User login
Evaluation of Spinal Epidural Abscess
Spinal epidural abscess (SEA) is caused by a suppurative infection in the epidural space. The mass effect of the abscess can compress and reduce blood flow to the spinal cord, conus medullaris, or cauda equina. Left untreated, the infection can lead to sensory loss, muscle weakness, visceral dysfunction, sepsis, and even death. Early diagnosis is essential to limit morbidity and neurologic injury. The classic triad of fever, axial pain, and neurological deficit occurs in as few as 13% of patients, highlighting the diagnostic challenge associated with SEA.[1]
This investigation reviews the current literature on SEA epidemiology, clinical findings, laboratory data, and treatment methods, with particular focus on nonsurgical versus surgical treatment. Our primary objective was to educate the clinician on when to suspect SEA and how to execute an appropriate diagnostic evaluation.
INCIDENCE AND EPIDEMIOLOGY
In 1975, the incidence of SEA was reported to be 0.2 to 2.0 per 10,000 hospital admissions.[2] Two decades later, a study of tertiary referral centers documented a much higher rate of 12.5 per 10,000 admissions.[3] The reported incidence continues to rise and has doubled in the past decade, with SEA currently representing approximately 10% of all primary spine infections.[4, 5] Potential explanations for this increasing incidence include aging of the population, an increasing prevalence of diabetes, increasing intravenous drug abuse, more widespread use of advanced immunosuppressive regimens, and an increased rate of invasive spinal procedures.[6, 7, 8] Other factors contributing to the rising incidence include increased detection due to the greater accessibility of magnetic resonance imaging (MRI) and increased reporting as a result of the concentration of cases at tertiary referral centers.
SEA is most common in patients older than 60 years[7] and those with multiple medical comorbidities. A review of over 30,000 patients found that the average number of comorbidities in patients who underwent surgical intervention for SEA was 6, ranging from 0 to 20.[5] The same study noted that diabetes was the most frequently associated disease (30% of patients), followed by chronic lung disease (19%), renal failure (13%), and obesity (13%) (Table 1).[5] A history of invasive spine interventions is an additional risk factor; between 14% and 22% occur as a result of spine surgery or percutaneous spine procedures (eg, epidural steroid injections).[8, 9] Regardless of pathogenesis, the rate of permanent neurologic injury after SEA is 30% to 50%, and the mortality rate ranges from 10% to 20%.[4, 5, 8, 10]
| Medical Comorbidity | Prevalence (%) |
|---|---|
| |
| Diabetes mellitus | 1546 |
| IV drug use | 437 |
| Spinal trauma | 533 |
| End‐stage renal disease | 213 |
| Immunosuppressant therapy | 716 |
| Cancer | 215 |
| HIV/AIDS | 29 |
MISSED DIAGNOSIS
Despite the availability of advanced imaging, rates of misdiagnosis at initial presentation remain substantial, with current estimates ranging from 11% to 75%.[4, 9] Back and neck pain symptoms are ubiquitous and nonspecific, often making the diagnosis difficult. Repeated emergency room visits for pain are common in patients who are eventually diagnosed with SEA. Davis et al. found that 51% of 63 patients present to the emergency room at least 2 or more times prior to diagnosis; 11% present 3 or more times.[1]
PATHOPHYSIOLOGY
Microbiology
Staphylococcus aureus, including methicillin resistant S aureus (MRSA), accounts for two‐thirds of all infections.[3] S aureus infection of the spine may occur in the setting of surgical intervention or concomitant skin infection, although it often occurs without an identifiable source. Historically, MRSA has been reported to be responsible for 15% of all staphylococcal infections of the epidural space; some institutions report MRSA rates as high as 40%.[4] S epidermidis is another common pathogen, which is most often encountered following spinal surgery, epidural catheter insertion, and spinal injections.[11] Gram‐negative infections are less common. Escherichia coli is characteristically isolated in patients with active urinary tract infections, and Pseudomonas aeruginosa is more common in intravenous (IV) drug users.[12] Rare causes of SEA include anaerobes such as Bacteroides,[13] various parasites, and fungal organisms such as actinomyces, nocardia, and mycobacteria.[8]
Mechanism of Inoculation
Infections may enter the epidural space by 4 mechanisms: hematogenous spread, direct extension, inoculation via spinal procedure, and trauma. Hematogenous spread from an existing infection is the most common mechanism.[14] Seeding of the epidural space from transient bacteremia after dental procedures has also been reported.[13]
The second most common mechanism of infection is direct spread from an infected component of the vertebral column or paraspinal soft tissues. Most commonly, this takes the form of an anterior SEA in association with vertebral body osteomyelitis. Septic arthritis of a facet joint can also cause a secondary infection of the posterior epidural space.[4, 15] Direct spread from other posterior structures (eg, retropharyngeal, psoas, or paraspinal muscle abscess) may cause SEA as well.[16]
Less‐frequent mechanisms include direct inoculation and trauma. Infection of the epidural space can occur in association with spinal surgery, placement of an epidural catheter, or spinal injections. Grewal et al. reported in 2006 that 1 in 1000 surgical and 1 in 2000 obstetric patients develop SEA following epidural nerve block.[17] Hematoma secondary to an osseous or ligamentous injury can become seeded by bacteria, leading to abscess formation.[18]
Development of Neurologic Symptoms
There are several proposed mechanisms by which SEA can produce neurologic dysfunction. The first theory is that the compressive effect of an expanding abscess decreases blood flow to the neuronal tissue.[4] Improvement of neurologic function following surgical decompression lends credence to this theory. A second potential mechanism is a loss of blood flow due to local vascular inflammation from the infection. Local arteritis may decrease inflow to the cord parenchyma. This theoretical mechanism offers an explanation for the rapid onset of profound neurologic compromise in some cases.[19, 20] Infection can also result in venous thrombophlebitis, which produces ischemic injury due to impaired outflow. Postmortem examination supports this hypothesis; autopsy has revealed local thrombosis of the leptomeningeal vessels adjacent to the level of SEA.[4] All of these mechanisms are probably involved to some degree in any given case of neurologic compromise.
PATIENT HISTORY
Patients present with a wide variety of complaints and confounding variables that complicate the diagnosis (eg, medical comorbidities, psychiatric disease, chronic pain, dementia, or preexistent nonambulatory status). Ninety‐five percent of patients with SEA have a chief complaint of axial spinal pain.[1] Approximately half of patients report fever, and 47% complain of weakness in either the upper or lower extremities.[9] The classic triad of fever, spine pain, and neurological deficits presents in only a minority of patients, with rates ranging from 13% to 37%.[1, 3] Additionally, 1 study found that the sensitivity of this triad was a mere 8%.[1]
The physician should inquire about comorbid conditions associated with SEA, including diabetes, kidney disease, and history of drug use. Any recent infection at a remote site, such as cellulitis or urinary tract infection, should also be investigated. As many as 44% of patients with vertebral osteomyelitis have an associated SEA; conversely, osteomyelitis may be present in up to 80% of patients with SEA.[2, 13] Spinal procedures such as epidural[13] or facet joint injections,[14] placement of hemodialysis catheters,[21] acupuncture,[22] and tattoos[23] have also been implicated as risk factors.
PHYSICAL EXAM
Physical exam findings range from subtle back tenderness to severe neurologic deficits and complete paralysis. Spinal tenderness is elicited in up to 75% of patients, with equal rates of focal and diffuse back tenderness.[1, 4] Radicular symptoms are evident in 12% to 47% of patients, presenting as weakness identified in 26% to 60% and altered sensation in up to 67% of patients.[1, 4, 8, 11, 19] One study revealed that 71% of patients have an abnormal neurologic exam at presentation, including paresthesias (39%), motor weakness (39%), and loss of bladder and bowel control (27%).[3] Thus, a thorough neurologic exam is vital in SEA evaluation.
Heusner staged patients based on their clinical findings (Table 2). Because the evolution of symptoms can be variable and rapidly progressive[4, 24] to stage 3 or 4, documenting subtle abnormalities on initial exam and monitoring for changes are important. Many patients initially present in stage 1 or 2 but remain undiagnosed until progression to stage 3 or 4.[24]
| Stage | Symptoms |
|---|---|
| I | Back pain |
| II | Radiculopathy, neck pain, reflex changes |
| III | Paresthesia, weakness, bladder symptoms |
| IV | Paralysis |
DIAGNOSTIC WORKUP
Laboratory Testing
Routine tests should include a white blood cell count (WBC), erythrocyte sedimentation rate (ESR), and C‐reactive protein (CRP). ESR has been shown to be the most sensitive and specific marker of SEA (Table 3). In a study of 63 patients matched with 126 controls, ESR was greater than 20 in 98% of cases and only 21% of controls.[1] Another study of 55 patients found the sensitivity and specificity of ESR to be 100% and 67%, respectively.[25] White count is less specific, with leukocytosis present in approximately two‐thirds of patients.[1, 25] CRP level rises faster than ESR in the setting of inflammation and also returns to baseline faster. As such, CRP is a useful means of monitoring the response to treatment.[1, 19]
|
| 1. Patient assessment |
| Evaluate for symptom triad (back pain, fever, neurologic dysfunction) |
| Assess for common comorbidities (diabetes, IV drug abuse, spinal trauma, ESRD, immunosuppressant therapy, recent spine procedure, systemic or local infection) |
| 2. Laboratory evaluation |
| Essential: ESR (most sensitive and specific), CRP, WBCs, blood cultures, UA |
| : echocardiogram, TB evaluation |
| No role: lumbar puncture |
| 3. Imaging |
| Gold standard: MRI w/gadolinium (90% sensitive) |
| If MRI contraindicated: CT myelogram |
| 4. Obtain tissue sample |
| Gold standard: open surgical biopsy, debridement fixation. |
| If patient unstable, diagnosis indeterminate, or no neuro symptoms: IR‐guided biopsy before surgery |
| 5. Antibiotics (once tissue sample obtained) |
| Empiric vancomycin plus third‐generation cephalosporin or aminoglycoside |
| Vancomycin only acceptable as monotherapy in patient with early diagnosis and no neurologic symptoms |
| 6. Surgical management (only if not done as step 4) |
| Early surgical consult recommended. Pursue surgical intervention if patient is stable for OR and neuro symptoms progressing or abscess refractory to antibiotics. Best outcomes occur with combined antibiotics and surgical debridement. |
Blood cultures are important to obtain at initial evaluation. Bacteremia from the offending organism may be present in up to 60% of cases; 60% to 90% of cases of bacteremia are caused by MRSA.[26] Urine culture should also be obtained in all patients. Sputum cultures should not be routinely obtained, but it may be beneficial in select patients with history of chronic obstructive pulmonary disease, new cough, or abnormality on chest radiographs. Transthoracic or transesophageal echocardiogram may be recommended for bacteremic patients, those with new heart murmur, or those with a history of IV drug abuse.
In patients in whom tuberculosis may be a potential risk, a tuberculin test with purified protein derivate can help rule out this organism. However, false‐negative testing may occur in up to 15% of patients, particularly the immunocompromised.[27] Patients with false positive results or prior vaccination with bacille Calmette‐Gurin (BCG) can be tested with QuantiFERON gold testing. These time‐consuming and expensive tests should not delay treatment with an empiric agent if tuberculosis is suspected due to patient risk factors or exposure, though an infectious disease specialist should be consulted before initiating treatment for tuberculosis.
Lumbar puncture should not be performed routinely in cases of suspected SEA, primarily due to the risk of bacterial contamination of the cerebrospinal fluid (CSF). Additionally, the increased protein and inflammatory cells seen in the CSF are nonspecific markers of parameningeal inflammation.[15]
Imaging
When the diagnosis of SEA is suspected based on clinical findings, MRI with gadolinium should be obtained first, as it is 90% sensitive for diagnosing SEA (Figure 1).[3] In patients unable to undergo MRI (eg, pacemaker), a computed tomography (CT) myelogram should be obtained instead. The study should be performed on an emergent basis, and the entire spine should be imaged due to the risk of noncontiguous lesions. Patients with plain radiograph and/or CT scan findings of bone lysis suggestive of vertebral osteomyelitis should also be evaluated with MRI if able.
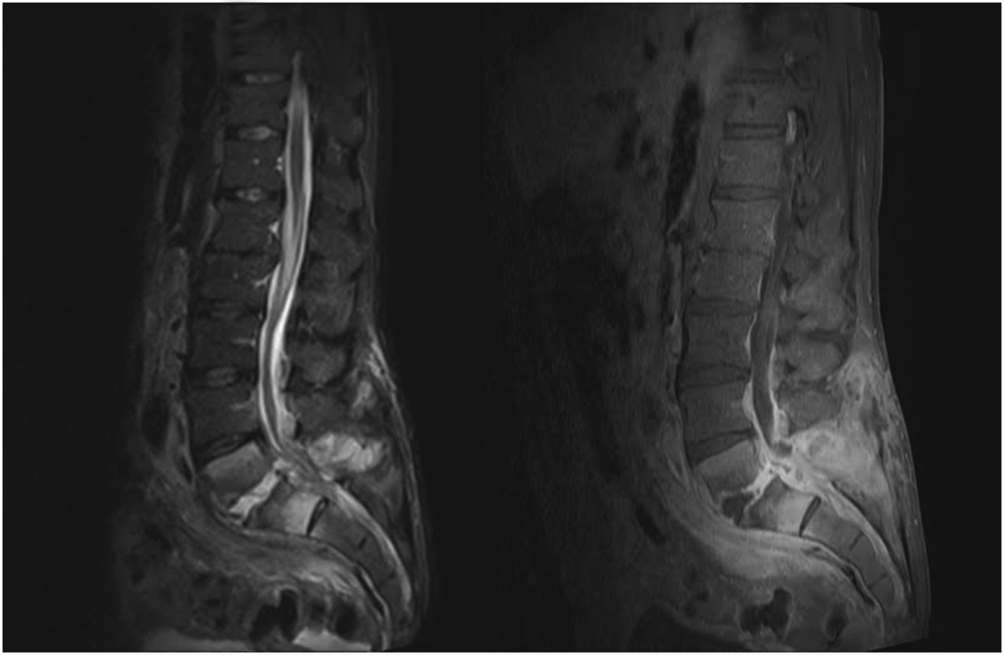
MRI with contrast can often differentiate SEA from malignancy and other space‐occupying lesions.[28] Plain radiography can rule out other causes of back pain, such as trauma and degenerative disc disease, but it cannot demonstrate SEA. One study found that x‐rays showed pathology in only 16.6% of patients with SEA.[29]
Patients with exam findings and laboratory studies concerning for SEA who present to a community hospital without MRI capability should be transferred to a tertiary care center for advanced imaging and potential emergent treatment.
Tissue Culture
Though MRI is essential to the workup of SEA, biopsy with cultures allows for a definitive diagnosis.[30] Cultures may be obtained in the operating room or interventional radiology (IR) suite via fine‐needle aspiration or core‐needle biopsy under CT guidance, if available.[30, 31] CT‐guided bone biopsy, which has a sensitivity of 81% and specificity of 100%, may also be performed when vertebral osteomyelitis is present.[32] Biopsy by IR should be considered before surgical intervention, when the patient has no evidence of progressive neurologic deficit, the diagnosis is unclear, or the patient is too high risk for surgical intervention. In very high‐risk surgical patients, IR aspiration may be curative. Lyu et al. describe a case of refractory SEA treated with percutaneous CT‐guided needle aspiration alone, though they note that surgical debridement is preferred when possible.[31]
NONSURGICAL VERSUS SURGICAL MANAGEMENT
SEA may be treated without surgery in carefully selected patients. Savage et al. studied 52 patients, and found that nonsurgical management was often effective in patients who were completely neurologically intact at initial presentation. In their study, only 3 patients needed to undergo surgery due to development of new neurologic symptoms.[33] However, other studies report neurologic symptoms in 71% of patients at initial diagnosis.[3]
Adogwa et al. reviewed surgical versus nonsurgical management in elderly patients (>50 years old) over 15 years.[10] Their study included 30 patients treated operatively and 52 who received antibiotics alone. The decision for surgical management was at the surgeons' discretion; however, most patients with grade 2 or 3 symptoms underwent surgery, and those with paraplegia or quadriplegia for >48 hours did not. The authors found no clinically significant difference in outcome between these 2 groups and cautioned against surgical intervention for elderly patients with multiple comorbidities. It is worth noting, however, that 7/30 (23%) patients treated with surgery versus 5/52 (10%) treated conservatively had improved neurological outcomes (P = 0.03).[10] Numerous other studies have also found that neurologic status at presentation is the most important predictor of surgical outcomes.[7, 15, 34]
Patel et al. performed a study analyzing risk factors for failure of medical management, and found that diabetes mellitus, CRP >115, WBC >12.5, and positive blood cultures were predictors of failure.[9] Patients with >3 of these risk factors required surgery 76.9% of the time compared to 40.2% with 2, 35.4% with 1, and 8.3% with none.[9] The authors also found that surgical patients experienced better mean improvement than patients who failed nonsurgical treatment and subsequently underwent surgical decompression.[9]
In a follow‐up study by Alton et al., 62 patients were treated with either nonsurgical or surgical management. Indications for surgery in their study included a neurologic deficit at initial presentation or the development of a new neurological deficit while undergoing treatment. Twenty‐four patients presented without deficits and underwent nonsurgical management, but only 6 (25%) were treated successfully, as defined by stable or improved neurological status following therapy. In contrast, none of the 38 patients who underwent therapy with both IV antibiotics and emergent surgical management within 24 hours experienced deterioration in the neurologic status. For the 18 patients who failed nonsurgical management, surgery was performed within an average of 7 days. This group experienced improvement following surgery, but their neurological status improved less than those who underwent early surgery.[35]
These recent studies demonstrate that medical management may be an option for patients who are diagnosed early and present without neurologic deficits, but surgery is the mainstay of treatment for most patients. Additionally, patients who experience a delay in operative management often do not recover function as well as those patients who undergo emergent surgical debridement.[35] A delay in diagnosis has also been shown to lead to increase in lawsuits against providers. French et al. found an increase in verdicts against the provider when a delay in diagnosis >48 hours was present, irrespective of the degree of permanent neurologic dysfunction.[36]
TREATMENT AND FOLLOW‐UP
Unless the patient is septic and hemodynamically unstable, antibiotics should be held until a tissue sample is obtained for speciation and culture, either in the IR suite or the operating room. Once cultures have been obtained, broad‐spectrum IV antibiotics, usually vancomycin for gram‐positive coverage in combination with a third‐generation cephalosporin or aminoglycoside for additional gram‐negative coverage, should be started until organism sensitivities are determined and specific antibiotics can be administered.[3] Antibiotic therapy should continue for 4 to 6 weeks, though therapy may be administered for 8 weeks or longer in patients with vertebral osteomyelitis or immunocompromised patients.[19] An infectious disease specialist should be involved to determine the duration of therapy and transition to oral antibiotics.
As noted earlier, biopsy by IR should be considered when a trial of nonsurgical management is planned for a patient with no evidence of neurologic involvement, when the diagnosis is unclear after imaging, or when the patient is too high risk for surgery. Otherwise, surgical management is standard and should involve extensive surgical debridement with possible fusion if there is considerable structural compromise to the spinal column. After definitive treatment, repeat MRI is not recommended in the follow‐up of SEA; however, it is useful if there is concern for recurrence, as indicated by new fevers or a rising leukocyte count or inflammatory markers, especially CRP. Inflammatory labs should be obtained every 1 to 2 weeks to monitor for resolution of the infectious process.
CONCLUSION
Spinal epidural abscess is a potentially devastating condition that can be difficult to diagnose. Although uncommon, the triad of axial spine pain, fever, and new‐onset neurologic dysfunction are concerning. Other factors that increase the likelihood of SEA include a history of diabetes, IV drug abuse, spinal trauma, end‐stage renal disease, immunosuppressant therapy, recent invasive spine procedures, and concurrent infection of the skin or urinary tract. In patients with suspected SEA, inflammatory laboratory studies should be obtained along with gadolinium‐enhanced MRI of the entire spinal axis. Once the diagnosis is established, spine surgery and infectious disease consultation is mandatory, and IR biopsy may be appropriate in some cases. The rapidity of diagnosis and initiation of treatment are critical factors in optimizing patient outcome.
Disclosures
Mark A. Palumbo, MD, receives research funding from Globus Medical and is a paid consultant for Stryker. Alan H. Daniels, MD, is a paid consultant for Stryker and Osseous. No funding was obtained in support of this work.
- , , , et al. The clinical presentation and impact of diagnostic delays on emergency department patients with spinal epidural abscess. J Emerg Med. 2004;26:285–291.
- , , , . Spinal epidural abscess. N Engl J Med. 1975;293:463–468.
- , , , et al. Spinal epidural abscess: contemporary trends in etiology, evaluation, and management. Surg Neurol. 1999;52:189–196; discussion 197.
- . Spinal epidural abscess. N Engl J Med. 2006;355:2012–2020.
- , . Mortality, complication risk, and total charges after the treatment of epidural abscess. Spine J. 2015;15:249–255.
- , . Spinal epidural abscess. Curr Infect Dis Rep. 2004;16:436.
- , , , . Spinal epidural abscess: a ten‐year perspective. Neurosurgery. 1980;27:177–184.
- , , . Spinal epidural abscess: a meta‐analysis of 915 patients. Neurosurg Rev. 2000;23:175–204; discussion 205.
- , , , , , . Spinal epidural abscesses: risk factors, medical versus surgical management, a retrospective review of 128 cases. Spine J. 2014;14:326–330.
- , , , et al. Spontaneous spinal epidural abscess in patients 50 years of age and older: a 15‐year institutional perspective and review of the literature: clinical article. J Neurosurg Spine. 2014;20:344–349.
- , . Spinal epidural abscesses: clinical manifestations, prognostic factors, and outcomes. Neurosurgery. 2002;51:79–85; discussion 86–87.
- , , . Infectious agents in spinal epidural abscesses. Neurology. 1980;30:844–850.
- , , , , , . Cervical epidural abscess after epidural steroid injection. Spine (Phila Pa 1976). 2004;29:E7–E9.
- , , . Spinal epidural abscesses in adults: review and report of iatrogenic cases. Scand J Infect Dis. 1990;22:249–257.
- , , , , . Bacterial spinal epidural abscess. Review of 43 cases and literature survey. Medicine (Baltimore). 1992;71:369–385.
- , , , , . Spinal epidural abscess: the importance of early diagnosis and treatment. J Neurol Neurosurg Psychiatry. 1998;65:209–212.
- , , . Epidural abscesses. Br J Anaesth. 2006;96:292–302.
- , . Spinal epidural abscess. Med Clin North Am. 1985;69;375–384.
- , , , . Spinal epidural abscess. J Emerg Med. 2010;39:384–390.
- , , . Spinal epidural abscess: clinical presentation, management and outcome [comment on Curry WT, Hoh BL, Hanjani SA, et al. Surg Neurol. 2005;63:364–371). Surg Neurol. 2005;64:279.
- , , , , . Routine replacement of tunneled, cuffed, hemodialysis catheters eliminates paraspinal/vertebral infections in patients with catheter‐associated bacteremia. Am J Nephrol. 2003;23:202–207.
- , . Paraplegia caused by spinal infection after acupuncture. Spinal Cord. 2006;44:258–259.
- , , , . Spinal epidural abscess after tattooing. Clin Infect Dis. 1999;29:225–226.
- . Nontuberculous spinal epidural infections. N Engl J Med. 1948;239:845–854.
- , , , . Prospective evaluation of a clinical decision guideline to diagnose spinal epidural abscess in patients who present to the emergency department with spine pain. J Neurosurg Spine. 2011;14:765–770.
- , , , . Spinal epidural abscess: clinical presentation, management, and outcome. Surg Neurol. 2005;63:364–371; discussion 371.
- , . Bone and joint tuberculosis. Eur Spine J. 2013;22:556–566.
- , . Surgical management of spinal epidural abscess: selection of approach based on MRI appearance. J Clin Neurosci. 2004;11:130–133.
- , . Infection as a cause of spinal cord compression: a review of 36 spinal epidural abscess cases. Acta Neurochir (Wien). 2000;142:17–23.
- , , , et al. Spondyloarthropathy from long‐term hemodialysis. Radiology. 1988;167:761–764.
- , , , . Spinal epidural abscess successfully treated with percutaneous, computed tomography‐guided, needle aspiration and parenteral antibiotic therapy: case report and review of the literature. Neurosurgery. 2002;51:509–512; discussion 512.
- , , , . CT‐guided core biopsy of subchondral bone and intervertebral space in suspected spondylodiskitis. AJR Am J Roentgenol. 2006;186:977–980.
- , , . Spinal epidural abscess: early clinical outcome in patients treated medically. Clin Orthop. 2005;439:56–60.
- , . Update on spinal epidural abscess: 35 cases and review of the literature. Rev Infect Dis. 1987;9:265–274.
- , , , , , . Is there a difference in neurologic outcome in medical versus early operative management of cervical epidural abscesses? Spine J. 2015;15:10–17.
- , , , . Medicolegal cases for spinal epidural hematoma and spinal epidural abscess. Orthopedics. 2013;36:48–53.
- , , , . Spinal epidural abscess—experience with 46 patients and evaluation of prognostic factors. J Infect. 2002;45:76–81.
Spinal epidural abscess (SEA) is caused by a suppurative infection in the epidural space. The mass effect of the abscess can compress and reduce blood flow to the spinal cord, conus medullaris, or cauda equina. Left untreated, the infection can lead to sensory loss, muscle weakness, visceral dysfunction, sepsis, and even death. Early diagnosis is essential to limit morbidity and neurologic injury. The classic triad of fever, axial pain, and neurological deficit occurs in as few as 13% of patients, highlighting the diagnostic challenge associated with SEA.[1]
This investigation reviews the current literature on SEA epidemiology, clinical findings, laboratory data, and treatment methods, with particular focus on nonsurgical versus surgical treatment. Our primary objective was to educate the clinician on when to suspect SEA and how to execute an appropriate diagnostic evaluation.
INCIDENCE AND EPIDEMIOLOGY
In 1975, the incidence of SEA was reported to be 0.2 to 2.0 per 10,000 hospital admissions.[2] Two decades later, a study of tertiary referral centers documented a much higher rate of 12.5 per 10,000 admissions.[3] The reported incidence continues to rise and has doubled in the past decade, with SEA currently representing approximately 10% of all primary spine infections.[4, 5] Potential explanations for this increasing incidence include aging of the population, an increasing prevalence of diabetes, increasing intravenous drug abuse, more widespread use of advanced immunosuppressive regimens, and an increased rate of invasive spinal procedures.[6, 7, 8] Other factors contributing to the rising incidence include increased detection due to the greater accessibility of magnetic resonance imaging (MRI) and increased reporting as a result of the concentration of cases at tertiary referral centers.
SEA is most common in patients older than 60 years[7] and those with multiple medical comorbidities. A review of over 30,000 patients found that the average number of comorbidities in patients who underwent surgical intervention for SEA was 6, ranging from 0 to 20.[5] The same study noted that diabetes was the most frequently associated disease (30% of patients), followed by chronic lung disease (19%), renal failure (13%), and obesity (13%) (Table 1).[5] A history of invasive spine interventions is an additional risk factor; between 14% and 22% occur as a result of spine surgery or percutaneous spine procedures (eg, epidural steroid injections).[8, 9] Regardless of pathogenesis, the rate of permanent neurologic injury after SEA is 30% to 50%, and the mortality rate ranges from 10% to 20%.[4, 5, 8, 10]
| Medical Comorbidity | Prevalence (%) |
|---|---|
| |
| Diabetes mellitus | 1546 |
| IV drug use | 437 |
| Spinal trauma | 533 |
| End‐stage renal disease | 213 |
| Immunosuppressant therapy | 716 |
| Cancer | 215 |
| HIV/AIDS | 29 |
MISSED DIAGNOSIS
Despite the availability of advanced imaging, rates of misdiagnosis at initial presentation remain substantial, with current estimates ranging from 11% to 75%.[4, 9] Back and neck pain symptoms are ubiquitous and nonspecific, often making the diagnosis difficult. Repeated emergency room visits for pain are common in patients who are eventually diagnosed with SEA. Davis et al. found that 51% of 63 patients present to the emergency room at least 2 or more times prior to diagnosis; 11% present 3 or more times.[1]
PATHOPHYSIOLOGY
Microbiology
Staphylococcus aureus, including methicillin resistant S aureus (MRSA), accounts for two‐thirds of all infections.[3] S aureus infection of the spine may occur in the setting of surgical intervention or concomitant skin infection, although it often occurs without an identifiable source. Historically, MRSA has been reported to be responsible for 15% of all staphylococcal infections of the epidural space; some institutions report MRSA rates as high as 40%.[4] S epidermidis is another common pathogen, which is most often encountered following spinal surgery, epidural catheter insertion, and spinal injections.[11] Gram‐negative infections are less common. Escherichia coli is characteristically isolated in patients with active urinary tract infections, and Pseudomonas aeruginosa is more common in intravenous (IV) drug users.[12] Rare causes of SEA include anaerobes such as Bacteroides,[13] various parasites, and fungal organisms such as actinomyces, nocardia, and mycobacteria.[8]
Mechanism of Inoculation
Infections may enter the epidural space by 4 mechanisms: hematogenous spread, direct extension, inoculation via spinal procedure, and trauma. Hematogenous spread from an existing infection is the most common mechanism.[14] Seeding of the epidural space from transient bacteremia after dental procedures has also been reported.[13]
The second most common mechanism of infection is direct spread from an infected component of the vertebral column or paraspinal soft tissues. Most commonly, this takes the form of an anterior SEA in association with vertebral body osteomyelitis. Septic arthritis of a facet joint can also cause a secondary infection of the posterior epidural space.[4, 15] Direct spread from other posterior structures (eg, retropharyngeal, psoas, or paraspinal muscle abscess) may cause SEA as well.[16]
Less‐frequent mechanisms include direct inoculation and trauma. Infection of the epidural space can occur in association with spinal surgery, placement of an epidural catheter, or spinal injections. Grewal et al. reported in 2006 that 1 in 1000 surgical and 1 in 2000 obstetric patients develop SEA following epidural nerve block.[17] Hematoma secondary to an osseous or ligamentous injury can become seeded by bacteria, leading to abscess formation.[18]
Development of Neurologic Symptoms
There are several proposed mechanisms by which SEA can produce neurologic dysfunction. The first theory is that the compressive effect of an expanding abscess decreases blood flow to the neuronal tissue.[4] Improvement of neurologic function following surgical decompression lends credence to this theory. A second potential mechanism is a loss of blood flow due to local vascular inflammation from the infection. Local arteritis may decrease inflow to the cord parenchyma. This theoretical mechanism offers an explanation for the rapid onset of profound neurologic compromise in some cases.[19, 20] Infection can also result in venous thrombophlebitis, which produces ischemic injury due to impaired outflow. Postmortem examination supports this hypothesis; autopsy has revealed local thrombosis of the leptomeningeal vessels adjacent to the level of SEA.[4] All of these mechanisms are probably involved to some degree in any given case of neurologic compromise.
PATIENT HISTORY
Patients present with a wide variety of complaints and confounding variables that complicate the diagnosis (eg, medical comorbidities, psychiatric disease, chronic pain, dementia, or preexistent nonambulatory status). Ninety‐five percent of patients with SEA have a chief complaint of axial spinal pain.[1] Approximately half of patients report fever, and 47% complain of weakness in either the upper or lower extremities.[9] The classic triad of fever, spine pain, and neurological deficits presents in only a minority of patients, with rates ranging from 13% to 37%.[1, 3] Additionally, 1 study found that the sensitivity of this triad was a mere 8%.[1]
The physician should inquire about comorbid conditions associated with SEA, including diabetes, kidney disease, and history of drug use. Any recent infection at a remote site, such as cellulitis or urinary tract infection, should also be investigated. As many as 44% of patients with vertebral osteomyelitis have an associated SEA; conversely, osteomyelitis may be present in up to 80% of patients with SEA.[2, 13] Spinal procedures such as epidural[13] or facet joint injections,[14] placement of hemodialysis catheters,[21] acupuncture,[22] and tattoos[23] have also been implicated as risk factors.
PHYSICAL EXAM
Physical exam findings range from subtle back tenderness to severe neurologic deficits and complete paralysis. Spinal tenderness is elicited in up to 75% of patients, with equal rates of focal and diffuse back tenderness.[1, 4] Radicular symptoms are evident in 12% to 47% of patients, presenting as weakness identified in 26% to 60% and altered sensation in up to 67% of patients.[1, 4, 8, 11, 19] One study revealed that 71% of patients have an abnormal neurologic exam at presentation, including paresthesias (39%), motor weakness (39%), and loss of bladder and bowel control (27%).[3] Thus, a thorough neurologic exam is vital in SEA evaluation.
Heusner staged patients based on their clinical findings (Table 2). Because the evolution of symptoms can be variable and rapidly progressive[4, 24] to stage 3 or 4, documenting subtle abnormalities on initial exam and monitoring for changes are important. Many patients initially present in stage 1 or 2 but remain undiagnosed until progression to stage 3 or 4.[24]
| Stage | Symptoms |
|---|---|
| I | Back pain |
| II | Radiculopathy, neck pain, reflex changes |
| III | Paresthesia, weakness, bladder symptoms |
| IV | Paralysis |
DIAGNOSTIC WORKUP
Laboratory Testing
Routine tests should include a white blood cell count (WBC), erythrocyte sedimentation rate (ESR), and C‐reactive protein (CRP). ESR has been shown to be the most sensitive and specific marker of SEA (Table 3). In a study of 63 patients matched with 126 controls, ESR was greater than 20 in 98% of cases and only 21% of controls.[1] Another study of 55 patients found the sensitivity and specificity of ESR to be 100% and 67%, respectively.[25] White count is less specific, with leukocytosis present in approximately two‐thirds of patients.[1, 25] CRP level rises faster than ESR in the setting of inflammation and also returns to baseline faster. As such, CRP is a useful means of monitoring the response to treatment.[1, 19]
|
| 1. Patient assessment |
| Evaluate for symptom triad (back pain, fever, neurologic dysfunction) |
| Assess for common comorbidities (diabetes, IV drug abuse, spinal trauma, ESRD, immunosuppressant therapy, recent spine procedure, systemic or local infection) |
| 2. Laboratory evaluation |
| Essential: ESR (most sensitive and specific), CRP, WBCs, blood cultures, UA |
| : echocardiogram, TB evaluation |
| No role: lumbar puncture |
| 3. Imaging |
| Gold standard: MRI w/gadolinium (90% sensitive) |
| If MRI contraindicated: CT myelogram |
| 4. Obtain tissue sample |
| Gold standard: open surgical biopsy, debridement fixation. |
| If patient unstable, diagnosis indeterminate, or no neuro symptoms: IR‐guided biopsy before surgery |
| 5. Antibiotics (once tissue sample obtained) |
| Empiric vancomycin plus third‐generation cephalosporin or aminoglycoside |
| Vancomycin only acceptable as monotherapy in patient with early diagnosis and no neurologic symptoms |
| 6. Surgical management (only if not done as step 4) |
| Early surgical consult recommended. Pursue surgical intervention if patient is stable for OR and neuro symptoms progressing or abscess refractory to antibiotics. Best outcomes occur with combined antibiotics and surgical debridement. |
Blood cultures are important to obtain at initial evaluation. Bacteremia from the offending organism may be present in up to 60% of cases; 60% to 90% of cases of bacteremia are caused by MRSA.[26] Urine culture should also be obtained in all patients. Sputum cultures should not be routinely obtained, but it may be beneficial in select patients with history of chronic obstructive pulmonary disease, new cough, or abnormality on chest radiographs. Transthoracic or transesophageal echocardiogram may be recommended for bacteremic patients, those with new heart murmur, or those with a history of IV drug abuse.
In patients in whom tuberculosis may be a potential risk, a tuberculin test with purified protein derivate can help rule out this organism. However, false‐negative testing may occur in up to 15% of patients, particularly the immunocompromised.[27] Patients with false positive results or prior vaccination with bacille Calmette‐Gurin (BCG) can be tested with QuantiFERON gold testing. These time‐consuming and expensive tests should not delay treatment with an empiric agent if tuberculosis is suspected due to patient risk factors or exposure, though an infectious disease specialist should be consulted before initiating treatment for tuberculosis.
Lumbar puncture should not be performed routinely in cases of suspected SEA, primarily due to the risk of bacterial contamination of the cerebrospinal fluid (CSF). Additionally, the increased protein and inflammatory cells seen in the CSF are nonspecific markers of parameningeal inflammation.[15]
Imaging
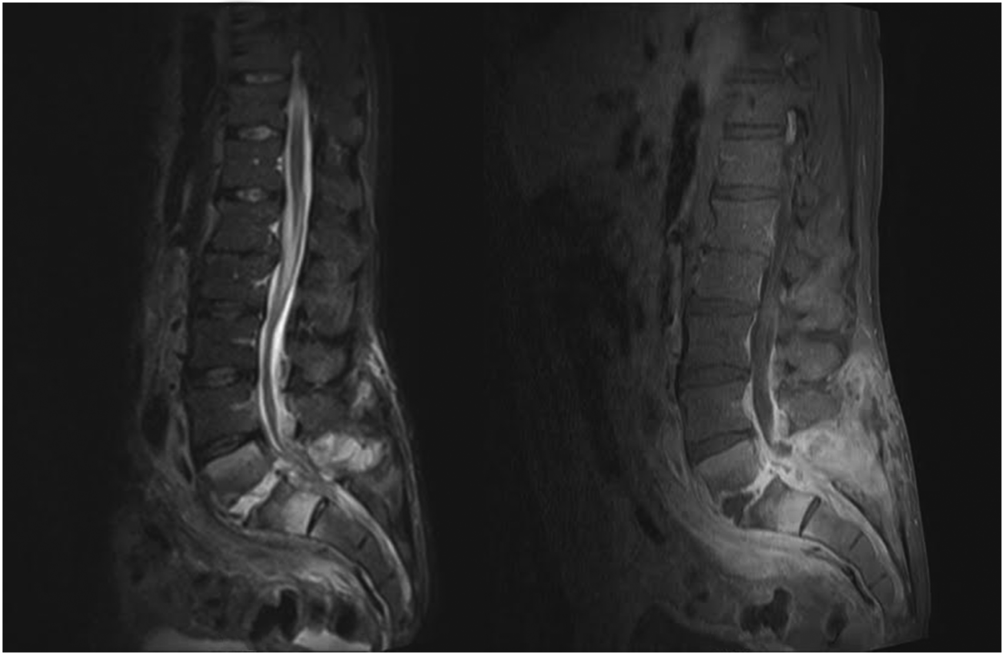
When the diagnosis of SEA is suspected based on clinical findings, MRI with gadolinium should be obtained first, as it is 90% sensitive for diagnosing SEA (Figure 1).[3] In patients unable to undergo MRI (eg, pacemaker), a computed tomography (CT) myelogram should be obtained instead. The study should be performed on an emergent basis, and the entire spine should be imaged due to the risk of noncontiguous lesions. Patients with plain radiograph and/or CT scan findings of bone lysis suggestive of vertebral osteomyelitis should also be evaluated with MRI if able.
MRI with contrast can often differentiate SEA from malignancy and other space‐occupying lesions.[28] Plain radiography can rule out other causes of back pain, such as trauma and degenerative disc disease, but it cannot demonstrate SEA. One study found that x‐rays showed pathology in only 16.6% of patients with SEA.[29]
Patients with exam findings and laboratory studies concerning for SEA who present to a community hospital without MRI capability should be transferred to a tertiary care center for advanced imaging and potential emergent treatment.
Tissue Culture
Though MRI is essential to the workup of SEA, biopsy with cultures allows for a definitive diagnosis.[30] Cultures may be obtained in the operating room or interventional radiology (IR) suite via fine‐needle aspiration or core‐needle biopsy under CT guidance, if available.[30, 31] CT‐guided bone biopsy, which has a sensitivity of 81% and specificity of 100%, may also be performed when vertebral osteomyelitis is present.[32] Biopsy by IR should be considered before surgical intervention, when the patient has no evidence of progressive neurologic deficit, the diagnosis is unclear, or the patient is too high risk for surgical intervention. In very high‐risk surgical patients, IR aspiration may be curative. Lyu et al. describe a case of refractory SEA treated with percutaneous CT‐guided needle aspiration alone, though they note that surgical debridement is preferred when possible.[31]
NONSURGICAL VERSUS SURGICAL MANAGEMENT
SEA may be treated without surgery in carefully selected patients. Savage et al. studied 52 patients, and found that nonsurgical management was often effective in patients who were completely neurologically intact at initial presentation. In their study, only 3 patients needed to undergo surgery due to development of new neurologic symptoms.[33] However, other studies report neurologic symptoms in 71% of patients at initial diagnosis.[3]
Adogwa et al. reviewed surgical versus nonsurgical management in elderly patients (>50 years old) over 15 years.[10] Their study included 30 patients treated operatively and 52 who received antibiotics alone. The decision for surgical management was at the surgeons' discretion; however, most patients with grade 2 or 3 symptoms underwent surgery, and those with paraplegia or quadriplegia for >48 hours did not. The authors found no clinically significant difference in outcome between these 2 groups and cautioned against surgical intervention for elderly patients with multiple comorbidities. It is worth noting, however, that 7/30 (23%) patients treated with surgery versus 5/52 (10%) treated conservatively had improved neurological outcomes (P = 0.03).[10] Numerous other studies have also found that neurologic status at presentation is the most important predictor of surgical outcomes.[7, 15, 34]
Patel et al. performed a study analyzing risk factors for failure of medical management, and found that diabetes mellitus, CRP >115, WBC >12.5, and positive blood cultures were predictors of failure.[9] Patients with >3 of these risk factors required surgery 76.9% of the time compared to 40.2% with 2, 35.4% with 1, and 8.3% with none.[9] The authors also found that surgical patients experienced better mean improvement than patients who failed nonsurgical treatment and subsequently underwent surgical decompression.[9]
In a follow‐up study by Alton et al., 62 patients were treated with either nonsurgical or surgical management. Indications for surgery in their study included a neurologic deficit at initial presentation or the development of a new neurological deficit while undergoing treatment. Twenty‐four patients presented without deficits and underwent nonsurgical management, but only 6 (25%) were treated successfully, as defined by stable or improved neurological status following therapy. In contrast, none of the 38 patients who underwent therapy with both IV antibiotics and emergent surgical management within 24 hours experienced deterioration in the neurologic status. For the 18 patients who failed nonsurgical management, surgery was performed within an average of 7 days. This group experienced improvement following surgery, but their neurological status improved less than those who underwent early surgery.[35]
These recent studies demonstrate that medical management may be an option for patients who are diagnosed early and present without neurologic deficits, but surgery is the mainstay of treatment for most patients. Additionally, patients who experience a delay in operative management often do not recover function as well as those patients who undergo emergent surgical debridement.[35] A delay in diagnosis has also been shown to lead to increase in lawsuits against providers. French et al. found an increase in verdicts against the provider when a delay in diagnosis >48 hours was present, irrespective of the degree of permanent neurologic dysfunction.[36]
TREATMENT AND FOLLOW‐UP
Unless the patient is septic and hemodynamically unstable, antibiotics should be held until a tissue sample is obtained for speciation and culture, either in the IR suite or the operating room. Once cultures have been obtained, broad‐spectrum IV antibiotics, usually vancomycin for gram‐positive coverage in combination with a third‐generation cephalosporin or aminoglycoside for additional gram‐negative coverage, should be started until organism sensitivities are determined and specific antibiotics can be administered.[3] Antibiotic therapy should continue for 4 to 6 weeks, though therapy may be administered for 8 weeks or longer in patients with vertebral osteomyelitis or immunocompromised patients.[19] An infectious disease specialist should be involved to determine the duration of therapy and transition to oral antibiotics.
As noted earlier, biopsy by IR should be considered when a trial of nonsurgical management is planned for a patient with no evidence of neurologic involvement, when the diagnosis is unclear after imaging, or when the patient is too high risk for surgery. Otherwise, surgical management is standard and should involve extensive surgical debridement with possible fusion if there is considerable structural compromise to the spinal column. After definitive treatment, repeat MRI is not recommended in the follow‐up of SEA; however, it is useful if there is concern for recurrence, as indicated by new fevers or a rising leukocyte count or inflammatory markers, especially CRP. Inflammatory labs should be obtained every 1 to 2 weeks to monitor for resolution of the infectious process.
CONCLUSION
Spinal epidural abscess is a potentially devastating condition that can be difficult to diagnose. Although uncommon, the triad of axial spine pain, fever, and new‐onset neurologic dysfunction are concerning. Other factors that increase the likelihood of SEA include a history of diabetes, IV drug abuse, spinal trauma, end‐stage renal disease, immunosuppressant therapy, recent invasive spine procedures, and concurrent infection of the skin or urinary tract. In patients with suspected SEA, inflammatory laboratory studies should be obtained along with gadolinium‐enhanced MRI of the entire spinal axis. Once the diagnosis is established, spine surgery and infectious disease consultation is mandatory, and IR biopsy may be appropriate in some cases. The rapidity of diagnosis and initiation of treatment are critical factors in optimizing patient outcome.
Disclosures
Mark A. Palumbo, MD, receives research funding from Globus Medical and is a paid consultant for Stryker. Alan H. Daniels, MD, is a paid consultant for Stryker and Osseous. No funding was obtained in support of this work.
Spinal epidural abscess (SEA) is caused by a suppurative infection in the epidural space. The mass effect of the abscess can compress and reduce blood flow to the spinal cord, conus medullaris, or cauda equina. Left untreated, the infection can lead to sensory loss, muscle weakness, visceral dysfunction, sepsis, and even death. Early diagnosis is essential to limit morbidity and neurologic injury. The classic triad of fever, axial pain, and neurological deficit occurs in as few as 13% of patients, highlighting the diagnostic challenge associated with SEA.[1]
This investigation reviews the current literature on SEA epidemiology, clinical findings, laboratory data, and treatment methods, with particular focus on nonsurgical versus surgical treatment. Our primary objective was to educate the clinician on when to suspect SEA and how to execute an appropriate diagnostic evaluation.
INCIDENCE AND EPIDEMIOLOGY
In 1975, the incidence of SEA was reported to be 0.2 to 2.0 per 10,000 hospital admissions.[2] Two decades later, a study of tertiary referral centers documented a much higher rate of 12.5 per 10,000 admissions.[3] The reported incidence continues to rise and has doubled in the past decade, with SEA currently representing approximately 10% of all primary spine infections.[4, 5] Potential explanations for this increasing incidence include aging of the population, an increasing prevalence of diabetes, increasing intravenous drug abuse, more widespread use of advanced immunosuppressive regimens, and an increased rate of invasive spinal procedures.[6, 7, 8] Other factors contributing to the rising incidence include increased detection due to the greater accessibility of magnetic resonance imaging (MRI) and increased reporting as a result of the concentration of cases at tertiary referral centers.
SEA is most common in patients older than 60 years[7] and those with multiple medical comorbidities. A review of over 30,000 patients found that the average number of comorbidities in patients who underwent surgical intervention for SEA was 6, ranging from 0 to 20.[5] The same study noted that diabetes was the most frequently associated disease (30% of patients), followed by chronic lung disease (19%), renal failure (13%), and obesity (13%) (Table 1).[5] A history of invasive spine interventions is an additional risk factor; between 14% and 22% occur as a result of spine surgery or percutaneous spine procedures (eg, epidural steroid injections).[8, 9] Regardless of pathogenesis, the rate of permanent neurologic injury after SEA is 30% to 50%, and the mortality rate ranges from 10% to 20%.[4, 5, 8, 10]
| Medical Comorbidity | Prevalence (%) |
|---|---|
| |
| Diabetes mellitus | 1546 |
| IV drug use | 437 |
| Spinal trauma | 533 |
| End‐stage renal disease | 213 |
| Immunosuppressant therapy | 716 |
| Cancer | 215 |
| HIV/AIDS | 29 |
MISSED DIAGNOSIS
Despite the availability of advanced imaging, rates of misdiagnosis at initial presentation remain substantial, with current estimates ranging from 11% to 75%.[4, 9] Back and neck pain symptoms are ubiquitous and nonspecific, often making the diagnosis difficult. Repeated emergency room visits for pain are common in patients who are eventually diagnosed with SEA. Davis et al. found that 51% of 63 patients present to the emergency room at least 2 or more times prior to diagnosis; 11% present 3 or more times.[1]
PATHOPHYSIOLOGY
Microbiology
Staphylococcus aureus, including methicillin resistant S aureus (MRSA), accounts for two‐thirds of all infections.[3] S aureus infection of the spine may occur in the setting of surgical intervention or concomitant skin infection, although it often occurs without an identifiable source. Historically, MRSA has been reported to be responsible for 15% of all staphylococcal infections of the epidural space; some institutions report MRSA rates as high as 40%.[4] S epidermidis is another common pathogen, which is most often encountered following spinal surgery, epidural catheter insertion, and spinal injections.[11] Gram‐negative infections are less common. Escherichia coli is characteristically isolated in patients with active urinary tract infections, and Pseudomonas aeruginosa is more common in intravenous (IV) drug users.[12] Rare causes of SEA include anaerobes such as Bacteroides,[13] various parasites, and fungal organisms such as actinomyces, nocardia, and mycobacteria.[8]
Mechanism of Inoculation
Infections may enter the epidural space by 4 mechanisms: hematogenous spread, direct extension, inoculation via spinal procedure, and trauma. Hematogenous spread from an existing infection is the most common mechanism.[14] Seeding of the epidural space from transient bacteremia after dental procedures has also been reported.[13]
The second most common mechanism of infection is direct spread from an infected component of the vertebral column or paraspinal soft tissues. Most commonly, this takes the form of an anterior SEA in association with vertebral body osteomyelitis. Septic arthritis of a facet joint can also cause a secondary infection of the posterior epidural space.[4, 15] Direct spread from other posterior structures (eg, retropharyngeal, psoas, or paraspinal muscle abscess) may cause SEA as well.[16]
Less‐frequent mechanisms include direct inoculation and trauma. Infection of the epidural space can occur in association with spinal surgery, placement of an epidural catheter, or spinal injections. Grewal et al. reported in 2006 that 1 in 1000 surgical and 1 in 2000 obstetric patients develop SEA following epidural nerve block.[17] Hematoma secondary to an osseous or ligamentous injury can become seeded by bacteria, leading to abscess formation.[18]
Development of Neurologic Symptoms
There are several proposed mechanisms by which SEA can produce neurologic dysfunction. The first theory is that the compressive effect of an expanding abscess decreases blood flow to the neuronal tissue.[4] Improvement of neurologic function following surgical decompression lends credence to this theory. A second potential mechanism is a loss of blood flow due to local vascular inflammation from the infection. Local arteritis may decrease inflow to the cord parenchyma. This theoretical mechanism offers an explanation for the rapid onset of profound neurologic compromise in some cases.[19, 20] Infection can also result in venous thrombophlebitis, which produces ischemic injury due to impaired outflow. Postmortem examination supports this hypothesis; autopsy has revealed local thrombosis of the leptomeningeal vessels adjacent to the level of SEA.[4] All of these mechanisms are probably involved to some degree in any given case of neurologic compromise.
PATIENT HISTORY
Patients present with a wide variety of complaints and confounding variables that complicate the diagnosis (eg, medical comorbidities, psychiatric disease, chronic pain, dementia, or preexistent nonambulatory status). Ninety‐five percent of patients with SEA have a chief complaint of axial spinal pain.[1] Approximately half of patients report fever, and 47% complain of weakness in either the upper or lower extremities.[9] The classic triad of fever, spine pain, and neurological deficits presents in only a minority of patients, with rates ranging from 13% to 37%.[1, 3] Additionally, 1 study found that the sensitivity of this triad was a mere 8%.[1]
The physician should inquire about comorbid conditions associated with SEA, including diabetes, kidney disease, and history of drug use. Any recent infection at a remote site, such as cellulitis or urinary tract infection, should also be investigated. As many as 44% of patients with vertebral osteomyelitis have an associated SEA; conversely, osteomyelitis may be present in up to 80% of patients with SEA.[2, 13] Spinal procedures such as epidural[13] or facet joint injections,[14] placement of hemodialysis catheters,[21] acupuncture,[22] and tattoos[23] have also been implicated as risk factors.
PHYSICAL EXAM
Physical exam findings range from subtle back tenderness to severe neurologic deficits and complete paralysis. Spinal tenderness is elicited in up to 75% of patients, with equal rates of focal and diffuse back tenderness.[1, 4] Radicular symptoms are evident in 12% to 47% of patients, presenting as weakness identified in 26% to 60% and altered sensation in up to 67% of patients.[1, 4, 8, 11, 19] One study revealed that 71% of patients have an abnormal neurologic exam at presentation, including paresthesias (39%), motor weakness (39%), and loss of bladder and bowel control (27%).[3] Thus, a thorough neurologic exam is vital in SEA evaluation.
Heusner staged patients based on their clinical findings (Table 2). Because the evolution of symptoms can be variable and rapidly progressive[4, 24] to stage 3 or 4, documenting subtle abnormalities on initial exam and monitoring for changes are important. Many patients initially present in stage 1 or 2 but remain undiagnosed until progression to stage 3 or 4.[24]
| Stage | Symptoms |
|---|---|
| I | Back pain |
| II | Radiculopathy, neck pain, reflex changes |
| III | Paresthesia, weakness, bladder symptoms |
| IV | Paralysis |
DIAGNOSTIC WORKUP
Laboratory Testing
Routine tests should include a white blood cell count (WBC), erythrocyte sedimentation rate (ESR), and C‐reactive protein (CRP). ESR has been shown to be the most sensitive and specific marker of SEA (Table 3). In a study of 63 patients matched with 126 controls, ESR was greater than 20 in 98% of cases and only 21% of controls.[1] Another study of 55 patients found the sensitivity and specificity of ESR to be 100% and 67%, respectively.[25] White count is less specific, with leukocytosis present in approximately two‐thirds of patients.[1, 25] CRP level rises faster than ESR in the setting of inflammation and also returns to baseline faster. As such, CRP is a useful means of monitoring the response to treatment.[1, 19]
|
| 1. Patient assessment |
| Evaluate for symptom triad (back pain, fever, neurologic dysfunction) |
| Assess for common comorbidities (diabetes, IV drug abuse, spinal trauma, ESRD, immunosuppressant therapy, recent spine procedure, systemic or local infection) |
| 2. Laboratory evaluation |
| Essential: ESR (most sensitive and specific), CRP, WBCs, blood cultures, UA |
| : echocardiogram, TB evaluation |
| No role: lumbar puncture |
| 3. Imaging |
| Gold standard: MRI w/gadolinium (90% sensitive) |
| If MRI contraindicated: CT myelogram |
| 4. Obtain tissue sample |
| Gold standard: open surgical biopsy, debridement fixation. |
| If patient unstable, diagnosis indeterminate, or no neuro symptoms: IR‐guided biopsy before surgery |
| 5. Antibiotics (once tissue sample obtained) |
| Empiric vancomycin plus third‐generation cephalosporin or aminoglycoside |
| Vancomycin only acceptable as monotherapy in patient with early diagnosis and no neurologic symptoms |
| 6. Surgical management (only if not done as step 4) |
| Early surgical consult recommended. Pursue surgical intervention if patient is stable for OR and neuro symptoms progressing or abscess refractory to antibiotics. Best outcomes occur with combined antibiotics and surgical debridement. |
Blood cultures are important to obtain at initial evaluation. Bacteremia from the offending organism may be present in up to 60% of cases; 60% to 90% of cases of bacteremia are caused by MRSA.[26] Urine culture should also be obtained in all patients. Sputum cultures should not be routinely obtained, but it may be beneficial in select patients with history of chronic obstructive pulmonary disease, new cough, or abnormality on chest radiographs. Transthoracic or transesophageal echocardiogram may be recommended for bacteremic patients, those with new heart murmur, or those with a history of IV drug abuse.
In patients in whom tuberculosis may be a potential risk, a tuberculin test with purified protein derivate can help rule out this organism. However, false‐negative testing may occur in up to 15% of patients, particularly the immunocompromised.[27] Patients with false positive results or prior vaccination with bacille Calmette‐Gurin (BCG) can be tested with QuantiFERON gold testing. These time‐consuming and expensive tests should not delay treatment with an empiric agent if tuberculosis is suspected due to patient risk factors or exposure, though an infectious disease specialist should be consulted before initiating treatment for tuberculosis.
Lumbar puncture should not be performed routinely in cases of suspected SEA, primarily due to the risk of bacterial contamination of the cerebrospinal fluid (CSF). Additionally, the increased protein and inflammatory cells seen in the CSF are nonspecific markers of parameningeal inflammation.[15]
Imaging
When the diagnosis of SEA is suspected based on clinical findings, MRI with gadolinium should be obtained first, as it is 90% sensitive for diagnosing SEA (Figure 1).[3] In patients unable to undergo MRI (eg, pacemaker), a computed tomography (CT) myelogram should be obtained instead. The study should be performed on an emergent basis, and the entire spine should be imaged due to the risk of noncontiguous lesions. Patients with plain radiograph and/or CT scan findings of bone lysis suggestive of vertebral osteomyelitis should also be evaluated with MRI if able.
MRI with contrast can often differentiate SEA from malignancy and other space‐occupying lesions.[28] Plain radiography can rule out other causes of back pain, such as trauma and degenerative disc disease, but it cannot demonstrate SEA. One study found that x‐rays showed pathology in only 16.6% of patients with SEA.[29]
Patients with exam findings and laboratory studies concerning for SEA who present to a community hospital without MRI capability should be transferred to a tertiary care center for advanced imaging and potential emergent treatment.
Tissue Culture
Though MRI is essential to the workup of SEA, biopsy with cultures allows for a definitive diagnosis.[30] Cultures may be obtained in the operating room or interventional radiology (IR) suite via fine‐needle aspiration or core‐needle biopsy under CT guidance, if available.[30, 31] CT‐guided bone biopsy, which has a sensitivity of 81% and specificity of 100%, may also be performed when vertebral osteomyelitis is present.[32] Biopsy by IR should be considered before surgical intervention, when the patient has no evidence of progressive neurologic deficit, the diagnosis is unclear, or the patient is too high risk for surgical intervention. In very high‐risk surgical patients, IR aspiration may be curative. Lyu et al. describe a case of refractory SEA treated with percutaneous CT‐guided needle aspiration alone, though they note that surgical debridement is preferred when possible.[31]
NONSURGICAL VERSUS SURGICAL MANAGEMENT
SEA may be treated without surgery in carefully selected patients. Savage et al. studied 52 patients, and found that nonsurgical management was often effective in patients who were completely neurologically intact at initial presentation. In their study, only 3 patients needed to undergo surgery due to development of new neurologic symptoms.[33] However, other studies report neurologic symptoms in 71% of patients at initial diagnosis.[3]
Adogwa et al. reviewed surgical versus nonsurgical management in elderly patients (>50 years old) over 15 years.[10] Their study included 30 patients treated operatively and 52 who received antibiotics alone. The decision for surgical management was at the surgeons' discretion; however, most patients with grade 2 or 3 symptoms underwent surgery, and those with paraplegia or quadriplegia for >48 hours did not. The authors found no clinically significant difference in outcome between these 2 groups and cautioned against surgical intervention for elderly patients with multiple comorbidities. It is worth noting, however, that 7/30 (23%) patients treated with surgery versus 5/52 (10%) treated conservatively had improved neurological outcomes (P = 0.03).[10] Numerous other studies have also found that neurologic status at presentation is the most important predictor of surgical outcomes.[7, 15, 34]
Patel et al. performed a study analyzing risk factors for failure of medical management, and found that diabetes mellitus, CRP >115, WBC >12.5, and positive blood cultures were predictors of failure.[9] Patients with >3 of these risk factors required surgery 76.9% of the time compared to 40.2% with 2, 35.4% with 1, and 8.3% with none.[9] The authors also found that surgical patients experienced better mean improvement than patients who failed nonsurgical treatment and subsequently underwent surgical decompression.[9]
In a follow‐up study by Alton et al., 62 patients were treated with either nonsurgical or surgical management. Indications for surgery in their study included a neurologic deficit at initial presentation or the development of a new neurological deficit while undergoing treatment. Twenty‐four patients presented without deficits and underwent nonsurgical management, but only 6 (25%) were treated successfully, as defined by stable or improved neurological status following therapy. In contrast, none of the 38 patients who underwent therapy with both IV antibiotics and emergent surgical management within 24 hours experienced deterioration in the neurologic status. For the 18 patients who failed nonsurgical management, surgery was performed within an average of 7 days. This group experienced improvement following surgery, but their neurological status improved less than those who underwent early surgery.[35]
These recent studies demonstrate that medical management may be an option for patients who are diagnosed early and present without neurologic deficits, but surgery is the mainstay of treatment for most patients. Additionally, patients who experience a delay in operative management often do not recover function as well as those patients who undergo emergent surgical debridement.[35] A delay in diagnosis has also been shown to lead to increase in lawsuits against providers. French et al. found an increase in verdicts against the provider when a delay in diagnosis >48 hours was present, irrespective of the degree of permanent neurologic dysfunction.[36]
TREATMENT AND FOLLOW‐UP
Unless the patient is septic and hemodynamically unstable, antibiotics should be held until a tissue sample is obtained for speciation and culture, either in the IR suite or the operating room. Once cultures have been obtained, broad‐spectrum IV antibiotics, usually vancomycin for gram‐positive coverage in combination with a third‐generation cephalosporin or aminoglycoside for additional gram‐negative coverage, should be started until organism sensitivities are determined and specific antibiotics can be administered.[3] Antibiotic therapy should continue for 4 to 6 weeks, though therapy may be administered for 8 weeks or longer in patients with vertebral osteomyelitis or immunocompromised patients.[19] An infectious disease specialist should be involved to determine the duration of therapy and transition to oral antibiotics.
As noted earlier, biopsy by IR should be considered when a trial of nonsurgical management is planned for a patient with no evidence of neurologic involvement, when the diagnosis is unclear after imaging, or when the patient is too high risk for surgery. Otherwise, surgical management is standard and should involve extensive surgical debridement with possible fusion if there is considerable structural compromise to the spinal column. After definitive treatment, repeat MRI is not recommended in the follow‐up of SEA; however, it is useful if there is concern for recurrence, as indicated by new fevers or a rising leukocyte count or inflammatory markers, especially CRP. Inflammatory labs should be obtained every 1 to 2 weeks to monitor for resolution of the infectious process.
CONCLUSION
Spinal epidural abscess is a potentially devastating condition that can be difficult to diagnose. Although uncommon, the triad of axial spine pain, fever, and new‐onset neurologic dysfunction are concerning. Other factors that increase the likelihood of SEA include a history of diabetes, IV drug abuse, spinal trauma, end‐stage renal disease, immunosuppressant therapy, recent invasive spine procedures, and concurrent infection of the skin or urinary tract. In patients with suspected SEA, inflammatory laboratory studies should be obtained along with gadolinium‐enhanced MRI of the entire spinal axis. Once the diagnosis is established, spine surgery and infectious disease consultation is mandatory, and IR biopsy may be appropriate in some cases. The rapidity of diagnosis and initiation of treatment are critical factors in optimizing patient outcome.
Disclosures
Mark A. Palumbo, MD, receives research funding from Globus Medical and is a paid consultant for Stryker. Alan H. Daniels, MD, is a paid consultant for Stryker and Osseous. No funding was obtained in support of this work.
- , , , et al. The clinical presentation and impact of diagnostic delays on emergency department patients with spinal epidural abscess. J Emerg Med. 2004;26:285–291.
- , , , . Spinal epidural abscess. N Engl J Med. 1975;293:463–468.
- , , , et al. Spinal epidural abscess: contemporary trends in etiology, evaluation, and management. Surg Neurol. 1999;52:189–196; discussion 197.
- . Spinal epidural abscess. N Engl J Med. 2006;355:2012–2020.
- , . Mortality, complication risk, and total charges after the treatment of epidural abscess. Spine J. 2015;15:249–255.
- , . Spinal epidural abscess. Curr Infect Dis Rep. 2004;16:436.
- , , , . Spinal epidural abscess: a ten‐year perspective. Neurosurgery. 1980;27:177–184.
- , , . Spinal epidural abscess: a meta‐analysis of 915 patients. Neurosurg Rev. 2000;23:175–204; discussion 205.
- , , , , , . Spinal epidural abscesses: risk factors, medical versus surgical management, a retrospective review of 128 cases. Spine J. 2014;14:326–330.
- , , , et al. Spontaneous spinal epidural abscess in patients 50 years of age and older: a 15‐year institutional perspective and review of the literature: clinical article. J Neurosurg Spine. 2014;20:344–349.
- , . Spinal epidural abscesses: clinical manifestations, prognostic factors, and outcomes. Neurosurgery. 2002;51:79–85; discussion 86–87.
- , , . Infectious agents in spinal epidural abscesses. Neurology. 1980;30:844–850.
- , , , , , . Cervical epidural abscess after epidural steroid injection. Spine (Phila Pa 1976). 2004;29:E7–E9.
- , , . Spinal epidural abscesses in adults: review and report of iatrogenic cases. Scand J Infect Dis. 1990;22:249–257.
- , , , , . Bacterial spinal epidural abscess. Review of 43 cases and literature survey. Medicine (Baltimore). 1992;71:369–385.
- , , , , . Spinal epidural abscess: the importance of early diagnosis and treatment. J Neurol Neurosurg Psychiatry. 1998;65:209–212.
- , , . Epidural abscesses. Br J Anaesth. 2006;96:292–302.
- , . Spinal epidural abscess. Med Clin North Am. 1985;69;375–384.
- , , , . Spinal epidural abscess. J Emerg Med. 2010;39:384–390.
- , , . Spinal epidural abscess: clinical presentation, management and outcome [comment on Curry WT, Hoh BL, Hanjani SA, et al. Surg Neurol. 2005;63:364–371). Surg Neurol. 2005;64:279.
- , , , , . Routine replacement of tunneled, cuffed, hemodialysis catheters eliminates paraspinal/vertebral infections in patients with catheter‐associated bacteremia. Am J Nephrol. 2003;23:202–207.
- , . Paraplegia caused by spinal infection after acupuncture. Spinal Cord. 2006;44:258–259.
- , , , . Spinal epidural abscess after tattooing. Clin Infect Dis. 1999;29:225–226.
- . Nontuberculous spinal epidural infections. N Engl J Med. 1948;239:845–854.
- , , , . Prospective evaluation of a clinical decision guideline to diagnose spinal epidural abscess in patients who present to the emergency department with spine pain. J Neurosurg Spine. 2011;14:765–770.
- , , , . Spinal epidural abscess: clinical presentation, management, and outcome. Surg Neurol. 2005;63:364–371; discussion 371.
- , . Bone and joint tuberculosis. Eur Spine J. 2013;22:556–566.
- , . Surgical management of spinal epidural abscess: selection of approach based on MRI appearance. J Clin Neurosci. 2004;11:130–133.
- , . Infection as a cause of spinal cord compression: a review of 36 spinal epidural abscess cases. Acta Neurochir (Wien). 2000;142:17–23.
- , , , et al. Spondyloarthropathy from long‐term hemodialysis. Radiology. 1988;167:761–764.
- , , , . Spinal epidural abscess successfully treated with percutaneous, computed tomography‐guided, needle aspiration and parenteral antibiotic therapy: case report and review of the literature. Neurosurgery. 2002;51:509–512; discussion 512.
- , , , . CT‐guided core biopsy of subchondral bone and intervertebral space in suspected spondylodiskitis. AJR Am J Roentgenol. 2006;186:977–980.
- , , . Spinal epidural abscess: early clinical outcome in patients treated medically. Clin Orthop. 2005;439:56–60.
- , . Update on spinal epidural abscess: 35 cases and review of the literature. Rev Infect Dis. 1987;9:265–274.
- , , , , , . Is there a difference in neurologic outcome in medical versus early operative management of cervical epidural abscesses? Spine J. 2015;15:10–17.
- , , , . Medicolegal cases for spinal epidural hematoma and spinal epidural abscess. Orthopedics. 2013;36:48–53.
- , , , . Spinal epidural abscess—experience with 46 patients and evaluation of prognostic factors. J Infect. 2002;45:76–81.
- , , , et al. The clinical presentation and impact of diagnostic delays on emergency department patients with spinal epidural abscess. J Emerg Med. 2004;26:285–291.
- , , , . Spinal epidural abscess. N Engl J Med. 1975;293:463–468.
- , , , et al. Spinal epidural abscess: contemporary trends in etiology, evaluation, and management. Surg Neurol. 1999;52:189–196; discussion 197.
- . Spinal epidural abscess. N Engl J Med. 2006;355:2012–2020.
- , . Mortality, complication risk, and total charges after the treatment of epidural abscess. Spine J. 2015;15:249–255.
- , . Spinal epidural abscess. Curr Infect Dis Rep. 2004;16:436.
- , , , . Spinal epidural abscess: a ten‐year perspective. Neurosurgery. 1980;27:177–184.
- , , . Spinal epidural abscess: a meta‐analysis of 915 patients. Neurosurg Rev. 2000;23:175–204; discussion 205.
- , , , , , . Spinal epidural abscesses: risk factors, medical versus surgical management, a retrospective review of 128 cases. Spine J. 2014;14:326–330.
- , , , et al. Spontaneous spinal epidural abscess in patients 50 years of age and older: a 15‐year institutional perspective and review of the literature: clinical article. J Neurosurg Spine. 2014;20:344–349.
- , . Spinal epidural abscesses: clinical manifestations, prognostic factors, and outcomes. Neurosurgery. 2002;51:79–85; discussion 86–87.
- , , . Infectious agents in spinal epidural abscesses. Neurology. 1980;30:844–850.
- , , , , , . Cervical epidural abscess after epidural steroid injection. Spine (Phila Pa 1976). 2004;29:E7–E9.
- , , . Spinal epidural abscesses in adults: review and report of iatrogenic cases. Scand J Infect Dis. 1990;22:249–257.
- , , , , . Bacterial spinal epidural abscess. Review of 43 cases and literature survey. Medicine (Baltimore). 1992;71:369–385.
- , , , , . Spinal epidural abscess: the importance of early diagnosis and treatment. J Neurol Neurosurg Psychiatry. 1998;65:209–212.
- , , . Epidural abscesses. Br J Anaesth. 2006;96:292–302.
- , . Spinal epidural abscess. Med Clin North Am. 1985;69;375–384.
- , , , . Spinal epidural abscess. J Emerg Med. 2010;39:384–390.
- , , . Spinal epidural abscess: clinical presentation, management and outcome [comment on Curry WT, Hoh BL, Hanjani SA, et al. Surg Neurol. 2005;63:364–371). Surg Neurol. 2005;64:279.
- , , , , . Routine replacement of tunneled, cuffed, hemodialysis catheters eliminates paraspinal/vertebral infections in patients with catheter‐associated bacteremia. Am J Nephrol. 2003;23:202–207.
- , . Paraplegia caused by spinal infection after acupuncture. Spinal Cord. 2006;44:258–259.
- , , , . Spinal epidural abscess after tattooing. Clin Infect Dis. 1999;29:225–226.
- . Nontuberculous spinal epidural infections. N Engl J Med. 1948;239:845–854.
- , , , . Prospective evaluation of a clinical decision guideline to diagnose spinal epidural abscess in patients who present to the emergency department with spine pain. J Neurosurg Spine. 2011;14:765–770.
- , , , . Spinal epidural abscess: clinical presentation, management, and outcome. Surg Neurol. 2005;63:364–371; discussion 371.
- , . Bone and joint tuberculosis. Eur Spine J. 2013;22:556–566.
- , . Surgical management of spinal epidural abscess: selection of approach based on MRI appearance. J Clin Neurosci. 2004;11:130–133.
- , . Infection as a cause of spinal cord compression: a review of 36 spinal epidural abscess cases. Acta Neurochir (Wien). 2000;142:17–23.
- , , , et al. Spondyloarthropathy from long‐term hemodialysis. Radiology. 1988;167:761–764.
- , , , . Spinal epidural abscess successfully treated with percutaneous, computed tomography‐guided, needle aspiration and parenteral antibiotic therapy: case report and review of the literature. Neurosurgery. 2002;51:509–512; discussion 512.
- , , , . CT‐guided core biopsy of subchondral bone and intervertebral space in suspected spondylodiskitis. AJR Am J Roentgenol. 2006;186:977–980.
- , , . Spinal epidural abscess: early clinical outcome in patients treated medically. Clin Orthop. 2005;439:56–60.
- , . Update on spinal epidural abscess: 35 cases and review of the literature. Rev Infect Dis. 1987;9:265–274.
- , , , , , . Is there a difference in neurologic outcome in medical versus early operative management of cervical epidural abscesses? Spine J. 2015;15:10–17.
- , , , . Medicolegal cases for spinal epidural hematoma and spinal epidural abscess. Orthopedics. 2013;36:48–53.
- , , , . Spinal epidural abscess—experience with 46 patients and evaluation of prognostic factors. J Infect. 2002;45:76–81.
ACOG plans consensus conference on uniform guidelines for breast cancer screening
The Susan G. Komen Foundation estimates that 84% of breast cancers are found through mammography.1 Clearly, the value of mammography is proven. But controversy and confusion abound on how much mammography, and beginning at what age, is best for women.
Currently, the United States Preventive Services Task Force (USPSTF), the American Cancer Society (ACS), and the American College of Obstetricians and Gynecologists (ACOG) all have differing recommendations about mammography and about the importance of clinical breast examinations. These inconsistencies largely are due to different interpretations of the same data, not the data itself, and tend to center on how harm is defined and measured. Importantly, these differences can wreak havoc on our patients’ confidence in our counsel and decision making, and can complicate women’s access to screening. Under the Affordable Care Act, women are guaranteed coverage of annual mammograms, but new USPSTF recommendations, due out soon, may undermine that guarantee.
On October 20, ACOG responded to the ACS’ new recommendations on breast cancer screening by emphasizing our continued advice that women should begin annual mammography screening at age 40, along with a clinical breast exam.2
Consensus conference plansIn an effort to address widespread confusion among patients, health care professionals, and payers, ACOG is convening a consensus conference in January 2016, with the goal of arriving at a consistent set of guidelines that can be agreed to, implemented clinically across the country, and hopefully adopted by insurers, as well. Major organizations and providers of women’s health care, including ACS, will gather to evaluate and interpret the data in greater detail and to consider the available data in the broader context of patient care.
Without doubt, guidelines and recommendations will need to evolve as new evidence emerges, but our hope is that scientific and medical organizations can look at the same evidence and speak with one voice on what is best for women’s health. Our patients would benefit from that alone.
ACOG’s recommendations, summarized
- Clinical breast examination every year for women aged 19 and older.
- Screening mammography every year for women aged 40 and older.
- Breast self-awareness has the potential to detect palpable breast cancer and can be recommended.2
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Susan G. Komen Web site. Accuracy of mammograms. http://ww5.komen.org/BreastCancer/AccuracyofMammograms.html. Updated June 26, 2015. Accessed October 30, 2015.
- ACOG Statement on Revised American Cancer Society Recommendations on Breast Cancer Screening. American College of Obstetricians and Gynecologists Web site. http://www.acog.org/About-ACOG/News-Room/Statements/2015/ACOG-Statement-on-Recommendations-on-Breast-Cancer-Screening. Published October 20, 2015. Accessed October 30, 2015.

Ms. DiVenere is Officer, Government and Political Affairs, at the American Congress of Obstetricians and Gynecologists, Washington, DC.
The author reports no financial relationships relevant to this article.
Ms. DiVenere is Officer, Government and Political Affairs, at the American Congress of Obstetricians and Gynecologists, Washington, DC.
The author reports no financial relationships relevant to this article.
Ms. DiVenere is Officer, Government and Political Affairs, at the American Congress of Obstetricians and Gynecologists, Washington, DC.
The author reports no financial relationships relevant to this article.
The Susan G. Komen Foundation estimates that 84% of breast cancers are found through mammography.1 Clearly, the value of mammography is proven. But controversy and confusion abound on how much mammography, and beginning at what age, is best for women.
Currently, the United States Preventive Services Task Force (USPSTF), the American Cancer Society (ACS), and the American College of Obstetricians and Gynecologists (ACOG) all have differing recommendations about mammography and about the importance of clinical breast examinations. These inconsistencies largely are due to different interpretations of the same data, not the data itself, and tend to center on how harm is defined and measured. Importantly, these differences can wreak havoc on our patients’ confidence in our counsel and decision making, and can complicate women’s access to screening. Under the Affordable Care Act, women are guaranteed coverage of annual mammograms, but new USPSTF recommendations, due out soon, may undermine that guarantee.
On October 20, ACOG responded to the ACS’ new recommendations on breast cancer screening by emphasizing our continued advice that women should begin annual mammography screening at age 40, along with a clinical breast exam.2
Consensus conference plansIn an effort to address widespread confusion among patients, health care professionals, and payers, ACOG is convening a consensus conference in January 2016, with the goal of arriving at a consistent set of guidelines that can be agreed to, implemented clinically across the country, and hopefully adopted by insurers, as well. Major organizations and providers of women’s health care, including ACS, will gather to evaluate and interpret the data in greater detail and to consider the available data in the broader context of patient care.
Without doubt, guidelines and recommendations will need to evolve as new evidence emerges, but our hope is that scientific and medical organizations can look at the same evidence and speak with one voice on what is best for women’s health. Our patients would benefit from that alone.
ACOG’s recommendations, summarized
- Clinical breast examination every year for women aged 19 and older.
- Screening mammography every year for women aged 40 and older.
- Breast self-awareness has the potential to detect palpable breast cancer and can be recommended.2
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
The Susan G. Komen Foundation estimates that 84% of breast cancers are found through mammography.1 Clearly, the value of mammography is proven. But controversy and confusion abound on how much mammography, and beginning at what age, is best for women.
Currently, the United States Preventive Services Task Force (USPSTF), the American Cancer Society (ACS), and the American College of Obstetricians and Gynecologists (ACOG) all have differing recommendations about mammography and about the importance of clinical breast examinations. These inconsistencies largely are due to different interpretations of the same data, not the data itself, and tend to center on how harm is defined and measured. Importantly, these differences can wreak havoc on our patients’ confidence in our counsel and decision making, and can complicate women’s access to screening. Under the Affordable Care Act, women are guaranteed coverage of annual mammograms, but new USPSTF recommendations, due out soon, may undermine that guarantee.
On October 20, ACOG responded to the ACS’ new recommendations on breast cancer screening by emphasizing our continued advice that women should begin annual mammography screening at age 40, along with a clinical breast exam.2
Consensus conference plansIn an effort to address widespread confusion among patients, health care professionals, and payers, ACOG is convening a consensus conference in January 2016, with the goal of arriving at a consistent set of guidelines that can be agreed to, implemented clinically across the country, and hopefully adopted by insurers, as well. Major organizations and providers of women’s health care, including ACS, will gather to evaluate and interpret the data in greater detail and to consider the available data in the broader context of patient care.
Without doubt, guidelines and recommendations will need to evolve as new evidence emerges, but our hope is that scientific and medical organizations can look at the same evidence and speak with one voice on what is best for women’s health. Our patients would benefit from that alone.
ACOG’s recommendations, summarized
- Clinical breast examination every year for women aged 19 and older.
- Screening mammography every year for women aged 40 and older.
- Breast self-awareness has the potential to detect palpable breast cancer and can be recommended.2
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Susan G. Komen Web site. Accuracy of mammograms. http://ww5.komen.org/BreastCancer/AccuracyofMammograms.html. Updated June 26, 2015. Accessed October 30, 2015.
- ACOG Statement on Revised American Cancer Society Recommendations on Breast Cancer Screening. American College of Obstetricians and Gynecologists Web site. http://www.acog.org/About-ACOG/News-Room/Statements/2015/ACOG-Statement-on-Recommendations-on-Breast-Cancer-Screening. Published October 20, 2015. Accessed October 30, 2015.
- Susan G. Komen Web site. Accuracy of mammograms. http://ww5.komen.org/BreastCancer/AccuracyofMammograms.html. Updated June 26, 2015. Accessed October 30, 2015.
- ACOG Statement on Revised American Cancer Society Recommendations on Breast Cancer Screening. American College of Obstetricians and Gynecologists Web site. http://www.acog.org/About-ACOG/News-Room/Statements/2015/ACOG-Statement-on-Recommendations-on-Breast-Cancer-Screening. Published October 20, 2015. Accessed October 30, 2015.
Adjuvant Systemic Therapy for Early-Stage Breast Cancer
Over the past 20 years, substantial progress has been achieved in our understanding of breast cancer and in breast cancer treatment, with mortality from breast cancer declining by more than 25% over this time. This progress has been characterized by a greater understanding of the molecular biology of breast cancer, rational drug design, development of agents with specific cellular targets and pathways, development of better prognostic and predictive multigene assays, and marked improvements in supportive care.
To read the full article in PDF:
Over the past 20 years, substantial progress has been achieved in our understanding of breast cancer and in breast cancer treatment, with mortality from breast cancer declining by more than 25% over this time. This progress has been characterized by a greater understanding of the molecular biology of breast cancer, rational drug design, development of agents with specific cellular targets and pathways, development of better prognostic and predictive multigene assays, and marked improvements in supportive care.
To read the full article in PDF:
Over the past 20 years, substantial progress has been achieved in our understanding of breast cancer and in breast cancer treatment, with mortality from breast cancer declining by more than 25% over this time. This progress has been characterized by a greater understanding of the molecular biology of breast cancer, rational drug design, development of agents with specific cellular targets and pathways, development of better prognostic and predictive multigene assays, and marked improvements in supportive care.
To read the full article in PDF:
Pancreas transplant for diabetes mellitus
Pancreas transplant is the only long-term diabetes treatment that consistently results in normal hemoglobin A1c levels without the risk of severe hypoglycemia. Additionally, pancreas transplant may prevent, halt, or even reverse the complications of diabetes.
Here, we explore the indications, options, and outcomes of pancreas transplant as a treatment for diabetes mellitus.
DIABETES IS COMMON, AND OFTEN NOT WELL CONTROLLED
Diabetes mellitus affects more than 25 million people in the United States (8.3% of the population) and is the leading cause of kidney failure, nontraumatic lower-limb amputation, and adult-onset blindness. In 2007, nearly $116 billion was spent on diabetes treatment, not counting another $58 billion in indirect costs such as disability, work loss, and premature death.1
Despite the tremendous expenditure in human, material, and financial resources, only about 50% of patients achieve their diabetes treatment goals. In 2013, a large US population-based study2 reported that 52.2% of patients were achieving the American Diabetes Association treatment goal of hemoglobin A1c lower than 7%. A similar study in South Korea3 found that 45.6% were at this goal.
Most of the patients in these studies had type 2 diabetes, and the data suggested that attaining glycemic goals is more difficult in insulin-treated patients. Studies of patients with type 1 diabetes found hemoglobin A1c levels lower than 7% in only 8.1% of hospitalized patients with type 1 diabetes, and in only 13% in an outpatient diabetes clinic.4,5
YET RATES OF PANCREAS TRANSPLANT ARE DECLINING
Pancreas transplant was first performed more than 40 years ago at the University of Minnesota.6 Since then, dramatic changes in immunosuppression, organ preservation, surgical technique, and donor and recipient selection have brought about significant progress.
Currently, more than 13,000 patients are alive with a functioning pancreas allograft. After reaching a peak in 2004, the annual number of pancreas transplants performed in the United States has declined steadily, whereas the procedure continues to increase in popularity outside North America.7 The primary reason for the decline is recognition of donor factors that lead to success—surgeons are refusing to transplant organs they might have accepted previously, because experience suggests they would yield poor results. In the United States, 1,043 pancreas transplants were performed in 2012, and more than 3,100 patients were on the waiting list.8
Islet cell transplant—a different procedure involving harvesting, encapsulating, and implanting insulin-producing beta cells—has not gained widespread application due to very low long-term success rates.
THREE CATEGORIES OF PANCREAS TRANSPLANT

Pancreas transplant can be categorized according to whether the patient is also receiving or has already received a kidney graft (Table 1).
Simultaneous kidney and pancreas transplant is performed in patients who have type 1 diabetes with advanced chronic kidney disease due to diabetic nephropathy. This remains the most commonly performed type, accounting for 79% of all pancreas transplants in 2012.8
Pancreas-after-kidney transplant is most often done after a living-donor kidney transplant. This procedure accounted for most of the increase in pancreas transplants during the first decade of the 2000s. However, the number of these procedures has steadily decreased since 2004, and in 2012 accounted for only 12% of pancreas transplants.8
Pancreas transplant alone is performed in nonuremic diabetic patients who have labile blood sugar control. Performed in patients with preserved renal function but severe complications of “brittle” diabetes, such as hypoglycemic unawareness, this type accounts for 8% of pancreas transplants.9
Indications for pancreas transplant
A small number of these procedures are done for indications unrelated to diabetes mellitus. In most of these cases, the pancreas is transplanted as part of a multivisceral transplant to facilitate the technical (surgical) aspect of the procedure—the pancreas, liver, stomach, gallbladder, and part of the intestines are transplanted en bloc to maintain the native vasculature. Very infrequently, pancreas transplant is done to replace exocrine pancreatic function.
A small, select group of patients with type 2 diabetes and low body mass index (BMI) may be eligible for pancreas transplant, and they accounted for 8.2% of active candidates in 2012.8 However, most pancreas transplants are performed in patients with type 1 diabetes.
WHAT MAKES A GOOD ALLOGRAFT?
Pancreas allografts are procured as whole organs from brain-dead organ donors. Relatively few pancreas allografts (3.1% in 2012) are from cardiac-death donors, because of concern about warm ischemic injury during the period of circulatory arrest.8

![]()
Proper donor selection is critical to the success of pancreas transplant, as donor factors including medical history, age, BMI, and cause of death can significantly affect the outcome. In general, transplant of a pancreas allograft from a young donor (age < 30) with excellent organ function, low BMI, and traumatic cause of death provides the best chance of success.
The Pancreas Donor Risk Index (PDRI)10 was developed after analysis of objective donor criteria, transplant type, and ischemic time in grafts transplanted between 2000 and 2006. One-year graft survival was directly related to the PDRI and ranged between 77% and 87% in recipients of “standard” pancreas allografts (PDRI score of 1.0). Use of grafts from the highest (worst) three quintiles of PDRI (PDRI score > 1.16) was associated with 1-year graft survival rates of 67% to 82%, significantly inferior to that seen with “higher- quality” grafts, again emphasizing the need for rigorous donor selection.10
In addition to these objective measures, visual assessment of pancreas quality at the time of procurement remains an equally important predictor of success. Determination of subjective features, such as fatty infiltration and glandular fibrosis, requires surgical experience developed over several years. In a 2010 analysis, dissatisfaction with the quality of the donor graft on inspection accounted for more than 80% of refusals of potential pancreas donors.11 These studies illustrate an ill-defined aspect of pancreas transplant, ie, even when the pancreas donor is perceived to be suitable, the outcome may be markedly different.
SURGICAL COMPLICATIONS
Surgical complications have long been considered a limiting factor in the growth of pancreas transplant. Technical failure or loss of the graft within 90 days is most commonly due to graft thrombosis, leakage of the enteric anastomosis, or severe peripancreatic infection. The rate of technical failure has declined across all recipient categories and is currently about 9%.8
DO RECIPIENT FACTORS AFFECT OUTCOMES?
As mentioned above, the PDRI identifies donor factors that influence the 1-year graft survival rate. Recipient factors are also thought to play a role, although the influence of these factors has not been consistently demonstrated.
Humar et al15 found that recipient obesity (defined in this study as BMI > 25 kg/m2) and donor age over 40 were risk factors for early laparotomy after pancreas transplant.15 Moreover, patients undergoing early laparotomy had poorer graft survival outcomes.
This finding was reinforced by an analysis of 5,725 primary simultaneous pancreas-kidney recipients between 2000 and 2007. Obesity (BMI 30 ≥ kg/m2) was associated with increased rates of patient death, pancreas graft loss, and kidney graft loss at 3 years.16
More recently, Finger et al17 did not find a statistically significant association between recipient BMI and technical failure, but they did notice a trend toward increased graft loss with a BMI greater than 25 kg/m2. Similarly, others have not found a clear adverse association between recipient BMI and pancreas graft survival.
Intuitively, obesity and other recipient factors such as age, vascular disease, duration of diabetes, and dialysis should influence pancreas graft survival but have not been shown in analyses to carry an adverse effect.18 The inability to consistently find adverse effects of recipient characteristics is most likely due to the relative similarity between the vast majority of pancreas transplant recipients and the relatively small numbers of adverse events. In 98 consecutive pancreas transplants at our center between 2009 and 2014, the technical loss rate was 1.8% (unpublished data).
Acute rejection most commonly occurs during the first year and is usually reversible. More than 1 year after transplant, graft loss is due to chronic rejection, and death is usually from underlying cardiovascular disease.
The immunosuppressive regimens used in pancreas transplant are similar to those in kidney transplant. Since the pancreas is considered to be more immunogenic than other organs, most centers employ a strategy of induction immunosuppression with T-cell–depleting or interleukin 2-receptor antibodies. Maintenance immunosuppression consists of a calcineurin inhibitor (tacrolimus or cyclosporine), an antimetabolite (mycophenolate), and a corticosteroid.8
Immunosuppressive complications occur at a rate similar to that seen in other solid-organ transplants and include an increased risk of opportunistic infection and malignancy. The risk of these complications must be balanced against the patient’s risk of health decline with dialysis and insulin-based therapies.
OVERALL OUTCOMES ARE GOOD
The success rate of pancreas transplant is currently at its highest since the inception of the procedure. The unadjusted patient survival rate for all groups is over 96% at 1 year, and over 80% at 5 years.8 One-year patient survival after pancreas transplant alone, at better than 96%, is the highest of all organ transplant procedures.9
Several recently published single-center reviews of pancreas transplant since 2000 report patient survival rates of 96% to 100% at 1 year and 88% to 100% at 5 years.19–22 This variability is likely closely linked to donor and recipient selection, as centers performing smaller numbers of transplants tend to be more selective and, in turn, report higher patient survival rates.19,21
Long-term patient survival outcomes can be gathered from larger, registry-based reviews, accepting limitations in assessing causes of patient death. Siskind et al23 analyzed the outcomes of 20,854 US pancreas transplants done between 1996 and 2012 and found the 10-year patient survival rate ranged from 43% to 77% and was highly dependent on patient age at the time of the procedure.23 Patient survival after transplant must be balanced against the generally poor long-term survival prospects of diabetic patients on dialysis.
By type of transplant, pancreas graft survival rates at 1 year are 89% for simultaneous pancreas-kidney transplant, 86% for pancreas-after-kidney transplant, and 84% for pancreas-alone transplant. Graft survival rates at 5 years are 71% for simultaneous pancreas-kidney transplant, 65% for pancreas-after-kidney transplant, and 58% for pancreas-alone transplant.8,9
Simultaneous pancreas-kidney transplant has been shown to improve the survival rate compared with cadaveric kidney transplant alone in patients with type 1 diabetes and chronic kidney disease.24,25 The survival benefit of isolated pancreas transplant (after kidney transplant and alone) is not evident at 4-year follow-up compared with patients on the waiting list. However, the benefit for the individual patient must be considered by weighing the incapacities experienced with insulin-based treatments against the risks of surgery and immunosuppression.26,27 For patients who have experienced frequent and significant hypoglycemic episodes, particularly those requiring third-party assistance, pancreas transplant can be a lifesaving procedure.
Effects on secondary diabetic complications
Notwithstanding the effect on the patient’s life span, data from several studies of long-term pancreas transplant recipients suggest that secondary diabetic complications can be halted or even improved. Most of these studies examined the effect of restoring euglycemia in nephropathy and the subsequent influence on renal function.
Effect on renal function. Kleinclauss et al28 examined renal allograft function in type 1 diabetic recipients of living-donor kidney transplants. Comparing kidney allograft survival and function in patients who received a subsequent pancreas-after-kidney transplant vs those who did not, graft survival was superior after 5 years, and the estimated glomerular filtration rate was 10 mL/min higher in pancreas-after-kidney recipients.28 This improvement in renal function was not seen immediately after the pancreas transplant but became evident more than 4 years after establishment of normoglycemia. Somewhat similarly, reversal of diabetic changes in native kidney biopsies has been seen 10 years after pancreas transplant.29
Effect on neuropathy. In other studies, reversal of autonomic neuropathy and hypoglycemic unawareness and improvements in peripheral sensory-motor neuropathy have also been observed.30–32
Effect on retinopathy. Improvements in early-stage nonproliferative diabetic retinopathy and laser-treated proliferative lesions have been seen, even within short periods of follow-up.33 Other groups have shown a significantly higher proportion of improvement or stability of advanced diabetic retinopathy at 3 years after simultaneous pancreas-kidney transplant, compared with kidney transplant alone in patients with type 1 diabetes.34
Effect on heart disease. Salutary effects on cardiovascular risk factors and amelioration of cardiac morphology and functional cardiac indices have been seen within the first posttransplant year.35 Moreover, with longer follow-up (nearly 4 years), simultaneous pancreas-kidney recipients with functioning pancreas grafts were found to have less progression of coronary atherosclerosis than simultaneous pancreas-kidney recipients with early pancreas graft loss.36 These data provide a potential pathophysiologic mechanism for the long-term survival advantage seen in uremic type 1 diabetic patients undergoing simultaneous pancreas-kidney transplant.
In the aggregate, these findings suggest that, in the absence of surgical and immunosuppression-related complications, a functioning pancreas allograft can alter the progress of diabetic complications. As an extension of these results, pancreas transplant done earlier in the course of diabetes may have an even greater impact.
- Centers for Disease Control and Prevention (CDC). National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed August 12, 2015.
- Ali MK, Bullard KM, Saaddine JB, Cowie CC, Imperatore G, Gregg EW. Achievement of goals in US diabetes care, 1999–2010. N Engl J Med 2013; 368:1613–1624.
- Jeon JY, Kim DJ, Ko SH, et al; Taskforce Team of Diabetes Fact Sheet of the Korean Diabetes Association. Current status of glycemic control of patients with diabetes in Korea: the fifth Korea national health and nutrition examination survey. Diabetes Metab J 2014; 38:197–203.
- Govan L, Wu O, Briggs A, et al; Scottish Diabetes Research Network Epidemiology Group. Achieved levels of HbA1c and likelihood of hospital admission in people with type 1 diabetes in the Scottish population: a study from the Scottish Diabetes Research Network Epidemiology Group. Diabetes Care 2011; 34:1992–1997.
- Bryant W, Greenfield JR, Chisholm DJ, Campbell LV. Diabetes guidelines: easier to preach than to practise? Med J Aust 2006; 185:305–309.
- Kelly WD, Lillehei RC, Merkel FK, Idezuki Y, Goetz FC. Allotransplantation of the pancreas and duodenum along with the kidney in diabetic nephropathy. Surgery 1967; 61:827–837.
- Gruessner AC, Gruessner RW. Pancreas transplant outcomes for United States and non United States cases as reported to the United Network for Organ Sharing and the International Pancreas Transplant Registry as of December 2011. Clin Transpl 2012: 23–40.
- Israni AK, Skeans MA, Gustafson SK, et al. OPTN/SRTR 2012 Annual Data Report: pancreas. Am J Transplant 2014; 14(suppl 1):45–68
- Gruessner RW, Gruessner AC. Pancreas transplant alone: a procedure coming of age. Diabetes Care 2013; 36:2440–2447.
- Axelrod DA, Sung RS, Meyer KH, Wolfe RA, Kaufman DB. Systematic evaluation of pancreas allograft quality, outcomes and geographic variation in utilization. Am J Transplant 2010; 10:837–845.
- Wiseman AC, Wainright JL, Sleeman E, et al. An analysis of the lack of donor pancreas utilization from younger adult organ donors. Transplantation 2010; 90:475–480.
- Gruessner RW, Gruessner AC. The current state of pancreas transplantation. Nat Rev Endocrinol 2013; 9:555–562.
- Gunasekaran G, Wee A, Rabets J, Winans C, Krishnamurthi V. Duodenoduodenostomy in pancreas transplantation. Clin Transplant 2012; 26:550–557.
- Sollinger HW, Odorico JS, Becker YT, D’Alessandro AM, Pirsch JD. One thousand simultaneous pancreas-kidney transplants at a single center with 22-year follow-up. Ann Surg 2009; 250:618–630.
- Humar A, Kandaswamy R, Granger D, Gruessner RW, Gruessner AC, Sutherland DE. Decreased surgical risks of pancreas transplantation in the modern era. Ann Surg 2000; 231:269–275.
- Sampaio MS, Reddy PN, Kuo HT, et al. Obesity was associated with inferior outcomes in simultaneous pancreas kidney transplant. Transplantation 2010; 89:1117–1125.
- Finger EB, Radosevich DM, Dunn TB, et al. A composite risk model for predicting technical failure in pancreas transplantation. Am J Transplant 2013; 13:1840–1849.
- Fridell JA, Mangus RS, Taber TE, et al. Growth of a nation part II: impact of recipient obesity on whole-organ pancreas transplantation. Clin Transplant 2011; 25:E366–E374.
- Tai DS, Hong J, Busuttil RW, Lipshutz GS. Low rates of short- and long-term graft loss after kidney-pancreas transplant from a single center. JAMA Surg 2013; 148:368–373.
- Bazerbachi F, Selzner M, Marquez MA, et al. Pancreas-after-kidney versus synchronous pancreas-kidney transplantation: comparison of intermediate-term results. Transplantation 2013; 95:489–494.
- Laftavi MR, Pankewycz O, Gruessner A, et al. Long-term outcomes of pancreas after kidney transplantation in small centers: is it justified? Transplant Proc 2014; 46:1920–1923.
- Stratta RJ, Farney AC, Orlando G, Farooq U, Al-Shraideh Y, Rogers J. Similar results with solitary pancreas transplantation compared with simultaneous pancreas-kidney transplantation in the new millennium. Transplant Proc 2014; 46:1924–1927.
- Siskind E, Maloney C, Akerman M, et al. An analysis of pancreas transplantation outcomes based on age groupings—an update of the UNOS database. Clin Transplant 2014; 28:990–994.
- Ojo AO, Meier-Kriesche HU, Hanson JA, et al. The impact of simultaneous pancreas-kidney transplantation on long-term patient survival. Transplantation 2001; 71:82–90.
- Reddy KS, Stablein D, Taranto S, et al. Long-term survival following simultaneous kidney-pancreas transplantation versus kidney transplantation alone in patients with type 1 diabetes mellitus and renal failure. Am J Kidney Dis 2003; 41:464–470.
- Venstrom JM, McBride MA, Rother KI, Hirshberg B, Orchard TJ, Harlan DM. Survival after pancreas transplantation in patients with diabetes and preserved kidney function. JAMA 2003; 290:2817–2823.
- Gruessner RW, Sutherland DE, Gruessner AC. Mortality assessment for pancreas transplants. Am J Transplant 2004; 4:2018–2026.
- Kleinclauss F, Fauda M, Sutherland DE, et al. Pancreas after living donor kidney transplants in diabetic patients: impact on long-term kidney graft function. Clin Transplant 2009; 23:437–446.
- Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med 1998; 339:69–75.
- Landgraf R. Impact of pancreas transplantation on diabetic secondary complications and quality of life. Diabetologia 1996; 39:1415–1424.
- Robertson RP. Update on transplanting beta cells for reversing type 1 diabetes. Endocrinol Metab Clin North Am 2010; 39:655–667.
- Robertson RP, Holohan TV, Genuth S. Therapeutic controversy: pancreas transplantation for type I diabetes. J Clin Endocrinol Metab 1998; 83:1868–1674.
- Giannarelli R, Coppelli A, Sartini MS, et al. Pancreas transplant alone has beneficial effects on retinopathy in type 1 diabetic patients. Diabetologia 2006; 49:2977–2982.
- Koznarová R, Saudek F, Sosna T, et al. Beneficial effect of pancreas and kidney transplantation on advanced diabetic retinopathy. Cell Transplant 2000; 9:903–908.
- Coppelli A, Giannarelli R, Mariotti R, et al. Pancreas transplant alone determines early improvement of cardiovascular risk factors and cardiac function in type 1 diabetic patients. Transplantation 2003; 76:974–976.
- Jukema JW, Smets YF, van der Pijl JW, et al. Impact of simultaneous pancreas and kidney transplantation on progression of coronary atherosclerosis in patients with end-stage renal failure due to type 1 diabetes. Diabetes Care 2002; 25:906–911.
Pancreas transplant is the only long-term diabetes treatment that consistently results in normal hemoglobin A1c levels without the risk of severe hypoglycemia. Additionally, pancreas transplant may prevent, halt, or even reverse the complications of diabetes.
Here, we explore the indications, options, and outcomes of pancreas transplant as a treatment for diabetes mellitus.
DIABETES IS COMMON, AND OFTEN NOT WELL CONTROLLED
Diabetes mellitus affects more than 25 million people in the United States (8.3% of the population) and is the leading cause of kidney failure, nontraumatic lower-limb amputation, and adult-onset blindness. In 2007, nearly $116 billion was spent on diabetes treatment, not counting another $58 billion in indirect costs such as disability, work loss, and premature death.1
Despite the tremendous expenditure in human, material, and financial resources, only about 50% of patients achieve their diabetes treatment goals. In 2013, a large US population-based study2 reported that 52.2% of patients were achieving the American Diabetes Association treatment goal of hemoglobin A1c lower than 7%. A similar study in South Korea3 found that 45.6% were at this goal.
Most of the patients in these studies had type 2 diabetes, and the data suggested that attaining glycemic goals is more difficult in insulin-treated patients. Studies of patients with type 1 diabetes found hemoglobin A1c levels lower than 7% in only 8.1% of hospitalized patients with type 1 diabetes, and in only 13% in an outpatient diabetes clinic.4,5
YET RATES OF PANCREAS TRANSPLANT ARE DECLINING
Pancreas transplant was first performed more than 40 years ago at the University of Minnesota.6 Since then, dramatic changes in immunosuppression, organ preservation, surgical technique, and donor and recipient selection have brought about significant progress.
Currently, more than 13,000 patients are alive with a functioning pancreas allograft. After reaching a peak in 2004, the annual number of pancreas transplants performed in the United States has declined steadily, whereas the procedure continues to increase in popularity outside North America.7 The primary reason for the decline is recognition of donor factors that lead to success—surgeons are refusing to transplant organs they might have accepted previously, because experience suggests they would yield poor results. In the United States, 1,043 pancreas transplants were performed in 2012, and more than 3,100 patients were on the waiting list.8
Islet cell transplant—a different procedure involving harvesting, encapsulating, and implanting insulin-producing beta cells—has not gained widespread application due to very low long-term success rates.
THREE CATEGORIES OF PANCREAS TRANSPLANT
Pancreas transplant can be categorized according to whether the patient is also receiving or has already received a kidney graft (Table 1).
Simultaneous kidney and pancreas transplant is performed in patients who have type 1 diabetes with advanced chronic kidney disease due to diabetic nephropathy. This remains the most commonly performed type, accounting for 79% of all pancreas transplants in 2012.8
Pancreas-after-kidney transplant is most often done after a living-donor kidney transplant. This procedure accounted for most of the increase in pancreas transplants during the first decade of the 2000s. However, the number of these procedures has steadily decreased since 2004, and in 2012 accounted for only 12% of pancreas transplants.8
Pancreas transplant alone is performed in nonuremic diabetic patients who have labile blood sugar control. Performed in patients with preserved renal function but severe complications of “brittle” diabetes, such as hypoglycemic unawareness, this type accounts for 8% of pancreas transplants.9
Indications for pancreas transplant
A small number of these procedures are done for indications unrelated to diabetes mellitus. In most of these cases, the pancreas is transplanted as part of a multivisceral transplant to facilitate the technical (surgical) aspect of the procedure—the pancreas, liver, stomach, gallbladder, and part of the intestines are transplanted en bloc to maintain the native vasculature. Very infrequently, pancreas transplant is done to replace exocrine pancreatic function.
A small, select group of patients with type 2 diabetes and low body mass index (BMI) may be eligible for pancreas transplant, and they accounted for 8.2% of active candidates in 2012.8 However, most pancreas transplants are performed in patients with type 1 diabetes.
WHAT MAKES A GOOD ALLOGRAFT?
Pancreas allografts are procured as whole organs from brain-dead organ donors. Relatively few pancreas allografts (3.1% in 2012) are from cardiac-death donors, because of concern about warm ischemic injury during the period of circulatory arrest.8
![]()
Proper donor selection is critical to the success of pancreas transplant, as donor factors including medical history, age, BMI, and cause of death can significantly affect the outcome. In general, transplant of a pancreas allograft from a young donor (age < 30) with excellent organ function, low BMI, and traumatic cause of death provides the best chance of success.
The Pancreas Donor Risk Index (PDRI)10 was developed after analysis of objective donor criteria, transplant type, and ischemic time in grafts transplanted between 2000 and 2006. One-year graft survival was directly related to the PDRI and ranged between 77% and 87% in recipients of “standard” pancreas allografts (PDRI score of 1.0). Use of grafts from the highest (worst) three quintiles of PDRI (PDRI score > 1.16) was associated with 1-year graft survival rates of 67% to 82%, significantly inferior to that seen with “higher- quality” grafts, again emphasizing the need for rigorous donor selection.10
In addition to these objective measures, visual assessment of pancreas quality at the time of procurement remains an equally important predictor of success. Determination of subjective features, such as fatty infiltration and glandular fibrosis, requires surgical experience developed over several years. In a 2010 analysis, dissatisfaction with the quality of the donor graft on inspection accounted for more than 80% of refusals of potential pancreas donors.11 These studies illustrate an ill-defined aspect of pancreas transplant, ie, even when the pancreas donor is perceived to be suitable, the outcome may be markedly different.
SURGICAL COMPLICATIONS
Surgical complications have long been considered a limiting factor in the growth of pancreas transplant. Technical failure or loss of the graft within 90 days is most commonly due to graft thrombosis, leakage of the enteric anastomosis, or severe peripancreatic infection. The rate of technical failure has declined across all recipient categories and is currently about 9%.8
DO RECIPIENT FACTORS AFFECT OUTCOMES?
As mentioned above, the PDRI identifies donor factors that influence the 1-year graft survival rate. Recipient factors are also thought to play a role, although the influence of these factors has not been consistently demonstrated.
Humar et al15 found that recipient obesity (defined in this study as BMI > 25 kg/m2) and donor age over 40 were risk factors for early laparotomy after pancreas transplant.15 Moreover, patients undergoing early laparotomy had poorer graft survival outcomes.
This finding was reinforced by an analysis of 5,725 primary simultaneous pancreas-kidney recipients between 2000 and 2007. Obesity (BMI 30 ≥ kg/m2) was associated with increased rates of patient death, pancreas graft loss, and kidney graft loss at 3 years.16
More recently, Finger et al17 did not find a statistically significant association between recipient BMI and technical failure, but they did notice a trend toward increased graft loss with a BMI greater than 25 kg/m2. Similarly, others have not found a clear adverse association between recipient BMI and pancreas graft survival.
Intuitively, obesity and other recipient factors such as age, vascular disease, duration of diabetes, and dialysis should influence pancreas graft survival but have not been shown in analyses to carry an adverse effect.18 The inability to consistently find adverse effects of recipient characteristics is most likely due to the relative similarity between the vast majority of pancreas transplant recipients and the relatively small numbers of adverse events. In 98 consecutive pancreas transplants at our center between 2009 and 2014, the technical loss rate was 1.8% (unpublished data).
Acute rejection most commonly occurs during the first year and is usually reversible. More than 1 year after transplant, graft loss is due to chronic rejection, and death is usually from underlying cardiovascular disease.
The immunosuppressive regimens used in pancreas transplant are similar to those in kidney transplant. Since the pancreas is considered to be more immunogenic than other organs, most centers employ a strategy of induction immunosuppression with T-cell–depleting or interleukin 2-receptor antibodies. Maintenance immunosuppression consists of a calcineurin inhibitor (tacrolimus or cyclosporine), an antimetabolite (mycophenolate), and a corticosteroid.8
Immunosuppressive complications occur at a rate similar to that seen in other solid-organ transplants and include an increased risk of opportunistic infection and malignancy. The risk of these complications must be balanced against the patient’s risk of health decline with dialysis and insulin-based therapies.
OVERALL OUTCOMES ARE GOOD
The success rate of pancreas transplant is currently at its highest since the inception of the procedure. The unadjusted patient survival rate for all groups is over 96% at 1 year, and over 80% at 5 years.8 One-year patient survival after pancreas transplant alone, at better than 96%, is the highest of all organ transplant procedures.9
Several recently published single-center reviews of pancreas transplant since 2000 report patient survival rates of 96% to 100% at 1 year and 88% to 100% at 5 years.19–22 This variability is likely closely linked to donor and recipient selection, as centers performing smaller numbers of transplants tend to be more selective and, in turn, report higher patient survival rates.19,21
Long-term patient survival outcomes can be gathered from larger, registry-based reviews, accepting limitations in assessing causes of patient death. Siskind et al23 analyzed the outcomes of 20,854 US pancreas transplants done between 1996 and 2012 and found the 10-year patient survival rate ranged from 43% to 77% and was highly dependent on patient age at the time of the procedure.23 Patient survival after transplant must be balanced against the generally poor long-term survival prospects of diabetic patients on dialysis.
By type of transplant, pancreas graft survival rates at 1 year are 89% for simultaneous pancreas-kidney transplant, 86% for pancreas-after-kidney transplant, and 84% for pancreas-alone transplant. Graft survival rates at 5 years are 71% for simultaneous pancreas-kidney transplant, 65% for pancreas-after-kidney transplant, and 58% for pancreas-alone transplant.8,9
Simultaneous pancreas-kidney transplant has been shown to improve the survival rate compared with cadaveric kidney transplant alone in patients with type 1 diabetes and chronic kidney disease.24,25 The survival benefit of isolated pancreas transplant (after kidney transplant and alone) is not evident at 4-year follow-up compared with patients on the waiting list. However, the benefit for the individual patient must be considered by weighing the incapacities experienced with insulin-based treatments against the risks of surgery and immunosuppression.26,27 For patients who have experienced frequent and significant hypoglycemic episodes, particularly those requiring third-party assistance, pancreas transplant can be a lifesaving procedure.
Effects on secondary diabetic complications
Notwithstanding the effect on the patient’s life span, data from several studies of long-term pancreas transplant recipients suggest that secondary diabetic complications can be halted or even improved. Most of these studies examined the effect of restoring euglycemia in nephropathy and the subsequent influence on renal function.
Effect on renal function. Kleinclauss et al28 examined renal allograft function in type 1 diabetic recipients of living-donor kidney transplants. Comparing kidney allograft survival and function in patients who received a subsequent pancreas-after-kidney transplant vs those who did not, graft survival was superior after 5 years, and the estimated glomerular filtration rate was 10 mL/min higher in pancreas-after-kidney recipients.28 This improvement in renal function was not seen immediately after the pancreas transplant but became evident more than 4 years after establishment of normoglycemia. Somewhat similarly, reversal of diabetic changes in native kidney biopsies has been seen 10 years after pancreas transplant.29
Effect on neuropathy. In other studies, reversal of autonomic neuropathy and hypoglycemic unawareness and improvements in peripheral sensory-motor neuropathy have also been observed.30–32
Effect on retinopathy. Improvements in early-stage nonproliferative diabetic retinopathy and laser-treated proliferative lesions have been seen, even within short periods of follow-up.33 Other groups have shown a significantly higher proportion of improvement or stability of advanced diabetic retinopathy at 3 years after simultaneous pancreas-kidney transplant, compared with kidney transplant alone in patients with type 1 diabetes.34
Effect on heart disease. Salutary effects on cardiovascular risk factors and amelioration of cardiac morphology and functional cardiac indices have been seen within the first posttransplant year.35 Moreover, with longer follow-up (nearly 4 years), simultaneous pancreas-kidney recipients with functioning pancreas grafts were found to have less progression of coronary atherosclerosis than simultaneous pancreas-kidney recipients with early pancreas graft loss.36 These data provide a potential pathophysiologic mechanism for the long-term survival advantage seen in uremic type 1 diabetic patients undergoing simultaneous pancreas-kidney transplant.
In the aggregate, these findings suggest that, in the absence of surgical and immunosuppression-related complications, a functioning pancreas allograft can alter the progress of diabetic complications. As an extension of these results, pancreas transplant done earlier in the course of diabetes may have an even greater impact.
Pancreas transplant is the only long-term diabetes treatment that consistently results in normal hemoglobin A1c levels without the risk of severe hypoglycemia. Additionally, pancreas transplant may prevent, halt, or even reverse the complications of diabetes.
Here, we explore the indications, options, and outcomes of pancreas transplant as a treatment for diabetes mellitus.
DIABETES IS COMMON, AND OFTEN NOT WELL CONTROLLED
Diabetes mellitus affects more than 25 million people in the United States (8.3% of the population) and is the leading cause of kidney failure, nontraumatic lower-limb amputation, and adult-onset blindness. In 2007, nearly $116 billion was spent on diabetes treatment, not counting another $58 billion in indirect costs such as disability, work loss, and premature death.1
Despite the tremendous expenditure in human, material, and financial resources, only about 50% of patients achieve their diabetes treatment goals. In 2013, a large US population-based study2 reported that 52.2% of patients were achieving the American Diabetes Association treatment goal of hemoglobin A1c lower than 7%. A similar study in South Korea3 found that 45.6% were at this goal.
Most of the patients in these studies had type 2 diabetes, and the data suggested that attaining glycemic goals is more difficult in insulin-treated patients. Studies of patients with type 1 diabetes found hemoglobin A1c levels lower than 7% in only 8.1% of hospitalized patients with type 1 diabetes, and in only 13% in an outpatient diabetes clinic.4,5
YET RATES OF PANCREAS TRANSPLANT ARE DECLINING
Pancreas transplant was first performed more than 40 years ago at the University of Minnesota.6 Since then, dramatic changes in immunosuppression, organ preservation, surgical technique, and donor and recipient selection have brought about significant progress.
Currently, more than 13,000 patients are alive with a functioning pancreas allograft. After reaching a peak in 2004, the annual number of pancreas transplants performed in the United States has declined steadily, whereas the procedure continues to increase in popularity outside North America.7 The primary reason for the decline is recognition of donor factors that lead to success—surgeons are refusing to transplant organs they might have accepted previously, because experience suggests they would yield poor results. In the United States, 1,043 pancreas transplants were performed in 2012, and more than 3,100 patients were on the waiting list.8
Islet cell transplant—a different procedure involving harvesting, encapsulating, and implanting insulin-producing beta cells—has not gained widespread application due to very low long-term success rates.
THREE CATEGORIES OF PANCREAS TRANSPLANT
Pancreas transplant can be categorized according to whether the patient is also receiving or has already received a kidney graft (Table 1).
Simultaneous kidney and pancreas transplant is performed in patients who have type 1 diabetes with advanced chronic kidney disease due to diabetic nephropathy. This remains the most commonly performed type, accounting for 79% of all pancreas transplants in 2012.8
Pancreas-after-kidney transplant is most often done after a living-donor kidney transplant. This procedure accounted for most of the increase in pancreas transplants during the first decade of the 2000s. However, the number of these procedures has steadily decreased since 2004, and in 2012 accounted for only 12% of pancreas transplants.8
Pancreas transplant alone is performed in nonuremic diabetic patients who have labile blood sugar control. Performed in patients with preserved renal function but severe complications of “brittle” diabetes, such as hypoglycemic unawareness, this type accounts for 8% of pancreas transplants.9
Indications for pancreas transplant
A small number of these procedures are done for indications unrelated to diabetes mellitus. In most of these cases, the pancreas is transplanted as part of a multivisceral transplant to facilitate the technical (surgical) aspect of the procedure—the pancreas, liver, stomach, gallbladder, and part of the intestines are transplanted en bloc to maintain the native vasculature. Very infrequently, pancreas transplant is done to replace exocrine pancreatic function.
A small, select group of patients with type 2 diabetes and low body mass index (BMI) may be eligible for pancreas transplant, and they accounted for 8.2% of active candidates in 2012.8 However, most pancreas transplants are performed in patients with type 1 diabetes.
WHAT MAKES A GOOD ALLOGRAFT?
Pancreas allografts are procured as whole organs from brain-dead organ donors. Relatively few pancreas allografts (3.1% in 2012) are from cardiac-death donors, because of concern about warm ischemic injury during the period of circulatory arrest.8
![]()
Proper donor selection is critical to the success of pancreas transplant, as donor factors including medical history, age, BMI, and cause of death can significantly affect the outcome. In general, transplant of a pancreas allograft from a young donor (age < 30) with excellent organ function, low BMI, and traumatic cause of death provides the best chance of success.
The Pancreas Donor Risk Index (PDRI)10 was developed after analysis of objective donor criteria, transplant type, and ischemic time in grafts transplanted between 2000 and 2006. One-year graft survival was directly related to the PDRI and ranged between 77% and 87% in recipients of “standard” pancreas allografts (PDRI score of 1.0). Use of grafts from the highest (worst) three quintiles of PDRI (PDRI score > 1.16) was associated with 1-year graft survival rates of 67% to 82%, significantly inferior to that seen with “higher- quality” grafts, again emphasizing the need for rigorous donor selection.10
In addition to these objective measures, visual assessment of pancreas quality at the time of procurement remains an equally important predictor of success. Determination of subjective features, such as fatty infiltration and glandular fibrosis, requires surgical experience developed over several years. In a 2010 analysis, dissatisfaction with the quality of the donor graft on inspection accounted for more than 80% of refusals of potential pancreas donors.11 These studies illustrate an ill-defined aspect of pancreas transplant, ie, even when the pancreas donor is perceived to be suitable, the outcome may be markedly different.
SURGICAL COMPLICATIONS
Surgical complications have long been considered a limiting factor in the growth of pancreas transplant. Technical failure or loss of the graft within 90 days is most commonly due to graft thrombosis, leakage of the enteric anastomosis, or severe peripancreatic infection. The rate of technical failure has declined across all recipient categories and is currently about 9%.8
DO RECIPIENT FACTORS AFFECT OUTCOMES?
As mentioned above, the PDRI identifies donor factors that influence the 1-year graft survival rate. Recipient factors are also thought to play a role, although the influence of these factors has not been consistently demonstrated.
Humar et al15 found that recipient obesity (defined in this study as BMI > 25 kg/m2) and donor age over 40 were risk factors for early laparotomy after pancreas transplant.15 Moreover, patients undergoing early laparotomy had poorer graft survival outcomes.
This finding was reinforced by an analysis of 5,725 primary simultaneous pancreas-kidney recipients between 2000 and 2007. Obesity (BMI 30 ≥ kg/m2) was associated with increased rates of patient death, pancreas graft loss, and kidney graft loss at 3 years.16
More recently, Finger et al17 did not find a statistically significant association between recipient BMI and technical failure, but they did notice a trend toward increased graft loss with a BMI greater than 25 kg/m2. Similarly, others have not found a clear adverse association between recipient BMI and pancreas graft survival.
Intuitively, obesity and other recipient factors such as age, vascular disease, duration of diabetes, and dialysis should influence pancreas graft survival but have not been shown in analyses to carry an adverse effect.18 The inability to consistently find adverse effects of recipient characteristics is most likely due to the relative similarity between the vast majority of pancreas transplant recipients and the relatively small numbers of adverse events. In 98 consecutive pancreas transplants at our center between 2009 and 2014, the technical loss rate was 1.8% (unpublished data).
Acute rejection most commonly occurs during the first year and is usually reversible. More than 1 year after transplant, graft loss is due to chronic rejection, and death is usually from underlying cardiovascular disease.
The immunosuppressive regimens used in pancreas transplant are similar to those in kidney transplant. Since the pancreas is considered to be more immunogenic than other organs, most centers employ a strategy of induction immunosuppression with T-cell–depleting or interleukin 2-receptor antibodies. Maintenance immunosuppression consists of a calcineurin inhibitor (tacrolimus or cyclosporine), an antimetabolite (mycophenolate), and a corticosteroid.8
Immunosuppressive complications occur at a rate similar to that seen in other solid-organ transplants and include an increased risk of opportunistic infection and malignancy. The risk of these complications must be balanced against the patient’s risk of health decline with dialysis and insulin-based therapies.
OVERALL OUTCOMES ARE GOOD
The success rate of pancreas transplant is currently at its highest since the inception of the procedure. The unadjusted patient survival rate for all groups is over 96% at 1 year, and over 80% at 5 years.8 One-year patient survival after pancreas transplant alone, at better than 96%, is the highest of all organ transplant procedures.9
Several recently published single-center reviews of pancreas transplant since 2000 report patient survival rates of 96% to 100% at 1 year and 88% to 100% at 5 years.19–22 This variability is likely closely linked to donor and recipient selection, as centers performing smaller numbers of transplants tend to be more selective and, in turn, report higher patient survival rates.19,21
Long-term patient survival outcomes can be gathered from larger, registry-based reviews, accepting limitations in assessing causes of patient death. Siskind et al23 analyzed the outcomes of 20,854 US pancreas transplants done between 1996 and 2012 and found the 10-year patient survival rate ranged from 43% to 77% and was highly dependent on patient age at the time of the procedure.23 Patient survival after transplant must be balanced against the generally poor long-term survival prospects of diabetic patients on dialysis.
By type of transplant, pancreas graft survival rates at 1 year are 89% for simultaneous pancreas-kidney transplant, 86% for pancreas-after-kidney transplant, and 84% for pancreas-alone transplant. Graft survival rates at 5 years are 71% for simultaneous pancreas-kidney transplant, 65% for pancreas-after-kidney transplant, and 58% for pancreas-alone transplant.8,9
Simultaneous pancreas-kidney transplant has been shown to improve the survival rate compared with cadaveric kidney transplant alone in patients with type 1 diabetes and chronic kidney disease.24,25 The survival benefit of isolated pancreas transplant (after kidney transplant and alone) is not evident at 4-year follow-up compared with patients on the waiting list. However, the benefit for the individual patient must be considered by weighing the incapacities experienced with insulin-based treatments against the risks of surgery and immunosuppression.26,27 For patients who have experienced frequent and significant hypoglycemic episodes, particularly those requiring third-party assistance, pancreas transplant can be a lifesaving procedure.
Effects on secondary diabetic complications
Notwithstanding the effect on the patient’s life span, data from several studies of long-term pancreas transplant recipients suggest that secondary diabetic complications can be halted or even improved. Most of these studies examined the effect of restoring euglycemia in nephropathy and the subsequent influence on renal function.
Effect on renal function. Kleinclauss et al28 examined renal allograft function in type 1 diabetic recipients of living-donor kidney transplants. Comparing kidney allograft survival and function in patients who received a subsequent pancreas-after-kidney transplant vs those who did not, graft survival was superior after 5 years, and the estimated glomerular filtration rate was 10 mL/min higher in pancreas-after-kidney recipients.28 This improvement in renal function was not seen immediately after the pancreas transplant but became evident more than 4 years after establishment of normoglycemia. Somewhat similarly, reversal of diabetic changes in native kidney biopsies has been seen 10 years after pancreas transplant.29
Effect on neuropathy. In other studies, reversal of autonomic neuropathy and hypoglycemic unawareness and improvements in peripheral sensory-motor neuropathy have also been observed.30–32
Effect on retinopathy. Improvements in early-stage nonproliferative diabetic retinopathy and laser-treated proliferative lesions have been seen, even within short periods of follow-up.33 Other groups have shown a significantly higher proportion of improvement or stability of advanced diabetic retinopathy at 3 years after simultaneous pancreas-kidney transplant, compared with kidney transplant alone in patients with type 1 diabetes.34
Effect on heart disease. Salutary effects on cardiovascular risk factors and amelioration of cardiac morphology and functional cardiac indices have been seen within the first posttransplant year.35 Moreover, with longer follow-up (nearly 4 years), simultaneous pancreas-kidney recipients with functioning pancreas grafts were found to have less progression of coronary atherosclerosis than simultaneous pancreas-kidney recipients with early pancreas graft loss.36 These data provide a potential pathophysiologic mechanism for the long-term survival advantage seen in uremic type 1 diabetic patients undergoing simultaneous pancreas-kidney transplant.
In the aggregate, these findings suggest that, in the absence of surgical and immunosuppression-related complications, a functioning pancreas allograft can alter the progress of diabetic complications. As an extension of these results, pancreas transplant done earlier in the course of diabetes may have an even greater impact.
- Centers for Disease Control and Prevention (CDC). National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed August 12, 2015.
- Ali MK, Bullard KM, Saaddine JB, Cowie CC, Imperatore G, Gregg EW. Achievement of goals in US diabetes care, 1999–2010. N Engl J Med 2013; 368:1613–1624.
- Jeon JY, Kim DJ, Ko SH, et al; Taskforce Team of Diabetes Fact Sheet of the Korean Diabetes Association. Current status of glycemic control of patients with diabetes in Korea: the fifth Korea national health and nutrition examination survey. Diabetes Metab J 2014; 38:197–203.
- Govan L, Wu O, Briggs A, et al; Scottish Diabetes Research Network Epidemiology Group. Achieved levels of HbA1c and likelihood of hospital admission in people with type 1 diabetes in the Scottish population: a study from the Scottish Diabetes Research Network Epidemiology Group. Diabetes Care 2011; 34:1992–1997.
- Bryant W, Greenfield JR, Chisholm DJ, Campbell LV. Diabetes guidelines: easier to preach than to practise? Med J Aust 2006; 185:305–309.
- Kelly WD, Lillehei RC, Merkel FK, Idezuki Y, Goetz FC. Allotransplantation of the pancreas and duodenum along with the kidney in diabetic nephropathy. Surgery 1967; 61:827–837.
- Gruessner AC, Gruessner RW. Pancreas transplant outcomes for United States and non United States cases as reported to the United Network for Organ Sharing and the International Pancreas Transplant Registry as of December 2011. Clin Transpl 2012: 23–40.
- Israni AK, Skeans MA, Gustafson SK, et al. OPTN/SRTR 2012 Annual Data Report: pancreas. Am J Transplant 2014; 14(suppl 1):45–68
- Gruessner RW, Gruessner AC. Pancreas transplant alone: a procedure coming of age. Diabetes Care 2013; 36:2440–2447.
- Axelrod DA, Sung RS, Meyer KH, Wolfe RA, Kaufman DB. Systematic evaluation of pancreas allograft quality, outcomes and geographic variation in utilization. Am J Transplant 2010; 10:837–845.
- Wiseman AC, Wainright JL, Sleeman E, et al. An analysis of the lack of donor pancreas utilization from younger adult organ donors. Transplantation 2010; 90:475–480.
- Gruessner RW, Gruessner AC. The current state of pancreas transplantation. Nat Rev Endocrinol 2013; 9:555–562.
- Gunasekaran G, Wee A, Rabets J, Winans C, Krishnamurthi V. Duodenoduodenostomy in pancreas transplantation. Clin Transplant 2012; 26:550–557.
- Sollinger HW, Odorico JS, Becker YT, D’Alessandro AM, Pirsch JD. One thousand simultaneous pancreas-kidney transplants at a single center with 22-year follow-up. Ann Surg 2009; 250:618–630.
- Humar A, Kandaswamy R, Granger D, Gruessner RW, Gruessner AC, Sutherland DE. Decreased surgical risks of pancreas transplantation in the modern era. Ann Surg 2000; 231:269–275.
- Sampaio MS, Reddy PN, Kuo HT, et al. Obesity was associated with inferior outcomes in simultaneous pancreas kidney transplant. Transplantation 2010; 89:1117–1125.
- Finger EB, Radosevich DM, Dunn TB, et al. A composite risk model for predicting technical failure in pancreas transplantation. Am J Transplant 2013; 13:1840–1849.
- Fridell JA, Mangus RS, Taber TE, et al. Growth of a nation part II: impact of recipient obesity on whole-organ pancreas transplantation. Clin Transplant 2011; 25:E366–E374.
- Tai DS, Hong J, Busuttil RW, Lipshutz GS. Low rates of short- and long-term graft loss after kidney-pancreas transplant from a single center. JAMA Surg 2013; 148:368–373.
- Bazerbachi F, Selzner M, Marquez MA, et al. Pancreas-after-kidney versus synchronous pancreas-kidney transplantation: comparison of intermediate-term results. Transplantation 2013; 95:489–494.
- Laftavi MR, Pankewycz O, Gruessner A, et al. Long-term outcomes of pancreas after kidney transplantation in small centers: is it justified? Transplant Proc 2014; 46:1920–1923.
- Stratta RJ, Farney AC, Orlando G, Farooq U, Al-Shraideh Y, Rogers J. Similar results with solitary pancreas transplantation compared with simultaneous pancreas-kidney transplantation in the new millennium. Transplant Proc 2014; 46:1924–1927.
- Siskind E, Maloney C, Akerman M, et al. An analysis of pancreas transplantation outcomes based on age groupings—an update of the UNOS database. Clin Transplant 2014; 28:990–994.
- Ojo AO, Meier-Kriesche HU, Hanson JA, et al. The impact of simultaneous pancreas-kidney transplantation on long-term patient survival. Transplantation 2001; 71:82–90.
- Reddy KS, Stablein D, Taranto S, et al. Long-term survival following simultaneous kidney-pancreas transplantation versus kidney transplantation alone in patients with type 1 diabetes mellitus and renal failure. Am J Kidney Dis 2003; 41:464–470.
- Venstrom JM, McBride MA, Rother KI, Hirshberg B, Orchard TJ, Harlan DM. Survival after pancreas transplantation in patients with diabetes and preserved kidney function. JAMA 2003; 290:2817–2823.
- Gruessner RW, Sutherland DE, Gruessner AC. Mortality assessment for pancreas transplants. Am J Transplant 2004; 4:2018–2026.
- Kleinclauss F, Fauda M, Sutherland DE, et al. Pancreas after living donor kidney transplants in diabetic patients: impact on long-term kidney graft function. Clin Transplant 2009; 23:437–446.
- Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med 1998; 339:69–75.
- Landgraf R. Impact of pancreas transplantation on diabetic secondary complications and quality of life. Diabetologia 1996; 39:1415–1424.
- Robertson RP. Update on transplanting beta cells for reversing type 1 diabetes. Endocrinol Metab Clin North Am 2010; 39:655–667.
- Robertson RP, Holohan TV, Genuth S. Therapeutic controversy: pancreas transplantation for type I diabetes. J Clin Endocrinol Metab 1998; 83:1868–1674.
- Giannarelli R, Coppelli A, Sartini MS, et al. Pancreas transplant alone has beneficial effects on retinopathy in type 1 diabetic patients. Diabetologia 2006; 49:2977–2982.
- Koznarová R, Saudek F, Sosna T, et al. Beneficial effect of pancreas and kidney transplantation on advanced diabetic retinopathy. Cell Transplant 2000; 9:903–908.
- Coppelli A, Giannarelli R, Mariotti R, et al. Pancreas transplant alone determines early improvement of cardiovascular risk factors and cardiac function in type 1 diabetic patients. Transplantation 2003; 76:974–976.
- Jukema JW, Smets YF, van der Pijl JW, et al. Impact of simultaneous pancreas and kidney transplantation on progression of coronary atherosclerosis in patients with end-stage renal failure due to type 1 diabetes. Diabetes Care 2002; 25:906–911.
- Centers for Disease Control and Prevention (CDC). National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed August 12, 2015.
- Ali MK, Bullard KM, Saaddine JB, Cowie CC, Imperatore G, Gregg EW. Achievement of goals in US diabetes care, 1999–2010. N Engl J Med 2013; 368:1613–1624.
- Jeon JY, Kim DJ, Ko SH, et al; Taskforce Team of Diabetes Fact Sheet of the Korean Diabetes Association. Current status of glycemic control of patients with diabetes in Korea: the fifth Korea national health and nutrition examination survey. Diabetes Metab J 2014; 38:197–203.
- Govan L, Wu O, Briggs A, et al; Scottish Diabetes Research Network Epidemiology Group. Achieved levels of HbA1c and likelihood of hospital admission in people with type 1 diabetes in the Scottish population: a study from the Scottish Diabetes Research Network Epidemiology Group. Diabetes Care 2011; 34:1992–1997.
- Bryant W, Greenfield JR, Chisholm DJ, Campbell LV. Diabetes guidelines: easier to preach than to practise? Med J Aust 2006; 185:305–309.
- Kelly WD, Lillehei RC, Merkel FK, Idezuki Y, Goetz FC. Allotransplantation of the pancreas and duodenum along with the kidney in diabetic nephropathy. Surgery 1967; 61:827–837.
- Gruessner AC, Gruessner RW. Pancreas transplant outcomes for United States and non United States cases as reported to the United Network for Organ Sharing and the International Pancreas Transplant Registry as of December 2011. Clin Transpl 2012: 23–40.
- Israni AK, Skeans MA, Gustafson SK, et al. OPTN/SRTR 2012 Annual Data Report: pancreas. Am J Transplant 2014; 14(suppl 1):45–68
- Gruessner RW, Gruessner AC. Pancreas transplant alone: a procedure coming of age. Diabetes Care 2013; 36:2440–2447.
- Axelrod DA, Sung RS, Meyer KH, Wolfe RA, Kaufman DB. Systematic evaluation of pancreas allograft quality, outcomes and geographic variation in utilization. Am J Transplant 2010; 10:837–845.
- Wiseman AC, Wainright JL, Sleeman E, et al. An analysis of the lack of donor pancreas utilization from younger adult organ donors. Transplantation 2010; 90:475–480.
- Gruessner RW, Gruessner AC. The current state of pancreas transplantation. Nat Rev Endocrinol 2013; 9:555–562.
- Gunasekaran G, Wee A, Rabets J, Winans C, Krishnamurthi V. Duodenoduodenostomy in pancreas transplantation. Clin Transplant 2012; 26:550–557.
- Sollinger HW, Odorico JS, Becker YT, D’Alessandro AM, Pirsch JD. One thousand simultaneous pancreas-kidney transplants at a single center with 22-year follow-up. Ann Surg 2009; 250:618–630.
- Humar A, Kandaswamy R, Granger D, Gruessner RW, Gruessner AC, Sutherland DE. Decreased surgical risks of pancreas transplantation in the modern era. Ann Surg 2000; 231:269–275.
- Sampaio MS, Reddy PN, Kuo HT, et al. Obesity was associated with inferior outcomes in simultaneous pancreas kidney transplant. Transplantation 2010; 89:1117–1125.
- Finger EB, Radosevich DM, Dunn TB, et al. A composite risk model for predicting technical failure in pancreas transplantation. Am J Transplant 2013; 13:1840–1849.
- Fridell JA, Mangus RS, Taber TE, et al. Growth of a nation part II: impact of recipient obesity on whole-organ pancreas transplantation. Clin Transplant 2011; 25:E366–E374.
- Tai DS, Hong J, Busuttil RW, Lipshutz GS. Low rates of short- and long-term graft loss after kidney-pancreas transplant from a single center. JAMA Surg 2013; 148:368–373.
- Bazerbachi F, Selzner M, Marquez MA, et al. Pancreas-after-kidney versus synchronous pancreas-kidney transplantation: comparison of intermediate-term results. Transplantation 2013; 95:489–494.
- Laftavi MR, Pankewycz O, Gruessner A, et al. Long-term outcomes of pancreas after kidney transplantation in small centers: is it justified? Transplant Proc 2014; 46:1920–1923.
- Stratta RJ, Farney AC, Orlando G, Farooq U, Al-Shraideh Y, Rogers J. Similar results with solitary pancreas transplantation compared with simultaneous pancreas-kidney transplantation in the new millennium. Transplant Proc 2014; 46:1924–1927.
- Siskind E, Maloney C, Akerman M, et al. An analysis of pancreas transplantation outcomes based on age groupings—an update of the UNOS database. Clin Transplant 2014; 28:990–994.
- Ojo AO, Meier-Kriesche HU, Hanson JA, et al. The impact of simultaneous pancreas-kidney transplantation on long-term patient survival. Transplantation 2001; 71:82–90.
- Reddy KS, Stablein D, Taranto S, et al. Long-term survival following simultaneous kidney-pancreas transplantation versus kidney transplantation alone in patients with type 1 diabetes mellitus and renal failure. Am J Kidney Dis 2003; 41:464–470.
- Venstrom JM, McBride MA, Rother KI, Hirshberg B, Orchard TJ, Harlan DM. Survival after pancreas transplantation in patients with diabetes and preserved kidney function. JAMA 2003; 290:2817–2823.
- Gruessner RW, Sutherland DE, Gruessner AC. Mortality assessment for pancreas transplants. Am J Transplant 2004; 4:2018–2026.
- Kleinclauss F, Fauda M, Sutherland DE, et al. Pancreas after living donor kidney transplants in diabetic patients: impact on long-term kidney graft function. Clin Transplant 2009; 23:437–446.
- Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med 1998; 339:69–75.
- Landgraf R. Impact of pancreas transplantation on diabetic secondary complications and quality of life. Diabetologia 1996; 39:1415–1424.
- Robertson RP. Update on transplanting beta cells for reversing type 1 diabetes. Endocrinol Metab Clin North Am 2010; 39:655–667.
- Robertson RP, Holohan TV, Genuth S. Therapeutic controversy: pancreas transplantation for type I diabetes. J Clin Endocrinol Metab 1998; 83:1868–1674.
- Giannarelli R, Coppelli A, Sartini MS, et al. Pancreas transplant alone has beneficial effects on retinopathy in type 1 diabetic patients. Diabetologia 2006; 49:2977–2982.
- Koznarová R, Saudek F, Sosna T, et al. Beneficial effect of pancreas and kidney transplantation on advanced diabetic retinopathy. Cell Transplant 2000; 9:903–908.
- Coppelli A, Giannarelli R, Mariotti R, et al. Pancreas transplant alone determines early improvement of cardiovascular risk factors and cardiac function in type 1 diabetic patients. Transplantation 2003; 76:974–976.
- Jukema JW, Smets YF, van der Pijl JW, et al. Impact of simultaneous pancreas and kidney transplantation on progression of coronary atherosclerosis in patients with end-stage renal failure due to type 1 diabetes. Diabetes Care 2002; 25:906–911.
KEY POINTS
- Current options are simultaneous pancreas-kidney transplant, pancreas-after-kidney transplant, and pancreas-alone transplant.
- Simultaneous pancreas-kidney transplant provides a significant survival benefit over insulin- and dialysis-based therapies.
- Isolated pancreas transplant for diabetic patients without uremia can prevent hypoglycemic unawareness.
Common infectious complications of liver transplant
The immunosuppressed state of liver transplant recipients makes them vulnerable to infections after surgery.1 These infections are directly correlated with the net state of immunosuppression. Higher levels of immunosuppression mean a higher risk of infection, with rates of infection typically highest in the early posttransplant period.
Common infections during this period include operative and perioperative nosocomial bacterial and fungal infections, reactivation of latent infections, and invasive fungal infections such as candidiasis, aspergillosis, and pneumocystosis. Donor-derived infections also must be considered. As time passes and the level of immunosuppression is reduced, liver recipients are less prone to infection.1
The risk of infection can be minimized by appropriate antimicrobial prophylaxis, strategies for safe living after transplant,2 vaccination,3 careful balancing of immunosuppressive therapy,4 and thoughtful donor selection.5 Drug-drug interactions are common and must be carefully considered to minimize the risk.
This review highlights common infectious complications encountered after liver transplant.
INTRA-ABDOMINAL INFECTIONS
Intra-abdominal infections are common in the early postoperative period.6,7
Risk factors include:
- Pretransplant ascites
- Posttransplant dialysis
- Wound infection
- Reoperation8
- Hepatic artery thrombosis
- Roux-en-Y choledochojejunostomy anastomosis.9
Signs that may indicate intra-abdominal infection include fever, abdominal pain, leukocytosis, and elevated liver enzymes. But because of their immunosuppressed state, transplant recipients may not manifest fever as readily as the general population. They should be evaluated for cholangitis, peritonitis, biloma, and intra-abdominal abscess.
Organisms. Intra-abdominal infections are often polymicrobial. Enterococci, Staphylococcus aureus, gram-negative species including Pseudomonas, Klebsiella, and Acinetobacter, and Candida species are the most common pathogens. Strains are often resistant to multiple drugs, especially in patients who received antibiotics in the weeks before transplant.8,10
Liver transplant recipients are also particularly susceptible to Clostridium difficile-associated colitis as a result of immunosuppression and frequent use of antibiotics perioperatively and postoperatively.11 The spectrum of C difficile infection ranges from mild diarrhea to life-threatening colitis, and the course in liver transplant patients tends to be more complicated than in immunocompetent patients.12
Diagnosis. Intra-abdominal infections should be looked for and treated promptly, as they are associated with a higher mortality rate, a greater risk of graft loss, and a higher incidence of retransplant.6,10 Abdominal ultrasonography or computed tomography (CT) can confirm the presence of fluid collections.
Treatment. Infected collections can be treated with percutaneous or surgical drainage and antimicrobial therapy. In the case of biliary tract complications, retransplant or surgical correction of biliary leakage or stenosis decreases the risk of death.6
Suspicion should be high for C difficile-associated colitis in cases of posttransplant diarrhea. C difficile toxin stool assays help confirm the diagnosis.12 Oral metronidazole is recommended in mild to moderate C difficile infection, with oral vancomycin and intravenous metronidazole reserved for severe cases. Colectomy may be necessary in patients with toxic megacolon.
CYTOMEGALOVIRUS INFECTION
Cytomegalovirus is an important opportunistic pathogen in liver transplant recipients.13 It causes a range of manifestations, from infection (viremia with or without symptoms) to cytomegalovirus syndrome (fever, malaise, and cell-line cytopenias) to tissue-invasive disease with end-organ disease.14 Without preventive measures and treatment, cytomegalovirus disease can increase the risk of morbidity, allograft loss and death.15,16

Risk factors for cytomegalovirus infection (Table 1) include:
- Discordant serostatus of the donor and recipient (the risk is highest in seronegative recipients of organs from seropositive donors)
- Higher levels of immunosuppression, especially when antilymphocyte antibodies are used
- Treatment of graft rejection
- Coinfection with other human herpesviruses, such as Epstein-Barr virus.4,17
Preventing cytomegalovirus infection

The strategy to prevent cytomegalovirus infection depends on the serologic status of the donor and recipient and may include antiviral prophylaxis or preemptive treatment (Table 2).18
Prophylaxis involves giving antiviral drugs during the early high-risk period, with the goal of preventing the development of cytomegalovirus viremia. The alternative preemptive strategy emphasizes serial testing for cytomegalovirus viremia, with the goal of intervening with antiviral medications while viremia is at a low level, thus avoiding potential progression to cytomegalovirus disease. Both strategies have pros and cons that should be considered by each transplant center when setting institutional policy.
A prophylactic approach seems very effective at preventing both infection and disease from cytomegalovirus and has been shown to reduce graft rejection and the risk of death.18 It is preferred in cytomegalovirus-negative recipients when the donor was cytomegalovirus-positive—a high-risk situation.19 However, these patients are also at higher risk of late-onset cytomegalovirus disease. Higher cost and potential drug toxicity, mainly neutropenia from ganciclovir-based regimens, are additional considerations.
Preemptive treatment, in contrast, reserves drug treatment for patients who are actually infected with cytomegalovirus, thus resulting in fewer adverse drug events and lower cost; but it requires regular monitoring. Preemptive methods, by definition, cannot prevent infection, and with this strategy tissue-invasive disease not associated with viremia does occasionally occur.20 As such, patients with a clinical presentation that suggests cytomegalovirus but have negative results on blood testing should be considered for tissue biopsy with culture and immunohistochemical stain.
The most commonly used regimens for antiviral prophylaxis and treatment in liver transplant recipients are intravenous ganciclovir and oral valganciclovir.21 Although valganciclovir is the most commonly used agent in this setting because of ease of administration, it has not been approved by the US Food and Drug Administration in liver transplant patients, as it was associated with higher rates of cytomegalovirus tissue-invasive disease.22–24 Additionally, drug-resistant cytomegalovirus strains have been associated with valganciclovir prophylaxis in cytomegalovirus-negative recipients of solid organs from cytomegalovirus-positive donors.25
Prophylaxis typically consists of therapy for 3 months from the time of transplant. In higher-risk patients (donor-positive, recipient-negative), longer courses of prophylaxis have been extrapolated from data in kidney transplant recipients.26 Extension or reinstitution of prophylaxis should also be considered in liver transplant patients receiving treatment for rejection with antilymphocyte therapy.
Routine screening for cytomegalovirus is not recommended while patients are receiving prophylaxis. High-risk patients who are not receiving prophylaxis should be monitored with nucleic acid or pp65 antigenemia testing as part of the preemptive strategy protocol.
Treatment of cytomegalovirus disease
Although no specific threshold has been established, treatment is generally indicated if a patient has a consistent clinical syndrome, evidence of tissue injury, and persistent or increasing viremia.
Treatment involves giving antiviral drugs and also reducing the level of immunosuppression, if possible, until symptoms and viremia have resolved.
The choice of antiviral therapy depends on the severity of disease. Intravenous ganciclovir (5 mg/kg twice daily adjusted for renal impairment) or oral valganciclovir (900 mg twice daily, also renally dose-adjusted when necessary) can be used for mild to moderate disease if no significant gastrointestinal involvement is reported. Intravenous ganciclovir is preferred for patients with more severe disease or gastrointestinal involvement. The minimum duration of treatment is 2 weeks and may need to be prolonged until both symptoms and viremia completely resolve.18
Drug resistance can occur and should be considered in patients who have a history of prolonged ganciclovir or valganciclovir exposure who do not clinically improve or have persistent or rising viremia. In such cases, genotype assays are helpful, and initiation of alternative therapy should be considered. Mutations conferring resistance to ganciclovir are often associated with cross-resistance to cidofovir. Cidofovir can therefore be considered only when genotype assays demonstrate specific mutations conferring an isolated resistance to ganciclovir.27 The addition of foscarnet to the ganciclovir regimen or substitution of foscarnet for ganciclovir are accepted approaches.
Although cytomegalovirus hyperimmunoglobulin has been used in prophylaxis and invasive disease treatment, its role in the management of ganciclovir-resistant cytomegalovirus infections remains controversial.28
EPSTEIN-BARR VIRUS POSTTRANSPLANT LYMPHOPROLIFERATIVE DISEASE
Epstein-Barr virus-associated posttransplant lymphoproliferative disease is a spectrum of disorders ranging from an infectious mononucleosis syndrome to aggressive malignancy with the potential for death and significant morbidity after liver transplant.29 The timeline of risk varies, but the disease is most common in the first year after transplant.
Risk factors for this disease (Table 1) are:
- Primary Epstein-Barr virus infection
- Cytomegalovirus donor-recipient mismatch
- Cytomegalovirus disease
- Higher levels of immunosuppression, especially with antilymphocyte antibodies.30
The likelihood of Epstein-Barr virus playing a contributing role is lower in later-onset posttransplant lymphoproliferative disease. Patients who are older at the time of transplant, who receive highly immunogenic allografts including a liver as a component of a multivisceral transplant, and who receive increased immunosuppression to treat rejection are at even greater risk of late posttransplant lymphoproliferative disease.31 This is in contrast to early posttransplant lymphoproliferative disease, which is seen more commonly in children as a result of primary Epstein-Barr virus infection.
Recognition and diagnosis. Heightened suspicion is required when considering posttransplant lymphoproliferative disease, and careful evaluation of consistent symptoms and allograft dysfunction are required.
Clinically, posttransplant lymphoproliferative disease should be suspected if a liver transplant recipient develops unexplained fever, weight loss, lymphadenopathy, or cell-line cytopenias.30,32 Other signs and symptoms may be related to the organ involved and may include evidence of hepatitis, pneumonitis, and gastrointestinal disease.31
Adjunctive diagnostic testing includes donor and recipient serology to characterize overall risk before transplantation and quantification of Epstein-Barr viral load, but confirmation relies on tissue histopathology.
Treatment focuses on reducing immunosuppression.30,32 Adding antiviral agents does not seem to improve outcome in all cases.33 Depending on clinical response and histologic classification, additional therapies such as anti-CD20 humanized chimeric monoclonal antibodies, surgery, radiation, and conventional chemotherapy may be required.34
Preventive approaches remain controversial. Chemoprophylaxis with an antiviral such as ganciclovir is occasionally used but has not been shown to consistently decrease rates of posttransplant lymphoproliferative disease. These agents may act in an indirect manner, leading to decreased rates of cytomegalovirus infection, a major cofactor for posttransplant lymphoproliferative disease.24
Passive immunoprophylaxis with immunoglobulin targeting cytomegalovirus has shown to decrease rates of non-Hodgkin lymphoma from posttransplant lymphoproliferative disease in renal transplant recipients in the first year after transplant,35 but data are lacking regarding its use in liver transplant recipients. Monitoring of the viral load and subsequent reduction of immunosuppression remain the most efficient measures to date.36
FUNGAL INFECTIONS
Candida species account for more than half of fungal infections in liver transplant recipients.37 However, a change has been noted in the past 20 years, with a decrease in Candida infections accompanied by an increase in Aspergillus infections.38 Endemic mycoses such as coccidioidomycosis, blastomycosis, and histoplasmosis should be considered with the appropriate epidemiologic history or if disease develops early after transplant and the donor came from a highly endemic region.39Cryptococcus may also be encountered.
Diagnosis. One of the most challenging aspects of fungal infection in liver transplant recipients is timely diagnosis. Heightened suspicion and early biopsy for pathological and microbiological confirmation are necessary. Although available noninvasive diagnostic tools often lack specificity, early detection of fungal markers may be of great use in guiding further diagnostic workup or empiric treatment in the critically ill.
Noninvasive tests include galactomannan, cryptococcal antigen, histoplasma antigen, (1-3)-beta-D-glucan assay and various antibody tests. Galactomannan testing has been widely used to aid in the diagnosis of invasive aspergillosis. Similarly, the (1-3)-beta-D-glucan assay is a non–culture-based tool for diagnosing and monitoring the treatment of invasive fungal infections. However, a definite diagnosis cannot be made on the basis of a positive test alone.40 The complementary diagnostic characteristics of combining noninvasive assays have yet to be fully elucidated.41 Cultures and tissue histopathology are also used when possible.
Treatment is based on targeted specific antifungal drug therapy and reduction of immunosuppressive therapy, when possible. The choice of antifungal agent varies with the pathogen, the site of involvement, and the severity of the disease. A focus on potential drug interactions, their management, and therapeutic drug monitoring when using antifungal medications is essential in the posttransplant period. Combination therapy can be considered in some situations to enhance synergy. The following sections discuss in greater detail Candida species, Aspergillus species, and Pneumocystis jirovecii infections.
Candida infections

Candidiasis after liver transplant is typically nosocomial, especially when diagnosed during the first 3 months (Table 3).37
Risk factors for invasive candidiasis include perioperative colonization, prolonged operative time, retransplant, greater transfusion requirements, and postoperative renal failure.37,42,43 Invasive candidiasis is of concern for its effects on morbidity, mortality, and cost of care.43–46
Organisms. The frequency of implicated species, in particular those with a natural resistance to fluconazole, differs in various reports.37,45,46Candida albicans remains the most commonly isolated pathogen; however, non-albicans species including those resistant to fluconazole have been reported more frequently and include Candida glabrata and Candida krusei.47,48
Signs and diagnosis. Invasive candidiasis in liver transplant recipients generally manifests itself in catheter-related blood stream infections, urinary tract infections, or intra-abdominal infections. Diagnosis can be made by isolating Candida from blood cultures, recovering organisms in culture of a normally sterile site, or finding direct microscopic evidence of the fungus on tissue specimens.49
Disseminated candidiasis refers to the involvement of distant anatomic sites. Clinical manifestations may cause vision changes, abdominal pain or skin nodules with findings of candidemia, hepatosplenic abscesses, or retinal exudates on funduscopy.49
Treatment of invasive candidiasis in liver recipients often involves antifungal therapy and reduction of immunosuppression. Broad-spectrum antifungals are initially advocated in an empirical approach to cover fluconazole-resistant strains of the non-albicans subgroups.50 Depending on antifungal susceptibility, treatment can later be adjusted.
Fluconazole remains the agent of choice in most C albicans infections.47 However, attention should be paid to the possibility of resistance in patients who have received fluconazole prophylaxis within the past 30 days. Additional agents used in treatment may include echinocandins, amphotericin, and additional azoles.
Antifungal prophylaxis is recommended in high-risk liver transplant patients, although its optimal duration remains undetermined.44 Antifungal prophylaxis has been associated with decreased incidence of both superficial and invasive candidiasis.51
Aspergillus infection
Aspergillus, the second most common fungal pathogen, has become a more common concern in liver transplant recipients. Aspergillus fumigatus is the most frequently encountered species.38,52
Risk factors. These infections typically occur in the first year, during intense immunosuppression. Retransplant, renal failure, and fulminant hepatic failure are major risk factors.52 In the presence of risk factors and a suggestive clinical setting, invasive aspergillosis should be considered and the diagnosis pursued.
Diagnosis is suggested by positive findings on CT accompanied by lower respiratory tract symptoms, focal lesions on neuroimaging, or demonstration of the fungus on cultures.49 However, Aspergillus is rarely grown in blood culture. The galactomannan antigen is a noninvasive test that can provide supporting evidence for the diagnosis.41,52 False-positive results do occur in the setting of certain antibiotics and cross-reacting fungi.53
Treatment consists of antifungal therapy and immunosuppression reduction.52
Voriconazole is the first-line agent for invasive aspergillosis. Monitoring for potential drug-drug interactions and side effects is required.54,55 Amphotericin B is considered a second-line choice due to toxicity and lack of an oral formulation. In refractory cases, combined antifungal therapy could be considered.52 The duration of treatment is generally a minimum of 12 weeks.
Prophylaxis. Specific prophylaxis against invasive aspergillosis is not currently recommended; however, some authors suggest a prophylactic approach using echinocandins or liposomal amphotericin B in high-risk patients.51,52 Aspergillosis is associated with a considerable increase in mortality in liver transplant recipients, which highlights the importance of timely management.52,56
Pneumocystis jirovecii
P jirovecii remains a common opportunistic pathogen in people with impaired immunity, including transplant and human immunodeficiency virus patients.
Prophylaxis. Widespread adoption of antimicrobial prophylaxis by transplant centers has decreased the rates of P jirovecii infection in liver transplant recipients.57,58 Commonly used prophylactic regimens after liver transplantation include a single-strength trimethoprim-sulfamethoxazole tablet daily or a double-strength tablet three times per week for a minimum of 6 to 12 months after transplant. Atovaquone and dapsone can be used as alternatives in cases of intolerance to trimethoprim-sulfamethoxazole (Table 2).
Inhaled pentamidine is clearly inferior and should be used only when the other medications are contraindicated.59
Signs and diagnosis. P jirovecii pneumonia is characterized by fever, cough, dyspnea, and chest pain. Insidious hypoxemia, abnormal chest examination, and bilateral interstitial pneumonia on chest radiography are common.
CT may be more sensitive than chest radiography.57 Findings suggestive of P jirovecii pneumonia on chest CT are extensive bilateral and symmetrical ground-glass attenuations. Other less-characteristic findings include upper lobar parenchymal opacities and spontaneous pneumothorax.57,60
The serum (1,3)-beta-D-glucan assay derived from major cell-wall components of P jirovecii might be helpful. Studies report a sensitivity for P jirovecii pneumonia as high as 96% and a negative predictive value of 99.8%.61,62
Definitive diagnosis requires identification of the pathogen. Routine expectorated sputum sampling is generally associated with a poor diagnostic yield. Bronchoscopy and bronchoalveolar lavage with silver or fluorescent antibody staining of samples, polymerase chain reaction testing, or both significantly improves diagnosis. Transbronchial or open lung biopsy are often unnecessary.57
Treatment. Trimethoprim-sulfamethoxazole is the first-line agent for treating P jirovecii pneumonia.57 The minimum duration of treatment is 14 days, with extended courses for severe infection.
Intravenous pentamidine or clindamycin plus primaquine are alternatives for patients who cannot tolerate trimethoprim-sulfamethoxazole. The major concern with intravenous pentamidine is renal dysfunction. Hypoglycemia or hyperglycemia, neutropenia, thrombocytopenia, nausea, dysgeusia, and pancreatitis may also occur.63
Atovaquone might also be beneficial in mild to moderate P jirovecii pneumonia. The main side effects include skin rashes, gastrointestinal intolerance, and elevation of transaminases.64
A corticosteroid (40–60 mg of prednisone or its equivalent) may be beneficial in conjunction with antimicrobial therapy in patients with significant hypoxia (partial pressure of arterial oxygen < 70 mm Hg on room air) in decreasing the risk of respiratory failure and need for intubation.
With appropriate and timely antimicrobial prophylaxis, cases of P jirovecii pneumonia should continue to decrease.
TUBERCULOSIS
Development of tuberculosis after transplantation is a catastrophic complication, with mortality rates of up to 30%.65 Most cases of posttransplant tuberculosis represent reactivation of latent disease.66 Screening with tuberculin skin tests or interferon-gamma-release assays is recommended in all liver transplant candidates. Chest radiography before transplant is necessary when assessing a positive screening test.67
The optimal management of latent tuberculosis in these cases remains controversial. Patients at high risk or those with positive screening results on chest radiography warrant treatment for latent tuberculosis infection with isoniazid unless contraindicated.67,68
The ideal time to initiate prophylactic isoniazid therapy is unclear. Some authors suggest delaying it, as it might be associated with poor tolerance and hepatotoxicity.69 Others have found that early isoniazid use was not associated with negative outcomes.70
Risk factors for symptomatic tuberculosis after liver transplant include previous infection with tuberculosis, intensified immunosuppression (especially anti-T-lymphocyte therapies), diabetes mellitus, and other co-infections (Table 1).71
The increased incidence of atypical presentations in recent years makes the diagnosis of active tuberculosis among liver transplant recipients challenging. Sputum smears can be negative due to low mycobacterial burdens, and tuberculin skin testing and interferon-gamma-release assays may be falsely negative due to immunosuppression.67
Treatment of active tuberculosis consists initially of a four-drug regimen using isoniazid, rifampin, pyrazinamide, and ethambutol for 2 months. Adjustments are made in accordance with culture and sensitivity results. Treatment can then be tapered to two drugs (isoniazid and rifampin) for a minimum of 4 additional months. Prolonged treatment may be required in instances of extrapulmonary or disseminated disease.65,72
Tuberculosis treatment can be complicated by hepatotoxicity in liver transplant recipients because of direct drug effects and drug-drug interactions with immunosuppressive agents. Close monitoring for rejection and hepatotoxicity is therefore imperative while liver transplant recipients are receiving antituberculosis therapy. Drug-drug interactions may also be responsible for marked reductions in immunosuppression levels, especially with regimens containing rifampin.71 Substitution of rifabutin for rifampin reduces the effect of drug interactions.66
VIRAL HEPATITIS
Hepatitis B virus
Hepatitis B virus-related end-stage liver disease and hepatocellular carcinoma are common indications for liver transplant in Asia. It is less common in the United States and Europe, accounting for less than 10% of all liver transplant cases. Prognosis is favorable in recipients undergoing liver transplant for hepatitis B virus, with excellent survival rates. Prevention of reinfection is crucial in these patients.
Treatment with combination antiviral agents and hepatitis B immunoglobulin (HBIG) is effective.73 Lamivudine was the first nucleoside analogue found to be effective against hepatitis B virus. Its low cost and relative safety are strong arguments in favor of its continued use in liver transplant recipients.74 In patients without evidence of hepatitis B viral replication at the time of transplant, monotherapy with lamivudine has led to low recurrence rates, and adefovir can be added to control resistant viral strains.75
The frequent emergence of resistance with lamivudine favors newer agents such as entecavir or tenofovir. These nucleoside and nucleotide analogues have a higher barrier to resistance, and thus resistance to them is rare. They are also more efficient, potentially allowing use of an HBIG-sparing protocol.76 However, they are associated with a higher risk of nephrotoxicity and require dose adjustments in renal insufficiency. Data directly comparing entecavir and tenofovir are scarce.
Prophylaxis. Most studies support an individualized approach for prevention of hepatitis B virus reinfection. High-risk patients, ie, those positive for HBe antigen or with high viral loads (> 100,000 copies/mL) are generally treated with both HBIG and antiviral agents.77 Low-risk patients are those with a negative HBe antigen, low hepatitis B virus DNA levels, hepatitis B virus-related acute liver failure, and cirrhosis resulting from coinfection with both hepatitis B and hepatitis D virus.75 In low-risk patients, discontinuation of HBIG after 1 to 2 years of treatment is appropriate, and long-term prophylaxis with antiviral agents alone is an option. However, levels of hepatitis B DNA should be monitored closely.78,79
Hepatitis C virus
Recurrence of hepatitis C virus infection is the rule among patients who are viremic at the time of liver transplant.80,81 Most of these patients will show histologic evidence of recurrent hepatitis within the first year after liver transplant. It is often difficult to distinguish between the histopathological appearance of a recurrent hepatitis C virus infection and acute cellular rejection.
Progression to fibrosis and subsequently cirrhosis and decompensation is highly variable in hepatitis C virus-infected liver transplant recipients. Diabetes, insulin resistance, and possibly hepatitis steatosis have been associated with a rapid progression to advanced fibrosis. The contribution of immunosuppression to the progression of hepatitis C virus remains an area of active study. Some studies point to antilymphocyte immunosuppressive agents as a potential cause.82 Liver biopsy is a useful tool in this situation. It allows monitoring of disease severity and progression and may distinguish recurrent hepatitis C virus disease from other causes of liver enzyme elevation.
The major concern with the recurrence of hepatitis C virus infection after liver transplant is allograft loss. Rates of patient and graft survival are reduced in infected patients compared with hepatitis C virus-negative patients.83,84 Prophylactic antiviral therapy has no current role in the management of hepatitis C virus disease. Those manifesting moderate to severe necroinflammation or mild to moderate fibrosis indicative of progressive disease should be treated.81,85
Sustained viral clearance with antiviral agents confers a graft survival benefit.
The combination of peg-interferon and weight-based ribavirin has been the standard of treatment but may be associated with increased rates of rejection.86,87 The sustained virologic response rates for hepatitis C virus range from 60% in genotypes 4, 5, and 6 after 48 weeks of treatment to 60% to 80% in genotypes 2 and 3 after 24 weeks, but only about 30% in genotype 1.88
Treatment with the newer agents, especially protease inhibitors, in genotype 1 (peg-interferon, ribavirin, and either telaprevir or boceprevir) has been evaluated. Success rates reaching 70% have been achieved.89 Adverse effects can be a major setback. Serious complications include severe anemia, renal dysfunction, increased risk of infection, and death.
Triple therapy should be carefully considered in liver transplant patients with genotype 1 hepatitis C virus.90 Significant drug-drug interactions are reported between hepatitis C virus protease inhibitors and immunosuppression regimens. Additional new oral direct- acting antivirals have been investigated. They bring promising advances in hepatitis C virus treatment and pave the way for interferon-free regimens with pangenotypic activity.
IMMUNIZATION
Immunization can decrease the risk of infectious complications in liver transplant recipients, as well as in close contacts and healthcare professionals.3
Influenza. Pretransplant influenza vaccine and posttransplant annual influenza vaccines are necessary.
Pneumococcal immunization should additionally be provided prior to transplant and repeated every 3 to 5 years thereafter.3,91
A number of other vaccinations should also be completed before transplant, including the hepatitis A and B vaccines and the tetanus/diphtheria/acellular pertussis vaccines. However, these vaccinations have not been shown to be detrimental to patients after transplant.91
Varicella and zoster vaccines should be given before liver transplant—zoster in patients over age 60, and varicella in patients with no immunity. Live vaccines, including varicella and zoster vaccines, are contraindicated after liver transplant.3
Human papillomavirus. The bivalent human papillomavirus vaccine can be given before transplant in females ages 9 to 26; the quadrivalent vaccine is beneficial in those ages 9 to 26 and in women under age 45.3,91
IMMUNOSUPPRESSION CARRIES RISK OF INFECTION
Most liver transplant patients require prolonged immunosuppressive therapy. This comes with an increased risk of new or recurrent infections, potentially causing death and significant morbidity.
Evaluation of existing risk factors, appropriate prophylaxis and immunization, timely diagnosis, and treatment of such infections are therefore essential steps for the successful management of liver transplant recipients.
- Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med 2007; 357:2601–2614.
- Avery RK, Michaels MG; AST Infectious Diseases Community of Practice. Strategies for safe living after solid organ transplantation. Am J Transplant 2013; 13(suppl 4):304–310.
- Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
- San Juan R, Aguado JM, Lumbreras C, et al; RESITRA Network, Spain. Incidence, clinical characteristics and risk factors of late infection in solid organ transplant recipients: data from the RESITRA study group. Am J Transplant 2007; 7:964–971.
- Ison MG, Grossi P; AST Infectious Diseases Community of Practice. Donor-derived infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):22–30.
- Kim YJ, Kim SI, Wie SH, et al. Infectious complications in living-donor liver transplant recipients: a 9-year single-center experience. Transpl Infect Dis 2008; 10:316–324.
- Arnow PM. Infections following orthotopic liver transplantation. HPB Surg 1991; 3:221–233.
- Reid GE, Grim SA, Sankary H, Benedetti E, Oberholzer J, Clark NM. Early intra-abdominal infections associated with orthotopic liver transplantation. Transplantation 2009; 87:1706–1711.
- Said A, Safdar N, Lucey MR, et al. Infected bilomas in liver transplant recipients, incidence, risk factors and implications for prevention. Am J Transplant 2004; 4:574–582.
- Safdar N, Said A, Lucey MR, et al. Infected bilomas in liver transplant recipients: clinical features, optimal management, and risk factors for mortality. Clin Infect Dis 2004; 39:517–525.
- Niemczyk M, Leszczyniski P, Wyzgał J, Paczek L, Krawczyk M, Luczak M. Infections caused by Clostridium difficile in kidney or liver graft recipients. Ann Transplant 2005; 10:70–74.
- Albright JB, Bonatti H, Mendez J, et al. Early and late onset Clostridium difficile-associated colitis following liver transplantation. Transpl Int 2007; 20:856–866.
- Lee SO, Razonable RR. Current concepts on cytomegalovirus infection after liver transplantation. World J Hepatol 2010; 2:325–336.
- Ljungman P, Griffiths P, Paya C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis 2002; 34:1094–1097.
- Beam E, Razonable RR. Cytomegalovirus in solid organ transplantation: epidemiology, prevention, and treatment. Curr Infect Dis Rep 2012; 14:633–641.
- Bodro M, Sabé N, Lladó L, et al. Prophylaxis versus preemptive therapy for cytomegalovirus disease in high-risk liver transplant recipients. Liver Transpl 2012; 18:1093–1099.
- Weigand K, Schnitzler P, Schmidt J, et al. Cytomegalovirus infection after liver transplantation incidence, risks, and benefits of prophylaxis. Transplant Proc 2010; 42:2634–2641.
- Razonable RR, Humar A; AST Infectious Diseases Community of Practice. Cytomegalovirus in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):93–106.
- Meije Y, Fortún J, Len Ó, et al; Spanish Network for Research on Infection in Transplantation (RESITRA) and the Spanish Network for Research on Infectious Diseases (REIPI). Prevention strategies for cytomegalovirus disease and long-term outcomes in the high-risk transplant patient (D+/R-): experience from the RESITRA-REIPI cohort. Transpl Infect Dis 2014; 16:387–396.
- Durand CM, Marr KA, Arnold CA, et al. Detection of cytomegalovirus DNA in plasma as an adjunct diagnostic for gastrointestinal tract disease in kidney and liver transplant recipients. Clin Infect Dis 2013; 57:1550–1559.
- Levitsky J, Singh N, Wagener MM, Stosor V, Abecassis M, Ison MG. A survey of CMV prevention strategies after liver transplantation. Am J Transplant 2008; 8:158–161.
- Marcelin JR, Beam E, Razonable RR. Cytomegalovirus infection in liver transplant recipients: updates on clinical management. World J Gastroenterol 2014; 20:10658–10667.
- Kalil AC, Freifeld AG, Lyden ER, Stoner JA. Valganciclovir for cytomegalovirus prevention in solid organ transplant patients: an evidence-based reassessment of safety and efficacy. PLoS One 2009; 4:e5512.
- Kalil AC, Mindru C, Botha JF, et al. Risk of cytomegalovirus disease in high-risk liver transplant recipients on valganciclovir prophylaxis: a systematic review and meta-analysis. Liver Transpl 2012; 18:1440–1447.
- Eid AJ, Arthurs SK, Deziel PJ, Wilhelm MP, Razonable RR. Emergence of drug-resistant cytomegalovirus in the era of valganciclovir prophylaxis: therapeutic implications and outcomes. Clin Transplant 2008; 22:162–170.
- Kumar D, Humar A. Cytomegalovirus prophylaxis: how long is enough? Nat Rev Nephrol 2010; 6:13–14.
- Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev 2010; 23:689–712.
- Torres-Madriz G, Boucher HW. Immunocompromised hosts: perspectives in the treatment and prophylaxis of cytomegalovirus disease in solid-organ transplant recipients. Clin Infect Dis 2008; 47:702–711.
- Burra P, Buda A, Livi U, et al. Occurrence of post-transplant lymphoproliferative disorders among over thousand adult recipients: any role for hepatitis C infection? Eur J Gastroenterol Hepatol 2006; 18:1065–1070.
- Jain A, Nalesnik M, Reyes J, et al. Posttransplant lymphoproliferative disorders in liver transplantation: a 20-year experience. Ann Surg 2002; 236:429–437.
- Allen UD, Preiksaitis JK; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):107–120.
- Allen U, Preiksaitis J; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S87–S96.
- Perrine SP, Hermine O, Small T, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 2007; 109:2571–2578.
- Jagadeesh D, Woda BA, Draper J, Evens AM. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol 2012; 13:122–136.
- Opelz G, Daniel V, Naujokat C, Fickenscher H, Döhler B. Effect of cytomegalovirus prophylaxis with immunoglobulin or with antiviral drugs on post-transplant non-Hodgkin lymphoma: a multicentre retrospective analysis. Lancet Oncol 2007; 8:212–218.
- Nowalk AJ, Green M. Epstein-Barr virus–associated posttransplant lymphoproliferative disorder: strategies for prevention and cure. Liver Transpl 2010; 16(suppl S2):S54–S59.
- Pappas PG, Silveira FP; AST Infectious Diseases Community of Practice. Candida in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S173–S179.
- Singh N, Wagener MM, Marino IR, Gayowski T. Trends in invasive fungal infections in liver transplant recipients: correlation with evolution in transplantation practices. Transplantation 2002; 73:63–67.
- Miller R, Assi M; AST Infectious Diseases Community of Practice. Endemic fungal infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):250–261.
- Fontana C, Gaziano R, Favaro M, Casalinuovo IA, Pistoia E, Di Francesco P. (1-3)-beta-D-glucan vs galactomannan antigen in diagnosing invasive fungal infections (IFIs). Open Microbiol J 2012; 6:70–73.
- Aydogan S, Kustimur S, Kalkancı A. Comparison of glucan and galactomannan tests with real-time PCR for diagnosis of invasive aspergillosis in a neutropenic rat model [Turkish]. Mikrobiyol Bul 2010; 44:441–452.
- Hadley S, Huckabee C, Pappas PG, et al. Outcomes of antifungal prophylaxis in high-risk liver transplant recipients. Transpl Infect Dis 2009; 11:40–48.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Person AK, Kontoyiannis DP, Alexander BD. Fungal infections in transplant and oncology patients. Infect Dis Clin North Am 2010; 24:439–459.
- Van Hal SJ, Marriott DJE, Chen SCA, et al; Australian Candidaemia Study. Candidemia following solid organ transplantation in the era of antifungal prophylaxis: the Australian experience. Transpl Infect Dis 2009; 11:122–127.
- Singh N. Fungal infections in the recipients of solid organ transplantation. Infect Dis Clin North Am 2003; 17:113–134,
- Liu X, Ling Z, Li L, Ruan B. Invasive fungal infections in liver transplantation. Int J Infect Dis 2011; 15:e298–e304.
- Raghuram A, Restrepo A, Safadjou S, et al. Invasive fungal infections following liver transplantation: incidence, risk factors, survival, and impact of fluconazole-resistant Candida parapsilosis (2003-2007). Liver Transpl 2012; 18:1100–1109.
- De Pauw B, Walsh TJ, Donnelly JP, et al; European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group; National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis 2008; 46:1813–1821.
- Moreno A, Cervera C, Gavaldá J, et al. Bloodstream infections among transplant recipients: results of a nationwide surveillance in Spain. Am J Transplant 2007; 7:2579–2586.
- Cruciani M, Mengoli C, Malena M, Bosco O, Serpelloni G, Grossi P. Antifungal prophylaxis in liver transplant patients: a systematic review and meta-analysis. Liver Transpl 2006; 12:850–858.
- Singh N, Husain S; AST Infectious Diseases Community of Practice. Invasive aspergillosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S180–S191.
- Fortún J, Martín-Dávila P, Alvarez ME, et al. False-positive results of Aspergillus galactomannan antigenemia in liver transplant recipients. Transplantation 2009; 87:256–260.
- Cherian T, Giakoustidis A, Yokoyama S, et al. Treatment of refractory cerebral aspergillosis in a liver transplant recipient with voriconazole: case report and review of the literature. Exp Clin Transplant 2012; 10:482–486.
- Luong ML, Hosseini-Moghaddam SM, Singer LG, et al. Risk factors for voriconazole hepatotoxicity at 12 weeks in lung transplant recipients. Am J Transplant 2012; 12:1929–1935.
- Neofytos D, Fishman JA, Horn D, et al. Epidemiology and outcome of invasive fungal infections in solid organ transplant recipients. Transpl Infect Dis 2010; 12:220–229.
- Martin SI, Fishman JA; AST Infectious Diseases Community of Practice. Pneumocystis pneumonia in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S227–S233.
- Levine SJ, Masur H, Gill VJ, et al. Effect of aerosolized pentamidine prophylaxis on the diagnosis of Pneumocystis carinii pneumonia by induced sputum examination in patients infected with the human immunodeficiency virus. Am Rev Respir Dis 1991; 144:760–764.
- Rodriguez M, Sifri CD, Fishman JA. Failure of low-dose atovaquone prophylaxis against Pneumocystis jiroveci infection in transplant recipients. Clin Infect Dis 2004; 38:e76–e78.
- Crans CA Jr, Boiselle PM. Imaging features of Pneumocystis carinii pneumonia. Crit Rev Diagn Imaging 1999; 40:251–284.
- Onishi A, Sugiyama D, Kogata Y, et al. Diagnostic accuracy of serum 1,3-beta-D-glucan for Pneumocystis jiroveci pneumonia, invasive candidiasis, and invasive aspergillosis: systematic review and meta-analysis. J Clin Microbiol 2012; 50:7–15.
- Held J, Koch MS, Reischl U, Danner T, Serr A. Serum (1→3)-ß-D-glucan measurement as an early indicator of Pneumocystis jirovecii pneumonia and evaluation of its prognostic value. Clin Microbiol Infect 2011; 17:595–602.
- Fishman JA. Prevention of infection caused by Pneumocystis carinii in transplant recipients. Clin Infect Dis 2001; 33:1397–1405.
- Colby C, McAfee S, Sackstein R, Finkelstein D, Fishman J, Spitzer T. A prospective randomized trial comparing the toxicity and safety of atovaquone with trimethoprim/sulfamethoxazole as Pneumocystis carinii pneumonia prophylaxis following autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 1999; 24:897–902.
- Subramanian A, Dorman S; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S57–S62.
- Subramanian AK, Morris MI; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):68–76.
- Horne DJ, Narita M, Spitters CL, Parimi S, Dodson S, Limaye AP. Challenging issues in tuberculosis in solid organ transplantation. Clin Infect Dis 2013; 57:1473–1482.
- Holty JE, Gould MK, Meinke L, Keeffe EB, Ruoss SJ. Tuberculosis in liver transplant recipients: a systematic review and meta-analysis of individual patient data. Liver Transpl 2009; 15:894–906.
- Jafri SM, Singal AG, Kaul D, Fontana RJ. Detection and management of latent tuberculosis in liver transplant patients. Liver Transpl 2011; 17:306–314.
- Fábrega E, Sampedro B, Cabezas J, et al. Chemoprophylaxis with isoniazid in liver transplant recipients. Liver Transpl 2012; 18:1110–1117.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis 2009; 48:1276–1284.
- Yehia BR, Blumberg EA. Mycobacterium tuberculosis infection in liver transplantation. Liver Transpl 2010; 16:1129–1135.
- Katz LH, Paul M, Guy DG, Tur-Kaspa R. Prevention of recurrent hepatitis B virus infection after liver transplantation: hepatitis B immunoglobulin, antiviral drugs, or both? Systematic review and meta-analysis. Transpl Infect Dis 2010; 12:292–308.
- Jiang L, Jiang LS, Cheng NS, Yan LN. Current prophylactic strategies against hepatitis B virus recurrence after liver transplantation. World J Gastroenterol 2009; 15:2489–2499.
- Riediger C, Berberat PO, Sauer P, et al. Prophylaxis and treatment of recurrent viral hepatitis after liver transplantation. Nephrol Dial Transplant 2007; 22(suppl 8):viii37–viii46.
- Cholongitas E, Vasiliadis T, Antoniadis N, Goulis I, Papanikolaou V, Akriviadis E. Hepatitis B prophylaxis post liver transplantation with newer nucleos(t)ide analogues after hepatitis B immunoglobulin discontinuation. Transpl Infect Dis 2012; 14:479–487.
- Fox AN, Terrault NA. Individualizing hepatitis B infection prophylaxis in liver transplant recipients. J Hepatol 2011; 55:507–509.
- Fox AN, Terrault NA. The option of HBIG-free prophylaxis against recurrent HBV. J Hepatol 2012; 56:1189–1197.
- Wesdorp DJ, Knoester M, Braat AE, et al. Nucleoside plus nucleotide analogs and cessation of hepatitis B immunoglobulin after liver transplantation in chronic hepatitis B is safe and effective. J Clin Virol 2013; 58:67–73.
- Terrault NA, Berenguer M. Treating hepatitis C infection in liver transplant recipients. Liver Transpl 2006; 12:1192–1204.
- Ciria R, Pleguezuelo M, Khorsandi SE, et al. Strategies to reduce hepatitis C virus recurrence after liver transplantation. World J Hepatol 2013; 5:237–250.
- Issa NC, Fishman JA. Infectious complications of antilymphocyte therapies in solid organ transplantation. Clin Infect Dis 2009; 48:772–786.
- Kalambokis G, Manousou P, Samonakis D, et al. Clinical outcome of HCV-related graft cirrhosis and prognostic value of hepatic venous pressure gradient. Transpl Int 2009; 22:172–181.
- Neumann UP, Berg T, Bahra M, et al. Long-term outcome of liver transplants for chronic hepatitis C: a 10-year follow-up. Transplantation 2004; 77:226–231.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first International Liver Transplantation Society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transpl 2003; 9:S1–S9.
- Dinges S, Morard I, Heim M, et al; Swiss Association for the Study of the Liver (SASL 17). Pegylated interferon-alpha2a/ribavirin treatment of recurrent hepatitis C after liver transplantation. Transpl Infect Dis 2009; 11:33–39.
- Veldt BJ, Poterucha JJ, Watt KD, et al. Impact of pegylated interferon and ribavirin treatment on graft survival in liver transplant patients with recurrent hepatitis C infection. Am J Transplant 2008; 8:2426–2433.
- Faisal N, Yoshida EM, Bilodeau M, et al. Protease inhibitor-based triple therapy is highly effective for hepatitis C recurrence after liver transplant: a multicenter experience. Ann Hepatol 2014; 13:525–532.
- Mariño Z, van Bömmel F, Forns X, Berg T. New concepts of sofosbuvir-based treatment regimens in patients with hepatitis C. Gut 2014; 63:207–215.
- Coilly A, Roche B, Dumortier J, et al. Safety and efficacy of protease inhibitors to treat hepatitis C after liver transplantation: a multicenter experience. J Hepatol 2014; 60:78–86.
- Lucey MR, Terrault N, Ojo L, et al. Long-term management of the successful adult liver transplant: 2012 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl 2013; 19:3–26.
The immunosuppressed state of liver transplant recipients makes them vulnerable to infections after surgery.1 These infections are directly correlated with the net state of immunosuppression. Higher levels of immunosuppression mean a higher risk of infection, with rates of infection typically highest in the early posttransplant period.
Common infections during this period include operative and perioperative nosocomial bacterial and fungal infections, reactivation of latent infections, and invasive fungal infections such as candidiasis, aspergillosis, and pneumocystosis. Donor-derived infections also must be considered. As time passes and the level of immunosuppression is reduced, liver recipients are less prone to infection.1
The risk of infection can be minimized by appropriate antimicrobial prophylaxis, strategies for safe living after transplant,2 vaccination,3 careful balancing of immunosuppressive therapy,4 and thoughtful donor selection.5 Drug-drug interactions are common and must be carefully considered to minimize the risk.
This review highlights common infectious complications encountered after liver transplant.
INTRA-ABDOMINAL INFECTIONS
Intra-abdominal infections are common in the early postoperative period.6,7
Risk factors include:
- Pretransplant ascites
- Posttransplant dialysis
- Wound infection
- Reoperation8
- Hepatic artery thrombosis
- Roux-en-Y choledochojejunostomy anastomosis.9
Signs that may indicate intra-abdominal infection include fever, abdominal pain, leukocytosis, and elevated liver enzymes. But because of their immunosuppressed state, transplant recipients may not manifest fever as readily as the general population. They should be evaluated for cholangitis, peritonitis, biloma, and intra-abdominal abscess.
Organisms. Intra-abdominal infections are often polymicrobial. Enterococci, Staphylococcus aureus, gram-negative species including Pseudomonas, Klebsiella, and Acinetobacter, and Candida species are the most common pathogens. Strains are often resistant to multiple drugs, especially in patients who received antibiotics in the weeks before transplant.8,10
Liver transplant recipients are also particularly susceptible to Clostridium difficile-associated colitis as a result of immunosuppression and frequent use of antibiotics perioperatively and postoperatively.11 The spectrum of C difficile infection ranges from mild diarrhea to life-threatening colitis, and the course in liver transplant patients tends to be more complicated than in immunocompetent patients.12
Diagnosis. Intra-abdominal infections should be looked for and treated promptly, as they are associated with a higher mortality rate, a greater risk of graft loss, and a higher incidence of retransplant.6,10 Abdominal ultrasonography or computed tomography (CT) can confirm the presence of fluid collections.
Treatment. Infected collections can be treated with percutaneous or surgical drainage and antimicrobial therapy. In the case of biliary tract complications, retransplant or surgical correction of biliary leakage or stenosis decreases the risk of death.6
Suspicion should be high for C difficile-associated colitis in cases of posttransplant diarrhea. C difficile toxin stool assays help confirm the diagnosis.12 Oral metronidazole is recommended in mild to moderate C difficile infection, with oral vancomycin and intravenous metronidazole reserved for severe cases. Colectomy may be necessary in patients with toxic megacolon.
CYTOMEGALOVIRUS INFECTION
Cytomegalovirus is an important opportunistic pathogen in liver transplant recipients.13 It causes a range of manifestations, from infection (viremia with or without symptoms) to cytomegalovirus syndrome (fever, malaise, and cell-line cytopenias) to tissue-invasive disease with end-organ disease.14 Without preventive measures and treatment, cytomegalovirus disease can increase the risk of morbidity, allograft loss and death.15,16
Risk factors for cytomegalovirus infection (Table 1) include:
- Discordant serostatus of the donor and recipient (the risk is highest in seronegative recipients of organs from seropositive donors)
- Higher levels of immunosuppression, especially when antilymphocyte antibodies are used
- Treatment of graft rejection
- Coinfection with other human herpesviruses, such as Epstein-Barr virus.4,17
Preventing cytomegalovirus infection
The strategy to prevent cytomegalovirus infection depends on the serologic status of the donor and recipient and may include antiviral prophylaxis or preemptive treatment (Table 2).18
Prophylaxis involves giving antiviral drugs during the early high-risk period, with the goal of preventing the development of cytomegalovirus viremia. The alternative preemptive strategy emphasizes serial testing for cytomegalovirus viremia, with the goal of intervening with antiviral medications while viremia is at a low level, thus avoiding potential progression to cytomegalovirus disease. Both strategies have pros and cons that should be considered by each transplant center when setting institutional policy.
A prophylactic approach seems very effective at preventing both infection and disease from cytomegalovirus and has been shown to reduce graft rejection and the risk of death.18 It is preferred in cytomegalovirus-negative recipients when the donor was cytomegalovirus-positive—a high-risk situation.19 However, these patients are also at higher risk of late-onset cytomegalovirus disease. Higher cost and potential drug toxicity, mainly neutropenia from ganciclovir-based regimens, are additional considerations.
Preemptive treatment, in contrast, reserves drug treatment for patients who are actually infected with cytomegalovirus, thus resulting in fewer adverse drug events and lower cost; but it requires regular monitoring. Preemptive methods, by definition, cannot prevent infection, and with this strategy tissue-invasive disease not associated with viremia does occasionally occur.20 As such, patients with a clinical presentation that suggests cytomegalovirus but have negative results on blood testing should be considered for tissue biopsy with culture and immunohistochemical stain.
The most commonly used regimens for antiviral prophylaxis and treatment in liver transplant recipients are intravenous ganciclovir and oral valganciclovir.21 Although valganciclovir is the most commonly used agent in this setting because of ease of administration, it has not been approved by the US Food and Drug Administration in liver transplant patients, as it was associated with higher rates of cytomegalovirus tissue-invasive disease.22–24 Additionally, drug-resistant cytomegalovirus strains have been associated with valganciclovir prophylaxis in cytomegalovirus-negative recipients of solid organs from cytomegalovirus-positive donors.25
Prophylaxis typically consists of therapy for 3 months from the time of transplant. In higher-risk patients (donor-positive, recipient-negative), longer courses of prophylaxis have been extrapolated from data in kidney transplant recipients.26 Extension or reinstitution of prophylaxis should also be considered in liver transplant patients receiving treatment for rejection with antilymphocyte therapy.
Routine screening for cytomegalovirus is not recommended while patients are receiving prophylaxis. High-risk patients who are not receiving prophylaxis should be monitored with nucleic acid or pp65 antigenemia testing as part of the preemptive strategy protocol.
Treatment of cytomegalovirus disease
Although no specific threshold has been established, treatment is generally indicated if a patient has a consistent clinical syndrome, evidence of tissue injury, and persistent or increasing viremia.
Treatment involves giving antiviral drugs and also reducing the level of immunosuppression, if possible, until symptoms and viremia have resolved.
The choice of antiviral therapy depends on the severity of disease. Intravenous ganciclovir (5 mg/kg twice daily adjusted for renal impairment) or oral valganciclovir (900 mg twice daily, also renally dose-adjusted when necessary) can be used for mild to moderate disease if no significant gastrointestinal involvement is reported. Intravenous ganciclovir is preferred for patients with more severe disease or gastrointestinal involvement. The minimum duration of treatment is 2 weeks and may need to be prolonged until both symptoms and viremia completely resolve.18
Drug resistance can occur and should be considered in patients who have a history of prolonged ganciclovir or valganciclovir exposure who do not clinically improve or have persistent or rising viremia. In such cases, genotype assays are helpful, and initiation of alternative therapy should be considered. Mutations conferring resistance to ganciclovir are often associated with cross-resistance to cidofovir. Cidofovir can therefore be considered only when genotype assays demonstrate specific mutations conferring an isolated resistance to ganciclovir.27 The addition of foscarnet to the ganciclovir regimen or substitution of foscarnet for ganciclovir are accepted approaches.
Although cytomegalovirus hyperimmunoglobulin has been used in prophylaxis and invasive disease treatment, its role in the management of ganciclovir-resistant cytomegalovirus infections remains controversial.28
EPSTEIN-BARR VIRUS POSTTRANSPLANT LYMPHOPROLIFERATIVE DISEASE
Epstein-Barr virus-associated posttransplant lymphoproliferative disease is a spectrum of disorders ranging from an infectious mononucleosis syndrome to aggressive malignancy with the potential for death and significant morbidity after liver transplant.29 The timeline of risk varies, but the disease is most common in the first year after transplant.
Risk factors for this disease (Table 1) are:
- Primary Epstein-Barr virus infection
- Cytomegalovirus donor-recipient mismatch
- Cytomegalovirus disease
- Higher levels of immunosuppression, especially with antilymphocyte antibodies.30
The likelihood of Epstein-Barr virus playing a contributing role is lower in later-onset posttransplant lymphoproliferative disease. Patients who are older at the time of transplant, who receive highly immunogenic allografts including a liver as a component of a multivisceral transplant, and who receive increased immunosuppression to treat rejection are at even greater risk of late posttransplant lymphoproliferative disease.31 This is in contrast to early posttransplant lymphoproliferative disease, which is seen more commonly in children as a result of primary Epstein-Barr virus infection.
Recognition and diagnosis. Heightened suspicion is required when considering posttransplant lymphoproliferative disease, and careful evaluation of consistent symptoms and allograft dysfunction are required.
Clinically, posttransplant lymphoproliferative disease should be suspected if a liver transplant recipient develops unexplained fever, weight loss, lymphadenopathy, or cell-line cytopenias.30,32 Other signs and symptoms may be related to the organ involved and may include evidence of hepatitis, pneumonitis, and gastrointestinal disease.31
Adjunctive diagnostic testing includes donor and recipient serology to characterize overall risk before transplantation and quantification of Epstein-Barr viral load, but confirmation relies on tissue histopathology.
Treatment focuses on reducing immunosuppression.30,32 Adding antiviral agents does not seem to improve outcome in all cases.33 Depending on clinical response and histologic classification, additional therapies such as anti-CD20 humanized chimeric monoclonal antibodies, surgery, radiation, and conventional chemotherapy may be required.34
Preventive approaches remain controversial. Chemoprophylaxis with an antiviral such as ganciclovir is occasionally used but has not been shown to consistently decrease rates of posttransplant lymphoproliferative disease. These agents may act in an indirect manner, leading to decreased rates of cytomegalovirus infection, a major cofactor for posttransplant lymphoproliferative disease.24
Passive immunoprophylaxis with immunoglobulin targeting cytomegalovirus has shown to decrease rates of non-Hodgkin lymphoma from posttransplant lymphoproliferative disease in renal transplant recipients in the first year after transplant,35 but data are lacking regarding its use in liver transplant recipients. Monitoring of the viral load and subsequent reduction of immunosuppression remain the most efficient measures to date.36
FUNGAL INFECTIONS
Candida species account for more than half of fungal infections in liver transplant recipients.37 However, a change has been noted in the past 20 years, with a decrease in Candida infections accompanied by an increase in Aspergillus infections.38 Endemic mycoses such as coccidioidomycosis, blastomycosis, and histoplasmosis should be considered with the appropriate epidemiologic history or if disease develops early after transplant and the donor came from a highly endemic region.39Cryptococcus may also be encountered.
Diagnosis. One of the most challenging aspects of fungal infection in liver transplant recipients is timely diagnosis. Heightened suspicion and early biopsy for pathological and microbiological confirmation are necessary. Although available noninvasive diagnostic tools often lack specificity, early detection of fungal markers may be of great use in guiding further diagnostic workup or empiric treatment in the critically ill.
Noninvasive tests include galactomannan, cryptococcal antigen, histoplasma antigen, (1-3)-beta-D-glucan assay and various antibody tests. Galactomannan testing has been widely used to aid in the diagnosis of invasive aspergillosis. Similarly, the (1-3)-beta-D-glucan assay is a non–culture-based tool for diagnosing and monitoring the treatment of invasive fungal infections. However, a definite diagnosis cannot be made on the basis of a positive test alone.40 The complementary diagnostic characteristics of combining noninvasive assays have yet to be fully elucidated.41 Cultures and tissue histopathology are also used when possible.
Treatment is based on targeted specific antifungal drug therapy and reduction of immunosuppressive therapy, when possible. The choice of antifungal agent varies with the pathogen, the site of involvement, and the severity of the disease. A focus on potential drug interactions, their management, and therapeutic drug monitoring when using antifungal medications is essential in the posttransplant period. Combination therapy can be considered in some situations to enhance synergy. The following sections discuss in greater detail Candida species, Aspergillus species, and Pneumocystis jirovecii infections.
Candida infections
Candidiasis after liver transplant is typically nosocomial, especially when diagnosed during the first 3 months (Table 3).37
Risk factors for invasive candidiasis include perioperative colonization, prolonged operative time, retransplant, greater transfusion requirements, and postoperative renal failure.37,42,43 Invasive candidiasis is of concern for its effects on morbidity, mortality, and cost of care.43–46
Organisms. The frequency of implicated species, in particular those with a natural resistance to fluconazole, differs in various reports.37,45,46Candida albicans remains the most commonly isolated pathogen; however, non-albicans species including those resistant to fluconazole have been reported more frequently and include Candida glabrata and Candida krusei.47,48
Signs and diagnosis. Invasive candidiasis in liver transplant recipients generally manifests itself in catheter-related blood stream infections, urinary tract infections, or intra-abdominal infections. Diagnosis can be made by isolating Candida from blood cultures, recovering organisms in culture of a normally sterile site, or finding direct microscopic evidence of the fungus on tissue specimens.49
Disseminated candidiasis refers to the involvement of distant anatomic sites. Clinical manifestations may cause vision changes, abdominal pain or skin nodules with findings of candidemia, hepatosplenic abscesses, or retinal exudates on funduscopy.49
Treatment of invasive candidiasis in liver recipients often involves antifungal therapy and reduction of immunosuppression. Broad-spectrum antifungals are initially advocated in an empirical approach to cover fluconazole-resistant strains of the non-albicans subgroups.50 Depending on antifungal susceptibility, treatment can later be adjusted.
Fluconazole remains the agent of choice in most C albicans infections.47 However, attention should be paid to the possibility of resistance in patients who have received fluconazole prophylaxis within the past 30 days. Additional agents used in treatment may include echinocandins, amphotericin, and additional azoles.
Antifungal prophylaxis is recommended in high-risk liver transplant patients, although its optimal duration remains undetermined.44 Antifungal prophylaxis has been associated with decreased incidence of both superficial and invasive candidiasis.51
Aspergillus infection
Aspergillus, the second most common fungal pathogen, has become a more common concern in liver transplant recipients. Aspergillus fumigatus is the most frequently encountered species.38,52
Risk factors. These infections typically occur in the first year, during intense immunosuppression. Retransplant, renal failure, and fulminant hepatic failure are major risk factors.52 In the presence of risk factors and a suggestive clinical setting, invasive aspergillosis should be considered and the diagnosis pursued.
Diagnosis is suggested by positive findings on CT accompanied by lower respiratory tract symptoms, focal lesions on neuroimaging, or demonstration of the fungus on cultures.49 However, Aspergillus is rarely grown in blood culture. The galactomannan antigen is a noninvasive test that can provide supporting evidence for the diagnosis.41,52 False-positive results do occur in the setting of certain antibiotics and cross-reacting fungi.53
Treatment consists of antifungal therapy and immunosuppression reduction.52
Voriconazole is the first-line agent for invasive aspergillosis. Monitoring for potential drug-drug interactions and side effects is required.54,55 Amphotericin B is considered a second-line choice due to toxicity and lack of an oral formulation. In refractory cases, combined antifungal therapy could be considered.52 The duration of treatment is generally a minimum of 12 weeks.
Prophylaxis. Specific prophylaxis against invasive aspergillosis is not currently recommended; however, some authors suggest a prophylactic approach using echinocandins or liposomal amphotericin B in high-risk patients.51,52 Aspergillosis is associated with a considerable increase in mortality in liver transplant recipients, which highlights the importance of timely management.52,56
Pneumocystis jirovecii
P jirovecii remains a common opportunistic pathogen in people with impaired immunity, including transplant and human immunodeficiency virus patients.
Prophylaxis. Widespread adoption of antimicrobial prophylaxis by transplant centers has decreased the rates of P jirovecii infection in liver transplant recipients.57,58 Commonly used prophylactic regimens after liver transplantation include a single-strength trimethoprim-sulfamethoxazole tablet daily or a double-strength tablet three times per week for a minimum of 6 to 12 months after transplant. Atovaquone and dapsone can be used as alternatives in cases of intolerance to trimethoprim-sulfamethoxazole (Table 2).
Inhaled pentamidine is clearly inferior and should be used only when the other medications are contraindicated.59
Signs and diagnosis. P jirovecii pneumonia is characterized by fever, cough, dyspnea, and chest pain. Insidious hypoxemia, abnormal chest examination, and bilateral interstitial pneumonia on chest radiography are common.
CT may be more sensitive than chest radiography.57 Findings suggestive of P jirovecii pneumonia on chest CT are extensive bilateral and symmetrical ground-glass attenuations. Other less-characteristic findings include upper lobar parenchymal opacities and spontaneous pneumothorax.57,60
The serum (1,3)-beta-D-glucan assay derived from major cell-wall components of P jirovecii might be helpful. Studies report a sensitivity for P jirovecii pneumonia as high as 96% and a negative predictive value of 99.8%.61,62
Definitive diagnosis requires identification of the pathogen. Routine expectorated sputum sampling is generally associated with a poor diagnostic yield. Bronchoscopy and bronchoalveolar lavage with silver or fluorescent antibody staining of samples, polymerase chain reaction testing, or both significantly improves diagnosis. Transbronchial or open lung biopsy are often unnecessary.57
Treatment. Trimethoprim-sulfamethoxazole is the first-line agent for treating P jirovecii pneumonia.57 The minimum duration of treatment is 14 days, with extended courses for severe infection.
Intravenous pentamidine or clindamycin plus primaquine are alternatives for patients who cannot tolerate trimethoprim-sulfamethoxazole. The major concern with intravenous pentamidine is renal dysfunction. Hypoglycemia or hyperglycemia, neutropenia, thrombocytopenia, nausea, dysgeusia, and pancreatitis may also occur.63
Atovaquone might also be beneficial in mild to moderate P jirovecii pneumonia. The main side effects include skin rashes, gastrointestinal intolerance, and elevation of transaminases.64
A corticosteroid (40–60 mg of prednisone or its equivalent) may be beneficial in conjunction with antimicrobial therapy in patients with significant hypoxia (partial pressure of arterial oxygen < 70 mm Hg on room air) in decreasing the risk of respiratory failure and need for intubation.
With appropriate and timely antimicrobial prophylaxis, cases of P jirovecii pneumonia should continue to decrease.
TUBERCULOSIS
Development of tuberculosis after transplantation is a catastrophic complication, with mortality rates of up to 30%.65 Most cases of posttransplant tuberculosis represent reactivation of latent disease.66 Screening with tuberculin skin tests or interferon-gamma-release assays is recommended in all liver transplant candidates. Chest radiography before transplant is necessary when assessing a positive screening test.67
The optimal management of latent tuberculosis in these cases remains controversial. Patients at high risk or those with positive screening results on chest radiography warrant treatment for latent tuberculosis infection with isoniazid unless contraindicated.67,68
The ideal time to initiate prophylactic isoniazid therapy is unclear. Some authors suggest delaying it, as it might be associated with poor tolerance and hepatotoxicity.69 Others have found that early isoniazid use was not associated with negative outcomes.70
Risk factors for symptomatic tuberculosis after liver transplant include previous infection with tuberculosis, intensified immunosuppression (especially anti-T-lymphocyte therapies), diabetes mellitus, and other co-infections (Table 1).71
The increased incidence of atypical presentations in recent years makes the diagnosis of active tuberculosis among liver transplant recipients challenging. Sputum smears can be negative due to low mycobacterial burdens, and tuberculin skin testing and interferon-gamma-release assays may be falsely negative due to immunosuppression.67
Treatment of active tuberculosis consists initially of a four-drug regimen using isoniazid, rifampin, pyrazinamide, and ethambutol for 2 months. Adjustments are made in accordance with culture and sensitivity results. Treatment can then be tapered to two drugs (isoniazid and rifampin) for a minimum of 4 additional months. Prolonged treatment may be required in instances of extrapulmonary or disseminated disease.65,72
Tuberculosis treatment can be complicated by hepatotoxicity in liver transplant recipients because of direct drug effects and drug-drug interactions with immunosuppressive agents. Close monitoring for rejection and hepatotoxicity is therefore imperative while liver transplant recipients are receiving antituberculosis therapy. Drug-drug interactions may also be responsible for marked reductions in immunosuppression levels, especially with regimens containing rifampin.71 Substitution of rifabutin for rifampin reduces the effect of drug interactions.66
VIRAL HEPATITIS
Hepatitis B virus
Hepatitis B virus-related end-stage liver disease and hepatocellular carcinoma are common indications for liver transplant in Asia. It is less common in the United States and Europe, accounting for less than 10% of all liver transplant cases. Prognosis is favorable in recipients undergoing liver transplant for hepatitis B virus, with excellent survival rates. Prevention of reinfection is crucial in these patients.
Treatment with combination antiviral agents and hepatitis B immunoglobulin (HBIG) is effective.73 Lamivudine was the first nucleoside analogue found to be effective against hepatitis B virus. Its low cost and relative safety are strong arguments in favor of its continued use in liver transplant recipients.74 In patients without evidence of hepatitis B viral replication at the time of transplant, monotherapy with lamivudine has led to low recurrence rates, and adefovir can be added to control resistant viral strains.75
The frequent emergence of resistance with lamivudine favors newer agents such as entecavir or tenofovir. These nucleoside and nucleotide analogues have a higher barrier to resistance, and thus resistance to them is rare. They are also more efficient, potentially allowing use of an HBIG-sparing protocol.76 However, they are associated with a higher risk of nephrotoxicity and require dose adjustments in renal insufficiency. Data directly comparing entecavir and tenofovir are scarce.
Prophylaxis. Most studies support an individualized approach for prevention of hepatitis B virus reinfection. High-risk patients, ie, those positive for HBe antigen or with high viral loads (> 100,000 copies/mL) are generally treated with both HBIG and antiviral agents.77 Low-risk patients are those with a negative HBe antigen, low hepatitis B virus DNA levels, hepatitis B virus-related acute liver failure, and cirrhosis resulting from coinfection with both hepatitis B and hepatitis D virus.75 In low-risk patients, discontinuation of HBIG after 1 to 2 years of treatment is appropriate, and long-term prophylaxis with antiviral agents alone is an option. However, levels of hepatitis B DNA should be monitored closely.78,79
Hepatitis C virus
Recurrence of hepatitis C virus infection is the rule among patients who are viremic at the time of liver transplant.80,81 Most of these patients will show histologic evidence of recurrent hepatitis within the first year after liver transplant. It is often difficult to distinguish between the histopathological appearance of a recurrent hepatitis C virus infection and acute cellular rejection.
Progression to fibrosis and subsequently cirrhosis and decompensation is highly variable in hepatitis C virus-infected liver transplant recipients. Diabetes, insulin resistance, and possibly hepatitis steatosis have been associated with a rapid progression to advanced fibrosis. The contribution of immunosuppression to the progression of hepatitis C virus remains an area of active study. Some studies point to antilymphocyte immunosuppressive agents as a potential cause.82 Liver biopsy is a useful tool in this situation. It allows monitoring of disease severity and progression and may distinguish recurrent hepatitis C virus disease from other causes of liver enzyme elevation.
The major concern with the recurrence of hepatitis C virus infection after liver transplant is allograft loss. Rates of patient and graft survival are reduced in infected patients compared with hepatitis C virus-negative patients.83,84 Prophylactic antiviral therapy has no current role in the management of hepatitis C virus disease. Those manifesting moderate to severe necroinflammation or mild to moderate fibrosis indicative of progressive disease should be treated.81,85
Sustained viral clearance with antiviral agents confers a graft survival benefit.
The combination of peg-interferon and weight-based ribavirin has been the standard of treatment but may be associated with increased rates of rejection.86,87 The sustained virologic response rates for hepatitis C virus range from 60% in genotypes 4, 5, and 6 after 48 weeks of treatment to 60% to 80% in genotypes 2 and 3 after 24 weeks, but only about 30% in genotype 1.88
Treatment with the newer agents, especially protease inhibitors, in genotype 1 (peg-interferon, ribavirin, and either telaprevir or boceprevir) has been evaluated. Success rates reaching 70% have been achieved.89 Adverse effects can be a major setback. Serious complications include severe anemia, renal dysfunction, increased risk of infection, and death.
Triple therapy should be carefully considered in liver transplant patients with genotype 1 hepatitis C virus.90 Significant drug-drug interactions are reported between hepatitis C virus protease inhibitors and immunosuppression regimens. Additional new oral direct- acting antivirals have been investigated. They bring promising advances in hepatitis C virus treatment and pave the way for interferon-free regimens with pangenotypic activity.
IMMUNIZATION
Immunization can decrease the risk of infectious complications in liver transplant recipients, as well as in close contacts and healthcare professionals.3
Influenza. Pretransplant influenza vaccine and posttransplant annual influenza vaccines are necessary.
Pneumococcal immunization should additionally be provided prior to transplant and repeated every 3 to 5 years thereafter.3,91
A number of other vaccinations should also be completed before transplant, including the hepatitis A and B vaccines and the tetanus/diphtheria/acellular pertussis vaccines. However, these vaccinations have not been shown to be detrimental to patients after transplant.91
Varicella and zoster vaccines should be given before liver transplant—zoster in patients over age 60, and varicella in patients with no immunity. Live vaccines, including varicella and zoster vaccines, are contraindicated after liver transplant.3
Human papillomavirus. The bivalent human papillomavirus vaccine can be given before transplant in females ages 9 to 26; the quadrivalent vaccine is beneficial in those ages 9 to 26 and in women under age 45.3,91
IMMUNOSUPPRESSION CARRIES RISK OF INFECTION
Most liver transplant patients require prolonged immunosuppressive therapy. This comes with an increased risk of new or recurrent infections, potentially causing death and significant morbidity.
Evaluation of existing risk factors, appropriate prophylaxis and immunization, timely diagnosis, and treatment of such infections are therefore essential steps for the successful management of liver transplant recipients.
The immunosuppressed state of liver transplant recipients makes them vulnerable to infections after surgery.1 These infections are directly correlated with the net state of immunosuppression. Higher levels of immunosuppression mean a higher risk of infection, with rates of infection typically highest in the early posttransplant period.
Common infections during this period include operative and perioperative nosocomial bacterial and fungal infections, reactivation of latent infections, and invasive fungal infections such as candidiasis, aspergillosis, and pneumocystosis. Donor-derived infections also must be considered. As time passes and the level of immunosuppression is reduced, liver recipients are less prone to infection.1
The risk of infection can be minimized by appropriate antimicrobial prophylaxis, strategies for safe living after transplant,2 vaccination,3 careful balancing of immunosuppressive therapy,4 and thoughtful donor selection.5 Drug-drug interactions are common and must be carefully considered to minimize the risk.
This review highlights common infectious complications encountered after liver transplant.
INTRA-ABDOMINAL INFECTIONS
Intra-abdominal infections are common in the early postoperative period.6,7
Risk factors include:
- Pretransplant ascites
- Posttransplant dialysis
- Wound infection
- Reoperation8
- Hepatic artery thrombosis
- Roux-en-Y choledochojejunostomy anastomosis.9
Signs that may indicate intra-abdominal infection include fever, abdominal pain, leukocytosis, and elevated liver enzymes. But because of their immunosuppressed state, transplant recipients may not manifest fever as readily as the general population. They should be evaluated for cholangitis, peritonitis, biloma, and intra-abdominal abscess.
Organisms. Intra-abdominal infections are often polymicrobial. Enterococci, Staphylococcus aureus, gram-negative species including Pseudomonas, Klebsiella, and Acinetobacter, and Candida species are the most common pathogens. Strains are often resistant to multiple drugs, especially in patients who received antibiotics in the weeks before transplant.8,10
Liver transplant recipients are also particularly susceptible to Clostridium difficile-associated colitis as a result of immunosuppression and frequent use of antibiotics perioperatively and postoperatively.11 The spectrum of C difficile infection ranges from mild diarrhea to life-threatening colitis, and the course in liver transplant patients tends to be more complicated than in immunocompetent patients.12
Diagnosis. Intra-abdominal infections should be looked for and treated promptly, as they are associated with a higher mortality rate, a greater risk of graft loss, and a higher incidence of retransplant.6,10 Abdominal ultrasonography or computed tomography (CT) can confirm the presence of fluid collections.
Treatment. Infected collections can be treated with percutaneous or surgical drainage and antimicrobial therapy. In the case of biliary tract complications, retransplant or surgical correction of biliary leakage or stenosis decreases the risk of death.6
Suspicion should be high for C difficile-associated colitis in cases of posttransplant diarrhea. C difficile toxin stool assays help confirm the diagnosis.12 Oral metronidazole is recommended in mild to moderate C difficile infection, with oral vancomycin and intravenous metronidazole reserved for severe cases. Colectomy may be necessary in patients with toxic megacolon.
CYTOMEGALOVIRUS INFECTION
Cytomegalovirus is an important opportunistic pathogen in liver transplant recipients.13 It causes a range of manifestations, from infection (viremia with or without symptoms) to cytomegalovirus syndrome (fever, malaise, and cell-line cytopenias) to tissue-invasive disease with end-organ disease.14 Without preventive measures and treatment, cytomegalovirus disease can increase the risk of morbidity, allograft loss and death.15,16
Risk factors for cytomegalovirus infection (Table 1) include:
- Discordant serostatus of the donor and recipient (the risk is highest in seronegative recipients of organs from seropositive donors)
- Higher levels of immunosuppression, especially when antilymphocyte antibodies are used
- Treatment of graft rejection
- Coinfection with other human herpesviruses, such as Epstein-Barr virus.4,17
Preventing cytomegalovirus infection
The strategy to prevent cytomegalovirus infection depends on the serologic status of the donor and recipient and may include antiviral prophylaxis or preemptive treatment (Table 2).18
Prophylaxis involves giving antiviral drugs during the early high-risk period, with the goal of preventing the development of cytomegalovirus viremia. The alternative preemptive strategy emphasizes serial testing for cytomegalovirus viremia, with the goal of intervening with antiviral medications while viremia is at a low level, thus avoiding potential progression to cytomegalovirus disease. Both strategies have pros and cons that should be considered by each transplant center when setting institutional policy.
A prophylactic approach seems very effective at preventing both infection and disease from cytomegalovirus and has been shown to reduce graft rejection and the risk of death.18 It is preferred in cytomegalovirus-negative recipients when the donor was cytomegalovirus-positive—a high-risk situation.19 However, these patients are also at higher risk of late-onset cytomegalovirus disease. Higher cost and potential drug toxicity, mainly neutropenia from ganciclovir-based regimens, are additional considerations.
Preemptive treatment, in contrast, reserves drug treatment for patients who are actually infected with cytomegalovirus, thus resulting in fewer adverse drug events and lower cost; but it requires regular monitoring. Preemptive methods, by definition, cannot prevent infection, and with this strategy tissue-invasive disease not associated with viremia does occasionally occur.20 As such, patients with a clinical presentation that suggests cytomegalovirus but have negative results on blood testing should be considered for tissue biopsy with culture and immunohistochemical stain.
The most commonly used regimens for antiviral prophylaxis and treatment in liver transplant recipients are intravenous ganciclovir and oral valganciclovir.21 Although valganciclovir is the most commonly used agent in this setting because of ease of administration, it has not been approved by the US Food and Drug Administration in liver transplant patients, as it was associated with higher rates of cytomegalovirus tissue-invasive disease.22–24 Additionally, drug-resistant cytomegalovirus strains have been associated with valganciclovir prophylaxis in cytomegalovirus-negative recipients of solid organs from cytomegalovirus-positive donors.25
Prophylaxis typically consists of therapy for 3 months from the time of transplant. In higher-risk patients (donor-positive, recipient-negative), longer courses of prophylaxis have been extrapolated from data in kidney transplant recipients.26 Extension or reinstitution of prophylaxis should also be considered in liver transplant patients receiving treatment for rejection with antilymphocyte therapy.
Routine screening for cytomegalovirus is not recommended while patients are receiving prophylaxis. High-risk patients who are not receiving prophylaxis should be monitored with nucleic acid or pp65 antigenemia testing as part of the preemptive strategy protocol.
Treatment of cytomegalovirus disease
Although no specific threshold has been established, treatment is generally indicated if a patient has a consistent clinical syndrome, evidence of tissue injury, and persistent or increasing viremia.
Treatment involves giving antiviral drugs and also reducing the level of immunosuppression, if possible, until symptoms and viremia have resolved.
The choice of antiviral therapy depends on the severity of disease. Intravenous ganciclovir (5 mg/kg twice daily adjusted for renal impairment) or oral valganciclovir (900 mg twice daily, also renally dose-adjusted when necessary) can be used for mild to moderate disease if no significant gastrointestinal involvement is reported. Intravenous ganciclovir is preferred for patients with more severe disease or gastrointestinal involvement. The minimum duration of treatment is 2 weeks and may need to be prolonged until both symptoms and viremia completely resolve.18
Drug resistance can occur and should be considered in patients who have a history of prolonged ganciclovir or valganciclovir exposure who do not clinically improve or have persistent or rising viremia. In such cases, genotype assays are helpful, and initiation of alternative therapy should be considered. Mutations conferring resistance to ganciclovir are often associated with cross-resistance to cidofovir. Cidofovir can therefore be considered only when genotype assays demonstrate specific mutations conferring an isolated resistance to ganciclovir.27 The addition of foscarnet to the ganciclovir regimen or substitution of foscarnet for ganciclovir are accepted approaches.
Although cytomegalovirus hyperimmunoglobulin has been used in prophylaxis and invasive disease treatment, its role in the management of ganciclovir-resistant cytomegalovirus infections remains controversial.28
EPSTEIN-BARR VIRUS POSTTRANSPLANT LYMPHOPROLIFERATIVE DISEASE
Epstein-Barr virus-associated posttransplant lymphoproliferative disease is a spectrum of disorders ranging from an infectious mononucleosis syndrome to aggressive malignancy with the potential for death and significant morbidity after liver transplant.29 The timeline of risk varies, but the disease is most common in the first year after transplant.
Risk factors for this disease (Table 1) are:
- Primary Epstein-Barr virus infection
- Cytomegalovirus donor-recipient mismatch
- Cytomegalovirus disease
- Higher levels of immunosuppression, especially with antilymphocyte antibodies.30
The likelihood of Epstein-Barr virus playing a contributing role is lower in later-onset posttransplant lymphoproliferative disease. Patients who are older at the time of transplant, who receive highly immunogenic allografts including a liver as a component of a multivisceral transplant, and who receive increased immunosuppression to treat rejection are at even greater risk of late posttransplant lymphoproliferative disease.31 This is in contrast to early posttransplant lymphoproliferative disease, which is seen more commonly in children as a result of primary Epstein-Barr virus infection.
Recognition and diagnosis. Heightened suspicion is required when considering posttransplant lymphoproliferative disease, and careful evaluation of consistent symptoms and allograft dysfunction are required.
Clinically, posttransplant lymphoproliferative disease should be suspected if a liver transplant recipient develops unexplained fever, weight loss, lymphadenopathy, or cell-line cytopenias.30,32 Other signs and symptoms may be related to the organ involved and may include evidence of hepatitis, pneumonitis, and gastrointestinal disease.31
Adjunctive diagnostic testing includes donor and recipient serology to characterize overall risk before transplantation and quantification of Epstein-Barr viral load, but confirmation relies on tissue histopathology.
Treatment focuses on reducing immunosuppression.30,32 Adding antiviral agents does not seem to improve outcome in all cases.33 Depending on clinical response and histologic classification, additional therapies such as anti-CD20 humanized chimeric monoclonal antibodies, surgery, radiation, and conventional chemotherapy may be required.34
Preventive approaches remain controversial. Chemoprophylaxis with an antiviral such as ganciclovir is occasionally used but has not been shown to consistently decrease rates of posttransplant lymphoproliferative disease. These agents may act in an indirect manner, leading to decreased rates of cytomegalovirus infection, a major cofactor for posttransplant lymphoproliferative disease.24
Passive immunoprophylaxis with immunoglobulin targeting cytomegalovirus has shown to decrease rates of non-Hodgkin lymphoma from posttransplant lymphoproliferative disease in renal transplant recipients in the first year after transplant,35 but data are lacking regarding its use in liver transplant recipients. Monitoring of the viral load and subsequent reduction of immunosuppression remain the most efficient measures to date.36
FUNGAL INFECTIONS
Candida species account for more than half of fungal infections in liver transplant recipients.37 However, a change has been noted in the past 20 years, with a decrease in Candida infections accompanied by an increase in Aspergillus infections.38 Endemic mycoses such as coccidioidomycosis, blastomycosis, and histoplasmosis should be considered with the appropriate epidemiologic history or if disease develops early after transplant and the donor came from a highly endemic region.39Cryptococcus may also be encountered.
Diagnosis. One of the most challenging aspects of fungal infection in liver transplant recipients is timely diagnosis. Heightened suspicion and early biopsy for pathological and microbiological confirmation are necessary. Although available noninvasive diagnostic tools often lack specificity, early detection of fungal markers may be of great use in guiding further diagnostic workup or empiric treatment in the critically ill.
Noninvasive tests include galactomannan, cryptococcal antigen, histoplasma antigen, (1-3)-beta-D-glucan assay and various antibody tests. Galactomannan testing has been widely used to aid in the diagnosis of invasive aspergillosis. Similarly, the (1-3)-beta-D-glucan assay is a non–culture-based tool for diagnosing and monitoring the treatment of invasive fungal infections. However, a definite diagnosis cannot be made on the basis of a positive test alone.40 The complementary diagnostic characteristics of combining noninvasive assays have yet to be fully elucidated.41 Cultures and tissue histopathology are also used when possible.
Treatment is based on targeted specific antifungal drug therapy and reduction of immunosuppressive therapy, when possible. The choice of antifungal agent varies with the pathogen, the site of involvement, and the severity of the disease. A focus on potential drug interactions, their management, and therapeutic drug monitoring when using antifungal medications is essential in the posttransplant period. Combination therapy can be considered in some situations to enhance synergy. The following sections discuss in greater detail Candida species, Aspergillus species, and Pneumocystis jirovecii infections.
Candida infections
Candidiasis after liver transplant is typically nosocomial, especially when diagnosed during the first 3 months (Table 3).37
Risk factors for invasive candidiasis include perioperative colonization, prolonged operative time, retransplant, greater transfusion requirements, and postoperative renal failure.37,42,43 Invasive candidiasis is of concern for its effects on morbidity, mortality, and cost of care.43–46
Organisms. The frequency of implicated species, in particular those with a natural resistance to fluconazole, differs in various reports.37,45,46Candida albicans remains the most commonly isolated pathogen; however, non-albicans species including those resistant to fluconazole have been reported more frequently and include Candida glabrata and Candida krusei.47,48
Signs and diagnosis. Invasive candidiasis in liver transplant recipients generally manifests itself in catheter-related blood stream infections, urinary tract infections, or intra-abdominal infections. Diagnosis can be made by isolating Candida from blood cultures, recovering organisms in culture of a normally sterile site, or finding direct microscopic evidence of the fungus on tissue specimens.49
Disseminated candidiasis refers to the involvement of distant anatomic sites. Clinical manifestations may cause vision changes, abdominal pain or skin nodules with findings of candidemia, hepatosplenic abscesses, or retinal exudates on funduscopy.49
Treatment of invasive candidiasis in liver recipients often involves antifungal therapy and reduction of immunosuppression. Broad-spectrum antifungals are initially advocated in an empirical approach to cover fluconazole-resistant strains of the non-albicans subgroups.50 Depending on antifungal susceptibility, treatment can later be adjusted.
Fluconazole remains the agent of choice in most C albicans infections.47 However, attention should be paid to the possibility of resistance in patients who have received fluconazole prophylaxis within the past 30 days. Additional agents used in treatment may include echinocandins, amphotericin, and additional azoles.
Antifungal prophylaxis is recommended in high-risk liver transplant patients, although its optimal duration remains undetermined.44 Antifungal prophylaxis has been associated with decreased incidence of both superficial and invasive candidiasis.51
Aspergillus infection
Aspergillus, the second most common fungal pathogen, has become a more common concern in liver transplant recipients. Aspergillus fumigatus is the most frequently encountered species.38,52
Risk factors. These infections typically occur in the first year, during intense immunosuppression. Retransplant, renal failure, and fulminant hepatic failure are major risk factors.52 In the presence of risk factors and a suggestive clinical setting, invasive aspergillosis should be considered and the diagnosis pursued.
Diagnosis is suggested by positive findings on CT accompanied by lower respiratory tract symptoms, focal lesions on neuroimaging, or demonstration of the fungus on cultures.49 However, Aspergillus is rarely grown in blood culture. The galactomannan antigen is a noninvasive test that can provide supporting evidence for the diagnosis.41,52 False-positive results do occur in the setting of certain antibiotics and cross-reacting fungi.53
Treatment consists of antifungal therapy and immunosuppression reduction.52
Voriconazole is the first-line agent for invasive aspergillosis. Monitoring for potential drug-drug interactions and side effects is required.54,55 Amphotericin B is considered a second-line choice due to toxicity and lack of an oral formulation. In refractory cases, combined antifungal therapy could be considered.52 The duration of treatment is generally a minimum of 12 weeks.
Prophylaxis. Specific prophylaxis against invasive aspergillosis is not currently recommended; however, some authors suggest a prophylactic approach using echinocandins or liposomal amphotericin B in high-risk patients.51,52 Aspergillosis is associated with a considerable increase in mortality in liver transplant recipients, which highlights the importance of timely management.52,56
Pneumocystis jirovecii
P jirovecii remains a common opportunistic pathogen in people with impaired immunity, including transplant and human immunodeficiency virus patients.
Prophylaxis. Widespread adoption of antimicrobial prophylaxis by transplant centers has decreased the rates of P jirovecii infection in liver transplant recipients.57,58 Commonly used prophylactic regimens after liver transplantation include a single-strength trimethoprim-sulfamethoxazole tablet daily or a double-strength tablet three times per week for a minimum of 6 to 12 months after transplant. Atovaquone and dapsone can be used as alternatives in cases of intolerance to trimethoprim-sulfamethoxazole (Table 2).
Inhaled pentamidine is clearly inferior and should be used only when the other medications are contraindicated.59
Signs and diagnosis. P jirovecii pneumonia is characterized by fever, cough, dyspnea, and chest pain. Insidious hypoxemia, abnormal chest examination, and bilateral interstitial pneumonia on chest radiography are common.
CT may be more sensitive than chest radiography.57 Findings suggestive of P jirovecii pneumonia on chest CT are extensive bilateral and symmetrical ground-glass attenuations. Other less-characteristic findings include upper lobar parenchymal opacities and spontaneous pneumothorax.57,60
The serum (1,3)-beta-D-glucan assay derived from major cell-wall components of P jirovecii might be helpful. Studies report a sensitivity for P jirovecii pneumonia as high as 96% and a negative predictive value of 99.8%.61,62
Definitive diagnosis requires identification of the pathogen. Routine expectorated sputum sampling is generally associated with a poor diagnostic yield. Bronchoscopy and bronchoalveolar lavage with silver or fluorescent antibody staining of samples, polymerase chain reaction testing, or both significantly improves diagnosis. Transbronchial or open lung biopsy are often unnecessary.57
Treatment. Trimethoprim-sulfamethoxazole is the first-line agent for treating P jirovecii pneumonia.57 The minimum duration of treatment is 14 days, with extended courses for severe infection.
Intravenous pentamidine or clindamycin plus primaquine are alternatives for patients who cannot tolerate trimethoprim-sulfamethoxazole. The major concern with intravenous pentamidine is renal dysfunction. Hypoglycemia or hyperglycemia, neutropenia, thrombocytopenia, nausea, dysgeusia, and pancreatitis may also occur.63
Atovaquone might also be beneficial in mild to moderate P jirovecii pneumonia. The main side effects include skin rashes, gastrointestinal intolerance, and elevation of transaminases.64
A corticosteroid (40–60 mg of prednisone or its equivalent) may be beneficial in conjunction with antimicrobial therapy in patients with significant hypoxia (partial pressure of arterial oxygen < 70 mm Hg on room air) in decreasing the risk of respiratory failure and need for intubation.
With appropriate and timely antimicrobial prophylaxis, cases of P jirovecii pneumonia should continue to decrease.
TUBERCULOSIS
Development of tuberculosis after transplantation is a catastrophic complication, with mortality rates of up to 30%.65 Most cases of posttransplant tuberculosis represent reactivation of latent disease.66 Screening with tuberculin skin tests or interferon-gamma-release assays is recommended in all liver transplant candidates. Chest radiography before transplant is necessary when assessing a positive screening test.67
The optimal management of latent tuberculosis in these cases remains controversial. Patients at high risk or those with positive screening results on chest radiography warrant treatment for latent tuberculosis infection with isoniazid unless contraindicated.67,68
The ideal time to initiate prophylactic isoniazid therapy is unclear. Some authors suggest delaying it, as it might be associated with poor tolerance and hepatotoxicity.69 Others have found that early isoniazid use was not associated with negative outcomes.70
Risk factors for symptomatic tuberculosis after liver transplant include previous infection with tuberculosis, intensified immunosuppression (especially anti-T-lymphocyte therapies), diabetes mellitus, and other co-infections (Table 1).71
The increased incidence of atypical presentations in recent years makes the diagnosis of active tuberculosis among liver transplant recipients challenging. Sputum smears can be negative due to low mycobacterial burdens, and tuberculin skin testing and interferon-gamma-release assays may be falsely negative due to immunosuppression.67
Treatment of active tuberculosis consists initially of a four-drug regimen using isoniazid, rifampin, pyrazinamide, and ethambutol for 2 months. Adjustments are made in accordance with culture and sensitivity results. Treatment can then be tapered to two drugs (isoniazid and rifampin) for a minimum of 4 additional months. Prolonged treatment may be required in instances of extrapulmonary or disseminated disease.65,72
Tuberculosis treatment can be complicated by hepatotoxicity in liver transplant recipients because of direct drug effects and drug-drug interactions with immunosuppressive agents. Close monitoring for rejection and hepatotoxicity is therefore imperative while liver transplant recipients are receiving antituberculosis therapy. Drug-drug interactions may also be responsible for marked reductions in immunosuppression levels, especially with regimens containing rifampin.71 Substitution of rifabutin for rifampin reduces the effect of drug interactions.66
VIRAL HEPATITIS
Hepatitis B virus
Hepatitis B virus-related end-stage liver disease and hepatocellular carcinoma are common indications for liver transplant in Asia. It is less common in the United States and Europe, accounting for less than 10% of all liver transplant cases. Prognosis is favorable in recipients undergoing liver transplant for hepatitis B virus, with excellent survival rates. Prevention of reinfection is crucial in these patients.
Treatment with combination antiviral agents and hepatitis B immunoglobulin (HBIG) is effective.73 Lamivudine was the first nucleoside analogue found to be effective against hepatitis B virus. Its low cost and relative safety are strong arguments in favor of its continued use in liver transplant recipients.74 In patients without evidence of hepatitis B viral replication at the time of transplant, monotherapy with lamivudine has led to low recurrence rates, and adefovir can be added to control resistant viral strains.75
The frequent emergence of resistance with lamivudine favors newer agents such as entecavir or tenofovir. These nucleoside and nucleotide analogues have a higher barrier to resistance, and thus resistance to them is rare. They are also more efficient, potentially allowing use of an HBIG-sparing protocol.76 However, they are associated with a higher risk of nephrotoxicity and require dose adjustments in renal insufficiency. Data directly comparing entecavir and tenofovir are scarce.
Prophylaxis. Most studies support an individualized approach for prevention of hepatitis B virus reinfection. High-risk patients, ie, those positive for HBe antigen or with high viral loads (> 100,000 copies/mL) are generally treated with both HBIG and antiviral agents.77 Low-risk patients are those with a negative HBe antigen, low hepatitis B virus DNA levels, hepatitis B virus-related acute liver failure, and cirrhosis resulting from coinfection with both hepatitis B and hepatitis D virus.75 In low-risk patients, discontinuation of HBIG after 1 to 2 years of treatment is appropriate, and long-term prophylaxis with antiviral agents alone is an option. However, levels of hepatitis B DNA should be monitored closely.78,79
Hepatitis C virus
Recurrence of hepatitis C virus infection is the rule among patients who are viremic at the time of liver transplant.80,81 Most of these patients will show histologic evidence of recurrent hepatitis within the first year after liver transplant. It is often difficult to distinguish between the histopathological appearance of a recurrent hepatitis C virus infection and acute cellular rejection.
Progression to fibrosis and subsequently cirrhosis and decompensation is highly variable in hepatitis C virus-infected liver transplant recipients. Diabetes, insulin resistance, and possibly hepatitis steatosis have been associated with a rapid progression to advanced fibrosis. The contribution of immunosuppression to the progression of hepatitis C virus remains an area of active study. Some studies point to antilymphocyte immunosuppressive agents as a potential cause.82 Liver biopsy is a useful tool in this situation. It allows monitoring of disease severity and progression and may distinguish recurrent hepatitis C virus disease from other causes of liver enzyme elevation.
The major concern with the recurrence of hepatitis C virus infection after liver transplant is allograft loss. Rates of patient and graft survival are reduced in infected patients compared with hepatitis C virus-negative patients.83,84 Prophylactic antiviral therapy has no current role in the management of hepatitis C virus disease. Those manifesting moderate to severe necroinflammation or mild to moderate fibrosis indicative of progressive disease should be treated.81,85
Sustained viral clearance with antiviral agents confers a graft survival benefit.
The combination of peg-interferon and weight-based ribavirin has been the standard of treatment but may be associated with increased rates of rejection.86,87 The sustained virologic response rates for hepatitis C virus range from 60% in genotypes 4, 5, and 6 after 48 weeks of treatment to 60% to 80% in genotypes 2 and 3 after 24 weeks, but only about 30% in genotype 1.88
Treatment with the newer agents, especially protease inhibitors, in genotype 1 (peg-interferon, ribavirin, and either telaprevir or boceprevir) has been evaluated. Success rates reaching 70% have been achieved.89 Adverse effects can be a major setback. Serious complications include severe anemia, renal dysfunction, increased risk of infection, and death.
Triple therapy should be carefully considered in liver transplant patients with genotype 1 hepatitis C virus.90 Significant drug-drug interactions are reported between hepatitis C virus protease inhibitors and immunosuppression regimens. Additional new oral direct- acting antivirals have been investigated. They bring promising advances in hepatitis C virus treatment and pave the way for interferon-free regimens with pangenotypic activity.
IMMUNIZATION
Immunization can decrease the risk of infectious complications in liver transplant recipients, as well as in close contacts and healthcare professionals.3
Influenza. Pretransplant influenza vaccine and posttransplant annual influenza vaccines are necessary.
Pneumococcal immunization should additionally be provided prior to transplant and repeated every 3 to 5 years thereafter.3,91
A number of other vaccinations should also be completed before transplant, including the hepatitis A and B vaccines and the tetanus/diphtheria/acellular pertussis vaccines. However, these vaccinations have not been shown to be detrimental to patients after transplant.91
Varicella and zoster vaccines should be given before liver transplant—zoster in patients over age 60, and varicella in patients with no immunity. Live vaccines, including varicella and zoster vaccines, are contraindicated after liver transplant.3
Human papillomavirus. The bivalent human papillomavirus vaccine can be given before transplant in females ages 9 to 26; the quadrivalent vaccine is beneficial in those ages 9 to 26 and in women under age 45.3,91
IMMUNOSUPPRESSION CARRIES RISK OF INFECTION
Most liver transplant patients require prolonged immunosuppressive therapy. This comes with an increased risk of new or recurrent infections, potentially causing death and significant morbidity.
Evaluation of existing risk factors, appropriate prophylaxis and immunization, timely diagnosis, and treatment of such infections are therefore essential steps for the successful management of liver transplant recipients.
- Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med 2007; 357:2601–2614.
- Avery RK, Michaels MG; AST Infectious Diseases Community of Practice. Strategies for safe living after solid organ transplantation. Am J Transplant 2013; 13(suppl 4):304–310.
- Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
- San Juan R, Aguado JM, Lumbreras C, et al; RESITRA Network, Spain. Incidence, clinical characteristics and risk factors of late infection in solid organ transplant recipients: data from the RESITRA study group. Am J Transplant 2007; 7:964–971.
- Ison MG, Grossi P; AST Infectious Diseases Community of Practice. Donor-derived infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):22–30.
- Kim YJ, Kim SI, Wie SH, et al. Infectious complications in living-donor liver transplant recipients: a 9-year single-center experience. Transpl Infect Dis 2008; 10:316–324.
- Arnow PM. Infections following orthotopic liver transplantation. HPB Surg 1991; 3:221–233.
- Reid GE, Grim SA, Sankary H, Benedetti E, Oberholzer J, Clark NM. Early intra-abdominal infections associated with orthotopic liver transplantation. Transplantation 2009; 87:1706–1711.
- Said A, Safdar N, Lucey MR, et al. Infected bilomas in liver transplant recipients, incidence, risk factors and implications for prevention. Am J Transplant 2004; 4:574–582.
- Safdar N, Said A, Lucey MR, et al. Infected bilomas in liver transplant recipients: clinical features, optimal management, and risk factors for mortality. Clin Infect Dis 2004; 39:517–525.
- Niemczyk M, Leszczyniski P, Wyzgał J, Paczek L, Krawczyk M, Luczak M. Infections caused by Clostridium difficile in kidney or liver graft recipients. Ann Transplant 2005; 10:70–74.
- Albright JB, Bonatti H, Mendez J, et al. Early and late onset Clostridium difficile-associated colitis following liver transplantation. Transpl Int 2007; 20:856–866.
- Lee SO, Razonable RR. Current concepts on cytomegalovirus infection after liver transplantation. World J Hepatol 2010; 2:325–336.
- Ljungman P, Griffiths P, Paya C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis 2002; 34:1094–1097.
- Beam E, Razonable RR. Cytomegalovirus in solid organ transplantation: epidemiology, prevention, and treatment. Curr Infect Dis Rep 2012; 14:633–641.
- Bodro M, Sabé N, Lladó L, et al. Prophylaxis versus preemptive therapy for cytomegalovirus disease in high-risk liver transplant recipients. Liver Transpl 2012; 18:1093–1099.
- Weigand K, Schnitzler P, Schmidt J, et al. Cytomegalovirus infection after liver transplantation incidence, risks, and benefits of prophylaxis. Transplant Proc 2010; 42:2634–2641.
- Razonable RR, Humar A; AST Infectious Diseases Community of Practice. Cytomegalovirus in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):93–106.
- Meije Y, Fortún J, Len Ó, et al; Spanish Network for Research on Infection in Transplantation (RESITRA) and the Spanish Network for Research on Infectious Diseases (REIPI). Prevention strategies for cytomegalovirus disease and long-term outcomes in the high-risk transplant patient (D+/R-): experience from the RESITRA-REIPI cohort. Transpl Infect Dis 2014; 16:387–396.
- Durand CM, Marr KA, Arnold CA, et al. Detection of cytomegalovirus DNA in plasma as an adjunct diagnostic for gastrointestinal tract disease in kidney and liver transplant recipients. Clin Infect Dis 2013; 57:1550–1559.
- Levitsky J, Singh N, Wagener MM, Stosor V, Abecassis M, Ison MG. A survey of CMV prevention strategies after liver transplantation. Am J Transplant 2008; 8:158–161.
- Marcelin JR, Beam E, Razonable RR. Cytomegalovirus infection in liver transplant recipients: updates on clinical management. World J Gastroenterol 2014; 20:10658–10667.
- Kalil AC, Freifeld AG, Lyden ER, Stoner JA. Valganciclovir for cytomegalovirus prevention in solid organ transplant patients: an evidence-based reassessment of safety and efficacy. PLoS One 2009; 4:e5512.
- Kalil AC, Mindru C, Botha JF, et al. Risk of cytomegalovirus disease in high-risk liver transplant recipients on valganciclovir prophylaxis: a systematic review and meta-analysis. Liver Transpl 2012; 18:1440–1447.
- Eid AJ, Arthurs SK, Deziel PJ, Wilhelm MP, Razonable RR. Emergence of drug-resistant cytomegalovirus in the era of valganciclovir prophylaxis: therapeutic implications and outcomes. Clin Transplant 2008; 22:162–170.
- Kumar D, Humar A. Cytomegalovirus prophylaxis: how long is enough? Nat Rev Nephrol 2010; 6:13–14.
- Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev 2010; 23:689–712.
- Torres-Madriz G, Boucher HW. Immunocompromised hosts: perspectives in the treatment and prophylaxis of cytomegalovirus disease in solid-organ transplant recipients. Clin Infect Dis 2008; 47:702–711.
- Burra P, Buda A, Livi U, et al. Occurrence of post-transplant lymphoproliferative disorders among over thousand adult recipients: any role for hepatitis C infection? Eur J Gastroenterol Hepatol 2006; 18:1065–1070.
- Jain A, Nalesnik M, Reyes J, et al. Posttransplant lymphoproliferative disorders in liver transplantation: a 20-year experience. Ann Surg 2002; 236:429–437.
- Allen UD, Preiksaitis JK; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):107–120.
- Allen U, Preiksaitis J; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S87–S96.
- Perrine SP, Hermine O, Small T, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 2007; 109:2571–2578.
- Jagadeesh D, Woda BA, Draper J, Evens AM. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol 2012; 13:122–136.
- Opelz G, Daniel V, Naujokat C, Fickenscher H, Döhler B. Effect of cytomegalovirus prophylaxis with immunoglobulin or with antiviral drugs on post-transplant non-Hodgkin lymphoma: a multicentre retrospective analysis. Lancet Oncol 2007; 8:212–218.
- Nowalk AJ, Green M. Epstein-Barr virus–associated posttransplant lymphoproliferative disorder: strategies for prevention and cure. Liver Transpl 2010; 16(suppl S2):S54–S59.
- Pappas PG, Silveira FP; AST Infectious Diseases Community of Practice. Candida in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S173–S179.
- Singh N, Wagener MM, Marino IR, Gayowski T. Trends in invasive fungal infections in liver transplant recipients: correlation with evolution in transplantation practices. Transplantation 2002; 73:63–67.
- Miller R, Assi M; AST Infectious Diseases Community of Practice. Endemic fungal infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):250–261.
- Fontana C, Gaziano R, Favaro M, Casalinuovo IA, Pistoia E, Di Francesco P. (1-3)-beta-D-glucan vs galactomannan antigen in diagnosing invasive fungal infections (IFIs). Open Microbiol J 2012; 6:70–73.
- Aydogan S, Kustimur S, Kalkancı A. Comparison of glucan and galactomannan tests with real-time PCR for diagnosis of invasive aspergillosis in a neutropenic rat model [Turkish]. Mikrobiyol Bul 2010; 44:441–452.
- Hadley S, Huckabee C, Pappas PG, et al. Outcomes of antifungal prophylaxis in high-risk liver transplant recipients. Transpl Infect Dis 2009; 11:40–48.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Person AK, Kontoyiannis DP, Alexander BD. Fungal infections in transplant and oncology patients. Infect Dis Clin North Am 2010; 24:439–459.
- Van Hal SJ, Marriott DJE, Chen SCA, et al; Australian Candidaemia Study. Candidemia following solid organ transplantation in the era of antifungal prophylaxis: the Australian experience. Transpl Infect Dis 2009; 11:122–127.
- Singh N. Fungal infections in the recipients of solid organ transplantation. Infect Dis Clin North Am 2003; 17:113–134,
- Liu X, Ling Z, Li L, Ruan B. Invasive fungal infections in liver transplantation. Int J Infect Dis 2011; 15:e298–e304.
- Raghuram A, Restrepo A, Safadjou S, et al. Invasive fungal infections following liver transplantation: incidence, risk factors, survival, and impact of fluconazole-resistant Candida parapsilosis (2003-2007). Liver Transpl 2012; 18:1100–1109.
- De Pauw B, Walsh TJ, Donnelly JP, et al; European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group; National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis 2008; 46:1813–1821.
- Moreno A, Cervera C, Gavaldá J, et al. Bloodstream infections among transplant recipients: results of a nationwide surveillance in Spain. Am J Transplant 2007; 7:2579–2586.
- Cruciani M, Mengoli C, Malena M, Bosco O, Serpelloni G, Grossi P. Antifungal prophylaxis in liver transplant patients: a systematic review and meta-analysis. Liver Transpl 2006; 12:850–858.
- Singh N, Husain S; AST Infectious Diseases Community of Practice. Invasive aspergillosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S180–S191.
- Fortún J, Martín-Dávila P, Alvarez ME, et al. False-positive results of Aspergillus galactomannan antigenemia in liver transplant recipients. Transplantation 2009; 87:256–260.
- Cherian T, Giakoustidis A, Yokoyama S, et al. Treatment of refractory cerebral aspergillosis in a liver transplant recipient with voriconazole: case report and review of the literature. Exp Clin Transplant 2012; 10:482–486.
- Luong ML, Hosseini-Moghaddam SM, Singer LG, et al. Risk factors for voriconazole hepatotoxicity at 12 weeks in lung transplant recipients. Am J Transplant 2012; 12:1929–1935.
- Neofytos D, Fishman JA, Horn D, et al. Epidemiology and outcome of invasive fungal infections in solid organ transplant recipients. Transpl Infect Dis 2010; 12:220–229.
- Martin SI, Fishman JA; AST Infectious Diseases Community of Practice. Pneumocystis pneumonia in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S227–S233.
- Levine SJ, Masur H, Gill VJ, et al. Effect of aerosolized pentamidine prophylaxis on the diagnosis of Pneumocystis carinii pneumonia by induced sputum examination in patients infected with the human immunodeficiency virus. Am Rev Respir Dis 1991; 144:760–764.
- Rodriguez M, Sifri CD, Fishman JA. Failure of low-dose atovaquone prophylaxis against Pneumocystis jiroveci infection in transplant recipients. Clin Infect Dis 2004; 38:e76–e78.
- Crans CA Jr, Boiselle PM. Imaging features of Pneumocystis carinii pneumonia. Crit Rev Diagn Imaging 1999; 40:251–284.
- Onishi A, Sugiyama D, Kogata Y, et al. Diagnostic accuracy of serum 1,3-beta-D-glucan for Pneumocystis jiroveci pneumonia, invasive candidiasis, and invasive aspergillosis: systematic review and meta-analysis. J Clin Microbiol 2012; 50:7–15.
- Held J, Koch MS, Reischl U, Danner T, Serr A. Serum (1→3)-ß-D-glucan measurement as an early indicator of Pneumocystis jirovecii pneumonia and evaluation of its prognostic value. Clin Microbiol Infect 2011; 17:595–602.
- Fishman JA. Prevention of infection caused by Pneumocystis carinii in transplant recipients. Clin Infect Dis 2001; 33:1397–1405.
- Colby C, McAfee S, Sackstein R, Finkelstein D, Fishman J, Spitzer T. A prospective randomized trial comparing the toxicity and safety of atovaquone with trimethoprim/sulfamethoxazole as Pneumocystis carinii pneumonia prophylaxis following autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 1999; 24:897–902.
- Subramanian A, Dorman S; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S57–S62.
- Subramanian AK, Morris MI; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):68–76.
- Horne DJ, Narita M, Spitters CL, Parimi S, Dodson S, Limaye AP. Challenging issues in tuberculosis in solid organ transplantation. Clin Infect Dis 2013; 57:1473–1482.
- Holty JE, Gould MK, Meinke L, Keeffe EB, Ruoss SJ. Tuberculosis in liver transplant recipients: a systematic review and meta-analysis of individual patient data. Liver Transpl 2009; 15:894–906.
- Jafri SM, Singal AG, Kaul D, Fontana RJ. Detection and management of latent tuberculosis in liver transplant patients. Liver Transpl 2011; 17:306–314.
- Fábrega E, Sampedro B, Cabezas J, et al. Chemoprophylaxis with isoniazid in liver transplant recipients. Liver Transpl 2012; 18:1110–1117.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis 2009; 48:1276–1284.
- Yehia BR, Blumberg EA. Mycobacterium tuberculosis infection in liver transplantation. Liver Transpl 2010; 16:1129–1135.
- Katz LH, Paul M, Guy DG, Tur-Kaspa R. Prevention of recurrent hepatitis B virus infection after liver transplantation: hepatitis B immunoglobulin, antiviral drugs, or both? Systematic review and meta-analysis. Transpl Infect Dis 2010; 12:292–308.
- Jiang L, Jiang LS, Cheng NS, Yan LN. Current prophylactic strategies against hepatitis B virus recurrence after liver transplantation. World J Gastroenterol 2009; 15:2489–2499.
- Riediger C, Berberat PO, Sauer P, et al. Prophylaxis and treatment of recurrent viral hepatitis after liver transplantation. Nephrol Dial Transplant 2007; 22(suppl 8):viii37–viii46.
- Cholongitas E, Vasiliadis T, Antoniadis N, Goulis I, Papanikolaou V, Akriviadis E. Hepatitis B prophylaxis post liver transplantation with newer nucleos(t)ide analogues after hepatitis B immunoglobulin discontinuation. Transpl Infect Dis 2012; 14:479–487.
- Fox AN, Terrault NA. Individualizing hepatitis B infection prophylaxis in liver transplant recipients. J Hepatol 2011; 55:507–509.
- Fox AN, Terrault NA. The option of HBIG-free prophylaxis against recurrent HBV. J Hepatol 2012; 56:1189–1197.
- Wesdorp DJ, Knoester M, Braat AE, et al. Nucleoside plus nucleotide analogs and cessation of hepatitis B immunoglobulin after liver transplantation in chronic hepatitis B is safe and effective. J Clin Virol 2013; 58:67–73.
- Terrault NA, Berenguer M. Treating hepatitis C infection in liver transplant recipients. Liver Transpl 2006; 12:1192–1204.
- Ciria R, Pleguezuelo M, Khorsandi SE, et al. Strategies to reduce hepatitis C virus recurrence after liver transplantation. World J Hepatol 2013; 5:237–250.
- Issa NC, Fishman JA. Infectious complications of antilymphocyte therapies in solid organ transplantation. Clin Infect Dis 2009; 48:772–786.
- Kalambokis G, Manousou P, Samonakis D, et al. Clinical outcome of HCV-related graft cirrhosis and prognostic value of hepatic venous pressure gradient. Transpl Int 2009; 22:172–181.
- Neumann UP, Berg T, Bahra M, et al. Long-term outcome of liver transplants for chronic hepatitis C: a 10-year follow-up. Transplantation 2004; 77:226–231.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first International Liver Transplantation Society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transpl 2003; 9:S1–S9.
- Dinges S, Morard I, Heim M, et al; Swiss Association for the Study of the Liver (SASL 17). Pegylated interferon-alpha2a/ribavirin treatment of recurrent hepatitis C after liver transplantation. Transpl Infect Dis 2009; 11:33–39.
- Veldt BJ, Poterucha JJ, Watt KD, et al. Impact of pegylated interferon and ribavirin treatment on graft survival in liver transplant patients with recurrent hepatitis C infection. Am J Transplant 2008; 8:2426–2433.
- Faisal N, Yoshida EM, Bilodeau M, et al. Protease inhibitor-based triple therapy is highly effective for hepatitis C recurrence after liver transplant: a multicenter experience. Ann Hepatol 2014; 13:525–532.
- Mariño Z, van Bömmel F, Forns X, Berg T. New concepts of sofosbuvir-based treatment regimens in patients with hepatitis C. Gut 2014; 63:207–215.
- Coilly A, Roche B, Dumortier J, et al. Safety and efficacy of protease inhibitors to treat hepatitis C after liver transplantation: a multicenter experience. J Hepatol 2014; 60:78–86.
- Lucey MR, Terrault N, Ojo L, et al. Long-term management of the successful adult liver transplant: 2012 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl 2013; 19:3–26.
- Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med 2007; 357:2601–2614.
- Avery RK, Michaels MG; AST Infectious Diseases Community of Practice. Strategies for safe living after solid organ transplantation. Am J Transplant 2013; 13(suppl 4):304–310.
- Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
- San Juan R, Aguado JM, Lumbreras C, et al; RESITRA Network, Spain. Incidence, clinical characteristics and risk factors of late infection in solid organ transplant recipients: data from the RESITRA study group. Am J Transplant 2007; 7:964–971.
- Ison MG, Grossi P; AST Infectious Diseases Community of Practice. Donor-derived infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):22–30.
- Kim YJ, Kim SI, Wie SH, et al. Infectious complications in living-donor liver transplant recipients: a 9-year single-center experience. Transpl Infect Dis 2008; 10:316–324.
- Arnow PM. Infections following orthotopic liver transplantation. HPB Surg 1991; 3:221–233.
- Reid GE, Grim SA, Sankary H, Benedetti E, Oberholzer J, Clark NM. Early intra-abdominal infections associated with orthotopic liver transplantation. Transplantation 2009; 87:1706–1711.
- Said A, Safdar N, Lucey MR, et al. Infected bilomas in liver transplant recipients, incidence, risk factors and implications for prevention. Am J Transplant 2004; 4:574–582.
- Safdar N, Said A, Lucey MR, et al. Infected bilomas in liver transplant recipients: clinical features, optimal management, and risk factors for mortality. Clin Infect Dis 2004; 39:517–525.
- Niemczyk M, Leszczyniski P, Wyzgał J, Paczek L, Krawczyk M, Luczak M. Infections caused by Clostridium difficile in kidney or liver graft recipients. Ann Transplant 2005; 10:70–74.
- Albright JB, Bonatti H, Mendez J, et al. Early and late onset Clostridium difficile-associated colitis following liver transplantation. Transpl Int 2007; 20:856–866.
- Lee SO, Razonable RR. Current concepts on cytomegalovirus infection after liver transplantation. World J Hepatol 2010; 2:325–336.
- Ljungman P, Griffiths P, Paya C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis 2002; 34:1094–1097.
- Beam E, Razonable RR. Cytomegalovirus in solid organ transplantation: epidemiology, prevention, and treatment. Curr Infect Dis Rep 2012; 14:633–641.
- Bodro M, Sabé N, Lladó L, et al. Prophylaxis versus preemptive therapy for cytomegalovirus disease in high-risk liver transplant recipients. Liver Transpl 2012; 18:1093–1099.
- Weigand K, Schnitzler P, Schmidt J, et al. Cytomegalovirus infection after liver transplantation incidence, risks, and benefits of prophylaxis. Transplant Proc 2010; 42:2634–2641.
- Razonable RR, Humar A; AST Infectious Diseases Community of Practice. Cytomegalovirus in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):93–106.
- Meije Y, Fortún J, Len Ó, et al; Spanish Network for Research on Infection in Transplantation (RESITRA) and the Spanish Network for Research on Infectious Diseases (REIPI). Prevention strategies for cytomegalovirus disease and long-term outcomes in the high-risk transplant patient (D+/R-): experience from the RESITRA-REIPI cohort. Transpl Infect Dis 2014; 16:387–396.
- Durand CM, Marr KA, Arnold CA, et al. Detection of cytomegalovirus DNA in plasma as an adjunct diagnostic for gastrointestinal tract disease in kidney and liver transplant recipients. Clin Infect Dis 2013; 57:1550–1559.
- Levitsky J, Singh N, Wagener MM, Stosor V, Abecassis M, Ison MG. A survey of CMV prevention strategies after liver transplantation. Am J Transplant 2008; 8:158–161.
- Marcelin JR, Beam E, Razonable RR. Cytomegalovirus infection in liver transplant recipients: updates on clinical management. World J Gastroenterol 2014; 20:10658–10667.
- Kalil AC, Freifeld AG, Lyden ER, Stoner JA. Valganciclovir for cytomegalovirus prevention in solid organ transplant patients: an evidence-based reassessment of safety and efficacy. PLoS One 2009; 4:e5512.
- Kalil AC, Mindru C, Botha JF, et al. Risk of cytomegalovirus disease in high-risk liver transplant recipients on valganciclovir prophylaxis: a systematic review and meta-analysis. Liver Transpl 2012; 18:1440–1447.
- Eid AJ, Arthurs SK, Deziel PJ, Wilhelm MP, Razonable RR. Emergence of drug-resistant cytomegalovirus in the era of valganciclovir prophylaxis: therapeutic implications and outcomes. Clin Transplant 2008; 22:162–170.
- Kumar D, Humar A. Cytomegalovirus prophylaxis: how long is enough? Nat Rev Nephrol 2010; 6:13–14.
- Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev 2010; 23:689–712.
- Torres-Madriz G, Boucher HW. Immunocompromised hosts: perspectives in the treatment and prophylaxis of cytomegalovirus disease in solid-organ transplant recipients. Clin Infect Dis 2008; 47:702–711.
- Burra P, Buda A, Livi U, et al. Occurrence of post-transplant lymphoproliferative disorders among over thousand adult recipients: any role for hepatitis C infection? Eur J Gastroenterol Hepatol 2006; 18:1065–1070.
- Jain A, Nalesnik M, Reyes J, et al. Posttransplant lymphoproliferative disorders in liver transplantation: a 20-year experience. Ann Surg 2002; 236:429–437.
- Allen UD, Preiksaitis JK; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):107–120.
- Allen U, Preiksaitis J; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S87–S96.
- Perrine SP, Hermine O, Small T, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 2007; 109:2571–2578.
- Jagadeesh D, Woda BA, Draper J, Evens AM. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol 2012; 13:122–136.
- Opelz G, Daniel V, Naujokat C, Fickenscher H, Döhler B. Effect of cytomegalovirus prophylaxis with immunoglobulin or with antiviral drugs on post-transplant non-Hodgkin lymphoma: a multicentre retrospective analysis. Lancet Oncol 2007; 8:212–218.
- Nowalk AJ, Green M. Epstein-Barr virus–associated posttransplant lymphoproliferative disorder: strategies for prevention and cure. Liver Transpl 2010; 16(suppl S2):S54–S59.
- Pappas PG, Silveira FP; AST Infectious Diseases Community of Practice. Candida in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S173–S179.
- Singh N, Wagener MM, Marino IR, Gayowski T. Trends in invasive fungal infections in liver transplant recipients: correlation with evolution in transplantation practices. Transplantation 2002; 73:63–67.
- Miller R, Assi M; AST Infectious Diseases Community of Practice. Endemic fungal infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):250–261.
- Fontana C, Gaziano R, Favaro M, Casalinuovo IA, Pistoia E, Di Francesco P. (1-3)-beta-D-glucan vs galactomannan antigen in diagnosing invasive fungal infections (IFIs). Open Microbiol J 2012; 6:70–73.
- Aydogan S, Kustimur S, Kalkancı A. Comparison of glucan and galactomannan tests with real-time PCR for diagnosis of invasive aspergillosis in a neutropenic rat model [Turkish]. Mikrobiyol Bul 2010; 44:441–452.
- Hadley S, Huckabee C, Pappas PG, et al. Outcomes of antifungal prophylaxis in high-risk liver transplant recipients. Transpl Infect Dis 2009; 11:40–48.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Person AK, Kontoyiannis DP, Alexander BD. Fungal infections in transplant and oncology patients. Infect Dis Clin North Am 2010; 24:439–459.
- Van Hal SJ, Marriott DJE, Chen SCA, et al; Australian Candidaemia Study. Candidemia following solid organ transplantation in the era of antifungal prophylaxis: the Australian experience. Transpl Infect Dis 2009; 11:122–127.
- Singh N. Fungal infections in the recipients of solid organ transplantation. Infect Dis Clin North Am 2003; 17:113–134,
- Liu X, Ling Z, Li L, Ruan B. Invasive fungal infections in liver transplantation. Int J Infect Dis 2011; 15:e298–e304.
- Raghuram A, Restrepo A, Safadjou S, et al. Invasive fungal infections following liver transplantation: incidence, risk factors, survival, and impact of fluconazole-resistant Candida parapsilosis (2003-2007). Liver Transpl 2012; 18:1100–1109.
- De Pauw B, Walsh TJ, Donnelly JP, et al; European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group; National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis 2008; 46:1813–1821.
- Moreno A, Cervera C, Gavaldá J, et al. Bloodstream infections among transplant recipients: results of a nationwide surveillance in Spain. Am J Transplant 2007; 7:2579–2586.
- Cruciani M, Mengoli C, Malena M, Bosco O, Serpelloni G, Grossi P. Antifungal prophylaxis in liver transplant patients: a systematic review and meta-analysis. Liver Transpl 2006; 12:850–858.
- Singh N, Husain S; AST Infectious Diseases Community of Practice. Invasive aspergillosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S180–S191.
- Fortún J, Martín-Dávila P, Alvarez ME, et al. False-positive results of Aspergillus galactomannan antigenemia in liver transplant recipients. Transplantation 2009; 87:256–260.
- Cherian T, Giakoustidis A, Yokoyama S, et al. Treatment of refractory cerebral aspergillosis in a liver transplant recipient with voriconazole: case report and review of the literature. Exp Clin Transplant 2012; 10:482–486.
- Luong ML, Hosseini-Moghaddam SM, Singer LG, et al. Risk factors for voriconazole hepatotoxicity at 12 weeks in lung transplant recipients. Am J Transplant 2012; 12:1929–1935.
- Neofytos D, Fishman JA, Horn D, et al. Epidemiology and outcome of invasive fungal infections in solid organ transplant recipients. Transpl Infect Dis 2010; 12:220–229.
- Martin SI, Fishman JA; AST Infectious Diseases Community of Practice. Pneumocystis pneumonia in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S227–S233.
- Levine SJ, Masur H, Gill VJ, et al. Effect of aerosolized pentamidine prophylaxis on the diagnosis of Pneumocystis carinii pneumonia by induced sputum examination in patients infected with the human immunodeficiency virus. Am Rev Respir Dis 1991; 144:760–764.
- Rodriguez M, Sifri CD, Fishman JA. Failure of low-dose atovaquone prophylaxis against Pneumocystis jiroveci infection in transplant recipients. Clin Infect Dis 2004; 38:e76–e78.
- Crans CA Jr, Boiselle PM. Imaging features of Pneumocystis carinii pneumonia. Crit Rev Diagn Imaging 1999; 40:251–284.
- Onishi A, Sugiyama D, Kogata Y, et al. Diagnostic accuracy of serum 1,3-beta-D-glucan for Pneumocystis jiroveci pneumonia, invasive candidiasis, and invasive aspergillosis: systematic review and meta-analysis. J Clin Microbiol 2012; 50:7–15.
- Held J, Koch MS, Reischl U, Danner T, Serr A. Serum (1→3)-ß-D-glucan measurement as an early indicator of Pneumocystis jirovecii pneumonia and evaluation of its prognostic value. Clin Microbiol Infect 2011; 17:595–602.
- Fishman JA. Prevention of infection caused by Pneumocystis carinii in transplant recipients. Clin Infect Dis 2001; 33:1397–1405.
- Colby C, McAfee S, Sackstein R, Finkelstein D, Fishman J, Spitzer T. A prospective randomized trial comparing the toxicity and safety of atovaquone with trimethoprim/sulfamethoxazole as Pneumocystis carinii pneumonia prophylaxis following autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 1999; 24:897–902.
- Subramanian A, Dorman S; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis in solid organ transplant recipients. Am J Transplant 2009; 9(suppl 4):S57–S62.
- Subramanian AK, Morris MI; AST Infectious Diseases Community of Practice. Mycobacterium tuberculosis infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):68–76.
- Horne DJ, Narita M, Spitters CL, Parimi S, Dodson S, Limaye AP. Challenging issues in tuberculosis in solid organ transplantation. Clin Infect Dis 2013; 57:1473–1482.
- Holty JE, Gould MK, Meinke L, Keeffe EB, Ruoss SJ. Tuberculosis in liver transplant recipients: a systematic review and meta-analysis of individual patient data. Liver Transpl 2009; 15:894–906.
- Jafri SM, Singal AG, Kaul D, Fontana RJ. Detection and management of latent tuberculosis in liver transplant patients. Liver Transpl 2011; 17:306–314.
- Fábrega E, Sampedro B, Cabezas J, et al. Chemoprophylaxis with isoniazid in liver transplant recipients. Liver Transpl 2012; 18:1110–1117.
- Aguado JM, Torre-Cisneros J, Fortún J, et al. Tuberculosis in solid-organ transplant recipients: consensus statement of the group for the study of infection in transplant recipients (GESITRA) of the Spanish Society of Infectious Diseases and Clinical Microbiology. Clin Infect Dis 2009; 48:1276–1284.
- Yehia BR, Blumberg EA. Mycobacterium tuberculosis infection in liver transplantation. Liver Transpl 2010; 16:1129–1135.
- Katz LH, Paul M, Guy DG, Tur-Kaspa R. Prevention of recurrent hepatitis B virus infection after liver transplantation: hepatitis B immunoglobulin, antiviral drugs, or both? Systematic review and meta-analysis. Transpl Infect Dis 2010; 12:292–308.
- Jiang L, Jiang LS, Cheng NS, Yan LN. Current prophylactic strategies against hepatitis B virus recurrence after liver transplantation. World J Gastroenterol 2009; 15:2489–2499.
- Riediger C, Berberat PO, Sauer P, et al. Prophylaxis and treatment of recurrent viral hepatitis after liver transplantation. Nephrol Dial Transplant 2007; 22(suppl 8):viii37–viii46.
- Cholongitas E, Vasiliadis T, Antoniadis N, Goulis I, Papanikolaou V, Akriviadis E. Hepatitis B prophylaxis post liver transplantation with newer nucleos(t)ide analogues after hepatitis B immunoglobulin discontinuation. Transpl Infect Dis 2012; 14:479–487.
- Fox AN, Terrault NA. Individualizing hepatitis B infection prophylaxis in liver transplant recipients. J Hepatol 2011; 55:507–509.
- Fox AN, Terrault NA. The option of HBIG-free prophylaxis against recurrent HBV. J Hepatol 2012; 56:1189–1197.
- Wesdorp DJ, Knoester M, Braat AE, et al. Nucleoside plus nucleotide analogs and cessation of hepatitis B immunoglobulin after liver transplantation in chronic hepatitis B is safe and effective. J Clin Virol 2013; 58:67–73.
- Terrault NA, Berenguer M. Treating hepatitis C infection in liver transplant recipients. Liver Transpl 2006; 12:1192–1204.
- Ciria R, Pleguezuelo M, Khorsandi SE, et al. Strategies to reduce hepatitis C virus recurrence after liver transplantation. World J Hepatol 2013; 5:237–250.
- Issa NC, Fishman JA. Infectious complications of antilymphocyte therapies in solid organ transplantation. Clin Infect Dis 2009; 48:772–786.
- Kalambokis G, Manousou P, Samonakis D, et al. Clinical outcome of HCV-related graft cirrhosis and prognostic value of hepatic venous pressure gradient. Transpl Int 2009; 22:172–181.
- Neumann UP, Berg T, Bahra M, et al. Long-term outcome of liver transplants for chronic hepatitis C: a 10-year follow-up. Transplantation 2004; 77:226–231.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first International Liver Transplantation Society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transpl 2003; 9:S1–S9.
- Dinges S, Morard I, Heim M, et al; Swiss Association for the Study of the Liver (SASL 17). Pegylated interferon-alpha2a/ribavirin treatment of recurrent hepatitis C after liver transplantation. Transpl Infect Dis 2009; 11:33–39.
- Veldt BJ, Poterucha JJ, Watt KD, et al. Impact of pegylated interferon and ribavirin treatment on graft survival in liver transplant patients with recurrent hepatitis C infection. Am J Transplant 2008; 8:2426–2433.
- Faisal N, Yoshida EM, Bilodeau M, et al. Protease inhibitor-based triple therapy is highly effective for hepatitis C recurrence after liver transplant: a multicenter experience. Ann Hepatol 2014; 13:525–532.
- Mariño Z, van Bömmel F, Forns X, Berg T. New concepts of sofosbuvir-based treatment regimens in patients with hepatitis C. Gut 2014; 63:207–215.
- Coilly A, Roche B, Dumortier J, et al. Safety and efficacy of protease inhibitors to treat hepatitis C after liver transplantation: a multicenter experience. J Hepatol 2014; 60:78–86.
- Lucey MR, Terrault N, Ojo L, et al. Long-term management of the successful adult liver transplant: 2012 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl 2013; 19:3–26.
KEY POINTS
- After liver transplant, the risk of infection and the likely causal organisms vary with the patient’s state of immunosuppression and the time of infection.
- Recurrent or newly acquired infections may jeopardize the survival of the graft and the recipient.
- Because infections with viruses, fungi, and atypical pathogens can alter the prognosis, they need to be prevented and carefully managed.
- An ongoing assessment of each patient’s risk of infection allows the clinician to constantly and efficiently adapt immunosuppressive, prophylactic, and therapeutic strategies.
Noncosmetic uses of botulinum toxin in otolaryngology
Botulinum toxin is commonly used to treat movement disorders of the head and neck. It was first used to treat focal eye dystonia (blepharospasm) and laryngeal dystonia (spasmodic dysphonia) and is now also used for other head and neck dystonias, movement disorders, and muscle spasticity or contraction.
This article reviews the use of botulinum toxin for primary disorders of the laryngopharynx—adductor and abductor spasmodic dysphonias, laryngopharyngeal tremor, and cricopharyngeus muscle dysfunction—and its efficacy and side effects for the different conditions.
ABNORMAL MUSCLE MOVEMENT
Dystonia is abnormal muscle movement characterized by repetitive involuntary contractions. Dystonic contractions are described as either sustained (tonic) or spasmodic (clonic) and are typically induced by conscious action to move the muscle group.1,2 Dystonia can be categorized according to the amount of muscle involvement: generalized (widespread muscle activity), segmental (involving neighboring groups of muscles), or focal (involving only one or a few local small muscles).3 Activity may be associated with gross posturing and disfigurement, depending on the size and location of the muscle contractions, although the muscle action is usually normal during rest.
The cause of dystonia has been the focus of much debate and investigation. Some types of dystonia have strong family inheritance patterns, but most are sporadic, possibly brought on by trauma or infection. In most cases, dystonia is idiopathic, although it may be associated with other muscle group dystonias, tremor, neurologic injury or insults, other neurologic diseases and neurodegenerative disorders, or tardive syndromes.1 Because of the relationship with other neurologic diseases, consultation with a neurologist should be considered.
Treatment of the muscle contractions of the various dystonias includes drug therapy and physical, occupational, and voice therapy. Botulinum toxin is a principal treatment for head and neck dystonias and works by blocking muscular contractions.4 It has the advantages of having few side effects and predictable results for many conditions, although repeat injections are usually required to achieve a sustained effect.
LARYNGEAL DYSTONIAS CAUSE VOICE ABNORMALITIES
The most common laryngeal dystonia is spasmodic dysphonia, a focal dystonia of the larynx. It is subdivided into two types according to whether spasm of the vocal folds occurs during adduction or abduction.
Adductor spasmodic dysphonia accounts for 80% to 90% of cases. It is characterized by irregular speech with pitch breaks and a strained or strangulated voice. It was formerly treated by resection of the nerve to the vocal folds, but results were neither consistent nor persistent. Currently, the primary treatment is injection of botulinum toxin, which has a high success rate,5 with patients reporting about a 90% return of normal function.
Abductor spasmodic dysphonia accounts for 10% to 20% of cases.6 Patients have a breathy quality to the voice with a short duration of vocalization due to excessive loss of air on phonation. This is especially noticeable when the patient speaks words that begin with a voiceless consonant followed by a vowel (eg, pat, puppy). Response to botulinum toxin injection is more variable,6 possibly because of the pathophysiology of the disorder or because of the technical challenges of administering the injection.
Fewer than 1% of patients have both abductor and adductor components, and their treatment can be particularly challenging.
Adductor spasmodic dysphonia: Treatment usually successful
Botulinum toxin can be injected for adductor spasmodic dysphonia via a number of approaches, the most common being through the cricothyroid membrane (Figure 1). Injections can be made into one or both vocal folds and can be performed under guidance with laryngeal electromyography or with a flexible laryngoscope to visualize the larynx.
Patients typically experience breathiness beginning 1 or 2 days after the injection, and this effect may last for up to 2 weeks. During that time, the patient may be more susceptible to aspiration of thin liquids and so is instructed to drink cautiously. Treatment benefits typically last for 3 to 6 months. As the botulinum toxin wears off, the patient notices a gradual increase in vocal straining and effort.
Dosages of botulinum toxin for subsequent treatments are adjusted by balancing the period of benefit with postinjection breathiness. The desire of the patient should be paramount. Some are willing to tolerate more side effects to avoid frequent injections, so they can be given a larger dose. Others cannot tolerate the breathiness but are willing to accept more frequent injections, so they should be given a smaller dose. In rare cases, patients have significant breathiness from even small doses; they may be helped by injecting into only one vocal fold or, alternatively, into a false vocal fold, allowing diffusion of the toxin down to the muscle of the true vocal fold.
Abductor spasmodic dysphonia: Treatment more challenging
The success of botulinum toxin treatment for abductor spasmodic dysphonia is more variable than for the adductor type. The injections are made into the posterior cricoarytenoid muscle (Figure 2); because this muscle cannot be directly visualized, this procedure requires guidance with laryngeal electromyography. Most patients note improvement, and about 20% have a good response.6 Most require a second injection about 1 month later, often on the other side. Bilateral injections at one sitting may compromise the airway, and vocal fold motion should be evaluated at the time of the contralateral injection to assess airway patency. Interest has increased in simultaneous bilateral injections with lower doses of botulinum toxin, and this approach has been shown to be safe.7
ESSENTIAL TREMOR OF THE VOICE
Essential tremor is an action tremor that can occur with voluntary movement. It can occur anywhere in the body, often the head or hand, but the voice can also be affected. About half of cases are hereditary. Essential tremor of the voice causes a rhythmic oscillation of pitch and intensity.
Consultation with a neurologist is recommended to evaluate the cause, although voice tremor is often idiopathic and occurs in about 30% of patients with essential tremor in the arms or legs, as well as in about 30% of patients with spasmodic dysphonia. Extremity tremor can usually be successfully managed medically, but this is not true for voice tremor.
Botulinum toxin injection is the mainstay of treatment for essential tremor of the voice, although its success is marginal. About two-thirds of patients have some degree of improvement from traditional botulinum toxin injections in the true vocal fold.8
The results of treatment are likely to be inconsistent because tremor tends to involve several different muscles used in voice production, commonly in the soft palate, tongue base, pharyngeal walls, strap muscles, false vocal folds, and true vocal folds. A location-oriented tremor scoring system9 can help identify the involved muscles to guide injections. Treatment is less likely to be successful in patients with multiple sites of voice tremor. Injection into the false vocal fold, true vocal fold, and interarytenoid muscle10 can safely be performed; injections into the palate, tongue base, and strap muscles are to be avoided because of the high risk of postinjection aspiration.
Patients who have good results can have repeat treatments as needed. The dosage of botulinum toxin is adjusted according to response, side effects (eg, breathy voice, dysphagia), and patient preference.
CRICOPHARYNGEUS MUSCLE DYSFUNCTION: TROUBLE SWALLOWING
Dysfunction of the cricopharyngeus muscle causes difficulty swallowing, especially swallowing solid foods. It can be attributed to a mechanical stricture or to hyperfunction (spasm).
Mechanical stricture at the esophageal inlet frequently occurs in patients who have had a total laryngectomy for advanced laryngeal cancer. Fibrosis tends to be worse in patients who have also undergone radiation therapy.
Stricture can be treated with botulinum toxin injections and dilation. Conservative treatment is preferred to surgical myotomy for patients with complex postlaryngectomy anatomy and scarring from radiation therapy.
Cricopharyngeus muscle spasm or hyperfunction can be an important cause of dysphagia, especially in the elderly. Patients should be evaluated with barium esophagography or a modified barium swallow. The finding of a cricopharyngeal “bar” provides evidence of contraction of the muscle that impedes the passage of food.
Botulinum toxin injections for cricopharyngeus muscle dysfunction (Figure 2) can be effective in some cases, especially if the toxin is injected bilaterally. However, because the cricopharyngeus muscle plays an important role in preventing esophageal reflux into the laryngopharynx, botulinum toxin injection in patients with substantial hiatal hernia or laryngopharyngeal reflux disease should only be done with caution. In addition, treatment of reflux disease should be considered in any patient undergoing botulinum toxin injection for cricopharyngeus muscle dysfunction.
Most patients require repeat injections when the toxin wears off, although occasionally one or two injections provide long-term or permanent relief. Dosages are adjusted for the patient’s age, the presence of other swallowing problems, and reflux. Patients may experience increased difficulty swallowing for 1 or 2 weeks after the procedure and so should be counseled to eat slowly and carefully.
- Cultrara A, Chitkara A, Blitzer A. Botulinum toxin injections for the treatment of oromandibular dystonia. Oper Tech Otolaryngol Head Neck Surg 2004; 15:97–102.
- Fahn S. The varied clinical expressions of dystonia. Neurol Clin 1984; 2:541–554.
- Fahn S. Concept and classification of dystonia. Adv Neurol 1988; 50:1–8.
- Benninger MS, Knott PD. Techniques of botulinum toxin injections in the head and neck. San Diego, CA: Plural Publishing, Inc; 2012.
- Benninger MS, Gardner G, Grywalski C. Outcomes of botulinum toxin treatment for spasmodic dysphonia. Arch Otolaryngol Head Neck Surg 2001; 127:1083–1085.
- Blitzer A, Brin MF, Stewart CF. Botulinum toxin management of spasmodic dysphonia (laryngeal dystonia): a 12-year experience in more than 900 patients. Laryngoscope 1998; 108:1435–1441.
- Klein AM, Stong BC, Wise J, DelGaudio JM, Hapner ER, Johns MM 3rd. Vocal outcome measures after bilateral posterior cricoarytenoid muscle botulinum toxin injections for abductor spasmodic dysphonia. Otolaryngol Head Neck Surg 2008; 139:421–423.
- Hertegård S, Granqvist S, Lindestad PA. Botulinum toxin injections for essential voice tremor. Ann Otol Rhinol Laryngol 2000; 109:204–209.
- Bové M, Daamen N, Rosen C, Wang CC, Sulica L, Gartner-Schmidt J. Development and validation of the vocal tremor scoring system. Laryngoscope 2006; 116:1662–1667.
- Kendall KA, Leonard RJ. Interarytenoid muscle Botox injection for treatment of adductor spasmodic dysphonia with vocal tremor. J Voice 2001; 25:114–119.
Botulinum toxin is commonly used to treat movement disorders of the head and neck. It was first used to treat focal eye dystonia (blepharospasm) and laryngeal dystonia (spasmodic dysphonia) and is now also used for other head and neck dystonias, movement disorders, and muscle spasticity or contraction.
This article reviews the use of botulinum toxin for primary disorders of the laryngopharynx—adductor and abductor spasmodic dysphonias, laryngopharyngeal tremor, and cricopharyngeus muscle dysfunction—and its efficacy and side effects for the different conditions.
ABNORMAL MUSCLE MOVEMENT
Dystonia is abnormal muscle movement characterized by repetitive involuntary contractions. Dystonic contractions are described as either sustained (tonic) or spasmodic (clonic) and are typically induced by conscious action to move the muscle group.1,2 Dystonia can be categorized according to the amount of muscle involvement: generalized (widespread muscle activity), segmental (involving neighboring groups of muscles), or focal (involving only one or a few local small muscles).3 Activity may be associated with gross posturing and disfigurement, depending on the size and location of the muscle contractions, although the muscle action is usually normal during rest.
The cause of dystonia has been the focus of much debate and investigation. Some types of dystonia have strong family inheritance patterns, but most are sporadic, possibly brought on by trauma or infection. In most cases, dystonia is idiopathic, although it may be associated with other muscle group dystonias, tremor, neurologic injury or insults, other neurologic diseases and neurodegenerative disorders, or tardive syndromes.1 Because of the relationship with other neurologic diseases, consultation with a neurologist should be considered.
Treatment of the muscle contractions of the various dystonias includes drug therapy and physical, occupational, and voice therapy. Botulinum toxin is a principal treatment for head and neck dystonias and works by blocking muscular contractions.4 It has the advantages of having few side effects and predictable results for many conditions, although repeat injections are usually required to achieve a sustained effect.
LARYNGEAL DYSTONIAS CAUSE VOICE ABNORMALITIES
The most common laryngeal dystonia is spasmodic dysphonia, a focal dystonia of the larynx. It is subdivided into two types according to whether spasm of the vocal folds occurs during adduction or abduction.
Adductor spasmodic dysphonia accounts for 80% to 90% of cases. It is characterized by irregular speech with pitch breaks and a strained or strangulated voice. It was formerly treated by resection of the nerve to the vocal folds, but results were neither consistent nor persistent. Currently, the primary treatment is injection of botulinum toxin, which has a high success rate,5 with patients reporting about a 90% return of normal function.
Abductor spasmodic dysphonia accounts for 10% to 20% of cases.6 Patients have a breathy quality to the voice with a short duration of vocalization due to excessive loss of air on phonation. This is especially noticeable when the patient speaks words that begin with a voiceless consonant followed by a vowel (eg, pat, puppy). Response to botulinum toxin injection is more variable,6 possibly because of the pathophysiology of the disorder or because of the technical challenges of administering the injection.
Fewer than 1% of patients have both abductor and adductor components, and their treatment can be particularly challenging.
Adductor spasmodic dysphonia: Treatment usually successful
Botulinum toxin can be injected for adductor spasmodic dysphonia via a number of approaches, the most common being through the cricothyroid membrane (Figure 1). Injections can be made into one or both vocal folds and can be performed under guidance with laryngeal electromyography or with a flexible laryngoscope to visualize the larynx.
Patients typically experience breathiness beginning 1 or 2 days after the injection, and this effect may last for up to 2 weeks. During that time, the patient may be more susceptible to aspiration of thin liquids and so is instructed to drink cautiously. Treatment benefits typically last for 3 to 6 months. As the botulinum toxin wears off, the patient notices a gradual increase in vocal straining and effort.
Dosages of botulinum toxin for subsequent treatments are adjusted by balancing the period of benefit with postinjection breathiness. The desire of the patient should be paramount. Some are willing to tolerate more side effects to avoid frequent injections, so they can be given a larger dose. Others cannot tolerate the breathiness but are willing to accept more frequent injections, so they should be given a smaller dose. In rare cases, patients have significant breathiness from even small doses; they may be helped by injecting into only one vocal fold or, alternatively, into a false vocal fold, allowing diffusion of the toxin down to the muscle of the true vocal fold.
Abductor spasmodic dysphonia: Treatment more challenging
The success of botulinum toxin treatment for abductor spasmodic dysphonia is more variable than for the adductor type. The injections are made into the posterior cricoarytenoid muscle (Figure 2); because this muscle cannot be directly visualized, this procedure requires guidance with laryngeal electromyography. Most patients note improvement, and about 20% have a good response.6 Most require a second injection about 1 month later, often on the other side. Bilateral injections at one sitting may compromise the airway, and vocal fold motion should be evaluated at the time of the contralateral injection to assess airway patency. Interest has increased in simultaneous bilateral injections with lower doses of botulinum toxin, and this approach has been shown to be safe.7
ESSENTIAL TREMOR OF THE VOICE
Essential tremor is an action tremor that can occur with voluntary movement. It can occur anywhere in the body, often the head or hand, but the voice can also be affected. About half of cases are hereditary. Essential tremor of the voice causes a rhythmic oscillation of pitch and intensity.
Consultation with a neurologist is recommended to evaluate the cause, although voice tremor is often idiopathic and occurs in about 30% of patients with essential tremor in the arms or legs, as well as in about 30% of patients with spasmodic dysphonia. Extremity tremor can usually be successfully managed medically, but this is not true for voice tremor.
Botulinum toxin injection is the mainstay of treatment for essential tremor of the voice, although its success is marginal. About two-thirds of patients have some degree of improvement from traditional botulinum toxin injections in the true vocal fold.8
The results of treatment are likely to be inconsistent because tremor tends to involve several different muscles used in voice production, commonly in the soft palate, tongue base, pharyngeal walls, strap muscles, false vocal folds, and true vocal folds. A location-oriented tremor scoring system9 can help identify the involved muscles to guide injections. Treatment is less likely to be successful in patients with multiple sites of voice tremor. Injection into the false vocal fold, true vocal fold, and interarytenoid muscle10 can safely be performed; injections into the palate, tongue base, and strap muscles are to be avoided because of the high risk of postinjection aspiration.
Patients who have good results can have repeat treatments as needed. The dosage of botulinum toxin is adjusted according to response, side effects (eg, breathy voice, dysphagia), and patient preference.
CRICOPHARYNGEUS MUSCLE DYSFUNCTION: TROUBLE SWALLOWING
Dysfunction of the cricopharyngeus muscle causes difficulty swallowing, especially swallowing solid foods. It can be attributed to a mechanical stricture or to hyperfunction (spasm).
Mechanical stricture at the esophageal inlet frequently occurs in patients who have had a total laryngectomy for advanced laryngeal cancer. Fibrosis tends to be worse in patients who have also undergone radiation therapy.
Stricture can be treated with botulinum toxin injections and dilation. Conservative treatment is preferred to surgical myotomy for patients with complex postlaryngectomy anatomy and scarring from radiation therapy.
Cricopharyngeus muscle spasm or hyperfunction can be an important cause of dysphagia, especially in the elderly. Patients should be evaluated with barium esophagography or a modified barium swallow. The finding of a cricopharyngeal “bar” provides evidence of contraction of the muscle that impedes the passage of food.
Botulinum toxin injections for cricopharyngeus muscle dysfunction (Figure 2) can be effective in some cases, especially if the toxin is injected bilaterally. However, because the cricopharyngeus muscle plays an important role in preventing esophageal reflux into the laryngopharynx, botulinum toxin injection in patients with substantial hiatal hernia or laryngopharyngeal reflux disease should only be done with caution. In addition, treatment of reflux disease should be considered in any patient undergoing botulinum toxin injection for cricopharyngeus muscle dysfunction.
Most patients require repeat injections when the toxin wears off, although occasionally one or two injections provide long-term or permanent relief. Dosages are adjusted for the patient’s age, the presence of other swallowing problems, and reflux. Patients may experience increased difficulty swallowing for 1 or 2 weeks after the procedure and so should be counseled to eat slowly and carefully.
Botulinum toxin is commonly used to treat movement disorders of the head and neck. It was first used to treat focal eye dystonia (blepharospasm) and laryngeal dystonia (spasmodic dysphonia) and is now also used for other head and neck dystonias, movement disorders, and muscle spasticity or contraction.
This article reviews the use of botulinum toxin for primary disorders of the laryngopharynx—adductor and abductor spasmodic dysphonias, laryngopharyngeal tremor, and cricopharyngeus muscle dysfunction—and its efficacy and side effects for the different conditions.
ABNORMAL MUSCLE MOVEMENT
Dystonia is abnormal muscle movement characterized by repetitive involuntary contractions. Dystonic contractions are described as either sustained (tonic) or spasmodic (clonic) and are typically induced by conscious action to move the muscle group.1,2 Dystonia can be categorized according to the amount of muscle involvement: generalized (widespread muscle activity), segmental (involving neighboring groups of muscles), or focal (involving only one or a few local small muscles).3 Activity may be associated with gross posturing and disfigurement, depending on the size and location of the muscle contractions, although the muscle action is usually normal during rest.
The cause of dystonia has been the focus of much debate and investigation. Some types of dystonia have strong family inheritance patterns, but most are sporadic, possibly brought on by trauma or infection. In most cases, dystonia is idiopathic, although it may be associated with other muscle group dystonias, tremor, neurologic injury or insults, other neurologic diseases and neurodegenerative disorders, or tardive syndromes.1 Because of the relationship with other neurologic diseases, consultation with a neurologist should be considered.
Treatment of the muscle contractions of the various dystonias includes drug therapy and physical, occupational, and voice therapy. Botulinum toxin is a principal treatment for head and neck dystonias and works by blocking muscular contractions.4 It has the advantages of having few side effects and predictable results for many conditions, although repeat injections are usually required to achieve a sustained effect.
LARYNGEAL DYSTONIAS CAUSE VOICE ABNORMALITIES
The most common laryngeal dystonia is spasmodic dysphonia, a focal dystonia of the larynx. It is subdivided into two types according to whether spasm of the vocal folds occurs during adduction or abduction.
Adductor spasmodic dysphonia accounts for 80% to 90% of cases. It is characterized by irregular speech with pitch breaks and a strained or strangulated voice. It was formerly treated by resection of the nerve to the vocal folds, but results were neither consistent nor persistent. Currently, the primary treatment is injection of botulinum toxin, which has a high success rate,5 with patients reporting about a 90% return of normal function.
Abductor spasmodic dysphonia accounts for 10% to 20% of cases.6 Patients have a breathy quality to the voice with a short duration of vocalization due to excessive loss of air on phonation. This is especially noticeable when the patient speaks words that begin with a voiceless consonant followed by a vowel (eg, pat, puppy). Response to botulinum toxin injection is more variable,6 possibly because of the pathophysiology of the disorder or because of the technical challenges of administering the injection.
Fewer than 1% of patients have both abductor and adductor components, and their treatment can be particularly challenging.
Adductor spasmodic dysphonia: Treatment usually successful
Botulinum toxin can be injected for adductor spasmodic dysphonia via a number of approaches, the most common being through the cricothyroid membrane (Figure 1). Injections can be made into one or both vocal folds and can be performed under guidance with laryngeal electromyography or with a flexible laryngoscope to visualize the larynx.
Patients typically experience breathiness beginning 1 or 2 days after the injection, and this effect may last for up to 2 weeks. During that time, the patient may be more susceptible to aspiration of thin liquids and so is instructed to drink cautiously. Treatment benefits typically last for 3 to 6 months. As the botulinum toxin wears off, the patient notices a gradual increase in vocal straining and effort.
Dosages of botulinum toxin for subsequent treatments are adjusted by balancing the period of benefit with postinjection breathiness. The desire of the patient should be paramount. Some are willing to tolerate more side effects to avoid frequent injections, so they can be given a larger dose. Others cannot tolerate the breathiness but are willing to accept more frequent injections, so they should be given a smaller dose. In rare cases, patients have significant breathiness from even small doses; they may be helped by injecting into only one vocal fold or, alternatively, into a false vocal fold, allowing diffusion of the toxin down to the muscle of the true vocal fold.
Abductor spasmodic dysphonia: Treatment more challenging
The success of botulinum toxin treatment for abductor spasmodic dysphonia is more variable than for the adductor type. The injections are made into the posterior cricoarytenoid muscle (Figure 2); because this muscle cannot be directly visualized, this procedure requires guidance with laryngeal electromyography. Most patients note improvement, and about 20% have a good response.6 Most require a second injection about 1 month later, often on the other side. Bilateral injections at one sitting may compromise the airway, and vocal fold motion should be evaluated at the time of the contralateral injection to assess airway patency. Interest has increased in simultaneous bilateral injections with lower doses of botulinum toxin, and this approach has been shown to be safe.7
ESSENTIAL TREMOR OF THE VOICE
Essential tremor is an action tremor that can occur with voluntary movement. It can occur anywhere in the body, often the head or hand, but the voice can also be affected. About half of cases are hereditary. Essential tremor of the voice causes a rhythmic oscillation of pitch and intensity.
Consultation with a neurologist is recommended to evaluate the cause, although voice tremor is often idiopathic and occurs in about 30% of patients with essential tremor in the arms or legs, as well as in about 30% of patients with spasmodic dysphonia. Extremity tremor can usually be successfully managed medically, but this is not true for voice tremor.
Botulinum toxin injection is the mainstay of treatment for essential tremor of the voice, although its success is marginal. About two-thirds of patients have some degree of improvement from traditional botulinum toxin injections in the true vocal fold.8
The results of treatment are likely to be inconsistent because tremor tends to involve several different muscles used in voice production, commonly in the soft palate, tongue base, pharyngeal walls, strap muscles, false vocal folds, and true vocal folds. A location-oriented tremor scoring system9 can help identify the involved muscles to guide injections. Treatment is less likely to be successful in patients with multiple sites of voice tremor. Injection into the false vocal fold, true vocal fold, and interarytenoid muscle10 can safely be performed; injections into the palate, tongue base, and strap muscles are to be avoided because of the high risk of postinjection aspiration.
Patients who have good results can have repeat treatments as needed. The dosage of botulinum toxin is adjusted according to response, side effects (eg, breathy voice, dysphagia), and patient preference.
CRICOPHARYNGEUS MUSCLE DYSFUNCTION: TROUBLE SWALLOWING
Dysfunction of the cricopharyngeus muscle causes difficulty swallowing, especially swallowing solid foods. It can be attributed to a mechanical stricture or to hyperfunction (spasm).
Mechanical stricture at the esophageal inlet frequently occurs in patients who have had a total laryngectomy for advanced laryngeal cancer. Fibrosis tends to be worse in patients who have also undergone radiation therapy.
Stricture can be treated with botulinum toxin injections and dilation. Conservative treatment is preferred to surgical myotomy for patients with complex postlaryngectomy anatomy and scarring from radiation therapy.
Cricopharyngeus muscle spasm or hyperfunction can be an important cause of dysphagia, especially in the elderly. Patients should be evaluated with barium esophagography or a modified barium swallow. The finding of a cricopharyngeal “bar” provides evidence of contraction of the muscle that impedes the passage of food.
Botulinum toxin injections for cricopharyngeus muscle dysfunction (Figure 2) can be effective in some cases, especially if the toxin is injected bilaterally. However, because the cricopharyngeus muscle plays an important role in preventing esophageal reflux into the laryngopharynx, botulinum toxin injection in patients with substantial hiatal hernia or laryngopharyngeal reflux disease should only be done with caution. In addition, treatment of reflux disease should be considered in any patient undergoing botulinum toxin injection for cricopharyngeus muscle dysfunction.
Most patients require repeat injections when the toxin wears off, although occasionally one or two injections provide long-term or permanent relief. Dosages are adjusted for the patient’s age, the presence of other swallowing problems, and reflux. Patients may experience increased difficulty swallowing for 1 or 2 weeks after the procedure and so should be counseled to eat slowly and carefully.
- Cultrara A, Chitkara A, Blitzer A. Botulinum toxin injections for the treatment of oromandibular dystonia. Oper Tech Otolaryngol Head Neck Surg 2004; 15:97–102.
- Fahn S. The varied clinical expressions of dystonia. Neurol Clin 1984; 2:541–554.
- Fahn S. Concept and classification of dystonia. Adv Neurol 1988; 50:1–8.
- Benninger MS, Knott PD. Techniques of botulinum toxin injections in the head and neck. San Diego, CA: Plural Publishing, Inc; 2012.
- Benninger MS, Gardner G, Grywalski C. Outcomes of botulinum toxin treatment for spasmodic dysphonia. Arch Otolaryngol Head Neck Surg 2001; 127:1083–1085.
- Blitzer A, Brin MF, Stewart CF. Botulinum toxin management of spasmodic dysphonia (laryngeal dystonia): a 12-year experience in more than 900 patients. Laryngoscope 1998; 108:1435–1441.
- Klein AM, Stong BC, Wise J, DelGaudio JM, Hapner ER, Johns MM 3rd. Vocal outcome measures after bilateral posterior cricoarytenoid muscle botulinum toxin injections for abductor spasmodic dysphonia. Otolaryngol Head Neck Surg 2008; 139:421–423.
- Hertegård S, Granqvist S, Lindestad PA. Botulinum toxin injections for essential voice tremor. Ann Otol Rhinol Laryngol 2000; 109:204–209.
- Bové M, Daamen N, Rosen C, Wang CC, Sulica L, Gartner-Schmidt J. Development and validation of the vocal tremor scoring system. Laryngoscope 2006; 116:1662–1667.
- Kendall KA, Leonard RJ. Interarytenoid muscle Botox injection for treatment of adductor spasmodic dysphonia with vocal tremor. J Voice 2001; 25:114–119.
- Cultrara A, Chitkara A, Blitzer A. Botulinum toxin injections for the treatment of oromandibular dystonia. Oper Tech Otolaryngol Head Neck Surg 2004; 15:97–102.
- Fahn S. The varied clinical expressions of dystonia. Neurol Clin 1984; 2:541–554.
- Fahn S. Concept and classification of dystonia. Adv Neurol 1988; 50:1–8.
- Benninger MS, Knott PD. Techniques of botulinum toxin injections in the head and neck. San Diego, CA: Plural Publishing, Inc; 2012.
- Benninger MS, Gardner G, Grywalski C. Outcomes of botulinum toxin treatment for spasmodic dysphonia. Arch Otolaryngol Head Neck Surg 2001; 127:1083–1085.
- Blitzer A, Brin MF, Stewart CF. Botulinum toxin management of spasmodic dysphonia (laryngeal dystonia): a 12-year experience in more than 900 patients. Laryngoscope 1998; 108:1435–1441.
- Klein AM, Stong BC, Wise J, DelGaudio JM, Hapner ER, Johns MM 3rd. Vocal outcome measures after bilateral posterior cricoarytenoid muscle botulinum toxin injections for abductor spasmodic dysphonia. Otolaryngol Head Neck Surg 2008; 139:421–423.
- Hertegård S, Granqvist S, Lindestad PA. Botulinum toxin injections for essential voice tremor. Ann Otol Rhinol Laryngol 2000; 109:204–209.
- Bové M, Daamen N, Rosen C, Wang CC, Sulica L, Gartner-Schmidt J. Development and validation of the vocal tremor scoring system. Laryngoscope 2006; 116:1662–1667.
- Kendall KA, Leonard RJ. Interarytenoid muscle Botox injection for treatment of adductor spasmodic dysphonia with vocal tremor. J Voice 2001; 25:114–119.
KEY POINTS
- Botulinum toxin can be injected with a variety of approaches directly into the affected muscle exhibiting abnormal contractions.
- Depending on the muscles involved, side effects may include breathiness or difficulty swallowing for a period soon after injection.
- Injections can be repeated as needed as the toxin wears off.
- Some conditions are more amenable to treatment than others. Benefit can be enhanced by altering the dosage or injection site.
Recreational cannabis use: Pleasures and pitfalls
Clinicians may be encountering more cannabis users than before, and may be encountering users with complications hitherto unseen. Several trends may explain this phenomenon: the legal status of cannabis is changing, cannabis today is more potent than in the past, and enthusiasts are conjuring new ways to enjoy this substance.
This article discusses the history, pharmacology, and potential complications of cannabis use.
A LONG AND TANGLED HISTORY
Cannabis is a broad term that refers to the cannabis plant and its preparations, such as marijuana and hashish, as well as to a family of more than 60 bioactive substances called cannabinoids. It is the most commonly used illegal drug in the world, with an estimated 160 million users. Each year, about 2.4 million people in the United States use it for the first time.1,2
Cannabis has been used throughout the world for recreational and spiritual purposes for nearly 5,000 years, beginning with the fabled Celestial Emperors of China. The tangled history of cannabis in America began in the 17th century, when farmers were required by law to grow it as a fiber crop. It later found its way into the US Pharmacopeia for a wide range of indications. During the long prelude to Prohibition in the latter half of the 19th century, the US government became increasingly suspicious of mind-altering substances and began restricting its prescription in 1934, culminating in its designation by the US Food and Drug Administration as a schedule I controlled substance in 1970.
Investigation into the potential medical uses for the different chemicals within cannabis is ongoing, as is debate over its changing legality and usefulness to society. The apparent cognitive dissonance surrounding the use and advocacy of medical marijuana is beyond the scope of this review,3 which will instead restrict itself to what is known of the cannabinoids and to the recreational use of cannabis.
THC IS THE PRINCIPAL PSYCHOACTIVE MOLECULE
Delta-9 tetrahydrocannabinol (THC), first isolated in 1964, was identified as the principal psychoactive constituent of cannabis in 2002.4
Two G-protein–linked cannabinoid receptors cloned in the 1990s—CB1 and CB2—were found to be a part of a system of endocannabinoid receptors present throughout the body, from the brain to the immune system to the vas deferens.5 Both receptors inhibit cellular excitation by activating inwardly rectifying potassium channels. These receptors are mostly absent in the brainstem, which may explain why cannabis use rarely causes life-threatening autonomic dysfunction. Although the intoxicating effects of marijuana are mediated by CB1 receptors, the specific mechanisms underlying the cannabis “high” are unclear.6
CANNABINOIDS ARE LIPID-SOLUBLE
The rate of absorption of cannabinoids depends on the route of administration and the type of cannabis product used. When cannabis products are smoked, up to 35% of THC is available, and the average time to peak serum concentration is 8 minutes.7 The peak concentration depends on the dose.
On the other hand, when cannabis products (eg, nabilone, dronabinol) are ingested, absorption is unpredictable because THC is unstable in gastric acid and undergoes first-pass metabolism in the liver, which reduces the drug’s bioavailability. Up to 20% of an ingested dose of THC is absorbed, and the time to peak serum concentration averages between 2 and 4 hours. Consequently, many users prefer to smoke cannabis as a means to control the desired effects.
Cannabinoids are lipid-soluble. They accumulate in fatty tissue in a biphasic pattern, initially moving into highly vascularized tissue such as the liver before accumulating in less well-vascularized tissue such as fat. They are then slowly released from fatty tissue as the fat turns over. THC itself has a volume of distribution of about 2.5 to 3.5 L/kg. It crosses the placenta and enters breast milk.8
THC is metabolized by the cytochrome P450 system, primarily by the enzymes CYP2C9 and CYP3A4. Its primary metabolite, 11-hydroxy-delta-9 THC, is also active, but subsequent metabolism produces many other inactive metabolites. THC is eliminated in feces and urine, and its half-life ranges from 2 to nearly 60 hours.8
A LITTLE ABOUT PLANTS AND STREET NAMES
The plant from which THC and nearly a hundred other chemicals, including cannabinoids, are derived has been called many things over the years:
Hemp is a tall fibrous plant grown for rope and fabric that was used as legal tender in early America. In the mid-19th century, there were over 16 million acres of hemp plantations. Hemp contains very low THC concentrations.
Cannabis is an annual flowering herb that is predominantly diecious (ie, there are male and female plants). After a centuries-long debate among taxonomists, the two principal species are considered to be C sativa and C indica, although today many cannabis cultivars are grown by a great number of breeding enthusiasts.
Concentrations of THC vary widely among cannabis cultivars, ranging historically from around 5% to today’s highly selectively bred species containing more than 30%. Concentrations in seized cannabis have been measured as high as 37%, although the average is around 11%.9 This concentration is defined by the percent of THC per dried mass of plant material tested, usually via gas chromatography.
Hashish is a solid or resinous preparation of the trichomes, or glandular hairs, that grow on the cannabis plant, chiefly on its flowers. Various methods to separate the trichomes from the rest of the plant result in a powder called kief that is then compressed into blocks or bricks. THC concentrations as high as 66% have been measured in nondomestic sources of hashish.9
Hash oil is a further purification, produced by using solvents to dissolve the resin and by filtering out remaining plant material. Evaporating the solvent produces hash oil, sometimes called butane hash oil or honey oil. This process has recently led to an increasing number of home explosions, as people attempt to make the product themselves but do not take suitable precautions when using flammable solvents such as butane. THC concentrations as high as 81% have been measured in nondomestic sources of hash oil.9
Other names for hash oil are dab, wax, and budder. Cannabis enthusiasts refer to the use of hash oil as dabbing, which involves heating a small amount (dab) of the product using a variety of paraphernalia and inhaling the vapor.
IT’S ALL ABOUT GETTING HIGH
For recreational users, the experience has always been about being intoxicated—getting high. The psychological effects range broadly from positive to negative and vary both within and between users, depending on the dose and route of administration. Additional factors that influence the psychological effects include the social and physical settings of drug use and even the user’s expectations. One user’s high is another user’s acute toxic effect.
Although subjective reports of the cannabis experience vary greatly, it typically begins with a feeling of dizziness or lightheadedness followed by a relaxed calm and a feeling of being somewhat “disconnected.” There is a quickening of the sense of humor, described by some as a fatuous euphoria; often there is silly giggling. Awareness of the senses and of music may be increased. Appetite increases, and time seems to pass quickly. Eventually, the user becomes drowsy and experiences decreased attention and difficulty maintaining a coherent conversation. Slowed reaction time and decreased psychomotor activity may also occur. The user may drift into daydreams and eventually fall asleep.
Common negative acute effects of getting high can include mild to severe anxiety and feeling tense or agitated. Clumsiness, headache, and confusion are also possible. Lingering effects the following day may include dry mouth, dry eyes, fatigue, slowed thinking, and slowed recall.6
ACUTE PHYSICAL EFFECTS
Acute physical effects of cannabis use include a rapid onset of increased airway conductance, decreased intraocular pressure, and conjunctival injection. A single cannabis cigarette can also induce cardiovascular effects including a dose-dependent increase in heart rate and blood pressure. Chronic users, however, can experience a decreased heart rate, lower blood pressure, and postural hypotension.
In a personal communication, colleagues in Colorado—where recreational use of cannabis was legalized in 2012—described a sharp increase (from virtually none) in the number of adults presenting to the emergency department with cannabis intoxication since 2012. Their patients experienced palpitations, light-headedness, and severe ataxia lasting as long as 12 hours, possibly reflecting the greater potency of current cannabis products. Most of these patients required only supportive care.
Other acute adverse cardiovascular reactions that have been reported include atrial fibrillation, ventricular tachycardia, and a fivefold increased risk of myocardial infarction in the 60 minutes following cannabis use, which subsequently drops sharply to baseline levels.10 Investigations into the cardiovascular effects of cannabis are often complicated by concurrent use of other drugs such as tobacco or cocaine. Possible mechanisms of injury include alterations in coronary microcirculation or slowed coronary flow. In fact, one author found that cannabis users with a history of myocardial infarction had a risk of death 4.2 times higher than users with no history of myocardial infarction.11,12
In children, acute toxicity has been reported from a variety of exposures to cannabis and hashish, including a report of an increase in pediatric cannabis exposures following the changes in Colorado state laws.13 Most of these patients had altered mental status ranging from drowsiness to coma; one report describes a child who experienced a first-time seizure. These patients unfortunately often underwent extensive evaluations such as brain imaging and lumbar puncture, and mechanical ventilation to protect the airway. Earlier consideration of cannabis exposure in these patients might have limited unnecessary testing. Supportive care is usually all that is needed, and most of these patients fully recover.13–17
CHRONIC EFFECTS
Cannabinoids cause a variety of adverse effects, but the ultimate risk these changes pose to human health has been difficult to calculate. Long-term studies are confounded by possible inaccuracies of patient self-reporting of cannabis use, poor control of covariates, and disparate methodologies.
For more than a century, cannabis use has been reported to cause both acute psychotic symptoms and persistent psychotic disorders.18 But the strength of this relationship is modest. Cannabis is more likely a component cause that, in addition to other factors (eg, specific genetic polymorphisms), contributes to the risk of schizophrenia. Individuals with prodromal symptoms and those who have experienced discrete episodes of psychosis related to cannabis use should be discouraged from using cannabis and cannabinoids.19–21
Mounting evidence implicates chronic cannabis use as a cause of long-term medical problems including chronic bronchitis,22 elevated rates of myocardial infarction and dysrhythmias,11 bone loss,23 and cancers at eight different sites including the lung, head, and neck.24 In view of these chronic effects, healthcare providers should caution their patients about cannabis use, as we do about other drugs such as tobacco.
WITHDRAWAL SYNDROME RECOGNIZED
Until recently, neither clinicians nor users recognized a withdrawal syndrome associated with chronic use of cannabis, probably because this syndrome is not as severe as withdrawal from other controlled substances such as opioids or sedative-hypnotics. A number of studies, however, have reported subtle cannabis withdrawal symptoms that are similar to those associated with tobacco withdrawal.
As such, the fifth and latest edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5)25 characterized withdrawal from cannabis use in 2013. The DSM-5 criteria require cessation of heavy or prolonged use of cannabis (ie, daily or almost daily over a period of at least a few months) and three or more of the following withdrawal symptoms:
- Irritability and anger
- Nervousness
- Sleep difficulty or insomnia
- Decreased appetite or weight loss
- Restlessness
- Depressed mood
- Physical symptoms causing discomfort.
Medical treatment of cannabis withdrawal has included a range of antidepressants, mood stabilizers, and alpha-2-adrenergic agonists, all of which have limited success.26 Symptoms of cannabis withdrawal tend to be most intense soon after cessation and decline over the next few weeks.27
CANNABINOID HYPEREMESIS SYNDROME
First reported in 2004,28 cannabinoid hyperemesis syndrome is a recurrent disorder, the pathophysiology of which is poorly understood. It has three phases.
The first phase is a prodrome that may last months or years and is characterized by morning nausea, fear of vomiting, and abdominal discomfort. During this phase, the patient maintains normal eating patterns and may well increase his or her cannabis use due to its well-known antiemetic effects.
The second phase is the hyperemetic phase, characterized by intense, incapacitating emesis with episodes of vomiting throughout the day. These symptoms can be relieved only with frequent hot baths, a feature that distinguishes cannabinoid hyperemesis syndrome from other vomiting syndromes. Hot-water bathing is reported to be a compulsive but learned behavior in which the patient learns that only hot water will provide relief. The extent of relief depends on the temperature of the water—the hotter, the better. Symptoms recur as the water cools.28 Patients often present to the emergency department repeatedly with recurrent symptoms and may remain misdiagnosed or subjected to repeated extensive evaluation including laboratory testing and imaging, which are usually not revealing. If the patient has not been accurately diagnosed, there may be reported weight loss of at least 5 kg.
The third phase, recovery, may take several months to complete, possibly because of the prolonged terminal elimination time of cannabinoids. Complete cessation of cannabis use, including synthetic cannabinoids, is usually necessary.29
Diagnostic criteria for cannabinoid hyperemesis syndrome have been suggested, based on a retrospective case series that included 98 patients.30 The most common features of these affected patients were:
- Severe cyclical vomiting, predominantly in the morning
- Resolution of symptoms with cessation of cannabis use
- Symptomatic relief with hot showers or baths
- Abdominal pain
- At least weekly use of cannabis.
Interestingly, long-term cannabis use has been cited as a critical identifying feature of these patients, with the duration of cannabis use ranging from 10 to 16 years.31,32 Other reports show greater variability in duration of cannabis use before the onset of cannabinoid hyperemesis syndrome. In the large study noted above,30 32% of users reported their duration of cannabis use to be less than 1 year, rendering this criterion less useful.
How can cannabis both cause and prevent vomiting?
The body controls nausea and vomiting via complex circuitry in the brain and gut that involves many neurotransmitters (eg, dopamine, serotonin, substance P) that interact with receptors such as CB1, 5-HT1–4, alpha adrenergic receptors, and mu receptors. Interestingly, cannabis use has antiemetic properties mediated by CB1 with a still unclear additional role of CB2 receptors. Data point to the existence of an underlying antiemetic tone mediated by the endocannabinoid system.
Unfortunately, the mechanism by which cannabinoid hyperemesis syndrome occurs is unknown and represents a paradoxical effect against the otherwise antiemetic effects of cannabis. Several theories have been proposed, including delayed gastric emptying, although only a third of patients demonstrated this on scintigraphy in one study.30 Other theories include disturbance of the hypothalamic-pituitary axis, a buildup of highly lipophilic THC in the brain, and a down-regulation of cannabinoid receptors that results from chronic exposure.30 Given that this syndrome has been recognized only relatively recently, one author has suggested the cause may be recent horticultural developments.5
Treating cannabinoid hyperemesis syndrome is difficult
Treatment of cannabinoid hyperemesis syndrome is notoriously difficult, with many authors reporting resistance to the usual first-line antiemetic drugs. Generally, treatment should include hydration and acid-suppression therapy because endoscopic evaluation of several patients has revealed varying degrees of esophagitis and gastritis.29
Antiemetic therapy should target receptors known to mediate nausea and vomiting. In some cases, antiemetic drugs are more effective when used in combination. Agents include the serotonergic receptor antagonists ondansetron and granisetron, the dopamine antagonists prochlorperazine and metoclopramide, and even haloperidol.33,34 Benzodiazepines may be effective by causing sedation, anxiolysis, and depression of the vomiting center.34,35 Two antihistamines—dimenhydrinate and diphenhydramine—have antiemetic effects, perhaps by inhibiting acetylcholine.34
Aprepitant is a neurokinin-1 antagonist that inhibits the action of substance P. When combined with a corticosteroid and a serotonin antagonist, it relieves nausea and vomiting in chemotherapy patients.34,36
Corticosteroids such as dexamethasone are potent antiemetics thought to inhibit prostaglandin synthesis.34
Capsaicin cream applied to the abdomen has also been reported to relieve symptoms, possibly through an interaction between the TRPv1 receptor and the endocannabinoid system.37,38
DIAGNOSTIC TESTING
Cannabinoids are detectable in plasma and urine, with urine testing being more common.
Common laboratory methods include the enzyme-multiplied immunoassay technique (EMIT) and radioimmunoassay. Gas chromatography coupled with mass spectrometry is the most specific assay; it is used for confirmation and is the reference method.
EMIT is a qualitative urine test that detects 9-carboxy-THC as well as other THC metabolites. These urine tests detect all metabolites, and the result is reported as positive if the total concentration is greater than or equal to a prespecified threshold level, such as 20 ng/mL or 50 ng/mL. A positive test does not denote intoxication, nor does the test identify the source of THC (eg, cannabis, dronabinol, butane hash oil). EMIT does not detect nabilone. The National Institute on Drug Abuse guidelines for urine testing specify a test threshold concentration of 50 ng/mL for screening and 15 ng/mL for confirmation.

Many factors affect the detection of THC metabolites and their presence and duration in urine: dose, duration of use, route of exposure, hydration status, urine volume and concentration, and urine pH. THC metabolites have been detected in urine using gas chromatography-mass spectrometry for up to 7 days after smoking one marijuana cigarette.7 Chronic users have also been reported to have positive urine EMIT tests for up to 46 days after cannabis cessation.39 Detection may be further complicated in chronic users: in one study, users produced both negative and positive specimens over 24 days, suggesting that diet and exercise may influence clearance.40 Also, many factors are known to produce false-positive and false-negative results for these immunoassays (Table 1).39,41
In the United States, penalties for driving under the influence of cannabis vary from state to state, and laws specify plasma testing for quantitative analysis. Some states use a threshold of 5 ng/mL in plasma to imply driving under the influence, whereas others use any detectable amount. Currently, there are no generally accepted guidelines for storage and testing of blood samples, despite the known instability of analytes.42
Saliva, hair, and sweat can also be used for cannabinoid testing. Saliva is easy to collect, can be tested for metabolites to rule out passive cannabis exposure, and can be positive for up to 1 day after exposure. Calculating a blood or plasma concentration from a saliva sample is not possible, however.
Hair testing can also rule out passive exposure, but THC binds very little to melanin, resulting in very low concentrations requiring sensitive tests, such as gas chromatography with tandem mass spectrometry.
Only one device is commercially available for sweat testing; further work is needed to elucidate sweat excretion pharmacokinetics and the limitations of the collection devices.43
CLINICAL MANAGEMENT IS GENERALLY SUPPORTIVE
Historically, clinical toxicity from recreational cannabis use is rarely serious or severe and generally responds to supportive care. Reports of cannabis exposure to poison centers are one-tenth of those reported for ethanol exposures annually.44 Gastrointestinal decontamination with activated charcoal is not recommended, even for orally administered cannabis, since the risks outweigh the expected benefits. Agitation or anxiety may be treated with benzodiazepines as needed. There is no antidote for cannabis toxicity. The ever-increasing availability of high-concentration THC preparations may prompt more aggressive supportive measures in the future.
SYNTHETIC MARIJUANA ALTERNATIVES
Available since the early 2000s, herbal marijuana alternatives are legally sold as incense or potpourri and are often labeled “not for human consumption.” They are known by such brand names as K2 and Spice and contain blends of herbs adulterated with synthetic cannabinoid chemicals developed by researchers exploring the receptor-ligand binding of the endocannabinoid system.
Clinical effects, generally psychiatric, include paranoia, anxiety, agitation, delusions, and psychosis. There are also reports of patients who arrive with sympathomimetic toxicity, some of whom develop bradycardia and hypotension, and some who progress to acute renal failure, seizures, and death. Detection of these products is difficult as they do not react on EMIT testing for THC metabolites and require either gas chromatography-mass spectrometry or liquid chromatography with tandem mass spectrometry.45–48
- Substance Abuse and Mental Health Services Administration. Results from the 2012 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-46, HHS Publication No. (SMA) 13-4795. www.samhsa.gov/data/sites/default/files/NSDUHresultsPDFWHTML2013/Web/NSDUHresults2013.pdf. Accessed October 2, 2015.
- United Nations Office on Drugs and Crime. 2008 World Drug Report. www.unodc.org/documents/wdr/WDR_2008/WDR_2008_eng_web.pdf. Accessed October 2, 2015.
- American Society of Addiction Medicine (ASAM). Public policy statement on medical marijuana. www.asam.org/docs/publicy-policy-statements/1medical-marijuana-4-10.pdf?sfvrsn=0. Accessed October 2, 2015.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002; 54:161–202.
- Sharkey KA, Darmani NA, Parker LA. Regulation of nausea and vomiting by cannabinoids and the endocannabinoid system. Eur J Pharmacol 2014; 722:134–146.
- Iversen L. Cannabis and the brain. Brain 2003; 126:1252–1270.
- Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol 1992; 16:276–282.
- Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet 2003; 42:327–360.
- Mehmedic Z, Chandra S, Slade D, et al. Potency trends of Δ9-THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. J Forensic Sci 2010; 55:1209–1217.
- Mittleman MA, Lewis RA, Maclure M, Sherwood JB, Muller JE. Triggering myocardial infarction by marijuana. Circulation 2001; 103:2805–2809.
- Mukamal KJ, Maclure M, Muller JE, Mittleman MA. An exploratory prospective study of marijuana use and mortality following acute myocardial infarction. Am Heart J 2008; 155:465–470.
- Thomas G, Kloner RA, Rezkalla S. Adverse cardiovascular, cerebrovascular, and peripheral vascular effects of marijuana inhalation: what cardiologists need to know. Am J Cardiol 2014; 113:187–190.
- Wang GS, Roosevelt G, Heard K. Pediatric marijuana exposures in a medical marijuana state. JAMA Pediatr 2013; 167:630–633.
- Carstairs SD, Fujinaka MK, Keeney GE, Ly BT. Prolonged coma in a child due to hashish ingestion with quantitation of THC metabolites in urine. J Emerg Med 2011; 41:e69–e71.
- Le Garrec S, Dauger S, Sachs P. Cannabis poisoning in children. Intensive Care Med 2014; 40:1394–1395.
- Ragab AR, Al-Mazroua MK. Passive cannabis smoking resulting in coma in a 16-month old infant. J Clin Case Rep 2012;2:237.
- Robinson K. Beyond resinable doubt? J Clin Forensic Med 2005;12:164–166.
- Burns JK. Pathways from cannabis to psychosis: a review of the evidence. Front Psychiatry 2013;4:128.
- Di Forti M, Sallis H, Allegri F, et al. Daily use, especially of high-potency cannabis, drives the earlier onset of psychosis in cannabis users. Schizophr Bull 2014; 40:1509–1517.
- Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328.
- Wilkinson ST, Radhakrishnan R, D'Souza DC. Impact of cannabis use on the development of psychotic disorders. Curr Addict Rep 2014;1:115–128.
- Aldington S, Williams M, Nowitz M, et al. Effects of cannabis on pulmonary structure, function and symptoms. Thorax 2007; 62:1058–1063.
- George KL, Saltman LH, Stein GS, Lian JB, Zurier RB. Ajulemic acid, a nonpsychoactive cannabinoid acid, suppresses osteoclastogenesis in mononuclear precursor cells and induces apoptosis in mature osteoclast-like cells. J Cell Physiol 2008; 214:714–720.
- Reece AS. Chronic toxicology of cannabis. Clin Toxicol (Phila) 2009; 47:517–524.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
- Allsop DJ, Copeland J, Lintzeris N, et al. Nabiximols as an agonist replacement therapy during cannabis withdrawal: a randomized clinical trial. JAMA Psychiatry 2014; 71:281–291.
- Hesse M, Thylstrup B. Time-course of the DSM-5 cannabis withdrawal symptoms in poly-substance abusers. BMC Psychiatry 2013; 13:258.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Galli JA, Sawaya RA, Friedenberg FK. Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev 2011; 4:241–249.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci 2010; 55:3113–3119.
- Wallace EA, Andrews SE, Garmany CL, Jelley MJ. Cannabinoid hyperemesis syndrome: literature review and proposed diagnosis and treatment algorithm. South Med J 2011; 104:659–664.
- Hickey JL, Witsil JC, Mycyk MB. Haloperidol for treatment of cannabinoid hyperemesis syndrome. Am J Emerg Med 2013; 31:1003.e5–1003.e6.
- Perwitasari DA, Gelderblom H, Atthobari J, et al. Anti-emetic drugs in oncology: pharmacology and individualization by pharmacogenetics. Int J Clin Pharm 2011; 33:33–43.
- Cox B, Chhabra A, Adler M, Simmons J, Randlett D. Cannabinoid hyperemesis syndrome: case report of a paradoxical reaction with heavy marijuana use. Case Rep Med 2012; 2012:757696.
- Sakurai M, Mori T, Kato J, et al. Efficacy of aprepitant in preventing nausea and vomiting due to high-dose melphalan-based conditioning for allogeneic hematopoietic stem cell transplantation. Int J Hematol 2014; 99:457–462.
- Lapoint J. Case series of patients treated for cannabinoid hyperemesis syndrome with capsaicin cream. Clin Tox 2014; 52:707. Abstract #53.
- Biary R, Oh A, Lapoint J, Nelson LS, Hoffman RS, Howland MA. Topical capsaicin cream used as a therapy for cannabinoid hyperemesis syndrome. Clin Tox 2014; 52:787. Abstract #232.
- Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc 2008; 83:66–76.
- Lowe RH, Abraham TT, Darwin WD, Herning R, Cadet JL, Huestis MA. Extended urinary delta9-tetrahydrocannabinol excretion in chronic cannabis users precludes use as a biomarker of new drug exposure. Drug Alcohol Depend 2009; 105:24–32.
- Paul BD, Jacobs A. Effects of oxidizing adulterants on detection of 11-nor-delta9-THC-9-carboxylic acid in urine. J Anal Toxicol 2002; 26:460–463.
- Schwope DM, Karschner EL, Gorelick DA, Huestis MA. Identification of recent cannabis use: whole-blood and plasma free and glucuronidated cannabinoid pharmacokinetics following controlled smoked cannabis administration. Clin Chem 2011; 57:1406-1414.
- Huestis MA, Smith ML. Cannabinoid pharmacokinetics and disposition in alternative matrices. In: Pertwee R, ed. Handbook of Cannabis. Oxford, United Kingdom: Oxford University Press; 2014:296–316.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila) 2013; 51:949–1229.
- Rosenbaum CD, Carreiro SP, Babu KM. Here today, gone tomorrow…and back again? A review of herbal marijuana alternatives (K2, Spice), synthetic cathinones (bath salts), kratom, Salvia divinorum, methoxetamine, and piperazines. J Med Toxicol 2012; 8:15–32.
- Gurney SMR, Scott KS, Kacinko SL, Presley BC, Logan BK. Pharmacology, toxicology, and adverse effects of synthetic cannabinoid drugs. Forensic Sci Rev 2014; 26:53–78.
- McKeever RG, Vearrier D, Jacobs D, LaSala G, Okaneku J, Greenberg MI. K2-not the spice of life; synthetic cannabinoids and ST elevation myocardial infarction: a case report. J Med Toxicol 2015; 11:129–131.
- Schneir AB, Baumbacher T. Convulsions associated with the use of a synthetic cannabinoid product. J Med Toxicol 2012; 8:62–64.
Clinicians may be encountering more cannabis users than before, and may be encountering users with complications hitherto unseen. Several trends may explain this phenomenon: the legal status of cannabis is changing, cannabis today is more potent than in the past, and enthusiasts are conjuring new ways to enjoy this substance.
This article discusses the history, pharmacology, and potential complications of cannabis use.
A LONG AND TANGLED HISTORY
Cannabis is a broad term that refers to the cannabis plant and its preparations, such as marijuana and hashish, as well as to a family of more than 60 bioactive substances called cannabinoids. It is the most commonly used illegal drug in the world, with an estimated 160 million users. Each year, about 2.4 million people in the United States use it for the first time.1,2
Cannabis has been used throughout the world for recreational and spiritual purposes for nearly 5,000 years, beginning with the fabled Celestial Emperors of China. The tangled history of cannabis in America began in the 17th century, when farmers were required by law to grow it as a fiber crop. It later found its way into the US Pharmacopeia for a wide range of indications. During the long prelude to Prohibition in the latter half of the 19th century, the US government became increasingly suspicious of mind-altering substances and began restricting its prescription in 1934, culminating in its designation by the US Food and Drug Administration as a schedule I controlled substance in 1970.
Investigation into the potential medical uses for the different chemicals within cannabis is ongoing, as is debate over its changing legality and usefulness to society. The apparent cognitive dissonance surrounding the use and advocacy of medical marijuana is beyond the scope of this review,3 which will instead restrict itself to what is known of the cannabinoids and to the recreational use of cannabis.
THC IS THE PRINCIPAL PSYCHOACTIVE MOLECULE
Delta-9 tetrahydrocannabinol (THC), first isolated in 1964, was identified as the principal psychoactive constituent of cannabis in 2002.4
Two G-protein–linked cannabinoid receptors cloned in the 1990s—CB1 and CB2—were found to be a part of a system of endocannabinoid receptors present throughout the body, from the brain to the immune system to the vas deferens.5 Both receptors inhibit cellular excitation by activating inwardly rectifying potassium channels. These receptors are mostly absent in the brainstem, which may explain why cannabis use rarely causes life-threatening autonomic dysfunction. Although the intoxicating effects of marijuana are mediated by CB1 receptors, the specific mechanisms underlying the cannabis “high” are unclear.6
CANNABINOIDS ARE LIPID-SOLUBLE
The rate of absorption of cannabinoids depends on the route of administration and the type of cannabis product used. When cannabis products are smoked, up to 35% of THC is available, and the average time to peak serum concentration is 8 minutes.7 The peak concentration depends on the dose.
On the other hand, when cannabis products (eg, nabilone, dronabinol) are ingested, absorption is unpredictable because THC is unstable in gastric acid and undergoes first-pass metabolism in the liver, which reduces the drug’s bioavailability. Up to 20% of an ingested dose of THC is absorbed, and the time to peak serum concentration averages between 2 and 4 hours. Consequently, many users prefer to smoke cannabis as a means to control the desired effects.
Cannabinoids are lipid-soluble. They accumulate in fatty tissue in a biphasic pattern, initially moving into highly vascularized tissue such as the liver before accumulating in less well-vascularized tissue such as fat. They are then slowly released from fatty tissue as the fat turns over. THC itself has a volume of distribution of about 2.5 to 3.5 L/kg. It crosses the placenta and enters breast milk.8
THC is metabolized by the cytochrome P450 system, primarily by the enzymes CYP2C9 and CYP3A4. Its primary metabolite, 11-hydroxy-delta-9 THC, is also active, but subsequent metabolism produces many other inactive metabolites. THC is eliminated in feces and urine, and its half-life ranges from 2 to nearly 60 hours.8
A LITTLE ABOUT PLANTS AND STREET NAMES
The plant from which THC and nearly a hundred other chemicals, including cannabinoids, are derived has been called many things over the years:
Hemp is a tall fibrous plant grown for rope and fabric that was used as legal tender in early America. In the mid-19th century, there were over 16 million acres of hemp plantations. Hemp contains very low THC concentrations.
Cannabis is an annual flowering herb that is predominantly diecious (ie, there are male and female plants). After a centuries-long debate among taxonomists, the two principal species are considered to be C sativa and C indica, although today many cannabis cultivars are grown by a great number of breeding enthusiasts.
Concentrations of THC vary widely among cannabis cultivars, ranging historically from around 5% to today’s highly selectively bred species containing more than 30%. Concentrations in seized cannabis have been measured as high as 37%, although the average is around 11%.9 This concentration is defined by the percent of THC per dried mass of plant material tested, usually via gas chromatography.
Hashish is a solid or resinous preparation of the trichomes, or glandular hairs, that grow on the cannabis plant, chiefly on its flowers. Various methods to separate the trichomes from the rest of the plant result in a powder called kief that is then compressed into blocks or bricks. THC concentrations as high as 66% have been measured in nondomestic sources of hashish.9
Hash oil is a further purification, produced by using solvents to dissolve the resin and by filtering out remaining plant material. Evaporating the solvent produces hash oil, sometimes called butane hash oil or honey oil. This process has recently led to an increasing number of home explosions, as people attempt to make the product themselves but do not take suitable precautions when using flammable solvents such as butane. THC concentrations as high as 81% have been measured in nondomestic sources of hash oil.9
Other names for hash oil are dab, wax, and budder. Cannabis enthusiasts refer to the use of hash oil as dabbing, which involves heating a small amount (dab) of the product using a variety of paraphernalia and inhaling the vapor.
IT’S ALL ABOUT GETTING HIGH
For recreational users, the experience has always been about being intoxicated—getting high. The psychological effects range broadly from positive to negative and vary both within and between users, depending on the dose and route of administration. Additional factors that influence the psychological effects include the social and physical settings of drug use and even the user’s expectations. One user’s high is another user’s acute toxic effect.
Although subjective reports of the cannabis experience vary greatly, it typically begins with a feeling of dizziness or lightheadedness followed by a relaxed calm and a feeling of being somewhat “disconnected.” There is a quickening of the sense of humor, described by some as a fatuous euphoria; often there is silly giggling. Awareness of the senses and of music may be increased. Appetite increases, and time seems to pass quickly. Eventually, the user becomes drowsy and experiences decreased attention and difficulty maintaining a coherent conversation. Slowed reaction time and decreased psychomotor activity may also occur. The user may drift into daydreams and eventually fall asleep.
Common negative acute effects of getting high can include mild to severe anxiety and feeling tense or agitated. Clumsiness, headache, and confusion are also possible. Lingering effects the following day may include dry mouth, dry eyes, fatigue, slowed thinking, and slowed recall.6
ACUTE PHYSICAL EFFECTS
Acute physical effects of cannabis use include a rapid onset of increased airway conductance, decreased intraocular pressure, and conjunctival injection. A single cannabis cigarette can also induce cardiovascular effects including a dose-dependent increase in heart rate and blood pressure. Chronic users, however, can experience a decreased heart rate, lower blood pressure, and postural hypotension.
In a personal communication, colleagues in Colorado—where recreational use of cannabis was legalized in 2012—described a sharp increase (from virtually none) in the number of adults presenting to the emergency department with cannabis intoxication since 2012. Their patients experienced palpitations, light-headedness, and severe ataxia lasting as long as 12 hours, possibly reflecting the greater potency of current cannabis products. Most of these patients required only supportive care.
Other acute adverse cardiovascular reactions that have been reported include atrial fibrillation, ventricular tachycardia, and a fivefold increased risk of myocardial infarction in the 60 minutes following cannabis use, which subsequently drops sharply to baseline levels.10 Investigations into the cardiovascular effects of cannabis are often complicated by concurrent use of other drugs such as tobacco or cocaine. Possible mechanisms of injury include alterations in coronary microcirculation or slowed coronary flow. In fact, one author found that cannabis users with a history of myocardial infarction had a risk of death 4.2 times higher than users with no history of myocardial infarction.11,12
In children, acute toxicity has been reported from a variety of exposures to cannabis and hashish, including a report of an increase in pediatric cannabis exposures following the changes in Colorado state laws.13 Most of these patients had altered mental status ranging from drowsiness to coma; one report describes a child who experienced a first-time seizure. These patients unfortunately often underwent extensive evaluations such as brain imaging and lumbar puncture, and mechanical ventilation to protect the airway. Earlier consideration of cannabis exposure in these patients might have limited unnecessary testing. Supportive care is usually all that is needed, and most of these patients fully recover.13–17
CHRONIC EFFECTS
Cannabinoids cause a variety of adverse effects, but the ultimate risk these changes pose to human health has been difficult to calculate. Long-term studies are confounded by possible inaccuracies of patient self-reporting of cannabis use, poor control of covariates, and disparate methodologies.
For more than a century, cannabis use has been reported to cause both acute psychotic symptoms and persistent psychotic disorders.18 But the strength of this relationship is modest. Cannabis is more likely a component cause that, in addition to other factors (eg, specific genetic polymorphisms), contributes to the risk of schizophrenia. Individuals with prodromal symptoms and those who have experienced discrete episodes of psychosis related to cannabis use should be discouraged from using cannabis and cannabinoids.19–21
Mounting evidence implicates chronic cannabis use as a cause of long-term medical problems including chronic bronchitis,22 elevated rates of myocardial infarction and dysrhythmias,11 bone loss,23 and cancers at eight different sites including the lung, head, and neck.24 In view of these chronic effects, healthcare providers should caution their patients about cannabis use, as we do about other drugs such as tobacco.
WITHDRAWAL SYNDROME RECOGNIZED
Until recently, neither clinicians nor users recognized a withdrawal syndrome associated with chronic use of cannabis, probably because this syndrome is not as severe as withdrawal from other controlled substances such as opioids or sedative-hypnotics. A number of studies, however, have reported subtle cannabis withdrawal symptoms that are similar to those associated with tobacco withdrawal.
As such, the fifth and latest edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5)25 characterized withdrawal from cannabis use in 2013. The DSM-5 criteria require cessation of heavy or prolonged use of cannabis (ie, daily or almost daily over a period of at least a few months) and three or more of the following withdrawal symptoms:
- Irritability and anger
- Nervousness
- Sleep difficulty or insomnia
- Decreased appetite or weight loss
- Restlessness
- Depressed mood
- Physical symptoms causing discomfort.
Medical treatment of cannabis withdrawal has included a range of antidepressants, mood stabilizers, and alpha-2-adrenergic agonists, all of which have limited success.26 Symptoms of cannabis withdrawal tend to be most intense soon after cessation and decline over the next few weeks.27
CANNABINOID HYPEREMESIS SYNDROME
First reported in 2004,28 cannabinoid hyperemesis syndrome is a recurrent disorder, the pathophysiology of which is poorly understood. It has three phases.
The first phase is a prodrome that may last months or years and is characterized by morning nausea, fear of vomiting, and abdominal discomfort. During this phase, the patient maintains normal eating patterns and may well increase his or her cannabis use due to its well-known antiemetic effects.
The second phase is the hyperemetic phase, characterized by intense, incapacitating emesis with episodes of vomiting throughout the day. These symptoms can be relieved only with frequent hot baths, a feature that distinguishes cannabinoid hyperemesis syndrome from other vomiting syndromes. Hot-water bathing is reported to be a compulsive but learned behavior in which the patient learns that only hot water will provide relief. The extent of relief depends on the temperature of the water—the hotter, the better. Symptoms recur as the water cools.28 Patients often present to the emergency department repeatedly with recurrent symptoms and may remain misdiagnosed or subjected to repeated extensive evaluation including laboratory testing and imaging, which are usually not revealing. If the patient has not been accurately diagnosed, there may be reported weight loss of at least 5 kg.
The third phase, recovery, may take several months to complete, possibly because of the prolonged terminal elimination time of cannabinoids. Complete cessation of cannabis use, including synthetic cannabinoids, is usually necessary.29
Diagnostic criteria for cannabinoid hyperemesis syndrome have been suggested, based on a retrospective case series that included 98 patients.30 The most common features of these affected patients were:
- Severe cyclical vomiting, predominantly in the morning
- Resolution of symptoms with cessation of cannabis use
- Symptomatic relief with hot showers or baths
- Abdominal pain
- At least weekly use of cannabis.
Interestingly, long-term cannabis use has been cited as a critical identifying feature of these patients, with the duration of cannabis use ranging from 10 to 16 years.31,32 Other reports show greater variability in duration of cannabis use before the onset of cannabinoid hyperemesis syndrome. In the large study noted above,30 32% of users reported their duration of cannabis use to be less than 1 year, rendering this criterion less useful.
How can cannabis both cause and prevent vomiting?
The body controls nausea and vomiting via complex circuitry in the brain and gut that involves many neurotransmitters (eg, dopamine, serotonin, substance P) that interact with receptors such as CB1, 5-HT1–4, alpha adrenergic receptors, and mu receptors. Interestingly, cannabis use has antiemetic properties mediated by CB1 with a still unclear additional role of CB2 receptors. Data point to the existence of an underlying antiemetic tone mediated by the endocannabinoid system.
Unfortunately, the mechanism by which cannabinoid hyperemesis syndrome occurs is unknown and represents a paradoxical effect against the otherwise antiemetic effects of cannabis. Several theories have been proposed, including delayed gastric emptying, although only a third of patients demonstrated this on scintigraphy in one study.30 Other theories include disturbance of the hypothalamic-pituitary axis, a buildup of highly lipophilic THC in the brain, and a down-regulation of cannabinoid receptors that results from chronic exposure.30 Given that this syndrome has been recognized only relatively recently, one author has suggested the cause may be recent horticultural developments.5
Treating cannabinoid hyperemesis syndrome is difficult
Treatment of cannabinoid hyperemesis syndrome is notoriously difficult, with many authors reporting resistance to the usual first-line antiemetic drugs. Generally, treatment should include hydration and acid-suppression therapy because endoscopic evaluation of several patients has revealed varying degrees of esophagitis and gastritis.29
Antiemetic therapy should target receptors known to mediate nausea and vomiting. In some cases, antiemetic drugs are more effective when used in combination. Agents include the serotonergic receptor antagonists ondansetron and granisetron, the dopamine antagonists prochlorperazine and metoclopramide, and even haloperidol.33,34 Benzodiazepines may be effective by causing sedation, anxiolysis, and depression of the vomiting center.34,35 Two antihistamines—dimenhydrinate and diphenhydramine—have antiemetic effects, perhaps by inhibiting acetylcholine.34
Aprepitant is a neurokinin-1 antagonist that inhibits the action of substance P. When combined with a corticosteroid and a serotonin antagonist, it relieves nausea and vomiting in chemotherapy patients.34,36
Corticosteroids such as dexamethasone are potent antiemetics thought to inhibit prostaglandin synthesis.34
Capsaicin cream applied to the abdomen has also been reported to relieve symptoms, possibly through an interaction between the TRPv1 receptor and the endocannabinoid system.37,38
DIAGNOSTIC TESTING
Cannabinoids are detectable in plasma and urine, with urine testing being more common.
Common laboratory methods include the enzyme-multiplied immunoassay technique (EMIT) and radioimmunoassay. Gas chromatography coupled with mass spectrometry is the most specific assay; it is used for confirmation and is the reference method.
EMIT is a qualitative urine test that detects 9-carboxy-THC as well as other THC metabolites. These urine tests detect all metabolites, and the result is reported as positive if the total concentration is greater than or equal to a prespecified threshold level, such as 20 ng/mL or 50 ng/mL. A positive test does not denote intoxication, nor does the test identify the source of THC (eg, cannabis, dronabinol, butane hash oil). EMIT does not detect nabilone. The National Institute on Drug Abuse guidelines for urine testing specify a test threshold concentration of 50 ng/mL for screening and 15 ng/mL for confirmation.
Many factors affect the detection of THC metabolites and their presence and duration in urine: dose, duration of use, route of exposure, hydration status, urine volume and concentration, and urine pH. THC metabolites have been detected in urine using gas chromatography-mass spectrometry for up to 7 days after smoking one marijuana cigarette.7 Chronic users have also been reported to have positive urine EMIT tests for up to 46 days after cannabis cessation.39 Detection may be further complicated in chronic users: in one study, users produced both negative and positive specimens over 24 days, suggesting that diet and exercise may influence clearance.40 Also, many factors are known to produce false-positive and false-negative results for these immunoassays (Table 1).39,41
In the United States, penalties for driving under the influence of cannabis vary from state to state, and laws specify plasma testing for quantitative analysis. Some states use a threshold of 5 ng/mL in plasma to imply driving under the influence, whereas others use any detectable amount. Currently, there are no generally accepted guidelines for storage and testing of blood samples, despite the known instability of analytes.42
Saliva, hair, and sweat can also be used for cannabinoid testing. Saliva is easy to collect, can be tested for metabolites to rule out passive cannabis exposure, and can be positive for up to 1 day after exposure. Calculating a blood or plasma concentration from a saliva sample is not possible, however.
Hair testing can also rule out passive exposure, but THC binds very little to melanin, resulting in very low concentrations requiring sensitive tests, such as gas chromatography with tandem mass spectrometry.
Only one device is commercially available for sweat testing; further work is needed to elucidate sweat excretion pharmacokinetics and the limitations of the collection devices.43
CLINICAL MANAGEMENT IS GENERALLY SUPPORTIVE
Historically, clinical toxicity from recreational cannabis use is rarely serious or severe and generally responds to supportive care. Reports of cannabis exposure to poison centers are one-tenth of those reported for ethanol exposures annually.44 Gastrointestinal decontamination with activated charcoal is not recommended, even for orally administered cannabis, since the risks outweigh the expected benefits. Agitation or anxiety may be treated with benzodiazepines as needed. There is no antidote for cannabis toxicity. The ever-increasing availability of high-concentration THC preparations may prompt more aggressive supportive measures in the future.
SYNTHETIC MARIJUANA ALTERNATIVES
Available since the early 2000s, herbal marijuana alternatives are legally sold as incense or potpourri and are often labeled “not for human consumption.” They are known by such brand names as K2 and Spice and contain blends of herbs adulterated with synthetic cannabinoid chemicals developed by researchers exploring the receptor-ligand binding of the endocannabinoid system.
Clinical effects, generally psychiatric, include paranoia, anxiety, agitation, delusions, and psychosis. There are also reports of patients who arrive with sympathomimetic toxicity, some of whom develop bradycardia and hypotension, and some who progress to acute renal failure, seizures, and death. Detection of these products is difficult as they do not react on EMIT testing for THC metabolites and require either gas chromatography-mass spectrometry or liquid chromatography with tandem mass spectrometry.45–48
Clinicians may be encountering more cannabis users than before, and may be encountering users with complications hitherto unseen. Several trends may explain this phenomenon: the legal status of cannabis is changing, cannabis today is more potent than in the past, and enthusiasts are conjuring new ways to enjoy this substance.
This article discusses the history, pharmacology, and potential complications of cannabis use.
A LONG AND TANGLED HISTORY
Cannabis is a broad term that refers to the cannabis plant and its preparations, such as marijuana and hashish, as well as to a family of more than 60 bioactive substances called cannabinoids. It is the most commonly used illegal drug in the world, with an estimated 160 million users. Each year, about 2.4 million people in the United States use it for the first time.1,2
Cannabis has been used throughout the world for recreational and spiritual purposes for nearly 5,000 years, beginning with the fabled Celestial Emperors of China. The tangled history of cannabis in America began in the 17th century, when farmers were required by law to grow it as a fiber crop. It later found its way into the US Pharmacopeia for a wide range of indications. During the long prelude to Prohibition in the latter half of the 19th century, the US government became increasingly suspicious of mind-altering substances and began restricting its prescription in 1934, culminating in its designation by the US Food and Drug Administration as a schedule I controlled substance in 1970.
Investigation into the potential medical uses for the different chemicals within cannabis is ongoing, as is debate over its changing legality and usefulness to society. The apparent cognitive dissonance surrounding the use and advocacy of medical marijuana is beyond the scope of this review,3 which will instead restrict itself to what is known of the cannabinoids and to the recreational use of cannabis.
THC IS THE PRINCIPAL PSYCHOACTIVE MOLECULE
Delta-9 tetrahydrocannabinol (THC), first isolated in 1964, was identified as the principal psychoactive constituent of cannabis in 2002.4
Two G-protein–linked cannabinoid receptors cloned in the 1990s—CB1 and CB2—were found to be a part of a system of endocannabinoid receptors present throughout the body, from the brain to the immune system to the vas deferens.5 Both receptors inhibit cellular excitation by activating inwardly rectifying potassium channels. These receptors are mostly absent in the brainstem, which may explain why cannabis use rarely causes life-threatening autonomic dysfunction. Although the intoxicating effects of marijuana are mediated by CB1 receptors, the specific mechanisms underlying the cannabis “high” are unclear.6
CANNABINOIDS ARE LIPID-SOLUBLE
The rate of absorption of cannabinoids depends on the route of administration and the type of cannabis product used. When cannabis products are smoked, up to 35% of THC is available, and the average time to peak serum concentration is 8 minutes.7 The peak concentration depends on the dose.
On the other hand, when cannabis products (eg, nabilone, dronabinol) are ingested, absorption is unpredictable because THC is unstable in gastric acid and undergoes first-pass metabolism in the liver, which reduces the drug’s bioavailability. Up to 20% of an ingested dose of THC is absorbed, and the time to peak serum concentration averages between 2 and 4 hours. Consequently, many users prefer to smoke cannabis as a means to control the desired effects.
Cannabinoids are lipid-soluble. They accumulate in fatty tissue in a biphasic pattern, initially moving into highly vascularized tissue such as the liver before accumulating in less well-vascularized tissue such as fat. They are then slowly released from fatty tissue as the fat turns over. THC itself has a volume of distribution of about 2.5 to 3.5 L/kg. It crosses the placenta and enters breast milk.8
THC is metabolized by the cytochrome P450 system, primarily by the enzymes CYP2C9 and CYP3A4. Its primary metabolite, 11-hydroxy-delta-9 THC, is also active, but subsequent metabolism produces many other inactive metabolites. THC is eliminated in feces and urine, and its half-life ranges from 2 to nearly 60 hours.8
A LITTLE ABOUT PLANTS AND STREET NAMES
The plant from which THC and nearly a hundred other chemicals, including cannabinoids, are derived has been called many things over the years:
Hemp is a tall fibrous plant grown for rope and fabric that was used as legal tender in early America. In the mid-19th century, there were over 16 million acres of hemp plantations. Hemp contains very low THC concentrations.
Cannabis is an annual flowering herb that is predominantly diecious (ie, there are male and female plants). After a centuries-long debate among taxonomists, the two principal species are considered to be C sativa and C indica, although today many cannabis cultivars are grown by a great number of breeding enthusiasts.
Concentrations of THC vary widely among cannabis cultivars, ranging historically from around 5% to today’s highly selectively bred species containing more than 30%. Concentrations in seized cannabis have been measured as high as 37%, although the average is around 11%.9 This concentration is defined by the percent of THC per dried mass of plant material tested, usually via gas chromatography.
Hashish is a solid or resinous preparation of the trichomes, or glandular hairs, that grow on the cannabis plant, chiefly on its flowers. Various methods to separate the trichomes from the rest of the plant result in a powder called kief that is then compressed into blocks or bricks. THC concentrations as high as 66% have been measured in nondomestic sources of hashish.9
Hash oil is a further purification, produced by using solvents to dissolve the resin and by filtering out remaining plant material. Evaporating the solvent produces hash oil, sometimes called butane hash oil or honey oil. This process has recently led to an increasing number of home explosions, as people attempt to make the product themselves but do not take suitable precautions when using flammable solvents such as butane. THC concentrations as high as 81% have been measured in nondomestic sources of hash oil.9
Other names for hash oil are dab, wax, and budder. Cannabis enthusiasts refer to the use of hash oil as dabbing, which involves heating a small amount (dab) of the product using a variety of paraphernalia and inhaling the vapor.
IT’S ALL ABOUT GETTING HIGH
For recreational users, the experience has always been about being intoxicated—getting high. The psychological effects range broadly from positive to negative and vary both within and between users, depending on the dose and route of administration. Additional factors that influence the psychological effects include the social and physical settings of drug use and even the user’s expectations. One user’s high is another user’s acute toxic effect.
Although subjective reports of the cannabis experience vary greatly, it typically begins with a feeling of dizziness or lightheadedness followed by a relaxed calm and a feeling of being somewhat “disconnected.” There is a quickening of the sense of humor, described by some as a fatuous euphoria; often there is silly giggling. Awareness of the senses and of music may be increased. Appetite increases, and time seems to pass quickly. Eventually, the user becomes drowsy and experiences decreased attention and difficulty maintaining a coherent conversation. Slowed reaction time and decreased psychomotor activity may also occur. The user may drift into daydreams and eventually fall asleep.
Common negative acute effects of getting high can include mild to severe anxiety and feeling tense or agitated. Clumsiness, headache, and confusion are also possible. Lingering effects the following day may include dry mouth, dry eyes, fatigue, slowed thinking, and slowed recall.6
ACUTE PHYSICAL EFFECTS
Acute physical effects of cannabis use include a rapid onset of increased airway conductance, decreased intraocular pressure, and conjunctival injection. A single cannabis cigarette can also induce cardiovascular effects including a dose-dependent increase in heart rate and blood pressure. Chronic users, however, can experience a decreased heart rate, lower blood pressure, and postural hypotension.
In a personal communication, colleagues in Colorado—where recreational use of cannabis was legalized in 2012—described a sharp increase (from virtually none) in the number of adults presenting to the emergency department with cannabis intoxication since 2012. Their patients experienced palpitations, light-headedness, and severe ataxia lasting as long as 12 hours, possibly reflecting the greater potency of current cannabis products. Most of these patients required only supportive care.
Other acute adverse cardiovascular reactions that have been reported include atrial fibrillation, ventricular tachycardia, and a fivefold increased risk of myocardial infarction in the 60 minutes following cannabis use, which subsequently drops sharply to baseline levels.10 Investigations into the cardiovascular effects of cannabis are often complicated by concurrent use of other drugs such as tobacco or cocaine. Possible mechanisms of injury include alterations in coronary microcirculation or slowed coronary flow. In fact, one author found that cannabis users with a history of myocardial infarction had a risk of death 4.2 times higher than users with no history of myocardial infarction.11,12
In children, acute toxicity has been reported from a variety of exposures to cannabis and hashish, including a report of an increase in pediatric cannabis exposures following the changes in Colorado state laws.13 Most of these patients had altered mental status ranging from drowsiness to coma; one report describes a child who experienced a first-time seizure. These patients unfortunately often underwent extensive evaluations such as brain imaging and lumbar puncture, and mechanical ventilation to protect the airway. Earlier consideration of cannabis exposure in these patients might have limited unnecessary testing. Supportive care is usually all that is needed, and most of these patients fully recover.13–17
CHRONIC EFFECTS
Cannabinoids cause a variety of adverse effects, but the ultimate risk these changes pose to human health has been difficult to calculate. Long-term studies are confounded by possible inaccuracies of patient self-reporting of cannabis use, poor control of covariates, and disparate methodologies.
For more than a century, cannabis use has been reported to cause both acute psychotic symptoms and persistent psychotic disorders.18 But the strength of this relationship is modest. Cannabis is more likely a component cause that, in addition to other factors (eg, specific genetic polymorphisms), contributes to the risk of schizophrenia. Individuals with prodromal symptoms and those who have experienced discrete episodes of psychosis related to cannabis use should be discouraged from using cannabis and cannabinoids.19–21
Mounting evidence implicates chronic cannabis use as a cause of long-term medical problems including chronic bronchitis,22 elevated rates of myocardial infarction and dysrhythmias,11 bone loss,23 and cancers at eight different sites including the lung, head, and neck.24 In view of these chronic effects, healthcare providers should caution their patients about cannabis use, as we do about other drugs such as tobacco.
WITHDRAWAL SYNDROME RECOGNIZED
Until recently, neither clinicians nor users recognized a withdrawal syndrome associated with chronic use of cannabis, probably because this syndrome is not as severe as withdrawal from other controlled substances such as opioids or sedative-hypnotics. A number of studies, however, have reported subtle cannabis withdrawal symptoms that are similar to those associated with tobacco withdrawal.
As such, the fifth and latest edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5)25 characterized withdrawal from cannabis use in 2013. The DSM-5 criteria require cessation of heavy or prolonged use of cannabis (ie, daily or almost daily over a period of at least a few months) and three or more of the following withdrawal symptoms:
- Irritability and anger
- Nervousness
- Sleep difficulty or insomnia
- Decreased appetite or weight loss
- Restlessness
- Depressed mood
- Physical symptoms causing discomfort.
Medical treatment of cannabis withdrawal has included a range of antidepressants, mood stabilizers, and alpha-2-adrenergic agonists, all of which have limited success.26 Symptoms of cannabis withdrawal tend to be most intense soon after cessation and decline over the next few weeks.27
CANNABINOID HYPEREMESIS SYNDROME
First reported in 2004,28 cannabinoid hyperemesis syndrome is a recurrent disorder, the pathophysiology of which is poorly understood. It has three phases.
The first phase is a prodrome that may last months or years and is characterized by morning nausea, fear of vomiting, and abdominal discomfort. During this phase, the patient maintains normal eating patterns and may well increase his or her cannabis use due to its well-known antiemetic effects.
The second phase is the hyperemetic phase, characterized by intense, incapacitating emesis with episodes of vomiting throughout the day. These symptoms can be relieved only with frequent hot baths, a feature that distinguishes cannabinoid hyperemesis syndrome from other vomiting syndromes. Hot-water bathing is reported to be a compulsive but learned behavior in which the patient learns that only hot water will provide relief. The extent of relief depends on the temperature of the water—the hotter, the better. Symptoms recur as the water cools.28 Patients often present to the emergency department repeatedly with recurrent symptoms and may remain misdiagnosed or subjected to repeated extensive evaluation including laboratory testing and imaging, which are usually not revealing. If the patient has not been accurately diagnosed, there may be reported weight loss of at least 5 kg.
The third phase, recovery, may take several months to complete, possibly because of the prolonged terminal elimination time of cannabinoids. Complete cessation of cannabis use, including synthetic cannabinoids, is usually necessary.29
Diagnostic criteria for cannabinoid hyperemesis syndrome have been suggested, based on a retrospective case series that included 98 patients.30 The most common features of these affected patients were:
- Severe cyclical vomiting, predominantly in the morning
- Resolution of symptoms with cessation of cannabis use
- Symptomatic relief with hot showers or baths
- Abdominal pain
- At least weekly use of cannabis.
Interestingly, long-term cannabis use has been cited as a critical identifying feature of these patients, with the duration of cannabis use ranging from 10 to 16 years.31,32 Other reports show greater variability in duration of cannabis use before the onset of cannabinoid hyperemesis syndrome. In the large study noted above,30 32% of users reported their duration of cannabis use to be less than 1 year, rendering this criterion less useful.
How can cannabis both cause and prevent vomiting?
The body controls nausea and vomiting via complex circuitry in the brain and gut that involves many neurotransmitters (eg, dopamine, serotonin, substance P) that interact with receptors such as CB1, 5-HT1–4, alpha adrenergic receptors, and mu receptors. Interestingly, cannabis use has antiemetic properties mediated by CB1 with a still unclear additional role of CB2 receptors. Data point to the existence of an underlying antiemetic tone mediated by the endocannabinoid system.
Unfortunately, the mechanism by which cannabinoid hyperemesis syndrome occurs is unknown and represents a paradoxical effect against the otherwise antiemetic effects of cannabis. Several theories have been proposed, including delayed gastric emptying, although only a third of patients demonstrated this on scintigraphy in one study.30 Other theories include disturbance of the hypothalamic-pituitary axis, a buildup of highly lipophilic THC in the brain, and a down-regulation of cannabinoid receptors that results from chronic exposure.30 Given that this syndrome has been recognized only relatively recently, one author has suggested the cause may be recent horticultural developments.5
Treating cannabinoid hyperemesis syndrome is difficult
Treatment of cannabinoid hyperemesis syndrome is notoriously difficult, with many authors reporting resistance to the usual first-line antiemetic drugs. Generally, treatment should include hydration and acid-suppression therapy because endoscopic evaluation of several patients has revealed varying degrees of esophagitis and gastritis.29
Antiemetic therapy should target receptors known to mediate nausea and vomiting. In some cases, antiemetic drugs are more effective when used in combination. Agents include the serotonergic receptor antagonists ondansetron and granisetron, the dopamine antagonists prochlorperazine and metoclopramide, and even haloperidol.33,34 Benzodiazepines may be effective by causing sedation, anxiolysis, and depression of the vomiting center.34,35 Two antihistamines—dimenhydrinate and diphenhydramine—have antiemetic effects, perhaps by inhibiting acetylcholine.34
Aprepitant is a neurokinin-1 antagonist that inhibits the action of substance P. When combined with a corticosteroid and a serotonin antagonist, it relieves nausea and vomiting in chemotherapy patients.34,36
Corticosteroids such as dexamethasone are potent antiemetics thought to inhibit prostaglandin synthesis.34
Capsaicin cream applied to the abdomen has also been reported to relieve symptoms, possibly through an interaction between the TRPv1 receptor and the endocannabinoid system.37,38
DIAGNOSTIC TESTING
Cannabinoids are detectable in plasma and urine, with urine testing being more common.
Common laboratory methods include the enzyme-multiplied immunoassay technique (EMIT) and radioimmunoassay. Gas chromatography coupled with mass spectrometry is the most specific assay; it is used for confirmation and is the reference method.
EMIT is a qualitative urine test that detects 9-carboxy-THC as well as other THC metabolites. These urine tests detect all metabolites, and the result is reported as positive if the total concentration is greater than or equal to a prespecified threshold level, such as 20 ng/mL or 50 ng/mL. A positive test does not denote intoxication, nor does the test identify the source of THC (eg, cannabis, dronabinol, butane hash oil). EMIT does not detect nabilone. The National Institute on Drug Abuse guidelines for urine testing specify a test threshold concentration of 50 ng/mL for screening and 15 ng/mL for confirmation.
Many factors affect the detection of THC metabolites and their presence and duration in urine: dose, duration of use, route of exposure, hydration status, urine volume and concentration, and urine pH. THC metabolites have been detected in urine using gas chromatography-mass spectrometry for up to 7 days after smoking one marijuana cigarette.7 Chronic users have also been reported to have positive urine EMIT tests for up to 46 days after cannabis cessation.39 Detection may be further complicated in chronic users: in one study, users produced both negative and positive specimens over 24 days, suggesting that diet and exercise may influence clearance.40 Also, many factors are known to produce false-positive and false-negative results for these immunoassays (Table 1).39,41
In the United States, penalties for driving under the influence of cannabis vary from state to state, and laws specify plasma testing for quantitative analysis. Some states use a threshold of 5 ng/mL in plasma to imply driving under the influence, whereas others use any detectable amount. Currently, there are no generally accepted guidelines for storage and testing of blood samples, despite the known instability of analytes.42
Saliva, hair, and sweat can also be used for cannabinoid testing. Saliva is easy to collect, can be tested for metabolites to rule out passive cannabis exposure, and can be positive for up to 1 day after exposure. Calculating a blood or plasma concentration from a saliva sample is not possible, however.
Hair testing can also rule out passive exposure, but THC binds very little to melanin, resulting in very low concentrations requiring sensitive tests, such as gas chromatography with tandem mass spectrometry.
Only one device is commercially available for sweat testing; further work is needed to elucidate sweat excretion pharmacokinetics and the limitations of the collection devices.43
CLINICAL MANAGEMENT IS GENERALLY SUPPORTIVE
Historically, clinical toxicity from recreational cannabis use is rarely serious or severe and generally responds to supportive care. Reports of cannabis exposure to poison centers are one-tenth of those reported for ethanol exposures annually.44 Gastrointestinal decontamination with activated charcoal is not recommended, even for orally administered cannabis, since the risks outweigh the expected benefits. Agitation or anxiety may be treated with benzodiazepines as needed. There is no antidote for cannabis toxicity. The ever-increasing availability of high-concentration THC preparations may prompt more aggressive supportive measures in the future.
SYNTHETIC MARIJUANA ALTERNATIVES
Available since the early 2000s, herbal marijuana alternatives are legally sold as incense or potpourri and are often labeled “not for human consumption.” They are known by such brand names as K2 and Spice and contain blends of herbs adulterated with synthetic cannabinoid chemicals developed by researchers exploring the receptor-ligand binding of the endocannabinoid system.
Clinical effects, generally psychiatric, include paranoia, anxiety, agitation, delusions, and psychosis. There are also reports of patients who arrive with sympathomimetic toxicity, some of whom develop bradycardia and hypotension, and some who progress to acute renal failure, seizures, and death. Detection of these products is difficult as they do not react on EMIT testing for THC metabolites and require either gas chromatography-mass spectrometry or liquid chromatography with tandem mass spectrometry.45–48
- Substance Abuse and Mental Health Services Administration. Results from the 2012 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-46, HHS Publication No. (SMA) 13-4795. www.samhsa.gov/data/sites/default/files/NSDUHresultsPDFWHTML2013/Web/NSDUHresults2013.pdf. Accessed October 2, 2015.
- United Nations Office on Drugs and Crime. 2008 World Drug Report. www.unodc.org/documents/wdr/WDR_2008/WDR_2008_eng_web.pdf. Accessed October 2, 2015.
- American Society of Addiction Medicine (ASAM). Public policy statement on medical marijuana. www.asam.org/docs/publicy-policy-statements/1medical-marijuana-4-10.pdf?sfvrsn=0. Accessed October 2, 2015.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002; 54:161–202.
- Sharkey KA, Darmani NA, Parker LA. Regulation of nausea and vomiting by cannabinoids and the endocannabinoid system. Eur J Pharmacol 2014; 722:134–146.
- Iversen L. Cannabis and the brain. Brain 2003; 126:1252–1270.
- Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol 1992; 16:276–282.
- Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet 2003; 42:327–360.
- Mehmedic Z, Chandra S, Slade D, et al. Potency trends of Δ9-THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. J Forensic Sci 2010; 55:1209–1217.
- Mittleman MA, Lewis RA, Maclure M, Sherwood JB, Muller JE. Triggering myocardial infarction by marijuana. Circulation 2001; 103:2805–2809.
- Mukamal KJ, Maclure M, Muller JE, Mittleman MA. An exploratory prospective study of marijuana use and mortality following acute myocardial infarction. Am Heart J 2008; 155:465–470.
- Thomas G, Kloner RA, Rezkalla S. Adverse cardiovascular, cerebrovascular, and peripheral vascular effects of marijuana inhalation: what cardiologists need to know. Am J Cardiol 2014; 113:187–190.
- Wang GS, Roosevelt G, Heard K. Pediatric marijuana exposures in a medical marijuana state. JAMA Pediatr 2013; 167:630–633.
- Carstairs SD, Fujinaka MK, Keeney GE, Ly BT. Prolonged coma in a child due to hashish ingestion with quantitation of THC metabolites in urine. J Emerg Med 2011; 41:e69–e71.
- Le Garrec S, Dauger S, Sachs P. Cannabis poisoning in children. Intensive Care Med 2014; 40:1394–1395.
- Ragab AR, Al-Mazroua MK. Passive cannabis smoking resulting in coma in a 16-month old infant. J Clin Case Rep 2012;2:237.
- Robinson K. Beyond resinable doubt? J Clin Forensic Med 2005;12:164–166.
- Burns JK. Pathways from cannabis to psychosis: a review of the evidence. Front Psychiatry 2013;4:128.
- Di Forti M, Sallis H, Allegri F, et al. Daily use, especially of high-potency cannabis, drives the earlier onset of psychosis in cannabis users. Schizophr Bull 2014; 40:1509–1517.
- Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328.
- Wilkinson ST, Radhakrishnan R, D'Souza DC. Impact of cannabis use on the development of psychotic disorders. Curr Addict Rep 2014;1:115–128.
- Aldington S, Williams M, Nowitz M, et al. Effects of cannabis on pulmonary structure, function and symptoms. Thorax 2007; 62:1058–1063.
- George KL, Saltman LH, Stein GS, Lian JB, Zurier RB. Ajulemic acid, a nonpsychoactive cannabinoid acid, suppresses osteoclastogenesis in mononuclear precursor cells and induces apoptosis in mature osteoclast-like cells. J Cell Physiol 2008; 214:714–720.
- Reece AS. Chronic toxicology of cannabis. Clin Toxicol (Phila) 2009; 47:517–524.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
- Allsop DJ, Copeland J, Lintzeris N, et al. Nabiximols as an agonist replacement therapy during cannabis withdrawal: a randomized clinical trial. JAMA Psychiatry 2014; 71:281–291.
- Hesse M, Thylstrup B. Time-course of the DSM-5 cannabis withdrawal symptoms in poly-substance abusers. BMC Psychiatry 2013; 13:258.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Galli JA, Sawaya RA, Friedenberg FK. Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev 2011; 4:241–249.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci 2010; 55:3113–3119.
- Wallace EA, Andrews SE, Garmany CL, Jelley MJ. Cannabinoid hyperemesis syndrome: literature review and proposed diagnosis and treatment algorithm. South Med J 2011; 104:659–664.
- Hickey JL, Witsil JC, Mycyk MB. Haloperidol for treatment of cannabinoid hyperemesis syndrome. Am J Emerg Med 2013; 31:1003.e5–1003.e6.
- Perwitasari DA, Gelderblom H, Atthobari J, et al. Anti-emetic drugs in oncology: pharmacology and individualization by pharmacogenetics. Int J Clin Pharm 2011; 33:33–43.
- Cox B, Chhabra A, Adler M, Simmons J, Randlett D. Cannabinoid hyperemesis syndrome: case report of a paradoxical reaction with heavy marijuana use. Case Rep Med 2012; 2012:757696.
- Sakurai M, Mori T, Kato J, et al. Efficacy of aprepitant in preventing nausea and vomiting due to high-dose melphalan-based conditioning for allogeneic hematopoietic stem cell transplantation. Int J Hematol 2014; 99:457–462.
- Lapoint J. Case series of patients treated for cannabinoid hyperemesis syndrome with capsaicin cream. Clin Tox 2014; 52:707. Abstract #53.
- Biary R, Oh A, Lapoint J, Nelson LS, Hoffman RS, Howland MA. Topical capsaicin cream used as a therapy for cannabinoid hyperemesis syndrome. Clin Tox 2014; 52:787. Abstract #232.
- Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc 2008; 83:66–76.
- Lowe RH, Abraham TT, Darwin WD, Herning R, Cadet JL, Huestis MA. Extended urinary delta9-tetrahydrocannabinol excretion in chronic cannabis users precludes use as a biomarker of new drug exposure. Drug Alcohol Depend 2009; 105:24–32.
- Paul BD, Jacobs A. Effects of oxidizing adulterants on detection of 11-nor-delta9-THC-9-carboxylic acid in urine. J Anal Toxicol 2002; 26:460–463.
- Schwope DM, Karschner EL, Gorelick DA, Huestis MA. Identification of recent cannabis use: whole-blood and plasma free and glucuronidated cannabinoid pharmacokinetics following controlled smoked cannabis administration. Clin Chem 2011; 57:1406-1414.
- Huestis MA, Smith ML. Cannabinoid pharmacokinetics and disposition in alternative matrices. In: Pertwee R, ed. Handbook of Cannabis. Oxford, United Kingdom: Oxford University Press; 2014:296–316.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila) 2013; 51:949–1229.
- Rosenbaum CD, Carreiro SP, Babu KM. Here today, gone tomorrow…and back again? A review of herbal marijuana alternatives (K2, Spice), synthetic cathinones (bath salts), kratom, Salvia divinorum, methoxetamine, and piperazines. J Med Toxicol 2012; 8:15–32.
- Gurney SMR, Scott KS, Kacinko SL, Presley BC, Logan BK. Pharmacology, toxicology, and adverse effects of synthetic cannabinoid drugs. Forensic Sci Rev 2014; 26:53–78.
- McKeever RG, Vearrier D, Jacobs D, LaSala G, Okaneku J, Greenberg MI. K2-not the spice of life; synthetic cannabinoids and ST elevation myocardial infarction: a case report. J Med Toxicol 2015; 11:129–131.
- Schneir AB, Baumbacher T. Convulsions associated with the use of a synthetic cannabinoid product. J Med Toxicol 2012; 8:62–64.
- Substance Abuse and Mental Health Services Administration. Results from the 2012 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-46, HHS Publication No. (SMA) 13-4795. www.samhsa.gov/data/sites/default/files/NSDUHresultsPDFWHTML2013/Web/NSDUHresults2013.pdf. Accessed October 2, 2015.
- United Nations Office on Drugs and Crime. 2008 World Drug Report. www.unodc.org/documents/wdr/WDR_2008/WDR_2008_eng_web.pdf. Accessed October 2, 2015.
- American Society of Addiction Medicine (ASAM). Public policy statement on medical marijuana. www.asam.org/docs/publicy-policy-statements/1medical-marijuana-4-10.pdf?sfvrsn=0. Accessed October 2, 2015.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002; 54:161–202.
- Sharkey KA, Darmani NA, Parker LA. Regulation of nausea and vomiting by cannabinoids and the endocannabinoid system. Eur J Pharmacol 2014; 722:134–146.
- Iversen L. Cannabis and the brain. Brain 2003; 126:1252–1270.
- Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol 1992; 16:276–282.
- Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet 2003; 42:327–360.
- Mehmedic Z, Chandra S, Slade D, et al. Potency trends of Δ9-THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. J Forensic Sci 2010; 55:1209–1217.
- Mittleman MA, Lewis RA, Maclure M, Sherwood JB, Muller JE. Triggering myocardial infarction by marijuana. Circulation 2001; 103:2805–2809.
- Mukamal KJ, Maclure M, Muller JE, Mittleman MA. An exploratory prospective study of marijuana use and mortality following acute myocardial infarction. Am Heart J 2008; 155:465–470.
- Thomas G, Kloner RA, Rezkalla S. Adverse cardiovascular, cerebrovascular, and peripheral vascular effects of marijuana inhalation: what cardiologists need to know. Am J Cardiol 2014; 113:187–190.
- Wang GS, Roosevelt G, Heard K. Pediatric marijuana exposures in a medical marijuana state. JAMA Pediatr 2013; 167:630–633.
- Carstairs SD, Fujinaka MK, Keeney GE, Ly BT. Prolonged coma in a child due to hashish ingestion with quantitation of THC metabolites in urine. J Emerg Med 2011; 41:e69–e71.
- Le Garrec S, Dauger S, Sachs P. Cannabis poisoning in children. Intensive Care Med 2014; 40:1394–1395.
- Ragab AR, Al-Mazroua MK. Passive cannabis smoking resulting in coma in a 16-month old infant. J Clin Case Rep 2012;2:237.
- Robinson K. Beyond resinable doubt? J Clin Forensic Med 2005;12:164–166.
- Burns JK. Pathways from cannabis to psychosis: a review of the evidence. Front Psychiatry 2013;4:128.
- Di Forti M, Sallis H, Allegri F, et al. Daily use, especially of high-potency cannabis, drives the earlier onset of psychosis in cannabis users. Schizophr Bull 2014; 40:1509–1517.
- Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328.
- Wilkinson ST, Radhakrishnan R, D'Souza DC. Impact of cannabis use on the development of psychotic disorders. Curr Addict Rep 2014;1:115–128.
- Aldington S, Williams M, Nowitz M, et al. Effects of cannabis on pulmonary structure, function and symptoms. Thorax 2007; 62:1058–1063.
- George KL, Saltman LH, Stein GS, Lian JB, Zurier RB. Ajulemic acid, a nonpsychoactive cannabinoid acid, suppresses osteoclastogenesis in mononuclear precursor cells and induces apoptosis in mature osteoclast-like cells. J Cell Physiol 2008; 214:714–720.
- Reece AS. Chronic toxicology of cannabis. Clin Toxicol (Phila) 2009; 47:517–524.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
- Allsop DJ, Copeland J, Lintzeris N, et al. Nabiximols as an agonist replacement therapy during cannabis withdrawal: a randomized clinical trial. JAMA Psychiatry 2014; 71:281–291.
- Hesse M, Thylstrup B. Time-course of the DSM-5 cannabis withdrawal symptoms in poly-substance abusers. BMC Psychiatry 2013; 13:258.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Galli JA, Sawaya RA, Friedenberg FK. Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev 2011; 4:241–249.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci 2010; 55:3113–3119.
- Wallace EA, Andrews SE, Garmany CL, Jelley MJ. Cannabinoid hyperemesis syndrome: literature review and proposed diagnosis and treatment algorithm. South Med J 2011; 104:659–664.
- Hickey JL, Witsil JC, Mycyk MB. Haloperidol for treatment of cannabinoid hyperemesis syndrome. Am J Emerg Med 2013; 31:1003.e5–1003.e6.
- Perwitasari DA, Gelderblom H, Atthobari J, et al. Anti-emetic drugs in oncology: pharmacology and individualization by pharmacogenetics. Int J Clin Pharm 2011; 33:33–43.
- Cox B, Chhabra A, Adler M, Simmons J, Randlett D. Cannabinoid hyperemesis syndrome: case report of a paradoxical reaction with heavy marijuana use. Case Rep Med 2012; 2012:757696.
- Sakurai M, Mori T, Kato J, et al. Efficacy of aprepitant in preventing nausea and vomiting due to high-dose melphalan-based conditioning for allogeneic hematopoietic stem cell transplantation. Int J Hematol 2014; 99:457–462.
- Lapoint J. Case series of patients treated for cannabinoid hyperemesis syndrome with capsaicin cream. Clin Tox 2014; 52:707. Abstract #53.
- Biary R, Oh A, Lapoint J, Nelson LS, Hoffman RS, Howland MA. Topical capsaicin cream used as a therapy for cannabinoid hyperemesis syndrome. Clin Tox 2014; 52:787. Abstract #232.
- Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc 2008; 83:66–76.
- Lowe RH, Abraham TT, Darwin WD, Herning R, Cadet JL, Huestis MA. Extended urinary delta9-tetrahydrocannabinol excretion in chronic cannabis users precludes use as a biomarker of new drug exposure. Drug Alcohol Depend 2009; 105:24–32.
- Paul BD, Jacobs A. Effects of oxidizing adulterants on detection of 11-nor-delta9-THC-9-carboxylic acid in urine. J Anal Toxicol 2002; 26:460–463.
- Schwope DM, Karschner EL, Gorelick DA, Huestis MA. Identification of recent cannabis use: whole-blood and plasma free and glucuronidated cannabinoid pharmacokinetics following controlled smoked cannabis administration. Clin Chem 2011; 57:1406-1414.
- Huestis MA, Smith ML. Cannabinoid pharmacokinetics and disposition in alternative matrices. In: Pertwee R, ed. Handbook of Cannabis. Oxford, United Kingdom: Oxford University Press; 2014:296–316.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila) 2013; 51:949–1229.
- Rosenbaum CD, Carreiro SP, Babu KM. Here today, gone tomorrow…and back again? A review of herbal marijuana alternatives (K2, Spice), synthetic cathinones (bath salts), kratom, Salvia divinorum, methoxetamine, and piperazines. J Med Toxicol 2012; 8:15–32.
- Gurney SMR, Scott KS, Kacinko SL, Presley BC, Logan BK. Pharmacology, toxicology, and adverse effects of synthetic cannabinoid drugs. Forensic Sci Rev 2014; 26:53–78.
- McKeever RG, Vearrier D, Jacobs D, LaSala G, Okaneku J, Greenberg MI. K2-not the spice of life; synthetic cannabinoids and ST elevation myocardial infarction: a case report. J Med Toxicol 2015; 11:129–131.
- Schneir AB, Baumbacher T. Convulsions associated with the use of a synthetic cannabinoid product. J Med Toxicol 2012; 8:62–64.
KEY POINTS
- Cannabis has been used throughout history and has become increasingly available for recreational purposes, despite its current classification as a schedule I controlled substance.
- Although severe acute toxicity has been reported, it is relatively rare, and most users’ casual experiences are benign.
- Internists are most likely to see complications such as cannabinoid hyperemesis syndrome and cardiovascular problems that cannot be resolved sufficiently in the emergency department.
- Screening urine testing is usually done by enzyme multiplied immunoassay, whereas confirmatory testing is done with gas chromatography-mass spectrometry, which is more specific.

Women’s health 2015: An update for the internist
Women's health encompasses a broad range of issues unique to the female patient, with a scope that has expanded beyond reproductive health. Providers who care for women must develop cross-disciplinary competencies and understand the complex role of sex and gender on disease expression and treatment outcomes. Staying current with the literature in this rapidly changing field can be challenging for the busy clinician.
This article reviews recent advances in the treatment of depression in pregnancy, nonhormonal therapies for menopausal symptoms, and heart failure therapy in women, highlighting notable studies published in 2014 and early 2015.
TREATMENT OF DEPRESSION IN PREGNANCY
A 32-year-old woman with well-controlled but recurrent depression presents to the clinic for preconception counseling. Her depression has been successfully managed with a selective serotonin reuptake inhibitor (SSRI). She and her husband would like to try to conceive soon, but she is worried that continuing on her current SSRI may harm her baby. How should you advise her?
Concern for teratogenic effects of SSRIs
Depression is common during pregnancy: 11.8% to 13.5% of pregnant women report symptoms of depression,1 and 7.5% of pregnant women take an antidepressant.2
SSRI use during pregnancy has drawn attention because of mixed reports of teratogenic effects on the newborn, such as omphalocele, congenital heart defects, and craniosynostosis.3 Previous observational studies have specifically linked paroxetine to small but significant increases in right ventricular outflow tract obstruction4,5 and have linked sertraline to ventricular septal defects.6
However, reports of associations of congenital malformations and SSRI use in pregnancy in observational studies have been questioned, with concern that these studies had low statistical power, self-reported data leading to recall bias, and limited assessment for confounding factors.3,7
Recent studies refute risk of cardiac malformations
Several newer studies have been published that further examine the association between SSRI use in pregnancy and congenital heart defects, and their findings suggest that once adjusted for confounding variables, SSRI use in pregnancy may not be associated with cardiac malformations.
Huybrechts et al,8 in a large study published in 2014, extracted data on 950,000 pregnant women from the Medicaid database over a 7-year period and examined it for SSRI use during the first 90 days of pregnancy. Though SSRI use was associated with cardiac malformations when unadjusted for confounding variables (unadjusted relative risk 1.25, 95% confidence interval [CI] 1.13–1.38), once the cohort was restricted to women with a diagnosis of only depression and was adjusted based on propensity scoring, the association was no longer statistically significant (adjusted relative risk 1.06, 95% CI 0.93–1.22).
Additionally, there was no association between sertraline and ventricular septal defects (63 cases in 14,040 women exposed to sertraline, adjusted relative risk 1.04, 95% CI 0.76–1.41), or between paroxetine and right ventricular outflow tract obstruction (93 cases in 11,126 women exposed to paroxetine, adjusted relative risk 1.07, 95% CI 0.59–1.93).8
Furu et al7 conducted a sibling-matched case-control comparison published in 2015, in which more than 2 million live births from five Nordic countries were examined in the full cohort study and 2,288 births in the sibling-matched case-control cohort. SSRI or venlafaxine use in the first 90 days of pregnancy was examined. There was a slightly higher rate of cardiac defects in infants born to SSRI or venlafaxine recipients in the cohort study (adjusted odds ratio 1.15, 95% CI 1.05–1.26). However, in the sibling-controlled analyses, neither an SSRI nor venlafaxine was associated with heart defects (adjusted odds ratio 0.92, 95% CI 0.72–1.17), leading the authors to conclude that there might be familial factors or other lifestyle factors that were not taken into consideration and that could have confounded the cohort results.
Bérard et al9 examined antidepressant use in the first trimester of pregnancy in a cohort of women in Canada and concluded that sertraline was associated with congenital atrial and ventricular defects (risk ratio 1.34; 95% CI 1.02–1.76).9 However, this association should be interpreted with caution, as the Canadian cohort was notably smaller than those in other studies we have discussed, with only 18,493 pregnancies in the total cohort, and this conclusion was drawn from 9 cases of ventricular or atrial septal defects in babies of 366 women exposed to sertraline.
Although at first glance SSRIs may appear to be associated with congenital heart defects, these recent studies are reassuring and suggest that the association may actually not be significant. As with any statistical analysis, thoughtful study design, adequate statistical power, and adjustment for confounding factors must be considered before drawing conclusions.
SSRIs, offspring psychiatric outcomes, and miscarriage rates
Clements et al10 studied a cohort extracted from Partners Healthcare consisting of newborns with autism spectrum disorder, newborns with attention-deficit hyperactivity disorder (ADHD), and healthy matched controls and found that SSRI use during pregnancy was not associated with offspring autism spectrum disorder (adjusted odds ratio 1.10, 95% CI 0.7–1.70). However, they did find an increased risk of ADHD with SSRI use during pregnancy (adjusted odds ratio 1.81, 95% CI 1.22–2.70).
Andersen et al11 examined more than 1 million pregnancies in Denmark and found no difference in risk of miscarriage between women who used an SSRI during pregnancy (adjusted hazard ratio 1.27) and women who discontinued their SSRI at least 3 months before pregnancy (adjusted hazard ratio 1.24, P = .47). The authors concluded that because of the similar rate of miscarriage in both groups, there was no association between SSRI use and miscarriage, and that the small increased risk of miscarriage in both groups could have been attributable to a confounding factor that was not measured.
Should our patient continue her SSRI through pregnancy?
Our patient has recurrent depression, and her risk of relapse with antidepressant cessation is high. Though previous, less well-done studies suggested a small risk of congenital heart defects, recent larger high-quality studies provide significant reassurance that SSRI use in pregnancy is not strongly associated with cardiac malformations. Recent studies also show no association with miscarriage or autism spectrum disorder, though there may be risk of offspring ADHD.
She can be counseled that she may continue on her SSRI during pregnancy and can be reassured that the risk to her baby is small compared with her risk of recurrent or postpartum depression.
NONHORMONAL TREATMENT FOR VASOMOTOR SYMPTOMS OF MENOPAUSE
You see a patient who is struggling with symptoms of menopause. She tells you she has terrible hot flashes day and night, and she would like to try drug therapy. She does not want hormone replacement therapy because she is worried about the risk of adverse events. Are there safe and effective nonhormonal pharmacologic treatments for her vasomotor symptoms?
Paroxetine 7.5 mg is approved for vasomotor symptoms of menopause
As many as 75% of menopausal women in the United States experience vasomotor symptoms related to menopause, or hot flashes and night sweats.12 These symptoms can disrupt sleep and negatively affect quality of life. Though previously thought to occur during a short and self-limited time period, a recently published large observational study reported the median duration of vasomotor symptoms was 7.4 years, and in African American women in the cohort the median duration of vasomotor symptoms was 10.1 years—an entire decade of life.13
In 2013, the US Food and Drug Administration (FDA) approved paroxetine 7.5 mg daily for treating moderate to severe hot flashes associated with menopause. It is the only approved nonhormonal treatment for vasomotor symptoms; the only other approved treatments are estrogen therapy for women who have had a hysterectomy and combination estrogen-progesterone therapy for women who have not had a hysterectomy.
Further studies of paroxetine for menopausal symptoms
Since its approval, further studies have been published supporting the use of paroxetine 7.5 mg in treating symptoms of menopause. In addition to reducing hot flashes, this treatment also improves sleep disturbance in women with menopause.14
Pinkerton et al,14 in a pooled analysis of the data from the phase 3 clinical trials of paroxetine 7.5 mg per day, found that participants in groups assigned to paroxetine reported a 62% reduction in nighttime awakenings due to hot flashes compared with a 43% reduction in the placebo group (P < .001). Those who took paroxetine also reported a statistically significantly greater increase in duration of sleep than those who took placebo (37 minutes in the treatment group vs 27 minutes in the placebo group, P = .03).
Some patients are hesitant to take an SSRI because of concerns about adverse effects when used for psychiatric conditions. However, the dose of paroxetine that was studied and approved for vasomotor symptoms is lower than doses used for psychiatric indications and does not appear to be associated with these adverse effects.
Portman et al15 in 2014 examined the effect of paroxetine 7.5 mg vs placebo on weight gain and sexual function in women with vasomotor symptoms of menopause and found no significant increase in weight or decrease in sexual function at 24 weeks of use. Participants were weighed during study visits, and those in the paroxetine group gained on average 0.48% from baseline at 24 weeks, compared with 0.09% in the placebo group (P = .29).
Sexual dysfunction was assessed using the Arizona Sexual Experience Scale, which has been validated in psychiatric patients using antidepressants, and there was no significant difference in symptoms such as sex drive, sexual arousal, vaginal lubrication, or ability to achieve orgasm between the treatment group and placebo group.15
Of note, paroxetine is a potent inhibitor of the cytochrome P-450 CYP2D6 enzyme, and concurrent use of paroxetine with tamoxifen decreases tamoxifen activity.12,16 Since women with a history of breast cancer who cannot use estrogen for hot flashes may be seeking nonhormonal treatment for their vasomotor symptoms, providers should perform careful medication reconciliation and be aware that concomitant use of paroxetine and tamoxifen is not recommended.
Other antidepressants show promise but are not approved for menopausal symptoms
In addition to paroxetine, other nonhormonal drugs have been studied for treating hot flashes, but they have been unable to secure FDA approval for this indication. One of these is the serotonin-norepinephrine reuptake inhibitor venlafaxine, and a 2014 study17 confirmed its efficacy in treating menopausal vasomotor symptoms.
Joffe et al17 performed a three-armed trial comparing venlafaxine 75 mg/day, estradiol 0.5 mg/day, and placebo and found that both of the active treatments were better than placebo at reducing vasomotor symptoms. Compared with each other, estradiol 0.5 mg/day reduced hot flash frequency by an additional 0.6 events per day compared with venlafaxine 75 mg/day (P = .09). Though this difference was statistically significant, the authors pointed out that the clinical significance of such a small absolute difference is questionable. Additionally, providers should be aware that venlafaxine has little or no effect on the metabolism of tamoxifen.16
Shams et al,18 in a meta-analysis published in 2014, concluded that SSRIs as a class are more effective than placebo in treating hot flashes, supporting their widespread off-label use for this purpose. Their analysis examined the results of 11 studies, which included more than 2,000 patients in total, and found that compared with placebo, SSRI use was associated with a significant decrease in hot flashes (mean difference –0.93 events per day, 95% CI –1.49 to –0.37). A mixed treatment comparison analysis was also performed to try to model performance of individual SSRIs based on the pooled data, and the model suggests that escitalopram may be the most efficacious SSRI at reducing hot flash severity.
These studies support the effectiveness of SSRIs18 and venlafaxine17 in reducing hot flashes compared with placebo, though providers should be aware that they are still not FDA-approved for this indication.
Nonhormonal therapy for our patient
We would recommend paroxetine 7.5 mg nightly to this patient, as it is an FDA-approved nonhormonal medication that has been shown to help patients with vasomotor symptoms of menopause as well as sleep disturbance, without sexual side effects or weight gain. If the patient cannot tolerate paroxetine, off-label use of another SSRI or venlafaxine is supported by the recent literature.
HEART DISEASE IN WOMEN: CARDIAC RESYNCHRONIZATION THERAPY
A 68-year-old woman with a history of nonischemic cardiomyopathy presents for routine follow-up in your office. Despite maximal medical therapy on a beta-blocker, an angiotensin II receptor blocker, and a diuretic, she has New York Heart Association (NYHA) class III symptoms. Her most recent studies showed an ejection fraction of 30% by echocardiography and left bundle-branch block on electrocardiography, with a QRS duration of 140 ms. She recently saw her cardiologist, who recommended cardiac resynchronization therapy, and she wants your opinion as to whether or not to proceed with this recommendation. How should you counsel her?
Which patients are candidates for cardiac resynchronization therapy?
Heart disease continues to be the number one cause of death in the United States for both men and women, and almost the same number of women and men die from heart disease every year.19 Though coronary artery disease accounts for most cases of cardiovascular disease in the United States, heart failure is a significant and growing contributor. Approximately 6.6 million adults had heart failure in 2010 in the United States, and an additional 3 million are projected to have heart failure by 2030.20 The burden of disease on our health system is high, with about 1 million hospitalizations and more than 3 million outpatient office visits attributable to heart failure yearly.20
Patients with heart failure may have symptoms of dyspnea, fatigue, orthopnea, and peripheral edema; laboratory and radiologic findings of pulmonary edema, renal insufficiency, and hyponatremia; and electrocardiographic findings of atrial fibrillation or prolonged QRS.21 Intraventricular conduction delay (QRS duration > 120 ms) is associated with dyssynchronous ventricular contraction and impaired pump function and is present in almost one-third of patients who have advanced heart failure.21
Cardiac resynchronization therapy, or biventricular pacing, can improve symptoms and pump function and has been shown to decrease rates of hospitalization and death in these patients.22 According to the joint 2012 guidelines of the American College of Cardiology Foundation, American Heart Association, and Heart Rhythm Society,22 it is indicated for patients with an ejection fraction of 35% or less, left bundle-branch block with QRS duration of 150 ms or more, and NYHA class II to IV symptoms who are in sinus rhythm (class I recommendation, level of evidence A).
Studies of cardiac resynchronization therapy in women
Recently published studies have suggested that women may derive greater benefit than men from cardiac resynchronization therapy.
Zusterzeel et al23 (2014) evaluated sex-specific data from the National Cardiovascular Data Registry, which contains data on all biventricular pacemaker and implantable cardioverter-defibrillator implantations from 80% of US hospitals.23 Of the 21,152 patients who had left bundle-branch block and received cardiac resynchronization therapy, women derived greater benefit in terms of death than men did, with a 21% lower risk of death than men (adjusted hazard ratio 0.79, 95% CI 0.74–0.84, P < .001). This study was also notable in that 36% of the patients were women, whereas in most earlier studies of cardiac resynchronization therapy women accounted for only 22% to 30% of the study population.22
Goldenberg et al24 (2014) performed a follow-up analysis of the Multicenter Automatic Defibrillator Implantation Trial With Cardiac Resynchronization Therapy. Subgroup analysis showed that although both men and women had a lower risk of death if they received cardiac resynchronization therapy compared with an implantable cardioverter-defibrillator only, the magnitude of benefit may be greater for women (hazard ratio 0.48, 95% CI 0.25–0.91, P = .03) than for men (hazard ratio 0.69, 95% CI 0.50–0.95, P = .02).
In addition to deriving greater mortality benefit, women may actually benefit from cardiac resynchronization therapy at shorter QRS durations than what is currently recommended. Women have a shorter baseline QRS than men, and a smaller left ventricular cavity.25 In an FDA meta-analysis published in August 2014, pooled data from more than 4,000 patients in three studies suggested that women with left bundle-branch block benefited from cardiac resynchronization therapy more than men with left bundle-branch block.26 Neither men nor women with left bundle-branch block benefited from it if their QRS duration was less than 130 ms, and both sexes benefited from it if they had left bundle-branch block and a QRS duration longer than 150 ms. However, women who received it who had left bundle-branch block and a QRS duration of 130 to 149 ms had a significant 76% reduction in the primary composite outcome of a heart failure event or death (hazard ratio 0.24, 95% CI 0.11–0.53, P < .001), while men in the same group did not derive significant benefit (hazard ratio 0.85, 95% CI 0.60–1.21, P = .38).
Despite the increasing evidence that there are sex-specific differences in the benefit from cardiac resynchronization therapy, what we know is limited by the low rates of female enrollment in most of the studies of this treatment. In a systematic review published in 2015, Herz et al27 found that 90% of the 183 studies they reviewed enrolled 35% women or less, and half of the studies enrolled less than 23% women. Furthermore, only 20 of the 183 studies reported baseline characteristics by sex.
Recognizing this lack of adequate data, in August 2014 the FDA issued an official guidance statement outlining its expectations regarding sex-specific patient recruitment, data analysis, and data reporting in future medical device studies.28 Hopefully, with this support for sex-specific research by the FDA, future studies will be able to identify therapeutic outcome differences that may exist between male and female patients.
Should our patient receive cardiac resynchronization therapy?
Regarding our patient with heart failure, the above studies suggest she will likely have a lower risk of death if she receives cardiac resynchronization therapy, even though her QRS interval is shorter than 150 ms. Providers who are aware of the emerging data regarding sex differences and treatment response can be powerful advocates for their patients, even in subspecialty areas, as highlighted by this case. We recommend counseling this patient to proceed with cardiac resynchronization therapy.
- Evans J, Heron J, Francomb H, Oke S, Golding J. Cohort study of depressed mood during pregnancy and after childbirth. BMJ 2001; 323:257–260.
- Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S; National Birth Defects Prevention Study. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol 2011; 205:51.e1–e8.
- Greene MF. Teratogenicity of SSRIs—serious concern or much ado about little? N Engl J Med 2007; 356:2732–2733.
- Louik C, Lin AE, Werler MM, Hernández-Díaz S, Mitchell AA. First-trimester use of selective serotonin-reuptake inhibitors and the risk of birth defects. N Engl J Med 2007; 356:2675–2683.
- Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM; National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med 2007; 356:2684–2692.
- Pedersen LH, Henriksen TB, Vestergaard M, Olsen J, Bech BH. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ 2009; 339:b3569.
- Furu K, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors and venlafaxine in early pregnancy and risk of birth defects: population based cohort study and sibling design. BMJ 2015; 350:h1798.
- Huybrechts KF, Palmsten K, Avorn J, et al. Antidepressant use in pregnancy and the risk of cardiac defects. N Engl J Med 2014; 370:2397–2407.
- Bérard A, Zhao J-P, Sheehy O. Sertraline use during pregnancy and the risk of major malformations. Am J Obstet Gynecol 2015; 212:795.e1–795.e12.
- Clements CC, Castro VM, Blumenthal SR, et al. Prenatal antidepressant exposure is associated with risk for attention-deficit hyperactivity disorder but not autism spectrum disorder in a large health system. Mol Psychiatry 2015; 20:727–734.
- Andersen JT, Andersen NL, Horwitz H, Poulsen HE, Jimenez-Solem E. Exposure to selective serotonin reuptake inhibitors in early pregnancy and the risk of miscarriage. Obstet Gynecol 2014; 124:655–661.
- Orleans RJ, Li L, Kim M-J, et al. FDA approval of paroxetine for menopausal hot flushes. N Engl J Med 2014; 370:1777–1779.
- Avis NE, Crawford SL, Greendale G, et al; Study of Women’s Health Across the Nation. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med 2015; 175:531–539.
- Pinkerton JV, Joffe H, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Low-dose paroxetine (7.5 mg) improves sleep in women with vasomotor symptoms associated with menopause. Menopause 2015; 22:50–58.
- Portman DJ, Kaunitz AM, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Effects of low-dose paroxetine 7.5 mg on weight and sexual function during treatment of vasomotor symptoms associated with menopause. Menopause 2014; 21:1082–1090.
- Desmarais JE, Looper KJ. Interactions between tamoxifen and antidepressants via cytochrome P450 2D6. J Clin Psychiatry 2009; 70:1688–1697.
- Joffe H, Guthrie KA, LaCroix AZ, et al. Low-dose estradiol and the serotonin-norepinephrine reuptake inhibitor venlafaxine for vasomotor symptoms: a randomized clinical trial. JAMA Intern Med 2014; 174:1058–1066.
- Shams T, Firwana B, Habib F, et al. SSRIs for hot flashes: a systematic review and meta-analysis of randomized trials. J Gen Intern Med 2014; 29:204–213.
- Kochanek KD, Xu J, Murphy SL, Minino AM, Kung H-C. Deaths: final data for 2009. Nat Vital Stat Rep 2012; 60(3):1–117.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—-2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
- McMurray JJV. Clinical practice. Systolic heart failure. N Engl J Med 2010; 362:228–238.
- Tracy CM, Epstein AE, Darbar D, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2013; 61:e6–e75.
- Zusterzeel R, Curtis JP, Canos DA, et al. Sex-specific mortality risk by QRS morphology and duration in patients receiving CRT. J Am Coll Cardiol 2014; 64:887–894.
- Goldenberg I, Kutyifa V, Klein HU, et al. Survival with cardiac-resynchronization therapy in mild heart failure. N Engl J Med 2014; 370:1694–1701.
- Dec GW. Leaning toward a better understanding of CRT in women. J Am Coll Cardiol 2014; 64:895–897.
- Zusterzeel R, Selzman KA, Sanders WE, et al. Cardiac resynchronization therapy in women: US Food and Drug Administration meta-analysis of patient-level data. JAMA Intern Med 2014; 174:1340–1348.
- Herz ND, Engeda J, Zusterzeel R, et al. Sex differences in device therapy for heart failure: utilization, outcomes, and adverse events. J Women’s Health 2015; 24:261–271.
- U.S. Department of Health and Human Services, Food and Drug Administration. Evaluation of sex-specific data in medical device clinical studies: guidance for industry and Food and Drug Administration staff. 2014; 1–30. www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM283707.pdf. Accessed October 1, 2015.
Women's health encompasses a broad range of issues unique to the female patient, with a scope that has expanded beyond reproductive health. Providers who care for women must develop cross-disciplinary competencies and understand the complex role of sex and gender on disease expression and treatment outcomes. Staying current with the literature in this rapidly changing field can be challenging for the busy clinician.
This article reviews recent advances in the treatment of depression in pregnancy, nonhormonal therapies for menopausal symptoms, and heart failure therapy in women, highlighting notable studies published in 2014 and early 2015.
TREATMENT OF DEPRESSION IN PREGNANCY
A 32-year-old woman with well-controlled but recurrent depression presents to the clinic for preconception counseling. Her depression has been successfully managed with a selective serotonin reuptake inhibitor (SSRI). She and her husband would like to try to conceive soon, but she is worried that continuing on her current SSRI may harm her baby. How should you advise her?
Concern for teratogenic effects of SSRIs
Depression is common during pregnancy: 11.8% to 13.5% of pregnant women report symptoms of depression,1 and 7.5% of pregnant women take an antidepressant.2
SSRI use during pregnancy has drawn attention because of mixed reports of teratogenic effects on the newborn, such as omphalocele, congenital heart defects, and craniosynostosis.3 Previous observational studies have specifically linked paroxetine to small but significant increases in right ventricular outflow tract obstruction4,5 and have linked sertraline to ventricular septal defects.6
However, reports of associations of congenital malformations and SSRI use in pregnancy in observational studies have been questioned, with concern that these studies had low statistical power, self-reported data leading to recall bias, and limited assessment for confounding factors.3,7
Recent studies refute risk of cardiac malformations
Several newer studies have been published that further examine the association between SSRI use in pregnancy and congenital heart defects, and their findings suggest that once adjusted for confounding variables, SSRI use in pregnancy may not be associated with cardiac malformations.
Huybrechts et al,8 in a large study published in 2014, extracted data on 950,000 pregnant women from the Medicaid database over a 7-year period and examined it for SSRI use during the first 90 days of pregnancy. Though SSRI use was associated with cardiac malformations when unadjusted for confounding variables (unadjusted relative risk 1.25, 95% confidence interval [CI] 1.13–1.38), once the cohort was restricted to women with a diagnosis of only depression and was adjusted based on propensity scoring, the association was no longer statistically significant (adjusted relative risk 1.06, 95% CI 0.93–1.22).
Additionally, there was no association between sertraline and ventricular septal defects (63 cases in 14,040 women exposed to sertraline, adjusted relative risk 1.04, 95% CI 0.76–1.41), or between paroxetine and right ventricular outflow tract obstruction (93 cases in 11,126 women exposed to paroxetine, adjusted relative risk 1.07, 95% CI 0.59–1.93).8
Furu et al7 conducted a sibling-matched case-control comparison published in 2015, in which more than 2 million live births from five Nordic countries were examined in the full cohort study and 2,288 births in the sibling-matched case-control cohort. SSRI or venlafaxine use in the first 90 days of pregnancy was examined. There was a slightly higher rate of cardiac defects in infants born to SSRI or venlafaxine recipients in the cohort study (adjusted odds ratio 1.15, 95% CI 1.05–1.26). However, in the sibling-controlled analyses, neither an SSRI nor venlafaxine was associated with heart defects (adjusted odds ratio 0.92, 95% CI 0.72–1.17), leading the authors to conclude that there might be familial factors or other lifestyle factors that were not taken into consideration and that could have confounded the cohort results.
Bérard et al9 examined antidepressant use in the first trimester of pregnancy in a cohort of women in Canada and concluded that sertraline was associated with congenital atrial and ventricular defects (risk ratio 1.34; 95% CI 1.02–1.76).9 However, this association should be interpreted with caution, as the Canadian cohort was notably smaller than those in other studies we have discussed, with only 18,493 pregnancies in the total cohort, and this conclusion was drawn from 9 cases of ventricular or atrial septal defects in babies of 366 women exposed to sertraline.
Although at first glance SSRIs may appear to be associated with congenital heart defects, these recent studies are reassuring and suggest that the association may actually not be significant. As with any statistical analysis, thoughtful study design, adequate statistical power, and adjustment for confounding factors must be considered before drawing conclusions.
SSRIs, offspring psychiatric outcomes, and miscarriage rates
Clements et al10 studied a cohort extracted from Partners Healthcare consisting of newborns with autism spectrum disorder, newborns with attention-deficit hyperactivity disorder (ADHD), and healthy matched controls and found that SSRI use during pregnancy was not associated with offspring autism spectrum disorder (adjusted odds ratio 1.10, 95% CI 0.7–1.70). However, they did find an increased risk of ADHD with SSRI use during pregnancy (adjusted odds ratio 1.81, 95% CI 1.22–2.70).
Andersen et al11 examined more than 1 million pregnancies in Denmark and found no difference in risk of miscarriage between women who used an SSRI during pregnancy (adjusted hazard ratio 1.27) and women who discontinued their SSRI at least 3 months before pregnancy (adjusted hazard ratio 1.24, P = .47). The authors concluded that because of the similar rate of miscarriage in both groups, there was no association between SSRI use and miscarriage, and that the small increased risk of miscarriage in both groups could have been attributable to a confounding factor that was not measured.
Should our patient continue her SSRI through pregnancy?
Our patient has recurrent depression, and her risk of relapse with antidepressant cessation is high. Though previous, less well-done studies suggested a small risk of congenital heart defects, recent larger high-quality studies provide significant reassurance that SSRI use in pregnancy is not strongly associated with cardiac malformations. Recent studies also show no association with miscarriage or autism spectrum disorder, though there may be risk of offspring ADHD.
She can be counseled that she may continue on her SSRI during pregnancy and can be reassured that the risk to her baby is small compared with her risk of recurrent or postpartum depression.
NONHORMONAL TREATMENT FOR VASOMOTOR SYMPTOMS OF MENOPAUSE
You see a patient who is struggling with symptoms of menopause. She tells you she has terrible hot flashes day and night, and she would like to try drug therapy. She does not want hormone replacement therapy because she is worried about the risk of adverse events. Are there safe and effective nonhormonal pharmacologic treatments for her vasomotor symptoms?
Paroxetine 7.5 mg is approved for vasomotor symptoms of menopause
As many as 75% of menopausal women in the United States experience vasomotor symptoms related to menopause, or hot flashes and night sweats.12 These symptoms can disrupt sleep and negatively affect quality of life. Though previously thought to occur during a short and self-limited time period, a recently published large observational study reported the median duration of vasomotor symptoms was 7.4 years, and in African American women in the cohort the median duration of vasomotor symptoms was 10.1 years—an entire decade of life.13
In 2013, the US Food and Drug Administration (FDA) approved paroxetine 7.5 mg daily for treating moderate to severe hot flashes associated with menopause. It is the only approved nonhormonal treatment for vasomotor symptoms; the only other approved treatments are estrogen therapy for women who have had a hysterectomy and combination estrogen-progesterone therapy for women who have not had a hysterectomy.
Further studies of paroxetine for menopausal symptoms
Since its approval, further studies have been published supporting the use of paroxetine 7.5 mg in treating symptoms of menopause. In addition to reducing hot flashes, this treatment also improves sleep disturbance in women with menopause.14
Pinkerton et al,14 in a pooled analysis of the data from the phase 3 clinical trials of paroxetine 7.5 mg per day, found that participants in groups assigned to paroxetine reported a 62% reduction in nighttime awakenings due to hot flashes compared with a 43% reduction in the placebo group (P < .001). Those who took paroxetine also reported a statistically significantly greater increase in duration of sleep than those who took placebo (37 minutes in the treatment group vs 27 minutes in the placebo group, P = .03).
Some patients are hesitant to take an SSRI because of concerns about adverse effects when used for psychiatric conditions. However, the dose of paroxetine that was studied and approved for vasomotor symptoms is lower than doses used for psychiatric indications and does not appear to be associated with these adverse effects.
Portman et al15 in 2014 examined the effect of paroxetine 7.5 mg vs placebo on weight gain and sexual function in women with vasomotor symptoms of menopause and found no significant increase in weight or decrease in sexual function at 24 weeks of use. Participants were weighed during study visits, and those in the paroxetine group gained on average 0.48% from baseline at 24 weeks, compared with 0.09% in the placebo group (P = .29).
Sexual dysfunction was assessed using the Arizona Sexual Experience Scale, which has been validated in psychiatric patients using antidepressants, and there was no significant difference in symptoms such as sex drive, sexual arousal, vaginal lubrication, or ability to achieve orgasm between the treatment group and placebo group.15
Of note, paroxetine is a potent inhibitor of the cytochrome P-450 CYP2D6 enzyme, and concurrent use of paroxetine with tamoxifen decreases tamoxifen activity.12,16 Since women with a history of breast cancer who cannot use estrogen for hot flashes may be seeking nonhormonal treatment for their vasomotor symptoms, providers should perform careful medication reconciliation and be aware that concomitant use of paroxetine and tamoxifen is not recommended.
Other antidepressants show promise but are not approved for menopausal symptoms
In addition to paroxetine, other nonhormonal drugs have been studied for treating hot flashes, but they have been unable to secure FDA approval for this indication. One of these is the serotonin-norepinephrine reuptake inhibitor venlafaxine, and a 2014 study17 confirmed its efficacy in treating menopausal vasomotor symptoms.
Joffe et al17 performed a three-armed trial comparing venlafaxine 75 mg/day, estradiol 0.5 mg/day, and placebo and found that both of the active treatments were better than placebo at reducing vasomotor symptoms. Compared with each other, estradiol 0.5 mg/day reduced hot flash frequency by an additional 0.6 events per day compared with venlafaxine 75 mg/day (P = .09). Though this difference was statistically significant, the authors pointed out that the clinical significance of such a small absolute difference is questionable. Additionally, providers should be aware that venlafaxine has little or no effect on the metabolism of tamoxifen.16
Shams et al,18 in a meta-analysis published in 2014, concluded that SSRIs as a class are more effective than placebo in treating hot flashes, supporting their widespread off-label use for this purpose. Their analysis examined the results of 11 studies, which included more than 2,000 patients in total, and found that compared with placebo, SSRI use was associated with a significant decrease in hot flashes (mean difference –0.93 events per day, 95% CI –1.49 to –0.37). A mixed treatment comparison analysis was also performed to try to model performance of individual SSRIs based on the pooled data, and the model suggests that escitalopram may be the most efficacious SSRI at reducing hot flash severity.
These studies support the effectiveness of SSRIs18 and venlafaxine17 in reducing hot flashes compared with placebo, though providers should be aware that they are still not FDA-approved for this indication.
Nonhormonal therapy for our patient
We would recommend paroxetine 7.5 mg nightly to this patient, as it is an FDA-approved nonhormonal medication that has been shown to help patients with vasomotor symptoms of menopause as well as sleep disturbance, without sexual side effects or weight gain. If the patient cannot tolerate paroxetine, off-label use of another SSRI or venlafaxine is supported by the recent literature.
HEART DISEASE IN WOMEN: CARDIAC RESYNCHRONIZATION THERAPY
A 68-year-old woman with a history of nonischemic cardiomyopathy presents for routine follow-up in your office. Despite maximal medical therapy on a beta-blocker, an angiotensin II receptor blocker, and a diuretic, she has New York Heart Association (NYHA) class III symptoms. Her most recent studies showed an ejection fraction of 30% by echocardiography and left bundle-branch block on electrocardiography, with a QRS duration of 140 ms. She recently saw her cardiologist, who recommended cardiac resynchronization therapy, and she wants your opinion as to whether or not to proceed with this recommendation. How should you counsel her?
Which patients are candidates for cardiac resynchronization therapy?
Heart disease continues to be the number one cause of death in the United States for both men and women, and almost the same number of women and men die from heart disease every year.19 Though coronary artery disease accounts for most cases of cardiovascular disease in the United States, heart failure is a significant and growing contributor. Approximately 6.6 million adults had heart failure in 2010 in the United States, and an additional 3 million are projected to have heart failure by 2030.20 The burden of disease on our health system is high, with about 1 million hospitalizations and more than 3 million outpatient office visits attributable to heart failure yearly.20
Patients with heart failure may have symptoms of dyspnea, fatigue, orthopnea, and peripheral edema; laboratory and radiologic findings of pulmonary edema, renal insufficiency, and hyponatremia; and electrocardiographic findings of atrial fibrillation or prolonged QRS.21 Intraventricular conduction delay (QRS duration > 120 ms) is associated with dyssynchronous ventricular contraction and impaired pump function and is present in almost one-third of patients who have advanced heart failure.21
Cardiac resynchronization therapy, or biventricular pacing, can improve symptoms and pump function and has been shown to decrease rates of hospitalization and death in these patients.22 According to the joint 2012 guidelines of the American College of Cardiology Foundation, American Heart Association, and Heart Rhythm Society,22 it is indicated for patients with an ejection fraction of 35% or less, left bundle-branch block with QRS duration of 150 ms or more, and NYHA class II to IV symptoms who are in sinus rhythm (class I recommendation, level of evidence A).
Studies of cardiac resynchronization therapy in women
Recently published studies have suggested that women may derive greater benefit than men from cardiac resynchronization therapy.
Zusterzeel et al23 (2014) evaluated sex-specific data from the National Cardiovascular Data Registry, which contains data on all biventricular pacemaker and implantable cardioverter-defibrillator implantations from 80% of US hospitals.23 Of the 21,152 patients who had left bundle-branch block and received cardiac resynchronization therapy, women derived greater benefit in terms of death than men did, with a 21% lower risk of death than men (adjusted hazard ratio 0.79, 95% CI 0.74–0.84, P < .001). This study was also notable in that 36% of the patients were women, whereas in most earlier studies of cardiac resynchronization therapy women accounted for only 22% to 30% of the study population.22
Goldenberg et al24 (2014) performed a follow-up analysis of the Multicenter Automatic Defibrillator Implantation Trial With Cardiac Resynchronization Therapy. Subgroup analysis showed that although both men and women had a lower risk of death if they received cardiac resynchronization therapy compared with an implantable cardioverter-defibrillator only, the magnitude of benefit may be greater for women (hazard ratio 0.48, 95% CI 0.25–0.91, P = .03) than for men (hazard ratio 0.69, 95% CI 0.50–0.95, P = .02).
In addition to deriving greater mortality benefit, women may actually benefit from cardiac resynchronization therapy at shorter QRS durations than what is currently recommended. Women have a shorter baseline QRS than men, and a smaller left ventricular cavity.25 In an FDA meta-analysis published in August 2014, pooled data from more than 4,000 patients in three studies suggested that women with left bundle-branch block benefited from cardiac resynchronization therapy more than men with left bundle-branch block.26 Neither men nor women with left bundle-branch block benefited from it if their QRS duration was less than 130 ms, and both sexes benefited from it if they had left bundle-branch block and a QRS duration longer than 150 ms. However, women who received it who had left bundle-branch block and a QRS duration of 130 to 149 ms had a significant 76% reduction in the primary composite outcome of a heart failure event or death (hazard ratio 0.24, 95% CI 0.11–0.53, P < .001), while men in the same group did not derive significant benefit (hazard ratio 0.85, 95% CI 0.60–1.21, P = .38).
Despite the increasing evidence that there are sex-specific differences in the benefit from cardiac resynchronization therapy, what we know is limited by the low rates of female enrollment in most of the studies of this treatment. In a systematic review published in 2015, Herz et al27 found that 90% of the 183 studies they reviewed enrolled 35% women or less, and half of the studies enrolled less than 23% women. Furthermore, only 20 of the 183 studies reported baseline characteristics by sex.
Recognizing this lack of adequate data, in August 2014 the FDA issued an official guidance statement outlining its expectations regarding sex-specific patient recruitment, data analysis, and data reporting in future medical device studies.28 Hopefully, with this support for sex-specific research by the FDA, future studies will be able to identify therapeutic outcome differences that may exist between male and female patients.
Should our patient receive cardiac resynchronization therapy?
Regarding our patient with heart failure, the above studies suggest she will likely have a lower risk of death if she receives cardiac resynchronization therapy, even though her QRS interval is shorter than 150 ms. Providers who are aware of the emerging data regarding sex differences and treatment response can be powerful advocates for their patients, even in subspecialty areas, as highlighted by this case. We recommend counseling this patient to proceed with cardiac resynchronization therapy.
Women's health encompasses a broad range of issues unique to the female patient, with a scope that has expanded beyond reproductive health. Providers who care for women must develop cross-disciplinary competencies and understand the complex role of sex and gender on disease expression and treatment outcomes. Staying current with the literature in this rapidly changing field can be challenging for the busy clinician.
This article reviews recent advances in the treatment of depression in pregnancy, nonhormonal therapies for menopausal symptoms, and heart failure therapy in women, highlighting notable studies published in 2014 and early 2015.
TREATMENT OF DEPRESSION IN PREGNANCY
A 32-year-old woman with well-controlled but recurrent depression presents to the clinic for preconception counseling. Her depression has been successfully managed with a selective serotonin reuptake inhibitor (SSRI). She and her husband would like to try to conceive soon, but she is worried that continuing on her current SSRI may harm her baby. How should you advise her?
Concern for teratogenic effects of SSRIs
Depression is common during pregnancy: 11.8% to 13.5% of pregnant women report symptoms of depression,1 and 7.5% of pregnant women take an antidepressant.2
SSRI use during pregnancy has drawn attention because of mixed reports of teratogenic effects on the newborn, such as omphalocele, congenital heart defects, and craniosynostosis.3 Previous observational studies have specifically linked paroxetine to small but significant increases in right ventricular outflow tract obstruction4,5 and have linked sertraline to ventricular septal defects.6
However, reports of associations of congenital malformations and SSRI use in pregnancy in observational studies have been questioned, with concern that these studies had low statistical power, self-reported data leading to recall bias, and limited assessment for confounding factors.3,7
Recent studies refute risk of cardiac malformations
Several newer studies have been published that further examine the association between SSRI use in pregnancy and congenital heart defects, and their findings suggest that once adjusted for confounding variables, SSRI use in pregnancy may not be associated with cardiac malformations.
Huybrechts et al,8 in a large study published in 2014, extracted data on 950,000 pregnant women from the Medicaid database over a 7-year period and examined it for SSRI use during the first 90 days of pregnancy. Though SSRI use was associated with cardiac malformations when unadjusted for confounding variables (unadjusted relative risk 1.25, 95% confidence interval [CI] 1.13–1.38), once the cohort was restricted to women with a diagnosis of only depression and was adjusted based on propensity scoring, the association was no longer statistically significant (adjusted relative risk 1.06, 95% CI 0.93–1.22).
Additionally, there was no association between sertraline and ventricular septal defects (63 cases in 14,040 women exposed to sertraline, adjusted relative risk 1.04, 95% CI 0.76–1.41), or between paroxetine and right ventricular outflow tract obstruction (93 cases in 11,126 women exposed to paroxetine, adjusted relative risk 1.07, 95% CI 0.59–1.93).8
Furu et al7 conducted a sibling-matched case-control comparison published in 2015, in which more than 2 million live births from five Nordic countries were examined in the full cohort study and 2,288 births in the sibling-matched case-control cohort. SSRI or venlafaxine use in the first 90 days of pregnancy was examined. There was a slightly higher rate of cardiac defects in infants born to SSRI or venlafaxine recipients in the cohort study (adjusted odds ratio 1.15, 95% CI 1.05–1.26). However, in the sibling-controlled analyses, neither an SSRI nor venlafaxine was associated with heart defects (adjusted odds ratio 0.92, 95% CI 0.72–1.17), leading the authors to conclude that there might be familial factors or other lifestyle factors that were not taken into consideration and that could have confounded the cohort results.
Bérard et al9 examined antidepressant use in the first trimester of pregnancy in a cohort of women in Canada and concluded that sertraline was associated with congenital atrial and ventricular defects (risk ratio 1.34; 95% CI 1.02–1.76).9 However, this association should be interpreted with caution, as the Canadian cohort was notably smaller than those in other studies we have discussed, with only 18,493 pregnancies in the total cohort, and this conclusion was drawn from 9 cases of ventricular or atrial septal defects in babies of 366 women exposed to sertraline.
Although at first glance SSRIs may appear to be associated with congenital heart defects, these recent studies are reassuring and suggest that the association may actually not be significant. As with any statistical analysis, thoughtful study design, adequate statistical power, and adjustment for confounding factors must be considered before drawing conclusions.
SSRIs, offspring psychiatric outcomes, and miscarriage rates
Clements et al10 studied a cohort extracted from Partners Healthcare consisting of newborns with autism spectrum disorder, newborns with attention-deficit hyperactivity disorder (ADHD), and healthy matched controls and found that SSRI use during pregnancy was not associated with offspring autism spectrum disorder (adjusted odds ratio 1.10, 95% CI 0.7–1.70). However, they did find an increased risk of ADHD with SSRI use during pregnancy (adjusted odds ratio 1.81, 95% CI 1.22–2.70).
Andersen et al11 examined more than 1 million pregnancies in Denmark and found no difference in risk of miscarriage between women who used an SSRI during pregnancy (adjusted hazard ratio 1.27) and women who discontinued their SSRI at least 3 months before pregnancy (adjusted hazard ratio 1.24, P = .47). The authors concluded that because of the similar rate of miscarriage in both groups, there was no association between SSRI use and miscarriage, and that the small increased risk of miscarriage in both groups could have been attributable to a confounding factor that was not measured.
Should our patient continue her SSRI through pregnancy?
Our patient has recurrent depression, and her risk of relapse with antidepressant cessation is high. Though previous, less well-done studies suggested a small risk of congenital heart defects, recent larger high-quality studies provide significant reassurance that SSRI use in pregnancy is not strongly associated with cardiac malformations. Recent studies also show no association with miscarriage or autism spectrum disorder, though there may be risk of offspring ADHD.
She can be counseled that she may continue on her SSRI during pregnancy and can be reassured that the risk to her baby is small compared with her risk of recurrent or postpartum depression.
NONHORMONAL TREATMENT FOR VASOMOTOR SYMPTOMS OF MENOPAUSE
You see a patient who is struggling with symptoms of menopause. She tells you she has terrible hot flashes day and night, and she would like to try drug therapy. She does not want hormone replacement therapy because she is worried about the risk of adverse events. Are there safe and effective nonhormonal pharmacologic treatments for her vasomotor symptoms?
Paroxetine 7.5 mg is approved for vasomotor symptoms of menopause
As many as 75% of menopausal women in the United States experience vasomotor symptoms related to menopause, or hot flashes and night sweats.12 These symptoms can disrupt sleep and negatively affect quality of life. Though previously thought to occur during a short and self-limited time period, a recently published large observational study reported the median duration of vasomotor symptoms was 7.4 years, and in African American women in the cohort the median duration of vasomotor symptoms was 10.1 years—an entire decade of life.13
In 2013, the US Food and Drug Administration (FDA) approved paroxetine 7.5 mg daily for treating moderate to severe hot flashes associated with menopause. It is the only approved nonhormonal treatment for vasomotor symptoms; the only other approved treatments are estrogen therapy for women who have had a hysterectomy and combination estrogen-progesterone therapy for women who have not had a hysterectomy.
Further studies of paroxetine for menopausal symptoms
Since its approval, further studies have been published supporting the use of paroxetine 7.5 mg in treating symptoms of menopause. In addition to reducing hot flashes, this treatment also improves sleep disturbance in women with menopause.14
Pinkerton et al,14 in a pooled analysis of the data from the phase 3 clinical trials of paroxetine 7.5 mg per day, found that participants in groups assigned to paroxetine reported a 62% reduction in nighttime awakenings due to hot flashes compared with a 43% reduction in the placebo group (P < .001). Those who took paroxetine also reported a statistically significantly greater increase in duration of sleep than those who took placebo (37 minutes in the treatment group vs 27 minutes in the placebo group, P = .03).
Some patients are hesitant to take an SSRI because of concerns about adverse effects when used for psychiatric conditions. However, the dose of paroxetine that was studied and approved for vasomotor symptoms is lower than doses used for psychiatric indications and does not appear to be associated with these adverse effects.
Portman et al15 in 2014 examined the effect of paroxetine 7.5 mg vs placebo on weight gain and sexual function in women with vasomotor symptoms of menopause and found no significant increase in weight or decrease in sexual function at 24 weeks of use. Participants were weighed during study visits, and those in the paroxetine group gained on average 0.48% from baseline at 24 weeks, compared with 0.09% in the placebo group (P = .29).
Sexual dysfunction was assessed using the Arizona Sexual Experience Scale, which has been validated in psychiatric patients using antidepressants, and there was no significant difference in symptoms such as sex drive, sexual arousal, vaginal lubrication, or ability to achieve orgasm between the treatment group and placebo group.15
Of note, paroxetine is a potent inhibitor of the cytochrome P-450 CYP2D6 enzyme, and concurrent use of paroxetine with tamoxifen decreases tamoxifen activity.12,16 Since women with a history of breast cancer who cannot use estrogen for hot flashes may be seeking nonhormonal treatment for their vasomotor symptoms, providers should perform careful medication reconciliation and be aware that concomitant use of paroxetine and tamoxifen is not recommended.
Other antidepressants show promise but are not approved for menopausal symptoms
In addition to paroxetine, other nonhormonal drugs have been studied for treating hot flashes, but they have been unable to secure FDA approval for this indication. One of these is the serotonin-norepinephrine reuptake inhibitor venlafaxine, and a 2014 study17 confirmed its efficacy in treating menopausal vasomotor symptoms.
Joffe et al17 performed a three-armed trial comparing venlafaxine 75 mg/day, estradiol 0.5 mg/day, and placebo and found that both of the active treatments were better than placebo at reducing vasomotor symptoms. Compared with each other, estradiol 0.5 mg/day reduced hot flash frequency by an additional 0.6 events per day compared with venlafaxine 75 mg/day (P = .09). Though this difference was statistically significant, the authors pointed out that the clinical significance of such a small absolute difference is questionable. Additionally, providers should be aware that venlafaxine has little or no effect on the metabolism of tamoxifen.16
Shams et al,18 in a meta-analysis published in 2014, concluded that SSRIs as a class are more effective than placebo in treating hot flashes, supporting their widespread off-label use for this purpose. Their analysis examined the results of 11 studies, which included more than 2,000 patients in total, and found that compared with placebo, SSRI use was associated with a significant decrease in hot flashes (mean difference –0.93 events per day, 95% CI –1.49 to –0.37). A mixed treatment comparison analysis was also performed to try to model performance of individual SSRIs based on the pooled data, and the model suggests that escitalopram may be the most efficacious SSRI at reducing hot flash severity.
These studies support the effectiveness of SSRIs18 and venlafaxine17 in reducing hot flashes compared with placebo, though providers should be aware that they are still not FDA-approved for this indication.
Nonhormonal therapy for our patient
We would recommend paroxetine 7.5 mg nightly to this patient, as it is an FDA-approved nonhormonal medication that has been shown to help patients with vasomotor symptoms of menopause as well as sleep disturbance, without sexual side effects or weight gain. If the patient cannot tolerate paroxetine, off-label use of another SSRI or venlafaxine is supported by the recent literature.
HEART DISEASE IN WOMEN: CARDIAC RESYNCHRONIZATION THERAPY
A 68-year-old woman with a history of nonischemic cardiomyopathy presents for routine follow-up in your office. Despite maximal medical therapy on a beta-blocker, an angiotensin II receptor blocker, and a diuretic, she has New York Heart Association (NYHA) class III symptoms. Her most recent studies showed an ejection fraction of 30% by echocardiography and left bundle-branch block on electrocardiography, with a QRS duration of 140 ms. She recently saw her cardiologist, who recommended cardiac resynchronization therapy, and she wants your opinion as to whether or not to proceed with this recommendation. How should you counsel her?
Which patients are candidates for cardiac resynchronization therapy?
Heart disease continues to be the number one cause of death in the United States for both men and women, and almost the same number of women and men die from heart disease every year.19 Though coronary artery disease accounts for most cases of cardiovascular disease in the United States, heart failure is a significant and growing contributor. Approximately 6.6 million adults had heart failure in 2010 in the United States, and an additional 3 million are projected to have heart failure by 2030.20 The burden of disease on our health system is high, with about 1 million hospitalizations and more than 3 million outpatient office visits attributable to heart failure yearly.20
Patients with heart failure may have symptoms of dyspnea, fatigue, orthopnea, and peripheral edema; laboratory and radiologic findings of pulmonary edema, renal insufficiency, and hyponatremia; and electrocardiographic findings of atrial fibrillation or prolonged QRS.21 Intraventricular conduction delay (QRS duration > 120 ms) is associated with dyssynchronous ventricular contraction and impaired pump function and is present in almost one-third of patients who have advanced heart failure.21
Cardiac resynchronization therapy, or biventricular pacing, can improve symptoms and pump function and has been shown to decrease rates of hospitalization and death in these patients.22 According to the joint 2012 guidelines of the American College of Cardiology Foundation, American Heart Association, and Heart Rhythm Society,22 it is indicated for patients with an ejection fraction of 35% or less, left bundle-branch block with QRS duration of 150 ms or more, and NYHA class II to IV symptoms who are in sinus rhythm (class I recommendation, level of evidence A).
Studies of cardiac resynchronization therapy in women
Recently published studies have suggested that women may derive greater benefit than men from cardiac resynchronization therapy.
Zusterzeel et al23 (2014) evaluated sex-specific data from the National Cardiovascular Data Registry, which contains data on all biventricular pacemaker and implantable cardioverter-defibrillator implantations from 80% of US hospitals.23 Of the 21,152 patients who had left bundle-branch block and received cardiac resynchronization therapy, women derived greater benefit in terms of death than men did, with a 21% lower risk of death than men (adjusted hazard ratio 0.79, 95% CI 0.74–0.84, P < .001). This study was also notable in that 36% of the patients were women, whereas in most earlier studies of cardiac resynchronization therapy women accounted for only 22% to 30% of the study population.22
Goldenberg et al24 (2014) performed a follow-up analysis of the Multicenter Automatic Defibrillator Implantation Trial With Cardiac Resynchronization Therapy. Subgroup analysis showed that although both men and women had a lower risk of death if they received cardiac resynchronization therapy compared with an implantable cardioverter-defibrillator only, the magnitude of benefit may be greater for women (hazard ratio 0.48, 95% CI 0.25–0.91, P = .03) than for men (hazard ratio 0.69, 95% CI 0.50–0.95, P = .02).
In addition to deriving greater mortality benefit, women may actually benefit from cardiac resynchronization therapy at shorter QRS durations than what is currently recommended. Women have a shorter baseline QRS than men, and a smaller left ventricular cavity.25 In an FDA meta-analysis published in August 2014, pooled data from more than 4,000 patients in three studies suggested that women with left bundle-branch block benefited from cardiac resynchronization therapy more than men with left bundle-branch block.26 Neither men nor women with left bundle-branch block benefited from it if their QRS duration was less than 130 ms, and both sexes benefited from it if they had left bundle-branch block and a QRS duration longer than 150 ms. However, women who received it who had left bundle-branch block and a QRS duration of 130 to 149 ms had a significant 76% reduction in the primary composite outcome of a heart failure event or death (hazard ratio 0.24, 95% CI 0.11–0.53, P < .001), while men in the same group did not derive significant benefit (hazard ratio 0.85, 95% CI 0.60–1.21, P = .38).
Despite the increasing evidence that there are sex-specific differences in the benefit from cardiac resynchronization therapy, what we know is limited by the low rates of female enrollment in most of the studies of this treatment. In a systematic review published in 2015, Herz et al27 found that 90% of the 183 studies they reviewed enrolled 35% women or less, and half of the studies enrolled less than 23% women. Furthermore, only 20 of the 183 studies reported baseline characteristics by sex.
Recognizing this lack of adequate data, in August 2014 the FDA issued an official guidance statement outlining its expectations regarding sex-specific patient recruitment, data analysis, and data reporting in future medical device studies.28 Hopefully, with this support for sex-specific research by the FDA, future studies will be able to identify therapeutic outcome differences that may exist between male and female patients.
Should our patient receive cardiac resynchronization therapy?
Regarding our patient with heart failure, the above studies suggest she will likely have a lower risk of death if she receives cardiac resynchronization therapy, even though her QRS interval is shorter than 150 ms. Providers who are aware of the emerging data regarding sex differences and treatment response can be powerful advocates for their patients, even in subspecialty areas, as highlighted by this case. We recommend counseling this patient to proceed with cardiac resynchronization therapy.
- Evans J, Heron J, Francomb H, Oke S, Golding J. Cohort study of depressed mood during pregnancy and after childbirth. BMJ 2001; 323:257–260.
- Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S; National Birth Defects Prevention Study. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol 2011; 205:51.e1–e8.
- Greene MF. Teratogenicity of SSRIs—serious concern or much ado about little? N Engl J Med 2007; 356:2732–2733.
- Louik C, Lin AE, Werler MM, Hernández-Díaz S, Mitchell AA. First-trimester use of selective serotonin-reuptake inhibitors and the risk of birth defects. N Engl J Med 2007; 356:2675–2683.
- Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM; National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med 2007; 356:2684–2692.
- Pedersen LH, Henriksen TB, Vestergaard M, Olsen J, Bech BH. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ 2009; 339:b3569.
- Furu K, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors and venlafaxine in early pregnancy and risk of birth defects: population based cohort study and sibling design. BMJ 2015; 350:h1798.
- Huybrechts KF, Palmsten K, Avorn J, et al. Antidepressant use in pregnancy and the risk of cardiac defects. N Engl J Med 2014; 370:2397–2407.
- Bérard A, Zhao J-P, Sheehy O. Sertraline use during pregnancy and the risk of major malformations. Am J Obstet Gynecol 2015; 212:795.e1–795.e12.
- Clements CC, Castro VM, Blumenthal SR, et al. Prenatal antidepressant exposure is associated with risk for attention-deficit hyperactivity disorder but not autism spectrum disorder in a large health system. Mol Psychiatry 2015; 20:727–734.
- Andersen JT, Andersen NL, Horwitz H, Poulsen HE, Jimenez-Solem E. Exposure to selective serotonin reuptake inhibitors in early pregnancy and the risk of miscarriage. Obstet Gynecol 2014; 124:655–661.
- Orleans RJ, Li L, Kim M-J, et al. FDA approval of paroxetine for menopausal hot flushes. N Engl J Med 2014; 370:1777–1779.
- Avis NE, Crawford SL, Greendale G, et al; Study of Women’s Health Across the Nation. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med 2015; 175:531–539.
- Pinkerton JV, Joffe H, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Low-dose paroxetine (7.5 mg) improves sleep in women with vasomotor symptoms associated with menopause. Menopause 2015; 22:50–58.
- Portman DJ, Kaunitz AM, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Effects of low-dose paroxetine 7.5 mg on weight and sexual function during treatment of vasomotor symptoms associated with menopause. Menopause 2014; 21:1082–1090.
- Desmarais JE, Looper KJ. Interactions between tamoxifen and antidepressants via cytochrome P450 2D6. J Clin Psychiatry 2009; 70:1688–1697.
- Joffe H, Guthrie KA, LaCroix AZ, et al. Low-dose estradiol and the serotonin-norepinephrine reuptake inhibitor venlafaxine for vasomotor symptoms: a randomized clinical trial. JAMA Intern Med 2014; 174:1058–1066.
- Shams T, Firwana B, Habib F, et al. SSRIs for hot flashes: a systematic review and meta-analysis of randomized trials. J Gen Intern Med 2014; 29:204–213.
- Kochanek KD, Xu J, Murphy SL, Minino AM, Kung H-C. Deaths: final data for 2009. Nat Vital Stat Rep 2012; 60(3):1–117.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—-2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
- McMurray JJV. Clinical practice. Systolic heart failure. N Engl J Med 2010; 362:228–238.
- Tracy CM, Epstein AE, Darbar D, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2013; 61:e6–e75.
- Zusterzeel R, Curtis JP, Canos DA, et al. Sex-specific mortality risk by QRS morphology and duration in patients receiving CRT. J Am Coll Cardiol 2014; 64:887–894.
- Goldenberg I, Kutyifa V, Klein HU, et al. Survival with cardiac-resynchronization therapy in mild heart failure. N Engl J Med 2014; 370:1694–1701.
- Dec GW. Leaning toward a better understanding of CRT in women. J Am Coll Cardiol 2014; 64:895–897.
- Zusterzeel R, Selzman KA, Sanders WE, et al. Cardiac resynchronization therapy in women: US Food and Drug Administration meta-analysis of patient-level data. JAMA Intern Med 2014; 174:1340–1348.
- Herz ND, Engeda J, Zusterzeel R, et al. Sex differences in device therapy for heart failure: utilization, outcomes, and adverse events. J Women’s Health 2015; 24:261–271.
- U.S. Department of Health and Human Services, Food and Drug Administration. Evaluation of sex-specific data in medical device clinical studies: guidance for industry and Food and Drug Administration staff. 2014; 1–30. www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM283707.pdf. Accessed October 1, 2015.
- Evans J, Heron J, Francomb H, Oke S, Golding J. Cohort study of depressed mood during pregnancy and after childbirth. BMJ 2001; 323:257–260.
- Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S; National Birth Defects Prevention Study. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol 2011; 205:51.e1–e8.
- Greene MF. Teratogenicity of SSRIs—serious concern or much ado about little? N Engl J Med 2007; 356:2732–2733.
- Louik C, Lin AE, Werler MM, Hernández-Díaz S, Mitchell AA. First-trimester use of selective serotonin-reuptake inhibitors and the risk of birth defects. N Engl J Med 2007; 356:2675–2683.
- Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM; National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med 2007; 356:2684–2692.
- Pedersen LH, Henriksen TB, Vestergaard M, Olsen J, Bech BH. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ 2009; 339:b3569.
- Furu K, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors and venlafaxine in early pregnancy and risk of birth defects: population based cohort study and sibling design. BMJ 2015; 350:h1798.
- Huybrechts KF, Palmsten K, Avorn J, et al. Antidepressant use in pregnancy and the risk of cardiac defects. N Engl J Med 2014; 370:2397–2407.
- Bérard A, Zhao J-P, Sheehy O. Sertraline use during pregnancy and the risk of major malformations. Am J Obstet Gynecol 2015; 212:795.e1–795.e12.
- Clements CC, Castro VM, Blumenthal SR, et al. Prenatal antidepressant exposure is associated with risk for attention-deficit hyperactivity disorder but not autism spectrum disorder in a large health system. Mol Psychiatry 2015; 20:727–734.
- Andersen JT, Andersen NL, Horwitz H, Poulsen HE, Jimenez-Solem E. Exposure to selective serotonin reuptake inhibitors in early pregnancy and the risk of miscarriage. Obstet Gynecol 2014; 124:655–661.
- Orleans RJ, Li L, Kim M-J, et al. FDA approval of paroxetine for menopausal hot flushes. N Engl J Med 2014; 370:1777–1779.
- Avis NE, Crawford SL, Greendale G, et al; Study of Women’s Health Across the Nation. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med 2015; 175:531–539.
- Pinkerton JV, Joffe H, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Low-dose paroxetine (7.5 mg) improves sleep in women with vasomotor symptoms associated with menopause. Menopause 2015; 22:50–58.
- Portman DJ, Kaunitz AM, Kazempour K, Mekonnen H, Bhaskar S, Lippman J. Effects of low-dose paroxetine 7.5 mg on weight and sexual function during treatment of vasomotor symptoms associated with menopause. Menopause 2014; 21:1082–1090.
- Desmarais JE, Looper KJ. Interactions between tamoxifen and antidepressants via cytochrome P450 2D6. J Clin Psychiatry 2009; 70:1688–1697.
- Joffe H, Guthrie KA, LaCroix AZ, et al. Low-dose estradiol and the serotonin-norepinephrine reuptake inhibitor venlafaxine for vasomotor symptoms: a randomized clinical trial. JAMA Intern Med 2014; 174:1058–1066.
- Shams T, Firwana B, Habib F, et al. SSRIs for hot flashes: a systematic review and meta-analysis of randomized trials. J Gen Intern Med 2014; 29:204–213.
- Kochanek KD, Xu J, Murphy SL, Minino AM, Kung H-C. Deaths: final data for 2009. Nat Vital Stat Rep 2012; 60(3):1–117.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—-2012 update: a report from the American Heart Association. Circulation 2012; 125:e2–e220.
- McMurray JJV. Clinical practice. Systolic heart failure. N Engl J Med 2010; 362:228–238.
- Tracy CM, Epstein AE, Darbar D, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2013; 61:e6–e75.
- Zusterzeel R, Curtis JP, Canos DA, et al. Sex-specific mortality risk by QRS morphology and duration in patients receiving CRT. J Am Coll Cardiol 2014; 64:887–894.
- Goldenberg I, Kutyifa V, Klein HU, et al. Survival with cardiac-resynchronization therapy in mild heart failure. N Engl J Med 2014; 370:1694–1701.
- Dec GW. Leaning toward a better understanding of CRT in women. J Am Coll Cardiol 2014; 64:895–897.
- Zusterzeel R, Selzman KA, Sanders WE, et al. Cardiac resynchronization therapy in women: US Food and Drug Administration meta-analysis of patient-level data. JAMA Intern Med 2014; 174:1340–1348.
- Herz ND, Engeda J, Zusterzeel R, et al. Sex differences in device therapy for heart failure: utilization, outcomes, and adverse events. J Women’s Health 2015; 24:261–271.
- U.S. Department of Health and Human Services, Food and Drug Administration. Evaluation of sex-specific data in medical device clinical studies: guidance for industry and Food and Drug Administration staff. 2014; 1–30. www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM283707.pdf. Accessed October 1, 2015.
KEY POINTS
- Earlier trials had raised concerns about possible teratogenic effects of selective serotonin reuptake inhibitors, but more recent trials have found no strong association between these drugs and congenital heart defects, and no association with miscarriage or autism spectrum disorder, though there may be a risk of attention deficit hyperactivity disorder in offspring.
- Paroxetine is approved for treating vasomotor symptoms of menopause, but in a lower dose (7.5 mg) than those used for depression and other psychiatric indications. Clinical trials have also shown good results with other antidepressants for treating hot flashes, but the drugs are not yet approved for this indication.
- Women with heart failure and left bundle-branch block can decrease their risk of death with cardiac resynchronization therapy more than men with the same condition. Moreover, women may benefit from this therapy even if their QRS duration is somewhat shorter than the established cutoff, ie, if it is in the range of 130 to 149 ms.

An elderly woman with ‘heart failure’: Cognitive biases and diagnostic error
An elderly Spanish-speaking woman with morbid obesity, diabetes, hypertension, and rheumatoid arthritis presents to the emergency department with worsening shortness of breath and cough. She speaks only Spanish, so her son provides the history without the aid of an interpreter.
Her shortness of breath is most noticeable with exertion and has increased gradually over the past 2 months. She has a nonproductive cough. Her son has noticed decreased oral intake and weight loss over the past few weeks. She has neither traveled recently nor been in contact with anyone known to have an infectious disease.
A review of systems is otherwise negative: specifically, she denies chest pain, fevers, or chills. She saw her primary care physician 3 weeks ago for these complaints and was prescribed a 3-day course of azithromycin with no improvement.
Her medications include lisinopril, atenolol, glipizide, and metformin; her son believes she may be taking others as well but is not sure. He is also unsure of what treatment his mother has received for her rheumatoid arthritis, and most of her medical records are within another health system.
On physical examination, the patient is coughing and appears ill. Her temperature is 99.9°F (37.7°C), heart rate 105 beats per minute, blood pressure 140/70 mm Hg, respiratory rate 24 per minute, and oxygen saturation by pulse oximetry 89% on room air. Heart sounds are normal, jugular venous pressure cannot be assessed because of her obese body habitus, pulmonary examination demonstrates crackles in all lung fields, and lower-extremity edema is not present. Her extremities are warm and well perfused. Musculoskeletal examination reveals deformities of the joints in both hands consistent with rheumatoid arthritis.
Laboratory data:
- White blood cell count 13.0 × 109/L (reference range 3.7–11.0)
- Hemoglobin level 10 g/dL (11.5–15)
- Serum creatinine 1.0 mg/dL (0.7–1.4)
- Pro-brain-type natriuretic peptide (pro-BNP) level greater than the upper limit of normal.
A chest radiograph is obtained, and the resident radiologist’s preliminary impression is that it is consistent with pulmonary vascular congestion.
The patient is admitted for further diagnostic evaluation. The emergency department resident orders intravenous furosemide and signs out to the night float medicine resident that this is an “elderly woman with hypertension, diabetes, and heart failure being admitted for a heart failure exacerbation.”
What is the accuracy of a physician’s initial working diagnosis?
Diagnostic accuracy requires both clinical knowledge and problem-solving skills.1
A decade ago, a National Patient Safety Foundation survey2 found that one in six patients had suffered a medical error related to misdiagnosis. In a large systematic review of autopsy-based diagnostic errors, the theorized rate of major errors ranged from 8.4% to as high as 24.4%.3 A study by Neale et al4 found that admitting diagnoses were incorrect in 6% of cases. In emergency departments, inaccuracy rates of up to 12% have been described.5
What factors influence the prevalence of diagnostic errors?
Initial empiric treatments, such as intravenous furosemide in the above scenario, add to the challenge of diagnosis in acute care settings and can influence clinical decisions made by subsequent providers.6
Nonspecific or vague symptoms make diagnosis especially challenging. Shortness of breath, for example, is a common chief complaint in medical patients, as in this case. Green et al7 found emergency department physicians reported clinical uncertainty for a diagnosis of heart failure in 31% of patients evaluated for “dyspnea.” Pulmonary embolism and pulmonary tuberculosis are also in the differential diagnosis for our patient, with studies reporting a misdiagnosis rate of 55% for pulmonary embolism8 and 50% for pulmonary tuberculosis.9
Hertwig et al,10 describing the diagnostic process in patients presenting to emergency departments with a nonspecific constellation of symptoms, found particularly low rates of agreement between the initial diagnostic impression and the final, correct one. In fact, the actual diagnosis was only in the physician’s initial “top three” differential diagnoses 29% to 83% of the time.
Atypical presentations of common diseases, initial nonspecific presentations of common diseases, and confounding comorbid conditions have also been associated with misdiagnosis.11 Our case scenario illustrates the frequent challenges physicians face when diagnosing patients who present with nonspecific symptoms and signs on a background of multiple, chronic comorbidities.
Contextual factors in the system and environment contribute to the potential for error.12 Examples include frequent interruptions, time pressure, poor handoffs, insufficient data, and multitasking.
In our scenario, incomplete data, time constraints, and multitasking in a busy work environment compelled the emergency department resident to rapidly synthesize information to establish a working diagnosis. Interpretations of radiographs by on-call radiology residents are similarly at risk of diagnostic error for the same reasons.13
Physician factors also influence diagnosis. Interestingly, physician certainty or uncertainty at the time of initial diagnosis does not uniformly appear to correlate with diagnostic accuracy. A recent study showed that physician confidence remained high regardless of the degree of difficulty in a given case, and degree of confidence also correlated poorly with whether the physician’s diagnosis was accurate.14
For patients admitted with a chief complaint of dyspnea, as in our scenario, Zwaan et al15 showed that “inappropriate selectivity” in reasoning contributed to an inaccurate diagnosis 23% of the time. Inappropriate selectivity, as defined by these authors, occurs when a probable diagnosis is not sufficiently considered and therefore is neither confirmed nor ruled out.
In our patient scenario, the failure to consider diagnoses other than heart failure and the inability to confirm a prior diagnosis of heart failure in the emergency department may contribute to a diagnostic error.
CASE CONTINUED: NO IMPROVEMENT OVER 3 DAYS
The night float resident, who has six other admissions this night, cannot ask the resident who evaluated this patient in the emergency department for further information because the shift has ended. The patient’s son left at the time of admission and is not available when the patient arrives on the medical ward.
The night float resident quickly examines the patient, enters admission orders, and signs the patient out to the intern and resident who will be caring for her during her hospitalization. The verbal handoff notes that the history was limited due to a language barrier. The initial problem list includes heart failure without a differential diagnosis, but notes that an elevated pro-BNP and chest radiograph confirm heart failure as the likely diagnosis.
Several hours after the night float resident has left, the resident presents this history to the attending physician, and together they decide to order her regular at-home medications, as well as deep vein thrombosis prophylaxis and echocardiography. In writing the orders, subcutaneous heparin once daily is erroneously entered instead of low-molecular-weight heparin daily, as this is the default in the medical record system. The tired resident fails to recognize this, and the pharmacist does not question it.
Over the next 2 days, the patient’s cough and shortness of breath persist.
On hospital day 3, two junior residents on the team (who finished their internship 2 weeks ago) review the attending radiologist’s interpretation of the chest radiograph. Unflagged, it confirms the resident’s interpretation but notes ill-defined, scattered, faint opacities. The residents believe that an interstitial pattern may be present and suggest that the patient may not have heart failure but rather a primary pulmonary disease. They bring this to the attention of their attending physician, who dismisses their concerns and comments that heart failure is a clinical diagnosis. The residents do not bring this idea up again to the attending physician.
That night, the float team is called by the nursing staff because of worsening oxygenation and cough. They add an intravenous corticosteroid, a broad-spectrum antibiotic, and an inhaled bronchodilator to the patient’s drug regimen.
How do cognitive errors predispose physicians to diagnostic errors?
When errors in diagnosis are reviewed retrospectively, cognitive or “thinking” errors are generally found, especially in nonprocedural or primary care specialties such as internal medicine, pediatrics, and emergency medicine.16,17
A widely accepted theory on how humans make decisions was described by the psychologists Tversky and Kahneman in 197418 and has been applied more recently to physicians’ diagnostic processes.19 Their dual process model theory states that persons with a requisite level of expertise use either the intuitive “system 1” process of thinking, based on pattern-recognition and heuristics, or the slower, more analytical “system 2” process.20 Experts disagree as to whether in medicine these processes represent a binary either-or model or a continuum21 with relative contributions of each process determined by the physician and the task.
What are some common types of cognitive error?
Experts agree that many diagnostic errors in medicine stem from decisions arrived at by inappropriate system 1 thinking due to biases. These biases have been identified and described as they relate to medicine, most notably by Croskerry.22
Several cognitive biases are illustrated in our clinical scenario:
The framing effect occurred when the emergency department resident listed the patient’s admitting diagnosis as heart failure during the clinical handoff of care.
Anchoring bias, as defined by Croskerry,22 is the tendency to lock onto salient features of the case too early in the diagnostic process and then to fail to adjust this initial diagnostic impression. This bias affected the admitting night float resident, primary intern, resident, and attending physician.
Diagnostic momentum, in turn, is a well-described phenomenon that clinical providers are especially vulnerable to in today’s environment of “copy-and-paste” medical records and numerous handovers of care as a consequence of residency duty-hour restrictions.23
Availability bias refers to commonly seen diagnoses like heart failure or recently seen diagnoses, which are more “available” to the human memory. These diagnoses, which spring to mind quickly, often trick providers into thinking that because they are more easily recalled, they are also more common or more likely.
Confirmation bias. The initial working diagnosis of heart failure may have led the medical team to place greater emphasis on the elevated pro-BNP and the chest radiograph to support the initial impression while ignoring findings such as weight loss that do not support this impression.
Blind obedience. Although the residents recognized the possibility of a primary pulmonary disease, they did not investigate this further. And when the attending physician dismissed their suggestion, they thus deferred to the person in authority or with a reputation of expertise.
Overconfidence bias. Despite minimal improvement in the patient’s clinical status after effective diuresis and the suggestion of alternative diagnoses by the residents, the attending physician remained confident—perhaps overconfident—in the diagnosis of heart failure and would not consider alternatives. Overconfidence bias has been well described and occurs when a medical provider believes too strongly in his or her ability to be correct and therefore fails to consider alternative diagnoses.24
Despite succumbing to overconfidence bias, the attending physician was able to overcome base-rate neglect, ie, failure to consider the prevalence of potential diagnoses in diagnostic reasoning.

Each of these biases, and others not mentioned, can lead to premature closure, which is the unfortunate root cause of many diagnostic errors and delays. We have illustrated several biases in our case scenario that led several physicians on the medical team to prematurely “close” on the diagnosis of heart failure (Table 1).
CASE CONTINUED: SURPRISES AND REASSESSMENT
On hospital day 4, the patient’s medication lists from her previous hospitalizations arrive, and the team is surprised to discover that she has been receiving infliximab for the past 3 to 4 months for her rheumatoid arthritis.
Additionally, an echocardiogram that was ordered on hospital day 1 but was lost in the cardiologist’s reading queue comes in and shows a normal ejection fraction with no evidence of elevated filling pressures.
Computed tomography of the chest reveals a reticular pattern with innumerable, tiny, 1- to 2-mm pulmonary nodules. The differential diagnosis is expanded to include hypersensitivity pneumonitis, lymphoma, fungal infection, and miliary tuberculosis.
How do faulty systems contribute to diagnostic error?
It is increasingly recognized that diagnostic errors can occur as a result of cognitive error, systems-based error, or quite commonly, both. Graber et al17 analyzed 100 cases of diagnostic error and determined that while cognitive errors did occur in most of them, nearly half the time both cognitive and systems-based errors contributed simultaneously.17 Observers have further delineated the importance of the systems context and how it affects our thinking.25
In this case, the language barrier, lack of availability of family, and inability to promptly utilize interpreter services contributed to early problems in acquiring a detailed history and a complete medication list that included the immunosuppressant infliximab. Later, a systems error led to a delay in the interpretation of an echocardiogram. Each of these factors, if prevented, would have presumably resulted in expansion of the differential diagnosis and earlier arrival at the correct diagnosis.
CASE CONTINUED: THE PATIENT DIES OF TUBERCULOSIS
The patient is moved to a negative pressure room, and the pulmonary consultants recommend bronchoscopy. During the procedure, the patient suffers acute respiratory failure, is intubated, and is transferred to the medical intensive care unit, where a saddle pulmonary embolism is diagnosed by computed tomographic angiography.
One day later, the sputum culture from the bronchoscopy returns as positive for acid-fast bacilli. A four-drug regimen for tuberculosis is started. The patient continues to have a downward course and expires 2 weeks later. Autopsy reveals miliary tuberculosis.
What is the frequency of diagnostic error in medicine?
Diagnostic error is estimated to have a frequency of 10% to 20%.24 Rates of diagnostic error are similar irrespective of method of determination, eg, from autopsy,3 standardized patients (ie, actors presenting with scripted scenarios),26 or case reviews.27 Patient surveys report patient-perceived harm from diagnostic error at a rate of 35% to 42%.28,29 The landmark Harvard Medical Practice Study found that 17% of all adverse events were attributable to diagnostic error.30
Diagnostic error is the most common type of medical error in nonprocedural medical fields.31 It causes a disproportionately large amount of morbidity and death.
Diagnostic error is the most common cause of malpractice claims in the United States. In inpatient and outpatient settings, for both medical and surgical patients, it accounted for 45.9% of all outpatient malpractice claims in 2009, making it the most common reason for medical malpractice litigation.32 A 2013 study indicated that diagnostic error is more common, more expensive, and two times more likely to result in death than any other category of error.33
CASE CONTINUED: MORBIDITY AND MORTALITY CONFERENCE
The patient’s case is brought to a morbidity and mortality conference for discussion. The systems issues in the case—including medication reconciliation, availability of interpreters, and timing and process of echocardiogram readings—are all discussed, but clinical reasoning and cognitive errors made in the case are avoided.
Why are cognitive errors often neglected in discussions of medical error?
Historically, openly discussing error in medicine has been difficult. Over the past decade, however, and fueled by the landmark Institute of Medicine report To Err is Human,34 the healthcare community has made substantial strides in identifying and talking about systems factors as a cause of preventable medical error.34,35
While systems contributions to medical error are inherently “external” to physicians and other healthcare providers, the cognitive contributions to error are inherently “internal” and are often considered personal. This has led to diagnostic error being kept out of many patient safety conversations. Further, while the solutions to systems errors are often tangible, such as implementing a fall prevention program or changing the physical packaging of a medication to reduce a medication dispensing or administration error, solutions to cognitive errors are generally considered more challenging to address by organizations trying to improve patient safety.
How can hospitals and department leaders do better?
Healthcare organizations and leaders of clinical teams or departments can implement several strategies.36
First, they can seek out and analyze the causes of diagnostic errors that are occurring locally in their institution and learn from their diagnostic errors, such as the one in our clinical scenario.
Second, they can promote a culture of open communication and questioning around diagnosis. Trainees, physicians, and nurses should be comfortable questioning each other, including those higher up in the hierarchy, by saying, “I’m not sure” or “What else could this be?” to help reduce cognitive bias and expand the diagnostic possibilities.
Similarly, developing strategies to promote feedback on diagnosis among physicians will allow us all to learn from our diagnostic mistakes.
Use of the electronic medical record to assist in follow-up of pending diagnostic studies and patient return visits is yet another strategy.
Finally, healthcare organizations can adopt strategies to promote patient involvement in diagnosis, such as providing patients with copies of their test results and discharge summaries, encouraging the use of electronic patient communication portals, and empowering patients to ask questions related to their diagnosis. Prioritizing potential solutions to reduce diagnostic errors may be helpful in situations, depending on the context and environment, in which all proposed interventions may not be possible.
CASE CONTINUED: LEARNING FROM MISTAKES
The attending physician and resident in the case meet after the conference to review their clinical decision-making. Both are interested in learning from this case and improving their diagnostic skills in the future.
What specific steps can clinicians take to mitigate cognitive bias in daily practice?
In addition to continuing to expand one’s medical knowledge and gain more clinical experience, we can suggest several small steps to busy clinicians, taken individually or in combination with others that may improve diagnostic skills by reducing the potential for biased thinking in clinical practice.

Think about your thinking. Our first recommendation would be to become more familiar with the dual process theory of clinical cognition (Figure 1).37,38 This theoretical framework may be very helpful as a foundation from which to build better thinking skills. Physicians, especially residents, and students can be taught these concepts and their potential to contribute to diagnostic errors, and can use these skills to recognize those contributions in others’ diagnostic practices and even in their own.39
Facilitating metacognition, or “thinking about one’s thinking,” may help clinicians catch themselves in thinking traps and provide the opportunity to reflect on biases retrospectively, as a double check or an opportunity to learn from a mistake.
Recognize your emotions. Gaining an understanding of the effect of one’s emotions on decision-making also can help clinicians free themselves of bias. As human beings, healthcare professionals are susceptible to emotion, and the best approach to mitigate the emotional influences may be to consciously name them and adjust for them.40
Because it is impractical to apply slow, analytical system 2 approaches to every case, skills that hone and develop more accurate, reliable system 1 thinking are crucial. Gaining broad exposure to increased numbers of cases may be the most reliable way to build an experiential repertoire of “illness scripts,” but there are ways to increase the experiential value of any case with a few techniques that have potential to promote better intuition.41
Embracing uncertainty in the early diagnostic process and envisioning the worst-case scenario in a case allows the consideration of additional diagnostic paths outside of the current working diagnosis, potentially priming the clinician to look for and recognize early warning signs that could argue against the initial diagnosis at a time when an adjustment could be made to prevent a bad outcome.
Practice progressive problem-solving,42 a technique in which the physician creates additional challenges to increase the cognitive burden of a “routine” case in an effort to train his or her mind and sharpen intuition. An example of this practice is contemplating a backup treatment plan in advance in the event of a poor response to or an adverse effect of treatment. Highly rated physicians and teachers perform this regularly.43,44 Other ways to maximize the learning value of an individual case include seeking feedback on patient outcomes, especially when a patient has been discharged or transferred to another provider’s care, or when the physician goes off service.
Simulation, traditionally used for procedural training, has potential as well. Cognitive simulation, such as case reports or virtual patient modules, have potential to enhance clinical reasoning skills as well, though possibly at greater cost of time and expense.
Decreased reliance on memory is likely to improve diagnostic reasoning. Systems tools such as checklists45 and health information technology46 have potential to reduce diagnostic errors, not by taking thinking away from the clinician but by relieving the cognitive load enough to facilitate greater effort toward reasoning.
Slow down. Finally, and perhaps most important, recent models of clinical expertise have suggested that mastery comes from having a robust intuitive method, with a sense of the limitations of the intuitive approach, an ability to recognize the need to perform more analytical reasoning in select cases, and the willingness to do so. In short, it may well be that the hallmark of a master clinician is the propensity to slow down when necessary.47
If one considers diagnosis a cognitive procedure, perhaps a brief “diagnostic time-out” for safety might afford an opportunity to recognize and mitigate biases and errors. There are likely many potential scripts for a good diagnostic time-out, but to be functional it should be brief and simple to facilitate consistent use. We have recommended the following four questions to our residents as a starting point, any of which could signal the need to switch to a slower, analytic approach.
Four-step diagnostic time-out
- What else can it be?
- Is there anything about the case that does not fit?
- Is it possible that multiple processes are going on?
- Do I need to slow down?
These questions can serve as a double check for an intuitively formed initial working diagnosis, incorporating many of the principles discussed above, in a way that would hopefully avoid undue burden on a busy clinician. These techniques, it must be acknowledged, have not yet been directly tied to reductions in diagnostic errors. However, diagnostic errors, as discussed, are very difficult to identify and study, and these techniques will serve mainly to improve habits that are likely to show benefits over much longer time periods than most studies can measure.
- Kassirer JP. Diagnostic reasoning. Ann Intern Med 1989; 110:893–900.
- Golodner L. How the public perceives patient safety. Newsletter of the National Patient Safety Foundation 2004; 1997:1–6.
- Shojania KG, Burton EC, McDonald KM, Goldman L. Changes in rates of autopsy-detected diagnostic errors over time: a systematic review. JAMA 2003; 289:2849–2856.
- Neale G, Woloshynowych M, Vincent C. Exploring the causes of adverse events in NHS hospital practice. J R Soc Med 2001; 94:322–330.
- Chellis M, Olson J, Augustine J, Hamilton G. Evaluation of missed diagnoses for patients admitted from the emergency department. Acad Emerg Med 2001; 8:125–130.
- Tallentire VR, Smith SE, Skinner J, Cameron HS. Exploring error in team-based acute care scenarios: an observational study from the United Kingdom. Acad Med 2012; 87:792–798.
- Green SM, Martinez-Rumayor A, Gregory SA, et al. Clinical uncertainty, diagnostic accuracy, and outcomes in emergency department patients presenting with dyspnea. Arch Intern Med 2008; 168:741–748.
- Pineda LA, Hathwar VS, Grant BJ. Clinical suspicion of fatal pulmonary embolism. Chest 2001; 120:791–795.
- Shojania KG, Burton EC, McDonald KM, Goldman L. The autopsy as an outcome and performance measure. Evid Rep Technol Assess (Summ) 2002; 58:1–5.
- Hertwig R, Meier N, Nickel C, et al. Correlates of diagnostic accuracy in patients with nonspecific complaints. Med Decis Making 2013; 33:533–543.
- Kostopoulou O, Delaney BC, Munro CW. Diagnostic difficulty and error in primary care—a systematic review. Fam Pract 2008; 25:400–413.
- Ogdie AR, Reilly JB, Pang WG, et al. Seen through their eyes: residents’ reflections on the cognitive and contextual components of diagnostic errors in medicine. Acad Med 2012; 87:1361–1367.
- Feldmann EJ, Jain VR, Rakoff S, Haramati LB. Radiology residents’ on-call interpretation of chest radiographs for congestive heart failure. Acad Radiol 2007; 14:1264–1270.
- Meyer AN, Payne VL, Meeks DW, Rao R, Singh H. Physicians’ diagnostic accuracy, confidence, and resource requests: a vignette study. JAMA Intern Med 2013; 173:1952–1958.
- Zwaan L, Thijs A, Wagner C, Timmermans DR. Does inappropriate selectivity in information use relate to diagnostic errors and patient harm? The diagnosis of patients with dyspnea. Soc Sci Med 2013; 91:32–38.
- Schiff GD, Hasan O, Kim S, et al. Diagnostic error in medicine: analysis of 583 physician-reported errors. Arch Intern Med 2009; 169:1881–1887.
- Graber ML, Franklin N, Gordon R. Diagnostic error in internal medicine. Arch Intern Med 2005; 165:1493–1499.
- Tversky A, Kahneman D. Judgment under uncertainty: heuristics and biases. Science 1974; 185:1124–1131.
- Kahneman D. Thinking, fast and slow. New York, NY: Farrar, Straus, and Giroux; 2011.
- Croskerry P. A universal model of diagnostic reasoning. Acad Med 2009; 84:1022–1028.
- Custers EJ. Medical education and cognitive continuum theory: an alternative perspective on medical problem solving and clinical reasoning. Acad Med 2013; 88:1074–1080.
- Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med 2003; 78:775–780.
- Hirschtick RE. A piece of my mind. Copy-and-paste. JAMA 2006; 295:2335–2336.
- Berner ES, Graber ML. Overconfidence as a cause of diagnostic error in medicine. Am J Med 2008;121(suppl 5):S2–S23.
- Henriksen K, Brady J. The pursuit of better diagnostic performance: a human factors perspective. BMJ Qual Saf 2013; 22(suppl 2):ii1–ii5.
- Peabody JW, Luck J, Jain S, Bertenthal D, Glassman P. Assessing the accuracy of administrative data in health information systems. Med Care 2004; 42:1066–1072.
- Hogan H, Healey F, Neale G, Thomson R, Vincent C, Black N. Preventable deaths due to problems in care in English acute hospitals: a retrospective case record review study. BMJ Qual Saf 2012; 21:737–745.
- Blendon RJ, DesRoches CM, Brodie M, et al. Views of practicing physicians and the public on medical errors. N Engl J Med 2002; 347:1933–1940.
- Burroughs TE, Waterman AD, Gallagher TH, et al. Patient concerns about medical errors in emergency departments. Acad Emerg Med 2005; 12:57–64.
- Leape LL, Brennan TA, Laird N, et al. The nature of adverse events in hospitalized patients. Results of the Harvard Medical Practice Study II. N Engl J Med 1991; 324:377–384.
- Thomas EJ, Studdert DM, Burstin HR, et al. Incidence and types of adverse events and negligent care in Utah and Colorado. Med Care 2000; 38:261–271.
- Bishop TF, Ryan AM, Casalino LP. Paid malpractice claims for adverse events in inpatient and outpatient settings. JAMA 2011; 305:2427–2431.
- Saber Tehrani AS, Lee H, Mathews SC, et al. 25-year summary of US malpractice claims for diagnostic errors 1986–2010: an analysis from the national practitioner data bank. BMJ Qual Saf 2013; 22:672–680.
- Kohn LT, Corrigan JM, Donaldson MS. To err is human: building a safer health system. Washington, DC: The National Academies Press; 2000.
- Singh H. Diagnostic errors: moving beyond ‘no respect’ and getting ready for prime time. BMJ Qual Saf 2013; 22:789–792.
- Graber ML, Trowbridge R, Myers JS, Umscheid CA, Strull W, Kanter MH. The next organizational challenge: finding and addressing diagnostic error. Jt Comm J Qual Patient Saf 2014; 40:102–110.
- Croskerry P. Clinical cognition and diagnostic error: applications of a dual process model of reasoning. Adv Health Sci Educ Theory Pract 2009; 14(suppl 1):27–35.
- Norman G. Dual processing and diagnostic errors. Adv Health Sci Educ Theory Pract 2009; 14(suppl 1):37–49.
- Reilly JB, Ogdie AR, Von Feldt JM, Myers JS. Teaching about how doctors think: a longitudinal curriculum in cognitive bias and diagnostic error for residents. BMJ Qual Saf 2013; 22:1044–1050.
- Croskerry P, Abbass A, Wu AW. Emotional influences in patient safety. J Patient Saf 2010; 6:199–205.
- Rajkomar A, Dhaliwal G. Improving diagnostic reasoning to improve patient safety. Perm J 2011; 15:68–73.
- Trowbridge RL, Dhaliwal G, Cosby KS. Educational agenda for diagnostic error reduction. BMJ Qual Saf 2013; 22(suppl 2):ii28–ii32.
- Sargeant J, Mann K, Sinclair D, et al. Learning in practice: experiences and perceptions of high-scoring physicians. Acad Med 2006; 81:655–660.
- Mylopoulos M, Lohfeld L, Norman GR, Dhaliwal G, Eva KW. Renowned physicians' perceptions of expert diagnostic practice. Acad Med 2012; 87:1413–1417.
- Sibbald M, de Bruin AB, van Merrienboer JJ. Checklists improve experts' diagnostic decisions. Med Educ 2013; 47:301–308.
- El-Kareh R, Hasan O, Schiff GD. Use of health information technology to reduce diagnostic errors. BMJ Qual Saf 2013; 22(suppl 2):ii40–ii51.
- Moulton CA, Regehr G, Mylopoulos M, MacRae HM. Slowing down when you should: a new model of expert judgment. Acad Med 2007; 82(suppl 10):S109–S116.
An elderly Spanish-speaking woman with morbid obesity, diabetes, hypertension, and rheumatoid arthritis presents to the emergency department with worsening shortness of breath and cough. She speaks only Spanish, so her son provides the history without the aid of an interpreter.
Her shortness of breath is most noticeable with exertion and has increased gradually over the past 2 months. She has a nonproductive cough. Her son has noticed decreased oral intake and weight loss over the past few weeks. She has neither traveled recently nor been in contact with anyone known to have an infectious disease.
A review of systems is otherwise negative: specifically, she denies chest pain, fevers, or chills. She saw her primary care physician 3 weeks ago for these complaints and was prescribed a 3-day course of azithromycin with no improvement.
Her medications include lisinopril, atenolol, glipizide, and metformin; her son believes she may be taking others as well but is not sure. He is also unsure of what treatment his mother has received for her rheumatoid arthritis, and most of her medical records are within another health system.
On physical examination, the patient is coughing and appears ill. Her temperature is 99.9°F (37.7°C), heart rate 105 beats per minute, blood pressure 140/70 mm Hg, respiratory rate 24 per minute, and oxygen saturation by pulse oximetry 89% on room air. Heart sounds are normal, jugular venous pressure cannot be assessed because of her obese body habitus, pulmonary examination demonstrates crackles in all lung fields, and lower-extremity edema is not present. Her extremities are warm and well perfused. Musculoskeletal examination reveals deformities of the joints in both hands consistent with rheumatoid arthritis.
Laboratory data:
- White blood cell count 13.0 × 109/L (reference range 3.7–11.0)
- Hemoglobin level 10 g/dL (11.5–15)
- Serum creatinine 1.0 mg/dL (0.7–1.4)
- Pro-brain-type natriuretic peptide (pro-BNP) level greater than the upper limit of normal.
A chest radiograph is obtained, and the resident radiologist’s preliminary impression is that it is consistent with pulmonary vascular congestion.
The patient is admitted for further diagnostic evaluation. The emergency department resident orders intravenous furosemide and signs out to the night float medicine resident that this is an “elderly woman with hypertension, diabetes, and heart failure being admitted for a heart failure exacerbation.”
What is the accuracy of a physician’s initial working diagnosis?
Diagnostic accuracy requires both clinical knowledge and problem-solving skills.1
A decade ago, a National Patient Safety Foundation survey2 found that one in six patients had suffered a medical error related to misdiagnosis. In a large systematic review of autopsy-based diagnostic errors, the theorized rate of major errors ranged from 8.4% to as high as 24.4%.3 A study by Neale et al4 found that admitting diagnoses were incorrect in 6% of cases. In emergency departments, inaccuracy rates of up to 12% have been described.5
What factors influence the prevalence of diagnostic errors?
Initial empiric treatments, such as intravenous furosemide in the above scenario, add to the challenge of diagnosis in acute care settings and can influence clinical decisions made by subsequent providers.6
Nonspecific or vague symptoms make diagnosis especially challenging. Shortness of breath, for example, is a common chief complaint in medical patients, as in this case. Green et al7 found emergency department physicians reported clinical uncertainty for a diagnosis of heart failure in 31% of patients evaluated for “dyspnea.” Pulmonary embolism and pulmonary tuberculosis are also in the differential diagnosis for our patient, with studies reporting a misdiagnosis rate of 55% for pulmonary embolism8 and 50% for pulmonary tuberculosis.9
Hertwig et al,10 describing the diagnostic process in patients presenting to emergency departments with a nonspecific constellation of symptoms, found particularly low rates of agreement between the initial diagnostic impression and the final, correct one. In fact, the actual diagnosis was only in the physician’s initial “top three” differential diagnoses 29% to 83% of the time.
Atypical presentations of common diseases, initial nonspecific presentations of common diseases, and confounding comorbid conditions have also been associated with misdiagnosis.11 Our case scenario illustrates the frequent challenges physicians face when diagnosing patients who present with nonspecific symptoms and signs on a background of multiple, chronic comorbidities.
Contextual factors in the system and environment contribute to the potential for error.12 Examples include frequent interruptions, time pressure, poor handoffs, insufficient data, and multitasking.
In our scenario, incomplete data, time constraints, and multitasking in a busy work environment compelled the emergency department resident to rapidly synthesize information to establish a working diagnosis. Interpretations of radiographs by on-call radiology residents are similarly at risk of diagnostic error for the same reasons.13
Physician factors also influence diagnosis. Interestingly, physician certainty or uncertainty at the time of initial diagnosis does not uniformly appear to correlate with diagnostic accuracy. A recent study showed that physician confidence remained high regardless of the degree of difficulty in a given case, and degree of confidence also correlated poorly with whether the physician’s diagnosis was accurate.14
For patients admitted with a chief complaint of dyspnea, as in our scenario, Zwaan et al15 showed that “inappropriate selectivity” in reasoning contributed to an inaccurate diagnosis 23% of the time. Inappropriate selectivity, as defined by these authors, occurs when a probable diagnosis is not sufficiently considered and therefore is neither confirmed nor ruled out.
In our patient scenario, the failure to consider diagnoses other than heart failure and the inability to confirm a prior diagnosis of heart failure in the emergency department may contribute to a diagnostic error.
CASE CONTINUED: NO IMPROVEMENT OVER 3 DAYS
The night float resident, who has six other admissions this night, cannot ask the resident who evaluated this patient in the emergency department for further information because the shift has ended. The patient’s son left at the time of admission and is not available when the patient arrives on the medical ward.
The night float resident quickly examines the patient, enters admission orders, and signs the patient out to the intern and resident who will be caring for her during her hospitalization. The verbal handoff notes that the history was limited due to a language barrier. The initial problem list includes heart failure without a differential diagnosis, but notes that an elevated pro-BNP and chest radiograph confirm heart failure as the likely diagnosis.
Several hours after the night float resident has left, the resident presents this history to the attending physician, and together they decide to order her regular at-home medications, as well as deep vein thrombosis prophylaxis and echocardiography. In writing the orders, subcutaneous heparin once daily is erroneously entered instead of low-molecular-weight heparin daily, as this is the default in the medical record system. The tired resident fails to recognize this, and the pharmacist does not question it.
Over the next 2 days, the patient’s cough and shortness of breath persist.
On hospital day 3, two junior residents on the team (who finished their internship 2 weeks ago) review the attending radiologist’s interpretation of the chest radiograph. Unflagged, it confirms the resident’s interpretation but notes ill-defined, scattered, faint opacities. The residents believe that an interstitial pattern may be present and suggest that the patient may not have heart failure but rather a primary pulmonary disease. They bring this to the attention of their attending physician, who dismisses their concerns and comments that heart failure is a clinical diagnosis. The residents do not bring this idea up again to the attending physician.
That night, the float team is called by the nursing staff because of worsening oxygenation and cough. They add an intravenous corticosteroid, a broad-spectrum antibiotic, and an inhaled bronchodilator to the patient’s drug regimen.
How do cognitive errors predispose physicians to diagnostic errors?
When errors in diagnosis are reviewed retrospectively, cognitive or “thinking” errors are generally found, especially in nonprocedural or primary care specialties such as internal medicine, pediatrics, and emergency medicine.16,17
A widely accepted theory on how humans make decisions was described by the psychologists Tversky and Kahneman in 197418 and has been applied more recently to physicians’ diagnostic processes.19 Their dual process model theory states that persons with a requisite level of expertise use either the intuitive “system 1” process of thinking, based on pattern-recognition and heuristics, or the slower, more analytical “system 2” process.20 Experts disagree as to whether in medicine these processes represent a binary either-or model or a continuum21 with relative contributions of each process determined by the physician and the task.
What are some common types of cognitive error?
Experts agree that many diagnostic errors in medicine stem from decisions arrived at by inappropriate system 1 thinking due to biases. These biases have been identified and described as they relate to medicine, most notably by Croskerry.22
Several cognitive biases are illustrated in our clinical scenario:
The framing effect occurred when the emergency department resident listed the patient’s admitting diagnosis as heart failure during the clinical handoff of care.
Anchoring bias, as defined by Croskerry,22 is the tendency to lock onto salient features of the case too early in the diagnostic process and then to fail to adjust this initial diagnostic impression. This bias affected the admitting night float resident, primary intern, resident, and attending physician.
Diagnostic momentum, in turn, is a well-described phenomenon that clinical providers are especially vulnerable to in today’s environment of “copy-and-paste” medical records and numerous handovers of care as a consequence of residency duty-hour restrictions.23
Availability bias refers to commonly seen diagnoses like heart failure or recently seen diagnoses, which are more “available” to the human memory. These diagnoses, which spring to mind quickly, often trick providers into thinking that because they are more easily recalled, they are also more common or more likely.
Confirmation bias. The initial working diagnosis of heart failure may have led the medical team to place greater emphasis on the elevated pro-BNP and the chest radiograph to support the initial impression while ignoring findings such as weight loss that do not support this impression.
Blind obedience. Although the residents recognized the possibility of a primary pulmonary disease, they did not investigate this further. And when the attending physician dismissed their suggestion, they thus deferred to the person in authority or with a reputation of expertise.
Overconfidence bias. Despite minimal improvement in the patient’s clinical status after effective diuresis and the suggestion of alternative diagnoses by the residents, the attending physician remained confident—perhaps overconfident—in the diagnosis of heart failure and would not consider alternatives. Overconfidence bias has been well described and occurs when a medical provider believes too strongly in his or her ability to be correct and therefore fails to consider alternative diagnoses.24
Despite succumbing to overconfidence bias, the attending physician was able to overcome base-rate neglect, ie, failure to consider the prevalence of potential diagnoses in diagnostic reasoning.
Each of these biases, and others not mentioned, can lead to premature closure, which is the unfortunate root cause of many diagnostic errors and delays. We have illustrated several biases in our case scenario that led several physicians on the medical team to prematurely “close” on the diagnosis of heart failure (Table 1).
CASE CONTINUED: SURPRISES AND REASSESSMENT
On hospital day 4, the patient’s medication lists from her previous hospitalizations arrive, and the team is surprised to discover that she has been receiving infliximab for the past 3 to 4 months for her rheumatoid arthritis.
Additionally, an echocardiogram that was ordered on hospital day 1 but was lost in the cardiologist’s reading queue comes in and shows a normal ejection fraction with no evidence of elevated filling pressures.
Computed tomography of the chest reveals a reticular pattern with innumerable, tiny, 1- to 2-mm pulmonary nodules. The differential diagnosis is expanded to include hypersensitivity pneumonitis, lymphoma, fungal infection, and miliary tuberculosis.
How do faulty systems contribute to diagnostic error?
It is increasingly recognized that diagnostic errors can occur as a result of cognitive error, systems-based error, or quite commonly, both. Graber et al17 analyzed 100 cases of diagnostic error and determined that while cognitive errors did occur in most of them, nearly half the time both cognitive and systems-based errors contributed simultaneously.17 Observers have further delineated the importance of the systems context and how it affects our thinking.25
In this case, the language barrier, lack of availability of family, and inability to promptly utilize interpreter services contributed to early problems in acquiring a detailed history and a complete medication list that included the immunosuppressant infliximab. Later, a systems error led to a delay in the interpretation of an echocardiogram. Each of these factors, if prevented, would have presumably resulted in expansion of the differential diagnosis and earlier arrival at the correct diagnosis.
CASE CONTINUED: THE PATIENT DIES OF TUBERCULOSIS
The patient is moved to a negative pressure room, and the pulmonary consultants recommend bronchoscopy. During the procedure, the patient suffers acute respiratory failure, is intubated, and is transferred to the medical intensive care unit, where a saddle pulmonary embolism is diagnosed by computed tomographic angiography.
One day later, the sputum culture from the bronchoscopy returns as positive for acid-fast bacilli. A four-drug regimen for tuberculosis is started. The patient continues to have a downward course and expires 2 weeks later. Autopsy reveals miliary tuberculosis.
What is the frequency of diagnostic error in medicine?
Diagnostic error is estimated to have a frequency of 10% to 20%.24 Rates of diagnostic error are similar irrespective of method of determination, eg, from autopsy,3 standardized patients (ie, actors presenting with scripted scenarios),26 or case reviews.27 Patient surveys report patient-perceived harm from diagnostic error at a rate of 35% to 42%.28,29 The landmark Harvard Medical Practice Study found that 17% of all adverse events were attributable to diagnostic error.30
Diagnostic error is the most common type of medical error in nonprocedural medical fields.31 It causes a disproportionately large amount of morbidity and death.
Diagnostic error is the most common cause of malpractice claims in the United States. In inpatient and outpatient settings, for both medical and surgical patients, it accounted for 45.9% of all outpatient malpractice claims in 2009, making it the most common reason for medical malpractice litigation.32 A 2013 study indicated that diagnostic error is more common, more expensive, and two times more likely to result in death than any other category of error.33
CASE CONTINUED: MORBIDITY AND MORTALITY CONFERENCE
The patient’s case is brought to a morbidity and mortality conference for discussion. The systems issues in the case—including medication reconciliation, availability of interpreters, and timing and process of echocardiogram readings—are all discussed, but clinical reasoning and cognitive errors made in the case are avoided.
Why are cognitive errors often neglected in discussions of medical error?
Historically, openly discussing error in medicine has been difficult. Over the past decade, however, and fueled by the landmark Institute of Medicine report To Err is Human,34 the healthcare community has made substantial strides in identifying and talking about systems factors as a cause of preventable medical error.34,35
While systems contributions to medical error are inherently “external” to physicians and other healthcare providers, the cognitive contributions to error are inherently “internal” and are often considered personal. This has led to diagnostic error being kept out of many patient safety conversations. Further, while the solutions to systems errors are often tangible, such as implementing a fall prevention program or changing the physical packaging of a medication to reduce a medication dispensing or administration error, solutions to cognitive errors are generally considered more challenging to address by organizations trying to improve patient safety.
How can hospitals and department leaders do better?
Healthcare organizations and leaders of clinical teams or departments can implement several strategies.36
First, they can seek out and analyze the causes of diagnostic errors that are occurring locally in their institution and learn from their diagnostic errors, such as the one in our clinical scenario.
Second, they can promote a culture of open communication and questioning around diagnosis. Trainees, physicians, and nurses should be comfortable questioning each other, including those higher up in the hierarchy, by saying, “I’m not sure” or “What else could this be?” to help reduce cognitive bias and expand the diagnostic possibilities.
Similarly, developing strategies to promote feedback on diagnosis among physicians will allow us all to learn from our diagnostic mistakes.
Use of the electronic medical record to assist in follow-up of pending diagnostic studies and patient return visits is yet another strategy.
Finally, healthcare organizations can adopt strategies to promote patient involvement in diagnosis, such as providing patients with copies of their test results and discharge summaries, encouraging the use of electronic patient communication portals, and empowering patients to ask questions related to their diagnosis. Prioritizing potential solutions to reduce diagnostic errors may be helpful in situations, depending on the context and environment, in which all proposed interventions may not be possible.
CASE CONTINUED: LEARNING FROM MISTAKES
The attending physician and resident in the case meet after the conference to review their clinical decision-making. Both are interested in learning from this case and improving their diagnostic skills in the future.
What specific steps can clinicians take to mitigate cognitive bias in daily practice?
In addition to continuing to expand one’s medical knowledge and gain more clinical experience, we can suggest several small steps to busy clinicians, taken individually or in combination with others that may improve diagnostic skills by reducing the potential for biased thinking in clinical practice.
Think about your thinking. Our first recommendation would be to become more familiar with the dual process theory of clinical cognition (Figure 1).37,38 This theoretical framework may be very helpful as a foundation from which to build better thinking skills. Physicians, especially residents, and students can be taught these concepts and their potential to contribute to diagnostic errors, and can use these skills to recognize those contributions in others’ diagnostic practices and even in their own.39
Facilitating metacognition, or “thinking about one’s thinking,” may help clinicians catch themselves in thinking traps and provide the opportunity to reflect on biases retrospectively, as a double check or an opportunity to learn from a mistake.
Recognize your emotions. Gaining an understanding of the effect of one’s emotions on decision-making also can help clinicians free themselves of bias. As human beings, healthcare professionals are susceptible to emotion, and the best approach to mitigate the emotional influences may be to consciously name them and adjust for them.40
Because it is impractical to apply slow, analytical system 2 approaches to every case, skills that hone and develop more accurate, reliable system 1 thinking are crucial. Gaining broad exposure to increased numbers of cases may be the most reliable way to build an experiential repertoire of “illness scripts,” but there are ways to increase the experiential value of any case with a few techniques that have potential to promote better intuition.41
Embracing uncertainty in the early diagnostic process and envisioning the worst-case scenario in a case allows the consideration of additional diagnostic paths outside of the current working diagnosis, potentially priming the clinician to look for and recognize early warning signs that could argue against the initial diagnosis at a time when an adjustment could be made to prevent a bad outcome.
Practice progressive problem-solving,42 a technique in which the physician creates additional challenges to increase the cognitive burden of a “routine” case in an effort to train his or her mind and sharpen intuition. An example of this practice is contemplating a backup treatment plan in advance in the event of a poor response to or an adverse effect of treatment. Highly rated physicians and teachers perform this regularly.43,44 Other ways to maximize the learning value of an individual case include seeking feedback on patient outcomes, especially when a patient has been discharged or transferred to another provider’s care, or when the physician goes off service.
Simulation, traditionally used for procedural training, has potential as well. Cognitive simulation, such as case reports or virtual patient modules, have potential to enhance clinical reasoning skills as well, though possibly at greater cost of time and expense.
Decreased reliance on memory is likely to improve diagnostic reasoning. Systems tools such as checklists45 and health information technology46 have potential to reduce diagnostic errors, not by taking thinking away from the clinician but by relieving the cognitive load enough to facilitate greater effort toward reasoning.
Slow down. Finally, and perhaps most important, recent models of clinical expertise have suggested that mastery comes from having a robust intuitive method, with a sense of the limitations of the intuitive approach, an ability to recognize the need to perform more analytical reasoning in select cases, and the willingness to do so. In short, it may well be that the hallmark of a master clinician is the propensity to slow down when necessary.47
If one considers diagnosis a cognitive procedure, perhaps a brief “diagnostic time-out” for safety might afford an opportunity to recognize and mitigate biases and errors. There are likely many potential scripts for a good diagnostic time-out, but to be functional it should be brief and simple to facilitate consistent use. We have recommended the following four questions to our residents as a starting point, any of which could signal the need to switch to a slower, analytic approach.
Four-step diagnostic time-out
- What else can it be?
- Is there anything about the case that does not fit?
- Is it possible that multiple processes are going on?
- Do I need to slow down?
These questions can serve as a double check for an intuitively formed initial working diagnosis, incorporating many of the principles discussed above, in a way that would hopefully avoid undue burden on a busy clinician. These techniques, it must be acknowledged, have not yet been directly tied to reductions in diagnostic errors. However, diagnostic errors, as discussed, are very difficult to identify and study, and these techniques will serve mainly to improve habits that are likely to show benefits over much longer time periods than most studies can measure.
An elderly Spanish-speaking woman with morbid obesity, diabetes, hypertension, and rheumatoid arthritis presents to the emergency department with worsening shortness of breath and cough. She speaks only Spanish, so her son provides the history without the aid of an interpreter.
Her shortness of breath is most noticeable with exertion and has increased gradually over the past 2 months. She has a nonproductive cough. Her son has noticed decreased oral intake and weight loss over the past few weeks. She has neither traveled recently nor been in contact with anyone known to have an infectious disease.
A review of systems is otherwise negative: specifically, she denies chest pain, fevers, or chills. She saw her primary care physician 3 weeks ago for these complaints and was prescribed a 3-day course of azithromycin with no improvement.
Her medications include lisinopril, atenolol, glipizide, and metformin; her son believes she may be taking others as well but is not sure. He is also unsure of what treatment his mother has received for her rheumatoid arthritis, and most of her medical records are within another health system.
On physical examination, the patient is coughing and appears ill. Her temperature is 99.9°F (37.7°C), heart rate 105 beats per minute, blood pressure 140/70 mm Hg, respiratory rate 24 per minute, and oxygen saturation by pulse oximetry 89% on room air. Heart sounds are normal, jugular venous pressure cannot be assessed because of her obese body habitus, pulmonary examination demonstrates crackles in all lung fields, and lower-extremity edema is not present. Her extremities are warm and well perfused. Musculoskeletal examination reveals deformities of the joints in both hands consistent with rheumatoid arthritis.
Laboratory data:
- White blood cell count 13.0 × 109/L (reference range 3.7–11.0)
- Hemoglobin level 10 g/dL (11.5–15)
- Serum creatinine 1.0 mg/dL (0.7–1.4)
- Pro-brain-type natriuretic peptide (pro-BNP) level greater than the upper limit of normal.
A chest radiograph is obtained, and the resident radiologist’s preliminary impression is that it is consistent with pulmonary vascular congestion.
The patient is admitted for further diagnostic evaluation. The emergency department resident orders intravenous furosemide and signs out to the night float medicine resident that this is an “elderly woman with hypertension, diabetes, and heart failure being admitted for a heart failure exacerbation.”
What is the accuracy of a physician’s initial working diagnosis?
Diagnostic accuracy requires both clinical knowledge and problem-solving skills.1
A decade ago, a National Patient Safety Foundation survey2 found that one in six patients had suffered a medical error related to misdiagnosis. In a large systematic review of autopsy-based diagnostic errors, the theorized rate of major errors ranged from 8.4% to as high as 24.4%.3 A study by Neale et al4 found that admitting diagnoses were incorrect in 6% of cases. In emergency departments, inaccuracy rates of up to 12% have been described.5
What factors influence the prevalence of diagnostic errors?
Initial empiric treatments, such as intravenous furosemide in the above scenario, add to the challenge of diagnosis in acute care settings and can influence clinical decisions made by subsequent providers.6
Nonspecific or vague symptoms make diagnosis especially challenging. Shortness of breath, for example, is a common chief complaint in medical patients, as in this case. Green et al7 found emergency department physicians reported clinical uncertainty for a diagnosis of heart failure in 31% of patients evaluated for “dyspnea.” Pulmonary embolism and pulmonary tuberculosis are also in the differential diagnosis for our patient, with studies reporting a misdiagnosis rate of 55% for pulmonary embolism8 and 50% for pulmonary tuberculosis.9
Hertwig et al,10 describing the diagnostic process in patients presenting to emergency departments with a nonspecific constellation of symptoms, found particularly low rates of agreement between the initial diagnostic impression and the final, correct one. In fact, the actual diagnosis was only in the physician’s initial “top three” differential diagnoses 29% to 83% of the time.
Atypical presentations of common diseases, initial nonspecific presentations of common diseases, and confounding comorbid conditions have also been associated with misdiagnosis.11 Our case scenario illustrates the frequent challenges physicians face when diagnosing patients who present with nonspecific symptoms and signs on a background of multiple, chronic comorbidities.
Contextual factors in the system and environment contribute to the potential for error.12 Examples include frequent interruptions, time pressure, poor handoffs, insufficient data, and multitasking.
In our scenario, incomplete data, time constraints, and multitasking in a busy work environment compelled the emergency department resident to rapidly synthesize information to establish a working diagnosis. Interpretations of radiographs by on-call radiology residents are similarly at risk of diagnostic error for the same reasons.13
Physician factors also influence diagnosis. Interestingly, physician certainty or uncertainty at the time of initial diagnosis does not uniformly appear to correlate with diagnostic accuracy. A recent study showed that physician confidence remained high regardless of the degree of difficulty in a given case, and degree of confidence also correlated poorly with whether the physician’s diagnosis was accurate.14
For patients admitted with a chief complaint of dyspnea, as in our scenario, Zwaan et al15 showed that “inappropriate selectivity” in reasoning contributed to an inaccurate diagnosis 23% of the time. Inappropriate selectivity, as defined by these authors, occurs when a probable diagnosis is not sufficiently considered and therefore is neither confirmed nor ruled out.
In our patient scenario, the failure to consider diagnoses other than heart failure and the inability to confirm a prior diagnosis of heart failure in the emergency department may contribute to a diagnostic error.
CASE CONTINUED: NO IMPROVEMENT OVER 3 DAYS
The night float resident, who has six other admissions this night, cannot ask the resident who evaluated this patient in the emergency department for further information because the shift has ended. The patient’s son left at the time of admission and is not available when the patient arrives on the medical ward.
The night float resident quickly examines the patient, enters admission orders, and signs the patient out to the intern and resident who will be caring for her during her hospitalization. The verbal handoff notes that the history was limited due to a language barrier. The initial problem list includes heart failure without a differential diagnosis, but notes that an elevated pro-BNP and chest radiograph confirm heart failure as the likely diagnosis.
Several hours after the night float resident has left, the resident presents this history to the attending physician, and together they decide to order her regular at-home medications, as well as deep vein thrombosis prophylaxis and echocardiography. In writing the orders, subcutaneous heparin once daily is erroneously entered instead of low-molecular-weight heparin daily, as this is the default in the medical record system. The tired resident fails to recognize this, and the pharmacist does not question it.
Over the next 2 days, the patient’s cough and shortness of breath persist.
On hospital day 3, two junior residents on the team (who finished their internship 2 weeks ago) review the attending radiologist’s interpretation of the chest radiograph. Unflagged, it confirms the resident’s interpretation but notes ill-defined, scattered, faint opacities. The residents believe that an interstitial pattern may be present and suggest that the patient may not have heart failure but rather a primary pulmonary disease. They bring this to the attention of their attending physician, who dismisses their concerns and comments that heart failure is a clinical diagnosis. The residents do not bring this idea up again to the attending physician.
That night, the float team is called by the nursing staff because of worsening oxygenation and cough. They add an intravenous corticosteroid, a broad-spectrum antibiotic, and an inhaled bronchodilator to the patient’s drug regimen.
How do cognitive errors predispose physicians to diagnostic errors?
When errors in diagnosis are reviewed retrospectively, cognitive or “thinking” errors are generally found, especially in nonprocedural or primary care specialties such as internal medicine, pediatrics, and emergency medicine.16,17
A widely accepted theory on how humans make decisions was described by the psychologists Tversky and Kahneman in 197418 and has been applied more recently to physicians’ diagnostic processes.19 Their dual process model theory states that persons with a requisite level of expertise use either the intuitive “system 1” process of thinking, based on pattern-recognition and heuristics, or the slower, more analytical “system 2” process.20 Experts disagree as to whether in medicine these processes represent a binary either-or model or a continuum21 with relative contributions of each process determined by the physician and the task.
What are some common types of cognitive error?
Experts agree that many diagnostic errors in medicine stem from decisions arrived at by inappropriate system 1 thinking due to biases. These biases have been identified and described as they relate to medicine, most notably by Croskerry.22
Several cognitive biases are illustrated in our clinical scenario:
The framing effect occurred when the emergency department resident listed the patient’s admitting diagnosis as heart failure during the clinical handoff of care.
Anchoring bias, as defined by Croskerry,22 is the tendency to lock onto salient features of the case too early in the diagnostic process and then to fail to adjust this initial diagnostic impression. This bias affected the admitting night float resident, primary intern, resident, and attending physician.
Diagnostic momentum, in turn, is a well-described phenomenon that clinical providers are especially vulnerable to in today’s environment of “copy-and-paste” medical records and numerous handovers of care as a consequence of residency duty-hour restrictions.23
Availability bias refers to commonly seen diagnoses like heart failure or recently seen diagnoses, which are more “available” to the human memory. These diagnoses, which spring to mind quickly, often trick providers into thinking that because they are more easily recalled, they are also more common or more likely.
Confirmation bias. The initial working diagnosis of heart failure may have led the medical team to place greater emphasis on the elevated pro-BNP and the chest radiograph to support the initial impression while ignoring findings such as weight loss that do not support this impression.
Blind obedience. Although the residents recognized the possibility of a primary pulmonary disease, they did not investigate this further. And when the attending physician dismissed their suggestion, they thus deferred to the person in authority or with a reputation of expertise.
Overconfidence bias. Despite minimal improvement in the patient’s clinical status after effective diuresis and the suggestion of alternative diagnoses by the residents, the attending physician remained confident—perhaps overconfident—in the diagnosis of heart failure and would not consider alternatives. Overconfidence bias has been well described and occurs when a medical provider believes too strongly in his or her ability to be correct and therefore fails to consider alternative diagnoses.24
Despite succumbing to overconfidence bias, the attending physician was able to overcome base-rate neglect, ie, failure to consider the prevalence of potential diagnoses in diagnostic reasoning.
Each of these biases, and others not mentioned, can lead to premature closure, which is the unfortunate root cause of many diagnostic errors and delays. We have illustrated several biases in our case scenario that led several physicians on the medical team to prematurely “close” on the diagnosis of heart failure (Table 1).
CASE CONTINUED: SURPRISES AND REASSESSMENT
On hospital day 4, the patient’s medication lists from her previous hospitalizations arrive, and the team is surprised to discover that she has been receiving infliximab for the past 3 to 4 months for her rheumatoid arthritis.
Additionally, an echocardiogram that was ordered on hospital day 1 but was lost in the cardiologist’s reading queue comes in and shows a normal ejection fraction with no evidence of elevated filling pressures.
Computed tomography of the chest reveals a reticular pattern with innumerable, tiny, 1- to 2-mm pulmonary nodules. The differential diagnosis is expanded to include hypersensitivity pneumonitis, lymphoma, fungal infection, and miliary tuberculosis.
How do faulty systems contribute to diagnostic error?
It is increasingly recognized that diagnostic errors can occur as a result of cognitive error, systems-based error, or quite commonly, both. Graber et al17 analyzed 100 cases of diagnostic error and determined that while cognitive errors did occur in most of them, nearly half the time both cognitive and systems-based errors contributed simultaneously.17 Observers have further delineated the importance of the systems context and how it affects our thinking.25
In this case, the language barrier, lack of availability of family, and inability to promptly utilize interpreter services contributed to early problems in acquiring a detailed history and a complete medication list that included the immunosuppressant infliximab. Later, a systems error led to a delay in the interpretation of an echocardiogram. Each of these factors, if prevented, would have presumably resulted in expansion of the differential diagnosis and earlier arrival at the correct diagnosis.
CASE CONTINUED: THE PATIENT DIES OF TUBERCULOSIS
The patient is moved to a negative pressure room, and the pulmonary consultants recommend bronchoscopy. During the procedure, the patient suffers acute respiratory failure, is intubated, and is transferred to the medical intensive care unit, where a saddle pulmonary embolism is diagnosed by computed tomographic angiography.
One day later, the sputum culture from the bronchoscopy returns as positive for acid-fast bacilli. A four-drug regimen for tuberculosis is started. The patient continues to have a downward course and expires 2 weeks later. Autopsy reveals miliary tuberculosis.
What is the frequency of diagnostic error in medicine?
Diagnostic error is estimated to have a frequency of 10% to 20%.24 Rates of diagnostic error are similar irrespective of method of determination, eg, from autopsy,3 standardized patients (ie, actors presenting with scripted scenarios),26 or case reviews.27 Patient surveys report patient-perceived harm from diagnostic error at a rate of 35% to 42%.28,29 The landmark Harvard Medical Practice Study found that 17% of all adverse events were attributable to diagnostic error.30
Diagnostic error is the most common type of medical error in nonprocedural medical fields.31 It causes a disproportionately large amount of morbidity and death.
Diagnostic error is the most common cause of malpractice claims in the United States. In inpatient and outpatient settings, for both medical and surgical patients, it accounted for 45.9% of all outpatient malpractice claims in 2009, making it the most common reason for medical malpractice litigation.32 A 2013 study indicated that diagnostic error is more common, more expensive, and two times more likely to result in death than any other category of error.33
CASE CONTINUED: MORBIDITY AND MORTALITY CONFERENCE
The patient’s case is brought to a morbidity and mortality conference for discussion. The systems issues in the case—including medication reconciliation, availability of interpreters, and timing and process of echocardiogram readings—are all discussed, but clinical reasoning and cognitive errors made in the case are avoided.
Why are cognitive errors often neglected in discussions of medical error?
Historically, openly discussing error in medicine has been difficult. Over the past decade, however, and fueled by the landmark Institute of Medicine report To Err is Human,34 the healthcare community has made substantial strides in identifying and talking about systems factors as a cause of preventable medical error.34,35
While systems contributions to medical error are inherently “external” to physicians and other healthcare providers, the cognitive contributions to error are inherently “internal” and are often considered personal. This has led to diagnostic error being kept out of many patient safety conversations. Further, while the solutions to systems errors are often tangible, such as implementing a fall prevention program or changing the physical packaging of a medication to reduce a medication dispensing or administration error, solutions to cognitive errors are generally considered more challenging to address by organizations trying to improve patient safety.
How can hospitals and department leaders do better?
Healthcare organizations and leaders of clinical teams or departments can implement several strategies.36
First, they can seek out and analyze the causes of diagnostic errors that are occurring locally in their institution and learn from their diagnostic errors, such as the one in our clinical scenario.
Second, they can promote a culture of open communication and questioning around diagnosis. Trainees, physicians, and nurses should be comfortable questioning each other, including those higher up in the hierarchy, by saying, “I’m not sure” or “What else could this be?” to help reduce cognitive bias and expand the diagnostic possibilities.
Similarly, developing strategies to promote feedback on diagnosis among physicians will allow us all to learn from our diagnostic mistakes.
Use of the electronic medical record to assist in follow-up of pending diagnostic studies and patient return visits is yet another strategy.
Finally, healthcare organizations can adopt strategies to promote patient involvement in diagnosis, such as providing patients with copies of their test results and discharge summaries, encouraging the use of electronic patient communication portals, and empowering patients to ask questions related to their diagnosis. Prioritizing potential solutions to reduce diagnostic errors may be helpful in situations, depending on the context and environment, in which all proposed interventions may not be possible.
CASE CONTINUED: LEARNING FROM MISTAKES
The attending physician and resident in the case meet after the conference to review their clinical decision-making. Both are interested in learning from this case and improving their diagnostic skills in the future.
What specific steps can clinicians take to mitigate cognitive bias in daily practice?
In addition to continuing to expand one’s medical knowledge and gain more clinical experience, we can suggest several small steps to busy clinicians, taken individually or in combination with others that may improve diagnostic skills by reducing the potential for biased thinking in clinical practice.
Think about your thinking. Our first recommendation would be to become more familiar with the dual process theory of clinical cognition (Figure 1).37,38 This theoretical framework may be very helpful as a foundation from which to build better thinking skills. Physicians, especially residents, and students can be taught these concepts and their potential to contribute to diagnostic errors, and can use these skills to recognize those contributions in others’ diagnostic practices and even in their own.39
Facilitating metacognition, or “thinking about one’s thinking,” may help clinicians catch themselves in thinking traps and provide the opportunity to reflect on biases retrospectively, as a double check or an opportunity to learn from a mistake.
Recognize your emotions. Gaining an understanding of the effect of one’s emotions on decision-making also can help clinicians free themselves of bias. As human beings, healthcare professionals are susceptible to emotion, and the best approach to mitigate the emotional influences may be to consciously name them and adjust for them.40
Because it is impractical to apply slow, analytical system 2 approaches to every case, skills that hone and develop more accurate, reliable system 1 thinking are crucial. Gaining broad exposure to increased numbers of cases may be the most reliable way to build an experiential repertoire of “illness scripts,” but there are ways to increase the experiential value of any case with a few techniques that have potential to promote better intuition.41
Embracing uncertainty in the early diagnostic process and envisioning the worst-case scenario in a case allows the consideration of additional diagnostic paths outside of the current working diagnosis, potentially priming the clinician to look for and recognize early warning signs that could argue against the initial diagnosis at a time when an adjustment could be made to prevent a bad outcome.
Practice progressive problem-solving,42 a technique in which the physician creates additional challenges to increase the cognitive burden of a “routine” case in an effort to train his or her mind and sharpen intuition. An example of this practice is contemplating a backup treatment plan in advance in the event of a poor response to or an adverse effect of treatment. Highly rated physicians and teachers perform this regularly.43,44 Other ways to maximize the learning value of an individual case include seeking feedback on patient outcomes, especially when a patient has been discharged or transferred to another provider’s care, or when the physician goes off service.
Simulation, traditionally used for procedural training, has potential as well. Cognitive simulation, such as case reports or virtual patient modules, have potential to enhance clinical reasoning skills as well, though possibly at greater cost of time and expense.
Decreased reliance on memory is likely to improve diagnostic reasoning. Systems tools such as checklists45 and health information technology46 have potential to reduce diagnostic errors, not by taking thinking away from the clinician but by relieving the cognitive load enough to facilitate greater effort toward reasoning.
Slow down. Finally, and perhaps most important, recent models of clinical expertise have suggested that mastery comes from having a robust intuitive method, with a sense of the limitations of the intuitive approach, an ability to recognize the need to perform more analytical reasoning in select cases, and the willingness to do so. In short, it may well be that the hallmark of a master clinician is the propensity to slow down when necessary.47
If one considers diagnosis a cognitive procedure, perhaps a brief “diagnostic time-out” for safety might afford an opportunity to recognize and mitigate biases and errors. There are likely many potential scripts for a good diagnostic time-out, but to be functional it should be brief and simple to facilitate consistent use. We have recommended the following four questions to our residents as a starting point, any of which could signal the need to switch to a slower, analytic approach.
Four-step diagnostic time-out
- What else can it be?
- Is there anything about the case that does not fit?
- Is it possible that multiple processes are going on?
- Do I need to slow down?
These questions can serve as a double check for an intuitively formed initial working diagnosis, incorporating many of the principles discussed above, in a way that would hopefully avoid undue burden on a busy clinician. These techniques, it must be acknowledged, have not yet been directly tied to reductions in diagnostic errors. However, diagnostic errors, as discussed, are very difficult to identify and study, and these techniques will serve mainly to improve habits that are likely to show benefits over much longer time periods than most studies can measure.
- Kassirer JP. Diagnostic reasoning. Ann Intern Med 1989; 110:893–900.
- Golodner L. How the public perceives patient safety. Newsletter of the National Patient Safety Foundation 2004; 1997:1–6.
- Shojania KG, Burton EC, McDonald KM, Goldman L. Changes in rates of autopsy-detected diagnostic errors over time: a systematic review. JAMA 2003; 289:2849–2856.
- Neale G, Woloshynowych M, Vincent C. Exploring the causes of adverse events in NHS hospital practice. J R Soc Med 2001; 94:322–330.
- Chellis M, Olson J, Augustine J, Hamilton G. Evaluation of missed diagnoses for patients admitted from the emergency department. Acad Emerg Med 2001; 8:125–130.
- Tallentire VR, Smith SE, Skinner J, Cameron HS. Exploring error in team-based acute care scenarios: an observational study from the United Kingdom. Acad Med 2012; 87:792–798.
- Green SM, Martinez-Rumayor A, Gregory SA, et al. Clinical uncertainty, diagnostic accuracy, and outcomes in emergency department patients presenting with dyspnea. Arch Intern Med 2008; 168:741–748.
- Pineda LA, Hathwar VS, Grant BJ. Clinical suspicion of fatal pulmonary embolism. Chest 2001; 120:791–795.
- Shojania KG, Burton EC, McDonald KM, Goldman L. The autopsy as an outcome and performance measure. Evid Rep Technol Assess (Summ) 2002; 58:1–5.
- Hertwig R, Meier N, Nickel C, et al. Correlates of diagnostic accuracy in patients with nonspecific complaints. Med Decis Making 2013; 33:533–543.
- Kostopoulou O, Delaney BC, Munro CW. Diagnostic difficulty and error in primary care—a systematic review. Fam Pract 2008; 25:400–413.
- Ogdie AR, Reilly JB, Pang WG, et al. Seen through their eyes: residents’ reflections on the cognitive and contextual components of diagnostic errors in medicine. Acad Med 2012; 87:1361–1367.
- Feldmann EJ, Jain VR, Rakoff S, Haramati LB. Radiology residents’ on-call interpretation of chest radiographs for congestive heart failure. Acad Radiol 2007; 14:1264–1270.
- Meyer AN, Payne VL, Meeks DW, Rao R, Singh H. Physicians’ diagnostic accuracy, confidence, and resource requests: a vignette study. JAMA Intern Med 2013; 173:1952–1958.
- Zwaan L, Thijs A, Wagner C, Timmermans DR. Does inappropriate selectivity in information use relate to diagnostic errors and patient harm? The diagnosis of patients with dyspnea. Soc Sci Med 2013; 91:32–38.
- Schiff GD, Hasan O, Kim S, et al. Diagnostic error in medicine: analysis of 583 physician-reported errors. Arch Intern Med 2009; 169:1881–1887.
- Graber ML, Franklin N, Gordon R. Diagnostic error in internal medicine. Arch Intern Med 2005; 165:1493–1499.
- Tversky A, Kahneman D. Judgment under uncertainty: heuristics and biases. Science 1974; 185:1124–1131.
- Kahneman D. Thinking, fast and slow. New York, NY: Farrar, Straus, and Giroux; 2011.
- Croskerry P. A universal model of diagnostic reasoning. Acad Med 2009; 84:1022–1028.
- Custers EJ. Medical education and cognitive continuum theory: an alternative perspective on medical problem solving and clinical reasoning. Acad Med 2013; 88:1074–1080.
- Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med 2003; 78:775–780.
- Hirschtick RE. A piece of my mind. Copy-and-paste. JAMA 2006; 295:2335–2336.
- Berner ES, Graber ML. Overconfidence as a cause of diagnostic error in medicine. Am J Med 2008;121(suppl 5):S2–S23.
- Henriksen K, Brady J. The pursuit of better diagnostic performance: a human factors perspective. BMJ Qual Saf 2013; 22(suppl 2):ii1–ii5.
- Peabody JW, Luck J, Jain S, Bertenthal D, Glassman P. Assessing the accuracy of administrative data in health information systems. Med Care 2004; 42:1066–1072.
- Hogan H, Healey F, Neale G, Thomson R, Vincent C, Black N. Preventable deaths due to problems in care in English acute hospitals: a retrospective case record review study. BMJ Qual Saf 2012; 21:737–745.
- Blendon RJ, DesRoches CM, Brodie M, et al. Views of practicing physicians and the public on medical errors. N Engl J Med 2002; 347:1933–1940.
- Burroughs TE, Waterman AD, Gallagher TH, et al. Patient concerns about medical errors in emergency departments. Acad Emerg Med 2005; 12:57–64.
- Leape LL, Brennan TA, Laird N, et al. The nature of adverse events in hospitalized patients. Results of the Harvard Medical Practice Study II. N Engl J Med 1991; 324:377–384.
- Thomas EJ, Studdert DM, Burstin HR, et al. Incidence and types of adverse events and negligent care in Utah and Colorado. Med Care 2000; 38:261–271.
- Bishop TF, Ryan AM, Casalino LP. Paid malpractice claims for adverse events in inpatient and outpatient settings. JAMA 2011; 305:2427–2431.
- Saber Tehrani AS, Lee H, Mathews SC, et al. 25-year summary of US malpractice claims for diagnostic errors 1986–2010: an analysis from the national practitioner data bank. BMJ Qual Saf 2013; 22:672–680.
- Kohn LT, Corrigan JM, Donaldson MS. To err is human: building a safer health system. Washington, DC: The National Academies Press; 2000.
- Singh H. Diagnostic errors: moving beyond ‘no respect’ and getting ready for prime time. BMJ Qual Saf 2013; 22:789–792.
- Graber ML, Trowbridge R, Myers JS, Umscheid CA, Strull W, Kanter MH. The next organizational challenge: finding and addressing diagnostic error. Jt Comm J Qual Patient Saf 2014; 40:102–110.
- Croskerry P. Clinical cognition and diagnostic error: applications of a dual process model of reasoning. Adv Health Sci Educ Theory Pract 2009; 14(suppl 1):27–35.
- Norman G. Dual processing and diagnostic errors. Adv Health Sci Educ Theory Pract 2009; 14(suppl 1):37–49.
- Reilly JB, Ogdie AR, Von Feldt JM, Myers JS. Teaching about how doctors think: a longitudinal curriculum in cognitive bias and diagnostic error for residents. BMJ Qual Saf 2013; 22:1044–1050.
- Croskerry P, Abbass A, Wu AW. Emotional influences in patient safety. J Patient Saf 2010; 6:199–205.
- Rajkomar A, Dhaliwal G. Improving diagnostic reasoning to improve patient safety. Perm J 2011; 15:68–73.
- Trowbridge RL, Dhaliwal G, Cosby KS. Educational agenda for diagnostic error reduction. BMJ Qual Saf 2013; 22(suppl 2):ii28–ii32.
- Sargeant J, Mann K, Sinclair D, et al. Learning in practice: experiences and perceptions of high-scoring physicians. Acad Med 2006; 81:655–660.
- Mylopoulos M, Lohfeld L, Norman GR, Dhaliwal G, Eva KW. Renowned physicians' perceptions of expert diagnostic practice. Acad Med 2012; 87:1413–1417.
- Sibbald M, de Bruin AB, van Merrienboer JJ. Checklists improve experts' diagnostic decisions. Med Educ 2013; 47:301–308.
- El-Kareh R, Hasan O, Schiff GD. Use of health information technology to reduce diagnostic errors. BMJ Qual Saf 2013; 22(suppl 2):ii40–ii51.
- Moulton CA, Regehr G, Mylopoulos M, MacRae HM. Slowing down when you should: a new model of expert judgment. Acad Med 2007; 82(suppl 10):S109–S116.
- Kassirer JP. Diagnostic reasoning. Ann Intern Med 1989; 110:893–900.
- Golodner L. How the public perceives patient safety. Newsletter of the National Patient Safety Foundation 2004; 1997:1–6.
- Shojania KG, Burton EC, McDonald KM, Goldman L. Changes in rates of autopsy-detected diagnostic errors over time: a systematic review. JAMA 2003; 289:2849–2856.
- Neale G, Woloshynowych M, Vincent C. Exploring the causes of adverse events in NHS hospital practice. J R Soc Med 2001; 94:322–330.
- Chellis M, Olson J, Augustine J, Hamilton G. Evaluation of missed diagnoses for patients admitted from the emergency department. Acad Emerg Med 2001; 8:125–130.
- Tallentire VR, Smith SE, Skinner J, Cameron HS. Exploring error in team-based acute care scenarios: an observational study from the United Kingdom. Acad Med 2012; 87:792–798.
- Green SM, Martinez-Rumayor A, Gregory SA, et al. Clinical uncertainty, diagnostic accuracy, and outcomes in emergency department patients presenting with dyspnea. Arch Intern Med 2008; 168:741–748.
- Pineda LA, Hathwar VS, Grant BJ. Clinical suspicion of fatal pulmonary embolism. Chest 2001; 120:791–795.
- Shojania KG, Burton EC, McDonald KM, Goldman L. The autopsy as an outcome and performance measure. Evid Rep Technol Assess (Summ) 2002; 58:1–5.
- Hertwig R, Meier N, Nickel C, et al. Correlates of diagnostic accuracy in patients with nonspecific complaints. Med Decis Making 2013; 33:533–543.
- Kostopoulou O, Delaney BC, Munro CW. Diagnostic difficulty and error in primary care—a systematic review. Fam Pract 2008; 25:400–413.
- Ogdie AR, Reilly JB, Pang WG, et al. Seen through their eyes: residents’ reflections on the cognitive and contextual components of diagnostic errors in medicine. Acad Med 2012; 87:1361–1367.
- Feldmann EJ, Jain VR, Rakoff S, Haramati LB. Radiology residents’ on-call interpretation of chest radiographs for congestive heart failure. Acad Radiol 2007; 14:1264–1270.
- Meyer AN, Payne VL, Meeks DW, Rao R, Singh H. Physicians’ diagnostic accuracy, confidence, and resource requests: a vignette study. JAMA Intern Med 2013; 173:1952–1958.
- Zwaan L, Thijs A, Wagner C, Timmermans DR. Does inappropriate selectivity in information use relate to diagnostic errors and patient harm? The diagnosis of patients with dyspnea. Soc Sci Med 2013; 91:32–38.
- Schiff GD, Hasan O, Kim S, et al. Diagnostic error in medicine: analysis of 583 physician-reported errors. Arch Intern Med 2009; 169:1881–1887.
- Graber ML, Franklin N, Gordon R. Diagnostic error in internal medicine. Arch Intern Med 2005; 165:1493–1499.
- Tversky A, Kahneman D. Judgment under uncertainty: heuristics and biases. Science 1974; 185:1124–1131.
- Kahneman D. Thinking, fast and slow. New York, NY: Farrar, Straus, and Giroux; 2011.
- Croskerry P. A universal model of diagnostic reasoning. Acad Med 2009; 84:1022–1028.
- Custers EJ. Medical education and cognitive continuum theory: an alternative perspective on medical problem solving and clinical reasoning. Acad Med 2013; 88:1074–1080.
- Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med 2003; 78:775–780.
- Hirschtick RE. A piece of my mind. Copy-and-paste. JAMA 2006; 295:2335–2336.
- Berner ES, Graber ML. Overconfidence as a cause of diagnostic error in medicine. Am J Med 2008;121(suppl 5):S2–S23.
- Henriksen K, Brady J. The pursuit of better diagnostic performance: a human factors perspective. BMJ Qual Saf 2013; 22(suppl 2):ii1–ii5.
- Peabody JW, Luck J, Jain S, Bertenthal D, Glassman P. Assessing the accuracy of administrative data in health information systems. Med Care 2004; 42:1066–1072.
- Hogan H, Healey F, Neale G, Thomson R, Vincent C, Black N. Preventable deaths due to problems in care in English acute hospitals: a retrospective case record review study. BMJ Qual Saf 2012; 21:737–745.
- Blendon RJ, DesRoches CM, Brodie M, et al. Views of practicing physicians and the public on medical errors. N Engl J Med 2002; 347:1933–1940.
- Burroughs TE, Waterman AD, Gallagher TH, et al. Patient concerns about medical errors in emergency departments. Acad Emerg Med 2005; 12:57–64.
- Leape LL, Brennan TA, Laird N, et al. The nature of adverse events in hospitalized patients. Results of the Harvard Medical Practice Study II. N Engl J Med 1991; 324:377–384.
- Thomas EJ, Studdert DM, Burstin HR, et al. Incidence and types of adverse events and negligent care in Utah and Colorado. Med Care 2000; 38:261–271.
- Bishop TF, Ryan AM, Casalino LP. Paid malpractice claims for adverse events in inpatient and outpatient settings. JAMA 2011; 305:2427–2431.
- Saber Tehrani AS, Lee H, Mathews SC, et al. 25-year summary of US malpractice claims for diagnostic errors 1986–2010: an analysis from the national practitioner data bank. BMJ Qual Saf 2013; 22:672–680.
- Kohn LT, Corrigan JM, Donaldson MS. To err is human: building a safer health system. Washington, DC: The National Academies Press; 2000.
- Singh H. Diagnostic errors: moving beyond ‘no respect’ and getting ready for prime time. BMJ Qual Saf 2013; 22:789–792.
- Graber ML, Trowbridge R, Myers JS, Umscheid CA, Strull W, Kanter MH. The next organizational challenge: finding and addressing diagnostic error. Jt Comm J Qual Patient Saf 2014; 40:102–110.
- Croskerry P. Clinical cognition and diagnostic error: applications of a dual process model of reasoning. Adv Health Sci Educ Theory Pract 2009; 14(suppl 1):27–35.
- Norman G. Dual processing and diagnostic errors. Adv Health Sci Educ Theory Pract 2009; 14(suppl 1):37–49.
- Reilly JB, Ogdie AR, Von Feldt JM, Myers JS. Teaching about how doctors think: a longitudinal curriculum in cognitive bias and diagnostic error for residents. BMJ Qual Saf 2013; 22:1044–1050.
- Croskerry P, Abbass A, Wu AW. Emotional influences in patient safety. J Patient Saf 2010; 6:199–205.
- Rajkomar A, Dhaliwal G. Improving diagnostic reasoning to improve patient safety. Perm J 2011; 15:68–73.
- Trowbridge RL, Dhaliwal G, Cosby KS. Educational agenda for diagnostic error reduction. BMJ Qual Saf 2013; 22(suppl 2):ii28–ii32.
- Sargeant J, Mann K, Sinclair D, et al. Learning in practice: experiences and perceptions of high-scoring physicians. Acad Med 2006; 81:655–660.
- Mylopoulos M, Lohfeld L, Norman GR, Dhaliwal G, Eva KW. Renowned physicians' perceptions of expert diagnostic practice. Acad Med 2012; 87:1413–1417.
- Sibbald M, de Bruin AB, van Merrienboer JJ. Checklists improve experts' diagnostic decisions. Med Educ 2013; 47:301–308.
- El-Kareh R, Hasan O, Schiff GD. Use of health information technology to reduce diagnostic errors. BMJ Qual Saf 2013; 22(suppl 2):ii40–ii51.
- Moulton CA, Regehr G, Mylopoulos M, MacRae HM. Slowing down when you should: a new model of expert judgment. Acad Med 2007; 82(suppl 10):S109–S116.
KEY POINTS
- Diagnostic errors are common and lead to bad outcomes.
- Factors that increase the risk of diagnostic error include initial empiric treatment, nonspecific or vague symptoms, atypical presentation, confounding comorbid conditions, contextual factors, and physician factors.
- Common types of cognitive error include the framing effect, anchoring bias, diagnostic momentum, availability bias, confirmation bias, blind obedience, overconfidence bias, base-rate neglect, and premature closure.
- Organizations and leaders can implement strategies to reduce diagnostic errors.

What you need to know (and do) to prescribe the new drug flibanserin
It was a long road to approval by the US Food and Drug Administration (FDA), but flibanserin (Addyi) got the nod on August 18, 2015. Its New Drug Application (NDA) originally was filed October 27, 2009. The drug launched October 17, 2015.
Although there has been a lot of fanfare about approval of this drug, most of the coverage has focused on its status as the “first female Viagra”—a less than accurate depiction. For a more realistic and practical assessment of the drug, OBG Management turned to Michael Krychman, MD, executive director of the Southern California Center for Sexual Health and Survivorship Medicine in Newport Beach, to determine the types of information clinicians need to know to begin prescribing flibanserin. This article highlights 11 questions (and answers) to help you get started.
1. How did the FDA arrive
at its approval?
In 2012, the agency determined that female sexual dysfunction was one of 20 disease areas that warranted focused attention. In October 2014, as part of its intensified look at female sexual dysfunction, the FDA convened a 2-day meeting “to advance our understanding,” reports Andrea Fischer, FDA press officer.
“During the first day of the meeting, the FDA solicited patients’ perspectives on their condition and its impact on daily life. While this meeting did not focus on flibanserin, it provided an opportunity for the FDA to hear directly from patients about the impact of their condition,” Ms. Fischer says. During the second day of the meeting, the FDA “discussed scientific issues and challenges with experts in sexual medicine.”
As a result, by the time of the FDA’s June 4, 2015 Advisory Committee meeting on the flibanserin NDA, FDA physician-scientists were well versed in many nuances of female sexual function. That meeting included an open public hearing “that provided an opportunity for members of the public, including patients, to provide input specifically on the flibanserin application,” Ms. Fischer notes.
Nuances of the deliberations
“The FDA’s regulatory decision making on any drug product is a science-based process that carefully weighs each drug in terms of its risks and benefits to the patient population for which the drug would be indicated,” says Ms. Fischer.
The challenge in the case of flibanserin was determining whether the drug provides “clinically meaningful” improvements in sexual activity and desire.
“For many conditions and diseases, what constitutes ‘clinically meaningful’ is well known and accepted,” Ms. Fischer notes, “such as when something is cured or a severe symptom that is life-altering resolves completely. For others, this is not the case. For example, a condition that has a wide range of degree of severity can offer challenges in assessing what constitutes a clinically meaningful treatment effect. Ascertaining this requires a comprehensive knowledge of the disease, affected patient population, management strategies and the drug in question, as well as an ability to look at the clinical trial data taking this all into account.”
“In clinical trials, an important method for assessing the impact of a treatment on a patient’s symptoms, mental state, or functional status is through direct self-report using well developed and thoughtfully integrated patient-reported outcome (PRO) assessments,” Ms. Fischer says. “PROs can provide valuable information on the patient perspective when determining whether benefits outweigh risks, and they also are used to support medical product labeling claims, which are a key source of information for both health care providers and patients. PROs have been and continue to be a high priority as part of FDA’s commitment to advance patient-focused drug development, and we fully expect this to continue. The clinical trials in the flibanserin NDA all utilized PRO assessments.”
Those assessments found that patients taking flibanserin had a significant increase in “sexually satisfying events.” Three 24-week randomized controlled trials explored this endpoint for flibanserin (studies 1–3).
As for improvements in desire, the first 2 trials utilized an e-diary to assess this aspect of sexual function, while the 3rd trial utilized the Female Sexual Function Index (FSFI).
Although the e-diary reflected no statistically significant improvement in desire in the first 2 trials, the FSFI did find significant improvement in the 3rd trial. In addition, when the FSFI was considered across all 3 trials, results in the desire domain were consistent. (The FSFI was used as a secondary tool in the first 2 trials.)
In addition, sexual distress, as measured by the Female Sexual Distress Scale (FSDS), was decreased in the trials with use of flibanserin, notes Dr. Krychman. The Advisory Committee determined that these findings were sufficient to demonstrate clinically meaningful improvements with use of the drug.
Although the drug was approved by the FDA, the agency was sufficiently concerned about some of its potential risks (see questions 4 and 5) that it implemented rigorous mitigation strategies (see question 7). Additional investigations were requested by the agency, including drug-drug interaction, alcohol challenge, and driving studies.
2. What are the indications?
Flibanserin is intended for use in premenopausal women who have acquired, generalized hypoactive sexual desire disorder (HSDD). That diagnosis no longer is included in the 5th edition of the Diagnostic and
Statistical Manual of Mental Disorders but is described in drug package labeling as “low sexual desire that causes marked distress or interpersonal difficulty and is not due to:
- a coexisting medical or psychiatric condition,
- problems within the relationship, or
- the effects of a medication or other drug substance.”1
- the effects of a medication or other drug substance.”
Although the drug has been tested in both premenopausal and postmenopausal women, it was approved for use only in premenopausal women. Also note inclusion of the term “acquired” before the diagnosis of HSDD, indicating that the drug is inappropriate for women who have never experienced a period of normal sexual desire.
3. How is HSDD diagnosed?
One of the best screening tools is the
Decreased Sexual Desire Screener, says
Dr. Krychman. It is available at http://obgynalliance.com/files/fsd/DSDS_Pocketcard.pdf. This tool is a validated instrument to help clinicians identify what HSDD is and is not.
4. Does the drug carry
any warnings?
Yes, it carries a black box warning about the risks of hypotension and syncope:
- when alcohol is consumed by users of the drug. (Alcohol use is contraindicated.)
- when the drug is taken in conjunction with moderate or strong CYP3A4 inhibitors or by patients with hepatic impairment. (The drug is contraindicated in both circumstances.) See question 9 for a list of drugs that are CYP3A4 inhibitors.
5. Are there any other risks worth noting?
The medication can increase the risks of hypotension and syncope even without concomitant use of alcohol. For example, in clinical trials, hypotension was reported in 0.2% of flibanserin-treated women versus less than 0.1% of placebo users. And syncope was reported in 0.4% of flibanserin users versus 0.2% of placebo-treated patients. Flibanserin is prescribed as a once-daily medication that is to be taken at bedtime; the risks of hypotension and syncope are increased if flibanserin is taken during waking hours.
The risk of adverse effects when flibanserin is taken with alcohol is highlighted by one case reported in package labeling: A 54-year-old postmenopausal woman died after taking flibanserin (100 mg daily at bedtime) for 14 days. This patient had a history of hypertension and hypercholesterolemia and consumed a baseline amount of 1 to 3 alcoholic beverages daily. She died of acute alcohol intoxication, with a blood alcohol concentration of 0.289 g/dL.1 Whether this patient’s death was related to flibanserin use is unknown.1
It is interesting to note that, in the studies of flibanserin leading up to the drug’s approval, alcohol use was not an exclusion, says Dr. Krychman. “Approximately 58% of women were self-described as mild to moderate drinkers. The clinical program was extremely large—more than 11,000 women were studied.”
Flibanserin is currently not approved for use in postmenopausal women, and concomitant alcohol consumption is contraindicated.
6. What is the dose?
The recommended dose is one tablet of
100 mg daily. The drug is to be taken at
bedtime to reduce the risks of hypotension, syncope, accidental injury, and central nervous system (CNS) depression, which can occur even in the absence of alcohol.
7. Are there any requirements for clinicians who want to prescribe the drug?
Yes. Because of the risks of hypotension, syncope, and CNS depression, the drug is subject to Risk Evaluation and Mitigation Strategies (REMS), as determined by the FDA. To prescribe the drug, providers must:
- review its prescribing information
- review the Provider and Pharmacy Training Program
- complete and submit the Knowledge Assessment Form
- enroll in REMS by completing and submitting the Prescriber Enrollment Form.
Before giving a patient her initial prescription, the provider must counsel her about the risks of hypotension and syncope and the interaction with alcohol using the Patient-Provider Agreement Form. The provider must then complete that form, provide a designated portion of it to the patient, and retain the remainder for the patient’s file.
For more information and to download the relevant forms, visit https://www.addyirems.com.
8. What are the most common
adverse reactions to the drug?
According to package labeling, the most common adverse reactions, with an incidence greater than 2%, are dizziness, somnolence, nausea, fatigue, insomnia, and dry mouth.
Less common reactions include anxiety, constipation, abdominal pain, rash, sedation, and vertigo.
In studies of the drug, appendicitis was reported among 0.2% of flibanserin-treated patients, compared with no reports of appendicitis among placebo-treated patients. The FDA has requested additional investigation of the association, if any, between flibanserin and appendicitis.
9. What drug interactions are notable?
As stated earlier, the concomitant use of flibanserin with alcohol or a moderate or strong CYP3A4 inhibitor can result in severe hypotension and syncope. Flibanserin also should not be prescribed for patients who use other CNS depressants such as diphenhydramine, opioids, benzodiazepines, and hypnotic agents.
Some examples of strong CYP3A4 inhibitors are ketoconazole, itraconazole, posaconazole, clarithromycin, nefazodone, ritonavir, saquinavir, nelfinavir, indinavir, boceprevir, telaprevir, telithromycin, and conivaptan.
Moderate CYP3A4 inhibitors include amprenavir, atazanavir, ciprofloxacin, diltiazem, erythromycin, fluconazole, fosamprenavir, verapamil, and grapefruit juice.
In addition, the concomitant use of flibanserin with multiple weak CYP3A4 inhibitors—which include herbal supplements such as ginkgo and resveratrol and nonprescription drugs such as cimetidine—also may increase the risks of hypotension and syncope.
The concomitant use of flibanserin with digoxin increases the digoxin concentration and may lead to toxicity.
10. Is the drug safe in pregnancy
and lactation?
There are currently no data on the use of flibanserin in human pregnancy. In animals, fetal toxicity occurred only in the presence of significant maternal toxicity. Adverse effects included decreased fetal weight, structural anomalies, and increases in fetal loss when exposure exceeded 15 times the recommended human dosage.
As for the advisability of using flibanserin during lactation, it is unknown whether the drug is excreted in human milk, whether it might have adverse effects in the breastfed infant, or whether it affects milk production. Package labeling states: “Because of the potential for serious adverse reactions, including sedation in a breastfed infant, breastfeeding is not recommended during treatment with [flibanserin].”1
11. When should the drug
be discontinued?
If there is no improvement in sexual desire after an 8-week trial of flibanserin, the drug should be
discontinued.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
Reference
- Addyi [package insert]. Raleigh, NC: Sprout Pharmaceuticals; 2015.
It was a long road to approval by the US Food and Drug Administration (FDA), but flibanserin (Addyi) got the nod on August 18, 2015. Its New Drug Application (NDA) originally was filed October 27, 2009. The drug launched October 17, 2015.
Although there has been a lot of fanfare about approval of this drug, most of the coverage has focused on its status as the “first female Viagra”—a less than accurate depiction. For a more realistic and practical assessment of the drug, OBG Management turned to Michael Krychman, MD, executive director of the Southern California Center for Sexual Health and Survivorship Medicine in Newport Beach, to determine the types of information clinicians need to know to begin prescribing flibanserin. This article highlights 11 questions (and answers) to help you get started.
1. How did the FDA arrive
at its approval?
In 2012, the agency determined that female sexual dysfunction was one of 20 disease areas that warranted focused attention. In October 2014, as part of its intensified look at female sexual dysfunction, the FDA convened a 2-day meeting “to advance our understanding,” reports Andrea Fischer, FDA press officer.
“During the first day of the meeting, the FDA solicited patients’ perspectives on their condition and its impact on daily life. While this meeting did not focus on flibanserin, it provided an opportunity for the FDA to hear directly from patients about the impact of their condition,” Ms. Fischer says. During the second day of the meeting, the FDA “discussed scientific issues and challenges with experts in sexual medicine.”
As a result, by the time of the FDA’s June 4, 2015 Advisory Committee meeting on the flibanserin NDA, FDA physician-scientists were well versed in many nuances of female sexual function. That meeting included an open public hearing “that provided an opportunity for members of the public, including patients, to provide input specifically on the flibanserin application,” Ms. Fischer notes.
Nuances of the deliberations
“The FDA’s regulatory decision making on any drug product is a science-based process that carefully weighs each drug in terms of its risks and benefits to the patient population for which the drug would be indicated,” says Ms. Fischer.
The challenge in the case of flibanserin was determining whether the drug provides “clinically meaningful” improvements in sexual activity and desire.
“For many conditions and diseases, what constitutes ‘clinically meaningful’ is well known and accepted,” Ms. Fischer notes, “such as when something is cured or a severe symptom that is life-altering resolves completely. For others, this is not the case. For example, a condition that has a wide range of degree of severity can offer challenges in assessing what constitutes a clinically meaningful treatment effect. Ascertaining this requires a comprehensive knowledge of the disease, affected patient population, management strategies and the drug in question, as well as an ability to look at the clinical trial data taking this all into account.”
“In clinical trials, an important method for assessing the impact of a treatment on a patient’s symptoms, mental state, or functional status is through direct self-report using well developed and thoughtfully integrated patient-reported outcome (PRO) assessments,” Ms. Fischer says. “PROs can provide valuable information on the patient perspective when determining whether benefits outweigh risks, and they also are used to support medical product labeling claims, which are a key source of information for both health care providers and patients. PROs have been and continue to be a high priority as part of FDA’s commitment to advance patient-focused drug development, and we fully expect this to continue. The clinical trials in the flibanserin NDA all utilized PRO assessments.”
Those assessments found that patients taking flibanserin had a significant increase in “sexually satisfying events.” Three 24-week randomized controlled trials explored this endpoint for flibanserin (studies 1–3).
As for improvements in desire, the first 2 trials utilized an e-diary to assess this aspect of sexual function, while the 3rd trial utilized the Female Sexual Function Index (FSFI).
Although the e-diary reflected no statistically significant improvement in desire in the first 2 trials, the FSFI did find significant improvement in the 3rd trial. In addition, when the FSFI was considered across all 3 trials, results in the desire domain were consistent. (The FSFI was used as a secondary tool in the first 2 trials.)
In addition, sexual distress, as measured by the Female Sexual Distress Scale (FSDS), was decreased in the trials with use of flibanserin, notes Dr. Krychman. The Advisory Committee determined that these findings were sufficient to demonstrate clinically meaningful improvements with use of the drug.
Although the drug was approved by the FDA, the agency was sufficiently concerned about some of its potential risks (see questions 4 and 5) that it implemented rigorous mitigation strategies (see question 7). Additional investigations were requested by the agency, including drug-drug interaction, alcohol challenge, and driving studies.
2. What are the indications?
Flibanserin is intended for use in premenopausal women who have acquired, generalized hypoactive sexual desire disorder (HSDD). That diagnosis no longer is included in the 5th edition of the Diagnostic and
Statistical Manual of Mental Disorders but is described in drug package labeling as “low sexual desire that causes marked distress or interpersonal difficulty and is not due to:
- a coexisting medical or psychiatric condition,
- problems within the relationship, or
- the effects of a medication or other drug substance.”1
- the effects of a medication or other drug substance.”
Although the drug has been tested in both premenopausal and postmenopausal women, it was approved for use only in premenopausal women. Also note inclusion of the term “acquired” before the diagnosis of HSDD, indicating that the drug is inappropriate for women who have never experienced a period of normal sexual desire.
3. How is HSDD diagnosed?
One of the best screening tools is the
Decreased Sexual Desire Screener, says
Dr. Krychman. It is available at http://obgynalliance.com/files/fsd/DSDS_Pocketcard.pdf. This tool is a validated instrument to help clinicians identify what HSDD is and is not.
4. Does the drug carry
any warnings?
Yes, it carries a black box warning about the risks of hypotension and syncope:
- when alcohol is consumed by users of the drug. (Alcohol use is contraindicated.)
- when the drug is taken in conjunction with moderate or strong CYP3A4 inhibitors or by patients with hepatic impairment. (The drug is contraindicated in both circumstances.) See question 9 for a list of drugs that are CYP3A4 inhibitors.
5. Are there any other risks worth noting?
The medication can increase the risks of hypotension and syncope even without concomitant use of alcohol. For example, in clinical trials, hypotension was reported in 0.2% of flibanserin-treated women versus less than 0.1% of placebo users. And syncope was reported in 0.4% of flibanserin users versus 0.2% of placebo-treated patients. Flibanserin is prescribed as a once-daily medication that is to be taken at bedtime; the risks of hypotension and syncope are increased if flibanserin is taken during waking hours.
The risk of adverse effects when flibanserin is taken with alcohol is highlighted by one case reported in package labeling: A 54-year-old postmenopausal woman died after taking flibanserin (100 mg daily at bedtime) for 14 days. This patient had a history of hypertension and hypercholesterolemia and consumed a baseline amount of 1 to 3 alcoholic beverages daily. She died of acute alcohol intoxication, with a blood alcohol concentration of 0.289 g/dL.1 Whether this patient’s death was related to flibanserin use is unknown.1
It is interesting to note that, in the studies of flibanserin leading up to the drug’s approval, alcohol use was not an exclusion, says Dr. Krychman. “Approximately 58% of women were self-described as mild to moderate drinkers. The clinical program was extremely large—more than 11,000 women were studied.”
Flibanserin is currently not approved for use in postmenopausal women, and concomitant alcohol consumption is contraindicated.
6. What is the dose?
The recommended dose is one tablet of
100 mg daily. The drug is to be taken at
bedtime to reduce the risks of hypotension, syncope, accidental injury, and central nervous system (CNS) depression, which can occur even in the absence of alcohol.
7. Are there any requirements for clinicians who want to prescribe the drug?
Yes. Because of the risks of hypotension, syncope, and CNS depression, the drug is subject to Risk Evaluation and Mitigation Strategies (REMS), as determined by the FDA. To prescribe the drug, providers must:
- review its prescribing information
- review the Provider and Pharmacy Training Program
- complete and submit the Knowledge Assessment Form
- enroll in REMS by completing and submitting the Prescriber Enrollment Form.
Before giving a patient her initial prescription, the provider must counsel her about the risks of hypotension and syncope and the interaction with alcohol using the Patient-Provider Agreement Form. The provider must then complete that form, provide a designated portion of it to the patient, and retain the remainder for the patient’s file.
For more information and to download the relevant forms, visit https://www.addyirems.com.
8. What are the most common
adverse reactions to the drug?
According to package labeling, the most common adverse reactions, with an incidence greater than 2%, are dizziness, somnolence, nausea, fatigue, insomnia, and dry mouth.
Less common reactions include anxiety, constipation, abdominal pain, rash, sedation, and vertigo.
In studies of the drug, appendicitis was reported among 0.2% of flibanserin-treated patients, compared with no reports of appendicitis among placebo-treated patients. The FDA has requested additional investigation of the association, if any, between flibanserin and appendicitis.
9. What drug interactions are notable?
As stated earlier, the concomitant use of flibanserin with alcohol or a moderate or strong CYP3A4 inhibitor can result in severe hypotension and syncope. Flibanserin also should not be prescribed for patients who use other CNS depressants such as diphenhydramine, opioids, benzodiazepines, and hypnotic agents.
Some examples of strong CYP3A4 inhibitors are ketoconazole, itraconazole, posaconazole, clarithromycin, nefazodone, ritonavir, saquinavir, nelfinavir, indinavir, boceprevir, telaprevir, telithromycin, and conivaptan.
Moderate CYP3A4 inhibitors include amprenavir, atazanavir, ciprofloxacin, diltiazem, erythromycin, fluconazole, fosamprenavir, verapamil, and grapefruit juice.
In addition, the concomitant use of flibanserin with multiple weak CYP3A4 inhibitors—which include herbal supplements such as ginkgo and resveratrol and nonprescription drugs such as cimetidine—also may increase the risks of hypotension and syncope.
The concomitant use of flibanserin with digoxin increases the digoxin concentration and may lead to toxicity.
10. Is the drug safe in pregnancy
and lactation?
There are currently no data on the use of flibanserin in human pregnancy. In animals, fetal toxicity occurred only in the presence of significant maternal toxicity. Adverse effects included decreased fetal weight, structural anomalies, and increases in fetal loss when exposure exceeded 15 times the recommended human dosage.
As for the advisability of using flibanserin during lactation, it is unknown whether the drug is excreted in human milk, whether it might have adverse effects in the breastfed infant, or whether it affects milk production. Package labeling states: “Because of the potential for serious adverse reactions, including sedation in a breastfed infant, breastfeeding is not recommended during treatment with [flibanserin].”1
11. When should the drug
be discontinued?
If there is no improvement in sexual desire after an 8-week trial of flibanserin, the drug should be
discontinued.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
It was a long road to approval by the US Food and Drug Administration (FDA), but flibanserin (Addyi) got the nod on August 18, 2015. Its New Drug Application (NDA) originally was filed October 27, 2009. The drug launched October 17, 2015.
Although there has been a lot of fanfare about approval of this drug, most of the coverage has focused on its status as the “first female Viagra”—a less than accurate depiction. For a more realistic and practical assessment of the drug, OBG Management turned to Michael Krychman, MD, executive director of the Southern California Center for Sexual Health and Survivorship Medicine in Newport Beach, to determine the types of information clinicians need to know to begin prescribing flibanserin. This article highlights 11 questions (and answers) to help you get started.
1. How did the FDA arrive
at its approval?
In 2012, the agency determined that female sexual dysfunction was one of 20 disease areas that warranted focused attention. In October 2014, as part of its intensified look at female sexual dysfunction, the FDA convened a 2-day meeting “to advance our understanding,” reports Andrea Fischer, FDA press officer.
“During the first day of the meeting, the FDA solicited patients’ perspectives on their condition and its impact on daily life. While this meeting did not focus on flibanserin, it provided an opportunity for the FDA to hear directly from patients about the impact of their condition,” Ms. Fischer says. During the second day of the meeting, the FDA “discussed scientific issues and challenges with experts in sexual medicine.”
As a result, by the time of the FDA’s June 4, 2015 Advisory Committee meeting on the flibanserin NDA, FDA physician-scientists were well versed in many nuances of female sexual function. That meeting included an open public hearing “that provided an opportunity for members of the public, including patients, to provide input specifically on the flibanserin application,” Ms. Fischer notes.
Nuances of the deliberations
“The FDA’s regulatory decision making on any drug product is a science-based process that carefully weighs each drug in terms of its risks and benefits to the patient population for which the drug would be indicated,” says Ms. Fischer.
The challenge in the case of flibanserin was determining whether the drug provides “clinically meaningful” improvements in sexual activity and desire.
“For many conditions and diseases, what constitutes ‘clinically meaningful’ is well known and accepted,” Ms. Fischer notes, “such as when something is cured or a severe symptom that is life-altering resolves completely. For others, this is not the case. For example, a condition that has a wide range of degree of severity can offer challenges in assessing what constitutes a clinically meaningful treatment effect. Ascertaining this requires a comprehensive knowledge of the disease, affected patient population, management strategies and the drug in question, as well as an ability to look at the clinical trial data taking this all into account.”
“In clinical trials, an important method for assessing the impact of a treatment on a patient’s symptoms, mental state, or functional status is through direct self-report using well developed and thoughtfully integrated patient-reported outcome (PRO) assessments,” Ms. Fischer says. “PROs can provide valuable information on the patient perspective when determining whether benefits outweigh risks, and they also are used to support medical product labeling claims, which are a key source of information for both health care providers and patients. PROs have been and continue to be a high priority as part of FDA’s commitment to advance patient-focused drug development, and we fully expect this to continue. The clinical trials in the flibanserin NDA all utilized PRO assessments.”
Those assessments found that patients taking flibanserin had a significant increase in “sexually satisfying events.” Three 24-week randomized controlled trials explored this endpoint for flibanserin (studies 1–3).
As for improvements in desire, the first 2 trials utilized an e-diary to assess this aspect of sexual function, while the 3rd trial utilized the Female Sexual Function Index (FSFI).
Although the e-diary reflected no statistically significant improvement in desire in the first 2 trials, the FSFI did find significant improvement in the 3rd trial. In addition, when the FSFI was considered across all 3 trials, results in the desire domain were consistent. (The FSFI was used as a secondary tool in the first 2 trials.)
In addition, sexual distress, as measured by the Female Sexual Distress Scale (FSDS), was decreased in the trials with use of flibanserin, notes Dr. Krychman. The Advisory Committee determined that these findings were sufficient to demonstrate clinically meaningful improvements with use of the drug.
Although the drug was approved by the FDA, the agency was sufficiently concerned about some of its potential risks (see questions 4 and 5) that it implemented rigorous mitigation strategies (see question 7). Additional investigations were requested by the agency, including drug-drug interaction, alcohol challenge, and driving studies.
2. What are the indications?
Flibanserin is intended for use in premenopausal women who have acquired, generalized hypoactive sexual desire disorder (HSDD). That diagnosis no longer is included in the 5th edition of the Diagnostic and
Statistical Manual of Mental Disorders but is described in drug package labeling as “low sexual desire that causes marked distress or interpersonal difficulty and is not due to:
- a coexisting medical or psychiatric condition,
- problems within the relationship, or
- the effects of a medication or other drug substance.”1
- the effects of a medication or other drug substance.”
Although the drug has been tested in both premenopausal and postmenopausal women, it was approved for use only in premenopausal women. Also note inclusion of the term “acquired” before the diagnosis of HSDD, indicating that the drug is inappropriate for women who have never experienced a period of normal sexual desire.
3. How is HSDD diagnosed?
One of the best screening tools is the
Decreased Sexual Desire Screener, says
Dr. Krychman. It is available at http://obgynalliance.com/files/fsd/DSDS_Pocketcard.pdf. This tool is a validated instrument to help clinicians identify what HSDD is and is not.
4. Does the drug carry
any warnings?
Yes, it carries a black box warning about the risks of hypotension and syncope:
- when alcohol is consumed by users of the drug. (Alcohol use is contraindicated.)
- when the drug is taken in conjunction with moderate or strong CYP3A4 inhibitors or by patients with hepatic impairment. (The drug is contraindicated in both circumstances.) See question 9 for a list of drugs that are CYP3A4 inhibitors.
5. Are there any other risks worth noting?
The medication can increase the risks of hypotension and syncope even without concomitant use of alcohol. For example, in clinical trials, hypotension was reported in 0.2% of flibanserin-treated women versus less than 0.1% of placebo users. And syncope was reported in 0.4% of flibanserin users versus 0.2% of placebo-treated patients. Flibanserin is prescribed as a once-daily medication that is to be taken at bedtime; the risks of hypotension and syncope are increased if flibanserin is taken during waking hours.
The risk of adverse effects when flibanserin is taken with alcohol is highlighted by one case reported in package labeling: A 54-year-old postmenopausal woman died after taking flibanserin (100 mg daily at bedtime) for 14 days. This patient had a history of hypertension and hypercholesterolemia and consumed a baseline amount of 1 to 3 alcoholic beverages daily. She died of acute alcohol intoxication, with a blood alcohol concentration of 0.289 g/dL.1 Whether this patient’s death was related to flibanserin use is unknown.1
It is interesting to note that, in the studies of flibanserin leading up to the drug’s approval, alcohol use was not an exclusion, says Dr. Krychman. “Approximately 58% of women were self-described as mild to moderate drinkers. The clinical program was extremely large—more than 11,000 women were studied.”
Flibanserin is currently not approved for use in postmenopausal women, and concomitant alcohol consumption is contraindicated.
6. What is the dose?
The recommended dose is one tablet of
100 mg daily. The drug is to be taken at
bedtime to reduce the risks of hypotension, syncope, accidental injury, and central nervous system (CNS) depression, which can occur even in the absence of alcohol.
7. Are there any requirements for clinicians who want to prescribe the drug?
Yes. Because of the risks of hypotension, syncope, and CNS depression, the drug is subject to Risk Evaluation and Mitigation Strategies (REMS), as determined by the FDA. To prescribe the drug, providers must:
- review its prescribing information
- review the Provider and Pharmacy Training Program
- complete and submit the Knowledge Assessment Form
- enroll in REMS by completing and submitting the Prescriber Enrollment Form.
Before giving a patient her initial prescription, the provider must counsel her about the risks of hypotension and syncope and the interaction with alcohol using the Patient-Provider Agreement Form. The provider must then complete that form, provide a designated portion of it to the patient, and retain the remainder for the patient’s file.
For more information and to download the relevant forms, visit https://www.addyirems.com.
8. What are the most common
adverse reactions to the drug?
According to package labeling, the most common adverse reactions, with an incidence greater than 2%, are dizziness, somnolence, nausea, fatigue, insomnia, and dry mouth.
Less common reactions include anxiety, constipation, abdominal pain, rash, sedation, and vertigo.
In studies of the drug, appendicitis was reported among 0.2% of flibanserin-treated patients, compared with no reports of appendicitis among placebo-treated patients. The FDA has requested additional investigation of the association, if any, between flibanserin and appendicitis.
9. What drug interactions are notable?
As stated earlier, the concomitant use of flibanserin with alcohol or a moderate or strong CYP3A4 inhibitor can result in severe hypotension and syncope. Flibanserin also should not be prescribed for patients who use other CNS depressants such as diphenhydramine, opioids, benzodiazepines, and hypnotic agents.
Some examples of strong CYP3A4 inhibitors are ketoconazole, itraconazole, posaconazole, clarithromycin, nefazodone, ritonavir, saquinavir, nelfinavir, indinavir, boceprevir, telaprevir, telithromycin, and conivaptan.
Moderate CYP3A4 inhibitors include amprenavir, atazanavir, ciprofloxacin, diltiazem, erythromycin, fluconazole, fosamprenavir, verapamil, and grapefruit juice.
In addition, the concomitant use of flibanserin with multiple weak CYP3A4 inhibitors—which include herbal supplements such as ginkgo and resveratrol and nonprescription drugs such as cimetidine—also may increase the risks of hypotension and syncope.
The concomitant use of flibanserin with digoxin increases the digoxin concentration and may lead to toxicity.
10. Is the drug safe in pregnancy
and lactation?
There are currently no data on the use of flibanserin in human pregnancy. In animals, fetal toxicity occurred only in the presence of significant maternal toxicity. Adverse effects included decreased fetal weight, structural anomalies, and increases in fetal loss when exposure exceeded 15 times the recommended human dosage.
As for the advisability of using flibanserin during lactation, it is unknown whether the drug is excreted in human milk, whether it might have adverse effects in the breastfed infant, or whether it affects milk production. Package labeling states: “Because of the potential for serious adverse reactions, including sedation in a breastfed infant, breastfeeding is not recommended during treatment with [flibanserin].”1
11. When should the drug
be discontinued?
If there is no improvement in sexual desire after an 8-week trial of flibanserin, the drug should be
discontinued.
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
Reference
- Addyi [package insert]. Raleigh, NC: Sprout Pharmaceuticals; 2015.
Reference
- Addyi [package insert]. Raleigh, NC: Sprout Pharmaceuticals; 2015.
In this Article
- How is HSDD diagnosed?
- What are clinicians required to do?
- Is the drug safe in pregnancy?
