User login
Woman, 57, With Painful, Swollen Ankle
IN THIS ARTICLE
- Diagnosis
- Treatment
- Care outcome
A 57-year-old horticulturist is working on a ladder leaned up against a tree trunk when the ladder slips, causing her to fall six feet onto concrete. Her right foot and ankle sustain the force of the fall; she is in excruciating pain and unable to bear weight on the foot. She is immediately transported to a local emergency department for evaluation.
Physical exam reveals a tearful middle-aged female in moderate distress and acute pain. There is moderate swelling of the right medial and lateral malleolus, as well as the midfoot, with blue and purple discoloration on the medial and lateral malleolus. Radiographs of the right ankle identify nondisplaced fractures of the distal fibula and tibia. Foot x-rays are unremarkable. A splint is ordered. The patient is given crutches (non-weight-bearing status), pain medication, and a referral to orthopedics.
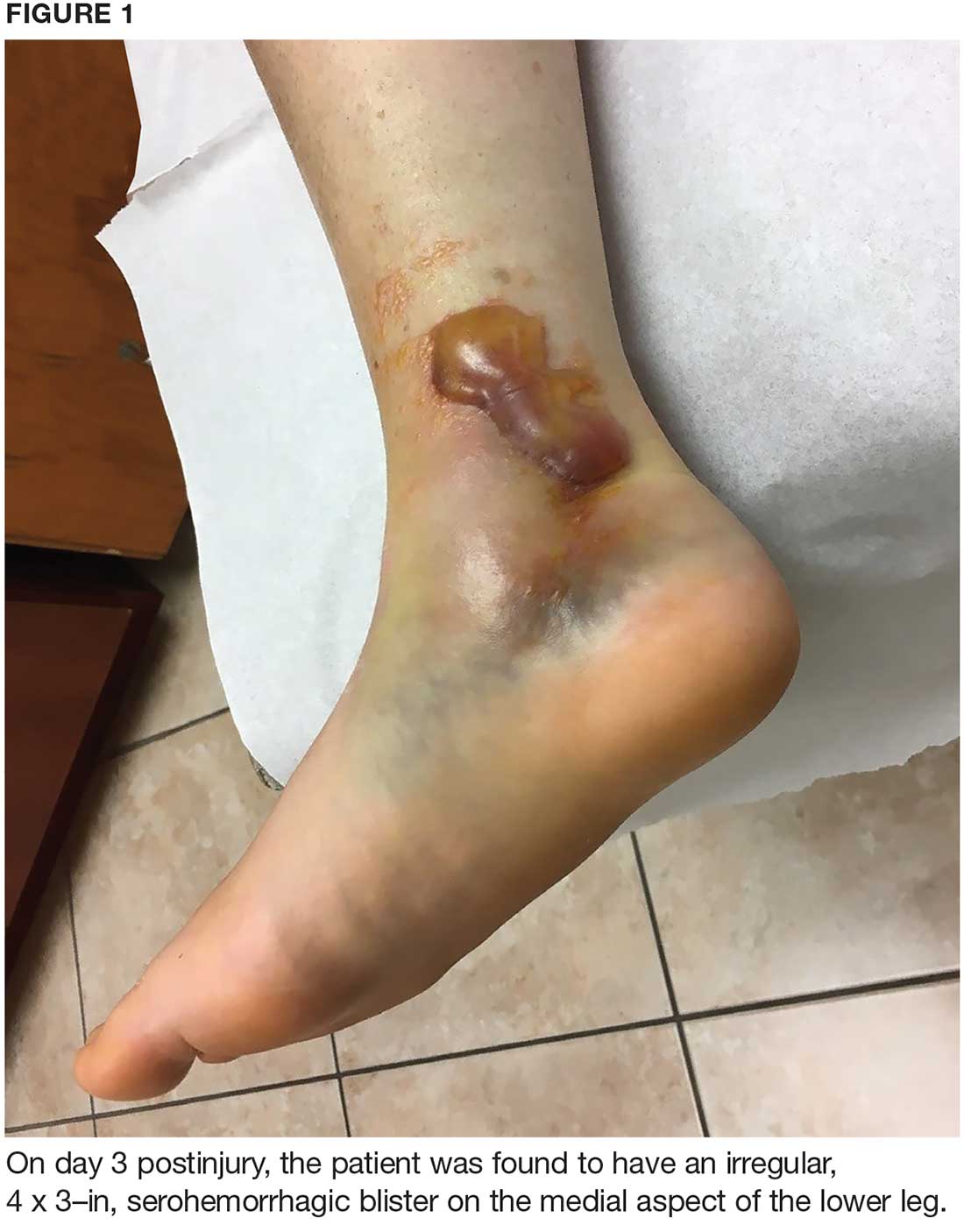
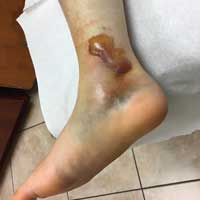
On day 3, the patient presents to orthopedics, where the splint is removed. An irregular, 4 × 3–in (at largest diameter), serohemorrhagic blister is discovered on the medial aspect of the lower leg, above the right malleolus (see Figure 1). Multiple 1- to 3-mm vesicles surround much of the anterior border. Moderate edema is noted from the top of the lesion to the midfoot, concentrated around the lateral and medial malleolus. Extensive blue, purple, and black discoloration is seen below the malleolus. The patient is diagnosed with a fracture blister.
DISCUSSION
Fracture blisters are taut, bullous, subepidermal vesicles that can accompany fractures or severe twisting injuries. They overlie markedly edematous soft tissue and histologically resemble a second-degree burn.1,2
Physiologically, blisters are caused by increased interstitial pressure due to swelling, with subsequent increased filtration pressure and colloid osmotic pressure in the epidermal gap.3 This causes a disruption that allows fluid to move into the weakened area.3 Areas most at risk for fracture blister formation are those with tight, closely adhered skin without muscle or enveloping fascia, where there is less soft tissue between the skin and bone prominences (eg, ankle, elbow, foot, distal tibia).2-4
Approximately 3% of all patients with acute fractures requiring hospitalization develop a fracture blister.4 Any condition that predisposes a patient to poor wound healing (eg, peripheral vascular disease, diabetes, hypertension) increases risk for a fracture blister.2 Recognizing which patients are at greatest risk is vital, as implementing prevention strategies and intervening when fracture blisters do form can help decrease complications—including infection and delayed surgery—and improve fracture resolution. In this patient’s case, the extent of the injury and force of the fall caused the fracture blister to form.
Diagnosis
Diagnosis of a fracture blister is based on clinical presentation. There are two types: hemorrhagic blisters and clear fluid-filled blisters. Hemorrhagic blisters indicate more severe injury and longer healing time (approximately 16 d), while clear fluid-filled blisters demonstrate minimal injury and therefore are quicker to heal.2,4
The differential diagnosis for fracture blisters includes friction blisters and disorders such as epidermolysis bullosa and bullous pemphigoid. Friction blisters form when the epidermis is subjected to repeated friction or shear forces (eg, from a cast or splint).5,6 These forces mechanically separate epidermal cells at the stratum spinosum layer.7 The pressure that moves across the skin forces fluid into the deeper open spaces, filling them but leaving the surface layer intact.1
Epidermolysis bullosa (EB) is a group of rare inherited cutaneous and mucus membrane disorders. EB involves fragility and detachment of subepithelial tissues, which results in blistering and erosions.8,9 The blisters tend to develop in areas subject to minor trauma, such as the extensor aspects of the elbows and the dorsal aspects of the hands and feet.9 They can also be triggered by exposure to heat, friction, scratching, and adhesive tape.10
Bullous pemphigoid, a chronic autoimmune skin disorder, is characterized by pruritic, bullous lesions. When IgG autoantibodies bind to certain hemidesmosomal antigens, complement activation causes a subepidermal blister.11While bullous pemphigoid most commonly affects those older than 60, it can also occur in children. Diagnosis is confirmed by skin biopsy and immunofluorescence testing.11
Treatment and management
Although several recommendations have been published, there is no gold standard and treatment of fracture blisters remains controversial. Early surgical intervention for fractures could decrease the incidence of fracture blisters.1,3
The goal of treatment is to achieve re-epithelialization of the dermis.3,12,13 Once a blister forms, management techniques vary. Some recommend keeping closed blisters covered with a dry dressing to protect them from damage.3 Strauss et al recommend unroofing to avoid traumatic rupture; however, this does increase risk for infection.12 Recommendations differ depending on provider preference and each patient’s individual situation.
Elective unroofing of a blister is typically followed with one of several treatment options. These include covering the open blister with a topical antibiotic cream (eg, silver sulfadiazine 2%); applying a nonadherent, occlusive bismuth-tribromophenate-petroleum gauze dressing; or elevating and immobilizing the affected extremity.12,13
Treatment of spontaneously ruptured fracture blisters entails
- Unroofing the blister completely and applying a topical antimicrobial (eg, silver sulfadiazine, polymyxin B, neomycin, bacitracin).
- Applying a hydrocolloid dressing to keep the environment moist.
- Using a first-aid gel containing melaleuca (tea tree) oil.
- Initiating prophylactic oral antibiotics.
- Using whirlpool treatments.
- Elevating and immobilizing the affected extremity.3,12,14
OUTCOME FOR THE CASE PATIENT
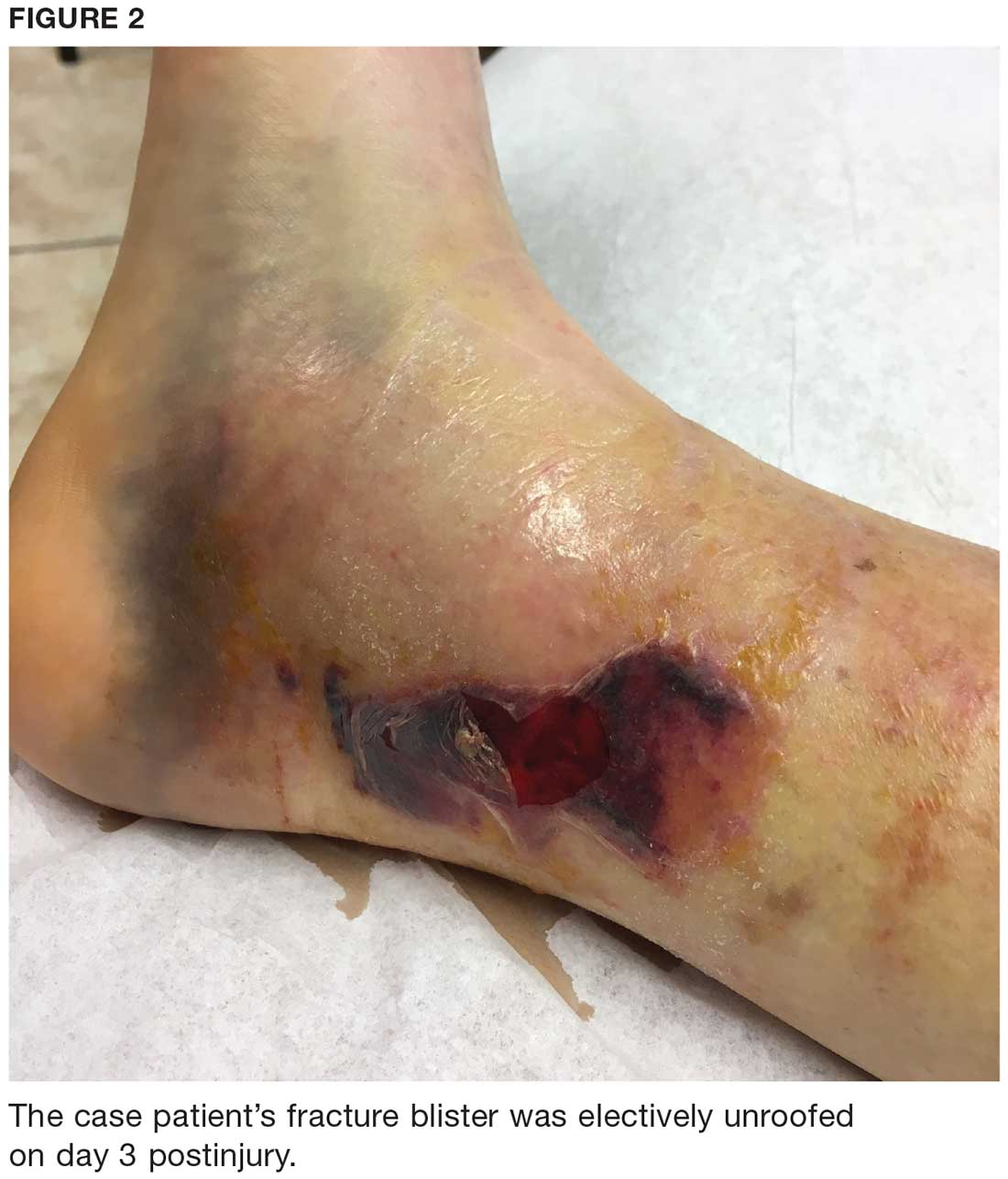
The fracture blister was electively unroofed (see Figure 2) based on provider preference. The patient was instructed to clean the wound daily and apply topical cream (silver sulfadiazine 2% bid) to the wound and cover it with gauze. The patient was made non-weight-bearing to the right lower extremity. Continuous elevation was highly encouraged except for bathing and restroom use, and an NSAID was recommended as needed for pain. She was reassessed the following day and, due to partial refilling, the blister required additional unroofing. The patient was instructed to resume previous wound care orders.
No surgical intervention was required. CT of the right foot and ankle without contrast (performed on day 4 postinjury) confirmed a nondisplaced transverse fracture of the medial malleolus and a sagittal avulsion fracture of the anterior-inferior lateral malleolus. Multiple smaller fracture fragments were noted posterior and medial to the medial malleolus as well as inferiorly along the course of the deltoid ligament. There was a small, nondisplaced avulsion fracture of the medial malleolus at the anterolateral and posterolateral tibial plafond.
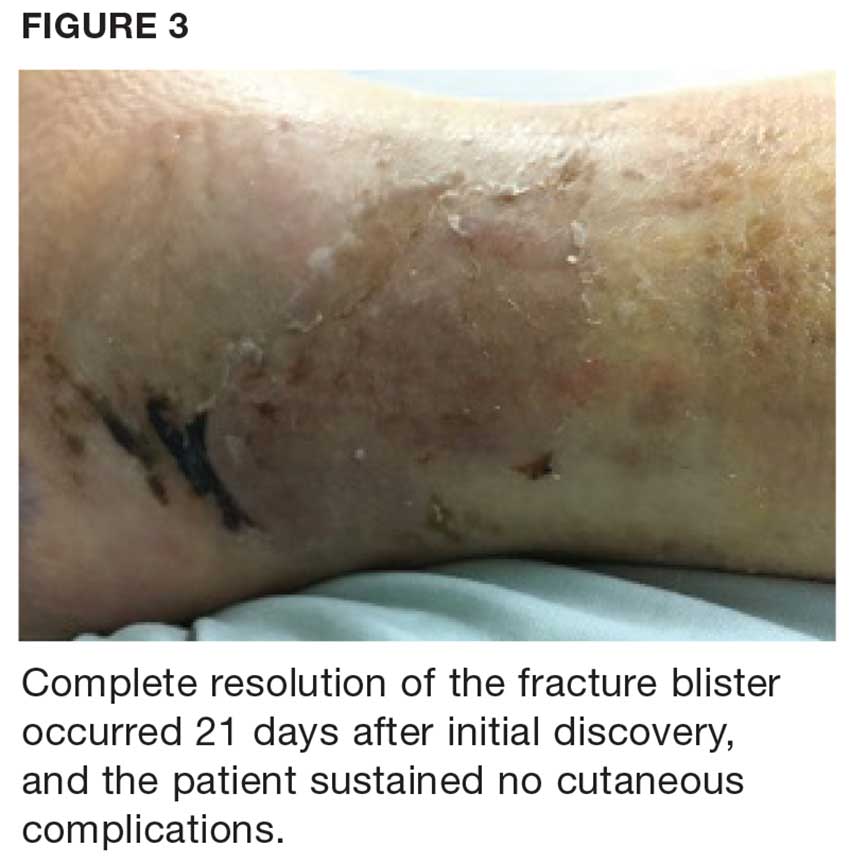
Due to the extent of the swelling, multiple fractures, and blister formation, the patient was essentially bed bound for the first three weeks; complete resolution of the fracture blister occurred 21 days after initial discovery (see Figure 3). The patient did not experience cutaneous complications. Her lower extremity was then casted in a short-leg removable cast for 10 weeks. She underwent physical therapy, and after 12 weeks, the patient was weight-bearing and was discharged from orthopedics. The patient reported refractory pain and swelling for an additional eight weeks following injury, warranting daily ibuprofen.
CONCLUSION
Fracture blisters are rare, and experience and knowledge about them in primary care is lacking. But clinicians need to be able to identify, diagnose, and refer at-risk patients to orthopedics in a timely manner.
Current management and treatment recommendations are inconsistent. Treatment varies depending on the site, severity, type, and status of the blister and the overall health of the patient. Fracture blisters may be left intact, electively unroofed, or treated after spontaneous rupture. More research is needed to clarify management recommendations, specifically regarding the decision to unroof a blister or leave it intact. Early surgical intervention may prevent the development of a fracture blister.
1. Wallace GF, Sullivan J. Fracture blisters. Clin Podiatr Med Surg. 1995;12(4):801-811.
2. Halawi MJ. Fracture blisters after primary total knee arthroplasty. Am J Orthop. 2015; 44(8):E291-E293.
3. McCann S, Gruen G. Fracture blisters: a review of the literature. Orthop Nurs. 1997; 16(2):17-24.
4. Uebbing CM, Walsh M, Miller JB, et al. Fracture blister. West J Emerg Med. 2011; 12(1):131-133.
5. Kirkham S, Lam S, Nester C, Hashmi F. The effect of hydration on the risk of friction blister formation on the heel of the foot. Skin Res Tech. 2014;20:246-253.
6. Boyd A, Benjamin H, Asplund C. Principles of casting and splinting. Am Fam Physician. 2009;79(1):16-24.
7. Knapik J, Reynolds K, Duplantis K, Jones B. Friction blisters. Pathophysiology, prevention and treatment. Sports Med. 1995; 20(3):136-147.
8. Iranzo P, Herrero-González JE, Mascaró-Galy JM, et al. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171(5):1022-1030.
9. Peraza DM. Epidermolysis bullosa acquisita. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/epidermolysis-bullosa-acquisita. Accessed January 26, 2018.
10. Lyons F, Ousley L. Dermatology for the Advanced Practice Nurse. New York, NY: Springer; 2015.
11. Peraza D. Bullous pemphigoid. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/bullous-pemphigoid. Accessed January 26, 2018.
12. Strauss EJ, Petrucelli G, Bong M, et al. Blisters associated with lower-extremity fracture: Results of a prospective treatment protocol. J Orthop Trauma. 2006;20(9): 618-622.
13. Tolpinrud WL, Rebolledo BJ, Lorich DG, Grossman ME. A case of extensive fracture bullae: a multidisciplinary approach for acute management. JAAD Case Rep. 2015;1(3):132-135.
14. Cox H, Nealon L. Case report: the use of Burnaid Gel on fracture blisters. Wound Practice and Research. 2008;16(1):32-36.
IN THIS ARTICLE
- Diagnosis
- Treatment
- Care outcome
A 57-year-old horticulturist is working on a ladder leaned up against a tree trunk when the ladder slips, causing her to fall six feet onto concrete. Her right foot and ankle sustain the force of the fall; she is in excruciating pain and unable to bear weight on the foot. She is immediately transported to a local emergency department for evaluation.
Physical exam reveals a tearful middle-aged female in moderate distress and acute pain. There is moderate swelling of the right medial and lateral malleolus, as well as the midfoot, with blue and purple discoloration on the medial and lateral malleolus. Radiographs of the right ankle identify nondisplaced fractures of the distal fibula and tibia. Foot x-rays are unremarkable. A splint is ordered. The patient is given crutches (non-weight-bearing status), pain medication, and a referral to orthopedics.
On day 3, the patient presents to orthopedics, where the splint is removed. An irregular, 4 × 3–in (at largest diameter), serohemorrhagic blister is discovered on the medial aspect of the lower leg, above the right malleolus (see Figure 1). Multiple 1- to 3-mm vesicles surround much of the anterior border. Moderate edema is noted from the top of the lesion to the midfoot, concentrated around the lateral and medial malleolus. Extensive blue, purple, and black discoloration is seen below the malleolus. The patient is diagnosed with a fracture blister.
DISCUSSION
Fracture blisters are taut, bullous, subepidermal vesicles that can accompany fractures or severe twisting injuries. They overlie markedly edematous soft tissue and histologically resemble a second-degree burn.1,2
Physiologically, blisters are caused by increased interstitial pressure due to swelling, with subsequent increased filtration pressure and colloid osmotic pressure in the epidermal gap.3 This causes a disruption that allows fluid to move into the weakened area.3 Areas most at risk for fracture blister formation are those with tight, closely adhered skin without muscle or enveloping fascia, where there is less soft tissue between the skin and bone prominences (eg, ankle, elbow, foot, distal tibia).2-4
Approximately 3% of all patients with acute fractures requiring hospitalization develop a fracture blister.4 Any condition that predisposes a patient to poor wound healing (eg, peripheral vascular disease, diabetes, hypertension) increases risk for a fracture blister.2 Recognizing which patients are at greatest risk is vital, as implementing prevention strategies and intervening when fracture blisters do form can help decrease complications—including infection and delayed surgery—and improve fracture resolution. In this patient’s case, the extent of the injury and force of the fall caused the fracture blister to form.
Diagnosis
Diagnosis of a fracture blister is based on clinical presentation. There are two types: hemorrhagic blisters and clear fluid-filled blisters. Hemorrhagic blisters indicate more severe injury and longer healing time (approximately 16 d), while clear fluid-filled blisters demonstrate minimal injury and therefore are quicker to heal.2,4
The differential diagnosis for fracture blisters includes friction blisters and disorders such as epidermolysis bullosa and bullous pemphigoid. Friction blisters form when the epidermis is subjected to repeated friction or shear forces (eg, from a cast or splint).5,6 These forces mechanically separate epidermal cells at the stratum spinosum layer.7 The pressure that moves across the skin forces fluid into the deeper open spaces, filling them but leaving the surface layer intact.1
Epidermolysis bullosa (EB) is a group of rare inherited cutaneous and mucus membrane disorders. EB involves fragility and detachment of subepithelial tissues, which results in blistering and erosions.8,9 The blisters tend to develop in areas subject to minor trauma, such as the extensor aspects of the elbows and the dorsal aspects of the hands and feet.9 They can also be triggered by exposure to heat, friction, scratching, and adhesive tape.10
Bullous pemphigoid, a chronic autoimmune skin disorder, is characterized by pruritic, bullous lesions. When IgG autoantibodies bind to certain hemidesmosomal antigens, complement activation causes a subepidermal blister.11While bullous pemphigoid most commonly affects those older than 60, it can also occur in children. Diagnosis is confirmed by skin biopsy and immunofluorescence testing.11
Treatment and management
Although several recommendations have been published, there is no gold standard and treatment of fracture blisters remains controversial. Early surgical intervention for fractures could decrease the incidence of fracture blisters.1,3
The goal of treatment is to achieve re-epithelialization of the dermis.3,12,13 Once a blister forms, management techniques vary. Some recommend keeping closed blisters covered with a dry dressing to protect them from damage.3 Strauss et al recommend unroofing to avoid traumatic rupture; however, this does increase risk for infection.12 Recommendations differ depending on provider preference and each patient’s individual situation.
Elective unroofing of a blister is typically followed with one of several treatment options. These include covering the open blister with a topical antibiotic cream (eg, silver sulfadiazine 2%); applying a nonadherent, occlusive bismuth-tribromophenate-petroleum gauze dressing; or elevating and immobilizing the affected extremity.12,13
Treatment of spontaneously ruptured fracture blisters entails
- Unroofing the blister completely and applying a topical antimicrobial (eg, silver sulfadiazine, polymyxin B, neomycin, bacitracin).
- Applying a hydrocolloid dressing to keep the environment moist.
- Using a first-aid gel containing melaleuca (tea tree) oil.
- Initiating prophylactic oral antibiotics.
- Using whirlpool treatments.
- Elevating and immobilizing the affected extremity.3,12,14
OUTCOME FOR THE CASE PATIENT
The fracture blister was electively unroofed (see Figure 2) based on provider preference. The patient was instructed to clean the wound daily and apply topical cream (silver sulfadiazine 2% bid) to the wound and cover it with gauze. The patient was made non-weight-bearing to the right lower extremity. Continuous elevation was highly encouraged except for bathing and restroom use, and an NSAID was recommended as needed for pain. She was reassessed the following day and, due to partial refilling, the blister required additional unroofing. The patient was instructed to resume previous wound care orders.
No surgical intervention was required. CT of the right foot and ankle without contrast (performed on day 4 postinjury) confirmed a nondisplaced transverse fracture of the medial malleolus and a sagittal avulsion fracture of the anterior-inferior lateral malleolus. Multiple smaller fracture fragments were noted posterior and medial to the medial malleolus as well as inferiorly along the course of the deltoid ligament. There was a small, nondisplaced avulsion fracture of the medial malleolus at the anterolateral and posterolateral tibial plafond.
Due to the extent of the swelling, multiple fractures, and blister formation, the patient was essentially bed bound for the first three weeks; complete resolution of the fracture blister occurred 21 days after initial discovery (see Figure 3). The patient did not experience cutaneous complications. Her lower extremity was then casted in a short-leg removable cast for 10 weeks. She underwent physical therapy, and after 12 weeks, the patient was weight-bearing and was discharged from orthopedics. The patient reported refractory pain and swelling for an additional eight weeks following injury, warranting daily ibuprofen.
CONCLUSION
Fracture blisters are rare, and experience and knowledge about them in primary care is lacking. But clinicians need to be able to identify, diagnose, and refer at-risk patients to orthopedics in a timely manner.
Current management and treatment recommendations are inconsistent. Treatment varies depending on the site, severity, type, and status of the blister and the overall health of the patient. Fracture blisters may be left intact, electively unroofed, or treated after spontaneous rupture. More research is needed to clarify management recommendations, specifically regarding the decision to unroof a blister or leave it intact. Early surgical intervention may prevent the development of a fracture blister.
IN THIS ARTICLE
- Diagnosis
- Treatment
- Care outcome
A 57-year-old horticulturist is working on a ladder leaned up against a tree trunk when the ladder slips, causing her to fall six feet onto concrete. Her right foot and ankle sustain the force of the fall; she is in excruciating pain and unable to bear weight on the foot. She is immediately transported to a local emergency department for evaluation.
Physical exam reveals a tearful middle-aged female in moderate distress and acute pain. There is moderate swelling of the right medial and lateral malleolus, as well as the midfoot, with blue and purple discoloration on the medial and lateral malleolus. Radiographs of the right ankle identify nondisplaced fractures of the distal fibula and tibia. Foot x-rays are unremarkable. A splint is ordered. The patient is given crutches (non-weight-bearing status), pain medication, and a referral to orthopedics.
On day 3, the patient presents to orthopedics, where the splint is removed. An irregular, 4 × 3–in (at largest diameter), serohemorrhagic blister is discovered on the medial aspect of the lower leg, above the right malleolus (see Figure 1). Multiple 1- to 3-mm vesicles surround much of the anterior border. Moderate edema is noted from the top of the lesion to the midfoot, concentrated around the lateral and medial malleolus. Extensive blue, purple, and black discoloration is seen below the malleolus. The patient is diagnosed with a fracture blister.
DISCUSSION
Fracture blisters are taut, bullous, subepidermal vesicles that can accompany fractures or severe twisting injuries. They overlie markedly edematous soft tissue and histologically resemble a second-degree burn.1,2
Physiologically, blisters are caused by increased interstitial pressure due to swelling, with subsequent increased filtration pressure and colloid osmotic pressure in the epidermal gap.3 This causes a disruption that allows fluid to move into the weakened area.3 Areas most at risk for fracture blister formation are those with tight, closely adhered skin without muscle or enveloping fascia, where there is less soft tissue between the skin and bone prominences (eg, ankle, elbow, foot, distal tibia).2-4
Approximately 3% of all patients with acute fractures requiring hospitalization develop a fracture blister.4 Any condition that predisposes a patient to poor wound healing (eg, peripheral vascular disease, diabetes, hypertension) increases risk for a fracture blister.2 Recognizing which patients are at greatest risk is vital, as implementing prevention strategies and intervening when fracture blisters do form can help decrease complications—including infection and delayed surgery—and improve fracture resolution. In this patient’s case, the extent of the injury and force of the fall caused the fracture blister to form.
Diagnosis
Diagnosis of a fracture blister is based on clinical presentation. There are two types: hemorrhagic blisters and clear fluid-filled blisters. Hemorrhagic blisters indicate more severe injury and longer healing time (approximately 16 d), while clear fluid-filled blisters demonstrate minimal injury and therefore are quicker to heal.2,4
The differential diagnosis for fracture blisters includes friction blisters and disorders such as epidermolysis bullosa and bullous pemphigoid. Friction blisters form when the epidermis is subjected to repeated friction or shear forces (eg, from a cast or splint).5,6 These forces mechanically separate epidermal cells at the stratum spinosum layer.7 The pressure that moves across the skin forces fluid into the deeper open spaces, filling them but leaving the surface layer intact.1
Epidermolysis bullosa (EB) is a group of rare inherited cutaneous and mucus membrane disorders. EB involves fragility and detachment of subepithelial tissues, which results in blistering and erosions.8,9 The blisters tend to develop in areas subject to minor trauma, such as the extensor aspects of the elbows and the dorsal aspects of the hands and feet.9 They can also be triggered by exposure to heat, friction, scratching, and adhesive tape.10
Bullous pemphigoid, a chronic autoimmune skin disorder, is characterized by pruritic, bullous lesions. When IgG autoantibodies bind to certain hemidesmosomal antigens, complement activation causes a subepidermal blister.11While bullous pemphigoid most commonly affects those older than 60, it can also occur in children. Diagnosis is confirmed by skin biopsy and immunofluorescence testing.11
Treatment and management
Although several recommendations have been published, there is no gold standard and treatment of fracture blisters remains controversial. Early surgical intervention for fractures could decrease the incidence of fracture blisters.1,3
The goal of treatment is to achieve re-epithelialization of the dermis.3,12,13 Once a blister forms, management techniques vary. Some recommend keeping closed blisters covered with a dry dressing to protect them from damage.3 Strauss et al recommend unroofing to avoid traumatic rupture; however, this does increase risk for infection.12 Recommendations differ depending on provider preference and each patient’s individual situation.
Elective unroofing of a blister is typically followed with one of several treatment options. These include covering the open blister with a topical antibiotic cream (eg, silver sulfadiazine 2%); applying a nonadherent, occlusive bismuth-tribromophenate-petroleum gauze dressing; or elevating and immobilizing the affected extremity.12,13
Treatment of spontaneously ruptured fracture blisters entails
- Unroofing the blister completely and applying a topical antimicrobial (eg, silver sulfadiazine, polymyxin B, neomycin, bacitracin).
- Applying a hydrocolloid dressing to keep the environment moist.
- Using a first-aid gel containing melaleuca (tea tree) oil.
- Initiating prophylactic oral antibiotics.
- Using whirlpool treatments.
- Elevating and immobilizing the affected extremity.3,12,14
OUTCOME FOR THE CASE PATIENT
The fracture blister was electively unroofed (see Figure 2) based on provider preference. The patient was instructed to clean the wound daily and apply topical cream (silver sulfadiazine 2% bid) to the wound and cover it with gauze. The patient was made non-weight-bearing to the right lower extremity. Continuous elevation was highly encouraged except for bathing and restroom use, and an NSAID was recommended as needed for pain. She was reassessed the following day and, due to partial refilling, the blister required additional unroofing. The patient was instructed to resume previous wound care orders.
No surgical intervention was required. CT of the right foot and ankle without contrast (performed on day 4 postinjury) confirmed a nondisplaced transverse fracture of the medial malleolus and a sagittal avulsion fracture of the anterior-inferior lateral malleolus. Multiple smaller fracture fragments were noted posterior and medial to the medial malleolus as well as inferiorly along the course of the deltoid ligament. There was a small, nondisplaced avulsion fracture of the medial malleolus at the anterolateral and posterolateral tibial plafond.
Due to the extent of the swelling, multiple fractures, and blister formation, the patient was essentially bed bound for the first three weeks; complete resolution of the fracture blister occurred 21 days after initial discovery (see Figure 3). The patient did not experience cutaneous complications. Her lower extremity was then casted in a short-leg removable cast for 10 weeks. She underwent physical therapy, and after 12 weeks, the patient was weight-bearing and was discharged from orthopedics. The patient reported refractory pain and swelling for an additional eight weeks following injury, warranting daily ibuprofen.
CONCLUSION
Fracture blisters are rare, and experience and knowledge about them in primary care is lacking. But clinicians need to be able to identify, diagnose, and refer at-risk patients to orthopedics in a timely manner.
Current management and treatment recommendations are inconsistent. Treatment varies depending on the site, severity, type, and status of the blister and the overall health of the patient. Fracture blisters may be left intact, electively unroofed, or treated after spontaneous rupture. More research is needed to clarify management recommendations, specifically regarding the decision to unroof a blister or leave it intact. Early surgical intervention may prevent the development of a fracture blister.
1. Wallace GF, Sullivan J. Fracture blisters. Clin Podiatr Med Surg. 1995;12(4):801-811.
2. Halawi MJ. Fracture blisters after primary total knee arthroplasty. Am J Orthop. 2015; 44(8):E291-E293.
3. McCann S, Gruen G. Fracture blisters: a review of the literature. Orthop Nurs. 1997; 16(2):17-24.
4. Uebbing CM, Walsh M, Miller JB, et al. Fracture blister. West J Emerg Med. 2011; 12(1):131-133.
5. Kirkham S, Lam S, Nester C, Hashmi F. The effect of hydration on the risk of friction blister formation on the heel of the foot. Skin Res Tech. 2014;20:246-253.
6. Boyd A, Benjamin H, Asplund C. Principles of casting and splinting. Am Fam Physician. 2009;79(1):16-24.
7. Knapik J, Reynolds K, Duplantis K, Jones B. Friction blisters. Pathophysiology, prevention and treatment. Sports Med. 1995; 20(3):136-147.
8. Iranzo P, Herrero-González JE, Mascaró-Galy JM, et al. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171(5):1022-1030.
9. Peraza DM. Epidermolysis bullosa acquisita. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/epidermolysis-bullosa-acquisita. Accessed January 26, 2018.
10. Lyons F, Ousley L. Dermatology for the Advanced Practice Nurse. New York, NY: Springer; 2015.
11. Peraza D. Bullous pemphigoid. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/bullous-pemphigoid. Accessed January 26, 2018.
12. Strauss EJ, Petrucelli G, Bong M, et al. Blisters associated with lower-extremity fracture: Results of a prospective treatment protocol. J Orthop Trauma. 2006;20(9): 618-622.
13. Tolpinrud WL, Rebolledo BJ, Lorich DG, Grossman ME. A case of extensive fracture bullae: a multidisciplinary approach for acute management. JAAD Case Rep. 2015;1(3):132-135.
14. Cox H, Nealon L. Case report: the use of Burnaid Gel on fracture blisters. Wound Practice and Research. 2008;16(1):32-36.
1. Wallace GF, Sullivan J. Fracture blisters. Clin Podiatr Med Surg. 1995;12(4):801-811.
2. Halawi MJ. Fracture blisters after primary total knee arthroplasty. Am J Orthop. 2015; 44(8):E291-E293.
3. McCann S, Gruen G. Fracture blisters: a review of the literature. Orthop Nurs. 1997; 16(2):17-24.
4. Uebbing CM, Walsh M, Miller JB, et al. Fracture blister. West J Emerg Med. 2011; 12(1):131-133.
5. Kirkham S, Lam S, Nester C, Hashmi F. The effect of hydration on the risk of friction blister formation on the heel of the foot. Skin Res Tech. 2014;20:246-253.
6. Boyd A, Benjamin H, Asplund C. Principles of casting and splinting. Am Fam Physician. 2009;79(1):16-24.
7. Knapik J, Reynolds K, Duplantis K, Jones B. Friction blisters. Pathophysiology, prevention and treatment. Sports Med. 1995; 20(3):136-147.
8. Iranzo P, Herrero-González JE, Mascaró-Galy JM, et al. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171(5):1022-1030.
9. Peraza DM. Epidermolysis bullosa acquisita. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/epidermolysis-bullosa-acquisita. Accessed January 26, 2018.
10. Lyons F, Ousley L. Dermatology for the Advanced Practice Nurse. New York, NY: Springer; 2015.
11. Peraza D. Bullous pemphigoid. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/bullous-pemphigoid. Accessed January 26, 2018.
12. Strauss EJ, Petrucelli G, Bong M, et al. Blisters associated with lower-extremity fracture: Results of a prospective treatment protocol. J Orthop Trauma. 2006;20(9): 618-622.
13. Tolpinrud WL, Rebolledo BJ, Lorich DG, Grossman ME. A case of extensive fracture bullae: a multidisciplinary approach for acute management. JAAD Case Rep. 2015;1(3):132-135.
14. Cox H, Nealon L. Case report: the use of Burnaid Gel on fracture blisters. Wound Practice and Research. 2008;16(1):32-36.
A 67-year-old woman with bilateral hand numbness
A 67-year-old woman presents to the emergency department after 8 weeks of progressive numbness and tingling in both hands, involving all fingers. The numbness has increased in severity in the last 3 days. She also has occasional numbness around her mouth. She reports no numbness in her feet.
She says she underwent thyroid surgery twice for thyroid cancer 10 years ago. Her medical history also includes type 2 diabetes mellitus (diagnosed 1 year ago), hypertension, dyslipidemia, and diastolic heart failure (diagnosed 5 years ago).
Her current medications are:
- Metformin 1 g twice a day
- Candesartan 16 mg once a day
- Atorvastatin 20 mg once a day
- Furosemide 40 mg twice a day
- Levothyroxine 100 μg per day
- Calcium carbonate 1,500 mg twice a day
- A vitamin D tablet twice a day, which she has not taken for the last 2 months.
She admits she has not been taking her medications regularly because she has been feeling depressed.
On physical examination, she is alert and oriented but appears anxious. She is not in respiratory distress. Her blood pressure is 150/90 mm Hg and her pulse is 92 beats per minute and regular. There is a thyroidectomy scar on the anterior neck. Her jugular venous pressure is not elevated. Her heart sounds are normal without extra sounds. She has no pulmonary rales and no lower-extremity edema.
The Phalen test and Tinel test for carpal tunnel syndrome are negative in both hands. Using a Katz hand diagram, the patient reports tingling and numbness in all fingers, both palms, and the dorsum of both hands. Tapping the area over the facial nerve does not elicit twitching of the facial muscles (ie, no Chvostek sign), but compression of the upper arm elicits carpal spasm (ie, positive Trousseau sign). There is no evidence of motor weakness in her hands. The rest of the physical examination is unremarkable.
POSSIBLE CAUSES OF NUMBNESS
1. Based on the initial evaluation, which of the following is the most likely cause of our patient’s bilateral hand numbness?
- Hypocalcemia due to primary hypoparathyroidism
- Carpal tunnel syndrome due to primary hypothyroidism
- Diabetic peripheral neuropathy
- Vitamin B12 deficiency due to metformin
- Hypocalcemia due to low serum calcitonin
All the conditions above except low serum calcitonin can cause bilateral hand paresthesia. Our patient most likely has hypocalcemia due to primary hypoparathyroidism.
Hypocalcemia
In our patient, bilateral hand numbness and perioral numbness after stopping vitamin D and a positive Trousseau sign strongly suggest hypocalcemia. The classic physical findings in patients with hypocalcemia are the Trousseau sign and the Chvostek sign. The Trousseau sign is elicited by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and observing for ischemia-induced carpopedal spasm, wrist and metacarpophalangeal joint flexion, thumb adduction, and interphalangeal joint extension. The Chvostek sign is elicited by tapping over the area of the facial nerve below the zygoma in front of the tragus, resulting in ipsilateral twitching of facial muscles.
Although the Trousseau sign is more sensitive and specific than the Chvostek sign, neither is pathognomonic for hypocalcemia.1 The Chvostek sign has been reported to be negative in 30% of patients with hypocalcemia and positive in 10% of normocalcemic individuals.1 The Trousseau sign, however, is present in 94% of hypocalcemic patients vs 1% of normocalcemic individuals.2
Primary hypoparathyroidism secondary to thyroidectomy. Postsurgical hypoparathyroidism is the most common cause of primary hypoparathyroidism. It results from ischemic injury or accidental removal of the parathyroid glands during anterior neck surgery.3,4 The consequent hypocalcemia can be transient, intermittent, or permanent. Permanent postsurgical hypoparathyroidism is defined as persistent hypocalcemia with insufficient parathyroid hormone (PTH) for more than 12 months after neck surgery; however, some consider 6 months to be enough to define the condition.5–7
The incidence of postsurgical hypoparathyroidism varies considerably with the extent of thyroid surgery and the experience of the surgeon.6,8 In the hands of experienced surgeons, permanent hypoparathyroidism occurs in fewer than 1% of patients after total thyroidectomy, whereas the rate may be higher than 6% with less-experienced surgeons.5,9 Other risk factors for postsurgical hypoparathyroidism include female sex, autoimmune thyroid disease, pregnancy, and lactation.5
Pseudohypoparathyroidism is a group of disorders characterized by renal resistance to PTH, leading to hypocalcemia, hyperphosphatemia, and elevated serum PTH. It is also associated with phenotypic features such as short stature and short fourth metacarpal bones.
Calcitonin deficiency. Calcitonin is a polypeptide hormone secreted from the parafollicular (C) cells of the thyroid gland. After total thyroidectomy, calcitonin levels are expected to be reduced. However, the role of calcitonin in humans is unclear. One study has shown that calcitonin is possibly a vestigial hormone, given that no calcitonin-related disorders (excess or deficiency) have been reported in humans.10
Carpal tunnel syndrome due to hypothyroidism
Our patient also could have primary hypothyroidism as a result of thyroidectomy. Hypothyroidism can cause bilateral hand numbness due to carpal tunnel syndrome, which is mediated by mucopolysaccharide deposition and synovial membrane swelling.11 One study reported that 29% of patients with hypothyroidism had carpal tunnel syndrome.12 Symptoms of carpal tunnel syndrome in hypothyroid patients may occur despite thyroid replacement therapy.13

Carpal tunnel syndrome is a clinical diagnosis. Patients usually experience hand paresthesia in the distribution of the median nerve. Provocative physical tests for carpal tunnel syndrome include the Tinel test, the Phalen test, and the Katz hand diagram, which is considered the best of the 3 tests.14,15 Based on how the patient marks the location and type of symptoms on the diagram, carpal tunnel syndrome is rated as classic, probable, possible, or unlikely (Table 1).14,16,17 The sensitivity of a classic or probable diagram ranges from 64% to 80%, while the specificity ranges from 73% to 90%.14,15
Carpal tunnel syndrome is less likely to be the cause of our patient’s symptoms, as her Katz hand diagram indicates only “possible” carpal tunnel syndrome. Her perioral numbness and positive Trousseau sign make hypocalcemia a more likely cause.
Diabetic peripheral neuropathy
Sensory peripheral neuropathy is a recognized complication of diabetes mellitus. However, neuropathy in diabetic patients most commonly manifests initially as distal symmetrical ascending neuropathy starting in the lower extremities.18 Therefore, diabetic peripheral neuropathy is less likely in this patient since her symptoms are limited to her hands.
Vitamin B12 deficiency
Metformin-induced vitamin B12 deficiency is another possible cause of peripheral neuropathy. It might be secondary to metformin-induced changes in intrinsic factor levels and small-intestine motility with resultant bacterial overgrowth, as well as inhibition of vitamin B12 absorption in the terminal ileum.19
However, metformin-induced vitamin B12 deficiency is not the most likely cause of our patient’s neuropathy, since she has been taking this drug for only 1 year. Vitamin B12 deficiency with consequent peripheral neuropathy is more likely in patients taking metformin in high doses for 10 or more years.20
Laboratory results and electrocardiography

Table 2 shows the patient’s initial laboratory results. Of note, her serum calcium level is 5.7 mg/dL (reference range 8.9–10.1). Electrocardiography in the emergency department shows:
- Prolonged PR interval (23 msec)
- Wide QRS complexes (13 msec)
- Flat T waves
- Prolonged corrected QT interval (475 msec)
- Occasional premature ventricular complexes.
CLINICAL MANIFESTATIONS OF HYPOCALCEMIA
2. Which of the following is not a manifestation of hypocalcemia?
- Tonic-clonic seizures
- Cyanosis
- Cardiac ventricular arrhythmias
- Acute pancreatitis
- Depression

Hypocalcemia can cause a wide range of clinical manifestations (Table 3), the extent and severity of which depend on the severity of hypocalcemia and how quickly it develops. The more acute the hypocalcemia, the more severe the manifestations.21
Tetany can cause seizures
Hypocalcemia is characterized by neuromuscular hyperexcitability, manifested clinically by tetany.22 Manifestations of tetany are numerous and include acral paresthesia, perioral numbness, muscle cramps, carpopedal spasm, and seizures. Tetany is the hallmark of hypocalcemia regardless of etiology. However, certain causes are associated with peculiar clinical manifestations. For example, chronic primary hypoparathyroidism may be associated with basal ganglia calcifications that can result in parkinsonism, other extrapyramidal disorders, and dementia (Table 4).6

Airway spasm can be fatal
A serious manifestation of acute severe hypocalcemia is spasm of the glottis muscles, which may cause cyanosis and, if untreated, death.21
Ventricular arrhythmias
Another potential fatal complication of acute severe hypocalcemia is polymorphic ventricular tachycardia due to prolongation of the QT interval, which is readily identified with electrocardiography.23
Hypocalcemia does not cause pancreatitis
Hypercalcemia, rather than hypocalcemia, may cause acute pancreatitis.24 Conversely, acute pancreatitis may cause hypocalcemia due to precipitation of calcium in the abdominal cavity.25
Psychiatric manifestations
In addition to depression, hypocalcemia is associated with psychiatric manifestations including anxiety, confusion, and emotional instability.
STEPS TO DIAGNOSIS OF HYPOCALCEMIA
First step: Confirm true hypocalcemia
Calcium circulates in the blood in 3 forms: bound to albumin (40% to 45%), bound to anions (10% to 15%), and free (ionized) (45%). Although ionized calcium is the active form, most laboratories report total serum calcium.
Since changes in serum albumin concentration affect the total serum calcium level, it is imperative to correct the measured serum calcium to the serum albumin concentration. Each 1-g/dL decrease in serum albumin lowers the total serum calcium by 0.8 mg/dL. Thus:
Corrected serum calcium (mg/dL) =
measured total serum calcium (mg/dL) +
0.8 (4 − serum albumin [g/dL]).
If the patient’s serum calcium level remains low when corrected for serum albumin, he or she has true hypocalcemia, which implies a low ionized serum calcium. Conversely, pseudohypocalcemia means that the measured calcium level is low but the corrected serum calcium is normal.
Using this formula, our patient’s corrected calcium level is calculated as 5.7 + 0.8 (4 – 3.2) = 6.3 mg/dL, indicating true hypocalcemia.
PHOSPHATE IS OFTEN HIGH WHEN CALCIUM IS LOW
In addition to hypocalcemia, our patient has an elevated phosphate level (Table 2).
3. Which of the following hypocalcemic disorders is not associated with hyperphosphatemia?
- End-stage renal disease
- Primary hypoparathyroidism
- Pseudohypoparathyroidism
- Vitamin D3 deficiency
- Rhabdomyolysis
Vitamin D deficiency is not associated with hyperphosphatemia.
Second step in evaluating hypocalcemia: Check phosphate, magnesium, creatinine

The major causes of hypocalcemia can be categorized according to the serum phosphate level: high vs normal or low (Table 5).
High-phosphate, low-calcium states. In the absence of concurrent end-stage renal disease and an excessive phosphate load, primary hypoparathyroidism is the most likely cause of hypocalcemia associated with hyperphosphatemia.
PTH increases serum ionized calcium by26,27:
- Increasing bone resorption
- Increasing reabsorption of calcium from the distal renal tubules
- Increasing the activity of 1-alpha-hydroxylase, responsible for conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (the most biologically active vitamin D metabolite); 1,25-dihydroxyvitamin D increases the absorption of calcium and phosphate from the intestine.
Conversely, PTH decreases reabsorption of phosphate from proximal renal tubules, resulting in hypophosphatemia. Therefore, low serum PTH (primary hypoparathyroidism) or a PTH-resistant state (pseudohypoparathyroidism) results in hypocalcemia and hyperphosphatemia.26,27
Both end-stage renal disease and rhabdomyolysis are associated with high serum phosphate levels. The kidney normally excretes excess dietary phosphate to maintain phosphate homeostasis; however, this is impaired in end-stage renal disease, leading to hyperphosphatemia. In rhabdomyolysis, it is mainly the transcellular shift of phosphate into the extracellular space from myocyte injury that raises phosphate levels.
Normal- or low-phosphate, low calcium states. Hypocalcemia can also result from vitamin D deficiency, but this cause is associated with a low or normal serum phosphate level. In such cases, hypocalcemia causes secondary hyperparathyroidism with consequent renal phosphate loss and, thus, hypophosphatemia.27
Third step: Check serum intact PTH and 25-hydroxyvitamin D levels
Hypocalcemia stimulates secretion of PTH. Therefore, hypocalcemia with elevated serum PTH is caused by disorders that do not impair PTH secretion, including chronic renal failure and vitamin D deficiency (Table 5). Conversely, hypocalcemia with low or normal serum PTH levels suggests primary hypoparathyroidism.
Our patient’s serum PTH level is 20 ng/mL, which is within the reference range. This does not discount the diagnosis of primary hypoparathyroidism. Although most patients with primary hypoparathyroidism have low or undetectable serum PTH levels, some have normal PTH levels if some degree of PTH production is preserved.5,7,28–30 In these patients, the remaining functioning parathyroid tissue is not enough to maintain a normal serum calcium level, resulting in hypocalcemia. As a result, hypocalcemia stimulates the remaining parathyroid tissue to its maximum output, producing PTH levels usually within the lower or middle-normal range.30 In such patients, the terms parathyroid insufficiency and relative primary hypoparathyroidism are more precise than primary hypoparathyroidism.
Postsurgical hypoparathyroidism with an inappropriately normal PTH level is usually seen in patients with disorders that impair intestinal calcium absorption or bone resorption.31 In our patient’s case, the “normal” serum PTH level is likely due to maximal stimulation of remaining functioning parathyroid tissue by severe hypocalcemia, which is a result of her discontinuation of calcium and calcitriol therapy and her vitamin D deficiency.
CASE RESUMED: NO RESPONSE TO INTRAVENOUS CALCIUM GLUCONATE
The patient is given 2 10-mL ampules of 10% calcium gluconate diluted in 100 mL of 5% dextrose in water over 20 minutes intravenously. Electrocardiographic monitoring is continued. Two hours later, her measured serum calcium is only 5.8 mg/dL, with no improvement in her symptoms.
A continuous infusion of calcium gluconate is started: 12 ampules of calcium gluconate are added to 380 mL of 5% dextrose in water and infused at 40 mL/hour (infused rate of elemental calcium = 1.3 mg/kg/hour); 3 hours later, her measured serum calcium level is still only 5.8 mg//dL; at 6 hours it is 5.9 mg/dL, and her symptoms have not improved.
4. Which of the following is the most appropriate next step?
- Change the calcium gluconate to calcium chloride
- Increase the infusion rate to 1.5 mg of elemental calcium/kg/hour
- Give a bolus of 2 10-mL ampules of 10% calcium gluconate intravenously over 1 minute
- Give additional oral calcium tablets
- Check the serum magnesium level
Treatment of hypocalcemia can involve intravenous or oral calcium therapy.
Intravenous calcium is indicated for patients with any of the following6,32:
- Moderate to severe neuromuscular irritability (eg, acral paresthesia, carpopedal spasm, prolonged QT interval, seizures, laryngospasm, bronchospasm)
- Acute hypocalcemia with corrected serum calcium level less than 7.6 mg/dL, even if the patient is asymptomatic
- Cardiac failure.
One 10-mL ampule of 10% calcium gluconate contains 93 mg of elemental calcium; 1 or 2 ampules are typically diluted in 50 to 100 mL of 5% dextrose in water and infused slowly over 15 to 20 minutes. Rapid administration of intravenous calcium is contraindicated, as it may produce cardiac arrhythmias and possibly cardiac arrest. Therefore, intravenous calcium should be given slowly while continuing electrocardiographic monitoring.33
Since the effect of 1 ampule of calcium gluconate lasts only 2 to 3 hours, most patients with symptomatic hypocalcemia require continuous intravenous calcium infusion. The recommended dose of infused elemental calcium is 0.5 to 1.5 mg/kg/hour.34 Several ampules are added to 500 to 1,000 mL of 5% dextrose in water or 0.9% normal saline and infused at a rate appropriate for the patient’s corrected calcium and symptoms.
Oral calcium and vitamin D supplements can be given initially to patients with a corrected serum calcium level of 7.6 mg/dL or greater, with or without mild symptoms, if they can tolerate oral intake. However, this is not the treatment of choice for resistant acute hypocalcemia, as in this case.
Calcium chloride has no advantages over calcium gluconate. Further, it can be associated with local irritation and may result in tissue necrosis if extravasation occurs.35
Increasing the infusion rate of calcium gluconate to the maximum recommended dose may improve the patient’s ionized calcium level and symptoms somewhat. However, it is not the best option for this patient, given that she did not respond to 2 ampules of calcium gluconate followed by continuous infusion of 1.3 mg/kg/hour for 6 hours.
Calcium gluconate bolus. Similarly, giving the patient an additional 2 ampules of calcium gluconate over 1 minute would not be recommended, as rapid administration of intravenous calcium gluconate (eg, over 1 minute) is contraindicated.
Check magnesium
If hypocalcemia persists despite intravenous calcium therapy, as in our patient, further investigation or action is required. An important cause of persistent hypocalcemia is severe hypomagnesemia. Severe hypomagnesemia (serum magnesium < 0.8 mg/dL) causes resistant hypocalcemia by several mechanisms:
- Inducing PTH resistance32,36,37
- Decreasing PTH secretion32,36
- Decreasing calcitriol production.
The decrease in calcitriol production is a direct effect of hypomagnesemia, but it is also an indirect effect of low PTH secretion, which inhibits the enzyme 1-alpha-hydroxylase. Thus, conversion of 25-hydroxyvitamin D3 to calcitriol is impaired, leading to low calcitriol production.
Our patient could have hypomagnesemia due to furosemide use and uncontrolled diabetes mellitus. Hypocalcemia resistant to calcium therapy may occasionally respond to magnesium therapy even if the serum magnesium level is normal. This may be due to depleted intracellular magnesium salt levels.6,38 Rarely, severe hypermagnesemia can also be associated with hypocalcemia due to inhibition of PTH secretion.37,39
CASE RESUMED
Our patient’s serum magnesium level is 0.6 mg/dL (reference range 1.7–2.4 mg/dL). She is given 2 g of magnesium sulfate in 60 mL of 0.9% normal saline infused over 1 hour, followed by a continuous infusion of magnesium sulfate (12 g diluted in 250 mL of 0.9% normal saline, infused over 24 hours). On repeat testing 4 hours later, her serum magnesium level is 0.7 mg/dL, and at 8 hours later it is 0.9 mg/dL. She is subsequently started on oral magnesium oxide 600 mg per day. The magnesium sulfate infusion is continued for another 24 hours.
PREVENTING HYPERCALCIURIA
Patients with low PTH (primary hypoparathyroidism) may have hypercalciuria due to decreased renal tubular calcium reabsorption. Two important measures can minimize hypercalciuria in such patients:
- Keeping the serum calcium level in the low-normal range4,5,40
- Giving a thiazide diuretic (eg, hydrochlorothiazide 12.5–50 mg daily) with a low-salt diet.41,42
A thiazide diuretic is usually started once the 24-hour urine calcium reaches 250 mg.6 Thiazides are thought to enhance both proximal and distal renal tubular calcium reabsorption.43,44
PRIMARY HYPOPARATHYROIDISM: LONG-TERM MANAGEMENT
Long-term management of primary hypoparathyroidism requires calcium and vitamin D supplementation.
Calcium supplements. The most commonly prescribed calcium preparations are calcium carbonate and calcium citrate (containing 40% and 20% elemental calcium, respectively). Calcium carbonate, which is less expensive than calcium citrate, binds with phosphate intake and requires an acidic environment for absorption, and so it is better absorbed when taken with meals. Because calcium citrate does not require an acidic environment for absorption, it is the calcium preparation of choice for patients on proton pump inhibitors, or patients with achlorhydria or constipation.45 Calcium doses vary widely, with most hypoparathyroid patients requiring 1 to 2 g of elemental calcium daily.6
Vitamin D supplements. To promote intestinal absorption, calcium is combined with vitamin D in a fixed-dose preparation given in divided doses.46 Calcitriol (1,25-dihydroxyvitamin D3) is the most active metabolite of vitamin D, with rapid onset and offset of action, and it is the preferred form of vitamin D therapy for patients with hypoparathyroidism. If calcitriol is not available or is not affordable, alphacalcidol (1-alpha-hydroxyvitamin D3) is another option. This is a synthetic analogue of vitamin D that is already hyroxylated at the C1 position. After oral intake, it is hydroxylated in the liver to form calcitriol.
Since renal production of calcitriol is PTH-dependent, in hypoparathyroidism the conversion of 25-hydroxyvitamin D3 to calcitriol is limited. Therefore, vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are not the preferred forms of vitamin D for such patients. However, either can be added to calcitriol, as they may have extraskeletal benefits.7
CASE CONCLUDED
Our patient presented with primary parathyroid insufficiency associated with vitamin D deficiency. Therefore, in addition to calcitriol and calcium combined with vitamin D in a fixed-dose preparation, her management included vitamin D3 for her vitamin D deficiency.
She was discharged on these medications. At a follow-up visit 3 weeks later, her measured serum calcium level was 8.6 mg/dL. She reported gradual resolution of her symptoms. She was also referred to a psychiatrist for her depression.
TAKE-HOME POINTS
- Hypocalcemia causes neuromuscular excitability, manifested clinically by tetany.
- Common causes of hypocalcemia include vitamin D deficiency, hypomagnesemia, renal failure, and primary hypoparathyroidism.
- The first step in evaluating hypocalcemia is to correct the measured serum calcium to the serum albumin concentration.
- Laboratory testing for hypocalcemia should include serum phosphorus, magnesium, creatinine, PTH, and 25-hydroxyvitamin D3.
- Primary hypoparathyroidism is characterized by hypocalcemia, hyperphosphatemia, and low serum PTH.
- Moderate to severe manifestations of hypo-
calcemia and acute hypocalcemia (< 7.6 mg/dL), even if asymptomatic, warrant intravenous calcium therapy. - Correction of hypomagnesemia is essential to treat hypocalcemia, especially if resistant to intravenous calcium therapy.
- The goal of chronic management of primary hypoparathyroidism includes correcting the serum calcium level to a low-normal range, the serum phosphorus level to an upper-normal range, and prevention of hypercalciuria.
Acknowledgments: The authors wish to thank Mr. Michael Edward Tierney of the School of Medicine, University of Sydney, Australia, for his linguistic editing of the manuscript.
- Jesus JE, Landry A. Images in clinical medicine. Chvostek’s and Trousseau’s signs. N Engl J Med 2012; 367:e15.
- Urbano FL. Signs of hypocalcemia: Chvostek’s and Trousseau’s. Hosp Physician 2000; 36:43–45.
- Chisthi MM, Nair RS, Kuttanchettiyar KG, Yadev I. Mechanisms behind post-thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin—a prospective study. J Invest Surg 2017; 30:217–225.
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 2016; 101:2300–2312.
- Stack BC Jr, Bimston DN, Bodenner DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: postoperative hypoparathyroidism—definitions and management. Endocr Pract 2015; 21:674–685.
- Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med 2008; 359:391–403.
- Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol (Lausanne) 2017; 7:172.
- Coimbra C, Monteiro F, Oliveira P, Ribeiro L, de Almeida MG, Condé A. Hypoparathyroidism following thyroidectomy: predictive factors. Acta Otorrinolaringol Esp 2017; 68:106–111.
- Thomusch O, Machens A, Sekulla C, Ukkat J, Brauckhoff M, Dralle H. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: a multivariate analysis of 5846 consecutive patients. Surgery 2003; 133:180–185.
- Hirsch PF, Lester GE, Talmage RV. Calcitonin, an enigmatic hormone: does it have a function? J Musculoskelet Neuronal Interact 2001; 1:299–305.
- Karne SS, Bhalerao NS. Carpal tunnel syndrome in hypothyroidism. J Clin Diagn Res 2016; 10:OC36–OC38.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Palumbo CF, Szabo RM, Olmsted SL. The effects of hypothyroidism and thyroid replacement on the development of carpal tunnel syndrome. J Hand Surg Am 2000; 25:734–739.
- Katz JN, Stirrat CR, Larson MG, Fossel AH, Eaton HM, Liang MH. A self-administered hand symptom diagram for the diagnosis and epidemiologic study of carpal tunnel syndrome. J Rheumatol 1990; 17:1495–1498.
- Katz JN, Stirrat CR. A self-administered hand diagram for the diagnosis of carpal tunnel syndrome. J Hand Surg Am 1990; 15:360–363.
- Calfee RP, Dale AM, Ryan D, Descatha A, Franzblau A, Evanoff B. Performance of simplified scoring systems for hand diagrams in carpal tunnel syndrome screening. J Hand Surg Am 2012; 37:10–17.
- D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? JAMA 2000; 283:3110–3117.
- Marchettini P, Lacerenza M, Mauri E, Marangoni C. Painful peripheral neuropathies. Curr Neuropharmacol 2006; 4:175–181.
- Kibirige D, Mwebaze R. Vitamin B12 deficiency among patients with diabetes mellitus: is routine screening and supplementation justified? J Diabetes Metab Disord 2013;12:17.
- Akinlade KS, Agbebaku SO, Rahamon SK, Balogun WO. Vitamin B12 levels in patients with type 2 diabetes mellitus on metformin. Ann Ib Postgrad Med 2015; 13:79–83.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Benoit SR, Mendelsohn AB, Nourjah P, Staffa JA, Graham DJ. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005; 12:363–368.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab 2009; 94:2115–2118.
- McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg 1994; 81:357–360.
- Talmage RV, Mobley HT. Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 2008; 156:1–8.
- Friedman PA, Gesek FA. Calcium transport in renal epithelial cells. Am J Physiol 1993; 264:F181–F198.
- Jensen PV, Jelstrup SM, Homøe P. Long-term outcomes after total thyroidectomy. Dan Med J 2015; 62:A5156.
- Ritter K, Elfenbein D, Schneider DF, Chen H, Sippel RS. Hypoparathyroidism after total thyroidectomy: incidence and resolution. J Surg Res 2015; 197:348–353.
- Promberger R, Ott J, Kober F, Karik M, Freissmuth M, Hermann M. Normal parathyroid hormone levels do not exclude permanent hypoparathyroidism after thyroidectomy. Thyroid 2011; 21:145–150.
- Lorente-Poch L, Sancho JJ, Muñoz-Nova JL, Sánchez-Velázquez P, Sitges-Serra A. Defining the syndromes of parathyroid failure after total thyroidectomy. Gland Surgery 2015; 4:82–90.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tohme JF, Bilezikian JP. Diagnosis and treatment of hypocalcemic emergencies. Endocrinologist 1996; 6:10–18.
- Carroll R, Matfin G. Endocrine and metabolic emergencies: hypocalcaemia. Ther Adv Endocrinol Metab 2010; 1:29–33.
- Kim MP, Raho VJ, Mak J, Kaynar AM. Skin and soft tissue necrosis from calcium chloride in a deicer. J Emerg Med 2007; 32:41–44.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Cholst IN, Steinberg SF, Tropper PJ, Fox HE, Segre GV, Bilezikian JP. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med 1984; 310:1221–1225.
- Ryzen E, Nelson TA, Rude RK. Low blood mononuclear cell magnesium content and hypocalcemia in normomagnesemic patients. West J Med 1987; 147:549–553.
- Koontz SL, Friedman SA, Schwartz ML. Symptomatic hypocalcemia after tocolytic therapy with magnesium sulfate and nifedipine. Am J Obstet Gynecol 2004; 190:1773–1776.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Porter RH, Cox BG, Heaney D, Hostetter TH, Stinebaugh BJ, Suki WN. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978; 298:577–581.
- Clarke BL, Brown EM, Collins MT, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metab 2016; 101:2284–2299.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005; 115:1651–1658.
- Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol 1985; 248:F527–F535.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Scotti A, Bianchini C, Abbiati G, Marzo A. Absorption of calcium administered alone or in fixed combination with vitamin D to healthy volunteers. Arzneimittelforschung 2001; 51:493–500.
A 67-year-old woman presents to the emergency department after 8 weeks of progressive numbness and tingling in both hands, involving all fingers. The numbness has increased in severity in the last 3 days. She also has occasional numbness around her mouth. She reports no numbness in her feet.
She says she underwent thyroid surgery twice for thyroid cancer 10 years ago. Her medical history also includes type 2 diabetes mellitus (diagnosed 1 year ago), hypertension, dyslipidemia, and diastolic heart failure (diagnosed 5 years ago).
Her current medications are:
- Metformin 1 g twice a day
- Candesartan 16 mg once a day
- Atorvastatin 20 mg once a day
- Furosemide 40 mg twice a day
- Levothyroxine 100 μg per day
- Calcium carbonate 1,500 mg twice a day
- A vitamin D tablet twice a day, which she has not taken for the last 2 months.
She admits she has not been taking her medications regularly because she has been feeling depressed.
On physical examination, she is alert and oriented but appears anxious. She is not in respiratory distress. Her blood pressure is 150/90 mm Hg and her pulse is 92 beats per minute and regular. There is a thyroidectomy scar on the anterior neck. Her jugular venous pressure is not elevated. Her heart sounds are normal without extra sounds. She has no pulmonary rales and no lower-extremity edema.
The Phalen test and Tinel test for carpal tunnel syndrome are negative in both hands. Using a Katz hand diagram, the patient reports tingling and numbness in all fingers, both palms, and the dorsum of both hands. Tapping the area over the facial nerve does not elicit twitching of the facial muscles (ie, no Chvostek sign), but compression of the upper arm elicits carpal spasm (ie, positive Trousseau sign). There is no evidence of motor weakness in her hands. The rest of the physical examination is unremarkable.
POSSIBLE CAUSES OF NUMBNESS
1. Based on the initial evaluation, which of the following is the most likely cause of our patient’s bilateral hand numbness?
- Hypocalcemia due to primary hypoparathyroidism
- Carpal tunnel syndrome due to primary hypothyroidism
- Diabetic peripheral neuropathy
- Vitamin B12 deficiency due to metformin
- Hypocalcemia due to low serum calcitonin
All the conditions above except low serum calcitonin can cause bilateral hand paresthesia. Our patient most likely has hypocalcemia due to primary hypoparathyroidism.
Hypocalcemia
In our patient, bilateral hand numbness and perioral numbness after stopping vitamin D and a positive Trousseau sign strongly suggest hypocalcemia. The classic physical findings in patients with hypocalcemia are the Trousseau sign and the Chvostek sign. The Trousseau sign is elicited by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and observing for ischemia-induced carpopedal spasm, wrist and metacarpophalangeal joint flexion, thumb adduction, and interphalangeal joint extension. The Chvostek sign is elicited by tapping over the area of the facial nerve below the zygoma in front of the tragus, resulting in ipsilateral twitching of facial muscles.
Although the Trousseau sign is more sensitive and specific than the Chvostek sign, neither is pathognomonic for hypocalcemia.1 The Chvostek sign has been reported to be negative in 30% of patients with hypocalcemia and positive in 10% of normocalcemic individuals.1 The Trousseau sign, however, is present in 94% of hypocalcemic patients vs 1% of normocalcemic individuals.2
Primary hypoparathyroidism secondary to thyroidectomy. Postsurgical hypoparathyroidism is the most common cause of primary hypoparathyroidism. It results from ischemic injury or accidental removal of the parathyroid glands during anterior neck surgery.3,4 The consequent hypocalcemia can be transient, intermittent, or permanent. Permanent postsurgical hypoparathyroidism is defined as persistent hypocalcemia with insufficient parathyroid hormone (PTH) for more than 12 months after neck surgery; however, some consider 6 months to be enough to define the condition.5–7
The incidence of postsurgical hypoparathyroidism varies considerably with the extent of thyroid surgery and the experience of the surgeon.6,8 In the hands of experienced surgeons, permanent hypoparathyroidism occurs in fewer than 1% of patients after total thyroidectomy, whereas the rate may be higher than 6% with less-experienced surgeons.5,9 Other risk factors for postsurgical hypoparathyroidism include female sex, autoimmune thyroid disease, pregnancy, and lactation.5
Pseudohypoparathyroidism is a group of disorders characterized by renal resistance to PTH, leading to hypocalcemia, hyperphosphatemia, and elevated serum PTH. It is also associated with phenotypic features such as short stature and short fourth metacarpal bones.
Calcitonin deficiency. Calcitonin is a polypeptide hormone secreted from the parafollicular (C) cells of the thyroid gland. After total thyroidectomy, calcitonin levels are expected to be reduced. However, the role of calcitonin in humans is unclear. One study has shown that calcitonin is possibly a vestigial hormone, given that no calcitonin-related disorders (excess or deficiency) have been reported in humans.10
Carpal tunnel syndrome due to hypothyroidism
Our patient also could have primary hypothyroidism as a result of thyroidectomy. Hypothyroidism can cause bilateral hand numbness due to carpal tunnel syndrome, which is mediated by mucopolysaccharide deposition and synovial membrane swelling.11 One study reported that 29% of patients with hypothyroidism had carpal tunnel syndrome.12 Symptoms of carpal tunnel syndrome in hypothyroid patients may occur despite thyroid replacement therapy.13
Carpal tunnel syndrome is a clinical diagnosis. Patients usually experience hand paresthesia in the distribution of the median nerve. Provocative physical tests for carpal tunnel syndrome include the Tinel test, the Phalen test, and the Katz hand diagram, which is considered the best of the 3 tests.14,15 Based on how the patient marks the location and type of symptoms on the diagram, carpal tunnel syndrome is rated as classic, probable, possible, or unlikely (Table 1).14,16,17 The sensitivity of a classic or probable diagram ranges from 64% to 80%, while the specificity ranges from 73% to 90%.14,15
Carpal tunnel syndrome is less likely to be the cause of our patient’s symptoms, as her Katz hand diagram indicates only “possible” carpal tunnel syndrome. Her perioral numbness and positive Trousseau sign make hypocalcemia a more likely cause.
Diabetic peripheral neuropathy
Sensory peripheral neuropathy is a recognized complication of diabetes mellitus. However, neuropathy in diabetic patients most commonly manifests initially as distal symmetrical ascending neuropathy starting in the lower extremities.18 Therefore, diabetic peripheral neuropathy is less likely in this patient since her symptoms are limited to her hands.
Vitamin B12 deficiency
Metformin-induced vitamin B12 deficiency is another possible cause of peripheral neuropathy. It might be secondary to metformin-induced changes in intrinsic factor levels and small-intestine motility with resultant bacterial overgrowth, as well as inhibition of vitamin B12 absorption in the terminal ileum.19
However, metformin-induced vitamin B12 deficiency is not the most likely cause of our patient’s neuropathy, since she has been taking this drug for only 1 year. Vitamin B12 deficiency with consequent peripheral neuropathy is more likely in patients taking metformin in high doses for 10 or more years.20
Laboratory results and electrocardiography
Table 2 shows the patient’s initial laboratory results. Of note, her serum calcium level is 5.7 mg/dL (reference range 8.9–10.1). Electrocardiography in the emergency department shows:
- Prolonged PR interval (23 msec)
- Wide QRS complexes (13 msec)
- Flat T waves
- Prolonged corrected QT interval (475 msec)
- Occasional premature ventricular complexes.
CLINICAL MANIFESTATIONS OF HYPOCALCEMIA
2. Which of the following is not a manifestation of hypocalcemia?
- Tonic-clonic seizures
- Cyanosis
- Cardiac ventricular arrhythmias
- Acute pancreatitis
- Depression
Hypocalcemia can cause a wide range of clinical manifestations (Table 3), the extent and severity of which depend on the severity of hypocalcemia and how quickly it develops. The more acute the hypocalcemia, the more severe the manifestations.21
Tetany can cause seizures
Hypocalcemia is characterized by neuromuscular hyperexcitability, manifested clinically by tetany.22 Manifestations of tetany are numerous and include acral paresthesia, perioral numbness, muscle cramps, carpopedal spasm, and seizures. Tetany is the hallmark of hypocalcemia regardless of etiology. However, certain causes are associated with peculiar clinical manifestations. For example, chronic primary hypoparathyroidism may be associated with basal ganglia calcifications that can result in parkinsonism, other extrapyramidal disorders, and dementia (Table 4).6
Airway spasm can be fatal
A serious manifestation of acute severe hypocalcemia is spasm of the glottis muscles, which may cause cyanosis and, if untreated, death.21
Ventricular arrhythmias
Another potential fatal complication of acute severe hypocalcemia is polymorphic ventricular tachycardia due to prolongation of the QT interval, which is readily identified with electrocardiography.23
Hypocalcemia does not cause pancreatitis
Hypercalcemia, rather than hypocalcemia, may cause acute pancreatitis.24 Conversely, acute pancreatitis may cause hypocalcemia due to precipitation of calcium in the abdominal cavity.25
Psychiatric manifestations
In addition to depression, hypocalcemia is associated with psychiatric manifestations including anxiety, confusion, and emotional instability.
STEPS TO DIAGNOSIS OF HYPOCALCEMIA
First step: Confirm true hypocalcemia
Calcium circulates in the blood in 3 forms: bound to albumin (40% to 45%), bound to anions (10% to 15%), and free (ionized) (45%). Although ionized calcium is the active form, most laboratories report total serum calcium.
Since changes in serum albumin concentration affect the total serum calcium level, it is imperative to correct the measured serum calcium to the serum albumin concentration. Each 1-g/dL decrease in serum albumin lowers the total serum calcium by 0.8 mg/dL. Thus:
Corrected serum calcium (mg/dL) =
measured total serum calcium (mg/dL) +
0.8 (4 − serum albumin [g/dL]).
If the patient’s serum calcium level remains low when corrected for serum albumin, he or she has true hypocalcemia, which implies a low ionized serum calcium. Conversely, pseudohypocalcemia means that the measured calcium level is low but the corrected serum calcium is normal.
Using this formula, our patient’s corrected calcium level is calculated as 5.7 + 0.8 (4 – 3.2) = 6.3 mg/dL, indicating true hypocalcemia.
PHOSPHATE IS OFTEN HIGH WHEN CALCIUM IS LOW
In addition to hypocalcemia, our patient has an elevated phosphate level (Table 2).
3. Which of the following hypocalcemic disorders is not associated with hyperphosphatemia?
- End-stage renal disease
- Primary hypoparathyroidism
- Pseudohypoparathyroidism
- Vitamin D3 deficiency
- Rhabdomyolysis
Vitamin D deficiency is not associated with hyperphosphatemia.
Second step in evaluating hypocalcemia: Check phosphate, magnesium, creatinine
The major causes of hypocalcemia can be categorized according to the serum phosphate level: high vs normal or low (Table 5).
High-phosphate, low-calcium states. In the absence of concurrent end-stage renal disease and an excessive phosphate load, primary hypoparathyroidism is the most likely cause of hypocalcemia associated with hyperphosphatemia.
PTH increases serum ionized calcium by26,27:
- Increasing bone resorption
- Increasing reabsorption of calcium from the distal renal tubules
- Increasing the activity of 1-alpha-hydroxylase, responsible for conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (the most biologically active vitamin D metabolite); 1,25-dihydroxyvitamin D increases the absorption of calcium and phosphate from the intestine.
Conversely, PTH decreases reabsorption of phosphate from proximal renal tubules, resulting in hypophosphatemia. Therefore, low serum PTH (primary hypoparathyroidism) or a PTH-resistant state (pseudohypoparathyroidism) results in hypocalcemia and hyperphosphatemia.26,27
Both end-stage renal disease and rhabdomyolysis are associated with high serum phosphate levels. The kidney normally excretes excess dietary phosphate to maintain phosphate homeostasis; however, this is impaired in end-stage renal disease, leading to hyperphosphatemia. In rhabdomyolysis, it is mainly the transcellular shift of phosphate into the extracellular space from myocyte injury that raises phosphate levels.
Normal- or low-phosphate, low calcium states. Hypocalcemia can also result from vitamin D deficiency, but this cause is associated with a low or normal serum phosphate level. In such cases, hypocalcemia causes secondary hyperparathyroidism with consequent renal phosphate loss and, thus, hypophosphatemia.27
Third step: Check serum intact PTH and 25-hydroxyvitamin D levels
Hypocalcemia stimulates secretion of PTH. Therefore, hypocalcemia with elevated serum PTH is caused by disorders that do not impair PTH secretion, including chronic renal failure and vitamin D deficiency (Table 5). Conversely, hypocalcemia with low or normal serum PTH levels suggests primary hypoparathyroidism.
Our patient’s serum PTH level is 20 ng/mL, which is within the reference range. This does not discount the diagnosis of primary hypoparathyroidism. Although most patients with primary hypoparathyroidism have low or undetectable serum PTH levels, some have normal PTH levels if some degree of PTH production is preserved.5,7,28–30 In these patients, the remaining functioning parathyroid tissue is not enough to maintain a normal serum calcium level, resulting in hypocalcemia. As a result, hypocalcemia stimulates the remaining parathyroid tissue to its maximum output, producing PTH levels usually within the lower or middle-normal range.30 In such patients, the terms parathyroid insufficiency and relative primary hypoparathyroidism are more precise than primary hypoparathyroidism.
Postsurgical hypoparathyroidism with an inappropriately normal PTH level is usually seen in patients with disorders that impair intestinal calcium absorption or bone resorption.31 In our patient’s case, the “normal” serum PTH level is likely due to maximal stimulation of remaining functioning parathyroid tissue by severe hypocalcemia, which is a result of her discontinuation of calcium and calcitriol therapy and her vitamin D deficiency.
CASE RESUMED: NO RESPONSE TO INTRAVENOUS CALCIUM GLUCONATE
The patient is given 2 10-mL ampules of 10% calcium gluconate diluted in 100 mL of 5% dextrose in water over 20 minutes intravenously. Electrocardiographic monitoring is continued. Two hours later, her measured serum calcium is only 5.8 mg/dL, with no improvement in her symptoms.
A continuous infusion of calcium gluconate is started: 12 ampules of calcium gluconate are added to 380 mL of 5% dextrose in water and infused at 40 mL/hour (infused rate of elemental calcium = 1.3 mg/kg/hour); 3 hours later, her measured serum calcium level is still only 5.8 mg//dL; at 6 hours it is 5.9 mg/dL, and her symptoms have not improved.
4. Which of the following is the most appropriate next step?
- Change the calcium gluconate to calcium chloride
- Increase the infusion rate to 1.5 mg of elemental calcium/kg/hour
- Give a bolus of 2 10-mL ampules of 10% calcium gluconate intravenously over 1 minute
- Give additional oral calcium tablets
- Check the serum magnesium level
Treatment of hypocalcemia can involve intravenous or oral calcium therapy.
Intravenous calcium is indicated for patients with any of the following6,32:
- Moderate to severe neuromuscular irritability (eg, acral paresthesia, carpopedal spasm, prolonged QT interval, seizures, laryngospasm, bronchospasm)
- Acute hypocalcemia with corrected serum calcium level less than 7.6 mg/dL, even if the patient is asymptomatic
- Cardiac failure.
One 10-mL ampule of 10% calcium gluconate contains 93 mg of elemental calcium; 1 or 2 ampules are typically diluted in 50 to 100 mL of 5% dextrose in water and infused slowly over 15 to 20 minutes. Rapid administration of intravenous calcium is contraindicated, as it may produce cardiac arrhythmias and possibly cardiac arrest. Therefore, intravenous calcium should be given slowly while continuing electrocardiographic monitoring.33
Since the effect of 1 ampule of calcium gluconate lasts only 2 to 3 hours, most patients with symptomatic hypocalcemia require continuous intravenous calcium infusion. The recommended dose of infused elemental calcium is 0.5 to 1.5 mg/kg/hour.34 Several ampules are added to 500 to 1,000 mL of 5% dextrose in water or 0.9% normal saline and infused at a rate appropriate for the patient’s corrected calcium and symptoms.
Oral calcium and vitamin D supplements can be given initially to patients with a corrected serum calcium level of 7.6 mg/dL or greater, with or without mild symptoms, if they can tolerate oral intake. However, this is not the treatment of choice for resistant acute hypocalcemia, as in this case.
Calcium chloride has no advantages over calcium gluconate. Further, it can be associated with local irritation and may result in tissue necrosis if extravasation occurs.35
Increasing the infusion rate of calcium gluconate to the maximum recommended dose may improve the patient’s ionized calcium level and symptoms somewhat. However, it is not the best option for this patient, given that she did not respond to 2 ampules of calcium gluconate followed by continuous infusion of 1.3 mg/kg/hour for 6 hours.
Calcium gluconate bolus. Similarly, giving the patient an additional 2 ampules of calcium gluconate over 1 minute would not be recommended, as rapid administration of intravenous calcium gluconate (eg, over 1 minute) is contraindicated.
Check magnesium
If hypocalcemia persists despite intravenous calcium therapy, as in our patient, further investigation or action is required. An important cause of persistent hypocalcemia is severe hypomagnesemia. Severe hypomagnesemia (serum magnesium < 0.8 mg/dL) causes resistant hypocalcemia by several mechanisms:
- Inducing PTH resistance32,36,37
- Decreasing PTH secretion32,36
- Decreasing calcitriol production.
The decrease in calcitriol production is a direct effect of hypomagnesemia, but it is also an indirect effect of low PTH secretion, which inhibits the enzyme 1-alpha-hydroxylase. Thus, conversion of 25-hydroxyvitamin D3 to calcitriol is impaired, leading to low calcitriol production.
Our patient could have hypomagnesemia due to furosemide use and uncontrolled diabetes mellitus. Hypocalcemia resistant to calcium therapy may occasionally respond to magnesium therapy even if the serum magnesium level is normal. This may be due to depleted intracellular magnesium salt levels.6,38 Rarely, severe hypermagnesemia can also be associated with hypocalcemia due to inhibition of PTH secretion.37,39
CASE RESUMED
Our patient’s serum magnesium level is 0.6 mg/dL (reference range 1.7–2.4 mg/dL). She is given 2 g of magnesium sulfate in 60 mL of 0.9% normal saline infused over 1 hour, followed by a continuous infusion of magnesium sulfate (12 g diluted in 250 mL of 0.9% normal saline, infused over 24 hours). On repeat testing 4 hours later, her serum magnesium level is 0.7 mg/dL, and at 8 hours later it is 0.9 mg/dL. She is subsequently started on oral magnesium oxide 600 mg per day. The magnesium sulfate infusion is continued for another 24 hours.
PREVENTING HYPERCALCIURIA
Patients with low PTH (primary hypoparathyroidism) may have hypercalciuria due to decreased renal tubular calcium reabsorption. Two important measures can minimize hypercalciuria in such patients:
- Keeping the serum calcium level in the low-normal range4,5,40
- Giving a thiazide diuretic (eg, hydrochlorothiazide 12.5–50 mg daily) with a low-salt diet.41,42
A thiazide diuretic is usually started once the 24-hour urine calcium reaches 250 mg.6 Thiazides are thought to enhance both proximal and distal renal tubular calcium reabsorption.43,44
PRIMARY HYPOPARATHYROIDISM: LONG-TERM MANAGEMENT
Long-term management of primary hypoparathyroidism requires calcium and vitamin D supplementation.
Calcium supplements. The most commonly prescribed calcium preparations are calcium carbonate and calcium citrate (containing 40% and 20% elemental calcium, respectively). Calcium carbonate, which is less expensive than calcium citrate, binds with phosphate intake and requires an acidic environment for absorption, and so it is better absorbed when taken with meals. Because calcium citrate does not require an acidic environment for absorption, it is the calcium preparation of choice for patients on proton pump inhibitors, or patients with achlorhydria or constipation.45 Calcium doses vary widely, with most hypoparathyroid patients requiring 1 to 2 g of elemental calcium daily.6
Vitamin D supplements. To promote intestinal absorption, calcium is combined with vitamin D in a fixed-dose preparation given in divided doses.46 Calcitriol (1,25-dihydroxyvitamin D3) is the most active metabolite of vitamin D, with rapid onset and offset of action, and it is the preferred form of vitamin D therapy for patients with hypoparathyroidism. If calcitriol is not available or is not affordable, alphacalcidol (1-alpha-hydroxyvitamin D3) is another option. This is a synthetic analogue of vitamin D that is already hyroxylated at the C1 position. After oral intake, it is hydroxylated in the liver to form calcitriol.
Since renal production of calcitriol is PTH-dependent, in hypoparathyroidism the conversion of 25-hydroxyvitamin D3 to calcitriol is limited. Therefore, vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are not the preferred forms of vitamin D for such patients. However, either can be added to calcitriol, as they may have extraskeletal benefits.7
CASE CONCLUDED
Our patient presented with primary parathyroid insufficiency associated with vitamin D deficiency. Therefore, in addition to calcitriol and calcium combined with vitamin D in a fixed-dose preparation, her management included vitamin D3 for her vitamin D deficiency.
She was discharged on these medications. At a follow-up visit 3 weeks later, her measured serum calcium level was 8.6 mg/dL. She reported gradual resolution of her symptoms. She was also referred to a psychiatrist for her depression.
TAKE-HOME POINTS
- Hypocalcemia causes neuromuscular excitability, manifested clinically by tetany.
- Common causes of hypocalcemia include vitamin D deficiency, hypomagnesemia, renal failure, and primary hypoparathyroidism.
- The first step in evaluating hypocalcemia is to correct the measured serum calcium to the serum albumin concentration.
- Laboratory testing for hypocalcemia should include serum phosphorus, magnesium, creatinine, PTH, and 25-hydroxyvitamin D3.
- Primary hypoparathyroidism is characterized by hypocalcemia, hyperphosphatemia, and low serum PTH.
- Moderate to severe manifestations of hypo-
calcemia and acute hypocalcemia (< 7.6 mg/dL), even if asymptomatic, warrant intravenous calcium therapy. - Correction of hypomagnesemia is essential to treat hypocalcemia, especially if resistant to intravenous calcium therapy.
- The goal of chronic management of primary hypoparathyroidism includes correcting the serum calcium level to a low-normal range, the serum phosphorus level to an upper-normal range, and prevention of hypercalciuria.
Acknowledgments: The authors wish to thank Mr. Michael Edward Tierney of the School of Medicine, University of Sydney, Australia, for his linguistic editing of the manuscript.
A 67-year-old woman presents to the emergency department after 8 weeks of progressive numbness and tingling in both hands, involving all fingers. The numbness has increased in severity in the last 3 days. She also has occasional numbness around her mouth. She reports no numbness in her feet.
She says she underwent thyroid surgery twice for thyroid cancer 10 years ago. Her medical history also includes type 2 diabetes mellitus (diagnosed 1 year ago), hypertension, dyslipidemia, and diastolic heart failure (diagnosed 5 years ago).
Her current medications are:
- Metformin 1 g twice a day
- Candesartan 16 mg once a day
- Atorvastatin 20 mg once a day
- Furosemide 40 mg twice a day
- Levothyroxine 100 μg per day
- Calcium carbonate 1,500 mg twice a day
- A vitamin D tablet twice a day, which she has not taken for the last 2 months.
She admits she has not been taking her medications regularly because she has been feeling depressed.
On physical examination, she is alert and oriented but appears anxious. She is not in respiratory distress. Her blood pressure is 150/90 mm Hg and her pulse is 92 beats per minute and regular. There is a thyroidectomy scar on the anterior neck. Her jugular venous pressure is not elevated. Her heart sounds are normal without extra sounds. She has no pulmonary rales and no lower-extremity edema.
The Phalen test and Tinel test for carpal tunnel syndrome are negative in both hands. Using a Katz hand diagram, the patient reports tingling and numbness in all fingers, both palms, and the dorsum of both hands. Tapping the area over the facial nerve does not elicit twitching of the facial muscles (ie, no Chvostek sign), but compression of the upper arm elicits carpal spasm (ie, positive Trousseau sign). There is no evidence of motor weakness in her hands. The rest of the physical examination is unremarkable.
POSSIBLE CAUSES OF NUMBNESS
1. Based on the initial evaluation, which of the following is the most likely cause of our patient’s bilateral hand numbness?
- Hypocalcemia due to primary hypoparathyroidism
- Carpal tunnel syndrome due to primary hypothyroidism
- Diabetic peripheral neuropathy
- Vitamin B12 deficiency due to metformin
- Hypocalcemia due to low serum calcitonin
All the conditions above except low serum calcitonin can cause bilateral hand paresthesia. Our patient most likely has hypocalcemia due to primary hypoparathyroidism.
Hypocalcemia
In our patient, bilateral hand numbness and perioral numbness after stopping vitamin D and a positive Trousseau sign strongly suggest hypocalcemia. The classic physical findings in patients with hypocalcemia are the Trousseau sign and the Chvostek sign. The Trousseau sign is elicited by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and observing for ischemia-induced carpopedal spasm, wrist and metacarpophalangeal joint flexion, thumb adduction, and interphalangeal joint extension. The Chvostek sign is elicited by tapping over the area of the facial nerve below the zygoma in front of the tragus, resulting in ipsilateral twitching of facial muscles.
Although the Trousseau sign is more sensitive and specific than the Chvostek sign, neither is pathognomonic for hypocalcemia.1 The Chvostek sign has been reported to be negative in 30% of patients with hypocalcemia and positive in 10% of normocalcemic individuals.1 The Trousseau sign, however, is present in 94% of hypocalcemic patients vs 1% of normocalcemic individuals.2
Primary hypoparathyroidism secondary to thyroidectomy. Postsurgical hypoparathyroidism is the most common cause of primary hypoparathyroidism. It results from ischemic injury or accidental removal of the parathyroid glands during anterior neck surgery.3,4 The consequent hypocalcemia can be transient, intermittent, or permanent. Permanent postsurgical hypoparathyroidism is defined as persistent hypocalcemia with insufficient parathyroid hormone (PTH) for more than 12 months after neck surgery; however, some consider 6 months to be enough to define the condition.5–7
The incidence of postsurgical hypoparathyroidism varies considerably with the extent of thyroid surgery and the experience of the surgeon.6,8 In the hands of experienced surgeons, permanent hypoparathyroidism occurs in fewer than 1% of patients after total thyroidectomy, whereas the rate may be higher than 6% with less-experienced surgeons.5,9 Other risk factors for postsurgical hypoparathyroidism include female sex, autoimmune thyroid disease, pregnancy, and lactation.5
Pseudohypoparathyroidism is a group of disorders characterized by renal resistance to PTH, leading to hypocalcemia, hyperphosphatemia, and elevated serum PTH. It is also associated with phenotypic features such as short stature and short fourth metacarpal bones.
Calcitonin deficiency. Calcitonin is a polypeptide hormone secreted from the parafollicular (C) cells of the thyroid gland. After total thyroidectomy, calcitonin levels are expected to be reduced. However, the role of calcitonin in humans is unclear. One study has shown that calcitonin is possibly a vestigial hormone, given that no calcitonin-related disorders (excess or deficiency) have been reported in humans.10
Carpal tunnel syndrome due to hypothyroidism
Our patient also could have primary hypothyroidism as a result of thyroidectomy. Hypothyroidism can cause bilateral hand numbness due to carpal tunnel syndrome, which is mediated by mucopolysaccharide deposition and synovial membrane swelling.11 One study reported that 29% of patients with hypothyroidism had carpal tunnel syndrome.12 Symptoms of carpal tunnel syndrome in hypothyroid patients may occur despite thyroid replacement therapy.13
Carpal tunnel syndrome is a clinical diagnosis. Patients usually experience hand paresthesia in the distribution of the median nerve. Provocative physical tests for carpal tunnel syndrome include the Tinel test, the Phalen test, and the Katz hand diagram, which is considered the best of the 3 tests.14,15 Based on how the patient marks the location and type of symptoms on the diagram, carpal tunnel syndrome is rated as classic, probable, possible, or unlikely (Table 1).14,16,17 The sensitivity of a classic or probable diagram ranges from 64% to 80%, while the specificity ranges from 73% to 90%.14,15
Carpal tunnel syndrome is less likely to be the cause of our patient’s symptoms, as her Katz hand diagram indicates only “possible” carpal tunnel syndrome. Her perioral numbness and positive Trousseau sign make hypocalcemia a more likely cause.
Diabetic peripheral neuropathy
Sensory peripheral neuropathy is a recognized complication of diabetes mellitus. However, neuropathy in diabetic patients most commonly manifests initially as distal symmetrical ascending neuropathy starting in the lower extremities.18 Therefore, diabetic peripheral neuropathy is less likely in this patient since her symptoms are limited to her hands.
Vitamin B12 deficiency
Metformin-induced vitamin B12 deficiency is another possible cause of peripheral neuropathy. It might be secondary to metformin-induced changes in intrinsic factor levels and small-intestine motility with resultant bacterial overgrowth, as well as inhibition of vitamin B12 absorption in the terminal ileum.19
However, metformin-induced vitamin B12 deficiency is not the most likely cause of our patient’s neuropathy, since she has been taking this drug for only 1 year. Vitamin B12 deficiency with consequent peripheral neuropathy is more likely in patients taking metformin in high doses for 10 or more years.20
Laboratory results and electrocardiography
Table 2 shows the patient’s initial laboratory results. Of note, her serum calcium level is 5.7 mg/dL (reference range 8.9–10.1). Electrocardiography in the emergency department shows:
- Prolonged PR interval (23 msec)
- Wide QRS complexes (13 msec)
- Flat T waves
- Prolonged corrected QT interval (475 msec)
- Occasional premature ventricular complexes.
CLINICAL MANIFESTATIONS OF HYPOCALCEMIA
2. Which of the following is not a manifestation of hypocalcemia?
- Tonic-clonic seizures
- Cyanosis
- Cardiac ventricular arrhythmias
- Acute pancreatitis
- Depression
Hypocalcemia can cause a wide range of clinical manifestations (Table 3), the extent and severity of which depend on the severity of hypocalcemia and how quickly it develops. The more acute the hypocalcemia, the more severe the manifestations.21
Tetany can cause seizures
Hypocalcemia is characterized by neuromuscular hyperexcitability, manifested clinically by tetany.22 Manifestations of tetany are numerous and include acral paresthesia, perioral numbness, muscle cramps, carpopedal spasm, and seizures. Tetany is the hallmark of hypocalcemia regardless of etiology. However, certain causes are associated with peculiar clinical manifestations. For example, chronic primary hypoparathyroidism may be associated with basal ganglia calcifications that can result in parkinsonism, other extrapyramidal disorders, and dementia (Table 4).6
Airway spasm can be fatal
A serious manifestation of acute severe hypocalcemia is spasm of the glottis muscles, which may cause cyanosis and, if untreated, death.21
Ventricular arrhythmias
Another potential fatal complication of acute severe hypocalcemia is polymorphic ventricular tachycardia due to prolongation of the QT interval, which is readily identified with electrocardiography.23
Hypocalcemia does not cause pancreatitis
Hypercalcemia, rather than hypocalcemia, may cause acute pancreatitis.24 Conversely, acute pancreatitis may cause hypocalcemia due to precipitation of calcium in the abdominal cavity.25
Psychiatric manifestations
In addition to depression, hypocalcemia is associated with psychiatric manifestations including anxiety, confusion, and emotional instability.
STEPS TO DIAGNOSIS OF HYPOCALCEMIA
First step: Confirm true hypocalcemia
Calcium circulates in the blood in 3 forms: bound to albumin (40% to 45%), bound to anions (10% to 15%), and free (ionized) (45%). Although ionized calcium is the active form, most laboratories report total serum calcium.
Since changes in serum albumin concentration affect the total serum calcium level, it is imperative to correct the measured serum calcium to the serum albumin concentration. Each 1-g/dL decrease in serum albumin lowers the total serum calcium by 0.8 mg/dL. Thus:
Corrected serum calcium (mg/dL) =
measured total serum calcium (mg/dL) +
0.8 (4 − serum albumin [g/dL]).
If the patient’s serum calcium level remains low when corrected for serum albumin, he or she has true hypocalcemia, which implies a low ionized serum calcium. Conversely, pseudohypocalcemia means that the measured calcium level is low but the corrected serum calcium is normal.
Using this formula, our patient’s corrected calcium level is calculated as 5.7 + 0.8 (4 – 3.2) = 6.3 mg/dL, indicating true hypocalcemia.
PHOSPHATE IS OFTEN HIGH WHEN CALCIUM IS LOW
In addition to hypocalcemia, our patient has an elevated phosphate level (Table 2).
3. Which of the following hypocalcemic disorders is not associated with hyperphosphatemia?
- End-stage renal disease
- Primary hypoparathyroidism
- Pseudohypoparathyroidism
- Vitamin D3 deficiency
- Rhabdomyolysis
Vitamin D deficiency is not associated with hyperphosphatemia.
Second step in evaluating hypocalcemia: Check phosphate, magnesium, creatinine
The major causes of hypocalcemia can be categorized according to the serum phosphate level: high vs normal or low (Table 5).
High-phosphate, low-calcium states. In the absence of concurrent end-stage renal disease and an excessive phosphate load, primary hypoparathyroidism is the most likely cause of hypocalcemia associated with hyperphosphatemia.
PTH increases serum ionized calcium by26,27:
- Increasing bone resorption
- Increasing reabsorption of calcium from the distal renal tubules
- Increasing the activity of 1-alpha-hydroxylase, responsible for conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (the most biologically active vitamin D metabolite); 1,25-dihydroxyvitamin D increases the absorption of calcium and phosphate from the intestine.
Conversely, PTH decreases reabsorption of phosphate from proximal renal tubules, resulting in hypophosphatemia. Therefore, low serum PTH (primary hypoparathyroidism) or a PTH-resistant state (pseudohypoparathyroidism) results in hypocalcemia and hyperphosphatemia.26,27
Both end-stage renal disease and rhabdomyolysis are associated with high serum phosphate levels. The kidney normally excretes excess dietary phosphate to maintain phosphate homeostasis; however, this is impaired in end-stage renal disease, leading to hyperphosphatemia. In rhabdomyolysis, it is mainly the transcellular shift of phosphate into the extracellular space from myocyte injury that raises phosphate levels.
Normal- or low-phosphate, low calcium states. Hypocalcemia can also result from vitamin D deficiency, but this cause is associated with a low or normal serum phosphate level. In such cases, hypocalcemia causes secondary hyperparathyroidism with consequent renal phosphate loss and, thus, hypophosphatemia.27
Third step: Check serum intact PTH and 25-hydroxyvitamin D levels
Hypocalcemia stimulates secretion of PTH. Therefore, hypocalcemia with elevated serum PTH is caused by disorders that do not impair PTH secretion, including chronic renal failure and vitamin D deficiency (Table 5). Conversely, hypocalcemia with low or normal serum PTH levels suggests primary hypoparathyroidism.
Our patient’s serum PTH level is 20 ng/mL, which is within the reference range. This does not discount the diagnosis of primary hypoparathyroidism. Although most patients with primary hypoparathyroidism have low or undetectable serum PTH levels, some have normal PTH levels if some degree of PTH production is preserved.5,7,28–30 In these patients, the remaining functioning parathyroid tissue is not enough to maintain a normal serum calcium level, resulting in hypocalcemia. As a result, hypocalcemia stimulates the remaining parathyroid tissue to its maximum output, producing PTH levels usually within the lower or middle-normal range.30 In such patients, the terms parathyroid insufficiency and relative primary hypoparathyroidism are more precise than primary hypoparathyroidism.
Postsurgical hypoparathyroidism with an inappropriately normal PTH level is usually seen in patients with disorders that impair intestinal calcium absorption or bone resorption.31 In our patient’s case, the “normal” serum PTH level is likely due to maximal stimulation of remaining functioning parathyroid tissue by severe hypocalcemia, which is a result of her discontinuation of calcium and calcitriol therapy and her vitamin D deficiency.
CASE RESUMED: NO RESPONSE TO INTRAVENOUS CALCIUM GLUCONATE
The patient is given 2 10-mL ampules of 10% calcium gluconate diluted in 100 mL of 5% dextrose in water over 20 minutes intravenously. Electrocardiographic monitoring is continued. Two hours later, her measured serum calcium is only 5.8 mg/dL, with no improvement in her symptoms.
A continuous infusion of calcium gluconate is started: 12 ampules of calcium gluconate are added to 380 mL of 5% dextrose in water and infused at 40 mL/hour (infused rate of elemental calcium = 1.3 mg/kg/hour); 3 hours later, her measured serum calcium level is still only 5.8 mg//dL; at 6 hours it is 5.9 mg/dL, and her symptoms have not improved.
4. Which of the following is the most appropriate next step?
- Change the calcium gluconate to calcium chloride
- Increase the infusion rate to 1.5 mg of elemental calcium/kg/hour
- Give a bolus of 2 10-mL ampules of 10% calcium gluconate intravenously over 1 minute
- Give additional oral calcium tablets
- Check the serum magnesium level
Treatment of hypocalcemia can involve intravenous or oral calcium therapy.
Intravenous calcium is indicated for patients with any of the following6,32:
- Moderate to severe neuromuscular irritability (eg, acral paresthesia, carpopedal spasm, prolonged QT interval, seizures, laryngospasm, bronchospasm)
- Acute hypocalcemia with corrected serum calcium level less than 7.6 mg/dL, even if the patient is asymptomatic
- Cardiac failure.
One 10-mL ampule of 10% calcium gluconate contains 93 mg of elemental calcium; 1 or 2 ampules are typically diluted in 50 to 100 mL of 5% dextrose in water and infused slowly over 15 to 20 minutes. Rapid administration of intravenous calcium is contraindicated, as it may produce cardiac arrhythmias and possibly cardiac arrest. Therefore, intravenous calcium should be given slowly while continuing electrocardiographic monitoring.33
Since the effect of 1 ampule of calcium gluconate lasts only 2 to 3 hours, most patients with symptomatic hypocalcemia require continuous intravenous calcium infusion. The recommended dose of infused elemental calcium is 0.5 to 1.5 mg/kg/hour.34 Several ampules are added to 500 to 1,000 mL of 5% dextrose in water or 0.9% normal saline and infused at a rate appropriate for the patient’s corrected calcium and symptoms.
Oral calcium and vitamin D supplements can be given initially to patients with a corrected serum calcium level of 7.6 mg/dL or greater, with or without mild symptoms, if they can tolerate oral intake. However, this is not the treatment of choice for resistant acute hypocalcemia, as in this case.
Calcium chloride has no advantages over calcium gluconate. Further, it can be associated with local irritation and may result in tissue necrosis if extravasation occurs.35
Increasing the infusion rate of calcium gluconate to the maximum recommended dose may improve the patient’s ionized calcium level and symptoms somewhat. However, it is not the best option for this patient, given that she did not respond to 2 ampules of calcium gluconate followed by continuous infusion of 1.3 mg/kg/hour for 6 hours.
Calcium gluconate bolus. Similarly, giving the patient an additional 2 ampules of calcium gluconate over 1 minute would not be recommended, as rapid administration of intravenous calcium gluconate (eg, over 1 minute) is contraindicated.
Check magnesium
If hypocalcemia persists despite intravenous calcium therapy, as in our patient, further investigation or action is required. An important cause of persistent hypocalcemia is severe hypomagnesemia. Severe hypomagnesemia (serum magnesium < 0.8 mg/dL) causes resistant hypocalcemia by several mechanisms:
- Inducing PTH resistance32,36,37
- Decreasing PTH secretion32,36
- Decreasing calcitriol production.
The decrease in calcitriol production is a direct effect of hypomagnesemia, but it is also an indirect effect of low PTH secretion, which inhibits the enzyme 1-alpha-hydroxylase. Thus, conversion of 25-hydroxyvitamin D3 to calcitriol is impaired, leading to low calcitriol production.
Our patient could have hypomagnesemia due to furosemide use and uncontrolled diabetes mellitus. Hypocalcemia resistant to calcium therapy may occasionally respond to magnesium therapy even if the serum magnesium level is normal. This may be due to depleted intracellular magnesium salt levels.6,38 Rarely, severe hypermagnesemia can also be associated with hypocalcemia due to inhibition of PTH secretion.37,39
CASE RESUMED
Our patient’s serum magnesium level is 0.6 mg/dL (reference range 1.7–2.4 mg/dL). She is given 2 g of magnesium sulfate in 60 mL of 0.9% normal saline infused over 1 hour, followed by a continuous infusion of magnesium sulfate (12 g diluted in 250 mL of 0.9% normal saline, infused over 24 hours). On repeat testing 4 hours later, her serum magnesium level is 0.7 mg/dL, and at 8 hours later it is 0.9 mg/dL. She is subsequently started on oral magnesium oxide 600 mg per day. The magnesium sulfate infusion is continued for another 24 hours.
PREVENTING HYPERCALCIURIA
Patients with low PTH (primary hypoparathyroidism) may have hypercalciuria due to decreased renal tubular calcium reabsorption. Two important measures can minimize hypercalciuria in such patients:
- Keeping the serum calcium level in the low-normal range4,5,40
- Giving a thiazide diuretic (eg, hydrochlorothiazide 12.5–50 mg daily) with a low-salt diet.41,42
A thiazide diuretic is usually started once the 24-hour urine calcium reaches 250 mg.6 Thiazides are thought to enhance both proximal and distal renal tubular calcium reabsorption.43,44
PRIMARY HYPOPARATHYROIDISM: LONG-TERM MANAGEMENT
Long-term management of primary hypoparathyroidism requires calcium and vitamin D supplementation.
Calcium supplements. The most commonly prescribed calcium preparations are calcium carbonate and calcium citrate (containing 40% and 20% elemental calcium, respectively). Calcium carbonate, which is less expensive than calcium citrate, binds with phosphate intake and requires an acidic environment for absorption, and so it is better absorbed when taken with meals. Because calcium citrate does not require an acidic environment for absorption, it is the calcium preparation of choice for patients on proton pump inhibitors, or patients with achlorhydria or constipation.45 Calcium doses vary widely, with most hypoparathyroid patients requiring 1 to 2 g of elemental calcium daily.6
Vitamin D supplements. To promote intestinal absorption, calcium is combined with vitamin D in a fixed-dose preparation given in divided doses.46 Calcitriol (1,25-dihydroxyvitamin D3) is the most active metabolite of vitamin D, with rapid onset and offset of action, and it is the preferred form of vitamin D therapy for patients with hypoparathyroidism. If calcitriol is not available or is not affordable, alphacalcidol (1-alpha-hydroxyvitamin D3) is another option. This is a synthetic analogue of vitamin D that is already hyroxylated at the C1 position. After oral intake, it is hydroxylated in the liver to form calcitriol.
Since renal production of calcitriol is PTH-dependent, in hypoparathyroidism the conversion of 25-hydroxyvitamin D3 to calcitriol is limited. Therefore, vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are not the preferred forms of vitamin D for such patients. However, either can be added to calcitriol, as they may have extraskeletal benefits.7
CASE CONCLUDED
Our patient presented with primary parathyroid insufficiency associated with vitamin D deficiency. Therefore, in addition to calcitriol and calcium combined with vitamin D in a fixed-dose preparation, her management included vitamin D3 for her vitamin D deficiency.
She was discharged on these medications. At a follow-up visit 3 weeks later, her measured serum calcium level was 8.6 mg/dL. She reported gradual resolution of her symptoms. She was also referred to a psychiatrist for her depression.
TAKE-HOME POINTS
- Hypocalcemia causes neuromuscular excitability, manifested clinically by tetany.
- Common causes of hypocalcemia include vitamin D deficiency, hypomagnesemia, renal failure, and primary hypoparathyroidism.
- The first step in evaluating hypocalcemia is to correct the measured serum calcium to the serum albumin concentration.
- Laboratory testing for hypocalcemia should include serum phosphorus, magnesium, creatinine, PTH, and 25-hydroxyvitamin D3.
- Primary hypoparathyroidism is characterized by hypocalcemia, hyperphosphatemia, and low serum PTH.
- Moderate to severe manifestations of hypo-
calcemia and acute hypocalcemia (< 7.6 mg/dL), even if asymptomatic, warrant intravenous calcium therapy. - Correction of hypomagnesemia is essential to treat hypocalcemia, especially if resistant to intravenous calcium therapy.
- The goal of chronic management of primary hypoparathyroidism includes correcting the serum calcium level to a low-normal range, the serum phosphorus level to an upper-normal range, and prevention of hypercalciuria.
Acknowledgments: The authors wish to thank Mr. Michael Edward Tierney of the School of Medicine, University of Sydney, Australia, for his linguistic editing of the manuscript.
- Jesus JE, Landry A. Images in clinical medicine. Chvostek’s and Trousseau’s signs. N Engl J Med 2012; 367:e15.
- Urbano FL. Signs of hypocalcemia: Chvostek’s and Trousseau’s. Hosp Physician 2000; 36:43–45.
- Chisthi MM, Nair RS, Kuttanchettiyar KG, Yadev I. Mechanisms behind post-thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin—a prospective study. J Invest Surg 2017; 30:217–225.
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 2016; 101:2300–2312.
- Stack BC Jr, Bimston DN, Bodenner DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: postoperative hypoparathyroidism—definitions and management. Endocr Pract 2015; 21:674–685.
- Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med 2008; 359:391–403.
- Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol (Lausanne) 2017; 7:172.
- Coimbra C, Monteiro F, Oliveira P, Ribeiro L, de Almeida MG, Condé A. Hypoparathyroidism following thyroidectomy: predictive factors. Acta Otorrinolaringol Esp 2017; 68:106–111.
- Thomusch O, Machens A, Sekulla C, Ukkat J, Brauckhoff M, Dralle H. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: a multivariate analysis of 5846 consecutive patients. Surgery 2003; 133:180–185.
- Hirsch PF, Lester GE, Talmage RV. Calcitonin, an enigmatic hormone: does it have a function? J Musculoskelet Neuronal Interact 2001; 1:299–305.
- Karne SS, Bhalerao NS. Carpal tunnel syndrome in hypothyroidism. J Clin Diagn Res 2016; 10:OC36–OC38.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Palumbo CF, Szabo RM, Olmsted SL. The effects of hypothyroidism and thyroid replacement on the development of carpal tunnel syndrome. J Hand Surg Am 2000; 25:734–739.
- Katz JN, Stirrat CR, Larson MG, Fossel AH, Eaton HM, Liang MH. A self-administered hand symptom diagram for the diagnosis and epidemiologic study of carpal tunnel syndrome. J Rheumatol 1990; 17:1495–1498.
- Katz JN, Stirrat CR. A self-administered hand diagram for the diagnosis of carpal tunnel syndrome. J Hand Surg Am 1990; 15:360–363.
- Calfee RP, Dale AM, Ryan D, Descatha A, Franzblau A, Evanoff B. Performance of simplified scoring systems for hand diagrams in carpal tunnel syndrome screening. J Hand Surg Am 2012; 37:10–17.
- D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? JAMA 2000; 283:3110–3117.
- Marchettini P, Lacerenza M, Mauri E, Marangoni C. Painful peripheral neuropathies. Curr Neuropharmacol 2006; 4:175–181.
- Kibirige D, Mwebaze R. Vitamin B12 deficiency among patients with diabetes mellitus: is routine screening and supplementation justified? J Diabetes Metab Disord 2013;12:17.
- Akinlade KS, Agbebaku SO, Rahamon SK, Balogun WO. Vitamin B12 levels in patients with type 2 diabetes mellitus on metformin. Ann Ib Postgrad Med 2015; 13:79–83.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Benoit SR, Mendelsohn AB, Nourjah P, Staffa JA, Graham DJ. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005; 12:363–368.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab 2009; 94:2115–2118.
- McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg 1994; 81:357–360.
- Talmage RV, Mobley HT. Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 2008; 156:1–8.
- Friedman PA, Gesek FA. Calcium transport in renal epithelial cells. Am J Physiol 1993; 264:F181–F198.
- Jensen PV, Jelstrup SM, Homøe P. Long-term outcomes after total thyroidectomy. Dan Med J 2015; 62:A5156.
- Ritter K, Elfenbein D, Schneider DF, Chen H, Sippel RS. Hypoparathyroidism after total thyroidectomy: incidence and resolution. J Surg Res 2015; 197:348–353.
- Promberger R, Ott J, Kober F, Karik M, Freissmuth M, Hermann M. Normal parathyroid hormone levels do not exclude permanent hypoparathyroidism after thyroidectomy. Thyroid 2011; 21:145–150.
- Lorente-Poch L, Sancho JJ, Muñoz-Nova JL, Sánchez-Velázquez P, Sitges-Serra A. Defining the syndromes of parathyroid failure after total thyroidectomy. Gland Surgery 2015; 4:82–90.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tohme JF, Bilezikian JP. Diagnosis and treatment of hypocalcemic emergencies. Endocrinologist 1996; 6:10–18.
- Carroll R, Matfin G. Endocrine and metabolic emergencies: hypocalcaemia. Ther Adv Endocrinol Metab 2010; 1:29–33.
- Kim MP, Raho VJ, Mak J, Kaynar AM. Skin and soft tissue necrosis from calcium chloride in a deicer. J Emerg Med 2007; 32:41–44.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Cholst IN, Steinberg SF, Tropper PJ, Fox HE, Segre GV, Bilezikian JP. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med 1984; 310:1221–1225.
- Ryzen E, Nelson TA, Rude RK. Low blood mononuclear cell magnesium content and hypocalcemia in normomagnesemic patients. West J Med 1987; 147:549–553.
- Koontz SL, Friedman SA, Schwartz ML. Symptomatic hypocalcemia after tocolytic therapy with magnesium sulfate and nifedipine. Am J Obstet Gynecol 2004; 190:1773–1776.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Porter RH, Cox BG, Heaney D, Hostetter TH, Stinebaugh BJ, Suki WN. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978; 298:577–581.
- Clarke BL, Brown EM, Collins MT, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metab 2016; 101:2284–2299.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005; 115:1651–1658.
- Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol 1985; 248:F527–F535.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Scotti A, Bianchini C, Abbiati G, Marzo A. Absorption of calcium administered alone or in fixed combination with vitamin D to healthy volunteers. Arzneimittelforschung 2001; 51:493–500.
- Jesus JE, Landry A. Images in clinical medicine. Chvostek’s and Trousseau’s signs. N Engl J Med 2012; 367:e15.
- Urbano FL. Signs of hypocalcemia: Chvostek’s and Trousseau’s. Hosp Physician 2000; 36:43–45.
- Chisthi MM, Nair RS, Kuttanchettiyar KG, Yadev I. Mechanisms behind post-thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin—a prospective study. J Invest Surg 2017; 30:217–225.
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 2016; 101:2300–2312.
- Stack BC Jr, Bimston DN, Bodenner DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: postoperative hypoparathyroidism—definitions and management. Endocr Pract 2015; 21:674–685.
- Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med 2008; 359:391–403.
- Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol (Lausanne) 2017; 7:172.
- Coimbra C, Monteiro F, Oliveira P, Ribeiro L, de Almeida MG, Condé A. Hypoparathyroidism following thyroidectomy: predictive factors. Acta Otorrinolaringol Esp 2017; 68:106–111.
- Thomusch O, Machens A, Sekulla C, Ukkat J, Brauckhoff M, Dralle H. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: a multivariate analysis of 5846 consecutive patients. Surgery 2003; 133:180–185.
- Hirsch PF, Lester GE, Talmage RV. Calcitonin, an enigmatic hormone: does it have a function? J Musculoskelet Neuronal Interact 2001; 1:299–305.
- Karne SS, Bhalerao NS. Carpal tunnel syndrome in hypothyroidism. J Clin Diagn Res 2016; 10:OC36–OC38.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Palumbo CF, Szabo RM, Olmsted SL. The effects of hypothyroidism and thyroid replacement on the development of carpal tunnel syndrome. J Hand Surg Am 2000; 25:734–739.
- Katz JN, Stirrat CR, Larson MG, Fossel AH, Eaton HM, Liang MH. A self-administered hand symptom diagram for the diagnosis and epidemiologic study of carpal tunnel syndrome. J Rheumatol 1990; 17:1495–1498.
- Katz JN, Stirrat CR. A self-administered hand diagram for the diagnosis of carpal tunnel syndrome. J Hand Surg Am 1990; 15:360–363.
- Calfee RP, Dale AM, Ryan D, Descatha A, Franzblau A, Evanoff B. Performance of simplified scoring systems for hand diagrams in carpal tunnel syndrome screening. J Hand Surg Am 2012; 37:10–17.
- D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? JAMA 2000; 283:3110–3117.
- Marchettini P, Lacerenza M, Mauri E, Marangoni C. Painful peripheral neuropathies. Curr Neuropharmacol 2006; 4:175–181.
- Kibirige D, Mwebaze R. Vitamin B12 deficiency among patients with diabetes mellitus: is routine screening and supplementation justified? J Diabetes Metab Disord 2013;12:17.
- Akinlade KS, Agbebaku SO, Rahamon SK, Balogun WO. Vitamin B12 levels in patients with type 2 diabetes mellitus on metformin. Ann Ib Postgrad Med 2015; 13:79–83.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Benoit SR, Mendelsohn AB, Nourjah P, Staffa JA, Graham DJ. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005; 12:363–368.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab 2009; 94:2115–2118.
- McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg 1994; 81:357–360.
- Talmage RV, Mobley HT. Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 2008; 156:1–8.
- Friedman PA, Gesek FA. Calcium transport in renal epithelial cells. Am J Physiol 1993; 264:F181–F198.
- Jensen PV, Jelstrup SM, Homøe P. Long-term outcomes after total thyroidectomy. Dan Med J 2015; 62:A5156.
- Ritter K, Elfenbein D, Schneider DF, Chen H, Sippel RS. Hypoparathyroidism after total thyroidectomy: incidence and resolution. J Surg Res 2015; 197:348–353.
- Promberger R, Ott J, Kober F, Karik M, Freissmuth M, Hermann M. Normal parathyroid hormone levels do not exclude permanent hypoparathyroidism after thyroidectomy. Thyroid 2011; 21:145–150.
- Lorente-Poch L, Sancho JJ, Muñoz-Nova JL, Sánchez-Velázquez P, Sitges-Serra A. Defining the syndromes of parathyroid failure after total thyroidectomy. Gland Surgery 2015; 4:82–90.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tohme JF, Bilezikian JP. Diagnosis and treatment of hypocalcemic emergencies. Endocrinologist 1996; 6:10–18.
- Carroll R, Matfin G. Endocrine and metabolic emergencies: hypocalcaemia. Ther Adv Endocrinol Metab 2010; 1:29–33.
- Kim MP, Raho VJ, Mak J, Kaynar AM. Skin and soft tissue necrosis from calcium chloride in a deicer. J Emerg Med 2007; 32:41–44.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Cholst IN, Steinberg SF, Tropper PJ, Fox HE, Segre GV, Bilezikian JP. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med 1984; 310:1221–1225.
- Ryzen E, Nelson TA, Rude RK. Low blood mononuclear cell magnesium content and hypocalcemia in normomagnesemic patients. West J Med 1987; 147:549–553.
- Koontz SL, Friedman SA, Schwartz ML. Symptomatic hypocalcemia after tocolytic therapy with magnesium sulfate and nifedipine. Am J Obstet Gynecol 2004; 190:1773–1776.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Porter RH, Cox BG, Heaney D, Hostetter TH, Stinebaugh BJ, Suki WN. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978; 298:577–581.
- Clarke BL, Brown EM, Collins MT, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metab 2016; 101:2284–2299.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005; 115:1651–1658.
- Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol 1985; 248:F527–F535.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Scotti A, Bianchini C, Abbiati G, Marzo A. Absorption of calcium administered alone or in fixed combination with vitamin D to healthy volunteers. Arzneimittelforschung 2001; 51:493–500.
The Deer Stand Strikes Back
ANSWER
The radiograph demonstrates a compression fracture of T12 with moderate loss of height approaching 50% anteriorly. In addition, there is a vertically oriented fracture within the middle of the vertebral body, causing the back portion to be posteriorly displaced.
This type of burst fracture is potentially unstable and should be treated as such. The patient was placed on strict spine precautions and transferred to a facility where trauma and neurosurgical services were available.
ANSWER
The radiograph demonstrates a compression fracture of T12 with moderate loss of height approaching 50% anteriorly. In addition, there is a vertically oriented fracture within the middle of the vertebral body, causing the back portion to be posteriorly displaced.
This type of burst fracture is potentially unstable and should be treated as such. The patient was placed on strict spine precautions and transferred to a facility where trauma and neurosurgical services were available.
ANSWER
The radiograph demonstrates a compression fracture of T12 with moderate loss of height approaching 50% anteriorly. In addition, there is a vertically oriented fracture within the middle of the vertebral body, causing the back portion to be posteriorly displaced.
This type of burst fracture is potentially unstable and should be treated as such. The patient was placed on strict spine precautions and transferred to a facility where trauma and neurosurgical services were available.
A 45-year-old man presents to your facility for evaluation of ongoing back pain. He reports that he fell out of a deer stand from an approximate height of 15 to 20 ft. He landed on his back but was able to get up, walk a short distance to his car, and drive home. Persistent pain is what brings him to the emergency department to seek treatment.
He denies any weakness or numbness in his lower extremities. There are no bowel or bladder issues. His medical history is unremarkable.
On physical examination, you note a moderately uncomfortable male whose vital signs are normal. He is able to move all four extremities well, and his strength is intact throughout. He does have moderate tenderness within the thoracolumbar region, with no step-off appreciated. The paraspinous muscles are tender as well.
You order lumbosacral radiographs (lateral view shown). What is your impression?
MedPAC: Medicare hospital readmissions program is working
WASHINGTON – The Medicare Hospital Readmissions Reduction Program is working, according to an original analysis of Medicare claims data presented at a meeting of the Medicare Payment Advisory Commission.
“First, readmissions declined,” MedPAC staff member Jeff Stensland, PhD, said during a congressionally mandated staff report to the commissioners. “Second, while observation stays increased, they did not fully offset the decrease in readmissions. Third, while [emergency department] visits also increased, those increases appear to largely be due to factors other than the readmission program. And fourth, in addition, all the evidence we examined suggests that the readmissions program did not result in increased mortality.”
including a reduction in readmissions and patients spending less time in the hospital with “at least equal outcomes,” Dr. Stensland said at the meeting.
Taxpayers benefited from a $2 billion reduction in spending on readmissions, which will “help extend the viability of the Medicare Trust Fund.” He noted that improvements to the program will be discussed at future MedPAC meetings.
Not all MedPAC commissioners agreed with the staff analysis.
David Nerenz, PhD, of the Henry Ford Health System, Detroit, also was not convinced the program was having an impact, noting that hospital readmissions began to decline even before the program started.
In looking at a graph presented that showed this trend, “I was impressed by the fact that the trend line started coming down all the way to the left side of the graph, and what my eye was impressed with was more just the continuation rather than a change, so I guess I feel cautious saying the program had certain effects because they certainly don’t jump off the graph visually,” Dr. Nerenz said. “I’m not disputing the numbers, but to say just as a clear unqualified conclusion the program reduced readmissions, I’m not so sure.”
It is likely premature to make any firm conclusions about how effectively this program decreases unnecessary utilization of hospitals. However, it is heartening to know that it did not increase mortality. The one variable that would best control readmissions is patient education. What constitutes an emergency requiring hospital evaluation and potential admission is often not explained to the patient by you and me. 
It is likely premature to make any firm conclusions about how effectively this program decreases unnecessary utilization of hospitals. However, it is heartening to know that it did not increase mortality. The one variable that would best control readmissions is patient education. What constitutes an emergency requiring hospital evaluation and potential admission is often not explained to the patient by you and me.
It is likely premature to make any firm conclusions about how effectively this program decreases unnecessary utilization of hospitals. However, it is heartening to know that it did not increase mortality. The one variable that would best control readmissions is patient education. What constitutes an emergency requiring hospital evaluation and potential admission is often not explained to the patient by you and me.
WASHINGTON – The Medicare Hospital Readmissions Reduction Program is working, according to an original analysis of Medicare claims data presented at a meeting of the Medicare Payment Advisory Commission.
“First, readmissions declined,” MedPAC staff member Jeff Stensland, PhD, said during a congressionally mandated staff report to the commissioners. “Second, while observation stays increased, they did not fully offset the decrease in readmissions. Third, while [emergency department] visits also increased, those increases appear to largely be due to factors other than the readmission program. And fourth, in addition, all the evidence we examined suggests that the readmissions program did not result in increased mortality.”
including a reduction in readmissions and patients spending less time in the hospital with “at least equal outcomes,” Dr. Stensland said at the meeting.
Taxpayers benefited from a $2 billion reduction in spending on readmissions, which will “help extend the viability of the Medicare Trust Fund.” He noted that improvements to the program will be discussed at future MedPAC meetings.
Not all MedPAC commissioners agreed with the staff analysis.
David Nerenz, PhD, of the Henry Ford Health System, Detroit, also was not convinced the program was having an impact, noting that hospital readmissions began to decline even before the program started.
In looking at a graph presented that showed this trend, “I was impressed by the fact that the trend line started coming down all the way to the left side of the graph, and what my eye was impressed with was more just the continuation rather than a change, so I guess I feel cautious saying the program had certain effects because they certainly don’t jump off the graph visually,” Dr. Nerenz said. “I’m not disputing the numbers, but to say just as a clear unqualified conclusion the program reduced readmissions, I’m not so sure.”
WASHINGTON – The Medicare Hospital Readmissions Reduction Program is working, according to an original analysis of Medicare claims data presented at a meeting of the Medicare Payment Advisory Commission.
“First, readmissions declined,” MedPAC staff member Jeff Stensland, PhD, said during a congressionally mandated staff report to the commissioners. “Second, while observation stays increased, they did not fully offset the decrease in readmissions. Third, while [emergency department] visits also increased, those increases appear to largely be due to factors other than the readmission program. And fourth, in addition, all the evidence we examined suggests that the readmissions program did not result in increased mortality.”
including a reduction in readmissions and patients spending less time in the hospital with “at least equal outcomes,” Dr. Stensland said at the meeting.
Taxpayers benefited from a $2 billion reduction in spending on readmissions, which will “help extend the viability of the Medicare Trust Fund.” He noted that improvements to the program will be discussed at future MedPAC meetings.
Not all MedPAC commissioners agreed with the staff analysis.
David Nerenz, PhD, of the Henry Ford Health System, Detroit, also was not convinced the program was having an impact, noting that hospital readmissions began to decline even before the program started.
In looking at a graph presented that showed this trend, “I was impressed by the fact that the trend line started coming down all the way to the left side of the graph, and what my eye was impressed with was more just the continuation rather than a change, so I guess I feel cautious saying the program had certain effects because they certainly don’t jump off the graph visually,” Dr. Nerenz said. “I’m not disputing the numbers, but to say just as a clear unqualified conclusion the program reduced readmissions, I’m not so sure.”
REPORTING FROM MEDPAC
Boy, 9, With Eye Pain, Blurred Vision, and Tearing
IN THIS ARTICLE
- Diagnosis
- Management
- Outcome for the case patient
An otherwise healthy 9-year-old boy is brought to the emergency department (ED) by his father for evaluation of severe pain, blurry vision, and four hours of tearing in his right eye. The patient was in school when he experienced sudden-onset irritation and scratching pain that caused him to rub his eye. He says it “feels like there is something in my eye,” but he denies any known substance or foreign body. He has no medical or surgical history, does not wear contact lenses or eyeglasses, and denies loss of vision. There is no history of recent illness or travel.
On evaluation, the patient is in no acute distress but is holding his right eye closed due to foreign-body sensation and increased photosensitivity and tearing. There is no obvious erythema or swelling in the upper or lower eyelids bilaterally. A visual acuity test with a Snellen eye chart shows 20/20 vision in the left eye and 20/50 in the right, secondary to pain, photophobia, and excessive tearing. The patient’s right sclera is significantly injected. Intraocular pressure, measured with a tonometer, is 12 to 14 mm Hg. A fluorescein stain of the eye yields no significant findings. The globe is intact.
At first glance, a slit-lamp exam shows no obvious signs of a foreign body. But much higher magnification reveals substantial conjunctival injection and numerous intracorneal linear foreign bodies in the right eye (see Figure 1 for example [not the case patient]). The anterior chamber shows no inflammatory reaction, and findings in the posterior segment are unremarkable.
The initial diagnosis is simple conjunctivitis—but closer examination reveals multiple fine, barbed hairs embedded in the patient’s right cornea. Upon further questioning, the patient reports that prior to symptom onset, he had been holding the classroom pet, a Chilean Rose tarantula, in the palm of his hands.
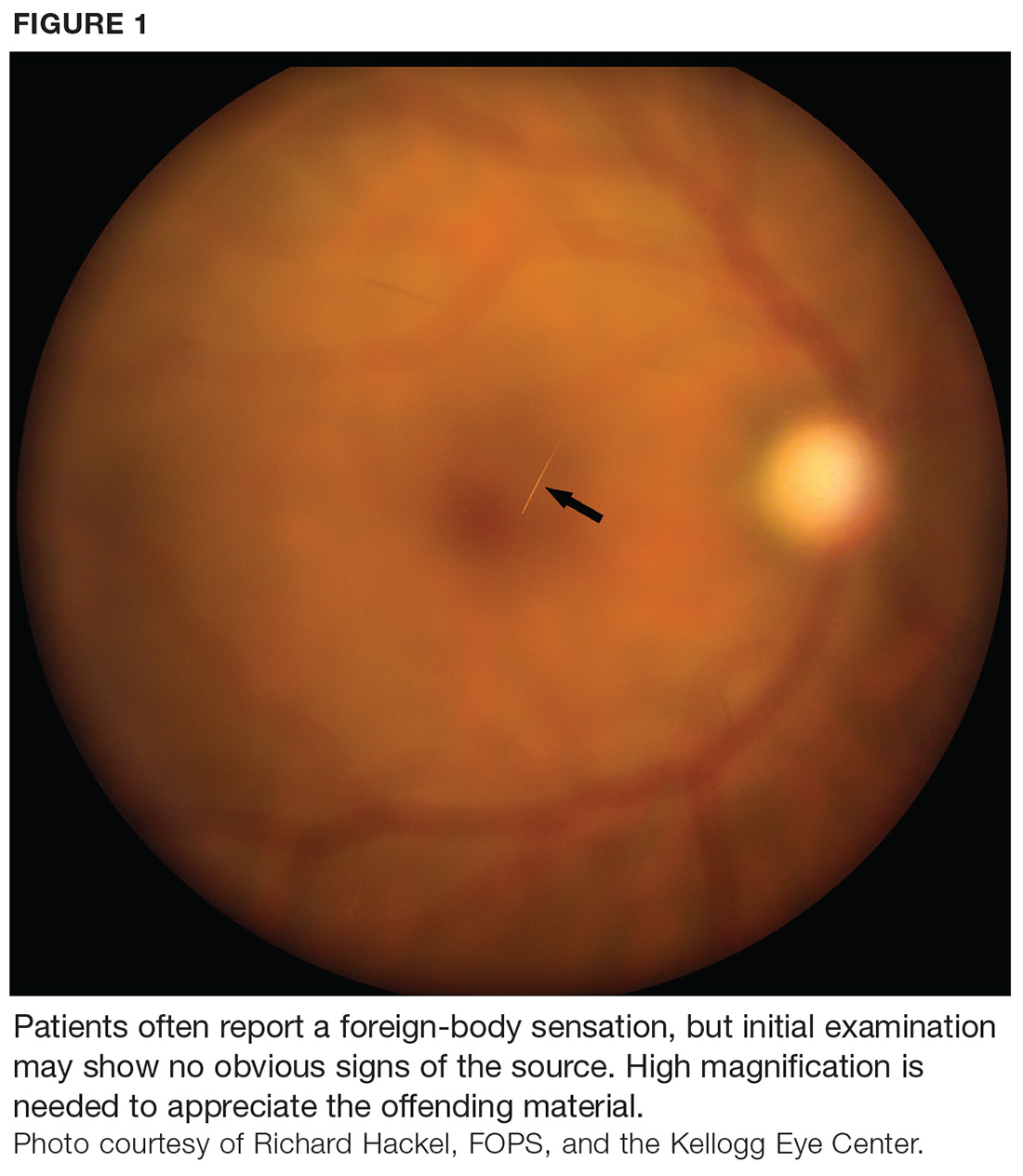
DISCUSSION
Foreign body injury is a common cause of ocular pain and corneal damage, which can lead to challenging complications. Ophthalmic emergencies account for 2% of ED visits in the US annually and are a major cause of visual impairment.1 But when a painful eye is the chief complaint, contact with insects, plants, or spiders is rarely included in the differential. Tarantulas are popular classroom and household pets, however, and ocular injury should be suspected in anyone who has been holding a tarantula prior to onset of pain.
Ophthalmia nodosa
Tarantulas are one of the most common arachnids known to cause ophthalmia nodosa—a granulomatous reaction of the conjunctiva or cornea to an implanted plant, insect, or spider hair that typically manifests with photophobia, irritation, and chemosis.2,3 Tarantulas, when scared or defending their eggs, shoot urticating setae at the threat—a defensive mechanism largely unknown to parents, tarantula owners, and medical professionals.
Urticating setae are found in roughly 90% of tarantula species throughout tropical and subtropical regions.4 Depending on the species, setae can be located on the distal prolateral surface of the palpal femur or the dorsum of the abdomen. They can be released when the tarantula scratches its legs against the abdominal urticating setae patch or scratches the palps against the chelicerae (appendages in front of the mouth), or when direct exterior contact is made with the abdominal setae.4
There are six types of urticating hairs. Each is attached to the spider’s cuticle by either a stalk (which represents the break-off region) or a socket.4 Tarantula hairs range in size from 0.1 mm to 0.3 mm and have a sharp, pointed head and numerous barbs, which help embed them in the target.5 They are long and thin, to facilitate deep tissue penetration, and can enter the eyes, lungs, or other body parts (see Figure 2).
Ocular injury from tarantula hairs commonly involves conjunctival injection, foreign body sensation, periorbital facial rash, photophobia, and tearing.3 When a tarantula’s cloud of barbed hairs is flicked into the eye and pierces the cornea, it can cause infection, irritation, scarring on the cornea, or vision loss. Eye movement or rubbing can cause the hairs—and their toxins—to migrate over time, traveling like an arrow (the tip and barbs resist backward movement) to the anterior chamber, lens, vitreous, and retina.6,7 This can cause corneal scars, cataracts, vitritis, or macular edema, and creates the possibility for acute or chronic conjunctivitis.7
Diagnosis and management
Ophthalmic emergencies can affect the visual system and, if left untreated, can lead to permanent vision loss. Affected patients require immediate medical attention and should be referred to an ophthalmologist for follow-up care.
Diagnosis. A thorough history and physical exam are of utmost importance; tiny setae can be easily overlooked if the examiner is not diligent, and the similar symptomatology can lead to misdiagnosis as simple conjunctivitis.3 A visual acuity test and slit-lamp exam are useful for confirmation.
Treatment. Once the diagnosis is confirmed, treatment should consist of mild topical antibiotics and steroids to effectively control infection and inflammation. While topical steroids may be appropriate, local adverse events associated with their use (eg, glaucoma, cataracts) can be problematic. Gentle eye irrigation has been noted by some researchers as contraindicated, while others find it useful to flush out some of the hairs.5,8,9
Most of the visible protruding tarantula hairs can and should be removed under microscopy during slit-lamp exam. Hairs that are buried in the cornea, however, are nearly impossible to remove and pose a threat of further complications, as described. Conservative management with careful observation is therefore recommended. If the patient develops a granuloma, excision—along with a course of systemic steroids and setae removal via vitrectomy—may be needed.9
The good news is that, in many cases, deeper hairs are absorbed without complication, making their removal unnecessary.5 Factors that encourage leaving the setae untouched include a large number of hairs, deep corneal penetration, lack of patient tolerance for the procedure, and risk for perforation.3
More invasive treatments (eg, laser photocoagulation, intraocular surgery) to remove offending hairs are possible, but literature on the outcome of these interventions is limited. One report to date used argon laser photocoagulation to treat endophthalmitis from vitreous hairs.10 The laser can fragment the hairs so that they lose their barbed characteristic and cannot penetrate deeper.6
Follow-up. Close follow-up is advised, and patients should be educated on the importance of medication compliance and return visits for reevaluation. Given the potential dangers of handling these spiders, tarantula owners should be advised to use protective gloving and goggles.2,5,8,9
OUTCOME FOR THE CASE PATIENT
The case patient was sent to an ophthalmologist on day 1. Proparacaine was placed in his right eye, and all of the superficial tarantula hairs were removed using 25- and 30-gauge needles with jeweler forceps under slit-lamp microscopy. Most of the hairs were removed from the superior cornea; fewer were found in the paracentral and inferior regions of the cornea. Approximately five hairs in the paracentral area of the cornea were embedded in the midstromal depth and could not be removed. One drop of ciprofloxacin was administered.
The patient was sent home with an eye shield and instructions to use tobramycin/dexamethasone eye drops (qid in his right eye) and avoid rubbing the eye. (The eye shield, though not technically necessary, was deemed beneficial to help the patient avoid touching the eye.) He was scheduled to return to the clinic one week later.
On follow-up, a careful exam performed under microscopy showed that the five tarantula hairs were still embedded, and an additional six hairs were found in the deep stroma. Superficial punctate keratitis—an eye disorder caused by epithelial cell death on the surface of the cornea—was noted, but no anterior chamber cells were seen. The patient was instructed to continue using the eye drops as prescribed until finished, then start using loteprednol (tid) and artificial lubricating tears (every 2 h).
He returned to the clinic every two weeks for a total of 10 visits. At the end of the treatment course, the remaining tarantula hairs were unable to be removed. The patient used tapering doses of topical eye steroids and antibiotic drops secondary to flare-up.
CONCLUSION
Determining the etiology of ophthalmic emergencies is essential to timely and appropriate management. In this case, a recognized but often overlooked cause, tarantula hairs, made the diagnosis more complicated than simple conjunctivitis. When ocular injury is suspected, the provider must obtain an accurate and detailed history along with a thorough physical exam. Since patients must comply with medication regimens to prevent acute and chronic infection, a clear treatment and follow-up plan should be established. With these in place, ophthalmia nodosa caused by urticating setae can be effectively managed.
1. Fitzpatrick J, Hickman R, Alfes CM. A Guide to Mastery in Clinical Nursing: The Comprehensive Reference. New York, NY: Springer; 2018:114.
2. Lambert SR, Lyons CJ. Taylor and Hoyt’s Pediatric Ophthalmology and Strabismus. 5th ed. New York, NY: Elsevier; 2017:138.
3. Stagg BC, Ambati BK. Tarantula hairs as corneal foreign bodies. Case Rep Ophthalmol. 2011;2(3):323-326.
4. Bertani R, Guadanucci JPL. Morphology, evolution, and usage of urticating setae by tarantulas (Araneae: Theraphosidae). Zoologia (Curitiba). 2013;30(4):403-418.
5. McAnena L, Murphy C, O’Connor J. Tarantula keratitis: a case report. Ir J Med Sci. 2013;182(3):349-350.
6. Yang Y, Christakis T, Mireskandari K. Acute conjunctivitis and corneal foreign bodies secondary to tarantula hairs. CMAJ. 2016;183(3):212-214.
7. Jain N, Soong HK, Gardner TW. Ophthalmia nodosa. EyeNet Magazine. November 2013. www.aao.org/eyenet/article/blink-mystery-image-17. Accessed January 24, 2018.
8. Choi JTL, Rauf A. Ophthalmia nodosa secondary to tarantula hairs. Eye (Lond). 2003;17(3):433-434.
9. Comez AT, Tufan HA, Gencer B. Ophthalmia nodosa as an occupational disease: is it unusual or is it casual? Ocul Immunol Inflamm. 2013;21(2):144-147.
10. Marti-Huguet T, Pujol O, Cabiro I, et al. Endophthalmos caused by intravitreal caterpillar hairs. Treatment by direct photocoagulation with argon laser [article in French]. J Fr Ophthalmol. 1987;10(10):559-564.
IN THIS ARTICLE
- Diagnosis
- Management
- Outcome for the case patient
An otherwise healthy 9-year-old boy is brought to the emergency department (ED) by his father for evaluation of severe pain, blurry vision, and four hours of tearing in his right eye. The patient was in school when he experienced sudden-onset irritation and scratching pain that caused him to rub his eye. He says it “feels like there is something in my eye,” but he denies any known substance or foreign body. He has no medical or surgical history, does not wear contact lenses or eyeglasses, and denies loss of vision. There is no history of recent illness or travel.
On evaluation, the patient is in no acute distress but is holding his right eye closed due to foreign-body sensation and increased photosensitivity and tearing. There is no obvious erythema or swelling in the upper or lower eyelids bilaterally. A visual acuity test with a Snellen eye chart shows 20/20 vision in the left eye and 20/50 in the right, secondary to pain, photophobia, and excessive tearing. The patient’s right sclera is significantly injected. Intraocular pressure, measured with a tonometer, is 12 to 14 mm Hg. A fluorescein stain of the eye yields no significant findings. The globe is intact.
At first glance, a slit-lamp exam shows no obvious signs of a foreign body. But much higher magnification reveals substantial conjunctival injection and numerous intracorneal linear foreign bodies in the right eye (see Figure 1 for example [not the case patient]). The anterior chamber shows no inflammatory reaction, and findings in the posterior segment are unremarkable.
The initial diagnosis is simple conjunctivitis—but closer examination reveals multiple fine, barbed hairs embedded in the patient’s right cornea. Upon further questioning, the patient reports that prior to symptom onset, he had been holding the classroom pet, a Chilean Rose tarantula, in the palm of his hands.
DISCUSSION
Foreign body injury is a common cause of ocular pain and corneal damage, which can lead to challenging complications. Ophthalmic emergencies account for 2% of ED visits in the US annually and are a major cause of visual impairment.1 But when a painful eye is the chief complaint, contact with insects, plants, or spiders is rarely included in the differential. Tarantulas are popular classroom and household pets, however, and ocular injury should be suspected in anyone who has been holding a tarantula prior to onset of pain.
Ophthalmia nodosa
Tarantulas are one of the most common arachnids known to cause ophthalmia nodosa—a granulomatous reaction of the conjunctiva or cornea to an implanted plant, insect, or spider hair that typically manifests with photophobia, irritation, and chemosis.2,3 Tarantulas, when scared or defending their eggs, shoot urticating setae at the threat—a defensive mechanism largely unknown to parents, tarantula owners, and medical professionals.
Urticating setae are found in roughly 90% of tarantula species throughout tropical and subtropical regions.4 Depending on the species, setae can be located on the distal prolateral surface of the palpal femur or the dorsum of the abdomen. They can be released when the tarantula scratches its legs against the abdominal urticating setae patch or scratches the palps against the chelicerae (appendages in front of the mouth), or when direct exterior contact is made with the abdominal setae.4
There are six types of urticating hairs. Each is attached to the spider’s cuticle by either a stalk (which represents the break-off region) or a socket.4 Tarantula hairs range in size from 0.1 mm to 0.3 mm and have a sharp, pointed head and numerous barbs, which help embed them in the target.5 They are long and thin, to facilitate deep tissue penetration, and can enter the eyes, lungs, or other body parts (see Figure 2).
Ocular injury from tarantula hairs commonly involves conjunctival injection, foreign body sensation, periorbital facial rash, photophobia, and tearing.3 When a tarantula’s cloud of barbed hairs is flicked into the eye and pierces the cornea, it can cause infection, irritation, scarring on the cornea, or vision loss. Eye movement or rubbing can cause the hairs—and their toxins—to migrate over time, traveling like an arrow (the tip and barbs resist backward movement) to the anterior chamber, lens, vitreous, and retina.6,7 This can cause corneal scars, cataracts, vitritis, or macular edema, and creates the possibility for acute or chronic conjunctivitis.7
Diagnosis and management
Ophthalmic emergencies can affect the visual system and, if left untreated, can lead to permanent vision loss. Affected patients require immediate medical attention and should be referred to an ophthalmologist for follow-up care.
Diagnosis. A thorough history and physical exam are of utmost importance; tiny setae can be easily overlooked if the examiner is not diligent, and the similar symptomatology can lead to misdiagnosis as simple conjunctivitis.3 A visual acuity test and slit-lamp exam are useful for confirmation.
Treatment. Once the diagnosis is confirmed, treatment should consist of mild topical antibiotics and steroids to effectively control infection and inflammation. While topical steroids may be appropriate, local adverse events associated with their use (eg, glaucoma, cataracts) can be problematic. Gentle eye irrigation has been noted by some researchers as contraindicated, while others find it useful to flush out some of the hairs.5,8,9
Most of the visible protruding tarantula hairs can and should be removed under microscopy during slit-lamp exam. Hairs that are buried in the cornea, however, are nearly impossible to remove and pose a threat of further complications, as described. Conservative management with careful observation is therefore recommended. If the patient develops a granuloma, excision—along with a course of systemic steroids and setae removal via vitrectomy—may be needed.9
The good news is that, in many cases, deeper hairs are absorbed without complication, making their removal unnecessary.5 Factors that encourage leaving the setae untouched include a large number of hairs, deep corneal penetration, lack of patient tolerance for the procedure, and risk for perforation.3
More invasive treatments (eg, laser photocoagulation, intraocular surgery) to remove offending hairs are possible, but literature on the outcome of these interventions is limited. One report to date used argon laser photocoagulation to treat endophthalmitis from vitreous hairs.10 The laser can fragment the hairs so that they lose their barbed characteristic and cannot penetrate deeper.6
Follow-up. Close follow-up is advised, and patients should be educated on the importance of medication compliance and return visits for reevaluation. Given the potential dangers of handling these spiders, tarantula owners should be advised to use protective gloving and goggles.2,5,8,9
OUTCOME FOR THE CASE PATIENT
The case patient was sent to an ophthalmologist on day 1. Proparacaine was placed in his right eye, and all of the superficial tarantula hairs were removed using 25- and 30-gauge needles with jeweler forceps under slit-lamp microscopy. Most of the hairs were removed from the superior cornea; fewer were found in the paracentral and inferior regions of the cornea. Approximately five hairs in the paracentral area of the cornea were embedded in the midstromal depth and could not be removed. One drop of ciprofloxacin was administered.
The patient was sent home with an eye shield and instructions to use tobramycin/dexamethasone eye drops (qid in his right eye) and avoid rubbing the eye. (The eye shield, though not technically necessary, was deemed beneficial to help the patient avoid touching the eye.) He was scheduled to return to the clinic one week later.
On follow-up, a careful exam performed under microscopy showed that the five tarantula hairs were still embedded, and an additional six hairs were found in the deep stroma. Superficial punctate keratitis—an eye disorder caused by epithelial cell death on the surface of the cornea—was noted, but no anterior chamber cells were seen. The patient was instructed to continue using the eye drops as prescribed until finished, then start using loteprednol (tid) and artificial lubricating tears (every 2 h).
He returned to the clinic every two weeks for a total of 10 visits. At the end of the treatment course, the remaining tarantula hairs were unable to be removed. The patient used tapering doses of topical eye steroids and antibiotic drops secondary to flare-up.
CONCLUSION
Determining the etiology of ophthalmic emergencies is essential to timely and appropriate management. In this case, a recognized but often overlooked cause, tarantula hairs, made the diagnosis more complicated than simple conjunctivitis. When ocular injury is suspected, the provider must obtain an accurate and detailed history along with a thorough physical exam. Since patients must comply with medication regimens to prevent acute and chronic infection, a clear treatment and follow-up plan should be established. With these in place, ophthalmia nodosa caused by urticating setae can be effectively managed.
IN THIS ARTICLE
- Diagnosis
- Management
- Outcome for the case patient
An otherwise healthy 9-year-old boy is brought to the emergency department (ED) by his father for evaluation of severe pain, blurry vision, and four hours of tearing in his right eye. The patient was in school when he experienced sudden-onset irritation and scratching pain that caused him to rub his eye. He says it “feels like there is something in my eye,” but he denies any known substance or foreign body. He has no medical or surgical history, does not wear contact lenses or eyeglasses, and denies loss of vision. There is no history of recent illness or travel.
On evaluation, the patient is in no acute distress but is holding his right eye closed due to foreign-body sensation and increased photosensitivity and tearing. There is no obvious erythema or swelling in the upper or lower eyelids bilaterally. A visual acuity test with a Snellen eye chart shows 20/20 vision in the left eye and 20/50 in the right, secondary to pain, photophobia, and excessive tearing. The patient’s right sclera is significantly injected. Intraocular pressure, measured with a tonometer, is 12 to 14 mm Hg. A fluorescein stain of the eye yields no significant findings. The globe is intact.
At first glance, a slit-lamp exam shows no obvious signs of a foreign body. But much higher magnification reveals substantial conjunctival injection and numerous intracorneal linear foreign bodies in the right eye (see Figure 1 for example [not the case patient]). The anterior chamber shows no inflammatory reaction, and findings in the posterior segment are unremarkable.
The initial diagnosis is simple conjunctivitis—but closer examination reveals multiple fine, barbed hairs embedded in the patient’s right cornea. Upon further questioning, the patient reports that prior to symptom onset, he had been holding the classroom pet, a Chilean Rose tarantula, in the palm of his hands.
DISCUSSION
Foreign body injury is a common cause of ocular pain and corneal damage, which can lead to challenging complications. Ophthalmic emergencies account for 2% of ED visits in the US annually and are a major cause of visual impairment.1 But when a painful eye is the chief complaint, contact with insects, plants, or spiders is rarely included in the differential. Tarantulas are popular classroom and household pets, however, and ocular injury should be suspected in anyone who has been holding a tarantula prior to onset of pain.
Ophthalmia nodosa
Tarantulas are one of the most common arachnids known to cause ophthalmia nodosa—a granulomatous reaction of the conjunctiva or cornea to an implanted plant, insect, or spider hair that typically manifests with photophobia, irritation, and chemosis.2,3 Tarantulas, when scared or defending their eggs, shoot urticating setae at the threat—a defensive mechanism largely unknown to parents, tarantula owners, and medical professionals.
Urticating setae are found in roughly 90% of tarantula species throughout tropical and subtropical regions.4 Depending on the species, setae can be located on the distal prolateral surface of the palpal femur or the dorsum of the abdomen. They can be released when the tarantula scratches its legs against the abdominal urticating setae patch or scratches the palps against the chelicerae (appendages in front of the mouth), or when direct exterior contact is made with the abdominal setae.4
There are six types of urticating hairs. Each is attached to the spider’s cuticle by either a stalk (which represents the break-off region) or a socket.4 Tarantula hairs range in size from 0.1 mm to 0.3 mm and have a sharp, pointed head and numerous barbs, which help embed them in the target.5 They are long and thin, to facilitate deep tissue penetration, and can enter the eyes, lungs, or other body parts (see Figure 2).
Ocular injury from tarantula hairs commonly involves conjunctival injection, foreign body sensation, periorbital facial rash, photophobia, and tearing.3 When a tarantula’s cloud of barbed hairs is flicked into the eye and pierces the cornea, it can cause infection, irritation, scarring on the cornea, or vision loss. Eye movement or rubbing can cause the hairs—and their toxins—to migrate over time, traveling like an arrow (the tip and barbs resist backward movement) to the anterior chamber, lens, vitreous, and retina.6,7 This can cause corneal scars, cataracts, vitritis, or macular edema, and creates the possibility for acute or chronic conjunctivitis.7
Diagnosis and management
Ophthalmic emergencies can affect the visual system and, if left untreated, can lead to permanent vision loss. Affected patients require immediate medical attention and should be referred to an ophthalmologist for follow-up care.
Diagnosis. A thorough history and physical exam are of utmost importance; tiny setae can be easily overlooked if the examiner is not diligent, and the similar symptomatology can lead to misdiagnosis as simple conjunctivitis.3 A visual acuity test and slit-lamp exam are useful for confirmation.
Treatment. Once the diagnosis is confirmed, treatment should consist of mild topical antibiotics and steroids to effectively control infection and inflammation. While topical steroids may be appropriate, local adverse events associated with their use (eg, glaucoma, cataracts) can be problematic. Gentle eye irrigation has been noted by some researchers as contraindicated, while others find it useful to flush out some of the hairs.5,8,9
Most of the visible protruding tarantula hairs can and should be removed under microscopy during slit-lamp exam. Hairs that are buried in the cornea, however, are nearly impossible to remove and pose a threat of further complications, as described. Conservative management with careful observation is therefore recommended. If the patient develops a granuloma, excision—along with a course of systemic steroids and setae removal via vitrectomy—may be needed.9
The good news is that, in many cases, deeper hairs are absorbed without complication, making their removal unnecessary.5 Factors that encourage leaving the setae untouched include a large number of hairs, deep corneal penetration, lack of patient tolerance for the procedure, and risk for perforation.3
More invasive treatments (eg, laser photocoagulation, intraocular surgery) to remove offending hairs are possible, but literature on the outcome of these interventions is limited. One report to date used argon laser photocoagulation to treat endophthalmitis from vitreous hairs.10 The laser can fragment the hairs so that they lose their barbed characteristic and cannot penetrate deeper.6
Follow-up. Close follow-up is advised, and patients should be educated on the importance of medication compliance and return visits for reevaluation. Given the potential dangers of handling these spiders, tarantula owners should be advised to use protective gloving and goggles.2,5,8,9
OUTCOME FOR THE CASE PATIENT
The case patient was sent to an ophthalmologist on day 1. Proparacaine was placed in his right eye, and all of the superficial tarantula hairs were removed using 25- and 30-gauge needles with jeweler forceps under slit-lamp microscopy. Most of the hairs were removed from the superior cornea; fewer were found in the paracentral and inferior regions of the cornea. Approximately five hairs in the paracentral area of the cornea were embedded in the midstromal depth and could not be removed. One drop of ciprofloxacin was administered.
The patient was sent home with an eye shield and instructions to use tobramycin/dexamethasone eye drops (qid in his right eye) and avoid rubbing the eye. (The eye shield, though not technically necessary, was deemed beneficial to help the patient avoid touching the eye.) He was scheduled to return to the clinic one week later.
On follow-up, a careful exam performed under microscopy showed that the five tarantula hairs were still embedded, and an additional six hairs were found in the deep stroma. Superficial punctate keratitis—an eye disorder caused by epithelial cell death on the surface of the cornea—was noted, but no anterior chamber cells were seen. The patient was instructed to continue using the eye drops as prescribed until finished, then start using loteprednol (tid) and artificial lubricating tears (every 2 h).
He returned to the clinic every two weeks for a total of 10 visits. At the end of the treatment course, the remaining tarantula hairs were unable to be removed. The patient used tapering doses of topical eye steroids and antibiotic drops secondary to flare-up.
CONCLUSION
Determining the etiology of ophthalmic emergencies is essential to timely and appropriate management. In this case, a recognized but often overlooked cause, tarantula hairs, made the diagnosis more complicated than simple conjunctivitis. When ocular injury is suspected, the provider must obtain an accurate and detailed history along with a thorough physical exam. Since patients must comply with medication regimens to prevent acute and chronic infection, a clear treatment and follow-up plan should be established. With these in place, ophthalmia nodosa caused by urticating setae can be effectively managed.
1. Fitzpatrick J, Hickman R, Alfes CM. A Guide to Mastery in Clinical Nursing: The Comprehensive Reference. New York, NY: Springer; 2018:114.
2. Lambert SR, Lyons CJ. Taylor and Hoyt’s Pediatric Ophthalmology and Strabismus. 5th ed. New York, NY: Elsevier; 2017:138.
3. Stagg BC, Ambati BK. Tarantula hairs as corneal foreign bodies. Case Rep Ophthalmol. 2011;2(3):323-326.
4. Bertani R, Guadanucci JPL. Morphology, evolution, and usage of urticating setae by tarantulas (Araneae: Theraphosidae). Zoologia (Curitiba). 2013;30(4):403-418.
5. McAnena L, Murphy C, O’Connor J. Tarantula keratitis: a case report. Ir J Med Sci. 2013;182(3):349-350.
6. Yang Y, Christakis T, Mireskandari K. Acute conjunctivitis and corneal foreign bodies secondary to tarantula hairs. CMAJ. 2016;183(3):212-214.
7. Jain N, Soong HK, Gardner TW. Ophthalmia nodosa. EyeNet Magazine. November 2013. www.aao.org/eyenet/article/blink-mystery-image-17. Accessed January 24, 2018.
8. Choi JTL, Rauf A. Ophthalmia nodosa secondary to tarantula hairs. Eye (Lond). 2003;17(3):433-434.
9. Comez AT, Tufan HA, Gencer B. Ophthalmia nodosa as an occupational disease: is it unusual or is it casual? Ocul Immunol Inflamm. 2013;21(2):144-147.
10. Marti-Huguet T, Pujol O, Cabiro I, et al. Endophthalmos caused by intravitreal caterpillar hairs. Treatment by direct photocoagulation with argon laser [article in French]. J Fr Ophthalmol. 1987;10(10):559-564.
1. Fitzpatrick J, Hickman R, Alfes CM. A Guide to Mastery in Clinical Nursing: The Comprehensive Reference. New York, NY: Springer; 2018:114.
2. Lambert SR, Lyons CJ. Taylor and Hoyt’s Pediatric Ophthalmology and Strabismus. 5th ed. New York, NY: Elsevier; 2017:138.
3. Stagg BC, Ambati BK. Tarantula hairs as corneal foreign bodies. Case Rep Ophthalmol. 2011;2(3):323-326.
4. Bertani R, Guadanucci JPL. Morphology, evolution, and usage of urticating setae by tarantulas (Araneae: Theraphosidae). Zoologia (Curitiba). 2013;30(4):403-418.
5. McAnena L, Murphy C, O’Connor J. Tarantula keratitis: a case report. Ir J Med Sci. 2013;182(3):349-350.
6. Yang Y, Christakis T, Mireskandari K. Acute conjunctivitis and corneal foreign bodies secondary to tarantula hairs. CMAJ. 2016;183(3):212-214.
7. Jain N, Soong HK, Gardner TW. Ophthalmia nodosa. EyeNet Magazine. November 2013. www.aao.org/eyenet/article/blink-mystery-image-17. Accessed January 24, 2018.
8. Choi JTL, Rauf A. Ophthalmia nodosa secondary to tarantula hairs. Eye (Lond). 2003;17(3):433-434.
9. Comez AT, Tufan HA, Gencer B. Ophthalmia nodosa as an occupational disease: is it unusual or is it casual? Ocul Immunol Inflamm. 2013;21(2):144-147.
10. Marti-Huguet T, Pujol O, Cabiro I, et al. Endophthalmos caused by intravitreal caterpillar hairs. Treatment by direct photocoagulation with argon laser [article in French]. J Fr Ophthalmol. 1987;10(10):559-564.
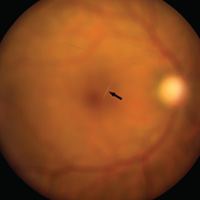
Gas under the right diaphragm
A 66-year-old man presented to the hospital with 3 days of nausea, vomiting, and abdominal pain. He had come to the emergency department several times during this period, but the cause of his symptoms had not been determined.
.")

The patient was successfully treated with urgent right hemicolectomy.
THE CHILAIDITI SIGN AND SYNDROME
The Chilaiditi sign is an infrequent anomaly found incidentally on chest or abdominal radiography as a colonic interposition between the liver and right hemidiaphragm.1 It is often asymptomatic but is sometimes accompanied by nausea, vomiting, abdominal pain, and constipation, ie, Chilaiditi syndrome.
Generally, after conservative treatment with fasting and pain control, symptoms may subside and follow-up should be sufficient. However, nasogastric decompression and laxatives are occasionally needed and are often effective in patients with Chilaiditi syndrome. Urgent abdominal surgery is indicated for patients with symptoms of volvulus of the colon, stomach, or small intestine.2
DISTINGUISHING CHARACTERISTICS
The Chilaiditi sign is often confused with pneumoperitoneum, which usually requires urgent abdominal surgery. But the presence of haustration or valvulae conniventes (folds in the small bowel mucosa) in the hepatodiaphragmatic space helps distinguish between intraluminal gas and free air. If the patient presents with abdominal pain without signs of peritonitis, and if imaging indicates the Chilaiditi sign, then supplementary imaging (eg, decubitus radiography, chest CT, abdominal CT) is recommended to make the definitive diagnosis and to avoid unnecessary surgery.
Gas under the diaphragm on standing chest radiography without signs of peritonitis may also be seen after laparotomy and after scuba diving as well as in cases of biliary enteric fistula, incompetent sphincter of Oddi, gallstone ileus, and pneumatosis cystoides intestinalis. The incidence rate of the Chilaiditi sign detected by radiography is between 0.025% and 0.28%.3
PREDISPOSING FACTORS
The cause of the Chilaiditi sign remains unknown. Predisposing factors can be categorized as diaphragmatic (diaphragmatic thinning, phrenic nerve injury, expanded thoracic cavity), intestinal (megacolon, increased intra-abdominal pressure), and hepatic (hepatic atrophy, cirrhosis, ascites).
In healthy people, Chilaiditi syndrome is usually attributed to a congenital abnormal lengthening of the colon or to undue looseness of ligaments of the colon and liver.
Recognizing the Chilaiditi sign is particularly important in patients scheduled to undergo a percutaneous transhepatic procedure or colonoscopic examination, as these procedures increase the risk of perforation.
- Chilaiditi D. Zur Frage der Hepatoptose und Ptose im allegemeinen im Anschluss an drei Fälle von temporärer, partieller Leberverlagerung. Fortschritte auf dem Gebiete der Röntgenstrahlen 1910; 16:173–208.
- Williams A, Cox R, Palaniappan B, Woodward A. Chilaiditi’s syndrome associated with colonic volvulus and intestinal malrotation—a rare case. Int J Surg Case Rep 2014; 5:335–338.
- Orangio GR, Fazio VW, Winkelman E, McGonagle BA. The Chilaiditi syndrome and associated volvulus of the transverse colon. An indication for surgical therapy. Dis Colon Rectum 1986; 29:653-656.
A 66-year-old man presented to the hospital with 3 days of nausea, vomiting, and abdominal pain. He had come to the emergency department several times during this period, but the cause of his symptoms had not been determined.
The patient was successfully treated with urgent right hemicolectomy.
THE CHILAIDITI SIGN AND SYNDROME
The Chilaiditi sign is an infrequent anomaly found incidentally on chest or abdominal radiography as a colonic interposition between the liver and right hemidiaphragm.1 It is often asymptomatic but is sometimes accompanied by nausea, vomiting, abdominal pain, and constipation, ie, Chilaiditi syndrome.
Generally, after conservative treatment with fasting and pain control, symptoms may subside and follow-up should be sufficient. However, nasogastric decompression and laxatives are occasionally needed and are often effective in patients with Chilaiditi syndrome. Urgent abdominal surgery is indicated for patients with symptoms of volvulus of the colon, stomach, or small intestine.2
DISTINGUISHING CHARACTERISTICS
The Chilaiditi sign is often confused with pneumoperitoneum, which usually requires urgent abdominal surgery. But the presence of haustration or valvulae conniventes (folds in the small bowel mucosa) in the hepatodiaphragmatic space helps distinguish between intraluminal gas and free air. If the patient presents with abdominal pain without signs of peritonitis, and if imaging indicates the Chilaiditi sign, then supplementary imaging (eg, decubitus radiography, chest CT, abdominal CT) is recommended to make the definitive diagnosis and to avoid unnecessary surgery.
Gas under the diaphragm on standing chest radiography without signs of peritonitis may also be seen after laparotomy and after scuba diving as well as in cases of biliary enteric fistula, incompetent sphincter of Oddi, gallstone ileus, and pneumatosis cystoides intestinalis. The incidence rate of the Chilaiditi sign detected by radiography is between 0.025% and 0.28%.3
PREDISPOSING FACTORS
The cause of the Chilaiditi sign remains unknown. Predisposing factors can be categorized as diaphragmatic (diaphragmatic thinning, phrenic nerve injury, expanded thoracic cavity), intestinal (megacolon, increased intra-abdominal pressure), and hepatic (hepatic atrophy, cirrhosis, ascites).
In healthy people, Chilaiditi syndrome is usually attributed to a congenital abnormal lengthening of the colon or to undue looseness of ligaments of the colon and liver.
Recognizing the Chilaiditi sign is particularly important in patients scheduled to undergo a percutaneous transhepatic procedure or colonoscopic examination, as these procedures increase the risk of perforation.
A 66-year-old man presented to the hospital with 3 days of nausea, vomiting, and abdominal pain. He had come to the emergency department several times during this period, but the cause of his symptoms had not been determined.
The patient was successfully treated with urgent right hemicolectomy.
THE CHILAIDITI SIGN AND SYNDROME
The Chilaiditi sign is an infrequent anomaly found incidentally on chest or abdominal radiography as a colonic interposition between the liver and right hemidiaphragm.1 It is often asymptomatic but is sometimes accompanied by nausea, vomiting, abdominal pain, and constipation, ie, Chilaiditi syndrome.
Generally, after conservative treatment with fasting and pain control, symptoms may subside and follow-up should be sufficient. However, nasogastric decompression and laxatives are occasionally needed and are often effective in patients with Chilaiditi syndrome. Urgent abdominal surgery is indicated for patients with symptoms of volvulus of the colon, stomach, or small intestine.2
DISTINGUISHING CHARACTERISTICS
The Chilaiditi sign is often confused with pneumoperitoneum, which usually requires urgent abdominal surgery. But the presence of haustration or valvulae conniventes (folds in the small bowel mucosa) in the hepatodiaphragmatic space helps distinguish between intraluminal gas and free air. If the patient presents with abdominal pain without signs of peritonitis, and if imaging indicates the Chilaiditi sign, then supplementary imaging (eg, decubitus radiography, chest CT, abdominal CT) is recommended to make the definitive diagnosis and to avoid unnecessary surgery.
Gas under the diaphragm on standing chest radiography without signs of peritonitis may also be seen after laparotomy and after scuba diving as well as in cases of biliary enteric fistula, incompetent sphincter of Oddi, gallstone ileus, and pneumatosis cystoides intestinalis. The incidence rate of the Chilaiditi sign detected by radiography is between 0.025% and 0.28%.3
PREDISPOSING FACTORS
The cause of the Chilaiditi sign remains unknown. Predisposing factors can be categorized as diaphragmatic (diaphragmatic thinning, phrenic nerve injury, expanded thoracic cavity), intestinal (megacolon, increased intra-abdominal pressure), and hepatic (hepatic atrophy, cirrhosis, ascites).
In healthy people, Chilaiditi syndrome is usually attributed to a congenital abnormal lengthening of the colon or to undue looseness of ligaments of the colon and liver.
Recognizing the Chilaiditi sign is particularly important in patients scheduled to undergo a percutaneous transhepatic procedure or colonoscopic examination, as these procedures increase the risk of perforation.
- Chilaiditi D. Zur Frage der Hepatoptose und Ptose im allegemeinen im Anschluss an drei Fälle von temporärer, partieller Leberverlagerung. Fortschritte auf dem Gebiete der Röntgenstrahlen 1910; 16:173–208.
- Williams A, Cox R, Palaniappan B, Woodward A. Chilaiditi’s syndrome associated with colonic volvulus and intestinal malrotation—a rare case. Int J Surg Case Rep 2014; 5:335–338.
- Orangio GR, Fazio VW, Winkelman E, McGonagle BA. The Chilaiditi syndrome and associated volvulus of the transverse colon. An indication for surgical therapy. Dis Colon Rectum 1986; 29:653-656.
- Chilaiditi D. Zur Frage der Hepatoptose und Ptose im allegemeinen im Anschluss an drei Fälle von temporärer, partieller Leberverlagerung. Fortschritte auf dem Gebiete der Röntgenstrahlen 1910; 16:173–208.
- Williams A, Cox R, Palaniappan B, Woodward A. Chilaiditi’s syndrome associated with colonic volvulus and intestinal malrotation—a rare case. Int J Surg Case Rep 2014; 5:335–338.
- Orangio GR, Fazio VW, Winkelman E, McGonagle BA. The Chilaiditi syndrome and associated volvulus of the transverse colon. An indication for surgical therapy. Dis Colon Rectum 1986; 29:653-656.
A 75-year-old with abdominal pain, hypoxia, and weak pulses in the left leg
A 75-year-old man presented to the emergency department for evaluation of abdominal pain. He had stage 3 chronic obstructive pulmonary disease (COPD), with a forced expiratory volume in 1 second of 33%.
PREVIOUS HOSPITALIZATION
Aside from his COPD, he had been healthy until 1 month earlier, when he had been hospitalized because of shortness of breath and chest pressure with exertion. His troponin T level had been elevated, peaking at 0.117 ng/mL (reference range 0–0.029).
Left heart catheterization had shown no significant coronary artery disease. A myocardial bridge of the distal left anterior descending coronary artery had been seen, so that the artery appeared to be narrowed by 50% to 60% with ventricular contraction. But this was not thought to have been the cause of his presentation.
On discharge, he required oxygen 4 L/min by nasal cannula. Previously, he had not needed supplemental oxygen.
CURRENT PRESENTATION
The patient described persistent and severe periumbilical abdominal pain during the previous day. It was not associated with eating, and he denied diarrhea, constipation, hematemesis, hematochezia, bright red blood per rectum, or melena. He continued to describe persistent shortness of breath and pleuritic chest pain. His vital signs were as follows:
- Heart rate 104 beats per minute
- Respiratory rate 16 to 20 breaths per minute
- Blood pressure 101–142/62–84 mm Hg
- Oxygen saturation 78% on room air.


His laboratory findings on presentation are shown in Table 1, and his electrocardiogram is shown in Figure 1.
WHAT DOES HIS ELECTROCARDIOGRAM SHOW?
1. Which of the following is the most accurate description of this patient’s electrocardiogram?
- Sinus tachycardia, peaked P waves (P pulmonale) in lead II, and T-wave inversions in the right precordial leads
- Sinus tachycardia and left bundle branch block
- Sinus tachycardia and poor R-wave progression
- Sinus tachycardia and ST elevation in the precordial leads
Our patient’s electrocardiogram shows sinus tachycardia, P pulmonale, T-wave inversion in the right precordial leads (V1–V3), and biphasic T waves in lead V4,, which suggest right ventricular strain.
The rhythm most commonly seen in patients with pulmonary embolism is sinus tachycardia, followed by nonspecific ST-segment or T-wave abnormalities. In one series of patients with acute pulmonary embolism, the classic findings of P pulmonale, right ventricular hypertrophy, right axis deviation, and right bundle branch block were rare (< 6%).1 Thus, these classic findings are not sensitive for the diagnosis of pulmonary embolism, and their absence does not rule it out.
Further studies for our patient
 with a chest pulmonary embolism protocol showed filling defects.")
Transthoracic echocardiography was performed to look for evidence of right ventricular strain secondary to the pulmonary embolism.
ECHOCARDIOGRAPHIC SIGNS OF PULMONARY EMBOLISM
2. Which of the following findings on transthoracic echocardiography would not suggest acute pulmonary embolism?
- Midright ventricular wall hypokinesis with apical sparing
- Severe tricuspid regurgitation
- Left ventricular dilation
- Lack of respiratory variation of the inferior vena cava
- Septal wall motion toward the left ventricle
Left ventricular dilation does not suggest acute pulmonary embolism. Echocardiograms of patients with acute submassive pulmonary embolism typically show evidence of right ventricular strain, such as the other entities listed above (midright ventricular hypokinesis with apical sparing, severe tricuspid regurgitation, lack of respiratory variation of the inferior vena cava, and septal wall motion toward the left ventricle).
The degree of right ventricular dysfunction is related to the extent of acute pulmonary vascular occlusion and aids in risk-stratification of patients with acute pulmonary embolism. Midright ventricular wall hypokinesis with apical sparing has been termed the McConnell sign.2
In our patient, transthoracic echocardiography showed:
- Normal left ventricular ejection fraction
- Mild diastolic dysfunction
- Right ventricular dilation with moderately decreased right ventricular systolic function and apical sparing
- Right ventricular systolic pressure 54 mm Hg, consistent with moderate pulmonary hypertension
- Right atrial pressure 10 mm Hg
- No inspiratory collapse of a dilated inferior vena cava
- Mild tricuspid valve regurgitation.
CLASSIFICATION OF ACUTE PULMONARY EMBOLISM
3. Given the above information, how would you classify the patient’s pulmonary embolism?
- Massive
- Submassive
- Low-risk
- Clinically stable
The patient’s pulmonary embolism is submassive.

Historically, the classification of pulmonary embolism was determined by the angiographic thrombus burden. However, this has limited utility because clinical factors (eg, hypotension on initial presentation) have been shown to be better predictors of short-term mortality risk.3
Our patient is characterized as having a submassive pulmonary embolism based on elevated biomarkers (troponin T, N-terminal pro-B-type natriuretic peptide) and right ventricular dysfunction in the absence of hypotension.
ULTRASONOGRAPHY FOR DIAGNOSIS OF DEEP VEIN THROMBOSIS

Venous duplex ultrasonography has become the standard for diagnosis of lower extremity deep vein thrombosis. However, its quality and diagnostic accuracy depend on the skill of the person performing the examination. It is further limited by certain patient characteristics, including severe obesity, edema, and wounds and dressings at the site being examined.5
Our patient underwent duplex ultrasonography of the lower extremities, which demonstrated acute proximal and calf deep vein thrombosis in the right femoral, popliteal, and peroneal veins and no deep vein thrombosis in the left leg.
RISK STRATIFICATION IN ACUTE PULMONARY EMBOLISM
Multiple models exist to estimate the risk of complications in patients with acute pulmonary embolism.
The Bova score6 is based on the following factors:
- Systolic blood pressure 90–100 mm Hg (2 points) (patients with systolic blood pressure lower than 90 mm Hg were excluded from the study from which this score was derived)
- Cardiac troponin elevation (2 points)
- Right ventricular dysfunction on echocardiography or computed tomography (2 points)
- Heart rate 100 beats/min or greater (1 point).
A total score of 0, 1, or 2 (stage I) denotes low risk, 3 or 4 points (stage II) intermediate risk, and more than 4 points (stage III) high risk.
The PESI score (Pulmonary Embolism Severity Index)7 is based on:
- Age (1 point per year)
- Sex (10 points for being male)
- Heart rate 110 per minute or greater (20 points)
- Cancer (30 points)
- Heart failure (10 points)
- Chronic lung disease (10 points)
- Systolic blood pressure less than 100 mm Hg (30 points)
- Respiratory rate at least 30 per minute (20 points)
- Temperature less than 36ºC (20 points)
- Altered mental status (60 points)
- Arterial oxygen saturation less than 90% (20 points).
The total score is broken down into 5 classes: I (< 65 points), II (65–85), III (86–105), IV (106–125), and V (> 126). Classes I and II are low risk, and the higher ones are high risk.
The simplified PESI score8 was developed to more rapidly risk-stratify patients and has been found to be similar to the PESI score in prognostic accuracy. Patients get 1 point for each of the following:
- Age over 80
- Cancer
- Chronic cardiopulmonary disease (heart failure or chronic lung disease)
- Heart rate 110 per minute or greater
- Systolic blood pressure less than 100 mm Hg
- Arterial oxygen saturation less than 90%.
A total score of 0 is low risk; anything higher is high risk.
Back to our patient
Our patient had proximal and calf deep vein thrombosis of the right leg, bilateral submassive pulmonary emboli with associated biomarker elevation and right ventricular dysfunction, and left renal artery thrombosis with infarction. Using the PESI score, his risk of death in the next 30 days was 13.7% and his 30-day risk of a complicated course was 27%. Using the Bova score, his 30-day risk of death was 15.5% and his 30-day risk of a complicated course was 29.2%.6,7
Notably, the patient’s right ventricular function had also been impaired on the echocardiogram performed during his admission 1 month previously. On transthoracic echocardiography during the current admission, the patient was found to have a similar degree of right ventricular dysfunction. This finding, along with the oxygen requirement that developed during the earlier admission, suggested that his pulmonary embolism may have been subacute and that the diagnosis may have been missed during the earlier hospital stay.
The patient was treated with unfractionated heparin. After the hospital’s multidisciplinary pulmonary embolism response team discussed and weighed the above factors, they recommended to not pursue thrombolytic therapy or inferior vena cava filter placement.
Of note, the patient’s pulses in the left lower extremity continued to be weak but palpable, and the left leg was cooler to touch than the right leg.
ASSESSING PERIPHERAL ARTERY DISEASE
4. How should the finding of weak pulses in this patient’s left leg be initially investigated?
- Computed tomographic angiography with runoff
- Ankle-brachial indices with pulse-volume recordings
- Arterial duplex ultrasonography
- Magnetic resonance angiography of the lower extremities
The ankle-brachial index is the initial diagnostic test for assessment of pulse abnormalities and for diagnosis of lower-extremity peripheral artery disease. It is calculated by dividing the higher of the ankle systolic pressures (posterior tibial or dorsalis pedis) by the higher of the 2 brachial pressures (left or right).9 Normal values are between 1.00 and 1.40.
Ankle-brachial indices in our patient
Our patient underwent measurement of his brachial, dorsalis pedis, and posterior tibial artery systolic pressures using blood pressure cuffs and continuous-wave Doppler. Ankle pulse-volume recordings were also obtained.

Given the patient’s poor renal function and concern for acute renal infarction, we thought it best to avoid iodinated or gadolinium contrast, such as with magnetic resonance or computed tomographic angiography.
Segmental leg pressures and pulse-volume recordings can be performed to help localize the level of arterial disease in the extremities, but were not done in this case because of the extensive deep vein thrombosis in the right leg.10,11
Arterial ultrasonography in our patient
Arterial duplex ultrasonography was performed to help determine the location of arterial disease. It showed patent arteries in the right leg. In the left lower extremity there was slow, monophasic blood flow in the distal superficial femoral artery. The popliteal artery was occluded. The posterior tibial artery was occluded at the origin, with reconstitution distally. The peroneal artery was occluded throughout. The anterior tibial artery was patent throughout. The ultrasonographic findings were thought to be suspicious for arterial thromboembolism.
WHAT CAN CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
5. Given that the patient had both arterial thrombosis (renal artery, lower-extremity arteries) and venous thromboembolism (deep vein thrombosis and pulmonary embolism), which of the following would be included in the differential diagnosis?
- Antiphospholipid antibody syndrome
- Protein C or protein S deficiency
- Malignancy
- Paradoxical embolization
- Factor V Leiden mutation
Correct answers include antiphospholipid antibody syndrome, malignancy, and paradoxical embolization.
The differential diagnosis for concomitant venous and arterial thrombosis is broad,12 and includes the following:
- Structural factors: patent foramen ovale, popliteal artery aneurysm
- Malignancy
- Inflammatory diseases: Behçet disease, Buerger disease, inflammatory bowel disease, antiphospholipid antibody syndrome, elevated lipoprotein(a), elevated homocysteine
- Hematologic diseases: myelodysplastic syndrome, disseminated intravascular coagulation, paroxysmal nocturnal hemoglobinuria, heparin-induced thrombocytopenia.
Traditional risk factors for venous thromboembolism include protein C deficiency, protein S deficiency, factor V Leiden mutation, the prothrombin G20210A gene mutation, and others. These are relatively minor risk factors for venous thrombosis and do not pose a risk for arterial thrombosis.12 In contrast, antiphospholipid antibody syndrome and malignancy pose a risk for both venous and arterial thrombosis. Paradoxical embolism is a mechanism by which arterial thrombosis (emboli) can develop in the setting of existing venous thrombosis.12
Our patient underwent testing for antiphospholipid antibodies and lupus anticoagulant, and he was encouraged to undergo age-appropriate cancer screening as an outpatient.12
ANTIPHOSPHOLIPID ANTIBODY SYNDROME
Antiphospholipid antibody syndrome is defined by both clinical and laboratory criteria. Clinical symptoms include vascular thrombosis (arterial, venous, or both) and pregnancy-related complications.13
Laboratory criteria require the presence of antiphospholipid antibodies or lupus anticoagulant. These must be confirmed with repeat testing in 12 weeks. Antiphospholipid antibodies are detected by an enzyme-linked immunosorbent assay; laboratory assessment for the presence of lupus anticoagulant is a stepwise process and relies on 4 criteria:
- There should be prolongation of a phospholipid-dependent clotting test (eg, activated partial thromboplastin time, dilute Russell viper venom time test).
- There must be evidence of an inhibitory activity with mixing study.
- The inhibitor must exhibit phospholipid dependence; that is, with more phospholipid there is shortening of clotting time.
- Specific inhibitors must be excluded, including factor VIII and anticoagulant drugs such as heparin.14–17

Clinically, one should consider antiphospholipid antibody syndrome in patients who have arterial thrombosis, a history of pregnancy morbidity, or unexplained prolongation of activated partial thromboplastin time.13
Antiphospholipid antibodies may be present in up to a quarter of patients with venous thromboembolism, but it is persistent positivity of antibody assays that is associated with increased future risk of venous thromboembolism.19 Of note, the risk of venous thromboembolism in patients with confirmed antiphospholipid antibody syndrome is 10 times higher than in the general population.20
ANTIPHOSPHOLIPID ANTIBODIES ARE NOT ALL THE SAME
6. Which of the following antiphospholipid antibodies have not been associated with an increased thrombotic risk?
- Anti-beta-2-glycoprotein I IgG
- Lupus anticoagulant
- Antiphosphatidylserine
- Anticardiolipin IgM
- Anticardiolipin IgG
The correct answer is antiphosphatidylserine.15
Antiphospholipid antibodies are directed against a portion of select plasma proteins that are uncovered upon phospholipid binding. While lupus anticoagulant, anti-beta-2-glycoprotein I, and anticardiolipin antibodies are associated with thrombosis, antiprothrombin antibodies (including antiprothrombin and antiphosphatidylserine antibodies) are not.15,21
PARADOXICAL EMBOLISM
Patent foramen ovale, a communication between the right and left atrium in the interatrial septum, is associated with an increased risk of paradoxical embolization. The prevalence of patent foramen ovale is estimated to be 27% to 29% in the general population.22 Noncerebral systemic paradoxical embolism occurs less frequently than cerebral embolism, accounting for approximately 5% to 10% of paradoxical emboli.22
To evaluate for patent foramen ovale, transthoracic echocardiography is performed with a bubble (agitated saline contrast) study to assess for interatrial shunting. Transesophageal echocardiography or transcranial Doppler bubble studies may also be performed.
Although patent foramen ovale is most commonly associated with cerebral embolism, peripheral emboli can occur. Some research suggests that this may be a more common cause of arterial thromboembolism in younger patients. There have also been reports of other sites of systemic embolization, including the renal artery.12
Back to our patient
Initial antiphospholipid antibody testing was positive for lupus anticoagulant. Anticardiolipin and anti-beta-2-glycoprotein I antibodies were not detected.
Transesophageal echocardiography revealed a patent foramen ovale with a highly mobile atrial septum (atrial septal aneurysm).
The patient was treated with intravenous unfractionated heparin with bridging to warfarin with a target international normalized ratio (INR) of 2 to 3. His renal artery infarction and his lower-extremity arterial thromboembolic event were conservatively managed. His respiratory status improved, and he no longer required supplemental oxygen. His creatinine peaked at 1.7 mg/dL during his admission and improved to 1.2 mg/dL before he was discharged.
At follow-up, repeat echocardiography showed that his right ventricular systolic pressure had improved (decreased) to 37 mm Hg from 54 mm Hg. Repeat confirmatory testing was positive for lupus anticoagulant 12 weeks later. He has been maintained on warfarin with an INR goal of 2 to 3 as well as low-dose aspirin with plans for long-term anticoagulation. We decided to keep the patient on anticoagulation indefinitely with warfarin; he was not a candidate for a direct oral anticoagulant, given limited data on the use of these agents in the setting of lupus anticoagulant and antiphospholipid antibody syndrome.
SUMMARY OF CASE
In summary, this patient was a 75-year-old man with COPD who presented with abdominal pain. He was noted to have a left renal infarction, extensive unprovoked lower-extremity deep vein thrombosis with pulmonary emboli, and lower limb arterial thromboembolism.
He also had an underlying hypercoagulable state—antiphospholipid antibody syndrome—that predisposed him to both arterial and venous thrombosis. He was ultimately found to have a patent foramen ovale, which further increased the risk of arterial thrombosis by facilitating paradoxical embolization of venous thrombi. It is not certain whether the renal infarction and leg artery thrombi were due to paradoxical embolism or to in situ thrombosis, but we believe that it was most likely paradoxical embolization.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Alsoos F, Khaddam A. Echocardiographic evaluation methods for right ventricular function. J Echocardiogr 2015; 13:43–51.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; American Heart Association Council on Peripheral Vascular Disease; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Heit JA, Silverstein MD, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med 2000; 160:809–815.
- Gornik HL, Sharma AM. Duplex ultrasound in the diagnosis of lower-extremity deep venous thrombosis. Circulation 2014; 129:917–921.
- Fernández C, Bova C, Sanchez O, et al. Validation of a model for identification of patients at intermediate to high risk for complications associated with acute symptomatic pulmonary embolism. Chest 2015; 148:211–218.
- Aujesky D, Perrier A, Roy PM, et al. Validation of a clinical prognostic model to identify low-risk patients with pulmonary embolism. J Intern Med 2007; 261:597–604.
- Jiménez D, Aujesky D, Moores L, et al; RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med 2010; 170:1383–1389.
- Kim ES, Wattanakit K, Gornik HL. Using the ankle-brachial index to diagnose peripheral artery disease and assess cardiovascular risk. Cleve Clin J Med 2012; 79:651–661.
- Jaff MR. Lower extremity arterial disease. Diagnostic aspects. Cardiol Clin 2002; 20:491–500.
- Rooke TW, Hirsch AT, Misra S, et al; American College of Cardiology Foundation Task Force; American Heart Association Task Force. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA Guideline Recommendations): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:1555–1570.
- Lichtin A, Bartholomew J. The coagulation consult: a case-based guide. New York, NY: Springer; 2014.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Miyakis S, Lockshin M, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Nichols WL, Kottke-Marchant K, Ledford-Kraemer MR, Homburger HA, Cardel LK. Lupus anticoagulants, antiphospholipid antibodies, and antiphospholipid syndrome. In: Kottke-Marchant K, Davis BH, editors. Laboratory Hematology Practice. Hoboken, New Jersey: Blackwell Publishing, Ltd.; 2012:509–525.
- Houghton DE, Moll S. Antiphospholipid antibodies. Vasc Med 2017; 22:545–550.
- Roldan V, Lecumberri R, Muñoz-Torrero JFS, et al; RIETE Investigators. Thrombophilia testing in patients with venous thromboembolism. Findings from the RIETE registry. Thromb Res 2009; 124:174–177.
- Wahl DG, Guillemin F, de Maistre E, Perret-Guillaume C, Lecompte T, Thibaut G. Meta-analysis of the risk of venous thrombosis in individuals with antiphospholipid antibodies without underlying autoimmune disease or previous thrombosis. Lupus 1998; 7:15–22.
- Love PE, Santoro SA. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders. Prevalence and clinical significance. Ann Intern Med 1990; 112:682–698.
- Thompson T, Evans W. Paradoxical embolism. QJM 1930; os-23:135–150.
A 75-year-old man presented to the emergency department for evaluation of abdominal pain. He had stage 3 chronic obstructive pulmonary disease (COPD), with a forced expiratory volume in 1 second of 33%.
PREVIOUS HOSPITALIZATION
Aside from his COPD, he had been healthy until 1 month earlier, when he had been hospitalized because of shortness of breath and chest pressure with exertion. His troponin T level had been elevated, peaking at 0.117 ng/mL (reference range 0–0.029).
Left heart catheterization had shown no significant coronary artery disease. A myocardial bridge of the distal left anterior descending coronary artery had been seen, so that the artery appeared to be narrowed by 50% to 60% with ventricular contraction. But this was not thought to have been the cause of his presentation.
On discharge, he required oxygen 4 L/min by nasal cannula. Previously, he had not needed supplemental oxygen.
CURRENT PRESENTATION
The patient described persistent and severe periumbilical abdominal pain during the previous day. It was not associated with eating, and he denied diarrhea, constipation, hematemesis, hematochezia, bright red blood per rectum, or melena. He continued to describe persistent shortness of breath and pleuritic chest pain. His vital signs were as follows:
- Heart rate 104 beats per minute
- Respiratory rate 16 to 20 breaths per minute
- Blood pressure 101–142/62–84 mm Hg
- Oxygen saturation 78% on room air.
His laboratory findings on presentation are shown in Table 1, and his electrocardiogram is shown in Figure 1.
WHAT DOES HIS ELECTROCARDIOGRAM SHOW?
1. Which of the following is the most accurate description of this patient’s electrocardiogram?
- Sinus tachycardia, peaked P waves (P pulmonale) in lead II, and T-wave inversions in the right precordial leads
- Sinus tachycardia and left bundle branch block
- Sinus tachycardia and poor R-wave progression
- Sinus tachycardia and ST elevation in the precordial leads
Our patient’s electrocardiogram shows sinus tachycardia, P pulmonale, T-wave inversion in the right precordial leads (V1–V3), and biphasic T waves in lead V4,, which suggest right ventricular strain.
The rhythm most commonly seen in patients with pulmonary embolism is sinus tachycardia, followed by nonspecific ST-segment or T-wave abnormalities. In one series of patients with acute pulmonary embolism, the classic findings of P pulmonale, right ventricular hypertrophy, right axis deviation, and right bundle branch block were rare (< 6%).1 Thus, these classic findings are not sensitive for the diagnosis of pulmonary embolism, and their absence does not rule it out.
Further studies for our patient
Transthoracic echocardiography was performed to look for evidence of right ventricular strain secondary to the pulmonary embolism.
ECHOCARDIOGRAPHIC SIGNS OF PULMONARY EMBOLISM
2. Which of the following findings on transthoracic echocardiography would not suggest acute pulmonary embolism?
- Midright ventricular wall hypokinesis with apical sparing
- Severe tricuspid regurgitation
- Left ventricular dilation
- Lack of respiratory variation of the inferior vena cava
- Septal wall motion toward the left ventricle
Left ventricular dilation does not suggest acute pulmonary embolism. Echocardiograms of patients with acute submassive pulmonary embolism typically show evidence of right ventricular strain, such as the other entities listed above (midright ventricular hypokinesis with apical sparing, severe tricuspid regurgitation, lack of respiratory variation of the inferior vena cava, and septal wall motion toward the left ventricle).
The degree of right ventricular dysfunction is related to the extent of acute pulmonary vascular occlusion and aids in risk-stratification of patients with acute pulmonary embolism. Midright ventricular wall hypokinesis with apical sparing has been termed the McConnell sign.2
In our patient, transthoracic echocardiography showed:
- Normal left ventricular ejection fraction
- Mild diastolic dysfunction
- Right ventricular dilation with moderately decreased right ventricular systolic function and apical sparing
- Right ventricular systolic pressure 54 mm Hg, consistent with moderate pulmonary hypertension
- Right atrial pressure 10 mm Hg
- No inspiratory collapse of a dilated inferior vena cava
- Mild tricuspid valve regurgitation.
CLASSIFICATION OF ACUTE PULMONARY EMBOLISM
3. Given the above information, how would you classify the patient’s pulmonary embolism?
- Massive
- Submassive
- Low-risk
- Clinically stable
The patient’s pulmonary embolism is submassive.
Historically, the classification of pulmonary embolism was determined by the angiographic thrombus burden. However, this has limited utility because clinical factors (eg, hypotension on initial presentation) have been shown to be better predictors of short-term mortality risk.3
Our patient is characterized as having a submassive pulmonary embolism based on elevated biomarkers (troponin T, N-terminal pro-B-type natriuretic peptide) and right ventricular dysfunction in the absence of hypotension.
ULTRASONOGRAPHY FOR DIAGNOSIS OF DEEP VEIN THROMBOSIS
Venous duplex ultrasonography has become the standard for diagnosis of lower extremity deep vein thrombosis. However, its quality and diagnostic accuracy depend on the skill of the person performing the examination. It is further limited by certain patient characteristics, including severe obesity, edema, and wounds and dressings at the site being examined.5
Our patient underwent duplex ultrasonography of the lower extremities, which demonstrated acute proximal and calf deep vein thrombosis in the right femoral, popliteal, and peroneal veins and no deep vein thrombosis in the left leg.
RISK STRATIFICATION IN ACUTE PULMONARY EMBOLISM
Multiple models exist to estimate the risk of complications in patients with acute pulmonary embolism.
The Bova score6 is based on the following factors:
- Systolic blood pressure 90–100 mm Hg (2 points) (patients with systolic blood pressure lower than 90 mm Hg were excluded from the study from which this score was derived)
- Cardiac troponin elevation (2 points)
- Right ventricular dysfunction on echocardiography or computed tomography (2 points)
- Heart rate 100 beats/min or greater (1 point).
A total score of 0, 1, or 2 (stage I) denotes low risk, 3 or 4 points (stage II) intermediate risk, and more than 4 points (stage III) high risk.
The PESI score (Pulmonary Embolism Severity Index)7 is based on:
- Age (1 point per year)
- Sex (10 points for being male)
- Heart rate 110 per minute or greater (20 points)
- Cancer (30 points)
- Heart failure (10 points)
- Chronic lung disease (10 points)
- Systolic blood pressure less than 100 mm Hg (30 points)
- Respiratory rate at least 30 per minute (20 points)
- Temperature less than 36ºC (20 points)
- Altered mental status (60 points)
- Arterial oxygen saturation less than 90% (20 points).
The total score is broken down into 5 classes: I (< 65 points), II (65–85), III (86–105), IV (106–125), and V (> 126). Classes I and II are low risk, and the higher ones are high risk.
The simplified PESI score8 was developed to more rapidly risk-stratify patients and has been found to be similar to the PESI score in prognostic accuracy. Patients get 1 point for each of the following:
- Age over 80
- Cancer
- Chronic cardiopulmonary disease (heart failure or chronic lung disease)
- Heart rate 110 per minute or greater
- Systolic blood pressure less than 100 mm Hg
- Arterial oxygen saturation less than 90%.
A total score of 0 is low risk; anything higher is high risk.
Back to our patient
Our patient had proximal and calf deep vein thrombosis of the right leg, bilateral submassive pulmonary emboli with associated biomarker elevation and right ventricular dysfunction, and left renal artery thrombosis with infarction. Using the PESI score, his risk of death in the next 30 days was 13.7% and his 30-day risk of a complicated course was 27%. Using the Bova score, his 30-day risk of death was 15.5% and his 30-day risk of a complicated course was 29.2%.6,7
Notably, the patient’s right ventricular function had also been impaired on the echocardiogram performed during his admission 1 month previously. On transthoracic echocardiography during the current admission, the patient was found to have a similar degree of right ventricular dysfunction. This finding, along with the oxygen requirement that developed during the earlier admission, suggested that his pulmonary embolism may have been subacute and that the diagnosis may have been missed during the earlier hospital stay.
The patient was treated with unfractionated heparin. After the hospital’s multidisciplinary pulmonary embolism response team discussed and weighed the above factors, they recommended to not pursue thrombolytic therapy or inferior vena cava filter placement.
Of note, the patient’s pulses in the left lower extremity continued to be weak but palpable, and the left leg was cooler to touch than the right leg.
ASSESSING PERIPHERAL ARTERY DISEASE
4. How should the finding of weak pulses in this patient’s left leg be initially investigated?
- Computed tomographic angiography with runoff
- Ankle-brachial indices with pulse-volume recordings
- Arterial duplex ultrasonography
- Magnetic resonance angiography of the lower extremities
The ankle-brachial index is the initial diagnostic test for assessment of pulse abnormalities and for diagnosis of lower-extremity peripheral artery disease. It is calculated by dividing the higher of the ankle systolic pressures (posterior tibial or dorsalis pedis) by the higher of the 2 brachial pressures (left or right).9 Normal values are between 1.00 and 1.40.
Ankle-brachial indices in our patient
Our patient underwent measurement of his brachial, dorsalis pedis, and posterior tibial artery systolic pressures using blood pressure cuffs and continuous-wave Doppler. Ankle pulse-volume recordings were also obtained.
Given the patient’s poor renal function and concern for acute renal infarction, we thought it best to avoid iodinated or gadolinium contrast, such as with magnetic resonance or computed tomographic angiography.
Segmental leg pressures and pulse-volume recordings can be performed to help localize the level of arterial disease in the extremities, but were not done in this case because of the extensive deep vein thrombosis in the right leg.10,11
Arterial ultrasonography in our patient
Arterial duplex ultrasonography was performed to help determine the location of arterial disease. It showed patent arteries in the right leg. In the left lower extremity there was slow, monophasic blood flow in the distal superficial femoral artery. The popliteal artery was occluded. The posterior tibial artery was occluded at the origin, with reconstitution distally. The peroneal artery was occluded throughout. The anterior tibial artery was patent throughout. The ultrasonographic findings were thought to be suspicious for arterial thromboembolism.
WHAT CAN CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
5. Given that the patient had both arterial thrombosis (renal artery, lower-extremity arteries) and venous thromboembolism (deep vein thrombosis and pulmonary embolism), which of the following would be included in the differential diagnosis?
- Antiphospholipid antibody syndrome
- Protein C or protein S deficiency
- Malignancy
- Paradoxical embolization
- Factor V Leiden mutation
Correct answers include antiphospholipid antibody syndrome, malignancy, and paradoxical embolization.
The differential diagnosis for concomitant venous and arterial thrombosis is broad,12 and includes the following:
- Structural factors: patent foramen ovale, popliteal artery aneurysm
- Malignancy
- Inflammatory diseases: Behçet disease, Buerger disease, inflammatory bowel disease, antiphospholipid antibody syndrome, elevated lipoprotein(a), elevated homocysteine
- Hematologic diseases: myelodysplastic syndrome, disseminated intravascular coagulation, paroxysmal nocturnal hemoglobinuria, heparin-induced thrombocytopenia.
Traditional risk factors for venous thromboembolism include protein C deficiency, protein S deficiency, factor V Leiden mutation, the prothrombin G20210A gene mutation, and others. These are relatively minor risk factors for venous thrombosis and do not pose a risk for arterial thrombosis.12 In contrast, antiphospholipid antibody syndrome and malignancy pose a risk for both venous and arterial thrombosis. Paradoxical embolism is a mechanism by which arterial thrombosis (emboli) can develop in the setting of existing venous thrombosis.12
Our patient underwent testing for antiphospholipid antibodies and lupus anticoagulant, and he was encouraged to undergo age-appropriate cancer screening as an outpatient.12
ANTIPHOSPHOLIPID ANTIBODY SYNDROME
Antiphospholipid antibody syndrome is defined by both clinical and laboratory criteria. Clinical symptoms include vascular thrombosis (arterial, venous, or both) and pregnancy-related complications.13
Laboratory criteria require the presence of antiphospholipid antibodies or lupus anticoagulant. These must be confirmed with repeat testing in 12 weeks. Antiphospholipid antibodies are detected by an enzyme-linked immunosorbent assay; laboratory assessment for the presence of lupus anticoagulant is a stepwise process and relies on 4 criteria:
- There should be prolongation of a phospholipid-dependent clotting test (eg, activated partial thromboplastin time, dilute Russell viper venom time test).
- There must be evidence of an inhibitory activity with mixing study.
- The inhibitor must exhibit phospholipid dependence; that is, with more phospholipid there is shortening of clotting time.
- Specific inhibitors must be excluded, including factor VIII and anticoagulant drugs such as heparin.14–17
Clinically, one should consider antiphospholipid antibody syndrome in patients who have arterial thrombosis, a history of pregnancy morbidity, or unexplained prolongation of activated partial thromboplastin time.13
Antiphospholipid antibodies may be present in up to a quarter of patients with venous thromboembolism, but it is persistent positivity of antibody assays that is associated with increased future risk of venous thromboembolism.19 Of note, the risk of venous thromboembolism in patients with confirmed antiphospholipid antibody syndrome is 10 times higher than in the general population.20
ANTIPHOSPHOLIPID ANTIBODIES ARE NOT ALL THE SAME
6. Which of the following antiphospholipid antibodies have not been associated with an increased thrombotic risk?
- Anti-beta-2-glycoprotein I IgG
- Lupus anticoagulant
- Antiphosphatidylserine
- Anticardiolipin IgM
- Anticardiolipin IgG
The correct answer is antiphosphatidylserine.15
Antiphospholipid antibodies are directed against a portion of select plasma proteins that are uncovered upon phospholipid binding. While lupus anticoagulant, anti-beta-2-glycoprotein I, and anticardiolipin antibodies are associated with thrombosis, antiprothrombin antibodies (including antiprothrombin and antiphosphatidylserine antibodies) are not.15,21
PARADOXICAL EMBOLISM
Patent foramen ovale, a communication between the right and left atrium in the interatrial septum, is associated with an increased risk of paradoxical embolization. The prevalence of patent foramen ovale is estimated to be 27% to 29% in the general population.22 Noncerebral systemic paradoxical embolism occurs less frequently than cerebral embolism, accounting for approximately 5% to 10% of paradoxical emboli.22
To evaluate for patent foramen ovale, transthoracic echocardiography is performed with a bubble (agitated saline contrast) study to assess for interatrial shunting. Transesophageal echocardiography or transcranial Doppler bubble studies may also be performed.
Although patent foramen ovale is most commonly associated with cerebral embolism, peripheral emboli can occur. Some research suggests that this may be a more common cause of arterial thromboembolism in younger patients. There have also been reports of other sites of systemic embolization, including the renal artery.12
Back to our patient
Initial antiphospholipid antibody testing was positive for lupus anticoagulant. Anticardiolipin and anti-beta-2-glycoprotein I antibodies were not detected.
Transesophageal echocardiography revealed a patent foramen ovale with a highly mobile atrial septum (atrial septal aneurysm).
The patient was treated with intravenous unfractionated heparin with bridging to warfarin with a target international normalized ratio (INR) of 2 to 3. His renal artery infarction and his lower-extremity arterial thromboembolic event were conservatively managed. His respiratory status improved, and he no longer required supplemental oxygen. His creatinine peaked at 1.7 mg/dL during his admission and improved to 1.2 mg/dL before he was discharged.
At follow-up, repeat echocardiography showed that his right ventricular systolic pressure had improved (decreased) to 37 mm Hg from 54 mm Hg. Repeat confirmatory testing was positive for lupus anticoagulant 12 weeks later. He has been maintained on warfarin with an INR goal of 2 to 3 as well as low-dose aspirin with plans for long-term anticoagulation. We decided to keep the patient on anticoagulation indefinitely with warfarin; he was not a candidate for a direct oral anticoagulant, given limited data on the use of these agents in the setting of lupus anticoagulant and antiphospholipid antibody syndrome.
SUMMARY OF CASE
In summary, this patient was a 75-year-old man with COPD who presented with abdominal pain. He was noted to have a left renal infarction, extensive unprovoked lower-extremity deep vein thrombosis with pulmonary emboli, and lower limb arterial thromboembolism.
He also had an underlying hypercoagulable state—antiphospholipid antibody syndrome—that predisposed him to both arterial and venous thrombosis. He was ultimately found to have a patent foramen ovale, which further increased the risk of arterial thrombosis by facilitating paradoxical embolization of venous thrombi. It is not certain whether the renal infarction and leg artery thrombi were due to paradoxical embolism or to in situ thrombosis, but we believe that it was most likely paradoxical embolization.
A 75-year-old man presented to the emergency department for evaluation of abdominal pain. He had stage 3 chronic obstructive pulmonary disease (COPD), with a forced expiratory volume in 1 second of 33%.
PREVIOUS HOSPITALIZATION
Aside from his COPD, he had been healthy until 1 month earlier, when he had been hospitalized because of shortness of breath and chest pressure with exertion. His troponin T level had been elevated, peaking at 0.117 ng/mL (reference range 0–0.029).
Left heart catheterization had shown no significant coronary artery disease. A myocardial bridge of the distal left anterior descending coronary artery had been seen, so that the artery appeared to be narrowed by 50% to 60% with ventricular contraction. But this was not thought to have been the cause of his presentation.
On discharge, he required oxygen 4 L/min by nasal cannula. Previously, he had not needed supplemental oxygen.
CURRENT PRESENTATION
The patient described persistent and severe periumbilical abdominal pain during the previous day. It was not associated with eating, and he denied diarrhea, constipation, hematemesis, hematochezia, bright red blood per rectum, or melena. He continued to describe persistent shortness of breath and pleuritic chest pain. His vital signs were as follows:
- Heart rate 104 beats per minute
- Respiratory rate 16 to 20 breaths per minute
- Blood pressure 101–142/62–84 mm Hg
- Oxygen saturation 78% on room air.
His laboratory findings on presentation are shown in Table 1, and his electrocardiogram is shown in Figure 1.
WHAT DOES HIS ELECTROCARDIOGRAM SHOW?
1. Which of the following is the most accurate description of this patient’s electrocardiogram?
- Sinus tachycardia, peaked P waves (P pulmonale) in lead II, and T-wave inversions in the right precordial leads
- Sinus tachycardia and left bundle branch block
- Sinus tachycardia and poor R-wave progression
- Sinus tachycardia and ST elevation in the precordial leads
Our patient’s electrocardiogram shows sinus tachycardia, P pulmonale, T-wave inversion in the right precordial leads (V1–V3), and biphasic T waves in lead V4,, which suggest right ventricular strain.
The rhythm most commonly seen in patients with pulmonary embolism is sinus tachycardia, followed by nonspecific ST-segment or T-wave abnormalities. In one series of patients with acute pulmonary embolism, the classic findings of P pulmonale, right ventricular hypertrophy, right axis deviation, and right bundle branch block were rare (< 6%).1 Thus, these classic findings are not sensitive for the diagnosis of pulmonary embolism, and their absence does not rule it out.
Further studies for our patient
Transthoracic echocardiography was performed to look for evidence of right ventricular strain secondary to the pulmonary embolism.
ECHOCARDIOGRAPHIC SIGNS OF PULMONARY EMBOLISM
2. Which of the following findings on transthoracic echocardiography would not suggest acute pulmonary embolism?
- Midright ventricular wall hypokinesis with apical sparing
- Severe tricuspid regurgitation
- Left ventricular dilation
- Lack of respiratory variation of the inferior vena cava
- Septal wall motion toward the left ventricle
Left ventricular dilation does not suggest acute pulmonary embolism. Echocardiograms of patients with acute submassive pulmonary embolism typically show evidence of right ventricular strain, such as the other entities listed above (midright ventricular hypokinesis with apical sparing, severe tricuspid regurgitation, lack of respiratory variation of the inferior vena cava, and septal wall motion toward the left ventricle).
The degree of right ventricular dysfunction is related to the extent of acute pulmonary vascular occlusion and aids in risk-stratification of patients with acute pulmonary embolism. Midright ventricular wall hypokinesis with apical sparing has been termed the McConnell sign.2
In our patient, transthoracic echocardiography showed:
- Normal left ventricular ejection fraction
- Mild diastolic dysfunction
- Right ventricular dilation with moderately decreased right ventricular systolic function and apical sparing
- Right ventricular systolic pressure 54 mm Hg, consistent with moderate pulmonary hypertension
- Right atrial pressure 10 mm Hg
- No inspiratory collapse of a dilated inferior vena cava
- Mild tricuspid valve regurgitation.
CLASSIFICATION OF ACUTE PULMONARY EMBOLISM
3. Given the above information, how would you classify the patient’s pulmonary embolism?
- Massive
- Submassive
- Low-risk
- Clinically stable
The patient’s pulmonary embolism is submassive.
Historically, the classification of pulmonary embolism was determined by the angiographic thrombus burden. However, this has limited utility because clinical factors (eg, hypotension on initial presentation) have been shown to be better predictors of short-term mortality risk.3
Our patient is characterized as having a submassive pulmonary embolism based on elevated biomarkers (troponin T, N-terminal pro-B-type natriuretic peptide) and right ventricular dysfunction in the absence of hypotension.
ULTRASONOGRAPHY FOR DIAGNOSIS OF DEEP VEIN THROMBOSIS
Venous duplex ultrasonography has become the standard for diagnosis of lower extremity deep vein thrombosis. However, its quality and diagnostic accuracy depend on the skill of the person performing the examination. It is further limited by certain patient characteristics, including severe obesity, edema, and wounds and dressings at the site being examined.5
Our patient underwent duplex ultrasonography of the lower extremities, which demonstrated acute proximal and calf deep vein thrombosis in the right femoral, popliteal, and peroneal veins and no deep vein thrombosis in the left leg.
RISK STRATIFICATION IN ACUTE PULMONARY EMBOLISM
Multiple models exist to estimate the risk of complications in patients with acute pulmonary embolism.
The Bova score6 is based on the following factors:
- Systolic blood pressure 90–100 mm Hg (2 points) (patients with systolic blood pressure lower than 90 mm Hg were excluded from the study from which this score was derived)
- Cardiac troponin elevation (2 points)
- Right ventricular dysfunction on echocardiography or computed tomography (2 points)
- Heart rate 100 beats/min or greater (1 point).
A total score of 0, 1, or 2 (stage I) denotes low risk, 3 or 4 points (stage II) intermediate risk, and more than 4 points (stage III) high risk.
The PESI score (Pulmonary Embolism Severity Index)7 is based on:
- Age (1 point per year)
- Sex (10 points for being male)
- Heart rate 110 per minute or greater (20 points)
- Cancer (30 points)
- Heart failure (10 points)
- Chronic lung disease (10 points)
- Systolic blood pressure less than 100 mm Hg (30 points)
- Respiratory rate at least 30 per minute (20 points)
- Temperature less than 36ºC (20 points)
- Altered mental status (60 points)
- Arterial oxygen saturation less than 90% (20 points).
The total score is broken down into 5 classes: I (< 65 points), II (65–85), III (86–105), IV (106–125), and V (> 126). Classes I and II are low risk, and the higher ones are high risk.
The simplified PESI score8 was developed to more rapidly risk-stratify patients and has been found to be similar to the PESI score in prognostic accuracy. Patients get 1 point for each of the following:
- Age over 80
- Cancer
- Chronic cardiopulmonary disease (heart failure or chronic lung disease)
- Heart rate 110 per minute or greater
- Systolic blood pressure less than 100 mm Hg
- Arterial oxygen saturation less than 90%.
A total score of 0 is low risk; anything higher is high risk.
Back to our patient
Our patient had proximal and calf deep vein thrombosis of the right leg, bilateral submassive pulmonary emboli with associated biomarker elevation and right ventricular dysfunction, and left renal artery thrombosis with infarction. Using the PESI score, his risk of death in the next 30 days was 13.7% and his 30-day risk of a complicated course was 27%. Using the Bova score, his 30-day risk of death was 15.5% and his 30-day risk of a complicated course was 29.2%.6,7
Notably, the patient’s right ventricular function had also been impaired on the echocardiogram performed during his admission 1 month previously. On transthoracic echocardiography during the current admission, the patient was found to have a similar degree of right ventricular dysfunction. This finding, along with the oxygen requirement that developed during the earlier admission, suggested that his pulmonary embolism may have been subacute and that the diagnosis may have been missed during the earlier hospital stay.
The patient was treated with unfractionated heparin. After the hospital’s multidisciplinary pulmonary embolism response team discussed and weighed the above factors, they recommended to not pursue thrombolytic therapy or inferior vena cava filter placement.
Of note, the patient’s pulses in the left lower extremity continued to be weak but palpable, and the left leg was cooler to touch than the right leg.
ASSESSING PERIPHERAL ARTERY DISEASE
4. How should the finding of weak pulses in this patient’s left leg be initially investigated?
- Computed tomographic angiography with runoff
- Ankle-brachial indices with pulse-volume recordings
- Arterial duplex ultrasonography
- Magnetic resonance angiography of the lower extremities
The ankle-brachial index is the initial diagnostic test for assessment of pulse abnormalities and for diagnosis of lower-extremity peripheral artery disease. It is calculated by dividing the higher of the ankle systolic pressures (posterior tibial or dorsalis pedis) by the higher of the 2 brachial pressures (left or right).9 Normal values are between 1.00 and 1.40.
Ankle-brachial indices in our patient
Our patient underwent measurement of his brachial, dorsalis pedis, and posterior tibial artery systolic pressures using blood pressure cuffs and continuous-wave Doppler. Ankle pulse-volume recordings were also obtained.
Given the patient’s poor renal function and concern for acute renal infarction, we thought it best to avoid iodinated or gadolinium contrast, such as with magnetic resonance or computed tomographic angiography.
Segmental leg pressures and pulse-volume recordings can be performed to help localize the level of arterial disease in the extremities, but were not done in this case because of the extensive deep vein thrombosis in the right leg.10,11
Arterial ultrasonography in our patient
Arterial duplex ultrasonography was performed to help determine the location of arterial disease. It showed patent arteries in the right leg. In the left lower extremity there was slow, monophasic blood flow in the distal superficial femoral artery. The popliteal artery was occluded. The posterior tibial artery was occluded at the origin, with reconstitution distally. The peroneal artery was occluded throughout. The anterior tibial artery was patent throughout. The ultrasonographic findings were thought to be suspicious for arterial thromboembolism.
WHAT CAN CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
5. Given that the patient had both arterial thrombosis (renal artery, lower-extremity arteries) and venous thromboembolism (deep vein thrombosis and pulmonary embolism), which of the following would be included in the differential diagnosis?
- Antiphospholipid antibody syndrome
- Protein C or protein S deficiency
- Malignancy
- Paradoxical embolization
- Factor V Leiden mutation
Correct answers include antiphospholipid antibody syndrome, malignancy, and paradoxical embolization.
The differential diagnosis for concomitant venous and arterial thrombosis is broad,12 and includes the following:
- Structural factors: patent foramen ovale, popliteal artery aneurysm
- Malignancy
- Inflammatory diseases: Behçet disease, Buerger disease, inflammatory bowel disease, antiphospholipid antibody syndrome, elevated lipoprotein(a), elevated homocysteine
- Hematologic diseases: myelodysplastic syndrome, disseminated intravascular coagulation, paroxysmal nocturnal hemoglobinuria, heparin-induced thrombocytopenia.
Traditional risk factors for venous thromboembolism include protein C deficiency, protein S deficiency, factor V Leiden mutation, the prothrombin G20210A gene mutation, and others. These are relatively minor risk factors for venous thrombosis and do not pose a risk for arterial thrombosis.12 In contrast, antiphospholipid antibody syndrome and malignancy pose a risk for both venous and arterial thrombosis. Paradoxical embolism is a mechanism by which arterial thrombosis (emboli) can develop in the setting of existing venous thrombosis.12
Our patient underwent testing for antiphospholipid antibodies and lupus anticoagulant, and he was encouraged to undergo age-appropriate cancer screening as an outpatient.12
ANTIPHOSPHOLIPID ANTIBODY SYNDROME
Antiphospholipid antibody syndrome is defined by both clinical and laboratory criteria. Clinical symptoms include vascular thrombosis (arterial, venous, or both) and pregnancy-related complications.13
Laboratory criteria require the presence of antiphospholipid antibodies or lupus anticoagulant. These must be confirmed with repeat testing in 12 weeks. Antiphospholipid antibodies are detected by an enzyme-linked immunosorbent assay; laboratory assessment for the presence of lupus anticoagulant is a stepwise process and relies on 4 criteria:
- There should be prolongation of a phospholipid-dependent clotting test (eg, activated partial thromboplastin time, dilute Russell viper venom time test).
- There must be evidence of an inhibitory activity with mixing study.
- The inhibitor must exhibit phospholipid dependence; that is, with more phospholipid there is shortening of clotting time.
- Specific inhibitors must be excluded, including factor VIII and anticoagulant drugs such as heparin.14–17
Clinically, one should consider antiphospholipid antibody syndrome in patients who have arterial thrombosis, a history of pregnancy morbidity, or unexplained prolongation of activated partial thromboplastin time.13
Antiphospholipid antibodies may be present in up to a quarter of patients with venous thromboembolism, but it is persistent positivity of antibody assays that is associated with increased future risk of venous thromboembolism.19 Of note, the risk of venous thromboembolism in patients with confirmed antiphospholipid antibody syndrome is 10 times higher than in the general population.20
ANTIPHOSPHOLIPID ANTIBODIES ARE NOT ALL THE SAME
6. Which of the following antiphospholipid antibodies have not been associated with an increased thrombotic risk?
- Anti-beta-2-glycoprotein I IgG
- Lupus anticoagulant
- Antiphosphatidylserine
- Anticardiolipin IgM
- Anticardiolipin IgG
The correct answer is antiphosphatidylserine.15
Antiphospholipid antibodies are directed against a portion of select plasma proteins that are uncovered upon phospholipid binding. While lupus anticoagulant, anti-beta-2-glycoprotein I, and anticardiolipin antibodies are associated with thrombosis, antiprothrombin antibodies (including antiprothrombin and antiphosphatidylserine antibodies) are not.15,21
PARADOXICAL EMBOLISM
Patent foramen ovale, a communication between the right and left atrium in the interatrial septum, is associated with an increased risk of paradoxical embolization. The prevalence of patent foramen ovale is estimated to be 27% to 29% in the general population.22 Noncerebral systemic paradoxical embolism occurs less frequently than cerebral embolism, accounting for approximately 5% to 10% of paradoxical emboli.22
To evaluate for patent foramen ovale, transthoracic echocardiography is performed with a bubble (agitated saline contrast) study to assess for interatrial shunting. Transesophageal echocardiography or transcranial Doppler bubble studies may also be performed.
Although patent foramen ovale is most commonly associated with cerebral embolism, peripheral emboli can occur. Some research suggests that this may be a more common cause of arterial thromboembolism in younger patients. There have also been reports of other sites of systemic embolization, including the renal artery.12
Back to our patient
Initial antiphospholipid antibody testing was positive for lupus anticoagulant. Anticardiolipin and anti-beta-2-glycoprotein I antibodies were not detected.
Transesophageal echocardiography revealed a patent foramen ovale with a highly mobile atrial septum (atrial septal aneurysm).
The patient was treated with intravenous unfractionated heparin with bridging to warfarin with a target international normalized ratio (INR) of 2 to 3. His renal artery infarction and his lower-extremity arterial thromboembolic event were conservatively managed. His respiratory status improved, and he no longer required supplemental oxygen. His creatinine peaked at 1.7 mg/dL during his admission and improved to 1.2 mg/dL before he was discharged.
At follow-up, repeat echocardiography showed that his right ventricular systolic pressure had improved (decreased) to 37 mm Hg from 54 mm Hg. Repeat confirmatory testing was positive for lupus anticoagulant 12 weeks later. He has been maintained on warfarin with an INR goal of 2 to 3 as well as low-dose aspirin with plans for long-term anticoagulation. We decided to keep the patient on anticoagulation indefinitely with warfarin; he was not a candidate for a direct oral anticoagulant, given limited data on the use of these agents in the setting of lupus anticoagulant and antiphospholipid antibody syndrome.
SUMMARY OF CASE
In summary, this patient was a 75-year-old man with COPD who presented with abdominal pain. He was noted to have a left renal infarction, extensive unprovoked lower-extremity deep vein thrombosis with pulmonary emboli, and lower limb arterial thromboembolism.
He also had an underlying hypercoagulable state—antiphospholipid antibody syndrome—that predisposed him to both arterial and venous thrombosis. He was ultimately found to have a patent foramen ovale, which further increased the risk of arterial thrombosis by facilitating paradoxical embolization of venous thrombi. It is not certain whether the renal infarction and leg artery thrombi were due to paradoxical embolism or to in situ thrombosis, but we believe that it was most likely paradoxical embolization.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Alsoos F, Khaddam A. Echocardiographic evaluation methods for right ventricular function. J Echocardiogr 2015; 13:43–51.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; American Heart Association Council on Peripheral Vascular Disease; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Heit JA, Silverstein MD, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med 2000; 160:809–815.
- Gornik HL, Sharma AM. Duplex ultrasound in the diagnosis of lower-extremity deep venous thrombosis. Circulation 2014; 129:917–921.
- Fernández C, Bova C, Sanchez O, et al. Validation of a model for identification of patients at intermediate to high risk for complications associated with acute symptomatic pulmonary embolism. Chest 2015; 148:211–218.
- Aujesky D, Perrier A, Roy PM, et al. Validation of a clinical prognostic model to identify low-risk patients with pulmonary embolism. J Intern Med 2007; 261:597–604.
- Jiménez D, Aujesky D, Moores L, et al; RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med 2010; 170:1383–1389.
- Kim ES, Wattanakit K, Gornik HL. Using the ankle-brachial index to diagnose peripheral artery disease and assess cardiovascular risk. Cleve Clin J Med 2012; 79:651–661.
- Jaff MR. Lower extremity arterial disease. Diagnostic aspects. Cardiol Clin 2002; 20:491–500.
- Rooke TW, Hirsch AT, Misra S, et al; American College of Cardiology Foundation Task Force; American Heart Association Task Force. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA Guideline Recommendations): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:1555–1570.
- Lichtin A, Bartholomew J. The coagulation consult: a case-based guide. New York, NY: Springer; 2014.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Miyakis S, Lockshin M, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Nichols WL, Kottke-Marchant K, Ledford-Kraemer MR, Homburger HA, Cardel LK. Lupus anticoagulants, antiphospholipid antibodies, and antiphospholipid syndrome. In: Kottke-Marchant K, Davis BH, editors. Laboratory Hematology Practice. Hoboken, New Jersey: Blackwell Publishing, Ltd.; 2012:509–525.
- Houghton DE, Moll S. Antiphospholipid antibodies. Vasc Med 2017; 22:545–550.
- Roldan V, Lecumberri R, Muñoz-Torrero JFS, et al; RIETE Investigators. Thrombophilia testing in patients with venous thromboembolism. Findings from the RIETE registry. Thromb Res 2009; 124:174–177.
- Wahl DG, Guillemin F, de Maistre E, Perret-Guillaume C, Lecompte T, Thibaut G. Meta-analysis of the risk of venous thrombosis in individuals with antiphospholipid antibodies without underlying autoimmune disease or previous thrombosis. Lupus 1998; 7:15–22.
- Love PE, Santoro SA. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders. Prevalence and clinical significance. Ann Intern Med 1990; 112:682–698.
- Thompson T, Evans W. Paradoxical embolism. QJM 1930; os-23:135–150.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Alsoos F, Khaddam A. Echocardiographic evaluation methods for right ventricular function. J Echocardiogr 2015; 13:43–51.
- Jaff MR, McMurtry MS, Archer SL, et al; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; American Heart Association Council on Peripheral Vascular Disease; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology. Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 2011; 123:1788–1830.
- Heit JA, Silverstein MD, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med 2000; 160:809–815.
- Gornik HL, Sharma AM. Duplex ultrasound in the diagnosis of lower-extremity deep venous thrombosis. Circulation 2014; 129:917–921.
- Fernández C, Bova C, Sanchez O, et al. Validation of a model for identification of patients at intermediate to high risk for complications associated with acute symptomatic pulmonary embolism. Chest 2015; 148:211–218.
- Aujesky D, Perrier A, Roy PM, et al. Validation of a clinical prognostic model to identify low-risk patients with pulmonary embolism. J Intern Med 2007; 261:597–604.
- Jiménez D, Aujesky D, Moores L, et al; RIETE Investigators. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med 2010; 170:1383–1389.
- Kim ES, Wattanakit K, Gornik HL. Using the ankle-brachial index to diagnose peripheral artery disease and assess cardiovascular risk. Cleve Clin J Med 2012; 79:651–661.
- Jaff MR. Lower extremity arterial disease. Diagnostic aspects. Cardiol Clin 2002; 20:491–500.
- Rooke TW, Hirsch AT, Misra S, et al; American College of Cardiology Foundation Task Force; American Heart Association Task Force. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA Guideline Recommendations): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:1555–1570.
- Lichtin A, Bartholomew J. The coagulation consult: a case-based guide. New York, NY: Springer; 2014.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Miyakis S, Lockshin M, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Nichols WL, Kottke-Marchant K, Ledford-Kraemer MR, Homburger HA, Cardel LK. Lupus anticoagulants, antiphospholipid antibodies, and antiphospholipid syndrome. In: Kottke-Marchant K, Davis BH, editors. Laboratory Hematology Practice. Hoboken, New Jersey: Blackwell Publishing, Ltd.; 2012:509–525.
- Houghton DE, Moll S. Antiphospholipid antibodies. Vasc Med 2017; 22:545–550.
- Roldan V, Lecumberri R, Muñoz-Torrero JFS, et al; RIETE Investigators. Thrombophilia testing in patients with venous thromboembolism. Findings from the RIETE registry. Thromb Res 2009; 124:174–177.
- Wahl DG, Guillemin F, de Maistre E, Perret-Guillaume C, Lecompte T, Thibaut G. Meta-analysis of the risk of venous thrombosis in individuals with antiphospholipid antibodies without underlying autoimmune disease or previous thrombosis. Lupus 1998; 7:15–22.
- Love PE, Santoro SA. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders. Prevalence and clinical significance. Ann Intern Med 1990; 112:682–698.
- Thompson T, Evans W. Paradoxical embolism. QJM 1930; os-23:135–150.
Ethanol Intoxication From Hand Sanitizer Ingestion
Case
A 29-year-old man presented to the ED several hours after ingesting what he described as a “hefty” bottle of hand sanitizer. The patient stated that he ingested such a considerable quantity of liquid hand sanitizer because he was unable to obtain beer or liquor. He further admitted to drinking two 40-ounce beers daily for the past several years, noting that he last consumed drinking alcohol the preceding day.
The patient denied any other coingestants. He also denied nausea, vomiting, abdominal pain, or other somatic complaints. The patient’s medical history was significant for hypertension and hepatitis C, and his social history was significant for daily alcohol consumption, tobacco abuse, and former benzodiazepine, marijuana, and intravenous heroin abuse. His psychiatric history was significant for borderline personality disorder, major depression, and bulimia. The patient’s home medications included a daily multivitamin, folate, thiamine, sertraline, mirtazapine, and prazosin.
Initial vital signs at presentation were: blood pressure, 124/77 mm Hg; heart rate, 86 beats/min; respiratory rate, 15 breaths/min; and temperature, 98.0°F. On physical examination, he was noted to have slurred speech and nystagmus. His pupils were equal and reactive, without scleral icterus. The abdomen was nontender and nondistended, with regular bowel sounds, and without ascites or varicosities visualized. The rest of the examination was unremarkable. The patient did express thoughts of suicidality, but denied any homicidal ideation.
Laboratory studies revealed a serum ethanol concentration of 446 mg/dL. The patient’s basic metabolic panel was unremarkable, and liver function test results showed mildly elevated enzymes. The coagulation panel was within normal limits.
Is alcohol-based hand sanitizer consumption an emerging public health concern?
Excessive alcohol consumption is a recognized public health problem in the United States and is associated with an average of 88,000 deaths per year.1 In a select population of patients, an untoward effect has developed from another public health target—that of hand hygiene.
Alcohol-based liquid hand sanitizers have become ubiquitous as a weapon in the antimicrobial arsenal with recommendations for its use as an alternative to soap and water in certain clinical settings. Liquid hand sanitizers are ideal for hospital or community use as they are faster, more effective, and less irritating to the skin than traditional hand-washing techniques.2
The downside to the widespread availability of hand sanitizers is that they offer easy access to individuals in search of clandestine sources of alcohol. Prior case reports have discussed the practice of consuming alcohol-based hand sanitizers for the purpose of intoxication in institutionalized persons, such as prisoners or patients in psychiatric facilities who are restricted to conventional sources of alcohol.
Children and confused elderly patients are also at risk for unintentional ingestions.3,4 An article reviewed exposures reported to the American Association of Poison Control Center’s National Poison Data System over a 5-year period from 2005 to 2009.3 Of the 68,712 reported cases in this cohort, 80.5% were in children younger than 6 years of age. The investigators also noted an increased incidence of exposure over this period with an average of 1,894 additional cases per year.3There were 17,154 children aged 12 years and younger reported in 2017 to poison centers with exposures to hand sanitizers. Young children may be enticed by the bright colorful packaging and similarity to food and candy smells.5
What are the clinical manifestations of alcohol-based hand sanitizer ingestion?
Significant hazards exist from ingesting liquid hand sanitizer, including the high alcohol content, which varies from 40% to 85%.2 Because isopropanol is commonly one of the components (if not the sole component) of many hand-sanitizer preparations, isopropanol toxicity may occur when ingested. The effects of isopropanol are similar to those of ethanol, with clinical effects reported after ingestion of as little as 100 mL of 70% isopropanol solution.4
Hand sanitizer formulations vary by manufacturer and contain different concentrations of ethanol and/or isopropanol, as well as additional potential inactive ingredients such as acetone, 1-propanol, 2-propanol, benzyl alcohol, hydrogen peroxide, glycerin, water, and different perfumes.3,4
Persons who consume hand sanitizers recreationally are often unaware of the large alcohol content by volume that they are consuming. Recreational ingestion of hand sanitizer is believed to be the cause of at least one case of lethal ethanol intoxication. An articlereported a case of a male patient who suffered respiratory arrest after consuming an ethanol-based hand sanitizer.6 This patient was noted to have a serum ethanol of 536 mg/dL after consuming an unknown quantity of a 354 mL container of a 62% ethanol by volume hand sanitizer.6
Institutionalized individuals seeking alcohol through this source have discovered novel ways to yield a stronger product. Through the use of table salt and a cotton sock, it is possible to extract a liquid from a gel hand sanitizer preparation, yielding an alcohol context 30% higher by volume than the parent mixture.7
Alcohol intoxication poses a host of health effects. In nonhabituated individuals, a lethal load of alcohol can be achieved by consuming a volume of as little as 400 mL of an 80% alcohol-based solution.4 Symptoms from ingestion of an alcohol-based liquid hand sanitizer typically appear 1 to 2 hours after ingestion and mirror that of the alcohol toxidrome. Most commonly, this includes nausea, vomiting, epigastric pain, and varying degrees of central nervous system (CNS) depression.4 The life-threatening clinical manifestation of alcohol intoxication includes severe CNS and respiratory depression resulting in respiratory arrest, hypothermia, cardiac dysrhythmias with possible cardiac arrest, hypoglycemia, ketoacidosis, and hypotension.3
How is alcohol-based hand sanitizer ingestion managed?
The management of patients with alcohol-based hand sanitizer ingestion is the same as the management of alcohol ingestion from more socially acceptable sources and is mainly supportive.3,4 These measures are directed at managing the patient’s airway with intubation and mechanical ventilation when appropriate, as well as supportive measures to address any underlying metabolic derangement or hypotension.2 While hemodialysis has been used in some patients who had severe organ dysfunction and did not respond to supportive measures, it is usually not necessary.1,3
Case Conclusion
The patient in this case was subsequently admitted to an intermediate level of care. He did not require intubation or further hemodynamic support during his initial acute intoxication. Later in the patient’s hospital course, he was noted to be in alcohol withdrawal, and proper management was initiated. He also required therapeutic one-to-one supervision after members of the nursing staff observed the patient consuming the hand sanitizer gel present in patient-care areas. He was later seen by psychiatry services. The psychiatrist recommended transfer to an inpatient psychiatric facility upon medical clearance for treatment of his psychiatric illness as well as alcohol dependence.
1. Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi:10.5888/pcd11.140329.
2. Pittet D, Boyce JM. Revolutionizing hand hygiene in health-care settings: guidelines revisted. Lancet Infect Dis. 2003;3(5):269-270.
3. Gormley NJ, Bronstein AC, Rasimas JJ, et al. The rising incidence of intentional ingestion of ethanol-containing hand sanitizers. Crit Care Med. 2012:40(1):290-294. doi:10.1097/CCM.0b013e31822f09c0.
4. Archer JR, Wood DM, Tizzard Z, Jones AL, Dargan PI. Alcohol hand rubs: hygiene and hazard. BMJ. 2007;335(7630):1154-1155.
5. Hand sanitizer. American Association of Poison Control Centers Web site. http://www.aapcc.org/alerts/hand-sanitizer/. Accessed December 27, 2017.
6. Schneir AB, Clark RF. Death caused by ingestion of an ethanol-based hand sanitizer. J Emerg Med. 2013;45(3):358-360. doi:10.1016/j.jemermed.2013.03.018.
7. Darracq MA, Ghafouri N, Pesce A, Cantrell FL. Hand sanitizer intoxication following a crude extraction method. Am J Drug Alcohol Abuse. 2013;39(3):217-218. doi:10.3109/00952990.2013.773335.
Case
A 29-year-old man presented to the ED several hours after ingesting what he described as a “hefty” bottle of hand sanitizer. The patient stated that he ingested such a considerable quantity of liquid hand sanitizer because he was unable to obtain beer or liquor. He further admitted to drinking two 40-ounce beers daily for the past several years, noting that he last consumed drinking alcohol the preceding day.
The patient denied any other coingestants. He also denied nausea, vomiting, abdominal pain, or other somatic complaints. The patient’s medical history was significant for hypertension and hepatitis C, and his social history was significant for daily alcohol consumption, tobacco abuse, and former benzodiazepine, marijuana, and intravenous heroin abuse. His psychiatric history was significant for borderline personality disorder, major depression, and bulimia. The patient’s home medications included a daily multivitamin, folate, thiamine, sertraline, mirtazapine, and prazosin.
Initial vital signs at presentation were: blood pressure, 124/77 mm Hg; heart rate, 86 beats/min; respiratory rate, 15 breaths/min; and temperature, 98.0°F. On physical examination, he was noted to have slurred speech and nystagmus. His pupils were equal and reactive, without scleral icterus. The abdomen was nontender and nondistended, with regular bowel sounds, and without ascites or varicosities visualized. The rest of the examination was unremarkable. The patient did express thoughts of suicidality, but denied any homicidal ideation.
Laboratory studies revealed a serum ethanol concentration of 446 mg/dL. The patient’s basic metabolic panel was unremarkable, and liver function test results showed mildly elevated enzymes. The coagulation panel was within normal limits.
Is alcohol-based hand sanitizer consumption an emerging public health concern?
Excessive alcohol consumption is a recognized public health problem in the United States and is associated with an average of 88,000 deaths per year.1 In a select population of patients, an untoward effect has developed from another public health target—that of hand hygiene.
Alcohol-based liquid hand sanitizers have become ubiquitous as a weapon in the antimicrobial arsenal with recommendations for its use as an alternative to soap and water in certain clinical settings. Liquid hand sanitizers are ideal for hospital or community use as they are faster, more effective, and less irritating to the skin than traditional hand-washing techniques.2
The downside to the widespread availability of hand sanitizers is that they offer easy access to individuals in search of clandestine sources of alcohol. Prior case reports have discussed the practice of consuming alcohol-based hand sanitizers for the purpose of intoxication in institutionalized persons, such as prisoners or patients in psychiatric facilities who are restricted to conventional sources of alcohol.
Children and confused elderly patients are also at risk for unintentional ingestions.3,4 An article reviewed exposures reported to the American Association of Poison Control Center’s National Poison Data System over a 5-year period from 2005 to 2009.3 Of the 68,712 reported cases in this cohort, 80.5% were in children younger than 6 years of age. The investigators also noted an increased incidence of exposure over this period with an average of 1,894 additional cases per year.3There were 17,154 children aged 12 years and younger reported in 2017 to poison centers with exposures to hand sanitizers. Young children may be enticed by the bright colorful packaging and similarity to food and candy smells.5
What are the clinical manifestations of alcohol-based hand sanitizer ingestion?
Significant hazards exist from ingesting liquid hand sanitizer, including the high alcohol content, which varies from 40% to 85%.2 Because isopropanol is commonly one of the components (if not the sole component) of many hand-sanitizer preparations, isopropanol toxicity may occur when ingested. The effects of isopropanol are similar to those of ethanol, with clinical effects reported after ingestion of as little as 100 mL of 70% isopropanol solution.4
Hand sanitizer formulations vary by manufacturer and contain different concentrations of ethanol and/or isopropanol, as well as additional potential inactive ingredients such as acetone, 1-propanol, 2-propanol, benzyl alcohol, hydrogen peroxide, glycerin, water, and different perfumes.3,4
Persons who consume hand sanitizers recreationally are often unaware of the large alcohol content by volume that they are consuming. Recreational ingestion of hand sanitizer is believed to be the cause of at least one case of lethal ethanol intoxication. An articlereported a case of a male patient who suffered respiratory arrest after consuming an ethanol-based hand sanitizer.6 This patient was noted to have a serum ethanol of 536 mg/dL after consuming an unknown quantity of a 354 mL container of a 62% ethanol by volume hand sanitizer.6
Institutionalized individuals seeking alcohol through this source have discovered novel ways to yield a stronger product. Through the use of table salt and a cotton sock, it is possible to extract a liquid from a gel hand sanitizer preparation, yielding an alcohol context 30% higher by volume than the parent mixture.7
Alcohol intoxication poses a host of health effects. In nonhabituated individuals, a lethal load of alcohol can be achieved by consuming a volume of as little as 400 mL of an 80% alcohol-based solution.4 Symptoms from ingestion of an alcohol-based liquid hand sanitizer typically appear 1 to 2 hours after ingestion and mirror that of the alcohol toxidrome. Most commonly, this includes nausea, vomiting, epigastric pain, and varying degrees of central nervous system (CNS) depression.4 The life-threatening clinical manifestation of alcohol intoxication includes severe CNS and respiratory depression resulting in respiratory arrest, hypothermia, cardiac dysrhythmias with possible cardiac arrest, hypoglycemia, ketoacidosis, and hypotension.3
How is alcohol-based hand sanitizer ingestion managed?
The management of patients with alcohol-based hand sanitizer ingestion is the same as the management of alcohol ingestion from more socially acceptable sources and is mainly supportive.3,4 These measures are directed at managing the patient’s airway with intubation and mechanical ventilation when appropriate, as well as supportive measures to address any underlying metabolic derangement or hypotension.2 While hemodialysis has been used in some patients who had severe organ dysfunction and did not respond to supportive measures, it is usually not necessary.1,3
Case Conclusion
The patient in this case was subsequently admitted to an intermediate level of care. He did not require intubation or further hemodynamic support during his initial acute intoxication. Later in the patient’s hospital course, he was noted to be in alcohol withdrawal, and proper management was initiated. He also required therapeutic one-to-one supervision after members of the nursing staff observed the patient consuming the hand sanitizer gel present in patient-care areas. He was later seen by psychiatry services. The psychiatrist recommended transfer to an inpatient psychiatric facility upon medical clearance for treatment of his psychiatric illness as well as alcohol dependence.
Case
A 29-year-old man presented to the ED several hours after ingesting what he described as a “hefty” bottle of hand sanitizer. The patient stated that he ingested such a considerable quantity of liquid hand sanitizer because he was unable to obtain beer or liquor. He further admitted to drinking two 40-ounce beers daily for the past several years, noting that he last consumed drinking alcohol the preceding day.
The patient denied any other coingestants. He also denied nausea, vomiting, abdominal pain, or other somatic complaints. The patient’s medical history was significant for hypertension and hepatitis C, and his social history was significant for daily alcohol consumption, tobacco abuse, and former benzodiazepine, marijuana, and intravenous heroin abuse. His psychiatric history was significant for borderline personality disorder, major depression, and bulimia. The patient’s home medications included a daily multivitamin, folate, thiamine, sertraline, mirtazapine, and prazosin.
Initial vital signs at presentation were: blood pressure, 124/77 mm Hg; heart rate, 86 beats/min; respiratory rate, 15 breaths/min; and temperature, 98.0°F. On physical examination, he was noted to have slurred speech and nystagmus. His pupils were equal and reactive, without scleral icterus. The abdomen was nontender and nondistended, with regular bowel sounds, and without ascites or varicosities visualized. The rest of the examination was unremarkable. The patient did express thoughts of suicidality, but denied any homicidal ideation.
Laboratory studies revealed a serum ethanol concentration of 446 mg/dL. The patient’s basic metabolic panel was unremarkable, and liver function test results showed mildly elevated enzymes. The coagulation panel was within normal limits.
Is alcohol-based hand sanitizer consumption an emerging public health concern?
Excessive alcohol consumption is a recognized public health problem in the United States and is associated with an average of 88,000 deaths per year.1 In a select population of patients, an untoward effect has developed from another public health target—that of hand hygiene.
Alcohol-based liquid hand sanitizers have become ubiquitous as a weapon in the antimicrobial arsenal with recommendations for its use as an alternative to soap and water in certain clinical settings. Liquid hand sanitizers are ideal for hospital or community use as they are faster, more effective, and less irritating to the skin than traditional hand-washing techniques.2
The downside to the widespread availability of hand sanitizers is that they offer easy access to individuals in search of clandestine sources of alcohol. Prior case reports have discussed the practice of consuming alcohol-based hand sanitizers for the purpose of intoxication in institutionalized persons, such as prisoners or patients in psychiatric facilities who are restricted to conventional sources of alcohol.
Children and confused elderly patients are also at risk for unintentional ingestions.3,4 An article reviewed exposures reported to the American Association of Poison Control Center’s National Poison Data System over a 5-year period from 2005 to 2009.3 Of the 68,712 reported cases in this cohort, 80.5% were in children younger than 6 years of age. The investigators also noted an increased incidence of exposure over this period with an average of 1,894 additional cases per year.3There were 17,154 children aged 12 years and younger reported in 2017 to poison centers with exposures to hand sanitizers. Young children may be enticed by the bright colorful packaging and similarity to food and candy smells.5
What are the clinical manifestations of alcohol-based hand sanitizer ingestion?
Significant hazards exist from ingesting liquid hand sanitizer, including the high alcohol content, which varies from 40% to 85%.2 Because isopropanol is commonly one of the components (if not the sole component) of many hand-sanitizer preparations, isopropanol toxicity may occur when ingested. The effects of isopropanol are similar to those of ethanol, with clinical effects reported after ingestion of as little as 100 mL of 70% isopropanol solution.4
Hand sanitizer formulations vary by manufacturer and contain different concentrations of ethanol and/or isopropanol, as well as additional potential inactive ingredients such as acetone, 1-propanol, 2-propanol, benzyl alcohol, hydrogen peroxide, glycerin, water, and different perfumes.3,4
Persons who consume hand sanitizers recreationally are often unaware of the large alcohol content by volume that they are consuming. Recreational ingestion of hand sanitizer is believed to be the cause of at least one case of lethal ethanol intoxication. An articlereported a case of a male patient who suffered respiratory arrest after consuming an ethanol-based hand sanitizer.6 This patient was noted to have a serum ethanol of 536 mg/dL after consuming an unknown quantity of a 354 mL container of a 62% ethanol by volume hand sanitizer.6
Institutionalized individuals seeking alcohol through this source have discovered novel ways to yield a stronger product. Through the use of table salt and a cotton sock, it is possible to extract a liquid from a gel hand sanitizer preparation, yielding an alcohol context 30% higher by volume than the parent mixture.7
Alcohol intoxication poses a host of health effects. In nonhabituated individuals, a lethal load of alcohol can be achieved by consuming a volume of as little as 400 mL of an 80% alcohol-based solution.4 Symptoms from ingestion of an alcohol-based liquid hand sanitizer typically appear 1 to 2 hours after ingestion and mirror that of the alcohol toxidrome. Most commonly, this includes nausea, vomiting, epigastric pain, and varying degrees of central nervous system (CNS) depression.4 The life-threatening clinical manifestation of alcohol intoxication includes severe CNS and respiratory depression resulting in respiratory arrest, hypothermia, cardiac dysrhythmias with possible cardiac arrest, hypoglycemia, ketoacidosis, and hypotension.3
How is alcohol-based hand sanitizer ingestion managed?
The management of patients with alcohol-based hand sanitizer ingestion is the same as the management of alcohol ingestion from more socially acceptable sources and is mainly supportive.3,4 These measures are directed at managing the patient’s airway with intubation and mechanical ventilation when appropriate, as well as supportive measures to address any underlying metabolic derangement or hypotension.2 While hemodialysis has been used in some patients who had severe organ dysfunction and did not respond to supportive measures, it is usually not necessary.1,3
Case Conclusion
The patient in this case was subsequently admitted to an intermediate level of care. He did not require intubation or further hemodynamic support during his initial acute intoxication. Later in the patient’s hospital course, he was noted to be in alcohol withdrawal, and proper management was initiated. He also required therapeutic one-to-one supervision after members of the nursing staff observed the patient consuming the hand sanitizer gel present in patient-care areas. He was later seen by psychiatry services. The psychiatrist recommended transfer to an inpatient psychiatric facility upon medical clearance for treatment of his psychiatric illness as well as alcohol dependence.
1. Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi:10.5888/pcd11.140329.
2. Pittet D, Boyce JM. Revolutionizing hand hygiene in health-care settings: guidelines revisted. Lancet Infect Dis. 2003;3(5):269-270.
3. Gormley NJ, Bronstein AC, Rasimas JJ, et al. The rising incidence of intentional ingestion of ethanol-containing hand sanitizers. Crit Care Med. 2012:40(1):290-294. doi:10.1097/CCM.0b013e31822f09c0.
4. Archer JR, Wood DM, Tizzard Z, Jones AL, Dargan PI. Alcohol hand rubs: hygiene and hazard. BMJ. 2007;335(7630):1154-1155.
5. Hand sanitizer. American Association of Poison Control Centers Web site. http://www.aapcc.org/alerts/hand-sanitizer/. Accessed December 27, 2017.
6. Schneir AB, Clark RF. Death caused by ingestion of an ethanol-based hand sanitizer. J Emerg Med. 2013;45(3):358-360. doi:10.1016/j.jemermed.2013.03.018.
7. Darracq MA, Ghafouri N, Pesce A, Cantrell FL. Hand sanitizer intoxication following a crude extraction method. Am J Drug Alcohol Abuse. 2013;39(3):217-218. doi:10.3109/00952990.2013.773335.
1. Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi:10.5888/pcd11.140329.
2. Pittet D, Boyce JM. Revolutionizing hand hygiene in health-care settings: guidelines revisted. Lancet Infect Dis. 2003;3(5):269-270.
3. Gormley NJ, Bronstein AC, Rasimas JJ, et al. The rising incidence of intentional ingestion of ethanol-containing hand sanitizers. Crit Care Med. 2012:40(1):290-294. doi:10.1097/CCM.0b013e31822f09c0.
4. Archer JR, Wood DM, Tizzard Z, Jones AL, Dargan PI. Alcohol hand rubs: hygiene and hazard. BMJ. 2007;335(7630):1154-1155.
5. Hand sanitizer. American Association of Poison Control Centers Web site. http://www.aapcc.org/alerts/hand-sanitizer/. Accessed December 27, 2017.
6. Schneir AB, Clark RF. Death caused by ingestion of an ethanol-based hand sanitizer. J Emerg Med. 2013;45(3):358-360. doi:10.1016/j.jemermed.2013.03.018.
7. Darracq MA, Ghafouri N, Pesce A, Cantrell FL. Hand sanitizer intoxication following a crude extraction method. Am J Drug Alcohol Abuse. 2013;39(3):217-218. doi:10.3109/00952990.2013.773335.

Hypothermia and severe first-degree heart block
A 96-year-old woman with hypertension, diabetes, and dementia was found unresponsive in her nursing home and was transferred to the hospital.
At presentation to the hospital, her blood pressure was 76/43 mm Hg, heart rate 42 beats per minute, rectal temperature 31.6°C (88.8°F), and blood glucose 36 mg/dL.
Causes of secondary hypothermia were sought. Blood and urine cultures were negative. Computed tomography of the head showed no acute intracranial abnormalities. Tests for adrenal insufficiency and hypothyroidism were negative.
HYPOTHERMIA AND THE ECG
Hypothermia can produce a number of changes on the ECG. At the start of hypothermia, a stress reaction is induced, resulting in sinus tachycardia. But when the temperature goes below 32°C, sinus bradycardia ensues,1 resulting in various degrees of heart block.2 In our patient, a severely prolonged PR interval resulted in first-degree heart block.
Other findings on ECG associated with hypothermia include atrial fibrillation, widening of the P and T waves, prolonging of the QT interval, and widening of the QRS interval. Progressive widening of the QRS interval can predispose to ventricular fibrillation.1,3
An Osborn or J wave is a wave found between the end of the QRS and the beginning of the ST segment and is usually seen on the inferior and lateral precordial leads. It is found in as many as 80% of patients when the body temperature is below 30°C.1,3,4
Although Osborn waves are a common finding in hypothermia, they are also seen in electrolyte imbalances such as hypercalcemia and in central nervous system diseases.5,6 Hypothermia-associated changes on ECG are usually readily reversible with rewarming.1
TAKE-HOME MESSAGES
The ECG should always be interpreted in the proper clinical context and, whenever possible, compared with a previous ECG. It is prudent to always consider potentially reversible triggers of hypothermia other than environmental exposure such as hypothyroidism, infection, adrenal insufficiency, ketoacidosis, medication side effects, and alcohol use.
Hypothermia, especially in elderly patients with multiple comorbidities, can lead to bradycardia and varying degrees of heart block.
- Alhaddad IA, Khalil M, Brown EJ Jr. Osborn waves of hypothermia. Circulation 2000; 101:E233–E244.
- Bashour TT, Gualberto A, Ryan C. Atrioventricular block in accidental hypothermia—a case report. Angiology 1989; 40:63–66.
- Okada M, Nishimura F, Yoshino H, Kimura M, Ogino T. The J wave in accidental hypothermia. J Electrocardiol 1983; 16:23–28.
- Kukla P, Baranchuk A, Jastrzebski M, Zabojszcz M, Bryniarski L. Electrocardiographic landmarks of hypothermia. Kardiol Pol 2013; 71:1188–1189.
- Maruyama M, Kobayashi Y, Kodani E, et al. Osborn waves: history and significance. Indian Pacing Electrophysiol J 2004; 4:33–39.
- Sheikh AM, Hurst JW. Osborn waves in the electrocardiogram, hypothermia not due to exposure, and death due to diabetic ketoacidosis. Clin Cardiol 2003; 26:555–560.
A 96-year-old woman with hypertension, diabetes, and dementia was found unresponsive in her nursing home and was transferred to the hospital.
At presentation to the hospital, her blood pressure was 76/43 mm Hg, heart rate 42 beats per minute, rectal temperature 31.6°C (88.8°F), and blood glucose 36 mg/dL.
Causes of secondary hypothermia were sought. Blood and urine cultures were negative. Computed tomography of the head showed no acute intracranial abnormalities. Tests for adrenal insufficiency and hypothyroidism were negative.
HYPOTHERMIA AND THE ECG
Hypothermia can produce a number of changes on the ECG. At the start of hypothermia, a stress reaction is induced, resulting in sinus tachycardia. But when the temperature goes below 32°C, sinus bradycardia ensues,1 resulting in various degrees of heart block.2 In our patient, a severely prolonged PR interval resulted in first-degree heart block.
Other findings on ECG associated with hypothermia include atrial fibrillation, widening of the P and T waves, prolonging of the QT interval, and widening of the QRS interval. Progressive widening of the QRS interval can predispose to ventricular fibrillation.1,3
An Osborn or J wave is a wave found between the end of the QRS and the beginning of the ST segment and is usually seen on the inferior and lateral precordial leads. It is found in as many as 80% of patients when the body temperature is below 30°C.1,3,4
Although Osborn waves are a common finding in hypothermia, they are also seen in electrolyte imbalances such as hypercalcemia and in central nervous system diseases.5,6 Hypothermia-associated changes on ECG are usually readily reversible with rewarming.1
TAKE-HOME MESSAGES
The ECG should always be interpreted in the proper clinical context and, whenever possible, compared with a previous ECG. It is prudent to always consider potentially reversible triggers of hypothermia other than environmental exposure such as hypothyroidism, infection, adrenal insufficiency, ketoacidosis, medication side effects, and alcohol use.
Hypothermia, especially in elderly patients with multiple comorbidities, can lead to bradycardia and varying degrees of heart block.
A 96-year-old woman with hypertension, diabetes, and dementia was found unresponsive in her nursing home and was transferred to the hospital.
At presentation to the hospital, her blood pressure was 76/43 mm Hg, heart rate 42 beats per minute, rectal temperature 31.6°C (88.8°F), and blood glucose 36 mg/dL.
Causes of secondary hypothermia were sought. Blood and urine cultures were negative. Computed tomography of the head showed no acute intracranial abnormalities. Tests for adrenal insufficiency and hypothyroidism were negative.
HYPOTHERMIA AND THE ECG
Hypothermia can produce a number of changes on the ECG. At the start of hypothermia, a stress reaction is induced, resulting in sinus tachycardia. But when the temperature goes below 32°C, sinus bradycardia ensues,1 resulting in various degrees of heart block.2 In our patient, a severely prolonged PR interval resulted in first-degree heart block.
Other findings on ECG associated with hypothermia include atrial fibrillation, widening of the P and T waves, prolonging of the QT interval, and widening of the QRS interval. Progressive widening of the QRS interval can predispose to ventricular fibrillation.1,3
An Osborn or J wave is a wave found between the end of the QRS and the beginning of the ST segment and is usually seen on the inferior and lateral precordial leads. It is found in as many as 80% of patients when the body temperature is below 30°C.1,3,4
Although Osborn waves are a common finding in hypothermia, they are also seen in electrolyte imbalances such as hypercalcemia and in central nervous system diseases.5,6 Hypothermia-associated changes on ECG are usually readily reversible with rewarming.1
TAKE-HOME MESSAGES
The ECG should always be interpreted in the proper clinical context and, whenever possible, compared with a previous ECG. It is prudent to always consider potentially reversible triggers of hypothermia other than environmental exposure such as hypothyroidism, infection, adrenal insufficiency, ketoacidosis, medication side effects, and alcohol use.
Hypothermia, especially in elderly patients with multiple comorbidities, can lead to bradycardia and varying degrees of heart block.
- Alhaddad IA, Khalil M, Brown EJ Jr. Osborn waves of hypothermia. Circulation 2000; 101:E233–E244.
- Bashour TT, Gualberto A, Ryan C. Atrioventricular block in accidental hypothermia—a case report. Angiology 1989; 40:63–66.
- Okada M, Nishimura F, Yoshino H, Kimura M, Ogino T. The J wave in accidental hypothermia. J Electrocardiol 1983; 16:23–28.
- Kukla P, Baranchuk A, Jastrzebski M, Zabojszcz M, Bryniarski L. Electrocardiographic landmarks of hypothermia. Kardiol Pol 2013; 71:1188–1189.
- Maruyama M, Kobayashi Y, Kodani E, et al. Osborn waves: history and significance. Indian Pacing Electrophysiol J 2004; 4:33–39.
- Sheikh AM, Hurst JW. Osborn waves in the electrocardiogram, hypothermia not due to exposure, and death due to diabetic ketoacidosis. Clin Cardiol 2003; 26:555–560.
- Alhaddad IA, Khalil M, Brown EJ Jr. Osborn waves of hypothermia. Circulation 2000; 101:E233–E244.
- Bashour TT, Gualberto A, Ryan C. Atrioventricular block in accidental hypothermia—a case report. Angiology 1989; 40:63–66.
- Okada M, Nishimura F, Yoshino H, Kimura M, Ogino T. The J wave in accidental hypothermia. J Electrocardiol 1983; 16:23–28.
- Kukla P, Baranchuk A, Jastrzebski M, Zabojszcz M, Bryniarski L. Electrocardiographic landmarks of hypothermia. Kardiol Pol 2013; 71:1188–1189.
- Maruyama M, Kobayashi Y, Kodani E, et al. Osborn waves: history and significance. Indian Pacing Electrophysiol J 2004; 4:33–39.
- Sheikh AM, Hurst JW. Osborn waves in the electrocardiogram, hypothermia not due to exposure, and death due to diabetic ketoacidosis. Clin Cardiol 2003; 26:555–560.
A 50-year-old woman with new-onset seizure
A 50-year-old woman presented to the emergency department after a witnessed loss of consciousness and seizurelike activity. She reported that she had been sitting outside her home, drinking coffee in the morning, but became very lightheaded when she went back into her house. At that time she felt could not focus and had a sense of impending doom. She sat down in a chair and her symptoms worsened.
According to her family, her eyes rolled back and she became rigid. The family helped her to the floor. Her body then made jerking movements that lasted for about 1 minute. She regained consciousness but was very confused for about 10 minutes until emergency medical services personnel arrived. She had no recollection of passing out. She said nothing like this had ever happened to her before.
On arrival in the emergency department, she complained of generalized headache and muscle soreness. She said the headache had been present for 1 week and was constant and dull. There were no aggravating or alleviating factors associated with the headache, and she denied fever, chills, nausea, numbness, tingling, incontinence, tongue biting, tremor, poor balance, ringing in ears, speech difficulty, or weakness.
Medical history: Multiple problems, medications
The patient’s medical history included depression, hypertension, anxiety, osteoarthritis, and asthma. She was allergic to penicillin. She had undergone carpal tunnel surgery on her right hand 5 years previously. She was perimenopausal with no children.
She denied using illicit drugs. She said she had smoked a half pack of cigarettes per day for more than 10 years and was a current smoker but was actively trying to quit. She said she occasionally used alcohol but had not consumed any alcohol in the last 2 weeks.
She had no history of central nervous system infection. She did report an episode of head trauma in grade school when a portable basketball hoop fell, striking her on the top of the head and causing her to briefly lose consciousness, but she did not seek medical attention.
She had no family history of seizure or neurologic disease.
Her current medications included atenolol, naproxen, gabapentin, venlafaxine, zolpidem, lorazepam, bupropion, and meloxicam. The bupropion and lorazepam had been prescribed recently for her anxiety. She reported that she had been given only 10 tablets of lorazepam and had taken the last tablet 48 hours previously. She had been taking the bupropion for 7 days. She reported an increase in stress lately and had been taking zolpidem due to an altered sleep pattern.
PHYSICAL EXAMINATION, INITIAL TESTS
On examination, the patient did not appear to be in acute distress. Her blood pressure was 107/22 mm Hg, pulse 100 beats per minute, respiratory rate 16 breaths per minute, temperature 37.1°C (98.8°F), and oxygen saturation 98% on room air.
Examination of her head, eyes, mouth, and neck were unremarkable. Cardiovascular, pulmonary, and abdominal examinations were normal. She had no neurologic deficits and was fully alert and oriented. She had no visible injuries.
Blood and urine samples were obtained about 15 minutes after her arrival to the emergency department. Results showed:
- Glucose 73 mg/dL (reference range 74–99)
- Sodium 142 mmol/L (136–144)
- Blood urea nitrogen 12 mg/dL (7–21)
- Creatinine 0.95 mg/dL (0.58–0.96)
- Chloride 97 mmol/L (97–105)
- Carbon dioxide (bicarbonate) 16 mmol/L (22–30)
- Prolactin 50.9 ng/mL (4.5–26.8)
- Anion gap 29 mmol/L (9–18)
- Ethanol undetectable
- White blood cell count 11.03 × 109/L (3.70–11.00)
- Creatine kinase 89 U/L (30–220)
- Urinalysis normal, specific gravity 1.010 (1.005–1.030), no detectable ketones, and no crystals seen on microscopic evaluation.
Electrocardiography showed normal sinus rhythm with no ectopy and no ST-segment changes. Chest radiography was negative for any acute process.
The patient was transferred to the 23-hour observation unit in stable condition for further evaluation, monitoring, and management.
SIGNS AND SYMPTOMS OF SEIZURE
1. What findings are consistent with seizure?
- Jerking movements
- Confusion following the event
- Tongue-biting
- Focal motor weakness
- Urinary incontinence
- Aura before the event
All of the above findings are consistent with seizure.
The first consideration in evaluating a patient who presents with a possible seizure is whether the patient’s recollections of the event—and those of the witnesses—are consistent with the symptoms of seizure.1
In generalized tonic-clonic or grand mal seizure, the patient may experience an aura or subjective sensations before the onset. These vary greatly among patients.2 There may be an initial vocalization at the onset of the seizure, such as crying out or unintelligible speech. The patient’s eyes may roll back in the head. This is followed by loss of muscle tone, and if the patient is standing, he or she may fall to the ground. The patient becomes unresponsive and may go into respiratory arrest. There is tonic stiffening of the limbs and body, followed by clonic movements typically lasting 1 to 2 minutes, or sometimes longer.1,3,4 The patient will then relax and experience a period of unconsciousness or confusion (postictal state).
Urinary incontinence and tongue-biting strongly suggest seizure activity, and turning the head to one side and posturing may also be seen.3,5 After the event, the patient may report headache, generalized muscle soreness, exhaustion, or periods of transient focal weakness, also known as Todd paralysis.2,5
Our patient had aura-like symptoms at the outset. She felt very lightheaded, had difficulty focusing, and felt a sense of impending doom. She did not make any vocalizations at the onset, but her eyes did roll backward and she became rigid (tonic). She then lost muscle tone and became unresponsive. Her family had to help her to the floor. Jerking (clonic) movements were witnessed.
She regained consciousness but was described as being confused (postictal) for 10 minutes. Additionally, she denied ever having had symptoms like this previously. On arrival in the emergency department, she reported generalized headache and muscle soreness, but no tongue-biting or urinary incontinence. Her event did not last for more than 1 to 2 minutes according to her family.
Her symptoms strongly suggest new-onset tonic-clonic or grand mal seizure, though this is not completely certain.
LABORATORY FINDINGS IN SEIZURES
2. What laboratory results are consistent with seizure?
- Prolactin elevation
- Anion gap acidosis
- Leukocytosis
As noted above, the patient had an elevated prolactin level and elevated anion gap. Both of these findings can be used, with caution, in evaluating seizure activity.
Prolactin testing is controversial
Prolactin testing in diagnosing seizure activity is controversial. The exact mechanism of prolactin release in seizures is not fully understood. Generalized tonic-clonic seizures and complex partial seizures have both been shown to elevate prolactin. Prolactin levels after these types of seizures should rise within 30 minutes of the event and normalize 1 hour later.6
However, other events and conditions that mimic seizure have been shown to cause a rise in prolactin; these include syncope, transient ischemic attack, cardiac dysrhythmia, migraine, and other epilepsy-like variants. This effect has not been adequately studied. Therefore, an elevated prolactin level alone cannot diagnose or exclude seizure.7
For the prolactin level to be helpful, the blood sample must be drawn within 10 to 20 minutes after a possible seizure. Even if the prolactin level remains normal, it does not rule out seizure. Prolactin levels should therefore be used in combination with other testing to make a definitive diagnosis or exclusion of seizure.8
Anion gap and Denver Seizure Score
The anion gap has also been shown to rise after generalized seizure due to the metabolic acidosis that occurs. With a bicarbonate level of 16 mmol/L, an elevated anion gap, and normal breathing, our patient very likely had metabolic acidosis.
It is sometimes difficult to differentiate syncope from seizure, as they share several features.
The Denver Seizure Score can help differentiate these two conditions. It is based on the patient’s anion gap and bicarbonate level and is calculated as follows:
(24 – bicarbonate) + [2 × (anion gap – 12)]
A score above 20 strongly indicates seizure activity. However, this is not a definitive tool for diagnosis. Like an elevated prolactin level, the Denver Seizure Score should be used in combination with other testing to move toward a definitive diagnosis.9
Our patient’s anion gap was 29 mmol/L and her bicarbonate level was 16 mmol/L. Her Denver Seizure Score was therefore 42, which supports this being an episode of generalized seizure activity.
Leukocytosis
The patient had a white blood cell count of 11.03 × 109/L, which was mildly elevated. She had no history of fever and no source of infection by history.
Leukocytosis is common following generalized tonic-clonic seizure. A fever may lower the seizure threshold; however, our patient was not febrile and clinically had no factors that raised concern for an underlying infection.
ANION GAP ACIDOSIS AND SEIZURE
3. Which of the following can cause both anion gap acidosis and seizure?
- Ethylene glycol
- Salicylate overdose
- Ethanol withdrawal without ketosis
- Alcoholic ketoacidosis
- Methanol
All of the above except for ethanol withdrawal without ketosis can cause both anion gap acidosis and seizure.
Ethylene glycol can cause seizure and an elevated anion gap acidosis. However, this patient had no history of ingesting antifreeze (the most common source of ethylene glycol in the home) and no evidence of calcium oxalate crystals in the urine, which would be a sign of ethylene glycol toxicity. Additional testing for ethylene glycol may include serum ethylene glycol levels and ultraviolet light testing of the urine to detect fluorescein, which is commonly added to automotive antifreeze to help mechanics find fluid leaks in engines.
Salicylate overdose can cause seizure and an elevated anion gap acidosis. However, this patient has no history of aspirin ingestion, and a serum aspirin level was later ordered and found to be negative. In addition, the acid-base disorder in salicylate overdose may be respiratory alkalosis from direct stimulation of respiratory centers in conjunction with metabolic acidosis.
Ethanol withdrawal can cause seizure and may result in ketoacidosis, which would appear as anion gap acidosis. The undetectable ethanol level in this patient would be consistent with withdrawal from ethanol, which may also lead to ketoacidosis.
Alcoholic ketoacidosis is a late finding in patients who have been drinking ethanol and is thus a possible cause of an elevated anion gap in this patient. However, the absence of ketones in her urine speaks against this diagnosis.
Methanol can cause seizure and acidosis, but laboratory testing would reveal a normal anion gap and an elevated osmolar gap. This was not likely in this patient.
The presence of anion gap acidosis is important in forming a differential diagnosis. Several causes of anion gap acidosis may also cause seizure. These include salicylates, ethanol withdrawal with ketosis, methanol, and isoniazid. None of these appears to be a factor in this patient’s case.
DIFFERENTIAL DIAGNOSIS IN OUR PATIENT
4. What is the most likely cause of this patient’s seizure?
- Bupropion side effect
- Benzodiazepine withdrawal
- Ethanol withdrawal
- Brain lesion
- Central nervous system infection
- Unprovoked seizure (new-onset epilepsy)
Bupropion, an inhibitor of neuronal reuptake of norepinephrine and dopamine, has been used in the United States since 1989 to treat major depression.10 At therapeutic doses, it lowers the seizure threshold; in cases of acute overdose, seizures typically occur within hours of the dose, or up to 24 hours in patients taking extended-release formulations.11
Bupropion should be used with caution or avoided in patients taking other medications that also lower the seizure threshold, or during withdrawal from alcohol, benzodiazepines, or barbiturates.10
Benzodiazepine withdrawal. Abrupt cessation of benzodiazepines also lowers the seizure threshold, and seizures are commonly seen in benzodiazepine withdrawal syndrome. The use of benzodiazepines is controversial in many situations, and discontinuing them may prove problematic for both the patient and physician, as the potential for abuse and addiction is significant.
Seizures have occurred during withdrawal from even short-term benzodiazepine use. Other factors, such as concomitant use of other medications that lower the seizure threshold, may play a more significant role in causing withdrawal seizures than the duration of benzodiazepine therapy.12
Medications shown to be useful in managing withdrawal from benzodiazepines include carbamazepine, imipramine, valproate, and trazodone. Paroxetine has also been shown to be helpful in patients with major depression who are being taken off a benzodiazepine.13
Ethanol withdrawal is common in patients presenting to emergency departments, and seizures are frequently seen in these patients. This patient reported social drinking but not drinking ethanol daily, although many patients are not forthcoming about alcohol or drug use when talking with a physician or other healthcare provider.
Alcohol withdrawal seizures may accompany delirium tremens or major withdrawal syndrome, but they are seen more often in the absence of major withdrawal or delirium tremens. Seizures are typically single or occur in a short grouping over a brief period of time and mostly occur in chronic alcoholism. The role of anticonvulsants in patients with alcohol withdrawal seizure has not been established.14
Brain lesion. A previously undiagnosed brain tumor is not a common cause of new-onset seizure, although it is not unusual for a brain tumor to cause new-onset seizure. In 1 study, 6% of patients with new-onset seizure had a clinically significant lesion on brain imaging.15 In addition, 15% to 30% of patients with a previously undiagnosed brain tumor present with seizure as the first symptom.16 Patients with abnormal findings on neurologic examination after the seizure activity are more likely to have a structural lesion that may be identified by computed tomography (CT) or magnetic resonance imaging. (MRI)15
Unprovoked seizure occurs without an identifiable precipitating factor, or from a central nervous system insult that occurred more than 7 days earlier. Patients who may have recurrent unprovoked seizure will likely be diagnosed with epilepsy.15 Patients with a first-time unprovoked seizure have a 30% or higher likelihood of having another unprovoked seizure within 5 years.17
It is most likely that bupropion is the key factor in lowering the seizure threshold in this patient. However, patients sometimes underreport the amount of alcohol they consume, and though less likely, our patient’s report of not drinking for 2 weeks may also be unreliable. Ethanol withdrawal, though unlikely, may also be a consideration with this case.
FURTHER TESTING FOR OUR PATIENT
5. Which tests may be helpful in this patient’s workup?
- CT of the brain
- Lumbar puncture for spinal fluid analysis
- MRI of the brain
- Electroencephalography (EEG)
This patient had had a headache for 1 week before presenting to the emergency department. Indications for neuroimaging in a patient with headache include sudden onset of severe headache, neurologic deficits, human immunodeficiency virus infection, loss of consciousness, immunosuppression, pregnancy, malignancy, and age over 50 with a new type of headache.18,19 Therefore, she should undergo some form of neuroimaging, either CT or MRI.
CT is the most readily available and fastest imaging study for the central nervous system available to emergency physicians. CT is limited, however, due to its decreased sensitivity in detecting some brain lesions. Therefore, many patients with first-time seizure may eventually require MRI.15 Furthermore, patients with focal onset of the seizure activity are more likely to have a structural lesion precipitating the seizure. MRI may have a higher yield than CT in these cases.15,20
Lumbar puncture for spinal fluid analysis is helpful in evaluating a patient with a suspected central nervous system infection such as meningitis or encephalitis, or subarachnoid hemorrhage.
This patient had a normal neurologic examination, no fever, and no meningeal signs, and central nervous system infection was very unlikely. Also, because she had had a headache for 1 week before the presentation with seizurelike activity, subarachnoid hemorrhage was very unlikely, and emergency lumbar puncture was not indicated.
MRI is less readily available than CT in a timely fashion in most emergency departments in the United States. It offers a higher yield than CT in diagnosing pathology such as acute stroke, brain tumor, and plaques seen in multiple sclerosis. CT is superior to MRI in diagnosing bony abnormalities and is very sensitive for detecting acute bleeding.
If MRI is performed, it should follow a specific protocol that includes high-resolution images for epilepsy evaluation rather than the more commonly ordered stroke protocol. The stroke protocol is more likely to be ordered in the emergency department.
EEG is well established in evaluating new-onset seizure in pediatric patients. Studies also demonstrate its utility in evaluating first-time seizure in adults, providing evidence that both epileptiform and nonepileptiform abnormalities seen on EEG are associated with a higher risk of recurrent seizure activity than in patients with normal findings on EEG.1
EEG may be difficult to interpret in adults. According to Benbadis,5 as many as one-third of adult patients diagnosed with epilepsy on EEG did not have epilepsy. This is because of normal variants, simple fluctuations of background rhythms, or fragmented alpha activity that can have a similar appearance to epileptiform patterns. EEG must always be interpreted in the context of the patient’s history and symptoms.5
Though EEG has limitations, it remains a crucial tool for identifying epilepsy. Following a single seizure, the decision to prescribe antiepileptic drugs is highly influenced by patterns on EEG associated with a risk of recurrence. In fact, a patient experiencing a single, idiopathic seizure and exhibiting an EEG pattern of spike wave discharges is likely to have recurrent seizure activity.21 Also, the appropriate use of EEG after even a single unprovoked seizure can identify patients with epilepsy and a risk of recurrent seizure greater than 60%.21,22
NO FURTHER SEIZURES
The patient was admitted to the observation unit from the emergency department after undergoing CT without intravenous contrast. While in observation, she had no additional episodes, and her vital signs remained within normal limits.
She underwent MRI and EEG as well as repeat laboratory studies and consultation by a neurologist. CT showed no structural abnormality, MRI results were read as normal, and EEG showed no epileptiform spikes or abnormal slow waves or other abnormality consistent with seizure. The repeat laboratory studies revealed normalization of the prolactin level at 11.3 ng/mL (reference range 2.0–17.4).
The final impression of the neurology consultant was that the patient had had a seizure that was most likely due to recently starting bupropion in combination with the withdrawal of the benzodiazepine, which lowered the seizure threshold. The neurologist also believed that our patient had no findings or symptoms other than the seizure that would suggest benzodiazepine withdrawal syndrome. According to the patient’s social history, it was unlikely that she had the pattern of alcohol consumption that would result in ethanol withdrawal seizure.
Seizures are common. In fact, every year, 180,000 US adults have their first seizure, and 10% of Americans will experience at least 1 seizure during their lifetime. However, only 20% to 25% of seizures are generalized tonic-clonic seizures as in our patient.23
As this patient had an identifiable cause for the seizure, there was no need to initiate anticonvulsant therapy at the time of discharge. She was discharged to home without any anticonvulsant, the bupropion was discontinued, and the lorazepam was not restarted. When contacted by telephone at 1 month and 18 months after discharge, she reported she had not experienced any additional seizures and has not needed antiepileptic medications.
- Seneviratne U. Management of the first seizure: an evidence based approach. Postgrad Med J 2009; 85:667–673.
- Krumholz A, Wiebe S, Gronseth G, et al; Quality Standards Subcommittee of the American Academy of Neurology; American Epilepsy Society. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 67:1996–2007.
- Gram L. Epileptic seizures and syndromes. Lancet 1990; 336:161–163.
- Smith PE, Cossburn MD. Seizures: assessment and management in the emergency unit. Clin Med (Lond) 2004; 4:118–122.
- Benbadis S. The differential diagnosis of epilepsy: a critical review. Epilepsy Behav 2009; 15:15–21.
- Lusic I, Pintaric I, Hozo I, Boic L, Capkun V. Serum prolactin levels after seizure and syncopal attacks. Seizure 1999; 8:218–222.
- Chen DK, So YT, Fisher RS; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Use of serum prolactin in diagnosing epileptic seizures: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2005; 65:668–675.
- Ben-Menachem E. Is prolactin a clinically useful measure of epilepsy? Epilepsy Curr 2006; 6:78–79.
- Bakes KM, Faragher J, Markovchick VJ, Donahoe K, Haukoos JS. The Denver Seizure Score: anion gap metabolic acidosis predicts generalized seizure. Am J Emerg Med 2011; 29:1097–1102.
- Jefferson JW, Pradok JF, Muir KT. Bupropion for major depressive disorder: pharmacokinetic and formulation considerations. Clin Ther 2005; 27:1685–1695.
- Stall N, Godwin J, Juurlink D. Bupropion abuse and overdose. CMAJ 2014; 186:1015.
- Fialip J, Aumaitre O, Eschalier A, Maradeix B, Dordain G, Lavarenne J. Benzodiazepine withdrawal seizures: analysis of 48 case reports. Clin Neuropharmacol 1987; 10:538–544.
- Lader M, Tylee A, Donoghue J. Withdrawing benzodiazepines in primary care. CNS Drugs 2009; 23:19–34.
- Chance JF. Emergency department treatment of alcohol withdrawal seizures with phenytoin. Ann Emerg Med 1991; 20:520–522.
- ACEP Clinical Policies Committee; Clinical Policies Subcommittee on Seizures. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with seizures. Ann Emerg Med 2004; 43:605–625.
- Sperling MR, Ko J. Seizures and brain tumors. Semin Oncol 2006; 33:333–341.
- Musicco M, Beghi E, Solari A, Viani F. Treatment of first tonic-clonic seizure does not improve the prognosis of epilepsy. First Seizure Trial Group (FIRST Group). Neurology 1997; 49:991–998.
- Edlow JA, Panagos PD, Godwin SA, Thomas TL, Decker WW; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with acute headache. Ann Emerg Med 2008; 52:407–436.
- Kaniecki R. Headache assessment and management. JAMA 2003; 289:1430–1433.
- Harden CL, Huff JS, Schwartz TH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Reassessment: neuroimaging in the emergency patient presenting with seizure (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2007; 69:1772–1780.
- Bergey GK. Management of a first seizure. Continuum (Minneap Minn) 2016; 22:38–50.
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014; 55:475–482.
- Ko DY. Generalized tonic-clonic seizures. Medscape. http://emedicine.medscape.com/article/1184608-overview. Accessed December 5, 2017.
A 50-year-old woman presented to the emergency department after a witnessed loss of consciousness and seizurelike activity. She reported that she had been sitting outside her home, drinking coffee in the morning, but became very lightheaded when she went back into her house. At that time she felt could not focus and had a sense of impending doom. She sat down in a chair and her symptoms worsened.
According to her family, her eyes rolled back and she became rigid. The family helped her to the floor. Her body then made jerking movements that lasted for about 1 minute. She regained consciousness but was very confused for about 10 minutes until emergency medical services personnel arrived. She had no recollection of passing out. She said nothing like this had ever happened to her before.
On arrival in the emergency department, she complained of generalized headache and muscle soreness. She said the headache had been present for 1 week and was constant and dull. There were no aggravating or alleviating factors associated with the headache, and she denied fever, chills, nausea, numbness, tingling, incontinence, tongue biting, tremor, poor balance, ringing in ears, speech difficulty, or weakness.
Medical history: Multiple problems, medications
The patient’s medical history included depression, hypertension, anxiety, osteoarthritis, and asthma. She was allergic to penicillin. She had undergone carpal tunnel surgery on her right hand 5 years previously. She was perimenopausal with no children.
She denied using illicit drugs. She said she had smoked a half pack of cigarettes per day for more than 10 years and was a current smoker but was actively trying to quit. She said she occasionally used alcohol but had not consumed any alcohol in the last 2 weeks.
She had no history of central nervous system infection. She did report an episode of head trauma in grade school when a portable basketball hoop fell, striking her on the top of the head and causing her to briefly lose consciousness, but she did not seek medical attention.
She had no family history of seizure or neurologic disease.
Her current medications included atenolol, naproxen, gabapentin, venlafaxine, zolpidem, lorazepam, bupropion, and meloxicam. The bupropion and lorazepam had been prescribed recently for her anxiety. She reported that she had been given only 10 tablets of lorazepam and had taken the last tablet 48 hours previously. She had been taking the bupropion for 7 days. She reported an increase in stress lately and had been taking zolpidem due to an altered sleep pattern.
PHYSICAL EXAMINATION, INITIAL TESTS
On examination, the patient did not appear to be in acute distress. Her blood pressure was 107/22 mm Hg, pulse 100 beats per minute, respiratory rate 16 breaths per minute, temperature 37.1°C (98.8°F), and oxygen saturation 98% on room air.
Examination of her head, eyes, mouth, and neck were unremarkable. Cardiovascular, pulmonary, and abdominal examinations were normal. She had no neurologic deficits and was fully alert and oriented. She had no visible injuries.
Blood and urine samples were obtained about 15 minutes after her arrival to the emergency department. Results showed:
- Glucose 73 mg/dL (reference range 74–99)
- Sodium 142 mmol/L (136–144)
- Blood urea nitrogen 12 mg/dL (7–21)
- Creatinine 0.95 mg/dL (0.58–0.96)
- Chloride 97 mmol/L (97–105)
- Carbon dioxide (bicarbonate) 16 mmol/L (22–30)
- Prolactin 50.9 ng/mL (4.5–26.8)
- Anion gap 29 mmol/L (9–18)
- Ethanol undetectable
- White blood cell count 11.03 × 109/L (3.70–11.00)
- Creatine kinase 89 U/L (30–220)
- Urinalysis normal, specific gravity 1.010 (1.005–1.030), no detectable ketones, and no crystals seen on microscopic evaluation.
Electrocardiography showed normal sinus rhythm with no ectopy and no ST-segment changes. Chest radiography was negative for any acute process.
The patient was transferred to the 23-hour observation unit in stable condition for further evaluation, monitoring, and management.
SIGNS AND SYMPTOMS OF SEIZURE
1. What findings are consistent with seizure?
- Jerking movements
- Confusion following the event
- Tongue-biting
- Focal motor weakness
- Urinary incontinence
- Aura before the event
All of the above findings are consistent with seizure.
The first consideration in evaluating a patient who presents with a possible seizure is whether the patient’s recollections of the event—and those of the witnesses—are consistent with the symptoms of seizure.1
In generalized tonic-clonic or grand mal seizure, the patient may experience an aura or subjective sensations before the onset. These vary greatly among patients.2 There may be an initial vocalization at the onset of the seizure, such as crying out or unintelligible speech. The patient’s eyes may roll back in the head. This is followed by loss of muscle tone, and if the patient is standing, he or she may fall to the ground. The patient becomes unresponsive and may go into respiratory arrest. There is tonic stiffening of the limbs and body, followed by clonic movements typically lasting 1 to 2 minutes, or sometimes longer.1,3,4 The patient will then relax and experience a period of unconsciousness or confusion (postictal state).
Urinary incontinence and tongue-biting strongly suggest seizure activity, and turning the head to one side and posturing may also be seen.3,5 After the event, the patient may report headache, generalized muscle soreness, exhaustion, or periods of transient focal weakness, also known as Todd paralysis.2,5
Our patient had aura-like symptoms at the outset. She felt very lightheaded, had difficulty focusing, and felt a sense of impending doom. She did not make any vocalizations at the onset, but her eyes did roll backward and she became rigid (tonic). She then lost muscle tone and became unresponsive. Her family had to help her to the floor. Jerking (clonic) movements were witnessed.
She regained consciousness but was described as being confused (postictal) for 10 minutes. Additionally, she denied ever having had symptoms like this previously. On arrival in the emergency department, she reported generalized headache and muscle soreness, but no tongue-biting or urinary incontinence. Her event did not last for more than 1 to 2 minutes according to her family.
Her symptoms strongly suggest new-onset tonic-clonic or grand mal seizure, though this is not completely certain.
LABORATORY FINDINGS IN SEIZURES
2. What laboratory results are consistent with seizure?
- Prolactin elevation
- Anion gap acidosis
- Leukocytosis
As noted above, the patient had an elevated prolactin level and elevated anion gap. Both of these findings can be used, with caution, in evaluating seizure activity.
Prolactin testing is controversial
Prolactin testing in diagnosing seizure activity is controversial. The exact mechanism of prolactin release in seizures is not fully understood. Generalized tonic-clonic seizures and complex partial seizures have both been shown to elevate prolactin. Prolactin levels after these types of seizures should rise within 30 minutes of the event and normalize 1 hour later.6
However, other events and conditions that mimic seizure have been shown to cause a rise in prolactin; these include syncope, transient ischemic attack, cardiac dysrhythmia, migraine, and other epilepsy-like variants. This effect has not been adequately studied. Therefore, an elevated prolactin level alone cannot diagnose or exclude seizure.7
For the prolactin level to be helpful, the blood sample must be drawn within 10 to 20 minutes after a possible seizure. Even if the prolactin level remains normal, it does not rule out seizure. Prolactin levels should therefore be used in combination with other testing to make a definitive diagnosis or exclusion of seizure.8
Anion gap and Denver Seizure Score
The anion gap has also been shown to rise after generalized seizure due to the metabolic acidosis that occurs. With a bicarbonate level of 16 mmol/L, an elevated anion gap, and normal breathing, our patient very likely had metabolic acidosis.
It is sometimes difficult to differentiate syncope from seizure, as they share several features.
The Denver Seizure Score can help differentiate these two conditions. It is based on the patient’s anion gap and bicarbonate level and is calculated as follows:
(24 – bicarbonate) + [2 × (anion gap – 12)]
A score above 20 strongly indicates seizure activity. However, this is not a definitive tool for diagnosis. Like an elevated prolactin level, the Denver Seizure Score should be used in combination with other testing to move toward a definitive diagnosis.9
Our patient’s anion gap was 29 mmol/L and her bicarbonate level was 16 mmol/L. Her Denver Seizure Score was therefore 42, which supports this being an episode of generalized seizure activity.
Leukocytosis
The patient had a white blood cell count of 11.03 × 109/L, which was mildly elevated. She had no history of fever and no source of infection by history.
Leukocytosis is common following generalized tonic-clonic seizure. A fever may lower the seizure threshold; however, our patient was not febrile and clinically had no factors that raised concern for an underlying infection.
ANION GAP ACIDOSIS AND SEIZURE
3. Which of the following can cause both anion gap acidosis and seizure?
- Ethylene glycol
- Salicylate overdose
- Ethanol withdrawal without ketosis
- Alcoholic ketoacidosis
- Methanol
All of the above except for ethanol withdrawal without ketosis can cause both anion gap acidosis and seizure.
Ethylene glycol can cause seizure and an elevated anion gap acidosis. However, this patient had no history of ingesting antifreeze (the most common source of ethylene glycol in the home) and no evidence of calcium oxalate crystals in the urine, which would be a sign of ethylene glycol toxicity. Additional testing for ethylene glycol may include serum ethylene glycol levels and ultraviolet light testing of the urine to detect fluorescein, which is commonly added to automotive antifreeze to help mechanics find fluid leaks in engines.
Salicylate overdose can cause seizure and an elevated anion gap acidosis. However, this patient has no history of aspirin ingestion, and a serum aspirin level was later ordered and found to be negative. In addition, the acid-base disorder in salicylate overdose may be respiratory alkalosis from direct stimulation of respiratory centers in conjunction with metabolic acidosis.
Ethanol withdrawal can cause seizure and may result in ketoacidosis, which would appear as anion gap acidosis. The undetectable ethanol level in this patient would be consistent with withdrawal from ethanol, which may also lead to ketoacidosis.
Alcoholic ketoacidosis is a late finding in patients who have been drinking ethanol and is thus a possible cause of an elevated anion gap in this patient. However, the absence of ketones in her urine speaks against this diagnosis.
Methanol can cause seizure and acidosis, but laboratory testing would reveal a normal anion gap and an elevated osmolar gap. This was not likely in this patient.
The presence of anion gap acidosis is important in forming a differential diagnosis. Several causes of anion gap acidosis may also cause seizure. These include salicylates, ethanol withdrawal with ketosis, methanol, and isoniazid. None of these appears to be a factor in this patient’s case.
DIFFERENTIAL DIAGNOSIS IN OUR PATIENT
4. What is the most likely cause of this patient’s seizure?
- Bupropion side effect
- Benzodiazepine withdrawal
- Ethanol withdrawal
- Brain lesion
- Central nervous system infection
- Unprovoked seizure (new-onset epilepsy)
Bupropion, an inhibitor of neuronal reuptake of norepinephrine and dopamine, has been used in the United States since 1989 to treat major depression.10 At therapeutic doses, it lowers the seizure threshold; in cases of acute overdose, seizures typically occur within hours of the dose, or up to 24 hours in patients taking extended-release formulations.11
Bupropion should be used with caution or avoided in patients taking other medications that also lower the seizure threshold, or during withdrawal from alcohol, benzodiazepines, or barbiturates.10
Benzodiazepine withdrawal. Abrupt cessation of benzodiazepines also lowers the seizure threshold, and seizures are commonly seen in benzodiazepine withdrawal syndrome. The use of benzodiazepines is controversial in many situations, and discontinuing them may prove problematic for both the patient and physician, as the potential for abuse and addiction is significant.
Seizures have occurred during withdrawal from even short-term benzodiazepine use. Other factors, such as concomitant use of other medications that lower the seizure threshold, may play a more significant role in causing withdrawal seizures than the duration of benzodiazepine therapy.12
Medications shown to be useful in managing withdrawal from benzodiazepines include carbamazepine, imipramine, valproate, and trazodone. Paroxetine has also been shown to be helpful in patients with major depression who are being taken off a benzodiazepine.13
Ethanol withdrawal is common in patients presenting to emergency departments, and seizures are frequently seen in these patients. This patient reported social drinking but not drinking ethanol daily, although many patients are not forthcoming about alcohol or drug use when talking with a physician or other healthcare provider.
Alcohol withdrawal seizures may accompany delirium tremens or major withdrawal syndrome, but they are seen more often in the absence of major withdrawal or delirium tremens. Seizures are typically single or occur in a short grouping over a brief period of time and mostly occur in chronic alcoholism. The role of anticonvulsants in patients with alcohol withdrawal seizure has not been established.14
Brain lesion. A previously undiagnosed brain tumor is not a common cause of new-onset seizure, although it is not unusual for a brain tumor to cause new-onset seizure. In 1 study, 6% of patients with new-onset seizure had a clinically significant lesion on brain imaging.15 In addition, 15% to 30% of patients with a previously undiagnosed brain tumor present with seizure as the first symptom.16 Patients with abnormal findings on neurologic examination after the seizure activity are more likely to have a structural lesion that may be identified by computed tomography (CT) or magnetic resonance imaging. (MRI)15
Unprovoked seizure occurs without an identifiable precipitating factor, or from a central nervous system insult that occurred more than 7 days earlier. Patients who may have recurrent unprovoked seizure will likely be diagnosed with epilepsy.15 Patients with a first-time unprovoked seizure have a 30% or higher likelihood of having another unprovoked seizure within 5 years.17
It is most likely that bupropion is the key factor in lowering the seizure threshold in this patient. However, patients sometimes underreport the amount of alcohol they consume, and though less likely, our patient’s report of not drinking for 2 weeks may also be unreliable. Ethanol withdrawal, though unlikely, may also be a consideration with this case.
FURTHER TESTING FOR OUR PATIENT
5. Which tests may be helpful in this patient’s workup?
- CT of the brain
- Lumbar puncture for spinal fluid analysis
- MRI of the brain
- Electroencephalography (EEG)
This patient had had a headache for 1 week before presenting to the emergency department. Indications for neuroimaging in a patient with headache include sudden onset of severe headache, neurologic deficits, human immunodeficiency virus infection, loss of consciousness, immunosuppression, pregnancy, malignancy, and age over 50 with a new type of headache.18,19 Therefore, she should undergo some form of neuroimaging, either CT or MRI.
CT is the most readily available and fastest imaging study for the central nervous system available to emergency physicians. CT is limited, however, due to its decreased sensitivity in detecting some brain lesions. Therefore, many patients with first-time seizure may eventually require MRI.15 Furthermore, patients with focal onset of the seizure activity are more likely to have a structural lesion precipitating the seizure. MRI may have a higher yield than CT in these cases.15,20
Lumbar puncture for spinal fluid analysis is helpful in evaluating a patient with a suspected central nervous system infection such as meningitis or encephalitis, or subarachnoid hemorrhage.
This patient had a normal neurologic examination, no fever, and no meningeal signs, and central nervous system infection was very unlikely. Also, because she had had a headache for 1 week before the presentation with seizurelike activity, subarachnoid hemorrhage was very unlikely, and emergency lumbar puncture was not indicated.
MRI is less readily available than CT in a timely fashion in most emergency departments in the United States. It offers a higher yield than CT in diagnosing pathology such as acute stroke, brain tumor, and plaques seen in multiple sclerosis. CT is superior to MRI in diagnosing bony abnormalities and is very sensitive for detecting acute bleeding.
If MRI is performed, it should follow a specific protocol that includes high-resolution images for epilepsy evaluation rather than the more commonly ordered stroke protocol. The stroke protocol is more likely to be ordered in the emergency department.
EEG is well established in evaluating new-onset seizure in pediatric patients. Studies also demonstrate its utility in evaluating first-time seizure in adults, providing evidence that both epileptiform and nonepileptiform abnormalities seen on EEG are associated with a higher risk of recurrent seizure activity than in patients with normal findings on EEG.1
EEG may be difficult to interpret in adults. According to Benbadis,5 as many as one-third of adult patients diagnosed with epilepsy on EEG did not have epilepsy. This is because of normal variants, simple fluctuations of background rhythms, or fragmented alpha activity that can have a similar appearance to epileptiform patterns. EEG must always be interpreted in the context of the patient’s history and symptoms.5
Though EEG has limitations, it remains a crucial tool for identifying epilepsy. Following a single seizure, the decision to prescribe antiepileptic drugs is highly influenced by patterns on EEG associated with a risk of recurrence. In fact, a patient experiencing a single, idiopathic seizure and exhibiting an EEG pattern of spike wave discharges is likely to have recurrent seizure activity.21 Also, the appropriate use of EEG after even a single unprovoked seizure can identify patients with epilepsy and a risk of recurrent seizure greater than 60%.21,22
NO FURTHER SEIZURES
The patient was admitted to the observation unit from the emergency department after undergoing CT without intravenous contrast. While in observation, she had no additional episodes, and her vital signs remained within normal limits.
She underwent MRI and EEG as well as repeat laboratory studies and consultation by a neurologist. CT showed no structural abnormality, MRI results were read as normal, and EEG showed no epileptiform spikes or abnormal slow waves or other abnormality consistent with seizure. The repeat laboratory studies revealed normalization of the prolactin level at 11.3 ng/mL (reference range 2.0–17.4).
The final impression of the neurology consultant was that the patient had had a seizure that was most likely due to recently starting bupropion in combination with the withdrawal of the benzodiazepine, which lowered the seizure threshold. The neurologist also believed that our patient had no findings or symptoms other than the seizure that would suggest benzodiazepine withdrawal syndrome. According to the patient’s social history, it was unlikely that she had the pattern of alcohol consumption that would result in ethanol withdrawal seizure.
Seizures are common. In fact, every year, 180,000 US adults have their first seizure, and 10% of Americans will experience at least 1 seizure during their lifetime. However, only 20% to 25% of seizures are generalized tonic-clonic seizures as in our patient.23
As this patient had an identifiable cause for the seizure, there was no need to initiate anticonvulsant therapy at the time of discharge. She was discharged to home without any anticonvulsant, the bupropion was discontinued, and the lorazepam was not restarted. When contacted by telephone at 1 month and 18 months after discharge, she reported she had not experienced any additional seizures and has not needed antiepileptic medications.
A 50-year-old woman presented to the emergency department after a witnessed loss of consciousness and seizurelike activity. She reported that she had been sitting outside her home, drinking coffee in the morning, but became very lightheaded when she went back into her house. At that time she felt could not focus and had a sense of impending doom. She sat down in a chair and her symptoms worsened.
According to her family, her eyes rolled back and she became rigid. The family helped her to the floor. Her body then made jerking movements that lasted for about 1 minute. She regained consciousness but was very confused for about 10 minutes until emergency medical services personnel arrived. She had no recollection of passing out. She said nothing like this had ever happened to her before.
On arrival in the emergency department, she complained of generalized headache and muscle soreness. She said the headache had been present for 1 week and was constant and dull. There were no aggravating or alleviating factors associated with the headache, and she denied fever, chills, nausea, numbness, tingling, incontinence, tongue biting, tremor, poor balance, ringing in ears, speech difficulty, or weakness.
Medical history: Multiple problems, medications
The patient’s medical history included depression, hypertension, anxiety, osteoarthritis, and asthma. She was allergic to penicillin. She had undergone carpal tunnel surgery on her right hand 5 years previously. She was perimenopausal with no children.
She denied using illicit drugs. She said she had smoked a half pack of cigarettes per day for more than 10 years and was a current smoker but was actively trying to quit. She said she occasionally used alcohol but had not consumed any alcohol in the last 2 weeks.
She had no history of central nervous system infection. She did report an episode of head trauma in grade school when a portable basketball hoop fell, striking her on the top of the head and causing her to briefly lose consciousness, but she did not seek medical attention.
She had no family history of seizure or neurologic disease.
Her current medications included atenolol, naproxen, gabapentin, venlafaxine, zolpidem, lorazepam, bupropion, and meloxicam. The bupropion and lorazepam had been prescribed recently for her anxiety. She reported that she had been given only 10 tablets of lorazepam and had taken the last tablet 48 hours previously. She had been taking the bupropion for 7 days. She reported an increase in stress lately and had been taking zolpidem due to an altered sleep pattern.
PHYSICAL EXAMINATION, INITIAL TESTS
On examination, the patient did not appear to be in acute distress. Her blood pressure was 107/22 mm Hg, pulse 100 beats per minute, respiratory rate 16 breaths per minute, temperature 37.1°C (98.8°F), and oxygen saturation 98% on room air.
Examination of her head, eyes, mouth, and neck were unremarkable. Cardiovascular, pulmonary, and abdominal examinations were normal. She had no neurologic deficits and was fully alert and oriented. She had no visible injuries.
Blood and urine samples were obtained about 15 minutes after her arrival to the emergency department. Results showed:
- Glucose 73 mg/dL (reference range 74–99)
- Sodium 142 mmol/L (136–144)
- Blood urea nitrogen 12 mg/dL (7–21)
- Creatinine 0.95 mg/dL (0.58–0.96)
- Chloride 97 mmol/L (97–105)
- Carbon dioxide (bicarbonate) 16 mmol/L (22–30)
- Prolactin 50.9 ng/mL (4.5–26.8)
- Anion gap 29 mmol/L (9–18)
- Ethanol undetectable
- White blood cell count 11.03 × 109/L (3.70–11.00)
- Creatine kinase 89 U/L (30–220)
- Urinalysis normal, specific gravity 1.010 (1.005–1.030), no detectable ketones, and no crystals seen on microscopic evaluation.
Electrocardiography showed normal sinus rhythm with no ectopy and no ST-segment changes. Chest radiography was negative for any acute process.
The patient was transferred to the 23-hour observation unit in stable condition for further evaluation, monitoring, and management.
SIGNS AND SYMPTOMS OF SEIZURE
1. What findings are consistent with seizure?
- Jerking movements
- Confusion following the event
- Tongue-biting
- Focal motor weakness
- Urinary incontinence
- Aura before the event
All of the above findings are consistent with seizure.
The first consideration in evaluating a patient who presents with a possible seizure is whether the patient’s recollections of the event—and those of the witnesses—are consistent with the symptoms of seizure.1
In generalized tonic-clonic or grand mal seizure, the patient may experience an aura or subjective sensations before the onset. These vary greatly among patients.2 There may be an initial vocalization at the onset of the seizure, such as crying out or unintelligible speech. The patient’s eyes may roll back in the head. This is followed by loss of muscle tone, and if the patient is standing, he or she may fall to the ground. The patient becomes unresponsive and may go into respiratory arrest. There is tonic stiffening of the limbs and body, followed by clonic movements typically lasting 1 to 2 minutes, or sometimes longer.1,3,4 The patient will then relax and experience a period of unconsciousness or confusion (postictal state).
Urinary incontinence and tongue-biting strongly suggest seizure activity, and turning the head to one side and posturing may also be seen.3,5 After the event, the patient may report headache, generalized muscle soreness, exhaustion, or periods of transient focal weakness, also known as Todd paralysis.2,5
Our patient had aura-like symptoms at the outset. She felt very lightheaded, had difficulty focusing, and felt a sense of impending doom. She did not make any vocalizations at the onset, but her eyes did roll backward and she became rigid (tonic). She then lost muscle tone and became unresponsive. Her family had to help her to the floor. Jerking (clonic) movements were witnessed.
She regained consciousness but was described as being confused (postictal) for 10 minutes. Additionally, she denied ever having had symptoms like this previously. On arrival in the emergency department, she reported generalized headache and muscle soreness, but no tongue-biting or urinary incontinence. Her event did not last for more than 1 to 2 minutes according to her family.
Her symptoms strongly suggest new-onset tonic-clonic or grand mal seizure, though this is not completely certain.
LABORATORY FINDINGS IN SEIZURES
2. What laboratory results are consistent with seizure?
- Prolactin elevation
- Anion gap acidosis
- Leukocytosis
As noted above, the patient had an elevated prolactin level and elevated anion gap. Both of these findings can be used, with caution, in evaluating seizure activity.
Prolactin testing is controversial
Prolactin testing in diagnosing seizure activity is controversial. The exact mechanism of prolactin release in seizures is not fully understood. Generalized tonic-clonic seizures and complex partial seizures have both been shown to elevate prolactin. Prolactin levels after these types of seizures should rise within 30 minutes of the event and normalize 1 hour later.6
However, other events and conditions that mimic seizure have been shown to cause a rise in prolactin; these include syncope, transient ischemic attack, cardiac dysrhythmia, migraine, and other epilepsy-like variants. This effect has not been adequately studied. Therefore, an elevated prolactin level alone cannot diagnose or exclude seizure.7
For the prolactin level to be helpful, the blood sample must be drawn within 10 to 20 minutes after a possible seizure. Even if the prolactin level remains normal, it does not rule out seizure. Prolactin levels should therefore be used in combination with other testing to make a definitive diagnosis or exclusion of seizure.8
Anion gap and Denver Seizure Score
The anion gap has also been shown to rise after generalized seizure due to the metabolic acidosis that occurs. With a bicarbonate level of 16 mmol/L, an elevated anion gap, and normal breathing, our patient very likely had metabolic acidosis.
It is sometimes difficult to differentiate syncope from seizure, as they share several features.
The Denver Seizure Score can help differentiate these two conditions. It is based on the patient’s anion gap and bicarbonate level and is calculated as follows:
(24 – bicarbonate) + [2 × (anion gap – 12)]
A score above 20 strongly indicates seizure activity. However, this is not a definitive tool for diagnosis. Like an elevated prolactin level, the Denver Seizure Score should be used in combination with other testing to move toward a definitive diagnosis.9
Our patient’s anion gap was 29 mmol/L and her bicarbonate level was 16 mmol/L. Her Denver Seizure Score was therefore 42, which supports this being an episode of generalized seizure activity.
Leukocytosis
The patient had a white blood cell count of 11.03 × 109/L, which was mildly elevated. She had no history of fever and no source of infection by history.
Leukocytosis is common following generalized tonic-clonic seizure. A fever may lower the seizure threshold; however, our patient was not febrile and clinically had no factors that raised concern for an underlying infection.
ANION GAP ACIDOSIS AND SEIZURE
3. Which of the following can cause both anion gap acidosis and seizure?
- Ethylene glycol
- Salicylate overdose
- Ethanol withdrawal without ketosis
- Alcoholic ketoacidosis
- Methanol
All of the above except for ethanol withdrawal without ketosis can cause both anion gap acidosis and seizure.
Ethylene glycol can cause seizure and an elevated anion gap acidosis. However, this patient had no history of ingesting antifreeze (the most common source of ethylene glycol in the home) and no evidence of calcium oxalate crystals in the urine, which would be a sign of ethylene glycol toxicity. Additional testing for ethylene glycol may include serum ethylene glycol levels and ultraviolet light testing of the urine to detect fluorescein, which is commonly added to automotive antifreeze to help mechanics find fluid leaks in engines.
Salicylate overdose can cause seizure and an elevated anion gap acidosis. However, this patient has no history of aspirin ingestion, and a serum aspirin level was later ordered and found to be negative. In addition, the acid-base disorder in salicylate overdose may be respiratory alkalosis from direct stimulation of respiratory centers in conjunction with metabolic acidosis.
Ethanol withdrawal can cause seizure and may result in ketoacidosis, which would appear as anion gap acidosis. The undetectable ethanol level in this patient would be consistent with withdrawal from ethanol, which may also lead to ketoacidosis.
Alcoholic ketoacidosis is a late finding in patients who have been drinking ethanol and is thus a possible cause of an elevated anion gap in this patient. However, the absence of ketones in her urine speaks against this diagnosis.
Methanol can cause seizure and acidosis, but laboratory testing would reveal a normal anion gap and an elevated osmolar gap. This was not likely in this patient.
The presence of anion gap acidosis is important in forming a differential diagnosis. Several causes of anion gap acidosis may also cause seizure. These include salicylates, ethanol withdrawal with ketosis, methanol, and isoniazid. None of these appears to be a factor in this patient’s case.
DIFFERENTIAL DIAGNOSIS IN OUR PATIENT
4. What is the most likely cause of this patient’s seizure?
- Bupropion side effect
- Benzodiazepine withdrawal
- Ethanol withdrawal
- Brain lesion
- Central nervous system infection
- Unprovoked seizure (new-onset epilepsy)
Bupropion, an inhibitor of neuronal reuptake of norepinephrine and dopamine, has been used in the United States since 1989 to treat major depression.10 At therapeutic doses, it lowers the seizure threshold; in cases of acute overdose, seizures typically occur within hours of the dose, or up to 24 hours in patients taking extended-release formulations.11
Bupropion should be used with caution or avoided in patients taking other medications that also lower the seizure threshold, or during withdrawal from alcohol, benzodiazepines, or barbiturates.10
Benzodiazepine withdrawal. Abrupt cessation of benzodiazepines also lowers the seizure threshold, and seizures are commonly seen in benzodiazepine withdrawal syndrome. The use of benzodiazepines is controversial in many situations, and discontinuing them may prove problematic for both the patient and physician, as the potential for abuse and addiction is significant.
Seizures have occurred during withdrawal from even short-term benzodiazepine use. Other factors, such as concomitant use of other medications that lower the seizure threshold, may play a more significant role in causing withdrawal seizures than the duration of benzodiazepine therapy.12
Medications shown to be useful in managing withdrawal from benzodiazepines include carbamazepine, imipramine, valproate, and trazodone. Paroxetine has also been shown to be helpful in patients with major depression who are being taken off a benzodiazepine.13
Ethanol withdrawal is common in patients presenting to emergency departments, and seizures are frequently seen in these patients. This patient reported social drinking but not drinking ethanol daily, although many patients are not forthcoming about alcohol or drug use when talking with a physician or other healthcare provider.
Alcohol withdrawal seizures may accompany delirium tremens or major withdrawal syndrome, but they are seen more often in the absence of major withdrawal or delirium tremens. Seizures are typically single or occur in a short grouping over a brief period of time and mostly occur in chronic alcoholism. The role of anticonvulsants in patients with alcohol withdrawal seizure has not been established.14
Brain lesion. A previously undiagnosed brain tumor is not a common cause of new-onset seizure, although it is not unusual for a brain tumor to cause new-onset seizure. In 1 study, 6% of patients with new-onset seizure had a clinically significant lesion on brain imaging.15 In addition, 15% to 30% of patients with a previously undiagnosed brain tumor present with seizure as the first symptom.16 Patients with abnormal findings on neurologic examination after the seizure activity are more likely to have a structural lesion that may be identified by computed tomography (CT) or magnetic resonance imaging. (MRI)15
Unprovoked seizure occurs without an identifiable precipitating factor, or from a central nervous system insult that occurred more than 7 days earlier. Patients who may have recurrent unprovoked seizure will likely be diagnosed with epilepsy.15 Patients with a first-time unprovoked seizure have a 30% or higher likelihood of having another unprovoked seizure within 5 years.17
It is most likely that bupropion is the key factor in lowering the seizure threshold in this patient. However, patients sometimes underreport the amount of alcohol they consume, and though less likely, our patient’s report of not drinking for 2 weeks may also be unreliable. Ethanol withdrawal, though unlikely, may also be a consideration with this case.
FURTHER TESTING FOR OUR PATIENT
5. Which tests may be helpful in this patient’s workup?
- CT of the brain
- Lumbar puncture for spinal fluid analysis
- MRI of the brain
- Electroencephalography (EEG)
This patient had had a headache for 1 week before presenting to the emergency department. Indications for neuroimaging in a patient with headache include sudden onset of severe headache, neurologic deficits, human immunodeficiency virus infection, loss of consciousness, immunosuppression, pregnancy, malignancy, and age over 50 with a new type of headache.18,19 Therefore, she should undergo some form of neuroimaging, either CT or MRI.
CT is the most readily available and fastest imaging study for the central nervous system available to emergency physicians. CT is limited, however, due to its decreased sensitivity in detecting some brain lesions. Therefore, many patients with first-time seizure may eventually require MRI.15 Furthermore, patients with focal onset of the seizure activity are more likely to have a structural lesion precipitating the seizure. MRI may have a higher yield than CT in these cases.15,20
Lumbar puncture for spinal fluid analysis is helpful in evaluating a patient with a suspected central nervous system infection such as meningitis or encephalitis, or subarachnoid hemorrhage.
This patient had a normal neurologic examination, no fever, and no meningeal signs, and central nervous system infection was very unlikely. Also, because she had had a headache for 1 week before the presentation with seizurelike activity, subarachnoid hemorrhage was very unlikely, and emergency lumbar puncture was not indicated.
MRI is less readily available than CT in a timely fashion in most emergency departments in the United States. It offers a higher yield than CT in diagnosing pathology such as acute stroke, brain tumor, and plaques seen in multiple sclerosis. CT is superior to MRI in diagnosing bony abnormalities and is very sensitive for detecting acute bleeding.
If MRI is performed, it should follow a specific protocol that includes high-resolution images for epilepsy evaluation rather than the more commonly ordered stroke protocol. The stroke protocol is more likely to be ordered in the emergency department.
EEG is well established in evaluating new-onset seizure in pediatric patients. Studies also demonstrate its utility in evaluating first-time seizure in adults, providing evidence that both epileptiform and nonepileptiform abnormalities seen on EEG are associated with a higher risk of recurrent seizure activity than in patients with normal findings on EEG.1
EEG may be difficult to interpret in adults. According to Benbadis,5 as many as one-third of adult patients diagnosed with epilepsy on EEG did not have epilepsy. This is because of normal variants, simple fluctuations of background rhythms, or fragmented alpha activity that can have a similar appearance to epileptiform patterns. EEG must always be interpreted in the context of the patient’s history and symptoms.5
Though EEG has limitations, it remains a crucial tool for identifying epilepsy. Following a single seizure, the decision to prescribe antiepileptic drugs is highly influenced by patterns on EEG associated with a risk of recurrence. In fact, a patient experiencing a single, idiopathic seizure and exhibiting an EEG pattern of spike wave discharges is likely to have recurrent seizure activity.21 Also, the appropriate use of EEG after even a single unprovoked seizure can identify patients with epilepsy and a risk of recurrent seizure greater than 60%.21,22
NO FURTHER SEIZURES
The patient was admitted to the observation unit from the emergency department after undergoing CT without intravenous contrast. While in observation, she had no additional episodes, and her vital signs remained within normal limits.
She underwent MRI and EEG as well as repeat laboratory studies and consultation by a neurologist. CT showed no structural abnormality, MRI results were read as normal, and EEG showed no epileptiform spikes or abnormal slow waves or other abnormality consistent with seizure. The repeat laboratory studies revealed normalization of the prolactin level at 11.3 ng/mL (reference range 2.0–17.4).
The final impression of the neurology consultant was that the patient had had a seizure that was most likely due to recently starting bupropion in combination with the withdrawal of the benzodiazepine, which lowered the seizure threshold. The neurologist also believed that our patient had no findings or symptoms other than the seizure that would suggest benzodiazepine withdrawal syndrome. According to the patient’s social history, it was unlikely that she had the pattern of alcohol consumption that would result in ethanol withdrawal seizure.
Seizures are common. In fact, every year, 180,000 US adults have their first seizure, and 10% of Americans will experience at least 1 seizure during their lifetime. However, only 20% to 25% of seizures are generalized tonic-clonic seizures as in our patient.23
As this patient had an identifiable cause for the seizure, there was no need to initiate anticonvulsant therapy at the time of discharge. She was discharged to home without any anticonvulsant, the bupropion was discontinued, and the lorazepam was not restarted. When contacted by telephone at 1 month and 18 months after discharge, she reported she had not experienced any additional seizures and has not needed antiepileptic medications.
- Seneviratne U. Management of the first seizure: an evidence based approach. Postgrad Med J 2009; 85:667–673.
- Krumholz A, Wiebe S, Gronseth G, et al; Quality Standards Subcommittee of the American Academy of Neurology; American Epilepsy Society. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 67:1996–2007.
- Gram L. Epileptic seizures and syndromes. Lancet 1990; 336:161–163.
- Smith PE, Cossburn MD. Seizures: assessment and management in the emergency unit. Clin Med (Lond) 2004; 4:118–122.
- Benbadis S. The differential diagnosis of epilepsy: a critical review. Epilepsy Behav 2009; 15:15–21.
- Lusic I, Pintaric I, Hozo I, Boic L, Capkun V. Serum prolactin levels after seizure and syncopal attacks. Seizure 1999; 8:218–222.
- Chen DK, So YT, Fisher RS; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Use of serum prolactin in diagnosing epileptic seizures: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2005; 65:668–675.
- Ben-Menachem E. Is prolactin a clinically useful measure of epilepsy? Epilepsy Curr 2006; 6:78–79.
- Bakes KM, Faragher J, Markovchick VJ, Donahoe K, Haukoos JS. The Denver Seizure Score: anion gap metabolic acidosis predicts generalized seizure. Am J Emerg Med 2011; 29:1097–1102.
- Jefferson JW, Pradok JF, Muir KT. Bupropion for major depressive disorder: pharmacokinetic and formulation considerations. Clin Ther 2005; 27:1685–1695.
- Stall N, Godwin J, Juurlink D. Bupropion abuse and overdose. CMAJ 2014; 186:1015.
- Fialip J, Aumaitre O, Eschalier A, Maradeix B, Dordain G, Lavarenne J. Benzodiazepine withdrawal seizures: analysis of 48 case reports. Clin Neuropharmacol 1987; 10:538–544.
- Lader M, Tylee A, Donoghue J. Withdrawing benzodiazepines in primary care. CNS Drugs 2009; 23:19–34.
- Chance JF. Emergency department treatment of alcohol withdrawal seizures with phenytoin. Ann Emerg Med 1991; 20:520–522.
- ACEP Clinical Policies Committee; Clinical Policies Subcommittee on Seizures. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with seizures. Ann Emerg Med 2004; 43:605–625.
- Sperling MR, Ko J. Seizures and brain tumors. Semin Oncol 2006; 33:333–341.
- Musicco M, Beghi E, Solari A, Viani F. Treatment of first tonic-clonic seizure does not improve the prognosis of epilepsy. First Seizure Trial Group (FIRST Group). Neurology 1997; 49:991–998.
- Edlow JA, Panagos PD, Godwin SA, Thomas TL, Decker WW; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with acute headache. Ann Emerg Med 2008; 52:407–436.
- Kaniecki R. Headache assessment and management. JAMA 2003; 289:1430–1433.
- Harden CL, Huff JS, Schwartz TH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Reassessment: neuroimaging in the emergency patient presenting with seizure (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2007; 69:1772–1780.
- Bergey GK. Management of a first seizure. Continuum (Minneap Minn) 2016; 22:38–50.
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014; 55:475–482.
- Ko DY. Generalized tonic-clonic seizures. Medscape. http://emedicine.medscape.com/article/1184608-overview. Accessed December 5, 2017.
- Seneviratne U. Management of the first seizure: an evidence based approach. Postgrad Med J 2009; 85:667–673.
- Krumholz A, Wiebe S, Gronseth G, et al; Quality Standards Subcommittee of the American Academy of Neurology; American Epilepsy Society. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 67:1996–2007.
- Gram L. Epileptic seizures and syndromes. Lancet 1990; 336:161–163.
- Smith PE, Cossburn MD. Seizures: assessment and management in the emergency unit. Clin Med (Lond) 2004; 4:118–122.
- Benbadis S. The differential diagnosis of epilepsy: a critical review. Epilepsy Behav 2009; 15:15–21.
- Lusic I, Pintaric I, Hozo I, Boic L, Capkun V. Serum prolactin levels after seizure and syncopal attacks. Seizure 1999; 8:218–222.
- Chen DK, So YT, Fisher RS; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Use of serum prolactin in diagnosing epileptic seizures: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2005; 65:668–675.
- Ben-Menachem E. Is prolactin a clinically useful measure of epilepsy? Epilepsy Curr 2006; 6:78–79.
- Bakes KM, Faragher J, Markovchick VJ, Donahoe K, Haukoos JS. The Denver Seizure Score: anion gap metabolic acidosis predicts generalized seizure. Am J Emerg Med 2011; 29:1097–1102.
- Jefferson JW, Pradok JF, Muir KT. Bupropion for major depressive disorder: pharmacokinetic and formulation considerations. Clin Ther 2005; 27:1685–1695.
- Stall N, Godwin J, Juurlink D. Bupropion abuse and overdose. CMAJ 2014; 186:1015.
- Fialip J, Aumaitre O, Eschalier A, Maradeix B, Dordain G, Lavarenne J. Benzodiazepine withdrawal seizures: analysis of 48 case reports. Clin Neuropharmacol 1987; 10:538–544.
- Lader M, Tylee A, Donoghue J. Withdrawing benzodiazepines in primary care. CNS Drugs 2009; 23:19–34.
- Chance JF. Emergency department treatment of alcohol withdrawal seizures with phenytoin. Ann Emerg Med 1991; 20:520–522.
- ACEP Clinical Policies Committee; Clinical Policies Subcommittee on Seizures. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with seizures. Ann Emerg Med 2004; 43:605–625.
- Sperling MR, Ko J. Seizures and brain tumors. Semin Oncol 2006; 33:333–341.
- Musicco M, Beghi E, Solari A, Viani F. Treatment of first tonic-clonic seizure does not improve the prognosis of epilepsy. First Seizure Trial Group (FIRST Group). Neurology 1997; 49:991–998.
- Edlow JA, Panagos PD, Godwin SA, Thomas TL, Decker WW; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of adult patients presenting to the emergency department with acute headache. Ann Emerg Med 2008; 52:407–436.
- Kaniecki R. Headache assessment and management. JAMA 2003; 289:1430–1433.
- Harden CL, Huff JS, Schwartz TH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Reassessment: neuroimaging in the emergency patient presenting with seizure (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2007; 69:1772–1780.
- Bergey GK. Management of a first seizure. Continuum (Minneap Minn) 2016; 22:38–50.
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014; 55:475–482.
- Ko DY. Generalized tonic-clonic seizures. Medscape. http://emedicine.medscape.com/article/1184608-overview. Accessed December 5, 2017.