User login
How Metals Affect the Brain
This transcript has been edited for clarity.
It has always amazed me that our bodies require these tiny amounts of incredibly rare substances to function. Sure, we need oxygen. We need water. But we also need molybdenum, which makes up just 1.2 parts per million of the Earth’s crust.
Without adequate molybdenum intake, we develop seizures, developmental delays, death. Fortunately, we need so little molybdenum that true molybdenum deficiency is incredibly rare — seen only in people on total parenteral nutrition without supplementation or those with certain rare genetic conditions. But still, molybdenum is necessary for life.
Many metals are. Figure 1 colors the essential minerals on the periodic table. You can see that to stay alive, we humans need not only things like sodium, but selenium, bromine, zinc, copper, and cobalt.

Some metals are very clearly not essential; we can all do without lead and mercury, and probably should.
But just because something is essential for life does not mean that more is better. The dose is the poison, as they say. And this week, we explore whether metals — even essential metals — might be adversely affecting our brains.
It’s not a stretch to think that metal intake could have weird effects on our nervous system. Lead exposure, primarily due to leaded gasoline, has been blamed for an average reduction of about 3 points in our national IQ, for example . But not all metals are created equal. Researchers set out to find out which might be more strongly associated with performance on cognitive tests and dementia, and reported their results in this study in JAMA Network Open.
To do this, they leveraged the MESA cohort study. This is a longitudinal study of a relatively diverse group of 6300 adults who were enrolled from 2000 to 2002 around the United States. At enrollment, they gave a urine sample and took a variety of cognitive tests. Important for this study was the digit symbol substitution test, where participants are provided a code and need to replace a list of numbers with symbols as per that code. Performance on this test worsens with age, depression, and cognitive impairment.
Participants were followed for more than a decade, and over that time, 559 (about 9%) were diagnosed with dementia.
Those baseline urine samples were assayed for a variety of metals — some essential, some very much not, as you can see in Figure 2.

Now, I have to put my kidney doctor hat on for a second and talk about urine measurement ... of anything. The problem with urine is that the concentration can change a lot — by more than 10-fold, in fact — based on how much water you drank recently. Researchers correct for this, and in the case of this study, they do what a lot of researchers do: divide the measured concentration by the urine creatinine level.

This introduces a bit of a problem. Take two people with exactly the same kidney function, who drank exactly the same water, whose urine is exactly the same concentration. The person with more muscle mass will have more creatinine in that urine sample, since creatinine is a byproduct of muscle metabolism. Because people with more muscle mass are generally healthier, when you divide your metal concentration by urine creatinine, you get a lower number, which might lead you to believe that lower levels of the metal in the urine are protective. But in fact, what you’re seeing is that higher levels of creatinine are protective. I see this issue all the time and it will always color results of studies like this.
Okay, I am doffing my kidney doctor hat now to show you the results.
The researchers first looked at the relationship between metal concentrations in the urine and performance on cognitive tests. The results were fairly equivocal, save for that digit substitution test which is shown in Figure 4.
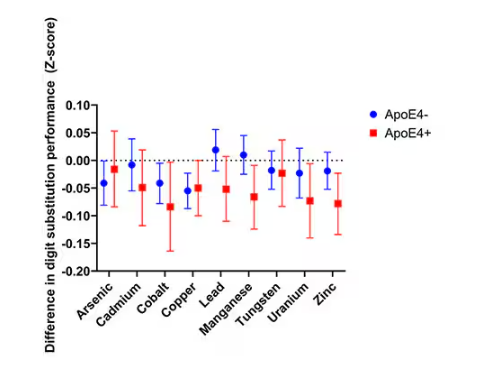
Even these results don’t ring major alarm bells for me. What you’re seeing here is the change in scores on the digit substitution test for each 25-percentile increase in urinary metal level — a pretty big change. And yet, you see really minor changes in the performance on the test. The digit substitution test is not an IQ test; but to give you a feeling for the magnitude of this change, if we looked at copper level, moving from the 25th to the 50th percentile would be associated with a loss of nine tenths of an IQ point.
You see two colors on the Figure 4 graph, by the way. That’s because the researchers stratified their findings based on whether the individual carried the ApoE4 gene allele, which is a risk factor for the development of dementia. There are reasons to believe that neurotoxic metals might be worse in this population, and I suppose you do see generally more adverse effects on scores in the red lines compared with the blue lines. But still, we’re not talking about a huge effect size here.
Let’s look at the relationship between these metals and the development of dementia itself, a clearly more important outcome than how well you can replace numeric digits with symbols. I’ll highlight a few of the results that are particularly telling.
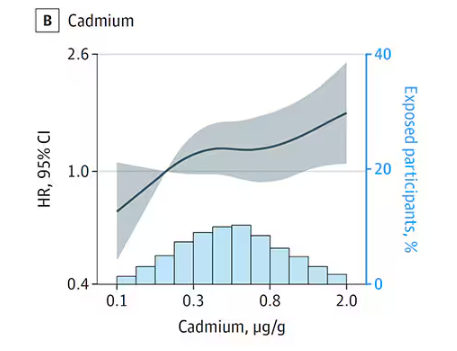
First, the nonessential mineral cadmium, which displays the type of relationship we would expect if the metal were neurotoxic: a clear, roughly linear increase in risk for dementia as urinary concentration increases.
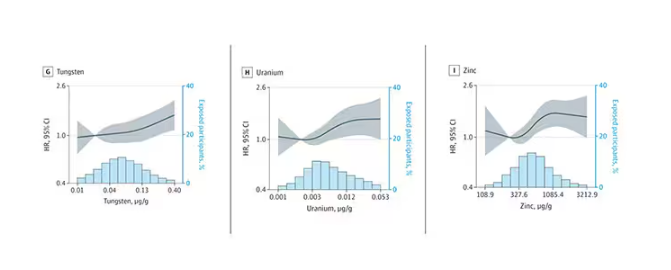
We see roughly similar patterns with the nonessential minerals tungsten and uranium, and the essential mineral zinc (beloved of respiratory-virus avoiders everywhere).
But it is very much not what we see for all metals. Strangest of all, look at lead, which shows basically no relationship with dementia.
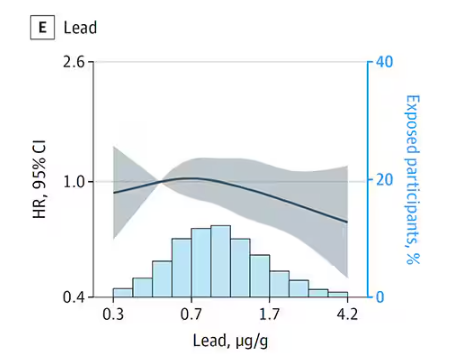
This concerns me a bit. Earlier, I discussed the issue of measuring stuff in urine and how standardizing levels to the urine creatinine level introduces a bias due to muscle mass. One way around this is to standardize urine levels to some other marker of urine dilution, like osmolality. But more fundamental than that, I like to see positive and negative controls in studies like this. For example, lead strikes me as a good positive control here. If the experimental framework were valid, I would think we’d see a relationship between lead level and dementia.
For a negative control? Well, something we are quite sure is not neurotoxic — something like sulfur, which is relatively ubiquitous, used in a variety of biological processes, and efficiently eliminated. We don’t have that in this study.
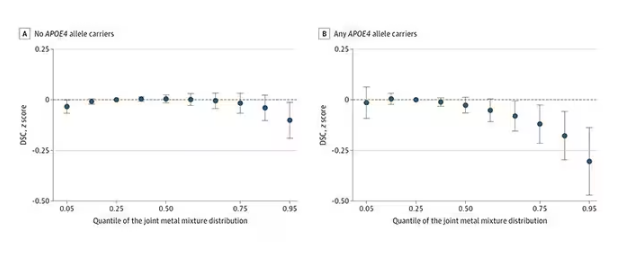
The authors close their case by creating a model that combines all the metal levels, asking the question of whether higher levels of metals in the urine in general worsen cognitive scores. And they find that the relationship exists, as you can see in Figure 8, both in carriers and noncarriers of ApoE4. But, to me, this is even more argument for the creatinine problem. If it’s not a specific metal but just the sort of general concentration of all metals, the risk for confounding by muscle mass is even higher.
So should we worry about ingesting metals? I suppose the answer is ... kind of.
I am sure we should be avoiding lead, despite the results of this study. It’s probably best to stay away from uranium too.
As for the essential metals, I’m sure there is some toxic dose; there’s a toxic dose for everything at some point. But I don’t see evidence in this study to make me worry that a significant chunk of the population is anywhere close to that.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Connecticut. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
It has always amazed me that our bodies require these tiny amounts of incredibly rare substances to function. Sure, we need oxygen. We need water. But we also need molybdenum, which makes up just 1.2 parts per million of the Earth’s crust.
Without adequate molybdenum intake, we develop seizures, developmental delays, death. Fortunately, we need so little molybdenum that true molybdenum deficiency is incredibly rare — seen only in people on total parenteral nutrition without supplementation or those with certain rare genetic conditions. But still, molybdenum is necessary for life.
Many metals are. Figure 1 colors the essential minerals on the periodic table. You can see that to stay alive, we humans need not only things like sodium, but selenium, bromine, zinc, copper, and cobalt.
Some metals are very clearly not essential; we can all do without lead and mercury, and probably should.
But just because something is essential for life does not mean that more is better. The dose is the poison, as they say. And this week, we explore whether metals — even essential metals — might be adversely affecting our brains.
It’s not a stretch to think that metal intake could have weird effects on our nervous system. Lead exposure, primarily due to leaded gasoline, has been blamed for an average reduction of about 3 points in our national IQ, for example . But not all metals are created equal. Researchers set out to find out which might be more strongly associated with performance on cognitive tests and dementia, and reported their results in this study in JAMA Network Open.
To do this, they leveraged the MESA cohort study. This is a longitudinal study of a relatively diverse group of 6300 adults who were enrolled from 2000 to 2002 around the United States. At enrollment, they gave a urine sample and took a variety of cognitive tests. Important for this study was the digit symbol substitution test, where participants are provided a code and need to replace a list of numbers with symbols as per that code. Performance on this test worsens with age, depression, and cognitive impairment.
Participants were followed for more than a decade, and over that time, 559 (about 9%) were diagnosed with dementia.
Those baseline urine samples were assayed for a variety of metals — some essential, some very much not, as you can see in Figure 2.
Now, I have to put my kidney doctor hat on for a second and talk about urine measurement ... of anything. The problem with urine is that the concentration can change a lot — by more than 10-fold, in fact — based on how much water you drank recently. Researchers correct for this, and in the case of this study, they do what a lot of researchers do: divide the measured concentration by the urine creatinine level.
This introduces a bit of a problem. Take two people with exactly the same kidney function, who drank exactly the same water, whose urine is exactly the same concentration. The person with more muscle mass will have more creatinine in that urine sample, since creatinine is a byproduct of muscle metabolism. Because people with more muscle mass are generally healthier, when you divide your metal concentration by urine creatinine, you get a lower number, which might lead you to believe that lower levels of the metal in the urine are protective. But in fact, what you’re seeing is that higher levels of creatinine are protective. I see this issue all the time and it will always color results of studies like this.
Okay, I am doffing my kidney doctor hat now to show you the results.
The researchers first looked at the relationship between metal concentrations in the urine and performance on cognitive tests. The results were fairly equivocal, save for that digit substitution test which is shown in Figure 4.
Even these results don’t ring major alarm bells for me. What you’re seeing here is the change in scores on the digit substitution test for each 25-percentile increase in urinary metal level — a pretty big change. And yet, you see really minor changes in the performance on the test. The digit substitution test is not an IQ test; but to give you a feeling for the magnitude of this change, if we looked at copper level, moving from the 25th to the 50th percentile would be associated with a loss of nine tenths of an IQ point.
You see two colors on the Figure 4 graph, by the way. That’s because the researchers stratified their findings based on whether the individual carried the ApoE4 gene allele, which is a risk factor for the development of dementia. There are reasons to believe that neurotoxic metals might be worse in this population, and I suppose you do see generally more adverse effects on scores in the red lines compared with the blue lines. But still, we’re not talking about a huge effect size here.
Let’s look at the relationship between these metals and the development of dementia itself, a clearly more important outcome than how well you can replace numeric digits with symbols. I’ll highlight a few of the results that are particularly telling.
First, the nonessential mineral cadmium, which displays the type of relationship we would expect if the metal were neurotoxic: a clear, roughly linear increase in risk for dementia as urinary concentration increases.
We see roughly similar patterns with the nonessential minerals tungsten and uranium, and the essential mineral zinc (beloved of respiratory-virus avoiders everywhere).
But it is very much not what we see for all metals. Strangest of all, look at lead, which shows basically no relationship with dementia.
This concerns me a bit. Earlier, I discussed the issue of measuring stuff in urine and how standardizing levels to the urine creatinine level introduces a bias due to muscle mass. One way around this is to standardize urine levels to some other marker of urine dilution, like osmolality. But more fundamental than that, I like to see positive and negative controls in studies like this. For example, lead strikes me as a good positive control here. If the experimental framework were valid, I would think we’d see a relationship between lead level and dementia.
For a negative control? Well, something we are quite sure is not neurotoxic — something like sulfur, which is relatively ubiquitous, used in a variety of biological processes, and efficiently eliminated. We don’t have that in this study.
The authors close their case by creating a model that combines all the metal levels, asking the question of whether higher levels of metals in the urine in general worsen cognitive scores. And they find that the relationship exists, as you can see in Figure 8, both in carriers and noncarriers of ApoE4. But, to me, this is even more argument for the creatinine problem. If it’s not a specific metal but just the sort of general concentration of all metals, the risk for confounding by muscle mass is even higher.
So should we worry about ingesting metals? I suppose the answer is ... kind of.
I am sure we should be avoiding lead, despite the results of this study. It’s probably best to stay away from uranium too.
As for the essential metals, I’m sure there is some toxic dose; there’s a toxic dose for everything at some point. But I don’t see evidence in this study to make me worry that a significant chunk of the population is anywhere close to that.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Connecticut. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
It has always amazed me that our bodies require these tiny amounts of incredibly rare substances to function. Sure, we need oxygen. We need water. But we also need molybdenum, which makes up just 1.2 parts per million of the Earth’s crust.
Without adequate molybdenum intake, we develop seizures, developmental delays, death. Fortunately, we need so little molybdenum that true molybdenum deficiency is incredibly rare — seen only in people on total parenteral nutrition without supplementation or those with certain rare genetic conditions. But still, molybdenum is necessary for life.
Many metals are. Figure 1 colors the essential minerals on the periodic table. You can see that to stay alive, we humans need not only things like sodium, but selenium, bromine, zinc, copper, and cobalt.
Some metals are very clearly not essential; we can all do without lead and mercury, and probably should.
But just because something is essential for life does not mean that more is better. The dose is the poison, as they say. And this week, we explore whether metals — even essential metals — might be adversely affecting our brains.
It’s not a stretch to think that metal intake could have weird effects on our nervous system. Lead exposure, primarily due to leaded gasoline, has been blamed for an average reduction of about 3 points in our national IQ, for example . But not all metals are created equal. Researchers set out to find out which might be more strongly associated with performance on cognitive tests and dementia, and reported their results in this study in JAMA Network Open.
To do this, they leveraged the MESA cohort study. This is a longitudinal study of a relatively diverse group of 6300 adults who were enrolled from 2000 to 2002 around the United States. At enrollment, they gave a urine sample and took a variety of cognitive tests. Important for this study was the digit symbol substitution test, where participants are provided a code and need to replace a list of numbers with symbols as per that code. Performance on this test worsens with age, depression, and cognitive impairment.
Participants were followed for more than a decade, and over that time, 559 (about 9%) were diagnosed with dementia.
Those baseline urine samples were assayed for a variety of metals — some essential, some very much not, as you can see in Figure 2.
Now, I have to put my kidney doctor hat on for a second and talk about urine measurement ... of anything. The problem with urine is that the concentration can change a lot — by more than 10-fold, in fact — based on how much water you drank recently. Researchers correct for this, and in the case of this study, they do what a lot of researchers do: divide the measured concentration by the urine creatinine level.
This introduces a bit of a problem. Take two people with exactly the same kidney function, who drank exactly the same water, whose urine is exactly the same concentration. The person with more muscle mass will have more creatinine in that urine sample, since creatinine is a byproduct of muscle metabolism. Because people with more muscle mass are generally healthier, when you divide your metal concentration by urine creatinine, you get a lower number, which might lead you to believe that lower levels of the metal in the urine are protective. But in fact, what you’re seeing is that higher levels of creatinine are protective. I see this issue all the time and it will always color results of studies like this.
Okay, I am doffing my kidney doctor hat now to show you the results.
The researchers first looked at the relationship between metal concentrations in the urine and performance on cognitive tests. The results were fairly equivocal, save for that digit substitution test which is shown in Figure 4.
Even these results don’t ring major alarm bells for me. What you’re seeing here is the change in scores on the digit substitution test for each 25-percentile increase in urinary metal level — a pretty big change. And yet, you see really minor changes in the performance on the test. The digit substitution test is not an IQ test; but to give you a feeling for the magnitude of this change, if we looked at copper level, moving from the 25th to the 50th percentile would be associated with a loss of nine tenths of an IQ point.
You see two colors on the Figure 4 graph, by the way. That’s because the researchers stratified their findings based on whether the individual carried the ApoE4 gene allele, which is a risk factor for the development of dementia. There are reasons to believe that neurotoxic metals might be worse in this population, and I suppose you do see generally more adverse effects on scores in the red lines compared with the blue lines. But still, we’re not talking about a huge effect size here.
Let’s look at the relationship between these metals and the development of dementia itself, a clearly more important outcome than how well you can replace numeric digits with symbols. I’ll highlight a few of the results that are particularly telling.
First, the nonessential mineral cadmium, which displays the type of relationship we would expect if the metal were neurotoxic: a clear, roughly linear increase in risk for dementia as urinary concentration increases.
We see roughly similar patterns with the nonessential minerals tungsten and uranium, and the essential mineral zinc (beloved of respiratory-virus avoiders everywhere).
But it is very much not what we see for all metals. Strangest of all, look at lead, which shows basically no relationship with dementia.
This concerns me a bit. Earlier, I discussed the issue of measuring stuff in urine and how standardizing levels to the urine creatinine level introduces a bias due to muscle mass. One way around this is to standardize urine levels to some other marker of urine dilution, like osmolality. But more fundamental than that, I like to see positive and negative controls in studies like this. For example, lead strikes me as a good positive control here. If the experimental framework were valid, I would think we’d see a relationship between lead level and dementia.
For a negative control? Well, something we are quite sure is not neurotoxic — something like sulfur, which is relatively ubiquitous, used in a variety of biological processes, and efficiently eliminated. We don’t have that in this study.
The authors close their case by creating a model that combines all the metal levels, asking the question of whether higher levels of metals in the urine in general worsen cognitive scores. And they find that the relationship exists, as you can see in Figure 8, both in carriers and noncarriers of ApoE4. But, to me, this is even more argument for the creatinine problem. If it’s not a specific metal but just the sort of general concentration of all metals, the risk for confounding by muscle mass is even higher.
So should we worry about ingesting metals? I suppose the answer is ... kind of.
I am sure we should be avoiding lead, despite the results of this study. It’s probably best to stay away from uranium too.
As for the essential metals, I’m sure there is some toxic dose; there’s a toxic dose for everything at some point. But I don’t see evidence in this study to make me worry that a significant chunk of the population is anywhere close to that.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Connecticut. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Six Updates on Stroke Management
This video transcript has been edited for clarity.
Dear colleagues, I am Christoph Diener, from the Faculty of Medicine at the University Duisburg-Essen in Germany. In this video, I would like to cover six publications on stroke, which were published this fall.
The Best Thrombolytic?
Let me start with systemic thrombolysis. We now have two thrombolytic agents available. One is the well-known alteplase, and newly approved for the treatment of stroke is tenecteplase. The ATTEST-2 study in the United Kingdom, published in The Lancet Neurology, compared tenecteplase 0.25 mg/kg body weight as a bolus with alteplase 0.9 mg/kg body weight as an infusion over 60 minutes in the 4.5-hour time window in 1777 patients with ischemic stroke.
There was no significant difference between the two thrombolytics for the primary endpoint of modified Rankin Scale score after 90 days. There was also no difference with respect to mortality, intracranial bleeding, or extracranial bleeding.
We finally have 11 randomized controlled trials that compared tenecteplase and alteplase in acute ischemic stroke. A meta-analysis of these randomized trials was published in Neurology. The analysis included 3700 patients treated with tenecteplase and 3700 patients treated with alteplase. For the primary endpoint, excellent functional outcome defined as modified Rankin Scale score 0-1 after 90 days, there was a significant benefit for tenecteplase (relative risk, 1.05), but the absolute difference was very small, at 3%. There was no difference in mortality or bleeding complications.
In conclusion, I think both substances are great. They are effective. Tenecteplase is most probably the drug which should be used in people who have to transfer from a primary stroke center to a dedicated stroke center that provides thrombectomy. Otherwise, I think it’s a choice of the physician as to which thrombolytic agent to use.
Mobile Stroke Units
A highly debated topic is mobile stroke units. These stroke units have a CT scanner and laboratory on board, and this makes it possible to perform thrombolysis on the way to the hospital. A retrospective, observational study collected data between 2018 and 2023, and included 19,400 patients with acute stroke, of whom 1237, or 6.4%, were treated in a mobile stroke unit. This study was published in JAMA Neurology.
The modified Rankin Scale score at the time of discharge was better in patients treated with a mobile stroke unit, but the absolute benefit was only 0.03 points on the modified Rankin Scale. The question is whether this is cost-effective, and can we really do this at times when there is a traumatic shortage of physicians and nursing staff in the hospital?
DOAC Reversal Agents
Oral anticoagulation, as you know, is usually considered a contraindication for systemic thrombolysis. Idarucizumab, a monoclonal antibody, was developed to reverse the biological activity of dabigatran and then allow systemic thrombolysis.
A recent publication in Neurology analyzed 13 cohort studies with 553 stroke patients on dabigatran who received idarucizumab prior to systemic thrombolysis, and the rate of intracranial hemorrhage was 4%. This means it’s obviously possible to perform thrombolysis when the activity of dabigatran is neutralized by idarucizumab.
Unfortunately, until today, we have no data on whether this can also be done with andexanet alfa in people who are treated with a factor Xa inhibitor like, for example, apixaban, rivaroxaban, or edoxaban.
Anticoagulation in ESUS
My next topic is ESUS, or embolic stroke of undetermined source. We have four large randomized trials and three smaller trials that compared antiplatelet therapy with DOACs in patients with ESUS. A group in Neurology published a meta-analysis of seven randomized controlled studies with, altogether, 14,800 patients with ESUS.
The comparison between antiplatelet therapy and anticoagulants showed no difference for recurrent ischemic stroke, and also not for major subgroups. This means that people with ESUS should receive antiplatelet therapy, most probably aspirin.
Anticoagulation Post–Ischemic Stroke With AF
My final topic is the optimal time to start anticoagulation in people with atrial fibrillation who suffer an ischemic stroke. The OPTIMAS study, published in The Lancet, randomized 3650 patients who were anticoagulated with DOACs early (which means less than 4 days) or delayed (between 7 and 14 days). There was no difference in the primary endpoint, which was recurrent ischemic stroke, intracranial hemorrhage, or systemic embolism at 90 days.
The conclusion is that, in most cases, we can probably initiate anticoagulation in people with ischemic stroke and atrial fibrillation within the first 4 days.
Dear colleagues, this is an exciting time for the stroke field. I presented six new studies that have impact, I think, on the management of patients with ischemic stroke.
Dr. Diener is a professor in the Department of Neurology, Stroke Center-Headache Center, University Duisburg-Essen in Germany. He reported conflicts of interest with Abbott, AbbVie, Boehringer Ingelheim, Lundbeck, Novartis, Orion Pharma, Teva, WebMD, and The German Research Council. He also serves on the editorial boards of Cephalalgia, Lancet Neurology, and Drugs.
A version of this article first appeared on Medscape.com.
This video transcript has been edited for clarity.
Dear colleagues, I am Christoph Diener, from the Faculty of Medicine at the University Duisburg-Essen in Germany. In this video, I would like to cover six publications on stroke, which were published this fall.
The Best Thrombolytic?
Let me start with systemic thrombolysis. We now have two thrombolytic agents available. One is the well-known alteplase, and newly approved for the treatment of stroke is tenecteplase. The ATTEST-2 study in the United Kingdom, published in The Lancet Neurology, compared tenecteplase 0.25 mg/kg body weight as a bolus with alteplase 0.9 mg/kg body weight as an infusion over 60 minutes in the 4.5-hour time window in 1777 patients with ischemic stroke.
There was no significant difference between the two thrombolytics for the primary endpoint of modified Rankin Scale score after 90 days. There was also no difference with respect to mortality, intracranial bleeding, or extracranial bleeding.
We finally have 11 randomized controlled trials that compared tenecteplase and alteplase in acute ischemic stroke. A meta-analysis of these randomized trials was published in Neurology. The analysis included 3700 patients treated with tenecteplase and 3700 patients treated with alteplase. For the primary endpoint, excellent functional outcome defined as modified Rankin Scale score 0-1 after 90 days, there was a significant benefit for tenecteplase (relative risk, 1.05), but the absolute difference was very small, at 3%. There was no difference in mortality or bleeding complications.
In conclusion, I think both substances are great. They are effective. Tenecteplase is most probably the drug which should be used in people who have to transfer from a primary stroke center to a dedicated stroke center that provides thrombectomy. Otherwise, I think it’s a choice of the physician as to which thrombolytic agent to use.
Mobile Stroke Units
A highly debated topic is mobile stroke units. These stroke units have a CT scanner and laboratory on board, and this makes it possible to perform thrombolysis on the way to the hospital. A retrospective, observational study collected data between 2018 and 2023, and included 19,400 patients with acute stroke, of whom 1237, or 6.4%, were treated in a mobile stroke unit. This study was published in JAMA Neurology.
The modified Rankin Scale score at the time of discharge was better in patients treated with a mobile stroke unit, but the absolute benefit was only 0.03 points on the modified Rankin Scale. The question is whether this is cost-effective, and can we really do this at times when there is a traumatic shortage of physicians and nursing staff in the hospital?
DOAC Reversal Agents
Oral anticoagulation, as you know, is usually considered a contraindication for systemic thrombolysis. Idarucizumab, a monoclonal antibody, was developed to reverse the biological activity of dabigatran and then allow systemic thrombolysis.
A recent publication in Neurology analyzed 13 cohort studies with 553 stroke patients on dabigatran who received idarucizumab prior to systemic thrombolysis, and the rate of intracranial hemorrhage was 4%. This means it’s obviously possible to perform thrombolysis when the activity of dabigatran is neutralized by idarucizumab.
Unfortunately, until today, we have no data on whether this can also be done with andexanet alfa in people who are treated with a factor Xa inhibitor like, for example, apixaban, rivaroxaban, or edoxaban.
Anticoagulation in ESUS
My next topic is ESUS, or embolic stroke of undetermined source. We have four large randomized trials and three smaller trials that compared antiplatelet therapy with DOACs in patients with ESUS. A group in Neurology published a meta-analysis of seven randomized controlled studies with, altogether, 14,800 patients with ESUS.
The comparison between antiplatelet therapy and anticoagulants showed no difference for recurrent ischemic stroke, and also not for major subgroups. This means that people with ESUS should receive antiplatelet therapy, most probably aspirin.
Anticoagulation Post–Ischemic Stroke With AF
My final topic is the optimal time to start anticoagulation in people with atrial fibrillation who suffer an ischemic stroke. The OPTIMAS study, published in The Lancet, randomized 3650 patients who were anticoagulated with DOACs early (which means less than 4 days) or delayed (between 7 and 14 days). There was no difference in the primary endpoint, which was recurrent ischemic stroke, intracranial hemorrhage, or systemic embolism at 90 days.
The conclusion is that, in most cases, we can probably initiate anticoagulation in people with ischemic stroke and atrial fibrillation within the first 4 days.
Dear colleagues, this is an exciting time for the stroke field. I presented six new studies that have impact, I think, on the management of patients with ischemic stroke.
Dr. Diener is a professor in the Department of Neurology, Stroke Center-Headache Center, University Duisburg-Essen in Germany. He reported conflicts of interest with Abbott, AbbVie, Boehringer Ingelheim, Lundbeck, Novartis, Orion Pharma, Teva, WebMD, and The German Research Council. He also serves on the editorial boards of Cephalalgia, Lancet Neurology, and Drugs.
A version of this article first appeared on Medscape.com.
This video transcript has been edited for clarity.
Dear colleagues, I am Christoph Diener, from the Faculty of Medicine at the University Duisburg-Essen in Germany. In this video, I would like to cover six publications on stroke, which were published this fall.
The Best Thrombolytic?
Let me start with systemic thrombolysis. We now have two thrombolytic agents available. One is the well-known alteplase, and newly approved for the treatment of stroke is tenecteplase. The ATTEST-2 study in the United Kingdom, published in The Lancet Neurology, compared tenecteplase 0.25 mg/kg body weight as a bolus with alteplase 0.9 mg/kg body weight as an infusion over 60 minutes in the 4.5-hour time window in 1777 patients with ischemic stroke.
There was no significant difference between the two thrombolytics for the primary endpoint of modified Rankin Scale score after 90 days. There was also no difference with respect to mortality, intracranial bleeding, or extracranial bleeding.
We finally have 11 randomized controlled trials that compared tenecteplase and alteplase in acute ischemic stroke. A meta-analysis of these randomized trials was published in Neurology. The analysis included 3700 patients treated with tenecteplase and 3700 patients treated with alteplase. For the primary endpoint, excellent functional outcome defined as modified Rankin Scale score 0-1 after 90 days, there was a significant benefit for tenecteplase (relative risk, 1.05), but the absolute difference was very small, at 3%. There was no difference in mortality or bleeding complications.
In conclusion, I think both substances are great. They are effective. Tenecteplase is most probably the drug which should be used in people who have to transfer from a primary stroke center to a dedicated stroke center that provides thrombectomy. Otherwise, I think it’s a choice of the physician as to which thrombolytic agent to use.
Mobile Stroke Units
A highly debated topic is mobile stroke units. These stroke units have a CT scanner and laboratory on board, and this makes it possible to perform thrombolysis on the way to the hospital. A retrospective, observational study collected data between 2018 and 2023, and included 19,400 patients with acute stroke, of whom 1237, or 6.4%, were treated in a mobile stroke unit. This study was published in JAMA Neurology.
The modified Rankin Scale score at the time of discharge was better in patients treated with a mobile stroke unit, but the absolute benefit was only 0.03 points on the modified Rankin Scale. The question is whether this is cost-effective, and can we really do this at times when there is a traumatic shortage of physicians and nursing staff in the hospital?
DOAC Reversal Agents
Oral anticoagulation, as you know, is usually considered a contraindication for systemic thrombolysis. Idarucizumab, a monoclonal antibody, was developed to reverse the biological activity of dabigatran and then allow systemic thrombolysis.
A recent publication in Neurology analyzed 13 cohort studies with 553 stroke patients on dabigatran who received idarucizumab prior to systemic thrombolysis, and the rate of intracranial hemorrhage was 4%. This means it’s obviously possible to perform thrombolysis when the activity of dabigatran is neutralized by idarucizumab.
Unfortunately, until today, we have no data on whether this can also be done with andexanet alfa in people who are treated with a factor Xa inhibitor like, for example, apixaban, rivaroxaban, or edoxaban.
Anticoagulation in ESUS
My next topic is ESUS, or embolic stroke of undetermined source. We have four large randomized trials and three smaller trials that compared antiplatelet therapy with DOACs in patients with ESUS. A group in Neurology published a meta-analysis of seven randomized controlled studies with, altogether, 14,800 patients with ESUS.
The comparison between antiplatelet therapy and anticoagulants showed no difference for recurrent ischemic stroke, and also not for major subgroups. This means that people with ESUS should receive antiplatelet therapy, most probably aspirin.
Anticoagulation Post–Ischemic Stroke With AF
My final topic is the optimal time to start anticoagulation in people with atrial fibrillation who suffer an ischemic stroke. The OPTIMAS study, published in The Lancet, randomized 3650 patients who were anticoagulated with DOACs early (which means less than 4 days) or delayed (between 7 and 14 days). There was no difference in the primary endpoint, which was recurrent ischemic stroke, intracranial hemorrhage, or systemic embolism at 90 days.
The conclusion is that, in most cases, we can probably initiate anticoagulation in people with ischemic stroke and atrial fibrillation within the first 4 days.
Dear colleagues, this is an exciting time for the stroke field. I presented six new studies that have impact, I think, on the management of patients with ischemic stroke.
Dr. Diener is a professor in the Department of Neurology, Stroke Center-Headache Center, University Duisburg-Essen in Germany. He reported conflicts of interest with Abbott, AbbVie, Boehringer Ingelheim, Lundbeck, Novartis, Orion Pharma, Teva, WebMD, and The German Research Council. He also serves on the editorial boards of Cephalalgia, Lancet Neurology, and Drugs.
A version of this article first appeared on Medscape.com.
Is There a Role for GLP-1s in Neurology and Psychiatry?
This transcript has been edited for clarity.
I usually report five or six studies in the field of neurology that were published in the last months, but July was a vacation month.
I decided to cover another topic, which is the role of glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) receptor agonists beyond diabetes and obesity, and in particular, for the field of neurology and psychiatry. Until a few years ago, the treatment of diabetes with traditional antidiabetic drugs was frustrating for vascular neurologists.
These drugs would lower glucose and had an impact on small-vessel disease, but they had no impact on large-vessel disease, stroke, and vascular mortality. This changed with the sodium-glucose cotransporter 2 antagonists because these drugs were not only effective for diabetes, but they also lowered cardiac mortality, in particular, in patients with cardiac failure.
The next generation of antidiabetic drugs were the GLP-1 receptor agonists and the combined GIP/GLP-1 receptor agonists. These two polypeptides and their receptors play a very important role in diabetes and in obesity. The receptors are found not only in the pancreas but also in the intestinal system, the liver, and the central nervous system.
We have a number of preclinical models, mostly in transgenic mice, which show that these drugs are not effective only in diabetes and obesity, but also in liver disease, kidney failure, and neurodegenerative diseases. GLP-1 receptor agonists also have powerful anti-inflammatory properties. These drugs reduce body weight, and they have positive effects on blood pressure and lipid metabolism.
In the studies on the use of GLP-1 receptor agonists in diabetes, a meta-analysis with more than 58,000 patients showed a significant risk reduction for stroke compared with placebo, and this risk reduction was in the range of 80%.
Stroke, Smoking, and Alcohol
A meta-analysis on the use of GLP-1 receptor agonists in over 30,000 nondiabetic patients with obesity found a significant reduction in blood pressure, mortality, and the risk of myocardial infarction. There was no significant decrease in the risk of stroke, but most probably this is due to the fact that strokes are much less frequent in obesity than in diabetes.
You all know that obesity is also a major risk factor for sleep apnea syndrome. Recently, two large studies with the GIP/GLP-1 receptor agonist tirzepatide found a significant improvement in sleep apnea syndrome compared to placebo, regardless of whether patients needed continuous positive airway pressure therapy or not.
In the therapy studies on diabetes and obesity, there were indications that some smokers in the studies stopped their nicotine consumption. A small pilot study with exenatide in 84 overweight patients who were smokers showed that 46% of patients on exenatide stopped smoking compared with 27% in the placebo group. This could be an indication that GLP-1 receptor agonists have activity on the reward system in the brain. Currently, there are a number of larger placebo-controlled trials ongoing.
Another aspect is alcohol consumption. An epidemiologic study in Denmark using data from the National Health Registry showed that the incidence of alcohol-related events decreased significantly in almost 40,000 patients with diabetes when they were treated with GLP-1 receptor agonists compared with other antidiabetic drugs.
A retrospective cohort study from the United States with over 80,000 patients with obesity showed that treatment with GLP-1 receptor agonists was associated with a 50%-60% lower risk for occurrence or recurrence of high alcohol consumption. There is only one small study with exenatide, which was not really informative.
There are a number of studies underway for GLP-1 receptor agonists compared with placebo in patients with alcohol dependence or alcohol consumption. Preclinical models also indicate that these drugs might be effective in cocaine abuse, and there is one placebo-controlled study ongoing.
Parkinson’s Disease
Let’s come to neurology. Preclinical models of Parkinson’s disease have shown neuroprotective activities of GLP-1. Until now, we have three randomized placebo-controlled trials with exenatide, NLY01, and lixisenatide. Two of these studies were positive, showing that the symptoms of Parkinson’s disease were stable over time and deteriorated with placebo. One study was neutral. This means we need more large-scale placebo-controlled studies in the early phases of Parkinson’s disease.
Another potential use of GIP/GLP-1 receptor agonists is in dementia. These substances, as you know, have positive effects on high blood pressure and vascular risk factors.
A working group in China analyzed 27 studies on the treatment of diabetes. A small number of randomized studies and a large number of cohort studies showed that modern antidiabetic drugs reduce the risk for dementia. The risk reduction for dementia for the GLP-1 receptor agonists was 75%. At the moment, there are only small prospective studies and they are not conclusive. Again, we need large-scale placebo-controlled studies.
The most important limitation at the moment beyond the cost is the other adverse drug reactions with the GLP-1 receptor agonists; these include nausea, vomiting, diarrhea, and constipation. There might be a slightly increased risk for pancreatitis. The US Food and Drug Administration recently reported there is no increased risk for suicide. Another potential adverse drug reaction is nonatherosclerotic anterior optic neuropathy.
These drugs, GLP-1 receptor agonists and GIP agonists, are also investigated in a variety of other non-neurologic diseases. The focus here is on metabolic liver disease, such as fatty liver and kidney diseases. Smaller, positive studies have been conducted in this area, and large placebo-controlled trials for both indications are currently underway.
If these diverse therapeutic properties would turn out to be really the case with GLP-1 receptor agonists, this would lead to a significant expansion of the range of indications. If we consider cost, this would be the end of our healthcare systems because we cannot afford this. In addition, the new antidiabetic drugs and the treatment of obesity are available only to a limited extent.
Finally, at least for neurology, it’s unclear whether the impact of these diseases is in the brain or whether it’s indirect, due to the effectiveness on vascular risk factors and concomitant diseases.
Dr. Diener is Professor in the Department of Neurology, Stroke Center-Headache Center, University Duisburg-Essen, Essen, Germany; he has disclosed conflicts of interest with numerous pharmaceutical companies.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
I usually report five or six studies in the field of neurology that were published in the last months, but July was a vacation month.
I decided to cover another topic, which is the role of glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) receptor agonists beyond diabetes and obesity, and in particular, for the field of neurology and psychiatry. Until a few years ago, the treatment of diabetes with traditional antidiabetic drugs was frustrating for vascular neurologists.
These drugs would lower glucose and had an impact on small-vessel disease, but they had no impact on large-vessel disease, stroke, and vascular mortality. This changed with the sodium-glucose cotransporter 2 antagonists because these drugs were not only effective for diabetes, but they also lowered cardiac mortality, in particular, in patients with cardiac failure.
The next generation of antidiabetic drugs were the GLP-1 receptor agonists and the combined GIP/GLP-1 receptor agonists. These two polypeptides and their receptors play a very important role in diabetes and in obesity. The receptors are found not only in the pancreas but also in the intestinal system, the liver, and the central nervous system.
We have a number of preclinical models, mostly in transgenic mice, which show that these drugs are not effective only in diabetes and obesity, but also in liver disease, kidney failure, and neurodegenerative diseases. GLP-1 receptor agonists also have powerful anti-inflammatory properties. These drugs reduce body weight, and they have positive effects on blood pressure and lipid metabolism.
In the studies on the use of GLP-1 receptor agonists in diabetes, a meta-analysis with more than 58,000 patients showed a significant risk reduction for stroke compared with placebo, and this risk reduction was in the range of 80%.
Stroke, Smoking, and Alcohol
A meta-analysis on the use of GLP-1 receptor agonists in over 30,000 nondiabetic patients with obesity found a significant reduction in blood pressure, mortality, and the risk of myocardial infarction. There was no significant decrease in the risk of stroke, but most probably this is due to the fact that strokes are much less frequent in obesity than in diabetes.
You all know that obesity is also a major risk factor for sleep apnea syndrome. Recently, two large studies with the GIP/GLP-1 receptor agonist tirzepatide found a significant improvement in sleep apnea syndrome compared to placebo, regardless of whether patients needed continuous positive airway pressure therapy or not.
In the therapy studies on diabetes and obesity, there were indications that some smokers in the studies stopped their nicotine consumption. A small pilot study with exenatide in 84 overweight patients who were smokers showed that 46% of patients on exenatide stopped smoking compared with 27% in the placebo group. This could be an indication that GLP-1 receptor agonists have activity on the reward system in the brain. Currently, there are a number of larger placebo-controlled trials ongoing.
Another aspect is alcohol consumption. An epidemiologic study in Denmark using data from the National Health Registry showed that the incidence of alcohol-related events decreased significantly in almost 40,000 patients with diabetes when they were treated with GLP-1 receptor agonists compared with other antidiabetic drugs.
A retrospective cohort study from the United States with over 80,000 patients with obesity showed that treatment with GLP-1 receptor agonists was associated with a 50%-60% lower risk for occurrence or recurrence of high alcohol consumption. There is only one small study with exenatide, which was not really informative.
There are a number of studies underway for GLP-1 receptor agonists compared with placebo in patients with alcohol dependence or alcohol consumption. Preclinical models also indicate that these drugs might be effective in cocaine abuse, and there is one placebo-controlled study ongoing.
Parkinson’s Disease
Let’s come to neurology. Preclinical models of Parkinson’s disease have shown neuroprotective activities of GLP-1. Until now, we have three randomized placebo-controlled trials with exenatide, NLY01, and lixisenatide. Two of these studies were positive, showing that the symptoms of Parkinson’s disease were stable over time and deteriorated with placebo. One study was neutral. This means we need more large-scale placebo-controlled studies in the early phases of Parkinson’s disease.
Another potential use of GIP/GLP-1 receptor agonists is in dementia. These substances, as you know, have positive effects on high blood pressure and vascular risk factors.
A working group in China analyzed 27 studies on the treatment of diabetes. A small number of randomized studies and a large number of cohort studies showed that modern antidiabetic drugs reduce the risk for dementia. The risk reduction for dementia for the GLP-1 receptor agonists was 75%. At the moment, there are only small prospective studies and they are not conclusive. Again, we need large-scale placebo-controlled studies.
The most important limitation at the moment beyond the cost is the other adverse drug reactions with the GLP-1 receptor agonists; these include nausea, vomiting, diarrhea, and constipation. There might be a slightly increased risk for pancreatitis. The US Food and Drug Administration recently reported there is no increased risk for suicide. Another potential adverse drug reaction is nonatherosclerotic anterior optic neuropathy.
These drugs, GLP-1 receptor agonists and GIP agonists, are also investigated in a variety of other non-neurologic diseases. The focus here is on metabolic liver disease, such as fatty liver and kidney diseases. Smaller, positive studies have been conducted in this area, and large placebo-controlled trials for both indications are currently underway.
If these diverse therapeutic properties would turn out to be really the case with GLP-1 receptor agonists, this would lead to a significant expansion of the range of indications. If we consider cost, this would be the end of our healthcare systems because we cannot afford this. In addition, the new antidiabetic drugs and the treatment of obesity are available only to a limited extent.
Finally, at least for neurology, it’s unclear whether the impact of these diseases is in the brain or whether it’s indirect, due to the effectiveness on vascular risk factors and concomitant diseases.
Dr. Diener is Professor in the Department of Neurology, Stroke Center-Headache Center, University Duisburg-Essen, Essen, Germany; he has disclosed conflicts of interest with numerous pharmaceutical companies.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
I usually report five or six studies in the field of neurology that were published in the last months, but July was a vacation month.
I decided to cover another topic, which is the role of glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) receptor agonists beyond diabetes and obesity, and in particular, for the field of neurology and psychiatry. Until a few years ago, the treatment of diabetes with traditional antidiabetic drugs was frustrating for vascular neurologists.
These drugs would lower glucose and had an impact on small-vessel disease, but they had no impact on large-vessel disease, stroke, and vascular mortality. This changed with the sodium-glucose cotransporter 2 antagonists because these drugs were not only effective for diabetes, but they also lowered cardiac mortality, in particular, in patients with cardiac failure.
The next generation of antidiabetic drugs were the GLP-1 receptor agonists and the combined GIP/GLP-1 receptor agonists. These two polypeptides and their receptors play a very important role in diabetes and in obesity. The receptors are found not only in the pancreas but also in the intestinal system, the liver, and the central nervous system.
We have a number of preclinical models, mostly in transgenic mice, which show that these drugs are not effective only in diabetes and obesity, but also in liver disease, kidney failure, and neurodegenerative diseases. GLP-1 receptor agonists also have powerful anti-inflammatory properties. These drugs reduce body weight, and they have positive effects on blood pressure and lipid metabolism.
In the studies on the use of GLP-1 receptor agonists in diabetes, a meta-analysis with more than 58,000 patients showed a significant risk reduction for stroke compared with placebo, and this risk reduction was in the range of 80%.
Stroke, Smoking, and Alcohol
A meta-analysis on the use of GLP-1 receptor agonists in over 30,000 nondiabetic patients with obesity found a significant reduction in blood pressure, mortality, and the risk of myocardial infarction. There was no significant decrease in the risk of stroke, but most probably this is due to the fact that strokes are much less frequent in obesity than in diabetes.
You all know that obesity is also a major risk factor for sleep apnea syndrome. Recently, two large studies with the GIP/GLP-1 receptor agonist tirzepatide found a significant improvement in sleep apnea syndrome compared to placebo, regardless of whether patients needed continuous positive airway pressure therapy or not.
In the therapy studies on diabetes and obesity, there were indications that some smokers in the studies stopped their nicotine consumption. A small pilot study with exenatide in 84 overweight patients who were smokers showed that 46% of patients on exenatide stopped smoking compared with 27% in the placebo group. This could be an indication that GLP-1 receptor agonists have activity on the reward system in the brain. Currently, there are a number of larger placebo-controlled trials ongoing.
Another aspect is alcohol consumption. An epidemiologic study in Denmark using data from the National Health Registry showed that the incidence of alcohol-related events decreased significantly in almost 40,000 patients with diabetes when they were treated with GLP-1 receptor agonists compared with other antidiabetic drugs.
A retrospective cohort study from the United States with over 80,000 patients with obesity showed that treatment with GLP-1 receptor agonists was associated with a 50%-60% lower risk for occurrence or recurrence of high alcohol consumption. There is only one small study with exenatide, which was not really informative.
There are a number of studies underway for GLP-1 receptor agonists compared with placebo in patients with alcohol dependence or alcohol consumption. Preclinical models also indicate that these drugs might be effective in cocaine abuse, and there is one placebo-controlled study ongoing.
Parkinson’s Disease
Let’s come to neurology. Preclinical models of Parkinson’s disease have shown neuroprotective activities of GLP-1. Until now, we have three randomized placebo-controlled trials with exenatide, NLY01, and lixisenatide. Two of these studies were positive, showing that the symptoms of Parkinson’s disease were stable over time and deteriorated with placebo. One study was neutral. This means we need more large-scale placebo-controlled studies in the early phases of Parkinson’s disease.
Another potential use of GIP/GLP-1 receptor agonists is in dementia. These substances, as you know, have positive effects on high blood pressure and vascular risk factors.
A working group in China analyzed 27 studies on the treatment of diabetes. A small number of randomized studies and a large number of cohort studies showed that modern antidiabetic drugs reduce the risk for dementia. The risk reduction for dementia for the GLP-1 receptor agonists was 75%. At the moment, there are only small prospective studies and they are not conclusive. Again, we need large-scale placebo-controlled studies.
The most important limitation at the moment beyond the cost is the other adverse drug reactions with the GLP-1 receptor agonists; these include nausea, vomiting, diarrhea, and constipation. There might be a slightly increased risk for pancreatitis. The US Food and Drug Administration recently reported there is no increased risk for suicide. Another potential adverse drug reaction is nonatherosclerotic anterior optic neuropathy.
These drugs, GLP-1 receptor agonists and GIP agonists, are also investigated in a variety of other non-neurologic diseases. The focus here is on metabolic liver disease, such as fatty liver and kidney diseases. Smaller, positive studies have been conducted in this area, and large placebo-controlled trials for both indications are currently underway.
If these diverse therapeutic properties would turn out to be really the case with GLP-1 receptor agonists, this would lead to a significant expansion of the range of indications. If we consider cost, this would be the end of our healthcare systems because we cannot afford this. In addition, the new antidiabetic drugs and the treatment of obesity are available only to a limited extent.
Finally, at least for neurology, it’s unclear whether the impact of these diseases is in the brain or whether it’s indirect, due to the effectiveness on vascular risk factors and concomitant diseases.
Dr. Diener is Professor in the Department of Neurology, Stroke Center-Headache Center, University Duisburg-Essen, Essen, Germany; he has disclosed conflicts of interest with numerous pharmaceutical companies.
A version of this article first appeared on Medscape.com.
New First-Line Therapies for Migraine Prevention
This transcript has been edited for clarity.
Today I am going to talk about the position statement from the American Headache Society (AHS) “Calcitonin gene-related peptide [CGRP]–targeting therapies are a first-line option for the prevention of migraine”. This update is of critical importance because about three fourths of people with migraine get their care from a primary care clinician, not from a neurologist or a headache specialist. CGRP-targeting therapies have transformed migraine care at the specialty level, but many in primary care are not yet familiar with this class of medicines. Until this new statement was released, CGRPs were not viewed as first-line agents for migraine. That has now changed.
Two main types of therapy for people with migraine headache are: (1) acute or abortive therapy (when a headache develops, it is treated), and (2) preventive therapy. Preventive therapy is typically used when the patient has headaches on 4 or more days per month. Preventive therapy is aimed at reducing the frequency and severity of headaches. About 40% of patients with migraine qualify for preventive therapy, but only a minority are receiving it.
The armamentarium for preventive therapy of migraines had not changed in a long time — until now. First-line preventive therapy has traditionally consisted of three classes of agents: beta-blockers, tricyclic antidepressants, and topiramate. These medicines were developed for different therapeutic purposes, yet they work for migraines. These drugs may have off-target effects that can make them difficult to tolerate.
Based on new evidence, candesartan — an angiotensin receptor blocker (ARB) — is now also a first-line drug for migraine. This is good news, because ARBs are a drug class that we have a lot of experience with, are easy to use, and could be an excellent choice for people with concomitant hypertension or chronic kidney disease. The serotonin-norepinephrine reuptake inhibitors (venlafaxine and duloxetine) are also considered first-line agents for migraine treatment.
In the AHS’s new position statement, the two main drug classes are small-molecule CGRP receptor antagonists and monoclonal antibodies.
The role of the neuropeptide CGRP in migraine was originally discovered after finding that blood levels of CGRP were elevated during migraine attacks. This led to the discovery of agents that blocked CGRP, initially for acute treatment of migraine, and then for preventive therapy. Multiple clinical studies show the CGRP targeting therapies to be as or even more effective than traditional first-line agents at decreasing the number of migraine days per month.
The efficacy and safety of these agents have been demonstrated in both randomized trials and in real-world studies. Other important positive endpoints include fewer days of migraine, reduced acute medication use, and improvements in many quality-of-life outcomes. Studies also have shown that CGRP-targeting therapies are well tolerated and safe, with very few serious adverse events.
Furthermore, studies have shown the CGRP targeting therapies are effective in individuals who have failed multiple other first-line therapies. They fit now both as first-line agents and as agents that can be used in difficult-to-treat patients as well as in patients who struggle with acute medication overuse, which is often very challenging.
To quote from the AHS statement,
Side effects are uncommon and can include hypertension, constipation, and Raynaud phenomenon.
The position statement is strong and is based on a lot of evidence and clinical experience. CGRP-targeting therapies are now first-line agents for the prevention of migraine headache. We should learn more about and begin to feel comfortable using this class of agents because they stand to benefit our patients greatly. I’d suggest looking at the table below and picking one new agent to become familiar with so that you can add that agent to your toolbox. 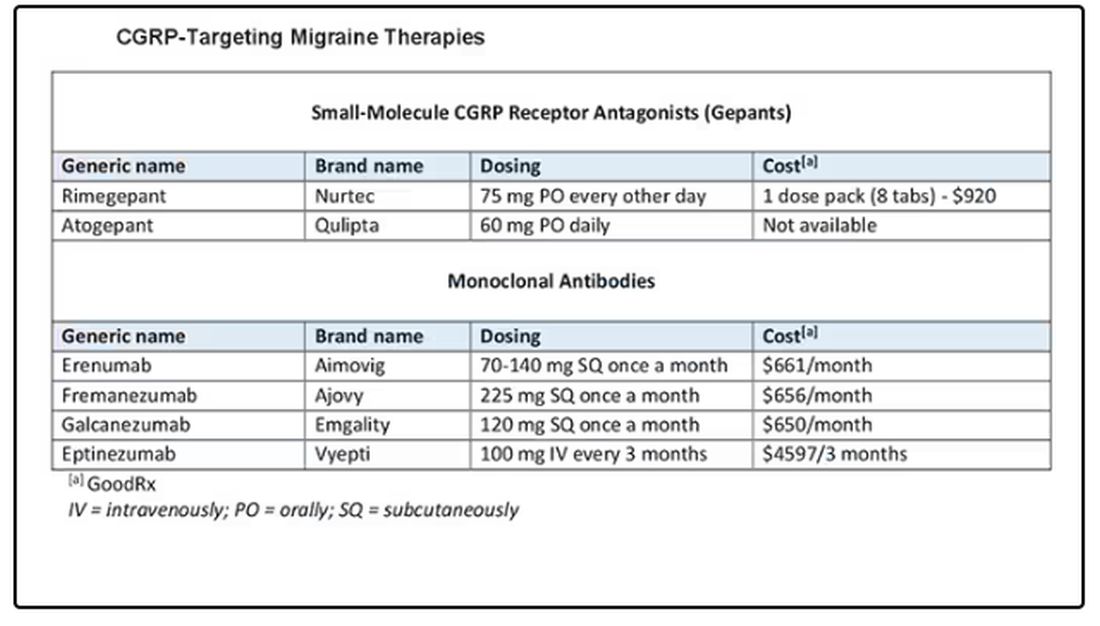
Dr. Skolnik, professor, Department of Family Medicine, Sidney Kimmel Medical College of Thomas Jefferson University, Philadelphia, Pennsylvania, and associate director, Department of Family Medicine, Abington Jefferson Health, Abington, Pennsylvania, disclosed ties with AstraZeneca, Teva, Eli Lilly, Boehringer Ingelheim, Sanofi, Sanofi Pasteur, GlaxoSmithKline, Merck, Bayer, and Teva.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Today I am going to talk about the position statement from the American Headache Society (AHS) “Calcitonin gene-related peptide [CGRP]–targeting therapies are a first-line option for the prevention of migraine”. This update is of critical importance because about three fourths of people with migraine get their care from a primary care clinician, not from a neurologist or a headache specialist. CGRP-targeting therapies have transformed migraine care at the specialty level, but many in primary care are not yet familiar with this class of medicines. Until this new statement was released, CGRPs were not viewed as first-line agents for migraine. That has now changed.
Two main types of therapy for people with migraine headache are: (1) acute or abortive therapy (when a headache develops, it is treated), and (2) preventive therapy. Preventive therapy is typically used when the patient has headaches on 4 or more days per month. Preventive therapy is aimed at reducing the frequency and severity of headaches. About 40% of patients with migraine qualify for preventive therapy, but only a minority are receiving it.
The armamentarium for preventive therapy of migraines had not changed in a long time — until now. First-line preventive therapy has traditionally consisted of three classes of agents: beta-blockers, tricyclic antidepressants, and topiramate. These medicines were developed for different therapeutic purposes, yet they work for migraines. These drugs may have off-target effects that can make them difficult to tolerate.
Based on new evidence, candesartan — an angiotensin receptor blocker (ARB) — is now also a first-line drug for migraine. This is good news, because ARBs are a drug class that we have a lot of experience with, are easy to use, and could be an excellent choice for people with concomitant hypertension or chronic kidney disease. The serotonin-norepinephrine reuptake inhibitors (venlafaxine and duloxetine) are also considered first-line agents for migraine treatment.
In the AHS’s new position statement, the two main drug classes are small-molecule CGRP receptor antagonists and monoclonal antibodies.
The role of the neuropeptide CGRP in migraine was originally discovered after finding that blood levels of CGRP were elevated during migraine attacks. This led to the discovery of agents that blocked CGRP, initially for acute treatment of migraine, and then for preventive therapy. Multiple clinical studies show the CGRP targeting therapies to be as or even more effective than traditional first-line agents at decreasing the number of migraine days per month.
The efficacy and safety of these agents have been demonstrated in both randomized trials and in real-world studies. Other important positive endpoints include fewer days of migraine, reduced acute medication use, and improvements in many quality-of-life outcomes. Studies also have shown that CGRP-targeting therapies are well tolerated and safe, with very few serious adverse events.
Furthermore, studies have shown the CGRP targeting therapies are effective in individuals who have failed multiple other first-line therapies. They fit now both as first-line agents and as agents that can be used in difficult-to-treat patients as well as in patients who struggle with acute medication overuse, which is often very challenging.
To quote from the AHS statement,
Side effects are uncommon and can include hypertension, constipation, and Raynaud phenomenon.
The position statement is strong and is based on a lot of evidence and clinical experience. CGRP-targeting therapies are now first-line agents for the prevention of migraine headache. We should learn more about and begin to feel comfortable using this class of agents because they stand to benefit our patients greatly. I’d suggest looking at the table below and picking one new agent to become familiar with so that you can add that agent to your toolbox.
Dr. Skolnik, professor, Department of Family Medicine, Sidney Kimmel Medical College of Thomas Jefferson University, Philadelphia, Pennsylvania, and associate director, Department of Family Medicine, Abington Jefferson Health, Abington, Pennsylvania, disclosed ties with AstraZeneca, Teva, Eli Lilly, Boehringer Ingelheim, Sanofi, Sanofi Pasteur, GlaxoSmithKline, Merck, Bayer, and Teva.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Today I am going to talk about the position statement from the American Headache Society (AHS) “Calcitonin gene-related peptide [CGRP]–targeting therapies are a first-line option for the prevention of migraine”. This update is of critical importance because about three fourths of people with migraine get their care from a primary care clinician, not from a neurologist or a headache specialist. CGRP-targeting therapies have transformed migraine care at the specialty level, but many in primary care are not yet familiar with this class of medicines. Until this new statement was released, CGRPs were not viewed as first-line agents for migraine. That has now changed.
Two main types of therapy for people with migraine headache are: (1) acute or abortive therapy (when a headache develops, it is treated), and (2) preventive therapy. Preventive therapy is typically used when the patient has headaches on 4 or more days per month. Preventive therapy is aimed at reducing the frequency and severity of headaches. About 40% of patients with migraine qualify for preventive therapy, but only a minority are receiving it.
The armamentarium for preventive therapy of migraines had not changed in a long time — until now. First-line preventive therapy has traditionally consisted of three classes of agents: beta-blockers, tricyclic antidepressants, and topiramate. These medicines were developed for different therapeutic purposes, yet they work for migraines. These drugs may have off-target effects that can make them difficult to tolerate.
Based on new evidence, candesartan — an angiotensin receptor blocker (ARB) — is now also a first-line drug for migraine. This is good news, because ARBs are a drug class that we have a lot of experience with, are easy to use, and could be an excellent choice for people with concomitant hypertension or chronic kidney disease. The serotonin-norepinephrine reuptake inhibitors (venlafaxine and duloxetine) are also considered first-line agents for migraine treatment.
In the AHS’s new position statement, the two main drug classes are small-molecule CGRP receptor antagonists and monoclonal antibodies.
The role of the neuropeptide CGRP in migraine was originally discovered after finding that blood levels of CGRP were elevated during migraine attacks. This led to the discovery of agents that blocked CGRP, initially for acute treatment of migraine, and then for preventive therapy. Multiple clinical studies show the CGRP targeting therapies to be as or even more effective than traditional first-line agents at decreasing the number of migraine days per month.
The efficacy and safety of these agents have been demonstrated in both randomized trials and in real-world studies. Other important positive endpoints include fewer days of migraine, reduced acute medication use, and improvements in many quality-of-life outcomes. Studies also have shown that CGRP-targeting therapies are well tolerated and safe, with very few serious adverse events.
Furthermore, studies have shown the CGRP targeting therapies are effective in individuals who have failed multiple other first-line therapies. They fit now both as first-line agents and as agents that can be used in difficult-to-treat patients as well as in patients who struggle with acute medication overuse, which is often very challenging.
To quote from the AHS statement,
Side effects are uncommon and can include hypertension, constipation, and Raynaud phenomenon.
The position statement is strong and is based on a lot of evidence and clinical experience. CGRP-targeting therapies are now first-line agents for the prevention of migraine headache. We should learn more about and begin to feel comfortable using this class of agents because they stand to benefit our patients greatly. I’d suggest looking at the table below and picking one new agent to become familiar with so that you can add that agent to your toolbox.
Dr. Skolnik, professor, Department of Family Medicine, Sidney Kimmel Medical College of Thomas Jefferson University, Philadelphia, Pennsylvania, and associate director, Department of Family Medicine, Abington Jefferson Health, Abington, Pennsylvania, disclosed ties with AstraZeneca, Teva, Eli Lilly, Boehringer Ingelheim, Sanofi, Sanofi Pasteur, GlaxoSmithKline, Merck, Bayer, and Teva.
A version of this article appeared on Medscape.com.
Is there a new role for metformin in the management of gestational diabetes?
Dunne F, Newman C, Alvarez-Iglesia A, et al. Early metformin in gestational diabetes: a randomized clinical trial. JAMA. 2023;330:1547-1556. doi:10.1001/jama .2023.19869
EXPERT COMMENTARY
Gestational diabetes mellitus occurs in 4% to 7% of pregnancies, and the prevalence is likely to continue to increase given the rising rates of hypertension, obesity, advanced maternal age, and other medical comorbidities in pregnant persons in the United States.1,2 Uncontrolled hyperglycemia in pregnancy is associated swith many adverse perinatal outcomes, including stillbirth, macrosomia, admission to the neonatal intensive care unit (NICU), development of hypertensive disorders, and cesarean deliveries. Hence, it is important to investigate and identify the optimal management of gestational diabetes.
Metformin, an oral biguanide, although studied for gestational diabetes treatment in phase 3 randomized clinical open-label trials, often is avoided in patients who are pregnant (with the exception of patients who have needle aversions, are financially unable to use insulin, or are unable to administer insulin safely).1,2 Metformin is a highly effective first-line agent in the management of both prediabetes and type 2 diabetes, which begs us to question if there is a role for it in the management of gestational diabetes.
Details about the study
The study by Dunne and colleagues was a randomized controlled trial (RCT) conducted in a 1:1 parallel fashion at two institutions in Ireland from 2017–2022. The primary outcome assessed if treatment with metformin would reduce fasting blood glucose levels and the initiation of insulin among women diagnosed with gestational diabetes. A total of 510 participants enrolled in the study, with 268 receiving metformin (up to a maximum dose of 2,500 mg) at diagnosis and 267 receiving an identical placebo. Blood sugar levels were monitored 7 times a day, and medication adherence was assessed every 4 weeks.
Results. At 32 or 38 weeks’ gestation, 56.8% of patients in the metformin arm, and 63.7% of patients in the placebo arm required insulin or had fasting blood glucose levels above 5.1 mmol/L (91.8mg/dL), which was a statistically insignificant difference (P = .13). Although there was similarly no difference in the total amount of insulin used in each study group, the percentage of patients who required insulin initiation was decreased in the metformin arm (38.4% vs 51.1%; P = .004).
Study strengths and weaknesses
The authors conducted a well-designed double-blinded RCT—in both rural and tertiary care settings. Additionally, the study had an impressive 90% patient adherence rate for home blood glucose monitoring 7 times per day. The study arms were balanced for body mass index, as obesity is a known contributor to the development of gestational diabetes and response to insulin.
This study findings’ generalizability is limited across subpopulations given the lack of ethnic and racial diversity—the study population was 80% White. Additionally, utilization of the World Health Organization guidelines for diagnosing gestational diabetes, although adopted by most associations across the world, limits its application to areas of the world that use the National Diabetes Data Group or the Carpenter-Coustan diagnosis guidelines.3,4 Furthermore, the diagnosis of gestational diabetes, which was based on 1 elevated value of a 2-hour glucose tolerance test, has limited scientific support, has not been proven to improve obstetric outcomes, and may increase health care costs when compared with the 2-step method.5 The criteria for insulin initiation in the trial was based on having 2 elevated measures of blood glucose during home glucose monitoring, a criteria that is much stricter than what is used in other countries or clinical practice. The trial authors concluded that use of metformin had a statistically significant reduction in neonates weighing > 4,000 g and > 90th% of weight, but they did not assess study group differences in neonatal skin fold thickness or anthropometric measurements, as reported in the Metformin in Gestational Diabetes trials.6 ●
The study findings by Dunne and colleagues reinforce the current standard practice for the management of gestational diabetes: prescribe medical nutrition therapy and exercise followed by insulin initiation in the setting of persistently elevated blood glucose levels. Knowing that metformin crosses the placenta, future studies should also address the long-term metabolic and health outcomes of fetuses exposed to metformin.
NKECHINYELUM OGU, MD; CHARLOTTE NIZNIK, APRN; MICHELLE A. KOMINIAREK, MD, MS
- Rowan JA, Hague WM, Gao W, et al. Metformin versus insulin for the treatment of gestational diabetes. N Engl J Med. 2008;358:2003-2015. doi: 10.1056/NEJMoa0707193
- American College of Obstetricians and Gynecologists. Gestational diabetes mellitus: Practice Bulletin No. 180. Obstet Gynecol. 2017;130:e17-31. doi: 10.1097/AOG.0000000000002159
- Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. National Diabetes Data Group. Diabetes. 1979;28:1039-1057. doi: 10.2337 /diab.28.12.1039
- Carpenter MW, Coustan DR. Criteria for screening tests for gestational diabetes. Am J Obstet Gynecol. 1982;144:768-773. doi: 10.1016/0002-9378(82)90349-0
- Vandorsten JP, Dodson WC, Espeland MA, et al. NIH consensus development conference: diagnosing gestational diabetes mellitus. NIH Consens State Sci Statements. 2013;29:1-31.
- Rowan JA, Rush EC, Obolonkin V, et al. Metformin in gestational diabetes: the offspring follow-up (MiG TOFU) body composition at 2 years of age. Diabetes Care. 2011;34:2279-2284. https://doi.org/10.2337/dc11-0660
Dunne F, Newman C, Alvarez-Iglesia A, et al. Early metformin in gestational diabetes: a randomized clinical trial. JAMA. 2023;330:1547-1556. doi:10.1001/jama .2023.19869
EXPERT COMMENTARY
Gestational diabetes mellitus occurs in 4% to 7% of pregnancies, and the prevalence is likely to continue to increase given the rising rates of hypertension, obesity, advanced maternal age, and other medical comorbidities in pregnant persons in the United States.1,2 Uncontrolled hyperglycemia in pregnancy is associated swith many adverse perinatal outcomes, including stillbirth, macrosomia, admission to the neonatal intensive care unit (NICU), development of hypertensive disorders, and cesarean deliveries. Hence, it is important to investigate and identify the optimal management of gestational diabetes.
Metformin, an oral biguanide, although studied for gestational diabetes treatment in phase 3 randomized clinical open-label trials, often is avoided in patients who are pregnant (with the exception of patients who have needle aversions, are financially unable to use insulin, or are unable to administer insulin safely).1,2 Metformin is a highly effective first-line agent in the management of both prediabetes and type 2 diabetes, which begs us to question if there is a role for it in the management of gestational diabetes.
Details about the study
The study by Dunne and colleagues was a randomized controlled trial (RCT) conducted in a 1:1 parallel fashion at two institutions in Ireland from 2017–2022. The primary outcome assessed if treatment with metformin would reduce fasting blood glucose levels and the initiation of insulin among women diagnosed with gestational diabetes. A total of 510 participants enrolled in the study, with 268 receiving metformin (up to a maximum dose of 2,500 mg) at diagnosis and 267 receiving an identical placebo. Blood sugar levels were monitored 7 times a day, and medication adherence was assessed every 4 weeks.
Results. At 32 or 38 weeks’ gestation, 56.8% of patients in the metformin arm, and 63.7% of patients in the placebo arm required insulin or had fasting blood glucose levels above 5.1 mmol/L (91.8mg/dL), which was a statistically insignificant difference (P = .13). Although there was similarly no difference in the total amount of insulin used in each study group, the percentage of patients who required insulin initiation was decreased in the metformin arm (38.4% vs 51.1%; P = .004).
Study strengths and weaknesses
The authors conducted a well-designed double-blinded RCT—in both rural and tertiary care settings. Additionally, the study had an impressive 90% patient adherence rate for home blood glucose monitoring 7 times per day. The study arms were balanced for body mass index, as obesity is a known contributor to the development of gestational diabetes and response to insulin.
This study findings’ generalizability is limited across subpopulations given the lack of ethnic and racial diversity—the study population was 80% White. Additionally, utilization of the World Health Organization guidelines for diagnosing gestational diabetes, although adopted by most associations across the world, limits its application to areas of the world that use the National Diabetes Data Group or the Carpenter-Coustan diagnosis guidelines.3,4 Furthermore, the diagnosis of gestational diabetes, which was based on 1 elevated value of a 2-hour glucose tolerance test, has limited scientific support, has not been proven to improve obstetric outcomes, and may increase health care costs when compared with the 2-step method.5 The criteria for insulin initiation in the trial was based on having 2 elevated measures of blood glucose during home glucose monitoring, a criteria that is much stricter than what is used in other countries or clinical practice. The trial authors concluded that use of metformin had a statistically significant reduction in neonates weighing > 4,000 g and > 90th% of weight, but they did not assess study group differences in neonatal skin fold thickness or anthropometric measurements, as reported in the Metformin in Gestational Diabetes trials.6 ●
The study findings by Dunne and colleagues reinforce the current standard practice for the management of gestational diabetes: prescribe medical nutrition therapy and exercise followed by insulin initiation in the setting of persistently elevated blood glucose levels. Knowing that metformin crosses the placenta, future studies should also address the long-term metabolic and health outcomes of fetuses exposed to metformin.
NKECHINYELUM OGU, MD; CHARLOTTE NIZNIK, APRN; MICHELLE A. KOMINIAREK, MD, MS
Dunne F, Newman C, Alvarez-Iglesia A, et al. Early metformin in gestational diabetes: a randomized clinical trial. JAMA. 2023;330:1547-1556. doi:10.1001/jama .2023.19869
EXPERT COMMENTARY
Gestational diabetes mellitus occurs in 4% to 7% of pregnancies, and the prevalence is likely to continue to increase given the rising rates of hypertension, obesity, advanced maternal age, and other medical comorbidities in pregnant persons in the United States.1,2 Uncontrolled hyperglycemia in pregnancy is associated swith many adverse perinatal outcomes, including stillbirth, macrosomia, admission to the neonatal intensive care unit (NICU), development of hypertensive disorders, and cesarean deliveries. Hence, it is important to investigate and identify the optimal management of gestational diabetes.
Metformin, an oral biguanide, although studied for gestational diabetes treatment in phase 3 randomized clinical open-label trials, often is avoided in patients who are pregnant (with the exception of patients who have needle aversions, are financially unable to use insulin, or are unable to administer insulin safely).1,2 Metformin is a highly effective first-line agent in the management of both prediabetes and type 2 diabetes, which begs us to question if there is a role for it in the management of gestational diabetes.
Details about the study
The study by Dunne and colleagues was a randomized controlled trial (RCT) conducted in a 1:1 parallel fashion at two institutions in Ireland from 2017–2022. The primary outcome assessed if treatment with metformin would reduce fasting blood glucose levels and the initiation of insulin among women diagnosed with gestational diabetes. A total of 510 participants enrolled in the study, with 268 receiving metformin (up to a maximum dose of 2,500 mg) at diagnosis and 267 receiving an identical placebo. Blood sugar levels were monitored 7 times a day, and medication adherence was assessed every 4 weeks.
Results. At 32 or 38 weeks’ gestation, 56.8% of patients in the metformin arm, and 63.7% of patients in the placebo arm required insulin or had fasting blood glucose levels above 5.1 mmol/L (91.8mg/dL), which was a statistically insignificant difference (P = .13). Although there was similarly no difference in the total amount of insulin used in each study group, the percentage of patients who required insulin initiation was decreased in the metformin arm (38.4% vs 51.1%; P = .004).
Study strengths and weaknesses
The authors conducted a well-designed double-blinded RCT—in both rural and tertiary care settings. Additionally, the study had an impressive 90% patient adherence rate for home blood glucose monitoring 7 times per day. The study arms were balanced for body mass index, as obesity is a known contributor to the development of gestational diabetes and response to insulin.
This study findings’ generalizability is limited across subpopulations given the lack of ethnic and racial diversity—the study population was 80% White. Additionally, utilization of the World Health Organization guidelines for diagnosing gestational diabetes, although adopted by most associations across the world, limits its application to areas of the world that use the National Diabetes Data Group or the Carpenter-Coustan diagnosis guidelines.3,4 Furthermore, the diagnosis of gestational diabetes, which was based on 1 elevated value of a 2-hour glucose tolerance test, has limited scientific support, has not been proven to improve obstetric outcomes, and may increase health care costs when compared with the 2-step method.5 The criteria for insulin initiation in the trial was based on having 2 elevated measures of blood glucose during home glucose monitoring, a criteria that is much stricter than what is used in other countries or clinical practice. The trial authors concluded that use of metformin had a statistically significant reduction in neonates weighing > 4,000 g and > 90th% of weight, but they did not assess study group differences in neonatal skin fold thickness or anthropometric measurements, as reported in the Metformin in Gestational Diabetes trials.6 ●
The study findings by Dunne and colleagues reinforce the current standard practice for the management of gestational diabetes: prescribe medical nutrition therapy and exercise followed by insulin initiation in the setting of persistently elevated blood glucose levels. Knowing that metformin crosses the placenta, future studies should also address the long-term metabolic and health outcomes of fetuses exposed to metformin.
NKECHINYELUM OGU, MD; CHARLOTTE NIZNIK, APRN; MICHELLE A. KOMINIAREK, MD, MS
- Rowan JA, Hague WM, Gao W, et al. Metformin versus insulin for the treatment of gestational diabetes. N Engl J Med. 2008;358:2003-2015. doi: 10.1056/NEJMoa0707193
- American College of Obstetricians and Gynecologists. Gestational diabetes mellitus: Practice Bulletin No. 180. Obstet Gynecol. 2017;130:e17-31. doi: 10.1097/AOG.0000000000002159
- Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. National Diabetes Data Group. Diabetes. 1979;28:1039-1057. doi: 10.2337 /diab.28.12.1039
- Carpenter MW, Coustan DR. Criteria for screening tests for gestational diabetes. Am J Obstet Gynecol. 1982;144:768-773. doi: 10.1016/0002-9378(82)90349-0
- Vandorsten JP, Dodson WC, Espeland MA, et al. NIH consensus development conference: diagnosing gestational diabetes mellitus. NIH Consens State Sci Statements. 2013;29:1-31.
- Rowan JA, Rush EC, Obolonkin V, et al. Metformin in gestational diabetes: the offspring follow-up (MiG TOFU) body composition at 2 years of age. Diabetes Care. 2011;34:2279-2284. https://doi.org/10.2337/dc11-0660
- Rowan JA, Hague WM, Gao W, et al. Metformin versus insulin for the treatment of gestational diabetes. N Engl J Med. 2008;358:2003-2015. doi: 10.1056/NEJMoa0707193
- American College of Obstetricians and Gynecologists. Gestational diabetes mellitus: Practice Bulletin No. 180. Obstet Gynecol. 2017;130:e17-31. doi: 10.1097/AOG.0000000000002159
- Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. National Diabetes Data Group. Diabetes. 1979;28:1039-1057. doi: 10.2337 /diab.28.12.1039
- Carpenter MW, Coustan DR. Criteria for screening tests for gestational diabetes. Am J Obstet Gynecol. 1982;144:768-773. doi: 10.1016/0002-9378(82)90349-0
- Vandorsten JP, Dodson WC, Espeland MA, et al. NIH consensus development conference: diagnosing gestational diabetes mellitus. NIH Consens State Sci Statements. 2013;29:1-31.
- Rowan JA, Rush EC, Obolonkin V, et al. Metformin in gestational diabetes: the offspring follow-up (MiG TOFU) body composition at 2 years of age. Diabetes Care. 2011;34:2279-2284. https://doi.org/10.2337/dc11-0660
Does taking an NSAID while on hormonal contraception increase VTE risk?
Meaidi A, Mascolo A, Sessa M, et al. Venous thromboembolism with use of hormonal contraception and non-steroidal anti-inflammatory drugs: nationwide cohort study. BMJ. 2023;382:e074450. doi:10.1136/bmj-2022-074450
EXPERT COMMENTARY
Combination (estrogen plus progestin) hormonal contraceptives as well as non–aspirin nonsteroidal anti-inflammatory drugs (NSAIDs) increase the risk of VTE events, including lower extremity clots and pulmonary embolism. Taking contraceptives formulated with ethinyl estradiol increases hepatic production of clotting factors on a dose-related basis. Newer progestins, including desogestrel and drospirenone, also may contribute to an elevated VTE risk, although this association is controversial.1 NSAIDs promote platelet aggregation, thereby activating the clotting system and formation of clots. Although studies that assessed the association between NSAID use and thrombosis have focused on arterial clots, a substantial literature suggests that NSAIDs, including older NSAIDs (such as ibuprofen, diclofenac, and naproxen), also increase VTE risk.2
Although combination contraceptives (oral contraceptives, patches, vaginal rings) and NSAIDs are both commonly used by reproductive-age women, little data have assessed the impact of concomitant use of these medications on VTE risk. Accordingly, investigators in Denmark, using national databases, conducted a retrospective cohort study to assess the impact that independent as well as concomitant use of these medications have on VTE risk.
Details of the study
Meaidi and colleagues included in the cohort reproductive-age women living in Denmark between 1996 and 2017 with no history of thrombosis, thrombophilia, cancer, tubal sterilization, hysterectomy, bilateral oophorectomy, or infertility treatment. National prescription data were used to assess exposure to hormonal contraception.
The investigators classified hormonal contraception into 3 VTE risk categories:
- high risk—estrogen-progestin patches and vaginal rings; oral contraceptives containing 50 µg of ethinyl estradiol; or the progestins desogestrel, drospirenone, gestodene, or cyproterone (with the latter 2 progestins not available in the United States)
- medium risk—all other combination oral contraceptives, including those formulated with the progestins norethindrone, norethindrone acetate, norgestrel, and levonorgestrel, as well as depot medroxyprogesterone acetate
- low/no risk—progestin-only pills, implants, and progestin-containing intrauterine devices (IUDs).
Because in Denmark NSAIDs are prescribed as a single package containing no more than 30 tablets, time exposed to non–aspirin NSAIDs was assumed to last 1 week from the prescription date.
The authors considered first-time diagnoses of lower limb venous thrombosis or pulmonary embolism that were made in hospitals to represent VTE. They also constructed a subgroup of VTE patients in whom the diagnosis was either confirmed with imaging or followed by prescription of an anticoagulant.
To address potential confounding, the authors adjusted their analysis based on age, calendar year, educational attainment, occurrence of pregnancy, surgery, hypertension, diabetes, polycystic ovary syndrome, endometriosis, migraine, systemic connective tissue diseases, inflammatory polyarthropathies, and use of tranexamic acid (a medication that may increase VTE risk). They also censored (temporarily excluded women from analysis) episodes associated with a transiently elevated risk of VTE: pregnancy and 6 months following delivery, 12 weeks after other pregnancy terminations, 8 weeks following any surgery involving hospital admission, and 8 weeks following prescription of tranexamic acid.
Continue to: VTEs associated with risk category of hormonal contraception used...
VTEs associated with risk category of hormonal contraception used
Results. The overall cohort included more than 2 million women who were followed for a median of 10 years. During 21.0 million person-years, 8,710 VTE events were diagnosed; almost one-third of these were pulmonary embolisms, with the remainder diagnosed as lower extremity VTE. Of these 8,710 women diagnosed with VTE, 7,043 (81%) were confirmed with either diagnostic imaging or prescription of an anticoagulant. Unfortunately, 228 women (2.6%) died within 30 days of the diagnosis of VTE.
The investigators identified concomitant use of hormonal contraception and NSAIDs in more than 500,000 women. Among women with such concomitant use, 58% were using contraceptives that were high risk while 23% used medium-risk and 19% used low/no-risk contraceptives. Ibuprofen (60%) was the most commonly used NSAID, followed by diclofenac (20%) and naproxen (6%). Between 97% and 98% of high-risk and medium-risk contraceptives were combination pills; 89% of low/no-risk contraceptives were progestin IUDs.
Compared with nonuse of both hormonal contraceptives and NSAIDs, incidence rate ratios of VTE adjusted for age, calendar year, and education were 8.1 (95% confidence interval [CI], 6.9–9.6) for use of NSAIDs only, 4.2 (95% CI, 4.0–4.4) for use of high-risk contraceptives only, 3.0 (95% CI, 2.8–3.2) for medium-risk contraceptive use, and 1.1 (95% CI, 1.0–1.3) for use of low/no-risk hormonal contraception. Risk of VTE was approximately twice as high with the use of diclofenac only compared with the risks associated with ibuprofen or naproxen use only.
With respect to concomitant use of NSAIDs and hormonal contraception, incidence rate ratios of VTE were 50.6 (95% CI, 44.2–57.8), 26.1 (95% CI, 19.6–34.7), and 5.7 (95% CI, 3.3–10.1), respectively, with use of high-risk, medium-risk, and low/no-risk hormonal contraceptives. Adjusting for time updated information on occurrences of migraine, connective tissue disorder, inflammatory polyarthropathies, endometriosis, polycystic ovary syndrome, hypertension, and diabetes did not materially affect these associations.
When analysis was limited to women without these occurring conditions, rate ratios were somewhat higher (5.7 and 4.1) for use of high-risk and medium-risk contraceptives only. Incidence rate ratios in this subcohort of healthier women were substantially higher for NSAID use only (15.0), and 111.7, 43.2, and 13.0, respectively, for concomitant use of NSAIDs with high-risk, medium-risk, and low/no-risk contraceptives. In this analysis of healthier women, diclofenac continued to be associated with substantially higher risks of VTE than ibuprofen or naproxen. When the stricter definition of VTE (confirmed cases) was used, adjusted rate ratios remained similar.
Absolute risks of VTE
Although some of the elevated rate ratios noted in this study might appear alarming, it is important to keep in mind that the baseline incidence of VTE in healthy reproductive-age women is low. Accordingly, as the authors pointed out, even among women who used NSAIDs concomitantly with high-risk combination hormonal contraceptives, the absolute risk of VTE was 2/10,000.
Study strengths and limitations
Strengths of this analysis by Meaidi and colleagues include the use of large, essentially all-inclusive national registries. In addition, nationwide Danish registry data that indicate a diagnosis of VTE have been found to have a high positive predictive value.3 Another strength is the large number of potentially confounding factors that the authors controlled for.
One potential limitation of their analysis is that the use of only prescribed NSAIDs was considered. Fortunately, however, the prevalence of over-the-counter ibuprofen use in Denmark is not high enough to materially affect the authors’ findings.4 Another potential limitation was that information on smoking and body mass index was not available for most of the women included in the study cohort. The authors countered this limitation by pointing out that, in Denmark, smoking and obesity are highly correlated with educational status, and that all analyses were adjusted for educational status. ●
It is important for clinicians and our patients to recognize that pregnancy—the condition prevented by hormonal contraception— is associated with far higher risks of VTE (10–14 VTE events per 10,000 deliveries) than the use of any modern hormonal contraceptive.5 Although concomitant use of combination contraceptives and NSAIDs increases VTE risk, the absolute risk is modest, particularly when the NSAID is ibuprofen or naproxen (these are the non–aspirin NSAIDs most commonly used in the United States6). Women who regularly take NSAIDs can minimize VTE risk by choosing hormonal contraceptives with little or no impact on the risk of VTE: the progestin implant, progestin IUDs, and progestinonly pills.
ANDREW M. KAUNITZ, MD, MSCP
- Reid RL. Oral hormonal contraception and venous thromboembolism (VTE). Contraception. 2014;89:235-236. doi:10.1016/j.contraception.2014.02.002
- Ungprasert P, Srivali N, Wijarnpreecha K, et al. Nonsteroidal anti-inflammatory drugs and risk of venous thromboembolism: a systematic review and meta-analysis. Rheumatology (Oxford). 2015;54:736-742. doi:10.1093 /rheumatology/keu408
- Sundbøll J, Adelborg K, Munch T, et al. Positive predictive value of cardiovascular diagnoses in the Danish National Patient Registry: a validation study. BMJ Open. 2016;6:e012832. doi:10.1136/bmjopen-2016-012832
- Gaster N, Hallas J, Pottegård A, et al. The validity of Danish prescription data to measure use of aspirin and other nonsteroidal anti-inflammatory drugs and quantification of bias due to non-prescription drug use. Clin Epidemiol. 2021;13:569-579. doi:10.2147/CLEP.S311450
- Maughan BC, Marin M, Han J, et al. Venous thromboembolism during pregnancy and the postpartum period: risk factors, diagnostic testing, and treatment. Obstet Gynecol Surv. 2022;77:433-444. doi:10.1097/OGX.0000000000001043
- Chu A. Ibuprofen, naproxen, and more: the 8 most common NSAIDs. GoodRx. July 20, 2023. Accessed October 4, 2023. https://www.goodrx.com/classes/nsaids/nsaid-list
Meaidi A, Mascolo A, Sessa M, et al. Venous thromboembolism with use of hormonal contraception and non-steroidal anti-inflammatory drugs: nationwide cohort study. BMJ. 2023;382:e074450. doi:10.1136/bmj-2022-074450
EXPERT COMMENTARY
Combination (estrogen plus progestin) hormonal contraceptives as well as non–aspirin nonsteroidal anti-inflammatory drugs (NSAIDs) increase the risk of VTE events, including lower extremity clots and pulmonary embolism. Taking contraceptives formulated with ethinyl estradiol increases hepatic production of clotting factors on a dose-related basis. Newer progestins, including desogestrel and drospirenone, also may contribute to an elevated VTE risk, although this association is controversial.1 NSAIDs promote platelet aggregation, thereby activating the clotting system and formation of clots. Although studies that assessed the association between NSAID use and thrombosis have focused on arterial clots, a substantial literature suggests that NSAIDs, including older NSAIDs (such as ibuprofen, diclofenac, and naproxen), also increase VTE risk.2
Although combination contraceptives (oral contraceptives, patches, vaginal rings) and NSAIDs are both commonly used by reproductive-age women, little data have assessed the impact of concomitant use of these medications on VTE risk. Accordingly, investigators in Denmark, using national databases, conducted a retrospective cohort study to assess the impact that independent as well as concomitant use of these medications have on VTE risk.
Details of the study
Meaidi and colleagues included in the cohort reproductive-age women living in Denmark between 1996 and 2017 with no history of thrombosis, thrombophilia, cancer, tubal sterilization, hysterectomy, bilateral oophorectomy, or infertility treatment. National prescription data were used to assess exposure to hormonal contraception.
The investigators classified hormonal contraception into 3 VTE risk categories:
- high risk—estrogen-progestin patches and vaginal rings; oral contraceptives containing 50 µg of ethinyl estradiol; or the progestins desogestrel, drospirenone, gestodene, or cyproterone (with the latter 2 progestins not available in the United States)
- medium risk—all other combination oral contraceptives, including those formulated with the progestins norethindrone, norethindrone acetate, norgestrel, and levonorgestrel, as well as depot medroxyprogesterone acetate
- low/no risk—progestin-only pills, implants, and progestin-containing intrauterine devices (IUDs).
Because in Denmark NSAIDs are prescribed as a single package containing no more than 30 tablets, time exposed to non–aspirin NSAIDs was assumed to last 1 week from the prescription date.
The authors considered first-time diagnoses of lower limb venous thrombosis or pulmonary embolism that were made in hospitals to represent VTE. They also constructed a subgroup of VTE patients in whom the diagnosis was either confirmed with imaging or followed by prescription of an anticoagulant.
To address potential confounding, the authors adjusted their analysis based on age, calendar year, educational attainment, occurrence of pregnancy, surgery, hypertension, diabetes, polycystic ovary syndrome, endometriosis, migraine, systemic connective tissue diseases, inflammatory polyarthropathies, and use of tranexamic acid (a medication that may increase VTE risk). They also censored (temporarily excluded women from analysis) episodes associated with a transiently elevated risk of VTE: pregnancy and 6 months following delivery, 12 weeks after other pregnancy terminations, 8 weeks following any surgery involving hospital admission, and 8 weeks following prescription of tranexamic acid.
Continue to: VTEs associated with risk category of hormonal contraception used...
VTEs associated with risk category of hormonal contraception used
Results. The overall cohort included more than 2 million women who were followed for a median of 10 years. During 21.0 million person-years, 8,710 VTE events were diagnosed; almost one-third of these were pulmonary embolisms, with the remainder diagnosed as lower extremity VTE. Of these 8,710 women diagnosed with VTE, 7,043 (81%) were confirmed with either diagnostic imaging or prescription of an anticoagulant. Unfortunately, 228 women (2.6%) died within 30 days of the diagnosis of VTE.
The investigators identified concomitant use of hormonal contraception and NSAIDs in more than 500,000 women. Among women with such concomitant use, 58% were using contraceptives that were high risk while 23% used medium-risk and 19% used low/no-risk contraceptives. Ibuprofen (60%) was the most commonly used NSAID, followed by diclofenac (20%) and naproxen (6%). Between 97% and 98% of high-risk and medium-risk contraceptives were combination pills; 89% of low/no-risk contraceptives were progestin IUDs.
Compared with nonuse of both hormonal contraceptives and NSAIDs, incidence rate ratios of VTE adjusted for age, calendar year, and education were 8.1 (95% confidence interval [CI], 6.9–9.6) for use of NSAIDs only, 4.2 (95% CI, 4.0–4.4) for use of high-risk contraceptives only, 3.0 (95% CI, 2.8–3.2) for medium-risk contraceptive use, and 1.1 (95% CI, 1.0–1.3) for use of low/no-risk hormonal contraception. Risk of VTE was approximately twice as high with the use of diclofenac only compared with the risks associated with ibuprofen or naproxen use only.
With respect to concomitant use of NSAIDs and hormonal contraception, incidence rate ratios of VTE were 50.6 (95% CI, 44.2–57.8), 26.1 (95% CI, 19.6–34.7), and 5.7 (95% CI, 3.3–10.1), respectively, with use of high-risk, medium-risk, and low/no-risk hormonal contraceptives. Adjusting for time updated information on occurrences of migraine, connective tissue disorder, inflammatory polyarthropathies, endometriosis, polycystic ovary syndrome, hypertension, and diabetes did not materially affect these associations.
When analysis was limited to women without these occurring conditions, rate ratios were somewhat higher (5.7 and 4.1) for use of high-risk and medium-risk contraceptives only. Incidence rate ratios in this subcohort of healthier women were substantially higher for NSAID use only (15.0), and 111.7, 43.2, and 13.0, respectively, for concomitant use of NSAIDs with high-risk, medium-risk, and low/no-risk contraceptives. In this analysis of healthier women, diclofenac continued to be associated with substantially higher risks of VTE than ibuprofen or naproxen. When the stricter definition of VTE (confirmed cases) was used, adjusted rate ratios remained similar.
Absolute risks of VTE
Although some of the elevated rate ratios noted in this study might appear alarming, it is important to keep in mind that the baseline incidence of VTE in healthy reproductive-age women is low. Accordingly, as the authors pointed out, even among women who used NSAIDs concomitantly with high-risk combination hormonal contraceptives, the absolute risk of VTE was 2/10,000.
Study strengths and limitations
Strengths of this analysis by Meaidi and colleagues include the use of large, essentially all-inclusive national registries. In addition, nationwide Danish registry data that indicate a diagnosis of VTE have been found to have a high positive predictive value.3 Another strength is the large number of potentially confounding factors that the authors controlled for.
One potential limitation of their analysis is that the use of only prescribed NSAIDs was considered. Fortunately, however, the prevalence of over-the-counter ibuprofen use in Denmark is not high enough to materially affect the authors’ findings.4 Another potential limitation was that information on smoking and body mass index was not available for most of the women included in the study cohort. The authors countered this limitation by pointing out that, in Denmark, smoking and obesity are highly correlated with educational status, and that all analyses were adjusted for educational status. ●
It is important for clinicians and our patients to recognize that pregnancy—the condition prevented by hormonal contraception— is associated with far higher risks of VTE (10–14 VTE events per 10,000 deliveries) than the use of any modern hormonal contraceptive.5 Although concomitant use of combination contraceptives and NSAIDs increases VTE risk, the absolute risk is modest, particularly when the NSAID is ibuprofen or naproxen (these are the non–aspirin NSAIDs most commonly used in the United States6). Women who regularly take NSAIDs can minimize VTE risk by choosing hormonal contraceptives with little or no impact on the risk of VTE: the progestin implant, progestin IUDs, and progestinonly pills.
ANDREW M. KAUNITZ, MD, MSCP
Meaidi A, Mascolo A, Sessa M, et al. Venous thromboembolism with use of hormonal contraception and non-steroidal anti-inflammatory drugs: nationwide cohort study. BMJ. 2023;382:e074450. doi:10.1136/bmj-2022-074450
EXPERT COMMENTARY
Combination (estrogen plus progestin) hormonal contraceptives as well as non–aspirin nonsteroidal anti-inflammatory drugs (NSAIDs) increase the risk of VTE events, including lower extremity clots and pulmonary embolism. Taking contraceptives formulated with ethinyl estradiol increases hepatic production of clotting factors on a dose-related basis. Newer progestins, including desogestrel and drospirenone, also may contribute to an elevated VTE risk, although this association is controversial.1 NSAIDs promote platelet aggregation, thereby activating the clotting system and formation of clots. Although studies that assessed the association between NSAID use and thrombosis have focused on arterial clots, a substantial literature suggests that NSAIDs, including older NSAIDs (such as ibuprofen, diclofenac, and naproxen), also increase VTE risk.2
Although combination contraceptives (oral contraceptives, patches, vaginal rings) and NSAIDs are both commonly used by reproductive-age women, little data have assessed the impact of concomitant use of these medications on VTE risk. Accordingly, investigators in Denmark, using national databases, conducted a retrospective cohort study to assess the impact that independent as well as concomitant use of these medications have on VTE risk.
Details of the study
Meaidi and colleagues included in the cohort reproductive-age women living in Denmark between 1996 and 2017 with no history of thrombosis, thrombophilia, cancer, tubal sterilization, hysterectomy, bilateral oophorectomy, or infertility treatment. National prescription data were used to assess exposure to hormonal contraception.
The investigators classified hormonal contraception into 3 VTE risk categories:
- high risk—estrogen-progestin patches and vaginal rings; oral contraceptives containing 50 µg of ethinyl estradiol; or the progestins desogestrel, drospirenone, gestodene, or cyproterone (with the latter 2 progestins not available in the United States)
- medium risk—all other combination oral contraceptives, including those formulated with the progestins norethindrone, norethindrone acetate, norgestrel, and levonorgestrel, as well as depot medroxyprogesterone acetate
- low/no risk—progestin-only pills, implants, and progestin-containing intrauterine devices (IUDs).
Because in Denmark NSAIDs are prescribed as a single package containing no more than 30 tablets, time exposed to non–aspirin NSAIDs was assumed to last 1 week from the prescription date.
The authors considered first-time diagnoses of lower limb venous thrombosis or pulmonary embolism that were made in hospitals to represent VTE. They also constructed a subgroup of VTE patients in whom the diagnosis was either confirmed with imaging or followed by prescription of an anticoagulant.
To address potential confounding, the authors adjusted their analysis based on age, calendar year, educational attainment, occurrence of pregnancy, surgery, hypertension, diabetes, polycystic ovary syndrome, endometriosis, migraine, systemic connective tissue diseases, inflammatory polyarthropathies, and use of tranexamic acid (a medication that may increase VTE risk). They also censored (temporarily excluded women from analysis) episodes associated with a transiently elevated risk of VTE: pregnancy and 6 months following delivery, 12 weeks after other pregnancy terminations, 8 weeks following any surgery involving hospital admission, and 8 weeks following prescription of tranexamic acid.
Continue to: VTEs associated with risk category of hormonal contraception used...
VTEs associated with risk category of hormonal contraception used
Results. The overall cohort included more than 2 million women who were followed for a median of 10 years. During 21.0 million person-years, 8,710 VTE events were diagnosed; almost one-third of these were pulmonary embolisms, with the remainder diagnosed as lower extremity VTE. Of these 8,710 women diagnosed with VTE, 7,043 (81%) were confirmed with either diagnostic imaging or prescription of an anticoagulant. Unfortunately, 228 women (2.6%) died within 30 days of the diagnosis of VTE.
The investigators identified concomitant use of hormonal contraception and NSAIDs in more than 500,000 women. Among women with such concomitant use, 58% were using contraceptives that were high risk while 23% used medium-risk and 19% used low/no-risk contraceptives. Ibuprofen (60%) was the most commonly used NSAID, followed by diclofenac (20%) and naproxen (6%). Between 97% and 98% of high-risk and medium-risk contraceptives were combination pills; 89% of low/no-risk contraceptives were progestin IUDs.
Compared with nonuse of both hormonal contraceptives and NSAIDs, incidence rate ratios of VTE adjusted for age, calendar year, and education were 8.1 (95% confidence interval [CI], 6.9–9.6) for use of NSAIDs only, 4.2 (95% CI, 4.0–4.4) for use of high-risk contraceptives only, 3.0 (95% CI, 2.8–3.2) for medium-risk contraceptive use, and 1.1 (95% CI, 1.0–1.3) for use of low/no-risk hormonal contraception. Risk of VTE was approximately twice as high with the use of diclofenac only compared with the risks associated with ibuprofen or naproxen use only.
With respect to concomitant use of NSAIDs and hormonal contraception, incidence rate ratios of VTE were 50.6 (95% CI, 44.2–57.8), 26.1 (95% CI, 19.6–34.7), and 5.7 (95% CI, 3.3–10.1), respectively, with use of high-risk, medium-risk, and low/no-risk hormonal contraceptives. Adjusting for time updated information on occurrences of migraine, connective tissue disorder, inflammatory polyarthropathies, endometriosis, polycystic ovary syndrome, hypertension, and diabetes did not materially affect these associations.
When analysis was limited to women without these occurring conditions, rate ratios were somewhat higher (5.7 and 4.1) for use of high-risk and medium-risk contraceptives only. Incidence rate ratios in this subcohort of healthier women were substantially higher for NSAID use only (15.0), and 111.7, 43.2, and 13.0, respectively, for concomitant use of NSAIDs with high-risk, medium-risk, and low/no-risk contraceptives. In this analysis of healthier women, diclofenac continued to be associated with substantially higher risks of VTE than ibuprofen or naproxen. When the stricter definition of VTE (confirmed cases) was used, adjusted rate ratios remained similar.
Absolute risks of VTE
Although some of the elevated rate ratios noted in this study might appear alarming, it is important to keep in mind that the baseline incidence of VTE in healthy reproductive-age women is low. Accordingly, as the authors pointed out, even among women who used NSAIDs concomitantly with high-risk combination hormonal contraceptives, the absolute risk of VTE was 2/10,000.
Study strengths and limitations
Strengths of this analysis by Meaidi and colleagues include the use of large, essentially all-inclusive national registries. In addition, nationwide Danish registry data that indicate a diagnosis of VTE have been found to have a high positive predictive value.3 Another strength is the large number of potentially confounding factors that the authors controlled for.
One potential limitation of their analysis is that the use of only prescribed NSAIDs was considered. Fortunately, however, the prevalence of over-the-counter ibuprofen use in Denmark is not high enough to materially affect the authors’ findings.4 Another potential limitation was that information on smoking and body mass index was not available for most of the women included in the study cohort. The authors countered this limitation by pointing out that, in Denmark, smoking and obesity are highly correlated with educational status, and that all analyses were adjusted for educational status. ●
It is important for clinicians and our patients to recognize that pregnancy—the condition prevented by hormonal contraception— is associated with far higher risks of VTE (10–14 VTE events per 10,000 deliveries) than the use of any modern hormonal contraceptive.5 Although concomitant use of combination contraceptives and NSAIDs increases VTE risk, the absolute risk is modest, particularly when the NSAID is ibuprofen or naproxen (these are the non–aspirin NSAIDs most commonly used in the United States6). Women who regularly take NSAIDs can minimize VTE risk by choosing hormonal contraceptives with little or no impact on the risk of VTE: the progestin implant, progestin IUDs, and progestinonly pills.
ANDREW M. KAUNITZ, MD, MSCP
- Reid RL. Oral hormonal contraception and venous thromboembolism (VTE). Contraception. 2014;89:235-236. doi:10.1016/j.contraception.2014.02.002
- Ungprasert P, Srivali N, Wijarnpreecha K, et al. Nonsteroidal anti-inflammatory drugs and risk of venous thromboembolism: a systematic review and meta-analysis. Rheumatology (Oxford). 2015;54:736-742. doi:10.1093 /rheumatology/keu408
- Sundbøll J, Adelborg K, Munch T, et al. Positive predictive value of cardiovascular diagnoses in the Danish National Patient Registry: a validation study. BMJ Open. 2016;6:e012832. doi:10.1136/bmjopen-2016-012832
- Gaster N, Hallas J, Pottegård A, et al. The validity of Danish prescription data to measure use of aspirin and other nonsteroidal anti-inflammatory drugs and quantification of bias due to non-prescription drug use. Clin Epidemiol. 2021;13:569-579. doi:10.2147/CLEP.S311450
- Maughan BC, Marin M, Han J, et al. Venous thromboembolism during pregnancy and the postpartum period: risk factors, diagnostic testing, and treatment. Obstet Gynecol Surv. 2022;77:433-444. doi:10.1097/OGX.0000000000001043
- Chu A. Ibuprofen, naproxen, and more: the 8 most common NSAIDs. GoodRx. July 20, 2023. Accessed October 4, 2023. https://www.goodrx.com/classes/nsaids/nsaid-list
- Reid RL. Oral hormonal contraception and venous thromboembolism (VTE). Contraception. 2014;89:235-236. doi:10.1016/j.contraception.2014.02.002
- Ungprasert P, Srivali N, Wijarnpreecha K, et al. Nonsteroidal anti-inflammatory drugs and risk of venous thromboembolism: a systematic review and meta-analysis. Rheumatology (Oxford). 2015;54:736-742. doi:10.1093 /rheumatology/keu408
- Sundbøll J, Adelborg K, Munch T, et al. Positive predictive value of cardiovascular diagnoses in the Danish National Patient Registry: a validation study. BMJ Open. 2016;6:e012832. doi:10.1136/bmjopen-2016-012832
- Gaster N, Hallas J, Pottegård A, et al. The validity of Danish prescription data to measure use of aspirin and other nonsteroidal anti-inflammatory drugs and quantification of bias due to non-prescription drug use. Clin Epidemiol. 2021;13:569-579. doi:10.2147/CLEP.S311450
- Maughan BC, Marin M, Han J, et al. Venous thromboembolism during pregnancy and the postpartum period: risk factors, diagnostic testing, and treatment. Obstet Gynecol Surv. 2022;77:433-444. doi:10.1097/OGX.0000000000001043
- Chu A. Ibuprofen, naproxen, and more: the 8 most common NSAIDs. GoodRx. July 20, 2023. Accessed October 4, 2023. https://www.goodrx.com/classes/nsaids/nsaid-list
Is the 9-valent HPV vaccine safe and effective long term?
Restrepo J, Herrera T, Samakoses R, et al. Ten-year follow-up of 9-valent human papillomavirus vaccine: immunogenicity, effectiveness, and safety. Pediatrics. 2023;152:e2022060993. doi:10.1542/peds.2022-060993
EXPERT COMMENTARY
Infection with human papillomavirus (HPV) is associated with nearly all cases of cervical cancer. Long-term safety and efficacy of the bivalent (Cervarix) and quadrivalent (Gardasil) vaccines have been demonstrated for up to 10 to 14 years.1-6 It is estimated that the 9-valent vaccine (Gardasil 9), which was licensed in 2014 and protects against HPV 16/18/31/33/45/52/58 and HPV 6/11, could prevent up to 90% of cervical cancer cases. The bivalent and quadrivalent vaccines could ideally prevent 70% of cases of cervical cancer. In a recent study, authors compared the efficacy and safety of the newer 9-valent vaccine at 10 years with long-term outcomes of previous vaccine studies.7
Details of the study
Study V503-002 conducted by Luxembourg and colleagues originally enrolled 1,935 boys and girls from 66 sites in Africa, Asia, Europe, Latin America, and North America to receive 3 doses of the 9-valent HPV vaccine, with follow-up for 12 to 36 months to monitor safety and immunogenicity.8 In an extension of this investigation, Restrepo and colleagues revisited 40 of these sites in 13 countries to gather 10 years of long-term follow-up data.7
The final long-term follow-up cohort included 971 girls and 301 boys aged 9 to 15 at vaccination.
Results. At month 126, participants continued to have very high seropositive rates (81%–100%, depending on assay sensitivity and HPV type). There were no cases of high-grade cervical, vaginal, or vulvar dysplasia related to HPV strains covered in the vaccine. Rates of infection in women with the vaccine-targeted HPV types were very low—54.6 per 10,000 person-years—compared with 927.4 per 10,000 person-years for HPV types not included in the vaccine. No adverse events attributable to the vaccine were reported.
Study strengths and limitations
Strengths of this study included the use of rigorous end points similar to those used in the initial efficacy studies for easy comparison. Limitations included the relatively small size, which precluded a robust assessment of adverse events, as well as the lack of controls. Furthermore, this study looked at children receiving 3 doses of HPV vaccine prior to the age of 15 and may not be generalizable to people who receive the vaccine at an older age or in fewer doses. ●
Previous studies have shown that the 9-valent HPV vaccine is effective and yields immunological responses within 4 weeks of receiving 3 doses, with sustained immunogenicity up to 36 months. The study by Restrepo and colleagues provides long-term follow-up data that demonstrated sustained immunological responses at 10 years following immunization, with no cases of high-grade intraepithelial neoplasia related to the covered HPV types and no adverse events. These results compare favorably with those of prior studies of the bivalent and quadrivalent HPV vaccines. The 9-valent HPV vaccine can be recommended for use in children aged 9 to 15 with excellent confidence regarding its safety and sustained effectiveness for at least 10 years after vaccination.
DIANA MIAO, MD; SARAH FELDMAN, MD, MPH
- Naud PS, Roteli-Martins CM, De Carvalho NS, et al. Sustained efficacy, immunogenicity, and safety of the HPV-16/18 AS04-adjuvanted vaccine: final analysis of a long-term follow-up study up to 9.4 years post-vaccination. Hum Vaccin Immunother. 2014;10:2147-2162. doi:10.4161/hv.29532
- Schwarz TF, Galaj A, Spaczynski M, et al. Ten-year immune persistence and safety of the HPV-16/18 AS04-adjuvanted vaccine in females vaccinated at 15–55 years of age. Cancer Med. 2017;6:2723-2731. doi:10.1002/cam4.1155
- Ferris DG, Samakoses R, Block SL, et al. 4-valent human papillomavirus (4vHPV) vaccine in preadolescents and adolescents after 10 years. Pediatrics. 2017;140:e20163947. doi:10.1542/peds.2016-3947
- Kjaer SK, Nygård M, Sundström K, et al. Final analysis of a 14-year long-term follow-up study of the effectiveness and immunogenicity of the quadrivalent human papillomavirus vaccine in women from four Nordic countries. EClinicalMedicine. 2020;23:100401. doi:10.1016 /j.eclinm.2020.100401
- Porras C, Tsang SH, Herrero R, et al; Costa Rica Vaccine Trial Group. Efficacy of the bivalent HPV vaccine against HPV 16/18-associated precancer: long-term follow-up results from the Costa Rica Vaccine Trial. Lancet Oncol. 2020;21:16431652. doi:10.1016/S1470-2045(20)30524-6
- Van Damme P, Olsson SE, Block S, et al. Immunogenicity and safety of a 9-valent HPV vaccine. Pediatrics. 2015;136:e28-e39. doi:10.1542/peds.2014-3745
- Restrepo J, Herrera T, Samakoses R, et al. Ten-year follow-up of 9-valent human papillomavirus vaccine: immunogenicity, effectiveness, and safety. Pediatrics. 2023;152:e2022060993. doi:10.1542/peds.2022-060993
- Luxembourg A, Moreira Jr ED, Samakoses R, et al. Phase III, randomized controlled trial in girls 9-15 years old to evaluate lot consistency of a novel nine-valent human papillomavirus L1 virus-like particle vaccine. Hum Vaccin Immunother. 11:1306-1312. doi:10.1080/21645515.2015.1009819
Restrepo J, Herrera T, Samakoses R, et al. Ten-year follow-up of 9-valent human papillomavirus vaccine: immunogenicity, effectiveness, and safety. Pediatrics. 2023;152:e2022060993. doi:10.1542/peds.2022-060993
EXPERT COMMENTARY
Infection with human papillomavirus (HPV) is associated with nearly all cases of cervical cancer. Long-term safety and efficacy of the bivalent (Cervarix) and quadrivalent (Gardasil) vaccines have been demonstrated for up to 10 to 14 years.1-6 It is estimated that the 9-valent vaccine (Gardasil 9), which was licensed in 2014 and protects against HPV 16/18/31/33/45/52/58 and HPV 6/11, could prevent up to 90% of cervical cancer cases. The bivalent and quadrivalent vaccines could ideally prevent 70% of cases of cervical cancer. In a recent study, authors compared the efficacy and safety of the newer 9-valent vaccine at 10 years with long-term outcomes of previous vaccine studies.7
Details of the study
Study V503-002 conducted by Luxembourg and colleagues originally enrolled 1,935 boys and girls from 66 sites in Africa, Asia, Europe, Latin America, and North America to receive 3 doses of the 9-valent HPV vaccine, with follow-up for 12 to 36 months to monitor safety and immunogenicity.8 In an extension of this investigation, Restrepo and colleagues revisited 40 of these sites in 13 countries to gather 10 years of long-term follow-up data.7
The final long-term follow-up cohort included 971 girls and 301 boys aged 9 to 15 at vaccination.
Results. At month 126, participants continued to have very high seropositive rates (81%–100%, depending on assay sensitivity and HPV type). There were no cases of high-grade cervical, vaginal, or vulvar dysplasia related to HPV strains covered in the vaccine. Rates of infection in women with the vaccine-targeted HPV types were very low—54.6 per 10,000 person-years—compared with 927.4 per 10,000 person-years for HPV types not included in the vaccine. No adverse events attributable to the vaccine were reported.
Study strengths and limitations
Strengths of this study included the use of rigorous end points similar to those used in the initial efficacy studies for easy comparison. Limitations included the relatively small size, which precluded a robust assessment of adverse events, as well as the lack of controls. Furthermore, this study looked at children receiving 3 doses of HPV vaccine prior to the age of 15 and may not be generalizable to people who receive the vaccine at an older age or in fewer doses. ●
Previous studies have shown that the 9-valent HPV vaccine is effective and yields immunological responses within 4 weeks of receiving 3 doses, with sustained immunogenicity up to 36 months. The study by Restrepo and colleagues provides long-term follow-up data that demonstrated sustained immunological responses at 10 years following immunization, with no cases of high-grade intraepithelial neoplasia related to the covered HPV types and no adverse events. These results compare favorably with those of prior studies of the bivalent and quadrivalent HPV vaccines. The 9-valent HPV vaccine can be recommended for use in children aged 9 to 15 with excellent confidence regarding its safety and sustained effectiveness for at least 10 years after vaccination.
DIANA MIAO, MD; SARAH FELDMAN, MD, MPH
Restrepo J, Herrera T, Samakoses R, et al. Ten-year follow-up of 9-valent human papillomavirus vaccine: immunogenicity, effectiveness, and safety. Pediatrics. 2023;152:e2022060993. doi:10.1542/peds.2022-060993
EXPERT COMMENTARY
Infection with human papillomavirus (HPV) is associated with nearly all cases of cervical cancer. Long-term safety and efficacy of the bivalent (Cervarix) and quadrivalent (Gardasil) vaccines have been demonstrated for up to 10 to 14 years.1-6 It is estimated that the 9-valent vaccine (Gardasil 9), which was licensed in 2014 and protects against HPV 16/18/31/33/45/52/58 and HPV 6/11, could prevent up to 90% of cervical cancer cases. The bivalent and quadrivalent vaccines could ideally prevent 70% of cases of cervical cancer. In a recent study, authors compared the efficacy and safety of the newer 9-valent vaccine at 10 years with long-term outcomes of previous vaccine studies.7
Details of the study
Study V503-002 conducted by Luxembourg and colleagues originally enrolled 1,935 boys and girls from 66 sites in Africa, Asia, Europe, Latin America, and North America to receive 3 doses of the 9-valent HPV vaccine, with follow-up for 12 to 36 months to monitor safety and immunogenicity.8 In an extension of this investigation, Restrepo and colleagues revisited 40 of these sites in 13 countries to gather 10 years of long-term follow-up data.7
The final long-term follow-up cohort included 971 girls and 301 boys aged 9 to 15 at vaccination.
Results. At month 126, participants continued to have very high seropositive rates (81%–100%, depending on assay sensitivity and HPV type). There were no cases of high-grade cervical, vaginal, or vulvar dysplasia related to HPV strains covered in the vaccine. Rates of infection in women with the vaccine-targeted HPV types were very low—54.6 per 10,000 person-years—compared with 927.4 per 10,000 person-years for HPV types not included in the vaccine. No adverse events attributable to the vaccine were reported.
Study strengths and limitations
Strengths of this study included the use of rigorous end points similar to those used in the initial efficacy studies for easy comparison. Limitations included the relatively small size, which precluded a robust assessment of adverse events, as well as the lack of controls. Furthermore, this study looked at children receiving 3 doses of HPV vaccine prior to the age of 15 and may not be generalizable to people who receive the vaccine at an older age or in fewer doses. ●
Previous studies have shown that the 9-valent HPV vaccine is effective and yields immunological responses within 4 weeks of receiving 3 doses, with sustained immunogenicity up to 36 months. The study by Restrepo and colleagues provides long-term follow-up data that demonstrated sustained immunological responses at 10 years following immunization, with no cases of high-grade intraepithelial neoplasia related to the covered HPV types and no adverse events. These results compare favorably with those of prior studies of the bivalent and quadrivalent HPV vaccines. The 9-valent HPV vaccine can be recommended for use in children aged 9 to 15 with excellent confidence regarding its safety and sustained effectiveness for at least 10 years after vaccination.
DIANA MIAO, MD; SARAH FELDMAN, MD, MPH
- Naud PS, Roteli-Martins CM, De Carvalho NS, et al. Sustained efficacy, immunogenicity, and safety of the HPV-16/18 AS04-adjuvanted vaccine: final analysis of a long-term follow-up study up to 9.4 years post-vaccination. Hum Vaccin Immunother. 2014;10:2147-2162. doi:10.4161/hv.29532
- Schwarz TF, Galaj A, Spaczynski M, et al. Ten-year immune persistence and safety of the HPV-16/18 AS04-adjuvanted vaccine in females vaccinated at 15–55 years of age. Cancer Med. 2017;6:2723-2731. doi:10.1002/cam4.1155
- Ferris DG, Samakoses R, Block SL, et al. 4-valent human papillomavirus (4vHPV) vaccine in preadolescents and adolescents after 10 years. Pediatrics. 2017;140:e20163947. doi:10.1542/peds.2016-3947
- Kjaer SK, Nygård M, Sundström K, et al. Final analysis of a 14-year long-term follow-up study of the effectiveness and immunogenicity of the quadrivalent human papillomavirus vaccine in women from four Nordic countries. EClinicalMedicine. 2020;23:100401. doi:10.1016 /j.eclinm.2020.100401
- Porras C, Tsang SH, Herrero R, et al; Costa Rica Vaccine Trial Group. Efficacy of the bivalent HPV vaccine against HPV 16/18-associated precancer: long-term follow-up results from the Costa Rica Vaccine Trial. Lancet Oncol. 2020;21:16431652. doi:10.1016/S1470-2045(20)30524-6
- Van Damme P, Olsson SE, Block S, et al. Immunogenicity and safety of a 9-valent HPV vaccine. Pediatrics. 2015;136:e28-e39. doi:10.1542/peds.2014-3745
- Restrepo J, Herrera T, Samakoses R, et al. Ten-year follow-up of 9-valent human papillomavirus vaccine: immunogenicity, effectiveness, and safety. Pediatrics. 2023;152:e2022060993. doi:10.1542/peds.2022-060993
- Luxembourg A, Moreira Jr ED, Samakoses R, et al. Phase III, randomized controlled trial in girls 9-15 years old to evaluate lot consistency of a novel nine-valent human papillomavirus L1 virus-like particle vaccine. Hum Vaccin Immunother. 11:1306-1312. doi:10.1080/21645515.2015.1009819
- Naud PS, Roteli-Martins CM, De Carvalho NS, et al. Sustained efficacy, immunogenicity, and safety of the HPV-16/18 AS04-adjuvanted vaccine: final analysis of a long-term follow-up study up to 9.4 years post-vaccination. Hum Vaccin Immunother. 2014;10:2147-2162. doi:10.4161/hv.29532
- Schwarz TF, Galaj A, Spaczynski M, et al. Ten-year immune persistence and safety of the HPV-16/18 AS04-adjuvanted vaccine in females vaccinated at 15–55 years of age. Cancer Med. 2017;6:2723-2731. doi:10.1002/cam4.1155
- Ferris DG, Samakoses R, Block SL, et al. 4-valent human papillomavirus (4vHPV) vaccine in preadolescents and adolescents after 10 years. Pediatrics. 2017;140:e20163947. doi:10.1542/peds.2016-3947
- Kjaer SK, Nygård M, Sundström K, et al. Final analysis of a 14-year long-term follow-up study of the effectiveness and immunogenicity of the quadrivalent human papillomavirus vaccine in women from four Nordic countries. EClinicalMedicine. 2020;23:100401. doi:10.1016 /j.eclinm.2020.100401
- Porras C, Tsang SH, Herrero R, et al; Costa Rica Vaccine Trial Group. Efficacy of the bivalent HPV vaccine against HPV 16/18-associated precancer: long-term follow-up results from the Costa Rica Vaccine Trial. Lancet Oncol. 2020;21:16431652. doi:10.1016/S1470-2045(20)30524-6
- Van Damme P, Olsson SE, Block S, et al. Immunogenicity and safety of a 9-valent HPV vaccine. Pediatrics. 2015;136:e28-e39. doi:10.1542/peds.2014-3745
- Restrepo J, Herrera T, Samakoses R, et al. Ten-year follow-up of 9-valent human papillomavirus vaccine: immunogenicity, effectiveness, and safety. Pediatrics. 2023;152:e2022060993. doi:10.1542/peds.2022-060993
- Luxembourg A, Moreira Jr ED, Samakoses R, et al. Phase III, randomized controlled trial in girls 9-15 years old to evaluate lot consistency of a novel nine-valent human papillomavirus L1 virus-like particle vaccine. Hum Vaccin Immunother. 11:1306-1312. doi:10.1080/21645515.2015.1009819

Do screening mammograms in women aged 70 and older improve stage at diagnosis or breast cancer–specific mortality?
Richman IB, Long JB, Soulos PR, et al. Estimating breast cancer overdiagnosis after screening mammography among older women in the United States. Ann Intern Med. 2023;176:1172-1180. doi:10.7326/M23-0133
EXPERT COMMENTARY
A screening test is performed to detect potential health disorders or diseases in people who do not have any symptoms of disease. The goal of screening is to detect the condition early enough to treat it most effectively, and ultimately to decrease morbidity and mortality related to the disease. Overdiagnosis refers to the finding of a cancer that would not have caused clinical problems during a person’s lifetime.
Current guidelines for the early detection of breast cancer vary considerably, including recommendations for what age to initiate screening, the cadence of screening (annual or biannual), the use of ancillary screening for people with dense breasts, and importantly the upper age limit for which screening is advised. The US Preventive Services Task Force recommends continuing screening to age 74. The American Cancer Society suggests ongoing screening if life expectancy is estimated at more than 10 years, and the American College of Physicians recommends stopping screening at age 75, or younger if life expectancy is less than 10 years. The American College of Obstetricians and Gynecologists states that women at average risk of breast cancer should continue screening mammography until at least age 75.
Overdiagnosis is a difficult concept for clinicians to understand let alone explain to our patients. Recently, Richman and colleagues published the results of their study aimed at estimating overdiagnosis associated with breast cancer screening among older women.1 As Dr. Otis Brawley, former Chief Medical and Scientific Officer of the American Cancer Society and current Distinguished Professor of Oncology and Epidemiology at Johns Hopkins University, states in the editorial that accompanies the study by Richman and colleagues, “Some tumors are not destined to grow, spread, and kill due to their genomics or their microenvironment. A second type of overdiagnosis involves small tumors that do have the potential to grow but will not grow fast enough to bother the patient within their natural lifetime.”2
Although screening mammography in older women results in frequent false positives that require additional imaging as well as biopsies, we have become more aware of the potential of overdiagnosis as an important downside of screening mammography in an elderly population.
Continue to: Details of the study...
Details of the study
Using the SEER registry to identify breast cancers linked to a 5% sample of Medicare beneficiaries, Richman and colleagues (funded by the National Cancer Institute and based at Yale University) conducted a retrospectivecohort study to estimate the likelihood of overdiagnosis associated with screening mammography among older women over 15 years of follow-up. Specifically, they assessed the difference in cumulative incidence of in situ and invasive breast cancer among women aged 70 years and older without a history of breast cancer when screened in 2002. During the subsequent 3 years, participants either continued screening (screened group) or did not (unscreened group). Women were followed through 2017.
Among almost 55,000 women followed, 88% were White, 6% were Black, and 3% were Hispanic. Mean follow-up was 13.7 years among women aged 70 to 74 years at baseline. For those aged 75 to 84 at baseline, mean follow-up was 10 years, and for those aged 85 years and older, mean follow-up was 5.7 years.
Estimated rates of overdiagnosis. Overall, among women aged 70 to 74 at baseline who were eventually diagnosed with breast cancer, the investigators estimated that 31% of these cancers were overdiagnosed. The corresponding percentage of breast cancers estimated to represent overdiagnosis climbed to 47% for those aged 75 to 84 years at baseline and to 54% for those aged 85 years and older at baseline.
The investigators assessed the impact of greater screening among women with a first-degree relative with a diagnosis of breast cancer and determined that this did not explain their results. With respect to cancer stage, the investigators noted that overdiagnosis was more prevalent among in situ and localized invasive cancers compared with those with regional or distant spread. Of note, the incidence of cancer with regional or distant spread was neither higher nor lower among those who were screened. Finally, the investigators did not observe significant differences in breast cancer–specific mortality by screening status.
The proportion of cancers that were overdiagnosed was particularly high among women with in situ as well as those with localized invasive disease. The investigators pointed out that as many as 90% of women aged 80 and older diagnosed with localized cancer undergo surgery, and almost two-thirds of those older than 70 years have radiation therapy for early-stage disease. In addition to the burdens associated with these treatments for overdiagnosed cancers in older women, simply being diagnosed with breast cancer profoundly affects the health and well-being of women, resulting in anxiety and substantial reductions in quality of life.
The authors also noted that some studies suggest that, among breast cancers diagnosed with screening, chemotherapy is less likely to be employed among older women, a screening benefit that must be weighed against the high likelihood of overdiagnosis. However, this benefit is unlikely to be meaningful for the majority of patients in this study who presented with in situ or early invasive lesions since chemotherapy often is not recommended for such women.
Study strengths and limitations
If screening mammography is effective, the incidence of advanced-stage tumors and breast cancer–specific mortality should be reduced in screened populations. Accordingly, in this large, long-term study using reliable sources of data, the findings that the incidence of advanced-stage disease as well as breast cancer–specific mortality were similar in the screened and unscreened cohorts provides powerful evidence that screening mammography is not effective in older women.3
As the authors pointed out, their findings regarding a high prevalence of overdiagnosis associated with screening mammography in older women are consistent with findings of other studies, some of which used different methodology.
The authors acknowledged that some women in their Medicare cohort who initially continued screening likely stopped screening subsequently, while some who initially did not continue screening might have been screened subsequently. They went on to indicate that if patients were completely adherent with subsequent screening (or not getting screened) the likelihood that cancers among screened women were overdiagnosed would be even higher.
Lead-time bias occurs when screening finds a cancer earlier than that cancer would have been diagnosed because of symptoms. This study followed the cohorts over a long timeframe to reduce the possibility that lead time was inappropriately identified as overdiagnosis. They also observed that, among women aged 85 and older, most cohort members had died by the end of study follow-up; accordingly, lead time is not likely to have explained their findings.
Limitations. The authors acknowledged that miscoding the mammogram type (screening vs diagnostic) could result in higher estimates of overdiagnosis. In their most conservative sensitivity analysis, the overdiagnosis rates could be as low as 15% for women aged 70 to 74, 36% for those aged 75 to 84, and 44% for people aged 85 and older.
Because this was an observational cohort study, unmeasured differences in breast cancer risk and underlying health factors may have been confounders. Specifically, people with severe life-threatening conditions that limited their expected life span may have chosen not to undergo regular screening. Although the authors did attempt to adjust for these factors, there may have been unrecognized confounders. This study was designed to estimate overdiagnosis, and therefore the specific benefits and harms of screening could not be addressed based on the data collected. ●
The high prevalence of overdiagnosis and lack of a breast cancer–specific mortality benefit among older women who undergo screening mammography is sobering. Clinician recommendations and shared decision making with our patients regarding screening mammography should take into consideration overdiagnosis and the considerable harms associated with overtreatment. Although we may recognize that overdiagnosed cancers are often indolent tumors with a long presymptomatic phase, in older women, even finding a biologically aggressive cancer may represent overdiagnosis if life expectancy is limited.
BARBARA LEVY, MD, MSCP; ANDREW M. KAUNITZ, MD, MSCP.
- Richman IB, Long JB, Soulos PR, et al. Estimating breast cancer overdiagnosis after screening mammography among older women in the United States. Ann Intern Med. 2023;176:1172-1180. doi:10.7326/M23-0133
- Brawley OW, Ramalingam R. Understanding the varying biological behaviors of breast and other types of cancer to avoid overdiagnosis. Ann Intern Med. 2023;176:1273-1274. doi:10.7326/M23-18953
- Welch HG, Gorski DH, Albertsen PC. Trends in metastatic breast and prostate cancer—lessons in cancer dynamics. N Engl J Med. 2015;373:1685-1687. doi:10.1056/NEJM p1510443
Richman IB, Long JB, Soulos PR, et al. Estimating breast cancer overdiagnosis after screening mammography among older women in the United States. Ann Intern Med. 2023;176:1172-1180. doi:10.7326/M23-0133
EXPERT COMMENTARY
A screening test is performed to detect potential health disorders or diseases in people who do not have any symptoms of disease. The goal of screening is to detect the condition early enough to treat it most effectively, and ultimately to decrease morbidity and mortality related to the disease. Overdiagnosis refers to the finding of a cancer that would not have caused clinical problems during a person’s lifetime.
Current guidelines for the early detection of breast cancer vary considerably, including recommendations for what age to initiate screening, the cadence of screening (annual or biannual), the use of ancillary screening for people with dense breasts, and importantly the upper age limit for which screening is advised. The US Preventive Services Task Force recommends continuing screening to age 74. The American Cancer Society suggests ongoing screening if life expectancy is estimated at more than 10 years, and the American College of Physicians recommends stopping screening at age 75, or younger if life expectancy is less than 10 years. The American College of Obstetricians and Gynecologists states that women at average risk of breast cancer should continue screening mammography until at least age 75.
Overdiagnosis is a difficult concept for clinicians to understand let alone explain to our patients. Recently, Richman and colleagues published the results of their study aimed at estimating overdiagnosis associated with breast cancer screening among older women.1 As Dr. Otis Brawley, former Chief Medical and Scientific Officer of the American Cancer Society and current Distinguished Professor of Oncology and Epidemiology at Johns Hopkins University, states in the editorial that accompanies the study by Richman and colleagues, “Some tumors are not destined to grow, spread, and kill due to their genomics or their microenvironment. A second type of overdiagnosis involves small tumors that do have the potential to grow but will not grow fast enough to bother the patient within their natural lifetime.”2
Although screening mammography in older women results in frequent false positives that require additional imaging as well as biopsies, we have become more aware of the potential of overdiagnosis as an important downside of screening mammography in an elderly population.
Continue to: Details of the study...
Details of the study
Using the SEER registry to identify breast cancers linked to a 5% sample of Medicare beneficiaries, Richman and colleagues (funded by the National Cancer Institute and based at Yale University) conducted a retrospectivecohort study to estimate the likelihood of overdiagnosis associated with screening mammography among older women over 15 years of follow-up. Specifically, they assessed the difference in cumulative incidence of in situ and invasive breast cancer among women aged 70 years and older without a history of breast cancer when screened in 2002. During the subsequent 3 years, participants either continued screening (screened group) or did not (unscreened group). Women were followed through 2017.
Among almost 55,000 women followed, 88% were White, 6% were Black, and 3% were Hispanic. Mean follow-up was 13.7 years among women aged 70 to 74 years at baseline. For those aged 75 to 84 at baseline, mean follow-up was 10 years, and for those aged 85 years and older, mean follow-up was 5.7 years.
Estimated rates of overdiagnosis. Overall, among women aged 70 to 74 at baseline who were eventually diagnosed with breast cancer, the investigators estimated that 31% of these cancers were overdiagnosed. The corresponding percentage of breast cancers estimated to represent overdiagnosis climbed to 47% for those aged 75 to 84 years at baseline and to 54% for those aged 85 years and older at baseline.
The investigators assessed the impact of greater screening among women with a first-degree relative with a diagnosis of breast cancer and determined that this did not explain their results. With respect to cancer stage, the investigators noted that overdiagnosis was more prevalent among in situ and localized invasive cancers compared with those with regional or distant spread. Of note, the incidence of cancer with regional or distant spread was neither higher nor lower among those who were screened. Finally, the investigators did not observe significant differences in breast cancer–specific mortality by screening status.
The proportion of cancers that were overdiagnosed was particularly high among women with in situ as well as those with localized invasive disease. The investigators pointed out that as many as 90% of women aged 80 and older diagnosed with localized cancer undergo surgery, and almost two-thirds of those older than 70 years have radiation therapy for early-stage disease. In addition to the burdens associated with these treatments for overdiagnosed cancers in older women, simply being diagnosed with breast cancer profoundly affects the health and well-being of women, resulting in anxiety and substantial reductions in quality of life.
The authors also noted that some studies suggest that, among breast cancers diagnosed with screening, chemotherapy is less likely to be employed among older women, a screening benefit that must be weighed against the high likelihood of overdiagnosis. However, this benefit is unlikely to be meaningful for the majority of patients in this study who presented with in situ or early invasive lesions since chemotherapy often is not recommended for such women.
Study strengths and limitations
If screening mammography is effective, the incidence of advanced-stage tumors and breast cancer–specific mortality should be reduced in screened populations. Accordingly, in this large, long-term study using reliable sources of data, the findings that the incidence of advanced-stage disease as well as breast cancer–specific mortality were similar in the screened and unscreened cohorts provides powerful evidence that screening mammography is not effective in older women.3
As the authors pointed out, their findings regarding a high prevalence of overdiagnosis associated with screening mammography in older women are consistent with findings of other studies, some of which used different methodology.
The authors acknowledged that some women in their Medicare cohort who initially continued screening likely stopped screening subsequently, while some who initially did not continue screening might have been screened subsequently. They went on to indicate that if patients were completely adherent with subsequent screening (or not getting screened) the likelihood that cancers among screened women were overdiagnosed would be even higher.
Lead-time bias occurs when screening finds a cancer earlier than that cancer would have been diagnosed because of symptoms. This study followed the cohorts over a long timeframe to reduce the possibility that lead time was inappropriately identified as overdiagnosis. They also observed that, among women aged 85 and older, most cohort members had died by the end of study follow-up; accordingly, lead time is not likely to have explained their findings.
Limitations. The authors acknowledged that miscoding the mammogram type (screening vs diagnostic) could result in higher estimates of overdiagnosis. In their most conservative sensitivity analysis, the overdiagnosis rates could be as low as 15% for women aged 70 to 74, 36% for those aged 75 to 84, and 44% for people aged 85 and older.
Because this was an observational cohort study, unmeasured differences in breast cancer risk and underlying health factors may have been confounders. Specifically, people with severe life-threatening conditions that limited their expected life span may have chosen not to undergo regular screening. Although the authors did attempt to adjust for these factors, there may have been unrecognized confounders. This study was designed to estimate overdiagnosis, and therefore the specific benefits and harms of screening could not be addressed based on the data collected. ●
The high prevalence of overdiagnosis and lack of a breast cancer–specific mortality benefit among older women who undergo screening mammography is sobering. Clinician recommendations and shared decision making with our patients regarding screening mammography should take into consideration overdiagnosis and the considerable harms associated with overtreatment. Although we may recognize that overdiagnosed cancers are often indolent tumors with a long presymptomatic phase, in older women, even finding a biologically aggressive cancer may represent overdiagnosis if life expectancy is limited.
BARBARA LEVY, MD, MSCP; ANDREW M. KAUNITZ, MD, MSCP.
Richman IB, Long JB, Soulos PR, et al. Estimating breast cancer overdiagnosis after screening mammography among older women in the United States. Ann Intern Med. 2023;176:1172-1180. doi:10.7326/M23-0133
EXPERT COMMENTARY
A screening test is performed to detect potential health disorders or diseases in people who do not have any symptoms of disease. The goal of screening is to detect the condition early enough to treat it most effectively, and ultimately to decrease morbidity and mortality related to the disease. Overdiagnosis refers to the finding of a cancer that would not have caused clinical problems during a person’s lifetime.
Current guidelines for the early detection of breast cancer vary considerably, including recommendations for what age to initiate screening, the cadence of screening (annual or biannual), the use of ancillary screening for people with dense breasts, and importantly the upper age limit for which screening is advised. The US Preventive Services Task Force recommends continuing screening to age 74. The American Cancer Society suggests ongoing screening if life expectancy is estimated at more than 10 years, and the American College of Physicians recommends stopping screening at age 75, or younger if life expectancy is less than 10 years. The American College of Obstetricians and Gynecologists states that women at average risk of breast cancer should continue screening mammography until at least age 75.
Overdiagnosis is a difficult concept for clinicians to understand let alone explain to our patients. Recently, Richman and colleagues published the results of their study aimed at estimating overdiagnosis associated with breast cancer screening among older women.1 As Dr. Otis Brawley, former Chief Medical and Scientific Officer of the American Cancer Society and current Distinguished Professor of Oncology and Epidemiology at Johns Hopkins University, states in the editorial that accompanies the study by Richman and colleagues, “Some tumors are not destined to grow, spread, and kill due to their genomics or their microenvironment. A second type of overdiagnosis involves small tumors that do have the potential to grow but will not grow fast enough to bother the patient within their natural lifetime.”2
Although screening mammography in older women results in frequent false positives that require additional imaging as well as biopsies, we have become more aware of the potential of overdiagnosis as an important downside of screening mammography in an elderly population.
Continue to: Details of the study...
Details of the study
Using the SEER registry to identify breast cancers linked to a 5% sample of Medicare beneficiaries, Richman and colleagues (funded by the National Cancer Institute and based at Yale University) conducted a retrospectivecohort study to estimate the likelihood of overdiagnosis associated with screening mammography among older women over 15 years of follow-up. Specifically, they assessed the difference in cumulative incidence of in situ and invasive breast cancer among women aged 70 years and older without a history of breast cancer when screened in 2002. During the subsequent 3 years, participants either continued screening (screened group) or did not (unscreened group). Women were followed through 2017.
Among almost 55,000 women followed, 88% were White, 6% were Black, and 3% were Hispanic. Mean follow-up was 13.7 years among women aged 70 to 74 years at baseline. For those aged 75 to 84 at baseline, mean follow-up was 10 years, and for those aged 85 years and older, mean follow-up was 5.7 years.
Estimated rates of overdiagnosis. Overall, among women aged 70 to 74 at baseline who were eventually diagnosed with breast cancer, the investigators estimated that 31% of these cancers were overdiagnosed. The corresponding percentage of breast cancers estimated to represent overdiagnosis climbed to 47% for those aged 75 to 84 years at baseline and to 54% for those aged 85 years and older at baseline.
The investigators assessed the impact of greater screening among women with a first-degree relative with a diagnosis of breast cancer and determined that this did not explain their results. With respect to cancer stage, the investigators noted that overdiagnosis was more prevalent among in situ and localized invasive cancers compared with those with regional or distant spread. Of note, the incidence of cancer with regional or distant spread was neither higher nor lower among those who were screened. Finally, the investigators did not observe significant differences in breast cancer–specific mortality by screening status.
The proportion of cancers that were overdiagnosed was particularly high among women with in situ as well as those with localized invasive disease. The investigators pointed out that as many as 90% of women aged 80 and older diagnosed with localized cancer undergo surgery, and almost two-thirds of those older than 70 years have radiation therapy for early-stage disease. In addition to the burdens associated with these treatments for overdiagnosed cancers in older women, simply being diagnosed with breast cancer profoundly affects the health and well-being of women, resulting in anxiety and substantial reductions in quality of life.
The authors also noted that some studies suggest that, among breast cancers diagnosed with screening, chemotherapy is less likely to be employed among older women, a screening benefit that must be weighed against the high likelihood of overdiagnosis. However, this benefit is unlikely to be meaningful for the majority of patients in this study who presented with in situ or early invasive lesions since chemotherapy often is not recommended for such women.
Study strengths and limitations
If screening mammography is effective, the incidence of advanced-stage tumors and breast cancer–specific mortality should be reduced in screened populations. Accordingly, in this large, long-term study using reliable sources of data, the findings that the incidence of advanced-stage disease as well as breast cancer–specific mortality were similar in the screened and unscreened cohorts provides powerful evidence that screening mammography is not effective in older women.3
As the authors pointed out, their findings regarding a high prevalence of overdiagnosis associated with screening mammography in older women are consistent with findings of other studies, some of which used different methodology.
The authors acknowledged that some women in their Medicare cohort who initially continued screening likely stopped screening subsequently, while some who initially did not continue screening might have been screened subsequently. They went on to indicate that if patients were completely adherent with subsequent screening (or not getting screened) the likelihood that cancers among screened women were overdiagnosed would be even higher.
Lead-time bias occurs when screening finds a cancer earlier than that cancer would have been diagnosed because of symptoms. This study followed the cohorts over a long timeframe to reduce the possibility that lead time was inappropriately identified as overdiagnosis. They also observed that, among women aged 85 and older, most cohort members had died by the end of study follow-up; accordingly, lead time is not likely to have explained their findings.
Limitations. The authors acknowledged that miscoding the mammogram type (screening vs diagnostic) could result in higher estimates of overdiagnosis. In their most conservative sensitivity analysis, the overdiagnosis rates could be as low as 15% for women aged 70 to 74, 36% for those aged 75 to 84, and 44% for people aged 85 and older.
Because this was an observational cohort study, unmeasured differences in breast cancer risk and underlying health factors may have been confounders. Specifically, people with severe life-threatening conditions that limited their expected life span may have chosen not to undergo regular screening. Although the authors did attempt to adjust for these factors, there may have been unrecognized confounders. This study was designed to estimate overdiagnosis, and therefore the specific benefits and harms of screening could not be addressed based on the data collected. ●
The high prevalence of overdiagnosis and lack of a breast cancer–specific mortality benefit among older women who undergo screening mammography is sobering. Clinician recommendations and shared decision making with our patients regarding screening mammography should take into consideration overdiagnosis and the considerable harms associated with overtreatment. Although we may recognize that overdiagnosed cancers are often indolent tumors with a long presymptomatic phase, in older women, even finding a biologically aggressive cancer may represent overdiagnosis if life expectancy is limited.
BARBARA LEVY, MD, MSCP; ANDREW M. KAUNITZ, MD, MSCP.
- Richman IB, Long JB, Soulos PR, et al. Estimating breast cancer overdiagnosis after screening mammography among older women in the United States. Ann Intern Med. 2023;176:1172-1180. doi:10.7326/M23-0133
- Brawley OW, Ramalingam R. Understanding the varying biological behaviors of breast and other types of cancer to avoid overdiagnosis. Ann Intern Med. 2023;176:1273-1274. doi:10.7326/M23-18953
- Welch HG, Gorski DH, Albertsen PC. Trends in metastatic breast and prostate cancer—lessons in cancer dynamics. N Engl J Med. 2015;373:1685-1687. doi:10.1056/NEJM p1510443
- Richman IB, Long JB, Soulos PR, et al. Estimating breast cancer overdiagnosis after screening mammography among older women in the United States. Ann Intern Med. 2023;176:1172-1180. doi:10.7326/M23-0133
- Brawley OW, Ramalingam R. Understanding the varying biological behaviors of breast and other types of cancer to avoid overdiagnosis. Ann Intern Med. 2023;176:1273-1274. doi:10.7326/M23-18953
- Welch HG, Gorski DH, Albertsen PC. Trends in metastatic breast and prostate cancer—lessons in cancer dynamics. N Engl J Med. 2015;373:1685-1687. doi:10.1056/NEJM p1510443
Can a novel, rapid-acting oral treatment effectively manage PPD?
Deligiannidis KM, Meltzer-Brody S, Maximos B, et al. Zuranolone for the treatment of postpartum depression. Am J Psychiatry. 2023;180:668-675. doi:10.1176/appi.ajp.20220785.
EXPERT COMMENTARY
Postpartum depression affects approximately 17.2% of patients in the peripartum period.1 Typical pharmacologic treatment of PPD includes selective serotonin reuptake inhibitors (SSRIs), which may take up to 12 weeks to take effect. Postpartum depression is thought to be secondary to maladaptation to hormonal fluctuations in the peripartum period, including allopregnanolone, a positive allosteric modulator of GABAA (γ-aminobutyric acid type A)receptors and a metabolite of progesterone, levels of which increase in pregnancy and abruptly decrease following delivery.1 In 2019, the GABAA receptor modulator brexanalone was approved by the US Food and Drug Administration (FDA) to treat PPD through continuous intravenous infusion over 60 hours in the hospital setting.
Zuranolone, an allosteric modulator of GABAA receptors, also has been studied as an investigational medication for rapid treatment of PPD. Prior studies demonstrated the efficacy of oral zuranolone 30 mg daily for the treatment of PPD2 and 50 mg for the treatment of major depression in nonpregnant patients.3 Deligiannidis and colleagues conducted a trial to investigate the 50-mg dose of zuranolone for the treatment of PPD. (Notably, in August 2023, the FDA approved oral zuranolone once daily for 14 days for the treatment of PPD.) Following the FDA approval, the American College of Obstetricians and Gynecologists (ACOG) released a Practice Advisory recommending consideration of zuranolone for PPD that takes into account balancing the benefits and risks, including known sedative effects, potential need for decreasing the dose due to adverse effects, lack of safety data in lactation, and unknown long-term efficacy.4
Details of the study
This randomized, double-blind, placebo-controlled study included 196 patients with an episode of major depression, characterized as a baseline score of 26 or greater on the Hamilton Depression Rating Scale (HAM-D) beginning in the third trimester or within the first 4 weeks postpartum. Patients were randomly assigned in a 1:1 ratio to receive zuranolone 50 mg daily or placebo, with stratification by stable concurrent antidepressant use. Treatment duration was for 14 days, with follow-up through day 45.
The study’s primary outcome was a change in the baseline HAM-D score at day 15. Changes in HAM-D score also were recorded at days 3, 28, and 45.
The 2 study groups were well balanced by demographic and baseline characteristics. In both groups, the majority of patients experienced the onset of their major depressive episodes within the first 4 weeks postpartum. Completion rates of the 14-day treatment course and 45-day follow-up were high and similar in both groups; 170 patients completed the study. The rate of concurrent psychiatric medications taken, most of which were SSRIs, was similar between the 2 groups at approximately 15% of patients.
Results. A statistically significant improvement in the primary outcome (the change in HAM-D score) at day 15 occurred in patients who received zuranolone versus placebo (P = .001). Additionally, there were statistically significant improvements in the secondary outcomes HAM-D scores at days 3, 28, and 45. Initial response, as measured by changes in HAM-D scores, occurred at a median duration of 9 days in the zuranolone group and 43 days in the placebo group. More patients in the zuranolone group achieved a reduction in HAM-D score at 15 days (57.0% vs 38.9%; P = .02). Zuranolone was associated with a higher rate of HAM-D remission at day 45 (44.0% vs 29.4%; P = .02).
With regard to safety, 16.3% of patients (17) in the zuranolone group (vs 1% in the placebo group) experienced an adverse event, most commonly somnolence, dizziness, and sedation, which led to a dose reduction. However, 15 of these 17 patients still completed the study, and there were no serious adverse events.
Study strengths and limitations
This study’s strengths include the double-blinded design that was continued throughout the duration of the follow-up. Additionally, the study population was heterogeneous andreflective of patients from diverse racial and ethnic backgrounds. Lastly, only minor and moderate adverse events were reported and, despite this, nearly all patients who experienced adverse events completed the study.
Limitations of the study include the lack of generalizability, as patients with bipolar disorder and mild or moderate PPD were excluded. Additionally, the majority of patients had depressive episodes within the first 4 weeks postpartum, thereby excluding patients with depressive episodes at other time points in the peripartum period. Further, as breastfeeding was prohibited, safety in lactating patients using zuranolone is unknown. Lastly, the study follow-up period was 45 days; therefore, the long-term efficacy of zuranolone treatment is unclear. ●
Zuranolone, a GABAA allosteric modulator, shows promise as an alternative to existing pharmacologic treatments for severe PPD that is orally administered and rapidly acting. While it is reasonable to consider its use in the specific patient population that benefited in this study, further studies are needed to determine its efficacy in other populations, the lowest effective dose for clinical improvement, and its interaction with other medications and breastfeeding. Additionally, the long-term remission rates of depressive symptoms in patients treated with zuranolone are unknown and warrant further study.
JAIMEY M. PAULI, MD; KENDALL CUNNINGHAM, MD
- Deligiannidis KM, Meltzer-Brody S, Maximos B, et al. Zuranolone for the treatment of postpartum depression. Am J Psychiatry. 2023;180:668-675. doi:10.1176/appi.ajp .20220785
- Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry. 2021;78:951-959. doi:10.1001/jamapsychiatry.2021.1559
- Clayton AH, Lasser R, Parikh SV, et al. Zuranolone for the treatment of adults with major depressive disorder: a randomized, placebo-controlled phase 3 trial. Am J Psychiatry. 2023;180:676-684. doi:10.1176/appi.ajp.20220459
- Zuranolone for the treatment of postpartum depression. Practice Advisory. American College of Obstetricians and Gynecologists. August 2023. Accessed September 18, 2023. https://www.acog.org/clinical/clinical-guidance/practice -advisory/articles/2023/08/zuranolone-for-the-treatment-of -postpartum-depression
Deligiannidis KM, Meltzer-Brody S, Maximos B, et al. Zuranolone for the treatment of postpartum depression. Am J Psychiatry. 2023;180:668-675. doi:10.1176/appi.ajp.20220785.
EXPERT COMMENTARY
Postpartum depression affects approximately 17.2% of patients in the peripartum period.1 Typical pharmacologic treatment of PPD includes selective serotonin reuptake inhibitors (SSRIs), which may take up to 12 weeks to take effect. Postpartum depression is thought to be secondary to maladaptation to hormonal fluctuations in the peripartum period, including allopregnanolone, a positive allosteric modulator of GABAA (γ-aminobutyric acid type A)receptors and a metabolite of progesterone, levels of which increase in pregnancy and abruptly decrease following delivery.1 In 2019, the GABAA receptor modulator brexanalone was approved by the US Food and Drug Administration (FDA) to treat PPD through continuous intravenous infusion over 60 hours in the hospital setting.
Zuranolone, an allosteric modulator of GABAA receptors, also has been studied as an investigational medication for rapid treatment of PPD. Prior studies demonstrated the efficacy of oral zuranolone 30 mg daily for the treatment of PPD2 and 50 mg for the treatment of major depression in nonpregnant patients.3 Deligiannidis and colleagues conducted a trial to investigate the 50-mg dose of zuranolone for the treatment of PPD. (Notably, in August 2023, the FDA approved oral zuranolone once daily for 14 days for the treatment of PPD.) Following the FDA approval, the American College of Obstetricians and Gynecologists (ACOG) released a Practice Advisory recommending consideration of zuranolone for PPD that takes into account balancing the benefits and risks, including known sedative effects, potential need for decreasing the dose due to adverse effects, lack of safety data in lactation, and unknown long-term efficacy.4
Details of the study
This randomized, double-blind, placebo-controlled study included 196 patients with an episode of major depression, characterized as a baseline score of 26 or greater on the Hamilton Depression Rating Scale (HAM-D) beginning in the third trimester or within the first 4 weeks postpartum. Patients were randomly assigned in a 1:1 ratio to receive zuranolone 50 mg daily or placebo, with stratification by stable concurrent antidepressant use. Treatment duration was for 14 days, with follow-up through day 45.
The study’s primary outcome was a change in the baseline HAM-D score at day 15. Changes in HAM-D score also were recorded at days 3, 28, and 45.
The 2 study groups were well balanced by demographic and baseline characteristics. In both groups, the majority of patients experienced the onset of their major depressive episodes within the first 4 weeks postpartum. Completion rates of the 14-day treatment course and 45-day follow-up were high and similar in both groups; 170 patients completed the study. The rate of concurrent psychiatric medications taken, most of which were SSRIs, was similar between the 2 groups at approximately 15% of patients.
Results. A statistically significant improvement in the primary outcome (the change in HAM-D score) at day 15 occurred in patients who received zuranolone versus placebo (P = .001). Additionally, there were statistically significant improvements in the secondary outcomes HAM-D scores at days 3, 28, and 45. Initial response, as measured by changes in HAM-D scores, occurred at a median duration of 9 days in the zuranolone group and 43 days in the placebo group. More patients in the zuranolone group achieved a reduction in HAM-D score at 15 days (57.0% vs 38.9%; P = .02). Zuranolone was associated with a higher rate of HAM-D remission at day 45 (44.0% vs 29.4%; P = .02).
With regard to safety, 16.3% of patients (17) in the zuranolone group (vs 1% in the placebo group) experienced an adverse event, most commonly somnolence, dizziness, and sedation, which led to a dose reduction. However, 15 of these 17 patients still completed the study, and there were no serious adverse events.
Study strengths and limitations
This study’s strengths include the double-blinded design that was continued throughout the duration of the follow-up. Additionally, the study population was heterogeneous andreflective of patients from diverse racial and ethnic backgrounds. Lastly, only minor and moderate adverse events were reported and, despite this, nearly all patients who experienced adverse events completed the study.
Limitations of the study include the lack of generalizability, as patients with bipolar disorder and mild or moderate PPD were excluded. Additionally, the majority of patients had depressive episodes within the first 4 weeks postpartum, thereby excluding patients with depressive episodes at other time points in the peripartum period. Further, as breastfeeding was prohibited, safety in lactating patients using zuranolone is unknown. Lastly, the study follow-up period was 45 days; therefore, the long-term efficacy of zuranolone treatment is unclear. ●
Zuranolone, a GABAA allosteric modulator, shows promise as an alternative to existing pharmacologic treatments for severe PPD that is orally administered and rapidly acting. While it is reasonable to consider its use in the specific patient population that benefited in this study, further studies are needed to determine its efficacy in other populations, the lowest effective dose for clinical improvement, and its interaction with other medications and breastfeeding. Additionally, the long-term remission rates of depressive symptoms in patients treated with zuranolone are unknown and warrant further study.
JAIMEY M. PAULI, MD; KENDALL CUNNINGHAM, MD
Deligiannidis KM, Meltzer-Brody S, Maximos B, et al. Zuranolone for the treatment of postpartum depression. Am J Psychiatry. 2023;180:668-675. doi:10.1176/appi.ajp.20220785.
EXPERT COMMENTARY
Postpartum depression affects approximately 17.2% of patients in the peripartum period.1 Typical pharmacologic treatment of PPD includes selective serotonin reuptake inhibitors (SSRIs), which may take up to 12 weeks to take effect. Postpartum depression is thought to be secondary to maladaptation to hormonal fluctuations in the peripartum period, including allopregnanolone, a positive allosteric modulator of GABAA (γ-aminobutyric acid type A)receptors and a metabolite of progesterone, levels of which increase in pregnancy and abruptly decrease following delivery.1 In 2019, the GABAA receptor modulator brexanalone was approved by the US Food and Drug Administration (FDA) to treat PPD through continuous intravenous infusion over 60 hours in the hospital setting.
Zuranolone, an allosteric modulator of GABAA receptors, also has been studied as an investigational medication for rapid treatment of PPD. Prior studies demonstrated the efficacy of oral zuranolone 30 mg daily for the treatment of PPD2 and 50 mg for the treatment of major depression in nonpregnant patients.3 Deligiannidis and colleagues conducted a trial to investigate the 50-mg dose of zuranolone for the treatment of PPD. (Notably, in August 2023, the FDA approved oral zuranolone once daily for 14 days for the treatment of PPD.) Following the FDA approval, the American College of Obstetricians and Gynecologists (ACOG) released a Practice Advisory recommending consideration of zuranolone for PPD that takes into account balancing the benefits and risks, including known sedative effects, potential need for decreasing the dose due to adverse effects, lack of safety data in lactation, and unknown long-term efficacy.4
Details of the study
This randomized, double-blind, placebo-controlled study included 196 patients with an episode of major depression, characterized as a baseline score of 26 or greater on the Hamilton Depression Rating Scale (HAM-D) beginning in the third trimester or within the first 4 weeks postpartum. Patients were randomly assigned in a 1:1 ratio to receive zuranolone 50 mg daily or placebo, with stratification by stable concurrent antidepressant use. Treatment duration was for 14 days, with follow-up through day 45.
The study’s primary outcome was a change in the baseline HAM-D score at day 15. Changes in HAM-D score also were recorded at days 3, 28, and 45.
The 2 study groups were well balanced by demographic and baseline characteristics. In both groups, the majority of patients experienced the onset of their major depressive episodes within the first 4 weeks postpartum. Completion rates of the 14-day treatment course and 45-day follow-up were high and similar in both groups; 170 patients completed the study. The rate of concurrent psychiatric medications taken, most of which were SSRIs, was similar between the 2 groups at approximately 15% of patients.
Results. A statistically significant improvement in the primary outcome (the change in HAM-D score) at day 15 occurred in patients who received zuranolone versus placebo (P = .001). Additionally, there were statistically significant improvements in the secondary outcomes HAM-D scores at days 3, 28, and 45. Initial response, as measured by changes in HAM-D scores, occurred at a median duration of 9 days in the zuranolone group and 43 days in the placebo group. More patients in the zuranolone group achieved a reduction in HAM-D score at 15 days (57.0% vs 38.9%; P = .02). Zuranolone was associated with a higher rate of HAM-D remission at day 45 (44.0% vs 29.4%; P = .02).
With regard to safety, 16.3% of patients (17) in the zuranolone group (vs 1% in the placebo group) experienced an adverse event, most commonly somnolence, dizziness, and sedation, which led to a dose reduction. However, 15 of these 17 patients still completed the study, and there were no serious adverse events.
Study strengths and limitations
This study’s strengths include the double-blinded design that was continued throughout the duration of the follow-up. Additionally, the study population was heterogeneous andreflective of patients from diverse racial and ethnic backgrounds. Lastly, only minor and moderate adverse events were reported and, despite this, nearly all patients who experienced adverse events completed the study.
Limitations of the study include the lack of generalizability, as patients with bipolar disorder and mild or moderate PPD were excluded. Additionally, the majority of patients had depressive episodes within the first 4 weeks postpartum, thereby excluding patients with depressive episodes at other time points in the peripartum period. Further, as breastfeeding was prohibited, safety in lactating patients using zuranolone is unknown. Lastly, the study follow-up period was 45 days; therefore, the long-term efficacy of zuranolone treatment is unclear. ●
Zuranolone, a GABAA allosteric modulator, shows promise as an alternative to existing pharmacologic treatments for severe PPD that is orally administered and rapidly acting. While it is reasonable to consider its use in the specific patient population that benefited in this study, further studies are needed to determine its efficacy in other populations, the lowest effective dose for clinical improvement, and its interaction with other medications and breastfeeding. Additionally, the long-term remission rates of depressive symptoms in patients treated with zuranolone are unknown and warrant further study.
JAIMEY M. PAULI, MD; KENDALL CUNNINGHAM, MD
- Deligiannidis KM, Meltzer-Brody S, Maximos B, et al. Zuranolone for the treatment of postpartum depression. Am J Psychiatry. 2023;180:668-675. doi:10.1176/appi.ajp .20220785
- Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry. 2021;78:951-959. doi:10.1001/jamapsychiatry.2021.1559
- Clayton AH, Lasser R, Parikh SV, et al. Zuranolone for the treatment of adults with major depressive disorder: a randomized, placebo-controlled phase 3 trial. Am J Psychiatry. 2023;180:676-684. doi:10.1176/appi.ajp.20220459
- Zuranolone for the treatment of postpartum depression. Practice Advisory. American College of Obstetricians and Gynecologists. August 2023. Accessed September 18, 2023. https://www.acog.org/clinical/clinical-guidance/practice -advisory/articles/2023/08/zuranolone-for-the-treatment-of -postpartum-depression
- Deligiannidis KM, Meltzer-Brody S, Maximos B, et al. Zuranolone for the treatment of postpartum depression. Am J Psychiatry. 2023;180:668-675. doi:10.1176/appi.ajp .20220785
- Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry. 2021;78:951-959. doi:10.1001/jamapsychiatry.2021.1559
- Clayton AH, Lasser R, Parikh SV, et al. Zuranolone for the treatment of adults with major depressive disorder: a randomized, placebo-controlled phase 3 trial. Am J Psychiatry. 2023;180:676-684. doi:10.1176/appi.ajp.20220459
- Zuranolone for the treatment of postpartum depression. Practice Advisory. American College of Obstetricians and Gynecologists. August 2023. Accessed September 18, 2023. https://www.acog.org/clinical/clinical-guidance/practice -advisory/articles/2023/08/zuranolone-for-the-treatment-of -postpartum-depression

Does remote blood pressure monitoring improve patient outcomes postpartum?
Hirshberg A, Zhu Y, Smith-McLallen A, et al. Association of a remote blood pressure monitoring program with postpartum adverse outcomes. Obstet Gynecol. 2023;141:1163-1170. doi:10.1097/AOG.0000000000005197.
EXPERT COMMENTARY
Courtney Bisson, MD, is a Maternal-Fetal Medicine Fellow, University of Chicago/NorthShore University HealthSystem, Chicago, Illinois.
Sarosh Rana, MD, MPH, is Professor of Obstetrics and Gynecology and Section Chief, Maternal-Fetal Medicine, University of Chicago.
Hypertensive disorders of pregnancy account for a significant amount of morbidity during pregnancy and postpartum. In the pregnant population, data have shown that the implementation of a standardized blood pressure education program, provision of a blood pressure cuff, and assistance with postpartum follow-up result in improved blood pressures and postpartum follow-up for up to 6 weeks. In the nonpregnant population, literature suggests that RBPM in patients with hypertension results in improved outcomes, although the long-term impact of RBPM in the postpartum population remains unclear.
Recently, Hirshberg and colleagues published the results of a retrospective cohort study that assessed the impact of RBPM with text message reminders for 10 days postpartum on a composite of adverse maternal outcomes, readmissions, and follow-up within 1 year postpartum.1

Details of the study
The retrospective cohort study was conducted during 2017–2021 based on insurance claims of patients with hypertensive disorders of pregnancy who were enrolled in a twice-daily text message–based RBPM program for 10 days postpartum.
Data from 1,700 patients enrolled in RBPM were compared with that of propensity score matched controls that included 2,297 women not enrolled in RBPM. Of these controls, 1,276 patients (cohort C) simultaneously received care at other institutions without RBPM, and 1,021 patients (cohort A) received care at the same institution prior to implementation of RBPM.
Results. Patients in the RBPM group were found to have a significantly lower rate of composite adverse maternal outcomes compared with their matched cohorts in the year after delivery. (Individual adverse outcomes included stroke, disseminated intravascular coagulation, eclampsia, pulmonary edema, renal injury or liver failure, HELLP [hemolysis, elevated liver enzymes, low platelet count] syndrome, myocardial infarction, and cardiomyopathy.) Rates were 2.9% versus 4.7% (odds ratio [OR], 0.61; 95% confidence interval [CI], 0.40–0.98) in the RBPM group compared with cohort A; rates in the RBPM group compared with cohort C were 3.2% versus 4.5% (OR, 0.71; 95% CI, 0.47–1.07).
Although not statistically significant, rates of emergency department visits and readmissions also were lower in the RBPM patients. Those enrolled in the RBPM program were more likely to have follow-up with cardiologists or specialist visits within 6 months postpartum. Fewer emergency department visits and readmissions resulted in lower health care utilization costs.
Study strengths and limitations
This study’s strength lies in its design and implementation of standardized protocols that allowed assessment of clinically meaningful outcomes postpartum. Although the program for RBPM was for only 10 days postpartum, it showed effects beyond the timeframe of the direct care. No such prior data exist evaluating a program’s effectiveness in improving postpartum clinical outcomes and costs through 1 year postdelivery.
Study limitations include residual bias from unobserved confounders, analysis of only 1 payer type, lack of patient level data, and evaluation of disparity. ●
Previous work by Suresh and colleagues illustrated that a standardized postpartum blood pressure monitoring quality improvement initiative resulted in better blood pressures, improved postpartum visit adherence, and reduced disparity.2 The study by Hirshberg and colleagues furthers these findings, illustrating how uniform protocols surrounding preeclampsia management in the postpartum setting could further improve morbidity and mortality in the year following childbirth. Such protocols should be incorporated hospital-wide in standard obstetrical management.
COURTNEY BISSON, MD; SAROSH RANA, MD, MPH
- Hirshberg A, Zhu Y, Smith-McLallen A, et al. Association of a remote blood pressure monitoring program with postpartum adverse outcomes. Obstet Gynecol. 2023;141:1163-1170. doi:10.1097/AOG.0000000000005197.
- Suresh SC, Duncan C, Kaur H, et al. Postpartum outcomes with systematic treatment and management of postpartum hypertension. Obstet Gynecol. 2021;138:777-787. doi:10.1097 /AOG.0000000000004574.
Hirshberg A, Zhu Y, Smith-McLallen A, et al. Association of a remote blood pressure monitoring program with postpartum adverse outcomes. Obstet Gynecol. 2023;141:1163-1170. doi:10.1097/AOG.0000000000005197.
EXPERT COMMENTARY
Courtney Bisson, MD, is a Maternal-Fetal Medicine Fellow, University of Chicago/NorthShore University HealthSystem, Chicago, Illinois.
Sarosh Rana, MD, MPH, is Professor of Obstetrics and Gynecology and Section Chief, Maternal-Fetal Medicine, University of Chicago.
Hypertensive disorders of pregnancy account for a significant amount of morbidity during pregnancy and postpartum. In the pregnant population, data have shown that the implementation of a standardized blood pressure education program, provision of a blood pressure cuff, and assistance with postpartum follow-up result in improved blood pressures and postpartum follow-up for up to 6 weeks. In the nonpregnant population, literature suggests that RBPM in patients with hypertension results in improved outcomes, although the long-term impact of RBPM in the postpartum population remains unclear.
Recently, Hirshberg and colleagues published the results of a retrospective cohort study that assessed the impact of RBPM with text message reminders for 10 days postpartum on a composite of adverse maternal outcomes, readmissions, and follow-up within 1 year postpartum.1
Details of the study
The retrospective cohort study was conducted during 2017–2021 based on insurance claims of patients with hypertensive disorders of pregnancy who were enrolled in a twice-daily text message–based RBPM program for 10 days postpartum.
Data from 1,700 patients enrolled in RBPM were compared with that of propensity score matched controls that included 2,297 women not enrolled in RBPM. Of these controls, 1,276 patients (cohort C) simultaneously received care at other institutions without RBPM, and 1,021 patients (cohort A) received care at the same institution prior to implementation of RBPM.
Results. Patients in the RBPM group were found to have a significantly lower rate of composite adverse maternal outcomes compared with their matched cohorts in the year after delivery. (Individual adverse outcomes included stroke, disseminated intravascular coagulation, eclampsia, pulmonary edema, renal injury or liver failure, HELLP [hemolysis, elevated liver enzymes, low platelet count] syndrome, myocardial infarction, and cardiomyopathy.) Rates were 2.9% versus 4.7% (odds ratio [OR], 0.61; 95% confidence interval [CI], 0.40–0.98) in the RBPM group compared with cohort A; rates in the RBPM group compared with cohort C were 3.2% versus 4.5% (OR, 0.71; 95% CI, 0.47–1.07).
Although not statistically significant, rates of emergency department visits and readmissions also were lower in the RBPM patients. Those enrolled in the RBPM program were more likely to have follow-up with cardiologists or specialist visits within 6 months postpartum. Fewer emergency department visits and readmissions resulted in lower health care utilization costs.
Study strengths and limitations
This study’s strength lies in its design and implementation of standardized protocols that allowed assessment of clinically meaningful outcomes postpartum. Although the program for RBPM was for only 10 days postpartum, it showed effects beyond the timeframe of the direct care. No such prior data exist evaluating a program’s effectiveness in improving postpartum clinical outcomes and costs through 1 year postdelivery.
Study limitations include residual bias from unobserved confounders, analysis of only 1 payer type, lack of patient level data, and evaluation of disparity. ●
Previous work by Suresh and colleagues illustrated that a standardized postpartum blood pressure monitoring quality improvement initiative resulted in better blood pressures, improved postpartum visit adherence, and reduced disparity.2 The study by Hirshberg and colleagues furthers these findings, illustrating how uniform protocols surrounding preeclampsia management in the postpartum setting could further improve morbidity and mortality in the year following childbirth. Such protocols should be incorporated hospital-wide in standard obstetrical management.
COURTNEY BISSON, MD; SAROSH RANA, MD, MPH
Hirshberg A, Zhu Y, Smith-McLallen A, et al. Association of a remote blood pressure monitoring program with postpartum adverse outcomes. Obstet Gynecol. 2023;141:1163-1170. doi:10.1097/AOG.0000000000005197.
EXPERT COMMENTARY
Courtney Bisson, MD, is a Maternal-Fetal Medicine Fellow, University of Chicago/NorthShore University HealthSystem, Chicago, Illinois.
Sarosh Rana, MD, MPH, is Professor of Obstetrics and Gynecology and Section Chief, Maternal-Fetal Medicine, University of Chicago.
Hypertensive disorders of pregnancy account for a significant amount of morbidity during pregnancy and postpartum. In the pregnant population, data have shown that the implementation of a standardized blood pressure education program, provision of a blood pressure cuff, and assistance with postpartum follow-up result in improved blood pressures and postpartum follow-up for up to 6 weeks. In the nonpregnant population, literature suggests that RBPM in patients with hypertension results in improved outcomes, although the long-term impact of RBPM in the postpartum population remains unclear.
Recently, Hirshberg and colleagues published the results of a retrospective cohort study that assessed the impact of RBPM with text message reminders for 10 days postpartum on a composite of adverse maternal outcomes, readmissions, and follow-up within 1 year postpartum.1
Details of the study
The retrospective cohort study was conducted during 2017–2021 based on insurance claims of patients with hypertensive disorders of pregnancy who were enrolled in a twice-daily text message–based RBPM program for 10 days postpartum.
Data from 1,700 patients enrolled in RBPM were compared with that of propensity score matched controls that included 2,297 women not enrolled in RBPM. Of these controls, 1,276 patients (cohort C) simultaneously received care at other institutions without RBPM, and 1,021 patients (cohort A) received care at the same institution prior to implementation of RBPM.
Results. Patients in the RBPM group were found to have a significantly lower rate of composite adverse maternal outcomes compared with their matched cohorts in the year after delivery. (Individual adverse outcomes included stroke, disseminated intravascular coagulation, eclampsia, pulmonary edema, renal injury or liver failure, HELLP [hemolysis, elevated liver enzymes, low platelet count] syndrome, myocardial infarction, and cardiomyopathy.) Rates were 2.9% versus 4.7% (odds ratio [OR], 0.61; 95% confidence interval [CI], 0.40–0.98) in the RBPM group compared with cohort A; rates in the RBPM group compared with cohort C were 3.2% versus 4.5% (OR, 0.71; 95% CI, 0.47–1.07).
Although not statistically significant, rates of emergency department visits and readmissions also were lower in the RBPM patients. Those enrolled in the RBPM program were more likely to have follow-up with cardiologists or specialist visits within 6 months postpartum. Fewer emergency department visits and readmissions resulted in lower health care utilization costs.
Study strengths and limitations
This study’s strength lies in its design and implementation of standardized protocols that allowed assessment of clinically meaningful outcomes postpartum. Although the program for RBPM was for only 10 days postpartum, it showed effects beyond the timeframe of the direct care. No such prior data exist evaluating a program’s effectiveness in improving postpartum clinical outcomes and costs through 1 year postdelivery.
Study limitations include residual bias from unobserved confounders, analysis of only 1 payer type, lack of patient level data, and evaluation of disparity. ●
Previous work by Suresh and colleagues illustrated that a standardized postpartum blood pressure monitoring quality improvement initiative resulted in better blood pressures, improved postpartum visit adherence, and reduced disparity.2 The study by Hirshberg and colleagues furthers these findings, illustrating how uniform protocols surrounding preeclampsia management in the postpartum setting could further improve morbidity and mortality in the year following childbirth. Such protocols should be incorporated hospital-wide in standard obstetrical management.
COURTNEY BISSON, MD; SAROSH RANA, MD, MPH
- Hirshberg A, Zhu Y, Smith-McLallen A, et al. Association of a remote blood pressure monitoring program with postpartum adverse outcomes. Obstet Gynecol. 2023;141:1163-1170. doi:10.1097/AOG.0000000000005197.
- Suresh SC, Duncan C, Kaur H, et al. Postpartum outcomes with systematic treatment and management of postpartum hypertension. Obstet Gynecol. 2021;138:777-787. doi:10.1097 /AOG.0000000000004574.
- Hirshberg A, Zhu Y, Smith-McLallen A, et al. Association of a remote blood pressure monitoring program with postpartum adverse outcomes. Obstet Gynecol. 2023;141:1163-1170. doi:10.1097/AOG.0000000000005197.
- Suresh SC, Duncan C, Kaur H, et al. Postpartum outcomes with systematic treatment and management of postpartum hypertension. Obstet Gynecol. 2021;138:777-787. doi:10.1097 /AOG.0000000000004574.
