User login
JUPITER to Earth: A statin helps people with normal LDL-C and high hs-CRP, but what does it mean?
The medical community has struggled with two important questions for the past 10 years: When it comes to the low-density lipoprotein cholesterol (LDL-C) level, how low should one go and at what cost? And are there other markers of risk that can identify a higher-risk subpopulation in relatively healthy people? The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) provided partial answers for these questions by finding that a highly potent statin lowered the risk of cardiovascular events in patients with “normal” LDL-C but elevated levels of high-sensitivity C-reactive protein (hs-CRP).1
In this article, we will critically evaluate the methods, results, and conclusions of the JUPITER trial. Additionally, we will discuss its limitations and areas of uncertainty.
BEFORE JUPITER
The LDL-C-lowering drugs called statins have revolutionized cardiovascular medicine.2 They are beneficial in both the primary prevention setting and in acute coronary syndromes, stable angina, and unstable angina and can halt the progression of coronary artery disease—in some cases even resulting in modest regression of plaque.3–6
Many experts have credited the reduction in LDL-C as being the sole factor responsible for the decrease in major adverse events seen with statin therapy.7 However, statins have other, non-lipid-lowering properties, including anti-inflammatory and antioxidant effects, that may also contribute to their benefits.8–15
One of the anti-inflammatory actions of statins is evidenced by lower levels of the acute-phase reactant CRP.10,11,15,16 Measuring systemic CRP levels with a highly sensitive assay (yielding the so-called high-sensitivity or hs-CRP level) provides significant clinical prognostic value across a spectrum of clinical situations, ranging from risk screening in apparently healthy people to stable and unstable angina.17–22 People with higher hs-CRP levels are, on average, at higher risk of adverse cardiovascular events. However, controversy remains as to whether hs-CRP plays a mechanistic role in plaque formation and acute complications. Indeed, recent genetic studies argue strongly that hs-CRP lies outside the mechanistic path of atherosclerosis.23 Nonetheless, an overwhelming amount of data indicates that hs-CRP serves as a marker of disease.17–21
Nissen et al10 showed that the rate of progression of atherosclerosis is lower when the levels of atherogenic lipoproteins and hs-CRP are both lowered with statin therapy. Simultaneously, Ridker et al11 showed that patients who have lower hs-CRP levels after statin therapy have better clinical outcomes than those with higher hs-CRP levels, regardless of their achieved level of LDL-C.
Collectively, these studies and others have led some to believe that, in people with relatively low LDL-C but persistently elevated hs-CRP, statin therapy may reduce the rate of events.15,24 The JUPITER trial was undertaken to test this hypothesis.
JUPITER STUDY DESIGN
JUPITER was designed to see whether highly potent statin therapy is beneficial in people with elevated hs-CRP who otherwise do not meet the criteria for lipid-lowering therapy. The study was conducted at 1,315 sites in 26 countries. It was sponsored by AstraZeneca, the maker of rosuvastatin (Crestor).
Inclusion and exclusion criteria
All participants had to be free of known cardiovascular disease, have an LDL-C level lower than 130 mg/dL, and have an hs-CRP level of 2.0 mg/L or greater. Patients were excluded if they were previous or current users of lipid-lowering drugs; had severe arthritis, lupus, or inflammatory bowel disease; or were taking immune-modulating drugs such as cyclosporine (Sandimmune, others), tacrolimus (Prograf), azathioprine (Azasan, Imuran), or long-term oral corticosteroids.
Rosuvastatin therapy
Participants were randomly assigned in a 1:1 ratio to receive rosuvastatin 20 mg daily or a matching placebo in a double-blind fashion.
End points
The primary end point was the composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from cardiovascular causes. Secondary end points were the individual components of the primary end point.
Statistical analysis
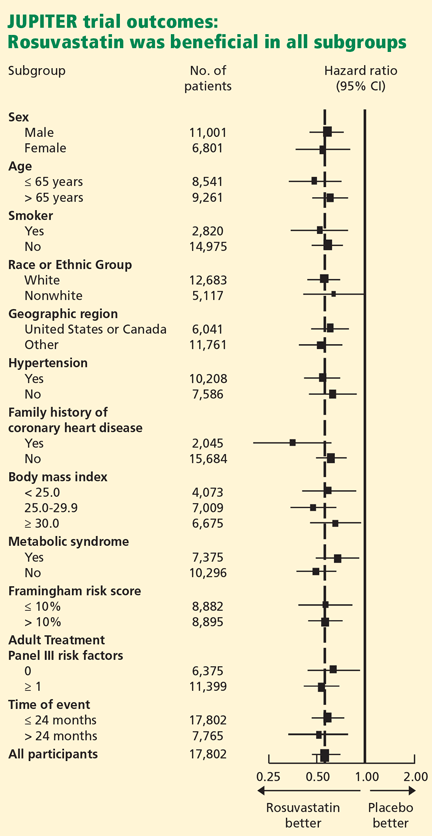
The study was powered to detect a 25% reduction in the primary end point among those treated with rosuvastatin. The trial was designed to run until 520 end point events had occurred. However, on March 29, 2008, after the first prespecified interim analysis, the Data and Safety Monitoring Board stopped the trial due to a significant reduction in the primary end point in the rosuvastatin group. As in most randomized clinical trials, all analyses were done on an intention-to-treat basis. Prespecified subgroup analyses were also performed.
STUDY RESULTS
Patient recruitment and eligibility
Between February 4, 2003, and December 15, 2006, a total of 89,890 people were screened. Of these, 17,802 met the inclusion and exclusion criteria and were included in the study. Of the 72,088 people who were excluded, 25,993 (36.1%) had an hs-CRP level below 2 mg/L and 37,611 (52.2%) had an LDL-C level of 130 mg/dL or higher.
A not-so-healthy population
The aim of the investigators was to include relatively healthy people. The median age was 66 years, about 16% of participants were current smokers, about 11% had a family history of heart disease, and about 41% met the criteria for metabolic syndrome, all conditions that are associated with elevated hs-CRP.25 Of note, the median hs-CRP level was 4.2 mg/L, a level indicating higher global risk according to the American College of Cardiology/American Heart Association consensus statement.26
Reduction in lipid levels and hs-CRP
By 12 months, in the rosuvastatin group, the median LDL-C level had fallen by 50% (from 108 to 55 mg/dL), and the median hs-CRP level had fallen by 37% (from 4.2 to 2.2 mg/L). Additionally, the triglyceride level had fallen by 17%. The high-density lipoprotein cholesterol levels did not change significantly.
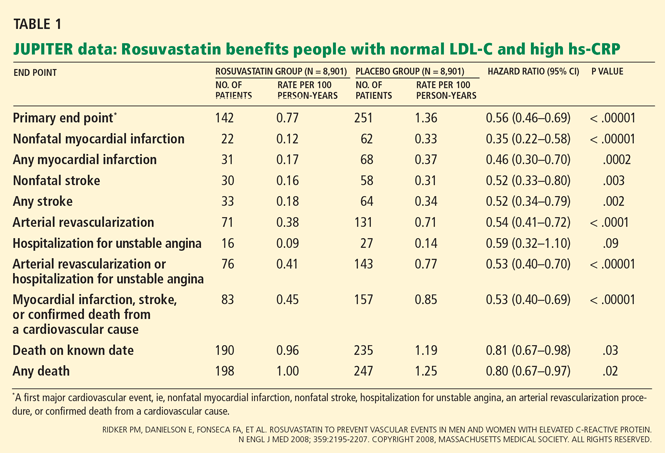
Impact on end points
Adverse events
WHAT DOES THIS MEAN?
Is lower LDL-C better?
The JUPITER trial is the latest of several statin trials that have shown significant reductions in major adverse cardiovascular events when LDL-C was lowered below what has been recommended by the current guidelines.27,28
In 2002, the Heart Protection Study29 showed a significant reduction in major adverse cardiovascular events in patients at high risk of coronary artery disease if they received simvastatin (Zocor), even if they had LDL-C levels lower than 100 mg/dL at baseline. Similarly, the Pravastatin or Atorvastatin Evaluation and Infection-Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial30 showed a 16% relative risk reduction in a composite end point in patients presenting with acute coronary syndrome if they received intensive statin therapy.
These two studies led to an update by the National Cholesterol Education Program (Adult Treatment Panel III), suggesting an optimal LDL-C goal of less than 70 mg/dL in those with coronary artery disease or its risk equivalent (ie, diabetes mellitus, peripheral vascular disease). Furthermore, in support of the “lower is better” theory, a number of studies that used intravascular ultrasonography have shown regression of coronary plaque with aggressive LDL-C lowering. Notably, in a Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (the ASTEROID trial),5 rosuvastatin 40 mg daily caused significant plaque regression while lowering LDL-C to 61 mg/dL over a 24-month period.
A number of high-dose statin trials have shown that lowering LDL-C to less than 70 mg/dL significantly reduces major adverse cardiovascular events.31–39 The JUPITER trial was unique in that it extended these findings to people without known coronary disease (ie, primary prevention) or elevated cholesterol but with elevated levels of a marker of inflammation—hs-CRP. In view of the JUPITER results and of studies using intravascular ultrasonography in the primary prevention setting, it seems clear that lowering LDL-C to levels less than 70 mg/dL also reduces both atherosclerotic plaque progression and the rate of first major adverse cardiovascular events in primary prevention in patients at higher global risk.
Did the study prove that reducing hs-CRP lowers risk?
Measuring hs-CRP levels has been extensively studied in apparently healthy populations, stable angina, unstable angina, and other cardiovascular settings.18,21,40–43 It has been shown to have significant prognostic implications in a number of primary and secondary trials.44 Additionally, those with elevated LDL-C and hs-CRP levels benefit the most from statin therapy.16,45,46 Animal studies have also provided some evidence that CRP may play a role in atherogenesis.47,48 However, recent clinical and genetic studies have raised doubt about the direct causal relationship between CRP and coronary artery disease,23,49,50 and epidemiologic studies have questioned its usefulness as a marker of risk.51,52
The JUPITER study adds little to clear up the controversy about whether hs-CRP is a mechanistic participant in atherosclerotic disease. However, it also shows that this issue is somewhat irrelevant, in that selection of patients for high-potency statin therapy solely on the basis of high hs-CRP without other indications for lipid-lowering therapy clearly reduces risk and improves survival.
JUPITER did not examine whether people with higher hs-CRP levels benefited more from statin therapy than those with lower levels. The hypothesis-generating data for JUPITER came from an analysis of changes in hs-CRP and LDL-C in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS).16 Thus, JUPITER did not include people with both low LDL-C and low hs-CRP because, in the AFCAPS/TexCAPS analysis, those with low LDL-C and low hs-CRP had extremely low event rates and no clinical efficacy of statin therapy, despite good LDL-C reduction. In marked contrast, those with low LDL-C but elevated hs-CRP had high event rates and large relative risk reductions— hence the need for JUPITER to prospectively test this hypothesis. Nevertheless, the initial results of JUPITER as presented do not yet make it clear that there is a dose-response relationship between higher levels of hs-CRP and a greater reduction in events, even in a cohort with elevated hs-CRP at baseline. This analysis will no doubt be forthcoming in another manuscript from Ridker and colleagues. Specifically, it will be of interest to examine whether those with the highest hs-CRP levels benefited the most from rosuvastatin on both an absolute and relative scale, and whether those with the greatest hs-CRP reduction also benefited more. With the present data available from JUPITER, a reasonable interpretation is that an elevated hs-CRP simply widens the inclusion criterion for those for whom high-potency statin therapy improves clinical outcomes.53
Better markers are needed
Even with a nonspecific marker such as hs-CRP, patients at higher global risk and with LDL-C below the recommended levels could be identified and treated aggressively. This benefit, however, required that approximately 100 people be treated with rosuvastatin for 2 years to prevent one event. Additionally, only 20% of all patients screened were eligible for the trial. Therefore, one could argue that its generalizability is limited.
Markers of risk that are more specific and sensitive are needed to identify people at higher global risk who would otherwise be considered to be at low risk with the current risk assessment tools. A number of such inflammatory and oxidative markers are under development.54–60
Absolute vs relative risk reduction and the public health burden
The 44% reduction in the number of primary end point events in the rosuvastatin group was considerable in relative terms. However, in absolute terms, 95 people had to be treated for up to 2 years in order to prevent one event.53 In making recommendations, the United States Department of Health and Human Services has to consider the clinical benefit of a test or a drug in light of its cost. With health care costs increasing, many agencies are refusing to pay for therapies on the basis of cost or small absolute benefit.
While we do not have the answer as to whether treating 95 people for 2 years to see one benefit is cost-effective, one thing is clear: the field of medicine is in desperate need of a better way to identify individuals who may benefit from a test or therapy.61 Additionally, we think it is important to note that the “numbers-needed-to-treat” (95 at 2 years and 25 at 5 years) derived from JUPITER are actually smaller than the values observed in the AFCAPS/TexCAPS and the West of Scotland Coronary Prevention Study.62,63 This suggests that statin therapy is at least as cost-effective in those with elevated hs-CRP as in those with elevated LDL-C. Even our most robust therapies are effective in only a minority of patients treated.61
Should ‘healthy’ people be tested for hs-CRP?
In 2003, we wrote in this journal21 that measuring hs-CRP may add to the current risk-prediction models by identifying people at increased risk who would otherwise not be considered as such by current risk models. The US Centers for Disease Control and Prevention and the American Heart Association have also stated that measuring hs-CRP in those at intermediate risk may be reasonable.26
The JUPITER investigators intended to study a relatively healthy population, but, as we mentioned, a close look at the cohort’s baseline characteristics indicates a substantial proportion met the criteria for metabolic syndrome. Therefore, one could challenge whether we really need hs-CRP in such a population to identify who will benefit from statin therapy.
We agree with the recommendation from the Centers for Disease Control and Prevention and the American Heart Association that measuring hs-CRP in people at intermediate risk is a reasonable option.26 We also believe that hs-CRP should be tested as a secondary risk factor, in combination with blood pressure, lipids, diabetes, smoking, serum creatinine, and fasting blood glucose. Factors such as obesity, sedentary lifestyle, family history of heart disease, and emotional and physical stress should also be considered.
Safety of high-dose statin therapy
High-dose statin therapy has been well tolerated in clinical trials, but rates of discontinuation have been higher (7%–10%) than with moderate-dose therapy (4%–5%).64 Fortunately, the rates of serious adverse events have in general been low. For example, with simvastatin 80 mg, the rates of myopathy and rhabdomyolysis were quite low.31
Rates of elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with high-dose statin therapy have been reported to be below 1.3%. Studies have shown that reducing LDL-C to below 100 mg/dL is associated with a higher incidence of ALT and AST elevations. However, these elevations have usually been benign and often return to normal when the drug is reduced in dose or withdrawn.
In previous studies of rosuvastatin,65 the incidence of myopathy and liver function abnormalities was less than 0.1%. Rates of proteinuria were similarly low, and in many patients renal function actually improved on rosuvastatin.66,67 Furthermore, rosuvastatin may have different pharmacokinetic properties than atorvastatin (Lipitor) and simvastatin, which may result in a lower incidence of musculoskeletal toxicity.68,69
In general, the incidence of cancer has been similar in those treated with high-dose statins and those treated with placebo. The Treating to New Targets trial70 suggested that the incidence of cancer was higher with atorvastatin 80 mg daily than with 20 mg daily. However, a meta-analysis of 14 trials of moderate-dose statin therapy did not show any evidence of increased cancer rates with these agents.70 Indeed, in JUPITER, there was a reduction in cancer-related mortality rates, which could have been due to chance.
The JUPITER trial also showed an increase in the physician-reported incidence of diabetes mellitus with rosuvastatin. This is an important finding, and it may be a class effect because modest increases have similarly been reported with other statins in other major trials, eg, with pravastatin (Pravachol) in PROSPER, simvastatin in the Heart Protection Study, and atorvastatin in PROVE-IT. However, even in those with diabetes or impaired fasting glucose, the reduction in the rate of major adverse events is significant. For example, in JUPITER, almost all of the cases of “incident diabetes” were in those with impaired fasting glucose at baseline, and this group had nearly a 50% reduction in rates of myocardial infarction, stroke, and cardiovascular death. Therefore, on balance, the modest risk of earlier diagnosis of diabetes with statin therapy seems substantially offset by the marked reduction in rates of major adverse cardiovascular events in people with diabetes and impaired fasting glucose on statin therapy.
TAKE-HOME POINTS
The JUPITER trial, like previous high-dose statin trials, calls into question whether current LDL-C guidelines are appropriate for people at higher global risk with otherwise “normal” LDL-C levels.27,28 This trial heralds a new era in preventive therapy because it extends beyond LDL-C as an indication for statin therapy within the primary prevention setting. Statins have revolutionized the therapy of cardiovascular disease, and they continue to show benefit even in the “healthy.”
Clearly, hs-CRP serves as a nonlipid marker to identify those who may benefit from statin therapy. Nonetheless, more specific and sensitive markers (or panels) of cardiovascular risk are necessary. In the future, we will need markers that not only identify people at higher global risk, but that also tell us who would benefit from certain medical or surgical therapies. Elevated hs-CRP in a patient who otherwise would not be a candidate for statin therapy should trigger a reassessment of the risks vs benefits of statin therapy—JUPITER teaches us that statin therapy will benefit these patients.
Aggressive lifestyle modification that encompasses a balanced diet, routine exercise, and smoking cessation should be applied in both primary and secondary prevention. Additionally, risk factors such as elevated blood pressure and hyperlipidemia should be aggressively treated with appropriate medications.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Topol EJ. Intensive statin therapy—a sea change in cardiovascular prevention. N Engl J Med 2004; 350:1562–1564.
- Cannon CP, Murphy SA, Braunwald E. Intensive lipid lowering with atorvastatin in coronary disease. N Engl J Med 2005; 353:93–96.
- Cohen DJ, Carrozza JP, Baim DS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med 1999; 341:1853–1854.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071–1080.
- Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005; 46:1855–1862.
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103:276–283.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol 2005; 96:24F–33F.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Shishehbor MH, Aviles RJ, Brennan ML, et al. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 2003; 289:1675–1680.
- Shishehbor MH, Brennan ML, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003; 108:426–431.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol 2001; 21:1712–1719.
- Shishehbor MH, Patel T, Bhatt DL. Using statins to treat inflammation in acute coronary syndromes: Are we there yet? Cleve Clin J Med 2006; 73:760–766.
- Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344:1959–1965.
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998; 98:731–733.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342:836–843.
- Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001; 285:2481–2485.
- Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem 2001; 47:403–411.
- Shishehbor MH, Bhatt DL, Topol EJ. Using C-reactive protein to assess cardiovascular disease risk. Cleve Clin J Med 2003; 70:634–640.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336:973–979.
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 2008; 359:1897–1908.
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep 2000; 2:269–273.
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003; 107:391–397.
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107:499–511.
- Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract 2002; 56:53–56.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Liem AH, van Boven AJ, Veeger NJ, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J 2002; 23:1931–1937.
- Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Pitt B, Waters D, Brown WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 1999; 341:70–76.
- Ray KK, Cannon CP, McCabe CH, et al. Early and late benefits of highdose atorvastatin in patients with acute coronary syndromes: results from the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2005; 46:1405–1410.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 287:3215–3222.
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J 2007; 28:664–672.
- Ridker PM. Novel risk factors and markers for coronary disease. Adv Intern Med 2000; 45:391–418.
- Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001; 103:1813–1818.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97:2007–2011.
- Cushman M, Arnold AM, Psaty BM, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 2005; 112:25–31.
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol 2007; 49:2129–2138.
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286:64–70.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100:230–235.
- Bisoendial RJ, Kastelein JJ, Levels JH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res 2005; 96:714–716.
- Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005; 112:1016–1023.
- Pfister R, Hellmich M. Multiple biomarkers and cardiovascular risk. N Engl J Med 2008; 359:760.
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis— chicken or egg? N Engl J Med 2008; 359:1953–1955.
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350:1387–1397.
- Kushner I, Sehgal AR. Is high-sensitivity C-reactive protein an effective screening test for cardiovascular risk? Arch Intern Med 2002; 162:867–869.
- Hlatky MA. Expanding the orbit of primary prevention—moving beyond JUPITER. N Engl J Med 2008; 359:2280–2282.
- Shishehbor MH, Hazen SL. Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr Atheroscler Rep 2004; 6:243–250.
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 2005; 25:1102–1111.
- Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008; 299:1265–1276.
- Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol 2008; 52:24–32.
- Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005; 293:2245–2256.
- Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol 2008; 51:1653–1662.
- Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2007; 27:134–140.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol 2003; 28:317–347
- West of Scotland Coronary Prevention Study: identification of highrisk groups and comparison with other cardiovascular intervention trials. Lancet 1996; 348:1339–1342.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007; 49:1753–1762.
- Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf 2004; 3:547–557.
- Kasiske BL, Wanner C, O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006; 97:82C–85C.
- McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87:28B–32B.
- Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006; 97:44C–51C.
- Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 2004; 94:1140–1146.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
The medical community has struggled with two important questions for the past 10 years: When it comes to the low-density lipoprotein cholesterol (LDL-C) level, how low should one go and at what cost? And are there other markers of risk that can identify a higher-risk subpopulation in relatively healthy people? The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) provided partial answers for these questions by finding that a highly potent statin lowered the risk of cardiovascular events in patients with “normal” LDL-C but elevated levels of high-sensitivity C-reactive protein (hs-CRP).1
In this article, we will critically evaluate the methods, results, and conclusions of the JUPITER trial. Additionally, we will discuss its limitations and areas of uncertainty.
BEFORE JUPITER
The LDL-C-lowering drugs called statins have revolutionized cardiovascular medicine.2 They are beneficial in both the primary prevention setting and in acute coronary syndromes, stable angina, and unstable angina and can halt the progression of coronary artery disease—in some cases even resulting in modest regression of plaque.3–6
Many experts have credited the reduction in LDL-C as being the sole factor responsible for the decrease in major adverse events seen with statin therapy.7 However, statins have other, non-lipid-lowering properties, including anti-inflammatory and antioxidant effects, that may also contribute to their benefits.8–15
One of the anti-inflammatory actions of statins is evidenced by lower levels of the acute-phase reactant CRP.10,11,15,16 Measuring systemic CRP levels with a highly sensitive assay (yielding the so-called high-sensitivity or hs-CRP level) provides significant clinical prognostic value across a spectrum of clinical situations, ranging from risk screening in apparently healthy people to stable and unstable angina.17–22 People with higher hs-CRP levels are, on average, at higher risk of adverse cardiovascular events. However, controversy remains as to whether hs-CRP plays a mechanistic role in plaque formation and acute complications. Indeed, recent genetic studies argue strongly that hs-CRP lies outside the mechanistic path of atherosclerosis.23 Nonetheless, an overwhelming amount of data indicates that hs-CRP serves as a marker of disease.17–21
Nissen et al10 showed that the rate of progression of atherosclerosis is lower when the levels of atherogenic lipoproteins and hs-CRP are both lowered with statin therapy. Simultaneously, Ridker et al11 showed that patients who have lower hs-CRP levels after statin therapy have better clinical outcomes than those with higher hs-CRP levels, regardless of their achieved level of LDL-C.
Collectively, these studies and others have led some to believe that, in people with relatively low LDL-C but persistently elevated hs-CRP, statin therapy may reduce the rate of events.15,24 The JUPITER trial was undertaken to test this hypothesis.
JUPITER STUDY DESIGN
JUPITER was designed to see whether highly potent statin therapy is beneficial in people with elevated hs-CRP who otherwise do not meet the criteria for lipid-lowering therapy. The study was conducted at 1,315 sites in 26 countries. It was sponsored by AstraZeneca, the maker of rosuvastatin (Crestor).
Inclusion and exclusion criteria
All participants had to be free of known cardiovascular disease, have an LDL-C level lower than 130 mg/dL, and have an hs-CRP level of 2.0 mg/L or greater. Patients were excluded if they were previous or current users of lipid-lowering drugs; had severe arthritis, lupus, or inflammatory bowel disease; or were taking immune-modulating drugs such as cyclosporine (Sandimmune, others), tacrolimus (Prograf), azathioprine (Azasan, Imuran), or long-term oral corticosteroids.
Rosuvastatin therapy
Participants were randomly assigned in a 1:1 ratio to receive rosuvastatin 20 mg daily or a matching placebo in a double-blind fashion.
End points
The primary end point was the composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from cardiovascular causes. Secondary end points were the individual components of the primary end point.
Statistical analysis
The study was powered to detect a 25% reduction in the primary end point among those treated with rosuvastatin. The trial was designed to run until 520 end point events had occurred. However, on March 29, 2008, after the first prespecified interim analysis, the Data and Safety Monitoring Board stopped the trial due to a significant reduction in the primary end point in the rosuvastatin group. As in most randomized clinical trials, all analyses were done on an intention-to-treat basis. Prespecified subgroup analyses were also performed.
STUDY RESULTS
Patient recruitment and eligibility
Between February 4, 2003, and December 15, 2006, a total of 89,890 people were screened. Of these, 17,802 met the inclusion and exclusion criteria and were included in the study. Of the 72,088 people who were excluded, 25,993 (36.1%) had an hs-CRP level below 2 mg/L and 37,611 (52.2%) had an LDL-C level of 130 mg/dL or higher.
A not-so-healthy population
The aim of the investigators was to include relatively healthy people. The median age was 66 years, about 16% of participants were current smokers, about 11% had a family history of heart disease, and about 41% met the criteria for metabolic syndrome, all conditions that are associated with elevated hs-CRP.25 Of note, the median hs-CRP level was 4.2 mg/L, a level indicating higher global risk according to the American College of Cardiology/American Heart Association consensus statement.26
Reduction in lipid levels and hs-CRP
By 12 months, in the rosuvastatin group, the median LDL-C level had fallen by 50% (from 108 to 55 mg/dL), and the median hs-CRP level had fallen by 37% (from 4.2 to 2.2 mg/L). Additionally, the triglyceride level had fallen by 17%. The high-density lipoprotein cholesterol levels did not change significantly.
Impact on end points
Adverse events
WHAT DOES THIS MEAN?
Is lower LDL-C better?
The JUPITER trial is the latest of several statin trials that have shown significant reductions in major adverse cardiovascular events when LDL-C was lowered below what has been recommended by the current guidelines.27,28
In 2002, the Heart Protection Study29 showed a significant reduction in major adverse cardiovascular events in patients at high risk of coronary artery disease if they received simvastatin (Zocor), even if they had LDL-C levels lower than 100 mg/dL at baseline. Similarly, the Pravastatin or Atorvastatin Evaluation and Infection-Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial30 showed a 16% relative risk reduction in a composite end point in patients presenting with acute coronary syndrome if they received intensive statin therapy.
These two studies led to an update by the National Cholesterol Education Program (Adult Treatment Panel III), suggesting an optimal LDL-C goal of less than 70 mg/dL in those with coronary artery disease or its risk equivalent (ie, diabetes mellitus, peripheral vascular disease). Furthermore, in support of the “lower is better” theory, a number of studies that used intravascular ultrasonography have shown regression of coronary plaque with aggressive LDL-C lowering. Notably, in a Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (the ASTEROID trial),5 rosuvastatin 40 mg daily caused significant plaque regression while lowering LDL-C to 61 mg/dL over a 24-month period.
A number of high-dose statin trials have shown that lowering LDL-C to less than 70 mg/dL significantly reduces major adverse cardiovascular events.31–39 The JUPITER trial was unique in that it extended these findings to people without known coronary disease (ie, primary prevention) or elevated cholesterol but with elevated levels of a marker of inflammation—hs-CRP. In view of the JUPITER results and of studies using intravascular ultrasonography in the primary prevention setting, it seems clear that lowering LDL-C to levels less than 70 mg/dL also reduces both atherosclerotic plaque progression and the rate of first major adverse cardiovascular events in primary prevention in patients at higher global risk.
Did the study prove that reducing hs-CRP lowers risk?
Measuring hs-CRP levels has been extensively studied in apparently healthy populations, stable angina, unstable angina, and other cardiovascular settings.18,21,40–43 It has been shown to have significant prognostic implications in a number of primary and secondary trials.44 Additionally, those with elevated LDL-C and hs-CRP levels benefit the most from statin therapy.16,45,46 Animal studies have also provided some evidence that CRP may play a role in atherogenesis.47,48 However, recent clinical and genetic studies have raised doubt about the direct causal relationship between CRP and coronary artery disease,23,49,50 and epidemiologic studies have questioned its usefulness as a marker of risk.51,52
The JUPITER study adds little to clear up the controversy about whether hs-CRP is a mechanistic participant in atherosclerotic disease. However, it also shows that this issue is somewhat irrelevant, in that selection of patients for high-potency statin therapy solely on the basis of high hs-CRP without other indications for lipid-lowering therapy clearly reduces risk and improves survival.
JUPITER did not examine whether people with higher hs-CRP levels benefited more from statin therapy than those with lower levels. The hypothesis-generating data for JUPITER came from an analysis of changes in hs-CRP and LDL-C in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS).16 Thus, JUPITER did not include people with both low LDL-C and low hs-CRP because, in the AFCAPS/TexCAPS analysis, those with low LDL-C and low hs-CRP had extremely low event rates and no clinical efficacy of statin therapy, despite good LDL-C reduction. In marked contrast, those with low LDL-C but elevated hs-CRP had high event rates and large relative risk reductions— hence the need for JUPITER to prospectively test this hypothesis. Nevertheless, the initial results of JUPITER as presented do not yet make it clear that there is a dose-response relationship between higher levels of hs-CRP and a greater reduction in events, even in a cohort with elevated hs-CRP at baseline. This analysis will no doubt be forthcoming in another manuscript from Ridker and colleagues. Specifically, it will be of interest to examine whether those with the highest hs-CRP levels benefited the most from rosuvastatin on both an absolute and relative scale, and whether those with the greatest hs-CRP reduction also benefited more. With the present data available from JUPITER, a reasonable interpretation is that an elevated hs-CRP simply widens the inclusion criterion for those for whom high-potency statin therapy improves clinical outcomes.53
Better markers are needed
Even with a nonspecific marker such as hs-CRP, patients at higher global risk and with LDL-C below the recommended levels could be identified and treated aggressively. This benefit, however, required that approximately 100 people be treated with rosuvastatin for 2 years to prevent one event. Additionally, only 20% of all patients screened were eligible for the trial. Therefore, one could argue that its generalizability is limited.
Markers of risk that are more specific and sensitive are needed to identify people at higher global risk who would otherwise be considered to be at low risk with the current risk assessment tools. A number of such inflammatory and oxidative markers are under development.54–60
Absolute vs relative risk reduction and the public health burden
The 44% reduction in the number of primary end point events in the rosuvastatin group was considerable in relative terms. However, in absolute terms, 95 people had to be treated for up to 2 years in order to prevent one event.53 In making recommendations, the United States Department of Health and Human Services has to consider the clinical benefit of a test or a drug in light of its cost. With health care costs increasing, many agencies are refusing to pay for therapies on the basis of cost or small absolute benefit.
While we do not have the answer as to whether treating 95 people for 2 years to see one benefit is cost-effective, one thing is clear: the field of medicine is in desperate need of a better way to identify individuals who may benefit from a test or therapy.61 Additionally, we think it is important to note that the “numbers-needed-to-treat” (95 at 2 years and 25 at 5 years) derived from JUPITER are actually smaller than the values observed in the AFCAPS/TexCAPS and the West of Scotland Coronary Prevention Study.62,63 This suggests that statin therapy is at least as cost-effective in those with elevated hs-CRP as in those with elevated LDL-C. Even our most robust therapies are effective in only a minority of patients treated.61
Should ‘healthy’ people be tested for hs-CRP?
In 2003, we wrote in this journal21 that measuring hs-CRP may add to the current risk-prediction models by identifying people at increased risk who would otherwise not be considered as such by current risk models. The US Centers for Disease Control and Prevention and the American Heart Association have also stated that measuring hs-CRP in those at intermediate risk may be reasonable.26
The JUPITER investigators intended to study a relatively healthy population, but, as we mentioned, a close look at the cohort’s baseline characteristics indicates a substantial proportion met the criteria for metabolic syndrome. Therefore, one could challenge whether we really need hs-CRP in such a population to identify who will benefit from statin therapy.
We agree with the recommendation from the Centers for Disease Control and Prevention and the American Heart Association that measuring hs-CRP in people at intermediate risk is a reasonable option.26 We also believe that hs-CRP should be tested as a secondary risk factor, in combination with blood pressure, lipids, diabetes, smoking, serum creatinine, and fasting blood glucose. Factors such as obesity, sedentary lifestyle, family history of heart disease, and emotional and physical stress should also be considered.
Safety of high-dose statin therapy
High-dose statin therapy has been well tolerated in clinical trials, but rates of discontinuation have been higher (7%–10%) than with moderate-dose therapy (4%–5%).64 Fortunately, the rates of serious adverse events have in general been low. For example, with simvastatin 80 mg, the rates of myopathy and rhabdomyolysis were quite low.31
Rates of elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with high-dose statin therapy have been reported to be below 1.3%. Studies have shown that reducing LDL-C to below 100 mg/dL is associated with a higher incidence of ALT and AST elevations. However, these elevations have usually been benign and often return to normal when the drug is reduced in dose or withdrawn.
In previous studies of rosuvastatin,65 the incidence of myopathy and liver function abnormalities was less than 0.1%. Rates of proteinuria were similarly low, and in many patients renal function actually improved on rosuvastatin.66,67 Furthermore, rosuvastatin may have different pharmacokinetic properties than atorvastatin (Lipitor) and simvastatin, which may result in a lower incidence of musculoskeletal toxicity.68,69
In general, the incidence of cancer has been similar in those treated with high-dose statins and those treated with placebo. The Treating to New Targets trial70 suggested that the incidence of cancer was higher with atorvastatin 80 mg daily than with 20 mg daily. However, a meta-analysis of 14 trials of moderate-dose statin therapy did not show any evidence of increased cancer rates with these agents.70 Indeed, in JUPITER, there was a reduction in cancer-related mortality rates, which could have been due to chance.
The JUPITER trial also showed an increase in the physician-reported incidence of diabetes mellitus with rosuvastatin. This is an important finding, and it may be a class effect because modest increases have similarly been reported with other statins in other major trials, eg, with pravastatin (Pravachol) in PROSPER, simvastatin in the Heart Protection Study, and atorvastatin in PROVE-IT. However, even in those with diabetes or impaired fasting glucose, the reduction in the rate of major adverse events is significant. For example, in JUPITER, almost all of the cases of “incident diabetes” were in those with impaired fasting glucose at baseline, and this group had nearly a 50% reduction in rates of myocardial infarction, stroke, and cardiovascular death. Therefore, on balance, the modest risk of earlier diagnosis of diabetes with statin therapy seems substantially offset by the marked reduction in rates of major adverse cardiovascular events in people with diabetes and impaired fasting glucose on statin therapy.
TAKE-HOME POINTS
The JUPITER trial, like previous high-dose statin trials, calls into question whether current LDL-C guidelines are appropriate for people at higher global risk with otherwise “normal” LDL-C levels.27,28 This trial heralds a new era in preventive therapy because it extends beyond LDL-C as an indication for statin therapy within the primary prevention setting. Statins have revolutionized the therapy of cardiovascular disease, and they continue to show benefit even in the “healthy.”
Clearly, hs-CRP serves as a nonlipid marker to identify those who may benefit from statin therapy. Nonetheless, more specific and sensitive markers (or panels) of cardiovascular risk are necessary. In the future, we will need markers that not only identify people at higher global risk, but that also tell us who would benefit from certain medical or surgical therapies. Elevated hs-CRP in a patient who otherwise would not be a candidate for statin therapy should trigger a reassessment of the risks vs benefits of statin therapy—JUPITER teaches us that statin therapy will benefit these patients.
Aggressive lifestyle modification that encompasses a balanced diet, routine exercise, and smoking cessation should be applied in both primary and secondary prevention. Additionally, risk factors such as elevated blood pressure and hyperlipidemia should be aggressively treated with appropriate medications.
The medical community has struggled with two important questions for the past 10 years: When it comes to the low-density lipoprotein cholesterol (LDL-C) level, how low should one go and at what cost? And are there other markers of risk that can identify a higher-risk subpopulation in relatively healthy people? The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) provided partial answers for these questions by finding that a highly potent statin lowered the risk of cardiovascular events in patients with “normal” LDL-C but elevated levels of high-sensitivity C-reactive protein (hs-CRP).1
In this article, we will critically evaluate the methods, results, and conclusions of the JUPITER trial. Additionally, we will discuss its limitations and areas of uncertainty.
BEFORE JUPITER
The LDL-C-lowering drugs called statins have revolutionized cardiovascular medicine.2 They are beneficial in both the primary prevention setting and in acute coronary syndromes, stable angina, and unstable angina and can halt the progression of coronary artery disease—in some cases even resulting in modest regression of plaque.3–6
Many experts have credited the reduction in LDL-C as being the sole factor responsible for the decrease in major adverse events seen with statin therapy.7 However, statins have other, non-lipid-lowering properties, including anti-inflammatory and antioxidant effects, that may also contribute to their benefits.8–15
One of the anti-inflammatory actions of statins is evidenced by lower levels of the acute-phase reactant CRP.10,11,15,16 Measuring systemic CRP levels with a highly sensitive assay (yielding the so-called high-sensitivity or hs-CRP level) provides significant clinical prognostic value across a spectrum of clinical situations, ranging from risk screening in apparently healthy people to stable and unstable angina.17–22 People with higher hs-CRP levels are, on average, at higher risk of adverse cardiovascular events. However, controversy remains as to whether hs-CRP plays a mechanistic role in plaque formation and acute complications. Indeed, recent genetic studies argue strongly that hs-CRP lies outside the mechanistic path of atherosclerosis.23 Nonetheless, an overwhelming amount of data indicates that hs-CRP serves as a marker of disease.17–21
Nissen et al10 showed that the rate of progression of atherosclerosis is lower when the levels of atherogenic lipoproteins and hs-CRP are both lowered with statin therapy. Simultaneously, Ridker et al11 showed that patients who have lower hs-CRP levels after statin therapy have better clinical outcomes than those with higher hs-CRP levels, regardless of their achieved level of LDL-C.
Collectively, these studies and others have led some to believe that, in people with relatively low LDL-C but persistently elevated hs-CRP, statin therapy may reduce the rate of events.15,24 The JUPITER trial was undertaken to test this hypothesis.
JUPITER STUDY DESIGN
JUPITER was designed to see whether highly potent statin therapy is beneficial in people with elevated hs-CRP who otherwise do not meet the criteria for lipid-lowering therapy. The study was conducted at 1,315 sites in 26 countries. It was sponsored by AstraZeneca, the maker of rosuvastatin (Crestor).
Inclusion and exclusion criteria
All participants had to be free of known cardiovascular disease, have an LDL-C level lower than 130 mg/dL, and have an hs-CRP level of 2.0 mg/L or greater. Patients were excluded if they were previous or current users of lipid-lowering drugs; had severe arthritis, lupus, or inflammatory bowel disease; or were taking immune-modulating drugs such as cyclosporine (Sandimmune, others), tacrolimus (Prograf), azathioprine (Azasan, Imuran), or long-term oral corticosteroids.
Rosuvastatin therapy
Participants were randomly assigned in a 1:1 ratio to receive rosuvastatin 20 mg daily or a matching placebo in a double-blind fashion.
End points
The primary end point was the composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from cardiovascular causes. Secondary end points were the individual components of the primary end point.
Statistical analysis
The study was powered to detect a 25% reduction in the primary end point among those treated with rosuvastatin. The trial was designed to run until 520 end point events had occurred. However, on March 29, 2008, after the first prespecified interim analysis, the Data and Safety Monitoring Board stopped the trial due to a significant reduction in the primary end point in the rosuvastatin group. As in most randomized clinical trials, all analyses were done on an intention-to-treat basis. Prespecified subgroup analyses were also performed.
STUDY RESULTS
Patient recruitment and eligibility
Between February 4, 2003, and December 15, 2006, a total of 89,890 people were screened. Of these, 17,802 met the inclusion and exclusion criteria and were included in the study. Of the 72,088 people who were excluded, 25,993 (36.1%) had an hs-CRP level below 2 mg/L and 37,611 (52.2%) had an LDL-C level of 130 mg/dL or higher.
A not-so-healthy population
The aim of the investigators was to include relatively healthy people. The median age was 66 years, about 16% of participants were current smokers, about 11% had a family history of heart disease, and about 41% met the criteria for metabolic syndrome, all conditions that are associated with elevated hs-CRP.25 Of note, the median hs-CRP level was 4.2 mg/L, a level indicating higher global risk according to the American College of Cardiology/American Heart Association consensus statement.26
Reduction in lipid levels and hs-CRP
By 12 months, in the rosuvastatin group, the median LDL-C level had fallen by 50% (from 108 to 55 mg/dL), and the median hs-CRP level had fallen by 37% (from 4.2 to 2.2 mg/L). Additionally, the triglyceride level had fallen by 17%. The high-density lipoprotein cholesterol levels did not change significantly.
Impact on end points
Adverse events
WHAT DOES THIS MEAN?
Is lower LDL-C better?
The JUPITER trial is the latest of several statin trials that have shown significant reductions in major adverse cardiovascular events when LDL-C was lowered below what has been recommended by the current guidelines.27,28
In 2002, the Heart Protection Study29 showed a significant reduction in major adverse cardiovascular events in patients at high risk of coronary artery disease if they received simvastatin (Zocor), even if they had LDL-C levels lower than 100 mg/dL at baseline. Similarly, the Pravastatin or Atorvastatin Evaluation and Infection-Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial30 showed a 16% relative risk reduction in a composite end point in patients presenting with acute coronary syndrome if they received intensive statin therapy.
These two studies led to an update by the National Cholesterol Education Program (Adult Treatment Panel III), suggesting an optimal LDL-C goal of less than 70 mg/dL in those with coronary artery disease or its risk equivalent (ie, diabetes mellitus, peripheral vascular disease). Furthermore, in support of the “lower is better” theory, a number of studies that used intravascular ultrasonography have shown regression of coronary plaque with aggressive LDL-C lowering. Notably, in a Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (the ASTEROID trial),5 rosuvastatin 40 mg daily caused significant plaque regression while lowering LDL-C to 61 mg/dL over a 24-month period.
A number of high-dose statin trials have shown that lowering LDL-C to less than 70 mg/dL significantly reduces major adverse cardiovascular events.31–39 The JUPITER trial was unique in that it extended these findings to people without known coronary disease (ie, primary prevention) or elevated cholesterol but with elevated levels of a marker of inflammation—hs-CRP. In view of the JUPITER results and of studies using intravascular ultrasonography in the primary prevention setting, it seems clear that lowering LDL-C to levels less than 70 mg/dL also reduces both atherosclerotic plaque progression and the rate of first major adverse cardiovascular events in primary prevention in patients at higher global risk.
Did the study prove that reducing hs-CRP lowers risk?
Measuring hs-CRP levels has been extensively studied in apparently healthy populations, stable angina, unstable angina, and other cardiovascular settings.18,21,40–43 It has been shown to have significant prognostic implications in a number of primary and secondary trials.44 Additionally, those with elevated LDL-C and hs-CRP levels benefit the most from statin therapy.16,45,46 Animal studies have also provided some evidence that CRP may play a role in atherogenesis.47,48 However, recent clinical and genetic studies have raised doubt about the direct causal relationship between CRP and coronary artery disease,23,49,50 and epidemiologic studies have questioned its usefulness as a marker of risk.51,52
The JUPITER study adds little to clear up the controversy about whether hs-CRP is a mechanistic participant in atherosclerotic disease. However, it also shows that this issue is somewhat irrelevant, in that selection of patients for high-potency statin therapy solely on the basis of high hs-CRP without other indications for lipid-lowering therapy clearly reduces risk and improves survival.
JUPITER did not examine whether people with higher hs-CRP levels benefited more from statin therapy than those with lower levels. The hypothesis-generating data for JUPITER came from an analysis of changes in hs-CRP and LDL-C in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS).16 Thus, JUPITER did not include people with both low LDL-C and low hs-CRP because, in the AFCAPS/TexCAPS analysis, those with low LDL-C and low hs-CRP had extremely low event rates and no clinical efficacy of statin therapy, despite good LDL-C reduction. In marked contrast, those with low LDL-C but elevated hs-CRP had high event rates and large relative risk reductions— hence the need for JUPITER to prospectively test this hypothesis. Nevertheless, the initial results of JUPITER as presented do not yet make it clear that there is a dose-response relationship between higher levels of hs-CRP and a greater reduction in events, even in a cohort with elevated hs-CRP at baseline. This analysis will no doubt be forthcoming in another manuscript from Ridker and colleagues. Specifically, it will be of interest to examine whether those with the highest hs-CRP levels benefited the most from rosuvastatin on both an absolute and relative scale, and whether those with the greatest hs-CRP reduction also benefited more. With the present data available from JUPITER, a reasonable interpretation is that an elevated hs-CRP simply widens the inclusion criterion for those for whom high-potency statin therapy improves clinical outcomes.53
Better markers are needed
Even with a nonspecific marker such as hs-CRP, patients at higher global risk and with LDL-C below the recommended levels could be identified and treated aggressively. This benefit, however, required that approximately 100 people be treated with rosuvastatin for 2 years to prevent one event. Additionally, only 20% of all patients screened were eligible for the trial. Therefore, one could argue that its generalizability is limited.
Markers of risk that are more specific and sensitive are needed to identify people at higher global risk who would otherwise be considered to be at low risk with the current risk assessment tools. A number of such inflammatory and oxidative markers are under development.54–60
Absolute vs relative risk reduction and the public health burden
The 44% reduction in the number of primary end point events in the rosuvastatin group was considerable in relative terms. However, in absolute terms, 95 people had to be treated for up to 2 years in order to prevent one event.53 In making recommendations, the United States Department of Health and Human Services has to consider the clinical benefit of a test or a drug in light of its cost. With health care costs increasing, many agencies are refusing to pay for therapies on the basis of cost or small absolute benefit.
While we do not have the answer as to whether treating 95 people for 2 years to see one benefit is cost-effective, one thing is clear: the field of medicine is in desperate need of a better way to identify individuals who may benefit from a test or therapy.61 Additionally, we think it is important to note that the “numbers-needed-to-treat” (95 at 2 years and 25 at 5 years) derived from JUPITER are actually smaller than the values observed in the AFCAPS/TexCAPS and the West of Scotland Coronary Prevention Study.62,63 This suggests that statin therapy is at least as cost-effective in those with elevated hs-CRP as in those with elevated LDL-C. Even our most robust therapies are effective in only a minority of patients treated.61
Should ‘healthy’ people be tested for hs-CRP?
In 2003, we wrote in this journal21 that measuring hs-CRP may add to the current risk-prediction models by identifying people at increased risk who would otherwise not be considered as such by current risk models. The US Centers for Disease Control and Prevention and the American Heart Association have also stated that measuring hs-CRP in those at intermediate risk may be reasonable.26
The JUPITER investigators intended to study a relatively healthy population, but, as we mentioned, a close look at the cohort’s baseline characteristics indicates a substantial proportion met the criteria for metabolic syndrome. Therefore, one could challenge whether we really need hs-CRP in such a population to identify who will benefit from statin therapy.
We agree with the recommendation from the Centers for Disease Control and Prevention and the American Heart Association that measuring hs-CRP in people at intermediate risk is a reasonable option.26 We also believe that hs-CRP should be tested as a secondary risk factor, in combination with blood pressure, lipids, diabetes, smoking, serum creatinine, and fasting blood glucose. Factors such as obesity, sedentary lifestyle, family history of heart disease, and emotional and physical stress should also be considered.
Safety of high-dose statin therapy
High-dose statin therapy has been well tolerated in clinical trials, but rates of discontinuation have been higher (7%–10%) than with moderate-dose therapy (4%–5%).64 Fortunately, the rates of serious adverse events have in general been low. For example, with simvastatin 80 mg, the rates of myopathy and rhabdomyolysis were quite low.31
Rates of elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with high-dose statin therapy have been reported to be below 1.3%. Studies have shown that reducing LDL-C to below 100 mg/dL is associated with a higher incidence of ALT and AST elevations. However, these elevations have usually been benign and often return to normal when the drug is reduced in dose or withdrawn.
In previous studies of rosuvastatin,65 the incidence of myopathy and liver function abnormalities was less than 0.1%. Rates of proteinuria were similarly low, and in many patients renal function actually improved on rosuvastatin.66,67 Furthermore, rosuvastatin may have different pharmacokinetic properties than atorvastatin (Lipitor) and simvastatin, which may result in a lower incidence of musculoskeletal toxicity.68,69
In general, the incidence of cancer has been similar in those treated with high-dose statins and those treated with placebo. The Treating to New Targets trial70 suggested that the incidence of cancer was higher with atorvastatin 80 mg daily than with 20 mg daily. However, a meta-analysis of 14 trials of moderate-dose statin therapy did not show any evidence of increased cancer rates with these agents.70 Indeed, in JUPITER, there was a reduction in cancer-related mortality rates, which could have been due to chance.
The JUPITER trial also showed an increase in the physician-reported incidence of diabetes mellitus with rosuvastatin. This is an important finding, and it may be a class effect because modest increases have similarly been reported with other statins in other major trials, eg, with pravastatin (Pravachol) in PROSPER, simvastatin in the Heart Protection Study, and atorvastatin in PROVE-IT. However, even in those with diabetes or impaired fasting glucose, the reduction in the rate of major adverse events is significant. For example, in JUPITER, almost all of the cases of “incident diabetes” were in those with impaired fasting glucose at baseline, and this group had nearly a 50% reduction in rates of myocardial infarction, stroke, and cardiovascular death. Therefore, on balance, the modest risk of earlier diagnosis of diabetes with statin therapy seems substantially offset by the marked reduction in rates of major adverse cardiovascular events in people with diabetes and impaired fasting glucose on statin therapy.
TAKE-HOME POINTS
The JUPITER trial, like previous high-dose statin trials, calls into question whether current LDL-C guidelines are appropriate for people at higher global risk with otherwise “normal” LDL-C levels.27,28 This trial heralds a new era in preventive therapy because it extends beyond LDL-C as an indication for statin therapy within the primary prevention setting. Statins have revolutionized the therapy of cardiovascular disease, and they continue to show benefit even in the “healthy.”
Clearly, hs-CRP serves as a nonlipid marker to identify those who may benefit from statin therapy. Nonetheless, more specific and sensitive markers (or panels) of cardiovascular risk are necessary. In the future, we will need markers that not only identify people at higher global risk, but that also tell us who would benefit from certain medical or surgical therapies. Elevated hs-CRP in a patient who otherwise would not be a candidate for statin therapy should trigger a reassessment of the risks vs benefits of statin therapy—JUPITER teaches us that statin therapy will benefit these patients.
Aggressive lifestyle modification that encompasses a balanced diet, routine exercise, and smoking cessation should be applied in both primary and secondary prevention. Additionally, risk factors such as elevated blood pressure and hyperlipidemia should be aggressively treated with appropriate medications.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Topol EJ. Intensive statin therapy—a sea change in cardiovascular prevention. N Engl J Med 2004; 350:1562–1564.
- Cannon CP, Murphy SA, Braunwald E. Intensive lipid lowering with atorvastatin in coronary disease. N Engl J Med 2005; 353:93–96.
- Cohen DJ, Carrozza JP, Baim DS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med 1999; 341:1853–1854.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071–1080.
- Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005; 46:1855–1862.
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103:276–283.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol 2005; 96:24F–33F.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Shishehbor MH, Aviles RJ, Brennan ML, et al. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 2003; 289:1675–1680.
- Shishehbor MH, Brennan ML, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003; 108:426–431.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol 2001; 21:1712–1719.
- Shishehbor MH, Patel T, Bhatt DL. Using statins to treat inflammation in acute coronary syndromes: Are we there yet? Cleve Clin J Med 2006; 73:760–766.
- Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344:1959–1965.
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998; 98:731–733.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342:836–843.
- Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001; 285:2481–2485.
- Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem 2001; 47:403–411.
- Shishehbor MH, Bhatt DL, Topol EJ. Using C-reactive protein to assess cardiovascular disease risk. Cleve Clin J Med 2003; 70:634–640.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336:973–979.
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 2008; 359:1897–1908.
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep 2000; 2:269–273.
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003; 107:391–397.
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107:499–511.
- Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract 2002; 56:53–56.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Liem AH, van Boven AJ, Veeger NJ, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J 2002; 23:1931–1937.
- Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Pitt B, Waters D, Brown WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 1999; 341:70–76.
- Ray KK, Cannon CP, McCabe CH, et al. Early and late benefits of highdose atorvastatin in patients with acute coronary syndromes: results from the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2005; 46:1405–1410.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 287:3215–3222.
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J 2007; 28:664–672.
- Ridker PM. Novel risk factors and markers for coronary disease. Adv Intern Med 2000; 45:391–418.
- Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001; 103:1813–1818.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97:2007–2011.
- Cushman M, Arnold AM, Psaty BM, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 2005; 112:25–31.
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol 2007; 49:2129–2138.
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286:64–70.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100:230–235.
- Bisoendial RJ, Kastelein JJ, Levels JH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res 2005; 96:714–716.
- Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005; 112:1016–1023.
- Pfister R, Hellmich M. Multiple biomarkers and cardiovascular risk. N Engl J Med 2008; 359:760.
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis— chicken or egg? N Engl J Med 2008; 359:1953–1955.
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350:1387–1397.
- Kushner I, Sehgal AR. Is high-sensitivity C-reactive protein an effective screening test for cardiovascular risk? Arch Intern Med 2002; 162:867–869.
- Hlatky MA. Expanding the orbit of primary prevention—moving beyond JUPITER. N Engl J Med 2008; 359:2280–2282.
- Shishehbor MH, Hazen SL. Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr Atheroscler Rep 2004; 6:243–250.
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 2005; 25:1102–1111.
- Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008; 299:1265–1276.
- Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol 2008; 52:24–32.
- Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005; 293:2245–2256.
- Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol 2008; 51:1653–1662.
- Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2007; 27:134–140.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol 2003; 28:317–347
- West of Scotland Coronary Prevention Study: identification of highrisk groups and comparison with other cardiovascular intervention trials. Lancet 1996; 348:1339–1342.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007; 49:1753–1762.
- Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf 2004; 3:547–557.
- Kasiske BL, Wanner C, O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006; 97:82C–85C.
- McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87:28B–32B.
- Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006; 97:44C–51C.
- Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 2004; 94:1140–1146.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Topol EJ. Intensive statin therapy—a sea change in cardiovascular prevention. N Engl J Med 2004; 350:1562–1564.
- Cannon CP, Murphy SA, Braunwald E. Intensive lipid lowering with atorvastatin in coronary disease. N Engl J Med 2005; 353:93–96.
- Cohen DJ, Carrozza JP, Baim DS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med 1999; 341:1853–1854.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071–1080.
- Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005; 46:1855–1862.
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103:276–283.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol 2005; 96:24F–33F.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Shishehbor MH, Aviles RJ, Brennan ML, et al. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 2003; 289:1675–1680.
- Shishehbor MH, Brennan ML, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003; 108:426–431.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol 2001; 21:1712–1719.
- Shishehbor MH, Patel T, Bhatt DL. Using statins to treat inflammation in acute coronary syndromes: Are we there yet? Cleve Clin J Med 2006; 73:760–766.
- Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344:1959–1965.
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998; 98:731–733.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342:836–843.
- Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001; 285:2481–2485.
- Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem 2001; 47:403–411.
- Shishehbor MH, Bhatt DL, Topol EJ. Using C-reactive protein to assess cardiovascular disease risk. Cleve Clin J Med 2003; 70:634–640.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336:973–979.
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 2008; 359:1897–1908.
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep 2000; 2:269–273.
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003; 107:391–397.
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107:499–511.
- Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract 2002; 56:53–56.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Liem AH, van Boven AJ, Veeger NJ, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J 2002; 23:1931–1937.
- Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Pitt B, Waters D, Brown WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 1999; 341:70–76.
- Ray KK, Cannon CP, McCabe CH, et al. Early and late benefits of highdose atorvastatin in patients with acute coronary syndromes: results from the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2005; 46:1405–1410.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 287:3215–3222.
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J 2007; 28:664–672.
- Ridker PM. Novel risk factors and markers for coronary disease. Adv Intern Med 2000; 45:391–418.
- Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001; 103:1813–1818.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97:2007–2011.
- Cushman M, Arnold AM, Psaty BM, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 2005; 112:25–31.
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol 2007; 49:2129–2138.
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286:64–70.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100:230–235.
- Bisoendial RJ, Kastelein JJ, Levels JH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res 2005; 96:714–716.
- Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005; 112:1016–1023.
- Pfister R, Hellmich M. Multiple biomarkers and cardiovascular risk. N Engl J Med 2008; 359:760.
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis— chicken or egg? N Engl J Med 2008; 359:1953–1955.
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350:1387–1397.
- Kushner I, Sehgal AR. Is high-sensitivity C-reactive protein an effective screening test for cardiovascular risk? Arch Intern Med 2002; 162:867–869.
- Hlatky MA. Expanding the orbit of primary prevention—moving beyond JUPITER. N Engl J Med 2008; 359:2280–2282.
- Shishehbor MH, Hazen SL. Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr Atheroscler Rep 2004; 6:243–250.
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 2005; 25:1102–1111.
- Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008; 299:1265–1276.
- Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol 2008; 52:24–32.
- Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005; 293:2245–2256.
- Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol 2008; 51:1653–1662.
- Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2007; 27:134–140.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol 2003; 28:317–347
- West of Scotland Coronary Prevention Study: identification of highrisk groups and comparison with other cardiovascular intervention trials. Lancet 1996; 348:1339–1342.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007; 49:1753–1762.
- Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf 2004; 3:547–557.
- Kasiske BL, Wanner C, O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006; 97:82C–85C.
- McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87:28B–32B.
- Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006; 97:44C–51C.
- Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 2004; 94:1140–1146.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
KEY POINTS
- LDL-C is the current gold standard diagnostic marker of risk, and elevated values should be aggressively treated in both primary and secondary prevention.
- The optional LDL-C goal of 70 mg/dL for patients at high risk may need to be extended to others at higher global risk, such as those with elevated hs-CRP.
- Although elevated hs-CRP may identify some people with low LDL-C who are nevertheless at higher global risk, more sensitive and specific markers of risk are needed.
Does intensive therapy of type 2 diabetes help or harm? Seeking accord on ACCORD
The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial1–5 was designed primarily to address, in patients with type 2 diabetes at high risk of cardiovascular events, whether intensive glucose control would result in a lower risk of atherosclerotic disease events or death than would standard treatment.
It was widely expected that intensive treatment would confer either modest benefit or, at worst, no benefit. However, the glucose-lowering arm of the trial was terminated early because of a higher mortality rate in the intensively treated group. (The ACCORD trial has two other arms, which concern blood pressure and lipid-lowering, and these are continuing.)
In earlier trials in type 2 diabetes, concerns had been raised about an increased risk of cardiovascular events and possibly death associated with glucose-lowering drugs, hypoglycemia itself, or both, and these were well known when ACCORD was convened. ACCORD was very carefully designed and included careful adjudication of each cardiovascular event and death, including whether hypoglycemia might have been a proximate cause of some sudden deaths.5
Therefore, the surprising result of the higher mortality rate with intensive glycemic control in ACCORD will be fodder for discussion in many arenas over the next several years, and it poses some challenges for physicians and patients in determining treatment goals, as well as for organizations that write clinical practice guidelines (and perhaps organizations involved in pay-for-performance based on these guidelines).
Still, I believe that the ACCORD results should not substantially change our approach to treatment goals in type 2 diabetes, although hemoglobin A1c targets below 6% may not have much added value for cardiovascular risk reduction. The low overall mortality rate in all the arms of the ACCORD trial emphasizes the importance of lifestyle modification, lipid and blood pressure therapy, and encouragement of aspirin use in all patients with type 2 diabetes.
This article reflects my views as a practicing diabetologist and clinical trialist (I was an investigator in the ACCORD trial) with a long-standing interest in clinical trials and in how the results influence clinical practice. The views I express herein may not reflect the views of other ACCORD investigators, the National Heart, Lung, and Blood Institute (NHLBI), the ACCORD trial coordinating center at Wake Forest University, or its data safety and monitoring board.
RISK OF CORONARY DISEASE INCREASES WITH GLUCOSE
Many observational studies6–10 have shown that the risk of cardiovascular disease, especially coronary heart disease, is two to five times higher in people with diabetes mellitus than in people without diabetes. The risk appears to be continuous, so the higher one’s glucose or hemoglobin A1c, the higher the risk.6 This risk even extends to glucose values well below the threshold values currently used to diagnose diabetes mellitus.6 Since there is no glucose threshold for coronary heart disease, the term dysglycemia (rather than hyperglycemia) has been proposed to note the relationship between glucose and coronary heart disease. (The glucose threshold for microvascular complications of diabetes, such as retinopathy and nephropathy, appears to be between 110 and 126 mg/dL).
The clustering of multiple coronary risk factors such as obesity, dyslipidemia, and hypertension has always raised the question of whether glucose is a culprit in coronary risk or whether it simply “runs in bad company.”
EARLIER CLINICAL TRIALS SUGGEST INTENSIVE TREATMENT RAISES RISK
Even though it has been widely believed that intensive glucose-lowering would reduce cardiovascular risk in type 2 diabetes, there have been hints in previous studies that some intensive-treatment regimens might increase risk.
Two large randomized clinical trials and one small one (discussed below) addressed whether glucose control would reduce the risk of atherosclerotic vascular disease events. In each of them, an increased risk of cardiovascular events and possibly of death was seen in at least one intensively treated group.
In the following discussion, I have calculated all of the death rates as the number of deaths per 1,000 patients per year, based on published study results. In this way, we can compare the rates in the various studies (including ACCORD), regardless of the trial duration.
The university group Diabetes Program: Controvery about tolbutamide therapy
The University Group Diabetes Program (UGDP)11–16 included about 1,000 participants randomized to five treatments: tolbutamide (Orinase, a sulfonylurea), insulin in a fixed dose based on body weight, insulin in adjusted doses based on fasting glucose levels, placebo, and (later) phenformin.
In the 1970s, when the UGDP was carried out, randomized clinical trials were uncommon. Like other trials from that era, the UGDP was underpowered by today’s standards and did not have a data safety and monitoring board.
Rates of cardiovascular events and deaths (per 1,000 patient-years):
- 25 (tolbutamide group)
- 12 (placebo group).
The two insulin groups did not differ from the placebo group in their rates of cardiovascular events or death.15 The tolbutamide arm was stopped, and the ensuing controversy about how to interpret the trial results lasted for more than a decade. It also resulted in a black-box warning for tolbutamide and all subsequent sulfonylureas.
United Kingdom Prospective Diabetes Study: Method of glucose-lowering an issue
The United Kingdom Prospective Diabetes Study (UKPDS)17–27 was launched in 1977. A cohort of 5,102 patients (mean age 54 years) with newly diagnosed type 2 diabetes mellitus followed a “prudent diet” for the first 3 to 4 months. Then, if their fasting glucose levels were in the range of 6.1 to 15 mmol/L (110–270 mg/dL), they were randomized to receive various treatments.
Patients who were not obese were randomized to receive either intensive treatment or conventional treatment. The intensive-treatment group received either insulin or a sulfonylurea (chlorpropamide [Diabinese], glibenclamide, or glipizide [Glucotrol]); the conventional-treatment group received diet therapy. The sulfonylurea arm was included partly to address the UGDP results.
Patients who were obese were randomized to receive one of three treatments: intensive treatment (with the agents listed above), conventional treatment, or metformin (Fortamet, Glucophage).
The mean in-trial hemoglobin A1c level in the intensive-treatment group was 7.0%, compared with 7.9% in the conventional-treatment group.
After a mean follow-up of more than 10 years, the incidence of myocardial infarction was 16% lower in the intensive-treatment group, but the difference was not statistically significant (P = .052).
Rates of death from all causes among nonobese subjects (per 1,000 patient-years):
- 18.2–20.5 (intensive-treatment group)
- 19.9 (conventional-treatment group).
In the obese patients who received metformin, the incidence of myocardial infarction was lower than in the conventional-treatment group but not the intensive-treatment group.
Rates of death among obese patients (per 1,000 patient-years):
- 13.5 (metformin group)
- 18.9 (intensive-treatment group)
- 20.6 (conventional-treatment group).
However, a small subset (n = 587) of the original group assigned to sulfonylurea therapy whose glycemic control deteriorated during the trial were rerandomized to continue to receive a sulfonylurea alone or to have metformin added. There was a statistically significantly higher rate of cardiovascular events and a nonsignificantly higher rate of total mortality in the metformin-plus-sulfonylurea group (30.3 per 1,000 patient-years) than in the sulfonylurea-only group (19.1 per 1,000 patient-years).
These data suggested that the way glucose-lowering was achieved might be as important as the glucose levels actually achieved. However, no definite conclusions could be drawn.
In an editorial on the UKPDS, Nathan26 made a comment that may have been prescient in terms of the ACCORD trial: “Professional organizations will now scramble to decide how to translate the UKPDS results … Whether the UKPDS firmly establishes the choice of any one therapy…or any combination of therapies for the long-term treatment of type 2 diabetes is more questionable.”26
Veterans Administration feasibility study
A Veterans Administration feasibility study28,29 included 153 men (mean age 60) with type 2 diabetes (mean duration 7.8 years) who received either conventional therapy (a single daily dose of insulin) or intensive therapy (multiple doses of insulin plus a sulfonylurea). Over a mean of 27 months, the intensive-therapy group achieved a hemoglobin A1c level that was 2 percentage points lower than in the conventional-therapy group.
At 2.25 years of follow-up, cardiovascular events had occurred in 24 (24%) of the intensive-therapy group and in 16 (20%) of the standard-therapy group (P = .10).
Rates of death from all causes (per 1,000 patient-years):
- 28.9 (intensive-treatment group)
- 17.5 (conventional-treatment group).
ACCORD TRIAL DESIGN
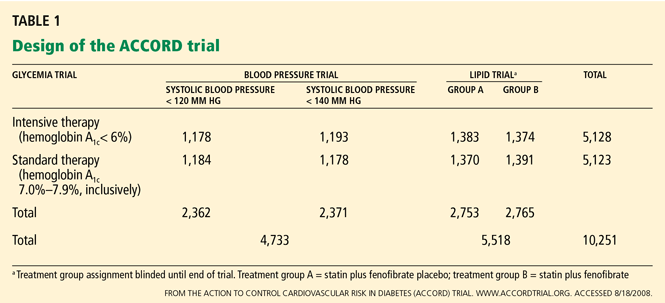
The primary outcome measured was the combined incidence of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes. Secondary outcomes included death from any cause. The study is also evaluating the effect of intensive treatment on microvascular disease, hypoglycemia, cognition, quality of life, and cost-effectiveness.
The ACCORD study was designed to have 89% power to detect a 15% treatment effect of intensive glycemic control compared with standard glycemic control for the primary end point.
ACCORD RESULTS
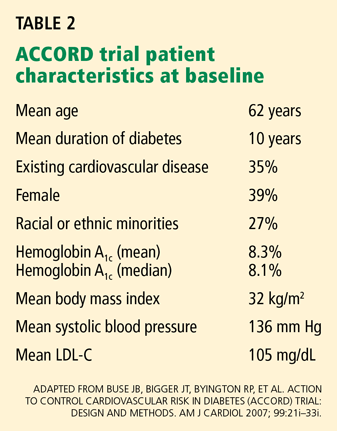
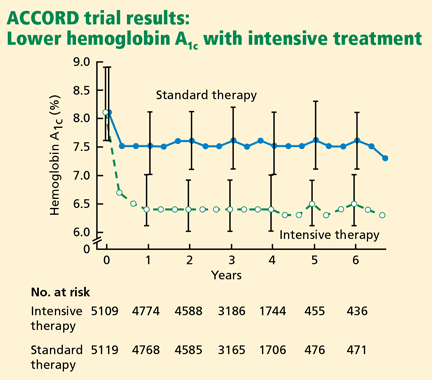
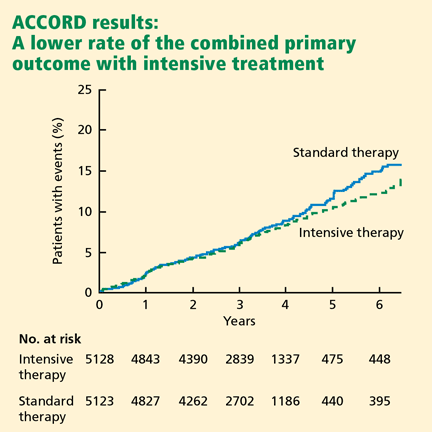
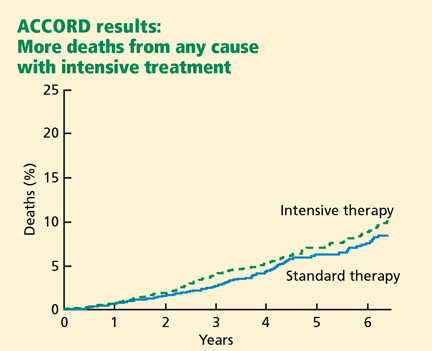
Rates of death from any cause (per 1,000 patient-years):
- 14 (intensive-treatment group)
- 11 (standard-treatment group).
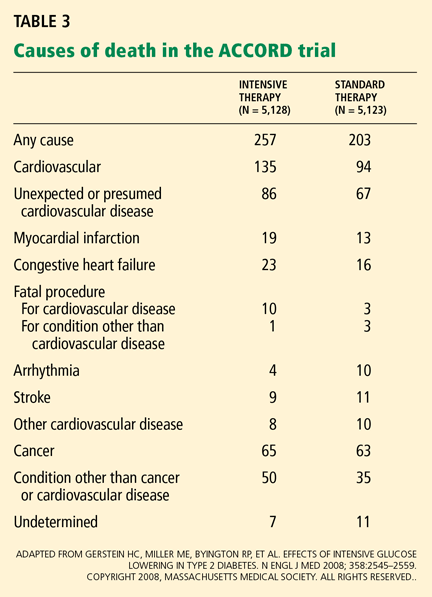
In the analyses available at the time that this study arm closed, the excess mortality was not attributable to any particular treatment regimen. In particular, rosiglitazone (Avandia) use did not contribute to the excess mortality. (Of note, 91.2% of the intensive-treatment group and 57.5% of the conventional-treatment group had been treated with rosiglitazone, with more than 19,000 patient-years of rosiglitazone exposure). The excess mortality was also not attributable to hypoglycemia immediately proximate to the death.
The ACCORD trial’s data safety and monitoring board recommended that this arm of the study be discontinued for safety reasons, and this recommendation was accepted by the NHLBI project office. All participants were notified by letter before the trial results were announced publicly, and all intensive-therapy group participants are now being treated by the protocol used in the standard-therapy group.1
FEWER DEATHS IN ACCORD THAN IN OTHER STUDIES IN DIABETES
The mortality rates in both arms of ACCORD were much lower than in other observational studies and clinical trials in type 2 diabetes.
The National Health and Nutrition Education Survey (NHANES),30 conducted from 1971 to 1975, included 14,374 people with diabetes between the ages of 25 and 74. Many of them were younger than the ACCORD patients, but two NHANES age-groups overlapped the ACCORD cohort. Rates of death from any cause at 22 years (per 1,000 patient-years):
- 39.7 (ages 45–64)
- 89.7 (ages 65–74).
The NHANES cohort would not have been treated as vigorously for coronary risk and other common causes of death.
UGDP, UKPDS. Death rates in the glucose-lowering trials of type 2 diabetes mellitus cited above were typically in the range of 20 deaths per 1,000 patient-years but were as high as 30 deaths per 1,000 patient-years in the UGDP tolbutamide group16 and the UK-PDS sulfonylurea-plus-metformin group.20,22,26
Steno-2.31 Half of 160 patients with type 2 diabetes were randomized to intensive strategies for controlling glucose, lipids, and blood pressure and for taking aspirin and angiotensin-converting enzyme inhibitors and following a healthy lifestyle. The other half received conventional therapy. Even in the intensive-treatment group, the mortality rate at 13 years was higher than in ACCORD. Rates of death from any cause (per 1,000 patient years):
- 22.5 (intensive-treatment group)
- 37.6 (conventional-treatment group).
After the ACCORD results were presented, two other trials addressing the question of whether lower hemoglobin A1c would reduce cardiovascular risk in type 2 diabetes have reported their outcomes:
The ADVANCE trial (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation),32,33 with 11,140 patients, had a target hemoglobin A1c of 6.5% in an intensive-treatment group and 7.3% in a usual-treatment group. The intensive-treatment group showed no difference in the rates of major macrovascular events (HR 0.94, 95% CI 0.84–1.06, P = .32) or all-cause mortality (HR 0.93, 95% CI 0.83–1.06, P = .32). The overall death rate in ADVANCE (about 18 deaths per 1,000 patient-years) was higher than in ACCORD.
The Veterans Administration Diabetes Trial included 1,791 patients.34 Like the ADVANCE trial, it also found no difference in major cardiovascular outcomes (HR 0.868, P = .11) or cardiovascular mortality rates (HR 1.258, P = .36) with intensive therapy vs conventional therapy, ie, achieved hemoglobin A1c levels of 6.9% vs 8.4% (presented at the American Diabetes Association 2008 Scientific Sessions). Hypoglycemia was associated with an increased risk of death in the standard-treatment group.
An analysis suggested that patients with a shorter duration of diabetes may have had cardiovascular benefit from intensive glucose-lowering, while those who had had it longer may have had increased risk associated with the more intensive therapy. The rate of death from all causes appears to have been higher than in ACCORD, but this could not be determined accurately from the presentations.
Comment. Thus, the ACCORD cohort as a whole has had strikingly lower death rates than in these other studies. The fact that all participants had lower glucose levels on therapy than at baseline may possibly contribute to these lower death rates. In addition, all ACCORD participants in the lipid arm received a statin; all participants in the blood pressure arm had their blood pressure lowered to levels below those commonly seen in clinical practice; participants were encouraged to exercise regularly; most participants were given diet instruction; and other healthy behaviors such as aspirin use, regular follow-up with primary care physicians, and recommendations about smoking were encouraged throughout the study. These comprehensive strategies may represent better care and thus result in lower death rates than in other studies.
POSSIBLE EXPLANATIONS FOR THE ACCORD OUTCOMES
The ACCORD trial has already stimulated fierce debate about the reasons for the higher mortality rate in the intensive-treatment group. With longer follow-up, some new risk factors for death may be identified that are not evident in the analyses of the current 460 deaths. What follows are some of my thoughts, with the caveat that they are not confirmed (supported statistically) by any currently available analyses from ACCORD.
It seems unlikely that lower glucose values as reflected by lower hemoglobin A1c values in the intensive-treatment group are an a priori explanation for the observed differences in mortality rates—especially since the mortality rates were lower than in the NHANES and clinical trial data sets cited above. If we assume that a type 1 statistical error (finding a difference where no difference actually exists) does not explain the findings, then at least four reasonable postulates exist:
Hypoglycemia may have some adverse effect, either acutely or from recurrent events that trigger a catecholamine response with associated risk for arrhythmia or increased coronary heart disease risk. However, the investigators analyzed each death to determine whether hypoglycemia was a contributing cause, and they found no statistically significant relationship between hypoglycemia and death in the intensive-treatment group.
Weight gain is common with intensive therapy. Obesity may be associated with greater cytokine production, higher concentrations of clotting factors, higher levels of free fatty acids, and other potential contributors to the risk of coronary heart disease and death. Currently, the ACCORD analyses do not suggest that weight gain explains the higher death rate.
Medications such as rosiglitazone, sulfonylureas, and the combination of a sulfonylurea plus metformin have been previously associated with increased death rates in some observational and intervention trials. These studies had some serious methodologic limitations (eg, absence of risk adjustment, events not adjudicated, small study cohorts, wide variation in study cohort characteristics) and small numbers of events.11–13,16,26,35 ACCORD analyses have not shown that any single glucose-lowering agent—including rosiglitazone—or combination of agents explains the death rates.
The stress of maintaining glycemic control has been speculated to have in some way contributed to an increased risk. To achieve intensive control, patients had to have frequent contact with their health care providers, they were often told that their hemoglobin A1c values were “too high” even when they were well below those in the American Diabetes Association guidelines, and they had to follow complex glucose-lowering regimens.
Semiquantitative measures of overall attitudes about health exist (eg, the “Feeling Thermometer” scale), but stress was not measured quantitatively in the ACCORD trial.
IMPLICATIONS OF ACCORD
In practice, most clinicians believe that the target glucose level in patients with type 2 diabetes should be as low as safely possible. This approach does not need to be modified on the basis of current information from ACCORD.
To be safe, regimens should be associated with a low risk of hypoglycemia and a low risk of weight gain. Use of combinations of medications that work by different mechanisms is still prudent. Agents should be used that may have favorable effects on other cardiovascular risk factors (eg, lipids, blood pressure, visceral fat).
Hemoglobin A1c targets below 7% are not precluded in all patients on the basis of the ACCORD results, though values lower than 6% may not have much added benefit for cardiovascular risk reduction. We should note that hemoglobin A1c was reduced in all ACCORD participants and that death rates were lower than in many other type 2 diabetic cohorts. Pending data on other outcomes in ACCORD (nephropathy, retinopathy, dementia, fracture risk), I believe it is premature for organizations to change their proposed hemoglobin A1c targets,36,37 as none have proposed values as low as the target in the ACCORD intensive-treatment group. At present, no class of glucose-lowering agents needs to be excluded from consideration on the basis of the ACCORD data.
The overall low rates of death in this population at high risk of coronary heart disease deserve comment. Not only are they lower than in other glucose-lowering trials, but they are also lower than in a number of studies of mortality in diabetes cohorts. As noted above, multiple risk factors for coronary heart disease and death were (and are) addressed in the ACCORD study participants, including repeated recommendation for lifestyle modification, intervention arms with lipid and blood pressure therapy, encouragement of aspirin use, and regular follow-up with health care providers for risk factors not managed by the ACCORD trial protocol. It is likely that multiple approaches to reducing the risk of cardiovascular disease contributed to this low mortality rate and that similar approaches will reduce the risk of coronary disease and death in regular clinical practice.
The ACCORD lipid and blood pressure arms are continuing, with results expected in 2010. The future results from ACCORD as well as from several glucose-lowering trials currently in progress (ADVANCE,32,33 Veteran’s Administration,34 Bypass Angioplasty Revascularization Investigation 2 Diabetes [BARI-2D]38) will likely help refine our understanding of the effects of glucose-lowering, glucose-lowering strategies and targets, and multiple interventions on coronary events and all-cause mortality.
For now, any strategy that lowers glucose and is associated with a low risk of hypoglycemia and does not cause excessive weight gain should be considered appropriate in patients with type 2 diabetes.
- Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545–2559.
- Goff DC, Gerstein HC, Ginsberg HN, et al. Prevention of cardiovascular disease in persons with type 2 diabetes mellitus: current knowledge and rationale for the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:4i–20i.
- Buse JB, Bigger JT, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol 2007; 99:21i–33i.
- Gerstein HC, Riddle MC, Kendall DM, et al. Glycemia treatment strategies in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:34i–43i.
- Bonds DE, Kurashige EM, Bergenstal R, et al. Severe hypoglycemia monitoring and risk management procedures in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:80i–89i.
- Gerstein HC. Dysglycemia, not just diabetes, is a continuous risk factor for cardiovascular disease. Evid Based Cardiovasc Med. 1997; 1:87–88.
- Gerstein HC, Pais P, Pogue J, Yusuf S. Relationship of glucose and insulin levels to the risk of myocardial infarction: a case-control study. J Am Coll Cardiol. 1999; 33:612–619.
- Gerstein HC, Capes SE. Dysglycemia: a key cardiovascular risk factor. Semin Vasc Med. 2002; 2:165–174.
- Gerstein HC, Santaguida P, Raina P, et al. Annual incidence and relative risk of diabetes in people with various categories of dysglycemia: a systematic overview and meta-analysis of prospective studies. Diabetes Res Clin Pract. 2007; 78:305–312.
- American Diabetes Association. Role of cardiovascular risk factors in prevention and treatment of macrovascular disease in diabetes. Diabetes Care. 1989; 12:573–579.
- Schor S. The University Group Diabetes Program. A statistician looks at the mortality results. JAMA. 1971; 217:1671–1675.
- Cornfield JThe University Group Diabetes Program. A further statistical analysis of the mortality findings. JAMA. 1971; 217:1676–1687.
- Feinstein AR. Clinical biostatistics. 8. An analytic appraisal of the University Group Diabetes Program (UGDP) study. Clin Pharmacol Ther. 1971; 12:167–191.
- The University Group Diabetes Program. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. V. Evaluation of pheniformin therapy. Diabetes 1975; 24( suppl 1):65–184.
- Knatterud GL, Klimt CR, Levin ME, Jacobson ME, Goldner MG. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA. 1978; 240:37–42.
- Schwartz TB, Meinert CL. The UGDP controversy: thirty-four years of contentious ambiguity laid to rest. Perspect Biol Med. 2004; 47:564–574.
- Turner RC, Holman RR. Lessons from UK Prospective Diabetes Study. Diabetes Res Clin Pract 1995; 28( suppl):S151–S157.
- UKPDS Research Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:854–865.
- UKPDS Study Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:837–853.
- UK Prospective Diabetes Study Group. UKPDS 28: a randomized trial of efficacy of early addition of metformin in sulfonylurea-treated type 2 diabetes. Diabetes Care. 1998; 21:87–92.
- Bretzel RG, Voigt K, Schatz H. The United Kingdom Prospective Diabetes Study (UKPDS) implications for the pharmacotherapy of type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 1998; 106:369–372.
- Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA. 1999; 281:2005–2012.
- Leslie RD. United Kingdom prospective diabetes study (UKPDS): what now or so what? Diabetes Metab Res Rev 1999; 15:65–71.
- Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000; 321:405–412.
- Mooradian AD, Chehade J. Implications of the UK Prospective Diabetes Study: questions answered and issues remaining. Drugs Aging. 2000; 16:159–164.
- Nathan DM. Some answers, more controversy, from UKPDS. United Kingdom Prospective Diabetes Study. Lancet. 1998; 352:832–833.
- Srimanunthiphol J, Beddow R, Arakaki R. A review of the United Kingdom Prospective Diabetes Study (UKPDS) and a discussion of the implications for patient care. Hawaii Med J. 2000; 59:295–298.
- Duckworth WC, McCarren M, Abraira C. Glucose control and cardiovascular complications: the VA Diabetes Trial. Diabetes Care. 2001; 24:942–945.
- Abraira C, Colwell JA, Nuttall FQ, et al. Veterans Affairs Cooperative Study on glycemic control and complications in type II diabetes (VA CSDM). Results of the feasibility trial. Veterans Affairs Cooperative Study in Type II Diabetes. Diabetes Care. 1995; 18:1113–1123.
- Gu K, Cowie CC, Harris MI. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971–1993. Diabetes Care. 1998; 21:1138–1145. NHANES
- Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008; 358:580–591.
- Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008; 358:2560–2572.
- Action in Diabetes and Vascular Disease: PreterAx and DiamicroN Modified-Release Controlled Evaluation. Rationale and design of the ADVANCE study: a randomised trial of blood pressure lowering and intensive glucose control in high-risk individuals with type 2 diabetes mellitus. J Hypertens 2001; 19(suppl):S21–S28.
- Abraira C, Duckworth W, McCarren M, et al. Design of the cooperative study on glycemic control and complications in diabetes mellitus type 2: Veterans Affairs Diabetes Trial. J Diabetes Complications. 2003; 17:314–322.
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007; 356:2457–2471.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract 2007; 13(suppl 1):1–68.
- American Diabetes Association. Standards of medical care in diabetes—2008. Diabetes Care 2008; 31(suppl 1):S12–S54.
- Magee MF, Isley WL. Rationale, design, and methods for glycemic control in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Trial. Am J Cardiol 2006; 97:20G–30G.
The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial1–5 was designed primarily to address, in patients with type 2 diabetes at high risk of cardiovascular events, whether intensive glucose control would result in a lower risk of atherosclerotic disease events or death than would standard treatment.
It was widely expected that intensive treatment would confer either modest benefit or, at worst, no benefit. However, the glucose-lowering arm of the trial was terminated early because of a higher mortality rate in the intensively treated group. (The ACCORD trial has two other arms, which concern blood pressure and lipid-lowering, and these are continuing.)
In earlier trials in type 2 diabetes, concerns had been raised about an increased risk of cardiovascular events and possibly death associated with glucose-lowering drugs, hypoglycemia itself, or both, and these were well known when ACCORD was convened. ACCORD was very carefully designed and included careful adjudication of each cardiovascular event and death, including whether hypoglycemia might have been a proximate cause of some sudden deaths.5
Therefore, the surprising result of the higher mortality rate with intensive glycemic control in ACCORD will be fodder for discussion in many arenas over the next several years, and it poses some challenges for physicians and patients in determining treatment goals, as well as for organizations that write clinical practice guidelines (and perhaps organizations involved in pay-for-performance based on these guidelines).
Still, I believe that the ACCORD results should not substantially change our approach to treatment goals in type 2 diabetes, although hemoglobin A1c targets below 6% may not have much added value for cardiovascular risk reduction. The low overall mortality rate in all the arms of the ACCORD trial emphasizes the importance of lifestyle modification, lipid and blood pressure therapy, and encouragement of aspirin use in all patients with type 2 diabetes.
This article reflects my views as a practicing diabetologist and clinical trialist (I was an investigator in the ACCORD trial) with a long-standing interest in clinical trials and in how the results influence clinical practice. The views I express herein may not reflect the views of other ACCORD investigators, the National Heart, Lung, and Blood Institute (NHLBI), the ACCORD trial coordinating center at Wake Forest University, or its data safety and monitoring board.
RISK OF CORONARY DISEASE INCREASES WITH GLUCOSE
Many observational studies6–10 have shown that the risk of cardiovascular disease, especially coronary heart disease, is two to five times higher in people with diabetes mellitus than in people without diabetes. The risk appears to be continuous, so the higher one’s glucose or hemoglobin A1c, the higher the risk.6 This risk even extends to glucose values well below the threshold values currently used to diagnose diabetes mellitus.6 Since there is no glucose threshold for coronary heart disease, the term dysglycemia (rather than hyperglycemia) has been proposed to note the relationship between glucose and coronary heart disease. (The glucose threshold for microvascular complications of diabetes, such as retinopathy and nephropathy, appears to be between 110 and 126 mg/dL).
The clustering of multiple coronary risk factors such as obesity, dyslipidemia, and hypertension has always raised the question of whether glucose is a culprit in coronary risk or whether it simply “runs in bad company.”
EARLIER CLINICAL TRIALS SUGGEST INTENSIVE TREATMENT RAISES RISK
Even though it has been widely believed that intensive glucose-lowering would reduce cardiovascular risk in type 2 diabetes, there have been hints in previous studies that some intensive-treatment regimens might increase risk.
Two large randomized clinical trials and one small one (discussed below) addressed whether glucose control would reduce the risk of atherosclerotic vascular disease events. In each of them, an increased risk of cardiovascular events and possibly of death was seen in at least one intensively treated group.
In the following discussion, I have calculated all of the death rates as the number of deaths per 1,000 patients per year, based on published study results. In this way, we can compare the rates in the various studies (including ACCORD), regardless of the trial duration.
The university group Diabetes Program: Controvery about tolbutamide therapy
The University Group Diabetes Program (UGDP)11–16 included about 1,000 participants randomized to five treatments: tolbutamide (Orinase, a sulfonylurea), insulin in a fixed dose based on body weight, insulin in adjusted doses based on fasting glucose levels, placebo, and (later) phenformin.
In the 1970s, when the UGDP was carried out, randomized clinical trials were uncommon. Like other trials from that era, the UGDP was underpowered by today’s standards and did not have a data safety and monitoring board.
Rates of cardiovascular events and deaths (per 1,000 patient-years):
- 25 (tolbutamide group)
- 12 (placebo group).
The two insulin groups did not differ from the placebo group in their rates of cardiovascular events or death.15 The tolbutamide arm was stopped, and the ensuing controversy about how to interpret the trial results lasted for more than a decade. It also resulted in a black-box warning for tolbutamide and all subsequent sulfonylureas.
United Kingdom Prospective Diabetes Study: Method of glucose-lowering an issue
The United Kingdom Prospective Diabetes Study (UKPDS)17–27 was launched in 1977. A cohort of 5,102 patients (mean age 54 years) with newly diagnosed type 2 diabetes mellitus followed a “prudent diet” for the first 3 to 4 months. Then, if their fasting glucose levels were in the range of 6.1 to 15 mmol/L (110–270 mg/dL), they were randomized to receive various treatments.
Patients who were not obese were randomized to receive either intensive treatment or conventional treatment. The intensive-treatment group received either insulin or a sulfonylurea (chlorpropamide [Diabinese], glibenclamide, or glipizide [Glucotrol]); the conventional-treatment group received diet therapy. The sulfonylurea arm was included partly to address the UGDP results.
Patients who were obese were randomized to receive one of three treatments: intensive treatment (with the agents listed above), conventional treatment, or metformin (Fortamet, Glucophage).
The mean in-trial hemoglobin A1c level in the intensive-treatment group was 7.0%, compared with 7.9% in the conventional-treatment group.
After a mean follow-up of more than 10 years, the incidence of myocardial infarction was 16% lower in the intensive-treatment group, but the difference was not statistically significant (P = .052).
Rates of death from all causes among nonobese subjects (per 1,000 patient-years):
- 18.2–20.5 (intensive-treatment group)
- 19.9 (conventional-treatment group).
In the obese patients who received metformin, the incidence of myocardial infarction was lower than in the conventional-treatment group but not the intensive-treatment group.
Rates of death among obese patients (per 1,000 patient-years):
- 13.5 (metformin group)
- 18.9 (intensive-treatment group)
- 20.6 (conventional-treatment group).
However, a small subset (n = 587) of the original group assigned to sulfonylurea therapy whose glycemic control deteriorated during the trial were rerandomized to continue to receive a sulfonylurea alone or to have metformin added. There was a statistically significantly higher rate of cardiovascular events and a nonsignificantly higher rate of total mortality in the metformin-plus-sulfonylurea group (30.3 per 1,000 patient-years) than in the sulfonylurea-only group (19.1 per 1,000 patient-years).
These data suggested that the way glucose-lowering was achieved might be as important as the glucose levels actually achieved. However, no definite conclusions could be drawn.
In an editorial on the UKPDS, Nathan26 made a comment that may have been prescient in terms of the ACCORD trial: “Professional organizations will now scramble to decide how to translate the UKPDS results … Whether the UKPDS firmly establishes the choice of any one therapy…or any combination of therapies for the long-term treatment of type 2 diabetes is more questionable.”26
Veterans Administration feasibility study
A Veterans Administration feasibility study28,29 included 153 men (mean age 60) with type 2 diabetes (mean duration 7.8 years) who received either conventional therapy (a single daily dose of insulin) or intensive therapy (multiple doses of insulin plus a sulfonylurea). Over a mean of 27 months, the intensive-therapy group achieved a hemoglobin A1c level that was 2 percentage points lower than in the conventional-therapy group.
At 2.25 years of follow-up, cardiovascular events had occurred in 24 (24%) of the intensive-therapy group and in 16 (20%) of the standard-therapy group (P = .10).
Rates of death from all causes (per 1,000 patient-years):
- 28.9 (intensive-treatment group)
- 17.5 (conventional-treatment group).
ACCORD TRIAL DESIGN
The primary outcome measured was the combined incidence of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes. Secondary outcomes included death from any cause. The study is also evaluating the effect of intensive treatment on microvascular disease, hypoglycemia, cognition, quality of life, and cost-effectiveness.
The ACCORD study was designed to have 89% power to detect a 15% treatment effect of intensive glycemic control compared with standard glycemic control for the primary end point.
ACCORD RESULTS
Rates of death from any cause (per 1,000 patient-years):
- 14 (intensive-treatment group)
- 11 (standard-treatment group).
In the analyses available at the time that this study arm closed, the excess mortality was not attributable to any particular treatment regimen. In particular, rosiglitazone (Avandia) use did not contribute to the excess mortality. (Of note, 91.2% of the intensive-treatment group and 57.5% of the conventional-treatment group had been treated with rosiglitazone, with more than 19,000 patient-years of rosiglitazone exposure). The excess mortality was also not attributable to hypoglycemia immediately proximate to the death.
The ACCORD trial’s data safety and monitoring board recommended that this arm of the study be discontinued for safety reasons, and this recommendation was accepted by the NHLBI project office. All participants were notified by letter before the trial results were announced publicly, and all intensive-therapy group participants are now being treated by the protocol used in the standard-therapy group.1
FEWER DEATHS IN ACCORD THAN IN OTHER STUDIES IN DIABETES
The mortality rates in both arms of ACCORD were much lower than in other observational studies and clinical trials in type 2 diabetes.
The National Health and Nutrition Education Survey (NHANES),30 conducted from 1971 to 1975, included 14,374 people with diabetes between the ages of 25 and 74. Many of them were younger than the ACCORD patients, but two NHANES age-groups overlapped the ACCORD cohort. Rates of death from any cause at 22 years (per 1,000 patient-years):
- 39.7 (ages 45–64)
- 89.7 (ages 65–74).
The NHANES cohort would not have been treated as vigorously for coronary risk and other common causes of death.
UGDP, UKPDS. Death rates in the glucose-lowering trials of type 2 diabetes mellitus cited above were typically in the range of 20 deaths per 1,000 patient-years but were as high as 30 deaths per 1,000 patient-years in the UGDP tolbutamide group16 and the UK-PDS sulfonylurea-plus-metformin group.20,22,26
Steno-2.31 Half of 160 patients with type 2 diabetes were randomized to intensive strategies for controlling glucose, lipids, and blood pressure and for taking aspirin and angiotensin-converting enzyme inhibitors and following a healthy lifestyle. The other half received conventional therapy. Even in the intensive-treatment group, the mortality rate at 13 years was higher than in ACCORD. Rates of death from any cause (per 1,000 patient years):
- 22.5 (intensive-treatment group)
- 37.6 (conventional-treatment group).
After the ACCORD results were presented, two other trials addressing the question of whether lower hemoglobin A1c would reduce cardiovascular risk in type 2 diabetes have reported their outcomes:
The ADVANCE trial (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation),32,33 with 11,140 patients, had a target hemoglobin A1c of 6.5% in an intensive-treatment group and 7.3% in a usual-treatment group. The intensive-treatment group showed no difference in the rates of major macrovascular events (HR 0.94, 95% CI 0.84–1.06, P = .32) or all-cause mortality (HR 0.93, 95% CI 0.83–1.06, P = .32). The overall death rate in ADVANCE (about 18 deaths per 1,000 patient-years) was higher than in ACCORD.
The Veterans Administration Diabetes Trial included 1,791 patients.34 Like the ADVANCE trial, it also found no difference in major cardiovascular outcomes (HR 0.868, P = .11) or cardiovascular mortality rates (HR 1.258, P = .36) with intensive therapy vs conventional therapy, ie, achieved hemoglobin A1c levels of 6.9% vs 8.4% (presented at the American Diabetes Association 2008 Scientific Sessions). Hypoglycemia was associated with an increased risk of death in the standard-treatment group.
An analysis suggested that patients with a shorter duration of diabetes may have had cardiovascular benefit from intensive glucose-lowering, while those who had had it longer may have had increased risk associated with the more intensive therapy. The rate of death from all causes appears to have been higher than in ACCORD, but this could not be determined accurately from the presentations.
Comment. Thus, the ACCORD cohort as a whole has had strikingly lower death rates than in these other studies. The fact that all participants had lower glucose levels on therapy than at baseline may possibly contribute to these lower death rates. In addition, all ACCORD participants in the lipid arm received a statin; all participants in the blood pressure arm had their blood pressure lowered to levels below those commonly seen in clinical practice; participants were encouraged to exercise regularly; most participants were given diet instruction; and other healthy behaviors such as aspirin use, regular follow-up with primary care physicians, and recommendations about smoking were encouraged throughout the study. These comprehensive strategies may represent better care and thus result in lower death rates than in other studies.
POSSIBLE EXPLANATIONS FOR THE ACCORD OUTCOMES
The ACCORD trial has already stimulated fierce debate about the reasons for the higher mortality rate in the intensive-treatment group. With longer follow-up, some new risk factors for death may be identified that are not evident in the analyses of the current 460 deaths. What follows are some of my thoughts, with the caveat that they are not confirmed (supported statistically) by any currently available analyses from ACCORD.
It seems unlikely that lower glucose values as reflected by lower hemoglobin A1c values in the intensive-treatment group are an a priori explanation for the observed differences in mortality rates—especially since the mortality rates were lower than in the NHANES and clinical trial data sets cited above. If we assume that a type 1 statistical error (finding a difference where no difference actually exists) does not explain the findings, then at least four reasonable postulates exist:
Hypoglycemia may have some adverse effect, either acutely or from recurrent events that trigger a catecholamine response with associated risk for arrhythmia or increased coronary heart disease risk. However, the investigators analyzed each death to determine whether hypoglycemia was a contributing cause, and they found no statistically significant relationship between hypoglycemia and death in the intensive-treatment group.
Weight gain is common with intensive therapy. Obesity may be associated with greater cytokine production, higher concentrations of clotting factors, higher levels of free fatty acids, and other potential contributors to the risk of coronary heart disease and death. Currently, the ACCORD analyses do not suggest that weight gain explains the higher death rate.
Medications such as rosiglitazone, sulfonylureas, and the combination of a sulfonylurea plus metformin have been previously associated with increased death rates in some observational and intervention trials. These studies had some serious methodologic limitations (eg, absence of risk adjustment, events not adjudicated, small study cohorts, wide variation in study cohort characteristics) and small numbers of events.11–13,16,26,35 ACCORD analyses have not shown that any single glucose-lowering agent—including rosiglitazone—or combination of agents explains the death rates.
The stress of maintaining glycemic control has been speculated to have in some way contributed to an increased risk. To achieve intensive control, patients had to have frequent contact with their health care providers, they were often told that their hemoglobin A1c values were “too high” even when they were well below those in the American Diabetes Association guidelines, and they had to follow complex glucose-lowering regimens.
Semiquantitative measures of overall attitudes about health exist (eg, the “Feeling Thermometer” scale), but stress was not measured quantitatively in the ACCORD trial.
IMPLICATIONS OF ACCORD
In practice, most clinicians believe that the target glucose level in patients with type 2 diabetes should be as low as safely possible. This approach does not need to be modified on the basis of current information from ACCORD.
To be safe, regimens should be associated with a low risk of hypoglycemia and a low risk of weight gain. Use of combinations of medications that work by different mechanisms is still prudent. Agents should be used that may have favorable effects on other cardiovascular risk factors (eg, lipids, blood pressure, visceral fat).
Hemoglobin A1c targets below 7% are not precluded in all patients on the basis of the ACCORD results, though values lower than 6% may not have much added benefit for cardiovascular risk reduction. We should note that hemoglobin A1c was reduced in all ACCORD participants and that death rates were lower than in many other type 2 diabetic cohorts. Pending data on other outcomes in ACCORD (nephropathy, retinopathy, dementia, fracture risk), I believe it is premature for organizations to change their proposed hemoglobin A1c targets,36,37 as none have proposed values as low as the target in the ACCORD intensive-treatment group. At present, no class of glucose-lowering agents needs to be excluded from consideration on the basis of the ACCORD data.
The overall low rates of death in this population at high risk of coronary heart disease deserve comment. Not only are they lower than in other glucose-lowering trials, but they are also lower than in a number of studies of mortality in diabetes cohorts. As noted above, multiple risk factors for coronary heart disease and death were (and are) addressed in the ACCORD study participants, including repeated recommendation for lifestyle modification, intervention arms with lipid and blood pressure therapy, encouragement of aspirin use, and regular follow-up with health care providers for risk factors not managed by the ACCORD trial protocol. It is likely that multiple approaches to reducing the risk of cardiovascular disease contributed to this low mortality rate and that similar approaches will reduce the risk of coronary disease and death in regular clinical practice.
The ACCORD lipid and blood pressure arms are continuing, with results expected in 2010. The future results from ACCORD as well as from several glucose-lowering trials currently in progress (ADVANCE,32,33 Veteran’s Administration,34 Bypass Angioplasty Revascularization Investigation 2 Diabetes [BARI-2D]38) will likely help refine our understanding of the effects of glucose-lowering, glucose-lowering strategies and targets, and multiple interventions on coronary events and all-cause mortality.
For now, any strategy that lowers glucose and is associated with a low risk of hypoglycemia and does not cause excessive weight gain should be considered appropriate in patients with type 2 diabetes.
The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial1–5 was designed primarily to address, in patients with type 2 diabetes at high risk of cardiovascular events, whether intensive glucose control would result in a lower risk of atherosclerotic disease events or death than would standard treatment.
It was widely expected that intensive treatment would confer either modest benefit or, at worst, no benefit. However, the glucose-lowering arm of the trial was terminated early because of a higher mortality rate in the intensively treated group. (The ACCORD trial has two other arms, which concern blood pressure and lipid-lowering, and these are continuing.)
In earlier trials in type 2 diabetes, concerns had been raised about an increased risk of cardiovascular events and possibly death associated with glucose-lowering drugs, hypoglycemia itself, or both, and these were well known when ACCORD was convened. ACCORD was very carefully designed and included careful adjudication of each cardiovascular event and death, including whether hypoglycemia might have been a proximate cause of some sudden deaths.5
Therefore, the surprising result of the higher mortality rate with intensive glycemic control in ACCORD will be fodder for discussion in many arenas over the next several years, and it poses some challenges for physicians and patients in determining treatment goals, as well as for organizations that write clinical practice guidelines (and perhaps organizations involved in pay-for-performance based on these guidelines).
Still, I believe that the ACCORD results should not substantially change our approach to treatment goals in type 2 diabetes, although hemoglobin A1c targets below 6% may not have much added value for cardiovascular risk reduction. The low overall mortality rate in all the arms of the ACCORD trial emphasizes the importance of lifestyle modification, lipid and blood pressure therapy, and encouragement of aspirin use in all patients with type 2 diabetes.
This article reflects my views as a practicing diabetologist and clinical trialist (I was an investigator in the ACCORD trial) with a long-standing interest in clinical trials and in how the results influence clinical practice. The views I express herein may not reflect the views of other ACCORD investigators, the National Heart, Lung, and Blood Institute (NHLBI), the ACCORD trial coordinating center at Wake Forest University, or its data safety and monitoring board.
RISK OF CORONARY DISEASE INCREASES WITH GLUCOSE
Many observational studies6–10 have shown that the risk of cardiovascular disease, especially coronary heart disease, is two to five times higher in people with diabetes mellitus than in people without diabetes. The risk appears to be continuous, so the higher one’s glucose or hemoglobin A1c, the higher the risk.6 This risk even extends to glucose values well below the threshold values currently used to diagnose diabetes mellitus.6 Since there is no glucose threshold for coronary heart disease, the term dysglycemia (rather than hyperglycemia) has been proposed to note the relationship between glucose and coronary heart disease. (The glucose threshold for microvascular complications of diabetes, such as retinopathy and nephropathy, appears to be between 110 and 126 mg/dL).
The clustering of multiple coronary risk factors such as obesity, dyslipidemia, and hypertension has always raised the question of whether glucose is a culprit in coronary risk or whether it simply “runs in bad company.”
EARLIER CLINICAL TRIALS SUGGEST INTENSIVE TREATMENT RAISES RISK
Even though it has been widely believed that intensive glucose-lowering would reduce cardiovascular risk in type 2 diabetes, there have been hints in previous studies that some intensive-treatment regimens might increase risk.
Two large randomized clinical trials and one small one (discussed below) addressed whether glucose control would reduce the risk of atherosclerotic vascular disease events. In each of them, an increased risk of cardiovascular events and possibly of death was seen in at least one intensively treated group.
In the following discussion, I have calculated all of the death rates as the number of deaths per 1,000 patients per year, based on published study results. In this way, we can compare the rates in the various studies (including ACCORD), regardless of the trial duration.
The university group Diabetes Program: Controvery about tolbutamide therapy
The University Group Diabetes Program (UGDP)11–16 included about 1,000 participants randomized to five treatments: tolbutamide (Orinase, a sulfonylurea), insulin in a fixed dose based on body weight, insulin in adjusted doses based on fasting glucose levels, placebo, and (later) phenformin.
In the 1970s, when the UGDP was carried out, randomized clinical trials were uncommon. Like other trials from that era, the UGDP was underpowered by today’s standards and did not have a data safety and monitoring board.
Rates of cardiovascular events and deaths (per 1,000 patient-years):
- 25 (tolbutamide group)
- 12 (placebo group).
The two insulin groups did not differ from the placebo group in their rates of cardiovascular events or death.15 The tolbutamide arm was stopped, and the ensuing controversy about how to interpret the trial results lasted for more than a decade. It also resulted in a black-box warning for tolbutamide and all subsequent sulfonylureas.
United Kingdom Prospective Diabetes Study: Method of glucose-lowering an issue
The United Kingdom Prospective Diabetes Study (UKPDS)17–27 was launched in 1977. A cohort of 5,102 patients (mean age 54 years) with newly diagnosed type 2 diabetes mellitus followed a “prudent diet” for the first 3 to 4 months. Then, if their fasting glucose levels were in the range of 6.1 to 15 mmol/L (110–270 mg/dL), they were randomized to receive various treatments.
Patients who were not obese were randomized to receive either intensive treatment or conventional treatment. The intensive-treatment group received either insulin or a sulfonylurea (chlorpropamide [Diabinese], glibenclamide, or glipizide [Glucotrol]); the conventional-treatment group received diet therapy. The sulfonylurea arm was included partly to address the UGDP results.
Patients who were obese were randomized to receive one of three treatments: intensive treatment (with the agents listed above), conventional treatment, or metformin (Fortamet, Glucophage).
The mean in-trial hemoglobin A1c level in the intensive-treatment group was 7.0%, compared with 7.9% in the conventional-treatment group.
After a mean follow-up of more than 10 years, the incidence of myocardial infarction was 16% lower in the intensive-treatment group, but the difference was not statistically significant (P = .052).
Rates of death from all causes among nonobese subjects (per 1,000 patient-years):
- 18.2–20.5 (intensive-treatment group)
- 19.9 (conventional-treatment group).
In the obese patients who received metformin, the incidence of myocardial infarction was lower than in the conventional-treatment group but not the intensive-treatment group.
Rates of death among obese patients (per 1,000 patient-years):
- 13.5 (metformin group)
- 18.9 (intensive-treatment group)
- 20.6 (conventional-treatment group).
However, a small subset (n = 587) of the original group assigned to sulfonylurea therapy whose glycemic control deteriorated during the trial were rerandomized to continue to receive a sulfonylurea alone or to have metformin added. There was a statistically significantly higher rate of cardiovascular events and a nonsignificantly higher rate of total mortality in the metformin-plus-sulfonylurea group (30.3 per 1,000 patient-years) than in the sulfonylurea-only group (19.1 per 1,000 patient-years).
These data suggested that the way glucose-lowering was achieved might be as important as the glucose levels actually achieved. However, no definite conclusions could be drawn.
In an editorial on the UKPDS, Nathan26 made a comment that may have been prescient in terms of the ACCORD trial: “Professional organizations will now scramble to decide how to translate the UKPDS results … Whether the UKPDS firmly establishes the choice of any one therapy…or any combination of therapies for the long-term treatment of type 2 diabetes is more questionable.”26
Veterans Administration feasibility study
A Veterans Administration feasibility study28,29 included 153 men (mean age 60) with type 2 diabetes (mean duration 7.8 years) who received either conventional therapy (a single daily dose of insulin) or intensive therapy (multiple doses of insulin plus a sulfonylurea). Over a mean of 27 months, the intensive-therapy group achieved a hemoglobin A1c level that was 2 percentage points lower than in the conventional-therapy group.
At 2.25 years of follow-up, cardiovascular events had occurred in 24 (24%) of the intensive-therapy group and in 16 (20%) of the standard-therapy group (P = .10).
Rates of death from all causes (per 1,000 patient-years):
- 28.9 (intensive-treatment group)
- 17.5 (conventional-treatment group).
ACCORD TRIAL DESIGN
The primary outcome measured was the combined incidence of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes. Secondary outcomes included death from any cause. The study is also evaluating the effect of intensive treatment on microvascular disease, hypoglycemia, cognition, quality of life, and cost-effectiveness.
The ACCORD study was designed to have 89% power to detect a 15% treatment effect of intensive glycemic control compared with standard glycemic control for the primary end point.
ACCORD RESULTS
Rates of death from any cause (per 1,000 patient-years):
- 14 (intensive-treatment group)
- 11 (standard-treatment group).
In the analyses available at the time that this study arm closed, the excess mortality was not attributable to any particular treatment regimen. In particular, rosiglitazone (Avandia) use did not contribute to the excess mortality. (Of note, 91.2% of the intensive-treatment group and 57.5% of the conventional-treatment group had been treated with rosiglitazone, with more than 19,000 patient-years of rosiglitazone exposure). The excess mortality was also not attributable to hypoglycemia immediately proximate to the death.
The ACCORD trial’s data safety and monitoring board recommended that this arm of the study be discontinued for safety reasons, and this recommendation was accepted by the NHLBI project office. All participants were notified by letter before the trial results were announced publicly, and all intensive-therapy group participants are now being treated by the protocol used in the standard-therapy group.1
FEWER DEATHS IN ACCORD THAN IN OTHER STUDIES IN DIABETES
The mortality rates in both arms of ACCORD were much lower than in other observational studies and clinical trials in type 2 diabetes.
The National Health and Nutrition Education Survey (NHANES),30 conducted from 1971 to 1975, included 14,374 people with diabetes between the ages of 25 and 74. Many of them were younger than the ACCORD patients, but two NHANES age-groups overlapped the ACCORD cohort. Rates of death from any cause at 22 years (per 1,000 patient-years):
- 39.7 (ages 45–64)
- 89.7 (ages 65–74).
The NHANES cohort would not have been treated as vigorously for coronary risk and other common causes of death.
UGDP, UKPDS. Death rates in the glucose-lowering trials of type 2 diabetes mellitus cited above were typically in the range of 20 deaths per 1,000 patient-years but were as high as 30 deaths per 1,000 patient-years in the UGDP tolbutamide group16 and the UK-PDS sulfonylurea-plus-metformin group.20,22,26
Steno-2.31 Half of 160 patients with type 2 diabetes were randomized to intensive strategies for controlling glucose, lipids, and blood pressure and for taking aspirin and angiotensin-converting enzyme inhibitors and following a healthy lifestyle. The other half received conventional therapy. Even in the intensive-treatment group, the mortality rate at 13 years was higher than in ACCORD. Rates of death from any cause (per 1,000 patient years):
- 22.5 (intensive-treatment group)
- 37.6 (conventional-treatment group).
After the ACCORD results were presented, two other trials addressing the question of whether lower hemoglobin A1c would reduce cardiovascular risk in type 2 diabetes have reported their outcomes:
The ADVANCE trial (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation),32,33 with 11,140 patients, had a target hemoglobin A1c of 6.5% in an intensive-treatment group and 7.3% in a usual-treatment group. The intensive-treatment group showed no difference in the rates of major macrovascular events (HR 0.94, 95% CI 0.84–1.06, P = .32) or all-cause mortality (HR 0.93, 95% CI 0.83–1.06, P = .32). The overall death rate in ADVANCE (about 18 deaths per 1,000 patient-years) was higher than in ACCORD.
The Veterans Administration Diabetes Trial included 1,791 patients.34 Like the ADVANCE trial, it also found no difference in major cardiovascular outcomes (HR 0.868, P = .11) or cardiovascular mortality rates (HR 1.258, P = .36) with intensive therapy vs conventional therapy, ie, achieved hemoglobin A1c levels of 6.9% vs 8.4% (presented at the American Diabetes Association 2008 Scientific Sessions). Hypoglycemia was associated with an increased risk of death in the standard-treatment group.
An analysis suggested that patients with a shorter duration of diabetes may have had cardiovascular benefit from intensive glucose-lowering, while those who had had it longer may have had increased risk associated with the more intensive therapy. The rate of death from all causes appears to have been higher than in ACCORD, but this could not be determined accurately from the presentations.
Comment. Thus, the ACCORD cohort as a whole has had strikingly lower death rates than in these other studies. The fact that all participants had lower glucose levels on therapy than at baseline may possibly contribute to these lower death rates. In addition, all ACCORD participants in the lipid arm received a statin; all participants in the blood pressure arm had their blood pressure lowered to levels below those commonly seen in clinical practice; participants were encouraged to exercise regularly; most participants were given diet instruction; and other healthy behaviors such as aspirin use, regular follow-up with primary care physicians, and recommendations about smoking were encouraged throughout the study. These comprehensive strategies may represent better care and thus result in lower death rates than in other studies.
POSSIBLE EXPLANATIONS FOR THE ACCORD OUTCOMES
The ACCORD trial has already stimulated fierce debate about the reasons for the higher mortality rate in the intensive-treatment group. With longer follow-up, some new risk factors for death may be identified that are not evident in the analyses of the current 460 deaths. What follows are some of my thoughts, with the caveat that they are not confirmed (supported statistically) by any currently available analyses from ACCORD.
It seems unlikely that lower glucose values as reflected by lower hemoglobin A1c values in the intensive-treatment group are an a priori explanation for the observed differences in mortality rates—especially since the mortality rates were lower than in the NHANES and clinical trial data sets cited above. If we assume that a type 1 statistical error (finding a difference where no difference actually exists) does not explain the findings, then at least four reasonable postulates exist:
Hypoglycemia may have some adverse effect, either acutely or from recurrent events that trigger a catecholamine response with associated risk for arrhythmia or increased coronary heart disease risk. However, the investigators analyzed each death to determine whether hypoglycemia was a contributing cause, and they found no statistically significant relationship between hypoglycemia and death in the intensive-treatment group.
Weight gain is common with intensive therapy. Obesity may be associated with greater cytokine production, higher concentrations of clotting factors, higher levels of free fatty acids, and other potential contributors to the risk of coronary heart disease and death. Currently, the ACCORD analyses do not suggest that weight gain explains the higher death rate.
Medications such as rosiglitazone, sulfonylureas, and the combination of a sulfonylurea plus metformin have been previously associated with increased death rates in some observational and intervention trials. These studies had some serious methodologic limitations (eg, absence of risk adjustment, events not adjudicated, small study cohorts, wide variation in study cohort characteristics) and small numbers of events.11–13,16,26,35 ACCORD analyses have not shown that any single glucose-lowering agent—including rosiglitazone—or combination of agents explains the death rates.
The stress of maintaining glycemic control has been speculated to have in some way contributed to an increased risk. To achieve intensive control, patients had to have frequent contact with their health care providers, they were often told that their hemoglobin A1c values were “too high” even when they were well below those in the American Diabetes Association guidelines, and they had to follow complex glucose-lowering regimens.
Semiquantitative measures of overall attitudes about health exist (eg, the “Feeling Thermometer” scale), but stress was not measured quantitatively in the ACCORD trial.
IMPLICATIONS OF ACCORD
In practice, most clinicians believe that the target glucose level in patients with type 2 diabetes should be as low as safely possible. This approach does not need to be modified on the basis of current information from ACCORD.
To be safe, regimens should be associated with a low risk of hypoglycemia and a low risk of weight gain. Use of combinations of medications that work by different mechanisms is still prudent. Agents should be used that may have favorable effects on other cardiovascular risk factors (eg, lipids, blood pressure, visceral fat).
Hemoglobin A1c targets below 7% are not precluded in all patients on the basis of the ACCORD results, though values lower than 6% may not have much added benefit for cardiovascular risk reduction. We should note that hemoglobin A1c was reduced in all ACCORD participants and that death rates were lower than in many other type 2 diabetic cohorts. Pending data on other outcomes in ACCORD (nephropathy, retinopathy, dementia, fracture risk), I believe it is premature for organizations to change their proposed hemoglobin A1c targets,36,37 as none have proposed values as low as the target in the ACCORD intensive-treatment group. At present, no class of glucose-lowering agents needs to be excluded from consideration on the basis of the ACCORD data.
The overall low rates of death in this population at high risk of coronary heart disease deserve comment. Not only are they lower than in other glucose-lowering trials, but they are also lower than in a number of studies of mortality in diabetes cohorts. As noted above, multiple risk factors for coronary heart disease and death were (and are) addressed in the ACCORD study participants, including repeated recommendation for lifestyle modification, intervention arms with lipid and blood pressure therapy, encouragement of aspirin use, and regular follow-up with health care providers for risk factors not managed by the ACCORD trial protocol. It is likely that multiple approaches to reducing the risk of cardiovascular disease contributed to this low mortality rate and that similar approaches will reduce the risk of coronary disease and death in regular clinical practice.
The ACCORD lipid and blood pressure arms are continuing, with results expected in 2010. The future results from ACCORD as well as from several glucose-lowering trials currently in progress (ADVANCE,32,33 Veteran’s Administration,34 Bypass Angioplasty Revascularization Investigation 2 Diabetes [BARI-2D]38) will likely help refine our understanding of the effects of glucose-lowering, glucose-lowering strategies and targets, and multiple interventions on coronary events and all-cause mortality.
For now, any strategy that lowers glucose and is associated with a low risk of hypoglycemia and does not cause excessive weight gain should be considered appropriate in patients with type 2 diabetes.
- Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545–2559.
- Goff DC, Gerstein HC, Ginsberg HN, et al. Prevention of cardiovascular disease in persons with type 2 diabetes mellitus: current knowledge and rationale for the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:4i–20i.
- Buse JB, Bigger JT, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol 2007; 99:21i–33i.
- Gerstein HC, Riddle MC, Kendall DM, et al. Glycemia treatment strategies in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:34i–43i.
- Bonds DE, Kurashige EM, Bergenstal R, et al. Severe hypoglycemia monitoring and risk management procedures in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:80i–89i.
- Gerstein HC. Dysglycemia, not just diabetes, is a continuous risk factor for cardiovascular disease. Evid Based Cardiovasc Med. 1997; 1:87–88.
- Gerstein HC, Pais P, Pogue J, Yusuf S. Relationship of glucose and insulin levels to the risk of myocardial infarction: a case-control study. J Am Coll Cardiol. 1999; 33:612–619.
- Gerstein HC, Capes SE. Dysglycemia: a key cardiovascular risk factor. Semin Vasc Med. 2002; 2:165–174.
- Gerstein HC, Santaguida P, Raina P, et al. Annual incidence and relative risk of diabetes in people with various categories of dysglycemia: a systematic overview and meta-analysis of prospective studies. Diabetes Res Clin Pract. 2007; 78:305–312.
- American Diabetes Association. Role of cardiovascular risk factors in prevention and treatment of macrovascular disease in diabetes. Diabetes Care. 1989; 12:573–579.
- Schor S. The University Group Diabetes Program. A statistician looks at the mortality results. JAMA. 1971; 217:1671–1675.
- Cornfield JThe University Group Diabetes Program. A further statistical analysis of the mortality findings. JAMA. 1971; 217:1676–1687.
- Feinstein AR. Clinical biostatistics. 8. An analytic appraisal of the University Group Diabetes Program (UGDP) study. Clin Pharmacol Ther. 1971; 12:167–191.
- The University Group Diabetes Program. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. V. Evaluation of pheniformin therapy. Diabetes 1975; 24( suppl 1):65–184.
- Knatterud GL, Klimt CR, Levin ME, Jacobson ME, Goldner MG. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA. 1978; 240:37–42.
- Schwartz TB, Meinert CL. The UGDP controversy: thirty-four years of contentious ambiguity laid to rest. Perspect Biol Med. 2004; 47:564–574.
- Turner RC, Holman RR. Lessons from UK Prospective Diabetes Study. Diabetes Res Clin Pract 1995; 28( suppl):S151–S157.
- UKPDS Research Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:854–865.
- UKPDS Study Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:837–853.
- UK Prospective Diabetes Study Group. UKPDS 28: a randomized trial of efficacy of early addition of metformin in sulfonylurea-treated type 2 diabetes. Diabetes Care. 1998; 21:87–92.
- Bretzel RG, Voigt K, Schatz H. The United Kingdom Prospective Diabetes Study (UKPDS) implications for the pharmacotherapy of type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 1998; 106:369–372.
- Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA. 1999; 281:2005–2012.
- Leslie RD. United Kingdom prospective diabetes study (UKPDS): what now or so what? Diabetes Metab Res Rev 1999; 15:65–71.
- Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000; 321:405–412.
- Mooradian AD, Chehade J. Implications of the UK Prospective Diabetes Study: questions answered and issues remaining. Drugs Aging. 2000; 16:159–164.
- Nathan DM. Some answers, more controversy, from UKPDS. United Kingdom Prospective Diabetes Study. Lancet. 1998; 352:832–833.
- Srimanunthiphol J, Beddow R, Arakaki R. A review of the United Kingdom Prospective Diabetes Study (UKPDS) and a discussion of the implications for patient care. Hawaii Med J. 2000; 59:295–298.
- Duckworth WC, McCarren M, Abraira C. Glucose control and cardiovascular complications: the VA Diabetes Trial. Diabetes Care. 2001; 24:942–945.
- Abraira C, Colwell JA, Nuttall FQ, et al. Veterans Affairs Cooperative Study on glycemic control and complications in type II diabetes (VA CSDM). Results of the feasibility trial. Veterans Affairs Cooperative Study in Type II Diabetes. Diabetes Care. 1995; 18:1113–1123.
- Gu K, Cowie CC, Harris MI. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971–1993. Diabetes Care. 1998; 21:1138–1145. NHANES
- Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008; 358:580–591.
- Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008; 358:2560–2572.
- Action in Diabetes and Vascular Disease: PreterAx and DiamicroN Modified-Release Controlled Evaluation. Rationale and design of the ADVANCE study: a randomised trial of blood pressure lowering and intensive glucose control in high-risk individuals with type 2 diabetes mellitus. J Hypertens 2001; 19(suppl):S21–S28.
- Abraira C, Duckworth W, McCarren M, et al. Design of the cooperative study on glycemic control and complications in diabetes mellitus type 2: Veterans Affairs Diabetes Trial. J Diabetes Complications. 2003; 17:314–322.
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007; 356:2457–2471.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract 2007; 13(suppl 1):1–68.
- American Diabetes Association. Standards of medical care in diabetes—2008. Diabetes Care 2008; 31(suppl 1):S12–S54.
- Magee MF, Isley WL. Rationale, design, and methods for glycemic control in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Trial. Am J Cardiol 2006; 97:20G–30G.
- Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358:2545–2559.
- Goff DC, Gerstein HC, Ginsberg HN, et al. Prevention of cardiovascular disease in persons with type 2 diabetes mellitus: current knowledge and rationale for the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:4i–20i.
- Buse JB, Bigger JT, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol 2007; 99:21i–33i.
- Gerstein HC, Riddle MC, Kendall DM, et al. Glycemia treatment strategies in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:34i–43i.
- Bonds DE, Kurashige EM, Bergenstal R, et al. Severe hypoglycemia monitoring and risk management procedures in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol 2007; 99:80i–89i.
- Gerstein HC. Dysglycemia, not just diabetes, is a continuous risk factor for cardiovascular disease. Evid Based Cardiovasc Med. 1997; 1:87–88.
- Gerstein HC, Pais P, Pogue J, Yusuf S. Relationship of glucose and insulin levels to the risk of myocardial infarction: a case-control study. J Am Coll Cardiol. 1999; 33:612–619.
- Gerstein HC, Capes SE. Dysglycemia: a key cardiovascular risk factor. Semin Vasc Med. 2002; 2:165–174.
- Gerstein HC, Santaguida P, Raina P, et al. Annual incidence and relative risk of diabetes in people with various categories of dysglycemia: a systematic overview and meta-analysis of prospective studies. Diabetes Res Clin Pract. 2007; 78:305–312.
- American Diabetes Association. Role of cardiovascular risk factors in prevention and treatment of macrovascular disease in diabetes. Diabetes Care. 1989; 12:573–579.
- Schor S. The University Group Diabetes Program. A statistician looks at the mortality results. JAMA. 1971; 217:1671–1675.
- Cornfield JThe University Group Diabetes Program. A further statistical analysis of the mortality findings. JAMA. 1971; 217:1676–1687.
- Feinstein AR. Clinical biostatistics. 8. An analytic appraisal of the University Group Diabetes Program (UGDP) study. Clin Pharmacol Ther. 1971; 12:167–191.
- The University Group Diabetes Program. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. V. Evaluation of pheniformin therapy. Diabetes 1975; 24( suppl 1):65–184.
- Knatterud GL, Klimt CR, Levin ME, Jacobson ME, Goldner MG. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. VII. Mortality and selected nonfatal events with insulin treatment. JAMA. 1978; 240:37–42.
- Schwartz TB, Meinert CL. The UGDP controversy: thirty-four years of contentious ambiguity laid to rest. Perspect Biol Med. 2004; 47:564–574.
- Turner RC, Holman RR. Lessons from UK Prospective Diabetes Study. Diabetes Res Clin Pract 1995; 28( suppl):S151–S157.
- UKPDS Research Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:854–865.
- UKPDS Study Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998; 352:837–853.
- UK Prospective Diabetes Study Group. UKPDS 28: a randomized trial of efficacy of early addition of metformin in sulfonylurea-treated type 2 diabetes. Diabetes Care. 1998; 21:87–92.
- Bretzel RG, Voigt K, Schatz H. The United Kingdom Prospective Diabetes Study (UKPDS) implications for the pharmacotherapy of type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 1998; 106:369–372.
- Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA. 1999; 281:2005–2012.
- Leslie RD. United Kingdom prospective diabetes study (UKPDS): what now or so what? Diabetes Metab Res Rev 1999; 15:65–71.
- Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000; 321:405–412.
- Mooradian AD, Chehade J. Implications of the UK Prospective Diabetes Study: questions answered and issues remaining. Drugs Aging. 2000; 16:159–164.
- Nathan DM. Some answers, more controversy, from UKPDS. United Kingdom Prospective Diabetes Study. Lancet. 1998; 352:832–833.
- Srimanunthiphol J, Beddow R, Arakaki R. A review of the United Kingdom Prospective Diabetes Study (UKPDS) and a discussion of the implications for patient care. Hawaii Med J. 2000; 59:295–298.
- Duckworth WC, McCarren M, Abraira C. Glucose control and cardiovascular complications: the VA Diabetes Trial. Diabetes Care. 2001; 24:942–945.
- Abraira C, Colwell JA, Nuttall FQ, et al. Veterans Affairs Cooperative Study on glycemic control and complications in type II diabetes (VA CSDM). Results of the feasibility trial. Veterans Affairs Cooperative Study in Type II Diabetes. Diabetes Care. 1995; 18:1113–1123.
- Gu K, Cowie CC, Harris MI. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971–1993. Diabetes Care. 1998; 21:1138–1145. NHANES
- Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008; 358:580–591.
- Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008; 358:2560–2572.
- Action in Diabetes and Vascular Disease: PreterAx and DiamicroN Modified-Release Controlled Evaluation. Rationale and design of the ADVANCE study: a randomised trial of blood pressure lowering and intensive glucose control in high-risk individuals with type 2 diabetes mellitus. J Hypertens 2001; 19(suppl):S21–S28.
- Abraira C, Duckworth W, McCarren M, et al. Design of the cooperative study on glycemic control and complications in diabetes mellitus type 2: Veterans Affairs Diabetes Trial. J Diabetes Complications. 2003; 17:314–322.
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007; 356:2457–2471.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract 2007; 13(suppl 1):1–68.
- American Diabetes Association. Standards of medical care in diabetes—2008. Diabetes Care 2008; 31(suppl 1):S12–S54.
- Magee MF, Isley WL. Rationale, design, and methods for glycemic control in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Trial. Am J Cardiol 2006; 97:20G–30G.
KEY POINTS
- No obvious cause, including hypoglycemia proximate to death or the use of any particular medication, clearly explained the excess deaths, although hypoglycemia occurred more often in intensively treated participants.
- The death rates in ACCORD were lower than in population studies and in other intervention trials. It is likely that multiple approaches to reducing the risk of cardiovascular disease contributed to this low mortality rate.
The Women’s Health Initiative: Implications for clinicians
More than 2 years have passed since we published the results of the Women’s Health Initiative (WHI), which caused a storm of information—and misinformation—about the effect of long-term dietary intervention on disease outcomes in postmenopausal women. Now that the dust has long settled, what have we learned from this landmark study?
The WHI results led to numerous additional analyses of all aspects of the study.1–7 What are the implications of all the analyses to clinical practice?
In this article, we summarize key aspects of the clinical trial, including study design, interventions, main results, and future plans. We also discuss potential clinical applications and practical considerations for public health efforts.
WHO WAS ELIGIBLE, WHO WAS NOT
A total of 48,835 postmenopausal women were randomly assigned to either no dietary intervention (n = 29,294) or a dietary intervention (n = 19,541) (see below).7 Participants were followed at 40 clinical centers between 1993 and 2005.4 Their mean age was 62.3 years; 18.6% were members of minorities.
Women were eligible if they were post-menopausal and had a daily dietary fat intake of at least 32% of total calories, based on assessment via a food-frequency questionnaire. They were excluded from the study if they had any of the following: a history of breast cancer, colorectal cancer, or other cancer except skin cancer during the past 10 years; type 1 diabetes; a medical condition in which the predicted survival was less than 3 years; and a potential barrier to adherence to the study regimen, including alcoholism or a lifestyle that involved often eating meals away from home.
THE WHI DIET: LESS FAT, BUT MORE FRUITS, VEGETABLES, GRAINS
The WHI dietary intervention was designed to prevent breast cancer, based on the evidence available when the study was planned. The targets included a total fat intake of less than 20% of energy (in kilocalories), increasing the intake of fruits and vegetables to at least five servings per day, and increasing the intake of grains to at least six servings per day.
Although reduction in saturated fat intake per se was not part of the WHI protocol, we assumed from previous pilot studies8 that the reduction of total fat intake would simultaneously produce a reduction in saturated fat intake to 7% of total calories.
A simpler dietary intervention
Unlike the 2006 American Heart Association guidelines and the US Department of Agriculture’s Dietary Guidelines for Americans 2005, the WHI dietary intervention had no specifications for dietary fiber, specific fatty acids (trans-fatty acids, omega-3 fatty acids, conjugated linoleic acid), complex carbohydrates, whole grains, vegetable protein, or other factors that have emerged as potential risk factors for cardiovascular and other chronic diseases since the study began. The WHI intervention also included no specific recommendation for total calorie intake, nor were patients in the intervention group encouraged to lose weight, as this could have confounded the results of the dietary intervention.
Education and encouragement
Those in the intervention group were each assigned a fat-gram goal, calculated on the basis of height. They were taught how to monitor their intake of total fat, fruits, vegetables, and grains. They attended intensive behavioral modification sessions to encourage them to keep to the dietary program: 18 group sessions in the first year and quarterly maintenance sessions thereafter, touching on a wide variety of nutrition- and behavior-related topics.7,9 Specially trained and certified nutritionists supervised the dietary intervention and the behavioral modification sessions according to the WHI study protocol.
Control-group participants received a copy of the US Department of Agriculture’s Dietary Guidelines for Americans10 and other health-related materials. They had no contact with the study nutritionists.
Other arms of the study
The WHI trial design included several arms,4,11–13 and many participants joined more than one arm: 20,592 postmenopausal women (42.2% of the total enrollment) chose dietary modification only, 8,050 (16.5%) chose diet plus hormone replacement therapy, 25,210 (51.6%) chose diet plus calcium and vitamin D supplementation, and 5,017 (10.3%) enrolled in all three.
Length of follow-up
Participants were followed from enrollment until they died, were lost to follow-up, or requested no further contact, or until the trial’s planned completion date, regardless of adherence to the dietary intervention, according to intention-to-treat analysis. All participants were contacted by clinic staff at 6-month intervals to provide updates on their health outcomes.
Factors assessed
Height, weight, waist circumference, and blood pressure were measured at annual visits using standardized procedures. Fasting blood samples were collected at baseline and at year 1 from all participants and from a subsample of 2,816 women (5.8% of the study population) at years 3 and 6. This subsample was randomly chosen with oversampling of minority women, for whom the odds for selection were six times higher than for white women.
Physical activity was assessed at baseline and at years 1, 3, 6, and 9. Walking and participation in sports and hours of activity per week were calculated for each participant. Physical activity was expressed as metabolic equivalent tasks per week for the analyses.
A food-frequency questionnaire6 to assess average dietary intake in the past 3 months was given at baseline and at year 1 for all participants. A third of all participants completed the questionnaire each year in a rotating sample. Completion rates were 100% at baseline and 81% thereafter. Follow-up data were collected from years 5 through 7. Also, 4-day food records were provided by all women before randomization.
HOW OUTCOMES WERE ASSESSED
The primary assessments of clinical outcome1–3 were mammographic screening, a self-reported medical history documented by a review of medical records, and electrocardiograms digitally obtained every 3 years. Mammograms and electrocardiograms were centrally adjudicated. The diagnosis of acute myocardial infarction was based on an algorithm that included cardiac pain, enzyme levels, and electrocardiographic readings.
OVERALL RESULTS
At 8.1 years, the incidence of breast cancer was 9% lower in the intervention group than in the comparison group (95% confidence interval [CI] = 0.83–1.01; P = .07, P = .09 weighted for length of follow-up).3 Subgroup analysis further showed that women who reported higher intakes of total dietary fat at baseline reduced their risk of breast cancer by 22% (95% CI = 0.64–0.96). Whether extended follow-up will show a significant association has yet to be determined.
Colon cancer rates did not differ between groups, but the number of polyps and adenomas reported was significantly lower in the dietary intervention group.1 The rate of colon cancer will also be included in the extended follow-up study of the WHI.
Risk factors for coronary heart disease in both groups—including levels of serum total cholesterol and serum low-density lipoprotein cholesterol, body weight, body mass index, diastolic blood pressure, and factor VIIc—improved slightly, but at year 3 of the trial, differences in overall rates of coronary heart disease and stroke in the two groups were not statistically significant.2 In addition, the low-fat diet intervention was associated with a reduction in blood estradiol concentrations between baseline and year 1.3 At the end of the study, however, differences in rates of breast cancer, colorectal cancer, and heart disease between the two groups were not statistically significant.
RESULTS OF DIETARY MODIFICATIONS
Fat as a percentage of total calories
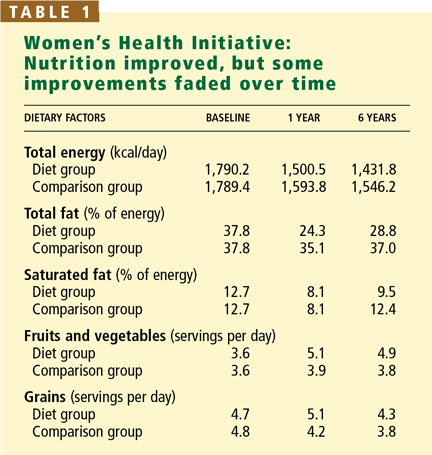
At the beginning of the WHI, all participants reported consuming an average of 35% of their caloric intake from fat (Table 1). At 1 year from baseline, the fat intake decreased to 24.3% in the intervention group (short of the study goal of 20%); this level had risen again to 26.7% by year 3 and to 28.8% at the end of the study. Stratified by quartile, women who achieved the greatest reductions in saturated and trans-fatty acids or the largest increases in their intake of fruits and vegetables appeared to have a moderate reduction in the risk of coronary heart disease.2 Women in the comparison group also decreased their fat intake initially, but to a lesser degree, and gradually increased it again thereafter. The mean net difference in self-reported total fat intake between the intervention group and the comparison group at 6 years was 8.2% (P < .001) (study goal, 13%).1–3
Intake of fruits, vegetables, and grains
At baseline, fruit and vegetable intake averaged 3.6 servings per day (Table 1). In the intervention group, this increased to 5.1 servings per day at year 1, and to 5.2 servings at year 3, but at the end of the study it had decreased to 4.9 servings.
Women in the intervention group were eating 4.7 servings of grains per day at baseline. This increased to 5.1 servings at year 1 and then decreased to 4.6 servings at year 3 and to 4.3 servings at the end of the study. It seems that as the women grew older their determination to increase servings of these foods diminished.
Proponents of some currently popular diets blame weight gain on a higher intake of carbohydrates, but the women following the WHI low-fat diet did not gain weight.2
Total fat vs saturated fat
Intake of total fat and saturated fat decreased in the intervention group during the study, but the difference between fat intake in the intervention group and that in the comparison group did not reach the degree expected.
At year 1, total fat as a percentage of total caloric intake was 10.8 percentage points below that of the comparison group, whereas the study expected difference was 13.0. At the end of the trial, the difference was only 8.2 percentage points, whereas the expected difference was 11.0.
Intake of all fatty acids (saturated and unsaturated) decreased at year 1, but then went back up slightly by the end of the trial but did not exceed baseline levels, and saturated fatty acids remained well below baseline levels: 9.5% vs 12.5% of caloric intake at baseline.4
INTERPRETING THE RESULTS
It might be tempting to dismiss the results of the WHI dietary intervention trial as not significant and therefore not meaningful. This would be unfortunate. The trial had some remarkable accomplishments and offers important lessons for future investigations.
The initial reductions in total fat intake were impressive, and women who had the highest total fat intake at baseline achieved the greatest reduction of total fat (to less than 22% of total calories).3 Nonetheless, the dietary intervention goal of less than 20% of calories from fat was not achieved despite intensive dietary counseling and a highly motivated study population. Thus, this dietary fat target may not be reasonable in the general population.
Also, despite the absence of targeted intervention on specific fatty acids, the observed blood cholesterol levels were as expected based on the well-known formula of Mensink and Katan,14 which incorporates information on changes in saturated fat, polyunsaturated fat, and dietary cholesterol intake. The predicted reduction in low-density lipoprotein cholesterol was 2.7 mg/dL; the observed reduction was 2.3 mg/dL.2 This illustrates that with greater modifications in specific known dietary risk factors for cardiovascular disease, such as saturated fatty acids, cholesterol, and unsaturated fatty acids, blood cholesterol levels respond in a predictable fashion. This was presumably not observed in WHI precisely because no goals and objectives were provided to participants for intake of saturated or polyunsaturated fatty acids.
Recent findings from the Optimal Macronutrient Intake Trial to Prevent Heart Disease (OmniHeart)15 further highlight differences in the total cholesterol response to diets of varying macronutrient (carbohydrate, protein, fat) content compared with the WHI dietary intervention.15 Participants in OmniHeart had reductions in levels of low-density lipoprotein cholesterol that were predictable from the changes reported in intake of saturated fatty acids. Presumably, the results of the WHI intervention would have been similar if the study had included this level of detail.
QUESTIONS REMAIN
Questions from the WHI that need consideration for future clinical applications include whether the study population may have already been “too old” to achieve a benefit from dietary modification, and whether the best timing for dietary intervention might be earlier adulthood with sustained changes in saturated fat, cholesterol, and unsaturated fat intake throughout life. Future subgroup analyses based on age at baseline will need to address these questions. Likewise, a longer follow-up period may be needed for a definitive evaluation of the impact of a regular low-fat diet on different health outcomes.
As reported by Patterson et al,16 the major contributors to total dietary fat intake at baseline were “added fats” such as sauces, gravies, butter, and margarines (25.1% of fat intake), followed by meats (20.9% of fat intake), and desserts (12.8% of fat intake). These findings highlight target areas for future interventions in women of this age group.
Another issue is how to standardize the dietary intervention from one clinical center to another—ie, to minimize differences in how each clinical center manages the study patients. Such differences were noted in WHI and other studies.17 Despite standardized training in delivering the dietary intervention, nutritionists encountered regional and cultural differences that required tailoring the dietary intervention to their patients’ needs. Staff turnover, an unavoidable phenomenon in long-term studies, has previously been reported to negatively influence dietary adherence.18
LIMITATIONS
A major limitation of diet modification research in general is the self-reporting of dietary intake, primarily by a food-frequency questionnaire. Although the use of a questionnaire is the most practical way to obtain dietary data for large studies, systematic biases may exist that obscure true nutrient-outcome relationships.19 Biomarker studies of energy balance suggest that people who are overweight or obese may under-report energy intake to a greater degree than people who are not overweight.20 Also, we still do not know how to get people to follow a healthy diet, although theories and models abound, such as social learning and cognitive-behavioral theory, and a lack of data limits our understanding of factors related to dietary adherence.21,22
FUTURE DIRECTIONS IN WHI
The WHI Extension Study is under way and has been funded through the year 2010. Outcomes ascertainment is the primary focus with no ongoing intervention, although the intervention group participants continue to receive a WHI newsletter that simply reiterates the importance of the study and encourages ongoing participation. As of 2006, an estimated 84% of the cohort, including both observational study and clinical trial participants, are involved. Efforts continue to recruit the remaining 16%, but many of these participants now consider themselves too old or too feeble to respond reliably.
In regard to breast cancer, the results published in 2006 are promising, albeit not statistically significant, and definitive statements cannot yet be made. However, postmenopausal women who are eating the diets highest in fat may have the greatest benefit from reductions in total fat.
Other considerations regarding the lack of statistically significant differences between groups may include the possibility that women in the intervention group may have been at lower risk for breast cancer at baseline. Likewise, although the results of the WHI dietary intervention do not include a statistically significant impact on colorectal cancer outcomes, the significant reduction in polyps and adenomas may later translate into a reduction in invasive cancer risk.
Finally, although no significant reduction was seen in the rate of death due to cardiovascular causes, greater reductions in saturated and trans-fatty acid intake were associated with greater reductions in blood cholesterol and cardiovascular risk.
Numerous subgroup analyses and ongoing assessments of the long-term impact of the diet modification are planned. Further associations are expected to emerge. The current and future results will continue to provide new insights that may lead to new clinical and public health recommendations in the future.
The WHI has raised additional issues that warrant further investigation:
- Will earlier dietary intervention, eg, during premenopausal years or even childhood, alter these results?
- Does the low-fat, high-carbohydrate diet used in WHI facilitate weight maintenance or even weight loss, as proposed by Howard et al23?
- Do quantitative changes in physical activity and weight control attenuate morbidity and mortality rates beyond changes in diet alone?
- Do vitamin and mineral supplements or hormone therapy alter disease outcomes or quality of life?
- Which behavioral approaches are best suited to the recruitment of patients for dietary intervention trials?
- Beresford SA, Johnson KC, Ritenbaugh C, et al. Low-fat dietary pattern and risk of colorectal cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:643–654.
- Howard BV, Van Horn L, Hsia J, et al. Low-fat dietary pattern and risk of cardiovascular disease: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:655–666.
- Prentice RL, Caan B, Chlebowski RT, et al. Low-fat dietary pattern and risk of invasive breast cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:629–642.
- The Women’s Health Initiative Study Group. Design of the Women’s Health Initiative clinical trial and observational study. Control Clin Trials 1998; 19:61–109.
- Ritenbaugh C, Patterson RE, Chlebowski RT, et al. The Women’s Health Initiative Dietary Modification trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S87–97.
- Patterson RE, Kristal AR, Tinker LF, Carter RA, Bolton MP, Agurs-Collins T. Measurement characteristics of the Women’s Health Initiative food frequency questionnaire. Ann Epidemiol 1999; 9:178–187.
- Tinker LF, Burrows ER, Henry H, Patterson RE, Rupp JW, Van Horn LV. The Women’s Health Initiative: overview of the nutrition components. In:Krummel DA, Kris-Etherton PM, editors. Nutrition in Women’s Health. Gaithersburg, MD: Aspen, 1996:510–542.
- Henderson MM, Kushi LH, Thompson DJ, et al. Feasibility of a randomized trial of a low-fat diet for the prevention of breast cancer: dietary compliance in the Women’s Health Trial Vanguard Study. Prev Med 1990; 19:115–133.
- Bowen D, Ehret C, Pedersen M, et al. Results of an adjunct dietary intervention program in the Women’s Health Initiative. J Am Diet Assoc 2002; 102:1631–1637.
- US Department of Agriculture. Dietary Guidelines for Americans. 6. Washington, DC: US Dept of Health and Human Services, 2005.
- Jackson RD, LaCroix AZ, Cauley JA, McGowan J. The Women’s Health Initiative calcium-vitamin D trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 Suppl:S98–106.
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- Stefanick ML, Cochrane BB, Hsia J, Barad DH, Liu JH, Johnson SR. The Women’s Health Initiative postmenopausal hormone trials: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S78–86.
- Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb 1992; 12:911–919.
- Appel LJ, Sacks FM, Carey VJ, et al. Effects of protein, monounsaturated fat, and carbohydrate intake on blood pressure and serum lipids: results of the OmniHeart randomized trial. JAMA 2005; 294:2455–2464.
- Patterson RE, Kristal A, Rodabough R, et al. Changes in food sources of dietary fat in response to an intensive low-fat dietary intervention: early results from the Women’s Health Initiative. J Am Diet Assoc 2003; 103:454–460.
- Lichtman JH, Roumanis SA, Radford MJ, Riedinger MS, Weingarten S, Krumholz HM. Can practice guidelines be transported effectively to different settings? Results from a multicenter interventional study. Jt Comm J Qual Improv 2001; 27:42–53.
- Jackson M, Berman N, Huber M, et al. Research staff turnover and participant adherence in the Women’s Health Initiative. Control Clin Trials 2003; 24:422–435.
- Willett W, Lenart E. Reproducibility and validity of food-frequency questionnaires. In:Willett W, ed. Nutritional Epidemiology. 2. New York: Oxford University Press, 1998:101–147.
- Subar AF, Kipnis V, Troiano RP, et al. Using intake biomarkers to evaluate the extent of dietary misreporting in a large sample of adults: the OPEN study. Am J Epidemiol 2003; 158:1–13.
- Bowen D, Raczynski J, George V, Feng Z, Fouad M. The role of participation in the women’s health trial: feasibility study in minority populations. Prev Med 2000; 31:474–480.
- Patterson RE, Kristal AR, White E. Do beliefs, knowledge, and perceived norms about diet and cancer predict dietary change? Am J Public Health 1996; 86:1394–1400.
- Howard BV, Manson JE, Stefanick ML, et al. Low-fat dietary pattern and weight change over 7 years: the Women’s Health Initiative Dietary Modification Trial. JAMA 2006; 295:35–49.
More than 2 years have passed since we published the results of the Women’s Health Initiative (WHI), which caused a storm of information—and misinformation—about the effect of long-term dietary intervention on disease outcomes in postmenopausal women. Now that the dust has long settled, what have we learned from this landmark study?
The WHI results led to numerous additional analyses of all aspects of the study.1–7 What are the implications of all the analyses to clinical practice?
In this article, we summarize key aspects of the clinical trial, including study design, interventions, main results, and future plans. We also discuss potential clinical applications and practical considerations for public health efforts.
WHO WAS ELIGIBLE, WHO WAS NOT
A total of 48,835 postmenopausal women were randomly assigned to either no dietary intervention (n = 29,294) or a dietary intervention (n = 19,541) (see below).7 Participants were followed at 40 clinical centers between 1993 and 2005.4 Their mean age was 62.3 years; 18.6% were members of minorities.
Women were eligible if they were post-menopausal and had a daily dietary fat intake of at least 32% of total calories, based on assessment via a food-frequency questionnaire. They were excluded from the study if they had any of the following: a history of breast cancer, colorectal cancer, or other cancer except skin cancer during the past 10 years; type 1 diabetes; a medical condition in which the predicted survival was less than 3 years; and a potential barrier to adherence to the study regimen, including alcoholism or a lifestyle that involved often eating meals away from home.
THE WHI DIET: LESS FAT, BUT MORE FRUITS, VEGETABLES, GRAINS
The WHI dietary intervention was designed to prevent breast cancer, based on the evidence available when the study was planned. The targets included a total fat intake of less than 20% of energy (in kilocalories), increasing the intake of fruits and vegetables to at least five servings per day, and increasing the intake of grains to at least six servings per day.
Although reduction in saturated fat intake per se was not part of the WHI protocol, we assumed from previous pilot studies8 that the reduction of total fat intake would simultaneously produce a reduction in saturated fat intake to 7% of total calories.
A simpler dietary intervention
Unlike the 2006 American Heart Association guidelines and the US Department of Agriculture’s Dietary Guidelines for Americans 2005, the WHI dietary intervention had no specifications for dietary fiber, specific fatty acids (trans-fatty acids, omega-3 fatty acids, conjugated linoleic acid), complex carbohydrates, whole grains, vegetable protein, or other factors that have emerged as potential risk factors for cardiovascular and other chronic diseases since the study began. The WHI intervention also included no specific recommendation for total calorie intake, nor were patients in the intervention group encouraged to lose weight, as this could have confounded the results of the dietary intervention.
Education and encouragement
Those in the intervention group were each assigned a fat-gram goal, calculated on the basis of height. They were taught how to monitor their intake of total fat, fruits, vegetables, and grains. They attended intensive behavioral modification sessions to encourage them to keep to the dietary program: 18 group sessions in the first year and quarterly maintenance sessions thereafter, touching on a wide variety of nutrition- and behavior-related topics.7,9 Specially trained and certified nutritionists supervised the dietary intervention and the behavioral modification sessions according to the WHI study protocol.
Control-group participants received a copy of the US Department of Agriculture’s Dietary Guidelines for Americans10 and other health-related materials. They had no contact with the study nutritionists.
Other arms of the study
The WHI trial design included several arms,4,11–13 and many participants joined more than one arm: 20,592 postmenopausal women (42.2% of the total enrollment) chose dietary modification only, 8,050 (16.5%) chose diet plus hormone replacement therapy, 25,210 (51.6%) chose diet plus calcium and vitamin D supplementation, and 5,017 (10.3%) enrolled in all three.
Length of follow-up
Participants were followed from enrollment until they died, were lost to follow-up, or requested no further contact, or until the trial’s planned completion date, regardless of adherence to the dietary intervention, according to intention-to-treat analysis. All participants were contacted by clinic staff at 6-month intervals to provide updates on their health outcomes.
Factors assessed
Height, weight, waist circumference, and blood pressure were measured at annual visits using standardized procedures. Fasting blood samples were collected at baseline and at year 1 from all participants and from a subsample of 2,816 women (5.8% of the study population) at years 3 and 6. This subsample was randomly chosen with oversampling of minority women, for whom the odds for selection were six times higher than for white women.
Physical activity was assessed at baseline and at years 1, 3, 6, and 9. Walking and participation in sports and hours of activity per week were calculated for each participant. Physical activity was expressed as metabolic equivalent tasks per week for the analyses.
A food-frequency questionnaire6 to assess average dietary intake in the past 3 months was given at baseline and at year 1 for all participants. A third of all participants completed the questionnaire each year in a rotating sample. Completion rates were 100% at baseline and 81% thereafter. Follow-up data were collected from years 5 through 7. Also, 4-day food records were provided by all women before randomization.
HOW OUTCOMES WERE ASSESSED
The primary assessments of clinical outcome1–3 were mammographic screening, a self-reported medical history documented by a review of medical records, and electrocardiograms digitally obtained every 3 years. Mammograms and electrocardiograms were centrally adjudicated. The diagnosis of acute myocardial infarction was based on an algorithm that included cardiac pain, enzyme levels, and electrocardiographic readings.
OVERALL RESULTS
At 8.1 years, the incidence of breast cancer was 9% lower in the intervention group than in the comparison group (95% confidence interval [CI] = 0.83–1.01; P = .07, P = .09 weighted for length of follow-up).3 Subgroup analysis further showed that women who reported higher intakes of total dietary fat at baseline reduced their risk of breast cancer by 22% (95% CI = 0.64–0.96). Whether extended follow-up will show a significant association has yet to be determined.
Colon cancer rates did not differ between groups, but the number of polyps and adenomas reported was significantly lower in the dietary intervention group.1 The rate of colon cancer will also be included in the extended follow-up study of the WHI.
Risk factors for coronary heart disease in both groups—including levels of serum total cholesterol and serum low-density lipoprotein cholesterol, body weight, body mass index, diastolic blood pressure, and factor VIIc—improved slightly, but at year 3 of the trial, differences in overall rates of coronary heart disease and stroke in the two groups were not statistically significant.2 In addition, the low-fat diet intervention was associated with a reduction in blood estradiol concentrations between baseline and year 1.3 At the end of the study, however, differences in rates of breast cancer, colorectal cancer, and heart disease between the two groups were not statistically significant.
RESULTS OF DIETARY MODIFICATIONS
Fat as a percentage of total calories
At the beginning of the WHI, all participants reported consuming an average of 35% of their caloric intake from fat (Table 1). At 1 year from baseline, the fat intake decreased to 24.3% in the intervention group (short of the study goal of 20%); this level had risen again to 26.7% by year 3 and to 28.8% at the end of the study. Stratified by quartile, women who achieved the greatest reductions in saturated and trans-fatty acids or the largest increases in their intake of fruits and vegetables appeared to have a moderate reduction in the risk of coronary heart disease.2 Women in the comparison group also decreased their fat intake initially, but to a lesser degree, and gradually increased it again thereafter. The mean net difference in self-reported total fat intake between the intervention group and the comparison group at 6 years was 8.2% (P < .001) (study goal, 13%).1–3
Intake of fruits, vegetables, and grains
At baseline, fruit and vegetable intake averaged 3.6 servings per day (Table 1). In the intervention group, this increased to 5.1 servings per day at year 1, and to 5.2 servings at year 3, but at the end of the study it had decreased to 4.9 servings.
Women in the intervention group were eating 4.7 servings of grains per day at baseline. This increased to 5.1 servings at year 1 and then decreased to 4.6 servings at year 3 and to 4.3 servings at the end of the study. It seems that as the women grew older their determination to increase servings of these foods diminished.
Proponents of some currently popular diets blame weight gain on a higher intake of carbohydrates, but the women following the WHI low-fat diet did not gain weight.2
Total fat vs saturated fat
Intake of total fat and saturated fat decreased in the intervention group during the study, but the difference between fat intake in the intervention group and that in the comparison group did not reach the degree expected.
At year 1, total fat as a percentage of total caloric intake was 10.8 percentage points below that of the comparison group, whereas the study expected difference was 13.0. At the end of the trial, the difference was only 8.2 percentage points, whereas the expected difference was 11.0.
Intake of all fatty acids (saturated and unsaturated) decreased at year 1, but then went back up slightly by the end of the trial but did not exceed baseline levels, and saturated fatty acids remained well below baseline levels: 9.5% vs 12.5% of caloric intake at baseline.4
INTERPRETING THE RESULTS
It might be tempting to dismiss the results of the WHI dietary intervention trial as not significant and therefore not meaningful. This would be unfortunate. The trial had some remarkable accomplishments and offers important lessons for future investigations.
The initial reductions in total fat intake were impressive, and women who had the highest total fat intake at baseline achieved the greatest reduction of total fat (to less than 22% of total calories).3 Nonetheless, the dietary intervention goal of less than 20% of calories from fat was not achieved despite intensive dietary counseling and a highly motivated study population. Thus, this dietary fat target may not be reasonable in the general population.
Also, despite the absence of targeted intervention on specific fatty acids, the observed blood cholesterol levels were as expected based on the well-known formula of Mensink and Katan,14 which incorporates information on changes in saturated fat, polyunsaturated fat, and dietary cholesterol intake. The predicted reduction in low-density lipoprotein cholesterol was 2.7 mg/dL; the observed reduction was 2.3 mg/dL.2 This illustrates that with greater modifications in specific known dietary risk factors for cardiovascular disease, such as saturated fatty acids, cholesterol, and unsaturated fatty acids, blood cholesterol levels respond in a predictable fashion. This was presumably not observed in WHI precisely because no goals and objectives were provided to participants for intake of saturated or polyunsaturated fatty acids.
Recent findings from the Optimal Macronutrient Intake Trial to Prevent Heart Disease (OmniHeart)15 further highlight differences in the total cholesterol response to diets of varying macronutrient (carbohydrate, protein, fat) content compared with the WHI dietary intervention.15 Participants in OmniHeart had reductions in levels of low-density lipoprotein cholesterol that were predictable from the changes reported in intake of saturated fatty acids. Presumably, the results of the WHI intervention would have been similar if the study had included this level of detail.
QUESTIONS REMAIN
Questions from the WHI that need consideration for future clinical applications include whether the study population may have already been “too old” to achieve a benefit from dietary modification, and whether the best timing for dietary intervention might be earlier adulthood with sustained changes in saturated fat, cholesterol, and unsaturated fat intake throughout life. Future subgroup analyses based on age at baseline will need to address these questions. Likewise, a longer follow-up period may be needed for a definitive evaluation of the impact of a regular low-fat diet on different health outcomes.
As reported by Patterson et al,16 the major contributors to total dietary fat intake at baseline were “added fats” such as sauces, gravies, butter, and margarines (25.1% of fat intake), followed by meats (20.9% of fat intake), and desserts (12.8% of fat intake). These findings highlight target areas for future interventions in women of this age group.
Another issue is how to standardize the dietary intervention from one clinical center to another—ie, to minimize differences in how each clinical center manages the study patients. Such differences were noted in WHI and other studies.17 Despite standardized training in delivering the dietary intervention, nutritionists encountered regional and cultural differences that required tailoring the dietary intervention to their patients’ needs. Staff turnover, an unavoidable phenomenon in long-term studies, has previously been reported to negatively influence dietary adherence.18
LIMITATIONS
A major limitation of diet modification research in general is the self-reporting of dietary intake, primarily by a food-frequency questionnaire. Although the use of a questionnaire is the most practical way to obtain dietary data for large studies, systematic biases may exist that obscure true nutrient-outcome relationships.19 Biomarker studies of energy balance suggest that people who are overweight or obese may under-report energy intake to a greater degree than people who are not overweight.20 Also, we still do not know how to get people to follow a healthy diet, although theories and models abound, such as social learning and cognitive-behavioral theory, and a lack of data limits our understanding of factors related to dietary adherence.21,22
FUTURE DIRECTIONS IN WHI
The WHI Extension Study is under way and has been funded through the year 2010. Outcomes ascertainment is the primary focus with no ongoing intervention, although the intervention group participants continue to receive a WHI newsletter that simply reiterates the importance of the study and encourages ongoing participation. As of 2006, an estimated 84% of the cohort, including both observational study and clinical trial participants, are involved. Efforts continue to recruit the remaining 16%, but many of these participants now consider themselves too old or too feeble to respond reliably.
In regard to breast cancer, the results published in 2006 are promising, albeit not statistically significant, and definitive statements cannot yet be made. However, postmenopausal women who are eating the diets highest in fat may have the greatest benefit from reductions in total fat.
Other considerations regarding the lack of statistically significant differences between groups may include the possibility that women in the intervention group may have been at lower risk for breast cancer at baseline. Likewise, although the results of the WHI dietary intervention do not include a statistically significant impact on colorectal cancer outcomes, the significant reduction in polyps and adenomas may later translate into a reduction in invasive cancer risk.
Finally, although no significant reduction was seen in the rate of death due to cardiovascular causes, greater reductions in saturated and trans-fatty acid intake were associated with greater reductions in blood cholesterol and cardiovascular risk.
Numerous subgroup analyses and ongoing assessments of the long-term impact of the diet modification are planned. Further associations are expected to emerge. The current and future results will continue to provide new insights that may lead to new clinical and public health recommendations in the future.
The WHI has raised additional issues that warrant further investigation:
- Will earlier dietary intervention, eg, during premenopausal years or even childhood, alter these results?
- Does the low-fat, high-carbohydrate diet used in WHI facilitate weight maintenance or even weight loss, as proposed by Howard et al23?
- Do quantitative changes in physical activity and weight control attenuate morbidity and mortality rates beyond changes in diet alone?
- Do vitamin and mineral supplements or hormone therapy alter disease outcomes or quality of life?
- Which behavioral approaches are best suited to the recruitment of patients for dietary intervention trials?
More than 2 years have passed since we published the results of the Women’s Health Initiative (WHI), which caused a storm of information—and misinformation—about the effect of long-term dietary intervention on disease outcomes in postmenopausal women. Now that the dust has long settled, what have we learned from this landmark study?
The WHI results led to numerous additional analyses of all aspects of the study.1–7 What are the implications of all the analyses to clinical practice?
In this article, we summarize key aspects of the clinical trial, including study design, interventions, main results, and future plans. We also discuss potential clinical applications and practical considerations for public health efforts.
WHO WAS ELIGIBLE, WHO WAS NOT
A total of 48,835 postmenopausal women were randomly assigned to either no dietary intervention (n = 29,294) or a dietary intervention (n = 19,541) (see below).7 Participants were followed at 40 clinical centers between 1993 and 2005.4 Their mean age was 62.3 years; 18.6% were members of minorities.
Women were eligible if they were post-menopausal and had a daily dietary fat intake of at least 32% of total calories, based on assessment via a food-frequency questionnaire. They were excluded from the study if they had any of the following: a history of breast cancer, colorectal cancer, or other cancer except skin cancer during the past 10 years; type 1 diabetes; a medical condition in which the predicted survival was less than 3 years; and a potential barrier to adherence to the study regimen, including alcoholism or a lifestyle that involved often eating meals away from home.
THE WHI DIET: LESS FAT, BUT MORE FRUITS, VEGETABLES, GRAINS
The WHI dietary intervention was designed to prevent breast cancer, based on the evidence available when the study was planned. The targets included a total fat intake of less than 20% of energy (in kilocalories), increasing the intake of fruits and vegetables to at least five servings per day, and increasing the intake of grains to at least six servings per day.
Although reduction in saturated fat intake per se was not part of the WHI protocol, we assumed from previous pilot studies8 that the reduction of total fat intake would simultaneously produce a reduction in saturated fat intake to 7% of total calories.
A simpler dietary intervention
Unlike the 2006 American Heart Association guidelines and the US Department of Agriculture’s Dietary Guidelines for Americans 2005, the WHI dietary intervention had no specifications for dietary fiber, specific fatty acids (trans-fatty acids, omega-3 fatty acids, conjugated linoleic acid), complex carbohydrates, whole grains, vegetable protein, or other factors that have emerged as potential risk factors for cardiovascular and other chronic diseases since the study began. The WHI intervention also included no specific recommendation for total calorie intake, nor were patients in the intervention group encouraged to lose weight, as this could have confounded the results of the dietary intervention.
Education and encouragement
Those in the intervention group were each assigned a fat-gram goal, calculated on the basis of height. They were taught how to monitor their intake of total fat, fruits, vegetables, and grains. They attended intensive behavioral modification sessions to encourage them to keep to the dietary program: 18 group sessions in the first year and quarterly maintenance sessions thereafter, touching on a wide variety of nutrition- and behavior-related topics.7,9 Specially trained and certified nutritionists supervised the dietary intervention and the behavioral modification sessions according to the WHI study protocol.
Control-group participants received a copy of the US Department of Agriculture’s Dietary Guidelines for Americans10 and other health-related materials. They had no contact with the study nutritionists.
Other arms of the study
The WHI trial design included several arms,4,11–13 and many participants joined more than one arm: 20,592 postmenopausal women (42.2% of the total enrollment) chose dietary modification only, 8,050 (16.5%) chose diet plus hormone replacement therapy, 25,210 (51.6%) chose diet plus calcium and vitamin D supplementation, and 5,017 (10.3%) enrolled in all three.
Length of follow-up
Participants were followed from enrollment until they died, were lost to follow-up, or requested no further contact, or until the trial’s planned completion date, regardless of adherence to the dietary intervention, according to intention-to-treat analysis. All participants were contacted by clinic staff at 6-month intervals to provide updates on their health outcomes.
Factors assessed
Height, weight, waist circumference, and blood pressure were measured at annual visits using standardized procedures. Fasting blood samples were collected at baseline and at year 1 from all participants and from a subsample of 2,816 women (5.8% of the study population) at years 3 and 6. This subsample was randomly chosen with oversampling of minority women, for whom the odds for selection were six times higher than for white women.
Physical activity was assessed at baseline and at years 1, 3, 6, and 9. Walking and participation in sports and hours of activity per week were calculated for each participant. Physical activity was expressed as metabolic equivalent tasks per week for the analyses.
A food-frequency questionnaire6 to assess average dietary intake in the past 3 months was given at baseline and at year 1 for all participants. A third of all participants completed the questionnaire each year in a rotating sample. Completion rates were 100% at baseline and 81% thereafter. Follow-up data were collected from years 5 through 7. Also, 4-day food records were provided by all women before randomization.
HOW OUTCOMES WERE ASSESSED
The primary assessments of clinical outcome1–3 were mammographic screening, a self-reported medical history documented by a review of medical records, and electrocardiograms digitally obtained every 3 years. Mammograms and electrocardiograms were centrally adjudicated. The diagnosis of acute myocardial infarction was based on an algorithm that included cardiac pain, enzyme levels, and electrocardiographic readings.
OVERALL RESULTS
At 8.1 years, the incidence of breast cancer was 9% lower in the intervention group than in the comparison group (95% confidence interval [CI] = 0.83–1.01; P = .07, P = .09 weighted for length of follow-up).3 Subgroup analysis further showed that women who reported higher intakes of total dietary fat at baseline reduced their risk of breast cancer by 22% (95% CI = 0.64–0.96). Whether extended follow-up will show a significant association has yet to be determined.
Colon cancer rates did not differ between groups, but the number of polyps and adenomas reported was significantly lower in the dietary intervention group.1 The rate of colon cancer will also be included in the extended follow-up study of the WHI.
Risk factors for coronary heart disease in both groups—including levels of serum total cholesterol and serum low-density lipoprotein cholesterol, body weight, body mass index, diastolic blood pressure, and factor VIIc—improved slightly, but at year 3 of the trial, differences in overall rates of coronary heart disease and stroke in the two groups were not statistically significant.2 In addition, the low-fat diet intervention was associated with a reduction in blood estradiol concentrations between baseline and year 1.3 At the end of the study, however, differences in rates of breast cancer, colorectal cancer, and heart disease between the two groups were not statistically significant.
RESULTS OF DIETARY MODIFICATIONS
Fat as a percentage of total calories
At the beginning of the WHI, all participants reported consuming an average of 35% of their caloric intake from fat (Table 1). At 1 year from baseline, the fat intake decreased to 24.3% in the intervention group (short of the study goal of 20%); this level had risen again to 26.7% by year 3 and to 28.8% at the end of the study. Stratified by quartile, women who achieved the greatest reductions in saturated and trans-fatty acids or the largest increases in their intake of fruits and vegetables appeared to have a moderate reduction in the risk of coronary heart disease.2 Women in the comparison group also decreased their fat intake initially, but to a lesser degree, and gradually increased it again thereafter. The mean net difference in self-reported total fat intake between the intervention group and the comparison group at 6 years was 8.2% (P < .001) (study goal, 13%).1–3
Intake of fruits, vegetables, and grains
At baseline, fruit and vegetable intake averaged 3.6 servings per day (Table 1). In the intervention group, this increased to 5.1 servings per day at year 1, and to 5.2 servings at year 3, but at the end of the study it had decreased to 4.9 servings.
Women in the intervention group were eating 4.7 servings of grains per day at baseline. This increased to 5.1 servings at year 1 and then decreased to 4.6 servings at year 3 and to 4.3 servings at the end of the study. It seems that as the women grew older their determination to increase servings of these foods diminished.
Proponents of some currently popular diets blame weight gain on a higher intake of carbohydrates, but the women following the WHI low-fat diet did not gain weight.2
Total fat vs saturated fat
Intake of total fat and saturated fat decreased in the intervention group during the study, but the difference between fat intake in the intervention group and that in the comparison group did not reach the degree expected.
At year 1, total fat as a percentage of total caloric intake was 10.8 percentage points below that of the comparison group, whereas the study expected difference was 13.0. At the end of the trial, the difference was only 8.2 percentage points, whereas the expected difference was 11.0.
Intake of all fatty acids (saturated and unsaturated) decreased at year 1, but then went back up slightly by the end of the trial but did not exceed baseline levels, and saturated fatty acids remained well below baseline levels: 9.5% vs 12.5% of caloric intake at baseline.4
INTERPRETING THE RESULTS
It might be tempting to dismiss the results of the WHI dietary intervention trial as not significant and therefore not meaningful. This would be unfortunate. The trial had some remarkable accomplishments and offers important lessons for future investigations.
The initial reductions in total fat intake were impressive, and women who had the highest total fat intake at baseline achieved the greatest reduction of total fat (to less than 22% of total calories).3 Nonetheless, the dietary intervention goal of less than 20% of calories from fat was not achieved despite intensive dietary counseling and a highly motivated study population. Thus, this dietary fat target may not be reasonable in the general population.
Also, despite the absence of targeted intervention on specific fatty acids, the observed blood cholesterol levels were as expected based on the well-known formula of Mensink and Katan,14 which incorporates information on changes in saturated fat, polyunsaturated fat, and dietary cholesterol intake. The predicted reduction in low-density lipoprotein cholesterol was 2.7 mg/dL; the observed reduction was 2.3 mg/dL.2 This illustrates that with greater modifications in specific known dietary risk factors for cardiovascular disease, such as saturated fatty acids, cholesterol, and unsaturated fatty acids, blood cholesterol levels respond in a predictable fashion. This was presumably not observed in WHI precisely because no goals and objectives were provided to participants for intake of saturated or polyunsaturated fatty acids.
Recent findings from the Optimal Macronutrient Intake Trial to Prevent Heart Disease (OmniHeart)15 further highlight differences in the total cholesterol response to diets of varying macronutrient (carbohydrate, protein, fat) content compared with the WHI dietary intervention.15 Participants in OmniHeart had reductions in levels of low-density lipoprotein cholesterol that were predictable from the changes reported in intake of saturated fatty acids. Presumably, the results of the WHI intervention would have been similar if the study had included this level of detail.
QUESTIONS REMAIN
Questions from the WHI that need consideration for future clinical applications include whether the study population may have already been “too old” to achieve a benefit from dietary modification, and whether the best timing for dietary intervention might be earlier adulthood with sustained changes in saturated fat, cholesterol, and unsaturated fat intake throughout life. Future subgroup analyses based on age at baseline will need to address these questions. Likewise, a longer follow-up period may be needed for a definitive evaluation of the impact of a regular low-fat diet on different health outcomes.
As reported by Patterson et al,16 the major contributors to total dietary fat intake at baseline were “added fats” such as sauces, gravies, butter, and margarines (25.1% of fat intake), followed by meats (20.9% of fat intake), and desserts (12.8% of fat intake). These findings highlight target areas for future interventions in women of this age group.
Another issue is how to standardize the dietary intervention from one clinical center to another—ie, to minimize differences in how each clinical center manages the study patients. Such differences were noted in WHI and other studies.17 Despite standardized training in delivering the dietary intervention, nutritionists encountered regional and cultural differences that required tailoring the dietary intervention to their patients’ needs. Staff turnover, an unavoidable phenomenon in long-term studies, has previously been reported to negatively influence dietary adherence.18
LIMITATIONS
A major limitation of diet modification research in general is the self-reporting of dietary intake, primarily by a food-frequency questionnaire. Although the use of a questionnaire is the most practical way to obtain dietary data for large studies, systematic biases may exist that obscure true nutrient-outcome relationships.19 Biomarker studies of energy balance suggest that people who are overweight or obese may under-report energy intake to a greater degree than people who are not overweight.20 Also, we still do not know how to get people to follow a healthy diet, although theories and models abound, such as social learning and cognitive-behavioral theory, and a lack of data limits our understanding of factors related to dietary adherence.21,22
FUTURE DIRECTIONS IN WHI
The WHI Extension Study is under way and has been funded through the year 2010. Outcomes ascertainment is the primary focus with no ongoing intervention, although the intervention group participants continue to receive a WHI newsletter that simply reiterates the importance of the study and encourages ongoing participation. As of 2006, an estimated 84% of the cohort, including both observational study and clinical trial participants, are involved. Efforts continue to recruit the remaining 16%, but many of these participants now consider themselves too old or too feeble to respond reliably.
In regard to breast cancer, the results published in 2006 are promising, albeit not statistically significant, and definitive statements cannot yet be made. However, postmenopausal women who are eating the diets highest in fat may have the greatest benefit from reductions in total fat.
Other considerations regarding the lack of statistically significant differences between groups may include the possibility that women in the intervention group may have been at lower risk for breast cancer at baseline. Likewise, although the results of the WHI dietary intervention do not include a statistically significant impact on colorectal cancer outcomes, the significant reduction in polyps and adenomas may later translate into a reduction in invasive cancer risk.
Finally, although no significant reduction was seen in the rate of death due to cardiovascular causes, greater reductions in saturated and trans-fatty acid intake were associated with greater reductions in blood cholesterol and cardiovascular risk.
Numerous subgroup analyses and ongoing assessments of the long-term impact of the diet modification are planned. Further associations are expected to emerge. The current and future results will continue to provide new insights that may lead to new clinical and public health recommendations in the future.
The WHI has raised additional issues that warrant further investigation:
- Will earlier dietary intervention, eg, during premenopausal years or even childhood, alter these results?
- Does the low-fat, high-carbohydrate diet used in WHI facilitate weight maintenance or even weight loss, as proposed by Howard et al23?
- Do quantitative changes in physical activity and weight control attenuate morbidity and mortality rates beyond changes in diet alone?
- Do vitamin and mineral supplements or hormone therapy alter disease outcomes or quality of life?
- Which behavioral approaches are best suited to the recruitment of patients for dietary intervention trials?
- Beresford SA, Johnson KC, Ritenbaugh C, et al. Low-fat dietary pattern and risk of colorectal cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:643–654.
- Howard BV, Van Horn L, Hsia J, et al. Low-fat dietary pattern and risk of cardiovascular disease: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:655–666.
- Prentice RL, Caan B, Chlebowski RT, et al. Low-fat dietary pattern and risk of invasive breast cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:629–642.
- The Women’s Health Initiative Study Group. Design of the Women’s Health Initiative clinical trial and observational study. Control Clin Trials 1998; 19:61–109.
- Ritenbaugh C, Patterson RE, Chlebowski RT, et al. The Women’s Health Initiative Dietary Modification trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S87–97.
- Patterson RE, Kristal AR, Tinker LF, Carter RA, Bolton MP, Agurs-Collins T. Measurement characteristics of the Women’s Health Initiative food frequency questionnaire. Ann Epidemiol 1999; 9:178–187.
- Tinker LF, Burrows ER, Henry H, Patterson RE, Rupp JW, Van Horn LV. The Women’s Health Initiative: overview of the nutrition components. In:Krummel DA, Kris-Etherton PM, editors. Nutrition in Women’s Health. Gaithersburg, MD: Aspen, 1996:510–542.
- Henderson MM, Kushi LH, Thompson DJ, et al. Feasibility of a randomized trial of a low-fat diet for the prevention of breast cancer: dietary compliance in the Women’s Health Trial Vanguard Study. Prev Med 1990; 19:115–133.
- Bowen D, Ehret C, Pedersen M, et al. Results of an adjunct dietary intervention program in the Women’s Health Initiative. J Am Diet Assoc 2002; 102:1631–1637.
- US Department of Agriculture. Dietary Guidelines for Americans. 6. Washington, DC: US Dept of Health and Human Services, 2005.
- Jackson RD, LaCroix AZ, Cauley JA, McGowan J. The Women’s Health Initiative calcium-vitamin D trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 Suppl:S98–106.
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- Stefanick ML, Cochrane BB, Hsia J, Barad DH, Liu JH, Johnson SR. The Women’s Health Initiative postmenopausal hormone trials: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S78–86.
- Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb 1992; 12:911–919.
- Appel LJ, Sacks FM, Carey VJ, et al. Effects of protein, monounsaturated fat, and carbohydrate intake on blood pressure and serum lipids: results of the OmniHeart randomized trial. JAMA 2005; 294:2455–2464.
- Patterson RE, Kristal A, Rodabough R, et al. Changes in food sources of dietary fat in response to an intensive low-fat dietary intervention: early results from the Women’s Health Initiative. J Am Diet Assoc 2003; 103:454–460.
- Lichtman JH, Roumanis SA, Radford MJ, Riedinger MS, Weingarten S, Krumholz HM. Can practice guidelines be transported effectively to different settings? Results from a multicenter interventional study. Jt Comm J Qual Improv 2001; 27:42–53.
- Jackson M, Berman N, Huber M, et al. Research staff turnover and participant adherence in the Women’s Health Initiative. Control Clin Trials 2003; 24:422–435.
- Willett W, Lenart E. Reproducibility and validity of food-frequency questionnaires. In:Willett W, ed. Nutritional Epidemiology. 2. New York: Oxford University Press, 1998:101–147.
- Subar AF, Kipnis V, Troiano RP, et al. Using intake biomarkers to evaluate the extent of dietary misreporting in a large sample of adults: the OPEN study. Am J Epidemiol 2003; 158:1–13.
- Bowen D, Raczynski J, George V, Feng Z, Fouad M. The role of participation in the women’s health trial: feasibility study in minority populations. Prev Med 2000; 31:474–480.
- Patterson RE, Kristal AR, White E. Do beliefs, knowledge, and perceived norms about diet and cancer predict dietary change? Am J Public Health 1996; 86:1394–1400.
- Howard BV, Manson JE, Stefanick ML, et al. Low-fat dietary pattern and weight change over 7 years: the Women’s Health Initiative Dietary Modification Trial. JAMA 2006; 295:35–49.
- Beresford SA, Johnson KC, Ritenbaugh C, et al. Low-fat dietary pattern and risk of colorectal cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:643–654.
- Howard BV, Van Horn L, Hsia J, et al. Low-fat dietary pattern and risk of cardiovascular disease: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:655–666.
- Prentice RL, Caan B, Chlebowski RT, et al. Low-fat dietary pattern and risk of invasive breast cancer: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA 2006; 295:629–642.
- The Women’s Health Initiative Study Group. Design of the Women’s Health Initiative clinical trial and observational study. Control Clin Trials 1998; 19:61–109.
- Ritenbaugh C, Patterson RE, Chlebowski RT, et al. The Women’s Health Initiative Dietary Modification trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S87–97.
- Patterson RE, Kristal AR, Tinker LF, Carter RA, Bolton MP, Agurs-Collins T. Measurement characteristics of the Women’s Health Initiative food frequency questionnaire. Ann Epidemiol 1999; 9:178–187.
- Tinker LF, Burrows ER, Henry H, Patterson RE, Rupp JW, Van Horn LV. The Women’s Health Initiative: overview of the nutrition components. In:Krummel DA, Kris-Etherton PM, editors. Nutrition in Women’s Health. Gaithersburg, MD: Aspen, 1996:510–542.
- Henderson MM, Kushi LH, Thompson DJ, et al. Feasibility of a randomized trial of a low-fat diet for the prevention of breast cancer: dietary compliance in the Women’s Health Trial Vanguard Study. Prev Med 1990; 19:115–133.
- Bowen D, Ehret C, Pedersen M, et al. Results of an adjunct dietary intervention program in the Women’s Health Initiative. J Am Diet Assoc 2002; 102:1631–1637.
- US Department of Agriculture. Dietary Guidelines for Americans. 6. Washington, DC: US Dept of Health and Human Services, 2005.
- Jackson RD, LaCroix AZ, Cauley JA, McGowan J. The Women’s Health Initiative calcium-vitamin D trial: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 Suppl:S98–106.
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- Stefanick ML, Cochrane BB, Hsia J, Barad DH, Liu JH, Johnson SR. The Women’s Health Initiative postmenopausal hormone trials: overview and baseline characteristics of participants. Ann Epidemiol 2003; 13 9 suppl:S78–86.
- Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb 1992; 12:911–919.
- Appel LJ, Sacks FM, Carey VJ, et al. Effects of protein, monounsaturated fat, and carbohydrate intake on blood pressure and serum lipids: results of the OmniHeart randomized trial. JAMA 2005; 294:2455–2464.
- Patterson RE, Kristal A, Rodabough R, et al. Changes in food sources of dietary fat in response to an intensive low-fat dietary intervention: early results from the Women’s Health Initiative. J Am Diet Assoc 2003; 103:454–460.
- Lichtman JH, Roumanis SA, Radford MJ, Riedinger MS, Weingarten S, Krumholz HM. Can practice guidelines be transported effectively to different settings? Results from a multicenter interventional study. Jt Comm J Qual Improv 2001; 27:42–53.
- Jackson M, Berman N, Huber M, et al. Research staff turnover and participant adherence in the Women’s Health Initiative. Control Clin Trials 2003; 24:422–435.
- Willett W, Lenart E. Reproducibility and validity of food-frequency questionnaires. In:Willett W, ed. Nutritional Epidemiology. 2. New York: Oxford University Press, 1998:101–147.
- Subar AF, Kipnis V, Troiano RP, et al. Using intake biomarkers to evaluate the extent of dietary misreporting in a large sample of adults: the OPEN study. Am J Epidemiol 2003; 158:1–13.
- Bowen D, Raczynski J, George V, Feng Z, Fouad M. The role of participation in the women’s health trial: feasibility study in minority populations. Prev Med 2000; 31:474–480.
- Patterson RE, Kristal AR, White E. Do beliefs, knowledge, and perceived norms about diet and cancer predict dietary change? Am J Public Health 1996; 86:1394–1400.
- Howard BV, Manson JE, Stefanick ML, et al. Low-fat dietary pattern and weight change over 7 years: the Women’s Health Initiative Dietary Modification Trial. JAMA 2006; 295:35–49.
KEY POINTS
- Colon cancer rates did not differ between the dietary intervention group and the comparison group, but the number of polyps and adenomas reported was significantly lower in the dietary intervention group.
- Risk factors for coronary heart disease improved slightly with the diet, but by trial year 3, differences in overall rates of coronary heart disease and stroke in the two groups were not statistically significant.
- When stratified by quartiles, those who reduced their intake of saturated and trans-fatty acids the most, or who increased their intake of fruits and vegetables the most, appeared to have a moderate reduction in the risk of coronary heart disease.
What is the role of dual antiplatelet therapy with clopidogrel and aspirin?
In patients at risk of myocardial infarction or stroke, two antiplatelet drugs are not always better than one. In a large recent trial,1,2 adding clopidogrel (Plavix) to aspirin therapy did not offer much benefit to a cohort of patients at risk of cardiovascular events, although a subgroup did appear to benefit: those at even higher risk because they already had a history of myocardial infarction, ischemic stroke, or peripheral arterial disease.
These were the principal findings in the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) study,1,2 in which one of us (D.L.B.) was principal investigator.
These findings further our understanding of who should receive dual antiplatelet therapy, and who would be better served with aspirin therapy alone. In this article, we discuss important studies that led up to the CHARISMA trial, review CHARISMA’s purpose and study design, and interpret its results.
PREVENTING ATHEROTHROMBOSIS BY BLOCKING PLATELETS
Platelets are key players in the atherothrom-botic process.3–5 The Antiplatelet Trialists’ Collaboration,6 in a meta-analysis of trials performed up to 1997, calculated that antiplatelet therapy (mostly with aspirin) reduced the vascular mortality rate by 15% in patients with acute or previous vascular disease or some other predisposing condition. Thus, aspirin has already been shown to be effective as primary prevention (ie, in patients at risk but without established vascular disease) and as secondary prevention (ie, in those with established disease).7,8
Yet many patients have significant vascular events in spite of taking aspirin.6 Aspirin failure is thought to be multifactorial, with causes that include weak platelet inhibition, noncompliance, discontinuation due to adverse effects (including severe bleeding), and drug interactions. In addition, aspirin resistance has been linked to worse prognosis and may prove to be another cause of aspirin failure.9–11
Clopidogrel, an adenosine diphosphate (ADP) receptor antagonist, has also been studied extensively as an antiplatelet agent.5,12 Several studies have indicated that clopidogrel and ticlopidine (Ticlid, a related drug) may be more potent than aspirin, both in the test tube and in real patients.13–15
KEY TRIALS LEADING TO CHARISMA

- Clopidogrel is more effective and slightly safer than aspirin as secondary prevention, as shown in the Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial.16–21
- The combination of clopidogrel plus aspirin is more beneficial than placebo plus aspirin in patients with acute coronary syndromes, as shown in the Clopidogrel in Unstable Angina to Prevent Recurrent Ischemic Events (CURE) trial,22–24 the Clopidogrel as Adjunctive Reperfusion Therapy-Thrombolysis in Myo-car-dial Infarction (CLARITY-TIMI 28) trial,25 and the Clopidogrel and Metoprolol in Myocardial Infarction Trial (COMMIT).26
- The combination of clopidogrel plus aspirin is beneficial in patients undergoing percutaneous coronary interventions, with or without drug-eluting stent placement,27–30 as shown in the Clopidogrel for the Reduction of Events During Observation (CREDO) trial,28 the Effect of Clopidogrel Pretreatment Before Percutaneous Coronary Intervention in Patients With ST-Elevation Myocardial Infarction With Fibrinolytics (PCI-CLARITY) study,29 and the Effects of Pre-treatment With Clopidogrel and Aspirin Followed by Long-term Therapy in Patients Undergoing Percutaneous Coronary Intervention (PCI-CURE) study.30 In fact, most patients undergoing percutaneous interventions now receive a loading dose of clopidogrel before the procedure and continue to take it for up to 1 year afterward. However, the ideal long-term duration of clopidogrel treatment is still under debate.
In view of these previous studies, we wanted to test dual antiplatelet therapy in a broader population at high risk of atherothrombosis, ie, in patients with either established vascular disease or with multiple risk factors for it.
CHARISMA STUDY DESIGN
CHARISMA was a prospective, randomized, double-blind, placebo-controlled study of the efficacy and safety of clopidogrel plus aspirin vs placebo plus aspirin in patients at high risk of cardiovascular events.
A total of 15,603 patients, all older than 45 years, were randomly assigned to receive clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin, in addition to standard therapy as directed by individual clinicians (eg, statins, beta-blockers). Patients were followed up at 1, 3, and 6 months and every 6 months thereafter until study completion, which occurred after 1,040 primary efficacy end points. The median duration of follow-up was 28 months.1

Patients had to have one of the following to be included: multiple atherothrombotic risk factors, documented coronary disease, documented cerebrovascular disease, or documented peripheral arterial disease (Table 2). Specific exclusion criteria included the use of oral antithrombotic or chronic nonsteroidal anti-inflammatory medications.1
End points
The primary end point was the combined incidence of the first episode of myocardial infarction or stroke, or death from cardiovascular causes.
The secondary end point was the combined incidence of myocardial infarction, stroke, death from cardiovascular causes, or hospitalization for unstable angina, a transient ischemic attack, or revascularization procedure.
The primary safety end point was severe bleeding, as defined in the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) study31 as intracranial hemorrhage, fatal bleeding, or bleeding leading to hemody-namic compromise. Moderate bleeding was defined as bleeding that required transfusion but did not meet the GUSTO definition of severe bleeding.
OVERALL, NO BENEFIT
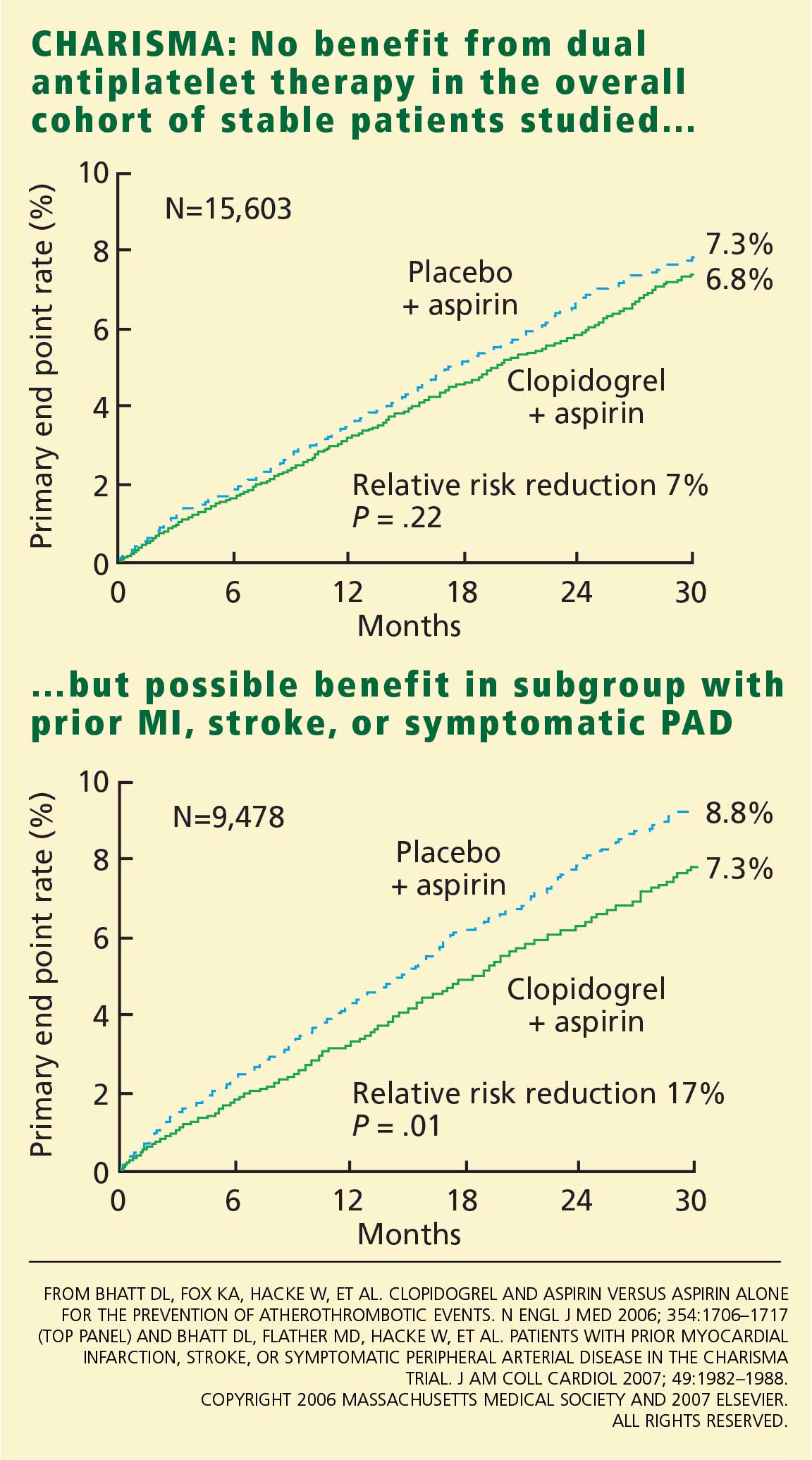
The rates of the secondary end point were 16.7% vs 17.9% (absolute risk reduction 1.2%; relative risk reduction 8%; P = .04).
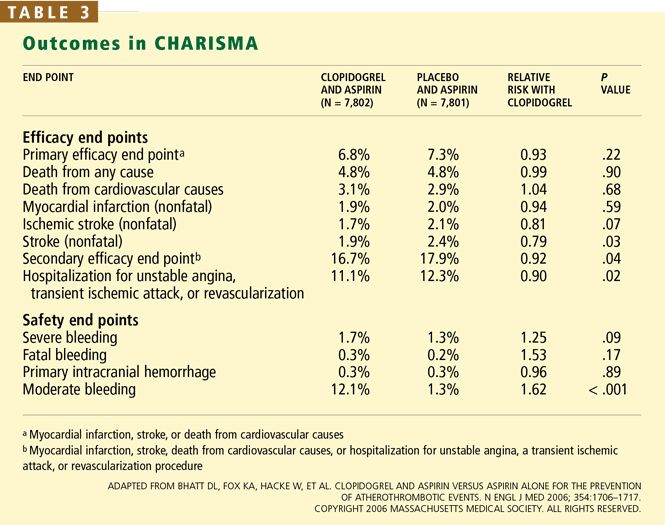
Possible benefit in symptomatic patients
In a prespecified analysis, patients were classified as being “symptomatic” (having documented cardiovascular disease, ie, coronary, cerebrovascular, or symptomatic peripheral arterial disease) or “asymptomatic” (having multiple risk factors without established cardiovascular disease).1
In the symptomatic group (n = 12,153), the primary end point was reached in 6.9% of patients treated with clopidogrel vs 7.9% with placebo (absolute risk reduction 1.0%; relative risk reduction 13%; P = .046). The 3,284 asymptomatic patients showed no benefit; the rate of the primary end point for the clopido-grel group was 6.6% vs 5.5% in the placebo group (P = .20).
In a post hoc analysis, we examined the data from 9,478 patients who were similar to those in the CAPRIE study (ie, with documented prior myocardial infarction, prior ischemic stroke, or symptomatic peripheral arterial disease). The rate of cardiovascular death, myocardial infarction, or stroke was 8.8% in the placebo-plus-aspirin group and 7.3% in the clopidogrel-plus-aspirin group (absolute risk reduction 1.5%; relative risk reduction 17%; P = .01; Figure 1).2
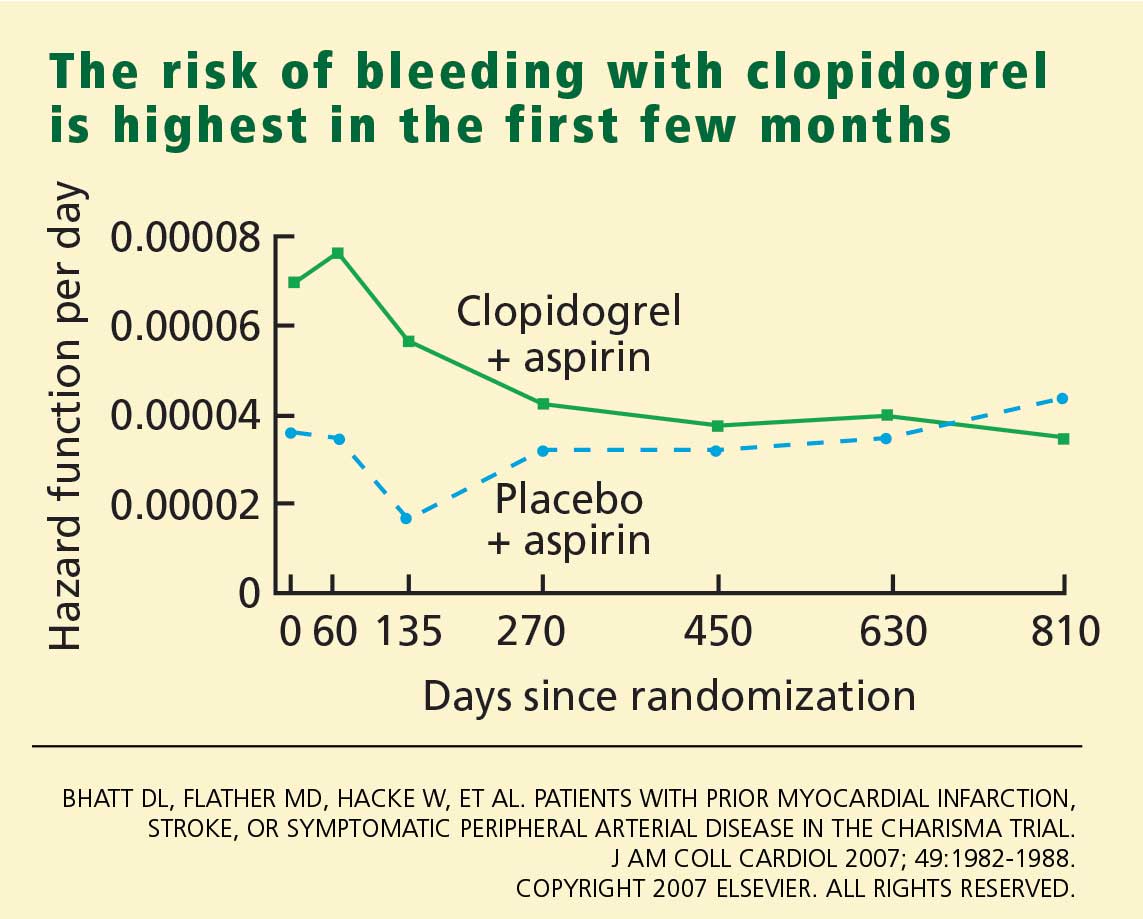
HOW SHOULD WE INTERPRET THESE FINDINGS?
CHARISMA was the first trial to evaluate whether adding clopidogrel to aspirin therapy would reduce the rates of vascular events and death from cardiovascular causes in stable patients at risk of ischemic events. As in other trials, the benefit of clopidogrel-plus-aspirin therapy was weighed against the risk of bleeding with this regimen. How are we to interpret the findings?
- In the group with multiple risk factors but without clearly documented cardiovascular disease, there was no benefit—and there was an increase in moderate bleeding. Given these findings, physicians should not prescribe dual antiplatelet therapy for primary prevention in patients without known vascular disease.
- A potential benefit was seen in a prespecified subgroup who had documented cardiovascular disease. Given the limitations of subgroup analysis, however, and given the increased risk of moderate bleeding, this positive result should be interpreted with some degree of caution.
- CHARISMA suggests that there may be benefit of protracted dual antiplatelet therapy in stable patients with documented prior ischemic events.
A possible reason for the observed lack of benefit in the overall cohort but the positive results in the subgroups with established vascular disease is that plaque rupture and thrombosis may be a precondition for dual antiplatelet therapy to work.
Another possibility is that, although we have been saying that diabetes mellitus (one of the possible entry criteria in CHARISMA) is a “coronary risk equivalent,” this may not be absolutely true. Although it had been demonstrated that patients with certain risk factors, such as diabetes, have an incidence of ischemic events similar to that in patients with prior MI and should be considered for antiplatelet therapy to prevent vascular events,32 more recent data have shown that patients with prior ischemic events are at much higher risk than patients without ischemic events, even if the latter have diabetes.33,34
- The observation in CHARISMA that the incremental bleeding risk of dual antiplatelet therapy vs aspirin does not persist beyond a year in patients who have tolerated therapy for a year without a bleeding event may affect the decision to continue clopidogrel beyond 1 year, such as in patients with acute coronary syndromes or patients who have received drug-eluting stents.35,36
- Another important consideration is cost-effectiveness. Several studies have analyzed the impact of cost and found clopidogrel to be cost-effective by preventing ischemic events and adding years of life.37,38 A recent analysis from CHARISMA also shows cost-effectiveness in the subgroup of patients enrolled with established cardiovascular disease.39 Once clopidogrel becomes generic, the cost-effectiveness will become even better.
Further studies should better define which stable patients with cardiovascular disease should be on more than aspirin alone.
- Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med 2002; 8:1227–1234.
- Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol 2005; 46:937–954.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sanmuganathan PS, Ghahramani P, Jackson PR, Wallis EJ, Ramsay LE. Aspirin for primary prevention of coronary heart disease: safety and absolute benefit related to coronary risk derived from meta-analysis of randomised trials. Heart 2001; 85:265–271.
- Hayden M, Pignone M, Phillips C, Mulrow C. Aspirin for the primary prevention of cardiovascular events: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2002; 136:161–172.
- Helgason CM, Bolin KM, Hoff JA, et al. Development of aspirin resistance in persons with previous ischemic stroke. Stroke 1994; 25:2331–2336.
- Helgason CM, Tortorice KL, Winkler SR, et al. Aspirin response and failure in cerebral infarction. Stroke 1993; 24:345–350.
- Gum PA, Kottke-Marchant K, Poggio ED, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol 2001; 88:230–235.
- Coukell AJ, Markham A. Clopidogrel. Drugs 1997; 54:745–750.
- Humbert M, Nurden P, Bihour C, et al. Ultrastructural studies of platelet aggregates from human subjects receiving clopidogrel and from a patient with an inherited defect of an ADP-dependent pathway of platelet activation. Arterioscler Thromb Vasc Biol 1996; 16:1532–1543.
- Hass WK, Easton JD, Adams HP, et al. A randomized trial comparing ticlopidine hydrochloride with aspirin for the prevention of stroke in high-risk patients. Ticlopidine Aspirin Stroke Study Group. N Engl J Med 1989; 321:501–507.
- Savi P, Bernat A, Dumas A, Ait-Chek L, Herbert JM. Effect of aspirin and clopidogrel on platelet-dependent tissue factor expression in endothelial cells. Thromb Res 1994; 73:117–124.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopido-grel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Bhatt DL, Marso SP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Amplified benefit of clopidogrel versus aspirin in patients with diabetes mellitus. Am J Cardiol 2002; 90:625–628.
- Bhatt DL, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Reduction in the need for hospitalization for recurrent ischemic events and bleeding with clopidogrel instead of aspirin. CAPRIE investigators. Am Heart J 2000; 140:67–73.
- Bhatt DL, Topol EJ. Antiplatelet and anticoagulant therapy in the secondary prevention of ischemic heart disease. Med Clin North Am 2000; 84 1:163–179.
- Ringleb PA, Bhatt DL, Hirsch AT, Topol EJ, Hacke W Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events Investigators. Benefit of clopidogrel over aspirin is amplified in patients with a history of ischemic events. Stroke 2004; 35:528–532.
- Bhatt DL, Chew DP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Superiority of clopidogrel versus aspirin in patients with prior cardiac surgery. Circulation 2001; 103:363–368.
- Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Budaj A, Yusuf S, Mehta SR, et al. Benefit of clopidogrel in patients with acute coronary syndromes without ST-segment elevation in various risk groups. Circulation 2002; 106:1622–1626.
- Fox KA, Mehta SR, Peters R, et al. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non–ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Sabatine MS, Cannon CP, Gibson CM, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 2005; 352:1179–1189.
- Chen ZM, Jiang LX, Chen YP, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Kapadia SR, Bajzer CT, et al. Dual antiplatelet therapy with clopidogrel and aspirin after carotid artery stenting. J Invasive Cardiol 2001; 13:767–771.
- Steinhubl SR, Berger PB, Mann JT, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Sabatine MS, Cannon CP, Gibson CM, et al. Effect of clopidogrel pre-treatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Bhatt DL, Steg PG, Ohman EM, et al. International prevalence, recognition, and treatment of cardiovascular risk factors in outpatients with atherothrombosis. JAMA 2006; 295:180–189.
- Steg PG, Bhatt DL, Wilson PW, et al. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007; 297:1197–1206.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Rabbat MG, Bavry AA, Bhatt DL, Ellis SG. Understanding and minimizing late thrombosis of drug-eluting stents. Cleve Clin J Med 2007; 74:129–136.
- Gaspoz JM, Coxson PG, Goldman PA, et al. Cost effectiveness of aspirin, clopidogrel, or both for secondary prevention of coronary heart disease. N Engl J Med 2002; 346:1800–1806.
- Beinart SC, Kolm P, Veledar E, et al. Longterm cost effectiveness of early and sustained dual oral antiplatelet therapy with clopidogrel given for up to one year after percutaneous coronary intervention results: from the Clopidogrel for the Reduction of Events During Observation (CREDO) trial. J Am Coll Cardiol 2005; 46:761–769.
- Chen J, Bhatt DL, Schneider E, et al. Cost-effectiveness of clopidogrel + aspirin vs. aspirin alone for secondary prevention of cardiovascular events: results from the CHARISMA Trial Session; APS.96.1; Presentation 3855; American Heart Association Scientific Sessions; Nov 12–15, 2006; Chicago IL.
In patients at risk of myocardial infarction or stroke, two antiplatelet drugs are not always better than one. In a large recent trial,1,2 adding clopidogrel (Plavix) to aspirin therapy did not offer much benefit to a cohort of patients at risk of cardiovascular events, although a subgroup did appear to benefit: those at even higher risk because they already had a history of myocardial infarction, ischemic stroke, or peripheral arterial disease.
These were the principal findings in the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) study,1,2 in which one of us (D.L.B.) was principal investigator.
These findings further our understanding of who should receive dual antiplatelet therapy, and who would be better served with aspirin therapy alone. In this article, we discuss important studies that led up to the CHARISMA trial, review CHARISMA’s purpose and study design, and interpret its results.
PREVENTING ATHEROTHROMBOSIS BY BLOCKING PLATELETS
Platelets are key players in the atherothrom-botic process.3–5 The Antiplatelet Trialists’ Collaboration,6 in a meta-analysis of trials performed up to 1997, calculated that antiplatelet therapy (mostly with aspirin) reduced the vascular mortality rate by 15% in patients with acute or previous vascular disease or some other predisposing condition. Thus, aspirin has already been shown to be effective as primary prevention (ie, in patients at risk but without established vascular disease) and as secondary prevention (ie, in those with established disease).7,8
Yet many patients have significant vascular events in spite of taking aspirin.6 Aspirin failure is thought to be multifactorial, with causes that include weak platelet inhibition, noncompliance, discontinuation due to adverse effects (including severe bleeding), and drug interactions. In addition, aspirin resistance has been linked to worse prognosis and may prove to be another cause of aspirin failure.9–11
Clopidogrel, an adenosine diphosphate (ADP) receptor antagonist, has also been studied extensively as an antiplatelet agent.5,12 Several studies have indicated that clopidogrel and ticlopidine (Ticlid, a related drug) may be more potent than aspirin, both in the test tube and in real patients.13–15
KEY TRIALS LEADING TO CHARISMA
- Clopidogrel is more effective and slightly safer than aspirin as secondary prevention, as shown in the Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial.16–21
- The combination of clopidogrel plus aspirin is more beneficial than placebo plus aspirin in patients with acute coronary syndromes, as shown in the Clopidogrel in Unstable Angina to Prevent Recurrent Ischemic Events (CURE) trial,22–24 the Clopidogrel as Adjunctive Reperfusion Therapy-Thrombolysis in Myo-car-dial Infarction (CLARITY-TIMI 28) trial,25 and the Clopidogrel and Metoprolol in Myocardial Infarction Trial (COMMIT).26
- The combination of clopidogrel plus aspirin is beneficial in patients undergoing percutaneous coronary interventions, with or without drug-eluting stent placement,27–30 as shown in the Clopidogrel for the Reduction of Events During Observation (CREDO) trial,28 the Effect of Clopidogrel Pretreatment Before Percutaneous Coronary Intervention in Patients With ST-Elevation Myocardial Infarction With Fibrinolytics (PCI-CLARITY) study,29 and the Effects of Pre-treatment With Clopidogrel and Aspirin Followed by Long-term Therapy in Patients Undergoing Percutaneous Coronary Intervention (PCI-CURE) study.30 In fact, most patients undergoing percutaneous interventions now receive a loading dose of clopidogrel before the procedure and continue to take it for up to 1 year afterward. However, the ideal long-term duration of clopidogrel treatment is still under debate.
In view of these previous studies, we wanted to test dual antiplatelet therapy in a broader population at high risk of atherothrombosis, ie, in patients with either established vascular disease or with multiple risk factors for it.
CHARISMA STUDY DESIGN
CHARISMA was a prospective, randomized, double-blind, placebo-controlled study of the efficacy and safety of clopidogrel plus aspirin vs placebo plus aspirin in patients at high risk of cardiovascular events.
A total of 15,603 patients, all older than 45 years, were randomly assigned to receive clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin, in addition to standard therapy as directed by individual clinicians (eg, statins, beta-blockers). Patients were followed up at 1, 3, and 6 months and every 6 months thereafter until study completion, which occurred after 1,040 primary efficacy end points. The median duration of follow-up was 28 months.1
Patients had to have one of the following to be included: multiple atherothrombotic risk factors, documented coronary disease, documented cerebrovascular disease, or documented peripheral arterial disease (Table 2). Specific exclusion criteria included the use of oral antithrombotic or chronic nonsteroidal anti-inflammatory medications.1
End points
The primary end point was the combined incidence of the first episode of myocardial infarction or stroke, or death from cardiovascular causes.
The secondary end point was the combined incidence of myocardial infarction, stroke, death from cardiovascular causes, or hospitalization for unstable angina, a transient ischemic attack, or revascularization procedure.
The primary safety end point was severe bleeding, as defined in the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) study31 as intracranial hemorrhage, fatal bleeding, or bleeding leading to hemody-namic compromise. Moderate bleeding was defined as bleeding that required transfusion but did not meet the GUSTO definition of severe bleeding.
OVERALL, NO BENEFIT
The rates of the secondary end point were 16.7% vs 17.9% (absolute risk reduction 1.2%; relative risk reduction 8%; P = .04).
Possible benefit in symptomatic patients
In a prespecified analysis, patients were classified as being “symptomatic” (having documented cardiovascular disease, ie, coronary, cerebrovascular, or symptomatic peripheral arterial disease) or “asymptomatic” (having multiple risk factors without established cardiovascular disease).1
In the symptomatic group (n = 12,153), the primary end point was reached in 6.9% of patients treated with clopidogrel vs 7.9% with placebo (absolute risk reduction 1.0%; relative risk reduction 13%; P = .046). The 3,284 asymptomatic patients showed no benefit; the rate of the primary end point for the clopido-grel group was 6.6% vs 5.5% in the placebo group (P = .20).
In a post hoc analysis, we examined the data from 9,478 patients who were similar to those in the CAPRIE study (ie, with documented prior myocardial infarction, prior ischemic stroke, or symptomatic peripheral arterial disease). The rate of cardiovascular death, myocardial infarction, or stroke was 8.8% in the placebo-plus-aspirin group and 7.3% in the clopidogrel-plus-aspirin group (absolute risk reduction 1.5%; relative risk reduction 17%; P = .01; Figure 1).2
HOW SHOULD WE INTERPRET THESE FINDINGS?
CHARISMA was the first trial to evaluate whether adding clopidogrel to aspirin therapy would reduce the rates of vascular events and death from cardiovascular causes in stable patients at risk of ischemic events. As in other trials, the benefit of clopidogrel-plus-aspirin therapy was weighed against the risk of bleeding with this regimen. How are we to interpret the findings?
- In the group with multiple risk factors but without clearly documented cardiovascular disease, there was no benefit—and there was an increase in moderate bleeding. Given these findings, physicians should not prescribe dual antiplatelet therapy for primary prevention in patients without known vascular disease.
- A potential benefit was seen in a prespecified subgroup who had documented cardiovascular disease. Given the limitations of subgroup analysis, however, and given the increased risk of moderate bleeding, this positive result should be interpreted with some degree of caution.
- CHARISMA suggests that there may be benefit of protracted dual antiplatelet therapy in stable patients with documented prior ischemic events.
A possible reason for the observed lack of benefit in the overall cohort but the positive results in the subgroups with established vascular disease is that plaque rupture and thrombosis may be a precondition for dual antiplatelet therapy to work.
Another possibility is that, although we have been saying that diabetes mellitus (one of the possible entry criteria in CHARISMA) is a “coronary risk equivalent,” this may not be absolutely true. Although it had been demonstrated that patients with certain risk factors, such as diabetes, have an incidence of ischemic events similar to that in patients with prior MI and should be considered for antiplatelet therapy to prevent vascular events,32 more recent data have shown that patients with prior ischemic events are at much higher risk than patients without ischemic events, even if the latter have diabetes.33,34
- The observation in CHARISMA that the incremental bleeding risk of dual antiplatelet therapy vs aspirin does not persist beyond a year in patients who have tolerated therapy for a year without a bleeding event may affect the decision to continue clopidogrel beyond 1 year, such as in patients with acute coronary syndromes or patients who have received drug-eluting stents.35,36
- Another important consideration is cost-effectiveness. Several studies have analyzed the impact of cost and found clopidogrel to be cost-effective by preventing ischemic events and adding years of life.37,38 A recent analysis from CHARISMA also shows cost-effectiveness in the subgroup of patients enrolled with established cardiovascular disease.39 Once clopidogrel becomes generic, the cost-effectiveness will become even better.
Further studies should better define which stable patients with cardiovascular disease should be on more than aspirin alone.
In patients at risk of myocardial infarction or stroke, two antiplatelet drugs are not always better than one. In a large recent trial,1,2 adding clopidogrel (Plavix) to aspirin therapy did not offer much benefit to a cohort of patients at risk of cardiovascular events, although a subgroup did appear to benefit: those at even higher risk because they already had a history of myocardial infarction, ischemic stroke, or peripheral arterial disease.
These were the principal findings in the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) study,1,2 in which one of us (D.L.B.) was principal investigator.
These findings further our understanding of who should receive dual antiplatelet therapy, and who would be better served with aspirin therapy alone. In this article, we discuss important studies that led up to the CHARISMA trial, review CHARISMA’s purpose and study design, and interpret its results.
PREVENTING ATHEROTHROMBOSIS BY BLOCKING PLATELETS
Platelets are key players in the atherothrom-botic process.3–5 The Antiplatelet Trialists’ Collaboration,6 in a meta-analysis of trials performed up to 1997, calculated that antiplatelet therapy (mostly with aspirin) reduced the vascular mortality rate by 15% in patients with acute or previous vascular disease or some other predisposing condition. Thus, aspirin has already been shown to be effective as primary prevention (ie, in patients at risk but without established vascular disease) and as secondary prevention (ie, in those with established disease).7,8
Yet many patients have significant vascular events in spite of taking aspirin.6 Aspirin failure is thought to be multifactorial, with causes that include weak platelet inhibition, noncompliance, discontinuation due to adverse effects (including severe bleeding), and drug interactions. In addition, aspirin resistance has been linked to worse prognosis and may prove to be another cause of aspirin failure.9–11
Clopidogrel, an adenosine diphosphate (ADP) receptor antagonist, has also been studied extensively as an antiplatelet agent.5,12 Several studies have indicated that clopidogrel and ticlopidine (Ticlid, a related drug) may be more potent than aspirin, both in the test tube and in real patients.13–15
KEY TRIALS LEADING TO CHARISMA
- Clopidogrel is more effective and slightly safer than aspirin as secondary prevention, as shown in the Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial.16–21
- The combination of clopidogrel plus aspirin is more beneficial than placebo plus aspirin in patients with acute coronary syndromes, as shown in the Clopidogrel in Unstable Angina to Prevent Recurrent Ischemic Events (CURE) trial,22–24 the Clopidogrel as Adjunctive Reperfusion Therapy-Thrombolysis in Myo-car-dial Infarction (CLARITY-TIMI 28) trial,25 and the Clopidogrel and Metoprolol in Myocardial Infarction Trial (COMMIT).26
- The combination of clopidogrel plus aspirin is beneficial in patients undergoing percutaneous coronary interventions, with or without drug-eluting stent placement,27–30 as shown in the Clopidogrel for the Reduction of Events During Observation (CREDO) trial,28 the Effect of Clopidogrel Pretreatment Before Percutaneous Coronary Intervention in Patients With ST-Elevation Myocardial Infarction With Fibrinolytics (PCI-CLARITY) study,29 and the Effects of Pre-treatment With Clopidogrel and Aspirin Followed by Long-term Therapy in Patients Undergoing Percutaneous Coronary Intervention (PCI-CURE) study.30 In fact, most patients undergoing percutaneous interventions now receive a loading dose of clopidogrel before the procedure and continue to take it for up to 1 year afterward. However, the ideal long-term duration of clopidogrel treatment is still under debate.
In view of these previous studies, we wanted to test dual antiplatelet therapy in a broader population at high risk of atherothrombosis, ie, in patients with either established vascular disease or with multiple risk factors for it.
CHARISMA STUDY DESIGN
CHARISMA was a prospective, randomized, double-blind, placebo-controlled study of the efficacy and safety of clopidogrel plus aspirin vs placebo plus aspirin in patients at high risk of cardiovascular events.
A total of 15,603 patients, all older than 45 years, were randomly assigned to receive clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin, in addition to standard therapy as directed by individual clinicians (eg, statins, beta-blockers). Patients were followed up at 1, 3, and 6 months and every 6 months thereafter until study completion, which occurred after 1,040 primary efficacy end points. The median duration of follow-up was 28 months.1
Patients had to have one of the following to be included: multiple atherothrombotic risk factors, documented coronary disease, documented cerebrovascular disease, or documented peripheral arterial disease (Table 2). Specific exclusion criteria included the use of oral antithrombotic or chronic nonsteroidal anti-inflammatory medications.1
End points
The primary end point was the combined incidence of the first episode of myocardial infarction or stroke, or death from cardiovascular causes.
The secondary end point was the combined incidence of myocardial infarction, stroke, death from cardiovascular causes, or hospitalization for unstable angina, a transient ischemic attack, or revascularization procedure.
The primary safety end point was severe bleeding, as defined in the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) study31 as intracranial hemorrhage, fatal bleeding, or bleeding leading to hemody-namic compromise. Moderate bleeding was defined as bleeding that required transfusion but did not meet the GUSTO definition of severe bleeding.
OVERALL, NO BENEFIT
The rates of the secondary end point were 16.7% vs 17.9% (absolute risk reduction 1.2%; relative risk reduction 8%; P = .04).
Possible benefit in symptomatic patients
In a prespecified analysis, patients were classified as being “symptomatic” (having documented cardiovascular disease, ie, coronary, cerebrovascular, or symptomatic peripheral arterial disease) or “asymptomatic” (having multiple risk factors without established cardiovascular disease).1
In the symptomatic group (n = 12,153), the primary end point was reached in 6.9% of patients treated with clopidogrel vs 7.9% with placebo (absolute risk reduction 1.0%; relative risk reduction 13%; P = .046). The 3,284 asymptomatic patients showed no benefit; the rate of the primary end point for the clopido-grel group was 6.6% vs 5.5% in the placebo group (P = .20).
In a post hoc analysis, we examined the data from 9,478 patients who were similar to those in the CAPRIE study (ie, with documented prior myocardial infarction, prior ischemic stroke, or symptomatic peripheral arterial disease). The rate of cardiovascular death, myocardial infarction, or stroke was 8.8% in the placebo-plus-aspirin group and 7.3% in the clopidogrel-plus-aspirin group (absolute risk reduction 1.5%; relative risk reduction 17%; P = .01; Figure 1).2
HOW SHOULD WE INTERPRET THESE FINDINGS?
CHARISMA was the first trial to evaluate whether adding clopidogrel to aspirin therapy would reduce the rates of vascular events and death from cardiovascular causes in stable patients at risk of ischemic events. As in other trials, the benefit of clopidogrel-plus-aspirin therapy was weighed against the risk of bleeding with this regimen. How are we to interpret the findings?
- In the group with multiple risk factors but without clearly documented cardiovascular disease, there was no benefit—and there was an increase in moderate bleeding. Given these findings, physicians should not prescribe dual antiplatelet therapy for primary prevention in patients without known vascular disease.
- A potential benefit was seen in a prespecified subgroup who had documented cardiovascular disease. Given the limitations of subgroup analysis, however, and given the increased risk of moderate bleeding, this positive result should be interpreted with some degree of caution.
- CHARISMA suggests that there may be benefit of protracted dual antiplatelet therapy in stable patients with documented prior ischemic events.
A possible reason for the observed lack of benefit in the overall cohort but the positive results in the subgroups with established vascular disease is that plaque rupture and thrombosis may be a precondition for dual antiplatelet therapy to work.
Another possibility is that, although we have been saying that diabetes mellitus (one of the possible entry criteria in CHARISMA) is a “coronary risk equivalent,” this may not be absolutely true. Although it had been demonstrated that patients with certain risk factors, such as diabetes, have an incidence of ischemic events similar to that in patients with prior MI and should be considered for antiplatelet therapy to prevent vascular events,32 more recent data have shown that patients with prior ischemic events are at much higher risk than patients without ischemic events, even if the latter have diabetes.33,34
- The observation in CHARISMA that the incremental bleeding risk of dual antiplatelet therapy vs aspirin does not persist beyond a year in patients who have tolerated therapy for a year without a bleeding event may affect the decision to continue clopidogrel beyond 1 year, such as in patients with acute coronary syndromes or patients who have received drug-eluting stents.35,36
- Another important consideration is cost-effectiveness. Several studies have analyzed the impact of cost and found clopidogrel to be cost-effective by preventing ischemic events and adding years of life.37,38 A recent analysis from CHARISMA also shows cost-effectiveness in the subgroup of patients enrolled with established cardiovascular disease.39 Once clopidogrel becomes generic, the cost-effectiveness will become even better.
Further studies should better define which stable patients with cardiovascular disease should be on more than aspirin alone.
- Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med 2002; 8:1227–1234.
- Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol 2005; 46:937–954.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sanmuganathan PS, Ghahramani P, Jackson PR, Wallis EJ, Ramsay LE. Aspirin for primary prevention of coronary heart disease: safety and absolute benefit related to coronary risk derived from meta-analysis of randomised trials. Heart 2001; 85:265–271.
- Hayden M, Pignone M, Phillips C, Mulrow C. Aspirin for the primary prevention of cardiovascular events: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2002; 136:161–172.
- Helgason CM, Bolin KM, Hoff JA, et al. Development of aspirin resistance in persons with previous ischemic stroke. Stroke 1994; 25:2331–2336.
- Helgason CM, Tortorice KL, Winkler SR, et al. Aspirin response and failure in cerebral infarction. Stroke 1993; 24:345–350.
- Gum PA, Kottke-Marchant K, Poggio ED, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol 2001; 88:230–235.
- Coukell AJ, Markham A. Clopidogrel. Drugs 1997; 54:745–750.
- Humbert M, Nurden P, Bihour C, et al. Ultrastructural studies of platelet aggregates from human subjects receiving clopidogrel and from a patient with an inherited defect of an ADP-dependent pathway of platelet activation. Arterioscler Thromb Vasc Biol 1996; 16:1532–1543.
- Hass WK, Easton JD, Adams HP, et al. A randomized trial comparing ticlopidine hydrochloride with aspirin for the prevention of stroke in high-risk patients. Ticlopidine Aspirin Stroke Study Group. N Engl J Med 1989; 321:501–507.
- Savi P, Bernat A, Dumas A, Ait-Chek L, Herbert JM. Effect of aspirin and clopidogrel on platelet-dependent tissue factor expression in endothelial cells. Thromb Res 1994; 73:117–124.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopido-grel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Bhatt DL, Marso SP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Amplified benefit of clopidogrel versus aspirin in patients with diabetes mellitus. Am J Cardiol 2002; 90:625–628.
- Bhatt DL, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Reduction in the need for hospitalization for recurrent ischemic events and bleeding with clopidogrel instead of aspirin. CAPRIE investigators. Am Heart J 2000; 140:67–73.
- Bhatt DL, Topol EJ. Antiplatelet and anticoagulant therapy in the secondary prevention of ischemic heart disease. Med Clin North Am 2000; 84 1:163–179.
- Ringleb PA, Bhatt DL, Hirsch AT, Topol EJ, Hacke W Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events Investigators. Benefit of clopidogrel over aspirin is amplified in patients with a history of ischemic events. Stroke 2004; 35:528–532.
- Bhatt DL, Chew DP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Superiority of clopidogrel versus aspirin in patients with prior cardiac surgery. Circulation 2001; 103:363–368.
- Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Budaj A, Yusuf S, Mehta SR, et al. Benefit of clopidogrel in patients with acute coronary syndromes without ST-segment elevation in various risk groups. Circulation 2002; 106:1622–1626.
- Fox KA, Mehta SR, Peters R, et al. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non–ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Sabatine MS, Cannon CP, Gibson CM, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 2005; 352:1179–1189.
- Chen ZM, Jiang LX, Chen YP, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Kapadia SR, Bajzer CT, et al. Dual antiplatelet therapy with clopidogrel and aspirin after carotid artery stenting. J Invasive Cardiol 2001; 13:767–771.
- Steinhubl SR, Berger PB, Mann JT, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Sabatine MS, Cannon CP, Gibson CM, et al. Effect of clopidogrel pre-treatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Bhatt DL, Steg PG, Ohman EM, et al. International prevalence, recognition, and treatment of cardiovascular risk factors in outpatients with atherothrombosis. JAMA 2006; 295:180–189.
- Steg PG, Bhatt DL, Wilson PW, et al. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007; 297:1197–1206.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Rabbat MG, Bavry AA, Bhatt DL, Ellis SG. Understanding and minimizing late thrombosis of drug-eluting stents. Cleve Clin J Med 2007; 74:129–136.
- Gaspoz JM, Coxson PG, Goldman PA, et al. Cost effectiveness of aspirin, clopidogrel, or both for secondary prevention of coronary heart disease. N Engl J Med 2002; 346:1800–1806.
- Beinart SC, Kolm P, Veledar E, et al. Longterm cost effectiveness of early and sustained dual oral antiplatelet therapy with clopidogrel given for up to one year after percutaneous coronary intervention results: from the Clopidogrel for the Reduction of Events During Observation (CREDO) trial. J Am Coll Cardiol 2005; 46:761–769.
- Chen J, Bhatt DL, Schneider E, et al. Cost-effectiveness of clopidogrel + aspirin vs. aspirin alone for secondary prevention of cardiovascular events: results from the CHARISMA Trial Session; APS.96.1; Presentation 3855; American Heart Association Scientific Sessions; Nov 12–15, 2006; Chicago IL.
- Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med 2002; 8:1227–1234.
- Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol 2005; 46:937–954.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Sanmuganathan PS, Ghahramani P, Jackson PR, Wallis EJ, Ramsay LE. Aspirin for primary prevention of coronary heart disease: safety and absolute benefit related to coronary risk derived from meta-analysis of randomised trials. Heart 2001; 85:265–271.
- Hayden M, Pignone M, Phillips C, Mulrow C. Aspirin for the primary prevention of cardiovascular events: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2002; 136:161–172.
- Helgason CM, Bolin KM, Hoff JA, et al. Development of aspirin resistance in persons with previous ischemic stroke. Stroke 1994; 25:2331–2336.
- Helgason CM, Tortorice KL, Winkler SR, et al. Aspirin response and failure in cerebral infarction. Stroke 1993; 24:345–350.
- Gum PA, Kottke-Marchant K, Poggio ED, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol 2001; 88:230–235.
- Coukell AJ, Markham A. Clopidogrel. Drugs 1997; 54:745–750.
- Humbert M, Nurden P, Bihour C, et al. Ultrastructural studies of platelet aggregates from human subjects receiving clopidogrel and from a patient with an inherited defect of an ADP-dependent pathway of platelet activation. Arterioscler Thromb Vasc Biol 1996; 16:1532–1543.
- Hass WK, Easton JD, Adams HP, et al. A randomized trial comparing ticlopidine hydrochloride with aspirin for the prevention of stroke in high-risk patients. Ticlopidine Aspirin Stroke Study Group. N Engl J Med 1989; 321:501–507.
- Savi P, Bernat A, Dumas A, Ait-Chek L, Herbert JM. Effect of aspirin and clopidogrel on platelet-dependent tissue factor expression in endothelial cells. Thromb Res 1994; 73:117–124.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopido-grel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Bhatt DL, Marso SP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Amplified benefit of clopidogrel versus aspirin in patients with diabetes mellitus. Am J Cardiol 2002; 90:625–628.
- Bhatt DL, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Reduction in the need for hospitalization for recurrent ischemic events and bleeding with clopidogrel instead of aspirin. CAPRIE investigators. Am Heart J 2000; 140:67–73.
- Bhatt DL, Topol EJ. Antiplatelet and anticoagulant therapy in the secondary prevention of ischemic heart disease. Med Clin North Am 2000; 84 1:163–179.
- Ringleb PA, Bhatt DL, Hirsch AT, Topol EJ, Hacke W Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events Investigators. Benefit of clopidogrel over aspirin is amplified in patients with a history of ischemic events. Stroke 2004; 35:528–532.
- Bhatt DL, Chew DP, Hirsch AT, Ringleb PA, Hacke W, Topol EJ. Superiority of clopidogrel versus aspirin in patients with prior cardiac surgery. Circulation 2001; 103:363–368.
- Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Budaj A, Yusuf S, Mehta SR, et al. Benefit of clopidogrel in patients with acute coronary syndromes without ST-segment elevation in various risk groups. Circulation 2002; 106:1622–1626.
- Fox KA, Mehta SR, Peters R, et al. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non–ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Sabatine MS, Cannon CP, Gibson CM, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med 2005; 352:1179–1189.
- Chen ZM, Jiang LX, Chen YP, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Kapadia SR, Bajzer CT, et al. Dual antiplatelet therapy with clopidogrel and aspirin after carotid artery stenting. J Invasive Cardiol 2001; 13:767–771.
- Steinhubl SR, Berger PB, Mann JT, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Sabatine MS, Cannon CP, Gibson CM, et al. Effect of clopidogrel pre-treatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Bhatt DL, Steg PG, Ohman EM, et al. International prevalence, recognition, and treatment of cardiovascular risk factors in outpatients with atherothrombosis. JAMA 2006; 295:180–189.
- Steg PG, Bhatt DL, Wilson PW, et al. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007; 297:1197–1206.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Rabbat MG, Bavry AA, Bhatt DL, Ellis SG. Understanding and minimizing late thrombosis of drug-eluting stents. Cleve Clin J Med 2007; 74:129–136.
- Gaspoz JM, Coxson PG, Goldman PA, et al. Cost effectiveness of aspirin, clopidogrel, or both for secondary prevention of coronary heart disease. N Engl J Med 2002; 346:1800–1806.
- Beinart SC, Kolm P, Veledar E, et al. Longterm cost effectiveness of early and sustained dual oral antiplatelet therapy with clopidogrel given for up to one year after percutaneous coronary intervention results: from the Clopidogrel for the Reduction of Events During Observation (CREDO) trial. J Am Coll Cardiol 2005; 46:761–769.
- Chen J, Bhatt DL, Schneider E, et al. Cost-effectiveness of clopidogrel + aspirin vs. aspirin alone for secondary prevention of cardiovascular events: results from the CHARISMA Trial Session; APS.96.1; Presentation 3855; American Heart Association Scientific Sessions; Nov 12–15, 2006; Chicago IL.
KEY POINTS
- Platelets are key players in atherothrombosis, and antiplatelet drugs such as aspirin and clopidogrel prevent events in patients at risk.
- In studies leading up to CHARISMA, the combination of clopidogrel and aspirin was found to be beneficial in patients with acute coronary syndromes and in those undergoing percutaneous coronary interventions.
- Clopidogrel should not be combined with aspirin as a primary preventive therapy (ie, for people without established vascular disease). How dual antiplatelet therapy should be used as secondary prevention in stable patients needs further study.
The STAR*D study: Treating depression in the real world
Depression can be treated successfully by primary care physicians under “real-world” conditions.
Furthermore, the particular drug or drugs used are not as important as following a rational plan: giving antidepressant medications in adequate doses, monitoring the patient’s symptoms and side effects and adjusting the regimen accordingly, and switching drugs or adding new drugs to the regimen only after an adequate trial.
These are among the lessons learned from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest prospective clinical trial of treatment of major depressive disorder ever conducted. It was funded by the National Institutes of Health and directed by A. John Rush, MD.
WHAT WERE THE AIMS OF STAR*D?
Depression, a common and debilitating condition, affects approximately one in eight people in the United States.1 It is expected2 to be the second-leading cause of disability in the world by the year 2020; today, it is the second-leading cause of disability-adjusted life years in those 15 to 44 years old.3
Nevertheless, the available evidence base for treatment is limited, since most participants in clinical trials are recruited by advertisement rather than from representative practices, and they are often selected to have few comorbid disorders, either medical or psychiatric. In addition, those with chronic depression or current suicidal ideation are excluded.1,4 These uncomplicated and “pristine” participants are unlike typical patients seen by primary care physicians or psychiatrists.
Similarly, the protocols used in these trials do not represent usual clinic practice.Patients in clinical trials undergo more assessment and more frequent follow-up than in real-world practice, they have no say in treatment decisions, the doses are fixed, and the patients and physicians are blinded to the intervention. Consequently, how to translate the results of these efficacy trials into practice is unclear.5
Further, even in relatively uncomplicated cases, only about one-half of outpatients with nonpsychotic major depressive disorder initially treated with a single medication or with psychotherapy will experience a clinically significant improvement in symptoms (ie, a response) during the 8 to 12 weeks of acute-phase treatment,6–10 and only 20% to 35% of patients will reach remission,9 the aim of treatment.8,11 The remission rates are even lower in treatment-resistant depression.12 How to manage most patients—those whose depression does not remit with the first, second, or third step of treatment—is unclear.
Accordingly, the overall objective of STAR*D was to develop and evaluate feasible treatment strategies to improve clinical outcomes for real-world patients with treatment-resistant depression, who were identified prospectively from a pool of patients in a current major depressive episode.13–15 Specifically, STAR*D aimed to determine prospectively which of several treatments is the most effective “next step” for patients who do not reach remission with an initial or subsequent treatment or who cannot tolerate the treatment.
WHY IS STAR*D RELEVANT FOR PRIMARY CARE?
Nearly 10% of all primary care office visits are depression-related.16 Primary care physicians provide nearly half the outpatient care for depressed patients.17 Indeed, primary care physicians log approximately as many outpatient visits for depression as psychiatrists do.18 Medical comorbidity is especially common in primary care settings.19 When to refer to a psychiatrist is not clear.
KEY FEATURES OF THE STUDY DESIGN
STAR*D involved a national consortium of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004.
Entry criteria were broad and inclusive
Patients had to:
- Be between 18 and 75 years of age
- Have a nonpsychotic major depressive disorder, identified by a clinician and confirmed with a symptom checklist based on the Diagnostic and Statistical Manual, fourth edition revised,20 and for which antidepressant treatment is recommended
- Score at least 14 on the 17-item Hamilton Rating Scale for Depression (HAMD17)21
- Not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder, which would require a different treatment strategy, or a seizure disorder (which would preclude bupropion as a second-step treatment).
Dosing recommendations were flexible but vigorous
Medications often were increased to maximally tolerated doses. For example, citalopram (Celexa) was started at 20 mg/day and increased by 20 mg every 2 to 4 weeks if the patient was tolerating it but had not achieved remission, to a maximum dose of 60 mg/day. Treatment could be given for up to 14 weeks, during which side effects22 and clinical ratings23 were assessed by both patients and study coordinators.
Measurement-based care
We used a systematic approach to treatment called “measurement-based care,”24 which involves routinely measuring symptoms23 and side effects22 and using this information to modify the medication doses at critical decision points. This algorithmic approach provided flexible treatment recommendations to ensure that the dosage and duration of antidepressant drug treatment were adequate.25
The severity of depression was assessed by the clinician-rated, 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16). The QIDS-SR16 (the self-report version) can substitute for the QIDS-C1623 to make this approach more feasible. Both tools are available at www.ids-qids.org.
This approach was easily worked into busy primary care and specialty care office workflows (clinic physicians, most with limited research experience, provided the treatment), and could be translated into primary care practice in the community as well.
Four-step protocol
A unique feature of the study design was that the patients, in consultation with their physicians, had some choice in the treatments they received. In this “equipoise-stratified randomized design,”26 at levels 2 and 3 the patient could choose either to switch therapies (stop the current drug and be randomized to receive one of several different treatments) or to augment their current therapy (by adding one of several treatments in a randomized fashion). Patients could decline certain strategies as long as there were at least two possible options to which one might be randomized.
At level 2, one of the options for both switching and augmentation was cognitive therapy, although patients could decline that option. Conversely, if they definitely wanted cognitive therapy, they could choose to be randomized to either cognitive therapy alone or to cognitive therapy added to citalopram. Also, anyone who received cognitive therapy in level 2 and failed to enter remission was additionally randomized to either bupropion or venlafaxine (level 2a) to ensure that all patients had failed trials on two medications before entering level 3.
When switching to medications other than a monoamine oxidase inhibitor (MAOI), the clinician could choose either to stop the current medication and immediately begin the next one, or to decrease the current medication while starting the new one at a low dose and then tapering and titrating over 1 week. (Switching to an MAOI, used only in the final level of treatment, required a 7- to 10-day washout period.)
Outcomes measured
Remission (complete recovery from the depressive episode), the primary study outcome, was defined as a HAM-D17 score of 7 or less, as assessed by treatment-blinded raters.A secondary remission outcome was a QIDS-SR16 score of 5 or less. Of note, the HAM-D17 remission rates were slightly lower than the rates based on the QIDS-SR16, since patients who did not have a HAM-D17 score measured at exit were defined as not being in remission a priori. Thus, the QIDS-SR16 rates might have been a slightly better reflection of actual remission rates.
Response, a secondary outcome, was defined as a reduction of at least 50% in the QIDS-SR16 score from baseline at the last assessment.
FEW DIFFERENCES BETWEEN PSYCHIATRIC, PRIMARY CARE PATIENTS
The patients seen in primary care clinics were surprisingly similar to those seen in psychiatric clinics.27,28 The two groups did not differ in severity of depression, distribution of severity scores, the likelihood of presenting with any of the nine core criteria of a major depressive episode, or the likelihood of having a concomitant axis I psychiatric disorder in addition to depression (about half of participants in each setting had an anxiety disorder).
Recurrent major depressive disorders were common in both groups, though more so in psychiatric patients (78% vs 69%, P < .001), while chronic depression was more common in primary care than in psychiatric patients (30% vs 21%, P < .001). Having either a chronic index episode (ie, lasting > 2 years) or a recurrent major depressive disorder was common in both groups (86% vs 83%, P = .0067).
That said, primary care patients were older (44 years vs 39 years, P < .001), more of them were Hispanic (18% vs 9%, P < .001), and more of them had public insurance (23% vs 9%, P < .001). Fewer of the primary care patients had completed college (20% vs 28%, P < .001), and the primary care patients tended to have greater medical comorbidity. Psychiatric patients were more likely to have attempted suicide in the past and to have had their first depressive illness before age 18.
LEVEL 1: WHAT CAN WE EXPECT FROM INITIAL TREATMENT?
At level 1, all the patients received citalopram. The mean dose was 40.6 ± 16.6 mg/day in the primary care clinics and 42.5 ± 16.8 mg/day in the psychiatric clinics, which are adequate, middle-range doses and higher than the average US dose.29
Approximately 30% of patients achieved remission: 27% as measured on the HAM-D17 and 33% on the QIDS-SR16. The response rate (on the QIDS-SR16) was 47%. There were no differences between primary and psychiatric care settings in remission or response rates.
Patients were more likely to achieve remission if they were white, female, employed, more educated, or wealthier. Longer current episodes, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), more general medical disorders, and lower baseline function and quality of life were each associated with lower remission rates.
What is an adequate trial?
Longer times than expected were needed to reach response or remission. The average duration required to achieve remission was almost 7 weeks (44 days in primary care; 49 days in psychiatric care). Further, approximately one-third of those who ultimately responded and half of those who entered remission did so after 6 weeks.30 Forty percent of those who entered remission required 8 or more weeks to do so.
These results suggest that longer treatment durations and more vigorous medication dosing than generally used are needed to achieve optimal remission rates. It is imprudent to stop a treatment that the patient is tolerating in a robust dose if the patient reports only partial benefit by 6 weeks; indeed, raising the dose, if tolerated, may help a substantial number of patients respond by 12 or 14 weeks. Instruments to monitor depression severity (eg, self-report measures) can be useful. At least 8 weeks with at least moderately vigorous dosing is recommended.
LEVEL 2: IF THE FIRST TREATMENT FAILS
When switching to a new drug, does it matter which one?
No.
In level 2, if patients had not achieved remission on citalopram alone, they had the choice of switching: stopping citalopram and being randomized to receive either sertraline (Zoloft, another SSRI), venlafaxine extended-release (XR) (Effexor XR, a serotonin and norepinephrine reuptake inhibitor), or bupropion sustained-release (SR) (Wellbutrin SR, a norepinephrine and dopamine reuptake inhibitor). At the last visit the mean daily doses were bupropion SR 282.7 mg/day, sertraline 135.5 mg/day, and venlafaxine-XR 193.6 mg/day.
The remission rate was approximately one-fourth with all three drugs31:
- With bupropion SR—21.3% by HAM-D17, 25.5% by QIDS-SR16
- With sertraline—17.6% by HAM-D17, 26.6% by QIDS-SR16
- With venlafaxine-XR—24.8% by HAM-D17, 25.0% by QIDS-SR16. The remission rates were neither statistically nor clinically different by either measure.
Though the types of side effects related to specific medications may have varied, the overall side-effect burden and the rate of serious adverse events did not differ significantly.
When adding a new drug, does it matter which one?
Again, no.
Instead of switching, patients in level 2 could choose to stay on citalopram and be randomized to add either bupropion SR or buspirone (BuSpar) to the regimen (augmentation). The mean daily doses at the end of level 2 were bupropion SR 267.5 mg and buspirone 40.9 mg.
Rates of remission32:
- With bupropion SR—29.7% on the HAMD-D17, 39.0% on the QIDS-SR16
- With buspirone—30.1% on the HAM-D17, 32.9% on the QIDS-SR16.
However, the QIDS-SR16 scores declined significantly more with bupropion SR than with buspirone (25.3% vs 17.1%, P < .04). The mean total QIDS-SR16 score at the last visit was lower with bupropion SR (8.0) than with buspirone (9.1, P < .02), and augmentation with bupropion SR was better tolerated (the dropout rate due to intolerance was 12.5% with bupropion-SR vs 20.6% with buspirone 20.6%; P < .009).
Can we directly compare the benefits of switching vs augmenting?
No.
Patients could choose whether to switch from citalopram to another drug or to add another drug at the second treatment level.33 Consequently, we could not ensure that the patient groups were equivalent at the point of randomization at the beginning of level 2, and, indeed, they were not.
Those who benefitted more from citalopram treatment and who better tolerated it preferred augmentation, while those who benefitted little or who could not tolerate it preferred to switch. Consequently, those in the augmentation group at level 2 were somewhat less depressed than those who switched. Whether augmentation is better even if the initial treatment is minimally effective could not be evaluated in STAR*D.
What about cognitive therapy?
There was no difference between cognitive therapy (either as a switch or as augmentation) and medication (as a switch or as augmentation).34 Adding another drug was more rapidly effective than adding cognitive therapy. Switching to cognitive therapy was better tolerated than switching to a different antidepressant.
Of note, fewer patients accepted cognitive therapy as a randomization option than we expected, so the sample sizes were small. Possible reasons were that all patients had to receive a medication at study entry (which may have biased selection towards those preferring medication), and cognitive therapy entailed additional copayments and visiting still another provider at another site.
After two levels of treatment, how many patients reach remission?
About 30% of patients in level 1 achieved remission, and of those progressing to level 2, another 30% achieved remission. Together, this adds up to about 50% of patients achieving remission if they remained in treatment (30% in level 1 plus 30% of the roughly 70% remaining in level 2).
IF A SECOND TREATMENT FAILS
If switching again to another drug, does it matter which one?
No.
In level 3, patients could choose to stop the drug they had been taking and be randomized to receive either mirtazapine (Remeron) or nortriptyline (Pamelor).
Switching medications was not as effective as a third step as it was as a second step.35
Remission rates:
- With mirtazapine—12.3% on the HAM-D17, 8.0% on the QIDS-SR16
- With nortriptyline—19.8% on the HAM-D17, 12.4% on the QIDS-SR16.
Response rates were 13.4% with mirtazapine and 16.5% with nortriptyline. Statistically, neither the response nor the remission rates differed by treatment, nor did these two treatments differ in tolerability or side-effect burden.
Does choice of augmentation agent matter: Lithium vs T3?
Similarly, after two failed medication treatments, medication augmentation was less effective than it was at the second step.36 The two augmentation options tested, lithium and T3 thyroid hormone (Cytomel), are commonly considered by psychiatrists but less commonly used by primary care doctors.
Lithium is believed to increase serotonergic function, which may have a synergistic effect on the mechanism of action of antidepressants; a meta-analysis of placebo-controlled studies supports lithium’s effectiveness as adjunctive treatment.37 Its side effects, however, must be closely monitored.38 The primary monitoring concern is the small difference between the therapeutic blood level (0.6–1.2 mEq/L) and potentially toxic blood levels (> 1.5 mEq/L).
Lithium was started at 450 mg/day, and at week 2 it was increased to the recommended dose of 900 mg/day (a dose below the target dose for bipolar disorder). If patients could not tolerate 450 mg/day, the initial dose was 225 mg/day for 1 week before being increased to 450 mg/day, still with the target dose of 900 mg/day. The mean exit dose was 859.9 mg/day, and the median blood level was 0.6 mEq/L.
Thyroid hormone augmentation using T3 is believed to work through both direct and indirect effects on the hypothalamic-pituitary-thyroid axis, which has a strong relationship with depression. The efficacy of T3 augmentation is supported by a meta-analysis of eight studies,39 and T3 is effective whether or not thyroid abnormalities are present.
In STAR*D, T3 was started at 25 μg/day for 1 week, than increased to the recommended dose of 50 μg/day. The mean exit dose was 45.2 μg/day.
Remission rates:
- With lithium augmentation—15.9% by the HAM-D17, 13.2% by the QIDS-SR16
- With T3 augmentation—24.7% by both measures.
Response rates were 16.2% with lithium augmentation and 23.3% with T3 augmentation.
While neither response nor remission rates were statistically significantly different by treatment, lithium was more frequently associated with side effects (P = .045), and more participants in the lithium group left treatment because of side effects (23.2% vs 9.6%; P = .027). These results suggest that in cases in which a clinician is considering an augmentation trial, T3 has slight advantages over lithium in effectiveness and tolerability. T3 also offers the advantages of being easy to use and not necessitating blood level monitoring. These latter benefits are especially relevant to the primary care physician. However, T3’s potential for long-term side effects (eg, osteoporosis, cardiovascular effects) were not examined, and it is not clear when to discontinue it.
LEVEL 4: AFTER THREE FAILURES, HOW SHOULD A CLINICIAN PROCEED?
Switch to mirtazapine plus venlafaxine XR or tranylcypromine?
Patients who reached level 4 were considered to have a highly treatment-resistant depressive illness, so treatments at this level were, by design, more aggressive. Accordingly, at level 4 we investigated treatments that might be considered more demanding than those a primary care physician would use. Approximately 40% of patients in each treatment group were from primary care settings.
Remission rates40:
- With the combination of mirtazapine (mean dose 35.7 mg/day) and venlafaxine XR (mean dose 210.3 mg/day)—13.7% by the HAM-D17 and 15.7% by the QIDS-SR16
- With the MAOI tranylcypromine (Parnate, mean dose 36.9 mg/day)—6.9% by the HAM-D17 and 13.8% by the QIDS-SR16. Response rates were 23.5% with the combination and 12.1% with tranylcypromine. Neither remission nor response rates differed significantly.
However, the percentage reduction in QIDS-SR16 score between baseline and exit was greater with the combination than with tranylcypromine. Further, more patients dropped out of treatment with tranylcypromine because of side effects (P < .03). Tranylcypromine also has the disadvantage of necessitating dietary restrictions.
A significant limitation of this comparison is that patients were less likely to get an adequate trial of tranylcypromine, an MAOI, than of the combination. When the 2-week washout period (required before switching to an MAOI) is subtracted from the total time in treatment, approximately 30% of participants in the tranylcypromine group had less than 2 weeks of treatment, and nearly half had less than 6 weeks of treatment.
Therefore, even though the remission and response rates were similar between groups, the combination of venlafaxine-XR plus mirtazapine therapy might have some advantages over tranylcypromine. These results provided the first evidence of tolerability and at least modest efficacy of this combination for treatment-resistant cases.
Overall, what was the cumulative remission rate?
The theoretical cumulative remission rate after four acute treatment steps was 67%. Remission was more likely to occur during the first two levels of treatment than during the last two. The cumulative remission rates for the first four steps were:
- Level 1—33%
- Level 2—57%
- Level 3—63%
- Level 4—67%.
RESULTS FROM LONG-TERM FOLLOW-UP AFTER REMISSION OR RESPONSE
Patients with a clinically meaningful response or, preferably, remission at any level could enter into a 12-month observational follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase.41 Further, if a patient achieved remission rather than just response to treatment, regardless of the treatment level, the prognosis at follow-up was better, confirming the importance of remission as the goal of treatment.
Results also provided a warning—the greater the number of treatment levels that a patient required, the more likely that patient and physician would settle for response. Whether the greater relapse rates reflect a harder-to-treat depression or the naturalistic design of the follow-up phase (with less control over dosing) is unclear.
WHAT DO THESE RESULTS MEAN FOR PRIMARY CARE PHYSICIANS?
- Measurement-based care is feasible in primary care. Primary care doctors can ensure vigorous but tolerable dosing using a self-report depression scale to monitor response, a side-effects tool to monitor tolerability, and medication adjustments at critical decision points guided by these two measures.
- Remission, ie, complete recovery from a depressive episode, rather than merely substantial improvement, is associated with a better prognosis and is the preferred goal of treatment.
- Pharmacologic differences between psychotropic medications did not translate into substantial clinical differences, although tolerability differed. These findings are consistent with a large-scale systematic evidence review recently completed by the Agency for Healthcare Research and Quality that compared the effectiveness of antidepressants.42 Given the difficulty in predicting what medication will be both efficacious for and tolerated by an individual patient, familiarity with a broad spectrum of antidepressants is prudent.
- Remission of depressive episodes will most likely require repeated trials of sufficiently sustained,vigorously dosed antidepressant medication. From treatment initiation, physicians should ensure maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- If a first treatment doesn’t work, either switching or augmenting it is a reasonable choice. Augmentation may be preferred if the patient is tolerating and receiving partial benefit from the initial medication choice. While bupropion SR and buspirone were not different as augmenters by the primary remission outcome measure, secondary measures (eg, tolerability, depressive symptom change over the course of treatment, clinician-rated Quick Inventory of Depressive Symptomatology) recommended bupropion-SR over buspirone.
- If physicians switch, either a within-class switch (eg, citalopram to sertraline) or an out-of-class switch (eg, citalopram to bupropion SR) is effective, as is a switch to a dual-action agent (eg, venlafaxine XR).
- The likelihood of improvement after two aggressive medication trials is very low and likely requires more complicated medication regimens, and the existing evidence base is quite thin. These primary care patients should likely be referred to psychiatrists for more aggressive and intensive treatment.
- For patients who present with major depressive disorder, STAR*D suggests that with persistence and aggressive yet feasible care, there is hope: after one round, approximately 30% will have a remission; after two rounds, 50%; after three rounds, 60%; and after four rounds, 70%.
- While STAR*D excluded depressed patients with bipolar disorder, a depressive episode in a patient with bipolar disorder can be difficult to distinguish from a depressive episode in a patient with major depressive disorder. Primary care physicians need to consider bipolar disorder both in patients presenting with a depressive episode and in those who fail an adequate trial.43
FUTURE CONSIDERATIONS
Subsequent STAR*D analyses will compare in greater depth outcomes in primary care vs psychiatric settings at each level of treatment. Given the greater risk of depression persistence associated with more successive levels of treatment, subsequent research will focus on ways to more successfully treat depression in the earlier stages, possibly through medication combinations earlier in treatment (somewhat analogous to a “broad-spectrum antibiotic” approach for infections).
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Zimmerman M, Chelminski I, Posternak MA. Generalizability of antidepressant efficacy trials: differences between depressed psychiatric outpatients who would or would not qualify for an efficacy trial. Am J Psychiatry 2005; 162:1370–1372.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply? Lancet 2005; 365:82–93.
- Depression Guideline Panel. Depression in primary care: Volume 1, diagnosis and detection. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Frank E, Karp J, Rush A. Efficacy of treatments for major depression. Psychopharmacol Bull 1993; 29:457–475.
- Depression Guideline Panel. Depression in primary care: Volume 2, Treatment of major depression. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19:179–200.
- Jarrett RB, Rush A. Short-term psychotherapy of depressive disorders: current status and future directions. Psychiatry: Interpers Biol Process 1994; 57:115–132.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depression (revision). Am J Psychiatry 2000; 157(suppl 4):1–45.
- Dunner DL, Rush AJ, Russell JM, et al. Prospective, long-term, multicenter study of the naturalistic outcomes of patients with treatment-resistant depression. J Clin Psychiatry 2006; 67:688–695.
- Fava M, Rush A, Trivedi M, et al. Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494.
- Gaynes B, Davis L, Rush A, Trivedi M, Fava M, Wisniewski S. The aims and design of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Prim Psychiatry 2005; 12:36–41.
- Rush A, Fava M, Wisniewski S, et al. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142.
- Stafford RS, Ausiello JC, Misra B, Saglam D. National Patterns o fDepression Treatment in Primary Care. Prim Care Companion J Clin Psychiatry 2000; 2:211–216.
- Regier D, Narrow W, Rae D, Mandersheid R, Locke B, Goodwin F. The de facto US mental and addictive disorders service system: epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry 1993; 50:85–94.
- Pincus H, Tanielian T, Marcus S, et al. Prescribing trends in psychotropic medications: primary care, psychiatry, and other medical specialities. JAMA 1998; 279:526–531.
- Vuorilehto M, Melartin T, Isometsa E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med 2005; 35:673–682.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–61.
- Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA for the STAR*D Investigators. Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatric Pract 2006; 12:71–79.
- Rush AJ, Bernstein IH, Trivedi MH, et al. An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial report. Biol Psychiatry 2006; 59:493–501.
- Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR*D measurement-based care. Neuropsychopharmacology 2007/04/04/online 2007.
- Kawamoto K, Houlihan CA, Balas EA, Lobach DF. Improving clinical practice using clinical decision support systems: a systematic review of trials to identify features critical to success. BMJ 2005; 330:765 e-pub March 14 2005.
- Lavori P, Rush A, Wisniewski S, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001; 50:792–801.
- Gaynes BN, Rush AJ, Trivedi MH, et al. Major depression symptoms in primary care and psychiatric care settings: a cross-sectional analysis. Ann Fam Med 2007; 5:126–134.
- Gaynes BN, Rush AJ, Trivedi M, et al. A direct comparison of presenting characteristics of depressed outpatients from primary vs. specialty care settings: preliminary findings from the STAR*D clinical trial. Gen Hosp Psychiatry 2005; 27:87–96.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004; 18:911–932.
- Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006; 354:1231–1242.
- Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006; 354:1243–1252.
- Wisniewski SR, Fava M, Trivedi MH, et al. Acceptability of second-step treatments to depressed outpatients: a STAR*D report. Am J Psychiatry 2007; 164:753–760.
- Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry 2007; 164:739–752.
- Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006; 163:1161–1172.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry 2006; 163:1519–1530.
- Bschor T, Lewitzka U, Sasse J, Adli M, Koberle U, Bauer M. Lithium augmentation in treatment-resistant depression: clinical evidence, serotonergic and endocrine mechanisms. Pharmacopsychiatry 2003; 36(suppl 3):S230–S234.
- Freeman MP, Freeman SA. Lithium: clinical considerations in internal medicine. Am J Med 2006; 119:478–481.
- Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996; 53:842–848.
- McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry 2006; 163:1531–1541.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Gartlehner G, Hansen R, Thieda P, et al. Comparative Effectiveness of Second-generation Antidepressants in the Pharmacologic Treatment of Depression. Agency for Healthcare Research and Quality. http://effectivehealthcare.ahrq.gov/reports/topic.cfm?topic=8&sid=39&rType=3. Accessed December 12, 2007.
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293:956–963.
Depression can be treated successfully by primary care physicians under “real-world” conditions.
Furthermore, the particular drug or drugs used are not as important as following a rational plan: giving antidepressant medications in adequate doses, monitoring the patient’s symptoms and side effects and adjusting the regimen accordingly, and switching drugs or adding new drugs to the regimen only after an adequate trial.
These are among the lessons learned from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest prospective clinical trial of treatment of major depressive disorder ever conducted. It was funded by the National Institutes of Health and directed by A. John Rush, MD.
WHAT WERE THE AIMS OF STAR*D?
Depression, a common and debilitating condition, affects approximately one in eight people in the United States.1 It is expected2 to be the second-leading cause of disability in the world by the year 2020; today, it is the second-leading cause of disability-adjusted life years in those 15 to 44 years old.3
Nevertheless, the available evidence base for treatment is limited, since most participants in clinical trials are recruited by advertisement rather than from representative practices, and they are often selected to have few comorbid disorders, either medical or psychiatric. In addition, those with chronic depression or current suicidal ideation are excluded.1,4 These uncomplicated and “pristine” participants are unlike typical patients seen by primary care physicians or psychiatrists.
Similarly, the protocols used in these trials do not represent usual clinic practice.Patients in clinical trials undergo more assessment and more frequent follow-up than in real-world practice, they have no say in treatment decisions, the doses are fixed, and the patients and physicians are blinded to the intervention. Consequently, how to translate the results of these efficacy trials into practice is unclear.5
Further, even in relatively uncomplicated cases, only about one-half of outpatients with nonpsychotic major depressive disorder initially treated with a single medication or with psychotherapy will experience a clinically significant improvement in symptoms (ie, a response) during the 8 to 12 weeks of acute-phase treatment,6–10 and only 20% to 35% of patients will reach remission,9 the aim of treatment.8,11 The remission rates are even lower in treatment-resistant depression.12 How to manage most patients—those whose depression does not remit with the first, second, or third step of treatment—is unclear.
Accordingly, the overall objective of STAR*D was to develop and evaluate feasible treatment strategies to improve clinical outcomes for real-world patients with treatment-resistant depression, who were identified prospectively from a pool of patients in a current major depressive episode.13–15 Specifically, STAR*D aimed to determine prospectively which of several treatments is the most effective “next step” for patients who do not reach remission with an initial or subsequent treatment or who cannot tolerate the treatment.
WHY IS STAR*D RELEVANT FOR PRIMARY CARE?
Nearly 10% of all primary care office visits are depression-related.16 Primary care physicians provide nearly half the outpatient care for depressed patients.17 Indeed, primary care physicians log approximately as many outpatient visits for depression as psychiatrists do.18 Medical comorbidity is especially common in primary care settings.19 When to refer to a psychiatrist is not clear.
KEY FEATURES OF THE STUDY DESIGN
STAR*D involved a national consortium of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004.
Entry criteria were broad and inclusive
Patients had to:
- Be between 18 and 75 years of age
- Have a nonpsychotic major depressive disorder, identified by a clinician and confirmed with a symptom checklist based on the Diagnostic and Statistical Manual, fourth edition revised,20 and for which antidepressant treatment is recommended
- Score at least 14 on the 17-item Hamilton Rating Scale for Depression (HAMD17)21
- Not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder, which would require a different treatment strategy, or a seizure disorder (which would preclude bupropion as a second-step treatment).
Dosing recommendations were flexible but vigorous
Medications often were increased to maximally tolerated doses. For example, citalopram (Celexa) was started at 20 mg/day and increased by 20 mg every 2 to 4 weeks if the patient was tolerating it but had not achieved remission, to a maximum dose of 60 mg/day. Treatment could be given for up to 14 weeks, during which side effects22 and clinical ratings23 were assessed by both patients and study coordinators.
Measurement-based care
We used a systematic approach to treatment called “measurement-based care,”24 which involves routinely measuring symptoms23 and side effects22 and using this information to modify the medication doses at critical decision points. This algorithmic approach provided flexible treatment recommendations to ensure that the dosage and duration of antidepressant drug treatment were adequate.25
The severity of depression was assessed by the clinician-rated, 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16). The QIDS-SR16 (the self-report version) can substitute for the QIDS-C1623 to make this approach more feasible. Both tools are available at www.ids-qids.org.
This approach was easily worked into busy primary care and specialty care office workflows (clinic physicians, most with limited research experience, provided the treatment), and could be translated into primary care practice in the community as well.
Four-step protocol
A unique feature of the study design was that the patients, in consultation with their physicians, had some choice in the treatments they received. In this “equipoise-stratified randomized design,”26 at levels 2 and 3 the patient could choose either to switch therapies (stop the current drug and be randomized to receive one of several different treatments) or to augment their current therapy (by adding one of several treatments in a randomized fashion). Patients could decline certain strategies as long as there were at least two possible options to which one might be randomized.
At level 2, one of the options for both switching and augmentation was cognitive therapy, although patients could decline that option. Conversely, if they definitely wanted cognitive therapy, they could choose to be randomized to either cognitive therapy alone or to cognitive therapy added to citalopram. Also, anyone who received cognitive therapy in level 2 and failed to enter remission was additionally randomized to either bupropion or venlafaxine (level 2a) to ensure that all patients had failed trials on two medications before entering level 3.
When switching to medications other than a monoamine oxidase inhibitor (MAOI), the clinician could choose either to stop the current medication and immediately begin the next one, or to decrease the current medication while starting the new one at a low dose and then tapering and titrating over 1 week. (Switching to an MAOI, used only in the final level of treatment, required a 7- to 10-day washout period.)
Outcomes measured
Remission (complete recovery from the depressive episode), the primary study outcome, was defined as a HAM-D17 score of 7 or less, as assessed by treatment-blinded raters.A secondary remission outcome was a QIDS-SR16 score of 5 or less. Of note, the HAM-D17 remission rates were slightly lower than the rates based on the QIDS-SR16, since patients who did not have a HAM-D17 score measured at exit were defined as not being in remission a priori. Thus, the QIDS-SR16 rates might have been a slightly better reflection of actual remission rates.
Response, a secondary outcome, was defined as a reduction of at least 50% in the QIDS-SR16 score from baseline at the last assessment.
FEW DIFFERENCES BETWEEN PSYCHIATRIC, PRIMARY CARE PATIENTS
The patients seen in primary care clinics were surprisingly similar to those seen in psychiatric clinics.27,28 The two groups did not differ in severity of depression, distribution of severity scores, the likelihood of presenting with any of the nine core criteria of a major depressive episode, or the likelihood of having a concomitant axis I psychiatric disorder in addition to depression (about half of participants in each setting had an anxiety disorder).
Recurrent major depressive disorders were common in both groups, though more so in psychiatric patients (78% vs 69%, P < .001), while chronic depression was more common in primary care than in psychiatric patients (30% vs 21%, P < .001). Having either a chronic index episode (ie, lasting > 2 years) or a recurrent major depressive disorder was common in both groups (86% vs 83%, P = .0067).
That said, primary care patients were older (44 years vs 39 years, P < .001), more of them were Hispanic (18% vs 9%, P < .001), and more of them had public insurance (23% vs 9%, P < .001). Fewer of the primary care patients had completed college (20% vs 28%, P < .001), and the primary care patients tended to have greater medical comorbidity. Psychiatric patients were more likely to have attempted suicide in the past and to have had their first depressive illness before age 18.
LEVEL 1: WHAT CAN WE EXPECT FROM INITIAL TREATMENT?
At level 1, all the patients received citalopram. The mean dose was 40.6 ± 16.6 mg/day in the primary care clinics and 42.5 ± 16.8 mg/day in the psychiatric clinics, which are adequate, middle-range doses and higher than the average US dose.29
Approximately 30% of patients achieved remission: 27% as measured on the HAM-D17 and 33% on the QIDS-SR16. The response rate (on the QIDS-SR16) was 47%. There were no differences between primary and psychiatric care settings in remission or response rates.
Patients were more likely to achieve remission if they were white, female, employed, more educated, or wealthier. Longer current episodes, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), more general medical disorders, and lower baseline function and quality of life were each associated with lower remission rates.
What is an adequate trial?
Longer times than expected were needed to reach response or remission. The average duration required to achieve remission was almost 7 weeks (44 days in primary care; 49 days in psychiatric care). Further, approximately one-third of those who ultimately responded and half of those who entered remission did so after 6 weeks.30 Forty percent of those who entered remission required 8 or more weeks to do so.
These results suggest that longer treatment durations and more vigorous medication dosing than generally used are needed to achieve optimal remission rates. It is imprudent to stop a treatment that the patient is tolerating in a robust dose if the patient reports only partial benefit by 6 weeks; indeed, raising the dose, if tolerated, may help a substantial number of patients respond by 12 or 14 weeks. Instruments to monitor depression severity (eg, self-report measures) can be useful. At least 8 weeks with at least moderately vigorous dosing is recommended.
LEVEL 2: IF THE FIRST TREATMENT FAILS
When switching to a new drug, does it matter which one?
No.
In level 2, if patients had not achieved remission on citalopram alone, they had the choice of switching: stopping citalopram and being randomized to receive either sertraline (Zoloft, another SSRI), venlafaxine extended-release (XR) (Effexor XR, a serotonin and norepinephrine reuptake inhibitor), or bupropion sustained-release (SR) (Wellbutrin SR, a norepinephrine and dopamine reuptake inhibitor). At the last visit the mean daily doses were bupropion SR 282.7 mg/day, sertraline 135.5 mg/day, and venlafaxine-XR 193.6 mg/day.
The remission rate was approximately one-fourth with all three drugs31:
- With bupropion SR—21.3% by HAM-D17, 25.5% by QIDS-SR16
- With sertraline—17.6% by HAM-D17, 26.6% by QIDS-SR16
- With venlafaxine-XR—24.8% by HAM-D17, 25.0% by QIDS-SR16. The remission rates were neither statistically nor clinically different by either measure.
Though the types of side effects related to specific medications may have varied, the overall side-effect burden and the rate of serious adverse events did not differ significantly.
When adding a new drug, does it matter which one?
Again, no.
Instead of switching, patients in level 2 could choose to stay on citalopram and be randomized to add either bupropion SR or buspirone (BuSpar) to the regimen (augmentation). The mean daily doses at the end of level 2 were bupropion SR 267.5 mg and buspirone 40.9 mg.
Rates of remission32:
- With bupropion SR—29.7% on the HAMD-D17, 39.0% on the QIDS-SR16
- With buspirone—30.1% on the HAM-D17, 32.9% on the QIDS-SR16.
However, the QIDS-SR16 scores declined significantly more with bupropion SR than with buspirone (25.3% vs 17.1%, P < .04). The mean total QIDS-SR16 score at the last visit was lower with bupropion SR (8.0) than with buspirone (9.1, P < .02), and augmentation with bupropion SR was better tolerated (the dropout rate due to intolerance was 12.5% with bupropion-SR vs 20.6% with buspirone 20.6%; P < .009).
Can we directly compare the benefits of switching vs augmenting?
No.
Patients could choose whether to switch from citalopram to another drug or to add another drug at the second treatment level.33 Consequently, we could not ensure that the patient groups were equivalent at the point of randomization at the beginning of level 2, and, indeed, they were not.
Those who benefitted more from citalopram treatment and who better tolerated it preferred augmentation, while those who benefitted little or who could not tolerate it preferred to switch. Consequently, those in the augmentation group at level 2 were somewhat less depressed than those who switched. Whether augmentation is better even if the initial treatment is minimally effective could not be evaluated in STAR*D.
What about cognitive therapy?
There was no difference between cognitive therapy (either as a switch or as augmentation) and medication (as a switch or as augmentation).34 Adding another drug was more rapidly effective than adding cognitive therapy. Switching to cognitive therapy was better tolerated than switching to a different antidepressant.
Of note, fewer patients accepted cognitive therapy as a randomization option than we expected, so the sample sizes were small. Possible reasons were that all patients had to receive a medication at study entry (which may have biased selection towards those preferring medication), and cognitive therapy entailed additional copayments and visiting still another provider at another site.
After two levels of treatment, how many patients reach remission?
About 30% of patients in level 1 achieved remission, and of those progressing to level 2, another 30% achieved remission. Together, this adds up to about 50% of patients achieving remission if they remained in treatment (30% in level 1 plus 30% of the roughly 70% remaining in level 2).
IF A SECOND TREATMENT FAILS
If switching again to another drug, does it matter which one?
No.
In level 3, patients could choose to stop the drug they had been taking and be randomized to receive either mirtazapine (Remeron) or nortriptyline (Pamelor).
Switching medications was not as effective as a third step as it was as a second step.35
Remission rates:
- With mirtazapine—12.3% on the HAM-D17, 8.0% on the QIDS-SR16
- With nortriptyline—19.8% on the HAM-D17, 12.4% on the QIDS-SR16.
Response rates were 13.4% with mirtazapine and 16.5% with nortriptyline. Statistically, neither the response nor the remission rates differed by treatment, nor did these two treatments differ in tolerability or side-effect burden.
Does choice of augmentation agent matter: Lithium vs T3?
Similarly, after two failed medication treatments, medication augmentation was less effective than it was at the second step.36 The two augmentation options tested, lithium and T3 thyroid hormone (Cytomel), are commonly considered by psychiatrists but less commonly used by primary care doctors.
Lithium is believed to increase serotonergic function, which may have a synergistic effect on the mechanism of action of antidepressants; a meta-analysis of placebo-controlled studies supports lithium’s effectiveness as adjunctive treatment.37 Its side effects, however, must be closely monitored.38 The primary monitoring concern is the small difference between the therapeutic blood level (0.6–1.2 mEq/L) and potentially toxic blood levels (> 1.5 mEq/L).
Lithium was started at 450 mg/day, and at week 2 it was increased to the recommended dose of 900 mg/day (a dose below the target dose for bipolar disorder). If patients could not tolerate 450 mg/day, the initial dose was 225 mg/day for 1 week before being increased to 450 mg/day, still with the target dose of 900 mg/day. The mean exit dose was 859.9 mg/day, and the median blood level was 0.6 mEq/L.
Thyroid hormone augmentation using T3 is believed to work through both direct and indirect effects on the hypothalamic-pituitary-thyroid axis, which has a strong relationship with depression. The efficacy of T3 augmentation is supported by a meta-analysis of eight studies,39 and T3 is effective whether or not thyroid abnormalities are present.
In STAR*D, T3 was started at 25 μg/day for 1 week, than increased to the recommended dose of 50 μg/day. The mean exit dose was 45.2 μg/day.
Remission rates:
- With lithium augmentation—15.9% by the HAM-D17, 13.2% by the QIDS-SR16
- With T3 augmentation—24.7% by both measures.
Response rates were 16.2% with lithium augmentation and 23.3% with T3 augmentation.
While neither response nor remission rates were statistically significantly different by treatment, lithium was more frequently associated with side effects (P = .045), and more participants in the lithium group left treatment because of side effects (23.2% vs 9.6%; P = .027). These results suggest that in cases in which a clinician is considering an augmentation trial, T3 has slight advantages over lithium in effectiveness and tolerability. T3 also offers the advantages of being easy to use and not necessitating blood level monitoring. These latter benefits are especially relevant to the primary care physician. However, T3’s potential for long-term side effects (eg, osteoporosis, cardiovascular effects) were not examined, and it is not clear when to discontinue it.
LEVEL 4: AFTER THREE FAILURES, HOW SHOULD A CLINICIAN PROCEED?
Switch to mirtazapine plus venlafaxine XR or tranylcypromine?
Patients who reached level 4 were considered to have a highly treatment-resistant depressive illness, so treatments at this level were, by design, more aggressive. Accordingly, at level 4 we investigated treatments that might be considered more demanding than those a primary care physician would use. Approximately 40% of patients in each treatment group were from primary care settings.
Remission rates40:
- With the combination of mirtazapine (mean dose 35.7 mg/day) and venlafaxine XR (mean dose 210.3 mg/day)—13.7% by the HAM-D17 and 15.7% by the QIDS-SR16
- With the MAOI tranylcypromine (Parnate, mean dose 36.9 mg/day)—6.9% by the HAM-D17 and 13.8% by the QIDS-SR16. Response rates were 23.5% with the combination and 12.1% with tranylcypromine. Neither remission nor response rates differed significantly.
However, the percentage reduction in QIDS-SR16 score between baseline and exit was greater with the combination than with tranylcypromine. Further, more patients dropped out of treatment with tranylcypromine because of side effects (P < .03). Tranylcypromine also has the disadvantage of necessitating dietary restrictions.
A significant limitation of this comparison is that patients were less likely to get an adequate trial of tranylcypromine, an MAOI, than of the combination. When the 2-week washout period (required before switching to an MAOI) is subtracted from the total time in treatment, approximately 30% of participants in the tranylcypromine group had less than 2 weeks of treatment, and nearly half had less than 6 weeks of treatment.
Therefore, even though the remission and response rates were similar between groups, the combination of venlafaxine-XR plus mirtazapine therapy might have some advantages over tranylcypromine. These results provided the first evidence of tolerability and at least modest efficacy of this combination for treatment-resistant cases.
Overall, what was the cumulative remission rate?
The theoretical cumulative remission rate after four acute treatment steps was 67%. Remission was more likely to occur during the first two levels of treatment than during the last two. The cumulative remission rates for the first four steps were:
- Level 1—33%
- Level 2—57%
- Level 3—63%
- Level 4—67%.
RESULTS FROM LONG-TERM FOLLOW-UP AFTER REMISSION OR RESPONSE
Patients with a clinically meaningful response or, preferably, remission at any level could enter into a 12-month observational follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase.41 Further, if a patient achieved remission rather than just response to treatment, regardless of the treatment level, the prognosis at follow-up was better, confirming the importance of remission as the goal of treatment.
Results also provided a warning—the greater the number of treatment levels that a patient required, the more likely that patient and physician would settle for response. Whether the greater relapse rates reflect a harder-to-treat depression or the naturalistic design of the follow-up phase (with less control over dosing) is unclear.
WHAT DO THESE RESULTS MEAN FOR PRIMARY CARE PHYSICIANS?
- Measurement-based care is feasible in primary care. Primary care doctors can ensure vigorous but tolerable dosing using a self-report depression scale to monitor response, a side-effects tool to monitor tolerability, and medication adjustments at critical decision points guided by these two measures.
- Remission, ie, complete recovery from a depressive episode, rather than merely substantial improvement, is associated with a better prognosis and is the preferred goal of treatment.
- Pharmacologic differences between psychotropic medications did not translate into substantial clinical differences, although tolerability differed. These findings are consistent with a large-scale systematic evidence review recently completed by the Agency for Healthcare Research and Quality that compared the effectiveness of antidepressants.42 Given the difficulty in predicting what medication will be both efficacious for and tolerated by an individual patient, familiarity with a broad spectrum of antidepressants is prudent.
- Remission of depressive episodes will most likely require repeated trials of sufficiently sustained,vigorously dosed antidepressant medication. From treatment initiation, physicians should ensure maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- If a first treatment doesn’t work, either switching or augmenting it is a reasonable choice. Augmentation may be preferred if the patient is tolerating and receiving partial benefit from the initial medication choice. While bupropion SR and buspirone were not different as augmenters by the primary remission outcome measure, secondary measures (eg, tolerability, depressive symptom change over the course of treatment, clinician-rated Quick Inventory of Depressive Symptomatology) recommended bupropion-SR over buspirone.
- If physicians switch, either a within-class switch (eg, citalopram to sertraline) or an out-of-class switch (eg, citalopram to bupropion SR) is effective, as is a switch to a dual-action agent (eg, venlafaxine XR).
- The likelihood of improvement after two aggressive medication trials is very low and likely requires more complicated medication regimens, and the existing evidence base is quite thin. These primary care patients should likely be referred to psychiatrists for more aggressive and intensive treatment.
- For patients who present with major depressive disorder, STAR*D suggests that with persistence and aggressive yet feasible care, there is hope: after one round, approximately 30% will have a remission; after two rounds, 50%; after three rounds, 60%; and after four rounds, 70%.
- While STAR*D excluded depressed patients with bipolar disorder, a depressive episode in a patient with bipolar disorder can be difficult to distinguish from a depressive episode in a patient with major depressive disorder. Primary care physicians need to consider bipolar disorder both in patients presenting with a depressive episode and in those who fail an adequate trial.43
FUTURE CONSIDERATIONS
Subsequent STAR*D analyses will compare in greater depth outcomes in primary care vs psychiatric settings at each level of treatment. Given the greater risk of depression persistence associated with more successive levels of treatment, subsequent research will focus on ways to more successfully treat depression in the earlier stages, possibly through medication combinations earlier in treatment (somewhat analogous to a “broad-spectrum antibiotic” approach for infections).
Depression can be treated successfully by primary care physicians under “real-world” conditions.
Furthermore, the particular drug or drugs used are not as important as following a rational plan: giving antidepressant medications in adequate doses, monitoring the patient’s symptoms and side effects and adjusting the regimen accordingly, and switching drugs or adding new drugs to the regimen only after an adequate trial.
These are among the lessons learned from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest prospective clinical trial of treatment of major depressive disorder ever conducted. It was funded by the National Institutes of Health and directed by A. John Rush, MD.
WHAT WERE THE AIMS OF STAR*D?
Depression, a common and debilitating condition, affects approximately one in eight people in the United States.1 It is expected2 to be the second-leading cause of disability in the world by the year 2020; today, it is the second-leading cause of disability-adjusted life years in those 15 to 44 years old.3
Nevertheless, the available evidence base for treatment is limited, since most participants in clinical trials are recruited by advertisement rather than from representative practices, and they are often selected to have few comorbid disorders, either medical or psychiatric. In addition, those with chronic depression or current suicidal ideation are excluded.1,4 These uncomplicated and “pristine” participants are unlike typical patients seen by primary care physicians or psychiatrists.
Similarly, the protocols used in these trials do not represent usual clinic practice.Patients in clinical trials undergo more assessment and more frequent follow-up than in real-world practice, they have no say in treatment decisions, the doses are fixed, and the patients and physicians are blinded to the intervention. Consequently, how to translate the results of these efficacy trials into practice is unclear.5
Further, even in relatively uncomplicated cases, only about one-half of outpatients with nonpsychotic major depressive disorder initially treated with a single medication or with psychotherapy will experience a clinically significant improvement in symptoms (ie, a response) during the 8 to 12 weeks of acute-phase treatment,6–10 and only 20% to 35% of patients will reach remission,9 the aim of treatment.8,11 The remission rates are even lower in treatment-resistant depression.12 How to manage most patients—those whose depression does not remit with the first, second, or third step of treatment—is unclear.
Accordingly, the overall objective of STAR*D was to develop and evaluate feasible treatment strategies to improve clinical outcomes for real-world patients with treatment-resistant depression, who were identified prospectively from a pool of patients in a current major depressive episode.13–15 Specifically, STAR*D aimed to determine prospectively which of several treatments is the most effective “next step” for patients who do not reach remission with an initial or subsequent treatment or who cannot tolerate the treatment.
WHY IS STAR*D RELEVANT FOR PRIMARY CARE?
Nearly 10% of all primary care office visits are depression-related.16 Primary care physicians provide nearly half the outpatient care for depressed patients.17 Indeed, primary care physicians log approximately as many outpatient visits for depression as psychiatrists do.18 Medical comorbidity is especially common in primary care settings.19 When to refer to a psychiatrist is not clear.
KEY FEATURES OF THE STUDY DESIGN
STAR*D involved a national consortium of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004.
Entry criteria were broad and inclusive
Patients had to:
- Be between 18 and 75 years of age
- Have a nonpsychotic major depressive disorder, identified by a clinician and confirmed with a symptom checklist based on the Diagnostic and Statistical Manual, fourth edition revised,20 and for which antidepressant treatment is recommended
- Score at least 14 on the 17-item Hamilton Rating Scale for Depression (HAMD17)21
- Not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder, which would require a different treatment strategy, or a seizure disorder (which would preclude bupropion as a second-step treatment).
Dosing recommendations were flexible but vigorous
Medications often were increased to maximally tolerated doses. For example, citalopram (Celexa) was started at 20 mg/day and increased by 20 mg every 2 to 4 weeks if the patient was tolerating it but had not achieved remission, to a maximum dose of 60 mg/day. Treatment could be given for up to 14 weeks, during which side effects22 and clinical ratings23 were assessed by both patients and study coordinators.
Measurement-based care
We used a systematic approach to treatment called “measurement-based care,”24 which involves routinely measuring symptoms23 and side effects22 and using this information to modify the medication doses at critical decision points. This algorithmic approach provided flexible treatment recommendations to ensure that the dosage and duration of antidepressant drug treatment were adequate.25
The severity of depression was assessed by the clinician-rated, 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16). The QIDS-SR16 (the self-report version) can substitute for the QIDS-C1623 to make this approach more feasible. Both tools are available at www.ids-qids.org.
This approach was easily worked into busy primary care and specialty care office workflows (clinic physicians, most with limited research experience, provided the treatment), and could be translated into primary care practice in the community as well.
Four-step protocol
A unique feature of the study design was that the patients, in consultation with their physicians, had some choice in the treatments they received. In this “equipoise-stratified randomized design,”26 at levels 2 and 3 the patient could choose either to switch therapies (stop the current drug and be randomized to receive one of several different treatments) or to augment their current therapy (by adding one of several treatments in a randomized fashion). Patients could decline certain strategies as long as there were at least two possible options to which one might be randomized.
At level 2, one of the options for both switching and augmentation was cognitive therapy, although patients could decline that option. Conversely, if they definitely wanted cognitive therapy, they could choose to be randomized to either cognitive therapy alone or to cognitive therapy added to citalopram. Also, anyone who received cognitive therapy in level 2 and failed to enter remission was additionally randomized to either bupropion or venlafaxine (level 2a) to ensure that all patients had failed trials on two medications before entering level 3.
When switching to medications other than a monoamine oxidase inhibitor (MAOI), the clinician could choose either to stop the current medication and immediately begin the next one, or to decrease the current medication while starting the new one at a low dose and then tapering and titrating over 1 week. (Switching to an MAOI, used only in the final level of treatment, required a 7- to 10-day washout period.)
Outcomes measured
Remission (complete recovery from the depressive episode), the primary study outcome, was defined as a HAM-D17 score of 7 or less, as assessed by treatment-blinded raters.A secondary remission outcome was a QIDS-SR16 score of 5 or less. Of note, the HAM-D17 remission rates were slightly lower than the rates based on the QIDS-SR16, since patients who did not have a HAM-D17 score measured at exit were defined as not being in remission a priori. Thus, the QIDS-SR16 rates might have been a slightly better reflection of actual remission rates.
Response, a secondary outcome, was defined as a reduction of at least 50% in the QIDS-SR16 score from baseline at the last assessment.
FEW DIFFERENCES BETWEEN PSYCHIATRIC, PRIMARY CARE PATIENTS
The patients seen in primary care clinics were surprisingly similar to those seen in psychiatric clinics.27,28 The two groups did not differ in severity of depression, distribution of severity scores, the likelihood of presenting with any of the nine core criteria of a major depressive episode, or the likelihood of having a concomitant axis I psychiatric disorder in addition to depression (about half of participants in each setting had an anxiety disorder).
Recurrent major depressive disorders were common in both groups, though more so in psychiatric patients (78% vs 69%, P < .001), while chronic depression was more common in primary care than in psychiatric patients (30% vs 21%, P < .001). Having either a chronic index episode (ie, lasting > 2 years) or a recurrent major depressive disorder was common in both groups (86% vs 83%, P = .0067).
That said, primary care patients were older (44 years vs 39 years, P < .001), more of them were Hispanic (18% vs 9%, P < .001), and more of them had public insurance (23% vs 9%, P < .001). Fewer of the primary care patients had completed college (20% vs 28%, P < .001), and the primary care patients tended to have greater medical comorbidity. Psychiatric patients were more likely to have attempted suicide in the past and to have had their first depressive illness before age 18.
LEVEL 1: WHAT CAN WE EXPECT FROM INITIAL TREATMENT?
At level 1, all the patients received citalopram. The mean dose was 40.6 ± 16.6 mg/day in the primary care clinics and 42.5 ± 16.8 mg/day in the psychiatric clinics, which are adequate, middle-range doses and higher than the average US dose.29
Approximately 30% of patients achieved remission: 27% as measured on the HAM-D17 and 33% on the QIDS-SR16. The response rate (on the QIDS-SR16) was 47%. There were no differences between primary and psychiatric care settings in remission or response rates.
Patients were more likely to achieve remission if they were white, female, employed, more educated, or wealthier. Longer current episodes, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), more general medical disorders, and lower baseline function and quality of life were each associated with lower remission rates.
What is an adequate trial?
Longer times than expected were needed to reach response or remission. The average duration required to achieve remission was almost 7 weeks (44 days in primary care; 49 days in psychiatric care). Further, approximately one-third of those who ultimately responded and half of those who entered remission did so after 6 weeks.30 Forty percent of those who entered remission required 8 or more weeks to do so.
These results suggest that longer treatment durations and more vigorous medication dosing than generally used are needed to achieve optimal remission rates. It is imprudent to stop a treatment that the patient is tolerating in a robust dose if the patient reports only partial benefit by 6 weeks; indeed, raising the dose, if tolerated, may help a substantial number of patients respond by 12 or 14 weeks. Instruments to monitor depression severity (eg, self-report measures) can be useful. At least 8 weeks with at least moderately vigorous dosing is recommended.
LEVEL 2: IF THE FIRST TREATMENT FAILS
When switching to a new drug, does it matter which one?
No.
In level 2, if patients had not achieved remission on citalopram alone, they had the choice of switching: stopping citalopram and being randomized to receive either sertraline (Zoloft, another SSRI), venlafaxine extended-release (XR) (Effexor XR, a serotonin and norepinephrine reuptake inhibitor), or bupropion sustained-release (SR) (Wellbutrin SR, a norepinephrine and dopamine reuptake inhibitor). At the last visit the mean daily doses were bupropion SR 282.7 mg/day, sertraline 135.5 mg/day, and venlafaxine-XR 193.6 mg/day.
The remission rate was approximately one-fourth with all three drugs31:
- With bupropion SR—21.3% by HAM-D17, 25.5% by QIDS-SR16
- With sertraline—17.6% by HAM-D17, 26.6% by QIDS-SR16
- With venlafaxine-XR—24.8% by HAM-D17, 25.0% by QIDS-SR16. The remission rates were neither statistically nor clinically different by either measure.
Though the types of side effects related to specific medications may have varied, the overall side-effect burden and the rate of serious adverse events did not differ significantly.
When adding a new drug, does it matter which one?
Again, no.
Instead of switching, patients in level 2 could choose to stay on citalopram and be randomized to add either bupropion SR or buspirone (BuSpar) to the regimen (augmentation). The mean daily doses at the end of level 2 were bupropion SR 267.5 mg and buspirone 40.9 mg.
Rates of remission32:
- With bupropion SR—29.7% on the HAMD-D17, 39.0% on the QIDS-SR16
- With buspirone—30.1% on the HAM-D17, 32.9% on the QIDS-SR16.
However, the QIDS-SR16 scores declined significantly more with bupropion SR than with buspirone (25.3% vs 17.1%, P < .04). The mean total QIDS-SR16 score at the last visit was lower with bupropion SR (8.0) than with buspirone (9.1, P < .02), and augmentation with bupropion SR was better tolerated (the dropout rate due to intolerance was 12.5% with bupropion-SR vs 20.6% with buspirone 20.6%; P < .009).
Can we directly compare the benefits of switching vs augmenting?
No.
Patients could choose whether to switch from citalopram to another drug or to add another drug at the second treatment level.33 Consequently, we could not ensure that the patient groups were equivalent at the point of randomization at the beginning of level 2, and, indeed, they were not.
Those who benefitted more from citalopram treatment and who better tolerated it preferred augmentation, while those who benefitted little or who could not tolerate it preferred to switch. Consequently, those in the augmentation group at level 2 were somewhat less depressed than those who switched. Whether augmentation is better even if the initial treatment is minimally effective could not be evaluated in STAR*D.
What about cognitive therapy?
There was no difference between cognitive therapy (either as a switch or as augmentation) and medication (as a switch or as augmentation).34 Adding another drug was more rapidly effective than adding cognitive therapy. Switching to cognitive therapy was better tolerated than switching to a different antidepressant.
Of note, fewer patients accepted cognitive therapy as a randomization option than we expected, so the sample sizes were small. Possible reasons were that all patients had to receive a medication at study entry (which may have biased selection towards those preferring medication), and cognitive therapy entailed additional copayments and visiting still another provider at another site.
After two levels of treatment, how many patients reach remission?
About 30% of patients in level 1 achieved remission, and of those progressing to level 2, another 30% achieved remission. Together, this adds up to about 50% of patients achieving remission if they remained in treatment (30% in level 1 plus 30% of the roughly 70% remaining in level 2).
IF A SECOND TREATMENT FAILS
If switching again to another drug, does it matter which one?
No.
In level 3, patients could choose to stop the drug they had been taking and be randomized to receive either mirtazapine (Remeron) or nortriptyline (Pamelor).
Switching medications was not as effective as a third step as it was as a second step.35
Remission rates:
- With mirtazapine—12.3% on the HAM-D17, 8.0% on the QIDS-SR16
- With nortriptyline—19.8% on the HAM-D17, 12.4% on the QIDS-SR16.
Response rates were 13.4% with mirtazapine and 16.5% with nortriptyline. Statistically, neither the response nor the remission rates differed by treatment, nor did these two treatments differ in tolerability or side-effect burden.
Does choice of augmentation agent matter: Lithium vs T3?
Similarly, after two failed medication treatments, medication augmentation was less effective than it was at the second step.36 The two augmentation options tested, lithium and T3 thyroid hormone (Cytomel), are commonly considered by psychiatrists but less commonly used by primary care doctors.
Lithium is believed to increase serotonergic function, which may have a synergistic effect on the mechanism of action of antidepressants; a meta-analysis of placebo-controlled studies supports lithium’s effectiveness as adjunctive treatment.37 Its side effects, however, must be closely monitored.38 The primary monitoring concern is the small difference between the therapeutic blood level (0.6–1.2 mEq/L) and potentially toxic blood levels (> 1.5 mEq/L).
Lithium was started at 450 mg/day, and at week 2 it was increased to the recommended dose of 900 mg/day (a dose below the target dose for bipolar disorder). If patients could not tolerate 450 mg/day, the initial dose was 225 mg/day for 1 week before being increased to 450 mg/day, still with the target dose of 900 mg/day. The mean exit dose was 859.9 mg/day, and the median blood level was 0.6 mEq/L.
Thyroid hormone augmentation using T3 is believed to work through both direct and indirect effects on the hypothalamic-pituitary-thyroid axis, which has a strong relationship with depression. The efficacy of T3 augmentation is supported by a meta-analysis of eight studies,39 and T3 is effective whether or not thyroid abnormalities are present.
In STAR*D, T3 was started at 25 μg/day for 1 week, than increased to the recommended dose of 50 μg/day. The mean exit dose was 45.2 μg/day.
Remission rates:
- With lithium augmentation—15.9% by the HAM-D17, 13.2% by the QIDS-SR16
- With T3 augmentation—24.7% by both measures.
Response rates were 16.2% with lithium augmentation and 23.3% with T3 augmentation.
While neither response nor remission rates were statistically significantly different by treatment, lithium was more frequently associated with side effects (P = .045), and more participants in the lithium group left treatment because of side effects (23.2% vs 9.6%; P = .027). These results suggest that in cases in which a clinician is considering an augmentation trial, T3 has slight advantages over lithium in effectiveness and tolerability. T3 also offers the advantages of being easy to use and not necessitating blood level monitoring. These latter benefits are especially relevant to the primary care physician. However, T3’s potential for long-term side effects (eg, osteoporosis, cardiovascular effects) were not examined, and it is not clear when to discontinue it.
LEVEL 4: AFTER THREE FAILURES, HOW SHOULD A CLINICIAN PROCEED?
Switch to mirtazapine plus venlafaxine XR or tranylcypromine?
Patients who reached level 4 were considered to have a highly treatment-resistant depressive illness, so treatments at this level were, by design, more aggressive. Accordingly, at level 4 we investigated treatments that might be considered more demanding than those a primary care physician would use. Approximately 40% of patients in each treatment group were from primary care settings.
Remission rates40:
- With the combination of mirtazapine (mean dose 35.7 mg/day) and venlafaxine XR (mean dose 210.3 mg/day)—13.7% by the HAM-D17 and 15.7% by the QIDS-SR16
- With the MAOI tranylcypromine (Parnate, mean dose 36.9 mg/day)—6.9% by the HAM-D17 and 13.8% by the QIDS-SR16. Response rates were 23.5% with the combination and 12.1% with tranylcypromine. Neither remission nor response rates differed significantly.
However, the percentage reduction in QIDS-SR16 score between baseline and exit was greater with the combination than with tranylcypromine. Further, more patients dropped out of treatment with tranylcypromine because of side effects (P < .03). Tranylcypromine also has the disadvantage of necessitating dietary restrictions.
A significant limitation of this comparison is that patients were less likely to get an adequate trial of tranylcypromine, an MAOI, than of the combination. When the 2-week washout period (required before switching to an MAOI) is subtracted from the total time in treatment, approximately 30% of participants in the tranylcypromine group had less than 2 weeks of treatment, and nearly half had less than 6 weeks of treatment.
Therefore, even though the remission and response rates were similar between groups, the combination of venlafaxine-XR plus mirtazapine therapy might have some advantages over tranylcypromine. These results provided the first evidence of tolerability and at least modest efficacy of this combination for treatment-resistant cases.
Overall, what was the cumulative remission rate?
The theoretical cumulative remission rate after four acute treatment steps was 67%. Remission was more likely to occur during the first two levels of treatment than during the last two. The cumulative remission rates for the first four steps were:
- Level 1—33%
- Level 2—57%
- Level 3—63%
- Level 4—67%.
RESULTS FROM LONG-TERM FOLLOW-UP AFTER REMISSION OR RESPONSE
Patients with a clinically meaningful response or, preferably, remission at any level could enter into a 12-month observational follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase.41 Further, if a patient achieved remission rather than just response to treatment, regardless of the treatment level, the prognosis at follow-up was better, confirming the importance of remission as the goal of treatment.
Results also provided a warning—the greater the number of treatment levels that a patient required, the more likely that patient and physician would settle for response. Whether the greater relapse rates reflect a harder-to-treat depression or the naturalistic design of the follow-up phase (with less control over dosing) is unclear.
WHAT DO THESE RESULTS MEAN FOR PRIMARY CARE PHYSICIANS?
- Measurement-based care is feasible in primary care. Primary care doctors can ensure vigorous but tolerable dosing using a self-report depression scale to monitor response, a side-effects tool to monitor tolerability, and medication adjustments at critical decision points guided by these two measures.
- Remission, ie, complete recovery from a depressive episode, rather than merely substantial improvement, is associated with a better prognosis and is the preferred goal of treatment.
- Pharmacologic differences between psychotropic medications did not translate into substantial clinical differences, although tolerability differed. These findings are consistent with a large-scale systematic evidence review recently completed by the Agency for Healthcare Research and Quality that compared the effectiveness of antidepressants.42 Given the difficulty in predicting what medication will be both efficacious for and tolerated by an individual patient, familiarity with a broad spectrum of antidepressants is prudent.
- Remission of depressive episodes will most likely require repeated trials of sufficiently sustained,vigorously dosed antidepressant medication. From treatment initiation, physicians should ensure maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- If a first treatment doesn’t work, either switching or augmenting it is a reasonable choice. Augmentation may be preferred if the patient is tolerating and receiving partial benefit from the initial medication choice. While bupropion SR and buspirone were not different as augmenters by the primary remission outcome measure, secondary measures (eg, tolerability, depressive symptom change over the course of treatment, clinician-rated Quick Inventory of Depressive Symptomatology) recommended bupropion-SR over buspirone.
- If physicians switch, either a within-class switch (eg, citalopram to sertraline) or an out-of-class switch (eg, citalopram to bupropion SR) is effective, as is a switch to a dual-action agent (eg, venlafaxine XR).
- The likelihood of improvement after two aggressive medication trials is very low and likely requires more complicated medication regimens, and the existing evidence base is quite thin. These primary care patients should likely be referred to psychiatrists for more aggressive and intensive treatment.
- For patients who present with major depressive disorder, STAR*D suggests that with persistence and aggressive yet feasible care, there is hope: after one round, approximately 30% will have a remission; after two rounds, 50%; after three rounds, 60%; and after four rounds, 70%.
- While STAR*D excluded depressed patients with bipolar disorder, a depressive episode in a patient with bipolar disorder can be difficult to distinguish from a depressive episode in a patient with major depressive disorder. Primary care physicians need to consider bipolar disorder both in patients presenting with a depressive episode and in those who fail an adequate trial.43
FUTURE CONSIDERATIONS
Subsequent STAR*D analyses will compare in greater depth outcomes in primary care vs psychiatric settings at each level of treatment. Given the greater risk of depression persistence associated with more successive levels of treatment, subsequent research will focus on ways to more successfully treat depression in the earlier stages, possibly through medication combinations earlier in treatment (somewhat analogous to a “broad-spectrum antibiotic” approach for infections).
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Zimmerman M, Chelminski I, Posternak MA. Generalizability of antidepressant efficacy trials: differences between depressed psychiatric outpatients who would or would not qualify for an efficacy trial. Am J Psychiatry 2005; 162:1370–1372.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply? Lancet 2005; 365:82–93.
- Depression Guideline Panel. Depression in primary care: Volume 1, diagnosis and detection. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Frank E, Karp J, Rush A. Efficacy of treatments for major depression. Psychopharmacol Bull 1993; 29:457–475.
- Depression Guideline Panel. Depression in primary care: Volume 2, Treatment of major depression. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19:179–200.
- Jarrett RB, Rush A. Short-term psychotherapy of depressive disorders: current status and future directions. Psychiatry: Interpers Biol Process 1994; 57:115–132.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depression (revision). Am J Psychiatry 2000; 157(suppl 4):1–45.
- Dunner DL, Rush AJ, Russell JM, et al. Prospective, long-term, multicenter study of the naturalistic outcomes of patients with treatment-resistant depression. J Clin Psychiatry 2006; 67:688–695.
- Fava M, Rush A, Trivedi M, et al. Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494.
- Gaynes B, Davis L, Rush A, Trivedi M, Fava M, Wisniewski S. The aims and design of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Prim Psychiatry 2005; 12:36–41.
- Rush A, Fava M, Wisniewski S, et al. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142.
- Stafford RS, Ausiello JC, Misra B, Saglam D. National Patterns o fDepression Treatment in Primary Care. Prim Care Companion J Clin Psychiatry 2000; 2:211–216.
- Regier D, Narrow W, Rae D, Mandersheid R, Locke B, Goodwin F. The de facto US mental and addictive disorders service system: epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry 1993; 50:85–94.
- Pincus H, Tanielian T, Marcus S, et al. Prescribing trends in psychotropic medications: primary care, psychiatry, and other medical specialities. JAMA 1998; 279:526–531.
- Vuorilehto M, Melartin T, Isometsa E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med 2005; 35:673–682.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–61.
- Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA for the STAR*D Investigators. Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatric Pract 2006; 12:71–79.
- Rush AJ, Bernstein IH, Trivedi MH, et al. An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial report. Biol Psychiatry 2006; 59:493–501.
- Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR*D measurement-based care. Neuropsychopharmacology 2007/04/04/online 2007.
- Kawamoto K, Houlihan CA, Balas EA, Lobach DF. Improving clinical practice using clinical decision support systems: a systematic review of trials to identify features critical to success. BMJ 2005; 330:765 e-pub March 14 2005.
- Lavori P, Rush A, Wisniewski S, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001; 50:792–801.
- Gaynes BN, Rush AJ, Trivedi MH, et al. Major depression symptoms in primary care and psychiatric care settings: a cross-sectional analysis. Ann Fam Med 2007; 5:126–134.
- Gaynes BN, Rush AJ, Trivedi M, et al. A direct comparison of presenting characteristics of depressed outpatients from primary vs. specialty care settings: preliminary findings from the STAR*D clinical trial. Gen Hosp Psychiatry 2005; 27:87–96.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004; 18:911–932.
- Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006; 354:1231–1242.
- Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006; 354:1243–1252.
- Wisniewski SR, Fava M, Trivedi MH, et al. Acceptability of second-step treatments to depressed outpatients: a STAR*D report. Am J Psychiatry 2007; 164:753–760.
- Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry 2007; 164:739–752.
- Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006; 163:1161–1172.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry 2006; 163:1519–1530.
- Bschor T, Lewitzka U, Sasse J, Adli M, Koberle U, Bauer M. Lithium augmentation in treatment-resistant depression: clinical evidence, serotonergic and endocrine mechanisms. Pharmacopsychiatry 2003; 36(suppl 3):S230–S234.
- Freeman MP, Freeman SA. Lithium: clinical considerations in internal medicine. Am J Med 2006; 119:478–481.
- Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996; 53:842–848.
- McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry 2006; 163:1531–1541.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Gartlehner G, Hansen R, Thieda P, et al. Comparative Effectiveness of Second-generation Antidepressants in the Pharmacologic Treatment of Depression. Agency for Healthcare Research and Quality. http://effectivehealthcare.ahrq.gov/reports/topic.cfm?topic=8&sid=39&rType=3. Accessed December 12, 2007.
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293:956–963.
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Zimmerman M, Chelminski I, Posternak MA. Generalizability of antidepressant efficacy trials: differences between depressed psychiatric outpatients who would or would not qualify for an efficacy trial. Am J Psychiatry 2005; 162:1370–1372.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply? Lancet 2005; 365:82–93.
- Depression Guideline Panel. Depression in primary care: Volume 1, diagnosis and detection. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Frank E, Karp J, Rush A. Efficacy of treatments for major depression. Psychopharmacol Bull 1993; 29:457–475.
- Depression Guideline Panel. Depression in primary care: Volume 2, Treatment of major depression. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19:179–200.
- Jarrett RB, Rush A. Short-term psychotherapy of depressive disorders: current status and future directions. Psychiatry: Interpers Biol Process 1994; 57:115–132.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depression (revision). Am J Psychiatry 2000; 157(suppl 4):1–45.
- Dunner DL, Rush AJ, Russell JM, et al. Prospective, long-term, multicenter study of the naturalistic outcomes of patients with treatment-resistant depression. J Clin Psychiatry 2006; 67:688–695.
- Fava M, Rush A, Trivedi M, et al. Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494.
- Gaynes B, Davis L, Rush A, Trivedi M, Fava M, Wisniewski S. The aims and design of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Prim Psychiatry 2005; 12:36–41.
- Rush A, Fava M, Wisniewski S, et al. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142.
- Stafford RS, Ausiello JC, Misra B, Saglam D. National Patterns o fDepression Treatment in Primary Care. Prim Care Companion J Clin Psychiatry 2000; 2:211–216.
- Regier D, Narrow W, Rae D, Mandersheid R, Locke B, Goodwin F. The de facto US mental and addictive disorders service system: epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry 1993; 50:85–94.
- Pincus H, Tanielian T, Marcus S, et al. Prescribing trends in psychotropic medications: primary care, psychiatry, and other medical specialities. JAMA 1998; 279:526–531.
- Vuorilehto M, Melartin T, Isometsa E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med 2005; 35:673–682.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–61.
- Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA for the STAR*D Investigators. Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatric Pract 2006; 12:71–79.
- Rush AJ, Bernstein IH, Trivedi MH, et al. An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial report. Biol Psychiatry 2006; 59:493–501.
- Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR*D measurement-based care. Neuropsychopharmacology 2007/04/04/online 2007.
- Kawamoto K, Houlihan CA, Balas EA, Lobach DF. Improving clinical practice using clinical decision support systems: a systematic review of trials to identify features critical to success. BMJ 2005; 330:765 e-pub March 14 2005.
- Lavori P, Rush A, Wisniewski S, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001; 50:792–801.
- Gaynes BN, Rush AJ, Trivedi MH, et al. Major depression symptoms in primary care and psychiatric care settings: a cross-sectional analysis. Ann Fam Med 2007; 5:126–134.
- Gaynes BN, Rush AJ, Trivedi M, et al. A direct comparison of presenting characteristics of depressed outpatients from primary vs. specialty care settings: preliminary findings from the STAR*D clinical trial. Gen Hosp Psychiatry 2005; 27:87–96.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004; 18:911–932.
- Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006; 354:1231–1242.
- Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006; 354:1243–1252.
- Wisniewski SR, Fava M, Trivedi MH, et al. Acceptability of second-step treatments to depressed outpatients: a STAR*D report. Am J Psychiatry 2007; 164:753–760.
- Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry 2007; 164:739–752.
- Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006; 163:1161–1172.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry 2006; 163:1519–1530.
- Bschor T, Lewitzka U, Sasse J, Adli M, Koberle U, Bauer M. Lithium augmentation in treatment-resistant depression: clinical evidence, serotonergic and endocrine mechanisms. Pharmacopsychiatry 2003; 36(suppl 3):S230–S234.
- Freeman MP, Freeman SA. Lithium: clinical considerations in internal medicine. Am J Med 2006; 119:478–481.
- Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996; 53:842–848.
- McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry 2006; 163:1531–1541.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Gartlehner G, Hansen R, Thieda P, et al. Comparative Effectiveness of Second-generation Antidepressants in the Pharmacologic Treatment of Depression. Agency for Healthcare Research and Quality. http://effectivehealthcare.ahrq.gov/reports/topic.cfm?topic=8&sid=39&rType=3. Accessed December 12, 2007.
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293:956–963.
KEY POINTS
- Remission (ie, complete relief from a depressive episode) rather than response (merely substantial improvement) should be the goal of treatment, as it is associated with a better prognosis and better function.
- Should the first treatment fail, either switching treat mentor augmenting the current treatment is reasonable.
- For most patients, remission will require repeated trials of sufficiently sustained, vigorously dosed antidepressant medication. Physicians should give maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- After two well-delivered medication trials, the likelihood of remission substantially decreases. Such patients likely require more complicated regimens. Given the thin existing database, these patients are best referred to a psychiatrist for more complex treatments.
- With persistent and vigorous treatment, most patients will enter remission: about 33% after one step, 50% after two steps, 60% after three steps, and 70% after four steps (assuming patients stay in treatment).