User login
‘Dry drowning’ and other myths
In June 2017, a 4-year-old boy died 1 week after being knocked over and briefly submerged while playing in knee-deep water. This story was widely reported as a case of a rare occurrence called “dry” or “secondary” drowning, depending on the source.1 The media accounts went viral, spreading fear in parents and others learning about these alleged conditions from the news and social media.
Many alleged cases of dry drowning are reported every year, but each has been found to have a recognized medical source that has a legitimate medically recognized diagnosis (which dry and secondary drowning are not).
Drowning is one of the most common causes of death in children, and so we ought to make sure that the information we share about it is accurate, as it is vital to effective prevention, rescue, and treatment.
Unfortunately, medical providers, medical journals, and the mass media continue to disseminate misinformation on drowning.2 These reports often prevail over updated information and hinder accurate understanding of the drowning problem and its solutions.
Every death is tragic, especially the death of a child, and our heartfelt sympathies go out to the family in this alleged drowning case, as well as to all families suffering the loss of a loved one to drowning. However, in the 2017 case, the cause of death was found on autopsy to be myocarditis not related in any way to drowning. As often happens in such situations, this clarification did not receive any media attention, despite the wide reporting and penetration of the original, erroneous story.
We hope our review will reduce misunderstanding among the public and healthcare providers, contribute to improved data collection, and help to promote interventions aimed at prevention, rescue, and mitigation of drowning incidents.
WHAT IS DROWNING?
A consensus committee of the World Health Organization defined drowning as “the process of experiencing respiratory impairment from submersion/immersion in liquid.”3 The process begins when the victim’s airway goes below the surface of the liquid (submersion) or when water splashes over the face (immersion). If the victim is rescued at any time, the process is interrupted, and this is termed a nonfatal drowning. If the victim dies at any time, this is a fatal drowning. Any water-distress incident without evidence of respiratory impairment (ie, without aspiration) should be considered a water rescue and not a drowning.
Rarely do minimally symptomatic cases progress to death, just as most cases of chest pain do not progress to cardiac arrest.4 Nonetheless, rescued drowning victims can deteriorate, which is why we encourage people to seek medical care immediately upon warning signs, as we do with chest pain. For drowning, such warning signs are any water distress followed by difficulty breathing, excessive coughing, foam in the mouth, or abnormal behavior.
A SERIOUS PUBLIC HEALTH ISSUE
Drowning is a serious and neglected public health issue, claiming the lives of 372,000 people a year worldwide.5 It is a leading cause of death in children ages 1 to 14. The toll continues largely unabated, and in low- and middle-income nations it does not attract the levels of funding that go to other forms of injury prevention, such as road safety.
Nonfatal drowning—with symptoms ranging from mild cough to severe pulmonary edema, and complications ranging from none to severe neurologic impairment—is far more common than fatal drowning.6 For every fatal drowning, there are at least 5 nonfatal drowning incidents in which medical care is needed, and 200 rescues are performed.7–10
In the United States, drowning accounts for almost 13,000 emergency department visits per year and about 3,500 deaths.7,8
In Brazil, with two-thirds the population of the United States, drowning accounts for far fewer hospital visits but about twice as many deaths. In Rio de Janeiro, where a highly effective and specialized prehospital service is provided at 3 drowning resuscitation centers staffed by medical doctors, an analysis of the 46,060 cases of rescue in 10 years from 1991 to 2000 showed that medical assistance was needed in only 930 cases (2%).10 The preventive and rescue actions of parents, bystanders, lifeguards, and prehospital rescue services significantly reduce the number of drowning deaths, but these groups do not consistently gather data on nonfatal drowning that can be included in a comprehensive database.
DROWNING IS A PROCESS
When a person in the water can no longer keep the airway clear, water that enters the mouth is voluntarily spit out or swallowed. Within a few seconds to minutes, the person can no longer clear the airways and water is aspirated, stimulating the cough reflex. Laryngospasm, another myth concerning drowning, is presumed to protect the airways but does not, as it is rare, occurring in less than 2% of cases.11,12
If the person is not rescued, aspiration of water continues, and hypoxemia leads to loss of consciousness and apnea within seconds to a few minutes, followed by cardiac arrest. As a consequence, hypoxemic cardiac arrest generally occurs after a period of tachycardia followed by bradycardia and pulseless electrical activity, usually leading to asystole.13,14
The entire drowning process, from water distress to cardiac arrest, usually takes a few minutes, but in rare situations, such as rapid hypothermia, it can go on for up to an hour.15 Most drowning patients have an otherwise healthy heart, and the apnea and hypoxemia precede the cardiac arrest by only a few seconds to minutes; thus, cardiac arrest is caused by the hypoxemic insult and not by ventricular dysrhythmias.6,16
Drowning can be interrupted at any point between distress and death. If the person is rescued early, the clinical picture is determined by the reactivity of the airway and the amount of water that has been aspirated, but not by the type of water (salt or fresh).
Another myth is that drowning in salt water is different from drowning in fresh water. Both salt water and fresh water cause similar surfactant destruction and washout and disrupt the alveolar-capillary membrane. Disruption of the alveolar-capillary membrane increases its permeability and exacerbates shifting of fluid, plasma, and electrolytes into the alveoli.13 The clinical picture of the damage is one of regional or generalized pulmonary edema, which interferes with gas exchange in the lungs.6,13,17
Animal studies by Modell et al showed that aspiration of just 2.2 mL of water per kilogram of body weight is sufficient to cause severe disturbances in oxygen exchange,17 reflected in a rise in arterial pH and a drop in partial pressure of oxygen. The situation must be similar in humans. In a 70-kg person, this is only about 154 mL of water—about two-thirds of a cup.
The combined effects of fluid in the lungs, the loss of surfactant, and the increase in capillary-alveolar permeability can result in decreased lung compliance, increased right-to-left shunting in the lungs, atelectasis, alveolitis, hypoxemia, and cerebral hypoxia.13
If the victim needs cardiopulmonary resuscitation, the possibility of neurologic damage is similar to that in other cardiac arrest situations, but exceptions exist. For example, in rare cases, hypothermia provides a protective mechanism that allows victims to survive prolonged submersion.4,15
The duration of submersion is the best predictor of death.18 Underwater, people are not taking in oxygen, and cerebral hypoxia causes both morbidity and death. For this reason, reversing cerebral hypoxia with effective ventilation, oxygen, and chest compression is the priority of treatment.
MYTHS AND SLOPPY TERMINOLOGY
“Near drowning,” “dry drowning,” “wet drowning,” “delayed drowning,” and “secondary drowning” are not medically accepted diagnoses,3,4,19 and many organizations and lifesaving institutions around the world discourage the use of these terms.19,20 Unfortunately, these terms still slip past the editors of medical journals and are thus perpetuated. The terms are most pervasive in the nonmedical media, where drowning seems to be synonymous with death.3,19,21 We urge all authors and stakeholders to abandon these terms in favor of understanding and communicating drowning as a process that can vary in severity and have a fatal or nonfatal outcome.
Near-drowning
Historically, drowning meant death, while near-drowning meant the victim survived, at least initially (usually for at least 24 hours).
Before 2002, there were 13 different published definitions of near-drowning.21,22 This variability has caused a great deal of confusion when trying to describe and monitor drowning.
A person can drown and survive, just as a person can have cardiac arrest and survive.4,21 Just as there is no recognized condition of “near-cardiac arrest,” there is also no condition of near-drowning. Using near-drowning as a medical diagnosis hides the true burden of drowning and consequently amplifies difficulties in developing effective prevention, rescue, and treatment programs.
Dry drowning
Dry drowning has never been an accepted medical term, although it has been used to describe different parts of the drowning process. While many authors use it as a synonym for secondary drowning (described below), in the past it was usually used in cases in which no water was found in the lungs at autopsy in persons who were found dead in the water.2–4,21 This occurred in about 10% to 15% of cases and was also called drowning “without water aspiration.”
Perhaps some victims suffer sudden cardiac death. It happens on land—why not in the water? Modell et al stated, “In the absence of the common finding of significant pulmonary edema in the victim’s respiratory system, to conclude his or her death was caused by ‘drowning without aspiration’ is unwise.”23
Laryngospasm is another proposed explanation. It could play a role in the fewer than 2% of cases in which no other cause of death is found on clinical examination or autopsy,11,12,19,23 but it does not occur in most cases of drowning, or it is brief and is terminated by the respiratory movements that allow the air in the lung to escape and water to be inhaled.
The problem with the term dry drowning is the harm caused by misdiagnosing cases of sudden death as drowning, when an alternative cause is present. Most importantly, the management is the same if small amounts of water are present or not; therefore, no clinical distinction is made between wet and dry drowning.
Secondary drowning
Secondary drowning, sometimes called delayed drowning, is another term that is not medically accepted. The historical use of this term reflects the reality that some patients may worsen due to pulmonary edema after aspirating small amounts of water.
Drowning starts with aspiration, and few or only mild symptoms may be present as soon as the person is removed from the water. Either the small amount of water in the lungs is absorbed and causes no complications or, rarely, the patient’s condition becomes progressively worse over the next few hours as the alveoli become inflamed and the alveolar-capillary membrane is disrupted. But people do not unexpectedly die of drowning days or weeks later with no preceding symptoms. The lungs and heart do not “fill up with water,” and water does not need to be pumped out of the lungs.
There has never been a case published in the medical literature of a patient who underwent clinical evaluation, was initially without symptoms, and later deteriorated and died more than 8 hours after the incident.6,10,21 People who have drowned and have minimal symptoms get better (usually) or worse (rarely) within 4 to 8 hours. In a study of more than 41,000 lifeguard rescues, only 0.5% of symptomatic patients died.6
Drowning secondary to injury or sudden illness
Any injury, trauma, or sudden illness that can cause loss of consciousness or mental or physical weakness can lead to drowning. Physicians need to recognize these situations to treat them appropriately. Drowning that is secondary to other primary insults can be classified as24:
- Drowning caused by injury or trauma (eg, a surfing, boating, or a hang-gliding accident)
- Drowning caused by a sudden illness such as cardiac disease (eg, myocardial ischemia, arrhythmias, prolonged QT syndrome, hypertrophic cardiomyopathy) or neurologic disease (eg, epilepsy, stroke)
- Diving disease (eg, decompression sickness, pulmonary overpressurization syndrome, compression barotrauma, narcosis [“rapture of the deep”], shallow water blackout, immersion pulmonary edema).

PREVENTION IS BEST
Drowning is a leading and preventable cause of death worldwide and for people of all ages. The danger is real, not esoteric or rare, and healthcare providers should use any opportunity to discuss with patients, parents, and the media the most important tool for treating drowning: primary prevention.
For example, small children should be continuously and uninterruptedly supervised within arm’s reach while in the water, even if a lifeguard is present. Other preventive measures are lifejackets, fences completely enclosing pools or ponds, and swimming and water safety lessons. Drowning often occurs in a deceptively pleasant environment that may not seem dangerous.
RECOGNIZE DISTRESS
When preventive measures fail, responders (usually a health professional is involved) need to be able to perform the necessary steps to interrupt the drowning process.
The first challenge is to recognize when someone in the water is at risk of drowning and needs to be rescued.25 Early self-rescue or rescue by others may stop the drowning process and prevent most cases of initial and subsequent water aspiration, respiratory distress, and medical complications.
DON’T BECOME A VICTIM
Rescuers must take care not to become victims themselves. Panicked swimmers can thrash about and injure the rescuer or clutch at anything they encounter, dragging the rescuer under. And the rescuer can succumb to the same hazards that got the victim into trouble, such as strong currents, deep water, or underwater hazards.
Certified lifeguards are trained to get victims out of the water safely. The American Red Cross slogan “Reach or throw, don’t go” means “Reach out with a pole or other object or throw something that floats; don’t get in the water yourself.”
WHAT TO TELL THE PUBLIC
While some journalists acknowledge that the terms dry drowning and secondary drowning are medically discredited, they still use them in their reports. The novelty of this story—and its appeal to media outlets—is precisely the unfamiliarity of these terms to the general public and the perceived mysterious, looming threat.
We often hear that these terms are more familiar to the public, which is likely true. More concerning, some physicians continue to use them (and older definitions of drowning that equate it with death) in media interviews, clinical care, and publications. The paradox is that we, the medical community, invented these terms, not patients or the media.
As clinicians and researchers, we should drive popular culture definitions, not the other way around. Rather than dismiss these terms as “semantics” or “technicalities,” we should take the opportunity to highlight the dangers of drowning and the importance of prevention, and to promote simpler language that is easier for us and our patients to understand.19,21
Healthcare providers should understand and share modern drowning science and best practices, which will reduce fear, improve resource utilization, and prevent potentially deadly consequences due to misunderstanding or misinterpretation of incorrect terminology.
WHEN PATIENTS SHOULD SEEK CARE
Anyone who experiences cough, breathlessness, or other worrisome symptoms such as abnormal mentation within 8 hours of a drowning incident (using the modern definition above) should seek medical advice immediately.
We tell people to seek care if symptoms seem any worse than the experience of a drink “going down the wrong pipe” at the dinner table.21 But symptoms can be minimal. Careful attention should be given to mild symptoms that get progressively worse during that time. These cases can rarely progress to acute respiratory distress syndrome.

Table 1 explores who needs further medical help after being rescued from the water.26
In most of these cases, it is most appropriate to call an ambulance, but care may involve seeing a doctor depending on the severity of the symptoms.6,21 Usually, drowning patients are observed for 4 to 8 hours in an emergency department and are discharged if normal. Symptoms that are more significant include persistent cough, foam at the mouth or nose, confusion, or abnormal behavior, and these require further medical evaluation.
Patients should also seek medical care even if they are 100% normal upon exiting the water but develop worrisome symptoms more than 8 hours later, and providers should consider diagnoses other than primary drowning. Spontaneous pneumothorax, chemical pneumonitis, bacterial or viral pneumonia, head injury, asthma, chest trauma, and acute respiratory distress syndrome have been mislabeled as delayed, dry, or secondary drowning.3,4,19,21
- Buffington B. Texas boy dies from ‘dry drowning’ days after swimming. USA Today, June 8, 2017. www.usatoday.com/story/news/nation-now/2017/06/08/texas-boy-dies-dry-drowning-days-after-swimming/379944001.
- Schmidt AC, Sempsrott JR, Szpilman D, et al. The use of non-uniform drowning terminology: a follow-up study. Scand J Trauma Resusc Emerg Med 2017; 25(1):72. doi:10.1186/s13049-017-0405-x
- van Beeck EF, Branche CM, Szpilman D, Modell JH, Bierens JJ. A new definition of drowning: towards documentation and prevention of a global public health problem. Bull World Health Organ 2005; 83(11):853–856. pmid:16302042
- Szpilman D, Bierens JJ, Handley AJ, Orlowski JP. Drowning. N Engl J Med 2012; 366(22):2102–2110. doi:10.1056/NEJMra1013317
- World Health Organization. Global report on drowning: preventing a leading killer. www.who.int/violence_injury_prevention/global_report_drowning/en. Accessed June 13, 2018.
- Szpilman D. Near-drowning and drowning classification: a proposal to stratify mortality based on the analysis of 1,831 cases. Chest 1997; 112(3):660–665. pmid:9315798
- Centers for Disease Control and Prevention. Welcome to WISQARS. www.cdc.gov/injury/wisqars. Accessed June 13, 2018.
- Centers for Disease Control and Prevention. WONDER. https://wonder.cdc.gov. Accessed June 13, 2018.
- Cummings P, Quan L. Trends in unintentional drowning: the role of alcohol and medical care. JAMA 1999; 281(23):2198–2202. pmid:10376572
- Szpilman D, Elmann J, Cruz-Filho FES. Drowning classification: a revalidation study based on the analysis of 930 cases over 10 years. World Congress on Drowning, Netherlands 2002. www.researchgate.net/publication/267981062_DROWNING_CLASSIFICATION_a_revalidation_study_based_on_the_analysis_of_930_cases_over_10_years. Accessed June 13, 2018.
- Szpilman D, Elmann J, Cruz-Filho FES. Dry-drowning—fact or myth? World Congress on Drowning. Netherlands, 2002. www.researchgate.net/publication/267981164_Dry-drowning_-Fact_or_Myth. Accessed June 13, 2018.
- Lunetta P, Modell JH, Sajantila A. What is the incidence and significance of "dry-lungs" in bodies found in water? Am J Forensic Med Pathol 2004; 25(4):291–301. pmid:15577518
- Orlowski JP, Abulleil MM, Phillips JM. The hemodynamic and cardiovascular effects of near-drowning in hypotonic, isotonic, or hypertonic solutions. Ann Emerg Med 1989; 18:1044–1049. pmid:2802278
- Grmec S, Strnad M, Podgorsek D. Comparison of the characteristics and outcome among patients suffering from out-of-hospital primary cardiac arrest and drowning victims in cardiac arrest. Int J Emerg Med 2009; 2(1):7–12. doi:10.1007/s12245-009-0084-0
- Tipton MJ, Golden FS. A proposed decision-making guide for the search, rescue and resuscitation of submersion (head under) victims based on expert opinion. Resuscitation 2011; 82(7):819–824. doi:10.1016/j.resuscitation.2011.02.021
- Orlowski JP, Szpilman D. Drowning. Rescue, resuscitation, and reanimation. Pediatr Clin North Am 2001; 48(3):627–646. pmid:11411297
- Modell JH, Moya F, Newby EJ, Ruiz BC, Showers AV. The effects of fluid volume in seawater drowning. Ann Intern Med 1967; 67(1):68–80. pmid:6028660
- Quan L, Wentz KR, Gore EJ, Copass MK. Outcome and predictors of outcome in pediatric submersion victims receiving prehospital care in King County, Washington. Pediatrics 1990; 86(4):586–593. pmid:2216625
- Szpilman D, Orlowski JP, Cruz-Filho FES. Hey “Near-drowning,” you’ve been messing up our minds! World Congress on Drowning. Amsterdam, 2002. www.researchgate.net/publication/267981173_HEY_Near-drowning_YOU%27VE_BEEN_MESSING_UP_OUR_MINDS. Accessed June 13, 2018.
- American College of Emergency Physicians. Death after swimming is extremely rare—and is not “dry drowning.” http://newsroom.acep.org/2017-07-11-Death-After-Swimming-Is-Extremely-Rare-And-Is-NOT-Dry-Drowning. Accessed June 13, 2018.
- Hawkins SC, Sempsrott J, Schmidt A. “Drowning” in a sea of misinformation. Emergency Medicine News 2017; 39*8):1. http://journals.lww.com/em-news/blog/BreakingNews/pages/post.aspx?PostID=377. Accessed June 5, 2018.
- Szpilman D, Tipton M, Sempsrott J, et al. Drowning timeline: a new systematic model of the drowning process. Am J Emerg Med 2016; 34(11):2224–2226. doi:10.1016/j.ajem.2016.07.063
- Modell JH, Bellefleur M, Davis JH. Drowning without aspiration: is this an appropriate diagnosis? J Forensic Sci 1999; 44(6):1119–1123. pmid:10582353
- Szpilman D, Orlowski JP. Sports related to drowning. Eur Respir Rev 2016; 25(141):348–359. doi:10.1183/16000617.0038-2016
- Szpilman D, Webber J, Quan L, et al. Creating a drowning chain of survival. Resuscitation 2014; 85(9):1149–1152. doi:10.1016/j.resuscitation.2014.05.034
- International Life Saving Federation. Who needs further medical help after rescue from the water. Medical Position Statement - MPS 06, 2016. www.ilsf.org/file/3916/download?token=pDnPDCrk. Accessed June 13, 2018.
In June 2017, a 4-year-old boy died 1 week after being knocked over and briefly submerged while playing in knee-deep water. This story was widely reported as a case of a rare occurrence called “dry” or “secondary” drowning, depending on the source.1 The media accounts went viral, spreading fear in parents and others learning about these alleged conditions from the news and social media.
Many alleged cases of dry drowning are reported every year, but each has been found to have a recognized medical source that has a legitimate medically recognized diagnosis (which dry and secondary drowning are not).
Drowning is one of the most common causes of death in children, and so we ought to make sure that the information we share about it is accurate, as it is vital to effective prevention, rescue, and treatment.
Unfortunately, medical providers, medical journals, and the mass media continue to disseminate misinformation on drowning.2 These reports often prevail over updated information and hinder accurate understanding of the drowning problem and its solutions.
Every death is tragic, especially the death of a child, and our heartfelt sympathies go out to the family in this alleged drowning case, as well as to all families suffering the loss of a loved one to drowning. However, in the 2017 case, the cause of death was found on autopsy to be myocarditis not related in any way to drowning. As often happens in such situations, this clarification did not receive any media attention, despite the wide reporting and penetration of the original, erroneous story.
We hope our review will reduce misunderstanding among the public and healthcare providers, contribute to improved data collection, and help to promote interventions aimed at prevention, rescue, and mitigation of drowning incidents.
WHAT IS DROWNING?
A consensus committee of the World Health Organization defined drowning as “the process of experiencing respiratory impairment from submersion/immersion in liquid.”3 The process begins when the victim’s airway goes below the surface of the liquid (submersion) or when water splashes over the face (immersion). If the victim is rescued at any time, the process is interrupted, and this is termed a nonfatal drowning. If the victim dies at any time, this is a fatal drowning. Any water-distress incident without evidence of respiratory impairment (ie, without aspiration) should be considered a water rescue and not a drowning.
Rarely do minimally symptomatic cases progress to death, just as most cases of chest pain do not progress to cardiac arrest.4 Nonetheless, rescued drowning victims can deteriorate, which is why we encourage people to seek medical care immediately upon warning signs, as we do with chest pain. For drowning, such warning signs are any water distress followed by difficulty breathing, excessive coughing, foam in the mouth, or abnormal behavior.
A SERIOUS PUBLIC HEALTH ISSUE
Drowning is a serious and neglected public health issue, claiming the lives of 372,000 people a year worldwide.5 It is a leading cause of death in children ages 1 to 14. The toll continues largely unabated, and in low- and middle-income nations it does not attract the levels of funding that go to other forms of injury prevention, such as road safety.
Nonfatal drowning—with symptoms ranging from mild cough to severe pulmonary edema, and complications ranging from none to severe neurologic impairment—is far more common than fatal drowning.6 For every fatal drowning, there are at least 5 nonfatal drowning incidents in which medical care is needed, and 200 rescues are performed.7–10
In the United States, drowning accounts for almost 13,000 emergency department visits per year and about 3,500 deaths.7,8
In Brazil, with two-thirds the population of the United States, drowning accounts for far fewer hospital visits but about twice as many deaths. In Rio de Janeiro, where a highly effective and specialized prehospital service is provided at 3 drowning resuscitation centers staffed by medical doctors, an analysis of the 46,060 cases of rescue in 10 years from 1991 to 2000 showed that medical assistance was needed in only 930 cases (2%).10 The preventive and rescue actions of parents, bystanders, lifeguards, and prehospital rescue services significantly reduce the number of drowning deaths, but these groups do not consistently gather data on nonfatal drowning that can be included in a comprehensive database.
DROWNING IS A PROCESS
When a person in the water can no longer keep the airway clear, water that enters the mouth is voluntarily spit out or swallowed. Within a few seconds to minutes, the person can no longer clear the airways and water is aspirated, stimulating the cough reflex. Laryngospasm, another myth concerning drowning, is presumed to protect the airways but does not, as it is rare, occurring in less than 2% of cases.11,12
If the person is not rescued, aspiration of water continues, and hypoxemia leads to loss of consciousness and apnea within seconds to a few minutes, followed by cardiac arrest. As a consequence, hypoxemic cardiac arrest generally occurs after a period of tachycardia followed by bradycardia and pulseless electrical activity, usually leading to asystole.13,14
The entire drowning process, from water distress to cardiac arrest, usually takes a few minutes, but in rare situations, such as rapid hypothermia, it can go on for up to an hour.15 Most drowning patients have an otherwise healthy heart, and the apnea and hypoxemia precede the cardiac arrest by only a few seconds to minutes; thus, cardiac arrest is caused by the hypoxemic insult and not by ventricular dysrhythmias.6,16
Drowning can be interrupted at any point between distress and death. If the person is rescued early, the clinical picture is determined by the reactivity of the airway and the amount of water that has been aspirated, but not by the type of water (salt or fresh).
Another myth is that drowning in salt water is different from drowning in fresh water. Both salt water and fresh water cause similar surfactant destruction and washout and disrupt the alveolar-capillary membrane. Disruption of the alveolar-capillary membrane increases its permeability and exacerbates shifting of fluid, plasma, and electrolytes into the alveoli.13 The clinical picture of the damage is one of regional or generalized pulmonary edema, which interferes with gas exchange in the lungs.6,13,17
Animal studies by Modell et al showed that aspiration of just 2.2 mL of water per kilogram of body weight is sufficient to cause severe disturbances in oxygen exchange,17 reflected in a rise in arterial pH and a drop in partial pressure of oxygen. The situation must be similar in humans. In a 70-kg person, this is only about 154 mL of water—about two-thirds of a cup.
The combined effects of fluid in the lungs, the loss of surfactant, and the increase in capillary-alveolar permeability can result in decreased lung compliance, increased right-to-left shunting in the lungs, atelectasis, alveolitis, hypoxemia, and cerebral hypoxia.13
If the victim needs cardiopulmonary resuscitation, the possibility of neurologic damage is similar to that in other cardiac arrest situations, but exceptions exist. For example, in rare cases, hypothermia provides a protective mechanism that allows victims to survive prolonged submersion.4,15
The duration of submersion is the best predictor of death.18 Underwater, people are not taking in oxygen, and cerebral hypoxia causes both morbidity and death. For this reason, reversing cerebral hypoxia with effective ventilation, oxygen, and chest compression is the priority of treatment.
MYTHS AND SLOPPY TERMINOLOGY
“Near drowning,” “dry drowning,” “wet drowning,” “delayed drowning,” and “secondary drowning” are not medically accepted diagnoses,3,4,19 and many organizations and lifesaving institutions around the world discourage the use of these terms.19,20 Unfortunately, these terms still slip past the editors of medical journals and are thus perpetuated. The terms are most pervasive in the nonmedical media, where drowning seems to be synonymous with death.3,19,21 We urge all authors and stakeholders to abandon these terms in favor of understanding and communicating drowning as a process that can vary in severity and have a fatal or nonfatal outcome.
Near-drowning
Historically, drowning meant death, while near-drowning meant the victim survived, at least initially (usually for at least 24 hours).
Before 2002, there were 13 different published definitions of near-drowning.21,22 This variability has caused a great deal of confusion when trying to describe and monitor drowning.
A person can drown and survive, just as a person can have cardiac arrest and survive.4,21 Just as there is no recognized condition of “near-cardiac arrest,” there is also no condition of near-drowning. Using near-drowning as a medical diagnosis hides the true burden of drowning and consequently amplifies difficulties in developing effective prevention, rescue, and treatment programs.
Dry drowning
Dry drowning has never been an accepted medical term, although it has been used to describe different parts of the drowning process. While many authors use it as a synonym for secondary drowning (described below), in the past it was usually used in cases in which no water was found in the lungs at autopsy in persons who were found dead in the water.2–4,21 This occurred in about 10% to 15% of cases and was also called drowning “without water aspiration.”
Perhaps some victims suffer sudden cardiac death. It happens on land—why not in the water? Modell et al stated, “In the absence of the common finding of significant pulmonary edema in the victim’s respiratory system, to conclude his or her death was caused by ‘drowning without aspiration’ is unwise.”23
Laryngospasm is another proposed explanation. It could play a role in the fewer than 2% of cases in which no other cause of death is found on clinical examination or autopsy,11,12,19,23 but it does not occur in most cases of drowning, or it is brief and is terminated by the respiratory movements that allow the air in the lung to escape and water to be inhaled.
The problem with the term dry drowning is the harm caused by misdiagnosing cases of sudden death as drowning, when an alternative cause is present. Most importantly, the management is the same if small amounts of water are present or not; therefore, no clinical distinction is made between wet and dry drowning.
Secondary drowning
Secondary drowning, sometimes called delayed drowning, is another term that is not medically accepted. The historical use of this term reflects the reality that some patients may worsen due to pulmonary edema after aspirating small amounts of water.
Drowning starts with aspiration, and few or only mild symptoms may be present as soon as the person is removed from the water. Either the small amount of water in the lungs is absorbed and causes no complications or, rarely, the patient’s condition becomes progressively worse over the next few hours as the alveoli become inflamed and the alveolar-capillary membrane is disrupted. But people do not unexpectedly die of drowning days or weeks later with no preceding symptoms. The lungs and heart do not “fill up with water,” and water does not need to be pumped out of the lungs.
There has never been a case published in the medical literature of a patient who underwent clinical evaluation, was initially without symptoms, and later deteriorated and died more than 8 hours after the incident.6,10,21 People who have drowned and have minimal symptoms get better (usually) or worse (rarely) within 4 to 8 hours. In a study of more than 41,000 lifeguard rescues, only 0.5% of symptomatic patients died.6
Drowning secondary to injury or sudden illness
Any injury, trauma, or sudden illness that can cause loss of consciousness or mental or physical weakness can lead to drowning. Physicians need to recognize these situations to treat them appropriately. Drowning that is secondary to other primary insults can be classified as24:
- Drowning caused by injury or trauma (eg, a surfing, boating, or a hang-gliding accident)
- Drowning caused by a sudden illness such as cardiac disease (eg, myocardial ischemia, arrhythmias, prolonged QT syndrome, hypertrophic cardiomyopathy) or neurologic disease (eg, epilepsy, stroke)
- Diving disease (eg, decompression sickness, pulmonary overpressurization syndrome, compression barotrauma, narcosis [“rapture of the deep”], shallow water blackout, immersion pulmonary edema).
PREVENTION IS BEST
Drowning is a leading and preventable cause of death worldwide and for people of all ages. The danger is real, not esoteric or rare, and healthcare providers should use any opportunity to discuss with patients, parents, and the media the most important tool for treating drowning: primary prevention.
For example, small children should be continuously and uninterruptedly supervised within arm’s reach while in the water, even if a lifeguard is present. Other preventive measures are lifejackets, fences completely enclosing pools or ponds, and swimming and water safety lessons. Drowning often occurs in a deceptively pleasant environment that may not seem dangerous.
RECOGNIZE DISTRESS
When preventive measures fail, responders (usually a health professional is involved) need to be able to perform the necessary steps to interrupt the drowning process.
The first challenge is to recognize when someone in the water is at risk of drowning and needs to be rescued.25 Early self-rescue or rescue by others may stop the drowning process and prevent most cases of initial and subsequent water aspiration, respiratory distress, and medical complications.
DON’T BECOME A VICTIM
Rescuers must take care not to become victims themselves. Panicked swimmers can thrash about and injure the rescuer or clutch at anything they encounter, dragging the rescuer under. And the rescuer can succumb to the same hazards that got the victim into trouble, such as strong currents, deep water, or underwater hazards.
Certified lifeguards are trained to get victims out of the water safely. The American Red Cross slogan “Reach or throw, don’t go” means “Reach out with a pole or other object or throw something that floats; don’t get in the water yourself.”
WHAT TO TELL THE PUBLIC
While some journalists acknowledge that the terms dry drowning and secondary drowning are medically discredited, they still use them in their reports. The novelty of this story—and its appeal to media outlets—is precisely the unfamiliarity of these terms to the general public and the perceived mysterious, looming threat.
We often hear that these terms are more familiar to the public, which is likely true. More concerning, some physicians continue to use them (and older definitions of drowning that equate it with death) in media interviews, clinical care, and publications. The paradox is that we, the medical community, invented these terms, not patients or the media.
As clinicians and researchers, we should drive popular culture definitions, not the other way around. Rather than dismiss these terms as “semantics” or “technicalities,” we should take the opportunity to highlight the dangers of drowning and the importance of prevention, and to promote simpler language that is easier for us and our patients to understand.19,21
Healthcare providers should understand and share modern drowning science and best practices, which will reduce fear, improve resource utilization, and prevent potentially deadly consequences due to misunderstanding or misinterpretation of incorrect terminology.
WHEN PATIENTS SHOULD SEEK CARE
Anyone who experiences cough, breathlessness, or other worrisome symptoms such as abnormal mentation within 8 hours of a drowning incident (using the modern definition above) should seek medical advice immediately.
We tell people to seek care if symptoms seem any worse than the experience of a drink “going down the wrong pipe” at the dinner table.21 But symptoms can be minimal. Careful attention should be given to mild symptoms that get progressively worse during that time. These cases can rarely progress to acute respiratory distress syndrome.
Table 1 explores who needs further medical help after being rescued from the water.26
In most of these cases, it is most appropriate to call an ambulance, but care may involve seeing a doctor depending on the severity of the symptoms.6,21 Usually, drowning patients are observed for 4 to 8 hours in an emergency department and are discharged if normal. Symptoms that are more significant include persistent cough, foam at the mouth or nose, confusion, or abnormal behavior, and these require further medical evaluation.
Patients should also seek medical care even if they are 100% normal upon exiting the water but develop worrisome symptoms more than 8 hours later, and providers should consider diagnoses other than primary drowning. Spontaneous pneumothorax, chemical pneumonitis, bacterial or viral pneumonia, head injury, asthma, chest trauma, and acute respiratory distress syndrome have been mislabeled as delayed, dry, or secondary drowning.3,4,19,21
In June 2017, a 4-year-old boy died 1 week after being knocked over and briefly submerged while playing in knee-deep water. This story was widely reported as a case of a rare occurrence called “dry” or “secondary” drowning, depending on the source.1 The media accounts went viral, spreading fear in parents and others learning about these alleged conditions from the news and social media.
Many alleged cases of dry drowning are reported every year, but each has been found to have a recognized medical source that has a legitimate medically recognized diagnosis (which dry and secondary drowning are not).
Drowning is one of the most common causes of death in children, and so we ought to make sure that the information we share about it is accurate, as it is vital to effective prevention, rescue, and treatment.
Unfortunately, medical providers, medical journals, and the mass media continue to disseminate misinformation on drowning.2 These reports often prevail over updated information and hinder accurate understanding of the drowning problem and its solutions.
Every death is tragic, especially the death of a child, and our heartfelt sympathies go out to the family in this alleged drowning case, as well as to all families suffering the loss of a loved one to drowning. However, in the 2017 case, the cause of death was found on autopsy to be myocarditis not related in any way to drowning. As often happens in such situations, this clarification did not receive any media attention, despite the wide reporting and penetration of the original, erroneous story.
We hope our review will reduce misunderstanding among the public and healthcare providers, contribute to improved data collection, and help to promote interventions aimed at prevention, rescue, and mitigation of drowning incidents.
WHAT IS DROWNING?
A consensus committee of the World Health Organization defined drowning as “the process of experiencing respiratory impairment from submersion/immersion in liquid.”3 The process begins when the victim’s airway goes below the surface of the liquid (submersion) or when water splashes over the face (immersion). If the victim is rescued at any time, the process is interrupted, and this is termed a nonfatal drowning. If the victim dies at any time, this is a fatal drowning. Any water-distress incident without evidence of respiratory impairment (ie, without aspiration) should be considered a water rescue and not a drowning.
Rarely do minimally symptomatic cases progress to death, just as most cases of chest pain do not progress to cardiac arrest.4 Nonetheless, rescued drowning victims can deteriorate, which is why we encourage people to seek medical care immediately upon warning signs, as we do with chest pain. For drowning, such warning signs are any water distress followed by difficulty breathing, excessive coughing, foam in the mouth, or abnormal behavior.
A SERIOUS PUBLIC HEALTH ISSUE
Drowning is a serious and neglected public health issue, claiming the lives of 372,000 people a year worldwide.5 It is a leading cause of death in children ages 1 to 14. The toll continues largely unabated, and in low- and middle-income nations it does not attract the levels of funding that go to other forms of injury prevention, such as road safety.
Nonfatal drowning—with symptoms ranging from mild cough to severe pulmonary edema, and complications ranging from none to severe neurologic impairment—is far more common than fatal drowning.6 For every fatal drowning, there are at least 5 nonfatal drowning incidents in which medical care is needed, and 200 rescues are performed.7–10
In the United States, drowning accounts for almost 13,000 emergency department visits per year and about 3,500 deaths.7,8
In Brazil, with two-thirds the population of the United States, drowning accounts for far fewer hospital visits but about twice as many deaths. In Rio de Janeiro, where a highly effective and specialized prehospital service is provided at 3 drowning resuscitation centers staffed by medical doctors, an analysis of the 46,060 cases of rescue in 10 years from 1991 to 2000 showed that medical assistance was needed in only 930 cases (2%).10 The preventive and rescue actions of parents, bystanders, lifeguards, and prehospital rescue services significantly reduce the number of drowning deaths, but these groups do not consistently gather data on nonfatal drowning that can be included in a comprehensive database.
DROWNING IS A PROCESS
When a person in the water can no longer keep the airway clear, water that enters the mouth is voluntarily spit out or swallowed. Within a few seconds to minutes, the person can no longer clear the airways and water is aspirated, stimulating the cough reflex. Laryngospasm, another myth concerning drowning, is presumed to protect the airways but does not, as it is rare, occurring in less than 2% of cases.11,12
If the person is not rescued, aspiration of water continues, and hypoxemia leads to loss of consciousness and apnea within seconds to a few minutes, followed by cardiac arrest. As a consequence, hypoxemic cardiac arrest generally occurs after a period of tachycardia followed by bradycardia and pulseless electrical activity, usually leading to asystole.13,14
The entire drowning process, from water distress to cardiac arrest, usually takes a few minutes, but in rare situations, such as rapid hypothermia, it can go on for up to an hour.15 Most drowning patients have an otherwise healthy heart, and the apnea and hypoxemia precede the cardiac arrest by only a few seconds to minutes; thus, cardiac arrest is caused by the hypoxemic insult and not by ventricular dysrhythmias.6,16
Drowning can be interrupted at any point between distress and death. If the person is rescued early, the clinical picture is determined by the reactivity of the airway and the amount of water that has been aspirated, but not by the type of water (salt or fresh).
Another myth is that drowning in salt water is different from drowning in fresh water. Both salt water and fresh water cause similar surfactant destruction and washout and disrupt the alveolar-capillary membrane. Disruption of the alveolar-capillary membrane increases its permeability and exacerbates shifting of fluid, plasma, and electrolytes into the alveoli.13 The clinical picture of the damage is one of regional or generalized pulmonary edema, which interferes with gas exchange in the lungs.6,13,17
Animal studies by Modell et al showed that aspiration of just 2.2 mL of water per kilogram of body weight is sufficient to cause severe disturbances in oxygen exchange,17 reflected in a rise in arterial pH and a drop in partial pressure of oxygen. The situation must be similar in humans. In a 70-kg person, this is only about 154 mL of water—about two-thirds of a cup.
The combined effects of fluid in the lungs, the loss of surfactant, and the increase in capillary-alveolar permeability can result in decreased lung compliance, increased right-to-left shunting in the lungs, atelectasis, alveolitis, hypoxemia, and cerebral hypoxia.13
If the victim needs cardiopulmonary resuscitation, the possibility of neurologic damage is similar to that in other cardiac arrest situations, but exceptions exist. For example, in rare cases, hypothermia provides a protective mechanism that allows victims to survive prolonged submersion.4,15
The duration of submersion is the best predictor of death.18 Underwater, people are not taking in oxygen, and cerebral hypoxia causes both morbidity and death. For this reason, reversing cerebral hypoxia with effective ventilation, oxygen, and chest compression is the priority of treatment.
MYTHS AND SLOPPY TERMINOLOGY
“Near drowning,” “dry drowning,” “wet drowning,” “delayed drowning,” and “secondary drowning” are not medically accepted diagnoses,3,4,19 and many organizations and lifesaving institutions around the world discourage the use of these terms.19,20 Unfortunately, these terms still slip past the editors of medical journals and are thus perpetuated. The terms are most pervasive in the nonmedical media, where drowning seems to be synonymous with death.3,19,21 We urge all authors and stakeholders to abandon these terms in favor of understanding and communicating drowning as a process that can vary in severity and have a fatal or nonfatal outcome.
Near-drowning
Historically, drowning meant death, while near-drowning meant the victim survived, at least initially (usually for at least 24 hours).
Before 2002, there were 13 different published definitions of near-drowning.21,22 This variability has caused a great deal of confusion when trying to describe and monitor drowning.
A person can drown and survive, just as a person can have cardiac arrest and survive.4,21 Just as there is no recognized condition of “near-cardiac arrest,” there is also no condition of near-drowning. Using near-drowning as a medical diagnosis hides the true burden of drowning and consequently amplifies difficulties in developing effective prevention, rescue, and treatment programs.
Dry drowning
Dry drowning has never been an accepted medical term, although it has been used to describe different parts of the drowning process. While many authors use it as a synonym for secondary drowning (described below), in the past it was usually used in cases in which no water was found in the lungs at autopsy in persons who were found dead in the water.2–4,21 This occurred in about 10% to 15% of cases and was also called drowning “without water aspiration.”
Perhaps some victims suffer sudden cardiac death. It happens on land—why not in the water? Modell et al stated, “In the absence of the common finding of significant pulmonary edema in the victim’s respiratory system, to conclude his or her death was caused by ‘drowning without aspiration’ is unwise.”23
Laryngospasm is another proposed explanation. It could play a role in the fewer than 2% of cases in which no other cause of death is found on clinical examination or autopsy,11,12,19,23 but it does not occur in most cases of drowning, or it is brief and is terminated by the respiratory movements that allow the air in the lung to escape and water to be inhaled.
The problem with the term dry drowning is the harm caused by misdiagnosing cases of sudden death as drowning, when an alternative cause is present. Most importantly, the management is the same if small amounts of water are present or not; therefore, no clinical distinction is made between wet and dry drowning.
Secondary drowning
Secondary drowning, sometimes called delayed drowning, is another term that is not medically accepted. The historical use of this term reflects the reality that some patients may worsen due to pulmonary edema after aspirating small amounts of water.
Drowning starts with aspiration, and few or only mild symptoms may be present as soon as the person is removed from the water. Either the small amount of water in the lungs is absorbed and causes no complications or, rarely, the patient’s condition becomes progressively worse over the next few hours as the alveoli become inflamed and the alveolar-capillary membrane is disrupted. But people do not unexpectedly die of drowning days or weeks later with no preceding symptoms. The lungs and heart do not “fill up with water,” and water does not need to be pumped out of the lungs.
There has never been a case published in the medical literature of a patient who underwent clinical evaluation, was initially without symptoms, and later deteriorated and died more than 8 hours after the incident.6,10,21 People who have drowned and have minimal symptoms get better (usually) or worse (rarely) within 4 to 8 hours. In a study of more than 41,000 lifeguard rescues, only 0.5% of symptomatic patients died.6
Drowning secondary to injury or sudden illness
Any injury, trauma, or sudden illness that can cause loss of consciousness or mental or physical weakness can lead to drowning. Physicians need to recognize these situations to treat them appropriately. Drowning that is secondary to other primary insults can be classified as24:
- Drowning caused by injury or trauma (eg, a surfing, boating, or a hang-gliding accident)
- Drowning caused by a sudden illness such as cardiac disease (eg, myocardial ischemia, arrhythmias, prolonged QT syndrome, hypertrophic cardiomyopathy) or neurologic disease (eg, epilepsy, stroke)
- Diving disease (eg, decompression sickness, pulmonary overpressurization syndrome, compression barotrauma, narcosis [“rapture of the deep”], shallow water blackout, immersion pulmonary edema).
PREVENTION IS BEST
Drowning is a leading and preventable cause of death worldwide and for people of all ages. The danger is real, not esoteric or rare, and healthcare providers should use any opportunity to discuss with patients, parents, and the media the most important tool for treating drowning: primary prevention.
For example, small children should be continuously and uninterruptedly supervised within arm’s reach while in the water, even if a lifeguard is present. Other preventive measures are lifejackets, fences completely enclosing pools or ponds, and swimming and water safety lessons. Drowning often occurs in a deceptively pleasant environment that may not seem dangerous.
RECOGNIZE DISTRESS
When preventive measures fail, responders (usually a health professional is involved) need to be able to perform the necessary steps to interrupt the drowning process.
The first challenge is to recognize when someone in the water is at risk of drowning and needs to be rescued.25 Early self-rescue or rescue by others may stop the drowning process and prevent most cases of initial and subsequent water aspiration, respiratory distress, and medical complications.
DON’T BECOME A VICTIM
Rescuers must take care not to become victims themselves. Panicked swimmers can thrash about and injure the rescuer or clutch at anything they encounter, dragging the rescuer under. And the rescuer can succumb to the same hazards that got the victim into trouble, such as strong currents, deep water, or underwater hazards.
Certified lifeguards are trained to get victims out of the water safely. The American Red Cross slogan “Reach or throw, don’t go” means “Reach out with a pole or other object or throw something that floats; don’t get in the water yourself.”
WHAT TO TELL THE PUBLIC
While some journalists acknowledge that the terms dry drowning and secondary drowning are medically discredited, they still use them in their reports. The novelty of this story—and its appeal to media outlets—is precisely the unfamiliarity of these terms to the general public and the perceived mysterious, looming threat.
We often hear that these terms are more familiar to the public, which is likely true. More concerning, some physicians continue to use them (and older definitions of drowning that equate it with death) in media interviews, clinical care, and publications. The paradox is that we, the medical community, invented these terms, not patients or the media.
As clinicians and researchers, we should drive popular culture definitions, not the other way around. Rather than dismiss these terms as “semantics” or “technicalities,” we should take the opportunity to highlight the dangers of drowning and the importance of prevention, and to promote simpler language that is easier for us and our patients to understand.19,21
Healthcare providers should understand and share modern drowning science and best practices, which will reduce fear, improve resource utilization, and prevent potentially deadly consequences due to misunderstanding or misinterpretation of incorrect terminology.
WHEN PATIENTS SHOULD SEEK CARE
Anyone who experiences cough, breathlessness, or other worrisome symptoms such as abnormal mentation within 8 hours of a drowning incident (using the modern definition above) should seek medical advice immediately.
We tell people to seek care if symptoms seem any worse than the experience of a drink “going down the wrong pipe” at the dinner table.21 But symptoms can be minimal. Careful attention should be given to mild symptoms that get progressively worse during that time. These cases can rarely progress to acute respiratory distress syndrome.
Table 1 explores who needs further medical help after being rescued from the water.26
In most of these cases, it is most appropriate to call an ambulance, but care may involve seeing a doctor depending on the severity of the symptoms.6,21 Usually, drowning patients are observed for 4 to 8 hours in an emergency department and are discharged if normal. Symptoms that are more significant include persistent cough, foam at the mouth or nose, confusion, or abnormal behavior, and these require further medical evaluation.
Patients should also seek medical care even if they are 100% normal upon exiting the water but develop worrisome symptoms more than 8 hours later, and providers should consider diagnoses other than primary drowning. Spontaneous pneumothorax, chemical pneumonitis, bacterial or viral pneumonia, head injury, asthma, chest trauma, and acute respiratory distress syndrome have been mislabeled as delayed, dry, or secondary drowning.3,4,19,21
- Buffington B. Texas boy dies from ‘dry drowning’ days after swimming. USA Today, June 8, 2017. www.usatoday.com/story/news/nation-now/2017/06/08/texas-boy-dies-dry-drowning-days-after-swimming/379944001.
- Schmidt AC, Sempsrott JR, Szpilman D, et al. The use of non-uniform drowning terminology: a follow-up study. Scand J Trauma Resusc Emerg Med 2017; 25(1):72. doi:10.1186/s13049-017-0405-x
- van Beeck EF, Branche CM, Szpilman D, Modell JH, Bierens JJ. A new definition of drowning: towards documentation and prevention of a global public health problem. Bull World Health Organ 2005; 83(11):853–856. pmid:16302042
- Szpilman D, Bierens JJ, Handley AJ, Orlowski JP. Drowning. N Engl J Med 2012; 366(22):2102–2110. doi:10.1056/NEJMra1013317
- World Health Organization. Global report on drowning: preventing a leading killer. www.who.int/violence_injury_prevention/global_report_drowning/en. Accessed June 13, 2018.
- Szpilman D. Near-drowning and drowning classification: a proposal to stratify mortality based on the analysis of 1,831 cases. Chest 1997; 112(3):660–665. pmid:9315798
- Centers for Disease Control and Prevention. Welcome to WISQARS. www.cdc.gov/injury/wisqars. Accessed June 13, 2018.
- Centers for Disease Control and Prevention. WONDER. https://wonder.cdc.gov. Accessed June 13, 2018.
- Cummings P, Quan L. Trends in unintentional drowning: the role of alcohol and medical care. JAMA 1999; 281(23):2198–2202. pmid:10376572
- Szpilman D, Elmann J, Cruz-Filho FES. Drowning classification: a revalidation study based on the analysis of 930 cases over 10 years. World Congress on Drowning, Netherlands 2002. www.researchgate.net/publication/267981062_DROWNING_CLASSIFICATION_a_revalidation_study_based_on_the_analysis_of_930_cases_over_10_years. Accessed June 13, 2018.
- Szpilman D, Elmann J, Cruz-Filho FES. Dry-drowning—fact or myth? World Congress on Drowning. Netherlands, 2002. www.researchgate.net/publication/267981164_Dry-drowning_-Fact_or_Myth. Accessed June 13, 2018.
- Lunetta P, Modell JH, Sajantila A. What is the incidence and significance of "dry-lungs" in bodies found in water? Am J Forensic Med Pathol 2004; 25(4):291–301. pmid:15577518
- Orlowski JP, Abulleil MM, Phillips JM. The hemodynamic and cardiovascular effects of near-drowning in hypotonic, isotonic, or hypertonic solutions. Ann Emerg Med 1989; 18:1044–1049. pmid:2802278
- Grmec S, Strnad M, Podgorsek D. Comparison of the characteristics and outcome among patients suffering from out-of-hospital primary cardiac arrest and drowning victims in cardiac arrest. Int J Emerg Med 2009; 2(1):7–12. doi:10.1007/s12245-009-0084-0
- Tipton MJ, Golden FS. A proposed decision-making guide for the search, rescue and resuscitation of submersion (head under) victims based on expert opinion. Resuscitation 2011; 82(7):819–824. doi:10.1016/j.resuscitation.2011.02.021
- Orlowski JP, Szpilman D. Drowning. Rescue, resuscitation, and reanimation. Pediatr Clin North Am 2001; 48(3):627–646. pmid:11411297
- Modell JH, Moya F, Newby EJ, Ruiz BC, Showers AV. The effects of fluid volume in seawater drowning. Ann Intern Med 1967; 67(1):68–80. pmid:6028660
- Quan L, Wentz KR, Gore EJ, Copass MK. Outcome and predictors of outcome in pediatric submersion victims receiving prehospital care in King County, Washington. Pediatrics 1990; 86(4):586–593. pmid:2216625
- Szpilman D, Orlowski JP, Cruz-Filho FES. Hey “Near-drowning,” you’ve been messing up our minds! World Congress on Drowning. Amsterdam, 2002. www.researchgate.net/publication/267981173_HEY_Near-drowning_YOU%27VE_BEEN_MESSING_UP_OUR_MINDS. Accessed June 13, 2018.
- American College of Emergency Physicians. Death after swimming is extremely rare—and is not “dry drowning.” http://newsroom.acep.org/2017-07-11-Death-After-Swimming-Is-Extremely-Rare-And-Is-NOT-Dry-Drowning. Accessed June 13, 2018.
- Hawkins SC, Sempsrott J, Schmidt A. “Drowning” in a sea of misinformation. Emergency Medicine News 2017; 39*8):1. http://journals.lww.com/em-news/blog/BreakingNews/pages/post.aspx?PostID=377. Accessed June 5, 2018.
- Szpilman D, Tipton M, Sempsrott J, et al. Drowning timeline: a new systematic model of the drowning process. Am J Emerg Med 2016; 34(11):2224–2226. doi:10.1016/j.ajem.2016.07.063
- Modell JH, Bellefleur M, Davis JH. Drowning without aspiration: is this an appropriate diagnosis? J Forensic Sci 1999; 44(6):1119–1123. pmid:10582353
- Szpilman D, Orlowski JP. Sports related to drowning. Eur Respir Rev 2016; 25(141):348–359. doi:10.1183/16000617.0038-2016
- Szpilman D, Webber J, Quan L, et al. Creating a drowning chain of survival. Resuscitation 2014; 85(9):1149–1152. doi:10.1016/j.resuscitation.2014.05.034
- International Life Saving Federation. Who needs further medical help after rescue from the water. Medical Position Statement - MPS 06, 2016. www.ilsf.org/file/3916/download?token=pDnPDCrk. Accessed June 13, 2018.
- Buffington B. Texas boy dies from ‘dry drowning’ days after swimming. USA Today, June 8, 2017. www.usatoday.com/story/news/nation-now/2017/06/08/texas-boy-dies-dry-drowning-days-after-swimming/379944001.
- Schmidt AC, Sempsrott JR, Szpilman D, et al. The use of non-uniform drowning terminology: a follow-up study. Scand J Trauma Resusc Emerg Med 2017; 25(1):72. doi:10.1186/s13049-017-0405-x
- van Beeck EF, Branche CM, Szpilman D, Modell JH, Bierens JJ. A new definition of drowning: towards documentation and prevention of a global public health problem. Bull World Health Organ 2005; 83(11):853–856. pmid:16302042
- Szpilman D, Bierens JJ, Handley AJ, Orlowski JP. Drowning. N Engl J Med 2012; 366(22):2102–2110. doi:10.1056/NEJMra1013317
- World Health Organization. Global report on drowning: preventing a leading killer. www.who.int/violence_injury_prevention/global_report_drowning/en. Accessed June 13, 2018.
- Szpilman D. Near-drowning and drowning classification: a proposal to stratify mortality based on the analysis of 1,831 cases. Chest 1997; 112(3):660–665. pmid:9315798
- Centers for Disease Control and Prevention. Welcome to WISQARS. www.cdc.gov/injury/wisqars. Accessed June 13, 2018.
- Centers for Disease Control and Prevention. WONDER. https://wonder.cdc.gov. Accessed June 13, 2018.
- Cummings P, Quan L. Trends in unintentional drowning: the role of alcohol and medical care. JAMA 1999; 281(23):2198–2202. pmid:10376572
- Szpilman D, Elmann J, Cruz-Filho FES. Drowning classification: a revalidation study based on the analysis of 930 cases over 10 years. World Congress on Drowning, Netherlands 2002. www.researchgate.net/publication/267981062_DROWNING_CLASSIFICATION_a_revalidation_study_based_on_the_analysis_of_930_cases_over_10_years. Accessed June 13, 2018.
- Szpilman D, Elmann J, Cruz-Filho FES. Dry-drowning—fact or myth? World Congress on Drowning. Netherlands, 2002. www.researchgate.net/publication/267981164_Dry-drowning_-Fact_or_Myth. Accessed June 13, 2018.
- Lunetta P, Modell JH, Sajantila A. What is the incidence and significance of "dry-lungs" in bodies found in water? Am J Forensic Med Pathol 2004; 25(4):291–301. pmid:15577518
- Orlowski JP, Abulleil MM, Phillips JM. The hemodynamic and cardiovascular effects of near-drowning in hypotonic, isotonic, or hypertonic solutions. Ann Emerg Med 1989; 18:1044–1049. pmid:2802278
- Grmec S, Strnad M, Podgorsek D. Comparison of the characteristics and outcome among patients suffering from out-of-hospital primary cardiac arrest and drowning victims in cardiac arrest. Int J Emerg Med 2009; 2(1):7–12. doi:10.1007/s12245-009-0084-0
- Tipton MJ, Golden FS. A proposed decision-making guide for the search, rescue and resuscitation of submersion (head under) victims based on expert opinion. Resuscitation 2011; 82(7):819–824. doi:10.1016/j.resuscitation.2011.02.021
- Orlowski JP, Szpilman D. Drowning. Rescue, resuscitation, and reanimation. Pediatr Clin North Am 2001; 48(3):627–646. pmid:11411297
- Modell JH, Moya F, Newby EJ, Ruiz BC, Showers AV. The effects of fluid volume in seawater drowning. Ann Intern Med 1967; 67(1):68–80. pmid:6028660
- Quan L, Wentz KR, Gore EJ, Copass MK. Outcome and predictors of outcome in pediatric submersion victims receiving prehospital care in King County, Washington. Pediatrics 1990; 86(4):586–593. pmid:2216625
- Szpilman D, Orlowski JP, Cruz-Filho FES. Hey “Near-drowning,” you’ve been messing up our minds! World Congress on Drowning. Amsterdam, 2002. www.researchgate.net/publication/267981173_HEY_Near-drowning_YOU%27VE_BEEN_MESSING_UP_OUR_MINDS. Accessed June 13, 2018.
- American College of Emergency Physicians. Death after swimming is extremely rare—and is not “dry drowning.” http://newsroom.acep.org/2017-07-11-Death-After-Swimming-Is-Extremely-Rare-And-Is-NOT-Dry-Drowning. Accessed June 13, 2018.
- Hawkins SC, Sempsrott J, Schmidt A. “Drowning” in a sea of misinformation. Emergency Medicine News 2017; 39*8):1. http://journals.lww.com/em-news/blog/BreakingNews/pages/post.aspx?PostID=377. Accessed June 5, 2018.
- Szpilman D, Tipton M, Sempsrott J, et al. Drowning timeline: a new systematic model of the drowning process. Am J Emerg Med 2016; 34(11):2224–2226. doi:10.1016/j.ajem.2016.07.063
- Modell JH, Bellefleur M, Davis JH. Drowning without aspiration: is this an appropriate diagnosis? J Forensic Sci 1999; 44(6):1119–1123. pmid:10582353
- Szpilman D, Orlowski JP. Sports related to drowning. Eur Respir Rev 2016; 25(141):348–359. doi:10.1183/16000617.0038-2016
- Szpilman D, Webber J, Quan L, et al. Creating a drowning chain of survival. Resuscitation 2014; 85(9):1149–1152. doi:10.1016/j.resuscitation.2014.05.034
- International Life Saving Federation. Who needs further medical help after rescue from the water. Medical Position Statement - MPS 06, 2016. www.ilsf.org/file/3916/download?token=pDnPDCrk. Accessed June 13, 2018.
KEY POINTS
- Drowning is a process of aspiration leading to hypoxia and eventually cardiac arrest. However, it is not synonymous with death: it can be interrupted.
- Patients who have been rescued from drowning and who have minimal symptoms generally get better within 4 to 8 hours of the event.
- Rescued victims should be warned that, although a rare condition, if they develop cough, breathlessness, or any other worrisome symptom within 8 hours of being in the water, they should seek medical attention immediately.

Wolff-Parkinson-White pattern unmasked by severe musculoskeletal pain
A 55-year-old man with no significant medical history presented to the emergency department with left-sided flank pain that had begun 3 days earlier. He described the pain as continuous, sharp, and aggravated by movement. He worked in construction, and before the pain started he had moved 8 sheets of drywall and lifted 5-gallon buckets of spackling compound. He denied any associated chest pain, palpitations, dyspnea, cough, or lightheadedness. His family history included sudden cardiac death in 2 second-degree relatives.
On arrival in the emergency department, his vital signs were normal, as were the rest of the findings on physical examination except for reproducible point tenderness below the left scapula.
Laboratory workup revealed normal blood cell counts, liver enzymes, and kidney function. His initial troponin test was negative.
.")
 and normal PR intervals.")
The patient was referred to an electrophysiologist for further evaluation, but he returned to his home country (Haiti) after discharge and was lost to follow-up.
WOLFF-PARKINSON-WHITE PATTERN VS SYNDROME
WPW syndrome is a disorder of the conduction system leading to preexcitation of the ventricles by an accessory pathway between the atria and ventricles. It is characterized by preexcitation manifested on electrocardiography and by symptomatic arrhythmias.
In contrast, the WPW pattern is defined only by preexcitation findings on electrocardiography without symptomatic arrhythmias. Patients with WPW syndrome can present with palpitation, dizziness, and syncope resulting from underlying arrhythmia.1 This is not seen in patients with the WPW pattern.
A short PR interval with or without delta waves can also be seen in the absence of an accessory pathway, eg, in hypoplastic left heart syndrome, atrioventricular canal defect, and Ebstein anomaly. These conditions are termed pseudopreexcitation syndrome.2
Our patient presented with severe musculoskeletal pain that precipitated the electrocardiographic changes of the WPW pattern and resolved with adequate pain control. The WPW pattern can be unmasked under different scenarios, including anesthesia, sympathomimetic drugs, and postoperatively.3–5
Catecholamine challenge has been used to unmask high-risk features in WPW syndrome.3 Our patient may have had a transient spike in catecholamine levels because of severe musculoskeletal pain, leading to unmasking of accessory pathways and resulting in the WPW pattern on electrocardiography.
Most patients with the WPW pattern experience no symptoms, but a small percentage develop arrhythmias.
In rare cases, sudden cardiac death can be the presenting feature of WPW syndrome. The estimated risk of sudden cardiac death in patients with the WPW pattern is 1.25 per 1,000 person-years; ventricular fibrillation is the underlying mechanism.6 As our patient had a family history of sudden cardiac death, he was considered at high risk and was therefore referred to an electrophysiologist.
- Munger TM, Packer DL, Hammill SC, et al. A population study of the natural history of Wolff-Parkinson-White syndrome in Olmsted County, Minnesota, 1953–1989. Circulation 1993; 87(3):866–873. pmid:8443907
- Carlson AM, Turek JW, Law IH, Von Bergen NH. Pseudo-preexcitation is prevalent among patients with repaired complex congenital heart disease. Pediatr Cardiol.2015; 36(1):8–13. doi:10.1007/s00246-014-0955-x
- Aleong RG, Singh SM, Levinson JR, Milan DJ. Catecholamine challenge unmasking high-risk features in the Wolff-Parkinson-White syndrome. Europace 2009; 11(10):1396–1398. doi:10.1093/europace/eup211
- Sahu S, Karna ST, Karna A, Lata I, Kapoor D. Anaesthetic management of Wolff-Parkinson-White syndrome for hysterectomy. Indian J Anaesth 2011; 55(4):378–380. doi:10.4103/0019-5049.84866
- Tseng ZH, Yadav AV, Scheinman MM. Catecholamine dependent accessory pathway automaticity. Pacing Clin Electrophysiol 2004; 27(7):1005–1007. doi:10.1111/j.1540-8159.2004.00574.x
- Obeyesekere MN, Leong-Sit P, Massel D, et al. Risk of arrhythmia and sudden death in patients with asymptomatic preexcitation: a meta-analysis. Circulation 2012; 125(19):2308–2315. doi:10.1161/CIRCULATIONAHA.111.055350
A 55-year-old man with no significant medical history presented to the emergency department with left-sided flank pain that had begun 3 days earlier. He described the pain as continuous, sharp, and aggravated by movement. He worked in construction, and before the pain started he had moved 8 sheets of drywall and lifted 5-gallon buckets of spackling compound. He denied any associated chest pain, palpitations, dyspnea, cough, or lightheadedness. His family history included sudden cardiac death in 2 second-degree relatives.
On arrival in the emergency department, his vital signs were normal, as were the rest of the findings on physical examination except for reproducible point tenderness below the left scapula.
Laboratory workup revealed normal blood cell counts, liver enzymes, and kidney function. His initial troponin test was negative.
The patient was referred to an electrophysiologist for further evaluation, but he returned to his home country (Haiti) after discharge and was lost to follow-up.
WOLFF-PARKINSON-WHITE PATTERN VS SYNDROME
WPW syndrome is a disorder of the conduction system leading to preexcitation of the ventricles by an accessory pathway between the atria and ventricles. It is characterized by preexcitation manifested on electrocardiography and by symptomatic arrhythmias.
In contrast, the WPW pattern is defined only by preexcitation findings on electrocardiography without symptomatic arrhythmias. Patients with WPW syndrome can present with palpitation, dizziness, and syncope resulting from underlying arrhythmia.1 This is not seen in patients with the WPW pattern.
A short PR interval with or without delta waves can also be seen in the absence of an accessory pathway, eg, in hypoplastic left heart syndrome, atrioventricular canal defect, and Ebstein anomaly. These conditions are termed pseudopreexcitation syndrome.2
Our patient presented with severe musculoskeletal pain that precipitated the electrocardiographic changes of the WPW pattern and resolved with adequate pain control. The WPW pattern can be unmasked under different scenarios, including anesthesia, sympathomimetic drugs, and postoperatively.3–5
Catecholamine challenge has been used to unmask high-risk features in WPW syndrome.3 Our patient may have had a transient spike in catecholamine levels because of severe musculoskeletal pain, leading to unmasking of accessory pathways and resulting in the WPW pattern on electrocardiography.
Most patients with the WPW pattern experience no symptoms, but a small percentage develop arrhythmias.
In rare cases, sudden cardiac death can be the presenting feature of WPW syndrome. The estimated risk of sudden cardiac death in patients with the WPW pattern is 1.25 per 1,000 person-years; ventricular fibrillation is the underlying mechanism.6 As our patient had a family history of sudden cardiac death, he was considered at high risk and was therefore referred to an electrophysiologist.
A 55-year-old man with no significant medical history presented to the emergency department with left-sided flank pain that had begun 3 days earlier. He described the pain as continuous, sharp, and aggravated by movement. He worked in construction, and before the pain started he had moved 8 sheets of drywall and lifted 5-gallon buckets of spackling compound. He denied any associated chest pain, palpitations, dyspnea, cough, or lightheadedness. His family history included sudden cardiac death in 2 second-degree relatives.
On arrival in the emergency department, his vital signs were normal, as were the rest of the findings on physical examination except for reproducible point tenderness below the left scapula.
Laboratory workup revealed normal blood cell counts, liver enzymes, and kidney function. His initial troponin test was negative.
The patient was referred to an electrophysiologist for further evaluation, but he returned to his home country (Haiti) after discharge and was lost to follow-up.
WOLFF-PARKINSON-WHITE PATTERN VS SYNDROME
WPW syndrome is a disorder of the conduction system leading to preexcitation of the ventricles by an accessory pathway between the atria and ventricles. It is characterized by preexcitation manifested on electrocardiography and by symptomatic arrhythmias.
In contrast, the WPW pattern is defined only by preexcitation findings on electrocardiography without symptomatic arrhythmias. Patients with WPW syndrome can present with palpitation, dizziness, and syncope resulting from underlying arrhythmia.1 This is not seen in patients with the WPW pattern.
A short PR interval with or without delta waves can also be seen in the absence of an accessory pathway, eg, in hypoplastic left heart syndrome, atrioventricular canal defect, and Ebstein anomaly. These conditions are termed pseudopreexcitation syndrome.2
Our patient presented with severe musculoskeletal pain that precipitated the electrocardiographic changes of the WPW pattern and resolved with adequate pain control. The WPW pattern can be unmasked under different scenarios, including anesthesia, sympathomimetic drugs, and postoperatively.3–5
Catecholamine challenge has been used to unmask high-risk features in WPW syndrome.3 Our patient may have had a transient spike in catecholamine levels because of severe musculoskeletal pain, leading to unmasking of accessory pathways and resulting in the WPW pattern on electrocardiography.
Most patients with the WPW pattern experience no symptoms, but a small percentage develop arrhythmias.
In rare cases, sudden cardiac death can be the presenting feature of WPW syndrome. The estimated risk of sudden cardiac death in patients with the WPW pattern is 1.25 per 1,000 person-years; ventricular fibrillation is the underlying mechanism.6 As our patient had a family history of sudden cardiac death, he was considered at high risk and was therefore referred to an electrophysiologist.
- Munger TM, Packer DL, Hammill SC, et al. A population study of the natural history of Wolff-Parkinson-White syndrome in Olmsted County, Minnesota, 1953–1989. Circulation 1993; 87(3):866–873. pmid:8443907
- Carlson AM, Turek JW, Law IH, Von Bergen NH. Pseudo-preexcitation is prevalent among patients with repaired complex congenital heart disease. Pediatr Cardiol.2015; 36(1):8–13. doi:10.1007/s00246-014-0955-x
- Aleong RG, Singh SM, Levinson JR, Milan DJ. Catecholamine challenge unmasking high-risk features in the Wolff-Parkinson-White syndrome. Europace 2009; 11(10):1396–1398. doi:10.1093/europace/eup211
- Sahu S, Karna ST, Karna A, Lata I, Kapoor D. Anaesthetic management of Wolff-Parkinson-White syndrome for hysterectomy. Indian J Anaesth 2011; 55(4):378–380. doi:10.4103/0019-5049.84866
- Tseng ZH, Yadav AV, Scheinman MM. Catecholamine dependent accessory pathway automaticity. Pacing Clin Electrophysiol 2004; 27(7):1005–1007. doi:10.1111/j.1540-8159.2004.00574.x
- Obeyesekere MN, Leong-Sit P, Massel D, et al. Risk of arrhythmia and sudden death in patients with asymptomatic preexcitation: a meta-analysis. Circulation 2012; 125(19):2308–2315. doi:10.1161/CIRCULATIONAHA.111.055350
- Munger TM, Packer DL, Hammill SC, et al. A population study of the natural history of Wolff-Parkinson-White syndrome in Olmsted County, Minnesota, 1953–1989. Circulation 1993; 87(3):866–873. pmid:8443907
- Carlson AM, Turek JW, Law IH, Von Bergen NH. Pseudo-preexcitation is prevalent among patients with repaired complex congenital heart disease. Pediatr Cardiol.2015; 36(1):8–13. doi:10.1007/s00246-014-0955-x
- Aleong RG, Singh SM, Levinson JR, Milan DJ. Catecholamine challenge unmasking high-risk features in the Wolff-Parkinson-White syndrome. Europace 2009; 11(10):1396–1398. doi:10.1093/europace/eup211
- Sahu S, Karna ST, Karna A, Lata I, Kapoor D. Anaesthetic management of Wolff-Parkinson-White syndrome for hysterectomy. Indian J Anaesth 2011; 55(4):378–380. doi:10.4103/0019-5049.84866
- Tseng ZH, Yadav AV, Scheinman MM. Catecholamine dependent accessory pathway automaticity. Pacing Clin Electrophysiol 2004; 27(7):1005–1007. doi:10.1111/j.1540-8159.2004.00574.x
- Obeyesekere MN, Leong-Sit P, Massel D, et al. Risk of arrhythmia and sudden death in patients with asymptomatic preexcitation: a meta-analysis. Circulation 2012; 125(19):2308–2315. doi:10.1161/CIRCULATIONAHA.111.055350
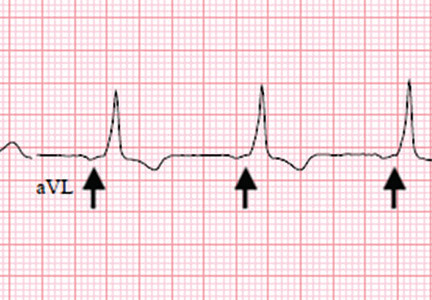
When the Poisoned Risk Poisoning Others: Fatal Sodium Azide Overdose
Case
A 24-year-old man in cardiac arrest was brought to the ED via emergency medical services (EMS). Unfortunately, resuscitation efforts were unsuccessful. Little was known about the patient, but the emergency physician was informed that the patient had ingested sodium azide (NaN3), which he had ordered online. The patient collapsed shortly after ingesting the sodium azide, approximately the same time police officers arrived at the patient’s home.
No specific details were known about the patient’s ingestion. Upon learning of the exposure to sodium azide, a member of the ED staff contacted the local poison control center for information on the proper course of action to ensure staff safety and limit exposure. Shortly thereafter, several of emergency medical technicians and police officers, who had responded to the emergency assistance call for this patient, presented to the ED with concerns of exposure.
What is sodium azide?
Sodium azide is a colorless, odorless crystalline water-soluble solid that has a pK of 4.8.1 When sodium azide is dissolved in an acid, it liberates hydrazoic acid (HN3), which has a pungent odor, high vapor pressure (484 mm Hg), and a relatively low-boiling point of 37°C (98°F).2
The most common industrial use of sodium azide is as a propellant in air bags. In this capacity, sodium azide rapidly decomposes to nitrogen gas when it reaches a temperature of 300°C (572°F), causing rapid expansion of the air bag. In addition to air bags, sodium azide is used in research laboratories as a preservative and in agriculture as a pesticide. The main nontoxicological concern with all azide agents is the potential for explosion when they react with metals, such as lead, copper, silver, and mercury, to form metal azides that are sensitive to shock.3 An example of the explosive nature of these azides was demonstrated in a report wherein diluted sodium azide was poured down a drain, causing an explosion as a worker was fixing the pipe.4
In addition to industrial and commercial use, sodium azide is occasionally used in suicide attempts because it is rapidly fatal, has no specific antidote, and can be purchased online.3
What is the toxicity of sodium azide?
The lethal dose for both oral and dermal exposure to sodium azide is approximately 10 to 20 mg/kg.3,5 Therefore, ingestion of 700 mg of sodium azide, a volume approximately the size of a penny, is likely to be fatal.3
Sodium azide is primarily a mitochondrial toxin, which binds the electron transport chain, inhibiting oxidative phosphorylation. The resulting reduction in adenosine triphosphate (ATP) production, even in the presence of oxygen, results in metabolic failure.6 This mechanism of action is similar to that of cyanide, although sodium azide causes more pronounced vasodilation due to the in vivo conversion of some azide to the vasodilator nitric oxide.7 Some reports suggest that azide lethality is due to enhanced excitatory transmission from nitric oxide in the central nervous system.8
What are the clinical manifestations of azide poisoning, and what is the treatment?
The early clinical findings of a patient with azide poisoning include hypotension, dizziness, headache, nausea, vomiting, palpitations, tachycardia, dyspnea, and restlessness. Inhalation of hydrazoic acid can also produce wheezing and coughing. The most common effect is hypotension, which can occur within 1 minute of exposure. Following depletion of cellular ATP, anaerobic glycolysis generates lactate and produces acidemia. More severe findings of azide poisoning include seizures, cardiac arrhythmia, loss of consciousness, pulmonary edema, and cardiopulmonary failure.3
Currently, there is no specific antidote for azide poisoning, and treatment mainly consists of supportive care. Cyanide antidote treatments are generally ineffective in reducing azide-related death in animal models.3,8Early aggressive supportive care can improve survival rates.9 Some authors suggest that administration of oral activated charcoal, orogastric lavage, hemodialysis, and plasma exchange reduce azide concentrations, while others believe these treatments have little effect.3,9 More research is needed to identify effective therapeutic measures and to control for dose, time, and patient population.
What are the safety concerns for emergency medical technicians and hospital staff following exposure to sodium azide?
The most probable routes of exposure for prehospital and hospital staff include dermal contact with sodium azide or inhalation of gaseous hydrazoic acid; inhalational exposure is most concerning.1 In one case, hospital-staff members developed headaches, light-headedness, and nausea while treating a patient for azide poisoning; however, staff exposure was not confirmed and no sequelae were evident.10
More objectively, workers at an azide plant exposed to azide concentrations above the occupational exposure limit developed headaches, hypotension, and palpitations.11 Another study found no evidence of kidney, heart, or liver damage after patients were given sodium azide for more than a year during a clinical trial.12 Not unexpectedly, there is little risk of exposure when proper safety precautions are taken.
Emergency response personnel should carefully inspect the scene for the presence of any sodium azide powder, and should also question bystanders and family members to determine if anyone performed mouth-to-mouth resuscitation on the patient. Standard universal precautions, along with attentiveness to one’s surroundings, should be sufficient to prevent dermal exposure. If small amounts of sodium azide residue are found on the patient, his or her clothes should be cautiously removed and placed in a plastic bag to prevent dispersion of particles. If large quantities of sodium azide are present on a patient, the hazardous materials response team should be called, in accordance with institutional and regional protocols. To avoid explosion, every attempt should be made to prevent azide salt (eg, from emesis) from contact with any metal surfaces (eg, oxygen tanks, metal stretcher).13Vomit from patients who have ingested sodium azide can cause liberation of hydrazoic acid, which can escape through the esophagus. A pungent ambient odor may provide a warning, which is particularly concerning in a confined space such as an ambulance. As a precaution, EMS personnel should open windows and maximize ventilation. After the call, EMS and hospital personnel should thoroughly wash their hands with soap and water, and change their uniform if they believe it has been contaminated. There is no risk of delayed exposure following exposure to hydrazoic acid.
During autopsy, medical examiners must exercise caution due to the potential for liberation of hydrazoic acids from the stomach.14Unless it is absolutely necessary, the medical examiner should avoid opening the stomach. If this is unavoidable, the autopsy should occur in a well-ventilated setting with the examiner wearing a supplied air respirator to limit exposure in a high-risk scenario.
Case Conclusion
None of the exposed first responders experienced dizziness, light-headedness, or irritation, and after a period of observation in the ED, they were discharged home without further sequelae. All hospital staff involved in the patient’s care, including those who performed cardiopulmonary resuscitation on the patient and cleaned his room, were advised to use protective equipment when handling the patient and bodily secretions. None of the health care workers developed abnormal clinical findings. Given the hazard in conducting a full postmortem examination, the medical examiner opted to send blood, bile, urine, and vitreous humor out for analysis, but did not conduct a full postmortem examination. Notably, the stomach was not opened, and its contents were not exposed.
1. Compound summary for CID 33557 (sodium azide). National Center for Biotechnology Information. PubChem Compound Database. https://pubchem.ncbi.nlm.nih.gov/compound/sodium_azide. Accessed May 10, 2018.
2. Compound summary for CID 24530 (hydrogen azide). National Center for Biotechnology Information. PubChem Compound Database. https://pubchem.ncbi.nlm.nih.gov/compound/hydrazoic_acid. Accessed May 10, 2018.
3. Chang S, Lamm SH. Human health effects of sodium azide exposure: a literature review and analysis. Int J Toxicol. 2003;22(3):175-186. doi:10.1080/10915810305109.
4. Sodium azide explosion hazard. Washington State Department of Labor & Industries. Division of Occupational Safety and Health. https://www.lni.wa.gov/safety/hazardalerts/SodiumAzide.pdf. August 11, 2011. Accessed May 10, 2018.
5. Safety data sheet: sodium azide. ThermoFischer Scientific. https://www.fishersci.com/store/msds?partNumber=S227I1&productDescription=SODIUM+AZIDE+GRAN+PURIF+1+KG&vendorId=VN00033897&countryCode=US&language=en. Updated January 17, 2018. Accessed May 10, 2018.
6. Bogucka K, Wojtczak L. Effect of sodium azide on oxidation and phosphorylation processes in rat-liver mitochondria. Biochim Biophys Acta. 1966;122(3):381-392. doi:10.1016/0926-6593(66)90031-2.
7. Kruszyna H, Kruszyna R, Smith RP, Wilcox DE. Red blood cells generate nitric oxide from directly acting, nitrogenous vasodilators. Toxicol Appl Pharmacol. 1987;91(3):429-438. doi:10.1016/0041-008x(87)90064-0.
8. Smith RP, Louis CA, Kruszyna R, Kruszyna H. Acute neurotoxicity of sodium azide and nitric oxide. Fundam Appl Toxicol. 1991;17(1):120-127. doi:10.1093/toxsci/17.1.120.
9. Watanabe K, Hirasawa H, Oda S, et al. A case of survival following high-dose sodium azide poisoning. Clin Toxicol (Phila). 2007;45(7):810-811.
10. Abrams J, el-Mallakh RS, Meyer R. Suicidal sodium azide ingestion. Ann Emerg Med. 1987;16(12):1378-1380. doi:10.1016/s0196-0644(87)80423-7
11. Trout D, Esswein EJ, Hales T, Brown K, Solomon G, Miller M. Exposures and health effects: an evaluation of workers at a sodium azide production plant. Am J Ind Med. 1996;30(3):343-350.
12. Black, MM, Zweifach BW, Speer FD. Comparison of hypotensive action of sodium azide in normotensive and hypertensive patients. Exper Biol Med. 1954;85(1):11-16. doi:10.3181/00379727-85-20770.
13. Emergency preparedness and response. Facts about sodium azide. Centers for Disease Control and Prevention. Office of Public Health Preparedness and Response. https://emergency.cdc.gov/agent/sodiumazide/basics/facts.asp. Updated April 10, 2018. Accessed May 10, 2018.
14. Le Blanc-Louvry I, Laburthe-Tolra P, Massol V, et al. Suicidal sodium azide intoxication: An analytical challenge based on a rare case. Forensic Sci Int. 2012;221(1-3):e17-20. doi:10.1016/j.forsciint.2012.04.006.
Case
A 24-year-old man in cardiac arrest was brought to the ED via emergency medical services (EMS). Unfortunately, resuscitation efforts were unsuccessful. Little was known about the patient, but the emergency physician was informed that the patient had ingested sodium azide (NaN3), which he had ordered online. The patient collapsed shortly after ingesting the sodium azide, approximately the same time police officers arrived at the patient’s home.
No specific details were known about the patient’s ingestion. Upon learning of the exposure to sodium azide, a member of the ED staff contacted the local poison control center for information on the proper course of action to ensure staff safety and limit exposure. Shortly thereafter, several of emergency medical technicians and police officers, who had responded to the emergency assistance call for this patient, presented to the ED with concerns of exposure.
What is sodium azide?
Sodium azide is a colorless, odorless crystalline water-soluble solid that has a pK of 4.8.1 When sodium azide is dissolved in an acid, it liberates hydrazoic acid (HN3), which has a pungent odor, high vapor pressure (484 mm Hg), and a relatively low-boiling point of 37°C (98°F).2
The most common industrial use of sodium azide is as a propellant in air bags. In this capacity, sodium azide rapidly decomposes to nitrogen gas when it reaches a temperature of 300°C (572°F), causing rapid expansion of the air bag. In addition to air bags, sodium azide is used in research laboratories as a preservative and in agriculture as a pesticide. The main nontoxicological concern with all azide agents is the potential for explosion when they react with metals, such as lead, copper, silver, and mercury, to form metal azides that are sensitive to shock.3 An example of the explosive nature of these azides was demonstrated in a report wherein diluted sodium azide was poured down a drain, causing an explosion as a worker was fixing the pipe.4
In addition to industrial and commercial use, sodium azide is occasionally used in suicide attempts because it is rapidly fatal, has no specific antidote, and can be purchased online.3
What is the toxicity of sodium azide?
The lethal dose for both oral and dermal exposure to sodium azide is approximately 10 to 20 mg/kg.3,5 Therefore, ingestion of 700 mg of sodium azide, a volume approximately the size of a penny, is likely to be fatal.3
Sodium azide is primarily a mitochondrial toxin, which binds the electron transport chain, inhibiting oxidative phosphorylation. The resulting reduction in adenosine triphosphate (ATP) production, even in the presence of oxygen, results in metabolic failure.6 This mechanism of action is similar to that of cyanide, although sodium azide causes more pronounced vasodilation due to the in vivo conversion of some azide to the vasodilator nitric oxide.7 Some reports suggest that azide lethality is due to enhanced excitatory transmission from nitric oxide in the central nervous system.8
What are the clinical manifestations of azide poisoning, and what is the treatment?
The early clinical findings of a patient with azide poisoning include hypotension, dizziness, headache, nausea, vomiting, palpitations, tachycardia, dyspnea, and restlessness. Inhalation of hydrazoic acid can also produce wheezing and coughing. The most common effect is hypotension, which can occur within 1 minute of exposure. Following depletion of cellular ATP, anaerobic glycolysis generates lactate and produces acidemia. More severe findings of azide poisoning include seizures, cardiac arrhythmia, loss of consciousness, pulmonary edema, and cardiopulmonary failure.3
Currently, there is no specific antidote for azide poisoning, and treatment mainly consists of supportive care. Cyanide antidote treatments are generally ineffective in reducing azide-related death in animal models.3,8Early aggressive supportive care can improve survival rates.9 Some authors suggest that administration of oral activated charcoal, orogastric lavage, hemodialysis, and plasma exchange reduce azide concentrations, while others believe these treatments have little effect.3,9 More research is needed to identify effective therapeutic measures and to control for dose, time, and patient population.
What are the safety concerns for emergency medical technicians and hospital staff following exposure to sodium azide?
The most probable routes of exposure for prehospital and hospital staff include dermal contact with sodium azide or inhalation of gaseous hydrazoic acid; inhalational exposure is most concerning.1 In one case, hospital-staff members developed headaches, light-headedness, and nausea while treating a patient for azide poisoning; however, staff exposure was not confirmed and no sequelae were evident.10
More objectively, workers at an azide plant exposed to azide concentrations above the occupational exposure limit developed headaches, hypotension, and palpitations.11 Another study found no evidence of kidney, heart, or liver damage after patients were given sodium azide for more than a year during a clinical trial.12 Not unexpectedly, there is little risk of exposure when proper safety precautions are taken.
Emergency response personnel should carefully inspect the scene for the presence of any sodium azide powder, and should also question bystanders and family members to determine if anyone performed mouth-to-mouth resuscitation on the patient. Standard universal precautions, along with attentiveness to one’s surroundings, should be sufficient to prevent dermal exposure. If small amounts of sodium azide residue are found on the patient, his or her clothes should be cautiously removed and placed in a plastic bag to prevent dispersion of particles. If large quantities of sodium azide are present on a patient, the hazardous materials response team should be called, in accordance with institutional and regional protocols. To avoid explosion, every attempt should be made to prevent azide salt (eg, from emesis) from contact with any metal surfaces (eg, oxygen tanks, metal stretcher).13Vomit from patients who have ingested sodium azide can cause liberation of hydrazoic acid, which can escape through the esophagus. A pungent ambient odor may provide a warning, which is particularly concerning in a confined space such as an ambulance. As a precaution, EMS personnel should open windows and maximize ventilation. After the call, EMS and hospital personnel should thoroughly wash their hands with soap and water, and change their uniform if they believe it has been contaminated. There is no risk of delayed exposure following exposure to hydrazoic acid.
During autopsy, medical examiners must exercise caution due to the potential for liberation of hydrazoic acids from the stomach.14Unless it is absolutely necessary, the medical examiner should avoid opening the stomach. If this is unavoidable, the autopsy should occur in a well-ventilated setting with the examiner wearing a supplied air respirator to limit exposure in a high-risk scenario.
Case Conclusion
None of the exposed first responders experienced dizziness, light-headedness, or irritation, and after a period of observation in the ED, they were discharged home without further sequelae. All hospital staff involved in the patient’s care, including those who performed cardiopulmonary resuscitation on the patient and cleaned his room, were advised to use protective equipment when handling the patient and bodily secretions. None of the health care workers developed abnormal clinical findings. Given the hazard in conducting a full postmortem examination, the medical examiner opted to send blood, bile, urine, and vitreous humor out for analysis, but did not conduct a full postmortem examination. Notably, the stomach was not opened, and its contents were not exposed.
Case
A 24-year-old man in cardiac arrest was brought to the ED via emergency medical services (EMS). Unfortunately, resuscitation efforts were unsuccessful. Little was known about the patient, but the emergency physician was informed that the patient had ingested sodium azide (NaN3), which he had ordered online. The patient collapsed shortly after ingesting the sodium azide, approximately the same time police officers arrived at the patient’s home.
No specific details were known about the patient’s ingestion. Upon learning of the exposure to sodium azide, a member of the ED staff contacted the local poison control center for information on the proper course of action to ensure staff safety and limit exposure. Shortly thereafter, several of emergency medical technicians and police officers, who had responded to the emergency assistance call for this patient, presented to the ED with concerns of exposure.
What is sodium azide?
Sodium azide is a colorless, odorless crystalline water-soluble solid that has a pK of 4.8.1 When sodium azide is dissolved in an acid, it liberates hydrazoic acid (HN3), which has a pungent odor, high vapor pressure (484 mm Hg), and a relatively low-boiling point of 37°C (98°F).2
The most common industrial use of sodium azide is as a propellant in air bags. In this capacity, sodium azide rapidly decomposes to nitrogen gas when it reaches a temperature of 300°C (572°F), causing rapid expansion of the air bag. In addition to air bags, sodium azide is used in research laboratories as a preservative and in agriculture as a pesticide. The main nontoxicological concern with all azide agents is the potential for explosion when they react with metals, such as lead, copper, silver, and mercury, to form metal azides that are sensitive to shock.3 An example of the explosive nature of these azides was demonstrated in a report wherein diluted sodium azide was poured down a drain, causing an explosion as a worker was fixing the pipe.4
In addition to industrial and commercial use, sodium azide is occasionally used in suicide attempts because it is rapidly fatal, has no specific antidote, and can be purchased online.3
What is the toxicity of sodium azide?
The lethal dose for both oral and dermal exposure to sodium azide is approximately 10 to 20 mg/kg.3,5 Therefore, ingestion of 700 mg of sodium azide, a volume approximately the size of a penny, is likely to be fatal.3
Sodium azide is primarily a mitochondrial toxin, which binds the electron transport chain, inhibiting oxidative phosphorylation. The resulting reduction in adenosine triphosphate (ATP) production, even in the presence of oxygen, results in metabolic failure.6 This mechanism of action is similar to that of cyanide, although sodium azide causes more pronounced vasodilation due to the in vivo conversion of some azide to the vasodilator nitric oxide.7 Some reports suggest that azide lethality is due to enhanced excitatory transmission from nitric oxide in the central nervous system.8
What are the clinical manifestations of azide poisoning, and what is the treatment?
The early clinical findings of a patient with azide poisoning include hypotension, dizziness, headache, nausea, vomiting, palpitations, tachycardia, dyspnea, and restlessness. Inhalation of hydrazoic acid can also produce wheezing and coughing. The most common effect is hypotension, which can occur within 1 minute of exposure. Following depletion of cellular ATP, anaerobic glycolysis generates lactate and produces acidemia. More severe findings of azide poisoning include seizures, cardiac arrhythmia, loss of consciousness, pulmonary edema, and cardiopulmonary failure.3
Currently, there is no specific antidote for azide poisoning, and treatment mainly consists of supportive care. Cyanide antidote treatments are generally ineffective in reducing azide-related death in animal models.3,8Early aggressive supportive care can improve survival rates.9 Some authors suggest that administration of oral activated charcoal, orogastric lavage, hemodialysis, and plasma exchange reduce azide concentrations, while others believe these treatments have little effect.3,9 More research is needed to identify effective therapeutic measures and to control for dose, time, and patient population.
What are the safety concerns for emergency medical technicians and hospital staff following exposure to sodium azide?
The most probable routes of exposure for prehospital and hospital staff include dermal contact with sodium azide or inhalation of gaseous hydrazoic acid; inhalational exposure is most concerning.1 In one case, hospital-staff members developed headaches, light-headedness, and nausea while treating a patient for azide poisoning; however, staff exposure was not confirmed and no sequelae were evident.10
More objectively, workers at an azide plant exposed to azide concentrations above the occupational exposure limit developed headaches, hypotension, and palpitations.11 Another study found no evidence of kidney, heart, or liver damage after patients were given sodium azide for more than a year during a clinical trial.12 Not unexpectedly, there is little risk of exposure when proper safety precautions are taken.
Emergency response personnel should carefully inspect the scene for the presence of any sodium azide powder, and should also question bystanders and family members to determine if anyone performed mouth-to-mouth resuscitation on the patient. Standard universal precautions, along with attentiveness to one’s surroundings, should be sufficient to prevent dermal exposure. If small amounts of sodium azide residue are found on the patient, his or her clothes should be cautiously removed and placed in a plastic bag to prevent dispersion of particles. If large quantities of sodium azide are present on a patient, the hazardous materials response team should be called, in accordance with institutional and regional protocols. To avoid explosion, every attempt should be made to prevent azide salt (eg, from emesis) from contact with any metal surfaces (eg, oxygen tanks, metal stretcher).13Vomit from patients who have ingested sodium azide can cause liberation of hydrazoic acid, which can escape through the esophagus. A pungent ambient odor may provide a warning, which is particularly concerning in a confined space such as an ambulance. As a precaution, EMS personnel should open windows and maximize ventilation. After the call, EMS and hospital personnel should thoroughly wash their hands with soap and water, and change their uniform if they believe it has been contaminated. There is no risk of delayed exposure following exposure to hydrazoic acid.
During autopsy, medical examiners must exercise caution due to the potential for liberation of hydrazoic acids from the stomach.14Unless it is absolutely necessary, the medical examiner should avoid opening the stomach. If this is unavoidable, the autopsy should occur in a well-ventilated setting with the examiner wearing a supplied air respirator to limit exposure in a high-risk scenario.
Case Conclusion
None of the exposed first responders experienced dizziness, light-headedness, or irritation, and after a period of observation in the ED, they were discharged home without further sequelae. All hospital staff involved in the patient’s care, including those who performed cardiopulmonary resuscitation on the patient and cleaned his room, were advised to use protective equipment when handling the patient and bodily secretions. None of the health care workers developed abnormal clinical findings. Given the hazard in conducting a full postmortem examination, the medical examiner opted to send blood, bile, urine, and vitreous humor out for analysis, but did not conduct a full postmortem examination. Notably, the stomach was not opened, and its contents were not exposed.
1. Compound summary for CID 33557 (sodium azide). National Center for Biotechnology Information. PubChem Compound Database. https://pubchem.ncbi.nlm.nih.gov/compound/sodium_azide. Accessed May 10, 2018.
2. Compound summary for CID 24530 (hydrogen azide). National Center for Biotechnology Information. PubChem Compound Database. https://pubchem.ncbi.nlm.nih.gov/compound/hydrazoic_acid. Accessed May 10, 2018.
3. Chang S, Lamm SH. Human health effects of sodium azide exposure: a literature review and analysis. Int J Toxicol. 2003;22(3):175-186. doi:10.1080/10915810305109.
4. Sodium azide explosion hazard. Washington State Department of Labor & Industries. Division of Occupational Safety and Health. https://www.lni.wa.gov/safety/hazardalerts/SodiumAzide.pdf. August 11, 2011. Accessed May 10, 2018.
5. Safety data sheet: sodium azide. ThermoFischer Scientific. https://www.fishersci.com/store/msds?partNumber=S227I1&productDescription=SODIUM+AZIDE+GRAN+PURIF+1+KG&vendorId=VN00033897&countryCode=US&language=en. Updated January 17, 2018. Accessed May 10, 2018.
6. Bogucka K, Wojtczak L. Effect of sodium azide on oxidation and phosphorylation processes in rat-liver mitochondria. Biochim Biophys Acta. 1966;122(3):381-392. doi:10.1016/0926-6593(66)90031-2.
7. Kruszyna H, Kruszyna R, Smith RP, Wilcox DE. Red blood cells generate nitric oxide from directly acting, nitrogenous vasodilators. Toxicol Appl Pharmacol. 1987;91(3):429-438. doi:10.1016/0041-008x(87)90064-0.
8. Smith RP, Louis CA, Kruszyna R, Kruszyna H. Acute neurotoxicity of sodium azide and nitric oxide. Fundam Appl Toxicol. 1991;17(1):120-127. doi:10.1093/toxsci/17.1.120.
9. Watanabe K, Hirasawa H, Oda S, et al. A case of survival following high-dose sodium azide poisoning. Clin Toxicol (Phila). 2007;45(7):810-811.
10. Abrams J, el-Mallakh RS, Meyer R. Suicidal sodium azide ingestion. Ann Emerg Med. 1987;16(12):1378-1380. doi:10.1016/s0196-0644(87)80423-7
11. Trout D, Esswein EJ, Hales T, Brown K, Solomon G, Miller M. Exposures and health effects: an evaluation of workers at a sodium azide production plant. Am J Ind Med. 1996;30(3):343-350.
12. Black, MM, Zweifach BW, Speer FD. Comparison of hypotensive action of sodium azide in normotensive and hypertensive patients. Exper Biol Med. 1954;85(1):11-16. doi:10.3181/00379727-85-20770.
13. Emergency preparedness and response. Facts about sodium azide. Centers for Disease Control and Prevention. Office of Public Health Preparedness and Response. https://emergency.cdc.gov/agent/sodiumazide/basics/facts.asp. Updated April 10, 2018. Accessed May 10, 2018.
14. Le Blanc-Louvry I, Laburthe-Tolra P, Massol V, et al. Suicidal sodium azide intoxication: An analytical challenge based on a rare case. Forensic Sci Int. 2012;221(1-3):e17-20. doi:10.1016/j.forsciint.2012.04.006.
1. Compound summary for CID 33557 (sodium azide). National Center for Biotechnology Information. PubChem Compound Database. https://pubchem.ncbi.nlm.nih.gov/compound/sodium_azide. Accessed May 10, 2018.
2. Compound summary for CID 24530 (hydrogen azide). National Center for Biotechnology Information. PubChem Compound Database. https://pubchem.ncbi.nlm.nih.gov/compound/hydrazoic_acid. Accessed May 10, 2018.
3. Chang S, Lamm SH. Human health effects of sodium azide exposure: a literature review and analysis. Int J Toxicol. 2003;22(3):175-186. doi:10.1080/10915810305109.
4. Sodium azide explosion hazard. Washington State Department of Labor & Industries. Division of Occupational Safety and Health. https://www.lni.wa.gov/safety/hazardalerts/SodiumAzide.pdf. August 11, 2011. Accessed May 10, 2018.
5. Safety data sheet: sodium azide. ThermoFischer Scientific. https://www.fishersci.com/store/msds?partNumber=S227I1&productDescription=SODIUM+AZIDE+GRAN+PURIF+1+KG&vendorId=VN00033897&countryCode=US&language=en. Updated January 17, 2018. Accessed May 10, 2018.
6. Bogucka K, Wojtczak L. Effect of sodium azide on oxidation and phosphorylation processes in rat-liver mitochondria. Biochim Biophys Acta. 1966;122(3):381-392. doi:10.1016/0926-6593(66)90031-2.
7. Kruszyna H, Kruszyna R, Smith RP, Wilcox DE. Red blood cells generate nitric oxide from directly acting, nitrogenous vasodilators. Toxicol Appl Pharmacol. 1987;91(3):429-438. doi:10.1016/0041-008x(87)90064-0.
8. Smith RP, Louis CA, Kruszyna R, Kruszyna H. Acute neurotoxicity of sodium azide and nitric oxide. Fundam Appl Toxicol. 1991;17(1):120-127. doi:10.1093/toxsci/17.1.120.
9. Watanabe K, Hirasawa H, Oda S, et al. A case of survival following high-dose sodium azide poisoning. Clin Toxicol (Phila). 2007;45(7):810-811.
10. Abrams J, el-Mallakh RS, Meyer R. Suicidal sodium azide ingestion. Ann Emerg Med. 1987;16(12):1378-1380. doi:10.1016/s0196-0644(87)80423-7
11. Trout D, Esswein EJ, Hales T, Brown K, Solomon G, Miller M. Exposures and health effects: an evaluation of workers at a sodium azide production plant. Am J Ind Med. 1996;30(3):343-350.
12. Black, MM, Zweifach BW, Speer FD. Comparison of hypotensive action of sodium azide in normotensive and hypertensive patients. Exper Biol Med. 1954;85(1):11-16. doi:10.3181/00379727-85-20770.
13. Emergency preparedness and response. Facts about sodium azide. Centers for Disease Control and Prevention. Office of Public Health Preparedness and Response. https://emergency.cdc.gov/agent/sodiumazide/basics/facts.asp. Updated April 10, 2018. Accessed May 10, 2018.
14. Le Blanc-Louvry I, Laburthe-Tolra P, Massol V, et al. Suicidal sodium azide intoxication: An analytical challenge based on a rare case. Forensic Sci Int. 2012;221(1-3):e17-20. doi:10.1016/j.forsciint.2012.04.006.
Getting Ahead of the Pain

ANSWER
The image reveals a hypodense extra-axial fluid collection in the right frontoparietal region, measuring 8 to 10 mm in diameter. There is some mass effect and evidence of right-to-left shift. These findings are consistent with a subacute subdural hematoma, possibly secondary to the patient’s anticoagulant use. (The patient later recalled bumping his head a couple of months prior—but that may have been incidental.)
Arrangements were made for him at a local hospital where neurosurgical services were available. He underwent successful evacuation and was subsequently symptom free.
ANSWER
The image reveals a hypodense extra-axial fluid collection in the right frontoparietal region, measuring 8 to 10 mm in diameter. There is some mass effect and evidence of right-to-left shift. These findings are consistent with a subacute subdural hematoma, possibly secondary to the patient’s anticoagulant use. (The patient later recalled bumping his head a couple of months prior—but that may have been incidental.)
Arrangements were made for him at a local hospital where neurosurgical services were available. He underwent successful evacuation and was subsequently symptom free.
ANSWER
The image reveals a hypodense extra-axial fluid collection in the right frontoparietal region, measuring 8 to 10 mm in diameter. There is some mass effect and evidence of right-to-left shift. These findings are consistent with a subacute subdural hematoma, possibly secondary to the patient’s anticoagulant use. (The patient later recalled bumping his head a couple of months prior—but that may have been incidental.)
Arrangements were made for him at a local hospital where neurosurgical services were available. He underwent successful evacuation and was subsequently symptom free.

An 80-year-old man presents to urgent care for intermittent severe headaches. The pain is reportedly bifrontal, slightly worse on the right side than the left. He denies any recent injury or trauma, as well as symptoms including fever, chills, nausea, vomiting, and visual disturbance.
His medical history is significant for hypertension and hyperlipidemia. His current medications include prasugrel and aspirin.
On examination, you note an elderly male who is awake, alert, and oriented x 3. His vital signs are normal. His physical exam is overall normal, with no focal findings or neurologic deficits.
Noncontrast CT of the head is obtained at a local hospital. As you review the images, you see the following cut (shown). What is your impression?

Electrocardiography: Flecainide Toxicity
Case
An 86-year-old woman, who recently had been seen in the same facility after a ground level fall, presented to the ED with to a 2- to 3-day history of vague abdominal pain, increasing weakness, nausea, and dry heaves.
Upon examination, the patient was unable to stand due to generalized weakness She arrived at the ED via emergency medical services. Her vital signs at presentation were significant for a systolic blood pressure (BP) of 90 mm Hg with a wide complex tachycardia concerning for ventricular tachycardia. The patient’s other vital signers were: heart rate, 136 beats/min; respiratory rate 20 breaths/min; and pulse oximetry was 94% on 4 liters/min of oxygen via nasal cannula.
The patient’s medical history was significant for atrial fibrillation and an indwelling pacemaker, for which she was chronically on flecai
The initial electrocardiogram (ECG) revealed a wide complex rhythm with pacemaker spikes (Figure 1). Based on these findings, electrodes were placed on the patient in the event she required cardioversion. The patient was started on an amiodarone intravenous (IV) drip for presumptive ventricular tachycardia.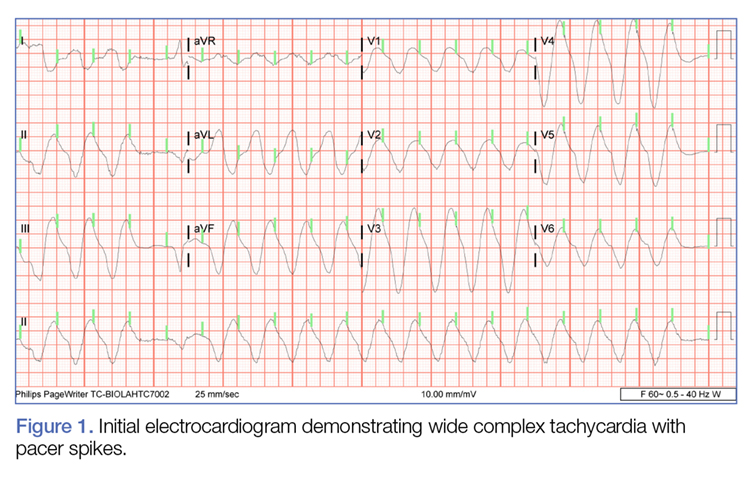
During the patient’s evaluation in the ED, she experienced transient drops in BP, which were responsive to an IV fluid bolus of normal saline, and the amiodarone drip was discontinued. The patient’s ECG findings were compared to previous ECG studies, as was her current medication list and prior health issues. After ruling-out other causes, flecainide toxicity was considered high in the differential, and she was given 1 ampule of bicarbonate IV, after which a second ECG showed heart rhythm converted from a wide-complex tachycardia to a paced rhythm, markedly improved from the initial ECG (Figure 2). Similarly, there was a marked improvement in BP.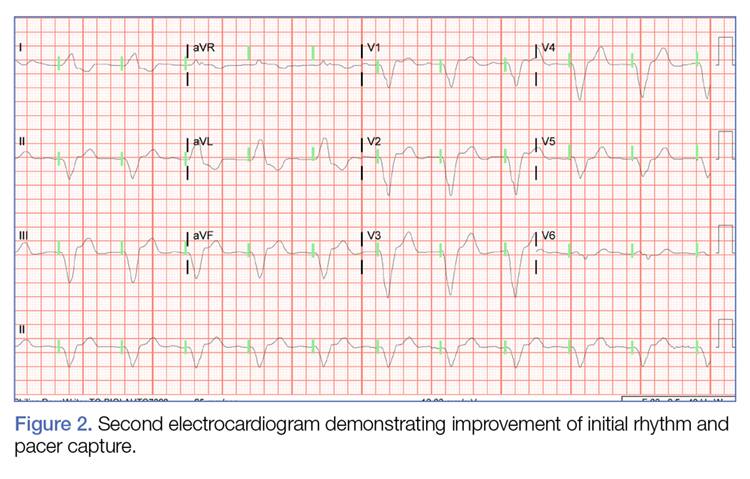
An interrogation of the patient’s pacemaker revealed an atrial flutter with a rate below detection for mode switch, with one-to-one tracking/pacing. The pacemaker was reprogrammed to divide the DDIR mode with detection rate at 120 mm Hg with mode switch activated. This was felt to be consistent with flecainide toxicity precipitating the cardiac conduction issues.
Laboratory studies showed an elevated flecainide level at 1.39 mcg/mL (upper limits of normal of 1 mcg/mL). Other studies showed worsening congestive heart failure, with a brain natriuretic peptide of 8,057 pg/mL and mild dehydration, with serum creatinine increased from her baseline of 0.9 to 1.38 mg/dL.
The patient’s abdominal pain was further evaluated and she was found to have acute cholecystitis. She was admitted to the intensive care unit with cardiology and general surgery consulting.
Discussion
Flecainide acetate was approved by the Food and Drug Administration in 1984.1It is a Vaughan-Williams class IC antiarrhythmic with a sodium channel blocker action used to treat supra ventricular arrhythmias. The CAST trial in 1989 investigated the efficacy of this class of antiarrhythmics, which resulted in a revision of its role.2 Based on this study, flecainide is not recommended for patients with structural heart disease or coronary artery disease.2,3 However, it is recommended as a first-line therapy for pharmacologic cardioversion and maintenance of normal sinus rhythm in patients with atrial fibrillation and supraventricular tachycardia4,5 without the above caveats.
Class IC agents produce a selective block at the sodium (Na+) channels, resulting in the slowing of cardiac conduction.6,7 This high affinity for Na+ channels combined with slow unbinding kinetics during diastole explain the slowing of recovery time and prolongation of the refractory period.6,8,9 These electrophysiologic properties all can increase the PR, QRS, and QT interval duration. The QT interval is not significantly affected, as most of the QT prolongation is due to the QRS widening.6,10,11 Widening of the QRS by greater than 25% as compared to the baseline value is used as the threshold to decrease dosing or discontinue the use of flecainide.3The toxic effects of flecainide on cardiac conduction can produce prolonged QRS duration of up to 50%, and PR interval up to 30%, especially in rapid heart rates. Signs of intoxication are difficult to discern owing to its nonspecific presentation. A well-documented, but under-recognized, presentation of flecainide toxicity is the transformation of atrial fibrillation to atrial flutter.5,7,9,11-13 The reported rate of this pro arrhythmic effect can be as high as 3.5% to 5%.14,15Flecainide toxicity can occur secondary to chronic ingestion and may be precipitated in mild renal failure. The majority of flecainide is renally excreted and the half-life is 20 hours. Maximum therapeutic effect is seen between levels of 0.2 to 1 mcg/mL with levels greater than 0.7 to 1 mcg/mL associated with adverse effects.9 Systemic effects include dizziness and visual disturbances. A high degree of suspicion for flecainide toxicity is required when the patient’s initial presentation is nonspecific. In this circumstance, real-time bedside interrogation of the pacemaker is invaluable. Early diagnosis and treatment minimizes the risk for adverse sequelae, including death. Treatment includes increasing the excretion of flecainide, symptomatic support (including pacemaker placement, intravenous fat emulsion, or extracorporeal circulatory support) and administration of sodium bicarbonate, to transiently reverse the effect of the sodium channel blockade, in severe cases.15-17
1. Hudak JM, Banitt EH, Schmid JR. Discovery and development of flecainide. Am J Cardiol. 1984;53(5):17B-20B.
2. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629.
3. Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7(2):76-85. doi:10.4330/wjc.v7.i2.76.
4. Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719-2747. doi:10.1093/eurheartj/ehs253.
5. Courand PY, Sibellas F, Ranc S, Mullier A, Kirkorian G, Bonnefoy E. Arrhythmogenic effect of flecainide toxicity. Cardiol J. 2013;20:203-205. doi:10.5603/CJ.2013.0035.
6. Holmes B, Heel RC. Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29(1):1-33.
7. Taylor R, Gandhi MM, Lloyd G. Tachycardia due to atrial flutter with rapid 1:1 conduction following treatment of atrial fibrillation with flecainide. Br Med J. 2010;340:b4684.
8. Roden DM, Woosley RL. Drug therapy. Flecainide. N Engl J Med. 1986;315(1):36-41.
9. Levis JT. ECG diagnosis: flecainide toxicity. Perm J. 2012;16(4):53.
10. Hellestrand KJ, Bexton RS, Nathan AW, Spurrell RA, Camm AJ. Acute electrophysiological effects of flecainide acetate on cardiac conduction and refractoriness in man. Br Heart J. 1982;48(2):140-148.
11. Rognoni A, Bertolazzi M, Peron M, et al. Electrocardiographic changes in a rare case of flecainide poisoning: a case report. Cases J. 2009;2:9137. doi:10.1186/1757-1626-2-9137.
12. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of “class IC atrial flutter” in patients with resistant atrial fibrillation. Am J Cardiol. 1999;83(5):785-787, A10.
13. Kola S, Mahata I, Kocheril AG. A case of flecainide toxicity. EP Lab Digest. 2015;15(5).
14. Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117(2):141-150.
15. Lloyd T, Zimmerman J, Griffin GD. Irreversible third-degree heart block and pacemaker implant in a case of flecainide toxicity. Am J Emerg Med. 2013;31(9):1418.e1-e2. doi:10.1016/j.ajem.2013.04.025.
16. Corkeron MA, van Heerden PV, Newman SM, Dusci L. Extracorporeal circulatory support in near-fatal flecainide overdose. Anaesth Intensive Care. 1999;27(4):405-408.
17. Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87-e89. doi:10.1111/j.1540-8159.2012.03485.x.
Case
An 86-year-old woman, who recently had been seen in the same facility after a ground level fall, presented to the ED with to a 2- to 3-day history of vague abdominal pain, increasing weakness, nausea, and dry heaves.
Upon examination, the patient was unable to stand due to generalized weakness She arrived at the ED via emergency medical services. Her vital signs at presentation were significant for a systolic blood pressure (BP) of 90 mm Hg with a wide complex tachycardia concerning for ventricular tachycardia. The patient’s other vital signers were: heart rate, 136 beats/min; respiratory rate 20 breaths/min; and pulse oximetry was 94% on 4 liters/min of oxygen via nasal cannula.
The patient’s medical history was significant for atrial fibrillation and an indwelling pacemaker, for which she was chronically on flecai
The initial electrocardiogram (ECG) revealed a wide complex rhythm with pacemaker spikes (Figure 1). Based on these findings, electrodes were placed on the patient in the event she required cardioversion. The patient was started on an amiodarone intravenous (IV) drip for presumptive ventricular tachycardia.
During the patient’s evaluation in the ED, she experienced transient drops in BP, which were responsive to an IV fluid bolus of normal saline, and the amiodarone drip was discontinued. The patient’s ECG findings were compared to previous ECG studies, as was her current medication list and prior health issues. After ruling-out other causes, flecainide toxicity was considered high in the differential, and she was given 1 ampule of bicarbonate IV, after which a second ECG showed heart rhythm converted from a wide-complex tachycardia to a paced rhythm, markedly improved from the initial ECG (Figure 2). Similarly, there was a marked improvement in BP.
An interrogation of the patient’s pacemaker revealed an atrial flutter with a rate below detection for mode switch, with one-to-one tracking/pacing. The pacemaker was reprogrammed to divide the DDIR mode with detection rate at 120 mm Hg with mode switch activated. This was felt to be consistent with flecainide toxicity precipitating the cardiac conduction issues.
Laboratory studies showed an elevated flecainide level at 1.39 mcg/mL (upper limits of normal of 1 mcg/mL). Other studies showed worsening congestive heart failure, with a brain natriuretic peptide of 8,057 pg/mL and mild dehydration, with serum creatinine increased from her baseline of 0.9 to 1.38 mg/dL.
The patient’s abdominal pain was further evaluated and she was found to have acute cholecystitis. She was admitted to the intensive care unit with cardiology and general surgery consulting.
Discussion
Flecainide acetate was approved by the Food and Drug Administration in 1984.1It is a Vaughan-Williams class IC antiarrhythmic with a sodium channel blocker action used to treat supra ventricular arrhythmias. The CAST trial in 1989 investigated the efficacy of this class of antiarrhythmics, which resulted in a revision of its role.2 Based on this study, flecainide is not recommended for patients with structural heart disease or coronary artery disease.2,3 However, it is recommended as a first-line therapy for pharmacologic cardioversion and maintenance of normal sinus rhythm in patients with atrial fibrillation and supraventricular tachycardia4,5 without the above caveats.
Class IC agents produce a selective block at the sodium (Na+) channels, resulting in the slowing of cardiac conduction.6,7 This high affinity for Na+ channels combined with slow unbinding kinetics during diastole explain the slowing of recovery time and prolongation of the refractory period.6,8,9 These electrophysiologic properties all can increase the PR, QRS, and QT interval duration. The QT interval is not significantly affected, as most of the QT prolongation is due to the QRS widening.6,10,11 Widening of the QRS by greater than 25% as compared to the baseline value is used as the threshold to decrease dosing or discontinue the use of flecainide.3The toxic effects of flecainide on cardiac conduction can produce prolonged QRS duration of up to 50%, and PR interval up to 30%, especially in rapid heart rates. Signs of intoxication are difficult to discern owing to its nonspecific presentation. A well-documented, but under-recognized, presentation of flecainide toxicity is the transformation of atrial fibrillation to atrial flutter.5,7,9,11-13 The reported rate of this pro arrhythmic effect can be as high as 3.5% to 5%.14,15Flecainide toxicity can occur secondary to chronic ingestion and may be precipitated in mild renal failure. The majority of flecainide is renally excreted and the half-life is 20 hours. Maximum therapeutic effect is seen between levels of 0.2 to 1 mcg/mL with levels greater than 0.7 to 1 mcg/mL associated with adverse effects.9 Systemic effects include dizziness and visual disturbances. A high degree of suspicion for flecainide toxicity is required when the patient’s initial presentation is nonspecific. In this circumstance, real-time bedside interrogation of the pacemaker is invaluable. Early diagnosis and treatment minimizes the risk for adverse sequelae, including death. Treatment includes increasing the excretion of flecainide, symptomatic support (including pacemaker placement, intravenous fat emulsion, or extracorporeal circulatory support) and administration of sodium bicarbonate, to transiently reverse the effect of the sodium channel blockade, in severe cases.15-17
Case
An 86-year-old woman, who recently had been seen in the same facility after a ground level fall, presented to the ED with to a 2- to 3-day history of vague abdominal pain, increasing weakness, nausea, and dry heaves.
Upon examination, the patient was unable to stand due to generalized weakness She arrived at the ED via emergency medical services. Her vital signs at presentation were significant for a systolic blood pressure (BP) of 90 mm Hg with a wide complex tachycardia concerning for ventricular tachycardia. The patient’s other vital signers were: heart rate, 136 beats/min; respiratory rate 20 breaths/min; and pulse oximetry was 94% on 4 liters/min of oxygen via nasal cannula.
The patient’s medical history was significant for atrial fibrillation and an indwelling pacemaker, for which she was chronically on flecai
The initial electrocardiogram (ECG) revealed a wide complex rhythm with pacemaker spikes (Figure 1). Based on these findings, electrodes were placed on the patient in the event she required cardioversion. The patient was started on an amiodarone intravenous (IV) drip for presumptive ventricular tachycardia.
During the patient’s evaluation in the ED, she experienced transient drops in BP, which were responsive to an IV fluid bolus of normal saline, and the amiodarone drip was discontinued. The patient’s ECG findings were compared to previous ECG studies, as was her current medication list and prior health issues. After ruling-out other causes, flecainide toxicity was considered high in the differential, and she was given 1 ampule of bicarbonate IV, after which a second ECG showed heart rhythm converted from a wide-complex tachycardia to a paced rhythm, markedly improved from the initial ECG (Figure 2). Similarly, there was a marked improvement in BP.
An interrogation of the patient’s pacemaker revealed an atrial flutter with a rate below detection for mode switch, with one-to-one tracking/pacing. The pacemaker was reprogrammed to divide the DDIR mode with detection rate at 120 mm Hg with mode switch activated. This was felt to be consistent with flecainide toxicity precipitating the cardiac conduction issues.
Laboratory studies showed an elevated flecainide level at 1.39 mcg/mL (upper limits of normal of 1 mcg/mL). Other studies showed worsening congestive heart failure, with a brain natriuretic peptide of 8,057 pg/mL and mild dehydration, with serum creatinine increased from her baseline of 0.9 to 1.38 mg/dL.
The patient’s abdominal pain was further evaluated and she was found to have acute cholecystitis. She was admitted to the intensive care unit with cardiology and general surgery consulting.
Discussion
Flecainide acetate was approved by the Food and Drug Administration in 1984.1It is a Vaughan-Williams class IC antiarrhythmic with a sodium channel blocker action used to treat supra ventricular arrhythmias. The CAST trial in 1989 investigated the efficacy of this class of antiarrhythmics, which resulted in a revision of its role.2 Based on this study, flecainide is not recommended for patients with structural heart disease or coronary artery disease.2,3 However, it is recommended as a first-line therapy for pharmacologic cardioversion and maintenance of normal sinus rhythm in patients with atrial fibrillation and supraventricular tachycardia4,5 without the above caveats.
Class IC agents produce a selective block at the sodium (Na+) channels, resulting in the slowing of cardiac conduction.6,7 This high affinity for Na+ channels combined with slow unbinding kinetics during diastole explain the slowing of recovery time and prolongation of the refractory period.6,8,9 These electrophysiologic properties all can increase the PR, QRS, and QT interval duration. The QT interval is not significantly affected, as most of the QT prolongation is due to the QRS widening.6,10,11 Widening of the QRS by greater than 25% as compared to the baseline value is used as the threshold to decrease dosing or discontinue the use of flecainide.3The toxic effects of flecainide on cardiac conduction can produce prolonged QRS duration of up to 50%, and PR interval up to 30%, especially in rapid heart rates. Signs of intoxication are difficult to discern owing to its nonspecific presentation. A well-documented, but under-recognized, presentation of flecainide toxicity is the transformation of atrial fibrillation to atrial flutter.5,7,9,11-13 The reported rate of this pro arrhythmic effect can be as high as 3.5% to 5%.14,15Flecainide toxicity can occur secondary to chronic ingestion and may be precipitated in mild renal failure. The majority of flecainide is renally excreted and the half-life is 20 hours. Maximum therapeutic effect is seen between levels of 0.2 to 1 mcg/mL with levels greater than 0.7 to 1 mcg/mL associated with adverse effects.9 Systemic effects include dizziness and visual disturbances. A high degree of suspicion for flecainide toxicity is required when the patient’s initial presentation is nonspecific. In this circumstance, real-time bedside interrogation of the pacemaker is invaluable. Early diagnosis and treatment minimizes the risk for adverse sequelae, including death. Treatment includes increasing the excretion of flecainide, symptomatic support (including pacemaker placement, intravenous fat emulsion, or extracorporeal circulatory support) and administration of sodium bicarbonate, to transiently reverse the effect of the sodium channel blockade, in severe cases.15-17
1. Hudak JM, Banitt EH, Schmid JR. Discovery and development of flecainide. Am J Cardiol. 1984;53(5):17B-20B.
2. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629.
3. Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7(2):76-85. doi:10.4330/wjc.v7.i2.76.
4. Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719-2747. doi:10.1093/eurheartj/ehs253.
5. Courand PY, Sibellas F, Ranc S, Mullier A, Kirkorian G, Bonnefoy E. Arrhythmogenic effect of flecainide toxicity. Cardiol J. 2013;20:203-205. doi:10.5603/CJ.2013.0035.
6. Holmes B, Heel RC. Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29(1):1-33.
7. Taylor R, Gandhi MM, Lloyd G. Tachycardia due to atrial flutter with rapid 1:1 conduction following treatment of atrial fibrillation with flecainide. Br Med J. 2010;340:b4684.
8. Roden DM, Woosley RL. Drug therapy. Flecainide. N Engl J Med. 1986;315(1):36-41.
9. Levis JT. ECG diagnosis: flecainide toxicity. Perm J. 2012;16(4):53.
10. Hellestrand KJ, Bexton RS, Nathan AW, Spurrell RA, Camm AJ. Acute electrophysiological effects of flecainide acetate on cardiac conduction and refractoriness in man. Br Heart J. 1982;48(2):140-148.
11. Rognoni A, Bertolazzi M, Peron M, et al. Electrocardiographic changes in a rare case of flecainide poisoning: a case report. Cases J. 2009;2:9137. doi:10.1186/1757-1626-2-9137.
12. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of “class IC atrial flutter” in patients with resistant atrial fibrillation. Am J Cardiol. 1999;83(5):785-787, A10.
13. Kola S, Mahata I, Kocheril AG. A case of flecainide toxicity. EP Lab Digest. 2015;15(5).
14. Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117(2):141-150.
15. Lloyd T, Zimmerman J, Griffin GD. Irreversible third-degree heart block and pacemaker implant in a case of flecainide toxicity. Am J Emerg Med. 2013;31(9):1418.e1-e2. doi:10.1016/j.ajem.2013.04.025.
16. Corkeron MA, van Heerden PV, Newman SM, Dusci L. Extracorporeal circulatory support in near-fatal flecainide overdose. Anaesth Intensive Care. 1999;27(4):405-408.
17. Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87-e89. doi:10.1111/j.1540-8159.2012.03485.x.
1. Hudak JM, Banitt EH, Schmid JR. Discovery and development of flecainide. Am J Cardiol. 1984;53(5):17B-20B.
2. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST). N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629.
3. Andrikopoulos GK, Pastromas S, Tzeis S. Flecainide: Current status and perspectives in arrhythmia management. World J Cardiol. 2015;7(2):76-85. doi:10.4330/wjc.v7.i2.76.
4. Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719-2747. doi:10.1093/eurheartj/ehs253.
5. Courand PY, Sibellas F, Ranc S, Mullier A, Kirkorian G, Bonnefoy E. Arrhythmogenic effect of flecainide toxicity. Cardiol J. 2013;20:203-205. doi:10.5603/CJ.2013.0035.
6. Holmes B, Heel RC. Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29(1):1-33.
7. Taylor R, Gandhi MM, Lloyd G. Tachycardia due to atrial flutter with rapid 1:1 conduction following treatment of atrial fibrillation with flecainide. Br Med J. 2010;340:b4684.
8. Roden DM, Woosley RL. Drug therapy. Flecainide. N Engl J Med. 1986;315(1):36-41.
9. Levis JT. ECG diagnosis: flecainide toxicity. Perm J. 2012;16(4):53.
10. Hellestrand KJ, Bexton RS, Nathan AW, Spurrell RA, Camm AJ. Acute electrophysiological effects of flecainide acetate on cardiac conduction and refractoriness in man. Br Heart J. 1982;48(2):140-148.
11. Rognoni A, Bertolazzi M, Peron M, et al. Electrocardiographic changes in a rare case of flecainide poisoning: a case report. Cases J. 2009;2:9137. doi:10.1186/1757-1626-2-9137.
12. Nabar A, Rodriguez LM, Timmermans C, Smeets JL, Wellens HJ. Radiofrequency ablation of “class IC atrial flutter” in patients with resistant atrial fibrillation. Am J Cardiol. 1999;83(5):785-787, A10.
13. Kola S, Mahata I, Kocheril AG. A case of flecainide toxicity. EP Lab Digest. 2015;15(5).
14. Falk RH. Proarrhythmia in patients treated for atrial fibrillation or flutter. Ann Intern Med. 1992;117(2):141-150.
15. Lloyd T, Zimmerman J, Griffin GD. Irreversible third-degree heart block and pacemaker implant in a case of flecainide toxicity. Am J Emerg Med. 2013;31(9):1418.e1-e2. doi:10.1016/j.ajem.2013.04.025.
16. Corkeron MA, van Heerden PV, Newman SM, Dusci L. Extracorporeal circulatory support in near-fatal flecainide overdose. Anaesth Intensive Care. 1999;27(4):405-408.
17. Ellsworth H, Stellpflug SJ, Cole JB, Dolan JA, Harris CR. A life-threatening flecainide overdose treated with intravenous fat emulsion. Pacing Clin Electrophysiol. 2013;36(3):e87-e89. doi:10.1111/j.1540-8159.2012.03485.x.
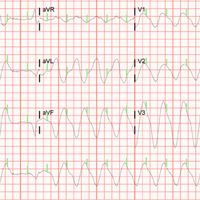
Aortic dissection presenting as ischemic limb
A 40-year-old man with a history of hypertension and alcohol abuse presented with acute onset of mild chest tightness, left leg pain, and increasing agitation, which prevented us from obtaining additional meaningful information from him.
On admission, his heart rate was 120 beats per minute, blood pressure 211/122 mm Hg, respiratory rate 18 per minute, and oxygen saturation 92% on room air. Given his history of alcohol abuse, we checked his blood ethanol level, which was less than 0.01%, well below the legal limit for intoxication.
We gave the patient intravenous lorazepam for possible alcohol withdrawal and started labetalol by intravenous infusion to lower his blood pressure.

On physical examination, his left lower extremity was cold and without pulses, including the femoral pulse. Suspecting acute arterial thrombosis, we ordered immediate computed tomographic (CT) angiography of the abdomen and pelvis with left lower extremity runoff. The images showed dissection of the abdominal aorta with extension to both the left and right common iliac arteries and the origin of the right external iliac artery. There was resultant occlusion of the left external iliac artery (Figure 1).

Immediate CT angiography of the chest was then performed, which revealed dissection of the thoracic aorta as well, starting superior to the aortic valve annulus and involving the ascending aorta, aortic arch, and the entire descending thoracic aorta (Figure 2).
The patient underwent emergency surgical repair of the aortic root, ascending aorta, and aortic arch. Residual dissection of the descending aorta was managed conservatively with blood pressure control using intravenous labetalol initially, which was then switched to oral carvedilol, and the pulses returned in his left lower extremity. He had an unremarkable postoperative recovery and was discharged after 1 week.
AORTIC DISSECTION AND MALPERFUSION SYNDROME
Aortic dissection is most often associated with acute onset of sharp chest pain and upper back pain. On rare occasions, it can have an atypical presentation such as stroke, paraplegia, mesenteric ischemia, or lower limb malperfusion.1
Extension of aortic dissection into the iliac and femoral arteries can cause impaired or absent blood flow to the lower extremity. These pulse deficits are a part of limb malperfusion syndrome. Symptoms of malperfusion syndrome vary greatly and depend on the vessels involved. Malperfusion of the branches of the aortic arch can result in stroke or altered sensorium. Compromise of intra-abdominal vessels due to dissection can involve the mesenteric bed, the renal arteries, or both, resulting in laboratory derangements such as lactic acidosis and renal failure.
How aortic dissection and malperfusion syndrome occur
Over time, shear forces on the aortic wall result in degeneration of the tunica intima and media. Dissection occurs when deterioration of the intima causes propagation of blood through a cleavage plane into the outer portion of the diseased media, forming a false lumen.
Anterograde or retrograde progression of dissection depends on the balance of the pressure gradient between true and false lumens.2 With every systolic ventricular contraction, a fluid and pressure wave travels down both lumens (true and false). However, the pressure gradient between the false and true lumens allows the more pliable intimal flap to bulge into the true lumen and ostia of branch vessels, resulting in static or dynamic obstruction.
Static obstruction occurs when the false lumen projects completely into the branch vessel and there is resultant thrombosis. As the name implies, dynamic obstruction is intermittent and is responsible for 80% of the cases of malperfusion syndrome.3 Dynamic obstruction has 2 distinct mechanisms: hypoperfusion through the true lumen due to impaired flow, and prolapse of the false lumen into a branch vessel.
Factors that exacerbate hypoperfusion through the true lumen and make obliteration by the false lumen more likely include large circumference of the dissected aorta, rapid heart rate, and high systolic pressure.4 Therefore, it is important to control the heart rate and blood pressure using beta-blockers in cases of aortic dissection with malperfusion syndrome. This treatment may resolve the dynamic obstruction through expansion and resumption of perfusion through the true lumen.5
MANAGEMENT OF MALPERFUSION SYNDROME
Aortic dissection can be classified as either Stanford type A (involving the ascending aorta) or type B (involving the descending aorta). Type B dissection associated with malperfusion syndrome is termed “complicated” type B aortic dissection. Our patient had both Stanford type A and complicated type B aortic dissection.
Unlike type A aortic dissection, which requires definitive open surgical repair, complicated type B aortic dissection occasionally responds to medical management alone. A plausible explanation for resolution of limb malperfusion with optimal blood pressure control is expansion of the true lumen and obliteration of the false lumen, as was likely the case in our patient.
In most cases, however, limb malperfusion persists despite optimal medical management. In such patients, endovascular graft stenting or open surgical repair may be needed. Open surgical repair procedures like bypass grafting or surgical fenestration are associated with significant rates of mortality and morbidity.5 Therefore, an endovascular approach rather than conventional surgical repair for complicated type B aortic dissection is advocated after optimal medical management.6 Endovascular repair also promotes favorable aortic remodeling without the morbidity associated with open surgical repair.
- Namana V, Balasubramanian R, Kariyanna PT, Sarasam R, Namana S, Shetty V. Aortic dissection with hemopericardium and thrombosed left common iliac artery presenting as acute limb ischemia: a case report and review. Am J Med Case Rep 2015; 3(10):338–343. doi:10.12691/ajmcr-3-10-9
- Crawford TC, Beaulieu RJ, Ehlert BA, Ratchford EV, Black JH 3rd. Malperfusion syndromes in aortic dissections. Vasc Med 2016; 21(3):264–273. doi:10.1177/1358863X15625371
- Williams DM, Lee DY, Hamilton BH, et al. The dissected aorta: percutaneous treatment of ischemic complications—principles and results. J Vasc Interv Radiol 1997; 8(4):605–625. pmid:9232578
- Chung JW, Elkins C, Sakai T, et al. True-lumen collapse in aortic dissection: part II. Evaluation of treatment methods in phantoms with pulsatile flow. Radiology 2000; 214(1):99–106. doi:10.1148/radiology.214.1.r00ja3499
- Gargiulo M, Bianchini Massoni C, Gallitto E, et al. Lower limb malperfusion in type B aortic dissection: a systematic review. Ann Cardiothorac Surg 2014; 3(4):351–367. doi:10.3978/j.issn.2225-319X.2014.07.05
- Dake MD, Kato N, Mitchell RS, et al. Endovascular stent-graft placement for the treatment of acute aortic dissection. N Engl J Med 1999; 340(20):1546–1552. doi:10.1056/NEJM199905203402004
A 40-year-old man with a history of hypertension and alcohol abuse presented with acute onset of mild chest tightness, left leg pain, and increasing agitation, which prevented us from obtaining additional meaningful information from him.
On admission, his heart rate was 120 beats per minute, blood pressure 211/122 mm Hg, respiratory rate 18 per minute, and oxygen saturation 92% on room air. Given his history of alcohol abuse, we checked his blood ethanol level, which was less than 0.01%, well below the legal limit for intoxication.
We gave the patient intravenous lorazepam for possible alcohol withdrawal and started labetalol by intravenous infusion to lower his blood pressure.
On physical examination, his left lower extremity was cold and without pulses, including the femoral pulse. Suspecting acute arterial thrombosis, we ordered immediate computed tomographic (CT) angiography of the abdomen and pelvis with left lower extremity runoff. The images showed dissection of the abdominal aorta with extension to both the left and right common iliac arteries and the origin of the right external iliac artery. There was resultant occlusion of the left external iliac artery (Figure 1).
Immediate CT angiography of the chest was then performed, which revealed dissection of the thoracic aorta as well, starting superior to the aortic valve annulus and involving the ascending aorta, aortic arch, and the entire descending thoracic aorta (Figure 2).
The patient underwent emergency surgical repair of the aortic root, ascending aorta, and aortic arch. Residual dissection of the descending aorta was managed conservatively with blood pressure control using intravenous labetalol initially, which was then switched to oral carvedilol, and the pulses returned in his left lower extremity. He had an unremarkable postoperative recovery and was discharged after 1 week.
AORTIC DISSECTION AND MALPERFUSION SYNDROME
Aortic dissection is most often associated with acute onset of sharp chest pain and upper back pain. On rare occasions, it can have an atypical presentation such as stroke, paraplegia, mesenteric ischemia, or lower limb malperfusion.1
Extension of aortic dissection into the iliac and femoral arteries can cause impaired or absent blood flow to the lower extremity. These pulse deficits are a part of limb malperfusion syndrome. Symptoms of malperfusion syndrome vary greatly and depend on the vessels involved. Malperfusion of the branches of the aortic arch can result in stroke or altered sensorium. Compromise of intra-abdominal vessels due to dissection can involve the mesenteric bed, the renal arteries, or both, resulting in laboratory derangements such as lactic acidosis and renal failure.
How aortic dissection and malperfusion syndrome occur
Over time, shear forces on the aortic wall result in degeneration of the tunica intima and media. Dissection occurs when deterioration of the intima causes propagation of blood through a cleavage plane into the outer portion of the diseased media, forming a false lumen.
Anterograde or retrograde progression of dissection depends on the balance of the pressure gradient between true and false lumens.2 With every systolic ventricular contraction, a fluid and pressure wave travels down both lumens (true and false). However, the pressure gradient between the false and true lumens allows the more pliable intimal flap to bulge into the true lumen and ostia of branch vessels, resulting in static or dynamic obstruction.
Static obstruction occurs when the false lumen projects completely into the branch vessel and there is resultant thrombosis. As the name implies, dynamic obstruction is intermittent and is responsible for 80% of the cases of malperfusion syndrome.3 Dynamic obstruction has 2 distinct mechanisms: hypoperfusion through the true lumen due to impaired flow, and prolapse of the false lumen into a branch vessel.
Factors that exacerbate hypoperfusion through the true lumen and make obliteration by the false lumen more likely include large circumference of the dissected aorta, rapid heart rate, and high systolic pressure.4 Therefore, it is important to control the heart rate and blood pressure using beta-blockers in cases of aortic dissection with malperfusion syndrome. This treatment may resolve the dynamic obstruction through expansion and resumption of perfusion through the true lumen.5
MANAGEMENT OF MALPERFUSION SYNDROME
Aortic dissection can be classified as either Stanford type A (involving the ascending aorta) or type B (involving the descending aorta). Type B dissection associated with malperfusion syndrome is termed “complicated” type B aortic dissection. Our patient had both Stanford type A and complicated type B aortic dissection.
Unlike type A aortic dissection, which requires definitive open surgical repair, complicated type B aortic dissection occasionally responds to medical management alone. A plausible explanation for resolution of limb malperfusion with optimal blood pressure control is expansion of the true lumen and obliteration of the false lumen, as was likely the case in our patient.
In most cases, however, limb malperfusion persists despite optimal medical management. In such patients, endovascular graft stenting or open surgical repair may be needed. Open surgical repair procedures like bypass grafting or surgical fenestration are associated with significant rates of mortality and morbidity.5 Therefore, an endovascular approach rather than conventional surgical repair for complicated type B aortic dissection is advocated after optimal medical management.6 Endovascular repair also promotes favorable aortic remodeling without the morbidity associated with open surgical repair.
A 40-year-old man with a history of hypertension and alcohol abuse presented with acute onset of mild chest tightness, left leg pain, and increasing agitation, which prevented us from obtaining additional meaningful information from him.
On admission, his heart rate was 120 beats per minute, blood pressure 211/122 mm Hg, respiratory rate 18 per minute, and oxygen saturation 92% on room air. Given his history of alcohol abuse, we checked his blood ethanol level, which was less than 0.01%, well below the legal limit for intoxication.
We gave the patient intravenous lorazepam for possible alcohol withdrawal and started labetalol by intravenous infusion to lower his blood pressure.
On physical examination, his left lower extremity was cold and without pulses, including the femoral pulse. Suspecting acute arterial thrombosis, we ordered immediate computed tomographic (CT) angiography of the abdomen and pelvis with left lower extremity runoff. The images showed dissection of the abdominal aorta with extension to both the left and right common iliac arteries and the origin of the right external iliac artery. There was resultant occlusion of the left external iliac artery (Figure 1).
Immediate CT angiography of the chest was then performed, which revealed dissection of the thoracic aorta as well, starting superior to the aortic valve annulus and involving the ascending aorta, aortic arch, and the entire descending thoracic aorta (Figure 2).
The patient underwent emergency surgical repair of the aortic root, ascending aorta, and aortic arch. Residual dissection of the descending aorta was managed conservatively with blood pressure control using intravenous labetalol initially, which was then switched to oral carvedilol, and the pulses returned in his left lower extremity. He had an unremarkable postoperative recovery and was discharged after 1 week.
AORTIC DISSECTION AND MALPERFUSION SYNDROME
Aortic dissection is most often associated with acute onset of sharp chest pain and upper back pain. On rare occasions, it can have an atypical presentation such as stroke, paraplegia, mesenteric ischemia, or lower limb malperfusion.1
Extension of aortic dissection into the iliac and femoral arteries can cause impaired or absent blood flow to the lower extremity. These pulse deficits are a part of limb malperfusion syndrome. Symptoms of malperfusion syndrome vary greatly and depend on the vessels involved. Malperfusion of the branches of the aortic arch can result in stroke or altered sensorium. Compromise of intra-abdominal vessels due to dissection can involve the mesenteric bed, the renal arteries, or both, resulting in laboratory derangements such as lactic acidosis and renal failure.
How aortic dissection and malperfusion syndrome occur
Over time, shear forces on the aortic wall result in degeneration of the tunica intima and media. Dissection occurs when deterioration of the intima causes propagation of blood through a cleavage plane into the outer portion of the diseased media, forming a false lumen.
Anterograde or retrograde progression of dissection depends on the balance of the pressure gradient between true and false lumens.2 With every systolic ventricular contraction, a fluid and pressure wave travels down both lumens (true and false). However, the pressure gradient between the false and true lumens allows the more pliable intimal flap to bulge into the true lumen and ostia of branch vessels, resulting in static or dynamic obstruction.
Static obstruction occurs when the false lumen projects completely into the branch vessel and there is resultant thrombosis. As the name implies, dynamic obstruction is intermittent and is responsible for 80% of the cases of malperfusion syndrome.3 Dynamic obstruction has 2 distinct mechanisms: hypoperfusion through the true lumen due to impaired flow, and prolapse of the false lumen into a branch vessel.
Factors that exacerbate hypoperfusion through the true lumen and make obliteration by the false lumen more likely include large circumference of the dissected aorta, rapid heart rate, and high systolic pressure.4 Therefore, it is important to control the heart rate and blood pressure using beta-blockers in cases of aortic dissection with malperfusion syndrome. This treatment may resolve the dynamic obstruction through expansion and resumption of perfusion through the true lumen.5
MANAGEMENT OF MALPERFUSION SYNDROME
Aortic dissection can be classified as either Stanford type A (involving the ascending aorta) or type B (involving the descending aorta). Type B dissection associated with malperfusion syndrome is termed “complicated” type B aortic dissection. Our patient had both Stanford type A and complicated type B aortic dissection.
Unlike type A aortic dissection, which requires definitive open surgical repair, complicated type B aortic dissection occasionally responds to medical management alone. A plausible explanation for resolution of limb malperfusion with optimal blood pressure control is expansion of the true lumen and obliteration of the false lumen, as was likely the case in our patient.
In most cases, however, limb malperfusion persists despite optimal medical management. In such patients, endovascular graft stenting or open surgical repair may be needed. Open surgical repair procedures like bypass grafting or surgical fenestration are associated with significant rates of mortality and morbidity.5 Therefore, an endovascular approach rather than conventional surgical repair for complicated type B aortic dissection is advocated after optimal medical management.6 Endovascular repair also promotes favorable aortic remodeling without the morbidity associated with open surgical repair.
- Namana V, Balasubramanian R, Kariyanna PT, Sarasam R, Namana S, Shetty V. Aortic dissection with hemopericardium and thrombosed left common iliac artery presenting as acute limb ischemia: a case report and review. Am J Med Case Rep 2015; 3(10):338–343. doi:10.12691/ajmcr-3-10-9
- Crawford TC, Beaulieu RJ, Ehlert BA, Ratchford EV, Black JH 3rd. Malperfusion syndromes in aortic dissections. Vasc Med 2016; 21(3):264–273. doi:10.1177/1358863X15625371
- Williams DM, Lee DY, Hamilton BH, et al. The dissected aorta: percutaneous treatment of ischemic complications—principles and results. J Vasc Interv Radiol 1997; 8(4):605–625. pmid:9232578
- Chung JW, Elkins C, Sakai T, et al. True-lumen collapse in aortic dissection: part II. Evaluation of treatment methods in phantoms with pulsatile flow. Radiology 2000; 214(1):99–106. doi:10.1148/radiology.214.1.r00ja3499
- Gargiulo M, Bianchini Massoni C, Gallitto E, et al. Lower limb malperfusion in type B aortic dissection: a systematic review. Ann Cardiothorac Surg 2014; 3(4):351–367. doi:10.3978/j.issn.2225-319X.2014.07.05
- Dake MD, Kato N, Mitchell RS, et al. Endovascular stent-graft placement for the treatment of acute aortic dissection. N Engl J Med 1999; 340(20):1546–1552. doi:10.1056/NEJM199905203402004
- Namana V, Balasubramanian R, Kariyanna PT, Sarasam R, Namana S, Shetty V. Aortic dissection with hemopericardium and thrombosed left common iliac artery presenting as acute limb ischemia: a case report and review. Am J Med Case Rep 2015; 3(10):338–343. doi:10.12691/ajmcr-3-10-9
- Crawford TC, Beaulieu RJ, Ehlert BA, Ratchford EV, Black JH 3rd. Malperfusion syndromes in aortic dissections. Vasc Med 2016; 21(3):264–273. doi:10.1177/1358863X15625371
- Williams DM, Lee DY, Hamilton BH, et al. The dissected aorta: percutaneous treatment of ischemic complications—principles and results. J Vasc Interv Radiol 1997; 8(4):605–625. pmid:9232578
- Chung JW, Elkins C, Sakai T, et al. True-lumen collapse in aortic dissection: part II. Evaluation of treatment methods in phantoms with pulsatile flow. Radiology 2000; 214(1):99–106. doi:10.1148/radiology.214.1.r00ja3499
- Gargiulo M, Bianchini Massoni C, Gallitto E, et al. Lower limb malperfusion in type B aortic dissection: a systematic review. Ann Cardiothorac Surg 2014; 3(4):351–367. doi:10.3978/j.issn.2225-319X.2014.07.05
- Dake MD, Kato N, Mitchell RS, et al. Endovascular stent-graft placement for the treatment of acute aortic dissection. N Engl J Med 1999; 340(20):1546–1552. doi:10.1056/NEJM199905203402004
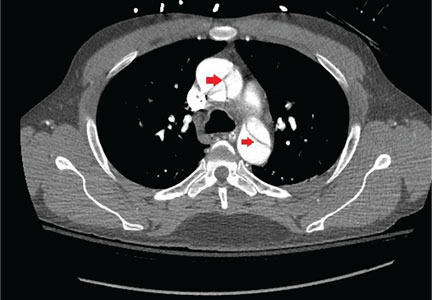
All-Terrain, No Control
ANSWER
The image shows a large, convex hyperdensity within the left parietal region. This is a textbook image of an acute epidural hematoma. There is considerable mass effect and evidence of left-to-right shift. Windowing shows an underlying fracture, which is typically associated with these types of hemorrhages.
There is also evidence of a right-side concave hyperdensity, consistent with an acute subdural hematoma. Typically, this is referred to as a contrecoup injury.
The patient was transported to the operating room for an emergent left craniotomy for epidural evacuation; he recovered uneventfully.
ANSWER
The image shows a large, convex hyperdensity within the left parietal region. This is a textbook image of an acute epidural hematoma. There is considerable mass effect and evidence of left-to-right shift. Windowing shows an underlying fracture, which is typically associated with these types of hemorrhages.
There is also evidence of a right-side concave hyperdensity, consistent with an acute subdural hematoma. Typically, this is referred to as a contrecoup injury.
The patient was transported to the operating room for an emergent left craniotomy for epidural evacuation; he recovered uneventfully.
ANSWER
The image shows a large, convex hyperdensity within the left parietal region. This is a textbook image of an acute epidural hematoma. There is considerable mass effect and evidence of left-to-right shift. Windowing shows an underlying fracture, which is typically associated with these types of hemorrhages.
There is also evidence of a right-side concave hyperdensity, consistent with an acute subdural hematoma. Typically, this is referred to as a contrecoup injury.
The patient was transported to the operating room for an emergent left craniotomy for epidural evacuation; he recovered uneventfully.
A 40-year-old man is brought to the emergency department (ED) with a suspected intracranial hemorrhage after being thrown off an all-terrain vehicle. He was reportedly riding the vehicle without a helmet when he somehow lost control; the accident itself was unwitnessed.
En route to the ED, he was reportedly confused but hemodynamically stable, with a Glasgow Coma Scale score of 13-14. He lost consciousness while in the CT scanner, requiring emergent intubation for airway protection.
When you arrive to assess him, you note an intubated male with stable vital signs. The pupils display slight anisocoria but equally react. The patient withdraws in all four extremities secondary to pain, with slight posturing.
Noncontrast CT of the head is obtained, a static image from which is shown. What is your impression?
Guiding Resuscitation in the Emergency Department
Resuscitation of critically ill patients in shock from cardiogenic, hypovolemic, obstructive, distributive, or neurogenic etiology is a cornerstone of the care delivered by emergency physicians (EPs).1 Regardless of the etiology, it is essential that the treating EP initiate resuscitative measures in a timely manner and closely trend the patient’s response to these interventions.
The early goal-directed therapy (EGDT) initially proposed by Rivers et al2 in 2001 demonstrated a bundled approach to fluid resuscitation by targeting end points for volume resuscitation, mean arterial blood pressure (MAP), oxygen (O2) delivery/extraction (mixed venous O2 saturation, [SvO2]), hemoglobin (Hgb) concentration, and cardiac contractility. Since then, advancements in laboratory testing and hemodynamic monitoring (HDM) devices further aid and guide resuscitative efforts, and are applicable to any etiology of shock.
In addition to these advancements, the growing evidence of the potential harm from improper fluid resuscitation, such as the administration of excessive intravascular fluid (IVF),3 underscores the importance of a precise, targeted, and individualized approach to care. This article reviews the background, benefits, and limitations of some of the common and readily available tools in the ED that the EP can employ to guide fluid resuscitation in critically ill patients.
Physical Examination
Background
The rapid recognition and treatment of septic shock in the ED is associated with lower rates of in-hospital morbidity and mortality.4 The physical examination by the EP begins immediately upon examining the patient. The acquisition of vital signs and recognition of physical examination findings suggestive of intravascular volume depletion allows the EP to initiate treatment immediately.
In this discussion, hypotension is defined as systolic blood pressure (SBP) of less than 95 mm Hg, MAP of less than 65 mm Hg, or a decrease in SBP of more than 40 mm Hg from baseline measurements. Subsequently, shock is defined as hypotension with evidence of tissue hypoperfusion-induced dysfunction.5,6 Although the use of findings from the physical examination to guide resuscitation allows for rapid patient assessment and treatment, the predictive value of the physical examination to assess hemodynamic status is limited.
Visual inspection of the patient’s skin and mucous membranes can serve as an indicator of volume status. The patient’s tongue should appear moist with engorged sublingual veins; a dry tongue and diminished veins may suggest the need for volume resuscitation. On examination of the skin, delayed capillary refill of the digits and cool, clammy extremities suggest the shunting of blood by systemic circulation from the skin to central circulation. Patients who progress to more severe peripheral vasoconstriction develop skin mottling, referred to as livedo reticularis (Figure 1).
Benefits
The major benefit of the physical examination as a tool to evaluate hemodynamic status is its ease and rapid acquisition. The patient’s vital signs and physical examination can be obtained in the matter of moments upon presentation, without the need to wait on results of laboratory evaluation or additional equipment. Additionally, serial examinations by the same physician can be helpful to monitor a patient’s response to resuscitative efforts. The negative predictive value (NPV) of the physical examination in evaluating for hypovolemia may be helpful, but only when it is taken in the appropriate clinical context and is used in conjunction with other diagnostic tools. The physical examination can exclude hypovolemic volume status with an NPV of approximately 70%.7
A constellation of findings from the physical examination may include altered mentation, hypotension, tachycardia, and decreased urinary output by 30% to 40% intravascular volume loss.8,9Findings from the physical examination to assess fluid status should be used with caution as interobserver reliability has proven to be poor and the prognostic value is limited.
Limitations
The literature shows the limited prognostic value of the physical examination in determining a patient’s volume status and whether fluid resuscitation is indicated. For example, in one meta-analysis,10 supine hypotension and tachycardia were frequently absent on examination—even in patients who underwent large volume phlebotomy.8 This study also showed postural dizziness to be of no prognostic value.
Another study by Saugel et al7 that compared the physical examination (skin assessment, lung auscultation, and percussion) to transpulmonary thermodilution measurements of the cardiac index, global end-diastolic volume index, and extravascular lung water index, found poor interobserver correlation and agreement among physicians.
The physical examination is also associated with weak predictive capabilities for the estimation of volume status compared to the device measurements. Another contemporary study by Saugel et al9 evaluated the predictive value of the physical examination to accurately identify volume responsiveness replicated these results, and reported poor interobserver correlation (κ coefficient 0.01; 95% caval index [CI] -0.39-0.42) among physical examination findings, with a sensitivity of only 71%, specificity of 23.5%, positive predictive value of 27.8%, and negative predictive value of 66.7%.9
Serum Lactate Levels
Background
In the 1843 book titled, Investigations of Pathological Substances Obtained During the Epidemic of Puerperal Fever, Johann Joseph Scherer described the cases of seven young peripartum female patients who died from a clinical picture of what is now understood to be septic shock.11 In his study of these cases, Scherer demonstrated the presence of lactic acid in patients with pathological conditions. Prior to this discovery, lactic acid had never been isolated in a healthy individual. These results were recreated in 1851 by Scherer and Virchow,11 who demonstrated the presence of lactic acid in the blood of a patient who died from leukemia. The inference based on Scherer and Virchow’s work correlated the presence of excessive lactic acid with bodily deterioration and severe disease. Since this finding, there has been a great deal of interest in measuring serum lactic acid as a means to identify and manage critical illness.
In a 2001 groundbreaking study of EGDT for severe sepsis and septic shock, Rivers et al2 studied lactic acid levels as a marker for severe disease. Likewise, years later, the 2014 Protocol-Based Care for Early Septic Shock (PROCESS), Prospective Multicenter Imaging Study for Evaluation of Chest Pain (PROMISE), and Australasian Resuscitation in Sepsis Evaluation (ARISE) trials used lactate levels in a similar manner to identify patients appropriate for randomization.12-14 While the purpose of measuring lactic acid was only employed in these studies to identify patients at risk for critical illness, the 2012 Surviving Sepsis Campaign Guidelines recommended serial measurement of lactate, based on the assumption that improved lactate levels signified better tissue perfusion.15
Although much of the studies on lactate levels appear to be based on the treatment and management of septic patients, findings can be applied to any etiology of shock. For example, a serum lactate level greater than 2 mmol/L is considered abnormal, and a serum lactate greater than 4 mmol/L indicates a significantly increased risk for in-hospital mortality.16
Benefits
It is now a widely accepted belief that the rapid identification, triage, and treatment of critically ill patients has a dramatic effect on morbidity and mortality.4 As previously noted, lactate has been extensively studied and identified as a marker of severe illness.17,18 A serum lactate level, which can be rapidly processed in the ED, can be easily obtained from a minimally invasive venous, arterial, or capillary blood draw.18 The only risk associated with serum lactate testing is that of any routine venipuncture; the test causes minimal, if any, patient discomfort.
Thanks to advances in point-of-care (POC) technology, the result of serum lactate assessment can be available within 10 minutes from blood draw. This technology is inexpensive and can be easily deployed in the prehospital setting or during the initial triage assessment of patients arriving at the ED.19 These POC instruments have been well correlated with whole blood measurements and permit for the rapid identification and treatment of at risk patients.
Limitations
The presence of elevated serum lactate levels is believed to represent the presence of cellular anaerobic metabolism due to impaired O2 delivery in the shock state. Abnormal measurements therefore prompt aggressive interventions aimed at maximizing O2 delivery to the tissues, such as intravenous fluid boluses, vasopressor therapy, or even blood product administration.
A return to a normalized serum lactate level is assumed to represent a transition back to aerobic metabolism. Lactate elevations, however, are not solely an indication of anaerobic metabolism and may only represent a small degree of lactate production.20 While the specific cellular mechanics are out of the scope of this article, it has been postulated that the increase in plasma lactate concentration is primarily driven by β-2 receptor stimulation from increased circulating catecholamines leading to increased aerobic glycolysis. Increased lactate levels could therefore be an adaptive mechanism of energy production—aggressive treatment and rapid clearance may, in fact, be harmful. Type A lactic acidosis is categorized as elevated serum levels due to tissue hypoperfusion.21
However, lactate elevations do not exclusively occur in severe illness. The use of β-2 receptor agonists such as continuous albuterol treatments or epinephrine may cause abnormal lactate levels.22 Other medications have also been associated with elevated serum lactate levels, including, but not limited to linezolid, metformin, and propofol.23-25 Additionally, lactate levels may be elevated after strenuous exercise, seizure activity, or in liver and kidney disease.26 These “secondary” causes of lactic acidosis that are not due to tissue hypoperfusion are referred to as type B lactic acidosis. Given these multiple etiologies and lack of specificity for this serum measurement, a failure to understand these limitations may result in over aggressive or unnecessary medical treatments.
Central Venous Pressure
Background
Central venous pressure (CVP) measurements can be obtained through a catheter, the distal tip of which transduces pressure of the superior vena cava at the entrance of the right atrium (RA). Thus, CVP is often used as a representation of RA pressure (RAP) and therefore an estimate of right ventricular (RV) preload. While CVP is used to diagnose and determine the etiology of shock, evidence and controversy regarding the use of CVP as a marker for resuscitation comes largely from sepsis-focused literature.5 Central venous pressure is meant to represent preload, which is essential for stroke volume as described by the Frank-Starling mechanism; however, its use as a target in distributive shock, a state in which it is difficult to determine a patient’s volume status, has been popularized by EGDT since 2001.2
Since the publication of the 2004 Surviving Sepsis Guidelines, CVP monitoring has been in the spotlight of sepsis resuscitation, albeit with some controversy.27 Included as the result of two studies, this recommendation has been removed in the most recent guidelines after 12 years of further study and scrutiny.2,27,28
Hypovolemic and hemorrhagic shock are usually diagnosed clinically and while a low CVP can be helpful in the diagnosis, the guidelines do not support CVP as a resuscitation endpoint. Obstructive and cardiogenic shock will both result in elevated CVP; however, treatment of obstructive shock is generally targeted at the underlying cause. While cardiogenic shock can be preload responsive, the mainstay of therapy in the ED is identification of patients for revascularization and inotropic support.29
Benefits
The CVP has been used as a surrogate for RV preload volume. If a patient’s preload volume is low, the treating physician can administer fluids to improve stroke volume and cardiac output (CO). Clinically, CVP measurements are easy to obtain provided a central venous line has been placed with the distal tip at the entrance to the RA. Central venous pressure is measured by transducing the pressure via manometry and connecting it to the patient’s bedside monitor. This provides an advantage of being able to provide serial or even continuous measurements. The “normal” RAP should be a low value (1-5 mm Hg, mean of 3 mm Hg), as this aids in the pressure gradient to drive blood from the higher pressures of the left ventricle (LV) and aorta through the circulation back to the low-pressure of the RA.30 The value of the CVP is meant to correspond to the physical examination findings of jugular venous distension.31,32 Thus, a low CVP may be “normal” and seen in patients with hypovolemic shock, whereas an elevated CVP can suggest volume overload or obstructive shock. However, this is of questionable value in distributive shock cases.
Aside from the two early studies on CVP monitoring during treatment of septic patients, there are few data to support the use of CVP measurement in the early resuscitation of patients with shock.2,28 More recent trials (PROMISE, ARISE, PROCESS) that compared protocolized sepsis care to standard care showed no benefit to bundles including CVP measurements.12-14 However, a subsequent, large observational trial spanning 7.5 years demonstrated improvements in sepsis-related mortality in patients who received a central venous catheter (CVC) and CVP-targeted therapy.33 Thus, it is possible that protocols including CVP are still beneficial in combination with other therapies even though CVP in isolation is not.
Limitations
The traditional two assumptions in CVP monitoring are CVP value represents the overall volume status of the patient, and the LV is able to utilize additional preload volume. The latter assumption, however, may be hampered by the presence of sepsis-induced myocardial dysfunction, which may be present in up to 40% of critically ill patients.34 The former assumption does not always hold true due to processes that change filling pressures independent of intravascular volume—eg, acute or chronic pulmonary hypertension, cardiac tamponade, intra-abdominal hypertension, or LV failure. Even before the landmark EGDT study, available data suggested that CVP was not a reliable marker for resuscitation management.35 A recent systematic review by Gottlieb and Hunter36 showed that the area under the receiver-operator curve for low, mid-range, or high CVPs was equivocal at best. In addition to its unreliability and lack of specificity, another significant drawback to using CVP to guide resuscitation therapy in the ED is that it necessitates placement of a CVC, which can be time-consuming and, if not otherwise indicated, lead to complications of infection, pneumothorax, and/or thrombosis.37
Mixed Venous Oxygen
Background
Most EPs are familiar with the use of ScvO2 in EGDT protocols to guide volume resuscitation of septic patients.2 A patient’s ScvO2 represents the O2 saturation of venous blood obtained via a CVC at the confluence of the superior vena cava and the RA, and thus it reflects tissue O2 consumption as a surrogate for tissue perfusion. The measurement parallels the SvO2 obtained from the pulmonary artery. In a healthy patient, SvO2 is around 65% to 70% and includes blood returning from both the superior and inferior vena cava (IVC). As such, ScvO2 values are typically 3% to 5% lower than SvO2 owing to the lower O2 extracted by tissues draining into the IVC compared to the mixed venous blood sampled from the pulmonary artery.38
Though a debate over the benefit of EGDT in treating sepsis continues, understanding the physiology of ScvO2 measurements is another potential tool the EP can use to guide the resuscitation of critically ill patients.39 A patient’s SvO2 and, by extension, ScvO2 represents the residual O2 saturation after the tissues have extracted the amount of O2 necessary to meet metabolic demands (Figure 2). 
Conversely, cellular dysfunction, which can occur in certain toxicities or in severe forms of sepsis, can lead to decreased tissue O2 consumption with a concomitant rise in ScvO2 to supernormal values.38 The EP should take care, however, to consider whether ScvO2 values exceeding 80% represent successful therapeutic intervention or impaired tissue O2 extraction and utilization. There are data from ED patients suggesting an increased risk of mortality with both extremely low and extremely high values of ScvO2.40
Benefits
A critically ill patient’s ScvO2 can potentially provide EPs with insight into the patient’s global tissue perfusion and the source of any mismatch between O2 delivery and consumption. Using additional tools and measurements (physical examination, serum Hgb levels, and pulse oximetry) in conjunction with an ScvO2 measurement, assists EPs in identifying targets for therapeutic intervention. The effectiveness of this intervention can then be assessed using serial ScvO2 measurements, as described in Rivers et al2 EGDT protocol. Importantly, EPs should take care to measure serial ScvO2 values to maximize its utility.38 Similar to a CVP measurement, ScvO2is easily obtained from blood samples for serial laboratory measurements, assuming the patient already has a CVC with the distal tip at the entrance to the RA (ScvO2) or a pulmonary artery catheter (PAC) (SvO2).
Limitations
Serial measurements provide the most reliable information, which may be more useful in patients who spend extended periods of their resuscitation in the ED. In comparison to other measures of global tissue hypoxia, work by Jones et al41 suggests non-inferiority of peripherally sampled, serial lactate measurements as an alternative to ScvO2. This, in conjunction with the requirement for an internal jugular CVC, subclavian CVC, or PAC with their associated risks, may make ScvO2 a less attractive guide for the resuscitation of critically ill patients in the ED.
Monitoring Devices
Background
As noted throughout this review, it is important not only to identify and rapidly treat shock, but to also correctly identify the type of shock, such that treatment can be appropriately directed at its underlying cause. However, prior work suggests that EPs are unable to grossly estimate CO or systemic vascular resistance when compared to objective measurements of these parameters.42 This is in agreement with the overall poor performance of physical examination and clinical evaluation as a means of predicting volume responsiveness or guiding resuscitation, as discussed previously. Fortunately, a wide variety of devices to objectively monitor hemodynamics are now available to the EP.
In 1970, Swan et al43 published their initial experience with pulmonary artery catheterization at the bedside, using a balloon-tipped, flow-guided PAC in lieu of fluoroscopy, which had been mandated by earlier techniques. The ability to measure CO, right heart pressures, pulmonary arterial pressures, and estimate LV end diastolic pressure ushered in an era of widespread PAC use, despite an absence of evidence for causation of improved patient outcomes. The utilization of PACs has fallen, as the literature suggests that the empiric placement of PACs in critically ill patients does not improve mortality, length of stay, or cost, and significant complication rates have been reported in large trials.44,45Subsequently, a number of non-invasive or less-invasive HDM devices have been developed. Amongst the more commonly encountered modern devices, the techniques utilized for providing hemodynamic assessments include thermodilution and pulse contour analysis (PiCCOTM), pulse contour analysis (FloTrac/VigileoTM), and lithium chemodilution with pulse power analysis (LiDCOplusTM).46 The primary utility of these devices for the EP lies in the ability to quantify CO, stroke volume, and stroke volume or pulse pressure variation (PPV) to predict or assess response to resuscitative interventions (volume administration, vasopressors, inotropes, etc).
Benefits
Many of these devices require placement of an arterial catheter. Some require the addition of a CVC. Both of these procedures are well within the clinical scope of the EP, and are performed with fair frequency on critically ill patients. This is a distinct advantage when compared to pulmonary artery catheterization, a higher risk procedure that is rarely performed outside of the intensive care unit or cardiac catheterization laboratory. In addition, all of the devices below present hemodynamic data in a graphical, easy-to-read format, in real time. All of the devices discussed report stroke volume variation (SVV) or PPV continuously.
Limitations
Though these measures have validated threshold values that predict volume responsiveness, they require the patient to be intubated with a set tidal volume of greater than or equal to 8 mL/kg without spontaneous respirations and cardiac arrhythmias, in order to accurately do so. All of the HDM devices that rely on pulse contour analysis as the primary means of CO measurement cannot be used in the presence of significant cardiac arrhythmias (ie, atrial fibrillation), or mechanical circulatory assistance devices (ie, intra-aortic balloon counterpulsation). None of these devices are capable of monitoring microcirculatory changes, felt to be of increasing clinical importance in the critically ill.
The use of HDM devices to monitor CO with a reasonable degree of accuracy, trend CO, and assess for volume responsiveness using a number of previously validated parameters such as SVV is now in little doubt. However, these devices are still invasive, if less so than a pulmonary artery. The crux of the discussion of HDM devices for use in ED resuscitation revolves around whether or not the use of such devices to drive previously validated, protocolized care results in better outcomes for patients. The EP can now have continuous knowledge of a large number of hemodynamic parameters at their fingertips with relatively minimal additional efforts. At the time of this writing, though, this is both untested and unproven, with respect to the ED population.
Point-of-Care Ultrasound
Background
Over the past two decades, ultrasound (US) has become an integral part of the practice of emergency medicine (EM), and is now included in all United States Accreditation Council for Graduate Medical Education Emergency Medicine Residency Programs.47,48 It has emerged as a very important bedside tool performed by the clinician to identify type of shock and guide resuscitation, and has been endorsed by both EM and critical care societies.49-51 This section reviews the utility of US as a modality in identifying shock and guiding resuscitation, in addition to the pitfalls and limitations of this important tool.
In 2010, Perera et al47 described in their landmark article the Rapid Ultrasound in SHock (RUSH) examination, which describes a stepwise (the pump, tank, pipes) approach to identify the type of shock (cardiogenic, hypovolemic, obstructive, or distributive) in the crashing, hypotensive ED patient. We do not describe the full RUSH examination in this review, but discuss key elements of it as examples of how POCUS can assist the EP to make a rapid diagnosis and aid in the management of patients in shock. The “pump” is the heart, which is assessed in four different views to identify a pericardial effusion and possible tamponade, assess contractility or ejection fraction of the LV (severely decreased, decreased, normal, or hyperdynamic), and right heart strain which is identified by an RV that is larger than the LV, indicative of a potential pulmonary embolus.
The “tank” is then assessed by visualizing the IVC in the subxiphoid plane, and is evaluated for respiratory collapsibility (CI) and maximum size. This has been quite the debated topic over the last two decades. In 1988, Simonson and Schiller52 were the first to describe a correlation in spontaneously breathing patients between IVC caliber (measured 2 cm from the cavoatrial junction) and variation and RAP, where a larger IVC diameter and less respiratory variation correlated with a high RAP. Kircher et al53 later went on to describe that a CI greater than 50% correlated with an RAP of less than 10 mm Hg and vice versa in spontaneously breathing patients. Since then there have been more studies attempting to verify these findings in both spontaneously breathing and mechanically ventilated patients.54-56 The purpose of performing these measurements is not to estimate CVP, but to assess fluid responsiveness (ie, a blood pressure response to a fluid challenge). It can be assumed in states of shock that a small IVC, or one with a high CI, in the presence of a hyperdynamic heart is indicative of an underfilled ventricle and fluid responsiveness, especially if the IVC size increases with fluid.55,57 However, there are several caveats to this. First, in mechanically ventilated patients, the IVC is already plethoric due to positive pressure ventilation, and increases in diameter with inspiration and decreases with expiration as compared to spontaneously breathing patients. Second, the CI value to predict volume responsiveness in ventilated patients is set at 15% instead of 50%.55 Third, it is important to always take the clinical scenario in context; a dilated IVC with small CI is not necessarily only due to volume overload and congestive heart failure, but can be due to elevated RAP from obstructive shock due to cardiac tamponade or massive pulmonary embolus, which is why it is important to assess the “pump” first.47,58 It is also crucial to not forget to assess the abdominal and thoracic cavities, as intraperitoneal or pleural fluid with a collapsed IVC can potentially make a diagnosis of hemorrhagic or hypovolemic shock depending on the clinical scenario.47 The final part of the RUSH protocol is to evaluate the “pipes,” inclusive of the lower extremity deep venous system for evaluation of potential thrombosis that could increase suspicion for a pulmonary embolism causing obstructive shock, and the aorta with the common iliac arteries if there is concern for aortic dissection or aneurysmal rupture.
Benefits
Some of the most significant advantages to the use of POCUS to guide resuscitation is that it is quick, non-invasive, does not use ionizing radiation, and can be easily repeated. As noted above, it is a requirement for EM residencies to teach its use, so that contemporary graduates are entering the specialty competent in applying it to the care of their patients. Furthermore, POCUS is done at the bedside, limiting the need to potentially transport unstable patients.
In the most basic applications, POCUS provides direct visualization of a patient’s cardiac function, presence or absence of lung sliding to suggest a pneumothorax, presence of pulmonary edema, assessment of CVP pressures or potential for fluid responsiveness, as well as identification of potential thoracic, peritoneal, or pelvic cavity fluid accumulation that may suggest hemorrhage. There is literature to support that these assessments performed by the EP have been shown to be comparable to those of cardiologists.59,60 With continued practice and additional training, it is possible for EPs to even perform more “advanced” hemodynamic assessments to both diagnose and guide therapy to patients in shock (Figures 3 and 4).61
Limitations
Although POCUS has been shown as a remarkable tool to help assist the EP in making rapid decisions regarding resuscitation, it is always important to remember its limitations. Most of the studies regarding its use are of very small sample sizes, and further prospective studies have to be performed in order for this modality to be fully relied on.62Compared to some of the previously mentioned HDM devices that may provide continuous data, POCUS needs to be performed by the treating physician, thereby occurring intermittently. Emergency physicians need to be aware of their own experience and limitations with this modality, as errors in misdiagnosis can lead to unnecessary procedures, with resulting significant morbidity and mortality. Blanco and Volpicelli63 describe several common errors that include misdiagnosing the stomach as a peritoneal effusion, assuming adequate volume resuscitation when the IVC is seen to be plethoric in the setting of cardiac tamponade, or mistaking IVC movement as indicative of collapsibility, amongst other described misinterpretations. Several other studies have shown that, despite adequate performance of EPs in POCUS, diagnostic sensitivities remained higher when performed by radiologists.64-67 Thus it remains important for the EPs to be vigilant and not anchor on a diagnosis when in doubt, and to consult early with radiology, particularly if there is any question, to avoid potential adverse patient outcomes.
Summary
There are several ways to diagnose and track resuscitation in the ED, which include physical examination, assessment of serum laboratory values, monitoring of hemodynamic status, and use of POCUS. Unfortunately, none of these methods provides a perfect assessment, and no method has been proven superior and effective over the others. Therefore, it is important for EPs treating patients in shock to be aware of the strengths and limitations of each assessment method (Table). 
1. Richards JB, Wilcox SR. Diagnosis and management of shock in the emergency department. Emerg Med Pract. 2014;16(3):1-22; quiz 22-23.
2. Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368-1377.
3. Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med. 2011;39:259-265. doi:10.1097/CCM.0b013e3181feeb15.
4. Seymour CW, Gesten F, Prescott HC, et al. Time to treatment and mortality during mandated emergency care for sepsis. N Engl J Med. 2017;376(23):2235-2244. doi:10.1056/NEJMoa1703058.
5. Cecconi M, De Backer D, Antonelli M, et al. Consensus on circulatory shock and hemodynamic monitoring. Task force of the European Society of Intensive Care Medicine. Intensive Care Med. 2014;40(12):1795-1815. doi:10.1007/s00134-014-3525-z.
6. Vincent JL, De Backer D. Circulatory shock. N Engl J Med. 2013;369(18):1726-1734. doi:10.1056/NEJMra1208943.
7. Saugel B, Ringmaier S, Holzapfel K, et al. Physical examination, central venous pressure, and chest radiography for the prediction of transpulmonary thermodilution-derived hemodynamic parameters in critically ill patients: a prospective trial. J Crit Care. 2011;26(4):402-410. doi:10.1016/j.jcrc.2010.11.001.
8. American College of Surgeons. Committee on Trauma. Shock. In: American College of Surgeons. Committee on Trauma, ed. Advanced Trauma Life Support: Student Course Manual. 9th ed. Chicago, IL: American College of Surgeons; 2012:69.
9. Saugel B, Kirsche SV, Hapfelmeier A, et al. Prediction of fluid responsiveness in patients admitted to the medical intensive care unit. J Crit Care. 2013:28(4):537.e1-e9. doi:10.1016/j.jcrc.2012.10.008.
10. McGee S, Abernethy WB 3rd, Simel DV. The rational clinical examination. Is this patient hypovolemic? JAMA. 1999;281(11):1022-1029.
11. Kompanje EJ, Jansen TC, van der Hoven B, Bakker J. The first demonstration of lactic acid in human blood in shock by Johann Joseph Scherer (1814-1869) in January 1843. Intensive Care Med. 2007;33(11):1967-1971. doi:10.1007/s00134-007-0788-7.
12. The ProCESS Investigators. A Randomized Trial of Protocol-Based Care for Early Septic Shock. N Engl J Med. 2014; 370:1683-1693. doi:10.1056/NEJMoa1401602.
13. Mouncey PR, Osborn TM, Power GS, et al. Protocolised Management In Sepsis (ProMISe): a multicentre randomised controlled trial of the clinical effectiveness and cost-effectiveness of early, goal-directed, protocolised resuscitation for emerging septic shock. Health Technol Assess. 2015;19(97):i-xxv, 1-150. doi:10.3310/hta19970.
14. ARISE Investigators; ANZICS Clinical Trials Group; Peake SL, Delaney A, Bailey M, et al. Goal-directed resuscitation for patients with early septic shock. N Engl J Med. 2014;371(16):1496-1506. doi:10.1056/NEJMoa1404380.
15. Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including The Pediatric Group. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013;39(2):165-228. doi:10.1007/s00134-012-2769-8.
16. Casserly B, Phillips GS, Schorr C, et al: Lactate measurements in sepsis-induced tissue hypoperfusion: results from the Surviving Sepsis Campaign database. Crit Care Med. 2015;43(3):567-573. doi:10.1097/CCM.0000000000000742.
17. Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care. 2013;3(1):12. doi:10.1186/2110-5820-3-12.
18. Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med. 2011;19:74. doi:10.1186/1757-7241-19-74.
19. Gaieski DF, Drumheller BC, Goyal M, Fuchs BD, Shofer FS, Zogby K. Accuracy of handheld point-of-care fingertip lactate measurement in the emergency department. West J Emerg Med. 2013;14(1):58-62. doi:10.5811/westjem.2011.5.6706.
20. Marik PE, Bellomo R. Lactate clearance as a target of therapy in sepsis: a flawed paradigm. OA Critical Care. 2013;1(1):3.
21. Kreisberg RA. Lactate homeostasis and lactic acidosis. Ann Intern Med. 1980;92(2 Pt 1):227-237.
22. Dodda VR, Spiro P. Can albuterol be blamed for lactic acidosis? Respir Care. 2012; 57(12):2115-2118. doi:10.4187/respcare.01810.
23. Scale T, Harvey JN. Diabetes, metformin and lactic acidosis. Clin Endocrinol (Oxf). 2011;74(2):191-196. doi:10.1111/j.1365-2265.2010.03891.x.
24. Velez JC, Janech MG. A case of lactic acidosis induced by linezolid. Nat Rev Nephrol. 2010;6(4):236-242. doi:10.1038/nrneph.2010.20.
25. Kam PC, Cardone D. Propofol infusion syndrome. Anaesthesia. 2007;62(7):690-701.
26. Griffith FR Jr, Lockwood JE, Emery FE. Adrenalin lactacidemia: proportionality with dose. Am J Physiol. 1939;127(3):415-421.
27. Rhodes A, Evans LE, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43(3):304-377. doi:10.1007/s00134-017-4683-6.
28. Early Goal-Directed Therapy Collaborative Group of Zhejiang Province. The effect of early goal-directed therapy on treatment of critical patients with severe sepsis/ septic shock: a multi-center, prospective, randomised, controlled study. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2010;22(6):331-334.
29. Thiele H, Ohman EM, Desch S, Eitel I, de Waha S. Management of cardiogenic shock. Eur Heart J. 2015;36(20):1223-1230. doi:10.1093/eurheartj/ehv051.
30. Lee M, Curley GF, Mustard M, Mazer CD. The Swan-Ganz catheter remains a critically important component of monitoring in cardiovascular critical care. Can J Cardiol. 2017;33(1):142-147. doi:10.1016/j.cjca.2016.10.026.
31. Morgan BC, Abel FL, Mullins GL, Guntheroth WG. Flow patterns in cavae, pulmonary artery, pulmonary vein, and aorta in intact dogs. Am J Physiol. 1966;210(4):903-909. doi:10.1152/ajplegacy.1966.210.4.903.
32. Brecher GA, Hubay CA. Pulmonary blood flow and venous return during spontaneous respiration. Circ Res. 1955;3(2):210-214.

33. Levy MM, Rhodes A, Phillips GS, et al. Surviving Sepsis Campaign: association between performance metrics and outcomes in a 7.5-year study. Intensive Care Med. 2014;40(11):1623-1633. doi:10.1007/s00134-014-3496-0.
34. Fernandes CJ Jr, Akamine N, Knobel E. Cardiac troponin: a new serum marker of myocardial injury in sepsis. Intensive Care Med. 1999;25(10):1165-1168. doi:10.1007/s001340051030.
35. Rady MY, Rivers EP, Nowak RM. Resuscitation of the critically III in the ED: responses of blood pressure, heart rate, shock index, central venous oxygen saturation, and lactate. Am J Emerg Med. 1996;14(2):218-225. doi:10.1016/s0735-6757(96)90136-9.
36. Gottlieb M, Hunter B. Utility of central venous pressure as a predictor of fluid responsiveness. Ann Emerg Med. 2016;68(1):114-116. doi:10.1016/j.annemergmed.2016.02.009.
37. Kornbau C, Lee KC, Hughes GD, Firstenberg MS. Central line complications. Int J Critical Illn Inj Sci. 2015;5(3):170-178. doi:10.4103/2229-5151.164940.
38. Walley KR. Use of central venous oxygen saturation to guide therapy. Am J Respir Crit Care Med. 2011;184(5):514-520. doi:10.1164/rccm.201010-1584CI.
39. PRISM Investigators, Rowan KM, Angus DC, et al. Early, goal-directed therapy for septic shock - a patient-level meta-analysis. N Engl J Med. 2017;376(23):2223-2234. doi:10.1056/NEJMoa1701380.
40. Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO(2)) as a predictor of mortality in patients with sepsis. Ann Emerg Med. 2010;55(1):40-46.e1. doi:10.1016/j.annemergmed.2009.08.014.
41. Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA. 2010;303(8):739-746. doi:10.1001/jama.2010.158.
42. Nowak RM, Sen A, Garcia, AJ, et al. The inability of emergency physicians to adequately clinically estimate the underlying hemodynamic profiles of acutely ill patients. Am J Emerg Med. 2012;30(6):954-960. doi:10.1016/j.ajem.2011.05.021.
43. Swan HJ, Ganz W, Forrester J, Marcus H, Diamond G, Chonette D. Catheterization of the heart in man with use of a flow-directed balloon-tipped catheter. N Engl J Med. 1970;283(9):447-451. doi:10.1056/NEJM197008272830902.
44. Hadian M, Pinsky MR. Evidence-based review of the use of the pulmonary artery catheter: impact data and complications. Crit Care. 2006;10 Suppl 3:S8.
45. Rajaram SS, Desai, NK, Kalra A, et al. Pulmonary artery catheters for adult patients in intensive care. Cochrane Database Syst Rev. 2013;(2):CD003408. doi:10.1002/14651858.CD003408.pub3.
46. Laher AE, Watermeyer MJ, Buchanan SK, et al. A review of hemodynamic monitoring techniques, methods and devices for the emergency physician. Am J Emerg Med. 2017;35(9):1335-1347. doi:10.1016/j.ajem.2017.03.036.
47. Perera P, Mailhot T, Riley D, Mandavia D. The RUSH exam: Rapid Ultrasound in SHock in the evaluation of the critically lll. Emerg Med Clin North Am. 2010;28(1):29-56, vii. doi:10.1016/j.emc.2009.09.010.
48. Heller MB, Mandavia D, Tayal VS, et al. Residency training in emergency ultrasound: fulfilling the mandate. Acad Emerg Med. 2002;9(8):835-839.
49. Ultrasound guidelines: emergency, point-of-care and clinical ultrasound guidelines in medicine. Ann Emerg Med. 2016;69(5):e27-e54. doi:10.1016/j.annemergmed.2016.08.457.
50. Expert Round Table on Ultrasound in ICU. International expert statement on training standards for critical care ultrasonography. Intensive Care Med. 2011;37(7):1077-1083. doi:10.1007/s00134-011-2246-9.
51. Neri L, Storti E, Lichtenstein D. Toward an ultrasound curriculum for critical care medicine. Crit Care Med. 2007;35(5 Suppl):S290-S304.
52. Simonson JS, Schiller NB. Sonospirometry: a new method for noninvasive estimation of mean right atrial pressure based on two-dimensional echographic measurements of the inferior vena cava during measured inspiration. J Am Coll Cardiol. 1988;11(3):557-564.
53. Kircher BJ, Himelman RB, Schiller NB. Noninvasive estimation of right atrial pressure from the inspiratory collapse of the inferior vena cava. Am J Cardiol. 1990;66(4):493-496.
54. Nagdev AD, Merchant RC, Tirado-Gonzalez A, Sisson CA, Murphy MC. Emergency department bedside ultrasonographic measurement of the caval index for noninvasive determination of low central venous pressure. Ann Emerg Med. 2010;55(3):290-295. doi:10.1016/j.annemergmed.2009.04.021.
55. Barbier C, Loubières Y, Schmit C, et al. Respiratory changes in inferior vena cava diameter are helpful in predicting fluid responsiveness in ventilated septic patients. Intensive Care Med. 2004;30(9):1740-1746.
56. Corl KA, George NR, Romanoff J, et al. Inferior vena cava collapsibility detects fluid responsiveness among spontaneously breathing critically-ill patients. J Crit Care. 2017;41:130-137. doi:10.1016/j.jcrc.2017.05.008.
57. Feissel M, Michard F, Faller JP, Teboul JL. The respiratory variation in inferior vena cava diameter as a guide to fluid therapy. Intensive Care Med. 2004;30(9):1834-1837.
58. Blehar DJ, Dickman E, Gaspari R. Identification of congestive heart failure via respiratory variation of inferior vena cava diameter. Am J Emerg Med. 2009;27(1):71-75. doi:10.1016/j.ajem.2008.01.002.
59. Moore CL, Rose GA, Tayal VS, Sullivan DM, Arrowood JA, Kline JA. Determination of left ventricular function by emergency physician echocardiography of hypotensive patients. Acad Emerg Med. 2002;9(3):186-193.
60. Mandavia DP, Hoffner RJ, Mahaney K, Henderson SO. Bedside echocardiography by emergency physicians. Ann Emerg Med. 2001;38(4):377-382.
61. Mosier JM, Martin J, Andrus P, et al. Advanced hemodynamic and cardiopulmonary ultrasound for critically ill patients in the emergency department. Emerg Med. 2018;50(1):17-34. doi:10.12788/emed.2018.0078.
62. Agarwal S, Swanson S, Murphy A, Yaeger K, Sharek P, Halamek LP. Comparing the utility of a standard pediatric resuscitation cart with a pediatric resuscitation cart based on the Broselow tape: a randomized, controlled, crossover trial involving simulated resuscitation scenarios. Pediatrics. 2005;116(3):e326-e333.
63. Blanco P, Volpicelli G. Common pitfalls in point-of-care ultrasound: a practical guide for emergency and critical care physicians. Crit Ultrasound J. 2016;8(1):15.
64. Tajoddini S, Shams Vahdati S. Ultrasonographic diagnosis of abdominal free fluid: accuracy comparison of emergency physicians and radiologists. Eur J Trauma Emerg Surg. 2013;39(1):9-13. doi:10.1007/s00068-012-0219-5.
65 Abbasi S, Bolverdi E, Zare MA, et al. Comparison of diagnostic value of conventional ultrasonography by emergency physicians with Doppler ultrasonography by radiology physicians for diagnosis of deep vein thrombosis. J Pak Med Assoc. 2012;62(5):461-465.
66. Arhami Dolatabadi A, Amini A, Hatamabadi H, et al. Comparison of the accuracy and reproducibility of focused abdominal sonography for trauma performed by emergency medicine and radiology residents. Ultrasound Med Biol. 2014;40(7):1476-1482. doi:10.1016/j.ultrasmedbio.2014.01.017.
67. Karimi E, Aminianfar M, Zarafshani K, Safaie A. The accuracy of emergency physicians in ultrasonographic screening of acute appendicitis; a cross sectional study. Emerg (Tehran). 2017;5(1):e22.
Resuscitation of critically ill patients in shock from cardiogenic, hypovolemic, obstructive, distributive, or neurogenic etiology is a cornerstone of the care delivered by emergency physicians (EPs).1 Regardless of the etiology, it is essential that the treating EP initiate resuscitative measures in a timely manner and closely trend the patient’s response to these interventions.
The early goal-directed therapy (EGDT) initially proposed by Rivers et al2 in 2001 demonstrated a bundled approach to fluid resuscitation by targeting end points for volume resuscitation, mean arterial blood pressure (MAP), oxygen (O2) delivery/extraction (mixed venous O2 saturation, [SvO2]), hemoglobin (Hgb) concentration, and cardiac contractility. Since then, advancements in laboratory testing and hemodynamic monitoring (HDM) devices further aid and guide resuscitative efforts, and are applicable to any etiology of shock.
In addition to these advancements, the growing evidence of the potential harm from improper fluid resuscitation, such as the administration of excessive intravascular fluid (IVF),3 underscores the importance of a precise, targeted, and individualized approach to care. This article reviews the background, benefits, and limitations of some of the common and readily available tools in the ED that the EP can employ to guide fluid resuscitation in critically ill patients.
Physical Examination
Background
The rapid recognition and treatment of septic shock in the ED is associated with lower rates of in-hospital morbidity and mortality.4 The physical examination by the EP begins immediately upon examining the patient. The acquisition of vital signs and recognition of physical examination findings suggestive of intravascular volume depletion allows the EP to initiate treatment immediately.
In this discussion, hypotension is defined as systolic blood pressure (SBP) of less than 95 mm Hg, MAP of less than 65 mm Hg, or a decrease in SBP of more than 40 mm Hg from baseline measurements. Subsequently, shock is defined as hypotension with evidence of tissue hypoperfusion-induced dysfunction.5,6 Although the use of findings from the physical examination to guide resuscitation allows for rapid patient assessment and treatment, the predictive value of the physical examination to assess hemodynamic status is limited.
Visual inspection of the patient’s skin and mucous membranes can serve as an indicator of volume status. The patient’s tongue should appear moist with engorged sublingual veins; a dry tongue and diminished veins may suggest the need for volume resuscitation. On examination of the skin, delayed capillary refill of the digits and cool, clammy extremities suggest the shunting of blood by systemic circulation from the skin to central circulation. Patients who progress to more severe peripheral vasoconstriction develop skin mottling, referred to as livedo reticularis (Figure 1).
Benefits
The major benefit of the physical examination as a tool to evaluate hemodynamic status is its ease and rapid acquisition. The patient’s vital signs and physical examination can be obtained in the matter of moments upon presentation, without the need to wait on results of laboratory evaluation or additional equipment. Additionally, serial examinations by the same physician can be helpful to monitor a patient’s response to resuscitative efforts. The negative predictive value (NPV) of the physical examination in evaluating for hypovolemia may be helpful, but only when it is taken in the appropriate clinical context and is used in conjunction with other diagnostic tools. The physical examination can exclude hypovolemic volume status with an NPV of approximately 70%.7
A constellation of findings from the physical examination may include altered mentation, hypotension, tachycardia, and decreased urinary output by 30% to 40% intravascular volume loss.8,9Findings from the physical examination to assess fluid status should be used with caution as interobserver reliability has proven to be poor and the prognostic value is limited.
Limitations
The literature shows the limited prognostic value of the physical examination in determining a patient’s volume status and whether fluid resuscitation is indicated. For example, in one meta-analysis,10 supine hypotension and tachycardia were frequently absent on examination—even in patients who underwent large volume phlebotomy.8 This study also showed postural dizziness to be of no prognostic value.
Another study by Saugel et al7 that compared the physical examination (skin assessment, lung auscultation, and percussion) to transpulmonary thermodilution measurements of the cardiac index, global end-diastolic volume index, and extravascular lung water index, found poor interobserver correlation and agreement among physicians.
The physical examination is also associated with weak predictive capabilities for the estimation of volume status compared to the device measurements. Another contemporary study by Saugel et al9 evaluated the predictive value of the physical examination to accurately identify volume responsiveness replicated these results, and reported poor interobserver correlation (κ coefficient 0.01; 95% caval index [CI] -0.39-0.42) among physical examination findings, with a sensitivity of only 71%, specificity of 23.5%, positive predictive value of 27.8%, and negative predictive value of 66.7%.9
Serum Lactate Levels
Background
In the 1843 book titled, Investigations of Pathological Substances Obtained During the Epidemic of Puerperal Fever, Johann Joseph Scherer described the cases of seven young peripartum female patients who died from a clinical picture of what is now understood to be septic shock.11 In his study of these cases, Scherer demonstrated the presence of lactic acid in patients with pathological conditions. Prior to this discovery, lactic acid had never been isolated in a healthy individual. These results were recreated in 1851 by Scherer and Virchow,11 who demonstrated the presence of lactic acid in the blood of a patient who died from leukemia. The inference based on Scherer and Virchow’s work correlated the presence of excessive lactic acid with bodily deterioration and severe disease. Since this finding, there has been a great deal of interest in measuring serum lactic acid as a means to identify and manage critical illness.
In a 2001 groundbreaking study of EGDT for severe sepsis and septic shock, Rivers et al2 studied lactic acid levels as a marker for severe disease. Likewise, years later, the 2014 Protocol-Based Care for Early Septic Shock (PROCESS), Prospective Multicenter Imaging Study for Evaluation of Chest Pain (PROMISE), and Australasian Resuscitation in Sepsis Evaluation (ARISE) trials used lactate levels in a similar manner to identify patients appropriate for randomization.12-14 While the purpose of measuring lactic acid was only employed in these studies to identify patients at risk for critical illness, the 2012 Surviving Sepsis Campaign Guidelines recommended serial measurement of lactate, based on the assumption that improved lactate levels signified better tissue perfusion.15
Although much of the studies on lactate levels appear to be based on the treatment and management of septic patients, findings can be applied to any etiology of shock. For example, a serum lactate level greater than 2 mmol/L is considered abnormal, and a serum lactate greater than 4 mmol/L indicates a significantly increased risk for in-hospital mortality.16
Benefits
It is now a widely accepted belief that the rapid identification, triage, and treatment of critically ill patients has a dramatic effect on morbidity and mortality.4 As previously noted, lactate has been extensively studied and identified as a marker of severe illness.17,18 A serum lactate level, which can be rapidly processed in the ED, can be easily obtained from a minimally invasive venous, arterial, or capillary blood draw.18 The only risk associated with serum lactate testing is that of any routine venipuncture; the test causes minimal, if any, patient discomfort.
Thanks to advances in point-of-care (POC) technology, the result of serum lactate assessment can be available within 10 minutes from blood draw. This technology is inexpensive and can be easily deployed in the prehospital setting or during the initial triage assessment of patients arriving at the ED.19 These POC instruments have been well correlated with whole blood measurements and permit for the rapid identification and treatment of at risk patients.
Limitations
The presence of elevated serum lactate levels is believed to represent the presence of cellular anaerobic metabolism due to impaired O2 delivery in the shock state. Abnormal measurements therefore prompt aggressive interventions aimed at maximizing O2 delivery to the tissues, such as intravenous fluid boluses, vasopressor therapy, or even blood product administration.
A return to a normalized serum lactate level is assumed to represent a transition back to aerobic metabolism. Lactate elevations, however, are not solely an indication of anaerobic metabolism and may only represent a small degree of lactate production.20 While the specific cellular mechanics are out of the scope of this article, it has been postulated that the increase in plasma lactate concentration is primarily driven by β-2 receptor stimulation from increased circulating catecholamines leading to increased aerobic glycolysis. Increased lactate levels could therefore be an adaptive mechanism of energy production—aggressive treatment and rapid clearance may, in fact, be harmful. Type A lactic acidosis is categorized as elevated serum levels due to tissue hypoperfusion.21
However, lactate elevations do not exclusively occur in severe illness. The use of β-2 receptor agonists such as continuous albuterol treatments or epinephrine may cause abnormal lactate levels.22 Other medications have also been associated with elevated serum lactate levels, including, but not limited to linezolid, metformin, and propofol.23-25 Additionally, lactate levels may be elevated after strenuous exercise, seizure activity, or in liver and kidney disease.26 These “secondary” causes of lactic acidosis that are not due to tissue hypoperfusion are referred to as type B lactic acidosis. Given these multiple etiologies and lack of specificity for this serum measurement, a failure to understand these limitations may result in over aggressive or unnecessary medical treatments.
Central Venous Pressure
Background
Central venous pressure (CVP) measurements can be obtained through a catheter, the distal tip of which transduces pressure of the superior vena cava at the entrance of the right atrium (RA). Thus, CVP is often used as a representation of RA pressure (RAP) and therefore an estimate of right ventricular (RV) preload. While CVP is used to diagnose and determine the etiology of shock, evidence and controversy regarding the use of CVP as a marker for resuscitation comes largely from sepsis-focused literature.5 Central venous pressure is meant to represent preload, which is essential for stroke volume as described by the Frank-Starling mechanism; however, its use as a target in distributive shock, a state in which it is difficult to determine a patient’s volume status, has been popularized by EGDT since 2001.2
Since the publication of the 2004 Surviving Sepsis Guidelines, CVP monitoring has been in the spotlight of sepsis resuscitation, albeit with some controversy.27 Included as the result of two studies, this recommendation has been removed in the most recent guidelines after 12 years of further study and scrutiny.2,27,28
Hypovolemic and hemorrhagic shock are usually diagnosed clinically and while a low CVP can be helpful in the diagnosis, the guidelines do not support CVP as a resuscitation endpoint. Obstructive and cardiogenic shock will both result in elevated CVP; however, treatment of obstructive shock is generally targeted at the underlying cause. While cardiogenic shock can be preload responsive, the mainstay of therapy in the ED is identification of patients for revascularization and inotropic support.29
Benefits
The CVP has been used as a surrogate for RV preload volume. If a patient’s preload volume is low, the treating physician can administer fluids to improve stroke volume and cardiac output (CO). Clinically, CVP measurements are easy to obtain provided a central venous line has been placed with the distal tip at the entrance to the RA. Central venous pressure is measured by transducing the pressure via manometry and connecting it to the patient’s bedside monitor. This provides an advantage of being able to provide serial or even continuous measurements. The “normal” RAP should be a low value (1-5 mm Hg, mean of 3 mm Hg), as this aids in the pressure gradient to drive blood from the higher pressures of the left ventricle (LV) and aorta through the circulation back to the low-pressure of the RA.30 The value of the CVP is meant to correspond to the physical examination findings of jugular venous distension.31,32 Thus, a low CVP may be “normal” and seen in patients with hypovolemic shock, whereas an elevated CVP can suggest volume overload or obstructive shock. However, this is of questionable value in distributive shock cases.
Aside from the two early studies on CVP monitoring during treatment of septic patients, there are few data to support the use of CVP measurement in the early resuscitation of patients with shock.2,28 More recent trials (PROMISE, ARISE, PROCESS) that compared protocolized sepsis care to standard care showed no benefit to bundles including CVP measurements.12-14 However, a subsequent, large observational trial spanning 7.5 years demonstrated improvements in sepsis-related mortality in patients who received a central venous catheter (CVC) and CVP-targeted therapy.33 Thus, it is possible that protocols including CVP are still beneficial in combination with other therapies even though CVP in isolation is not.
Limitations
The traditional two assumptions in CVP monitoring are CVP value represents the overall volume status of the patient, and the LV is able to utilize additional preload volume. The latter assumption, however, may be hampered by the presence of sepsis-induced myocardial dysfunction, which may be present in up to 40% of critically ill patients.34 The former assumption does not always hold true due to processes that change filling pressures independent of intravascular volume—eg, acute or chronic pulmonary hypertension, cardiac tamponade, intra-abdominal hypertension, or LV failure. Even before the landmark EGDT study, available data suggested that CVP was not a reliable marker for resuscitation management.35 A recent systematic review by Gottlieb and Hunter36 showed that the area under the receiver-operator curve for low, mid-range, or high CVPs was equivocal at best. In addition to its unreliability and lack of specificity, another significant drawback to using CVP to guide resuscitation therapy in the ED is that it necessitates placement of a CVC, which can be time-consuming and, if not otherwise indicated, lead to complications of infection, pneumothorax, and/or thrombosis.37
Mixed Venous Oxygen
Background
Most EPs are familiar with the use of ScvO2 in EGDT protocols to guide volume resuscitation of septic patients.2 A patient’s ScvO2 represents the O2 saturation of venous blood obtained via a CVC at the confluence of the superior vena cava and the RA, and thus it reflects tissue O2 consumption as a surrogate for tissue perfusion. The measurement parallels the SvO2 obtained from the pulmonary artery. In a healthy patient, SvO2 is around 65% to 70% and includes blood returning from both the superior and inferior vena cava (IVC). As such, ScvO2 values are typically 3% to 5% lower than SvO2 owing to the lower O2 extracted by tissues draining into the IVC compared to the mixed venous blood sampled from the pulmonary artery.38
Though a debate over the benefit of EGDT in treating sepsis continues, understanding the physiology of ScvO2 measurements is another potential tool the EP can use to guide the resuscitation of critically ill patients.39 A patient’s SvO2 and, by extension, ScvO2 represents the residual O2 saturation after the tissues have extracted the amount of O2 necessary to meet metabolic demands (Figure 2).
Conversely, cellular dysfunction, which can occur in certain toxicities or in severe forms of sepsis, can lead to decreased tissue O2 consumption with a concomitant rise in ScvO2 to supernormal values.38 The EP should take care, however, to consider whether ScvO2 values exceeding 80% represent successful therapeutic intervention or impaired tissue O2 extraction and utilization. There are data from ED patients suggesting an increased risk of mortality with both extremely low and extremely high values of ScvO2.40
Benefits
A critically ill patient’s ScvO2 can potentially provide EPs with insight into the patient’s global tissue perfusion and the source of any mismatch between O2 delivery and consumption. Using additional tools and measurements (physical examination, serum Hgb levels, and pulse oximetry) in conjunction with an ScvO2 measurement, assists EPs in identifying targets for therapeutic intervention. The effectiveness of this intervention can then be assessed using serial ScvO2 measurements, as described in Rivers et al2 EGDT protocol. Importantly, EPs should take care to measure serial ScvO2 values to maximize its utility.38 Similar to a CVP measurement, ScvO2is easily obtained from blood samples for serial laboratory measurements, assuming the patient already has a CVC with the distal tip at the entrance to the RA (ScvO2) or a pulmonary artery catheter (PAC) (SvO2).
Limitations
Serial measurements provide the most reliable information, which may be more useful in patients who spend extended periods of their resuscitation in the ED. In comparison to other measures of global tissue hypoxia, work by Jones et al41 suggests non-inferiority of peripherally sampled, serial lactate measurements as an alternative to ScvO2. This, in conjunction with the requirement for an internal jugular CVC, subclavian CVC, or PAC with their associated risks, may make ScvO2 a less attractive guide for the resuscitation of critically ill patients in the ED.
Monitoring Devices
Background
As noted throughout this review, it is important not only to identify and rapidly treat shock, but to also correctly identify the type of shock, such that treatment can be appropriately directed at its underlying cause. However, prior work suggests that EPs are unable to grossly estimate CO or systemic vascular resistance when compared to objective measurements of these parameters.42 This is in agreement with the overall poor performance of physical examination and clinical evaluation as a means of predicting volume responsiveness or guiding resuscitation, as discussed previously. Fortunately, a wide variety of devices to objectively monitor hemodynamics are now available to the EP.
In 1970, Swan et al43 published their initial experience with pulmonary artery catheterization at the bedside, using a balloon-tipped, flow-guided PAC in lieu of fluoroscopy, which had been mandated by earlier techniques. The ability to measure CO, right heart pressures, pulmonary arterial pressures, and estimate LV end diastolic pressure ushered in an era of widespread PAC use, despite an absence of evidence for causation of improved patient outcomes. The utilization of PACs has fallen, as the literature suggests that the empiric placement of PACs in critically ill patients does not improve mortality, length of stay, or cost, and significant complication rates have been reported in large trials.44,45Subsequently, a number of non-invasive or less-invasive HDM devices have been developed. Amongst the more commonly encountered modern devices, the techniques utilized for providing hemodynamic assessments include thermodilution and pulse contour analysis (PiCCOTM), pulse contour analysis (FloTrac/VigileoTM), and lithium chemodilution with pulse power analysis (LiDCOplusTM).46 The primary utility of these devices for the EP lies in the ability to quantify CO, stroke volume, and stroke volume or pulse pressure variation (PPV) to predict or assess response to resuscitative interventions (volume administration, vasopressors, inotropes, etc).
Benefits
Many of these devices require placement of an arterial catheter. Some require the addition of a CVC. Both of these procedures are well within the clinical scope of the EP, and are performed with fair frequency on critically ill patients. This is a distinct advantage when compared to pulmonary artery catheterization, a higher risk procedure that is rarely performed outside of the intensive care unit or cardiac catheterization laboratory. In addition, all of the devices below present hemodynamic data in a graphical, easy-to-read format, in real time. All of the devices discussed report stroke volume variation (SVV) or PPV continuously.
Limitations
Though these measures have validated threshold values that predict volume responsiveness, they require the patient to be intubated with a set tidal volume of greater than or equal to 8 mL/kg without spontaneous respirations and cardiac arrhythmias, in order to accurately do so. All of the HDM devices that rely on pulse contour analysis as the primary means of CO measurement cannot be used in the presence of significant cardiac arrhythmias (ie, atrial fibrillation), or mechanical circulatory assistance devices (ie, intra-aortic balloon counterpulsation). None of these devices are capable of monitoring microcirculatory changes, felt to be of increasing clinical importance in the critically ill.
The use of HDM devices to monitor CO with a reasonable degree of accuracy, trend CO, and assess for volume responsiveness using a number of previously validated parameters such as SVV is now in little doubt. However, these devices are still invasive, if less so than a pulmonary artery. The crux of the discussion of HDM devices for use in ED resuscitation revolves around whether or not the use of such devices to drive previously validated, protocolized care results in better outcomes for patients. The EP can now have continuous knowledge of a large number of hemodynamic parameters at their fingertips with relatively minimal additional efforts. At the time of this writing, though, this is both untested and unproven, with respect to the ED population.
Point-of-Care Ultrasound
Background
Over the past two decades, ultrasound (US) has become an integral part of the practice of emergency medicine (EM), and is now included in all United States Accreditation Council for Graduate Medical Education Emergency Medicine Residency Programs.47,48 It has emerged as a very important bedside tool performed by the clinician to identify type of shock and guide resuscitation, and has been endorsed by both EM and critical care societies.49-51 This section reviews the utility of US as a modality in identifying shock and guiding resuscitation, in addition to the pitfalls and limitations of this important tool.
In 2010, Perera et al47 described in their landmark article the Rapid Ultrasound in SHock (RUSH) examination, which describes a stepwise (the pump, tank, pipes) approach to identify the type of shock (cardiogenic, hypovolemic, obstructive, or distributive) in the crashing, hypotensive ED patient. We do not describe the full RUSH examination in this review, but discuss key elements of it as examples of how POCUS can assist the EP to make a rapid diagnosis and aid in the management of patients in shock. The “pump” is the heart, which is assessed in four different views to identify a pericardial effusion and possible tamponade, assess contractility or ejection fraction of the LV (severely decreased, decreased, normal, or hyperdynamic), and right heart strain which is identified by an RV that is larger than the LV, indicative of a potential pulmonary embolus.
The “tank” is then assessed by visualizing the IVC in the subxiphoid plane, and is evaluated for respiratory collapsibility (CI) and maximum size. This has been quite the debated topic over the last two decades. In 1988, Simonson and Schiller52 were the first to describe a correlation in spontaneously breathing patients between IVC caliber (measured 2 cm from the cavoatrial junction) and variation and RAP, where a larger IVC diameter and less respiratory variation correlated with a high RAP. Kircher et al53 later went on to describe that a CI greater than 50% correlated with an RAP of less than 10 mm Hg and vice versa in spontaneously breathing patients. Since then there have been more studies attempting to verify these findings in both spontaneously breathing and mechanically ventilated patients.54-56 The purpose of performing these measurements is not to estimate CVP, but to assess fluid responsiveness (ie, a blood pressure response to a fluid challenge). It can be assumed in states of shock that a small IVC, or one with a high CI, in the presence of a hyperdynamic heart is indicative of an underfilled ventricle and fluid responsiveness, especially if the IVC size increases with fluid.55,57 However, there are several caveats to this. First, in mechanically ventilated patients, the IVC is already plethoric due to positive pressure ventilation, and increases in diameter with inspiration and decreases with expiration as compared to spontaneously breathing patients. Second, the CI value to predict volume responsiveness in ventilated patients is set at 15% instead of 50%.55 Third, it is important to always take the clinical scenario in context; a dilated IVC with small CI is not necessarily only due to volume overload and congestive heart failure, but can be due to elevated RAP from obstructive shock due to cardiac tamponade or massive pulmonary embolus, which is why it is important to assess the “pump” first.47,58 It is also crucial to not forget to assess the abdominal and thoracic cavities, as intraperitoneal or pleural fluid with a collapsed IVC can potentially make a diagnosis of hemorrhagic or hypovolemic shock depending on the clinical scenario.47 The final part of the RUSH protocol is to evaluate the “pipes,” inclusive of the lower extremity deep venous system for evaluation of potential thrombosis that could increase suspicion for a pulmonary embolism causing obstructive shock, and the aorta with the common iliac arteries if there is concern for aortic dissection or aneurysmal rupture.
Benefits
Some of the most significant advantages to the use of POCUS to guide resuscitation is that it is quick, non-invasive, does not use ionizing radiation, and can be easily repeated. As noted above, it is a requirement for EM residencies to teach its use, so that contemporary graduates are entering the specialty competent in applying it to the care of their patients. Furthermore, POCUS is done at the bedside, limiting the need to potentially transport unstable patients.
In the most basic applications, POCUS provides direct visualization of a patient’s cardiac function, presence or absence of lung sliding to suggest a pneumothorax, presence of pulmonary edema, assessment of CVP pressures or potential for fluid responsiveness, as well as identification of potential thoracic, peritoneal, or pelvic cavity fluid accumulation that may suggest hemorrhage. There is literature to support that these assessments performed by the EP have been shown to be comparable to those of cardiologists.59,60 With continued practice and additional training, it is possible for EPs to even perform more “advanced” hemodynamic assessments to both diagnose and guide therapy to patients in shock (Figures 3 and 4).61
Limitations
Although POCUS has been shown as a remarkable tool to help assist the EP in making rapid decisions regarding resuscitation, it is always important to remember its limitations. Most of the studies regarding its use are of very small sample sizes, and further prospective studies have to be performed in order for this modality to be fully relied on.62Compared to some of the previously mentioned HDM devices that may provide continuous data, POCUS needs to be performed by the treating physician, thereby occurring intermittently. Emergency physicians need to be aware of their own experience and limitations with this modality, as errors in misdiagnosis can lead to unnecessary procedures, with resulting significant morbidity and mortality. Blanco and Volpicelli63 describe several common errors that include misdiagnosing the stomach as a peritoneal effusion, assuming adequate volume resuscitation when the IVC is seen to be plethoric in the setting of cardiac tamponade, or mistaking IVC movement as indicative of collapsibility, amongst other described misinterpretations. Several other studies have shown that, despite adequate performance of EPs in POCUS, diagnostic sensitivities remained higher when performed by radiologists.64-67 Thus it remains important for the EPs to be vigilant and not anchor on a diagnosis when in doubt, and to consult early with radiology, particularly if there is any question, to avoid potential adverse patient outcomes.
Summary
There are several ways to diagnose and track resuscitation in the ED, which include physical examination, assessment of serum laboratory values, monitoring of hemodynamic status, and use of POCUS. Unfortunately, none of these methods provides a perfect assessment, and no method has been proven superior and effective over the others. Therefore, it is important for EPs treating patients in shock to be aware of the strengths and limitations of each assessment method (Table).
Resuscitation of critically ill patients in shock from cardiogenic, hypovolemic, obstructive, distributive, or neurogenic etiology is a cornerstone of the care delivered by emergency physicians (EPs).1 Regardless of the etiology, it is essential that the treating EP initiate resuscitative measures in a timely manner and closely trend the patient’s response to these interventions.
The early goal-directed therapy (EGDT) initially proposed by Rivers et al2 in 2001 demonstrated a bundled approach to fluid resuscitation by targeting end points for volume resuscitation, mean arterial blood pressure (MAP), oxygen (O2) delivery/extraction (mixed venous O2 saturation, [SvO2]), hemoglobin (Hgb) concentration, and cardiac contractility. Since then, advancements in laboratory testing and hemodynamic monitoring (HDM) devices further aid and guide resuscitative efforts, and are applicable to any etiology of shock.
In addition to these advancements, the growing evidence of the potential harm from improper fluid resuscitation, such as the administration of excessive intravascular fluid (IVF),3 underscores the importance of a precise, targeted, and individualized approach to care. This article reviews the background, benefits, and limitations of some of the common and readily available tools in the ED that the EP can employ to guide fluid resuscitation in critically ill patients.
Physical Examination
Background
The rapid recognition and treatment of septic shock in the ED is associated with lower rates of in-hospital morbidity and mortality.4 The physical examination by the EP begins immediately upon examining the patient. The acquisition of vital signs and recognition of physical examination findings suggestive of intravascular volume depletion allows the EP to initiate treatment immediately.
In this discussion, hypotension is defined as systolic blood pressure (SBP) of less than 95 mm Hg, MAP of less than 65 mm Hg, or a decrease in SBP of more than 40 mm Hg from baseline measurements. Subsequently, shock is defined as hypotension with evidence of tissue hypoperfusion-induced dysfunction.5,6 Although the use of findings from the physical examination to guide resuscitation allows for rapid patient assessment and treatment, the predictive value of the physical examination to assess hemodynamic status is limited.
Visual inspection of the patient’s skin and mucous membranes can serve as an indicator of volume status. The patient’s tongue should appear moist with engorged sublingual veins; a dry tongue and diminished veins may suggest the need for volume resuscitation. On examination of the skin, delayed capillary refill of the digits and cool, clammy extremities suggest the shunting of blood by systemic circulation from the skin to central circulation. Patients who progress to more severe peripheral vasoconstriction develop skin mottling, referred to as livedo reticularis (Figure 1).
Benefits
The major benefit of the physical examination as a tool to evaluate hemodynamic status is its ease and rapid acquisition. The patient’s vital signs and physical examination can be obtained in the matter of moments upon presentation, without the need to wait on results of laboratory evaluation or additional equipment. Additionally, serial examinations by the same physician can be helpful to monitor a patient’s response to resuscitative efforts. The negative predictive value (NPV) of the physical examination in evaluating for hypovolemia may be helpful, but only when it is taken in the appropriate clinical context and is used in conjunction with other diagnostic tools. The physical examination can exclude hypovolemic volume status with an NPV of approximately 70%.7
A constellation of findings from the physical examination may include altered mentation, hypotension, tachycardia, and decreased urinary output by 30% to 40% intravascular volume loss.8,9Findings from the physical examination to assess fluid status should be used with caution as interobserver reliability has proven to be poor and the prognostic value is limited.
Limitations
The literature shows the limited prognostic value of the physical examination in determining a patient’s volume status and whether fluid resuscitation is indicated. For example, in one meta-analysis,10 supine hypotension and tachycardia were frequently absent on examination—even in patients who underwent large volume phlebotomy.8 This study also showed postural dizziness to be of no prognostic value.
Another study by Saugel et al7 that compared the physical examination (skin assessment, lung auscultation, and percussion) to transpulmonary thermodilution measurements of the cardiac index, global end-diastolic volume index, and extravascular lung water index, found poor interobserver correlation and agreement among physicians.
The physical examination is also associated with weak predictive capabilities for the estimation of volume status compared to the device measurements. Another contemporary study by Saugel et al9 evaluated the predictive value of the physical examination to accurately identify volume responsiveness replicated these results, and reported poor interobserver correlation (κ coefficient 0.01; 95% caval index [CI] -0.39-0.42) among physical examination findings, with a sensitivity of only 71%, specificity of 23.5%, positive predictive value of 27.8%, and negative predictive value of 66.7%.9
Serum Lactate Levels
Background
In the 1843 book titled, Investigations of Pathological Substances Obtained During the Epidemic of Puerperal Fever, Johann Joseph Scherer described the cases of seven young peripartum female patients who died from a clinical picture of what is now understood to be septic shock.11 In his study of these cases, Scherer demonstrated the presence of lactic acid in patients with pathological conditions. Prior to this discovery, lactic acid had never been isolated in a healthy individual. These results were recreated in 1851 by Scherer and Virchow,11 who demonstrated the presence of lactic acid in the blood of a patient who died from leukemia. The inference based on Scherer and Virchow’s work correlated the presence of excessive lactic acid with bodily deterioration and severe disease. Since this finding, there has been a great deal of interest in measuring serum lactic acid as a means to identify and manage critical illness.
In a 2001 groundbreaking study of EGDT for severe sepsis and septic shock, Rivers et al2 studied lactic acid levels as a marker for severe disease. Likewise, years later, the 2014 Protocol-Based Care for Early Septic Shock (PROCESS), Prospective Multicenter Imaging Study for Evaluation of Chest Pain (PROMISE), and Australasian Resuscitation in Sepsis Evaluation (ARISE) trials used lactate levels in a similar manner to identify patients appropriate for randomization.12-14 While the purpose of measuring lactic acid was only employed in these studies to identify patients at risk for critical illness, the 2012 Surviving Sepsis Campaign Guidelines recommended serial measurement of lactate, based on the assumption that improved lactate levels signified better tissue perfusion.15
Although much of the studies on lactate levels appear to be based on the treatment and management of septic patients, findings can be applied to any etiology of shock. For example, a serum lactate level greater than 2 mmol/L is considered abnormal, and a serum lactate greater than 4 mmol/L indicates a significantly increased risk for in-hospital mortality.16
Benefits
It is now a widely accepted belief that the rapid identification, triage, and treatment of critically ill patients has a dramatic effect on morbidity and mortality.4 As previously noted, lactate has been extensively studied and identified as a marker of severe illness.17,18 A serum lactate level, which can be rapidly processed in the ED, can be easily obtained from a minimally invasive venous, arterial, or capillary blood draw.18 The only risk associated with serum lactate testing is that of any routine venipuncture; the test causes minimal, if any, patient discomfort.
Thanks to advances in point-of-care (POC) technology, the result of serum lactate assessment can be available within 10 minutes from blood draw. This technology is inexpensive and can be easily deployed in the prehospital setting or during the initial triage assessment of patients arriving at the ED.19 These POC instruments have been well correlated with whole blood measurements and permit for the rapid identification and treatment of at risk patients.
Limitations
The presence of elevated serum lactate levels is believed to represent the presence of cellular anaerobic metabolism due to impaired O2 delivery in the shock state. Abnormal measurements therefore prompt aggressive interventions aimed at maximizing O2 delivery to the tissues, such as intravenous fluid boluses, vasopressor therapy, or even blood product administration.
A return to a normalized serum lactate level is assumed to represent a transition back to aerobic metabolism. Lactate elevations, however, are not solely an indication of anaerobic metabolism and may only represent a small degree of lactate production.20 While the specific cellular mechanics are out of the scope of this article, it has been postulated that the increase in plasma lactate concentration is primarily driven by β-2 receptor stimulation from increased circulating catecholamines leading to increased aerobic glycolysis. Increased lactate levels could therefore be an adaptive mechanism of energy production—aggressive treatment and rapid clearance may, in fact, be harmful. Type A lactic acidosis is categorized as elevated serum levels due to tissue hypoperfusion.21
However, lactate elevations do not exclusively occur in severe illness. The use of β-2 receptor agonists such as continuous albuterol treatments or epinephrine may cause abnormal lactate levels.22 Other medications have also been associated with elevated serum lactate levels, including, but not limited to linezolid, metformin, and propofol.23-25 Additionally, lactate levels may be elevated after strenuous exercise, seizure activity, or in liver and kidney disease.26 These “secondary” causes of lactic acidosis that are not due to tissue hypoperfusion are referred to as type B lactic acidosis. Given these multiple etiologies and lack of specificity for this serum measurement, a failure to understand these limitations may result in over aggressive or unnecessary medical treatments.
Central Venous Pressure
Background
Central venous pressure (CVP) measurements can be obtained through a catheter, the distal tip of which transduces pressure of the superior vena cava at the entrance of the right atrium (RA). Thus, CVP is often used as a representation of RA pressure (RAP) and therefore an estimate of right ventricular (RV) preload. While CVP is used to diagnose and determine the etiology of shock, evidence and controversy regarding the use of CVP as a marker for resuscitation comes largely from sepsis-focused literature.5 Central venous pressure is meant to represent preload, which is essential for stroke volume as described by the Frank-Starling mechanism; however, its use as a target in distributive shock, a state in which it is difficult to determine a patient’s volume status, has been popularized by EGDT since 2001.2
Since the publication of the 2004 Surviving Sepsis Guidelines, CVP monitoring has been in the spotlight of sepsis resuscitation, albeit with some controversy.27 Included as the result of two studies, this recommendation has been removed in the most recent guidelines after 12 years of further study and scrutiny.2,27,28
Hypovolemic and hemorrhagic shock are usually diagnosed clinically and while a low CVP can be helpful in the diagnosis, the guidelines do not support CVP as a resuscitation endpoint. Obstructive and cardiogenic shock will both result in elevated CVP; however, treatment of obstructive shock is generally targeted at the underlying cause. While cardiogenic shock can be preload responsive, the mainstay of therapy in the ED is identification of patients for revascularization and inotropic support.29
Benefits
The CVP has been used as a surrogate for RV preload volume. If a patient’s preload volume is low, the treating physician can administer fluids to improve stroke volume and cardiac output (CO). Clinically, CVP measurements are easy to obtain provided a central venous line has been placed with the distal tip at the entrance to the RA. Central venous pressure is measured by transducing the pressure via manometry and connecting it to the patient’s bedside monitor. This provides an advantage of being able to provide serial or even continuous measurements. The “normal” RAP should be a low value (1-5 mm Hg, mean of 3 mm Hg), as this aids in the pressure gradient to drive blood from the higher pressures of the left ventricle (LV) and aorta through the circulation back to the low-pressure of the RA.30 The value of the CVP is meant to correspond to the physical examination findings of jugular venous distension.31,32 Thus, a low CVP may be “normal” and seen in patients with hypovolemic shock, whereas an elevated CVP can suggest volume overload or obstructive shock. However, this is of questionable value in distributive shock cases.
Aside from the two early studies on CVP monitoring during treatment of septic patients, there are few data to support the use of CVP measurement in the early resuscitation of patients with shock.2,28 More recent trials (PROMISE, ARISE, PROCESS) that compared protocolized sepsis care to standard care showed no benefit to bundles including CVP measurements.12-14 However, a subsequent, large observational trial spanning 7.5 years demonstrated improvements in sepsis-related mortality in patients who received a central venous catheter (CVC) and CVP-targeted therapy.33 Thus, it is possible that protocols including CVP are still beneficial in combination with other therapies even though CVP in isolation is not.
Limitations
The traditional two assumptions in CVP monitoring are CVP value represents the overall volume status of the patient, and the LV is able to utilize additional preload volume. The latter assumption, however, may be hampered by the presence of sepsis-induced myocardial dysfunction, which may be present in up to 40% of critically ill patients.34 The former assumption does not always hold true due to processes that change filling pressures independent of intravascular volume—eg, acute or chronic pulmonary hypertension, cardiac tamponade, intra-abdominal hypertension, or LV failure. Even before the landmark EGDT study, available data suggested that CVP was not a reliable marker for resuscitation management.35 A recent systematic review by Gottlieb and Hunter36 showed that the area under the receiver-operator curve for low, mid-range, or high CVPs was equivocal at best. In addition to its unreliability and lack of specificity, another significant drawback to using CVP to guide resuscitation therapy in the ED is that it necessitates placement of a CVC, which can be time-consuming and, if not otherwise indicated, lead to complications of infection, pneumothorax, and/or thrombosis.37
Mixed Venous Oxygen
Background
Most EPs are familiar with the use of ScvO2 in EGDT protocols to guide volume resuscitation of septic patients.2 A patient’s ScvO2 represents the O2 saturation of venous blood obtained via a CVC at the confluence of the superior vena cava and the RA, and thus it reflects tissue O2 consumption as a surrogate for tissue perfusion. The measurement parallels the SvO2 obtained from the pulmonary artery. In a healthy patient, SvO2 is around 65% to 70% and includes blood returning from both the superior and inferior vena cava (IVC). As such, ScvO2 values are typically 3% to 5% lower than SvO2 owing to the lower O2 extracted by tissues draining into the IVC compared to the mixed venous blood sampled from the pulmonary artery.38
Though a debate over the benefit of EGDT in treating sepsis continues, understanding the physiology of ScvO2 measurements is another potential tool the EP can use to guide the resuscitation of critically ill patients.39 A patient’s SvO2 and, by extension, ScvO2 represents the residual O2 saturation after the tissues have extracted the amount of O2 necessary to meet metabolic demands (Figure 2).
Conversely, cellular dysfunction, which can occur in certain toxicities or in severe forms of sepsis, can lead to decreased tissue O2 consumption with a concomitant rise in ScvO2 to supernormal values.38 The EP should take care, however, to consider whether ScvO2 values exceeding 80% represent successful therapeutic intervention or impaired tissue O2 extraction and utilization. There are data from ED patients suggesting an increased risk of mortality with both extremely low and extremely high values of ScvO2.40
Benefits
A critically ill patient’s ScvO2 can potentially provide EPs with insight into the patient’s global tissue perfusion and the source of any mismatch between O2 delivery and consumption. Using additional tools and measurements (physical examination, serum Hgb levels, and pulse oximetry) in conjunction with an ScvO2 measurement, assists EPs in identifying targets for therapeutic intervention. The effectiveness of this intervention can then be assessed using serial ScvO2 measurements, as described in Rivers et al2 EGDT protocol. Importantly, EPs should take care to measure serial ScvO2 values to maximize its utility.38 Similar to a CVP measurement, ScvO2is easily obtained from blood samples for serial laboratory measurements, assuming the patient already has a CVC with the distal tip at the entrance to the RA (ScvO2) or a pulmonary artery catheter (PAC) (SvO2).
Limitations
Serial measurements provide the most reliable information, which may be more useful in patients who spend extended periods of their resuscitation in the ED. In comparison to other measures of global tissue hypoxia, work by Jones et al41 suggests non-inferiority of peripherally sampled, serial lactate measurements as an alternative to ScvO2. This, in conjunction with the requirement for an internal jugular CVC, subclavian CVC, or PAC with their associated risks, may make ScvO2 a less attractive guide for the resuscitation of critically ill patients in the ED.
Monitoring Devices
Background
As noted throughout this review, it is important not only to identify and rapidly treat shock, but to also correctly identify the type of shock, such that treatment can be appropriately directed at its underlying cause. However, prior work suggests that EPs are unable to grossly estimate CO or systemic vascular resistance when compared to objective measurements of these parameters.42 This is in agreement with the overall poor performance of physical examination and clinical evaluation as a means of predicting volume responsiveness or guiding resuscitation, as discussed previously. Fortunately, a wide variety of devices to objectively monitor hemodynamics are now available to the EP.
In 1970, Swan et al43 published their initial experience with pulmonary artery catheterization at the bedside, using a balloon-tipped, flow-guided PAC in lieu of fluoroscopy, which had been mandated by earlier techniques. The ability to measure CO, right heart pressures, pulmonary arterial pressures, and estimate LV end diastolic pressure ushered in an era of widespread PAC use, despite an absence of evidence for causation of improved patient outcomes. The utilization of PACs has fallen, as the literature suggests that the empiric placement of PACs in critically ill patients does not improve mortality, length of stay, or cost, and significant complication rates have been reported in large trials.44,45Subsequently, a number of non-invasive or less-invasive HDM devices have been developed. Amongst the more commonly encountered modern devices, the techniques utilized for providing hemodynamic assessments include thermodilution and pulse contour analysis (PiCCOTM), pulse contour analysis (FloTrac/VigileoTM), and lithium chemodilution with pulse power analysis (LiDCOplusTM).46 The primary utility of these devices for the EP lies in the ability to quantify CO, stroke volume, and stroke volume or pulse pressure variation (PPV) to predict or assess response to resuscitative interventions (volume administration, vasopressors, inotropes, etc).
Benefits
Many of these devices require placement of an arterial catheter. Some require the addition of a CVC. Both of these procedures are well within the clinical scope of the EP, and are performed with fair frequency on critically ill patients. This is a distinct advantage when compared to pulmonary artery catheterization, a higher risk procedure that is rarely performed outside of the intensive care unit or cardiac catheterization laboratory. In addition, all of the devices below present hemodynamic data in a graphical, easy-to-read format, in real time. All of the devices discussed report stroke volume variation (SVV) or PPV continuously.
Limitations
Though these measures have validated threshold values that predict volume responsiveness, they require the patient to be intubated with a set tidal volume of greater than or equal to 8 mL/kg without spontaneous respirations and cardiac arrhythmias, in order to accurately do so. All of the HDM devices that rely on pulse contour analysis as the primary means of CO measurement cannot be used in the presence of significant cardiac arrhythmias (ie, atrial fibrillation), or mechanical circulatory assistance devices (ie, intra-aortic balloon counterpulsation). None of these devices are capable of monitoring microcirculatory changes, felt to be of increasing clinical importance in the critically ill.
The use of HDM devices to monitor CO with a reasonable degree of accuracy, trend CO, and assess for volume responsiveness using a number of previously validated parameters such as SVV is now in little doubt. However, these devices are still invasive, if less so than a pulmonary artery. The crux of the discussion of HDM devices for use in ED resuscitation revolves around whether or not the use of such devices to drive previously validated, protocolized care results in better outcomes for patients. The EP can now have continuous knowledge of a large number of hemodynamic parameters at their fingertips with relatively minimal additional efforts. At the time of this writing, though, this is both untested and unproven, with respect to the ED population.
Point-of-Care Ultrasound
Background
Over the past two decades, ultrasound (US) has become an integral part of the practice of emergency medicine (EM), and is now included in all United States Accreditation Council for Graduate Medical Education Emergency Medicine Residency Programs.47,48 It has emerged as a very important bedside tool performed by the clinician to identify type of shock and guide resuscitation, and has been endorsed by both EM and critical care societies.49-51 This section reviews the utility of US as a modality in identifying shock and guiding resuscitation, in addition to the pitfalls and limitations of this important tool.
In 2010, Perera et al47 described in their landmark article the Rapid Ultrasound in SHock (RUSH) examination, which describes a stepwise (the pump, tank, pipes) approach to identify the type of shock (cardiogenic, hypovolemic, obstructive, or distributive) in the crashing, hypotensive ED patient. We do not describe the full RUSH examination in this review, but discuss key elements of it as examples of how POCUS can assist the EP to make a rapid diagnosis and aid in the management of patients in shock. The “pump” is the heart, which is assessed in four different views to identify a pericardial effusion and possible tamponade, assess contractility or ejection fraction of the LV (severely decreased, decreased, normal, or hyperdynamic), and right heart strain which is identified by an RV that is larger than the LV, indicative of a potential pulmonary embolus.
The “tank” is then assessed by visualizing the IVC in the subxiphoid plane, and is evaluated for respiratory collapsibility (CI) and maximum size. This has been quite the debated topic over the last two decades. In 1988, Simonson and Schiller52 were the first to describe a correlation in spontaneously breathing patients between IVC caliber (measured 2 cm from the cavoatrial junction) and variation and RAP, where a larger IVC diameter and less respiratory variation correlated with a high RAP. Kircher et al53 later went on to describe that a CI greater than 50% correlated with an RAP of less than 10 mm Hg and vice versa in spontaneously breathing patients. Since then there have been more studies attempting to verify these findings in both spontaneously breathing and mechanically ventilated patients.54-56 The purpose of performing these measurements is not to estimate CVP, but to assess fluid responsiveness (ie, a blood pressure response to a fluid challenge). It can be assumed in states of shock that a small IVC, or one with a high CI, in the presence of a hyperdynamic heart is indicative of an underfilled ventricle and fluid responsiveness, especially if the IVC size increases with fluid.55,57 However, there are several caveats to this. First, in mechanically ventilated patients, the IVC is already plethoric due to positive pressure ventilation, and increases in diameter with inspiration and decreases with expiration as compared to spontaneously breathing patients. Second, the CI value to predict volume responsiveness in ventilated patients is set at 15% instead of 50%.55 Third, it is important to always take the clinical scenario in context; a dilated IVC with small CI is not necessarily only due to volume overload and congestive heart failure, but can be due to elevated RAP from obstructive shock due to cardiac tamponade or massive pulmonary embolus, which is why it is important to assess the “pump” first.47,58 It is also crucial to not forget to assess the abdominal and thoracic cavities, as intraperitoneal or pleural fluid with a collapsed IVC can potentially make a diagnosis of hemorrhagic or hypovolemic shock depending on the clinical scenario.47 The final part of the RUSH protocol is to evaluate the “pipes,” inclusive of the lower extremity deep venous system for evaluation of potential thrombosis that could increase suspicion for a pulmonary embolism causing obstructive shock, and the aorta with the common iliac arteries if there is concern for aortic dissection or aneurysmal rupture.
Benefits
Some of the most significant advantages to the use of POCUS to guide resuscitation is that it is quick, non-invasive, does not use ionizing radiation, and can be easily repeated. As noted above, it is a requirement for EM residencies to teach its use, so that contemporary graduates are entering the specialty competent in applying it to the care of their patients. Furthermore, POCUS is done at the bedside, limiting the need to potentially transport unstable patients.
In the most basic applications, POCUS provides direct visualization of a patient’s cardiac function, presence or absence of lung sliding to suggest a pneumothorax, presence of pulmonary edema, assessment of CVP pressures or potential for fluid responsiveness, as well as identification of potential thoracic, peritoneal, or pelvic cavity fluid accumulation that may suggest hemorrhage. There is literature to support that these assessments performed by the EP have been shown to be comparable to those of cardiologists.59,60 With continued practice and additional training, it is possible for EPs to even perform more “advanced” hemodynamic assessments to both diagnose and guide therapy to patients in shock (Figures 3 and 4).61
Limitations
Although POCUS has been shown as a remarkable tool to help assist the EP in making rapid decisions regarding resuscitation, it is always important to remember its limitations. Most of the studies regarding its use are of very small sample sizes, and further prospective studies have to be performed in order for this modality to be fully relied on.62Compared to some of the previously mentioned HDM devices that may provide continuous data, POCUS needs to be performed by the treating physician, thereby occurring intermittently. Emergency physicians need to be aware of their own experience and limitations with this modality, as errors in misdiagnosis can lead to unnecessary procedures, with resulting significant morbidity and mortality. Blanco and Volpicelli63 describe several common errors that include misdiagnosing the stomach as a peritoneal effusion, assuming adequate volume resuscitation when the IVC is seen to be plethoric in the setting of cardiac tamponade, or mistaking IVC movement as indicative of collapsibility, amongst other described misinterpretations. Several other studies have shown that, despite adequate performance of EPs in POCUS, diagnostic sensitivities remained higher when performed by radiologists.64-67 Thus it remains important for the EPs to be vigilant and not anchor on a diagnosis when in doubt, and to consult early with radiology, particularly if there is any question, to avoid potential adverse patient outcomes.
Summary
There are several ways to diagnose and track resuscitation in the ED, which include physical examination, assessment of serum laboratory values, monitoring of hemodynamic status, and use of POCUS. Unfortunately, none of these methods provides a perfect assessment, and no method has been proven superior and effective over the others. Therefore, it is important for EPs treating patients in shock to be aware of the strengths and limitations of each assessment method (Table).
1. Richards JB, Wilcox SR. Diagnosis and management of shock in the emergency department. Emerg Med Pract. 2014;16(3):1-22; quiz 22-23.
2. Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368-1377.
3. Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med. 2011;39:259-265. doi:10.1097/CCM.0b013e3181feeb15.
4. Seymour CW, Gesten F, Prescott HC, et al. Time to treatment and mortality during mandated emergency care for sepsis. N Engl J Med. 2017;376(23):2235-2244. doi:10.1056/NEJMoa1703058.
5. Cecconi M, De Backer D, Antonelli M, et al. Consensus on circulatory shock and hemodynamic monitoring. Task force of the European Society of Intensive Care Medicine. Intensive Care Med. 2014;40(12):1795-1815. doi:10.1007/s00134-014-3525-z.
6. Vincent JL, De Backer D. Circulatory shock. N Engl J Med. 2013;369(18):1726-1734. doi:10.1056/NEJMra1208943.
7. Saugel B, Ringmaier S, Holzapfel K, et al. Physical examination, central venous pressure, and chest radiography for the prediction of transpulmonary thermodilution-derived hemodynamic parameters in critically ill patients: a prospective trial. J Crit Care. 2011;26(4):402-410. doi:10.1016/j.jcrc.2010.11.001.
8. American College of Surgeons. Committee on Trauma. Shock. In: American College of Surgeons. Committee on Trauma, ed. Advanced Trauma Life Support: Student Course Manual. 9th ed. Chicago, IL: American College of Surgeons; 2012:69.
9. Saugel B, Kirsche SV, Hapfelmeier A, et al. Prediction of fluid responsiveness in patients admitted to the medical intensive care unit. J Crit Care. 2013:28(4):537.e1-e9. doi:10.1016/j.jcrc.2012.10.008.
10. McGee S, Abernethy WB 3rd, Simel DV. The rational clinical examination. Is this patient hypovolemic? JAMA. 1999;281(11):1022-1029.
11. Kompanje EJ, Jansen TC, van der Hoven B, Bakker J. The first demonstration of lactic acid in human blood in shock by Johann Joseph Scherer (1814-1869) in January 1843. Intensive Care Med. 2007;33(11):1967-1971. doi:10.1007/s00134-007-0788-7.
12. The ProCESS Investigators. A Randomized Trial of Protocol-Based Care for Early Septic Shock. N Engl J Med. 2014; 370:1683-1693. doi:10.1056/NEJMoa1401602.
13. Mouncey PR, Osborn TM, Power GS, et al. Protocolised Management In Sepsis (ProMISe): a multicentre randomised controlled trial of the clinical effectiveness and cost-effectiveness of early, goal-directed, protocolised resuscitation for emerging septic shock. Health Technol Assess. 2015;19(97):i-xxv, 1-150. doi:10.3310/hta19970.
14. ARISE Investigators; ANZICS Clinical Trials Group; Peake SL, Delaney A, Bailey M, et al. Goal-directed resuscitation for patients with early septic shock. N Engl J Med. 2014;371(16):1496-1506. doi:10.1056/NEJMoa1404380.
15. Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including The Pediatric Group. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013;39(2):165-228. doi:10.1007/s00134-012-2769-8.
16. Casserly B, Phillips GS, Schorr C, et al: Lactate measurements in sepsis-induced tissue hypoperfusion: results from the Surviving Sepsis Campaign database. Crit Care Med. 2015;43(3):567-573. doi:10.1097/CCM.0000000000000742.
17. Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care. 2013;3(1):12. doi:10.1186/2110-5820-3-12.
18. Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med. 2011;19:74. doi:10.1186/1757-7241-19-74.
19. Gaieski DF, Drumheller BC, Goyal M, Fuchs BD, Shofer FS, Zogby K. Accuracy of handheld point-of-care fingertip lactate measurement in the emergency department. West J Emerg Med. 2013;14(1):58-62. doi:10.5811/westjem.2011.5.6706.
20. Marik PE, Bellomo R. Lactate clearance as a target of therapy in sepsis: a flawed paradigm. OA Critical Care. 2013;1(1):3.
21. Kreisberg RA. Lactate homeostasis and lactic acidosis. Ann Intern Med. 1980;92(2 Pt 1):227-237.
22. Dodda VR, Spiro P. Can albuterol be blamed for lactic acidosis? Respir Care. 2012; 57(12):2115-2118. doi:10.4187/respcare.01810.
23. Scale T, Harvey JN. Diabetes, metformin and lactic acidosis. Clin Endocrinol (Oxf). 2011;74(2):191-196. doi:10.1111/j.1365-2265.2010.03891.x.
24. Velez JC, Janech MG. A case of lactic acidosis induced by linezolid. Nat Rev Nephrol. 2010;6(4):236-242. doi:10.1038/nrneph.2010.20.
25. Kam PC, Cardone D. Propofol infusion syndrome. Anaesthesia. 2007;62(7):690-701.
26. Griffith FR Jr, Lockwood JE, Emery FE. Adrenalin lactacidemia: proportionality with dose. Am J Physiol. 1939;127(3):415-421.
27. Rhodes A, Evans LE, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43(3):304-377. doi:10.1007/s00134-017-4683-6.
28. Early Goal-Directed Therapy Collaborative Group of Zhejiang Province. The effect of early goal-directed therapy on treatment of critical patients with severe sepsis/ septic shock: a multi-center, prospective, randomised, controlled study. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2010;22(6):331-334.
29. Thiele H, Ohman EM, Desch S, Eitel I, de Waha S. Management of cardiogenic shock. Eur Heart J. 2015;36(20):1223-1230. doi:10.1093/eurheartj/ehv051.
30. Lee M, Curley GF, Mustard M, Mazer CD. The Swan-Ganz catheter remains a critically important component of monitoring in cardiovascular critical care. Can J Cardiol. 2017;33(1):142-147. doi:10.1016/j.cjca.2016.10.026.
31. Morgan BC, Abel FL, Mullins GL, Guntheroth WG. Flow patterns in cavae, pulmonary artery, pulmonary vein, and aorta in intact dogs. Am J Physiol. 1966;210(4):903-909. doi:10.1152/ajplegacy.1966.210.4.903.
32. Brecher GA, Hubay CA. Pulmonary blood flow and venous return during spontaneous respiration. Circ Res. 1955;3(2):210-214.
33. Levy MM, Rhodes A, Phillips GS, et al. Surviving Sepsis Campaign: association between performance metrics and outcomes in a 7.5-year study. Intensive Care Med. 2014;40(11):1623-1633. doi:10.1007/s00134-014-3496-0.
34. Fernandes CJ Jr, Akamine N, Knobel E. Cardiac troponin: a new serum marker of myocardial injury in sepsis. Intensive Care Med. 1999;25(10):1165-1168. doi:10.1007/s001340051030.
35. Rady MY, Rivers EP, Nowak RM. Resuscitation of the critically III in the ED: responses of blood pressure, heart rate, shock index, central venous oxygen saturation, and lactate. Am J Emerg Med. 1996;14(2):218-225. doi:10.1016/s0735-6757(96)90136-9.
36. Gottlieb M, Hunter B. Utility of central venous pressure as a predictor of fluid responsiveness. Ann Emerg Med. 2016;68(1):114-116. doi:10.1016/j.annemergmed.2016.02.009.
37. Kornbau C, Lee KC, Hughes GD, Firstenberg MS. Central line complications. Int J Critical Illn Inj Sci. 2015;5(3):170-178. doi:10.4103/2229-5151.164940.
38. Walley KR. Use of central venous oxygen saturation to guide therapy. Am J Respir Crit Care Med. 2011;184(5):514-520. doi:10.1164/rccm.201010-1584CI.
39. PRISM Investigators, Rowan KM, Angus DC, et al. Early, goal-directed therapy for septic shock - a patient-level meta-analysis. N Engl J Med. 2017;376(23):2223-2234. doi:10.1056/NEJMoa1701380.
40. Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO(2)) as a predictor of mortality in patients with sepsis. Ann Emerg Med. 2010;55(1):40-46.e1. doi:10.1016/j.annemergmed.2009.08.014.
41. Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA. 2010;303(8):739-746. doi:10.1001/jama.2010.158.
42. Nowak RM, Sen A, Garcia, AJ, et al. The inability of emergency physicians to adequately clinically estimate the underlying hemodynamic profiles of acutely ill patients. Am J Emerg Med. 2012;30(6):954-960. doi:10.1016/j.ajem.2011.05.021.
43. Swan HJ, Ganz W, Forrester J, Marcus H, Diamond G, Chonette D. Catheterization of the heart in man with use of a flow-directed balloon-tipped catheter. N Engl J Med. 1970;283(9):447-451. doi:10.1056/NEJM197008272830902.
44. Hadian M, Pinsky MR. Evidence-based review of the use of the pulmonary artery catheter: impact data and complications. Crit Care. 2006;10 Suppl 3:S8.
45. Rajaram SS, Desai, NK, Kalra A, et al. Pulmonary artery catheters for adult patients in intensive care. Cochrane Database Syst Rev. 2013;(2):CD003408. doi:10.1002/14651858.CD003408.pub3.
46. Laher AE, Watermeyer MJ, Buchanan SK, et al. A review of hemodynamic monitoring techniques, methods and devices for the emergency physician. Am J Emerg Med. 2017;35(9):1335-1347. doi:10.1016/j.ajem.2017.03.036.
47. Perera P, Mailhot T, Riley D, Mandavia D. The RUSH exam: Rapid Ultrasound in SHock in the evaluation of the critically lll. Emerg Med Clin North Am. 2010;28(1):29-56, vii. doi:10.1016/j.emc.2009.09.010.
48. Heller MB, Mandavia D, Tayal VS, et al. Residency training in emergency ultrasound: fulfilling the mandate. Acad Emerg Med. 2002;9(8):835-839.
49. Ultrasound guidelines: emergency, point-of-care and clinical ultrasound guidelines in medicine. Ann Emerg Med. 2016;69(5):e27-e54. doi:10.1016/j.annemergmed.2016.08.457.
50. Expert Round Table on Ultrasound in ICU. International expert statement on training standards for critical care ultrasonography. Intensive Care Med. 2011;37(7):1077-1083. doi:10.1007/s00134-011-2246-9.
51. Neri L, Storti E, Lichtenstein D. Toward an ultrasound curriculum for critical care medicine. Crit Care Med. 2007;35(5 Suppl):S290-S304.
52. Simonson JS, Schiller NB. Sonospirometry: a new method for noninvasive estimation of mean right atrial pressure based on two-dimensional echographic measurements of the inferior vena cava during measured inspiration. J Am Coll Cardiol. 1988;11(3):557-564.
53. Kircher BJ, Himelman RB, Schiller NB. Noninvasive estimation of right atrial pressure from the inspiratory collapse of the inferior vena cava. Am J Cardiol. 1990;66(4):493-496.
54. Nagdev AD, Merchant RC, Tirado-Gonzalez A, Sisson CA, Murphy MC. Emergency department bedside ultrasonographic measurement of the caval index for noninvasive determination of low central venous pressure. Ann Emerg Med. 2010;55(3):290-295. doi:10.1016/j.annemergmed.2009.04.021.
55. Barbier C, Loubières Y, Schmit C, et al. Respiratory changes in inferior vena cava diameter are helpful in predicting fluid responsiveness in ventilated septic patients. Intensive Care Med. 2004;30(9):1740-1746.
56. Corl KA, George NR, Romanoff J, et al. Inferior vena cava collapsibility detects fluid responsiveness among spontaneously breathing critically-ill patients. J Crit Care. 2017;41:130-137. doi:10.1016/j.jcrc.2017.05.008.
57. Feissel M, Michard F, Faller JP, Teboul JL. The respiratory variation in inferior vena cava diameter as a guide to fluid therapy. Intensive Care Med. 2004;30(9):1834-1837.
58. Blehar DJ, Dickman E, Gaspari R. Identification of congestive heart failure via respiratory variation of inferior vena cava diameter. Am J Emerg Med. 2009;27(1):71-75. doi:10.1016/j.ajem.2008.01.002.
59. Moore CL, Rose GA, Tayal VS, Sullivan DM, Arrowood JA, Kline JA. Determination of left ventricular function by emergency physician echocardiography of hypotensive patients. Acad Emerg Med. 2002;9(3):186-193.
60. Mandavia DP, Hoffner RJ, Mahaney K, Henderson SO. Bedside echocardiography by emergency physicians. Ann Emerg Med. 2001;38(4):377-382.
61. Mosier JM, Martin J, Andrus P, et al. Advanced hemodynamic and cardiopulmonary ultrasound for critically ill patients in the emergency department. Emerg Med. 2018;50(1):17-34. doi:10.12788/emed.2018.0078.
62. Agarwal S, Swanson S, Murphy A, Yaeger K, Sharek P, Halamek LP. Comparing the utility of a standard pediatric resuscitation cart with a pediatric resuscitation cart based on the Broselow tape: a randomized, controlled, crossover trial involving simulated resuscitation scenarios. Pediatrics. 2005;116(3):e326-e333.
63. Blanco P, Volpicelli G. Common pitfalls in point-of-care ultrasound: a practical guide for emergency and critical care physicians. Crit Ultrasound J. 2016;8(1):15.
64. Tajoddini S, Shams Vahdati S. Ultrasonographic diagnosis of abdominal free fluid: accuracy comparison of emergency physicians and radiologists. Eur J Trauma Emerg Surg. 2013;39(1):9-13. doi:10.1007/s00068-012-0219-5.
65 Abbasi S, Bolverdi E, Zare MA, et al. Comparison of diagnostic value of conventional ultrasonography by emergency physicians with Doppler ultrasonography by radiology physicians for diagnosis of deep vein thrombosis. J Pak Med Assoc. 2012;62(5):461-465.
66. Arhami Dolatabadi A, Amini A, Hatamabadi H, et al. Comparison of the accuracy and reproducibility of focused abdominal sonography for trauma performed by emergency medicine and radiology residents. Ultrasound Med Biol. 2014;40(7):1476-1482. doi:10.1016/j.ultrasmedbio.2014.01.017.
67. Karimi E, Aminianfar M, Zarafshani K, Safaie A. The accuracy of emergency physicians in ultrasonographic screening of acute appendicitis; a cross sectional study. Emerg (Tehran). 2017;5(1):e22.
1. Richards JB, Wilcox SR. Diagnosis and management of shock in the emergency department. Emerg Med Pract. 2014;16(3):1-22; quiz 22-23.
2. Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368-1377.
3. Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med. 2011;39:259-265. doi:10.1097/CCM.0b013e3181feeb15.
4. Seymour CW, Gesten F, Prescott HC, et al. Time to treatment and mortality during mandated emergency care for sepsis. N Engl J Med. 2017;376(23):2235-2244. doi:10.1056/NEJMoa1703058.
5. Cecconi M, De Backer D, Antonelli M, et al. Consensus on circulatory shock and hemodynamic monitoring. Task force of the European Society of Intensive Care Medicine. Intensive Care Med. 2014;40(12):1795-1815. doi:10.1007/s00134-014-3525-z.
6. Vincent JL, De Backer D. Circulatory shock. N Engl J Med. 2013;369(18):1726-1734. doi:10.1056/NEJMra1208943.
7. Saugel B, Ringmaier S, Holzapfel K, et al. Physical examination, central venous pressure, and chest radiography for the prediction of transpulmonary thermodilution-derived hemodynamic parameters in critically ill patients: a prospective trial. J Crit Care. 2011;26(4):402-410. doi:10.1016/j.jcrc.2010.11.001.
8. American College of Surgeons. Committee on Trauma. Shock. In: American College of Surgeons. Committee on Trauma, ed. Advanced Trauma Life Support: Student Course Manual. 9th ed. Chicago, IL: American College of Surgeons; 2012:69.
9. Saugel B, Kirsche SV, Hapfelmeier A, et al. Prediction of fluid responsiveness in patients admitted to the medical intensive care unit. J Crit Care. 2013:28(4):537.e1-e9. doi:10.1016/j.jcrc.2012.10.008.
10. McGee S, Abernethy WB 3rd, Simel DV. The rational clinical examination. Is this patient hypovolemic? JAMA. 1999;281(11):1022-1029.
11. Kompanje EJ, Jansen TC, van der Hoven B, Bakker J. The first demonstration of lactic acid in human blood in shock by Johann Joseph Scherer (1814-1869) in January 1843. Intensive Care Med. 2007;33(11):1967-1971. doi:10.1007/s00134-007-0788-7.
12. The ProCESS Investigators. A Randomized Trial of Protocol-Based Care for Early Septic Shock. N Engl J Med. 2014; 370:1683-1693. doi:10.1056/NEJMoa1401602.
13. Mouncey PR, Osborn TM, Power GS, et al. Protocolised Management In Sepsis (ProMISe): a multicentre randomised controlled trial of the clinical effectiveness and cost-effectiveness of early, goal-directed, protocolised resuscitation for emerging septic shock. Health Technol Assess. 2015;19(97):i-xxv, 1-150. doi:10.3310/hta19970.
14. ARISE Investigators; ANZICS Clinical Trials Group; Peake SL, Delaney A, Bailey M, et al. Goal-directed resuscitation for patients with early septic shock. N Engl J Med. 2014;371(16):1496-1506. doi:10.1056/NEJMoa1404380.
15. Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including The Pediatric Group. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013;39(2):165-228. doi:10.1007/s00134-012-2769-8.
16. Casserly B, Phillips GS, Schorr C, et al: Lactate measurements in sepsis-induced tissue hypoperfusion: results from the Surviving Sepsis Campaign database. Crit Care Med. 2015;43(3):567-573. doi:10.1097/CCM.0000000000000742.
17. Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care. 2013;3(1):12. doi:10.1186/2110-5820-3-12.
18. Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med. 2011;19:74. doi:10.1186/1757-7241-19-74.
19. Gaieski DF, Drumheller BC, Goyal M, Fuchs BD, Shofer FS, Zogby K. Accuracy of handheld point-of-care fingertip lactate measurement in the emergency department. West J Emerg Med. 2013;14(1):58-62. doi:10.5811/westjem.2011.5.6706.
20. Marik PE, Bellomo R. Lactate clearance as a target of therapy in sepsis: a flawed paradigm. OA Critical Care. 2013;1(1):3.
21. Kreisberg RA. Lactate homeostasis and lactic acidosis. Ann Intern Med. 1980;92(2 Pt 1):227-237.
22. Dodda VR, Spiro P. Can albuterol be blamed for lactic acidosis? Respir Care. 2012; 57(12):2115-2118. doi:10.4187/respcare.01810.
23. Scale T, Harvey JN. Diabetes, metformin and lactic acidosis. Clin Endocrinol (Oxf). 2011;74(2):191-196. doi:10.1111/j.1365-2265.2010.03891.x.
24. Velez JC, Janech MG. A case of lactic acidosis induced by linezolid. Nat Rev Nephrol. 2010;6(4):236-242. doi:10.1038/nrneph.2010.20.
25. Kam PC, Cardone D. Propofol infusion syndrome. Anaesthesia. 2007;62(7):690-701.
26. Griffith FR Jr, Lockwood JE, Emery FE. Adrenalin lactacidemia: proportionality with dose. Am J Physiol. 1939;127(3):415-421.
27. Rhodes A, Evans LE, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43(3):304-377. doi:10.1007/s00134-017-4683-6.
28. Early Goal-Directed Therapy Collaborative Group of Zhejiang Province. The effect of early goal-directed therapy on treatment of critical patients with severe sepsis/ septic shock: a multi-center, prospective, randomised, controlled study. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2010;22(6):331-334.
29. Thiele H, Ohman EM, Desch S, Eitel I, de Waha S. Management of cardiogenic shock. Eur Heart J. 2015;36(20):1223-1230. doi:10.1093/eurheartj/ehv051.
30. Lee M, Curley GF, Mustard M, Mazer CD. The Swan-Ganz catheter remains a critically important component of monitoring in cardiovascular critical care. Can J Cardiol. 2017;33(1):142-147. doi:10.1016/j.cjca.2016.10.026.
31. Morgan BC, Abel FL, Mullins GL, Guntheroth WG. Flow patterns in cavae, pulmonary artery, pulmonary vein, and aorta in intact dogs. Am J Physiol. 1966;210(4):903-909. doi:10.1152/ajplegacy.1966.210.4.903.
32. Brecher GA, Hubay CA. Pulmonary blood flow and venous return during spontaneous respiration. Circ Res. 1955;3(2):210-214.
33. Levy MM, Rhodes A, Phillips GS, et al. Surviving Sepsis Campaign: association between performance metrics and outcomes in a 7.5-year study. Intensive Care Med. 2014;40(11):1623-1633. doi:10.1007/s00134-014-3496-0.
34. Fernandes CJ Jr, Akamine N, Knobel E. Cardiac troponin: a new serum marker of myocardial injury in sepsis. Intensive Care Med. 1999;25(10):1165-1168. doi:10.1007/s001340051030.
35. Rady MY, Rivers EP, Nowak RM. Resuscitation of the critically III in the ED: responses of blood pressure, heart rate, shock index, central venous oxygen saturation, and lactate. Am J Emerg Med. 1996;14(2):218-225. doi:10.1016/s0735-6757(96)90136-9.
36. Gottlieb M, Hunter B. Utility of central venous pressure as a predictor of fluid responsiveness. Ann Emerg Med. 2016;68(1):114-116. doi:10.1016/j.annemergmed.2016.02.009.
37. Kornbau C, Lee KC, Hughes GD, Firstenberg MS. Central line complications. Int J Critical Illn Inj Sci. 2015;5(3):170-178. doi:10.4103/2229-5151.164940.
38. Walley KR. Use of central venous oxygen saturation to guide therapy. Am J Respir Crit Care Med. 2011;184(5):514-520. doi:10.1164/rccm.201010-1584CI.
39. PRISM Investigators, Rowan KM, Angus DC, et al. Early, goal-directed therapy for septic shock - a patient-level meta-analysis. N Engl J Med. 2017;376(23):2223-2234. doi:10.1056/NEJMoa1701380.
40. Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO(2)) as a predictor of mortality in patients with sepsis. Ann Emerg Med. 2010;55(1):40-46.e1. doi:10.1016/j.annemergmed.2009.08.014.
41. Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA. 2010;303(8):739-746. doi:10.1001/jama.2010.158.
42. Nowak RM, Sen A, Garcia, AJ, et al. The inability of emergency physicians to adequately clinically estimate the underlying hemodynamic profiles of acutely ill patients. Am J Emerg Med. 2012;30(6):954-960. doi:10.1016/j.ajem.2011.05.021.
43. Swan HJ, Ganz W, Forrester J, Marcus H, Diamond G, Chonette D. Catheterization of the heart in man with use of a flow-directed balloon-tipped catheter. N Engl J Med. 1970;283(9):447-451. doi:10.1056/NEJM197008272830902.
44. Hadian M, Pinsky MR. Evidence-based review of the use of the pulmonary artery catheter: impact data and complications. Crit Care. 2006;10 Suppl 3:S8.
45. Rajaram SS, Desai, NK, Kalra A, et al. Pulmonary artery catheters for adult patients in intensive care. Cochrane Database Syst Rev. 2013;(2):CD003408. doi:10.1002/14651858.CD003408.pub3.
46. Laher AE, Watermeyer MJ, Buchanan SK, et al. A review of hemodynamic monitoring techniques, methods and devices for the emergency physician. Am J Emerg Med. 2017;35(9):1335-1347. doi:10.1016/j.ajem.2017.03.036.
47. Perera P, Mailhot T, Riley D, Mandavia D. The RUSH exam: Rapid Ultrasound in SHock in the evaluation of the critically lll. Emerg Med Clin North Am. 2010;28(1):29-56, vii. doi:10.1016/j.emc.2009.09.010.
48. Heller MB, Mandavia D, Tayal VS, et al. Residency training in emergency ultrasound: fulfilling the mandate. Acad Emerg Med. 2002;9(8):835-839.
49. Ultrasound guidelines: emergency, point-of-care and clinical ultrasound guidelines in medicine. Ann Emerg Med. 2016;69(5):e27-e54. doi:10.1016/j.annemergmed.2016.08.457.
50. Expert Round Table on Ultrasound in ICU. International expert statement on training standards for critical care ultrasonography. Intensive Care Med. 2011;37(7):1077-1083. doi:10.1007/s00134-011-2246-9.
51. Neri L, Storti E, Lichtenstein D. Toward an ultrasound curriculum for critical care medicine. Crit Care Med. 2007;35(5 Suppl):S290-S304.
52. Simonson JS, Schiller NB. Sonospirometry: a new method for noninvasive estimation of mean right atrial pressure based on two-dimensional echographic measurements of the inferior vena cava during measured inspiration. J Am Coll Cardiol. 1988;11(3):557-564.
53. Kircher BJ, Himelman RB, Schiller NB. Noninvasive estimation of right atrial pressure from the inspiratory collapse of the inferior vena cava. Am J Cardiol. 1990;66(4):493-496.
54. Nagdev AD, Merchant RC, Tirado-Gonzalez A, Sisson CA, Murphy MC. Emergency department bedside ultrasonographic measurement of the caval index for noninvasive determination of low central venous pressure. Ann Emerg Med. 2010;55(3):290-295. doi:10.1016/j.annemergmed.2009.04.021.
55. Barbier C, Loubières Y, Schmit C, et al. Respiratory changes in inferior vena cava diameter are helpful in predicting fluid responsiveness in ventilated septic patients. Intensive Care Med. 2004;30(9):1740-1746.
56. Corl KA, George NR, Romanoff J, et al. Inferior vena cava collapsibility detects fluid responsiveness among spontaneously breathing critically-ill patients. J Crit Care. 2017;41:130-137. doi:10.1016/j.jcrc.2017.05.008.
57. Feissel M, Michard F, Faller JP, Teboul JL. The respiratory variation in inferior vena cava diameter as a guide to fluid therapy. Intensive Care Med. 2004;30(9):1834-1837.
58. Blehar DJ, Dickman E, Gaspari R. Identification of congestive heart failure via respiratory variation of inferior vena cava diameter. Am J Emerg Med. 2009;27(1):71-75. doi:10.1016/j.ajem.2008.01.002.
59. Moore CL, Rose GA, Tayal VS, Sullivan DM, Arrowood JA, Kline JA. Determination of left ventricular function by emergency physician echocardiography of hypotensive patients. Acad Emerg Med. 2002;9(3):186-193.
60. Mandavia DP, Hoffner RJ, Mahaney K, Henderson SO. Bedside echocardiography by emergency physicians. Ann Emerg Med. 2001;38(4):377-382.
61. Mosier JM, Martin J, Andrus P, et al. Advanced hemodynamic and cardiopulmonary ultrasound for critically ill patients in the emergency department. Emerg Med. 2018;50(1):17-34. doi:10.12788/emed.2018.0078.
62. Agarwal S, Swanson S, Murphy A, Yaeger K, Sharek P, Halamek LP. Comparing the utility of a standard pediatric resuscitation cart with a pediatric resuscitation cart based on the Broselow tape: a randomized, controlled, crossover trial involving simulated resuscitation scenarios. Pediatrics. 2005;116(3):e326-e333.
63. Blanco P, Volpicelli G. Common pitfalls in point-of-care ultrasound: a practical guide for emergency and critical care physicians. Crit Ultrasound J. 2016;8(1):15.
64. Tajoddini S, Shams Vahdati S. Ultrasonographic diagnosis of abdominal free fluid: accuracy comparison of emergency physicians and radiologists. Eur J Trauma Emerg Surg. 2013;39(1):9-13. doi:10.1007/s00068-012-0219-5.
65 Abbasi S, Bolverdi E, Zare MA, et al. Comparison of diagnostic value of conventional ultrasonography by emergency physicians with Doppler ultrasonography by radiology physicians for diagnosis of deep vein thrombosis. J Pak Med Assoc. 2012;62(5):461-465.
66. Arhami Dolatabadi A, Amini A, Hatamabadi H, et al. Comparison of the accuracy and reproducibility of focused abdominal sonography for trauma performed by emergency medicine and radiology residents. Ultrasound Med Biol. 2014;40(7):1476-1482. doi:10.1016/j.ultrasmedbio.2014.01.017.
67. Karimi E, Aminianfar M, Zarafshani K, Safaie A. The accuracy of emergency physicians in ultrasonographic screening of acute appendicitis; a cross sectional study. Emerg (Tehran). 2017;5(1):e22.
Eyes of the mimicker
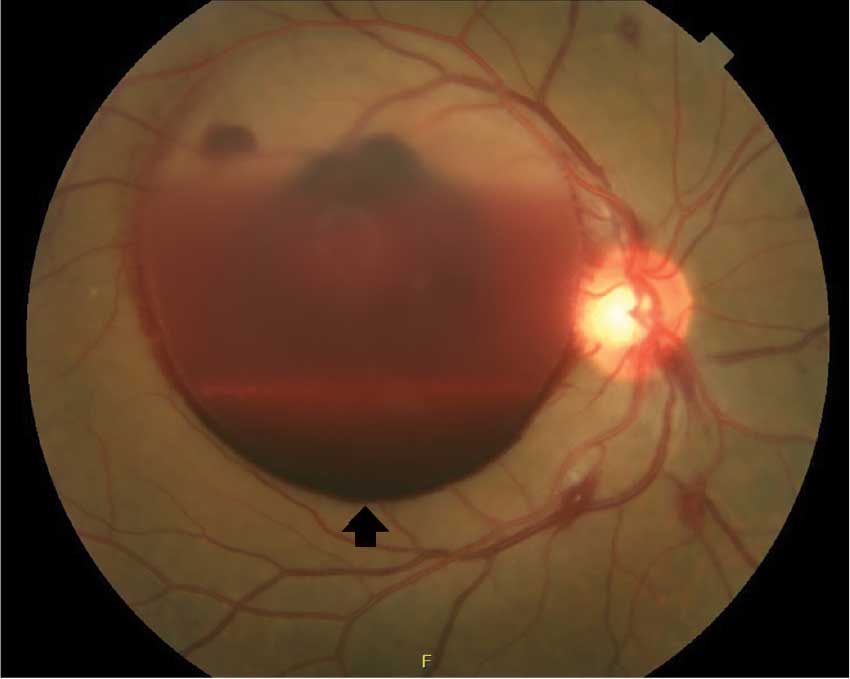
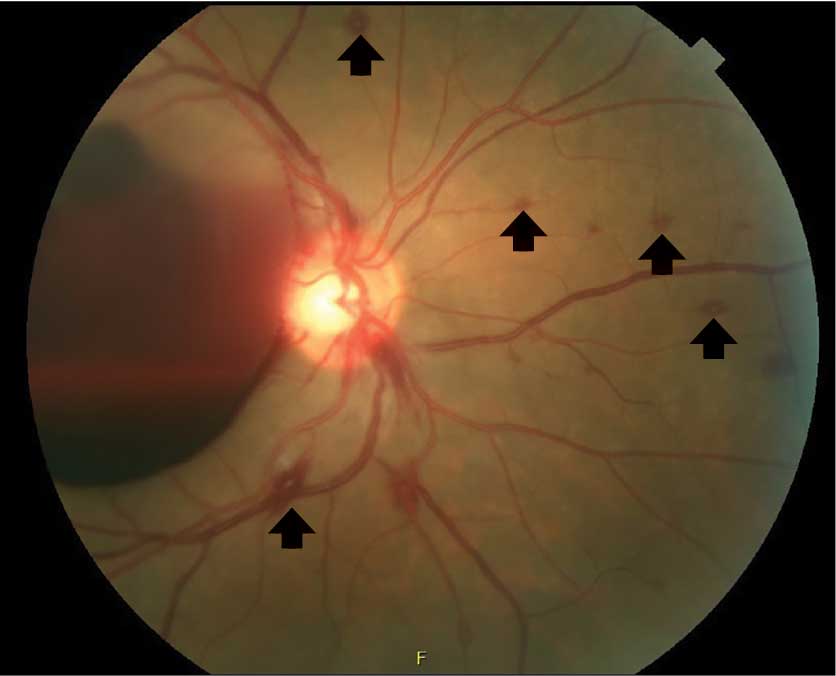
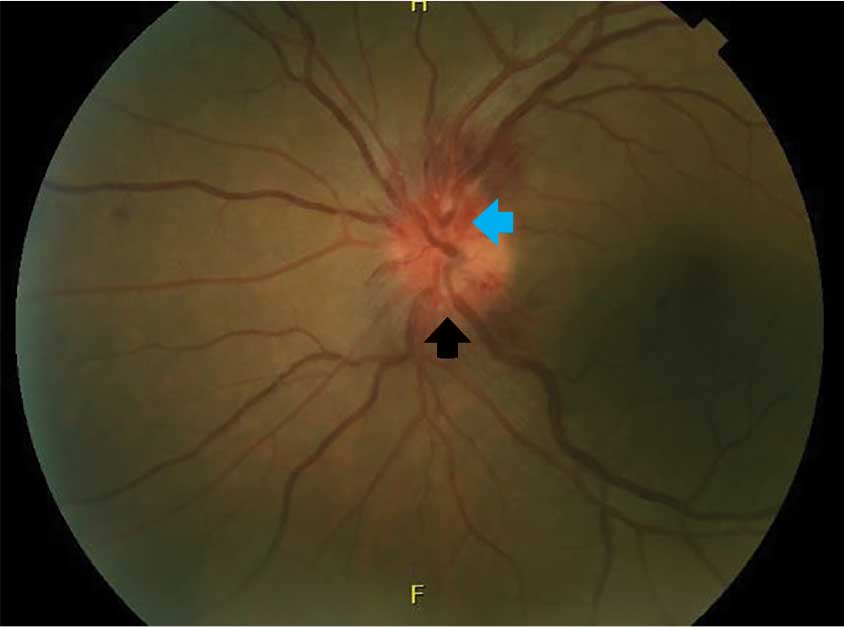
Lumbar puncture study revealed 34 nucleated cells/µL (94% lymphocytes), protein 58 mg/dL, and glucose 62 mg/dL. Cerebrospinal fluid Venereal Disease Research Laboratory and fluorescent treponemal antibody absorption tests were reactive, confirming a diagnosis of ocular syphilis.
The patient was admitted to the hospital for treatment with intravenous penicillin G. After 5 days, he was discharged with instructions to complete a 10-day course of intravenous ceftriaxone (chosen for its ease of administration), for a total of 14 days of antibiotic therapy. His vision improved with treatment.
He continued to follow up with ophthalmology and infectious disease. Subsequent dilated fundus examinations showed resolution of pathology in the left eye, resolution of Roth spots in the right eye, and resolution of the subhyaloid hemorrhage. Repeat cerebrospinal fluid study examination was planned if the serum rapid plasma reagin had not become nonreactive 24 months after treatment.
RECOGNIZING AND MANAGING OCULAR SYPHILIS AND NEUROSYPHILIS
In addition to ocular syphilis and neurosyphilis, the differential diagnosis for Roth spots and disc edema on dilated funduscopy includes endocarditis, viral retinitis, and autoimmune or inflammatory conditions such as sarcoidosis and vasculitis.
In our patient, infectious endocarditis was considered, given his history of intermittent fevers and rigors, but it was ultimately ruled out by negative blood cultures and the absence of valvular vegetations on echocardiography.
The large subhyaloid hemorrhage raised suspicion of leukemia, but this was ruled out by the normal total white blood cell count and differential. HIV, herpetic retinitis, and toxoplasmosis were also considered, but laboratory tests for these infections were negative.
Typically, retinal precipitates are more characteristic of syphilitic retinitis and distinguish it from other infectious causes such as herpetic retinitis and toxoplasmosis.1 Additionally, ocular syphilis more commonly manifests as uveitis or panuveitis.1,2 Our patient’s ocular syphilis presented with white-centered retinal hemorrhages, subhyaloid hemorrhage, and optic disc edema.
Who is at highest risk?
About 90% of syphilis cases occur in men, and 81% occur in men who have sex with men. The US Centers for Disease Control and Prevention (CDC) thus recommends annual syphilis testing for men who have sex with men.3
Classically, syphilis was called “the great imitator” because it mimicked manifestations of other diseases. Patients with ocular manifestations of syphilis may not have other neurologic symptoms.4,5 Nevertheless, cerebrospinal fluid examination should be done in all instances of ocular syphilis, as many patients with ocular syphilis have evidence of neurosyphilis on testing.2 The CDC also recommends follow-up cerebrospinal fluid analysis to assess treatment response.2 This was planned in our patient.
- Fu EX, Geraets RL, Dodds EM, et al. Superficial retinal precipitates in patients with syphilitic retinitis. Retina 2010; 30(7):1135–1143. doi:10.1097/IAE.0b013e3181cdf3ae
- US Centers for Disease Control and Prevention. Sexually Transmitted Diseases. Clinical Advisory: Ocular Syphilis in the United States, March 24, 2016. www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Accessed March 28, 2018.
- US Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance, 2015. www.cdc.gov/std/stats15/std-surveillance-2015-print.pdf. Accessed March 28, 2018.
- Rishi E, Govindarajan MV, Biswas J, Agarwal M, Sudharshan S, Rishi P. Syphilitic uveitis as the presenting feature of HIV. Indian J Ophthalmol 2016; 64(2):149–150. doi:10.4103/0301-4738.179714
- Zhang R, Qian J, Guo J, et al. Clinical manifestations and treatment outcomes of syphilitic uveitis in a Chinese population. J Ophthalmol 2016; 2016:2797028. doi:10.1155/2016/2797028
Lumbar puncture study revealed 34 nucleated cells/µL (94% lymphocytes), protein 58 mg/dL, and glucose 62 mg/dL. Cerebrospinal fluid Venereal Disease Research Laboratory and fluorescent treponemal antibody absorption tests were reactive, confirming a diagnosis of ocular syphilis.
The patient was admitted to the hospital for treatment with intravenous penicillin G. After 5 days, he was discharged with instructions to complete a 10-day course of intravenous ceftriaxone (chosen for its ease of administration), for a total of 14 days of antibiotic therapy. His vision improved with treatment.
He continued to follow up with ophthalmology and infectious disease. Subsequent dilated fundus examinations showed resolution of pathology in the left eye, resolution of Roth spots in the right eye, and resolution of the subhyaloid hemorrhage. Repeat cerebrospinal fluid study examination was planned if the serum rapid plasma reagin had not become nonreactive 24 months after treatment.
RECOGNIZING AND MANAGING OCULAR SYPHILIS AND NEUROSYPHILIS
In addition to ocular syphilis and neurosyphilis, the differential diagnosis for Roth spots and disc edema on dilated funduscopy includes endocarditis, viral retinitis, and autoimmune or inflammatory conditions such as sarcoidosis and vasculitis.
In our patient, infectious endocarditis was considered, given his history of intermittent fevers and rigors, but it was ultimately ruled out by negative blood cultures and the absence of valvular vegetations on echocardiography.
The large subhyaloid hemorrhage raised suspicion of leukemia, but this was ruled out by the normal total white blood cell count and differential. HIV, herpetic retinitis, and toxoplasmosis were also considered, but laboratory tests for these infections were negative.
Typically, retinal precipitates are more characteristic of syphilitic retinitis and distinguish it from other infectious causes such as herpetic retinitis and toxoplasmosis.1 Additionally, ocular syphilis more commonly manifests as uveitis or panuveitis.1,2 Our patient’s ocular syphilis presented with white-centered retinal hemorrhages, subhyaloid hemorrhage, and optic disc edema.
Who is at highest risk?
About 90% of syphilis cases occur in men, and 81% occur in men who have sex with men. The US Centers for Disease Control and Prevention (CDC) thus recommends annual syphilis testing for men who have sex with men.3
Classically, syphilis was called “the great imitator” because it mimicked manifestations of other diseases. Patients with ocular manifestations of syphilis may not have other neurologic symptoms.4,5 Nevertheless, cerebrospinal fluid examination should be done in all instances of ocular syphilis, as many patients with ocular syphilis have evidence of neurosyphilis on testing.2 The CDC also recommends follow-up cerebrospinal fluid analysis to assess treatment response.2 This was planned in our patient.
Lumbar puncture study revealed 34 nucleated cells/µL (94% lymphocytes), protein 58 mg/dL, and glucose 62 mg/dL. Cerebrospinal fluid Venereal Disease Research Laboratory and fluorescent treponemal antibody absorption tests were reactive, confirming a diagnosis of ocular syphilis.
The patient was admitted to the hospital for treatment with intravenous penicillin G. After 5 days, he was discharged with instructions to complete a 10-day course of intravenous ceftriaxone (chosen for its ease of administration), for a total of 14 days of antibiotic therapy. His vision improved with treatment.
He continued to follow up with ophthalmology and infectious disease. Subsequent dilated fundus examinations showed resolution of pathology in the left eye, resolution of Roth spots in the right eye, and resolution of the subhyaloid hemorrhage. Repeat cerebrospinal fluid study examination was planned if the serum rapid plasma reagin had not become nonreactive 24 months after treatment.
RECOGNIZING AND MANAGING OCULAR SYPHILIS AND NEUROSYPHILIS
In addition to ocular syphilis and neurosyphilis, the differential diagnosis for Roth spots and disc edema on dilated funduscopy includes endocarditis, viral retinitis, and autoimmune or inflammatory conditions such as sarcoidosis and vasculitis.
In our patient, infectious endocarditis was considered, given his history of intermittent fevers and rigors, but it was ultimately ruled out by negative blood cultures and the absence of valvular vegetations on echocardiography.
The large subhyaloid hemorrhage raised suspicion of leukemia, but this was ruled out by the normal total white blood cell count and differential. HIV, herpetic retinitis, and toxoplasmosis were also considered, but laboratory tests for these infections were negative.
Typically, retinal precipitates are more characteristic of syphilitic retinitis and distinguish it from other infectious causes such as herpetic retinitis and toxoplasmosis.1 Additionally, ocular syphilis more commonly manifests as uveitis or panuveitis.1,2 Our patient’s ocular syphilis presented with white-centered retinal hemorrhages, subhyaloid hemorrhage, and optic disc edema.
Who is at highest risk?
About 90% of syphilis cases occur in men, and 81% occur in men who have sex with men. The US Centers for Disease Control and Prevention (CDC) thus recommends annual syphilis testing for men who have sex with men.3
Classically, syphilis was called “the great imitator” because it mimicked manifestations of other diseases. Patients with ocular manifestations of syphilis may not have other neurologic symptoms.4,5 Nevertheless, cerebrospinal fluid examination should be done in all instances of ocular syphilis, as many patients with ocular syphilis have evidence of neurosyphilis on testing.2 The CDC also recommends follow-up cerebrospinal fluid analysis to assess treatment response.2 This was planned in our patient.
- Fu EX, Geraets RL, Dodds EM, et al. Superficial retinal precipitates in patients with syphilitic retinitis. Retina 2010; 30(7):1135–1143. doi:10.1097/IAE.0b013e3181cdf3ae
- US Centers for Disease Control and Prevention. Sexually Transmitted Diseases. Clinical Advisory: Ocular Syphilis in the United States, March 24, 2016. www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Accessed March 28, 2018.
- US Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance, 2015. www.cdc.gov/std/stats15/std-surveillance-2015-print.pdf. Accessed March 28, 2018.
- Rishi E, Govindarajan MV, Biswas J, Agarwal M, Sudharshan S, Rishi P. Syphilitic uveitis as the presenting feature of HIV. Indian J Ophthalmol 2016; 64(2):149–150. doi:10.4103/0301-4738.179714
- Zhang R, Qian J, Guo J, et al. Clinical manifestations and treatment outcomes of syphilitic uveitis in a Chinese population. J Ophthalmol 2016; 2016:2797028. doi:10.1155/2016/2797028
- Fu EX, Geraets RL, Dodds EM, et al. Superficial retinal precipitates in patients with syphilitic retinitis. Retina 2010; 30(7):1135–1143. doi:10.1097/IAE.0b013e3181cdf3ae
- US Centers for Disease Control and Prevention. Sexually Transmitted Diseases. Clinical Advisory: Ocular Syphilis in the United States, March 24, 2016. www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Accessed March 28, 2018.
- US Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance, 2015. www.cdc.gov/std/stats15/std-surveillance-2015-print.pdf. Accessed March 28, 2018.
- Rishi E, Govindarajan MV, Biswas J, Agarwal M, Sudharshan S, Rishi P. Syphilitic uveitis as the presenting feature of HIV. Indian J Ophthalmol 2016; 64(2):149–150. doi:10.4103/0301-4738.179714
- Zhang R, Qian J, Guo J, et al. Clinical manifestations and treatment outcomes of syphilitic uveitis in a Chinese population. J Ophthalmol 2016; 2016:2797028. doi:10.1155/2016/2797028
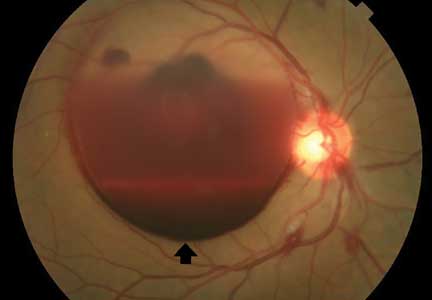
The algorithm less traveled

To this day I remain impressed by the algorithmic nature of trauma management. A routine that to the internist could appear mindless and slavish was to the trauma physician a protocol designed to take no chances on missing a life-threatening complication in the heat of the moment. The trauma physician cannot afford to wait for a cognitively derived epiphany in a clinical setting that often rapidly unfolds as a series of “never-miss” scenarios. The appropriate algorithm, rigorously followed, offers the best chance of avoiding a catastrophe of omission. This was long before Atul Gawande published his Checklist Manifesto.
Reviewing the article by Sussman et al, “Eyes of the mimicker,” in this issue of the Journal got me thinking about the power of algorithmic thinking and practice in internal medicine, how the patient they describe specifically relates to my practice experiences over the years, and how important the context of where we practice and who we treat informs (and can misinform) our clinical reasoning. When I was a medical student at Bellevue Hospital in New York City (in the pre-HIV era), the rapid plasma reagin (RPR) was a routine blood test, as syphilis routinely earned its moniker as the “great imitator.” When I did my residency at the Hospital of the University of Pennsylvania, my ingrained habit of ordering this test was extinguished, along with my also previously learned habit of obtaining blood cultures in all patients who presented with new heart failure that was not explained by the electrocardiogram. These habits disappeared not because of arguments steeped in evidence-based medicine or an emphasis on Bayesian test-ordering, but because in Philadelphia at that time we were not seeing patients with occult syphilis and endocarditis with the same frequency as at Bellevue. Context can and should play a role in our diagnostic reasoning.
But I still remember the patient I saw in the Philadelphia emergency room, a second visit for a man in his 20s with a diffuse, mostly macular rash on his trunk, palms, and soles (visible when the light was turned up in his darkened room, as he felt uncomfortable with bright light), diffuse adenopathy, and enlarged doughy and minimally tender wrists and finger (metacarpophalangeal) joints. I recall wondering why no one had thought to obtain an RPR test on him the first time he had presented to the emergency room; if he had been at Bellevue, the test results would already have returned.
Without appropriate algorithms, things get missed. But using algorithms indiscriminately is cost-ineffective and can lead to cascades of inappropriate tests and interventions. Striking the appropriate balance is part of what comprises the writing of useful clinical care paths.
As I read the article by Sussman et al I wondered who first looked at the patient’s retinas and what initially prompted the testing that was ordered. The presentation was not typical of ocular syphilis, and I would guess that an ophthalmologist or infectious disease consultant evaluating the blurred vision observed the retinal findings, suspected the diagnosis, and ordered serologies, as well as other studies searching for infections and systemic autoimmune disorders that can also cause Roth spots. Gone are the days when internists (and residents) routinely examine the eyes as part of a full physical examination. I am certain an evidence-based study of this practice would find it time-ineffective and with inappropriately low sensitivity.
I don’t think the retinal examination will return to the internist’s checklist. Yet that is where the algorithm that led to this patient’s diagnosis likely began. One can “google” the causes of Roth spots, but as yet there is no app for demonstrating that they are present.
To this day I remain impressed by the algorithmic nature of trauma management. A routine that to the internist could appear mindless and slavish was to the trauma physician a protocol designed to take no chances on missing a life-threatening complication in the heat of the moment. The trauma physician cannot afford to wait for a cognitively derived epiphany in a clinical setting that often rapidly unfolds as a series of “never-miss” scenarios. The appropriate algorithm, rigorously followed, offers the best chance of avoiding a catastrophe of omission. This was long before Atul Gawande published his Checklist Manifesto.
Reviewing the article by Sussman et al, “Eyes of the mimicker,” in this issue of the Journal got me thinking about the power of algorithmic thinking and practice in internal medicine, how the patient they describe specifically relates to my practice experiences over the years, and how important the context of where we practice and who we treat informs (and can misinform) our clinical reasoning. When I was a medical student at Bellevue Hospital in New York City (in the pre-HIV era), the rapid plasma reagin (RPR) was a routine blood test, as syphilis routinely earned its moniker as the “great imitator.” When I did my residency at the Hospital of the University of Pennsylvania, my ingrained habit of ordering this test was extinguished, along with my also previously learned habit of obtaining blood cultures in all patients who presented with new heart failure that was not explained by the electrocardiogram. These habits disappeared not because of arguments steeped in evidence-based medicine or an emphasis on Bayesian test-ordering, but because in Philadelphia at that time we were not seeing patients with occult syphilis and endocarditis with the same frequency as at Bellevue. Context can and should play a role in our diagnostic reasoning.
But I still remember the patient I saw in the Philadelphia emergency room, a second visit for a man in his 20s with a diffuse, mostly macular rash on his trunk, palms, and soles (visible when the light was turned up in his darkened room, as he felt uncomfortable with bright light), diffuse adenopathy, and enlarged doughy and minimally tender wrists and finger (metacarpophalangeal) joints. I recall wondering why no one had thought to obtain an RPR test on him the first time he had presented to the emergency room; if he had been at Bellevue, the test results would already have returned.
Without appropriate algorithms, things get missed. But using algorithms indiscriminately is cost-ineffective and can lead to cascades of inappropriate tests and interventions. Striking the appropriate balance is part of what comprises the writing of useful clinical care paths.
As I read the article by Sussman et al I wondered who first looked at the patient’s retinas and what initially prompted the testing that was ordered. The presentation was not typical of ocular syphilis, and I would guess that an ophthalmologist or infectious disease consultant evaluating the blurred vision observed the retinal findings, suspected the diagnosis, and ordered serologies, as well as other studies searching for infections and systemic autoimmune disorders that can also cause Roth spots. Gone are the days when internists (and residents) routinely examine the eyes as part of a full physical examination. I am certain an evidence-based study of this practice would find it time-ineffective and with inappropriately low sensitivity.
I don’t think the retinal examination will return to the internist’s checklist. Yet that is where the algorithm that led to this patient’s diagnosis likely began. One can “google” the causes of Roth spots, but as yet there is no app for demonstrating that they are present.
To this day I remain impressed by the algorithmic nature of trauma management. A routine that to the internist could appear mindless and slavish was to the trauma physician a protocol designed to take no chances on missing a life-threatening complication in the heat of the moment. The trauma physician cannot afford to wait for a cognitively derived epiphany in a clinical setting that often rapidly unfolds as a series of “never-miss” scenarios. The appropriate algorithm, rigorously followed, offers the best chance of avoiding a catastrophe of omission. This was long before Atul Gawande published his Checklist Manifesto.
Reviewing the article by Sussman et al, “Eyes of the mimicker,” in this issue of the Journal got me thinking about the power of algorithmic thinking and practice in internal medicine, how the patient they describe specifically relates to my practice experiences over the years, and how important the context of where we practice and who we treat informs (and can misinform) our clinical reasoning. When I was a medical student at Bellevue Hospital in New York City (in the pre-HIV era), the rapid plasma reagin (RPR) was a routine blood test, as syphilis routinely earned its moniker as the “great imitator.” When I did my residency at the Hospital of the University of Pennsylvania, my ingrained habit of ordering this test was extinguished, along with my also previously learned habit of obtaining blood cultures in all patients who presented with new heart failure that was not explained by the electrocardiogram. These habits disappeared not because of arguments steeped in evidence-based medicine or an emphasis on Bayesian test-ordering, but because in Philadelphia at that time we were not seeing patients with occult syphilis and endocarditis with the same frequency as at Bellevue. Context can and should play a role in our diagnostic reasoning.
But I still remember the patient I saw in the Philadelphia emergency room, a second visit for a man in his 20s with a diffuse, mostly macular rash on his trunk, palms, and soles (visible when the light was turned up in his darkened room, as he felt uncomfortable with bright light), diffuse adenopathy, and enlarged doughy and minimally tender wrists and finger (metacarpophalangeal) joints. I recall wondering why no one had thought to obtain an RPR test on him the first time he had presented to the emergency room; if he had been at Bellevue, the test results would already have returned.
Without appropriate algorithms, things get missed. But using algorithms indiscriminately is cost-ineffective and can lead to cascades of inappropriate tests and interventions. Striking the appropriate balance is part of what comprises the writing of useful clinical care paths.
As I read the article by Sussman et al I wondered who first looked at the patient’s retinas and what initially prompted the testing that was ordered. The presentation was not typical of ocular syphilis, and I would guess that an ophthalmologist or infectious disease consultant evaluating the blurred vision observed the retinal findings, suspected the diagnosis, and ordered serologies, as well as other studies searching for infections and systemic autoimmune disorders that can also cause Roth spots. Gone are the days when internists (and residents) routinely examine the eyes as part of a full physical examination. I am certain an evidence-based study of this practice would find it time-ineffective and with inappropriately low sensitivity.
I don’t think the retinal examination will return to the internist’s checklist. Yet that is where the algorithm that led to this patient’s diagnosis likely began. One can “google” the causes of Roth spots, but as yet there is no app for demonstrating that they are present.
