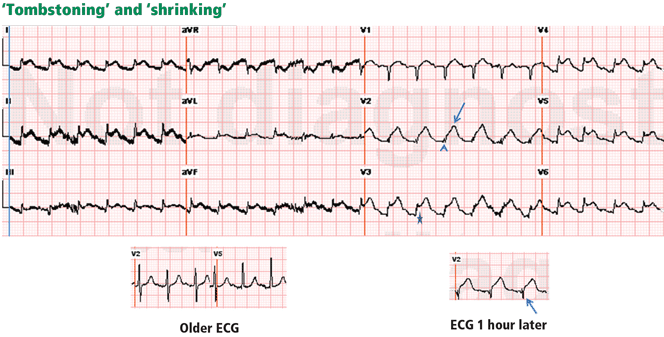
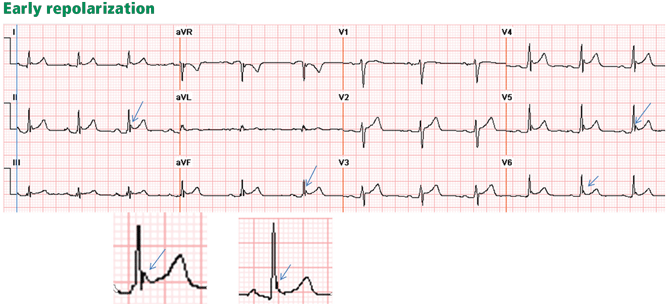
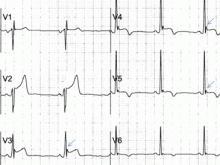
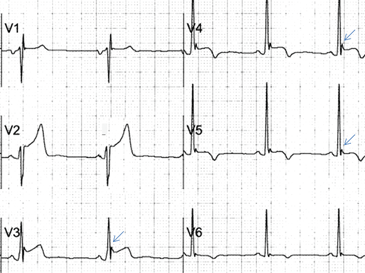
Psychiatric providers often encounter older adult patients who report difficulty with memory and express the fear they are “developing dementia.” Often, after a thorough evaluation of the reported deficits and history, we find that a serious or progressive neurocognitive disorder is unlikely. However, such occasions are an opportunity to discuss lifestyle changes that may help prevent, or at least slow, development of later-life cognitive decline.
Although I inform my patients that the body of evidence supporting many of these preventive measures still is evolving, I suggest the following approach that may provide a DEFENSE against future cognitive disability.
Diet options that are “heart healthy” seem to be “brain healthy” as well. This may be due, in part, to the antioxidant and anti-inflammatory effects of particular foods.1 Therefore, I suggest patients try to implement a Mediterranean-type diet that emphasizes fish (especially those rich in omega-3 fats, such as salmon and tuna), poultry, fresh fruit, and vegetables, as well as legumes.
ETOH has been shown, in a moderate amount (eg, 1 drink a day for women and 1 to 2 drinks for men), to be brain protective because of the antioxidants found in the alcohol or the direct relaxation effects that are produced—or both. Although red wine often is recommended, recent studies have shown that those who enjoyed an active life into their 70s and 80s had consumed a moderate amount of alcohol over their lifetime regardless of the type of spirit (eg, 12 oz of beer, 4 oz of wine, 1 oz of hard liquor).2
Friends contribute to an active, stimulating, and emotionally supported life. Having a strong social network, an antidote to loneliness and depression, has been shown to reduce the risk of “turning on” specific genes that stimulate an inflammatory process that can lead to brain cell death and neural damage.3
Exercise might be the most important ingredient for a longer, healthier, and more cognitively intact life. Moderate exercise, several times a week, increases blood flow to the brain and, subsequently, stimulates neuronal synapses and the hippocampus.4 The forms of exercise include walking, biking, swimming, resistance training, and even gardening.
No tobacco! It is known that smoking leads to accelerated aging for the heart and brain, so it is our responsibility to remain vigilant in promoting smoking cessation strategies.
Sleep has received increased attention, with recent studies providing evidence that the brain uses that time to “flush out” neurotoxic by-products of cognitive activity that have accumulated throughout the day.5 As evidence continues to be examined on this process, it is reasonable to recommend adequate sleep and a consistent sleep pattern as possible defenses against brain cell insult.
Engagement in tasks that are cognitively stimulating has been promoted as potential “brain exercises” to stave off future memory loss. For example, computer games that are mentally challenging; lively and frequent conversations; and learning a language all appear to increase neural activation and communication throughout the brain.6
As brain research continues to expand, providers will become more knowledgeable and aware of the steps our patients can take when they discuss concerns about their risk of progressive cognitive disability and memory loss. For now, however, it is important to describe what we do know based on current research and help our patients develop the best defense they can against age-related cognitive decline.
Disclosure The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Gu Y, Nieves JW, Stern Y, et al. Food combination and Alzheimer disease risk: a protective diet. Arch Neurol. 2010;67(6):699-706. 2. Paganini-Hill A, Kawas CH, Corrada MM. Type of alcohol consumed, changes in intake over time, and mortality: the Leisure World Cohort Study. Age Ageing. 2007;36(2):203-209. 3. Cole SW, Hawkley LC, Arevelo JM, et al. Transcript origin analysis identifies antigen-presenting cells as primary targets of socially regulated gene expression in leukocytes. Proc Natl Acad Sci U S A. 2011;108(7):3080-3085. 4. Small G, Vorgan G. The Alzheimer’s Prevention Program: keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc; 2011:71. 5. Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373-377. 6. Hall CB, Liptor RB, Sliwinski M, et al. Cognitive activities delay onset of memory decline in persons who develop dementia. Neurology. 2009;73(5):356-361.
Psychiatric providers often encounter older adult patients who report difficulty with memory and express the fear they are “developing dementia.” Often, after a thorough evaluation of the reported deficits and history, we find that a serious or progressive neurocognitive disorder is unlikely. However, such occasions are an opportunity to discuss lifestyle changes that may help prevent, or at least slow, development of later-life cognitive decline.
Although I inform my patients that the body of evidence supporting many of these preventive measures still is evolving, I suggest the following approach that may provide a DEFENSE against future cognitive disability.
Diet options that are “heart healthy” seem to be “brain healthy” as well. This may be due, in part, to the antioxidant and anti-inflammatory effects of particular foods.1 Therefore, I suggest patients try to implement a Mediterranean-type diet that emphasizes fish (especially those rich in omega-3 fats, such as salmon and tuna), poultry, fresh fruit, and vegetables, as well as legumes.
ETOH has been shown, in a moderate amount (eg, 1 drink a day for women and 1 to 2 drinks for men), to be brain protective because of the antioxidants found in the alcohol or the direct relaxation effects that are produced—or both. Although red wine often is recommended, recent studies have shown that those who enjoyed an active life into their 70s and 80s had consumed a moderate amount of alcohol over their lifetime regardless of the type of spirit (eg, 12 oz of beer, 4 oz of wine, 1 oz of hard liquor).2
Friends contribute to an active, stimulating, and emotionally supported life. Having a strong social network, an antidote to loneliness and depression, has been shown to reduce the risk of “turning on” specific genes that stimulate an inflammatory process that can lead to brain cell death and neural damage.3
Exercise might be the most important ingredient for a longer, healthier, and more cognitively intact life. Moderate exercise, several times a week, increases blood flow to the brain and, subsequently, stimulates neuronal synapses and the hippocampus.4 The forms of exercise include walking, biking, swimming, resistance training, and even gardening.
No tobacco! It is known that smoking leads to accelerated aging for the heart and brain, so it is our responsibility to remain vigilant in promoting smoking cessation strategies.
Sleep has received increased attention, with recent studies providing evidence that the brain uses that time to “flush out” neurotoxic by-products of cognitive activity that have accumulated throughout the day.5 As evidence continues to be examined on this process, it is reasonable to recommend adequate sleep and a consistent sleep pattern as possible defenses against brain cell insult.
Engagement in tasks that are cognitively stimulating has been promoted as potential “brain exercises” to stave off future memory loss. For example, computer games that are mentally challenging; lively and frequent conversations; and learning a language all appear to increase neural activation and communication throughout the brain.6
As brain research continues to expand, providers will become more knowledgeable and aware of the steps our patients can take when they discuss concerns about their risk of progressive cognitive disability and memory loss. For now, however, it is important to describe what we do know based on current research and help our patients develop the best defense they can against age-related cognitive decline.
Disclosure The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Psychiatric providers often encounter older adult patients who report difficulty with memory and express the fear they are “developing dementia.” Often, after a thorough evaluation of the reported deficits and history, we find that a serious or progressive neurocognitive disorder is unlikely. However, such occasions are an opportunity to discuss lifestyle changes that may help prevent, or at least slow, development of later-life cognitive decline.
Although I inform my patients that the body of evidence supporting many of these preventive measures still is evolving, I suggest the following approach that may provide a DEFENSE against future cognitive disability.
Diet options that are “heart healthy” seem to be “brain healthy” as well. This may be due, in part, to the antioxidant and anti-inflammatory effects of particular foods.1 Therefore, I suggest patients try to implement a Mediterranean-type diet that emphasizes fish (especially those rich in omega-3 fats, such as salmon and tuna), poultry, fresh fruit, and vegetables, as well as legumes.
ETOH has been shown, in a moderate amount (eg, 1 drink a day for women and 1 to 2 drinks for men), to be brain protective because of the antioxidants found in the alcohol or the direct relaxation effects that are produced—or both. Although red wine often is recommended, recent studies have shown that those who enjoyed an active life into their 70s and 80s had consumed a moderate amount of alcohol over their lifetime regardless of the type of spirit (eg, 12 oz of beer, 4 oz of wine, 1 oz of hard liquor).2
Friends contribute to an active, stimulating, and emotionally supported life. Having a strong social network, an antidote to loneliness and depression, has been shown to reduce the risk of “turning on” specific genes that stimulate an inflammatory process that can lead to brain cell death and neural damage.3
Exercise might be the most important ingredient for a longer, healthier, and more cognitively intact life. Moderate exercise, several times a week, increases blood flow to the brain and, subsequently, stimulates neuronal synapses and the hippocampus.4 The forms of exercise include walking, biking, swimming, resistance training, and even gardening.
No tobacco! It is known that smoking leads to accelerated aging for the heart and brain, so it is our responsibility to remain vigilant in promoting smoking cessation strategies.
Sleep has received increased attention, with recent studies providing evidence that the brain uses that time to “flush out” neurotoxic by-products of cognitive activity that have accumulated throughout the day.5 As evidence continues to be examined on this process, it is reasonable to recommend adequate sleep and a consistent sleep pattern as possible defenses against brain cell insult.
Engagement in tasks that are cognitively stimulating has been promoted as potential “brain exercises” to stave off future memory loss. For example, computer games that are mentally challenging; lively and frequent conversations; and learning a language all appear to increase neural activation and communication throughout the brain.6
As brain research continues to expand, providers will become more knowledgeable and aware of the steps our patients can take when they discuss concerns about their risk of progressive cognitive disability and memory loss. For now, however, it is important to describe what we do know based on current research and help our patients develop the best defense they can against age-related cognitive decline.
Disclosure The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Gu Y, Nieves JW, Stern Y, et al. Food combination and Alzheimer disease risk: a protective diet. Arch Neurol. 2010;67(6):699-706. 2. Paganini-Hill A, Kawas CH, Corrada MM. Type of alcohol consumed, changes in intake over time, and mortality: the Leisure World Cohort Study. Age Ageing. 2007;36(2):203-209. 3. Cole SW, Hawkley LC, Arevelo JM, et al. Transcript origin analysis identifies antigen-presenting cells as primary targets of socially regulated gene expression in leukocytes. Proc Natl Acad Sci U S A. 2011;108(7):3080-3085. 4. Small G, Vorgan G. The Alzheimer’s Prevention Program: keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc; 2011:71. 5. Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373-377. 6. Hall CB, Liptor RB, Sliwinski M, et al. Cognitive activities delay onset of memory decline in persons who develop dementia. Neurology. 2009;73(5):356-361.
References
1. Gu Y, Nieves JW, Stern Y, et al. Food combination and Alzheimer disease risk: a protective diet. Arch Neurol. 2010;67(6):699-706. 2. Paganini-Hill A, Kawas CH, Corrada MM. Type of alcohol consumed, changes in intake over time, and mortality: the Leisure World Cohort Study. Age Ageing. 2007;36(2):203-209. 3. Cole SW, Hawkley LC, Arevelo JM, et al. Transcript origin analysis identifies antigen-presenting cells as primary targets of socially regulated gene expression in leukocytes. Proc Natl Acad Sci U S A. 2011;108(7):3080-3085. 4. Small G, Vorgan G. The Alzheimer’s Prevention Program: keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc; 2011:71. 5. Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373-377. 6. Hall CB, Liptor RB, Sliwinski M, et al. Cognitive activities delay onset of memory decline in persons who develop dementia. Neurology. 2009;73(5):356-361.
First-episode psychosis (FEP) in schizophrenia is characterized by high response rates to antipsychotic therapy, followed by frequent antipsychotic discontinuation and elevated relapse rates soon after maintenance treatment begins.1,2 With subsequent episodes, time to response progressively increases and likelihood of response decreases.3,4
To address these issues, this article—the second of 2 parts5—describes the rationale and evidence for using nonstandard first-line antipsychotic therapies to manage FEP. Specifically, we discuss when clinicians might consider monotherapy exceeding FDA-approved maximum dosages, combination therapy, long-acting injectable antipsychotics (LAIA), or clozapine.
Monotherapy beyond FDA-approved dosages Treatment guidelines for FEP recommend oral antipsychotic dosages in the lower half of the treatment range and lower than those that are required for multi-episode schizophrenia.6-16 Ultimately, clinicians prescribe individualized dosages for their patients based on symptom improvement and tolerability. The optimal dosage at which to achieve a favorable D2 receptor occupancy likely will vary from patient to patient.17
To control symptoms, higher dosages may be needed than those used in FEP clinical trials, recommended by guidelines for FEP or multi-episode patients, or approved by the FDA. Patients seen in everyday practice may be more complicated (eg, have a comorbid condition or history of nonresponse) than study populations. Higher dosages also may be reasonable to overcome drug−drug interactions (eg, cigarette smoking-mediated cytochrome P450 1A2 induction, resulting in increased olanzapine metabolism),18 or to establish antipsychotic failure if adequate trials at lower dosages have resulted in a suboptimal response and the patient is not experiencing tolerability or safety concerns.
In a study of low-, full-, and high-dosage antipsychotic therapy in FEP, an additional 15% of patients responded to higher dosages of olanzapine and risperidone after failing to respond to a standard dosage.19 A study of data from the Recovery After an Initial Schizophrenia Episode Project’s Early Treatment Program (RAISE-ETP) found that, of participants identified who may benefit from therapy modification, 8.8% were prescribed an antipsychotic (often, olanzapine, risperidone, and haloperidol) at a higher-than-recommended dosage.20 Of note, only olanzapine was prescribed at higher than FDA-approved dosages.
Antipsychotic combination therapy Prescribing combinations of antipsychotics—antipsychotic polypharmacy (APP)— has a negative connotation because of limited efficacy and safety data,21 and limited endorsement in schizophrenia treatment guidelines.9,13 Caution with APP is warranted; a complex medication regimen may increase the potential for adverse effects, poorer adherence, and adverse drug-drug interactions.9 APP has been shown to independently predict both shorter treatment duration and discontinuation before 1 year.22
Nonetheless, the clinician and patient may share the decision to implement APP and observe whether benefits outweigh risks in situations such as: • to optimize neuroreceptor occupancy and targets (eg, attempting to achieve adequate D2 receptor blockade while minimizing side effects secondary to binding other receptors) • to manage co-existing symptom domains (eg, mood changes, aggression, negative symptoms, disorganization, and cognitive deficits) • to mitigate antipsychotic-induced side effects (eg, initiating aripiprazole to treat hyperprolactinemia induced by another antipsychotic to which the patient has achieved a favorable response).23
Clinicians report using APP to treat as many as 50% of patients with a history of multiple psychotic episodes.23 For FEP patients, 23% of participants in the RAISE-ETP trial who were identified as possibly benefiting from therapy modification were prescribed APP.20 Regrettably, researchers have not found evidence to support a reported rationale for using APP—that lower dosages of individual antipsychotics when used in combination may avoid high-dosage prescriptions.24
Before implementing APP, thoroughly explore and manage reasons for a patient’s suboptimal response to monotherapy.25 An adequate trial with any antipsychotic should be at the highest tolerated dosage for 12 to 16 weeks. Be mindful that response to an APP trial may be the result of additional time on the original antipsychotic.
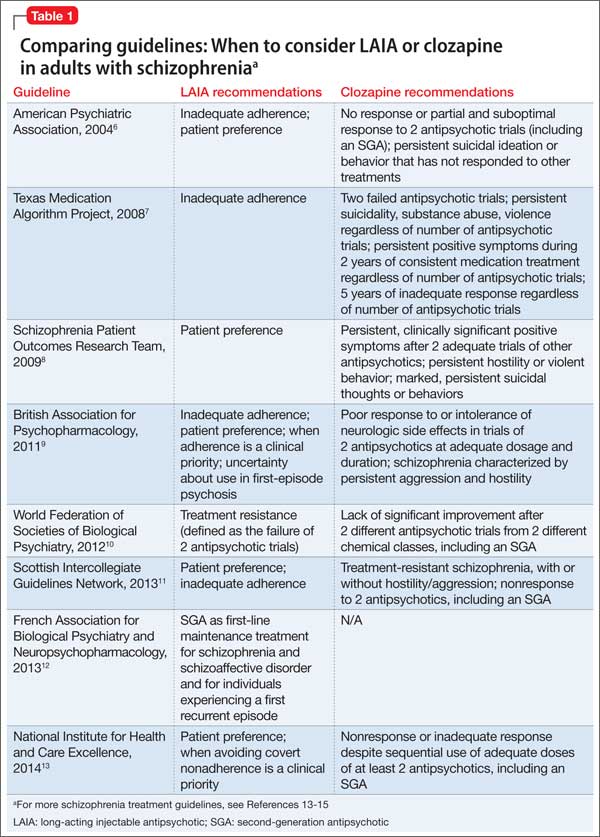
Long-acting injectable antipsychotics in FEP Guideline recommendations. Most older guidelines for schizophrenia treatment suggest LAIA after multiple relapses related to medication nonadherence or when a patient prefers injected medication (Table 1).6-13 Expert consensus guidelines also recommend considering LAIA in patients who lack insight into their illness. The Texas Medication Algorithm Project (TMAP) guidelines7 state LAIA can be considered for inadequate adherence at any stage, whereas the 2010 British Association for Psychopharmacology (BAP) guidelines9 express uncertainty about their use in FEP, because of limited evidence. Both the BAP and National Institute for Health and Care Excellence guidelines13 urge clinicians to consider LAIA when avoiding nonadherence is a treatment priority.
Recently, the French Association for Biological Psychiatry and Neuro-psychopharmacology (AFPBN) created expert consensus guidelines12 on using LAIA in practice. They recommend long-acting injectable second-generation antipsychotics (SGAs) as first-line maintenance treatment for schizophrenia and schizoaffective disorder and for individuals experiencing a first recurrent episode. The World Federation of Societies of Biological Psychiatry guidelines contain LAIA dosage recommendations for FEP (Table 2).10
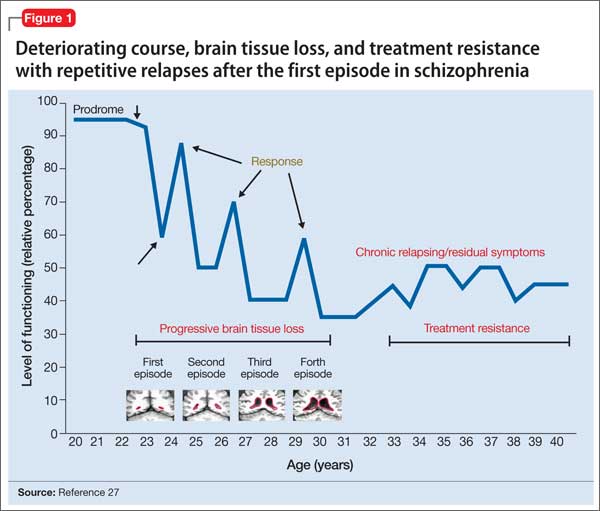
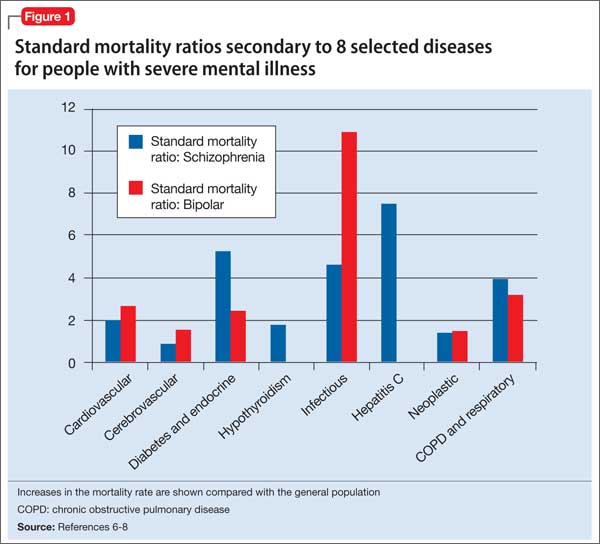
Advances have been made in understanding the serious neurobiological adverse effects of psychotic relapses, including neuroinflammation and oxidative stress, that may explain the atrophic changes observed with psychotic episodes starting with the FEP. Protecting the patient from a second episode has become a vital therapeutic management goal26 (Figure 127).
Concerns. Compared with oral antipsychotics, LAIA offers clinical advantages: • improved pharmacokinetic profile (lower “peaks” and higher “valleys”) • more consistent plasma concentrations (no variability related to administration timing or food effects) • no first-pass metabolism, which can ease the process of finding the lowest effective and safe dosage • reduced administration burden and objective tracking of adherence with typical dosing every 2 to 4 weeks • less stigmatizing than oral medication for FEP patients, such as college students living in a dormitory.28,29
Barriers to LAIA use include: • slow dosage titration and increased time to reach steady state drug level • oral supplementation for some (eg, risperidone microspheres and aripiprazole long-acting injectable) • logistical challenges for some (eg, 3-hour post-injection monitoring for delirium sedation syndrome with olanzapine pamoate) • additional planning to coordinate care for scheduled injections • higher expenses up front • local injection site reactions • dosage adjustment difficulties if adverse effects occur.28,29
Adoption rates of LAIA are low, especially for FEP.30 Most surveys indicate that (1) physicians believe LAIA treatment is ineffective for FEP31 and (2) patients do not prefer injectable to oral antipsychotics,32 despite evidence to the contrary.33,34 A survey of 198 psychiatrists identified 3 factors that influenced their decisions against using LAIA patients with FEP: • limited availability of SGA depot formulations (4, to date, in the United States) • frequent rejection by the patient when LAIA is offered without adequate explanation or encouragement • skepticism of FEP patients (and their family) who lack experience with relapse.35
In reality, when SGA depots were introduced in the United Kingdom, prescribing rates of LAIA did not increase. As for patient rejection being a major reason for not prescribing LAIA, few patients (5% to 36%) are offered depot injections, particularly in FEP.29 Most patients using LAIA are chronic, multi-episode, violent people who are receiving medications involuntarily.29 Interestingly, this survey did not find 2 factors to be influential in psychiatrists’ decision not to use LAIA in FEP: • guidelines do not explicitly recommend depot treatment in FEP • treatment in FEP may be limited to 1 year, therefore depot administration is not worthwhile.35
Preliminary evidence. At least a dozen studies have explored LAIA treatment for FEP, with the use of fluphenazine decanoate,36 perphenazine enanthate37 (discontinued), and risperidone microspheres.37-48 The research demonstrates the efficacy and safety of LAIA in FEP as measured by these endpoints: • improved symptom control38,40-43,46,48 • adherence43,44,48 • reduced relapse rates37,43 and rehospitalizations37,47 • lesser reductions in white matter brain volume45 • no differences in extrapyramidal side effects or prolactin-associated adverse effects.48
A few small studies demonstrate significant differences in outcomes between risperidone LAIA and oral comparator groups (Table 3).43-45 Ongoing studies of LAIA use in FEP are comparing paliperidone palmitate with risperidone microspheres and other oral antipsychotics.49-51 No studies are examining olanzapine pamoate in FEP, likely because several guidelines do not recommended its use. No studies have been published regarding aripiprazole long-acting injectable in FEP. This LAIA formulation was approved in February 2013, and robust studies of the oral formulation in FEP are limited.52
Discussion and recommendations. Psychiatrists relying on subjective measures of antipsychotic adherence may inaccurately assess whether patients meet this criterion for LAIA use.53 LAIA could combat the high relapse rate in FEP, yet depot antipsychotics are prescribed infrequently for FEP patients (eg, for only 9.5% of participants in the RAISE-ETP study).20 Most schizophrenia treatment guidelines do not discuss LAIA use specifically in FEP, although the AFPBN expert consensus guidelines published in 2013 do recommend SGA depot formulations in FEP.12 SGA LAIA may be preferable, given its neuroprotective effects, in contrast to the neurotoxicity concerns of FGA LAIA.54,55
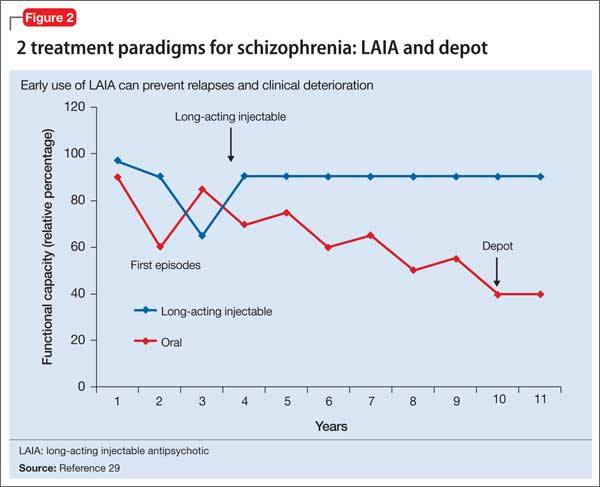
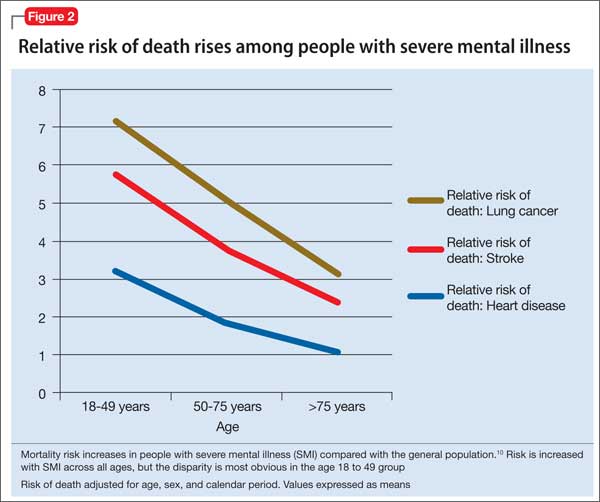
Relapses begin within a few months of illness stabilization after FEP, and >50% of patients relapse within 1 or 2 years2—the recommended minimum treatment duration for FEP.8,9,13 The use of LAIA is advisable in any patient with schizophrenia for whom long-term antipsychotic therapy is indicated.56 LAIA administration requirements objectively track medication adherence, which allows clinicians to be proactive in relapse prevention. Not using an intervention in FEP that improves adherence and decreases relapse rates contradicts our goal of instituting early, effective treatment to improve long-term functional outcomes (Figure 2).29
Considering clozapine in FEP Guideline recommendations. Schizo-phrenia treatment guidelines and FDA labeling57 reserve clozapine for third-line treatment of refractory schizophrenia after 2 adequate antipsychotic trials have failed despite optimal dosing (Table 1).6-13 Some guidelines specify 1 of the 2 failed antipsychotic trials must include an SGA.6,7,10,11,13-16 Most say clozapine may be considered in patients with chronic aggression or hostility,7-9,14,16 or suicidal thoughts and behaviors.6-8,14,16 TMAP guidelines recommend a clozapine trial with concomitant substance abuse, persistent positive symptoms during 2 years of consistent medication treatment, and after 5 years of inadequate response (“treatment resistance”), regardless of the number of antipsychotic trials.7
Rationale and concerns. Clozapine is a superior choice for treatment-refractory delusions or hallucinations of schizophrenia, because it markedly enhances the response rate to antipsychotic therapy.58 Researchers therefore have investigated whether clozapine, compared with other antipsychotics, would yield more favorable initial and long-term outcomes when used first-line in FEP.
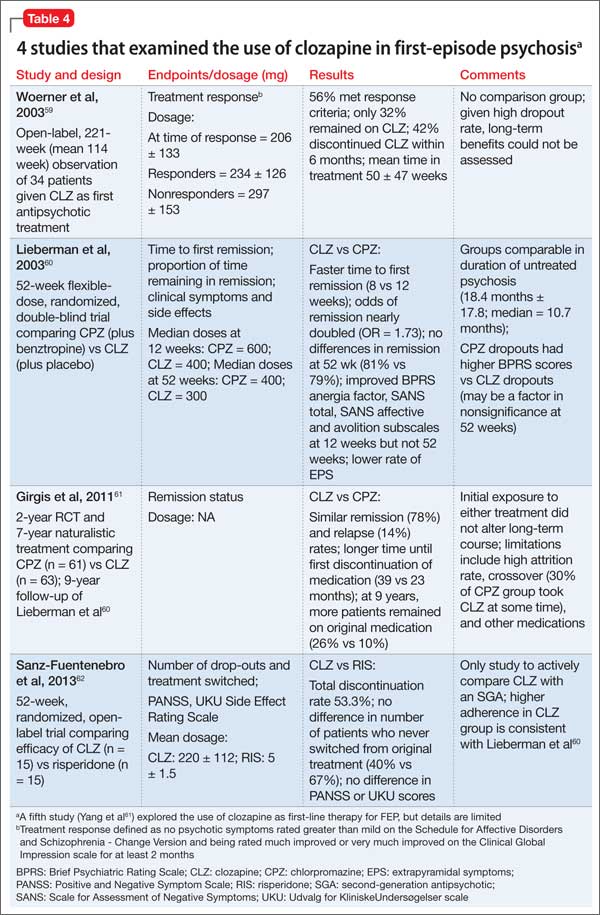
Preliminary evidence. Five studies have explored the use of clozapine as first-line therapy in FEP (Table 4).59-63 Interpreting the results is difficult because clozapine trials may be brief (mostly, 12 to 52 weeks); lack a comparator arm; suffer from a high attrition rate; enroll few patients; and lack potentially important outcome measures such as negative symptoms, suicidality, and functional assessment.
Overall, these studies demonstrate clozapine is as efficacious in this patient population as chlorpromazine (no difference in remission at 1-year, although clozapine-treated patients remitted faster and stayed in remission longer)60,61 or risperidone (no difference in Positive and Negative Syndrome Scale scores).62
At present, clozapine has not been shown superior to other antipsychotics as a first-line treatment for FEP. Research does underscore the importance of a clozapine trial as third-line treatment for FEP patients who have not responded well to 2 SGA trials.63 Many of these nonresponders (77%) have demonstrated a favorable response when promptly switched to clozapine.64
Discussion and recommendations. The limited evidence argues against using clozapine earlier than as third-line treatment in FEP. Perhaps the high treatment response that characterizes FEP creates a ceiling effect that obscures differences in antipsychotic efficacy at this stage.65 Clozapine use as first-line treatment should be re-evaluated with more robust methodology. One approach could be to assess its benefit in FEP by the duration of untreated psychosis.
The odds of achieving remission have been shown to decrease by 15% for each year that psychosis has not been treated.59 Studies exploring the use of clozapine as a second-line agent for FEP also are warranted, as antipsychotic response during subsequent trials is substantially reduced. In fact, the Scottish Intercollegiate Guidelines Network guidelines recommend this as an area for future research.11
For now, clozapine should continue to be reserved as second- or third-line treatment in a patient with FEP. The risks of clozapine’s potentially serious adverse effects (eg, agranulocytosis, seizures, obesity, diabetes, dyslipidemia, myocarditis, pancreatitis, hypotension, sialorrhea, severe sedation, ileus) can be justified only in the treatment of severe and persistent psychotic symptoms.57
Bottom Line Nonstandard use of antipsychotic monotherapy dosages beyond the approved FDA limit and combination antipsychotic therapy may be reasonable for select first-episode psychosis (FEP) patients. Strongly consider long-acting injectable antipsychotics in FEP to proactively combat the high relapse rate and more easily identify antipsychotic failure. Continue to use clozapine as second- or third-line therapy in FEP: Studies have not found that it is more efficacious than other antipsychotics for first-line use.
Related Resource • Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
Disclosures Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products. Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
References
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804. 2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283. 3. Lieberman JA, Koreen AR, Chakos M, et al. Factors influencing treatment response and outcome of first-episode schizophrenia: implications for understanding the pathophysiology of schizophrenia. J Clin Psychiatry. 1996;57(suppl 9):5-9. 4. Agid O, Arenovich T, Sajeev G, et al. An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J Clin Psychiatry. 2011;72(11):1439-1444. 5. Gardner KN, Nasrallah HA. Managing first-episode psychosis. An early stage of schizophrenia with distinct treatment needs. Current Psychiatry. 2015;14(5):32-34,36-40,42. 6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56. 7. Texas Department of State Health Services. Texas Medication Algorithm Project (TMAP) Procedural Manual. Schizophrenia Treatment Algorithms. http://www.jpshealthnet.org/sites/default/files/ tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed June 11, 2015. 8. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93. 9. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620. 10. Hasan A, Falkai P, Wobrok T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44. 11. Scottish Intercollegiate Guidelines Network. SIGN 131: Management of schizophrenia. http://www.sign.ac.uk/ pdf/sign131.pdf. Published March 2013. Accessed June 11, 2015. 12. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serous mental illness. BMC Psychiatry. 2013;13:340. 13. National Institute for Health and Care Excellence. NICE clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. https://www.nice.org. uk/guidance/cg178/resources/guidance-psychosis-and-schizophrenia-in-adults-treatment-and-management-pdf. Updated March 2014. Accessed June 16, 2015. 14. Canadian Psychiatric Association. Clinical practice guidelines. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S. 15. McEvoy JP, Scheifler PL, Frances A. The expert consensus guideline series: treatment of schizophrenia. J Clin Psychiatry. 1999;60(suppl 11):3-80. 16. Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16. 17. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520. 18. Fankhauser MP. Drug interactions with tobacco smoke: implications for patient care. Current Psychiatry. 2013;12(1):12-16. 19. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022. 20. Robinson DG, Schooler NR, John M, et al. Prescription practices in the treatment of first-episode schizophrenia spectrum disorders: data from the national RAISE-ETP study. Am J Psychiatry. 2015;172(3):237-248. 21. Correll CU, Rummel-Kluge C, Corves C, et al. Antipsychotic combinations vs monotherapy in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Bull. 2009;35(2):443-457. 22. Fisher MD, Reilly K, Isenberg K, et al. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry. 2014;14(1):341. 23. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399. 24. John AP, Dragovic M. Antipsychotic polypharmacy is not associated with reduced dose of individual antipsychotics in schizophrenia. J Clin Psychopharmacol. 2015;35(2):193-195. 25. Nasrallah HA. Treatment-resistant schizophrenia. Current Psychiatry. http://www.currentpsychiatry.com/specialty-focus/schizophrenia-other-psychotic-disorders/article/ treatment-resistant-schizophrenia/9be7bba3713d4a4cd68aa 8c92b79e5b1.html. Accessed June 16, 2015. 26. Alvarez-Jiménez M, Parker AG, Hetrick SE, et al. Preventing the second episode: a systematic review and meta-analysis of psychosocial and pharmacological trials in first-episode psychosis. Schizophr Bull. 2011;37(3):619-630. 27. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. 2nd ed. Newton, PA: Handbooks in Health Care Co; 2011. 28. McEvoy JP. Risks versus benefits of different types of long-acting injectable antipsychotics. J Clin Psychiatry. 2006;67(suppl 5):15-18. 29. Agid O, Foussias G, Remington G. Long-acting injectable antipsychotics in the treatment of schizophrenia: their role in relapse prevention. Expert Opin Pharmacother. 2010;11(14):2301-2317. 30. Kirschner M, Theodoridou A, Fusar-Poli P, et al. Patients’ and clinicians’ attitude towards long-acting depot antipsychotics in subjects with a first episode psychosis. Ther Adv Psychophamacol. 2013;3(2):89-99. 31. Heres S, Hamann J, Mendel R, et al. Identifying the profile of optimal candidates for antipsychotic depot therapy: A cluster analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1987-1993. 32. Heres S, Lambert M, Vauth R. Treatment of early episode in patents with schizophrenia: the role of long acting antipsychotics. Eur Psychiatry. 2014;29(suppl 2):1409-1413. 33. Heres S, Schmitz FS, Leucht S, et al. The attitude of patients towards antipsychotic depot treatment. Int Clin Psychopharmacol. 2007;22(5):275-282. 34. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406. 35. Heres S, Reichhart T, Hamann J, et al. Psychiatrists’ attitude to antipsychotic depot treatment in patients with first-episode schizophrenia. Eur Psychiatry. 2011;26(5):297-301. 36. Kane JM, Rifkin A, Quitkin F, et al. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Arch Gen Psychiatry. 1982;39(1):70-73. 37. Tiihonen J, Wahlbeck K, Lönnqvist J, et al. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in a community care after first hospitalization due to schizophrenia and schizoaffective disorder: observational follow-up study. BMJ. 2006;333(7561):224. 38. Parellada E, Andrezina R, Milanova V, et al. Patients in the early phases of schizophrenia and schizoaffective disorders effectively treated with risperidone long-acting injectable. J Psychopharmacol. 2005;19(suppl 5):5-14. 39. Malla A, Binder C, Chue P. Comparison of long-acting injectable risperidone and oral novel antipsychotic drugs for treatment in early phase of schizophrenia spectrum psychosis. Proceedings of the 61st Annual Convention Society of Biological Psychiatry; Toronto, Canada; 2006. 40. Lasser RA, Bossie CA, Zhu Y, et al. Long-acting risperidone in young adults with early schizophrenia or schizoaffective illness. Ann Clin Psychiatry. 2007;19(2):65-71. 41. Emsley R, Oosthuizen P, Koen L, et al. Remission in patients with first-episode schizophrenia receiving assured antipsychotic medication: a study with risperidone long-acting injection. Int Clin Psychopharmacol. 2008;23(6):325-331. 42. Emsley R, Oosthuizen P, Koen L, et al. Oral versus injectable antipsychotic treatment in early psychosis: post hoc comparison of two studies. Clin Ther. 2008;30(12):2378-2386. 43. Kim B, Lee SH, Choi TK, et al. Effectiveness of risperidone long-acting injection in first-episode schizophrenia: in naturalistic setting. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1231-1235. 44. Weiden PJ, Schooler NJ, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406. 45. Bartzokis G, Lu PH, Amar CP, et al. Long acting injection versus oral risperidone in first-episode schizophrenia: differential impact on white matter myelination trajectory. Schizophr Res. 2011;132(1):35-41. 46. Napryeyenko O, Burba B, Martinez G, et al. Risperidone long-acting injectable in recent-onset schizophrenia examined with clinician and patient self-report measures. J Clin Psychopharmacol. 2010;30(2):200-202. 47. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. Am J Psychiatry. 2011;168(6):603-609. 48. Dubois V, Megens J, Mertens C, et al. Long-acting risperidone in early-episode schizophrenia. Acta Psychiatrica Belgica. 2011;111(1):9-21. 49. ClinicalTrials.gov. Oral risperidone versus injectable paliperidone palmitate for treating first-episode schizophrenia. https://clinicaltrials.gov/ct2/show/ NCT01451736. Accessed June 16, 2015. 50. ClinicalTrials.gov. Brain myelination effects of paliperidone palmitate versus oral risperidone in first episode schizophrenia. https://clinicaltrials.gov/ct2/ show/NCT01458379. Accessed June 16, 2015. 51. ClinicalTrials.gov. Effects of paliperidone palmitate versus oral antipsychotics on clinical outcomes and MRI measures. https://clinicaltrials.gov/ct2/show/NCT01359293. Accessed June 16, 2016. 52. U.S. Food and Drug Administration. Drugs@FDA. http:// www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed January 11, 2015. 53. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1-46; quiz 47-48. 54. Nandra KS, Agius M. The difference between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99. 55. Nasrallah HA. Haloperidol is clearly neurotoxic. Should it be banned? Current Psychiatry. 2013;12(7):7-8. 56. Kane JM, Garcia-Ribora C. Clinical guideline recommendations for antipsychotic long-acting injections. Br J Psychiatry. 2009;52:S63-S67. 57. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014. 58. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796. 59. Woerner MG, Robinson DG, Alvir JMJ, et al. Clozapine as a first treatment for schizophrenia. Am J Psychiatry. 2003;160(8):1514-1516. 60. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003. 61. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288. 62. Sanz-Fuentenebro J, Taboada D, Palomo T, et al. Randomized trial of clozapine vs. risperidone in treatment-naïve first-episode schizophrenia: results after one year. Schizophr Res. 2013;149(1-3):156-161. 63. Yang PD, Ji Z. The efficacy and related factors of clozapine on first-episode schizophrenia. Chin J Nerv Ment Dis. 1997;23:155-158. 64. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022. 65. Remington G, Agid O, Foussias G, et al. Clozapine’s role in the treatment of first-episode schizophrenia. Am J Psychiatry. 2013;170(2):146-151.
Kristen N. Gardner, PharmD PGY-2 Psychiatric Pharmacy Resident Western Missouri Psychiatric Pharmacy Residency Program Kansas City, Missouri
Henry A. Nasrallah, MD Sydney W. Souers Endowed Chair and Professor Department of Neurology and Psychiatry Saint Louis University School of Medicine St. Louis, Missouri
first-line treatments for schizophrenia, first line treatments for schizophrenia, first-line treatment for schizophrenia, first line treatment for schizophrenia, schizophrenia, psychosis, psychotic disorders, first episode psychosis, first-episode psychosis, clozapien, injectable antipsychotics, long acting injectable antipsychotics, long-acting injectable antipsychotics, monotherapy
Kristen N. Gardner, PharmD PGY-2 Psychiatric Pharmacy Resident Western Missouri Psychiatric Pharmacy Residency Program Kansas City, Missouri
Henry A. Nasrallah, MD Sydney W. Souers Endowed Chair and Professor Department of Neurology and Psychiatry Saint Louis University School of Medicine St. Louis, Missouri
Author and Disclosure Information
Kristen N. Gardner, PharmD PGY-2 Psychiatric Pharmacy Resident Western Missouri Psychiatric Pharmacy Residency Program Kansas City, Missouri
Henry A. Nasrallah, MD Sydney W. Souers Endowed Chair and Professor Department of Neurology and Psychiatry Saint Louis University School of Medicine St. Louis, Missouri
First-episode psychosis (FEP) in schizophrenia is characterized by high response rates to antipsychotic therapy, followed by frequent antipsychotic discontinuation and elevated relapse rates soon after maintenance treatment begins.1,2 With subsequent episodes, time to response progressively increases and likelihood of response decreases.3,4
To address these issues, this article—the second of 2 parts5—describes the rationale and evidence for using nonstandard first-line antipsychotic therapies to manage FEP. Specifically, we discuss when clinicians might consider monotherapy exceeding FDA-approved maximum dosages, combination therapy, long-acting injectable antipsychotics (LAIA), or clozapine.
Monotherapy beyond FDA-approved dosages Treatment guidelines for FEP recommend oral antipsychotic dosages in the lower half of the treatment range and lower than those that are required for multi-episode schizophrenia.6-16 Ultimately, clinicians prescribe individualized dosages for their patients based on symptom improvement and tolerability. The optimal dosage at which to achieve a favorable D2 receptor occupancy likely will vary from patient to patient.17
To control symptoms, higher dosages may be needed than those used in FEP clinical trials, recommended by guidelines for FEP or multi-episode patients, or approved by the FDA. Patients seen in everyday practice may be more complicated (eg, have a comorbid condition or history of nonresponse) than study populations. Higher dosages also may be reasonable to overcome drug−drug interactions (eg, cigarette smoking-mediated cytochrome P450 1A2 induction, resulting in increased olanzapine metabolism),18 or to establish antipsychotic failure if adequate trials at lower dosages have resulted in a suboptimal response and the patient is not experiencing tolerability or safety concerns.
In a study of low-, full-, and high-dosage antipsychotic therapy in FEP, an additional 15% of patients responded to higher dosages of olanzapine and risperidone after failing to respond to a standard dosage.19 A study of data from the Recovery After an Initial Schizophrenia Episode Project’s Early Treatment Program (RAISE-ETP) found that, of participants identified who may benefit from therapy modification, 8.8% were prescribed an antipsychotic (often, olanzapine, risperidone, and haloperidol) at a higher-than-recommended dosage.20 Of note, only olanzapine was prescribed at higher than FDA-approved dosages.
Antipsychotic combination therapy Prescribing combinations of antipsychotics—antipsychotic polypharmacy (APP)— has a negative connotation because of limited efficacy and safety data,21 and limited endorsement in schizophrenia treatment guidelines.9,13 Caution with APP is warranted; a complex medication regimen may increase the potential for adverse effects, poorer adherence, and adverse drug-drug interactions.9 APP has been shown to independently predict both shorter treatment duration and discontinuation before 1 year.22
Nonetheless, the clinician and patient may share the decision to implement APP and observe whether benefits outweigh risks in situations such as: • to optimize neuroreceptor occupancy and targets (eg, attempting to achieve adequate D2 receptor blockade while minimizing side effects secondary to binding other receptors) • to manage co-existing symptom domains (eg, mood changes, aggression, negative symptoms, disorganization, and cognitive deficits) • to mitigate antipsychotic-induced side effects (eg, initiating aripiprazole to treat hyperprolactinemia induced by another antipsychotic to which the patient has achieved a favorable response).23
Clinicians report using APP to treat as many as 50% of patients with a history of multiple psychotic episodes.23 For FEP patients, 23% of participants in the RAISE-ETP trial who were identified as possibly benefiting from therapy modification were prescribed APP.20 Regrettably, researchers have not found evidence to support a reported rationale for using APP—that lower dosages of individual antipsychotics when used in combination may avoid high-dosage prescriptions.24
Before implementing APP, thoroughly explore and manage reasons for a patient’s suboptimal response to monotherapy.25 An adequate trial with any antipsychotic should be at the highest tolerated dosage for 12 to 16 weeks. Be mindful that response to an APP trial may be the result of additional time on the original antipsychotic.
Long-acting injectable antipsychotics in FEP Guideline recommendations. Most older guidelines for schizophrenia treatment suggest LAIA after multiple relapses related to medication nonadherence or when a patient prefers injected medication (Table 1).6-13 Expert consensus guidelines also recommend considering LAIA in patients who lack insight into their illness. The Texas Medication Algorithm Project (TMAP) guidelines7 state LAIA can be considered for inadequate adherence at any stage, whereas the 2010 British Association for Psychopharmacology (BAP) guidelines9 express uncertainty about their use in FEP, because of limited evidence. Both the BAP and National Institute for Health and Care Excellence guidelines13 urge clinicians to consider LAIA when avoiding nonadherence is a treatment priority.
Recently, the French Association for Biological Psychiatry and Neuro-psychopharmacology (AFPBN) created expert consensus guidelines12 on using LAIA in practice. They recommend long-acting injectable second-generation antipsychotics (SGAs) as first-line maintenance treatment for schizophrenia and schizoaffective disorder and for individuals experiencing a first recurrent episode. The World Federation of Societies of Biological Psychiatry guidelines contain LAIA dosage recommendations for FEP (Table 2).10
Advances have been made in understanding the serious neurobiological adverse effects of psychotic relapses, including neuroinflammation and oxidative stress, that may explain the atrophic changes observed with psychotic episodes starting with the FEP. Protecting the patient from a second episode has become a vital therapeutic management goal26 (Figure 127).
Concerns. Compared with oral antipsychotics, LAIA offers clinical advantages: • improved pharmacokinetic profile (lower “peaks” and higher “valleys”) • more consistent plasma concentrations (no variability related to administration timing or food effects) • no first-pass metabolism, which can ease the process of finding the lowest effective and safe dosage • reduced administration burden and objective tracking of adherence with typical dosing every 2 to 4 weeks • less stigmatizing than oral medication for FEP patients, such as college students living in a dormitory.28,29
Barriers to LAIA use include: • slow dosage titration and increased time to reach steady state drug level • oral supplementation for some (eg, risperidone microspheres and aripiprazole long-acting injectable) • logistical challenges for some (eg, 3-hour post-injection monitoring for delirium sedation syndrome with olanzapine pamoate) • additional planning to coordinate care for scheduled injections • higher expenses up front • local injection site reactions • dosage adjustment difficulties if adverse effects occur.28,29
Adoption rates of LAIA are low, especially for FEP.30 Most surveys indicate that (1) physicians believe LAIA treatment is ineffective for FEP31 and (2) patients do not prefer injectable to oral antipsychotics,32 despite evidence to the contrary.33,34 A survey of 198 psychiatrists identified 3 factors that influenced their decisions against using LAIA patients with FEP: • limited availability of SGA depot formulations (4, to date, in the United States) • frequent rejection by the patient when LAIA is offered without adequate explanation or encouragement • skepticism of FEP patients (and their family) who lack experience with relapse.35
In reality, when SGA depots were introduced in the United Kingdom, prescribing rates of LAIA did not increase. As for patient rejection being a major reason for not prescribing LAIA, few patients (5% to 36%) are offered depot injections, particularly in FEP.29 Most patients using LAIA are chronic, multi-episode, violent people who are receiving medications involuntarily.29 Interestingly, this survey did not find 2 factors to be influential in psychiatrists’ decision not to use LAIA in FEP: • guidelines do not explicitly recommend depot treatment in FEP • treatment in FEP may be limited to 1 year, therefore depot administration is not worthwhile.35
Preliminary evidence. At least a dozen studies have explored LAIA treatment for FEP, with the use of fluphenazine decanoate,36 perphenazine enanthate37 (discontinued), and risperidone microspheres.37-48 The research demonstrates the efficacy and safety of LAIA in FEP as measured by these endpoints: • improved symptom control38,40-43,46,48 • adherence43,44,48 • reduced relapse rates37,43 and rehospitalizations37,47 • lesser reductions in white matter brain volume45 • no differences in extrapyramidal side effects or prolactin-associated adverse effects.48
A few small studies demonstrate significant differences in outcomes between risperidone LAIA and oral comparator groups (Table 3).43-45 Ongoing studies of LAIA use in FEP are comparing paliperidone palmitate with risperidone microspheres and other oral antipsychotics.49-51 No studies are examining olanzapine pamoate in FEP, likely because several guidelines do not recommended its use. No studies have been published regarding aripiprazole long-acting injectable in FEP. This LAIA formulation was approved in February 2013, and robust studies of the oral formulation in FEP are limited.52
Discussion and recommendations. Psychiatrists relying on subjective measures of antipsychotic adherence may inaccurately assess whether patients meet this criterion for LAIA use.53 LAIA could combat the high relapse rate in FEP, yet depot antipsychotics are prescribed infrequently for FEP patients (eg, for only 9.5% of participants in the RAISE-ETP study).20 Most schizophrenia treatment guidelines do not discuss LAIA use specifically in FEP, although the AFPBN expert consensus guidelines published in 2013 do recommend SGA depot formulations in FEP.12 SGA LAIA may be preferable, given its neuroprotective effects, in contrast to the neurotoxicity concerns of FGA LAIA.54,55
Relapses begin within a few months of illness stabilization after FEP, and >50% of patients relapse within 1 or 2 years2—the recommended minimum treatment duration for FEP.8,9,13 The use of LAIA is advisable in any patient with schizophrenia for whom long-term antipsychotic therapy is indicated.56 LAIA administration requirements objectively track medication adherence, which allows clinicians to be proactive in relapse prevention. Not using an intervention in FEP that improves adherence and decreases relapse rates contradicts our goal of instituting early, effective treatment to improve long-term functional outcomes (Figure 2).29
Considering clozapine in FEP Guideline recommendations. Schizo-phrenia treatment guidelines and FDA labeling57 reserve clozapine for third-line treatment of refractory schizophrenia after 2 adequate antipsychotic trials have failed despite optimal dosing (Table 1).6-13 Some guidelines specify 1 of the 2 failed antipsychotic trials must include an SGA.6,7,10,11,13-16 Most say clozapine may be considered in patients with chronic aggression or hostility,7-9,14,16 or suicidal thoughts and behaviors.6-8,14,16 TMAP guidelines recommend a clozapine trial with concomitant substance abuse, persistent positive symptoms during 2 years of consistent medication treatment, and after 5 years of inadequate response (“treatment resistance”), regardless of the number of antipsychotic trials.7
Rationale and concerns. Clozapine is a superior choice for treatment-refractory delusions or hallucinations of schizophrenia, because it markedly enhances the response rate to antipsychotic therapy.58 Researchers therefore have investigated whether clozapine, compared with other antipsychotics, would yield more favorable initial and long-term outcomes when used first-line in FEP.
Preliminary evidence. Five studies have explored the use of clozapine as first-line therapy in FEP (Table 4).59-63 Interpreting the results is difficult because clozapine trials may be brief (mostly, 12 to 52 weeks); lack a comparator arm; suffer from a high attrition rate; enroll few patients; and lack potentially important outcome measures such as negative symptoms, suicidality, and functional assessment.
Overall, these studies demonstrate clozapine is as efficacious in this patient population as chlorpromazine (no difference in remission at 1-year, although clozapine-treated patients remitted faster and stayed in remission longer)60,61 or risperidone (no difference in Positive and Negative Syndrome Scale scores).62
At present, clozapine has not been shown superior to other antipsychotics as a first-line treatment for FEP. Research does underscore the importance of a clozapine trial as third-line treatment for FEP patients who have not responded well to 2 SGA trials.63 Many of these nonresponders (77%) have demonstrated a favorable response when promptly switched to clozapine.64
Discussion and recommendations. The limited evidence argues against using clozapine earlier than as third-line treatment in FEP. Perhaps the high treatment response that characterizes FEP creates a ceiling effect that obscures differences in antipsychotic efficacy at this stage.65 Clozapine use as first-line treatment should be re-evaluated with more robust methodology. One approach could be to assess its benefit in FEP by the duration of untreated psychosis.
The odds of achieving remission have been shown to decrease by 15% for each year that psychosis has not been treated.59 Studies exploring the use of clozapine as a second-line agent for FEP also are warranted, as antipsychotic response during subsequent trials is substantially reduced. In fact, the Scottish Intercollegiate Guidelines Network guidelines recommend this as an area for future research.11
For now, clozapine should continue to be reserved as second- or third-line treatment in a patient with FEP. The risks of clozapine’s potentially serious adverse effects (eg, agranulocytosis, seizures, obesity, diabetes, dyslipidemia, myocarditis, pancreatitis, hypotension, sialorrhea, severe sedation, ileus) can be justified only in the treatment of severe and persistent psychotic symptoms.57
Bottom Line Nonstandard use of antipsychotic monotherapy dosages beyond the approved FDA limit and combination antipsychotic therapy may be reasonable for select first-episode psychosis (FEP) patients. Strongly consider long-acting injectable antipsychotics in FEP to proactively combat the high relapse rate and more easily identify antipsychotic failure. Continue to use clozapine as second- or third-line therapy in FEP: Studies have not found that it is more efficacious than other antipsychotics for first-line use.
Related Resource • Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
Disclosures Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products. Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
First-episode psychosis (FEP) in schizophrenia is characterized by high response rates to antipsychotic therapy, followed by frequent antipsychotic discontinuation and elevated relapse rates soon after maintenance treatment begins.1,2 With subsequent episodes, time to response progressively increases and likelihood of response decreases.3,4
To address these issues, this article—the second of 2 parts5—describes the rationale and evidence for using nonstandard first-line antipsychotic therapies to manage FEP. Specifically, we discuss when clinicians might consider monotherapy exceeding FDA-approved maximum dosages, combination therapy, long-acting injectable antipsychotics (LAIA), or clozapine.
Monotherapy beyond FDA-approved dosages Treatment guidelines for FEP recommend oral antipsychotic dosages in the lower half of the treatment range and lower than those that are required for multi-episode schizophrenia.6-16 Ultimately, clinicians prescribe individualized dosages for their patients based on symptom improvement and tolerability. The optimal dosage at which to achieve a favorable D2 receptor occupancy likely will vary from patient to patient.17
To control symptoms, higher dosages may be needed than those used in FEP clinical trials, recommended by guidelines for FEP or multi-episode patients, or approved by the FDA. Patients seen in everyday practice may be more complicated (eg, have a comorbid condition or history of nonresponse) than study populations. Higher dosages also may be reasonable to overcome drug−drug interactions (eg, cigarette smoking-mediated cytochrome P450 1A2 induction, resulting in increased olanzapine metabolism),18 or to establish antipsychotic failure if adequate trials at lower dosages have resulted in a suboptimal response and the patient is not experiencing tolerability or safety concerns.
In a study of low-, full-, and high-dosage antipsychotic therapy in FEP, an additional 15% of patients responded to higher dosages of olanzapine and risperidone after failing to respond to a standard dosage.19 A study of data from the Recovery After an Initial Schizophrenia Episode Project’s Early Treatment Program (RAISE-ETP) found that, of participants identified who may benefit from therapy modification, 8.8% were prescribed an antipsychotic (often, olanzapine, risperidone, and haloperidol) at a higher-than-recommended dosage.20 Of note, only olanzapine was prescribed at higher than FDA-approved dosages.
Antipsychotic combination therapy Prescribing combinations of antipsychotics—antipsychotic polypharmacy (APP)— has a negative connotation because of limited efficacy and safety data,21 and limited endorsement in schizophrenia treatment guidelines.9,13 Caution with APP is warranted; a complex medication regimen may increase the potential for adverse effects, poorer adherence, and adverse drug-drug interactions.9 APP has been shown to independently predict both shorter treatment duration and discontinuation before 1 year.22
Nonetheless, the clinician and patient may share the decision to implement APP and observe whether benefits outweigh risks in situations such as: • to optimize neuroreceptor occupancy and targets (eg, attempting to achieve adequate D2 receptor blockade while minimizing side effects secondary to binding other receptors) • to manage co-existing symptom domains (eg, mood changes, aggression, negative symptoms, disorganization, and cognitive deficits) • to mitigate antipsychotic-induced side effects (eg, initiating aripiprazole to treat hyperprolactinemia induced by another antipsychotic to which the patient has achieved a favorable response).23
Clinicians report using APP to treat as many as 50% of patients with a history of multiple psychotic episodes.23 For FEP patients, 23% of participants in the RAISE-ETP trial who were identified as possibly benefiting from therapy modification were prescribed APP.20 Regrettably, researchers have not found evidence to support a reported rationale for using APP—that lower dosages of individual antipsychotics when used in combination may avoid high-dosage prescriptions.24
Before implementing APP, thoroughly explore and manage reasons for a patient’s suboptimal response to monotherapy.25 An adequate trial with any antipsychotic should be at the highest tolerated dosage for 12 to 16 weeks. Be mindful that response to an APP trial may be the result of additional time on the original antipsychotic.
Long-acting injectable antipsychotics in FEP Guideline recommendations. Most older guidelines for schizophrenia treatment suggest LAIA after multiple relapses related to medication nonadherence or when a patient prefers injected medication (Table 1).6-13 Expert consensus guidelines also recommend considering LAIA in patients who lack insight into their illness. The Texas Medication Algorithm Project (TMAP) guidelines7 state LAIA can be considered for inadequate adherence at any stage, whereas the 2010 British Association for Psychopharmacology (BAP) guidelines9 express uncertainty about their use in FEP, because of limited evidence. Both the BAP and National Institute for Health and Care Excellence guidelines13 urge clinicians to consider LAIA when avoiding nonadherence is a treatment priority.
Recently, the French Association for Biological Psychiatry and Neuro-psychopharmacology (AFPBN) created expert consensus guidelines12 on using LAIA in practice. They recommend long-acting injectable second-generation antipsychotics (SGAs) as first-line maintenance treatment for schizophrenia and schizoaffective disorder and for individuals experiencing a first recurrent episode. The World Federation of Societies of Biological Psychiatry guidelines contain LAIA dosage recommendations for FEP (Table 2).10
Advances have been made in understanding the serious neurobiological adverse effects of psychotic relapses, including neuroinflammation and oxidative stress, that may explain the atrophic changes observed with psychotic episodes starting with the FEP. Protecting the patient from a second episode has become a vital therapeutic management goal26 (Figure 127).
Concerns. Compared with oral antipsychotics, LAIA offers clinical advantages: • improved pharmacokinetic profile (lower “peaks” and higher “valleys”) • more consistent plasma concentrations (no variability related to administration timing or food effects) • no first-pass metabolism, which can ease the process of finding the lowest effective and safe dosage • reduced administration burden and objective tracking of adherence with typical dosing every 2 to 4 weeks • less stigmatizing than oral medication for FEP patients, such as college students living in a dormitory.28,29
Barriers to LAIA use include: • slow dosage titration and increased time to reach steady state drug level • oral supplementation for some (eg, risperidone microspheres and aripiprazole long-acting injectable) • logistical challenges for some (eg, 3-hour post-injection monitoring for delirium sedation syndrome with olanzapine pamoate) • additional planning to coordinate care for scheduled injections • higher expenses up front • local injection site reactions • dosage adjustment difficulties if adverse effects occur.28,29
Adoption rates of LAIA are low, especially for FEP.30 Most surveys indicate that (1) physicians believe LAIA treatment is ineffective for FEP31 and (2) patients do not prefer injectable to oral antipsychotics,32 despite evidence to the contrary.33,34 A survey of 198 psychiatrists identified 3 factors that influenced their decisions against using LAIA patients with FEP: • limited availability of SGA depot formulations (4, to date, in the United States) • frequent rejection by the patient when LAIA is offered without adequate explanation or encouragement • skepticism of FEP patients (and their family) who lack experience with relapse.35
In reality, when SGA depots were introduced in the United Kingdom, prescribing rates of LAIA did not increase. As for patient rejection being a major reason for not prescribing LAIA, few patients (5% to 36%) are offered depot injections, particularly in FEP.29 Most patients using LAIA are chronic, multi-episode, violent people who are receiving medications involuntarily.29 Interestingly, this survey did not find 2 factors to be influential in psychiatrists’ decision not to use LAIA in FEP: • guidelines do not explicitly recommend depot treatment in FEP • treatment in FEP may be limited to 1 year, therefore depot administration is not worthwhile.35
Preliminary evidence. At least a dozen studies have explored LAIA treatment for FEP, with the use of fluphenazine decanoate,36 perphenazine enanthate37 (discontinued), and risperidone microspheres.37-48 The research demonstrates the efficacy and safety of LAIA in FEP as measured by these endpoints: • improved symptom control38,40-43,46,48 • adherence43,44,48 • reduced relapse rates37,43 and rehospitalizations37,47 • lesser reductions in white matter brain volume45 • no differences in extrapyramidal side effects or prolactin-associated adverse effects.48
A few small studies demonstrate significant differences in outcomes between risperidone LAIA and oral comparator groups (Table 3).43-45 Ongoing studies of LAIA use in FEP are comparing paliperidone palmitate with risperidone microspheres and other oral antipsychotics.49-51 No studies are examining olanzapine pamoate in FEP, likely because several guidelines do not recommended its use. No studies have been published regarding aripiprazole long-acting injectable in FEP. This LAIA formulation was approved in February 2013, and robust studies of the oral formulation in FEP are limited.52
Discussion and recommendations. Psychiatrists relying on subjective measures of antipsychotic adherence may inaccurately assess whether patients meet this criterion for LAIA use.53 LAIA could combat the high relapse rate in FEP, yet depot antipsychotics are prescribed infrequently for FEP patients (eg, for only 9.5% of participants in the RAISE-ETP study).20 Most schizophrenia treatment guidelines do not discuss LAIA use specifically in FEP, although the AFPBN expert consensus guidelines published in 2013 do recommend SGA depot formulations in FEP.12 SGA LAIA may be preferable, given its neuroprotective effects, in contrast to the neurotoxicity concerns of FGA LAIA.54,55
Relapses begin within a few months of illness stabilization after FEP, and >50% of patients relapse within 1 or 2 years2—the recommended minimum treatment duration for FEP.8,9,13 The use of LAIA is advisable in any patient with schizophrenia for whom long-term antipsychotic therapy is indicated.56 LAIA administration requirements objectively track medication adherence, which allows clinicians to be proactive in relapse prevention. Not using an intervention in FEP that improves adherence and decreases relapse rates contradicts our goal of instituting early, effective treatment to improve long-term functional outcomes (Figure 2).29
Considering clozapine in FEP Guideline recommendations. Schizo-phrenia treatment guidelines and FDA labeling57 reserve clozapine for third-line treatment of refractory schizophrenia after 2 adequate antipsychotic trials have failed despite optimal dosing (Table 1).6-13 Some guidelines specify 1 of the 2 failed antipsychotic trials must include an SGA.6,7,10,11,13-16 Most say clozapine may be considered in patients with chronic aggression or hostility,7-9,14,16 or suicidal thoughts and behaviors.6-8,14,16 TMAP guidelines recommend a clozapine trial with concomitant substance abuse, persistent positive symptoms during 2 years of consistent medication treatment, and after 5 years of inadequate response (“treatment resistance”), regardless of the number of antipsychotic trials.7
Rationale and concerns. Clozapine is a superior choice for treatment-refractory delusions or hallucinations of schizophrenia, because it markedly enhances the response rate to antipsychotic therapy.58 Researchers therefore have investigated whether clozapine, compared with other antipsychotics, would yield more favorable initial and long-term outcomes when used first-line in FEP.
Preliminary evidence. Five studies have explored the use of clozapine as first-line therapy in FEP (Table 4).59-63 Interpreting the results is difficult because clozapine trials may be brief (mostly, 12 to 52 weeks); lack a comparator arm; suffer from a high attrition rate; enroll few patients; and lack potentially important outcome measures such as negative symptoms, suicidality, and functional assessment.
Overall, these studies demonstrate clozapine is as efficacious in this patient population as chlorpromazine (no difference in remission at 1-year, although clozapine-treated patients remitted faster and stayed in remission longer)60,61 or risperidone (no difference in Positive and Negative Syndrome Scale scores).62
At present, clozapine has not been shown superior to other antipsychotics as a first-line treatment for FEP. Research does underscore the importance of a clozapine trial as third-line treatment for FEP patients who have not responded well to 2 SGA trials.63 Many of these nonresponders (77%) have demonstrated a favorable response when promptly switched to clozapine.64
Discussion and recommendations. The limited evidence argues against using clozapine earlier than as third-line treatment in FEP. Perhaps the high treatment response that characterizes FEP creates a ceiling effect that obscures differences in antipsychotic efficacy at this stage.65 Clozapine use as first-line treatment should be re-evaluated with more robust methodology. One approach could be to assess its benefit in FEP by the duration of untreated psychosis.
The odds of achieving remission have been shown to decrease by 15% for each year that psychosis has not been treated.59 Studies exploring the use of clozapine as a second-line agent for FEP also are warranted, as antipsychotic response during subsequent trials is substantially reduced. In fact, the Scottish Intercollegiate Guidelines Network guidelines recommend this as an area for future research.11
For now, clozapine should continue to be reserved as second- or third-line treatment in a patient with FEP. The risks of clozapine’s potentially serious adverse effects (eg, agranulocytosis, seizures, obesity, diabetes, dyslipidemia, myocarditis, pancreatitis, hypotension, sialorrhea, severe sedation, ileus) can be justified only in the treatment of severe and persistent psychotic symptoms.57
Bottom Line Nonstandard use of antipsychotic monotherapy dosages beyond the approved FDA limit and combination antipsychotic therapy may be reasonable for select first-episode psychosis (FEP) patients. Strongly consider long-acting injectable antipsychotics in FEP to proactively combat the high relapse rate and more easily identify antipsychotic failure. Continue to use clozapine as second- or third-line therapy in FEP: Studies have not found that it is more efficacious than other antipsychotics for first-line use.
Related Resource • Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
Disclosures Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products. Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
References
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804. 2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283. 3. Lieberman JA, Koreen AR, Chakos M, et al. Factors influencing treatment response and outcome of first-episode schizophrenia: implications for understanding the pathophysiology of schizophrenia. J Clin Psychiatry. 1996;57(suppl 9):5-9. 4. Agid O, Arenovich T, Sajeev G, et al. An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J Clin Psychiatry. 2011;72(11):1439-1444. 5. Gardner KN, Nasrallah HA. Managing first-episode psychosis. An early stage of schizophrenia with distinct treatment needs. Current Psychiatry. 2015;14(5):32-34,36-40,42. 6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56. 7. Texas Department of State Health Services. Texas Medication Algorithm Project (TMAP) Procedural Manual. Schizophrenia Treatment Algorithms. http://www.jpshealthnet.org/sites/default/files/ tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed June 11, 2015. 8. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93. 9. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620. 10. Hasan A, Falkai P, Wobrok T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44. 11. Scottish Intercollegiate Guidelines Network. SIGN 131: Management of schizophrenia. http://www.sign.ac.uk/ pdf/sign131.pdf. Published March 2013. Accessed June 11, 2015. 12. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serous mental illness. BMC Psychiatry. 2013;13:340. 13. National Institute for Health and Care Excellence. NICE clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. https://www.nice.org. uk/guidance/cg178/resources/guidance-psychosis-and-schizophrenia-in-adults-treatment-and-management-pdf. Updated March 2014. Accessed June 16, 2015. 14. Canadian Psychiatric Association. Clinical practice guidelines. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S. 15. McEvoy JP, Scheifler PL, Frances A. The expert consensus guideline series: treatment of schizophrenia. J Clin Psychiatry. 1999;60(suppl 11):3-80. 16. Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16. 17. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520. 18. Fankhauser MP. Drug interactions with tobacco smoke: implications for patient care. Current Psychiatry. 2013;12(1):12-16. 19. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022. 20. Robinson DG, Schooler NR, John M, et al. Prescription practices in the treatment of first-episode schizophrenia spectrum disorders: data from the national RAISE-ETP study. Am J Psychiatry. 2015;172(3):237-248. 21. Correll CU, Rummel-Kluge C, Corves C, et al. Antipsychotic combinations vs monotherapy in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Bull. 2009;35(2):443-457. 22. Fisher MD, Reilly K, Isenberg K, et al. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry. 2014;14(1):341. 23. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399. 24. John AP, Dragovic M. Antipsychotic polypharmacy is not associated with reduced dose of individual antipsychotics in schizophrenia. J Clin Psychopharmacol. 2015;35(2):193-195. 25. Nasrallah HA. Treatment-resistant schizophrenia. Current Psychiatry. http://www.currentpsychiatry.com/specialty-focus/schizophrenia-other-psychotic-disorders/article/ treatment-resistant-schizophrenia/9be7bba3713d4a4cd68aa 8c92b79e5b1.html. Accessed June 16, 2015. 26. Alvarez-Jiménez M, Parker AG, Hetrick SE, et al. Preventing the second episode: a systematic review and meta-analysis of psychosocial and pharmacological trials in first-episode psychosis. Schizophr Bull. 2011;37(3):619-630. 27. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. 2nd ed. Newton, PA: Handbooks in Health Care Co; 2011. 28. McEvoy JP. Risks versus benefits of different types of long-acting injectable antipsychotics. J Clin Psychiatry. 2006;67(suppl 5):15-18. 29. Agid O, Foussias G, Remington G. Long-acting injectable antipsychotics in the treatment of schizophrenia: their role in relapse prevention. Expert Opin Pharmacother. 2010;11(14):2301-2317. 30. Kirschner M, Theodoridou A, Fusar-Poli P, et al. Patients’ and clinicians’ attitude towards long-acting depot antipsychotics in subjects with a first episode psychosis. Ther Adv Psychophamacol. 2013;3(2):89-99. 31. Heres S, Hamann J, Mendel R, et al. Identifying the profile of optimal candidates for antipsychotic depot therapy: A cluster analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1987-1993. 32. Heres S, Lambert M, Vauth R. Treatment of early episode in patents with schizophrenia: the role of long acting antipsychotics. Eur Psychiatry. 2014;29(suppl 2):1409-1413. 33. Heres S, Schmitz FS, Leucht S, et al. The attitude of patients towards antipsychotic depot treatment. Int Clin Psychopharmacol. 2007;22(5):275-282. 34. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406. 35. Heres S, Reichhart T, Hamann J, et al. Psychiatrists’ attitude to antipsychotic depot treatment in patients with first-episode schizophrenia. Eur Psychiatry. 2011;26(5):297-301. 36. Kane JM, Rifkin A, Quitkin F, et al. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Arch Gen Psychiatry. 1982;39(1):70-73. 37. Tiihonen J, Wahlbeck K, Lönnqvist J, et al. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in a community care after first hospitalization due to schizophrenia and schizoaffective disorder: observational follow-up study. BMJ. 2006;333(7561):224. 38. Parellada E, Andrezina R, Milanova V, et al. Patients in the early phases of schizophrenia and schizoaffective disorders effectively treated with risperidone long-acting injectable. J Psychopharmacol. 2005;19(suppl 5):5-14. 39. Malla A, Binder C, Chue P. Comparison of long-acting injectable risperidone and oral novel antipsychotic drugs for treatment in early phase of schizophrenia spectrum psychosis. Proceedings of the 61st Annual Convention Society of Biological Psychiatry; Toronto, Canada; 2006. 40. Lasser RA, Bossie CA, Zhu Y, et al. Long-acting risperidone in young adults with early schizophrenia or schizoaffective illness. Ann Clin Psychiatry. 2007;19(2):65-71. 41. Emsley R, Oosthuizen P, Koen L, et al. Remission in patients with first-episode schizophrenia receiving assured antipsychotic medication: a study with risperidone long-acting injection. Int Clin Psychopharmacol. 2008;23(6):325-331. 42. Emsley R, Oosthuizen P, Koen L, et al. Oral versus injectable antipsychotic treatment in early psychosis: post hoc comparison of two studies. Clin Ther. 2008;30(12):2378-2386. 43. Kim B, Lee SH, Choi TK, et al. Effectiveness of risperidone long-acting injection in first-episode schizophrenia: in naturalistic setting. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1231-1235. 44. Weiden PJ, Schooler NJ, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406. 45. Bartzokis G, Lu PH, Amar CP, et al. Long acting injection versus oral risperidone in first-episode schizophrenia: differential impact on white matter myelination trajectory. Schizophr Res. 2011;132(1):35-41. 46. Napryeyenko O, Burba B, Martinez G, et al. Risperidone long-acting injectable in recent-onset schizophrenia examined with clinician and patient self-report measures. J Clin Psychopharmacol. 2010;30(2):200-202. 47. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. Am J Psychiatry. 2011;168(6):603-609. 48. Dubois V, Megens J, Mertens C, et al. Long-acting risperidone in early-episode schizophrenia. Acta Psychiatrica Belgica. 2011;111(1):9-21. 49. ClinicalTrials.gov. Oral risperidone versus injectable paliperidone palmitate for treating first-episode schizophrenia. https://clinicaltrials.gov/ct2/show/ NCT01451736. Accessed June 16, 2015. 50. ClinicalTrials.gov. Brain myelination effects of paliperidone palmitate versus oral risperidone in first episode schizophrenia. https://clinicaltrials.gov/ct2/ show/NCT01458379. Accessed June 16, 2015. 51. ClinicalTrials.gov. Effects of paliperidone palmitate versus oral antipsychotics on clinical outcomes and MRI measures. https://clinicaltrials.gov/ct2/show/NCT01359293. Accessed June 16, 2016. 52. U.S. Food and Drug Administration. Drugs@FDA. http:// www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed January 11, 2015. 53. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1-46; quiz 47-48. 54. Nandra KS, Agius M. The difference between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99. 55. Nasrallah HA. Haloperidol is clearly neurotoxic. Should it be banned? Current Psychiatry. 2013;12(7):7-8. 56. Kane JM, Garcia-Ribora C. Clinical guideline recommendations for antipsychotic long-acting injections. Br J Psychiatry. 2009;52:S63-S67. 57. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014. 58. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796. 59. Woerner MG, Robinson DG, Alvir JMJ, et al. Clozapine as a first treatment for schizophrenia. Am J Psychiatry. 2003;160(8):1514-1516. 60. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003. 61. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288. 62. Sanz-Fuentenebro J, Taboada D, Palomo T, et al. Randomized trial of clozapine vs. risperidone in treatment-naïve first-episode schizophrenia: results after one year. Schizophr Res. 2013;149(1-3):156-161. 63. Yang PD, Ji Z. The efficacy and related factors of clozapine on first-episode schizophrenia. Chin J Nerv Ment Dis. 1997;23:155-158. 64. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022. 65. Remington G, Agid O, Foussias G, et al. Clozapine’s role in the treatment of first-episode schizophrenia. Am J Psychiatry. 2013;170(2):146-151.
References
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804. 2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283. 3. Lieberman JA, Koreen AR, Chakos M, et al. Factors influencing treatment response and outcome of first-episode schizophrenia: implications for understanding the pathophysiology of schizophrenia. J Clin Psychiatry. 1996;57(suppl 9):5-9. 4. Agid O, Arenovich T, Sajeev G, et al. An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J Clin Psychiatry. 2011;72(11):1439-1444. 5. Gardner KN, Nasrallah HA. Managing first-episode psychosis. An early stage of schizophrenia with distinct treatment needs. Current Psychiatry. 2015;14(5):32-34,36-40,42. 6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56. 7. Texas Department of State Health Services. Texas Medication Algorithm Project (TMAP) Procedural Manual. Schizophrenia Treatment Algorithms. http://www.jpshealthnet.org/sites/default/files/ tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed June 11, 2015. 8. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93. 9. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620. 10. Hasan A, Falkai P, Wobrok T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44. 11. Scottish Intercollegiate Guidelines Network. SIGN 131: Management of schizophrenia. http://www.sign.ac.uk/ pdf/sign131.pdf. Published March 2013. Accessed June 11, 2015. 12. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serous mental illness. BMC Psychiatry. 2013;13:340. 13. National Institute for Health and Care Excellence. NICE clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. https://www.nice.org. uk/guidance/cg178/resources/guidance-psychosis-and-schizophrenia-in-adults-treatment-and-management-pdf. Updated March 2014. Accessed June 16, 2015. 14. Canadian Psychiatric Association. Clinical practice guidelines. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S. 15. McEvoy JP, Scheifler PL, Frances A. The expert consensus guideline series: treatment of schizophrenia. J Clin Psychiatry. 1999;60(suppl 11):3-80. 16. Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16. 17. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520. 18. Fankhauser MP. Drug interactions with tobacco smoke: implications for patient care. Current Psychiatry. 2013;12(1):12-16. 19. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022. 20. Robinson DG, Schooler NR, John M, et al. Prescription practices in the treatment of first-episode schizophrenia spectrum disorders: data from the national RAISE-ETP study. Am J Psychiatry. 2015;172(3):237-248. 21. Correll CU, Rummel-Kluge C, Corves C, et al. Antipsychotic combinations vs monotherapy in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Bull. 2009;35(2):443-457. 22. Fisher MD, Reilly K, Isenberg K, et al. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry. 2014;14(1):341. 23. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399. 24. John AP, Dragovic M. Antipsychotic polypharmacy is not associated with reduced dose of individual antipsychotics in schizophrenia. J Clin Psychopharmacol. 2015;35(2):193-195. 25. Nasrallah HA. Treatment-resistant schizophrenia. Current Psychiatry. http://www.currentpsychiatry.com/specialty-focus/schizophrenia-other-psychotic-disorders/article/ treatment-resistant-schizophrenia/9be7bba3713d4a4cd68aa 8c92b79e5b1.html. Accessed June 16, 2015. 26. Alvarez-Jiménez M, Parker AG, Hetrick SE, et al. Preventing the second episode: a systematic review and meta-analysis of psychosocial and pharmacological trials in first-episode psychosis. Schizophr Bull. 2011;37(3):619-630. 27. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. 2nd ed. Newton, PA: Handbooks in Health Care Co; 2011. 28. McEvoy JP. Risks versus benefits of different types of long-acting injectable antipsychotics. J Clin Psychiatry. 2006;67(suppl 5):15-18. 29. Agid O, Foussias G, Remington G. Long-acting injectable antipsychotics in the treatment of schizophrenia: their role in relapse prevention. Expert Opin Pharmacother. 2010;11(14):2301-2317. 30. Kirschner M, Theodoridou A, Fusar-Poli P, et al. Patients’ and clinicians’ attitude towards long-acting depot antipsychotics in subjects with a first episode psychosis. Ther Adv Psychophamacol. 2013;3(2):89-99. 31. Heres S, Hamann J, Mendel R, et al. Identifying the profile of optimal candidates for antipsychotic depot therapy: A cluster analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1987-1993. 32. Heres S, Lambert M, Vauth R. Treatment of early episode in patents with schizophrenia: the role of long acting antipsychotics. Eur Psychiatry. 2014;29(suppl 2):1409-1413. 33. Heres S, Schmitz FS, Leucht S, et al. The attitude of patients towards antipsychotic depot treatment. Int Clin Psychopharmacol. 2007;22(5):275-282. 34. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406. 35. Heres S, Reichhart T, Hamann J, et al. Psychiatrists’ attitude to antipsychotic depot treatment in patients with first-episode schizophrenia. Eur Psychiatry. 2011;26(5):297-301. 36. Kane JM, Rifkin A, Quitkin F, et al. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Arch Gen Psychiatry. 1982;39(1):70-73. 37. Tiihonen J, Wahlbeck K, Lönnqvist J, et al. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in a community care after first hospitalization due to schizophrenia and schizoaffective disorder: observational follow-up study. BMJ. 2006;333(7561):224. 38. Parellada E, Andrezina R, Milanova V, et al. Patients in the early phases of schizophrenia and schizoaffective disorders effectively treated with risperidone long-acting injectable. J Psychopharmacol. 2005;19(suppl 5):5-14. 39. Malla A, Binder C, Chue P. Comparison of long-acting injectable risperidone and oral novel antipsychotic drugs for treatment in early phase of schizophrenia spectrum psychosis. Proceedings of the 61st Annual Convention Society of Biological Psychiatry; Toronto, Canada; 2006. 40. Lasser RA, Bossie CA, Zhu Y, et al. Long-acting risperidone in young adults with early schizophrenia or schizoaffective illness. Ann Clin Psychiatry. 2007;19(2):65-71. 41. Emsley R, Oosthuizen P, Koen L, et al. Remission in patients with first-episode schizophrenia receiving assured antipsychotic medication: a study with risperidone long-acting injection. Int Clin Psychopharmacol. 2008;23(6):325-331. 42. Emsley R, Oosthuizen P, Koen L, et al. Oral versus injectable antipsychotic treatment in early psychosis: post hoc comparison of two studies. Clin Ther. 2008;30(12):2378-2386. 43. Kim B, Lee SH, Choi TK, et al. Effectiveness of risperidone long-acting injection in first-episode schizophrenia: in naturalistic setting. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1231-1235. 44. Weiden PJ, Schooler NJ, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406. 45. Bartzokis G, Lu PH, Amar CP, et al. Long acting injection versus oral risperidone in first-episode schizophrenia: differential impact on white matter myelination trajectory. Schizophr Res. 2011;132(1):35-41. 46. Napryeyenko O, Burba B, Martinez G, et al. Risperidone long-acting injectable in recent-onset schizophrenia examined with clinician and patient self-report measures. J Clin Psychopharmacol. 2010;30(2):200-202. 47. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. Am J Psychiatry. 2011;168(6):603-609. 48. Dubois V, Megens J, Mertens C, et al. Long-acting risperidone in early-episode schizophrenia. Acta Psychiatrica Belgica. 2011;111(1):9-21. 49. ClinicalTrials.gov. Oral risperidone versus injectable paliperidone palmitate for treating first-episode schizophrenia. https://clinicaltrials.gov/ct2/show/ NCT01451736. Accessed June 16, 2015. 50. ClinicalTrials.gov. Brain myelination effects of paliperidone palmitate versus oral risperidone in first episode schizophrenia. https://clinicaltrials.gov/ct2/ show/NCT01458379. Accessed June 16, 2015. 51. ClinicalTrials.gov. Effects of paliperidone palmitate versus oral antipsychotics on clinical outcomes and MRI measures. https://clinicaltrials.gov/ct2/show/NCT01359293. Accessed June 16, 2016. 52. U.S. Food and Drug Administration. Drugs@FDA. http:// www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed January 11, 2015. 53. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1-46; quiz 47-48. 54. Nandra KS, Agius M. The difference between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99. 55. Nasrallah HA. Haloperidol is clearly neurotoxic. Should it be banned? Current Psychiatry. 2013;12(7):7-8. 56. Kane JM, Garcia-Ribora C. Clinical guideline recommendations for antipsychotic long-acting injections. Br J Psychiatry. 2009;52:S63-S67. 57. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014. 58. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796. 59. Woerner MG, Robinson DG, Alvir JMJ, et al. Clozapine as a first treatment for schizophrenia. Am J Psychiatry. 2003;160(8):1514-1516. 60. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003. 61. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288. 62. Sanz-Fuentenebro J, Taboada D, Palomo T, et al. Randomized trial of clozapine vs. risperidone in treatment-naïve first-episode schizophrenia: results after one year. Schizophr Res. 2013;149(1-3):156-161. 63. Yang PD, Ji Z. The efficacy and related factors of clozapine on first-episode schizophrenia. Chin J Nerv Ment Dis. 1997;23:155-158. 64. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022. 65. Remington G, Agid O, Foussias G, et al. Clozapine’s role in the treatment of first-episode schizophrenia. Am J Psychiatry. 2013;170(2):146-151.
Managing first-episode psychosis: Rationale and evidence for nonstandard first-line treatments for schizophrenia
Display Headline
Managing first-episode psychosis: Rationale and evidence for nonstandard first-line treatments for schizophrenia
Legacy Keywords
first-line treatments for schizophrenia, first line treatments for schizophrenia, first-line treatment for schizophrenia, first line treatment for schizophrenia, schizophrenia, psychosis, psychotic disorders, first episode psychosis, first-episode psychosis, clozapien, injectable antipsychotics, long acting injectable antipsychotics, long-acting injectable antipsychotics, monotherapy
Legacy Keywords
first-line treatments for schizophrenia, first line treatments for schizophrenia, first-line treatment for schizophrenia, first line treatment for schizophrenia, schizophrenia, psychosis, psychotic disorders, first episode psychosis, first-episode psychosis, clozapien, injectable antipsychotics, long acting injectable antipsychotics, long-acting injectable antipsychotics, monotherapy
People with serious mental illness (SMI) have a life expectancy that is 25 years less than the general population, according to the Centers for Disease Control and Prevention.1 This disparity is partially a consequence of the lack of primary and preventive medical care for those with psychiatric illness. Decades of research have shown that people with SMI experience higher medical morbidity and mortality in addition to facing the stigma of mental illness.
This article aims to advance the idea that longitudinal “cross education” between primary care providers (PCPs) and behavioral health providers (BHPs) is essential in addressing this problem. BHPs include psychiatry clinics, which often are part of a university or large health systems; county-based community mental health programs; and independent mental health clinics that contract with public and private health plans to provide mental health services.
Although suicide and injury account for 40% of the excess mortality in schizophrenia, 60% can be attributed to cardiovascular disease, diabetes, respiratory diseases, and infection.2 Patients with SMI have 2 to 3 times the risk of diabetes, dyslipidemia, hypertension, and obesity.3,4 Furthermore, those with SMI consume more than one-third of tobacco products,5 and 50% to 80% of people with SMI smoke tobacco, an important reversible risk factor for cardiovascular disease.
Figure 1 shows that people with SMI are at higher risk of dying from a chronic medical condition, such as cardiovascular disease, diabetes, chronic obstructive pulmonary disease, and hepatitis C6-8—many of which can be managed by primary and preventive medical interventions. These and other conditions often are not diagnosed or effectively managed in patients with SMI.
The high prevalence of metabolic syndrome and tobacco dependence among people with SMI accelerates development of cardiovascular disease, as shown by several studies. Bobes et al9 found that the prevalence of metabolic syndrome and cardiovascular risk among patients with SMI is similar to what is found in the general population at 10 to 15 years of greater age. Osborn et al10 demonstrated that people with SMI age 18 to 49 had a higher relative risk of death from coronary heart disease, stroke, and lung cancer than age-matched controls (Figure 2).
It can be said, therefore, that patients with SMI seem to “age” and die prematurely. To reduce this disparity, primary and preventive medical care—especially for cardiovascular disease—must be delivered earlier in life for those with SMI.
Iatrogenic causes of morbidity Many psychiatric medications, especially second-generation antipsychotics (SGAs), could exacerbate cardiovascular and metabolic conditions by increasing the risk of weight gain, insulin resistance, and dyslipidemia. Antipsychotics that generally are considered to be more effective for refractory psychotic illness (eg, clozapine and olanzapine) are associated with the highest risk of metabolic syndrome. Simon et al11 found a dose-response relationship between olanzapine and clozapine serum concentrations and worsening metabolic outcomes. Valproic acid also can cause significant weight gain and could require monitoring similar to what is done with to SGAs, although there has been less clinical and research attention to this mood stabilizer.
The American Diabetes Association et al12 have published guidelines on monitoring antipsychotic-induced obesity and diabetes, but adoption of these guidelines has been slow. Mackin et al13 found that providers are slow to recognize the elevated rate of obesity and dyslipidemia among psychiatric patients, possibly because of “an alarmingly poor rate of monitoring of metabolic parameters.”
Treating adverse metabolic outcomes also seems to lag behind. The same study13 found that physical health parameters among psychiatric patients continue to become worse even when appropriate health care professionals were notified. Rates of nontreatment for diabetes, dyslipidemia, and hypertension were 30%, 60%, and 88% respectively, according to Nasrallah et al.14
Randomized controlled studies have shown that obesity and metabolic syndrome can be effectively managed using lifestyle and pharmacotherapeutic approaches,15,16 but more research is needed to test long-term outcomes and how to best incorporate these interventions. Newcomer et al17 found that gradually switching an antipsychotic with high risk of metabolic adverse effects to one with lower risk could reduce adverse metabolic outcomes; however, some patients returned to their prior antipsychotic because other medications did not effectively treat their schizophrenia symptoms. Therefore, physicians must pay careful attention to the trade-off between benefits and risks of antipsychotics and make treatment decisions on an individual basis.
Barriers to medical care Research has demonstrated that patients with SMI receive less screening and fewer preventive medical services, especially blood pressure monitoring, vaccinations, mammography, lipid monitoring, and osteoporosis screening, compared with the general population (Table).18 Some barriers to preventive services could exist because of demographic factors and medical insurance coverage19 or medical providers’ discomfort with symptoms of SMI,20 although Mitchell et al21 found that disparities in mammography screening could not be explained by the presence of emotional distress in women with SMI.
DiMatteo et al22 reported that patients with SMI are 3 times more likely to be noncompliant with medical treatment. These patients also are less likely to receive sec ondary preventive medical care and invasive medical procedures. Those with SMI who experience acute myocardial infarction are less likely to receive drug therapy, such as a thrombolytic, aspirin, beta blocker, or angiotensin-converting enzyme inhibitor.23 They also are less likely to receive invasive cardiovascular procedures, including cardiac catheterization, angioplasty, and coronary artery bypass grafting.24
Therefore, not only are patients with SMI less likely to receive preventive care, they are also less likely to receive potentially lifesaving treatments for SMI. Because those with SMI might not be able to advocate for themselves in these matters, psychiatric clinicians can improve their patients’ lives by advocating for appropriate medical care despite multiple barriers.
Bridging the gap: Managing mental health in primary care Research from the 1970s and 1980s demonstrated that most persons who sought help for depression or anxiety received treatment from their PCP, many of whom felt limited by their lack of behavioral health training. Moreover, many patients failed to receive a psychiatric diagnosis or adequate treatment, despite efforts to educate primary care physicians on appropriate diagnosis and treatment of mental illness.
Katon et al25 at the University of Washington developed the collaborative care model in the early 1990s to help improve treatment of depression in primary care settings. This model involved: • case load review by psychiatrists • use of nurses and other support staff to help monitor patients’ adherence and treatment response • use of standardized tools such as the Patient Health Questionnaire to monitor symptoms • enhancement of patient education with pamphlets or classes.
Studies evaluating the success of collaborative care models found overall improved outcomes, making it the only evidence-based model for integration of behavioral health and primary care.26 As a result, the collaborative care model has been implemented across the United States in primary care clinics and specialty care settings, such as obstetrics and gynecology.27
Regrettably, access to primary care has been hampered by: • population growth • a shortage of PCPs • enrollment of a flood of new patients into the health care marketplace as a result of mandates of the Affordable Care Act (ACA).
In many settings, a psychiatrist might be the patient’s only consistent care provider, and could be thought of as a “primary care psychiatrist.”
To resolve this predicament, mental health professionals need to recognize the unique medical conditions faced by people with SMI, and also might need to provide treatment of common medical conditions, either directly or through collaborative arrangements. Psychiatrists who are capable of managing core medical issues likely will witness improved psychiatric and overall health outcomes in their patients. Consequently, psychiatrists and mental health professionals are increasingly called on to be advocates to improve access to medical services in patients with SMI and to participate in health systems reform.
Managing medical conditions in mental health settings Although traditional collaborative care involves mental health providers working at primary care sites, other models have emerged that manage chronic disease in behavioral health settings. Federally funded grants for primary behavioral health care integration have allowed community mental health centers to partner with federally qualified health centers to provide on-site primary care services.28
In these models, care managers in mental health clinics: • link patients to primary care services • encourage lifestyle changes to improve their overall health • identify and overcome barriers to receiving care • track clinical outcomes in a registry format.
Currently, 126 mental health sites in the United States have received these grants and are working toward greater integration of primary care.
In addition, the ACA provided funding for “health homes” in non-primary care settings, which includes SMI. These health homes cannot provide direct primary care, but can deliver comprehensive care management, care coordination, health promotion, comprehensive transitional care services between facilities, individual and family support, and referral to community social support services. In these health homes, a PCP can act as a consultant to help establish priorities for disease management and improving health status.29 The PCP consultant also can support psychiatric staff and collaborate with providers who want to provide some direct care of medical conditions.30
Last, some behavioral health sites are choosing to apply for Federally Qualified Health Clinic status or add primary care services to their clinics, with the hope that sustainable funding will become available. Without additional funding to cover the limited reimbursement provided by public payers, such as Medicaid and Medicare, these models might be unsustainable. Current innovations in health care funding reform hopefully will offer solutions for sites to provide medical care in the natural “medical home” of the SMI population.
Bottom Line Psychiatric providers are in a favorable position to develop and oversee a partnership with primary care physicians with the goal of addressing significant and often lethal health disparities among those with mental illness. Psychiatric providers must use evidence-based practices that include assessment and prevention of cardiopulmonary, metabolic, infectious, and oncologic disorders. True primary care–behavioral health integration must include longitudinal “cross education” and changes in health care policy, with an emphasis on decreasing morbidity and mortality in psychiatric patients.
Disclosure The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42. 2. Parks J, Svendsen D, Singer P, et al, eds. Morbidity and mortality in people with serious mental illness. Alexandria, VA: National Association of State Mental Health Program Directors (NASMHPD) Medical Directors Council; 2006. 3. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796. 4. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trails of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19-32. 5. Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harv Rev Psychiatry. 2006;14(2):212-222. 6. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131. 7. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156. 8. Carney CP, Jones L, Woolson RF. Medical comorbidity in women and men with schizophrenia: a population-based study. J Gen Intern Med. 2006;21(11):1133-1137. 9. Bobes J, Arango C, Aranda P, et al; CLAMORS Study Collaborative Group. Cardiovascular and metabolic risk in outpatients with schizoaffective disorder treated with antipsychotics; results from the CLAMORS study. Eur Psychiatry. 2012;27(4):267-274. 10. Osborn DP, Levy G, Nazareth I, et al. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s General Practice Research Database [Erratum in: Arch Gen Psychiatry. 2007;64(6):736]. Arch Gen Psychiatry. 2007;64(2):242-249. 11. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050. 12. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601. 13. Mackin P, Bishop DR, Watkinson HM. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry. 2007;7:28. 14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22. 15. Alvarez-Jiménez M, Hetrick SE, González-Blanch C, et al. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomized controlled trials. Br J Psychiatry. 2008; 193(2):101-107. 16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medication used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology. 2010;35(7):1520-1530. 17. Newcomer JW, Weiden PJ, Buchanan RW. Switching antipsychotic medications to reduce adverse event burden in schizophrenia: establishing evidence-based practice. J Clin Psychiatry. 2013;74(11):1108-1120. 18. Lord O, Malone D, Mitchell AJ. Receipt of preventive medical care and medical screening for patients with mental illness: a comparative analysis. Gen Hosp Psychiatry. 2010;32(5):519-543. 19. Xiong GL, Iosif AM, Bermudes RA, et al. Preventive medical services use among community mental health patients with severe mental illness: the influence of gender and insurance coverage. Prim Care Companion J Clin Psychiatry. 2010;12(5). doi: 10.4088/PCC.09m00927gre. 20. Daub S. Turning toward treating the seriously mentally ill in primary care. Fam Syst Health. 2014;32(1):12-13. 21. Mitchell A, Pereira IE, Yadegarfar M, et al. Breast cancer screening in women with mental illness: comparative meta-analysis of mammography uptake. Br J Psychiatry. 2014;205(6):428-435. 22. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107. 23. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572. 24. Druss BG, Bradford DW, Rosenheck RA, et al. Mental disorders and use of cardiovascular procedures after myocardial infarction. JAMA. 2000;283(4):506-511. 25. Katon W, Unützer J, Wells K, et al. Collaborative depression care: history, evolution and ways to enhance dissemination and sustainability. Gen Hosp Psychiatry. 2010;32(5):456-464. 26. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525. 27. Katon W, Russo J, Reed SD, et al. A randomized trial of collaborative depression care in obstetrics and gynecology clinics: socioeconomic disadvantage and treatment response. Am J Psychiatry. 2015;172(1):32-40. 28. Substance Abuse and Mental Health Services Administration. Request for Applications (RFA) No. SM- 09-011. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2009. 29. Parks J. Behavioral health homes. In: Integrated care: working at the interface of primary care and behavioral health. Raney LE, ed. Arlington, VA: American Psychiatric Publishing; 2015:195. 30. Raney L. Integrated care: the evolving role of psychiatry in the era of health care reform. Psychiatr Serv. 2013;64(11):1076-1078.
People with serious mental illness (SMI) have a life expectancy that is 25 years less than the general population, according to the Centers for Disease Control and Prevention.1 This disparity is partially a consequence of the lack of primary and preventive medical care for those with psychiatric illness. Decades of research have shown that people with SMI experience higher medical morbidity and mortality in addition to facing the stigma of mental illness.
This article aims to advance the idea that longitudinal “cross education” between primary care providers (PCPs) and behavioral health providers (BHPs) is essential in addressing this problem. BHPs include psychiatry clinics, which often are part of a university or large health systems; county-based community mental health programs; and independent mental health clinics that contract with public and private health plans to provide mental health services.
Although suicide and injury account for 40% of the excess mortality in schizophrenia, 60% can be attributed to cardiovascular disease, diabetes, respiratory diseases, and infection.2 Patients with SMI have 2 to 3 times the risk of diabetes, dyslipidemia, hypertension, and obesity.3,4 Furthermore, those with SMI consume more than one-third of tobacco products,5 and 50% to 80% of people with SMI smoke tobacco, an important reversible risk factor for cardiovascular disease.
Figure 1 shows that people with SMI are at higher risk of dying from a chronic medical condition, such as cardiovascular disease, diabetes, chronic obstructive pulmonary disease, and hepatitis C6-8—many of which can be managed by primary and preventive medical interventions. These and other conditions often are not diagnosed or effectively managed in patients with SMI.
The high prevalence of metabolic syndrome and tobacco dependence among people with SMI accelerates development of cardiovascular disease, as shown by several studies. Bobes et al9 found that the prevalence of metabolic syndrome and cardiovascular risk among patients with SMI is similar to what is found in the general population at 10 to 15 years of greater age. Osborn et al10 demonstrated that people with SMI age 18 to 49 had a higher relative risk of death from coronary heart disease, stroke, and lung cancer than age-matched controls (Figure 2).
It can be said, therefore, that patients with SMI seem to “age” and die prematurely. To reduce this disparity, primary and preventive medical care—especially for cardiovascular disease—must be delivered earlier in life for those with SMI.
Iatrogenic causes of morbidity Many psychiatric medications, especially second-generation antipsychotics (SGAs), could exacerbate cardiovascular and metabolic conditions by increasing the risk of weight gain, insulin resistance, and dyslipidemia. Antipsychotics that generally are considered to be more effective for refractory psychotic illness (eg, clozapine and olanzapine) are associated with the highest risk of metabolic syndrome. Simon et al11 found a dose-response relationship between olanzapine and clozapine serum concentrations and worsening metabolic outcomes. Valproic acid also can cause significant weight gain and could require monitoring similar to what is done with to SGAs, although there has been less clinical and research attention to this mood stabilizer.
The American Diabetes Association et al12 have published guidelines on monitoring antipsychotic-induced obesity and diabetes, but adoption of these guidelines has been slow. Mackin et al13 found that providers are slow to recognize the elevated rate of obesity and dyslipidemia among psychiatric patients, possibly because of “an alarmingly poor rate of monitoring of metabolic parameters.”
Treating adverse metabolic outcomes also seems to lag behind. The same study13 found that physical health parameters among psychiatric patients continue to become worse even when appropriate health care professionals were notified. Rates of nontreatment for diabetes, dyslipidemia, and hypertension were 30%, 60%, and 88% respectively, according to Nasrallah et al.14
Randomized controlled studies have shown that obesity and metabolic syndrome can be effectively managed using lifestyle and pharmacotherapeutic approaches,15,16 but more research is needed to test long-term outcomes and how to best incorporate these interventions. Newcomer et al17 found that gradually switching an antipsychotic with high risk of metabolic adverse effects to one with lower risk could reduce adverse metabolic outcomes; however, some patients returned to their prior antipsychotic because other medications did not effectively treat their schizophrenia symptoms. Therefore, physicians must pay careful attention to the trade-off between benefits and risks of antipsychotics and make treatment decisions on an individual basis.
Barriers to medical care Research has demonstrated that patients with SMI receive less screening and fewer preventive medical services, especially blood pressure monitoring, vaccinations, mammography, lipid monitoring, and osteoporosis screening, compared with the general population (Table).18 Some barriers to preventive services could exist because of demographic factors and medical insurance coverage19 or medical providers’ discomfort with symptoms of SMI,20 although Mitchell et al21 found that disparities in mammography screening could not be explained by the presence of emotional distress in women with SMI.
DiMatteo et al22 reported that patients with SMI are 3 times more likely to be noncompliant with medical treatment. These patients also are less likely to receive sec ondary preventive medical care and invasive medical procedures. Those with SMI who experience acute myocardial infarction are less likely to receive drug therapy, such as a thrombolytic, aspirin, beta blocker, or angiotensin-converting enzyme inhibitor.23 They also are less likely to receive invasive cardiovascular procedures, including cardiac catheterization, angioplasty, and coronary artery bypass grafting.24
Therefore, not only are patients with SMI less likely to receive preventive care, they are also less likely to receive potentially lifesaving treatments for SMI. Because those with SMI might not be able to advocate for themselves in these matters, psychiatric clinicians can improve their patients’ lives by advocating for appropriate medical care despite multiple barriers.
Bridging the gap: Managing mental health in primary care Research from the 1970s and 1980s demonstrated that most persons who sought help for depression or anxiety received treatment from their PCP, many of whom felt limited by their lack of behavioral health training. Moreover, many patients failed to receive a psychiatric diagnosis or adequate treatment, despite efforts to educate primary care physicians on appropriate diagnosis and treatment of mental illness.
Katon et al25 at the University of Washington developed the collaborative care model in the early 1990s to help improve treatment of depression in primary care settings. This model involved: • case load review by psychiatrists • use of nurses and other support staff to help monitor patients’ adherence and treatment response • use of standardized tools such as the Patient Health Questionnaire to monitor symptoms • enhancement of patient education with pamphlets or classes.
Studies evaluating the success of collaborative care models found overall improved outcomes, making it the only evidence-based model for integration of behavioral health and primary care.26 As a result, the collaborative care model has been implemented across the United States in primary care clinics and specialty care settings, such as obstetrics and gynecology.27
Regrettably, access to primary care has been hampered by: • population growth • a shortage of PCPs • enrollment of a flood of new patients into the health care marketplace as a result of mandates of the Affordable Care Act (ACA).
In many settings, a psychiatrist might be the patient’s only consistent care provider, and could be thought of as a “primary care psychiatrist.”
To resolve this predicament, mental health professionals need to recognize the unique medical conditions faced by people with SMI, and also might need to provide treatment of common medical conditions, either directly or through collaborative arrangements. Psychiatrists who are capable of managing core medical issues likely will witness improved psychiatric and overall health outcomes in their patients. Consequently, psychiatrists and mental health professionals are increasingly called on to be advocates to improve access to medical services in patients with SMI and to participate in health systems reform.
Managing medical conditions in mental health settings Although traditional collaborative care involves mental health providers working at primary care sites, other models have emerged that manage chronic disease in behavioral health settings. Federally funded grants for primary behavioral health care integration have allowed community mental health centers to partner with federally qualified health centers to provide on-site primary care services.28
In these models, care managers in mental health clinics: • link patients to primary care services • encourage lifestyle changes to improve their overall health • identify and overcome barriers to receiving care • track clinical outcomes in a registry format.
Currently, 126 mental health sites in the United States have received these grants and are working toward greater integration of primary care.
In addition, the ACA provided funding for “health homes” in non-primary care settings, which includes SMI. These health homes cannot provide direct primary care, but can deliver comprehensive care management, care coordination, health promotion, comprehensive transitional care services between facilities, individual and family support, and referral to community social support services. In these health homes, a PCP can act as a consultant to help establish priorities for disease management and improving health status.29 The PCP consultant also can support psychiatric staff and collaborate with providers who want to provide some direct care of medical conditions.30
Last, some behavioral health sites are choosing to apply for Federally Qualified Health Clinic status or add primary care services to their clinics, with the hope that sustainable funding will become available. Without additional funding to cover the limited reimbursement provided by public payers, such as Medicaid and Medicare, these models might be unsustainable. Current innovations in health care funding reform hopefully will offer solutions for sites to provide medical care in the natural “medical home” of the SMI population.
Bottom Line Psychiatric providers are in a favorable position to develop and oversee a partnership with primary care physicians with the goal of addressing significant and often lethal health disparities among those with mental illness. Psychiatric providers must use evidence-based practices that include assessment and prevention of cardiopulmonary, metabolic, infectious, and oncologic disorders. True primary care–behavioral health integration must include longitudinal “cross education” and changes in health care policy, with an emphasis on decreasing morbidity and mortality in psychiatric patients.
Disclosure The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
People with serious mental illness (SMI) have a life expectancy that is 25 years less than the general population, according to the Centers for Disease Control and Prevention.1 This disparity is partially a consequence of the lack of primary and preventive medical care for those with psychiatric illness. Decades of research have shown that people with SMI experience higher medical morbidity and mortality in addition to facing the stigma of mental illness.
This article aims to advance the idea that longitudinal “cross education” between primary care providers (PCPs) and behavioral health providers (BHPs) is essential in addressing this problem. BHPs include psychiatry clinics, which often are part of a university or large health systems; county-based community mental health programs; and independent mental health clinics that contract with public and private health plans to provide mental health services.
Although suicide and injury account for 40% of the excess mortality in schizophrenia, 60% can be attributed to cardiovascular disease, diabetes, respiratory diseases, and infection.2 Patients with SMI have 2 to 3 times the risk of diabetes, dyslipidemia, hypertension, and obesity.3,4 Furthermore, those with SMI consume more than one-third of tobacco products,5 and 50% to 80% of people with SMI smoke tobacco, an important reversible risk factor for cardiovascular disease.
Figure 1 shows that people with SMI are at higher risk of dying from a chronic medical condition, such as cardiovascular disease, diabetes, chronic obstructive pulmonary disease, and hepatitis C6-8—many of which can be managed by primary and preventive medical interventions. These and other conditions often are not diagnosed or effectively managed in patients with SMI.
The high prevalence of metabolic syndrome and tobacco dependence among people with SMI accelerates development of cardiovascular disease, as shown by several studies. Bobes et al9 found that the prevalence of metabolic syndrome and cardiovascular risk among patients with SMI is similar to what is found in the general population at 10 to 15 years of greater age. Osborn et al10 demonstrated that people with SMI age 18 to 49 had a higher relative risk of death from coronary heart disease, stroke, and lung cancer than age-matched controls (Figure 2).
It can be said, therefore, that patients with SMI seem to “age” and die prematurely. To reduce this disparity, primary and preventive medical care—especially for cardiovascular disease—must be delivered earlier in life for those with SMI.
Iatrogenic causes of morbidity Many psychiatric medications, especially second-generation antipsychotics (SGAs), could exacerbate cardiovascular and metabolic conditions by increasing the risk of weight gain, insulin resistance, and dyslipidemia. Antipsychotics that generally are considered to be more effective for refractory psychotic illness (eg, clozapine and olanzapine) are associated with the highest risk of metabolic syndrome. Simon et al11 found a dose-response relationship between olanzapine and clozapine serum concentrations and worsening metabolic outcomes. Valproic acid also can cause significant weight gain and could require monitoring similar to what is done with to SGAs, although there has been less clinical and research attention to this mood stabilizer.
The American Diabetes Association et al12 have published guidelines on monitoring antipsychotic-induced obesity and diabetes, but adoption of these guidelines has been slow. Mackin et al13 found that providers are slow to recognize the elevated rate of obesity and dyslipidemia among psychiatric patients, possibly because of “an alarmingly poor rate of monitoring of metabolic parameters.”
Treating adverse metabolic outcomes also seems to lag behind. The same study13 found that physical health parameters among psychiatric patients continue to become worse even when appropriate health care professionals were notified. Rates of nontreatment for diabetes, dyslipidemia, and hypertension were 30%, 60%, and 88% respectively, according to Nasrallah et al.14
Randomized controlled studies have shown that obesity and metabolic syndrome can be effectively managed using lifestyle and pharmacotherapeutic approaches,15,16 but more research is needed to test long-term outcomes and how to best incorporate these interventions. Newcomer et al17 found that gradually switching an antipsychotic with high risk of metabolic adverse effects to one with lower risk could reduce adverse metabolic outcomes; however, some patients returned to their prior antipsychotic because other medications did not effectively treat their schizophrenia symptoms. Therefore, physicians must pay careful attention to the trade-off between benefits and risks of antipsychotics and make treatment decisions on an individual basis.
Barriers to medical care Research has demonstrated that patients with SMI receive less screening and fewer preventive medical services, especially blood pressure monitoring, vaccinations, mammography, lipid monitoring, and osteoporosis screening, compared with the general population (Table).18 Some barriers to preventive services could exist because of demographic factors and medical insurance coverage19 or medical providers’ discomfort with symptoms of SMI,20 although Mitchell et al21 found that disparities in mammography screening could not be explained by the presence of emotional distress in women with SMI.
DiMatteo et al22 reported that patients with SMI are 3 times more likely to be noncompliant with medical treatment. These patients also are less likely to receive sec ondary preventive medical care and invasive medical procedures. Those with SMI who experience acute myocardial infarction are less likely to receive drug therapy, such as a thrombolytic, aspirin, beta blocker, or angiotensin-converting enzyme inhibitor.23 They also are less likely to receive invasive cardiovascular procedures, including cardiac catheterization, angioplasty, and coronary artery bypass grafting.24
Therefore, not only are patients with SMI less likely to receive preventive care, they are also less likely to receive potentially lifesaving treatments for SMI. Because those with SMI might not be able to advocate for themselves in these matters, psychiatric clinicians can improve their patients’ lives by advocating for appropriate medical care despite multiple barriers.
Bridging the gap: Managing mental health in primary care Research from the 1970s and 1980s demonstrated that most persons who sought help for depression or anxiety received treatment from their PCP, many of whom felt limited by their lack of behavioral health training. Moreover, many patients failed to receive a psychiatric diagnosis or adequate treatment, despite efforts to educate primary care physicians on appropriate diagnosis and treatment of mental illness.
Katon et al25 at the University of Washington developed the collaborative care model in the early 1990s to help improve treatment of depression in primary care settings. This model involved: • case load review by psychiatrists • use of nurses and other support staff to help monitor patients’ adherence and treatment response • use of standardized tools such as the Patient Health Questionnaire to monitor symptoms • enhancement of patient education with pamphlets or classes.
Studies evaluating the success of collaborative care models found overall improved outcomes, making it the only evidence-based model for integration of behavioral health and primary care.26 As a result, the collaborative care model has been implemented across the United States in primary care clinics and specialty care settings, such as obstetrics and gynecology.27
Regrettably, access to primary care has been hampered by: • population growth • a shortage of PCPs • enrollment of a flood of new patients into the health care marketplace as a result of mandates of the Affordable Care Act (ACA).
In many settings, a psychiatrist might be the patient’s only consistent care provider, and could be thought of as a “primary care psychiatrist.”
To resolve this predicament, mental health professionals need to recognize the unique medical conditions faced by people with SMI, and also might need to provide treatment of common medical conditions, either directly or through collaborative arrangements. Psychiatrists who are capable of managing core medical issues likely will witness improved psychiatric and overall health outcomes in their patients. Consequently, psychiatrists and mental health professionals are increasingly called on to be advocates to improve access to medical services in patients with SMI and to participate in health systems reform.
Managing medical conditions in mental health settings Although traditional collaborative care involves mental health providers working at primary care sites, other models have emerged that manage chronic disease in behavioral health settings. Federally funded grants for primary behavioral health care integration have allowed community mental health centers to partner with federally qualified health centers to provide on-site primary care services.28
In these models, care managers in mental health clinics: • link patients to primary care services • encourage lifestyle changes to improve their overall health • identify and overcome barriers to receiving care • track clinical outcomes in a registry format.
Currently, 126 mental health sites in the United States have received these grants and are working toward greater integration of primary care.
In addition, the ACA provided funding for “health homes” in non-primary care settings, which includes SMI. These health homes cannot provide direct primary care, but can deliver comprehensive care management, care coordination, health promotion, comprehensive transitional care services between facilities, individual and family support, and referral to community social support services. In these health homes, a PCP can act as a consultant to help establish priorities for disease management and improving health status.29 The PCP consultant also can support psychiatric staff and collaborate with providers who want to provide some direct care of medical conditions.30
Last, some behavioral health sites are choosing to apply for Federally Qualified Health Clinic status or add primary care services to their clinics, with the hope that sustainable funding will become available. Without additional funding to cover the limited reimbursement provided by public payers, such as Medicaid and Medicare, these models might be unsustainable. Current innovations in health care funding reform hopefully will offer solutions for sites to provide medical care in the natural “medical home” of the SMI population.
Bottom Line Psychiatric providers are in a favorable position to develop and oversee a partnership with primary care physicians with the goal of addressing significant and often lethal health disparities among those with mental illness. Psychiatric providers must use evidence-based practices that include assessment and prevention of cardiopulmonary, metabolic, infectious, and oncologic disorders. True primary care–behavioral health integration must include longitudinal “cross education” and changes in health care policy, with an emphasis on decreasing morbidity and mortality in psychiatric patients.
Disclosure The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42. 2. Parks J, Svendsen D, Singer P, et al, eds. Morbidity and mortality in people with serious mental illness. Alexandria, VA: National Association of State Mental Health Program Directors (NASMHPD) Medical Directors Council; 2006. 3. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796. 4. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trails of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19-32. 5. Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harv Rev Psychiatry. 2006;14(2):212-222. 6. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131. 7. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156. 8. Carney CP, Jones L, Woolson RF. Medical comorbidity in women and men with schizophrenia: a population-based study. J Gen Intern Med. 2006;21(11):1133-1137. 9. Bobes J, Arango C, Aranda P, et al; CLAMORS Study Collaborative Group. Cardiovascular and metabolic risk in outpatients with schizoaffective disorder treated with antipsychotics; results from the CLAMORS study. Eur Psychiatry. 2012;27(4):267-274. 10. Osborn DP, Levy G, Nazareth I, et al. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s General Practice Research Database [Erratum in: Arch Gen Psychiatry. 2007;64(6):736]. Arch Gen Psychiatry. 2007;64(2):242-249. 11. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050. 12. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601. 13. Mackin P, Bishop DR, Watkinson HM. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry. 2007;7:28. 14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22. 15. Alvarez-Jiménez M, Hetrick SE, González-Blanch C, et al. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomized controlled trials. Br J Psychiatry. 2008; 193(2):101-107. 16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medication used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology. 2010;35(7):1520-1530. 17. Newcomer JW, Weiden PJ, Buchanan RW. Switching antipsychotic medications to reduce adverse event burden in schizophrenia: establishing evidence-based practice. J Clin Psychiatry. 2013;74(11):1108-1120. 18. Lord O, Malone D, Mitchell AJ. Receipt of preventive medical care and medical screening for patients with mental illness: a comparative analysis. Gen Hosp Psychiatry. 2010;32(5):519-543. 19. Xiong GL, Iosif AM, Bermudes RA, et al. Preventive medical services use among community mental health patients with severe mental illness: the influence of gender and insurance coverage. Prim Care Companion J Clin Psychiatry. 2010;12(5). doi: 10.4088/PCC.09m00927gre. 20. Daub S. Turning toward treating the seriously mentally ill in primary care. Fam Syst Health. 2014;32(1):12-13. 21. Mitchell A, Pereira IE, Yadegarfar M, et al. Breast cancer screening in women with mental illness: comparative meta-analysis of mammography uptake. Br J Psychiatry. 2014;205(6):428-435. 22. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107. 23. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572. 24. Druss BG, Bradford DW, Rosenheck RA, et al. Mental disorders and use of cardiovascular procedures after myocardial infarction. JAMA. 2000;283(4):506-511. 25. Katon W, Unützer J, Wells K, et al. Collaborative depression care: history, evolution and ways to enhance dissemination and sustainability. Gen Hosp Psychiatry. 2010;32(5):456-464. 26. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525. 27. Katon W, Russo J, Reed SD, et al. A randomized trial of collaborative depression care in obstetrics and gynecology clinics: socioeconomic disadvantage and treatment response. Am J Psychiatry. 2015;172(1):32-40. 28. Substance Abuse and Mental Health Services Administration. Request for Applications (RFA) No. SM- 09-011. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2009. 29. Parks J. Behavioral health homes. In: Integrated care: working at the interface of primary care and behavioral health. Raney LE, ed. Arlington, VA: American Psychiatric Publishing; 2015:195. 30. Raney L. Integrated care: the evolving role of psychiatry in the era of health care reform. Psychiatr Serv. 2013;64(11):1076-1078.
References
1. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chronic Dis. 2006;3(2):A42. 2. Parks J, Svendsen D, Singer P, et al, eds. Morbidity and mortality in people with serious mental illness. Alexandria, VA: National Association of State Mental Health Program Directors (NASMHPD) Medical Directors Council; 2006. 3. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796. 4. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trails of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19-32. 5. Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harv Rev Psychiatry. 2006;14(2):212-222. 6. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry. 2007;64(10):1123-1131. 7. Roshanaei-Moghaddam B, Katon W. Premature mortality from general medical illnesses among persons with bipolar disorder: a review. Psychiatr Serv. 2009;60(2):147-156. 8. Carney CP, Jones L, Woolson RF. Medical comorbidity in women and men with schizophrenia: a population-based study. J Gen Intern Med. 2006;21(11):1133-1137. 9. Bobes J, Arango C, Aranda P, et al; CLAMORS Study Collaborative Group. Cardiovascular and metabolic risk in outpatients with schizoaffective disorder treated with antipsychotics; results from the CLAMORS study. Eur Psychiatry. 2012;27(4):267-274. 10. Osborn DP, Levy G, Nazareth I, et al. Relative risk of cardiovascular and cancer mortality in people with severe mental illness from the United Kingdom’s General Practice Research Database [Erratum in: Arch Gen Psychiatry. 2007;64(6):736]. Arch Gen Psychiatry. 2007;64(2):242-249. 11. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050. 12. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601. 13. Mackin P, Bishop DR, Watkinson HM. A prospective study of monitoring practices for metabolic disease in antipsychotic-treated community psychiatric patients. BMC Psychiatry. 2007;7:28. 14. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22. 15. Alvarez-Jiménez M, Hetrick SE, González-Blanch C, et al. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomized controlled trials. Br J Psychiatry. 2008; 193(2):101-107. 16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medication used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology. 2010;35(7):1520-1530. 17. Newcomer JW, Weiden PJ, Buchanan RW. Switching antipsychotic medications to reduce adverse event burden in schizophrenia: establishing evidence-based practice. J Clin Psychiatry. 2013;74(11):1108-1120. 18. Lord O, Malone D, Mitchell AJ. Receipt of preventive medical care and medical screening for patients with mental illness: a comparative analysis. Gen Hosp Psychiatry. 2010;32(5):519-543. 19. Xiong GL, Iosif AM, Bermudes RA, et al. Preventive medical services use among community mental health patients with severe mental illness: the influence of gender and insurance coverage. Prim Care Companion J Clin Psychiatry. 2010;12(5). doi: 10.4088/PCC.09m00927gre. 20. Daub S. Turning toward treating the seriously mentally ill in primary care. Fam Syst Health. 2014;32(1):12-13. 21. Mitchell A, Pereira IE, Yadegarfar M, et al. Breast cancer screening in women with mental illness: comparative meta-analysis of mammography uptake. Br J Psychiatry. 2014;205(6):428-435. 22. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107. 23. Druss BG, Bradford WD, Rosenheck RA, et al. Quality of medical care and excess mortality in older patients with mental disorders. Arch Gen Psychiatry. 2001;58(6):565-572. 24. Druss BG, Bradford DW, Rosenheck RA, et al. Mental disorders and use of cardiovascular procedures after myocardial infarction. JAMA. 2000;283(4):506-511. 25. Katon W, Unützer J, Wells K, et al. Collaborative depression care: history, evolution and ways to enhance dissemination and sustainability. Gen Hosp Psychiatry. 2010;32(5):456-464. 26. Archer J, Bower P, Gilbody S, et al. Collaborative care for depression and anxiety problems. Cochrane Database Syst Rev. 2012;10:CD006525. 27. Katon W, Russo J, Reed SD, et al. A randomized trial of collaborative depression care in obstetrics and gynecology clinics: socioeconomic disadvantage and treatment response. Am J Psychiatry. 2015;172(1):32-40. 28. Substance Abuse and Mental Health Services Administration. Request for Applications (RFA) No. SM- 09-011. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2009. 29. Parks J. Behavioral health homes. In: Integrated care: working at the interface of primary care and behavioral health. Raney LE, ed. Arlington, VA: American Psychiatric Publishing; 2015:195. 30. Raney L. Integrated care: the evolving role of psychiatry in the era of health care reform. Psychiatr Serv. 2013;64(11):1076-1078.
Figure 1.When the ST segment is elevated on the electrocardiogram, our first concern is whether the patient is having an ST-segment elevation myocardial infarction (STEMI). However, a number of other conditions can cause ST elevation, and to complicate matters, some of these can coexist with STEMI.
Nevertheless, careful attention to the ST-T and QRS-complex configurations often allows diagnosis of the cause of ST elevation (Figure 1, Table 1). This paper discusses the differential diagnosis of ST elevation.
MEASURED AT THE J POINT OR LATER
ST-segment deviation is usually measured at its junction with the end of the QRS complex, ie, the J point, and is referenced against the TP or PR segment.1 Some authors prefer measuring the magnitude of the ST deviation 40 to 80 msec after the J point, when all myocardial fibers are expected to have reached the same level of membrane potential and to form an isoelectric ST segment.2,3
ST-SEGMENT ELEVATION MYOCARDIAL INFARCTION
A diagnosis of STEMI that mandates emergency reperfusion requires ST elevation equaling or exceeding the following cut-points, in at least two contiguous leads (using the standardization of 1.0 mV = 10 mm)4,5:
1 mm in all standard leads other than V2 and V3
2.5 mm in leads V2 and V3 in men younger than age 40, 2 mm in leads V2 and V3 in men age 40 and older, and 1.5 mm in these leads in women
0.5 mm in the posterior chest leads V7 to V9; ST elevation is attenuated in the posterior leads because of their greater distance from the heart, explaining the lower cut-point.6
While ST elevation that falls below these cut-points may be a normal variant, any ST elevation or depression (≥ 0.5 mm) may be abnormal and may necessitate further evaluation for ischemia, particularly when the clinical setting or the ST morphology suggests ischemia or when other signs of ischemia such as T-wave abnormalities, Q waves, or reciprocal ST-segment changes are also present on the electrocardiogram.
Conversely, ST elevation that exceeds these cut-points may not represent STEMI. In an analysis of patients with chest pain manifesting ST elevation, only 15% were eventually diagnosed with STEMI.7 In addition to size, careful attention to the morphology of the ST segment and the associated features is critical (Figure 1).
Other features of STEMI
Figure 2. Diffuse ST-segment elevation with ST-segment depression in lead aVR. This initially suggests pericarditis. PR depression in leads II, aVF, V5, and V6 further suggests pericarditis. But the presence of features of pericarditis does not necessarily rule out STEMI. The five STEMI features must be ruled out. In this case, the ST-segment morphology and the abnormally wide T wave are features of STEMI. The ST elevation has an upwardly convex shape with a wide and high T wave fused with the ST segment, typical of STEMI (leads V2–V4, arrows). Also, the size of the ST elevation (ie, > 5 mm in V2–V4 and larger than the QRS complex in V4, a feature called “tombstoning”) is more consistent with STEMI than with pericarditis. In this patient, the left anterior descending artery was found to be occluded on coronary arteriography.In STEMI, the ST elevation is typically a convex or a straight oblique line, blending with a wide T wave to form a dome.8 But ST elevation may be concave in up to 40% of anterior STEMIs, especially in the early stage.3,9,10 The nonconcave morphology is highly specific but not sensitive for the diagnosis of anterior STEMI.3,8,9
Four other features characteristic of STEMI may be present (Figures 2 and 3):
Concomitant T-wave abnormalities (wide, ample, or inverted T waves)
Q waves
ST depression in the reciprocal leads. Reciprocal ST depression is seen in all inferior STEMIs and in 70% of anterior STEMIs.11,12 Diffuse ST elevation mimicking pericarditis may be seen with midvessel occlusion of a left anterior descending artery that wraps around the apex and supplies part of the inferior wall.
Figure 3. In a patient with lung cancer, sinus tachycardia is seen with diffuse ST-segment elevation, along with ST-segment depression in aVR. The QRS voltage is low, particularly when compared with the electrocardio-gram recorded a few days earlier (left lower panel). PR depression is seen in lead II. The combination of these findings may suggest pericarditis with a pericardial effusion. However, the ST-T morphology in lead V2, where the ST and T are blended to form one dome, is characteristic of STEMI (top arrow). Moreover, the ST elevation and T wave in leads V2–V4 are larger than the QRS, the QRS voltage is “shrinking” (arrowhead), and the R wave is pulled up by the ST segment (star); this is called “tombstoning.” All these features are characteristic of STEMI, wherein the R wave and the QRS complex shrink before forming a deep Q wave. In fact, an electrocardiogram recorded 1 hour later (right lower panel) shows a fully developed Q wave in lead V2 (bottom arrow).ST or T-wave amplitude may approximate or exceed the QRS amplitude in at least one lead.3,13,14 This finding is characteristic of STEMI, in which the QRS “shrinks” as the infarcted area becomes electrically neutral, whereas the ST-T segments become ample.3,13 In fact, early STEMI may be characterized by a small R wave that seems to be “pulled up” by the elevated ST segment. A small or absent R wave along with an ample, convex ST segment that fuses with the T wave and exceeds the height of the remaining R wave is called “tombstoning” (Figure 3). Tombstoning is most commonly seen with anterior infarction and implies more extensive myocardial damage and a worse prognosis than STEMI without tombstoning.15
Note that ST elevation may not be acute STEMI but an old STEMI with a chronically dysfunctional myocardium (dyskinetic or aneurysmal myocardium). In fact, an old STEMI may manifest as a chronic, persistent ST elevation along with Q waves, and T waves may be inverted or upright, but not ample.14 A history of an old MI, old electrocardiograms, if available, and quick bedside echocardiography may allow the diagnosis. In the case of an old dyskinetic infarct, echocardiography shows a thin, bright (scarred), and possibly aneurysmal myocardium, whereas in acute STEMI, the myocardium is neither thin nor scarred yet. If the patient does not report a history of MI, if the T wave is ample (> 75% the size of QRS), or if the patient presents with atypical ongoing angina, presume it is acute STEMI.
EARLY REPOLARIZATION
Early repolarization is a normal variant of ST elevation that equals or exceeds 1 mm (measured at the J point). It is highly prevalent in people under age 40 and remains prevalent in middle-aged people.
Two distinct and sometimes coexistent forms of early repolarization have been described: (1) ST elevation in the anterior leads V1 to V3,16–19 and (2) ST elevation in the lateral leads (V4 to V6, I, aVL) or inferior leads.18–22 The prevalence of the first form—ie, ST elevation of 1 mm or more in any of the leads V1 through V3—is 60% to 90% in men age 45 and younger, 20% to 40% in men over age 45, and about 10% in women of any age.16 Thus, this form of early repolarization is called “normal male pattern.”
Even early repolarization that involves the lateral or inferior leads is common, with a prevalence of about 15% in people ages 30 to 40 and about 5% to 10% in those 40 to 65.20–23 It is two to four times more prevalent in men and three times more prevalent in African Americans. It is also highly prevalent in athletes younger than 25 (about 30% to 40%).22
Figure 4. Early repolarization with ST-segment elevation is seen in the inferior leads and in the anterolateral leads V2 to V6. ST elevation is most prominent in lead V4 and lead II, with a concavely upward ST morphology and a notch at the J point (arrows and left magnified image). In half of early repolarization cases, the J point is smooth but well demarcated (right magnified image). Note the slight PR depression in leads II and V5. Slight PR depression may be seen in normal individuals and corresponds to the normal atrial repolarization.Either way, early repolarization closely resembles the ST elevation of pericarditis and has the following features (Figure 4):
The ST segment is concave upward, and the J point is well demarcated and may be notched or slurred (Figure 1).
ST elevation is usually no more than 3 mm.
ST elevation may be limited to the anterior leads or, in many instances, may extend to the inferior or lateral leads. Early repolarization is very rarely limited to the limb leads, and involvement of some precordial leads is the rule.18,19 The ST segment is depressed in lead aVR in 50% of patients.18,19
Figure 5. Early repolarization with a normal variant T-wave inversion in a 33-year-old black man. The ST segment is elevated with a notched J point in leads V2 to V5The T wave is usually ample and may be more than 10 mm tallin the precordial leads in one-third of patients,17 but as opposed to the ample T wave of STEMI, it is not broad and remains smaller than the QRS complex. The ample T wave distinguishes early repolarization from pericarditis, and explains the low ST-T ratio in lead V6. In up to 10% of young black men, the T wave has a terminal inversion in leads V3 to V5, and occasionally in V1 and V2, mimicking infarction(Figure 5).24
The QRS complex tends to have prominent precordial voltage, in sharp contrast to STEMI, in which QRS shrinking occurs.3,17,22
The early repolarization pattern may be intermittent, may vary among serial electrocardiograms, may decrease with a rise in sympathetic tone, as observed during exercise, and may increase with a rise in vagal tone.18,19,25,26 Although it is usually a benign finding, the early repolarization pattern in leads other than V1 to V3 has been associated with an increased risk of sudden death, particularly when the ST elevation is horizontal-descending rather than upsloping and, possibly, when early repolarization involves the inferior leads with a J point that is notched or elevated 2 mm or more.20,22
PERICARDITIS
Figure 6. Diffuse ST-segment elevation in most leads, with ST depression in lead aVR and an isoelectric ST segment in V1. None of the STEMI features are present: ST elevation is concave upward, no reciprocal ST depression is seen except in lead aVR; the T wave is not wide, inverted, or ample (in relation to the QRS complex); and no Q wave is seen. Furthermore, ST elevation does not exceed 5 mm; ST and T heights are smaller than QRS height; and PR depression is present (circled areas). As opposed to early repolarization, the ratio of ST to T in leads V5 and V6 exceeds 25%. This is consistent with pericarditis, and the hospital course of this patient confirmed this diagnosis.In pericarditis, ST elevation is concave upward and is widespread to more than one region without reciprocal ST depression, except for the frequent ST depression in leads aVR and V1 (64%)27; ST elevation is seldom greater than 4 to 5 mm (Figure 6).27,28 Since the subepicardial injury is diffuse in pericarditis, the axis of the ST segment follows the anatomic axis of the heart and is generally +45° in the frontal plane. Thus, ST depression is seen in leads aVR and V1; ST elevation is highest in leads II, V5, and V6 and is less in leads III and aVL, where the ST segment may occasionally be depressed.29
Transient PR depression greater than 1 mm is often seen, particularly in leads II, aVF, and V4 to V6, and represents atrial subepicardial injury. PR depression in those leads is always associated with PR elevation in lead aVR and sometimes V1. PR changes often coexist with ST changes but may be isolated and may precede ST changes.30 PR depression is characteristic of pericarditis but may be seen in early repolarization, where it is less marked than in pericarditis (< 0.8 mm) and implies early repolarization of the atrial tissue,31 and in MI, where it implies atrial infarction with atrial injury pattern.
Classically, it is said that in pericarditis, unlike in STEMI, the T wave does not invert until the ST elevation subsides. In reality, up to 40% of patients develop a notched or biphasic positive-negative T wave before full return of the ST segment to the baseline.27,32 And if T-wave inversion antedates pericarditis, concomitant ST elevation and T-wave inversion may be seen once pericarditis develops. However, the T wave inverts less deeply and less completely than in STEMI, and the corrected QT interval remains normal even when the T wave inverts.
Three criteria distinguish pericarditis from early repolarization (but not from STEMI):
PR depression greater than 1 mm
ST-segment depression in lead V1
A ratio of ST-segment height to T-wave height of at least 25% in lead V6, V5, V4, or I. This feature distinguishes pericarditis from early repolarization with a high sensitivity and specificity. In pericarditis, the T waves have normal or reduced amplitude, and the ST-T ratio is therefore high,33 whereas in early repolarization the T waves are tall, so the ST-T ratio is less than 25%.
Widespread ST elevation may be seen with both pericarditis and early repolarization. ST elevation limited to the anterior leads is more likely to be early repolarization than pericarditis.
LEFT BUNDLE BRANCH BLOCK
Figure 7. Supraventricular tachycardia with a typical left bundle branch block pattern in leads I and aVL. Concordant ST-segment elevation is seen in leads I and aVL, while concordant ST depression is seen in the inferior leads (arrows). The ST elevation in lead V2 is discordant but is disproportionately high in relation to the QRS (well above 25% of the QRS height). All these features are diagnostic of STEMI.In left bundle branch block, a deep and wide S wave is seen in leads V1 to V3 and sometimes in the inferior leads, with ST elevation and T waves that are discordant with the QRS complex—ie, directed opposite to the QRS (Figures 7–9). The ST elevation is typically concave upward.8,34 Occasionally, ST elevation may be straight or convex, mimicking the dome of STEMI. In the lateral leads, the discordant ST segment is depressed, mimicking a reciprocal ST change.
The following findings imply MI:
Figure 8. Left bundle branch block with discordant ST-segment changes. However, the T wave is wide and fused with the ST segment in a domed morphology, and the T wave is larger than the QRS in leads V4, V5, and II (arrows). This implies the diagnosis of STEMI with hyperacute T waves. This patient had an occluded left anterior descending coronary artery.ST elevation or depression that is concordant with the QRS complex. Moreover, since ST deviation is mandatory with left bundle branch block, a “normal-looking” ST segment implies ischemia.
Inverted T waves concordant with the QRS in more than one lead, or biphasic T waves in more than one lead (eg, V1 to V3). Across the precordial leads, T waves may transition from positive to negative one lead earlier or later than the QRS and ST transition. Therefore, even in the absence of ischemia, the T wave may be inverted in lead V3, in which the QRS is deeply negative and the ST is still elevated (negative T-wave concordance in one lead). Also, the T wave may be upright in leads V5, V6, and I where QRS is upright and the ST segment is depressed (positive T-wave concordance does not imply ischemia).
Figure 9. Left bundle branch block with abnormal T waves. Panels A and B show discordant ST-segment elevation in V1 to V3 but concordant T wave inversion (A) or biphasic T wave (B). This is consistent with an anterior injury pattern. Panel C shows concordant T-wave inversion in the inferior leads, consistent with inferior injury. Panel D shows a large concordant T wave in lead V6, larger than the QRS, consistent with injury.In addition to concordance, a discordant ST segment or T wave that is very large may imply ischemia. For example, a discordant ST segment or T wave that is larger than the QRS height implies ischemia. A discordant ST elevation greater than 5 mm has been suggested by Sgarbossa et al35 as a diagnostic feature of STEMI; however, this feature is seen in 10% of control patients with left bundle branch block and no STEMI, and it is thus poorly specific and also poorly sensitive, frequently missing STEMI.35–37 Smith et al36 have suggested that a discordant ST elevation of at least 25% of the S-wave depth is a far more sensitive and accurate feature but one that may still be found in up to 10% of control patients.36
LEFT VENTRICULAR HYPERTROPHY
In left ventricular hypertrophy, a deep S wave is seen in leads V1 to V3, with ST elevation and T waves that are discordant with the QRS complex. Rarely, ST elevation may be straight or convex. The following findings imply MI:
ST elevation or depression that is concordant with the QRS.
Inverted T waves that are concordant with the QRS in more than one lead, or biphasic T waves in more than one lead (eg, V1 to V3).
A discordant ST segment or a T wave that is very large may imply ischemia. In left ventricular hypertrophy, ST elevation is usually less than 2.5 mm in leads V1 to V3 and is rarely seen in the inferior leads, where it would be less than 1 mm.34 When ST elevation is seen in leads V1 to V3 in left ventricular hypertrophy, an ST magnitude of 25% or more of the total QRS voltage has a 91% specificity for STEMI.34
On another note, right ventricular hypertrophy and right bundle branch block may lead to ST-segment depression and T-wave inversion, but not to ST elevation. Thus, ST elevation occurring with right ventricular hypertrophy or right bundle branch block implies STEMI. While only left bundle branch block poses a diagnostic challenge, both types of bundle branch block, if secondary to STEMI, represent equally high-risk categories.38
PREEXCITATION
Figure 10. At first glance, it seems there is ST-segment elevation in the inferior leads II, III, and aVF, with a wide Q wave. Moreover, there is a wide and tall R wave in lead V1 suggesting an associated posterior infarction. All this is consistent with acute inferoposterior STEMI. On further analysis, however, a slur is seen on the upslope of QRS in leads V1 to V6 (arrows), and the P wave is “riding” this slur. In the inferior leads, the P wave is riding the Q wave, which is in fact a negative delta wave. Thus, this electrocardiogram represents preexcitation. The ST deviations are secondary to the preexcitation and have an orientation opposite to the delta wave.Preexcitation may be associated with negative delta waves that mimic Q waves, and with ST elevation in the leads where the negative delta waves are seen, ie, ST elevation discordant with the delta wave (Figure 10). The QRS morphology and the delta wave allow preexcitation to be distinguished from STEMI.
HYPERKALEMIA
Figure 11. There are ST-segment elevations in leads V1–V4, ST-segment depressions in the inferior leads, and peaked T waves in leads V3–V5. These T waves have a narrow base and seem to “pull” the ST segment, creating ST elevation in the anterior leads and ST depression in the inferior leads (arrows). This shape is consistent with hyperkalemia. In addition, the downsloping ST elevation seen in V1 and V2 is consistent with hyperkalemia (arrowhead). Occasionally, STEMI may have a similar ST-T shape. An rSR’ pattern is seen in V1–V2; this is consistent with STEMI but also with hyperkalemia, in which conduction blocks are common. The serum potassium level was 7.4 mmol/L (normal 3.5–5), and coronary angiography revealed normal coronary arteries.The most common finding in hyperkalemia is a peaked, narrow-based T wave that is usually, but not necessarily, tall. ST elevation may be evident in leads V1 to V3 (Figure 11). In contrast with hyperkalemia, the T wave of STEMI is typically wide.
OTHER CAUSES OF ST-SEGMENT ELEVATION
Takotsubo cardiomyopathy
Takotsubo cardiomyopathy mimics all electrocardiographic features of anteroapical STEMI. ST elevation may extend to the inferior leads but cannot be isolated in the inferior leads.39 As in apical STEMI, reciprocal ST depression is uncommon. Within 24 to 48 hours, ST elevation evolves into deep anterior T-wave inversion and a prolonged QT interval. Transient Q waves may be seen.
Myocarditis
Myocarditis may have one of two electrocardiographic patterns: a pericarditis pattern, or a typical STEMI pattern with Q waves sometimes localized to one area.40
Atrial flutter waves
Figure 12. Atrial flutter that simulates ST-segment elevation. An “F” indicates the negative flutter wave; an asterisk indicates the upslope of the flutter wave that is superimposed on the ST segment, mimicking ST elevation.Atrial flutter waves, particularly of 2:1 atrial flutter, may deform the ST segment so that it mimics an injury pattern on the electrocardiogram. Flutter waves may mimic ST elevation or ST depression (Figure 12).
Large pulmonary embolism
A large pulmonary embolism may be associated with T-wave inversion in the anterior leads or the inferior leads, or both, reflective of cor pulmonale. Less commonly, ST elevation in the anterior or inferior leads is seen. In fact, changes of both anterior and inferior ischemia should always suggest a pulmonary embolism.41,42
Brugada syndrome
Figure 13. Type 1 Brugada pattern in V1 and V1, with a downsloping ST-segment elevation that creates a pseudo-R’ wave (pseudo-right bundle branch block). The QRS does not have a right bundle branch block morphology in leads V5 and V6.Brugada syndrome is characterized by ST elevation and a right bundle branch block or pseudo-right bundle branch block pattern in at least two of the leads V1 to V3. In pseudo-right bundle branch block, the QRS adopts an rSR morphology in the anterior leads but is normal in the lateral leads. Type 1 Brugada pattern, the pattern that is most specifically associated with sudden death, is characterized by a coved, downsloping ST elevation of 2 mm or more with T-wave inversion (Figure 13).43 The Brugada pattern can be transient, triggered by fever, cocaine, or class I antiarrhythmic drugs.
Hyperkalemia, Brugada syndrome, and sometimes pulmonary embolism are characterized by an ST elevation that slopes downward (Figures 11 and 13), which contrasts with the upsloping, convex ST elevation of STEMI.
References
Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST-segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
Surawicz B, Knilans TK. Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric. 5th ed. Philadelphia, PA: WB Saunders; 2001:194–207.
Smith SW, Khalil A, Henry TD, et al. Electrocardiographic differentiation of early repolarization from subtle anterior ST-segment elevation myocardial infarction. Ann Emerg Med 2012; 60:45–56.e2.
American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
Thygesen K, Alpert JS, Jaffe AS, et al; Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction. Third universal definition of myocardial infarction. Circulation 2012; 126:2020–2035.
Matetzky S, Freimark D, Chouraqui P, et al. Significance of ST segment elevations in posterior chest leads (V7 to V9) in patients with acute inferior myocardial infarction: application for thrombolytic therapy. J Am Coll Cardiol 1998; 31:506–511.
Brady WJ, Perron AD, Martin ML, Beagle C, Aufderheide TP. Cause of ST-segment abnormality in ED chest pain patients. Am J Emerg Med 2001; 19:25–28.
Brady WJ, Syverud SA, Beagle C, et al. Electrocardiographic ST-segment elevation: the diagnosis of acute myocardial infarction by morphologic analysis of the ST segment. Acad Emerg Med 2001; 8:961–967.
Smith SW. Upwardly concave ST-segment morphology is common in acute left anterior descending coronary occlusion. J Emerg Med 2006; 31:69–77.
Kosuge M, Kimura K, Ishikawa T, et al. Value of ST-segment elevation pattern in predicting infarct size and left ventricular function at discharge in patients with reperfused acute anterior myocardial infarction. Am Heart J 1999; 137:522–527.
Birnbaum Y, Sclarovsky S, Mager A, Strasberg B, Rechavia E. ST segment depression in a VL: a sensitive marker for acute inferior myocardial infarction. Eur Heart J 1993; 14:4–7.
Engelen DJ, Gorgels AP, Cheriex EC, et al. Value of the electrocardiogram in localizing the occlusion site in the left anterior descending coronary artery in acute anterior myocardial infarction. J Am Coll Cardiol 1999; 34:389–395.
Collins MS, Carter JE, Dougherty JM, Majercik SM, Hodsden JE, Logue EE. Hyperacute T-wave criteria using computer ECG analysis. Ann Emerg Med 1990; 19:114–120.
Smith SW. T/QRS ratio best distinguishes ventricular aneurysm from anterior myocardial infarction. Am J Emerg Med 2005; 23:279–287.
Surawicz B, Parikh SR. Prevalence of male and female patterns of early ventricular repolarization in the normal ECG of males and females from childhood to old age. J Am Coll Cardiol 2002; 40:1870–1876.
Klatsky AL, Oehm R, Cooper RA, Udaltsova N, Armstrong MA. The early repolarization normal variant electrocardiogram: correlates and consequences. Am J Med 2003; 115:171–177.
Mehta M, Jain AC, Mehta A. Early repolarization. Clin Cardiol 1999; 22:59–65.
Mehta MC, Jain AC. Early repolarization on scalar electrocardiogram. Am J Med Sci 1995; 309:305–311.
Rollin A, Maury P, Bongard V, et al. Prevalence, prognosis, and identification of the malignant form of early repolarization pattern in a population-based study. Am J Cardiol 2012; 110:1302–1308.
Tikkanen JT, Anttonen O, Junttila MJ, et al. Long-term outcome associated with early repolarization on electrocardiography. N Engl J Med 2009; 361:2529–2537.
Tikkanen JT, Junttila MJ, Anttonen O, et al. Early repolarization: electrocardiographic phenotypes associated with favorable long-term outcome. Circulation 2011; 123:2666–2673.
Noseworthy PA, Tikkanen JT, Porthan K, et al. The early repolarization pattern in the general population: clinical correlates and heritability. J Am Coll Cardiol 2011; 57:2284–2289.
Wasserburger RH. Observations on the juvenile pattern of adult negro males. Am J Med 1955; 18:428–437.
Kralios FA, Martin L, Burgess MJ, Millar K. Local ventricular repolarization changes due to sympathetic nerve-branch stimulation. Am J Physiol 1975; 228:1621–1626.
Spratt KA, Borans SM, Michelson EL. Early repolarization: normalization of the electrocardiogram with exercise as a clinically useful diagnostic feature. J Invasive Cardiol 1995; 7:238–242.
Surawicz B, Lasseter KC. Electrocardiogram in pericarditis. Am J Cardiol 1970; 26:471–474.
Hull E. The electrocardiogram in pericarditis. Am J Cardiol 1961; 7:21–32.
Spodick DH. Diagnostic electrocardiographic sequences in acute pericarditis. Significance of PR segment and PR vector changes. Circulation 1973; 48:575–580.
Spodick DH. Acute pericarditis: current concepts and practice. JAMA 2003; 289:1150–1153.
Charles MA, Bensinger TA, Glasser SP. Atrial injury current in pericarditis. Arch Intern Med 1973; 131:657–662.
Noth PH, Barnes HR. Electrocardiographic changes associated with pericarditis. Arch Intern Med 1940; 65:291–320.
Ginzton LE, Laks MM. The differential diagnosis of acute pericarditis from the normal variant: new electrocardiographic criteria. Circulation 1982; 65:1004–1009.
Armstrong EJ, Kulkarni AR, Bhave PD, et al. Electrocardiographic criteria for ST-elevation myocardial infarction in patients with left ventricular hypertrophy. Am J Cardiol 2012; 110:977–983.
Sgarbossa EB, Pinski SL, Barbagelata A, et al. Electrocardiographic diagnosis of evolving acute myocardial infarction in the presence of left bundle-branch block. GUSTO-1 (Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries) Investigators. N Engl J Med 1996; 334:481–487.
Smith SW, Dodd KW, Henry TD, Dvorak DM, Pearce LA. Diagnosis of ST-elevation myocardial infarction in the presence of left bundle branch block with the ST-elevation to S-wave ratio in a modified Sgarbossa rule. Ann Emerg Med 2012; 60:766–776.
Madias JE, Sinha A, Agarwal H, Ashtiani R. ST-segment elevation in leads V1-V3 in patients with LBBB. J Electrocardiol 2001; 34:87–88.
Sgarbossa EB, Pinski SL, Topol EJ, et al. Acute myocardial infarction and complete bundle branch block at hospital admission: clinical characteristics and outcome in the thrombolytic era. GUSTO-I Investigators. Global Utilization of Streptokinase and t-PA [tissue-type plasminogen activator] for Occluded Coronary Arteries. J Am Coll Cardiol 1998; 31:105–110.
Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
Glancy DL, Mikdadi GM. Syncope in a 67-year-old man. Proc (Bayl Univ Med Cent) 2005; 18:74–75.
Wilde AA, Antzelevitch C, Borggrefe M, et al; Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002; 106:2514–2519.
Elias B. Hanna, MD Assistant Professor of Medicine, Department of Medicine, Cardiovascular Section, Louisiana State University Health Sciences Center, New Orleans
David Luke Glancy, MD Emeritus Professor of Medicine, Department of Medicine, Cardiovascular Section, Louisiana State University Health Sciences Center, New Orleans
Address: Elias B. Hanna, MD, Department of Medicine, Cardiovascular Section, Louisiana State University Health Sciences Center, 1542 Tulane Avenue, 3rd Floor, Room 323, New Orleans, LA, 70112; e-mail: ehanna@lsuhsc.edu
ST, ST segment, ST segment elevation, ST elevation myocardial infarction, STEMI, early repolarization, pericarditis, left bundle branch block, hyperkalemia, Elias Hanna, David Glancy
Elias B. Hanna, MD Assistant Professor of Medicine, Department of Medicine, Cardiovascular Section, Louisiana State University Health Sciences Center, New Orleans
David Luke Glancy, MD Emeritus Professor of Medicine, Department of Medicine, Cardiovascular Section, Louisiana State University Health Sciences Center, New Orleans
Address: Elias B. Hanna, MD, Department of Medicine, Cardiovascular Section, Louisiana State University Health Sciences Center, 1542 Tulane Avenue, 3rd Floor, Room 323, New Orleans, LA, 70112; e-mail: ehanna@lsuhsc.edu
Author and Disclosure Information
Elias B. Hanna, MD Assistant Professor of Medicine, Department of Medicine, Cardiovascular Section, Louisiana State University Health Sciences Center, New Orleans
David Luke Glancy, MD Emeritus Professor of Medicine, Department of Medicine, Cardiovascular Section, Louisiana State University Health Sciences Center, New Orleans
Address: Elias B. Hanna, MD, Department of Medicine, Cardiovascular Section, Louisiana State University Health Sciences Center, 1542 Tulane Avenue, 3rd Floor, Room 323, New Orleans, LA, 70112; e-mail: ehanna@lsuhsc.edu
Figure 1.When the ST segment is elevated on the electrocardiogram, our first concern is whether the patient is having an ST-segment elevation myocardial infarction (STEMI). However, a number of other conditions can cause ST elevation, and to complicate matters, some of these can coexist with STEMI.
Nevertheless, careful attention to the ST-T and QRS-complex configurations often allows diagnosis of the cause of ST elevation (Figure 1, Table 1). This paper discusses the differential diagnosis of ST elevation.
MEASURED AT THE J POINT OR LATER
ST-segment deviation is usually measured at its junction with the end of the QRS complex, ie, the J point, and is referenced against the TP or PR segment.1 Some authors prefer measuring the magnitude of the ST deviation 40 to 80 msec after the J point, when all myocardial fibers are expected to have reached the same level of membrane potential and to form an isoelectric ST segment.2,3
ST-SEGMENT ELEVATION MYOCARDIAL INFARCTION
A diagnosis of STEMI that mandates emergency reperfusion requires ST elevation equaling or exceeding the following cut-points, in at least two contiguous leads (using the standardization of 1.0 mV = 10 mm)4,5:
1 mm in all standard leads other than V2 and V3
2.5 mm in leads V2 and V3 in men younger than age 40, 2 mm in leads V2 and V3 in men age 40 and older, and 1.5 mm in these leads in women
0.5 mm in the posterior chest leads V7 to V9; ST elevation is attenuated in the posterior leads because of their greater distance from the heart, explaining the lower cut-point.6
While ST elevation that falls below these cut-points may be a normal variant, any ST elevation or depression (≥ 0.5 mm) may be abnormal and may necessitate further evaluation for ischemia, particularly when the clinical setting or the ST morphology suggests ischemia or when other signs of ischemia such as T-wave abnormalities, Q waves, or reciprocal ST-segment changes are also present on the electrocardiogram.
Conversely, ST elevation that exceeds these cut-points may not represent STEMI. In an analysis of patients with chest pain manifesting ST elevation, only 15% were eventually diagnosed with STEMI.7 In addition to size, careful attention to the morphology of the ST segment and the associated features is critical (Figure 1).
Other features of STEMI
Figure 2. Diffuse ST-segment elevation with ST-segment depression in lead aVR. This initially suggests pericarditis. PR depression in leads II, aVF, V5, and V6 further suggests pericarditis. But the presence of features of pericarditis does not necessarily rule out STEMI. The five STEMI features must be ruled out. In this case, the ST-segment morphology and the abnormally wide T wave are features of STEMI. The ST elevation has an upwardly convex shape with a wide and high T wave fused with the ST segment, typical of STEMI (leads V2–V4, arrows). Also, the size of the ST elevation (ie, > 5 mm in V2–V4 and larger than the QRS complex in V4, a feature called “tombstoning”) is more consistent with STEMI than with pericarditis. In this patient, the left anterior descending artery was found to be occluded on coronary arteriography.In STEMI, the ST elevation is typically a convex or a straight oblique line, blending with a wide T wave to form a dome.8 But ST elevation may be concave in up to 40% of anterior STEMIs, especially in the early stage.3,9,10 The nonconcave morphology is highly specific but not sensitive for the diagnosis of anterior STEMI.3,8,9
Four other features characteristic of STEMI may be present (Figures 2 and 3):
Concomitant T-wave abnormalities (wide, ample, or inverted T waves)
Q waves
ST depression in the reciprocal leads. Reciprocal ST depression is seen in all inferior STEMIs and in 70% of anterior STEMIs.11,12 Diffuse ST elevation mimicking pericarditis may be seen with midvessel occlusion of a left anterior descending artery that wraps around the apex and supplies part of the inferior wall.
Figure 3. In a patient with lung cancer, sinus tachycardia is seen with diffuse ST-segment elevation, along with ST-segment depression in aVR. The QRS voltage is low, particularly when compared with the electrocardio-gram recorded a few days earlier (left lower panel). PR depression is seen in lead II. The combination of these findings may suggest pericarditis with a pericardial effusion. However, the ST-T morphology in lead V2, where the ST and T are blended to form one dome, is characteristic of STEMI (top arrow). Moreover, the ST elevation and T wave in leads V2–V4 are larger than the QRS, the QRS voltage is “shrinking” (arrowhead), and the R wave is pulled up by the ST segment (star); this is called “tombstoning.” All these features are characteristic of STEMI, wherein the R wave and the QRS complex shrink before forming a deep Q wave. In fact, an electrocardiogram recorded 1 hour later (right lower panel) shows a fully developed Q wave in lead V2 (bottom arrow).ST or T-wave amplitude may approximate or exceed the QRS amplitude in at least one lead.3,13,14 This finding is characteristic of STEMI, in which the QRS “shrinks” as the infarcted area becomes electrically neutral, whereas the ST-T segments become ample.3,13 In fact, early STEMI may be characterized by a small R wave that seems to be “pulled up” by the elevated ST segment. A small or absent R wave along with an ample, convex ST segment that fuses with the T wave and exceeds the height of the remaining R wave is called “tombstoning” (Figure 3). Tombstoning is most commonly seen with anterior infarction and implies more extensive myocardial damage and a worse prognosis than STEMI without tombstoning.15
Note that ST elevation may not be acute STEMI but an old STEMI with a chronically dysfunctional myocardium (dyskinetic or aneurysmal myocardium). In fact, an old STEMI may manifest as a chronic, persistent ST elevation along with Q waves, and T waves may be inverted or upright, but not ample.14 A history of an old MI, old electrocardiograms, if available, and quick bedside echocardiography may allow the diagnosis. In the case of an old dyskinetic infarct, echocardiography shows a thin, bright (scarred), and possibly aneurysmal myocardium, whereas in acute STEMI, the myocardium is neither thin nor scarred yet. If the patient does not report a history of MI, if the T wave is ample (> 75% the size of QRS), or if the patient presents with atypical ongoing angina, presume it is acute STEMI.
EARLY REPOLARIZATION
Early repolarization is a normal variant of ST elevation that equals or exceeds 1 mm (measured at the J point). It is highly prevalent in people under age 40 and remains prevalent in middle-aged people.
Two distinct and sometimes coexistent forms of early repolarization have been described: (1) ST elevation in the anterior leads V1 to V3,16–19 and (2) ST elevation in the lateral leads (V4 to V6, I, aVL) or inferior leads.18–22 The prevalence of the first form—ie, ST elevation of 1 mm or more in any of the leads V1 through V3—is 60% to 90% in men age 45 and younger, 20% to 40% in men over age 45, and about 10% in women of any age.16 Thus, this form of early repolarization is called “normal male pattern.”
Even early repolarization that involves the lateral or inferior leads is common, with a prevalence of about 15% in people ages 30 to 40 and about 5% to 10% in those 40 to 65.20–23 It is two to four times more prevalent in men and three times more prevalent in African Americans. It is also highly prevalent in athletes younger than 25 (about 30% to 40%).22
Figure 4. Early repolarization with ST-segment elevation is seen in the inferior leads and in the anterolateral leads V2 to V6. ST elevation is most prominent in lead V4 and lead II, with a concavely upward ST morphology and a notch at the J point (arrows and left magnified image). In half of early repolarization cases, the J point is smooth but well demarcated (right magnified image). Note the slight PR depression in leads II and V5. Slight PR depression may be seen in normal individuals and corresponds to the normal atrial repolarization.Either way, early repolarization closely resembles the ST elevation of pericarditis and has the following features (Figure 4):
The ST segment is concave upward, and the J point is well demarcated and may be notched or slurred (Figure 1).
ST elevation is usually no more than 3 mm.
ST elevation may be limited to the anterior leads or, in many instances, may extend to the inferior or lateral leads. Early repolarization is very rarely limited to the limb leads, and involvement of some precordial leads is the rule.18,19 The ST segment is depressed in lead aVR in 50% of patients.18,19
Figure 5. Early repolarization with a normal variant T-wave inversion in a 33-year-old black man. The ST segment is elevated with a notched J point in leads V2 to V5The T wave is usually ample and may be more than 10 mm tallin the precordial leads in one-third of patients,17 but as opposed to the ample T wave of STEMI, it is not broad and remains smaller than the QRS complex. The ample T wave distinguishes early repolarization from pericarditis, and explains the low ST-T ratio in lead V6. In up to 10% of young black men, the T wave has a terminal inversion in leads V3 to V5, and occasionally in V1 and V2, mimicking infarction(Figure 5).24
The QRS complex tends to have prominent precordial voltage, in sharp contrast to STEMI, in which QRS shrinking occurs.3,17,22
The early repolarization pattern may be intermittent, may vary among serial electrocardiograms, may decrease with a rise in sympathetic tone, as observed during exercise, and may increase with a rise in vagal tone.18,19,25,26 Although it is usually a benign finding, the early repolarization pattern in leads other than V1 to V3 has been associated with an increased risk of sudden death, particularly when the ST elevation is horizontal-descending rather than upsloping and, possibly, when early repolarization involves the inferior leads with a J point that is notched or elevated 2 mm or more.20,22
PERICARDITIS
Figure 6. Diffuse ST-segment elevation in most leads, with ST depression in lead aVR and an isoelectric ST segment in V1. None of the STEMI features are present: ST elevation is concave upward, no reciprocal ST depression is seen except in lead aVR; the T wave is not wide, inverted, or ample (in relation to the QRS complex); and no Q wave is seen. Furthermore, ST elevation does not exceed 5 mm; ST and T heights are smaller than QRS height; and PR depression is present (circled areas). As opposed to early repolarization, the ratio of ST to T in leads V5 and V6 exceeds 25%. This is consistent with pericarditis, and the hospital course of this patient confirmed this diagnosis.In pericarditis, ST elevation is concave upward and is widespread to more than one region without reciprocal ST depression, except for the frequent ST depression in leads aVR and V1 (64%)27; ST elevation is seldom greater than 4 to 5 mm (Figure 6).27,28 Since the subepicardial injury is diffuse in pericarditis, the axis of the ST segment follows the anatomic axis of the heart and is generally +45° in the frontal plane. Thus, ST depression is seen in leads aVR and V1; ST elevation is highest in leads II, V5, and V6 and is less in leads III and aVL, where the ST segment may occasionally be depressed.29
Transient PR depression greater than 1 mm is often seen, particularly in leads II, aVF, and V4 to V6, and represents atrial subepicardial injury. PR depression in those leads is always associated with PR elevation in lead aVR and sometimes V1. PR changes often coexist with ST changes but may be isolated and may precede ST changes.30 PR depression is characteristic of pericarditis but may be seen in early repolarization, where it is less marked than in pericarditis (< 0.8 mm) and implies early repolarization of the atrial tissue,31 and in MI, where it implies atrial infarction with atrial injury pattern.
Classically, it is said that in pericarditis, unlike in STEMI, the T wave does not invert until the ST elevation subsides. In reality, up to 40% of patients develop a notched or biphasic positive-negative T wave before full return of the ST segment to the baseline.27,32 And if T-wave inversion antedates pericarditis, concomitant ST elevation and T-wave inversion may be seen once pericarditis develops. However, the T wave inverts less deeply and less completely than in STEMI, and the corrected QT interval remains normal even when the T wave inverts.
Three criteria distinguish pericarditis from early repolarization (but not from STEMI):
PR depression greater than 1 mm
ST-segment depression in lead V1
A ratio of ST-segment height to T-wave height of at least 25% in lead V6, V5, V4, or I. This feature distinguishes pericarditis from early repolarization with a high sensitivity and specificity. In pericarditis, the T waves have normal or reduced amplitude, and the ST-T ratio is therefore high,33 whereas in early repolarization the T waves are tall, so the ST-T ratio is less than 25%.
Widespread ST elevation may be seen with both pericarditis and early repolarization. ST elevation limited to the anterior leads is more likely to be early repolarization than pericarditis.
LEFT BUNDLE BRANCH BLOCK
Figure 7. Supraventricular tachycardia with a typical left bundle branch block pattern in leads I and aVL. Concordant ST-segment elevation is seen in leads I and aVL, while concordant ST depression is seen in the inferior leads (arrows). The ST elevation in lead V2 is discordant but is disproportionately high in relation to the QRS (well above 25% of the QRS height). All these features are diagnostic of STEMI.In left bundle branch block, a deep and wide S wave is seen in leads V1 to V3 and sometimes in the inferior leads, with ST elevation and T waves that are discordant with the QRS complex—ie, directed opposite to the QRS (Figures 7–9). The ST elevation is typically concave upward.8,34 Occasionally, ST elevation may be straight or convex, mimicking the dome of STEMI. In the lateral leads, the discordant ST segment is depressed, mimicking a reciprocal ST change.
The following findings imply MI:
Figure 8. Left bundle branch block with discordant ST-segment changes. However, the T wave is wide and fused with the ST segment in a domed morphology, and the T wave is larger than the QRS in leads V4, V5, and II (arrows). This implies the diagnosis of STEMI with hyperacute T waves. This patient had an occluded left anterior descending coronary artery.ST elevation or depression that is concordant with the QRS complex. Moreover, since ST deviation is mandatory with left bundle branch block, a “normal-looking” ST segment implies ischemia.
Inverted T waves concordant with the QRS in more than one lead, or biphasic T waves in more than one lead (eg, V1 to V3). Across the precordial leads, T waves may transition from positive to negative one lead earlier or later than the QRS and ST transition. Therefore, even in the absence of ischemia, the T wave may be inverted in lead V3, in which the QRS is deeply negative and the ST is still elevated (negative T-wave concordance in one lead). Also, the T wave may be upright in leads V5, V6, and I where QRS is upright and the ST segment is depressed (positive T-wave concordance does not imply ischemia).
Figure 9. Left bundle branch block with abnormal T waves. Panels A and B show discordant ST-segment elevation in V1 to V3 but concordant T wave inversion (A) or biphasic T wave (B). This is consistent with an anterior injury pattern. Panel C shows concordant T-wave inversion in the inferior leads, consistent with inferior injury. Panel D shows a large concordant T wave in lead V6, larger than the QRS, consistent with injury.In addition to concordance, a discordant ST segment or T wave that is very large may imply ischemia. For example, a discordant ST segment or T wave that is larger than the QRS height implies ischemia. A discordant ST elevation greater than 5 mm has been suggested by Sgarbossa et al35 as a diagnostic feature of STEMI; however, this feature is seen in 10% of control patients with left bundle branch block and no STEMI, and it is thus poorly specific and also poorly sensitive, frequently missing STEMI.35–37 Smith et al36 have suggested that a discordant ST elevation of at least 25% of the S-wave depth is a far more sensitive and accurate feature but one that may still be found in up to 10% of control patients.36
LEFT VENTRICULAR HYPERTROPHY
In left ventricular hypertrophy, a deep S wave is seen in leads V1 to V3, with ST elevation and T waves that are discordant with the QRS complex. Rarely, ST elevation may be straight or convex. The following findings imply MI:
ST elevation or depression that is concordant with the QRS.
Inverted T waves that are concordant with the QRS in more than one lead, or biphasic T waves in more than one lead (eg, V1 to V3).
A discordant ST segment or a T wave that is very large may imply ischemia. In left ventricular hypertrophy, ST elevation is usually less than 2.5 mm in leads V1 to V3 and is rarely seen in the inferior leads, where it would be less than 1 mm.34 When ST elevation is seen in leads V1 to V3 in left ventricular hypertrophy, an ST magnitude of 25% or more of the total QRS voltage has a 91% specificity for STEMI.34
On another note, right ventricular hypertrophy and right bundle branch block may lead to ST-segment depression and T-wave inversion, but not to ST elevation. Thus, ST elevation occurring with right ventricular hypertrophy or right bundle branch block implies STEMI. While only left bundle branch block poses a diagnostic challenge, both types of bundle branch block, if secondary to STEMI, represent equally high-risk categories.38
PREEXCITATION
Figure 10. At first glance, it seems there is ST-segment elevation in the inferior leads II, III, and aVF, with a wide Q wave. Moreover, there is a wide and tall R wave in lead V1 suggesting an associated posterior infarction. All this is consistent with acute inferoposterior STEMI. On further analysis, however, a slur is seen on the upslope of QRS in leads V1 to V6 (arrows), and the P wave is “riding” this slur. In the inferior leads, the P wave is riding the Q wave, which is in fact a negative delta wave. Thus, this electrocardiogram represents preexcitation. The ST deviations are secondary to the preexcitation and have an orientation opposite to the delta wave.Preexcitation may be associated with negative delta waves that mimic Q waves, and with ST elevation in the leads where the negative delta waves are seen, ie, ST elevation discordant with the delta wave (Figure 10). The QRS morphology and the delta wave allow preexcitation to be distinguished from STEMI.
HYPERKALEMIA
Figure 11. There are ST-segment elevations in leads V1–V4, ST-segment depressions in the inferior leads, and peaked T waves in leads V3–V5. These T waves have a narrow base and seem to “pull” the ST segment, creating ST elevation in the anterior leads and ST depression in the inferior leads (arrows). This shape is consistent with hyperkalemia. In addition, the downsloping ST elevation seen in V1 and V2 is consistent with hyperkalemia (arrowhead). Occasionally, STEMI may have a similar ST-T shape. An rSR’ pattern is seen in V1–V2; this is consistent with STEMI but also with hyperkalemia, in which conduction blocks are common. The serum potassium level was 7.4 mmol/L (normal 3.5–5), and coronary angiography revealed normal coronary arteries.The most common finding in hyperkalemia is a peaked, narrow-based T wave that is usually, but not necessarily, tall. ST elevation may be evident in leads V1 to V3 (Figure 11). In contrast with hyperkalemia, the T wave of STEMI is typically wide.
OTHER CAUSES OF ST-SEGMENT ELEVATION
Takotsubo cardiomyopathy
Takotsubo cardiomyopathy mimics all electrocardiographic features of anteroapical STEMI. ST elevation may extend to the inferior leads but cannot be isolated in the inferior leads.39 As in apical STEMI, reciprocal ST depression is uncommon. Within 24 to 48 hours, ST elevation evolves into deep anterior T-wave inversion and a prolonged QT interval. Transient Q waves may be seen.
Myocarditis
Myocarditis may have one of two electrocardiographic patterns: a pericarditis pattern, or a typical STEMI pattern with Q waves sometimes localized to one area.40
Atrial flutter waves
Figure 12. Atrial flutter that simulates ST-segment elevation. An “F” indicates the negative flutter wave; an asterisk indicates the upslope of the flutter wave that is superimposed on the ST segment, mimicking ST elevation.Atrial flutter waves, particularly of 2:1 atrial flutter, may deform the ST segment so that it mimics an injury pattern on the electrocardiogram. Flutter waves may mimic ST elevation or ST depression (Figure 12).
Large pulmonary embolism
A large pulmonary embolism may be associated with T-wave inversion in the anterior leads or the inferior leads, or both, reflective of cor pulmonale. Less commonly, ST elevation in the anterior or inferior leads is seen. In fact, changes of both anterior and inferior ischemia should always suggest a pulmonary embolism.41,42
Brugada syndrome
Figure 13. Type 1 Brugada pattern in V1 and V1, with a downsloping ST-segment elevation that creates a pseudo-R’ wave (pseudo-right bundle branch block). The QRS does not have a right bundle branch block morphology in leads V5 and V6.Brugada syndrome is characterized by ST elevation and a right bundle branch block or pseudo-right bundle branch block pattern in at least two of the leads V1 to V3. In pseudo-right bundle branch block, the QRS adopts an rSR morphology in the anterior leads but is normal in the lateral leads. Type 1 Brugada pattern, the pattern that is most specifically associated with sudden death, is characterized by a coved, downsloping ST elevation of 2 mm or more with T-wave inversion (Figure 13).43 The Brugada pattern can be transient, triggered by fever, cocaine, or class I antiarrhythmic drugs.
Hyperkalemia, Brugada syndrome, and sometimes pulmonary embolism are characterized by an ST elevation that slopes downward (Figures 11 and 13), which contrasts with the upsloping, convex ST elevation of STEMI.
Figure 1.When the ST segment is elevated on the electrocardiogram, our first concern is whether the patient is having an ST-segment elevation myocardial infarction (STEMI). However, a number of other conditions can cause ST elevation, and to complicate matters, some of these can coexist with STEMI.
Nevertheless, careful attention to the ST-T and QRS-complex configurations often allows diagnosis of the cause of ST elevation (Figure 1, Table 1). This paper discusses the differential diagnosis of ST elevation.
MEASURED AT THE J POINT OR LATER
ST-segment deviation is usually measured at its junction with the end of the QRS complex, ie, the J point, and is referenced against the TP or PR segment.1 Some authors prefer measuring the magnitude of the ST deviation 40 to 80 msec after the J point, when all myocardial fibers are expected to have reached the same level of membrane potential and to form an isoelectric ST segment.2,3
ST-SEGMENT ELEVATION MYOCARDIAL INFARCTION
A diagnosis of STEMI that mandates emergency reperfusion requires ST elevation equaling or exceeding the following cut-points, in at least two contiguous leads (using the standardization of 1.0 mV = 10 mm)4,5:
1 mm in all standard leads other than V2 and V3
2.5 mm in leads V2 and V3 in men younger than age 40, 2 mm in leads V2 and V3 in men age 40 and older, and 1.5 mm in these leads in women
0.5 mm in the posterior chest leads V7 to V9; ST elevation is attenuated in the posterior leads because of their greater distance from the heart, explaining the lower cut-point.6
While ST elevation that falls below these cut-points may be a normal variant, any ST elevation or depression (≥ 0.5 mm) may be abnormal and may necessitate further evaluation for ischemia, particularly when the clinical setting or the ST morphology suggests ischemia or when other signs of ischemia such as T-wave abnormalities, Q waves, or reciprocal ST-segment changes are also present on the electrocardiogram.
Conversely, ST elevation that exceeds these cut-points may not represent STEMI. In an analysis of patients with chest pain manifesting ST elevation, only 15% were eventually diagnosed with STEMI.7 In addition to size, careful attention to the morphology of the ST segment and the associated features is critical (Figure 1).
Other features of STEMI
Figure 2. Diffuse ST-segment elevation with ST-segment depression in lead aVR. This initially suggests pericarditis. PR depression in leads II, aVF, V5, and V6 further suggests pericarditis. But the presence of features of pericarditis does not necessarily rule out STEMI. The five STEMI features must be ruled out. In this case, the ST-segment morphology and the abnormally wide T wave are features of STEMI. The ST elevation has an upwardly convex shape with a wide and high T wave fused with the ST segment, typical of STEMI (leads V2–V4, arrows). Also, the size of the ST elevation (ie, > 5 mm in V2–V4 and larger than the QRS complex in V4, a feature called “tombstoning”) is more consistent with STEMI than with pericarditis. In this patient, the left anterior descending artery was found to be occluded on coronary arteriography.In STEMI, the ST elevation is typically a convex or a straight oblique line, blending with a wide T wave to form a dome.8 But ST elevation may be concave in up to 40% of anterior STEMIs, especially in the early stage.3,9,10 The nonconcave morphology is highly specific but not sensitive for the diagnosis of anterior STEMI.3,8,9
Four other features characteristic of STEMI may be present (Figures 2 and 3):
Concomitant T-wave abnormalities (wide, ample, or inverted T waves)
Q waves
ST depression in the reciprocal leads. Reciprocal ST depression is seen in all inferior STEMIs and in 70% of anterior STEMIs.11,12 Diffuse ST elevation mimicking pericarditis may be seen with midvessel occlusion of a left anterior descending artery that wraps around the apex and supplies part of the inferior wall.
Figure 3. In a patient with lung cancer, sinus tachycardia is seen with diffuse ST-segment elevation, along with ST-segment depression in aVR. The QRS voltage is low, particularly when compared with the electrocardio-gram recorded a few days earlier (left lower panel). PR depression is seen in lead II. The combination of these findings may suggest pericarditis with a pericardial effusion. However, the ST-T morphology in lead V2, where the ST and T are blended to form one dome, is characteristic of STEMI (top arrow). Moreover, the ST elevation and T wave in leads V2–V4 are larger than the QRS, the QRS voltage is “shrinking” (arrowhead), and the R wave is pulled up by the ST segment (star); this is called “tombstoning.” All these features are characteristic of STEMI, wherein the R wave and the QRS complex shrink before forming a deep Q wave. In fact, an electrocardiogram recorded 1 hour later (right lower panel) shows a fully developed Q wave in lead V2 (bottom arrow).ST or T-wave amplitude may approximate or exceed the QRS amplitude in at least one lead.3,13,14 This finding is characteristic of STEMI, in which the QRS “shrinks” as the infarcted area becomes electrically neutral, whereas the ST-T segments become ample.3,13 In fact, early STEMI may be characterized by a small R wave that seems to be “pulled up” by the elevated ST segment. A small or absent R wave along with an ample, convex ST segment that fuses with the T wave and exceeds the height of the remaining R wave is called “tombstoning” (Figure 3). Tombstoning is most commonly seen with anterior infarction and implies more extensive myocardial damage and a worse prognosis than STEMI without tombstoning.15
Note that ST elevation may not be acute STEMI but an old STEMI with a chronically dysfunctional myocardium (dyskinetic or aneurysmal myocardium). In fact, an old STEMI may manifest as a chronic, persistent ST elevation along with Q waves, and T waves may be inverted or upright, but not ample.14 A history of an old MI, old electrocardiograms, if available, and quick bedside echocardiography may allow the diagnosis. In the case of an old dyskinetic infarct, echocardiography shows a thin, bright (scarred), and possibly aneurysmal myocardium, whereas in acute STEMI, the myocardium is neither thin nor scarred yet. If the patient does not report a history of MI, if the T wave is ample (> 75% the size of QRS), or if the patient presents with atypical ongoing angina, presume it is acute STEMI.
EARLY REPOLARIZATION
Early repolarization is a normal variant of ST elevation that equals or exceeds 1 mm (measured at the J point). It is highly prevalent in people under age 40 and remains prevalent in middle-aged people.
Two distinct and sometimes coexistent forms of early repolarization have been described: (1) ST elevation in the anterior leads V1 to V3,16–19 and (2) ST elevation in the lateral leads (V4 to V6, I, aVL) or inferior leads.18–22 The prevalence of the first form—ie, ST elevation of 1 mm or more in any of the leads V1 through V3—is 60% to 90% in men age 45 and younger, 20% to 40% in men over age 45, and about 10% in women of any age.16 Thus, this form of early repolarization is called “normal male pattern.”
Even early repolarization that involves the lateral or inferior leads is common, with a prevalence of about 15% in people ages 30 to 40 and about 5% to 10% in those 40 to 65.20–23 It is two to four times more prevalent in men and three times more prevalent in African Americans. It is also highly prevalent in athletes younger than 25 (about 30% to 40%).22
Figure 4. Early repolarization with ST-segment elevation is seen in the inferior leads and in the anterolateral leads V2 to V6. ST elevation is most prominent in lead V4 and lead II, with a concavely upward ST morphology and a notch at the J point (arrows and left magnified image). In half of early repolarization cases, the J point is smooth but well demarcated (right magnified image). Note the slight PR depression in leads II and V5. Slight PR depression may be seen in normal individuals and corresponds to the normal atrial repolarization.Either way, early repolarization closely resembles the ST elevation of pericarditis and has the following features (Figure 4):
The ST segment is concave upward, and the J point is well demarcated and may be notched or slurred (Figure 1).
ST elevation is usually no more than 3 mm.
ST elevation may be limited to the anterior leads or, in many instances, may extend to the inferior or lateral leads. Early repolarization is very rarely limited to the limb leads, and involvement of some precordial leads is the rule.18,19 The ST segment is depressed in lead aVR in 50% of patients.18,19
Figure 5. Early repolarization with a normal variant T-wave inversion in a 33-year-old black man. The ST segment is elevated with a notched J point in leads V2 to V5The T wave is usually ample and may be more than 10 mm tallin the precordial leads in one-third of patients,17 but as opposed to the ample T wave of STEMI, it is not broad and remains smaller than the QRS complex. The ample T wave distinguishes early repolarization from pericarditis, and explains the low ST-T ratio in lead V6. In up to 10% of young black men, the T wave has a terminal inversion in leads V3 to V5, and occasionally in V1 and V2, mimicking infarction(Figure 5).24
The QRS complex tends to have prominent precordial voltage, in sharp contrast to STEMI, in which QRS shrinking occurs.3,17,22
The early repolarization pattern may be intermittent, may vary among serial electrocardiograms, may decrease with a rise in sympathetic tone, as observed during exercise, and may increase with a rise in vagal tone.18,19,25,26 Although it is usually a benign finding, the early repolarization pattern in leads other than V1 to V3 has been associated with an increased risk of sudden death, particularly when the ST elevation is horizontal-descending rather than upsloping and, possibly, when early repolarization involves the inferior leads with a J point that is notched or elevated 2 mm or more.20,22
PERICARDITIS
Figure 6. Diffuse ST-segment elevation in most leads, with ST depression in lead aVR and an isoelectric ST segment in V1. None of the STEMI features are present: ST elevation is concave upward, no reciprocal ST depression is seen except in lead aVR; the T wave is not wide, inverted, or ample (in relation to the QRS complex); and no Q wave is seen. Furthermore, ST elevation does not exceed 5 mm; ST and T heights are smaller than QRS height; and PR depression is present (circled areas). As opposed to early repolarization, the ratio of ST to T in leads V5 and V6 exceeds 25%. This is consistent with pericarditis, and the hospital course of this patient confirmed this diagnosis.In pericarditis, ST elevation is concave upward and is widespread to more than one region without reciprocal ST depression, except for the frequent ST depression in leads aVR and V1 (64%)27; ST elevation is seldom greater than 4 to 5 mm (Figure 6).27,28 Since the subepicardial injury is diffuse in pericarditis, the axis of the ST segment follows the anatomic axis of the heart and is generally +45° in the frontal plane. Thus, ST depression is seen in leads aVR and V1; ST elevation is highest in leads II, V5, and V6 and is less in leads III and aVL, where the ST segment may occasionally be depressed.29
Transient PR depression greater than 1 mm is often seen, particularly in leads II, aVF, and V4 to V6, and represents atrial subepicardial injury. PR depression in those leads is always associated with PR elevation in lead aVR and sometimes V1. PR changes often coexist with ST changes but may be isolated and may precede ST changes.30 PR depression is characteristic of pericarditis but may be seen in early repolarization, where it is less marked than in pericarditis (< 0.8 mm) and implies early repolarization of the atrial tissue,31 and in MI, where it implies atrial infarction with atrial injury pattern.
Classically, it is said that in pericarditis, unlike in STEMI, the T wave does not invert until the ST elevation subsides. In reality, up to 40% of patients develop a notched or biphasic positive-negative T wave before full return of the ST segment to the baseline.27,32 And if T-wave inversion antedates pericarditis, concomitant ST elevation and T-wave inversion may be seen once pericarditis develops. However, the T wave inverts less deeply and less completely than in STEMI, and the corrected QT interval remains normal even when the T wave inverts.
Three criteria distinguish pericarditis from early repolarization (but not from STEMI):
PR depression greater than 1 mm
ST-segment depression in lead V1
A ratio of ST-segment height to T-wave height of at least 25% in lead V6, V5, V4, or I. This feature distinguishes pericarditis from early repolarization with a high sensitivity and specificity. In pericarditis, the T waves have normal or reduced amplitude, and the ST-T ratio is therefore high,33 whereas in early repolarization the T waves are tall, so the ST-T ratio is less than 25%.
Widespread ST elevation may be seen with both pericarditis and early repolarization. ST elevation limited to the anterior leads is more likely to be early repolarization than pericarditis.
LEFT BUNDLE BRANCH BLOCK
Figure 7. Supraventricular tachycardia with a typical left bundle branch block pattern in leads I and aVL. Concordant ST-segment elevation is seen in leads I and aVL, while concordant ST depression is seen in the inferior leads (arrows). The ST elevation in lead V2 is discordant but is disproportionately high in relation to the QRS (well above 25% of the QRS height). All these features are diagnostic of STEMI.In left bundle branch block, a deep and wide S wave is seen in leads V1 to V3 and sometimes in the inferior leads, with ST elevation and T waves that are discordant with the QRS complex—ie, directed opposite to the QRS (Figures 7–9). The ST elevation is typically concave upward.8,34 Occasionally, ST elevation may be straight or convex, mimicking the dome of STEMI. In the lateral leads, the discordant ST segment is depressed, mimicking a reciprocal ST change.
The following findings imply MI:
Figure 8. Left bundle branch block with discordant ST-segment changes. However, the T wave is wide and fused with the ST segment in a domed morphology, and the T wave is larger than the QRS in leads V4, V5, and II (arrows). This implies the diagnosis of STEMI with hyperacute T waves. This patient had an occluded left anterior descending coronary artery.ST elevation or depression that is concordant with the QRS complex. Moreover, since ST deviation is mandatory with left bundle branch block, a “normal-looking” ST segment implies ischemia.
Inverted T waves concordant with the QRS in more than one lead, or biphasic T waves in more than one lead (eg, V1 to V3). Across the precordial leads, T waves may transition from positive to negative one lead earlier or later than the QRS and ST transition. Therefore, even in the absence of ischemia, the T wave may be inverted in lead V3, in which the QRS is deeply negative and the ST is still elevated (negative T-wave concordance in one lead). Also, the T wave may be upright in leads V5, V6, and I where QRS is upright and the ST segment is depressed (positive T-wave concordance does not imply ischemia).
Figure 9. Left bundle branch block with abnormal T waves. Panels A and B show discordant ST-segment elevation in V1 to V3 but concordant T wave inversion (A) or biphasic T wave (B). This is consistent with an anterior injury pattern. Panel C shows concordant T-wave inversion in the inferior leads, consistent with inferior injury. Panel D shows a large concordant T wave in lead V6, larger than the QRS, consistent with injury.In addition to concordance, a discordant ST segment or T wave that is very large may imply ischemia. For example, a discordant ST segment or T wave that is larger than the QRS height implies ischemia. A discordant ST elevation greater than 5 mm has been suggested by Sgarbossa et al35 as a diagnostic feature of STEMI; however, this feature is seen in 10% of control patients with left bundle branch block and no STEMI, and it is thus poorly specific and also poorly sensitive, frequently missing STEMI.35–37 Smith et al36 have suggested that a discordant ST elevation of at least 25% of the S-wave depth is a far more sensitive and accurate feature but one that may still be found in up to 10% of control patients.36
LEFT VENTRICULAR HYPERTROPHY
In left ventricular hypertrophy, a deep S wave is seen in leads V1 to V3, with ST elevation and T waves that are discordant with the QRS complex. Rarely, ST elevation may be straight or convex. The following findings imply MI:
ST elevation or depression that is concordant with the QRS.
Inverted T waves that are concordant with the QRS in more than one lead, or biphasic T waves in more than one lead (eg, V1 to V3).
A discordant ST segment or a T wave that is very large may imply ischemia. In left ventricular hypertrophy, ST elevation is usually less than 2.5 mm in leads V1 to V3 and is rarely seen in the inferior leads, where it would be less than 1 mm.34 When ST elevation is seen in leads V1 to V3 in left ventricular hypertrophy, an ST magnitude of 25% or more of the total QRS voltage has a 91% specificity for STEMI.34
On another note, right ventricular hypertrophy and right bundle branch block may lead to ST-segment depression and T-wave inversion, but not to ST elevation. Thus, ST elevation occurring with right ventricular hypertrophy or right bundle branch block implies STEMI. While only left bundle branch block poses a diagnostic challenge, both types of bundle branch block, if secondary to STEMI, represent equally high-risk categories.38
PREEXCITATION
Figure 10. At first glance, it seems there is ST-segment elevation in the inferior leads II, III, and aVF, with a wide Q wave. Moreover, there is a wide and tall R wave in lead V1 suggesting an associated posterior infarction. All this is consistent with acute inferoposterior STEMI. On further analysis, however, a slur is seen on the upslope of QRS in leads V1 to V6 (arrows), and the P wave is “riding” this slur. In the inferior leads, the P wave is riding the Q wave, which is in fact a negative delta wave. Thus, this electrocardiogram represents preexcitation. The ST deviations are secondary to the preexcitation and have an orientation opposite to the delta wave.Preexcitation may be associated with negative delta waves that mimic Q waves, and with ST elevation in the leads where the negative delta waves are seen, ie, ST elevation discordant with the delta wave (Figure 10). The QRS morphology and the delta wave allow preexcitation to be distinguished from STEMI.
HYPERKALEMIA
Figure 11. There are ST-segment elevations in leads V1–V4, ST-segment depressions in the inferior leads, and peaked T waves in leads V3–V5. These T waves have a narrow base and seem to “pull” the ST segment, creating ST elevation in the anterior leads and ST depression in the inferior leads (arrows). This shape is consistent with hyperkalemia. In addition, the downsloping ST elevation seen in V1 and V2 is consistent with hyperkalemia (arrowhead). Occasionally, STEMI may have a similar ST-T shape. An rSR’ pattern is seen in V1–V2; this is consistent with STEMI but also with hyperkalemia, in which conduction blocks are common. The serum potassium level was 7.4 mmol/L (normal 3.5–5), and coronary angiography revealed normal coronary arteries.The most common finding in hyperkalemia is a peaked, narrow-based T wave that is usually, but not necessarily, tall. ST elevation may be evident in leads V1 to V3 (Figure 11). In contrast with hyperkalemia, the T wave of STEMI is typically wide.
OTHER CAUSES OF ST-SEGMENT ELEVATION
Takotsubo cardiomyopathy
Takotsubo cardiomyopathy mimics all electrocardiographic features of anteroapical STEMI. ST elevation may extend to the inferior leads but cannot be isolated in the inferior leads.39 As in apical STEMI, reciprocal ST depression is uncommon. Within 24 to 48 hours, ST elevation evolves into deep anterior T-wave inversion and a prolonged QT interval. Transient Q waves may be seen.
Myocarditis
Myocarditis may have one of two electrocardiographic patterns: a pericarditis pattern, or a typical STEMI pattern with Q waves sometimes localized to one area.40
Atrial flutter waves
Figure 12. Atrial flutter that simulates ST-segment elevation. An “F” indicates the negative flutter wave; an asterisk indicates the upslope of the flutter wave that is superimposed on the ST segment, mimicking ST elevation.Atrial flutter waves, particularly of 2:1 atrial flutter, may deform the ST segment so that it mimics an injury pattern on the electrocardiogram. Flutter waves may mimic ST elevation or ST depression (Figure 12).
Large pulmonary embolism
A large pulmonary embolism may be associated with T-wave inversion in the anterior leads or the inferior leads, or both, reflective of cor pulmonale. Less commonly, ST elevation in the anterior or inferior leads is seen. In fact, changes of both anterior and inferior ischemia should always suggest a pulmonary embolism.41,42
Brugada syndrome
Figure 13. Type 1 Brugada pattern in V1 and V1, with a downsloping ST-segment elevation that creates a pseudo-R’ wave (pseudo-right bundle branch block). The QRS does not have a right bundle branch block morphology in leads V5 and V6.Brugada syndrome is characterized by ST elevation and a right bundle branch block or pseudo-right bundle branch block pattern in at least two of the leads V1 to V3. In pseudo-right bundle branch block, the QRS adopts an rSR morphology in the anterior leads but is normal in the lateral leads. Type 1 Brugada pattern, the pattern that is most specifically associated with sudden death, is characterized by a coved, downsloping ST elevation of 2 mm or more with T-wave inversion (Figure 13).43 The Brugada pattern can be transient, triggered by fever, cocaine, or class I antiarrhythmic drugs.
Hyperkalemia, Brugada syndrome, and sometimes pulmonary embolism are characterized by an ST elevation that slopes downward (Figures 11 and 13), which contrasts with the upsloping, convex ST elevation of STEMI.
References
Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST-segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
Surawicz B, Knilans TK. Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric. 5th ed. Philadelphia, PA: WB Saunders; 2001:194–207.
Smith SW, Khalil A, Henry TD, et al. Electrocardiographic differentiation of early repolarization from subtle anterior ST-segment elevation myocardial infarction. Ann Emerg Med 2012; 60:45–56.e2.
American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
Thygesen K, Alpert JS, Jaffe AS, et al; Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction. Third universal definition of myocardial infarction. Circulation 2012; 126:2020–2035.
Matetzky S, Freimark D, Chouraqui P, et al. Significance of ST segment elevations in posterior chest leads (V7 to V9) in patients with acute inferior myocardial infarction: application for thrombolytic therapy. J Am Coll Cardiol 1998; 31:506–511.
Brady WJ, Perron AD, Martin ML, Beagle C, Aufderheide TP. Cause of ST-segment abnormality in ED chest pain patients. Am J Emerg Med 2001; 19:25–28.
Brady WJ, Syverud SA, Beagle C, et al. Electrocardiographic ST-segment elevation: the diagnosis of acute myocardial infarction by morphologic analysis of the ST segment. Acad Emerg Med 2001; 8:961–967.
Smith SW. Upwardly concave ST-segment morphology is common in acute left anterior descending coronary occlusion. J Emerg Med 2006; 31:69–77.
Kosuge M, Kimura K, Ishikawa T, et al. Value of ST-segment elevation pattern in predicting infarct size and left ventricular function at discharge in patients with reperfused acute anterior myocardial infarction. Am Heart J 1999; 137:522–527.
Birnbaum Y, Sclarovsky S, Mager A, Strasberg B, Rechavia E. ST segment depression in a VL: a sensitive marker for acute inferior myocardial infarction. Eur Heart J 1993; 14:4–7.
Engelen DJ, Gorgels AP, Cheriex EC, et al. Value of the electrocardiogram in localizing the occlusion site in the left anterior descending coronary artery in acute anterior myocardial infarction. J Am Coll Cardiol 1999; 34:389–395.
Collins MS, Carter JE, Dougherty JM, Majercik SM, Hodsden JE, Logue EE. Hyperacute T-wave criteria using computer ECG analysis. Ann Emerg Med 1990; 19:114–120.
Smith SW. T/QRS ratio best distinguishes ventricular aneurysm from anterior myocardial infarction. Am J Emerg Med 2005; 23:279–287.
Surawicz B, Parikh SR. Prevalence of male and female patterns of early ventricular repolarization in the normal ECG of males and females from childhood to old age. J Am Coll Cardiol 2002; 40:1870–1876.
Klatsky AL, Oehm R, Cooper RA, Udaltsova N, Armstrong MA. The early repolarization normal variant electrocardiogram: correlates and consequences. Am J Med 2003; 115:171–177.
Mehta M, Jain AC, Mehta A. Early repolarization. Clin Cardiol 1999; 22:59–65.
Mehta MC, Jain AC. Early repolarization on scalar electrocardiogram. Am J Med Sci 1995; 309:305–311.
Rollin A, Maury P, Bongard V, et al. Prevalence, prognosis, and identification of the malignant form of early repolarization pattern in a population-based study. Am J Cardiol 2012; 110:1302–1308.
Tikkanen JT, Anttonen O, Junttila MJ, et al. Long-term outcome associated with early repolarization on electrocardiography. N Engl J Med 2009; 361:2529–2537.
Tikkanen JT, Junttila MJ, Anttonen O, et al. Early repolarization: electrocardiographic phenotypes associated with favorable long-term outcome. Circulation 2011; 123:2666–2673.
Noseworthy PA, Tikkanen JT, Porthan K, et al. The early repolarization pattern in the general population: clinical correlates and heritability. J Am Coll Cardiol 2011; 57:2284–2289.
Wasserburger RH. Observations on the juvenile pattern of adult negro males. Am J Med 1955; 18:428–437.
Kralios FA, Martin L, Burgess MJ, Millar K. Local ventricular repolarization changes due to sympathetic nerve-branch stimulation. Am J Physiol 1975; 228:1621–1626.
Spratt KA, Borans SM, Michelson EL. Early repolarization: normalization of the electrocardiogram with exercise as a clinically useful diagnostic feature. J Invasive Cardiol 1995; 7:238–242.
Surawicz B, Lasseter KC. Electrocardiogram in pericarditis. Am J Cardiol 1970; 26:471–474.
Hull E. The electrocardiogram in pericarditis. Am J Cardiol 1961; 7:21–32.
Spodick DH. Diagnostic electrocardiographic sequences in acute pericarditis. Significance of PR segment and PR vector changes. Circulation 1973; 48:575–580.
Spodick DH. Acute pericarditis: current concepts and practice. JAMA 2003; 289:1150–1153.
Charles MA, Bensinger TA, Glasser SP. Atrial injury current in pericarditis. Arch Intern Med 1973; 131:657–662.
Noth PH, Barnes HR. Electrocardiographic changes associated with pericarditis. Arch Intern Med 1940; 65:291–320.
Ginzton LE, Laks MM. The differential diagnosis of acute pericarditis from the normal variant: new electrocardiographic criteria. Circulation 1982; 65:1004–1009.
Armstrong EJ, Kulkarni AR, Bhave PD, et al. Electrocardiographic criteria for ST-elevation myocardial infarction in patients with left ventricular hypertrophy. Am J Cardiol 2012; 110:977–983.
Sgarbossa EB, Pinski SL, Barbagelata A, et al. Electrocardiographic diagnosis of evolving acute myocardial infarction in the presence of left bundle-branch block. GUSTO-1 (Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries) Investigators. N Engl J Med 1996; 334:481–487.
Smith SW, Dodd KW, Henry TD, Dvorak DM, Pearce LA. Diagnosis of ST-elevation myocardial infarction in the presence of left bundle branch block with the ST-elevation to S-wave ratio in a modified Sgarbossa rule. Ann Emerg Med 2012; 60:766–776.
Madias JE, Sinha A, Agarwal H, Ashtiani R. ST-segment elevation in leads V1-V3 in patients with LBBB. J Electrocardiol 2001; 34:87–88.
Sgarbossa EB, Pinski SL, Topol EJ, et al. Acute myocardial infarction and complete bundle branch block at hospital admission: clinical characteristics and outcome in the thrombolytic era. GUSTO-I Investigators. Global Utilization of Streptokinase and t-PA [tissue-type plasminogen activator] for Occluded Coronary Arteries. J Am Coll Cardiol 1998; 31:105–110.
Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
Glancy DL, Mikdadi GM. Syncope in a 67-year-old man. Proc (Bayl Univ Med Cent) 2005; 18:74–75.
Wilde AA, Antzelevitch C, Borggrefe M, et al; Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002; 106:2514–2519.
References
Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST-segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
Surawicz B, Knilans TK. Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric. 5th ed. Philadelphia, PA: WB Saunders; 2001:194–207.
Smith SW, Khalil A, Henry TD, et al. Electrocardiographic differentiation of early repolarization from subtle anterior ST-segment elevation myocardial infarction. Ann Emerg Med 2012; 60:45–56.e2.
American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
Thygesen K, Alpert JS, Jaffe AS, et al; Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction. Third universal definition of myocardial infarction. Circulation 2012; 126:2020–2035.
Matetzky S, Freimark D, Chouraqui P, et al. Significance of ST segment elevations in posterior chest leads (V7 to V9) in patients with acute inferior myocardial infarction: application for thrombolytic therapy. J Am Coll Cardiol 1998; 31:506–511.
Brady WJ, Perron AD, Martin ML, Beagle C, Aufderheide TP. Cause of ST-segment abnormality in ED chest pain patients. Am J Emerg Med 2001; 19:25–28.
Brady WJ, Syverud SA, Beagle C, et al. Electrocardiographic ST-segment elevation: the diagnosis of acute myocardial infarction by morphologic analysis of the ST segment. Acad Emerg Med 2001; 8:961–967.
Smith SW. Upwardly concave ST-segment morphology is common in acute left anterior descending coronary occlusion. J Emerg Med 2006; 31:69–77.
Kosuge M, Kimura K, Ishikawa T, et al. Value of ST-segment elevation pattern in predicting infarct size and left ventricular function at discharge in patients with reperfused acute anterior myocardial infarction. Am Heart J 1999; 137:522–527.
Birnbaum Y, Sclarovsky S, Mager A, Strasberg B, Rechavia E. ST segment depression in a VL: a sensitive marker for acute inferior myocardial infarction. Eur Heart J 1993; 14:4–7.
Engelen DJ, Gorgels AP, Cheriex EC, et al. Value of the electrocardiogram in localizing the occlusion site in the left anterior descending coronary artery in acute anterior myocardial infarction. J Am Coll Cardiol 1999; 34:389–395.
Collins MS, Carter JE, Dougherty JM, Majercik SM, Hodsden JE, Logue EE. Hyperacute T-wave criteria using computer ECG analysis. Ann Emerg Med 1990; 19:114–120.
Smith SW. T/QRS ratio best distinguishes ventricular aneurysm from anterior myocardial infarction. Am J Emerg Med 2005; 23:279–287.
Surawicz B, Parikh SR. Prevalence of male and female patterns of early ventricular repolarization in the normal ECG of males and females from childhood to old age. J Am Coll Cardiol 2002; 40:1870–1876.
Klatsky AL, Oehm R, Cooper RA, Udaltsova N, Armstrong MA. The early repolarization normal variant electrocardiogram: correlates and consequences. Am J Med 2003; 115:171–177.
Mehta M, Jain AC, Mehta A. Early repolarization. Clin Cardiol 1999; 22:59–65.
Mehta MC, Jain AC. Early repolarization on scalar electrocardiogram. Am J Med Sci 1995; 309:305–311.
Rollin A, Maury P, Bongard V, et al. Prevalence, prognosis, and identification of the malignant form of early repolarization pattern in a population-based study. Am J Cardiol 2012; 110:1302–1308.
Tikkanen JT, Anttonen O, Junttila MJ, et al. Long-term outcome associated with early repolarization on electrocardiography. N Engl J Med 2009; 361:2529–2537.
Tikkanen JT, Junttila MJ, Anttonen O, et al. Early repolarization: electrocardiographic phenotypes associated with favorable long-term outcome. Circulation 2011; 123:2666–2673.
Noseworthy PA, Tikkanen JT, Porthan K, et al. The early repolarization pattern in the general population: clinical correlates and heritability. J Am Coll Cardiol 2011; 57:2284–2289.
Wasserburger RH. Observations on the juvenile pattern of adult negro males. Am J Med 1955; 18:428–437.
Kralios FA, Martin L, Burgess MJ, Millar K. Local ventricular repolarization changes due to sympathetic nerve-branch stimulation. Am J Physiol 1975; 228:1621–1626.
Spratt KA, Borans SM, Michelson EL. Early repolarization: normalization of the electrocardiogram with exercise as a clinically useful diagnostic feature. J Invasive Cardiol 1995; 7:238–242.
Surawicz B, Lasseter KC. Electrocardiogram in pericarditis. Am J Cardiol 1970; 26:471–474.
Hull E. The electrocardiogram in pericarditis. Am J Cardiol 1961; 7:21–32.
Spodick DH. Diagnostic electrocardiographic sequences in acute pericarditis. Significance of PR segment and PR vector changes. Circulation 1973; 48:575–580.
Spodick DH. Acute pericarditis: current concepts and practice. JAMA 2003; 289:1150–1153.
Charles MA, Bensinger TA, Glasser SP. Atrial injury current in pericarditis. Arch Intern Med 1973; 131:657–662.
Noth PH, Barnes HR. Electrocardiographic changes associated with pericarditis. Arch Intern Med 1940; 65:291–320.
Ginzton LE, Laks MM. The differential diagnosis of acute pericarditis from the normal variant: new electrocardiographic criteria. Circulation 1982; 65:1004–1009.
Armstrong EJ, Kulkarni AR, Bhave PD, et al. Electrocardiographic criteria for ST-elevation myocardial infarction in patients with left ventricular hypertrophy. Am J Cardiol 2012; 110:977–983.
Sgarbossa EB, Pinski SL, Barbagelata A, et al. Electrocardiographic diagnosis of evolving acute myocardial infarction in the presence of left bundle-branch block. GUSTO-1 (Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries) Investigators. N Engl J Med 1996; 334:481–487.
Smith SW, Dodd KW, Henry TD, Dvorak DM, Pearce LA. Diagnosis of ST-elevation myocardial infarction in the presence of left bundle branch block with the ST-elevation to S-wave ratio in a modified Sgarbossa rule. Ann Emerg Med 2012; 60:766–776.
Madias JE, Sinha A, Agarwal H, Ashtiani R. ST-segment elevation in leads V1-V3 in patients with LBBB. J Electrocardiol 2001; 34:87–88.
Sgarbossa EB, Pinski SL, Topol EJ, et al. Acute myocardial infarction and complete bundle branch block at hospital admission: clinical characteristics and outcome in the thrombolytic era. GUSTO-I Investigators. Global Utilization of Streptokinase and t-PA [tissue-type plasminogen activator] for Occluded Coronary Arteries. J Am Coll Cardiol 1998; 31:105–110.
Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
Glancy DL, Mikdadi GM. Syncope in a 67-year-old man. Proc (Bayl Univ Med Cent) 2005; 18:74–75.
Wilde AA, Antzelevitch C, Borggrefe M, et al; Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002; 106:2514–2519.
ST, ST segment, ST segment elevation, ST elevation myocardial infarction, STEMI, early repolarization, pericarditis, left bundle branch block, hyperkalemia, Elias Hanna, David Glancy
Legacy Keywords
ST, ST segment, ST segment elevation, ST elevation myocardial infarction, STEMI, early repolarization, pericarditis, left bundle branch block, hyperkalemia, Elias Hanna, David Glancy
Features of STEMI: (1) ST elevation that is straight or convex upward and blends with T to form a dome; (2) wide upright T or inverted T waves; (3) Q waves; (4) ST elevation or T waves that may approximate or exceed QRS height; and (5) reciprocal ST depression.
Features of early repolarization include a notched J point and ST elevation not exceeding 3 mm.
Features of pericarditis include PR depression greater than 1 mm and ST elevation less than 5 mm.
Features of left bundle branch block, left ventricular hypertrophy, and preexcitation: both ST and T are discordant to QRS; ST elevation is less than 25% of QRS height (and less than 2.5 mm in left ventricular hypertrophy); and delta waves, short PR, and pseudo-Q waves are seen in preexcitation.
Features of hyperkalemia include narrow-based, peaked T waves “pulling” the ST segment.
Disallow All Ads
Alternative CME
Consolidated Pubs: Do Not Show Source Publication Logo
Since 1963, when Starzl et al performed the first successful liver transplantation,1 outcomes of this life-saving procedure have continued to improve. Long-term survival rates have increased markedly: the current 5-year rate is 73.8% and the 10-year rate is 60%.2
This success means that internists will be caring for a greater number of liver transplant recipients and managing their long-term problems, such as hypertension, diabetes mellitus, dyslipidemia, obesity, metabolic syndrome, cardiovascular disease, renal insufficiency, osteoporosis, cancer, and gout.
This review will discuss these complications, focusing on the role the primary care physician assumes beyond the first year after transplantation.
ROLE OF THE PRIMARY CARE PHYSICIAN
Hepatologists, primary care physicians, and surgeons share the care of transplant recipients. The first several weeks after transplantation require close follow-up by the hepatologist and transplantation team, with particular attention paid to the patient’s overall health and well-being, medication compliance, and biochemical and immunosuppression monitoring.
After the first year, the primary care physician assumes a greater role, becoming the main provider of the patient’s care.3,4 Good communication between the transplant center and the primary care physician should lead to a smooth transition.4 Although the hepatologist continues to manage immunosuppressive drugs, allograft rejections, and biliary complications, the primary care physician manages most of the long-term complications and thus needs to be aware of the common ones and feel comfortable managing them. Aims during visits are to screen for and detect common complications and manage them appropriately, in addition to performing annual physical examinations and routine health care. A reasonable interval for liver transplant recipients to visit their primary care physician is every 6 months.
IMMUNOSUPPRESSANT MEDICATIONS
Multiple agents are used for immunosuppression after liver transplantation:
Calcineurin inhibitors (cyclosporine and tacrolimus)
Antimetabolites (mycophenolate mofetil, azathioprine, and mycophenolate sodium)
Mammalian target of rapamycin (mTOR) inhibitors (sirolimus and everolimus)
Corticosteroids.
Table 1 lists their common side effects.
Most centers use a combination of two to four immunosuppressants as induction therapy in the immediate posttransplant period, then taper the doses and eliminate all but a calcineurin inhibitor and an antimetabolite. For example, some start with a combination of tacrolimus, mycophenolate mofetil, and a corticosteroid. The choice in the immediate posttransplant period is frequently made by the transplant center in cooperation with the hepatologist. By the time primary care physicians see these patients, they usually are on a calcineurin inhibitor alone or a calcineurin inhibitor plus mycophenolate mofetil.
Calcineurin inhibitors
Cyclosporine is metabolized by the cytochrome CYP3A4 pathway. With an average half-life of 15 hours, it is given orally, usually every 12 hours.
The dosage is adjusted according to the trough level. Higher levels are needed in the initial posttransplant period to prevent graft rejection, whereas lower levels are preferred later to decrease the occurrence and severity of adverse effects. Typical long-term trough levels are 50 to 100 ng/mL. Levels should be checked more often if an acute illness develops or the patient starts taking a potentially interfering drug.
Of importance: the dosage should be based on trough levels and not on random levels. Levels are often falsely high if blood samples are not drawn at the trough level. Repeating the measurement and making sure the sample is drawn at the trough level,ie, 12 hours after the last dose, is advised in this condition.
Cyclosporine causes widespread vasoconstriction resulting in decreased renal blood flow and systemic hypertension, often within a few days of starting it. Other important adverse effects include renal insufficiency, dyslipidemia, neurotoxicity (headache, tremor, seizure), and diabetes.
Tacrolimus is superior to cyclosporine in terms of survival, graft loss, acute rejection, and steroid-resistant rejection in the first year.5 Currently, it is the agent used most often for maintenance immunosuppression after liver transplantation.
Like cyclosporine, tacrolimus is metabolized in the liver by CYP3A4. Satisfactory troughlevels after 1 year are 4 to 6 ng/mL.
The adverse effects of tacrolimus are similar to those of cyclosporine, but diabetes mellitus is more common with tacrolimus. Bone marrow suppression may occur more often with tacrolimus as well.
Antimetabolites
Antimetabolites are generally not potent enough to be used alone.
Mycophenolate mofetil causes adverse effects that include bone marrow suppression and gastrointestinal symptoms such as gastritis, diarrhea, and abdominal pain.
Azathioprine, infrequently used in transplantation in the United States, is nevertheless sometimes substituted for mycophenolate mofetil in pregnant women, as it seems safer for use in pregnancy.
Serum levels of azathioprine and mycophenolate mofetil are not routinely monitored.
mTOR inhibitors
Sirolimus and everolimus are mTOR inhibitors, inhibiting proliferation of lymphocytes.6,7
Unlike calcineurin inhibitors, mTOR inhibitors are not associated with nephrotoxicity, neurotoxicity, renal dysfunction, hypertension, or diabetes. Sirolimus is considered an alternative to calcineurin inhibitors or, in some instances, used as add-on-therapy to lower the dose of the calcineurin inhibitor.
Sirolimus carries a black-box warning about hepatic artery thrombosis
However, sirolimus carries a potential risk of hepatic artery thrombosis, a life-threatening complication.8 This has led the US Food and Drug Administration (FDA) to require sirolimus to carry a black-box warning, and most transplant centers avoid using it in the first 30 days after transplantation.
Dyslipidemia is perhaps the most common adverse effect of sirolimus. Others include dose-related cytopenia and wound dehiscence.9
Everolimus has yet to be established for use in liver transplantation, although safety trials have been published.10,11 The FDA currently recommends against using it in the first 30 days after liver transplantation.
Both sirolimus and everolimus are metabolized by CYP3A4, which is the same metabolic pathway used by cyclosporine and tacrolimus. Hence, drugs that inhibit CYP3A4 may significantly impair clearance of both sirolimus and everolimus.
Corticosteroids
Corticosteroids have been the cornerstone of immunosuppression and remain the first line of treatment for acute allograft rejection. High intravenous doses of corticosteroids are usually started in the peritransplant period and are then switched to oral doses, which are tapered and continued with a fixed dose such as 20 mg of prednisone daily for 3 to 6 months after transplantation. However, some transplant centers keep patients on prednisone 5 mg/day indefinitely.
Adverse effects of corticosteroids include diabetes, salt and fluid retention, hypertension, hyperlipidemia, cosmetic changes (acne, cervical fat pad or “buffalo hump”), delayed wound healing, susceptibility to infection, cataracts, osteopenia, and potential adrenal suppression.12 There is concern that the use of these drugs may increase hepatitis C virus replication in patients who received a liver transplant for hepatitis C cirrhosis. Randomized trials have yielded conflicting results.13–15
Drug interactions
Certain drugs can affect the metabolism of calcineurin inhibitors and mTOR inhibitors by inducing CYP3A4, which results in decreasing the levels of the immunosuppressive drugs, or by inhibiting CYP3A4, which has the opposite effect.
Medications that can decrease the levels of calcineurin inhibitors and mTOR inhibitors:
Selected antibiotics are generally well tolerated, such as penicillins, cephalosporins, quinolones, sulfonamides, and topical antifungal agents.
LONG-TERM COMPLICATIONS
Figure 1.
Figure 1 summarizes the common long-term complications of liver transplantation.
Hypertension
The prevalence of hypertension after liver transplantation is 40% to 85%, which is markedly higher than in patients with chronic liver disease before liver transplantation.17,18
One of the factors contributing to this increase is the use of immunosuppressive medications. Of these drugs, cyclosporine seems to be the one that most often causes an increase in both the incidence and the severity of hypertension, as it produces widespread vasoconstriction.19 Corticosteroids cause hypertension through their mineralocorticoid effects.
The diagnostic cutoffs for hypertension (ie, 140/90 mm Hg) and the treatment goals in posttransplant patients are similar to those in the general population. However, at our institution we target a blood pressure of less than 130/80 mm Hg in transplant patients because they have a high prevalence of other cardiovascular risk factors such as diabetes, obesity, and renal insufficiency.20
Dihydropyridine calcium channel blockers such as amlodipine and nifedipine are considered the best first-line agents because they dilate renal afferent arterioles, an effect that may counteract the vasoconstriction mediated by calcineurin inhibitors. Nondihydropyridine calcium channel blockers such as diltiazem and verapamil tend to have more marked negative inotropic effects and are not recommended in liver transplant recipients because they increase the levels of calcineurin inhibitors.21
Diuretics (eg, furosemide) might be the second-line agents, especially in patients with peripheral edema.16 One should be vigilant for hyperuricemia if thiazide agents are used.
Angiotensin-converting-enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are typically avoided in the early posttransplant period, but they can be started later and have additional benefits in patients with diabetes and congestive heart failure. Starting ACE inhibitors is acceptable in these patients unless there is a contraindication such as allergy to ACE inhibitors, hypotension, history of bilateral renal artery stenosis, significant hyperkalemia, or acute kidney injury. Monitor the serum potassium level closely for hyperkalemia in patients concurrently using calcineurin inhibitors.
Alpha-blockers and beta-blockers can be used as add-on therapy in patients with uncontrolled hypertension with the exception of carvedilol, because it increases the levels of calcineurin inhibitors.22
Blood pressure monitoring by the primary care physician is recommended every 6 months after the early posttransplant period, or more frequently when changes in treatment are being considered.
If hypertension continues to be inadequately controlled despite treatment, changing the immunosuppressive drugs or decreasing the doses can be considered, but the transplant hepatologist must be involved in this decision.23,24
Diabetes mellitus
The prevalence of diabetes mellitus is higher in liver transplant recipients than in the general population, reaching 30% to 40%.17,25 In addition to preexisting diabetes, 15% of liver transplant recipients develop new-onset diabetes.26,27
Risk factors for developing diabetes after liver transplantation include African American or Hispanic ethnicity, obesity, family history, pretransplant diabetes, hepatitis C virus infection, use of corticosteroids, and use of calcineurin inhibitors (tacrolimus more than cyclosporine) and sirolimus.26
In addition to increasing the risk of cardiovascular disease and other diseases, diabetes decreases both patient and graft survival after liver transplantation.28
The management of diabetes and the treatment target after transplantation should follow the American Diabetes Association guidelines for the treatment of type 2 diabetes mellitus.29 Lifestyle modifications, diet, and exercise are as important for transplant patients as for the nontransplant population. Insulin therapy is usually needed in the early posttransplant period to control blood glucose levels well, especially with the high doses of corticosteroids used during the first few weeks. No trials to date have compared oral agents in posttransplant patients. Therefore, the choice of oral hypoglycemic agents should be individualized on the basis of the patient’s characteristics and comorbidities.
Screen all liver transplant recipients for diabetes regardless of their pretransplant status
We recommend that primary care providers screen all liver transplant recipients for diabetes regardless of their pretransplant status. This can be done by obtaining regular fasting blood glucose levels or a hemoglobin A1c level every 6 months. Additionally, liver transplant recipients diagnosed with diabetes require annual eye examinations to look for cataracts and diabetes-related changes.30
Dyslipidemia
On November 12, 2013, the American College of Cardiology and the American Heart Association (ACC/AHA) released new clinical practice guidelines for treating blood cholesterol levels.31,32 According to these new guidelines, there are four groups of patients for whom treatment with statins is clearly indicated:
Patients with cardiovascular disease
Patients with low-density lipoprotein cholesterol (LDL-C) levels ≥ 190 mg/dL
Patients 40 to 75 years old with type 2 diabetes
Patients 40 to 75 years old with an estimated 10-year risk of cardiovascular disease of 7.5% or greater.
Liver transplant recipients should be evaluated on an individual basis to see if they fit in any of the four groups and if statin treatment therefore needs to be initiated.
A few things need to be kept in mind. First, the incidence of dyslipidemia after liver transplantation is estimated to be 45% to 69%. Risk factors include obesity, diabetes mellitus, cholestatic liver disease, and immunosuppressant medications.33 Sirolimus has a significant and well-documented association with dyslipidemia. Cyclosporine and corticosteroids are also strongly associated with dyslipidemia. Tacrolimus has a minor effect, and mycophenolate mofetil and azathioprine have no significant effects on serum lipid levels.16
Second, of the seven currently marketed statins, pravastatin and fluvastatin are preferred in liver transplant recipients because they are not metabolized by the same cytochrome CYP3A4 pathway that metabolizes calcineurin inhibitors and sirolimus.34 The doses of 40 to 80 mg daily of pravastatin or 40 mg twice daily of fluvastatin lower low-density lipoprotein cholesterol (LDL-C) levels by approximately 30% to 35%. However, these two agents are considered “moderate-intensity” statins according to the new ACC/AHA guidelines. The only two “high-intensity” statins are atorvastatin (40–80 mg) and rosuvastatin (20–40 mg), but they are both metabolized by CYP3A4. Therefore it is prudent to avoid them with the concurrent use of a calcineurin inhibitor or tacrolimus.
Gemfibrozil does not lower LDL-C and should not be used concomitantly with statins due to unacceptable risk of rhabdomyolysis and myopathy. Fenofibrates are usually avoided due to potential nephrotoxicity in patients receiving cyclosporine. Bile acid sequestrants (cholestyramine, colestipol, colesevelam) can decrease plasma mycophenolate mofetil levels by 35%.16,35 Thus, these agents should be avoided if mycophenolate mofetil is used.
It is reasonable to screen all liver transplant recipients with a fasting lipid profile at 3, 6, and 12 months after transplantation and annually thereafter. Creatine kinase should be measured if the patient complains of severe muscle pain or weakness but not on a routine basis.
Obesity
Approximately one-third of patients who are of normal weight at the time of transplantation will become obese afterward.18,25 Corticosteroid use is an important risk factor for posttransplant obesity, and tapering these drugs helps reduce weight.36 Patients treated with cyclosporine are more likely to gain weight than those who receive tacrolimus.37
Of importance: nonalcoholic fatty liver disease, currently the most common cause of chronic liver disease in adults, is rapidly increasing as an indication for liver transplantation. In fact, the proportion of liver transplantation procedures for nonalcoholic steatohepatitis-related cirrhosis increased from 1.2% in 2001 to 9.7% in 2009, and nonalcoholic steatohepatitis is expected to become the leading indication for liver transplantation in the next 20 years. And because nonalcoholic fatty liver disease is directly linked to obesity, the prevalence of obesity as a complication of liver transplantation will most likely increase in the near future.
Overweight liver transplant recipients may have great difficulty losing weight. Treatment starts with patient education on caloric restriction and exercise. If traditional measures fail to result in adequate weight loss, additional options include switching from cyclosporine to tacrolimus.23
Bariatric surgery may become an option for posttransplant patients. In a recent case series from the University of Minnesota, Al-Nowaylati et al38 reported their experience with seven patients who underwent orthotopic liver transplantation and then open Roux-en-Y gastric bypass. After bariatric surgery, the patients’ mean body mass index declined significantly, and glycemic control and high-density lipoprotein cholesterol (HDL-C) levels improved. However, one patient died of multiple organ failure, to which the bariatric surgery might have contributed.38
Heimbach et al39 conducted a study in patients referred for liver transplantation for whom a rigorous noninvasive weight-loss program before transplantation had failed. The researchers performed combined liver transplantation and sleeve gastrectomy in seven carefully selected patients who had failed to achieve weight loss to a body mass index less than 35 kg/m2 before transplantation. All seven patients lost weight, decreasing their mean body mass index from 49 kg/m2 before the procedure to 29 kg/m2 at last follow-up, and none of them developed posttransplant diabetes or steatosis.
At this time, there is not enough evidence to recommend concurrent orthotopic liver transplantation plus bariatric surgery, or combined orthotopic liver transplantation and sleeve gastrectomy. More study is needed to further evaluate these advanced approaches.
Posttransplant metabolic syndrome
Metabolic syndrome is common after liver transplantation and is strongly associated with increased morbidity in this patient population.40,41 The general definition of metabolic syndrome includes a combination of at least three of the following: hypertension, insulin resistance, hypertriglyceridemia, low HDL-C, and obesity.
The prevalence of metabolic syndrome is higher in patients after liver transplantation than in nontransplant patients. In a review of 252 liver transplant recipients, 52% were diagnosed with posttransplant metabolic syndrome, but only 5% had had it pretransplant.42
Careful screening for posttransplant metabolic syndrome and early recognition of risk factors are important. Nevertheless, the treatment of this condition depends on treating its components according to recommended guidelines.41
Cardiovascular disease
The incidence of cardiovascular morbidity and death is increased after liver transplantation.24 In addition, after liver transplantation, cardiovascular disease is a major cause of death unrelated to liver disease. It accounts for 12% to 16% of deaths and is the third most common cause of late mortality after liver transplantation.43 Of note, a recent study by our group demonstrated that patients undergoing liver transplantation for nonalcoholic steatohepatitis had a significantly higher risk of a cardiovascular event during the 3 years after transplantation than patients undergoing liver transplantation for cholestatic liver disease.44
Risk factors for cardiovascular disease after liver transplantation include older age at transplantation, male sex, posttransplant diabetes, posttransplant hypertension, and the use of mycophenolate mofetil.44 Modifying the risk factors is essential in decreasing the risk of cardiovascular events.
It is reasonable to perform dobutamine stress testing every 3 to 5 years in patients with multiple risk factors for cardiovascular disease, or more frequently in those with preexisting coronary artery disease.45,46
Malignancy
The risk of several malignancies increases after liver transplantation. Liver transplant recipients have an incidence of cancer 2.1 to 4.3 times greater than age- and sex-matched controls.24,47–49
Skin cancers are the most common and account for almost 40% of malignancies in organ transplant recipients.50 Whereas basal cell carcinoma is more common in the general population, squamous cell carcinoma is equally common in liver transplant recipients.
Multiple clinical studies have linked calcineurin inhibitors and azathioprine to the development of skin cancer. Annual skin examinations in addition to avoiding other risk factors such as smoking and sun exposure are generally recommended. Changing the immunosuppressants to sirolimus in high-risk patients may lower their chance of developing skin cancer.51,52
Patients with ulcerative colitis who undergo liver transplantation because of sclerosing cholangitis are at higher risk of colon cancer and require annual colonoscopy with surveillance biopsies. Patients who undergo transplantation for alcoholic liver disease seem to have a higher risk of pulmonary and oropharyngeal cancers.53,54
It is important that transplant patients adhere to recommended cancer screening guidelines, in view of their increased risk. Studies have shown improved overall survival in liver transplant recipients who underwent intensive cancer surveillance.55
Renal insufficiency
Renal insufficiency is a well-recognized complication of liver transplantation and is associated with an increased long-term death rate.56,57
The incidence of renal insufficiency increases dramatically over time. Ojo et al,57 in a study of almost 37,000 liver transplant recipients, found that the incidence of chronic kidney disease (defined as an estimated glomerular filtration rate < 30 mL/min/1.73 m2) was 13.9% at 3 years, 18% at 5 years, and approximately 26% at 10 years.
Risk factors include the use of calcineurin inhibitors (both cyclosporine and tacrolimus), older age, female sex, lower pretransplant glomerular filtration rate, postoperative acute renal failure, diabetes, hypertension, hepatitis C virus infection, and transplantation before 1998.58,59 Replacing a calcineurin inhibitor with mycophenolate mofetil or sirolimus may be considered with communication with the transplant center, as mycophenolate mofetil or sirolimus are associated with a lower risk of renal injury.60–64
All liver transplant recipients should avoid nonsteroidal anti-inflammatory drugs and nephrotoxic medications
Starting 1 year after liver transplantation, primary care providers should screen for renal dysfunction by obtaining kidney function tests every 6 months, including urinalysis and microalbuminuria assessment. Equations for estimating the glomerular filtration rate used in practice, such as the Modification of Diet in Renal Disease Study equation, rely mainly on serum creatinine, which may lead to overestimating renal function in some circumstances. Therefore, other equations can be used to confirm the estimated glomerular filtration rate measured by creatinine clearance, and to more accurately evaluate kidney function. Calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.
All liver transplant recipients should avoid nonsteroidal anti-inflammatory drugs and nephrotoxic medications, and should have their hypertension and diabetes adequately controlled.
Bone diseases
Osteopenia is another major complication of liver transplantation. One-third of liver transplant recipients have a bone mineral density below the fracture threshold.65
Multiple factors contribute to increased bone loss after transplantation, including use of corticosteroids, use of calcineurin inhibitors (cyclosporine, tacrolimus), poor nutrition, vitamin D deficiency, immobility, sarcopenia (reduced muscle mass), hypogonadism, smoking, and alcohol abuse.66 Even at low doses of less than 7.5 mg per day, corticosteroids inhibit osteoblast activity and increase bone resorption.
Studies have reported rapid bone loss at around 6 months after transplantation.67–69 However, long-term follow-up of bone mineral density up to 15 years after transplantation revealed an improvement mainly in the 2nd postoperative year, with no deterioration afterward.65
High-risk patients need to be identified early with appropriate screening and evaluation. Evaluation includes dual-energy x-ray absorptiometry and serum levels of calcium, phosphorous, parathyroid hormone, testosterone (men), estradiol (women), and alpha-25-hydroxyvitamin D. These tests are typically done before transplantation and then every other year afterward.
We recommend a daily dose of calcitriol and a calcium supplement to all our liver transplant patients.70 If osteoporosis (a T-score 2.5 or more standard deviations below the mean) or a fragility fracture occurs, then the patient may benefit from an oral bisphosphonate. Calcitonin has also been shown to improve bone mineral density in patients with osteoporosis after liver transplantation.71
Hyperuricemia and gout
Although hyperuricemia is common in liver transplant recipients (reported in approximately 47%), the development of clinical gout is less common (6%).72 Asymptomatic hyperuricemia requires observation only and is not usually treated in liver transplant recipients.
Acute attacks of gout are typically managed with colchicine 0.6 mg every 2 hours, up to five doses. Prednisone can be considered if symptoms persist despite treatment with colchicine. Allopurinol in an initial dose of 100 mg daily is used as maintenance therapy to reduce production of uric acid.73 However, because of the potential for drug interactions, the combination of azathioprine and allopurinol should be avoided.
Psychiatric complications and quality of life
Depression is common in liver transplant recipients, significantly more so in patients who received a transplant because of hepatitis C.74 The type of immunosuppressant is not associated with the incidence of depression. When indicated, the internist may start the patient on a selective serotonin reuptake inhibitor such as citalopram 20 mg daily, as these medications are usually effective and well tolerated in liver transplant patients.73
Liver transplantation has a major positive effect on quality of life. Most patients with end-stage liver disease have poor quality of life before transplantation, but this seems to improve notably afterward. A meta-analysis showed significant improvement in posttransplant physical health, sexual functioning, daily activities, and social functioning compared with before transplantation.75
Alcohol abuse and smoking
Patients who underwent liver transplantation because of alcoholic liver disease should be advised to abstain from alcohol.19 Patients who underwent the procedure for a different indication are advised to avoid excessive alcohol intake, as it is proven to lower the survival rate.20 Alcohol recidivism and smoking (including marijuana) are major problems, and internists are best positioned to address these issues and treat them.
Vaccinations
All liver transplant recipients should be vaccinated against influenza, pneumococcal infection, and tetanus. Hepatitis A and B vaccines are typically given before transplantation. In general, live vaccines such as measles-mumps-rubella and varicella are not recommended after any solid organ transplant.76
A study in Germany showed that immunization rates were too low in solid-organ transplant recipients, and almost 90% of patients were not adequately informed about immunizations.77 Hence, there may be room for improvement, and primary care providers should take the lead toward better outcomes in this regard.
Recurrence of the primary liver disease after transplantation
Different primary liver diseases recur with different frequencies.
Hepatitis C has the highest rate of recurrence of the liver diseases.78,79 Reinfection with hepatitis C virus after liver transplantation is almost universal and can follow different patterns. One of the most aggressive patterns is fibrosing cholestatic hepatitis, which frequently leads to graft failure and death, and hence necessitates urgent detection and treatment.
Hepatocellular carcinoma also has a high recurrence rate.80 Surveillance with liver ultrasonography or computed tomography is required every 6 months for the first 5 years after liver transplantation.
Other liver diseases. Nonalcoholic steatohepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, and hepatitis B infection also tend to recur after liver transplantation.46,81 On the other hand, alpha-1 antitrypsin deficiency, Wilson disease, hemochromatosis, and metabolic disorders are “cured” after liver transplantation.
It is important to detect any increase in liver enzymes above baseline. An elevation of 1.5 times the upper limit of normal or more should trigger further investigation.
Allograft dysfunction
A number of complications can develop in the liver allograft and result in abnormal liver function tests and, if not treated, graft failure. The most common causes of late graft dysfunction include recurrence of primary liver disease, biliary complications, and chronic rejection.46
Vascular complications include hepatic artery thrombosis and stenosis and are usually evaluated by liver ultrasonography and Doppler scan of the hepatic artery and venous structures.24
Biliary strictures give a cholestatic picture, with elevated bilirubin and greater elevation of alkaline phosphatase than of alanine aminotransferase and aspartate aminotransferase. Strictures are usually treated by endoscopic dilation and stenting, but they may eventually require surgery.
Late acute cellular rejections occur in 10% to 20% of cases and are a risk factor for chronic rejection. Liver biopsy is needed to make the diagnosis, and pulsed doses of corticosteroids remain the backbone of treatment therapy.
Chronic rejection is not common, occurring in 3% to 4% of liver transplant recipients.46 Treatment is based on increasing immunosuppression and ensuring compliance with prescribed medications. However, chronic rejection may not respond well, and repeat transplantation may be the last resort for some patients.
WHEN TO REFER TO THE HEPATOLOGIST
Some situations require referral to the hepatologist or the transplant center. In general, the following are best managed by a hepatologist: adjustment of immunosuppressive drugs and dosages, allograft dysfunction, vascular and biliary complications, progressing renal dysfunction, and recurrence of primary liver disease. Early communication with a hepatologist and the transplant center is recommended in these cases.
References
Starzl TE, Marchioro TL, Vonkaulla KN, Hermann G, Brittain RS, Waddell WR. Homotransplantation of the liver in humans. Surg Gynecol Obstet 1963; 117:659–676.
Matas AJ, Smith JM, Skeans MA, et al. OPTN/SRTR 2011 annual data report: kidney. Am J Transplant 2013; 13(suppl 1):11–46.
McCashland TM. Posttransplantation care: role of the primary care physician versus transplant center. Liver Transpl 2001; 7(suppl 1):S2–S12.
Heller JC, Prochazka AV, Everson GT, Forman LM. Long-term management after liver transplantation: primary care physician versus hepatologist. Liver Transpl 2009; 15:1330–1335.
McAlister VC, Haddad E, Renouf E, Malthaner RA, Kjaer MS, Gluud LL. Cyclosporin versus tacrolimus as primary immunosuppressant after liver transplantation: a meta-analysis. Am J Transplant 2006; 6:1578–1585.
Neuhaus P, Klupp J, Langrehr JM. mTOR inhibitors: an overview. Liver Transpl 2001; 7:473–484.
Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem 1998; 31:335–340.
Asrani SK, Wiesner RH, Trotter JF, et al. De novo sirolimus and reduced-dose tacrolimus versus standard-dose tacrolimus after liver transplantation: the 2000-2003 phase II prospective randomized trial. Am J Transplant 2014; 14:356–366.
Montalbano M, Neff GW, Yamashiki N, et al. A retrospective review of liver transplant patients treated with sirolimus from a single center: an analysis of sirolimus-related complications. Transplantation 2004; 78:264–268.
Everson GT. Everolimus and mTOR inhibitors in liver transplantation: opening the “box.” Liver Transpl 2006; 12:1571–1573.
Levy G, Schmidli H, Punch J, et al. Safety, tolerability, and efficacy of everolimus in de novo liver transplant recipients: 12- and 36-month results. Liver Transpl 2006; 12:1640–1648.
Toniutto P, Fabris C, Fumolo E, et al. Prevalence and risk factors for delayed adrenal insufficiency after liver transplantation. Liver Transpl 2008; 14:1014–1019.
Klintmalm GB, Davis GL, Teperman L, et al. A randomized, multicenter study comparing steroid-free immunosuppression and standard immunosuppression for liver transplant recipients with chronic hepatitis C. Liver Transpl 2011; 17:1394–1403.
Llado L, Fabregat J, Castellote J, et al. Impact of immunosuppression without steroids on rejection and hepatitis C virus evolution after liver transplantation: results of a prospective randomized study. Liver Transpl 2008; 14:1752–1760.
Lake JR. Immunosuppression and outcomes of patients transplanted for hepatitis C. J Hepatol 2006; 44:627–629.
Sohn AJ, Jeon H, Ahn J. Primary care of the liver transplant recipient. Prim Care 2011; 38:499–514.
Laish I, Braun M, Mor E, Sulkes J, Harif Y, Ben Ari Z. Metabolic syndrome in liver transplant recipients: prevalence, risk factors, and association with cardiovascular events. Liver Transpl 2011; 17:15–22.
Stegall MD, Everson G, Schroter G, Bilir B, Karrer F, Kam I. Metabolic complications after liver transplantation. Diabetes, hypercholesterolemia, hypertension, and obesity. Transplantation 1995; 60:1057–1060.
Textor SC, Canzanello VJ, Taler SJ, et al. Cyclosporine-induced hypertension after transplantation. Mayo Clin Proc 1994; 69:1182–1193.
Prevention, detection, evaluation, and treatment of hypertension. The Sixth Report of the Joint National Committee. National Institutes of Health-National Heart, Lung, and Blood Institute. National High Blood Pressure Education Programme. Indian Heart J 1999; 51:381–396.
Frishman WH. Calcium channel blockers: differences between subclasses. Am J Cardiovasc Drugs 2007; 7(suppl 1):17–23.
Galioto A, Semplicini A, Zanus G, et al. Nifedipine versus carvedilol in the treatment of de novo arterial hypertension after liver transplantation: results of a controlled clinical trial. Liver Transpl 2008; 14:1020–1028.
Neal DA, Gimson AE, Gibbs P, Alexander GJ. Beneficial effects of converting liver transplant recipients from cyclosporine to tacrolimus on blood pressure, serum lipids, and weight. Liver Transpl 2001; 7:533–539.
Singh S, Watt KD. Long-term medical management of the liver transplant recipient: what the primary care physician needs to know. Mayo Clin Proc 2012; 87:779–790.
Bianchi G, Marchesini G, Marzocchi R, Pinna AD, Zoli M. Metabolic syndrome in liver transplantation: relation to etiology and immunosuppression. Liver Transpl 2008; 14:1648–1654.
Lane JT, Dagogo-Jack S. Approach to the patient with new-onset diabetes after transplant (NODAT). J Clin Endocrinol Metab 2011; 96:3289–3297.
Wilkinson A, Davidson J, Dotta F, et al. Guidelines for the treatment and management of new-onset diabetes after transplantation. Clin Transplant 2005; 19:291–298.
Moon JI, Barbeito R, Faradji RN, Gaynor JJ, Tzakis AG. Negative impact of new-onset diabetes mellitus on patient and graft survival after liver transplantation: long-term follow up. Transplantation 2006; 82:1625–1628.
American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63.
Marchetti P. New-onset diabetes after liver transplantation: from pathogenesis to management. Liver Transpl 2005; 11:612–620.
Stone NJ, Robinson J, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl 2):S1–S45.
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
Gisbert C, Prieto M, Berenguer M, et al. Hyperlipidemia in liver transplant recipients: prevalence and risk factors. Liver Transpl Surg 1997; 3:416–422.
Asberg A. Interactions between cyclosporin and lipid-lowering drugs: implications for organ transplant recipients. Drugs 2003; 63:367–378.
Bullingham RE, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet 1998; 34:429–455.
Everhart JE, Lombardero M, Lake JR, Wiesner RH, Zetterman RK, Hoofnagle JH. Weight change and obesity after liver transplantation: incidence and risk factors. Liver Transpl Surg 1998; 4:285–296.
Canzanello VJ, Schwartz L, Taler SJ, et al. Evolution of cardiovascular risk after liver transplantation: a comparison of cyclosporine A and tacrolimus (FK506). Liver Transpl Surg 1997; 3:1–9.
Al-Nowaylati AR, Al-Haddad BJ, Dorman RB, et al. Gastric bypass after liver transplantation. Liver Transpl 2013; 19:1324–1329.
Heimbach JK, Watt KD, Poterucha JJ, et al. Combined liver transplantation and gastric sleeve resection for patients with medically complicated obesity and end-stage liver disease. Am J Transplant 2013; 13:363–368.
Watt KD, Charlton MR. Metabolic syndrome and liver transplantation: a review and guide to management. J Hepatol 2010; 53:199–206.
Pagadala M, Dasarathy S, Eghtesad B, McCullough AJ. Posttransplant metabolic syndrome: an epidemic waiting to happen. Liver Transpl 2009; 15:1662–1670.
Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009; 120:1640–1645.
Watt KD, Pedersen RA, Kremers WK, Heimbach JK, Charlton MR. Evolution of causes and risk factors for mortality post-liver transplant: results of the NIDDK long-term follow-up study. Am J Transplant 2010; 10:1420–1427.
Albeldawi M, Aggarwal A, Madhwal S, et al. Cumulative risk of cardiovascular events after orthotopic liver transplantation. Liver Transpl 2012; 18:370–375.
Gibbons RJ, Balady GJ, Bricker JT, et al. ACC/AHA 2002 guideline update for exercise testing: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1997 Exercise Testing Guidelines). Circulation 2002; 106:1883–1892.
Aberg F, Isoniemi H, Höckerstedt K. Long-term results of liver transplantation. Scand J Surg 2011; 100:14–21.
Aberg F, Pukkala E, Höckerstedt K, Sankila R, Isoniemi H. Risk of malignant neoplasms after liver transplantation: a population-based study. Liver Transpl 2008; 14:1428–1436.
Mells G, Neuberger J. Long-term care of the liver allograft recipient. Semin Liver Dis 2009; 29:102–120.
Herrero JI. De novo malignancies following liver transplantation: impact and recommendations. Liver Transpl 2009; 15(suppl 2):S90–S94.
Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ transplantation. N Engl J Med 2003; 348:1681–1691.
Euvrard S, Morelon E, Rostaing L, et al; TUMORAPA Study Group. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med 2012; 367:329–339.
Salgo R, Gossmann J, Schofer H, et al. Switch to a sirolimus-based immunosuppression in long-term renal transplant recipients: reduced rate of (pre-)malignancies and nonmelanoma skin cancer in a prospective, randomized, assessor-blinded, controlled clinical trial. Am J Transplant 2010; 10:1385–1393.
Narumi S, Roberts JP, Emond JC, Lake J, Ascher NL. Liver transplantation for sclerosing cholangitis. Hepatology 1995; 22:451–457.
Knechtle SJ, D’Alessandro AM, Harms BA, Pirsch JD, Belzer FO, Kalayoglu M. Relationships between sclerosing cholangitis, inflammatory bowel disease, and cancer in patients undergoing liver transplantation. Surgery 1995; 118:615–620.
Finkenstedt A, Graziadei IW, Oberaigner W, et al. Extensive surveillance promotes early diagnosis and improved survival of de novo malignancies in liver transplant recipients. Am J Transplant 2009; 9:2355–2361.
Fisher NC, Nightingale PG, Gunson BK, Lipkin GW, Neuberger JM. Chronic renal failure following liver transplantation: a retrospective analysis. Transplantation 1998; 66:59–66.
Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
Klintmalm GB, Gonwa TA. Nephrotoxicity associated with cyclosporine and FK506. Liver Transpl Surg 1995; 1:11–19.
A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression in liver transplantation. The US Multicenter FK506 Liver Study Group. N Engl J Med 1994; 331:1110–1115.
Neff GW, Montalbano M, Slapak-Green G, et al. Sirolimus therapy in orthotopic liver transplant recipients with calcineurin inhibitor related chronic renal insufficiency. Transplant Proc 2003; 35:3029–3031.
Cotterell AH, Fisher RA, King AL, et al. Calcineurin inhibitor-induced chronic nephrotoxicity in liver transplant patients is reversible using rapamycin as the primary immunosuppressive agent. Clin Transplant 2002; 16(suppl 7):49–51.
Manzia TM, De Liguori Carino N, Orlando G, et al. Use of mycophenolate mofetil in liver transplantation: a literature review. Transplant Proc 2005; 37:2616–2617.
Schlitt HJ, Barkmann A, Boker KH, et al. Replacement of calcineurin inhibitors with mycophenolate mofetil in liver-transplant patients with renal dysfunction: a randomised controlled study. Lancet 2001; 357:587–591.
Hodge EE, Reich DJ, Clavien PA, Kim-Schluger L. Use of mycophenolate mofetil in liver transplant recipients experiencing renal dysfunction on cyclosporine or tacrolimus-randomized, prospective, multicenter study results. Transplant Proc 2002; 34:1546–1547.
Hamburg SM, Piers DA, van den Berg AP, Slooff MJ, Haagsma EB. Bone mineral density in the long term after liver transplantation. Osteoporos Int 2000; 11:600–606.
Maalouf NM, Shane E. Osteoporosis after solid organ transplantation. J Clin Endocrinol Metab 2005; 90:2456–2465.
Crosbie OM, Freaney R, McKenna MJ, Curry MP, Hegarty JE. Predicting bone loss following orthotopic liver transplantation. Gut 1999; 44:430–434.
Giannini S, Nobile M, Ciuffreda M, et al. Long-term persistence of low bone density in orthotopic liver transplantation. Osteoporos Int 2000; 11:417–424.
Monegal A, Navasa M, Guanabens N, et al. Bone disease after liver transplantation: a long-term prospective study of bone mass changes, hormonal status and histomorphometric characteristics. Osteoporos Int 2001; 12:484–492.
Neuhaus R, Lohmann R, Platz KP, et al. Treatment of osteoporosis after liver transplantation. Transplant Proc 1995; 27:1226–1227.
Valero MA, Loinaz C, Larrodera L, Leon M, Moreno E, Hawkins F. Calcitonin and bisphosphonates treatment in bone loss after liver transplantation. Calcif Tissue Int 1995; 57:15–19.
Neal DA, Tom BD, Gimson AE, Gibbs P, Alexander GJ. Hyperuricemia, gout, and renal function after liver transplantation. Transplantation 2001; 72:1689–1691.
Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver. 10th ed. Philadephia, PA: Lippincott Williams & Wilkins; 2007.
Tombazzi CR, Waters B, Shokouh-Amiri MH, Vera SR, Riely CA. Neuropsychiatric complications after liver transplantation: role of immunosuppression and hepatitis C. Dig Dis Sci 2006; 51:1079–1081.
Bravata DM, Olkin I, Barnato AE, Keeffe EB, Owens DK. Health-related quality of life after liver transplantation: a meta-analysis. Liver Transpl Surg 1999; 5:318–331.
Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
Chesi C, Gunther M, Huzly D, et al. Immunization of liver and renal transplant recipients: a seroepidemiological and sociodemographic survey. Transpl Infect Dis 2009; 11:507–512.
Berenguer M, Lopez-Labrador FX, Wright TL. Hepatitis C and liver transplantation. J Hepatol 2001; 35:666–678.
Forman LM, Lewis JD, Berlin JA, Feldman HI, Lucey MR. The association between hepatitis C infection and survival after orthotopic liver transplantation. Gastroenterology 2002; 122:889–896.
Benten D, Staufer K, Sterneck M. Orthotopic liver transplantation and what to do during follow-up: recommendations for the practitioner. Nat Clin Pract Gastroenterol Hepatol 2009; 6:23–36.
Kotlyar DS, Campbell MS, Reddy KR. Recurrence of diseases following orthotopic liver transplantation. Am J Gastroenterol 2006; 101:1370–1378.
Since 1963, when Starzl et al performed the first successful liver transplantation,1 outcomes of this life-saving procedure have continued to improve. Long-term survival rates have increased markedly: the current 5-year rate is 73.8% and the 10-year rate is 60%.2
This success means that internists will be caring for a greater number of liver transplant recipients and managing their long-term problems, such as hypertension, diabetes mellitus, dyslipidemia, obesity, metabolic syndrome, cardiovascular disease, renal insufficiency, osteoporosis, cancer, and gout.
This review will discuss these complications, focusing on the role the primary care physician assumes beyond the first year after transplantation.
ROLE OF THE PRIMARY CARE PHYSICIAN
Hepatologists, primary care physicians, and surgeons share the care of transplant recipients. The first several weeks after transplantation require close follow-up by the hepatologist and transplantation team, with particular attention paid to the patient’s overall health and well-being, medication compliance, and biochemical and immunosuppression monitoring.
After the first year, the primary care physician assumes a greater role, becoming the main provider of the patient’s care.3,4 Good communication between the transplant center and the primary care physician should lead to a smooth transition.4 Although the hepatologist continues to manage immunosuppressive drugs, allograft rejections, and biliary complications, the primary care physician manages most of the long-term complications and thus needs to be aware of the common ones and feel comfortable managing them. Aims during visits are to screen for and detect common complications and manage them appropriately, in addition to performing annual physical examinations and routine health care. A reasonable interval for liver transplant recipients to visit their primary care physician is every 6 months.
IMMUNOSUPPRESSANT MEDICATIONS
Multiple agents are used for immunosuppression after liver transplantation:
Calcineurin inhibitors (cyclosporine and tacrolimus)
Antimetabolites (mycophenolate mofetil, azathioprine, and mycophenolate sodium)
Mammalian target of rapamycin (mTOR) inhibitors (sirolimus and everolimus)
Corticosteroids.
Table 1 lists their common side effects.
Most centers use a combination of two to four immunosuppressants as induction therapy in the immediate posttransplant period, then taper the doses and eliminate all but a calcineurin inhibitor and an antimetabolite. For example, some start with a combination of tacrolimus, mycophenolate mofetil, and a corticosteroid. The choice in the immediate posttransplant period is frequently made by the transplant center in cooperation with the hepatologist. By the time primary care physicians see these patients, they usually are on a calcineurin inhibitor alone or a calcineurin inhibitor plus mycophenolate mofetil.
Calcineurin inhibitors
Cyclosporine is metabolized by the cytochrome CYP3A4 pathway. With an average half-life of 15 hours, it is given orally, usually every 12 hours.
The dosage is adjusted according to the trough level. Higher levels are needed in the initial posttransplant period to prevent graft rejection, whereas lower levels are preferred later to decrease the occurrence and severity of adverse effects. Typical long-term trough levels are 50 to 100 ng/mL. Levels should be checked more often if an acute illness develops or the patient starts taking a potentially interfering drug.
Of importance: the dosage should be based on trough levels and not on random levels. Levels are often falsely high if blood samples are not drawn at the trough level. Repeating the measurement and making sure the sample is drawn at the trough level,ie, 12 hours after the last dose, is advised in this condition.
Cyclosporine causes widespread vasoconstriction resulting in decreased renal blood flow and systemic hypertension, often within a few days of starting it. Other important adverse effects include renal insufficiency, dyslipidemia, neurotoxicity (headache, tremor, seizure), and diabetes.
Tacrolimus is superior to cyclosporine in terms of survival, graft loss, acute rejection, and steroid-resistant rejection in the first year.5 Currently, it is the agent used most often for maintenance immunosuppression after liver transplantation.
Like cyclosporine, tacrolimus is metabolized in the liver by CYP3A4. Satisfactory troughlevels after 1 year are 4 to 6 ng/mL.
The adverse effects of tacrolimus are similar to those of cyclosporine, but diabetes mellitus is more common with tacrolimus. Bone marrow suppression may occur more often with tacrolimus as well.
Antimetabolites
Antimetabolites are generally not potent enough to be used alone.
Mycophenolate mofetil causes adverse effects that include bone marrow suppression and gastrointestinal symptoms such as gastritis, diarrhea, and abdominal pain.
Azathioprine, infrequently used in transplantation in the United States, is nevertheless sometimes substituted for mycophenolate mofetil in pregnant women, as it seems safer for use in pregnancy.
Serum levels of azathioprine and mycophenolate mofetil are not routinely monitored.
mTOR inhibitors
Sirolimus and everolimus are mTOR inhibitors, inhibiting proliferation of lymphocytes.6,7
Unlike calcineurin inhibitors, mTOR inhibitors are not associated with nephrotoxicity, neurotoxicity, renal dysfunction, hypertension, or diabetes. Sirolimus is considered an alternative to calcineurin inhibitors or, in some instances, used as add-on-therapy to lower the dose of the calcineurin inhibitor.
Sirolimus carries a black-box warning about hepatic artery thrombosis
However, sirolimus carries a potential risk of hepatic artery thrombosis, a life-threatening complication.8 This has led the US Food and Drug Administration (FDA) to require sirolimus to carry a black-box warning, and most transplant centers avoid using it in the first 30 days after transplantation.
Dyslipidemia is perhaps the most common adverse effect of sirolimus. Others include dose-related cytopenia and wound dehiscence.9
Everolimus has yet to be established for use in liver transplantation, although safety trials have been published.10,11 The FDA currently recommends against using it in the first 30 days after liver transplantation.
Both sirolimus and everolimus are metabolized by CYP3A4, which is the same metabolic pathway used by cyclosporine and tacrolimus. Hence, drugs that inhibit CYP3A4 may significantly impair clearance of both sirolimus and everolimus.
Corticosteroids
Corticosteroids have been the cornerstone of immunosuppression and remain the first line of treatment for acute allograft rejection. High intravenous doses of corticosteroids are usually started in the peritransplant period and are then switched to oral doses, which are tapered and continued with a fixed dose such as 20 mg of prednisone daily for 3 to 6 months after transplantation. However, some transplant centers keep patients on prednisone 5 mg/day indefinitely.
Adverse effects of corticosteroids include diabetes, salt and fluid retention, hypertension, hyperlipidemia, cosmetic changes (acne, cervical fat pad or “buffalo hump”), delayed wound healing, susceptibility to infection, cataracts, osteopenia, and potential adrenal suppression.12 There is concern that the use of these drugs may increase hepatitis C virus replication in patients who received a liver transplant for hepatitis C cirrhosis. Randomized trials have yielded conflicting results.13–15
Drug interactions
Certain drugs can affect the metabolism of calcineurin inhibitors and mTOR inhibitors by inducing CYP3A4, which results in decreasing the levels of the immunosuppressive drugs, or by inhibiting CYP3A4, which has the opposite effect.
Medications that can decrease the levels of calcineurin inhibitors and mTOR inhibitors:
Selected antibiotics are generally well tolerated, such as penicillins, cephalosporins, quinolones, sulfonamides, and topical antifungal agents.
LONG-TERM COMPLICATIONS
Figure 1.
Figure 1 summarizes the common long-term complications of liver transplantation.
Hypertension
The prevalence of hypertension after liver transplantation is 40% to 85%, which is markedly higher than in patients with chronic liver disease before liver transplantation.17,18
One of the factors contributing to this increase is the use of immunosuppressive medications. Of these drugs, cyclosporine seems to be the one that most often causes an increase in both the incidence and the severity of hypertension, as it produces widespread vasoconstriction.19 Corticosteroids cause hypertension through their mineralocorticoid effects.
The diagnostic cutoffs for hypertension (ie, 140/90 mm Hg) and the treatment goals in posttransplant patients are similar to those in the general population. However, at our institution we target a blood pressure of less than 130/80 mm Hg in transplant patients because they have a high prevalence of other cardiovascular risk factors such as diabetes, obesity, and renal insufficiency.20
Dihydropyridine calcium channel blockers such as amlodipine and nifedipine are considered the best first-line agents because they dilate renal afferent arterioles, an effect that may counteract the vasoconstriction mediated by calcineurin inhibitors. Nondihydropyridine calcium channel blockers such as diltiazem and verapamil tend to have more marked negative inotropic effects and are not recommended in liver transplant recipients because they increase the levels of calcineurin inhibitors.21
Diuretics (eg, furosemide) might be the second-line agents, especially in patients with peripheral edema.16 One should be vigilant for hyperuricemia if thiazide agents are used.
Angiotensin-converting-enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are typically avoided in the early posttransplant period, but they can be started later and have additional benefits in patients with diabetes and congestive heart failure. Starting ACE inhibitors is acceptable in these patients unless there is a contraindication such as allergy to ACE inhibitors, hypotension, history of bilateral renal artery stenosis, significant hyperkalemia, or acute kidney injury. Monitor the serum potassium level closely for hyperkalemia in patients concurrently using calcineurin inhibitors.
Alpha-blockers and beta-blockers can be used as add-on therapy in patients with uncontrolled hypertension with the exception of carvedilol, because it increases the levels of calcineurin inhibitors.22
Blood pressure monitoring by the primary care physician is recommended every 6 months after the early posttransplant period, or more frequently when changes in treatment are being considered.
If hypertension continues to be inadequately controlled despite treatment, changing the immunosuppressive drugs or decreasing the doses can be considered, but the transplant hepatologist must be involved in this decision.23,24
Diabetes mellitus
The prevalence of diabetes mellitus is higher in liver transplant recipients than in the general population, reaching 30% to 40%.17,25 In addition to preexisting diabetes, 15% of liver transplant recipients develop new-onset diabetes.26,27
Risk factors for developing diabetes after liver transplantation include African American or Hispanic ethnicity, obesity, family history, pretransplant diabetes, hepatitis C virus infection, use of corticosteroids, and use of calcineurin inhibitors (tacrolimus more than cyclosporine) and sirolimus.26
In addition to increasing the risk of cardiovascular disease and other diseases, diabetes decreases both patient and graft survival after liver transplantation.28
The management of diabetes and the treatment target after transplantation should follow the American Diabetes Association guidelines for the treatment of type 2 diabetes mellitus.29 Lifestyle modifications, diet, and exercise are as important for transplant patients as for the nontransplant population. Insulin therapy is usually needed in the early posttransplant period to control blood glucose levels well, especially with the high doses of corticosteroids used during the first few weeks. No trials to date have compared oral agents in posttransplant patients. Therefore, the choice of oral hypoglycemic agents should be individualized on the basis of the patient’s characteristics and comorbidities.
Screen all liver transplant recipients for diabetes regardless of their pretransplant status
We recommend that primary care providers screen all liver transplant recipients for diabetes regardless of their pretransplant status. This can be done by obtaining regular fasting blood glucose levels or a hemoglobin A1c level every 6 months. Additionally, liver transplant recipients diagnosed with diabetes require annual eye examinations to look for cataracts and diabetes-related changes.30
Dyslipidemia
On November 12, 2013, the American College of Cardiology and the American Heart Association (ACC/AHA) released new clinical practice guidelines for treating blood cholesterol levels.31,32 According to these new guidelines, there are four groups of patients for whom treatment with statins is clearly indicated:
Patients with cardiovascular disease
Patients with low-density lipoprotein cholesterol (LDL-C) levels ≥ 190 mg/dL
Patients 40 to 75 years old with type 2 diabetes
Patients 40 to 75 years old with an estimated 10-year risk of cardiovascular disease of 7.5% or greater.
Liver transplant recipients should be evaluated on an individual basis to see if they fit in any of the four groups and if statin treatment therefore needs to be initiated.
A few things need to be kept in mind. First, the incidence of dyslipidemia after liver transplantation is estimated to be 45% to 69%. Risk factors include obesity, diabetes mellitus, cholestatic liver disease, and immunosuppressant medications.33 Sirolimus has a significant and well-documented association with dyslipidemia. Cyclosporine and corticosteroids are also strongly associated with dyslipidemia. Tacrolimus has a minor effect, and mycophenolate mofetil and azathioprine have no significant effects on serum lipid levels.16
Second, of the seven currently marketed statins, pravastatin and fluvastatin are preferred in liver transplant recipients because they are not metabolized by the same cytochrome CYP3A4 pathway that metabolizes calcineurin inhibitors and sirolimus.34 The doses of 40 to 80 mg daily of pravastatin or 40 mg twice daily of fluvastatin lower low-density lipoprotein cholesterol (LDL-C) levels by approximately 30% to 35%. However, these two agents are considered “moderate-intensity” statins according to the new ACC/AHA guidelines. The only two “high-intensity” statins are atorvastatin (40–80 mg) and rosuvastatin (20–40 mg), but they are both metabolized by CYP3A4. Therefore it is prudent to avoid them with the concurrent use of a calcineurin inhibitor or tacrolimus.
Gemfibrozil does not lower LDL-C and should not be used concomitantly with statins due to unacceptable risk of rhabdomyolysis and myopathy. Fenofibrates are usually avoided due to potential nephrotoxicity in patients receiving cyclosporine. Bile acid sequestrants (cholestyramine, colestipol, colesevelam) can decrease plasma mycophenolate mofetil levels by 35%.16,35 Thus, these agents should be avoided if mycophenolate mofetil is used.
It is reasonable to screen all liver transplant recipients with a fasting lipid profile at 3, 6, and 12 months after transplantation and annually thereafter. Creatine kinase should be measured if the patient complains of severe muscle pain or weakness but not on a routine basis.
Obesity
Approximately one-third of patients who are of normal weight at the time of transplantation will become obese afterward.18,25 Corticosteroid use is an important risk factor for posttransplant obesity, and tapering these drugs helps reduce weight.36 Patients treated with cyclosporine are more likely to gain weight than those who receive tacrolimus.37
Of importance: nonalcoholic fatty liver disease, currently the most common cause of chronic liver disease in adults, is rapidly increasing as an indication for liver transplantation. In fact, the proportion of liver transplantation procedures for nonalcoholic steatohepatitis-related cirrhosis increased from 1.2% in 2001 to 9.7% in 2009, and nonalcoholic steatohepatitis is expected to become the leading indication for liver transplantation in the next 20 years. And because nonalcoholic fatty liver disease is directly linked to obesity, the prevalence of obesity as a complication of liver transplantation will most likely increase in the near future.
Overweight liver transplant recipients may have great difficulty losing weight. Treatment starts with patient education on caloric restriction and exercise. If traditional measures fail to result in adequate weight loss, additional options include switching from cyclosporine to tacrolimus.23
Bariatric surgery may become an option for posttransplant patients. In a recent case series from the University of Minnesota, Al-Nowaylati et al38 reported their experience with seven patients who underwent orthotopic liver transplantation and then open Roux-en-Y gastric bypass. After bariatric surgery, the patients’ mean body mass index declined significantly, and glycemic control and high-density lipoprotein cholesterol (HDL-C) levels improved. However, one patient died of multiple organ failure, to which the bariatric surgery might have contributed.38
Heimbach et al39 conducted a study in patients referred for liver transplantation for whom a rigorous noninvasive weight-loss program before transplantation had failed. The researchers performed combined liver transplantation and sleeve gastrectomy in seven carefully selected patients who had failed to achieve weight loss to a body mass index less than 35 kg/m2 before transplantation. All seven patients lost weight, decreasing their mean body mass index from 49 kg/m2 before the procedure to 29 kg/m2 at last follow-up, and none of them developed posttransplant diabetes or steatosis.
At this time, there is not enough evidence to recommend concurrent orthotopic liver transplantation plus bariatric surgery, or combined orthotopic liver transplantation and sleeve gastrectomy. More study is needed to further evaluate these advanced approaches.
Posttransplant metabolic syndrome
Metabolic syndrome is common after liver transplantation and is strongly associated with increased morbidity in this patient population.40,41 The general definition of metabolic syndrome includes a combination of at least three of the following: hypertension, insulin resistance, hypertriglyceridemia, low HDL-C, and obesity.
The prevalence of metabolic syndrome is higher in patients after liver transplantation than in nontransplant patients. In a review of 252 liver transplant recipients, 52% were diagnosed with posttransplant metabolic syndrome, but only 5% had had it pretransplant.42
Careful screening for posttransplant metabolic syndrome and early recognition of risk factors are important. Nevertheless, the treatment of this condition depends on treating its components according to recommended guidelines.41
Cardiovascular disease
The incidence of cardiovascular morbidity and death is increased after liver transplantation.24 In addition, after liver transplantation, cardiovascular disease is a major cause of death unrelated to liver disease. It accounts for 12% to 16% of deaths and is the third most common cause of late mortality after liver transplantation.43 Of note, a recent study by our group demonstrated that patients undergoing liver transplantation for nonalcoholic steatohepatitis had a significantly higher risk of a cardiovascular event during the 3 years after transplantation than patients undergoing liver transplantation for cholestatic liver disease.44
Risk factors for cardiovascular disease after liver transplantation include older age at transplantation, male sex, posttransplant diabetes, posttransplant hypertension, and the use of mycophenolate mofetil.44 Modifying the risk factors is essential in decreasing the risk of cardiovascular events.
It is reasonable to perform dobutamine stress testing every 3 to 5 years in patients with multiple risk factors for cardiovascular disease, or more frequently in those with preexisting coronary artery disease.45,46
Malignancy
The risk of several malignancies increases after liver transplantation. Liver transplant recipients have an incidence of cancer 2.1 to 4.3 times greater than age- and sex-matched controls.24,47–49
Skin cancers are the most common and account for almost 40% of malignancies in organ transplant recipients.50 Whereas basal cell carcinoma is more common in the general population, squamous cell carcinoma is equally common in liver transplant recipients.
Multiple clinical studies have linked calcineurin inhibitors and azathioprine to the development of skin cancer. Annual skin examinations in addition to avoiding other risk factors such as smoking and sun exposure are generally recommended. Changing the immunosuppressants to sirolimus in high-risk patients may lower their chance of developing skin cancer.51,52
Patients with ulcerative colitis who undergo liver transplantation because of sclerosing cholangitis are at higher risk of colon cancer and require annual colonoscopy with surveillance biopsies. Patients who undergo transplantation for alcoholic liver disease seem to have a higher risk of pulmonary and oropharyngeal cancers.53,54
It is important that transplant patients adhere to recommended cancer screening guidelines, in view of their increased risk. Studies have shown improved overall survival in liver transplant recipients who underwent intensive cancer surveillance.55
Renal insufficiency
Renal insufficiency is a well-recognized complication of liver transplantation and is associated with an increased long-term death rate.56,57
The incidence of renal insufficiency increases dramatically over time. Ojo et al,57 in a study of almost 37,000 liver transplant recipients, found that the incidence of chronic kidney disease (defined as an estimated glomerular filtration rate < 30 mL/min/1.73 m2) was 13.9% at 3 years, 18% at 5 years, and approximately 26% at 10 years.
Risk factors include the use of calcineurin inhibitors (both cyclosporine and tacrolimus), older age, female sex, lower pretransplant glomerular filtration rate, postoperative acute renal failure, diabetes, hypertension, hepatitis C virus infection, and transplantation before 1998.58,59 Replacing a calcineurin inhibitor with mycophenolate mofetil or sirolimus may be considered with communication with the transplant center, as mycophenolate mofetil or sirolimus are associated with a lower risk of renal injury.60–64
All liver transplant recipients should avoid nonsteroidal anti-inflammatory drugs and nephrotoxic medications
Starting 1 year after liver transplantation, primary care providers should screen for renal dysfunction by obtaining kidney function tests every 6 months, including urinalysis and microalbuminuria assessment. Equations for estimating the glomerular filtration rate used in practice, such as the Modification of Diet in Renal Disease Study equation, rely mainly on serum creatinine, which may lead to overestimating renal function in some circumstances. Therefore, other equations can be used to confirm the estimated glomerular filtration rate measured by creatinine clearance, and to more accurately evaluate kidney function. Calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.
All liver transplant recipients should avoid nonsteroidal anti-inflammatory drugs and nephrotoxic medications, and should have their hypertension and diabetes adequately controlled.
Bone diseases
Osteopenia is another major complication of liver transplantation. One-third of liver transplant recipients have a bone mineral density below the fracture threshold.65
Multiple factors contribute to increased bone loss after transplantation, including use of corticosteroids, use of calcineurin inhibitors (cyclosporine, tacrolimus), poor nutrition, vitamin D deficiency, immobility, sarcopenia (reduced muscle mass), hypogonadism, smoking, and alcohol abuse.66 Even at low doses of less than 7.5 mg per day, corticosteroids inhibit osteoblast activity and increase bone resorption.
Studies have reported rapid bone loss at around 6 months after transplantation.67–69 However, long-term follow-up of bone mineral density up to 15 years after transplantation revealed an improvement mainly in the 2nd postoperative year, with no deterioration afterward.65
High-risk patients need to be identified early with appropriate screening and evaluation. Evaluation includes dual-energy x-ray absorptiometry and serum levels of calcium, phosphorous, parathyroid hormone, testosterone (men), estradiol (women), and alpha-25-hydroxyvitamin D. These tests are typically done before transplantation and then every other year afterward.
We recommend a daily dose of calcitriol and a calcium supplement to all our liver transplant patients.70 If osteoporosis (a T-score 2.5 or more standard deviations below the mean) or a fragility fracture occurs, then the patient may benefit from an oral bisphosphonate. Calcitonin has also been shown to improve bone mineral density in patients with osteoporosis after liver transplantation.71
Hyperuricemia and gout
Although hyperuricemia is common in liver transplant recipients (reported in approximately 47%), the development of clinical gout is less common (6%).72 Asymptomatic hyperuricemia requires observation only and is not usually treated in liver transplant recipients.
Acute attacks of gout are typically managed with colchicine 0.6 mg every 2 hours, up to five doses. Prednisone can be considered if symptoms persist despite treatment with colchicine. Allopurinol in an initial dose of 100 mg daily is used as maintenance therapy to reduce production of uric acid.73 However, because of the potential for drug interactions, the combination of azathioprine and allopurinol should be avoided.
Psychiatric complications and quality of life
Depression is common in liver transplant recipients, significantly more so in patients who received a transplant because of hepatitis C.74 The type of immunosuppressant is not associated with the incidence of depression. When indicated, the internist may start the patient on a selective serotonin reuptake inhibitor such as citalopram 20 mg daily, as these medications are usually effective and well tolerated in liver transplant patients.73
Liver transplantation has a major positive effect on quality of life. Most patients with end-stage liver disease have poor quality of life before transplantation, but this seems to improve notably afterward. A meta-analysis showed significant improvement in posttransplant physical health, sexual functioning, daily activities, and social functioning compared with before transplantation.75
Alcohol abuse and smoking
Patients who underwent liver transplantation because of alcoholic liver disease should be advised to abstain from alcohol.19 Patients who underwent the procedure for a different indication are advised to avoid excessive alcohol intake, as it is proven to lower the survival rate.20 Alcohol recidivism and smoking (including marijuana) are major problems, and internists are best positioned to address these issues and treat them.
Vaccinations
All liver transplant recipients should be vaccinated against influenza, pneumococcal infection, and tetanus. Hepatitis A and B vaccines are typically given before transplantation. In general, live vaccines such as measles-mumps-rubella and varicella are not recommended after any solid organ transplant.76
A study in Germany showed that immunization rates were too low in solid-organ transplant recipients, and almost 90% of patients were not adequately informed about immunizations.77 Hence, there may be room for improvement, and primary care providers should take the lead toward better outcomes in this regard.
Recurrence of the primary liver disease after transplantation
Different primary liver diseases recur with different frequencies.
Hepatitis C has the highest rate of recurrence of the liver diseases.78,79 Reinfection with hepatitis C virus after liver transplantation is almost universal and can follow different patterns. One of the most aggressive patterns is fibrosing cholestatic hepatitis, which frequently leads to graft failure and death, and hence necessitates urgent detection and treatment.
Hepatocellular carcinoma also has a high recurrence rate.80 Surveillance with liver ultrasonography or computed tomography is required every 6 months for the first 5 years after liver transplantation.
Other liver diseases. Nonalcoholic steatohepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, and hepatitis B infection also tend to recur after liver transplantation.46,81 On the other hand, alpha-1 antitrypsin deficiency, Wilson disease, hemochromatosis, and metabolic disorders are “cured” after liver transplantation.
It is important to detect any increase in liver enzymes above baseline. An elevation of 1.5 times the upper limit of normal or more should trigger further investigation.
Allograft dysfunction
A number of complications can develop in the liver allograft and result in abnormal liver function tests and, if not treated, graft failure. The most common causes of late graft dysfunction include recurrence of primary liver disease, biliary complications, and chronic rejection.46
Vascular complications include hepatic artery thrombosis and stenosis and are usually evaluated by liver ultrasonography and Doppler scan of the hepatic artery and venous structures.24
Biliary strictures give a cholestatic picture, with elevated bilirubin and greater elevation of alkaline phosphatase than of alanine aminotransferase and aspartate aminotransferase. Strictures are usually treated by endoscopic dilation and stenting, but they may eventually require surgery.
Late acute cellular rejections occur in 10% to 20% of cases and are a risk factor for chronic rejection. Liver biopsy is needed to make the diagnosis, and pulsed doses of corticosteroids remain the backbone of treatment therapy.
Chronic rejection is not common, occurring in 3% to 4% of liver transplant recipients.46 Treatment is based on increasing immunosuppression and ensuring compliance with prescribed medications. However, chronic rejection may not respond well, and repeat transplantation may be the last resort for some patients.
WHEN TO REFER TO THE HEPATOLOGIST
Some situations require referral to the hepatologist or the transplant center. In general, the following are best managed by a hepatologist: adjustment of immunosuppressive drugs and dosages, allograft dysfunction, vascular and biliary complications, progressing renal dysfunction, and recurrence of primary liver disease. Early communication with a hepatologist and the transplant center is recommended in these cases.
Since 1963, when Starzl et al performed the first successful liver transplantation,1 outcomes of this life-saving procedure have continued to improve. Long-term survival rates have increased markedly: the current 5-year rate is 73.8% and the 10-year rate is 60%.2
This success means that internists will be caring for a greater number of liver transplant recipients and managing their long-term problems, such as hypertension, diabetes mellitus, dyslipidemia, obesity, metabolic syndrome, cardiovascular disease, renal insufficiency, osteoporosis, cancer, and gout.
This review will discuss these complications, focusing on the role the primary care physician assumes beyond the first year after transplantation.
ROLE OF THE PRIMARY CARE PHYSICIAN
Hepatologists, primary care physicians, and surgeons share the care of transplant recipients. The first several weeks after transplantation require close follow-up by the hepatologist and transplantation team, with particular attention paid to the patient’s overall health and well-being, medication compliance, and biochemical and immunosuppression monitoring.
After the first year, the primary care physician assumes a greater role, becoming the main provider of the patient’s care.3,4 Good communication between the transplant center and the primary care physician should lead to a smooth transition.4 Although the hepatologist continues to manage immunosuppressive drugs, allograft rejections, and biliary complications, the primary care physician manages most of the long-term complications and thus needs to be aware of the common ones and feel comfortable managing them. Aims during visits are to screen for and detect common complications and manage them appropriately, in addition to performing annual physical examinations and routine health care. A reasonable interval for liver transplant recipients to visit their primary care physician is every 6 months.
IMMUNOSUPPRESSANT MEDICATIONS
Multiple agents are used for immunosuppression after liver transplantation:
Calcineurin inhibitors (cyclosporine and tacrolimus)
Antimetabolites (mycophenolate mofetil, azathioprine, and mycophenolate sodium)
Mammalian target of rapamycin (mTOR) inhibitors (sirolimus and everolimus)
Corticosteroids.
Table 1 lists their common side effects.
Most centers use a combination of two to four immunosuppressants as induction therapy in the immediate posttransplant period, then taper the doses and eliminate all but a calcineurin inhibitor and an antimetabolite. For example, some start with a combination of tacrolimus, mycophenolate mofetil, and a corticosteroid. The choice in the immediate posttransplant period is frequently made by the transplant center in cooperation with the hepatologist. By the time primary care physicians see these patients, they usually are on a calcineurin inhibitor alone or a calcineurin inhibitor plus mycophenolate mofetil.
Calcineurin inhibitors
Cyclosporine is metabolized by the cytochrome CYP3A4 pathway. With an average half-life of 15 hours, it is given orally, usually every 12 hours.
The dosage is adjusted according to the trough level. Higher levels are needed in the initial posttransplant period to prevent graft rejection, whereas lower levels are preferred later to decrease the occurrence and severity of adverse effects. Typical long-term trough levels are 50 to 100 ng/mL. Levels should be checked more often if an acute illness develops or the patient starts taking a potentially interfering drug.
Of importance: the dosage should be based on trough levels and not on random levels. Levels are often falsely high if blood samples are not drawn at the trough level. Repeating the measurement and making sure the sample is drawn at the trough level,ie, 12 hours after the last dose, is advised in this condition.
Cyclosporine causes widespread vasoconstriction resulting in decreased renal blood flow and systemic hypertension, often within a few days of starting it. Other important adverse effects include renal insufficiency, dyslipidemia, neurotoxicity (headache, tremor, seizure), and diabetes.
Tacrolimus is superior to cyclosporine in terms of survival, graft loss, acute rejection, and steroid-resistant rejection in the first year.5 Currently, it is the agent used most often for maintenance immunosuppression after liver transplantation.
Like cyclosporine, tacrolimus is metabolized in the liver by CYP3A4. Satisfactory troughlevels after 1 year are 4 to 6 ng/mL.
The adverse effects of tacrolimus are similar to those of cyclosporine, but diabetes mellitus is more common with tacrolimus. Bone marrow suppression may occur more often with tacrolimus as well.
Antimetabolites
Antimetabolites are generally not potent enough to be used alone.
Mycophenolate mofetil causes adverse effects that include bone marrow suppression and gastrointestinal symptoms such as gastritis, diarrhea, and abdominal pain.
Azathioprine, infrequently used in transplantation in the United States, is nevertheless sometimes substituted for mycophenolate mofetil in pregnant women, as it seems safer for use in pregnancy.
Serum levels of azathioprine and mycophenolate mofetil are not routinely monitored.
mTOR inhibitors
Sirolimus and everolimus are mTOR inhibitors, inhibiting proliferation of lymphocytes.6,7
Unlike calcineurin inhibitors, mTOR inhibitors are not associated with nephrotoxicity, neurotoxicity, renal dysfunction, hypertension, or diabetes. Sirolimus is considered an alternative to calcineurin inhibitors or, in some instances, used as add-on-therapy to lower the dose of the calcineurin inhibitor.
Sirolimus carries a black-box warning about hepatic artery thrombosis
However, sirolimus carries a potential risk of hepatic artery thrombosis, a life-threatening complication.8 This has led the US Food and Drug Administration (FDA) to require sirolimus to carry a black-box warning, and most transplant centers avoid using it in the first 30 days after transplantation.
Dyslipidemia is perhaps the most common adverse effect of sirolimus. Others include dose-related cytopenia and wound dehiscence.9
Everolimus has yet to be established for use in liver transplantation, although safety trials have been published.10,11 The FDA currently recommends against using it in the first 30 days after liver transplantation.
Both sirolimus and everolimus are metabolized by CYP3A4, which is the same metabolic pathway used by cyclosporine and tacrolimus. Hence, drugs that inhibit CYP3A4 may significantly impair clearance of both sirolimus and everolimus.
Corticosteroids
Corticosteroids have been the cornerstone of immunosuppression and remain the first line of treatment for acute allograft rejection. High intravenous doses of corticosteroids are usually started in the peritransplant period and are then switched to oral doses, which are tapered and continued with a fixed dose such as 20 mg of prednisone daily for 3 to 6 months after transplantation. However, some transplant centers keep patients on prednisone 5 mg/day indefinitely.
Adverse effects of corticosteroids include diabetes, salt and fluid retention, hypertension, hyperlipidemia, cosmetic changes (acne, cervical fat pad or “buffalo hump”), delayed wound healing, susceptibility to infection, cataracts, osteopenia, and potential adrenal suppression.12 There is concern that the use of these drugs may increase hepatitis C virus replication in patients who received a liver transplant for hepatitis C cirrhosis. Randomized trials have yielded conflicting results.13–15
Drug interactions
Certain drugs can affect the metabolism of calcineurin inhibitors and mTOR inhibitors by inducing CYP3A4, which results in decreasing the levels of the immunosuppressive drugs, or by inhibiting CYP3A4, which has the opposite effect.
Medications that can decrease the levels of calcineurin inhibitors and mTOR inhibitors:
Selected antibiotics are generally well tolerated, such as penicillins, cephalosporins, quinolones, sulfonamides, and topical antifungal agents.
LONG-TERM COMPLICATIONS
Figure 1.
Figure 1 summarizes the common long-term complications of liver transplantation.
Hypertension
The prevalence of hypertension after liver transplantation is 40% to 85%, which is markedly higher than in patients with chronic liver disease before liver transplantation.17,18
One of the factors contributing to this increase is the use of immunosuppressive medications. Of these drugs, cyclosporine seems to be the one that most often causes an increase in both the incidence and the severity of hypertension, as it produces widespread vasoconstriction.19 Corticosteroids cause hypertension through their mineralocorticoid effects.
The diagnostic cutoffs for hypertension (ie, 140/90 mm Hg) and the treatment goals in posttransplant patients are similar to those in the general population. However, at our institution we target a blood pressure of less than 130/80 mm Hg in transplant patients because they have a high prevalence of other cardiovascular risk factors such as diabetes, obesity, and renal insufficiency.20
Dihydropyridine calcium channel blockers such as amlodipine and nifedipine are considered the best first-line agents because they dilate renal afferent arterioles, an effect that may counteract the vasoconstriction mediated by calcineurin inhibitors. Nondihydropyridine calcium channel blockers such as diltiazem and verapamil tend to have more marked negative inotropic effects and are not recommended in liver transplant recipients because they increase the levels of calcineurin inhibitors.21
Diuretics (eg, furosemide) might be the second-line agents, especially in patients with peripheral edema.16 One should be vigilant for hyperuricemia if thiazide agents are used.
Angiotensin-converting-enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are typically avoided in the early posttransplant period, but they can be started later and have additional benefits in patients with diabetes and congestive heart failure. Starting ACE inhibitors is acceptable in these patients unless there is a contraindication such as allergy to ACE inhibitors, hypotension, history of bilateral renal artery stenosis, significant hyperkalemia, or acute kidney injury. Monitor the serum potassium level closely for hyperkalemia in patients concurrently using calcineurin inhibitors.
Alpha-blockers and beta-blockers can be used as add-on therapy in patients with uncontrolled hypertension with the exception of carvedilol, because it increases the levels of calcineurin inhibitors.22
Blood pressure monitoring by the primary care physician is recommended every 6 months after the early posttransplant period, or more frequently when changes in treatment are being considered.
If hypertension continues to be inadequately controlled despite treatment, changing the immunosuppressive drugs or decreasing the doses can be considered, but the transplant hepatologist must be involved in this decision.23,24
Diabetes mellitus
The prevalence of diabetes mellitus is higher in liver transplant recipients than in the general population, reaching 30% to 40%.17,25 In addition to preexisting diabetes, 15% of liver transplant recipients develop new-onset diabetes.26,27
Risk factors for developing diabetes after liver transplantation include African American or Hispanic ethnicity, obesity, family history, pretransplant diabetes, hepatitis C virus infection, use of corticosteroids, and use of calcineurin inhibitors (tacrolimus more than cyclosporine) and sirolimus.26
In addition to increasing the risk of cardiovascular disease and other diseases, diabetes decreases both patient and graft survival after liver transplantation.28
The management of diabetes and the treatment target after transplantation should follow the American Diabetes Association guidelines for the treatment of type 2 diabetes mellitus.29 Lifestyle modifications, diet, and exercise are as important for transplant patients as for the nontransplant population. Insulin therapy is usually needed in the early posttransplant period to control blood glucose levels well, especially with the high doses of corticosteroids used during the first few weeks. No trials to date have compared oral agents in posttransplant patients. Therefore, the choice of oral hypoglycemic agents should be individualized on the basis of the patient’s characteristics and comorbidities.
Screen all liver transplant recipients for diabetes regardless of their pretransplant status
We recommend that primary care providers screen all liver transplant recipients for diabetes regardless of their pretransplant status. This can be done by obtaining regular fasting blood glucose levels or a hemoglobin A1c level every 6 months. Additionally, liver transplant recipients diagnosed with diabetes require annual eye examinations to look for cataracts and diabetes-related changes.30
Dyslipidemia
On November 12, 2013, the American College of Cardiology and the American Heart Association (ACC/AHA) released new clinical practice guidelines for treating blood cholesterol levels.31,32 According to these new guidelines, there are four groups of patients for whom treatment with statins is clearly indicated:
Patients with cardiovascular disease
Patients with low-density lipoprotein cholesterol (LDL-C) levels ≥ 190 mg/dL
Patients 40 to 75 years old with type 2 diabetes
Patients 40 to 75 years old with an estimated 10-year risk of cardiovascular disease of 7.5% or greater.
Liver transplant recipients should be evaluated on an individual basis to see if they fit in any of the four groups and if statin treatment therefore needs to be initiated.
A few things need to be kept in mind. First, the incidence of dyslipidemia after liver transplantation is estimated to be 45% to 69%. Risk factors include obesity, diabetes mellitus, cholestatic liver disease, and immunosuppressant medications.33 Sirolimus has a significant and well-documented association with dyslipidemia. Cyclosporine and corticosteroids are also strongly associated with dyslipidemia. Tacrolimus has a minor effect, and mycophenolate mofetil and azathioprine have no significant effects on serum lipid levels.16
Second, of the seven currently marketed statins, pravastatin and fluvastatin are preferred in liver transplant recipients because they are not metabolized by the same cytochrome CYP3A4 pathway that metabolizes calcineurin inhibitors and sirolimus.34 The doses of 40 to 80 mg daily of pravastatin or 40 mg twice daily of fluvastatin lower low-density lipoprotein cholesterol (LDL-C) levels by approximately 30% to 35%. However, these two agents are considered “moderate-intensity” statins according to the new ACC/AHA guidelines. The only two “high-intensity” statins are atorvastatin (40–80 mg) and rosuvastatin (20–40 mg), but they are both metabolized by CYP3A4. Therefore it is prudent to avoid them with the concurrent use of a calcineurin inhibitor or tacrolimus.
Gemfibrozil does not lower LDL-C and should not be used concomitantly with statins due to unacceptable risk of rhabdomyolysis and myopathy. Fenofibrates are usually avoided due to potential nephrotoxicity in patients receiving cyclosporine. Bile acid sequestrants (cholestyramine, colestipol, colesevelam) can decrease plasma mycophenolate mofetil levels by 35%.16,35 Thus, these agents should be avoided if mycophenolate mofetil is used.
It is reasonable to screen all liver transplant recipients with a fasting lipid profile at 3, 6, and 12 months after transplantation and annually thereafter. Creatine kinase should be measured if the patient complains of severe muscle pain or weakness but not on a routine basis.
Obesity
Approximately one-third of patients who are of normal weight at the time of transplantation will become obese afterward.18,25 Corticosteroid use is an important risk factor for posttransplant obesity, and tapering these drugs helps reduce weight.36 Patients treated with cyclosporine are more likely to gain weight than those who receive tacrolimus.37
Of importance: nonalcoholic fatty liver disease, currently the most common cause of chronic liver disease in adults, is rapidly increasing as an indication for liver transplantation. In fact, the proportion of liver transplantation procedures for nonalcoholic steatohepatitis-related cirrhosis increased from 1.2% in 2001 to 9.7% in 2009, and nonalcoholic steatohepatitis is expected to become the leading indication for liver transplantation in the next 20 years. And because nonalcoholic fatty liver disease is directly linked to obesity, the prevalence of obesity as a complication of liver transplantation will most likely increase in the near future.
Overweight liver transplant recipients may have great difficulty losing weight. Treatment starts with patient education on caloric restriction and exercise. If traditional measures fail to result in adequate weight loss, additional options include switching from cyclosporine to tacrolimus.23
Bariatric surgery may become an option for posttransplant patients. In a recent case series from the University of Minnesota, Al-Nowaylati et al38 reported their experience with seven patients who underwent orthotopic liver transplantation and then open Roux-en-Y gastric bypass. After bariatric surgery, the patients’ mean body mass index declined significantly, and glycemic control and high-density lipoprotein cholesterol (HDL-C) levels improved. However, one patient died of multiple organ failure, to which the bariatric surgery might have contributed.38
Heimbach et al39 conducted a study in patients referred for liver transplantation for whom a rigorous noninvasive weight-loss program before transplantation had failed. The researchers performed combined liver transplantation and sleeve gastrectomy in seven carefully selected patients who had failed to achieve weight loss to a body mass index less than 35 kg/m2 before transplantation. All seven patients lost weight, decreasing their mean body mass index from 49 kg/m2 before the procedure to 29 kg/m2 at last follow-up, and none of them developed posttransplant diabetes or steatosis.
At this time, there is not enough evidence to recommend concurrent orthotopic liver transplantation plus bariatric surgery, or combined orthotopic liver transplantation and sleeve gastrectomy. More study is needed to further evaluate these advanced approaches.
Posttransplant metabolic syndrome
Metabolic syndrome is common after liver transplantation and is strongly associated with increased morbidity in this patient population.40,41 The general definition of metabolic syndrome includes a combination of at least three of the following: hypertension, insulin resistance, hypertriglyceridemia, low HDL-C, and obesity.
The prevalence of metabolic syndrome is higher in patients after liver transplantation than in nontransplant patients. In a review of 252 liver transplant recipients, 52% were diagnosed with posttransplant metabolic syndrome, but only 5% had had it pretransplant.42
Careful screening for posttransplant metabolic syndrome and early recognition of risk factors are important. Nevertheless, the treatment of this condition depends on treating its components according to recommended guidelines.41
Cardiovascular disease
The incidence of cardiovascular morbidity and death is increased after liver transplantation.24 In addition, after liver transplantation, cardiovascular disease is a major cause of death unrelated to liver disease. It accounts for 12% to 16% of deaths and is the third most common cause of late mortality after liver transplantation.43 Of note, a recent study by our group demonstrated that patients undergoing liver transplantation for nonalcoholic steatohepatitis had a significantly higher risk of a cardiovascular event during the 3 years after transplantation than patients undergoing liver transplantation for cholestatic liver disease.44
Risk factors for cardiovascular disease after liver transplantation include older age at transplantation, male sex, posttransplant diabetes, posttransplant hypertension, and the use of mycophenolate mofetil.44 Modifying the risk factors is essential in decreasing the risk of cardiovascular events.
It is reasonable to perform dobutamine stress testing every 3 to 5 years in patients with multiple risk factors for cardiovascular disease, or more frequently in those with preexisting coronary artery disease.45,46
Malignancy
The risk of several malignancies increases after liver transplantation. Liver transplant recipients have an incidence of cancer 2.1 to 4.3 times greater than age- and sex-matched controls.24,47–49
Skin cancers are the most common and account for almost 40% of malignancies in organ transplant recipients.50 Whereas basal cell carcinoma is more common in the general population, squamous cell carcinoma is equally common in liver transplant recipients.
Multiple clinical studies have linked calcineurin inhibitors and azathioprine to the development of skin cancer. Annual skin examinations in addition to avoiding other risk factors such as smoking and sun exposure are generally recommended. Changing the immunosuppressants to sirolimus in high-risk patients may lower their chance of developing skin cancer.51,52
Patients with ulcerative colitis who undergo liver transplantation because of sclerosing cholangitis are at higher risk of colon cancer and require annual colonoscopy with surveillance biopsies. Patients who undergo transplantation for alcoholic liver disease seem to have a higher risk of pulmonary and oropharyngeal cancers.53,54
It is important that transplant patients adhere to recommended cancer screening guidelines, in view of their increased risk. Studies have shown improved overall survival in liver transplant recipients who underwent intensive cancer surveillance.55
Renal insufficiency
Renal insufficiency is a well-recognized complication of liver transplantation and is associated with an increased long-term death rate.56,57
The incidence of renal insufficiency increases dramatically over time. Ojo et al,57 in a study of almost 37,000 liver transplant recipients, found that the incidence of chronic kidney disease (defined as an estimated glomerular filtration rate < 30 mL/min/1.73 m2) was 13.9% at 3 years, 18% at 5 years, and approximately 26% at 10 years.
Risk factors include the use of calcineurin inhibitors (both cyclosporine and tacrolimus), older age, female sex, lower pretransplant glomerular filtration rate, postoperative acute renal failure, diabetes, hypertension, hepatitis C virus infection, and transplantation before 1998.58,59 Replacing a calcineurin inhibitor with mycophenolate mofetil or sirolimus may be considered with communication with the transplant center, as mycophenolate mofetil or sirolimus are associated with a lower risk of renal injury.60–64
All liver transplant recipients should avoid nonsteroidal anti-inflammatory drugs and nephrotoxic medications
Starting 1 year after liver transplantation, primary care providers should screen for renal dysfunction by obtaining kidney function tests every 6 months, including urinalysis and microalbuminuria assessment. Equations for estimating the glomerular filtration rate used in practice, such as the Modification of Diet in Renal Disease Study equation, rely mainly on serum creatinine, which may lead to overestimating renal function in some circumstances. Therefore, other equations can be used to confirm the estimated glomerular filtration rate measured by creatinine clearance, and to more accurately evaluate kidney function. Calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.
All liver transplant recipients should avoid nonsteroidal anti-inflammatory drugs and nephrotoxic medications, and should have their hypertension and diabetes adequately controlled.
Bone diseases
Osteopenia is another major complication of liver transplantation. One-third of liver transplant recipients have a bone mineral density below the fracture threshold.65
Multiple factors contribute to increased bone loss after transplantation, including use of corticosteroids, use of calcineurin inhibitors (cyclosporine, tacrolimus), poor nutrition, vitamin D deficiency, immobility, sarcopenia (reduced muscle mass), hypogonadism, smoking, and alcohol abuse.66 Even at low doses of less than 7.5 mg per day, corticosteroids inhibit osteoblast activity and increase bone resorption.
Studies have reported rapid bone loss at around 6 months after transplantation.67–69 However, long-term follow-up of bone mineral density up to 15 years after transplantation revealed an improvement mainly in the 2nd postoperative year, with no deterioration afterward.65
High-risk patients need to be identified early with appropriate screening and evaluation. Evaluation includes dual-energy x-ray absorptiometry and serum levels of calcium, phosphorous, parathyroid hormone, testosterone (men), estradiol (women), and alpha-25-hydroxyvitamin D. These tests are typically done before transplantation and then every other year afterward.
We recommend a daily dose of calcitriol and a calcium supplement to all our liver transplant patients.70 If osteoporosis (a T-score 2.5 or more standard deviations below the mean) or a fragility fracture occurs, then the patient may benefit from an oral bisphosphonate. Calcitonin has also been shown to improve bone mineral density in patients with osteoporosis after liver transplantation.71
Hyperuricemia and gout
Although hyperuricemia is common in liver transplant recipients (reported in approximately 47%), the development of clinical gout is less common (6%).72 Asymptomatic hyperuricemia requires observation only and is not usually treated in liver transplant recipients.
Acute attacks of gout are typically managed with colchicine 0.6 mg every 2 hours, up to five doses. Prednisone can be considered if symptoms persist despite treatment with colchicine. Allopurinol in an initial dose of 100 mg daily is used as maintenance therapy to reduce production of uric acid.73 However, because of the potential for drug interactions, the combination of azathioprine and allopurinol should be avoided.
Psychiatric complications and quality of life
Depression is common in liver transplant recipients, significantly more so in patients who received a transplant because of hepatitis C.74 The type of immunosuppressant is not associated with the incidence of depression. When indicated, the internist may start the patient on a selective serotonin reuptake inhibitor such as citalopram 20 mg daily, as these medications are usually effective and well tolerated in liver transplant patients.73
Liver transplantation has a major positive effect on quality of life. Most patients with end-stage liver disease have poor quality of life before transplantation, but this seems to improve notably afterward. A meta-analysis showed significant improvement in posttransplant physical health, sexual functioning, daily activities, and social functioning compared with before transplantation.75
Alcohol abuse and smoking
Patients who underwent liver transplantation because of alcoholic liver disease should be advised to abstain from alcohol.19 Patients who underwent the procedure for a different indication are advised to avoid excessive alcohol intake, as it is proven to lower the survival rate.20 Alcohol recidivism and smoking (including marijuana) are major problems, and internists are best positioned to address these issues and treat them.
Vaccinations
All liver transplant recipients should be vaccinated against influenza, pneumococcal infection, and tetanus. Hepatitis A and B vaccines are typically given before transplantation. In general, live vaccines such as measles-mumps-rubella and varicella are not recommended after any solid organ transplant.76
A study in Germany showed that immunization rates were too low in solid-organ transplant recipients, and almost 90% of patients were not adequately informed about immunizations.77 Hence, there may be room for improvement, and primary care providers should take the lead toward better outcomes in this regard.
Recurrence of the primary liver disease after transplantation
Different primary liver diseases recur with different frequencies.
Hepatitis C has the highest rate of recurrence of the liver diseases.78,79 Reinfection with hepatitis C virus after liver transplantation is almost universal and can follow different patterns. One of the most aggressive patterns is fibrosing cholestatic hepatitis, which frequently leads to graft failure and death, and hence necessitates urgent detection and treatment.
Hepatocellular carcinoma also has a high recurrence rate.80 Surveillance with liver ultrasonography or computed tomography is required every 6 months for the first 5 years after liver transplantation.
Other liver diseases. Nonalcoholic steatohepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, and hepatitis B infection also tend to recur after liver transplantation.46,81 On the other hand, alpha-1 antitrypsin deficiency, Wilson disease, hemochromatosis, and metabolic disorders are “cured” after liver transplantation.
It is important to detect any increase in liver enzymes above baseline. An elevation of 1.5 times the upper limit of normal or more should trigger further investigation.
Allograft dysfunction
A number of complications can develop in the liver allograft and result in abnormal liver function tests and, if not treated, graft failure. The most common causes of late graft dysfunction include recurrence of primary liver disease, biliary complications, and chronic rejection.46
Vascular complications include hepatic artery thrombosis and stenosis and are usually evaluated by liver ultrasonography and Doppler scan of the hepatic artery and venous structures.24
Biliary strictures give a cholestatic picture, with elevated bilirubin and greater elevation of alkaline phosphatase than of alanine aminotransferase and aspartate aminotransferase. Strictures are usually treated by endoscopic dilation and stenting, but they may eventually require surgery.
Late acute cellular rejections occur in 10% to 20% of cases and are a risk factor for chronic rejection. Liver biopsy is needed to make the diagnosis, and pulsed doses of corticosteroids remain the backbone of treatment therapy.
Chronic rejection is not common, occurring in 3% to 4% of liver transplant recipients.46 Treatment is based on increasing immunosuppression and ensuring compliance with prescribed medications. However, chronic rejection may not respond well, and repeat transplantation may be the last resort for some patients.
WHEN TO REFER TO THE HEPATOLOGIST
Some situations require referral to the hepatologist or the transplant center. In general, the following are best managed by a hepatologist: adjustment of immunosuppressive drugs and dosages, allograft dysfunction, vascular and biliary complications, progressing renal dysfunction, and recurrence of primary liver disease. Early communication with a hepatologist and the transplant center is recommended in these cases.
References
Starzl TE, Marchioro TL, Vonkaulla KN, Hermann G, Brittain RS, Waddell WR. Homotransplantation of the liver in humans. Surg Gynecol Obstet 1963; 117:659–676.
Matas AJ, Smith JM, Skeans MA, et al. OPTN/SRTR 2011 annual data report: kidney. Am J Transplant 2013; 13(suppl 1):11–46.
McCashland TM. Posttransplantation care: role of the primary care physician versus transplant center. Liver Transpl 2001; 7(suppl 1):S2–S12.
Heller JC, Prochazka AV, Everson GT, Forman LM. Long-term management after liver transplantation: primary care physician versus hepatologist. Liver Transpl 2009; 15:1330–1335.
McAlister VC, Haddad E, Renouf E, Malthaner RA, Kjaer MS, Gluud LL. Cyclosporin versus tacrolimus as primary immunosuppressant after liver transplantation: a meta-analysis. Am J Transplant 2006; 6:1578–1585.
Neuhaus P, Klupp J, Langrehr JM. mTOR inhibitors: an overview. Liver Transpl 2001; 7:473–484.
Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem 1998; 31:335–340.
Asrani SK, Wiesner RH, Trotter JF, et al. De novo sirolimus and reduced-dose tacrolimus versus standard-dose tacrolimus after liver transplantation: the 2000-2003 phase II prospective randomized trial. Am J Transplant 2014; 14:356–366.
Montalbano M, Neff GW, Yamashiki N, et al. A retrospective review of liver transplant patients treated with sirolimus from a single center: an analysis of sirolimus-related complications. Transplantation 2004; 78:264–268.
Everson GT. Everolimus and mTOR inhibitors in liver transplantation: opening the “box.” Liver Transpl 2006; 12:1571–1573.
Levy G, Schmidli H, Punch J, et al. Safety, tolerability, and efficacy of everolimus in de novo liver transplant recipients: 12- and 36-month results. Liver Transpl 2006; 12:1640–1648.
Toniutto P, Fabris C, Fumolo E, et al. Prevalence and risk factors for delayed adrenal insufficiency after liver transplantation. Liver Transpl 2008; 14:1014–1019.
Klintmalm GB, Davis GL, Teperman L, et al. A randomized, multicenter study comparing steroid-free immunosuppression and standard immunosuppression for liver transplant recipients with chronic hepatitis C. Liver Transpl 2011; 17:1394–1403.
Llado L, Fabregat J, Castellote J, et al. Impact of immunosuppression without steroids on rejection and hepatitis C virus evolution after liver transplantation: results of a prospective randomized study. Liver Transpl 2008; 14:1752–1760.
Lake JR. Immunosuppression and outcomes of patients transplanted for hepatitis C. J Hepatol 2006; 44:627–629.
Sohn AJ, Jeon H, Ahn J. Primary care of the liver transplant recipient. Prim Care 2011; 38:499–514.
Laish I, Braun M, Mor E, Sulkes J, Harif Y, Ben Ari Z. Metabolic syndrome in liver transplant recipients: prevalence, risk factors, and association with cardiovascular events. Liver Transpl 2011; 17:15–22.
Stegall MD, Everson G, Schroter G, Bilir B, Karrer F, Kam I. Metabolic complications after liver transplantation. Diabetes, hypercholesterolemia, hypertension, and obesity. Transplantation 1995; 60:1057–1060.
Textor SC, Canzanello VJ, Taler SJ, et al. Cyclosporine-induced hypertension after transplantation. Mayo Clin Proc 1994; 69:1182–1193.
Prevention, detection, evaluation, and treatment of hypertension. The Sixth Report of the Joint National Committee. National Institutes of Health-National Heart, Lung, and Blood Institute. National High Blood Pressure Education Programme. Indian Heart J 1999; 51:381–396.
Frishman WH. Calcium channel blockers: differences between subclasses. Am J Cardiovasc Drugs 2007; 7(suppl 1):17–23.
Galioto A, Semplicini A, Zanus G, et al. Nifedipine versus carvedilol in the treatment of de novo arterial hypertension after liver transplantation: results of a controlled clinical trial. Liver Transpl 2008; 14:1020–1028.
Neal DA, Gimson AE, Gibbs P, Alexander GJ. Beneficial effects of converting liver transplant recipients from cyclosporine to tacrolimus on blood pressure, serum lipids, and weight. Liver Transpl 2001; 7:533–539.
Singh S, Watt KD. Long-term medical management of the liver transplant recipient: what the primary care physician needs to know. Mayo Clin Proc 2012; 87:779–790.
Bianchi G, Marchesini G, Marzocchi R, Pinna AD, Zoli M. Metabolic syndrome in liver transplantation: relation to etiology and immunosuppression. Liver Transpl 2008; 14:1648–1654.
Lane JT, Dagogo-Jack S. Approach to the patient with new-onset diabetes after transplant (NODAT). J Clin Endocrinol Metab 2011; 96:3289–3297.
Wilkinson A, Davidson J, Dotta F, et al. Guidelines for the treatment and management of new-onset diabetes after transplantation. Clin Transplant 2005; 19:291–298.
Moon JI, Barbeito R, Faradji RN, Gaynor JJ, Tzakis AG. Negative impact of new-onset diabetes mellitus on patient and graft survival after liver transplantation: long-term follow up. Transplantation 2006; 82:1625–1628.
American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63.
Marchetti P. New-onset diabetes after liver transplantation: from pathogenesis to management. Liver Transpl 2005; 11:612–620.
Stone NJ, Robinson J, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl 2):S1–S45.
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
Gisbert C, Prieto M, Berenguer M, et al. Hyperlipidemia in liver transplant recipients: prevalence and risk factors. Liver Transpl Surg 1997; 3:416–422.
Asberg A. Interactions between cyclosporin and lipid-lowering drugs: implications for organ transplant recipients. Drugs 2003; 63:367–378.
Bullingham RE, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet 1998; 34:429–455.
Everhart JE, Lombardero M, Lake JR, Wiesner RH, Zetterman RK, Hoofnagle JH. Weight change and obesity after liver transplantation: incidence and risk factors. Liver Transpl Surg 1998; 4:285–296.
Canzanello VJ, Schwartz L, Taler SJ, et al. Evolution of cardiovascular risk after liver transplantation: a comparison of cyclosporine A and tacrolimus (FK506). Liver Transpl Surg 1997; 3:1–9.
Al-Nowaylati AR, Al-Haddad BJ, Dorman RB, et al. Gastric bypass after liver transplantation. Liver Transpl 2013; 19:1324–1329.
Heimbach JK, Watt KD, Poterucha JJ, et al. Combined liver transplantation and gastric sleeve resection for patients with medically complicated obesity and end-stage liver disease. Am J Transplant 2013; 13:363–368.
Watt KD, Charlton MR. Metabolic syndrome and liver transplantation: a review and guide to management. J Hepatol 2010; 53:199–206.
Pagadala M, Dasarathy S, Eghtesad B, McCullough AJ. Posttransplant metabolic syndrome: an epidemic waiting to happen. Liver Transpl 2009; 15:1662–1670.
Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009; 120:1640–1645.
Watt KD, Pedersen RA, Kremers WK, Heimbach JK, Charlton MR. Evolution of causes and risk factors for mortality post-liver transplant: results of the NIDDK long-term follow-up study. Am J Transplant 2010; 10:1420–1427.
Albeldawi M, Aggarwal A, Madhwal S, et al. Cumulative risk of cardiovascular events after orthotopic liver transplantation. Liver Transpl 2012; 18:370–375.
Gibbons RJ, Balady GJ, Bricker JT, et al. ACC/AHA 2002 guideline update for exercise testing: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1997 Exercise Testing Guidelines). Circulation 2002; 106:1883–1892.
Aberg F, Isoniemi H, Höckerstedt K. Long-term results of liver transplantation. Scand J Surg 2011; 100:14–21.
Aberg F, Pukkala E, Höckerstedt K, Sankila R, Isoniemi H. Risk of malignant neoplasms after liver transplantation: a population-based study. Liver Transpl 2008; 14:1428–1436.
Mells G, Neuberger J. Long-term care of the liver allograft recipient. Semin Liver Dis 2009; 29:102–120.
Herrero JI. De novo malignancies following liver transplantation: impact and recommendations. Liver Transpl 2009; 15(suppl 2):S90–S94.
Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ transplantation. N Engl J Med 2003; 348:1681–1691.
Euvrard S, Morelon E, Rostaing L, et al; TUMORAPA Study Group. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med 2012; 367:329–339.
Salgo R, Gossmann J, Schofer H, et al. Switch to a sirolimus-based immunosuppression in long-term renal transplant recipients: reduced rate of (pre-)malignancies and nonmelanoma skin cancer in a prospective, randomized, assessor-blinded, controlled clinical trial. Am J Transplant 2010; 10:1385–1393.
Narumi S, Roberts JP, Emond JC, Lake J, Ascher NL. Liver transplantation for sclerosing cholangitis. Hepatology 1995; 22:451–457.
Knechtle SJ, D’Alessandro AM, Harms BA, Pirsch JD, Belzer FO, Kalayoglu M. Relationships between sclerosing cholangitis, inflammatory bowel disease, and cancer in patients undergoing liver transplantation. Surgery 1995; 118:615–620.
Finkenstedt A, Graziadei IW, Oberaigner W, et al. Extensive surveillance promotes early diagnosis and improved survival of de novo malignancies in liver transplant recipients. Am J Transplant 2009; 9:2355–2361.
Fisher NC, Nightingale PG, Gunson BK, Lipkin GW, Neuberger JM. Chronic renal failure following liver transplantation: a retrospective analysis. Transplantation 1998; 66:59–66.
Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
Klintmalm GB, Gonwa TA. Nephrotoxicity associated with cyclosporine and FK506. Liver Transpl Surg 1995; 1:11–19.
A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression in liver transplantation. The US Multicenter FK506 Liver Study Group. N Engl J Med 1994; 331:1110–1115.
Neff GW, Montalbano M, Slapak-Green G, et al. Sirolimus therapy in orthotopic liver transplant recipients with calcineurin inhibitor related chronic renal insufficiency. Transplant Proc 2003; 35:3029–3031.
Cotterell AH, Fisher RA, King AL, et al. Calcineurin inhibitor-induced chronic nephrotoxicity in liver transplant patients is reversible using rapamycin as the primary immunosuppressive agent. Clin Transplant 2002; 16(suppl 7):49–51.
Manzia TM, De Liguori Carino N, Orlando G, et al. Use of mycophenolate mofetil in liver transplantation: a literature review. Transplant Proc 2005; 37:2616–2617.
Schlitt HJ, Barkmann A, Boker KH, et al. Replacement of calcineurin inhibitors with mycophenolate mofetil in liver-transplant patients with renal dysfunction: a randomised controlled study. Lancet 2001; 357:587–591.
Hodge EE, Reich DJ, Clavien PA, Kim-Schluger L. Use of mycophenolate mofetil in liver transplant recipients experiencing renal dysfunction on cyclosporine or tacrolimus-randomized, prospective, multicenter study results. Transplant Proc 2002; 34:1546–1547.
Hamburg SM, Piers DA, van den Berg AP, Slooff MJ, Haagsma EB. Bone mineral density in the long term after liver transplantation. Osteoporos Int 2000; 11:600–606.
Maalouf NM, Shane E. Osteoporosis after solid organ transplantation. J Clin Endocrinol Metab 2005; 90:2456–2465.
Crosbie OM, Freaney R, McKenna MJ, Curry MP, Hegarty JE. Predicting bone loss following orthotopic liver transplantation. Gut 1999; 44:430–434.
Giannini S, Nobile M, Ciuffreda M, et al. Long-term persistence of low bone density in orthotopic liver transplantation. Osteoporos Int 2000; 11:417–424.
Monegal A, Navasa M, Guanabens N, et al. Bone disease after liver transplantation: a long-term prospective study of bone mass changes, hormonal status and histomorphometric characteristics. Osteoporos Int 2001; 12:484–492.
Neuhaus R, Lohmann R, Platz KP, et al. Treatment of osteoporosis after liver transplantation. Transplant Proc 1995; 27:1226–1227.
Valero MA, Loinaz C, Larrodera L, Leon M, Moreno E, Hawkins F. Calcitonin and bisphosphonates treatment in bone loss after liver transplantation. Calcif Tissue Int 1995; 57:15–19.
Neal DA, Tom BD, Gimson AE, Gibbs P, Alexander GJ. Hyperuricemia, gout, and renal function after liver transplantation. Transplantation 2001; 72:1689–1691.
Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver. 10th ed. Philadephia, PA: Lippincott Williams & Wilkins; 2007.
Tombazzi CR, Waters B, Shokouh-Amiri MH, Vera SR, Riely CA. Neuropsychiatric complications after liver transplantation: role of immunosuppression and hepatitis C. Dig Dis Sci 2006; 51:1079–1081.
Bravata DM, Olkin I, Barnato AE, Keeffe EB, Owens DK. Health-related quality of life after liver transplantation: a meta-analysis. Liver Transpl Surg 1999; 5:318–331.
Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
Chesi C, Gunther M, Huzly D, et al. Immunization of liver and renal transplant recipients: a seroepidemiological and sociodemographic survey. Transpl Infect Dis 2009; 11:507–512.
Berenguer M, Lopez-Labrador FX, Wright TL. Hepatitis C and liver transplantation. J Hepatol 2001; 35:666–678.
Forman LM, Lewis JD, Berlin JA, Feldman HI, Lucey MR. The association between hepatitis C infection and survival after orthotopic liver transplantation. Gastroenterology 2002; 122:889–896.
Benten D, Staufer K, Sterneck M. Orthotopic liver transplantation and what to do during follow-up: recommendations for the practitioner. Nat Clin Pract Gastroenterol Hepatol 2009; 6:23–36.
Kotlyar DS, Campbell MS, Reddy KR. Recurrence of diseases following orthotopic liver transplantation. Am J Gastroenterol 2006; 101:1370–1378.
References
Starzl TE, Marchioro TL, Vonkaulla KN, Hermann G, Brittain RS, Waddell WR. Homotransplantation of the liver in humans. Surg Gynecol Obstet 1963; 117:659–676.
Matas AJ, Smith JM, Skeans MA, et al. OPTN/SRTR 2011 annual data report: kidney. Am J Transplant 2013; 13(suppl 1):11–46.
McCashland TM. Posttransplantation care: role of the primary care physician versus transplant center. Liver Transpl 2001; 7(suppl 1):S2–S12.
Heller JC, Prochazka AV, Everson GT, Forman LM. Long-term management after liver transplantation: primary care physician versus hepatologist. Liver Transpl 2009; 15:1330–1335.
McAlister VC, Haddad E, Renouf E, Malthaner RA, Kjaer MS, Gluud LL. Cyclosporin versus tacrolimus as primary immunosuppressant after liver transplantation: a meta-analysis. Am J Transplant 2006; 6:1578–1585.
Neuhaus P, Klupp J, Langrehr JM. mTOR inhibitors: an overview. Liver Transpl 2001; 7:473–484.
Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem 1998; 31:335–340.
Asrani SK, Wiesner RH, Trotter JF, et al. De novo sirolimus and reduced-dose tacrolimus versus standard-dose tacrolimus after liver transplantation: the 2000-2003 phase II prospective randomized trial. Am J Transplant 2014; 14:356–366.
Montalbano M, Neff GW, Yamashiki N, et al. A retrospective review of liver transplant patients treated with sirolimus from a single center: an analysis of sirolimus-related complications. Transplantation 2004; 78:264–268.
Everson GT. Everolimus and mTOR inhibitors in liver transplantation: opening the “box.” Liver Transpl 2006; 12:1571–1573.
Levy G, Schmidli H, Punch J, et al. Safety, tolerability, and efficacy of everolimus in de novo liver transplant recipients: 12- and 36-month results. Liver Transpl 2006; 12:1640–1648.
Toniutto P, Fabris C, Fumolo E, et al. Prevalence and risk factors for delayed adrenal insufficiency after liver transplantation. Liver Transpl 2008; 14:1014–1019.
Klintmalm GB, Davis GL, Teperman L, et al. A randomized, multicenter study comparing steroid-free immunosuppression and standard immunosuppression for liver transplant recipients with chronic hepatitis C. Liver Transpl 2011; 17:1394–1403.
Llado L, Fabregat J, Castellote J, et al. Impact of immunosuppression without steroids on rejection and hepatitis C virus evolution after liver transplantation: results of a prospective randomized study. Liver Transpl 2008; 14:1752–1760.
Lake JR. Immunosuppression and outcomes of patients transplanted for hepatitis C. J Hepatol 2006; 44:627–629.
Sohn AJ, Jeon H, Ahn J. Primary care of the liver transplant recipient. Prim Care 2011; 38:499–514.
Laish I, Braun M, Mor E, Sulkes J, Harif Y, Ben Ari Z. Metabolic syndrome in liver transplant recipients: prevalence, risk factors, and association with cardiovascular events. Liver Transpl 2011; 17:15–22.
Stegall MD, Everson G, Schroter G, Bilir B, Karrer F, Kam I. Metabolic complications after liver transplantation. Diabetes, hypercholesterolemia, hypertension, and obesity. Transplantation 1995; 60:1057–1060.
Textor SC, Canzanello VJ, Taler SJ, et al. Cyclosporine-induced hypertension after transplantation. Mayo Clin Proc 1994; 69:1182–1193.
Prevention, detection, evaluation, and treatment of hypertension. The Sixth Report of the Joint National Committee. National Institutes of Health-National Heart, Lung, and Blood Institute. National High Blood Pressure Education Programme. Indian Heart J 1999; 51:381–396.
Frishman WH. Calcium channel blockers: differences between subclasses. Am J Cardiovasc Drugs 2007; 7(suppl 1):17–23.
Galioto A, Semplicini A, Zanus G, et al. Nifedipine versus carvedilol in the treatment of de novo arterial hypertension after liver transplantation: results of a controlled clinical trial. Liver Transpl 2008; 14:1020–1028.
Neal DA, Gimson AE, Gibbs P, Alexander GJ. Beneficial effects of converting liver transplant recipients from cyclosporine to tacrolimus on blood pressure, serum lipids, and weight. Liver Transpl 2001; 7:533–539.
Singh S, Watt KD. Long-term medical management of the liver transplant recipient: what the primary care physician needs to know. Mayo Clin Proc 2012; 87:779–790.
Bianchi G, Marchesini G, Marzocchi R, Pinna AD, Zoli M. Metabolic syndrome in liver transplantation: relation to etiology and immunosuppression. Liver Transpl 2008; 14:1648–1654.
Lane JT, Dagogo-Jack S. Approach to the patient with new-onset diabetes after transplant (NODAT). J Clin Endocrinol Metab 2011; 96:3289–3297.
Wilkinson A, Davidson J, Dotta F, et al. Guidelines for the treatment and management of new-onset diabetes after transplantation. Clin Transplant 2005; 19:291–298.
Moon JI, Barbeito R, Faradji RN, Gaynor JJ, Tzakis AG. Negative impact of new-onset diabetes mellitus on patient and graft survival after liver transplantation: long-term follow up. Transplantation 2006; 82:1625–1628.
American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63.
Marchetti P. New-onset diabetes after liver transplantation: from pathogenesis to management. Liver Transpl 2005; 11:612–620.
Stone NJ, Robinson J, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl 2):S1–S45.
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
Gisbert C, Prieto M, Berenguer M, et al. Hyperlipidemia in liver transplant recipients: prevalence and risk factors. Liver Transpl Surg 1997; 3:416–422.
Asberg A. Interactions between cyclosporin and lipid-lowering drugs: implications for organ transplant recipients. Drugs 2003; 63:367–378.
Bullingham RE, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet 1998; 34:429–455.
Everhart JE, Lombardero M, Lake JR, Wiesner RH, Zetterman RK, Hoofnagle JH. Weight change and obesity after liver transplantation: incidence and risk factors. Liver Transpl Surg 1998; 4:285–296.
Canzanello VJ, Schwartz L, Taler SJ, et al. Evolution of cardiovascular risk after liver transplantation: a comparison of cyclosporine A and tacrolimus (FK506). Liver Transpl Surg 1997; 3:1–9.
Al-Nowaylati AR, Al-Haddad BJ, Dorman RB, et al. Gastric bypass after liver transplantation. Liver Transpl 2013; 19:1324–1329.
Heimbach JK, Watt KD, Poterucha JJ, et al. Combined liver transplantation and gastric sleeve resection for patients with medically complicated obesity and end-stage liver disease. Am J Transplant 2013; 13:363–368.
Watt KD, Charlton MR. Metabolic syndrome and liver transplantation: a review and guide to management. J Hepatol 2010; 53:199–206.
Pagadala M, Dasarathy S, Eghtesad B, McCullough AJ. Posttransplant metabolic syndrome: an epidemic waiting to happen. Liver Transpl 2009; 15:1662–1670.
Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009; 120:1640–1645.
Watt KD, Pedersen RA, Kremers WK, Heimbach JK, Charlton MR. Evolution of causes and risk factors for mortality post-liver transplant: results of the NIDDK long-term follow-up study. Am J Transplant 2010; 10:1420–1427.
Albeldawi M, Aggarwal A, Madhwal S, et al. Cumulative risk of cardiovascular events after orthotopic liver transplantation. Liver Transpl 2012; 18:370–375.
Gibbons RJ, Balady GJ, Bricker JT, et al. ACC/AHA 2002 guideline update for exercise testing: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1997 Exercise Testing Guidelines). Circulation 2002; 106:1883–1892.
Aberg F, Isoniemi H, Höckerstedt K. Long-term results of liver transplantation. Scand J Surg 2011; 100:14–21.
Aberg F, Pukkala E, Höckerstedt K, Sankila R, Isoniemi H. Risk of malignant neoplasms after liver transplantation: a population-based study. Liver Transpl 2008; 14:1428–1436.
Mells G, Neuberger J. Long-term care of the liver allograft recipient. Semin Liver Dis 2009; 29:102–120.
Herrero JI. De novo malignancies following liver transplantation: impact and recommendations. Liver Transpl 2009; 15(suppl 2):S90–S94.
Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ transplantation. N Engl J Med 2003; 348:1681–1691.
Euvrard S, Morelon E, Rostaing L, et al; TUMORAPA Study Group. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med 2012; 367:329–339.
Salgo R, Gossmann J, Schofer H, et al. Switch to a sirolimus-based immunosuppression in long-term renal transplant recipients: reduced rate of (pre-)malignancies and nonmelanoma skin cancer in a prospective, randomized, assessor-blinded, controlled clinical trial. Am J Transplant 2010; 10:1385–1393.
Narumi S, Roberts JP, Emond JC, Lake J, Ascher NL. Liver transplantation for sclerosing cholangitis. Hepatology 1995; 22:451–457.
Knechtle SJ, D’Alessandro AM, Harms BA, Pirsch JD, Belzer FO, Kalayoglu M. Relationships between sclerosing cholangitis, inflammatory bowel disease, and cancer in patients undergoing liver transplantation. Surgery 1995; 118:615–620.
Finkenstedt A, Graziadei IW, Oberaigner W, et al. Extensive surveillance promotes early diagnosis and improved survival of de novo malignancies in liver transplant recipients. Am J Transplant 2009; 9:2355–2361.
Fisher NC, Nightingale PG, Gunson BK, Lipkin GW, Neuberger JM. Chronic renal failure following liver transplantation: a retrospective analysis. Transplantation 1998; 66:59–66.
Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
Klintmalm GB, Gonwa TA. Nephrotoxicity associated with cyclosporine and FK506. Liver Transpl Surg 1995; 1:11–19.
A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression in liver transplantation. The US Multicenter FK506 Liver Study Group. N Engl J Med 1994; 331:1110–1115.
Neff GW, Montalbano M, Slapak-Green G, et al. Sirolimus therapy in orthotopic liver transplant recipients with calcineurin inhibitor related chronic renal insufficiency. Transplant Proc 2003; 35:3029–3031.
Cotterell AH, Fisher RA, King AL, et al. Calcineurin inhibitor-induced chronic nephrotoxicity in liver transplant patients is reversible using rapamycin as the primary immunosuppressive agent. Clin Transplant 2002; 16(suppl 7):49–51.
Manzia TM, De Liguori Carino N, Orlando G, et al. Use of mycophenolate mofetil in liver transplantation: a literature review. Transplant Proc 2005; 37:2616–2617.
Schlitt HJ, Barkmann A, Boker KH, et al. Replacement of calcineurin inhibitors with mycophenolate mofetil in liver-transplant patients with renal dysfunction: a randomised controlled study. Lancet 2001; 357:587–591.
Hodge EE, Reich DJ, Clavien PA, Kim-Schluger L. Use of mycophenolate mofetil in liver transplant recipients experiencing renal dysfunction on cyclosporine or tacrolimus-randomized, prospective, multicenter study results. Transplant Proc 2002; 34:1546–1547.
Hamburg SM, Piers DA, van den Berg AP, Slooff MJ, Haagsma EB. Bone mineral density in the long term after liver transplantation. Osteoporos Int 2000; 11:600–606.
Maalouf NM, Shane E. Osteoporosis after solid organ transplantation. J Clin Endocrinol Metab 2005; 90:2456–2465.
Crosbie OM, Freaney R, McKenna MJ, Curry MP, Hegarty JE. Predicting bone loss following orthotopic liver transplantation. Gut 1999; 44:430–434.
Giannini S, Nobile M, Ciuffreda M, et al. Long-term persistence of low bone density in orthotopic liver transplantation. Osteoporos Int 2000; 11:417–424.
Monegal A, Navasa M, Guanabens N, et al. Bone disease after liver transplantation: a long-term prospective study of bone mass changes, hormonal status and histomorphometric characteristics. Osteoporos Int 2001; 12:484–492.
Neuhaus R, Lohmann R, Platz KP, et al. Treatment of osteoporosis after liver transplantation. Transplant Proc 1995; 27:1226–1227.
Valero MA, Loinaz C, Larrodera L, Leon M, Moreno E, Hawkins F. Calcitonin and bisphosphonates treatment in bone loss after liver transplantation. Calcif Tissue Int 1995; 57:15–19.
Neal DA, Tom BD, Gimson AE, Gibbs P, Alexander GJ. Hyperuricemia, gout, and renal function after liver transplantation. Transplantation 2001; 72:1689–1691.
Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver. 10th ed. Philadephia, PA: Lippincott Williams & Wilkins; 2007.
Tombazzi CR, Waters B, Shokouh-Amiri MH, Vera SR, Riely CA. Neuropsychiatric complications after liver transplantation: role of immunosuppression and hepatitis C. Dig Dis Sci 2006; 51:1079–1081.
Bravata DM, Olkin I, Barnato AE, Keeffe EB, Owens DK. Health-related quality of life after liver transplantation: a meta-analysis. Liver Transpl Surg 1999; 5:318–331.
Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
Chesi C, Gunther M, Huzly D, et al. Immunization of liver and renal transplant recipients: a seroepidemiological and sociodemographic survey. Transpl Infect Dis 2009; 11:507–512.
Berenguer M, Lopez-Labrador FX, Wright TL. Hepatitis C and liver transplantation. J Hepatol 2001; 35:666–678.
Forman LM, Lewis JD, Berlin JA, Feldman HI, Lucey MR. The association between hepatitis C infection and survival after orthotopic liver transplantation. Gastroenterology 2002; 122:889–896.
Benten D, Staufer K, Sterneck M. Orthotopic liver transplantation and what to do during follow-up: recommendations for the practitioner. Nat Clin Pract Gastroenterol Hepatol 2009; 6:23–36.
Kotlyar DS, Campbell MS, Reddy KR. Recurrence of diseases following orthotopic liver transplantation. Am J Gastroenterol 2006; 101:1370–1378.
Tacrolimus and cyclosporine are the most commonly used immunosuppressive agents in liver transplant recipients. Adverse effects include hypertension, hypercholesterolemia, diabetes (more common with tacrolimus), renal insufficiency, and osteoporosis.
Hypertension affects 40% to 85% of liver transplant patients. Dihydropyridine calcium channel blockers (eg, amlodipine, nifedipine) are the first-line agents.
Cardiovascular disease is the third most common cause of death after liver transplantation. Modifying risk factors is essential.
Skin cancers account for 40% of all cancers after liver transplantation. Intensive screening is required and has been proven to lower the risk of death.
Any time patients enter or leave the hospital, they risk being harmed by errors in their medications.1 Adverse events from medication errors during transitions of care are common but often preventable. One key approach is to systematically review every patient’s medication list on admission and discharge and resolve any discrepancies. These transitions are also an opportunity to address other medication-related problems, such as adherence, drug interactions, and clinical appropriateness.
This article summarizes the types and prevalence of medication problems that occur during hospital-based transitions of care, and suggests strategies to decrease the risk of medication errors, focusing on medication reconciliation and related interventions that clinicians can use at the bedside to improve medication safety.
DEFINING TERMS
A medication discrepancy is any variance noted in a patient’s documented medication regimen across different medication lists or sites of care. While some differences reflect intentional and clinically appropriate changes to the regimen, others are unintentional and reflect inaccurate or incomplete information. These unintentional discrepancies are medication errors.
Depending on the clinical circumstances and medications involved, such errors may lead to an adverse drug event (ADE), defined as actual harm or injury resulting from a medication. Sometimes a medication error does not cause harm immediately but could if left uncorrected; this is called a potential ADE.
An important goal during transitions of care is to reduce unintentional medication discrepancies, thereby reducing potential and actual ADEs.
ERRORS ARISE AT DISCHARGE—AND EVEN MORE AT ADMISSION
Hospital discharge is a widely recognized transition in which patient harm occurs. As many as 70% of patients may have an unintentional medication discrepancy at hospital discharge, with many of those discrepancies having potential for harm.2 Indeed, during the first few weeks after discharge, 50% of patients have a clinically important medication error,3 and 20% experience an adverse event, most commonly an ADE.4 ADEs are associated with excess health care utilization,5–7 and many are preventable through strategies such as medication reconciliation.5,8
Importantly, more errors arise at hospital admission than at other times.9,10
Errors in medication histories are the most common source of discrepancies, affecting up to two-thirds of admitted patients.11,12 More than one-quarter of hospital prescribing errors can be attributed to incomplete medication histories at the time of admission,13 and nearly three times as many clinically important medication discrepancies are related to history-taking errors on admission rather than reconciliation errors at discharge.9
Most discrepancies occurring at the time of admission have the potential to cause harm, particularly if the errors persist beyond discharge.14 Therefore, taking a complete and accurate medication history on hospital admission is critical to ensuring safe care transitions.
MEDICATION RECONCILIATION: BARRIERS AND FACILITATORS
Medication reconciliation is a strategy for reducing medication discrepancies in patients moving across care settings. Simply put, it is the process by which a patient’s medication list is obtained, compared, and clarified across different sites of care.15 It has consistently been shown to decrease medication errors compared with usual care,16 and it is strongly supported by national and international organizations.17–21
In clinical practice, many physicians and institutions have found medication reconciliation difficult to implement, owing to barriers at the level of the patient, provider, and system (Table 1). In response to these challenges, two initiatives have synthesized best practices and offer toolkits that hospitals and clinicians can use: the Medications at Transitions and Clinical Handoffs (MATCH) program22 and the Multi-Center Medication Reconciliation Quality Improvement Study (MARQUIS).23
Lack of resources is a widely acknowledged challenge. Thus, the MARQUIS investigators23 suggested focusing on the admission history, where most errors occur, and applying the most resource-intensive interventions in patients at highest risk of ADEs, ie, those who are elderly, have multiple comorbid conditions, or take numerous medications.16
Although the risk of an ADE increases with the number of medications a patient takes,4 the exact number of drugs that defines high risk has not been well established. Targeting patients who take 10 or more maintenance medications is a reasonable initial approach,24 but institutions should tailor risk stratification to their patient populations and available resources. Patients taking high-risk medications such as anticoagulants and insulin could also be prioritized for review, since these medications are more likely to cause serious patient harm when used without appropriate clinical oversight.7
Using pharmacy staff to perform medication history-taking, reconciliation, and patient counseling has been shown to produce favorable patient outcomes, particularly for higher-risk patients.16,23
The MARQUIS investigators found they could boost the chances of success by sharing stories of patient harm to foster “buy-in” among frontline staff, providing formal training to clinicians on how to take a medication history, and obtaining the support of nursing leaders to champion improvement efforts.23
Additionally, patients should be empowered to maintain an accurate medication list. We address strategies for improving patient engagement and adherence in a later section.
BEST PRACTICES FOR IMPROVING MEDICATION SAFETY
Medication reconciliation is one of several measures necessary to optimize medication safety during transitions of care. It typically includes the following actions:
Interview the patient or caregiver to determine the list of medications the patient is currently taking (or supposed to be taking); consult other sources if needed.
List medications that are being ordered during the clinical encounter.
Compare these two lists, making note of medications that are stopped, changed, or newly prescribed.
Resolve any discrepancies.
Communicate the reconciled list to the patient, appropriate caregivers, and providers of follow-up care.
At a rudimentary level, medication reconciliation encompasses medication list management along the continuum of care. However, we recommend leveraging medication reconciliation as an opportunity to further enhance medication safety by reviewing the appropriateness of each medication, seizing opportunities to streamline or simplify the regimen, assessing patient and caregiver understanding of medication instructions and potential ADEs, and delivering appropriate counseling to enhance medication use. Table 2 outlines our framework for medication management during hospital-based transitions.
STEP 1: OBTAIN A COMPLETE PREADMISSION MEDICATION LIST
The “best-possible medication history” is obtained in a systematic process of interviewing the patient or caregiver plus reviewing at least one other reliable source.23 The resulting list should include all medications the patient is taking (prescription and nonprescription), doses, directions for use, formulations if applicable, indications, start and stop dates, and medication allergies and reactions.
Review existing information. Before eliciting a history from the patient, review his or her recorded medical history and existing medication lists (eg, prior discharge summaries, records from other facilities, records from outpatient visits, pharmacy fill data). This will provide context about the regimen and help identify issues and questions that can be addressed during the history-taking.
Ask open-ended questions. Instead of just asking the patient to confirm the accuracy of the existing medication list, we recommend actively obtaining the full medication list from the patient or caregiver. The conversation should begin with an open-ended question such as, “What medications do you take at home?” This approach will also allow the clinician to gauge the patient’s level of understanding of each medication’s indication and dosing instructions. Using a series of prompts such as those recommended in Table 3 will elicit a best-possible medical history, while verifying all of the medications on the existing list.
Clarify discrepancies. Resolve differences between the existing medication lists and the patient’s or caregiver’s report during the preadmission interview. Examples include errors of omission (a medication is missing), errors of commission (an additional medication is present), and discrepancies in the strength, formulation, dosing instructions, and indications for the drugs. If necessary, other sources of information should be consulted, such as the patient’s medication bottles, pharmacy or pharmacies, primary care physician, and a family member or caregiver.
Assess adherence. The extent to which patients take their medications as directed is an important component of the history, but is often left out. Medication nonadherence rates in the United States are 40% to 70%,25 contributing to poor patient outcomes and imposing extraordinary costs on the health care system.26
Asking open-ended, nonjudgmental questions at the time of hospital admission will help to uncover medication-taking behaviors as well as barriers to adherence (Table 3). The patient’s responses should be taken into account when determining the treatment plan.
Document your findings. After completing the medication history and clarifying discrepancies, document the preadmission list in the medical record. All members of the health care team should have access to view and update the same list, as new information about the preadmission medications may be uncovered after the initial history.
Make clinical decisions. Complete the admission medication reconciliation by deciding whether each medication on the list should be continued, changed, held, or discontinued on the basis of the patient’s clinical condition. Well-designed information technology applications enable the provider to document each action and the rationale for it, as well as carry that information into the order-entry system. Medications marked as held or discontinued on admission should be revisited as the patient’s clinical condition changes and at discharge.
STEP 2: AVOID RECONCILIATION ERRORS
Reconciliation errors reflect discrepancies between the medication history and the medications that are ordered after admission.
Reconciliation errors are less common than medication history errors, accounting for approximately one-third of potentially harmful medication errors in hospitalized medical patients.9 These include errors of omission (a medication is omitted from the orders), errors of commission (a medication is prescribed with no indication for continuation), and therapeutic duplication.
Preventing errors of omission
Medications are often held at transition points for appropriate clinical reasons. Examples include holding anticoagulants and antiplatelet agents in patients who have gastrointestinal bleeding or an upcoming procedure, antihypertensives in patients with hemodynamic instability, and other chronic medications in patients with an acute illness.
Poor documentation and communication of these decisions can lead to a failure to resume the medications—an error of omission—at hospital discharge.
Hospitalized patients are at risk of unintentional discontinuation of their chronic medications, including antiplatelet drugs, anticoagulants, statins, and thyroid replacement, particularly if admitted to the intensive care unit.12 These errors can be minimized by a standardized medication reconciliation process at each transition and clear documentation of the medication plan.
Communication among providers can be improved if the admitting clinician documents clearly whether each preadmission medication is being continued, changed, or stopped, along with the reason for doing so, and makes this information available throughout the hospital stay. Upon transfer to another unit and at discharge, the physician should review each; preadmission medication that was held and the patient’s current clinical status and, based on that information, decide whether medications that were held should be resumed. If a medication will be restarted later, specific instructions should be documented and communicated to the patient and the physicians who are taking over his or her care.
Preventing errors of commission
Failure to perform a complete reconciliation at each transition of care and match each medication with an appropriate indication can lead to errors of commission.
One study showed that 44% of patients were prescribed at least one unnecessary drug at hospital discharge, one-fourth of which were started during the hospitalization.27 Commonly prescribed unnecessarily were gastrointestinal agents, central nervous system drugs, nutrients, and supplements.
It is critical to assess each medication’s ongoing need, appropriateness, and risk-benefit ratio at every transition. Medications no longer indicated should be discontinued in order to simplify the regimen, avoid unnecessary drug exposure, and prevent ADEs.
For example, proton pump inhibitors or histamine 2 receptor blockers are often started in the hospital for stress ulcer prophylaxis. One-third of patients are then discharged home on the medication, and 6 months later half of those patients are still taking the unnecessary drug.28 This situation can be avoided by limiting use of these medications to appropriate circumstances, clearly marking the indication as stress ulcer prophylaxis (as opposed to an ongoing condition that will require continuing it after discharge), and discontinuing the agent when appropriate.
All drugs, even common and seemingly benign ones, carry some risk and should be discontinued when no longer needed. Thus, medications added during the hospitalization to control acute symptoms should also be reviewed at each transition to prevent inappropriate continuation when symptoms have resolved.
One study, for example, found that many patients were discharged with inappropriate prescriptions for atypical antipsychotics after receiving them in the intensive care unit, likely for delirium.29 Documenting that an acute issue such as delirium has resolved should prompt the discontinuation of therapy.
Preventing therapeutic duplication
Therapeutic duplication occurs in about 8% of discharges.1 These errors often result from formulary substitutions or altering the dosage form in the acute setting. For example, patients who receive a prescription for the substituted agent at discharge and also resume their prehospitalization medications end up with duplicate therapy.
Therapeutic substitution is common at the time of admission to the hospital as a result of formulary restrictions. Drug classes that are frequently substituted include statins, antihypertensives, urinary antispasmodics, and proton pump inhibitors. Physicians should be familiar with the preferred agents on the hospital formulary and make careful note when a substitution occurs. Furthermore, hospital systems should be developed to remind the physician to switch back to the outpatient medication at discharge.
Similar problems occur when home medications are replaced with different dosage forms with different pharmacokinetic properties. For example, a long-acting medication may be temporarily replaced with an intravenous solution or immediate-release tablet for several reasons, including nothing-by-mouth status, unstable clinical condition, need for titration, and need to crush the tablet to give the drug per tube. The differing formulations must be reconciled throughout the patient’s hospital course and at discharge to avoid therapeutic duplication and serious medication errors. Deliberate changes to the dosage form should be clearly communicated in the discharge medication list so that patients and other clinicians are aware.
Hospital systems should also have the capability to identify duplications in the medication list and to warn prescribers of these errors. The ability to group medications by drug class or sort the medication list alphabetically by generic name can help uncover duplication errors.
STEP 3: REVIEW THE LIST IN VIEW OF THE CLINICAL PICTURE
Transitions of care should prompt providers to review the medication list for possible drug-disease interactions, confirm compliance with evidence-based guidelines, and evaluate the risks and benefits of each medication in the context of the patient’s age and acute and chronic medical issues. This is also an opportunity to screen the full list for potentially inappropriate medications and high-alert drugs such as insulin or anticoagulants, which are more likely than other drugs to cause severe harm when used in error.
Acute kidney injury. New drug-disease interactions can arise during a hospitalization and can affect dosing and the choice of drug. The onset of acute kidney injury, for example, often necessitates adjusting or discontinuing nephrotoxic and renally excreted medications. ADEs or potential ADEs have been reported in 43% of hospitalized patients with acute kidney injury.30
Because acute kidney injury is often transient, medications may need to be held or adjusted several times until renal function stabilizes. This can be challenging across the continuum of care and requires close monitoring of the serum creatinine level and associated drug doses and levels, if applicable. Well-designed clinical decision support tools can integrate laboratory data and alert the prescriber to a clinically important increase or decrease in serum creatinine that may warrant a change in therapy. Modifications to the regimen and a plan for timely follow-up of the serum creatinine level should be clearly documented in the discharge plan.
Liver disease. Similar attention should be given to drugs that are hepatically metabolized if a patient has acute or chronic liver impairment.
Geriatric patients, particularly those who present with altered mental status, falls, or urinary retention, should have their medication list reviewed for potentially inappropriate medications, which are drugs that pose increased risk of poor outcomes in older adults.31,32 Patients and providers may have been willing to accept the risk of medications such as anticholinergics or sedative-hypnotics when the drugs were initiated, but circumstances can change over time, especially in this patient population. Hospitalization is a prime opportunity to screen for medications that meet the Beers criteria31 for agents to avoid or use with caution in older adults.
As-needed medications. Medications prescribed on an as-needed basis in the hospital should be reviewed for continuation or discontinuation at discharge. How often the medication was given can inform this decision.
For example, if as-needed opioids were used frequently, failure to develop a plan of care for pain can lead to persistent symptoms and, possibly, to readmission.33,34 Similar scenarios occur with use of as-needed blood pressure medications, laxatives, and correction-dose insulin.
If an as-needed medication was used consistently during hospitalization, the physician should consider whether a regularly scheduled medication is needed. Conversely, if the medication was not used during the inpatient admission, it can likely be discontinued.
STEP 4: PREPARE THE PATIENT AND FOLLOW-UP PROVIDER
Once a clinician has performed medication reconciliation, including obtaining a best-possible medical history and carefully reviewing the medication list and orders for errors and clinical appropriateness, the next steps are to ensure the patient understands what he or she needs to do and to confirm that suitable follow-up plans are in place. These measures should be taken at all transitions of care but are critically important at hospital discharge.
Preparing the patient and caregiver
An accurate, reconciled medication list should be given to the patient, caregiver, or both, and should be reviewed before discharge.17
Approximately one-third of Americans have low health literacy skills, so medication lists and associated materials should be easy to understand.35 Medication lists should be written in plain language and formatted for optimal readability (Table 4), clearly stating which medications to continue, change, hold temporarily, and stop.
Patients recall and comprehend about half of the information provided during a medical encounter.36 Thus, medication teaching should focus on key points including changes or additions to the regimen, specific instructions for follow-up and monitoring, and how to handle common and serious side effects.
To confirm patient understanding, clinicians should use “teach-back,” ie, provide the patient with information and then ask him or her to repeat back key points.37,38 The patient and family should also be encouraged to ask questions before discharge.
If not already addressed during the hospital stay, barriers to medication adherence and ability to obtain the medications should be attended to at this time (Table 5). Also, the plan to pick up the medications should be verified with the patient and caregiver. Verify that there is transportation to a particular pharmacy that is open at the time of discharge, and that the patient can afford the medications.
Ensuring appropriate follow-up
Studies have shown that timely in-home or telephone follow-up after discharge can decrease adverse events and postdischarge health care utilization.39,40 Telephone follow-up that includes thorough medication reconciliation can help detect and resolve medication issues early after discharge and can close gaps related to monitoring and follow-up.
Medication reconciliation by telephone can be time-consuming. Depending on the number of medications that need to be reviewed, calls can take between 10 and 60 minutes. Postdischarge phone calls should be performed by clinical personnel who are able to identify medication-related problems. A pharmacist should be an available resource to assist with complex regimens, to help resolve medication discrepancies, and to address patient concerns. Table 6 provides tips for conducting follow-up phone calls.
Resolving discrepancies identified during follow-up calls can be difficult, as changes to the medication regimen are often not communicated effectively to other members of the care team. Physicians should document the complete medication list and plan in the discharge summary, and there should be a method for the caller to record updates to the medication list in the medical record so that they are apparent at the outpatient follow-up visit.
An additional challenge is that it is frequently unclear which physician “owns” which medications. Therefore, designating a contact person for each medication until follow-up can be very valuable. At a minimum, a “physician owner” for high-alert medications such as insulin, anticoagulants, and diuretics should be identified to provide close follow-up, titration, and monitoring.
There should also be a plan for the patient to obtain refills of essential long-term medications, such as antiplatelet agents following stent placement.
SUMMARY AND RECOMMENDATIONS
Medication-related problems during hospital admission and discharge are common and range from minor discrepancies in the medication list to errors in history-taking, prescribing, and reconciliation that can lead to potential or actual patient harm. Putting systems in place to facilitate medication reconciliation can decrease the occurrence of medication discrepancies and ADEs, thereby improving patient safety during these critical transitions between care settings and providers. Institutional medication reconciliation programs should focus resources on the admission history-taking step, target the highest-risk patients for the most intensive interventions, and involve pharmacy personnel when possible.
On an individual level, clinicians can incorporate additional interventions into their workflows to optimize medication safety for hospitalized patients. Using a structured approach to obtain a complete and accurate medication list at the time of hospital admission will help providers identify medication-related problems and prevent the propagation of errors throughout the hospital stay and at discharge. Focusing additional time and effort on a comprehensive review of the medication list for errors of omission and commission, patient-specific needs, and high-alert drugs will further decrease the risk of medication errors. Finally, providing discharge counseling targeting patient barriers to adherence and ensuring a proper handover of medication information and rationale for medication changes to outpatient providers will improve the chances of a safe transition.
References
Coleman EA, Smith JD, Raha D, Min SJ. Posthospital medication discrepancies: prevalence and contributing factors. Arch Intern Med 2005; 165:1842–1847.
Wong JD, Bajcar JM, Wong GG, et al. Medication reconciliation at hospital discharge: evaluating discrepancies. Ann Pharmacother 2008; 42:1373–1379.
Kripalani S, Roumie CL, Dalal AK, et al; PILL-CVD (Pharmacist Intervention for Low Literacy in Cardiovascular Disease) Study Group. Effect of a pharmacist intervention on clinically important medication errors after hospital discharge: a randomized trial. Ann Intern Med 2012; 157:1-10.
Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med 2003; 138:161–167.
Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. Adverse drug events occurring following hospital discharge. J Gen Intern Med 2005; 20:317–323.
Johnson JA, Bootman JL. Drug-related morbidity and mortality. A cost-of-illness model. Arch Intern Med 1995; 155:1949–1956.
Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 2011; 365:2002–2012.
Bates DW, Boyle DL, Vander Vliet MB, Schneider J, Leape L. Relationship between medication errors and adverse drug events. J Gen Intern Med 1995;10:199–205.
Pippins JR, Gandhi TK, Hamann C, et al. Classifying and predicting errors of inpatient medication reconciliation. J Gen Intern Med 2008; 23:1414–1422.
Lau HS, Florax C, Porsius AJ, De Boer A.The completeness of medication histories in hospital medical records of patients admitted to general internal medicine wards. Br J Clin Pharmacol 2000; 49:597–603.
Tam VC, Knowles SR, Cornish PL, Fine N, Marchesano R, Etchells EE. Frequency, type and clinical importance of medication history errors at admission to hospital: a systematic review. CMAJ 2005; 173:510–515.
Bell CM, Brener SS, Gunraj N, et al. Association of ICU or hospital admission with unintentional discontinuation of medications for chronic diseases. JAMA 2011; 306:840–847.
Dobranski S, Hammond I, Khan G, Holdsworth H. The nature of hospital prescribing errors. Br J Clin Governance 2002; 7:187–193.
Gleason KM, Groszek JM, Sullivan C, Rooney D, Barnard C, Noskin GA. Reconciliation of discrepancies in medication histories and admission orders of newly hospitalized patients. Am J Health Syst Pharm 2004; 61:1689–1695.
Rozich JD, Resar KR. Medication safety: one organization’s approach to the challenge. J Clin Outcomes Manage 2001; 8:27–34.
Mueller SK, Sponsler KC, Kripalani S, Schnipper JL. Hospital-based medication reconciliation practices: a systematic review. Arch Intern Med 2012; 172:1057–1069.
Greenwald JL, Halasyamani L, Greene J, et al. Making inpatient medication reconciliation patient centered, clinically relevant and implementable: a consensus statement on key principles and necessary first steps. J Hosp Med 2010; 5:477–485.
Berwick DM, Calkins DR, McCannon CJ, Hackbarth AD. The 100,000 lives campaign: setting a goal and a deadline for improving health care quality. JAMA 2006; 295:324–327.
McCannon CJ, Hackbarth AD, Griffin FA. Miles to go: an introduction to the 5 Million Lives Campaign. Jt Comm J Qual Patient Saf 2007; 33:477–484.
Leotsakos A, Caisley L, Karga M, Kelly E, O’Leary D, Timmons K. High 5s: addressing excellence in patient safety. World Hosp Health Serv 2009; 45:19–22.
Mueller SK, Kripalani S, Stein J, et al. A toolkit to disseminate best practices in inpatient medication reconciliation: multi-center medication reconciliation quality improvement study (MARQUIS). Jt Comm J Qual Patient Saf 2013; 39:371–382.
Pal A, Babbott S, Wilkinson ST. Can the targeted use of a discharge pharmacist significantly decrease 30-day readmissions? Hosp Pharm 2013; 48:380–388.
Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther 2001; 23:1296–1310.
Osterberg L, Blaschke T. Adherence to medication. N Engl J Med 2005; 353:487–497.
Hajjar ER, Hanlon JT, Sloane RJ, et al. Unnecessary drug use in frail older people at hospital discharge. J Am Geriatr Soc 2005; 53:1518–1523.
Zink DA, Pohlman M, Barnes M, Cannon ME. Long-term use of acid suppression started inappropriately during hospitalization. Aliment Pharmacol Ther 2005; 21:1203–1209.
Morandi A, Vasilevskis E, Pandharipande PP, et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J Am Geriatr Soc 2013; 61:1128–1134.
Cox ZL, McCoy AB, Matheny ME, et al. Adverse drug events during AKI and its recovery. Clin J Am Soc Nephrol 2013; 8:1070–1078.
American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
Gallagher P, O’Mahony D. STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions): application to acutely ill elderly patients and comparison with Beers’ criteria. Age Ageing 2008; 37:673–679.
Tjia J, Bonner A, Briesacher BA, McGee S, Terrill E, Miller K. Medication discrepancies upon hospital to skilled nursing facility transitions. J Gen Intern Med 2009; 24:630–635.
Boockvar K, Fishman E, Kyriacou CK, Monias A, Gavi S, Cortes T. Adverse events due to discontinuations in drug use and dose changes in patients transferred between acute and long-term care facilities. Arch Intern Med 2004; 164:545–550.
Kutner M, Greenberg E, Jin Y, Paulsen C. The Health Literacy of America’s Adults: Results From the 2003 National Assessment of Adult Literacy. http://nces.ed.gov/pubs2006/2006483_1.pdf. Accessed March 31, 2015.
Crane JA. Patient comprehension of doctor-patient communication on discharge from the emergency department. J Emerg Med 1997; 15:1–7.
Schillinger D, Piette J, Grumbach K, et al. Closing the loop: physician communication with diabetic patients who have low health literacy. Arch Intern Med 2003; 163:83–90.
Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
Jack BW, Chetty VK, Anthony D, et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med 2009; 150:178–187.
Kelly C. Sponsler, MD Assistant Professor, Section of Hospital Medicine, Division of General Internal Medicine and Public Health, Department of Medicine, Vanderbilt University, Nashville, TN; Staff Physician, VA Tennessee Valley Medical Center, Nashville, TN
Erin B. Neal, PharmD Clinical Pharmacist, Department of Pharmaceutical Services, Vanderbilt University; Vanderbilt Health Affiliated Network, Nashville, TN
Sunil Kripalani, MD, MSc Associate Professor, Section of Hospital Medicine, Division of General Internal Medicine and Public Health, Department of Medicine, Vanderbilt University, Nashville, TN; Center for Clinical Quality and Implementation Research; Center for Effective Health Communication, Nashville, TN
Address: Kelly C. Sponsler, MD, Assistant Professor, Section of Hospital Medicine, Department of Medicine, Vanderbilt University, 1215 21st Avenue South, Suite 6000 Medical Center East, North Tower, Nashville, TN 37232; e-mail: kelly.e.cunningham@Vanderbilt.Edu
Kelly C. Sponsler, MD Assistant Professor, Section of Hospital Medicine, Division of General Internal Medicine and Public Health, Department of Medicine, Vanderbilt University, Nashville, TN; Staff Physician, VA Tennessee Valley Medical Center, Nashville, TN
Erin B. Neal, PharmD Clinical Pharmacist, Department of Pharmaceutical Services, Vanderbilt University; Vanderbilt Health Affiliated Network, Nashville, TN
Sunil Kripalani, MD, MSc Associate Professor, Section of Hospital Medicine, Division of General Internal Medicine and Public Health, Department of Medicine, Vanderbilt University, Nashville, TN; Center for Clinical Quality and Implementation Research; Center for Effective Health Communication, Nashville, TN
Address: Kelly C. Sponsler, MD, Assistant Professor, Section of Hospital Medicine, Department of Medicine, Vanderbilt University, 1215 21st Avenue South, Suite 6000 Medical Center East, North Tower, Nashville, TN 37232; e-mail: kelly.e.cunningham@Vanderbilt.Edu
Author and Disclosure Information
Kelly C. Sponsler, MD Assistant Professor, Section of Hospital Medicine, Division of General Internal Medicine and Public Health, Department of Medicine, Vanderbilt University, Nashville, TN; Staff Physician, VA Tennessee Valley Medical Center, Nashville, TN
Erin B. Neal, PharmD Clinical Pharmacist, Department of Pharmaceutical Services, Vanderbilt University; Vanderbilt Health Affiliated Network, Nashville, TN
Sunil Kripalani, MD, MSc Associate Professor, Section of Hospital Medicine, Division of General Internal Medicine and Public Health, Department of Medicine, Vanderbilt University, Nashville, TN; Center for Clinical Quality and Implementation Research; Center for Effective Health Communication, Nashville, TN
Address: Kelly C. Sponsler, MD, Assistant Professor, Section of Hospital Medicine, Department of Medicine, Vanderbilt University, 1215 21st Avenue South, Suite 6000 Medical Center East, North Tower, Nashville, TN 37232; e-mail: kelly.e.cunningham@Vanderbilt.Edu
Any time patients enter or leave the hospital, they risk being harmed by errors in their medications.1 Adverse events from medication errors during transitions of care are common but often preventable. One key approach is to systematically review every patient’s medication list on admission and discharge and resolve any discrepancies. These transitions are also an opportunity to address other medication-related problems, such as adherence, drug interactions, and clinical appropriateness.
This article summarizes the types and prevalence of medication problems that occur during hospital-based transitions of care, and suggests strategies to decrease the risk of medication errors, focusing on medication reconciliation and related interventions that clinicians can use at the bedside to improve medication safety.
DEFINING TERMS
A medication discrepancy is any variance noted in a patient’s documented medication regimen across different medication lists or sites of care. While some differences reflect intentional and clinically appropriate changes to the regimen, others are unintentional and reflect inaccurate or incomplete information. These unintentional discrepancies are medication errors.
Depending on the clinical circumstances and medications involved, such errors may lead to an adverse drug event (ADE), defined as actual harm or injury resulting from a medication. Sometimes a medication error does not cause harm immediately but could if left uncorrected; this is called a potential ADE.
An important goal during transitions of care is to reduce unintentional medication discrepancies, thereby reducing potential and actual ADEs.
ERRORS ARISE AT DISCHARGE—AND EVEN MORE AT ADMISSION
Hospital discharge is a widely recognized transition in which patient harm occurs. As many as 70% of patients may have an unintentional medication discrepancy at hospital discharge, with many of those discrepancies having potential for harm.2 Indeed, during the first few weeks after discharge, 50% of patients have a clinically important medication error,3 and 20% experience an adverse event, most commonly an ADE.4 ADEs are associated with excess health care utilization,5–7 and many are preventable through strategies such as medication reconciliation.5,8
Importantly, more errors arise at hospital admission than at other times.9,10
Errors in medication histories are the most common source of discrepancies, affecting up to two-thirds of admitted patients.11,12 More than one-quarter of hospital prescribing errors can be attributed to incomplete medication histories at the time of admission,13 and nearly three times as many clinically important medication discrepancies are related to history-taking errors on admission rather than reconciliation errors at discharge.9
Most discrepancies occurring at the time of admission have the potential to cause harm, particularly if the errors persist beyond discharge.14 Therefore, taking a complete and accurate medication history on hospital admission is critical to ensuring safe care transitions.
MEDICATION RECONCILIATION: BARRIERS AND FACILITATORS
Medication reconciliation is a strategy for reducing medication discrepancies in patients moving across care settings. Simply put, it is the process by which a patient’s medication list is obtained, compared, and clarified across different sites of care.15 It has consistently been shown to decrease medication errors compared with usual care,16 and it is strongly supported by national and international organizations.17–21
In clinical practice, many physicians and institutions have found medication reconciliation difficult to implement, owing to barriers at the level of the patient, provider, and system (Table 1). In response to these challenges, two initiatives have synthesized best practices and offer toolkits that hospitals and clinicians can use: the Medications at Transitions and Clinical Handoffs (MATCH) program22 and the Multi-Center Medication Reconciliation Quality Improvement Study (MARQUIS).23
Lack of resources is a widely acknowledged challenge. Thus, the MARQUIS investigators23 suggested focusing on the admission history, where most errors occur, and applying the most resource-intensive interventions in patients at highest risk of ADEs, ie, those who are elderly, have multiple comorbid conditions, or take numerous medications.16
Although the risk of an ADE increases with the number of medications a patient takes,4 the exact number of drugs that defines high risk has not been well established. Targeting patients who take 10 or more maintenance medications is a reasonable initial approach,24 but institutions should tailor risk stratification to their patient populations and available resources. Patients taking high-risk medications such as anticoagulants and insulin could also be prioritized for review, since these medications are more likely to cause serious patient harm when used without appropriate clinical oversight.7
Using pharmacy staff to perform medication history-taking, reconciliation, and patient counseling has been shown to produce favorable patient outcomes, particularly for higher-risk patients.16,23
The MARQUIS investigators found they could boost the chances of success by sharing stories of patient harm to foster “buy-in” among frontline staff, providing formal training to clinicians on how to take a medication history, and obtaining the support of nursing leaders to champion improvement efforts.23
Additionally, patients should be empowered to maintain an accurate medication list. We address strategies for improving patient engagement and adherence in a later section.
BEST PRACTICES FOR IMPROVING MEDICATION SAFETY
Medication reconciliation is one of several measures necessary to optimize medication safety during transitions of care. It typically includes the following actions:
Interview the patient or caregiver to determine the list of medications the patient is currently taking (or supposed to be taking); consult other sources if needed.
List medications that are being ordered during the clinical encounter.
Compare these two lists, making note of medications that are stopped, changed, or newly prescribed.
Resolve any discrepancies.
Communicate the reconciled list to the patient, appropriate caregivers, and providers of follow-up care.
At a rudimentary level, medication reconciliation encompasses medication list management along the continuum of care. However, we recommend leveraging medication reconciliation as an opportunity to further enhance medication safety by reviewing the appropriateness of each medication, seizing opportunities to streamline or simplify the regimen, assessing patient and caregiver understanding of medication instructions and potential ADEs, and delivering appropriate counseling to enhance medication use. Table 2 outlines our framework for medication management during hospital-based transitions.
STEP 1: OBTAIN A COMPLETE PREADMISSION MEDICATION LIST
The “best-possible medication history” is obtained in a systematic process of interviewing the patient or caregiver plus reviewing at least one other reliable source.23 The resulting list should include all medications the patient is taking (prescription and nonprescription), doses, directions for use, formulations if applicable, indications, start and stop dates, and medication allergies and reactions.
Review existing information. Before eliciting a history from the patient, review his or her recorded medical history and existing medication lists (eg, prior discharge summaries, records from other facilities, records from outpatient visits, pharmacy fill data). This will provide context about the regimen and help identify issues and questions that can be addressed during the history-taking.
Ask open-ended questions. Instead of just asking the patient to confirm the accuracy of the existing medication list, we recommend actively obtaining the full medication list from the patient or caregiver. The conversation should begin with an open-ended question such as, “What medications do you take at home?” This approach will also allow the clinician to gauge the patient’s level of understanding of each medication’s indication and dosing instructions. Using a series of prompts such as those recommended in Table 3 will elicit a best-possible medical history, while verifying all of the medications on the existing list.
Clarify discrepancies. Resolve differences between the existing medication lists and the patient’s or caregiver’s report during the preadmission interview. Examples include errors of omission (a medication is missing), errors of commission (an additional medication is present), and discrepancies in the strength, formulation, dosing instructions, and indications for the drugs. If necessary, other sources of information should be consulted, such as the patient’s medication bottles, pharmacy or pharmacies, primary care physician, and a family member or caregiver.
Assess adherence. The extent to which patients take their medications as directed is an important component of the history, but is often left out. Medication nonadherence rates in the United States are 40% to 70%,25 contributing to poor patient outcomes and imposing extraordinary costs on the health care system.26
Asking open-ended, nonjudgmental questions at the time of hospital admission will help to uncover medication-taking behaviors as well as barriers to adherence (Table 3). The patient’s responses should be taken into account when determining the treatment plan.
Document your findings. After completing the medication history and clarifying discrepancies, document the preadmission list in the medical record. All members of the health care team should have access to view and update the same list, as new information about the preadmission medications may be uncovered after the initial history.
Make clinical decisions. Complete the admission medication reconciliation by deciding whether each medication on the list should be continued, changed, held, or discontinued on the basis of the patient’s clinical condition. Well-designed information technology applications enable the provider to document each action and the rationale for it, as well as carry that information into the order-entry system. Medications marked as held or discontinued on admission should be revisited as the patient’s clinical condition changes and at discharge.
STEP 2: AVOID RECONCILIATION ERRORS
Reconciliation errors reflect discrepancies between the medication history and the medications that are ordered after admission.
Reconciliation errors are less common than medication history errors, accounting for approximately one-third of potentially harmful medication errors in hospitalized medical patients.9 These include errors of omission (a medication is omitted from the orders), errors of commission (a medication is prescribed with no indication for continuation), and therapeutic duplication.
Preventing errors of omission
Medications are often held at transition points for appropriate clinical reasons. Examples include holding anticoagulants and antiplatelet agents in patients who have gastrointestinal bleeding or an upcoming procedure, antihypertensives in patients with hemodynamic instability, and other chronic medications in patients with an acute illness.
Poor documentation and communication of these decisions can lead to a failure to resume the medications—an error of omission—at hospital discharge.
Hospitalized patients are at risk of unintentional discontinuation of their chronic medications, including antiplatelet drugs, anticoagulants, statins, and thyroid replacement, particularly if admitted to the intensive care unit.12 These errors can be minimized by a standardized medication reconciliation process at each transition and clear documentation of the medication plan.
Communication among providers can be improved if the admitting clinician documents clearly whether each preadmission medication is being continued, changed, or stopped, along with the reason for doing so, and makes this information available throughout the hospital stay. Upon transfer to another unit and at discharge, the physician should review each; preadmission medication that was held and the patient’s current clinical status and, based on that information, decide whether medications that were held should be resumed. If a medication will be restarted later, specific instructions should be documented and communicated to the patient and the physicians who are taking over his or her care.
Preventing errors of commission
Failure to perform a complete reconciliation at each transition of care and match each medication with an appropriate indication can lead to errors of commission.
One study showed that 44% of patients were prescribed at least one unnecessary drug at hospital discharge, one-fourth of which were started during the hospitalization.27 Commonly prescribed unnecessarily were gastrointestinal agents, central nervous system drugs, nutrients, and supplements.
It is critical to assess each medication’s ongoing need, appropriateness, and risk-benefit ratio at every transition. Medications no longer indicated should be discontinued in order to simplify the regimen, avoid unnecessary drug exposure, and prevent ADEs.
For example, proton pump inhibitors or histamine 2 receptor blockers are often started in the hospital for stress ulcer prophylaxis. One-third of patients are then discharged home on the medication, and 6 months later half of those patients are still taking the unnecessary drug.28 This situation can be avoided by limiting use of these medications to appropriate circumstances, clearly marking the indication as stress ulcer prophylaxis (as opposed to an ongoing condition that will require continuing it after discharge), and discontinuing the agent when appropriate.
All drugs, even common and seemingly benign ones, carry some risk and should be discontinued when no longer needed. Thus, medications added during the hospitalization to control acute symptoms should also be reviewed at each transition to prevent inappropriate continuation when symptoms have resolved.
One study, for example, found that many patients were discharged with inappropriate prescriptions for atypical antipsychotics after receiving them in the intensive care unit, likely for delirium.29 Documenting that an acute issue such as delirium has resolved should prompt the discontinuation of therapy.
Preventing therapeutic duplication
Therapeutic duplication occurs in about 8% of discharges.1 These errors often result from formulary substitutions or altering the dosage form in the acute setting. For example, patients who receive a prescription for the substituted agent at discharge and also resume their prehospitalization medications end up with duplicate therapy.
Therapeutic substitution is common at the time of admission to the hospital as a result of formulary restrictions. Drug classes that are frequently substituted include statins, antihypertensives, urinary antispasmodics, and proton pump inhibitors. Physicians should be familiar with the preferred agents on the hospital formulary and make careful note when a substitution occurs. Furthermore, hospital systems should be developed to remind the physician to switch back to the outpatient medication at discharge.
Similar problems occur when home medications are replaced with different dosage forms with different pharmacokinetic properties. For example, a long-acting medication may be temporarily replaced with an intravenous solution or immediate-release tablet for several reasons, including nothing-by-mouth status, unstable clinical condition, need for titration, and need to crush the tablet to give the drug per tube. The differing formulations must be reconciled throughout the patient’s hospital course and at discharge to avoid therapeutic duplication and serious medication errors. Deliberate changes to the dosage form should be clearly communicated in the discharge medication list so that patients and other clinicians are aware.
Hospital systems should also have the capability to identify duplications in the medication list and to warn prescribers of these errors. The ability to group medications by drug class or sort the medication list alphabetically by generic name can help uncover duplication errors.
STEP 3: REVIEW THE LIST IN VIEW OF THE CLINICAL PICTURE
Transitions of care should prompt providers to review the medication list for possible drug-disease interactions, confirm compliance with evidence-based guidelines, and evaluate the risks and benefits of each medication in the context of the patient’s age and acute and chronic medical issues. This is also an opportunity to screen the full list for potentially inappropriate medications and high-alert drugs such as insulin or anticoagulants, which are more likely than other drugs to cause severe harm when used in error.
Acute kidney injury. New drug-disease interactions can arise during a hospitalization and can affect dosing and the choice of drug. The onset of acute kidney injury, for example, often necessitates adjusting or discontinuing nephrotoxic and renally excreted medications. ADEs or potential ADEs have been reported in 43% of hospitalized patients with acute kidney injury.30
Because acute kidney injury is often transient, medications may need to be held or adjusted several times until renal function stabilizes. This can be challenging across the continuum of care and requires close monitoring of the serum creatinine level and associated drug doses and levels, if applicable. Well-designed clinical decision support tools can integrate laboratory data and alert the prescriber to a clinically important increase or decrease in serum creatinine that may warrant a change in therapy. Modifications to the regimen and a plan for timely follow-up of the serum creatinine level should be clearly documented in the discharge plan.
Liver disease. Similar attention should be given to drugs that are hepatically metabolized if a patient has acute or chronic liver impairment.
Geriatric patients, particularly those who present with altered mental status, falls, or urinary retention, should have their medication list reviewed for potentially inappropriate medications, which are drugs that pose increased risk of poor outcomes in older adults.31,32 Patients and providers may have been willing to accept the risk of medications such as anticholinergics or sedative-hypnotics when the drugs were initiated, but circumstances can change over time, especially in this patient population. Hospitalization is a prime opportunity to screen for medications that meet the Beers criteria31 for agents to avoid or use with caution in older adults.
As-needed medications. Medications prescribed on an as-needed basis in the hospital should be reviewed for continuation or discontinuation at discharge. How often the medication was given can inform this decision.
For example, if as-needed opioids were used frequently, failure to develop a plan of care for pain can lead to persistent symptoms and, possibly, to readmission.33,34 Similar scenarios occur with use of as-needed blood pressure medications, laxatives, and correction-dose insulin.
If an as-needed medication was used consistently during hospitalization, the physician should consider whether a regularly scheduled medication is needed. Conversely, if the medication was not used during the inpatient admission, it can likely be discontinued.
STEP 4: PREPARE THE PATIENT AND FOLLOW-UP PROVIDER
Once a clinician has performed medication reconciliation, including obtaining a best-possible medical history and carefully reviewing the medication list and orders for errors and clinical appropriateness, the next steps are to ensure the patient understands what he or she needs to do and to confirm that suitable follow-up plans are in place. These measures should be taken at all transitions of care but are critically important at hospital discharge.
Preparing the patient and caregiver
An accurate, reconciled medication list should be given to the patient, caregiver, or both, and should be reviewed before discharge.17
Approximately one-third of Americans have low health literacy skills, so medication lists and associated materials should be easy to understand.35 Medication lists should be written in plain language and formatted for optimal readability (Table 4), clearly stating which medications to continue, change, hold temporarily, and stop.
Patients recall and comprehend about half of the information provided during a medical encounter.36 Thus, medication teaching should focus on key points including changes or additions to the regimen, specific instructions for follow-up and monitoring, and how to handle common and serious side effects.
To confirm patient understanding, clinicians should use “teach-back,” ie, provide the patient with information and then ask him or her to repeat back key points.37,38 The patient and family should also be encouraged to ask questions before discharge.
If not already addressed during the hospital stay, barriers to medication adherence and ability to obtain the medications should be attended to at this time (Table 5). Also, the plan to pick up the medications should be verified with the patient and caregiver. Verify that there is transportation to a particular pharmacy that is open at the time of discharge, and that the patient can afford the medications.
Ensuring appropriate follow-up
Studies have shown that timely in-home or telephone follow-up after discharge can decrease adverse events and postdischarge health care utilization.39,40 Telephone follow-up that includes thorough medication reconciliation can help detect and resolve medication issues early after discharge and can close gaps related to monitoring and follow-up.
Medication reconciliation by telephone can be time-consuming. Depending on the number of medications that need to be reviewed, calls can take between 10 and 60 minutes. Postdischarge phone calls should be performed by clinical personnel who are able to identify medication-related problems. A pharmacist should be an available resource to assist with complex regimens, to help resolve medication discrepancies, and to address patient concerns. Table 6 provides tips for conducting follow-up phone calls.
Resolving discrepancies identified during follow-up calls can be difficult, as changes to the medication regimen are often not communicated effectively to other members of the care team. Physicians should document the complete medication list and plan in the discharge summary, and there should be a method for the caller to record updates to the medication list in the medical record so that they are apparent at the outpatient follow-up visit.
An additional challenge is that it is frequently unclear which physician “owns” which medications. Therefore, designating a contact person for each medication until follow-up can be very valuable. At a minimum, a “physician owner” for high-alert medications such as insulin, anticoagulants, and diuretics should be identified to provide close follow-up, titration, and monitoring.
There should also be a plan for the patient to obtain refills of essential long-term medications, such as antiplatelet agents following stent placement.
SUMMARY AND RECOMMENDATIONS
Medication-related problems during hospital admission and discharge are common and range from minor discrepancies in the medication list to errors in history-taking, prescribing, and reconciliation that can lead to potential or actual patient harm. Putting systems in place to facilitate medication reconciliation can decrease the occurrence of medication discrepancies and ADEs, thereby improving patient safety during these critical transitions between care settings and providers. Institutional medication reconciliation programs should focus resources on the admission history-taking step, target the highest-risk patients for the most intensive interventions, and involve pharmacy personnel when possible.
On an individual level, clinicians can incorporate additional interventions into their workflows to optimize medication safety for hospitalized patients. Using a structured approach to obtain a complete and accurate medication list at the time of hospital admission will help providers identify medication-related problems and prevent the propagation of errors throughout the hospital stay and at discharge. Focusing additional time and effort on a comprehensive review of the medication list for errors of omission and commission, patient-specific needs, and high-alert drugs will further decrease the risk of medication errors. Finally, providing discharge counseling targeting patient barriers to adherence and ensuring a proper handover of medication information and rationale for medication changes to outpatient providers will improve the chances of a safe transition.
Any time patients enter or leave the hospital, they risk being harmed by errors in their medications.1 Adverse events from medication errors during transitions of care are common but often preventable. One key approach is to systematically review every patient’s medication list on admission and discharge and resolve any discrepancies. These transitions are also an opportunity to address other medication-related problems, such as adherence, drug interactions, and clinical appropriateness.
This article summarizes the types and prevalence of medication problems that occur during hospital-based transitions of care, and suggests strategies to decrease the risk of medication errors, focusing on medication reconciliation and related interventions that clinicians can use at the bedside to improve medication safety.
DEFINING TERMS
A medication discrepancy is any variance noted in a patient’s documented medication regimen across different medication lists or sites of care. While some differences reflect intentional and clinically appropriate changes to the regimen, others are unintentional and reflect inaccurate or incomplete information. These unintentional discrepancies are medication errors.
Depending on the clinical circumstances and medications involved, such errors may lead to an adverse drug event (ADE), defined as actual harm or injury resulting from a medication. Sometimes a medication error does not cause harm immediately but could if left uncorrected; this is called a potential ADE.
An important goal during transitions of care is to reduce unintentional medication discrepancies, thereby reducing potential and actual ADEs.
ERRORS ARISE AT DISCHARGE—AND EVEN MORE AT ADMISSION
Hospital discharge is a widely recognized transition in which patient harm occurs. As many as 70% of patients may have an unintentional medication discrepancy at hospital discharge, with many of those discrepancies having potential for harm.2 Indeed, during the first few weeks after discharge, 50% of patients have a clinically important medication error,3 and 20% experience an adverse event, most commonly an ADE.4 ADEs are associated with excess health care utilization,5–7 and many are preventable through strategies such as medication reconciliation.5,8
Importantly, more errors arise at hospital admission than at other times.9,10
Errors in medication histories are the most common source of discrepancies, affecting up to two-thirds of admitted patients.11,12 More than one-quarter of hospital prescribing errors can be attributed to incomplete medication histories at the time of admission,13 and nearly three times as many clinically important medication discrepancies are related to history-taking errors on admission rather than reconciliation errors at discharge.9
Most discrepancies occurring at the time of admission have the potential to cause harm, particularly if the errors persist beyond discharge.14 Therefore, taking a complete and accurate medication history on hospital admission is critical to ensuring safe care transitions.
MEDICATION RECONCILIATION: BARRIERS AND FACILITATORS
Medication reconciliation is a strategy for reducing medication discrepancies in patients moving across care settings. Simply put, it is the process by which a patient’s medication list is obtained, compared, and clarified across different sites of care.15 It has consistently been shown to decrease medication errors compared with usual care,16 and it is strongly supported by national and international organizations.17–21
In clinical practice, many physicians and institutions have found medication reconciliation difficult to implement, owing to barriers at the level of the patient, provider, and system (Table 1). In response to these challenges, two initiatives have synthesized best practices and offer toolkits that hospitals and clinicians can use: the Medications at Transitions and Clinical Handoffs (MATCH) program22 and the Multi-Center Medication Reconciliation Quality Improvement Study (MARQUIS).23
Lack of resources is a widely acknowledged challenge. Thus, the MARQUIS investigators23 suggested focusing on the admission history, where most errors occur, and applying the most resource-intensive interventions in patients at highest risk of ADEs, ie, those who are elderly, have multiple comorbid conditions, or take numerous medications.16
Although the risk of an ADE increases with the number of medications a patient takes,4 the exact number of drugs that defines high risk has not been well established. Targeting patients who take 10 or more maintenance medications is a reasonable initial approach,24 but institutions should tailor risk stratification to their patient populations and available resources. Patients taking high-risk medications such as anticoagulants and insulin could also be prioritized for review, since these medications are more likely to cause serious patient harm when used without appropriate clinical oversight.7
Using pharmacy staff to perform medication history-taking, reconciliation, and patient counseling has been shown to produce favorable patient outcomes, particularly for higher-risk patients.16,23
The MARQUIS investigators found they could boost the chances of success by sharing stories of patient harm to foster “buy-in” among frontline staff, providing formal training to clinicians on how to take a medication history, and obtaining the support of nursing leaders to champion improvement efforts.23
Additionally, patients should be empowered to maintain an accurate medication list. We address strategies for improving patient engagement and adherence in a later section.
BEST PRACTICES FOR IMPROVING MEDICATION SAFETY
Medication reconciliation is one of several measures necessary to optimize medication safety during transitions of care. It typically includes the following actions:
Interview the patient or caregiver to determine the list of medications the patient is currently taking (or supposed to be taking); consult other sources if needed.
List medications that are being ordered during the clinical encounter.
Compare these two lists, making note of medications that are stopped, changed, or newly prescribed.
Resolve any discrepancies.
Communicate the reconciled list to the patient, appropriate caregivers, and providers of follow-up care.
At a rudimentary level, medication reconciliation encompasses medication list management along the continuum of care. However, we recommend leveraging medication reconciliation as an opportunity to further enhance medication safety by reviewing the appropriateness of each medication, seizing opportunities to streamline or simplify the regimen, assessing patient and caregiver understanding of medication instructions and potential ADEs, and delivering appropriate counseling to enhance medication use. Table 2 outlines our framework for medication management during hospital-based transitions.
STEP 1: OBTAIN A COMPLETE PREADMISSION MEDICATION LIST
The “best-possible medication history” is obtained in a systematic process of interviewing the patient or caregiver plus reviewing at least one other reliable source.23 The resulting list should include all medications the patient is taking (prescription and nonprescription), doses, directions for use, formulations if applicable, indications, start and stop dates, and medication allergies and reactions.
Review existing information. Before eliciting a history from the patient, review his or her recorded medical history and existing medication lists (eg, prior discharge summaries, records from other facilities, records from outpatient visits, pharmacy fill data). This will provide context about the regimen and help identify issues and questions that can be addressed during the history-taking.
Ask open-ended questions. Instead of just asking the patient to confirm the accuracy of the existing medication list, we recommend actively obtaining the full medication list from the patient or caregiver. The conversation should begin with an open-ended question such as, “What medications do you take at home?” This approach will also allow the clinician to gauge the patient’s level of understanding of each medication’s indication and dosing instructions. Using a series of prompts such as those recommended in Table 3 will elicit a best-possible medical history, while verifying all of the medications on the existing list.
Clarify discrepancies. Resolve differences between the existing medication lists and the patient’s or caregiver’s report during the preadmission interview. Examples include errors of omission (a medication is missing), errors of commission (an additional medication is present), and discrepancies in the strength, formulation, dosing instructions, and indications for the drugs. If necessary, other sources of information should be consulted, such as the patient’s medication bottles, pharmacy or pharmacies, primary care physician, and a family member or caregiver.
Assess adherence. The extent to which patients take their medications as directed is an important component of the history, but is often left out. Medication nonadherence rates in the United States are 40% to 70%,25 contributing to poor patient outcomes and imposing extraordinary costs on the health care system.26
Asking open-ended, nonjudgmental questions at the time of hospital admission will help to uncover medication-taking behaviors as well as barriers to adherence (Table 3). The patient’s responses should be taken into account when determining the treatment plan.
Document your findings. After completing the medication history and clarifying discrepancies, document the preadmission list in the medical record. All members of the health care team should have access to view and update the same list, as new information about the preadmission medications may be uncovered after the initial history.
Make clinical decisions. Complete the admission medication reconciliation by deciding whether each medication on the list should be continued, changed, held, or discontinued on the basis of the patient’s clinical condition. Well-designed information technology applications enable the provider to document each action and the rationale for it, as well as carry that information into the order-entry system. Medications marked as held or discontinued on admission should be revisited as the patient’s clinical condition changes and at discharge.
STEP 2: AVOID RECONCILIATION ERRORS
Reconciliation errors reflect discrepancies between the medication history and the medications that are ordered after admission.
Reconciliation errors are less common than medication history errors, accounting for approximately one-third of potentially harmful medication errors in hospitalized medical patients.9 These include errors of omission (a medication is omitted from the orders), errors of commission (a medication is prescribed with no indication for continuation), and therapeutic duplication.
Preventing errors of omission
Medications are often held at transition points for appropriate clinical reasons. Examples include holding anticoagulants and antiplatelet agents in patients who have gastrointestinal bleeding or an upcoming procedure, antihypertensives in patients with hemodynamic instability, and other chronic medications in patients with an acute illness.
Poor documentation and communication of these decisions can lead to a failure to resume the medications—an error of omission—at hospital discharge.
Hospitalized patients are at risk of unintentional discontinuation of their chronic medications, including antiplatelet drugs, anticoagulants, statins, and thyroid replacement, particularly if admitted to the intensive care unit.12 These errors can be minimized by a standardized medication reconciliation process at each transition and clear documentation of the medication plan.
Communication among providers can be improved if the admitting clinician documents clearly whether each preadmission medication is being continued, changed, or stopped, along with the reason for doing so, and makes this information available throughout the hospital stay. Upon transfer to another unit and at discharge, the physician should review each; preadmission medication that was held and the patient’s current clinical status and, based on that information, decide whether medications that were held should be resumed. If a medication will be restarted later, specific instructions should be documented and communicated to the patient and the physicians who are taking over his or her care.
Preventing errors of commission
Failure to perform a complete reconciliation at each transition of care and match each medication with an appropriate indication can lead to errors of commission.
One study showed that 44% of patients were prescribed at least one unnecessary drug at hospital discharge, one-fourth of which were started during the hospitalization.27 Commonly prescribed unnecessarily were gastrointestinal agents, central nervous system drugs, nutrients, and supplements.
It is critical to assess each medication’s ongoing need, appropriateness, and risk-benefit ratio at every transition. Medications no longer indicated should be discontinued in order to simplify the regimen, avoid unnecessary drug exposure, and prevent ADEs.
For example, proton pump inhibitors or histamine 2 receptor blockers are often started in the hospital for stress ulcer prophylaxis. One-third of patients are then discharged home on the medication, and 6 months later half of those patients are still taking the unnecessary drug.28 This situation can be avoided by limiting use of these medications to appropriate circumstances, clearly marking the indication as stress ulcer prophylaxis (as opposed to an ongoing condition that will require continuing it after discharge), and discontinuing the agent when appropriate.
All drugs, even common and seemingly benign ones, carry some risk and should be discontinued when no longer needed. Thus, medications added during the hospitalization to control acute symptoms should also be reviewed at each transition to prevent inappropriate continuation when symptoms have resolved.
One study, for example, found that many patients were discharged with inappropriate prescriptions for atypical antipsychotics after receiving them in the intensive care unit, likely for delirium.29 Documenting that an acute issue such as delirium has resolved should prompt the discontinuation of therapy.
Preventing therapeutic duplication
Therapeutic duplication occurs in about 8% of discharges.1 These errors often result from formulary substitutions or altering the dosage form in the acute setting. For example, patients who receive a prescription for the substituted agent at discharge and also resume their prehospitalization medications end up with duplicate therapy.
Therapeutic substitution is common at the time of admission to the hospital as a result of formulary restrictions. Drug classes that are frequently substituted include statins, antihypertensives, urinary antispasmodics, and proton pump inhibitors. Physicians should be familiar with the preferred agents on the hospital formulary and make careful note when a substitution occurs. Furthermore, hospital systems should be developed to remind the physician to switch back to the outpatient medication at discharge.
Similar problems occur when home medications are replaced with different dosage forms with different pharmacokinetic properties. For example, a long-acting medication may be temporarily replaced with an intravenous solution or immediate-release tablet for several reasons, including nothing-by-mouth status, unstable clinical condition, need for titration, and need to crush the tablet to give the drug per tube. The differing formulations must be reconciled throughout the patient’s hospital course and at discharge to avoid therapeutic duplication and serious medication errors. Deliberate changes to the dosage form should be clearly communicated in the discharge medication list so that patients and other clinicians are aware.
Hospital systems should also have the capability to identify duplications in the medication list and to warn prescribers of these errors. The ability to group medications by drug class or sort the medication list alphabetically by generic name can help uncover duplication errors.
STEP 3: REVIEW THE LIST IN VIEW OF THE CLINICAL PICTURE
Transitions of care should prompt providers to review the medication list for possible drug-disease interactions, confirm compliance with evidence-based guidelines, and evaluate the risks and benefits of each medication in the context of the patient’s age and acute and chronic medical issues. This is also an opportunity to screen the full list for potentially inappropriate medications and high-alert drugs such as insulin or anticoagulants, which are more likely than other drugs to cause severe harm when used in error.
Acute kidney injury. New drug-disease interactions can arise during a hospitalization and can affect dosing and the choice of drug. The onset of acute kidney injury, for example, often necessitates adjusting or discontinuing nephrotoxic and renally excreted medications. ADEs or potential ADEs have been reported in 43% of hospitalized patients with acute kidney injury.30
Because acute kidney injury is often transient, medications may need to be held or adjusted several times until renal function stabilizes. This can be challenging across the continuum of care and requires close monitoring of the serum creatinine level and associated drug doses and levels, if applicable. Well-designed clinical decision support tools can integrate laboratory data and alert the prescriber to a clinically important increase or decrease in serum creatinine that may warrant a change in therapy. Modifications to the regimen and a plan for timely follow-up of the serum creatinine level should be clearly documented in the discharge plan.
Liver disease. Similar attention should be given to drugs that are hepatically metabolized if a patient has acute or chronic liver impairment.
Geriatric patients, particularly those who present with altered mental status, falls, or urinary retention, should have their medication list reviewed for potentially inappropriate medications, which are drugs that pose increased risk of poor outcomes in older adults.31,32 Patients and providers may have been willing to accept the risk of medications such as anticholinergics or sedative-hypnotics when the drugs were initiated, but circumstances can change over time, especially in this patient population. Hospitalization is a prime opportunity to screen for medications that meet the Beers criteria31 for agents to avoid or use with caution in older adults.
As-needed medications. Medications prescribed on an as-needed basis in the hospital should be reviewed for continuation or discontinuation at discharge. How often the medication was given can inform this decision.
For example, if as-needed opioids were used frequently, failure to develop a plan of care for pain can lead to persistent symptoms and, possibly, to readmission.33,34 Similar scenarios occur with use of as-needed blood pressure medications, laxatives, and correction-dose insulin.
If an as-needed medication was used consistently during hospitalization, the physician should consider whether a regularly scheduled medication is needed. Conversely, if the medication was not used during the inpatient admission, it can likely be discontinued.
STEP 4: PREPARE THE PATIENT AND FOLLOW-UP PROVIDER
Once a clinician has performed medication reconciliation, including obtaining a best-possible medical history and carefully reviewing the medication list and orders for errors and clinical appropriateness, the next steps are to ensure the patient understands what he or she needs to do and to confirm that suitable follow-up plans are in place. These measures should be taken at all transitions of care but are critically important at hospital discharge.
Preparing the patient and caregiver
An accurate, reconciled medication list should be given to the patient, caregiver, or both, and should be reviewed before discharge.17
Approximately one-third of Americans have low health literacy skills, so medication lists and associated materials should be easy to understand.35 Medication lists should be written in plain language and formatted for optimal readability (Table 4), clearly stating which medications to continue, change, hold temporarily, and stop.
Patients recall and comprehend about half of the information provided during a medical encounter.36 Thus, medication teaching should focus on key points including changes or additions to the regimen, specific instructions for follow-up and monitoring, and how to handle common and serious side effects.
To confirm patient understanding, clinicians should use “teach-back,” ie, provide the patient with information and then ask him or her to repeat back key points.37,38 The patient and family should also be encouraged to ask questions before discharge.
If not already addressed during the hospital stay, barriers to medication adherence and ability to obtain the medications should be attended to at this time (Table 5). Also, the plan to pick up the medications should be verified with the patient and caregiver. Verify that there is transportation to a particular pharmacy that is open at the time of discharge, and that the patient can afford the medications.
Ensuring appropriate follow-up
Studies have shown that timely in-home or telephone follow-up after discharge can decrease adverse events and postdischarge health care utilization.39,40 Telephone follow-up that includes thorough medication reconciliation can help detect and resolve medication issues early after discharge and can close gaps related to monitoring and follow-up.
Medication reconciliation by telephone can be time-consuming. Depending on the number of medications that need to be reviewed, calls can take between 10 and 60 minutes. Postdischarge phone calls should be performed by clinical personnel who are able to identify medication-related problems. A pharmacist should be an available resource to assist with complex regimens, to help resolve medication discrepancies, and to address patient concerns. Table 6 provides tips for conducting follow-up phone calls.
Resolving discrepancies identified during follow-up calls can be difficult, as changes to the medication regimen are often not communicated effectively to other members of the care team. Physicians should document the complete medication list and plan in the discharge summary, and there should be a method for the caller to record updates to the medication list in the medical record so that they are apparent at the outpatient follow-up visit.
An additional challenge is that it is frequently unclear which physician “owns” which medications. Therefore, designating a contact person for each medication until follow-up can be very valuable. At a minimum, a “physician owner” for high-alert medications such as insulin, anticoagulants, and diuretics should be identified to provide close follow-up, titration, and monitoring.
There should also be a plan for the patient to obtain refills of essential long-term medications, such as antiplatelet agents following stent placement.
SUMMARY AND RECOMMENDATIONS
Medication-related problems during hospital admission and discharge are common and range from minor discrepancies in the medication list to errors in history-taking, prescribing, and reconciliation that can lead to potential or actual patient harm. Putting systems in place to facilitate medication reconciliation can decrease the occurrence of medication discrepancies and ADEs, thereby improving patient safety during these critical transitions between care settings and providers. Institutional medication reconciliation programs should focus resources on the admission history-taking step, target the highest-risk patients for the most intensive interventions, and involve pharmacy personnel when possible.
On an individual level, clinicians can incorporate additional interventions into their workflows to optimize medication safety for hospitalized patients. Using a structured approach to obtain a complete and accurate medication list at the time of hospital admission will help providers identify medication-related problems and prevent the propagation of errors throughout the hospital stay and at discharge. Focusing additional time and effort on a comprehensive review of the medication list for errors of omission and commission, patient-specific needs, and high-alert drugs will further decrease the risk of medication errors. Finally, providing discharge counseling targeting patient barriers to adherence and ensuring a proper handover of medication information and rationale for medication changes to outpatient providers will improve the chances of a safe transition.
References
Coleman EA, Smith JD, Raha D, Min SJ. Posthospital medication discrepancies: prevalence and contributing factors. Arch Intern Med 2005; 165:1842–1847.
Wong JD, Bajcar JM, Wong GG, et al. Medication reconciliation at hospital discharge: evaluating discrepancies. Ann Pharmacother 2008; 42:1373–1379.
Kripalani S, Roumie CL, Dalal AK, et al; PILL-CVD (Pharmacist Intervention for Low Literacy in Cardiovascular Disease) Study Group. Effect of a pharmacist intervention on clinically important medication errors after hospital discharge: a randomized trial. Ann Intern Med 2012; 157:1-10.
Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med 2003; 138:161–167.
Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. Adverse drug events occurring following hospital discharge. J Gen Intern Med 2005; 20:317–323.
Johnson JA, Bootman JL. Drug-related morbidity and mortality. A cost-of-illness model. Arch Intern Med 1995; 155:1949–1956.
Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 2011; 365:2002–2012.
Bates DW, Boyle DL, Vander Vliet MB, Schneider J, Leape L. Relationship between medication errors and adverse drug events. J Gen Intern Med 1995;10:199–205.
Pippins JR, Gandhi TK, Hamann C, et al. Classifying and predicting errors of inpatient medication reconciliation. J Gen Intern Med 2008; 23:1414–1422.
Lau HS, Florax C, Porsius AJ, De Boer A.The completeness of medication histories in hospital medical records of patients admitted to general internal medicine wards. Br J Clin Pharmacol 2000; 49:597–603.
Tam VC, Knowles SR, Cornish PL, Fine N, Marchesano R, Etchells EE. Frequency, type and clinical importance of medication history errors at admission to hospital: a systematic review. CMAJ 2005; 173:510–515.
Bell CM, Brener SS, Gunraj N, et al. Association of ICU or hospital admission with unintentional discontinuation of medications for chronic diseases. JAMA 2011; 306:840–847.
Dobranski S, Hammond I, Khan G, Holdsworth H. The nature of hospital prescribing errors. Br J Clin Governance 2002; 7:187–193.
Gleason KM, Groszek JM, Sullivan C, Rooney D, Barnard C, Noskin GA. Reconciliation of discrepancies in medication histories and admission orders of newly hospitalized patients. Am J Health Syst Pharm 2004; 61:1689–1695.
Rozich JD, Resar KR. Medication safety: one organization’s approach to the challenge. J Clin Outcomes Manage 2001; 8:27–34.
Mueller SK, Sponsler KC, Kripalani S, Schnipper JL. Hospital-based medication reconciliation practices: a systematic review. Arch Intern Med 2012; 172:1057–1069.
Greenwald JL, Halasyamani L, Greene J, et al. Making inpatient medication reconciliation patient centered, clinically relevant and implementable: a consensus statement on key principles and necessary first steps. J Hosp Med 2010; 5:477–485.
Berwick DM, Calkins DR, McCannon CJ, Hackbarth AD. The 100,000 lives campaign: setting a goal and a deadline for improving health care quality. JAMA 2006; 295:324–327.
McCannon CJ, Hackbarth AD, Griffin FA. Miles to go: an introduction to the 5 Million Lives Campaign. Jt Comm J Qual Patient Saf 2007; 33:477–484.
Leotsakos A, Caisley L, Karga M, Kelly E, O’Leary D, Timmons K. High 5s: addressing excellence in patient safety. World Hosp Health Serv 2009; 45:19–22.
Mueller SK, Kripalani S, Stein J, et al. A toolkit to disseminate best practices in inpatient medication reconciliation: multi-center medication reconciliation quality improvement study (MARQUIS). Jt Comm J Qual Patient Saf 2013; 39:371–382.
Pal A, Babbott S, Wilkinson ST. Can the targeted use of a discharge pharmacist significantly decrease 30-day readmissions? Hosp Pharm 2013; 48:380–388.
Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther 2001; 23:1296–1310.
Osterberg L, Blaschke T. Adherence to medication. N Engl J Med 2005; 353:487–497.
Hajjar ER, Hanlon JT, Sloane RJ, et al. Unnecessary drug use in frail older people at hospital discharge. J Am Geriatr Soc 2005; 53:1518–1523.
Zink DA, Pohlman M, Barnes M, Cannon ME. Long-term use of acid suppression started inappropriately during hospitalization. Aliment Pharmacol Ther 2005; 21:1203–1209.
Morandi A, Vasilevskis E, Pandharipande PP, et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J Am Geriatr Soc 2013; 61:1128–1134.
Cox ZL, McCoy AB, Matheny ME, et al. Adverse drug events during AKI and its recovery. Clin J Am Soc Nephrol 2013; 8:1070–1078.
American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
Gallagher P, O’Mahony D. STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions): application to acutely ill elderly patients and comparison with Beers’ criteria. Age Ageing 2008; 37:673–679.
Tjia J, Bonner A, Briesacher BA, McGee S, Terrill E, Miller K. Medication discrepancies upon hospital to skilled nursing facility transitions. J Gen Intern Med 2009; 24:630–635.
Boockvar K, Fishman E, Kyriacou CK, Monias A, Gavi S, Cortes T. Adverse events due to discontinuations in drug use and dose changes in patients transferred between acute and long-term care facilities. Arch Intern Med 2004; 164:545–550.
Kutner M, Greenberg E, Jin Y, Paulsen C. The Health Literacy of America’s Adults: Results From the 2003 National Assessment of Adult Literacy. http://nces.ed.gov/pubs2006/2006483_1.pdf. Accessed March 31, 2015.
Crane JA. Patient comprehension of doctor-patient communication on discharge from the emergency department. J Emerg Med 1997; 15:1–7.
Schillinger D, Piette J, Grumbach K, et al. Closing the loop: physician communication with diabetic patients who have low health literacy. Arch Intern Med 2003; 163:83–90.
Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
Jack BW, Chetty VK, Anthony D, et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med 2009; 150:178–187.
References
Coleman EA, Smith JD, Raha D, Min SJ. Posthospital medication discrepancies: prevalence and contributing factors. Arch Intern Med 2005; 165:1842–1847.
Wong JD, Bajcar JM, Wong GG, et al. Medication reconciliation at hospital discharge: evaluating discrepancies. Ann Pharmacother 2008; 42:1373–1379.
Kripalani S, Roumie CL, Dalal AK, et al; PILL-CVD (Pharmacist Intervention for Low Literacy in Cardiovascular Disease) Study Group. Effect of a pharmacist intervention on clinically important medication errors after hospital discharge: a randomized trial. Ann Intern Med 2012; 157:1-10.
Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med 2003; 138:161–167.
Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. Adverse drug events occurring following hospital discharge. J Gen Intern Med 2005; 20:317–323.
Johnson JA, Bootman JL. Drug-related morbidity and mortality. A cost-of-illness model. Arch Intern Med 1995; 155:1949–1956.
Budnitz DS, Lovegrove MC, Shehab N, Richards CL. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med 2011; 365:2002–2012.
Bates DW, Boyle DL, Vander Vliet MB, Schneider J, Leape L. Relationship between medication errors and adverse drug events. J Gen Intern Med 1995;10:199–205.
Pippins JR, Gandhi TK, Hamann C, et al. Classifying and predicting errors of inpatient medication reconciliation. J Gen Intern Med 2008; 23:1414–1422.
Lau HS, Florax C, Porsius AJ, De Boer A.The completeness of medication histories in hospital medical records of patients admitted to general internal medicine wards. Br J Clin Pharmacol 2000; 49:597–603.
Tam VC, Knowles SR, Cornish PL, Fine N, Marchesano R, Etchells EE. Frequency, type and clinical importance of medication history errors at admission to hospital: a systematic review. CMAJ 2005; 173:510–515.
Bell CM, Brener SS, Gunraj N, et al. Association of ICU or hospital admission with unintentional discontinuation of medications for chronic diseases. JAMA 2011; 306:840–847.
Dobranski S, Hammond I, Khan G, Holdsworth H. The nature of hospital prescribing errors. Br J Clin Governance 2002; 7:187–193.
Gleason KM, Groszek JM, Sullivan C, Rooney D, Barnard C, Noskin GA. Reconciliation of discrepancies in medication histories and admission orders of newly hospitalized patients. Am J Health Syst Pharm 2004; 61:1689–1695.
Rozich JD, Resar KR. Medication safety: one organization’s approach to the challenge. J Clin Outcomes Manage 2001; 8:27–34.
Mueller SK, Sponsler KC, Kripalani S, Schnipper JL. Hospital-based medication reconciliation practices: a systematic review. Arch Intern Med 2012; 172:1057–1069.
Greenwald JL, Halasyamani L, Greene J, et al. Making inpatient medication reconciliation patient centered, clinically relevant and implementable: a consensus statement on key principles and necessary first steps. J Hosp Med 2010; 5:477–485.
Berwick DM, Calkins DR, McCannon CJ, Hackbarth AD. The 100,000 lives campaign: setting a goal and a deadline for improving health care quality. JAMA 2006; 295:324–327.
McCannon CJ, Hackbarth AD, Griffin FA. Miles to go: an introduction to the 5 Million Lives Campaign. Jt Comm J Qual Patient Saf 2007; 33:477–484.
Leotsakos A, Caisley L, Karga M, Kelly E, O’Leary D, Timmons K. High 5s: addressing excellence in patient safety. World Hosp Health Serv 2009; 45:19–22.
Mueller SK, Kripalani S, Stein J, et al. A toolkit to disseminate best practices in inpatient medication reconciliation: multi-center medication reconciliation quality improvement study (MARQUIS). Jt Comm J Qual Patient Saf 2013; 39:371–382.
Pal A, Babbott S, Wilkinson ST. Can the targeted use of a discharge pharmacist significantly decrease 30-day readmissions? Hosp Pharm 2013; 48:380–388.
Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther 2001; 23:1296–1310.
Osterberg L, Blaschke T. Adherence to medication. N Engl J Med 2005; 353:487–497.
Hajjar ER, Hanlon JT, Sloane RJ, et al. Unnecessary drug use in frail older people at hospital discharge. J Am Geriatr Soc 2005; 53:1518–1523.
Zink DA, Pohlman M, Barnes M, Cannon ME. Long-term use of acid suppression started inappropriately during hospitalization. Aliment Pharmacol Ther 2005; 21:1203–1209.
Morandi A, Vasilevskis E, Pandharipande PP, et al. Inappropriate medication prescriptions in elderly adults surviving an intensive care unit hospitalization. J Am Geriatr Soc 2013; 61:1128–1134.
Cox ZL, McCoy AB, Matheny ME, et al. Adverse drug events during AKI and its recovery. Clin J Am Soc Nephrol 2013; 8:1070–1078.
American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
Gallagher P, O’Mahony D. STOPP (Screening Tool of Older Persons’ potentially inappropriate Prescriptions): application to acutely ill elderly patients and comparison with Beers’ criteria. Age Ageing 2008; 37:673–679.
Tjia J, Bonner A, Briesacher BA, McGee S, Terrill E, Miller K. Medication discrepancies upon hospital to skilled nursing facility transitions. J Gen Intern Med 2009; 24:630–635.
Boockvar K, Fishman E, Kyriacou CK, Monias A, Gavi S, Cortes T. Adverse events due to discontinuations in drug use and dose changes in patients transferred between acute and long-term care facilities. Arch Intern Med 2004; 164:545–550.
Kutner M, Greenberg E, Jin Y, Paulsen C. The Health Literacy of America’s Adults: Results From the 2003 National Assessment of Adult Literacy. http://nces.ed.gov/pubs2006/2006483_1.pdf. Accessed March 31, 2015.
Crane JA. Patient comprehension of doctor-patient communication on discharge from the emergency department. J Emerg Med 1997; 15:1–7.
Schillinger D, Piette J, Grumbach K, et al. Closing the loop: physician communication with diabetic patients who have low health literacy. Arch Intern Med 2003; 163:83–90.
Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
Jack BW, Chetty VK, Anthony D, et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med 2009; 150:178–187.
Institutional medication reconciliation programs should include taking a best-possible medication history at admission, intervening when patients are at high risk, and involving pharmacy staff when possible.
Clinicians can incorporate additional interventions into their workflows to optimize medication safety for hospitalized patients.
Reviewing the medication list for errors of omission and commission, patient-specific needs, and “high-alert” drugs further decreases the risk of medication errors.
At discharge, patients should receive counseling to ensure understanding of medications and follow-up plans. Hospital physicians should communicate with outpatient providers about medications and rationales for medication changes.
Most vaccinations are given during childhood, but some require boosting during adulthood or are indicated for specific patient populations such as international travelers or those with certain medical conditions. Although generally safe, some vaccines contain live, attenuated organisms that can cause disease in immunocompromised patients. Thus, knowledge of the indications for and contraindications to specific vaccinations is critical to protect adults in special circumstances who are at risk.
Vaccines have helped eliminate or significantly reduce the burden of more than a dozen illnesses.1–3 The Advisory Committee on Immunization Practices (ACIP) of the US Centers for Disease Control and Prevention (CDC) makes recommendations about vaccinations for normal adults and children as well as for certain groups at high risk of vaccine-preventable infections.4Tables 1 and 2 summarize the recommendations for vaccination by medical condition.4 In addition, several applications are available online, including downloadable apps from the (www.cdc.gov/vaccines/schedules/Schedulers/adult-scheduler.html) and the American College of Physicians (http://immunization.acponline.org/app/).
HUMANITY’S GREATEST ADVANCES IN PREVENTING INFECTIOUS DISEASE
Immunization and improved sanitation are humanity’s greatest advances in preventing sickness and death from infectious diseases. Since Jenner’s discovery in 1796 that milkmaids who had contracted cowpox (vaccinia) were immune to smallpox, vaccination has eliminated smallpox, markedly decreased the incidence of many infectious diseases, and, most recently, shown efficacy in preventing cervical cancer (with the human papillomavirus vaccine) and hepatocellular cancer (with the hepatitis B vaccine).1–3
Unfortunately, vaccination rates remain low for most routine vaccinations indicated for adults. For example, about 60% of adults over age 65 receive pneumococcal vaccination, and fewer than 10% of black patients over age 60 receive zoster vaccination.5 Various factors may account for these low rates, including financial disincentives.6
Nevertheless, vaccination remains one of medicine’s most effective defenses against infectious diseases and is especially important in the special populations discussed below. By being steadfast proponents of vaccination, especially for the most vulnerable patients, physicians can help ensure the optimum protection for their patients.
VACCINATING PREGNANT PATIENTS
When considering vaccination during pregnancy, one must consider the risk and benefit of the vaccine and the risk of the disease in both the mother and the child.
In general, if a pregnant woman is at high risk of exposure to a particular infection, the benefits of vaccinating her against it outweigh the risks. Vaccinating the mother can also protect against certain infections in early infancy through transfer of vaccine-induced immunoglobin G (IgG) across the placenta.7 In general, inactivated vaccines are considered safe in pregnancy, while live-attenuated vaccines are contraindicated.4 Special considerations for pregnant women include:
Tetanus, diphtheria, and acellular pertussis (Tdap). One dose of Tdap vaccine should be given during each pregnancy, preferably at 27 to 36 weeks of gestation, regardless of when the patient received a previous dose.8
Inactivated influenza vaccine should be given as early as possible during the influenza season (October to March) to all pregnant women, regardless of trimester.
Inactivated polio vaccine may be considered for pregnant women with known exposure to polio or travel to endemic areas.
Hepatitis A, hepatitis B, pneumococcal polysaccharide, meningococcal conjugate, and meningococcal polysaccharide vaccines can be given to women at risk of these infections. If a pregnant patient requires pneumococcal polysaccharide vaccine, it should be given during the second or third trimester, as the safety of this vaccine during the first trimester has not been established.9
Smallpox, measles-mumps-rubella, and varicella-containing vaccines are contraindicated in pregnancy. Household contacts of a pregnant woman should not receive smallpox vaccine, as it is the only vaccine known to cause harm to the fetus.10
Human papillomavirus vaccination is not recommended during pregnancy.
Yellow fever live-attenuated vaccine. The safety of this vaccine during pregnancy has not been established,and it is in the US Food and Drug Administration (FDA) pregnancy category C. However, this vaccine is required for entry into certain countries, and it may be offered if the patient is truly at risk of contracting yellow fever. Because pregnancy may affect immunologic response, serologic testing is recommended to document an immune response. If the patient’s itinerary puts her at low risk of yellow fever, then writing her a vaccine waiver letter can be considered.11
VACCINATING IMMUNOCOMPROMISED PATIENTS (NON-HIV)
People who do not have human immunodeficiency virus (HIV) but have a condition such as functional asplenia (sickle cell disease), anatomic asplenia, or complement component deficiency are at higher risk of infection with the encapsulated bacteria Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type b.
Corticosteroids, chemotherapy, radiation for hematologic or solid-organ malignancies, and immune modulators can alter the immune system and pose a risk with the use of live-attenuated vaccines. A corticosteroid dosage equivalent to 2 mg/kg of body weight per day or higher or 20 mg/day of prednisone or higher is generally considered immunosuppressive.
Candidates for organ transplantation should receive vaccinations as early as possible during the disease course leading to transplantation. Vaccinations should be given as soon as the decision is made that the patient is a candidate for transplantation, which could be years or months before the patient actually receives the transplant. In addition to reviewing previously administered vaccinations, pretransplant serologic testing for hepatitis B, varicella, measles, mumps, and rubella antibodieshelps to evaluate the need for vaccination.12
Recipients of hematopoietic stem cell transplantation are at risk of infections with encapsulated bacteria and certain other vaccine-preventable infections. Antibody titers are significantly reduced after stem cell transplantation because of ablation of bone marrow, and thus certain vaccines should be readministered 3 to 6 months after transplantation (eg, influenza, pneumococcal, and H influenzae vaccines). If the recipient is presumed to be immunocompetent, then varicella or measles-mumps-rubella vaccine can be given 24 months after transplantation.13
Apart from adhering to the routine vaccination schedule and avoiding live-attenuated vaccines, specific recommendations apply to persons with immunocompromising conditions14:
Quadrivalent meningococcal conjugate vaccine should be given to adults of all ages with asplenia or complement component deficiency. The schedule includes two doses at least 2 months apart initially and then revaccination every 5 years.
H influenzae type b vaccine should be given to people with asplenia and recipients of hematopoietic stem cells. One dose is recommended for those with asplenia (functional, anatomic, or elective splenectomy) or sickle cell disease if they have not already received it. A three-dose schedule is considered for hematopoietic stem cell transplant recipients 6 to 12 months after successful transplantation.
Pneumococcal conjugate (PCV13) and pneumococcal polysaccharide (PPSV23) vaccinations are recommended for people who have immunocompromising conditions. PCV13, the newer pneumococcal vaccine, was approved by the FDA in 2010 for use in children and was recommended by the ACIP in 2012 for adults age 19 and older with immunocompromising conditions.
People who have not previously received either of these vaccines and are age 19 or older with immunocompromising conditions including asplenia, chronic renal failure, nephrotic syndrome, cerebrospinal fluid leakage, or cochlear implant should receive a single dose of PCV13 followed by a dose of PPSV23 at least 8 weeks later. One-time revaccination 5 years after the first dose of PPSV23 is recommended for patients with immunocompromising conditions.
For those who have previously been vaccinated with PPSV23, a dose of PCV13 can be given 1 or more years after the last dose of PPSV23. These dosing intervals are important, as lower opsonophagocytic antibody responses have been noted if repeat doses of either pneumococcal vaccine are given sooner than the recommended interval.15
Inactivated influenza vaccine is recommended annually, except for patients who are unlikely to respond or those who have received anti-B-cell antibodies within 6 months. Live-attenuated influenza vaccine should not be given to immunocompromised patients.
VACCINATING PATIENTS WHO HAVE HIV
People with HIV should be routinely screened for immunity against certain infections and should be offered vaccination if not immune. The response to vaccines may vary depending on the CD4 count, with a good response in patients whose infection is well controlled with antiretroviral agents and with a preserved CD4 count.16 Special considerations for HIV patients include the following:
Hepatitis A vaccine may be offered to all HIV patients who have no evidence of immunity against hepatitis A, with negative antihepatitis A total and IgG antibodies.
Human papillomavirus vaccine is recommended for men and women with HIV through age 26.
Varicella and measles-mumps-rubella are live-attenuated vaccines and may be considered in patients who are nonimmune and with CD4 counts of 200 cells/µL or higher. However, the ACIP does not make a recommendation regarding the zoster vaccine in HIV patients with CD4 cell counts of 200 cells/µL or higher. In general, live-attenuated vaccines should be avoided in patients with CD4 counts less than 200 or with severe immunocompromised status because of risk of acquiring severe, life-threatening infections.
Pneumococcal vaccine should be given to HIV patients if they have not received it before. The schedule is one dose of PCV13, followed by a dose of PPSV23 at least 8 weeks later. If a patient has been previously vaccinated with PPSV23, then PCV13 is recommended at least 1 year after PPSV23.
Inactivated influenza vaccine is recommended annually. Live-attenuated influenza vaccine should not be given.
Hepatitis B vaccine should be given to nonimmune patients without past or present hepatitis B infection. These patients require higher doses of hepatitis B vaccine (40 μg/mL) than immunocompetent patients, who receive 20 μg/mL. The options include Recombivax HB 40 μg/mL given on a three-dose schedule at 0, 1, and 6 months, and Engerix B, two 20-μg/mL injections given simultaneously on a four-dose schedule at 0, 1, 2, and 6 months.
Meningococcal vaccine. HIV infection is not an indication for meningococcal vaccineunless the patient has other risk factors, such as anatomic or functional asplenia, persistent complement component deficiency, occupational exposure, and travel to endemic areas.
VACCINATING PATIENTS WHO ARE OLDER THAN 60
The immune system deteriorates with age, as does immunity gained from previous vaccinations. Vaccination in this age group reduces the risk of illness and death.17
Zoster vaccine should be offered to people age 60 and older regardless of previous episodes of herpes zoster unless there is a contraindication such as severe immunodeficiency. The zoster vaccine can reduce the incidence of postherpetic neuralgia by 66.5% and herpes zoster by 51% in patients over age 60.18
Pneumococcal conjugate vaccine. PCV13should be offered to all adults age 65 or older. If a person age 65 or older has not received any pneumococcal vaccine before then, PCV13 should be given first, followed by a dose of PPSV23 at least 6 to 12 months after PCV13.
Pneumococcal polysaccharide vaccine. If PPSV23 was given before age 65 for another indication, a dose of PCV 13 should be given at age 65 or later, as long as 6 to 12 months have passed since the previous dose of PPSV 23. The patient should receive the last dose of PPSV23 vaccine 5 years after the first dose of PPSV23.4
Influenza vaccine. People 65 or older are at higher risk of complications from influenza, and vaccine should be offered annually. High-dose inactivated influenza vaccine can be used in this age group.4
Tdap. If never given before, Tdap is recommended regardless of the interval since the most recent Td vaccination, followed by a Td booster every 10 years.
VACCINATING PATIENTS WHO HAVE CHRONIC KIDNEY DISEASE
Patients with chronic kidney disease are at risk of certain infections, so vaccination is an important preventive measure.19 Immunizations should be offered to all patients with chronic kidney disease regardless of the disease stage, but they are recommended during the early stages of progressive renal disease to increase the likelihood of vaccine-induced immunity.20
Pneumococcal conjugate vaccine. PCV13 is recommended for adults 19 or older with chronic renal disease or nephrotic syndrome. One dose of PCV13 should be given, followed by a dose of PPSV23 at least 8 weeks later. If the patient has been previously vaccinated with PPSV23, then PCV13 at least 1 year after PPSV23 is recommended.
Hepatitis B vaccine should be given to nonimmune patients without past or present hepatitis B infection. Adult patients on hemodialysis require higher doses of hepatitis B vaccine. The options include Recombivax HB 40 μg/mL given on a three-dose schedule at 0, 1, and 6 months, and Engerix B, two 20-μg/mL injections given simultaneously on a four-dose schedule at 0, 1, 2, and 6 months.
Influenza vaccine should be offered annually to patients with chronic kidney disease.
VACCINATING IMMUNOCOMPROMISED INTERNATIONAL TRAVELERS
International travel for business or pleasure is increasingly common, and immunocompromised patients require specific attention as they may face unanticipated pathogens or have special requirements. Transplant recipients should ideally receive routine and travel-related vaccines as early as possible before transplantation. Vaccination is generally avoided in the first 6 months after organ transplantation to avoid confusion with early graft dysfunction or rejection.21 However, it should be considered as soon as a patient develops an illness that might lead to transplantation.
Evaluation of patients for vaccination should include an assessment of the travel-specific epidemiologic risk, the nature of the vaccine (live-attenuated or other), and the immune status. As discussed above, live-attenuated vaccines should be avoided in immunocompromised patients, and thus the injectable typhoid vaccine should be given in lieu of the attenuated oral vaccine.
Yellow fever vaccine is required before entrance to certain countries but should not be given to immunocompromised patients, although it can probably be given to asymptomatic HIV-infected adults with a CD4 count higher than 200 cells/μL who are exposed to substantial risk.22 For patients who cannot receive the vaccine, some governments will accept a physician’s letter stating the patient has a contraindication to vaccination.
VACCINATING HOUSEHOLD MEMBERS OF IMMUNOCOMPROMISED PATIENTS
Protecting immunocompromised patients from infectious diseases involves vaccinating not only the patient but also household members so that they do not acquire infections and then bring them into the household. Immunocompetent members of a household can receive inactivated vaccines based on the recommended ACIP schedule.
Annual inactivated influenza vaccination is recommended, although the live-attenuated influenza virus vaccine can be substituted if the immunocompromised patient is not within 2 months of hematopoietic stem cell transplantation, does not have graft-vs-host disease, and does not have severe combined immune deficiency.
Other live-attenuated vaccines can usually be given if indicated, including measles-mumps-rubella vaccine, rotavirus vaccine in infants, varicella vaccine, and zoster vaccine.14
References
Crosignani P, De Stefani A, Fara GM, et al. Towards the eradication of HPV infection through universal specific vaccination. BMC Public Health 2013;13:642.
Plotkin SL, Plotkin SA. A short history of vaccination. In: Plotkin, SA, Orenstein W, Offit PA, editors. Vaccines, 5th ed. Philadelphia, PA: Elsevier Health Sciences; 2008:1–16.
Wong VW, Chan HL. Prevention of hepatocellular carcinoma: a concise review of contemporary issues. Ann Hepatol 2012; 11:284–293.
Kim DK, Bridges CB, Harriman K; Centers for Disease Control and Prevention (CDC). Advisory Committee on Immunization Practices. Advisory Committee on Immunization Practices recommended immunization schedule for adults aged 19 years or older: United States, 2015. Ann Intern Med 2015; 162:214–223.
Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Noninfluenza vaccination coverage among adults—United States, 2012. MMWR Morb Mortal Wkly Rep 2014; 63:95-102.
Hurley LP, Bridges CB, Harpaz R, et al. US physicians’ perspective of adult vaccine delivery. Ann Intern Med 2014; 160:161.
Lindsey B, Kampmann B, Jones C. Maternal immunization as a strategy to decrease susceptibility to infection in newborn infants. Curr Opin Infect Dis 2013; 26:248–253.
Centers for Disease Control and Prevention (CDC). Updated recommendations for use of tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccine (Tdap) in pregnant women—Advisory Committee on Immunization Practices (ACIP), 2012. MMWR Morb Mortal Wkly Rep 2013; 62:131–135.
Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997; 46:1–24.
Wharton M, Strikas RA, Harpaz R, et al; Advisory Committee on Immunization Practices; Healthcare Infection Control Practices Advisory Committee. Recommendations for using smallpox vaccine in a pre-event vaccination program. Supplemental recommendations of the Advisory Committee on Immunization Practices (ACIP) and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm Rep 2003; 52:1–16.
Staples JE, Gershman M, Fischer M; Centers for Disease Control and Prevention (CDC). Yellow fever vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2010; 59:1–27.
Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2011; 60:1–64.
Rubin LG, Levin MJ, Ljungman P, et al. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis 2014; 58:309–318.
Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine for adults with immunocompromising conditions: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2012; 61:816–819.
Aberg JA, Gallant JE, Ghanem KG, Emmanuel P, Zingman BS, Horberg MA, Infectious Diseases Society of America. Primary care guidelines for the management of persons infected with HIV: 2013 update by the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis 2014; 58:1–10.
Eilers R, Krabbe PF, van Essen TG, Suijkerbuijk A, van Lier A, de Melker HE. Assessment of vaccine candidates for persons aged 50 and older: a review. BMC Geriatr 2013; 13:32.
Oxman MN, Levin MJ; Shingles Prevention Study Group. Vaccination against Herpes Zoster and Postherpetic Neuralgia. J Infect Dis 2008; 197(suppl 2):S228–S236.
Soni R, Horowitz B, Unruh M. Immunization in end-stage renal disease: opportunity to improve outcomes. Semin Dial 2013; 26:416–426.
Faria Farhat, MD, FACP Section of Infectious Diseases, Department of Internal Medicine, MedStar Washington Hospital Center, Washington, DC
Glenn Wortmann, MD, FIDSA, FACP Chief, Section of Infectious Diseases, Department of Internal Medicine, MedStar Washington Hospital Center, Washington, DC
Address: Faria Farhat, MD, FACP, Department of Internal Medicine, Section of Infectious Diseases, MedStar Washington Hospital Center, 110 Irving Street, NW, Washington, DC 20010; e-mail: faria.farhat@medstar.net
Faria Farhat, MD, FACP Section of Infectious Diseases, Department of Internal Medicine, MedStar Washington Hospital Center, Washington, DC
Glenn Wortmann, MD, FIDSA, FACP Chief, Section of Infectious Diseases, Department of Internal Medicine, MedStar Washington Hospital Center, Washington, DC
Address: Faria Farhat, MD, FACP, Department of Internal Medicine, Section of Infectious Diseases, MedStar Washington Hospital Center, 110 Irving Street, NW, Washington, DC 20010; e-mail: faria.farhat@medstar.net
Author and Disclosure Information
Faria Farhat, MD, FACP Section of Infectious Diseases, Department of Internal Medicine, MedStar Washington Hospital Center, Washington, DC
Glenn Wortmann, MD, FIDSA, FACP Chief, Section of Infectious Diseases, Department of Internal Medicine, MedStar Washington Hospital Center, Washington, DC
Address: Faria Farhat, MD, FACP, Department of Internal Medicine, Section of Infectious Diseases, MedStar Washington Hospital Center, 110 Irving Street, NW, Washington, DC 20010; e-mail: faria.farhat@medstar.net
Most vaccinations are given during childhood, but some require boosting during adulthood or are indicated for specific patient populations such as international travelers or those with certain medical conditions. Although generally safe, some vaccines contain live, attenuated organisms that can cause disease in immunocompromised patients. Thus, knowledge of the indications for and contraindications to specific vaccinations is critical to protect adults in special circumstances who are at risk.
Vaccines have helped eliminate or significantly reduce the burden of more than a dozen illnesses.1–3 The Advisory Committee on Immunization Practices (ACIP) of the US Centers for Disease Control and Prevention (CDC) makes recommendations about vaccinations for normal adults and children as well as for certain groups at high risk of vaccine-preventable infections.4Tables 1 and 2 summarize the recommendations for vaccination by medical condition.4 In addition, several applications are available online, including downloadable apps from the (www.cdc.gov/vaccines/schedules/Schedulers/adult-scheduler.html) and the American College of Physicians (http://immunization.acponline.org/app/).
HUMANITY’S GREATEST ADVANCES IN PREVENTING INFECTIOUS DISEASE
Immunization and improved sanitation are humanity’s greatest advances in preventing sickness and death from infectious diseases. Since Jenner’s discovery in 1796 that milkmaids who had contracted cowpox (vaccinia) were immune to smallpox, vaccination has eliminated smallpox, markedly decreased the incidence of many infectious diseases, and, most recently, shown efficacy in preventing cervical cancer (with the human papillomavirus vaccine) and hepatocellular cancer (with the hepatitis B vaccine).1–3
Unfortunately, vaccination rates remain low for most routine vaccinations indicated for adults. For example, about 60% of adults over age 65 receive pneumococcal vaccination, and fewer than 10% of black patients over age 60 receive zoster vaccination.5 Various factors may account for these low rates, including financial disincentives.6
Nevertheless, vaccination remains one of medicine’s most effective defenses against infectious diseases and is especially important in the special populations discussed below. By being steadfast proponents of vaccination, especially for the most vulnerable patients, physicians can help ensure the optimum protection for their patients.
VACCINATING PREGNANT PATIENTS
When considering vaccination during pregnancy, one must consider the risk and benefit of the vaccine and the risk of the disease in both the mother and the child.
In general, if a pregnant woman is at high risk of exposure to a particular infection, the benefits of vaccinating her against it outweigh the risks. Vaccinating the mother can also protect against certain infections in early infancy through transfer of vaccine-induced immunoglobin G (IgG) across the placenta.7 In general, inactivated vaccines are considered safe in pregnancy, while live-attenuated vaccines are contraindicated.4 Special considerations for pregnant women include:
Tetanus, diphtheria, and acellular pertussis (Tdap). One dose of Tdap vaccine should be given during each pregnancy, preferably at 27 to 36 weeks of gestation, regardless of when the patient received a previous dose.8
Inactivated influenza vaccine should be given as early as possible during the influenza season (October to March) to all pregnant women, regardless of trimester.
Inactivated polio vaccine may be considered for pregnant women with known exposure to polio or travel to endemic areas.
Hepatitis A, hepatitis B, pneumococcal polysaccharide, meningococcal conjugate, and meningococcal polysaccharide vaccines can be given to women at risk of these infections. If a pregnant patient requires pneumococcal polysaccharide vaccine, it should be given during the second or third trimester, as the safety of this vaccine during the first trimester has not been established.9
Smallpox, measles-mumps-rubella, and varicella-containing vaccines are contraindicated in pregnancy. Household contacts of a pregnant woman should not receive smallpox vaccine, as it is the only vaccine known to cause harm to the fetus.10
Human papillomavirus vaccination is not recommended during pregnancy.
Yellow fever live-attenuated vaccine. The safety of this vaccine during pregnancy has not been established,and it is in the US Food and Drug Administration (FDA) pregnancy category C. However, this vaccine is required for entry into certain countries, and it may be offered if the patient is truly at risk of contracting yellow fever. Because pregnancy may affect immunologic response, serologic testing is recommended to document an immune response. If the patient’s itinerary puts her at low risk of yellow fever, then writing her a vaccine waiver letter can be considered.11
VACCINATING IMMUNOCOMPROMISED PATIENTS (NON-HIV)
People who do not have human immunodeficiency virus (HIV) but have a condition such as functional asplenia (sickle cell disease), anatomic asplenia, or complement component deficiency are at higher risk of infection with the encapsulated bacteria Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type b.
Corticosteroids, chemotherapy, radiation for hematologic or solid-organ malignancies, and immune modulators can alter the immune system and pose a risk with the use of live-attenuated vaccines. A corticosteroid dosage equivalent to 2 mg/kg of body weight per day or higher or 20 mg/day of prednisone or higher is generally considered immunosuppressive.
Candidates for organ transplantation should receive vaccinations as early as possible during the disease course leading to transplantation. Vaccinations should be given as soon as the decision is made that the patient is a candidate for transplantation, which could be years or months before the patient actually receives the transplant. In addition to reviewing previously administered vaccinations, pretransplant serologic testing for hepatitis B, varicella, measles, mumps, and rubella antibodieshelps to evaluate the need for vaccination.12
Recipients of hematopoietic stem cell transplantation are at risk of infections with encapsulated bacteria and certain other vaccine-preventable infections. Antibody titers are significantly reduced after stem cell transplantation because of ablation of bone marrow, and thus certain vaccines should be readministered 3 to 6 months after transplantation (eg, influenza, pneumococcal, and H influenzae vaccines). If the recipient is presumed to be immunocompetent, then varicella or measles-mumps-rubella vaccine can be given 24 months after transplantation.13
Apart from adhering to the routine vaccination schedule and avoiding live-attenuated vaccines, specific recommendations apply to persons with immunocompromising conditions14:
Quadrivalent meningococcal conjugate vaccine should be given to adults of all ages with asplenia or complement component deficiency. The schedule includes two doses at least 2 months apart initially and then revaccination every 5 years.
H influenzae type b vaccine should be given to people with asplenia and recipients of hematopoietic stem cells. One dose is recommended for those with asplenia (functional, anatomic, or elective splenectomy) or sickle cell disease if they have not already received it. A three-dose schedule is considered for hematopoietic stem cell transplant recipients 6 to 12 months after successful transplantation.
Pneumococcal conjugate (PCV13) and pneumococcal polysaccharide (PPSV23) vaccinations are recommended for people who have immunocompromising conditions. PCV13, the newer pneumococcal vaccine, was approved by the FDA in 2010 for use in children and was recommended by the ACIP in 2012 for adults age 19 and older with immunocompromising conditions.
People who have not previously received either of these vaccines and are age 19 or older with immunocompromising conditions including asplenia, chronic renal failure, nephrotic syndrome, cerebrospinal fluid leakage, or cochlear implant should receive a single dose of PCV13 followed by a dose of PPSV23 at least 8 weeks later. One-time revaccination 5 years after the first dose of PPSV23 is recommended for patients with immunocompromising conditions.
For those who have previously been vaccinated with PPSV23, a dose of PCV13 can be given 1 or more years after the last dose of PPSV23. These dosing intervals are important, as lower opsonophagocytic antibody responses have been noted if repeat doses of either pneumococcal vaccine are given sooner than the recommended interval.15
Inactivated influenza vaccine is recommended annually, except for patients who are unlikely to respond or those who have received anti-B-cell antibodies within 6 months. Live-attenuated influenza vaccine should not be given to immunocompromised patients.
VACCINATING PATIENTS WHO HAVE HIV
People with HIV should be routinely screened for immunity against certain infections and should be offered vaccination if not immune. The response to vaccines may vary depending on the CD4 count, with a good response in patients whose infection is well controlled with antiretroviral agents and with a preserved CD4 count.16 Special considerations for HIV patients include the following:
Hepatitis A vaccine may be offered to all HIV patients who have no evidence of immunity against hepatitis A, with negative antihepatitis A total and IgG antibodies.
Human papillomavirus vaccine is recommended for men and women with HIV through age 26.
Varicella and measles-mumps-rubella are live-attenuated vaccines and may be considered in patients who are nonimmune and with CD4 counts of 200 cells/µL or higher. However, the ACIP does not make a recommendation regarding the zoster vaccine in HIV patients with CD4 cell counts of 200 cells/µL or higher. In general, live-attenuated vaccines should be avoided in patients with CD4 counts less than 200 or with severe immunocompromised status because of risk of acquiring severe, life-threatening infections.
Pneumococcal vaccine should be given to HIV patients if they have not received it before. The schedule is one dose of PCV13, followed by a dose of PPSV23 at least 8 weeks later. If a patient has been previously vaccinated with PPSV23, then PCV13 is recommended at least 1 year after PPSV23.
Inactivated influenza vaccine is recommended annually. Live-attenuated influenza vaccine should not be given.
Hepatitis B vaccine should be given to nonimmune patients without past or present hepatitis B infection. These patients require higher doses of hepatitis B vaccine (40 μg/mL) than immunocompetent patients, who receive 20 μg/mL. The options include Recombivax HB 40 μg/mL given on a three-dose schedule at 0, 1, and 6 months, and Engerix B, two 20-μg/mL injections given simultaneously on a four-dose schedule at 0, 1, 2, and 6 months.
Meningococcal vaccine. HIV infection is not an indication for meningococcal vaccineunless the patient has other risk factors, such as anatomic or functional asplenia, persistent complement component deficiency, occupational exposure, and travel to endemic areas.
VACCINATING PATIENTS WHO ARE OLDER THAN 60
The immune system deteriorates with age, as does immunity gained from previous vaccinations. Vaccination in this age group reduces the risk of illness and death.17
Zoster vaccine should be offered to people age 60 and older regardless of previous episodes of herpes zoster unless there is a contraindication such as severe immunodeficiency. The zoster vaccine can reduce the incidence of postherpetic neuralgia by 66.5% and herpes zoster by 51% in patients over age 60.18
Pneumococcal conjugate vaccine. PCV13should be offered to all adults age 65 or older. If a person age 65 or older has not received any pneumococcal vaccine before then, PCV13 should be given first, followed by a dose of PPSV23 at least 6 to 12 months after PCV13.
Pneumococcal polysaccharide vaccine. If PPSV23 was given before age 65 for another indication, a dose of PCV 13 should be given at age 65 or later, as long as 6 to 12 months have passed since the previous dose of PPSV 23. The patient should receive the last dose of PPSV23 vaccine 5 years after the first dose of PPSV23.4
Influenza vaccine. People 65 or older are at higher risk of complications from influenza, and vaccine should be offered annually. High-dose inactivated influenza vaccine can be used in this age group.4
Tdap. If never given before, Tdap is recommended regardless of the interval since the most recent Td vaccination, followed by a Td booster every 10 years.
VACCINATING PATIENTS WHO HAVE CHRONIC KIDNEY DISEASE
Patients with chronic kidney disease are at risk of certain infections, so vaccination is an important preventive measure.19 Immunizations should be offered to all patients with chronic kidney disease regardless of the disease stage, but they are recommended during the early stages of progressive renal disease to increase the likelihood of vaccine-induced immunity.20
Pneumococcal conjugate vaccine. PCV13 is recommended for adults 19 or older with chronic renal disease or nephrotic syndrome. One dose of PCV13 should be given, followed by a dose of PPSV23 at least 8 weeks later. If the patient has been previously vaccinated with PPSV23, then PCV13 at least 1 year after PPSV23 is recommended.
Hepatitis B vaccine should be given to nonimmune patients without past or present hepatitis B infection. Adult patients on hemodialysis require higher doses of hepatitis B vaccine. The options include Recombivax HB 40 μg/mL given on a three-dose schedule at 0, 1, and 6 months, and Engerix B, two 20-μg/mL injections given simultaneously on a four-dose schedule at 0, 1, 2, and 6 months.
Influenza vaccine should be offered annually to patients with chronic kidney disease.
VACCINATING IMMUNOCOMPROMISED INTERNATIONAL TRAVELERS
International travel for business or pleasure is increasingly common, and immunocompromised patients require specific attention as they may face unanticipated pathogens or have special requirements. Transplant recipients should ideally receive routine and travel-related vaccines as early as possible before transplantation. Vaccination is generally avoided in the first 6 months after organ transplantation to avoid confusion with early graft dysfunction or rejection.21 However, it should be considered as soon as a patient develops an illness that might lead to transplantation.
Evaluation of patients for vaccination should include an assessment of the travel-specific epidemiologic risk, the nature of the vaccine (live-attenuated or other), and the immune status. As discussed above, live-attenuated vaccines should be avoided in immunocompromised patients, and thus the injectable typhoid vaccine should be given in lieu of the attenuated oral vaccine.
Yellow fever vaccine is required before entrance to certain countries but should not be given to immunocompromised patients, although it can probably be given to asymptomatic HIV-infected adults with a CD4 count higher than 200 cells/μL who are exposed to substantial risk.22 For patients who cannot receive the vaccine, some governments will accept a physician’s letter stating the patient has a contraindication to vaccination.
VACCINATING HOUSEHOLD MEMBERS OF IMMUNOCOMPROMISED PATIENTS
Protecting immunocompromised patients from infectious diseases involves vaccinating not only the patient but also household members so that they do not acquire infections and then bring them into the household. Immunocompetent members of a household can receive inactivated vaccines based on the recommended ACIP schedule.
Annual inactivated influenza vaccination is recommended, although the live-attenuated influenza virus vaccine can be substituted if the immunocompromised patient is not within 2 months of hematopoietic stem cell transplantation, does not have graft-vs-host disease, and does not have severe combined immune deficiency.
Other live-attenuated vaccines can usually be given if indicated, including measles-mumps-rubella vaccine, rotavirus vaccine in infants, varicella vaccine, and zoster vaccine.14
Most vaccinations are given during childhood, but some require boosting during adulthood or are indicated for specific patient populations such as international travelers or those with certain medical conditions. Although generally safe, some vaccines contain live, attenuated organisms that can cause disease in immunocompromised patients. Thus, knowledge of the indications for and contraindications to specific vaccinations is critical to protect adults in special circumstances who are at risk.
Vaccines have helped eliminate or significantly reduce the burden of more than a dozen illnesses.1–3 The Advisory Committee on Immunization Practices (ACIP) of the US Centers for Disease Control and Prevention (CDC) makes recommendations about vaccinations for normal adults and children as well as for certain groups at high risk of vaccine-preventable infections.4Tables 1 and 2 summarize the recommendations for vaccination by medical condition.4 In addition, several applications are available online, including downloadable apps from the (www.cdc.gov/vaccines/schedules/Schedulers/adult-scheduler.html) and the American College of Physicians (http://immunization.acponline.org/app/).
HUMANITY’S GREATEST ADVANCES IN PREVENTING INFECTIOUS DISEASE
Immunization and improved sanitation are humanity’s greatest advances in preventing sickness and death from infectious diseases. Since Jenner’s discovery in 1796 that milkmaids who had contracted cowpox (vaccinia) were immune to smallpox, vaccination has eliminated smallpox, markedly decreased the incidence of many infectious diseases, and, most recently, shown efficacy in preventing cervical cancer (with the human papillomavirus vaccine) and hepatocellular cancer (with the hepatitis B vaccine).1–3
Unfortunately, vaccination rates remain low for most routine vaccinations indicated for adults. For example, about 60% of adults over age 65 receive pneumococcal vaccination, and fewer than 10% of black patients over age 60 receive zoster vaccination.5 Various factors may account for these low rates, including financial disincentives.6
Nevertheless, vaccination remains one of medicine’s most effective defenses against infectious diseases and is especially important in the special populations discussed below. By being steadfast proponents of vaccination, especially for the most vulnerable patients, physicians can help ensure the optimum protection for their patients.
VACCINATING PREGNANT PATIENTS
When considering vaccination during pregnancy, one must consider the risk and benefit of the vaccine and the risk of the disease in both the mother and the child.
In general, if a pregnant woman is at high risk of exposure to a particular infection, the benefits of vaccinating her against it outweigh the risks. Vaccinating the mother can also protect against certain infections in early infancy through transfer of vaccine-induced immunoglobin G (IgG) across the placenta.7 In general, inactivated vaccines are considered safe in pregnancy, while live-attenuated vaccines are contraindicated.4 Special considerations for pregnant women include:
Tetanus, diphtheria, and acellular pertussis (Tdap). One dose of Tdap vaccine should be given during each pregnancy, preferably at 27 to 36 weeks of gestation, regardless of when the patient received a previous dose.8
Inactivated influenza vaccine should be given as early as possible during the influenza season (October to March) to all pregnant women, regardless of trimester.
Inactivated polio vaccine may be considered for pregnant women with known exposure to polio or travel to endemic areas.
Hepatitis A, hepatitis B, pneumococcal polysaccharide, meningococcal conjugate, and meningococcal polysaccharide vaccines can be given to women at risk of these infections. If a pregnant patient requires pneumococcal polysaccharide vaccine, it should be given during the second or third trimester, as the safety of this vaccine during the first trimester has not been established.9
Smallpox, measles-mumps-rubella, and varicella-containing vaccines are contraindicated in pregnancy. Household contacts of a pregnant woman should not receive smallpox vaccine, as it is the only vaccine known to cause harm to the fetus.10
Human papillomavirus vaccination is not recommended during pregnancy.
Yellow fever live-attenuated vaccine. The safety of this vaccine during pregnancy has not been established,and it is in the US Food and Drug Administration (FDA) pregnancy category C. However, this vaccine is required for entry into certain countries, and it may be offered if the patient is truly at risk of contracting yellow fever. Because pregnancy may affect immunologic response, serologic testing is recommended to document an immune response. If the patient’s itinerary puts her at low risk of yellow fever, then writing her a vaccine waiver letter can be considered.11
VACCINATING IMMUNOCOMPROMISED PATIENTS (NON-HIV)
People who do not have human immunodeficiency virus (HIV) but have a condition such as functional asplenia (sickle cell disease), anatomic asplenia, or complement component deficiency are at higher risk of infection with the encapsulated bacteria Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type b.
Corticosteroids, chemotherapy, radiation for hematologic or solid-organ malignancies, and immune modulators can alter the immune system and pose a risk with the use of live-attenuated vaccines. A corticosteroid dosage equivalent to 2 mg/kg of body weight per day or higher or 20 mg/day of prednisone or higher is generally considered immunosuppressive.
Candidates for organ transplantation should receive vaccinations as early as possible during the disease course leading to transplantation. Vaccinations should be given as soon as the decision is made that the patient is a candidate for transplantation, which could be years or months before the patient actually receives the transplant. In addition to reviewing previously administered vaccinations, pretransplant serologic testing for hepatitis B, varicella, measles, mumps, and rubella antibodieshelps to evaluate the need for vaccination.12
Recipients of hematopoietic stem cell transplantation are at risk of infections with encapsulated bacteria and certain other vaccine-preventable infections. Antibody titers are significantly reduced after stem cell transplantation because of ablation of bone marrow, and thus certain vaccines should be readministered 3 to 6 months after transplantation (eg, influenza, pneumococcal, and H influenzae vaccines). If the recipient is presumed to be immunocompetent, then varicella or measles-mumps-rubella vaccine can be given 24 months after transplantation.13
Apart from adhering to the routine vaccination schedule and avoiding live-attenuated vaccines, specific recommendations apply to persons with immunocompromising conditions14:
Quadrivalent meningococcal conjugate vaccine should be given to adults of all ages with asplenia or complement component deficiency. The schedule includes two doses at least 2 months apart initially and then revaccination every 5 years.
H influenzae type b vaccine should be given to people with asplenia and recipients of hematopoietic stem cells. One dose is recommended for those with asplenia (functional, anatomic, or elective splenectomy) or sickle cell disease if they have not already received it. A three-dose schedule is considered for hematopoietic stem cell transplant recipients 6 to 12 months after successful transplantation.
Pneumococcal conjugate (PCV13) and pneumococcal polysaccharide (PPSV23) vaccinations are recommended for people who have immunocompromising conditions. PCV13, the newer pneumococcal vaccine, was approved by the FDA in 2010 for use in children and was recommended by the ACIP in 2012 for adults age 19 and older with immunocompromising conditions.
People who have not previously received either of these vaccines and are age 19 or older with immunocompromising conditions including asplenia, chronic renal failure, nephrotic syndrome, cerebrospinal fluid leakage, or cochlear implant should receive a single dose of PCV13 followed by a dose of PPSV23 at least 8 weeks later. One-time revaccination 5 years after the first dose of PPSV23 is recommended for patients with immunocompromising conditions.
For those who have previously been vaccinated with PPSV23, a dose of PCV13 can be given 1 or more years after the last dose of PPSV23. These dosing intervals are important, as lower opsonophagocytic antibody responses have been noted if repeat doses of either pneumococcal vaccine are given sooner than the recommended interval.15
Inactivated influenza vaccine is recommended annually, except for patients who are unlikely to respond or those who have received anti-B-cell antibodies within 6 months. Live-attenuated influenza vaccine should not be given to immunocompromised patients.
VACCINATING PATIENTS WHO HAVE HIV
People with HIV should be routinely screened for immunity against certain infections and should be offered vaccination if not immune. The response to vaccines may vary depending on the CD4 count, with a good response in patients whose infection is well controlled with antiretroviral agents and with a preserved CD4 count.16 Special considerations for HIV patients include the following:
Hepatitis A vaccine may be offered to all HIV patients who have no evidence of immunity against hepatitis A, with negative antihepatitis A total and IgG antibodies.
Human papillomavirus vaccine is recommended for men and women with HIV through age 26.
Varicella and measles-mumps-rubella are live-attenuated vaccines and may be considered in patients who are nonimmune and with CD4 counts of 200 cells/µL or higher. However, the ACIP does not make a recommendation regarding the zoster vaccine in HIV patients with CD4 cell counts of 200 cells/µL or higher. In general, live-attenuated vaccines should be avoided in patients with CD4 counts less than 200 or with severe immunocompromised status because of risk of acquiring severe, life-threatening infections.
Pneumococcal vaccine should be given to HIV patients if they have not received it before. The schedule is one dose of PCV13, followed by a dose of PPSV23 at least 8 weeks later. If a patient has been previously vaccinated with PPSV23, then PCV13 is recommended at least 1 year after PPSV23.
Inactivated influenza vaccine is recommended annually. Live-attenuated influenza vaccine should not be given.
Hepatitis B vaccine should be given to nonimmune patients without past or present hepatitis B infection. These patients require higher doses of hepatitis B vaccine (40 μg/mL) than immunocompetent patients, who receive 20 μg/mL. The options include Recombivax HB 40 μg/mL given on a three-dose schedule at 0, 1, and 6 months, and Engerix B, two 20-μg/mL injections given simultaneously on a four-dose schedule at 0, 1, 2, and 6 months.
Meningococcal vaccine. HIV infection is not an indication for meningococcal vaccineunless the patient has other risk factors, such as anatomic or functional asplenia, persistent complement component deficiency, occupational exposure, and travel to endemic areas.
VACCINATING PATIENTS WHO ARE OLDER THAN 60
The immune system deteriorates with age, as does immunity gained from previous vaccinations. Vaccination in this age group reduces the risk of illness and death.17
Zoster vaccine should be offered to people age 60 and older regardless of previous episodes of herpes zoster unless there is a contraindication such as severe immunodeficiency. The zoster vaccine can reduce the incidence of postherpetic neuralgia by 66.5% and herpes zoster by 51% in patients over age 60.18
Pneumococcal conjugate vaccine. PCV13should be offered to all adults age 65 or older. If a person age 65 or older has not received any pneumococcal vaccine before then, PCV13 should be given first, followed by a dose of PPSV23 at least 6 to 12 months after PCV13.
Pneumococcal polysaccharide vaccine. If PPSV23 was given before age 65 for another indication, a dose of PCV 13 should be given at age 65 or later, as long as 6 to 12 months have passed since the previous dose of PPSV 23. The patient should receive the last dose of PPSV23 vaccine 5 years after the first dose of PPSV23.4
Influenza vaccine. People 65 or older are at higher risk of complications from influenza, and vaccine should be offered annually. High-dose inactivated influenza vaccine can be used in this age group.4
Tdap. If never given before, Tdap is recommended regardless of the interval since the most recent Td vaccination, followed by a Td booster every 10 years.
VACCINATING PATIENTS WHO HAVE CHRONIC KIDNEY DISEASE
Patients with chronic kidney disease are at risk of certain infections, so vaccination is an important preventive measure.19 Immunizations should be offered to all patients with chronic kidney disease regardless of the disease stage, but they are recommended during the early stages of progressive renal disease to increase the likelihood of vaccine-induced immunity.20
Pneumococcal conjugate vaccine. PCV13 is recommended for adults 19 or older with chronic renal disease or nephrotic syndrome. One dose of PCV13 should be given, followed by a dose of PPSV23 at least 8 weeks later. If the patient has been previously vaccinated with PPSV23, then PCV13 at least 1 year after PPSV23 is recommended.
Hepatitis B vaccine should be given to nonimmune patients without past or present hepatitis B infection. Adult patients on hemodialysis require higher doses of hepatitis B vaccine. The options include Recombivax HB 40 μg/mL given on a three-dose schedule at 0, 1, and 6 months, and Engerix B, two 20-μg/mL injections given simultaneously on a four-dose schedule at 0, 1, 2, and 6 months.
Influenza vaccine should be offered annually to patients with chronic kidney disease.
VACCINATING IMMUNOCOMPROMISED INTERNATIONAL TRAVELERS
International travel for business or pleasure is increasingly common, and immunocompromised patients require specific attention as they may face unanticipated pathogens or have special requirements. Transplant recipients should ideally receive routine and travel-related vaccines as early as possible before transplantation. Vaccination is generally avoided in the first 6 months after organ transplantation to avoid confusion with early graft dysfunction or rejection.21 However, it should be considered as soon as a patient develops an illness that might lead to transplantation.
Evaluation of patients for vaccination should include an assessment of the travel-specific epidemiologic risk, the nature of the vaccine (live-attenuated or other), and the immune status. As discussed above, live-attenuated vaccines should be avoided in immunocompromised patients, and thus the injectable typhoid vaccine should be given in lieu of the attenuated oral vaccine.
Yellow fever vaccine is required before entrance to certain countries but should not be given to immunocompromised patients, although it can probably be given to asymptomatic HIV-infected adults with a CD4 count higher than 200 cells/μL who are exposed to substantial risk.22 For patients who cannot receive the vaccine, some governments will accept a physician’s letter stating the patient has a contraindication to vaccination.
VACCINATING HOUSEHOLD MEMBERS OF IMMUNOCOMPROMISED PATIENTS
Protecting immunocompromised patients from infectious diseases involves vaccinating not only the patient but also household members so that they do not acquire infections and then bring them into the household. Immunocompetent members of a household can receive inactivated vaccines based on the recommended ACIP schedule.
Annual inactivated influenza vaccination is recommended, although the live-attenuated influenza virus vaccine can be substituted if the immunocompromised patient is not within 2 months of hematopoietic stem cell transplantation, does not have graft-vs-host disease, and does not have severe combined immune deficiency.
Other live-attenuated vaccines can usually be given if indicated, including measles-mumps-rubella vaccine, rotavirus vaccine in infants, varicella vaccine, and zoster vaccine.14
References
Crosignani P, De Stefani A, Fara GM, et al. Towards the eradication of HPV infection through universal specific vaccination. BMC Public Health 2013;13:642.
Plotkin SL, Plotkin SA. A short history of vaccination. In: Plotkin, SA, Orenstein W, Offit PA, editors. Vaccines, 5th ed. Philadelphia, PA: Elsevier Health Sciences; 2008:1–16.
Wong VW, Chan HL. Prevention of hepatocellular carcinoma: a concise review of contemporary issues. Ann Hepatol 2012; 11:284–293.
Kim DK, Bridges CB, Harriman K; Centers for Disease Control and Prevention (CDC). Advisory Committee on Immunization Practices. Advisory Committee on Immunization Practices recommended immunization schedule for adults aged 19 years or older: United States, 2015. Ann Intern Med 2015; 162:214–223.
Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Noninfluenza vaccination coverage among adults—United States, 2012. MMWR Morb Mortal Wkly Rep 2014; 63:95-102.
Hurley LP, Bridges CB, Harpaz R, et al. US physicians’ perspective of adult vaccine delivery. Ann Intern Med 2014; 160:161.
Lindsey B, Kampmann B, Jones C. Maternal immunization as a strategy to decrease susceptibility to infection in newborn infants. Curr Opin Infect Dis 2013; 26:248–253.
Centers for Disease Control and Prevention (CDC). Updated recommendations for use of tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccine (Tdap) in pregnant women—Advisory Committee on Immunization Practices (ACIP), 2012. MMWR Morb Mortal Wkly Rep 2013; 62:131–135.
Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997; 46:1–24.
Wharton M, Strikas RA, Harpaz R, et al; Advisory Committee on Immunization Practices; Healthcare Infection Control Practices Advisory Committee. Recommendations for using smallpox vaccine in a pre-event vaccination program. Supplemental recommendations of the Advisory Committee on Immunization Practices (ACIP) and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm Rep 2003; 52:1–16.
Staples JE, Gershman M, Fischer M; Centers for Disease Control and Prevention (CDC). Yellow fever vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2010; 59:1–27.
Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2011; 60:1–64.
Rubin LG, Levin MJ, Ljungman P, et al. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis 2014; 58:309–318.
Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine for adults with immunocompromising conditions: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2012; 61:816–819.
Aberg JA, Gallant JE, Ghanem KG, Emmanuel P, Zingman BS, Horberg MA, Infectious Diseases Society of America. Primary care guidelines for the management of persons infected with HIV: 2013 update by the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis 2014; 58:1–10.
Eilers R, Krabbe PF, van Essen TG, Suijkerbuijk A, van Lier A, de Melker HE. Assessment of vaccine candidates for persons aged 50 and older: a review. BMC Geriatr 2013; 13:32.
Oxman MN, Levin MJ; Shingles Prevention Study Group. Vaccination against Herpes Zoster and Postherpetic Neuralgia. J Infect Dis 2008; 197(suppl 2):S228–S236.
Soni R, Horowitz B, Unruh M. Immunization in end-stage renal disease: opportunity to improve outcomes. Semin Dial 2013; 26:416–426.
Kotton CN, Ryan ET, Fishman JA. Prevention of infection in adult travelers after solid organ transplantation. Am J Transplant 2005; 5:8–14.
Castelli F, Patroni A. The human immunodeficiency virus-infected traveler. Clin Infect Dis 2000; 31:1403–1408.
References
Crosignani P, De Stefani A, Fara GM, et al. Towards the eradication of HPV infection through universal specific vaccination. BMC Public Health 2013;13:642.
Plotkin SL, Plotkin SA. A short history of vaccination. In: Plotkin, SA, Orenstein W, Offit PA, editors. Vaccines, 5th ed. Philadelphia, PA: Elsevier Health Sciences; 2008:1–16.
Wong VW, Chan HL. Prevention of hepatocellular carcinoma: a concise review of contemporary issues. Ann Hepatol 2012; 11:284–293.
Kim DK, Bridges CB, Harriman K; Centers for Disease Control and Prevention (CDC). Advisory Committee on Immunization Practices. Advisory Committee on Immunization Practices recommended immunization schedule for adults aged 19 years or older: United States, 2015. Ann Intern Med 2015; 162:214–223.
Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Noninfluenza vaccination coverage among adults—United States, 2012. MMWR Morb Mortal Wkly Rep 2014; 63:95-102.
Hurley LP, Bridges CB, Harpaz R, et al. US physicians’ perspective of adult vaccine delivery. Ann Intern Med 2014; 160:161.
Lindsey B, Kampmann B, Jones C. Maternal immunization as a strategy to decrease susceptibility to infection in newborn infants. Curr Opin Infect Dis 2013; 26:248–253.
Centers for Disease Control and Prevention (CDC). Updated recommendations for use of tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccine (Tdap) in pregnant women—Advisory Committee on Immunization Practices (ACIP), 2012. MMWR Morb Mortal Wkly Rep 2013; 62:131–135.
Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997; 46:1–24.
Wharton M, Strikas RA, Harpaz R, et al; Advisory Committee on Immunization Practices; Healthcare Infection Control Practices Advisory Committee. Recommendations for using smallpox vaccine in a pre-event vaccination program. Supplemental recommendations of the Advisory Committee on Immunization Practices (ACIP) and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm Rep 2003; 52:1–16.
Staples JE, Gershman M, Fischer M; Centers for Disease Control and Prevention (CDC). Yellow fever vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2010; 59:1–27.
Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2011; 60:1–64.
Rubin LG, Levin MJ, Ljungman P, et al. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis 2014; 58:309–318.
Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine for adults with immunocompromising conditions: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2012; 61:816–819.
Aberg JA, Gallant JE, Ghanem KG, Emmanuel P, Zingman BS, Horberg MA, Infectious Diseases Society of America. Primary care guidelines for the management of persons infected with HIV: 2013 update by the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis 2014; 58:1–10.
Eilers R, Krabbe PF, van Essen TG, Suijkerbuijk A, van Lier A, de Melker HE. Assessment of vaccine candidates for persons aged 50 and older: a review. BMC Geriatr 2013; 13:32.
Oxman MN, Levin MJ; Shingles Prevention Study Group. Vaccination against Herpes Zoster and Postherpetic Neuralgia. J Infect Dis 2008; 197(suppl 2):S228–S236.
Soni R, Horowitz B, Unruh M. Immunization in end-stage renal disease: opportunity to improve outcomes. Semin Dial 2013; 26:416–426.
Avoid live-attenuated vaccines (influenza, varicella, zoster, measles-mumps-rubella, and yellow fever) in immunocompromised patients.
Tetanus, diphtheria, and acellular pertussis (Tdap) vaccine is now recommended for pregnant women during each pregnancy, preferably at 27 to 36 weeks of gestation.
Zoster vaccine is recommended for patients age 60 and older, regardless of earlier episodes of herpes zoster.
The use of aripiprazole in the management of bipolar disorder during pregnancy
"This patient had presented 2-weeks postpartum in a manic state with psycotic features. She was screened by Ob-Gyn who collaborated with her care while she was admitted to the psychiatric inpatient unit. Patient had been non-compliant with prescribed medications prior to admission and she was started on aripiprazole from day one and the dose was tapered up to 15 mg BID by day 5. Patient's manic symptoms improved slowly as the days progressed by day 8 psychotic symptoms started to subside. As delivery was imminent, patient was transferred to Ob-Gyn service. She delivered a healthy but premature child via csection on day 12. Child did not exhibit any gross or anatomic malformations. She was continued on aripiprazole 15 mg BID after discharge and was seen weeks later in outpatient psychiatry."
"This patient had presented 2-weeks postpartum in a manic state with psycotic features. She was screened by Ob-Gyn who collaborated with her care while she was admitted to the psychiatric inpatient unit. Patient had been non-compliant with prescribed medications prior to admission and she was started on aripiprazole from day one and the dose was tapered up to 15 mg BID by day 5. Patient's manic symptoms improved slowly as the days progressed by day 8 psychotic symptoms started to subside. As delivery was imminent, patient was transferred to Ob-Gyn service. She delivered a healthy but premature child via csection on day 12. Child did not exhibit any gross or anatomic malformations. She was continued on aripiprazole 15 mg BID after discharge and was seen weeks later in outpatient psychiatry."
"This patient had presented 2-weeks postpartum in a manic state with psycotic features. She was screened by Ob-Gyn who collaborated with her care while she was admitted to the psychiatric inpatient unit. Patient had been non-compliant with prescribed medications prior to admission and she was started on aripiprazole from day one and the dose was tapered up to 15 mg BID by day 5. Patient's manic symptoms improved slowly as the days progressed by day 8 psychotic symptoms started to subside. As delivery was imminent, patient was transferred to Ob-Gyn service. She delivered a healthy but premature child via csection on day 12. Child did not exhibit any gross or anatomic malformations. She was continued on aripiprazole 15 mg BID after discharge and was seen weeks later in outpatient psychiatry."
College students with depressive symptoms with and without fatigue: Differences in functioning, suicidality, anxiety, and depressive severity
Nyer et al examined whether fatigue was associated with greater symptomatic burden and functional impairment in 287 college students with depressive symptoms using data from the self-report Beck Depression Inventory (BDI). Students endorsing significant symptoms of depression (BDI score ≥13) were grouped into 3 levels: no fatigue, mild fatigue, or moderate/severe fatigue. Researchers compared the 3 levels of fatigue across a battery of psychiatric and functional outcome measures.
The study found that depressed college students with symptoms of fatigue demonstrated functional impairment and symptomatic burden that worsened with increasing levels of fatigue. The authors call for more attention to assessing and treating symptoms of fatigue within this population.
References
Author and Disclosure Information
Maren Nyer, PhD, David Mischoulon, MD, PhD, Jonathan E. Alpert, MD, PhD, Daphne J. Holt, MD, PhD, Charlotte D. Brill, MBA, Albert Yeung, MD, Paola Pedrelli, PhD, Lee Baer, PhD, Christina Dording, MD, Ilana Huz, BA, Lauren Fisher, PhD, Maurizio Fava, MD, and Amy Farabaugh, PhD
Maren Nyer, PhD, David Mischoulon, MD, PhD, Jonathan E. Alpert, MD, PhD, Daphne J. Holt, MD, PhD, Charlotte D. Brill, MBA, Albert Yeung, MD, Paola Pedrelli, PhD, Lee Baer, PhD, Christina Dording, MD, Ilana Huz, BA, Lauren Fisher, PhD, Maurizio Fava, MD, and Amy Farabaugh, PhD
Author and Disclosure Information
Maren Nyer, PhD, David Mischoulon, MD, PhD, Jonathan E. Alpert, MD, PhD, Daphne J. Holt, MD, PhD, Charlotte D. Brill, MBA, Albert Yeung, MD, Paola Pedrelli, PhD, Lee Baer, PhD, Christina Dording, MD, Ilana Huz, BA, Lauren Fisher, PhD, Maurizio Fava, MD, and Amy Farabaugh, PhD
Nyer et al examined whether fatigue was associated with greater symptomatic burden and functional impairment in 287 college students with depressive symptoms using data from the self-report Beck Depression Inventory (BDI). Students endorsing significant symptoms of depression (BDI score ≥13) were grouped into 3 levels: no fatigue, mild fatigue, or moderate/severe fatigue. Researchers compared the 3 levels of fatigue across a battery of psychiatric and functional outcome measures.
The study found that depressed college students with symptoms of fatigue demonstrated functional impairment and symptomatic burden that worsened with increasing levels of fatigue. The authors call for more attention to assessing and treating symptoms of fatigue within this population.
Nyer et al examined whether fatigue was associated with greater symptomatic burden and functional impairment in 287 college students with depressive symptoms using data from the self-report Beck Depression Inventory (BDI). Students endorsing significant symptoms of depression (BDI score ≥13) were grouped into 3 levels: no fatigue, mild fatigue, or moderate/severe fatigue. Researchers compared the 3 levels of fatigue across a battery of psychiatric and functional outcome measures.
The study found that depressed college students with symptoms of fatigue demonstrated functional impairment and symptomatic burden that worsened with increasing levels of fatigue. The authors call for more attention to assessing and treating symptoms of fatigue within this population.
Congratulations, OBG Management readers! After years of hard work and collective advocacy on your part, the US Congress finally passed, and President Barack Obama quickly signed into law, a permanent repeal of the Medicare Sustainable Growth Rate (SGR) physician payment system. Yes, celebrations are in order.
The US House of Representatives passed the bill, HR 2, the Medicare Access and CHIP Reauthorization Act of 2015 (MACRA), sponsored by American College of Obstetricians and Gynecologists (ACOG) Fellow and US Rep. Michael Burgess (R-TX), on March 26, with 382 Republicans and Democrats voting “Yes.” The Senate followed, on April 14, and agreed with the House to repeal, forever, the Medicare SGR, passing the Burgess bill without amendment, on a bipartisan vote of 92–8. With only hours to go before the scheduled 21.2% cut took effect, the President signed the bill, now Public Law (PL) 114-10, on April 16. The President noted that he was “proud to sign the bill into law.” ACOG is proud to have been such an important part of this landmark moment.
SGR: the perennial nemesis of physicians The SGR has wreaked havoc on medicine and patient care for 15 years or more. Approximately 30,000 ObGyns participate in Medicare, and many private health insurers use Medicare payment policies, as does TriCare, the nation’s health care coverage for military members and their families. The SGR’s effect was felt widely across medicine, making it nearly impossible for physician practices to invest in health information technology and other patient safety advances, or even to plan for the next year or continue accepting Medicare patients.
When it was introduced last year, HR 2 was supported by more than 600 national and state medical societies and specialty organizations, plus patient and provider organizations, policy think tanks, and advocacy groups across the political spectrum.
ACOG Fellows petitioned their members of Congress with incredible passion, perseverance, and commitment to put an end to the SGR wrecking ball. Hundreds flew into Washington, DC, sent thousands of emails, made phone calls, wrote letters, and personally lobbied at home and in the halls of Congress.
Special kudos, too, to our champions in Congress, and there are many, led by ACOG Fellows and US Reps. Dr. Burgess and Phil Roe, MD (R-TN). Burgess wrote the House bill and, together with Roe, pushed nonstop to get this bill over the finish line. It wouldn’t have happened without them.
ACOG worked tirelessly on its own and in coalition with the American Medical Association, surgical groups, and many other partners. We were able to win important provisions in the statute that we anticipate will greatly help ObGyns successfully transition to this new payment system.
PL114-10 replaces the SGR with a new payment system intended to promote care coordination and quality improvement and lead to better health for our nation’s seniors. Congress developed this new payment plan with the physician community, rather than imposing it on us. That’s why throughout the statute, we see repeated requirements that the Secretary of Health and Human Services must develop quality measures, alternative payment models, and a host of key aspects with input from and in consultation with physicians and the relevant medical specialties, ensuring that physicians retain their preeminent roles in these areas. Funding is provided for quality measure development at $15 million per year from 2015 to 2019.
This law will likely change physician practices more than the ACA ever will, and Congress agreed that physicians should be integral to its development to ensure that they can continue to thrive and provide high-quality care and access for their patients.
Let’s take a closer look at the new Medicare payment system—especially what it will mean for your practice.
What the new law does Important provisions
MACRA retains the fee-for-service payment model, now called the Merit-based Incentive Payment System, or MIPS. Physician participation in the Advanced Payment Models (APMs) is entirely voluntary. But physicians who participate in APMs and who score better each year will earn more.
All physician types are treated equally. Congress didn’t pick specialty winners and losers.
The new payment system rewards physicians for continuous improvement. You can determine how financially well you do.
Beginning in 2019, Medicare physician payments will reflect each individual physician’s performance, based on a range of measures developed by the relevant medical specialty that will give individuals options that best reflect their practices.
Individual physicians will receive confidential quarterly feedback on their performance.
Technical support is provided for smaller practices, funded at $20 million per year from 2016 to 2020, to help them transition to MIPS and APMs. And physicians in small practices can opt to join a “virtual MIPS group,” associating with other practices or hospitals in the same geographic region or by specialty types.
The law protects physicians from liability from federal or state standards of care. No health care guideline or other standard developed under federal or state requirements associated with this law may be used as a standard of care or duty of care owed by a health care professional to a patient in a medical liability lawsuit.
MACRA stabilizes the Medicare payment system by permanently repealing the SGR and scheduling payments into the future:
through June 2015: Stable payments with no cuts
July 2015–2019: 0.5% annual payment increases to all Medicare physicians
2020–2025: No automatic annual payment changes but opportunities for payment increases based on individual performance
2026 and beyond: 0.75% annual payment increases for qualifying APMs, 0.25% for MIPS providers, with opportunities in both systems for higher payments based on individual performance.
Top ACOG wins
Among the most meaningful accomplishments achieved by ACOG in its work to repeal the sustainable growth rate are:
Reliable payment increases for the first 5 years. The law ensures a period of stability with modest Medicare payment in- creases for 5 years and no cuts, with opportunity for payment increases for the next 5 years. This 5-year period gives physicians time to get ready for the new payment systems.
Protection for low-Medicare–volume physician practices. ObGyns and other physicians with a small Medicare patient population are exempt from many program requirements and penalties.
Stops the Centers for Medicare and Medicaid Services (CMS) policy on global surgical codes, reinstating 10-day and 90-day global payment bundles for surgical services. This directly helps ObGyn subspecialists, including urogynecologists and gynecologic oncologists.
Physician liability protections. The law ensures that federal quality measurements cannot be used to imply medical negligence and generate lawsuits.
Protection for ultrasound. There are no cuts to ultrasound reimbursement.
An end, in 2018, to penalties related to electronic health record (EHR) meaningful use, Physician Quality Reporting Systems, and the use of the value-based modifier.
APM bonus payments. Bonus eligibility for Alternative Payment Model (APM) participation is based on patient volume, not just revenue, to make it easier for ObGyns to qualify.
2-year extension of the Children’s Health Insurance Program (CHIP), which provides comprehensive coverage to 8 million children, adolescents, and pregnant women across the country.
Quality-measure development. The law helps professional organizations, such as ACOG, develop quality measures for the Merit-Based Incentive Payment System (MIPS) rather than allow these measures to be developed by a federal agency, ensuring that this new program works for physicians and our patients.
Two payment system options reward continuous quality improvement Option 1: MIPS. MACRA consolidates and expands pay-for-performance incentives within the old SGR fee-for-service system, creating the new MIPS. Under MIPS, the Physician Quality Reporting System (PQRS), electronic health record (EHR) meaningful use incentive program, and physician value-based modifiers (VBMs) become a single program. In 2019, a physician’s individual score on these measures will be used to adjust his or her Medicare payments, and the penalties previously associated with these programs come to an end.
MACRA creates 4 categories of measures that are weighted to calculate an individual physician’s MIPS score:
Quality (50% of total adjustment in 2019, shrinking to 30% of total adjustment in 2021). Quality measures currently in use in the PQRS, VBM, and EHR meaningful use programs will continue to be used. The Secretary of Health and Human Services must fund and work with specialty societies to develop any additional measures, and measures utilized in clinical data registries can be used for this category as well. Measures will be updated annually, and ACOG and other specialties can submit measures directly for approval, rather than rely on an outside entity.
Resource use (10% of total adjustment in 2019, growing to 30% of total adjustment by 2021). Resource use measures are risk-adjusted and include those already used in the VBM program; others must be developed with physicians, reflecting both the physician’s role in treating the patient (eg, primary or specialty care) and the type of treatment (eg, chronic or acute).
EHR use (25% of total adjustment). Current meaningful use systems will qualify for this category. The law also requires EHR interoperability by 2018 and prohibits the blocking of information sharing between EHR vendors.
Clinical improvement (15% of total adjustment). This is a new component of physician measurement, intended to give physicians credit for working to improve their practices and help them participate in APMs, which have higher reimbursement potential. This menu of qualifying activities—including 24-hour availability, safety, and patient satisfaction—must be developed with physicians and must be attainable by all specialties and practice types, including small practices and those in rural and underserved areas. Maintenance of certification can be used to qual-ify for a high score.
Physicians will only be assessed on the categories, measures, and activities that apply to them. A physician’s composite score (0–100) will be compared with a performance threshold that reflects all physicians. Those who score above the threshold will receive increased payments; those who score below the threshold will receive reduced payments. Physicians will know these thresholds in advance and will know the score they must reach to avoid penalties and win higher reimbursements in each performance period.
As physicians as a whole improve their performance, the threshold will move with them. So each year, physicians will have the incentive to keep improving their quality, resource use, clinical improvement, and EHR use. A physician’s payment adjustment in one year will not affect his or her payment adjustment in the next year.
The range of potential payment adjustments based on MIPS performance measures increases each year through 2022. Providers who have high scores are rewarded with a 4% increase in 2019. By 2022, the reward is 9%. The program is budget-neutral, so total positive adjustments across all providers will equal total negative adjustments across all providers to poor performers. Separate funds are set aside to reward the highest performers, who will earn bonuses of up to 10% of their fee-for-service payment rate from 2019 through 2024, as well as to help low performers improve and qualify for increased payments from 2016 through 2020.
Help for physicians includes:
flexibility to participate in a way that best reflects their practice, using risk-adjusted clinical outcome measures
option to participate in a virtual MIPS group rather than go it alone
technical assistance to practices with 15 or fewer professionals, $20 million annually from 2016 through 2020, with preference to practices with low MIPS scores and those in rural and underserved areas
quarterly confidential feedback on performance in the quality and resource use categories
advance notification to each physician of the score needed to reach higher payment levels
exclusion from MIPS of physicians who treat few Medicare patients, as well as those who receive a significant portion of their revenues from APMs.
Option 2: APMs. Physicians can earn higher fees by opting out of MIPS fee for service and participating in APMs. The law defines qualifying APMs as those that require participating providers to take on “more than nominal” financial risk, report quality measures, and use certified EHR technology.
APMs will cover multiple services, show that they can limit the growth of spending, and use performance-based methods of compensation. These and other provisions will likely continue the trend away from physicians practicing in solo or small-group fee-for-service practices into risk-based multispecialty settings that are subject to increased management and oversight.
From 2019 to 2024, qualified APM physicians will receive a 5% annual lump sum bonus based on their prior year’s physician fee-schedule payments plus shared savings from participation. This bonus is based on patient volume, not just revenue, to make it easier for ObGyns to qualify. To make the bonus widely available, the Secretary of Health and Human Services must test APMs designed for specific specialties and physicians in small practices. As in MIPS, top APM performers will also receive an additional bonus.
To qualify, physicians must meet increasing thresholds for the percentage of their revenue that they receive through APMs. Those who are below but near the required level of APM revenue can be exempted from MIPS adjustments.
2019–2020: 25% of Medicare revenue must be received through APMs.
2021–2022: 50% of Medicare revenue or 50% of all-payer revenue along with 25% of Medicare revenue must be received through APMs.
2023 and beyond: 75% of Medicare revenue or 75% of all-payer revenue along with 25% of Medicare revenue must be received through APMs.
Who pays the bill? Medicare beneficiaries pay more The new law increases the percentage of Medicare Parts B and D premiums that high-income beneficiaries must pay beginning in 2018:
Single seniors reporting income of more than $133,500 and married couples with income of more than $267,000 will see their share of premiums rise from 50% to 65%.
Single seniors reporting income above $160,000 and married couples with income above $320,000 will see their premium share rise from 65% to 80%.
This change will affect about 2% of Medicare beneficiaries; half of all Medicare beneficiaries currently have annual incomes below $26,000.1
Medigap “first-dollar coverage” will end Many Medigap plans on the market today provide “first-dollar coverage” for beneficiaries, which means that the plans pay the deductibles and copayments so that the beneficiaries have no out-of-pocket costs. Beginning in 2020, Medigap plans will only be available to cover costs above the Medicare Part B deductible, currently $147 per year, for new Medigap enrollees. Many lawmakers thought it was important for Medicare beneficiaries to have “skin in the game.”
The law cuts payments for some providers To partially offset the cost of repealing the SGR, MACRA cuts Medicare payments to hospitals and postacute providers. It:
delays Disproportionate Share Hospital (DSH) cuts scheduled to begin in 2017 by a year and extends them through 2025
requires an increase in payments to hospitals scheduled for 2018 to instead be phased in over 6 years
limits the 2018 payment update for post-acute providers to 1%.
The law extends many programs These programs are vital to support the future ObGyn workforce and access to health care. Among these programs are:
a halt to the Centers for Medicare and Medicaid Services (CMS) policy on global surgical codes. The law reinstates 10-day and 90-day global payment bundles for surgical services. This directly helps ObGyn subspecialists, such as urogynecologists and gynecologic oncologists.
renewal of the Children’s Health Insurance Program (CHIP), which provides comprehensive coverage to 8 million children, adolescents, and pregnant women across the country
establishment of a Medicaid/CHIP Pediatric Quality Measures Program, supporting the development and physician adoption of quality measures, including for prenatal and preconception care
funding for the Maternal, Infant, and Early Childhood Home Visiting Program, helping at-risk pregnant women and their families to promote healthy births and early childhood development
funding for community health centers, an important source of care for 13 million women and girls in all 50 states and the District of Columbia
funding for the National Health Service Corps, bringing ObGyns and other primary care providers to underserved rural and urban areas through scholarships and loan repayment programs
funding for the Teaching Health Center Graduate Medical Education Payment Program, enhancing training for ObGyns and other primary care providers in community-based settings
extending the Medicare Geographic Practice Cost Index floor, helping ensure access to care for women in rural areas
extending the Personal Responsibility Education Program to help prevent teen pregnancies and sexually transmitted infections.
Next steps It’s very important that ObGyns and other physicians use these early years to understand and get ready for the new payment systems. ACOG is developing educational material for our members, and will work closely with our colleague medical organizations and the Department of Health and Human Services to develop key aspects of the law and ensure that it is properly implemented to work for physicians and patients.
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
References
Reference 1. Aaron HJ. Three cheers for log-rolling: The demise of the SGR. Brookings Health360. http://www.brookings.edu/blogs/health360/posts/2015/04/22-medicare-sgr-repeal-doc-fix-aaron. Published April 22, 2015. Accessed May 12, 2015.
Lucia DiVenere MA, Medicare, Sustainable Growth Rate, SGR, US Congress, President Barack Obama, US House of Representatives, HR 2, Medicare Access and CHIP reauthorization Act of 2015, MACRA, American College of Obstetricians and Gynecologists, ACOG, Michael Burgess, US Senate, Medicare SGR, Burgess bill, TriCare, ACOG Fellows, Phil Roe, American Medical Association, AMA, PL114-10, ACA, Affordable Care Act, fee-for-service payment model, Merit-based Incentive Payment System, MIPS, Advanced Payment Models, APMs, Physician Quality Reporting System, PQRS, electronic health record, EHR, value-based modifiers, VBMs, clinical improvement, CMS, Centers for Medicare and Medicaid Services, Children’s Health Insurance Program, CHIP,
Congratulations, OBG Management readers! After years of hard work and collective advocacy on your part, the US Congress finally passed, and President Barack Obama quickly signed into law, a permanent repeal of the Medicare Sustainable Growth Rate (SGR) physician payment system. Yes, celebrations are in order.
The US House of Representatives passed the bill, HR 2, the Medicare Access and CHIP Reauthorization Act of 2015 (MACRA), sponsored by American College of Obstetricians and Gynecologists (ACOG) Fellow and US Rep. Michael Burgess (R-TX), on March 26, with 382 Republicans and Democrats voting “Yes.” The Senate followed, on April 14, and agreed with the House to repeal, forever, the Medicare SGR, passing the Burgess bill without amendment, on a bipartisan vote of 92–8. With only hours to go before the scheduled 21.2% cut took effect, the President signed the bill, now Public Law (PL) 114-10, on April 16. The President noted that he was “proud to sign the bill into law.” ACOG is proud to have been such an important part of this landmark moment.
SGR: the perennial nemesis of physicians The SGR has wreaked havoc on medicine and patient care for 15 years or more. Approximately 30,000 ObGyns participate in Medicare, and many private health insurers use Medicare payment policies, as does TriCare, the nation’s health care coverage for military members and their families. The SGR’s effect was felt widely across medicine, making it nearly impossible for physician practices to invest in health information technology and other patient safety advances, or even to plan for the next year or continue accepting Medicare patients.
When it was introduced last year, HR 2 was supported by more than 600 national and state medical societies and specialty organizations, plus patient and provider organizations, policy think tanks, and advocacy groups across the political spectrum.
ACOG Fellows petitioned their members of Congress with incredible passion, perseverance, and commitment to put an end to the SGR wrecking ball. Hundreds flew into Washington, DC, sent thousands of emails, made phone calls, wrote letters, and personally lobbied at home and in the halls of Congress.
Special kudos, too, to our champions in Congress, and there are many, led by ACOG Fellows and US Reps. Dr. Burgess and Phil Roe, MD (R-TN). Burgess wrote the House bill and, together with Roe, pushed nonstop to get this bill over the finish line. It wouldn’t have happened without them.
ACOG worked tirelessly on its own and in coalition with the American Medical Association, surgical groups, and many other partners. We were able to win important provisions in the statute that we anticipate will greatly help ObGyns successfully transition to this new payment system.
PL114-10 replaces the SGR with a new payment system intended to promote care coordination and quality improvement and lead to better health for our nation’s seniors. Congress developed this new payment plan with the physician community, rather than imposing it on us. That’s why throughout the statute, we see repeated requirements that the Secretary of Health and Human Services must develop quality measures, alternative payment models, and a host of key aspects with input from and in consultation with physicians and the relevant medical specialties, ensuring that physicians retain their preeminent roles in these areas. Funding is provided for quality measure development at $15 million per year from 2015 to 2019.
This law will likely change physician practices more than the ACA ever will, and Congress agreed that physicians should be integral to its development to ensure that they can continue to thrive and provide high-quality care and access for their patients.
Let’s take a closer look at the new Medicare payment system—especially what it will mean for your practice.
What the new law does Important provisions
MACRA retains the fee-for-service payment model, now called the Merit-based Incentive Payment System, or MIPS. Physician participation in the Advanced Payment Models (APMs) is entirely voluntary. But physicians who participate in APMs and who score better each year will earn more.
All physician types are treated equally. Congress didn’t pick specialty winners and losers.
The new payment system rewards physicians for continuous improvement. You can determine how financially well you do.
Beginning in 2019, Medicare physician payments will reflect each individual physician’s performance, based on a range of measures developed by the relevant medical specialty that will give individuals options that best reflect their practices.
Individual physicians will receive confidential quarterly feedback on their performance.
Technical support is provided for smaller practices, funded at $20 million per year from 2016 to 2020, to help them transition to MIPS and APMs. And physicians in small practices can opt to join a “virtual MIPS group,” associating with other practices or hospitals in the same geographic region or by specialty types.
The law protects physicians from liability from federal or state standards of care. No health care guideline or other standard developed under federal or state requirements associated with this law may be used as a standard of care or duty of care owed by a health care professional to a patient in a medical liability lawsuit.
MACRA stabilizes the Medicare payment system by permanently repealing the SGR and scheduling payments into the future:
through June 2015: Stable payments with no cuts
July 2015–2019: 0.5% annual payment increases to all Medicare physicians
2020–2025: No automatic annual payment changes but opportunities for payment increases based on individual performance
2026 and beyond: 0.75% annual payment increases for qualifying APMs, 0.25% for MIPS providers, with opportunities in both systems for higher payments based on individual performance.
Top ACOG wins
Among the most meaningful accomplishments achieved by ACOG in its work to repeal the sustainable growth rate are:
Reliable payment increases for the first 5 years. The law ensures a period of stability with modest Medicare payment in- creases for 5 years and no cuts, with opportunity for payment increases for the next 5 years. This 5-year period gives physicians time to get ready for the new payment systems.
Protection for low-Medicare–volume physician practices. ObGyns and other physicians with a small Medicare patient population are exempt from many program requirements and penalties.
Stops the Centers for Medicare and Medicaid Services (CMS) policy on global surgical codes, reinstating 10-day and 90-day global payment bundles for surgical services. This directly helps ObGyn subspecialists, including urogynecologists and gynecologic oncologists.
Physician liability protections. The law ensures that federal quality measurements cannot be used to imply medical negligence and generate lawsuits.
Protection for ultrasound. There are no cuts to ultrasound reimbursement.
An end, in 2018, to penalties related to electronic health record (EHR) meaningful use, Physician Quality Reporting Systems, and the use of the value-based modifier.
APM bonus payments. Bonus eligibility for Alternative Payment Model (APM) participation is based on patient volume, not just revenue, to make it easier for ObGyns to qualify.
2-year extension of the Children’s Health Insurance Program (CHIP), which provides comprehensive coverage to 8 million children, adolescents, and pregnant women across the country.
Quality-measure development. The law helps professional organizations, such as ACOG, develop quality measures for the Merit-Based Incentive Payment System (MIPS) rather than allow these measures to be developed by a federal agency, ensuring that this new program works for physicians and our patients.
Two payment system options reward continuous quality improvement Option 1: MIPS. MACRA consolidates and expands pay-for-performance incentives within the old SGR fee-for-service system, creating the new MIPS. Under MIPS, the Physician Quality Reporting System (PQRS), electronic health record (EHR) meaningful use incentive program, and physician value-based modifiers (VBMs) become a single program. In 2019, a physician’s individual score on these measures will be used to adjust his or her Medicare payments, and the penalties previously associated with these programs come to an end.
MACRA creates 4 categories of measures that are weighted to calculate an individual physician’s MIPS score:
Quality (50% of total adjustment in 2019, shrinking to 30% of total adjustment in 2021). Quality measures currently in use in the PQRS, VBM, and EHR meaningful use programs will continue to be used. The Secretary of Health and Human Services must fund and work with specialty societies to develop any additional measures, and measures utilized in clinical data registries can be used for this category as well. Measures will be updated annually, and ACOG and other specialties can submit measures directly for approval, rather than rely on an outside entity.
Resource use (10% of total adjustment in 2019, growing to 30% of total adjustment by 2021). Resource use measures are risk-adjusted and include those already used in the VBM program; others must be developed with physicians, reflecting both the physician’s role in treating the patient (eg, primary or specialty care) and the type of treatment (eg, chronic or acute).
EHR use (25% of total adjustment). Current meaningful use systems will qualify for this category. The law also requires EHR interoperability by 2018 and prohibits the blocking of information sharing between EHR vendors.
Clinical improvement (15% of total adjustment). This is a new component of physician measurement, intended to give physicians credit for working to improve their practices and help them participate in APMs, which have higher reimbursement potential. This menu of qualifying activities—including 24-hour availability, safety, and patient satisfaction—must be developed with physicians and must be attainable by all specialties and practice types, including small practices and those in rural and underserved areas. Maintenance of certification can be used to qual-ify for a high score.
Physicians will only be assessed on the categories, measures, and activities that apply to them. A physician’s composite score (0–100) will be compared with a performance threshold that reflects all physicians. Those who score above the threshold will receive increased payments; those who score below the threshold will receive reduced payments. Physicians will know these thresholds in advance and will know the score they must reach to avoid penalties and win higher reimbursements in each performance period.
As physicians as a whole improve their performance, the threshold will move with them. So each year, physicians will have the incentive to keep improving their quality, resource use, clinical improvement, and EHR use. A physician’s payment adjustment in one year will not affect his or her payment adjustment in the next year.
The range of potential payment adjustments based on MIPS performance measures increases each year through 2022. Providers who have high scores are rewarded with a 4% increase in 2019. By 2022, the reward is 9%. The program is budget-neutral, so total positive adjustments across all providers will equal total negative adjustments across all providers to poor performers. Separate funds are set aside to reward the highest performers, who will earn bonuses of up to 10% of their fee-for-service payment rate from 2019 through 2024, as well as to help low performers improve and qualify for increased payments from 2016 through 2020.
Help for physicians includes:
flexibility to participate in a way that best reflects their practice, using risk-adjusted clinical outcome measures
option to participate in a virtual MIPS group rather than go it alone
technical assistance to practices with 15 or fewer professionals, $20 million annually from 2016 through 2020, with preference to practices with low MIPS scores and those in rural and underserved areas
quarterly confidential feedback on performance in the quality and resource use categories
advance notification to each physician of the score needed to reach higher payment levels
exclusion from MIPS of physicians who treat few Medicare patients, as well as those who receive a significant portion of their revenues from APMs.
Option 2: APMs. Physicians can earn higher fees by opting out of MIPS fee for service and participating in APMs. The law defines qualifying APMs as those that require participating providers to take on “more than nominal” financial risk, report quality measures, and use certified EHR technology.
APMs will cover multiple services, show that they can limit the growth of spending, and use performance-based methods of compensation. These and other provisions will likely continue the trend away from physicians practicing in solo or small-group fee-for-service practices into risk-based multispecialty settings that are subject to increased management and oversight.
From 2019 to 2024, qualified APM physicians will receive a 5% annual lump sum bonus based on their prior year’s physician fee-schedule payments plus shared savings from participation. This bonus is based on patient volume, not just revenue, to make it easier for ObGyns to qualify. To make the bonus widely available, the Secretary of Health and Human Services must test APMs designed for specific specialties and physicians in small practices. As in MIPS, top APM performers will also receive an additional bonus.
To qualify, physicians must meet increasing thresholds for the percentage of their revenue that they receive through APMs. Those who are below but near the required level of APM revenue can be exempted from MIPS adjustments.
2019–2020: 25% of Medicare revenue must be received through APMs.
2021–2022: 50% of Medicare revenue or 50% of all-payer revenue along with 25% of Medicare revenue must be received through APMs.
2023 and beyond: 75% of Medicare revenue or 75% of all-payer revenue along with 25% of Medicare revenue must be received through APMs.
Who pays the bill? Medicare beneficiaries pay more The new law increases the percentage of Medicare Parts B and D premiums that high-income beneficiaries must pay beginning in 2018:
Single seniors reporting income of more than $133,500 and married couples with income of more than $267,000 will see their share of premiums rise from 50% to 65%.
Single seniors reporting income above $160,000 and married couples with income above $320,000 will see their premium share rise from 65% to 80%.
This change will affect about 2% of Medicare beneficiaries; half of all Medicare beneficiaries currently have annual incomes below $26,000.1
Medigap “first-dollar coverage” will end Many Medigap plans on the market today provide “first-dollar coverage” for beneficiaries, which means that the plans pay the deductibles and copayments so that the beneficiaries have no out-of-pocket costs. Beginning in 2020, Medigap plans will only be available to cover costs above the Medicare Part B deductible, currently $147 per year, for new Medigap enrollees. Many lawmakers thought it was important for Medicare beneficiaries to have “skin in the game.”
The law cuts payments for some providers To partially offset the cost of repealing the SGR, MACRA cuts Medicare payments to hospitals and postacute providers. It:
delays Disproportionate Share Hospital (DSH) cuts scheduled to begin in 2017 by a year and extends them through 2025
requires an increase in payments to hospitals scheduled for 2018 to instead be phased in over 6 years
limits the 2018 payment update for post-acute providers to 1%.
The law extends many programs These programs are vital to support the future ObGyn workforce and access to health care. Among these programs are:
a halt to the Centers for Medicare and Medicaid Services (CMS) policy on global surgical codes. The law reinstates 10-day and 90-day global payment bundles for surgical services. This directly helps ObGyn subspecialists, such as urogynecologists and gynecologic oncologists.
renewal of the Children’s Health Insurance Program (CHIP), which provides comprehensive coverage to 8 million children, adolescents, and pregnant women across the country
establishment of a Medicaid/CHIP Pediatric Quality Measures Program, supporting the development and physician adoption of quality measures, including for prenatal and preconception care
funding for the Maternal, Infant, and Early Childhood Home Visiting Program, helping at-risk pregnant women and their families to promote healthy births and early childhood development
funding for community health centers, an important source of care for 13 million women and girls in all 50 states and the District of Columbia
funding for the National Health Service Corps, bringing ObGyns and other primary care providers to underserved rural and urban areas through scholarships and loan repayment programs
funding for the Teaching Health Center Graduate Medical Education Payment Program, enhancing training for ObGyns and other primary care providers in community-based settings
extending the Medicare Geographic Practice Cost Index floor, helping ensure access to care for women in rural areas
extending the Personal Responsibility Education Program to help prevent teen pregnancies and sexually transmitted infections.
Next steps It’s very important that ObGyns and other physicians use these early years to understand and get ready for the new payment systems. ACOG is developing educational material for our members, and will work closely with our colleague medical organizations and the Department of Health and Human Services to develop key aspects of the law and ensure that it is properly implemented to work for physicians and patients.
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
Congratulations, OBG Management readers! After years of hard work and collective advocacy on your part, the US Congress finally passed, and President Barack Obama quickly signed into law, a permanent repeal of the Medicare Sustainable Growth Rate (SGR) physician payment system. Yes, celebrations are in order.
The US House of Representatives passed the bill, HR 2, the Medicare Access and CHIP Reauthorization Act of 2015 (MACRA), sponsored by American College of Obstetricians and Gynecologists (ACOG) Fellow and US Rep. Michael Burgess (R-TX), on March 26, with 382 Republicans and Democrats voting “Yes.” The Senate followed, on April 14, and agreed with the House to repeal, forever, the Medicare SGR, passing the Burgess bill without amendment, on a bipartisan vote of 92–8. With only hours to go before the scheduled 21.2% cut took effect, the President signed the bill, now Public Law (PL) 114-10, on April 16. The President noted that he was “proud to sign the bill into law.” ACOG is proud to have been such an important part of this landmark moment.
SGR: the perennial nemesis of physicians The SGR has wreaked havoc on medicine and patient care for 15 years or more. Approximately 30,000 ObGyns participate in Medicare, and many private health insurers use Medicare payment policies, as does TriCare, the nation’s health care coverage for military members and their families. The SGR’s effect was felt widely across medicine, making it nearly impossible for physician practices to invest in health information technology and other patient safety advances, or even to plan for the next year or continue accepting Medicare patients.
When it was introduced last year, HR 2 was supported by more than 600 national and state medical societies and specialty organizations, plus patient and provider organizations, policy think tanks, and advocacy groups across the political spectrum.
ACOG Fellows petitioned their members of Congress with incredible passion, perseverance, and commitment to put an end to the SGR wrecking ball. Hundreds flew into Washington, DC, sent thousands of emails, made phone calls, wrote letters, and personally lobbied at home and in the halls of Congress.
Special kudos, too, to our champions in Congress, and there are many, led by ACOG Fellows and US Reps. Dr. Burgess and Phil Roe, MD (R-TN). Burgess wrote the House bill and, together with Roe, pushed nonstop to get this bill over the finish line. It wouldn’t have happened without them.
ACOG worked tirelessly on its own and in coalition with the American Medical Association, surgical groups, and many other partners. We were able to win important provisions in the statute that we anticipate will greatly help ObGyns successfully transition to this new payment system.
PL114-10 replaces the SGR with a new payment system intended to promote care coordination and quality improvement and lead to better health for our nation’s seniors. Congress developed this new payment plan with the physician community, rather than imposing it on us. That’s why throughout the statute, we see repeated requirements that the Secretary of Health and Human Services must develop quality measures, alternative payment models, and a host of key aspects with input from and in consultation with physicians and the relevant medical specialties, ensuring that physicians retain their preeminent roles in these areas. Funding is provided for quality measure development at $15 million per year from 2015 to 2019.
This law will likely change physician practices more than the ACA ever will, and Congress agreed that physicians should be integral to its development to ensure that they can continue to thrive and provide high-quality care and access for their patients.
Let’s take a closer look at the new Medicare payment system—especially what it will mean for your practice.
What the new law does Important provisions
MACRA retains the fee-for-service payment model, now called the Merit-based Incentive Payment System, or MIPS. Physician participation in the Advanced Payment Models (APMs) is entirely voluntary. But physicians who participate in APMs and who score better each year will earn more.
All physician types are treated equally. Congress didn’t pick specialty winners and losers.
The new payment system rewards physicians for continuous improvement. You can determine how financially well you do.
Beginning in 2019, Medicare physician payments will reflect each individual physician’s performance, based on a range of measures developed by the relevant medical specialty that will give individuals options that best reflect their practices.
Individual physicians will receive confidential quarterly feedback on their performance.
Technical support is provided for smaller practices, funded at $20 million per year from 2016 to 2020, to help them transition to MIPS and APMs. And physicians in small practices can opt to join a “virtual MIPS group,” associating with other practices or hospitals in the same geographic region or by specialty types.
The law protects physicians from liability from federal or state standards of care. No health care guideline or other standard developed under federal or state requirements associated with this law may be used as a standard of care or duty of care owed by a health care professional to a patient in a medical liability lawsuit.
MACRA stabilizes the Medicare payment system by permanently repealing the SGR and scheduling payments into the future:
through June 2015: Stable payments with no cuts
July 2015–2019: 0.5% annual payment increases to all Medicare physicians
2020–2025: No automatic annual payment changes but opportunities for payment increases based on individual performance
2026 and beyond: 0.75% annual payment increases for qualifying APMs, 0.25% for MIPS providers, with opportunities in both systems for higher payments based on individual performance.
Top ACOG wins
Among the most meaningful accomplishments achieved by ACOG in its work to repeal the sustainable growth rate are:
Reliable payment increases for the first 5 years. The law ensures a period of stability with modest Medicare payment in- creases for 5 years and no cuts, with opportunity for payment increases for the next 5 years. This 5-year period gives physicians time to get ready for the new payment systems.
Protection for low-Medicare–volume physician practices. ObGyns and other physicians with a small Medicare patient population are exempt from many program requirements and penalties.
Stops the Centers for Medicare and Medicaid Services (CMS) policy on global surgical codes, reinstating 10-day and 90-day global payment bundles for surgical services. This directly helps ObGyn subspecialists, including urogynecologists and gynecologic oncologists.
Physician liability protections. The law ensures that federal quality measurements cannot be used to imply medical negligence and generate lawsuits.
Protection for ultrasound. There are no cuts to ultrasound reimbursement.
An end, in 2018, to penalties related to electronic health record (EHR) meaningful use, Physician Quality Reporting Systems, and the use of the value-based modifier.
APM bonus payments. Bonus eligibility for Alternative Payment Model (APM) participation is based on patient volume, not just revenue, to make it easier for ObGyns to qualify.
2-year extension of the Children’s Health Insurance Program (CHIP), which provides comprehensive coverage to 8 million children, adolescents, and pregnant women across the country.
Quality-measure development. The law helps professional organizations, such as ACOG, develop quality measures for the Merit-Based Incentive Payment System (MIPS) rather than allow these measures to be developed by a federal agency, ensuring that this new program works for physicians and our patients.
Two payment system options reward continuous quality improvement Option 1: MIPS. MACRA consolidates and expands pay-for-performance incentives within the old SGR fee-for-service system, creating the new MIPS. Under MIPS, the Physician Quality Reporting System (PQRS), electronic health record (EHR) meaningful use incentive program, and physician value-based modifiers (VBMs) become a single program. In 2019, a physician’s individual score on these measures will be used to adjust his or her Medicare payments, and the penalties previously associated with these programs come to an end.
MACRA creates 4 categories of measures that are weighted to calculate an individual physician’s MIPS score:
Quality (50% of total adjustment in 2019, shrinking to 30% of total adjustment in 2021). Quality measures currently in use in the PQRS, VBM, and EHR meaningful use programs will continue to be used. The Secretary of Health and Human Services must fund and work with specialty societies to develop any additional measures, and measures utilized in clinical data registries can be used for this category as well. Measures will be updated annually, and ACOG and other specialties can submit measures directly for approval, rather than rely on an outside entity.
Resource use (10% of total adjustment in 2019, growing to 30% of total adjustment by 2021). Resource use measures are risk-adjusted and include those already used in the VBM program; others must be developed with physicians, reflecting both the physician’s role in treating the patient (eg, primary or specialty care) and the type of treatment (eg, chronic or acute).
EHR use (25% of total adjustment). Current meaningful use systems will qualify for this category. The law also requires EHR interoperability by 2018 and prohibits the blocking of information sharing between EHR vendors.
Clinical improvement (15% of total adjustment). This is a new component of physician measurement, intended to give physicians credit for working to improve their practices and help them participate in APMs, which have higher reimbursement potential. This menu of qualifying activities—including 24-hour availability, safety, and patient satisfaction—must be developed with physicians and must be attainable by all specialties and practice types, including small practices and those in rural and underserved areas. Maintenance of certification can be used to qual-ify for a high score.
Physicians will only be assessed on the categories, measures, and activities that apply to them. A physician’s composite score (0–100) will be compared with a performance threshold that reflects all physicians. Those who score above the threshold will receive increased payments; those who score below the threshold will receive reduced payments. Physicians will know these thresholds in advance and will know the score they must reach to avoid penalties and win higher reimbursements in each performance period.
As physicians as a whole improve their performance, the threshold will move with them. So each year, physicians will have the incentive to keep improving their quality, resource use, clinical improvement, and EHR use. A physician’s payment adjustment in one year will not affect his or her payment adjustment in the next year.
The range of potential payment adjustments based on MIPS performance measures increases each year through 2022. Providers who have high scores are rewarded with a 4% increase in 2019. By 2022, the reward is 9%. The program is budget-neutral, so total positive adjustments across all providers will equal total negative adjustments across all providers to poor performers. Separate funds are set aside to reward the highest performers, who will earn bonuses of up to 10% of their fee-for-service payment rate from 2019 through 2024, as well as to help low performers improve and qualify for increased payments from 2016 through 2020.
Help for physicians includes:
flexibility to participate in a way that best reflects their practice, using risk-adjusted clinical outcome measures
option to participate in a virtual MIPS group rather than go it alone
technical assistance to practices with 15 or fewer professionals, $20 million annually from 2016 through 2020, with preference to practices with low MIPS scores and those in rural and underserved areas
quarterly confidential feedback on performance in the quality and resource use categories
advance notification to each physician of the score needed to reach higher payment levels
exclusion from MIPS of physicians who treat few Medicare patients, as well as those who receive a significant portion of their revenues from APMs.
Option 2: APMs. Physicians can earn higher fees by opting out of MIPS fee for service and participating in APMs. The law defines qualifying APMs as those that require participating providers to take on “more than nominal” financial risk, report quality measures, and use certified EHR technology.
APMs will cover multiple services, show that they can limit the growth of spending, and use performance-based methods of compensation. These and other provisions will likely continue the trend away from physicians practicing in solo or small-group fee-for-service practices into risk-based multispecialty settings that are subject to increased management and oversight.
From 2019 to 2024, qualified APM physicians will receive a 5% annual lump sum bonus based on their prior year’s physician fee-schedule payments plus shared savings from participation. This bonus is based on patient volume, not just revenue, to make it easier for ObGyns to qualify. To make the bonus widely available, the Secretary of Health and Human Services must test APMs designed for specific specialties and physicians in small practices. As in MIPS, top APM performers will also receive an additional bonus.
To qualify, physicians must meet increasing thresholds for the percentage of their revenue that they receive through APMs. Those who are below but near the required level of APM revenue can be exempted from MIPS adjustments.
2019–2020: 25% of Medicare revenue must be received through APMs.
2021–2022: 50% of Medicare revenue or 50% of all-payer revenue along with 25% of Medicare revenue must be received through APMs.
2023 and beyond: 75% of Medicare revenue or 75% of all-payer revenue along with 25% of Medicare revenue must be received through APMs.
Who pays the bill? Medicare beneficiaries pay more The new law increases the percentage of Medicare Parts B and D premiums that high-income beneficiaries must pay beginning in 2018:
Single seniors reporting income of more than $133,500 and married couples with income of more than $267,000 will see their share of premiums rise from 50% to 65%.
Single seniors reporting income above $160,000 and married couples with income above $320,000 will see their premium share rise from 65% to 80%.
This change will affect about 2% of Medicare beneficiaries; half of all Medicare beneficiaries currently have annual incomes below $26,000.1
Medigap “first-dollar coverage” will end Many Medigap plans on the market today provide “first-dollar coverage” for beneficiaries, which means that the plans pay the deductibles and copayments so that the beneficiaries have no out-of-pocket costs. Beginning in 2020, Medigap plans will only be available to cover costs above the Medicare Part B deductible, currently $147 per year, for new Medigap enrollees. Many lawmakers thought it was important for Medicare beneficiaries to have “skin in the game.”
The law cuts payments for some providers To partially offset the cost of repealing the SGR, MACRA cuts Medicare payments to hospitals and postacute providers. It:
delays Disproportionate Share Hospital (DSH) cuts scheduled to begin in 2017 by a year and extends them through 2025
requires an increase in payments to hospitals scheduled for 2018 to instead be phased in over 6 years
limits the 2018 payment update for post-acute providers to 1%.
The law extends many programs These programs are vital to support the future ObGyn workforce and access to health care. Among these programs are:
a halt to the Centers for Medicare and Medicaid Services (CMS) policy on global surgical codes. The law reinstates 10-day and 90-day global payment bundles for surgical services. This directly helps ObGyn subspecialists, such as urogynecologists and gynecologic oncologists.
renewal of the Children’s Health Insurance Program (CHIP), which provides comprehensive coverage to 8 million children, adolescents, and pregnant women across the country
establishment of a Medicaid/CHIP Pediatric Quality Measures Program, supporting the development and physician adoption of quality measures, including for prenatal and preconception care
funding for the Maternal, Infant, and Early Childhood Home Visiting Program, helping at-risk pregnant women and their families to promote healthy births and early childhood development
funding for community health centers, an important source of care for 13 million women and girls in all 50 states and the District of Columbia
funding for the National Health Service Corps, bringing ObGyns and other primary care providers to underserved rural and urban areas through scholarships and loan repayment programs
funding for the Teaching Health Center Graduate Medical Education Payment Program, enhancing training for ObGyns and other primary care providers in community-based settings
extending the Medicare Geographic Practice Cost Index floor, helping ensure access to care for women in rural areas
extending the Personal Responsibility Education Program to help prevent teen pregnancies and sexually transmitted infections.
Next steps It’s very important that ObGyns and other physicians use these early years to understand and get ready for the new payment systems. ACOG is developing educational material for our members, and will work closely with our colleague medical organizations and the Department of Health and Human Services to develop key aspects of the law and ensure that it is properly implemented to work for physicians and patients.
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
References
Reference 1. Aaron HJ. Three cheers for log-rolling: The demise of the SGR. Brookings Health360. http://www.brookings.edu/blogs/health360/posts/2015/04/22-medicare-sgr-repeal-doc-fix-aaron. Published April 22, 2015. Accessed May 12, 2015.
References
Reference 1. Aaron HJ. Three cheers for log-rolling: The demise of the SGR. Brookings Health360. http://www.brookings.edu/blogs/health360/posts/2015/04/22-medicare-sgr-repeal-doc-fix-aaron. Published April 22, 2015. Accessed May 12, 2015.
Lucia DiVenere MA, Medicare, Sustainable Growth Rate, SGR, US Congress, President Barack Obama, US House of Representatives, HR 2, Medicare Access and CHIP reauthorization Act of 2015, MACRA, American College of Obstetricians and Gynecologists, ACOG, Michael Burgess, US Senate, Medicare SGR, Burgess bill, TriCare, ACOG Fellows, Phil Roe, American Medical Association, AMA, PL114-10, ACA, Affordable Care Act, fee-for-service payment model, Merit-based Incentive Payment System, MIPS, Advanced Payment Models, APMs, Physician Quality Reporting System, PQRS, electronic health record, EHR, value-based modifiers, VBMs, clinical improvement, CMS, Centers for Medicare and Medicaid Services, Children’s Health Insurance Program, CHIP,
Legacy Keywords
Lucia DiVenere MA, Medicare, Sustainable Growth Rate, SGR, US Congress, President Barack Obama, US House of Representatives, HR 2, Medicare Access and CHIP reauthorization Act of 2015, MACRA, American College of Obstetricians and Gynecologists, ACOG, Michael Burgess, US Senate, Medicare SGR, Burgess bill, TriCare, ACOG Fellows, Phil Roe, American Medical Association, AMA, PL114-10, ACA, Affordable Care Act, fee-for-service payment model, Merit-based Incentive Payment System, MIPS, Advanced Payment Models, APMs, Physician Quality Reporting System, PQRS, electronic health record, EHR, value-based modifiers, VBMs, clinical improvement, CMS, Centers for Medicare and Medicaid Services, Children’s Health Insurance Program, CHIP,