User login
How soon should patients with infective endocarditis be referred for valve surgery?
WHAT IS ‘EARLY’ SURGERY?
More than 50% of patients with infective endocarditis undergo cardiac surgery during their initial presentation.1
The 2017 guidelines of the American Association for Thoracic Surgery (AATS) recommend surgery once a surgical indication has been established and effective antimicrobial therapy has been started.2
The American Heart Association/American College of Cardiology (ACC/AHA) guidelines recommend surgery during the initial hospitalization before completion of a full course of antibiotics.3
The European Society of Cardiology guidelines define surgery according to the time since the patient received intravenous antibiotic therapy: emergency surgery is performed within 24 hours of therapy, urgent surgery is performed within a few days, and elective surgery is performed after at least 1 to 2 weeks.4
These slight differences are due to the dearth of large randomized trials addressing this question.
INDICATIONS FOR EARLY SURGERY
Left ventricular dysfunction and heart failure
Of all the complications of infectious endocarditis, concomitant heart failure has the greatest impact on prognosis5 and is one of the most frequent indications for surgery.6
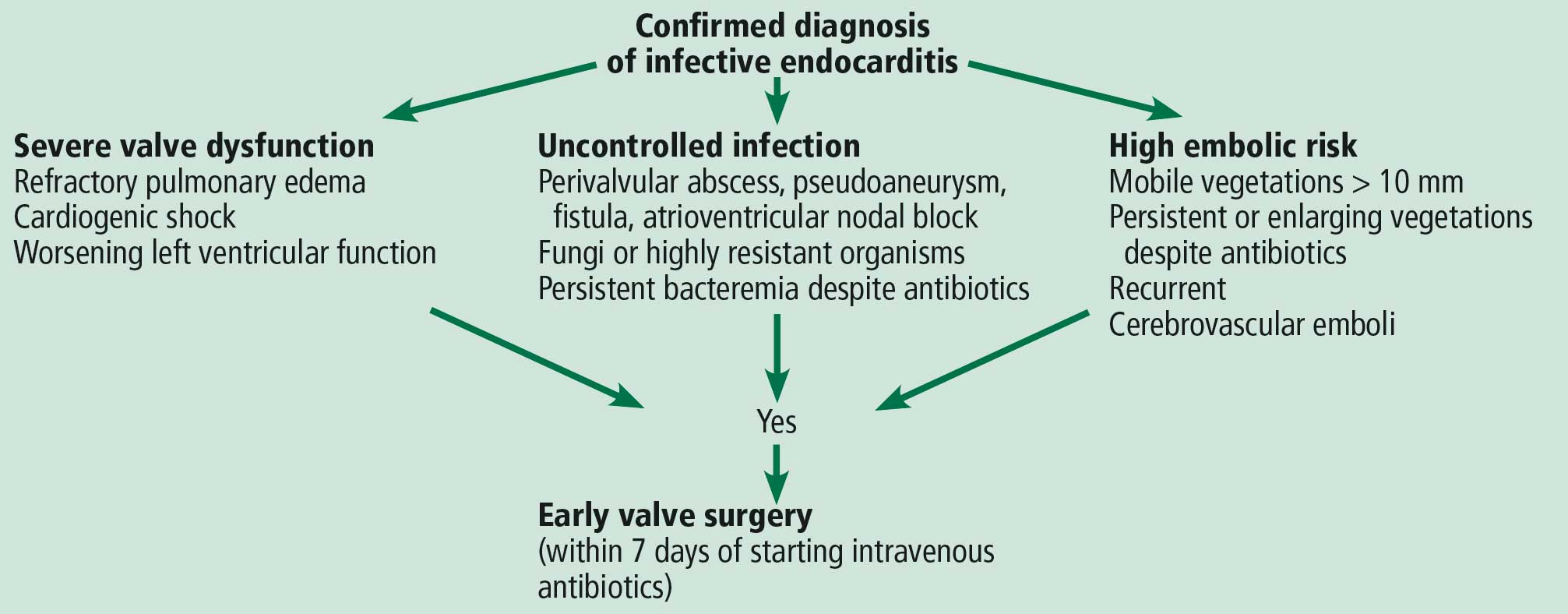
The guidelines recommend emergency surgery during the initial hospitalization for all patients with infective endocarditis who present with refractory pulmonary edema, worsening left ventricular dysfunction, or cardiogenic shock, regardless of whether they have completed a full course of antibiotics. This applies to both native valve endocarditis and prosthetic valve endocarditis.
Uncontrolled persistent infection
Persistent infection is defined as fever and positive cultures persisting after 1 week of appropriate antibiotic treatment.4 However, 1 week is a long time. Persistence of positive blood cultures more than 48 to 72 hours after starting antibiotic therapy is associated with poor outcome and is an independent predictor of in-hospital mortality.7
The ACC/AHA guidelines recommend early surgery in patients with left-sided infective endocarditis caused by fungi or highly resistant organisms such as vancomycin-resistant enterococci or multidrug-resistant gram-negative bacilli.3 Nonetheless, antibiotic resistance is an unusual reason for expediting surgery unless there are additional indications for it.
Extension of the infection beyond the valve annulus, which occurs in about 30% of cases of native valve endocarditis and 50% of cases of prosthetic valve endocarditis,8 is considered a more valid reason to expedite surgery. Similarly, urgent surgery should be considered if there is any evidence of locally uncontrolled infection causing perivalvular abscess, fistula, pseudoaneurysm, or conduction system abnormalities causing atrioventricular nodal block.2–4
Some authors suggest reviewing the surgical pathology and microbial sequencing of excised cardiac valves after surgery to confirm the diagnosis and identify the culprit pathogen.9,10
Right-sided infective endocarditis
Right-sided infective endocarditis has a more favorable prognosis than left-sided infective endocarditis and usually responds well to medical therapy.11
Nevertheless, surgery for right-sided infective endocarditis should be expedited in patients with right heart failure secondary to severe tricuspid regurgitation with poor response to medical therapy or in the case of large tricuspid valve vegetations.12 Likewise, recurrent septic pulmonary emboli can be encountered in the setting of right-sided infective endocarditis and are an indication for early surgery.4,12
Since many patients with right-sided infective endocarditis acquire the infection by intravenous drug use, there is often a reluctance to recommend surgery, given the risk of prosthetic valve infection if they continue to use intravenous drugs.4,12 One study showed that the risk of death or reoperation between 3 and 6 months after surgery for infective endocarditis was 10 times higher in intravenous drug users. Yet their survival after surgery beyond this period was similar to that of patients with endocarditis who did not inject drugs.13 Therefore, the AATS guidelines recommend applying normal indications for surgery to those patients, with emphasis on the need for strict follow-up aimed at addiction treatment.2
Prevention of embolic events
Neurologic embolic events are a frequent complication of infective endocarditis, with the highest risk during the first few days after antibiotics are started. However, this risk decreases significantly after 2 weeks.14
The timing of surgery largely depends on whether the patient has had previous neurologic embolic events and on the size and mobility of the vegetation. The current guidelines recommend early surgery for recurrent emboli and persistent or enlarging vegetations despite appropriate antibiotic therapy, or in case of large vegetations (> 10 mm) on a native valve even in the absence of embolic events.4
A randomized trial by Kang et al15 demonstrated that, compared with conventional care, early surgery (within 48 hours of diagnosis) in patients with native valve endocarditis with large vegetations (> 10 mm) and severe valve dysfunction was associated with a significant reduction in the risk of death and embolic events.
Timing of surgery after a neurologic complication
Determining the right time for surgery is challenging in patients with infective endocarditis who have had neurologic complications, given the risk of hemorrhagic conversion of existing stroke with anticoagulation or exacerbation of cerebral ischemia in case of intraoperative hypotension. The decision should take into account the severity of cardiac decompensation, weighed against the severity of neurologic symptoms.
In general, surgery should be postponed for at least 4 weeks after intracerebral hemorrhage. However, it should be expedited in the event of silent cerebral embolism or transient ischemic attack, or in patients with infective endocarditis with stroke who have other indications for early surgery, as long as cerebral hemorrhage has been excluded by appropriate imaging.4
Early surgery for prosthetic valve endocarditis
The timing of surgery for prosthetic valve endocarditis follows the same general principles as for native valve endocarditis.2–4,12
One study showed that early surgery for prosthetic valve endocarditis was not associated with lower in-hospital and 1-year mortality rates compared with medical therapy.16 On the other hand, a subgroup analysis demonstrated surgery to be significantly beneficial in those with the strongest indications for surgery, including severe valve regurgitation, heart failure, paravalvular abscess, fistula, or prosthetic valve dehiscence.
The decision to proceed with surgery in prosthetic valve endocarditis should be weighed carefully, taking into consideration the patient’s overall clinical condition and estimated surgical risk.16
COLLABORATION IS HELPFUL
Early surgery is indicated for infective endocarditis patients presenting with:
- Refractory heart failure symptoms
- Persistent infection
- Large vegetations with a high risk of embolism.
Expeditious and successful treatment entails multidisciplinary collaboration among experts in cardiology and infectious diseases with access to cardiac surgery input early in the evaluation.
- Lalani T, Cabell CH, Benjamin DK, et al; International Collaboration on Endocarditis-Prospective Cohort Study (ICE-PCS) Investigators. Analysis of the impact of early surgery on in-hospital mortality of native valve endocarditis: use of propensity score and instrumental variable methods to adjust for treatment-selection bias. Circulation 2010; 121(8):1005–1013. doi:10.1161/CIRCULATIONAHA.109.864488
- AATS Surgical Treatment of Infective Endocarditis Consensus Guidelines Writing Committee Chairs; Pettersson GB, Coselli JS; Writing Committee, et al. 2016 The American Association for Thoracic Surgery (AATS) consensus guidelines: surgical treatment of infective endocarditis: executive summary. J Thorac Cardiovasc Surg 2017; 153(6):1241–1258.e29. doi:10.1016/j.jtcvs.2016.09.093
- Nishimura RA, Otto CM, Bonow RO, et al; ACC/AHA Task Force Members. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(23):2440–2492. doi:10.1161/CIR.0000000000000029
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC guidelines for the management of infective endocarditis. Eur Heart J 2015; 36(44):3075–3128. doi:10.1093/eurheartj/ehv319
- Prendergast BD, Tornos P. Surgery for infective endocarditis. Who and when? Circulation 2010; 121(9):1141–1152. doi:10.1161/CIRCULATIONAHA.108.773598
- Tornos P, Iung B, Permanyer-Miralda G, et al. Infective endocarditis in Europe: lessons from the Euro heart survey. Heart 2005; 91(5):571–575. doi:10.1136/hrt.2003.032128
- López J, Sevilla T, Vilacosta I, et al. Prognostic role of persistent positive blood cultures after initiation of antibiotic therapy in left-sided infective endocarditis. Eur Heart J 2013; 34(23):1749–1754. doi:10.1093/eurheartj/ehs379
- Graupner C, Vilacosta I, SanRoman J, et al. Periannular extension of infective endocarditis. J Am Coll Cardiol 2002; 39(7):1204–1211. doi:10.1016/S0735-1097(02)01747-3
- Shrestha NK, Ledtke CS, Wang H, et al. Heart valve culture and sequencing to identify the infective endocarditis pathogen in surgically treated patients. Ann Thorac Surg 2015; 99(1):33–37. doi:10.1016/j.athoracsur.2014.07.028
- Shapira N, Merin O, Rosenmann E, et al. Latent infective endocarditis: epidemiology and clinical characteristics of patients with unsuspected endocarditis detected after elective valve replacement. Ann Thorac Surg 2004; 78(5):1623–1629. doi:10.1016/j.athoracsur.2004.05.052
- Hecht SR, Berger M. Right-sided endocarditis in intravenous drug users. Prognostic features in 102 episodes. Ann Intern Med 1992; 117(7):560–566. doi:10.7326/0003-4819-117-7-560
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132(15):1435–1486. doi:10.1161/CIR.0000000000000296
- Shrestha NK, Jue J, Hussain ST, et al. Injection drug use and outcomes after surgical intervention for infective endocarditis. Ann Thorac Surg 2015; 100(3):875–882. doi:10.1016/j.athoracsur.2015.03.019
- Garcia-Cabrera E, Fernandez-Hidalgo N, Almirante B, et al. Neurological complications of infective endocarditis: risk factors, outcome, and impact of cardiac surgery: a multicenter observational study. Circulation 2013; 127(23):2272–2284. doi:10.1161/CIRCULATIONAHA.112.000813
- Kang DH, Kim YJ, Kim SH, et al. Early surgery versus conventional treatment for infective endocarditis. N Engl J Med 2012; 366(26):2466–2473. doi:10.1056/NEJMoa1112843
- Lalani T, Chu VH, Park LP, et al; International Collaboration on Endocarditis–Prospective Cohort Study Investigators. In-hospital and 1-year mortality in patients undergoing early surgery for prosthetic valve endocarditis. JAMA Intern Med 2013; 173(16):1495–1504. doi:10.1001/jamainternmed.2013.8203
WHAT IS ‘EARLY’ SURGERY?
More than 50% of patients with infective endocarditis undergo cardiac surgery during their initial presentation.1
The 2017 guidelines of the American Association for Thoracic Surgery (AATS) recommend surgery once a surgical indication has been established and effective antimicrobial therapy has been started.2
The American Heart Association/American College of Cardiology (ACC/AHA) guidelines recommend surgery during the initial hospitalization before completion of a full course of antibiotics.3
The European Society of Cardiology guidelines define surgery according to the time since the patient received intravenous antibiotic therapy: emergency surgery is performed within 24 hours of therapy, urgent surgery is performed within a few days, and elective surgery is performed after at least 1 to 2 weeks.4
These slight differences are due to the dearth of large randomized trials addressing this question.
INDICATIONS FOR EARLY SURGERY
Left ventricular dysfunction and heart failure
Of all the complications of infectious endocarditis, concomitant heart failure has the greatest impact on prognosis5 and is one of the most frequent indications for surgery.6
The guidelines recommend emergency surgery during the initial hospitalization for all patients with infective endocarditis who present with refractory pulmonary edema, worsening left ventricular dysfunction, or cardiogenic shock, regardless of whether they have completed a full course of antibiotics. This applies to both native valve endocarditis and prosthetic valve endocarditis.
Uncontrolled persistent infection
Persistent infection is defined as fever and positive cultures persisting after 1 week of appropriate antibiotic treatment.4 However, 1 week is a long time. Persistence of positive blood cultures more than 48 to 72 hours after starting antibiotic therapy is associated with poor outcome and is an independent predictor of in-hospital mortality.7
The ACC/AHA guidelines recommend early surgery in patients with left-sided infective endocarditis caused by fungi or highly resistant organisms such as vancomycin-resistant enterococci or multidrug-resistant gram-negative bacilli.3 Nonetheless, antibiotic resistance is an unusual reason for expediting surgery unless there are additional indications for it.
Extension of the infection beyond the valve annulus, which occurs in about 30% of cases of native valve endocarditis and 50% of cases of prosthetic valve endocarditis,8 is considered a more valid reason to expedite surgery. Similarly, urgent surgery should be considered if there is any evidence of locally uncontrolled infection causing perivalvular abscess, fistula, pseudoaneurysm, or conduction system abnormalities causing atrioventricular nodal block.2–4
Some authors suggest reviewing the surgical pathology and microbial sequencing of excised cardiac valves after surgery to confirm the diagnosis and identify the culprit pathogen.9,10
Right-sided infective endocarditis
Right-sided infective endocarditis has a more favorable prognosis than left-sided infective endocarditis and usually responds well to medical therapy.11
Nevertheless, surgery for right-sided infective endocarditis should be expedited in patients with right heart failure secondary to severe tricuspid regurgitation with poor response to medical therapy or in the case of large tricuspid valve vegetations.12 Likewise, recurrent septic pulmonary emboli can be encountered in the setting of right-sided infective endocarditis and are an indication for early surgery.4,12
Since many patients with right-sided infective endocarditis acquire the infection by intravenous drug use, there is often a reluctance to recommend surgery, given the risk of prosthetic valve infection if they continue to use intravenous drugs.4,12 One study showed that the risk of death or reoperation between 3 and 6 months after surgery for infective endocarditis was 10 times higher in intravenous drug users. Yet their survival after surgery beyond this period was similar to that of patients with endocarditis who did not inject drugs.13 Therefore, the AATS guidelines recommend applying normal indications for surgery to those patients, with emphasis on the need for strict follow-up aimed at addiction treatment.2
Prevention of embolic events
Neurologic embolic events are a frequent complication of infective endocarditis, with the highest risk during the first few days after antibiotics are started. However, this risk decreases significantly after 2 weeks.14
The timing of surgery largely depends on whether the patient has had previous neurologic embolic events and on the size and mobility of the vegetation. The current guidelines recommend early surgery for recurrent emboli and persistent or enlarging vegetations despite appropriate antibiotic therapy, or in case of large vegetations (> 10 mm) on a native valve even in the absence of embolic events.4
A randomized trial by Kang et al15 demonstrated that, compared with conventional care, early surgery (within 48 hours of diagnosis) in patients with native valve endocarditis with large vegetations (> 10 mm) and severe valve dysfunction was associated with a significant reduction in the risk of death and embolic events.
Timing of surgery after a neurologic complication
Determining the right time for surgery is challenging in patients with infective endocarditis who have had neurologic complications, given the risk of hemorrhagic conversion of existing stroke with anticoagulation or exacerbation of cerebral ischemia in case of intraoperative hypotension. The decision should take into account the severity of cardiac decompensation, weighed against the severity of neurologic symptoms.
In general, surgery should be postponed for at least 4 weeks after intracerebral hemorrhage. However, it should be expedited in the event of silent cerebral embolism or transient ischemic attack, or in patients with infective endocarditis with stroke who have other indications for early surgery, as long as cerebral hemorrhage has been excluded by appropriate imaging.4
Early surgery for prosthetic valve endocarditis
The timing of surgery for prosthetic valve endocarditis follows the same general principles as for native valve endocarditis.2–4,12
One study showed that early surgery for prosthetic valve endocarditis was not associated with lower in-hospital and 1-year mortality rates compared with medical therapy.16 On the other hand, a subgroup analysis demonstrated surgery to be significantly beneficial in those with the strongest indications for surgery, including severe valve regurgitation, heart failure, paravalvular abscess, fistula, or prosthetic valve dehiscence.
The decision to proceed with surgery in prosthetic valve endocarditis should be weighed carefully, taking into consideration the patient’s overall clinical condition and estimated surgical risk.16
COLLABORATION IS HELPFUL
Early surgery is indicated for infective endocarditis patients presenting with:
- Refractory heart failure symptoms
- Persistent infection
- Large vegetations with a high risk of embolism.
Expeditious and successful treatment entails multidisciplinary collaboration among experts in cardiology and infectious diseases with access to cardiac surgery input early in the evaluation.
WHAT IS ‘EARLY’ SURGERY?
More than 50% of patients with infective endocarditis undergo cardiac surgery during their initial presentation.1
The 2017 guidelines of the American Association for Thoracic Surgery (AATS) recommend surgery once a surgical indication has been established and effective antimicrobial therapy has been started.2
The American Heart Association/American College of Cardiology (ACC/AHA) guidelines recommend surgery during the initial hospitalization before completion of a full course of antibiotics.3
The European Society of Cardiology guidelines define surgery according to the time since the patient received intravenous antibiotic therapy: emergency surgery is performed within 24 hours of therapy, urgent surgery is performed within a few days, and elective surgery is performed after at least 1 to 2 weeks.4
These slight differences are due to the dearth of large randomized trials addressing this question.
INDICATIONS FOR EARLY SURGERY
Left ventricular dysfunction and heart failure
Of all the complications of infectious endocarditis, concomitant heart failure has the greatest impact on prognosis5 and is one of the most frequent indications for surgery.6
The guidelines recommend emergency surgery during the initial hospitalization for all patients with infective endocarditis who present with refractory pulmonary edema, worsening left ventricular dysfunction, or cardiogenic shock, regardless of whether they have completed a full course of antibiotics. This applies to both native valve endocarditis and prosthetic valve endocarditis.
Uncontrolled persistent infection
Persistent infection is defined as fever and positive cultures persisting after 1 week of appropriate antibiotic treatment.4 However, 1 week is a long time. Persistence of positive blood cultures more than 48 to 72 hours after starting antibiotic therapy is associated with poor outcome and is an independent predictor of in-hospital mortality.7
The ACC/AHA guidelines recommend early surgery in patients with left-sided infective endocarditis caused by fungi or highly resistant organisms such as vancomycin-resistant enterococci or multidrug-resistant gram-negative bacilli.3 Nonetheless, antibiotic resistance is an unusual reason for expediting surgery unless there are additional indications for it.
Extension of the infection beyond the valve annulus, which occurs in about 30% of cases of native valve endocarditis and 50% of cases of prosthetic valve endocarditis,8 is considered a more valid reason to expedite surgery. Similarly, urgent surgery should be considered if there is any evidence of locally uncontrolled infection causing perivalvular abscess, fistula, pseudoaneurysm, or conduction system abnormalities causing atrioventricular nodal block.2–4
Some authors suggest reviewing the surgical pathology and microbial sequencing of excised cardiac valves after surgery to confirm the diagnosis and identify the culprit pathogen.9,10
Right-sided infective endocarditis
Right-sided infective endocarditis has a more favorable prognosis than left-sided infective endocarditis and usually responds well to medical therapy.11
Nevertheless, surgery for right-sided infective endocarditis should be expedited in patients with right heart failure secondary to severe tricuspid regurgitation with poor response to medical therapy or in the case of large tricuspid valve vegetations.12 Likewise, recurrent septic pulmonary emboli can be encountered in the setting of right-sided infective endocarditis and are an indication for early surgery.4,12
Since many patients with right-sided infective endocarditis acquire the infection by intravenous drug use, there is often a reluctance to recommend surgery, given the risk of prosthetic valve infection if they continue to use intravenous drugs.4,12 One study showed that the risk of death or reoperation between 3 and 6 months after surgery for infective endocarditis was 10 times higher in intravenous drug users. Yet their survival after surgery beyond this period was similar to that of patients with endocarditis who did not inject drugs.13 Therefore, the AATS guidelines recommend applying normal indications for surgery to those patients, with emphasis on the need for strict follow-up aimed at addiction treatment.2
Prevention of embolic events
Neurologic embolic events are a frequent complication of infective endocarditis, with the highest risk during the first few days after antibiotics are started. However, this risk decreases significantly after 2 weeks.14
The timing of surgery largely depends on whether the patient has had previous neurologic embolic events and on the size and mobility of the vegetation. The current guidelines recommend early surgery for recurrent emboli and persistent or enlarging vegetations despite appropriate antibiotic therapy, or in case of large vegetations (> 10 mm) on a native valve even in the absence of embolic events.4
A randomized trial by Kang et al15 demonstrated that, compared with conventional care, early surgery (within 48 hours of diagnosis) in patients with native valve endocarditis with large vegetations (> 10 mm) and severe valve dysfunction was associated with a significant reduction in the risk of death and embolic events.
Timing of surgery after a neurologic complication
Determining the right time for surgery is challenging in patients with infective endocarditis who have had neurologic complications, given the risk of hemorrhagic conversion of existing stroke with anticoagulation or exacerbation of cerebral ischemia in case of intraoperative hypotension. The decision should take into account the severity of cardiac decompensation, weighed against the severity of neurologic symptoms.
In general, surgery should be postponed for at least 4 weeks after intracerebral hemorrhage. However, it should be expedited in the event of silent cerebral embolism or transient ischemic attack, or in patients with infective endocarditis with stroke who have other indications for early surgery, as long as cerebral hemorrhage has been excluded by appropriate imaging.4
Early surgery for prosthetic valve endocarditis
The timing of surgery for prosthetic valve endocarditis follows the same general principles as for native valve endocarditis.2–4,12
One study showed that early surgery for prosthetic valve endocarditis was not associated with lower in-hospital and 1-year mortality rates compared with medical therapy.16 On the other hand, a subgroup analysis demonstrated surgery to be significantly beneficial in those with the strongest indications for surgery, including severe valve regurgitation, heart failure, paravalvular abscess, fistula, or prosthetic valve dehiscence.
The decision to proceed with surgery in prosthetic valve endocarditis should be weighed carefully, taking into consideration the patient’s overall clinical condition and estimated surgical risk.16
COLLABORATION IS HELPFUL
Early surgery is indicated for infective endocarditis patients presenting with:
- Refractory heart failure symptoms
- Persistent infection
- Large vegetations with a high risk of embolism.
Expeditious and successful treatment entails multidisciplinary collaboration among experts in cardiology and infectious diseases with access to cardiac surgery input early in the evaluation.
- Lalani T, Cabell CH, Benjamin DK, et al; International Collaboration on Endocarditis-Prospective Cohort Study (ICE-PCS) Investigators. Analysis of the impact of early surgery on in-hospital mortality of native valve endocarditis: use of propensity score and instrumental variable methods to adjust for treatment-selection bias. Circulation 2010; 121(8):1005–1013. doi:10.1161/CIRCULATIONAHA.109.864488
- AATS Surgical Treatment of Infective Endocarditis Consensus Guidelines Writing Committee Chairs; Pettersson GB, Coselli JS; Writing Committee, et al. 2016 The American Association for Thoracic Surgery (AATS) consensus guidelines: surgical treatment of infective endocarditis: executive summary. J Thorac Cardiovasc Surg 2017; 153(6):1241–1258.e29. doi:10.1016/j.jtcvs.2016.09.093
- Nishimura RA, Otto CM, Bonow RO, et al; ACC/AHA Task Force Members. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(23):2440–2492. doi:10.1161/CIR.0000000000000029
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC guidelines for the management of infective endocarditis. Eur Heart J 2015; 36(44):3075–3128. doi:10.1093/eurheartj/ehv319
- Prendergast BD, Tornos P. Surgery for infective endocarditis. Who and when? Circulation 2010; 121(9):1141–1152. doi:10.1161/CIRCULATIONAHA.108.773598
- Tornos P, Iung B, Permanyer-Miralda G, et al. Infective endocarditis in Europe: lessons from the Euro heart survey. Heart 2005; 91(5):571–575. doi:10.1136/hrt.2003.032128
- López J, Sevilla T, Vilacosta I, et al. Prognostic role of persistent positive blood cultures after initiation of antibiotic therapy in left-sided infective endocarditis. Eur Heart J 2013; 34(23):1749–1754. doi:10.1093/eurheartj/ehs379
- Graupner C, Vilacosta I, SanRoman J, et al. Periannular extension of infective endocarditis. J Am Coll Cardiol 2002; 39(7):1204–1211. doi:10.1016/S0735-1097(02)01747-3
- Shrestha NK, Ledtke CS, Wang H, et al. Heart valve culture and sequencing to identify the infective endocarditis pathogen in surgically treated patients. Ann Thorac Surg 2015; 99(1):33–37. doi:10.1016/j.athoracsur.2014.07.028
- Shapira N, Merin O, Rosenmann E, et al. Latent infective endocarditis: epidemiology and clinical characteristics of patients with unsuspected endocarditis detected after elective valve replacement. Ann Thorac Surg 2004; 78(5):1623–1629. doi:10.1016/j.athoracsur.2004.05.052
- Hecht SR, Berger M. Right-sided endocarditis in intravenous drug users. Prognostic features in 102 episodes. Ann Intern Med 1992; 117(7):560–566. doi:10.7326/0003-4819-117-7-560
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132(15):1435–1486. doi:10.1161/CIR.0000000000000296
- Shrestha NK, Jue J, Hussain ST, et al. Injection drug use and outcomes after surgical intervention for infective endocarditis. Ann Thorac Surg 2015; 100(3):875–882. doi:10.1016/j.athoracsur.2015.03.019
- Garcia-Cabrera E, Fernandez-Hidalgo N, Almirante B, et al. Neurological complications of infective endocarditis: risk factors, outcome, and impact of cardiac surgery: a multicenter observational study. Circulation 2013; 127(23):2272–2284. doi:10.1161/CIRCULATIONAHA.112.000813
- Kang DH, Kim YJ, Kim SH, et al. Early surgery versus conventional treatment for infective endocarditis. N Engl J Med 2012; 366(26):2466–2473. doi:10.1056/NEJMoa1112843
- Lalani T, Chu VH, Park LP, et al; International Collaboration on Endocarditis–Prospective Cohort Study Investigators. In-hospital and 1-year mortality in patients undergoing early surgery for prosthetic valve endocarditis. JAMA Intern Med 2013; 173(16):1495–1504. doi:10.1001/jamainternmed.2013.8203
- Lalani T, Cabell CH, Benjamin DK, et al; International Collaboration on Endocarditis-Prospective Cohort Study (ICE-PCS) Investigators. Analysis of the impact of early surgery on in-hospital mortality of native valve endocarditis: use of propensity score and instrumental variable methods to adjust for treatment-selection bias. Circulation 2010; 121(8):1005–1013. doi:10.1161/CIRCULATIONAHA.109.864488
- AATS Surgical Treatment of Infective Endocarditis Consensus Guidelines Writing Committee Chairs; Pettersson GB, Coselli JS; Writing Committee, et al. 2016 The American Association for Thoracic Surgery (AATS) consensus guidelines: surgical treatment of infective endocarditis: executive summary. J Thorac Cardiovasc Surg 2017; 153(6):1241–1258.e29. doi:10.1016/j.jtcvs.2016.09.093
- Nishimura RA, Otto CM, Bonow RO, et al; ACC/AHA Task Force Members. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(23):2440–2492. doi:10.1161/CIR.0000000000000029
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC guidelines for the management of infective endocarditis. Eur Heart J 2015; 36(44):3075–3128. doi:10.1093/eurheartj/ehv319
- Prendergast BD, Tornos P. Surgery for infective endocarditis. Who and when? Circulation 2010; 121(9):1141–1152. doi:10.1161/CIRCULATIONAHA.108.773598
- Tornos P, Iung B, Permanyer-Miralda G, et al. Infective endocarditis in Europe: lessons from the Euro heart survey. Heart 2005; 91(5):571–575. doi:10.1136/hrt.2003.032128
- López J, Sevilla T, Vilacosta I, et al. Prognostic role of persistent positive blood cultures after initiation of antibiotic therapy in left-sided infective endocarditis. Eur Heart J 2013; 34(23):1749–1754. doi:10.1093/eurheartj/ehs379
- Graupner C, Vilacosta I, SanRoman J, et al. Periannular extension of infective endocarditis. J Am Coll Cardiol 2002; 39(7):1204–1211. doi:10.1016/S0735-1097(02)01747-3
- Shrestha NK, Ledtke CS, Wang H, et al. Heart valve culture and sequencing to identify the infective endocarditis pathogen in surgically treated patients. Ann Thorac Surg 2015; 99(1):33–37. doi:10.1016/j.athoracsur.2014.07.028
- Shapira N, Merin O, Rosenmann E, et al. Latent infective endocarditis: epidemiology and clinical characteristics of patients with unsuspected endocarditis detected after elective valve replacement. Ann Thorac Surg 2004; 78(5):1623–1629. doi:10.1016/j.athoracsur.2004.05.052
- Hecht SR, Berger M. Right-sided endocarditis in intravenous drug users. Prognostic features in 102 episodes. Ann Intern Med 1992; 117(7):560–566. doi:10.7326/0003-4819-117-7-560
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132(15):1435–1486. doi:10.1161/CIR.0000000000000296
- Shrestha NK, Jue J, Hussain ST, et al. Injection drug use and outcomes after surgical intervention for infective endocarditis. Ann Thorac Surg 2015; 100(3):875–882. doi:10.1016/j.athoracsur.2015.03.019
- Garcia-Cabrera E, Fernandez-Hidalgo N, Almirante B, et al. Neurological complications of infective endocarditis: risk factors, outcome, and impact of cardiac surgery: a multicenter observational study. Circulation 2013; 127(23):2272–2284. doi:10.1161/CIRCULATIONAHA.112.000813
- Kang DH, Kim YJ, Kim SH, et al. Early surgery versus conventional treatment for infective endocarditis. N Engl J Med 2012; 366(26):2466–2473. doi:10.1056/NEJMoa1112843
- Lalani T, Chu VH, Park LP, et al; International Collaboration on Endocarditis–Prospective Cohort Study Investigators. In-hospital and 1-year mortality in patients undergoing early surgery for prosthetic valve endocarditis. JAMA Intern Med 2013; 173(16):1495–1504. doi:10.1001/jamainternmed.2013.8203
Infective endocarditis: Refer for expert team care as soon as possible
In this issue of the Journal, Soud et al discuss the timing of referral of patients with infective endocarditis to surgery.1 When having this discussion, it is important to understand the nature of the disease and the role of surgery in its treatment.
Unless successfully treated and cured, infective endocarditis is fatal. It is associated with septic embolism (systemic with left-sided infective endocarditis and pulmonary with right-sided infective endocarditis), destruction of valve tissue, and invasion outside the aortic root or into the atrioventricular groove. Antimicrobials kill sensitive and exposed organisms but cannot reach those hiding in vegetations or biofilm, on foreign material, or in invaded extravascular tissue.
The objectives of surgery are to eliminate the source of embolism, debride and remove infected tissue and foreign material, expose and make residual organisms vulnerable to antimicrobials, and restore functional valves and cardiac integrity. Surgery to treat infective endocarditis is difficult and high-risk and requires an experienced surgeon. But final cure of the infection is still by antimicrobial treatment.
INFECTIVE ENDOCARDITIS NEEDS MULTIDISCIPLINARY CARE
Every aspect of infective endocarditis—diagnosis, medical management, management of complications, and surgery—is difficult. Recent guidelines2–6 therefore favor care by a multidisciplinary team that includes an infectious disease specialist, cardiologist, and cardiac surgeon from the very beginning, with access to any other needed discipline, often including neurology, neurosurgery, nephrology, and dependence specialists. Patients with infective endocarditis should be referred early to a center with access to a full endocarditis treatment team. The need for surgery and the optimal timing of it are team decisions. The American Association for Thoracic Surgery infective endocarditis guidelines are question-based and address most aspects that surgeons must consider before, during, and after operation.2
IF SURGERY IS INDICATED, IT IS BEST DONE SOONER
Once there is an indication to operate, the operation should be expedited. Delays mean continued risk of disease progression, invasion, heart block, and embolic events. Determining the timing of surgery is difficult in patients who have suffered an embolic stroke—nonhemorrhagic or hemorrhagic—or who have suffered brain bleeding; management of these issues has recently triggered expert opinion and review articles.7,8 The recommendation for early surgery is based on the conviction that once the patient has been stabilized (or has overwhelming mechanical hemodynamic problems requiring emergency surgery) and adequate antimicrobial coverage is on board, there are no additional benefits to delaying surgery.9 When the indication to operate is large mobile vegetations associated with a high risk of stroke, surgery before another event can make all the difference.
In the operating room, the first aspect addressed is adequate debridement. There is wide agreement that repair is preferable to replacement for the mitral and tricuspid valves, but there is no agreement that an allograft (although favored by our team) is the best replacement alternative for a destroyed aortic root. The key is that surgeons and their surgical teams must have the experience and tools that work for them.
Our recommendation is to refer all patients with infective endocarditis to a center with access to a full team of experienced experts able to address all aspects of the disease and its complications.
- Soud M, Pacha HM, Alraies MC. How soon should patients with infective endocarditis be referred for valve surgery? Cleve Clin J Med 2018; 85(5):362–364. doi:10.3949/ccjm.85a:17052
- Pettersson GB, Coselli JS, Pettersson GB, et al. 2016 The American Association for Thoracic Surgery (AATS) consensus guidelines: surgical treatment of infective endocarditis: executive summary. J Thorac Cardiovasc Surg 2017; 153(6):1241–1258.e29. doi:10.1016/j.jtcvs.2016.09.093
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132(15):1435–1486. doi:10.1161/CIR.0000000000000296
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J 2015; 36(44):3075–3128. doi:10.1093/eurheartj/ehv319
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease:executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129(23):2440–2492. doi:10.1161/CIR.0000000000000029
- Byrne JG, Rezai K, Sanchez JA, et al. Surgical management of endocarditis: the Society of Thoracic Surgeons clinical practice guideline. Ann Thorac Surg 2011; 91(6):2012–2019. doi:10.1016/j.athoracsur.2011.01.106
- Yanagawa B, Pettersson GB, Habib G, et al. Surgical management of infective endocarditis complicated by embolic stroke: practical recommendations for clinicians. Circulation 2016; 134(17):1280–1292. doi:10.1161/CIRCULATIONAHA.116.024156
- Cahill TJ , Baddour LM, Habib G, et al. Challenges in infective endocarditis. J Am Coll Cardiol 2017; 69(3):325–344. doi:10.1016/j.jacc.2016.10.066
- Kang DH, Kim YJ, Kim SH, et al. Early surgery versus conventional treatment for infective endocarditis. N Engl J Med 2012; 366(26):2466–2473. doi:10.1056/NEJMoa1112843
In this issue of the Journal, Soud et al discuss the timing of referral of patients with infective endocarditis to surgery.1 When having this discussion, it is important to understand the nature of the disease and the role of surgery in its treatment.
Unless successfully treated and cured, infective endocarditis is fatal. It is associated with septic embolism (systemic with left-sided infective endocarditis and pulmonary with right-sided infective endocarditis), destruction of valve tissue, and invasion outside the aortic root or into the atrioventricular groove. Antimicrobials kill sensitive and exposed organisms but cannot reach those hiding in vegetations or biofilm, on foreign material, or in invaded extravascular tissue.
The objectives of surgery are to eliminate the source of embolism, debride and remove infected tissue and foreign material, expose and make residual organisms vulnerable to antimicrobials, and restore functional valves and cardiac integrity. Surgery to treat infective endocarditis is difficult and high-risk and requires an experienced surgeon. But final cure of the infection is still by antimicrobial treatment.
INFECTIVE ENDOCARDITIS NEEDS MULTIDISCIPLINARY CARE
Every aspect of infective endocarditis—diagnosis, medical management, management of complications, and surgery—is difficult. Recent guidelines2–6 therefore favor care by a multidisciplinary team that includes an infectious disease specialist, cardiologist, and cardiac surgeon from the very beginning, with access to any other needed discipline, often including neurology, neurosurgery, nephrology, and dependence specialists. Patients with infective endocarditis should be referred early to a center with access to a full endocarditis treatment team. The need for surgery and the optimal timing of it are team decisions. The American Association for Thoracic Surgery infective endocarditis guidelines are question-based and address most aspects that surgeons must consider before, during, and after operation.2
IF SURGERY IS INDICATED, IT IS BEST DONE SOONER
Once there is an indication to operate, the operation should be expedited. Delays mean continued risk of disease progression, invasion, heart block, and embolic events. Determining the timing of surgery is difficult in patients who have suffered an embolic stroke—nonhemorrhagic or hemorrhagic—or who have suffered brain bleeding; management of these issues has recently triggered expert opinion and review articles.7,8 The recommendation for early surgery is based on the conviction that once the patient has been stabilized (or has overwhelming mechanical hemodynamic problems requiring emergency surgery) and adequate antimicrobial coverage is on board, there are no additional benefits to delaying surgery.9 When the indication to operate is large mobile vegetations associated with a high risk of stroke, surgery before another event can make all the difference.
In the operating room, the first aspect addressed is adequate debridement. There is wide agreement that repair is preferable to replacement for the mitral and tricuspid valves, but there is no agreement that an allograft (although favored by our team) is the best replacement alternative for a destroyed aortic root. The key is that surgeons and their surgical teams must have the experience and tools that work for them.
Our recommendation is to refer all patients with infective endocarditis to a center with access to a full team of experienced experts able to address all aspects of the disease and its complications.
In this issue of the Journal, Soud et al discuss the timing of referral of patients with infective endocarditis to surgery.1 When having this discussion, it is important to understand the nature of the disease and the role of surgery in its treatment.
Unless successfully treated and cured, infective endocarditis is fatal. It is associated with septic embolism (systemic with left-sided infective endocarditis and pulmonary with right-sided infective endocarditis), destruction of valve tissue, and invasion outside the aortic root or into the atrioventricular groove. Antimicrobials kill sensitive and exposed organisms but cannot reach those hiding in vegetations or biofilm, on foreign material, or in invaded extravascular tissue.
The objectives of surgery are to eliminate the source of embolism, debride and remove infected tissue and foreign material, expose and make residual organisms vulnerable to antimicrobials, and restore functional valves and cardiac integrity. Surgery to treat infective endocarditis is difficult and high-risk and requires an experienced surgeon. But final cure of the infection is still by antimicrobial treatment.
INFECTIVE ENDOCARDITIS NEEDS MULTIDISCIPLINARY CARE
Every aspect of infective endocarditis—diagnosis, medical management, management of complications, and surgery—is difficult. Recent guidelines2–6 therefore favor care by a multidisciplinary team that includes an infectious disease specialist, cardiologist, and cardiac surgeon from the very beginning, with access to any other needed discipline, often including neurology, neurosurgery, nephrology, and dependence specialists. Patients with infective endocarditis should be referred early to a center with access to a full endocarditis treatment team. The need for surgery and the optimal timing of it are team decisions. The American Association for Thoracic Surgery infective endocarditis guidelines are question-based and address most aspects that surgeons must consider before, during, and after operation.2
IF SURGERY IS INDICATED, IT IS BEST DONE SOONER
Once there is an indication to operate, the operation should be expedited. Delays mean continued risk of disease progression, invasion, heart block, and embolic events. Determining the timing of surgery is difficult in patients who have suffered an embolic stroke—nonhemorrhagic or hemorrhagic—or who have suffered brain bleeding; management of these issues has recently triggered expert opinion and review articles.7,8 The recommendation for early surgery is based on the conviction that once the patient has been stabilized (or has overwhelming mechanical hemodynamic problems requiring emergency surgery) and adequate antimicrobial coverage is on board, there are no additional benefits to delaying surgery.9 When the indication to operate is large mobile vegetations associated with a high risk of stroke, surgery before another event can make all the difference.
In the operating room, the first aspect addressed is adequate debridement. There is wide agreement that repair is preferable to replacement for the mitral and tricuspid valves, but there is no agreement that an allograft (although favored by our team) is the best replacement alternative for a destroyed aortic root. The key is that surgeons and their surgical teams must have the experience and tools that work for them.
Our recommendation is to refer all patients with infective endocarditis to a center with access to a full team of experienced experts able to address all aspects of the disease and its complications.
- Soud M, Pacha HM, Alraies MC. How soon should patients with infective endocarditis be referred for valve surgery? Cleve Clin J Med 2018; 85(5):362–364. doi:10.3949/ccjm.85a:17052
- Pettersson GB, Coselli JS, Pettersson GB, et al. 2016 The American Association for Thoracic Surgery (AATS) consensus guidelines: surgical treatment of infective endocarditis: executive summary. J Thorac Cardiovasc Surg 2017; 153(6):1241–1258.e29. doi:10.1016/j.jtcvs.2016.09.093
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132(15):1435–1486. doi:10.1161/CIR.0000000000000296
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J 2015; 36(44):3075–3128. doi:10.1093/eurheartj/ehv319
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease:executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129(23):2440–2492. doi:10.1161/CIR.0000000000000029
- Byrne JG, Rezai K, Sanchez JA, et al. Surgical management of endocarditis: the Society of Thoracic Surgeons clinical practice guideline. Ann Thorac Surg 2011; 91(6):2012–2019. doi:10.1016/j.athoracsur.2011.01.106
- Yanagawa B, Pettersson GB, Habib G, et al. Surgical management of infective endocarditis complicated by embolic stroke: practical recommendations for clinicians. Circulation 2016; 134(17):1280–1292. doi:10.1161/CIRCULATIONAHA.116.024156
- Cahill TJ , Baddour LM, Habib G, et al. Challenges in infective endocarditis. J Am Coll Cardiol 2017; 69(3):325–344. doi:10.1016/j.jacc.2016.10.066
- Kang DH, Kim YJ, Kim SH, et al. Early surgery versus conventional treatment for infective endocarditis. N Engl J Med 2012; 366(26):2466–2473. doi:10.1056/NEJMoa1112843
- Soud M, Pacha HM, Alraies MC. How soon should patients with infective endocarditis be referred for valve surgery? Cleve Clin J Med 2018; 85(5):362–364. doi:10.3949/ccjm.85a:17052
- Pettersson GB, Coselli JS, Pettersson GB, et al. 2016 The American Association for Thoracic Surgery (AATS) consensus guidelines: surgical treatment of infective endocarditis: executive summary. J Thorac Cardiovasc Surg 2017; 153(6):1241–1258.e29. doi:10.1016/j.jtcvs.2016.09.093
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132(15):1435–1486. doi:10.1161/CIR.0000000000000296
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J 2015; 36(44):3075–3128. doi:10.1093/eurheartj/ehv319
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease:executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129(23):2440–2492. doi:10.1161/CIR.0000000000000029
- Byrne JG, Rezai K, Sanchez JA, et al. Surgical management of endocarditis: the Society of Thoracic Surgeons clinical practice guideline. Ann Thorac Surg 2011; 91(6):2012–2019. doi:10.1016/j.athoracsur.2011.01.106
- Yanagawa B, Pettersson GB, Habib G, et al. Surgical management of infective endocarditis complicated by embolic stroke: practical recommendations for clinicians. Circulation 2016; 134(17):1280–1292. doi:10.1161/CIRCULATIONAHA.116.024156
- Cahill TJ , Baddour LM, Habib G, et al. Challenges in infective endocarditis. J Am Coll Cardiol 2017; 69(3):325–344. doi:10.1016/j.jacc.2016.10.066
- Kang DH, Kim YJ, Kim SH, et al. Early surgery versus conventional treatment for infective endocarditis. N Engl J Med 2012; 366(26):2466–2473. doi:10.1056/NEJMoa1112843

Cardiorenal syndrome
To the Editor: I read with interest the thoughtful review of cardiorenal syndrome by Drs. Thind, Loehrke, and Wilt1 and the accompanying editorial by Dr. Grodin.2 These articles certainly add to our growing knowledge of the syndrome and the importance of treating volume overload in these complex patients.
Indeed, we and others have stressed the primary importance of renal dysfunction in patients with volume overload and acute decompensated heart failure.3,4 We have learned that even small rises in serum creatinine predict poor outcomes in these patients. And even if the serum creatinine level comes back down during hospitalization, acute kidney injury (AKI) is still associated with risk.5
Nevertheless, clinicians remain frustrated with the practical management of patients with volume overload and worsening AKI. When faced with a rising serum creatinine level in a patient being treated for decompensated heart failure with signs or symptoms of volume overload, I suggest the following:
Perform careful bedside and chart review searching for evidence of AKI related to causes other than cardiorenal syndrome. Ask whether the rise in serum creatinine could be caused by new obstruction (eg, urinary retention, upper urinary tract obstruction), a nephrotoxin (eg, nonsteroidal anti-inflammatory drugs), a primary tubulointerstitial or glomerular process (eg, drug-induced acute interstitial nephritis, acute glomerulonephritis), acute tubular necrosis, or a new hemodynamic event threatening renal perfusion (eg, hypotension, a new arrhythmia). It is often best to arrive at a diagnosis of AKI due to cardiorenal dysfunction by exclusion, much like the working definitions of hepatorenal syndrome.6 This requires review of the urine sediment (looking for evidence of granular casts of acute tubular necrosis, or evidence of glomerulonephritis or interstitial nephritis), electronic medical record, vital signs, telemetry, and perhaps renal ultrasonography.
In the absence of frank evidence of “overdiuresis” such as worsening hypernatremia, with dropping blood pressure, clinical hypoperfusion, and contraction alkalosis, avoid the temptation to suspend diuretics. Alternatively, an increase in diuretic dose, or addition of a distal diuretic (ie, metolazone) may be needed to address persistent renal venous congestion as the cause of the AKI.3 In this situation, be sure to monitor electrolytes, volume status, and renal function closely while diuretic treatment is augmented. In many such cases, the serum creatinine may actually start to decrease after a more robust diuresis is generated. In these patients, it may also be prudent to temporarily suspend antagonists of the renin-angiotensin-aldosterone system, although this remains controversial.
Management of such patients should be done collaboratively with cardiologists well versed in the treatment of cardiorenal syndrome. It may be possible that the worsening renal function in these patients represents important changes in cardiac rhythm or function (eg, low cardiac output state, new or worsening valvular disease, ongoing myocardial ischemia, cardiac tamponade, uncontrolled bradycardia or tachyarrythmia). Interventions aimed at reversing such perturbations could be the most important steps in improving cardiorenal function and reversing AKI.
- Thind GS, Loehrke M, Wilt JL. Acute cardiorenal syndrome: mechanisms and clinical implications. Cleve Clin J Med 2018; 85(3):231–239. doi:10.3949/ccjm.85a.17019
- Grodin JL. Hemodynamically, the kidney is at the heart of cardiorenal syndrome. Cleve Clin J Med 2018; 85(3):240–242. doi:10.3949/ccjm.85a.17126
- Freda BJ, Slawsky M, Mallidi J, Braden GL. Decongestive treatment of acute decompensated heart failure: cardiorenal implications of ultrafiltration and diuretics. Am J Kid Dis 2011; 58(6):1005–1017. doi:10.1053/j.ajkd.2011.07.023
- Tang WH, Kitai T. Intrarenal blood flow: a window into the congestive kidney failure phenotype of heart failure? JACC Heart Fail 2016; 4(8):683–686. doi:10.1016/j.jchf.2016.05.009
- Freda BJ, Knee AB, Braden GL, Visintainer PF, Thakaer CV. Effect of transient and sustained acute kidney injury on readmissions in acute decompensated heart failure. Am J Cardiol 2017; 119(11):1809–1814. doi:10.1016/j.amjcard.2017.02.044
- Bucsics T, Krones E. Renal dysfunction in cirrhosis: acute kidney injury and the hepatorenal syndrome. Gastroenterol Rep (Oxf) 2017; 5(2):127–137. doi:10.1093/gastro/gox009
To the Editor: I read with interest the thoughtful review of cardiorenal syndrome by Drs. Thind, Loehrke, and Wilt1 and the accompanying editorial by Dr. Grodin.2 These articles certainly add to our growing knowledge of the syndrome and the importance of treating volume overload in these complex patients.
Indeed, we and others have stressed the primary importance of renal dysfunction in patients with volume overload and acute decompensated heart failure.3,4 We have learned that even small rises in serum creatinine predict poor outcomes in these patients. And even if the serum creatinine level comes back down during hospitalization, acute kidney injury (AKI) is still associated with risk.5
Nevertheless, clinicians remain frustrated with the practical management of patients with volume overload and worsening AKI. When faced with a rising serum creatinine level in a patient being treated for decompensated heart failure with signs or symptoms of volume overload, I suggest the following:
Perform careful bedside and chart review searching for evidence of AKI related to causes other than cardiorenal syndrome. Ask whether the rise in serum creatinine could be caused by new obstruction (eg, urinary retention, upper urinary tract obstruction), a nephrotoxin (eg, nonsteroidal anti-inflammatory drugs), a primary tubulointerstitial or glomerular process (eg, drug-induced acute interstitial nephritis, acute glomerulonephritis), acute tubular necrosis, or a new hemodynamic event threatening renal perfusion (eg, hypotension, a new arrhythmia). It is often best to arrive at a diagnosis of AKI due to cardiorenal dysfunction by exclusion, much like the working definitions of hepatorenal syndrome.6 This requires review of the urine sediment (looking for evidence of granular casts of acute tubular necrosis, or evidence of glomerulonephritis or interstitial nephritis), electronic medical record, vital signs, telemetry, and perhaps renal ultrasonography.
In the absence of frank evidence of “overdiuresis” such as worsening hypernatremia, with dropping blood pressure, clinical hypoperfusion, and contraction alkalosis, avoid the temptation to suspend diuretics. Alternatively, an increase in diuretic dose, or addition of a distal diuretic (ie, metolazone) may be needed to address persistent renal venous congestion as the cause of the AKI.3 In this situation, be sure to monitor electrolytes, volume status, and renal function closely while diuretic treatment is augmented. In many such cases, the serum creatinine may actually start to decrease after a more robust diuresis is generated. In these patients, it may also be prudent to temporarily suspend antagonists of the renin-angiotensin-aldosterone system, although this remains controversial.
Management of such patients should be done collaboratively with cardiologists well versed in the treatment of cardiorenal syndrome. It may be possible that the worsening renal function in these patients represents important changes in cardiac rhythm or function (eg, low cardiac output state, new or worsening valvular disease, ongoing myocardial ischemia, cardiac tamponade, uncontrolled bradycardia or tachyarrythmia). Interventions aimed at reversing such perturbations could be the most important steps in improving cardiorenal function and reversing AKI.
To the Editor: I read with interest the thoughtful review of cardiorenal syndrome by Drs. Thind, Loehrke, and Wilt1 and the accompanying editorial by Dr. Grodin.2 These articles certainly add to our growing knowledge of the syndrome and the importance of treating volume overload in these complex patients.
Indeed, we and others have stressed the primary importance of renal dysfunction in patients with volume overload and acute decompensated heart failure.3,4 We have learned that even small rises in serum creatinine predict poor outcomes in these patients. And even if the serum creatinine level comes back down during hospitalization, acute kidney injury (AKI) is still associated with risk.5
Nevertheless, clinicians remain frustrated with the practical management of patients with volume overload and worsening AKI. When faced with a rising serum creatinine level in a patient being treated for decompensated heart failure with signs or symptoms of volume overload, I suggest the following:
Perform careful bedside and chart review searching for evidence of AKI related to causes other than cardiorenal syndrome. Ask whether the rise in serum creatinine could be caused by new obstruction (eg, urinary retention, upper urinary tract obstruction), a nephrotoxin (eg, nonsteroidal anti-inflammatory drugs), a primary tubulointerstitial or glomerular process (eg, drug-induced acute interstitial nephritis, acute glomerulonephritis), acute tubular necrosis, or a new hemodynamic event threatening renal perfusion (eg, hypotension, a new arrhythmia). It is often best to arrive at a diagnosis of AKI due to cardiorenal dysfunction by exclusion, much like the working definitions of hepatorenal syndrome.6 This requires review of the urine sediment (looking for evidence of granular casts of acute tubular necrosis, or evidence of glomerulonephritis or interstitial nephritis), electronic medical record, vital signs, telemetry, and perhaps renal ultrasonography.
In the absence of frank evidence of “overdiuresis” such as worsening hypernatremia, with dropping blood pressure, clinical hypoperfusion, and contraction alkalosis, avoid the temptation to suspend diuretics. Alternatively, an increase in diuretic dose, or addition of a distal diuretic (ie, metolazone) may be needed to address persistent renal venous congestion as the cause of the AKI.3 In this situation, be sure to monitor electrolytes, volume status, and renal function closely while diuretic treatment is augmented. In many such cases, the serum creatinine may actually start to decrease after a more robust diuresis is generated. In these patients, it may also be prudent to temporarily suspend antagonists of the renin-angiotensin-aldosterone system, although this remains controversial.
Management of such patients should be done collaboratively with cardiologists well versed in the treatment of cardiorenal syndrome. It may be possible that the worsening renal function in these patients represents important changes in cardiac rhythm or function (eg, low cardiac output state, new or worsening valvular disease, ongoing myocardial ischemia, cardiac tamponade, uncontrolled bradycardia or tachyarrythmia). Interventions aimed at reversing such perturbations could be the most important steps in improving cardiorenal function and reversing AKI.
- Thind GS, Loehrke M, Wilt JL. Acute cardiorenal syndrome: mechanisms and clinical implications. Cleve Clin J Med 2018; 85(3):231–239. doi:10.3949/ccjm.85a.17019
- Grodin JL. Hemodynamically, the kidney is at the heart of cardiorenal syndrome. Cleve Clin J Med 2018; 85(3):240–242. doi:10.3949/ccjm.85a.17126
- Freda BJ, Slawsky M, Mallidi J, Braden GL. Decongestive treatment of acute decompensated heart failure: cardiorenal implications of ultrafiltration and diuretics. Am J Kid Dis 2011; 58(6):1005–1017. doi:10.1053/j.ajkd.2011.07.023
- Tang WH, Kitai T. Intrarenal blood flow: a window into the congestive kidney failure phenotype of heart failure? JACC Heart Fail 2016; 4(8):683–686. doi:10.1016/j.jchf.2016.05.009
- Freda BJ, Knee AB, Braden GL, Visintainer PF, Thakaer CV. Effect of transient and sustained acute kidney injury on readmissions in acute decompensated heart failure. Am J Cardiol 2017; 119(11):1809–1814. doi:10.1016/j.amjcard.2017.02.044
- Bucsics T, Krones E. Renal dysfunction in cirrhosis: acute kidney injury and the hepatorenal syndrome. Gastroenterol Rep (Oxf) 2017; 5(2):127–137. doi:10.1093/gastro/gox009
- Thind GS, Loehrke M, Wilt JL. Acute cardiorenal syndrome: mechanisms and clinical implications. Cleve Clin J Med 2018; 85(3):231–239. doi:10.3949/ccjm.85a.17019
- Grodin JL. Hemodynamically, the kidney is at the heart of cardiorenal syndrome. Cleve Clin J Med 2018; 85(3):240–242. doi:10.3949/ccjm.85a.17126
- Freda BJ, Slawsky M, Mallidi J, Braden GL. Decongestive treatment of acute decompensated heart failure: cardiorenal implications of ultrafiltration and diuretics. Am J Kid Dis 2011; 58(6):1005–1017. doi:10.1053/j.ajkd.2011.07.023
- Tang WH, Kitai T. Intrarenal blood flow: a window into the congestive kidney failure phenotype of heart failure? JACC Heart Fail 2016; 4(8):683–686. doi:10.1016/j.jchf.2016.05.009
- Freda BJ, Knee AB, Braden GL, Visintainer PF, Thakaer CV. Effect of transient and sustained acute kidney injury on readmissions in acute decompensated heart failure. Am J Cardiol 2017; 119(11):1809–1814. doi:10.1016/j.amjcard.2017.02.044
- Bucsics T, Krones E. Renal dysfunction in cirrhosis: acute kidney injury and the hepatorenal syndrome. Gastroenterol Rep (Oxf) 2017; 5(2):127–137. doi:10.1093/gastro/gox009

In reply: Cardiorenal syndrome
In Reply: We thank Dr. Freda for his remarks and observations. Certainly, the clinical importance of this entity and the challenge it poses to clinicians cannot be overemphasized. We concur with the overall message and reply to his specific comments:
We completely agree that clinical data-gathering is of paramount importance. This includes careful history-taking, physical examination, electronic medical record review, laboratory data review, and imaging. As discussed in our article, renal electrolytes will reveal a prerenal state in acute cardiorenal syndrome, and other causes of prerenal acute kidney injury (AKI) should be ruled out. The role of point-of-care ultrasonography (eg, to measure the size and respirophasic variation of the inferior vena cava) as a vital diagnostic tool has been well described, and we endorse it.1 Moreover, apart from snapshot values, trends are also very important. This is especially pertinent when the patient care is being transferred to a new service (eg, from hospitalist service to the critical care service). In this case, careful review of diuretic dosage, renal function trend, intake and output, and weight trend would help in the diagnosis.
Inadequate diuretic therapy is perhaps one of the most common errors made in the management of patients with acute cardiorenal syndrome. As mentioned in our article, diuretics should be correctly dosed based on the patient’s renal function. It is a common misconception that diuretics are nephrotoxic: in reality, there is no direct renal toxicity from the drug itself. Certainly, overdiuresis may lead to AKI, but this is not a valid concern in patients with acute cardiorenal syndrome, who are fluid-overloaded by definition.
Another challenging clinical scenario is when a patient is diagnosed with acute cardiorenal syndrome but renal function worsens with diuretic therapy. In our experience, this is a paradoxical situation and often stems from misinterpretation of clinical data. The most common example is diuretic underdosage leading to inadequate diuretic response. Renal function will continue to decline in these patients, as renal congestion has not yet been relieved. This reiterates the importance of paying close attention to urine output and intake-output data. When the diuretic regimen is strengthened and a robust diuretic response is achieved, renal function should improve as systemic congestion diminishes.
Acute cardiorenal syndrome stems from hemodynamic derangements, and a multidisciplinary approach may certainly lead to better outcomes. Although we described the general theme of hemodynamic disturbances, patients with acute cardiorenal syndrome may have certain unique and complex hemodynamic “phenotypes” that we did not discuss due to the limited scope of the paper. One such phenotype worth mentioning is decompensated right heart failure, as seen in patients with severe pulmonary hypertension. Acute cardiorenal syndrome due to renal congestion is often seen in these patients, but they also have certain other unique characteristics such as ventricular interdependence.2 Giving intravenous fluids to these patients not only will worsen renal function but can also cause catastrophic reduction in cardiac output and blood pressure due to worsening interventricular septal bowing. Certain treatments (eg, pulmonary vasodilators) are unique to this patient population, and these patients should hence be managed by experienced clinicians.
- Blehar DJ, Dickman E, Gaspari R. Identification of congestive heart failure via respiratory variation of inferior vena cava diameter. Am J Emerg Med 2009; 27(1):71–75. doi:10.1016/j.ajem.2008.01.002
- Piazza G, Goldhaber SZ. The acutely decompensated right ventricle: pathways for diagnosis and management. Chest 2005128(3):1836–1852. doi:10.1378/chest.128.3.1836
In Reply: We thank Dr. Freda for his remarks and observations. Certainly, the clinical importance of this entity and the challenge it poses to clinicians cannot be overemphasized. We concur with the overall message and reply to his specific comments:
We completely agree that clinical data-gathering is of paramount importance. This includes careful history-taking, physical examination, electronic medical record review, laboratory data review, and imaging. As discussed in our article, renal electrolytes will reveal a prerenal state in acute cardiorenal syndrome, and other causes of prerenal acute kidney injury (AKI) should be ruled out. The role of point-of-care ultrasonography (eg, to measure the size and respirophasic variation of the inferior vena cava) as a vital diagnostic tool has been well described, and we endorse it.1 Moreover, apart from snapshot values, trends are also very important. This is especially pertinent when the patient care is being transferred to a new service (eg, from hospitalist service to the critical care service). In this case, careful review of diuretic dosage, renal function trend, intake and output, and weight trend would help in the diagnosis.
Inadequate diuretic therapy is perhaps one of the most common errors made in the management of patients with acute cardiorenal syndrome. As mentioned in our article, diuretics should be correctly dosed based on the patient’s renal function. It is a common misconception that diuretics are nephrotoxic: in reality, there is no direct renal toxicity from the drug itself. Certainly, overdiuresis may lead to AKI, but this is not a valid concern in patients with acute cardiorenal syndrome, who are fluid-overloaded by definition.
Another challenging clinical scenario is when a patient is diagnosed with acute cardiorenal syndrome but renal function worsens with diuretic therapy. In our experience, this is a paradoxical situation and often stems from misinterpretation of clinical data. The most common example is diuretic underdosage leading to inadequate diuretic response. Renal function will continue to decline in these patients, as renal congestion has not yet been relieved. This reiterates the importance of paying close attention to urine output and intake-output data. When the diuretic regimen is strengthened and a robust diuretic response is achieved, renal function should improve as systemic congestion diminishes.
Acute cardiorenal syndrome stems from hemodynamic derangements, and a multidisciplinary approach may certainly lead to better outcomes. Although we described the general theme of hemodynamic disturbances, patients with acute cardiorenal syndrome may have certain unique and complex hemodynamic “phenotypes” that we did not discuss due to the limited scope of the paper. One such phenotype worth mentioning is decompensated right heart failure, as seen in patients with severe pulmonary hypertension. Acute cardiorenal syndrome due to renal congestion is often seen in these patients, but they also have certain other unique characteristics such as ventricular interdependence.2 Giving intravenous fluids to these patients not only will worsen renal function but can also cause catastrophic reduction in cardiac output and blood pressure due to worsening interventricular septal bowing. Certain treatments (eg, pulmonary vasodilators) are unique to this patient population, and these patients should hence be managed by experienced clinicians.
In Reply: We thank Dr. Freda for his remarks and observations. Certainly, the clinical importance of this entity and the challenge it poses to clinicians cannot be overemphasized. We concur with the overall message and reply to his specific comments:
We completely agree that clinical data-gathering is of paramount importance. This includes careful history-taking, physical examination, electronic medical record review, laboratory data review, and imaging. As discussed in our article, renal electrolytes will reveal a prerenal state in acute cardiorenal syndrome, and other causes of prerenal acute kidney injury (AKI) should be ruled out. The role of point-of-care ultrasonography (eg, to measure the size and respirophasic variation of the inferior vena cava) as a vital diagnostic tool has been well described, and we endorse it.1 Moreover, apart from snapshot values, trends are also very important. This is especially pertinent when the patient care is being transferred to a new service (eg, from hospitalist service to the critical care service). In this case, careful review of diuretic dosage, renal function trend, intake and output, and weight trend would help in the diagnosis.
Inadequate diuretic therapy is perhaps one of the most common errors made in the management of patients with acute cardiorenal syndrome. As mentioned in our article, diuretics should be correctly dosed based on the patient’s renal function. It is a common misconception that diuretics are nephrotoxic: in reality, there is no direct renal toxicity from the drug itself. Certainly, overdiuresis may lead to AKI, but this is not a valid concern in patients with acute cardiorenal syndrome, who are fluid-overloaded by definition.
Another challenging clinical scenario is when a patient is diagnosed with acute cardiorenal syndrome but renal function worsens with diuretic therapy. In our experience, this is a paradoxical situation and often stems from misinterpretation of clinical data. The most common example is diuretic underdosage leading to inadequate diuretic response. Renal function will continue to decline in these patients, as renal congestion has not yet been relieved. This reiterates the importance of paying close attention to urine output and intake-output data. When the diuretic regimen is strengthened and a robust diuretic response is achieved, renal function should improve as systemic congestion diminishes.
Acute cardiorenal syndrome stems from hemodynamic derangements, and a multidisciplinary approach may certainly lead to better outcomes. Although we described the general theme of hemodynamic disturbances, patients with acute cardiorenal syndrome may have certain unique and complex hemodynamic “phenotypes” that we did not discuss due to the limited scope of the paper. One such phenotype worth mentioning is decompensated right heart failure, as seen in patients with severe pulmonary hypertension. Acute cardiorenal syndrome due to renal congestion is often seen in these patients, but they also have certain other unique characteristics such as ventricular interdependence.2 Giving intravenous fluids to these patients not only will worsen renal function but can also cause catastrophic reduction in cardiac output and blood pressure due to worsening interventricular septal bowing. Certain treatments (eg, pulmonary vasodilators) are unique to this patient population, and these patients should hence be managed by experienced clinicians.
- Blehar DJ, Dickman E, Gaspari R. Identification of congestive heart failure via respiratory variation of inferior vena cava diameter. Am J Emerg Med 2009; 27(1):71–75. doi:10.1016/j.ajem.2008.01.002
- Piazza G, Goldhaber SZ. The acutely decompensated right ventricle: pathways for diagnosis and management. Chest 2005128(3):1836–1852. doi:10.1378/chest.128.3.1836
- Blehar DJ, Dickman E, Gaspari R. Identification of congestive heart failure via respiratory variation of inferior vena cava diameter. Am J Emerg Med 2009; 27(1):71–75. doi:10.1016/j.ajem.2008.01.002
- Piazza G, Goldhaber SZ. The acutely decompensated right ventricle: pathways for diagnosis and management. Chest 2005128(3):1836–1852. doi:10.1378/chest.128.3.1836

Rapid Development of Life-Threatening Emamectin Benzoate Poisoning
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).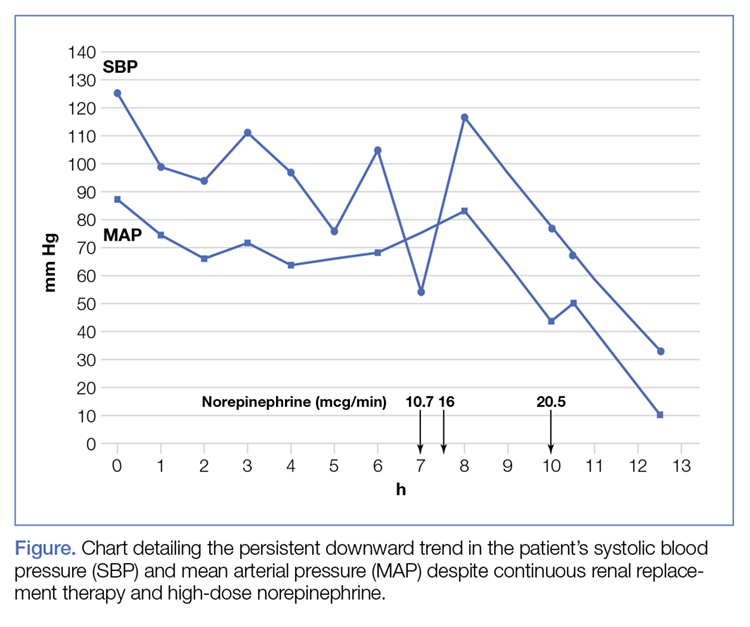
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
From Local Bar to Police Car
ANSWER
The radiograph demonstrates no acute injury to the wrist. There is, however, a subtle nondisplaced fracture of the distal fifth metacarpal joint. The patient was given a wrist splint for symptomatic relief, and orthopedic follow-up was coordinated.
ANSWER
The radiograph demonstrates no acute injury to the wrist. There is, however, a subtle nondisplaced fracture of the distal fifth metacarpal joint. The patient was given a wrist splint for symptomatic relief, and orthopedic follow-up was coordinated.
ANSWER
The radiograph demonstrates no acute injury to the wrist. There is, however, a subtle nondisplaced fracture of the distal fifth metacarpal joint. The patient was given a wrist splint for symptomatic relief, and orthopedic follow-up was coordinated.
A 31-year-old man is brought to your facility by local law enforcement for evaluation of left wrist pain following an altercation. The patient was reportedly at a local bar; as he was leaving, he started arguing with some other patrons. A fight ensued.
The patient believes he was struck by something on his left wrist. He denies any other injuries or complaints. His medical history is unremarkable, and vital signs are stable.
Physical examination of his left wrist shows no obvious deformity. There is some mild swelling over the dorsal aspect of his wrist. Range of motion is painful and limited. Good capillary refill is noted in the fingers, and sensation is intact. Good pulses are present.
Radiograph of the left wrist is obtained. What is your impression?
Are serum troponin levels elevated in conditions other than acute coronary syndrome?
Yes. Sepsis, stroke, chronic kidney disease, pulmonary disease, chemotherapy, heart failure, and stress cardiomyopathy can all raise serum troponin concentrations, and in some cases the elevations are prognostically important. Careful clinical assessment, serial monitoring of troponin levels, and other supportive tests are usually necessary to tell whether troponin elevations are due to acute coronary syndrome or to these other causes.
NOT ONLY A MARKER OF MYOCARDIAL DAMAGE
Troponin, an intracellular protein found in skeletal and cardiac muscle cells, is essential for muscle contraction. Troponin T and troponin I are clinically equivalent, and both are biomarkers of myocardial damage.
A troponin assay is ordered when patients present with sudden onset of symptoms of acute coronary syndrome such as chest pain, dyspnea, diaphoresis, and electrocardiographic abnormalities. The assay is positive when the manufacturer-specified threshold corresponding to a concentration above the 99th percentile is detected.
Serial testing of serum biomarkers of acute myocardial damage is essential to confirm the diagnosis of myocardial infarction. Because of their higher sensitivity and specificity compared with creatine kinase-MB and other markers, troponins are the preferred biomarker in diagnosing acute coronary syndrome.
In 1984, Piper et al1 reported that free cytosolic pools of cardiac enzymes could be released after reversible myocardial injury as a result of temporary disruption of the cell membrane. This upended the previous assumption that troponin was released only after irreversible myocardial necrosis, and it provided an explanation for troponin elevations observed in conditions with no evidence of epicardial coronary artery disease or permanent myocardial damage.1
SEPSIS
Studies of patients with sepsis, severe sepsis, and septic shock have shown troponin elevations with no evidence of acute coronary syndrome.2 In sepsis, troponin elevations are presumed to be caused by a combination of events. Renal dysfunction leads to decreased clearance of troponin fragments by the kidneys. The massive inflammatory response in septic shock results in cytokine-induced cardiac damage, and increased levels of endogenous and exogenous catecholamines damage cardiac myocytes.3
Studies of the prognostic value of these elevations have produced mixed and contradictory results. But a 2013 meta-analysis4 showed that patients with a troponin elevation at the time of diagnosis of sepsis had a risk of death almost twice that of patients without a troponin elevation (relative risk 1.91, 95% confidence interval [CI] 1.63–2.24).
STROKE
Acute ischemic stroke can trigger troponin elevations in several ways. Since the risk factors for acute ischemic stroke and coronary stenosis are similar, patients who have an ischemic stroke have a higher risk of coronary atherosclerosis and coronary stenosis than the general population.5
Stroke can cause a variety of cardiovascular and respiratory responses (eg, tachyarrhythmia, hypertensive crisis, respiratory failure) that increase the stress on the myocardium. In patients with stroke and concurrent coronary artery stenosis, the increased metabolic demand can exceed the oxygen supply capacity, resulting in myocardial ischemia, which can manifest as increased levels of serum troponin.5
Stroke can also cause troponin elevation through neurogenic myocardial damage. Ischemic stroke and intracranial hemorrhage can trigger alterations in autonomic control. Sometimes this results in increased sympathetic activity with concomitant catecholamine surge, leading to contraction band necrosis and other forms of myocardial damage and, as a result, troponin elevation.5,6 This may explain troponin elevation in patients with acute ischemic stroke in the absence of concomitant coronary artery disease. Recent evidence suggests that patients with acute ischemic stroke and elevated troponin had significantly less angiographic evidence of coronary artery disease than matched patients with non-ST-elevation myocardial infarction.7
CHRONIC KIDNEY DISEASE
Cardiac troponins may be elevated in chronic kidney disease. Explanations for this include the theory that troponin is broken down into fragments that are cleared by the kidney.8 Therefore, decreased renal function leads to an increase in troponin fragments measured with troponin assays. Other explanations are chronic volume overload, chronic elevation of proinflammatory cytokines, and associated comorbidities such as hypertension.
Troponin elevations can have prognostic significance in chronic kidney disease. In a meta-analysis of 98 studies of patients with chronic kidney disease and no symptoms of acute coronary syndrome, troponin elevation was associated with 2- to 4-fold higher rates of all-cause mortality, cardiovascular mortality, and major acute coronary events in both dialysis-dependent and nondialysis patients.8 Thus, troponin is a unique factor in risk-stratification in patients with chronic kidney disease and could affect how it is managed in the future.
To determine if an acute coronary syndrome is taking place when evaluating patients with chronic kidney disease and elevated troponins, physicians must use other evidence—for example, serial measurements of troponin levels showing continued troponin elevation, elevations in troponin from the patient’s baseline, elevated creatine kinase-MB levels, electrocardiographic changes, and clinical symptoms.
PULMONARY DISEASE
Troponin elevation can signify right heart strain in a variety of pulmonary diseases.
Pulmonary embolism. Troponin elevation is a marker of right ventricular dysfunction in patients with moderate to large pulmonary embolism.
In a study of normotensive patients with acute pulmonary embolism, 52% had elevated serum troponin, and they had a higher risk of an adverse outcome (death, recurrent pulmonary embolism, or major bleeding) within 30 days (odds ratio 4.97, 95% CI 1.71–14.43) and a lower probability of 6-month survival.9 Troponin elevation in pulmonary embolism is not helpful in confirming the diagnosis but is primarily useful in prognosis.
Pulmonary arterial hypertension. Cardiac troponin elevations can also indicate severe disease and poor outcomes in patients with pulmonary arterial hypertension. A study by Heresi et al10 confirmed this, even in patients with only slight elevations in troponin levels. Troponin was detected in 17 (25%) of 68 patients with pulmonary arterial hypertension diagnostic category 1. Further, patients with detectable troponin had more advanced functional class symptoms, a shorter 6-minute walk distance, more pericardial effusions, larger right atrial area, and higher B-type natriuretic peptide and C-reactive protein levels.10
Measuring troponins in the setting of pulmonary hypertension allows clinicians to identify high-risk patients and may help guide the management of these patients.
Chronic obstructive pulmonary disease. Elevation of serum troponins is also reported in patients with acute exacerbation of chronic obstructive pulmonary disease and has been correlated with increased all-cause mortality rates in these patients.11
CHEMOTHERAPY
Chemotherapy-induced cardiotoxicity may result in troponin elevations. Chemotherapy causes cardiac toxicity by several mechanisms, including production of oxygen free radicals, disturbance of mitochondrial energy metabolism, intracellular calcium overload, and increased lipid peroxidation. Chemotherapeutic agents associated with cardiotoxicity include anthracyclines, trastuzumab, chlormethine, and mitomycin.
Chemotherapy-induced left ventricular deterioration is often irreversible. Monitoring troponin levels can help identify problems before cardiac dysfunction becomes clinically evident during the weeks and months after the start of high-dose chemotherapy.
Cardinale et al12 found marked myocardial depression 7 months after the start of high-dose chemotherapy. They reported a close relationship between short-term troponin elevation and the greatest reduction in left-ventricular ejection fraction (r = −0.87; P < .0001). Normal troponin values after high-dose chemotherapy also seemed to identify patients at lower risk, with either no cardiac damage or only transient subclinical left-ventricular dysfunction.12
HEART FAILURE
Heart failure leads to release of cardiac troponins through myocardial strain and myocardial death. Volume and pressure overload of the ventricles causes excessive wall tension, resulting in myofibrillar damage. Measuring troponins is an effective way to detect cardiac myolysis in heart failure, independent of the presence of coronary artery disease.
In heart failure, elevated troponins correlate with adverse outcome in both hospitalized and stable patients. In addition, elevation of both troponins and B-type natriuretic peptide is associated with worse heart failure outcomes than elevation of either marker alone.
A prospective study13 of patients with New York Heart Association class III or IV heart failure showed that an increase in troponin concentration from normal baseline was associated with a risk of death, cardiac transplant, or hospitalization that was 3.4 to 5.09 times higher. Further elevations in B-type natriuretic peptide during the study period were associated with a poor outcome (hazard ratio 5.09; P < .001). Combined elevations of troponin and B-type natriuretic peptide defined the group at highest risk (hazard ratio 8.58; P < .001).
Increased myocardial wall stress may lead to decreased subendocardial perfusion, with resulting troponin elevation and decline in left ventricular systolic function. Further, in vitro experiments with myocytes established a link between myocardial wall stretch and programmed cell death, which may contribute to troponin elevations.14
STRESS CARDIOMYOPATHY
Profound reversible myocardial depression and troponin elevation are seen after sudden emotional stress, a condition called stress-induced or takotsubo cardiomyopathy. While the exact mechanism of stress-induced cardiomyopathy remains unclear, it is thought to be due to sudden supraphysiologic elevation of catecholamines and related neuropeptides. Although vasospasm in the epicardial and microvascular circulation has been suggested as the possible mechanism of left ventricular systolic dysfunction and troponin elevation, cardiac myocyte injury from catecholamine- induced cyclic AMP-mediated calcium overload and oxygen-derived free radicals appears to be a more likely mechanism.15
PSEUDOELEVATIONS OF TROPONIN
In rare cases, endogenous antibodies (eg, heterophilic antibodies) in the blood specimen can interfere with the processing of the troponin immunoassay in the laboratory, causing a false-positive assay. This can occur with samples from patients with a viral infection or autoimmune condition as well as with samples from patients treated with intravenous immunoglobulin (Ig). Heterophilic antibodies can bind to the Fc region of the test antibodies in certain troponin assays, leading to false-positive elevations.16 Macrotroponin, a molecule found in patients with autoantibodies against troponin I, is composed of troponin I fragments and IgG antibodies and can also cause a false-positive troponin immunoassay.16
In patients with seropositive rheumatoid arthritis, a false-positive troponin I assay was associated with a high concentration of IgM rheumatoid factor with the use of certain immunoassay techniques.17 In patients with acute skeletal muscle injury, the first-generation troponin T assay was found to be falsely positive due to the nonspecific binding of skeletal muscle troponin T to the walls of the test tube used for the assay. When the second-generation troponin T assay was used, troponin T levels were found to be slightly more positive than troponin I levels (1.7 vs 1.5 times the upper limit of normal), especially in patients with renal failure.18
Troponin may also be falsely elevated in hemolyzed blood samples. This has to be taken into consideration in interpreting the results of troponin testing in severely hemolyzed blood samples. However, Puelacher et al19 suggested that the presence of hemolysis did not appear to interfere with clinical value of the test.
- Piper HM, Schwartz P, Spahr R, Hütter J, Spieckermann P. Early enzyme release from myocardial cells is not due to irreversible cell damage. J Mol Cell Cardiol 1984; 16(4):385–388. doi:10.1016/S0022-2828(84)80609-4
- Ammann P, Fehr T, Minder EI, Günter C, Bertel O. Elevation of troponin I in sepsis and septic shock. Intensive Care Med 2001; 27(6):965–969.
- Landesberg G, Jaffe AS, Gilon D, et al. Troponin elevation in severe sepsis and septic shock. Crit Care Med 2014; 42(4):790–800. doi:10.1097/CCM.0000000000000107
- Bessière F, Khenifer S, Dubourg J, Durieu I, Lega JC. Prognostic value of troponins in sepsis: a meta-analysis. Intensive Care Med 2013; 39(7):1181–1189. doi:10.1007/s00134-013-2902-3
- Scheitz JF, Nolte CH, Laufs U, Endres M. Application and interpretation of high-sensitivity cardiac troponin assays in patients with acute ischemic stroke. Stroke 2015; 46(4):1132–1140. doi:10.1161/STROKEAHA.114.007858
- Naidech AM, Kreiter KT, Janjua N, et al. Cardiac troponin elevation, cardiovascular morbidity, and outcome after subarachnoid hemorrhage. Circulation 2005; 112(18):2851–2656. doi:10.1161/CIRCULATIONAHA.105.533620
- Mochmann HC, Scheitz JF, Petzold GC, et al; TRELAS Study Group. Coronary angiographic findings in acute ischemic stroke patients with elevated cardiac troponin: the Troponin Elevation in Acute Ischemic Stroke (TRELAS) Study. Circulation 2016; 133(13):1264–1271. doi:10.1161/CIRCULATIONAHA.115.018547
- Michos ED, Wilson LM, Yeh HC, et al. Prognostic value of cardiac troponin in patients with chronic kidney disease without suspected acute coronary syndrome. Ann Intern Med 2014; 161(7):491–501. doi:10.7326/M14-0743
- Lankeit M, Jiménez D, Kostrubiec M, et al. Predictive value of the high-sensitivity troponin T assay and the simplified pulmonary embolism severity index in hemodynamically stable patients with acute pulmonary embolism: a prospective validation study. Circulation 2011; 124(24):2716–2724. doi:10.1161/CIRCULATIONAHA.111.051177
- Heresi GA, Tang WH, Aytekin M, Hammel J, Hazen SL, Dweik RA. Sensitive cardiac troponin I predicts poor outcomes in pulmonary arterial hypertension. Eur Respir J 2012; 39(4)939–944. doi:10.1183/09031936.00067011
- Pavasini R, d’Ascenzo F, Campo G, et al. Cardiac troponin elevation predicts all-cause mortality in patients with acute exacerbation of chronic obstructive pulmonary disease: systematic review and meta-analysis. Int J Cardiol 2015; 191:187–193. doi:10.1016/j.ijcard.2015.05.006
- Cardinale D, Sandri MT, Martinoni A, et al. Left ventricular dysfunction predicted by early troponin I release after high-dose chemotherapy. J Am Coll Cardiol 2000; 36(2):517–522.
- Miller WL, Hartman KA, Burritt MF, et al. Serial biomarker measurements in ambulatory patients with chronic heart failure: the importance of change over time. Circulation 2007; 116(3):249–257. doi:10.1161/CIRCULATIONAHA.107.694562
- Logeart D, Beyne P, Cusson C, et al. Evidence of cardiac myolysis in severe nonischemic heart failure and the potential role of increased wall strain. Am Heart J 2001; 141(2):247–253. doi:10.1067/mhj.2001.111767
- Whittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352(6):539–548. doi:10.1056/NEJMoa043046
- McClennen S, Halamka JD, Horowitz GL, Kannam JP, Ho KK. Clinical prevalence and ramifications of false-positive cardiac troponin I elevations from the Abbott AxSYM Analyzer. Am J Cardiol 2003; 91(9):1125–1127.
- Bradham WS, Bian A, Oeser A, et al. High-sensitivity cardiac troponin-I is elevated in patients with rheumatoid arthritis, independent of cardiovascular risk factors and inflammation. PLoS One 2012; 7(6):e38930. doi:10.1371/journal.pone.0038930
- Li SF, Zapata J, Tillem E. The prevalence of false-positive cardiac troponin I in ED patients with rhabdomyolysis. Am J Emerg Med 2005; 23(7):860–863. doi:10.1016/j.ajem.2005.05.008
- Puelacher C, Twerenbold R, Mosimann T, et al. Effects of hemolysis on the diagnostic accuracy of cardiac troponin I for the diagnosis of myocardial infarction. Int J Cardiol 2015; 187:313–315. doi:10.1016/j.ijcard.2015.03.378
Yes. Sepsis, stroke, chronic kidney disease, pulmonary disease, chemotherapy, heart failure, and stress cardiomyopathy can all raise serum troponin concentrations, and in some cases the elevations are prognostically important. Careful clinical assessment, serial monitoring of troponin levels, and other supportive tests are usually necessary to tell whether troponin elevations are due to acute coronary syndrome or to these other causes.
NOT ONLY A MARKER OF MYOCARDIAL DAMAGE
Troponin, an intracellular protein found in skeletal and cardiac muscle cells, is essential for muscle contraction. Troponin T and troponin I are clinically equivalent, and both are biomarkers of myocardial damage.
A troponin assay is ordered when patients present with sudden onset of symptoms of acute coronary syndrome such as chest pain, dyspnea, diaphoresis, and electrocardiographic abnormalities. The assay is positive when the manufacturer-specified threshold corresponding to a concentration above the 99th percentile is detected.
Serial testing of serum biomarkers of acute myocardial damage is essential to confirm the diagnosis of myocardial infarction. Because of their higher sensitivity and specificity compared with creatine kinase-MB and other markers, troponins are the preferred biomarker in diagnosing acute coronary syndrome.
In 1984, Piper et al1 reported that free cytosolic pools of cardiac enzymes could be released after reversible myocardial injury as a result of temporary disruption of the cell membrane. This upended the previous assumption that troponin was released only after irreversible myocardial necrosis, and it provided an explanation for troponin elevations observed in conditions with no evidence of epicardial coronary artery disease or permanent myocardial damage.1
SEPSIS
Studies of patients with sepsis, severe sepsis, and septic shock have shown troponin elevations with no evidence of acute coronary syndrome.2 In sepsis, troponin elevations are presumed to be caused by a combination of events. Renal dysfunction leads to decreased clearance of troponin fragments by the kidneys. The massive inflammatory response in septic shock results in cytokine-induced cardiac damage, and increased levels of endogenous and exogenous catecholamines damage cardiac myocytes.3
Studies of the prognostic value of these elevations have produced mixed and contradictory results. But a 2013 meta-analysis4 showed that patients with a troponin elevation at the time of diagnosis of sepsis had a risk of death almost twice that of patients without a troponin elevation (relative risk 1.91, 95% confidence interval [CI] 1.63–2.24).
STROKE
Acute ischemic stroke can trigger troponin elevations in several ways. Since the risk factors for acute ischemic stroke and coronary stenosis are similar, patients who have an ischemic stroke have a higher risk of coronary atherosclerosis and coronary stenosis than the general population.5
Stroke can cause a variety of cardiovascular and respiratory responses (eg, tachyarrhythmia, hypertensive crisis, respiratory failure) that increase the stress on the myocardium. In patients with stroke and concurrent coronary artery stenosis, the increased metabolic demand can exceed the oxygen supply capacity, resulting in myocardial ischemia, which can manifest as increased levels of serum troponin.5
Stroke can also cause troponin elevation through neurogenic myocardial damage. Ischemic stroke and intracranial hemorrhage can trigger alterations in autonomic control. Sometimes this results in increased sympathetic activity with concomitant catecholamine surge, leading to contraction band necrosis and other forms of myocardial damage and, as a result, troponin elevation.5,6 This may explain troponin elevation in patients with acute ischemic stroke in the absence of concomitant coronary artery disease. Recent evidence suggests that patients with acute ischemic stroke and elevated troponin had significantly less angiographic evidence of coronary artery disease than matched patients with non-ST-elevation myocardial infarction.7
CHRONIC KIDNEY DISEASE
Cardiac troponins may be elevated in chronic kidney disease. Explanations for this include the theory that troponin is broken down into fragments that are cleared by the kidney.8 Therefore, decreased renal function leads to an increase in troponin fragments measured with troponin assays. Other explanations are chronic volume overload, chronic elevation of proinflammatory cytokines, and associated comorbidities such as hypertension.
Troponin elevations can have prognostic significance in chronic kidney disease. In a meta-analysis of 98 studies of patients with chronic kidney disease and no symptoms of acute coronary syndrome, troponin elevation was associated with 2- to 4-fold higher rates of all-cause mortality, cardiovascular mortality, and major acute coronary events in both dialysis-dependent and nondialysis patients.8 Thus, troponin is a unique factor in risk-stratification in patients with chronic kidney disease and could affect how it is managed in the future.
To determine if an acute coronary syndrome is taking place when evaluating patients with chronic kidney disease and elevated troponins, physicians must use other evidence—for example, serial measurements of troponin levels showing continued troponin elevation, elevations in troponin from the patient’s baseline, elevated creatine kinase-MB levels, electrocardiographic changes, and clinical symptoms.
PULMONARY DISEASE
Troponin elevation can signify right heart strain in a variety of pulmonary diseases.
Pulmonary embolism. Troponin elevation is a marker of right ventricular dysfunction in patients with moderate to large pulmonary embolism.
In a study of normotensive patients with acute pulmonary embolism, 52% had elevated serum troponin, and they had a higher risk of an adverse outcome (death, recurrent pulmonary embolism, or major bleeding) within 30 days (odds ratio 4.97, 95% CI 1.71–14.43) and a lower probability of 6-month survival.9 Troponin elevation in pulmonary embolism is not helpful in confirming the diagnosis but is primarily useful in prognosis.
Pulmonary arterial hypertension. Cardiac troponin elevations can also indicate severe disease and poor outcomes in patients with pulmonary arterial hypertension. A study by Heresi et al10 confirmed this, even in patients with only slight elevations in troponin levels. Troponin was detected in 17 (25%) of 68 patients with pulmonary arterial hypertension diagnostic category 1. Further, patients with detectable troponin had more advanced functional class symptoms, a shorter 6-minute walk distance, more pericardial effusions, larger right atrial area, and higher B-type natriuretic peptide and C-reactive protein levels.10
Measuring troponins in the setting of pulmonary hypertension allows clinicians to identify high-risk patients and may help guide the management of these patients.
Chronic obstructive pulmonary disease. Elevation of serum troponins is also reported in patients with acute exacerbation of chronic obstructive pulmonary disease and has been correlated with increased all-cause mortality rates in these patients.11
CHEMOTHERAPY
Chemotherapy-induced cardiotoxicity may result in troponin elevations. Chemotherapy causes cardiac toxicity by several mechanisms, including production of oxygen free radicals, disturbance of mitochondrial energy metabolism, intracellular calcium overload, and increased lipid peroxidation. Chemotherapeutic agents associated with cardiotoxicity include anthracyclines, trastuzumab, chlormethine, and mitomycin.
Chemotherapy-induced left ventricular deterioration is often irreversible. Monitoring troponin levels can help identify problems before cardiac dysfunction becomes clinically evident during the weeks and months after the start of high-dose chemotherapy.
Cardinale et al12 found marked myocardial depression 7 months after the start of high-dose chemotherapy. They reported a close relationship between short-term troponin elevation and the greatest reduction in left-ventricular ejection fraction (r = −0.87; P < .0001). Normal troponin values after high-dose chemotherapy also seemed to identify patients at lower risk, with either no cardiac damage or only transient subclinical left-ventricular dysfunction.12
HEART FAILURE
Heart failure leads to release of cardiac troponins through myocardial strain and myocardial death. Volume and pressure overload of the ventricles causes excessive wall tension, resulting in myofibrillar damage. Measuring troponins is an effective way to detect cardiac myolysis in heart failure, independent of the presence of coronary artery disease.
In heart failure, elevated troponins correlate with adverse outcome in both hospitalized and stable patients. In addition, elevation of both troponins and B-type natriuretic peptide is associated with worse heart failure outcomes than elevation of either marker alone.
A prospective study13 of patients with New York Heart Association class III or IV heart failure showed that an increase in troponin concentration from normal baseline was associated with a risk of death, cardiac transplant, or hospitalization that was 3.4 to 5.09 times higher. Further elevations in B-type natriuretic peptide during the study period were associated with a poor outcome (hazard ratio 5.09; P < .001). Combined elevations of troponin and B-type natriuretic peptide defined the group at highest risk (hazard ratio 8.58; P < .001).
Increased myocardial wall stress may lead to decreased subendocardial perfusion, with resulting troponin elevation and decline in left ventricular systolic function. Further, in vitro experiments with myocytes established a link between myocardial wall stretch and programmed cell death, which may contribute to troponin elevations.14
STRESS CARDIOMYOPATHY
Profound reversible myocardial depression and troponin elevation are seen after sudden emotional stress, a condition called stress-induced or takotsubo cardiomyopathy. While the exact mechanism of stress-induced cardiomyopathy remains unclear, it is thought to be due to sudden supraphysiologic elevation of catecholamines and related neuropeptides. Although vasospasm in the epicardial and microvascular circulation has been suggested as the possible mechanism of left ventricular systolic dysfunction and troponin elevation, cardiac myocyte injury from catecholamine- induced cyclic AMP-mediated calcium overload and oxygen-derived free radicals appears to be a more likely mechanism.15
PSEUDOELEVATIONS OF TROPONIN
In rare cases, endogenous antibodies (eg, heterophilic antibodies) in the blood specimen can interfere with the processing of the troponin immunoassay in the laboratory, causing a false-positive assay. This can occur with samples from patients with a viral infection or autoimmune condition as well as with samples from patients treated with intravenous immunoglobulin (Ig). Heterophilic antibodies can bind to the Fc region of the test antibodies in certain troponin assays, leading to false-positive elevations.16 Macrotroponin, a molecule found in patients with autoantibodies against troponin I, is composed of troponin I fragments and IgG antibodies and can also cause a false-positive troponin immunoassay.16
In patients with seropositive rheumatoid arthritis, a false-positive troponin I assay was associated with a high concentration of IgM rheumatoid factor with the use of certain immunoassay techniques.17 In patients with acute skeletal muscle injury, the first-generation troponin T assay was found to be falsely positive due to the nonspecific binding of skeletal muscle troponin T to the walls of the test tube used for the assay. When the second-generation troponin T assay was used, troponin T levels were found to be slightly more positive than troponin I levels (1.7 vs 1.5 times the upper limit of normal), especially in patients with renal failure.18
Troponin may also be falsely elevated in hemolyzed blood samples. This has to be taken into consideration in interpreting the results of troponin testing in severely hemolyzed blood samples. However, Puelacher et al19 suggested that the presence of hemolysis did not appear to interfere with clinical value of the test.
Yes. Sepsis, stroke, chronic kidney disease, pulmonary disease, chemotherapy, heart failure, and stress cardiomyopathy can all raise serum troponin concentrations, and in some cases the elevations are prognostically important. Careful clinical assessment, serial monitoring of troponin levels, and other supportive tests are usually necessary to tell whether troponin elevations are due to acute coronary syndrome or to these other causes.
NOT ONLY A MARKER OF MYOCARDIAL DAMAGE
Troponin, an intracellular protein found in skeletal and cardiac muscle cells, is essential for muscle contraction. Troponin T and troponin I are clinically equivalent, and both are biomarkers of myocardial damage.
A troponin assay is ordered when patients present with sudden onset of symptoms of acute coronary syndrome such as chest pain, dyspnea, diaphoresis, and electrocardiographic abnormalities. The assay is positive when the manufacturer-specified threshold corresponding to a concentration above the 99th percentile is detected.
Serial testing of serum biomarkers of acute myocardial damage is essential to confirm the diagnosis of myocardial infarction. Because of their higher sensitivity and specificity compared with creatine kinase-MB and other markers, troponins are the preferred biomarker in diagnosing acute coronary syndrome.
In 1984, Piper et al1 reported that free cytosolic pools of cardiac enzymes could be released after reversible myocardial injury as a result of temporary disruption of the cell membrane. This upended the previous assumption that troponin was released only after irreversible myocardial necrosis, and it provided an explanation for troponin elevations observed in conditions with no evidence of epicardial coronary artery disease or permanent myocardial damage.1
SEPSIS
Studies of patients with sepsis, severe sepsis, and septic shock have shown troponin elevations with no evidence of acute coronary syndrome.2 In sepsis, troponin elevations are presumed to be caused by a combination of events. Renal dysfunction leads to decreased clearance of troponin fragments by the kidneys. The massive inflammatory response in septic shock results in cytokine-induced cardiac damage, and increased levels of endogenous and exogenous catecholamines damage cardiac myocytes.3
Studies of the prognostic value of these elevations have produced mixed and contradictory results. But a 2013 meta-analysis4 showed that patients with a troponin elevation at the time of diagnosis of sepsis had a risk of death almost twice that of patients without a troponin elevation (relative risk 1.91, 95% confidence interval [CI] 1.63–2.24).
STROKE
Acute ischemic stroke can trigger troponin elevations in several ways. Since the risk factors for acute ischemic stroke and coronary stenosis are similar, patients who have an ischemic stroke have a higher risk of coronary atherosclerosis and coronary stenosis than the general population.5
Stroke can cause a variety of cardiovascular and respiratory responses (eg, tachyarrhythmia, hypertensive crisis, respiratory failure) that increase the stress on the myocardium. In patients with stroke and concurrent coronary artery stenosis, the increased metabolic demand can exceed the oxygen supply capacity, resulting in myocardial ischemia, which can manifest as increased levels of serum troponin.5
Stroke can also cause troponin elevation through neurogenic myocardial damage. Ischemic stroke and intracranial hemorrhage can trigger alterations in autonomic control. Sometimes this results in increased sympathetic activity with concomitant catecholamine surge, leading to contraction band necrosis and other forms of myocardial damage and, as a result, troponin elevation.5,6 This may explain troponin elevation in patients with acute ischemic stroke in the absence of concomitant coronary artery disease. Recent evidence suggests that patients with acute ischemic stroke and elevated troponin had significantly less angiographic evidence of coronary artery disease than matched patients with non-ST-elevation myocardial infarction.7
CHRONIC KIDNEY DISEASE
Cardiac troponins may be elevated in chronic kidney disease. Explanations for this include the theory that troponin is broken down into fragments that are cleared by the kidney.8 Therefore, decreased renal function leads to an increase in troponin fragments measured with troponin assays. Other explanations are chronic volume overload, chronic elevation of proinflammatory cytokines, and associated comorbidities such as hypertension.
Troponin elevations can have prognostic significance in chronic kidney disease. In a meta-analysis of 98 studies of patients with chronic kidney disease and no symptoms of acute coronary syndrome, troponin elevation was associated with 2- to 4-fold higher rates of all-cause mortality, cardiovascular mortality, and major acute coronary events in both dialysis-dependent and nondialysis patients.8 Thus, troponin is a unique factor in risk-stratification in patients with chronic kidney disease and could affect how it is managed in the future.
To determine if an acute coronary syndrome is taking place when evaluating patients with chronic kidney disease and elevated troponins, physicians must use other evidence—for example, serial measurements of troponin levels showing continued troponin elevation, elevations in troponin from the patient’s baseline, elevated creatine kinase-MB levels, electrocardiographic changes, and clinical symptoms.
PULMONARY DISEASE
Troponin elevation can signify right heart strain in a variety of pulmonary diseases.
Pulmonary embolism. Troponin elevation is a marker of right ventricular dysfunction in patients with moderate to large pulmonary embolism.
In a study of normotensive patients with acute pulmonary embolism, 52% had elevated serum troponin, and they had a higher risk of an adverse outcome (death, recurrent pulmonary embolism, or major bleeding) within 30 days (odds ratio 4.97, 95% CI 1.71–14.43) and a lower probability of 6-month survival.9 Troponin elevation in pulmonary embolism is not helpful in confirming the diagnosis but is primarily useful in prognosis.
Pulmonary arterial hypertension. Cardiac troponin elevations can also indicate severe disease and poor outcomes in patients with pulmonary arterial hypertension. A study by Heresi et al10 confirmed this, even in patients with only slight elevations in troponin levels. Troponin was detected in 17 (25%) of 68 patients with pulmonary arterial hypertension diagnostic category 1. Further, patients with detectable troponin had more advanced functional class symptoms, a shorter 6-minute walk distance, more pericardial effusions, larger right atrial area, and higher B-type natriuretic peptide and C-reactive protein levels.10
Measuring troponins in the setting of pulmonary hypertension allows clinicians to identify high-risk patients and may help guide the management of these patients.
Chronic obstructive pulmonary disease. Elevation of serum troponins is also reported in patients with acute exacerbation of chronic obstructive pulmonary disease and has been correlated with increased all-cause mortality rates in these patients.11
CHEMOTHERAPY
Chemotherapy-induced cardiotoxicity may result in troponin elevations. Chemotherapy causes cardiac toxicity by several mechanisms, including production of oxygen free radicals, disturbance of mitochondrial energy metabolism, intracellular calcium overload, and increased lipid peroxidation. Chemotherapeutic agents associated with cardiotoxicity include anthracyclines, trastuzumab, chlormethine, and mitomycin.
Chemotherapy-induced left ventricular deterioration is often irreversible. Monitoring troponin levels can help identify problems before cardiac dysfunction becomes clinically evident during the weeks and months after the start of high-dose chemotherapy.
Cardinale et al12 found marked myocardial depression 7 months after the start of high-dose chemotherapy. They reported a close relationship between short-term troponin elevation and the greatest reduction in left-ventricular ejection fraction (r = −0.87; P < .0001). Normal troponin values after high-dose chemotherapy also seemed to identify patients at lower risk, with either no cardiac damage or only transient subclinical left-ventricular dysfunction.12
HEART FAILURE
Heart failure leads to release of cardiac troponins through myocardial strain and myocardial death. Volume and pressure overload of the ventricles causes excessive wall tension, resulting in myofibrillar damage. Measuring troponins is an effective way to detect cardiac myolysis in heart failure, independent of the presence of coronary artery disease.
In heart failure, elevated troponins correlate with adverse outcome in both hospitalized and stable patients. In addition, elevation of both troponins and B-type natriuretic peptide is associated with worse heart failure outcomes than elevation of either marker alone.
A prospective study13 of patients with New York Heart Association class III or IV heart failure showed that an increase in troponin concentration from normal baseline was associated with a risk of death, cardiac transplant, or hospitalization that was 3.4 to 5.09 times higher. Further elevations in B-type natriuretic peptide during the study period were associated with a poor outcome (hazard ratio 5.09; P < .001). Combined elevations of troponin and B-type natriuretic peptide defined the group at highest risk (hazard ratio 8.58; P < .001).
Increased myocardial wall stress may lead to decreased subendocardial perfusion, with resulting troponin elevation and decline in left ventricular systolic function. Further, in vitro experiments with myocytes established a link between myocardial wall stretch and programmed cell death, which may contribute to troponin elevations.14
STRESS CARDIOMYOPATHY
Profound reversible myocardial depression and troponin elevation are seen after sudden emotional stress, a condition called stress-induced or takotsubo cardiomyopathy. While the exact mechanism of stress-induced cardiomyopathy remains unclear, it is thought to be due to sudden supraphysiologic elevation of catecholamines and related neuropeptides. Although vasospasm in the epicardial and microvascular circulation has been suggested as the possible mechanism of left ventricular systolic dysfunction and troponin elevation, cardiac myocyte injury from catecholamine- induced cyclic AMP-mediated calcium overload and oxygen-derived free radicals appears to be a more likely mechanism.15
PSEUDOELEVATIONS OF TROPONIN
In rare cases, endogenous antibodies (eg, heterophilic antibodies) in the blood specimen can interfere with the processing of the troponin immunoassay in the laboratory, causing a false-positive assay. This can occur with samples from patients with a viral infection or autoimmune condition as well as with samples from patients treated with intravenous immunoglobulin (Ig). Heterophilic antibodies can bind to the Fc region of the test antibodies in certain troponin assays, leading to false-positive elevations.16 Macrotroponin, a molecule found in patients with autoantibodies against troponin I, is composed of troponin I fragments and IgG antibodies and can also cause a false-positive troponin immunoassay.16
In patients with seropositive rheumatoid arthritis, a false-positive troponin I assay was associated with a high concentration of IgM rheumatoid factor with the use of certain immunoassay techniques.17 In patients with acute skeletal muscle injury, the first-generation troponin T assay was found to be falsely positive due to the nonspecific binding of skeletal muscle troponin T to the walls of the test tube used for the assay. When the second-generation troponin T assay was used, troponin T levels were found to be slightly more positive than troponin I levels (1.7 vs 1.5 times the upper limit of normal), especially in patients with renal failure.18
Troponin may also be falsely elevated in hemolyzed blood samples. This has to be taken into consideration in interpreting the results of troponin testing in severely hemolyzed blood samples. However, Puelacher et al19 suggested that the presence of hemolysis did not appear to interfere with clinical value of the test.
- Piper HM, Schwartz P, Spahr R, Hütter J, Spieckermann P. Early enzyme release from myocardial cells is not due to irreversible cell damage. J Mol Cell Cardiol 1984; 16(4):385–388. doi:10.1016/S0022-2828(84)80609-4
- Ammann P, Fehr T, Minder EI, Günter C, Bertel O. Elevation of troponin I in sepsis and septic shock. Intensive Care Med 2001; 27(6):965–969.
- Landesberg G, Jaffe AS, Gilon D, et al. Troponin elevation in severe sepsis and septic shock. Crit Care Med 2014; 42(4):790–800. doi:10.1097/CCM.0000000000000107
- Bessière F, Khenifer S, Dubourg J, Durieu I, Lega JC. Prognostic value of troponins in sepsis: a meta-analysis. Intensive Care Med 2013; 39(7):1181–1189. doi:10.1007/s00134-013-2902-3
- Scheitz JF, Nolte CH, Laufs U, Endres M. Application and interpretation of high-sensitivity cardiac troponin assays in patients with acute ischemic stroke. Stroke 2015; 46(4):1132–1140. doi:10.1161/STROKEAHA.114.007858
- Naidech AM, Kreiter KT, Janjua N, et al. Cardiac troponin elevation, cardiovascular morbidity, and outcome after subarachnoid hemorrhage. Circulation 2005; 112(18):2851–2656. doi:10.1161/CIRCULATIONAHA.105.533620
- Mochmann HC, Scheitz JF, Petzold GC, et al; TRELAS Study Group. Coronary angiographic findings in acute ischemic stroke patients with elevated cardiac troponin: the Troponin Elevation in Acute Ischemic Stroke (TRELAS) Study. Circulation 2016; 133(13):1264–1271. doi:10.1161/CIRCULATIONAHA.115.018547
- Michos ED, Wilson LM, Yeh HC, et al. Prognostic value of cardiac troponin in patients with chronic kidney disease without suspected acute coronary syndrome. Ann Intern Med 2014; 161(7):491–501. doi:10.7326/M14-0743
- Lankeit M, Jiménez D, Kostrubiec M, et al. Predictive value of the high-sensitivity troponin T assay and the simplified pulmonary embolism severity index in hemodynamically stable patients with acute pulmonary embolism: a prospective validation study. Circulation 2011; 124(24):2716–2724. doi:10.1161/CIRCULATIONAHA.111.051177
- Heresi GA, Tang WH, Aytekin M, Hammel J, Hazen SL, Dweik RA. Sensitive cardiac troponin I predicts poor outcomes in pulmonary arterial hypertension. Eur Respir J 2012; 39(4)939–944. doi:10.1183/09031936.00067011
- Pavasini R, d’Ascenzo F, Campo G, et al. Cardiac troponin elevation predicts all-cause mortality in patients with acute exacerbation of chronic obstructive pulmonary disease: systematic review and meta-analysis. Int J Cardiol 2015; 191:187–193. doi:10.1016/j.ijcard.2015.05.006
- Cardinale D, Sandri MT, Martinoni A, et al. Left ventricular dysfunction predicted by early troponin I release after high-dose chemotherapy. J Am Coll Cardiol 2000; 36(2):517–522.
- Miller WL, Hartman KA, Burritt MF, et al. Serial biomarker measurements in ambulatory patients with chronic heart failure: the importance of change over time. Circulation 2007; 116(3):249–257. doi:10.1161/CIRCULATIONAHA.107.694562
- Logeart D, Beyne P, Cusson C, et al. Evidence of cardiac myolysis in severe nonischemic heart failure and the potential role of increased wall strain. Am Heart J 2001; 141(2):247–253. doi:10.1067/mhj.2001.111767
- Whittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352(6):539–548. doi:10.1056/NEJMoa043046
- McClennen S, Halamka JD, Horowitz GL, Kannam JP, Ho KK. Clinical prevalence and ramifications of false-positive cardiac troponin I elevations from the Abbott AxSYM Analyzer. Am J Cardiol 2003; 91(9):1125–1127.
- Bradham WS, Bian A, Oeser A, et al. High-sensitivity cardiac troponin-I is elevated in patients with rheumatoid arthritis, independent of cardiovascular risk factors and inflammation. PLoS One 2012; 7(6):e38930. doi:10.1371/journal.pone.0038930
- Li SF, Zapata J, Tillem E. The prevalence of false-positive cardiac troponin I in ED patients with rhabdomyolysis. Am J Emerg Med 2005; 23(7):860–863. doi:10.1016/j.ajem.2005.05.008
- Puelacher C, Twerenbold R, Mosimann T, et al. Effects of hemolysis on the diagnostic accuracy of cardiac troponin I for the diagnosis of myocardial infarction. Int J Cardiol 2015; 187:313–315. doi:10.1016/j.ijcard.2015.03.378
- Piper HM, Schwartz P, Spahr R, Hütter J, Spieckermann P. Early enzyme release from myocardial cells is not due to irreversible cell damage. J Mol Cell Cardiol 1984; 16(4):385–388. doi:10.1016/S0022-2828(84)80609-4
- Ammann P, Fehr T, Minder EI, Günter C, Bertel O. Elevation of troponin I in sepsis and septic shock. Intensive Care Med 2001; 27(6):965–969.
- Landesberg G, Jaffe AS, Gilon D, et al. Troponin elevation in severe sepsis and septic shock. Crit Care Med 2014; 42(4):790–800. doi:10.1097/CCM.0000000000000107
- Bessière F, Khenifer S, Dubourg J, Durieu I, Lega JC. Prognostic value of troponins in sepsis: a meta-analysis. Intensive Care Med 2013; 39(7):1181–1189. doi:10.1007/s00134-013-2902-3
- Scheitz JF, Nolte CH, Laufs U, Endres M. Application and interpretation of high-sensitivity cardiac troponin assays in patients with acute ischemic stroke. Stroke 2015; 46(4):1132–1140. doi:10.1161/STROKEAHA.114.007858
- Naidech AM, Kreiter KT, Janjua N, et al. Cardiac troponin elevation, cardiovascular morbidity, and outcome after subarachnoid hemorrhage. Circulation 2005; 112(18):2851–2656. doi:10.1161/CIRCULATIONAHA.105.533620
- Mochmann HC, Scheitz JF, Petzold GC, et al; TRELAS Study Group. Coronary angiographic findings in acute ischemic stroke patients with elevated cardiac troponin: the Troponin Elevation in Acute Ischemic Stroke (TRELAS) Study. Circulation 2016; 133(13):1264–1271. doi:10.1161/CIRCULATIONAHA.115.018547
- Michos ED, Wilson LM, Yeh HC, et al. Prognostic value of cardiac troponin in patients with chronic kidney disease without suspected acute coronary syndrome. Ann Intern Med 2014; 161(7):491–501. doi:10.7326/M14-0743
- Lankeit M, Jiménez D, Kostrubiec M, et al. Predictive value of the high-sensitivity troponin T assay and the simplified pulmonary embolism severity index in hemodynamically stable patients with acute pulmonary embolism: a prospective validation study. Circulation 2011; 124(24):2716–2724. doi:10.1161/CIRCULATIONAHA.111.051177
- Heresi GA, Tang WH, Aytekin M, Hammel J, Hazen SL, Dweik RA. Sensitive cardiac troponin I predicts poor outcomes in pulmonary arterial hypertension. Eur Respir J 2012; 39(4)939–944. doi:10.1183/09031936.00067011
- Pavasini R, d’Ascenzo F, Campo G, et al. Cardiac troponin elevation predicts all-cause mortality in patients with acute exacerbation of chronic obstructive pulmonary disease: systematic review and meta-analysis. Int J Cardiol 2015; 191:187–193. doi:10.1016/j.ijcard.2015.05.006
- Cardinale D, Sandri MT, Martinoni A, et al. Left ventricular dysfunction predicted by early troponin I release after high-dose chemotherapy. J Am Coll Cardiol 2000; 36(2):517–522.
- Miller WL, Hartman KA, Burritt MF, et al. Serial biomarker measurements in ambulatory patients with chronic heart failure: the importance of change over time. Circulation 2007; 116(3):249–257. doi:10.1161/CIRCULATIONAHA.107.694562
- Logeart D, Beyne P, Cusson C, et al. Evidence of cardiac myolysis in severe nonischemic heart failure and the potential role of increased wall strain. Am Heart J 2001; 141(2):247–253. doi:10.1067/mhj.2001.111767
- Whittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352(6):539–548. doi:10.1056/NEJMoa043046
- McClennen S, Halamka JD, Horowitz GL, Kannam JP, Ho KK. Clinical prevalence and ramifications of false-positive cardiac troponin I elevations from the Abbott AxSYM Analyzer. Am J Cardiol 2003; 91(9):1125–1127.
- Bradham WS, Bian A, Oeser A, et al. High-sensitivity cardiac troponin-I is elevated in patients with rheumatoid arthritis, independent of cardiovascular risk factors and inflammation. PLoS One 2012; 7(6):e38930. doi:10.1371/journal.pone.0038930
- Li SF, Zapata J, Tillem E. The prevalence of false-positive cardiac troponin I in ED patients with rhabdomyolysis. Am J Emerg Med 2005; 23(7):860–863. doi:10.1016/j.ajem.2005.05.008
- Puelacher C, Twerenbold R, Mosimann T, et al. Effects of hemolysis on the diagnostic accuracy of cardiac troponin I for the diagnosis of myocardial infarction. Int J Cardiol 2015; 187:313–315. doi:10.1016/j.ijcard.2015.03.378
A 71-year-old woman with shock and a high INR
A 71-year-old woman is brought to the emergency department by her neighbor after complaining of fatigue and light-headedness for the last 8 hours. The patient lives alone and was feeling well when she woke up this morning, but then began to feel nauseated and vomited twice.
The patient appears drowsy and confused and cannot provide any further history. Her medical records show that she was seen in the cardiology clinic 6 months ago but has not kept her appointments since then.
Her medical history includes atrial fibrillation, hypertension, type 2 diabetes mellitus, and osteoarthritis. Her medications are daily warfarin, atenolol, aspirin, candesartan, and metformin, and she takes acetaminophen as needed. She is neither a smoker nor a drug user, but she drinks alcohol occasionally. Her family history is significant for her mother’s death from breast cancer at age 55.
The neighbor confirms that the patient appeared well this morning and has not had any recent illnesses except for a minor cold last week that improved over 5 days with acetaminophen only.
INITIAL EVALUATION AND MANAGEMENT
Physical examination
On physical examination, her blood pressure is 80/40 mm Hg, respiratory rate 25 breaths per minute, oral temperature 38.3°C (100.9°F), and heart rate 130 beats per minute and irregular.
Her neck veins are flat, and her chest is clear to auscultation with normal heart sounds. Abdominal palpation elicits discomfort in the middle segments, voluntary withdrawal, and abdominal wall rigidity. Her skin feels dry and cool, with decreased turgor.
Initial treatment
The patient is given 1 L of 0.9% saline intravenously over the first hour and then is transferred to the intensive care unit, where a norepinephrine drip is started to treat her ongoing hypotension. Normal saline is continued at a rate of 500 mL per hour for the next 4 hours.
Cardiac monitoring and 12-lead electrocardiography show atrial fibrillation with a rapid ventricular response of 138 beats per minute, but electrical cardioversion is not done.
Initial laboratory tests

Of note, her international normalized ratio (INR) is 6.13, while the therapeutic range for a patient taking warfarin because of atrial fibrillation is 2.0 to 3.0.
Her blood pH is 7.34 (reference range 7.35–7.45), and her bicarbonate level is 18 mmol/L (22–26); a low pH and low bicarbonate together indicate metabolic acidosis. Her sodium level is 128 mmol/L (135–145), her chloride level is 100 mmol/L (97–107), and, as mentioned, her bicarbonate level is 18 mmol/L; therefore, her anion gap is 128 – (100 + 18) = 10 mmol/L, which is normal (≤ 10).1
Her serum creatinine level is 1.3 mg/dL (0.5–1.1), and her blood urea nitrogen level is 35 mg/dL (7–20).
Her potassium level is 5.8 mmol/L, which is consistent with hyperkalemia (reference range 3.5–5.2).
DIFFERENTIAL DIAGNOSIS
1. Which of the following is the most likely cause of this patient’s symptoms?
- Adrenal crisis
- Cardiogenic shock due to decreased cardiac contractility
- Intracranial hemorrhage
- Acute abdomen due to small bowel obstruction
- Septic shock due to bacterial toxin-induced loss of vascular tone
Our patient is presenting with shock. Given our inability to obtain a meaningful history, the differential diagnosis is broad and includes all of the above.
Adrenal crisis
The sudden onset and laboratory results that include hyperkalemia, hyponatremia, and normal anion gap metabolic acidosis raise suspicion of adrenal crisis resulting in acute mineralocorticoid and glucocorticoid insufficiency.1
The patient’s elevated serum creatinine and high blood urea nitrogen-to-creatinine ratio of 26.9 (reference range 10–20) also suggest intravascular volume contraction. Her low hemoglobin level and supratherapeutic INR, possibly due to an interaction between warfarin and acetaminophen combined with poor medical follow-up, raise suspicion of acute bilateral adrenal necrosis due to hemorrhage.

Bilateral adrenal hemorrhage is one cause of adrenal crisis resulting in bilateral adrenal necrosis. Risk factors for adrenal hemorrhage include anticoagulation therapy, underlying coagulopathy, postoperative states, and certain infections such as meningococcemia and Haemophilus influenzae infection.2–5 Nevertheless, in most cases the INR is in the therapeutic range and the patient has no bleeding elsewhere.4 Other causes of adrenal necrosis include emboli, sepsis, and blunt trauma.6,7
Other causes of adrenal crisis are listed in Table 3.
Cardiogenic shock

Intracranial hemorrhage
Intracranial hemorrhage can present with a decreased level of consciousness, but it is less likely to cause hypotension, as the cranial space is limited. If massive intracranial hemorrhage would occur, the increase in intracranial pressure would more likely cause hypertension by the Cushing reflex than hypotension.
Acute abdomen
Abdominal pain and rigidity along with fever can be presenting symptoms of both adrenal insufficiency and an acute abdomen due to intestinal obstruction.4 However, intestinal obstruction typically causes a high anion gap metabolic acidosis due to lactic acidosis, instead of the normal anion gap metabolic acidosis present in this patient.8 Moreover, her deranged electrolytes, supratherapeutic INR, and absence of previous gastroenterologic conditions make adrenal crisis a more likely diagnosis.
Septic shock
Septic shock would also cause fever and hypotension as bacterial toxins induce a pyrexic response and vasodilation. However, at such an early stage of sepsis, the patient would be expected to be warm and hyperemic, whereas this patient’s skin is cool and dry due to volume depletion secondary to adrenal insufficiency.9 Sepsis would also cause a high anion gap metabolic acidosis due to lactic acidosis, as opposed to this patient’s normal anion gap metabolic acidosis. These findings, along with the metabolic derangements and the absence of a focus of infection, make sepsis a less likely possibility.
CASE CONTINUED: CARDIOMEGALY, PERSISTENT HYPOTENSION
Blood is drawn for cultures and measurement of troponins and lactic acid, and urine samples are taken for culture and biochemical analysis. Chest radiography shows mild cardiomegaly. The patient is started empirically on vancomycin and cefepime, and her warfarin is discontinued.
Five hours after presenting to the emergency department, her blood pressure remains at 80/40 mm Hg even after receiving 3 L of normal saline intravenously.
PROMPT MANAGEMENT OF ADRENAL CRISIS
2. Which of the following is the most appropriate next step in managing this patient?
- Draw samples for serum cortisol and plasma adrenocorticotropic hormone (ACTH) levels, then give hydrocortisone 100 mg intravenously
- Perform abdominal computed tomography (CT) without contrast
- Perform transthoracic echocardiography
- Increase the norepinephrine infusion
- Immediately give fludrocortisone
First give fluids
The first step in managing a patient with suspected adrenal crisis is liberal intravenous fluid administration to replenish the depleted intravascular space. The amount and choice of fluid is empiric, but a recommendation is 1 L of normal saline or dextrose 5% in normal saline, infused quickly over the first hour and then titrated according to the patient’s fluid status.10
Measure cortisol and ACTH; start corticosteroids immediately
Immediate therapy with an appropriate stress dose of intravenous corticosteroids (eg, hydrocortisone 100 mg) is essential. However, this should be done after drawing blood for cortisol and ACTH measurements.10
Do not delay corticosteroid therapy while awaiting the results of the diagnostic tests.

In addition, in the early phase of evolving primary adrenal insufficiency, measurement of plasma renin and aldosterone levels may be beneficial, as mineralocorticoid deficiency may predominate.10,12,13
One of the most important aims of early corticosteroid supplementation is to prevent further hyponatremia by reducing a reactive increase in antidiuretic hormone secretion caused by cortisol deficiency. Corticosteroids also help to restore normal blood pressure by increasing vascular tone, as glucocorticoid receptor activation potentiates the vasoconstrictor actions of norepinephrine, angiotensin II, and other vasoconstrictors.14,15
Which corticosteroid to use?
Which corticosteroid to use in previously undiagnosed adrenal insufficiency is controversial. The Endocrine Society10 and Japan Endocrine Society16 clinical practice guidelines recommend hydrocortisone in a 100-mg intravenous bolus followed by 200 mg over 24 hours.
The choice of hydrocortisone is justified by its superior mineralocorticoid activity.10,16 Further, hydrocortisone is preferred over dexamethasone if the patient is known to have primary adrenal insufficiency, or if the serum potassium level is higher than 6.0 mmol/L.
Some clinicians, on the other hand, recommend dexamethasone, given as a 4-mg intravenous bolus followed by 4-mg boluses every 12 hours. Their rationale is that dexamethasone, unlike hydrocortisone, does not interfere with subsequent serum cortisol assays if the patient later undergoes ACTH stimulation testing.17 Dexamethasone may also be preferred to minimize unwanted mineralocorticoid effects, such as in neurosurgical patients at risk of brain edema.
If hydrocortisone is used, ACTH stimulation testing can be done after withholding hydrocortisone for 24 hours once the patient is stable. (It should be restarted after the test if the results are abnormal.)
Other possible steps
Abdominal CT should be done in our patient to address the possibility of bilateral adrenal hemorrhage. However, it is preferable to wait until the patient is stabilized.
Echocardiography. Our patient is likely to have an element of cardiac failure, given her hypertension and cardiomegaly. However, decompensated heart failure is probably not the cause of her presentation. Thus, the first priority is to treat her adrenal crisis, and echocardiography should be deferred.
Increasing the norepinephrine infusion is unlikely to improve her blood pressure very much, as she is significantly volume-depleted. Further, low cortisol decreases the vascular response to norepinephrine.15
Mineralocorticoids such as fludrocortisone are used to treat primary adrenal insufficiency. However, they are not required during acute management of adrenal crisis, as 40 mg of hydrocortisone offers mineralocorticoid activity equivalent to 100 µg of fludrocortisone. Thus, the high doses of hydrocortisone used to treat adrenal crisis provide adequate mineralocorticoid therapy.10,18
If dexamethasone is used, its effect along with normal saline supplementation would be sufficient to replete the intravenous space and bring the sodium level back up to normal in the acute setting.
CASE RESUMED: IMPROVEMENT WITH HYDROCORTISONE
The patient’s blood is drawn for serum cortisol and plasma ACTH measurements. A 100-mg intravenous bolus of hydrocortisone is given, followed by a 50-mg bolus every 6 hours until the patient stabilizes.
Twenty-four hours later, the patient states that she has more energy, and her appetite has improved. The norepinephrine infusion is stopped 48 hours after presentation, at which time her blood pressure is 120/70 mm Hg, heart rate 85 beats per minute and irregular, and temperature 36.7°C (98.1°F). Her current laboratory values include the following:
- Serum sodium 137 mmolL
- Serum potassium 4.3 mmol/L
- Hemoglobin 9.3 g/dL
- Serum cortisol (random) 7.2 μg/dL
- Plasma ACTH 752 pg/mL (10–60 pg/mL).
ESTABLISHING THE DIAGNOSIS OF ADRENAL INSUFFICIENCY
3. Which of the following is the most appropriate test to establish the diagnosis of adrenal insufficiency?
- 7 am total serum cortisol measurement
- Random serum cortisol measurement
- 7 am salivary cortisol measurement
- 24-hour urinary free cortisol measurement
- ACTH stimulation test for cortisol
- Insulin tolerance test for cortisol

These tests also help determine the type of adrenal insufficiency (primary, secondary, or tertiary) and guide further management. Secondary adrenal insufficiency is caused by inadequate pituitary ACTH secretion and subsequent inadequate cortisol production, whereas tertiary adrenal insufficiency is caused by inadequate hypothalamic corticotropin-releasing hormone secretion and subsequent inadequate ACTH and cortisol production. The diagnosis of adrenal insufficiency relies first on demonstrating inappropriately low total serum cortisol production. Subsequently, serum ACTH helps to differentiate between primary (high ACTH) and secondary or tertiary (low or inappropriately normal ACTH) adrenal insufficiency.
Each test listed above may demonstrate a low cortisol level. However, in a nonacute setting, safety concerns (especially regarding insulin tolerance testing), poor diagnostic value, feasibility (ie, the difficulty of 24-hour tests), and poor sensitivity of 7 am cortisol make the ACTH stimulation test the most appropriate test in clinical practice to establish the diagnosis of adrenal insufficiency.
7 am serum cortisol measurement
Measuring the serum cortisol level early in the morning in the nonacute setting could be of diagnostic value, as an extremely low value (< 3–5 μg/dL) is almost 100% specific for adrenal insufficiency in the absence of concurrent exogenous steroid intake. However, the very low cutoff for this test causes poor sensitivity (about 33%), as many patients have partial adrenal insufficiency and hence have higher serum cortisol levels that may even be in the normal physiologic range.19–22
Random serum cortisol measurements
Random serum cortisol measurements are not very useful in a nonacute setting, since cortisol levels are affected by factors such as stress and hydration status. Moreover, they fluctuate during the day in a circadian rhythm.
On the other hand, random serum cortisol is a very good test to evaluate for adrenal insufficiency in the acute setting. A random value higher than 15 to 18 μg/dL is almost always associated with adequate adrenal function and generally rules out adrenal insufficiency.11,23,24
7 am salivary cortisol measurement
The same principle applies to early morning salivary cortisol. Only extremely low values (< 2.65 ng/mL) may distinguish patients with adrenal insufficiency from healthy individuals, with 97.1% sensitivity and 93.3% specificity.25
Of note, early morning salivary cortisol is not routinely measured in most clinical practices for evaluation of adrenal function. Hence, morning serum and morning salivary cortisol are useful screening tools and have meaningful results when their values are in the extremes of the spectrum, but they are not reliable as a single test, as they may overlook patients with partial adrenal insufficiency.
Urinary cortisol measurement
Urinary cortisol measurement is not used to diagnose adrenal insufficiency, as values can be normal in patients with partial adrenal insufficiency.
The ACTH stimulation test
The ACTH stimulation test involves an intramuscular or intravenous injection of cosyntropin (a synthetic analogue of ACTH fragment 1–24 that has the full activity of native ACTH) and measuring total serum cortisol at baseline, 30 minutes, and 60 minutes to assess the response of the adrenal glands.
The test can be done using a high or low dose of cosyntropin. The Endocrine Society’s 2016 guidelines recommend the high dose (250 μg) for most patients.10 The standard high-dose stimulation test can be done at any time during the day.26 If the cosyntropin is injected intravenously, any value higher than 18 to 20 μg/dL indicates normal adrenal function and excludes adrenal insufficiency.27,28 If intramuscular injection is used, any value higher than 16 to 18 μg/dL at 30 minutes post-consyntropin excludes adrenal insufficiency.29
The ACTH stimulation test may not exclude acute secondary or tertiary adrenal insufficiency.
Insulin tolerance testing
Insulin tolerance testing remains the gold standard for diagnosing adrenal insufficiency and assessing the integrity of the pituitary-adrenal axis. However, given its difficulty to perform, safety concerns, and the availability of other reliable tests, its use in clinical practice is limited. It is nonetheless useful in assessing patients with recent onset of ACTH deficiency.30,31
CASE RESUMED: PATIENT DISCHARGED, LOST TO FOLLOW-UP
Abdominal CT without contrast is done and demonstrates bilateral adrenal hemorrhage. Thus, the patient is diagnosed with primary acute adrenal insufficiency due to adrenal necrosis.
She is started on oral hydrocortisone and fludrocortisone after intravenous hydrocortisone is discontinued. She is counseled about adhering to medications, wearing a medical alert bracelet, giving herself emergency cortisol injections, taking higher doses of hydrocortisone if she is ill, and monitoring her INR. She is discharged home after her symptoms resolve.
The patient does not keep her scheduled appointment and is lost to follow-up. She returns 2 years later complaining of fatigue and feeling unwell. She admits that she stopped taking hydrocortisone 1 year ago after reading an online article about corticosteroid side effects. She has continued to take fludrocortisone.
MINERALOCORTICOID VS CORTICOSTEROID DEFICIENCY

4. Which of the following is least likely to be present in this patient at this time?
- Intravascular volume depletion
- Hyponatremia
- Skin hyperpigmentation
- Normokalemia
- Elevated serum ACTH level
Intravascular volume depletion
Intravascular volume depletion is the least likely to be present. This is because intravascular volume depletion is mainly secondary to mineralocorticoid deficiency rather than corticosteroid deficiency, which is not present in this patient, as she is compliant with her mineralocorticoid replacement therapy.32,33 However, even with sufficient mineralocorticoid replacement, mild hypotension may be present in this patient due to corticosteroid deficiency-induced loss of vascular tone.
Hyponatremia
Hyponatremia in adrenal insufficiency is not due only to mineralocorticoid deficiency. Patients with secondary or tertiary adrenal insufficiency may also exhibit hyponatremia.34 ACTH deficiency in such patients is not expected to cause mineralocorticoid deficiency, as ACTH has only a minor role in aldosterone production.
It has been proposed that hyponatremia in secondary adrenal insufficiency is due to cortisol deficiency resulting in an increase of antidiuretic hormone secretion.35,36 The mechanisms for increased antidiuretic hormone include cortisol deficiency resulting in an increased corticotropin-releasing hormone level, which acts as an antidiuretic hormone secretagogue,37,38 and cortisol directly suppressing antidiuretic hormone secretion.39
In our patient, volume expansion and hyponatremia are expected due to increased antidiuretic hormone secretion as a result of corticosteroid insufficiency.
Hyperpigmentation
Hyperpigmentation of the skin is present only in long-standing primary adrenal insufficiency. This is due to chronic cortisol deficiency causing an increased secretion of pro-opiomelanocortin, a prohormone that is cleaved into ACTH, melanocyte-stimulating hormone, and other hormones. Melanocyte-stimulating hormone causes skin hyperpigmentation due to increased melanin synthesis.40 The hyperpigmentation is seen in sun-exposed areas, pressure areas, palmar creases, nipples, and mucous membranes.
This patient has long-standing corticosteroid deficiency due to noncompliance and primary adrenal insufficiency, and as a result she is expected to have elevated serum ACTH and hyperpigmentation.
Normokalemia
Mineralocorticoid deficiency results in hyperkalemia and metabolic acidosis by impairing renal excretion of potassium and acid.41 This patient is compliant with her mineralocorticoid replacement regimen; thus, potassium levels and pH are expected to be normal.
TAKE-HOME POINTS
- Suspect adrenal crisis in any patient who presents with shock.
- Acute abdomen or unexplained fever could be among the manifestations.
- Initial management requires liberal normal saline intravenous fluid administration to replete the intravascular space.
- Draw blood samples for serum chemistry, cortisol, and ACTH, followed immediately by intravenous hydrocortisone supplementation.
- In critically ill patients, evaluate adrenal function with random serum cortisol; in a nonacute setting use the ACTH stimulation test.
- Chronic management of primary adrenal insufficiency requires corticosteroid and mineralocorticoid therapy.
- Mani S, Rutecki GW. A patient with altered mental status and an acid-base disturbance. Cleve Clin J Med 2017; 84(1):27–34. doi:10.3949/ccjm.84a.16042
- Almiani M, Gorthi J, Subbiah S, Firoz M. Quiz page November 2012: an unusual case of acute hyponatremia and normal anion gap metabolic acidosis. Am J Kidney Dis 2012; 60(5):xxxiii–xxxvi. doi:10.1053/j.ajkd.2012.05.026
- Migeon CJ, Kenny FM, Hung W, Voorhess ML. Study of adrenal function in children with meningitis. Pediatrics 1967; 40(2):163–183.
- Rao RH, Vagnucci AH, Amico JA. Bilateral massive adrenal hemorrhage: early recognition and treatment. Ann Intern Med 1989; 110(3):227–235.
- Shimizu S, Tahara Y, Atsumi T, et al. Waterhouse-Friderichsen syndrome caused by invasive Haemophilus influenzae type B infection in a previously healthy young man. Anaesth Intensive Care 2010; 38(1):214–215.
- Castaldo ET, Guillamondegui OD, Greco JA 3rd, Feurer ID, Miller RS, Morris JA Jr. Are adrenal injuries predictive of adrenal insufficiency in patients sustaining blunt trauma? Am Surg 2008; 74(3):262–266.
- Xarli VP, Steele AA, Davis PJ, Buescher ES, Rios CN, Garcia-Bunuel R. Adrenal hemorrhage in the adult. Medicine (Baltimore) 1978; 57(3):211–221.
- Takeuchi K, Tsuzuki Y, Ando T, et al. Clinical studies of strangulating small bowel obstruction. Am Surg 2004; 70(1):40–44.
- MacKenzie IM. The haemodynamics of human septic shock. Anaesthesia 2001; 56(2):130–144.
- Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2016; 101(2):364–389. doi:10.1210/jc.2015-1710
- Hamrahian AH, Fleseriu M; AACE Adrenal Scientific Committee. Evaluation and management of adrenal insufficiency in critically ill patients: disease state review. Endocr Pract 2017; 23(6):716–725. doi:10.4158/EP161720.RA
- Saenger P, Levine LS, Irvine WJ, et al. Progressive adrenal failure in polyglandular autoimmune disease. J Clin Endocrinol Metab 1982; 54(4):863–867.
- Coco G, Dal Pra C, Presotto F, et al. Estimated risk for developing autoimmune Addison's disease in patients with adrenal cortex autoantibodies. J Clin Endocrinol Metab 2006; 91(5):1637–1645. doi:10.1210/jc.2005-0860
- Ullian ME. The role of corticosteroids in the regulation of vascular tone. Cardiovasc Res 1999; 41(1):55–64.
- Yang S, Zhang L. Glucocorticoids and vascular reactivity. Curr Vasc Pharmacol 2004; 2(1):1–12.
- Yanase T, Tajima T, Katabami T, et al. Diagnosis and treatment of adrenal insufficiency including adrenal crisis: a Japan Endocrine Society clinical practice guideline [Opinion]. Endocr J 2016; 63(9):765–784. doi:10.1507/endocrj.EJ16-0242
- Taylor RL, Grebe SK, Singh RJ. Quantitative, highly sensitive liquid chromatography-tandem mass spectrometry method for detection of synthetic corticosteroids. Clin Chem 2004; 50(10):2345–2352. doi:10.1373/clinchem.2004.033605
- Goldfien A, Laidlaw JC, Haydar NA, Renold AE, Thorn GW. Fluorohydrocortisone and chlorohydrocortisone, highly potent derivatives of compound F. N Engl J Med 1955; 252(11):415–421. doi:10.1056/NEJM195503172521101
- Jenkins D, Forsham PH, Laidlaw JC, Reddy WJ, Thorn GW. Use of ACTH in the diagnosis of adrenal cortical insufficiency. Am J Med 1955; 18(1):3–14.
- Hägg E, Asplund K, Lithner F. Value of basal plasma cortisol assays in the assessment of pituitary-adrenal insufficiency. Clin Endocrinol (Oxf) 1987; 26(2):221–226.
- Deutschbein T, Unger N, Mann K, Petersenn S. Diagnosis of secondary adrenal insufficiency: unstimulated early morning cortisol in saliva and serum in comparison with the insulin tolerance test. Horm Metab Res 2009; 41(4):834–839. doi:10.1055/s-0029-1225630
- Erturk E, Jaffe CA, Barkan AL. Evaluation of the integrity of the hypothalamic-pituitary-adrenal axis by insulin hypoglycemia test. J Clin Endocrinol Metab 1998; 83(7):2350–2354.
- Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med 2003; 348(8):727–734. doi:10.1056/NEJMra020529
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 2013; 39(2):165–228. doi:10.1007/s00134-012-2769-8
- Ceccato F, Barbot M, Zilio M, et al. Performance of salivary cortisol in the diagnosis of Cushing's syndrome, adrenal incidentaloma, and adrenal insufficiency. Eur J Endocrinol 2013; 169(1):31–36. doi:10.1530/EJE-13-0159
- Dickstein G, Shechner C, Nicholson WE, et al. Adrenocorticotropin stimulation test: effects of basal cortisol level, time of day, and suggested new sensitive low dose test. J Clin Endocrinol Metab 1991; 72(4):773–778. doi:10.1210/jcem-72-4-773
- May ME, Carey RM. Rapid adrenocorticotropic hormone test in practice. Retrospective review. Am J Med 1985; 79(6):679–884.
- Speckart PF, Nicoloff JT, Bethune JE. Screening for adrenocortical insufficiency with cosyntropin (synthetic ACTH). Arch Intern Med 1971; 128(5):761–763.
- Peechakara S, Bena J, Clarke NJ, et al. Total and free cortisol levels during 1 μg, 25 μg, and 250 μg cosyntropin stimulation tests compared to insulin tolerance test: results of a randomized, prospective, pilot study. Endocrine 2017; 57(3):388–393. doi:10.1007/s12020-017-1371-9
- Finucane FM, Liew A, Thornton E, Rogers B, Tormey W, Agha A. Clinical insights into the safety and utility of the insulin tolerance test (ITT) in the assessment of the hypothalamo-pituitary-adrenal axis. Clin Endocrinol (Oxf) 2008; 69(4):603–607. doi:10.1111/j.1365-2265.2008.03240.x
- Lindholm J, Kehlet H. Re-evaluation of the clinical value of the 30 min ACTH test in assessing the hypothalamic-pituitary-adrenocortical function. Clin Endocrinol (Oxf) 1987; 26(1):53–59.
- Charmandari E, Nicolaides NC, Chrousos GP. Adrenal insufficiency. Lancet 2014; 383(9935):2152–2167. doi:10.1016/S0140-6736(13)61684-0
- Burke CW. Adrenocortical insufficiency. Clin Endocrinol Metab 1985; 14(4):947–976.
- Jessani N, Jehangir W, Behman D, Yousif A, Spiler IJ. Secondary adrenal insufficiency: an overlooked cause of hyponatremia. J Clin Med Res 2015; 7(4):286–288. doi:10.14740/jocmr2041w
- Oelkers W. Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism. N Engl J Med 1989; 321(8):492–496. doi:10.1056/NEJM198908243210802
- Ishikawa S, Schrier RW. Effect of arginine vasopressin antagonist on renal water excretion in glucocorticoid and mineralocorticoid deficient rats. Kidney Int 1982; 22(6):587–593.
- Wolfson B, Manning RW, Davis LG, Arentzen R, Baldino F Jr. Co-localization of corticotropin releasing factor and vasopressin mRNA in neurones after adrenalectomy. Nature 1985; 315(6014):59–61.
- Kalogeras KT, Nieman LK, Friedman TC, et al. Inferior petrosal sinus sampling in healthy subjects reveals a unilateral corticotropin-releasing hormone-induced arginine vasopressin release associated with ipsilateral adrenocorticotropin secretion. J Clin Invest 1996; 97:2045–2050.
- Kovacs KJ, Foldes A, Sawchenko PE. Glucocorticoid negative feedback selectively targets vasopressin transcription in parvocellular neurosecretory neurons. J Neurosci 2000; 20:3843–3852.
- Sarkar SB, Sarkar S, Ghosh S, Bandyopadhyay S. Addison's disease. Contemp Clin Dent 2012; 3(4):484–486. doi:10.4103/0976-237X.107450
- Szylman P, Better OS, Chaimowitz C, Rosler A. Role of hyperkalemia in the metabolic acidosis of isolated hypoaldosteronism. N Engl J Med 1976; 294(7):361–365. doi:10.1056/NEJM197602122940703
A 71-year-old woman is brought to the emergency department by her neighbor after complaining of fatigue and light-headedness for the last 8 hours. The patient lives alone and was feeling well when she woke up this morning, but then began to feel nauseated and vomited twice.
The patient appears drowsy and confused and cannot provide any further history. Her medical records show that she was seen in the cardiology clinic 6 months ago but has not kept her appointments since then.
Her medical history includes atrial fibrillation, hypertension, type 2 diabetes mellitus, and osteoarthritis. Her medications are daily warfarin, atenolol, aspirin, candesartan, and metformin, and she takes acetaminophen as needed. She is neither a smoker nor a drug user, but she drinks alcohol occasionally. Her family history is significant for her mother’s death from breast cancer at age 55.
The neighbor confirms that the patient appeared well this morning and has not had any recent illnesses except for a minor cold last week that improved over 5 days with acetaminophen only.
INITIAL EVALUATION AND MANAGEMENT
Physical examination
On physical examination, her blood pressure is 80/40 mm Hg, respiratory rate 25 breaths per minute, oral temperature 38.3°C (100.9°F), and heart rate 130 beats per minute and irregular.
Her neck veins are flat, and her chest is clear to auscultation with normal heart sounds. Abdominal palpation elicits discomfort in the middle segments, voluntary withdrawal, and abdominal wall rigidity. Her skin feels dry and cool, with decreased turgor.
Initial treatment
The patient is given 1 L of 0.9% saline intravenously over the first hour and then is transferred to the intensive care unit, where a norepinephrine drip is started to treat her ongoing hypotension. Normal saline is continued at a rate of 500 mL per hour for the next 4 hours.
Cardiac monitoring and 12-lead electrocardiography show atrial fibrillation with a rapid ventricular response of 138 beats per minute, but electrical cardioversion is not done.
Initial laboratory tests
Of note, her international normalized ratio (INR) is 6.13, while the therapeutic range for a patient taking warfarin because of atrial fibrillation is 2.0 to 3.0.
Her blood pH is 7.34 (reference range 7.35–7.45), and her bicarbonate level is 18 mmol/L (22–26); a low pH and low bicarbonate together indicate metabolic acidosis. Her sodium level is 128 mmol/L (135–145), her chloride level is 100 mmol/L (97–107), and, as mentioned, her bicarbonate level is 18 mmol/L; therefore, her anion gap is 128 – (100 + 18) = 10 mmol/L, which is normal (≤ 10).1
Her serum creatinine level is 1.3 mg/dL (0.5–1.1), and her blood urea nitrogen level is 35 mg/dL (7–20).
Her potassium level is 5.8 mmol/L, which is consistent with hyperkalemia (reference range 3.5–5.2).
DIFFERENTIAL DIAGNOSIS
1. Which of the following is the most likely cause of this patient’s symptoms?
- Adrenal crisis
- Cardiogenic shock due to decreased cardiac contractility
- Intracranial hemorrhage
- Acute abdomen due to small bowel obstruction
- Septic shock due to bacterial toxin-induced loss of vascular tone
Our patient is presenting with shock. Given our inability to obtain a meaningful history, the differential diagnosis is broad and includes all of the above.
Adrenal crisis
The sudden onset and laboratory results that include hyperkalemia, hyponatremia, and normal anion gap metabolic acidosis raise suspicion of adrenal crisis resulting in acute mineralocorticoid and glucocorticoid insufficiency.1
The patient’s elevated serum creatinine and high blood urea nitrogen-to-creatinine ratio of 26.9 (reference range 10–20) also suggest intravascular volume contraction. Her low hemoglobin level and supratherapeutic INR, possibly due to an interaction between warfarin and acetaminophen combined with poor medical follow-up, raise suspicion of acute bilateral adrenal necrosis due to hemorrhage.
Bilateral adrenal hemorrhage is one cause of adrenal crisis resulting in bilateral adrenal necrosis. Risk factors for adrenal hemorrhage include anticoagulation therapy, underlying coagulopathy, postoperative states, and certain infections such as meningococcemia and Haemophilus influenzae infection.2–5 Nevertheless, in most cases the INR is in the therapeutic range and the patient has no bleeding elsewhere.4 Other causes of adrenal necrosis include emboli, sepsis, and blunt trauma.6,7
Other causes of adrenal crisis are listed in Table 3.
Cardiogenic shock
Intracranial hemorrhage
Intracranial hemorrhage can present with a decreased level of consciousness, but it is less likely to cause hypotension, as the cranial space is limited. If massive intracranial hemorrhage would occur, the increase in intracranial pressure would more likely cause hypertension by the Cushing reflex than hypotension.
Acute abdomen
Abdominal pain and rigidity along with fever can be presenting symptoms of both adrenal insufficiency and an acute abdomen due to intestinal obstruction.4 However, intestinal obstruction typically causes a high anion gap metabolic acidosis due to lactic acidosis, instead of the normal anion gap metabolic acidosis present in this patient.8 Moreover, her deranged electrolytes, supratherapeutic INR, and absence of previous gastroenterologic conditions make adrenal crisis a more likely diagnosis.
Septic shock
Septic shock would also cause fever and hypotension as bacterial toxins induce a pyrexic response and vasodilation. However, at such an early stage of sepsis, the patient would be expected to be warm and hyperemic, whereas this patient’s skin is cool and dry due to volume depletion secondary to adrenal insufficiency.9 Sepsis would also cause a high anion gap metabolic acidosis due to lactic acidosis, as opposed to this patient’s normal anion gap metabolic acidosis. These findings, along with the metabolic derangements and the absence of a focus of infection, make sepsis a less likely possibility.
CASE CONTINUED: CARDIOMEGALY, PERSISTENT HYPOTENSION
Blood is drawn for cultures and measurement of troponins and lactic acid, and urine samples are taken for culture and biochemical analysis. Chest radiography shows mild cardiomegaly. The patient is started empirically on vancomycin and cefepime, and her warfarin is discontinued.
Five hours after presenting to the emergency department, her blood pressure remains at 80/40 mm Hg even after receiving 3 L of normal saline intravenously.
PROMPT MANAGEMENT OF ADRENAL CRISIS
2. Which of the following is the most appropriate next step in managing this patient?
- Draw samples for serum cortisol and plasma adrenocorticotropic hormone (ACTH) levels, then give hydrocortisone 100 mg intravenously
- Perform abdominal computed tomography (CT) without contrast
- Perform transthoracic echocardiography
- Increase the norepinephrine infusion
- Immediately give fludrocortisone
First give fluids
The first step in managing a patient with suspected adrenal crisis is liberal intravenous fluid administration to replenish the depleted intravascular space. The amount and choice of fluid is empiric, but a recommendation is 1 L of normal saline or dextrose 5% in normal saline, infused quickly over the first hour and then titrated according to the patient’s fluid status.10
Measure cortisol and ACTH; start corticosteroids immediately
Immediate therapy with an appropriate stress dose of intravenous corticosteroids (eg, hydrocortisone 100 mg) is essential. However, this should be done after drawing blood for cortisol and ACTH measurements.10
Do not delay corticosteroid therapy while awaiting the results of the diagnostic tests.
In addition, in the early phase of evolving primary adrenal insufficiency, measurement of plasma renin and aldosterone levels may be beneficial, as mineralocorticoid deficiency may predominate.10,12,13
One of the most important aims of early corticosteroid supplementation is to prevent further hyponatremia by reducing a reactive increase in antidiuretic hormone secretion caused by cortisol deficiency. Corticosteroids also help to restore normal blood pressure by increasing vascular tone, as glucocorticoid receptor activation potentiates the vasoconstrictor actions of norepinephrine, angiotensin II, and other vasoconstrictors.14,15
Which corticosteroid to use?
Which corticosteroid to use in previously undiagnosed adrenal insufficiency is controversial. The Endocrine Society10 and Japan Endocrine Society16 clinical practice guidelines recommend hydrocortisone in a 100-mg intravenous bolus followed by 200 mg over 24 hours.
The choice of hydrocortisone is justified by its superior mineralocorticoid activity.10,16 Further, hydrocortisone is preferred over dexamethasone if the patient is known to have primary adrenal insufficiency, or if the serum potassium level is higher than 6.0 mmol/L.
Some clinicians, on the other hand, recommend dexamethasone, given as a 4-mg intravenous bolus followed by 4-mg boluses every 12 hours. Their rationale is that dexamethasone, unlike hydrocortisone, does not interfere with subsequent serum cortisol assays if the patient later undergoes ACTH stimulation testing.17 Dexamethasone may also be preferred to minimize unwanted mineralocorticoid effects, such as in neurosurgical patients at risk of brain edema.
If hydrocortisone is used, ACTH stimulation testing can be done after withholding hydrocortisone for 24 hours once the patient is stable. (It should be restarted after the test if the results are abnormal.)
Other possible steps
Abdominal CT should be done in our patient to address the possibility of bilateral adrenal hemorrhage. However, it is preferable to wait until the patient is stabilized.
Echocardiography. Our patient is likely to have an element of cardiac failure, given her hypertension and cardiomegaly. However, decompensated heart failure is probably not the cause of her presentation. Thus, the first priority is to treat her adrenal crisis, and echocardiography should be deferred.
Increasing the norepinephrine infusion is unlikely to improve her blood pressure very much, as she is significantly volume-depleted. Further, low cortisol decreases the vascular response to norepinephrine.15
Mineralocorticoids such as fludrocortisone are used to treat primary adrenal insufficiency. However, they are not required during acute management of adrenal crisis, as 40 mg of hydrocortisone offers mineralocorticoid activity equivalent to 100 µg of fludrocortisone. Thus, the high doses of hydrocortisone used to treat adrenal crisis provide adequate mineralocorticoid therapy.10,18
If dexamethasone is used, its effect along with normal saline supplementation would be sufficient to replete the intravenous space and bring the sodium level back up to normal in the acute setting.
CASE RESUMED: IMPROVEMENT WITH HYDROCORTISONE
The patient’s blood is drawn for serum cortisol and plasma ACTH measurements. A 100-mg intravenous bolus of hydrocortisone is given, followed by a 50-mg bolus every 6 hours until the patient stabilizes.
Twenty-four hours later, the patient states that she has more energy, and her appetite has improved. The norepinephrine infusion is stopped 48 hours after presentation, at which time her blood pressure is 120/70 mm Hg, heart rate 85 beats per minute and irregular, and temperature 36.7°C (98.1°F). Her current laboratory values include the following:
- Serum sodium 137 mmolL
- Serum potassium 4.3 mmol/L
- Hemoglobin 9.3 g/dL
- Serum cortisol (random) 7.2 μg/dL
- Plasma ACTH 752 pg/mL (10–60 pg/mL).
ESTABLISHING THE DIAGNOSIS OF ADRENAL INSUFFICIENCY
3. Which of the following is the most appropriate test to establish the diagnosis of adrenal insufficiency?
- 7 am total serum cortisol measurement
- Random serum cortisol measurement
- 7 am salivary cortisol measurement
- 24-hour urinary free cortisol measurement
- ACTH stimulation test for cortisol
- Insulin tolerance test for cortisol
These tests also help determine the type of adrenal insufficiency (primary, secondary, or tertiary) and guide further management. Secondary adrenal insufficiency is caused by inadequate pituitary ACTH secretion and subsequent inadequate cortisol production, whereas tertiary adrenal insufficiency is caused by inadequate hypothalamic corticotropin-releasing hormone secretion and subsequent inadequate ACTH and cortisol production. The diagnosis of adrenal insufficiency relies first on demonstrating inappropriately low total serum cortisol production. Subsequently, serum ACTH helps to differentiate between primary (high ACTH) and secondary or tertiary (low or inappropriately normal ACTH) adrenal insufficiency.
Each test listed above may demonstrate a low cortisol level. However, in a nonacute setting, safety concerns (especially regarding insulin tolerance testing), poor diagnostic value, feasibility (ie, the difficulty of 24-hour tests), and poor sensitivity of 7 am cortisol make the ACTH stimulation test the most appropriate test in clinical practice to establish the diagnosis of adrenal insufficiency.
7 am serum cortisol measurement
Measuring the serum cortisol level early in the morning in the nonacute setting could be of diagnostic value, as an extremely low value (< 3–5 μg/dL) is almost 100% specific for adrenal insufficiency in the absence of concurrent exogenous steroid intake. However, the very low cutoff for this test causes poor sensitivity (about 33%), as many patients have partial adrenal insufficiency and hence have higher serum cortisol levels that may even be in the normal physiologic range.19–22
Random serum cortisol measurements
Random serum cortisol measurements are not very useful in a nonacute setting, since cortisol levels are affected by factors such as stress and hydration status. Moreover, they fluctuate during the day in a circadian rhythm.
On the other hand, random serum cortisol is a very good test to evaluate for adrenal insufficiency in the acute setting. A random value higher than 15 to 18 μg/dL is almost always associated with adequate adrenal function and generally rules out adrenal insufficiency.11,23,24
7 am salivary cortisol measurement
The same principle applies to early morning salivary cortisol. Only extremely low values (< 2.65 ng/mL) may distinguish patients with adrenal insufficiency from healthy individuals, with 97.1% sensitivity and 93.3% specificity.25
Of note, early morning salivary cortisol is not routinely measured in most clinical practices for evaluation of adrenal function. Hence, morning serum and morning salivary cortisol are useful screening tools and have meaningful results when their values are in the extremes of the spectrum, but they are not reliable as a single test, as they may overlook patients with partial adrenal insufficiency.
Urinary cortisol measurement
Urinary cortisol measurement is not used to diagnose adrenal insufficiency, as values can be normal in patients with partial adrenal insufficiency.
The ACTH stimulation test
The ACTH stimulation test involves an intramuscular or intravenous injection of cosyntropin (a synthetic analogue of ACTH fragment 1–24 that has the full activity of native ACTH) and measuring total serum cortisol at baseline, 30 minutes, and 60 minutes to assess the response of the adrenal glands.
The test can be done using a high or low dose of cosyntropin. The Endocrine Society’s 2016 guidelines recommend the high dose (250 μg) for most patients.10 The standard high-dose stimulation test can be done at any time during the day.26 If the cosyntropin is injected intravenously, any value higher than 18 to 20 μg/dL indicates normal adrenal function and excludes adrenal insufficiency.27,28 If intramuscular injection is used, any value higher than 16 to 18 μg/dL at 30 minutes post-consyntropin excludes adrenal insufficiency.29
The ACTH stimulation test may not exclude acute secondary or tertiary adrenal insufficiency.
Insulin tolerance testing
Insulin tolerance testing remains the gold standard for diagnosing adrenal insufficiency and assessing the integrity of the pituitary-adrenal axis. However, given its difficulty to perform, safety concerns, and the availability of other reliable tests, its use in clinical practice is limited. It is nonetheless useful in assessing patients with recent onset of ACTH deficiency.30,31
CASE RESUMED: PATIENT DISCHARGED, LOST TO FOLLOW-UP
Abdominal CT without contrast is done and demonstrates bilateral adrenal hemorrhage. Thus, the patient is diagnosed with primary acute adrenal insufficiency due to adrenal necrosis.
She is started on oral hydrocortisone and fludrocortisone after intravenous hydrocortisone is discontinued. She is counseled about adhering to medications, wearing a medical alert bracelet, giving herself emergency cortisol injections, taking higher doses of hydrocortisone if she is ill, and monitoring her INR. She is discharged home after her symptoms resolve.
The patient does not keep her scheduled appointment and is lost to follow-up. She returns 2 years later complaining of fatigue and feeling unwell. She admits that she stopped taking hydrocortisone 1 year ago after reading an online article about corticosteroid side effects. She has continued to take fludrocortisone.
MINERALOCORTICOID VS CORTICOSTEROID DEFICIENCY
4. Which of the following is least likely to be present in this patient at this time?
- Intravascular volume depletion
- Hyponatremia
- Skin hyperpigmentation
- Normokalemia
- Elevated serum ACTH level
Intravascular volume depletion
Intravascular volume depletion is the least likely to be present. This is because intravascular volume depletion is mainly secondary to mineralocorticoid deficiency rather than corticosteroid deficiency, which is not present in this patient, as she is compliant with her mineralocorticoid replacement therapy.32,33 However, even with sufficient mineralocorticoid replacement, mild hypotension may be present in this patient due to corticosteroid deficiency-induced loss of vascular tone.
Hyponatremia
Hyponatremia in adrenal insufficiency is not due only to mineralocorticoid deficiency. Patients with secondary or tertiary adrenal insufficiency may also exhibit hyponatremia.34 ACTH deficiency in such patients is not expected to cause mineralocorticoid deficiency, as ACTH has only a minor role in aldosterone production.
It has been proposed that hyponatremia in secondary adrenal insufficiency is due to cortisol deficiency resulting in an increase of antidiuretic hormone secretion.35,36 The mechanisms for increased antidiuretic hormone include cortisol deficiency resulting in an increased corticotropin-releasing hormone level, which acts as an antidiuretic hormone secretagogue,37,38 and cortisol directly suppressing antidiuretic hormone secretion.39
In our patient, volume expansion and hyponatremia are expected due to increased antidiuretic hormone secretion as a result of corticosteroid insufficiency.
Hyperpigmentation
Hyperpigmentation of the skin is present only in long-standing primary adrenal insufficiency. This is due to chronic cortisol deficiency causing an increased secretion of pro-opiomelanocortin, a prohormone that is cleaved into ACTH, melanocyte-stimulating hormone, and other hormones. Melanocyte-stimulating hormone causes skin hyperpigmentation due to increased melanin synthesis.40 The hyperpigmentation is seen in sun-exposed areas, pressure areas, palmar creases, nipples, and mucous membranes.
This patient has long-standing corticosteroid deficiency due to noncompliance and primary adrenal insufficiency, and as a result she is expected to have elevated serum ACTH and hyperpigmentation.
Normokalemia
Mineralocorticoid deficiency results in hyperkalemia and metabolic acidosis by impairing renal excretion of potassium and acid.41 This patient is compliant with her mineralocorticoid replacement regimen; thus, potassium levels and pH are expected to be normal.
TAKE-HOME POINTS
- Suspect adrenal crisis in any patient who presents with shock.
- Acute abdomen or unexplained fever could be among the manifestations.
- Initial management requires liberal normal saline intravenous fluid administration to replete the intravascular space.
- Draw blood samples for serum chemistry, cortisol, and ACTH, followed immediately by intravenous hydrocortisone supplementation.
- In critically ill patients, evaluate adrenal function with random serum cortisol; in a nonacute setting use the ACTH stimulation test.
- Chronic management of primary adrenal insufficiency requires corticosteroid and mineralocorticoid therapy.
A 71-year-old woman is brought to the emergency department by her neighbor after complaining of fatigue and light-headedness for the last 8 hours. The patient lives alone and was feeling well when she woke up this morning, but then began to feel nauseated and vomited twice.
The patient appears drowsy and confused and cannot provide any further history. Her medical records show that she was seen in the cardiology clinic 6 months ago but has not kept her appointments since then.
Her medical history includes atrial fibrillation, hypertension, type 2 diabetes mellitus, and osteoarthritis. Her medications are daily warfarin, atenolol, aspirin, candesartan, and metformin, and she takes acetaminophen as needed. She is neither a smoker nor a drug user, but she drinks alcohol occasionally. Her family history is significant for her mother’s death from breast cancer at age 55.
The neighbor confirms that the patient appeared well this morning and has not had any recent illnesses except for a minor cold last week that improved over 5 days with acetaminophen only.
INITIAL EVALUATION AND MANAGEMENT
Physical examination
On physical examination, her blood pressure is 80/40 mm Hg, respiratory rate 25 breaths per minute, oral temperature 38.3°C (100.9°F), and heart rate 130 beats per minute and irregular.
Her neck veins are flat, and her chest is clear to auscultation with normal heart sounds. Abdominal palpation elicits discomfort in the middle segments, voluntary withdrawal, and abdominal wall rigidity. Her skin feels dry and cool, with decreased turgor.
Initial treatment
The patient is given 1 L of 0.9% saline intravenously over the first hour and then is transferred to the intensive care unit, where a norepinephrine drip is started to treat her ongoing hypotension. Normal saline is continued at a rate of 500 mL per hour for the next 4 hours.
Cardiac monitoring and 12-lead electrocardiography show atrial fibrillation with a rapid ventricular response of 138 beats per minute, but electrical cardioversion is not done.
Initial laboratory tests
Of note, her international normalized ratio (INR) is 6.13, while the therapeutic range for a patient taking warfarin because of atrial fibrillation is 2.0 to 3.0.
Her blood pH is 7.34 (reference range 7.35–7.45), and her bicarbonate level is 18 mmol/L (22–26); a low pH and low bicarbonate together indicate metabolic acidosis. Her sodium level is 128 mmol/L (135–145), her chloride level is 100 mmol/L (97–107), and, as mentioned, her bicarbonate level is 18 mmol/L; therefore, her anion gap is 128 – (100 + 18) = 10 mmol/L, which is normal (≤ 10).1
Her serum creatinine level is 1.3 mg/dL (0.5–1.1), and her blood urea nitrogen level is 35 mg/dL (7–20).
Her potassium level is 5.8 mmol/L, which is consistent with hyperkalemia (reference range 3.5–5.2).
DIFFERENTIAL DIAGNOSIS
1. Which of the following is the most likely cause of this patient’s symptoms?
- Adrenal crisis
- Cardiogenic shock due to decreased cardiac contractility
- Intracranial hemorrhage
- Acute abdomen due to small bowel obstruction
- Septic shock due to bacterial toxin-induced loss of vascular tone
Our patient is presenting with shock. Given our inability to obtain a meaningful history, the differential diagnosis is broad and includes all of the above.
Adrenal crisis
The sudden onset and laboratory results that include hyperkalemia, hyponatremia, and normal anion gap metabolic acidosis raise suspicion of adrenal crisis resulting in acute mineralocorticoid and glucocorticoid insufficiency.1
The patient’s elevated serum creatinine and high blood urea nitrogen-to-creatinine ratio of 26.9 (reference range 10–20) also suggest intravascular volume contraction. Her low hemoglobin level and supratherapeutic INR, possibly due to an interaction between warfarin and acetaminophen combined with poor medical follow-up, raise suspicion of acute bilateral adrenal necrosis due to hemorrhage.
Bilateral adrenal hemorrhage is one cause of adrenal crisis resulting in bilateral adrenal necrosis. Risk factors for adrenal hemorrhage include anticoagulation therapy, underlying coagulopathy, postoperative states, and certain infections such as meningococcemia and Haemophilus influenzae infection.2–5 Nevertheless, in most cases the INR is in the therapeutic range and the patient has no bleeding elsewhere.4 Other causes of adrenal necrosis include emboli, sepsis, and blunt trauma.6,7
Other causes of adrenal crisis are listed in Table 3.
Cardiogenic shock
Intracranial hemorrhage
Intracranial hemorrhage can present with a decreased level of consciousness, but it is less likely to cause hypotension, as the cranial space is limited. If massive intracranial hemorrhage would occur, the increase in intracranial pressure would more likely cause hypertension by the Cushing reflex than hypotension.
Acute abdomen
Abdominal pain and rigidity along with fever can be presenting symptoms of both adrenal insufficiency and an acute abdomen due to intestinal obstruction.4 However, intestinal obstruction typically causes a high anion gap metabolic acidosis due to lactic acidosis, instead of the normal anion gap metabolic acidosis present in this patient.8 Moreover, her deranged electrolytes, supratherapeutic INR, and absence of previous gastroenterologic conditions make adrenal crisis a more likely diagnosis.
Septic shock
Septic shock would also cause fever and hypotension as bacterial toxins induce a pyrexic response and vasodilation. However, at such an early stage of sepsis, the patient would be expected to be warm and hyperemic, whereas this patient’s skin is cool and dry due to volume depletion secondary to adrenal insufficiency.9 Sepsis would also cause a high anion gap metabolic acidosis due to lactic acidosis, as opposed to this patient’s normal anion gap metabolic acidosis. These findings, along with the metabolic derangements and the absence of a focus of infection, make sepsis a less likely possibility.
CASE CONTINUED: CARDIOMEGALY, PERSISTENT HYPOTENSION
Blood is drawn for cultures and measurement of troponins and lactic acid, and urine samples are taken for culture and biochemical analysis. Chest radiography shows mild cardiomegaly. The patient is started empirically on vancomycin and cefepime, and her warfarin is discontinued.
Five hours after presenting to the emergency department, her blood pressure remains at 80/40 mm Hg even after receiving 3 L of normal saline intravenously.
PROMPT MANAGEMENT OF ADRENAL CRISIS
2. Which of the following is the most appropriate next step in managing this patient?
- Draw samples for serum cortisol and plasma adrenocorticotropic hormone (ACTH) levels, then give hydrocortisone 100 mg intravenously
- Perform abdominal computed tomography (CT) without contrast
- Perform transthoracic echocardiography
- Increase the norepinephrine infusion
- Immediately give fludrocortisone
First give fluids
The first step in managing a patient with suspected adrenal crisis is liberal intravenous fluid administration to replenish the depleted intravascular space. The amount and choice of fluid is empiric, but a recommendation is 1 L of normal saline or dextrose 5% in normal saline, infused quickly over the first hour and then titrated according to the patient’s fluid status.10
Measure cortisol and ACTH; start corticosteroids immediately
Immediate therapy with an appropriate stress dose of intravenous corticosteroids (eg, hydrocortisone 100 mg) is essential. However, this should be done after drawing blood for cortisol and ACTH measurements.10
Do not delay corticosteroid therapy while awaiting the results of the diagnostic tests.
In addition, in the early phase of evolving primary adrenal insufficiency, measurement of plasma renin and aldosterone levels may be beneficial, as mineralocorticoid deficiency may predominate.10,12,13
One of the most important aims of early corticosteroid supplementation is to prevent further hyponatremia by reducing a reactive increase in antidiuretic hormone secretion caused by cortisol deficiency. Corticosteroids also help to restore normal blood pressure by increasing vascular tone, as glucocorticoid receptor activation potentiates the vasoconstrictor actions of norepinephrine, angiotensin II, and other vasoconstrictors.14,15
Which corticosteroid to use?
Which corticosteroid to use in previously undiagnosed adrenal insufficiency is controversial. The Endocrine Society10 and Japan Endocrine Society16 clinical practice guidelines recommend hydrocortisone in a 100-mg intravenous bolus followed by 200 mg over 24 hours.
The choice of hydrocortisone is justified by its superior mineralocorticoid activity.10,16 Further, hydrocortisone is preferred over dexamethasone if the patient is known to have primary adrenal insufficiency, or if the serum potassium level is higher than 6.0 mmol/L.
Some clinicians, on the other hand, recommend dexamethasone, given as a 4-mg intravenous bolus followed by 4-mg boluses every 12 hours. Their rationale is that dexamethasone, unlike hydrocortisone, does not interfere with subsequent serum cortisol assays if the patient later undergoes ACTH stimulation testing.17 Dexamethasone may also be preferred to minimize unwanted mineralocorticoid effects, such as in neurosurgical patients at risk of brain edema.
If hydrocortisone is used, ACTH stimulation testing can be done after withholding hydrocortisone for 24 hours once the patient is stable. (It should be restarted after the test if the results are abnormal.)
Other possible steps
Abdominal CT should be done in our patient to address the possibility of bilateral adrenal hemorrhage. However, it is preferable to wait until the patient is stabilized.
Echocardiography. Our patient is likely to have an element of cardiac failure, given her hypertension and cardiomegaly. However, decompensated heart failure is probably not the cause of her presentation. Thus, the first priority is to treat her adrenal crisis, and echocardiography should be deferred.
Increasing the norepinephrine infusion is unlikely to improve her blood pressure very much, as she is significantly volume-depleted. Further, low cortisol decreases the vascular response to norepinephrine.15
Mineralocorticoids such as fludrocortisone are used to treat primary adrenal insufficiency. However, they are not required during acute management of adrenal crisis, as 40 mg of hydrocortisone offers mineralocorticoid activity equivalent to 100 µg of fludrocortisone. Thus, the high doses of hydrocortisone used to treat adrenal crisis provide adequate mineralocorticoid therapy.10,18
If dexamethasone is used, its effect along with normal saline supplementation would be sufficient to replete the intravenous space and bring the sodium level back up to normal in the acute setting.
CASE RESUMED: IMPROVEMENT WITH HYDROCORTISONE
The patient’s blood is drawn for serum cortisol and plasma ACTH measurements. A 100-mg intravenous bolus of hydrocortisone is given, followed by a 50-mg bolus every 6 hours until the patient stabilizes.
Twenty-four hours later, the patient states that she has more energy, and her appetite has improved. The norepinephrine infusion is stopped 48 hours after presentation, at which time her blood pressure is 120/70 mm Hg, heart rate 85 beats per minute and irregular, and temperature 36.7°C (98.1°F). Her current laboratory values include the following:
- Serum sodium 137 mmolL
- Serum potassium 4.3 mmol/L
- Hemoglobin 9.3 g/dL
- Serum cortisol (random) 7.2 μg/dL
- Plasma ACTH 752 pg/mL (10–60 pg/mL).
ESTABLISHING THE DIAGNOSIS OF ADRENAL INSUFFICIENCY
3. Which of the following is the most appropriate test to establish the diagnosis of adrenal insufficiency?
- 7 am total serum cortisol measurement
- Random serum cortisol measurement
- 7 am salivary cortisol measurement
- 24-hour urinary free cortisol measurement
- ACTH stimulation test for cortisol
- Insulin tolerance test for cortisol
These tests also help determine the type of adrenal insufficiency (primary, secondary, or tertiary) and guide further management. Secondary adrenal insufficiency is caused by inadequate pituitary ACTH secretion and subsequent inadequate cortisol production, whereas tertiary adrenal insufficiency is caused by inadequate hypothalamic corticotropin-releasing hormone secretion and subsequent inadequate ACTH and cortisol production. The diagnosis of adrenal insufficiency relies first on demonstrating inappropriately low total serum cortisol production. Subsequently, serum ACTH helps to differentiate between primary (high ACTH) and secondary or tertiary (low or inappropriately normal ACTH) adrenal insufficiency.
Each test listed above may demonstrate a low cortisol level. However, in a nonacute setting, safety concerns (especially regarding insulin tolerance testing), poor diagnostic value, feasibility (ie, the difficulty of 24-hour tests), and poor sensitivity of 7 am cortisol make the ACTH stimulation test the most appropriate test in clinical practice to establish the diagnosis of adrenal insufficiency.
7 am serum cortisol measurement
Measuring the serum cortisol level early in the morning in the nonacute setting could be of diagnostic value, as an extremely low value (< 3–5 μg/dL) is almost 100% specific for adrenal insufficiency in the absence of concurrent exogenous steroid intake. However, the very low cutoff for this test causes poor sensitivity (about 33%), as many patients have partial adrenal insufficiency and hence have higher serum cortisol levels that may even be in the normal physiologic range.19–22
Random serum cortisol measurements
Random serum cortisol measurements are not very useful in a nonacute setting, since cortisol levels are affected by factors such as stress and hydration status. Moreover, they fluctuate during the day in a circadian rhythm.
On the other hand, random serum cortisol is a very good test to evaluate for adrenal insufficiency in the acute setting. A random value higher than 15 to 18 μg/dL is almost always associated with adequate adrenal function and generally rules out adrenal insufficiency.11,23,24
7 am salivary cortisol measurement
The same principle applies to early morning salivary cortisol. Only extremely low values (< 2.65 ng/mL) may distinguish patients with adrenal insufficiency from healthy individuals, with 97.1% sensitivity and 93.3% specificity.25
Of note, early morning salivary cortisol is not routinely measured in most clinical practices for evaluation of adrenal function. Hence, morning serum and morning salivary cortisol are useful screening tools and have meaningful results when their values are in the extremes of the spectrum, but they are not reliable as a single test, as they may overlook patients with partial adrenal insufficiency.
Urinary cortisol measurement
Urinary cortisol measurement is not used to diagnose adrenal insufficiency, as values can be normal in patients with partial adrenal insufficiency.
The ACTH stimulation test
The ACTH stimulation test involves an intramuscular or intravenous injection of cosyntropin (a synthetic analogue of ACTH fragment 1–24 that has the full activity of native ACTH) and measuring total serum cortisol at baseline, 30 minutes, and 60 minutes to assess the response of the adrenal glands.
The test can be done using a high or low dose of cosyntropin. The Endocrine Society’s 2016 guidelines recommend the high dose (250 μg) for most patients.10 The standard high-dose stimulation test can be done at any time during the day.26 If the cosyntropin is injected intravenously, any value higher than 18 to 20 μg/dL indicates normal adrenal function and excludes adrenal insufficiency.27,28 If intramuscular injection is used, any value higher than 16 to 18 μg/dL at 30 minutes post-consyntropin excludes adrenal insufficiency.29
The ACTH stimulation test may not exclude acute secondary or tertiary adrenal insufficiency.
Insulin tolerance testing
Insulin tolerance testing remains the gold standard for diagnosing adrenal insufficiency and assessing the integrity of the pituitary-adrenal axis. However, given its difficulty to perform, safety concerns, and the availability of other reliable tests, its use in clinical practice is limited. It is nonetheless useful in assessing patients with recent onset of ACTH deficiency.30,31
CASE RESUMED: PATIENT DISCHARGED, LOST TO FOLLOW-UP
Abdominal CT without contrast is done and demonstrates bilateral adrenal hemorrhage. Thus, the patient is diagnosed with primary acute adrenal insufficiency due to adrenal necrosis.
She is started on oral hydrocortisone and fludrocortisone after intravenous hydrocortisone is discontinued. She is counseled about adhering to medications, wearing a medical alert bracelet, giving herself emergency cortisol injections, taking higher doses of hydrocortisone if she is ill, and monitoring her INR. She is discharged home after her symptoms resolve.
The patient does not keep her scheduled appointment and is lost to follow-up. She returns 2 years later complaining of fatigue and feeling unwell. She admits that she stopped taking hydrocortisone 1 year ago after reading an online article about corticosteroid side effects. She has continued to take fludrocortisone.
MINERALOCORTICOID VS CORTICOSTEROID DEFICIENCY
4. Which of the following is least likely to be present in this patient at this time?
- Intravascular volume depletion
- Hyponatremia
- Skin hyperpigmentation
- Normokalemia
- Elevated serum ACTH level
Intravascular volume depletion
Intravascular volume depletion is the least likely to be present. This is because intravascular volume depletion is mainly secondary to mineralocorticoid deficiency rather than corticosteroid deficiency, which is not present in this patient, as she is compliant with her mineralocorticoid replacement therapy.32,33 However, even with sufficient mineralocorticoid replacement, mild hypotension may be present in this patient due to corticosteroid deficiency-induced loss of vascular tone.
Hyponatremia
Hyponatremia in adrenal insufficiency is not due only to mineralocorticoid deficiency. Patients with secondary or tertiary adrenal insufficiency may also exhibit hyponatremia.34 ACTH deficiency in such patients is not expected to cause mineralocorticoid deficiency, as ACTH has only a minor role in aldosterone production.
It has been proposed that hyponatremia in secondary adrenal insufficiency is due to cortisol deficiency resulting in an increase of antidiuretic hormone secretion.35,36 The mechanisms for increased antidiuretic hormone include cortisol deficiency resulting in an increased corticotropin-releasing hormone level, which acts as an antidiuretic hormone secretagogue,37,38 and cortisol directly suppressing antidiuretic hormone secretion.39
In our patient, volume expansion and hyponatremia are expected due to increased antidiuretic hormone secretion as a result of corticosteroid insufficiency.
Hyperpigmentation
Hyperpigmentation of the skin is present only in long-standing primary adrenal insufficiency. This is due to chronic cortisol deficiency causing an increased secretion of pro-opiomelanocortin, a prohormone that is cleaved into ACTH, melanocyte-stimulating hormone, and other hormones. Melanocyte-stimulating hormone causes skin hyperpigmentation due to increased melanin synthesis.40 The hyperpigmentation is seen in sun-exposed areas, pressure areas, palmar creases, nipples, and mucous membranes.
This patient has long-standing corticosteroid deficiency due to noncompliance and primary adrenal insufficiency, and as a result she is expected to have elevated serum ACTH and hyperpigmentation.
Normokalemia
Mineralocorticoid deficiency results in hyperkalemia and metabolic acidosis by impairing renal excretion of potassium and acid.41 This patient is compliant with her mineralocorticoid replacement regimen; thus, potassium levels and pH are expected to be normal.
TAKE-HOME POINTS
- Suspect adrenal crisis in any patient who presents with shock.
- Acute abdomen or unexplained fever could be among the manifestations.
- Initial management requires liberal normal saline intravenous fluid administration to replete the intravascular space.
- Draw blood samples for serum chemistry, cortisol, and ACTH, followed immediately by intravenous hydrocortisone supplementation.
- In critically ill patients, evaluate adrenal function with random serum cortisol; in a nonacute setting use the ACTH stimulation test.
- Chronic management of primary adrenal insufficiency requires corticosteroid and mineralocorticoid therapy.
- Mani S, Rutecki GW. A patient with altered mental status and an acid-base disturbance. Cleve Clin J Med 2017; 84(1):27–34. doi:10.3949/ccjm.84a.16042
- Almiani M, Gorthi J, Subbiah S, Firoz M. Quiz page November 2012: an unusual case of acute hyponatremia and normal anion gap metabolic acidosis. Am J Kidney Dis 2012; 60(5):xxxiii–xxxvi. doi:10.1053/j.ajkd.2012.05.026
- Migeon CJ, Kenny FM, Hung W, Voorhess ML. Study of adrenal function in children with meningitis. Pediatrics 1967; 40(2):163–183.
- Rao RH, Vagnucci AH, Amico JA. Bilateral massive adrenal hemorrhage: early recognition and treatment. Ann Intern Med 1989; 110(3):227–235.
- Shimizu S, Tahara Y, Atsumi T, et al. Waterhouse-Friderichsen syndrome caused by invasive Haemophilus influenzae type B infection in a previously healthy young man. Anaesth Intensive Care 2010; 38(1):214–215.
- Castaldo ET, Guillamondegui OD, Greco JA 3rd, Feurer ID, Miller RS, Morris JA Jr. Are adrenal injuries predictive of adrenal insufficiency in patients sustaining blunt trauma? Am Surg 2008; 74(3):262–266.
- Xarli VP, Steele AA, Davis PJ, Buescher ES, Rios CN, Garcia-Bunuel R. Adrenal hemorrhage in the adult. Medicine (Baltimore) 1978; 57(3):211–221.
- Takeuchi K, Tsuzuki Y, Ando T, et al. Clinical studies of strangulating small bowel obstruction. Am Surg 2004; 70(1):40–44.
- MacKenzie IM. The haemodynamics of human septic shock. Anaesthesia 2001; 56(2):130–144.
- Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2016; 101(2):364–389. doi:10.1210/jc.2015-1710
- Hamrahian AH, Fleseriu M; AACE Adrenal Scientific Committee. Evaluation and management of adrenal insufficiency in critically ill patients: disease state review. Endocr Pract 2017; 23(6):716–725. doi:10.4158/EP161720.RA
- Saenger P, Levine LS, Irvine WJ, et al. Progressive adrenal failure in polyglandular autoimmune disease. J Clin Endocrinol Metab 1982; 54(4):863–867.
- Coco G, Dal Pra C, Presotto F, et al. Estimated risk for developing autoimmune Addison's disease in patients with adrenal cortex autoantibodies. J Clin Endocrinol Metab 2006; 91(5):1637–1645. doi:10.1210/jc.2005-0860
- Ullian ME. The role of corticosteroids in the regulation of vascular tone. Cardiovasc Res 1999; 41(1):55–64.
- Yang S, Zhang L. Glucocorticoids and vascular reactivity. Curr Vasc Pharmacol 2004; 2(1):1–12.
- Yanase T, Tajima T, Katabami T, et al. Diagnosis and treatment of adrenal insufficiency including adrenal crisis: a Japan Endocrine Society clinical practice guideline [Opinion]. Endocr J 2016; 63(9):765–784. doi:10.1507/endocrj.EJ16-0242
- Taylor RL, Grebe SK, Singh RJ. Quantitative, highly sensitive liquid chromatography-tandem mass spectrometry method for detection of synthetic corticosteroids. Clin Chem 2004; 50(10):2345–2352. doi:10.1373/clinchem.2004.033605
- Goldfien A, Laidlaw JC, Haydar NA, Renold AE, Thorn GW. Fluorohydrocortisone and chlorohydrocortisone, highly potent derivatives of compound F. N Engl J Med 1955; 252(11):415–421. doi:10.1056/NEJM195503172521101
- Jenkins D, Forsham PH, Laidlaw JC, Reddy WJ, Thorn GW. Use of ACTH in the diagnosis of adrenal cortical insufficiency. Am J Med 1955; 18(1):3–14.
- Hägg E, Asplund K, Lithner F. Value of basal plasma cortisol assays in the assessment of pituitary-adrenal insufficiency. Clin Endocrinol (Oxf) 1987; 26(2):221–226.
- Deutschbein T, Unger N, Mann K, Petersenn S. Diagnosis of secondary adrenal insufficiency: unstimulated early morning cortisol in saliva and serum in comparison with the insulin tolerance test. Horm Metab Res 2009; 41(4):834–839. doi:10.1055/s-0029-1225630
- Erturk E, Jaffe CA, Barkan AL. Evaluation of the integrity of the hypothalamic-pituitary-adrenal axis by insulin hypoglycemia test. J Clin Endocrinol Metab 1998; 83(7):2350–2354.
- Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med 2003; 348(8):727–734. doi:10.1056/NEJMra020529
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 2013; 39(2):165–228. doi:10.1007/s00134-012-2769-8
- Ceccato F, Barbot M, Zilio M, et al. Performance of salivary cortisol in the diagnosis of Cushing's syndrome, adrenal incidentaloma, and adrenal insufficiency. Eur J Endocrinol 2013; 169(1):31–36. doi:10.1530/EJE-13-0159
- Dickstein G, Shechner C, Nicholson WE, et al. Adrenocorticotropin stimulation test: effects of basal cortisol level, time of day, and suggested new sensitive low dose test. J Clin Endocrinol Metab 1991; 72(4):773–778. doi:10.1210/jcem-72-4-773
- May ME, Carey RM. Rapid adrenocorticotropic hormone test in practice. Retrospective review. Am J Med 1985; 79(6):679–884.
- Speckart PF, Nicoloff JT, Bethune JE. Screening for adrenocortical insufficiency with cosyntropin (synthetic ACTH). Arch Intern Med 1971; 128(5):761–763.
- Peechakara S, Bena J, Clarke NJ, et al. Total and free cortisol levels during 1 μg, 25 μg, and 250 μg cosyntropin stimulation tests compared to insulin tolerance test: results of a randomized, prospective, pilot study. Endocrine 2017; 57(3):388–393. doi:10.1007/s12020-017-1371-9
- Finucane FM, Liew A, Thornton E, Rogers B, Tormey W, Agha A. Clinical insights into the safety and utility of the insulin tolerance test (ITT) in the assessment of the hypothalamo-pituitary-adrenal axis. Clin Endocrinol (Oxf) 2008; 69(4):603–607. doi:10.1111/j.1365-2265.2008.03240.x
- Lindholm J, Kehlet H. Re-evaluation of the clinical value of the 30 min ACTH test in assessing the hypothalamic-pituitary-adrenocortical function. Clin Endocrinol (Oxf) 1987; 26(1):53–59.
- Charmandari E, Nicolaides NC, Chrousos GP. Adrenal insufficiency. Lancet 2014; 383(9935):2152–2167. doi:10.1016/S0140-6736(13)61684-0
- Burke CW. Adrenocortical insufficiency. Clin Endocrinol Metab 1985; 14(4):947–976.
- Jessani N, Jehangir W, Behman D, Yousif A, Spiler IJ. Secondary adrenal insufficiency: an overlooked cause of hyponatremia. J Clin Med Res 2015; 7(4):286–288. doi:10.14740/jocmr2041w
- Oelkers W. Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism. N Engl J Med 1989; 321(8):492–496. doi:10.1056/NEJM198908243210802
- Ishikawa S, Schrier RW. Effect of arginine vasopressin antagonist on renal water excretion in glucocorticoid and mineralocorticoid deficient rats. Kidney Int 1982; 22(6):587–593.
- Wolfson B, Manning RW, Davis LG, Arentzen R, Baldino F Jr. Co-localization of corticotropin releasing factor and vasopressin mRNA in neurones after adrenalectomy. Nature 1985; 315(6014):59–61.
- Kalogeras KT, Nieman LK, Friedman TC, et al. Inferior petrosal sinus sampling in healthy subjects reveals a unilateral corticotropin-releasing hormone-induced arginine vasopressin release associated with ipsilateral adrenocorticotropin secretion. J Clin Invest 1996; 97:2045–2050.
- Kovacs KJ, Foldes A, Sawchenko PE. Glucocorticoid negative feedback selectively targets vasopressin transcription in parvocellular neurosecretory neurons. J Neurosci 2000; 20:3843–3852.
- Sarkar SB, Sarkar S, Ghosh S, Bandyopadhyay S. Addison's disease. Contemp Clin Dent 2012; 3(4):484–486. doi:10.4103/0976-237X.107450
- Szylman P, Better OS, Chaimowitz C, Rosler A. Role of hyperkalemia in the metabolic acidosis of isolated hypoaldosteronism. N Engl J Med 1976; 294(7):361–365. doi:10.1056/NEJM197602122940703
- Mani S, Rutecki GW. A patient with altered mental status and an acid-base disturbance. Cleve Clin J Med 2017; 84(1):27–34. doi:10.3949/ccjm.84a.16042
- Almiani M, Gorthi J, Subbiah S, Firoz M. Quiz page November 2012: an unusual case of acute hyponatremia and normal anion gap metabolic acidosis. Am J Kidney Dis 2012; 60(5):xxxiii–xxxvi. doi:10.1053/j.ajkd.2012.05.026
- Migeon CJ, Kenny FM, Hung W, Voorhess ML. Study of adrenal function in children with meningitis. Pediatrics 1967; 40(2):163–183.
- Rao RH, Vagnucci AH, Amico JA. Bilateral massive adrenal hemorrhage: early recognition and treatment. Ann Intern Med 1989; 110(3):227–235.
- Shimizu S, Tahara Y, Atsumi T, et al. Waterhouse-Friderichsen syndrome caused by invasive Haemophilus influenzae type B infection in a previously healthy young man. Anaesth Intensive Care 2010; 38(1):214–215.
- Castaldo ET, Guillamondegui OD, Greco JA 3rd, Feurer ID, Miller RS, Morris JA Jr. Are adrenal injuries predictive of adrenal insufficiency in patients sustaining blunt trauma? Am Surg 2008; 74(3):262–266.
- Xarli VP, Steele AA, Davis PJ, Buescher ES, Rios CN, Garcia-Bunuel R. Adrenal hemorrhage in the adult. Medicine (Baltimore) 1978; 57(3):211–221.
- Takeuchi K, Tsuzuki Y, Ando T, et al. Clinical studies of strangulating small bowel obstruction. Am Surg 2004; 70(1):40–44.
- MacKenzie IM. The haemodynamics of human septic shock. Anaesthesia 2001; 56(2):130–144.
- Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2016; 101(2):364–389. doi:10.1210/jc.2015-1710
- Hamrahian AH, Fleseriu M; AACE Adrenal Scientific Committee. Evaluation and management of adrenal insufficiency in critically ill patients: disease state review. Endocr Pract 2017; 23(6):716–725. doi:10.4158/EP161720.RA
- Saenger P, Levine LS, Irvine WJ, et al. Progressive adrenal failure in polyglandular autoimmune disease. J Clin Endocrinol Metab 1982; 54(4):863–867.
- Coco G, Dal Pra C, Presotto F, et al. Estimated risk for developing autoimmune Addison's disease in patients with adrenal cortex autoantibodies. J Clin Endocrinol Metab 2006; 91(5):1637–1645. doi:10.1210/jc.2005-0860
- Ullian ME. The role of corticosteroids in the regulation of vascular tone. Cardiovasc Res 1999; 41(1):55–64.
- Yang S, Zhang L. Glucocorticoids and vascular reactivity. Curr Vasc Pharmacol 2004; 2(1):1–12.
- Yanase T, Tajima T, Katabami T, et al. Diagnosis and treatment of adrenal insufficiency including adrenal crisis: a Japan Endocrine Society clinical practice guideline [Opinion]. Endocr J 2016; 63(9):765–784. doi:10.1507/endocrj.EJ16-0242
- Taylor RL, Grebe SK, Singh RJ. Quantitative, highly sensitive liquid chromatography-tandem mass spectrometry method for detection of synthetic corticosteroids. Clin Chem 2004; 50(10):2345–2352. doi:10.1373/clinchem.2004.033605
- Goldfien A, Laidlaw JC, Haydar NA, Renold AE, Thorn GW. Fluorohydrocortisone and chlorohydrocortisone, highly potent derivatives of compound F. N Engl J Med 1955; 252(11):415–421. doi:10.1056/NEJM195503172521101
- Jenkins D, Forsham PH, Laidlaw JC, Reddy WJ, Thorn GW. Use of ACTH in the diagnosis of adrenal cortical insufficiency. Am J Med 1955; 18(1):3–14.
- Hägg E, Asplund K, Lithner F. Value of basal plasma cortisol assays in the assessment of pituitary-adrenal insufficiency. Clin Endocrinol (Oxf) 1987; 26(2):221–226.
- Deutschbein T, Unger N, Mann K, Petersenn S. Diagnosis of secondary adrenal insufficiency: unstimulated early morning cortisol in saliva and serum in comparison with the insulin tolerance test. Horm Metab Res 2009; 41(4):834–839. doi:10.1055/s-0029-1225630
- Erturk E, Jaffe CA, Barkan AL. Evaluation of the integrity of the hypothalamic-pituitary-adrenal axis by insulin hypoglycemia test. J Clin Endocrinol Metab 1998; 83(7):2350–2354.
- Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med 2003; 348(8):727–734. doi:10.1056/NEJMra020529
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 2013; 39(2):165–228. doi:10.1007/s00134-012-2769-8
- Ceccato F, Barbot M, Zilio M, et al. Performance of salivary cortisol in the diagnosis of Cushing's syndrome, adrenal incidentaloma, and adrenal insufficiency. Eur J Endocrinol 2013; 169(1):31–36. doi:10.1530/EJE-13-0159
- Dickstein G, Shechner C, Nicholson WE, et al. Adrenocorticotropin stimulation test: effects of basal cortisol level, time of day, and suggested new sensitive low dose test. J Clin Endocrinol Metab 1991; 72(4):773–778. doi:10.1210/jcem-72-4-773
- May ME, Carey RM. Rapid adrenocorticotropic hormone test in practice. Retrospective review. Am J Med 1985; 79(6):679–884.
- Speckart PF, Nicoloff JT, Bethune JE. Screening for adrenocortical insufficiency with cosyntropin (synthetic ACTH). Arch Intern Med 1971; 128(5):761–763.
- Peechakara S, Bena J, Clarke NJ, et al. Total and free cortisol levels during 1 μg, 25 μg, and 250 μg cosyntropin stimulation tests compared to insulin tolerance test: results of a randomized, prospective, pilot study. Endocrine 2017; 57(3):388–393. doi:10.1007/s12020-017-1371-9
- Finucane FM, Liew A, Thornton E, Rogers B, Tormey W, Agha A. Clinical insights into the safety and utility of the insulin tolerance test (ITT) in the assessment of the hypothalamo-pituitary-adrenal axis. Clin Endocrinol (Oxf) 2008; 69(4):603–607. doi:10.1111/j.1365-2265.2008.03240.x
- Lindholm J, Kehlet H. Re-evaluation of the clinical value of the 30 min ACTH test in assessing the hypothalamic-pituitary-adrenocortical function. Clin Endocrinol (Oxf) 1987; 26(1):53–59.
- Charmandari E, Nicolaides NC, Chrousos GP. Adrenal insufficiency. Lancet 2014; 383(9935):2152–2167. doi:10.1016/S0140-6736(13)61684-0
- Burke CW. Adrenocortical insufficiency. Clin Endocrinol Metab 1985; 14(4):947–976.
- Jessani N, Jehangir W, Behman D, Yousif A, Spiler IJ. Secondary adrenal insufficiency: an overlooked cause of hyponatremia. J Clin Med Res 2015; 7(4):286–288. doi:10.14740/jocmr2041w
- Oelkers W. Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism. N Engl J Med 1989; 321(8):492–496. doi:10.1056/NEJM198908243210802
- Ishikawa S, Schrier RW. Effect of arginine vasopressin antagonist on renal water excretion in glucocorticoid and mineralocorticoid deficient rats. Kidney Int 1982; 22(6):587–593.
- Wolfson B, Manning RW, Davis LG, Arentzen R, Baldino F Jr. Co-localization of corticotropin releasing factor and vasopressin mRNA in neurones after adrenalectomy. Nature 1985; 315(6014):59–61.
- Kalogeras KT, Nieman LK, Friedman TC, et al. Inferior petrosal sinus sampling in healthy subjects reveals a unilateral corticotropin-releasing hormone-induced arginine vasopressin release associated with ipsilateral adrenocorticotropin secretion. J Clin Invest 1996; 97:2045–2050.
- Kovacs KJ, Foldes A, Sawchenko PE. Glucocorticoid negative feedback selectively targets vasopressin transcription in parvocellular neurosecretory neurons. J Neurosci 2000; 20:3843–3852.
- Sarkar SB, Sarkar S, Ghosh S, Bandyopadhyay S. Addison's disease. Contemp Clin Dent 2012; 3(4):484–486. doi:10.4103/0976-237X.107450
- Szylman P, Better OS, Chaimowitz C, Rosler A. Role of hyperkalemia in the metabolic acidosis of isolated hypoaldosteronism. N Engl J Med 1976; 294(7):361–365. doi:10.1056/NEJM197602122940703
Single-Dose Niacin-Induced Hepatitis
Niacin, also known as vitamin B3, is an important cofactor in many metabolic processes necessary to life. Over the past 15 to 20 years, niacin has been prescribed to patients with hyperlipidemia to increase high-density lipoprotein and lower low-density lipoprotein.1 As a naturally occurring vitamin, niacin is also available over-the-counter (OTC) as a dietary supplement, and is also a common ingredient in energy drinks and multivitamins.2
In addition to treating hyperlipidemia and as a nutritional supplement, some anecdotal reports amongst lay-persons suggests that niacin offers other health benefits, such as promoting weight loss and expediting the elimination of alcohol and illicit drugs from one’s system (eg, marijuana).3,4 The increased use of niacin supplementation in the general population for all of the aforementioned reasons has resulted in an increased incidence of niacin toxicity.
Formulations
Niacin is available in three formulations: extended-release (ER, also referred to as intermediate-release), immediate-release (IR), and sustained-release (SR).
The ER formulations of niacin are typically prescribed to treat hyperlipidemia. Patients are usually started on ER niacin at an initial dose of 250 mg once daily. The dose is gradually increased, as tolerated or necessary, to 2 g per day, taken in three doses. It is not uncommon for patients with hyperlipidemia to take more than 1 g of niacin per day after titration by their primary physicians.
Side Effects
Since niacin increases the release of arachidonic acid from cell membranes that metabolizes into prostaglandins, specifically prostaglandins E2 and D2, many patients taking niacin experience uncomfortable flushing and itching.5 Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent this side effect by inhibiting the metabolism of arachidonic acid into those vasodilatory prostaglandins. The newer ER and SR formulations of niacin, which are approved for OTC use as a dietary supplement, are less likely to cause flushing.5
Extended-release niacin, however, is associated with a higher incidence of hepatotoxicity than the other prescription formulations of niacin.6 Toxicity has been well recognized in patients taking niacin chronically for hyperlipidemia, with reports of such cases dating back to the 1980s.7,8 We report a unique case of niacin toxicity following a single-dose ingestion in a young man.
Case
A 22-year-old man presented to the ED for evaluation of a 2-week history of intermittent periumbilical abdominal pain. This visit represented the patient’s second visit to the ED over the past week for the same complaint.
Upon presentation the patient’s vital signs were: blood pressure (BP), 113/64 mm Hg; heart rate, 82 beats/min; respiratory rate, 16 breaths/min; and temperature 36.6°C. Oxygen saturation was 100% on room air. The patient was otherwise healthy and had no significant recent or remote medical history. He denied taking any medications prior to his initial presentation, and reported only occasional alcohol use.
At the patient’s initial presentation 1 week earlier, he was diagnosed with acute gastroenteritis and treated with famotidine and ondansetron in the ED. The patient appeared well clinically at this visit, and laboratory values were within normal limits, including normal blood glucose and urinalysis.
The patient was discharged home from this first visit with prescriptions of famotidine and ondansetron, and was advised to follow up with his primary care physician in 1 week. Throughout the week after discharge from the ED, the patient experienced worsening abdominal pain, and he developed frequent nonbloody emesis, prompting his second presentation to the ED. At this second visit, the patient stated that he had taken one dose of ondansetron at home, without effect. He also noted subjective fevers, but had no diarrhea or melena.
Vital signs remained within normal limits with BPs ranging from 115 to 130 mm Hg systolic and 50 to 89 mm Hg diastolic. The patient was never tachycardic, tachypneic, febrile, or hypoxic. Physical examination was remarkable for periumbilical tenderness. The patient had no jaundice. A more thorough laboratory evaluation revealed elevated anion gap and blood urea nitrogen/creatinine values, and leukocytosis. The patient’s hepatic enzymes were also elevated, with aspartate aminotransferase (AST) over 2,000 U/L and alanine aminotransferase (ALT) of 1,698 U/L. Lipase, bilirubin, and alkaline phosphatase were all within normal limits. The patient’s prothrombin time (PT) was elevated at 14 seconds, and the international normalized ratio (INR) was elevated at 1.28. Laboratory analysis for acetaminophen and alcohol was negative.
A computed tomography (CT) scan of the abdomen/pelvis with intravenous (IV) contrast was unremarkable, demonstrating a liver devoid of any masses, portal or biliary dilation, or cirrhotic changes.
The patient received IV famotidine and ondansetron, and morphine for pain control, and was admitted to the general medical floor for hepatitis of uncertain etiology. A viral hepatitis panel was negative.
On the recommendation of the toxicology service, the patient was given N-acetylcysteine (NAC), and his hepatic enzymes trended down to an AST of 642 U/L and an ALT of 456 U/L by hospital day 2. (The patient essentially completed a positive dechallenge test).9
A gastroenterology consult was ordered, during which additional history-taking and chart review noted that the patient admitted to taking one or two tablets of OTC niacin as a dietary supplement the day before his initial presentation. Although the patient could not recall the exact dosage, he stated that he had been taking supplemental niacin approximately once a month over the past several years without any issues. Since OTC niacin is most commonly available in 500-mg tablets, this suggested the patient’s recent one-time ingested dose was approximately 500 to 1,000 mg.
Based on the patient’s admission to niacin use, additional studies were ordered, including an abdominal ultrasound and a urine drug screen. Ultrasound findings were unremarkable for portal venous thrombosis. The urine drug screen, however, was positive for marijuana and opiates. While the patient denied any history of opioid use, the positive opiate assay could have been attributed to the morphine given in the ED.
Throughout the patient’s hospital course, he remained normotensive and had no change in mental status. His liver enzymes, PT, and INR continued to normalize, and he was discharged home after 3 days, with instructions to follow up with the gastrointestinal clinic within 11 days. An appointment was made for him, which he did not attend.
Given the patient’s negative autoimmune and viral workup, and rapid resolution of symptoms after discontinuing niacin use, it is believed that he had an acute drug-induced hepatitis due to niacin ingestion. Regarding any coingestants that could have contributed to the hepatitis, the patient denied taking other common coingestants such as alcohol and acetaminophen; this assertion was supported by laboratory results.
Since we were unable to attain a qualitative measurement of the patient’s niacin concentration, our diagnosis was primarily based on the patient’s reported history.10 It is possible the patient had been taking more niacin than that to which he admitted, or that he was taking another hepatotoxic substance not detected on our toxicology workup. As previously noted, there are many medications and/or dietary supplements that could cause or contribute to a synergistic effect of drug-induced hepatitis for which the patient was not tested at his initial presentation. The patient could have co-ingested this large dose of niacin with acetaminophen and/or other supplements, energy drinks, or alcohol. A combination such as this could have contributed to his hepatitis, and the metabolites of these other substances would have been eliminated by the time of his second ED presentation.
Discussion
There are over 900 different drugs, toxins, and supplements known to cause hepatic injury.11,12 Clinical manifestations of toxicity range from asymptomatic incidental elevations in transaminases to fulminant liver failure causing mortality. Ingestion of commonly used medications such as statins (although not in overdose quantities) can cause transient asymptomatic transaminitis.13 These elevations are usually mild—ie, less than twice the upper limit of normal. Patients who experience such elevations can usually continue to take the medications with frequent and vigilant monitoring of hepatic function.
Signs and Symptoms
Acute Liver Injury. Acute liver injury is diagnosed when AST and ALT levels are greater than twice the upper limit of normal. Patients also typically have mild-to-moderate abdominal findings, such as pain, nausea, and vomiting—as was experienced by our patient. Along with niacin, angiotensin-converting enzyme inhibitors, NSAIDs, and antifungal medications are examples of other medications that can cause this degree of drug-induced hepatitis.
Severe Liver Injury. Severe liver injury features elevations in not only AST and ALT, but also alkaline phosphate and bilirubin. Patients with severe hepatic injury appear clinically ill and may exhibit altered mental status and jaundice. This type of subfulminant hepatic failure commonly results from acetaminophen toxicity, anesthetic gases, iron toxicity, phosphorus toxicity, and cocaine toxicity. Examples of drugs that result in massive liver necrosis and fulminant hepatitis are acetaminophen, isoniazid, phenelzine, phenytoin, propylthiouracil, and sertraline. Patients with massive hepatic necrosis and hepatitis may require liver transplantation.
Etiology
Identifying the etiology of liver injury is made largely through the patient’s history because there are simply too many possible hepatotoxic agents to test for them all. Diagnostic suspicion of hepatic toxicity should be increased with signs of more serious disease; however, drug-induced liver injury should be included in the differential diagnosis for all cases of abdominal pain.
With respect to the patient in our case, obtaining a more complete history involving supplement and vitamin use would have allowed us to make the diagnosis in the ED. Unfortunately, these subtle aspects of a patient’s history are often overlooked in the emergent care setting.
Treatment
The treatment of niacin-induced liver injury is similar to the guidelines for treating most other drug-induced pathology.14 Removal of the offending agent and providing supportive care is the primary treatment modality.15 In addition, it is important that the clinician exclude and rule-out other causes of hepatitis such as those of viral, autoimmune, or ischemic etiology.
N-acetylcysteine. A medication classically used in patients with acetaminophen overdose, NAC is a safe and effective treatment for non-acetaminophen-induced liver injury, and was given to treat our patient.16L-carnitine. L-carnitine has been shown to be effective in cases of chronic steatosis from hepatitis C and in valproic acid induced hepatitis.17Since L-carnitine is not included on our hospital’s formulary, it was not a treatment option for our patient.Glucocorticoid Therapy. Although glucocorticoids are occasionally given to patients with systemic symptoms of drug reactions, its effectiveness has not been adequately studied.18
Prognosis
The prognosis of patients with acute drug-induced hepatitis is generally good, and most patients fully recover once the offending agent is removed. Poor prognostic factors include the presence of jaundice, requirement for dialysis, underlying chronic liver conditions, or elevated serum creatinine. While most patients will experience a complete recovery, approximately 5% to 10% will develop chronic hepatitis and/or cirrhosis.
Conclusion
Niacin is now available as prescription and OTC formulations and is a potentially hepatotoxic medication and dietary supplement. Niacin can cause an acute hepatitis, especially when taken in conjunction with other hepatotoxic substances. Drug-induced liver injury from niacin ingestion will improve quickly following removal, and the prognosis in otherwise healthy individuals is good.
Patients, especially young, healthy patients who present with symptoms concerning for hepatitis, should be asked specifically about any nutritional, herbal, or other supplement usage. During the history intake, many patients do not consider vitamins or other nutritional or herbal supplements as “medication” or as being significant, and only report prescription and OTC medications.
1. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969-2989. doi:10.1210/jc.2011-3213.
2. Vivekanandarajah A, Ni S, Waked A. Acute hepatitis in a woman following excessive ingestion of an energy drink: a case report. J Med Case Rep. 2011;5:227. doi:10.1186/1752-1947-5-227.
3. Niacin. U.S. National Library of Medicine. https://medlineplus.gov/druginfo/meds/a682518.html. Updated July 15, 2017. Accessed February 21, 2018.
4. Addiction Resource. Niacin flush for drug detox. https://addictionresource.com/drug-testing/niacin-drug-test/. Accessed February 20, 2018.
5. Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract. 2009;63(9):1369-1377. doi:10.1111/j.1742-1241.2009.02099.x.
6. Etchason JA, Miller TD, Squires RW, et al. Niacin-induced hepatitis: a potential side effect with low-dose time-release niacin. Mayo Clin Proc. 1991;66(1):23-28.
7. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J. 1983;76(2):239-241.
8. Ferenchick G, Rovner D. Hepatitis and hematemesis complicating nicotinic acid use. Am J Med Sci. 1989;298(3):191-193.
9. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA. 1990;264(2):241-243.
10. Barritt AS 4th, Lee J, Hayashi PH. Detective work in drug-induced liver injury: sometimes it is all about interviewing the right witness. Clin Gastroenterol Hepatol. 2010;8(7):635-637. doi:10.1016/j.cgh.2010.03.020.
11. Chitturi S, Teoh NC, Farrell GC. Hepatic drug metabolism and liver disease caused by drugs. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:1442-1477.
12. National Institutes of Health. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Niacin. https://livertox.nih.gov/Niacin.htm. Updated January 18, 2018. Accessed February 21, 2018.
13. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41(4):690-695.
14. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966. doi:10.1038/ajg.2014.131.
15. Larson AM. Hepatotoxicity due to herbal medication and dietary supplements. UpToDate Web site. https://www.uptodate.com/contents/hepatotoxicity-due-to-herbal-medications-and-dietary-supplements?source=search_result&search=niacin%20induced%20hepatitis&selectedTitle=3~150. Updated December 21, 2017. Accessed February 21, 2018.
16. Mumtaz K, Azam Z, Hamid S, et al. Role of N-acetylcysteine in adults with non-acetaminophen-induced acute liver failure in a center without the facility of liver transplantation. Hepatol Int. 2009;3(4):563-570. doi:10.1007/s12072-009-9151-0.
17. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293. doi:10.1345/aph.1P135.
18. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439-445.
Niacin, also known as vitamin B3, is an important cofactor in many metabolic processes necessary to life. Over the past 15 to 20 years, niacin has been prescribed to patients with hyperlipidemia to increase high-density lipoprotein and lower low-density lipoprotein.1 As a naturally occurring vitamin, niacin is also available over-the-counter (OTC) as a dietary supplement, and is also a common ingredient in energy drinks and multivitamins.2
In addition to treating hyperlipidemia and as a nutritional supplement, some anecdotal reports amongst lay-persons suggests that niacin offers other health benefits, such as promoting weight loss and expediting the elimination of alcohol and illicit drugs from one’s system (eg, marijuana).3,4 The increased use of niacin supplementation in the general population for all of the aforementioned reasons has resulted in an increased incidence of niacin toxicity.
Formulations
Niacin is available in three formulations: extended-release (ER, also referred to as intermediate-release), immediate-release (IR), and sustained-release (SR).
The ER formulations of niacin are typically prescribed to treat hyperlipidemia. Patients are usually started on ER niacin at an initial dose of 250 mg once daily. The dose is gradually increased, as tolerated or necessary, to 2 g per day, taken in three doses. It is not uncommon for patients with hyperlipidemia to take more than 1 g of niacin per day after titration by their primary physicians.
Side Effects
Since niacin increases the release of arachidonic acid from cell membranes that metabolizes into prostaglandins, specifically prostaglandins E2 and D2, many patients taking niacin experience uncomfortable flushing and itching.5 Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent this side effect by inhibiting the metabolism of arachidonic acid into those vasodilatory prostaglandins. The newer ER and SR formulations of niacin, which are approved for OTC use as a dietary supplement, are less likely to cause flushing.5
Extended-release niacin, however, is associated with a higher incidence of hepatotoxicity than the other prescription formulations of niacin.6 Toxicity has been well recognized in patients taking niacin chronically for hyperlipidemia, with reports of such cases dating back to the 1980s.7,8 We report a unique case of niacin toxicity following a single-dose ingestion in a young man.
Case
A 22-year-old man presented to the ED for evaluation of a 2-week history of intermittent periumbilical abdominal pain. This visit represented the patient’s second visit to the ED over the past week for the same complaint.
Upon presentation the patient’s vital signs were: blood pressure (BP), 113/64 mm Hg; heart rate, 82 beats/min; respiratory rate, 16 breaths/min; and temperature 36.6°C. Oxygen saturation was 100% on room air. The patient was otherwise healthy and had no significant recent or remote medical history. He denied taking any medications prior to his initial presentation, and reported only occasional alcohol use.
At the patient’s initial presentation 1 week earlier, he was diagnosed with acute gastroenteritis and treated with famotidine and ondansetron in the ED. The patient appeared well clinically at this visit, and laboratory values were within normal limits, including normal blood glucose and urinalysis.
The patient was discharged home from this first visit with prescriptions of famotidine and ondansetron, and was advised to follow up with his primary care physician in 1 week. Throughout the week after discharge from the ED, the patient experienced worsening abdominal pain, and he developed frequent nonbloody emesis, prompting his second presentation to the ED. At this second visit, the patient stated that he had taken one dose of ondansetron at home, without effect. He also noted subjective fevers, but had no diarrhea or melena.
Vital signs remained within normal limits with BPs ranging from 115 to 130 mm Hg systolic and 50 to 89 mm Hg diastolic. The patient was never tachycardic, tachypneic, febrile, or hypoxic. Physical examination was remarkable for periumbilical tenderness. The patient had no jaundice. A more thorough laboratory evaluation revealed elevated anion gap and blood urea nitrogen/creatinine values, and leukocytosis. The patient’s hepatic enzymes were also elevated, with aspartate aminotransferase (AST) over 2,000 U/L and alanine aminotransferase (ALT) of 1,698 U/L. Lipase, bilirubin, and alkaline phosphatase were all within normal limits. The patient’s prothrombin time (PT) was elevated at 14 seconds, and the international normalized ratio (INR) was elevated at 1.28. Laboratory analysis for acetaminophen and alcohol was negative.
A computed tomography (CT) scan of the abdomen/pelvis with intravenous (IV) contrast was unremarkable, demonstrating a liver devoid of any masses, portal or biliary dilation, or cirrhotic changes.
The patient received IV famotidine and ondansetron, and morphine for pain control, and was admitted to the general medical floor for hepatitis of uncertain etiology. A viral hepatitis panel was negative.
On the recommendation of the toxicology service, the patient was given N-acetylcysteine (NAC), and his hepatic enzymes trended down to an AST of 642 U/L and an ALT of 456 U/L by hospital day 2. (The patient essentially completed a positive dechallenge test).9
A gastroenterology consult was ordered, during which additional history-taking and chart review noted that the patient admitted to taking one or two tablets of OTC niacin as a dietary supplement the day before his initial presentation. Although the patient could not recall the exact dosage, he stated that he had been taking supplemental niacin approximately once a month over the past several years without any issues. Since OTC niacin is most commonly available in 500-mg tablets, this suggested the patient’s recent one-time ingested dose was approximately 500 to 1,000 mg.
Based on the patient’s admission to niacin use, additional studies were ordered, including an abdominal ultrasound and a urine drug screen. Ultrasound findings were unremarkable for portal venous thrombosis. The urine drug screen, however, was positive for marijuana and opiates. While the patient denied any history of opioid use, the positive opiate assay could have been attributed to the morphine given in the ED.
Throughout the patient’s hospital course, he remained normotensive and had no change in mental status. His liver enzymes, PT, and INR continued to normalize, and he was discharged home after 3 days, with instructions to follow up with the gastrointestinal clinic within 11 days. An appointment was made for him, which he did not attend.
Given the patient’s negative autoimmune and viral workup, and rapid resolution of symptoms after discontinuing niacin use, it is believed that he had an acute drug-induced hepatitis due to niacin ingestion. Regarding any coingestants that could have contributed to the hepatitis, the patient denied taking other common coingestants such as alcohol and acetaminophen; this assertion was supported by laboratory results.
Since we were unable to attain a qualitative measurement of the patient’s niacin concentration, our diagnosis was primarily based on the patient’s reported history.10 It is possible the patient had been taking more niacin than that to which he admitted, or that he was taking another hepatotoxic substance not detected on our toxicology workup. As previously noted, there are many medications and/or dietary supplements that could cause or contribute to a synergistic effect of drug-induced hepatitis for which the patient was not tested at his initial presentation. The patient could have co-ingested this large dose of niacin with acetaminophen and/or other supplements, energy drinks, or alcohol. A combination such as this could have contributed to his hepatitis, and the metabolites of these other substances would have been eliminated by the time of his second ED presentation.
Discussion
There are over 900 different drugs, toxins, and supplements known to cause hepatic injury.11,12 Clinical manifestations of toxicity range from asymptomatic incidental elevations in transaminases to fulminant liver failure causing mortality. Ingestion of commonly used medications such as statins (although not in overdose quantities) can cause transient asymptomatic transaminitis.13 These elevations are usually mild—ie, less than twice the upper limit of normal. Patients who experience such elevations can usually continue to take the medications with frequent and vigilant monitoring of hepatic function.
Signs and Symptoms
Acute Liver Injury. Acute liver injury is diagnosed when AST and ALT levels are greater than twice the upper limit of normal. Patients also typically have mild-to-moderate abdominal findings, such as pain, nausea, and vomiting—as was experienced by our patient. Along with niacin, angiotensin-converting enzyme inhibitors, NSAIDs, and antifungal medications are examples of other medications that can cause this degree of drug-induced hepatitis.
Severe Liver Injury. Severe liver injury features elevations in not only AST and ALT, but also alkaline phosphate and bilirubin. Patients with severe hepatic injury appear clinically ill and may exhibit altered mental status and jaundice. This type of subfulminant hepatic failure commonly results from acetaminophen toxicity, anesthetic gases, iron toxicity, phosphorus toxicity, and cocaine toxicity. Examples of drugs that result in massive liver necrosis and fulminant hepatitis are acetaminophen, isoniazid, phenelzine, phenytoin, propylthiouracil, and sertraline. Patients with massive hepatic necrosis and hepatitis may require liver transplantation.
Etiology
Identifying the etiology of liver injury is made largely through the patient’s history because there are simply too many possible hepatotoxic agents to test for them all. Diagnostic suspicion of hepatic toxicity should be increased with signs of more serious disease; however, drug-induced liver injury should be included in the differential diagnosis for all cases of abdominal pain.
With respect to the patient in our case, obtaining a more complete history involving supplement and vitamin use would have allowed us to make the diagnosis in the ED. Unfortunately, these subtle aspects of a patient’s history are often overlooked in the emergent care setting.
Treatment
The treatment of niacin-induced liver injury is similar to the guidelines for treating most other drug-induced pathology.14 Removal of the offending agent and providing supportive care is the primary treatment modality.15 In addition, it is important that the clinician exclude and rule-out other causes of hepatitis such as those of viral, autoimmune, or ischemic etiology.
N-acetylcysteine. A medication classically used in patients with acetaminophen overdose, NAC is a safe and effective treatment for non-acetaminophen-induced liver injury, and was given to treat our patient.16L-carnitine. L-carnitine has been shown to be effective in cases of chronic steatosis from hepatitis C and in valproic acid induced hepatitis.17Since L-carnitine is not included on our hospital’s formulary, it was not a treatment option for our patient.Glucocorticoid Therapy. Although glucocorticoids are occasionally given to patients with systemic symptoms of drug reactions, its effectiveness has not been adequately studied.18
Prognosis
The prognosis of patients with acute drug-induced hepatitis is generally good, and most patients fully recover once the offending agent is removed. Poor prognostic factors include the presence of jaundice, requirement for dialysis, underlying chronic liver conditions, or elevated serum creatinine. While most patients will experience a complete recovery, approximately 5% to 10% will develop chronic hepatitis and/or cirrhosis.
Conclusion
Niacin is now available as prescription and OTC formulations and is a potentially hepatotoxic medication and dietary supplement. Niacin can cause an acute hepatitis, especially when taken in conjunction with other hepatotoxic substances. Drug-induced liver injury from niacin ingestion will improve quickly following removal, and the prognosis in otherwise healthy individuals is good.
Patients, especially young, healthy patients who present with symptoms concerning for hepatitis, should be asked specifically about any nutritional, herbal, or other supplement usage. During the history intake, many patients do not consider vitamins or other nutritional or herbal supplements as “medication” or as being significant, and only report prescription and OTC medications.
Niacin, also known as vitamin B3, is an important cofactor in many metabolic processes necessary to life. Over the past 15 to 20 years, niacin has been prescribed to patients with hyperlipidemia to increase high-density lipoprotein and lower low-density lipoprotein.1 As a naturally occurring vitamin, niacin is also available over-the-counter (OTC) as a dietary supplement, and is also a common ingredient in energy drinks and multivitamins.2
In addition to treating hyperlipidemia and as a nutritional supplement, some anecdotal reports amongst lay-persons suggests that niacin offers other health benefits, such as promoting weight loss and expediting the elimination of alcohol and illicit drugs from one’s system (eg, marijuana).3,4 The increased use of niacin supplementation in the general population for all of the aforementioned reasons has resulted in an increased incidence of niacin toxicity.
Formulations
Niacin is available in three formulations: extended-release (ER, also referred to as intermediate-release), immediate-release (IR), and sustained-release (SR).
The ER formulations of niacin are typically prescribed to treat hyperlipidemia. Patients are usually started on ER niacin at an initial dose of 250 mg once daily. The dose is gradually increased, as tolerated or necessary, to 2 g per day, taken in three doses. It is not uncommon for patients with hyperlipidemia to take more than 1 g of niacin per day after titration by their primary physicians.
Side Effects
Since niacin increases the release of arachidonic acid from cell membranes that metabolizes into prostaglandins, specifically prostaglandins E2 and D2, many patients taking niacin experience uncomfortable flushing and itching.5 Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent this side effect by inhibiting the metabolism of arachidonic acid into those vasodilatory prostaglandins. The newer ER and SR formulations of niacin, which are approved for OTC use as a dietary supplement, are less likely to cause flushing.5
Extended-release niacin, however, is associated with a higher incidence of hepatotoxicity than the other prescription formulations of niacin.6 Toxicity has been well recognized in patients taking niacin chronically for hyperlipidemia, with reports of such cases dating back to the 1980s.7,8 We report a unique case of niacin toxicity following a single-dose ingestion in a young man.
Case
A 22-year-old man presented to the ED for evaluation of a 2-week history of intermittent periumbilical abdominal pain. This visit represented the patient’s second visit to the ED over the past week for the same complaint.
Upon presentation the patient’s vital signs were: blood pressure (BP), 113/64 mm Hg; heart rate, 82 beats/min; respiratory rate, 16 breaths/min; and temperature 36.6°C. Oxygen saturation was 100% on room air. The patient was otherwise healthy and had no significant recent or remote medical history. He denied taking any medications prior to his initial presentation, and reported only occasional alcohol use.
At the patient’s initial presentation 1 week earlier, he was diagnosed with acute gastroenteritis and treated with famotidine and ondansetron in the ED. The patient appeared well clinically at this visit, and laboratory values were within normal limits, including normal blood glucose and urinalysis.
The patient was discharged home from this first visit with prescriptions of famotidine and ondansetron, and was advised to follow up with his primary care physician in 1 week. Throughout the week after discharge from the ED, the patient experienced worsening abdominal pain, and he developed frequent nonbloody emesis, prompting his second presentation to the ED. At this second visit, the patient stated that he had taken one dose of ondansetron at home, without effect. He also noted subjective fevers, but had no diarrhea or melena.
Vital signs remained within normal limits with BPs ranging from 115 to 130 mm Hg systolic and 50 to 89 mm Hg diastolic. The patient was never tachycardic, tachypneic, febrile, or hypoxic. Physical examination was remarkable for periumbilical tenderness. The patient had no jaundice. A more thorough laboratory evaluation revealed elevated anion gap and blood urea nitrogen/creatinine values, and leukocytosis. The patient’s hepatic enzymes were also elevated, with aspartate aminotransferase (AST) over 2,000 U/L and alanine aminotransferase (ALT) of 1,698 U/L. Lipase, bilirubin, and alkaline phosphatase were all within normal limits. The patient’s prothrombin time (PT) was elevated at 14 seconds, and the international normalized ratio (INR) was elevated at 1.28. Laboratory analysis for acetaminophen and alcohol was negative.
A computed tomography (CT) scan of the abdomen/pelvis with intravenous (IV) contrast was unremarkable, demonstrating a liver devoid of any masses, portal or biliary dilation, or cirrhotic changes.
The patient received IV famotidine and ondansetron, and morphine for pain control, and was admitted to the general medical floor for hepatitis of uncertain etiology. A viral hepatitis panel was negative.
On the recommendation of the toxicology service, the patient was given N-acetylcysteine (NAC), and his hepatic enzymes trended down to an AST of 642 U/L and an ALT of 456 U/L by hospital day 2. (The patient essentially completed a positive dechallenge test).9
A gastroenterology consult was ordered, during which additional history-taking and chart review noted that the patient admitted to taking one or two tablets of OTC niacin as a dietary supplement the day before his initial presentation. Although the patient could not recall the exact dosage, he stated that he had been taking supplemental niacin approximately once a month over the past several years without any issues. Since OTC niacin is most commonly available in 500-mg tablets, this suggested the patient’s recent one-time ingested dose was approximately 500 to 1,000 mg.
Based on the patient’s admission to niacin use, additional studies were ordered, including an abdominal ultrasound and a urine drug screen. Ultrasound findings were unremarkable for portal venous thrombosis. The urine drug screen, however, was positive for marijuana and opiates. While the patient denied any history of opioid use, the positive opiate assay could have been attributed to the morphine given in the ED.
Throughout the patient’s hospital course, he remained normotensive and had no change in mental status. His liver enzymes, PT, and INR continued to normalize, and he was discharged home after 3 days, with instructions to follow up with the gastrointestinal clinic within 11 days. An appointment was made for him, which he did not attend.
Given the patient’s negative autoimmune and viral workup, and rapid resolution of symptoms after discontinuing niacin use, it is believed that he had an acute drug-induced hepatitis due to niacin ingestion. Regarding any coingestants that could have contributed to the hepatitis, the patient denied taking other common coingestants such as alcohol and acetaminophen; this assertion was supported by laboratory results.
Since we were unable to attain a qualitative measurement of the patient’s niacin concentration, our diagnosis was primarily based on the patient’s reported history.10 It is possible the patient had been taking more niacin than that to which he admitted, or that he was taking another hepatotoxic substance not detected on our toxicology workup. As previously noted, there are many medications and/or dietary supplements that could cause or contribute to a synergistic effect of drug-induced hepatitis for which the patient was not tested at his initial presentation. The patient could have co-ingested this large dose of niacin with acetaminophen and/or other supplements, energy drinks, or alcohol. A combination such as this could have contributed to his hepatitis, and the metabolites of these other substances would have been eliminated by the time of his second ED presentation.
Discussion
There are over 900 different drugs, toxins, and supplements known to cause hepatic injury.11,12 Clinical manifestations of toxicity range from asymptomatic incidental elevations in transaminases to fulminant liver failure causing mortality. Ingestion of commonly used medications such as statins (although not in overdose quantities) can cause transient asymptomatic transaminitis.13 These elevations are usually mild—ie, less than twice the upper limit of normal. Patients who experience such elevations can usually continue to take the medications with frequent and vigilant monitoring of hepatic function.
Signs and Symptoms
Acute Liver Injury. Acute liver injury is diagnosed when AST and ALT levels are greater than twice the upper limit of normal. Patients also typically have mild-to-moderate abdominal findings, such as pain, nausea, and vomiting—as was experienced by our patient. Along with niacin, angiotensin-converting enzyme inhibitors, NSAIDs, and antifungal medications are examples of other medications that can cause this degree of drug-induced hepatitis.
Severe Liver Injury. Severe liver injury features elevations in not only AST and ALT, but also alkaline phosphate and bilirubin. Patients with severe hepatic injury appear clinically ill and may exhibit altered mental status and jaundice. This type of subfulminant hepatic failure commonly results from acetaminophen toxicity, anesthetic gases, iron toxicity, phosphorus toxicity, and cocaine toxicity. Examples of drugs that result in massive liver necrosis and fulminant hepatitis are acetaminophen, isoniazid, phenelzine, phenytoin, propylthiouracil, and sertraline. Patients with massive hepatic necrosis and hepatitis may require liver transplantation.
Etiology
Identifying the etiology of liver injury is made largely through the patient’s history because there are simply too many possible hepatotoxic agents to test for them all. Diagnostic suspicion of hepatic toxicity should be increased with signs of more serious disease; however, drug-induced liver injury should be included in the differential diagnosis for all cases of abdominal pain.
With respect to the patient in our case, obtaining a more complete history involving supplement and vitamin use would have allowed us to make the diagnosis in the ED. Unfortunately, these subtle aspects of a patient’s history are often overlooked in the emergent care setting.
Treatment
The treatment of niacin-induced liver injury is similar to the guidelines for treating most other drug-induced pathology.14 Removal of the offending agent and providing supportive care is the primary treatment modality.15 In addition, it is important that the clinician exclude and rule-out other causes of hepatitis such as those of viral, autoimmune, or ischemic etiology.
N-acetylcysteine. A medication classically used in patients with acetaminophen overdose, NAC is a safe and effective treatment for non-acetaminophen-induced liver injury, and was given to treat our patient.16L-carnitine. L-carnitine has been shown to be effective in cases of chronic steatosis from hepatitis C and in valproic acid induced hepatitis.17Since L-carnitine is not included on our hospital’s formulary, it was not a treatment option for our patient.Glucocorticoid Therapy. Although glucocorticoids are occasionally given to patients with systemic symptoms of drug reactions, its effectiveness has not been adequately studied.18
Prognosis
The prognosis of patients with acute drug-induced hepatitis is generally good, and most patients fully recover once the offending agent is removed. Poor prognostic factors include the presence of jaundice, requirement for dialysis, underlying chronic liver conditions, or elevated serum creatinine. While most patients will experience a complete recovery, approximately 5% to 10% will develop chronic hepatitis and/or cirrhosis.
Conclusion
Niacin is now available as prescription and OTC formulations and is a potentially hepatotoxic medication and dietary supplement. Niacin can cause an acute hepatitis, especially when taken in conjunction with other hepatotoxic substances. Drug-induced liver injury from niacin ingestion will improve quickly following removal, and the prognosis in otherwise healthy individuals is good.
Patients, especially young, healthy patients who present with symptoms concerning for hepatitis, should be asked specifically about any nutritional, herbal, or other supplement usage. During the history intake, many patients do not consider vitamins or other nutritional or herbal supplements as “medication” or as being significant, and only report prescription and OTC medications.
1. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969-2989. doi:10.1210/jc.2011-3213.
2. Vivekanandarajah A, Ni S, Waked A. Acute hepatitis in a woman following excessive ingestion of an energy drink: a case report. J Med Case Rep. 2011;5:227. doi:10.1186/1752-1947-5-227.
3. Niacin. U.S. National Library of Medicine. https://medlineplus.gov/druginfo/meds/a682518.html. Updated July 15, 2017. Accessed February 21, 2018.
4. Addiction Resource. Niacin flush for drug detox. https://addictionresource.com/drug-testing/niacin-drug-test/. Accessed February 20, 2018.
5. Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract. 2009;63(9):1369-1377. doi:10.1111/j.1742-1241.2009.02099.x.
6. Etchason JA, Miller TD, Squires RW, et al. Niacin-induced hepatitis: a potential side effect with low-dose time-release niacin. Mayo Clin Proc. 1991;66(1):23-28.
7. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J. 1983;76(2):239-241.
8. Ferenchick G, Rovner D. Hepatitis and hematemesis complicating nicotinic acid use. Am J Med Sci. 1989;298(3):191-193.
9. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA. 1990;264(2):241-243.
10. Barritt AS 4th, Lee J, Hayashi PH. Detective work in drug-induced liver injury: sometimes it is all about interviewing the right witness. Clin Gastroenterol Hepatol. 2010;8(7):635-637. doi:10.1016/j.cgh.2010.03.020.
11. Chitturi S, Teoh NC, Farrell GC. Hepatic drug metabolism and liver disease caused by drugs. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:1442-1477.
12. National Institutes of Health. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Niacin. https://livertox.nih.gov/Niacin.htm. Updated January 18, 2018. Accessed February 21, 2018.
13. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41(4):690-695.
14. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966. doi:10.1038/ajg.2014.131.
15. Larson AM. Hepatotoxicity due to herbal medication and dietary supplements. UpToDate Web site. https://www.uptodate.com/contents/hepatotoxicity-due-to-herbal-medications-and-dietary-supplements?source=search_result&search=niacin%20induced%20hepatitis&selectedTitle=3~150. Updated December 21, 2017. Accessed February 21, 2018.
16. Mumtaz K, Azam Z, Hamid S, et al. Role of N-acetylcysteine in adults with non-acetaminophen-induced acute liver failure in a center without the facility of liver transplantation. Hepatol Int. 2009;3(4):563-570. doi:10.1007/s12072-009-9151-0.
17. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293. doi:10.1345/aph.1P135.
18. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439-445.
1. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969-2989. doi:10.1210/jc.2011-3213.
2. Vivekanandarajah A, Ni S, Waked A. Acute hepatitis in a woman following excessive ingestion of an energy drink: a case report. J Med Case Rep. 2011;5:227. doi:10.1186/1752-1947-5-227.
3. Niacin. U.S. National Library of Medicine. https://medlineplus.gov/druginfo/meds/a682518.html. Updated July 15, 2017. Accessed February 21, 2018.
4. Addiction Resource. Niacin flush for drug detox. https://addictionresource.com/drug-testing/niacin-drug-test/. Accessed February 20, 2018.
5. Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract. 2009;63(9):1369-1377. doi:10.1111/j.1742-1241.2009.02099.x.
6. Etchason JA, Miller TD, Squires RW, et al. Niacin-induced hepatitis: a potential side effect with low-dose time-release niacin. Mayo Clin Proc. 1991;66(1):23-28.
7. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J. 1983;76(2):239-241.
8. Ferenchick G, Rovner D. Hepatitis and hematemesis complicating nicotinic acid use. Am J Med Sci. 1989;298(3):191-193.
9. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA. 1990;264(2):241-243.
10. Barritt AS 4th, Lee J, Hayashi PH. Detective work in drug-induced liver injury: sometimes it is all about interviewing the right witness. Clin Gastroenterol Hepatol. 2010;8(7):635-637. doi:10.1016/j.cgh.2010.03.020.
11. Chitturi S, Teoh NC, Farrell GC. Hepatic drug metabolism and liver disease caused by drugs. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:1442-1477.
12. National Institutes of Health. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Niacin. https://livertox.nih.gov/Niacin.htm. Updated January 18, 2018. Accessed February 21, 2018.
13. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41(4):690-695.
14. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966. doi:10.1038/ajg.2014.131.
15. Larson AM. Hepatotoxicity due to herbal medication and dietary supplements. UpToDate Web site. https://www.uptodate.com/contents/hepatotoxicity-due-to-herbal-medications-and-dietary-supplements?source=search_result&search=niacin%20induced%20hepatitis&selectedTitle=3~150. Updated December 21, 2017. Accessed February 21, 2018.
16. Mumtaz K, Azam Z, Hamid S, et al. Role of N-acetylcysteine in adults with non-acetaminophen-induced acute liver failure in a center without the facility of liver transplantation. Hepatol Int. 2009;3(4):563-570. doi:10.1007/s12072-009-9151-0.
17. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293. doi:10.1345/aph.1P135.
18. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439-445.
His Old Pain Is Back
ANSWER
The radiograph demonstrates a fracture within the femoral neck. These types of fractures, which are slightly angulated and located at the base of the neck, are typically referred to as basicervical femur fractures.
Prompt orthopedic surgery consultation was obtained, and the patient underwent an open reduction and internal fixation of his fracture.
ANSWER
The radiograph demonstrates a fracture within the femoral neck. These types of fractures, which are slightly angulated and located at the base of the neck, are typically referred to as basicervical femur fractures.
Prompt orthopedic surgery consultation was obtained, and the patient underwent an open reduction and internal fixation of his fracture.
ANSWER
The radiograph demonstrates a fracture within the femoral neck. These types of fractures, which are slightly angulated and located at the base of the neck, are typically referred to as basicervical femur fractures.
Prompt orthopedic surgery consultation was obtained, and the patient underwent an open reduction and internal fixation of his fracture.
You are called in for a neurosurgical consult of a 60-year-old man who underwent a lumbar decompression approximately 18 months ago. For the past several weeks, he has been experiencing worsening back pain that radiates down his left leg; at times, he has to use his cane and walker. The patient denies any acute injury or trauma preceding the pain; however, last night he slipped and fell in his bedroom, which made it much worse.
Medical history is significant for hypertension and hyperlipidemia. The patient responded well to his previous back surgery. The emergency department clinician is concerned that his symptoms may be caused by a new lumbar stenosis at an adjacent level.
On exam, you note a mildly obese male who appears uncomfortable but is in no obvious distress. His vital signs are stable. He has some tenderness within his left hip. Distally, his strength is good, and there is no evidence of neurovascular compromise. Internal and external rotation causes some discomfort.
A portable pelvic radiograph is obtained (shown). What is your impression?