic cell populations using a revised histological staging of morphoea. Br J Dermatol. 2000;143:1183-1192.
19. Sayama K, Chen M, Shiraishi S, et al. Morphea profunda. Int J Dermatol. 1991;30:873-875.
20. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
21. Bischoff L, Derk CT. Eosinophilic fasciitis: demographics disease pattern and response to treatment: report of 12 cases and review of the literature. Int J Dermatol. 2008;47:29-35.
22. Roldan R, Morote G, Castro Mdel C, et al. Efficacy of bosentan in treatment of unresponsive cutaneous ulceration in disabling pansclerotic morphea in children. J Rheumatol. 2006;33:2538-2540.
23. Falanga V, Medsger TA Jr. D-penicillamine in the treatment of localized scleroderma. Arch Dermatol. 1990;126:609-612.
24. Breuckmann F, Gambichler T, Altmeyer P, et al. UVA/UVA1 phototherapy and PUVA photochemotherapy in connective tissue diseases and related disorders: a research based review. BMC Dermatol. 2004;4:11.
25. Kreuter A, Hyun J, Stücker M, et al. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma [published online ahead of print January 30, 2006]. J Am Acad Dermatol. 2006;54:440-447.
26. Ozdemir M, Engin B, Toy H, et al. Treatment of plaque-type localized scleroderma with retinoic acid and ultraviolet A plus the photosensitizer psoralen: a case series. J Eur Acad Dermatol Venereol. 2008;22:519-521.
27. Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141:847-852.
28. Kroft EB, Creemers MC, van den Hoogen FH, et al. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study [published online ahead of print February 4, 2009]. Br J Dermatol. 2009;160:1075-1082.
29. Weibel L, Sampaio MC, Visentin MT, et al. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. 2006;155:1013-1020.
30. Kreuter A, Gambichler T, Avermaete A, et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet A1 phototherapy in childhood morphea. Pediatr Dermatol. 2001;18:241-245.
31. Elst EF, Van Suijlekom-Smit LW, Oranje AP. Treatment of linear scleroderma with oral 1,25-dihydroxy vitamin D3 (calcitriol) in seven children. Pediatr Dermatol. 1999;16:53-58.
32. Crespo MP, Mas IB, Díaz JM, et al. Rapid response to cyclosporine and maintenance with methotrexate in linear scleroderma in a young girl. Pediatr Dermatol. 2009;26:118-120.
33. Martini G, Ramanan AV, Falcini F, et al. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil [published online ahead of print August 27, 2009]. Rheumatology (Oxford). 2009;48:1410-1413.
34. Neustadter JH, Samarin F, Carlson KR, et al. Extracorporeal photochemotherapy for generalized deep morphea. Arch Dermatol. 2009;145:127-130.
35. Hulshof MM, Bouwes Bavinck JN, Bergman W, et al. Double-blind, placebo-controlled study of oral calcitriol for the treatment of localized and systemic scleroderma. J Am Acad Dermatol. 2000;43:1017-1023.
36. Toledano C, Rabhi S, Kettaneh A, et al. Localized scleroderma: a series of 52 patients [published online ahead of print September 17, 2008]. Eur J Intern Med. 2009;20:331-336.
1. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
2. Nguyen JV, Werth VP, Fett N. Morphea treatment & management. Medscape Web site. http://emedicine.medscape.com/article/1065782-treatment. Updated July 21, 2014. Accessed December 16, 2104.
3. Melani L, Cardinali C, Giomi B, et al. Case study: periodic follow-up is necessary in morphea profunda to identify systemic evolution. Skinmed. 2005;4:188-190.
4. Azad J, Dawn G, Shaffrali FC, et al. Does solitary morphoea profunda progress? Clin Exp Dermatol. 2004;29:25-27.
5. Su WP, Person JR. Morphea profunda. a new concept and a histopathologic study of 23 cases. Am J Dermatopathol. 1981;3:251-260.
6. Whittaker SJ, Smith NP, Jones RR. Solitary morphoea profunda. Br J Dermatol. 1989;120:431-440.
7. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
8. Torrelo A, Suárez J, Colmenero I, et al. Deep morphea after vaccination in two young children. Pediatr Dermatol. 2006;23:484-487.
9. Kreft B, Wohlrab J, Radant K, et al. Unrecognized radiation-induced localized scleroderma: a cause of postoperative wound-healing disorder [published online ahead of print June 22, 2009]. Clin Exp Dermatol. 2009;34:e383-e384.
10. Braun-Falco O, Plewig G, Wolff HH, et al, eds. Dermatology. Berlin, Germany: Springer; 2002.
11. Prinz JC, Kutasi Z, Weisenseel P, et al. “Borrelia-associated early-onset morphea”: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? results of a cohort analysis and presentation of three cases [published online ahead of print November 20, 2008]. J Am Acad Dermatol. 2009;60:248-255.
12. Wojas-Pelc A, Wielowieyska-Szybińska D, Kiełtyka A. Presence of the antinuclear antibodies and antibodies to Borrelia burgdorferi among patients with morphea en plaque, deep linear scleroderma and atrophoderma Pasini-Pierini [in Polish]. Przegl Lek. 2002;59:898-902.
13. Falanga V, Medsger TA Jr, Reichlin M, et al. Linear scleroderma. clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med. 1986;104:849-857.
14. Kikuchi K, Sato S, Kadono T, et al. Serum concentration of procollagen type I carboxyterminal propeptide in localized scleroderma. Arch Dermatol. 1994;130:1269-1272.
15. Arnett FC, Tan FK, Uziel Y, et al. Autoantibodies to the extracellular matrix microfibrillar protein, fibrilin 1, in patients with localized scleroderma. Arthritis Rheum. 1999;42:2656-2659.
16. Savoia P, Zaccagna A, Bernengo MG. Guess what? inflammatory disseminated morphea profunda. Eur J Dermatol. 1999;9:654-656.
17. Igarashi A, Nashiro K, Kikuchi K, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J Invest Dermatol. 1996;106:729-733.
18. Gilmour TK, Wilkinson B, Breit SN, et al. Analysis of dendritic cell populations using a revised histological staging of morphoea. Br J Dermatol. 2000;143:1183-1192.
19. Sayama K, Chen M, Shiraishi S, et al. Morphea profunda. Int J Dermatol. 1991;30:873-875.
20. Bielsa I, Ariza A. Deep morphea. Semin Cutan Med Surg. 2007;26:90-95.
21. Bischoff L, Derk CT. Eosinophilic fasciitis: demographics disease pattern and response to treatment: report of 12 cases and review of the literature. Int J Dermatol. 2008;47:29-35.
22. Roldan R, Morote G, Castro Mdel C, et al. Efficacy of bosentan in treatment of unresponsive cutaneous ulceration in disabling pansclerotic morphea in children. J Rheumatol. 2006;33:2538-2540.
23. Falanga V, Medsger TA Jr. D-penicillamine in the treatment of localized scleroderma. Arch Dermatol. 1990;126:609-612.
24. Breuckmann F, Gambichler T, Altmeyer P, et al. UVA/UVA1 phototherapy and PUVA photochemotherapy in connective tissue diseases and related disorders: a research based review. BMC Dermatol. 2004;4:11.
25. Kreuter A, Hyun J, Stücker M, et al. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma [published online ahead of print January 30, 2006]. J Am Acad Dermatol. 2006;54:440-447.
26. Ozdemir M, Engin B, Toy H, et al. Treatment of plaque-type localized scleroderma with retinoic acid and ultraviolet A plus the photosensitizer psoralen: a case series. J Eur Acad Dermatol Venereol. 2008;22:519-521.
27. Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141:847-852.
28. Kroft EB, Creemers MC, van den Hoogen FH, et al. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study [published online ahead of print February 4, 2009]. Br J Dermatol. 2009;160:1075-1082.
29. Weibel L, Sampaio MC, Visentin MT, et al. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. 2006;155:1013-1020.
30. Kreuter A, Gambichler T, Avermaete A, et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet A1 phototherapy in childhood morphea. Pediatr Dermatol. 2001;18:241-245.
31. Elst EF, Van Suijlekom-Smit LW, Oranje AP. Treatment of linear scleroderma with oral 1,25-dihydroxy vitamin D3 (calcitriol) in seven children. Pediatr Dermatol. 1999;16:53-58.
32. Crespo MP, Mas IB, Díaz JM, et al. Rapid response to cyclosporine and maintenance with methotrexate in linear scleroderma in a young girl. Pediatr Dermatol. 2009;26:118-120.
33. Martini G, Ramanan AV, Falcini F, et al. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil [published online ahead of print August 27, 2009]. Rheumatology (Oxford). 2009;48:1410-1413.
34. Neustadter JH, Samarin F, Carlson KR, et al. Extracorporeal photochemotherapy for generalized deep morphea. Arch Dermatol. 2009;145:127-130.
35. Hulshof MM, Bouwes Bavinck JN, Bergman W, et al. Double-blind, placebo-controlled study of oral calcitriol for the treatment of localized and systemic scleroderma. J Am Acad Dermatol. 2000;43:1017-1023.
36. Toledano C, Rabhi S, Kettaneh A, et al. Localized scleroderma: a series of 52 patients [published online ahead of print September 17, 2008]. Eur J Intern Med. 2009;20:331-336.
Practice Points
- Localized trauma to the skin may be an inciting event to trigger morphea.
- Morphea is a clinical diagnosis but should be confirmed through biopsy to differentiate it from other similar entities.
Cold Panniculitis: Delayed Onset in an Adult
The panniculitides can be a complex dermatologic entity for both dermatologists and dermatopathologists. The history, clinical examination, and histology need to be correlated to arrive at a differential diagnosis that will ultimately provide a diagnosis for the subcutaneous lesions. Panniculitis is an inflammation of the subcutaneous adipose tissue and can be associated with systemic diseases. According to Peters and Su,1 “Anatomic location of lesions, presence or absence of ulceration, occurrence of lipoatrophy, history of trauma, association with immunologic or metabolic disorders, and age of the patient are important clinical data to consider in conjunction with the microscopic features.” The panniculitides histologic differences may be subtle because they all include septal and lobular components, but one is usually more dominant in leading to a diagnosis along with the clinical findings.2
Cold panniculitis is a form of traumatic panniculitis. We present a unique case of this condition that was caused by use of a cold therapy unit following surgery to relieve pain.
Case Report
A 37-year-old woman presented for a routine postoperative visit 15 days following arthroscopic repair of a superior labrum anterior posterior tear in the left shoulder with a single suture anchor. The patient reported a rash that had developed 10 days postoperatively on the left upper arm. The rash started as red dots that progressively became larger, painful, and warm to the touch. The rash did not spread anywhere else on the patient’s body, and she denied fever, chills, and pruritus. She had tried using diphenhydramine without relief. The only new medication the patient had started prior to the eruption was oxycodone, which was initiated immediately following surgery. Prior to surgery, the entire left upper extremity including the shoulder had been prepared with a preoperative surgical skin antiseptic. There were no visible signs of the antiseptic on the skin at the time of presentation. The patient reported that she had applied a cold therapy unit to the left upper arm over her clothing for 1 hour every night since surgery. The cold therapy unit frequently is used to help decrease postoperative pain, swelling, inflammation, and narcotic use following surgical procedures.
Physical examination revealed multiple well-defined, erythematous, tender, indurated, warm nodules on the lateral aspect of the left upper arm (Figure 1). No other areas of eruption were noted on the body, and there was no swelling of the left elbow, forearm, wrist, or hand. The left upper extremity demonstrated intact sensation, rapid capillary refill, and a palpable radial pulse. Her weight was 230.1 lb with a body mass index of 35.
|
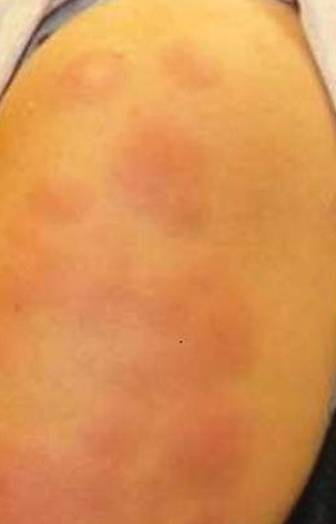
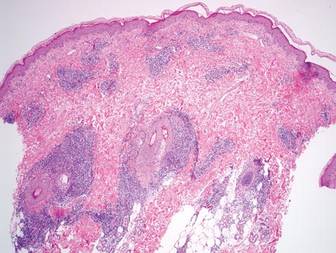
A 5-mm punch biopsy from a nodule on the left upper arm was performed, and pathology demonstrated vacuolar interface changes with patchy parakeratosis, spongiosis, and dyskeratosis on staining with hematoxylin and eosin. Pandermal and subcutaneous perivascular, periadnexal, and mild interstitial lymphohistiocytic infiltrate with occasional neutrophils and eosinophils were noted (Figure 2). The inflammation extended to the subcutaneous fat involving both septae and lobules with a primarily lobular distribution.
Clinical and pathologic correlation was required to arrive at a definitive diagnosis of cold panniculitis. The epidermal and dermal changes were consistent with a pernio or chilblains type of insult, and the septal and lobular panniculitis was indicative of cold panniculitis. The patient was advised to discontinue use of the cold therapy device as well as any other form of icing of the left shoulder or arm. She continued the oxycodone for pain control. Four weeks postoperatively, only desquamation remained where the nodules had previously appeared, which also eventually resolved.
Comment
Infants and small children are more predisposed to cold panniculitis than adults. In their 2008 review, Quesada-Cortés et al3 found the first report of cold panniculitis by Hochsinger in 1902 in a German pediatric journal, followed by reports from Lemez in 1928 and Haxthausen in 1941, which subsequently described similar cases in infants. Adult cases were not reported until 1963 by Solomon and Beerman4 and then in 1980 by Beacham et al.5
Etiologies for children have included popsicles, ice packs applied to the face to control supraventricular tachycardia or to the lower extremities after vaccinations, and cold weather exposure.6 The chemical composition of fat tissue plays a role in pediatric patients. According to Quesada-Cortés et al,3 subcutaneous fat in newborns is rich in saturated oils such as palmitic and stearic acids that have a higher solidification point. A small decrease in an infant’s temperature may result in crystallization of fat. The subcutaneous fat tends to become more unsaturated with aging with more oleic acid, and the solidification temperature diminishes.7
Cryoglobulins and cold agglutinins have not been demonstrated to be a cause of cold panniculitis in infants.7 Severe cold exposure or predisposition to certain conditions such as cryofibrinogenemia may occur in some adult patients. Gender does not seem to be a factor in children; however, in adults, women tend to be more predisposed to cold panniculitis secondary to obesity and participation in activities such as cycling, motorcycling, or horseback riding in cold conditions.3
On clinical examination, cold panniculitis features erythematous, firm, tender nodules on the cheeks and chin in infants and small children.2 These areas often are exposed to cold weather or wind because they typically are not covered with protective clothing.3 Nodules generally occur 1 to 3 days following exposure to cold and usually resolve spontaneously within 2 weeks.8 Popsicle panniculitis is characterized by a reddish discoloration on both cheeks 1 or 2 days after sucking on popsicles or ice cubes. This reaction can be reproduced in a half day by applying an ice cube to the volar forearm for 2 minutes, which can help diagnose and differentiate this subset of cold panniuculits.3 The red area in cold panniculitis eventually turns purple, becomes less indurated, and fades in approximately 3 months, but occasionally residual hyperpigmentation will last for a few months. Ice packs used as treatment of congenital cardiac arrhythmias in some cardiac surgeries and as surface cooling for management of birth asphyxia can produce a similar physical presentation.3
Equestrian panniculitis is characterized by erythematous, violaceous, tender plaques on the upper lateral thighs of young females who participate in horseback riding in the winter while wearing tight-fitting pants.2,5 These plaques typically occur within several hours and over the next week become painful, violaceous, and indurated or develop red nodules or plaques that can ulcerate or become crusted.3 These lesions usually will spontaneously resolve within 3 weeks, but new areas may occur again during the winter on further exposure with occasional persistent hyperpigmentation. These areas usually disappear at the end of winter with warmer weather or when horseback riding is discontinued. Perniosis also needs to be considered in the differential diagnosis due to the location and appearance of the lesions.3
It is important to obtain the correct specimen for biopsy. According to Peters and Su,1 a deep excisional biopsy that includes multiple fat lobules in addition to dermis and epidermis is critical. On histology, cold panniculitis usually demonstrates a primarily lobular inflammation. There typically is a superficial and deep perivascular lymphocytic infiltrate in the papillary dermis with edema noted in the connective tissue around the eccrine glands that can appear similar to perniosis on histopathology.9 Deposition of mucin, focal panniculitis surrounded by fatty tissue without inflammatory changes within the same field, and fat necrosis with pseudocysts and numerous lipophages also are characteristic features of cold panniculitis.10 Needlelike clefts are not present in cold panniculitis but appear in subcutaneous fat necrosis of the newborn.1
Different treatments have been tried, but no substantial impact on the rate of dissipation of the lesions has been noted. The plaques slowly resolve without scarring over 2 to 3 weeks if the cold source is removed.2 Application of a heating pad to the affected area has been used with limited success. Vasodilators such as nifedipine have been used but have not been found to be effective.3 Antihistamines also have failed to control the lesions.11
Treatment of cold panniculitis is based on the prevention of further insult versus trying to cure the condition. Avoidance of cold and wind exposure as well as direct contact with ice are key methods in preventing cold panniculitis.
Our patient’s presentation of this condition was unique. Although cold panniculitis lesions usually develop 1 to 3 days after cold exposure, our patient did not develop lesions until 10 days following surgery. The cold therapy unit used by our patient was evaluated in our office and also by the manufacturer and was found to be functioning normally with no defects. The late onset of the lesions was attributed to limited application of the cold therapy unit; our patient used it for only 1 hour every night, whereas application for 6 to 8 hours continuously is normally recommended. The lesions may have occurred sooner had the patient been using a solid ice pack versus the continuous cold circulating water of the cold therapy unit. Pathology was consistent with the patient’s history and physical examination indicating a diagnosis of cold panniculitis. The challenge of treatment was to alleviate the pain of the lesions as well as the postoperative shoulder pain without the aid of any form of cold therapy. The patient only needed a tincture of time, as the lesions resolved after 4 weeks. Patient education was provided on future prevention of this condition by avoiding exposure to cold or applying cold packs directly to the skin.
Acknowledgment
The authors thank the staff at the Office of Scientific Writing and Publication at the Marshfield Clinic Research Foundation, Wisconsin, for their editorial assistance in the preparation of this manuscript.
1. Peters MS, Su WP. Panniculitis. Dermatol Clin. 1992;10:37-57.
2. Patterson JW. Panniculitis. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. St. Louis, MO: Mosby Elsevier; 2008:1515-1530.
3. Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489.
4. Solomon LM, Beerman H. Cold panniculitis. Arch Dermatol. 1963;88:897-900.
5. Beacham BE, Cooper PH, Buchanan CS, et al. Equestrian cold panniculitis in women. Arch Dermatol. 1980;116:1025-1027.
6. Ter Poorten MC, Thiers BH. Panniculitis. Dermatol Clin. 2002;20:421-433.
7. Ter Poorten JC, Hebert AA, Ilkiw R. Cold panniculitis in a neonate. J Am Acad Dermatol. 1995;33(2, pt 2):383-385.
8. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
9. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
10. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22:530-549.
11. Duncan WC, Freeman RG, Heaton CL. Cold panniculitis. Arch Dermatol. 1966;94:722-724.
The panniculitides can be a complex dermatologic entity for both dermatologists and dermatopathologists. The history, clinical examination, and histology need to be correlated to arrive at a differential diagnosis that will ultimately provide a diagnosis for the subcutaneous lesions. Panniculitis is an inflammation of the subcutaneous adipose tissue and can be associated with systemic diseases. According to Peters and Su,1 “Anatomic location of lesions, presence or absence of ulceration, occurrence of lipoatrophy, history of trauma, association with immunologic or metabolic disorders, and age of the patient are important clinical data to consider in conjunction with the microscopic features.” The panniculitides histologic differences may be subtle because they all include septal and lobular components, but one is usually more dominant in leading to a diagnosis along with the clinical findings.2
Cold panniculitis is a form of traumatic panniculitis. We present a unique case of this condition that was caused by use of a cold therapy unit following surgery to relieve pain.
Case Report
A 37-year-old woman presented for a routine postoperative visit 15 days following arthroscopic repair of a superior labrum anterior posterior tear in the left shoulder with a single suture anchor. The patient reported a rash that had developed 10 days postoperatively on the left upper arm. The rash started as red dots that progressively became larger, painful, and warm to the touch. The rash did not spread anywhere else on the patient’s body, and she denied fever, chills, and pruritus. She had tried using diphenhydramine without relief. The only new medication the patient had started prior to the eruption was oxycodone, which was initiated immediately following surgery. Prior to surgery, the entire left upper extremity including the shoulder had been prepared with a preoperative surgical skin antiseptic. There were no visible signs of the antiseptic on the skin at the time of presentation. The patient reported that she had applied a cold therapy unit to the left upper arm over her clothing for 1 hour every night since surgery. The cold therapy unit frequently is used to help decrease postoperative pain, swelling, inflammation, and narcotic use following surgical procedures.
Physical examination revealed multiple well-defined, erythematous, tender, indurated, warm nodules on the lateral aspect of the left upper arm (Figure 1). No other areas of eruption were noted on the body, and there was no swelling of the left elbow, forearm, wrist, or hand. The left upper extremity demonstrated intact sensation, rapid capillary refill, and a palpable radial pulse. Her weight was 230.1 lb with a body mass index of 35.
|
A 5-mm punch biopsy from a nodule on the left upper arm was performed, and pathology demonstrated vacuolar interface changes with patchy parakeratosis, spongiosis, and dyskeratosis on staining with hematoxylin and eosin. Pandermal and subcutaneous perivascular, periadnexal, and mild interstitial lymphohistiocytic infiltrate with occasional neutrophils and eosinophils were noted (Figure 2). The inflammation extended to the subcutaneous fat involving both septae and lobules with a primarily lobular distribution.
Clinical and pathologic correlation was required to arrive at a definitive diagnosis of cold panniculitis. The epidermal and dermal changes were consistent with a pernio or chilblains type of insult, and the septal and lobular panniculitis was indicative of cold panniculitis. The patient was advised to discontinue use of the cold therapy device as well as any other form of icing of the left shoulder or arm. She continued the oxycodone for pain control. Four weeks postoperatively, only desquamation remained where the nodules had previously appeared, which also eventually resolved.
Comment
Infants and small children are more predisposed to cold panniculitis than adults. In their 2008 review, Quesada-Cortés et al3 found the first report of cold panniculitis by Hochsinger in 1902 in a German pediatric journal, followed by reports from Lemez in 1928 and Haxthausen in 1941, which subsequently described similar cases in infants. Adult cases were not reported until 1963 by Solomon and Beerman4 and then in 1980 by Beacham et al.5
Etiologies for children have included popsicles, ice packs applied to the face to control supraventricular tachycardia or to the lower extremities after vaccinations, and cold weather exposure.6 The chemical composition of fat tissue plays a role in pediatric patients. According to Quesada-Cortés et al,3 subcutaneous fat in newborns is rich in saturated oils such as palmitic and stearic acids that have a higher solidification point. A small decrease in an infant’s temperature may result in crystallization of fat. The subcutaneous fat tends to become more unsaturated with aging with more oleic acid, and the solidification temperature diminishes.7
Cryoglobulins and cold agglutinins have not been demonstrated to be a cause of cold panniculitis in infants.7 Severe cold exposure or predisposition to certain conditions such as cryofibrinogenemia may occur in some adult patients. Gender does not seem to be a factor in children; however, in adults, women tend to be more predisposed to cold panniculitis secondary to obesity and participation in activities such as cycling, motorcycling, or horseback riding in cold conditions.3
On clinical examination, cold panniculitis features erythematous, firm, tender nodules on the cheeks and chin in infants and small children.2 These areas often are exposed to cold weather or wind because they typically are not covered with protective clothing.3 Nodules generally occur 1 to 3 days following exposure to cold and usually resolve spontaneously within 2 weeks.8 Popsicle panniculitis is characterized by a reddish discoloration on both cheeks 1 or 2 days after sucking on popsicles or ice cubes. This reaction can be reproduced in a half day by applying an ice cube to the volar forearm for 2 minutes, which can help diagnose and differentiate this subset of cold panniuculits.3 The red area in cold panniculitis eventually turns purple, becomes less indurated, and fades in approximately 3 months, but occasionally residual hyperpigmentation will last for a few months. Ice packs used as treatment of congenital cardiac arrhythmias in some cardiac surgeries and as surface cooling for management of birth asphyxia can produce a similar physical presentation.3
Equestrian panniculitis is characterized by erythematous, violaceous, tender plaques on the upper lateral thighs of young females who participate in horseback riding in the winter while wearing tight-fitting pants.2,5 These plaques typically occur within several hours and over the next week become painful, violaceous, and indurated or develop red nodules or plaques that can ulcerate or become crusted.3 These lesions usually will spontaneously resolve within 3 weeks, but new areas may occur again during the winter on further exposure with occasional persistent hyperpigmentation. These areas usually disappear at the end of winter with warmer weather or when horseback riding is discontinued. Perniosis also needs to be considered in the differential diagnosis due to the location and appearance of the lesions.3
It is important to obtain the correct specimen for biopsy. According to Peters and Su,1 a deep excisional biopsy that includes multiple fat lobules in addition to dermis and epidermis is critical. On histology, cold panniculitis usually demonstrates a primarily lobular inflammation. There typically is a superficial and deep perivascular lymphocytic infiltrate in the papillary dermis with edema noted in the connective tissue around the eccrine glands that can appear similar to perniosis on histopathology.9 Deposition of mucin, focal panniculitis surrounded by fatty tissue without inflammatory changes within the same field, and fat necrosis with pseudocysts and numerous lipophages also are characteristic features of cold panniculitis.10 Needlelike clefts are not present in cold panniculitis but appear in subcutaneous fat necrosis of the newborn.1
Different treatments have been tried, but no substantial impact on the rate of dissipation of the lesions has been noted. The plaques slowly resolve without scarring over 2 to 3 weeks if the cold source is removed.2 Application of a heating pad to the affected area has been used with limited success. Vasodilators such as nifedipine have been used but have not been found to be effective.3 Antihistamines also have failed to control the lesions.11
Treatment of cold panniculitis is based on the prevention of further insult versus trying to cure the condition. Avoidance of cold and wind exposure as well as direct contact with ice are key methods in preventing cold panniculitis.
Our patient’s presentation of this condition was unique. Although cold panniculitis lesions usually develop 1 to 3 days after cold exposure, our patient did not develop lesions until 10 days following surgery. The cold therapy unit used by our patient was evaluated in our office and also by the manufacturer and was found to be functioning normally with no defects. The late onset of the lesions was attributed to limited application of the cold therapy unit; our patient used it for only 1 hour every night, whereas application for 6 to 8 hours continuously is normally recommended. The lesions may have occurred sooner had the patient been using a solid ice pack versus the continuous cold circulating water of the cold therapy unit. Pathology was consistent with the patient’s history and physical examination indicating a diagnosis of cold panniculitis. The challenge of treatment was to alleviate the pain of the lesions as well as the postoperative shoulder pain without the aid of any form of cold therapy. The patient only needed a tincture of time, as the lesions resolved after 4 weeks. Patient education was provided on future prevention of this condition by avoiding exposure to cold or applying cold packs directly to the skin.
Acknowledgment
The authors thank the staff at the Office of Scientific Writing and Publication at the Marshfield Clinic Research Foundation, Wisconsin, for their editorial assistance in the preparation of this manuscript.
The panniculitides can be a complex dermatologic entity for both dermatologists and dermatopathologists. The history, clinical examination, and histology need to be correlated to arrive at a differential diagnosis that will ultimately provide a diagnosis for the subcutaneous lesions. Panniculitis is an inflammation of the subcutaneous adipose tissue and can be associated with systemic diseases. According to Peters and Su,1 “Anatomic location of lesions, presence or absence of ulceration, occurrence of lipoatrophy, history of trauma, association with immunologic or metabolic disorders, and age of the patient are important clinical data to consider in conjunction with the microscopic features.” The panniculitides histologic differences may be subtle because they all include septal and lobular components, but one is usually more dominant in leading to a diagnosis along with the clinical findings.2
Cold panniculitis is a form of traumatic panniculitis. We present a unique case of this condition that was caused by use of a cold therapy unit following surgery to relieve pain.
Case Report
A 37-year-old woman presented for a routine postoperative visit 15 days following arthroscopic repair of a superior labrum anterior posterior tear in the left shoulder with a single suture anchor. The patient reported a rash that had developed 10 days postoperatively on the left upper arm. The rash started as red dots that progressively became larger, painful, and warm to the touch. The rash did not spread anywhere else on the patient’s body, and she denied fever, chills, and pruritus. She had tried using diphenhydramine without relief. The only new medication the patient had started prior to the eruption was oxycodone, which was initiated immediately following surgery. Prior to surgery, the entire left upper extremity including the shoulder had been prepared with a preoperative surgical skin antiseptic. There were no visible signs of the antiseptic on the skin at the time of presentation. The patient reported that she had applied a cold therapy unit to the left upper arm over her clothing for 1 hour every night since surgery. The cold therapy unit frequently is used to help decrease postoperative pain, swelling, inflammation, and narcotic use following surgical procedures.
Physical examination revealed multiple well-defined, erythematous, tender, indurated, warm nodules on the lateral aspect of the left upper arm (Figure 1). No other areas of eruption were noted on the body, and there was no swelling of the left elbow, forearm, wrist, or hand. The left upper extremity demonstrated intact sensation, rapid capillary refill, and a palpable radial pulse. Her weight was 230.1 lb with a body mass index of 35.
|
A 5-mm punch biopsy from a nodule on the left upper arm was performed, and pathology demonstrated vacuolar interface changes with patchy parakeratosis, spongiosis, and dyskeratosis on staining with hematoxylin and eosin. Pandermal and subcutaneous perivascular, periadnexal, and mild interstitial lymphohistiocytic infiltrate with occasional neutrophils and eosinophils were noted (Figure 2). The inflammation extended to the subcutaneous fat involving both septae and lobules with a primarily lobular distribution.
Clinical and pathologic correlation was required to arrive at a definitive diagnosis of cold panniculitis. The epidermal and dermal changes were consistent with a pernio or chilblains type of insult, and the septal and lobular panniculitis was indicative of cold panniculitis. The patient was advised to discontinue use of the cold therapy device as well as any other form of icing of the left shoulder or arm. She continued the oxycodone for pain control. Four weeks postoperatively, only desquamation remained where the nodules had previously appeared, which also eventually resolved.
Comment
Infants and small children are more predisposed to cold panniculitis than adults. In their 2008 review, Quesada-Cortés et al3 found the first report of cold panniculitis by Hochsinger in 1902 in a German pediatric journal, followed by reports from Lemez in 1928 and Haxthausen in 1941, which subsequently described similar cases in infants. Adult cases were not reported until 1963 by Solomon and Beerman4 and then in 1980 by Beacham et al.5
Etiologies for children have included popsicles, ice packs applied to the face to control supraventricular tachycardia or to the lower extremities after vaccinations, and cold weather exposure.6 The chemical composition of fat tissue plays a role in pediatric patients. According to Quesada-Cortés et al,3 subcutaneous fat in newborns is rich in saturated oils such as palmitic and stearic acids that have a higher solidification point. A small decrease in an infant’s temperature may result in crystallization of fat. The subcutaneous fat tends to become more unsaturated with aging with more oleic acid, and the solidification temperature diminishes.7
Cryoglobulins and cold agglutinins have not been demonstrated to be a cause of cold panniculitis in infants.7 Severe cold exposure or predisposition to certain conditions such as cryofibrinogenemia may occur in some adult patients. Gender does not seem to be a factor in children; however, in adults, women tend to be more predisposed to cold panniculitis secondary to obesity and participation in activities such as cycling, motorcycling, or horseback riding in cold conditions.3
On clinical examination, cold panniculitis features erythematous, firm, tender nodules on the cheeks and chin in infants and small children.2 These areas often are exposed to cold weather or wind because they typically are not covered with protective clothing.3 Nodules generally occur 1 to 3 days following exposure to cold and usually resolve spontaneously within 2 weeks.8 Popsicle panniculitis is characterized by a reddish discoloration on both cheeks 1 or 2 days after sucking on popsicles or ice cubes. This reaction can be reproduced in a half day by applying an ice cube to the volar forearm for 2 minutes, which can help diagnose and differentiate this subset of cold panniuculits.3 The red area in cold panniculitis eventually turns purple, becomes less indurated, and fades in approximately 3 months, but occasionally residual hyperpigmentation will last for a few months. Ice packs used as treatment of congenital cardiac arrhythmias in some cardiac surgeries and as surface cooling for management of birth asphyxia can produce a similar physical presentation.3
Equestrian panniculitis is characterized by erythematous, violaceous, tender plaques on the upper lateral thighs of young females who participate in horseback riding in the winter while wearing tight-fitting pants.2,5 These plaques typically occur within several hours and over the next week become painful, violaceous, and indurated or develop red nodules or plaques that can ulcerate or become crusted.3 These lesions usually will spontaneously resolve within 3 weeks, but new areas may occur again during the winter on further exposure with occasional persistent hyperpigmentation. These areas usually disappear at the end of winter with warmer weather or when horseback riding is discontinued. Perniosis also needs to be considered in the differential diagnosis due to the location and appearance of the lesions.3
It is important to obtain the correct specimen for biopsy. According to Peters and Su,1 a deep excisional biopsy that includes multiple fat lobules in addition to dermis and epidermis is critical. On histology, cold panniculitis usually demonstrates a primarily lobular inflammation. There typically is a superficial and deep perivascular lymphocytic infiltrate in the papillary dermis with edema noted in the connective tissue around the eccrine glands that can appear similar to perniosis on histopathology.9 Deposition of mucin, focal panniculitis surrounded by fatty tissue without inflammatory changes within the same field, and fat necrosis with pseudocysts and numerous lipophages also are characteristic features of cold panniculitis.10 Needlelike clefts are not present in cold panniculitis but appear in subcutaneous fat necrosis of the newborn.1
Different treatments have been tried, but no substantial impact on the rate of dissipation of the lesions has been noted. The plaques slowly resolve without scarring over 2 to 3 weeks if the cold source is removed.2 Application of a heating pad to the affected area has been used with limited success. Vasodilators such as nifedipine have been used but have not been found to be effective.3 Antihistamines also have failed to control the lesions.11
Treatment of cold panniculitis is based on the prevention of further insult versus trying to cure the condition. Avoidance of cold and wind exposure as well as direct contact with ice are key methods in preventing cold panniculitis.
Our patient’s presentation of this condition was unique. Although cold panniculitis lesions usually develop 1 to 3 days after cold exposure, our patient did not develop lesions until 10 days following surgery. The cold therapy unit used by our patient was evaluated in our office and also by the manufacturer and was found to be functioning normally with no defects. The late onset of the lesions was attributed to limited application of the cold therapy unit; our patient used it for only 1 hour every night, whereas application for 6 to 8 hours continuously is normally recommended. The lesions may have occurred sooner had the patient been using a solid ice pack versus the continuous cold circulating water of the cold therapy unit. Pathology was consistent with the patient’s history and physical examination indicating a diagnosis of cold panniculitis. The challenge of treatment was to alleviate the pain of the lesions as well as the postoperative shoulder pain without the aid of any form of cold therapy. The patient only needed a tincture of time, as the lesions resolved after 4 weeks. Patient education was provided on future prevention of this condition by avoiding exposure to cold or applying cold packs directly to the skin.
Acknowledgment
The authors thank the staff at the Office of Scientific Writing and Publication at the Marshfield Clinic Research Foundation, Wisconsin, for their editorial assistance in the preparation of this manuscript.
1. Peters MS, Su WP. Panniculitis. Dermatol Clin. 1992;10:37-57.
2. Patterson JW. Panniculitis. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. St. Louis, MO: Mosby Elsevier; 2008:1515-1530.
3. Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489.
4. Solomon LM, Beerman H. Cold panniculitis. Arch Dermatol. 1963;88:897-900.
5. Beacham BE, Cooper PH, Buchanan CS, et al. Equestrian cold panniculitis in women. Arch Dermatol. 1980;116:1025-1027.
6. Ter Poorten MC, Thiers BH. Panniculitis. Dermatol Clin. 2002;20:421-433.
7. Ter Poorten JC, Hebert AA, Ilkiw R. Cold panniculitis in a neonate. J Am Acad Dermatol. 1995;33(2, pt 2):383-385.
8. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
9. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
10. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22:530-549.
11. Duncan WC, Freeman RG, Heaton CL. Cold panniculitis. Arch Dermatol. 1966;94:722-724.
1. Peters MS, Su WP. Panniculitis. Dermatol Clin. 1992;10:37-57.
2. Patterson JW. Panniculitis. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. St. Louis, MO: Mosby Elsevier; 2008:1515-1530.
3. Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489.
4. Solomon LM, Beerman H. Cold panniculitis. Arch Dermatol. 1963;88:897-900.
5. Beacham BE, Cooper PH, Buchanan CS, et al. Equestrian cold panniculitis in women. Arch Dermatol. 1980;116:1025-1027.
6. Ter Poorten MC, Thiers BH. Panniculitis. Dermatol Clin. 2002;20:421-433.
7. Ter Poorten JC, Hebert AA, Ilkiw R. Cold panniculitis in a neonate. J Am Acad Dermatol. 1995;33(2, pt 2):383-385.
8. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
9. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
10. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22:530-549.
11. Duncan WC, Freeman RG, Heaton CL. Cold panniculitis. Arch Dermatol. 1966;94:722-724.
Practice Points
- Cold panniculitis is a form of traumatic panniculitis.
- Cold panniculitis often appears on the cheeks and chin, areas that are exposed to cold weather or wind because they are not covered with protective clothing, in infants and small children.
- Treatment of cold panniculitis is based on the prevention of further insult.
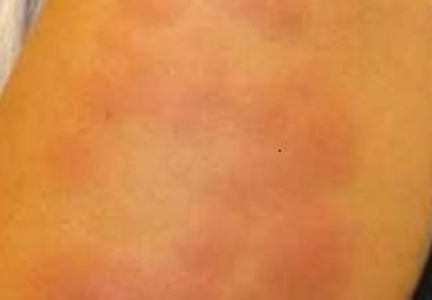
Autosomal-Dominant Familial Angiolipomatosis
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
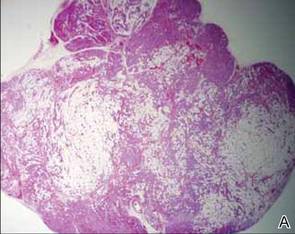
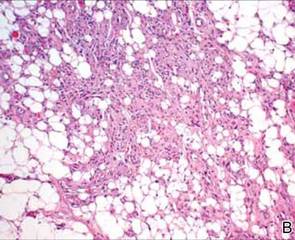
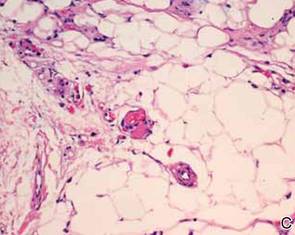
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Practice Points
- Dermatologists should be familiar with the clinical and histological features of angiolipomas along with their potential inheritance patterns.
- Familial angiolipomatosis is a rare condition characterized by multiple angiolipomas that has been described as having an autosomal-recessive transmission pattern. Autosomal-dominant inheritance also may occur, as illustrated in the current case report.
- Awareness of the autosomal-dominant form of this entity is important to prevent its misdiagnosis as
neurofibromatosis type I, which has a similar family history and clinical presentation.
Easy bruising • low platelet count • recent cold-like illness • Dx?
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
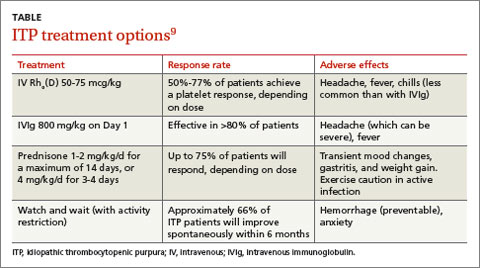
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
Hyperthyroidism • myalgia • rapidly progressing paralysis • Dx?
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
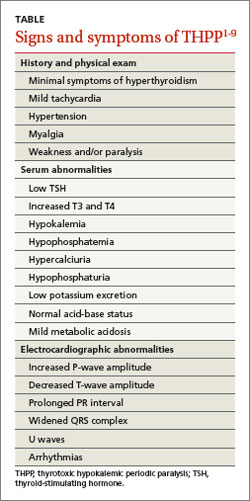
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.

Case Report: Conus Medullaris Syndrome From Spinal Metastasis
Case
A 46-year-old white woman with sudden onset of numbness in her lower extremities and inability to ambulate was transported to the ED via emergency medical services. At the onset of symptoms, the patient reported a feeling of “heaviness” in her lower extremities, which was greater on the left side than the right. After an unsuccessful attempt at ambulation, she subsequently presented to a community hospital where she could no longer move her left lower extremity. Upon evaluation, the patient was found to have progressive neurological deficits and was transferred by ambulance to the authors’ tertiary medical center for definitive management.
A review of the patient’s recent symptoms indicated that she had also experienced lower abdominal paresthesias of 5 days’ duration. She described this sensation as sharp, numb, and constant since its onset and unrelieved with the use of a muscle relaxant at home. She further noted that the pain became worse with movement, having no palliative modifying factors. Upon further questioning, the patient acknowledged recent urinary incontinence of unknown duration, nausea, and current menstruation. She denied any recent injury or illness.
Her past medical history was unknown, and she stated that she had not seen a physician in several years. The patient’s surgical history included a tonsillectomy and an appendectomy at a young age. She had no known drug allergies. Although she denied the use of medications, electronic medical records show that the patient had been prescribed baclofen, hydrochlorothiazide, metoprolol, and tramadol. She was unaware of her family’s medical history and denied use of tobacco, alcohol, or illicit drugs.
Upon physical examination, the patient’s vital signs were: blood pressure, 161/99 mm Hg; heart rate, 103 beats/minute; respiratory rate, 16 breaths/minute; oxygen saturation, 97% on room air; and temperature, 97.0°F. She appeared to be a middle-aged obese woman in no apparent distress and was alert with normal mentation, lying comfortably on the gurney.
The head and neck examinations were normal. Lung auscultation demonstrated equal and unlabored breath sounds bilaterally with no adventitious sounds. Incidentally, it was noted at this time that the left breast had a significantly large fungating mass about the areola and within the deep tissue that was visually evidenced by prominent erythema and classic peau d’orange skin. The right breast had minimal skin involvement with a smaller palpable mass below the dermal surface. Both breast masses and enlarged axillary lymph nodes on the left were nontender. The cardiovascular examination demonstrated mild tachycardia with normal heart sounds, no extremity edema, and normal pulses throughout. The gastrointestinal examination had normal borborygmus with mild infraabdominal tenderness to palpation superficially over a nondistended abdomen. Neither organomegaly, hernia, nor masses were appreciated. In addition to urinary incontinence, the patient also had fecal incontinence, which correlated with diminished tone on digital rectal examination.

Neurological sensation was intact in all extremities and no deficits were noted in the cranial nerves. Patellar and ankle tendon-testing demonstrated left-sided hyperreflexia with ipsilateral Babinski reflex exhibiting up-going toes. Musculoskeletal weakness was grossly noted in the left lower extremity to be +2/5, whereas the right lower limb had +4/5 strength. Palpation of the thoracic and lumbar spines did not elicit tenderness. Aside from the aforementioned observations, no additional integumentary findings were noted.
The patient was given oxygen by nasal cannula, connected to cardiac monitoring and pulse oximetry. A urinary catheter was inserted, and she was given parenteral dexamethasone,3 morphine sulfate, ondansetron, and normal saline. An electrocardiogram showed a normal sinus rhythm. A chest X-ray and basic blood analysis were ordered in preparation for the likelihood of surgical management. Neurosurgery and radiology were consulted. Emergent magnetic resonance imaging (MRI) of the cervical, thoracic, and lumbar spine with and without contrast was obtained to rule out SCC.
The MRI of the spine revealed pathologic fractures leading to cord compression at T9 and spinal stenosis at the L2 segment (Figure 1); diffuse bone metastasis of the spine was also observed. Subsequent surgical decompressive laminectomy from T7 to L3 was performed without complication. Despite the reportedly poor outcome in CMS,2,4-6 the patient demonstrated a moderate return of strength, sensation, and function within the first month of postoperative follow-up. At 3 months, she had minimal subjective and objective deficits and was ambulating without difficulty. She denied urinary and fecal incontinence during these periods. The biopsied breast mass was determined to be stage IV infiltrating ductal carcinoma mucinous type, for which she was followed by an oncologist and received radiation and chemotherapy.
Discussion
The patient’s chief complaint of lower extremity muscle weakness was a clinical emergency that merited thorough investigation in a timely manner to preserve limb function. Since her medical history did not provide pathologic insight concerning her condition, physical examination by emergency personnel served as the founding evidence for this patient’s diagnosis. Decreased muscle tone of the lower extremities and rectal sphincter raised suspicion for a neurological etiology. These symptoms, along with hyperreflexia, the presence of a Babinski sign, and dual-system incontinence, were suggestive of an underlying central nervous system lesion. Of note, urinary complaints commonly result from retention leading to overflow incontinence, a time-dependent symptom that may not be experienced before presentation to medical personnel. Urinary retention is one of the most consistent findings in patients with CMS and SCC, with a relative prevalence of 90%.4,7,8
For providers not familiar with CMS presentation, preserved tactile sensation, normoreflexia, and lack of a Babinski sign and/or incontinence are not sufficient indicators to discontinue the consideration of spinal cord lesions in the differential diagnosis and may in fact be misleading.6,9,10 Although the patient’s deficits were not symmetrical as is commonly reported, this did not rule out the diagnosis.
Appropriate diagnosis and treatment of such a rare entity in the emergency setting consists of a high clinical suspicion, MRI of the spine, urgent consultations, and early treatment with parenteral corticosteroids.3,4 The patient did not have a previous diagnosis of breast carcinoma; however, once discovered on examination, the condition became suspect as approximately 80% of patients with SCC have a preexisting cancer. The peak incidence of SCC is in the sixth and seventh decades of life. The most common primary cancers metastasizing to bone are breast, prostate, and lung. When found to affect the spine, roughly 60% will be located in the thoracic spine, 30% at the lumbosacral level, and 10% in the cervical spine.
As demonstrated in this case presentation, a thorough examination cannot be stressed enough in emergent situations. The patient’s dermatological findings and nontender lymphadenopathy were adequately significant to consider the possibility of a metastatic process as the underlying etiology. Although discouraged due to the fast-paced environment of the ED, patients are frequently assessed and examined in street clothing, which in this case, may have masked the underlying cause of the patient’s neurological deficits. As a result, imaging studies, corticosteroid treatment, consultations, and surgical management may have been delayed, leading to a nonreversible outcome for the patient.
Central and Peripheral Nervous System Structures and Deficits
Central and peripheral nervous system structures animate the body through coordinated signaling of upper and lower motor neurons respectively. In most adults, the distal spinal cord terminates at the level of the first or second lumbar vertebrae where the conus medullaris is found, giving rise to S2, S3 and S4 functionality. Lesions at this level exhibit lower motor neuron deficits of the bladder and rectum resulting in incontinence and sexual dysfunction. Deficits of sensorium such as saddle anesthesia or upper motor neuron lesions as evidenced by increased motor tone and abnormal reflexes are not uncommon.1 Branches of the cauda equina extend caudally from the epiconus, a structure proximal to the conus medullaris, as peripheral nervous system branches that innervate spinal cord segments L4 through S1 (Figure 2). Lesions of the epiconus are clinically distinguished by lower motor neuron deficits wherein muscles of the lower extremities are often weakened with potential sparing of the bulbocavernosus and micturition reflexes.2
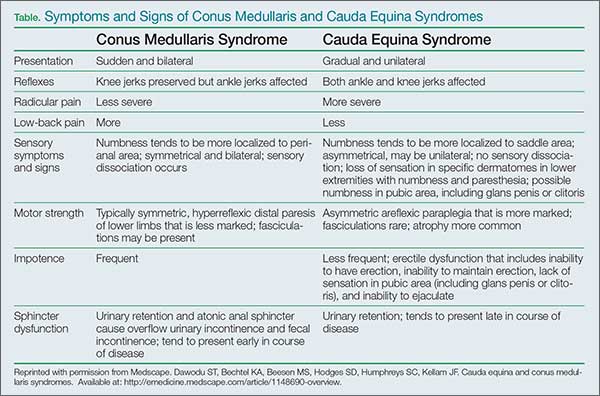
Conclusion
While many EPs are cognizant of cauda equina syndrome and its presentation, CMS is less well known and not commonly documented. Due to symptomatic overlap and epidemiological rarity of these conditions, most of the literature describing these entities combines their discussion. This case contributes to the growing body of literature to assist clinicians in the evaluation and management of CMS.
Dr Batt is an emergency medicine resident, Arrowhead Regional Medical Center, Colton, California. Dr Stone is the emergency medical services director, Travis Air Force Base, Fairfield, California.
- Lewandrowski KU, McLain RF, Lieberman I, Orr D. Cord and cauda equina injury complicating elective orthopedic surgery. Spine (Phila Pa 1976). 2006;31(9):1056-1059.
- Kirshblum S, Anderson K, Krassioukov A, Donovan W. Assessment and classification of traumatic spinal cord injury. In: Kirshblum S, Campagnolo DI, eds. Spinal Cord Medicine. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Ruckdeschel JC. Early detection and treatment of spinal cord compression. Oncology (Williston Park). 2005;19(1):81-86.
- Perron AD, Huff JS. Spinal cord disorders. In: Marx JA, Hockberger RS, Walls RM, et al. Rosen’s Emergency Medicine: Concepts and Clinical Practice. 8th ed. Vol 2. Philadelphia: Mosby/Elsevier, 2013; 1419-1427.
- Wagner R, Jagoda A. Spinal cord syndromes. Emerg Med Clin North Am. 1997;15(3):699-711.
- Sciubba DM, Gokaslan ZL. Diagnosis and management of metastatic spine disease. Surg Oncol. 2006;15(3):141-151.
- Jalloh I, Minhas P. Delays in the treatment of cauda equina syndrome due to its variable clinical features in patients presenting to the emergency department. Emerg Med J. 2007;24(1):33-34.
- Korse NS, Jacobs WCH, Elzevier HW, Vieggeert-Lankamp CL. Complaints of micturition, defecation and sexual function in cauda equina syndrome due to lumbar disk herniation: a systematic review. Eur Spine J. 2013;22(5):1019-1029.
- Dawodu ST, Bechtel KA, Beeson MS, et al. Cauda equina and conus medullaris syndromes. Medscape Web site. http://emedicine.medscape.com/article/1148690-clinical. Accessed September 1, 2014.
- Glick TH, Workman TP, Gaufberg SV. Spinal cord emergencies: false reassurance from reflexes. Acad Emerg Med. 1998;5(10):1041-1043.
Case
A 46-year-old white woman with sudden onset of numbness in her lower extremities and inability to ambulate was transported to the ED via emergency medical services. At the onset of symptoms, the patient reported a feeling of “heaviness” in her lower extremities, which was greater on the left side than the right. After an unsuccessful attempt at ambulation, she subsequently presented to a community hospital where she could no longer move her left lower extremity. Upon evaluation, the patient was found to have progressive neurological deficits and was transferred by ambulance to the authors’ tertiary medical center for definitive management.
A review of the patient’s recent symptoms indicated that she had also experienced lower abdominal paresthesias of 5 days’ duration. She described this sensation as sharp, numb, and constant since its onset and unrelieved with the use of a muscle relaxant at home. She further noted that the pain became worse with movement, having no palliative modifying factors. Upon further questioning, the patient acknowledged recent urinary incontinence of unknown duration, nausea, and current menstruation. She denied any recent injury or illness.
Her past medical history was unknown, and she stated that she had not seen a physician in several years. The patient’s surgical history included a tonsillectomy and an appendectomy at a young age. She had no known drug allergies. Although she denied the use of medications, electronic medical records show that the patient had been prescribed baclofen, hydrochlorothiazide, metoprolol, and tramadol. She was unaware of her family’s medical history and denied use of tobacco, alcohol, or illicit drugs.
Upon physical examination, the patient’s vital signs were: blood pressure, 161/99 mm Hg; heart rate, 103 beats/minute; respiratory rate, 16 breaths/minute; oxygen saturation, 97% on room air; and temperature, 97.0°F. She appeared to be a middle-aged obese woman in no apparent distress and was alert with normal mentation, lying comfortably on the gurney.
The head and neck examinations were normal. Lung auscultation demonstrated equal and unlabored breath sounds bilaterally with no adventitious sounds. Incidentally, it was noted at this time that the left breast had a significantly large fungating mass about the areola and within the deep tissue that was visually evidenced by prominent erythema and classic peau d’orange skin. The right breast had minimal skin involvement with a smaller palpable mass below the dermal surface. Both breast masses and enlarged axillary lymph nodes on the left were nontender. The cardiovascular examination demonstrated mild tachycardia with normal heart sounds, no extremity edema, and normal pulses throughout. The gastrointestinal examination had normal borborygmus with mild infraabdominal tenderness to palpation superficially over a nondistended abdomen. Neither organomegaly, hernia, nor masses were appreciated. In addition to urinary incontinence, the patient also had fecal incontinence, which correlated with diminished tone on digital rectal examination.
Neurological sensation was intact in all extremities and no deficits were noted in the cranial nerves. Patellar and ankle tendon-testing demonstrated left-sided hyperreflexia with ipsilateral Babinski reflex exhibiting up-going toes. Musculoskeletal weakness was grossly noted in the left lower extremity to be +2/5, whereas the right lower limb had +4/5 strength. Palpation of the thoracic and lumbar spines did not elicit tenderness. Aside from the aforementioned observations, no additional integumentary findings were noted.
The patient was given oxygen by nasal cannula, connected to cardiac monitoring and pulse oximetry. A urinary catheter was inserted, and she was given parenteral dexamethasone,3 morphine sulfate, ondansetron, and normal saline. An electrocardiogram showed a normal sinus rhythm. A chest X-ray and basic blood analysis were ordered in preparation for the likelihood of surgical management. Neurosurgery and radiology were consulted. Emergent magnetic resonance imaging (MRI) of the cervical, thoracic, and lumbar spine with and without contrast was obtained to rule out SCC.
The MRI of the spine revealed pathologic fractures leading to cord compression at T9 and spinal stenosis at the L2 segment (Figure 1); diffuse bone metastasis of the spine was also observed. Subsequent surgical decompressive laminectomy from T7 to L3 was performed without complication. Despite the reportedly poor outcome in CMS,2,4-6 the patient demonstrated a moderate return of strength, sensation, and function within the first month of postoperative follow-up. At 3 months, she had minimal subjective and objective deficits and was ambulating without difficulty. She denied urinary and fecal incontinence during these periods. The biopsied breast mass was determined to be stage IV infiltrating ductal carcinoma mucinous type, for which she was followed by an oncologist and received radiation and chemotherapy.
Discussion
The patient’s chief complaint of lower extremity muscle weakness was a clinical emergency that merited thorough investigation in a timely manner to preserve limb function. Since her medical history did not provide pathologic insight concerning her condition, physical examination by emergency personnel served as the founding evidence for this patient’s diagnosis. Decreased muscle tone of the lower extremities and rectal sphincter raised suspicion for a neurological etiology. These symptoms, along with hyperreflexia, the presence of a Babinski sign, and dual-system incontinence, were suggestive of an underlying central nervous system lesion. Of note, urinary complaints commonly result from retention leading to overflow incontinence, a time-dependent symptom that may not be experienced before presentation to medical personnel. Urinary retention is one of the most consistent findings in patients with CMS and SCC, with a relative prevalence of 90%.4,7,8
For providers not familiar with CMS presentation, preserved tactile sensation, normoreflexia, and lack of a Babinski sign and/or incontinence are not sufficient indicators to discontinue the consideration of spinal cord lesions in the differential diagnosis and may in fact be misleading.6,9,10 Although the patient’s deficits were not symmetrical as is commonly reported, this did not rule out the diagnosis.
Appropriate diagnosis and treatment of such a rare entity in the emergency setting consists of a high clinical suspicion, MRI of the spine, urgent consultations, and early treatment with parenteral corticosteroids.3,4 The patient did not have a previous diagnosis of breast carcinoma; however, once discovered on examination, the condition became suspect as approximately 80% of patients with SCC have a preexisting cancer. The peak incidence of SCC is in the sixth and seventh decades of life. The most common primary cancers metastasizing to bone are breast, prostate, and lung. When found to affect the spine, roughly 60% will be located in the thoracic spine, 30% at the lumbosacral level, and 10% in the cervical spine.
As demonstrated in this case presentation, a thorough examination cannot be stressed enough in emergent situations. The patient’s dermatological findings and nontender lymphadenopathy were adequately significant to consider the possibility of a metastatic process as the underlying etiology. Although discouraged due to the fast-paced environment of the ED, patients are frequently assessed and examined in street clothing, which in this case, may have masked the underlying cause of the patient’s neurological deficits. As a result, imaging studies, corticosteroid treatment, consultations, and surgical management may have been delayed, leading to a nonreversible outcome for the patient.
Central and Peripheral Nervous System Structures and Deficits
Central and peripheral nervous system structures animate the body through coordinated signaling of upper and lower motor neurons respectively. In most adults, the distal spinal cord terminates at the level of the first or second lumbar vertebrae where the conus medullaris is found, giving rise to S2, S3 and S4 functionality. Lesions at this level exhibit lower motor neuron deficits of the bladder and rectum resulting in incontinence and sexual dysfunction. Deficits of sensorium such as saddle anesthesia or upper motor neuron lesions as evidenced by increased motor tone and abnormal reflexes are not uncommon.1 Branches of the cauda equina extend caudally from the epiconus, a structure proximal to the conus medullaris, as peripheral nervous system branches that innervate spinal cord segments L4 through S1 (Figure 2). Lesions of the epiconus are clinically distinguished by lower motor neuron deficits wherein muscles of the lower extremities are often weakened with potential sparing of the bulbocavernosus and micturition reflexes.2
Conclusion
While many EPs are cognizant of cauda equina syndrome and its presentation, CMS is less well known and not commonly documented. Due to symptomatic overlap and epidemiological rarity of these conditions, most of the literature describing these entities combines their discussion. This case contributes to the growing body of literature to assist clinicians in the evaluation and management of CMS.
Dr Batt is an emergency medicine resident, Arrowhead Regional Medical Center, Colton, California. Dr Stone is the emergency medical services director, Travis Air Force Base, Fairfield, California.
Case
A 46-year-old white woman with sudden onset of numbness in her lower extremities and inability to ambulate was transported to the ED via emergency medical services. At the onset of symptoms, the patient reported a feeling of “heaviness” in her lower extremities, which was greater on the left side than the right. After an unsuccessful attempt at ambulation, she subsequently presented to a community hospital where she could no longer move her left lower extremity. Upon evaluation, the patient was found to have progressive neurological deficits and was transferred by ambulance to the authors’ tertiary medical center for definitive management.
A review of the patient’s recent symptoms indicated that she had also experienced lower abdominal paresthesias of 5 days’ duration. She described this sensation as sharp, numb, and constant since its onset and unrelieved with the use of a muscle relaxant at home. She further noted that the pain became worse with movement, having no palliative modifying factors. Upon further questioning, the patient acknowledged recent urinary incontinence of unknown duration, nausea, and current menstruation. She denied any recent injury or illness.
Her past medical history was unknown, and she stated that she had not seen a physician in several years. The patient’s surgical history included a tonsillectomy and an appendectomy at a young age. She had no known drug allergies. Although she denied the use of medications, electronic medical records show that the patient had been prescribed baclofen, hydrochlorothiazide, metoprolol, and tramadol. She was unaware of her family’s medical history and denied use of tobacco, alcohol, or illicit drugs.
Upon physical examination, the patient’s vital signs were: blood pressure, 161/99 mm Hg; heart rate, 103 beats/minute; respiratory rate, 16 breaths/minute; oxygen saturation, 97% on room air; and temperature, 97.0°F. She appeared to be a middle-aged obese woman in no apparent distress and was alert with normal mentation, lying comfortably on the gurney.
The head and neck examinations were normal. Lung auscultation demonstrated equal and unlabored breath sounds bilaterally with no adventitious sounds. Incidentally, it was noted at this time that the left breast had a significantly large fungating mass about the areola and within the deep tissue that was visually evidenced by prominent erythema and classic peau d’orange skin. The right breast had minimal skin involvement with a smaller palpable mass below the dermal surface. Both breast masses and enlarged axillary lymph nodes on the left were nontender. The cardiovascular examination demonstrated mild tachycardia with normal heart sounds, no extremity edema, and normal pulses throughout. The gastrointestinal examination had normal borborygmus with mild infraabdominal tenderness to palpation superficially over a nondistended abdomen. Neither organomegaly, hernia, nor masses were appreciated. In addition to urinary incontinence, the patient also had fecal incontinence, which correlated with diminished tone on digital rectal examination.
Neurological sensation was intact in all extremities and no deficits were noted in the cranial nerves. Patellar and ankle tendon-testing demonstrated left-sided hyperreflexia with ipsilateral Babinski reflex exhibiting up-going toes. Musculoskeletal weakness was grossly noted in the left lower extremity to be +2/5, whereas the right lower limb had +4/5 strength. Palpation of the thoracic and lumbar spines did not elicit tenderness. Aside from the aforementioned observations, no additional integumentary findings were noted.
The patient was given oxygen by nasal cannula, connected to cardiac monitoring and pulse oximetry. A urinary catheter was inserted, and she was given parenteral dexamethasone,3 morphine sulfate, ondansetron, and normal saline. An electrocardiogram showed a normal sinus rhythm. A chest X-ray and basic blood analysis were ordered in preparation for the likelihood of surgical management. Neurosurgery and radiology were consulted. Emergent magnetic resonance imaging (MRI) of the cervical, thoracic, and lumbar spine with and without contrast was obtained to rule out SCC.
The MRI of the spine revealed pathologic fractures leading to cord compression at T9 and spinal stenosis at the L2 segment (Figure 1); diffuse bone metastasis of the spine was also observed. Subsequent surgical decompressive laminectomy from T7 to L3 was performed without complication. Despite the reportedly poor outcome in CMS,2,4-6 the patient demonstrated a moderate return of strength, sensation, and function within the first month of postoperative follow-up. At 3 months, she had minimal subjective and objective deficits and was ambulating without difficulty. She denied urinary and fecal incontinence during these periods. The biopsied breast mass was determined to be stage IV infiltrating ductal carcinoma mucinous type, for which she was followed by an oncologist and received radiation and chemotherapy.
Discussion
The patient’s chief complaint of lower extremity muscle weakness was a clinical emergency that merited thorough investigation in a timely manner to preserve limb function. Since her medical history did not provide pathologic insight concerning her condition, physical examination by emergency personnel served as the founding evidence for this patient’s diagnosis. Decreased muscle tone of the lower extremities and rectal sphincter raised suspicion for a neurological etiology. These symptoms, along with hyperreflexia, the presence of a Babinski sign, and dual-system incontinence, were suggestive of an underlying central nervous system lesion. Of note, urinary complaints commonly result from retention leading to overflow incontinence, a time-dependent symptom that may not be experienced before presentation to medical personnel. Urinary retention is one of the most consistent findings in patients with CMS and SCC, with a relative prevalence of 90%.4,7,8
For providers not familiar with CMS presentation, preserved tactile sensation, normoreflexia, and lack of a Babinski sign and/or incontinence are not sufficient indicators to discontinue the consideration of spinal cord lesions in the differential diagnosis and may in fact be misleading.6,9,10 Although the patient’s deficits were not symmetrical as is commonly reported, this did not rule out the diagnosis.
Appropriate diagnosis and treatment of such a rare entity in the emergency setting consists of a high clinical suspicion, MRI of the spine, urgent consultations, and early treatment with parenteral corticosteroids.3,4 The patient did not have a previous diagnosis of breast carcinoma; however, once discovered on examination, the condition became suspect as approximately 80% of patients with SCC have a preexisting cancer. The peak incidence of SCC is in the sixth and seventh decades of life. The most common primary cancers metastasizing to bone are breast, prostate, and lung. When found to affect the spine, roughly 60% will be located in the thoracic spine, 30% at the lumbosacral level, and 10% in the cervical spine.
As demonstrated in this case presentation, a thorough examination cannot be stressed enough in emergent situations. The patient’s dermatological findings and nontender lymphadenopathy were adequately significant to consider the possibility of a metastatic process as the underlying etiology. Although discouraged due to the fast-paced environment of the ED, patients are frequently assessed and examined in street clothing, which in this case, may have masked the underlying cause of the patient’s neurological deficits. As a result, imaging studies, corticosteroid treatment, consultations, and surgical management may have been delayed, leading to a nonreversible outcome for the patient.
Central and Peripheral Nervous System Structures and Deficits
Central and peripheral nervous system structures animate the body through coordinated signaling of upper and lower motor neurons respectively. In most adults, the distal spinal cord terminates at the level of the first or second lumbar vertebrae where the conus medullaris is found, giving rise to S2, S3 and S4 functionality. Lesions at this level exhibit lower motor neuron deficits of the bladder and rectum resulting in incontinence and sexual dysfunction. Deficits of sensorium such as saddle anesthesia or upper motor neuron lesions as evidenced by increased motor tone and abnormal reflexes are not uncommon.1 Branches of the cauda equina extend caudally from the epiconus, a structure proximal to the conus medullaris, as peripheral nervous system branches that innervate spinal cord segments L4 through S1 (Figure 2). Lesions of the epiconus are clinically distinguished by lower motor neuron deficits wherein muscles of the lower extremities are often weakened with potential sparing of the bulbocavernosus and micturition reflexes.2
Conclusion
While many EPs are cognizant of cauda equina syndrome and its presentation, CMS is less well known and not commonly documented. Due to symptomatic overlap and epidemiological rarity of these conditions, most of the literature describing these entities combines their discussion. This case contributes to the growing body of literature to assist clinicians in the evaluation and management of CMS.
Dr Batt is an emergency medicine resident, Arrowhead Regional Medical Center, Colton, California. Dr Stone is the emergency medical services director, Travis Air Force Base, Fairfield, California.
- Lewandrowski KU, McLain RF, Lieberman I, Orr D. Cord and cauda equina injury complicating elective orthopedic surgery. Spine (Phila Pa 1976). 2006;31(9):1056-1059.
- Kirshblum S, Anderson K, Krassioukov A, Donovan W. Assessment and classification of traumatic spinal cord injury. In: Kirshblum S, Campagnolo DI, eds. Spinal Cord Medicine. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Ruckdeschel JC. Early detection and treatment of spinal cord compression. Oncology (Williston Park). 2005;19(1):81-86.
- Perron AD, Huff JS. Spinal cord disorders. In: Marx JA, Hockberger RS, Walls RM, et al. Rosen’s Emergency Medicine: Concepts and Clinical Practice. 8th ed. Vol 2. Philadelphia: Mosby/Elsevier, 2013; 1419-1427.
- Wagner R, Jagoda A. Spinal cord syndromes. Emerg Med Clin North Am. 1997;15(3):699-711.
- Sciubba DM, Gokaslan ZL. Diagnosis and management of metastatic spine disease. Surg Oncol. 2006;15(3):141-151.
- Jalloh I, Minhas P. Delays in the treatment of cauda equina syndrome due to its variable clinical features in patients presenting to the emergency department. Emerg Med J. 2007;24(1):33-34.
- Korse NS, Jacobs WCH, Elzevier HW, Vieggeert-Lankamp CL. Complaints of micturition, defecation and sexual function in cauda equina syndrome due to lumbar disk herniation: a systematic review. Eur Spine J. 2013;22(5):1019-1029.
- Dawodu ST, Bechtel KA, Beeson MS, et al. Cauda equina and conus medullaris syndromes. Medscape Web site. http://emedicine.medscape.com/article/1148690-clinical. Accessed September 1, 2014.
- Glick TH, Workman TP, Gaufberg SV. Spinal cord emergencies: false reassurance from reflexes. Acad Emerg Med. 1998;5(10):1041-1043.
- Lewandrowski KU, McLain RF, Lieberman I, Orr D. Cord and cauda equina injury complicating elective orthopedic surgery. Spine (Phila Pa 1976). 2006;31(9):1056-1059.
- Kirshblum S, Anderson K, Krassioukov A, Donovan W. Assessment and classification of traumatic spinal cord injury. In: Kirshblum S, Campagnolo DI, eds. Spinal Cord Medicine. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Ruckdeschel JC. Early detection and treatment of spinal cord compression. Oncology (Williston Park). 2005;19(1):81-86.
- Perron AD, Huff JS. Spinal cord disorders. In: Marx JA, Hockberger RS, Walls RM, et al. Rosen’s Emergency Medicine: Concepts and Clinical Practice. 8th ed. Vol 2. Philadelphia: Mosby/Elsevier, 2013; 1419-1427.
- Wagner R, Jagoda A. Spinal cord syndromes. Emerg Med Clin North Am. 1997;15(3):699-711.
- Sciubba DM, Gokaslan ZL. Diagnosis and management of metastatic spine disease. Surg Oncol. 2006;15(3):141-151.
- Jalloh I, Minhas P. Delays in the treatment of cauda equina syndrome due to its variable clinical features in patients presenting to the emergency department. Emerg Med J. 2007;24(1):33-34.
- Korse NS, Jacobs WCH, Elzevier HW, Vieggeert-Lankamp CL. Complaints of micturition, defecation and sexual function in cauda equina syndrome due to lumbar disk herniation: a systematic review. Eur Spine J. 2013;22(5):1019-1029.
- Dawodu ST, Bechtel KA, Beeson MS, et al. Cauda equina and conus medullaris syndromes. Medscape Web site. http://emedicine.medscape.com/article/1148690-clinical. Accessed September 1, 2014.
- Glick TH, Workman TP, Gaufberg SV. Spinal cord emergencies: false reassurance from reflexes. Acad Emerg Med. 1998;5(10):1041-1043.

Case Report: Rapidly Ascending Weakness in a 22-year-old Man
Case
A 22-year-old white man presented to the ED after waking from sleep and experiencing painless bilateral lower extremity weakness that ascended to the right upper extremity. This weakness left the patient unable to walk. He denied incontinence and was otherwise healthy with no prior history of similar symptoms. A family history of the presenting symptomatology was denied, and there was no family history of diabetes or kidney disease. The patient did state he had upper respiratory symptoms with a sore throat and runny nose 2 weeks earlier. He denied use of tobacco, alcohol, and illicit drugs.
Physical examination revealed abnormal deep tendon reflexes, absent patellar and achilles reflexes on the right side, reduced patellar and achilles reflexes of the left side, and diminished bilateral brachioradialis reflexes. Examination of the lower extremities revealed motor strength at 2/5 on the left and 1/5 on the right. His upper extremities demonstrated bilateral 4/5 motor strength. No ptosis was observed. All pulses of the extremities were normal with good capillary refill. His vital signs were: blood pressure, 131/75 mm Hg; pulse, 75 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 97.2°F
Brain and cervical spine magnetic resonance imaging, computed tomography of the head, and blood cultures were ordered; the results of each were unremarkable. A lumbar puncture (LP) was attempted in the ED, but was unsuccessful due to difficulty in patient positioning secondary to profound weakness. An LP was later succesfully performed under fluoroscopy and revealed no abnormal findings. An electrocardiogram (ECG) demonstrated normal sinus rhythm with ST-segment flattening. The differential diagnosis for rapidly ascending weakness included Guillain-Barré syndrome, transverse myelitis, toxic metabolic syndromes, and myelopathies.
A basic metabolic panel showed that the patient was profoundly hypokalemic, with a potassium level of 1.9 mEq/L. After further blood analysis, mild hypomagnesemia was observed, with a level of 1.4 mEq/L. Whle in the ED, he was given 40 mEq potassium chloride intravenously (IV) and orally, and 2 g IV magnesium sulfate in the ED. The patient was evaluated by a neurologist in the ED. No respiratory compromise was present and the patient was admitted to neurology service with consultations from both nephrology and endocrinology.
Within 12 hours of starting potassium and magnesium supplementation, the patient’s potassium and magnesium levels returned to normal and his weakness concurrently subsided. The final diagnosis of the patient was hypokalemic periodic paralysis.
Discussion
Hypokalemic periodic paralysis (HPP), a rare autosomal dominant disorder with a prevalence of one in 100,000, is characterized by muscle weakness with accompanying hypokalemia. The first onset of symptoms is usually in the first or second decade of life, with a quarter of cases presenting before the age 10 years, and half before age 16 years. Being a typically-inherited autosomal dominant disorder, approximately two-thirds of cases present with a family history—unlike this patient case in which there was no family history of the disease. It is three to four times more common in men than women.
Hypokalemic periodic paralysis typically occurs upon waking from sleep or during rest following exercise. It may also be triggered by high-carbohydrate or high-salt meals, or from alcohol consumption. The muscles of the extremeties are usually affected, while those of the eyes, face, and sphincters are typically spared. If untreated, an attack can last from several hours to several days. Tendon and cutaneous reflexes can also be reduced or absent.1 The manifestation of HPP can, at first impression, lead the differential diagnosis in the direction of the many other conditions that cause weakness of the extremities. Guillain-Barré syndrome and transverse myelitis are the more common diagnoses for which HPP can be mistaken. It is therefore important that the EP consider HPP in the differential so that an unusual presentation such as the one in this case may be diagnosed.
When presented with extraneous complications (eg, when the condition is encountered perioperatively or suffered comorbidly with similar conditions such as Guillain-Barré syndrome), HPP can be especially difficult to recognize.2,3 Although a relatively uncommon condition, it can be potentially life-threatening if not recognized and treated appropriately. Severe hypokalemia may cause sequela such as respiratory failure and cardiac arhythmia. Thus, ECG and cardiorespiratory monitoring are essential in patients with HPP and hypokalemia as very severe hypokalemia may also cause paralysis of bulbar and cranial nerve musculature. Electocardiographic changes are common, and may include ST-segment sagging and flattening, U waves, and T-wave inversion. Other causes in patients presenting with HPP must also be considered, as hypokalemic paralysis of a sporadic nature may be associated with conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies, and gastrointestinal potassium losses.4
The pathogenesis of HPP is not completely understood; however, alteration in potassium regulation are well documented. Treatment of HPP includes both oral and IV potassium supplementation. Prophylaxis against recurrent attacks has been successful with various modalities including spiranolactone daily and acetazolamide.4 Care must be taken to consider thyrotoxic periodic paralysis, which most commonly presents in Asian men, as hyperthyroid symptoms may be subtle. Treatment focuses on reversal of the thyrotoxic state. β-Adrenergic blocking agents reduce the frequency and severity of attacks and should be started while measures to control thyrotoxicosis are being instituted.4
Before a diagnosis of HPP is made, other causes of hypokalemic paralysis must first be excluded.
Conclusion
This case is important because it demonstrates an unusual presentation of HPP in an emergency setting. This perspective of HPP can help the EP in recognizing and differentiating the condsition from similar disorders.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Ropper AH, Samuels MA. The myotonias, periodic paralyses, cramps, spasms, and states of persistent muscle fiber activity. In: Ropper AH, Samuels MA, Klein JP, eds. Adams & Victor’s Principles of Neurology. 10th ed. New York, NY: McGraw-Hill Education; 2014:1490-1508.
- Abbas H, Kothari N, Bogra J. Hypokalemic periodic paralysis. Natl J Maxillofac Surg. 2012;3(2):220-221.
- Saroja AO, Naik KR, Khanpet MS. Uncommon dyselectrolytemia complicating Guillain-Barré syndrome. J Neurosci Rural Pract. 2013;4(3):328-330.
- Ahlawat SK, Sachdev A. Hypokalemic paralysis. Postgrad Med J. 1999;75(882):193-197.
Case
A 22-year-old white man presented to the ED after waking from sleep and experiencing painless bilateral lower extremity weakness that ascended to the right upper extremity. This weakness left the patient unable to walk. He denied incontinence and was otherwise healthy with no prior history of similar symptoms. A family history of the presenting symptomatology was denied, and there was no family history of diabetes or kidney disease. The patient did state he had upper respiratory symptoms with a sore throat and runny nose 2 weeks earlier. He denied use of tobacco, alcohol, and illicit drugs.
Physical examination revealed abnormal deep tendon reflexes, absent patellar and achilles reflexes on the right side, reduced patellar and achilles reflexes of the left side, and diminished bilateral brachioradialis reflexes. Examination of the lower extremities revealed motor strength at 2/5 on the left and 1/5 on the right. His upper extremities demonstrated bilateral 4/5 motor strength. No ptosis was observed. All pulses of the extremities were normal with good capillary refill. His vital signs were: blood pressure, 131/75 mm Hg; pulse, 75 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 97.2°F
Brain and cervical spine magnetic resonance imaging, computed tomography of the head, and blood cultures were ordered; the results of each were unremarkable. A lumbar puncture (LP) was attempted in the ED, but was unsuccessful due to difficulty in patient positioning secondary to profound weakness. An LP was later succesfully performed under fluoroscopy and revealed no abnormal findings. An electrocardiogram (ECG) demonstrated normal sinus rhythm with ST-segment flattening. The differential diagnosis for rapidly ascending weakness included Guillain-Barré syndrome, transverse myelitis, toxic metabolic syndromes, and myelopathies.
A basic metabolic panel showed that the patient was profoundly hypokalemic, with a potassium level of 1.9 mEq/L. After further blood analysis, mild hypomagnesemia was observed, with a level of 1.4 mEq/L. Whle in the ED, he was given 40 mEq potassium chloride intravenously (IV) and orally, and 2 g IV magnesium sulfate in the ED. The patient was evaluated by a neurologist in the ED. No respiratory compromise was present and the patient was admitted to neurology service with consultations from both nephrology and endocrinology.
Within 12 hours of starting potassium and magnesium supplementation, the patient’s potassium and magnesium levels returned to normal and his weakness concurrently subsided. The final diagnosis of the patient was hypokalemic periodic paralysis.
Discussion
Hypokalemic periodic paralysis (HPP), a rare autosomal dominant disorder with a prevalence of one in 100,000, is characterized by muscle weakness with accompanying hypokalemia. The first onset of symptoms is usually in the first or second decade of life, with a quarter of cases presenting before the age 10 years, and half before age 16 years. Being a typically-inherited autosomal dominant disorder, approximately two-thirds of cases present with a family history—unlike this patient case in which there was no family history of the disease. It is three to four times more common in men than women.
Hypokalemic periodic paralysis typically occurs upon waking from sleep or during rest following exercise. It may also be triggered by high-carbohydrate or high-salt meals, or from alcohol consumption. The muscles of the extremeties are usually affected, while those of the eyes, face, and sphincters are typically spared. If untreated, an attack can last from several hours to several days. Tendon and cutaneous reflexes can also be reduced or absent.1 The manifestation of HPP can, at first impression, lead the differential diagnosis in the direction of the many other conditions that cause weakness of the extremities. Guillain-Barré syndrome and transverse myelitis are the more common diagnoses for which HPP can be mistaken. It is therefore important that the EP consider HPP in the differential so that an unusual presentation such as the one in this case may be diagnosed.
When presented with extraneous complications (eg, when the condition is encountered perioperatively or suffered comorbidly with similar conditions such as Guillain-Barré syndrome), HPP can be especially difficult to recognize.2,3 Although a relatively uncommon condition, it can be potentially life-threatening if not recognized and treated appropriately. Severe hypokalemia may cause sequela such as respiratory failure and cardiac arhythmia. Thus, ECG and cardiorespiratory monitoring are essential in patients with HPP and hypokalemia as very severe hypokalemia may also cause paralysis of bulbar and cranial nerve musculature. Electocardiographic changes are common, and may include ST-segment sagging and flattening, U waves, and T-wave inversion. Other causes in patients presenting with HPP must also be considered, as hypokalemic paralysis of a sporadic nature may be associated with conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies, and gastrointestinal potassium losses.4
The pathogenesis of HPP is not completely understood; however, alteration in potassium regulation are well documented. Treatment of HPP includes both oral and IV potassium supplementation. Prophylaxis against recurrent attacks has been successful with various modalities including spiranolactone daily and acetazolamide.4 Care must be taken to consider thyrotoxic periodic paralysis, which most commonly presents in Asian men, as hyperthyroid symptoms may be subtle. Treatment focuses on reversal of the thyrotoxic state. β-Adrenergic blocking agents reduce the frequency and severity of attacks and should be started while measures to control thyrotoxicosis are being instituted.4
Before a diagnosis of HPP is made, other causes of hypokalemic paralysis must first be excluded.
Conclusion
This case is important because it demonstrates an unusual presentation of HPP in an emergency setting. This perspective of HPP can help the EP in recognizing and differentiating the condsition from similar disorders.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
Case
A 22-year-old white man presented to the ED after waking from sleep and experiencing painless bilateral lower extremity weakness that ascended to the right upper extremity. This weakness left the patient unable to walk. He denied incontinence and was otherwise healthy with no prior history of similar symptoms. A family history of the presenting symptomatology was denied, and there was no family history of diabetes or kidney disease. The patient did state he had upper respiratory symptoms with a sore throat and runny nose 2 weeks earlier. He denied use of tobacco, alcohol, and illicit drugs.
Physical examination revealed abnormal deep tendon reflexes, absent patellar and achilles reflexes on the right side, reduced patellar and achilles reflexes of the left side, and diminished bilateral brachioradialis reflexes. Examination of the lower extremities revealed motor strength at 2/5 on the left and 1/5 on the right. His upper extremities demonstrated bilateral 4/5 motor strength. No ptosis was observed. All pulses of the extremities were normal with good capillary refill. His vital signs were: blood pressure, 131/75 mm Hg; pulse, 75 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 97.2°F
Brain and cervical spine magnetic resonance imaging, computed tomography of the head, and blood cultures were ordered; the results of each were unremarkable. A lumbar puncture (LP) was attempted in the ED, but was unsuccessful due to difficulty in patient positioning secondary to profound weakness. An LP was later succesfully performed under fluoroscopy and revealed no abnormal findings. An electrocardiogram (ECG) demonstrated normal sinus rhythm with ST-segment flattening. The differential diagnosis for rapidly ascending weakness included Guillain-Barré syndrome, transverse myelitis, toxic metabolic syndromes, and myelopathies.
A basic metabolic panel showed that the patient was profoundly hypokalemic, with a potassium level of 1.9 mEq/L. After further blood analysis, mild hypomagnesemia was observed, with a level of 1.4 mEq/L. Whle in the ED, he was given 40 mEq potassium chloride intravenously (IV) and orally, and 2 g IV magnesium sulfate in the ED. The patient was evaluated by a neurologist in the ED. No respiratory compromise was present and the patient was admitted to neurology service with consultations from both nephrology and endocrinology.
Within 12 hours of starting potassium and magnesium supplementation, the patient’s potassium and magnesium levels returned to normal and his weakness concurrently subsided. The final diagnosis of the patient was hypokalemic periodic paralysis.
Discussion
Hypokalemic periodic paralysis (HPP), a rare autosomal dominant disorder with a prevalence of one in 100,000, is characterized by muscle weakness with accompanying hypokalemia. The first onset of symptoms is usually in the first or second decade of life, with a quarter of cases presenting before the age 10 years, and half before age 16 years. Being a typically-inherited autosomal dominant disorder, approximately two-thirds of cases present with a family history—unlike this patient case in which there was no family history of the disease. It is three to four times more common in men than women.
Hypokalemic periodic paralysis typically occurs upon waking from sleep or during rest following exercise. It may also be triggered by high-carbohydrate or high-salt meals, or from alcohol consumption. The muscles of the extremeties are usually affected, while those of the eyes, face, and sphincters are typically spared. If untreated, an attack can last from several hours to several days. Tendon and cutaneous reflexes can also be reduced or absent.1 The manifestation of HPP can, at first impression, lead the differential diagnosis in the direction of the many other conditions that cause weakness of the extremities. Guillain-Barré syndrome and transverse myelitis are the more common diagnoses for which HPP can be mistaken. It is therefore important that the EP consider HPP in the differential so that an unusual presentation such as the one in this case may be diagnosed.
When presented with extraneous complications (eg, when the condition is encountered perioperatively or suffered comorbidly with similar conditions such as Guillain-Barré syndrome), HPP can be especially difficult to recognize.2,3 Although a relatively uncommon condition, it can be potentially life-threatening if not recognized and treated appropriately. Severe hypokalemia may cause sequela such as respiratory failure and cardiac arhythmia. Thus, ECG and cardiorespiratory monitoring are essential in patients with HPP and hypokalemia as very severe hypokalemia may also cause paralysis of bulbar and cranial nerve musculature. Electocardiographic changes are common, and may include ST-segment sagging and flattening, U waves, and T-wave inversion. Other causes in patients presenting with HPP must also be considered, as hypokalemic paralysis of a sporadic nature may be associated with conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies, and gastrointestinal potassium losses.4
The pathogenesis of HPP is not completely understood; however, alteration in potassium regulation are well documented. Treatment of HPP includes both oral and IV potassium supplementation. Prophylaxis against recurrent attacks has been successful with various modalities including spiranolactone daily and acetazolamide.4 Care must be taken to consider thyrotoxic periodic paralysis, which most commonly presents in Asian men, as hyperthyroid symptoms may be subtle. Treatment focuses on reversal of the thyrotoxic state. β-Adrenergic blocking agents reduce the frequency and severity of attacks and should be started while measures to control thyrotoxicosis are being instituted.4
Before a diagnosis of HPP is made, other causes of hypokalemic paralysis must first be excluded.
Conclusion
This case is important because it demonstrates an unusual presentation of HPP in an emergency setting. This perspective of HPP can help the EP in recognizing and differentiating the condsition from similar disorders.
Dr Orlik is a resident, department of emergency medicine, Akron General Medical Center, Ohio. Mr Kovacs is a student and summer research fellow, department of emergency medicine, Akron General Medical Center, Ohio. Dr Simon is the emergency medicine research director, department of emergency medicine, Akron General Medical Center, Northeast Ohio Medical University.
- Ropper AH, Samuels MA. The myotonias, periodic paralyses, cramps, spasms, and states of persistent muscle fiber activity. In: Ropper AH, Samuels MA, Klein JP, eds. Adams & Victor’s Principles of Neurology. 10th ed. New York, NY: McGraw-Hill Education; 2014:1490-1508.
- Abbas H, Kothari N, Bogra J. Hypokalemic periodic paralysis. Natl J Maxillofac Surg. 2012;3(2):220-221.
- Saroja AO, Naik KR, Khanpet MS. Uncommon dyselectrolytemia complicating Guillain-Barré syndrome. J Neurosci Rural Pract. 2013;4(3):328-330.
- Ahlawat SK, Sachdev A. Hypokalemic paralysis. Postgrad Med J. 1999;75(882):193-197.
- Ropper AH, Samuels MA. The myotonias, periodic paralyses, cramps, spasms, and states of persistent muscle fiber activity. In: Ropper AH, Samuels MA, Klein JP, eds. Adams & Victor’s Principles of Neurology. 10th ed. New York, NY: McGraw-Hill Education; 2014:1490-1508.
- Abbas H, Kothari N, Bogra J. Hypokalemic periodic paralysis. Natl J Maxillofac Surg. 2012;3(2):220-221.
- Saroja AO, Naik KR, Khanpet MS. Uncommon dyselectrolytemia complicating Guillain-Barré syndrome. J Neurosci Rural Pract. 2013;4(3):328-330.
- Ahlawat SK, Sachdev A. Hypokalemic paralysis. Postgrad Med J. 1999;75(882):193-197.
Case Studies in Toxicology: You Can’t See Dragonfly or Hear NBOMe, but They Can Still Hurt You
Case
A 24-year-old man was brought to the ED by emergency medical services (EMS) for altered mental status. The EMS crew reported they had picked up the patient at a nearby arts festival and concert series. A bystander at the event reported that the patient had taken something called “dragonfly.”
Initial assessment revealed the patient to be disoriented, with nonlinear thought patterns and an inability to follow commands. His vital signs were: blood pressure, 160/100 mm Hg; heart rate, 120 beats/minute; respiratory rate, 24 breaths/minute; and temperature, 102.2˚F. Oxygen saturation was 99% on room air. He was diaphoretic and agitated, and the nursing staff was concerned he would become aggressive and potentially violent. A quick Web search revealed that the agent the bystander mentioned was most likely Bromo-DragonFLY (BDF).
What is Bromo-DragonFLY?
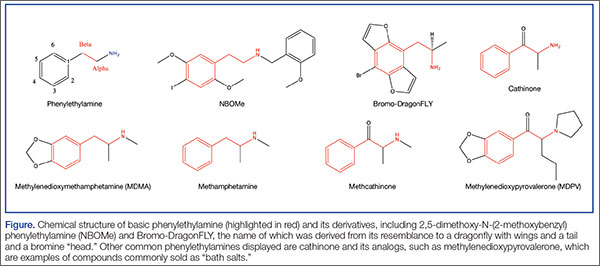
In the 1960s, an American chemist named Alexander Shulgin ushered in a new era of psychedelic drug use by establishing a simple synthesis of 3,4-methylenedioxy-methamphetamine (MDMA). Following this discovery, he suggested a therapist friend use the drug therapeutically.1 Shulgin then began a process of homologation (ie, creating novel compounds by slightly altering existing ones in an organized fashion) and developed systems for rating the drug experiences and naming the drugs in shorthand, both of which are still in use. The chemical structure common to nearly all of the drugs he studied is phenylethylamine. The Figure shows the structures of several phenylethylamine derivatives that were created by adding functional groups to the phenylethylamine backbone. Although the popularity of psychedelic drugs surged during this time period, 2,5-dimethoxy-N-(2-methoxybenzyl)phenylethylamine) (NBOMe), one of a number of newly popular psychedelics, only became available in 2003.
What is known about the pharmacology of Bromo-DragonFLY and NBOMe?
The major target of psychedelic drugs is the serotonin (5-HT2) receptor, specifically the central 5-HT2A subtype. Bromo-DragonFLY is a classic example of designer pharmacology in that the it was intended to potently exert its effect at this specific receptor site.
As its name suggests, BDF adds the “wings of the fly” to the phenylethylamine backbone furanyl rings at positions 2 and 5, and a halogen (bromine) at position 4. The furanyl ring impairs enzymatic clearance of the drug,2 resulting in a duration of action of up to 3 days.3 The addition of halogens increases drug potency, but the mechanism is not clear. The psychedelic agent NBOMe results from chemical additions of methoxy groups at position 2 and 5, and the halogen moiety (iodine in this case) at position 4 of the phenyl ring of the phenylethylamine structure.4
Through the work of Shulgin, some of his colleagues, and many disparate street chemists, a vast family of substituted phenylethylamines have been synthesized and used. Shulgin’s semiautobiographical book PiHKAL: A Chemical Love Story includes his laboratory notes for the synthesis and initial test-dose experience of 179 compounds1; this does not include research done by others or any work since its publication in 1995.
Notable popular drugs chemically similar to NBOMe and BDF are mescaline (found in peyote), cathinones (“bath salts”), and MDMA (found in ecstasy) (Figure). Naturally occurring (and more complex) compounds with similar effects include ayahuasca, a plant-derived beverage consisting of Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana from the Brazilian rainforest (see Emerg Med. 2014;46[12]:553-556); psilocybin (“magic mushrooms”); and lysergic acid diethylamide.
How are these drugs used and what are their clinical effects?
Most phenylethylamine compounds are well absorbed across the buccal mucosa, which is why BDF and NBOMe are commonly used in liquid form or on blotter paper. Dosing guides also exist for insufflation and claim equipotent dosing for this route.5 Regardless of delivery route, given the high potency, inadvertent exposures to these drugs should be expected.
Users simply seeking to hallucinate may not be aware of the significant risks associated with these potent serotonergic agents, which include both life- and limb-threatening effects.6 The high 5-HT2A potency results both in vasoconstriction and promotion of clot formation due to the presence of 5HT2A receptors on small blood vessels and platelets, respectively. Ergotism, historically called Saint Anthony’s fire, is an example of serotonergic vasoconstriction and hallucination.7 Chronic users of substituted amphetamines can develop necrotic ulcers in distal vascular beds such as the hands and feet; these ulcers may progress to amputation despite treatment attempts with vasodilators.
In addition to the vasoconstrictive properties, there are multiple reports of serotonin toxicity (serotonin syndrome) associated with use of these designer serotonergic amphetamines. This syndrome includes severe psychomotor agitation that can lead to personal injury, along with muscle rigidity, tremor, hyperthermia, rhabdomyolysis, and seizures.8
How are patients with phenylethylamine exposures managed?
Management of a patient with a substituted phenylethylamine exposure is similar to management of those with cocaine overdose. Attention to the life-threatening clinical effects of psychomotor agitation, hyperthermia, and seizures is paramount. Appropriate supportive care includes intravenous (IV) benzodiazepines to control agitation and muscle rigidity, replacement of lost volume with crystalloids, and active cooling measures. Failure of benzodiazepines (preferably in conjunction with continuous electroencephalogram monitoring) to control rigidity may lead to the need for propofol and/or result in paralysis. Similar to patients with cocaine intoxication, some may experience ischemic chest pain, and the usual protocol of sedation, nitroglycerin, morphine, and an antiplatelet drug is appropriate.
Identification of phenylethylamines typically requires specialized laboratory testing since most will not trigger a positive result on a standard urine immunoassay. Many specialized laboratories have test catalogs on their Web sites listing under the “stimulants panel” which drugs can be identified. However, none of these assays is likely truly comprehensive, and minor alterations or substitutions to the compounds result in new analogs that may not be in the reference laboratory’s identification library.
The patient was initially restrained and given 5 mg IV diazepam, which was followed by escalating doses every 5 minutes to a total of 35 mg for effect. He had a rectal temperature of 102.5˚F and was externally cooled after sedation. After 20 minutes, he had a generalized convulsion; an additional 10 mg of IV diazepam terminated the seizure, but he remained hyperthermic at 104˚F. The patient was intubated, placed on a propofol infusion, and admitted to the intensive care unit where his temperature was carefully monitored. The following day his temperature had normalized and he was weaned from the ventilator and discharged to the floor for monitoring. On hospital day 3, he was discharged in stable condition.
Mr Waldrop is a fourth-year medical student at the State University of New York, Upstate Medical University, Syracuse. Dr Nacca is a fellow in medical toxicology, department of emergency medicine, State University of New York, Upstate Medical University, Syracuse. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine, and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Shulgin A, Shulgin A. PiHKAL: A Chemical Love Story. Berkeley, CA: Transform Press; 1995.
- Andreasen MF, Telving R, Birkler RI, Schumacher B, Johannsen M. A fatal poisoning involving bromo-dragonfly. Forensic Sci Int. 2009;183(1-3):91-96.
- Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
- Gentry CL, Egleton RD, Gillespie T, et al. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides. 1999;20(10):1229-1238.
- Erowid. Bromo-Dragonfly Dosage. http://www.erowid.org/chemicals/bromo_dragonfly/bromo_dragonfly_dose.shtml. Accessed January 14, 2015.
- Baumann MH, Ayestas MA Jr, Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37(5):1192-1203.
- Walterscheid JP, Phillips GT, Lopez AE, Gonsoulin ML, Chen HH, Sanchez LA. Pathological findings in 2 cases of fatal 25I-NBOMe toxicity. Am J Forensic Med Pathol. 2014;35(1):20-25.
- Wood DM, Looker JJ, Shaikh L, et al. Delayed onset of seizures and toxicity associated with recreational use of Bromo-dragonFLY. J Med Toxicol. 2009;5(4):226-229.
Case
A 24-year-old man was brought to the ED by emergency medical services (EMS) for altered mental status. The EMS crew reported they had picked up the patient at a nearby arts festival and concert series. A bystander at the event reported that the patient had taken something called “dragonfly.”
Initial assessment revealed the patient to be disoriented, with nonlinear thought patterns and an inability to follow commands. His vital signs were: blood pressure, 160/100 mm Hg; heart rate, 120 beats/minute; respiratory rate, 24 breaths/minute; and temperature, 102.2˚F. Oxygen saturation was 99% on room air. He was diaphoretic and agitated, and the nursing staff was concerned he would become aggressive and potentially violent. A quick Web search revealed that the agent the bystander mentioned was most likely Bromo-DragonFLY (BDF).
What is Bromo-DragonFLY?
In the 1960s, an American chemist named Alexander Shulgin ushered in a new era of psychedelic drug use by establishing a simple synthesis of 3,4-methylenedioxy-methamphetamine (MDMA). Following this discovery, he suggested a therapist friend use the drug therapeutically.1 Shulgin then began a process of homologation (ie, creating novel compounds by slightly altering existing ones in an organized fashion) and developed systems for rating the drug experiences and naming the drugs in shorthand, both of which are still in use. The chemical structure common to nearly all of the drugs he studied is phenylethylamine. The Figure shows the structures of several phenylethylamine derivatives that were created by adding functional groups to the phenylethylamine backbone. Although the popularity of psychedelic drugs surged during this time period, 2,5-dimethoxy-N-(2-methoxybenzyl)phenylethylamine) (NBOMe), one of a number of newly popular psychedelics, only became available in 2003.
What is known about the pharmacology of Bromo-DragonFLY and NBOMe?
The major target of psychedelic drugs is the serotonin (5-HT2) receptor, specifically the central 5-HT2A subtype. Bromo-DragonFLY is a classic example of designer pharmacology in that the it was intended to potently exert its effect at this specific receptor site.
As its name suggests, BDF adds the “wings of the fly” to the phenylethylamine backbone furanyl rings at positions 2 and 5, and a halogen (bromine) at position 4. The furanyl ring impairs enzymatic clearance of the drug,2 resulting in a duration of action of up to 3 days.3 The addition of halogens increases drug potency, but the mechanism is not clear. The psychedelic agent NBOMe results from chemical additions of methoxy groups at position 2 and 5, and the halogen moiety (iodine in this case) at position 4 of the phenyl ring of the phenylethylamine structure.4
Through the work of Shulgin, some of his colleagues, and many disparate street chemists, a vast family of substituted phenylethylamines have been synthesized and used. Shulgin’s semiautobiographical book PiHKAL: A Chemical Love Story includes his laboratory notes for the synthesis and initial test-dose experience of 179 compounds1; this does not include research done by others or any work since its publication in 1995.
Notable popular drugs chemically similar to NBOMe and BDF are mescaline (found in peyote), cathinones (“bath salts”), and MDMA (found in ecstasy) (Figure). Naturally occurring (and more complex) compounds with similar effects include ayahuasca, a plant-derived beverage consisting of Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana from the Brazilian rainforest (see Emerg Med. 2014;46[12]:553-556); psilocybin (“magic mushrooms”); and lysergic acid diethylamide.
How are these drugs used and what are their clinical effects?
Most phenylethylamine compounds are well absorbed across the buccal mucosa, which is why BDF and NBOMe are commonly used in liquid form or on blotter paper. Dosing guides also exist for insufflation and claim equipotent dosing for this route.5 Regardless of delivery route, given the high potency, inadvertent exposures to these drugs should be expected.
Users simply seeking to hallucinate may not be aware of the significant risks associated with these potent serotonergic agents, which include both life- and limb-threatening effects.6 The high 5-HT2A potency results both in vasoconstriction and promotion of clot formation due to the presence of 5HT2A receptors on small blood vessels and platelets, respectively. Ergotism, historically called Saint Anthony’s fire, is an example of serotonergic vasoconstriction and hallucination.7 Chronic users of substituted amphetamines can develop necrotic ulcers in distal vascular beds such as the hands and feet; these ulcers may progress to amputation despite treatment attempts with vasodilators.
In addition to the vasoconstrictive properties, there are multiple reports of serotonin toxicity (serotonin syndrome) associated with use of these designer serotonergic amphetamines. This syndrome includes severe psychomotor agitation that can lead to personal injury, along with muscle rigidity, tremor, hyperthermia, rhabdomyolysis, and seizures.8
How are patients with phenylethylamine exposures managed?
Management of a patient with a substituted phenylethylamine exposure is similar to management of those with cocaine overdose. Attention to the life-threatening clinical effects of psychomotor agitation, hyperthermia, and seizures is paramount. Appropriate supportive care includes intravenous (IV) benzodiazepines to control agitation and muscle rigidity, replacement of lost volume with crystalloids, and active cooling measures. Failure of benzodiazepines (preferably in conjunction with continuous electroencephalogram monitoring) to control rigidity may lead to the need for propofol and/or result in paralysis. Similar to patients with cocaine intoxication, some may experience ischemic chest pain, and the usual protocol of sedation, nitroglycerin, morphine, and an antiplatelet drug is appropriate.
Identification of phenylethylamines typically requires specialized laboratory testing since most will not trigger a positive result on a standard urine immunoassay. Many specialized laboratories have test catalogs on their Web sites listing under the “stimulants panel” which drugs can be identified. However, none of these assays is likely truly comprehensive, and minor alterations or substitutions to the compounds result in new analogs that may not be in the reference laboratory’s identification library.
The patient was initially restrained and given 5 mg IV diazepam, which was followed by escalating doses every 5 minutes to a total of 35 mg for effect. He had a rectal temperature of 102.5˚F and was externally cooled after sedation. After 20 minutes, he had a generalized convulsion; an additional 10 mg of IV diazepam terminated the seizure, but he remained hyperthermic at 104˚F. The patient was intubated, placed on a propofol infusion, and admitted to the intensive care unit where his temperature was carefully monitored. The following day his temperature had normalized and he was weaned from the ventilator and discharged to the floor for monitoring. On hospital day 3, he was discharged in stable condition.
Mr Waldrop is a fourth-year medical student at the State University of New York, Upstate Medical University, Syracuse. Dr Nacca is a fellow in medical toxicology, department of emergency medicine, State University of New York, Upstate Medical University, Syracuse. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine, and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 24-year-old man was brought to the ED by emergency medical services (EMS) for altered mental status. The EMS crew reported they had picked up the patient at a nearby arts festival and concert series. A bystander at the event reported that the patient had taken something called “dragonfly.”
Initial assessment revealed the patient to be disoriented, with nonlinear thought patterns and an inability to follow commands. His vital signs were: blood pressure, 160/100 mm Hg; heart rate, 120 beats/minute; respiratory rate, 24 breaths/minute; and temperature, 102.2˚F. Oxygen saturation was 99% on room air. He was diaphoretic and agitated, and the nursing staff was concerned he would become aggressive and potentially violent. A quick Web search revealed that the agent the bystander mentioned was most likely Bromo-DragonFLY (BDF).
What is Bromo-DragonFLY?
In the 1960s, an American chemist named Alexander Shulgin ushered in a new era of psychedelic drug use by establishing a simple synthesis of 3,4-methylenedioxy-methamphetamine (MDMA). Following this discovery, he suggested a therapist friend use the drug therapeutically.1 Shulgin then began a process of homologation (ie, creating novel compounds by slightly altering existing ones in an organized fashion) and developed systems for rating the drug experiences and naming the drugs in shorthand, both of which are still in use. The chemical structure common to nearly all of the drugs he studied is phenylethylamine. The Figure shows the structures of several phenylethylamine derivatives that were created by adding functional groups to the phenylethylamine backbone. Although the popularity of psychedelic drugs surged during this time period, 2,5-dimethoxy-N-(2-methoxybenzyl)phenylethylamine) (NBOMe), one of a number of newly popular psychedelics, only became available in 2003.
What is known about the pharmacology of Bromo-DragonFLY and NBOMe?
The major target of psychedelic drugs is the serotonin (5-HT2) receptor, specifically the central 5-HT2A subtype. Bromo-DragonFLY is a classic example of designer pharmacology in that the it was intended to potently exert its effect at this specific receptor site.
As its name suggests, BDF adds the “wings of the fly” to the phenylethylamine backbone furanyl rings at positions 2 and 5, and a halogen (bromine) at position 4. The furanyl ring impairs enzymatic clearance of the drug,2 resulting in a duration of action of up to 3 days.3 The addition of halogens increases drug potency, but the mechanism is not clear. The psychedelic agent NBOMe results from chemical additions of methoxy groups at position 2 and 5, and the halogen moiety (iodine in this case) at position 4 of the phenyl ring of the phenylethylamine structure.4
Through the work of Shulgin, some of his colleagues, and many disparate street chemists, a vast family of substituted phenylethylamines have been synthesized and used. Shulgin’s semiautobiographical book PiHKAL: A Chemical Love Story includes his laboratory notes for the synthesis and initial test-dose experience of 179 compounds1; this does not include research done by others or any work since its publication in 1995.
Notable popular drugs chemically similar to NBOMe and BDF are mescaline (found in peyote), cathinones (“bath salts”), and MDMA (found in ecstasy) (Figure). Naturally occurring (and more complex) compounds with similar effects include ayahuasca, a plant-derived beverage consisting of Banisteriopsis caapi and either Psychotria viridis or Diplopterys cabrerana from the Brazilian rainforest (see Emerg Med. 2014;46[12]:553-556); psilocybin (“magic mushrooms”); and lysergic acid diethylamide.
How are these drugs used and what are their clinical effects?
Most phenylethylamine compounds are well absorbed across the buccal mucosa, which is why BDF and NBOMe are commonly used in liquid form or on blotter paper. Dosing guides also exist for insufflation and claim equipotent dosing for this route.5 Regardless of delivery route, given the high potency, inadvertent exposures to these drugs should be expected.
Users simply seeking to hallucinate may not be aware of the significant risks associated with these potent serotonergic agents, which include both life- and limb-threatening effects.6 The high 5-HT2A potency results both in vasoconstriction and promotion of clot formation due to the presence of 5HT2A receptors on small blood vessels and platelets, respectively. Ergotism, historically called Saint Anthony’s fire, is an example of serotonergic vasoconstriction and hallucination.7 Chronic users of substituted amphetamines can develop necrotic ulcers in distal vascular beds such as the hands and feet; these ulcers may progress to amputation despite treatment attempts with vasodilators.
In addition to the vasoconstrictive properties, there are multiple reports of serotonin toxicity (serotonin syndrome) associated with use of these designer serotonergic amphetamines. This syndrome includes severe psychomotor agitation that can lead to personal injury, along with muscle rigidity, tremor, hyperthermia, rhabdomyolysis, and seizures.8
How are patients with phenylethylamine exposures managed?
Management of a patient with a substituted phenylethylamine exposure is similar to management of those with cocaine overdose. Attention to the life-threatening clinical effects of psychomotor agitation, hyperthermia, and seizures is paramount. Appropriate supportive care includes intravenous (IV) benzodiazepines to control agitation and muscle rigidity, replacement of lost volume with crystalloids, and active cooling measures. Failure of benzodiazepines (preferably in conjunction with continuous electroencephalogram monitoring) to control rigidity may lead to the need for propofol and/or result in paralysis. Similar to patients with cocaine intoxication, some may experience ischemic chest pain, and the usual protocol of sedation, nitroglycerin, morphine, and an antiplatelet drug is appropriate.
Identification of phenylethylamines typically requires specialized laboratory testing since most will not trigger a positive result on a standard urine immunoassay. Many specialized laboratories have test catalogs on their Web sites listing under the “stimulants panel” which drugs can be identified. However, none of these assays is likely truly comprehensive, and minor alterations or substitutions to the compounds result in new analogs that may not be in the reference laboratory’s identification library.
The patient was initially restrained and given 5 mg IV diazepam, which was followed by escalating doses every 5 minutes to a total of 35 mg for effect. He had a rectal temperature of 102.5˚F and was externally cooled after sedation. After 20 minutes, he had a generalized convulsion; an additional 10 mg of IV diazepam terminated the seizure, but he remained hyperthermic at 104˚F. The patient was intubated, placed on a propofol infusion, and admitted to the intensive care unit where his temperature was carefully monitored. The following day his temperature had normalized and he was weaned from the ventilator and discharged to the floor for monitoring. On hospital day 3, he was discharged in stable condition.
Mr Waldrop is a fourth-year medical student at the State University of New York, Upstate Medical University, Syracuse. Dr Nacca is a fellow in medical toxicology, department of emergency medicine, State University of New York, Upstate Medical University, Syracuse. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine, and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Shulgin A, Shulgin A. PiHKAL: A Chemical Love Story. Berkeley, CA: Transform Press; 1995.
- Andreasen MF, Telving R, Birkler RI, Schumacher B, Johannsen M. A fatal poisoning involving bromo-dragonfly. Forensic Sci Int. 2009;183(1-3):91-96.
- Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
- Gentry CL, Egleton RD, Gillespie T, et al. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides. 1999;20(10):1229-1238.
- Erowid. Bromo-Dragonfly Dosage. http://www.erowid.org/chemicals/bromo_dragonfly/bromo_dragonfly_dose.shtml. Accessed January 14, 2015.
- Baumann MH, Ayestas MA Jr, Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37(5):1192-1203.
- Walterscheid JP, Phillips GT, Lopez AE, Gonsoulin ML, Chen HH, Sanchez LA. Pathological findings in 2 cases of fatal 25I-NBOMe toxicity. Am J Forensic Med Pathol. 2014;35(1):20-25.
- Wood DM, Looker JJ, Shaikh L, et al. Delayed onset of seizures and toxicity associated with recreational use of Bromo-dragonFLY. J Med Toxicol. 2009;5(4):226-229.
- Shulgin A, Shulgin A. PiHKAL: A Chemical Love Story. Berkeley, CA: Transform Press; 1995.
- Andreasen MF, Telving R, Birkler RI, Schumacher B, Johannsen M. A fatal poisoning involving bromo-dragonfly. Forensic Sci Int. 2009;183(1-3):91-96.
- Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila). 2011;49(8):705-719.
- Gentry CL, Egleton RD, Gillespie T, et al. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides. 1999;20(10):1229-1238.
- Erowid. Bromo-Dragonfly Dosage. http://www.erowid.org/chemicals/bromo_dragonfly/bromo_dragonfly_dose.shtml. Accessed January 14, 2015.
- Baumann MH, Ayestas MA Jr, Partilla JS, et al. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37(5):1192-1203.
- Walterscheid JP, Phillips GT, Lopez AE, Gonsoulin ML, Chen HH, Sanchez LA. Pathological findings in 2 cases of fatal 25I-NBOMe toxicity. Am J Forensic Med Pathol. 2014;35(1):20-25.
- Wood DM, Looker JJ, Shaikh L, et al. Delayed onset of seizures and toxicity associated with recreational use of Bromo-dragonFLY. J Med Toxicol. 2009;5(4):226-229.
