User login
Widespread Skin Thickening
The Diagnosis: Scleromyxedema
Scleromyxedema is a rare skin disorder characterized by a diffuse eruption of small waxy papules that are linearly arranged and closely spaced together. As the papular lesions coalesce, the skin thickens. Firm induration of the skin is widespread and--unlike the distribution in scleroderma and scleredema--amplified over the facial convexities, especially the glabella and ears. Histopathology reveals the classic triad of mucin accumulation, proliferation of fibroblasts, and collagen deposition with associated fibrotic changes.1
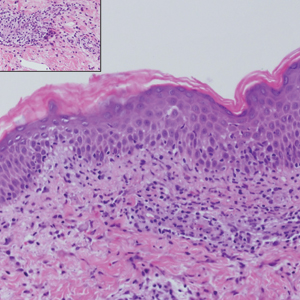
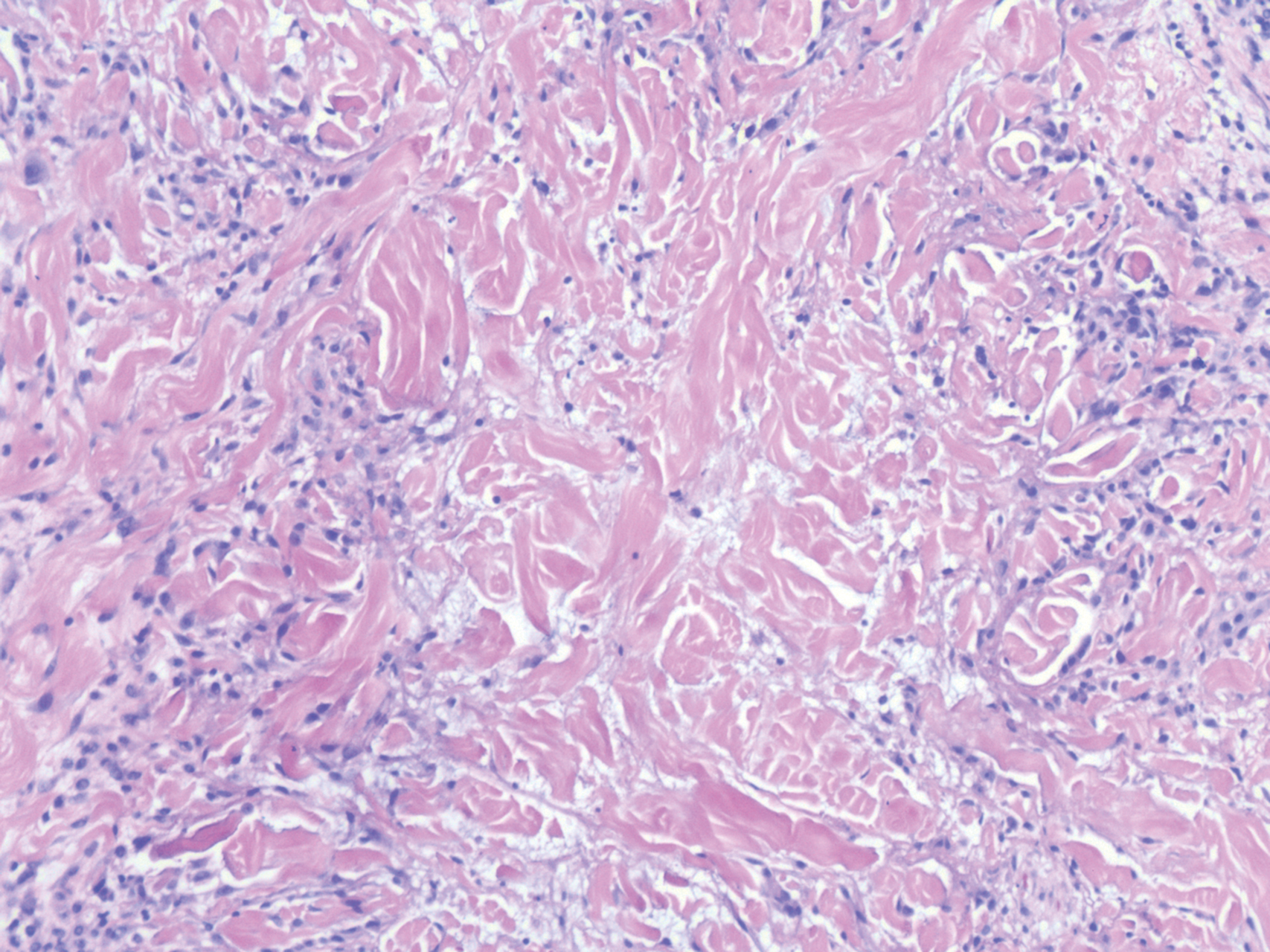
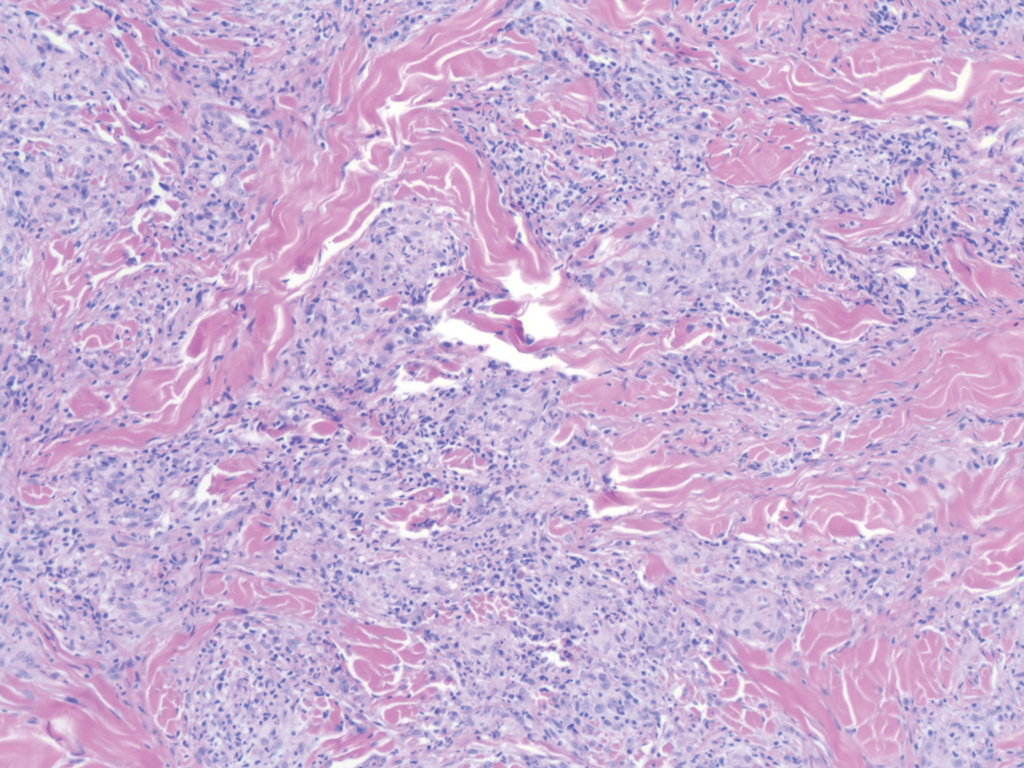
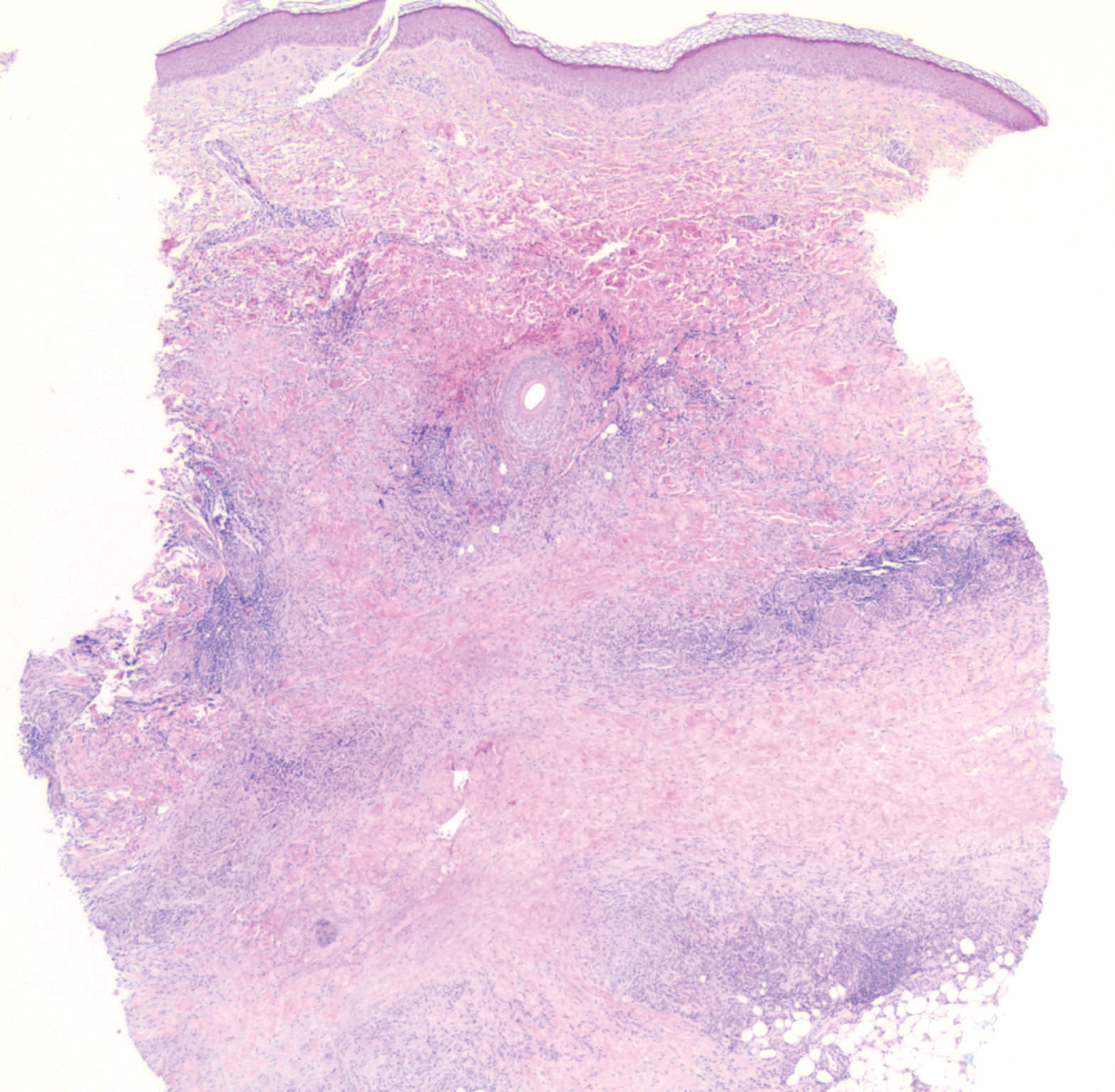
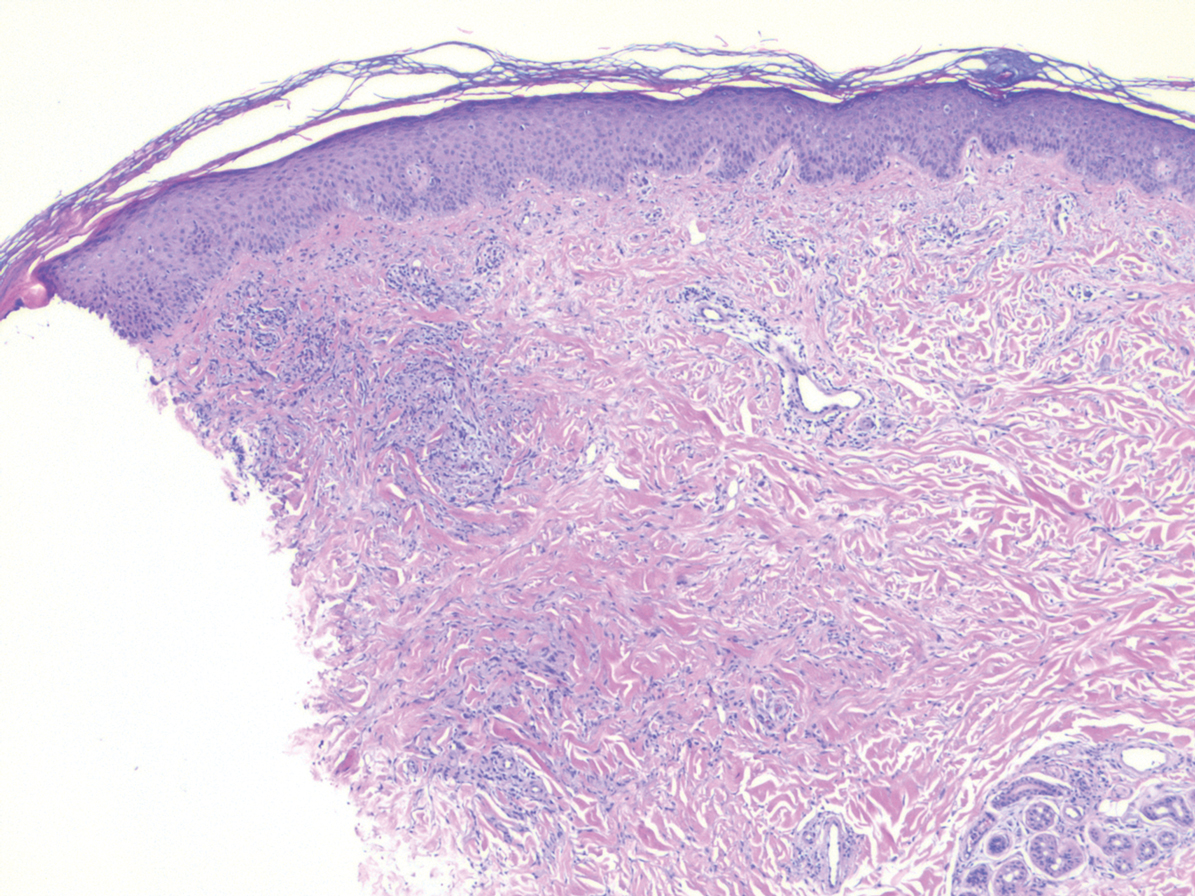
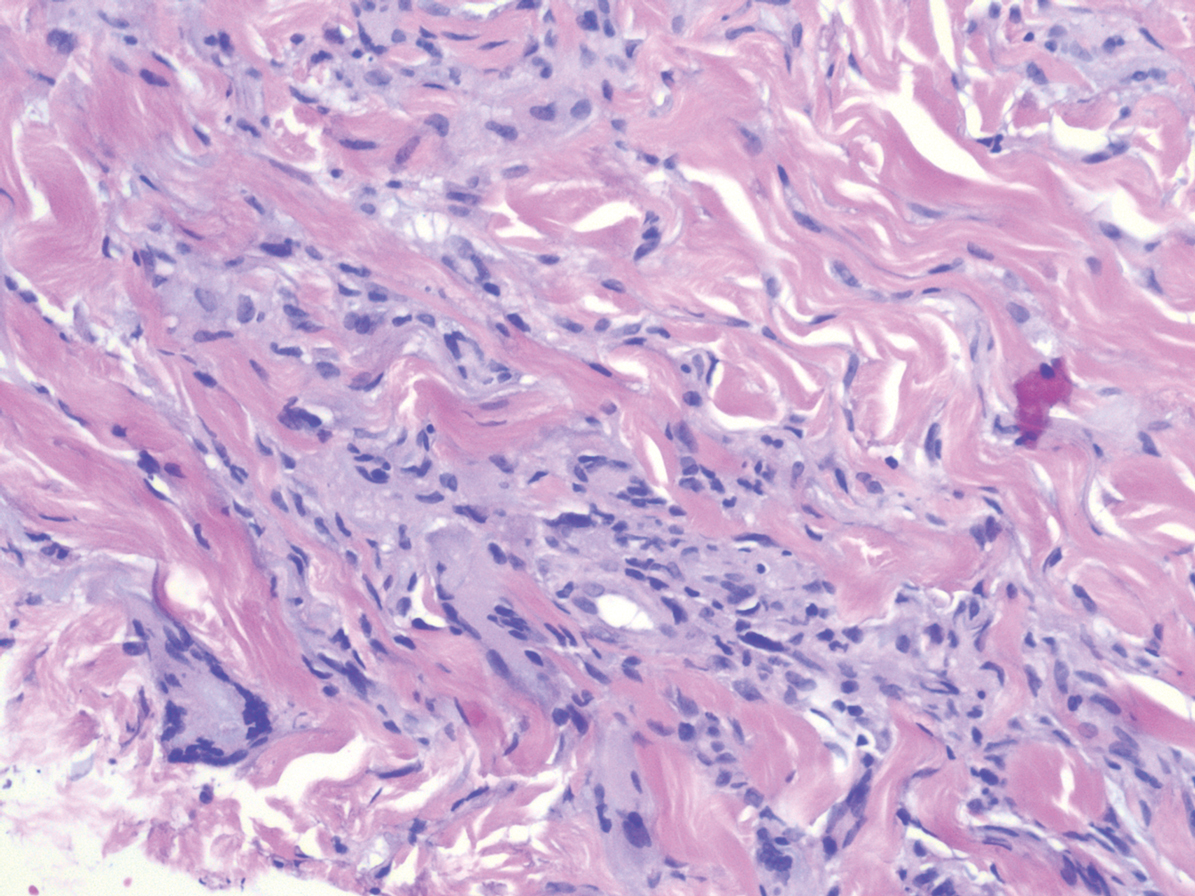
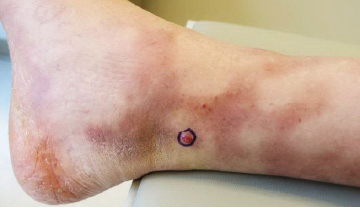
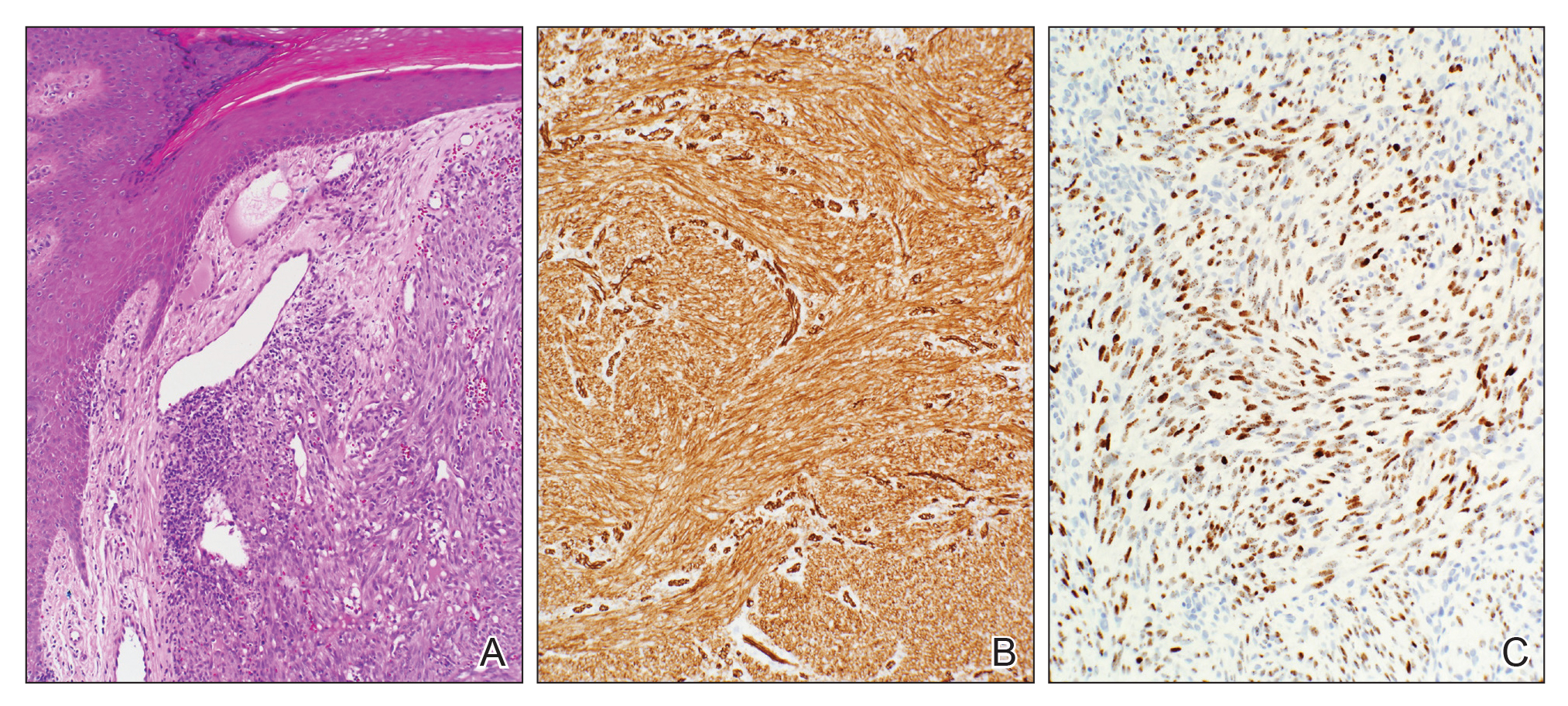
In this case, the patient exhibited the characteristic doughnut sign over the second interphalangeal joint of the right hand due to thickening and dimpling of the skin (quiz image). Histopathology from the right second finger showed the dermis with a spindled histiocytic infiltrate, fibroblasts, fibrosis, and increased interstitial mucin deposition highlighted with colloidal iron stain (Figures 1 and 2). Multinucleated giant cells also were seen in the dermis (Figure 3), and a Shikata orcein stain illustrated decreased elastic fibers (stained black) in the superficial dermis within areas of increased collagen deposition (Figure 4).
Scleromyxedema is strongly associated with a monoclonal gammopathy, which is generally mild and, in almost 80% of cases, related to λ IgG. However, the clinical implications are unclear.2,3 Scleromyxedema also is accompanied by systemic features, most commonly dysphagia, myalgias, and arthralgias, which distinguishes it from other cutaneous mucinoses. Notable morbidity and mortality are associated with involvement of the central nervous system, which can manifest as encephalopathy, seizures, or coma. Mucin deposition may be found in the myocardium and the coronary and pulmonary vasculature, resulting in rare cases of cardiac and pulmonary disease.4,5
Clinically, scleroderma also demonstrates progressive generalized skin tightening. Histopathology reveals hyalinization and thickening of the connective tissue of the deep dermis, subcutaneous fat, and muscular fascia. These changes are accompanied by a perivascular and focal interstitial lymphocytic and plasma cell infiltrate with prominent myofibroblast proliferation. In this case, the normal nail fold capillaries and absence of Raynaud phenomenon help to distinguish the diagnosis from scleroderma. In limited scleroderma, common findings include calcinosis, esophageal dysmotility, telangiectasia, and pulmonary hypertension, while systemic sclerosis can involve the internal organs, with the greatest morbidity stemming from interstitial lung disease.3,6,7
Nephrogenic systemic fibrosis is another disorder of skin thickening that involves the extremities and trunk. It is a rare complication of exposure to gadolinium contrast media in patients with advanced renal disease and those undergoing dialysis; use of gadolinium is now strictly avoided in patients with at least moderately impaired glomerular filtration rate.8 Although nephrogenic systemic fibrosis histologically looks similar to scleromyxedema, it involves deeper tissues. There is fibrosis with haphazard collagen bundle deposition in the deep dermis and subcutaneous septa. Fibroblasts and histiocytes are increased between the collagen bundles, with surrounding edema and mucin buildup. Multinucleated giant cells may or may not be present.9
Patients with scleredema present with nonpitting edema and dermal hardening over the neck, face, upper trunk, and arms. Lesional skin appears shiny and indurated. The skin is characteristically difficult to wrinkle, and the face may appear expressionless. Skin biopsy will show a thickened reticular dermis with mucin deposition and eccrine glands appearing in the upper third or mid dermis. Fibroblasts are normal in number. Scleredema generally is associated with poorly controlled diabetes, a monoclonal gammopathy, or as the aftermath of an acute infection, especially streptococcal pharyngitis. The condition may develop at any age, though nearly 50% of cases occur in children and adolescents.3,10
Interstitial granuloma annulare (GA) is a common, benign, and self-limiting dermatosis. Distinct from other skin-thickening disorders, GA is composed of smooth annular papules and plaques that do not result in skin hardening. Although GA may manifest in a generalized distribution, it is more frequently localized to the distal extremities.11 The presence of an inflammatory infiltrate surrounded by abundant mucin and collagen resembles scleromyxedema. However, GA is distinguished by palisading granulomas of histiocytes, fibroblasts, and lymphocytes lining a necrobiotic center of collagen, mucin, and fibrin.12
This patient's skin showed an immediate and marked response to intravenous immunoglobulin with dramatic softening. Such a response is well reported in the literature, and intravenous immunoglobulin is considered first-line therapy for scleromyxedema, typically in conjunction with steroids.5 Other immunosuppressive agents have been employed with reported efficacy, as have several treatments used for multiple myeloma, including thalidomide and lenalidomide.
Although spontaneous improvement infrequently occurs, a chronic progressive course is far more common with scleromyxedema. Studies are ongoing to elucidate the etiopathogenesis of scleromyxedema, scleroderma, and similar disorders to uncover the triggers that provoke the underlying dysregulation of dermal fibroblast activation and proliferation, which could offer a more precise target for effective treatments.5,13-15
- Crowe DR. Scleromyxedema. In: Crowe DR, Morgan M, Somach S, Trapp K, eds. Deadly Dermatologic Diseases: Clinicopathologic Atlas and Text. Cham, Switzerland: Springer International Publishing; 2016:139-142.
- Farmer ER, Hambrick GW Jr, Shulman LE. Papular mucinosis: a clinicopathologic study of four patients. Arch Dermatol. 1982;118:9-13.
- Rongioletti F. Mucinoses. In: Smoller BR, Rongioletti F, eds. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer New York; 2010:139-152.
- Gabriel PH, Oleson GB, Bowles GA. Scleromyxoedema: a scleroderma-like disorder with systemic manifestations. Medicine. 1988;67:58-65.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Hamodat M. Scleroderma. PathologyOutlines.com. http://www.pathologyoutlines.com/topic/skinnontumorscleroderma.html. Published August 1, 2011. Updated March 29, 2019. Accessed December 11, 2019.
- Van Praet JT, Smith V, Haspeslagh M, et al. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthritis Res Ther. 2011;13:R35.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am. 2008;34:199-ix.
- Plaza JA, Prieto VG. Inflammatory Skin Conditions. In: Modern Surgical Pathology. 2nd ed. Philadelphia, PA: Saunders; 2009:1843-1889.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine. 2008;87:10-20.
- Caradonna S, Jacobe H. Thalidomide as a potential treatment for scleromyxedema. Arch Dermatol. 2004;140:277-280.
- Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J Haematol. 2012;157:411.
The Diagnosis: Scleromyxedema
Scleromyxedema is a rare skin disorder characterized by a diffuse eruption of small waxy papules that are linearly arranged and closely spaced together. As the papular lesions coalesce, the skin thickens. Firm induration of the skin is widespread and--unlike the distribution in scleroderma and scleredema--amplified over the facial convexities, especially the glabella and ears. Histopathology reveals the classic triad of mucin accumulation, proliferation of fibroblasts, and collagen deposition with associated fibrotic changes.1
In this case, the patient exhibited the characteristic doughnut sign over the second interphalangeal joint of the right hand due to thickening and dimpling of the skin (quiz image). Histopathology from the right second finger showed the dermis with a spindled histiocytic infiltrate, fibroblasts, fibrosis, and increased interstitial mucin deposition highlighted with colloidal iron stain (Figures 1 and 2). Multinucleated giant cells also were seen in the dermis (Figure 3), and a Shikata orcein stain illustrated decreased elastic fibers (stained black) in the superficial dermis within areas of increased collagen deposition (Figure 4).
Scleromyxedema is strongly associated with a monoclonal gammopathy, which is generally mild and, in almost 80% of cases, related to λ IgG. However, the clinical implications are unclear.2,3 Scleromyxedema also is accompanied by systemic features, most commonly dysphagia, myalgias, and arthralgias, which distinguishes it from other cutaneous mucinoses. Notable morbidity and mortality are associated with involvement of the central nervous system, which can manifest as encephalopathy, seizures, or coma. Mucin deposition may be found in the myocardium and the coronary and pulmonary vasculature, resulting in rare cases of cardiac and pulmonary disease.4,5
Clinically, scleroderma also demonstrates progressive generalized skin tightening. Histopathology reveals hyalinization and thickening of the connective tissue of the deep dermis, subcutaneous fat, and muscular fascia. These changes are accompanied by a perivascular and focal interstitial lymphocytic and plasma cell infiltrate with prominent myofibroblast proliferation. In this case, the normal nail fold capillaries and absence of Raynaud phenomenon help to distinguish the diagnosis from scleroderma. In limited scleroderma, common findings include calcinosis, esophageal dysmotility, telangiectasia, and pulmonary hypertension, while systemic sclerosis can involve the internal organs, with the greatest morbidity stemming from interstitial lung disease.3,6,7
Nephrogenic systemic fibrosis is another disorder of skin thickening that involves the extremities and trunk. It is a rare complication of exposure to gadolinium contrast media in patients with advanced renal disease and those undergoing dialysis; use of gadolinium is now strictly avoided in patients with at least moderately impaired glomerular filtration rate.8 Although nephrogenic systemic fibrosis histologically looks similar to scleromyxedema, it involves deeper tissues. There is fibrosis with haphazard collagen bundle deposition in the deep dermis and subcutaneous septa. Fibroblasts and histiocytes are increased between the collagen bundles, with surrounding edema and mucin buildup. Multinucleated giant cells may or may not be present.9
Patients with scleredema present with nonpitting edema and dermal hardening over the neck, face, upper trunk, and arms. Lesional skin appears shiny and indurated. The skin is characteristically difficult to wrinkle, and the face may appear expressionless. Skin biopsy will show a thickened reticular dermis with mucin deposition and eccrine glands appearing in the upper third or mid dermis. Fibroblasts are normal in number. Scleredema generally is associated with poorly controlled diabetes, a monoclonal gammopathy, or as the aftermath of an acute infection, especially streptococcal pharyngitis. The condition may develop at any age, though nearly 50% of cases occur in children and adolescents.3,10
Interstitial granuloma annulare (GA) is a common, benign, and self-limiting dermatosis. Distinct from other skin-thickening disorders, GA is composed of smooth annular papules and plaques that do not result in skin hardening. Although GA may manifest in a generalized distribution, it is more frequently localized to the distal extremities.11 The presence of an inflammatory infiltrate surrounded by abundant mucin and collagen resembles scleromyxedema. However, GA is distinguished by palisading granulomas of histiocytes, fibroblasts, and lymphocytes lining a necrobiotic center of collagen, mucin, and fibrin.12
This patient's skin showed an immediate and marked response to intravenous immunoglobulin with dramatic softening. Such a response is well reported in the literature, and intravenous immunoglobulin is considered first-line therapy for scleromyxedema, typically in conjunction with steroids.5 Other immunosuppressive agents have been employed with reported efficacy, as have several treatments used for multiple myeloma, including thalidomide and lenalidomide.
Although spontaneous improvement infrequently occurs, a chronic progressive course is far more common with scleromyxedema. Studies are ongoing to elucidate the etiopathogenesis of scleromyxedema, scleroderma, and similar disorders to uncover the triggers that provoke the underlying dysregulation of dermal fibroblast activation and proliferation, which could offer a more precise target for effective treatments.5,13-15
The Diagnosis: Scleromyxedema
Scleromyxedema is a rare skin disorder characterized by a diffuse eruption of small waxy papules that are linearly arranged and closely spaced together. As the papular lesions coalesce, the skin thickens. Firm induration of the skin is widespread and--unlike the distribution in scleroderma and scleredema--amplified over the facial convexities, especially the glabella and ears. Histopathology reveals the classic triad of mucin accumulation, proliferation of fibroblasts, and collagen deposition with associated fibrotic changes.1
In this case, the patient exhibited the characteristic doughnut sign over the second interphalangeal joint of the right hand due to thickening and dimpling of the skin (quiz image). Histopathology from the right second finger showed the dermis with a spindled histiocytic infiltrate, fibroblasts, fibrosis, and increased interstitial mucin deposition highlighted with colloidal iron stain (Figures 1 and 2). Multinucleated giant cells also were seen in the dermis (Figure 3), and a Shikata orcein stain illustrated decreased elastic fibers (stained black) in the superficial dermis within areas of increased collagen deposition (Figure 4).
Scleromyxedema is strongly associated with a monoclonal gammopathy, which is generally mild and, in almost 80% of cases, related to λ IgG. However, the clinical implications are unclear.2,3 Scleromyxedema also is accompanied by systemic features, most commonly dysphagia, myalgias, and arthralgias, which distinguishes it from other cutaneous mucinoses. Notable morbidity and mortality are associated with involvement of the central nervous system, which can manifest as encephalopathy, seizures, or coma. Mucin deposition may be found in the myocardium and the coronary and pulmonary vasculature, resulting in rare cases of cardiac and pulmonary disease.4,5
Clinically, scleroderma also demonstrates progressive generalized skin tightening. Histopathology reveals hyalinization and thickening of the connective tissue of the deep dermis, subcutaneous fat, and muscular fascia. These changes are accompanied by a perivascular and focal interstitial lymphocytic and plasma cell infiltrate with prominent myofibroblast proliferation. In this case, the normal nail fold capillaries and absence of Raynaud phenomenon help to distinguish the diagnosis from scleroderma. In limited scleroderma, common findings include calcinosis, esophageal dysmotility, telangiectasia, and pulmonary hypertension, while systemic sclerosis can involve the internal organs, with the greatest morbidity stemming from interstitial lung disease.3,6,7
Nephrogenic systemic fibrosis is another disorder of skin thickening that involves the extremities and trunk. It is a rare complication of exposure to gadolinium contrast media in patients with advanced renal disease and those undergoing dialysis; use of gadolinium is now strictly avoided in patients with at least moderately impaired glomerular filtration rate.8 Although nephrogenic systemic fibrosis histologically looks similar to scleromyxedema, it involves deeper tissues. There is fibrosis with haphazard collagen bundle deposition in the deep dermis and subcutaneous septa. Fibroblasts and histiocytes are increased between the collagen bundles, with surrounding edema and mucin buildup. Multinucleated giant cells may or may not be present.9
Patients with scleredema present with nonpitting edema and dermal hardening over the neck, face, upper trunk, and arms. Lesional skin appears shiny and indurated. The skin is characteristically difficult to wrinkle, and the face may appear expressionless. Skin biopsy will show a thickened reticular dermis with mucin deposition and eccrine glands appearing in the upper third or mid dermis. Fibroblasts are normal in number. Scleredema generally is associated with poorly controlled diabetes, a monoclonal gammopathy, or as the aftermath of an acute infection, especially streptococcal pharyngitis. The condition may develop at any age, though nearly 50% of cases occur in children and adolescents.3,10
Interstitial granuloma annulare (GA) is a common, benign, and self-limiting dermatosis. Distinct from other skin-thickening disorders, GA is composed of smooth annular papules and plaques that do not result in skin hardening. Although GA may manifest in a generalized distribution, it is more frequently localized to the distal extremities.11 The presence of an inflammatory infiltrate surrounded by abundant mucin and collagen resembles scleromyxedema. However, GA is distinguished by palisading granulomas of histiocytes, fibroblasts, and lymphocytes lining a necrobiotic center of collagen, mucin, and fibrin.12
This patient's skin showed an immediate and marked response to intravenous immunoglobulin with dramatic softening. Such a response is well reported in the literature, and intravenous immunoglobulin is considered first-line therapy for scleromyxedema, typically in conjunction with steroids.5 Other immunosuppressive agents have been employed with reported efficacy, as have several treatments used for multiple myeloma, including thalidomide and lenalidomide.
Although spontaneous improvement infrequently occurs, a chronic progressive course is far more common with scleromyxedema. Studies are ongoing to elucidate the etiopathogenesis of scleromyxedema, scleroderma, and similar disorders to uncover the triggers that provoke the underlying dysregulation of dermal fibroblast activation and proliferation, which could offer a more precise target for effective treatments.5,13-15
- Crowe DR. Scleromyxedema. In: Crowe DR, Morgan M, Somach S, Trapp K, eds. Deadly Dermatologic Diseases: Clinicopathologic Atlas and Text. Cham, Switzerland: Springer International Publishing; 2016:139-142.
- Farmer ER, Hambrick GW Jr, Shulman LE. Papular mucinosis: a clinicopathologic study of four patients. Arch Dermatol. 1982;118:9-13.
- Rongioletti F. Mucinoses. In: Smoller BR, Rongioletti F, eds. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer New York; 2010:139-152.
- Gabriel PH, Oleson GB, Bowles GA. Scleromyxoedema: a scleroderma-like disorder with systemic manifestations. Medicine. 1988;67:58-65.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Hamodat M. Scleroderma. PathologyOutlines.com. http://www.pathologyoutlines.com/topic/skinnontumorscleroderma.html. Published August 1, 2011. Updated March 29, 2019. Accessed December 11, 2019.
- Van Praet JT, Smith V, Haspeslagh M, et al. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthritis Res Ther. 2011;13:R35.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am. 2008;34:199-ix.
- Plaza JA, Prieto VG. Inflammatory Skin Conditions. In: Modern Surgical Pathology. 2nd ed. Philadelphia, PA: Saunders; 2009:1843-1889.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine. 2008;87:10-20.
- Caradonna S, Jacobe H. Thalidomide as a potential treatment for scleromyxedema. Arch Dermatol. 2004;140:277-280.
- Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J Haematol. 2012;157:411.
- Crowe DR. Scleromyxedema. In: Crowe DR, Morgan M, Somach S, Trapp K, eds. Deadly Dermatologic Diseases: Clinicopathologic Atlas and Text. Cham, Switzerland: Springer International Publishing; 2016:139-142.
- Farmer ER, Hambrick GW Jr, Shulman LE. Papular mucinosis: a clinicopathologic study of four patients. Arch Dermatol. 1982;118:9-13.
- Rongioletti F. Mucinoses. In: Smoller BR, Rongioletti F, eds. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer New York; 2010:139-152.
- Gabriel PH, Oleson GB, Bowles GA. Scleromyxoedema: a scleroderma-like disorder with systemic manifestations. Medicine. 1988;67:58-65.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Hamodat M. Scleroderma. PathologyOutlines.com. http://www.pathologyoutlines.com/topic/skinnontumorscleroderma.html. Published August 1, 2011. Updated March 29, 2019. Accessed December 11, 2019.
- Van Praet JT, Smith V, Haspeslagh M, et al. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthritis Res Ther. 2011;13:R35.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am. 2008;34:199-ix.
- Plaza JA, Prieto VG. Inflammatory Skin Conditions. In: Modern Surgical Pathology. 2nd ed. Philadelphia, PA: Saunders; 2009:1843-1889.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine. 2008;87:10-20.
- Caradonna S, Jacobe H. Thalidomide as a potential treatment for scleromyxedema. Arch Dermatol. 2004;140:277-280.
- Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J Haematol. 2012;157:411.
![]()
A 62-year-old woman presented with widespread skin thickening and tightness that progressed over 2 months. Physical examination showed generalized, nonpruritic, nonpainful, flesh-colored papules along the fingers, bilateral arms and legs, chest, neck, forehead, chin, and ears. She reported mild acid reflux on review of systems. She denied a history of Raynaud phenomenon and had normal-appearing nail beds on capillaroscopy. Laboratory studies including autoimmune serologies were normal, aside from a mildly elevated monoclonal IgG spike.
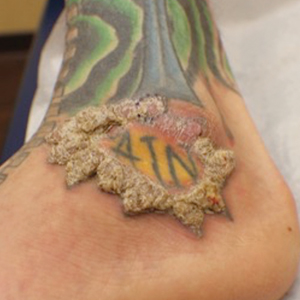
Verruciform Plaques Within a Tattoo of an HIV-Positive Patient
The Diagnosis: Lichenoid Reaction With Pseudoepitheliomatous Hyperplasia
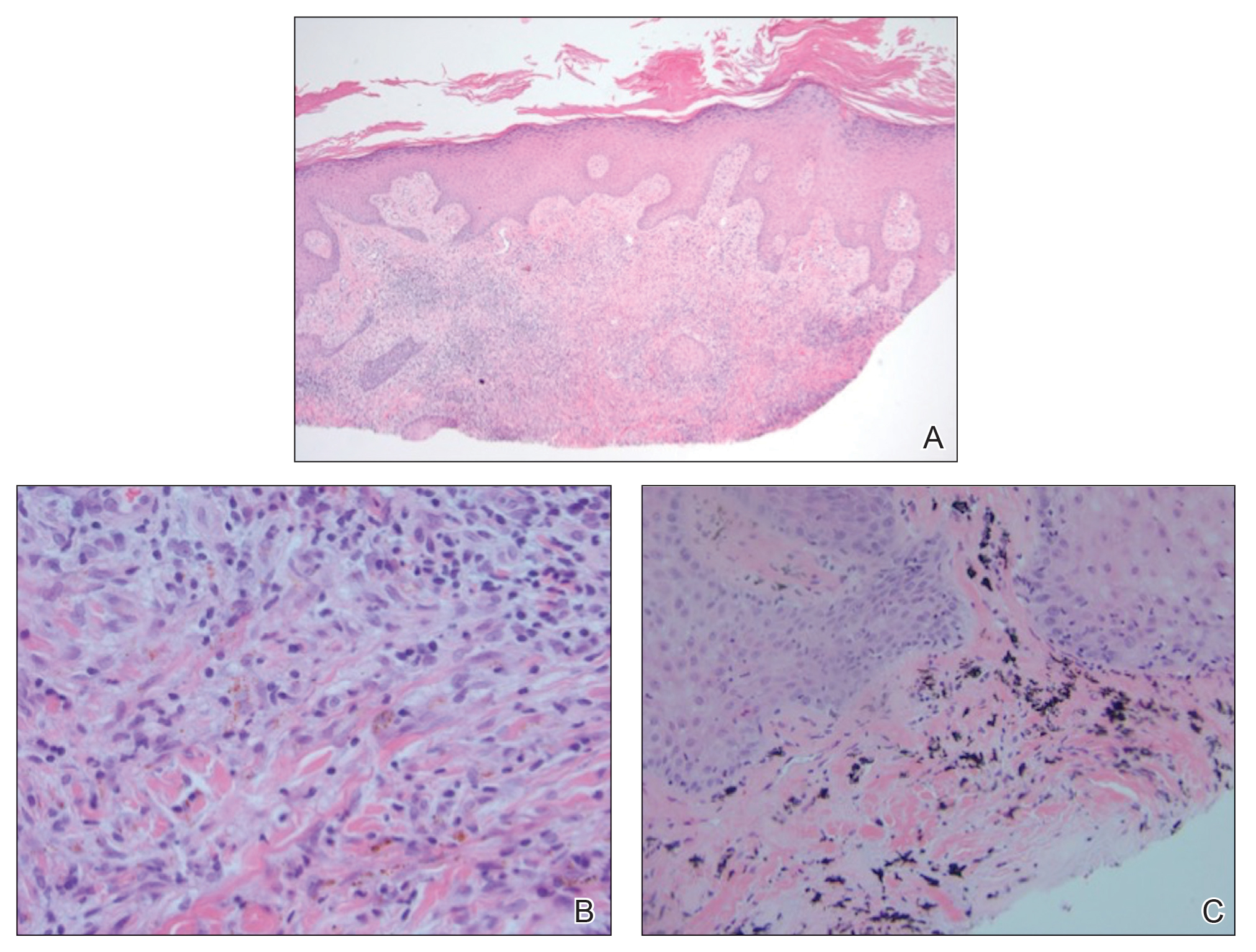
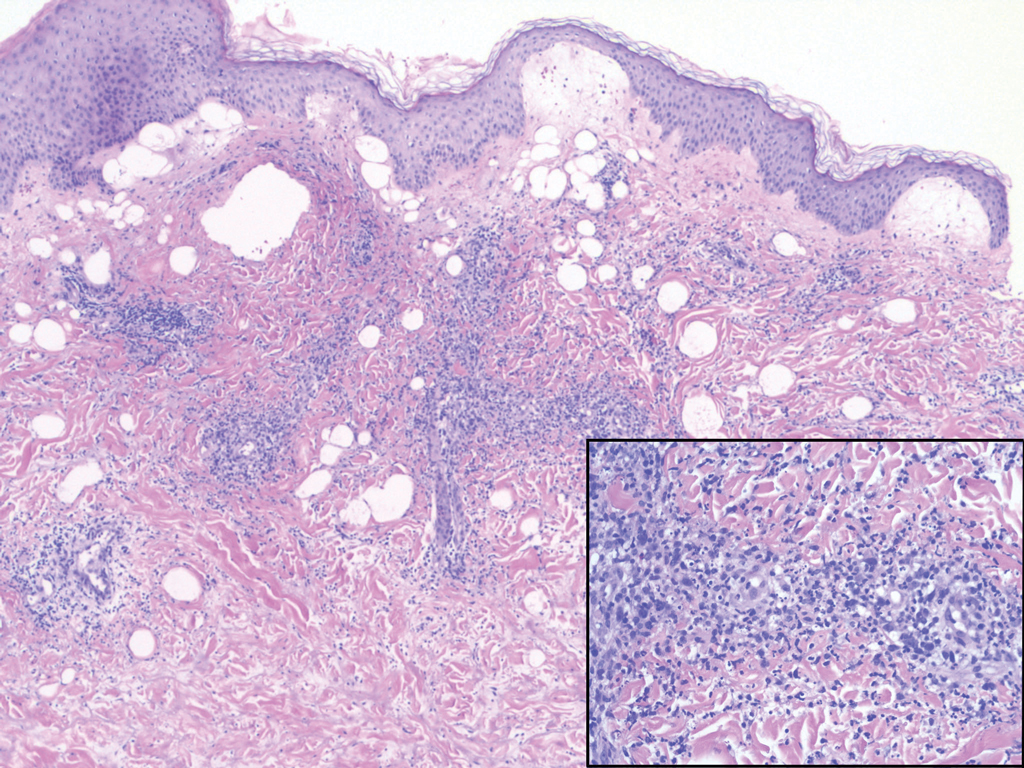
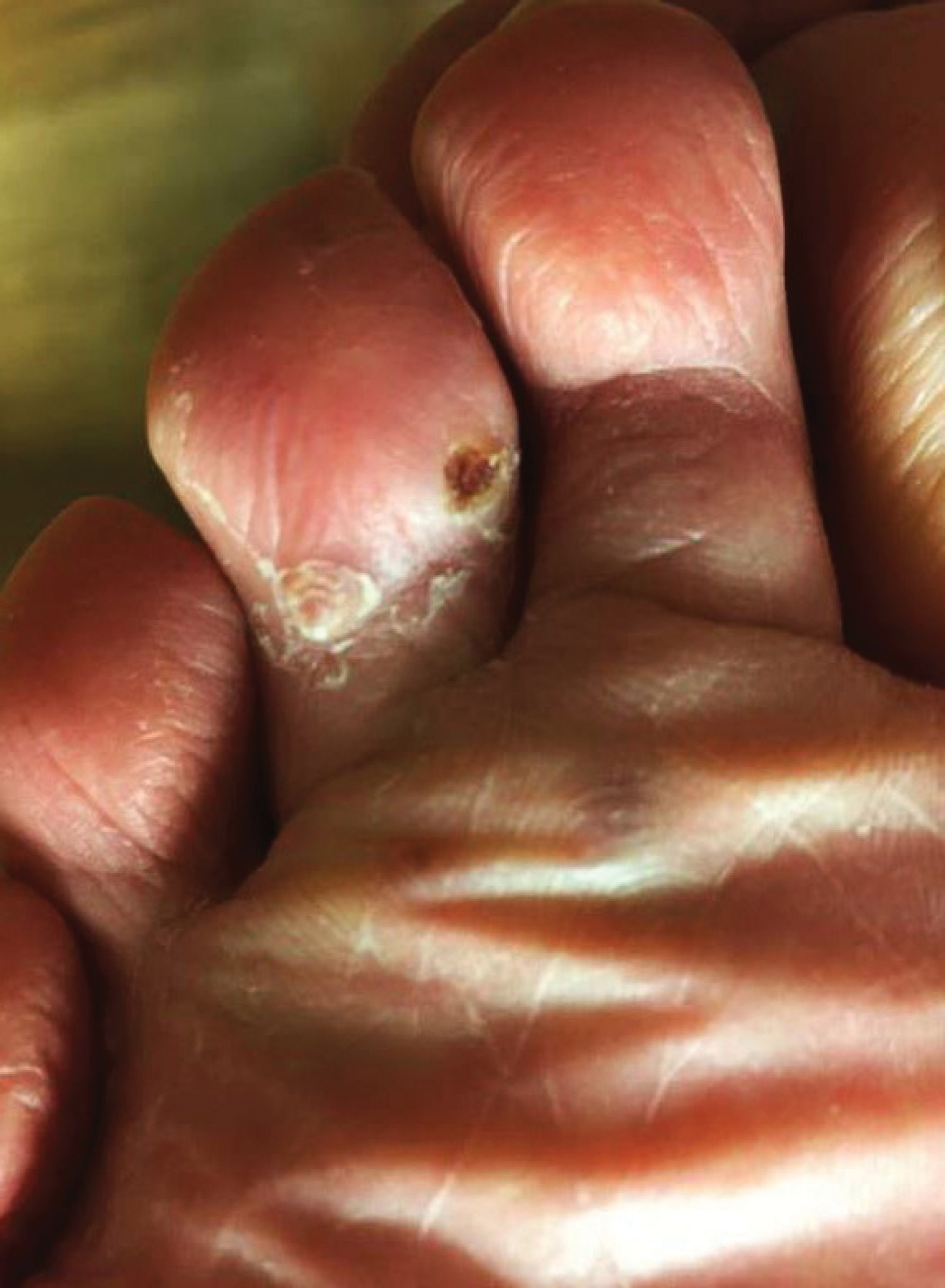
A shave biopsy of the left ankle and a punch biopsy of the left medial calf were performed and sent for histologic examination and acid-fast stain. Bacterial, fungal, and mycobacterial tissue cultures also were sent for testing. The findings from direct examination were negative, and tissue cultures exhibited no growth. The shave and punch biopsies and histology revealed pseudoepitheliomatous hyperplasia (PEH) with keratinocyte necrosis, satellitosis, and areas of acute folliculitis (Figure 1). A lichenoid hypersensitivity mixed infiltrate that included histiocytes admixed with anthracotic and red-orange pigment, lymphocytes, plasma cells, neutrophils, and rare eosinophils was noted in the dermis. Given these clinical and histopathologic findings, the patient was diagnosed with red-pigment tattoo lichenoid reaction with PEH.
Tattoo-related inflammatory reactions can manifest clinically as allergic contact dermatitis, photodermatitis, infection, malignancy, foreign body granulomas, and delayed hypersensitivity reactions with myriad associated histopathologic patterns including spongiotic, psoriasiform, granulomatous, and lichenoid (as seen in our patient). Lichenoid tattoo reactions are the most common histopathologic variants of delayed hypersensitivity seen, mostly with cinnabar or red dye.1 However, there is a paucity of cases in the literature of PEH following tattooing with red dye. Interestingly, lichenoid tissue reaction accompanies PEH in all reported cases.2
Pseudoepitheliomatous hyperplasia can mimic squamous cell carcinoma and keratoacanthoma (KA) both clinically and histologically. All 3 conditions may exhibit epithelial hyperplasia with prominent dilated hyperplastic infundibula. In a case series of 11 presumed KAs within tattoos, Fraga and Prossick2 reported 82% (9/11) of the lesions were located strictly in areas with red pigment, and many were associated with a lichenoid tissue reaction. Kazlouskaya and Junkins-Hopkins3 previously described cases of KAs in tattoos that may represent PEH.
When treating lesions with this histologic appearance, consider the clinical and histologic overlap between KAs and PEH. Our patient was managed with clobetasol ointment 0.05% under occlusion followed by intralesional triamcinolone acetonide with notable improvement of the verrucous plaques on the left lateral malleolus (Figure 2). He also noted near resolution of the papules on the pretibial shin and complete resolution of all associated pruritus and burning. Calcineurin inhibitors, photochemotherapy, CO2 laser, excimer laser, and surgical removal with interval grafting also were considered.

It is important to recognize PEH in the differential of eruptions occurring within tattoos to avoid unnecessary invasive surgical procedures such as complete surgical excision of a KA to avoid malignant transformation.
- Mortimer NJ, Chave TA, Johnston GA. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
The Diagnosis: Lichenoid Reaction With Pseudoepitheliomatous Hyperplasia
A shave biopsy of the left ankle and a punch biopsy of the left medial calf were performed and sent for histologic examination and acid-fast stain. Bacterial, fungal, and mycobacterial tissue cultures also were sent for testing. The findings from direct examination were negative, and tissue cultures exhibited no growth. The shave and punch biopsies and histology revealed pseudoepitheliomatous hyperplasia (PEH) with keratinocyte necrosis, satellitosis, and areas of acute folliculitis (Figure 1). A lichenoid hypersensitivity mixed infiltrate that included histiocytes admixed with anthracotic and red-orange pigment, lymphocytes, plasma cells, neutrophils, and rare eosinophils was noted in the dermis. Given these clinical and histopathologic findings, the patient was diagnosed with red-pigment tattoo lichenoid reaction with PEH.
Tattoo-related inflammatory reactions can manifest clinically as allergic contact dermatitis, photodermatitis, infection, malignancy, foreign body granulomas, and delayed hypersensitivity reactions with myriad associated histopathologic patterns including spongiotic, psoriasiform, granulomatous, and lichenoid (as seen in our patient). Lichenoid tattoo reactions are the most common histopathologic variants of delayed hypersensitivity seen, mostly with cinnabar or red dye.1 However, there is a paucity of cases in the literature of PEH following tattooing with red dye. Interestingly, lichenoid tissue reaction accompanies PEH in all reported cases.2
Pseudoepitheliomatous hyperplasia can mimic squamous cell carcinoma and keratoacanthoma (KA) both clinically and histologically. All 3 conditions may exhibit epithelial hyperplasia with prominent dilated hyperplastic infundibula. In a case series of 11 presumed KAs within tattoos, Fraga and Prossick2 reported 82% (9/11) of the lesions were located strictly in areas with red pigment, and many were associated with a lichenoid tissue reaction. Kazlouskaya and Junkins-Hopkins3 previously described cases of KAs in tattoos that may represent PEH.
When treating lesions with this histologic appearance, consider the clinical and histologic overlap between KAs and PEH. Our patient was managed with clobetasol ointment 0.05% under occlusion followed by intralesional triamcinolone acetonide with notable improvement of the verrucous plaques on the left lateral malleolus (Figure 2). He also noted near resolution of the papules on the pretibial shin and complete resolution of all associated pruritus and burning. Calcineurin inhibitors, photochemotherapy, CO2 laser, excimer laser, and surgical removal with interval grafting also were considered.
It is important to recognize PEH in the differential of eruptions occurring within tattoos to avoid unnecessary invasive surgical procedures such as complete surgical excision of a KA to avoid malignant transformation.
The Diagnosis: Lichenoid Reaction With Pseudoepitheliomatous Hyperplasia
A shave biopsy of the left ankle and a punch biopsy of the left medial calf were performed and sent for histologic examination and acid-fast stain. Bacterial, fungal, and mycobacterial tissue cultures also were sent for testing. The findings from direct examination were negative, and tissue cultures exhibited no growth. The shave and punch biopsies and histology revealed pseudoepitheliomatous hyperplasia (PEH) with keratinocyte necrosis, satellitosis, and areas of acute folliculitis (Figure 1). A lichenoid hypersensitivity mixed infiltrate that included histiocytes admixed with anthracotic and red-orange pigment, lymphocytes, plasma cells, neutrophils, and rare eosinophils was noted in the dermis. Given these clinical and histopathologic findings, the patient was diagnosed with red-pigment tattoo lichenoid reaction with PEH.
Tattoo-related inflammatory reactions can manifest clinically as allergic contact dermatitis, photodermatitis, infection, malignancy, foreign body granulomas, and delayed hypersensitivity reactions with myriad associated histopathologic patterns including spongiotic, psoriasiform, granulomatous, and lichenoid (as seen in our patient). Lichenoid tattoo reactions are the most common histopathologic variants of delayed hypersensitivity seen, mostly with cinnabar or red dye.1 However, there is a paucity of cases in the literature of PEH following tattooing with red dye. Interestingly, lichenoid tissue reaction accompanies PEH in all reported cases.2
Pseudoepitheliomatous hyperplasia can mimic squamous cell carcinoma and keratoacanthoma (KA) both clinically and histologically. All 3 conditions may exhibit epithelial hyperplasia with prominent dilated hyperplastic infundibula. In a case series of 11 presumed KAs within tattoos, Fraga and Prossick2 reported 82% (9/11) of the lesions were located strictly in areas with red pigment, and many were associated with a lichenoid tissue reaction. Kazlouskaya and Junkins-Hopkins3 previously described cases of KAs in tattoos that may represent PEH.
When treating lesions with this histologic appearance, consider the clinical and histologic overlap between KAs and PEH. Our patient was managed with clobetasol ointment 0.05% under occlusion followed by intralesional triamcinolone acetonide with notable improvement of the verrucous plaques on the left lateral malleolus (Figure 2). He also noted near resolution of the papules on the pretibial shin and complete resolution of all associated pruritus and burning. Calcineurin inhibitors, photochemotherapy, CO2 laser, excimer laser, and surgical removal with interval grafting also were considered.
It is important to recognize PEH in the differential of eruptions occurring within tattoos to avoid unnecessary invasive surgical procedures such as complete surgical excision of a KA to avoid malignant transformation.
- Mortimer NJ, Chave TA, Johnston GA. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
- Mortimer NJ, Chave TA, Johnston GA. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
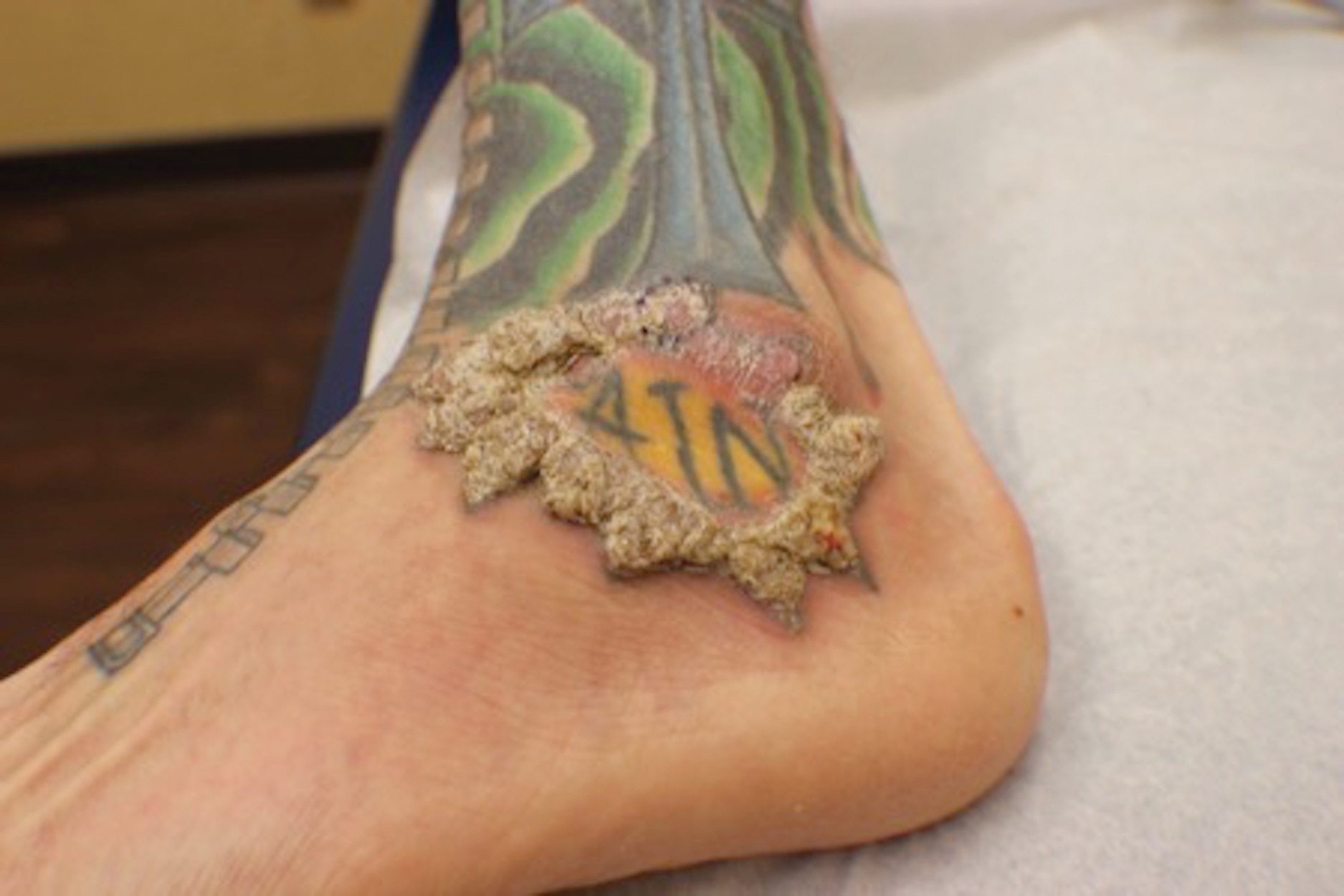
A 40-year-old man with a medical history of human immunodeficiency virus infection managed with highly active antiretroviral therapy (CD4 count, 888 cells/mm3 and an undetectable viral load), psoriasis, and recurrent condyloma acuminatum presented with exophytic, annular, hyperkeratotic, verrucous plaques on the left lateral malleolus with multiple erythematous hyperkeratotic papules on the pretibial shin of the left leg of 6 months' duration. These plaques and papules were localized to areas where red dye was used in a tattoo the patient had received 2 years prior to presentation. There was no associated fluctuance or drainage. The patient reported paroxysmal pruritus and burning pain.
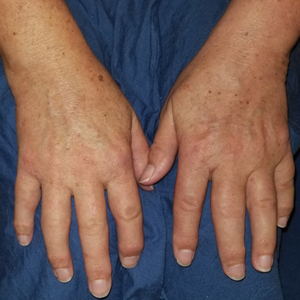
Tender Papules on the Bilateral Dorsal Hands
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
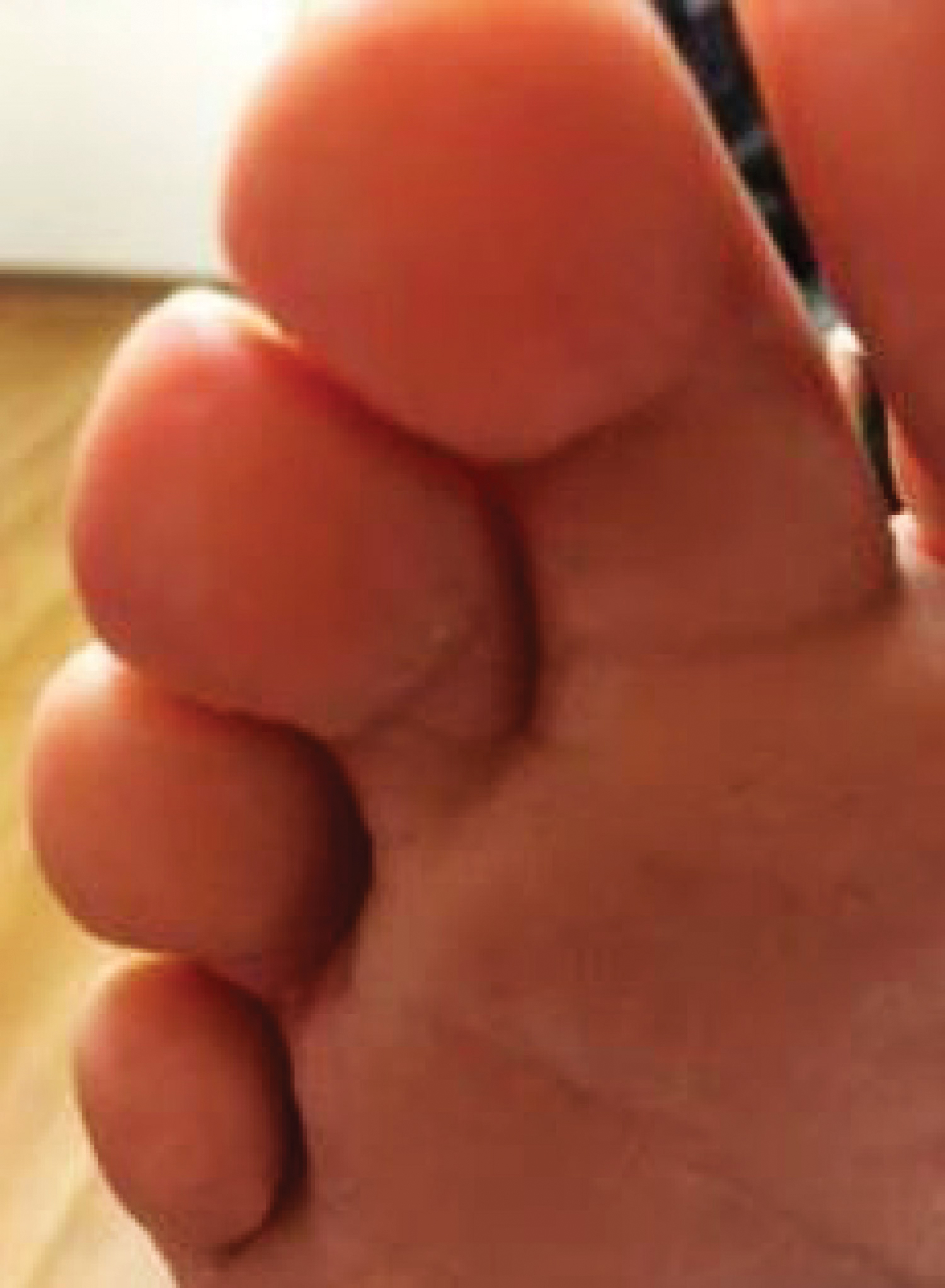
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
A 58-year-old woman with a medical history of asthma, hypertension, hypothyroidism, and hyperlipidemia presented with a painful rash of 10 days' duration. The rash was associated with fever at home (temperature, 38.5.2 °C), and a review of systems was positive for joint pain. Physical examination revealed numerous 8- to 10-mm, erythematous, discus-shaped papules on the bilateral dorsal hands, bilateral palms, right knee, and right dorsal foot with slight tenderness to palpation. A papule on the right dorsal hand was biopsied.
Kaposi Sarcoma in a Patient With Postpolio Syndrome
Kaposi sarcoma (KS) is a low-grade vascular tumor that is rare among the general US population, with an incidence rate of less than 1 per 100,000.1 The tumor is more common among certain groups of individuals due to geographic differences in the prevalence of KS-associated herpesvirus (also referred to as human herpesvirus 8) as well as host immune factors.2 Kaposi sarcoma often is defined by the patient's predisposing characteristics yielding the following distinct epidemiologic subtypes: (1) classic KS is a rare disease affecting older men of Mediterranean descent; (2) African KS is an endemic cancer with male predominance in sub-Saharan Africa; (3) AIDS-associated KS is an often aggressive AIDS-defining illness; and (4) iatrogenic KS occurs in patients on immunosuppressive therapy.3 When evaluating a patient without any of these risk factors, the clinical suspicion for KS may be low. We report a patient with postpolio syndrome (PPS) who presented with KS of the right leg, ankle, and foot.
A 77-year-old man with a distant history of paralytic poliomyelitis presented for an annual skin examination with concern for a new lesion on the right ankle. The patient had a history of PPS primarily affecting the right leg. Physical examination revealed residual weakness in an atrophic right lower extremity with a mottled appearance and mild pitting edema to the knee. Two red, dome-shaped, vascular papules were appreciated on the medial aspect of the right ankle (Figure 1), and a shave biopsy of the larger papule was performed. Microscopic examination of the biopsy specimen was consistent with KS (Figure 2). This patient had no history of human immunodeficiency virus or immunosuppressive therapy and was not of Mediterranean descent.
Because KS is a radiosensitive vascular neoplasm and radiation therapy (RT) alone can achieve local control,4 the patient was treated with 6 megaelectron-volt electron-beam RT. He received 30 Gy in 10 fractions to the affected area of the medial ankle. The patient tolerated RT well. Three weeks after completing treatment, he was found to have mild lichenification on the right medial ankle with no clinical evidence of disease. Four months later, he presented with multiple additional vascular papules on the right third toe and in the interdigital web space (Figure 3). Shave biopsy of one of these lesions was consistent with KS. Contrast computed tomography of the chest, abdomen, and pelvis was performed, revealing no evidence of metastatic disease. The patient was treated with 30 Gy in 15 fractions using opposed lateral 6 megaelectron-volt photon fields to the entire right lower extremity below the knee to treat all of the skin affected by the PPS. His posttreatment course was complicated by edema in the affected leg that resolved after daily pneumatic compression. He had no evidence of residual or recurrent disease 6 months after completing RT (Figure 4).
Cutaneous KS is a human herpesvirus 8-positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.4 Our patient did not have any classic risk factors for KS, but his disease did arise in the setting of a right lower extremity that was notably affected by PPS. Postpolio syndrome is characterized by muscle atrophy due to denervation of the motor unit.5 Bruno et al6 found that such deficits in motor innervation could lead to impairments in venous outflow causing cutaneous venous congestion. Acroangiodermatitis clinically resembles KS but is a benign reactive vasoproliferative disorder and is well known to occur in the lower extremities as a sequela of chronic venous insufficiency.7 A case of bilateral lower extremity pseudo-KS was reported in a patient with notable PPS.8 A report of 2 patients describes KS arising in the setting of chronic venous insufficiency without any classic risk factors.9 Therefore, patients with PPS characterized by venous insufficiency may represent a population at increased risk for KS.
- Surveillance, Epidemiology, and End Results (SEER) Program. US Population Data--1969-2017. https://seer.cancer.gov/popdata/. Published January 2019. Accessed November 25, 2019.
- Uldrick TS, Whitby D. Update on KSHV epidemiology, kaposi sarcoma pathogenesis, and treatment of saposi sarcoma. Cancer Lett. 2011;305:150-162.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
- Arnold HL, Odom RB, James WD, et al. Andrews' Diseases of the Skin: Clinical Dermatology. Philadelphia, PA: Saunders; 1990.
- Boyer FV, Tiffreau V, Rapin A, et al. Post-polio syndrome: pathophysiological hypotheses, diagnosis criteria, drug therapy. Ann Phys Rehabil Med. 2010;53:34-41.
- Bruno RL, Johnson JC, Berman WS. Vasomotor abnormalities as post-polio sequelae: functional and clinical implications. Orthopedics. 1985;8:865-869.
- Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86:239-240.
- Rotbart G. Kaposi's disease and venous insufficiency. Phlebologie. 1978;31:439-443.
- Que SK, DeFelice T, Abdulla FR, et al. Non-HIV-related kaposi sarcoma in 2 Hispanic patients arising in the setting of chronic venous insufficiency. Cutis. 2015;95:E30-E33.
Kaposi sarcoma (KS) is a low-grade vascular tumor that is rare among the general US population, with an incidence rate of less than 1 per 100,000.1 The tumor is more common among certain groups of individuals due to geographic differences in the prevalence of KS-associated herpesvirus (also referred to as human herpesvirus 8) as well as host immune factors.2 Kaposi sarcoma often is defined by the patient's predisposing characteristics yielding the following distinct epidemiologic subtypes: (1) classic KS is a rare disease affecting older men of Mediterranean descent; (2) African KS is an endemic cancer with male predominance in sub-Saharan Africa; (3) AIDS-associated KS is an often aggressive AIDS-defining illness; and (4) iatrogenic KS occurs in patients on immunosuppressive therapy.3 When evaluating a patient without any of these risk factors, the clinical suspicion for KS may be low. We report a patient with postpolio syndrome (PPS) who presented with KS of the right leg, ankle, and foot.
A 77-year-old man with a distant history of paralytic poliomyelitis presented for an annual skin examination with concern for a new lesion on the right ankle. The patient had a history of PPS primarily affecting the right leg. Physical examination revealed residual weakness in an atrophic right lower extremity with a mottled appearance and mild pitting edema to the knee. Two red, dome-shaped, vascular papules were appreciated on the medial aspect of the right ankle (Figure 1), and a shave biopsy of the larger papule was performed. Microscopic examination of the biopsy specimen was consistent with KS (Figure 2). This patient had no history of human immunodeficiency virus or immunosuppressive therapy and was not of Mediterranean descent.
Because KS is a radiosensitive vascular neoplasm and radiation therapy (RT) alone can achieve local control,4 the patient was treated with 6 megaelectron-volt electron-beam RT. He received 30 Gy in 10 fractions to the affected area of the medial ankle. The patient tolerated RT well. Three weeks after completing treatment, he was found to have mild lichenification on the right medial ankle with no clinical evidence of disease. Four months later, he presented with multiple additional vascular papules on the right third toe and in the interdigital web space (Figure 3). Shave biopsy of one of these lesions was consistent with KS. Contrast computed tomography of the chest, abdomen, and pelvis was performed, revealing no evidence of metastatic disease. The patient was treated with 30 Gy in 15 fractions using opposed lateral 6 megaelectron-volt photon fields to the entire right lower extremity below the knee to treat all of the skin affected by the PPS. His posttreatment course was complicated by edema in the affected leg that resolved after daily pneumatic compression. He had no evidence of residual or recurrent disease 6 months after completing RT (Figure 4).
Cutaneous KS is a human herpesvirus 8-positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.4 Our patient did not have any classic risk factors for KS, but his disease did arise in the setting of a right lower extremity that was notably affected by PPS. Postpolio syndrome is characterized by muscle atrophy due to denervation of the motor unit.5 Bruno et al6 found that such deficits in motor innervation could lead to impairments in venous outflow causing cutaneous venous congestion. Acroangiodermatitis clinically resembles KS but is a benign reactive vasoproliferative disorder and is well known to occur in the lower extremities as a sequela of chronic venous insufficiency.7 A case of bilateral lower extremity pseudo-KS was reported in a patient with notable PPS.8 A report of 2 patients describes KS arising in the setting of chronic venous insufficiency without any classic risk factors.9 Therefore, patients with PPS characterized by venous insufficiency may represent a population at increased risk for KS.
Kaposi sarcoma (KS) is a low-grade vascular tumor that is rare among the general US population, with an incidence rate of less than 1 per 100,000.1 The tumor is more common among certain groups of individuals due to geographic differences in the prevalence of KS-associated herpesvirus (also referred to as human herpesvirus 8) as well as host immune factors.2 Kaposi sarcoma often is defined by the patient's predisposing characteristics yielding the following distinct epidemiologic subtypes: (1) classic KS is a rare disease affecting older men of Mediterranean descent; (2) African KS is an endemic cancer with male predominance in sub-Saharan Africa; (3) AIDS-associated KS is an often aggressive AIDS-defining illness; and (4) iatrogenic KS occurs in patients on immunosuppressive therapy.3 When evaluating a patient without any of these risk factors, the clinical suspicion for KS may be low. We report a patient with postpolio syndrome (PPS) who presented with KS of the right leg, ankle, and foot.
A 77-year-old man with a distant history of paralytic poliomyelitis presented for an annual skin examination with concern for a new lesion on the right ankle. The patient had a history of PPS primarily affecting the right leg. Physical examination revealed residual weakness in an atrophic right lower extremity with a mottled appearance and mild pitting edema to the knee. Two red, dome-shaped, vascular papules were appreciated on the medial aspect of the right ankle (Figure 1), and a shave biopsy of the larger papule was performed. Microscopic examination of the biopsy specimen was consistent with KS (Figure 2). This patient had no history of human immunodeficiency virus or immunosuppressive therapy and was not of Mediterranean descent.
Because KS is a radiosensitive vascular neoplasm and radiation therapy (RT) alone can achieve local control,4 the patient was treated with 6 megaelectron-volt electron-beam RT. He received 30 Gy in 10 fractions to the affected area of the medial ankle. The patient tolerated RT well. Three weeks after completing treatment, he was found to have mild lichenification on the right medial ankle with no clinical evidence of disease. Four months later, he presented with multiple additional vascular papules on the right third toe and in the interdigital web space (Figure 3). Shave biopsy of one of these lesions was consistent with KS. Contrast computed tomography of the chest, abdomen, and pelvis was performed, revealing no evidence of metastatic disease. The patient was treated with 30 Gy in 15 fractions using opposed lateral 6 megaelectron-volt photon fields to the entire right lower extremity below the knee to treat all of the skin affected by the PPS. His posttreatment course was complicated by edema in the affected leg that resolved after daily pneumatic compression. He had no evidence of residual or recurrent disease 6 months after completing RT (Figure 4).
Cutaneous KS is a human herpesvirus 8-positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.4 Our patient did not have any classic risk factors for KS, but his disease did arise in the setting of a right lower extremity that was notably affected by PPS. Postpolio syndrome is characterized by muscle atrophy due to denervation of the motor unit.5 Bruno et al6 found that such deficits in motor innervation could lead to impairments in venous outflow causing cutaneous venous congestion. Acroangiodermatitis clinically resembles KS but is a benign reactive vasoproliferative disorder and is well known to occur in the lower extremities as a sequela of chronic venous insufficiency.7 A case of bilateral lower extremity pseudo-KS was reported in a patient with notable PPS.8 A report of 2 patients describes KS arising in the setting of chronic venous insufficiency without any classic risk factors.9 Therefore, patients with PPS characterized by venous insufficiency may represent a population at increased risk for KS.
- Surveillance, Epidemiology, and End Results (SEER) Program. US Population Data--1969-2017. https://seer.cancer.gov/popdata/. Published January 2019. Accessed November 25, 2019.
- Uldrick TS, Whitby D. Update on KSHV epidemiology, kaposi sarcoma pathogenesis, and treatment of saposi sarcoma. Cancer Lett. 2011;305:150-162.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
- Arnold HL, Odom RB, James WD, et al. Andrews' Diseases of the Skin: Clinical Dermatology. Philadelphia, PA: Saunders; 1990.
- Boyer FV, Tiffreau V, Rapin A, et al. Post-polio syndrome: pathophysiological hypotheses, diagnosis criteria, drug therapy. Ann Phys Rehabil Med. 2010;53:34-41.
- Bruno RL, Johnson JC, Berman WS. Vasomotor abnormalities as post-polio sequelae: functional and clinical implications. Orthopedics. 1985;8:865-869.
- Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86:239-240.
- Rotbart G. Kaposi's disease and venous insufficiency. Phlebologie. 1978;31:439-443.
- Que SK, DeFelice T, Abdulla FR, et al. Non-HIV-related kaposi sarcoma in 2 Hispanic patients arising in the setting of chronic venous insufficiency. Cutis. 2015;95:E30-E33.
- Surveillance, Epidemiology, and End Results (SEER) Program. US Population Data--1969-2017. https://seer.cancer.gov/popdata/. Published January 2019. Accessed November 25, 2019.
- Uldrick TS, Whitby D. Update on KSHV epidemiology, kaposi sarcoma pathogenesis, and treatment of saposi sarcoma. Cancer Lett. 2011;305:150-162.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
- Arnold HL, Odom RB, James WD, et al. Andrews' Diseases of the Skin: Clinical Dermatology. Philadelphia, PA: Saunders; 1990.
- Boyer FV, Tiffreau V, Rapin A, et al. Post-polio syndrome: pathophysiological hypotheses, diagnosis criteria, drug therapy. Ann Phys Rehabil Med. 2010;53:34-41.
- Bruno RL, Johnson JC, Berman WS. Vasomotor abnormalities as post-polio sequelae: functional and clinical implications. Orthopedics. 1985;8:865-869.
- Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86:239-240.
- Rotbart G. Kaposi's disease and venous insufficiency. Phlebologie. 1978;31:439-443.
- Que SK, DeFelice T, Abdulla FR, et al. Non-HIV-related kaposi sarcoma in 2 Hispanic patients arising in the setting of chronic venous insufficiency. Cutis. 2015;95:E30-E33.
Practice Points
- Cutaneous Kaposi sarcoma (KS) is a human herpesvirus 8–positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.
- In addition, patients with postpolio syndrome characterized by venous insufficiency may represent a population at increased risk for KS.
- Kaposi sarcoma is a radiosensitive vascular neoplasm, and radiation therapy can achieve local control.
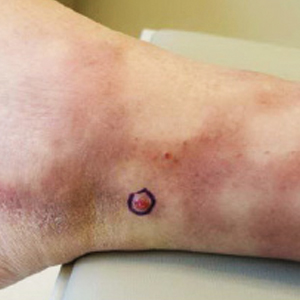
Multiple Facial Papules
The Diagnosis: Birt-Hogg-Dubé Syndrome
Histopathologic examination revealed a collection of bland spindle cells with perifollicular fibrosis consistent with a fibrofolliculoma, confirming the diagnosis of Birt-Hogg-Dubé syndrome (Figure). Cosmetic treatment with ablative therapy was offered, but the patient declined.
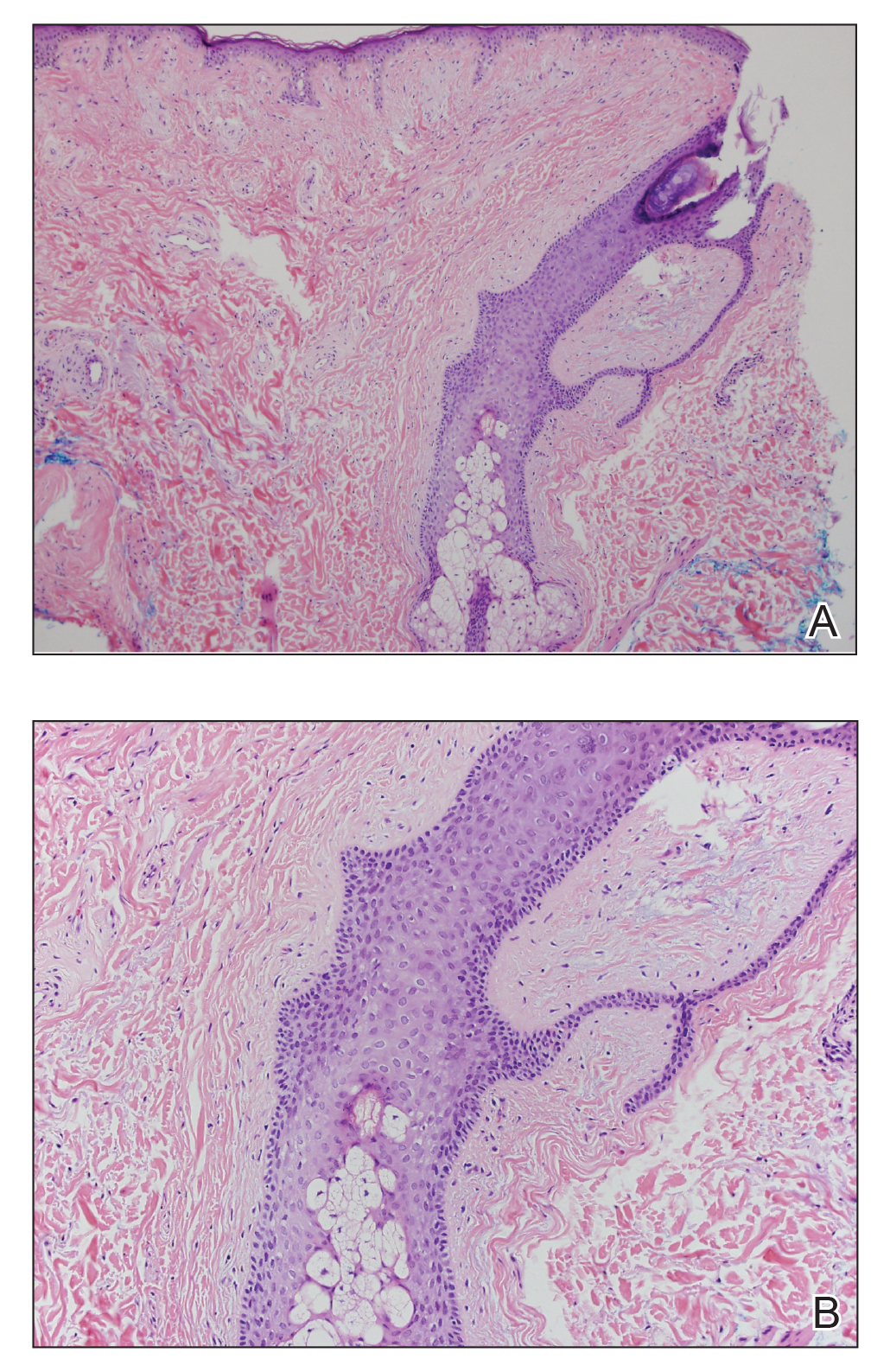
Birt-Hogg-Dubé syndrome is an autosomal-dominant genodermatosis caused by a loss-of-function mutation in the folliculin gene, FLCN, on chromosome arm 17p11.2.1 Cutaneous findings include benign follicular hamartomas, such as fibrofolliculomas and trichodiscomas. Angiofibromas, perifollicular fibromas, oral papillomas, and acrochordons also can be present.1 Cutaneous lesions usually appear on the head and neck in the third decade of life.
Patients with Birt-Hogg-Dubé syndrome are at an increased risk for pneumothorax and renal cancer, specifically hybrid oncocytic-chromophobe renal cell carcinomas.2 In a study of 89 patients with a FLCN mutation, 90% (80/89) of patients had cutaneous lesions, 84% (34/89) had pulmonary cysts, and 34% (30/89) had kidney tumors. Affected individuals were at a higher risk for pneumothorax and kidney tumors if there was a family history of these tumors.2
Proposed diagnostic criteria include any 1 of the following: 2 or more skin lesions clinically consistent with fibrofolliculomas and 1 histologically confirmed fibrofolliculoma; multiple bilateral pulmonary cysts in the basilar lung with or without pneumothorax before 40 years of age; bilateral multifocal chromophobe renal carcinomas or hybrid oncocytic tumors; combination of cutaneous, pulmonary, or renal manifestation in the patient and family; or a FLCN mutation.3
Current recommendations for the workup of a patient with Birt-Hogg-Dubé syndrome include referral to genetic counseling for the patient and family, a baseline computed tomography of the chest to evaluate for pulmonary cysts, and gadolinium-enhanced abdominal magnetic resonance imaging starting at 20 years of age and repeated every 3 to 4 years to screen for renal tumors.1 Pulmonary function tests can be considered if the patient is symptomatic or has a high cyst burden. Patients should be advised against smoking and scuba diving.
The differential diagnosis of multiple facial papules includes Cowden syndrome, tuberous sclerosis, Brooke-Spiegler syndrome, and Muir-Torre syndrome. Cowden syndrome is caused by a mutation in the protein tyrosine phosphatase gene, PTEN.4 The characteristic cutaneous findings on the face are trichilemmomas, which appear as flesh-colored papules that may have a verrucous surface.
Tuberous sclerosis is caused by mutations in hamartin (TSC1) or tuberin (TSC2). Angiofibromas are most commonly found on the face and appear as flesh-colored to red-brown papules. Fibrous plaques, periungual fibromas, gingival fibromas, hypopigmented macules, and connective tissue nevi also are found in tuberous sclerosis.5
Brooke-Spiegler syndrome is caused by a mutation in the CYLD lysine 63 deubiquitinase gene, CYLD. Trichoepitheliomas, cylindromas, and spiradenomas are caused by the CYLD mutation and appear on the head and neck. Trichoepitheliomas are flesh-colored to pink papules found on the face, often concentrated in the nasolabial folds.6 Cylindromas and spiradenomas are flesh-colored to pink papules or nodules most commonly found on the scalp.6
Muir-Torre syndrome is caused by a mutation in DNA mismatch repair genes MSH2 and/or MLH1.7 Sebaceous neoplasms, including sebaceous adenomas, sebaceomas, and less frequently sebaceous carcinomas, are characteristic cutaneous findings and appear as pink to yellow papules commonly found on the head and neck.
Careful history taking, physical examination, and histopathologic analysis are important in recognizing the features of Birt-Hogg-Dubé syndrome. Accurate and timely diagnosis is essential for the appropriate care of patients and their families, given the syndrome's systemic implications.
- Gupta N, Sunwoo BY, Kotloff RM. Birt-Hogg-Dubé syndrome. Clin Chest Med. 2016;37:475-486.
- Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321-331.
- Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol. 2015;12:558-569.
- Marsh D, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461-1472.
- Wataya-Kaneda M, Uemura M, Fujita K, et al. Tuberous sclerosis complex: recent advances in manifestations and therapy. Int J Urol. 2017;24:681-691.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Mahalingam M. MSH6, Past and present and Muir-Torre syndrome--connecting the dots. Am J Dermatopathol. 2017;39:239-249.
The Diagnosis: Birt-Hogg-Dubé Syndrome
Histopathologic examination revealed a collection of bland spindle cells with perifollicular fibrosis consistent with a fibrofolliculoma, confirming the diagnosis of Birt-Hogg-Dubé syndrome (Figure). Cosmetic treatment with ablative therapy was offered, but the patient declined.
Birt-Hogg-Dubé syndrome is an autosomal-dominant genodermatosis caused by a loss-of-function mutation in the folliculin gene, FLCN, on chromosome arm 17p11.2.1 Cutaneous findings include benign follicular hamartomas, such as fibrofolliculomas and trichodiscomas. Angiofibromas, perifollicular fibromas, oral papillomas, and acrochordons also can be present.1 Cutaneous lesions usually appear on the head and neck in the third decade of life.
Patients with Birt-Hogg-Dubé syndrome are at an increased risk for pneumothorax and renal cancer, specifically hybrid oncocytic-chromophobe renal cell carcinomas.2 In a study of 89 patients with a FLCN mutation, 90% (80/89) of patients had cutaneous lesions, 84% (34/89) had pulmonary cysts, and 34% (30/89) had kidney tumors. Affected individuals were at a higher risk for pneumothorax and kidney tumors if there was a family history of these tumors.2
Proposed diagnostic criteria include any 1 of the following: 2 or more skin lesions clinically consistent with fibrofolliculomas and 1 histologically confirmed fibrofolliculoma; multiple bilateral pulmonary cysts in the basilar lung with or without pneumothorax before 40 years of age; bilateral multifocal chromophobe renal carcinomas or hybrid oncocytic tumors; combination of cutaneous, pulmonary, or renal manifestation in the patient and family; or a FLCN mutation.3
Current recommendations for the workup of a patient with Birt-Hogg-Dubé syndrome include referral to genetic counseling for the patient and family, a baseline computed tomography of the chest to evaluate for pulmonary cysts, and gadolinium-enhanced abdominal magnetic resonance imaging starting at 20 years of age and repeated every 3 to 4 years to screen for renal tumors.1 Pulmonary function tests can be considered if the patient is symptomatic or has a high cyst burden. Patients should be advised against smoking and scuba diving.
The differential diagnosis of multiple facial papules includes Cowden syndrome, tuberous sclerosis, Brooke-Spiegler syndrome, and Muir-Torre syndrome. Cowden syndrome is caused by a mutation in the protein tyrosine phosphatase gene, PTEN.4 The characteristic cutaneous findings on the face are trichilemmomas, which appear as flesh-colored papules that may have a verrucous surface.
Tuberous sclerosis is caused by mutations in hamartin (TSC1) or tuberin (TSC2). Angiofibromas are most commonly found on the face and appear as flesh-colored to red-brown papules. Fibrous plaques, periungual fibromas, gingival fibromas, hypopigmented macules, and connective tissue nevi also are found in tuberous sclerosis.5
Brooke-Spiegler syndrome is caused by a mutation in the CYLD lysine 63 deubiquitinase gene, CYLD. Trichoepitheliomas, cylindromas, and spiradenomas are caused by the CYLD mutation and appear on the head and neck. Trichoepitheliomas are flesh-colored to pink papules found on the face, often concentrated in the nasolabial folds.6 Cylindromas and spiradenomas are flesh-colored to pink papules or nodules most commonly found on the scalp.6
Muir-Torre syndrome is caused by a mutation in DNA mismatch repair genes MSH2 and/or MLH1.7 Sebaceous neoplasms, including sebaceous adenomas, sebaceomas, and less frequently sebaceous carcinomas, are characteristic cutaneous findings and appear as pink to yellow papules commonly found on the head and neck.
Careful history taking, physical examination, and histopathologic analysis are important in recognizing the features of Birt-Hogg-Dubé syndrome. Accurate and timely diagnosis is essential for the appropriate care of patients and their families, given the syndrome's systemic implications.
The Diagnosis: Birt-Hogg-Dubé Syndrome
Histopathologic examination revealed a collection of bland spindle cells with perifollicular fibrosis consistent with a fibrofolliculoma, confirming the diagnosis of Birt-Hogg-Dubé syndrome (Figure). Cosmetic treatment with ablative therapy was offered, but the patient declined.
Birt-Hogg-Dubé syndrome is an autosomal-dominant genodermatosis caused by a loss-of-function mutation in the folliculin gene, FLCN, on chromosome arm 17p11.2.1 Cutaneous findings include benign follicular hamartomas, such as fibrofolliculomas and trichodiscomas. Angiofibromas, perifollicular fibromas, oral papillomas, and acrochordons also can be present.1 Cutaneous lesions usually appear on the head and neck in the third decade of life.
Patients with Birt-Hogg-Dubé syndrome are at an increased risk for pneumothorax and renal cancer, specifically hybrid oncocytic-chromophobe renal cell carcinomas.2 In a study of 89 patients with a FLCN mutation, 90% (80/89) of patients had cutaneous lesions, 84% (34/89) had pulmonary cysts, and 34% (30/89) had kidney tumors. Affected individuals were at a higher risk for pneumothorax and kidney tumors if there was a family history of these tumors.2
Proposed diagnostic criteria include any 1 of the following: 2 or more skin lesions clinically consistent with fibrofolliculomas and 1 histologically confirmed fibrofolliculoma; multiple bilateral pulmonary cysts in the basilar lung with or without pneumothorax before 40 years of age; bilateral multifocal chromophobe renal carcinomas or hybrid oncocytic tumors; combination of cutaneous, pulmonary, or renal manifestation in the patient and family; or a FLCN mutation.3
Current recommendations for the workup of a patient with Birt-Hogg-Dubé syndrome include referral to genetic counseling for the patient and family, a baseline computed tomography of the chest to evaluate for pulmonary cysts, and gadolinium-enhanced abdominal magnetic resonance imaging starting at 20 years of age and repeated every 3 to 4 years to screen for renal tumors.1 Pulmonary function tests can be considered if the patient is symptomatic or has a high cyst burden. Patients should be advised against smoking and scuba diving.
The differential diagnosis of multiple facial papules includes Cowden syndrome, tuberous sclerosis, Brooke-Spiegler syndrome, and Muir-Torre syndrome. Cowden syndrome is caused by a mutation in the protein tyrosine phosphatase gene, PTEN.4 The characteristic cutaneous findings on the face are trichilemmomas, which appear as flesh-colored papules that may have a verrucous surface.
Tuberous sclerosis is caused by mutations in hamartin (TSC1) or tuberin (TSC2). Angiofibromas are most commonly found on the face and appear as flesh-colored to red-brown papules. Fibrous plaques, periungual fibromas, gingival fibromas, hypopigmented macules, and connective tissue nevi also are found in tuberous sclerosis.5
Brooke-Spiegler syndrome is caused by a mutation in the CYLD lysine 63 deubiquitinase gene, CYLD. Trichoepitheliomas, cylindromas, and spiradenomas are caused by the CYLD mutation and appear on the head and neck. Trichoepitheliomas are flesh-colored to pink papules found on the face, often concentrated in the nasolabial folds.6 Cylindromas and spiradenomas are flesh-colored to pink papules or nodules most commonly found on the scalp.6
Muir-Torre syndrome is caused by a mutation in DNA mismatch repair genes MSH2 and/or MLH1.7 Sebaceous neoplasms, including sebaceous adenomas, sebaceomas, and less frequently sebaceous carcinomas, are characteristic cutaneous findings and appear as pink to yellow papules commonly found on the head and neck.
Careful history taking, physical examination, and histopathologic analysis are important in recognizing the features of Birt-Hogg-Dubé syndrome. Accurate and timely diagnosis is essential for the appropriate care of patients and their families, given the syndrome's systemic implications.
- Gupta N, Sunwoo BY, Kotloff RM. Birt-Hogg-Dubé syndrome. Clin Chest Med. 2016;37:475-486.
- Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321-331.
- Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol. 2015;12:558-569.
- Marsh D, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461-1472.
- Wataya-Kaneda M, Uemura M, Fujita K, et al. Tuberous sclerosis complex: recent advances in manifestations and therapy. Int J Urol. 2017;24:681-691.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Mahalingam M. MSH6, Past and present and Muir-Torre syndrome--connecting the dots. Am J Dermatopathol. 2017;39:239-249.
- Gupta N, Sunwoo BY, Kotloff RM. Birt-Hogg-Dubé syndrome. Clin Chest Med. 2016;37:475-486.
- Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321-331.
- Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol. 2015;12:558-569.
- Marsh D, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461-1472.
- Wataya-Kaneda M, Uemura M, Fujita K, et al. Tuberous sclerosis complex: recent advances in manifestations and therapy. Int J Urol. 2017;24:681-691.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Mahalingam M. MSH6, Past and present and Muir-Torre syndrome--connecting the dots. Am J Dermatopathol. 2017;39:239-249.

A 50-year-old man presented with facial papules on the cheeks that had appeared approximately 1.5 years prior and gradually spread over the face and neck. They were occasionally pruritic but otherwise were asymptomatic. His mother and brother reportedly had similar clinical findings. Family history was notable for a maternal uncle who had died in his 30s of an unknown type of renal cancer. Physical examination revealed innumerable white-gray papules that measured 1 to 5 mm and were scattered across the face and neck. Punch biopsies were obtained. Computed tomography of the chest showed multiple bibasilar pulmonary cysts. Magnetic resonance imaging was negative for renal tumors.
Unilateral Papules on the Face
The Diagnosis: Mosaic Tuberous Sclerosis
A punch biopsy of the facial lesion was stained with hematoxylin and eosin, which demonstrated spindled and stellate fibroblasts with dilated blood vessels (Figure), consistent with an angiofibroma. Given the clinical presentation and histologic findings, there was concern for a diagnosis of tuberous sclerosis (TSC). The patient was referred for genetic workup but tested negative for mutations of the TSC genes in the blood. Because the patient had only unilateral facial lesions, a possible cortical tuber, no other symptoms, and tested negative for TSC gene mutations, mosaic TSC was considered a likely diagnosis. Her facial lesions were treated with pulsed dye vascular laser therapy.
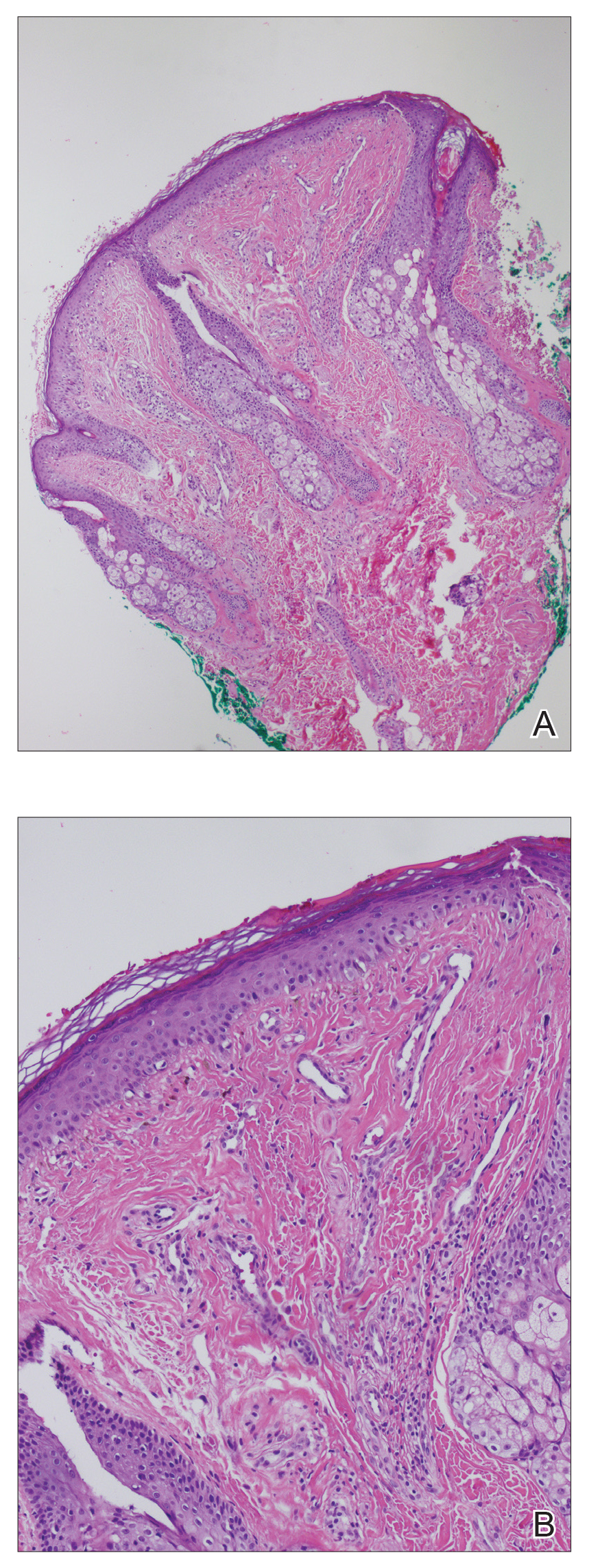
Tuberous sclerosis is an autosomal-dominant neurocutaneous disorder caused by inactivation of the genes TSC1 (encoding hamartin) and TSC2 (encoding tuberin). Mutation results in overactivation of the downstream mTOR (mammalian target of rapamycin) pathway, resulting in abnormal cellular proliferation and hamartomas. These benign tumors can be found in nearly every organ, most often in the central nervous system and skin, and they provide for a highly variable presentation of the disease.1
Tuberous sclerosis affects 1 in 6000 to 10,000 live births and has a prevalence of 1 in 20,000 individuals. Of these individuals, 75% carry sporadic mutations, and 75% to 90% eventually test positive for a TSC gene mutation.2 Genetic mosaicism has been reported in 28% of cases affected by large deletions1 and as few as 1% of cases involving small mutations.3
The dermatologic manifestation of mosaic TSC most often includes unilateral angiofibromas, whereas in nonmosaic cases, angiofibromas cover both cheeks, the forehead, and the eyelids. The other skin lesions of TSC--shagreen patches, forehead plaques, hypomelanotic macules, and ungual fibromas--are seen less frequently.4-6 Additionally, neurologic disease in mosaic patients is notably milder, with 57% of mosaic patients found to have epilepsy compared to 91% of nonmosaic patients.7 Our patient had both unilateral facial angiofibromas and a cortical lesion suspicious for a tuber, prompting a suspected diagnosis of mosaic TSC.
The methods of diagnosis outlined by the International Tuberous Sclerosis Complex Consensus Group pose a challenge in diagnosing mosaic TSC. The clinical criteria require 2 major (eg, multiple angiofibromas, angiomyolipomas, a shagreen patch) and 1 minor feature (eg, dental enamel pit, renal cyst).2 However, case reports detailing unilateral facial angiofibromas have described patients with isolated dermatologic findings.5,6 Further, it has been demonstrated that genetic studies in mosaic TSC can be unreliable depending on the tissue sampled.8 Thus, for patients who have mosaic TSC, establishing a definitive diagnosis is not always possible and may rely solely on the clinical picture.
Considering the differential diagnosis, benign cephalic histiocytosis usually would present with small red-brown macules and papules symmetrically located on the head and neck. The lesions occur at a younger age, usually in the first year or two of life. Fibrofolliculomas present as multiple whitish, slightly larger papules found on the central face. They are a marker for Birt-Hogg-Dubé syndrome, which also is associated with spontaneous pneumothorax.
Agminated means clustering or grouping of lesions. Agminated melanocytic nevi and agminated Spitz nevi are clusters of nevi. These lesions can vary in size and color. They may be elevated or flat. Melanocytic nevi usually are tan-brown or black. Spitz nevi may be pink or pigmented brown or black. To definitively differentiate between these 2 diagnoses and this patient's diagnosis of angiofibroma, a biopsy is needed.
- Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259-273.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. Weinham, Germany: Wiley-Blackwell; 2011.
- Alshaiji JM, Spock CR, Connelly EA, et al. Facial angiofibromas in a mosaic pattern tuberous sclerosis: a case report. Dermatol Online J. 2012;18:8.
- Gutte R, Khopkar U. Unilateral multiple facial angiofibromas: a case report with brief review of literature. Indian J Dermatol. 2013;58:159.
- Silvestre JF, Bañuls J, Ramón R, et al. Unilateral multiple facial angiofibromas: a mosaic form of TSC. J Am Acad Dermatol. 2000;43(1, pt 1):127-129.
- Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389-400.
- Kwiatkowska J, Wigowska-Sowinska J, Napierala D, et al. Mosaicism in TSC as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703-707.
The Diagnosis: Mosaic Tuberous Sclerosis
A punch biopsy of the facial lesion was stained with hematoxylin and eosin, which demonstrated spindled and stellate fibroblasts with dilated blood vessels (Figure), consistent with an angiofibroma. Given the clinical presentation and histologic findings, there was concern for a diagnosis of tuberous sclerosis (TSC). The patient was referred for genetic workup but tested negative for mutations of the TSC genes in the blood. Because the patient had only unilateral facial lesions, a possible cortical tuber, no other symptoms, and tested negative for TSC gene mutations, mosaic TSC was considered a likely diagnosis. Her facial lesions were treated with pulsed dye vascular laser therapy.
Tuberous sclerosis is an autosomal-dominant neurocutaneous disorder caused by inactivation of the genes TSC1 (encoding hamartin) and TSC2 (encoding tuberin). Mutation results in overactivation of the downstream mTOR (mammalian target of rapamycin) pathway, resulting in abnormal cellular proliferation and hamartomas. These benign tumors can be found in nearly every organ, most often in the central nervous system and skin, and they provide for a highly variable presentation of the disease.1
Tuberous sclerosis affects 1 in 6000 to 10,000 live births and has a prevalence of 1 in 20,000 individuals. Of these individuals, 75% carry sporadic mutations, and 75% to 90% eventually test positive for a TSC gene mutation.2 Genetic mosaicism has been reported in 28% of cases affected by large deletions1 and as few as 1% of cases involving small mutations.3
The dermatologic manifestation of mosaic TSC most often includes unilateral angiofibromas, whereas in nonmosaic cases, angiofibromas cover both cheeks, the forehead, and the eyelids. The other skin lesions of TSC--shagreen patches, forehead plaques, hypomelanotic macules, and ungual fibromas--are seen less frequently.4-6 Additionally, neurologic disease in mosaic patients is notably milder, with 57% of mosaic patients found to have epilepsy compared to 91% of nonmosaic patients.7 Our patient had both unilateral facial angiofibromas and a cortical lesion suspicious for a tuber, prompting a suspected diagnosis of mosaic TSC.
The methods of diagnosis outlined by the International Tuberous Sclerosis Complex Consensus Group pose a challenge in diagnosing mosaic TSC. The clinical criteria require 2 major (eg, multiple angiofibromas, angiomyolipomas, a shagreen patch) and 1 minor feature (eg, dental enamel pit, renal cyst).2 However, case reports detailing unilateral facial angiofibromas have described patients with isolated dermatologic findings.5,6 Further, it has been demonstrated that genetic studies in mosaic TSC can be unreliable depending on the tissue sampled.8 Thus, for patients who have mosaic TSC, establishing a definitive diagnosis is not always possible and may rely solely on the clinical picture.
Considering the differential diagnosis, benign cephalic histiocytosis usually would present with small red-brown macules and papules symmetrically located on the head and neck. The lesions occur at a younger age, usually in the first year or two of life. Fibrofolliculomas present as multiple whitish, slightly larger papules found on the central face. They are a marker for Birt-Hogg-Dubé syndrome, which also is associated with spontaneous pneumothorax.
Agminated means clustering or grouping of lesions. Agminated melanocytic nevi and agminated Spitz nevi are clusters of nevi. These lesions can vary in size and color. They may be elevated or flat. Melanocytic nevi usually are tan-brown or black. Spitz nevi may be pink or pigmented brown or black. To definitively differentiate between these 2 diagnoses and this patient's diagnosis of angiofibroma, a biopsy is needed.
The Diagnosis: Mosaic Tuberous Sclerosis
A punch biopsy of the facial lesion was stained with hematoxylin and eosin, which demonstrated spindled and stellate fibroblasts with dilated blood vessels (Figure), consistent with an angiofibroma. Given the clinical presentation and histologic findings, there was concern for a diagnosis of tuberous sclerosis (TSC). The patient was referred for genetic workup but tested negative for mutations of the TSC genes in the blood. Because the patient had only unilateral facial lesions, a possible cortical tuber, no other symptoms, and tested negative for TSC gene mutations, mosaic TSC was considered a likely diagnosis. Her facial lesions were treated with pulsed dye vascular laser therapy.
Tuberous sclerosis is an autosomal-dominant neurocutaneous disorder caused by inactivation of the genes TSC1 (encoding hamartin) and TSC2 (encoding tuberin). Mutation results in overactivation of the downstream mTOR (mammalian target of rapamycin) pathway, resulting in abnormal cellular proliferation and hamartomas. These benign tumors can be found in nearly every organ, most often in the central nervous system and skin, and they provide for a highly variable presentation of the disease.1
Tuberous sclerosis affects 1 in 6000 to 10,000 live births and has a prevalence of 1 in 20,000 individuals. Of these individuals, 75% carry sporadic mutations, and 75% to 90% eventually test positive for a TSC gene mutation.2 Genetic mosaicism has been reported in 28% of cases affected by large deletions1 and as few as 1% of cases involving small mutations.3
The dermatologic manifestation of mosaic TSC most often includes unilateral angiofibromas, whereas in nonmosaic cases, angiofibromas cover both cheeks, the forehead, and the eyelids. The other skin lesions of TSC--shagreen patches, forehead plaques, hypomelanotic macules, and ungual fibromas--are seen less frequently.4-6 Additionally, neurologic disease in mosaic patients is notably milder, with 57% of mosaic patients found to have epilepsy compared to 91% of nonmosaic patients.7 Our patient had both unilateral facial angiofibromas and a cortical lesion suspicious for a tuber, prompting a suspected diagnosis of mosaic TSC.
The methods of diagnosis outlined by the International Tuberous Sclerosis Complex Consensus Group pose a challenge in diagnosing mosaic TSC. The clinical criteria require 2 major (eg, multiple angiofibromas, angiomyolipomas, a shagreen patch) and 1 minor feature (eg, dental enamel pit, renal cyst).2 However, case reports detailing unilateral facial angiofibromas have described patients with isolated dermatologic findings.5,6 Further, it has been demonstrated that genetic studies in mosaic TSC can be unreliable depending on the tissue sampled.8 Thus, for patients who have mosaic TSC, establishing a definitive diagnosis is not always possible and may rely solely on the clinical picture.
Considering the differential diagnosis, benign cephalic histiocytosis usually would present with small red-brown macules and papules symmetrically located on the head and neck. The lesions occur at a younger age, usually in the first year or two of life. Fibrofolliculomas present as multiple whitish, slightly larger papules found on the central face. They are a marker for Birt-Hogg-Dubé syndrome, which also is associated with spontaneous pneumothorax.
Agminated means clustering or grouping of lesions. Agminated melanocytic nevi and agminated Spitz nevi are clusters of nevi. These lesions can vary in size and color. They may be elevated or flat. Melanocytic nevi usually are tan-brown or black. Spitz nevi may be pink or pigmented brown or black. To definitively differentiate between these 2 diagnoses and this patient's diagnosis of angiofibroma, a biopsy is needed.
- Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259-273.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. Weinham, Germany: Wiley-Blackwell; 2011.
- Alshaiji JM, Spock CR, Connelly EA, et al. Facial angiofibromas in a mosaic pattern tuberous sclerosis: a case report. Dermatol Online J. 2012;18:8.
- Gutte R, Khopkar U. Unilateral multiple facial angiofibromas: a case report with brief review of literature. Indian J Dermatol. 2013;58:159.
- Silvestre JF, Bañuls J, Ramón R, et al. Unilateral multiple facial angiofibromas: a mosaic form of TSC. J Am Acad Dermatol. 2000;43(1, pt 1):127-129.
- Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389-400.
- Kwiatkowska J, Wigowska-Sowinska J, Napierala D, et al. Mosaicism in TSC as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703-707.
- Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259-273.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. Weinham, Germany: Wiley-Blackwell; 2011.
- Alshaiji JM, Spock CR, Connelly EA, et al. Facial angiofibromas in a mosaic pattern tuberous sclerosis: a case report. Dermatol Online J. 2012;18:8.
- Gutte R, Khopkar U. Unilateral multiple facial angiofibromas: a case report with brief review of literature. Indian J Dermatol. 2013;58:159.
- Silvestre JF, Bañuls J, Ramón R, et al. Unilateral multiple facial angiofibromas: a mosaic form of TSC. J Am Acad Dermatol. 2000;43(1, pt 1):127-129.
- Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389-400.
- Kwiatkowska J, Wigowska-Sowinska J, Napierala D, et al. Mosaicism in TSC as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703-707.
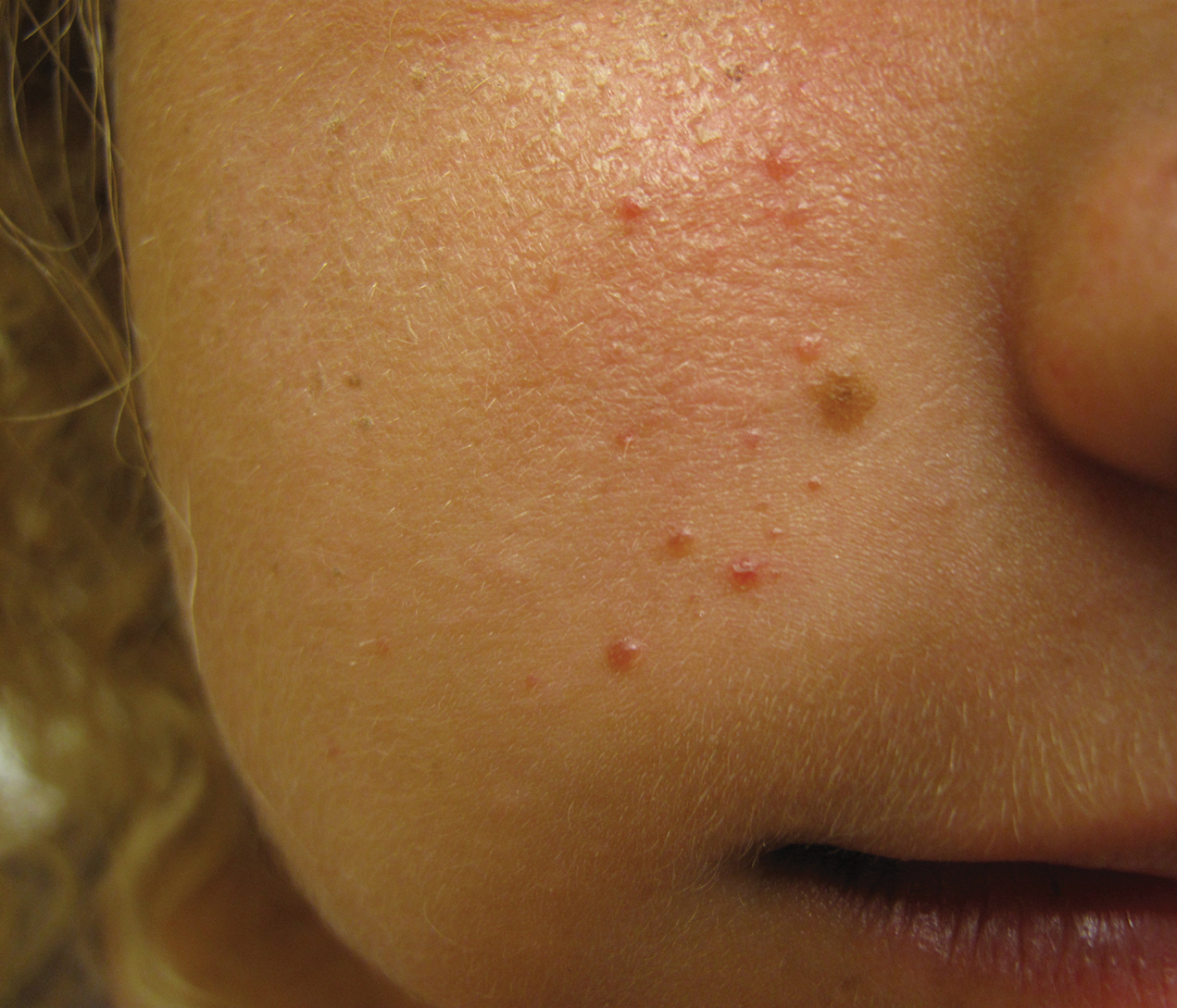
An 18-year-old woman presented with a progressive appearance of firm, red-brown, asymptomatic, 1- to 3-mm, dome-shaped papules on the right cheek that developed over the course of 2 years. She had 10 lesions that covered a 2.2 ×4-cm area on the right medial cheek. No similar-appearing lesions were detectable on a full-body skin examination, and no periungual tumors, café au lait macules, or shagreen patches were noted. A full-body skin examination using a Wood lamp revealed 1 small hypopigmented macule on the right second finger. The patient had a history of treatment-refractory migraines; magnetic resonance imaging 5 years prior to the current presentation revealed a nonspecific lesion in the left parietal gyrus. There was no personal or family history of seizures, cognitive delay, kidney disease, or ocular disease. Punch biopsy of a facial lesion was performed for histopathologic correlation.
Solitary Papule on the Nose
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
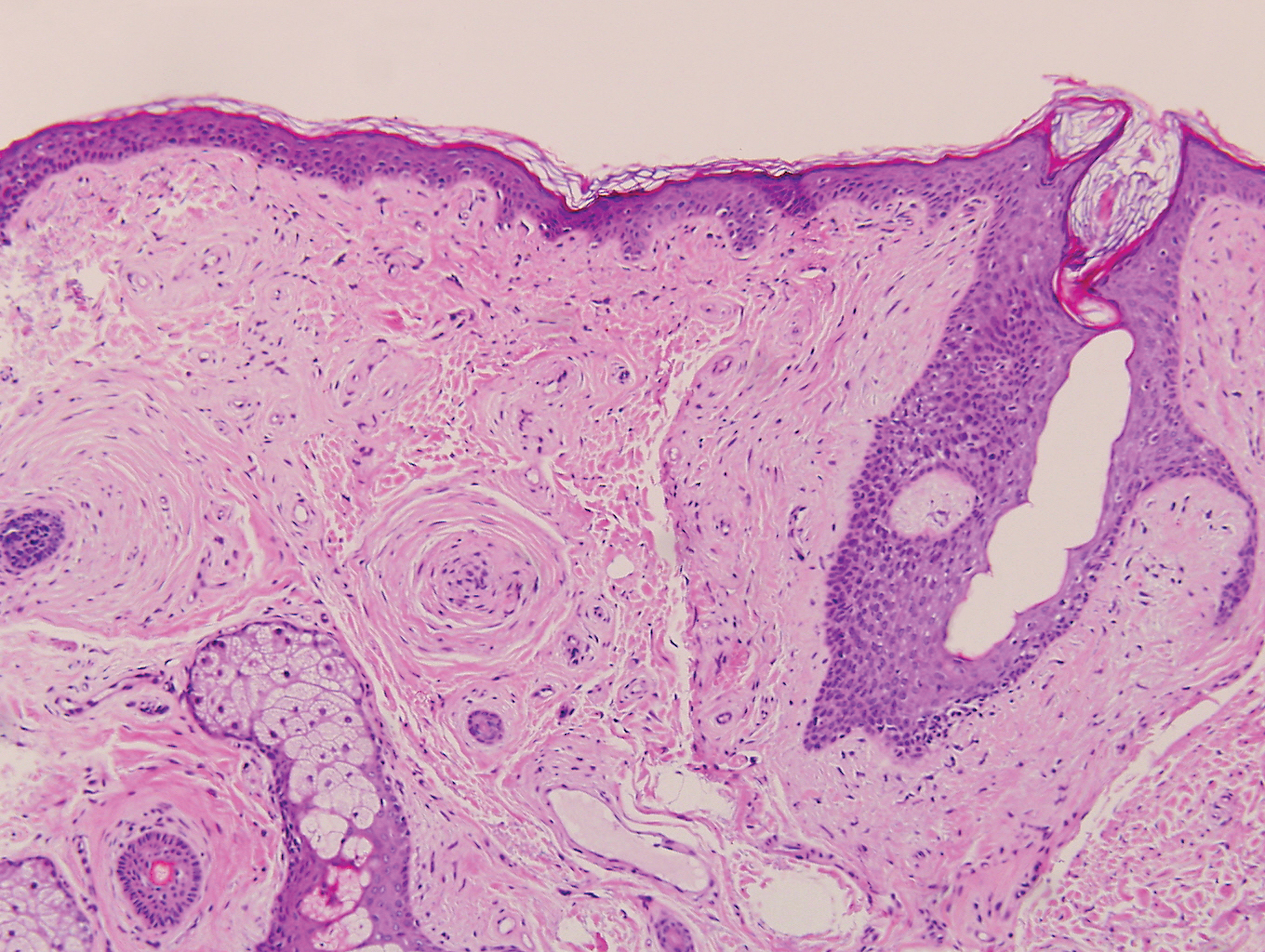
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
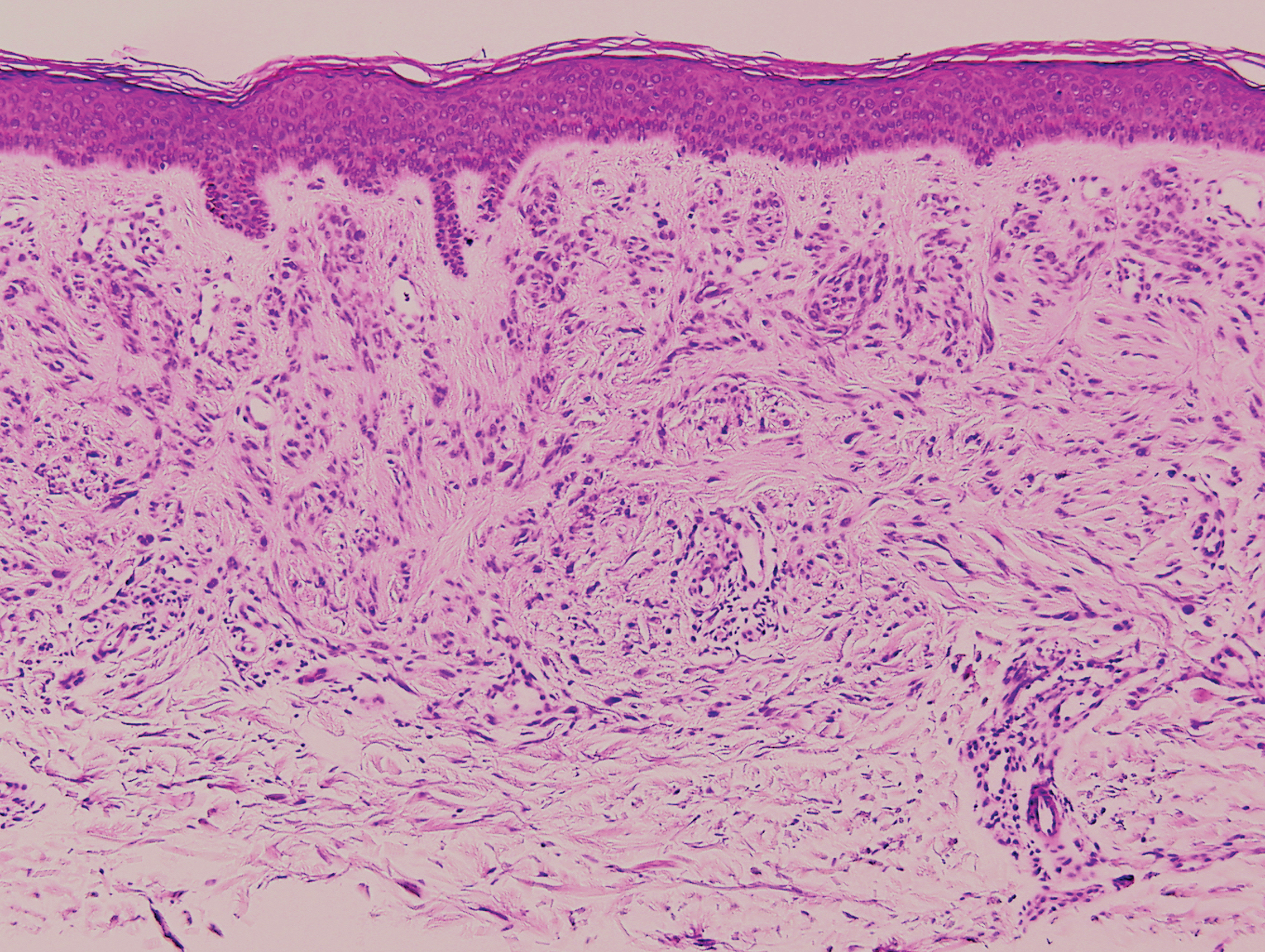
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
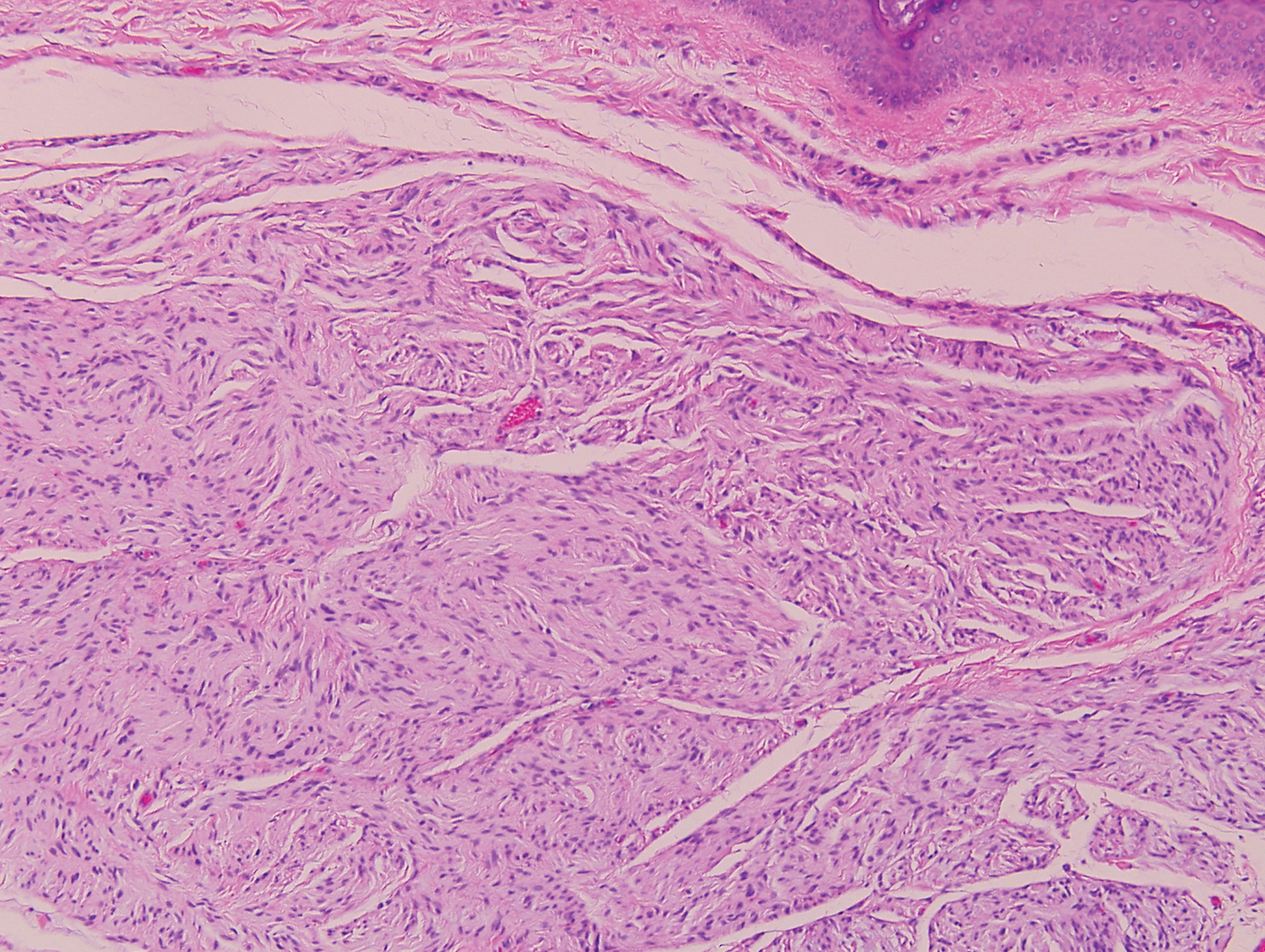
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
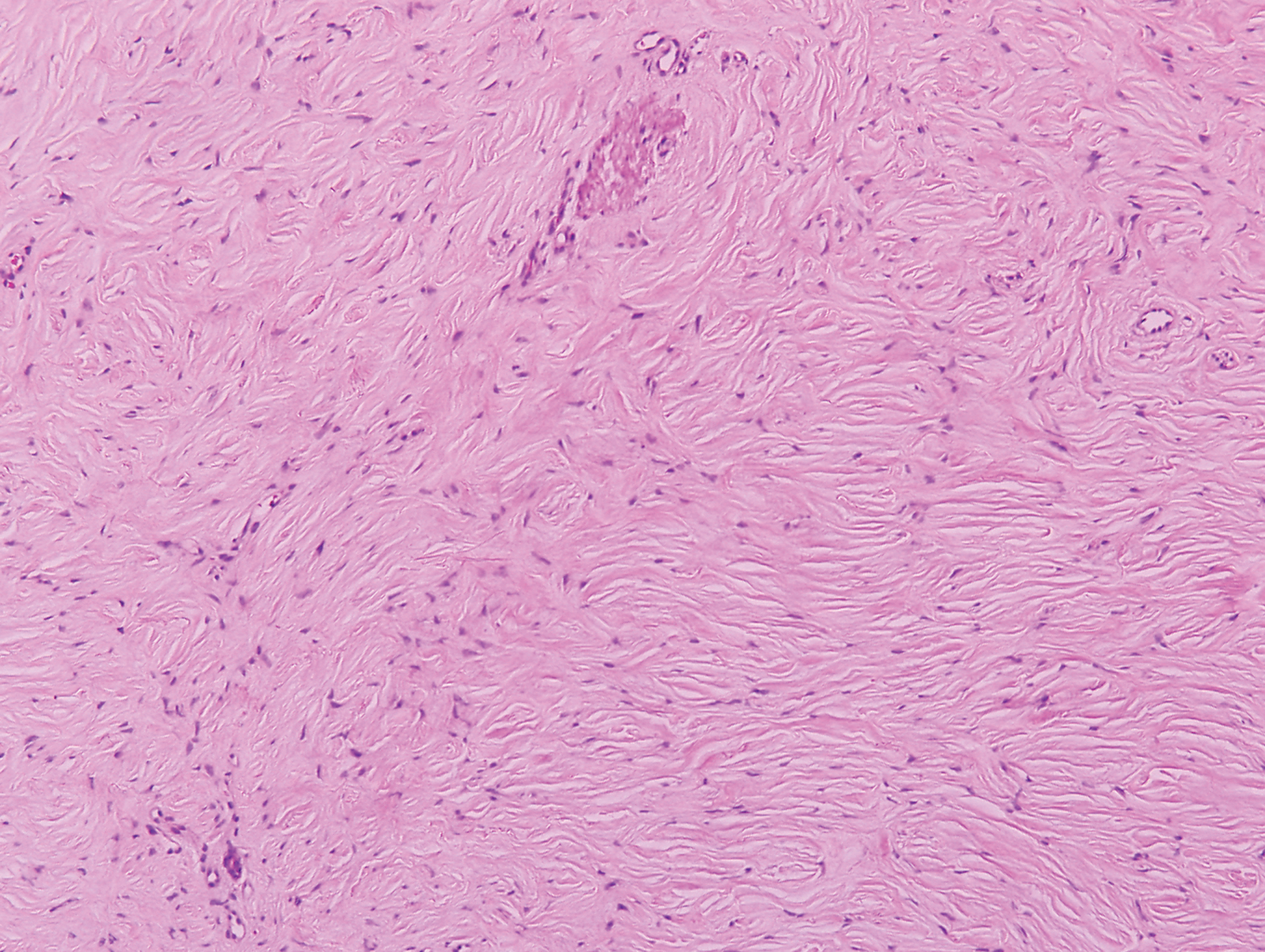
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
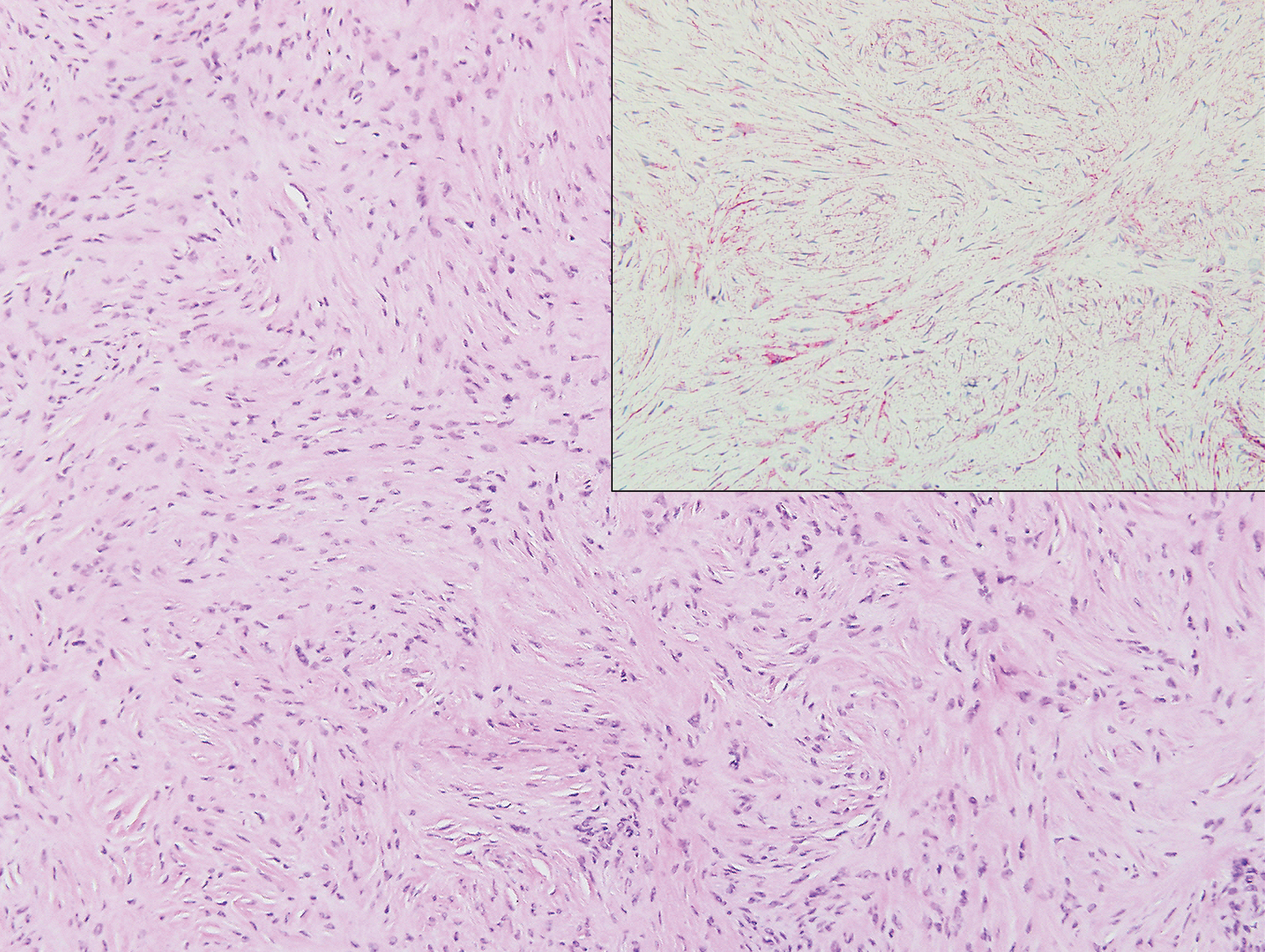
A 25-year-old man presented with a flesh-colored papule on the left side of the nose of 2 years' duration.
Systemic Epstein-Barr Virus–Positive T-cell Lymphoma of Childhood
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
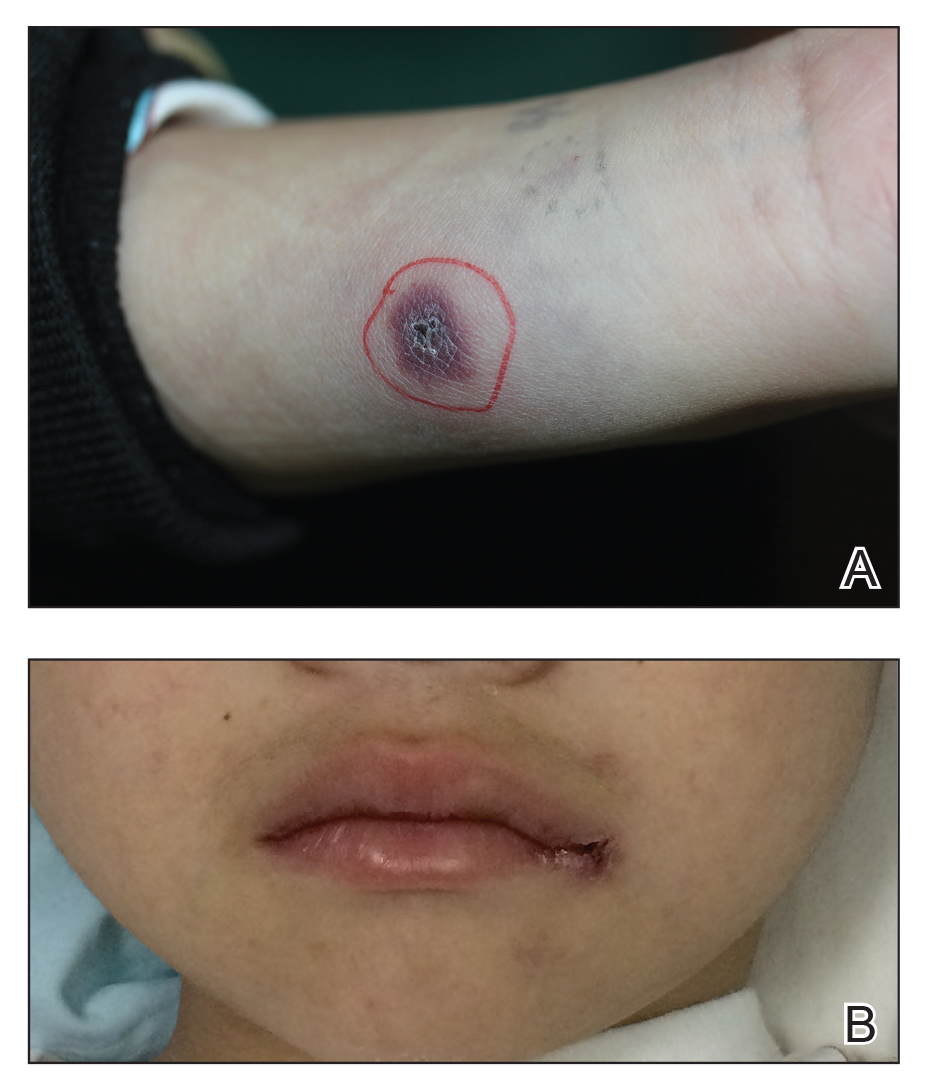
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
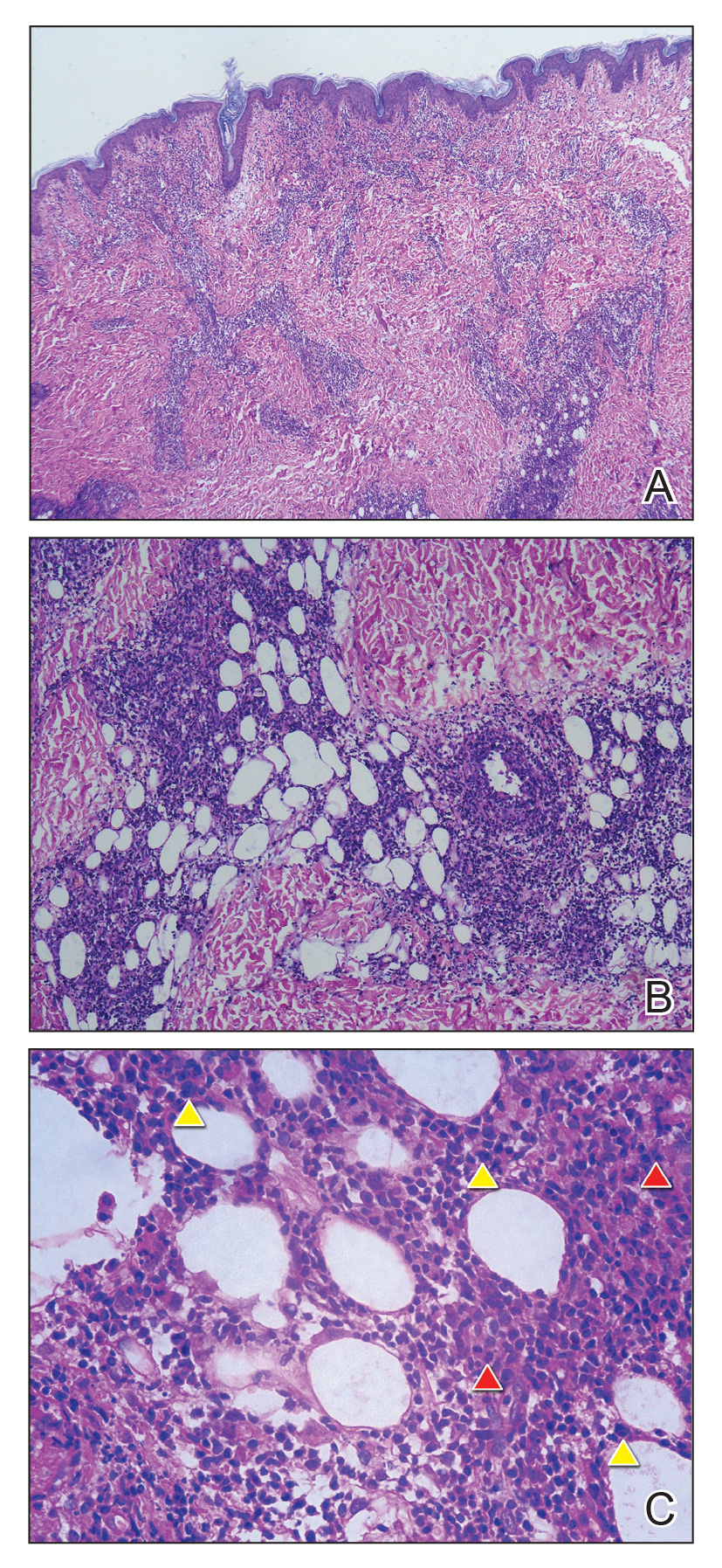
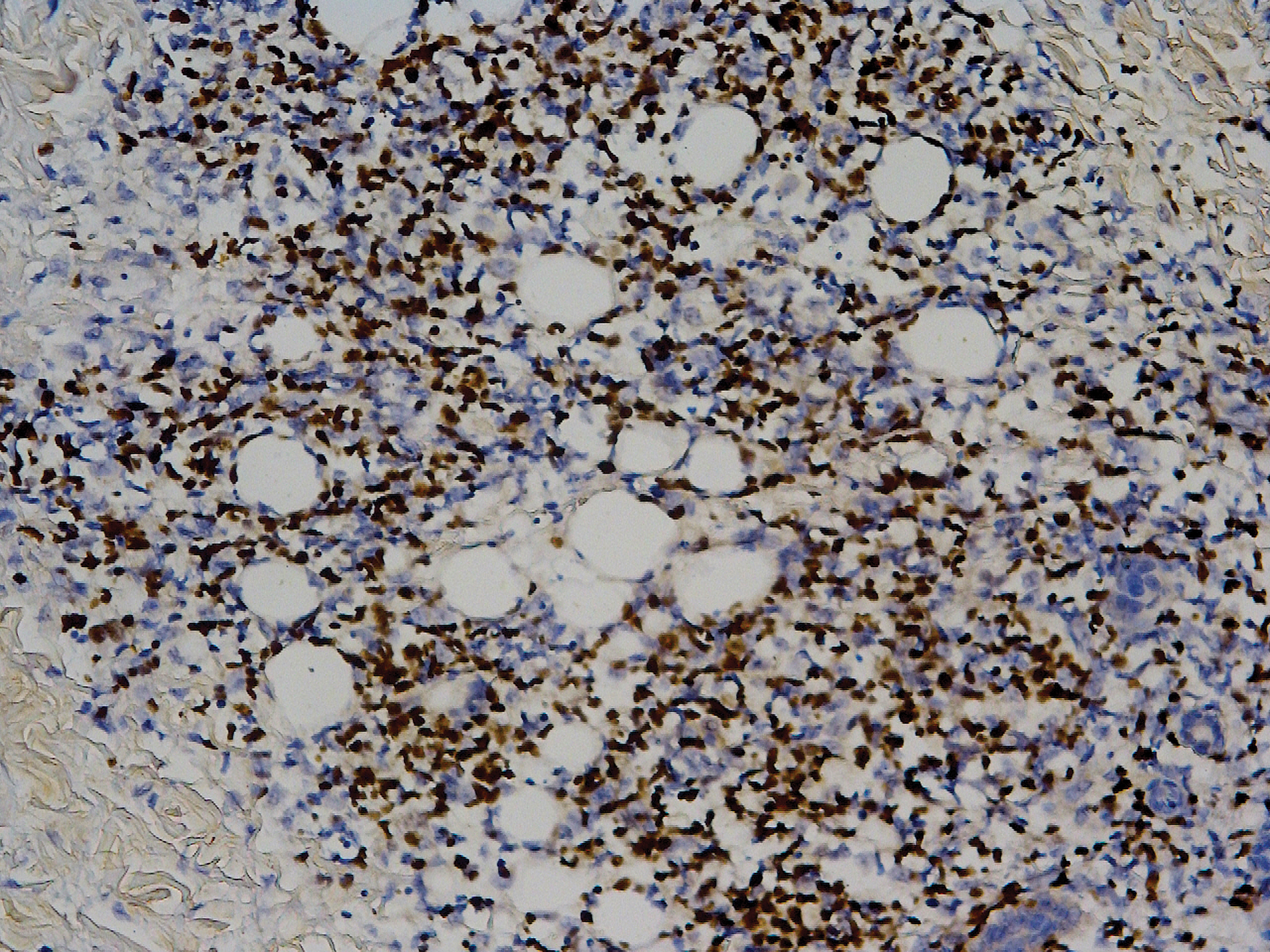
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Practice Points
- Systemic Epstein-Barr virus (EBV)–positive T-cell lymphoma of childhood is a fulminant illness with a predilection for Asians and indigenous populations from Mexico and Central and South America. In most patients, the disease has an aggressive clinical course with high mortality.
- The disease often is complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. When these severe complications are absent, the prognosis might be better.
- In situ hybridization for EBV-encoded RNA and for T-cell receptor gene rearrangements is an important tool to establish the diagnosis as well as for treatment options and predicting the prognosis.
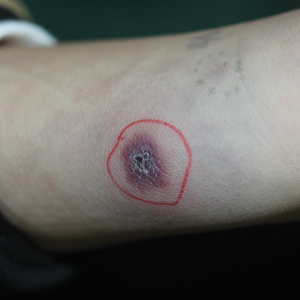
Erythematous Plaques and Nodules on the Abdomen and Groin
The Diagnosis: Inflammatory Urothelial Carcinoma
Microscopic examination revealed metastatic carcinoma with extensive dermal lymphatic invasion (Figure). Immunohistochemical stains were positive for p63 and GATA3, markers for urothelial carcinomas, and negative for S-100 and Melan-A, markers for melanoma. Thus, the biopsy was compatible with a diagnosis of urothelial carcinoma. Gram and Grocott-Gomori methenamine-silver stains were negative for bacterial or fungal organisms. An additional 4-mm punch biopsy was performed of the left thigh at the distal-most aspect of the eruption to determine the extent of cutaneous metastasis. Pathology again showed metastatic urothelial carcinoma with extensive dermal lymphatic involvement and overlying epidermal spongiosis.
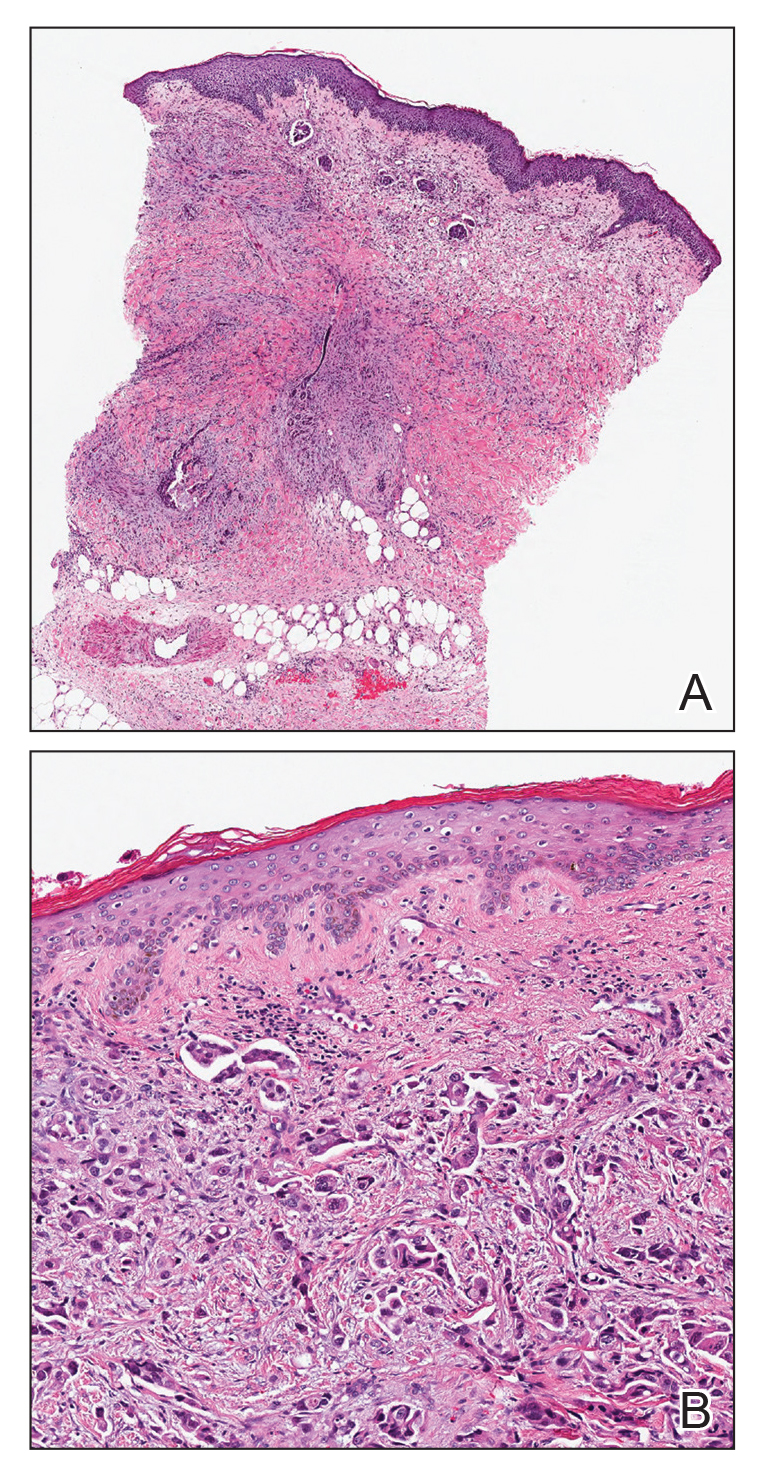
The patient had a history of bladder cancer diagnosed 1.5 years prior to presentation. It was a high-grade (World Health Organization) urothelial carcinoma that penetrated the bladder muscular wall, focally infiltrating into pericystic fat with multifocal seeding of pericystic lymphatics. It was unresponsive to bacillus Calmette-Guérin therapy. He underwent a cystoprostatectomy and bilateral staging lymph node dissection with clear surgical margins without adjuvant chemotherapy or radiation. He also reported a history of 2 prior cutaneous melanomas that were excised without sentinel lymph node biopsy.
Four months prior to presentation, he developed a mildly pruritic cutaneous eruption on the abdomen that was treated with topical miconazole for presumed tinea cruris without improvement. He also was previously diagnosed with candidiasis of his urostomy and was taking oral fluconazole. The patient was admitted for the abdominal pain and distension, and computed tomography of the abdomen and pelvis revealed peritoneal carcinomatosis resulting in mechanical small bowel obstruction as well as enlarged pelvic and retroperitoneal lymph nodes. Confirmation of metastatic disease via skin biopsy avoided an invasive peritoneal biopsy. He was treated with triamcinolone acetonide ointment 0.1% with moderate relief of pruritus, and a palliative percutaneous endoscopic gastrostomy tube was placed for bowel decompression. The patient's hospital course was complicated by Proteus mirabilis bacteremia requiring cefepime. He was transitioned to home hospice and died 1 month after presentation.
Inflammatory carcinoma, also called carcinoma erysipeloides, is a type of cutaneous metastasis most commonly seen in breast adenocarcinoma. Reported cases secondary to urothelial carcinoma are rare and most often involve the abdomen, groin, and lower extremities.1-5 Clinically, inflammatory carcinoma presents as erythematous indurated patches or plaques with well-defined borders, often with edema, warmth, and tenderness. Its morphologic appearance is due to the obstruction of lymphatic vessels by tumor cells and the release of inflammatory cytokines. Its presentation can mimic other dermatoses such as cellulitis, erysipelas, fungal infection, radiation dermatitis, Majocchi granuloma, or contact dermatitis.6 Cutaneous metastases may be the first clinical manifestations of metastatic disease, and they may occur due to hematogenous and lymphatic spread, direct contiguous tissue invasion, or iatrogenic implantation following surgical excision of the primary tumor. Histologically, nuclear markers GATA3 and p63 stain positively in urothelial carcinomas and are negative in prostatic adenocarcinomas.7,8 Other markers may be used such as cytokeratins 7 and 20, which are cytoplasmic epithelial markers that both stain positive in urothelial neoplasms.9
Inflammatory carcinoma may be treated with radiation or systemic chemotherapy depending on the extent of systemic involvement in the patient; however, its presence portends a poor prognosis. Less than 1% of genitourinary malignancies have cutaneous involvement, and median disease-specific survival is less than 6 months from presentation of the cutaneous metastasis.10 Clinicians faced with a recalcitrant inflammatory cutaneous eruption should maintain a high index of suspicion for cutaneous metastases, particularly in patients with a history of cancer. Early dermatology referral may help establish the diagnosis and guide disease-targeted therapy or goals of care discussions.
- Grace SA, Livingood MR, Boyd AS. Metastatic urothelial carcinoma presenting as carcinoma erysipeloides. J Cutan Pathol. 2017;44:513-515.
- Zangrilli A, Saraceno R, Sarmati L, et al. Erysipeloid cutaneous metastasis from bladder carcinoma. Eur J Dermatol. 2007;17:534-536.
- Chang CP, Lee Y, Shih HJ. Unusual presentation of cutaneous metastasis from bladder urothelial carcinoma. Chin J Cancer Res. 2013;25:362-365.
- Aloi F, Solaroli C, Paradiso M, et al. Inflammatory type cutaneous metastasis of bladder neoplasm: erysipeloid carcinoma [in Italian]. Minerva Urol Nefrol. 1998;50:205-208.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Al Ameer A, Imran M, Kaliyadan F, et al. Carcinoma erysipeloides as a presenting feature of breast carcinoma: a case report and brief review of literature. Indian Dermatol Online J. 2015;6:396-398.
- Chang A, Amin A, Gabrielson E, et al. Utility of GATA3 immunohistochemistry in differentiating urothelial carcinoma from prostate adenocarcinoma and squamous cell carcinomas of the uterine cervix, anus, and lung. Am J Surg Pathol. 2012;36:1472-1476.
- Ud Din N, Qureshi A, Mansoor S. Utility of p63 immunohistochemical stain in differentiating urothelial carcinomas from adenocarcinomas of prostate. Indian J Pathol Microbiol. 2011;54:59-62.
- Bassily NH, Vallorosi CJ, Akdas G, et al. Coordinate expression of cytokeratins 7 and 20 in prostate adenocarcinoma and bladder urothelial carcinoma. Am J Clin Pathol. 2000;113:383-388.
- Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026.
The Diagnosis: Inflammatory Urothelial Carcinoma
Microscopic examination revealed metastatic carcinoma with extensive dermal lymphatic invasion (Figure). Immunohistochemical stains were positive for p63 and GATA3, markers for urothelial carcinomas, and negative for S-100 and Melan-A, markers for melanoma. Thus, the biopsy was compatible with a diagnosis of urothelial carcinoma. Gram and Grocott-Gomori methenamine-silver stains were negative for bacterial or fungal organisms. An additional 4-mm punch biopsy was performed of the left thigh at the distal-most aspect of the eruption to determine the extent of cutaneous metastasis. Pathology again showed metastatic urothelial carcinoma with extensive dermal lymphatic involvement and overlying epidermal spongiosis.
The patient had a history of bladder cancer diagnosed 1.5 years prior to presentation. It was a high-grade (World Health Organization) urothelial carcinoma that penetrated the bladder muscular wall, focally infiltrating into pericystic fat with multifocal seeding of pericystic lymphatics. It was unresponsive to bacillus Calmette-Guérin therapy. He underwent a cystoprostatectomy and bilateral staging lymph node dissection with clear surgical margins without adjuvant chemotherapy or radiation. He also reported a history of 2 prior cutaneous melanomas that were excised without sentinel lymph node biopsy.
Four months prior to presentation, he developed a mildly pruritic cutaneous eruption on the abdomen that was treated with topical miconazole for presumed tinea cruris without improvement. He also was previously diagnosed with candidiasis of his urostomy and was taking oral fluconazole. The patient was admitted for the abdominal pain and distension, and computed tomography of the abdomen and pelvis revealed peritoneal carcinomatosis resulting in mechanical small bowel obstruction as well as enlarged pelvic and retroperitoneal lymph nodes. Confirmation of metastatic disease via skin biopsy avoided an invasive peritoneal biopsy. He was treated with triamcinolone acetonide ointment 0.1% with moderate relief of pruritus, and a palliative percutaneous endoscopic gastrostomy tube was placed for bowel decompression. The patient's hospital course was complicated by Proteus mirabilis bacteremia requiring cefepime. He was transitioned to home hospice and died 1 month after presentation.
Inflammatory carcinoma, also called carcinoma erysipeloides, is a type of cutaneous metastasis most commonly seen in breast adenocarcinoma. Reported cases secondary to urothelial carcinoma are rare and most often involve the abdomen, groin, and lower extremities.1-5 Clinically, inflammatory carcinoma presents as erythematous indurated patches or plaques with well-defined borders, often with edema, warmth, and tenderness. Its morphologic appearance is due to the obstruction of lymphatic vessels by tumor cells and the release of inflammatory cytokines. Its presentation can mimic other dermatoses such as cellulitis, erysipelas, fungal infection, radiation dermatitis, Majocchi granuloma, or contact dermatitis.6 Cutaneous metastases may be the first clinical manifestations of metastatic disease, and they may occur due to hematogenous and lymphatic spread, direct contiguous tissue invasion, or iatrogenic implantation following surgical excision of the primary tumor. Histologically, nuclear markers GATA3 and p63 stain positively in urothelial carcinomas and are negative in prostatic adenocarcinomas.7,8 Other markers may be used such as cytokeratins 7 and 20, which are cytoplasmic epithelial markers that both stain positive in urothelial neoplasms.9
Inflammatory carcinoma may be treated with radiation or systemic chemotherapy depending on the extent of systemic involvement in the patient; however, its presence portends a poor prognosis. Less than 1% of genitourinary malignancies have cutaneous involvement, and median disease-specific survival is less than 6 months from presentation of the cutaneous metastasis.10 Clinicians faced with a recalcitrant inflammatory cutaneous eruption should maintain a high index of suspicion for cutaneous metastases, particularly in patients with a history of cancer. Early dermatology referral may help establish the diagnosis and guide disease-targeted therapy or goals of care discussions.
The Diagnosis: Inflammatory Urothelial Carcinoma
Microscopic examination revealed metastatic carcinoma with extensive dermal lymphatic invasion (Figure). Immunohistochemical stains were positive for p63 and GATA3, markers for urothelial carcinomas, and negative for S-100 and Melan-A, markers for melanoma. Thus, the biopsy was compatible with a diagnosis of urothelial carcinoma. Gram and Grocott-Gomori methenamine-silver stains were negative for bacterial or fungal organisms. An additional 4-mm punch biopsy was performed of the left thigh at the distal-most aspect of the eruption to determine the extent of cutaneous metastasis. Pathology again showed metastatic urothelial carcinoma with extensive dermal lymphatic involvement and overlying epidermal spongiosis.
The patient had a history of bladder cancer diagnosed 1.5 years prior to presentation. It was a high-grade (World Health Organization) urothelial carcinoma that penetrated the bladder muscular wall, focally infiltrating into pericystic fat with multifocal seeding of pericystic lymphatics. It was unresponsive to bacillus Calmette-Guérin therapy. He underwent a cystoprostatectomy and bilateral staging lymph node dissection with clear surgical margins without adjuvant chemotherapy or radiation. He also reported a history of 2 prior cutaneous melanomas that were excised without sentinel lymph node biopsy.
Four months prior to presentation, he developed a mildly pruritic cutaneous eruption on the abdomen that was treated with topical miconazole for presumed tinea cruris without improvement. He also was previously diagnosed with candidiasis of his urostomy and was taking oral fluconazole. The patient was admitted for the abdominal pain and distension, and computed tomography of the abdomen and pelvis revealed peritoneal carcinomatosis resulting in mechanical small bowel obstruction as well as enlarged pelvic and retroperitoneal lymph nodes. Confirmation of metastatic disease via skin biopsy avoided an invasive peritoneal biopsy. He was treated with triamcinolone acetonide ointment 0.1% with moderate relief of pruritus, and a palliative percutaneous endoscopic gastrostomy tube was placed for bowel decompression. The patient's hospital course was complicated by Proteus mirabilis bacteremia requiring cefepime. He was transitioned to home hospice and died 1 month after presentation.
Inflammatory carcinoma, also called carcinoma erysipeloides, is a type of cutaneous metastasis most commonly seen in breast adenocarcinoma. Reported cases secondary to urothelial carcinoma are rare and most often involve the abdomen, groin, and lower extremities.1-5 Clinically, inflammatory carcinoma presents as erythematous indurated patches or plaques with well-defined borders, often with edema, warmth, and tenderness. Its morphologic appearance is due to the obstruction of lymphatic vessels by tumor cells and the release of inflammatory cytokines. Its presentation can mimic other dermatoses such as cellulitis, erysipelas, fungal infection, radiation dermatitis, Majocchi granuloma, or contact dermatitis.6 Cutaneous metastases may be the first clinical manifestations of metastatic disease, and they may occur due to hematogenous and lymphatic spread, direct contiguous tissue invasion, or iatrogenic implantation following surgical excision of the primary tumor. Histologically, nuclear markers GATA3 and p63 stain positively in urothelial carcinomas and are negative in prostatic adenocarcinomas.7,8 Other markers may be used such as cytokeratins 7 and 20, which are cytoplasmic epithelial markers that both stain positive in urothelial neoplasms.9
Inflammatory carcinoma may be treated with radiation or systemic chemotherapy depending on the extent of systemic involvement in the patient; however, its presence portends a poor prognosis. Less than 1% of genitourinary malignancies have cutaneous involvement, and median disease-specific survival is less than 6 months from presentation of the cutaneous metastasis.10 Clinicians faced with a recalcitrant inflammatory cutaneous eruption should maintain a high index of suspicion for cutaneous metastases, particularly in patients with a history of cancer. Early dermatology referral may help establish the diagnosis and guide disease-targeted therapy or goals of care discussions.
- Grace SA, Livingood MR, Boyd AS. Metastatic urothelial carcinoma presenting as carcinoma erysipeloides. J Cutan Pathol. 2017;44:513-515.
- Zangrilli A, Saraceno R, Sarmati L, et al. Erysipeloid cutaneous metastasis from bladder carcinoma. Eur J Dermatol. 2007;17:534-536.
- Chang CP, Lee Y, Shih HJ. Unusual presentation of cutaneous metastasis from bladder urothelial carcinoma. Chin J Cancer Res. 2013;25:362-365.
- Aloi F, Solaroli C, Paradiso M, et al. Inflammatory type cutaneous metastasis of bladder neoplasm: erysipeloid carcinoma [in Italian]. Minerva Urol Nefrol. 1998;50:205-208.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Al Ameer A, Imran M, Kaliyadan F, et al. Carcinoma erysipeloides as a presenting feature of breast carcinoma: a case report and brief review of literature. Indian Dermatol Online J. 2015;6:396-398.
- Chang A, Amin A, Gabrielson E, et al. Utility of GATA3 immunohistochemistry in differentiating urothelial carcinoma from prostate adenocarcinoma and squamous cell carcinomas of the uterine cervix, anus, and lung. Am J Surg Pathol. 2012;36:1472-1476.
- Ud Din N, Qureshi A, Mansoor S. Utility of p63 immunohistochemical stain in differentiating urothelial carcinomas from adenocarcinomas of prostate. Indian J Pathol Microbiol. 2011;54:59-62.
- Bassily NH, Vallorosi CJ, Akdas G, et al. Coordinate expression of cytokeratins 7 and 20 in prostate adenocarcinoma and bladder urothelial carcinoma. Am J Clin Pathol. 2000;113:383-388.
- Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026.
- Grace SA, Livingood MR, Boyd AS. Metastatic urothelial carcinoma presenting as carcinoma erysipeloides. J Cutan Pathol. 2017;44:513-515.
- Zangrilli A, Saraceno R, Sarmati L, et al. Erysipeloid cutaneous metastasis from bladder carcinoma. Eur J Dermatol. 2007;17:534-536.
- Chang CP, Lee Y, Shih HJ. Unusual presentation of cutaneous metastasis from bladder urothelial carcinoma. Chin J Cancer Res. 2013;25:362-365.
- Aloi F, Solaroli C, Paradiso M, et al. Inflammatory type cutaneous metastasis of bladder neoplasm: erysipeloid carcinoma [in Italian]. Minerva Urol Nefrol. 1998;50:205-208.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Al Ameer A, Imran M, Kaliyadan F, et al. Carcinoma erysipeloides as a presenting feature of breast carcinoma: a case report and brief review of literature. Indian Dermatol Online J. 2015;6:396-398.
- Chang A, Amin A, Gabrielson E, et al. Utility of GATA3 immunohistochemistry in differentiating urothelial carcinoma from prostate adenocarcinoma and squamous cell carcinomas of the uterine cervix, anus, and lung. Am J Surg Pathol. 2012;36:1472-1476.
- Ud Din N, Qureshi A, Mansoor S. Utility of p63 immunohistochemical stain in differentiating urothelial carcinomas from adenocarcinomas of prostate. Indian J Pathol Microbiol. 2011;54:59-62.
- Bassily NH, Vallorosi CJ, Akdas G, et al. Coordinate expression of cytokeratins 7 and 20 in prostate adenocarcinoma and bladder urothelial carcinoma. Am J Clin Pathol. 2000;113:383-388.
- Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026.
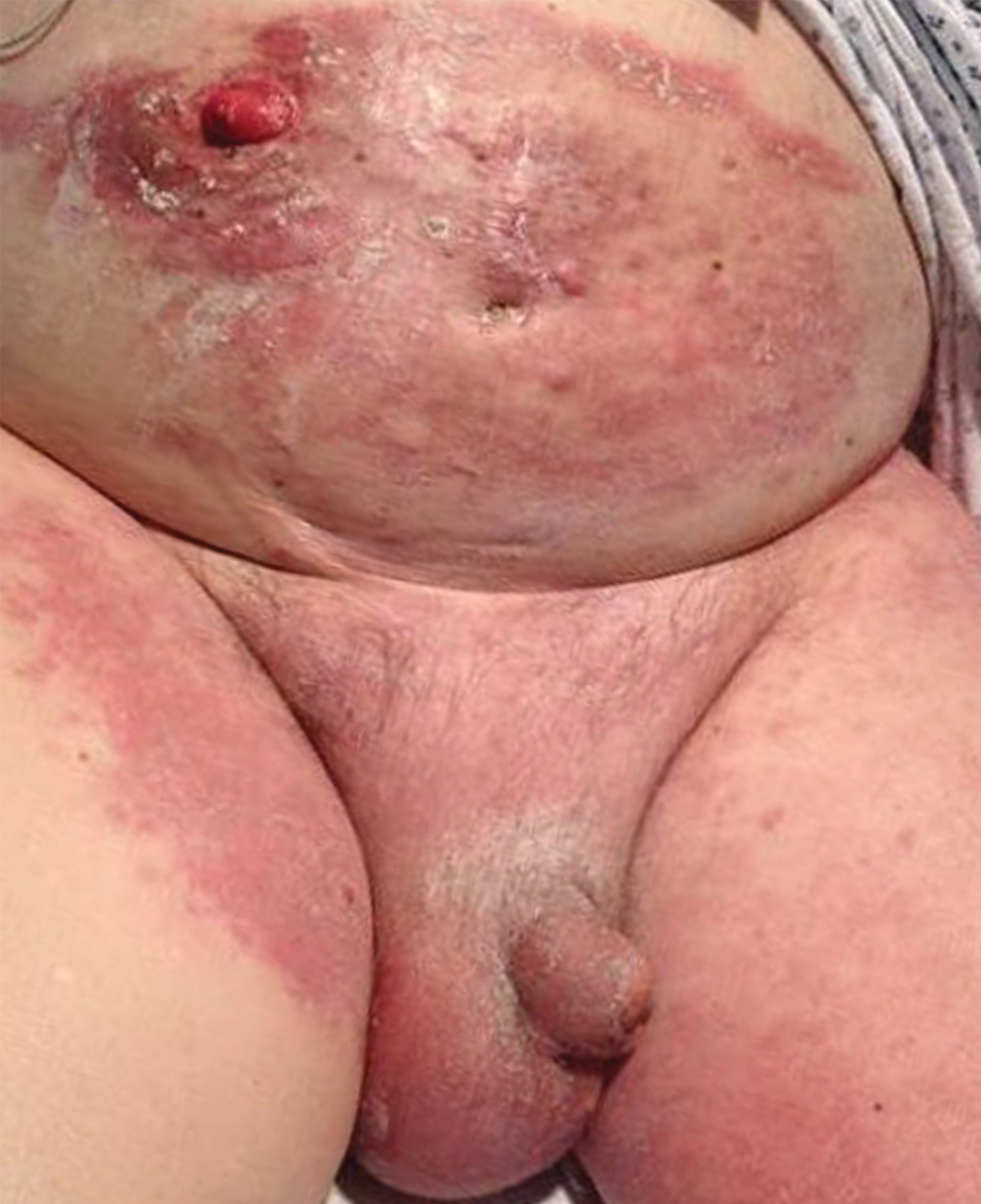
An 82-year-old man presented with acute abdominal pain and distension as well as an abdominal rash of 4 months' duration that was expanding despite treatment with topical miconazole. He had a history of melanoma and bladder cancer treated with cystoprostatectomy. He previously was diagnosed with candidiasis of his urostomy and was taking oral fluconazole. Physical examination revealed a large, well-demarcated, erythematous, smooth plaque covering the entire abdomen, scrotum, penis, inguinal folds, and bilateral upper thighs, with several satellite plaques and firm nodules clustered around the umbilicus. An 8-mm punch biopsy of a periumbilical nodule was performed.
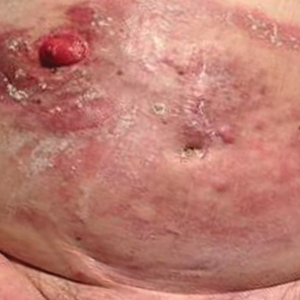
Erythematous Papules on the Scrotum, Trunk, and Extremities
The Diagnosis: Lichenoid and Granulomatous Dermatitis in the Setting of Secondary Syphilis
Syphilis, an infectious disease that has risen in incidence and is most commonly reported in men who have sex with men, involves a vast array of clinical and histologic presentations.1 Clinically, secondary syphilis involves an erythematous maculopapular eruption on the face, trunk, palms, soles, or genital area.2 The characteristic histologic features for secondary syphilis include endothelial swelling, interstitial inflammatory array, irregular acanthosis, elongated rete ridges, and vacuolar interface dermatitis with lymphocytes and plasma cells.1 Syphilitic infection has been associated with lichenoid and granulomatous dermatitis, which is an inflammatory skin disease described by Magro and Crowson.3 Lichenoid and granulomatous dermatitis has been linked to various systemic disorders, including chronic hepatitis C, Crohn disease, rheumatoid arthritis, endocrinopathy, subacute cutaneous lupus erythematosus, secondary syphilis, prior herpes infection, tuberculoid leprosy, mycobacterial infection, and human immunodeficiency virus infection.3-7 For this patient, given histopathology findings, clinical presentation, and positive rapid plasma reagin serologies, a diagnosis of lichenoid and granulomatous dermatitis in the setting of a secondary syphilis infection was established. A comprehensive investigation should be conducted to consider secondary syphilis or other systemic diseases in patients with a histologic finding of lichenoid and granulomatous dermatitis.
Histologically, lichenoid and granulomatous dermatitis cases show a bandlike infiltrate of lymphocytes with neighboring histiocytes along the dermoepidermal junction, accompanied by epithelial changes of dyskeratosis, vasculopathy, and colloid body formation, in addition to a dermal histiocytic component.3 Our patient's biopsy showed a lichenoid reaction pattern with vacuolar interface changes, dyskeratosis, plump endothelial cells, and small collections of plasma cells. Additionally, there was a granulomatous component in the dermis with histiocytes admixed with lymphocytes and plasma cells. The presence of spirochetes was confirmed with antitreponemal immunohistochemical stain (Figure 1). Quantitative rapid plasma reagin was 1:64 (reference range, <1:1) and Treponema pallidum antibody was reactive.
Interstitial granulomatous dermatitis has a variable clinical presentation, often with red-purple annular plaques, hyperpigmented papules, and nodules frequently in a linear arrangement and predominantly on the trunk, thighs, groin, or buttocks.8,9 On histopathology, there are histiocytes in the reticular dermis and/or a macrophage infiltrate in the mid to deep dermis with collections of degenerated collagen (Figure 2).8,10 An interstitial infiltrate of eosinophils and neutrophils also may be appreciated, but mucin generally is absent.8,11 This condition often coexists with rheumatic and systemic autoimmune diseases.8-10
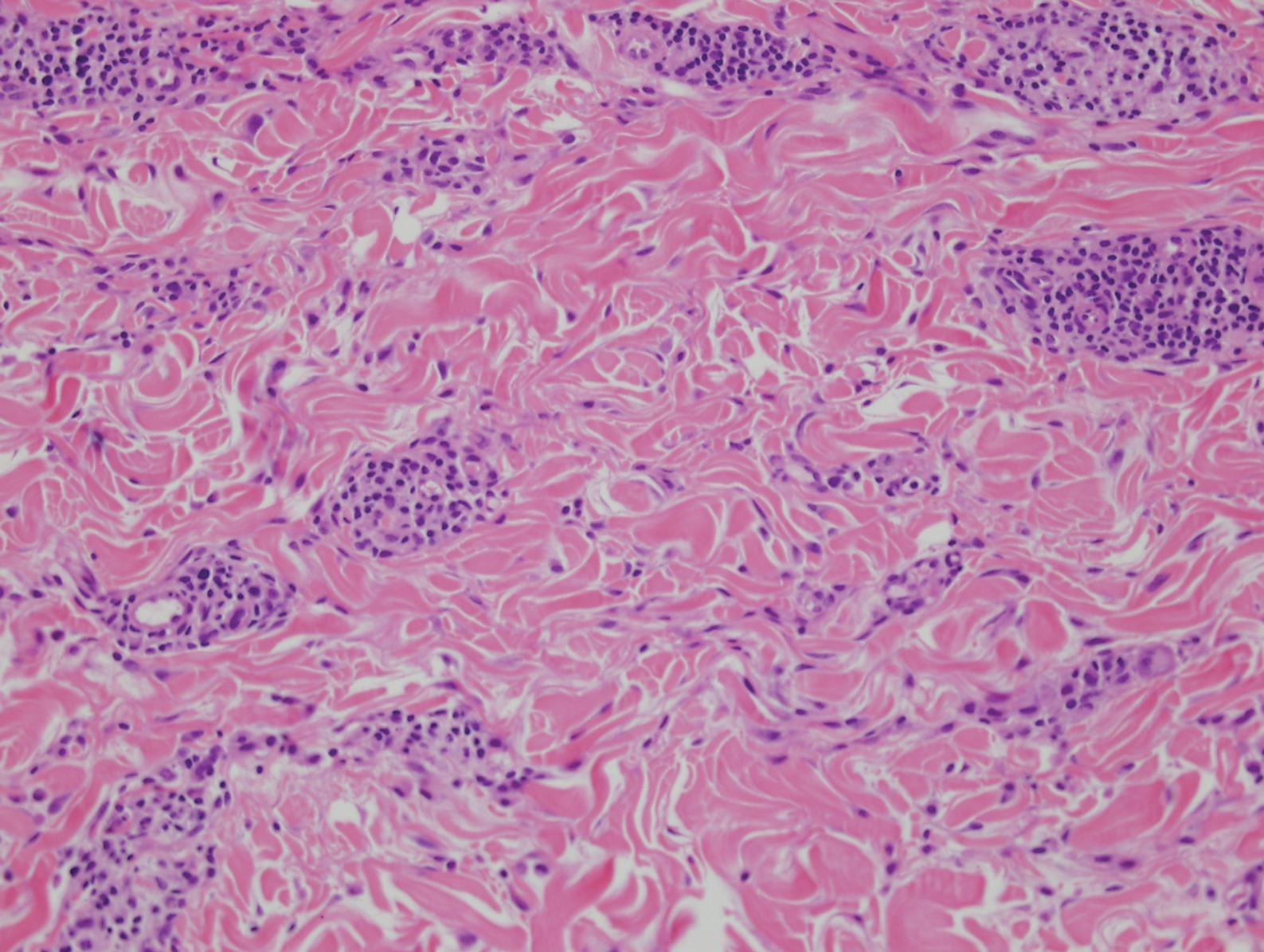
Interstitial granuloma annulare is a noninfectious granulomatous skin condition that often presents clinically as asymptomatic annular red-brown patches, usually on the extremities.11-13 On histopathology, an interstitial or palisaded inflammatory infiltrate with histiocytes and multinucleated giant cells may be seen along with collagen degeneration or collagen bundles without necrosis (Figure 3).9 Mucin often is associated with the histiocytes.11 Of note, our patient's skin biopsy shows interface dermatitis, differentiating it from both interstitial granuloma annulare and interstitial granulomatous dermatitis.
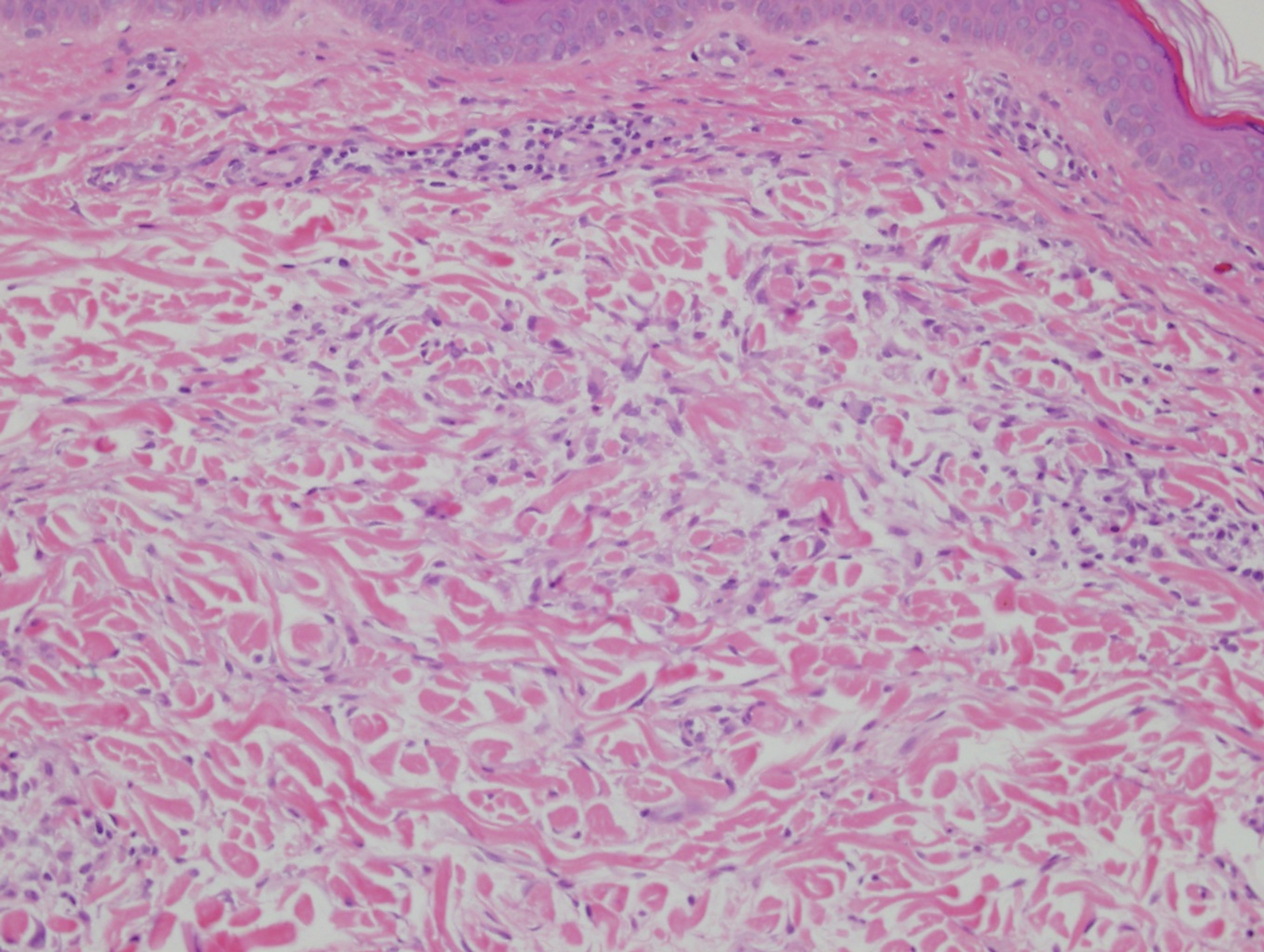
Postviral granulomatous reactions are the most frequently reported types of reactions to occur at the location of herpes zoster infection up to years after the initial disease. Wolf isotopic reaction encompasses skin reactions in the body region of formerly resolved skin disease, commonly herpesvirus infection.14,15 This manifestation may occur due to a hypersensitivity reaction from enduring viral proteins, resident memory T cells, or local neuroimmune imbalance from herpesvirus-induced injury to dermal sensory nerve fibers.14-17 Clinically, patients present with red-purple pruritic papules and plaques in a bandlike unilateral pattern, usually in the same region as the prior herpes infection and often accompanied by postherpetic neuralgia.16-19 Of note, our patient's clinical findings were more diffuse than the frequently localized and often linear distribution seen in postherpetic granulomatous reaction. On histopathology, granulomatous or lichenoid tissue reaction most commonly is appreciated.15 Specifically, interstitial granulomatous dermatitis with histiocytes, lymphocytes, and multinucleated giant cells showing elastophagocytosis and an inflammatory infiltrate with lymphocytes and plasma cells around vasculature, eccrine glands, and nerves can be noted (Figure 4).19

Lupus erythematosus is an autoimmune condition with a wide array of clinical features, including skin manifestations and systemic symptoms. Specifically, discoid lupus erythematosus presents with clearly outlined, red-pink macules or papules with scaling. Histologic features include keratotic follicular plugging, acanthosis, dermal mucin, thickening of the basement membrane zone, and dense lymphocytic infiltrate (Figure 5).20
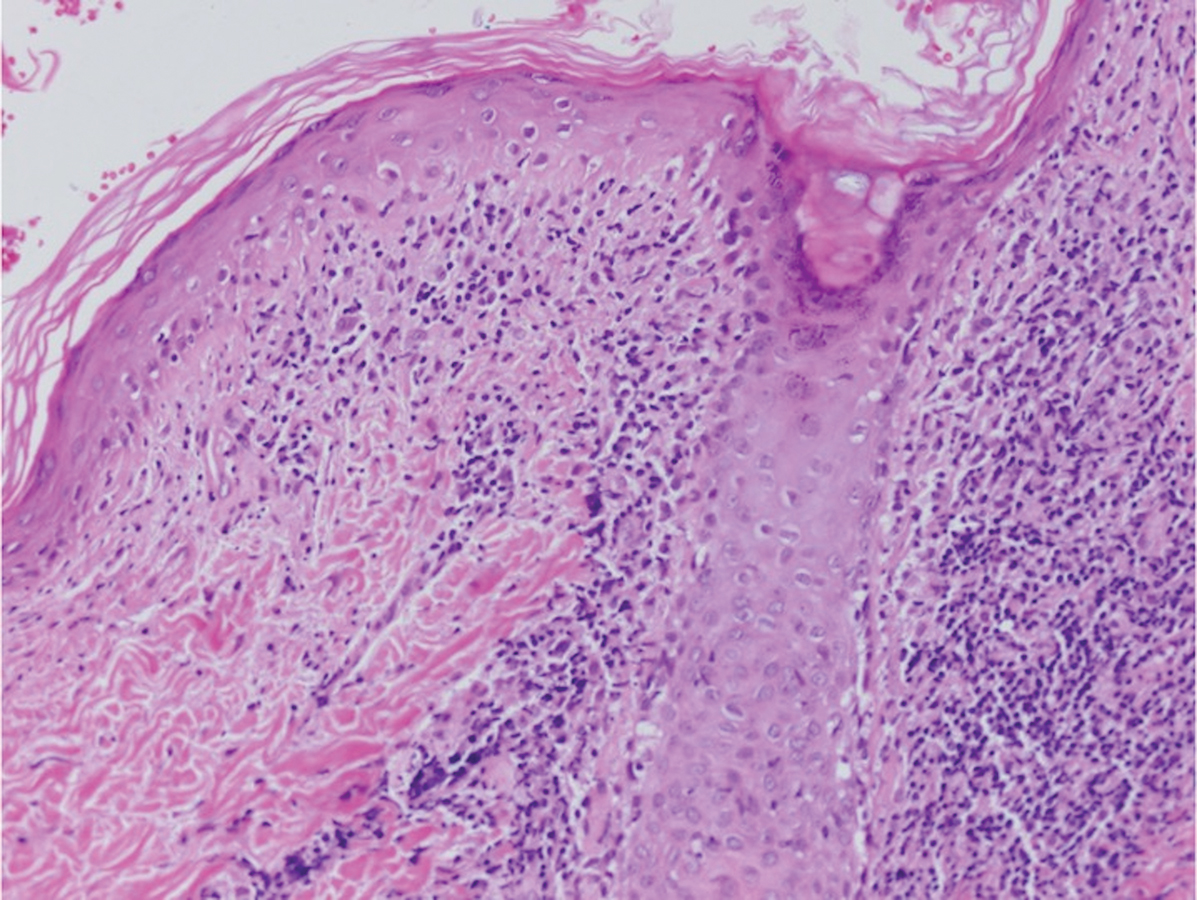
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:325-330.
- Zeltser R, Kurban AK. Syphilis. Clin Dermatol. 2004;22:461-468.
- Magro CM, Crowson AN. Lichenoid and granulomatous dermatitis. Int J Dermatol. 2000;39:12-33.
- S Breza T Jr, Magro CM. Lichenoid and granulomatous dermatitis associated with atypical mycobacterium infections. J Cutan Pathol. 2006;33:512-515.
- Granel B, Serratrice J, Rey J, et al. Chronic hepatitis C virus infection associated with a generalized granuloma annulare. J Am Acad Dermatol. 2000;43(5, pt 2):918-919.
- Jorizzo JL, Gonzalez EB, Apisarnthanarax P, et al. Pigmented purpuric eruption in a patient with rheumatoid arthritis. Arch Intern Med. 1982;142:2184-2185.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol. 1996;27:50-56.
- Błażewicz I, Szczerkowska-Dobosz A, Pęksa R, et al. Interstitial granulomatous dermatitis: a characteristic histological pattern with variable clinical manifestations. Postepy Dermatol Alergol. 2015;32:475-477.
- Sezer E, Luzar B, Calonje E. Secondary syphilis with an interstitial granuloma annulare-like histopathologic pattern. J Cutan Pathol. 2011;38:439-442.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Sakiyama T, Hirai I, Konohana A, et al. Interstitial-type granuloma annulare associated with Sjögren syndrome. J Dtsch Dermatol Ges. 2014;12:415-416.
- Spring P, Vernez M, Maniu CM, et al. Localized interstitial granuloma annulare induced by subcutaneous injections for desensitization. Dermatol Online J. 2013;19:18572.
- Kluger N, Moguelet P, Chaslin-Ferbus D, et al. Generalized interstitial granuloma annulare induced by pegylated interferon-alpha. Dermatology. 2006;213:248-249.
- Ruocco E, Baroni A, Cutrì FT, et al. Granuloma annulare in a site of healed herpes zoster: Wolf's isotopic response. J Eur Acad Dermatol Venereol. 2003;17:686-688.
- Ise M, Tanese K, Adachi T, et al. Postherpetic Wolf's isotopic response: possible contribution of resident memory T cells to the pathogenesis of lichenoid reaction. Br J Dermatol. 2015;173:1331-1334.
- Lora V, Cota C, Kanitakis J. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Dermatol Online J. 2014;20. pii:13030/qt5vf99178.
- Lin CH, Chen HC, Gao HW, et al. Wolf's post-herpetic isotopic response to tocilizumab for rheumatoid arthritis. Australas J Dermatol. 2018;59:E135-E137.
- Melgar E, Henry J, Valois A, et al. Extra-facial Lever granuloma on a herpes zoster scar: Wolf's isotopic response. Ann Dermatol Venereol. 2018;145:354-358.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Li Q, Wu H, Liao W, et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J Autoimmun. 2018;93:1-15.
The Diagnosis: Lichenoid and Granulomatous Dermatitis in the Setting of Secondary Syphilis
Syphilis, an infectious disease that has risen in incidence and is most commonly reported in men who have sex with men, involves a vast array of clinical and histologic presentations.1 Clinically, secondary syphilis involves an erythematous maculopapular eruption on the face, trunk, palms, soles, or genital area.2 The characteristic histologic features for secondary syphilis include endothelial swelling, interstitial inflammatory array, irregular acanthosis, elongated rete ridges, and vacuolar interface dermatitis with lymphocytes and plasma cells.1 Syphilitic infection has been associated with lichenoid and granulomatous dermatitis, which is an inflammatory skin disease described by Magro and Crowson.3 Lichenoid and granulomatous dermatitis has been linked to various systemic disorders, including chronic hepatitis C, Crohn disease, rheumatoid arthritis, endocrinopathy, subacute cutaneous lupus erythematosus, secondary syphilis, prior herpes infection, tuberculoid leprosy, mycobacterial infection, and human immunodeficiency virus infection.3-7 For this patient, given histopathology findings, clinical presentation, and positive rapid plasma reagin serologies, a diagnosis of lichenoid and granulomatous dermatitis in the setting of a secondary syphilis infection was established. A comprehensive investigation should be conducted to consider secondary syphilis or other systemic diseases in patients with a histologic finding of lichenoid and granulomatous dermatitis.
Histologically, lichenoid and granulomatous dermatitis cases show a bandlike infiltrate of lymphocytes with neighboring histiocytes along the dermoepidermal junction, accompanied by epithelial changes of dyskeratosis, vasculopathy, and colloid body formation, in addition to a dermal histiocytic component.3 Our patient's biopsy showed a lichenoid reaction pattern with vacuolar interface changes, dyskeratosis, plump endothelial cells, and small collections of plasma cells. Additionally, there was a granulomatous component in the dermis with histiocytes admixed with lymphocytes and plasma cells. The presence of spirochetes was confirmed with antitreponemal immunohistochemical stain (Figure 1). Quantitative rapid plasma reagin was 1:64 (reference range, <1:1) and Treponema pallidum antibody was reactive.
Interstitial granulomatous dermatitis has a variable clinical presentation, often with red-purple annular plaques, hyperpigmented papules, and nodules frequently in a linear arrangement and predominantly on the trunk, thighs, groin, or buttocks.8,9 On histopathology, there are histiocytes in the reticular dermis and/or a macrophage infiltrate in the mid to deep dermis with collections of degenerated collagen (Figure 2).8,10 An interstitial infiltrate of eosinophils and neutrophils also may be appreciated, but mucin generally is absent.8,11 This condition often coexists with rheumatic and systemic autoimmune diseases.8-10
Interstitial granuloma annulare is a noninfectious granulomatous skin condition that often presents clinically as asymptomatic annular red-brown patches, usually on the extremities.11-13 On histopathology, an interstitial or palisaded inflammatory infiltrate with histiocytes and multinucleated giant cells may be seen along with collagen degeneration or collagen bundles without necrosis (Figure 3).9 Mucin often is associated with the histiocytes.11 Of note, our patient's skin biopsy shows interface dermatitis, differentiating it from both interstitial granuloma annulare and interstitial granulomatous dermatitis.
Postviral granulomatous reactions are the most frequently reported types of reactions to occur at the location of herpes zoster infection up to years after the initial disease. Wolf isotopic reaction encompasses skin reactions in the body region of formerly resolved skin disease, commonly herpesvirus infection.14,15 This manifestation may occur due to a hypersensitivity reaction from enduring viral proteins, resident memory T cells, or local neuroimmune imbalance from herpesvirus-induced injury to dermal sensory nerve fibers.14-17 Clinically, patients present with red-purple pruritic papules and plaques in a bandlike unilateral pattern, usually in the same region as the prior herpes infection and often accompanied by postherpetic neuralgia.16-19 Of note, our patient's clinical findings were more diffuse than the frequently localized and often linear distribution seen in postherpetic granulomatous reaction. On histopathology, granulomatous or lichenoid tissue reaction most commonly is appreciated.15 Specifically, interstitial granulomatous dermatitis with histiocytes, lymphocytes, and multinucleated giant cells showing elastophagocytosis and an inflammatory infiltrate with lymphocytes and plasma cells around vasculature, eccrine glands, and nerves can be noted (Figure 4).19
Lupus erythematosus is an autoimmune condition with a wide array of clinical features, including skin manifestations and systemic symptoms. Specifically, discoid lupus erythematosus presents with clearly outlined, red-pink macules or papules with scaling. Histologic features include keratotic follicular plugging, acanthosis, dermal mucin, thickening of the basement membrane zone, and dense lymphocytic infiltrate (Figure 5).20
The Diagnosis: Lichenoid and Granulomatous Dermatitis in the Setting of Secondary Syphilis
Syphilis, an infectious disease that has risen in incidence and is most commonly reported in men who have sex with men, involves a vast array of clinical and histologic presentations.1 Clinically, secondary syphilis involves an erythematous maculopapular eruption on the face, trunk, palms, soles, or genital area.2 The characteristic histologic features for secondary syphilis include endothelial swelling, interstitial inflammatory array, irregular acanthosis, elongated rete ridges, and vacuolar interface dermatitis with lymphocytes and plasma cells.1 Syphilitic infection has been associated with lichenoid and granulomatous dermatitis, which is an inflammatory skin disease described by Magro and Crowson.3 Lichenoid and granulomatous dermatitis has been linked to various systemic disorders, including chronic hepatitis C, Crohn disease, rheumatoid arthritis, endocrinopathy, subacute cutaneous lupus erythematosus, secondary syphilis, prior herpes infection, tuberculoid leprosy, mycobacterial infection, and human immunodeficiency virus infection.3-7 For this patient, given histopathology findings, clinical presentation, and positive rapid plasma reagin serologies, a diagnosis of lichenoid and granulomatous dermatitis in the setting of a secondary syphilis infection was established. A comprehensive investigation should be conducted to consider secondary syphilis or other systemic diseases in patients with a histologic finding of lichenoid and granulomatous dermatitis.
Histologically, lichenoid and granulomatous dermatitis cases show a bandlike infiltrate of lymphocytes with neighboring histiocytes along the dermoepidermal junction, accompanied by epithelial changes of dyskeratosis, vasculopathy, and colloid body formation, in addition to a dermal histiocytic component.3 Our patient's biopsy showed a lichenoid reaction pattern with vacuolar interface changes, dyskeratosis, plump endothelial cells, and small collections of plasma cells. Additionally, there was a granulomatous component in the dermis with histiocytes admixed with lymphocytes and plasma cells. The presence of spirochetes was confirmed with antitreponemal immunohistochemical stain (Figure 1). Quantitative rapid plasma reagin was 1:64 (reference range, <1:1) and Treponema pallidum antibody was reactive.
Interstitial granulomatous dermatitis has a variable clinical presentation, often with red-purple annular plaques, hyperpigmented papules, and nodules frequently in a linear arrangement and predominantly on the trunk, thighs, groin, or buttocks.8,9 On histopathology, there are histiocytes in the reticular dermis and/or a macrophage infiltrate in the mid to deep dermis with collections of degenerated collagen (Figure 2).8,10 An interstitial infiltrate of eosinophils and neutrophils also may be appreciated, but mucin generally is absent.8,11 This condition often coexists with rheumatic and systemic autoimmune diseases.8-10
Interstitial granuloma annulare is a noninfectious granulomatous skin condition that often presents clinically as asymptomatic annular red-brown patches, usually on the extremities.11-13 On histopathology, an interstitial or palisaded inflammatory infiltrate with histiocytes and multinucleated giant cells may be seen along with collagen degeneration or collagen bundles without necrosis (Figure 3).9 Mucin often is associated with the histiocytes.11 Of note, our patient's skin biopsy shows interface dermatitis, differentiating it from both interstitial granuloma annulare and interstitial granulomatous dermatitis.
Postviral granulomatous reactions are the most frequently reported types of reactions to occur at the location of herpes zoster infection up to years after the initial disease. Wolf isotopic reaction encompasses skin reactions in the body region of formerly resolved skin disease, commonly herpesvirus infection.14,15 This manifestation may occur due to a hypersensitivity reaction from enduring viral proteins, resident memory T cells, or local neuroimmune imbalance from herpesvirus-induced injury to dermal sensory nerve fibers.14-17 Clinically, patients present with red-purple pruritic papules and plaques in a bandlike unilateral pattern, usually in the same region as the prior herpes infection and often accompanied by postherpetic neuralgia.16-19 Of note, our patient's clinical findings were more diffuse than the frequently localized and often linear distribution seen in postherpetic granulomatous reaction. On histopathology, granulomatous or lichenoid tissue reaction most commonly is appreciated.15 Specifically, interstitial granulomatous dermatitis with histiocytes, lymphocytes, and multinucleated giant cells showing elastophagocytosis and an inflammatory infiltrate with lymphocytes and plasma cells around vasculature, eccrine glands, and nerves can be noted (Figure 4).19
Lupus erythematosus is an autoimmune condition with a wide array of clinical features, including skin manifestations and systemic symptoms. Specifically, discoid lupus erythematosus presents with clearly outlined, red-pink macules or papules with scaling. Histologic features include keratotic follicular plugging, acanthosis, dermal mucin, thickening of the basement membrane zone, and dense lymphocytic infiltrate (Figure 5).20
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:325-330.
- Zeltser R, Kurban AK. Syphilis. Clin Dermatol. 2004;22:461-468.
- Magro CM, Crowson AN. Lichenoid and granulomatous dermatitis. Int J Dermatol. 2000;39:12-33.
- S Breza T Jr, Magro CM. Lichenoid and granulomatous dermatitis associated with atypical mycobacterium infections. J Cutan Pathol. 2006;33:512-515.
- Granel B, Serratrice J, Rey J, et al. Chronic hepatitis C virus infection associated with a generalized granuloma annulare. J Am Acad Dermatol. 2000;43(5, pt 2):918-919.
- Jorizzo JL, Gonzalez EB, Apisarnthanarax P, et al. Pigmented purpuric eruption in a patient with rheumatoid arthritis. Arch Intern Med. 1982;142:2184-2185.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol. 1996;27:50-56.
- Błażewicz I, Szczerkowska-Dobosz A, Pęksa R, et al. Interstitial granulomatous dermatitis: a characteristic histological pattern with variable clinical manifestations. Postepy Dermatol Alergol. 2015;32:475-477.
- Sezer E, Luzar B, Calonje E. Secondary syphilis with an interstitial granuloma annulare-like histopathologic pattern. J Cutan Pathol. 2011;38:439-442.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Sakiyama T, Hirai I, Konohana A, et al. Interstitial-type granuloma annulare associated with Sjögren syndrome. J Dtsch Dermatol Ges. 2014;12:415-416.
- Spring P, Vernez M, Maniu CM, et al. Localized interstitial granuloma annulare induced by subcutaneous injections for desensitization. Dermatol Online J. 2013;19:18572.
- Kluger N, Moguelet P, Chaslin-Ferbus D, et al. Generalized interstitial granuloma annulare induced by pegylated interferon-alpha. Dermatology. 2006;213:248-249.
- Ruocco E, Baroni A, Cutrì FT, et al. Granuloma annulare in a site of healed herpes zoster: Wolf's isotopic response. J Eur Acad Dermatol Venereol. 2003;17:686-688.
- Ise M, Tanese K, Adachi T, et al. Postherpetic Wolf's isotopic response: possible contribution of resident memory T cells to the pathogenesis of lichenoid reaction. Br J Dermatol. 2015;173:1331-1334.
- Lora V, Cota C, Kanitakis J. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Dermatol Online J. 2014;20. pii:13030/qt5vf99178.
- Lin CH, Chen HC, Gao HW, et al. Wolf's post-herpetic isotopic response to tocilizumab for rheumatoid arthritis. Australas J Dermatol. 2018;59:E135-E137.
- Melgar E, Henry J, Valois A, et al. Extra-facial Lever granuloma on a herpes zoster scar: Wolf's isotopic response. Ann Dermatol Venereol. 2018;145:354-358.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Li Q, Wu H, Liao W, et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J Autoimmun. 2018;93:1-15.
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:325-330.
- Zeltser R, Kurban AK. Syphilis. Clin Dermatol. 2004;22:461-468.
- Magro CM, Crowson AN. Lichenoid and granulomatous dermatitis. Int J Dermatol. 2000;39:12-33.
- S Breza T Jr, Magro CM. Lichenoid and granulomatous dermatitis associated with atypical mycobacterium infections. J Cutan Pathol. 2006;33:512-515.
- Granel B, Serratrice J, Rey J, et al. Chronic hepatitis C virus infection associated with a generalized granuloma annulare. J Am Acad Dermatol. 2000;43(5, pt 2):918-919.
- Jorizzo JL, Gonzalez EB, Apisarnthanarax P, et al. Pigmented purpuric eruption in a patient with rheumatoid arthritis. Arch Intern Med. 1982;142:2184-2185.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol. 1996;27:50-56.
- Błażewicz I, Szczerkowska-Dobosz A, Pęksa R, et al. Interstitial granulomatous dermatitis: a characteristic histological pattern with variable clinical manifestations. Postepy Dermatol Alergol. 2015;32:475-477.
- Sezer E, Luzar B, Calonje E. Secondary syphilis with an interstitial granuloma annulare-like histopathologic pattern. J Cutan Pathol. 2011;38:439-442.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Sakiyama T, Hirai I, Konohana A, et al. Interstitial-type granuloma annulare associated with Sjögren syndrome. J Dtsch Dermatol Ges. 2014;12:415-416.
- Spring P, Vernez M, Maniu CM, et al. Localized interstitial granuloma annulare induced by subcutaneous injections for desensitization. Dermatol Online J. 2013;19:18572.
- Kluger N, Moguelet P, Chaslin-Ferbus D, et al. Generalized interstitial granuloma annulare induced by pegylated interferon-alpha. Dermatology. 2006;213:248-249.
- Ruocco E, Baroni A, Cutrì FT, et al. Granuloma annulare in a site of healed herpes zoster: Wolf's isotopic response. J Eur Acad Dermatol Venereol. 2003;17:686-688.
- Ise M, Tanese K, Adachi T, et al. Postherpetic Wolf's isotopic response: possible contribution of resident memory T cells to the pathogenesis of lichenoid reaction. Br J Dermatol. 2015;173:1331-1334.
- Lora V, Cota C, Kanitakis J. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Dermatol Online J. 2014;20. pii:13030/qt5vf99178.
- Lin CH, Chen HC, Gao HW, et al. Wolf's post-herpetic isotopic response to tocilizumab for rheumatoid arthritis. Australas J Dermatol. 2018;59:E135-E137.
- Melgar E, Henry J, Valois A, et al. Extra-facial Lever granuloma on a herpes zoster scar: Wolf's isotopic response. Ann Dermatol Venereol. 2018;145:354-358.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Li Q, Wu H, Liao W, et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J Autoimmun. 2018;93:1-15.
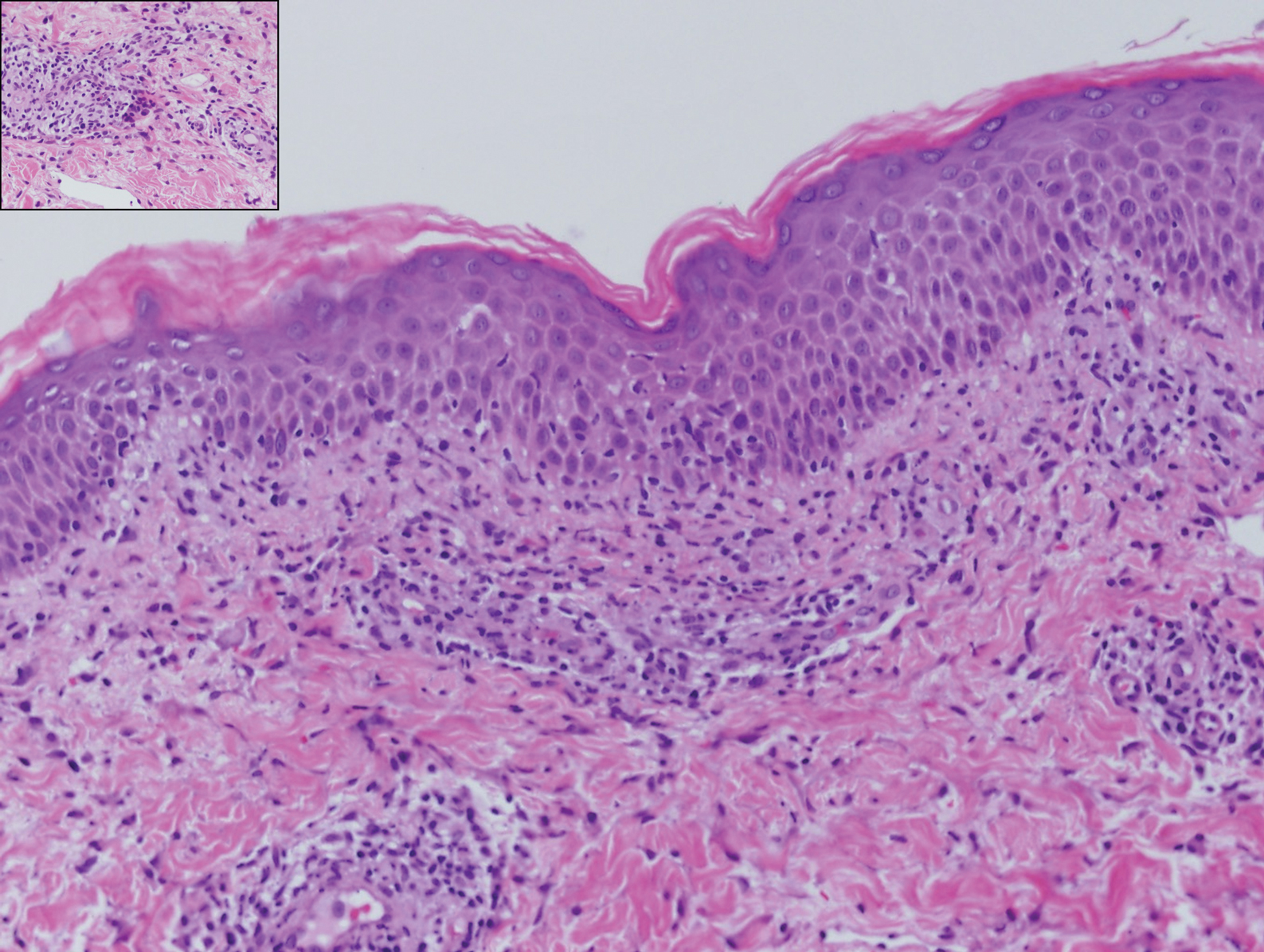
A 54-year-old man presented with painful, nonpruritic, erythematous papules that began on the scrotum. The eruption progressed to involve the trunk, arms, and legs.